
エネルギー代謝の極性代謝物を分析するための方法
本発明は、極性代謝物の分析に関するものであり、また極性代謝物を分析するための方法であって、生物学的試料に含まれる細胞の即時破壊を可能にする条件下で相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、その抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および分離した極性代謝物を分析することを含む上記方法を提供するものである。さらには、細胞性物質を含む生物学的試料をクエンチするための方法を意図したものでもある。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、極性代謝物の分析に関するものであり、また極性代謝物を分析するための方法であって、生物学的試料に含まれる細胞の即時破壊を可能にする条件下で相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、その抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および分離した極性代謝物を分析することを含む上記方法を提供するものである。さらには、細胞性物質を含む生物学的試料をクエンチするための方法を意図したものである。
【背景技術】
【0002】
カルボキシル化またはリン酸化された代謝物などの極性代謝物は、細胞のメタボロームのうち最高で90%を占める。従って、メタボロミクス、すなわち、メタボロームの系統立った比較による分析は、極性代謝物の分析と大いに関係がある。
【0003】
メタボロミクスに関して現在利用できる分析技術は、本質的には、液体もしくはガスクロマトグラフィーをベースとする質量分析法、または液体もしくはガスクロマトグラフィーをベースとするNMR分光法のいずれかに基づくものである。
【0004】
メタボロミクスにガスクロマトグラフィーを適用する際の欠点は、代謝物を、その分子を破壊することなく気相に移行させなければならないということである。この変換を改善するために誘導体化を利用する。しかし、実際の代謝物と殆ど区別できない誘導体化人工物を生じることが時々あるため、誘導体化にもある程度の欠点がある。
【0005】
液体クロマトグラフィーは気相への変換も誘導体化も必要としない。しかし、極性代謝物の分離という点での有効性は低い。さらに、生物学的試料中にタンパク質、ペプチドおよび無機塩が過剰量で存在すると、大抵の場合、極性代謝物に対する分離能力が著しく低下する。
【0006】
従って、ガスクロマトグラフィーの特徴である精密な分離と、液体クロマトグラフィーに適用しうる穏やかな条件とを組み合わせることが望ましい。とは言え、前述の欠点は避けなければならない。
【発明の概要】
【発明が解決しようとする課題】
【0007】
こうした理由から、代謝物の効率的な抽出および分析のための方法は、まだ得られていないものの強く必要とされている。
【課題を解決するための手段】
【0008】
本発明は、極性代謝物を分析するための方法であって、以下:
i)生物学的試料に含まれる細胞の即時破壊を可能にする条件下で相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、
ii)工程i)で得た抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および
iii)工程ii)で得た、分離した極性代謝物を分析すること
を含む上記方法に関する。
【図面の簡単な説明】
【0009】
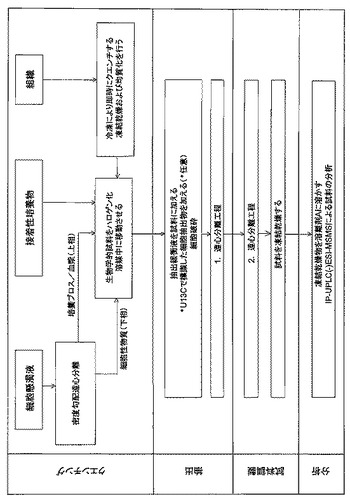
【図1】図1は、生物学的試料からの極性代謝物を分析するための方法のワークフローを示したものである。
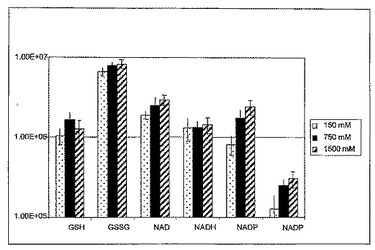
【図2】図2は、使用した酢酸アンモニウム緩衝液の濃度を等張条件から1500 mMに高めた際の、ADPおよびATPの抽出収率における2倍の増大、ならびに補酵素Aおよびアセチル補酵素Aの抽出収率における5〜12倍の増大をそれぞれ示したものであり、ニコチンアミドジヌクレオチドについては最大で3倍の増大が示されている。
【図3A】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。上部最初のクロマトグラム(3A)は、オーバービューXIC(-MRMのXIC(80ペア):87.0/43.0 amu 予想RT:4.9 ID:肝臓26の試料21(10uL-C4)からのPYR neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
【図3B】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。2番目のクロマトグラム(3B)は、レドックス(Redox)およびエネルギー(-MRMのXIC(80ペア):305.8/143.0 amu 予想RT:4.7 ID:肝臓26の試料21(10uL-C4)からのGSH neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
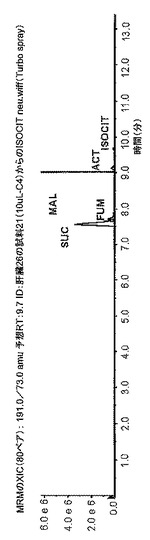
【図3C】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。3番目のクロマトグラム(3C)は、TCA(クレブス回路)(-MRMのXIC(80ペア):191.0/73.0 amu 予想RT:9.7 ID:肝臓26の試料21(10uL-C4)からのISOCIT neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
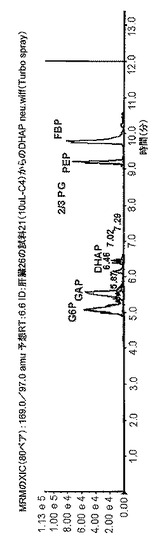
【図3D】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。4番目のクロマトグラム(3D)は、解糖(-MRMのXIC(80ペア):169.0/97.0 amu 予想RT:6.6 ID:肝臓26の試料21(10uL-C4)からのDHAP neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
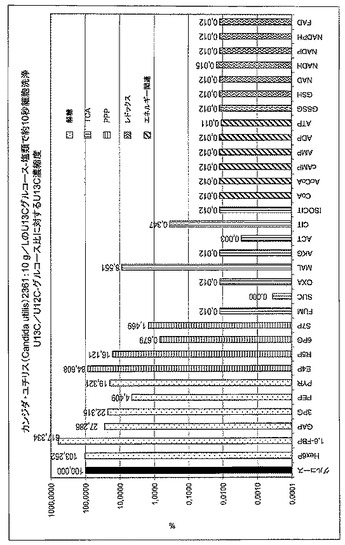
【図4A】図4は、細胞内U13Cグルコース/U12C-グルコースについて見出されている比を基準とした、極性代謝物の抽出物におけるU13Cの相対濃縮度(relative enrichment)を示したものである。(A)従来のクエンチング方法。
【図4B】図4は、細胞内U13Cグルコース/U12C-グルコースについて見出されている比を基準とした、極性代謝物の抽出物におけるU13Cの相対濃縮度(relative enrichment)を示したものである。(B)密度遠心分離によるクエンチング。
【発明を実施するための形態】
【0010】
本明細書中で使用する用語「生物学的試料」は、あらゆる生物学的起源からの生物学的物質を含む試料を指す。該生物学的物質が代謝物を含むことは理解されよう。該生物学的試料に含まれる好適な物質は、細胞または細胞画分などの細胞性物質である。従って、好ましくは、該生物学的試料は、懸濁細胞、接着細胞もしくは組織またはこれらのいずれかの画分を含む。好適な生物学的試料は、体液試料、好ましくは、血液、血漿、血清、リンパ液、汗、唾液、涙、精液、膣液、糞便、尿もしくは脳脊髄液の試料、または例えば、生検により、細胞、組織もしくは臓器から得られる試料を含む臨床サンプルからの生物学的物質を含む。また好ましくは、該生物学的試料はある生物の種々の状態のうちの1つを表しており、例えば、健康なまたは罹患した生物、薬物で処理した生物または未処理の生物などに由来するものでありうる。細菌または真菌などの微生物あるいは植物、植物の一部(例えば、葉、茎、根、もしくは花)または植物の種子を含む生物学的試料もまた包含される。
【0011】
本明細書中で使用する用語「代謝物」は、代謝経路の酵素に対する基質、かかる経路の中間生成物または代謝経路により得られる生成物などの小分子化合物を指す。代謝経路は当分野で周知であるが、種によって異なる場合がある。好ましくは、該経路として少なくとも、クエン酸回路、呼吸鎖、光合成、光呼吸、解糖、糖新生、ヘキソース一リン酸経路、酸化的ペントースリン酸経路、脂肪酸の生成およびβ-酸化、尿素回路、アミノ酸生合成経路、タンパク質分解経路、例えばプロテアソーム分解、アミノ酸分解経路、以下のもの:脂質、ポリケチド(例えば、フラボノイドおよびイソフラボノイドが挙げられる)、イソプレノイド(例えば、テルペン、ステロール、ステロイド、カロテノイド、キサントフィルが挙げられる)、炭水化物、フェニルプロパノイドおよび誘導体、アルカロイド、ベンゼノイド、インドール、インドール-硫黄化合物、ポルフィリン、アントシアン、ホルモン、ビタミン、補欠分子族または電子伝達体などの補因子、リグニン、グルコシノレート、プリン類、ピリミジン類、ヌクレオシド、ヌクレオチドならびにtRNA、マイクロRNA(miRNA)またはmRNAなど関連分子、の生合成または分解が挙げられる。従って、小分子化合物代謝物は、好ましくは、以下の化合物群:アルコール、アルカン、アルケン、アルキン、芳香族化合物、ケトン、アルデヒド、カルボン酸、エステル、アミン、イミン、アミド、シアン化物、アミノ酸、ペプチド、チオール、チオエステル、リン酸エステル、硫酸エステル、チオエーテル、スルホキシド、エーテル、または前述の化合物の組み合わせもしくは誘導体から構成される。代謝物の中でもかかる小分子は正常な細胞機能、臓器機能または動物の成長、発達もしくは健康ならびに植物の成長に必要とされる一次代謝物でありうる。加えて、小分子代謝物は、必須の生態学的機能を有する二次代謝物、例えば、生物をその環境に適応させる代謝物をさらに含む。さらに言うならば、代謝物は該一次および二次代謝物に限定されるものではなく、人工小分子化合物をさらに包含する。該人工小分子化合物は、投与されるかまたは生物に取り込まれるが先に定義した一次または二次代謝物ではない、細胞外に供給される小分子に由来する。例えば、人工小分子化合物は、動物の代謝経路によって薬物から得られる代謝産物でありうる。加えて、代謝物としてさらに、ペプチド、オリゴペプチド、ポリペプチド、オリゴヌクレオチドおよびポリヌクレオチド、例えば、RNAまたはDNAが挙げられる。より好ましくは、代謝物は、50 Da(ダルトン)〜30,000 Da、最も好ましくは30,000 Da未満、20,000 Da未満、15,000 Da未満、10,000 Da未満、8,000 Da未満、7,000 Da未満、6,000 Da未満、5,000 Da未満、4,000 Da未満、3,000 Da未満、2,000 Da未満、1,000 Da未満、500 Da未満、300 Da未満、200 Da未満、100 Da未満の分子量を有する。しかし好ましくは、代謝物は少なくとも50 Daの分子量を有する。最も好ましくは、本発明による代謝物は、50 Daから最大で1,500 Daまでの分子量を有する。該代謝物について、本発明では極性代謝物を想定している、すなわち、小分子化合物は極性であると想定している。
【0012】
生物学的試料からの極性代謝物などの分子の抽出は、複数分子の溶解度の差を利用することに基づいた周知の手順である。対象分子を抽出緩衝液中に抽出する、すなわち溶解させることで、生物学的起源から取り出してもよい。抽出緩衝液の化学的性質を基に、異なる種類の分子を溶解させて生物学的試料物質から分離しうることは理解されよう。本発明の方法は、特に、生物学的試料からの極性代謝物の抽出を想定したものである。抽出を連続工程として、またはバッチ式で行ってもよい。生物学的試料の場合の抽出工程は、好ましくは、該生物学的試料に含まれる生物学的物質を機械的に破壊することにより支援する。かかる支援により、好ましくは40秒未満、30秒未満、20秒未満という短期かつ効率的な抽出時間が可能となることが好ましい。これは、例えば、すりつぶし(pestilling)またはビーズミリング(bead milling)、例えばFastPrep(MP Biomedicals、ドイツ)による細胞破砕などの周知の均質化技術により達成することができる。
【0013】
本発明の方法に適用される前記抽出緩衝液は、相分離剤、すなわち、極性と無極性の分子の相などの異なる相への分子の分離を促進および/または改善する能力がある化合物を含む。
【0014】
好ましくは、本発明の方法に従って使用される前記相分離剤はハロゲン化溶媒を含む。好ましくは、該ハロゲン化溶媒は、1.01 g/cm3(20℃)より大きな密度を有する。より好ましくは、該ハロゲン化溶媒は、ジクロロメタン、クロロホルム、またはパークロルエチレンからなる群より選択され、最も好ましくは、該ハロゲン化溶媒はジクロロメタンである。疎水性が高く、かつ好ましくは水より密度が高い相分離剤、例えばジクロロメタンは、本発明の方法において好ましい。これらの相分離剤は試料中のタンパク質を沈殿させることが可能であり、それゆえに、除去しておかなければ前記代謝物のその後の分析を妨げるであろうタンパク質混入物の、好ましくは遠心分離をベースとする除去を容易にする。さらに、水溶液への有機溶媒の溶解度は高塩混入により低下するため、該相分離剤による相分離は都合が良い。
【0015】
前記ハロゲン化溶媒の他に、前記相分離剤は、好ましくは、低級アルカンのアルコールを含む。好ましくは、該アルコールは、エタノール、メタノールまたはイソプロパノールであり、また最も好ましくはエタノールである。
【0016】
前記のハロゲン化溶媒および低級アルカンのアルコールは、相分離剤中に、好ましくは、3:1〜1:1の比率で、また好ましくは2:1の比率で存在する。しかし、該相分離剤が本質的に前記ハロゲン化溶媒からなる場合も同様に好ましい。
【0017】
前記抽出緩衝液はさらに、揮発性中性アンモニウム塩を含む。本明細書中で揮発性中性アンモニウム塩と称されているのは、凍結乾燥手順により昇華させうるアンモニウム塩である。好ましくは、該揮発性中性アンモニウム塩は、酢酸アンモニウム、ギ酸アンモニウム、または炭酸水素アンモニウムである。最も好ましくは、該中性アンモニウム塩は酢酸アンモニウムである。具体的に述べると、極性代謝物だけを本発明の酢酸アンモニウム含有抽出緩衝液に溶解させることができるため、酢酸アンモニウムは極性代謝物と無極性代謝物との分離を促進する。該代謝物を分析するために行う分析の種類によっては、無極性代謝物から極性のものを分離させることが必要であるかまたは有益であることがあろう。これは特に、タンパク質含有試料を分析する場合に当てはまる。好ましくは、前記抽出緩衝液は、前記揮発性中性アンモニウム塩を少なくとも0.5 Mの濃度で含む。より好ましくは0.75 M〜1.8 Mの濃度、また最も好ましくは1.5 Mの濃度が想定される。該濃度は、好ましくはプラスマイナス15%、10%または5%の範囲内の、わずかな偏差を含みうることは理解されよう。炭酸アンモニウムなどの塩基性揮発性アンモニウム塩は、例えば、CoA-エステルなどのリン酸化代謝物を切断することにより試料の代謝組成を変えてしまう場合があるため、本発明の方法における中性(すなわち非塩基性)揮発性アンモニウム塩の使用はこうした塩基性塩よりも有利である。本発明の方法において使用する揮発性中性アンモニウム塩は、好ましくは、イオン代謝物とタンパク質とを含む複合体に対する競合物質として作用する。ある特定の高濃度の中性アンモニウム塩は、このアンモニウム塩のイオン交換特性によって前記のイオン代謝物-タンパク質複合体を解離させることが可能であり、またその結果、イオン性、すなわち極性の代謝物の収率を高める。
【0018】
前記抽出緩衝液は、好ましくは、溶媒、安定剤などに加えて他の成分、または前記抽出物のさらに別の用途によっては、測定のための標準物質を含む。該抽出緩衝液に使用される好適な標準物質は、本明細書中の他の箇所においてより詳細に開示されている。
【0019】
本発明が、先に明記した抽出緩衝液もまた意図したものであることは理解されよう。
【0020】
工程i)における生物学的試料の抽出は、原則として、前記抽出緩衝液の沸点より低い任意の温度で実施することができる。好ましくは95℃未満の温度が想定されるが、より好ましくは、前記抽出を、酵素活性を無効にするかもしくは低下させるために、または試料中の代謝物を化学的に変化させる(例えば、分解する)化学反応を低減するために、-20℃未満という低温で実施する。好ましくは、抽出を抽出温度-20〜-80℃で実施する。特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0021】
前記の抽出した極性代謝物を、本発明の方法では従来のクロマトグラフ技術により分離する。好適な技術としては、キャピラリー電気泳動(CE)、陽イオン、陰イオン交換クロマトグラフィー、サイズ排除クロマトグラフィー、逆相クロマトグラフィー、順相クロマトグラフィー、親水性液体相互作用クロマトグラフィー、免疫親和性クロマトグラフィー、液体(LC)またはガスクロマトグラフィー(GC)ならびに高圧(HPLC)または超高圧液体クロマトグラフィー(UPLC)が挙げられる。最も好ましくは、分離をUPLCまたはHPLCによって行う。UPLCを適用する場合、径<2.0μmのC18粒子を、好ましくは作動圧力約400〜1000バール、より好ましくは580〜750バールで使用する。UPLCを適用することで、より高い感度での迅速な分離(30分未満)と通常のHPLC手順よりも多くの異性体の分離が可能となる。さらに、無機塩はクロマトグラフィーにより所望の極性代謝物からより良く分離される。さらなる利点は、特にケト炭酸またはポリヒドロキシ酸のクロマトグラフピークにおけるテーリングの低減、およびより良いシグナル 対 ノイズ比による検出限界の改善である。
【0022】
次いで、それらの化学的性質を調べる、および/またはそれらの構造を決定するため、前記の分離した極性代謝物を分析する。かかる分析は、該極性代謝物の物理的または化学的特性を調べることにより実施することが可能であり、その後かかる特性を既知分子の同特性と比較することができる。この比較を基に、既知分子との特性の合致により、代謝物を同定することができる。また、試料中に存在する代謝物の量は、本発明の方法を適用することにより定量的または半定量的に測定することができる。
【0023】
前記極性代謝物を分析するための好適な技術は、質量分析ベースまたはNMRベースの技術である。好ましくは、質量分析、特に、ガスクロマトグラフィー質量分析(GC-MS)、液体クロマトグラフィー質量分析(LC-MS)、直接注入(direct infusion)質量分析またはフーリエ変換イオン-サイクロトロン-共鳴質量分析(FT-ICR-MS)、キャピラリー電気泳動質量分析(CE-MS)、高速液体クロマトグラフィー結合質量分析(HPLC-MS)、四重極質量分析、任意の連続結合(sequentially coupled)質量分析、例えばESI-MS-MSもしくはMS-MS-MS、誘導結合プラズマ質量分析(ICP-MS)、熱分解質量分析(Py-MS)、イオン移動度質量分析または飛行時間型質量分析(TOF)が用いられる。かかる技術は、例えば、Nissen, Journal of Chromatography A, 703, 1995:37-57,米国第4,540,884号または米国第5,397,894号に開示されており、これらの文献の開示内容は参照により本明細書中に組み込まれるものとする。最も好ましくは、UPLCおよびESI-MS/MSを使用する。
【0024】
質量分析技術の代替技術として、または質量分析技術に加えて、以下の技術を代謝物測定に利用してもよい:核磁気共鳴(NMR)、核磁気共鳴画像法(MRI)、フーリエ変換赤外分析(FT-IR)、紫外(UV)分光法、屈折率(RI)、蛍光検出、放射化学的検出、電気化学的検出、光散乱(LS)、分散ラマン分光法またはフレームイオン化検出(FID)。これらの技術は当業者に周知であり、さらなる困難を伴わずに適用することができる。
【0025】
さらに、前記極性代謝物は、特定の化学的または生物学的アッセイにより分析することもできる。該アッセイには、試料中の代謝物の特異的な検出を可能にする手段が含まれるものとする。好ましくは、該手段は、代謝物の化学構造を特異的に認識できるものであるか、または代謝物を、他の化合物と反応するその能力もしくは生物学的読出システムにおける応答(例えば、レポーター遺伝子の誘導)を引き出すその能力に基づいて特異的に同定できるものである。代謝物の化学構造を特異的に認識できる手段は、好ましくは、抗体、または化学構造と特異的に相互作用する他のタンパク質、例えば受容体もしくは酵素である。特異的抗体は、例えば、当業者に周知の方法により代謝物を抗原として用いて得ることができる。本明細書中に記載する抗体としては、ポリクローナルおよびモノクローナル抗体の両方、ならびに抗原またはハプテンと結合できるそのフラグメント、例えばFv、FabおよびF(ab)2フラグメントが挙げられる。また本発明には、所望の抗原特異性を呈する非ヒトドナー抗体のアミノ酸配列をヒトアクセプター抗体の配列とつないだヒト化ハイブリッド抗体も含まれる。さらに、一本鎖抗体も包含される。ドナー配列は通常は少なくともドナーの抗原結合性アミノ酸残基を含むが、ドナー抗体の他の構造上および/または機能上関連のあるアミノ酸残基を同様に含んでいてもよい。かかるハイブリッドは、当分野で周知の幾つかの方法により調製することができる。代謝物を特異的に認識できる適切なタンパク質は、好ましくは、該代謝物の代謝変換に関与する酵素である。該酵素は、代謝物を基質として利用するものであっても、または基質を代謝物に変換するものであってもよい。さらに、前記抗体を基礎として使用することにより、代謝物を特異的に認識するオリゴペプチドを作成してもよい。これらのオリゴペプチドは、例えば、前記代謝物に対する酵素の結合ドメインまたはポケットを含むものとする。適切な抗体および/または酵素ベースのアッセイは、RIA(ラジオイムノアッセイ)、ELISA(固相酵素免疫測定法)、サンドイッチ酵素免疫試験、電気化学発光サンドイッチ免疫測定法(ECLIA)、解離促進ランタニド蛍光イムノアッセイ(DELFIA)または固相免疫試験でありうる。また、前記代謝物を、他の化合物と反応するその能力に基づいて、すなわち特異的化学反応により、同定してもよい。さらに、前記代謝物を、生物学的読出システムにおける応答を引き出すその能力により分析してもよい。この生物学的応答は、試料に含まれる代謝物の存在および/または量を示す読出し情報として検出されるものとする。該生物学的応答は、例えば、遺伝子発現の誘導または細胞もしくは生物の表現型応答でありうる。
【0026】
本発明の方法は、好ましくは、自動化により支援されているものとする。例えば、抽出および/または分離の際の試料の処理または前処理は、ロボットにより自動化することができる。データの処理および比較は、好ましくは、適切なコンピュータプログラムおよびデータベースにより支援されている。先に本明細書中に記載した自動化により、ハイスループットアプローチにおいて本発明の方法を用いることが可能となる。
【0027】
有利なことに、本発明の基礎を成す研究において、本発明の方法により生物学的試料からの極性代謝物の効率的な抽出が可能となることが明らかとなった。特に、低温で抽出を実施すると、化学的および/または生物学的な反応性が全体的に低下することで、例えば、酵素が不活性なままとなることで、生物学的分子の分解が阻止される。先に明記した相分離剤を抽出緩衝液中に使用すると、水相の凝固点の降下、タンパク質の変性および効率的な沈殿ならびに疎水性酵素と親水性(極性)代謝物の別々の相への分離が生じる。さらには、驚いたことに、揮発性中性アンモニウム塩を高濃度で加えると、そのイオン交換特性のおかげで抽出率が有意に上昇することが明らかとなった。さらに、該揮発性中性アンモニウム塩は後の段階で該抽出物から除去することが可能であり、そのため極性代謝物の分析に影響を与えない。
【0028】
本発明の方法の好適な実施形態では、前記抽出物を、工程ii)に先立って以下の工程:
a)水相のスピン遠心分離(spin centrifugation)、
b)工程a)で得た上清の凍結乾燥、および
c)工程b)で得た凍結乾燥物の、イオン対形成剤(ion pair generating agent)を含む分析緩衝液への溶解
により処理する。
【0029】
特に、先に明記したように前記極性代謝物のMSベースの分析が想定される場合には、前述の工程a)〜c)を実施することが好ましい。水相のスピン遠心分離により、該水相の残りの極性代謝物から細胞残屑を効率的に除去する。好ましくは、スピン遠心分離は、公称カットオフ0.2μm〜5μmのメンブランフィルターを使用してスピンろ過により実施する。極性代謝物と抽出緩衝液の極性成分、例えば揮発性中性アンモニウム塩とを含む該スピン遠心分離工程の上清またはろ液を、その後標準的な手順で凍結乾燥する。この処理で揮発性中性アンモニウム塩もまた昇華により除去されるため、該極性代謝物をクロマトグラフ分離または分析するその後の工程に揮発性中性アンモニウム塩が影響を与えることはない。得られた凍結乾燥物は、該代謝物のクロマトグラフ分離および分析のための分析緩衝液に溶かす。
【0030】
本発明の方法に従って使用される分析緩衝液は、イオン対形成剤を含むものとする。好ましくは、この薬剤は、トリブチルアンモニウム陽イオン、トリエチルアンモニウム陽イオン、トリプロピルアンモニウム陽イオンまたはn-ヘキシルアンモニウム陽イオンである。これらの薬剤は遊離アミンとして得ることが可能であり、その後該遊離アミンを質量分析適合性の酸、例えば酢酸またはギ酸によって活性化する。
【0031】
好ましくは、前記分析緩衝液は中性〜弱酸性のpH、より好ましくは、6.0〜6.8、最も好ましくは、6.4〜6.8のpHを有する。
【0032】
以下に、前述の方法の好適な実施形態の特定の態様について記載する。
【0033】
本発明の方法の好適な実施形態では、前記生物学的試料は細胞懸濁液である。
【0034】
本発明の方法の実施に先立って(すなわち、工程i)に先立って)、好ましくは、生物学的試料をクエンチすること、すなわち、酵素活性を即時阻害すること、および、例えば酸化による、化学修飾を抑制することが想定される。しかし、特に細胞内代謝物を分析する場合には、クエンチング手順が細胞内からの細胞内代謝物の「流出(bleeding)」を引き起こすことにより代謝組成に影響を与えないことが望ましい。特に、懸濁細胞の場合、クエンチング手順は非常に重要である。
【0035】
従って、本発明はまた、細胞性物質を含む生物学的試料をクエンチするための方法であって、以下の工程:
a)プロパンジオールまたはエタンジオールを含む予冷したクエンチング溶液からなる下相、不活性相分離剤からなる疎水性中間相、および生物学的試料からなる上相を含む遠心分離バイアルの提供、ただし該下相は該中間相よりも密度が高く、かつ該中間相は該上相よりも密度が高いものとする、ならびに
b)生物学的試料に含まれる細胞性物質を前記上相から前記下相へ移動させるための、前記バイアルの遠心分離
を含む上記方法にも関する。
【0036】
好ましくは、前記プロパンジオールは、プロパン1,2ジオールであるかまたはこれを含む。また好ましくは、前記不活性相分離剤は1.01<rho<1.04(20℃)という密度を有する疎水性の流体であり、より好ましくはシリコーン油であるかまたはこれを含む。
【0037】
遠心分離の結果、懸濁細胞は前記上相(例えば、培養培地または緩衝液)から、予冷したクエンチング溶液が存在する前記下相へと即時移動する。前記疎水性中間相は上相または下相のいずれか一方とは非混和性であるため、両相を分離した状態に保ち、拡散現象を最小限に抑え、そして上相と下相を混じり合った状態にする。
【0038】
好ましくは、前記下相は、-20〜-80℃の温度を有する。さらに、特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、より好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0039】
また、前述の方法のさらに別の好適な実施形態では、前記の上部水相を遠心分離後に前記疎水性中間相から除去し、かつ同位体で標識した標準物質を前記下相に加える。この同位体で標識した標準物質は、本発明の方法を伴う個々の実験の回収率測定および比較のために使用される。好ましくは、同位体で標識した標準物質は、同位体で標識した基質および/または同位体で標識したアンモニウム塩の存在下で合成培地上で増殖させた、好ましくは微生物細胞の細胞抽出物、より好ましくは酵母または細菌培養物からの細胞抽出物である。好ましくは前記基質は13Cで標識されている。好ましくは、前記の同位体で標識した基質は均一に標識したU13C-グルコースまたはU13C-グルタメートであり、一方のアンモニウム源は15N塩化アンモニウムまたは15N硫酸アンモニウムである。前記抽出物は、生物学的に重要な種々のクラスの分子がその中に集められているが、それらは前記生物学的試料に由来する非標識形態と区別しうる同位体特性を備えているため特に有利である。従って、分子の様々なクラスを考慮に入れた標準物質の高価かつ煩雑な作成を、低コストで効率的に回避することができる。前記水相と前記クエンチング溶液とを分離した状態に保つために使用した添加疎水性中間相は、工程i)に従って取り除く。
【0040】
前述のクエンチするための方法は、好ましくは、特に懸濁細胞を生物学的試料として調べる際に、先に記載した極性代謝物を分析するための方法に適用しうることが理解されよう。従って、細胞懸濁液を、極性代謝物を分析するための本発明の方法の工程i)に先立って前述の方法に従いクエンチする。
【0041】
本発明の方法の別の好適な実施形態では、前記生物学的試料は膜上で増殖させた接着細胞の培養物である。
【0042】
本明細書中で使用する用語「膜(membrane)」は、異なるバイアル間で移動させることが可能であり、かつ細胞がその上に接着して増殖しうる任意の固体支持体を指す。適切な膜は当分野で周知であり、その膜としてトラックエッジ膜(track edge membranes)および接着細胞を培養するためのあらゆる種類の親水性または疎水性支持体が挙げられる。より好ましくは、本発明の方法に従って使用される膜はトラックエッジ膜である。有利なことに、該膜は抽出処理の際に破壊されるため、効率的なクエンチングを妨げかねない解体(scrapping)、退屈な消化、または他の手順を必要とせずに接着細胞が放出される。
【0043】
好ましくは、前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、先に定義した-20〜-80℃の温度を有する抽出緩衝液に該膜を即時移動させることによりクエンチする。該抽出緩衝液の特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0044】
また好ましくは、前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、液体窒素中に該膜を即時移動させることによりクエンチする。かかる移動により、約-196℃の温度が即時適用される。該移動は、好ましくは、容器内に該膜を移動させて液体窒素中で該膜を冷凍し、その結果として約-196℃の温度を適用することにより達成することができる。あるいは、工程i)に先立って接着細胞の培養物を含む該膜を洗浄用緩衝液ですすぐことにより、培地由来の細胞外成分を該膜から取り除く。その後、該膜を先に記載した通りにクエンチする。
【0045】
本発明の方法のさらに別の好適な実施形態では、前記生物学的試料は組織試料である。好ましくは、該組織も同様に、工程i)に先立って該組織を-20〜-80℃の温度まで即時冷凍することによりクエンチする。クエンチするための特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。また好ましくは、工程i)に先立ってのクエンチングは、液体窒素中で該組織を即時冷凍することにより、例えば、約-196℃の温度を適用することにより達成することができる。
【0046】
上述した全ての参考文献は、その全開示内容ならびに上記説明箇所において明確に言及したその具体的な開示内容に関して参照により本明細書に含まれるものとする。
【0047】
実施例
【実施例1】
【0048】
実施例1:クエンチングおよび抽出
a)細胞懸濁液
350μLのプロパン1,2-ジオールを、5個のスチールビーズを予め充填した多数の抽出バイアルに加え、-80℃まで冷却した。その後、350μLのシリコン油を各バイアルに加えてから、次回使用時まで-80℃で保存した。1ミリリットルという典型的な容量の水性細胞懸濁液を油層の上に注意深く加え、この試料を即時15000×gで2分間、0℃にて遠心分離した。続いて、該試料を即時ドライアイス上に置き、上相を除去した。その後、1.5 M酢酸アンモニウム水溶液(4℃)、同位体で標識した細胞抽出物(4℃)、およびジクロロメタン(-80℃)を含む抽出緩衝液を加えた。細胞破壊、タンパク質変性、および代謝物抽出を、極低温条件下でビーズミリング処理を介して20秒以内に1つの工程で達成した(FastPrep24装置、MP Biomedicals Inc.を使用)。相分離は前述の条件下で遠心分離により行った。あらゆる細胞残屑を完全に除去するため、上部極性相を、前述の条件を用いたスピンろ過により、0.2μmのろ材に対してろ過した。得られたろ液のアリコートを水で希釈し、-80℃で冷凍し、その後凍結乾燥した。
【0049】
b)接着細胞培養物
メンブレンを底に張ったインサートを備えた培養皿を使用して接着細胞系を培養した(例えば、トラックエッジ膜、Nunc Inc.)。細胞をサンプリングするために、該膜を切り取り、5個のスチールビーズとジクロロメタンおよびエタノールの2:1混合物(-80℃)とを予め充填した抽出バイアルに移した。抽出のために、1.5 M酢酸アンモニウム水溶液(4℃)および同位体で標識した細胞抽出物(4℃)を加えた。a)に記載したものと同じビーズミリング手順を用いて、細胞と膜を完全に分解した。a)に記載した遠心分離条件を適用してタンパク質、細胞残屑および膜残留物を除去し、試料を上部極性相と下部無極性相とに分離した。上相を集めてから、a)で記載した通りにさらに処理した。
【0050】
c)組織試料
組織をサンプリングするために、該組織を液体窒素の種々の温度で即時冷凍し、その後凍結乾燥した。続いて、3〜5 mgの乾燥組織をジクロロメタンおよびエタノールの2:1混合物(-80℃)で覆った。最後に、1.5 M酢酸アンモニウム水溶液(4℃)および同位体で標識した細胞抽出物(4℃)を加えた。この試料を、b)に記載した通りにさらに処理した。
【実施例2】
【0051】
実施例2:同位体で標識した細胞抽出物の作成
カンジダ・ユチリス(Candida utilis)(DSMZ sp. 2361として寄託されている)の酵母培養物を、振とうフラスコ内で、アミノ酸を含まないYNB最少培地(Sigma)中のU13C-D-グルコース(10 g/L)上で増殖させた。好気条件を維持するため、該フラスコをオービタルシェーカーを使用して180 rpm、28℃で揺り動かした。細胞の採取は、200 mLの酵母培養物を2分間、1000×gかつ4℃で遠心分離(Falconチューブ)することにより実施した。続いて該細胞を、0.15 M酢酸アンモニウムおよび10 g/L U13C-D-グルコースからなる洗浄用緩衝液と連続した遠心分離を利用して2回洗浄した。得られた細胞ペレットを、7.5 mLの2:1(v/v)ジクロロメタン/エタノール溶液(-80℃)を使用してクエンチした。抽出は、このクエンチ済み試料に1.5 M酢酸アンモニウム水溶液2.5 mLを加えることにより、a)に記載した通りに極低温条件下で実施した。抽出後、試料をa)に記載した条件下で遠心分離してから、U13C-標識代謝物を含む上相を集めて次回使用時まで-80℃で保存した。
【実施例3】
【0052】
実施例3:クロマトグラフ分離
リン酸化および/またはカルボキシル化極性代謝物のクロマトグラフ分離のために、超高圧イオン対形成液体クロマトグラフィーIP-UPLCを適用した。溶媒A(脱イオン水)と溶媒B(50%アセトニトリル、50%水(v/v))の間のクロマトグラフ勾配処理を行ったが、一方で一定のカラム流量0.4 mL/分およびカラムオーブン温度45℃を維持した。揮発性添加剤としてトリブチルアミンを両溶離剤に加え、氷酢酸を使用してpH値をpH値6.2に調整することにより、イオン対を形成する(ion pairing)トリブチルアンモニウム陽イオンを生成させた。前記の凍結乾燥試料を低用量の溶離剤Aに溶かした後、前記抽出物1〜20μLを注入した。
【実施例4】
【0053】
実施例4:質量分析
負モードのエレクトロスプレー・タンデム質量分析(-ESI-MSMS)を利用して、UPLCにより分離した極性代謝物を評価した。タンデムMSMSはいわゆる計画(scheduled)または選択(selected)多重反応モニタリングモード(sMRM)で操作したが、単位分解能でユニークな質量調整を規定した。個々の代謝物の同位体標識形態および非標識形態は、種々の質量トレース(mass traces)により区別した。
【実施例5】
【0054】
実施例5:高濃度で適用した揮発性抽出緩衝液による抽出収率の上昇
実施例5は、細胞性組織を抽出する際に揮発性酢酸アンモニウムを高濃度で使用したことによる、数種の代謝物の抽出収率の上昇を例示するものである。加えた揮発性緩衝液はその後の凍結乾燥工程中に除去されたが、揮発性が低いリン酸化代謝物は保持された。従って、細胞性組織を高塩濃度緩衝液にさらすと、従来の条件下よりもかなり高い程度まで多数のタンパク質結合代謝物が放出される。この知見は、試料中に少ししか存在しない代謝物にとって特に重要である。さらに、ニコチンアミドの抽出収率の上昇はNADPHの酸化型と還元型の比率に強く影響を与え、このことは細胞または試料における還元当量の入手性を示している(図2)。
【実施例6】
【0055】
実施例6:典型的なクロマトグラム
実施例6は、凍結乾燥組織の典型的なクロマトグラムを示す。5 mgの乾燥試料は、極低温条件下での抽出前にU13C-標識酵母抽出物を用いて補正(amended)した。図3は、計画的多重反応モニタリングを利用して負モードのESI-MSMSにより測定した70の質量トレース(標識/非標識)のクロマトグラムを示したものである。試料または標識酵母における天然存在度に応じて、質量存在度(mass abundance)には3桁の幅が生じた。オーバービューモード(XIC)の他に、3つの包括的な質量エクストラクト(comprehensive mass extracts)についても図示したが、これらはそれぞれエネルギー代謝、クレブス回路(TCA)、および解糖を表している。
【実施例7】
【0056】
実施例7:密度勾配遠心分離を介したクエンチングによる酵素活性の改善された阻害
カンジダ・ユチリスsp.(Candida utilis sp.)2361の酵母培養物を、10 g L-1の非標識D-グルコースを使用してYNB培地(Sigma)で増殖させてから、光学密度4.0(600 nm、1 cm光路長)で回収した。クエンチング法によって生じるバイアスを評価するため、該培養物を、
(A)低温溶媒によるクエンチング前に、0.2 μmフィルターに素早く通し、10 g L-1のU13C-標識D-グルコースを含有する150 mMの酢酸アンモニウム緩衝液で洗浄(最大10秒)し、そして
(B)密度勾配遠心分離法を利用して即時クエンチしたが、プロパン1,2-ジオール相は同量のU13C-D-グルコースで補正(amended)した。
【0057】
標準的なクエンチング法(例えば、ろ過および迅速な細胞洗浄)を用いたところ、解糖中間体、2種のクレブス回路の酸、ならびにペントースリン酸経路の代謝物は有意な13C-濃縮を呈した。反対に、酵母細胞を新しい密度勾配法によりクエンチすると、解糖中間体および他の中間体における13Cの濃縮はみられず、そのことは、解糖酵素の完全な不活性化を本質的に証明している(図4)。
【技術分野】
【0001】
本発明は、極性代謝物の分析に関するものであり、また極性代謝物を分析するための方法であって、生物学的試料に含まれる細胞の即時破壊を可能にする条件下で相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、その抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および分離した極性代謝物を分析することを含む上記方法を提供するものである。さらには、細胞性物質を含む生物学的試料をクエンチするための方法を意図したものである。
【背景技術】
【0002】
カルボキシル化またはリン酸化された代謝物などの極性代謝物は、細胞のメタボロームのうち最高で90%を占める。従って、メタボロミクス、すなわち、メタボロームの系統立った比較による分析は、極性代謝物の分析と大いに関係がある。
【0003】
メタボロミクスに関して現在利用できる分析技術は、本質的には、液体もしくはガスクロマトグラフィーをベースとする質量分析法、または液体もしくはガスクロマトグラフィーをベースとするNMR分光法のいずれかに基づくものである。
【0004】
メタボロミクスにガスクロマトグラフィーを適用する際の欠点は、代謝物を、その分子を破壊することなく気相に移行させなければならないということである。この変換を改善するために誘導体化を利用する。しかし、実際の代謝物と殆ど区別できない誘導体化人工物を生じることが時々あるため、誘導体化にもある程度の欠点がある。
【0005】
液体クロマトグラフィーは気相への変換も誘導体化も必要としない。しかし、極性代謝物の分離という点での有効性は低い。さらに、生物学的試料中にタンパク質、ペプチドおよび無機塩が過剰量で存在すると、大抵の場合、極性代謝物に対する分離能力が著しく低下する。
【0006】
従って、ガスクロマトグラフィーの特徴である精密な分離と、液体クロマトグラフィーに適用しうる穏やかな条件とを組み合わせることが望ましい。とは言え、前述の欠点は避けなければならない。
【発明の概要】
【発明が解決しようとする課題】
【0007】
こうした理由から、代謝物の効率的な抽出および分析のための方法は、まだ得られていないものの強く必要とされている。
【課題を解決するための手段】
【0008】
本発明は、極性代謝物を分析するための方法であって、以下:
i)生物学的試料に含まれる細胞の即時破壊を可能にする条件下で相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、
ii)工程i)で得た抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および
iii)工程ii)で得た、分離した極性代謝物を分析すること
を含む上記方法に関する。
【図面の簡単な説明】
【0009】
【図1】図1は、生物学的試料からの極性代謝物を分析するための方法のワークフローを示したものである。
【図2】図2は、使用した酢酸アンモニウム緩衝液の濃度を等張条件から1500 mMに高めた際の、ADPおよびATPの抽出収率における2倍の増大、ならびに補酵素Aおよびアセチル補酵素Aの抽出収率における5〜12倍の増大をそれぞれ示したものであり、ニコチンアミドジヌクレオチドについては最大で3倍の増大が示されている。
【図3A】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。上部最初のクロマトグラム(3A)は、オーバービューXIC(-MRMのXIC(80ペア):87.0/43.0 amu 予想RT:4.9 ID:肝臓26の試料21(10uL-C4)からのPYR neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
【図3B】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。2番目のクロマトグラム(3B)は、レドックス(Redox)およびエネルギー(-MRMのXIC(80ペア):305.8/143.0 amu 予想RT:4.7 ID:肝臓26の試料21(10uL-C4)からのGSH neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
【図3C】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。3番目のクロマトグラム(3C)は、TCA(クレブス回路)(-MRMのXIC(80ペア):191.0/73.0 amu 予想RT:9.7 ID:肝臓26の試料21(10uL-C4)からのISOCIT neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
【図3D】図3は、組織試料(ヒト膵臓)、5 mgの重さの試料の凍結乾燥抽出物の典型的なクロマトグラムを示したものである。4番目のクロマトグラム(3D)は、解糖(-MRMのXIC(80ペア):169.0/97.0 amu 予想RT:6.6 ID:肝臓26の試料21(10uL-C4)からのDHAP neu wiff(Turbo spray))を示したものである。強度(cps)をy軸上に表示し、分単位の保持時間(分)をX軸上に表示した。
【図4A】図4は、細胞内U13Cグルコース/U12C-グルコースについて見出されている比を基準とした、極性代謝物の抽出物におけるU13Cの相対濃縮度(relative enrichment)を示したものである。(A)従来のクエンチング方法。
【図4B】図4は、細胞内U13Cグルコース/U12C-グルコースについて見出されている比を基準とした、極性代謝物の抽出物におけるU13Cの相対濃縮度(relative enrichment)を示したものである。(B)密度遠心分離によるクエンチング。
【発明を実施するための形態】
【0010】
本明細書中で使用する用語「生物学的試料」は、あらゆる生物学的起源からの生物学的物質を含む試料を指す。該生物学的物質が代謝物を含むことは理解されよう。該生物学的試料に含まれる好適な物質は、細胞または細胞画分などの細胞性物質である。従って、好ましくは、該生物学的試料は、懸濁細胞、接着細胞もしくは組織またはこれらのいずれかの画分を含む。好適な生物学的試料は、体液試料、好ましくは、血液、血漿、血清、リンパ液、汗、唾液、涙、精液、膣液、糞便、尿もしくは脳脊髄液の試料、または例えば、生検により、細胞、組織もしくは臓器から得られる試料を含む臨床サンプルからの生物学的物質を含む。また好ましくは、該生物学的試料はある生物の種々の状態のうちの1つを表しており、例えば、健康なまたは罹患した生物、薬物で処理した生物または未処理の生物などに由来するものでありうる。細菌または真菌などの微生物あるいは植物、植物の一部(例えば、葉、茎、根、もしくは花)または植物の種子を含む生物学的試料もまた包含される。
【0011】
本明細書中で使用する用語「代謝物」は、代謝経路の酵素に対する基質、かかる経路の中間生成物または代謝経路により得られる生成物などの小分子化合物を指す。代謝経路は当分野で周知であるが、種によって異なる場合がある。好ましくは、該経路として少なくとも、クエン酸回路、呼吸鎖、光合成、光呼吸、解糖、糖新生、ヘキソース一リン酸経路、酸化的ペントースリン酸経路、脂肪酸の生成およびβ-酸化、尿素回路、アミノ酸生合成経路、タンパク質分解経路、例えばプロテアソーム分解、アミノ酸分解経路、以下のもの:脂質、ポリケチド(例えば、フラボノイドおよびイソフラボノイドが挙げられる)、イソプレノイド(例えば、テルペン、ステロール、ステロイド、カロテノイド、キサントフィルが挙げられる)、炭水化物、フェニルプロパノイドおよび誘導体、アルカロイド、ベンゼノイド、インドール、インドール-硫黄化合物、ポルフィリン、アントシアン、ホルモン、ビタミン、補欠分子族または電子伝達体などの補因子、リグニン、グルコシノレート、プリン類、ピリミジン類、ヌクレオシド、ヌクレオチドならびにtRNA、マイクロRNA(miRNA)またはmRNAなど関連分子、の生合成または分解が挙げられる。従って、小分子化合物代謝物は、好ましくは、以下の化合物群:アルコール、アルカン、アルケン、アルキン、芳香族化合物、ケトン、アルデヒド、カルボン酸、エステル、アミン、イミン、アミド、シアン化物、アミノ酸、ペプチド、チオール、チオエステル、リン酸エステル、硫酸エステル、チオエーテル、スルホキシド、エーテル、または前述の化合物の組み合わせもしくは誘導体から構成される。代謝物の中でもかかる小分子は正常な細胞機能、臓器機能または動物の成長、発達もしくは健康ならびに植物の成長に必要とされる一次代謝物でありうる。加えて、小分子代謝物は、必須の生態学的機能を有する二次代謝物、例えば、生物をその環境に適応させる代謝物をさらに含む。さらに言うならば、代謝物は該一次および二次代謝物に限定されるものではなく、人工小分子化合物をさらに包含する。該人工小分子化合物は、投与されるかまたは生物に取り込まれるが先に定義した一次または二次代謝物ではない、細胞外に供給される小分子に由来する。例えば、人工小分子化合物は、動物の代謝経路によって薬物から得られる代謝産物でありうる。加えて、代謝物としてさらに、ペプチド、オリゴペプチド、ポリペプチド、オリゴヌクレオチドおよびポリヌクレオチド、例えば、RNAまたはDNAが挙げられる。より好ましくは、代謝物は、50 Da(ダルトン)〜30,000 Da、最も好ましくは30,000 Da未満、20,000 Da未満、15,000 Da未満、10,000 Da未満、8,000 Da未満、7,000 Da未満、6,000 Da未満、5,000 Da未満、4,000 Da未満、3,000 Da未満、2,000 Da未満、1,000 Da未満、500 Da未満、300 Da未満、200 Da未満、100 Da未満の分子量を有する。しかし好ましくは、代謝物は少なくとも50 Daの分子量を有する。最も好ましくは、本発明による代謝物は、50 Daから最大で1,500 Daまでの分子量を有する。該代謝物について、本発明では極性代謝物を想定している、すなわち、小分子化合物は極性であると想定している。
【0012】
生物学的試料からの極性代謝物などの分子の抽出は、複数分子の溶解度の差を利用することに基づいた周知の手順である。対象分子を抽出緩衝液中に抽出する、すなわち溶解させることで、生物学的起源から取り出してもよい。抽出緩衝液の化学的性質を基に、異なる種類の分子を溶解させて生物学的試料物質から分離しうることは理解されよう。本発明の方法は、特に、生物学的試料からの極性代謝物の抽出を想定したものである。抽出を連続工程として、またはバッチ式で行ってもよい。生物学的試料の場合の抽出工程は、好ましくは、該生物学的試料に含まれる生物学的物質を機械的に破壊することにより支援する。かかる支援により、好ましくは40秒未満、30秒未満、20秒未満という短期かつ効率的な抽出時間が可能となることが好ましい。これは、例えば、すりつぶし(pestilling)またはビーズミリング(bead milling)、例えばFastPrep(MP Biomedicals、ドイツ)による細胞破砕などの周知の均質化技術により達成することができる。
【0013】
本発明の方法に適用される前記抽出緩衝液は、相分離剤、すなわち、極性と無極性の分子の相などの異なる相への分子の分離を促進および/または改善する能力がある化合物を含む。
【0014】
好ましくは、本発明の方法に従って使用される前記相分離剤はハロゲン化溶媒を含む。好ましくは、該ハロゲン化溶媒は、1.01 g/cm3(20℃)より大きな密度を有する。より好ましくは、該ハロゲン化溶媒は、ジクロロメタン、クロロホルム、またはパークロルエチレンからなる群より選択され、最も好ましくは、該ハロゲン化溶媒はジクロロメタンである。疎水性が高く、かつ好ましくは水より密度が高い相分離剤、例えばジクロロメタンは、本発明の方法において好ましい。これらの相分離剤は試料中のタンパク質を沈殿させることが可能であり、それゆえに、除去しておかなければ前記代謝物のその後の分析を妨げるであろうタンパク質混入物の、好ましくは遠心分離をベースとする除去を容易にする。さらに、水溶液への有機溶媒の溶解度は高塩混入により低下するため、該相分離剤による相分離は都合が良い。
【0015】
前記ハロゲン化溶媒の他に、前記相分離剤は、好ましくは、低級アルカンのアルコールを含む。好ましくは、該アルコールは、エタノール、メタノールまたはイソプロパノールであり、また最も好ましくはエタノールである。
【0016】
前記のハロゲン化溶媒および低級アルカンのアルコールは、相分離剤中に、好ましくは、3:1〜1:1の比率で、また好ましくは2:1の比率で存在する。しかし、該相分離剤が本質的に前記ハロゲン化溶媒からなる場合も同様に好ましい。
【0017】
前記抽出緩衝液はさらに、揮発性中性アンモニウム塩を含む。本明細書中で揮発性中性アンモニウム塩と称されているのは、凍結乾燥手順により昇華させうるアンモニウム塩である。好ましくは、該揮発性中性アンモニウム塩は、酢酸アンモニウム、ギ酸アンモニウム、または炭酸水素アンモニウムである。最も好ましくは、該中性アンモニウム塩は酢酸アンモニウムである。具体的に述べると、極性代謝物だけを本発明の酢酸アンモニウム含有抽出緩衝液に溶解させることができるため、酢酸アンモニウムは極性代謝物と無極性代謝物との分離を促進する。該代謝物を分析するために行う分析の種類によっては、無極性代謝物から極性のものを分離させることが必要であるかまたは有益であることがあろう。これは特に、タンパク質含有試料を分析する場合に当てはまる。好ましくは、前記抽出緩衝液は、前記揮発性中性アンモニウム塩を少なくとも0.5 Mの濃度で含む。より好ましくは0.75 M〜1.8 Mの濃度、また最も好ましくは1.5 Mの濃度が想定される。該濃度は、好ましくはプラスマイナス15%、10%または5%の範囲内の、わずかな偏差を含みうることは理解されよう。炭酸アンモニウムなどの塩基性揮発性アンモニウム塩は、例えば、CoA-エステルなどのリン酸化代謝物を切断することにより試料の代謝組成を変えてしまう場合があるため、本発明の方法における中性(すなわち非塩基性)揮発性アンモニウム塩の使用はこうした塩基性塩よりも有利である。本発明の方法において使用する揮発性中性アンモニウム塩は、好ましくは、イオン代謝物とタンパク質とを含む複合体に対する競合物質として作用する。ある特定の高濃度の中性アンモニウム塩は、このアンモニウム塩のイオン交換特性によって前記のイオン代謝物-タンパク質複合体を解離させることが可能であり、またその結果、イオン性、すなわち極性の代謝物の収率を高める。
【0018】
前記抽出緩衝液は、好ましくは、溶媒、安定剤などに加えて他の成分、または前記抽出物のさらに別の用途によっては、測定のための標準物質を含む。該抽出緩衝液に使用される好適な標準物質は、本明細書中の他の箇所においてより詳細に開示されている。
【0019】
本発明が、先に明記した抽出緩衝液もまた意図したものであることは理解されよう。
【0020】
工程i)における生物学的試料の抽出は、原則として、前記抽出緩衝液の沸点より低い任意の温度で実施することができる。好ましくは95℃未満の温度が想定されるが、より好ましくは、前記抽出を、酵素活性を無効にするかもしくは低下させるために、または試料中の代謝物を化学的に変化させる(例えば、分解する)化学反応を低減するために、-20℃未満という低温で実施する。好ましくは、抽出を抽出温度-20〜-80℃で実施する。特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0021】
前記の抽出した極性代謝物を、本発明の方法では従来のクロマトグラフ技術により分離する。好適な技術としては、キャピラリー電気泳動(CE)、陽イオン、陰イオン交換クロマトグラフィー、サイズ排除クロマトグラフィー、逆相クロマトグラフィー、順相クロマトグラフィー、親水性液体相互作用クロマトグラフィー、免疫親和性クロマトグラフィー、液体(LC)またはガスクロマトグラフィー(GC)ならびに高圧(HPLC)または超高圧液体クロマトグラフィー(UPLC)が挙げられる。最も好ましくは、分離をUPLCまたはHPLCによって行う。UPLCを適用する場合、径<2.0μmのC18粒子を、好ましくは作動圧力約400〜1000バール、より好ましくは580〜750バールで使用する。UPLCを適用することで、より高い感度での迅速な分離(30分未満)と通常のHPLC手順よりも多くの異性体の分離が可能となる。さらに、無機塩はクロマトグラフィーにより所望の極性代謝物からより良く分離される。さらなる利点は、特にケト炭酸またはポリヒドロキシ酸のクロマトグラフピークにおけるテーリングの低減、およびより良いシグナル 対 ノイズ比による検出限界の改善である。
【0022】
次いで、それらの化学的性質を調べる、および/またはそれらの構造を決定するため、前記の分離した極性代謝物を分析する。かかる分析は、該極性代謝物の物理的または化学的特性を調べることにより実施することが可能であり、その後かかる特性を既知分子の同特性と比較することができる。この比較を基に、既知分子との特性の合致により、代謝物を同定することができる。また、試料中に存在する代謝物の量は、本発明の方法を適用することにより定量的または半定量的に測定することができる。
【0023】
前記極性代謝物を分析するための好適な技術は、質量分析ベースまたはNMRベースの技術である。好ましくは、質量分析、特に、ガスクロマトグラフィー質量分析(GC-MS)、液体クロマトグラフィー質量分析(LC-MS)、直接注入(direct infusion)質量分析またはフーリエ変換イオン-サイクロトロン-共鳴質量分析(FT-ICR-MS)、キャピラリー電気泳動質量分析(CE-MS)、高速液体クロマトグラフィー結合質量分析(HPLC-MS)、四重極質量分析、任意の連続結合(sequentially coupled)質量分析、例えばESI-MS-MSもしくはMS-MS-MS、誘導結合プラズマ質量分析(ICP-MS)、熱分解質量分析(Py-MS)、イオン移動度質量分析または飛行時間型質量分析(TOF)が用いられる。かかる技術は、例えば、Nissen, Journal of Chromatography A, 703, 1995:37-57,米国第4,540,884号または米国第5,397,894号に開示されており、これらの文献の開示内容は参照により本明細書中に組み込まれるものとする。最も好ましくは、UPLCおよびESI-MS/MSを使用する。
【0024】
質量分析技術の代替技術として、または質量分析技術に加えて、以下の技術を代謝物測定に利用してもよい:核磁気共鳴(NMR)、核磁気共鳴画像法(MRI)、フーリエ変換赤外分析(FT-IR)、紫外(UV)分光法、屈折率(RI)、蛍光検出、放射化学的検出、電気化学的検出、光散乱(LS)、分散ラマン分光法またはフレームイオン化検出(FID)。これらの技術は当業者に周知であり、さらなる困難を伴わずに適用することができる。
【0025】
さらに、前記極性代謝物は、特定の化学的または生物学的アッセイにより分析することもできる。該アッセイには、試料中の代謝物の特異的な検出を可能にする手段が含まれるものとする。好ましくは、該手段は、代謝物の化学構造を特異的に認識できるものであるか、または代謝物を、他の化合物と反応するその能力もしくは生物学的読出システムにおける応答(例えば、レポーター遺伝子の誘導)を引き出すその能力に基づいて特異的に同定できるものである。代謝物の化学構造を特異的に認識できる手段は、好ましくは、抗体、または化学構造と特異的に相互作用する他のタンパク質、例えば受容体もしくは酵素である。特異的抗体は、例えば、当業者に周知の方法により代謝物を抗原として用いて得ることができる。本明細書中に記載する抗体としては、ポリクローナルおよびモノクローナル抗体の両方、ならびに抗原またはハプテンと結合できるそのフラグメント、例えばFv、FabおよびF(ab)2フラグメントが挙げられる。また本発明には、所望の抗原特異性を呈する非ヒトドナー抗体のアミノ酸配列をヒトアクセプター抗体の配列とつないだヒト化ハイブリッド抗体も含まれる。さらに、一本鎖抗体も包含される。ドナー配列は通常は少なくともドナーの抗原結合性アミノ酸残基を含むが、ドナー抗体の他の構造上および/または機能上関連のあるアミノ酸残基を同様に含んでいてもよい。かかるハイブリッドは、当分野で周知の幾つかの方法により調製することができる。代謝物を特異的に認識できる適切なタンパク質は、好ましくは、該代謝物の代謝変換に関与する酵素である。該酵素は、代謝物を基質として利用するものであっても、または基質を代謝物に変換するものであってもよい。さらに、前記抗体を基礎として使用することにより、代謝物を特異的に認識するオリゴペプチドを作成してもよい。これらのオリゴペプチドは、例えば、前記代謝物に対する酵素の結合ドメインまたはポケットを含むものとする。適切な抗体および/または酵素ベースのアッセイは、RIA(ラジオイムノアッセイ)、ELISA(固相酵素免疫測定法)、サンドイッチ酵素免疫試験、電気化学発光サンドイッチ免疫測定法(ECLIA)、解離促進ランタニド蛍光イムノアッセイ(DELFIA)または固相免疫試験でありうる。また、前記代謝物を、他の化合物と反応するその能力に基づいて、すなわち特異的化学反応により、同定してもよい。さらに、前記代謝物を、生物学的読出システムにおける応答を引き出すその能力により分析してもよい。この生物学的応答は、試料に含まれる代謝物の存在および/または量を示す読出し情報として検出されるものとする。該生物学的応答は、例えば、遺伝子発現の誘導または細胞もしくは生物の表現型応答でありうる。
【0026】
本発明の方法は、好ましくは、自動化により支援されているものとする。例えば、抽出および/または分離の際の試料の処理または前処理は、ロボットにより自動化することができる。データの処理および比較は、好ましくは、適切なコンピュータプログラムおよびデータベースにより支援されている。先に本明細書中に記載した自動化により、ハイスループットアプローチにおいて本発明の方法を用いることが可能となる。
【0027】
有利なことに、本発明の基礎を成す研究において、本発明の方法により生物学的試料からの極性代謝物の効率的な抽出が可能となることが明らかとなった。特に、低温で抽出を実施すると、化学的および/または生物学的な反応性が全体的に低下することで、例えば、酵素が不活性なままとなることで、生物学的分子の分解が阻止される。先に明記した相分離剤を抽出緩衝液中に使用すると、水相の凝固点の降下、タンパク質の変性および効率的な沈殿ならびに疎水性酵素と親水性(極性)代謝物の別々の相への分離が生じる。さらには、驚いたことに、揮発性中性アンモニウム塩を高濃度で加えると、そのイオン交換特性のおかげで抽出率が有意に上昇することが明らかとなった。さらに、該揮発性中性アンモニウム塩は後の段階で該抽出物から除去することが可能であり、そのため極性代謝物の分析に影響を与えない。
【0028】
本発明の方法の好適な実施形態では、前記抽出物を、工程ii)に先立って以下の工程:
a)水相のスピン遠心分離(spin centrifugation)、
b)工程a)で得た上清の凍結乾燥、および
c)工程b)で得た凍結乾燥物の、イオン対形成剤(ion pair generating agent)を含む分析緩衝液への溶解
により処理する。
【0029】
特に、先に明記したように前記極性代謝物のMSベースの分析が想定される場合には、前述の工程a)〜c)を実施することが好ましい。水相のスピン遠心分離により、該水相の残りの極性代謝物から細胞残屑を効率的に除去する。好ましくは、スピン遠心分離は、公称カットオフ0.2μm〜5μmのメンブランフィルターを使用してスピンろ過により実施する。極性代謝物と抽出緩衝液の極性成分、例えば揮発性中性アンモニウム塩とを含む該スピン遠心分離工程の上清またはろ液を、その後標準的な手順で凍結乾燥する。この処理で揮発性中性アンモニウム塩もまた昇華により除去されるため、該極性代謝物をクロマトグラフ分離または分析するその後の工程に揮発性中性アンモニウム塩が影響を与えることはない。得られた凍結乾燥物は、該代謝物のクロマトグラフ分離および分析のための分析緩衝液に溶かす。
【0030】
本発明の方法に従って使用される分析緩衝液は、イオン対形成剤を含むものとする。好ましくは、この薬剤は、トリブチルアンモニウム陽イオン、トリエチルアンモニウム陽イオン、トリプロピルアンモニウム陽イオンまたはn-ヘキシルアンモニウム陽イオンである。これらの薬剤は遊離アミンとして得ることが可能であり、その後該遊離アミンを質量分析適合性の酸、例えば酢酸またはギ酸によって活性化する。
【0031】
好ましくは、前記分析緩衝液は中性〜弱酸性のpH、より好ましくは、6.0〜6.8、最も好ましくは、6.4〜6.8のpHを有する。
【0032】
以下に、前述の方法の好適な実施形態の特定の態様について記載する。
【0033】
本発明の方法の好適な実施形態では、前記生物学的試料は細胞懸濁液である。
【0034】
本発明の方法の実施に先立って(すなわち、工程i)に先立って)、好ましくは、生物学的試料をクエンチすること、すなわち、酵素活性を即時阻害すること、および、例えば酸化による、化学修飾を抑制することが想定される。しかし、特に細胞内代謝物を分析する場合には、クエンチング手順が細胞内からの細胞内代謝物の「流出(bleeding)」を引き起こすことにより代謝組成に影響を与えないことが望ましい。特に、懸濁細胞の場合、クエンチング手順は非常に重要である。
【0035】
従って、本発明はまた、細胞性物質を含む生物学的試料をクエンチするための方法であって、以下の工程:
a)プロパンジオールまたはエタンジオールを含む予冷したクエンチング溶液からなる下相、不活性相分離剤からなる疎水性中間相、および生物学的試料からなる上相を含む遠心分離バイアルの提供、ただし該下相は該中間相よりも密度が高く、かつ該中間相は該上相よりも密度が高いものとする、ならびに
b)生物学的試料に含まれる細胞性物質を前記上相から前記下相へ移動させるための、前記バイアルの遠心分離
を含む上記方法にも関する。
【0036】
好ましくは、前記プロパンジオールは、プロパン1,2ジオールであるかまたはこれを含む。また好ましくは、前記不活性相分離剤は1.01<rho<1.04(20℃)という密度を有する疎水性の流体であり、より好ましくはシリコーン油であるかまたはこれを含む。
【0037】
遠心分離の結果、懸濁細胞は前記上相(例えば、培養培地または緩衝液)から、予冷したクエンチング溶液が存在する前記下相へと即時移動する。前記疎水性中間相は上相または下相のいずれか一方とは非混和性であるため、両相を分離した状態に保ち、拡散現象を最小限に抑え、そして上相と下相を混じり合った状態にする。
【0038】
好ましくは、前記下相は、-20〜-80℃の温度を有する。さらに、特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、より好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0039】
また、前述の方法のさらに別の好適な実施形態では、前記の上部水相を遠心分離後に前記疎水性中間相から除去し、かつ同位体で標識した標準物質を前記下相に加える。この同位体で標識した標準物質は、本発明の方法を伴う個々の実験の回収率測定および比較のために使用される。好ましくは、同位体で標識した標準物質は、同位体で標識した基質および/または同位体で標識したアンモニウム塩の存在下で合成培地上で増殖させた、好ましくは微生物細胞の細胞抽出物、より好ましくは酵母または細菌培養物からの細胞抽出物である。好ましくは前記基質は13Cで標識されている。好ましくは、前記の同位体で標識した基質は均一に標識したU13C-グルコースまたはU13C-グルタメートであり、一方のアンモニウム源は15N塩化アンモニウムまたは15N硫酸アンモニウムである。前記抽出物は、生物学的に重要な種々のクラスの分子がその中に集められているが、それらは前記生物学的試料に由来する非標識形態と区別しうる同位体特性を備えているため特に有利である。従って、分子の様々なクラスを考慮に入れた標準物質の高価かつ煩雑な作成を、低コストで効率的に回避することができる。前記水相と前記クエンチング溶液とを分離した状態に保つために使用した添加疎水性中間相は、工程i)に従って取り除く。
【0040】
前述のクエンチするための方法は、好ましくは、特に懸濁細胞を生物学的試料として調べる際に、先に記載した極性代謝物を分析するための方法に適用しうることが理解されよう。従って、細胞懸濁液を、極性代謝物を分析するための本発明の方法の工程i)に先立って前述の方法に従いクエンチする。
【0041】
本発明の方法の別の好適な実施形態では、前記生物学的試料は膜上で増殖させた接着細胞の培養物である。
【0042】
本明細書中で使用する用語「膜(membrane)」は、異なるバイアル間で移動させることが可能であり、かつ細胞がその上に接着して増殖しうる任意の固体支持体を指す。適切な膜は当分野で周知であり、その膜としてトラックエッジ膜(track edge membranes)および接着細胞を培養するためのあらゆる種類の親水性または疎水性支持体が挙げられる。より好ましくは、本発明の方法に従って使用される膜はトラックエッジ膜である。有利なことに、該膜は抽出処理の際に破壊されるため、効率的なクエンチングを妨げかねない解体(scrapping)、退屈な消化、または他の手順を必要とせずに接着細胞が放出される。
【0043】
好ましくは、前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、先に定義した-20〜-80℃の温度を有する抽出緩衝液に該膜を即時移動させることによりクエンチする。該抽出緩衝液の特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。
【0044】
また好ましくは、前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、液体窒素中に該膜を即時移動させることによりクエンチする。かかる移動により、約-196℃の温度が即時適用される。該移動は、好ましくは、容器内に該膜を移動させて液体窒素中で該膜を冷凍し、その結果として約-196℃の温度を適用することにより達成することができる。あるいは、工程i)に先立って接着細胞の培養物を含む該膜を洗浄用緩衝液ですすぐことにより、培地由来の細胞外成分を該膜から取り除く。その後、該膜を先に記載した通りにクエンチする。
【0045】
本発明の方法のさらに別の好適な実施形態では、前記生物学的試料は組織試料である。好ましくは、該組織も同様に、工程i)に先立って該組織を-20〜-80℃の温度まで即時冷凍することによりクエンチする。クエンチするための特に好適な温度は、-25〜-80℃、-30℃〜-80℃、-35℃〜-80℃、-40℃〜-80℃、-45℃〜-80℃、-50℃〜-80℃、-55℃〜-80℃、-60℃〜-80℃、-65℃〜-80℃、-70℃〜-80℃の範囲内であり、またより好ましくは、該温度は-70℃、-71℃、-71℃、-73℃、-74℃、-75℃、-76℃、-77℃、-78℃、-79℃、または-80℃である。また好ましくは、工程i)に先立ってのクエンチングは、液体窒素中で該組織を即時冷凍することにより、例えば、約-196℃の温度を適用することにより達成することができる。
【0046】
上述した全ての参考文献は、その全開示内容ならびに上記説明箇所において明確に言及したその具体的な開示内容に関して参照により本明細書に含まれるものとする。
【0047】
実施例
【実施例1】
【0048】
実施例1:クエンチングおよび抽出
a)細胞懸濁液
350μLのプロパン1,2-ジオールを、5個のスチールビーズを予め充填した多数の抽出バイアルに加え、-80℃まで冷却した。その後、350μLのシリコン油を各バイアルに加えてから、次回使用時まで-80℃で保存した。1ミリリットルという典型的な容量の水性細胞懸濁液を油層の上に注意深く加え、この試料を即時15000×gで2分間、0℃にて遠心分離した。続いて、該試料を即時ドライアイス上に置き、上相を除去した。その後、1.5 M酢酸アンモニウム水溶液(4℃)、同位体で標識した細胞抽出物(4℃)、およびジクロロメタン(-80℃)を含む抽出緩衝液を加えた。細胞破壊、タンパク質変性、および代謝物抽出を、極低温条件下でビーズミリング処理を介して20秒以内に1つの工程で達成した(FastPrep24装置、MP Biomedicals Inc.を使用)。相分離は前述の条件下で遠心分離により行った。あらゆる細胞残屑を完全に除去するため、上部極性相を、前述の条件を用いたスピンろ過により、0.2μmのろ材に対してろ過した。得られたろ液のアリコートを水で希釈し、-80℃で冷凍し、その後凍結乾燥した。
【0049】
b)接着細胞培養物
メンブレンを底に張ったインサートを備えた培養皿を使用して接着細胞系を培養した(例えば、トラックエッジ膜、Nunc Inc.)。細胞をサンプリングするために、該膜を切り取り、5個のスチールビーズとジクロロメタンおよびエタノールの2:1混合物(-80℃)とを予め充填した抽出バイアルに移した。抽出のために、1.5 M酢酸アンモニウム水溶液(4℃)および同位体で標識した細胞抽出物(4℃)を加えた。a)に記載したものと同じビーズミリング手順を用いて、細胞と膜を完全に分解した。a)に記載した遠心分離条件を適用してタンパク質、細胞残屑および膜残留物を除去し、試料を上部極性相と下部無極性相とに分離した。上相を集めてから、a)で記載した通りにさらに処理した。
【0050】
c)組織試料
組織をサンプリングするために、該組織を液体窒素の種々の温度で即時冷凍し、その後凍結乾燥した。続いて、3〜5 mgの乾燥組織をジクロロメタンおよびエタノールの2:1混合物(-80℃)で覆った。最後に、1.5 M酢酸アンモニウム水溶液(4℃)および同位体で標識した細胞抽出物(4℃)を加えた。この試料を、b)に記載した通りにさらに処理した。
【実施例2】
【0051】
実施例2:同位体で標識した細胞抽出物の作成
カンジダ・ユチリス(Candida utilis)(DSMZ sp. 2361として寄託されている)の酵母培養物を、振とうフラスコ内で、アミノ酸を含まないYNB最少培地(Sigma)中のU13C-D-グルコース(10 g/L)上で増殖させた。好気条件を維持するため、該フラスコをオービタルシェーカーを使用して180 rpm、28℃で揺り動かした。細胞の採取は、200 mLの酵母培養物を2分間、1000×gかつ4℃で遠心分離(Falconチューブ)することにより実施した。続いて該細胞を、0.15 M酢酸アンモニウムおよび10 g/L U13C-D-グルコースからなる洗浄用緩衝液と連続した遠心分離を利用して2回洗浄した。得られた細胞ペレットを、7.5 mLの2:1(v/v)ジクロロメタン/エタノール溶液(-80℃)を使用してクエンチした。抽出は、このクエンチ済み試料に1.5 M酢酸アンモニウム水溶液2.5 mLを加えることにより、a)に記載した通りに極低温条件下で実施した。抽出後、試料をa)に記載した条件下で遠心分離してから、U13C-標識代謝物を含む上相を集めて次回使用時まで-80℃で保存した。
【実施例3】
【0052】
実施例3:クロマトグラフ分離
リン酸化および/またはカルボキシル化極性代謝物のクロマトグラフ分離のために、超高圧イオン対形成液体クロマトグラフィーIP-UPLCを適用した。溶媒A(脱イオン水)と溶媒B(50%アセトニトリル、50%水(v/v))の間のクロマトグラフ勾配処理を行ったが、一方で一定のカラム流量0.4 mL/分およびカラムオーブン温度45℃を維持した。揮発性添加剤としてトリブチルアミンを両溶離剤に加え、氷酢酸を使用してpH値をpH値6.2に調整することにより、イオン対を形成する(ion pairing)トリブチルアンモニウム陽イオンを生成させた。前記の凍結乾燥試料を低用量の溶離剤Aに溶かした後、前記抽出物1〜20μLを注入した。
【実施例4】
【0053】
実施例4:質量分析
負モードのエレクトロスプレー・タンデム質量分析(-ESI-MSMS)を利用して、UPLCにより分離した極性代謝物を評価した。タンデムMSMSはいわゆる計画(scheduled)または選択(selected)多重反応モニタリングモード(sMRM)で操作したが、単位分解能でユニークな質量調整を規定した。個々の代謝物の同位体標識形態および非標識形態は、種々の質量トレース(mass traces)により区別した。
【実施例5】
【0054】
実施例5:高濃度で適用した揮発性抽出緩衝液による抽出収率の上昇
実施例5は、細胞性組織を抽出する際に揮発性酢酸アンモニウムを高濃度で使用したことによる、数種の代謝物の抽出収率の上昇を例示するものである。加えた揮発性緩衝液はその後の凍結乾燥工程中に除去されたが、揮発性が低いリン酸化代謝物は保持された。従って、細胞性組織を高塩濃度緩衝液にさらすと、従来の条件下よりもかなり高い程度まで多数のタンパク質結合代謝物が放出される。この知見は、試料中に少ししか存在しない代謝物にとって特に重要である。さらに、ニコチンアミドの抽出収率の上昇はNADPHの酸化型と還元型の比率に強く影響を与え、このことは細胞または試料における還元当量の入手性を示している(図2)。
【実施例6】
【0055】
実施例6:典型的なクロマトグラム
実施例6は、凍結乾燥組織の典型的なクロマトグラムを示す。5 mgの乾燥試料は、極低温条件下での抽出前にU13C-標識酵母抽出物を用いて補正(amended)した。図3は、計画的多重反応モニタリングを利用して負モードのESI-MSMSにより測定した70の質量トレース(標識/非標識)のクロマトグラムを示したものである。試料または標識酵母における天然存在度に応じて、質量存在度(mass abundance)には3桁の幅が生じた。オーバービューモード(XIC)の他に、3つの包括的な質量エクストラクト(comprehensive mass extracts)についても図示したが、これらはそれぞれエネルギー代謝、クレブス回路(TCA)、および解糖を表している。
【実施例7】
【0056】
実施例7:密度勾配遠心分離を介したクエンチングによる酵素活性の改善された阻害
カンジダ・ユチリスsp.(Candida utilis sp.)2361の酵母培養物を、10 g L-1の非標識D-グルコースを使用してYNB培地(Sigma)で増殖させてから、光学密度4.0(600 nm、1 cm光路長)で回収した。クエンチング法によって生じるバイアスを評価するため、該培養物を、
(A)低温溶媒によるクエンチング前に、0.2 μmフィルターに素早く通し、10 g L-1のU13C-標識D-グルコースを含有する150 mMの酢酸アンモニウム緩衝液で洗浄(最大10秒)し、そして
(B)密度勾配遠心分離法を利用して即時クエンチしたが、プロパン1,2-ジオール相は同量のU13C-D-グルコースで補正(amended)した。
【0057】
標準的なクエンチング法(例えば、ろ過および迅速な細胞洗浄)を用いたところ、解糖中間体、2種のクレブス回路の酸、ならびにペントースリン酸経路の代謝物は有意な13C-濃縮を呈した。反対に、酵母細胞を新しい密度勾配法によりクエンチすると、解糖中間体および他の中間体における13Cの濃縮はみられず、そのことは、解糖酵素の完全な不活性化を本質的に証明している(図4)。
【特許請求の範囲】
【請求項1】
極性代謝物を分析するための方法であって、以下:
i)生物学的試料に含まれる細胞の即時破壊を可能にする条件下で、相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、
ii)工程i)で得た抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および
iii)工程ii)で得た、分離した極性代謝物を分析すること
を含む上記方法。
【請求項2】
前記相分離剤がジクロロメタンを含む、請求項1記載の方法。
【請求項3】
前記揮発性アンモニウム塩が酢酸アンモニウム、ギ酸アンモニウム、または炭酸水素アンモニウムである、請求項1または2記載の方法。
【請求項4】
前記揮発性中性アンモニウム塩の濃度が少なくとも0.5 Mである、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記の工程i)における生物学的試料の抽出を抽出温度-20〜-80℃で実施する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記抽出物を、工程ii)に先立って以下の工程:
a)水相のスピン遠心分離、
b)工程a)で得た上清の凍結乾燥、および
c)工程b)で得た前記凍結乾燥物の、イオン対形成剤を含む分析緩衝液への溶解
により処理する、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記イオン対形成剤が、トリブチルアンモニウム陽イオン、トリエチルアンモニウム陽イオン、トリプロピル-アンモニウム陽イオンまたはn-ヘキシルアンモニウム陽イオンである、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記クロマトグラフィーがUPLCまたはHPLCである、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記の工程iii)で得た分離した極性代謝物の分析が質量分析を含む、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記質量分析の方法がESI-MS/MSである、請求項9記載の方法。
【請求項11】
前記生物学的試料が細胞懸濁液である、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
細胞性物質を含む生物学的試料をクエンチするための方法であって、以下の工程:
a)プロパンジオールまたはエタンジオールを含むクエンチング溶液からなる下相、不活性相分離剤からなる中間相、および生物学的試料からなる上相を含む遠心分離バイアルを提供する工程、ただし該下相は該中間相よりも高い密度を有し、かつ該中間相は該上相よりも高い密度を有する、ならびに
b)前記生物学的試料に含まれる細胞性物質を前記上相から前記下相へ移動させるために、前記バイアルを遠心分離する工程
を含む上記方法。
【請求項13】
前記下相が-20〜-80℃の温度を有する、請求項12記載の方法。
【請求項14】
上清を遠心分離後に前記下相から除去し、かつ同位体で標識した標準物質をこの下相に加える、請求項12または13記載の方法。
【請求項15】
前記細胞懸濁液を、工程i)に先立って請求項12〜14のいずれか1項に記載した通りにクエンチングに供する、請求項11記載の方法。
【請求項16】
前記生物学的試料が膜上で増殖させた接着細胞の培養物である、請求項1〜10のいずれか1項に記載の方法。
【請求項17】
前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、-20〜-80℃の温度を有する請求項1〜4のいずれか1項に記載した抽出緩衝液に該膜を即時移動させることによりクエンチングに供する、請求項16記載の方法。
【請求項18】
前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、液体窒素中に該膜を即時移動させることによりクエンチングに供する、請求項16記載の方法。
【請求項19】
前記生物学的試料が組織試料である、請求項1〜10のいずれか1項に記載の方法。
【請求項20】
前記組織を、工程i)に先立って、該組織を-20〜-80℃の温度まで即時冷凍することにより、または液体窒素を適用することによりクエンチングに供する、請求項18記載の方法。
【請求項1】
極性代謝物を分析するための方法であって、以下:
i)生物学的試料に含まれる細胞の即時破壊を可能にする条件下で、相分離剤と揮発性中性アンモニウム塩とを含む抽出緩衝液を用いて該生物学的試料を抽出すること、
ii)工程i)で得た抽出物に含まれる極性代謝物をクロマトグラフィーにより分離すること、および
iii)工程ii)で得た、分離した極性代謝物を分析すること
を含む上記方法。
【請求項2】
前記相分離剤がジクロロメタンを含む、請求項1記載の方法。
【請求項3】
前記揮発性アンモニウム塩が酢酸アンモニウム、ギ酸アンモニウム、または炭酸水素アンモニウムである、請求項1または2記載の方法。
【請求項4】
前記揮発性中性アンモニウム塩の濃度が少なくとも0.5 Mである、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記の工程i)における生物学的試料の抽出を抽出温度-20〜-80℃で実施する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記抽出物を、工程ii)に先立って以下の工程:
a)水相のスピン遠心分離、
b)工程a)で得た上清の凍結乾燥、および
c)工程b)で得た前記凍結乾燥物の、イオン対形成剤を含む分析緩衝液への溶解
により処理する、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記イオン対形成剤が、トリブチルアンモニウム陽イオン、トリエチルアンモニウム陽イオン、トリプロピル-アンモニウム陽イオンまたはn-ヘキシルアンモニウム陽イオンである、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記クロマトグラフィーがUPLCまたはHPLCである、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記の工程iii)で得た分離した極性代謝物の分析が質量分析を含む、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記質量分析の方法がESI-MS/MSである、請求項9記載の方法。
【請求項11】
前記生物学的試料が細胞懸濁液である、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
細胞性物質を含む生物学的試料をクエンチするための方法であって、以下の工程:
a)プロパンジオールまたはエタンジオールを含むクエンチング溶液からなる下相、不活性相分離剤からなる中間相、および生物学的試料からなる上相を含む遠心分離バイアルを提供する工程、ただし該下相は該中間相よりも高い密度を有し、かつ該中間相は該上相よりも高い密度を有する、ならびに
b)前記生物学的試料に含まれる細胞性物質を前記上相から前記下相へ移動させるために、前記バイアルを遠心分離する工程
を含む上記方法。
【請求項13】
前記下相が-20〜-80℃の温度を有する、請求項12記載の方法。
【請求項14】
上清を遠心分離後に前記下相から除去し、かつ同位体で標識した標準物質をこの下相に加える、請求項12または13記載の方法。
【請求項15】
前記細胞懸濁液を、工程i)に先立って請求項12〜14のいずれか1項に記載した通りにクエンチングに供する、請求項11記載の方法。
【請求項16】
前記生物学的試料が膜上で増殖させた接着細胞の培養物である、請求項1〜10のいずれか1項に記載の方法。
【請求項17】
前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、-20〜-80℃の温度を有する請求項1〜4のいずれか1項に記載した抽出緩衝液に該膜を即時移動させることによりクエンチングに供する、請求項16記載の方法。
【請求項18】
前記の膜上で増殖させた接着細胞の培養物を、工程i)に先立って、液体窒素中に該膜を即時移動させることによりクエンチングに供する、請求項16記載の方法。
【請求項19】
前記生物学的試料が組織試料である、請求項1〜10のいずれか1項に記載の方法。
【請求項20】
前記組織を、工程i)に先立って、該組織を-20〜-80℃の温度まで即時冷凍することにより、または液体窒素を適用することによりクエンチングに供する、請求項18記載の方法。
【図1】


【図2】
【図3A】

【図3B】

【図3C】
【図3D】
【図4A】
【図4B】

【図2】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【公表番号】特表2012−533053(P2012−533053A)
【公表日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2012−518980(P2012−518980)
【出願日】平成22年7月7日(2010.7.7)
【国際出願番号】PCT/EP2010/059740
【国際公開番号】WO2011/003945
【国際公開日】平成23年1月13日(2011.1.13)
【出願人】(512005634)ビーエーエスエフ プラント サイエンス カンパニー ゲーエムベーハー (8)
【Fターム(参考)】
【公表日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成22年7月7日(2010.7.7)
【国際出願番号】PCT/EP2010/059740
【国際公開番号】WO2011/003945
【国際公開日】平成23年1月13日(2011.1.13)
【出願人】(512005634)ビーエーエスエフ プラント サイエンス カンパニー ゲーエムベーハー (8)
【Fターム(参考)】
[ Back to top ]