
エノイル−アシルキャリアプロテインレダクターゼ(FabK)の結晶
【課題】
創薬において重要であるFabKの立体構造情報が存在せず効率的な阻害剤の設計が困難である。X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計等を可能とする高品質のFabK結晶を提供する。
【解決手段】肺炎連鎖球菌(Streptococcus pneumoniae)由来FabKを調製し、膨大な数の結晶化条件を鋭意検討した結果、高品質で再現性良く得られるFabK蛋白質の結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功した。
創薬において重要であるFabKの立体構造情報が存在せず効率的な阻害剤の設計が困難である。X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計等を可能とする高品質のFabK結晶を提供する。
【解決手段】肺炎連鎖球菌(Streptococcus pneumoniae)由来FabKを調製し、膨大な数の結晶化条件を鋭意検討した結果、高品質で再現性良く得られるFabK蛋白質の結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、X線結晶構造解析および阻害剤の設計等に利用するためのエノイル−アシルキャリアプロテイン(FabK)の結晶に関する。
【背景技術】
【0002】
細菌における脂肪酸の生合成はタイプIIの脂肪酸シンターゼ(FAS)系と呼ばれる一連の酵素群により行われている。一方、哺乳動物などの真核生物においてはタイプIのFAS系と呼ばれる、単一の多機能酵素により触媒されており、細菌の脂肪酸生合成系とは大きく異なるため、タイプIIのFAS系酵素を阻害する化合物は、選択的かつ新規な抗菌剤としての可能性が示唆される(非特許文献1参照)。
【0003】
細菌の脂肪酸生合成系では4つの酵素反応が順次進行してサイクルを形成し、複数回のサイクルを経て、長鎖の飽和脂肪酸が生合成される。サイクルの第1工程はβ−ケトアシル−ACPシンターゼによる縮合反応で、はじめのサイクルではβ−ケトアシル−ACPシンターゼIII(FabH)がマロニル−ACPとアセチル−CoAを縮合する。2回目以降のサイクルではβ−ケトアシル−ACPシンターゼIまたはII(FabBまたはFabF)がマロニル−ACPとアシル−ACPを縮合する。第2工程ではNADPH−依存性β−ケトアシル−ACPレダクターゼ(FabG)による還元反応が起こる。第3工程でβ−ヒドロキシアシル−ACPデヒドラーゼ(FabAまたはFabZ)により脱水され、トランス−2−エノイル−ACPが得られる。第4工程において、エノイル−ACPレダクターゼ(FabIまたはFabKまたはFabL)により還元されて、アシル−ACPを生ずる。サイクル当たり2個の炭素原子が付加され、最終的にパルミトイル−ACP(16C)が得られるが、このサイクルは主にパルミトイル−ACPによるエノイル―ACPレダクターゼのフィードバック阻害を介して停止する(非特許文献2、3参照)。すなわち、エノイル―ACPレダクターゼが細菌の脂肪酸合成経路の律速酵素であり、細菌における脂肪酸生合成全体の重要な調整ポイントである。また、大腸菌(Escherichia coli)や黄色ブドウ球菌(Staphyrococcus aureus)などに存在するエノイル―ACPレダクターゼであるFabIは生育に必須な酵素であることが大腸菌のFabI温度感受性変異株の解析から明らかにされた(非特許文献4)。したがって、エノイル−ACPレダクターゼを阻害することにより抗菌作用が発現されると考えられるため、抗菌剤の標的として重要な蛋白質であるといえる。
【0004】
また、FabIが広域スペクトル抗生物質トリクロサンの標的であることも明らかにされている。NADおよびトリクロサンと大腸菌(Escherichia coli)由来FabIとの複合体結晶構造についても研究されており、その天然基質を模倣することで、トリクロサンが特異性の高いFabIの阻害剤として作用することが示されている。FabIとNADおよびトリクロサンの複合体に関する立体構造データは特異的な阻害剤設計において重要な情報を提供する。この情報に基づいて設計した阻害剤が、細菌感染症の治療に有用である可能性が見出されている(非特許文献5、6、7、8参照)。
【0005】
ゲノム解析研究により、大腸菌や黄色ブドウ球菌などのほとんどの細菌はエノイル―ACPレダクターゼとしてFabIを有していることが知られているが、いくつかの菌種ではFabIのかわりにFabKが存在していることが明らかになっている。中でも臨床上重要な病原菌である肺炎連鎖球菌(Streptococcus pneumoniae)はエノイル―ACPレダクターゼとしてFabKのみを有しており、緑膿菌(Pseudomonas aeruginosa)、腸球菌(Enterococcus feacalis)ではFabIとFabKの両方を有していることが明らかになっている(非特許文献9参照)。そのため、より広範な抗菌活性を有する抗菌剤を創出するには、FabIだけでなくFabKを阻害する必要がある。
【0006】
FabKは肺炎連鎖球菌において生育に必須であり、補酵素としてFMNを1:1のモル濃度比で有するフラビン蛋白質であることがわかっている(非特許文献10参照)。その他、FabKを弱いながらも阻害する化合物の報告はあるが、FabKを強く阻害する化合物の報告やFabKの立体構造に関する報告はない。
【0007】
FabKを阻害する化合物を効率的に設計するにはFabKの立体構造情報が有用である。FabKの結晶構造を明らかにすることで、FabK特異的な阻害剤が設計できれば、代表的な薬剤耐性菌であるペニシリン耐性肺炎連鎖球菌(PRSP)を含む肺炎連鎖球菌に対する特効薬、またはFabIおよびFabK両方に特異的な阻害剤を設計できれば、メチシリン耐性黄色ブドウ球菌(MRSA)や多剤耐性緑膿菌を対象に含む、より広範な抗菌剤の創出に非常に有用であることが考えられる。そのため、X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計を可能とする高品質のFabK結晶の創出が望まれている。
【非特許文献1】Prog. Lipid Res.(2001)、40、467−497
【非特許文献2】Escherichia coli and Salmonella :Cellular and Molecular Biology(1996)、612−636、American Society for Microbiology、Washington D.C.
【非特許文献3】J.Biol.Chem.(1996)、271、1833−1836
【非特許文献4】J.Biol.Chem.(1995)、270、26538−26542
【非特許文献5】Nature(1998)394、531−532
【非特許文献6】J.Biol.Chem.(1998)、273、30316−30320
【非特許文献7】Nature(1999)398、383−384
【非特許文献8】J.Med.Chem.(2003)、46、1627−1635
【非特許文献9】Nature (2000)、406、145−146
【非特許文献10】Biochem.J(2003)、370、1055−1062
【発明の開示】
【発明が解決しようとする課題】
【0008】
これまでFabKの結晶および結晶構造解析の報告はない。そのため、創薬において重要であるFabKの立体構造情報が存在せず効率的な阻害剤の設計が困難である。
【課題を解決するための手段】
【0009】
本発明は、X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計等を可能とする高品質のFabK結晶を提供することで上記課題を解決する。
本発明者は、肺炎連鎖球菌(Streptococcus pneumoniae)由来FabKを調製し、膨大な数の結晶化条件を鋭意研究した結果、高品質で再現性よく得られるFabK蛋白質の結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功し、創薬研究に重要なFabKの立体構造を決定した。すなわち、本発明はX線結晶構造解析および阻害剤の設計等に利用するためのFabK結晶に関する。また、前記結晶における空間群はP21、P212121、I222、C2221である。
【0010】
本発明は以下の発明を含有する。
(1)エノイル―アシルキャリアプロテインレダクターゼ(FabK)の結晶。
(2)ストレプトコッカス属(Streptococcus)由来である(1)記載の結晶。
(3)肺炎連鎖球菌(Streptococcus pneumoniae)由来で(1)または(2)記載の結晶。
(4)空間群がP21である(1)〜(3)のいずれか一項に記載の結晶。
(5)空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である(1)〜(3)のいずれか一項に記載の結晶。
(6)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21である(1)〜(3)のいずれか一項に記載の結晶。
(7)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である(1)〜(3)のいずれか一項に記載の結晶。
(8)空間群がP212121である(1)〜(3)のいずれか一項に記載の結晶。
(9)空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(10)空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(11)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121である(1)〜(3)のいずれか一項に記載の結晶。
(12)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(13)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(14)空間群がI222である(1)〜(3)のいずれか一項に記載の結晶。
(15)空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(16)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222である(1)〜(3)のいずれか一項に記載の結晶。
(17)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(18)空間群がC2221である(1)〜(3)のいずれか一項に記載の結晶。
(19)空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(20)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221である(1)〜(3)のいずれか一項に記載の結晶。
(21)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126
±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(22)(1)〜(21)のいずれか一項に記載の結晶であって、該結晶を構成する蛋白質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
(23)(1)〜(22)のいずれか一項に記載の結晶であって、該結晶がリガンドと複合体を形成している結晶。
【発明の効果】
【0011】
本発明のFabK結晶は高品質で再現性良く調製可能であるため、X線結晶構造解析に適しており、これまでに全く報告の無かったFabKの立体構造解析が初めて可能となった。本発明のFabK結晶を利用することにより、FabKを阻害する化合物の効率的な設計等に役立つ立体構造情報を取得できるため、創薬において極めて有用である。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。
<<FabKの発現>>
FabKは、それをコードするDNA断片を、宿主細胞内で複製可能でかつ同遺伝子が発現可能な状態で含むDNA分子、特にDNA発現ベクターの形態とし、それによって宿主細胞の形質転換を行い、その形質転換体を培養することによって得られる。このDNA分子は、ベクター分子にFabKをコードするDNA断片を組み込むことによって得ることができる。本発明の好ましい態様によれば、このベクターはプラスミドである。本発明において利用されるベクターは、使用する宿主細胞の種類を勘案して、ウィルス、プラスミド、コスミドベクターなどから適宜選択することができる。例えば宿主細胞が大腸菌の場合はpUC、pBR系のプラスミド、枯草菌の場合はpUB系のプラスミド、酵母の場合はYEp、YRp、YCp系のプラスミドベクターが挙げられる。宿主細胞としては、宿主−ベクター系が確立されているものであれば利用可能であり、好ましくは大腸菌が挙げられる。宿主細胞の形質転換により得られた形質転換体は、適当な条件で培養し、得られた形質転換体の細胞抽出液を慣用の方法に従い回収することができる。ここで、FabKに付加的ポリペプチド、例えばグルタチオン−S−トランスフェラーゼ(GST)またはヒスチジン残基に富むポリペプチド(His−tag)を融合タンパク質として発現することも慣用の技術により可能である。
【0013】
<<FabKの精製>>
His−tagを融合して大腸菌で発現させたFabK(His−FabK)は、金属イオン、好ましくはニッケルイオンを配位させたアフィニティーカラムを用いた慣用の技術により容易に精製が可能である。精製したHis−FabKを50mM トリス塩酸 pH 8.0、50mM 塩化アンモニウム、5mM ジチオスレイトール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン溶液に対して透析した後、陰イオン交換カラム、疎水性相互作用カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号1、配列番号3に示されるアミノ酸配列を有するHis−FabKを各々分取することができる。また、His−tagを融合させずに大腸菌で発現させたFabKは陰イオン交換カラム、疎水性相互作用カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号2、配列番号4に示されるアミノ酸配列を有するFabKを各々分取することができる。
【0014】
<<FabKの結晶化>>
一般的に蛋白質の結晶化は容易ではない。結晶化できる条件が比較的容易に見出される場合もあるが、通常は種々の結晶化条件をスクリーニングしなければならない。さらに沈殿剤の濃度、pH、塩の種類と濃度、添加剤などの条件を最適化することによって、X線結晶構造解析に供することができる高品質の結晶が得られる。また、いかなる条件でも結
晶化しない蛋白質も存在すると考えられている。蛋白質の結晶が得られなければX線結晶構造解析を行うことができない。さらに、X線結晶構造解析に用いる蛋白質の結晶は、結晶であればいかなるものでも解析可能であるわけではない。実際にX線を照射した際に、高分解能の回折強度データが得られることが必要である。
本発明におけるFabK結晶化の方法は、ハンギングドロップ法、シッティングドロップ法、マイクロバッチ法などのいかなる方法を用いてもよいが、好ましくは蒸気拡散法であるハンギングドロップ法が用いられる。しかしながら通常の方法で作製したFabKの結晶はクラスター化する傾向にあるため、シーディング法により良質な単結晶を得ることが好ましい。結晶化に用いるタンパク質としては、配列番号1、配列番号2に示されるアミノ酸配列を有するHis−FabKおよびFabKが望ましく、精製の簡便さから配列番号1に示されるアミノ酸配列を有するFabKが最も望ましい。結晶化に用いる精製タンパク質の濃度は、10mg/ml程度であることが望ましい。そして、蛋白質溶液に沈殿剤、塩類、緩衝液、添加剤などを適当量加えて、結晶化を行う。 本発明のFabKの結晶化に用いられる沈殿剤としては、ポリエチレングリコールなどの水溶性高分子、イソプロパノールや2−メチル−2,4−ペンタンジオール(MPD)などの有機溶媒などが挙げられる。また、緩衝剤としては公知の緩衝剤のいずれも用いることができるが、MES(pH 5.5−7.0)を用いるのが好ましい。沈殿剤の組み合わせについては(1)8−15% ポリエチレングリコール1000、8−15% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(2)20−25% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(3)2.5−10% ポリエチレングリコール8000、2.5−10% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.0−6.5の3通りが挙げられるが、中でも(1)の組み合わせが最も再現性良く、高品質の結晶を得ることができる。この条件は当初実施した通常市販されている結晶化条件スクリーニングキットには含まれておらず、スクリーニングキットにはあまり含まれない組み合わせであるポリエチレングリコールとMPDを組み合わせた上で、塩化アンモニウムや塩化カルシウム、ジチオスレイトール等を加えた独自の条件である。また、(1)の条件で得られる結晶はミクロシーディング法により、クラスター化を抑制しなければ、高分解能でのX線結晶構造解析に利用するのは困難である。
【0015】
<<FabK結晶のX線回折強度データの収集>>
上記のようにして得られる結晶の外観、単位格子の種類・大きさなどはその結晶に固有のものであり、良質の結晶を用いれば、X線回折実験により高分解能の回折強度データを取得することができる。結晶を多価アルコールを含有する抗凍結溶液中に浸した後凍結させ、凍結状態でX線回折データを収集し、X線回折像を得る。ここで、多価アルコールとしては、スクロース、トレハロース、グリセロールなどが挙げられる。これらの中で特にグリセロールが好ましい。抗凍結溶液中の多価アルコールの濃度は、好ましくは10%〜20%である。X線を照射して回折強度データを取得するには、いかなるX線発生装置も用いることができるが、好ましくは、(財)高輝度光科学研究センターの大型放射光施設SPring−8などの第3世代の放射光施設を利用することができる。得られたX線回折強度データを処理することにより、結晶学的パラメータを特定することが可能である。例えば、配列番号1のアミノ酸配列を有する蛋白質から構成されるHis−FabKの結晶としては、空間群がP21であり、格子定数はa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°である結晶などが挙げられる。
【0016】
<<FabKの結晶構造の決定>>
上記のように得られたデータを用いてFabKの結晶構造の決定が可能となる。一般に位相決定には、分子置換法、重原子同型置換法、多波長異常分散法(MAD法)などが用
いられるが、目的蛋白質に対する類縁蛋白質の立体構造が未知であり、その立体構造を利用した分子置換法による構造解析が不可能な本発明のような場合には、重原子同型置換法またはMAD法などが用いられる。すなわち、ネイティブ結晶および重原子誘導体結晶の回折データ間の回折強度差、あるいは異なる波長間で測定した回折データの回折強度差から、電子密度を計算するための初期位相を求めることにより、立体構造を決定することが可能である。本発明では波長を変えることのできる大型放射光施設SPring−8を利用可能であったため、セレノメチオニンを導入した結晶を用いたMAD法によるX線結晶構造解析を実施し、その後の解析では実験で得られたFabKの立体構造をサーチモデルとした分子置換法による構造決定が可能であった。
【0017】
以下、実施例により本発明をさらに詳細に説明するが、本発明の技術的範囲は実施例によって限定されるものではない。また、各種ベクターの作製、蛋白質の発現等は、特に記載のない限り、Molecular Cloning,A Laboratory Manual,3rd edition(Sambrook and Russell著,Cold Spring Harbor laboratory Press刊(2001))などに記載の公知の手法に従って実施できる。
【実施例1】
【0018】
<<肺炎連鎖球菌由来FabKの大腸菌発現系の構築>>
(1)肺炎連鎖球菌R6株由来のゲノムDNAの単離
肺炎連鎖球菌(Streptococcus pneumoniae)R6株(ATCC49619)を血液寒天基礎培地(ベクトン・ディッキンソン社製)で二酸化炭素5%、37℃の条件下で18時間培養し、低速遠心によって集菌した。得られた菌体からDNeasy Tissue Kit(QIAGEN社製)を用い、添付の説明書記載の方法に従って、ゲノムDNAを調製した。
【0019】
(2)肺炎連鎖球菌R6株由来FabK発現ベクターの構築
FabKをコードするDNAを単離するため以下のプライマーを設計した。
【0020】
FabK−F.P.:5’−GGAATTCCATATGAAAACGCGTATTACAGAA−3’(配列番号5)
His−FabK−R.P.:5’−CCGCTCGAGGTCATTTCTTACAACTCCTGT−3’(配列番号6)
FabK−R.P.:5’−CCGCTCGAGTTAGTCATTTCTTACAACTCC−3’(配列番号7)
【0021】
上記(1)で単離したゲノムDNAを鋳型に、前記プライマーを用いて、Pyrobest DNA Polymerase(タカラバイオ社製)でポリメラーゼ連鎖反応(PCR)を添付の説明書に従い実施した。His−tag付のFabK(His−FabK)を作製する場合には配列番号5および配列番号6のプライマーを、タグなしのFabKを作製する場合には配列番号5および配列番号7のプライマーを使用した。増幅したDNA断片はNdeIおよびXhoIで消化した。この断片を予めNdeIおよびXhoIで切断したベクターpET−21b(+)(Novagen社製)にDNA Ligation Kit ver.2(タカラバイオ社製)を添付の説明書の方法に従い用い、サブクローニングした。得られた組換えプラスミドを大腸菌COMPETENT high DH5α(TOYOBO社製)に添付の説明書の方法に従い導入し、形質転換体を得た。形質転換体を、50μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。取得したコロニーから組換えプラスミド(His−FabK/pET−21b(+)およびFabK/pET−21b(+))を調製しDNA配列を確認し、配列番号1および2に記載のSWISSPROT
Accession番号Q8DR17に登録されている肺炎連鎖球菌R6株由来のFabKをコードすることを確認した。
【実施例2】
【0022】
<<FabK(His−FabKおよびFabK)の発現>>
実施例1の(2)で得られたHis−FabK/pET−21b(+)およびFabK/pET−21b(+)の組み換えプラスミドを大腸菌BL21(DE3)(Novagen社製)に形質転換し、得られた形質転換体を100μg/mlのアンピシリンを含むSB培地(1.2%(w/v)Bacto Tryptone、2.4%(w/v)Yeast Extract、0.5%(v/v)グリセロール、0.072M リン酸水素二カリウム、0.028M リン酸二水素カリウム)5L中で600nmにおけるO.D.が0.6−1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)で3時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体をHis−FabK/pET−21b(+)の組み換え大腸菌の場合には菌体破砕バッファーA(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、5mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール、0.1mg/mL リゾチーム)、FabK/pET−21b(+)の組み換え大腸菌の場合には菌体破砕バッファーB(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール、0.1mg/mL リゾチーム)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を5−7回施し、細胞を破砕した。遠心分離(30分間、15000rpm)とそれに続く0.22μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例3】
【0023】
<<His−FabKの精製>>
(1)アフィニティーカラムによる精製
以下の精製操作はすべて4℃で行った。His−FabK/pET−21b(+)の組み換え大腸菌から実施例2の方法で得られた細胞抽出液をアフィニティークロマトグラフィーで精製した。アフィニティークロマトグラフィーカラムは、Ni−NTA(QIAGEN社製)を担体として用い、マニュアル記載の方法でカラム容量30mlのカラムを作製した。平衡化バッファー(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、5mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で平衡化した後、細胞抽出液を流速1mL/minでアプライし、3カラム容量の洗浄バッファー(50mM
リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、20mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で洗浄した後、溶出バッファー(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、200mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要なフラクションをまとめて回収した。
【0024】
(2)陰イオン交換カラムによる精製
上記(1)で得られたHis−FabK溶液を透析バッファー(50mM トリス塩酸緩衝液 pH 8.0、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で2時間透析した後に、陰イオン交換カラムであるPOROS HQ/20(PerSeptive Bio
systems社製)を用いて精製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラムに、流速10mL/minでHis−FabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−500mM 塩化カリウムの直線勾配(25カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要な画分をまとめ、以後の精製を実施した。
【0025】
(3)疎水性相互作用クロマトグラフィーによる精製
上記(2)で得られたHis−FabK溶液を、50mM トリス塩酸緩衝液 pH 7.5、3.5M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液と混合した後、硫酸アンモニウムの終濃度が2Mになるように調製した。調製したHis−FabK溶液を平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、2M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化した疎水性相互作用カラムであるRESOURCE PHE 6ml(アマシャムバイオサイエンス社製)に、流速5ml/minでアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の2−0M 硫酸アンモニウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要な画分をまとめ、以後の精製を実施した。
【0026】
(4)ゲルろ過カラムによる精製
上記(3)で得られたHis−FabK溶液を保持分子量30000以上の限外ろ過膜であるAmicon Ultra−4 30kDa NMWL(ミリポア社製)を用いて1mLまで濃縮し、ゲルろ過バッファー(50mM トリス塩酸緩衝液 pH 7.5、150mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload
16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1ml/minでアプライした。SDS−PAGEにより主要な画分を各々まとめ、上記記載の限外ろ過膜を用いて濃縮した。その後溶液交換のため、10mM
トリス塩酸緩衝液 pH 7.5、100mM 塩化アンモニウム、1mM ジチオスレイトール溶液で希釈し、再度濃縮する操作を3度繰り返した後、10mg/mlになるまで濃縮し、終濃度5mMになるようにβ−ニコチンアミドアデニンジヌクレオチド二ナトリウム(還元型)を添加して、結晶化サンプルとした。サンプルは液体窒素中で凍結し、−80℃で保存した。
【実施例4】
【0027】
<<FabK(タグなし)の精製>>
(1)陰イオン交換カラムによる精製
以下の精製操作はすべて4℃で行った。FabK/pET−21b(+)の組み換え大腸菌から実施例2の方法で得られた細胞抽出液を陰イオン交換カラムクロマトグラフィーで精製した。陰イオン交換カラムは、Q―Sepharose High Performance(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量114mlのカラムを作製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラ
ムに、流速5mL/minでFabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−1M 塩化カリウムの直線勾配(5カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、以後の精製を実施した。
【0028】
(2)疎水性相互作用クロマトグラフィーによる精製
上記(1)で得られたFabK溶液を、50mM トリス塩酸緩衝液 pH 7.5、3.5M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液と混合した後、硫酸アンモニウムの終濃度が2Mになるように調製した。調製したFabK溶液を平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、2M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化した疎水性相互作用カラムであるRESOURCE PHE 6ml(アマシャムバイオサイエンス社製)に、流速5ml/minでアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の2−0M
硫酸アンモニウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、以後の精製を実施した。
【0029】
(3)ゲルろ過カラムによる精製
上記(2)で得られたFabK溶液を保持分子量30000以上の限外ろ過膜であるAmicon Ultra−4 30kDa NMWL(ミリポア社製)を用いて1mLまで濃縮し、ゲルろ過バッファー(50mM トリス塩酸緩衝液 pH 7.5、150mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1ml/minでアプライした。SDS−PAGEにより主要な画分を各々まとめ、以後の精製を実施した。
【0030】
(4)陰イオン交換カラムによる精製
上記(3)で得られたFabK溶液を透析バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で2時間透析した後に、陰イオン交換カラムクロマトグラフィーであるMonoQ HR10/10(アマシャムバイオサイエンス社製)で精製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH
7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラムに、流速3mL/minでFabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM
フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−1M 塩化カリウムの直線勾配(20カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、上記記載の限外ろ過膜を用いて濃縮した。その後溶液交換のため、10mM トリス塩酸緩衝液 pH 7.5、100mM 塩化アンモニウム、1mM ジチオスレイトール溶液で希釈し、再度濃縮する操作を3度繰り返した後、10mg/mlになるまで濃縮し、終濃度5mMになるようにβ−ニコチンアミドアデニンジヌクレオチド二ナトリウム(還元型)を添加して、結晶化サンプルとした。サンプルは液体窒素中で凍結し、−80℃で保存した。
【実施例5】
【0031】
<<セレノメチオニン置換FabK(Se−His−FabKおよびSe−FabK)の発現>>
MAD法によるX線結晶構造解析に使用する重原子誘導体結晶を作製するために、メチオニンの硫黄原子が重原子であるセレンに置換されたセレノメチオニン(Se−Met)置換His−FabK(Se−His−FabK)およびFabK(Se−FabK)を作製した。Se−His−FabKおよびSe−FabKは実施例1の(2)に記載した各々の組み換えプラスミドをメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mlのアンピシリンを含むLB培地100mlにて、37℃で一晩培養し、遠心分離機で集菌後、100μg/mLのアンピシリンを含む100mlのLeMaster培地(組成は表1に示す。)で懸濁し、再度、遠心分離機によって集菌し、100μg/mLのアンピシリンを含むLeMaster培地100mlに再懸濁した。この懸濁液を100μg/mLのアンピシリンを含む5LのLeMaster培地に接種し、600nmにおけるO.D.が0.6−1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)で3時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。以下、Se−His−FabKおよびSe−FabKは天然型と同様の方法で精製した。
【0032】
【表1】

【実施例6】
【0033】
<<A141S変異FabK(A141S His−FabKおよびA141S FabK)の作製>>
実施例1の(2)で作製した組み換えプラスミドHis−FabK/pET−21b(+)およびFabK/pET−21b(+)を利用して、141番目のアミノ酸残基アラニンをセリンに置換した点変異体(A141S His−FabKおよびA141S FabK)をコードする組み換えプラスミドを作製した。点変異導入にはStratagene社のQuickChange Site−Directed Mutagenesis Kitを利用して1塩基の点変異を導入した。まず、目的とする変異を中央にしてその前後に10−15塩基ずつ付加したプライマー、およびそれに相補的な下記プライマーを用意した。
FabK−Mutant−F.P.:5’−GGGATAATCGTTATTCCTGT
CGTTCCTAGTG−3’(配列番号8)
FabK−Mutant−R.P.:5’−CACTAGGAACGACAGGAATAACGATTATCCC−3’(配列番号9)
上記プライマーを用い、添付の説明書の方法に従ってA141S His−FabKおよびA141S FabK点変異体発現プラスミドを作製した。得られたプラスミドについては、目的とした塩基以外に変異が入っていないことをDNA配列解析により確認し、発現ベクターに挿入されているDNAが配列番号3および4をコードすることを確認した。作製した発現ベクターを大腸菌BL21(DE3)に形質転換し、天然型と同様の方法で、発現および精製した。
【実施例7】
【0034】
<<FabKの結晶化>>
(7−1)Free体結晶の作製
His−FabKおよびFabKの結晶化において市販のスクリーニングキットやその組み合わせ、類似蛋白質の結晶化条件等、膨大な数の結晶化条件の検討の末、X線結晶構造解析可能な結晶を取得した。
結晶が得られた沈殿剤の組み合わせとしては(1)8−15% ポリエチレングリコール1000、8−15% 2−メチル−2,4−ペンタンジオール(MPD)、0.1M
塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(2)20−25% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(3)2.5−10% ポリエチレングリコール8000、2.5−10% ポリエチレングリコール1000、0.1M
塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.0−6.5の3通りが挙げられるが、中でも(1)の組み合わせが再現性良く、高品質の結晶を得ることができた。
MAD法によるX線結晶構造解析に利用した結晶は実施例5で精製したSe−His−FabKを用いて作製した。10mg/mlのSe−His−FabK溶液2.0μLと結晶化剤(10% ポリエチレングリコール1000、10% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)2.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを4℃の恒温槽内に静置すると数日でクラスター結晶が得られた。得られたクラスター結晶を用いてミクロシーディング法により、再度結晶化することでX線結晶構造解析可能な結晶が作製可能であった。得られたSe−His−FabK結晶の顕微鏡写真を図1に示す。この結晶は空間群がP21であり、格子定数がa=50.26Å、b=126.69Å、c=53.63Å、α=γ=90°、β=112.46°の結晶である。
また、結晶化条件(1)において実施例3で精製したHis−FabKを使用した場合にも同様の結晶が得られ、空間群がP21であり、格子定数がa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°であった。
さらに結晶化条件(1)では、His−FabKを利用した場合、空間群がP212121であり、格子定数がa=58.92Å、b=73.89Å、c=126.50Å、α=β=γ=90°の結晶も作製可能であった。
また、A141S His−FabKを使用した場合、結晶化条件(1)においては空間群がC2221であり、格子定数がa=58.99Å、b=84.60Å、c=126.17Å、α=β=γ=90°の結晶が得られた。
その他、結晶化条件(2)においてSe−FabKを使用した場合、空間群がP212121であり、格子定数がa=86.99Å、b=160.41Å、c=183.25Å
、α=β=γ=90°の結晶が得られ、結晶化条件(3)においてはHis−FabKを使用した場合、空間群がI222であり、格子定数がa=117.20Å、b=186.58Å、c=214.79Å、α=β=γ=90°の結晶が得られ、Se−His−FabKを使用した場合には空間群がI222であり、格子定数がa=118.38Å、b=190.30Å、c=213.04Å、α=β=γ=90°の結晶が得られた。上記記載のすべての結晶はハンギングドロップ蒸気拡散法、シッティングドロップ蒸気拡散法のいずれの方法で結晶化した場合でも、同様の結晶が得られた。
【0035】
(7−2)化合物複合体結晶の作製
当社で見出した2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸ナトリウム塩(特願2006−21372記載の実施例31化合物)は、FabKの酵素阻害活性を有する化合物であり、当社研究所で合成し、使用した。実施例3で精製したHis−FabKを用いて結晶化条件(1)で作製した結晶を、50mM 2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸ナトリウム塩含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)中に移し、4℃で一晩反応させることにより、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体結晶を調製した。
【実施例8】
【0036】
<<Se−His−FabKのX線回折強度データの収集および結晶構造の決定>>
実施例7(7−1)の結晶化条件(1)で得られたSe−His−FabK結晶を、15%グリセロール含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)に移し、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しCCDカメラを検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97932、0.97888および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。各データは回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、分解能2.0Åで処理し、回折強度を数値化し、結晶構造因子を求めた。
【0037】
その結果、空間群がP21であり、格子定数がa=50.26Å、b=126.69Å、c=53.63Å、α=γ=90°、β=112.46°の結晶であることがわかった。得られたデータのCompletenessはエッジ、ピーク、リモートの測定波長でそれぞれ97.2%、96.1%、95.4%、Rmergeはそれぞれ6.2%、6.4%、8.6%であった。結晶の空間群、格子定数、Se−His−FabKの分子量から、結晶の非対称単位にSe−His−FabK分子が2分子存在していることが明らかになった。これらのデータ収集の結果を下記の表2にまとめた。
【0038】
【表2】
【0039】
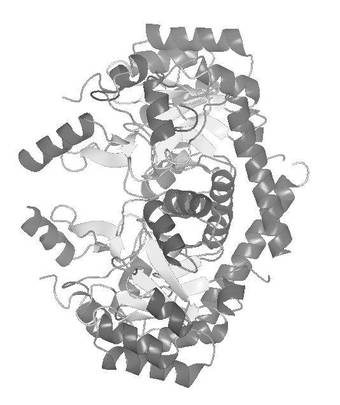
次に、プログラムSOLVE(Terwillinger, T.C. & Berendzen,J. ActaCrystallogr. D 55,849−861(1999))を用いてセレン原子の位置を決定し初期位相を計算した。位相の改良及び初期モデルの構築はプログラムRESOLVE(Terwilliger, T.C. Acta Crystallogr. D56,965−972(2000))を用いた。次いで、プログラムTurbo Frodo(AFMB−CNRS製)、プログラムRefmac5(Collaborative Computational Project Number 4(ccp4))によるモデルの構築および修正を繰り返し実施し、Se−His−FabKの結晶構造を決定した。構造精密化は分解能2.2Åで実施した。決定した結晶構造の正確さの指標であるR因子は、0.220であった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.282であった。また、決定したFabKの非対称単位中の構造を図2に示した。
【実施例9】
【0040】
<<A141S His−FabKのX線回折強度データの収集および結晶構造の決定>>
実施例7(7−1)の結晶化条件(1)で得られたA141S His−FabK結晶を、15%グリセロール含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.0)に移し、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しCCDカメラを検出装置としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、A141S His−FabK結晶を分解能1.8Åで処理し、回折強度を数値化し、結晶構造因子を求めた。
【0041】
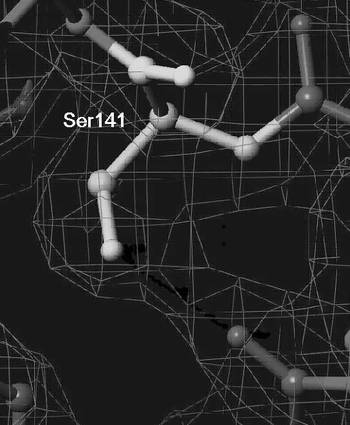
その結果、A141S His−FabK結晶は、空間群がC2221であり、格子定数がa=58.99Å、b=84.60Å、c=126.17Å、α=β=γ=90°であった。得られたデータのCompletenessは99.8%、Rmergeは6.6%であった。プログラムMolrep(CCP4)を用いた分子置換法により位相を決定し、プログラムRefmac5(CCP4)を用いて構造を精密化した結果、R因子は、0.244となった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.282となり、構造決定に成功した。また、点変異部分の電子密度図とモデルを図3に示した。
【実施例10】
【0042】
<<化合物複合体のX線回折強度データの収集および結晶構造の決定>>
実施例7(7−2)で得られた複合体結晶を、実施例9と同様の方法でX線回折測定およびデータ処理を実施し、分解能2.3Åで、回折強度を数値化し、結晶構造因子を求めた。
【0043】
その結果、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体結晶は空間群がP21であり、格子定数がa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°であった。得られたデータのCompletenessは94.0%、Rmergeは11.0%であった。プログラムMolrep(CCP4)を用いた分子置換法により位相を決定し、プログラムRefmac5(CCP4)を用いて構造を精密化した結果、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸に相当する電子密度が観測された(図4)。決定した結晶構造の正確さの指標であるR因子は、0.226となった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.316となり、構造決定に成功した。
【産業上の利用可能性】
【0044】
本発明のFabK結晶は、X線結晶構造解析可能な新規な結晶であり、解析に成功した立体構造情報を利用することによりFabKの特異的阻害剤の効率的な設計等が可能となるため、新規作用機序の新薬創出に活用でき、産業上極めて有用である。
【図面の簡単な説明】
【0045】
【図1】本発明のSe−His−FabK結晶の顕微鏡写真である。
【図2】決定したFabKの非対称単位中の構造をリボン図で示す。
【図3】点変異部分(A141S)の電子密度図とモデルを示す。
【図4】His−FabK/2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体における薬物結合部位近傍の図である。蛋白質をCαモデルで表示し、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸を除いたモデルから計算したFo−Fc電子密度図および2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸を重ねて表示している。
【技術分野】
【0001】
本発明は、X線結晶構造解析および阻害剤の設計等に利用するためのエノイル−アシルキャリアプロテイン(FabK)の結晶に関する。
【背景技術】
【0002】
細菌における脂肪酸の生合成はタイプIIの脂肪酸シンターゼ(FAS)系と呼ばれる一連の酵素群により行われている。一方、哺乳動物などの真核生物においてはタイプIのFAS系と呼ばれる、単一の多機能酵素により触媒されており、細菌の脂肪酸生合成系とは大きく異なるため、タイプIIのFAS系酵素を阻害する化合物は、選択的かつ新規な抗菌剤としての可能性が示唆される(非特許文献1参照)。
【0003】
細菌の脂肪酸生合成系では4つの酵素反応が順次進行してサイクルを形成し、複数回のサイクルを経て、長鎖の飽和脂肪酸が生合成される。サイクルの第1工程はβ−ケトアシル−ACPシンターゼによる縮合反応で、はじめのサイクルではβ−ケトアシル−ACPシンターゼIII(FabH)がマロニル−ACPとアセチル−CoAを縮合する。2回目以降のサイクルではβ−ケトアシル−ACPシンターゼIまたはII(FabBまたはFabF)がマロニル−ACPとアシル−ACPを縮合する。第2工程ではNADPH−依存性β−ケトアシル−ACPレダクターゼ(FabG)による還元反応が起こる。第3工程でβ−ヒドロキシアシル−ACPデヒドラーゼ(FabAまたはFabZ)により脱水され、トランス−2−エノイル−ACPが得られる。第4工程において、エノイル−ACPレダクターゼ(FabIまたはFabKまたはFabL)により還元されて、アシル−ACPを生ずる。サイクル当たり2個の炭素原子が付加され、最終的にパルミトイル−ACP(16C)が得られるが、このサイクルは主にパルミトイル−ACPによるエノイル―ACPレダクターゼのフィードバック阻害を介して停止する(非特許文献2、3参照)。すなわち、エノイル―ACPレダクターゼが細菌の脂肪酸合成経路の律速酵素であり、細菌における脂肪酸生合成全体の重要な調整ポイントである。また、大腸菌(Escherichia coli)や黄色ブドウ球菌(Staphyrococcus aureus)などに存在するエノイル―ACPレダクターゼであるFabIは生育に必須な酵素であることが大腸菌のFabI温度感受性変異株の解析から明らかにされた(非特許文献4)。したがって、エノイル−ACPレダクターゼを阻害することにより抗菌作用が発現されると考えられるため、抗菌剤の標的として重要な蛋白質であるといえる。
【0004】
また、FabIが広域スペクトル抗生物質トリクロサンの標的であることも明らかにされている。NADおよびトリクロサンと大腸菌(Escherichia coli)由来FabIとの複合体結晶構造についても研究されており、その天然基質を模倣することで、トリクロサンが特異性の高いFabIの阻害剤として作用することが示されている。FabIとNADおよびトリクロサンの複合体に関する立体構造データは特異的な阻害剤設計において重要な情報を提供する。この情報に基づいて設計した阻害剤が、細菌感染症の治療に有用である可能性が見出されている(非特許文献5、6、7、8参照)。
【0005】
ゲノム解析研究により、大腸菌や黄色ブドウ球菌などのほとんどの細菌はエノイル―ACPレダクターゼとしてFabIを有していることが知られているが、いくつかの菌種ではFabIのかわりにFabKが存在していることが明らかになっている。中でも臨床上重要な病原菌である肺炎連鎖球菌(Streptococcus pneumoniae)はエノイル―ACPレダクターゼとしてFabKのみを有しており、緑膿菌(Pseudomonas aeruginosa)、腸球菌(Enterococcus feacalis)ではFabIとFabKの両方を有していることが明らかになっている(非特許文献9参照)。そのため、より広範な抗菌活性を有する抗菌剤を創出するには、FabIだけでなくFabKを阻害する必要がある。
【0006】
FabKは肺炎連鎖球菌において生育に必須であり、補酵素としてFMNを1:1のモル濃度比で有するフラビン蛋白質であることがわかっている(非特許文献10参照)。その他、FabKを弱いながらも阻害する化合物の報告はあるが、FabKを強く阻害する化合物の報告やFabKの立体構造に関する報告はない。
【0007】
FabKを阻害する化合物を効率的に設計するにはFabKの立体構造情報が有用である。FabKの結晶構造を明らかにすることで、FabK特異的な阻害剤が設計できれば、代表的な薬剤耐性菌であるペニシリン耐性肺炎連鎖球菌(PRSP)を含む肺炎連鎖球菌に対する特効薬、またはFabIおよびFabK両方に特異的な阻害剤を設計できれば、メチシリン耐性黄色ブドウ球菌(MRSA)や多剤耐性緑膿菌を対象に含む、より広範な抗菌剤の創出に非常に有用であることが考えられる。そのため、X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計を可能とする高品質のFabK結晶の創出が望まれている。
【非特許文献1】Prog. Lipid Res.(2001)、40、467−497
【非特許文献2】Escherichia coli and Salmonella :Cellular and Molecular Biology(1996)、612−636、American Society for Microbiology、Washington D.C.
【非特許文献3】J.Biol.Chem.(1996)、271、1833−1836
【非特許文献4】J.Biol.Chem.(1995)、270、26538−26542
【非特許文献5】Nature(1998)394、531−532
【非特許文献6】J.Biol.Chem.(1998)、273、30316−30320
【非特許文献7】Nature(1999)398、383−384
【非特許文献8】J.Med.Chem.(2003)、46、1627−1635
【非特許文献9】Nature (2000)、406、145−146
【非特許文献10】Biochem.J(2003)、370、1055−1062
【発明の開示】
【発明が解決しようとする課題】
【0008】
これまでFabKの結晶および結晶構造解析の報告はない。そのため、創薬において重要であるFabKの立体構造情報が存在せず効率的な阻害剤の設計が困難である。
【課題を解決するための手段】
【0009】
本発明は、X線結晶構造解析によりFabKの立体構造を決定し、効率的な阻害剤の設計等を可能とする高品質のFabK結晶を提供することで上記課題を解決する。
本発明者は、肺炎連鎖球菌(Streptococcus pneumoniae)由来FabKを調製し、膨大な数の結晶化条件を鋭意研究した結果、高品質で再現性よく得られるFabK蛋白質の結晶を取得した。この結晶を用いて、多波長異常分散法(MAD法)により分解能2.2Åの結晶構造解析に成功し、創薬研究に重要なFabKの立体構造を決定した。すなわち、本発明はX線結晶構造解析および阻害剤の設計等に利用するためのFabK結晶に関する。また、前記結晶における空間群はP21、P212121、I222、C2221である。
【0010】
本発明は以下の発明を含有する。
(1)エノイル―アシルキャリアプロテインレダクターゼ(FabK)の結晶。
(2)ストレプトコッカス属(Streptococcus)由来である(1)記載の結晶。
(3)肺炎連鎖球菌(Streptococcus pneumoniae)由来で(1)または(2)記載の結晶。
(4)空間群がP21である(1)〜(3)のいずれか一項に記載の結晶。
(5)空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である(1)〜(3)のいずれか一項に記載の結晶。
(6)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21である(1)〜(3)のいずれか一項に記載の結晶。
(7)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である(1)〜(3)のいずれか一項に記載の結晶。
(8)空間群がP212121である(1)〜(3)のいずれか一項に記載の結晶。
(9)空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(10)空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(11)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121である(1)〜(3)のいずれか一項に記載の結晶。
(12)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(13)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(14)空間群がI222である(1)〜(3)のいずれか一項に記載の結晶。
(15)空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(16)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222である(1)〜(3)のいずれか一項に記載の結晶。
(17)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(18)空間群がC2221である(1)〜(3)のいずれか一項に記載の結晶。
(19)空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(20)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221である(1)〜(3)のいずれか一項に記載の結晶。
(21)配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126
±13Å、α=β=γ=90°である(1)〜(3)のいずれか一項に記載の結晶。
(22)(1)〜(21)のいずれか一項に記載の結晶であって、該結晶を構成する蛋白質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
(23)(1)〜(22)のいずれか一項に記載の結晶であって、該結晶がリガンドと複合体を形成している結晶。
【発明の効果】
【0011】
本発明のFabK結晶は高品質で再現性良く調製可能であるため、X線結晶構造解析に適しており、これまでに全く報告の無かったFabKの立体構造解析が初めて可能となった。本発明のFabK結晶を利用することにより、FabKを阻害する化合物の効率的な設計等に役立つ立体構造情報を取得できるため、創薬において極めて有用である。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。
<<FabKの発現>>
FabKは、それをコードするDNA断片を、宿主細胞内で複製可能でかつ同遺伝子が発現可能な状態で含むDNA分子、特にDNA発現ベクターの形態とし、それによって宿主細胞の形質転換を行い、その形質転換体を培養することによって得られる。このDNA分子は、ベクター分子にFabKをコードするDNA断片を組み込むことによって得ることができる。本発明の好ましい態様によれば、このベクターはプラスミドである。本発明において利用されるベクターは、使用する宿主細胞の種類を勘案して、ウィルス、プラスミド、コスミドベクターなどから適宜選択することができる。例えば宿主細胞が大腸菌の場合はpUC、pBR系のプラスミド、枯草菌の場合はpUB系のプラスミド、酵母の場合はYEp、YRp、YCp系のプラスミドベクターが挙げられる。宿主細胞としては、宿主−ベクター系が確立されているものであれば利用可能であり、好ましくは大腸菌が挙げられる。宿主細胞の形質転換により得られた形質転換体は、適当な条件で培養し、得られた形質転換体の細胞抽出液を慣用の方法に従い回収することができる。ここで、FabKに付加的ポリペプチド、例えばグルタチオン−S−トランスフェラーゼ(GST)またはヒスチジン残基に富むポリペプチド(His−tag)を融合タンパク質として発現することも慣用の技術により可能である。
【0013】
<<FabKの精製>>
His−tagを融合して大腸菌で発現させたFabK(His−FabK)は、金属イオン、好ましくはニッケルイオンを配位させたアフィニティーカラムを用いた慣用の技術により容易に精製が可能である。精製したHis−FabKを50mM トリス塩酸 pH 8.0、50mM 塩化アンモニウム、5mM ジチオスレイトール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン溶液に対して透析した後、陰イオン交換カラム、疎水性相互作用カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号1、配列番号3に示されるアミノ酸配列を有するHis−FabKを各々分取することができる。また、His−tagを融合させずに大腸菌で発現させたFabKは陰イオン交換カラム、疎水性相互作用カラム、ゲルろ過カラムを用いた一般的なクロマトグラフィーの手法を組み合わせて精製することが可能であり、配列番号2、配列番号4に示されるアミノ酸配列を有するFabKを各々分取することができる。
【0014】
<<FabKの結晶化>>
一般的に蛋白質の結晶化は容易ではない。結晶化できる条件が比較的容易に見出される場合もあるが、通常は種々の結晶化条件をスクリーニングしなければならない。さらに沈殿剤の濃度、pH、塩の種類と濃度、添加剤などの条件を最適化することによって、X線結晶構造解析に供することができる高品質の結晶が得られる。また、いかなる条件でも結
晶化しない蛋白質も存在すると考えられている。蛋白質の結晶が得られなければX線結晶構造解析を行うことができない。さらに、X線結晶構造解析に用いる蛋白質の結晶は、結晶であればいかなるものでも解析可能であるわけではない。実際にX線を照射した際に、高分解能の回折強度データが得られることが必要である。
本発明におけるFabK結晶化の方法は、ハンギングドロップ法、シッティングドロップ法、マイクロバッチ法などのいかなる方法を用いてもよいが、好ましくは蒸気拡散法であるハンギングドロップ法が用いられる。しかしながら通常の方法で作製したFabKの結晶はクラスター化する傾向にあるため、シーディング法により良質な単結晶を得ることが好ましい。結晶化に用いるタンパク質としては、配列番号1、配列番号2に示されるアミノ酸配列を有するHis−FabKおよびFabKが望ましく、精製の簡便さから配列番号1に示されるアミノ酸配列を有するFabKが最も望ましい。結晶化に用いる精製タンパク質の濃度は、10mg/ml程度であることが望ましい。そして、蛋白質溶液に沈殿剤、塩類、緩衝液、添加剤などを適当量加えて、結晶化を行う。 本発明のFabKの結晶化に用いられる沈殿剤としては、ポリエチレングリコールなどの水溶性高分子、イソプロパノールや2−メチル−2,4−ペンタンジオール(MPD)などの有機溶媒などが挙げられる。また、緩衝剤としては公知の緩衝剤のいずれも用いることができるが、MES(pH 5.5−7.0)を用いるのが好ましい。沈殿剤の組み合わせについては(1)8−15% ポリエチレングリコール1000、8−15% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(2)20−25% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(3)2.5−10% ポリエチレングリコール8000、2.5−10% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.0−6.5の3通りが挙げられるが、中でも(1)の組み合わせが最も再現性良く、高品質の結晶を得ることができる。この条件は当初実施した通常市販されている結晶化条件スクリーニングキットには含まれておらず、スクリーニングキットにはあまり含まれない組み合わせであるポリエチレングリコールとMPDを組み合わせた上で、塩化アンモニウムや塩化カルシウム、ジチオスレイトール等を加えた独自の条件である。また、(1)の条件で得られる結晶はミクロシーディング法により、クラスター化を抑制しなければ、高分解能でのX線結晶構造解析に利用するのは困難である。
【0015】
<<FabK結晶のX線回折強度データの収集>>
上記のようにして得られる結晶の外観、単位格子の種類・大きさなどはその結晶に固有のものであり、良質の結晶を用いれば、X線回折実験により高分解能の回折強度データを取得することができる。結晶を多価アルコールを含有する抗凍結溶液中に浸した後凍結させ、凍結状態でX線回折データを収集し、X線回折像を得る。ここで、多価アルコールとしては、スクロース、トレハロース、グリセロールなどが挙げられる。これらの中で特にグリセロールが好ましい。抗凍結溶液中の多価アルコールの濃度は、好ましくは10%〜20%である。X線を照射して回折強度データを取得するには、いかなるX線発生装置も用いることができるが、好ましくは、(財)高輝度光科学研究センターの大型放射光施設SPring−8などの第3世代の放射光施設を利用することができる。得られたX線回折強度データを処理することにより、結晶学的パラメータを特定することが可能である。例えば、配列番号1のアミノ酸配列を有する蛋白質から構成されるHis−FabKの結晶としては、空間群がP21であり、格子定数はa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°である結晶などが挙げられる。
【0016】
<<FabKの結晶構造の決定>>
上記のように得られたデータを用いてFabKの結晶構造の決定が可能となる。一般に位相決定には、分子置換法、重原子同型置換法、多波長異常分散法(MAD法)などが用
いられるが、目的蛋白質に対する類縁蛋白質の立体構造が未知であり、その立体構造を利用した分子置換法による構造解析が不可能な本発明のような場合には、重原子同型置換法またはMAD法などが用いられる。すなわち、ネイティブ結晶および重原子誘導体結晶の回折データ間の回折強度差、あるいは異なる波長間で測定した回折データの回折強度差から、電子密度を計算するための初期位相を求めることにより、立体構造を決定することが可能である。本発明では波長を変えることのできる大型放射光施設SPring−8を利用可能であったため、セレノメチオニンを導入した結晶を用いたMAD法によるX線結晶構造解析を実施し、その後の解析では実験で得られたFabKの立体構造をサーチモデルとした分子置換法による構造決定が可能であった。
【0017】
以下、実施例により本発明をさらに詳細に説明するが、本発明の技術的範囲は実施例によって限定されるものではない。また、各種ベクターの作製、蛋白質の発現等は、特に記載のない限り、Molecular Cloning,A Laboratory Manual,3rd edition(Sambrook and Russell著,Cold Spring Harbor laboratory Press刊(2001))などに記載の公知の手法に従って実施できる。
【実施例1】
【0018】
<<肺炎連鎖球菌由来FabKの大腸菌発現系の構築>>
(1)肺炎連鎖球菌R6株由来のゲノムDNAの単離
肺炎連鎖球菌(Streptococcus pneumoniae)R6株(ATCC49619)を血液寒天基礎培地(ベクトン・ディッキンソン社製)で二酸化炭素5%、37℃の条件下で18時間培養し、低速遠心によって集菌した。得られた菌体からDNeasy Tissue Kit(QIAGEN社製)を用い、添付の説明書記載の方法に従って、ゲノムDNAを調製した。
【0019】
(2)肺炎連鎖球菌R6株由来FabK発現ベクターの構築
FabKをコードするDNAを単離するため以下のプライマーを設計した。
【0020】
FabK−F.P.:5’−GGAATTCCATATGAAAACGCGTATTACAGAA−3’(配列番号5)
His−FabK−R.P.:5’−CCGCTCGAGGTCATTTCTTACAACTCCTGT−3’(配列番号6)
FabK−R.P.:5’−CCGCTCGAGTTAGTCATTTCTTACAACTCC−3’(配列番号7)
【0021】
上記(1)で単離したゲノムDNAを鋳型に、前記プライマーを用いて、Pyrobest DNA Polymerase(タカラバイオ社製)でポリメラーゼ連鎖反応(PCR)を添付の説明書に従い実施した。His−tag付のFabK(His−FabK)を作製する場合には配列番号5および配列番号6のプライマーを、タグなしのFabKを作製する場合には配列番号5および配列番号7のプライマーを使用した。増幅したDNA断片はNdeIおよびXhoIで消化した。この断片を予めNdeIおよびXhoIで切断したベクターpET−21b(+)(Novagen社製)にDNA Ligation Kit ver.2(タカラバイオ社製)を添付の説明書の方法に従い用い、サブクローニングした。得られた組換えプラスミドを大腸菌COMPETENT high DH5α(TOYOBO社製)に添付の説明書の方法に従い導入し、形質転換体を得た。形質転換体を、50μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。取得したコロニーから組換えプラスミド(His−FabK/pET−21b(+)およびFabK/pET−21b(+))を調製しDNA配列を確認し、配列番号1および2に記載のSWISSPROT
Accession番号Q8DR17に登録されている肺炎連鎖球菌R6株由来のFabKをコードすることを確認した。
【実施例2】
【0022】
<<FabK(His−FabKおよびFabK)の発現>>
実施例1の(2)で得られたHis−FabK/pET−21b(+)およびFabK/pET−21b(+)の組み換えプラスミドを大腸菌BL21(DE3)(Novagen社製)に形質転換し、得られた形質転換体を100μg/mlのアンピシリンを含むSB培地(1.2%(w/v)Bacto Tryptone、2.4%(w/v)Yeast Extract、0.5%(v/v)グリセロール、0.072M リン酸水素二カリウム、0.028M リン酸二水素カリウム)5L中で600nmにおけるO.D.が0.6−1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)で3時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。菌体をHis−FabK/pET−21b(+)の組み換え大腸菌の場合には菌体破砕バッファーA(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、5mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール、0.1mg/mL リゾチーム)、FabK/pET−21b(+)の組み換え大腸菌の場合には菌体破砕バッファーB(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール、0.1mg/mL リゾチーム)に懸濁し、氷上で30分インキュベートした。次に1分間の超音波処理を5−7回施し、細胞を破砕した。遠心分離(30分間、15000rpm)とそれに続く0.22μmのフィルターによって超音波処理の残渣を除去し、細胞抽出液を得た。
【実施例3】
【0023】
<<His−FabKの精製>>
(1)アフィニティーカラムによる精製
以下の精製操作はすべて4℃で行った。His−FabK/pET−21b(+)の組み換え大腸菌から実施例2の方法で得られた細胞抽出液をアフィニティークロマトグラフィーで精製した。アフィニティークロマトグラフィーカラムは、Ni−NTA(QIAGEN社製)を担体として用い、マニュアル記載の方法でカラム容量30mlのカラムを作製した。平衡化バッファー(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、5mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で平衡化した後、細胞抽出液を流速1mL/minでアプライし、3カラム容量の洗浄バッファー(50mM
リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、20mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で洗浄した後、溶出バッファー(50mM リン酸ナトリウム緩衝液 pH 8.0、300mM 塩化ナトリウム、200mM イミダゾール、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM β―メルカプトエタノール)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要なフラクションをまとめて回収した。
【0024】
(2)陰イオン交換カラムによる精製
上記(1)で得られたHis−FabK溶液を透析バッファー(50mM トリス塩酸緩衝液 pH 8.0、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で2時間透析した後に、陰イオン交換カラムであるPOROS HQ/20(PerSeptive Bio
systems社製)を用いて精製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラムに、流速10mL/minでHis−FabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−500mM 塩化カリウムの直線勾配(25カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要な画分をまとめ、以後の精製を実施した。
【0025】
(3)疎水性相互作用クロマトグラフィーによる精製
上記(2)で得られたHis−FabK溶液を、50mM トリス塩酸緩衝液 pH 7.5、3.5M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液と混合した後、硫酸アンモニウムの終濃度が2Mになるように調製した。調製したHis−FabK溶液を平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、2M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化した疎水性相互作用カラムであるRESOURCE PHE 6ml(アマシャムバイオサイエンス社製)に、流速5ml/minでアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の2−0M 硫酸アンモニウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、His−FabKの主要な画分をまとめ、以後の精製を実施した。
【0026】
(4)ゲルろ過カラムによる精製
上記(3)で得られたHis−FabK溶液を保持分子量30000以上の限外ろ過膜であるAmicon Ultra−4 30kDa NMWL(ミリポア社製)を用いて1mLまで濃縮し、ゲルろ過バッファー(50mM トリス塩酸緩衝液 pH 7.5、150mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload
16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1ml/minでアプライした。SDS−PAGEにより主要な画分を各々まとめ、上記記載の限外ろ過膜を用いて濃縮した。その後溶液交換のため、10mM
トリス塩酸緩衝液 pH 7.5、100mM 塩化アンモニウム、1mM ジチオスレイトール溶液で希釈し、再度濃縮する操作を3度繰り返した後、10mg/mlになるまで濃縮し、終濃度5mMになるようにβ−ニコチンアミドアデニンジヌクレオチド二ナトリウム(還元型)を添加して、結晶化サンプルとした。サンプルは液体窒素中で凍結し、−80℃で保存した。
【実施例4】
【0027】
<<FabK(タグなし)の精製>>
(1)陰イオン交換カラムによる精製
以下の精製操作はすべて4℃で行った。FabK/pET−21b(+)の組み換え大腸菌から実施例2の方法で得られた細胞抽出液を陰イオン交換カラムクロマトグラフィーで精製した。陰イオン交換カラムは、Q―Sepharose High Performance(アマシャムバイオサイエンス社製)を担体として用い、マニュアル記載の方法でカラム容量114mlのカラムを作製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラ
ムに、流速5mL/minでFabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−1M 塩化カリウムの直線勾配(5カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、以後の精製を実施した。
【0028】
(2)疎水性相互作用クロマトグラフィーによる精製
上記(1)で得られたFabK溶液を、50mM トリス塩酸緩衝液 pH 7.5、3.5M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液と混合した後、硫酸アンモニウムの終濃度が2Mになるように調製した。調製したFabK溶液を平衡化バッファー(50mM トリス塩酸緩衝液 pH 7.5、2M 硫酸アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化した疎水性相互作用カラムであるRESOURCE PHE 6ml(アマシャムバイオサイエンス社製)に、流速5ml/minでアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の2−0M
硫酸アンモニウムの直線勾配(15カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、以後の精製を実施した。
【0029】
(3)ゲルろ過カラムによる精製
上記(2)で得られたFabK溶液を保持分子量30000以上の限外ろ過膜であるAmicon Ultra−4 30kDa NMWL(ミリポア社製)を用いて1mLまで濃縮し、ゲルろ過バッファー(50mM トリス塩酸緩衝液 pH 7.5、150mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したゲルろ過カラムHiload 16/60 Superdex75 prep grade(アマシャムバイオサイエンス社製)に流速1ml/minでアプライした。SDS−PAGEにより主要な画分を各々まとめ、以後の精製を実施した。
【0030】
(4)陰イオン交換カラムによる精製
上記(3)で得られたFabK溶液を透析バッファー(50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で2時間透析した後に、陰イオン交換カラムクロマトグラフィーであるMonoQ HR10/10(アマシャムバイオサイエンス社製)で精製した。平衡化バッファー(50mM トリス塩酸緩衝液 pH
7.5、50mM 塩化アンモニウム、1mM フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール)で平衡化したカラムに、流速3mL/minでFabK溶液をアプライした。3カラム容量の平衡化バッファーで洗浄した後、50mM トリス塩酸緩衝液 pH 7.5、50mM 塩化アンモニウム、1mM
フッ化フェニルメチルスルホニル、1mM ベンズアミジン、1mM ジチオスレイトール溶液中の0−1M 塩化カリウムの直線勾配(20カラム容量)で溶出した。SDS−PAGEにより溶出画分を確認し、FabKの主要な画分をまとめ、上記記載の限外ろ過膜を用いて濃縮した。その後溶液交換のため、10mM トリス塩酸緩衝液 pH 7.5、100mM 塩化アンモニウム、1mM ジチオスレイトール溶液で希釈し、再度濃縮する操作を3度繰り返した後、10mg/mlになるまで濃縮し、終濃度5mMになるようにβ−ニコチンアミドアデニンジヌクレオチド二ナトリウム(還元型)を添加して、結晶化サンプルとした。サンプルは液体窒素中で凍結し、−80℃で保存した。
【実施例5】
【0031】
<<セレノメチオニン置換FabK(Se−His−FabKおよびSe−FabK)の発現>>
MAD法によるX線結晶構造解析に使用する重原子誘導体結晶を作製するために、メチオニンの硫黄原子が重原子であるセレンに置換されたセレノメチオニン(Se−Met)置換His−FabK(Se−His−FabK)およびFabK(Se−FabK)を作製した。Se−His−FabKおよびSe−FabKは実施例1の(2)に記載した各々の組み換えプラスミドをメチオニン要求株である大腸菌B834(DE3)(Novagen社製)に形質転換し、100μg/mLのアンピシリンを含むLB agarプレート上にて、37℃で一晩培養し、アンピシリン耐性コロニーを取得した。得られた形質転換体は100μg/mlのアンピシリンを含むLB培地100mlにて、37℃で一晩培養し、遠心分離機で集菌後、100μg/mLのアンピシリンを含む100mlのLeMaster培地(組成は表1に示す。)で懸濁し、再度、遠心分離機によって集菌し、100μg/mLのアンピシリンを含むLeMaster培地100mlに再懸濁した。この懸濁液を100μg/mLのアンピシリンを含む5LのLeMaster培地に接種し、600nmにおけるO.D.が0.6−1.0に達するまで増殖させた。終濃度1mMのイソプロピル−β−D−チオガラクトピラノシド(IPTG)で3時間誘導後、遠心分離機によって集菌し、菌体をリン酸緩衝食塩水(PBS)200mLに懸濁した後、再度、遠心分離機によって集菌し、−20℃で凍結保存した。以下、Se−His−FabKおよびSe−FabKは天然型と同様の方法で精製した。
【0032】
【表1】
【実施例6】
【0033】
<<A141S変異FabK(A141S His−FabKおよびA141S FabK)の作製>>
実施例1の(2)で作製した組み換えプラスミドHis−FabK/pET−21b(+)およびFabK/pET−21b(+)を利用して、141番目のアミノ酸残基アラニンをセリンに置換した点変異体(A141S His−FabKおよびA141S FabK)をコードする組み換えプラスミドを作製した。点変異導入にはStratagene社のQuickChange Site−Directed Mutagenesis Kitを利用して1塩基の点変異を導入した。まず、目的とする変異を中央にしてその前後に10−15塩基ずつ付加したプライマー、およびそれに相補的な下記プライマーを用意した。
FabK−Mutant−F.P.:5’−GGGATAATCGTTATTCCTGT
CGTTCCTAGTG−3’(配列番号8)
FabK−Mutant−R.P.:5’−CACTAGGAACGACAGGAATAACGATTATCCC−3’(配列番号9)
上記プライマーを用い、添付の説明書の方法に従ってA141S His−FabKおよびA141S FabK点変異体発現プラスミドを作製した。得られたプラスミドについては、目的とした塩基以外に変異が入っていないことをDNA配列解析により確認し、発現ベクターに挿入されているDNAが配列番号3および4をコードすることを確認した。作製した発現ベクターを大腸菌BL21(DE3)に形質転換し、天然型と同様の方法で、発現および精製した。
【実施例7】
【0034】
<<FabKの結晶化>>
(7−1)Free体結晶の作製
His−FabKおよびFabKの結晶化において市販のスクリーニングキットやその組み合わせ、類似蛋白質の結晶化条件等、膨大な数の結晶化条件の検討の末、X線結晶構造解析可能な結晶を取得した。
結晶が得られた沈殿剤の組み合わせとしては(1)8−15% ポリエチレングリコール1000、8−15% 2−メチル−2,4−ペンタンジオール(MPD)、0.1M
塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(2)20−25% ポリエチレングリコール1000、0.1M 塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.5−7.0、(3)2.5−10% ポリエチレングリコール8000、2.5−10% ポリエチレングリコール1000、0.1M
塩化アンモニウム、0.2M 酢酸アンモニウム、5mM ジチオスレイトール、0.1M MES pH 5.0−6.5の3通りが挙げられるが、中でも(1)の組み合わせが再現性良く、高品質の結晶を得ることができた。
MAD法によるX線結晶構造解析に利用した結晶は実施例5で精製したSe−His−FabKを用いて作製した。10mg/mlのSe−His−FabK溶液2.0μLと結晶化剤(10% ポリエチレングリコール1000、10% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)2.0μLをカバーガラス上で混合し結晶化ドロップとした。リザーバー溶液として上記結晶化剤500μLをVDXプレート(ハンプトンリサーチ社製)のウェルに分注し、ウェルの淵に高真空グリースを塗り、カバーガラスをドロップが内側になるように被せ密閉した。プレートを4℃の恒温槽内に静置すると数日でクラスター結晶が得られた。得られたクラスター結晶を用いてミクロシーディング法により、再度結晶化することでX線結晶構造解析可能な結晶が作製可能であった。得られたSe−His−FabK結晶の顕微鏡写真を図1に示す。この結晶は空間群がP21であり、格子定数がa=50.26Å、b=126.69Å、c=53.63Å、α=γ=90°、β=112.46°の結晶である。
また、結晶化条件(1)において実施例3で精製したHis−FabKを使用した場合にも同様の結晶が得られ、空間群がP21であり、格子定数がa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°であった。
さらに結晶化条件(1)では、His−FabKを利用した場合、空間群がP212121であり、格子定数がa=58.92Å、b=73.89Å、c=126.50Å、α=β=γ=90°の結晶も作製可能であった。
また、A141S His−FabKを使用した場合、結晶化条件(1)においては空間群がC2221であり、格子定数がa=58.99Å、b=84.60Å、c=126.17Å、α=β=γ=90°の結晶が得られた。
その他、結晶化条件(2)においてSe−FabKを使用した場合、空間群がP212121であり、格子定数がa=86.99Å、b=160.41Å、c=183.25Å
、α=β=γ=90°の結晶が得られ、結晶化条件(3)においてはHis−FabKを使用した場合、空間群がI222であり、格子定数がa=117.20Å、b=186.58Å、c=214.79Å、α=β=γ=90°の結晶が得られ、Se−His−FabKを使用した場合には空間群がI222であり、格子定数がa=118.38Å、b=190.30Å、c=213.04Å、α=β=γ=90°の結晶が得られた。上記記載のすべての結晶はハンギングドロップ蒸気拡散法、シッティングドロップ蒸気拡散法のいずれの方法で結晶化した場合でも、同様の結晶が得られた。
【0035】
(7−2)化合物複合体結晶の作製
当社で見出した2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸ナトリウム塩(特願2006−21372記載の実施例31化合物)は、FabKの酵素阻害活性を有する化合物であり、当社研究所で合成し、使用した。実施例3で精製したHis−FabKを用いて結晶化条件(1)で作製した結晶を、50mM 2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸ナトリウム塩含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)中に移し、4℃で一晩反応させることにより、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体結晶を調製した。
【実施例8】
【0036】
<<Se−His−FabKのX線回折強度データの収集および結晶構造の決定>>
実施例7(7−1)の結晶化条件(1)で得られたSe−His−FabK結晶を、15%グリセロール含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.5)に移し、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しCCDカメラを検出装置としてX線回折データを振動法にて測定した。このとき、セレンの異常散乱効果を用いて構造解析を行うために、波長は0.97932、0.97888および0.90000Åの3波長、即ち、吸収端のエッジ、ピーク、リモートの3カ所でMADデータを収集した。振動角は1°/フレームであった。各データは回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、分解能2.0Åで処理し、回折強度を数値化し、結晶構造因子を求めた。
【0037】
その結果、空間群がP21であり、格子定数がa=50.26Å、b=126.69Å、c=53.63Å、α=γ=90°、β=112.46°の結晶であることがわかった。得られたデータのCompletenessはエッジ、ピーク、リモートの測定波長でそれぞれ97.2%、96.1%、95.4%、Rmergeはそれぞれ6.2%、6.4%、8.6%であった。結晶の空間群、格子定数、Se−His−FabKの分子量から、結晶の非対称単位にSe−His−FabK分子が2分子存在していることが明らかになった。これらのデータ収集の結果を下記の表2にまとめた。
【0038】
【表2】
【0039】
次に、プログラムSOLVE(Terwillinger, T.C. & Berendzen,J. ActaCrystallogr. D 55,849−861(1999))を用いてセレン原子の位置を決定し初期位相を計算した。位相の改良及び初期モデルの構築はプログラムRESOLVE(Terwilliger, T.C. Acta Crystallogr. D56,965−972(2000))を用いた。次いで、プログラムTurbo Frodo(AFMB−CNRS製)、プログラムRefmac5(Collaborative Computational Project Number 4(ccp4))によるモデルの構築および修正を繰り返し実施し、Se−His−FabKの結晶構造を決定した。構造精密化は分解能2.2Åで実施した。決定した結晶構造の正確さの指標であるR因子は、0.220であった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.282であった。また、決定したFabKの非対称単位中の構造を図2に示した。
【実施例9】
【0040】
<<A141S His−FabKのX線回折強度データの収集および結晶構造の決定>>
実施例7(7−1)の結晶化条件(1)で得られたA141S His−FabK結晶を、15%グリセロール含有の安定化母液(12.5% ポリエチレングリコール1000、12.5% MPD、0.1M 塩化アンモニウム、0.2M 塩化カルシウム、5mM ジチオスレイトール、0.1M MES pH 6.0)に移し、−170℃の窒素ガス気流にて急速凍結した。これを(財)高輝度光科学研究センターの大型放射光施設SPring−8の創薬産業ビームラインBL32B2を利用しCCDカメラを検出装置としてX線回折データを振動法にて測定した。X線の波長は1Å、振動角は1°/フレームであった。次に、回折強度データ処理プログラムCrystalClear(リガク社製)を使用して、A141S His−FabK結晶を分解能1.8Åで処理し、回折強度を数値化し、結晶構造因子を求めた。
【0041】
その結果、A141S His−FabK結晶は、空間群がC2221であり、格子定数がa=58.99Å、b=84.60Å、c=126.17Å、α=β=γ=90°であった。得られたデータのCompletenessは99.8%、Rmergeは6.6%であった。プログラムMolrep(CCP4)を用いた分子置換法により位相を決定し、プログラムRefmac5(CCP4)を用いて構造を精密化した結果、R因子は、0.244となった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.282となり、構造決定に成功した。また、点変異部分の電子密度図とモデルを図3に示した。
【実施例10】
【0042】
<<化合物複合体のX線回折強度データの収集および結晶構造の決定>>
実施例7(7−2)で得られた複合体結晶を、実施例9と同様の方法でX線回折測定およびデータ処理を実施し、分解能2.3Åで、回折強度を数値化し、結晶構造因子を求めた。
【0043】
その結果、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体結晶は空間群がP21であり、格子定数がa=50.61Å、b=125.95Å、c=53.01Å、α=γ=90°、β=112.78°であった。得られたデータのCompletenessは94.0%、Rmergeは11.0%であった。プログラムMolrep(CCP4)を用いた分子置換法により位相を決定し、プログラムRefmac5(CCP4)を用いて構造を精密化した結果、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸に相当する電子密度が観測された(図4)。決定した結晶構造の正確さの指標であるR因子は、0.226となった。さらに、精密化の段階で計算に入れなかった全反射の5%に相当する構造因子から計算されるRfree因子は0.316となり、構造決定に成功した。
【産業上の利用可能性】
【0044】
本発明のFabK結晶は、X線結晶構造解析可能な新規な結晶であり、解析に成功した立体構造情報を利用することによりFabKの特異的阻害剤の効率的な設計等が可能となるため、新規作用機序の新薬創出に活用でき、産業上極めて有用である。
【図面の簡単な説明】
【0045】
【図1】本発明のSe−His−FabK結晶の顕微鏡写真である。
【図2】決定したFabKの非対称単位中の構造をリボン図で示す。
【図3】点変異部分(A141S)の電子密度図とモデルを示す。
【図4】His−FabK/2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸複合体における薬物結合部位近傍の図である。蛋白質をCαモデルで表示し、2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸を除いたモデルから計算したFo−Fc電子密度図および2−(4−(2−((3−(5−(ピリジン−2−イルチオ)チアゾール−2−イル)ウレイド)メチル)−1H−イミダゾール−4−イル)フェノキシ)酢酸を重ねて表示している。
【特許請求の範囲】
【請求項1】
エノイル―アシルキャリアプロテインレダクターゼ(FabK)の結晶。
【請求項2】
ストレプトコッカス属(Streptococcus)由来である請求項1記載の結晶。
【請求項3】
肺炎連鎖球菌(Streptococcus pneumoniae)由来である請求項1または2記載の結晶。
【請求項4】
空間群がP21である請求項1〜3のいずれか一項に記載の結晶。
【請求項5】
空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である請求項1〜3のいずれか一項に記載の結晶。
【請求項6】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21である請求項1〜3のいずれか一項に記載の結晶。
【請求項7】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である請求項1〜3のいずれか一項に記載の結晶。
【請求項8】
空間群がP212121である請求項1〜3のいずれか一項に記載の結晶。
【請求項9】
空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項10】
空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項11】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121である請求項1〜3のいずれか一項に記載の結晶。
【請求項12】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項13】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項14】
空間群がI222である請求項1〜3のいずれか一項に記載の結晶。
【請求項15】
空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項16】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222である請求項1〜3のいずれか一項に記載の結晶。
【請求項17】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項18】
空間群がC2221である請求項1〜3のいずれか一項に記載の結晶。
【請求項19】
空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項20】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221である請求項1〜3のいずれか一項に記載の結晶。
【請求項21】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構
成され、空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項22】
請求項1〜21のいずれか一項に記載の結晶であって、該結晶を構成する蛋白質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
【請求項23】
請求項1〜22のいずれか一項に記載の結晶であって、該結晶がリガンドと複合体を形成している結晶。
【請求項1】
エノイル―アシルキャリアプロテインレダクターゼ(FabK)の結晶。
【請求項2】
ストレプトコッカス属(Streptococcus)由来である請求項1記載の結晶。
【請求項3】
肺炎連鎖球菌(Streptococcus pneumoniae)由来である請求項1または2記載の結晶。
【請求項4】
空間群がP21である請求項1〜3のいずれか一項に記載の結晶。
【請求項5】
空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である請求項1〜3のいずれか一項に記載の結晶。
【請求項6】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21である請求項1〜3のいずれか一項に記載の結晶。
【請求項7】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP21であり、格子定数がa=50±5Å、b=126±13Å、c=53±5Å、α=γ=90°、β=113±11°である請求項1〜3のいずれか一項に記載の結晶。
【請求項8】
空間群がP212121である請求項1〜3のいずれか一項に記載の結晶。
【請求項9】
空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項10】
空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項11】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121である請求項1〜3のいずれか一項に記載の結晶。
【請求項12】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=59±6Å、b=74±7Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項13】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がP212121であり、格子定数がa=87±9Å、b=160±16Å、c=183±18Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項14】
空間群がI222である請求項1〜3のいずれか一項に記載の結晶。
【請求項15】
空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項16】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222である請求項1〜3のいずれか一項に記載の結晶。
【請求項17】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がI222であり、格子定数がa=117±12Å、b=187±19Å、c=215±22Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項18】
空間群がC2221である請求項1〜3のいずれか一項に記載の結晶。
【請求項19】
空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項20】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構成され、空間群がC2221である請求項1〜3のいずれか一項に記載の結晶。
【請求項21】
配列番号1〜4のいずれかに示されるアミノ酸配列を有する蛋白質から構
成され、空間群がC2221であり、格子定数がa=59±6Å、b=85±9Å、c=126±13Å、α=β=γ=90°である請求項1〜3のいずれか一項に記載の結晶。
【請求項22】
請求項1〜21のいずれか一項に記載の結晶であって、該結晶を構成する蛋白質中の少なくとも1つのメチオニン残基がセレノメチオニンに置換されている結晶。
【請求項23】
請求項1〜22のいずれか一項に記載の結晶であって、該結晶がリガンドと複合体を形成している結晶。
【図1】


【図2】
【図3】
【図4】

【図2】
【図3】
【図4】
【公開番号】特開2009−89602(P2009−89602A)
【公開日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願番号】特願2006−21384(P2006−21384)
【出願日】平成18年1月30日(2006.1.30)
【出願人】(000006091)明治製菓株式会社 (180)
【Fターム(参考)】
【公開日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願日】平成18年1月30日(2006.1.30)
【出願人】(000006091)明治製菓株式会社 (180)
【Fターム(参考)】
[ Back to top ]