
エプスタイン−バールウイルス感染細胞を特異的に攻撃する細胞傷害性T細胞エピトープペプチド及びその用途
【課題】 本発明は、エプスタイン-バールウイルス(以下EBVと記載する)に特異的な細胞傷害性T細胞エピトープペプチド、該ペプチドを用いたEBVの感染および同ウイルス陽性の癌を治療又は予防するワクチン、EBVに対する受動免疫療法剤、およびEBVに特異的な細胞傷害性T細胞の定量方法の提供を目的とする。
【解決手段】 本発明者らは、EBV関連蛋白質であるLMP1およびEBNA1のmRNAを抗原提示細胞に導入し、該細胞がEBVに特異的な細胞傷害性T細胞を誘導することを明らかにした。また、該細胞傷害性T細胞は、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子に提示されているエピトープペプチドを認識し、さらにEBVが感染しているB細胞の成長を阻害し、EBVが感染しているNKリンパ腫またはNK細胞を溶解することを明らかにした。
【解決手段】 本発明者らは、EBV関連蛋白質であるLMP1およびEBNA1のmRNAを抗原提示細胞に導入し、該細胞がEBVに特異的な細胞傷害性T細胞を誘導することを明らかにした。また、該細胞傷害性T細胞は、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子に提示されているエピトープペプチドを認識し、さらにEBVが感染しているB細胞の成長を阻害し、EBVが感染しているNKリンパ腫またはNK細胞を溶解することを明らかにした。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド、該ペプチドを用いたエプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するワクチン、エプスタイン-バールウイルスに対する受動免疫療法剤に関する。
【背景技術】
【0002】
エプスタイン-バールウイルス(記憶B細胞に長期間潜伏するヘルペスγウイルス、以下EBVと標記することもある)は、多くの悪性腫瘍に関連している。例えば、バーキットリンパ腫(BL)、ホジキン病(HD)、鼻咽頭癌(NPC)、または移植後リンパ増殖性疾患(PTLD)などである(非特許文献1)。潜伏感染では、ウイルス由来のタンパク質の発現は抑制されている。全てのEBV陽性の悪性細胞は、以下の3つの潜在タイプのうち一つを示し、発現されたEBV抗原のパターンによって互いに区別される(非特許文献2)。
潜伏感染様式1:バーキットリンパ腫では、EBVの核抗原(EBNA)1だけが発現される。
潜伏感染様式2:ホジキン病や鼻咽頭癌では、潜在膜蛋白1(以後LMP1と標記する)、LMP2、およびEBNA1が発現される。
潜伏感染様式3:移植後リンパ増殖性疾患では、EBVの潜在蛋白即ちEBNA1、2、3A、3B、3C、leader protein、LMP1およびLMP2が全て発現される。
【0003】
EBV関連悪性腫瘍の免疫治療方法に対する関心は増え続けており、インビトロで活性化されたEBV特異的な細胞傷害性T細胞(以後CTLと標記することもある)を用いた養子免疫療法は、造血幹細胞移植や臓器移植後におけるEBV関連リンパ増殖性疾患の予防や治療に効果があることが証明されている(非特許文献3〜7)。ホジキン病(非特許文献8)、鼻咽頭癌(非特許文献9)等のEBVが関連している悪性腫瘍への同様の戦略の応用は、何人かの患者で有効であることが報告されている。しかしながら、それらの研究で使用された、リンパ芽球細胞株(以後LCLと標記する)によって活性化されたCTLは主にEBNA3A、EBNA3BやEBNA3Cを標的としており、これらの抗原はホジキン病や鼻咽頭癌のような悪性腫瘍では発現されていない。LCLに活性化されたCTLの一部はLMP2由来のペプチドを認識し、患者の免疫治療効果に貢献した可能性がある(非特許文献8,9)。しかし、LMP1ペプチドを標的とするT細胞は非常にまれであり、それはLMP1ペプチド特異的なCTL先駆細胞の低い頻度を反映している(非特許文献10)。サブドミナントなEBV抗原に特異的なT細胞を選択的に活性化させるために、LinらはLMP2由来ペプチドでパルスされた単球由来の樹状細胞(DC)を利用してNPC患者を免疫した(非特許文献11)。また、LMP1特異的なCTLを活性化するためにいくつかの方法が報告されている。KhannaらはHLA-A2拘束性のLMP1エピトープと、該ペプチドでパルスした抗原提示細胞(APC)を用いたCTLの誘導について最初に報告している(非特許文献10)。また、彼らは複数のLMP1エピトープをコードする複製能力のないアデノウイルスや組み換えワクシニアウイルスを利用して、HLA-A2トランスジェニックマウスを免疫することに成功し、LMP1遺伝子導入細胞の成長を阻害できたことを報告している(非特許文献12,13)。Gottschalkらは、N末端を切断された毒性のない変種LMP1を発現している組み換えアデノウイルスを感染させたDCを利用して、ポリクローナルなLMP1特異的CTLの誘導を報告している(非特許文献14)。
【0004】
EBVが関連している悪性腫瘍の一つのカテゴリーとして、NK/T細胞におけるEBV感染が挙げられる(非特許文献1)。慢性活動性EBV感染(CAEBV)もEBVが主にNK/T細胞に感染し、生命に危険を及ぼすリンパ球増殖を引き起こす疾患である(非特許文献15)。EBV陽性のNK/T細胞悪性腫瘍は潜在的なCTLの標的としてEBNA1とLMP1を発現している(非特許文献15〜17)。
【0005】
LMP1は抗アポトーシス遺伝子のアップレギュレーションを通じて細胞の生存を促進する膜貫通癌蛋白質である(非特許文献2)。LMP1の発現はヒトBリンパ球の成長変化に不可欠であり、EBVが感染したヒト単核細胞の増殖に必要である(非特許文献2)。LMP1はまた、マウス細胞株BALB/c3T3やB細胞リンパ腫一般において癌原性の形質転換を誘導することが知られている。さらに、LMP1の発現はEBV感染NK細胞の増殖能に重要である可能性がある(非特許文献18)。しかしながら、NK/T細胞がLMP1を処理して、HLA拘束性のエピトープを産生できることは証明されていない。
【0006】
一方、EBNA1は、EBV-形質転換細胞におけるウイルスプラスミドの維持と複製に必要である(非特許文献19)。EBNA1はEBVが関連している全ての腫瘍で発現されることから、免疫治療のための魅力的な標的である。しかしながら、CD8+CTLの応答は潜伏感染抗原のうちEBNA3A,3Bおよび3Cに優先的に向けられ、EBNA1はCTLに認識されず、免疫学的には検出できないものと信じられていた(非特許文献20〜23)。EBNA1中のグリシンアラニン反復配列(GAr)はCTL認識のための抗原処理を妨げることがわかっている(非特許文献24)。このGArの存在は、MHCクラスIエピトープを産生する主要な触媒機構である、プロテアソームによる処理を妨げることが判明している(非特許文献25、26)。さらに、全く同じドメインが、EBNA1 mRNAの翻訳を妨げることが明らかとなっている(非特許文献27)。
【0007】
EBV特異的なCD4+T細胞の応答が調べられ、EBNA1特異的CD4+T細胞応答は主にヘルパーT細胞タイプ1で見られ、直接的にEBVが感染した細胞を認識することがわかった。数個のMHCクラスII拘束性のEBNA1エピトープが同定され(非特許文献28〜32)、これらのことから、EBNA1特異的CD4+T細胞が生体内で腫瘍の成長制御の役割を担っている可能性が示唆された。また、最近の研究では、驚くべきことに、EBNA1特異的CD8+CTLが、EBVが感染しているリンパ芽球様細胞(LCLs)を穏やかに溶解し、生体内でのLCLの成長を抑制されることが示されている(非特許文献33〜35)。しかしながら、CD8+CTLを活性化させる、EBNA1エピトープについてはこれまで少数が同定されたのみである。
【0008】
極少数の前駆細胞からCTLを誘導させる試みが、腫瘍関連抗原を標的にした免疫治療を望んでいる臨床医や免疫学者により行われてきた。特定の抗原をコードし、インビトロにおいて転写されたmRNAを導入した抗原提示細胞は、癌関連抗原または免疫寛容を超えて自己抗原に特異的なCTLを誘導する性質を持つ(非特許文献36〜40)。上記の手法には、以下の利点がある。
1)ベクターの主要な配列に対する免疫性の完全な欠失
2)in vitroでの転写による再現性の高い収率
3)エレクトロポレーションを利用した遺伝子導入の効率が高いこと
【0009】
すなわち、エプスタイン-バールウイルス抗原(エピトープ)のmRNAが導入された抗原提示細胞は、エプスタイン-バールウイルス特異的なCTLを誘導する上で適切な方法だと考えられる。上記の方法を実現するために、エプスタイン-バールウイルスに特異的なCTLエピトープペプチドの同定が求められてきた。
【0010】
なお、本出願の発明に関連する先行技術文献情報を以下に示す。
【先行技術文献】
【非特許文献】
【0011】
【非特許文献1】Babcock GJ., et al., Immunity, 13,497-506 (2000)
【非特許文献2】Rickinson AB., et al., In: Knipe DM, Howley PM, eds. Fields Virology (ed Fourth Edition), Philadelphia, Lippincott Williams & Wilkins, 2575-2628 (2001)
【非特許文献3】Rooney, CM., et al., Lancet, 345, 9-13 (1995)
【非特許文献4】Heslop, HE., et al., Nat Med., 2, 551-555 (1996)
【非特許文献5】Rooney, CM., et al., Blood, 92, 1549-1555 (1998)
【非特許文献6】Khanna, R., et al., Proc Natl Acad Sci U S A, 96, 10391-10396 (1999)
【非特許文献7】Comoli, P., et al., Blood, 99, 2592- 2598 (2002)
【非特許文献8】Bollard, CM., et al., J Exp Med, 200, 1623-1633 (2004)
【非特許文献9】Straathof, KC., et al., Blood 105, 1898-1904 (2005)
【非特許文献10】Khanna, R., et al., Eur J Immunol, 28, 451-458 (1998)
【非特許文献11】Lin, CL., et al., Cancer Res., 62, 6952-6958 (2002)
【非特許文献12】Duraiswamy, J., et al., Blood, 101, 3150-3156 (2003)
【非特許文献13】Duraiswamy, J., et al., Cancer Res., 64, 1483-1489 (2004)
【非特許文献14】Gottschalk, S., et al., Blood, 101, 1905-1912 (2003)
【非特許文献15】Kimura, H., et al., Blood, 98, 280-286 (2001)
【非特許文献16】Nagata, H., et al., Blood, 97, 708-713 (2001)
【非特許文献17】Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003)
【非特許文献18】Demachi, A., et al., Microbiol Immunol., 47, 543-52 (2003)
【非特許文献19】Kieff E., et al., In: Knipe DM, Howley PM, eds. Fields Virology (ed Fourth Edition) , Philadelphia, Lippincott Williams & Wilkins, 2511-2574 (2001)
【非特許文献20】Khanna R., et al., J Exp Med., 176, 169-176 (1992)
【非特許文献21】Murray RJ., et al., J Exp Med., 176, 157-168 (1992)
【非特許文献22】Steven NM., et al., J Exp Med., 184, 1801-1813 (1996)
【非特許文献23】Callan MF., et al., J Exp Med., 187, 1395-1402 (1998)
【非特許文献24】Levitskaya J., et al., Nature, 375, 685-688 (1995)
【非特許文献25】Blake N., et al., Immunity, 7, 791-802 (1997)
【非特許文献26】Levitskaya J., et al., Proc Natl Acad Sci U S A, 94, 12616-12621 (1997)
【非特許文献27】Yin Y., et al., Science, 301, 1371-1374 (2003)
【非特許文献28】Khanna R., et al., Virology, 214, 633-637 (1995)
【非特許文献29】Leen A., et al., J Virol., 75, 8649-8659 (2001)
【非特許文献30】Paludan C., et al., J Immunol., 169, 1593-1603 (2002)
【非特許文献31】Voo KS., et al., Cancer Res., 62, 7195-7199 (2002)
【非特許文献32】Kruger S., et al., J Immunother, 26, 212-221 (2003)
【非特許文献33】Lee SP., et al., J Exp Med., 199, 1409-1420 (2004)
【非特許文献34】Tellam J., et al., J Exp Med., 199, 1421-1431 (2004)
【非特許文献35】Voo KS., et al., J Exp Med., 199, 459-470 (2004)
【非特許文献36】Kuzushima, K., et al., Blood, 94, 3094-3100 (1999)
【非特許文献37】Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998)
【非特許文献38】Akatsuka, Y., et al., Tissue Antigens, 59, 502-511 (2002)
【非特許文献39】Kondo, E., et al., J Immunol., 169, 2164-2171 (2002)
【非特許文献40】Dauer, M., et al., J Immunol., 170, 4069-4076 (2003)
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明はこのような状況に鑑みてなされたものであり、その目的は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド、該ペプチドを用いたエプスタイン-バールウイルスの感染を治療又は予防するワクチン、エプスタイン-バールウイルスに対する受動免疫療法剤、およびエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明者らは、上記の課題を解決するために、エプスタイン-バールウイルス関連蛋白質であるLMP1およびEBNA1のmRNAを抗原提示細胞に導入し、該抗原提示細胞がエプスタイン-バールウイルスに特異的な細胞傷害性T細胞(CTL)を誘導する能力があるかを検討した。
【0014】
その結果、エプスタイン-バールウイルス関連蛋白質を発現させた抗原提示細胞は、健常人由来のCTLを刺激し、該CTLはEBVが感染しているB細胞の成長を阻害し、EBVが感染しているNKリンパ腫またはNK細胞を溶解することが明らかとなった。さらに、該CTLは、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子に提示されているエピトープペプチドを認識することが明らかとなった。
【0015】
即ち、本発明者らは、エプスタイン-バールウイルスに特異的なCTLエピトープペプチドを同定することに成功し、これにより本発明を完成するに至った。
【0016】
本発明は、より具体的には以下の(1)〜(16)を提供するものである。
(1) エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド。
(2) エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むものである、(1)に記載のペプチド。
(3) 配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、(1)に記載のペプチド。
(4) HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子の拘束性抗原ペプチドであって、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、(1)〜(3)のいずれかに記載のペプチド。
(5) (1)〜(4)のいずれかに記載のペプチドをコードする核酸。
(6) (1)〜(4)のいずれかに記載のペプチドを有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(7) (5)に記載の核酸を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(8) (1)〜(4)のいずれかに記載のペプチドをHLAに提示した抗原提示細胞を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(9) (1)〜(4)のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
(10) (1)〜(4)のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
(11) (1)〜(4)のいずれかに記載のペプチドを用いて細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導方法。
(12) (1)〜(4)のいずれかに記載のペプチドと末梢血単核球を、血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的細胞傷害性T細胞を誘導する、(11)に記載の誘導方法。
(13) (1)〜(4)のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
(14) (1)〜(4)のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
(15) (1)〜(4)のいずれかに記載のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
(16) (1)〜(4)のいずれかに記載のペプチドから主要組織適合性抗原複合体-テトラマーを調製し、主要組織適合性抗原複合体-テトラマーと末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【発明の効果】
【0017】
本発明により、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドを同定することに成功した。該ペプチドは、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞(CTL)を効率的に誘導し得る機能を有する。従って該ペプチド及び該ペプチドをコードする核酸は、エプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するためのワクチン(能動免疫療法剤)として有用である。本発明で提供されるエピトープペプチドは、ワクチンとして用いる事で、エプスタイン-バールウイルス特異的CTLを生体内で誘導し、エプスタイン-バールウイルス感染に対して免疫力を保持させることができる。生体外において末梢血等の生体試料に対してもエプスタイン-バールウイルスを感染させる事無く、安全かつ効率的にエプスタイン-バールウイルス特異的CTLを人為的に誘導増殖させ細胞免疫療法に用いる事ができる。さらに、エプスタイン-バールウイルスに対する免疫力の有無の診断に用いる事ができ、エプスタイン-バールウイルス感染症に対して有効な治療法と診断方法を提供する。
【0018】
また、本発明のこれらのエピトープペプチドによって誘導されるCTLは、エプスタイン-バールウイルス感染細胞を特異的に溶解し、かつ該細胞の成長を阻害する機能を有し、受動免疫療法剤の成分として非常に有用である。
【0019】
さらに、該エピトープペプチドを用いることにより、エプスタイン-バールウイルスに特異的なCTLを定量することが可能である。エプスタイン-バールウイルスに特異的なCTLが、ハイリスクの患者の末梢血に存在するか否かを知ることは、抗ウイルス剤や免疫抑制剤の適正な使用を含め、これらの感染症管理の上で重要な情報である。
【図面の簡単な説明】
【0020】
【図1】樹状細胞またはCD40-B細胞へのΔLMP1mRNAの導入、およびΔLMP1の発現、CTLの誘導を示す図である。A:樹状細胞(DC)およびCD40-B細胞(CD40-B)において、ΔLMP1-mRNAを導入した後、フローサイトメトリーでΔLMP1の発現を解析した結果を示す図である。点線が遺伝子を導入しなかった細胞を示し、実線はΔLMP1を導入した細胞を示す。B:末梢血由来CD8+T細胞を、ΔLMP1-mRNA導入し、放射線を当てた自己の抗原提示細胞で3回刺激した後、ΔLMP1-mRNA導入あるいは非遺伝子導入CD40-B細胞を抗原提示細胞として用いたELISPOTアッセイを行った結果を示す図である。データはCD8+T細胞500個あたりのスポットの数を示す。C:HLA完全ミスマッチのLCL細胞に、ドナー由来の各HLA遺伝子を導入した6種類の抗原提示細胞を作成した。CTLクローンH7を刺激し、IFNγの産生をELISPOT法で測定した結果を示す図である。1ウェル当たり1,000個のH7細胞を入れた。
【図2】CTLクローンH7が認識するLMP1エピトープペプチドを示す図である。A:C-端末を削った一連のΔLMP1-mRNAを、インビトロ転写で作製した。各mRNAのスタートコドンとして、アミノ酸位置44番にあるメチオニンを利用した。C-端末を削った各ΔLMP1-mRNAを導入したCD40-B細胞を、ELISPOTアッセイにおける抗原提示細胞として使用した。白枠で示されたmRNA断片はH7細胞に認識されたが、黒枠で示されたmRNA断片はH7細胞に認識されなかった。B:一連のC-端末およびN-端末を削った断片はPCRによって増幅されて、pcDNA3.1(+)ベクターに導入された。導入したDNAによりコードされるアミノ酸配列が示されている。各プラスミドを導入したA0206-293T細胞に対する、H7細胞の認識をELISPOT法(ウェル当たりのH7細胞1,000個)により測定し、IFNγのスポット数によって以下の二つのパターンに分類した。(+)IFNγスポット数50以上;(-)IFNγスポット数10未満。C:各遺伝子を導入したA0206-293T細胞により刺激されたH7細胞の、IFNγスポット数を示す図である。各バーはH7細胞1,000個あたりのスポットの数を表す。D:様々な濃度の合成のペプチドでパルスされたA0206-293T細胞を用いて、ELISPOT測定を行った結果を示す図である。データはH7細胞500個あたりのスポットの数を示す。
【図3】LMP1エピトープ処理におけるip-LMP7の重要性を検討した結果を示す図である。A:コントロールsiRNA、ip-LMP2 siRNAまたはip-LMP7siRNAを、レトロウイルスを用いて、自己のLCL細胞に導入した。細胞を14日間プロマイシンで選別し、ウェスターンブロット解析を行った:ip-LMP2(上側のパネル);ip-LMP7(下側パネル)。B:ip-LMP2やip-LMP7遺伝子の発現を抑制した自己のLCLを用いたELISPOT測定の結果を示す図である。H7のIFNγスポットの産生を評価した。各バーはH7細胞5,000個あたりのスポット数を表す。
【図4】LMP1特異的CTLクローンH7の細胞傷害活性を示す図である。A:標的細胞として自己のA*0206を共有する細胞と、完全にHLAがミスマッチしているLCL細胞を用いた、16時間CTLアッセイの結果を示す図である。B:ΔLMP1あるいはEGFPを導入したLCLを標的細胞にした4時間CTLアッセイの結果を示す図である。各バーは3ウェルの細胞傷害活性の平均を示す。
【図5】CTLはEBV陽性NK細胞株を特異的に溶解する。A:H7 CTLクローンのEBV陽性NK細胞株に対する細胞傷害活性を、16時間CTLアッセイで計測した結果を示す図である。2つのHLA-A*0206陽性NK細胞株(SNK-6とSNK-10)、及び1つのHLA-A*0206陰性NK細胞株(HANK-1)のデータをそれぞれ、○、△及び□で示す。100nMのエピトープペプチドをパルスしたSNK-6細胞を使った4時間CTLアッセイで測定された細胞傷害活性を、●で示す。B:HLA-A*0206(◇)とA*2402(*)遺伝子導入HANK-1細胞を用いた、16時間CTLアッセイの結果を示す図である。
【図6】インビトロで転写された全長EBNA1 mRNAにより形質転換された細胞中でのEBNA1の発現を示す図である。A:インビトロで転写された全長EBNA1 mRNAを、EBNA1-cDNAプラスミドから作成した。EBNA1 mRNAをエチジウムブロマイドで染色し、その後ゲル電気泳動によって確認した。B:EBNA1-mRNAを導入したCD40-B細胞におけるEBNA1タンパク質の発現を示す図である。CD40-B細胞に全長EBNA1 mRNAをエレクトロポレーションによって導入し、EBNA1タンパク質の細胞内染色を行い、フローサイトメトリーで確認した。
【図7】EBNA1-mRNAを導入した樹状細胞を用いた培養液中における抗EBNA1−T細胞の存在を示す図である。健康なドナーからのCD8+T細胞を、インビトロで転写されたEBNA1 mRNAで形質転換した自己の樹状細胞で刺激した。一週間間隔で三回刺激した後、限界希釈法によって2つの陽性となった培地から、ポリクローナルなCD8+T細胞をクローン化した。次に、確立したクローンのB5とC6が、EBNA1-mRNAで形質転換した自己のCD40-B細胞、及び自己のLCLによって認識されるかどうかをELISPOTアッセイによって検討した。5,000個のCTLが各ウェルに蒔かれた。2つの実験のうち代表的な1つのデータを示した。
【図8】EBNA1特異的CTLクローンに提示されているHLA分子の同定の結果を示す図である。A:CTLクローンC6に対する拘束分子として、HLA-B*3501分子が機能することを示す図である。B:CTLクローンB5対する拘束分子として、HLA-Cw*0303とCw*0304が機能することを示す図である。自己及び同種のLCLを、B5またはC6クローンがIFNγスポットを生産するための抗原提示細胞として使用した。各LCLを、CTL(5x103)と共に20時間培養した。各バーは2つのウェルにおけるスポットの平均数を表わす。
【図9】EBNA1特異的CTLクローンによって認識されるオーバーラップしたペプチドの同定の結果を示す図である。GArドメインを除いたEBNA1タンパク質の全てのアミノ酸配列を網羅する、オーバーラップする20残基のペプチドのセット(各10μg/ml)で、自己のCD40-B細胞(1x105/ウエル)をパルスし、5x102のCTLクローンB5若しくはC6と共培養した。IFNγを産生するスポットを、ELISPOTアッセイで測定した。
【図10A−B】EBNA1特異的CTLクローンによって認識される最適のEBNA1抗原ペプチドの同定の結果を示す図である。A:HLA-B*3501拘束性のクローンC6によって認識される、オーバーラップするペプチドのアミノ酸配列を示す図である。既知のエピトープHPVGEADYFEY(配列番号:28)を、太字とアンダーラインで示す。配列の下の数字はEBNA1タンパク質におけるアミノ酸の番号を示す。B:クローンB5によって認識される2つの連続した、オーバーラップするペプチドアミノ酸配列および最適のエピトープ配列を示す図である。#38と#39ペプチドの間でオーバーラップする配列を、アンダーラインで示す。矢印はプログラムSYFPEITHIによって予測されたHLA-Cw*0303が固定するための第一かつ補助のアンカーを示す。配列の下の数字はEBNA1タンパク質におけるアミノ酸位置の数字を示す。
【図10C】EBNA1から誘導された合成のペプチドの力価測定の結果を示す図である。自己のCD40-B細胞を、合成ペプチド507-526(#39 20残基)、507-517(11残基)、508-517(10残基)、および509-517(9残基)の10倍ごとの連続希釈液と、1時間インキュベートした。次いでCTLクローンB5(200細胞/ウェル)を加えて、20時間培養した。各記号は2ウェルでの観察されたスポットの平均の数を示す。
【図11】HLA-Cw*0303とCw*0304のテトラマーの、B5 CTLクローンへの特異的結合を示す図である。HLA-Cw*0303-拘束性のEBNA1特異的CTLクローンB5は、PE標識したHLA-Cw*0303-FVYGGSKTSL(配列番号:3)、HLA-Cw*0303-VFVYGGSKTSL(配列番号:2)若しくはHLA-Cw*0304―FVYGGSKTSL(配列番号:3)テトラマー複合体及びFITC標識した抗CD8抗体で染色し、フローサイトメトリーで分析した。
【図12】EBNA1特異的CTLクローンの、HLAが一致するLCLへのインビトロでの増殖阻害を示す図である。標的となるLCL(2×104)を、丸底96ウェルのプレートの三つのウェルにおいて、EBNA1特異的CTLクローン(1×104)または培地のみ(陰性コントロール)と培養し、4週間後に細胞の成長を評価した。増殖した細胞がLCLであることは、CD19の発現で確認した。2つの実験のうちの代表的な1つのデータを示す。
【発明を実施するための形態】
【0021】
本発明は、エプスタイン-バールウイルスに特異的に細胞傷害性T細胞を誘導し得る機能を有するT細胞エピトープペプチドに関する。従って本発明は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドを提供する。該エピトープペプチドは、本明細書において「エピトープペプチド」、あるいは単に「本発明のペプチド」と記載する場合がある。
【0022】
本発明において「ペプチド」とは、生理活性を有し、隣接するアミノ酸残基のα−アミノ基とカルボキシル基間のペプチド結合により相互に結合した線状のアミノ酸の分子鎖を意味する。ペプチドは特定長のものを意味するものではなく、種々の長さであり得る。従って、本発明のペプチドには、所謂「オリゴペプチド」、「ポリペプチド」も含まれる。また、無電荷又は塩の形態であってもよく、場合によっては、グリコシル化、アミド化、ホスホリル化、カルボキシル化、リン酸化等により修飾されていてもよい。
【0023】
以下に、エピトープの候補ペプチドの選択方法についてその一例を記載する。
1.コンピュータを用いた解析
本発明のエプスタイン-バールウイルスに特異的なCTLエピトープペプチドは、エプスタイン-バールウイルス関連タンパク質のアミノ酸配列について、目的とするHLA分子の各結合モチーフを有する8〜11個のアミノ酸よりなるエピトープペプチドを検索し得る、インターネット上に公開されている複数のソフトウェア(Pingping G., et al., Nucleic Acids Res., 31, 3621-3624(2003))に照合してCTLエピトープの候補ペプチドを選択する事ができる。本発明において、エプスタイン-バールウイルス関連タンパク質としては、EBNA1、2、3A、3B、3C、LMP1、LMP2、またはleader protein等を挙げることができ、より好ましくは、EBNA1またはLMP1を挙げることが出来る。EBNA1のアミノ酸配列およびDNA配列を配列番号:36および37に、N末端が欠損したLMP1のアミノ酸配列およびDNA配列を配列番号:34および35に示す。
【0024】
2.アンカーモチーフを用いた検討
HLAクラスI分子は、主としてHLA-A、HLA-B、HLA-Cがあり、これらに結合して提示されるエピトープペプチドは、8〜11個のアミノ酸からなる。エピトープペプチドのN末端側から2番目と、9あるいは10番目のアミノ酸はHLA分子との結合に対して最も重要なアミノ酸であり、アンカーモチーフと呼ばれている。このアンカーモチーフは、各々のHLA分子の種類によって異なることが報告されている。例えば、HLA-A2分子に結合するペプチドとしては、N末端より2番目の位置にLeuが配置され、9あるいは10番目の位置にLeu又はValが配置されたペプチドであって、9〜10個のアミノ酸残基からなるペプチドが最も良く知られている(Sudo T., et al., J. Immunol., 155, 4749-4756 (1995))。また、日本人を含むアジアの人種に多いHLA-A24に結合するペプチドとしては、N末端より2番目の位置にTyr、Phe、Met又はTrpのいずれかが配置され、9あるいは10番目の位置にLeu、Ile、Trp又はPheのいずれかが配置されたペプチドであって、9〜10個のアミノ酸からなるペプチドが最もよく知られている(Kondo A., J. Immunol., 155, 4307-4312 (1995))。蛋白質のアミノ酸配列中からこのアンカーモチーフを有する配列を検索し、CTLエピトープの候補ペプチドを選択する事ができる。
【0025】
3.ペプチドライブラリーの作製
エプスタイン-バールウイルス関連タンパク質の蛋白質全体を網羅する20個前後のアミノ酸よりなるペプチドライブラリーを合成する。20個前後のアミノ酸のうち、前後10個程度(例えば、7,8,9,10,11,12,13個)のアミノ酸は、前後のペプチド配列と重複するようにライブラリーを作製する。これによって、蛋白質全体を網羅的に検索する事ができる。
【0026】
なお、前述の方法にて選択されたエピトープ候補ペプチドは必ずしもCTLエピトープになり得る訳ではなく、以下に示す検討を経て初めてエプスタイン-バールウイルス特異的CTLエピトープになり得る。以下に、エピトープペプチドの決定方法について記載するが、これらの方法に限定されるものではない。
【0027】
(1)エピトープペプチド決定方法1
エプスタイン-バールウイルス感染歴がある人から分離した末梢血単核球(peripheral blood mononuclear cells;PBMC)、あるいはPBMCから分離したT細胞を適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させる。これに同じ人からあらかじめ分離培養しておいたエプスタイン-バールウイルス感染細胞1×105/mLを加え、5%炭酸ガス(CO2)恒温槽にて37℃で7日間培養する。培養7日後にエプスタイン-バールウイルス感染細胞とインターロイキン2(IL-2)を添加し、以後、エプスタイン-バールウイルス感染細胞とIL-2による刺激を毎週繰返すことによりCTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、MHC−テトラマー法、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する(Current Protocols in Immunology, Edited by: John E. Coligan, Ada M. Kruisbeek, David H. Margulies, Ethan M. Shevach, Warren Strober, 6.19 ELISPOT Assay to Detect Cytokine-Secreting Murine and Human Cells, 6.24 Detection of Intracellular Cytokines by Flow Cytometry, published by John Wiley & Sons, Inc.)。
【0028】
(2)エピトープペプチド決定方法2
エプスタイン-バールウイルス感染歴がある人から分離したPBMCを適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させ、これにエピトープ候補ペプチドの任意の1種を0.01〜100μg/mLの濃度で加える。5%CO2恒温槽にて37℃で培養し、2日後にIL-2を添加する。以後、前記ペプチドとIL-2による刺激を週に1度あるいは2週間に1度繰返すことによりCTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、MHC−テトラマー法、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する。
【0029】
(3)エピトープペプチド決定方法3
エプスタイン-バールウイルス感染歴がある人から分離したPBMCを適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させ、これに合成したペプチドライブラリーを適当な数(例えば10種類ずつ)にプールした物を加える。5%CO2恒温槽にて37℃で培養し、2日後にIL-2を添加する。以後、プールペプチドとIL-2による刺激を週に1度あるいは2週間に1度繰返すことにより、CTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する。良好な結果を示したプールペプチドに対しては、1種類ずつペプチドを加えて上記実験を繰返せば、CTL誘導能を有するペプチドを選択する事が可能である。反応したペプチドについて順次短くし、基幹のペプチドを得て本発明のエピトープペプチドとする。一般的に、エピトープペプチドは最終的に8〜11個のアミノ酸までに短くすることができる。
【0030】
本発明のペプチドとして具体的には、配列番号:1〜3のいずれかに記載の塩基配列からなるペプチドを例示することができる。
【0031】
また本発明のペプチドは、配列番号:1〜3のいずれかに記載のアミノ酸領域を含むペプチドであってもよい。即ち、本発明のペプチドの好ましい態様としては、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むペプチドである。
【0032】
さらに、本発明のエピトープペプチドは、生理活性及び免疫活性を実質的に改変せず、投与した場合に有害な活性を有するものでない限り、上記のペプチドの改変体であってもよい。
【0033】
即ち本発明の好ましい態様としては、配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、改変前の配列番号:1〜3のいずれかに記載のペプチドと同等の機能を有するペプチドを挙げることができる。
【0034】
上記「同等の機能」としては、例えば、(1)エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能、(2)ヒト主要組織適合性抗原等、特にHLA-A*0206分子、またはHLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞(CTL)を誘導し得る機能、等を挙げることができる。
【0035】
改変されたペプチドが、上記の機能を有するか否かは、例えば、後述の実施例に記載の方法、あるいは該方法を適宜改変した方法によって、評価することが可能である。
【0036】
本発明のペプチドは、天然に存在する状態から修飾されていないもの、及び修飾されているものの双方を含む。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチド又はヌクレオチド誘導体の共有結合、脂質又は脂質誘導体の共有結合、ホスファチジルイノシトールの共有結合、架橋、環化、ジスルフィド結合の形成、メチル化、脱メチル化、共有架橋の形成、シスチンの形成、ピログルタメートの形成、ホルミル化、Y-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリストイル化、酸化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、アルギニル化のようなタンパク質へのアミノ酸の転移RNA媒介付加、ユビキチン化等が含まれる。また、本発明のペプチドは、N末端又はC末端に付加的アミノ酸配列が介在するものであってもよい。また、本発明のペプチドは、糖類、ポリエチレングリコール、脂質等が付加された複合体、放射性同位元素等による誘導体、あるいは重合体等の形態として用いることができる。また、本発明におけるアミノ酸には、所謂「アミノ酸アナログ」が含まれる。該アミノ酸アナログとしては、例えば、種々のアミノ酸のN-アシル化物、O-アシル化物、エステル化物、酸アミド化物、アルキル化物等が挙げられる。
【0037】
また、本発明のペプチドと該HLA分子との複合体が、該複合体を細胞表面に提示する細胞を特異的に認識しうるTCRを有するCTLによって認識されるものである限り、本発明のペプチドのN末端や遊離のアミノ基には、ホルミル基、アセチル基、t-ブトキシカルボニル(t-Boc)基等が結合していてもよく、本発明のペプチドのC末端や遊離のカルボキシル基には、メチル基、エチル基、t-ブチル基、ベンジル基等が結合していてもよい。
【0038】
また、本発明のペプチドは、生体内への導入を容易にしうる各種修飾を施されたものであってもよい。生体内への導入を容易にしうる各種修飾の例としては、PT(Protein Transduction)ドメインが有名である。HIV(human immunodeficiency virus)のPTドメインは、Tatタンパク質の49〜57番目のアミノ酸(Arg Lys Lys Arg Arg Gln Arg Arg Arg/配列番号:38)で構成されたペプチドで、このペプチド配列を目的とするタンパク質あるいはペプチド配列の前後、又はいずれかに付加することで、容易に細胞内に導入できることが報告されている(Ryu J., Mol Cells., 16, 385-391 (2003), Kim DT., J Immunol., 159:1666-1668 (1997))。
【0039】
MHCクラスI分子を介して提示されるほとんどの抗原は、細胞質内のプロテアソームにより分解された後、TAP(transporter in antigen processing)へと移送され、粗面小胞体内においてTAPに会合しているMHCクラスI分子とβ2-ミクログロブリンの複合体と結合し、ゴルジ装置を経てエクソサイトーシスにより細胞表面へと運搬される。これら一連の抗原提示経路にて作用するシャペロンであるHSP(heart shock prtein)70やHSP90、又はgp96と目的とするペプチドやタンパク質を融合させることで、効果的な抗原提示がなされることが報告されており(Basu S., Immunity., 14, 303-313 (2001))、本発明のペプチドに上記PTドメインまたはシャペロンを融合させたタンパクは本発明の一態様を成す。
【0040】
また、本発明のペプチドは、従来の各種のペプチド合成方法によって調製され得る。例えば、固相ペプチド合成法等の有機化学的合成法、あるいは、ペプチドをコードする核酸を調製し、組換えDNA技術を用いて調製することも可能である。また、市販の化学合成装置(例えば、アプライドバイオシステムズ社のペプチド合成装置)による合成も可能である。
【0041】
即ち本発明のペプチドは、そのアミノ酸配列に従って、一般的な化学合成法により製造することが可能であり、該方法には、通常の液相法及び固相法によるペプチド合成法が包含される。該ペプチド合成法は、より詳しくはアミノ酸配列の情報に基づいて、各アミノ酸を1個ずつ逐次合成させて鎖を延長していくステップワイズエロンゲーション法と、アミノ酸数個からなるフラグメントを予め合成し、次いで各フラグメントをカップリング反応させるフラグメント・コンデンセーション法を包含し、本発明のペプチドの合成は、いずれの方法を用いてもよい。
【0042】
このようなペプチド合成法にて用いられる縮合法も、各種方法に従って行うことができる。その具体例としては、例えばアジド法、混合酸無水物法、DCC法、活性エステル法、酸化還元法、DPPA(ジフェニルホスホリルアジド)法、ウッドワード法等を例示できる。
【0043】
これら各種方法に利用できる溶媒もまた、一般的に使用されるものを適宜利用することができる。その例としては、例えばジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、ヘキサホスホロアミド、ジオキサン、テトラヒドロフラン(THF)、酢酸エチル等及びこれらの混合溶媒等を挙げることができる。なお、上記ペプチド合成反応に際して、反応に関与しないアミノ酸及びペプチドにおけるカルボキシル基は、一般にはエステル化により、例えばメチルエステル、エチルエステル、第三級ブチルエステル等の低級アルキルエステル、例えばベンジルエステル、P−メトキシベンジルエステル、P−ニトロベンジルエステルアラルキルエステル等として保護することができる。また、側鎖に官能基を有するアミノ酸、例えばTyrの水酸基は、アセチル基、ベンジル基、ベンジルオキシカルボニル基、第三級ブチル基等で保護されてもよいが、必ずしもかかる保護は必須ではない。また、例えば、Argのグアニジノ基は、ニトロ基、トシル基、2−メトキシベンゼンスルホニル基、メチシレン−2−スルホニル基、ベンジルオキシカルボニル基、イソボルニルオキシカルボニル基、アダマンチルオキシカルボニル基等の適当な保護基により保護することも可能である。
【0044】
上記のようにして得ることが可能な本発明のペプチドは、通常の方法に従って、例えばイオン交換樹脂、分配クロマトグラフィー、ゲルクロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィー(HPLC)、向流分配法等のペプチド化学の分野で汎用されている方法に従って、適宜、精製を行うことができる。
【0045】
本発明のペプチドは、本発明のペプチドをコードするDNA核酸分子を合成し、適当な発現ベクターへ導入した後、宿主細胞内において発現させる遺伝子工学的手法によっても取得することができる。
【0046】
一例を示せば、まず配列番号:1〜3のいずれかに記載されたアミノ酸配列をコードする核酸を合成する。該方法としては、リン酸トリエステル法やリン酸アミダイト法などの化学合成手段〔Letsinger, RL., et al., J. Am.Chem. Soc., 89, 4801 (1967);Letsinger, RL., et al., J. Am.Chem. Soc., 91, 3350-3355 (1969);Merrifield, R. B., et al., Science, 150, 178-185 (1968);Beaucage, SL and Caruthers, MH., Tetrahedron Lett., 22, 1859-1862 (1981); McBride, LJ and Caruthers, MH., Tetrahedron Lett., 24, 245 (1983)〕及びそれらの組合せ方法などが例示できる。より具体的には、DNAの合成は、ホスホルアミダイト法又はトリエステル法による化学合成により行うこともでき、市販されている自動ポリヌクレオチド合成装置上で行うこともできる。二本鎖断片は、相補鎖を合成し、適当な条件下で該鎖を共にアニーリングさせるか、又は適当なプライマー配列と共にDNAポリメラーゼを用い相補鎖を付加するかによって、化学合成した一本鎖生成物から得ることもできる。
【0047】
本発明のペプチドには、上述のように、本発明者らによって同定されたエピトープペプチド(配列番号:1〜3)と機能的に同等なペプチドが含まれる。
【0048】
あるペプチドと機能的に同等なペプチドを調製するための、当業者によく知られた方法としては、例えばペプチド中のアミノ酸配列に変異を導入する方法が挙げられる。具体的には当業者であれば部位特異的変異誘発法(Hashimoto-Gotoh, T. et al., Gene, 152, 271-275 (1995)、Zoller, MJ, and Smith, M. Methods Enzymol. 100, 468-500 (1983)、Kramer, W. et al., Nucleic Acids Res. 12, 9441-9456 (1984)、Kramer W, and Fritz HJ., Methods. Enzymol., 154, 350-367 (1987)、Kunkel,TA., Proc Natl Acad Sci USA., 82, 488-492 (1985)、Kunkel, Methods Enzymol., 85, 2763-2766 (1988))などを用いて、配列番号:1〜3のいずれかに記載のアミノ酸配列に適宜変異を導入することにより、該ペプチドと機能的に同等なペプチドを調製することができる。また、ペプチド中のアミノ酸の変異は自然に生じることもある。このように、人工的か自然に生じたものかを問わず、本発明者らにより同定されたエピトープペプチド(配列番号:1〜3)のアミノ酸配列において1もしくは複数のアミノ酸配列が変異したアミノ酸配列を有し、該ペプチドと機能的に同等なペプチドは、本発明のペプチドに含まれる。
【0049】
上述のようなアミノ酸の変更(改変)目的は、例えば、
1.HLAとの親和性を高める為の変更(Rosenberg SA., et al., Nat Med., 4, 321-327 (1998)、Berzofsky JA., et al., Nat Rev Immunol., 1, 209-219 (2001))、
2.TCRの認識性を向上させるための変更(Fong L., et al., Proc Natl Acad Sci U S A., 98, 8809-8814 (2001), Rivoltini L., et al., Cancer Res., 59, 301-306 (1999))、
3.血清中のペプチド分解酵素等による代謝を回避する為の変更(Berzofsky JA., et al., Nat Rev Immunol., 1, 209-219 (2001), Parmiani G., et al., J Natl Cancer Inst., 94, 805-818 (2002), Brinckerhoff LH., et al., Int J Cancer., 83, 326-334 (1999))等が挙げられる。
【0050】
上記変異体における、変異(置換、挿入、欠失等)するアミノ酸数は、本発明のペプチドの有する機能が保持される限り制限はないが、通常5アミノ酸以内であり、好ましくは4アミノ酸以内であり、より好ましくは3アミノ酸以内であり、さらに好ましくは1〜2アミノ酸である。また、上記改変が、例えば、配列番号:1〜3のいずれかに記載のペプチドの末端へのアミノ酸残基の付加である場合には、付加されるアミノ酸数は本発明のペプチドの有する機能が保持される限り制限はないが、通常、20アミノ酸以内であり、好ましくは10アミノ酸以内であり、より好ましくは5アミノ酸以内であり、さらに好ましくは3アミノ酸以内であり、最も好ましくは1〜2アミノ酸である。
【0051】
変異するアミノ酸残基の種類としては、改変前のアミノ酸の性質が保存されている他のアミノ酸(改変前のアミノ酸と類似のアミノ酸)に変異されることが望ましい。例えばアミノ酸側鎖の性質としては、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ酸(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)を挙げることができる(括弧内はいずれもアミノ酸の一文字表記を表す)。
【0052】
あるアミノ酸配列に対する1又は複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するペプチドがその生物学的機能(活性)を維持し得ることはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA., 81, 5662-5666 (1984)、Zoller, M. J. & Smith, M. Nucleic Acids Research, 10, 6487-6500 (1982)、Wang, A. et al., Science, 224, 1431-1433 (1984)、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA., 79, 6409-6413 (1982))。
【0053】
本発明のペプチドのアミノ酸配列に複数個のアミノ酸残基が付加されたペプチドには、これらペプチドを含む融合ペプチドが含まれる。融合ペプチドは、本発明のペプチドと他のペプチドとが融合したものである。融合ペプチドを作製する方法は、本発明のペプチド(例えば、配列番号:1〜3に記載のペプチド)をコードするポリヌクレオチドと他のペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。
【0054】
本発明のペプチドとの融合に付される他のペプチドは、CTLの誘導目的以外にも、例えば、該ペプチドの単離・精製、あるいは応用研究等の種々の目的に応じて、適宜選択することができる。例えばFLAG(Hopp, T. P. et al., BioTechnology, 6, 1204-1210 (1988))、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein Cの断片等の公知のペプチドを使用することができる。また、本発明のペプチドとの融合に付される他のタンパク質としては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチド又はペプチドをコードするポリヌクレオチドを本発明のペプチドをコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ペプチドを調製することができる。
【0055】
また、本発明のペプチドの融合体を作製する場合、融合箇所にFactorXa,エンテロキナーゼまたはトロンビンのようなプロテアーゼ切断部位ペプチド配列を挿入することもできる。このような場合、上記GST、MBP、あるいはFLAGタグのような精製用タグを付加させたペプチド融合体として発現させ、精製した後、必要に応じてペプチド融合体のうち、本発明の目的のペプチド以外の領域を、対応するFactorXa,エンテロキナーゼまたはトロンビンのようなプロテアーゼなどにより切断、除去することも可能であり、有用である。
【0056】
また、あるペプチドと機能的に同等なペプチドを調製する当業者によく知られた他の方法としては、ハイブリダイゼーション技術(Sambrook,J et al., Molecular Cloning 2nd ed., 9.47-9.58, Cold Spring Harbor Lab. press, (1989))を利用する方法が挙げられる。即ち、当業者であれば、本発明のペプチドをコードするポリヌクレオチドもしくはその一部をもとに、種々の生物由来(例えば、エプスタイン-バールウイルス由来)のポリヌクレオチド試料、あるいは人工的に合成されたペプチドライブラリー等から、これと相同性の高いポリヌクレオチドを単離して、該ポリヌクレオチドから本発明のペプチドと機能的に同等なペプチドを単離することも通常行いうることである。
【0057】
本発明には、本発明の配列番号:1〜3に記載のペプチドをコードするポリヌクレオチドとハイブリダイズするポリヌクレオチドによってコードされるペプチドであって、配列番号:1〜3に記載のペプチドと機能的に同等なペプチドが含まれる。
【0058】
配列番号:1〜3に記載のペプチドと機能的に同等なペプチドをコードするポリヌクレオチドを単離するためのハイブリダイゼーションの条件は、当業者であれば適宜選択することができる。ハイブリダイゼーションの条件としては、例えば、低ストリンジェントな条件が挙げられる。低ストリンジェントな条件とは、ハイブリダイゼーション後の洗浄において、例えば42℃、0.1×SSC、0.1%SDSの条件であり、好ましくは50℃、0.1×SSC、0.1%SDSの条件である。より好ましいハイブリダイゼーションの条件としては、高ストリンジェントな条件が挙げられる。高ストリンジェントな条件とは、例えば65℃、5×SSC及び0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。但し、ハイブリダイゼーションのストリンジェンシーに影響を及ぼす要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。
【0059】
また、ハイブリダイゼーションにかえて、遺伝子増幅技術(PCR)(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley&Sons Section 6.1-6.4)を用いて、本発明者のペプチド(配列番号:1〜3)をコードするポリヌクレオチドの一部を基にプライマーを設計し、本発明者らにより同定されたペプチドをコードするポリヌクレオチドと相同性の高いポリヌクレオチド断片を単離し、該ポリヌクレオチドを基に本発明者らにより同定されたペプチドと機能的に同等なペプチドを取得することも可能である。
【0060】
上述のハイブリダイゼーション技術や遺伝子増幅技術により単離されるポリヌクレオチドがコードする、配列番号:1〜3に記載のペプチドと機能的に同等なペプチドは、通常、該ペプチドとアミノ酸配列において、通常高い相同性を有する。本発明のペプチドには、配列番号:1〜3に記載のペプチドと機能的に同等であり、かつ該ペプチドのアミノ酸配列と高い相同性を有するペプチドも含まれる。高い相同性とは、アミノ酸レベルにおいて、通常、少なくとも50%以上の同一性、好ましくは75%以上の同一性、さらに好ましくは85%以上の同一性、さらに好ましくは95%以上(例えば、96%以上、97%以上、98%以上、99%以上)の同一性を指す。ペプチドの相同性を決定するには、文献(Wilbur, W. J. and Lipman, D. J. Proc. Natl. Acad. Sci. USA, 80, 726-730 (1983))に記載のアルゴリズムに従えばよい。
【0061】
アミノ酸配列の同一性は、例えば、Karlin and Altschul によるアルゴリズムBLAST (Proc. Natl. Acad. Sci. USA 87:2264-2268 (1990)、Proc. Natl. Acad. Sci. USA 90:5873-5877 (1993))によって決定することができる。このアルゴリズムに基づいて、BLASTXと呼ばれるプログラムが開発されている(Altschul et al. J. Mol. Biol.215: 403-410 (1990))。BLASTXによってアミノ酸配列を解析する場合には、パラメーターは例えば、score = 50、wordlength = 3とする。BLASTとGapped BLASTプログラムを用いる場合には、各プログラムのデフォルトパラメーターを用いる。これらの解析方法の具体的な手法は公知である(http://www.ncbi.nlm.nih.gov)。
【0062】
本発明のペプチドは、当業者に公知の方法により、組み換えペプチドとして、また天然のペプチドとして調製することが可能である。組み換えペプチドであれば、例えば、本発明のペプチドをコードするポリヌクレオチドを、適当な発現ベクターに組み込み、これを適当な宿主細胞に導入して得た形質転換体を回収し、抽出物を得た後、イオン交換、逆相、ゲル濾過などのクロマトグラフィー、あるいは本発明のペプチドに対する抗体をカラムに固定したアフィニティークロマトグラフィーにかけることにより、または、さらにこれらのカラムを複数組み合わせることにより精製し、調製することが可能である。
【0063】
精製用タグ(例えばヒスチジンタグ)を融合させた組み換えペプチドとして宿主細胞(例えば、動物細胞や大腸菌など)内で発現させた場合には、発現させた融合ペプチドは、市販の、融合したタグに対応した精製カラム(ヒスチジンタグの場合ニッケルカラム)を用いて精製することができる。
【0064】
天然のペプチドであれば、当業者に周知の方法、例えば本発明のペプチドを発現している組織や細胞の抽出物に対し、本発明のペプチドに結合する抗体が結合したアフィニティーカラムを作用させて精製することにより単離することができる。抗体はポリクローナル抗体であっても、モノクローナル抗体であってもよい。
【0065】
本発明のペプチドをコードする上記核酸もしくはベクターもまた、本発明に含まれる。
【0066】
本発明の核酸(ポリヌクレオチド)は、本発明のペプチドをコードし得るものであればいかなる形態でもよい。即ち、mRNAから合成されたcDNAであるか、ゲノムDNAであるか、化学合成DNAであるかなどを問わない。また、本発明のペプチドをコードしうる限り、遺伝暗号の縮重に基づく任意の塩基配列を有するポリヌクレオチドが含まれる。
【0067】
本発明の核酸(ポリヌクレオチド)は、当業者に公知の方法により取得することができる。例えば、市販の核酸合成装置を用いて、適宜作製することができる。作製したポリヌクレオチドの塩基配列は、公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法等により確認することができる。
【0068】
本発明のペプチドをコードする核酸は、例えば、遺伝子組換え技術を用いて、本発明のペプチドを宿主内で産生させる為に重要である。この場合、宿主間でアミノ酸コドンの使用頻度(codon usage)が異なる為、産生させる宿主のcodon usageに適合するようアミノ酸のコドンを変更することが望ましい。本発明のペプチドをコードする核酸は、ワクチンとしても利用可能で、むき出しの核酸として移送することもできるし、または適切なウイルスもしくは細菌ベクターを用いて移送することもできる(Berzofsky JA., et al., J Clin Invest., 114, 450-462 (2004), Berzofsky JA., et al., J Clin Invest., 113, 1515-1525 (2004))。適切な細菌ベクターとしては、サルモネラ属亜種の細菌ベクターを挙げることができる。適切なウイルスベクターとしては、レトロウイルスベクター、アデノウイルスベクター、センダイウイルスベクター、レンチウイルスベクター、ワクシニアベクター等を例示することができる。適切なワクシニアベクターの1例は、改変ワクシニア・アンカラベクターである。
【0069】
本発明の核酸の好ましい態様としては、本発明のペプチドを発現し得るベクターを例示することができる。該ベクターは、通常、プロモーターの下流に本発明の核酸が機能的に連結された構造のDNA構築物を担持するベクターである。該ベクターとしては、通常、プラスミドもしくはウイルスベクター等が一般的である。
【0070】
当業者においては、所望のDNAを有するベクターを、一般的な遺伝子工学技術によって、適宜、作製することが可能である。通常、市販の種々の発現ベクターを利用することができる。
【0071】
本発明のベクターは、宿主細胞内において本発明のポリヌクレオチドを保持したり、本発明のペプチドを発現させるためにも有用である。本発明の核酸(ポリヌクレオチド)は、通常、適当なベクターへ担持(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入したDNAを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明のペプチドを生産する目的としてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター(GenBank Accession No. AB009864)、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などを例示することができる。ベクターへの本発明の核酸の挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる。
【0072】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)及び植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法(GIBCO-BRL社製)、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0073】
宿主細胞において発現させたペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のペプチドに組み込むことができる。これらのシグナルは目的のペプチドに対して内因性であっても、異種シグナルであってもよい。
【0074】
上記製造方法におけるペプチドの回収は、本発明のペプチドが培地に分泌される場合は、培地を回収する。本発明のペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にペプチドを回収する。
【0075】
組換え細胞培養物から本発明のペプチドを回収し精製するには、硫酸アンモニウム又はエタノール沈殿、酸抽出、アニオン又はカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィー及びレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0076】
また、動物の生体内で本発明のポリヌクレオチド(核酸)を発現させる方法としては、本発明の核酸を適当なベクターに組み込み、例えば、レトロウィルス法、リポソーム法、カチオニックリポソーム法、アデノウイルス法などにより生体内に導入する方法などが挙げられる。これにより、免疫療法を行うことが可能である。用いられるベクターとしては、例えば、レトロウイルスベクターなどが挙げられるが、これらに制限されない。ベクターへの本発明のDNAの挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning ,5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0077】
本発明のペプチド(エプスタイン-バールウイルス特異的CTLエピトープペプチド)は、能動免疫療法においてペプチドワクチンとして用いることができる。即ち、本発明のCTLエピトープペプチドを含んでなるワクチンを健常人あるいは患者に投与し、エプスタイン-バールウイルスに特異的なCTLを体内で増殖させ、疾患に対する予防あるいは治療に役立てることができる。使用するエピトープペプチドは1種のみの使用であっても、あるいはワクチンの使用目的に応じて2種以上のペプチドを組み合わせ、混合して使用することもできる。
【0078】
従って本発明は、本発明のペプチドもしくは本発明の核酸を有効成分として含む、エプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するためのワクチンを提供する。
【0079】
なお、本発明の「ワクチン」は、「能動免疫療法剤」、「免疫治療剤」、「エプスタイン-バールウイルス関連疾患治療剤」と表現することも可能である。また、本発明において「治療剤」は、「医薬品」、「医薬組成物」、「治療用医薬」等と表現することもできる。
【0080】
さらに、本発明のCTLエピトープペプチドが提示された抗原提示細胞は、能動免疫療法においてワクチンとして用いることができる。CTLエピトープペプチドが提示された抗原提示細胞とは、
1. 適当な培養液中で、抗原提示細胞とCTLエピトープペプチドを30分から1時間混合したエピトープペプチドパルス抗原提示細胞、
2. CTLエピトープペプチドをコードする核酸を用い、遺伝子導入等で抗原提示細胞にCTLエピトープペプチドを提示された細胞、
3. 人工的に作製した抗原提示能を有する人工抗原提示細胞、等を意味する。
【0081】
抗原提示細胞とは、例えば、樹状細胞、B細胞、マクロファージ、ある種のT細胞等を示すが、該ペプチドが結合し得るHLAをその表面上に発現する細胞であって、CTL刺激能を有するものを意味する。人工的に作製した抗原提示能を有する人工抗原提示細胞とは、例えば脂質2重膜やプラスティックあるいはラテックス等のビーズにHLAとCTLエピトープペプチドとβ2-ミクログロブリンとの3者複合体を固定し、CTLを刺激し得るCD80、CD83、CD86等の共刺激分子を固定するか、もしくは、共刺激分子と結合するT細胞側のリガンドであるCD28に対してアゴニスティックに作用する抗体等を固定することで作製可能である(Oelke M., et al., Nat Med., 9, 619-624 (2003), Walter S., et al., J Immunol., 171, 4974-4978 (2003), Oosten LE., et al., Blood, 104, 224-226 (2004))。
従って本発明の好ましい態様としては、本発明のペプチドをHLAに提示した抗原提示細胞を有効成分とするワクチンを提供する。
【0082】
本発明の核酸は、能動免疫療法においてDNAワクチンや組換えウイルスベクターワクチン等に用いる事ができる。この場合、CTLエピトープペプチドの核酸配列は、組換えワクチンや、組換えウイルスワクチンを産生させる宿主に適合したcodon usageに変更する事が望ましい(Casimiro, D.R., et al., J. Virol., 77, 6305-6313 (2003), Berzofsky JA, et al., J Clin Invest., 114, 450-462 (2004))。
【0083】
本発明のペプチド、又は本発明のペプチドが提示された抗原提示細胞を含んでなるワクチンは、当分野において公知の方法を用いて調製することができる。例えば、かかるワクチンの一例としては、本発明のペプチドを有効成分として含有する注射剤又は固形剤等の薬剤が挙げられる。
【0084】
本発明のペプチドは、エプスタイン-バールウイルスに対する受動免疫治療剤の調製に用いることができる。下記のようにして得られたエプスタイン-バールウイルスに特異的なCTLはヒトアルブミン含有PBS等に懸濁させて、エプスタイン-バールウイルスに対する受動免疫療法剤とすることができる。受動免疫療法剤に含まれるエプスタイン-バールウイルスに特異的なCTLは、以下のような調製方法によって得ることができ、CTLの純度を高める為に精製して用いる事も可能である。
以下にCTLの調製方法の具体例を記載するが、必ずしもこれらの方法に制限されない。
【0085】
(a)CTL調製方法1
PBMCと、適当な濃度のエプスタイン-バールウイルス特異的エピトープペプチド−テトラマー試薬を反応させる。エピトープペプチド−テトラマー試薬と結合したエプスタイン-バールウイルス特異的CTLは標識色素により染色されるので、セルソーター、顕微鏡などを用いて染色されたCTLのみを単離する。このようにして単離されたエプスタイン-バールウイルスに特異的なCTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤や、X照射あるいはマイトマイシン処理等で増殖能を損失させた抗原提示細胞で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0086】
(b)CTL調製方法2
エプスタイン-バールウイルス特異的エピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマー試薬を無菌プレートなどに固相化し、PBMCを固相化プレートで培養する。プレートに固相化されたエピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマーに結合したエプスタイン-バールウイルス特異的CTLを単離するためには、結合せずに浮遊している他の細胞を洗い流した後に、プレート上に残った抗原特異的CTLだけを新しい培地に懸濁する。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤や、X照射あるいはマイトマイシン処理等で増殖能を損失させた抗原提示細胞で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0087】
(c)CTL調製方法3
エプスタイン-バールウイルス特異的エピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマー試薬と、CD80、CD83、CD86等の共刺激分子か、もしくは、共刺激分子と結合するT細胞側のリガンドであるCD28に対してアゴニスティックに作用する抗体等を無菌プレートなどに固相化し、PBMCを固相化プレートで培養する。2日後にIL-2を培地に添加し5%CO2恒温槽にて37℃で7〜10日培養する。培養した細胞を回収し新たな固相化プレート上で培養を続ける。この操作を繰り返す事で受動免疫療法に必要な細胞数のCTLを確保する。
【0088】
(d)CTL調製方法4
PBMCあるいはT細胞を本発明のCTLエピトープペプチドで直接刺激するか、該ペプチドをパルスした抗原提示細胞、遺伝子導入した抗原提示細胞、又は人工的に作製した抗原提示能を有する人工抗原提示細胞で刺激する。刺激によって誘導されたCTLを5%CO2恒温槽にて37℃で7〜10日培養する。CTLエピトープペプチドとIL-2、又は抗原提示細胞とIL-2による刺激を週に1度繰り返す事で受動免疫療法に必要な細胞数のCTLを確保する。
【0089】
なお、本発明のペプチド(エプスタイン-バールウイルス特異的CTLエピトープペプチド)を使用したエピトープペプチド−モノマー及びエピトープペプチド−テトラマーは、公知の方法(US Patent Number 5,635,363, Inventors : J. D. Altman, M. G. McHeyzer-Williams, Mark M. Davis, Compositions and methods for the detection, quantitation and purification of antigen-specific T cells, French Application Number FR9911133, Inventor : M. Bonneville, et al., Means for detecting and for purifying CD8+ T-lymphocyte populations specific for peptides presented in the HLA context)により調製することができる。タンパク質発現用の遺伝子組換え宿主から精製したHLAクラスI分子、β2−ミクログロブリン及び本発明のCTLエピトープペプチドの複合体である、MHC−モノマーをバッファー内で形成させる。組換えHLAクラスI分子のC末端には予めビオチン結合部位を付加しておき、エピトープペプチド−モノマー形成後この部位にビオチンを付加する。市販の色素標識されたストレプトアビジンとビオチン化エピトープペプチド−モノマーをモル比1:4で混合することによってエピトープペプチド−テトラマーを作製することができる。
【0090】
また、CTL調製方法において、特異的CTLの割合が低い場合は、随時以下の方法を用いる事でCTLを高純度で回収する事が可能である。
【0091】
(a)エピトープペプチド−テトラマー試薬による精製
エプスタイン-バールウイルス特異的エピトープペプチド−テトラマー試薬と、CTL調製方法にて誘導されたCTLを反応させ、エピトープペプチド−テトラマーを標識している標識色素に対する抗体等を磁気標識した2次抗体を用いて分離する事が可能である。このような磁気標識した2次抗体と、磁気細胞分離装置は例えば、Dynal社やMiltenyi Biotec GmbH社から入手可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0092】
(b)分泌されるサイトカインによる精製
エプスタイン-バールウイルス特異的CTLが、放出するサイトカイン等を利用して、エプスタイン-バールウイルスに特異的なCTLを精製する事ができる。例えば、Miltenyi Biotec GmbH社から入手可能なキットを用いる事で、CTLから放出されるサイトカインを細胞表面で特異抗体により補足し、サイトカイン特異的な標識抗体で染色し、続いて磁気標識した標識物質特異的な抗体で染色後、磁気標識細胞分離装置を用いて精製する事も可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0093】
(c)細胞表面蛋白質特異的抗体を用いた精製
特異的CTLの細胞表面では、特異的なペプチドの刺激により発現が増強する細胞表面蛋白質(例えばCD107a、CD107b、CD63、CD69など)が報告されている(Betts MR., et al., J Immunol Methods., 281, 65-78 (2003), Trimble LA., et al., J Virol., 74, 7320-7330 (2000))。このような蛋白質の特異抗体を磁気標識する事で、磁気分離装置等を用いてCTLを精製する事が可能である。また、このような特異抗体に対する抗IgG抗体等を磁気標識する事でも同様にCTLの精製が可能である。あるいは、これら抗体を培養用のプラスティックプレートにコートし、このプレートを用いて刺激を加えたPBMCを培養し、プレートに結合しなかった細胞集団を洗い流す事で特異的なCTLを精製することも可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0094】
本発明は、上述のようにして取得・精製された細胞傷害性T細胞(CTL)を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤(免疫治療剤)を提供する。
なお、本発明の「受動免疫療法剤」は、「免疫治療剤」、「エプスタイン-バールウイルス関連疾患治療剤」、又は「CTL誘導剤」と表現することも可能である。
本発明の受動免疫療法剤の好ましい態様としては、本発明のペプチド、もしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤である。
【0095】
また、別の態様としては、本発明のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤である。
【0096】
本発明の薬剤(本発明のワクチンもしくは受動免疫療法剤等)は、生理学的に許容される担体、賦形剤、あるいは希釈剤等と混合し、医薬組成物として経口、あるいは非経口的に投与することができる。経口剤としては、顆粒剤、散剤、錠剤、カプセル剤、溶剤、乳剤、あるいは懸濁剤等の剤型とすることができる。非経口剤としては、注射剤、点滴剤、外用薬剤、あるいは座剤等の剤型を選択することができる。注射剤には、皮下注射剤、筋肉注射剤、あるいは腹腔内注射剤等を示すことができる。外用薬剤には、経鼻投与剤、あるいは軟膏剤等を示すことができる。主成分である本発明の薬剤を含むように、上記の剤型とする製剤技術は公知である。
【0097】
例えば、経口投与用の錠剤は、本発明の薬剤に賦形剤、崩壊剤、結合剤、及び滑沢剤等を加えて混合し、圧縮整形することにより製造することができる。
【0098】
例えば、本発明のペプチドが提示された抗原提示細胞は、製薬上許容され、該ペプチド又は該細胞の活性と相容性を有する賦形剤、例えば、水、食塩水、デキストロース、エタノール、グリセロール、DMSO(dimethyl sulphoxide)、及びその他のアジュバント等、又はこれらの組み合わせと混合して用いることができる。さらに、必要に応じて、アルブミン、湿潤剤、乳化剤等の補助剤を添加してもよい。崩壊剤としては、炭酸カルシウムやカルボキシメチルセルロースカルシウム等が一般的に用いられる。結合剤には、アラビアゴム、カルボキシメチルセルロース、あるいはポリビニルピロリドンが用いられる。滑沢剤としては、タルクやステアリン酸マグネシウム等が公知である。
【0099】
本発明の薬剤を含む錠剤は、マスキングや、腸溶性製剤とするために、公知のコーティングを施すことができる。コーティング剤には、エチルセルロースやポリオキシエチレングリコール等を用いることができる。
【0100】
また注射剤は、主成分である本発明の薬剤を適当な分散剤とともに溶解、分散媒に溶解、あるいは分散させることにより得ることができる。分散媒の選択により、水性溶剤と油性溶剤のいずれの剤型とすることもできる。水性溶剤とするには、蒸留水、生理食塩水、あるいはリンゲル液等を分散媒とする。油性溶剤では、各種植物油やプロピレングリコール等を分散媒に利用する。このとき、必要に応じてパラベン等の保存剤を添加することもできる。また注射剤中には、塩化ナトリウムやブドウ糖等の公知の等張化剤を加えることができる。更に、塩化ベンザルコニウムや塩酸プロカインのような無痛化剤を添加することができる。
【0101】
また、本発明の薬剤を固形、液状、あるいは半固形状の組成物とすることにより外用剤とすることができる。固形、あるいは液状の組成物については、先に述べたものと同様の組成物とすることで外用剤とすることができる。半固形状の組成物は、適当な溶剤に必要に応じて増粘剤を加えて調製することができる。溶剤には、水、エチルアルコール、あるいはポリエチレングリコール等を用いることができる。増粘剤には、一般にベントナイト、ポリビニルアルコール、アクリル酸、メタクリル酸、あるいはポリビニルピロリドン等が用いられる。この組成物には、塩化ベンザルコニウム等の保存剤を加えることができる。また、担体としてカカオ脂のような油性基材、あるいはセルロース誘導体のような水性ゲル基材を組み合わせることにより、座剤とすることもできる。
【0102】
また、本発明のペプチドは、中性又は塩の形態で処方することができ、例えば、薬学上許容され得る塩としては、塩酸、リン酸などの無機塩、又は、酢酸、酒石酸などの有機酸が挙げられる。
【0103】
本発明の薬剤を遺伝子治療剤として使用する場合は、本発明の薬剤を注射により直接投与する方法のほか、核酸が組込まれたベクターを投与する方法が挙げられる。上記ベクターとしては、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、レトロウイルスベクター、レンチウイルスベクター等が挙げられ、これらのウイルスベクターを用いることにより効率よく投与することができる。
【0104】
また、本発明の薬剤をリポソームなどのリン脂質小胞体に導入し、その小胞体を投与することも可能である。例えば、本発明のペプチドもしくはベクターを保持させた小胞体をリポフェクション法により所定の細胞に導入する。そして、得られる細胞を例えば静脈内、動脈内等に全身投与する。
【0105】
本発明の薬剤は、非経口投与及び経口投与により投与することができるが、該薬剤がペプチドを主成分とする場合には、一般的に非経口投与が好ましい。非経口投与としては経鼻投与や皮下注射、筋肉内注射、静脈内注射等の注射剤、座薬等がある。また、経口投与としては、スターチ、マンニトール、ラクトース、ステアリン酸マグネシウム、セルロース等の賦形剤との混合物として調製することができる。
【0106】
本発明のワクチンは、治療上有効な量で投与する。投与される量は、治療対象、免疫系に依存し、必要とする投与量は臨床医の判断により決定される。通常、適当な投与量は、患者一人当たり、本発明のペプチド(エピトープペプチド)では1〜100mg、エピトープペプチドパルス細胞では106〜109個の含有量とする。また、投与間隔は、対象、目的により設定することができる。
【0107】
また本発明の薬剤によって予防もしくは治療効果が期待されるエプスタイン-バールウイルス関連疾患としては、例えば、バーキットリンパ腫(BL)、ホジキン病(HD)、鼻咽頭癌(NPC)、または移植後リンパ増殖性疾患(PTLD)等の悪性腫瘍を挙げることができる。
【0108】
本発明の薬剤(ワクチン等)が接種可能な動物としては、免疫系を有し、かつエプスタイン-バールウイルスに感染し得る生物であれば特に制限されないが、好ましくは、ヒトである。
【0109】
さらに本発明は、本発明のペプチドを用いて、細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導(活性化)方法を提供する。
【0110】
本発明の誘導方法の好ましい態様としては、本発明のペプチド(例えば、HLA-A24等拘束性抗原ペプチド)と、末梢血単核球を血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的な細胞傷害性T細胞(CTL)を誘導する方法に関する。
【0111】
また、上記方法によって誘導されたエプスタイン-バールウイルス特異的CTLを検出及び定量する方法もまた本発明に含まれる。
【0112】
また、上述の方法によって誘導されたエプスタイン-バールウイルス特異的な細胞傷害性T細胞は、エプスタイン-バールウイルスに対する受動免疫療法剤の成分となり得る。従って、上述の方法によってエプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し、該T細胞を取得することによって、受動免疫療法剤を製造することが可能である。
【0113】
本発明は、本発明のエプスタイン-バールウイルス特異的な細胞傷害性T細胞の誘導方法を用いた受動免疫療法剤の製造方法を提供する。
【0114】
本製造方法の好ましい態様としては、本発明のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞によって末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む方法である。
【0115】
また好ましい別の態様においては、本発明のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法である。
【0116】
上記製造方法には、上述の工程に加えて、必要に応じて、「取得される細胞傷害性T細胞へ薬学的に許容される担体を混合する工程」を含んでいてもよい。
【0117】
さらに本発明のペプチドを用い作製した、MHC−テトラマー試薬は、特異的なCTLの定量だけでなく細胞内サイトカイン産生細胞定量法や細胞表面蛋白質に対する特異抗体と組み合わせることで、特異的CTLの分化段階や機能性も同時に判定する事が可能である。
本発明はさらに、本発明のペプチドを利用したエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量(検出・測定)方法を提供する。
【0118】
エプスタイン-バールウイルスに特異的なCTLが、ハイリスクの患者(何らかの原因により免疫能が低下した人、先天性免疫不全症患者、又は骨髄移植、造血幹細胞移植、臍帯血移植、固形臓器移植を受けて拒絶予防のために免疫抑制剤の投与を受けている患者、慢性ウイルス感染症患者、エイズ患者、高齢者、幼小児、妊婦等)の末梢血に存在するか否かを知ることは、抗ウイルス剤や免疫抑制剤の適正な使用を含め、これらの感染症管理の上で重要な情報である。エプスタイン-バールウイルスに特異的なCTLの定量は、例えば、本発明のペプチド(CTLエピトープペプチド)を用いた以下の3つの方法によって行うことができるが、必ずしもこれらの方法に制限されない。
【0119】
(a)定量方法1
本発明のCTLエピトープペプチドを使用して作製したMHC−テトラマー試薬を用いて、末梢血中のエプスタイン-バールウイルスに特異的なCTLを定量することができる。定量は、例えば、以下のようにして実施することができる。末梢血あるいはPBMCを、適当な濃度のエピトープペプチド−テトラマー試薬と反応させる。該エピトープペプチド−テトラマー試薬と結合したCTLは標識色素により染色されるので、フローサイトメーター、顕微鏡等を用いてカウントする。エピトープペプチド−テトラマー試薬と反応させる時に、エピトープペプチド−テトラマー試薬と異なる色素で標識された抗CD3抗体、抗CD4抗体、抗CD8抗体等を反応させる事で、エプスタイン-バールウイルスに特異的なCTLのT細胞サブセットも同時に判定できる。
【0120】
(b)定量方法2
PBMCを本発明のCTLエピトープペプチドで刺激することによってCTLが産生するIFNIY(interferon gamma)、TNF(tumor necrosis factor)、インターロイキン等のサイトカイン及び/又はケモカインを定量する方法である。以下にIFNYを例にとり具体的に方法を示す。
【0121】
b−1 サイトカイン定量による方法1(細胞内IFNY産生細胞定量)
PBMCを適当な培地におよそ2×106/mLの細胞濃度で浮遊させ、本発明のCTLエピトープペプチドを加える。さらに細胞内蛋白輸送阻止剤(例えば、Brefeldin AやMonensin等)を加え、5%CO2恒温槽にて37℃で5〜16時間培養する。培養後、T細胞マーカー抗体(抗CD3抗体、抗CD4抗体、抗CD8抗体)あるいは/または、エピトープペプチド−テトラマー試薬と反応させ、細胞を固定後、膜透過処理を行い、色素標識抗IFNY抗体を反応させる。フローサイトメーター等を用いて解析し、全細胞中、T細胞中あるいはエピトープペプチド−テトラマー陽性細胞中のIFNY陽性細胞率を定量する。
【0122】
b−2 サイトカイン定量による方法2(エリスポットアッセイ)
抗IFNY抗体を固相化した96穴MultiScreen-HAプレート(Millipore社)にPBMCをまく。エピトープペプチドを各穴に入れ37℃の5%CO2恒温槽培養器にて20時間培養する。翌日、プレートを洗浄し、抗IFNY抗体、ペルオキシダーゼ標識抗IgG抗体の順で反応させる。さらにペルオキシダーゼの基質を加え、発色によりIFNYスポットを可視化し、実体顕微鏡でカウントする。
【0123】
b−3 サイトカイン定量による方法3(培養上清中に分泌されたIFNYを定量する方法)
PBMCを適当な培地におよそ2×106/mLの細胞濃度で浮遊させ、本発明のCTLエピトープペプチドを加える。5%CO2恒温槽にて37℃で24〜48時間培養する。培養後、上清を回収し、その中に含まれるIFNY濃度を市販のELISAキット(例えばMBL社のHUMAN IFN gamma ELISA)を使用して定量する。
【0124】
(c)定量方法3
細胞表面蛋白質特異的抗体を用いて定量を行う。特異的CTLは、特異的なペプチドの刺激により細胞表面に発現が増強する細胞表面蛋白質(例えばCD107a、CD107b、CD63、CD69など)が報告されている。このような蛋白質を特異的に認識する標識抗体と、ペプチド刺激したPBMC等を混合する事で、標識抗体と結合したCTLは標識色素により染色されるので、フローサイトメーター、顕微鏡等を用いてカウントする。標識抗体と反応させる時に、同時にあるいは、順番に、標識抗体と異なる色素で標識された抗CD3抗体、抗CD4抗体、抗CD8抗体等を反応させる事で、エプスタイン-バールウイルスに特異的なCTLのT細胞サブセットも同時に判定できる。
【0125】
本発明の定量方法の好ましい態様として、具体的には、上述の(a)〜(c)の定量方法を挙げることができる。
従って本発明の定量方法の好ましい態様は、本発明のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法に関する。
【0126】
また、MHC−テトラマー(またはモノマーもしくは多量体)試薬は、前述のように特異的なCTLの分離精製特異的なCTLの定量だけでなく、特異的なCTLの定量に用いる事ができ、別の態様としては、本発明のペプチドからMHC−テトラマー試薬を調製し、MHC−テトラマー試薬と末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法に関する。
上記方法においては、被検者から予め取得・単離された末梢血試料を用いることができる。
【実施例】
【0127】
以下に実施例を示し、本発明を具体的に説明するが、本発明はこれらに限定されるものではない。
【0128】
〔実施例1〕細胞株および樹状細胞の調製
本発明者らは、まずEBV関連タンパク質のうち、LMP1に特異的なCTLクローンの誘導を行い、LMP1由来のエピトープの同定を行った。
LMP1由来のエピトープの同定に関連する実験(実施例1〜8)に用いられた、細胞株および樹状細胞の調製方法について以下に示す。
【0129】
愛知県がんセンター(日本)の倫理審査委員会によって承認された研究デザインと目的は、すべてのドナーに十分に説明され、インフォームド・コンセントを得た。
【0130】
EBVが感染したLCLを、公知の方法で作製し(Kuzushima, K., et al., Blood, 94, 3094-3100 (1999))、10%ウシ胎仔血清(PAA Laboratories、Pasching、オーストリア)、2mM L-グルタミン、50U/mLのペニシリン、50μg/mLストレプトマイシンと50μg/mLのカナマイシンを加えたRPMI1640(Sigma Chemical Co., 米国St. Lois、MO)で培養した。
【0131】
EBVを保持しているNK細胞株、SNK-6(Nagata, H., et al., Blood, 97, 708-713 (2001))とSNK-10(Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003))は清水博士(東京医科歯科大学、東京、日本)から供与された。別のEBVを保持しているNK細胞株であるHANK-1(Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998))は鏡味博士(愛知県がんセンター病院、名古屋、日本)から供与された。この3つの細胞株は当業者に公知の方法で培養した(Nagata, H., Blood, 97, 708-713 (2001))。
【0132】
HEK-293T細胞(American Type Culture Collection、米国マナッサス(ヴァージニア))とPhoenix-GALV細胞(Akatsuka, Y., et al., Tissue Antigens, 59, 502-511 (2002))(Kiem博士、フレッドハッチンソン癌研究センター、およびNolan博士、スタンフォード大学、米国スタンフォード、カリフォルニアから供与された)を、公知の方法で培養した。HLA遺伝子のレトロウイルスへの導入は、公知の方法で行った(Kondo, E., et al., J Immunol., 169, 2164-2171 (2002))。
【0133】
また、樹状細胞を公知の方法で調製した(Romani N, et al., J Exp Med. 1994;180:83-93, Sallusto F, et al., J Exp Med. 1994;179:1109-1118, Dauer M, et al., J Immunol. 2003;170:4069-4076)。CD8+T細胞を、PBMCsからCD8 MicroBeads(Miltenyi Biotec、ベルギッシュグラートバハ(ドイツ))を用いて分離し、−135℃で保存した。
【0134】
CD8を除いたPBMCを、5%のヒトの血清(MP Biomedicals、Aurora、OH)と2mMのL-グルタミン、50U/mLのペニシリン、50μg/mLのストレプトマイシン、50μg/mLのカナマイシンを含む4mLのRPMI1640培地(DC培地)に懸濁し、6ウエルプレートの一つのプレート中で37℃、2時間インキュベートした。
【0135】
非接着性細胞を穏やかにピペッティングすることで取り除き、接着細胞を50ng/mLの顆粒球マクロファージコロニー刺激因子(GM-CSF、Osteogenetics、Wuerzburg、ドイツ)と10ng/mL IL-4(Osteogenetics)を含むDC培地で培養した。2日目と4日目に、半分の培地を、GM-CSFとIL-4を含んでいる新鮮なDC培地に交換した。6日目に、樹状細胞を集め、mRNAを導入するため、エレクトロポレーションを行なった。
【0136】
CD40刺激で活性化されたB細胞(CD-40B)を、米国ボストン、MAにあるDana-Farber癌研究所のFreeman博士から供与された3T3NIH/ヒトCD40リガンド細胞を利用して、公知の方法に従って作成した(Kondo, E., et al.,J Immunol., 169, 2164-2171 (2002))。
【0137】
〔実施例2〕欠失変異体ΔLMP1 mRNAの作成
欠失変異体△LMP1を発現している抗原提示細胞を作製するために、本発明者らは、pcDNA/ΔLMP1プラスミドからインビトロ転写システムを用いて、欠失変異体ΔLMP1 mRNAを産生した。
【0138】
N末から43アミノ酸を削った欠失変異体ΔLMP1(Gottschalk, S., Blood, 101, 1905-1912 (2003))を作製する為に、EBV株B95-8(NCBI accession no.V01555)のcDNAをテンプレート、5'-AAGCTTGCCACCATGAGTGACTGGACTGGA-3'(配列番号:23)をセンスプライマー、5'--TTGAATTCTTAGTCATAGTAGCTTAGCTGA-3'(配列番号:24)をアンチセンスプライマーとして、PCRを行った。欠失変異体ΔLMP1のアミノ酸配列およびDNA配列を、配列番号:34および35に示す。出来上がったDNA断片を、HindIIIとEcoRI制限酵素認識部位を使って、pcDNA3.1(+) (Invitrogen、米国カールスバッド(CA))に挿入した(pcDNA/ΔLMP1)。ΔLMP1から更にN末端及びC末端のアミノ酸を削った欠失変異体を作るために、切断断片をpcDNA/ΔLMP1をテンプレートとしてPCRで作製し、pcDNA3.1(+)に挿入した。幾つかの短いLMP1ペプチド断片をコードするプラスミドを構築するために、各相補的塩基配列オリゴヌクレオチドのペアをアニールし、制限酵素で切断したpcDNA3.1(+)に挿入した。
【0139】
ΔLMP1と蛍光蛋白EGFP(Kondo, E., et al., J Immunol., 169, 2164-2171 (2002))のcDNAをpMSCVpuroというレトロウイルスベクター(BD Biosciences Clontech、米国パロアルト(CA))に挿入し、pMSCVpuro/ΔLMP1及びpMSCVpuro/EGFPを作製した。ショートヘアピンRNA(shRNA)干渉レトロウイルスベクターを、RNAi-Ready pSIREN-RetroQベクター(BD Biosciences Clontech)を用いて構築した。
【0140】
レトロウイルス生産細胞を確立するために、pMSCVpuro/ΔLMP1、pMSCVpuro/EGFPとRNAi-Ready pSIREN-RetroQ系の様々なベクターをLipofectamine2000(Invitrogen)を利用してPT67細胞(BD Biosciences Clontech)の中に組み込んだ。LCL細胞を、8mg/L polybrene(Sigma Chemical Co.)を含むレトロウイルス培養上清で感染させ、1時間32℃、1000xgで遠心分離し、2日間37℃で培養した。その後、このLCL細胞を、プロマイシン0.8 μg/mLの存在下で更に14日間培養した。EGFPとLMP1の発現をフローサイトメトリーで解析した。
【0141】
〔実施例3〕欠失変異体ΔLMP1 mRNAの細胞への導入および、細胞におけるΔLMP1の発現確認
T7プロモータ領域とΔLMP1をコードする領域を含む断片を、pcDNA/ΔLMP1をテンプレートとしてPCRで作製した。増幅されたDNAは、5’-capped mRNAのインビトロ転写の際のテンプレートとして利用された。5’-capped mRNAのインビトロ転写を、mMESSAGE mMACHINEキット(Ambion,米国 Austin, TX)を用いて行った。
【0142】
次に、polyAポリメラーゼ(Ambion)を用い3’polyA末端を追加し、RNeasyキット(QIAGEN、日本、東京)によって精製した。
【0143】
エレクトロポレーションに先立ち、樹状細胞とCD40-B細胞を、無血清のRPMI1640を用いて二回洗浄し、最終的に2.5×107細胞/mLとなるように調整した。40μLのRPMI1640培地中で20μgのmRNAと混合した後に、2mmのセルの中に入れElectro Square Porator ECM830(ハーバードApparatus、ホリストン(MA))を用いてエレクトロポレーションを行った。条件設定は樹状細胞に対しては450V、500μS、CD40-B細胞に対しては350V、350μSに設定した。
【0144】
エレクトロポレーションの後、樹状細胞の場合はIL-4とGM-CSFを追加したDC培地の中で3時間培養し、成熟させるためにTNF-α(PeproTech,米国Rocky Hill, NJ)、IL-1β(PeproTech)とPGE2(Cayman Chemical,米国 Ann Arbor, MI)を加えた。CD40-B細胞を、直ちに放射線照射したNIH/3T3-ヒトCD40リガンド細胞の上に播いて、36-48時間後抗原提示細胞として使用した。
【0145】
LMP1抗原の細胞内染色はわずかな変更以外、公知の方法により行われた(Gottschalk, S., et al., Blood, 101, 1905-1912 (2003))。エレクトロポレーションで遺伝子導入された細胞を集め、10分間室温で、4%パラホルムアルデヒドを加えたPBS中で固定した。PBSで洗浄してから、細胞はIC Perm (BioSource International、キャマリロ、米国カリフォルニア州)で膜透過処理され、30分間4℃でLMP-1のC末端を認識するモノクローン抗体(CS1-4、DAKO Cytomation、Glostrup、デンマーク)と反応させた。PBSで洗浄した後、細胞はフルオレセインイソチオシアネート(FITC)標識抗マウスIgG(H+L)(Immunotech、マルセイユ(フランス))で30分間4℃染色した。染色した細胞を、CellQUESTソフトウェア(BD Biosciences、米国サンノゼ、カリフォルニア州)を使用したFACSCallibur(BD Biosciences)によって分析した。
【0146】
フローサイトメトリーでΔLMP1の発現を解析した結果、樹状細胞とCD40-B細胞の両方の70%以上がΔLMP1陽性であることが明らかとなった(図1A)。エレクトロポレーション後36-48時間で、生存細胞は8割以上であった(データは示さない)。
これらの細胞を、抗原提示細胞として、血清反応陽性献血者5人からLMP1特異的なT細胞の誘導を試みた。
【0147】
〔実施例4〕ΔLMP1 mRNAを導入した樹状細胞を用いた、ΔLMP1特異的CTLクローンの誘導
次に、ΔLMP1特異的CTLクローンの誘導を以下の工程により行った。
【0148】
保存されたCD8+T細胞を、解凍し洗浄してから、10%ヒト血清、2mM L-グルタミン、50U/mLペニシリン、50μg/mLストレプトマイシン、50μg/mLカナマイシン、25ng/mL IL-7(R&D Systems、ミネアポリス(米国ミネソタ州))と5ng/mL IL-12(R&D Systems)を加えたRPMI1640倍地2mLにおいて、5%CO2加湿インキュベーターの中で、33Gyの放射線を当てた自己のΔLMP1-mRNA導入樹状細胞と共培養した。8日目と15日目に、ΔLMP1-mRNAを導入しγ-放射線を照射した樹状細胞及びCD40-B細胞で、T細胞を再び刺激した。各刺激の1日後に、IL-2(塩野義、大阪、日本)を20U/mLの濃度になるように加えた。T細胞クローンを確立するために、公知の方法で(Kuzushima, K., et al., Blood, 98, 1872-1881 (2001))、96穴丸底ウェルのを用いてポリクローンCTLの限界希釈法を実施した。
【0149】
2週間の培養後、増殖しているウェルを3つに分けて、その中の一部ずつをエフェクター細胞として、それぞれΔLMP1-mRNAやEGFP-mRNAを導入した自己のCD40-B細胞のCTLアッセイに用いた。LMP1-mRNAを導入したCD40-B細胞の一分あたりの放射カウントが、EGFP-mRNAを導入したCD40-B細胞の一分あたりの放射カウントの平均の標準偏差の三倍を超えた場合に、そのウェルを陽性として記録した。陽性ウェルをフラスコに移して、公知の方法によって増殖させた(Kuzushima, K., et al., Blood, 98, 1872-1881 (2001))。
【0150】
ΔLMP1-mRNAを導入しγ-放射線を照射した樹状細胞及びCD40-B細胞について3回の刺激の後、T細胞株の特異性を調べる為にELISPOTアッセイを行った。
ELISPOTアッセイは、公知の方法によって実施した(Kuzushima, K., et al., Blood, 94, 3094-3100 (1999), Kondo, E., et al., J Immunol., 169, 2164-2171 (2002), Kuzushima, K., et al., Blood, 98, 1872-1881(2001))。CD8+T細胞を、抗ヒトインターフェロンγ(IFNγ)モノクローン抗体(Pierce Biotechnology、フィラデルフィア(PA))でコーティングされたMultiscreen-HAプレート(MAHA S4510; Millipore、米国ビルリカ(MA州))のウェルの中で、様々な刺激因子と共に培養した。
【0151】
刺激因子として、Lipofectamin 2000 (Invitrogen)を用いてプラスミドを導入したHLA-A*0206陽性あるいは陰性LCL細胞(1ウェル当たりに10万細胞)、又はHLA-A*0206を発現するHEK-293T細胞(A0206-295T)(1ウェル当たり5万細胞)を、T細胞を加える48時間前に各ウェルに播いた。ペプチドタイトレーションアッセイを行うために、いくつかの濃度の合成ペプチド(Greiner, Frickenhausen, Germany)を1時間室温でA0206-295T細胞にパルスした。抗ウサギIFNγポリクローナル抗体(Pierce Biotechnology)でプローブした後、ペルオキシダーゼ標識抗ウサギIgG抗体と反応させて(Genzyme、米国ケンブリッジ、MA州)スポットを視覚化し、プレートを洗浄し乾燥させた。IFNγのスポットを、実体顕微鏡を用いて計測した。
【0152】
上記の結果、5人のドナーのうちの1人由来のポリクローナルなT細胞は、ΔLMP1-mRNAを導入した自己のCD40-B細胞に反応して、特異的にIFN-γを分泌したが、非変異導入CD40-B細胞に対してはIFN-γを分泌しないことが明らかとなった(図1B)。
【0153】
バルクCTL株から、限界希釈法を用いて、H7と名前を付けたT細胞クローンを確立した。H7はΔLMP1-mRNAを導入した自己のCD40-B細胞を溶解したが、EGFP-mRNAを導入したCD40-B細胞を溶解しなかった(データは示さない)。血液ドナーの遺伝子型(HLA)を調べた所、HLA-A*0206、A*2402、B*0702、B*4801、Cw*0304、およびCw*0702であることが明らかになった。CTLエピトープを提示するHLA分子を特定するために、レトロウイルスベクターを用いて各HLA遺伝子を導入し、完全にHLAをミスマッチしたLCLが利用された。H7はHLA-A*0206を導入したLCLと培養したときに、IFN-γスポットを作ったので、HLA-A*0206がエピトープを提示する分子であることを示している(図1C)。
【0154】
〔実施例5〕LMP1エピトープの同定
HLA-A2スーパータイプに存在するペプチドYLLEMLWRL(配列番号:25)を除き、HLA-A*0206拘束性のLMP1由来エピトープはこれまでに報告されていない。H7はペプチドYLLEMLWRL(配列番号:25)でIFN-γスポットを作らなかった(データは示さない)ことから、H7が認識するエピトープの同定を行った。
C末端側を削ったいくつかのΔLMP1-mRNAをエレクトロポレーションで自己のCD40-B細胞に導入し、H7に認識されるかどうかをELISPOTアッセイで検討した。
図2Aに示されるように、抗原性はアミノ酸残基70と77の間のC-末端トランケーションで失われたことから、エピトープのC-末端はアミノ酸残基71と77の間に位置することが明らかとなった。
【0155】
プラスミドを導入したA0206-293T細胞のほうがCD40-B細胞およびmRNAより調製が簡便であることから、以降の検討においては、C末端側を削ったΔLMP1-mRNAを含んだプラスミドが導入されたA0206-293T細胞を用いた。A0206-293T細胞を抗原提示細胞として用いた際には、C-末端トランケーションがアミノ酸残基77と88の間に及んだときに、抗原性が失われた(データは示していない)。CD40-B細胞を使ったデータとの不一致の理由は不明である。ここでは、アミノ酸残基88でC-端末を削ったLMP1を使用した。N-末端側に更に削除を加えた一連のプラスミドを用意し、分析した(図2B)。最も短い刺激断片はアミノ酸残基64-83と特定された。N-末端とC-末端を正確に特定する為に、更に近辺のトランケーションを行った。
【0156】
図2Cに示されているように、アミノ酸残基64-71(IIILIIFI、配列番号:18)をコードするプラスミドが最も強い抗原性を示したが、アミノ酸残基64か71を削除すると抗原性は完全に失われた。
【0157】
アミノ酸残基64-71はH7の最短エピトープを構成する可能性があるが、発現ベクターのスタートコドンによってコードされるN-末端のメチオニンが、63位におけるイソロイシンの代用となり、MHC結合とH7認識のための構造的な必要性を満たしている可能性がある。この点を明確にするために、合成した8アミノ酸ペプチド(アミノ酸残基64-71:IIILIIFI、配列番号:18)と9アミノ酸ペプチド(アミノ酸残基63-71:IIIILIIFI、配列番号:1)をA0206-293T細胞にパルスし、H7の反応性をELISPOTアッセイで検討した。その結果、図2Dで示されるように、9アミノ酸ペプチドだけがH7によって認識され、最短のエピトープは位置63のイソロイシンから始まることが示された。
【0158】
〔実施例6〕免疫プロテアソームサブユニットip-LMP7のLMP1 63-71エピトープの産生における重要性についての検討
LMP1導入細胞の抗原処理に関して、ELISPOTアッセイで2つの不一致が観察された。
(1)H7は全長ΔLMP1-mRNAを導入したCD40-Bを認識した(図2A)が、同じ配列を発現しているプラスミドを導入したA0206-293T細胞を認識しなかった。
(2)H7はアミノ酸残基44−77をコードするトランケートされたΔLMP1-mRNAを導入したCD40-Bを認識した(図2A)が、同じ配列を発現しているプラスミドを導入したA0206-293T細胞を認識しなかった。
【0159】
ここで本発明者らは、抗原処理機構の違いがLMP1エピトープ産生における不一致の原因であるという仮説を立てた。A0206-293T細胞では主に標準プロテアソームが発現しているが、CD40-B細胞とLCL細胞の場合は免疫プロテアゾームが優勢である(Frisan, T., et al., Int J Cancer, 2000; 88: 881-888, Morel, S., et al., Immunity 2000; 12: 107-117)。標準プロテアゾームは抗原処理経路に重要な役割を持ち、細胞は免疫反応中にIFN-γに晒されると、低分子重蛋白2(ip-LMP2)や低分子重蛋白7(ip-LMP7)のような新しく合成されたの免疫プロテアゾームβサブユニットの誘導により、免疫プロテアゾームが構築され、プロテアゾーム活性は量的にも質的にも変化する。
【0160】
免疫プロテアゾームの効果がエピトープ処理に必要であるかどうかを調べるために、免疫プロテアゾームのサブユニットの発現が抑制されているLCL細胞を用いた。ip-LMP7とip-LMP2は、膜貫通タンパク質(例えば、EBV-LMP2(Lautscham, G., et al., J Virol., 77: 2757-2761(2003)), MAGE-3(Levitskaya, J., et al., Nature 375: 685-688 (1995))のエピトープ産生に関して不可欠な分子として知られているので、この二つをsiRNAの標的として利用した。
【0161】
本発明においては、以下のsiRNA標的を選択した。即ち、ip-LMP2については、AAGUGAAGGAGGUCAGGUAUA(配列番号:26)、 ip-LMP7についてはAGAUUAACCCUUACCUGCUTT(配列番号:27)を選択した。
shRNA構造物は、その後に5T終止シグナルが続く、センスとアンチセンス配列を分かつTTCAAGAGA-ループを含んでいる。これらの構造物は、2つの相補的DNAオリゴヌクレオチドとして合成され、アニールされ、ベクターのBamHIとEcoRI制限酵素認識部位の間に挿入された。更に、ネガティブコントロールsiRNA(BD Biosciences Clontech)が同じベクターに挿入され、コントロールとして使われた。クローン化された遺伝子は配列を解析してその同一性を確認した。
ip-LMP2とip-LMP7の発現を、公知の方法(Schwarz, K., et al., J Immunol., 165, 768-778(2000), Tajima, K., et al., Int J Cancer, 110, 403-412 (2004))、すなわちウェスターンブロッティング法により評価を行った。
【0162】
その結果、図3Aに示すように、対応するshRNA-ベクターを導入したLCL細胞では、ip-LMP7あるいはip-LMP2の発現は有意に減少していた。遺伝子抑制によるLMP1エピトープ産生における遺伝子抑制の効果は、ELISPOTアッセイで評価した。興味深いことに、H7クローンによるIFN-γのスポットの産生はip-LMP7を発現抑制した LCLで刺激したときには有意に減少したが、ip-LMP2の発現抑制はほとんど効果を示さなかった(図3B)。
上記の結果より、LMP1エピトープの処理と提示には、ip-LMP7が不可欠であることが明らかとなった。
【0163】
〔実施例7〕LMP1特異的CTLクローンのLCL細胞に対して細胞傷害性活性の測定
次に、H7のLCLに対する傷害活性を調べた。CTLアッセイは以下の工程により行った。標的細胞を37℃で1.5時間50μCiクロム(51Cr)でラベルし洗浄して、指示されたエフェクターと目的の割合になるように96穴プレート中でCTLと混合した。37℃で4時間または16時間培養した後に、上清の放射活性をγ計測器で数えた。特異的51Cr放出の割合は次のように計算した。:100x(実験的な放出-自然発生的な放出)/(最大放出-最小放出)
【0164】
標準的なCTLアッセイにより、H7は4時間のインキュベーションではHLA-A0206陽性LCL細胞を効果的に溶解できない(データは示さない)が、16時間後に自己及びHLA-A0206陽性の同種LCL細胞を溶解することが示された(図4A)。このことは、H7を介した細胞溶解のためのLCLにおけるLMP1の発現が、4時間でのCTLアッセイにおいては、不十分であることを示唆している。
そこで、図4Bに示すように、標的細胞としてΔLMP1を強制発現させたLCLで実験を行ったところ、H7は4時間CTLアッセイでΔLMP1を発現させたLCLを特異的に溶解したが、EGFPを発現させたLCLは溶解しなかった。
【0165】
〔実施例8〕EBVが感染したNK細胞株に対するLMP1特異的CTLクローンの細胞傷害性活性の検討
EBV LMP1は他の蛋白と共に潜伏感染様式IIIとしてLCL細胞に発現され、潜伏感染様式IIとしてNK/T細胞に発現される(Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003))。そこで、EBV潜伏感染様式II型の悪性腫瘍の代表として、EBV陽性NK細胞株(Nagata, H., et al., Blood 2001; 97: 708-713, Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003), Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998))に対するH7細胞の傷害活性を検討した。検討したLMP1を発現している3つのNK細胞株の内、2つはHLA-A*0206陽性であった。図5Aに示したように、H7細胞は一つのHLA-A*0206陽性株(SNK-10)を溶解したが、他の細胞(SNK-6)、あるいはHLA-A*0206陰性株(HANK-1)は溶解しなかった。HLA-A*0206を導入したHANK-1細胞はH7細胞によって特異的に溶解された(図5B)。エピトープペプチドでパルスされたSNK-6細胞はH7によって特異的に溶解された(図5A)ため、SNK-6細胞はLMP1エピトープによって突然変異を生じた可能性が考えられる。さらに、本発明者らはLMP1エピトープに結合するゲノムDNAをシークエンスした。その結果、3つのEBV陽性NK細胞株のすべてがアミノ酸残基55-80に影響を与えない同一の突然変異を保有していることが明らかとなった(データは示さない)。
【0166】
〔実施例9〕細胞株および樹状細胞の調製
本発明者らは、次にEBV関連タンパク質のうち、EBNA1に特異的なCTLクローンの誘導を行い、EBNA1由来のエピトープの同定を行った。
EBNA1由来のエピトープの同定に関連する実験(実施例9〜16)において用いられた、細胞株および樹状細胞の調製方法について以下に示す。
【0167】
愛知県がんセンター(日本)の倫理審査委員会によって承認された研究デザインと目的は、すべての献血者に対して十分に説明され、インフォームド・コンセントを得た。
CD40で活性化されたB細胞(CD40-B)は公知の方法に基づいて、献血者のPBMCから産生された(Schultze JL, et al., J Clin Invest. 100, 2757-2765 (1997), Kondo E, et al., J Immunol., 169, 2164-2171 (2002))。簡単に述べると、PBMCを、放射線照射されCD40Lで形質転換したNIH3T3細胞 (以下t-CD40Lと記載する。BostonにあるDana-Farber Cancer InstituteのDr. Gordon Freemanから提供されたものである)と、リコンビナントIL-4(Genzyme, Cambridge, MA)、およびシクロスポリンA (Sandoz, Basel, Switzerland)と共に培養液中で培養した。増殖したCD40-Bに、週に2回刺激を加えた。
EBVを保持したLCLは、公知の方法(Kuzushima K, et al., Blood, 94, 3094-3100 (1999))に基づき、B95-8細胞培養上清を用いて、PBMCを分化させることによって調製した。そして、10%ウシ胎仔血清、2mM L-グルタミン、50U/mLのペニシリン、50μg/mLストレプトマイシンと50μg/mLのカナマイシンを含むRPMI1640培地(シグマケミカル社、セントルイス、MO)中で培養した。
【0168】
また、樹状細胞を公知の方法で調製した(Romani N, et al., J Exp Med., 180, 83-93 (1994), Sallusto F, et al., J Exp Med., 179, 1109-1118 (1994), Dauer M, et al., J Immunol., 170, 4069-4076 (2003))。CD8+T細胞を、PBMCsからCD8 MicroBeads(Miltenyi Biotec、ベルギッシュグラートバハ(ドイツ))を用いて分離し、−135℃で保存した。
【0169】
CD8を除いたPBMCを、5%のヒトの血清(MP Biomedicals、Aurora、OH)と2mMのL-グルタミン、50U/mLのペニシリン、50μg/mLのストレプトマイシン、50μg/mLのカナマイシンを含む4mLのRPMI1640培地(DC培地)に懸濁し、6ウェルプレートの一つのプレート中で37℃、2時間インキュベートした。
【0170】
非接着性細胞を穏やかにピペッティングすることで取り除き、接着細胞を50ng/mLの顆粒球マクロファージコロニー刺激因子(GM-CSF、Osteogenetics、Wuerzburg、ドイツ)と10ng/mL IL-4(Osteogenetics)を含むDC培地で培養した。2日目と4日目に、半分の培地を、GM-CSFとIL-4を含んでいる新鮮なDC培地に交換した。6日目に、樹状細胞を集め、mRNAを導入するため、エレクトロポレーションを行なった。
【0171】
〔実施例10〕全長EBNA1 mRNAの作成
EBNA1を発現している抗原提示細胞を作製するために、本発明者らは、pcDNA/EBNA1プラスミドからインビトロ転写システムを用いて、ポリA鎖を持つ全長EBNA1 mRNAを産生した。
インビトロで転写されたEBNA1 mRNAを作製するために、まずpcDNA/EBNA1ベクターを構築した。RNeasy Kit(Qiagen、Hilden (ドイツ))を用いて、上記のB95.8で形質転換したLCLから全てのRNAの抽出し、EBNA1をコードする配列を得た。EBNA1のアミノ酸配列およびDNA配列を配列番号:36および37に示す。そして、逆転写の後に、以下の特異的プライマーを用いたPCR法によってEBNA1 cDNAを増幅した。
フォワードプライマー
5'-AAGCTTGCCACCATGTCTGACGAGGGGCCAGGTACAG (配列番号:28)
リバースプライマー
5'-GAATTCTCACTCCTGCCCTTCCTCACCCTC (配列番号:29)
【0172】
全長のEBNA1断片を、HindIIIとEcoRIサイトを用いてpcDNA3.1(+)(Invitrogen, Carlsba樹状細胞A)に挿入し、pcDNA/EBNA1を構築した。EBNA1が導入されたことを確認するために、配列決定を行った。得られたプラスミドDNAを、mMESSAGEとmMACHINEキット(Ambion, Austin, TX)を用いて線状化し、インビトロで転写した。ポリAポリメラーゼ(Ambion)を用いて、3'末端のポリA鎖をEBNA1 mRNAに付加した後、RNeasyキットを用いて精製した。精製したmRNAをReliantRNAゲルシステム(Cambrex、ロックランド(ME))を使用して確認した。
【0173】
Capped-mRNAの収量は低いことが明らかとなったが、これはおそらくGCを多く含む配列からなるGAr領域が原因であり、反応温度の変化や反応混合物に一本鎖結合タンパク質を加えても、収量の問題は完全には解決できなかった(データは示さない)。しかしながら、mRNAの量は十分であり、ゲル上でも単一のバンドとして観察された(図6A)。
【0174】
〔実施例11〕全長EBNA1 mRNAの細胞への導入および、細胞におけるEBNA1の発現確認
次に、樹状細胞とCD40-B細胞に、エレクトロポレーションによって全長EBNA1mRNAを導入した。まず、無血清のRPMI1640を用いて二度洗浄し、最終濃度が2.5×107細胞/mLとなるように懸濁した。40μLのRPMI1640培地中で20μgのmRNAと混合した後に、2mmのセルの中に入れElectro Square Porator ECM830(ハーバードApparatus、ホリストン(MA))を用いてエレクトロポレーションを行った。条件設定は樹状細胞に対しては450V、500μS、CD40-B細胞に対しては350V、350μSに設定した。
【0175】
エレクトロポレーションの後、樹状細胞の場合はIL-4とGM-CSFを追加したDC培地の中で3時間培養し、成熟させるためにTNF-α(PeproTech,米国Rocky Hill, NJ)、IL-1β(PeproTech)とPGE2(Cayman Chemical,米国 Ann Arbor, MI)を加えた。CD40-B細胞を、放射線照射したNIH/3T3-ヒトCD40リガンド細胞の上に直ちに播いて、36-48時間後抗原提示細胞として使用した。
【0176】
次に、以下の工程でEBNA1の染色を行い、細胞内におけるEBNA1の発現を確認した。EBNA1 mRNAを導入したCD40-B細胞を集め、10分間室温で4%のパラホルムアルデヒドを含むリン酸緩衝生理食塩水(PBS)に定着させた。PBSで洗浄した後に、細胞を30分間4℃で0.5%のTween-20を含むPBSで浸透化し、抗EBNA1ウサギポリクローナル抗体(鶴見達也博士から提供、愛知がんセンター研究所、名古屋、日本)と反応させた。PBSで洗浄した後に、細胞をFITC標識したヤギの抗ウサギIgG(Beckman Coulter、Fullerton(カリフォルニア))で30分間4℃染色した。染色した細胞は、CellQUESTソフトウェア(BD Biosciences)を用い、FACSCallibur(BD Biosciences、サンノゼ(カリフォルニア))で分析した。
【0177】
上記の結果、EBNA1の発現は、平均蛍光強度は低いように見えたが、ほとんどのCD40-B細胞の上に検出された(図6B)。樹状細胞は、ドナーのPBMCsから得られた細胞数が限られていたために、本分析では利用できなかった。
【0178】
〔実施例12〕EBNA1 mRNAを導入した樹状細胞を用いた、EBNA1特異的CTLクローンの誘導
健康なドナーの単核球由来の樹状細胞に、全長EBNA1 mRNAをエレクトロポレーションによって導入し、炎症性サイトカインの混合物の添加することにより、樹状細胞の成熟化を誘導した。
EBNA1特異的CTLクローンの誘導は以下の工程により行った。
保存されていたCD8+T細胞を解凍し、10%ヒト血清、2mM L-グルタミン、50U/mLペニシリン、50μg/mLストレプトマイシン、および50μg/mLカナマイシン(CTL培地という)を含み、さらに5ng/mL IL-7(R&D Systems, Minneapolis, MN) と5ng/mL IL-12 (R&D systems) 5ng/mLを加えた200μL のRPMI1640倍地中で、5%CO2濃度の下、湿潤インキュベーターで培養した。8、16、および23日目に、EBNA1-mRNAを導入し、γ線を照射した樹状細胞によって、T細胞を刺激した。各再刺激の1日後に、最終的に20U/mLの濃度になるように、IL-2(塩野義、大阪、日本)を加えた。T細胞クローンを確立するため、ポリクローナルなCTLの限界希釈法を実施した(Kuzushima K, et al., Blood. 2001;98:1872-1881)。
【0179】
多クローンのCD8+T細胞を、ガンマ線などを照射された1×105のPBMCs(33Gy)、および2×104LCLs(55Gy)に、CD3(30ng/mL、Ortho Biotech、ブリッジウォーター、ニュージャージー)特異的なモノクロナール抗体(mAb)を含むCTL培地を入れた、丸底の96ウェルのプレートの1ウェルに1細胞となるように蒔いた。翌日、IL-2を各ウエル(50U/mL)に加えた。2週間の培養後、良好に増殖しているウェルを二つに分け、ELISPOTアッセイで自己のEBNA1 mRNAで形質転換したCD40-B細胞、または自己のLCLに対するエフェクターとして使用した。
【0180】
刺激を三回行った後に、各微量培養液の一定量について、EBNA1mRNAを導入した自己のCD40-B細胞と接触させ、ELISPOTアッセイで特異的にIFNγを分泌する能力をテストした。
ELISPOTアッセイは、公知の方法で行った(Kuzushima K, et al., Blood, 101, 1460-1468 (2003))。CD8+T細胞を、抗ヒトインターフェロンγ(IFNγ)モノクローナル抗体(ピアスBiotechnology、フィラデルフィア(PA))でコーティングされたMultiscreen-HAプレート(Millipore、ビルリカ(MA))のウェル中で各種刺激因子と共に培養した。刺激因子として、(A)自己のEBNA1 mRNAを導入したCD40-B細胞若しくは導入しなかったCD40のB細胞と、(B)自己若しくは同種のLCL(1×105の細胞/ウィル)を各ウェルに蒔いた。(以下に説明するペプチドタイトレーションとオバーラッピングアッセイにおいては、濃度をふった合成ペプチドを室温で1時間、自己のCD40-B細胞に加えた。)
【0181】
その後、抗ヒトIFNγウサギポリクローナル抗体(ピアスBiotechnology)と反応させた後、ペルオキシダーゼでラベルした抗ウサギの免疫グロブリン抗体(ゲンザイム、ケンブリッジ、MA)と基質を加えて反応後、プレートを洗浄し乾燥させた。IFNγスポットは実体顕微鏡の下で数えた。
【0182】
上記の結果、36のウェルのうち、32ウェルの細胞中にEBNA1特異的CTLが存在した(データ示さず)。CTLクローンB5とC6を限界希釈法により樹立した。クローンは抗CD3モノクローナル抗体、照射されたフィーダー細胞およびIL-2と供に増殖させた。
これらのクローンは、EBNA1-mRNAを導入した自己のCD40-B細胞とLCLを認識するが、EBNA1-mRNAを導入しなかった自己CD40B細胞やHLAが不一致の同種LCLは認識しなかった(図7)。
【0183】
〔実施例13〕抗原を提示しているHLA分子の同定
献血者はHLA-A*2402、A*3101、B*1507、B*3501、およびCw*0303として遺伝学的に分類された。抗原を提示しているHLA分子を同定するため、HLAが部分的に一致しているLCLのパネルを、クローンB5若しくはC6を刺激してIFNγを生産するために用いた。自己のLCLに加えて、HLA-B*3501を発現している同種LCLが、CTLクローンC6によって認識された(図8A)。そして、HLA-Cw*0304または、HLA-Cw*0303を持つLCLが、クローンB5(図8B)によって認識された。このことは、HLA-B*3501がクローンC6認識のための推定上の制限因子であり、一方、HLA-Cw*0303とHLA-Cw*0304はクローンB5の制限因子として働いていることを示している。
【0184】
〔実施例14〕EBNA1抗原ペプチドの識別
エピトープ領域を特定するために、クローンB5およびC6を、20個のアミノ酸を持つペプチドのセットと共に自己のCD40-B細胞において刺激した。本発明に使用されたペプチドは、Bio-Synthesis, Inc. Lewisville, TX.から購入した。GArの一次構造はMHCクラスIエピトープを含みそうにないことから、本発明者らはエピトープソースとしてこの部分を除いた。アミノ酸セット(合計56のペプチド)は13残基のオーバーラップによって、GArドメイン除く完全なEBNA1タンパク質配列をカバーしている。
【0185】
上記の結果、ペプチド#24はクローンC6によって認識された(図9)。HLA-B*3501拘束性のエピトープに関し、HPVGEADYFEY(配列番号:30)という配列が以前に報告されている(Blake N, et al., Immunity. 1997;7:791-802)。
【0186】
このエピトープ配列はペプチド#24(402-421)の中央に位置していることから(図10A)、本発明者らは、クローンC6が、HPVGEADYFEY(配列番号:30)をエピトープペプチドとして認識しているだろうと考えた。
【0187】
クローンB5の場合では、2つの重複するペプチド#38(500-519)と#39(507-526)が、認識され(図9)、これらは図10Bでアンダーラインを引いた13アミノ酸配列VFVYGGSKTSLYN(配列番号:31)を共有している。HLA-Cw*0303に結合する最適のエピトープを予測するために、プログラムSYFPEITHIを用いた(http://www.syfpeithi.de/ Rammensee H, et al., Immunogenetics. 1999;50:213-219)。そして、プログラムSYFPEITHIによる予測に基づき、エピトープとなりうる候補のペプチド、VYGGSKTSL(509-517)(配列番号:22)、FVYGGSKTSL(508-517)(配列番号:3)、およびVFVYGGSKTSL(507-517)(配列番号:2)を合成した。
【0188】
C-末端にあるアンカーのロイシンとエピトープの三番目の部分にある補助的アンカーのバリンとチロシンはプログラムで予測されていたことから、発明者らは11mer(VFVYGGSKTSL、配列番号:2)と10mer(FVYGGSKTSL、配列番号:3)(図10B)について調べた。
【0189】
ペプチドを加えた標的細胞のHalf Maximal Recognitionはそれぞれ5-10nM の10残基ペプチドと1-5nM の11残基ペプチドで得られた(図10C)。9残基(VYGGSKTSL、配列番号:22)ははるかに高い濃度においても認識されなかった。
【0190】
ペプチド希釈アッセイによってもクローンB5(図10C)の最適なエピトープの長さは明らかにならなかったことから、本発明者らは、同じ疑問を解決するのに構造面からのアプローチを用いることに決めた。このために、本発明者らは10残基ペプチドFVYGGSKTSL(配列番号:3)若しくは11mer VFVYGGSKTSL(配列番号:2)を導入し、蛍光ラベルしたテトラマーを作製した。
【0191】
テトラマーの作製と染色は以下の工程により行った。
HLA-Cw*0303とCw*0304のcDNAクローンは、プライマーであるC03F(5'-AACCATGGGCAGCCATTCTATGCGCTATTTTTACACCGCTGTGTCCCGGCC-3'、配列番号:32)及びC03R(5'-AAGGATCCTGGCTCCCATCTCAGGGTGAGG-3'、配列番号:33)プライマーを用いて、HLA-Cw*0303とCw*0304の重鎖の細胞外ドメインをコードする配列をPCRで増やすためのテンプレート(鋳型)として使用された。
【0192】
C03FはE.coli BL21(DE3)pLysSでタンパク質発現を最適化するように設計された数個の塩基の置換を含んでいる。PCR産物は、NcoIとBamHIとで切断され、ベクターに組み込まれた。このベクターは、HLA配列の3'末端にBirA biotinylationサイトを含んでいる。
【0193】
組換えHLA-Cw*0303とCw*0304の分子はβ2-ミクログロブリンとペプチドFVYGGSKTSL(配列番号:3)若しくはVFVYGGSKTSL(配列番号:2)と共にインビトロで組み込まれた。ゲル濾過で精製された可溶性複合体は、BirA酵素(vidity LCC、デンバー(CO))を用いてビオチン化された。
【0194】
フィコエリスリン(PE)をラベルされたテトラマーは、PEでラベルされたストレプトアビジン(Molecular Probes、Carlsbad (カリフォルニア))にこれらのビオチン化複合体を混合して作製した。
【0195】
テトラマーでの染色は以下の通りに行われた。CTLクローン(2x105)を、15分間4℃でFITCラベルした抗CD8モノクローナル抗体(Caltag、バーリンゲーム(カリフォルニア))と、20μg/mLのテトラマーでインキュベートして染色した。二度洗浄した後、染色した細胞を、0.5%のパラホルムアルデヒドで固定し、フローサイトメトリーにより分析した。
【0196】
上記の分析の結果、図11に示されるように、これらのテトラマーは特異的にCTLクローンB5に結合した。しかしながら、10残基ペプチドを導入したテトラマーはB5クローンに対して、11残基ペプチドを導入したものより高い親和力を示し、このことは10残基ペプチドFVYGGSKTSL(配列番号:3)がCTLのための最小の、そして、最適のエピトープであること示唆している。さらに、クローンB5は、結果が図8Aのデータと同様に、10残基を組み込んだHLA-Cw*0304テトラマーに強く結合した。
【0197】
〔実施例16〕EBNA1特異的CTLsのEBVが感染したB細胞への成長抑制効果の検討
クローンのB5とC6は、クロムリリースアッセイにおいては、自己のLCLを溶解しなかった(データは示さない)。ここで、これらのEBNA1特異的CTLが、EBNA1を発現しているLCLの長期的成長と生存に影響を及ぼすか否かを検討した。
【0198】
拘束性HLA分子の存在下若しくは非存在下において、自己及び同種のLCLが、反応するCTLの存在下若しくは非存在下において96ウェルプレートに蒔かれた。
【0199】
成長抑制アッセイは、公知の方法を多少修正して実施した(Lee SP, et al., J Exp Med., 199, 1409-1420 (2004))。標的となるLCL細胞は2×104細胞/ウェルの濃度で3ウェルずつ丸底の96ウェルのプレートに蒔いた。 EBNA1特異的CTLクローン(1×104の細胞/ウェル)若しくはコントロールとしてのCTL培地を標的細胞の培地に加えた。すべての培地を週ごとに半分を替えることによって維持し、PE-cyanin5でラベルされた抗CD19と、PEによってラベルされた抗CD8モノクローナル抗体(Beckman Coulter)で染色し、フローサイトメトリーによる分析でB細胞の特性を確認した。さらに、培養液は4週間後にLCLがどれだけ増えたかを確認した。
最終的にLCLがどれだけ増えたかを、顕微鏡観察によって評価し、フローサイトメトリーによるCD19の発現で確認した。
【0200】
図12に示されるように、両方のCTLクローンは明確に自己のLCLだけではなく、拘束性HLAを持つ同種のLCLについて増殖を抑制した。このことは、CTLクローンが潜伏タイプIIIであるEBV-陽性細胞の成長を阻害する能力を持つことを示唆している。
【技術分野】
【0001】
本発明は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド、該ペプチドを用いたエプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するワクチン、エプスタイン-バールウイルスに対する受動免疫療法剤に関する。
【背景技術】
【0002】
エプスタイン-バールウイルス(記憶B細胞に長期間潜伏するヘルペスγウイルス、以下EBVと標記することもある)は、多くの悪性腫瘍に関連している。例えば、バーキットリンパ腫(BL)、ホジキン病(HD)、鼻咽頭癌(NPC)、または移植後リンパ増殖性疾患(PTLD)などである(非特許文献1)。潜伏感染では、ウイルス由来のタンパク質の発現は抑制されている。全てのEBV陽性の悪性細胞は、以下の3つの潜在タイプのうち一つを示し、発現されたEBV抗原のパターンによって互いに区別される(非特許文献2)。
潜伏感染様式1:バーキットリンパ腫では、EBVの核抗原(EBNA)1だけが発現される。
潜伏感染様式2:ホジキン病や鼻咽頭癌では、潜在膜蛋白1(以後LMP1と標記する)、LMP2、およびEBNA1が発現される。
潜伏感染様式3:移植後リンパ増殖性疾患では、EBVの潜在蛋白即ちEBNA1、2、3A、3B、3C、leader protein、LMP1およびLMP2が全て発現される。
【0003】
EBV関連悪性腫瘍の免疫治療方法に対する関心は増え続けており、インビトロで活性化されたEBV特異的な細胞傷害性T細胞(以後CTLと標記することもある)を用いた養子免疫療法は、造血幹細胞移植や臓器移植後におけるEBV関連リンパ増殖性疾患の予防や治療に効果があることが証明されている(非特許文献3〜7)。ホジキン病(非特許文献8)、鼻咽頭癌(非特許文献9)等のEBVが関連している悪性腫瘍への同様の戦略の応用は、何人かの患者で有効であることが報告されている。しかしながら、それらの研究で使用された、リンパ芽球細胞株(以後LCLと標記する)によって活性化されたCTLは主にEBNA3A、EBNA3BやEBNA3Cを標的としており、これらの抗原はホジキン病や鼻咽頭癌のような悪性腫瘍では発現されていない。LCLに活性化されたCTLの一部はLMP2由来のペプチドを認識し、患者の免疫治療効果に貢献した可能性がある(非特許文献8,9)。しかし、LMP1ペプチドを標的とするT細胞は非常にまれであり、それはLMP1ペプチド特異的なCTL先駆細胞の低い頻度を反映している(非特許文献10)。サブドミナントなEBV抗原に特異的なT細胞を選択的に活性化させるために、LinらはLMP2由来ペプチドでパルスされた単球由来の樹状細胞(DC)を利用してNPC患者を免疫した(非特許文献11)。また、LMP1特異的なCTLを活性化するためにいくつかの方法が報告されている。KhannaらはHLA-A2拘束性のLMP1エピトープと、該ペプチドでパルスした抗原提示細胞(APC)を用いたCTLの誘導について最初に報告している(非特許文献10)。また、彼らは複数のLMP1エピトープをコードする複製能力のないアデノウイルスや組み換えワクシニアウイルスを利用して、HLA-A2トランスジェニックマウスを免疫することに成功し、LMP1遺伝子導入細胞の成長を阻害できたことを報告している(非特許文献12,13)。Gottschalkらは、N末端を切断された毒性のない変種LMP1を発現している組み換えアデノウイルスを感染させたDCを利用して、ポリクローナルなLMP1特異的CTLの誘導を報告している(非特許文献14)。
【0004】
EBVが関連している悪性腫瘍の一つのカテゴリーとして、NK/T細胞におけるEBV感染が挙げられる(非特許文献1)。慢性活動性EBV感染(CAEBV)もEBVが主にNK/T細胞に感染し、生命に危険を及ぼすリンパ球増殖を引き起こす疾患である(非特許文献15)。EBV陽性のNK/T細胞悪性腫瘍は潜在的なCTLの標的としてEBNA1とLMP1を発現している(非特許文献15〜17)。
【0005】
LMP1は抗アポトーシス遺伝子のアップレギュレーションを通じて細胞の生存を促進する膜貫通癌蛋白質である(非特許文献2)。LMP1の発現はヒトBリンパ球の成長変化に不可欠であり、EBVが感染したヒト単核細胞の増殖に必要である(非特許文献2)。LMP1はまた、マウス細胞株BALB/c3T3やB細胞リンパ腫一般において癌原性の形質転換を誘導することが知られている。さらに、LMP1の発現はEBV感染NK細胞の増殖能に重要である可能性がある(非特許文献18)。しかしながら、NK/T細胞がLMP1を処理して、HLA拘束性のエピトープを産生できることは証明されていない。
【0006】
一方、EBNA1は、EBV-形質転換細胞におけるウイルスプラスミドの維持と複製に必要である(非特許文献19)。EBNA1はEBVが関連している全ての腫瘍で発現されることから、免疫治療のための魅力的な標的である。しかしながら、CD8+CTLの応答は潜伏感染抗原のうちEBNA3A,3Bおよび3Cに優先的に向けられ、EBNA1はCTLに認識されず、免疫学的には検出できないものと信じられていた(非特許文献20〜23)。EBNA1中のグリシンアラニン反復配列(GAr)はCTL認識のための抗原処理を妨げることがわかっている(非特許文献24)。このGArの存在は、MHCクラスIエピトープを産生する主要な触媒機構である、プロテアソームによる処理を妨げることが判明している(非特許文献25、26)。さらに、全く同じドメインが、EBNA1 mRNAの翻訳を妨げることが明らかとなっている(非特許文献27)。
【0007】
EBV特異的なCD4+T細胞の応答が調べられ、EBNA1特異的CD4+T細胞応答は主にヘルパーT細胞タイプ1で見られ、直接的にEBVが感染した細胞を認識することがわかった。数個のMHCクラスII拘束性のEBNA1エピトープが同定され(非特許文献28〜32)、これらのことから、EBNA1特異的CD4+T細胞が生体内で腫瘍の成長制御の役割を担っている可能性が示唆された。また、最近の研究では、驚くべきことに、EBNA1特異的CD8+CTLが、EBVが感染しているリンパ芽球様細胞(LCLs)を穏やかに溶解し、生体内でのLCLの成長を抑制されることが示されている(非特許文献33〜35)。しかしながら、CD8+CTLを活性化させる、EBNA1エピトープについてはこれまで少数が同定されたのみである。
【0008】
極少数の前駆細胞からCTLを誘導させる試みが、腫瘍関連抗原を標的にした免疫治療を望んでいる臨床医や免疫学者により行われてきた。特定の抗原をコードし、インビトロにおいて転写されたmRNAを導入した抗原提示細胞は、癌関連抗原または免疫寛容を超えて自己抗原に特異的なCTLを誘導する性質を持つ(非特許文献36〜40)。上記の手法には、以下の利点がある。
1)ベクターの主要な配列に対する免疫性の完全な欠失
2)in vitroでの転写による再現性の高い収率
3)エレクトロポレーションを利用した遺伝子導入の効率が高いこと
【0009】
すなわち、エプスタイン-バールウイルス抗原(エピトープ)のmRNAが導入された抗原提示細胞は、エプスタイン-バールウイルス特異的なCTLを誘導する上で適切な方法だと考えられる。上記の方法を実現するために、エプスタイン-バールウイルスに特異的なCTLエピトープペプチドの同定が求められてきた。
【0010】
なお、本出願の発明に関連する先行技術文献情報を以下に示す。
【先行技術文献】
【非特許文献】
【0011】
【非特許文献1】Babcock GJ., et al., Immunity, 13,497-506 (2000)
【非特許文献2】Rickinson AB., et al., In: Knipe DM, Howley PM, eds. Fields Virology (ed Fourth Edition), Philadelphia, Lippincott Williams & Wilkins, 2575-2628 (2001)
【非特許文献3】Rooney, CM., et al., Lancet, 345, 9-13 (1995)
【非特許文献4】Heslop, HE., et al., Nat Med., 2, 551-555 (1996)
【非特許文献5】Rooney, CM., et al., Blood, 92, 1549-1555 (1998)
【非特許文献6】Khanna, R., et al., Proc Natl Acad Sci U S A, 96, 10391-10396 (1999)
【非特許文献7】Comoli, P., et al., Blood, 99, 2592- 2598 (2002)
【非特許文献8】Bollard, CM., et al., J Exp Med, 200, 1623-1633 (2004)
【非特許文献9】Straathof, KC., et al., Blood 105, 1898-1904 (2005)
【非特許文献10】Khanna, R., et al., Eur J Immunol, 28, 451-458 (1998)
【非特許文献11】Lin, CL., et al., Cancer Res., 62, 6952-6958 (2002)
【非特許文献12】Duraiswamy, J., et al., Blood, 101, 3150-3156 (2003)
【非特許文献13】Duraiswamy, J., et al., Cancer Res., 64, 1483-1489 (2004)
【非特許文献14】Gottschalk, S., et al., Blood, 101, 1905-1912 (2003)
【非特許文献15】Kimura, H., et al., Blood, 98, 280-286 (2001)
【非特許文献16】Nagata, H., et al., Blood, 97, 708-713 (2001)
【非特許文献17】Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003)
【非特許文献18】Demachi, A., et al., Microbiol Immunol., 47, 543-52 (2003)
【非特許文献19】Kieff E., et al., In: Knipe DM, Howley PM, eds. Fields Virology (ed Fourth Edition) , Philadelphia, Lippincott Williams & Wilkins, 2511-2574 (2001)
【非特許文献20】Khanna R., et al., J Exp Med., 176, 169-176 (1992)
【非特許文献21】Murray RJ., et al., J Exp Med., 176, 157-168 (1992)
【非特許文献22】Steven NM., et al., J Exp Med., 184, 1801-1813 (1996)
【非特許文献23】Callan MF., et al., J Exp Med., 187, 1395-1402 (1998)
【非特許文献24】Levitskaya J., et al., Nature, 375, 685-688 (1995)
【非特許文献25】Blake N., et al., Immunity, 7, 791-802 (1997)
【非特許文献26】Levitskaya J., et al., Proc Natl Acad Sci U S A, 94, 12616-12621 (1997)
【非特許文献27】Yin Y., et al., Science, 301, 1371-1374 (2003)
【非特許文献28】Khanna R., et al., Virology, 214, 633-637 (1995)
【非特許文献29】Leen A., et al., J Virol., 75, 8649-8659 (2001)
【非特許文献30】Paludan C., et al., J Immunol., 169, 1593-1603 (2002)
【非特許文献31】Voo KS., et al., Cancer Res., 62, 7195-7199 (2002)
【非特許文献32】Kruger S., et al., J Immunother, 26, 212-221 (2003)
【非特許文献33】Lee SP., et al., J Exp Med., 199, 1409-1420 (2004)
【非特許文献34】Tellam J., et al., J Exp Med., 199, 1421-1431 (2004)
【非特許文献35】Voo KS., et al., J Exp Med., 199, 459-470 (2004)
【非特許文献36】Kuzushima, K., et al., Blood, 94, 3094-3100 (1999)
【非特許文献37】Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998)
【非特許文献38】Akatsuka, Y., et al., Tissue Antigens, 59, 502-511 (2002)
【非特許文献39】Kondo, E., et al., J Immunol., 169, 2164-2171 (2002)
【非特許文献40】Dauer, M., et al., J Immunol., 170, 4069-4076 (2003)
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明はこのような状況に鑑みてなされたものであり、その目的は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド、該ペプチドを用いたエプスタイン-バールウイルスの感染を治療又は予防するワクチン、エプスタイン-バールウイルスに対する受動免疫療法剤、およびエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明者らは、上記の課題を解決するために、エプスタイン-バールウイルス関連蛋白質であるLMP1およびEBNA1のmRNAを抗原提示細胞に導入し、該抗原提示細胞がエプスタイン-バールウイルスに特異的な細胞傷害性T細胞(CTL)を誘導する能力があるかを検討した。
【0014】
その結果、エプスタイン-バールウイルス関連蛋白質を発現させた抗原提示細胞は、健常人由来のCTLを刺激し、該CTLはEBVが感染しているB細胞の成長を阻害し、EBVが感染しているNKリンパ腫またはNK細胞を溶解することが明らかとなった。さらに、該CTLは、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子に提示されているエピトープペプチドを認識することが明らかとなった。
【0015】
即ち、本発明者らは、エプスタイン-バールウイルスに特異的なCTLエピトープペプチドを同定することに成功し、これにより本発明を完成するに至った。
【0016】
本発明は、より具体的には以下の(1)〜(16)を提供するものである。
(1) エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド。
(2) エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むものである、(1)に記載のペプチド。
(3) 配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、(1)に記載のペプチド。
(4) HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子の拘束性抗原ペプチドであって、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、(1)〜(3)のいずれかに記載のペプチド。
(5) (1)〜(4)のいずれかに記載のペプチドをコードする核酸。
(6) (1)〜(4)のいずれかに記載のペプチドを有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(7) (5)に記載の核酸を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(8) (1)〜(4)のいずれかに記載のペプチドをHLAに提示した抗原提示細胞を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
(9) (1)〜(4)のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
(10) (1)〜(4)のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
(11) (1)〜(4)のいずれかに記載のペプチドを用いて細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導方法。
(12) (1)〜(4)のいずれかに記載のペプチドと末梢血単核球を、血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的細胞傷害性T細胞を誘導する、(11)に記載の誘導方法。
(13) (1)〜(4)のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
(14) (1)〜(4)のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
(15) (1)〜(4)のいずれかに記載のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
(16) (1)〜(4)のいずれかに記載のペプチドから主要組織適合性抗原複合体-テトラマーを調製し、主要組織適合性抗原複合体-テトラマーと末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【発明の効果】
【0017】
本発明により、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドを同定することに成功した。該ペプチドは、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞(CTL)を効率的に誘導し得る機能を有する。従って該ペプチド及び該ペプチドをコードする核酸は、エプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するためのワクチン(能動免疫療法剤)として有用である。本発明で提供されるエピトープペプチドは、ワクチンとして用いる事で、エプスタイン-バールウイルス特異的CTLを生体内で誘導し、エプスタイン-バールウイルス感染に対して免疫力を保持させることができる。生体外において末梢血等の生体試料に対してもエプスタイン-バールウイルスを感染させる事無く、安全かつ効率的にエプスタイン-バールウイルス特異的CTLを人為的に誘導増殖させ細胞免疫療法に用いる事ができる。さらに、エプスタイン-バールウイルスに対する免疫力の有無の診断に用いる事ができ、エプスタイン-バールウイルス感染症に対して有効な治療法と診断方法を提供する。
【0018】
また、本発明のこれらのエピトープペプチドによって誘導されるCTLは、エプスタイン-バールウイルス感染細胞を特異的に溶解し、かつ該細胞の成長を阻害する機能を有し、受動免疫療法剤の成分として非常に有用である。
【0019】
さらに、該エピトープペプチドを用いることにより、エプスタイン-バールウイルスに特異的なCTLを定量することが可能である。エプスタイン-バールウイルスに特異的なCTLが、ハイリスクの患者の末梢血に存在するか否かを知ることは、抗ウイルス剤や免疫抑制剤の適正な使用を含め、これらの感染症管理の上で重要な情報である。
【図面の簡単な説明】
【0020】
【図1】樹状細胞またはCD40-B細胞へのΔLMP1mRNAの導入、およびΔLMP1の発現、CTLの誘導を示す図である。A:樹状細胞(DC)およびCD40-B細胞(CD40-B)において、ΔLMP1-mRNAを導入した後、フローサイトメトリーでΔLMP1の発現を解析した結果を示す図である。点線が遺伝子を導入しなかった細胞を示し、実線はΔLMP1を導入した細胞を示す。B:末梢血由来CD8+T細胞を、ΔLMP1-mRNA導入し、放射線を当てた自己の抗原提示細胞で3回刺激した後、ΔLMP1-mRNA導入あるいは非遺伝子導入CD40-B細胞を抗原提示細胞として用いたELISPOTアッセイを行った結果を示す図である。データはCD8+T細胞500個あたりのスポットの数を示す。C:HLA完全ミスマッチのLCL細胞に、ドナー由来の各HLA遺伝子を導入した6種類の抗原提示細胞を作成した。CTLクローンH7を刺激し、IFNγの産生をELISPOT法で測定した結果を示す図である。1ウェル当たり1,000個のH7細胞を入れた。
【図2】CTLクローンH7が認識するLMP1エピトープペプチドを示す図である。A:C-端末を削った一連のΔLMP1-mRNAを、インビトロ転写で作製した。各mRNAのスタートコドンとして、アミノ酸位置44番にあるメチオニンを利用した。C-端末を削った各ΔLMP1-mRNAを導入したCD40-B細胞を、ELISPOTアッセイにおける抗原提示細胞として使用した。白枠で示されたmRNA断片はH7細胞に認識されたが、黒枠で示されたmRNA断片はH7細胞に認識されなかった。B:一連のC-端末およびN-端末を削った断片はPCRによって増幅されて、pcDNA3.1(+)ベクターに導入された。導入したDNAによりコードされるアミノ酸配列が示されている。各プラスミドを導入したA0206-293T細胞に対する、H7細胞の認識をELISPOT法(ウェル当たりのH7細胞1,000個)により測定し、IFNγのスポット数によって以下の二つのパターンに分類した。(+)IFNγスポット数50以上;(-)IFNγスポット数10未満。C:各遺伝子を導入したA0206-293T細胞により刺激されたH7細胞の、IFNγスポット数を示す図である。各バーはH7細胞1,000個あたりのスポットの数を表す。D:様々な濃度の合成のペプチドでパルスされたA0206-293T細胞を用いて、ELISPOT測定を行った結果を示す図である。データはH7細胞500個あたりのスポットの数を示す。
【図3】LMP1エピトープ処理におけるip-LMP7の重要性を検討した結果を示す図である。A:コントロールsiRNA、ip-LMP2 siRNAまたはip-LMP7siRNAを、レトロウイルスを用いて、自己のLCL細胞に導入した。細胞を14日間プロマイシンで選別し、ウェスターンブロット解析を行った:ip-LMP2(上側のパネル);ip-LMP7(下側パネル)。B:ip-LMP2やip-LMP7遺伝子の発現を抑制した自己のLCLを用いたELISPOT測定の結果を示す図である。H7のIFNγスポットの産生を評価した。各バーはH7細胞5,000個あたりのスポット数を表す。
【図4】LMP1特異的CTLクローンH7の細胞傷害活性を示す図である。A:標的細胞として自己のA*0206を共有する細胞と、完全にHLAがミスマッチしているLCL細胞を用いた、16時間CTLアッセイの結果を示す図である。B:ΔLMP1あるいはEGFPを導入したLCLを標的細胞にした4時間CTLアッセイの結果を示す図である。各バーは3ウェルの細胞傷害活性の平均を示す。
【図5】CTLはEBV陽性NK細胞株を特異的に溶解する。A:H7 CTLクローンのEBV陽性NK細胞株に対する細胞傷害活性を、16時間CTLアッセイで計測した結果を示す図である。2つのHLA-A*0206陽性NK細胞株(SNK-6とSNK-10)、及び1つのHLA-A*0206陰性NK細胞株(HANK-1)のデータをそれぞれ、○、△及び□で示す。100nMのエピトープペプチドをパルスしたSNK-6細胞を使った4時間CTLアッセイで測定された細胞傷害活性を、●で示す。B:HLA-A*0206(◇)とA*2402(*)遺伝子導入HANK-1細胞を用いた、16時間CTLアッセイの結果を示す図である。
【図6】インビトロで転写された全長EBNA1 mRNAにより形質転換された細胞中でのEBNA1の発現を示す図である。A:インビトロで転写された全長EBNA1 mRNAを、EBNA1-cDNAプラスミドから作成した。EBNA1 mRNAをエチジウムブロマイドで染色し、その後ゲル電気泳動によって確認した。B:EBNA1-mRNAを導入したCD40-B細胞におけるEBNA1タンパク質の発現を示す図である。CD40-B細胞に全長EBNA1 mRNAをエレクトロポレーションによって導入し、EBNA1タンパク質の細胞内染色を行い、フローサイトメトリーで確認した。
【図7】EBNA1-mRNAを導入した樹状細胞を用いた培養液中における抗EBNA1−T細胞の存在を示す図である。健康なドナーからのCD8+T細胞を、インビトロで転写されたEBNA1 mRNAで形質転換した自己の樹状細胞で刺激した。一週間間隔で三回刺激した後、限界希釈法によって2つの陽性となった培地から、ポリクローナルなCD8+T細胞をクローン化した。次に、確立したクローンのB5とC6が、EBNA1-mRNAで形質転換した自己のCD40-B細胞、及び自己のLCLによって認識されるかどうかをELISPOTアッセイによって検討した。5,000個のCTLが各ウェルに蒔かれた。2つの実験のうち代表的な1つのデータを示した。
【図8】EBNA1特異的CTLクローンに提示されているHLA分子の同定の結果を示す図である。A:CTLクローンC6に対する拘束分子として、HLA-B*3501分子が機能することを示す図である。B:CTLクローンB5対する拘束分子として、HLA-Cw*0303とCw*0304が機能することを示す図である。自己及び同種のLCLを、B5またはC6クローンがIFNγスポットを生産するための抗原提示細胞として使用した。各LCLを、CTL(5x103)と共に20時間培養した。各バーは2つのウェルにおけるスポットの平均数を表わす。
【図9】EBNA1特異的CTLクローンによって認識されるオーバーラップしたペプチドの同定の結果を示す図である。GArドメインを除いたEBNA1タンパク質の全てのアミノ酸配列を網羅する、オーバーラップする20残基のペプチドのセット(各10μg/ml)で、自己のCD40-B細胞(1x105/ウエル)をパルスし、5x102のCTLクローンB5若しくはC6と共培養した。IFNγを産生するスポットを、ELISPOTアッセイで測定した。
【図10A−B】EBNA1特異的CTLクローンによって認識される最適のEBNA1抗原ペプチドの同定の結果を示す図である。A:HLA-B*3501拘束性のクローンC6によって認識される、オーバーラップするペプチドのアミノ酸配列を示す図である。既知のエピトープHPVGEADYFEY(配列番号:28)を、太字とアンダーラインで示す。配列の下の数字はEBNA1タンパク質におけるアミノ酸の番号を示す。B:クローンB5によって認識される2つの連続した、オーバーラップするペプチドアミノ酸配列および最適のエピトープ配列を示す図である。#38と#39ペプチドの間でオーバーラップする配列を、アンダーラインで示す。矢印はプログラムSYFPEITHIによって予測されたHLA-Cw*0303が固定するための第一かつ補助のアンカーを示す。配列の下の数字はEBNA1タンパク質におけるアミノ酸位置の数字を示す。
【図10C】EBNA1から誘導された合成のペプチドの力価測定の結果を示す図である。自己のCD40-B細胞を、合成ペプチド507-526(#39 20残基)、507-517(11残基)、508-517(10残基)、および509-517(9残基)の10倍ごとの連続希釈液と、1時間インキュベートした。次いでCTLクローンB5(200細胞/ウェル)を加えて、20時間培養した。各記号は2ウェルでの観察されたスポットの平均の数を示す。
【図11】HLA-Cw*0303とCw*0304のテトラマーの、B5 CTLクローンへの特異的結合を示す図である。HLA-Cw*0303-拘束性のEBNA1特異的CTLクローンB5は、PE標識したHLA-Cw*0303-FVYGGSKTSL(配列番号:3)、HLA-Cw*0303-VFVYGGSKTSL(配列番号:2)若しくはHLA-Cw*0304―FVYGGSKTSL(配列番号:3)テトラマー複合体及びFITC標識した抗CD8抗体で染色し、フローサイトメトリーで分析した。
【図12】EBNA1特異的CTLクローンの、HLAが一致するLCLへのインビトロでの増殖阻害を示す図である。標的となるLCL(2×104)を、丸底96ウェルのプレートの三つのウェルにおいて、EBNA1特異的CTLクローン(1×104)または培地のみ(陰性コントロール)と培養し、4週間後に細胞の成長を評価した。増殖した細胞がLCLであることは、CD19の発現で確認した。2つの実験のうちの代表的な1つのデータを示す。
【発明を実施するための形態】
【0021】
本発明は、エプスタイン-バールウイルスに特異的に細胞傷害性T細胞を誘導し得る機能を有するT細胞エピトープペプチドに関する。従って本発明は、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドを提供する。該エピトープペプチドは、本明細書において「エピトープペプチド」、あるいは単に「本発明のペプチド」と記載する場合がある。
【0022】
本発明において「ペプチド」とは、生理活性を有し、隣接するアミノ酸残基のα−アミノ基とカルボキシル基間のペプチド結合により相互に結合した線状のアミノ酸の分子鎖を意味する。ペプチドは特定長のものを意味するものではなく、種々の長さであり得る。従って、本発明のペプチドには、所謂「オリゴペプチド」、「ポリペプチド」も含まれる。また、無電荷又は塩の形態であってもよく、場合によっては、グリコシル化、アミド化、ホスホリル化、カルボキシル化、リン酸化等により修飾されていてもよい。
【0023】
以下に、エピトープの候補ペプチドの選択方法についてその一例を記載する。
1.コンピュータを用いた解析
本発明のエプスタイン-バールウイルスに特異的なCTLエピトープペプチドは、エプスタイン-バールウイルス関連タンパク質のアミノ酸配列について、目的とするHLA分子の各結合モチーフを有する8〜11個のアミノ酸よりなるエピトープペプチドを検索し得る、インターネット上に公開されている複数のソフトウェア(Pingping G., et al., Nucleic Acids Res., 31, 3621-3624(2003))に照合してCTLエピトープの候補ペプチドを選択する事ができる。本発明において、エプスタイン-バールウイルス関連タンパク質としては、EBNA1、2、3A、3B、3C、LMP1、LMP2、またはleader protein等を挙げることができ、より好ましくは、EBNA1またはLMP1を挙げることが出来る。EBNA1のアミノ酸配列およびDNA配列を配列番号:36および37に、N末端が欠損したLMP1のアミノ酸配列およびDNA配列を配列番号:34および35に示す。
【0024】
2.アンカーモチーフを用いた検討
HLAクラスI分子は、主としてHLA-A、HLA-B、HLA-Cがあり、これらに結合して提示されるエピトープペプチドは、8〜11個のアミノ酸からなる。エピトープペプチドのN末端側から2番目と、9あるいは10番目のアミノ酸はHLA分子との結合に対して最も重要なアミノ酸であり、アンカーモチーフと呼ばれている。このアンカーモチーフは、各々のHLA分子の種類によって異なることが報告されている。例えば、HLA-A2分子に結合するペプチドとしては、N末端より2番目の位置にLeuが配置され、9あるいは10番目の位置にLeu又はValが配置されたペプチドであって、9〜10個のアミノ酸残基からなるペプチドが最も良く知られている(Sudo T., et al., J. Immunol., 155, 4749-4756 (1995))。また、日本人を含むアジアの人種に多いHLA-A24に結合するペプチドとしては、N末端より2番目の位置にTyr、Phe、Met又はTrpのいずれかが配置され、9あるいは10番目の位置にLeu、Ile、Trp又はPheのいずれかが配置されたペプチドであって、9〜10個のアミノ酸からなるペプチドが最もよく知られている(Kondo A., J. Immunol., 155, 4307-4312 (1995))。蛋白質のアミノ酸配列中からこのアンカーモチーフを有する配列を検索し、CTLエピトープの候補ペプチドを選択する事ができる。
【0025】
3.ペプチドライブラリーの作製
エプスタイン-バールウイルス関連タンパク質の蛋白質全体を網羅する20個前後のアミノ酸よりなるペプチドライブラリーを合成する。20個前後のアミノ酸のうち、前後10個程度(例えば、7,8,9,10,11,12,13個)のアミノ酸は、前後のペプチド配列と重複するようにライブラリーを作製する。これによって、蛋白質全体を網羅的に検索する事ができる。
【0026】
なお、前述の方法にて選択されたエピトープ候補ペプチドは必ずしもCTLエピトープになり得る訳ではなく、以下に示す検討を経て初めてエプスタイン-バールウイルス特異的CTLエピトープになり得る。以下に、エピトープペプチドの決定方法について記載するが、これらの方法に限定されるものではない。
【0027】
(1)エピトープペプチド決定方法1
エプスタイン-バールウイルス感染歴がある人から分離した末梢血単核球(peripheral blood mononuclear cells;PBMC)、あるいはPBMCから分離したT細胞を適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させる。これに同じ人からあらかじめ分離培養しておいたエプスタイン-バールウイルス感染細胞1×105/mLを加え、5%炭酸ガス(CO2)恒温槽にて37℃で7日間培養する。培養7日後にエプスタイン-バールウイルス感染細胞とインターロイキン2(IL-2)を添加し、以後、エプスタイン-バールウイルス感染細胞とIL-2による刺激を毎週繰返すことによりCTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、MHC−テトラマー法、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する(Current Protocols in Immunology, Edited by: John E. Coligan, Ada M. Kruisbeek, David H. Margulies, Ethan M. Shevach, Warren Strober, 6.19 ELISPOT Assay to Detect Cytokine-Secreting Murine and Human Cells, 6.24 Detection of Intracellular Cytokines by Flow Cytometry, published by John Wiley & Sons, Inc.)。
【0028】
(2)エピトープペプチド決定方法2
エプスタイン-バールウイルス感染歴がある人から分離したPBMCを適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させ、これにエピトープ候補ペプチドの任意の1種を0.01〜100μg/mLの濃度で加える。5%CO2恒温槽にて37℃で培養し、2日後にIL-2を添加する。以後、前記ペプチドとIL-2による刺激を週に1度あるいは2週間に1度繰返すことによりCTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、MHC−テトラマー法、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する。
【0029】
(3)エピトープペプチド決定方法3
エプスタイン-バールウイルス感染歴がある人から分離したPBMCを適切な培地に0.1〜2×106/mLの細胞濃度で浮遊させ、これに合成したペプチドライブラリーを適当な数(例えば10種類ずつ)にプールした物を加える。5%CO2恒温槽にて37℃で培養し、2日後にIL-2を添加する。以後、プールペプチドとIL-2による刺激を週に1度あるいは2週間に1度繰返すことにより、CTLを誘導する。このようにして誘導したCTLがエピトープ候補ペプチドに対して特異性があるかどうかの検討は、エリスポットアッセイ、クロムリリースアッセイ、細胞内サイトカイン染色法等で判定する。良好な結果を示したプールペプチドに対しては、1種類ずつペプチドを加えて上記実験を繰返せば、CTL誘導能を有するペプチドを選択する事が可能である。反応したペプチドについて順次短くし、基幹のペプチドを得て本発明のエピトープペプチドとする。一般的に、エピトープペプチドは最終的に8〜11個のアミノ酸までに短くすることができる。
【0030】
本発明のペプチドとして具体的には、配列番号:1〜3のいずれかに記載の塩基配列からなるペプチドを例示することができる。
【0031】
また本発明のペプチドは、配列番号:1〜3のいずれかに記載のアミノ酸領域を含むペプチドであってもよい。即ち、本発明のペプチドの好ましい態様としては、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むペプチドである。
【0032】
さらに、本発明のエピトープペプチドは、生理活性及び免疫活性を実質的に改変せず、投与した場合に有害な活性を有するものでない限り、上記のペプチドの改変体であってもよい。
【0033】
即ち本発明の好ましい態様としては、配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、改変前の配列番号:1〜3のいずれかに記載のペプチドと同等の機能を有するペプチドを挙げることができる。
【0034】
上記「同等の機能」としては、例えば、(1)エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能、(2)ヒト主要組織適合性抗原等、特にHLA-A*0206分子、またはHLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞(CTL)を誘導し得る機能、等を挙げることができる。
【0035】
改変されたペプチドが、上記の機能を有するか否かは、例えば、後述の実施例に記載の方法、あるいは該方法を適宜改変した方法によって、評価することが可能である。
【0036】
本発明のペプチドは、天然に存在する状態から修飾されていないもの、及び修飾されているものの双方を含む。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチド又はヌクレオチド誘導体の共有結合、脂質又は脂質誘導体の共有結合、ホスファチジルイノシトールの共有結合、架橋、環化、ジスルフィド結合の形成、メチル化、脱メチル化、共有架橋の形成、シスチンの形成、ピログルタメートの形成、ホルミル化、Y-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリストイル化、酸化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、アルギニル化のようなタンパク質へのアミノ酸の転移RNA媒介付加、ユビキチン化等が含まれる。また、本発明のペプチドは、N末端又はC末端に付加的アミノ酸配列が介在するものであってもよい。また、本発明のペプチドは、糖類、ポリエチレングリコール、脂質等が付加された複合体、放射性同位元素等による誘導体、あるいは重合体等の形態として用いることができる。また、本発明におけるアミノ酸には、所謂「アミノ酸アナログ」が含まれる。該アミノ酸アナログとしては、例えば、種々のアミノ酸のN-アシル化物、O-アシル化物、エステル化物、酸アミド化物、アルキル化物等が挙げられる。
【0037】
また、本発明のペプチドと該HLA分子との複合体が、該複合体を細胞表面に提示する細胞を特異的に認識しうるTCRを有するCTLによって認識されるものである限り、本発明のペプチドのN末端や遊離のアミノ基には、ホルミル基、アセチル基、t-ブトキシカルボニル(t-Boc)基等が結合していてもよく、本発明のペプチドのC末端や遊離のカルボキシル基には、メチル基、エチル基、t-ブチル基、ベンジル基等が結合していてもよい。
【0038】
また、本発明のペプチドは、生体内への導入を容易にしうる各種修飾を施されたものであってもよい。生体内への導入を容易にしうる各種修飾の例としては、PT(Protein Transduction)ドメインが有名である。HIV(human immunodeficiency virus)のPTドメインは、Tatタンパク質の49〜57番目のアミノ酸(Arg Lys Lys Arg Arg Gln Arg Arg Arg/配列番号:38)で構成されたペプチドで、このペプチド配列を目的とするタンパク質あるいはペプチド配列の前後、又はいずれかに付加することで、容易に細胞内に導入できることが報告されている(Ryu J., Mol Cells., 16, 385-391 (2003), Kim DT., J Immunol., 159:1666-1668 (1997))。
【0039】
MHCクラスI分子を介して提示されるほとんどの抗原は、細胞質内のプロテアソームにより分解された後、TAP(transporter in antigen processing)へと移送され、粗面小胞体内においてTAPに会合しているMHCクラスI分子とβ2-ミクログロブリンの複合体と結合し、ゴルジ装置を経てエクソサイトーシスにより細胞表面へと運搬される。これら一連の抗原提示経路にて作用するシャペロンであるHSP(heart shock prtein)70やHSP90、又はgp96と目的とするペプチドやタンパク質を融合させることで、効果的な抗原提示がなされることが報告されており(Basu S., Immunity., 14, 303-313 (2001))、本発明のペプチドに上記PTドメインまたはシャペロンを融合させたタンパクは本発明の一態様を成す。
【0040】
また、本発明のペプチドは、従来の各種のペプチド合成方法によって調製され得る。例えば、固相ペプチド合成法等の有機化学的合成法、あるいは、ペプチドをコードする核酸を調製し、組換えDNA技術を用いて調製することも可能である。また、市販の化学合成装置(例えば、アプライドバイオシステムズ社のペプチド合成装置)による合成も可能である。
【0041】
即ち本発明のペプチドは、そのアミノ酸配列に従って、一般的な化学合成法により製造することが可能であり、該方法には、通常の液相法及び固相法によるペプチド合成法が包含される。該ペプチド合成法は、より詳しくはアミノ酸配列の情報に基づいて、各アミノ酸を1個ずつ逐次合成させて鎖を延長していくステップワイズエロンゲーション法と、アミノ酸数個からなるフラグメントを予め合成し、次いで各フラグメントをカップリング反応させるフラグメント・コンデンセーション法を包含し、本発明のペプチドの合成は、いずれの方法を用いてもよい。
【0042】
このようなペプチド合成法にて用いられる縮合法も、各種方法に従って行うことができる。その具体例としては、例えばアジド法、混合酸無水物法、DCC法、活性エステル法、酸化還元法、DPPA(ジフェニルホスホリルアジド)法、ウッドワード法等を例示できる。
【0043】
これら各種方法に利用できる溶媒もまた、一般的に使用されるものを適宜利用することができる。その例としては、例えばジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、ヘキサホスホロアミド、ジオキサン、テトラヒドロフラン(THF)、酢酸エチル等及びこれらの混合溶媒等を挙げることができる。なお、上記ペプチド合成反応に際して、反応に関与しないアミノ酸及びペプチドにおけるカルボキシル基は、一般にはエステル化により、例えばメチルエステル、エチルエステル、第三級ブチルエステル等の低級アルキルエステル、例えばベンジルエステル、P−メトキシベンジルエステル、P−ニトロベンジルエステルアラルキルエステル等として保護することができる。また、側鎖に官能基を有するアミノ酸、例えばTyrの水酸基は、アセチル基、ベンジル基、ベンジルオキシカルボニル基、第三級ブチル基等で保護されてもよいが、必ずしもかかる保護は必須ではない。また、例えば、Argのグアニジノ基は、ニトロ基、トシル基、2−メトキシベンゼンスルホニル基、メチシレン−2−スルホニル基、ベンジルオキシカルボニル基、イソボルニルオキシカルボニル基、アダマンチルオキシカルボニル基等の適当な保護基により保護することも可能である。
【0044】
上記のようにして得ることが可能な本発明のペプチドは、通常の方法に従って、例えばイオン交換樹脂、分配クロマトグラフィー、ゲルクロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィー(HPLC)、向流分配法等のペプチド化学の分野で汎用されている方法に従って、適宜、精製を行うことができる。
【0045】
本発明のペプチドは、本発明のペプチドをコードするDNA核酸分子を合成し、適当な発現ベクターへ導入した後、宿主細胞内において発現させる遺伝子工学的手法によっても取得することができる。
【0046】
一例を示せば、まず配列番号:1〜3のいずれかに記載されたアミノ酸配列をコードする核酸を合成する。該方法としては、リン酸トリエステル法やリン酸アミダイト法などの化学合成手段〔Letsinger, RL., et al., J. Am.Chem. Soc., 89, 4801 (1967);Letsinger, RL., et al., J. Am.Chem. Soc., 91, 3350-3355 (1969);Merrifield, R. B., et al., Science, 150, 178-185 (1968);Beaucage, SL and Caruthers, MH., Tetrahedron Lett., 22, 1859-1862 (1981); McBride, LJ and Caruthers, MH., Tetrahedron Lett., 24, 245 (1983)〕及びそれらの組合せ方法などが例示できる。より具体的には、DNAの合成は、ホスホルアミダイト法又はトリエステル法による化学合成により行うこともでき、市販されている自動ポリヌクレオチド合成装置上で行うこともできる。二本鎖断片は、相補鎖を合成し、適当な条件下で該鎖を共にアニーリングさせるか、又は適当なプライマー配列と共にDNAポリメラーゼを用い相補鎖を付加するかによって、化学合成した一本鎖生成物から得ることもできる。
【0047】
本発明のペプチドには、上述のように、本発明者らによって同定されたエピトープペプチド(配列番号:1〜3)と機能的に同等なペプチドが含まれる。
【0048】
あるペプチドと機能的に同等なペプチドを調製するための、当業者によく知られた方法としては、例えばペプチド中のアミノ酸配列に変異を導入する方法が挙げられる。具体的には当業者であれば部位特異的変異誘発法(Hashimoto-Gotoh, T. et al., Gene, 152, 271-275 (1995)、Zoller, MJ, and Smith, M. Methods Enzymol. 100, 468-500 (1983)、Kramer, W. et al., Nucleic Acids Res. 12, 9441-9456 (1984)、Kramer W, and Fritz HJ., Methods. Enzymol., 154, 350-367 (1987)、Kunkel,TA., Proc Natl Acad Sci USA., 82, 488-492 (1985)、Kunkel, Methods Enzymol., 85, 2763-2766 (1988))などを用いて、配列番号:1〜3のいずれかに記載のアミノ酸配列に適宜変異を導入することにより、該ペプチドと機能的に同等なペプチドを調製することができる。また、ペプチド中のアミノ酸の変異は自然に生じることもある。このように、人工的か自然に生じたものかを問わず、本発明者らにより同定されたエピトープペプチド(配列番号:1〜3)のアミノ酸配列において1もしくは複数のアミノ酸配列が変異したアミノ酸配列を有し、該ペプチドと機能的に同等なペプチドは、本発明のペプチドに含まれる。
【0049】
上述のようなアミノ酸の変更(改変)目的は、例えば、
1.HLAとの親和性を高める為の変更(Rosenberg SA., et al., Nat Med., 4, 321-327 (1998)、Berzofsky JA., et al., Nat Rev Immunol., 1, 209-219 (2001))、
2.TCRの認識性を向上させるための変更(Fong L., et al., Proc Natl Acad Sci U S A., 98, 8809-8814 (2001), Rivoltini L., et al., Cancer Res., 59, 301-306 (1999))、
3.血清中のペプチド分解酵素等による代謝を回避する為の変更(Berzofsky JA., et al., Nat Rev Immunol., 1, 209-219 (2001), Parmiani G., et al., J Natl Cancer Inst., 94, 805-818 (2002), Brinckerhoff LH., et al., Int J Cancer., 83, 326-334 (1999))等が挙げられる。
【0050】
上記変異体における、変異(置換、挿入、欠失等)するアミノ酸数は、本発明のペプチドの有する機能が保持される限り制限はないが、通常5アミノ酸以内であり、好ましくは4アミノ酸以内であり、より好ましくは3アミノ酸以内であり、さらに好ましくは1〜2アミノ酸である。また、上記改変が、例えば、配列番号:1〜3のいずれかに記載のペプチドの末端へのアミノ酸残基の付加である場合には、付加されるアミノ酸数は本発明のペプチドの有する機能が保持される限り制限はないが、通常、20アミノ酸以内であり、好ましくは10アミノ酸以内であり、より好ましくは5アミノ酸以内であり、さらに好ましくは3アミノ酸以内であり、最も好ましくは1〜2アミノ酸である。
【0051】
変異するアミノ酸残基の種類としては、改変前のアミノ酸の性質が保存されている他のアミノ酸(改変前のアミノ酸と類似のアミノ酸)に変異されることが望ましい。例えばアミノ酸側鎖の性質としては、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ酸(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)を挙げることができる(括弧内はいずれもアミノ酸の一文字表記を表す)。
【0052】
あるアミノ酸配列に対する1又は複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するペプチドがその生物学的機能(活性)を維持し得ることはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA., 81, 5662-5666 (1984)、Zoller, M. J. & Smith, M. Nucleic Acids Research, 10, 6487-6500 (1982)、Wang, A. et al., Science, 224, 1431-1433 (1984)、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA., 79, 6409-6413 (1982))。
【0053】
本発明のペプチドのアミノ酸配列に複数個のアミノ酸残基が付加されたペプチドには、これらペプチドを含む融合ペプチドが含まれる。融合ペプチドは、本発明のペプチドと他のペプチドとが融合したものである。融合ペプチドを作製する方法は、本発明のペプチド(例えば、配列番号:1〜3に記載のペプチド)をコードするポリヌクレオチドと他のペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。
【0054】
本発明のペプチドとの融合に付される他のペプチドは、CTLの誘導目的以外にも、例えば、該ペプチドの単離・精製、あるいは応用研究等の種々の目的に応じて、適宜選択することができる。例えばFLAG(Hopp, T. P. et al., BioTechnology, 6, 1204-1210 (1988))、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein Cの断片等の公知のペプチドを使用することができる。また、本発明のペプチドとの融合に付される他のタンパク質としては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチド又はペプチドをコードするポリヌクレオチドを本発明のペプチドをコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ペプチドを調製することができる。
【0055】
また、本発明のペプチドの融合体を作製する場合、融合箇所にFactorXa,エンテロキナーゼまたはトロンビンのようなプロテアーゼ切断部位ペプチド配列を挿入することもできる。このような場合、上記GST、MBP、あるいはFLAGタグのような精製用タグを付加させたペプチド融合体として発現させ、精製した後、必要に応じてペプチド融合体のうち、本発明の目的のペプチド以外の領域を、対応するFactorXa,エンテロキナーゼまたはトロンビンのようなプロテアーゼなどにより切断、除去することも可能であり、有用である。
【0056】
また、あるペプチドと機能的に同等なペプチドを調製する当業者によく知られた他の方法としては、ハイブリダイゼーション技術(Sambrook,J et al., Molecular Cloning 2nd ed., 9.47-9.58, Cold Spring Harbor Lab. press, (1989))を利用する方法が挙げられる。即ち、当業者であれば、本発明のペプチドをコードするポリヌクレオチドもしくはその一部をもとに、種々の生物由来(例えば、エプスタイン-バールウイルス由来)のポリヌクレオチド試料、あるいは人工的に合成されたペプチドライブラリー等から、これと相同性の高いポリヌクレオチドを単離して、該ポリヌクレオチドから本発明のペプチドと機能的に同等なペプチドを単離することも通常行いうることである。
【0057】
本発明には、本発明の配列番号:1〜3に記載のペプチドをコードするポリヌクレオチドとハイブリダイズするポリヌクレオチドによってコードされるペプチドであって、配列番号:1〜3に記載のペプチドと機能的に同等なペプチドが含まれる。
【0058】
配列番号:1〜3に記載のペプチドと機能的に同等なペプチドをコードするポリヌクレオチドを単離するためのハイブリダイゼーションの条件は、当業者であれば適宜選択することができる。ハイブリダイゼーションの条件としては、例えば、低ストリンジェントな条件が挙げられる。低ストリンジェントな条件とは、ハイブリダイゼーション後の洗浄において、例えば42℃、0.1×SSC、0.1%SDSの条件であり、好ましくは50℃、0.1×SSC、0.1%SDSの条件である。より好ましいハイブリダイゼーションの条件としては、高ストリンジェントな条件が挙げられる。高ストリンジェントな条件とは、例えば65℃、5×SSC及び0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。但し、ハイブリダイゼーションのストリンジェンシーに影響を及ぼす要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。
【0059】
また、ハイブリダイゼーションにかえて、遺伝子増幅技術(PCR)(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley&Sons Section 6.1-6.4)を用いて、本発明者のペプチド(配列番号:1〜3)をコードするポリヌクレオチドの一部を基にプライマーを設計し、本発明者らにより同定されたペプチドをコードするポリヌクレオチドと相同性の高いポリヌクレオチド断片を単離し、該ポリヌクレオチドを基に本発明者らにより同定されたペプチドと機能的に同等なペプチドを取得することも可能である。
【0060】
上述のハイブリダイゼーション技術や遺伝子増幅技術により単離されるポリヌクレオチドがコードする、配列番号:1〜3に記載のペプチドと機能的に同等なペプチドは、通常、該ペプチドとアミノ酸配列において、通常高い相同性を有する。本発明のペプチドには、配列番号:1〜3に記載のペプチドと機能的に同等であり、かつ該ペプチドのアミノ酸配列と高い相同性を有するペプチドも含まれる。高い相同性とは、アミノ酸レベルにおいて、通常、少なくとも50%以上の同一性、好ましくは75%以上の同一性、さらに好ましくは85%以上の同一性、さらに好ましくは95%以上(例えば、96%以上、97%以上、98%以上、99%以上)の同一性を指す。ペプチドの相同性を決定するには、文献(Wilbur, W. J. and Lipman, D. J. Proc. Natl. Acad. Sci. USA, 80, 726-730 (1983))に記載のアルゴリズムに従えばよい。
【0061】
アミノ酸配列の同一性は、例えば、Karlin and Altschul によるアルゴリズムBLAST (Proc. Natl. Acad. Sci. USA 87:2264-2268 (1990)、Proc. Natl. Acad. Sci. USA 90:5873-5877 (1993))によって決定することができる。このアルゴリズムに基づいて、BLASTXと呼ばれるプログラムが開発されている(Altschul et al. J. Mol. Biol.215: 403-410 (1990))。BLASTXによってアミノ酸配列を解析する場合には、パラメーターは例えば、score = 50、wordlength = 3とする。BLASTとGapped BLASTプログラムを用いる場合には、各プログラムのデフォルトパラメーターを用いる。これらの解析方法の具体的な手法は公知である(http://www.ncbi.nlm.nih.gov)。
【0062】
本発明のペプチドは、当業者に公知の方法により、組み換えペプチドとして、また天然のペプチドとして調製することが可能である。組み換えペプチドであれば、例えば、本発明のペプチドをコードするポリヌクレオチドを、適当な発現ベクターに組み込み、これを適当な宿主細胞に導入して得た形質転換体を回収し、抽出物を得た後、イオン交換、逆相、ゲル濾過などのクロマトグラフィー、あるいは本発明のペプチドに対する抗体をカラムに固定したアフィニティークロマトグラフィーにかけることにより、または、さらにこれらのカラムを複数組み合わせることにより精製し、調製することが可能である。
【0063】
精製用タグ(例えばヒスチジンタグ)を融合させた組み換えペプチドとして宿主細胞(例えば、動物細胞や大腸菌など)内で発現させた場合には、発現させた融合ペプチドは、市販の、融合したタグに対応した精製カラム(ヒスチジンタグの場合ニッケルカラム)を用いて精製することができる。
【0064】
天然のペプチドであれば、当業者に周知の方法、例えば本発明のペプチドを発現している組織や細胞の抽出物に対し、本発明のペプチドに結合する抗体が結合したアフィニティーカラムを作用させて精製することにより単離することができる。抗体はポリクローナル抗体であっても、モノクローナル抗体であってもよい。
【0065】
本発明のペプチドをコードする上記核酸もしくはベクターもまた、本発明に含まれる。
【0066】
本発明の核酸(ポリヌクレオチド)は、本発明のペプチドをコードし得るものであればいかなる形態でもよい。即ち、mRNAから合成されたcDNAであるか、ゲノムDNAであるか、化学合成DNAであるかなどを問わない。また、本発明のペプチドをコードしうる限り、遺伝暗号の縮重に基づく任意の塩基配列を有するポリヌクレオチドが含まれる。
【0067】
本発明の核酸(ポリヌクレオチド)は、当業者に公知の方法により取得することができる。例えば、市販の核酸合成装置を用いて、適宜作製することができる。作製したポリヌクレオチドの塩基配列は、公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法等により確認することができる。
【0068】
本発明のペプチドをコードする核酸は、例えば、遺伝子組換え技術を用いて、本発明のペプチドを宿主内で産生させる為に重要である。この場合、宿主間でアミノ酸コドンの使用頻度(codon usage)が異なる為、産生させる宿主のcodon usageに適合するようアミノ酸のコドンを変更することが望ましい。本発明のペプチドをコードする核酸は、ワクチンとしても利用可能で、むき出しの核酸として移送することもできるし、または適切なウイルスもしくは細菌ベクターを用いて移送することもできる(Berzofsky JA., et al., J Clin Invest., 114, 450-462 (2004), Berzofsky JA., et al., J Clin Invest., 113, 1515-1525 (2004))。適切な細菌ベクターとしては、サルモネラ属亜種の細菌ベクターを挙げることができる。適切なウイルスベクターとしては、レトロウイルスベクター、アデノウイルスベクター、センダイウイルスベクター、レンチウイルスベクター、ワクシニアベクター等を例示することができる。適切なワクシニアベクターの1例は、改変ワクシニア・アンカラベクターである。
【0069】
本発明の核酸の好ましい態様としては、本発明のペプチドを発現し得るベクターを例示することができる。該ベクターは、通常、プロモーターの下流に本発明の核酸が機能的に連結された構造のDNA構築物を担持するベクターである。該ベクターとしては、通常、プラスミドもしくはウイルスベクター等が一般的である。
【0070】
当業者においては、所望のDNAを有するベクターを、一般的な遺伝子工学技術によって、適宜、作製することが可能である。通常、市販の種々の発現ベクターを利用することができる。
【0071】
本発明のベクターは、宿主細胞内において本発明のポリヌクレオチドを保持したり、本発明のペプチドを発現させるためにも有用である。本発明の核酸(ポリヌクレオチド)は、通常、適当なベクターへ担持(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入したDNAを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明のペプチドを生産する目的としてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター(GenBank Accession No. AB009864)、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などを例示することができる。ベクターへの本発明の核酸の挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる。
【0072】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)及び植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法(GIBCO-BRL社製)、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0073】
宿主細胞において発現させたペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のペプチドに組み込むことができる。これらのシグナルは目的のペプチドに対して内因性であっても、異種シグナルであってもよい。
【0074】
上記製造方法におけるペプチドの回収は、本発明のペプチドが培地に分泌される場合は、培地を回収する。本発明のペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にペプチドを回収する。
【0075】
組換え細胞培養物から本発明のペプチドを回収し精製するには、硫酸アンモニウム又はエタノール沈殿、酸抽出、アニオン又はカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィー及びレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0076】
また、動物の生体内で本発明のポリヌクレオチド(核酸)を発現させる方法としては、本発明の核酸を適当なベクターに組み込み、例えば、レトロウィルス法、リポソーム法、カチオニックリポソーム法、アデノウイルス法などにより生体内に導入する方法などが挙げられる。これにより、免疫療法を行うことが可能である。用いられるベクターとしては、例えば、レトロウイルスベクターなどが挙げられるが、これらに制限されない。ベクターへの本発明のDNAの挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning ,5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0077】
本発明のペプチド(エプスタイン-バールウイルス特異的CTLエピトープペプチド)は、能動免疫療法においてペプチドワクチンとして用いることができる。即ち、本発明のCTLエピトープペプチドを含んでなるワクチンを健常人あるいは患者に投与し、エプスタイン-バールウイルスに特異的なCTLを体内で増殖させ、疾患に対する予防あるいは治療に役立てることができる。使用するエピトープペプチドは1種のみの使用であっても、あるいはワクチンの使用目的に応じて2種以上のペプチドを組み合わせ、混合して使用することもできる。
【0078】
従って本発明は、本発明のペプチドもしくは本発明の核酸を有効成分として含む、エプスタイン-バールウイルスの感染および同ウイルス陽性の癌を治療又は予防するためのワクチンを提供する。
【0079】
なお、本発明の「ワクチン」は、「能動免疫療法剤」、「免疫治療剤」、「エプスタイン-バールウイルス関連疾患治療剤」と表現することも可能である。また、本発明において「治療剤」は、「医薬品」、「医薬組成物」、「治療用医薬」等と表現することもできる。
【0080】
さらに、本発明のCTLエピトープペプチドが提示された抗原提示細胞は、能動免疫療法においてワクチンとして用いることができる。CTLエピトープペプチドが提示された抗原提示細胞とは、
1. 適当な培養液中で、抗原提示細胞とCTLエピトープペプチドを30分から1時間混合したエピトープペプチドパルス抗原提示細胞、
2. CTLエピトープペプチドをコードする核酸を用い、遺伝子導入等で抗原提示細胞にCTLエピトープペプチドを提示された細胞、
3. 人工的に作製した抗原提示能を有する人工抗原提示細胞、等を意味する。
【0081】
抗原提示細胞とは、例えば、樹状細胞、B細胞、マクロファージ、ある種のT細胞等を示すが、該ペプチドが結合し得るHLAをその表面上に発現する細胞であって、CTL刺激能を有するものを意味する。人工的に作製した抗原提示能を有する人工抗原提示細胞とは、例えば脂質2重膜やプラスティックあるいはラテックス等のビーズにHLAとCTLエピトープペプチドとβ2-ミクログロブリンとの3者複合体を固定し、CTLを刺激し得るCD80、CD83、CD86等の共刺激分子を固定するか、もしくは、共刺激分子と結合するT細胞側のリガンドであるCD28に対してアゴニスティックに作用する抗体等を固定することで作製可能である(Oelke M., et al., Nat Med., 9, 619-624 (2003), Walter S., et al., J Immunol., 171, 4974-4978 (2003), Oosten LE., et al., Blood, 104, 224-226 (2004))。
従って本発明の好ましい態様としては、本発明のペプチドをHLAに提示した抗原提示細胞を有効成分とするワクチンを提供する。
【0082】
本発明の核酸は、能動免疫療法においてDNAワクチンや組換えウイルスベクターワクチン等に用いる事ができる。この場合、CTLエピトープペプチドの核酸配列は、組換えワクチンや、組換えウイルスワクチンを産生させる宿主に適合したcodon usageに変更する事が望ましい(Casimiro, D.R., et al., J. Virol., 77, 6305-6313 (2003), Berzofsky JA, et al., J Clin Invest., 114, 450-462 (2004))。
【0083】
本発明のペプチド、又は本発明のペプチドが提示された抗原提示細胞を含んでなるワクチンは、当分野において公知の方法を用いて調製することができる。例えば、かかるワクチンの一例としては、本発明のペプチドを有効成分として含有する注射剤又は固形剤等の薬剤が挙げられる。
【0084】
本発明のペプチドは、エプスタイン-バールウイルスに対する受動免疫治療剤の調製に用いることができる。下記のようにして得られたエプスタイン-バールウイルスに特異的なCTLはヒトアルブミン含有PBS等に懸濁させて、エプスタイン-バールウイルスに対する受動免疫療法剤とすることができる。受動免疫療法剤に含まれるエプスタイン-バールウイルスに特異的なCTLは、以下のような調製方法によって得ることができ、CTLの純度を高める為に精製して用いる事も可能である。
以下にCTLの調製方法の具体例を記載するが、必ずしもこれらの方法に制限されない。
【0085】
(a)CTL調製方法1
PBMCと、適当な濃度のエプスタイン-バールウイルス特異的エピトープペプチド−テトラマー試薬を反応させる。エピトープペプチド−テトラマー試薬と結合したエプスタイン-バールウイルス特異的CTLは標識色素により染色されるので、セルソーター、顕微鏡などを用いて染色されたCTLのみを単離する。このようにして単離されたエプスタイン-バールウイルスに特異的なCTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤や、X照射あるいはマイトマイシン処理等で増殖能を損失させた抗原提示細胞で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0086】
(b)CTL調製方法2
エプスタイン-バールウイルス特異的エピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマー試薬を無菌プレートなどに固相化し、PBMCを固相化プレートで培養する。プレートに固相化されたエピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマーに結合したエプスタイン-バールウイルス特異的CTLを単離するためには、結合せずに浮遊している他の細胞を洗い流した後に、プレート上に残った抗原特異的CTLだけを新しい培地に懸濁する。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤や、X照射あるいはマイトマイシン処理等で増殖能を損失させた抗原提示細胞で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0087】
(c)CTL調製方法3
エプスタイン-バールウイルス特異的エピトープペプチド−モノマー及び/又はエピトープペプチド−テトラマー試薬と、CD80、CD83、CD86等の共刺激分子か、もしくは、共刺激分子と結合するT細胞側のリガンドであるCD28に対してアゴニスティックに作用する抗体等を無菌プレートなどに固相化し、PBMCを固相化プレートで培養する。2日後にIL-2を培地に添加し5%CO2恒温槽にて37℃で7〜10日培養する。培養した細胞を回収し新たな固相化プレート上で培養を続ける。この操作を繰り返す事で受動免疫療法に必要な細胞数のCTLを確保する。
【0088】
(d)CTL調製方法4
PBMCあるいはT細胞を本発明のCTLエピトープペプチドで直接刺激するか、該ペプチドをパルスした抗原提示細胞、遺伝子導入した抗原提示細胞、又は人工的に作製した抗原提示能を有する人工抗原提示細胞で刺激する。刺激によって誘導されたCTLを5%CO2恒温槽にて37℃で7〜10日培養する。CTLエピトープペプチドとIL-2、又は抗原提示細胞とIL-2による刺激を週に1度繰り返す事で受動免疫療法に必要な細胞数のCTLを確保する。
【0089】
なお、本発明のペプチド(エプスタイン-バールウイルス特異的CTLエピトープペプチド)を使用したエピトープペプチド−モノマー及びエピトープペプチド−テトラマーは、公知の方法(US Patent Number 5,635,363, Inventors : J. D. Altman, M. G. McHeyzer-Williams, Mark M. Davis, Compositions and methods for the detection, quantitation and purification of antigen-specific T cells, French Application Number FR9911133, Inventor : M. Bonneville, et al., Means for detecting and for purifying CD8+ T-lymphocyte populations specific for peptides presented in the HLA context)により調製することができる。タンパク質発現用の遺伝子組換え宿主から精製したHLAクラスI分子、β2−ミクログロブリン及び本発明のCTLエピトープペプチドの複合体である、MHC−モノマーをバッファー内で形成させる。組換えHLAクラスI分子のC末端には予めビオチン結合部位を付加しておき、エピトープペプチド−モノマー形成後この部位にビオチンを付加する。市販の色素標識されたストレプトアビジンとビオチン化エピトープペプチド−モノマーをモル比1:4で混合することによってエピトープペプチド−テトラマーを作製することができる。
【0090】
また、CTL調製方法において、特異的CTLの割合が低い場合は、随時以下の方法を用いる事でCTLを高純度で回収する事が可能である。
【0091】
(a)エピトープペプチド−テトラマー試薬による精製
エプスタイン-バールウイルス特異的エピトープペプチド−テトラマー試薬と、CTL調製方法にて誘導されたCTLを反応させ、エピトープペプチド−テトラマーを標識している標識色素に対する抗体等を磁気標識した2次抗体を用いて分離する事が可能である。このような磁気標識した2次抗体と、磁気細胞分離装置は例えば、Dynal社やMiltenyi Biotec GmbH社から入手可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0092】
(b)分泌されるサイトカインによる精製
エプスタイン-バールウイルス特異的CTLが、放出するサイトカイン等を利用して、エプスタイン-バールウイルスに特異的なCTLを精製する事ができる。例えば、Miltenyi Biotec GmbH社から入手可能なキットを用いる事で、CTLから放出されるサイトカインを細胞表面で特異抗体により補足し、サイトカイン特異的な標識抗体で染色し、続いて磁気標識した標識物質特異的な抗体で染色後、磁気標識細胞分離装置を用いて精製する事も可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0093】
(c)細胞表面蛋白質特異的抗体を用いた精製
特異的CTLの細胞表面では、特異的なペプチドの刺激により発現が増強する細胞表面蛋白質(例えばCD107a、CD107b、CD63、CD69など)が報告されている(Betts MR., et al., J Immunol Methods., 281, 65-78 (2003), Trimble LA., et al., J Virol., 74, 7320-7330 (2000))。このような蛋白質の特異抗体を磁気標識する事で、磁気分離装置等を用いてCTLを精製する事が可能である。また、このような特異抗体に対する抗IgG抗体等を磁気標識する事でも同様にCTLの精製が可能である。あるいは、これら抗体を培養用のプラスティックプレートにコートし、このプレートを用いて刺激を加えたPBMCを培養し、プレートに結合しなかった細胞集団を洗い流す事で特異的なCTLを精製することも可能である。このようにして単離されたエプスタイン-バールウイルス特異的CTLは、抗CD3抗体、PHA、IL-2等のT細胞刺激薬剤で刺激増殖させ、受動免疫療法に必要な細胞数を確保する。
【0094】
本発明は、上述のようにして取得・精製された細胞傷害性T細胞(CTL)を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤(免疫治療剤)を提供する。
なお、本発明の「受動免疫療法剤」は、「免疫治療剤」、「エプスタイン-バールウイルス関連疾患治療剤」、又は「CTL誘導剤」と表現することも可能である。
本発明の受動免疫療法剤の好ましい態様としては、本発明のペプチド、もしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤である。
【0095】
また、別の態様としては、本発明のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤である。
【0096】
本発明の薬剤(本発明のワクチンもしくは受動免疫療法剤等)は、生理学的に許容される担体、賦形剤、あるいは希釈剤等と混合し、医薬組成物として経口、あるいは非経口的に投与することができる。経口剤としては、顆粒剤、散剤、錠剤、カプセル剤、溶剤、乳剤、あるいは懸濁剤等の剤型とすることができる。非経口剤としては、注射剤、点滴剤、外用薬剤、あるいは座剤等の剤型を選択することができる。注射剤には、皮下注射剤、筋肉注射剤、あるいは腹腔内注射剤等を示すことができる。外用薬剤には、経鼻投与剤、あるいは軟膏剤等を示すことができる。主成分である本発明の薬剤を含むように、上記の剤型とする製剤技術は公知である。
【0097】
例えば、経口投与用の錠剤は、本発明の薬剤に賦形剤、崩壊剤、結合剤、及び滑沢剤等を加えて混合し、圧縮整形することにより製造することができる。
【0098】
例えば、本発明のペプチドが提示された抗原提示細胞は、製薬上許容され、該ペプチド又は該細胞の活性と相容性を有する賦形剤、例えば、水、食塩水、デキストロース、エタノール、グリセロール、DMSO(dimethyl sulphoxide)、及びその他のアジュバント等、又はこれらの組み合わせと混合して用いることができる。さらに、必要に応じて、アルブミン、湿潤剤、乳化剤等の補助剤を添加してもよい。崩壊剤としては、炭酸カルシウムやカルボキシメチルセルロースカルシウム等が一般的に用いられる。結合剤には、アラビアゴム、カルボキシメチルセルロース、あるいはポリビニルピロリドンが用いられる。滑沢剤としては、タルクやステアリン酸マグネシウム等が公知である。
【0099】
本発明の薬剤を含む錠剤は、マスキングや、腸溶性製剤とするために、公知のコーティングを施すことができる。コーティング剤には、エチルセルロースやポリオキシエチレングリコール等を用いることができる。
【0100】
また注射剤は、主成分である本発明の薬剤を適当な分散剤とともに溶解、分散媒に溶解、あるいは分散させることにより得ることができる。分散媒の選択により、水性溶剤と油性溶剤のいずれの剤型とすることもできる。水性溶剤とするには、蒸留水、生理食塩水、あるいはリンゲル液等を分散媒とする。油性溶剤では、各種植物油やプロピレングリコール等を分散媒に利用する。このとき、必要に応じてパラベン等の保存剤を添加することもできる。また注射剤中には、塩化ナトリウムやブドウ糖等の公知の等張化剤を加えることができる。更に、塩化ベンザルコニウムや塩酸プロカインのような無痛化剤を添加することができる。
【0101】
また、本発明の薬剤を固形、液状、あるいは半固形状の組成物とすることにより外用剤とすることができる。固形、あるいは液状の組成物については、先に述べたものと同様の組成物とすることで外用剤とすることができる。半固形状の組成物は、適当な溶剤に必要に応じて増粘剤を加えて調製することができる。溶剤には、水、エチルアルコール、あるいはポリエチレングリコール等を用いることができる。増粘剤には、一般にベントナイト、ポリビニルアルコール、アクリル酸、メタクリル酸、あるいはポリビニルピロリドン等が用いられる。この組成物には、塩化ベンザルコニウム等の保存剤を加えることができる。また、担体としてカカオ脂のような油性基材、あるいはセルロース誘導体のような水性ゲル基材を組み合わせることにより、座剤とすることもできる。
【0102】
また、本発明のペプチドは、中性又は塩の形態で処方することができ、例えば、薬学上許容され得る塩としては、塩酸、リン酸などの無機塩、又は、酢酸、酒石酸などの有機酸が挙げられる。
【0103】
本発明の薬剤を遺伝子治療剤として使用する場合は、本発明の薬剤を注射により直接投与する方法のほか、核酸が組込まれたベクターを投与する方法が挙げられる。上記ベクターとしては、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、レトロウイルスベクター、レンチウイルスベクター等が挙げられ、これらのウイルスベクターを用いることにより効率よく投与することができる。
【0104】
また、本発明の薬剤をリポソームなどのリン脂質小胞体に導入し、その小胞体を投与することも可能である。例えば、本発明のペプチドもしくはベクターを保持させた小胞体をリポフェクション法により所定の細胞に導入する。そして、得られる細胞を例えば静脈内、動脈内等に全身投与する。
【0105】
本発明の薬剤は、非経口投与及び経口投与により投与することができるが、該薬剤がペプチドを主成分とする場合には、一般的に非経口投与が好ましい。非経口投与としては経鼻投与や皮下注射、筋肉内注射、静脈内注射等の注射剤、座薬等がある。また、経口投与としては、スターチ、マンニトール、ラクトース、ステアリン酸マグネシウム、セルロース等の賦形剤との混合物として調製することができる。
【0106】
本発明のワクチンは、治療上有効な量で投与する。投与される量は、治療対象、免疫系に依存し、必要とする投与量は臨床医の判断により決定される。通常、適当な投与量は、患者一人当たり、本発明のペプチド(エピトープペプチド)では1〜100mg、エピトープペプチドパルス細胞では106〜109個の含有量とする。また、投与間隔は、対象、目的により設定することができる。
【0107】
また本発明の薬剤によって予防もしくは治療効果が期待されるエプスタイン-バールウイルス関連疾患としては、例えば、バーキットリンパ腫(BL)、ホジキン病(HD)、鼻咽頭癌(NPC)、または移植後リンパ増殖性疾患(PTLD)等の悪性腫瘍を挙げることができる。
【0108】
本発明の薬剤(ワクチン等)が接種可能な動物としては、免疫系を有し、かつエプスタイン-バールウイルスに感染し得る生物であれば特に制限されないが、好ましくは、ヒトである。
【0109】
さらに本発明は、本発明のペプチドを用いて、細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導(活性化)方法を提供する。
【0110】
本発明の誘導方法の好ましい態様としては、本発明のペプチド(例えば、HLA-A24等拘束性抗原ペプチド)と、末梢血単核球を血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的な細胞傷害性T細胞(CTL)を誘導する方法に関する。
【0111】
また、上記方法によって誘導されたエプスタイン-バールウイルス特異的CTLを検出及び定量する方法もまた本発明に含まれる。
【0112】
また、上述の方法によって誘導されたエプスタイン-バールウイルス特異的な細胞傷害性T細胞は、エプスタイン-バールウイルスに対する受動免疫療法剤の成分となり得る。従って、上述の方法によってエプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し、該T細胞を取得することによって、受動免疫療法剤を製造することが可能である。
【0113】
本発明は、本発明のエプスタイン-バールウイルス特異的な細胞傷害性T細胞の誘導方法を用いた受動免疫療法剤の製造方法を提供する。
【0114】
本製造方法の好ましい態様としては、本発明のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞によって末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む方法である。
【0115】
また好ましい別の態様においては、本発明のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法である。
【0116】
上記製造方法には、上述の工程に加えて、必要に応じて、「取得される細胞傷害性T細胞へ薬学的に許容される担体を混合する工程」を含んでいてもよい。
【0117】
さらに本発明のペプチドを用い作製した、MHC−テトラマー試薬は、特異的なCTLの定量だけでなく細胞内サイトカイン産生細胞定量法や細胞表面蛋白質に対する特異抗体と組み合わせることで、特異的CTLの分化段階や機能性も同時に判定する事が可能である。
本発明はさらに、本発明のペプチドを利用したエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量(検出・測定)方法を提供する。
【0118】
エプスタイン-バールウイルスに特異的なCTLが、ハイリスクの患者(何らかの原因により免疫能が低下した人、先天性免疫不全症患者、又は骨髄移植、造血幹細胞移植、臍帯血移植、固形臓器移植を受けて拒絶予防のために免疫抑制剤の投与を受けている患者、慢性ウイルス感染症患者、エイズ患者、高齢者、幼小児、妊婦等)の末梢血に存在するか否かを知ることは、抗ウイルス剤や免疫抑制剤の適正な使用を含め、これらの感染症管理の上で重要な情報である。エプスタイン-バールウイルスに特異的なCTLの定量は、例えば、本発明のペプチド(CTLエピトープペプチド)を用いた以下の3つの方法によって行うことができるが、必ずしもこれらの方法に制限されない。
【0119】
(a)定量方法1
本発明のCTLエピトープペプチドを使用して作製したMHC−テトラマー試薬を用いて、末梢血中のエプスタイン-バールウイルスに特異的なCTLを定量することができる。定量は、例えば、以下のようにして実施することができる。末梢血あるいはPBMCを、適当な濃度のエピトープペプチド−テトラマー試薬と反応させる。該エピトープペプチド−テトラマー試薬と結合したCTLは標識色素により染色されるので、フローサイトメーター、顕微鏡等を用いてカウントする。エピトープペプチド−テトラマー試薬と反応させる時に、エピトープペプチド−テトラマー試薬と異なる色素で標識された抗CD3抗体、抗CD4抗体、抗CD8抗体等を反応させる事で、エプスタイン-バールウイルスに特異的なCTLのT細胞サブセットも同時に判定できる。
【0120】
(b)定量方法2
PBMCを本発明のCTLエピトープペプチドで刺激することによってCTLが産生するIFNIY(interferon gamma)、TNF(tumor necrosis factor)、インターロイキン等のサイトカイン及び/又はケモカインを定量する方法である。以下にIFNYを例にとり具体的に方法を示す。
【0121】
b−1 サイトカイン定量による方法1(細胞内IFNY産生細胞定量)
PBMCを適当な培地におよそ2×106/mLの細胞濃度で浮遊させ、本発明のCTLエピトープペプチドを加える。さらに細胞内蛋白輸送阻止剤(例えば、Brefeldin AやMonensin等)を加え、5%CO2恒温槽にて37℃で5〜16時間培養する。培養後、T細胞マーカー抗体(抗CD3抗体、抗CD4抗体、抗CD8抗体)あるいは/または、エピトープペプチド−テトラマー試薬と反応させ、細胞を固定後、膜透過処理を行い、色素標識抗IFNY抗体を反応させる。フローサイトメーター等を用いて解析し、全細胞中、T細胞中あるいはエピトープペプチド−テトラマー陽性細胞中のIFNY陽性細胞率を定量する。
【0122】
b−2 サイトカイン定量による方法2(エリスポットアッセイ)
抗IFNY抗体を固相化した96穴MultiScreen-HAプレート(Millipore社)にPBMCをまく。エピトープペプチドを各穴に入れ37℃の5%CO2恒温槽培養器にて20時間培養する。翌日、プレートを洗浄し、抗IFNY抗体、ペルオキシダーゼ標識抗IgG抗体の順で反応させる。さらにペルオキシダーゼの基質を加え、発色によりIFNYスポットを可視化し、実体顕微鏡でカウントする。
【0123】
b−3 サイトカイン定量による方法3(培養上清中に分泌されたIFNYを定量する方法)
PBMCを適当な培地におよそ2×106/mLの細胞濃度で浮遊させ、本発明のCTLエピトープペプチドを加える。5%CO2恒温槽にて37℃で24〜48時間培養する。培養後、上清を回収し、その中に含まれるIFNY濃度を市販のELISAキット(例えばMBL社のHUMAN IFN gamma ELISA)を使用して定量する。
【0124】
(c)定量方法3
細胞表面蛋白質特異的抗体を用いて定量を行う。特異的CTLは、特異的なペプチドの刺激により細胞表面に発現が増強する細胞表面蛋白質(例えばCD107a、CD107b、CD63、CD69など)が報告されている。このような蛋白質を特異的に認識する標識抗体と、ペプチド刺激したPBMC等を混合する事で、標識抗体と結合したCTLは標識色素により染色されるので、フローサイトメーター、顕微鏡等を用いてカウントする。標識抗体と反応させる時に、同時にあるいは、順番に、標識抗体と異なる色素で標識された抗CD3抗体、抗CD4抗体、抗CD8抗体等を反応させる事で、エプスタイン-バールウイルスに特異的なCTLのT細胞サブセットも同時に判定できる。
【0125】
本発明の定量方法の好ましい態様として、具体的には、上述の(a)〜(c)の定量方法を挙げることができる。
従って本発明の定量方法の好ましい態様は、本発明のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法に関する。
【0126】
また、MHC−テトラマー(またはモノマーもしくは多量体)試薬は、前述のように特異的なCTLの分離精製特異的なCTLの定量だけでなく、特異的なCTLの定量に用いる事ができ、別の態様としては、本発明のペプチドからMHC−テトラマー試薬を調製し、MHC−テトラマー試薬と末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法に関する。
上記方法においては、被検者から予め取得・単離された末梢血試料を用いることができる。
【実施例】
【0127】
以下に実施例を示し、本発明を具体的に説明するが、本発明はこれらに限定されるものではない。
【0128】
〔実施例1〕細胞株および樹状細胞の調製
本発明者らは、まずEBV関連タンパク質のうち、LMP1に特異的なCTLクローンの誘導を行い、LMP1由来のエピトープの同定を行った。
LMP1由来のエピトープの同定に関連する実験(実施例1〜8)に用いられた、細胞株および樹状細胞の調製方法について以下に示す。
【0129】
愛知県がんセンター(日本)の倫理審査委員会によって承認された研究デザインと目的は、すべてのドナーに十分に説明され、インフォームド・コンセントを得た。
【0130】
EBVが感染したLCLを、公知の方法で作製し(Kuzushima, K., et al., Blood, 94, 3094-3100 (1999))、10%ウシ胎仔血清(PAA Laboratories、Pasching、オーストリア)、2mM L-グルタミン、50U/mLのペニシリン、50μg/mLストレプトマイシンと50μg/mLのカナマイシンを加えたRPMI1640(Sigma Chemical Co., 米国St. Lois、MO)で培養した。
【0131】
EBVを保持しているNK細胞株、SNK-6(Nagata, H., et al., Blood, 97, 708-713 (2001))とSNK-10(Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003))は清水博士(東京医科歯科大学、東京、日本)から供与された。別のEBVを保持しているNK細胞株であるHANK-1(Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998))は鏡味博士(愛知県がんセンター病院、名古屋、日本)から供与された。この3つの細胞株は当業者に公知の方法で培養した(Nagata, H., Blood, 97, 708-713 (2001))。
【0132】
HEK-293T細胞(American Type Culture Collection、米国マナッサス(ヴァージニア))とPhoenix-GALV細胞(Akatsuka, Y., et al., Tissue Antigens, 59, 502-511 (2002))(Kiem博士、フレッドハッチンソン癌研究センター、およびNolan博士、スタンフォード大学、米国スタンフォード、カリフォルニアから供与された)を、公知の方法で培養した。HLA遺伝子のレトロウイルスへの導入は、公知の方法で行った(Kondo, E., et al., J Immunol., 169, 2164-2171 (2002))。
【0133】
また、樹状細胞を公知の方法で調製した(Romani N, et al., J Exp Med. 1994;180:83-93, Sallusto F, et al., J Exp Med. 1994;179:1109-1118, Dauer M, et al., J Immunol. 2003;170:4069-4076)。CD8+T細胞を、PBMCsからCD8 MicroBeads(Miltenyi Biotec、ベルギッシュグラートバハ(ドイツ))を用いて分離し、−135℃で保存した。
【0134】
CD8を除いたPBMCを、5%のヒトの血清(MP Biomedicals、Aurora、OH)と2mMのL-グルタミン、50U/mLのペニシリン、50μg/mLのストレプトマイシン、50μg/mLのカナマイシンを含む4mLのRPMI1640培地(DC培地)に懸濁し、6ウエルプレートの一つのプレート中で37℃、2時間インキュベートした。
【0135】
非接着性細胞を穏やかにピペッティングすることで取り除き、接着細胞を50ng/mLの顆粒球マクロファージコロニー刺激因子(GM-CSF、Osteogenetics、Wuerzburg、ドイツ)と10ng/mL IL-4(Osteogenetics)を含むDC培地で培養した。2日目と4日目に、半分の培地を、GM-CSFとIL-4を含んでいる新鮮なDC培地に交換した。6日目に、樹状細胞を集め、mRNAを導入するため、エレクトロポレーションを行なった。
【0136】
CD40刺激で活性化されたB細胞(CD-40B)を、米国ボストン、MAにあるDana-Farber癌研究所のFreeman博士から供与された3T3NIH/ヒトCD40リガンド細胞を利用して、公知の方法に従って作成した(Kondo, E., et al.,J Immunol., 169, 2164-2171 (2002))。
【0137】
〔実施例2〕欠失変異体ΔLMP1 mRNAの作成
欠失変異体△LMP1を発現している抗原提示細胞を作製するために、本発明者らは、pcDNA/ΔLMP1プラスミドからインビトロ転写システムを用いて、欠失変異体ΔLMP1 mRNAを産生した。
【0138】
N末から43アミノ酸を削った欠失変異体ΔLMP1(Gottschalk, S., Blood, 101, 1905-1912 (2003))を作製する為に、EBV株B95-8(NCBI accession no.V01555)のcDNAをテンプレート、5'-AAGCTTGCCACCATGAGTGACTGGACTGGA-3'(配列番号:23)をセンスプライマー、5'--TTGAATTCTTAGTCATAGTAGCTTAGCTGA-3'(配列番号:24)をアンチセンスプライマーとして、PCRを行った。欠失変異体ΔLMP1のアミノ酸配列およびDNA配列を、配列番号:34および35に示す。出来上がったDNA断片を、HindIIIとEcoRI制限酵素認識部位を使って、pcDNA3.1(+) (Invitrogen、米国カールスバッド(CA))に挿入した(pcDNA/ΔLMP1)。ΔLMP1から更にN末端及びC末端のアミノ酸を削った欠失変異体を作るために、切断断片をpcDNA/ΔLMP1をテンプレートとしてPCRで作製し、pcDNA3.1(+)に挿入した。幾つかの短いLMP1ペプチド断片をコードするプラスミドを構築するために、各相補的塩基配列オリゴヌクレオチドのペアをアニールし、制限酵素で切断したpcDNA3.1(+)に挿入した。
【0139】
ΔLMP1と蛍光蛋白EGFP(Kondo, E., et al., J Immunol., 169, 2164-2171 (2002))のcDNAをpMSCVpuroというレトロウイルスベクター(BD Biosciences Clontech、米国パロアルト(CA))に挿入し、pMSCVpuro/ΔLMP1及びpMSCVpuro/EGFPを作製した。ショートヘアピンRNA(shRNA)干渉レトロウイルスベクターを、RNAi-Ready pSIREN-RetroQベクター(BD Biosciences Clontech)を用いて構築した。
【0140】
レトロウイルス生産細胞を確立するために、pMSCVpuro/ΔLMP1、pMSCVpuro/EGFPとRNAi-Ready pSIREN-RetroQ系の様々なベクターをLipofectamine2000(Invitrogen)を利用してPT67細胞(BD Biosciences Clontech)の中に組み込んだ。LCL細胞を、8mg/L polybrene(Sigma Chemical Co.)を含むレトロウイルス培養上清で感染させ、1時間32℃、1000xgで遠心分離し、2日間37℃で培養した。その後、このLCL細胞を、プロマイシン0.8 μg/mLの存在下で更に14日間培養した。EGFPとLMP1の発現をフローサイトメトリーで解析した。
【0141】
〔実施例3〕欠失変異体ΔLMP1 mRNAの細胞への導入および、細胞におけるΔLMP1の発現確認
T7プロモータ領域とΔLMP1をコードする領域を含む断片を、pcDNA/ΔLMP1をテンプレートとしてPCRで作製した。増幅されたDNAは、5’-capped mRNAのインビトロ転写の際のテンプレートとして利用された。5’-capped mRNAのインビトロ転写を、mMESSAGE mMACHINEキット(Ambion,米国 Austin, TX)を用いて行った。
【0142】
次に、polyAポリメラーゼ(Ambion)を用い3’polyA末端を追加し、RNeasyキット(QIAGEN、日本、東京)によって精製した。
【0143】
エレクトロポレーションに先立ち、樹状細胞とCD40-B細胞を、無血清のRPMI1640を用いて二回洗浄し、最終的に2.5×107細胞/mLとなるように調整した。40μLのRPMI1640培地中で20μgのmRNAと混合した後に、2mmのセルの中に入れElectro Square Porator ECM830(ハーバードApparatus、ホリストン(MA))を用いてエレクトロポレーションを行った。条件設定は樹状細胞に対しては450V、500μS、CD40-B細胞に対しては350V、350μSに設定した。
【0144】
エレクトロポレーションの後、樹状細胞の場合はIL-4とGM-CSFを追加したDC培地の中で3時間培養し、成熟させるためにTNF-α(PeproTech,米国Rocky Hill, NJ)、IL-1β(PeproTech)とPGE2(Cayman Chemical,米国 Ann Arbor, MI)を加えた。CD40-B細胞を、直ちに放射線照射したNIH/3T3-ヒトCD40リガンド細胞の上に播いて、36-48時間後抗原提示細胞として使用した。
【0145】
LMP1抗原の細胞内染色はわずかな変更以外、公知の方法により行われた(Gottschalk, S., et al., Blood, 101, 1905-1912 (2003))。エレクトロポレーションで遺伝子導入された細胞を集め、10分間室温で、4%パラホルムアルデヒドを加えたPBS中で固定した。PBSで洗浄してから、細胞はIC Perm (BioSource International、キャマリロ、米国カリフォルニア州)で膜透過処理され、30分間4℃でLMP-1のC末端を認識するモノクローン抗体(CS1-4、DAKO Cytomation、Glostrup、デンマーク)と反応させた。PBSで洗浄した後、細胞はフルオレセインイソチオシアネート(FITC)標識抗マウスIgG(H+L)(Immunotech、マルセイユ(フランス))で30分間4℃染色した。染色した細胞を、CellQUESTソフトウェア(BD Biosciences、米国サンノゼ、カリフォルニア州)を使用したFACSCallibur(BD Biosciences)によって分析した。
【0146】
フローサイトメトリーでΔLMP1の発現を解析した結果、樹状細胞とCD40-B細胞の両方の70%以上がΔLMP1陽性であることが明らかとなった(図1A)。エレクトロポレーション後36-48時間で、生存細胞は8割以上であった(データは示さない)。
これらの細胞を、抗原提示細胞として、血清反応陽性献血者5人からLMP1特異的なT細胞の誘導を試みた。
【0147】
〔実施例4〕ΔLMP1 mRNAを導入した樹状細胞を用いた、ΔLMP1特異的CTLクローンの誘導
次に、ΔLMP1特異的CTLクローンの誘導を以下の工程により行った。
【0148】
保存されたCD8+T細胞を、解凍し洗浄してから、10%ヒト血清、2mM L-グルタミン、50U/mLペニシリン、50μg/mLストレプトマイシン、50μg/mLカナマイシン、25ng/mL IL-7(R&D Systems、ミネアポリス(米国ミネソタ州))と5ng/mL IL-12(R&D Systems)を加えたRPMI1640倍地2mLにおいて、5%CO2加湿インキュベーターの中で、33Gyの放射線を当てた自己のΔLMP1-mRNA導入樹状細胞と共培養した。8日目と15日目に、ΔLMP1-mRNAを導入しγ-放射線を照射した樹状細胞及びCD40-B細胞で、T細胞を再び刺激した。各刺激の1日後に、IL-2(塩野義、大阪、日本)を20U/mLの濃度になるように加えた。T細胞クローンを確立するために、公知の方法で(Kuzushima, K., et al., Blood, 98, 1872-1881 (2001))、96穴丸底ウェルのを用いてポリクローンCTLの限界希釈法を実施した。
【0149】
2週間の培養後、増殖しているウェルを3つに分けて、その中の一部ずつをエフェクター細胞として、それぞれΔLMP1-mRNAやEGFP-mRNAを導入した自己のCD40-B細胞のCTLアッセイに用いた。LMP1-mRNAを導入したCD40-B細胞の一分あたりの放射カウントが、EGFP-mRNAを導入したCD40-B細胞の一分あたりの放射カウントの平均の標準偏差の三倍を超えた場合に、そのウェルを陽性として記録した。陽性ウェルをフラスコに移して、公知の方法によって増殖させた(Kuzushima, K., et al., Blood, 98, 1872-1881 (2001))。
【0150】
ΔLMP1-mRNAを導入しγ-放射線を照射した樹状細胞及びCD40-B細胞について3回の刺激の後、T細胞株の特異性を調べる為にELISPOTアッセイを行った。
ELISPOTアッセイは、公知の方法によって実施した(Kuzushima, K., et al., Blood, 94, 3094-3100 (1999), Kondo, E., et al., J Immunol., 169, 2164-2171 (2002), Kuzushima, K., et al., Blood, 98, 1872-1881(2001))。CD8+T細胞を、抗ヒトインターフェロンγ(IFNγ)モノクローン抗体(Pierce Biotechnology、フィラデルフィア(PA))でコーティングされたMultiscreen-HAプレート(MAHA S4510; Millipore、米国ビルリカ(MA州))のウェルの中で、様々な刺激因子と共に培養した。
【0151】
刺激因子として、Lipofectamin 2000 (Invitrogen)を用いてプラスミドを導入したHLA-A*0206陽性あるいは陰性LCL細胞(1ウェル当たりに10万細胞)、又はHLA-A*0206を発現するHEK-293T細胞(A0206-295T)(1ウェル当たり5万細胞)を、T細胞を加える48時間前に各ウェルに播いた。ペプチドタイトレーションアッセイを行うために、いくつかの濃度の合成ペプチド(Greiner, Frickenhausen, Germany)を1時間室温でA0206-295T細胞にパルスした。抗ウサギIFNγポリクローナル抗体(Pierce Biotechnology)でプローブした後、ペルオキシダーゼ標識抗ウサギIgG抗体と反応させて(Genzyme、米国ケンブリッジ、MA州)スポットを視覚化し、プレートを洗浄し乾燥させた。IFNγのスポットを、実体顕微鏡を用いて計測した。
【0152】
上記の結果、5人のドナーのうちの1人由来のポリクローナルなT細胞は、ΔLMP1-mRNAを導入した自己のCD40-B細胞に反応して、特異的にIFN-γを分泌したが、非変異導入CD40-B細胞に対してはIFN-γを分泌しないことが明らかとなった(図1B)。
【0153】
バルクCTL株から、限界希釈法を用いて、H7と名前を付けたT細胞クローンを確立した。H7はΔLMP1-mRNAを導入した自己のCD40-B細胞を溶解したが、EGFP-mRNAを導入したCD40-B細胞を溶解しなかった(データは示さない)。血液ドナーの遺伝子型(HLA)を調べた所、HLA-A*0206、A*2402、B*0702、B*4801、Cw*0304、およびCw*0702であることが明らかになった。CTLエピトープを提示するHLA分子を特定するために、レトロウイルスベクターを用いて各HLA遺伝子を導入し、完全にHLAをミスマッチしたLCLが利用された。H7はHLA-A*0206を導入したLCLと培養したときに、IFN-γスポットを作ったので、HLA-A*0206がエピトープを提示する分子であることを示している(図1C)。
【0154】
〔実施例5〕LMP1エピトープの同定
HLA-A2スーパータイプに存在するペプチドYLLEMLWRL(配列番号:25)を除き、HLA-A*0206拘束性のLMP1由来エピトープはこれまでに報告されていない。H7はペプチドYLLEMLWRL(配列番号:25)でIFN-γスポットを作らなかった(データは示さない)ことから、H7が認識するエピトープの同定を行った。
C末端側を削ったいくつかのΔLMP1-mRNAをエレクトロポレーションで自己のCD40-B細胞に導入し、H7に認識されるかどうかをELISPOTアッセイで検討した。
図2Aに示されるように、抗原性はアミノ酸残基70と77の間のC-末端トランケーションで失われたことから、エピトープのC-末端はアミノ酸残基71と77の間に位置することが明らかとなった。
【0155】
プラスミドを導入したA0206-293T細胞のほうがCD40-B細胞およびmRNAより調製が簡便であることから、以降の検討においては、C末端側を削ったΔLMP1-mRNAを含んだプラスミドが導入されたA0206-293T細胞を用いた。A0206-293T細胞を抗原提示細胞として用いた際には、C-末端トランケーションがアミノ酸残基77と88の間に及んだときに、抗原性が失われた(データは示していない)。CD40-B細胞を使ったデータとの不一致の理由は不明である。ここでは、アミノ酸残基88でC-端末を削ったLMP1を使用した。N-末端側に更に削除を加えた一連のプラスミドを用意し、分析した(図2B)。最も短い刺激断片はアミノ酸残基64-83と特定された。N-末端とC-末端を正確に特定する為に、更に近辺のトランケーションを行った。
【0156】
図2Cに示されているように、アミノ酸残基64-71(IIILIIFI、配列番号:18)をコードするプラスミドが最も強い抗原性を示したが、アミノ酸残基64か71を削除すると抗原性は完全に失われた。
【0157】
アミノ酸残基64-71はH7の最短エピトープを構成する可能性があるが、発現ベクターのスタートコドンによってコードされるN-末端のメチオニンが、63位におけるイソロイシンの代用となり、MHC結合とH7認識のための構造的な必要性を満たしている可能性がある。この点を明確にするために、合成した8アミノ酸ペプチド(アミノ酸残基64-71:IIILIIFI、配列番号:18)と9アミノ酸ペプチド(アミノ酸残基63-71:IIIILIIFI、配列番号:1)をA0206-293T細胞にパルスし、H7の反応性をELISPOTアッセイで検討した。その結果、図2Dで示されるように、9アミノ酸ペプチドだけがH7によって認識され、最短のエピトープは位置63のイソロイシンから始まることが示された。
【0158】
〔実施例6〕免疫プロテアソームサブユニットip-LMP7のLMP1 63-71エピトープの産生における重要性についての検討
LMP1導入細胞の抗原処理に関して、ELISPOTアッセイで2つの不一致が観察された。
(1)H7は全長ΔLMP1-mRNAを導入したCD40-Bを認識した(図2A)が、同じ配列を発現しているプラスミドを導入したA0206-293T細胞を認識しなかった。
(2)H7はアミノ酸残基44−77をコードするトランケートされたΔLMP1-mRNAを導入したCD40-Bを認識した(図2A)が、同じ配列を発現しているプラスミドを導入したA0206-293T細胞を認識しなかった。
【0159】
ここで本発明者らは、抗原処理機構の違いがLMP1エピトープ産生における不一致の原因であるという仮説を立てた。A0206-293T細胞では主に標準プロテアソームが発現しているが、CD40-B細胞とLCL細胞の場合は免疫プロテアゾームが優勢である(Frisan, T., et al., Int J Cancer, 2000; 88: 881-888, Morel, S., et al., Immunity 2000; 12: 107-117)。標準プロテアゾームは抗原処理経路に重要な役割を持ち、細胞は免疫反応中にIFN-γに晒されると、低分子重蛋白2(ip-LMP2)や低分子重蛋白7(ip-LMP7)のような新しく合成されたの免疫プロテアゾームβサブユニットの誘導により、免疫プロテアゾームが構築され、プロテアゾーム活性は量的にも質的にも変化する。
【0160】
免疫プロテアゾームの効果がエピトープ処理に必要であるかどうかを調べるために、免疫プロテアゾームのサブユニットの発現が抑制されているLCL細胞を用いた。ip-LMP7とip-LMP2は、膜貫通タンパク質(例えば、EBV-LMP2(Lautscham, G., et al., J Virol., 77: 2757-2761(2003)), MAGE-3(Levitskaya, J., et al., Nature 375: 685-688 (1995))のエピトープ産生に関して不可欠な分子として知られているので、この二つをsiRNAの標的として利用した。
【0161】
本発明においては、以下のsiRNA標的を選択した。即ち、ip-LMP2については、AAGUGAAGGAGGUCAGGUAUA(配列番号:26)、 ip-LMP7についてはAGAUUAACCCUUACCUGCUTT(配列番号:27)を選択した。
shRNA構造物は、その後に5T終止シグナルが続く、センスとアンチセンス配列を分かつTTCAAGAGA-ループを含んでいる。これらの構造物は、2つの相補的DNAオリゴヌクレオチドとして合成され、アニールされ、ベクターのBamHIとEcoRI制限酵素認識部位の間に挿入された。更に、ネガティブコントロールsiRNA(BD Biosciences Clontech)が同じベクターに挿入され、コントロールとして使われた。クローン化された遺伝子は配列を解析してその同一性を確認した。
ip-LMP2とip-LMP7の発現を、公知の方法(Schwarz, K., et al., J Immunol., 165, 768-778(2000), Tajima, K., et al., Int J Cancer, 110, 403-412 (2004))、すなわちウェスターンブロッティング法により評価を行った。
【0162】
その結果、図3Aに示すように、対応するshRNA-ベクターを導入したLCL細胞では、ip-LMP7あるいはip-LMP2の発現は有意に減少していた。遺伝子抑制によるLMP1エピトープ産生における遺伝子抑制の効果は、ELISPOTアッセイで評価した。興味深いことに、H7クローンによるIFN-γのスポットの産生はip-LMP7を発現抑制した LCLで刺激したときには有意に減少したが、ip-LMP2の発現抑制はほとんど効果を示さなかった(図3B)。
上記の結果より、LMP1エピトープの処理と提示には、ip-LMP7が不可欠であることが明らかとなった。
【0163】
〔実施例7〕LMP1特異的CTLクローンのLCL細胞に対して細胞傷害性活性の測定
次に、H7のLCLに対する傷害活性を調べた。CTLアッセイは以下の工程により行った。標的細胞を37℃で1.5時間50μCiクロム(51Cr)でラベルし洗浄して、指示されたエフェクターと目的の割合になるように96穴プレート中でCTLと混合した。37℃で4時間または16時間培養した後に、上清の放射活性をγ計測器で数えた。特異的51Cr放出の割合は次のように計算した。:100x(実験的な放出-自然発生的な放出)/(最大放出-最小放出)
【0164】
標準的なCTLアッセイにより、H7は4時間のインキュベーションではHLA-A0206陽性LCL細胞を効果的に溶解できない(データは示さない)が、16時間後に自己及びHLA-A0206陽性の同種LCL細胞を溶解することが示された(図4A)。このことは、H7を介した細胞溶解のためのLCLにおけるLMP1の発現が、4時間でのCTLアッセイにおいては、不十分であることを示唆している。
そこで、図4Bに示すように、標的細胞としてΔLMP1を強制発現させたLCLで実験を行ったところ、H7は4時間CTLアッセイでΔLMP1を発現させたLCLを特異的に溶解したが、EGFPを発現させたLCLは溶解しなかった。
【0165】
〔実施例8〕EBVが感染したNK細胞株に対するLMP1特異的CTLクローンの細胞傷害性活性の検討
EBV LMP1は他の蛋白と共に潜伏感染様式IIIとしてLCL細胞に発現され、潜伏感染様式IIとしてNK/T細胞に発現される(Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003))。そこで、EBV潜伏感染様式II型の悪性腫瘍の代表として、EBV陽性NK細胞株(Nagata, H., et al., Blood 2001; 97: 708-713, Zhang, Y., et al., Br J Haematol, 121, 805-814 (2003), Kagami, Y., et al., Br J Haematol, 103, 669-677 (1998))に対するH7細胞の傷害活性を検討した。検討したLMP1を発現している3つのNK細胞株の内、2つはHLA-A*0206陽性であった。図5Aに示したように、H7細胞は一つのHLA-A*0206陽性株(SNK-10)を溶解したが、他の細胞(SNK-6)、あるいはHLA-A*0206陰性株(HANK-1)は溶解しなかった。HLA-A*0206を導入したHANK-1細胞はH7細胞によって特異的に溶解された(図5B)。エピトープペプチドでパルスされたSNK-6細胞はH7によって特異的に溶解された(図5A)ため、SNK-6細胞はLMP1エピトープによって突然変異を生じた可能性が考えられる。さらに、本発明者らはLMP1エピトープに結合するゲノムDNAをシークエンスした。その結果、3つのEBV陽性NK細胞株のすべてがアミノ酸残基55-80に影響を与えない同一の突然変異を保有していることが明らかとなった(データは示さない)。
【0166】
〔実施例9〕細胞株および樹状細胞の調製
本発明者らは、次にEBV関連タンパク質のうち、EBNA1に特異的なCTLクローンの誘導を行い、EBNA1由来のエピトープの同定を行った。
EBNA1由来のエピトープの同定に関連する実験(実施例9〜16)において用いられた、細胞株および樹状細胞の調製方法について以下に示す。
【0167】
愛知県がんセンター(日本)の倫理審査委員会によって承認された研究デザインと目的は、すべての献血者に対して十分に説明され、インフォームド・コンセントを得た。
CD40で活性化されたB細胞(CD40-B)は公知の方法に基づいて、献血者のPBMCから産生された(Schultze JL, et al., J Clin Invest. 100, 2757-2765 (1997), Kondo E, et al., J Immunol., 169, 2164-2171 (2002))。簡単に述べると、PBMCを、放射線照射されCD40Lで形質転換したNIH3T3細胞 (以下t-CD40Lと記載する。BostonにあるDana-Farber Cancer InstituteのDr. Gordon Freemanから提供されたものである)と、リコンビナントIL-4(Genzyme, Cambridge, MA)、およびシクロスポリンA (Sandoz, Basel, Switzerland)と共に培養液中で培養した。増殖したCD40-Bに、週に2回刺激を加えた。
EBVを保持したLCLは、公知の方法(Kuzushima K, et al., Blood, 94, 3094-3100 (1999))に基づき、B95-8細胞培養上清を用いて、PBMCを分化させることによって調製した。そして、10%ウシ胎仔血清、2mM L-グルタミン、50U/mLのペニシリン、50μg/mLストレプトマイシンと50μg/mLのカナマイシンを含むRPMI1640培地(シグマケミカル社、セントルイス、MO)中で培養した。
【0168】
また、樹状細胞を公知の方法で調製した(Romani N, et al., J Exp Med., 180, 83-93 (1994), Sallusto F, et al., J Exp Med., 179, 1109-1118 (1994), Dauer M, et al., J Immunol., 170, 4069-4076 (2003))。CD8+T細胞を、PBMCsからCD8 MicroBeads(Miltenyi Biotec、ベルギッシュグラートバハ(ドイツ))を用いて分離し、−135℃で保存した。
【0169】
CD8を除いたPBMCを、5%のヒトの血清(MP Biomedicals、Aurora、OH)と2mMのL-グルタミン、50U/mLのペニシリン、50μg/mLのストレプトマイシン、50μg/mLのカナマイシンを含む4mLのRPMI1640培地(DC培地)に懸濁し、6ウェルプレートの一つのプレート中で37℃、2時間インキュベートした。
【0170】
非接着性細胞を穏やかにピペッティングすることで取り除き、接着細胞を50ng/mLの顆粒球マクロファージコロニー刺激因子(GM-CSF、Osteogenetics、Wuerzburg、ドイツ)と10ng/mL IL-4(Osteogenetics)を含むDC培地で培養した。2日目と4日目に、半分の培地を、GM-CSFとIL-4を含んでいる新鮮なDC培地に交換した。6日目に、樹状細胞を集め、mRNAを導入するため、エレクトロポレーションを行なった。
【0171】
〔実施例10〕全長EBNA1 mRNAの作成
EBNA1を発現している抗原提示細胞を作製するために、本発明者らは、pcDNA/EBNA1プラスミドからインビトロ転写システムを用いて、ポリA鎖を持つ全長EBNA1 mRNAを産生した。
インビトロで転写されたEBNA1 mRNAを作製するために、まずpcDNA/EBNA1ベクターを構築した。RNeasy Kit(Qiagen、Hilden (ドイツ))を用いて、上記のB95.8で形質転換したLCLから全てのRNAの抽出し、EBNA1をコードする配列を得た。EBNA1のアミノ酸配列およびDNA配列を配列番号:36および37に示す。そして、逆転写の後に、以下の特異的プライマーを用いたPCR法によってEBNA1 cDNAを増幅した。
フォワードプライマー
5'-AAGCTTGCCACCATGTCTGACGAGGGGCCAGGTACAG (配列番号:28)
リバースプライマー
5'-GAATTCTCACTCCTGCCCTTCCTCACCCTC (配列番号:29)
【0172】
全長のEBNA1断片を、HindIIIとEcoRIサイトを用いてpcDNA3.1(+)(Invitrogen, Carlsba樹状細胞A)に挿入し、pcDNA/EBNA1を構築した。EBNA1が導入されたことを確認するために、配列決定を行った。得られたプラスミドDNAを、mMESSAGEとmMACHINEキット(Ambion, Austin, TX)を用いて線状化し、インビトロで転写した。ポリAポリメラーゼ(Ambion)を用いて、3'末端のポリA鎖をEBNA1 mRNAに付加した後、RNeasyキットを用いて精製した。精製したmRNAをReliantRNAゲルシステム(Cambrex、ロックランド(ME))を使用して確認した。
【0173】
Capped-mRNAの収量は低いことが明らかとなったが、これはおそらくGCを多く含む配列からなるGAr領域が原因であり、反応温度の変化や反応混合物に一本鎖結合タンパク質を加えても、収量の問題は完全には解決できなかった(データは示さない)。しかしながら、mRNAの量は十分であり、ゲル上でも単一のバンドとして観察された(図6A)。
【0174】
〔実施例11〕全長EBNA1 mRNAの細胞への導入および、細胞におけるEBNA1の発現確認
次に、樹状細胞とCD40-B細胞に、エレクトロポレーションによって全長EBNA1mRNAを導入した。まず、無血清のRPMI1640を用いて二度洗浄し、最終濃度が2.5×107細胞/mLとなるように懸濁した。40μLのRPMI1640培地中で20μgのmRNAと混合した後に、2mmのセルの中に入れElectro Square Porator ECM830(ハーバードApparatus、ホリストン(MA))を用いてエレクトロポレーションを行った。条件設定は樹状細胞に対しては450V、500μS、CD40-B細胞に対しては350V、350μSに設定した。
【0175】
エレクトロポレーションの後、樹状細胞の場合はIL-4とGM-CSFを追加したDC培地の中で3時間培養し、成熟させるためにTNF-α(PeproTech,米国Rocky Hill, NJ)、IL-1β(PeproTech)とPGE2(Cayman Chemical,米国 Ann Arbor, MI)を加えた。CD40-B細胞を、放射線照射したNIH/3T3-ヒトCD40リガンド細胞の上に直ちに播いて、36-48時間後抗原提示細胞として使用した。
【0176】
次に、以下の工程でEBNA1の染色を行い、細胞内におけるEBNA1の発現を確認した。EBNA1 mRNAを導入したCD40-B細胞を集め、10分間室温で4%のパラホルムアルデヒドを含むリン酸緩衝生理食塩水(PBS)に定着させた。PBSで洗浄した後に、細胞を30分間4℃で0.5%のTween-20を含むPBSで浸透化し、抗EBNA1ウサギポリクローナル抗体(鶴見達也博士から提供、愛知がんセンター研究所、名古屋、日本)と反応させた。PBSで洗浄した後に、細胞をFITC標識したヤギの抗ウサギIgG(Beckman Coulter、Fullerton(カリフォルニア))で30分間4℃染色した。染色した細胞は、CellQUESTソフトウェア(BD Biosciences)を用い、FACSCallibur(BD Biosciences、サンノゼ(カリフォルニア))で分析した。
【0177】
上記の結果、EBNA1の発現は、平均蛍光強度は低いように見えたが、ほとんどのCD40-B細胞の上に検出された(図6B)。樹状細胞は、ドナーのPBMCsから得られた細胞数が限られていたために、本分析では利用できなかった。
【0178】
〔実施例12〕EBNA1 mRNAを導入した樹状細胞を用いた、EBNA1特異的CTLクローンの誘導
健康なドナーの単核球由来の樹状細胞に、全長EBNA1 mRNAをエレクトロポレーションによって導入し、炎症性サイトカインの混合物の添加することにより、樹状細胞の成熟化を誘導した。
EBNA1特異的CTLクローンの誘導は以下の工程により行った。
保存されていたCD8+T細胞を解凍し、10%ヒト血清、2mM L-グルタミン、50U/mLペニシリン、50μg/mLストレプトマイシン、および50μg/mLカナマイシン(CTL培地という)を含み、さらに5ng/mL IL-7(R&D Systems, Minneapolis, MN) と5ng/mL IL-12 (R&D systems) 5ng/mLを加えた200μL のRPMI1640倍地中で、5%CO2濃度の下、湿潤インキュベーターで培養した。8、16、および23日目に、EBNA1-mRNAを導入し、γ線を照射した樹状細胞によって、T細胞を刺激した。各再刺激の1日後に、最終的に20U/mLの濃度になるように、IL-2(塩野義、大阪、日本)を加えた。T細胞クローンを確立するため、ポリクローナルなCTLの限界希釈法を実施した(Kuzushima K, et al., Blood. 2001;98:1872-1881)。
【0179】
多クローンのCD8+T細胞を、ガンマ線などを照射された1×105のPBMCs(33Gy)、および2×104LCLs(55Gy)に、CD3(30ng/mL、Ortho Biotech、ブリッジウォーター、ニュージャージー)特異的なモノクロナール抗体(mAb)を含むCTL培地を入れた、丸底の96ウェルのプレートの1ウェルに1細胞となるように蒔いた。翌日、IL-2を各ウエル(50U/mL)に加えた。2週間の培養後、良好に増殖しているウェルを二つに分け、ELISPOTアッセイで自己のEBNA1 mRNAで形質転換したCD40-B細胞、または自己のLCLに対するエフェクターとして使用した。
【0180】
刺激を三回行った後に、各微量培養液の一定量について、EBNA1mRNAを導入した自己のCD40-B細胞と接触させ、ELISPOTアッセイで特異的にIFNγを分泌する能力をテストした。
ELISPOTアッセイは、公知の方法で行った(Kuzushima K, et al., Blood, 101, 1460-1468 (2003))。CD8+T細胞を、抗ヒトインターフェロンγ(IFNγ)モノクローナル抗体(ピアスBiotechnology、フィラデルフィア(PA))でコーティングされたMultiscreen-HAプレート(Millipore、ビルリカ(MA))のウェル中で各種刺激因子と共に培養した。刺激因子として、(A)自己のEBNA1 mRNAを導入したCD40-B細胞若しくは導入しなかったCD40のB細胞と、(B)自己若しくは同種のLCL(1×105の細胞/ウィル)を各ウェルに蒔いた。(以下に説明するペプチドタイトレーションとオバーラッピングアッセイにおいては、濃度をふった合成ペプチドを室温で1時間、自己のCD40-B細胞に加えた。)
【0181】
その後、抗ヒトIFNγウサギポリクローナル抗体(ピアスBiotechnology)と反応させた後、ペルオキシダーゼでラベルした抗ウサギの免疫グロブリン抗体(ゲンザイム、ケンブリッジ、MA)と基質を加えて反応後、プレートを洗浄し乾燥させた。IFNγスポットは実体顕微鏡の下で数えた。
【0182】
上記の結果、36のウェルのうち、32ウェルの細胞中にEBNA1特異的CTLが存在した(データ示さず)。CTLクローンB5とC6を限界希釈法により樹立した。クローンは抗CD3モノクローナル抗体、照射されたフィーダー細胞およびIL-2と供に増殖させた。
これらのクローンは、EBNA1-mRNAを導入した自己のCD40-B細胞とLCLを認識するが、EBNA1-mRNAを導入しなかった自己CD40B細胞やHLAが不一致の同種LCLは認識しなかった(図7)。
【0183】
〔実施例13〕抗原を提示しているHLA分子の同定
献血者はHLA-A*2402、A*3101、B*1507、B*3501、およびCw*0303として遺伝学的に分類された。抗原を提示しているHLA分子を同定するため、HLAが部分的に一致しているLCLのパネルを、クローンB5若しくはC6を刺激してIFNγを生産するために用いた。自己のLCLに加えて、HLA-B*3501を発現している同種LCLが、CTLクローンC6によって認識された(図8A)。そして、HLA-Cw*0304または、HLA-Cw*0303を持つLCLが、クローンB5(図8B)によって認識された。このことは、HLA-B*3501がクローンC6認識のための推定上の制限因子であり、一方、HLA-Cw*0303とHLA-Cw*0304はクローンB5の制限因子として働いていることを示している。
【0184】
〔実施例14〕EBNA1抗原ペプチドの識別
エピトープ領域を特定するために、クローンB5およびC6を、20個のアミノ酸を持つペプチドのセットと共に自己のCD40-B細胞において刺激した。本発明に使用されたペプチドは、Bio-Synthesis, Inc. Lewisville, TX.から購入した。GArの一次構造はMHCクラスIエピトープを含みそうにないことから、本発明者らはエピトープソースとしてこの部分を除いた。アミノ酸セット(合計56のペプチド)は13残基のオーバーラップによって、GArドメイン除く完全なEBNA1タンパク質配列をカバーしている。
【0185】
上記の結果、ペプチド#24はクローンC6によって認識された(図9)。HLA-B*3501拘束性のエピトープに関し、HPVGEADYFEY(配列番号:30)という配列が以前に報告されている(Blake N, et al., Immunity. 1997;7:791-802)。
【0186】
このエピトープ配列はペプチド#24(402-421)の中央に位置していることから(図10A)、本発明者らは、クローンC6が、HPVGEADYFEY(配列番号:30)をエピトープペプチドとして認識しているだろうと考えた。
【0187】
クローンB5の場合では、2つの重複するペプチド#38(500-519)と#39(507-526)が、認識され(図9)、これらは図10Bでアンダーラインを引いた13アミノ酸配列VFVYGGSKTSLYN(配列番号:31)を共有している。HLA-Cw*0303に結合する最適のエピトープを予測するために、プログラムSYFPEITHIを用いた(http://www.syfpeithi.de/ Rammensee H, et al., Immunogenetics. 1999;50:213-219)。そして、プログラムSYFPEITHIによる予測に基づき、エピトープとなりうる候補のペプチド、VYGGSKTSL(509-517)(配列番号:22)、FVYGGSKTSL(508-517)(配列番号:3)、およびVFVYGGSKTSL(507-517)(配列番号:2)を合成した。
【0188】
C-末端にあるアンカーのロイシンとエピトープの三番目の部分にある補助的アンカーのバリンとチロシンはプログラムで予測されていたことから、発明者らは11mer(VFVYGGSKTSL、配列番号:2)と10mer(FVYGGSKTSL、配列番号:3)(図10B)について調べた。
【0189】
ペプチドを加えた標的細胞のHalf Maximal Recognitionはそれぞれ5-10nM の10残基ペプチドと1-5nM の11残基ペプチドで得られた(図10C)。9残基(VYGGSKTSL、配列番号:22)ははるかに高い濃度においても認識されなかった。
【0190】
ペプチド希釈アッセイによってもクローンB5(図10C)の最適なエピトープの長さは明らかにならなかったことから、本発明者らは、同じ疑問を解決するのに構造面からのアプローチを用いることに決めた。このために、本発明者らは10残基ペプチドFVYGGSKTSL(配列番号:3)若しくは11mer VFVYGGSKTSL(配列番号:2)を導入し、蛍光ラベルしたテトラマーを作製した。
【0191】
テトラマーの作製と染色は以下の工程により行った。
HLA-Cw*0303とCw*0304のcDNAクローンは、プライマーであるC03F(5'-AACCATGGGCAGCCATTCTATGCGCTATTTTTACACCGCTGTGTCCCGGCC-3'、配列番号:32)及びC03R(5'-AAGGATCCTGGCTCCCATCTCAGGGTGAGG-3'、配列番号:33)プライマーを用いて、HLA-Cw*0303とCw*0304の重鎖の細胞外ドメインをコードする配列をPCRで増やすためのテンプレート(鋳型)として使用された。
【0192】
C03FはE.coli BL21(DE3)pLysSでタンパク質発現を最適化するように設計された数個の塩基の置換を含んでいる。PCR産物は、NcoIとBamHIとで切断され、ベクターに組み込まれた。このベクターは、HLA配列の3'末端にBirA biotinylationサイトを含んでいる。
【0193】
組換えHLA-Cw*0303とCw*0304の分子はβ2-ミクログロブリンとペプチドFVYGGSKTSL(配列番号:3)若しくはVFVYGGSKTSL(配列番号:2)と共にインビトロで組み込まれた。ゲル濾過で精製された可溶性複合体は、BirA酵素(vidity LCC、デンバー(CO))を用いてビオチン化された。
【0194】
フィコエリスリン(PE)をラベルされたテトラマーは、PEでラベルされたストレプトアビジン(Molecular Probes、Carlsbad (カリフォルニア))にこれらのビオチン化複合体を混合して作製した。
【0195】
テトラマーでの染色は以下の通りに行われた。CTLクローン(2x105)を、15分間4℃でFITCラベルした抗CD8モノクローナル抗体(Caltag、バーリンゲーム(カリフォルニア))と、20μg/mLのテトラマーでインキュベートして染色した。二度洗浄した後、染色した細胞を、0.5%のパラホルムアルデヒドで固定し、フローサイトメトリーにより分析した。
【0196】
上記の分析の結果、図11に示されるように、これらのテトラマーは特異的にCTLクローンB5に結合した。しかしながら、10残基ペプチドを導入したテトラマーはB5クローンに対して、11残基ペプチドを導入したものより高い親和力を示し、このことは10残基ペプチドFVYGGSKTSL(配列番号:3)がCTLのための最小の、そして、最適のエピトープであること示唆している。さらに、クローンB5は、結果が図8Aのデータと同様に、10残基を組み込んだHLA-Cw*0304テトラマーに強く結合した。
【0197】
〔実施例16〕EBNA1特異的CTLsのEBVが感染したB細胞への成長抑制効果の検討
クローンのB5とC6は、クロムリリースアッセイにおいては、自己のLCLを溶解しなかった(データは示さない)。ここで、これらのEBNA1特異的CTLが、EBNA1を発現しているLCLの長期的成長と生存に影響を及ぼすか否かを検討した。
【0198】
拘束性HLA分子の存在下若しくは非存在下において、自己及び同種のLCLが、反応するCTLの存在下若しくは非存在下において96ウェルプレートに蒔かれた。
【0199】
成長抑制アッセイは、公知の方法を多少修正して実施した(Lee SP, et al., J Exp Med., 199, 1409-1420 (2004))。標的となるLCL細胞は2×104細胞/ウェルの濃度で3ウェルずつ丸底の96ウェルのプレートに蒔いた。 EBNA1特異的CTLクローン(1×104の細胞/ウェル)若しくはコントロールとしてのCTL培地を標的細胞の培地に加えた。すべての培地を週ごとに半分を替えることによって維持し、PE-cyanin5でラベルされた抗CD19と、PEによってラベルされた抗CD8モノクローナル抗体(Beckman Coulter)で染色し、フローサイトメトリーによる分析でB細胞の特性を確認した。さらに、培養液は4週間後にLCLがどれだけ増えたかを確認した。
最終的にLCLがどれだけ増えたかを、顕微鏡観察によって評価し、フローサイトメトリーによるCD19の発現で確認した。
【0200】
図12に示されるように、両方のCTLクローンは明確に自己のLCLだけではなく、拘束性HLAを持つ同種のLCLについて増殖を抑制した。このことは、CTLクローンが潜伏タイプIIIであるEBV-陽性細胞の成長を阻害する能力を持つことを示唆している。
【特許請求の範囲】
【請求項1】
エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド。
【請求項2】
エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むものである、請求項1に記載のペプチド。
【請求項3】
配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、請求項1に記載のペプチド。
【請求項4】
HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子の拘束性抗原ペプチドであって、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、請求項1〜3のいずれかに記載のペプチド。
【請求項5】
請求項1〜4のいずれかに記載のペプチドをコードする核酸。
【請求項6】
請求項1〜4のいずれかに記載のペプチドを有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項7】
請求項5に記載の核酸を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項8】
請求項1〜4のいずれかに記載のペプチドをHLAに提示した抗原提示細胞を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項9】
請求項1〜4のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
【請求項10】
請求項1〜4のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
【請求項11】
請求項1〜4のいずれかに記載のペプチドを用いて細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導方法。
【請求項12】
請求項1〜4のいずれかに記載のペプチドと末梢血単核球を、血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的細胞傷害性T細胞を誘導する、請求項11に記載の誘導方法。
【請求項13】
請求項1〜4のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
【請求項14】
請求項1〜4のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
【請求項15】
請求項1〜4のいずれかに記載のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【請求項16】
請求項1〜4のいずれかに記載のペプチドから主要組織適合性抗原複合体-テトラマーを調製し、主要組織適合性抗原複合体-テトラマーと末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【請求項1】
エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチド。
【請求項2】
エプスタイン-バールウイルスに特異的な細胞傷害性T細胞エピトープペプチドが配列番号:1〜3からなる群から選択される少なくとも1つのアミノ酸配列を含むものである、請求項1に記載のペプチド。
【請求項3】
配列番号:1〜3のいずれかに記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入及び/又は付加されたアミノ酸配列からなるペプチドであって、エプスタイン-バールウイルス特異的な細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、請求項1に記載のペプチド。
【請求項4】
HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子の拘束性抗原ペプチドであって、HLA-A*0206分子、HLA-Cw*0303分子またはHLA-Cw*0304分子との複合体を細胞表面に提示する細胞を特異的に認識しうるT細胞レセプターを有する細胞傷害性T細胞を誘導し得る機能を有することを特徴とする、請求項1〜3のいずれかに記載のペプチド。
【請求項5】
請求項1〜4のいずれかに記載のペプチドをコードする核酸。
【請求項6】
請求項1〜4のいずれかに記載のペプチドを有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項7】
請求項5に記載の核酸を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項8】
請求項1〜4のいずれかに記載のペプチドをHLAに提示した抗原提示細胞を有効成分として含む、エプスタイン-バールウイルスの感染を治療又は予防するためのワクチン。
【請求項9】
請求項1〜4のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激して得られるエプスタイン-バールウイルス特異的な細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
【請求項10】
請求項1〜4のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を有効成分として含む、エプスタイン-バールウイルスに対する受動免疫療法剤。
【請求項11】
請求項1〜4のいずれかに記載のペプチドを用いて細胞傷害性T細胞を誘導することを特徴とする、細胞傷害性T細胞の誘導方法。
【請求項12】
請求項1〜4のいずれかに記載のペプチドと末梢血単核球を、血漿を含む培地中で接触させることにより、エプスタイン-バールウイルス特異的細胞傷害性T細胞を誘導する、請求項11に記載の誘導方法。
【請求項13】
請求項1〜4のいずれかに記載のペプチドもしくは該ペプチドをHLAに提示した抗原提示細胞により末梢血リンパ球を刺激してエプスタイン-バールウイルス特異的な細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
【請求項14】
請求項1〜4のいずれかに記載のペプチドから調製した主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーと末梢血リンパ球とを反応させ、該主要組織適合性抗原複合体及び/又は主要組織適合性抗原複合体-テトラマーに細胞傷害性T細胞が結合した結合体を形成させ、該結合体から単離して得られる細胞傷害性T細胞を取得する工程を含む、エプスタイン-バールウイルスに対する受動免疫療法剤の製造方法。
【請求項15】
請求項1〜4のいずれかに記載のペプチドで末梢血を刺激し、該ウイルスに特異的な細胞傷害性T細胞を取得し、細胞傷害性T細胞が産生するサイトカイン及び/又はケモカイン及び/又は細胞表面分子を測定することを特徴とする、エプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【請求項16】
請求項1〜4のいずれかに記載のペプチドから主要組織適合性抗原複合体-テトラマーを調製し、主要組織適合性抗原複合体-テトラマーと末梢血とを反応させることを特徴とする、該末梢血中のエプスタイン-バールウイルスに特異的な細胞傷害性T細胞の定量方法。
【図1】


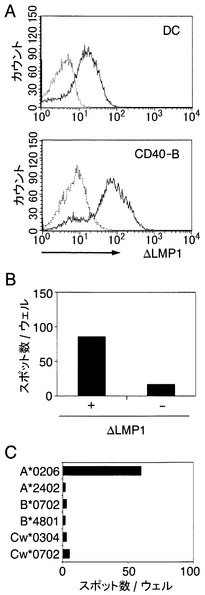
【図2】
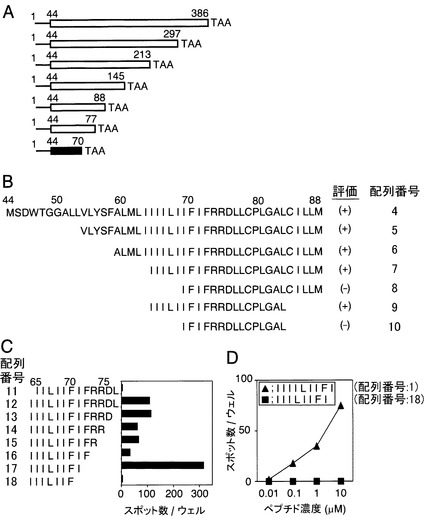
【図4】
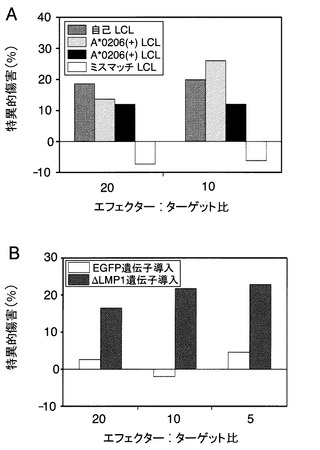
【図5】
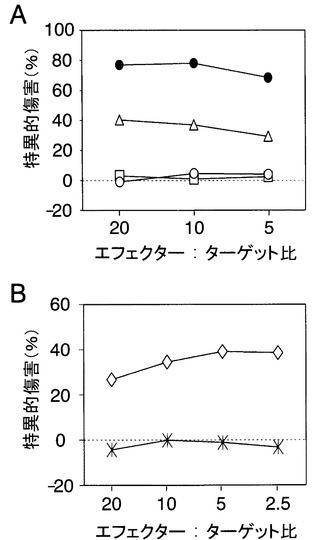
【図7】
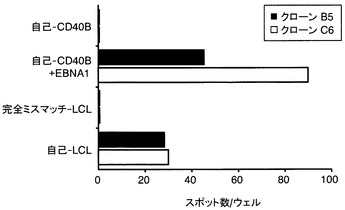
【図8】
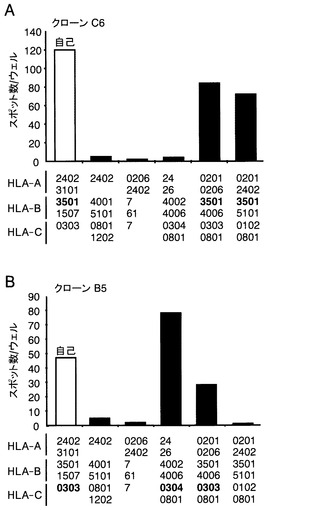
【図9】
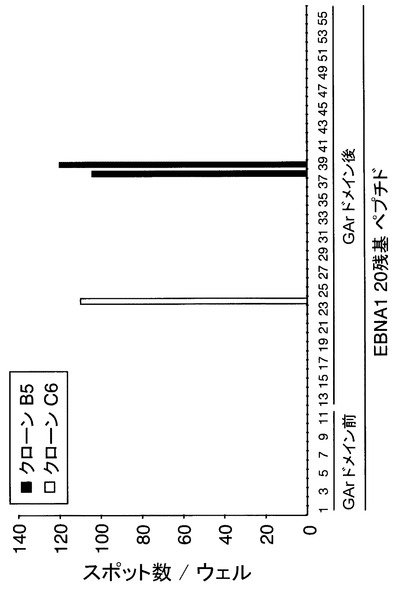
【図10A−B】

【図10C】
【図11】
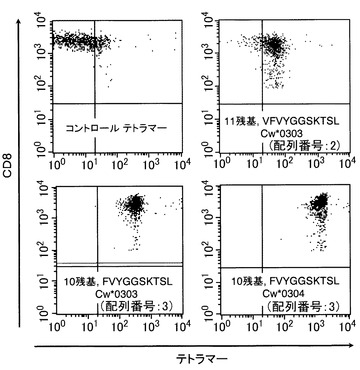
【図12】
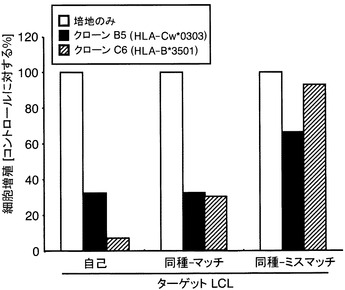
【図3】
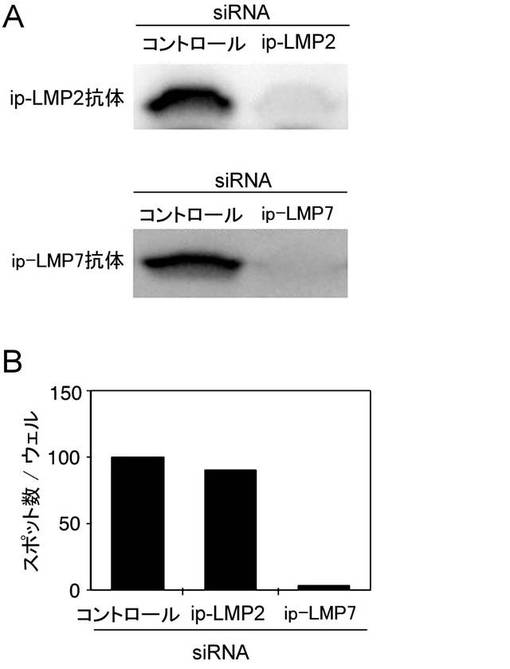
【図6】
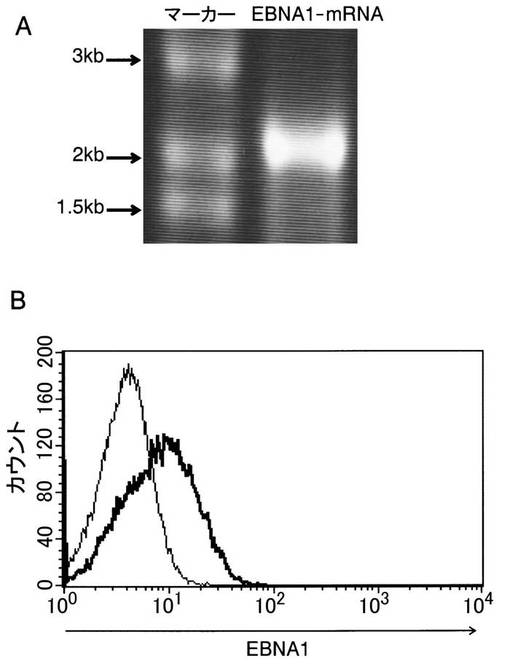
【図2】
【図4】
【図5】
【図7】
【図8】
【図9】
【図10A−B】
【図10C】
【図11】
【図12】
【図3】
【図6】
【公開番号】特開2011−239787(P2011−239787A)
【公開日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願番号】特願2011−177487(P2011−177487)
【出願日】平成23年8月15日(2011.8.15)
【分割の表示】特願2005−315306(P2005−315306)の分割
【原出願日】平成17年10月28日(2005.10.28)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【出願人】(304031427)愛知県 (36)
【Fターム(参考)】
【公開日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願日】平成23年8月15日(2011.8.15)
【分割の表示】特願2005−315306(P2005−315306)の分割
【原出願日】平成17年10月28日(2005.10.28)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【出願人】(304031427)愛知県 (36)
【Fターム(参考)】
[ Back to top ]