
エポキシ樹脂組成物
【課題】高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物を提供する。
【解決手段】ポットライフが長いエポキシ樹脂硬化剤とモノアミン縮合ホウ酸塩とを併用することによって、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物が得られることを見出した。即ち、(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤と、(D)溶剤とを含有するエポキシ樹脂組成物により上記課題を解決した。
【解決手段】ポットライフが長いエポキシ樹脂硬化剤とモノアミン縮合ホウ酸塩とを併用することによって、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物が得られることを見出した。即ち、(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤と、(D)溶剤とを含有するエポキシ樹脂組成物により上記課題を解決した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エポキシ樹脂とモノアミン縮合ホウ酸塩を含有するエポキシ樹脂組成物に関する。より詳しくは、硬化物に高いガラス転移温度を与え、且つ優れた保存安定性を有するエポキシ樹脂組成物及びその製造方法に関する。
【背景技術】
【0002】
プリント配線板に用いられている銅張り積層板用接着剤やICパッケージの封止に用いられている半導体封止材料としては、これまでに主にエポキシ樹脂が使われている。近年、鉛フリー半田を用いているのに伴い、半田接合時の加熱温度が高くなり、銅張り積層板や半導体封止材料用エポキシ樹脂組成物から得られる硬化物には、更なる高ガラス転移温度、高弾性率化や低線膨張率などの耐熱性が強く求められている。従来、電子材料用エポキシ樹脂組成物は、ジシアンジアミドやノボラックフェノール樹脂などの硬化剤を配合したものが主に使用されている。しかし、得られた樹脂硬化物のガラス転移温度が低く、耐熱性に劣るという問題があった。
【0003】
耐熱性を向上させるためにノボラック型エポキシ樹脂を配合する方法がよく行われている(特許文献1参照)。しかしながら、この方法では樹脂硬化物のガラス転移温度が数度しか上がらず十分ではない。
【0004】
更に、エポキシ樹脂硬化物の耐熱性を向上させる方法として、エポキシ樹脂にポリアミンホウ酸塩を必須成分として添加することが報告され(特許文献2参照)、得られたエポキシ樹脂組成物の硬化物が高いガラス転移温度を有することが示されている。しかし、ポリアミンホウ酸塩を含有するエポキシ樹脂組成物の殆どはポットライフが短く、保存安定性に劣る欠点を有する。
【0005】
上述のポリアミンホウ酸塩含有エポキシ樹脂組成物の保存安定性を改良する方法として、ポリアミンホウ酸塩の添加量を抑え、潜在硬化性エポキシ樹脂の硬化剤と併用することが特許文献3に提案されている。しかし、ポリアミンホウ酸塩の添加量が少ないため、耐熱性を損なう場合があり、特に高エポキシ当量のエポキシ樹脂を用いた場合、ガラス転移温度の向上は十分ではない。
【0006】
また、エポキシ樹脂硬化物のガラス転移点を向上させることを目的として、エポキシ樹脂とピペリジン縮合ホウ酸塩を含有するエポキシ樹脂組成物に関する技術が知られている(特許文献4参照)。しかしながら、この技術は、ポットライフが短く、保存安定性に劣るものであった。
【0007】
【特許文献1】特公平7-60920号公報
【特許文献2】特開2005-68417号公報
【特許文献3】特開2005-264106号公報
【特許文献4】特開2005-89317号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記の目的を達成しようと鋭意研究を重ねた結果、ポットライフが長いエポキシ樹脂硬化剤とモノアミン縮合ホウ酸塩とを併用することによって、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物が得られることを見出し、本発明を完成させたものである。
【0010】
即ち、本発明は、(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤とを含有するエポキシ樹脂組成物を提供する。
【発明の効果】
【0011】
本発明のエポキシ樹脂組成物は高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れており、銅張積層板や半導体封止材料など電子材料として好適に用いられる。
【発明を実施するための最良の形態】
【0012】
エポキシ樹脂(A)としては、1分子内に平均2個以上のエポキシ基を有する慣用のエポキシ樹脂であり、その種類は特に限定されない。例えば、次に掲げる各種のエポキシ樹脂を単独又は2種以上組み合わせて使用できる。
(1) フェノール系グリシジルエーテル型エポキシ樹脂
ビスフェノールーA、ビスフェノールーF、テトラブロモビスフェノールーA、テトラフェニロールエタン、フェニールノボラック、クレゾールノボラックなどのフェノール化合物とエピクロルヒドリンとの反応により得られるフェノール系グリシジルエーテル型エポキシ樹脂。
(2) アルコール系グリシジルエーテル型エポキシ樹脂:
(a) ビスフェノールーA、ビスフェノールーF、テトラブロモビスフェノールーA、テトラフェニロールエタンなどのフェノール化合物とアルキレンオキサイドとの付加反応により得られるポリオール、又は水添ビスフェノールAなどのポリオールと、(b) エピクロルヒドリンとの反応により得られるアルコール系グリシジルエーテル型エポキシ樹脂。
(3) グリシジルエステル型エポキシ樹脂:
ヘキサヒドロフタル酸ジグリシジルエステル、ダイマー酸グリシジルエステル等のジグリシジルエステル型エポキシ樹脂。
(4) グリシジルアミン型エポキシ樹脂:
1,3-ジグリシジルヒダントイン、トリグリシジルイソシアヌレート、テトラグリシジルジアミノジフェニルメタン、トリグリシジルーパラアミノフェノール等のグリシジルアミン型エポキシ樹脂。
(5) 混合型エポキシ樹脂:
アミノフェノール又はオキシ安息香酸とエピクロルヒドリンとの反応により得られるエポキシ樹脂、シクロペタジエンやジシクロペンタジエン骨格を有する脂環式型エポキシ樹脂、臭素化エポキシ樹脂等の混合型エポキシ樹脂。
【0013】
本発明に於いて、エポキシ樹脂組成物の硬化物が充分高いガラス転移温度と優れた力学物性を獲得する為に、エポキシ樹脂のエポキシ基当量は、好ましくは800以下、より好ましくは600以下、特に好ましくは400以下である。エポキシ当量が800以上になると、得られる硬化物が脆くなり、好ましくない。
【0014】
本発明で用いるモノアミン縮合ホウ酸塩(B)としては、通常モノアミン化合物とホウ酸系化合物とを反応させて得られるものである。かかるモノアミン化合物は、通常エポキシ樹脂の硬化促進剤として用いる2級、3級アミン及びイミタゾール化合物が好適である。具体的にはピペリジン、N-メチルピペラジン、ヒドロキシエチルピペラジン、ピロリジン、モルホリン、トリフェニルアミン、トリメチルアミン、トリエチルアミン、トリプロピルアミン、トリエタノールアミン、トリイソプロパノールアミン、ベンジルジメチルアミン、2-(ジメチルアミノメチル)フェノール、2,4,6-トリス(ジアミノメチル)フェノール、2-メチルイミダゾール、2-エチル-4-メチルイミダゾール、2-ウンデシルイミダゾール、2-ヘプタデシルイミダゾール、2-フェニルイミダゾール、1-ベンジル-2-メチルイミダゾール、1-シアノエチル-2-メチルイミダゾールなどが挙げられる。
本発明でのホウ酸系化合物としては、ホウ酸およびホウ酸エステルが用いられる。ホウ酸およびホウ酸エステルとしては、代表的なものとしては一般式(1)
【0015】
【化1】

(式中、nは0〜3までの整数、RはCmH2m+1のアルキル基であり、mは1〜10の整数を表す。)で表わされる。ホウ酸の具体的なものとしては、例えばオルトホウ酸、メタホウ酸、四ホウ酸、およびそれらの混合物であり、また、ホウ酸エステルの具体的なものとしては、例えばホウ酸トリメチル、ホウ酸トリエチル、ホウ酸トリプロピル、ホウ酸トリブチル等が挙げられる。これらのホウ酸及びホウ酸エステルは、単独又は2種以上組み合わせて使用できる。上記の中ではホウ酸が最も好ましく用いられる。
【0016】
本発明で用いるピペリジン縮合ホウ酸塩の結晶構造は、M.Wiebckeらによって明らかにされた(Chem.Sci.,48(7),978-85 1993)。一般式(2)に示したように、ピペリジン縮合ホウ酸塩は、五核の縮合ホウ酸塩であり、つまり、一分子のピペリジンカチオンに対して、五分子のホウ酸が自己縮合した形でピペリジンとイオン結合しているピペリジン五ホウ酸縮合塩である。
【0017】
【化2】
また、本発明で用いる他の2級、3級モノアミン及びイミタゾール化合物とホウ酸系化合物とを反応して得られたアミンホウ酸塩は、そのホウ素含有率が単核ホウ酸塩より高いことから、縮合ホウ酸塩が形成されていると結論した。更に、これらのアミン縮合ホウ酸塩のFT-IR及び11B-NMRを測定したところ、ピペリジン五ホウ酸縮合塩と比べて、両者のピーク形状と強度が極めて類似していることから、本発明で得られたモノアミン縮合ホウ酸塩は、五ホウ酸縮合塩またはそれを含むものと推定された。
【0018】
本発明のモノアミン五ホウ酸縮合塩では、エポキシ樹脂の硬化触媒になるモノアミンカチオンを持ち、そのモノアミンカチオンはエポキシ樹脂との相互作用によって、モノアミン五ホウ酸縮合塩を分子レベルでエポキシ樹脂に均一に分散することができる。また、一般式2に示したように、五ホウ酸縮合塩基は反応性の高い複数の水酸基を持ち、その水酸基はエポキシ樹脂の水酸基との縮合反応によって、ホウ酸エステル結合ができて、エポキシ樹脂の中に無機架橋が形成されていると推定する。即ち、エポキシ樹脂に分子レベルに分散したモノアミン五ホウ酸縮合塩のホウ酸塩基が無機架橋剤として働き、エポキシ樹脂のガラス転移温度の大幅向上に寄与したと考えられる。
【0019】
本発明で用いるモノアミン縮合ホウ酸塩(B)の合成は、例えば次のようにして行うことができる。即ち、有機溶剤または水にホウ酸を溶解させて攪拌しながら、モノアミン化合物溶液を滴下する。場合によっては、添加順序を逆にしてモノアミン化合物の有機溶剤溶液または水溶液を攪拌しながら、ホウ酸溶液を滴下することもある。続いて、室温または加熱下、一定時間において反応を行う。得たモノアミン縮合ホウ酸塩が析出した場合、吸引濾過により沈殿物を回収する。一方、反応生成物のモノアミン縮合ホウ酸塩が反応溶媒に溶けている場合、エパポレーターにより溶媒を留去する。このようにして得られた反応生成物をアセトンで数回繰り返し洗浄した後、真空乾燥により白色粉末のモノアミン縮合ホウ酸塩が得られる。
【0020】
上記アミンホウ酸塩の合成溶媒としては、ホウ酸またはモノアミン化合物の少なくとも一種を溶解するようなものが必要である。例えば、メタノール、エタノール、イソプロパノールなどの低級アルコール、アセトン、メチルエチルケトン、テトラヒドロフラン、N,N-ジメチルホルムアミド、N-メチルピロリドン、N,N-ジメチルアセトアミド、ジメチルスルホキシド、水などが挙げられ、これらは単独又は二種以上の混合で使用できる。その中では、特にN,N-ジメチルホルムアミド、水を用いることが好ましい。溶媒の使用量は、ホウ酸およびモノアミン化合物の合計100質量部に対して溶媒が300〜1500質量部となるように用いることが好ましい。
【0021】
上記モノアミン縮合ホウ酸塩(B)の合成条件として、モノアミン化合物の中のアミノ基およびイミノ基とホウ酸とのモル比及び反応温度と時間が重要である。ホウ酸の比率を増やすと、多核縮合ホウ酸塩が形成しやすく、高いホウ素含有量のアミン縮合ホウ酸塩が得られる。これに対してモノアミン化合物の比率を増やすと、より低いホウ素含有量のアミン縮合ホウ酸塩を得ることができる。一般的にモノアミン化合物の中のアミノ基およびイミノ基の合計1モルに対して、ホウ酸が0.25〜10モルが好ましく、より好ましくは0.5〜8モル、特に好ましくは1〜6モル、最も好ましくは5モルである。アミノ基1モルに対して、5モルのホウ酸を用いることによって、最も安定なアミン五ホウ酸縮合塩が形成され、好ましい。一方、0.5モル未満又は10モルを超える場合、アミンホウ酸塩の収率が低く、経済的に不利になり、好ましくない。また、反応温度について、用いるアミン系化合物の種類によっては異なるが、一般的に15℃〜100℃が好ましく、より好ましくは20℃〜80℃であり、特に好ましくは25℃〜50℃である。反応時間は反応温度にもよるが、通常0.1〜10時間である。
【0022】
上記モノアミン縮合ホウ酸塩(B)がアミン特有の刺激臭が殆どない固形状粉末であり、水またはメタノールやメチルセロソルプなどのアルコール性有機溶剤によく溶ける性質を持っており、エポキシ樹脂の硬化剤又は硬化促進剤として用いられる。該アミン縮合ホウ酸塩中のホウ素含有量は、使用するモノアミン化合物の種類及び添加量により一定ではないが、好ましくは5〜25質量%、より好ましくは8〜20質量%である。
【0023】
本発明で用いるアミンホウ酸塩(B)の使用量は、エポキシ樹脂に対して5〜40質量%で配合することが好ましい。かかる使用量であれば、得られる組成物の耐熱性が良好であり、組成物の機械特性も優れるため、好ましい。
【0024】
上述のモノアミン縮合ホウ酸塩では、嵩高い縮合ホウ酸塩基の立体障害で、モノアミンカチオンの触媒活性が低下させられて、単独でエポキシ樹脂の硬化剤として用いると、硬化不十分になる傾向があり、硬化物の耐熱性を損なう。従って、他のエポキシ樹脂の硬化剤と併用することが必要である。本発明でアミンホウ酸塩(B)と併用するエポキシ樹脂硬化剤(C)としては、エポキシ樹脂に対するポットライフが室温で10日以上である積層板用のエポキシ樹脂硬化剤が用いられる。かかるポットライフは、具体的にはエポキシ樹脂、エポキシ樹脂硬化剤及び溶剤からなる組成物を室温で保存して脂触によりゲル化(硬化)するまでに要した時間であり、該硬化剤がエポキシ樹脂と反応する硬化剤タイプについてはエポキシ樹脂1当量に対して1当量用いて測定することを基準とし、又該硬化剤が触媒タイプについてはエポキシ樹脂100質量部に対して2質量部を用いて測定することを基準としている。
【0025】
本発明で用いられる上述のエポキシ樹脂硬化剤(C)は、優れた組成物の保存安定性をもたらした潜在硬化性やポットライフの長いエポキシ樹脂硬化剤である。例えば、ジシアンジアミド、フェノールノボラック樹脂、メタフェニレンジアミンなどのエポキシ樹脂と反応する硬化剤タイプや3級アミン、イミダゾール化合物などの触媒タイプが挙げられる。これらの硬化剤の使用量は、併用するアミンホウ酸塩の添加量によって異なるが、エポキシ樹脂と反応する硬化剤タイプの場合はエポキシ樹脂1当量に対して0.1〜0.95当量で配合することが好ましく、触媒タイプの場合はエポキシ樹脂に対して1〜3phrで配合することが好ましい。
【0026】
本発明におけるエポキシ樹脂組成物の有機溶媒としては、エポキシ樹脂、モノアミン縮合ホウ酸塩及びエポキシ樹脂硬化剤を均一に溶解できる有機溶媒が使用される。エポキシ樹脂、モノアミン縮合ホウ酸塩及びエポキシ樹脂硬化剤を溶解できる有機溶媒としては、通常低級アルコ−ルを含有するもの又はセロソルプ類が使用される。例えば、アセトン、メチルエチルケトン、テトラヒドロフラン、N,N-ジメチルホルムアミド、N-メチルピロリドン、N,N-ジメチルアセトアミド、ジメチルスルホキシドなど有機溶媒にメタノールを添加した有機溶媒の混合物が挙げられる。また、メチルセロソルプなどのセロソルプ類は単独でも用いられる。
【0027】
本発明のエポキシ樹脂組成物の製造方法に於いては、エポキシ樹脂およびモノアミン縮合ホウ酸塩を有機溶媒に均一に溶解した溶液状態での加熱処理を行うことは均一透明なエポキシ樹脂組成物を得るために極めて重要である。溶液状態での加熱処理とは、実質的に溶媒を除去することなく、即ちクローズドシステムで加熱し、しかも樹脂がゲル化しないように行うことを云う。それを行うことにより、エポキシ樹脂とモノアミン縮合ホウ酸塩との相互作用を持たせることにより、モノアミン縮合ホウ酸塩がエポキシ樹脂溶液中に分子レベルで均一に相溶する。得られた溶液を脱溶剤して得られる組成物はモノアミン縮合ホウ酸塩の凝集がなく均一透明となる。
【0028】
溶液状態での加熱処理の条件は、用いるモノアミン縮合ホウ酸塩とエポキシ樹脂との反応の容易さにより異なる。基本的には加熱処理の下限及び上限としては、その後の脱溶剤によりモノアミン縮合ホウ酸塩が析出しなくなる範囲(下限)、且つ溶液がゲル化しない範囲(上限)で加熱処理を行うことが重要である。加熱処理を過度に行った場合、組成物が増粘またはゲル化してしまい、実用性の点から好ましくない。
【0029】
溶液状態での加熱処理温度は、好ましくは25〜100℃、より好ましくは30〜90℃、特に好ましくは40〜80℃で行える。加熱処理時間は加熱処理温度により異なるが、0.2〜20時間が好ましく、より好ましくは0.5〜15時間、特に好ましくは1〜10時間である。
【0030】
上述により得られるエポキシ樹脂組成物から、硬化反応を進めないようにして溶媒を除去することにより、無溶媒のエポキシ樹脂組成物が製造できる。脱溶媒温度は用いるアミンホウ酸塩及びエポキシ樹脂硬化剤により異なるが、100℃以下が好ましく、より好ましくは80℃以下、特に好ましくは60℃以下である。
【0031】
而して製造される溶液状のエポキシ樹脂組成物は、積層板用樹脂材料として利用できるほか、金属、セラミック、耐熱性プラスチックなどの基材に対する耐熱性塗料としても利用することができる。
【0032】
例えば、上記エポキシ樹脂組成物を耐熱性基材に塗布又は含浸させた後、乾燥してプリプレグとする。このプリプレグに銅箔を重ね合わせ、加熱圧縮することにより銅張積層板を製造することができる。かかる耐熱性基材は、ガラス繊維が一般的であるが、他に芳香族ポリアミド繊維を用いることもできる。
【0033】
また、上記エポキシ樹脂組成物を直接銅箔に塗布し、乾燥した後、それをエポキシ樹脂ガラスクロス板と重ね合わせ、加熱圧縮することにより銅張積層板を製造することもできる。
【0034】
上述の方法により製造される銅張り積層板は、耐熱性に優れ、電子部品として好適に用いられる。
【実施例】
【0035】
次に、本発明を合成例、実施例によって具体的に説明する。
【0036】
また、以下の合成例において、核磁気共鳴スペクトル(NMR)の測定は日本電子(株)製 JNM-LA-300を用いた。11B-NMRスペクトルは重水中のホウ酸ピークを基準とした。
粉末X線回折の測定は理学電機(株)製X線回折装置 RINT ULTIMA+ を使用した。
ホウ素含有量はPerkn Elmer社製 Optima 3300DVを用いて、ICPの測定を行い、予めホウ酸を用いて作成しておいた検量線により定量した。
アミンの定量は1H-NMRの内部標準法により行った。すなわち、試料に一定量のベンゼンを内部標準として添加し、そのピークと一定量のアミンホウ酸塩のアミンの特定ピークとの面積比により定量した。
【0037】
なお、本発明の合成例及び実施例について次の試薬を使用した。
(1)モノアミン化合物
ベンジルジメチルアミン(BDMA):和光純薬工業株式会社製、試薬特級
2-エチル-4-メチルイミダゾール(2E4MZ) :和光純薬工業株式会社製、試薬特級
ピペリジン:和光純薬工業株式会社製、試薬特級
(2) ホウ酸: 和光純薬工業株式会社製、試薬特級
(3) 溶剤
N,N-ジメチルホルムアミド(DMF):和光純薬工業株式会社製、試薬特級
アセトン:和光純薬工業株式会社製、試薬1級
メチルエチルケトン(MEK) :和光純薬工業株式会社製、試薬特級
メタノール:和光純薬工業株式会社製、試薬特級
メチルセロソルプ: 和光純薬工業株式会社製、試薬特級
(4) 硬化剤
ジシアンジアミド(DICY) :和光純薬工業株式会社製、試薬特級
2-エチル-4-メチルイミダゾール(2E4MZ) :和光純薬工業株式会社製、試薬特級
【0038】
(合成例1) [ベンジルジメチルアミン五ホウ酸縮合塩の合成]
ホウ酸34.3g(0.555mol)をDMF 350gに溶かした溶液を攪拌しながら、ベンジルジメチルアミンBDMA 15g(0.111mol)を滴下した。白色沈殿が直ちに析出した。室温で18時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトン二回洗浄し、70℃、3時間真空乾燥により、26.1gの白色粉末1aを得た。
【0039】
(合成例2) [2-エチル-4-メチルイミダゾール五ホウ酸縮合塩の合成]
ホウ酸42.1g(0.68mol)をDMF 260gに溶かした溶液を攪拌しながら、2-エチル-4-メチルイミダゾール2E4MZ 15g(0.136mol)を加えた。白色沈殿が析出した。室温で13時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトンで二回洗浄し、70℃、4時間真空乾燥により、反応生成物の白色粉末1b 34.5gを得た。
【0040】
(合成例3) [ピペリジン五ホウ酸縮合塩の合成]
ホウ酸30g(0.485mol)をDMF 120gに溶かした溶液を攪拌しながら、ピペリジン 41.34g(0.485mol)を滴下した。白色沈殿が直ちに析出した。室温で24時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトン二回洗浄し、50℃、8時間真空乾燥により、19.1gの白色粉末1cを得た。
【0041】
【表1】
【0042】
【表2】
【0043】
以下の実施例および比較例において、光透過率は日本電色工業株式会社製NDH-300Aを用いて、厚さ300μmのフィルムの平行透過率を測定した。ガラス転移温度(Tg)及び貯蔵弾性率(E')は、固体動的粘弾性測定装置(エスアイアイ・ナノテクノロジー株式会社製DMS6100)を用い、測定周波数1Hz、昇温速度3℃/分で測定した。なお、ガラス転移温度はtanδピーク温度(tanδmax)とした。また、熱線膨張係数は、エスアイアイ・ナノテクノロジー株式会社製TMA/SS6100を用いて、窒素雰囲気下、引張りモート、昇温速度10℃/分で測定した。
【0044】
(実施例1及び比較例1,2)
ベンジルジメチルアミン五ホウ酸縮合塩(1a) 20gをメチルセロソルプ160gに溶かした透明溶液にビスフェノールA型エポキシ樹脂 エピクロン850(大日本インキ化学工業株式会社製、エポキシ当量188g/eq)100gを加え、攪拌混合した後、50℃で4時間の溶液状態での加熱処理を行った。次に、ジシアンジアミド(DICY) 5.6gを加え、攪拌溶解させて、エポキシ樹脂組成物溶液を得た。得られた組成物は50℃にて20日放置したところ、溶液が増粘せず、優れた保存安定性を示した。続いて該溶液を清浄なアルミ箔上に塗布し、50℃で8時間溶媒キャストを行った後、80℃、3時間更に乾燥した。続いて、150℃、180℃で各2時間の熱処理を行い、該エポキシ樹脂組成物の硬化物を得た。
【0045】
上記の硬化物は透明性に優れ、可視光域での光透過率は91%(100μm厚み換算)であり、クラックやしわ、気泡なども無く、良好な表面形態を示した。また、比較例1においてジシアンジアミドを使用せずベンジルジメチルアミン五ホウ酸縮合塩のみを用いること、比較例2においてベンジルジメチルアミン五ホウ酸縮合塩を使用せずジシアンジアミドのみを用いること、それ以外に実施例1と同様にしてエポキシ樹脂の硬化物を作製した。
【0046】
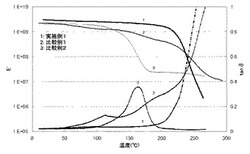
実施例及び比較例で得られた厚み約300μmの硬化フィルムを用いて動的粘弾性測定(周波数1Hz)を行った。表3及び図1に示したように、実施例1のtanδのピーク温度(Tg)は250℃を超えた。これに対して、比較例1では140℃にtanδのサブピークがあり、比較例2ではtanδのピーク温度(Tg)は150℃であった。アミン縮合ホウ酸塩とジシアンジアミドを併用したエポキシ樹脂硬化物のガラス転移温度が大きく向上していることが明らかである。また、表5に示したように、実施例1の線膨張係数は、比較例1及び比較例2より低くなった。
【0047】
(実施例2)
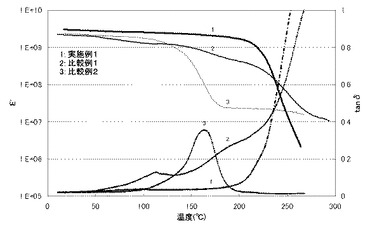
DICYの変わりに、2E4MZを用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4及び図2に示す。
【0048】
(実施例3)
BDMA五ホウ酸縮合塩の変わりに、2E4MZ五ホウ酸縮合塩を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4、表5及び図2に示す。
【0049】
(実施例4)
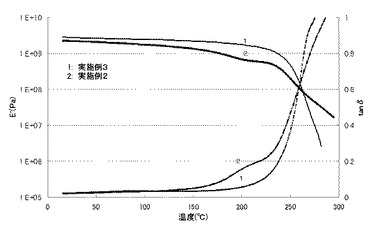
エポキシ樹脂850の変わりに、HP-4032D(大日本インキ化学工業株式会社製、エポキシ当量142g/eq)を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4及び図3に示す。
【0050】
(実施例5)
BDMA五ホウ酸縮合塩の変わりに、ピペリジン五ホウ酸縮合塩を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4、表5及び図3に示す。
【0051】
(実施例6)
ベンジルジメチルアミン五ホウ酸縮合塩(1a) 20g、ジシアンジアミド(DICY) 5.6gをメチルセロソルプ160gに溶かした透明溶液にビスフェノールA型エポキシ樹脂 エピクロン850(大日本インキ化学工業株式会社製、エポキシ当量188g/eq)100gを加え、攪拌混合した後、50℃で4時間の溶液状態での加熱処理を行った。得られた組成物は50℃にて20日放置したところ、溶液が増粘せず、優れた保存安定性を示した。続いて該溶液を60℃にて減圧によってメチルセロソルプを除き、均一透明な無溶剤一液性エポキシ樹脂組成物を得た。更に、エチルメチルケトン150gを加え、攪拌により均一透明な低粘度一液性エポキシ樹脂組成物を得た。続いて該組成物溶液を清浄なアルミ箔上に塗布し、25℃で8時間溶媒キャストを行った後、80℃、3時間更に乾燥した。続いて、150℃、180℃で各2時間の熱処理を行い、該エポキシ樹脂組成物の硬化物を得た。得られた硬化物は透明性に優れ、クラックやしわ、気泡なども無く、良好な表面形態を示した。動的粘弾性を測定したところ、tanδのピーク温度(Tg)は250℃を超え、実施例1と同等の耐熱性を示した。
【0052】
【表3】
【0053】
【表4】
【0054】
【表5】
【図面の簡単な説明】
【0055】
【図1】実施例1及び比較例1,2で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【図2】実施例2,3で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【図3】実施例4,5で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【技術分野】
【0001】
本発明は、エポキシ樹脂とモノアミン縮合ホウ酸塩を含有するエポキシ樹脂組成物に関する。より詳しくは、硬化物に高いガラス転移温度を与え、且つ優れた保存安定性を有するエポキシ樹脂組成物及びその製造方法に関する。
【背景技術】
【0002】
プリント配線板に用いられている銅張り積層板用接着剤やICパッケージの封止に用いられている半導体封止材料としては、これまでに主にエポキシ樹脂が使われている。近年、鉛フリー半田を用いているのに伴い、半田接合時の加熱温度が高くなり、銅張り積層板や半導体封止材料用エポキシ樹脂組成物から得られる硬化物には、更なる高ガラス転移温度、高弾性率化や低線膨張率などの耐熱性が強く求められている。従来、電子材料用エポキシ樹脂組成物は、ジシアンジアミドやノボラックフェノール樹脂などの硬化剤を配合したものが主に使用されている。しかし、得られた樹脂硬化物のガラス転移温度が低く、耐熱性に劣るという問題があった。
【0003】
耐熱性を向上させるためにノボラック型エポキシ樹脂を配合する方法がよく行われている(特許文献1参照)。しかしながら、この方法では樹脂硬化物のガラス転移温度が数度しか上がらず十分ではない。
【0004】
更に、エポキシ樹脂硬化物の耐熱性を向上させる方法として、エポキシ樹脂にポリアミンホウ酸塩を必須成分として添加することが報告され(特許文献2参照)、得られたエポキシ樹脂組成物の硬化物が高いガラス転移温度を有することが示されている。しかし、ポリアミンホウ酸塩を含有するエポキシ樹脂組成物の殆どはポットライフが短く、保存安定性に劣る欠点を有する。
【0005】
上述のポリアミンホウ酸塩含有エポキシ樹脂組成物の保存安定性を改良する方法として、ポリアミンホウ酸塩の添加量を抑え、潜在硬化性エポキシ樹脂の硬化剤と併用することが特許文献3に提案されている。しかし、ポリアミンホウ酸塩の添加量が少ないため、耐熱性を損なう場合があり、特に高エポキシ当量のエポキシ樹脂を用いた場合、ガラス転移温度の向上は十分ではない。
【0006】
また、エポキシ樹脂硬化物のガラス転移点を向上させることを目的として、エポキシ樹脂とピペリジン縮合ホウ酸塩を含有するエポキシ樹脂組成物に関する技術が知られている(特許文献4参照)。しかしながら、この技術は、ポットライフが短く、保存安定性に劣るものであった。
【0007】
【特許文献1】特公平7-60920号公報
【特許文献2】特開2005-68417号公報
【特許文献3】特開2005-264106号公報
【特許文献4】特開2005-89317号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記の目的を達成しようと鋭意研究を重ねた結果、ポットライフが長いエポキシ樹脂硬化剤とモノアミン縮合ホウ酸塩とを併用することによって、高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れたエポキシ樹脂組成物が得られることを見出し、本発明を完成させたものである。
【0010】
即ち、本発明は、(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤とを含有するエポキシ樹脂組成物を提供する。
【発明の効果】
【0011】
本発明のエポキシ樹脂組成物は高いガラス転移温度と低線膨張係数を有し、且つ保存安定性に優れており、銅張積層板や半導体封止材料など電子材料として好適に用いられる。
【発明を実施するための最良の形態】
【0012】
エポキシ樹脂(A)としては、1分子内に平均2個以上のエポキシ基を有する慣用のエポキシ樹脂であり、その種類は特に限定されない。例えば、次に掲げる各種のエポキシ樹脂を単独又は2種以上組み合わせて使用できる。
(1) フェノール系グリシジルエーテル型エポキシ樹脂
ビスフェノールーA、ビスフェノールーF、テトラブロモビスフェノールーA、テトラフェニロールエタン、フェニールノボラック、クレゾールノボラックなどのフェノール化合物とエピクロルヒドリンとの反応により得られるフェノール系グリシジルエーテル型エポキシ樹脂。
(2) アルコール系グリシジルエーテル型エポキシ樹脂:
(a) ビスフェノールーA、ビスフェノールーF、テトラブロモビスフェノールーA、テトラフェニロールエタンなどのフェノール化合物とアルキレンオキサイドとの付加反応により得られるポリオール、又は水添ビスフェノールAなどのポリオールと、(b) エピクロルヒドリンとの反応により得られるアルコール系グリシジルエーテル型エポキシ樹脂。
(3) グリシジルエステル型エポキシ樹脂:
ヘキサヒドロフタル酸ジグリシジルエステル、ダイマー酸グリシジルエステル等のジグリシジルエステル型エポキシ樹脂。
(4) グリシジルアミン型エポキシ樹脂:
1,3-ジグリシジルヒダントイン、トリグリシジルイソシアヌレート、テトラグリシジルジアミノジフェニルメタン、トリグリシジルーパラアミノフェノール等のグリシジルアミン型エポキシ樹脂。
(5) 混合型エポキシ樹脂:
アミノフェノール又はオキシ安息香酸とエピクロルヒドリンとの反応により得られるエポキシ樹脂、シクロペタジエンやジシクロペンタジエン骨格を有する脂環式型エポキシ樹脂、臭素化エポキシ樹脂等の混合型エポキシ樹脂。
【0013】
本発明に於いて、エポキシ樹脂組成物の硬化物が充分高いガラス転移温度と優れた力学物性を獲得する為に、エポキシ樹脂のエポキシ基当量は、好ましくは800以下、より好ましくは600以下、特に好ましくは400以下である。エポキシ当量が800以上になると、得られる硬化物が脆くなり、好ましくない。
【0014】
本発明で用いるモノアミン縮合ホウ酸塩(B)としては、通常モノアミン化合物とホウ酸系化合物とを反応させて得られるものである。かかるモノアミン化合物は、通常エポキシ樹脂の硬化促進剤として用いる2級、3級アミン及びイミタゾール化合物が好適である。具体的にはピペリジン、N-メチルピペラジン、ヒドロキシエチルピペラジン、ピロリジン、モルホリン、トリフェニルアミン、トリメチルアミン、トリエチルアミン、トリプロピルアミン、トリエタノールアミン、トリイソプロパノールアミン、ベンジルジメチルアミン、2-(ジメチルアミノメチル)フェノール、2,4,6-トリス(ジアミノメチル)フェノール、2-メチルイミダゾール、2-エチル-4-メチルイミダゾール、2-ウンデシルイミダゾール、2-ヘプタデシルイミダゾール、2-フェニルイミダゾール、1-ベンジル-2-メチルイミダゾール、1-シアノエチル-2-メチルイミダゾールなどが挙げられる。
本発明でのホウ酸系化合物としては、ホウ酸およびホウ酸エステルが用いられる。ホウ酸およびホウ酸エステルとしては、代表的なものとしては一般式(1)
【0015】
【化1】
(式中、nは0〜3までの整数、RはCmH2m+1のアルキル基であり、mは1〜10の整数を表す。)で表わされる。ホウ酸の具体的なものとしては、例えばオルトホウ酸、メタホウ酸、四ホウ酸、およびそれらの混合物であり、また、ホウ酸エステルの具体的なものとしては、例えばホウ酸トリメチル、ホウ酸トリエチル、ホウ酸トリプロピル、ホウ酸トリブチル等が挙げられる。これらのホウ酸及びホウ酸エステルは、単独又は2種以上組み合わせて使用できる。上記の中ではホウ酸が最も好ましく用いられる。
【0016】
本発明で用いるピペリジン縮合ホウ酸塩の結晶構造は、M.Wiebckeらによって明らかにされた(Chem.Sci.,48(7),978-85 1993)。一般式(2)に示したように、ピペリジン縮合ホウ酸塩は、五核の縮合ホウ酸塩であり、つまり、一分子のピペリジンカチオンに対して、五分子のホウ酸が自己縮合した形でピペリジンとイオン結合しているピペリジン五ホウ酸縮合塩である。
【0017】
【化2】
また、本発明で用いる他の2級、3級モノアミン及びイミタゾール化合物とホウ酸系化合物とを反応して得られたアミンホウ酸塩は、そのホウ素含有率が単核ホウ酸塩より高いことから、縮合ホウ酸塩が形成されていると結論した。更に、これらのアミン縮合ホウ酸塩のFT-IR及び11B-NMRを測定したところ、ピペリジン五ホウ酸縮合塩と比べて、両者のピーク形状と強度が極めて類似していることから、本発明で得られたモノアミン縮合ホウ酸塩は、五ホウ酸縮合塩またはそれを含むものと推定された。
【0018】
本発明のモノアミン五ホウ酸縮合塩では、エポキシ樹脂の硬化触媒になるモノアミンカチオンを持ち、そのモノアミンカチオンはエポキシ樹脂との相互作用によって、モノアミン五ホウ酸縮合塩を分子レベルでエポキシ樹脂に均一に分散することができる。また、一般式2に示したように、五ホウ酸縮合塩基は反応性の高い複数の水酸基を持ち、その水酸基はエポキシ樹脂の水酸基との縮合反応によって、ホウ酸エステル結合ができて、エポキシ樹脂の中に無機架橋が形成されていると推定する。即ち、エポキシ樹脂に分子レベルに分散したモノアミン五ホウ酸縮合塩のホウ酸塩基が無機架橋剤として働き、エポキシ樹脂のガラス転移温度の大幅向上に寄与したと考えられる。
【0019】
本発明で用いるモノアミン縮合ホウ酸塩(B)の合成は、例えば次のようにして行うことができる。即ち、有機溶剤または水にホウ酸を溶解させて攪拌しながら、モノアミン化合物溶液を滴下する。場合によっては、添加順序を逆にしてモノアミン化合物の有機溶剤溶液または水溶液を攪拌しながら、ホウ酸溶液を滴下することもある。続いて、室温または加熱下、一定時間において反応を行う。得たモノアミン縮合ホウ酸塩が析出した場合、吸引濾過により沈殿物を回収する。一方、反応生成物のモノアミン縮合ホウ酸塩が反応溶媒に溶けている場合、エパポレーターにより溶媒を留去する。このようにして得られた反応生成物をアセトンで数回繰り返し洗浄した後、真空乾燥により白色粉末のモノアミン縮合ホウ酸塩が得られる。
【0020】
上記アミンホウ酸塩の合成溶媒としては、ホウ酸またはモノアミン化合物の少なくとも一種を溶解するようなものが必要である。例えば、メタノール、エタノール、イソプロパノールなどの低級アルコール、アセトン、メチルエチルケトン、テトラヒドロフラン、N,N-ジメチルホルムアミド、N-メチルピロリドン、N,N-ジメチルアセトアミド、ジメチルスルホキシド、水などが挙げられ、これらは単独又は二種以上の混合で使用できる。その中では、特にN,N-ジメチルホルムアミド、水を用いることが好ましい。溶媒の使用量は、ホウ酸およびモノアミン化合物の合計100質量部に対して溶媒が300〜1500質量部となるように用いることが好ましい。
【0021】
上記モノアミン縮合ホウ酸塩(B)の合成条件として、モノアミン化合物の中のアミノ基およびイミノ基とホウ酸とのモル比及び反応温度と時間が重要である。ホウ酸の比率を増やすと、多核縮合ホウ酸塩が形成しやすく、高いホウ素含有量のアミン縮合ホウ酸塩が得られる。これに対してモノアミン化合物の比率を増やすと、より低いホウ素含有量のアミン縮合ホウ酸塩を得ることができる。一般的にモノアミン化合物の中のアミノ基およびイミノ基の合計1モルに対して、ホウ酸が0.25〜10モルが好ましく、より好ましくは0.5〜8モル、特に好ましくは1〜6モル、最も好ましくは5モルである。アミノ基1モルに対して、5モルのホウ酸を用いることによって、最も安定なアミン五ホウ酸縮合塩が形成され、好ましい。一方、0.5モル未満又は10モルを超える場合、アミンホウ酸塩の収率が低く、経済的に不利になり、好ましくない。また、反応温度について、用いるアミン系化合物の種類によっては異なるが、一般的に15℃〜100℃が好ましく、より好ましくは20℃〜80℃であり、特に好ましくは25℃〜50℃である。反応時間は反応温度にもよるが、通常0.1〜10時間である。
【0022】
上記モノアミン縮合ホウ酸塩(B)がアミン特有の刺激臭が殆どない固形状粉末であり、水またはメタノールやメチルセロソルプなどのアルコール性有機溶剤によく溶ける性質を持っており、エポキシ樹脂の硬化剤又は硬化促進剤として用いられる。該アミン縮合ホウ酸塩中のホウ素含有量は、使用するモノアミン化合物の種類及び添加量により一定ではないが、好ましくは5〜25質量%、より好ましくは8〜20質量%である。
【0023】
本発明で用いるアミンホウ酸塩(B)の使用量は、エポキシ樹脂に対して5〜40質量%で配合することが好ましい。かかる使用量であれば、得られる組成物の耐熱性が良好であり、組成物の機械特性も優れるため、好ましい。
【0024】
上述のモノアミン縮合ホウ酸塩では、嵩高い縮合ホウ酸塩基の立体障害で、モノアミンカチオンの触媒活性が低下させられて、単独でエポキシ樹脂の硬化剤として用いると、硬化不十分になる傾向があり、硬化物の耐熱性を損なう。従って、他のエポキシ樹脂の硬化剤と併用することが必要である。本発明でアミンホウ酸塩(B)と併用するエポキシ樹脂硬化剤(C)としては、エポキシ樹脂に対するポットライフが室温で10日以上である積層板用のエポキシ樹脂硬化剤が用いられる。かかるポットライフは、具体的にはエポキシ樹脂、エポキシ樹脂硬化剤及び溶剤からなる組成物を室温で保存して脂触によりゲル化(硬化)するまでに要した時間であり、該硬化剤がエポキシ樹脂と反応する硬化剤タイプについてはエポキシ樹脂1当量に対して1当量用いて測定することを基準とし、又該硬化剤が触媒タイプについてはエポキシ樹脂100質量部に対して2質量部を用いて測定することを基準としている。
【0025】
本発明で用いられる上述のエポキシ樹脂硬化剤(C)は、優れた組成物の保存安定性をもたらした潜在硬化性やポットライフの長いエポキシ樹脂硬化剤である。例えば、ジシアンジアミド、フェノールノボラック樹脂、メタフェニレンジアミンなどのエポキシ樹脂と反応する硬化剤タイプや3級アミン、イミダゾール化合物などの触媒タイプが挙げられる。これらの硬化剤の使用量は、併用するアミンホウ酸塩の添加量によって異なるが、エポキシ樹脂と反応する硬化剤タイプの場合はエポキシ樹脂1当量に対して0.1〜0.95当量で配合することが好ましく、触媒タイプの場合はエポキシ樹脂に対して1〜3phrで配合することが好ましい。
【0026】
本発明におけるエポキシ樹脂組成物の有機溶媒としては、エポキシ樹脂、モノアミン縮合ホウ酸塩及びエポキシ樹脂硬化剤を均一に溶解できる有機溶媒が使用される。エポキシ樹脂、モノアミン縮合ホウ酸塩及びエポキシ樹脂硬化剤を溶解できる有機溶媒としては、通常低級アルコ−ルを含有するもの又はセロソルプ類が使用される。例えば、アセトン、メチルエチルケトン、テトラヒドロフラン、N,N-ジメチルホルムアミド、N-メチルピロリドン、N,N-ジメチルアセトアミド、ジメチルスルホキシドなど有機溶媒にメタノールを添加した有機溶媒の混合物が挙げられる。また、メチルセロソルプなどのセロソルプ類は単独でも用いられる。
【0027】
本発明のエポキシ樹脂組成物の製造方法に於いては、エポキシ樹脂およびモノアミン縮合ホウ酸塩を有機溶媒に均一に溶解した溶液状態での加熱処理を行うことは均一透明なエポキシ樹脂組成物を得るために極めて重要である。溶液状態での加熱処理とは、実質的に溶媒を除去することなく、即ちクローズドシステムで加熱し、しかも樹脂がゲル化しないように行うことを云う。それを行うことにより、エポキシ樹脂とモノアミン縮合ホウ酸塩との相互作用を持たせることにより、モノアミン縮合ホウ酸塩がエポキシ樹脂溶液中に分子レベルで均一に相溶する。得られた溶液を脱溶剤して得られる組成物はモノアミン縮合ホウ酸塩の凝集がなく均一透明となる。
【0028】
溶液状態での加熱処理の条件は、用いるモノアミン縮合ホウ酸塩とエポキシ樹脂との反応の容易さにより異なる。基本的には加熱処理の下限及び上限としては、その後の脱溶剤によりモノアミン縮合ホウ酸塩が析出しなくなる範囲(下限)、且つ溶液がゲル化しない範囲(上限)で加熱処理を行うことが重要である。加熱処理を過度に行った場合、組成物が増粘またはゲル化してしまい、実用性の点から好ましくない。
【0029】
溶液状態での加熱処理温度は、好ましくは25〜100℃、より好ましくは30〜90℃、特に好ましくは40〜80℃で行える。加熱処理時間は加熱処理温度により異なるが、0.2〜20時間が好ましく、より好ましくは0.5〜15時間、特に好ましくは1〜10時間である。
【0030】
上述により得られるエポキシ樹脂組成物から、硬化反応を進めないようにして溶媒を除去することにより、無溶媒のエポキシ樹脂組成物が製造できる。脱溶媒温度は用いるアミンホウ酸塩及びエポキシ樹脂硬化剤により異なるが、100℃以下が好ましく、より好ましくは80℃以下、特に好ましくは60℃以下である。
【0031】
而して製造される溶液状のエポキシ樹脂組成物は、積層板用樹脂材料として利用できるほか、金属、セラミック、耐熱性プラスチックなどの基材に対する耐熱性塗料としても利用することができる。
【0032】
例えば、上記エポキシ樹脂組成物を耐熱性基材に塗布又は含浸させた後、乾燥してプリプレグとする。このプリプレグに銅箔を重ね合わせ、加熱圧縮することにより銅張積層板を製造することができる。かかる耐熱性基材は、ガラス繊維が一般的であるが、他に芳香族ポリアミド繊維を用いることもできる。
【0033】
また、上記エポキシ樹脂組成物を直接銅箔に塗布し、乾燥した後、それをエポキシ樹脂ガラスクロス板と重ね合わせ、加熱圧縮することにより銅張積層板を製造することもできる。
【0034】
上述の方法により製造される銅張り積層板は、耐熱性に優れ、電子部品として好適に用いられる。
【実施例】
【0035】
次に、本発明を合成例、実施例によって具体的に説明する。
【0036】
また、以下の合成例において、核磁気共鳴スペクトル(NMR)の測定は日本電子(株)製 JNM-LA-300を用いた。11B-NMRスペクトルは重水中のホウ酸ピークを基準とした。
粉末X線回折の測定は理学電機(株)製X線回折装置 RINT ULTIMA+ を使用した。
ホウ素含有量はPerkn Elmer社製 Optima 3300DVを用いて、ICPの測定を行い、予めホウ酸を用いて作成しておいた検量線により定量した。
アミンの定量は1H-NMRの内部標準法により行った。すなわち、試料に一定量のベンゼンを内部標準として添加し、そのピークと一定量のアミンホウ酸塩のアミンの特定ピークとの面積比により定量した。
【0037】
なお、本発明の合成例及び実施例について次の試薬を使用した。
(1)モノアミン化合物
ベンジルジメチルアミン(BDMA):和光純薬工業株式会社製、試薬特級
2-エチル-4-メチルイミダゾール(2E4MZ) :和光純薬工業株式会社製、試薬特級
ピペリジン:和光純薬工業株式会社製、試薬特級
(2) ホウ酸: 和光純薬工業株式会社製、試薬特級
(3) 溶剤
N,N-ジメチルホルムアミド(DMF):和光純薬工業株式会社製、試薬特級
アセトン:和光純薬工業株式会社製、試薬1級
メチルエチルケトン(MEK) :和光純薬工業株式会社製、試薬特級
メタノール:和光純薬工業株式会社製、試薬特級
メチルセロソルプ: 和光純薬工業株式会社製、試薬特級
(4) 硬化剤
ジシアンジアミド(DICY) :和光純薬工業株式会社製、試薬特級
2-エチル-4-メチルイミダゾール(2E4MZ) :和光純薬工業株式会社製、試薬特級
【0038】
(合成例1) [ベンジルジメチルアミン五ホウ酸縮合塩の合成]
ホウ酸34.3g(0.555mol)をDMF 350gに溶かした溶液を攪拌しながら、ベンジルジメチルアミンBDMA 15g(0.111mol)を滴下した。白色沈殿が直ちに析出した。室温で18時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトン二回洗浄し、70℃、3時間真空乾燥により、26.1gの白色粉末1aを得た。
【0039】
(合成例2) [2-エチル-4-メチルイミダゾール五ホウ酸縮合塩の合成]
ホウ酸42.1g(0.68mol)をDMF 260gに溶かした溶液を攪拌しながら、2-エチル-4-メチルイミダゾール2E4MZ 15g(0.136mol)を加えた。白色沈殿が析出した。室温で13時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトンで二回洗浄し、70℃、4時間真空乾燥により、反応生成物の白色粉末1b 34.5gを得た。
【0040】
(合成例3) [ピペリジン五ホウ酸縮合塩の合成]
ホウ酸30g(0.485mol)をDMF 120gに溶かした溶液を攪拌しながら、ピペリジン 41.34g(0.485mol)を滴下した。白色沈殿が直ちに析出した。室温で24時間攪拌して、吸引濾過により沈殿物を回収した。続いて、得た沈殿物をアセトン二回洗浄し、50℃、8時間真空乾燥により、19.1gの白色粉末1cを得た。
【0041】
【表1】
【0042】
【表2】
【0043】
以下の実施例および比較例において、光透過率は日本電色工業株式会社製NDH-300Aを用いて、厚さ300μmのフィルムの平行透過率を測定した。ガラス転移温度(Tg)及び貯蔵弾性率(E')は、固体動的粘弾性測定装置(エスアイアイ・ナノテクノロジー株式会社製DMS6100)を用い、測定周波数1Hz、昇温速度3℃/分で測定した。なお、ガラス転移温度はtanδピーク温度(tanδmax)とした。また、熱線膨張係数は、エスアイアイ・ナノテクノロジー株式会社製TMA/SS6100を用いて、窒素雰囲気下、引張りモート、昇温速度10℃/分で測定した。
【0044】
(実施例1及び比較例1,2)
ベンジルジメチルアミン五ホウ酸縮合塩(1a) 20gをメチルセロソルプ160gに溶かした透明溶液にビスフェノールA型エポキシ樹脂 エピクロン850(大日本インキ化学工業株式会社製、エポキシ当量188g/eq)100gを加え、攪拌混合した後、50℃で4時間の溶液状態での加熱処理を行った。次に、ジシアンジアミド(DICY) 5.6gを加え、攪拌溶解させて、エポキシ樹脂組成物溶液を得た。得られた組成物は50℃にて20日放置したところ、溶液が増粘せず、優れた保存安定性を示した。続いて該溶液を清浄なアルミ箔上に塗布し、50℃で8時間溶媒キャストを行った後、80℃、3時間更に乾燥した。続いて、150℃、180℃で各2時間の熱処理を行い、該エポキシ樹脂組成物の硬化物を得た。
【0045】
上記の硬化物は透明性に優れ、可視光域での光透過率は91%(100μm厚み換算)であり、クラックやしわ、気泡なども無く、良好な表面形態を示した。また、比較例1においてジシアンジアミドを使用せずベンジルジメチルアミン五ホウ酸縮合塩のみを用いること、比較例2においてベンジルジメチルアミン五ホウ酸縮合塩を使用せずジシアンジアミドのみを用いること、それ以外に実施例1と同様にしてエポキシ樹脂の硬化物を作製した。
【0046】
実施例及び比較例で得られた厚み約300μmの硬化フィルムを用いて動的粘弾性測定(周波数1Hz)を行った。表3及び図1に示したように、実施例1のtanδのピーク温度(Tg)は250℃を超えた。これに対して、比較例1では140℃にtanδのサブピークがあり、比較例2ではtanδのピーク温度(Tg)は150℃であった。アミン縮合ホウ酸塩とジシアンジアミドを併用したエポキシ樹脂硬化物のガラス転移温度が大きく向上していることが明らかである。また、表5に示したように、実施例1の線膨張係数は、比較例1及び比較例2より低くなった。
【0047】
(実施例2)
DICYの変わりに、2E4MZを用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4及び図2に示す。
【0048】
(実施例3)
BDMA五ホウ酸縮合塩の変わりに、2E4MZ五ホウ酸縮合塩を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4、表5及び図2に示す。
【0049】
(実施例4)
エポキシ樹脂850の変わりに、HP-4032D(大日本インキ化学工業株式会社製、エポキシ当量142g/eq)を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4及び図3に示す。
【0050】
(実施例5)
BDMA五ホウ酸縮合塩の変わりに、ピペリジン五ホウ酸縮合塩を用いたこと以外に、実施例1と同様にしてエポキシ樹脂の硬化物を作製した。測定結果を表4、表5及び図3に示す。
【0051】
(実施例6)
ベンジルジメチルアミン五ホウ酸縮合塩(1a) 20g、ジシアンジアミド(DICY) 5.6gをメチルセロソルプ160gに溶かした透明溶液にビスフェノールA型エポキシ樹脂 エピクロン850(大日本インキ化学工業株式会社製、エポキシ当量188g/eq)100gを加え、攪拌混合した後、50℃で4時間の溶液状態での加熱処理を行った。得られた組成物は50℃にて20日放置したところ、溶液が増粘せず、優れた保存安定性を示した。続いて該溶液を60℃にて減圧によってメチルセロソルプを除き、均一透明な無溶剤一液性エポキシ樹脂組成物を得た。更に、エチルメチルケトン150gを加え、攪拌により均一透明な低粘度一液性エポキシ樹脂組成物を得た。続いて該組成物溶液を清浄なアルミ箔上に塗布し、25℃で8時間溶媒キャストを行った後、80℃、3時間更に乾燥した。続いて、150℃、180℃で各2時間の熱処理を行い、該エポキシ樹脂組成物の硬化物を得た。得られた硬化物は透明性に優れ、クラックやしわ、気泡なども無く、良好な表面形態を示した。動的粘弾性を測定したところ、tanδのピーク温度(Tg)は250℃を超え、実施例1と同等の耐熱性を示した。
【0052】
【表3】
【0053】
【表4】
【0054】
【表5】
【図面の簡単な説明】
【0055】
【図1】実施例1及び比較例1,2で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【図2】実施例2,3で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【図3】実施例4,5で得られたエポキシ樹脂硬化物の貯蔵弾性率(E')とtanδの温度分散を示す図である。なお、実線は貯蔵弾性率(E')を表し、点線はtanδを表す。
【特許請求の範囲】
【請求項1】
(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤と、(D)溶剤とを含有するエポキシ樹脂組成物。
【請求項2】
前記モノアミン縮合ホウ酸塩が、2級アミン化合物、3級アミン化合物又はイミタゾール化合物と、ホウ酸系化合物とを反応して得られるアミン縮合ホウ酸塩である請求項1に記載のエポキシ樹脂組成物。
【請求項3】
前記ホウ酸系化合物がホウ酸又はメタホウ酸またはホウ酸エステルであることを特徴とする請求項2に記載のエポキシ樹脂組成物。
【請求項4】
前記エポキシ樹脂硬化剤がジシアンジアミド、フェノールノボラック樹脂、芳香族ポリアミンおよびイミタゾール化合物から選ばれる一種以上の硬化剤である請求項1〜3のいずれか一つに記載のエポキシ樹脂組成物。
【請求項5】
前記溶剤がアルコール系溶剤を含有する請求項1〜4のいずれか一つに記載のエポキシ樹脂組成物。
【請求項6】
(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤とを、(D)溶剤に均一に溶解した樹脂材料を加熱処理することを特徴とする非ゲル状1液性エポキシ樹脂組成物の製造方法。
【請求項1】
(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤と、(D)溶剤とを含有するエポキシ樹脂組成物。
【請求項2】
前記モノアミン縮合ホウ酸塩が、2級アミン化合物、3級アミン化合物又はイミタゾール化合物と、ホウ酸系化合物とを反応して得られるアミン縮合ホウ酸塩である請求項1に記載のエポキシ樹脂組成物。
【請求項3】
前記ホウ酸系化合物がホウ酸又はメタホウ酸またはホウ酸エステルであることを特徴とする請求項2に記載のエポキシ樹脂組成物。
【請求項4】
前記エポキシ樹脂硬化剤がジシアンジアミド、フェノールノボラック樹脂、芳香族ポリアミンおよびイミタゾール化合物から選ばれる一種以上の硬化剤である請求項1〜3のいずれか一つに記載のエポキシ樹脂組成物。
【請求項5】
前記溶剤がアルコール系溶剤を含有する請求項1〜4のいずれか一つに記載のエポキシ樹脂組成物。
【請求項6】
(A)エポキシ樹脂と、(B)モノアミン縮合ホウ酸塩と、(C)エポキシ樹脂に対するポットライフが室温で10日以上であるエポキシ樹脂硬化剤とを、(D)溶剤に均一に溶解した樹脂材料を加熱処理することを特徴とする非ゲル状1液性エポキシ樹脂組成物の製造方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2008−222910(P2008−222910A)
【公開日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願番号】特願2007−64982(P2007−64982)
【出願日】平成19年3月14日(2007.3.14)
【出願人】(000173751)財団法人川村理化学研究所 (206)
【Fターム(参考)】
【公開日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願日】平成19年3月14日(2007.3.14)
【出願人】(000173751)財団法人川村理化学研究所 (206)
【Fターム(参考)】
[ Back to top ]