
エマルジョン由来(emulsion−derived)粒子

架橋剤によって架橋されたポリマー鎖で成る格子と、前記格子を成すポリマー鎖に隣接および囲まれる形で存在する開口領域とを有するエマルジョン由来粒子。格子上には官能基が配されており、タンパク質または修飾タンパク質はこれらと反応することで、格子に結合され、固定化される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はエマルジョン由来粒子に関する。また、本発明は当該粒子の製造方法に関する。
【背景技術】
【0002】
固定化酵素を含んだ粒子が通常用いられるのは、バイオ触媒や診断などの用途である。しかしながら、出願人の知る粒子は、充分な酵素固定化を実現するには表面積が足りないという問題を抱えている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】欧州特許第0 534 057号明細書
【特許文献2】国際公開第02/29406号明細書
【発明の概要】
【発明が解決しようとする課題】
【0004】
そこで、本発明が目的とするのは、この問題を少なくとも軽減できる粒子を提供すること、および、タンパク質に対する結合容量が大きくタンパク質を固定化できる粒子の製造方法を提供することである。
【課題を解決するための手段】
【0005】
そこで、本発明の第1の様態として、架橋剤によって架橋されたポリマー鎖で成る格子と、前記格子を成すポリマー鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有するエマルジョン由来粒子であって、タンパク質または修飾タンパク質と反応することで、当該タンパク質または修飾タンパク質を格子に結合させ、固定化する、というエマルジョン由来粒子を提供する。
【0006】
「エマルジョン由来」とは、粒子がエマルジョン技術を用いて製造または形成されたことを意味し、当該エマルジョン技術としては、本発明の第2、第3の様態によるエマルジョンベース(emulsion based)の手法がある(ただし、これらに限定はされない)。
「修飾(modified)タンパク質」は、化学的手段(例えばジアルデヒドの追加)によって修飾されたタンパク質、あるいは、遺伝子レベルで(例えばhisタグを用いて)修飾されたタンパク質を意味する。
【0007】
従って、本発明の粒子は、鎖状(strand)あるいは繊維状(fiber)のポリマーに、または架橋剤に官能基を含み、タンパク質または修飾タンパク質を官能基と反応させることができる。より具体的には、官能基は、鎖状あるいは繊維状のポリマーに存在しており、タンパク質または修飾タンパク質のポリマーへの結合が、ポリマー上の官能基を変更する形で共有結合、イオン結合、疎水結合、親和結合のうち1または複数で成されるように選択される。
【0008】
そうして、粒子は、官能基によってポリマーに結合され、それによって固定化されるタンパク質または修飾タンパク質を少なくとも1つ含む。タンパク質については、単一の酵素または酵素の混合物、抗体または抗体の混合物、抗原または抗原の混合物、または、機能的特性または構造的特性を備えた他のタンパク質とすればよい。すなわち、必要に応じて、複数の異なるタンパク質や複数の異なる修飾タンパク質を、粒子内部で固定化できる。タンパク質が酵素である場合、本粒子は、酵素の最適pHを酸性域またはアルカリ性域にシフトすることを可能にする手段を提供する。これは粒子内での酵素の固定化による。
【0009】
タンパク質または修飾タンパク質をポリマーに共有結合する場合、これは、例えば、エポキシド、アルデヒドをタンパク質または修飾タンパク質のアミン基と相互作用させることで達成される。
タンパク質または修飾タンパク質をポリマーにイオン結合する場合、これは、ポリマー上で正または負に帯電した官能基を、タンパク質または修飾タンパク質上で前記官能基と反対に帯電されたアミノ酸残基とイオン結合させることで達成される。
【0010】
タンパク質をポリマーに疎水結合する場合、これは、ポリマー上の芳香族アルカンまたは長鎖アルカンの疎水基とタンパク質上の疎水性アミノ酸とが結合することで達成される。
タンパク質をポリマーに親和性結合する場合、これは、二価金属またはアビディンといった親和性タグが、ヒスチジンまたはビオチン化タンパク質を結合することで達成される。
【0011】
当然のことながら、粒子には、タンパク質のより効率的な結合のために、必要に応じて上記の種類またはカテゴリーの官能基を複数含ませてもよい。
鎖状または繊維状のポリマーは、ホモポリマーとしてよいし、ポリエチレンイミンとしてもよい。
架橋剤は、グルタルアルデヒドまたは他のアルデヒド、エポキシド、あるいは、複数の官能基を有する他の適当な化合物とすればよい。
【0012】
粒子は、格子の開口領域や空間の内部に捕捉された補助剤を含む。補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択すればよい。適当な酵素または基質を補助剤として粒子に含ませれば、反応に用いる補因子の連続的な再生を達成でき、それによって、反応を平衡状態に到達させたり、あるいは反応を完了させたりすることができる。補助剤として磁鉄鉱または磁性物質を含ませれば、形成液状媒体からの粒子の回収は、磁気分離を用いて速やかに行える。
【0013】
また、本発明の第2の様態として、第1の液相の液滴が第2の液相内に分散したエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中には溶解したポリマーおよび架橋剤が含まれるという状態で製造する処理と、架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、を特徴とする粒子生成方法を提供する。
【0014】
第1の液相は水相であり、従って第2の液相は油相となる。よって、エマルジョンは油中水分散型(water(w)-in-oil(o))のエマルジョン(すなわちw/oエマルジョン)となる。しかし、本発明の別の実施の形態では、エマルジョンを、水中油分散型(oil-in-water(o/w))エマルジョン、水中油分中に水が分散した型(water-in-oil-in-water(w/o/w))のエマルジョン、あるいは、油中水分中に油が分散した型(oil-in-water-in-oil(o/w/o))のエマルジョンとすることもできる。
【0015】
本エマルジョンは、第1のエマルジョン(水滴で成り、その中にポリマーが溶けて油相で分散した状態で含まれるもの)を、第2のエマルジョン(水滴で成り、その中に架橋剤が溶けて油相で分散した状態で含まれるもの)と混合する方法で形成できる。
また、本発明の第3の様態として、第1の液相の液滴が第2の液相内に分散した第1のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解したポリマーが含まれているという状態で製造する処理と、第1の液相の液滴が第2の液相内に分散した第2のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解した架橋剤が含まれるという状態で、第1のエマルジョンと併せる処理と、架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、を特徴とする粒子生成方法を提供する。
【0016】
相のうち少なくとも1つは、界面活性剤(detergent、surfactant)を含む。界面活性剤は、両性イオン界面活性剤、中性界面活性剤、荷電界面活性剤またはポリマー界面活性剤から選択すればよい。陰イオン界面活性剤としては、アルキル硫酸塩(ラウリル硫酸ナトリウム、ラウレス硫酸ナトリウム、アルキルエーテル硫酸など)がある。陽イオン界面活性剤としては、セントリモニウムクロリド(centrimonium chloride)がある。非イオン物質の界面活性剤としては、エトキシ化アルキルフェノール(ポリオキシエチレン(10)イソオクチルシクロヘキシルエーテル(Triton X100)、ポリオキシエチレン(9)ノニルフェニルエーテル(ノノキシノール-9)など)がある。両性または両親媒性の界面活性剤としては、デシルベタイン(decyl betaine)がある。ポリマー界面活性剤としては、ソルビトール−(エチレンオキシド)80、エチレンオキシド−プロピレンオキシド−エチレンオキシドのトリブロックコポリマー(これは、ポロキサマーとしても知られ、Pluronicの商品名でBASF社から入手できるものなどがある)、そして、プロピレンオキシド−エチレンオキシド−プロピレンオキシドのトリブロックコポリマー(メロキサポールとしても知られるもの)がある。
【0017】
油相の油は、少なくとも原則上は、水と混和しない何らかの適当な有機溶剤、植物油、鉱物油、石炭または原油由来の油相成分(oily component)、または合成油であればよい。しかしながら、鉱物油、パラフィン、溶剤(イソオクタンなど)から選択するのが好ましい。
これまでに述べた通り、ポリマーはポリエチレンイミン(PEI)であり、架橋剤は、二官能基または多官能基のアルデヒド(グルタルアルデヒド、スクシンアルデヒド、デキストランアルデヒド、ヘキサメチレンジイソシアネート、グリオキサールなど)である。他の適当な架橋剤を、PEIまたは誘導化PEI、あるいは他のポリマーに対して使用する場合もある。イソシアネート(ヘキサメチレンジイソシアネートまたはトルエンジイソシアネート、またはイソチオシアネートを含む);エポキシド(2-クロロメチルオキシランなど);無水物;エピクロルヒドリン,1-エチル-3,3-ジメチルアミノプロピルカルボジイミド;エチルクロロアセテート、または、それに類するものが挙げられる。タンパク質または修飾タンパク質の固定化には未反応の官能基が用いられるので、架橋剤は、ポリマー修飾または架橋後(post cross-linking)修飾のための誘導(derivitization)剤とも見なされるであろう。他のポリマーまたはコポリマーを用いることもでき、そうしてものとしては、ポリビニルアルコール、ナイロン、アルギネート、その他タンパク質(アルブミン、コラーゲン、それに類するもの)、およびそれに類するもの(修飾されたもの、修飾されていないもの)がある。
【0018】
本方法には、タンパク質(酵素、抗体または抗原、または修飾タンパク質)を粒子内または粒子上に導入する処理が含まれる場合があり、それによって、タンパク質または修飾タンパク質は、上述したように、ポリマー鎖または繊維において、または架橋剤において官能基と反応し、その結果、鎖状または繊維状のポリマーに、または架橋剤に結合されることで固定化される。
【0019】
本方法は、相の1つに補助剤を加えて、補助剤を粒子の格子の内部に捕捉させる処理を含む場合がある。また、格子へのタンパク質の結合の前または後に補助剤を更に追加する場合がある。上述したように、補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択される。
また、本方法には、粒子を油相から回収する処理が含まれる場合がある。具体的には、粒子の回収は物理的な分離手段(遠心分離や濾過)で実行する。
【0020】
また、本方法には、回収した粒子の乾燥処理が含まれる場合がある。粒子の乾燥処理としては、アセトン脱水、空気乾燥、噴霧乾燥、そして、好ましいものとして、凍結乾燥または真空乾燥がある。
なお、上述したとおり、本方法は、回収した粒子に乾燥処理に先立って補助剤を加える処理を含む場合があり、そうすると、補助剤は粒子の格子の内部に捕捉されることになる。
【0021】
回収した粒子の乾燥処理は、タンパク質固定化の前後いずれであっても、タンパク質および粒子の安定化促進の達成、または、天然補因子や修飾補因子といった添加物の捕捉に関して効果がある。乾燥処理によって更に、多点結合によるタンパク質または修飾タンパク質の架橋状態の改善という結果が得られる。そして、これにより、酵素などのタンパク質の安定性が向上することになる。
【0022】
また、本発明の粒子には多様な用法や用途があると考えられる。例えば、生体触媒用、酵素ベースの生物機能を利用した環境修復、診断に用いられる。また、膜反応器やタンパク質固定化マトリックスといった固体支持体の表面に結合させて表面積を大きくするのに用いることができる。
【図面の簡単な説明】
【0023】
【図1】例1によるグルタルアルデヒド架橋PEIポリマー粒子の顕微鏡写真であり、ポリマー鎖/繊維のPEI支持体格子(support lattice)または網目主鎖(network backbone)を示す図である。
【図2】例2による、繊維格子に結合するラッカーゼに関して得られた結果を示すグラフであり、pHプロフィール(profile)のシフトに関して結合効率の補正を行った場合の図である。
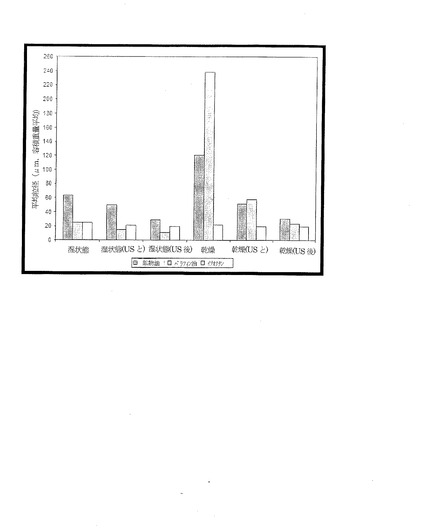
【図3】各種の油相を用いて製造した粒子の粒径分布(平均粒径)の分析結果を示す図であり、粒子を乾燥させなかった場合(湿った状態)、乾燥させた場合(凍結乾燥を採用)、インライン(in-line)超音波照射(US)を伴う場合、超音波照射で前処理した後に測定した場合、について示す図である。
【図4】各種の界面活性剤を用いて製造した粒子の粒径分布の分析結果を示す図であり、そのままの状態で光散乱によって測定した場合と、超音波照射後(US後)の分析で求めた場合とを示す図である。
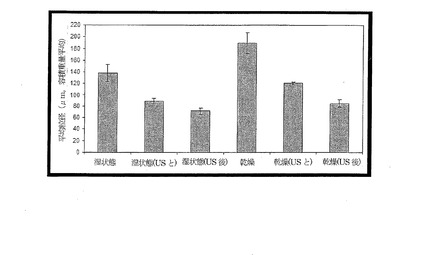
【図5】単一エマルジョンを用いて製造した乾湿両粒子について粒径分布を示す図であり、結果は3回繰り返して行った実験の標準偏差を示しており、そのままの状態での光散乱によって測定した場合と、インライン超音波照射を伴う場合と(「USと共に」)、サンプルに事前超音波照射した後の分析で求めた場合(US後)とを示す図である。
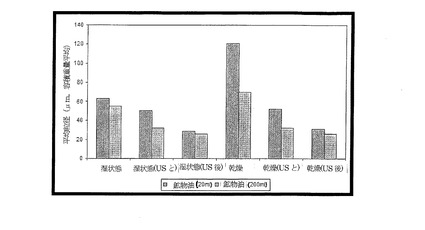
【図6】20mlの量の鉱物油と200mlの量の鉱物油とで製造した乾湿両粒子について粒径分布を示す図である。
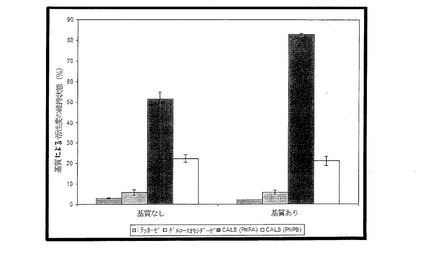
【図7】潜在的な保護剤としての基質を有する場合と有していない場合とで、粒子に結合された各種酵素の活性度の維持状態を示す図である。
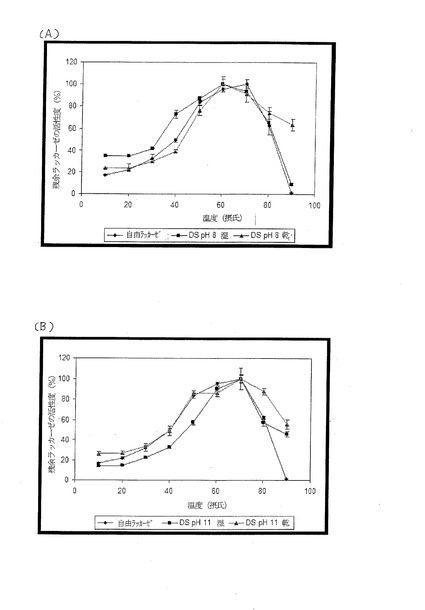
【図8】自由(free)ラッカーゼ結合粒子および乾湿両ラッカーゼ結合粒子に関する温度最適条件を示す図であり、(A)はPEIがpH8で製造された場合、(B)はPEIがpH11で製造された場合を示す図である。
【図9】PEIがpH8、ph11で製造された乾湿両方の粒子結合ラッカーゼおよび自由ラッカーゼについてpH安定性(6時間)を示す図である。
【図10】(A)は、例14による、後処理していない繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、ポリエチレンイミン(‘PEI’)濃度の変化がpHプロフィールのシフトに与える影響を表す図であり、(B)は、例14による、後処理していない繊維格子または網目における固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、グルタルアルデヒド濃度の変化がpHプロフィールのシフトに与える影響を表す図である。
【図11】(A)は、グルタルアルデヒド後処理した繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、PEI濃度の変化がpHプロフィールのシフトに与える影響を表す図であり、(B)は、例14により、グルタルアルデヒド後処理した繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、グルタルアルデヒド濃度の変化がpHプロフィールシフトに与える影響を表す図である。
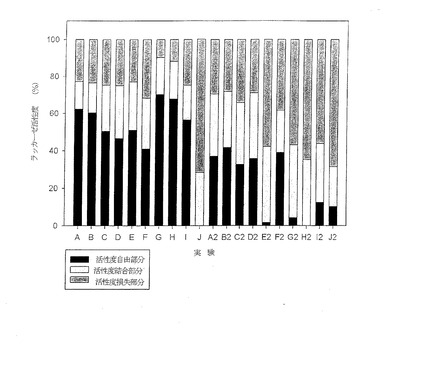
【図12】実験A〜Fに関する粒子のペルオキシダーゼ活動度について得られた結果を示す図である。
【発明を実施するための形態】
【0024】
ここから、本発明について、後述の実施の形態および添付図面を参照しながら、より詳細に説明する。
(例1:ポリマー鎖/繊維の網目または格子からなる粒子の製造方法)
本方法で形成される油中水分散エマルジョンは、その内部で、ポリアミンポリマー(ポリエチレンイミン)を含むエマルジョンと別の主要なアミン架橋剤(グルタルアルデヒド)とが組み合わされる。2つの試薬が反応して、微細な粒子または粒の形をしたポリマーを形成する。
(使用化学物質)
グルタルアルデヒド(25%の水溶液)は、Acros Organics社(Geel West Zone 2, Jansen Pharmaceuticalaan 3a, 2440 ゲール, ベルギー)から入手した。ポリエチレンイミン(PEI)(50%の水溶液、カタログ番号P−3143、Mw750,000およびMn60,000)は、Sigma-Aldrich社(セントルイス、ミズーリ州 63178)から取得した。鉱物油(医薬用ホワイトオイル、48031)は、Castrol社(8 ジャンクションアベニュー、パークタウン、2193 ヨハネスバーグ、南アフリカ)から購入した。
(粒子の製造方法)
(エマルジョンAの組成)
・10mlの鉱物油(油相)
・0.05mlのノノキシノール−4(界面活性剤)
・0.5mlのポリエチレンイミン(ポリアミン)、(10%m/v水溶液)、pH11
700rpmで30分間、電磁撹拌器を用いて撹拌した。
(エマルジョンBの組成)
・10mlの鉱物油(油相)
・0.05mlのノノキシノール−4(界面活性剤)
・0.5mlのグルタルアルデヒド、(25%m/v、等級(grade)II)
700rpmで30分間、電磁撹拌器を用いて撹拌した。
【0025】
2つのエマルジョン(A、B)を組み合わせると、ポリマー架橋反応を生じさせることができた。そして、電磁撹拌器を用いて700rpmで30分〜1時間撹拌することで、エマルジョンの状態は確実に維持された。その後、JA20.1ロータを備えたBeckman-Coulter J2-21MEに入れて10分間、エマルジョンを3000rpmで遠心分離し、形成された粒子を回収した。ペレットは、脱イオン水に再懸濁して10〜40mlまで希釈した後、再び遠心分離した。そして、この洗浄工程を更に2回繰り返した。最終的に得られた上澄みは澄んでいた(clear)。そして最終的に残ったペレットは、10mlのTris-Cl緩衝液(0.05M、pH8.0)に懸濁した。
(結果)
全てのエマルジョン調製で得られる材料を、遠心分離によって回収し、光学顕微鏡検査によって視覚化した。図1に示すのは格子形成の結果であり、おおよそ球状の粒子が形成されたことを示している。
(PEI対グルタルアルデヒドの濃度比が粒子形成に与える影響)
グルタルアルデヒドに対するPEIの比率が粒子形成に与える影響を調査した。サンプルは表1に従って調製した。
【0026】
乾燥重量の測定は、繊維主鎖格子/支持体の凍結乾燥および重量計測という手順で行った。この実験の結果は表1に示してあり、幅広い反応体の組合せで粒子が形成されることを示している。
表1:繊維ポリマー主鎖の製造評価に使用するPEIおよびグルタルアルデヒドの量
【0027】
【表1】

(例2:ポリマー鎖/繊維のPEI支持体格子または網目主鎖へのラッカーゼの結合)
(酵素)
DeniLite(登録商標)(ラッカーゼ)は、Novozymes社(Novozymes A/S、Krogshoejvej 36、2880 バウスベア、デンマーク)から取得した。
(酵素洗浄)
ラッカーゼは、DeniLiteから部分的に精製した。その方法は、2回蒸留した100mlのH2Oに5gのDeniLite II Baseを溶かしながら、4℃で1時間、200rpmで撹拌するというものであった。懸濁物質は、Beckman-Coulter J2-21ME遠心分離機のJA14ロータを用い、4℃で1時間、10000rpmで遠心分離を行って除去した。上澄みを取り除き、10kDaカットオフ機能を備えたSnakeSkin(登録商標)(Pierce社)透析管を用いて、4℃の水5Lを3回交換して透析した。最初の2回の交換までの透析は2時間継続し、最後の透析は12時間継続した。酵素は、液体窒素の中で凍結させて凍結乾燥した。その後、このラッカーゼは、必要になるまで4℃で保管した。
(ラッカーゼの分析評価)
ラッカーゼの分析評価は、ラッカーゼを支持体に結合した後の遠心分離による上澄みと、支持体に固定化されたラッカーゼとを対象に実施し、結合時の活性度の維持状態を測定した。ラッカーゼ試薬は、基質としての1mMのグアヤコールを、100mMのサクシネートラクテート緩衝液(pH4.5)の中に含むものとした(Jordaan J,Pletschke B,Leukes W(著)、2004年、(題名)“Purification and partial characterization of a thermostable laccase from an unidentified basidiomycete.”(掲載誌)Enz. Microb. Technol. 34:635-641)。分析評価は、5 200M−1.cm−1の減衰係数で、450nmにおいて3回繰り返して実施した。分析評価の実施には、PowerWave HT Microtitreプレートリーダを用いた。酵素の単位は、1分につき1μmolの基質を酸化させるのに必要な酵素量を1単位として定めた。
(タンパク質の測定)
タンパク質担持(protein loading)後に、ラッカーゼを基準タンパク質とし、280nmでの光吸光を利用して溶液中のタンパク質濃度を測定した。溶液中の残留タンパク質をタンパク質全体から差し引いたものを、結合されたタンパク質と見なした。
(酵素固定化)
室温で30分間緩やかに揺動して、酵素(ラッカーゼ)(10mg.ml−1溶液で1ml)を支持体に結合させた。このとき、酵素は、表2に示す乾燥重量を持つ主鎖と結合した。ラッカーゼと結合した粒子は、700×gで5分間遠心分離を行って回収した。固定化された酵素は、50mlの水で5回洗浄し、上述したような遠心分離を行って回収した。
【0028】
タンパク質(この場合は酵素であるラッカーゼ)は、緩衝液の形でPEI-グルタルアルデヒド粒子(誘導化したもの、あるいは、誘導化していないもの)に加えた。粒子は例1で示した形で調製した。粒子をタンパク質と反応させて、上述したように、タンパク質をポリマーに固定化することができた。粒子は、遠心分離による回収から使用時まで、乾燥させないようにした。
【0029】
様々な形で製造したタンパク質支持体にタンパク質を結合した結果を、下の表2に示す。一方、ラッカーゼ活性度に関する結果は図2に示してある。
(結果)
表2:主鎖支持体へのラッカーゼの結合効率
【0030】
【表2】
別の実験用セットを表1に示す通りに調製した。しかし、本例での粒子はグルタルアルデヒドで後処理してあり、数字の2で区別している(すなわち、A2−J2)。調製に続き、上述した方法に従ってラッカーゼを粒子に結合した。ラッカーゼが結合した粒子を、700×gで5分間遠心分離して回収した。
【0031】
ポリマー支持体に担持された酵素の活性に関する結果を図2に示す。
本調査が示しているのは、繊維格子または網目をタンパク質固定化の支持体として用いた場合、タンパク質結合容量が大きいと同時に、タンパク質の機能活性は保たれる、ということである。
(例3:各種の油を用いたポリマー鎖/繊維のPEI支持体格子または網目主鎖の製造方法)
エマルジョンの油相の違いが与える影響を調査した。粒子の製造にあたって、先ず2種類の異なるエマルジョンA,Bを調製した。
(エマルジョンAの組成)
・10mlの鉱物油、パラフィン油またはイソオクタン(油相)
・0.1mlのノノキシノール-4(界面活性剤)
・0.5mlのポリエチレンイミン(ポリアミン)、(10%m/v水溶液)、pH11
25℃において、500rpmで30分間、電磁撹拌器を用いて撹拌した。
(エマルジョンBの組成)
・上述したのと同じ油相10ml
・0.1mlのノノキシノール-4(界面活性剤)
・0.5mlのグルタルアルデヒド、(25%m/v、等級II)
25℃において、500rpmで30分間、電磁撹拌器を用いて撹拌した。
【0032】
続いて、エマルジョンAを短時間でエマルジョンBに加え、更に1時間撹拌した(700rpm)。そして、Sorvall benchtop遠心分離機によって3000×gで10分間遠心分離した後、10mlの量の脱イオン水で6回洗浄するという手順で、エマルジョンから粒子を回収した。洗浄後、最終的に得られた粒子を脱イオン水で20mlに再懸濁し、その半分を凍結乾燥によって乾燥させた。湿潤粒子(wet particle)および乾燥粒子の両方について、粒径分布を分析した(Malvern Mastersizer 2000使用)。凝塊の存在を調査する目的で、粒径の測定を、インライン超音波処理の前後、そして同処理と共に行った。また、凍結乾燥後の回収量を質量で測定した。
(結果)
異なる油相で製造した粒子(各々の総量は20ml)の質量での回収量は、鉱物油、パラフィン油、イソオクタンでそれぞれ111mg、90mg、79mgと測定された。異なる油相で製造し、超音波処理を行わなかった湿潤粒子と乾燥粒子とを比較した結果、鉱物油およびパラフィン油のサンプルについては、乾燥後に粒径の平均値が大きくなったことが分かった(図3)。イソオクタンで製造された粒子は、乾燥処理にもかかわらず比較的変化がなかった。インライン超音波処理およびプレ超音波処理(pre-sonication)では、鉱物油およびパラフィン油で製造し乾燥した粒子の粒径分布に大幅な低下が見られた。それは、乾燥処理した粒子には凝塊が生じるが(図3)、超音波処理によって分解することができた、ということを示している。また、鉱物油およびパラフィン油で作られた湿潤粒子の粒径の分析でも、粒子の超音波処理に伴って粒径の平均値が下がることが示されたが、その幅は乾燥した粒子の場合(図3)に比べれば小さい。イソオクタンで製造した粒子は、乾燥処理や超音波処理にかかわりなく、比較的変化がなかった。
【0033】
結論としては、様々な油相を用いて粒子を製造することが可能である。また、異なる油相を用いること、ならびに乾燥処理や超音波処理などの各種後処理の技術を用いることで、粒径を操作することができる(油相の違いはエマルジョンの液滴サイズに影響すると推測される)。
(例4:各種の界面活性剤を用いたポリマー鎖/繊維のPEI支持体格子または網目主鎖の製造方法)
粒子の合成では、界面活性剤の種類が影響を与える場合がある。これを調査した。
【0034】
粒子は、ほぼ例3に従って調製したが、油相としては鉱物油のみを用い、界面活性剤の種類を変えた。
(結果)
鉱物油に各種の界面活性剤を用いて製造した粒子(総量20ml)の質量での回収量は、ノノキシノール-4、CHAPS、Triton X-100のそれぞれについて、111mg、72mg、92mgと測定された。CHAPSやTriton X100を用いた場合、異なる界面活性剤で製造した粒子を乾燥処理しても、粒径に実際的な差は表れなかった(図4)。ノノキシノール-4を界面活性剤として製造した粒子を乾燥処理すると、粒径は約50%大きくなった(図4)。さらに、超音波処理後の粒径分析は、テスト対象の全ての界面活性剤で製造した粒子で、そろって粒径が小さくなったことを示した。超音波処理した粒子の粒径を乾燥処理の前後で分析すると、結果は類似したものとなった。
【0035】
結論としては、様々な界面活性剤を用いて粒子を製造することが可能であった。さらに、粒径は、界面活性剤の種類および後処理によって操作することができた。
(例5:ポリマー架橋剤として各種ポリアルデヒドを用いた粒子合成)
粒子の合成に用いる架橋剤アルデヒドは、様々に変更することができる。架橋剤としては、グルタルアルデヒド、デキストランアルデヒド、ヘキサメチレンジイソシアネートが挙げられる。
【0036】
粒子を作る方法は、以下の点を除けば、例3と同じである。すなわち、ある場合では、グルタルアルデヒドをデキストランアルデヒド(15mg/mlのものを1ml)と置き換えた。また、別の場合では、0.5mlのヘキサメチレンジイソシアネート(25%v/v)と置き換えた。
5mlの粒子懸濁液に、Tris-Cl緩衝液(0.05M、pH8.0)に入れた5mg.ml−1の精製カンジダアンタークティカリパーゼB(CALB)を6ml加え、25℃で1時間緩やかに撹拌した。その後、懸濁液を遠心分離し、4℃の緩衝液10mlを用いて2度洗浄した。それから、懸濁液を遠心分離し、ペレットを10mlのTris-Cl緩衝液に再懸濁した。そして、p-ニトロフェニルブチラートを使って懸濁液(10μl)を分析評価した。また、比較のために、Tris-Cl緩衝液(0.05M、pH8.0)に0.5mg-per-mlの精製カンジダアンタークティカリパーゼBを10μl入れた場合についても分析したが、これには、p-ニトロフェニルブチラートの加水分解に基づく分析評価と、それに続く分光測光法による分析とを用いた(表3)。
(リパーゼ活性の分析評価)
リパーゼの活性により、p-ニトロフェニルエステル(p-ニトロフェニルブチラート(PNPB))はp-ニトロフェノールおよびブチル酸へと加水分解された。p-ニトロフェノールの放出によって黄色が生じ、この黄色は、UV/Vis分光光度計を用いれば410nmで測定される。活性度の測定は3回繰り返した。溶液の調製は以下のように行う。すなわち、溶液Aは、8mlのプロパン-2-オールに溶けた形で酵素基質を含むものとした。一方、溶液Bは、50mMのTris-緩衝液(pH8.0)にデオキシコール酸ナトリウム267mgを溶かした後に、66.7mgのアラビアゴムを溶解させたものとした。動的(Kinetic)分析評価を、PowerWave microtitreプレートリーダ(BioTek Instruments社)を用いて25℃で実行し、上述した溶液の1対10(A:B)の混合物240μlと固定化リパーゼ懸濁溶液または自由酵素10μlとを用いた。
(結果)
(表3:各種のアルデヒド架橋剤を用いて作った粒子の比較−CALBの活性度)
【0037】
【表3】
この方法は最適化されていないが、ある程度の範囲のポリアルデヒド合成物が使用して粒子を生成できることを示している。
【0038】
本材料は、0.45μmフィルタ(Sartorius社)で回収および再利用することができた。5回の再利用を終えて、グルタルアルデヒド粒子は7.05U.ml−1の平均値を示し、活性度は当初の81%であった。
類似の実験を、蛍光菌(PFL:Pseudomonas fluorescens)由来のリパーゼを用いて実施した。酵素活性度の分析評価には、基質として、ブチル酸のp-ニトロフェニルエステル(PNPB)とパルミチン酸のそれ(PNPP)とを用いた。
(表4:各種のアルデヒド架橋剤で作られた粒子の比較−PFLの活性度)
【0039】
【表4】
各種のアルデヒドを用いることで、粒子形成のための効果的な架橋の実現と、タンパク質を粒子に架橋するための官能基の提供との両方が可能となった。架橋剤の選択は、酵素変性の程度や基質および生成物に対する粒子の接触可能性(accessibility)の程度によって、酵素の活性度に影響を与える可能性がある。最適な架橋剤は、酵素および反応基質に基づいて選択すればよい。
(例6:2官能基エポキシド架橋剤を用いた粒子製造)
粒子の合成に使用する架橋剤アルデヒドは、ジエポキシド(例:1,4-ブタンジオールジグリシジルエーテル)などの他の架橋剤と置き換えることができる。これを(グルタルアルデヒドの代わりに)、50%v/v、25%v/v、12.5%v/vの薄めていない(neat)0.5mlの溶液の形で、架橋剤エマルジョン(B)に加え、そして、40℃で2時間反応させた(例3の場合と同様)。エマルジョンからの粒子の回収には、3000×gの遠心分離(Sorvall RT7 benchtop遠心分離機使用)を10分間行い、その後、10mlの量の脱イオン水で6回洗浄した。洗浄後、最終的に得られた粒子を脱イオン水で20mlに再懸濁した。12.5%v/vまたは25%v/vのエポキシド溶液を用いて上記のやり方で調製したものは、形状の点で最も均一であったのに対し、50%のエポキシドを薄めずに用いて形成したものは、大きく、しかもばらつきがあった。
【0040】
タンパク質の結合は、(前出の例に従い)CALBを使って調査した。それを一晩(overnight)透析し、エポキシドベースの粒子(12.5%v/vエポキシド溶液を用いて上述の通り調製したもの)と、緩やかに撹拌しながら1時間反応させた。例5で述べたようにリパーゼ結合粒子を回収および洗浄した後、酵素の活性度を分析評価した。全ての分析評価はPNPBを基質として実施した。
(結果)
ブタンジオールジグリシジルエーテルを架橋剤として使用した結果、直径が約0.1mmの白色粒子が形成された。しかしながら、この粒子は形状がいくぶん不規則であり、これは、架橋または後続のクリーニングの手順で粒子の凝塊が生じたことを示す。
【0041】
12.5%v/vエポキシド溶液で製造した粒子を、酵素が存在する状態で一晩インキュベート(incubate)した。これらの粒子は、0.28U.ml−1の活性度を示した。これは、エポキシド架橋剤を用いた粒子の製造が可能であることを示す。
(例7:単一のエマルジョンで製造したポリマー鎖/繊維のPEI支持体格子または網目主鎖)
粒子の形成方法は、必要に応じて調整することができる。例えば、単一のエマルジョンを用いて粒子を形成することもできる。
【0042】
ポリマーと架橋剤とは、混合チャンバに2重注入を行う装置を用いれば、水相で即座に混合することができる。こうした装置の最も単純な型は、同じポイントで共通のラインに注入を行う2つのシリンジで構成されたものであろう。これは油相への直接注入が可能である。一例として、20mlの鉱物油に0.2mlのノノキシノール-4を約5分間溶解させ、さらに500rpmで電磁撹拌器による撹拌を行う、という設定で評価を行った。一方のシリンジには0.5mlのPEI(10%)を入れ、他方には0.5mlのグルタルアルデヒド(20%、等級II)を入れた。両シリンジを同時に押し込んで、鉱物油に直接注入した。結果として得られたエマルジョンを1時間撹拌し、その後、粒子を例3と同じやり方で回収した。実験は3回繰り返して実施した。粒子は2等分し、その一方は冷凍乾燥し(Virtis使用)、元の量に再懸濁した後、粒径分布についての分析評価を行った(Malvern Mastersizer 2000使用)。サンプルに対し、乾燥後、回収量(質量)に関する分析評価を行った。
(結果)
単一のエマルジョンを用いて製造した粒子の回収量(質量)は152±12mgと算出されたが、その値は、2エマルジョン方式(例3を参照)を用いて得られるものに比べて約1.4倍大きかった。
【0043】
単一のエマルジョンを用いた粒子形成は可能であり、現行の製造法(図5)を用いて再現可能であった。粒子は、2種のエマルジョンを用いて形成したものに比べ、粒径分布がより広いことが分かった(図5)。単一エマルジョン粒子に関して得られた乾湿の粒径分布は、2エマルジョン実験の場合に得られるもの(図3)に比べ、サイズで、50〜70%大きいことが分かった。
(例8:ポリマー鎖/繊維のPEI支持体網目または格子の拡大規模での製造)
ここでの目的は、粒子製造の規模を線形的に10倍に拡大して粒径を評価することであった。別個の2バッチの粒子を、例3で概説した標準的製造方法にほぼ従って調製したが、異なる点として、第2のバッチは必要な構成成分の各々について10倍の量を含むものとした。両バッチに対しては、規模に応じた形で、例3と同様の粒子回収処理を行った。
(結果)
20ml、200mlの鉱物油量で製造した粒子の回収量は、質量でそれぞれ0.111g、1.11gと算出され、ちょうど10倍の差があった。鉱物油量が20mlでも200mlでも粒子を製造することは可能であり、より大きな規模で製造した粒子の粒径は、20mlの規模で得られるものに比べ、一貫してわずかに小さいだけであった(図6)。興味深い点は、より大きい規模で製造し、超音波処理していない乾燥粒子のサイズが、より小さい規模で製造した乾燥粒子と比べて約50%小さかったことである(図6)。
【0044】
回収量(質量)および粒径分析に基づいて言えば、標準的条件での粒子の製造法は、少なくとも10倍の規模拡大が可能であった。
(例9:ポリマー鎖/繊維のPEI支持体網目または格子を用いた広範囲の酵素群の固定化)
ここでの目的は、異なる酵素を粒子主鎖に結合し、それらの結合率(%)と酵素活性度の維持の程度とを算出することであった。
【0045】
例3に従って製造した粒子を使用した。検査対象の酵素は、ラッカーゼ(Novozymes 51009, ミセリオフトラサーモフィラ)、グルコースオキシダーゼ(Seravac Pty Ltd, アスペルギルスニゲル)、そしてリパーゼ(CALB, Novozymes のカンジダアンタークチカ)であった。各々の実験について、各酵素5mg(10mg.ml−1で0.5ml)を14mg(28mg.ml−1で0.5ml)の粒子に結合し、25℃で2時間緩やかに振盪させた。各サンプルは、Allegra X22R遠心分離機を用いて10分間遠心分離した(2000×g)。粒子は、連続して6回洗浄し、各回2mlの脱イオン水を用いた。混ぜ合わせて生じた上澄み部分を分析して、総タンパク質を求めた。個別の酵素結合実験を、各々、潜在的(potential)保護剤として特定の基質を含む場合と含まない場合とで実施した。ラッカーゼの場合、市販の伝達物質Denillite II Assist(Novozymes)を潜在的保護剤とした(100mg.ml−1 (pH6.8に調整)のものを50μl)。グルコースオキシダーゼの場合、グルコースを潜在的保護剤とした(10%m/vグルコース一水和物を50μl)。そして、CALBの場合、2-イソプロピル-5-メチルシクロヘキサール(メントール)の8−ジアステレオ異性体混合物を使用した(50μl)。粒子は、脱イオン水で2mlに再懸濁した。粒子を分析し、それぞれの酵素の活性度を求めた。全ての分析評価を3回繰り返した。
(総タンパク質)
総タンパク質分析評価は、Bio-Rad Total Protein Assay kit(カタログ番号:500-0006)というキットを用い、個別の酵素を各々基準として用いて実施した。
【0046】
ラッカーゼの活性度を測定するために、100mMのサクシネートラクテート緩衝液(pH4.5)に入れた1mMの2,2’-アジノビス(3-エチルベンゾチアゾリン-6-スルホン酸)ジアンモニウム塩(ABTS)を基質として使用した。溶液の光学濃度は420nmで測定した。これらの分析評価は、180μlのABTS試薬にサンプル20μlを加えて実行した。活性度の調査は、PowerWave HT(Biotek Instruments社)を用い、25℃でインキュベートして、420nmで分光測光法によって行った。
(グルコースオキシダーゼ)
グルコースオキシダーゼ(GOX)の活性度は、ホースラディッシュペルオキシダーゼ(HRP)によるo-ジアニシジンの間接酸化を利用して計測した。分析評価は、Bergmeyer(その他)による方法(1988年)に従って実施した。調剤した試薬は以下の通り:(試薬A)o-ジアニシジン-2HCL(0.006%)を含む0.1Mのリン酸カリウム緩衝液(pH7);(試薬B)D−グルコースの10%の水溶液(使用前に1時間変旋光(mutarotate)を生じさせたもの);(試薬C)60Uml−1HRP水溶液。試薬A、B、Cは、それぞれを24:5:1の割合で、グルコースオキシダーゼの分析評価の直前に混合した。反応試薬0.3mlを入れて反応させており、サンプルを10μl加えることで反応が開始された。反応結果は、25℃において、436nmで動力学的に計測した(Powerwave HT microtitreプレートリーダ使用)。グルコースオキシダーゼの活性度の単位は、25℃およびpH7で、1分につきに1μmoleのβ-D-グルコースをD-グルコノラクトンとH2O2とに転換するだけの触媒作用を生じる酵素の量を1単位と定めた。
(リパーゼ)
リパーゼの活性により、p-ニトロフェニルエステルはp-ニトロフェノールおよび脂肪族カルボン酸に加水分解される。p-ニトロフェノールの放出によって黄色が生じ、この黄色はUV/Vis光度計では410nmで計測される。2種のp-ニトロフェノールエステル(p-ニトロフェニルアセテート(PNPA)およびp-ニトロフェニルブチレート(PNPB))を使用し、活性度の測定は3回繰り返した。溶液は以下のように調製した:溶液Aは、酵素基質(11.6mgのPNPAまたは24mgのPNPP)を8mlのプロパン-2-オールに溶かされた状態で含むものであり;一方の溶液Bは、267mgのデオキシコール酸ナトリウムを含み、これを50mMのTris-緩衝液(pH8.0)に溶かした後に、66.7mgのアラビアゴムを加えたものである。動力学的分析評価を、25℃で、PowerWave microtitreプレートリーダ(BioTek社)を使って実施し、上述した複数の溶液の混合物240μlと球体(sphere)酵素または自由リパーゼの溶液10μlとを用いた。
(結果)
粒子への結合について検査した全ての酵素で、タンパク質担持の率は30〜36%の範囲となった(表5)。
(表5:粒子主鎖支持体への各種酵素の結合効率)
【0047】
【表5】
(*)活性部位の保護のため酵素の固定化中に基質を追加した(上述の方法を参照)。
PNPAに対するCALBの活性度の維持には、メントール基質を含ませることが効果的であり、約30%高い活性度(83%)が保たれた(図7)。
【0048】
ラッカーゼ、グルコースオキシダーゼ、CALBは全て、活性度を保ちながら粒子に結合することに成功した。それは、広範囲のタンパク質を、タンパク質担持が高い状態で、粒子に効果的に結合できることを示す。更に、他の酵素(ホースラディッシュペルオキシダーゼ、プロテアーゼ、デヒドロゲナーゼなど)も、粒子に結合することが示された(下の例を参照)。
(例10:ポリマー鎖/繊維のPEI支持体網目または格子;複数の酵素が結合した粒子)
本調査は、複数の酵素を、両酵素に対する活性度を保った状態で粒子主鎖に結合できることを示す意図で行った。グルコースオキシダーゼとホースラディッシュペルオキシダーゼ系とを選択した。
【0049】
粒子は、例3に従って製造した。調査した酵素は、グルコースオキシダーゼ(Seravac Pty Ltdのアスペルゲルスニゲル)およびホースラディッシュペルオキシダーゼ(Serevac Pty Ltd)であった。一例として、5mg(10mg.ml−1で0.5ml)のグルコースオキシダーゼと10mg(10mg.ml−1で1ml)のホースラディッシュペルオキシダーゼとを、25℃で2時間緩やかに振盪して、28mg(28mg.ml−1で1ml)の粒子主鎖に結合した。各サンプルを、Allegra X22遠心分離機(200×g)を用い、10分間遠心分離した。粒子は、連続して6回、各回2mlの脱イオン水を用いて洗浄した。結合後の上澄み部分を、総タンパク質について分析した。粒子の活性度は、例9のグルコースオキシダーゼ用の分析評価方法に従って測定したが、分析評価試薬にホースラディッシュペルオキシダーゼは含まれていない。
(結果)
グルコースオキシダーゼおよびホースラディッシュペルオキシダーゼの粒子主鎖への結合は成功し、3μmole.min−1の率でグルコースを変換することができた(表6)。このことは、分析評価で検出した活性度と同様、グルコースオキシダーゼとホースラディッシュペルオキシダーゼとが結合され、活性度を維持していたことを示す。すなわち、粒子を用いれば、複数の酵素を、両方の酵素が活性度を保った状態で結合することができる。
(表6:グルコースオキシダーゼおよびホースラディッシュペルオキシダーゼの2種酵素粒子に関する結合効率および活性度)
【0050】
【表6】
(例11:各種乾燥方法を用いて製造したポリマー鎖/繊維のPEI支持体格子または網目)
ここでの目的は、粒子を製造した後に、様々な方法(凍結乾燥、真空乾燥、アセトン乾燥など)を用いて粒子を乾燥させることであった。
【0051】
粒子は、例1で概説した標準的な条件で製造した。そして、洗浄した粒子を、凍結乾燥(Virtis Genesis凍結乾燥機使用)、真空濃縮機(Savant RVT100蒸気収集機を備えたSavant SpeedVac SC110)を用いた真空乾燥、または、アセトンを用いた脱水によって乾燥させ、その後さらに、25℃で12時間空気乾燥させた。アセトン乾燥した粒子は、凝塊したため水媒体に再懸濁することができず、そのため、それ以上の考察は行っていない。
【0052】
しかし、真空乾燥と凍結乾燥とは、粒子を乾燥させる技術として両方とも成功であり、乾燥後の粒子を用いて、広範囲の酵素やタンパク質を付着させることができた(表7)。5mg per mlの精製酵素6mlをTris-Cl緩衝液(0.05M、pH8.0)に入れて、粒子懸濁液5mlに加えた。そして、25℃で1時間緩やかに撹拌した。懸濁液は、その後遠心分離してから、4℃の緩衝液10mlを用いて2度洗浄した。さらにその後、懸濁液を遠心分離し、ペレットを10mlのTris-Cl緩衝液(0.05M、pH8.0)で再懸濁した。粒径は、Malvern Mastersizerを用いて測定した。
(結果)
(表7:各種乾燥処置後の平均粒径)
【0053】
【表7】
本例が示すのは、乾燥処理、特に凍結乾燥した場合、酵素と結合した粒子が大きく凝塊することはない、ということである。
(例12:様々なpHでPEIから調製したポリマー鎖/繊維の支持体網目または格子へのラッカーゼ結合の特性評価)
乾燥処理が、結合後のラッカーゼ結合粒子の特性、酵素の性質に与える影響を判定した。
【0054】
粒子は、例3で概説した標準の製造方法にほぼ従って製造したが、pH11で調製したPEI(10%)に加えて、pH8に調整して調製したものについても評価を行った。さらにラッカーゼを主鎖に固定化し、その方法は例9で述べたものとほぼ同じであるが、以下の点が異なる。すなわち、1mlのラッカーゼ(Novozyme 51004,50mg.ml−1)を6.25mlの粒子(16mg.ml−1)と反応させた。従って、100mgの粒子について50mgのラッカーゼが結合したことになる。ラッカーゼ結合粒子は、脱イオン水12.5mlで洗浄し、さらに、脱イオン水を用いて20mlに再懸濁した。粒子は10mlずつ2等分した。そして、そのサンプルの1つを、回収量(質量)測定のために凍結乾燥し、乾湿両方のラッカーゼ結合粒子の特徴を調査した。乾燥させたサンプルは、脱イオン水で元の量に再懸濁した。全ての分析評価は3回繰り返した。
【0055】
ラッカーゼの上澄みのサンプルの総タンパク質を、Bio-Rad Total Protein Assay kit(カタログ番号:500-0006)というキットを用い、ラッカーゼをタンパク質の基準として測定した。ラッカーゼの活性度は例9に従って測定した。
(結果)
凍結乾燥によるラッカーゼ結合粒子の乾燥処理では、粒子製造の際にPEIがpH8であった場合の方が、粒子に結合されるラッカーゼタンパク質は多いものの、粒子製造の際にPEIがpH11であった場合の方が、回収できる量(質量)は大きいことが分かった(表8)。ABTSに対しての活性度を最も高く保つことができたのは、pH8のPEIを用いて製造し、凍結乾燥しなかった粒子であったが(16%)、凍結乾燥後の同じサンプルはラッカーゼ活性度を3%しか保持していなかった。注目すべき点として、pH8のPEIを用いて製造された粒子は乾燥後に再懸濁するのが難しく、そのため凝塊する可能性があるが、凝塊すれば表面対面積(surface to area)の比率が下がり、従って、基質および生成物への分散制約(diffusional constraint)が原因で活性度が下がることになる。pH11のPEIを用いて生成された粒子については、活性度に関して同様の影響は観察されなかった(表8)。
【0056】
表8:凍結乾燥を行う場合と行わない場合とで、PEIのpH値を様々に変えながら製造したラッカーゼ結合粒子の結合効率および活性度保持の度合
【0057】
【表8】
この実験が示すのは、粒子はPEIのpHを変えて調製することができ、これによって酵素に対する粒子の結合特性が変わる、ということである。
(例13:pH8またはpH11で製造および凍結乾燥し、ポリマー鎖/繊維のPEI支持体網目または格子に結合されたラッカーゼの安定性)
pH8またはpH11で製造および凍結乾燥され、ポリマー鎖/繊維のPEI支持体網目または格子に結合されたラッカーゼの熱安定性およびpH安定性を測定した(ラッカーゼ結合粒子は例12に記述した形で製造した)。
(温度最適条件)
自由ラッカーゼおよび粒子結合ラッカーゼ(タンパク質担持は均等)の温度最適条件の解析(profile)を、100mMのサクシネートラクテート緩衝液(pH4.5)に基質として入れた1mMのABTSを用いて実行した。ウォーターバス内で、正確な温度に前平衡(pre-equilibrated)しておいた1.9mlの基質に、サンプル(100μl)を加えた。分析評価は、ペルティエ温度コントローラを備えたDU800分光光度計(Beckman-Coulter社、420nm)を用いて実施した。分光光度計を対象の温度に設定し、キュベットは、ウォーターバス内の平衡化した試薬を追加する前に、5分間平衡化させた。
(pH安定性)
自由ラッカーゼおよび粒子結合ラッカーゼ(タンパク質担持は均等)のpH安定化は、100mMのサクシネートラクテート緩衝液(pH4.5)に入れたABTS(1mM)を用いて行った。各ラッカーゼサンプルは、pH2.5、pH3、pHにおいて、Britton-Robinson汎用緩衝液内でインキュベートした。サンプル(20μl)を、定期的に取り出し、25℃でインキュベートすると共に、PowerWave HT(Biotek Instruments社)を用いて420nmで分析評価した(230μlの分析評価試薬)。6時間の時点での本実験の結果を図9に示す。
(結果)
結果を図8(A)、(B)、図9に示す。
【0058】
粒子結合ラッカーゼと自由(固定化されていない)ラッカーゼとは両方とも、70℃で活性度が最高になった(図8)。しかし、粒子結合ラッカーゼは、90℃では、自由ラッカーゼ(活性度0%)と比較して熱安定性の向上(活性度55〜65%)を示した(分析評価用サンプルの調製に要する時間で)。また、乾燥粒子を使用した場合も小幅な向上が見られ、乾燥による効果があることを示唆している。これは、水が除去されるにつれて、形成されるタンパク質−粒子連結が多くなることによるかもしれない。これによってタンパク質または酵素の複数点(multi-point)共有結合が増加するが、それは、熱安定性の向上などの形で安定性を高めることが知られている [“Improvement of enzyme activity, stability and selectivity via immobilization techniques”、Mateo, C, Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M., Fernandez-Lafuente, R.(著)、2007年、(誌名)“Enzyme and Microbial Technology 40(6)”、ページ1451-1463]。
【0059】
また、ラッカーゼ結合粒子および自由酵素のpH安定性を、pH2.5、pH3、pH6で測定した。pH2.5およびpH3では、ラッカーゼ結合粒子のサンプル全てが、80〜110%の活性度を保ったが、自由酵素は、それぞれのpHで活性度を約40〜70%失っていた(図9)。すなわち、固定化によってpH安定性は向上する。自由酵素を含むサンプルの全ては、6時間後にpH6で安定していた。
【0060】
従って、ポリマー鎖/繊維のPEI支持体網目または格子の粒子に酵素を固定化することにより、pHの両極での安定性を更に高めることができる。
(例14:ポリマー鎖/繊維のPEI支持体網目または格子への固定化に伴う酵素最適pHのシフト)
酵素は活性に最適のpHを有する。いくつかの場合、酵素の最適pHは、他の反応にとっての最適pHと一致しない。たとえば、酵素基質/反応物は別のpH値で最も溶解しやすい、という場合がある。もう1つの例として、複数の酵素を複数ステップのワンポット(one-pot)反応に用いる場合、それらの最適pHは一致しないことがある。それゆえ、固定化処理中に酵素の最適pHを変化させることができれば、これによって経済的および技術的な効果が得られるだろう。そこで、ラッカーゼの最適pHを、粒子上に固定化する場合と固定化しない場合とで測定した。
(酵素分析評価)
ラッカーゼ試薬は、50mMのBritton-Robinson汎用緩衝液の中に1mMのグアヤコールを含んでおり、当該緩衝液は目標とするpH値に調整してあった(Davies TJ, Banks CE, Nuthakki B, Rusling JF, France RR, Wadhawana JD, Compton RG(著)、2002年。(題名)“Surfactant-free emulsion electrosynthesis via power ultrasound: electrocatalytic formation of carbon-carbon bonds”、(誌名)Green Chem. 4:570-577)。汎用緩衝液を用いたのは、同じ緩衝液系が存在すること、そして広範囲のpHにわたって効果的に緩衝できることを確実にするためである。分析評価は、5 200M−1.cm−1の吸光係数で、450nmにおいて3回繰り返した。自由酵素ならびに支持体網目または格子の粒子に固定化されたラッカーゼについて、pH値プロフィールを実験で測定した。分析評価の実施には、PowerWave HT Microtitreプレートリーダを用いた。酵素の単位については、1分につき1μmolの基質を酸化させるのに必要な酵素の量を1単位とした。
(pH値プロフィールのシフト)
pHプロフィールのシフトは固定化処理中に起こることは公知であるため、繊維格子または網目主鎖支持体に結合されたラッカーゼについて、pHプロフィールを測定した。粒子は例3に従って作った。グルタルアルデヒドおよびPEIの濃度の変化が最適pHのシフトに与える影響についても、グルタルアルデヒド後処置(例2、A2−J2)の影響と同様に調査した。
(結果)
結果を図10(A)、(B)、図11(A)、(B)に示す。
【0061】
pH値プロフィールのシフトの全体的な傾向は、中性から弱アルカリpHに向かっている。本例が示すのは、酵素のpHプロフィールが、粒子への固定化によってシフトするということである。
(例15:ポリマー鎖/繊維のPEI網目または支持体格子の官能性)
粒子主鎖の官能性を変えることで、タンパク質の疎水性結合、イオン結合、そして親和性をベースとした結合を示すものにすることができる。
【0062】
粒子は、0.1mlのノノキシノール-4と0.5mlのポリエチレンイミン溶液(pH11)とを含む鉱物油10mlのエマルジョンと、0.1mlのノノキシノール-4と0.5mlのグルタルアルデヒド(Sigma 等級II)とを含む鉱物油10mlのエマルジョンとを混合することで製造した。混合前に、両エマルジョンを500rpmで30分間揺動した。そして、一緒にしたエマルジョンを500rpmで30分間揺動した。これにより、装飾されていない粒子が得られ、それを上述したように遠心分離処理で回収した。
(イオン結合)
イオン性基は、上記の修飾されていない粒子で生成され、その方法は、グルタルアルデヒドを用いて官能化し(25%の水溶液200μl、20mlの脱イオン水で3回洗浄)、その後、エチレンジアミン(0.33Mを1ml)を用いた処理を行うことで、自由なアルデヒド残基と反応させる(定期的にひっくり返しながら室温で1時間)、というものであった。粒子は、充分な量の脱イオン水で繰り返し洗浄し、2000×gで10分間遠心分離を行って回収した。ラッカーゼ(2.5mg)を5mgの修飾された粒子と、充分な混合が確実に行われるように定期的にひっくり返しながら、4℃で30分間インキュベートした。イオン結合を促進するために、1.0MのNaClを(対イオンとして)ラッカーゼ結合粒子に加え、ひっくり返して5分間混合した。粒子は、上述したのと同様に遠心分離によって回収し、活性度を測定した。
(疎水性結合)
修飾されていない粒子(5mg)をエポキシオクタン(0.1ml)と共に25℃で4時間インキュベートするやり方で、疎水基を粒子に追加した。その後、これらを充分な量の水で繰り返し洗浄し、上述したように遠心分離によって回収した。疎水結合されたタンパク質(蛍光菌リパーゼ、5mg)は、界面活性剤(1%、デオキシコレート)の追加によって取り出すことができた。280nmでのタンパク質吸光度を測るやり方で測定を行った。
(親和性結合)
タンパク質の親和性結合については例17に示す。
(結果)
(イオン結合)
修飾された粒子には、追加したラッカーゼの45%が結合された(Bio-Rad protein assay dye reagent method(500-0006)によって求めた値、メーカ側の記録による)。この45%のラッカーゼのうち76.9%は、対イオンとして塩(1.0M、NaCl)を追加することで取り出すことができた。これに対し、修飾されていない粒子はラッカーゼの14%としか結合せず、塩溶液によって取り出せたのは、その52.6%だけであった。
(疎水性結合)
修飾された粒子にタンパク質を結合した。修飾された粒子に結合されたタンパク質の約28%は、デオキシコレートを追加することで取り出されたが、これは、結合された酵素のうちの疎水性結合部分である。
【0063】
本例が示すのは、粒子マトリックスの官能性を変更することで、粒子マトリックスへのタンパク質結合の別メカニズムを実現できる、ということである。
(例16:化学伝達物質および補因子の共捕捉(co-entrapment))
補因子(または修飾された補因子や化学伝達物質)を含ませることで、架橋後、酵素と補因子との共補足が可能になる。処理条件を選択することにより、反応体(例:酵素基質、共基質、生成物、共生成物)などの小分子の出入りを許す一方で補因子を保持するよう、粒子の多孔度を決めることができる。
【0064】
アミノ酸デヒドロゲナーゼ(AADH)、ギ酸塩デヒドロゲナーゼ(凍結乾燥したもの)は、Biocatalytics社(米国)から購入した。PEG20000-NADHは、Julich Fine Chemicals社(ドイツ)から取得した。鉱物油は、Castrol社(ドイツ)から取得した。ノノキシノール-4は、ICI社(英国)から取得した。グルタルアルデヒド(Glut)(25%水溶液)は、Acros Organics社(ベルギー)から取得した。ギ酸は、Merck社(ドイツ)から取得した。エチレンジアミン(EDA)、ポリエチレンイミン(PEI)、3-メチル-2-オキソ酪酸(2-ケトバリン)、DL-バリン、NADHそしてNAD+は、Sigma Aldrich社から取得した。
【0065】
ポリエチレンイミン(PEI)をグルタルアルデヒドと架橋することで、ポリマー粒子を製造した。ポリエチレンイミンの油中水エマルジョンは、予め溶かした状態の200μlのノノキシノール-4を含む鉱物油40mlに10%のPEIを800μl乳化する方法で調製した(500rpmで撹拌する20mmの電磁撹拌器を用い、100mlビーカー内で20分間電磁撹拌した)。第2の油中水エマルジョンも同様に、20%のグルタルアルデヒド溶液を用いて調製した。2つのエマルジョンは、高速(700rpm)で撹拌しているポリエチレンイミンエマルジョンにグルタルアルデヒドエマルジョンを加えるやり方で混合した。これにより、連続的な撹拌で30分間反応させることができた。
【0066】
ポリマー粒子は、2000×gで10分間の遠心分離を行って回収した(Sorvall, RT7)。そのポリマー粒子を45mlの脱イオン水で4回洗浄した。洗浄作業の間に、先に述べた例と同様の遠心分離によって回収を実行した。そして、生成物は、10mlに再懸濁した。この溶液(1ml)をエッペンドルフチューブに等分して、以降の実験に使用した。
【0067】
10mg.ml−1を含んだ各タンパク質(ギ酸塩デヒドロゲナーゼ、バリンデヒドロゲナーゼ)のタンパク質溶液を調製した。その後、これら2つの溶液(各溶液200μl)を混合し、ポリマー材料とインキュベートした。この溶液は、逆さまにして混合し、緩やかに揺動しながら30分間反応させた。固定化酵素を含む粒子を、後述の方法を用いて、活性度測定のために分析評価した。
【0068】
粒子は、脱イオン水で洗浄し、上述したやり方で回収した。粒子は、100μlの PEG20000-NADH(ドイツ、ユーリッヒのJulich Fine Chemicals社から取得)と混合し、緩やかに揺動しながら室温で2時間インキュベートした。その後、この溶液を凍結乾燥した。凍結乾燥による生成物を、2mlの水で2回洗浄し、遠心分離によって回収した。それから、粒子を、100mgのリジンを含んだ100mMのTris-Cl緩衝液(pH8.0)1mlに再懸濁して、粒子上の余剰のアルデヒド官能基を抑制(quench)し、室温で1時間インキュベートした。そして、粒子を2mlのTris-Cl緩衝液(20mM、pH8.0)で5回洗浄した。その後、このサンプルに対しては、1mlの再利用試薬を用い、再利用能力に関する検査をした。サンプルは、サイクルとサイクルとの合間に2ml量の水で3回洗浄した。そしてサンプルを分析したが、これは、後述するやり方でアミノ酸TLCとHPLCとからバリンを製造するためである。
【0069】
これらの粒子を反応させ、再利用して、後続の反応のための新しい反応媒体とした。
(試薬の再利用)
バリン生成を目的としたPEG-20000-NADHの再利用のための試薬は、50mMのギ酸塩(ナトリウムギ酸塩1M株(stock)から出たもの、pH8.0)、50mMのTris-Cl緩衝液(pH8.0)、50mMの酒石酸アンモニウム、そして10mMの2-ケトバリンから成るものとした。この試薬の組成を、PEG NADHが再利用された場合に確実にバリンのみが生成されるように公式化(formulated)した。
(分析法)
アミノ酸TLCを、F254シリカゲルプレート(Merck社)で実施した。使用した移動相は、氷酢酸に対してエタノール9:1であった。アミノ酸(バリン)は、アセトン中にニンヒドリン2%の溶液を用いて染色した。バリン(Rf0.49)とアンモニア(Rf0.31)とが適当に分離したことが明らかに見て取れるまで、プレートを120℃で加熱した。
【0070】
HPLCは、アミノ酸の測定のために、OPA誘導化法(o-フタルアルデヒド試薬)を用いて実行した。サンプルは、インライン誘導化した。また、バリン標準(valine standards)を含めた。
(結果)
反応および再利用は、TLCデータによれば成功であり、連続的な粒子触媒反応の全てについてバリンスポット(valine spot)が形成された。6回の連続的な16時間反応サイクルのそれぞれにおいて、粒子として、10mMのケトバリンを48、35、59、43、33、29%のいずれかの割合でバリンに変換したものを用いた。これによって肯定的なTLC結果が確認され、HPLC OPA法を用いて定量化できた。
【0071】
この例が示すのは、酵素粒子マトリックスでの補因子の捕捉によって、酵素は補因子を利用して官能基を維持でき、そして、捕捉された補因子は再利用できる、ということである。
(例17:ポリマー鎖/繊維のPEI支持体網目または格子を有する粒子による抗体の結合)
本例で示すのは、粒子への抗体や抗原の結合である。抗体や抗原が結合された場合には、固定化された抗体または抗原を介したタンパク質の親和性結合に対する粒子の適合性が示される。さらに、診断用途(例:ELISA)に対する粒子の信頼性が示される。
【0072】
ポリエチレンイミン(P3143;50%水溶液)、等級IIのグルタルアルデヒド(G6257;25%水溶液)、鉱物油(M8410)、抗原マウスインターロイキン2(I0523−20UG;SL06092)、そして、Streptomyces avidinii由来のマーカー酵素ストレプトアビジン−ペロキシダーゼ(S5512−250UG;SL05181)は、Sigma-Aldrich社から取得した。主要な抗体であるラットアンチマウス(rat anti-mouse)インターロイキン2(IL−2)MAB(I7663−27K1; L6080801)と、補助的な抗体であるラットアンチマウスインターロイキン2(IL−2)ビオチンMAB(I7663−27M5;L6080803)とは、USBiologicals社から取得した。塩化ナトリウム(S7653)、二塩基のリン酸ナトリウム(S0876)、一塩基のリン酸ナトリウム(S0757)、過酸化水素(21676−3)、ツイン20(P9416)、そして塩酸(H1758)は、Signa Aldrich社から取得した。2,2'-アジノ-ビス(3-エチルベンズチアゾリン-6-スルホン酸)(10102946001)はRoche社から取得した。
(粒子の調製)
架橋ポリエチレンイミン粒子は、例2(サンプルG)に従って調製した。試薬に対して唯一行った調整は、ポリエチレンイミン溶液を10%に希薄する前にHCIでpH9にしたことである。実験は4倍に直接拡大して行った。従って、乳化した反応体それぞれ800μlと共に溶解したNP4を200μl含んだ20mlの鉱物油が必要であった。結果として生じる架橋ポリエチレンイミン粒子を、50mlの脱イオン水で6回洗浄し、洗浄と洗浄との合間に5000×gで5分間遠心分離する、というやり方で鉱物油から回収した。粒子ペレットは、その後、脱イオン水で10mlの量に再懸濁し、実験A〜Fに500μlずつ使用した。
(タンパク質の固定化)
各実験には、下の表9に示すように文字を割り当てた。各種のタンパク質の追加は、表9で実験の下側の欄に示す手順で実行した。各実験について、第1のタンパク質成分の固定化は、10分毎にサンプルを逆さまにして充分な混合が行われることを確実にしつつ、4℃の脱イオン水で1時間かけて実施した(表9の順次追加ステップの1)。粒子は、1mlの脱イオン水で3回洗浄し、上述したやり方で回収した。
【0073】
それに続くタンパク質結合、タンパク質処理(表9の第2列から第5列の処理)は、150mMの塩化ナトリウム(結合緩衝液)を含んだ10mMのリン酸塩緩衝液(pH6.8)で実施した。この結合は、充分な混合を確実にするために5分ごとにひっくり返しながら、37℃で30分間実行した。タンパク質結合が終わる度に、サンプルは、10分間緩やかに揺動させながら(IKA Vortex Genius使用、設定2)、ツイン20を0.05%含んだ1mlの結合緩衝液で2回洗浄し、非特異的なタンパク質相互作用を抑制した。順次行うステップの合間々々に、5000×gで5分間遠心分離を行って粒子を回収した。タンパク質処理の各ステップ(表9)に関して追加したタンパク質の量は、以下の通りである。アルブミンは5mg;ラットアンチマウスインターロイキン2MAB(A-IL2-MA)は50μg;ラットアンチマウスIL2MABビオチン(B-A-IL2-MA)は25μg;インターロイキン2(IL-2)は2ug;ストレプトアビジンペルオキシダーゼ(strep-perox)は10μg。これらの量のタンパク質を1mlの結合緩衝液で調製した。
(表9:実験A〜Fのタンパク質結合の手順)
【0074】
【表9】
(分析評価)
ペルオキシダーゼ分析評価試薬は、150mM塩化ナトリウムを含む10mMリン酸塩緩衝液(pH6.8)に、2mMのABTSと2mMの過酸化水素とを含んで成るものとした。分析評価値は3回繰り返して測定し、1ウェル(well)につき200μlの試薬と50μlの粒子懸濁液とを用い、30℃において、Powerwave HT マイクロプレート分光光度計(Biotek Instruments社)で420nmの値を計測した。
(結果)
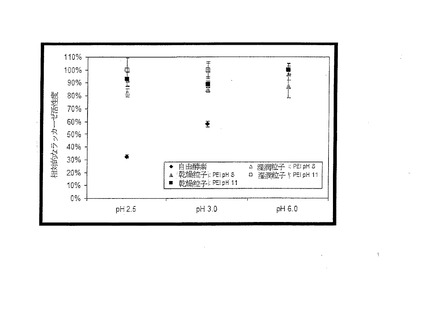
抗原に対する抗体の結合は、実験B(図12)で評価した。この実験では、粒子表面への主要な抗体(A-IL2-MA)の結合、それに続く抗原(IL-2)の結合、粒子への補助的な抗体(B-A-IL2-MA)およびストレプトアビジン−ペロキシダーゼの結合について、結合が成功したと解釈しうる肯定的な反応が示された。これは、マイクロプレートのウェルなどの表面で実行されるsandwich-ELISAに類似している。実験Cは、抗原が粒子の表面に結合され、その後、抗体結合のための認識要素(recognition element)として用いられることを示す。これらの実験は、各種の対照例(control)と比較して見た場合、粒子を抗体または抗原の結合に用いることが可能であることを示している。
【0075】
実験Aは、主要な抗体が固定化された状態の粒子に対し、補助的な抗体(B-A-IL2-MA)またはストレプトアビジンペルオキシダーゼが非特異結合することを示すための対照例である。Bと比較して反応が低いことは、粒子に対する補助的な抗体の結合を抗原(IL-2)が強化することを示す。実験D,Eは、アルブミンを抑制処理(quenching)した後の粒子に対し、主要な抗体(A-IL2-MA)や補助的な抗体(B-A-IL2-MA)が非特異性結合することを示すための対照例である。実験Fは、非処理の架橋ポリエチレンイミン粒子に関するものであり、分析評価の対照用に用いた。
【0076】
本例は、粒子が、抗原または抗体にとって適当な固定化支持体であることを示す。更に強調しておきたい点は、抗原に対して抗体が、抗体に対して抗原が、それぞれ親和相互作用によって固定化されることである。従って、本例は、親和クロマトグラフィおよび診断法(酵素結合免疫吸着検査法など)に、当該支持体が適用できることを示している。
(例18:ポリマー鎖/繊維のPEI支持体網目または格子の粒子への磁鉄鉱の組み入れ)
化学伝達物質および補因子を粒子に含ませる処理についてはすでに示した。本例では、粒子に磁性粒子を含ませる処理について示す。
(粒子の調製)
粒子は、上記の例17に従って調製し、各エマルジョンについて鉱物油が25mlになるよう線形的に数値を大きくした。乳化の前に、10%のポリエチレンイミン溶液(pH9.0)に磁鉄鉱(250mg)を含ませた。架橋したポリエチレンイミン粒子を、50mlの量の脱イオン水で6回洗浄し、洗浄ステップの合間に、5000×gで5分間遠心分離して鉱物油から回収した。続いて、粒子ペレットを脱イオン水で10mlの量に再懸濁し、500μlのエチレンジアミンと30分間反応させることで、陰イオン交換のために官能化した。次いで、球体を、50mlに3等分した脱イオン水で洗浄し、上述したように遠心分離を行って回収した。最終的に得られたペレットは、脱イオン水で10mlに再懸濁し、後述するアルブミン結合実験に使用した。粒子の乾燥重量は、10mlの粒子懸濁液を1mlずつに等分したものを凍結乾燥および重量測定するやり方で、3回繰り返して測定した。
(タンパク質の結合および定量化)
上記の等分した懸濁液から得た粒子を、磁気スタンド(Magnetic Separation Stand (Promega社; Z5332))で磁気分離して回収し、液体は除去した。その粒子を2mlのTris-Cl緩衝液(pH7.4)(50mM)で2回洗浄して平衡化した。そして、粒子にウシ血清アルブミン(BSA)を加えて、最終的な濃度を20mg.ml-1とした。タンパク質のイオン結合を、室温において30分間、回転混合することで生じさせた。混合物は磁気スタンド内に配置して、磁化された樹脂を反応管の側壁に保持させた状態で、サンプルからの液体が除去されるようにした。樹脂は、50mMの Tris-Cl(pH7.4)1ml で5回洗浄し、前述した磁気保持器(retainer)によって回収した。50mM のTris-Cl(pH7.4)に入れた1MのNaCl500μlを2回追加することで、イオン結合されたBSAを樹脂から抽出した。
(タンパク質定量化)
溶出した分のタンパク質は、メーカの指示に従って、Qubit Fluorometer(Invitrogen社)で、Quant-iT分析評価を用いて定量化した(表10)。
(結果)
(表10:磁鉄鉱含有粒子へのアルブミンの結合(3回分データの平均))
【0077】
【表10】
この結果が示すのは、磁鉄鉱を粒子に取り込めば、粒子の磁気特性を利用して粒子を懸濁液から効果的に分離できる、ということである。更に、上記の結果は、本支持体が効率の良いイオン交換樹脂として使用できることを示している。ここで提示する例では、アミン(エチレンジアミン)などの正荷電分子で粒子マトリックスを修飾することで、粒子を陰イオン交換樹脂として使用することができる。また、カルボキシル含有分子などの負荷電分子を用いれば、陽イオン交換樹脂としての使用が可能である。
【0078】
本例は更に、格子内に磁性粒子を含ませる方式の別の回収方法の例を提供する。その方法では、磁場の利用によって粒子を引きつけることができる。
本発明の粒子は、架橋剤によって架橋されたポリマー鎖または繊維の格子と、隣接して繊維を囲む格子間の開口領域または空間とを有する。言い換えると、本発明が提供するのは、繊維相互貫入網目粒子であり、好ましい構成として、グルタルアルデヒド架橋したポリエチレンイミンから構成または製造されている。この粒子は、酵素固定化マトリックスとして利用できる。
【0079】
粒子は、本発明の第2、第3の様態によるエマルジョンベースのテクノロジーまたは技術を用いて製造するのが好ましい。エマルジョンベースのテクノロジーを用いれば、粒子のサイズに関する制御(例えば、粒子における体積と表面積との比率)や所定の粒径分布を実現できる利点があると共に、粒子を単一ステップで合成できるという効果もある。本粒子は、固定化に利用できる表面積が大きくなるため、大小の基質の生体触媒にも利用できる(ただし、それに限定はされない)。この酵素固定化マトリックスは、固定化効率が高く、固定後の酵素の活性度も高く保たれる。
【0080】
樹枝状粒子の繊維の性質として、酵素の結合に役立つ内部表面積が大きくなる。また、酵素サブユニットごとの利用可能な結合点の数が大きいため、タンパク質の安定化は、主鎖支持体の材料(例えば、PEIそれ自体)と比較した場合、大幅に改善する可能性がある。これを、ゆるい格子または網目と組み合わせた場合、生体触媒付加後の重量に対する活性度の比率を高めることができるが、それは、固定化に有用な露出表面積が大きいこと、そして、大小の基質について分散制約が限られていることによる。更に、粒径の制御によって、基質の分散制約をより小さくすることができる(それが本固定化マトリックスの障害となる場合)。
【0081】
粒子の調製方法には、同相で架橋剤が存在する場合、存在しない場合について、主鎖支持体または格子を乳化する処理が含まれる。上述の2エマルジョンシステムを製造に用いるのが望ましい。架橋剤が第1のエマルジョンに含まれない場合、架橋剤は、油相に溶かしてもよいし、前記架橋剤を含んだ第2のエマルジョンを混合する形で取り込んでもよい。
【0082】
好ましい製造方法には、エマルジョンの中で主鎖ポリマー(ポリエチレンイミン)を別個に乳化する処理が含まれるが、その場合は、第2のエマルジョンに少なくとも2官能基の架橋化学物質(グルタルアルデヒド)を入れておく。ポリマーと架橋剤との間の自然反応の結果、繊維の格子または網目が生じるが、この場合、余剰のアルデヒド官能基が含まれている。続いて、その余剰のアルデヒド官能基を用いて、アミンアルデヒド交差反応性を介したタンパク質の格子または支持体への自然発生的な共有結合連結が実現される。このアルデヒドの官能基については更に、別の化合物と連結すること、あるいは、疎水性などの他の特性をもたらすことにすれば、より広い範囲のタンパク質(例:疎水性タンパク質)の結合にまで用途を拡大するものと推測される。
【0083】
マトリックスにタンパク質を固定化することにより、酵素の溶媒安定性、熱安定性、pH安定性は高まる。こうした安定は、樹枝状支持体へのタンパク質固定化の後で支持体を乾燥させれば、更に強化される。この乾燥処理によって、交差反応性を有する化学官能基が近接する状況が少なくなるので、その結果、「タンパク質−主鎖」および「主鎖−主鎖」の更なる自然発生的な化学結合(chemical coupling)が引き起こされると考えられる。さらに、乾燥処理の間に添加物がマトリックスに捕捉されるが、これは、孔サイズの制御や、機能分子または補助剤の捕捉に役立つ可能性がある。本発明の粒子であれば、タンパク質担持を高めることができる(すなわち、小さな粒子体積に対して比較的大量のタンパク質を担持することができる)。
【技術分野】
【0001】
本発明はエマルジョン由来粒子に関する。また、本発明は当該粒子の製造方法に関する。
【背景技術】
【0002】
固定化酵素を含んだ粒子が通常用いられるのは、バイオ触媒や診断などの用途である。しかしながら、出願人の知る粒子は、充分な酵素固定化を実現するには表面積が足りないという問題を抱えている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】欧州特許第0 534 057号明細書
【特許文献2】国際公開第02/29406号明細書
【発明の概要】
【発明が解決しようとする課題】
【0004】
そこで、本発明が目的とするのは、この問題を少なくとも軽減できる粒子を提供すること、および、タンパク質に対する結合容量が大きくタンパク質を固定化できる粒子の製造方法を提供することである。
【課題を解決するための手段】
【0005】
そこで、本発明の第1の様態として、架橋剤によって架橋されたポリマー鎖で成る格子と、前記格子を成すポリマー鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有するエマルジョン由来粒子であって、タンパク質または修飾タンパク質と反応することで、当該タンパク質または修飾タンパク質を格子に結合させ、固定化する、というエマルジョン由来粒子を提供する。
【0006】
「エマルジョン由来」とは、粒子がエマルジョン技術を用いて製造または形成されたことを意味し、当該エマルジョン技術としては、本発明の第2、第3の様態によるエマルジョンベース(emulsion based)の手法がある(ただし、これらに限定はされない)。
「修飾(modified)タンパク質」は、化学的手段(例えばジアルデヒドの追加)によって修飾されたタンパク質、あるいは、遺伝子レベルで(例えばhisタグを用いて)修飾されたタンパク質を意味する。
【0007】
従って、本発明の粒子は、鎖状(strand)あるいは繊維状(fiber)のポリマーに、または架橋剤に官能基を含み、タンパク質または修飾タンパク質を官能基と反応させることができる。より具体的には、官能基は、鎖状あるいは繊維状のポリマーに存在しており、タンパク質または修飾タンパク質のポリマーへの結合が、ポリマー上の官能基を変更する形で共有結合、イオン結合、疎水結合、親和結合のうち1または複数で成されるように選択される。
【0008】
そうして、粒子は、官能基によってポリマーに結合され、それによって固定化されるタンパク質または修飾タンパク質を少なくとも1つ含む。タンパク質については、単一の酵素または酵素の混合物、抗体または抗体の混合物、抗原または抗原の混合物、または、機能的特性または構造的特性を備えた他のタンパク質とすればよい。すなわち、必要に応じて、複数の異なるタンパク質や複数の異なる修飾タンパク質を、粒子内部で固定化できる。タンパク質が酵素である場合、本粒子は、酵素の最適pHを酸性域またはアルカリ性域にシフトすることを可能にする手段を提供する。これは粒子内での酵素の固定化による。
【0009】
タンパク質または修飾タンパク質をポリマーに共有結合する場合、これは、例えば、エポキシド、アルデヒドをタンパク質または修飾タンパク質のアミン基と相互作用させることで達成される。
タンパク質または修飾タンパク質をポリマーにイオン結合する場合、これは、ポリマー上で正または負に帯電した官能基を、タンパク質または修飾タンパク質上で前記官能基と反対に帯電されたアミノ酸残基とイオン結合させることで達成される。
【0010】
タンパク質をポリマーに疎水結合する場合、これは、ポリマー上の芳香族アルカンまたは長鎖アルカンの疎水基とタンパク質上の疎水性アミノ酸とが結合することで達成される。
タンパク質をポリマーに親和性結合する場合、これは、二価金属またはアビディンといった親和性タグが、ヒスチジンまたはビオチン化タンパク質を結合することで達成される。
【0011】
当然のことながら、粒子には、タンパク質のより効率的な結合のために、必要に応じて上記の種類またはカテゴリーの官能基を複数含ませてもよい。
鎖状または繊維状のポリマーは、ホモポリマーとしてよいし、ポリエチレンイミンとしてもよい。
架橋剤は、グルタルアルデヒドまたは他のアルデヒド、エポキシド、あるいは、複数の官能基を有する他の適当な化合物とすればよい。
【0012】
粒子は、格子の開口領域や空間の内部に捕捉された補助剤を含む。補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択すればよい。適当な酵素または基質を補助剤として粒子に含ませれば、反応に用いる補因子の連続的な再生を達成でき、それによって、反応を平衡状態に到達させたり、あるいは反応を完了させたりすることができる。補助剤として磁鉄鉱または磁性物質を含ませれば、形成液状媒体からの粒子の回収は、磁気分離を用いて速やかに行える。
【0013】
また、本発明の第2の様態として、第1の液相の液滴が第2の液相内に分散したエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中には溶解したポリマーおよび架橋剤が含まれるという状態で製造する処理と、架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、を特徴とする粒子生成方法を提供する。
【0014】
第1の液相は水相であり、従って第2の液相は油相となる。よって、エマルジョンは油中水分散型(water(w)-in-oil(o))のエマルジョン(すなわちw/oエマルジョン)となる。しかし、本発明の別の実施の形態では、エマルジョンを、水中油分散型(oil-in-water(o/w))エマルジョン、水中油分中に水が分散した型(water-in-oil-in-water(w/o/w))のエマルジョン、あるいは、油中水分中に油が分散した型(oil-in-water-in-oil(o/w/o))のエマルジョンとすることもできる。
【0015】
本エマルジョンは、第1のエマルジョン(水滴で成り、その中にポリマーが溶けて油相で分散した状態で含まれるもの)を、第2のエマルジョン(水滴で成り、その中に架橋剤が溶けて油相で分散した状態で含まれるもの)と混合する方法で形成できる。
また、本発明の第3の様態として、第1の液相の液滴が第2の液相内に分散した第1のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解したポリマーが含まれているという状態で製造する処理と、第1の液相の液滴が第2の液相内に分散した第2のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解した架橋剤が含まれるという状態で、第1のエマルジョンと併せる処理と、架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、を特徴とする粒子生成方法を提供する。
【0016】
相のうち少なくとも1つは、界面活性剤(detergent、surfactant)を含む。界面活性剤は、両性イオン界面活性剤、中性界面活性剤、荷電界面活性剤またはポリマー界面活性剤から選択すればよい。陰イオン界面活性剤としては、アルキル硫酸塩(ラウリル硫酸ナトリウム、ラウレス硫酸ナトリウム、アルキルエーテル硫酸など)がある。陽イオン界面活性剤としては、セントリモニウムクロリド(centrimonium chloride)がある。非イオン物質の界面活性剤としては、エトキシ化アルキルフェノール(ポリオキシエチレン(10)イソオクチルシクロヘキシルエーテル(Triton X100)、ポリオキシエチレン(9)ノニルフェニルエーテル(ノノキシノール-9)など)がある。両性または両親媒性の界面活性剤としては、デシルベタイン(decyl betaine)がある。ポリマー界面活性剤としては、ソルビトール−(エチレンオキシド)80、エチレンオキシド−プロピレンオキシド−エチレンオキシドのトリブロックコポリマー(これは、ポロキサマーとしても知られ、Pluronicの商品名でBASF社から入手できるものなどがある)、そして、プロピレンオキシド−エチレンオキシド−プロピレンオキシドのトリブロックコポリマー(メロキサポールとしても知られるもの)がある。
【0017】
油相の油は、少なくとも原則上は、水と混和しない何らかの適当な有機溶剤、植物油、鉱物油、石炭または原油由来の油相成分(oily component)、または合成油であればよい。しかしながら、鉱物油、パラフィン、溶剤(イソオクタンなど)から選択するのが好ましい。
これまでに述べた通り、ポリマーはポリエチレンイミン(PEI)であり、架橋剤は、二官能基または多官能基のアルデヒド(グルタルアルデヒド、スクシンアルデヒド、デキストランアルデヒド、ヘキサメチレンジイソシアネート、グリオキサールなど)である。他の適当な架橋剤を、PEIまたは誘導化PEI、あるいは他のポリマーに対して使用する場合もある。イソシアネート(ヘキサメチレンジイソシアネートまたはトルエンジイソシアネート、またはイソチオシアネートを含む);エポキシド(2-クロロメチルオキシランなど);無水物;エピクロルヒドリン,1-エチル-3,3-ジメチルアミノプロピルカルボジイミド;エチルクロロアセテート、または、それに類するものが挙げられる。タンパク質または修飾タンパク質の固定化には未反応の官能基が用いられるので、架橋剤は、ポリマー修飾または架橋後(post cross-linking)修飾のための誘導(derivitization)剤とも見なされるであろう。他のポリマーまたはコポリマーを用いることもでき、そうしてものとしては、ポリビニルアルコール、ナイロン、アルギネート、その他タンパク質(アルブミン、コラーゲン、それに類するもの)、およびそれに類するもの(修飾されたもの、修飾されていないもの)がある。
【0018】
本方法には、タンパク質(酵素、抗体または抗原、または修飾タンパク質)を粒子内または粒子上に導入する処理が含まれる場合があり、それによって、タンパク質または修飾タンパク質は、上述したように、ポリマー鎖または繊維において、または架橋剤において官能基と反応し、その結果、鎖状または繊維状のポリマーに、または架橋剤に結合されることで固定化される。
【0019】
本方法は、相の1つに補助剤を加えて、補助剤を粒子の格子の内部に捕捉させる処理を含む場合がある。また、格子へのタンパク質の結合の前または後に補助剤を更に追加する場合がある。上述したように、補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択される。
また、本方法には、粒子を油相から回収する処理が含まれる場合がある。具体的には、粒子の回収は物理的な分離手段(遠心分離や濾過)で実行する。
【0020】
また、本方法には、回収した粒子の乾燥処理が含まれる場合がある。粒子の乾燥処理としては、アセトン脱水、空気乾燥、噴霧乾燥、そして、好ましいものとして、凍結乾燥または真空乾燥がある。
なお、上述したとおり、本方法は、回収した粒子に乾燥処理に先立って補助剤を加える処理を含む場合があり、そうすると、補助剤は粒子の格子の内部に捕捉されることになる。
【0021】
回収した粒子の乾燥処理は、タンパク質固定化の前後いずれであっても、タンパク質および粒子の安定化促進の達成、または、天然補因子や修飾補因子といった添加物の捕捉に関して効果がある。乾燥処理によって更に、多点結合によるタンパク質または修飾タンパク質の架橋状態の改善という結果が得られる。そして、これにより、酵素などのタンパク質の安定性が向上することになる。
【0022】
また、本発明の粒子には多様な用法や用途があると考えられる。例えば、生体触媒用、酵素ベースの生物機能を利用した環境修復、診断に用いられる。また、膜反応器やタンパク質固定化マトリックスといった固体支持体の表面に結合させて表面積を大きくするのに用いることができる。
【図面の簡単な説明】
【0023】
【図1】例1によるグルタルアルデヒド架橋PEIポリマー粒子の顕微鏡写真であり、ポリマー鎖/繊維のPEI支持体格子(support lattice)または網目主鎖(network backbone)を示す図である。
【図2】例2による、繊維格子に結合するラッカーゼに関して得られた結果を示すグラフであり、pHプロフィール(profile)のシフトに関して結合効率の補正を行った場合の図である。
【図3】各種の油相を用いて製造した粒子の粒径分布(平均粒径)の分析結果を示す図であり、粒子を乾燥させなかった場合(湿った状態)、乾燥させた場合(凍結乾燥を採用)、インライン(in-line)超音波照射(US)を伴う場合、超音波照射で前処理した後に測定した場合、について示す図である。
【図4】各種の界面活性剤を用いて製造した粒子の粒径分布の分析結果を示す図であり、そのままの状態で光散乱によって測定した場合と、超音波照射後(US後)の分析で求めた場合とを示す図である。
【図5】単一エマルジョンを用いて製造した乾湿両粒子について粒径分布を示す図であり、結果は3回繰り返して行った実験の標準偏差を示しており、そのままの状態での光散乱によって測定した場合と、インライン超音波照射を伴う場合と(「USと共に」)、サンプルに事前超音波照射した後の分析で求めた場合(US後)とを示す図である。
【図6】20mlの量の鉱物油と200mlの量の鉱物油とで製造した乾湿両粒子について粒径分布を示す図である。
【図7】潜在的な保護剤としての基質を有する場合と有していない場合とで、粒子に結合された各種酵素の活性度の維持状態を示す図である。
【図8】自由(free)ラッカーゼ結合粒子および乾湿両ラッカーゼ結合粒子に関する温度最適条件を示す図であり、(A)はPEIがpH8で製造された場合、(B)はPEIがpH11で製造された場合を示す図である。
【図9】PEIがpH8、ph11で製造された乾湿両方の粒子結合ラッカーゼおよび自由ラッカーゼについてpH安定性(6時間)を示す図である。
【図10】(A)は、例14による、後処理していない繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、ポリエチレンイミン(‘PEI’)濃度の変化がpHプロフィールのシフトに与える影響を表す図であり、(B)は、例14による、後処理していない繊維格子または網目における固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、グルタルアルデヒド濃度の変化がpHプロフィールのシフトに与える影響を表す図である。
【図11】(A)は、グルタルアルデヒド後処理した繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、PEI濃度の変化がpHプロフィールのシフトに与える影響を表す図であり、(B)は、例14により、グルタルアルデヒド後処理した繊維格子または網目における、固定化酵素および自由酵素のラッカーゼpHプロフィールを示しており、グルタルアルデヒド濃度の変化がpHプロフィールシフトに与える影響を表す図である。
【図12】実験A〜Fに関する粒子のペルオキシダーゼ活動度について得られた結果を示す図である。
【発明を実施するための形態】
【0024】
ここから、本発明について、後述の実施の形態および添付図面を参照しながら、より詳細に説明する。
(例1:ポリマー鎖/繊維の網目または格子からなる粒子の製造方法)
本方法で形成される油中水分散エマルジョンは、その内部で、ポリアミンポリマー(ポリエチレンイミン)を含むエマルジョンと別の主要なアミン架橋剤(グルタルアルデヒド)とが組み合わされる。2つの試薬が反応して、微細な粒子または粒の形をしたポリマーを形成する。
(使用化学物質)
グルタルアルデヒド(25%の水溶液)は、Acros Organics社(Geel West Zone 2, Jansen Pharmaceuticalaan 3a, 2440 ゲール, ベルギー)から入手した。ポリエチレンイミン(PEI)(50%の水溶液、カタログ番号P−3143、Mw750,000およびMn60,000)は、Sigma-Aldrich社(セントルイス、ミズーリ州 63178)から取得した。鉱物油(医薬用ホワイトオイル、48031)は、Castrol社(8 ジャンクションアベニュー、パークタウン、2193 ヨハネスバーグ、南アフリカ)から購入した。
(粒子の製造方法)
(エマルジョンAの組成)
・10mlの鉱物油(油相)
・0.05mlのノノキシノール−4(界面活性剤)
・0.5mlのポリエチレンイミン(ポリアミン)、(10%m/v水溶液)、pH11
700rpmで30分間、電磁撹拌器を用いて撹拌した。
(エマルジョンBの組成)
・10mlの鉱物油(油相)
・0.05mlのノノキシノール−4(界面活性剤)
・0.5mlのグルタルアルデヒド、(25%m/v、等級(grade)II)
700rpmで30分間、電磁撹拌器を用いて撹拌した。
【0025】
2つのエマルジョン(A、B)を組み合わせると、ポリマー架橋反応を生じさせることができた。そして、電磁撹拌器を用いて700rpmで30分〜1時間撹拌することで、エマルジョンの状態は確実に維持された。その後、JA20.1ロータを備えたBeckman-Coulter J2-21MEに入れて10分間、エマルジョンを3000rpmで遠心分離し、形成された粒子を回収した。ペレットは、脱イオン水に再懸濁して10〜40mlまで希釈した後、再び遠心分離した。そして、この洗浄工程を更に2回繰り返した。最終的に得られた上澄みは澄んでいた(clear)。そして最終的に残ったペレットは、10mlのTris-Cl緩衝液(0.05M、pH8.0)に懸濁した。
(結果)
全てのエマルジョン調製で得られる材料を、遠心分離によって回収し、光学顕微鏡検査によって視覚化した。図1に示すのは格子形成の結果であり、おおよそ球状の粒子が形成されたことを示している。
(PEI対グルタルアルデヒドの濃度比が粒子形成に与える影響)
グルタルアルデヒドに対するPEIの比率が粒子形成に与える影響を調査した。サンプルは表1に従って調製した。
【0026】
乾燥重量の測定は、繊維主鎖格子/支持体の凍結乾燥および重量計測という手順で行った。この実験の結果は表1に示してあり、幅広い反応体の組合せで粒子が形成されることを示している。
表1:繊維ポリマー主鎖の製造評価に使用するPEIおよびグルタルアルデヒドの量
【0027】
【表1】
(例2:ポリマー鎖/繊維のPEI支持体格子または網目主鎖へのラッカーゼの結合)
(酵素)
DeniLite(登録商標)(ラッカーゼ)は、Novozymes社(Novozymes A/S、Krogshoejvej 36、2880 バウスベア、デンマーク)から取得した。
(酵素洗浄)
ラッカーゼは、DeniLiteから部分的に精製した。その方法は、2回蒸留した100mlのH2Oに5gのDeniLite II Baseを溶かしながら、4℃で1時間、200rpmで撹拌するというものであった。懸濁物質は、Beckman-Coulter J2-21ME遠心分離機のJA14ロータを用い、4℃で1時間、10000rpmで遠心分離を行って除去した。上澄みを取り除き、10kDaカットオフ機能を備えたSnakeSkin(登録商標)(Pierce社)透析管を用いて、4℃の水5Lを3回交換して透析した。最初の2回の交換までの透析は2時間継続し、最後の透析は12時間継続した。酵素は、液体窒素の中で凍結させて凍結乾燥した。その後、このラッカーゼは、必要になるまで4℃で保管した。
(ラッカーゼの分析評価)
ラッカーゼの分析評価は、ラッカーゼを支持体に結合した後の遠心分離による上澄みと、支持体に固定化されたラッカーゼとを対象に実施し、結合時の活性度の維持状態を測定した。ラッカーゼ試薬は、基質としての1mMのグアヤコールを、100mMのサクシネートラクテート緩衝液(pH4.5)の中に含むものとした(Jordaan J,Pletschke B,Leukes W(著)、2004年、(題名)“Purification and partial characterization of a thermostable laccase from an unidentified basidiomycete.”(掲載誌)Enz. Microb. Technol. 34:635-641)。分析評価は、5 200M−1.cm−1の減衰係数で、450nmにおいて3回繰り返して実施した。分析評価の実施には、PowerWave HT Microtitreプレートリーダを用いた。酵素の単位は、1分につき1μmolの基質を酸化させるのに必要な酵素量を1単位として定めた。
(タンパク質の測定)
タンパク質担持(protein loading)後に、ラッカーゼを基準タンパク質とし、280nmでの光吸光を利用して溶液中のタンパク質濃度を測定した。溶液中の残留タンパク質をタンパク質全体から差し引いたものを、結合されたタンパク質と見なした。
(酵素固定化)
室温で30分間緩やかに揺動して、酵素(ラッカーゼ)(10mg.ml−1溶液で1ml)を支持体に結合させた。このとき、酵素は、表2に示す乾燥重量を持つ主鎖と結合した。ラッカーゼと結合した粒子は、700×gで5分間遠心分離を行って回収した。固定化された酵素は、50mlの水で5回洗浄し、上述したような遠心分離を行って回収した。
【0028】
タンパク質(この場合は酵素であるラッカーゼ)は、緩衝液の形でPEI-グルタルアルデヒド粒子(誘導化したもの、あるいは、誘導化していないもの)に加えた。粒子は例1で示した形で調製した。粒子をタンパク質と反応させて、上述したように、タンパク質をポリマーに固定化することができた。粒子は、遠心分離による回収から使用時まで、乾燥させないようにした。
【0029】
様々な形で製造したタンパク質支持体にタンパク質を結合した結果を、下の表2に示す。一方、ラッカーゼ活性度に関する結果は図2に示してある。
(結果)
表2:主鎖支持体へのラッカーゼの結合効率
【0030】
【表2】
別の実験用セットを表1に示す通りに調製した。しかし、本例での粒子はグルタルアルデヒドで後処理してあり、数字の2で区別している(すなわち、A2−J2)。調製に続き、上述した方法に従ってラッカーゼを粒子に結合した。ラッカーゼが結合した粒子を、700×gで5分間遠心分離して回収した。
【0031】
ポリマー支持体に担持された酵素の活性に関する結果を図2に示す。
本調査が示しているのは、繊維格子または網目をタンパク質固定化の支持体として用いた場合、タンパク質結合容量が大きいと同時に、タンパク質の機能活性は保たれる、ということである。
(例3:各種の油を用いたポリマー鎖/繊維のPEI支持体格子または網目主鎖の製造方法)
エマルジョンの油相の違いが与える影響を調査した。粒子の製造にあたって、先ず2種類の異なるエマルジョンA,Bを調製した。
(エマルジョンAの組成)
・10mlの鉱物油、パラフィン油またはイソオクタン(油相)
・0.1mlのノノキシノール-4(界面活性剤)
・0.5mlのポリエチレンイミン(ポリアミン)、(10%m/v水溶液)、pH11
25℃において、500rpmで30分間、電磁撹拌器を用いて撹拌した。
(エマルジョンBの組成)
・上述したのと同じ油相10ml
・0.1mlのノノキシノール-4(界面活性剤)
・0.5mlのグルタルアルデヒド、(25%m/v、等級II)
25℃において、500rpmで30分間、電磁撹拌器を用いて撹拌した。
【0032】
続いて、エマルジョンAを短時間でエマルジョンBに加え、更に1時間撹拌した(700rpm)。そして、Sorvall benchtop遠心分離機によって3000×gで10分間遠心分離した後、10mlの量の脱イオン水で6回洗浄するという手順で、エマルジョンから粒子を回収した。洗浄後、最終的に得られた粒子を脱イオン水で20mlに再懸濁し、その半分を凍結乾燥によって乾燥させた。湿潤粒子(wet particle)および乾燥粒子の両方について、粒径分布を分析した(Malvern Mastersizer 2000使用)。凝塊の存在を調査する目的で、粒径の測定を、インライン超音波処理の前後、そして同処理と共に行った。また、凍結乾燥後の回収量を質量で測定した。
(結果)
異なる油相で製造した粒子(各々の総量は20ml)の質量での回収量は、鉱物油、パラフィン油、イソオクタンでそれぞれ111mg、90mg、79mgと測定された。異なる油相で製造し、超音波処理を行わなかった湿潤粒子と乾燥粒子とを比較した結果、鉱物油およびパラフィン油のサンプルについては、乾燥後に粒径の平均値が大きくなったことが分かった(図3)。イソオクタンで製造された粒子は、乾燥処理にもかかわらず比較的変化がなかった。インライン超音波処理およびプレ超音波処理(pre-sonication)では、鉱物油およびパラフィン油で製造し乾燥した粒子の粒径分布に大幅な低下が見られた。それは、乾燥処理した粒子には凝塊が生じるが(図3)、超音波処理によって分解することができた、ということを示している。また、鉱物油およびパラフィン油で作られた湿潤粒子の粒径の分析でも、粒子の超音波処理に伴って粒径の平均値が下がることが示されたが、その幅は乾燥した粒子の場合(図3)に比べれば小さい。イソオクタンで製造した粒子は、乾燥処理や超音波処理にかかわりなく、比較的変化がなかった。
【0033】
結論としては、様々な油相を用いて粒子を製造することが可能である。また、異なる油相を用いること、ならびに乾燥処理や超音波処理などの各種後処理の技術を用いることで、粒径を操作することができる(油相の違いはエマルジョンの液滴サイズに影響すると推測される)。
(例4:各種の界面活性剤を用いたポリマー鎖/繊維のPEI支持体格子または網目主鎖の製造方法)
粒子の合成では、界面活性剤の種類が影響を与える場合がある。これを調査した。
【0034】
粒子は、ほぼ例3に従って調製したが、油相としては鉱物油のみを用い、界面活性剤の種類を変えた。
(結果)
鉱物油に各種の界面活性剤を用いて製造した粒子(総量20ml)の質量での回収量は、ノノキシノール-4、CHAPS、Triton X-100のそれぞれについて、111mg、72mg、92mgと測定された。CHAPSやTriton X100を用いた場合、異なる界面活性剤で製造した粒子を乾燥処理しても、粒径に実際的な差は表れなかった(図4)。ノノキシノール-4を界面活性剤として製造した粒子を乾燥処理すると、粒径は約50%大きくなった(図4)。さらに、超音波処理後の粒径分析は、テスト対象の全ての界面活性剤で製造した粒子で、そろって粒径が小さくなったことを示した。超音波処理した粒子の粒径を乾燥処理の前後で分析すると、結果は類似したものとなった。
【0035】
結論としては、様々な界面活性剤を用いて粒子を製造することが可能であった。さらに、粒径は、界面活性剤の種類および後処理によって操作することができた。
(例5:ポリマー架橋剤として各種ポリアルデヒドを用いた粒子合成)
粒子の合成に用いる架橋剤アルデヒドは、様々に変更することができる。架橋剤としては、グルタルアルデヒド、デキストランアルデヒド、ヘキサメチレンジイソシアネートが挙げられる。
【0036】
粒子を作る方法は、以下の点を除けば、例3と同じである。すなわち、ある場合では、グルタルアルデヒドをデキストランアルデヒド(15mg/mlのものを1ml)と置き換えた。また、別の場合では、0.5mlのヘキサメチレンジイソシアネート(25%v/v)と置き換えた。
5mlの粒子懸濁液に、Tris-Cl緩衝液(0.05M、pH8.0)に入れた5mg.ml−1の精製カンジダアンタークティカリパーゼB(CALB)を6ml加え、25℃で1時間緩やかに撹拌した。その後、懸濁液を遠心分離し、4℃の緩衝液10mlを用いて2度洗浄した。それから、懸濁液を遠心分離し、ペレットを10mlのTris-Cl緩衝液に再懸濁した。そして、p-ニトロフェニルブチラートを使って懸濁液(10μl)を分析評価した。また、比較のために、Tris-Cl緩衝液(0.05M、pH8.0)に0.5mg-per-mlの精製カンジダアンタークティカリパーゼBを10μl入れた場合についても分析したが、これには、p-ニトロフェニルブチラートの加水分解に基づく分析評価と、それに続く分光測光法による分析とを用いた(表3)。
(リパーゼ活性の分析評価)
リパーゼの活性により、p-ニトロフェニルエステル(p-ニトロフェニルブチラート(PNPB))はp-ニトロフェノールおよびブチル酸へと加水分解された。p-ニトロフェノールの放出によって黄色が生じ、この黄色は、UV/Vis分光光度計を用いれば410nmで測定される。活性度の測定は3回繰り返した。溶液の調製は以下のように行う。すなわち、溶液Aは、8mlのプロパン-2-オールに溶けた形で酵素基質を含むものとした。一方、溶液Bは、50mMのTris-緩衝液(pH8.0)にデオキシコール酸ナトリウム267mgを溶かした後に、66.7mgのアラビアゴムを溶解させたものとした。動的(Kinetic)分析評価を、PowerWave microtitreプレートリーダ(BioTek Instruments社)を用いて25℃で実行し、上述した溶液の1対10(A:B)の混合物240μlと固定化リパーゼ懸濁溶液または自由酵素10μlとを用いた。
(結果)
(表3:各種のアルデヒド架橋剤を用いて作った粒子の比較−CALBの活性度)
【0037】
【表3】
この方法は最適化されていないが、ある程度の範囲のポリアルデヒド合成物が使用して粒子を生成できることを示している。
【0038】
本材料は、0.45μmフィルタ(Sartorius社)で回収および再利用することができた。5回の再利用を終えて、グルタルアルデヒド粒子は7.05U.ml−1の平均値を示し、活性度は当初の81%であった。
類似の実験を、蛍光菌(PFL:Pseudomonas fluorescens)由来のリパーゼを用いて実施した。酵素活性度の分析評価には、基質として、ブチル酸のp-ニトロフェニルエステル(PNPB)とパルミチン酸のそれ(PNPP)とを用いた。
(表4:各種のアルデヒド架橋剤で作られた粒子の比較−PFLの活性度)
【0039】
【表4】
各種のアルデヒドを用いることで、粒子形成のための効果的な架橋の実現と、タンパク質を粒子に架橋するための官能基の提供との両方が可能となった。架橋剤の選択は、酵素変性の程度や基質および生成物に対する粒子の接触可能性(accessibility)の程度によって、酵素の活性度に影響を与える可能性がある。最適な架橋剤は、酵素および反応基質に基づいて選択すればよい。
(例6:2官能基エポキシド架橋剤を用いた粒子製造)
粒子の合成に使用する架橋剤アルデヒドは、ジエポキシド(例:1,4-ブタンジオールジグリシジルエーテル)などの他の架橋剤と置き換えることができる。これを(グルタルアルデヒドの代わりに)、50%v/v、25%v/v、12.5%v/vの薄めていない(neat)0.5mlの溶液の形で、架橋剤エマルジョン(B)に加え、そして、40℃で2時間反応させた(例3の場合と同様)。エマルジョンからの粒子の回収には、3000×gの遠心分離(Sorvall RT7 benchtop遠心分離機使用)を10分間行い、その後、10mlの量の脱イオン水で6回洗浄した。洗浄後、最終的に得られた粒子を脱イオン水で20mlに再懸濁した。12.5%v/vまたは25%v/vのエポキシド溶液を用いて上記のやり方で調製したものは、形状の点で最も均一であったのに対し、50%のエポキシドを薄めずに用いて形成したものは、大きく、しかもばらつきがあった。
【0040】
タンパク質の結合は、(前出の例に従い)CALBを使って調査した。それを一晩(overnight)透析し、エポキシドベースの粒子(12.5%v/vエポキシド溶液を用いて上述の通り調製したもの)と、緩やかに撹拌しながら1時間反応させた。例5で述べたようにリパーゼ結合粒子を回収および洗浄した後、酵素の活性度を分析評価した。全ての分析評価はPNPBを基質として実施した。
(結果)
ブタンジオールジグリシジルエーテルを架橋剤として使用した結果、直径が約0.1mmの白色粒子が形成された。しかしながら、この粒子は形状がいくぶん不規則であり、これは、架橋または後続のクリーニングの手順で粒子の凝塊が生じたことを示す。
【0041】
12.5%v/vエポキシド溶液で製造した粒子を、酵素が存在する状態で一晩インキュベート(incubate)した。これらの粒子は、0.28U.ml−1の活性度を示した。これは、エポキシド架橋剤を用いた粒子の製造が可能であることを示す。
(例7:単一のエマルジョンで製造したポリマー鎖/繊維のPEI支持体格子または網目主鎖)
粒子の形成方法は、必要に応じて調整することができる。例えば、単一のエマルジョンを用いて粒子を形成することもできる。
【0042】
ポリマーと架橋剤とは、混合チャンバに2重注入を行う装置を用いれば、水相で即座に混合することができる。こうした装置の最も単純な型は、同じポイントで共通のラインに注入を行う2つのシリンジで構成されたものであろう。これは油相への直接注入が可能である。一例として、20mlの鉱物油に0.2mlのノノキシノール-4を約5分間溶解させ、さらに500rpmで電磁撹拌器による撹拌を行う、という設定で評価を行った。一方のシリンジには0.5mlのPEI(10%)を入れ、他方には0.5mlのグルタルアルデヒド(20%、等級II)を入れた。両シリンジを同時に押し込んで、鉱物油に直接注入した。結果として得られたエマルジョンを1時間撹拌し、その後、粒子を例3と同じやり方で回収した。実験は3回繰り返して実施した。粒子は2等分し、その一方は冷凍乾燥し(Virtis使用)、元の量に再懸濁した後、粒径分布についての分析評価を行った(Malvern Mastersizer 2000使用)。サンプルに対し、乾燥後、回収量(質量)に関する分析評価を行った。
(結果)
単一のエマルジョンを用いて製造した粒子の回収量(質量)は152±12mgと算出されたが、その値は、2エマルジョン方式(例3を参照)を用いて得られるものに比べて約1.4倍大きかった。
【0043】
単一のエマルジョンを用いた粒子形成は可能であり、現行の製造法(図5)を用いて再現可能であった。粒子は、2種のエマルジョンを用いて形成したものに比べ、粒径分布がより広いことが分かった(図5)。単一エマルジョン粒子に関して得られた乾湿の粒径分布は、2エマルジョン実験の場合に得られるもの(図3)に比べ、サイズで、50〜70%大きいことが分かった。
(例8:ポリマー鎖/繊維のPEI支持体網目または格子の拡大規模での製造)
ここでの目的は、粒子製造の規模を線形的に10倍に拡大して粒径を評価することであった。別個の2バッチの粒子を、例3で概説した標準的製造方法にほぼ従って調製したが、異なる点として、第2のバッチは必要な構成成分の各々について10倍の量を含むものとした。両バッチに対しては、規模に応じた形で、例3と同様の粒子回収処理を行った。
(結果)
20ml、200mlの鉱物油量で製造した粒子の回収量は、質量でそれぞれ0.111g、1.11gと算出され、ちょうど10倍の差があった。鉱物油量が20mlでも200mlでも粒子を製造することは可能であり、より大きな規模で製造した粒子の粒径は、20mlの規模で得られるものに比べ、一貫してわずかに小さいだけであった(図6)。興味深い点は、より大きい規模で製造し、超音波処理していない乾燥粒子のサイズが、より小さい規模で製造した乾燥粒子と比べて約50%小さかったことである(図6)。
【0044】
回収量(質量)および粒径分析に基づいて言えば、標準的条件での粒子の製造法は、少なくとも10倍の規模拡大が可能であった。
(例9:ポリマー鎖/繊維のPEI支持体網目または格子を用いた広範囲の酵素群の固定化)
ここでの目的は、異なる酵素を粒子主鎖に結合し、それらの結合率(%)と酵素活性度の維持の程度とを算出することであった。
【0045】
例3に従って製造した粒子を使用した。検査対象の酵素は、ラッカーゼ(Novozymes 51009, ミセリオフトラサーモフィラ)、グルコースオキシダーゼ(Seravac Pty Ltd, アスペルギルスニゲル)、そしてリパーゼ(CALB, Novozymes のカンジダアンタークチカ)であった。各々の実験について、各酵素5mg(10mg.ml−1で0.5ml)を14mg(28mg.ml−1で0.5ml)の粒子に結合し、25℃で2時間緩やかに振盪させた。各サンプルは、Allegra X22R遠心分離機を用いて10分間遠心分離した(2000×g)。粒子は、連続して6回洗浄し、各回2mlの脱イオン水を用いた。混ぜ合わせて生じた上澄み部分を分析して、総タンパク質を求めた。個別の酵素結合実験を、各々、潜在的(potential)保護剤として特定の基質を含む場合と含まない場合とで実施した。ラッカーゼの場合、市販の伝達物質Denillite II Assist(Novozymes)を潜在的保護剤とした(100mg.ml−1 (pH6.8に調整)のものを50μl)。グルコースオキシダーゼの場合、グルコースを潜在的保護剤とした(10%m/vグルコース一水和物を50μl)。そして、CALBの場合、2-イソプロピル-5-メチルシクロヘキサール(メントール)の8−ジアステレオ異性体混合物を使用した(50μl)。粒子は、脱イオン水で2mlに再懸濁した。粒子を分析し、それぞれの酵素の活性度を求めた。全ての分析評価を3回繰り返した。
(総タンパク質)
総タンパク質分析評価は、Bio-Rad Total Protein Assay kit(カタログ番号:500-0006)というキットを用い、個別の酵素を各々基準として用いて実施した。
【0046】
ラッカーゼの活性度を測定するために、100mMのサクシネートラクテート緩衝液(pH4.5)に入れた1mMの2,2’-アジノビス(3-エチルベンゾチアゾリン-6-スルホン酸)ジアンモニウム塩(ABTS)を基質として使用した。溶液の光学濃度は420nmで測定した。これらの分析評価は、180μlのABTS試薬にサンプル20μlを加えて実行した。活性度の調査は、PowerWave HT(Biotek Instruments社)を用い、25℃でインキュベートして、420nmで分光測光法によって行った。
(グルコースオキシダーゼ)
グルコースオキシダーゼ(GOX)の活性度は、ホースラディッシュペルオキシダーゼ(HRP)によるo-ジアニシジンの間接酸化を利用して計測した。分析評価は、Bergmeyer(その他)による方法(1988年)に従って実施した。調剤した試薬は以下の通り:(試薬A)o-ジアニシジン-2HCL(0.006%)を含む0.1Mのリン酸カリウム緩衝液(pH7);(試薬B)D−グルコースの10%の水溶液(使用前に1時間変旋光(mutarotate)を生じさせたもの);(試薬C)60Uml−1HRP水溶液。試薬A、B、Cは、それぞれを24:5:1の割合で、グルコースオキシダーゼの分析評価の直前に混合した。反応試薬0.3mlを入れて反応させており、サンプルを10μl加えることで反応が開始された。反応結果は、25℃において、436nmで動力学的に計測した(Powerwave HT microtitreプレートリーダ使用)。グルコースオキシダーゼの活性度の単位は、25℃およびpH7で、1分につきに1μmoleのβ-D-グルコースをD-グルコノラクトンとH2O2とに転換するだけの触媒作用を生じる酵素の量を1単位と定めた。
(リパーゼ)
リパーゼの活性により、p-ニトロフェニルエステルはp-ニトロフェノールおよび脂肪族カルボン酸に加水分解される。p-ニトロフェノールの放出によって黄色が生じ、この黄色はUV/Vis光度計では410nmで計測される。2種のp-ニトロフェノールエステル(p-ニトロフェニルアセテート(PNPA)およびp-ニトロフェニルブチレート(PNPB))を使用し、活性度の測定は3回繰り返した。溶液は以下のように調製した:溶液Aは、酵素基質(11.6mgのPNPAまたは24mgのPNPP)を8mlのプロパン-2-オールに溶かされた状態で含むものであり;一方の溶液Bは、267mgのデオキシコール酸ナトリウムを含み、これを50mMのTris-緩衝液(pH8.0)に溶かした後に、66.7mgのアラビアゴムを加えたものである。動力学的分析評価を、25℃で、PowerWave microtitreプレートリーダ(BioTek社)を使って実施し、上述した複数の溶液の混合物240μlと球体(sphere)酵素または自由リパーゼの溶液10μlとを用いた。
(結果)
粒子への結合について検査した全ての酵素で、タンパク質担持の率は30〜36%の範囲となった(表5)。
(表5:粒子主鎖支持体への各種酵素の結合効率)
【0047】
【表5】
(*)活性部位の保護のため酵素の固定化中に基質を追加した(上述の方法を参照)。
PNPAに対するCALBの活性度の維持には、メントール基質を含ませることが効果的であり、約30%高い活性度(83%)が保たれた(図7)。
【0048】
ラッカーゼ、グルコースオキシダーゼ、CALBは全て、活性度を保ちながら粒子に結合することに成功した。それは、広範囲のタンパク質を、タンパク質担持が高い状態で、粒子に効果的に結合できることを示す。更に、他の酵素(ホースラディッシュペルオキシダーゼ、プロテアーゼ、デヒドロゲナーゼなど)も、粒子に結合することが示された(下の例を参照)。
(例10:ポリマー鎖/繊維のPEI支持体網目または格子;複数の酵素が結合した粒子)
本調査は、複数の酵素を、両酵素に対する活性度を保った状態で粒子主鎖に結合できることを示す意図で行った。グルコースオキシダーゼとホースラディッシュペルオキシダーゼ系とを選択した。
【0049】
粒子は、例3に従って製造した。調査した酵素は、グルコースオキシダーゼ(Seravac Pty Ltdのアスペルゲルスニゲル)およびホースラディッシュペルオキシダーゼ(Serevac Pty Ltd)であった。一例として、5mg(10mg.ml−1で0.5ml)のグルコースオキシダーゼと10mg(10mg.ml−1で1ml)のホースラディッシュペルオキシダーゼとを、25℃で2時間緩やかに振盪して、28mg(28mg.ml−1で1ml)の粒子主鎖に結合した。各サンプルを、Allegra X22遠心分離機(200×g)を用い、10分間遠心分離した。粒子は、連続して6回、各回2mlの脱イオン水を用いて洗浄した。結合後の上澄み部分を、総タンパク質について分析した。粒子の活性度は、例9のグルコースオキシダーゼ用の分析評価方法に従って測定したが、分析評価試薬にホースラディッシュペルオキシダーゼは含まれていない。
(結果)
グルコースオキシダーゼおよびホースラディッシュペルオキシダーゼの粒子主鎖への結合は成功し、3μmole.min−1の率でグルコースを変換することができた(表6)。このことは、分析評価で検出した活性度と同様、グルコースオキシダーゼとホースラディッシュペルオキシダーゼとが結合され、活性度を維持していたことを示す。すなわち、粒子を用いれば、複数の酵素を、両方の酵素が活性度を保った状態で結合することができる。
(表6:グルコースオキシダーゼおよびホースラディッシュペルオキシダーゼの2種酵素粒子に関する結合効率および活性度)
【0050】
【表6】
(例11:各種乾燥方法を用いて製造したポリマー鎖/繊維のPEI支持体格子または網目)
ここでの目的は、粒子を製造した後に、様々な方法(凍結乾燥、真空乾燥、アセトン乾燥など)を用いて粒子を乾燥させることであった。
【0051】
粒子は、例1で概説した標準的な条件で製造した。そして、洗浄した粒子を、凍結乾燥(Virtis Genesis凍結乾燥機使用)、真空濃縮機(Savant RVT100蒸気収集機を備えたSavant SpeedVac SC110)を用いた真空乾燥、または、アセトンを用いた脱水によって乾燥させ、その後さらに、25℃で12時間空気乾燥させた。アセトン乾燥した粒子は、凝塊したため水媒体に再懸濁することができず、そのため、それ以上の考察は行っていない。
【0052】
しかし、真空乾燥と凍結乾燥とは、粒子を乾燥させる技術として両方とも成功であり、乾燥後の粒子を用いて、広範囲の酵素やタンパク質を付着させることができた(表7)。5mg per mlの精製酵素6mlをTris-Cl緩衝液(0.05M、pH8.0)に入れて、粒子懸濁液5mlに加えた。そして、25℃で1時間緩やかに撹拌した。懸濁液は、その後遠心分離してから、4℃の緩衝液10mlを用いて2度洗浄した。さらにその後、懸濁液を遠心分離し、ペレットを10mlのTris-Cl緩衝液(0.05M、pH8.0)で再懸濁した。粒径は、Malvern Mastersizerを用いて測定した。
(結果)
(表7:各種乾燥処置後の平均粒径)
【0053】
【表7】
本例が示すのは、乾燥処理、特に凍結乾燥した場合、酵素と結合した粒子が大きく凝塊することはない、ということである。
(例12:様々なpHでPEIから調製したポリマー鎖/繊維の支持体網目または格子へのラッカーゼ結合の特性評価)
乾燥処理が、結合後のラッカーゼ結合粒子の特性、酵素の性質に与える影響を判定した。
【0054】
粒子は、例3で概説した標準の製造方法にほぼ従って製造したが、pH11で調製したPEI(10%)に加えて、pH8に調整して調製したものについても評価を行った。さらにラッカーゼを主鎖に固定化し、その方法は例9で述べたものとほぼ同じであるが、以下の点が異なる。すなわち、1mlのラッカーゼ(Novozyme 51004,50mg.ml−1)を6.25mlの粒子(16mg.ml−1)と反応させた。従って、100mgの粒子について50mgのラッカーゼが結合したことになる。ラッカーゼ結合粒子は、脱イオン水12.5mlで洗浄し、さらに、脱イオン水を用いて20mlに再懸濁した。粒子は10mlずつ2等分した。そして、そのサンプルの1つを、回収量(質量)測定のために凍結乾燥し、乾湿両方のラッカーゼ結合粒子の特徴を調査した。乾燥させたサンプルは、脱イオン水で元の量に再懸濁した。全ての分析評価は3回繰り返した。
【0055】
ラッカーゼの上澄みのサンプルの総タンパク質を、Bio-Rad Total Protein Assay kit(カタログ番号:500-0006)というキットを用い、ラッカーゼをタンパク質の基準として測定した。ラッカーゼの活性度は例9に従って測定した。
(結果)
凍結乾燥によるラッカーゼ結合粒子の乾燥処理では、粒子製造の際にPEIがpH8であった場合の方が、粒子に結合されるラッカーゼタンパク質は多いものの、粒子製造の際にPEIがpH11であった場合の方が、回収できる量(質量)は大きいことが分かった(表8)。ABTSに対しての活性度を最も高く保つことができたのは、pH8のPEIを用いて製造し、凍結乾燥しなかった粒子であったが(16%)、凍結乾燥後の同じサンプルはラッカーゼ活性度を3%しか保持していなかった。注目すべき点として、pH8のPEIを用いて製造された粒子は乾燥後に再懸濁するのが難しく、そのため凝塊する可能性があるが、凝塊すれば表面対面積(surface to area)の比率が下がり、従って、基質および生成物への分散制約(diffusional constraint)が原因で活性度が下がることになる。pH11のPEIを用いて生成された粒子については、活性度に関して同様の影響は観察されなかった(表8)。
【0056】
表8:凍結乾燥を行う場合と行わない場合とで、PEIのpH値を様々に変えながら製造したラッカーゼ結合粒子の結合効率および活性度保持の度合
【0057】
【表8】
この実験が示すのは、粒子はPEIのpHを変えて調製することができ、これによって酵素に対する粒子の結合特性が変わる、ということである。
(例13:pH8またはpH11で製造および凍結乾燥し、ポリマー鎖/繊維のPEI支持体網目または格子に結合されたラッカーゼの安定性)
pH8またはpH11で製造および凍結乾燥され、ポリマー鎖/繊維のPEI支持体網目または格子に結合されたラッカーゼの熱安定性およびpH安定性を測定した(ラッカーゼ結合粒子は例12に記述した形で製造した)。
(温度最適条件)
自由ラッカーゼおよび粒子結合ラッカーゼ(タンパク質担持は均等)の温度最適条件の解析(profile)を、100mMのサクシネートラクテート緩衝液(pH4.5)に基質として入れた1mMのABTSを用いて実行した。ウォーターバス内で、正確な温度に前平衡(pre-equilibrated)しておいた1.9mlの基質に、サンプル(100μl)を加えた。分析評価は、ペルティエ温度コントローラを備えたDU800分光光度計(Beckman-Coulter社、420nm)を用いて実施した。分光光度計を対象の温度に設定し、キュベットは、ウォーターバス内の平衡化した試薬を追加する前に、5分間平衡化させた。
(pH安定性)
自由ラッカーゼおよび粒子結合ラッカーゼ(タンパク質担持は均等)のpH安定化は、100mMのサクシネートラクテート緩衝液(pH4.5)に入れたABTS(1mM)を用いて行った。各ラッカーゼサンプルは、pH2.5、pH3、pHにおいて、Britton-Robinson汎用緩衝液内でインキュベートした。サンプル(20μl)を、定期的に取り出し、25℃でインキュベートすると共に、PowerWave HT(Biotek Instruments社)を用いて420nmで分析評価した(230μlの分析評価試薬)。6時間の時点での本実験の結果を図9に示す。
(結果)
結果を図8(A)、(B)、図9に示す。
【0058】
粒子結合ラッカーゼと自由(固定化されていない)ラッカーゼとは両方とも、70℃で活性度が最高になった(図8)。しかし、粒子結合ラッカーゼは、90℃では、自由ラッカーゼ(活性度0%)と比較して熱安定性の向上(活性度55〜65%)を示した(分析評価用サンプルの調製に要する時間で)。また、乾燥粒子を使用した場合も小幅な向上が見られ、乾燥による効果があることを示唆している。これは、水が除去されるにつれて、形成されるタンパク質−粒子連結が多くなることによるかもしれない。これによってタンパク質または酵素の複数点(multi-point)共有結合が増加するが、それは、熱安定性の向上などの形で安定性を高めることが知られている [“Improvement of enzyme activity, stability and selectivity via immobilization techniques”、Mateo, C, Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M., Fernandez-Lafuente, R.(著)、2007年、(誌名)“Enzyme and Microbial Technology 40(6)”、ページ1451-1463]。
【0059】
また、ラッカーゼ結合粒子および自由酵素のpH安定性を、pH2.5、pH3、pH6で測定した。pH2.5およびpH3では、ラッカーゼ結合粒子のサンプル全てが、80〜110%の活性度を保ったが、自由酵素は、それぞれのpHで活性度を約40〜70%失っていた(図9)。すなわち、固定化によってpH安定性は向上する。自由酵素を含むサンプルの全ては、6時間後にpH6で安定していた。
【0060】
従って、ポリマー鎖/繊維のPEI支持体網目または格子の粒子に酵素を固定化することにより、pHの両極での安定性を更に高めることができる。
(例14:ポリマー鎖/繊維のPEI支持体網目または格子への固定化に伴う酵素最適pHのシフト)
酵素は活性に最適のpHを有する。いくつかの場合、酵素の最適pHは、他の反応にとっての最適pHと一致しない。たとえば、酵素基質/反応物は別のpH値で最も溶解しやすい、という場合がある。もう1つの例として、複数の酵素を複数ステップのワンポット(one-pot)反応に用いる場合、それらの最適pHは一致しないことがある。それゆえ、固定化処理中に酵素の最適pHを変化させることができれば、これによって経済的および技術的な効果が得られるだろう。そこで、ラッカーゼの最適pHを、粒子上に固定化する場合と固定化しない場合とで測定した。
(酵素分析評価)
ラッカーゼ試薬は、50mMのBritton-Robinson汎用緩衝液の中に1mMのグアヤコールを含んでおり、当該緩衝液は目標とするpH値に調整してあった(Davies TJ, Banks CE, Nuthakki B, Rusling JF, France RR, Wadhawana JD, Compton RG(著)、2002年。(題名)“Surfactant-free emulsion electrosynthesis via power ultrasound: electrocatalytic formation of carbon-carbon bonds”、(誌名)Green Chem. 4:570-577)。汎用緩衝液を用いたのは、同じ緩衝液系が存在すること、そして広範囲のpHにわたって効果的に緩衝できることを確実にするためである。分析評価は、5 200M−1.cm−1の吸光係数で、450nmにおいて3回繰り返した。自由酵素ならびに支持体網目または格子の粒子に固定化されたラッカーゼについて、pH値プロフィールを実験で測定した。分析評価の実施には、PowerWave HT Microtitreプレートリーダを用いた。酵素の単位については、1分につき1μmolの基質を酸化させるのに必要な酵素の量を1単位とした。
(pH値プロフィールのシフト)
pHプロフィールのシフトは固定化処理中に起こることは公知であるため、繊維格子または網目主鎖支持体に結合されたラッカーゼについて、pHプロフィールを測定した。粒子は例3に従って作った。グルタルアルデヒドおよびPEIの濃度の変化が最適pHのシフトに与える影響についても、グルタルアルデヒド後処置(例2、A2−J2)の影響と同様に調査した。
(結果)
結果を図10(A)、(B)、図11(A)、(B)に示す。
【0061】
pH値プロフィールのシフトの全体的な傾向は、中性から弱アルカリpHに向かっている。本例が示すのは、酵素のpHプロフィールが、粒子への固定化によってシフトするということである。
(例15:ポリマー鎖/繊維のPEI網目または支持体格子の官能性)
粒子主鎖の官能性を変えることで、タンパク質の疎水性結合、イオン結合、そして親和性をベースとした結合を示すものにすることができる。
【0062】
粒子は、0.1mlのノノキシノール-4と0.5mlのポリエチレンイミン溶液(pH11)とを含む鉱物油10mlのエマルジョンと、0.1mlのノノキシノール-4と0.5mlのグルタルアルデヒド(Sigma 等級II)とを含む鉱物油10mlのエマルジョンとを混合することで製造した。混合前に、両エマルジョンを500rpmで30分間揺動した。そして、一緒にしたエマルジョンを500rpmで30分間揺動した。これにより、装飾されていない粒子が得られ、それを上述したように遠心分離処理で回収した。
(イオン結合)
イオン性基は、上記の修飾されていない粒子で生成され、その方法は、グルタルアルデヒドを用いて官能化し(25%の水溶液200μl、20mlの脱イオン水で3回洗浄)、その後、エチレンジアミン(0.33Mを1ml)を用いた処理を行うことで、自由なアルデヒド残基と反応させる(定期的にひっくり返しながら室温で1時間)、というものであった。粒子は、充分な量の脱イオン水で繰り返し洗浄し、2000×gで10分間遠心分離を行って回収した。ラッカーゼ(2.5mg)を5mgの修飾された粒子と、充分な混合が確実に行われるように定期的にひっくり返しながら、4℃で30分間インキュベートした。イオン結合を促進するために、1.0MのNaClを(対イオンとして)ラッカーゼ結合粒子に加え、ひっくり返して5分間混合した。粒子は、上述したのと同様に遠心分離によって回収し、活性度を測定した。
(疎水性結合)
修飾されていない粒子(5mg)をエポキシオクタン(0.1ml)と共に25℃で4時間インキュベートするやり方で、疎水基を粒子に追加した。その後、これらを充分な量の水で繰り返し洗浄し、上述したように遠心分離によって回収した。疎水結合されたタンパク質(蛍光菌リパーゼ、5mg)は、界面活性剤(1%、デオキシコレート)の追加によって取り出すことができた。280nmでのタンパク質吸光度を測るやり方で測定を行った。
(親和性結合)
タンパク質の親和性結合については例17に示す。
(結果)
(イオン結合)
修飾された粒子には、追加したラッカーゼの45%が結合された(Bio-Rad protein assay dye reagent method(500-0006)によって求めた値、メーカ側の記録による)。この45%のラッカーゼのうち76.9%は、対イオンとして塩(1.0M、NaCl)を追加することで取り出すことができた。これに対し、修飾されていない粒子はラッカーゼの14%としか結合せず、塩溶液によって取り出せたのは、その52.6%だけであった。
(疎水性結合)
修飾された粒子にタンパク質を結合した。修飾された粒子に結合されたタンパク質の約28%は、デオキシコレートを追加することで取り出されたが、これは、結合された酵素のうちの疎水性結合部分である。
【0063】
本例が示すのは、粒子マトリックスの官能性を変更することで、粒子マトリックスへのタンパク質結合の別メカニズムを実現できる、ということである。
(例16:化学伝達物質および補因子の共捕捉(co-entrapment))
補因子(または修飾された補因子や化学伝達物質)を含ませることで、架橋後、酵素と補因子との共補足が可能になる。処理条件を選択することにより、反応体(例:酵素基質、共基質、生成物、共生成物)などの小分子の出入りを許す一方で補因子を保持するよう、粒子の多孔度を決めることができる。
【0064】
アミノ酸デヒドロゲナーゼ(AADH)、ギ酸塩デヒドロゲナーゼ(凍結乾燥したもの)は、Biocatalytics社(米国)から購入した。PEG20000-NADHは、Julich Fine Chemicals社(ドイツ)から取得した。鉱物油は、Castrol社(ドイツ)から取得した。ノノキシノール-4は、ICI社(英国)から取得した。グルタルアルデヒド(Glut)(25%水溶液)は、Acros Organics社(ベルギー)から取得した。ギ酸は、Merck社(ドイツ)から取得した。エチレンジアミン(EDA)、ポリエチレンイミン(PEI)、3-メチル-2-オキソ酪酸(2-ケトバリン)、DL-バリン、NADHそしてNAD+は、Sigma Aldrich社から取得した。
【0065】
ポリエチレンイミン(PEI)をグルタルアルデヒドと架橋することで、ポリマー粒子を製造した。ポリエチレンイミンの油中水エマルジョンは、予め溶かした状態の200μlのノノキシノール-4を含む鉱物油40mlに10%のPEIを800μl乳化する方法で調製した(500rpmで撹拌する20mmの電磁撹拌器を用い、100mlビーカー内で20分間電磁撹拌した)。第2の油中水エマルジョンも同様に、20%のグルタルアルデヒド溶液を用いて調製した。2つのエマルジョンは、高速(700rpm)で撹拌しているポリエチレンイミンエマルジョンにグルタルアルデヒドエマルジョンを加えるやり方で混合した。これにより、連続的な撹拌で30分間反応させることができた。
【0066】
ポリマー粒子は、2000×gで10分間の遠心分離を行って回収した(Sorvall, RT7)。そのポリマー粒子を45mlの脱イオン水で4回洗浄した。洗浄作業の間に、先に述べた例と同様の遠心分離によって回収を実行した。そして、生成物は、10mlに再懸濁した。この溶液(1ml)をエッペンドルフチューブに等分して、以降の実験に使用した。
【0067】
10mg.ml−1を含んだ各タンパク質(ギ酸塩デヒドロゲナーゼ、バリンデヒドロゲナーゼ)のタンパク質溶液を調製した。その後、これら2つの溶液(各溶液200μl)を混合し、ポリマー材料とインキュベートした。この溶液は、逆さまにして混合し、緩やかに揺動しながら30分間反応させた。固定化酵素を含む粒子を、後述の方法を用いて、活性度測定のために分析評価した。
【0068】
粒子は、脱イオン水で洗浄し、上述したやり方で回収した。粒子は、100μlの PEG20000-NADH(ドイツ、ユーリッヒのJulich Fine Chemicals社から取得)と混合し、緩やかに揺動しながら室温で2時間インキュベートした。その後、この溶液を凍結乾燥した。凍結乾燥による生成物を、2mlの水で2回洗浄し、遠心分離によって回収した。それから、粒子を、100mgのリジンを含んだ100mMのTris-Cl緩衝液(pH8.0)1mlに再懸濁して、粒子上の余剰のアルデヒド官能基を抑制(quench)し、室温で1時間インキュベートした。そして、粒子を2mlのTris-Cl緩衝液(20mM、pH8.0)で5回洗浄した。その後、このサンプルに対しては、1mlの再利用試薬を用い、再利用能力に関する検査をした。サンプルは、サイクルとサイクルとの合間に2ml量の水で3回洗浄した。そしてサンプルを分析したが、これは、後述するやり方でアミノ酸TLCとHPLCとからバリンを製造するためである。
【0069】
これらの粒子を反応させ、再利用して、後続の反応のための新しい反応媒体とした。
(試薬の再利用)
バリン生成を目的としたPEG-20000-NADHの再利用のための試薬は、50mMのギ酸塩(ナトリウムギ酸塩1M株(stock)から出たもの、pH8.0)、50mMのTris-Cl緩衝液(pH8.0)、50mMの酒石酸アンモニウム、そして10mMの2-ケトバリンから成るものとした。この試薬の組成を、PEG NADHが再利用された場合に確実にバリンのみが生成されるように公式化(formulated)した。
(分析法)
アミノ酸TLCを、F254シリカゲルプレート(Merck社)で実施した。使用した移動相は、氷酢酸に対してエタノール9:1であった。アミノ酸(バリン)は、アセトン中にニンヒドリン2%の溶液を用いて染色した。バリン(Rf0.49)とアンモニア(Rf0.31)とが適当に分離したことが明らかに見て取れるまで、プレートを120℃で加熱した。
【0070】
HPLCは、アミノ酸の測定のために、OPA誘導化法(o-フタルアルデヒド試薬)を用いて実行した。サンプルは、インライン誘導化した。また、バリン標準(valine standards)を含めた。
(結果)
反応および再利用は、TLCデータによれば成功であり、連続的な粒子触媒反応の全てについてバリンスポット(valine spot)が形成された。6回の連続的な16時間反応サイクルのそれぞれにおいて、粒子として、10mMのケトバリンを48、35、59、43、33、29%のいずれかの割合でバリンに変換したものを用いた。これによって肯定的なTLC結果が確認され、HPLC OPA法を用いて定量化できた。
【0071】
この例が示すのは、酵素粒子マトリックスでの補因子の捕捉によって、酵素は補因子を利用して官能基を維持でき、そして、捕捉された補因子は再利用できる、ということである。
(例17:ポリマー鎖/繊維のPEI支持体網目または格子を有する粒子による抗体の結合)
本例で示すのは、粒子への抗体や抗原の結合である。抗体や抗原が結合された場合には、固定化された抗体または抗原を介したタンパク質の親和性結合に対する粒子の適合性が示される。さらに、診断用途(例:ELISA)に対する粒子の信頼性が示される。
【0072】
ポリエチレンイミン(P3143;50%水溶液)、等級IIのグルタルアルデヒド(G6257;25%水溶液)、鉱物油(M8410)、抗原マウスインターロイキン2(I0523−20UG;SL06092)、そして、Streptomyces avidinii由来のマーカー酵素ストレプトアビジン−ペロキシダーゼ(S5512−250UG;SL05181)は、Sigma-Aldrich社から取得した。主要な抗体であるラットアンチマウス(rat anti-mouse)インターロイキン2(IL−2)MAB(I7663−27K1; L6080801)と、補助的な抗体であるラットアンチマウスインターロイキン2(IL−2)ビオチンMAB(I7663−27M5;L6080803)とは、USBiologicals社から取得した。塩化ナトリウム(S7653)、二塩基のリン酸ナトリウム(S0876)、一塩基のリン酸ナトリウム(S0757)、過酸化水素(21676−3)、ツイン20(P9416)、そして塩酸(H1758)は、Signa Aldrich社から取得した。2,2'-アジノ-ビス(3-エチルベンズチアゾリン-6-スルホン酸)(10102946001)はRoche社から取得した。
(粒子の調製)
架橋ポリエチレンイミン粒子は、例2(サンプルG)に従って調製した。試薬に対して唯一行った調整は、ポリエチレンイミン溶液を10%に希薄する前にHCIでpH9にしたことである。実験は4倍に直接拡大して行った。従って、乳化した反応体それぞれ800μlと共に溶解したNP4を200μl含んだ20mlの鉱物油が必要であった。結果として生じる架橋ポリエチレンイミン粒子を、50mlの脱イオン水で6回洗浄し、洗浄と洗浄との合間に5000×gで5分間遠心分離する、というやり方で鉱物油から回収した。粒子ペレットは、その後、脱イオン水で10mlの量に再懸濁し、実験A〜Fに500μlずつ使用した。
(タンパク質の固定化)
各実験には、下の表9に示すように文字を割り当てた。各種のタンパク質の追加は、表9で実験の下側の欄に示す手順で実行した。各実験について、第1のタンパク質成分の固定化は、10分毎にサンプルを逆さまにして充分な混合が行われることを確実にしつつ、4℃の脱イオン水で1時間かけて実施した(表9の順次追加ステップの1)。粒子は、1mlの脱イオン水で3回洗浄し、上述したやり方で回収した。
【0073】
それに続くタンパク質結合、タンパク質処理(表9の第2列から第5列の処理)は、150mMの塩化ナトリウム(結合緩衝液)を含んだ10mMのリン酸塩緩衝液(pH6.8)で実施した。この結合は、充分な混合を確実にするために5分ごとにひっくり返しながら、37℃で30分間実行した。タンパク質結合が終わる度に、サンプルは、10分間緩やかに揺動させながら(IKA Vortex Genius使用、設定2)、ツイン20を0.05%含んだ1mlの結合緩衝液で2回洗浄し、非特異的なタンパク質相互作用を抑制した。順次行うステップの合間々々に、5000×gで5分間遠心分離を行って粒子を回収した。タンパク質処理の各ステップ(表9)に関して追加したタンパク質の量は、以下の通りである。アルブミンは5mg;ラットアンチマウスインターロイキン2MAB(A-IL2-MA)は50μg;ラットアンチマウスIL2MABビオチン(B-A-IL2-MA)は25μg;インターロイキン2(IL-2)は2ug;ストレプトアビジンペルオキシダーゼ(strep-perox)は10μg。これらの量のタンパク質を1mlの結合緩衝液で調製した。
(表9:実験A〜Fのタンパク質結合の手順)
【0074】
【表9】
(分析評価)
ペルオキシダーゼ分析評価試薬は、150mM塩化ナトリウムを含む10mMリン酸塩緩衝液(pH6.8)に、2mMのABTSと2mMの過酸化水素とを含んで成るものとした。分析評価値は3回繰り返して測定し、1ウェル(well)につき200μlの試薬と50μlの粒子懸濁液とを用い、30℃において、Powerwave HT マイクロプレート分光光度計(Biotek Instruments社)で420nmの値を計測した。
(結果)
抗原に対する抗体の結合は、実験B(図12)で評価した。この実験では、粒子表面への主要な抗体(A-IL2-MA)の結合、それに続く抗原(IL-2)の結合、粒子への補助的な抗体(B-A-IL2-MA)およびストレプトアビジン−ペロキシダーゼの結合について、結合が成功したと解釈しうる肯定的な反応が示された。これは、マイクロプレートのウェルなどの表面で実行されるsandwich-ELISAに類似している。実験Cは、抗原が粒子の表面に結合され、その後、抗体結合のための認識要素(recognition element)として用いられることを示す。これらの実験は、各種の対照例(control)と比較して見た場合、粒子を抗体または抗原の結合に用いることが可能であることを示している。
【0075】
実験Aは、主要な抗体が固定化された状態の粒子に対し、補助的な抗体(B-A-IL2-MA)またはストレプトアビジンペルオキシダーゼが非特異結合することを示すための対照例である。Bと比較して反応が低いことは、粒子に対する補助的な抗体の結合を抗原(IL-2)が強化することを示す。実験D,Eは、アルブミンを抑制処理(quenching)した後の粒子に対し、主要な抗体(A-IL2-MA)や補助的な抗体(B-A-IL2-MA)が非特異性結合することを示すための対照例である。実験Fは、非処理の架橋ポリエチレンイミン粒子に関するものであり、分析評価の対照用に用いた。
【0076】
本例は、粒子が、抗原または抗体にとって適当な固定化支持体であることを示す。更に強調しておきたい点は、抗原に対して抗体が、抗体に対して抗原が、それぞれ親和相互作用によって固定化されることである。従って、本例は、親和クロマトグラフィおよび診断法(酵素結合免疫吸着検査法など)に、当該支持体が適用できることを示している。
(例18:ポリマー鎖/繊維のPEI支持体網目または格子の粒子への磁鉄鉱の組み入れ)
化学伝達物質および補因子を粒子に含ませる処理についてはすでに示した。本例では、粒子に磁性粒子を含ませる処理について示す。
(粒子の調製)
粒子は、上記の例17に従って調製し、各エマルジョンについて鉱物油が25mlになるよう線形的に数値を大きくした。乳化の前に、10%のポリエチレンイミン溶液(pH9.0)に磁鉄鉱(250mg)を含ませた。架橋したポリエチレンイミン粒子を、50mlの量の脱イオン水で6回洗浄し、洗浄ステップの合間に、5000×gで5分間遠心分離して鉱物油から回収した。続いて、粒子ペレットを脱イオン水で10mlの量に再懸濁し、500μlのエチレンジアミンと30分間反応させることで、陰イオン交換のために官能化した。次いで、球体を、50mlに3等分した脱イオン水で洗浄し、上述したように遠心分離を行って回収した。最終的に得られたペレットは、脱イオン水で10mlに再懸濁し、後述するアルブミン結合実験に使用した。粒子の乾燥重量は、10mlの粒子懸濁液を1mlずつに等分したものを凍結乾燥および重量測定するやり方で、3回繰り返して測定した。
(タンパク質の結合および定量化)
上記の等分した懸濁液から得た粒子を、磁気スタンド(Magnetic Separation Stand (Promega社; Z5332))で磁気分離して回収し、液体は除去した。その粒子を2mlのTris-Cl緩衝液(pH7.4)(50mM)で2回洗浄して平衡化した。そして、粒子にウシ血清アルブミン(BSA)を加えて、最終的な濃度を20mg.ml-1とした。タンパク質のイオン結合を、室温において30分間、回転混合することで生じさせた。混合物は磁気スタンド内に配置して、磁化された樹脂を反応管の側壁に保持させた状態で、サンプルからの液体が除去されるようにした。樹脂は、50mMの Tris-Cl(pH7.4)1ml で5回洗浄し、前述した磁気保持器(retainer)によって回収した。50mM のTris-Cl(pH7.4)に入れた1MのNaCl500μlを2回追加することで、イオン結合されたBSAを樹脂から抽出した。
(タンパク質定量化)
溶出した分のタンパク質は、メーカの指示に従って、Qubit Fluorometer(Invitrogen社)で、Quant-iT分析評価を用いて定量化した(表10)。
(結果)
(表10:磁鉄鉱含有粒子へのアルブミンの結合(3回分データの平均))
【0077】
【表10】
この結果が示すのは、磁鉄鉱を粒子に取り込めば、粒子の磁気特性を利用して粒子を懸濁液から効果的に分離できる、ということである。更に、上記の結果は、本支持体が効率の良いイオン交換樹脂として使用できることを示している。ここで提示する例では、アミン(エチレンジアミン)などの正荷電分子で粒子マトリックスを修飾することで、粒子を陰イオン交換樹脂として使用することができる。また、カルボキシル含有分子などの負荷電分子を用いれば、陽イオン交換樹脂としての使用が可能である。
【0078】
本例は更に、格子内に磁性粒子を含ませる方式の別の回収方法の例を提供する。その方法では、磁場の利用によって粒子を引きつけることができる。
本発明の粒子は、架橋剤によって架橋されたポリマー鎖または繊維の格子と、隣接して繊維を囲む格子間の開口領域または空間とを有する。言い換えると、本発明が提供するのは、繊維相互貫入網目粒子であり、好ましい構成として、グルタルアルデヒド架橋したポリエチレンイミンから構成または製造されている。この粒子は、酵素固定化マトリックスとして利用できる。
【0079】
粒子は、本発明の第2、第3の様態によるエマルジョンベースのテクノロジーまたは技術を用いて製造するのが好ましい。エマルジョンベースのテクノロジーを用いれば、粒子のサイズに関する制御(例えば、粒子における体積と表面積との比率)や所定の粒径分布を実現できる利点があると共に、粒子を単一ステップで合成できるという効果もある。本粒子は、固定化に利用できる表面積が大きくなるため、大小の基質の生体触媒にも利用できる(ただし、それに限定はされない)。この酵素固定化マトリックスは、固定化効率が高く、固定後の酵素の活性度も高く保たれる。
【0080】
樹枝状粒子の繊維の性質として、酵素の結合に役立つ内部表面積が大きくなる。また、酵素サブユニットごとの利用可能な結合点の数が大きいため、タンパク質の安定化は、主鎖支持体の材料(例えば、PEIそれ自体)と比較した場合、大幅に改善する可能性がある。これを、ゆるい格子または網目と組み合わせた場合、生体触媒付加後の重量に対する活性度の比率を高めることができるが、それは、固定化に有用な露出表面積が大きいこと、そして、大小の基質について分散制約が限られていることによる。更に、粒径の制御によって、基質の分散制約をより小さくすることができる(それが本固定化マトリックスの障害となる場合)。
【0081】
粒子の調製方法には、同相で架橋剤が存在する場合、存在しない場合について、主鎖支持体または格子を乳化する処理が含まれる。上述の2エマルジョンシステムを製造に用いるのが望ましい。架橋剤が第1のエマルジョンに含まれない場合、架橋剤は、油相に溶かしてもよいし、前記架橋剤を含んだ第2のエマルジョンを混合する形で取り込んでもよい。
【0082】
好ましい製造方法には、エマルジョンの中で主鎖ポリマー(ポリエチレンイミン)を別個に乳化する処理が含まれるが、その場合は、第2のエマルジョンに少なくとも2官能基の架橋化学物質(グルタルアルデヒド)を入れておく。ポリマーと架橋剤との間の自然反応の結果、繊維の格子または網目が生じるが、この場合、余剰のアルデヒド官能基が含まれている。続いて、その余剰のアルデヒド官能基を用いて、アミンアルデヒド交差反応性を介したタンパク質の格子または支持体への自然発生的な共有結合連結が実現される。このアルデヒドの官能基については更に、別の化合物と連結すること、あるいは、疎水性などの他の特性をもたらすことにすれば、より広い範囲のタンパク質(例:疎水性タンパク質)の結合にまで用途を拡大するものと推測される。
【0083】
マトリックスにタンパク質を固定化することにより、酵素の溶媒安定性、熱安定性、pH安定性は高まる。こうした安定は、樹枝状支持体へのタンパク質固定化の後で支持体を乾燥させれば、更に強化される。この乾燥処理によって、交差反応性を有する化学官能基が近接する状況が少なくなるので、その結果、「タンパク質−主鎖」および「主鎖−主鎖」の更なる自然発生的な化学結合(chemical coupling)が引き起こされると考えられる。さらに、乾燥処理の間に添加物がマトリックスに捕捉されるが、これは、孔サイズの制御や、機能分子または補助剤の捕捉に役立つ可能性がある。本発明の粒子であれば、タンパク質担持を高めることができる(すなわち、小さな粒子体積に対して比較的大量のタンパク質を担持することができる)。
【特許請求の範囲】
【請求項1】
架橋剤によって架橋されたポリマー鎖で成る格子と、前記格子を成すポリマー鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有するエマルジョン由来粒子であって、
タンパク質または修飾タンパク質と反応することで、当該タンパク質または修飾タンパク質を格子に結合させ、固定化すること、
を特徴とするエマルジョン由来粒子。
【請求項2】
官能基は、前記格子を成すポリマー鎖に存在しており、タンパク質または修飾タンパク質のポリマーへの結合が、格子修飾を通して官能基を付与するように実現することができる共有結合、イオン結合、疎水結合、親和結合のうち1または複数で成されるように選択されたものであること、
を特徴とする請求項1に記載のエマルジョン由来粒子。
【請求項3】
官能基によって格子に結合され、それによって固定化されたタンパク質または修飾タンパク質を少なくとも1つ含むこと、
を特徴とする請求項1または2に記載のエマルジョン由来粒子。
【請求項4】
複数の異なるタンパク質または複数の異なる修飾タンパク質が内部で固定化されていること、
を特徴とする請求項3に記載のエマルジョン由来粒子。
【請求項5】
鎖状または繊維状のポリマーはポリエチレンイミンであること、
を特徴とする請求項1乃至4のいずれかに記載のエマルジョン由来粒子。
【請求項6】
格子内に捕捉された補助剤を含むこと、
を特徴とする請求項1乃至5のいずれかに記載のエマルジョン由来粒子。
【請求項7】
補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択されること、
を特徴とする請求項6に記載のエマルジョン由来粒子。
【請求項8】
第1の液相の液滴が第2の液相内に分散したエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中には溶解したポリマーおよび架橋剤が含まれるという状態で製造する処理と、
架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、
当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、
を特徴とする粒子生成方法。
【請求項9】
第1の液相の液滴が第2の液相内に分散した第1のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解したポリマーが含まれているという状態で製造する処理と、
第1の液相の液滴が第2の液相内に分散した第2のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解した架橋剤が含まれるという状態で、第1のエマルジョンと併せる処理と、
架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、
当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、
を特徴とする粒子生成方法。
【請求項10】
ポリマーはポリエチレンイミンであること、
を特徴とする請求項8または9に記載の粒子生成方法。
【請求項11】
液相の少なくとも1つに補助剤を加え、当該補助剤が粒子の格子内に捕捉された状態にする処理を有すること、
を特徴とする請求項8乃至10のいずれかに記載の粒子生成方法。
【請求項12】
粒子を回収し、回収した粒子を乾燥させる処理を有すること、
を特徴とする請求項8乃至11のいずれかに記載の粒子生成方法。
【請求項13】
回収された粒子に対し、当該粒子の乾燥処理の前に補助剤を加え、当該補助剤が粒子の格子内に捕捉された状態にする処理を有すること、
を特徴とする請求項12に記載の粒子生成方法。
【請求項14】
補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択されること、
を特徴とする請求項11乃至13のいずれかに記載の粒子生成方法。
【請求項1】
架橋剤によって架橋されたポリマー鎖で成る格子と、前記格子を成すポリマー鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有するエマルジョン由来粒子であって、
タンパク質または修飾タンパク質と反応することで、当該タンパク質または修飾タンパク質を格子に結合させ、固定化すること、
を特徴とするエマルジョン由来粒子。
【請求項2】
官能基は、前記格子を成すポリマー鎖に存在しており、タンパク質または修飾タンパク質のポリマーへの結合が、格子修飾を通して官能基を付与するように実現することができる共有結合、イオン結合、疎水結合、親和結合のうち1または複数で成されるように選択されたものであること、
を特徴とする請求項1に記載のエマルジョン由来粒子。
【請求項3】
官能基によって格子に結合され、それによって固定化されたタンパク質または修飾タンパク質を少なくとも1つ含むこと、
を特徴とする請求項1または2に記載のエマルジョン由来粒子。
【請求項4】
複数の異なるタンパク質または複数の異なる修飾タンパク質が内部で固定化されていること、
を特徴とする請求項3に記載のエマルジョン由来粒子。
【請求項5】
鎖状または繊維状のポリマーはポリエチレンイミンであること、
を特徴とする請求項1乃至4のいずれかに記載のエマルジョン由来粒子。
【請求項6】
格子内に捕捉された補助剤を含むこと、
を特徴とする請求項1乃至5のいずれかに記載のエマルジョン由来粒子。
【請求項7】
補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択されること、
を特徴とする請求項6に記載のエマルジョン由来粒子。
【請求項8】
第1の液相の液滴が第2の液相内に分散したエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中には溶解したポリマーおよび架橋剤が含まれるという状態で製造する処理と、
架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、
当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、
を特徴とする粒子生成方法。
【請求項9】
第1の液相の液滴が第2の液相内に分散した第1のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解したポリマーが含まれているという状態で製造する処理と、
第1の液相の液滴が第2の液相内に分散した第2のエマルジョンを、一方の液相は水相であり、他方の液相は油相であり、水相中に溶解した架橋剤が含まれるという状態で、第1のエマルジョンと併せる処理と、
架橋剤にポリマーの鎖を架橋させて粒子を形成する処理と、を有し、
当該粒子の各々は、架橋剤によって架橋されたポリマーの鎖で成る格子と、前記格子を成すポリマーの鎖に隣接および囲まれる形で存在する開口領域と、格子上に配された官能基とを有し、タンパク質または修飾タンパク質が当該粒子と反応し、それによって格子に結合され、固定化されること、
を特徴とする粒子生成方法。
【請求項10】
ポリマーはポリエチレンイミンであること、
を特徴とする請求項8または9に記載の粒子生成方法。
【請求項11】
液相の少なくとも1つに補助剤を加え、当該補助剤が粒子の格子内に捕捉された状態にする処理を有すること、
を特徴とする請求項8乃至10のいずれかに記載の粒子生成方法。
【請求項12】
粒子を回収し、回収した粒子を乾燥させる処理を有すること、
を特徴とする請求項8乃至11のいずれかに記載の粒子生成方法。
【請求項13】
回収された粒子に対し、当該粒子の乾燥処理の前に補助剤を加え、当該補助剤が粒子の格子内に捕捉された状態にする処理を有すること、
を特徴とする請求項12に記載の粒子生成方法。
【請求項14】
補助剤は、補因子、修飾補因子、化学伝達物質、磁鉄鉱または磁性物質から選択されること、
を特徴とする請求項11乃至13のいずれかに記載の粒子生成方法。
【図1】


【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図10】

【図11】

【図12】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2011−500099(P2011−500099A)
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願番号】特願2010−531623(P2010−531623)
【出願日】平成20年10月29日(2008.10.29)
【国際出願番号】PCT/IB2008/054458
【国際公開番号】WO2009/057049
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(506257272)
【氏名又は名称原語表記】CSIR
【住所又は居所原語表記】Scientia,0002 PRETORIA(ZA)
【Fターム(参考)】
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願日】平成20年10月29日(2008.10.29)
【国際出願番号】PCT/IB2008/054458
【国際公開番号】WO2009/057049
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(506257272)
【氏名又は名称原語表記】CSIR
【住所又は居所原語表記】Scientia,0002 PRETORIA(ZA)
【Fターム(参考)】
[ Back to top ]