
エリスロポエチンの多糖誘導体
【課題】EPO(エリスロポエチン)の安定性、薬物動態、及び薬力学の改善に有用である、新規なEPOの多糖誘導体を提供する。
【解決手段】EPO又はEPO様タンパク質の多糖誘導体である化合物であって、多糖がアニオン性であり、且つ2個〜200個の糖ユニットを含む、化合物。新規化合物を含む医薬組成物、及び新規化合物の製造方法。
【解決手段】EPO又はEPO様タンパク質の多糖誘導体である化合物であって、多糖がアニオン性であり、且つ2個〜200個の糖ユニットを含む、化合物。新規化合物を含む医薬組成物、及び新規化合物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なEPOの多糖誘導体及びかかる誘導体の製造方法に関する。誘導体は、EPOの安定性、薬物動態(pharmacokinetics)及び薬力学(pharmacodynamics)の改善に有用である。
【背景技術】
【0002】
エリスロポエチンは糖タンパク質ホルモンであると共に、骨髄の赤血球(erythrocyte
(red blood cell))前駆細胞に対するサイトカインである。ヘマトポエチン又はヘモポエチンとも呼ばれ、腎臓によって産生され、赤血球産生を調節する。
【0003】
エリスロポエチンは、身体における赤血球造血(erythropoiesis:赤血球形成)の主要な調節因子である(Martindale、1996)。組換えヒトエリスロポエチンは、エポエチンα及びエポエチンβとして知られる形態で市販されている。これらは、慢性腎不全患者における貧血の管理で使用される。エポエチンγも利用可能である。全て、同一の165アミノ酸配列を有するが、そのグリコシル化パターンが異なる。
【0004】
組換えヒトエリスロポエチンは、高血圧患者、発作歴のある患者、血小板増加症患者、慢性肝不全患者、虚血性血管疾患患者又は悪性腫瘍患者においては、慎重に使用するべきである。エポエチンα及びエポエチンβは、場合によってはグリコシル化及び市販製剤の配合の違いに起因して、その薬物動態が幾分異なる。エポエチンαは、皮下注射後の吸収が緩徐且つ不完全であり、静脈内投与に対する相対的バイオアベイラビリティは約10%〜50%と報告されている。エポエチンβも吸収が緩徐且つ不完全であり、その絶対的バイオアベイラビリティはおよそ40%と報告されている。
【0005】
EPOを誘導体化してその薬物動態特性を改善させる試みが為されている。EPOのポリエチレングリコール誘導体である、CERA(持続性エリスロポエチン受容体活性化剤(Constant Erythropoiesis Receptor Activator))として知られる、Rocheにより開発
中の製品がある。これは、EPOよりも長い半減期を有することが示されており、頻繁な注射の必要性を低減させる。さらなる新規赤血球造血刺激剤は、慢性腎疾患及び癌に随伴する貧血を治療するための新規PEG化合成ペプチド、Hematideである。これはFan他(2006)によってさらに記載されている。
【0006】
静脈内投与後の最終相半減期が組換えヒトEPO及び天然ホルモンよりもおよそ3倍長い、組換えヒトエリスロポエチンの高度にグリコシル化されたアナログである、Darbepoetin等、EPOの他の形態も開発されている。
【0007】
特許文献1には、循環における血漿中半減期が長期化した、微生物から製造されたEPOの修飾変異タンパク質が記載されている。無細胞タンパク質合成技法を使用することにより、PEG又は多糖等の修飾因子と反応し得る非天然アミノ酸を有する、EPOの変異タンパク質が製造される。概して、PEGは、EPOの変異タンパク質中の遊離のスルフヒドリル基と結合する。
【0008】
特許文献2は、EPOとPEGとのN末端複合体を対象とする。複合体は、長い循環半減期及び血漿中滞留時間を有する。
【0009】
特許文献3には、PEGと複合体を形成したEPOの製造方法の改善が記載されている。本発明者らは、rhEPO1分子当たり1個〜3個の直鎖PEG分子を有する複合体の
組成物が最も持続的な効力を示すことを見出した。
【0010】
特許文献4も、生物学的に活性なEPO複合体組成物を提供するという課題に取り組んでいる。EPOは、組成物の循環血清中半減期を増大させる有機分子と共有結合した非抗原性の親水性ポリマーと共有結合で複合体を形成する。水溶性ポリマーは、とりわけ、ポリアルキレンオキシド、ポリアミド又は糖質であり得る。
【0011】
しかしながら、ポリシアル酸(PSA)等のアニオン性多糖を用いたEPOの誘導体化について記載する公表された著作は今日まで全く存在しない。
【0012】
ポリシアル酸(PSA)は、或る特定の菌株により、また哺乳類の或る特定の細胞で産生されるシアル酸の天然非分岐ポリマーである。ポリシアル酸は、天然の細菌由来型ポリマーの、限定(limited)酸加水分解若しくはノイラミニダーゼによる消化、又は分画に
よって、n=約80以上のシアル酸残基から、最小でn=2のシアル酸残基までの各種重合度で製造することができる。
【0013】
近年、ポリシアル酸の生物学的特性、特にα−2,8結合ポリシアル酸ホモポリマーの生物学的特性が、タンパク質及び低分子量薬物分子の薬物動態特性を修飾するために活用されている。ポリシアル酸誘導体化により、多数の治療用タンパク質、例えば、カタラーゼ及びアスパラギナーゼの循環半減期が劇的に改善し、治療用タンパク質に対する先の曝露の望ましくない(且つ時には不可避の)結果として、既存抗体が増加するにもかかわらず、かかるタンパク質を使用することが可能である。α−2,8結合ポリシアル酸はPEGの魅力的な代替物であり、元々人体の一部であり、且つ組織ノイラミニダーゼにより、無毒性の糖であるシアル酸に分解する、免疫学的には不可視の生分解性ポリマーである。
【0014】
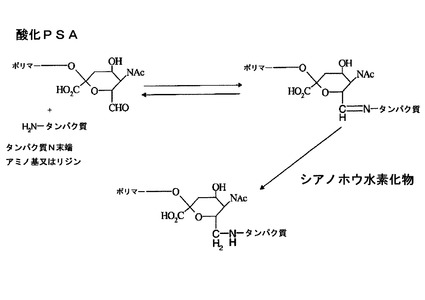
本発明者らは以前に、多糖(特にPSA)をタンパク質等の治療薬に結合させる方法を記載している[特許文献5、特許文献6]。これらの方法のうちの幾つかは、ポリマーの「非還元」末端を化学的に誘導体化して、第1級アミン基で反応するタンパク質反応性のアルデヒド部分を作り出すことに基づいている。非還元シアル酸末端ユニットは、隣接ジオールを含有するため、過ヨウ素酸塩を用いて容易に(且つ選択的に)酸化することにより、タンパク質に対する反応性が遥かに高く、且つ還元的アミノ化及び他の化学反応(chemistries:ケミストリー)によるタンパク質の結合に好適な反応要素を含む、モノアル
デヒド体を得ることができる。この反応を図1及び図2に示す。ここで、
図1は、過ヨウ素酸ナトリウムを用いたコロミン酸(大腸菌由来のα−2,8結合ポリシアル酸)の酸化により、非還元末端でタンパク質反応性アルデヒドが形成されることを示し、
図2は、シアノ水素化ホウ素ナトリウムを用いたシッフ塩基の選択的還元により、タンパク質のアミノ基との安定で不可逆的な共有結合が形成されることを示す。
【0015】
上記従来の複合体形成反応中に、例えば、コロミン酸がアミノ酸の側鎖と反応することによって、意図しない副生成物が生成する可能性がある。これは、ヒト及び動物の治療用途で監督機関によって要求される既知組成の複合体の製造において十分に厄介なものとなる可能性がある。
【0016】
反応生成物の殆どの物理化学的特性が類似しているため、意図する反応生成物(例えば、モノポリシアル化体)を各種の意図しない生成物と切り離して精製することは容易ではない。このことは、イオン交換クロマトグラフィ及びゲル浸透クロマトグラフィ(それぞれ電荷及びサイズに基づいて分離する)等の技法によっては、好ましい精製プロファイルが得られないことを意味する。この問題は、複合体形成反応における生成物の複雑度を低減させることによって克服することができる。本発明者らは、多糖とタンパク質との複合
体を形成する新規な方法であって、それによりタンパク質のN末端の高い反応性を利用することができ、且つタンパク質と過ヨウ素酸塩酸化した天然のコロミン酸との還元的アミノ化に関する確立された方法(図1及び図2)を使用して得られる生成物の複雑度を回避する、新規な方法を開発した。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】欧州特許第1219636号
【特許文献2】米国特許第7,128,913号
【特許文献3】米国特許出願公開第2004/0082765号
【特許文献4】米国特許第7,074,755号
【特許文献5】米国特許第5,846,951号
【特許文献6】国際公開第WO−A−0187922号
【発明の概要】
【発明が解決しようとする課題】
【0018】
従来技術を鑑みて、ヒト及び動物の治療で使用することができ、且つ安定性、半減期が最適化され、毒性の低い、改善されたEPO誘導体の提供が必要とされている。本発明者らは、PSA等の多糖をEPOと結合させることで、かかる性質が付与されることを見出し、それにより本発明に到達した。アニオン性多糖と結合したEPOを記載したのはこれが初めてである。
【課題を解決するための手段】
【0019】
本発明の第1の態様により、本発明者らは、EPO又はEPO様タンパク質の多糖誘導体である化合物であって、多糖がアニオン性であり、且つ2個〜200個の糖ユニットを含む、化合物を提供する。
【0020】
以下、「EPO」という用語を使用する場合、本発明者らは、EPO様タンパク質を包含することも意図している。EPO様タンパク質とは、EPOの活性と同等の活性を有するタンパク質を意味する。EPOは、上記で詳述したように赤血球産生を調節する。EPO又はEPO様タンパク質の活性は、Krystal(1983)に記載の標準的なアッセイを使用
して測定することができる。マウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料の活性を測定する。マウスは、事前にフェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしている。アッセイにおいて、EPOを赤血球前駆細胞に加え、DNA複製速度を、3H−チミジン取り込み速度を求めることによって測定する。タンパク質は、NIBSC由来の標準EPOと比較して、10%〜200%の複製速度を誘導する場合、「EPO様」として分類される。典型的には、EPO様タンパク質は、標準EPOの活性の少なくとも35%、好ましくは、標準EPOの活性の少なくとも50%を有する。
【0021】
上記で詳述した、必要な活性を有するEPOの変異体も使用し得る。「EPO様」タンパク質は、「EPOホモログ(homologue)」とも称され得る。2個の配列が相同である
かどうかは、当該技術分野において既知の用語である、%類似性又は%同一性を使用してルーチン的に計算される。配列は、スイスプロット(swissprot)アクセッション番号P
O1588のヒトEPO前駆体である、配列番号1と比較するものとする。活性EPOは、この配列の28残基〜193残基である。EPOホモログ配列は、配列番号1の全体と比較してもよく、又はその28残基〜193残基と比較してもよい。好ましくは、EPOホモログ配列は、活性EPO、すなわち、28残基〜193残基と比較される。
【0022】
本発明において、ホモログは、50%以上の類似性又は同一性(核酸レベル又はアミノ
酸レベル)、好ましくは60%、70%、80%以上、より好ましくは90%以上、例えば、95%又は99%の同一性又は類似性(アミノ酸レベル)を有する。類似性又は同一性の計算には多数のプログラムが利用可能であるが、好ましいプログラムは、BLASTnプログラム、BLASTpプログラム及びBLASTxプログラムであり、初期設定パラメータで実行され、www.ncbi.nlm.nih.govで利用可能である。例えば、2個のアミノ酸配列は、初期設定パラメータ(スコア=100、ワード長=11、期待値=11、低複雑度フィルタリング=オン)でBLASTnプログラムを使用して比較し得る。上記の相同性のレベルは、これらの初期設定パラメータを使用して計算し得る。
【0023】
EPOは、グリコシル化されてもよく、又はグリコシル化されなくてもよい。EPOがグリコシル化される場合、化合物は、典型的には2個〜100個の糖ユニットを含む。より典型的には、化合物は、10個〜80個の糖ユニット、好ましくは20個〜60個の糖ユニット、最も好ましくは40個〜50個の糖ユニットを含む。
【0024】
EPOがグリコシル化されない場合、化合物は、典型的には80個〜180個の糖ユニット、好ましくは100個〜150個の糖ユニット、より好ましくは120個〜145個の糖ユニット、最も好ましくは130個〜140個の糖ユニットを含む。
【0025】
好ましくは、アニオン性多糖は、少なくとも2個、より好ましくは少なくとも5個、最も好ましくは少なくとも10個、例えば少なくとも50個の糖ユニットを有する。
【0026】
アニオン性多糖は、好ましくは、ポリシアル酸、ヘパリン、ヒアルロン酸及びコンドロイチン硫酸から選択される。好ましくは、多糖はポリシアル酸であり、実質的にシアル酸ユニットのみから成る。しかしながら、多糖は、分子中にシアル酸以外のユニットを有し得る。例えば、シアル酸ユニットを他の糖ユニットと交互にしてもよい。しかしながら、好ましくは、多糖は、実質的にシアル酸のユニットから成る。
【0027】
好ましくは、化合物は、EPO又はEPO様タンパク質のN末端誘導体である。すなわち、多糖は、そのN末端でEPOと結合する。本明細書中において、N末端での誘導体化とは、EPOのN末端アミン基での誘導体化を意味する。しかしながら、代替的には、多糖は、中間鎖(mid-chain)アミノ酸、例えば、リジン、システイン、アスパラギン酸、
アルギニン、グルタミン、チロシン、グルタミン酸又はヒスチジンの側鎖で、EPO又はEPO様タンパク質と結合し得る。典型的には、側鎖は、リジンアミノ酸又はシステインアミノ酸の側鎖である。
【0028】
好ましくは、多糖は末端シアル酸基を有し、上記で詳述したように、より好ましくは、α−2,8結合又はα−2,9結合を通じて相互に結合した少なくとも2個のシアル酸ユニットを含む多糖である、ポリシアル酸である。好適なポリシアル酸の重量平均分子量は2kDa〜50kDaの範囲、好ましくは5kDa〜50kDaの範囲である。最も好ましくは、ポリシアル酸は、細菌源、例えば、大腸菌KI、N.meningitidis、Maraxella liquefaciens若しくはPasteurella aeruginosaの多糖B、又は大腸菌K92株由来のK92多糖に由来する。最も好ましくは、大腸菌K1に由来のコロミン酸である。
【0029】
ポリシアル酸は、塩又は遊離酸の形態であり得る。細菌源からの回収後に分子量が低下した、加水分解形態であってもよい。
【0030】
好ましくは、ポリシアル酸である多糖は、分子量が広範な(例えば多分散度が1.3超、例えば2以上にもなる)物質であり得る。好ましくは分子量の多分散度は1.3未満、より好ましくは1.2未満、例えば1.1未満である。多分散度は低いと1.01にもな
り得る。
【0031】
EPOは、2個以上のアニオン性多糖を用いて誘導体化され得る。例えば、EPOは、そのN末端及び内部のアミノ酸側鎖の両方で誘導体化され得る。例えば、リジン、システイン、アスパラギン酸、アルギニン、グルタミン、チロシン、グルタミン酸、セリン及びヒスチジンの側鎖が、アニオン性多糖によって誘導体化され得る。EPOはグリコン(glycon)ユニットでも誘導体化され得る。しかしながら、本発明の好ましい実施形態において、EPOはそのN末端のみで誘導体化される。
【0032】
本明細書中において、N末端での誘導体化とは、EPOのN末端アミン基での誘導体化を意味する。
【0033】
本発明の第1の態様による化合物は、EPOとアニオン性多糖とが共有結合した複合体であり得る。多糖とEPOとの間を結び付ける他の手段としては静電引力が挙げられる。しかしながら、共有結合が好ましい。EPOは、そのN末端アミノ酸で多糖と共有結合し得る。共有結合は、カルボキシル基とアミン基との間のアミド結合であり得る。EPOが多糖と共有結合することができる別の結合は、シッフ塩基を介する。アミンとの複合体形成に好適な基は、国際公開第WO2006/016168号にさらに記載されている。
【0034】
本発明において、多糖は、天然多糖又は天然多糖の誘導体、例えば、糖残基上での1個又は複数の活性基の反応によって誘導体化されているか、又は多糖鎖の末端で誘導体化基と共有結合している多糖であり得る。
【0035】
多糖は、その還元末端ユニット又は非還元末端ユニットのいずれかを介してEPOと結合し得る。このことは、1個の多糖鎖が、2個のEPOタンパク質と結合し得る、すなわち、その還元末端及び非還元末端の両方で誘導体化され得ることを意味する。
【0036】
多糖をタンパク質と結合させる方法は、当該技術分野において既知であり、国際公開第WO92/22331号及び特許文献6において、より詳細に記載されている。本発明における好ましい方法を以下でより詳細に説明する。方法は、本出願の図1及び図2にも記載されている。
【0037】
多糖は、EPO又はEPO様タンパク質と直接、すなわち、図1及び図2に示すように、又はリンカーを介して結合し得る。好適なリンカーは、N−マレイミド、ビニルスルホン、N−ヨードアセトアミド、オルトピリジル又はN−ヒドロキシコハク酸イミドを含有する試薬に由来する。リンカーはまた、生物学的に安定である(biostable)か、又は生
分解性であり、例えば、ポリペプチド又は合成オリゴマーを含み得る。リンカーは、国際公開第WO2005/016973号でさらに記載されるように、二官能性部分に由来し得る。好適な二官能性試薬は、例えば、Bis−NHSである。試薬は、一般式Z−R1−Z(式中、各Zは官能基であり、同一であってもよく、又は異なっていてもよく、且つR1は二官能性有機基である)を有し得る。好ましくは、R1は、アルカンジイル、アリーレン、アルカリーレン、ヘテロアリーレン及びアルキルヘテロアリーレン(これらは任意でカルボニル結合、エステル結合、スルフィド結合、エーテル結合、アミド結合及び/又はアミン結合によって置換及び/又は介在され得る)から成る群より選択される。特に好ましくは、C3〜C6アルカンジイルである。最も好ましくは、R1は、好適な二官能性試薬の適切な部分に相当する。
【0038】
本発明の好ましい化合物は、一般式(I):
【0039】
【化1】

【0040】
(式中、mは、少なくとも1であり、
XBは、EPO又はEPO様タンパク質であるB−XHに由来し(ここで、XHは、NH2又はSHである)、
Lは、結合、連結基であるか、又はポリペプチド若しくは合成オリゴマーを含み、
GlyOは、アニオン性糖ユニットであり、
結合基は、存在すれば、一般式−Y−C(O)−R1−C(O)−であり、Yは、NR2又はNR2−NR2であり、且つR1は、上記で規定した二官能性有機基であり、且つR2は、H又はC1−6アルキルである)
のものである。
【0041】
本発明の本態様において、EPOは、多糖の非還元末端と結合する。末端多糖ユニットはシアル酸ユニットである。多糖中の他の糖ユニットは、GlyOで表され、同一であってもよく、又は異なっていてもよい。好適な糖ユニットとしては、ヘパリン、ヒアルロン酸及びコンドロイチン硫酸が挙げられる。
【0042】
EPOが多糖と直接結合する場合、L基は結合である。しかしながら、L基は代替的には、N−マレイミド、ビニルスルホン、N−ヨードアセトアミド、オルトピリジル又はN−ヒドロキシコハク酸イミドを含有する試薬に由来し得る。試薬は、上記で規定したように、一般式Z−R1−Zを有し得る。本実施形態において、Lは典型的には、基
【0043】
【化2】
【0044】
である。
【0045】
好ましくは、XHはNH2であり、且つEPO又はEPO様タンパク質のN末端である。代替的には、NH2はリジンアミノ酸側鎖の第1級アミンである。異なる実施形態において、XHはシステインアミノ酸の側鎖のチオール基、SHである。
【0046】
本発明の別の態様は、上記で規定したような新規化合物及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物である。
【0047】
医薬組成物は、水性懸濁液の形態であってもよい。水性懸濁液は、水性懸濁液の製造に好適な賦形剤と混合した新規化合物を含有する。医薬組成物は、無菌の注射可能な水性懸濁液又は均質懸濁液の形態であってもよい。懸濁液は、好適な分散剤又は湿潤剤及び懸濁剤を使用する既知の技術に従って製剤化され得る。
【0048】
医薬組成物は、ヒト用途又は家畜(veterinary)用途で、経口投与、静脈内投与、腹腔内投与、筋肉内投与、皮下投与、鼻腔内投与、皮内投与、局所投与又は気管内投与され得る。
【0049】
組成物は製剤添加物をさらに含み得る。製剤添加物とは、Wang他(1999)に記載されている、内的に又は外的にEPOを安定化させることが可能な賦形剤を意味する。賦形剤は、安定化剤、可溶化剤又は金属イオンであり得る。製剤添加物の好適例としては、1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸が挙げられる。これらは単独で使用してもよく、又は組み合わせて使用してもよい。
【0050】
安定化剤は、典型的には、タンパク質のアンフォールディングに対するギブス自由エネルギー変化の増大を引き起こすタンパク質の変性状態の不安定化により作用する。安定化剤は、好ましくは糖又はポリオール、例えばスクロース、ソルビトール、トレハロース、グリセロール、マンニトール、ラクトース及びエチレングリコールである。安定化バッファーはリン酸ナトリウムである。
【0051】
可溶化剤は、好ましくは界面活性剤、好ましくは非イオン性界面活性剤である。好適例としては、Tween 80、Tween 20、Tween 40、Pluoronic F68、Brij 35及びTriton X100が挙げられる。
【0052】
金属イオンは、好ましくは二価である。好適な金属イオンとしては、Zn2+、Ni2+、Co2+、Sr2+、Cu2+及びFe2+が挙げられる。
【0053】
製剤添加物はまた、ヒト血清アルブミン、PSA、PEG又はヒドロキシ−β−シクロデキストリンから選択されるポリマーであり得る。
【0054】
製剤添加物としての使用に好適なアミノ酸及びアミノ酸誘導体としては、ヒスチジン、グリシン、他の類似のアミノ酸及びアスパラギン酸ナトリウムが挙げられる。
【0055】
本発明の別の態様は、EPO又はEPO様タンパク質のアニオン性多糖誘導体の集団を含む、組成物であって、誘導体が2個〜125個の糖ユニットを含み、且つ集団が実質的にタンパク質のN末端誘導体のみから成る、組成物である。「集団」とは、組成物中に2個以上の多糖誘導体が存在することを意味する。誘導体は、同一であるか又は異なる数の糖ユニットを含み得る。好ましくは、組成物中の多糖の多分散度は、1.3未満、より好ましくは1.1未満である。好ましい多糖は、上記本発明の他の態様に関して詳述した通りである。
【0056】
集団において、EPOの実質的に全てがN末端のみで誘導体化される。このことは、集団内のタンパク質の85%、好ましくは少なくとも90%、最も好ましくは少なくとも95%がPSAによりN末端のみで誘導体化されることを意味する。
【0057】
N末端での誘導体化の程度は、当該技術分野において既知の技法、例えばペプチドマッピング又はエドマン分解によって求められ得る。
【0058】
本発明のさらなる態様は、治療で使用するための上記化合物である。
【0059】
本発明の最後の態様により、本発明者らは、EPO又はEPO様タンパク質の多糖誘導体の製造方法であって、2個〜200個の糖ユニットを含むアニオン性多糖が、EPO又はEPO様タンパク質と化学的に反応する、製造方法を提供する。
【0060】
なお、本発明の本態様において、多糖は、EPO又はEPO様タンパク質上の任意の基で反応し得る。例えば、多糖は、アミン基、ヒドロキシル基、カルボキシル基又はスルフ
ヒドリル基と反応し得る。好ましくは、基は、アミン基、より好ましくは末端アミン基である。アミンは、代替的には、アミノ酸、例えばリジンアミノ酸のアミン側鎖であり得る。多糖はまた、EPO上の任意の糖質残基、例えばペンダントグリコン基で反応し得る。
【0061】
多糖は、当該技術分野で既知の方法によってアミノ酸側鎖と結合し得る。例えば、多糖は、in vitroでのカップリングによってAsp又はGluのC末端、−COOH又はカルボキシル側鎖とカップリングし得る。システインアミノ酸のチオール基もin vitroでのカップリングによって多糖と結合し得る。これらの方法は、国際公開第WO03/055526号、特に第6頁及び第7頁の表にさらに記載されている。この参考文献において、in vitroでのカップリングは、オリゴ糖部分とGlnの側鎖上のアミド基との結合にも使用されている。in vitroでのArg残基及びHis残基のイミダゾール基もそれぞれ記載されている。これらの方法をそれぞれ使用して、本発明のEPOを誘導体化し得る。
【0062】
多糖はEPOの修飾形態とも反応し得る。例えば、EPO上の1個又は複数の基は、例えば、還元又は酸化による化学変換を受けた可能性がある。例えば酸化条件を使用して、EPOの末端アミノ基の代わりに反応性カルボニルを生成することができる。
【0063】
本発明の方法での使用に好適な多糖は、新規化合物について先に記載した通りである。
【0064】
本発明の化合物は、従来技術で記載した好適な方法のいずれかによって製造し得る。例えば、典型的な方法は、本発明者らの先の国際特許出願第WO92/22331号に記載されている。
【0065】
典型的には、アニオン性多糖は、EPOに対する誘導体化の前に活性化されている。例えば、反応性アルデヒド基を有する可能性があり、誘導体化反応が還元条件下で実行され得る。反応性アルデヒド基は、多糖のヒドロキシル基の制御酸化によって生成し得る。最も好ましくは、この反応性アルデヒドは予備段階(ここで多糖を、例えば、水溶液中で過ヨウ素酸ナトリウムを使用する制御酸化条件下で反応させる)で生じる。好ましくは、酸化は化学酸化であるが、この段階を実行することが可能な酵素も使用され得る。反応性アルデヒド基は、多糖の非還元末端又は還元末端に存在し得る。その後、EPO、典型的にはN末端が反応性アルデヒド基と反応することにより、還元時にEPOのN末端誘導体を生成する付加体を得ることができる。
【0066】
多糖の活性化は、好ましくは多糖の中間鎖切断が実質的に無い、すなわち、実質的に分子量が低減しない条件下で実行されるべきである。酸化剤は、好適には過ルテニウム酸塩、又は好ましくは過ヨウ素酸塩である。酸化は、1mM〜1Mの範囲の濃度の過ヨウ素酸塩を用い、3〜10の範囲のpH、0℃〜60℃の範囲の温度で、1分〜48時間の範囲の時間、実行され得る。
【0067】
誘導体化反応に好適な還元条件は、水素と触媒、又は好ましくはホウ水素化物(borohydride)等の水素化物を利用し得る。これらは固定化してもよい(例えば、Amberli
te(商標)−担持型ホウ水素化物)。好ましくは、水素化ホウ素ナトリウム等のアルカリ金属水素化物が、1μM〜0.1Mの範囲の濃度、5.0〜10の範囲のpH、0℃〜60℃の範囲の温度、及び1分〜48時間の範囲の期間で、還元剤として使用される。出発原料上のペンダントカルボキシル基が還元されない反応条件が選択される。他の好適な還元剤は、酸性条件下でのシアノホウ水素化物(cyanoborohydride)、例えば、ポリマー担持型シアノホウ水素化物又はアルカリ金属シアノホウ水素化物、L−アスコルビン酸、メタ重亜硫酸ナトリウム、L−セレクトリド、トリアセトキシホウ水素化物等である。
【0068】
多糖の他の活性化誘導体、例えば、本発明者らの先の国際特許出願第WO06/00540号に記載されている、NHS等のペンダント官能基を有するものは、本発明において有用であり得る。
【0069】
一実施形態において、反応性アルデヒドは、多糖の還元末端に存在し、非還元末端は、EPO上のペンダント基と反応しないように不動態化(passivated)されている。
【0070】
コロミン酸の還元末端の反応性は、タンパク質標的に対しては弱いものの、化学的に規定された複合体の製造においては十分に厄介である。
【0071】
多糖の還元末端に反応性アルデヒドを有する多糖を調製するのに好適な化学反応は、本発明者らの先の国際特許出願第WO05/016974号に記載されている。この工程は、予備的な選択的酸化段階、続いての還元、及びその後のさらなる酸化により、還元末端にアルデヒドを有し、且つ非還元末端が不動態化された化合物を得ることを包含する。
【0072】
国際公開第WO2005/016973号には、タンパク質との複合体形成に有用なポリシアル酸誘導体、特に遊離のスルフヒドリル薬物を有するものが記載されている。ポ0リシアル酸化合物をヘテロ二官能性試薬と反応させることにより、スルフヒドリル基との部位特異的複合体形成のためのペンダント官能基を導入する。本発明で使用するアニオン性多糖も、このようにヘテロ二官能性試薬を用いて誘導体化され得る。
【0073】
多糖は、EPOと反応する前に誘導体化され得る。例えば、多糖は二官能性試薬と反応し得る。
【0074】
多糖は、国際公開第WO2006/016168号でさらに記載されているように、予備反応段階(ここで、第1級アミン基、第2級アミン基及びヒドラジンから選択される基が、好ましくはシアル酸である末端糖上に形成される)と、続く反応段階(ここで、これが二官能性試薬と反応することにより、反応中間体が形成される)に付され得る。次いで、中間体はEPOと反応し得る。二官能性試薬は、先に規定したように、一般式Z−R1−Zを有し得る。
【0075】
本発明者らは、或る特定の反応条件が、EPOのN末端での選択的誘導体化を促進することを見出した。N末端での選択的反応を促進するために、誘導体化反応は、酸性pHの第1の水溶液中で実行すべきであり、次いで得られた多糖誘導体は、第1の水溶液よりも高いpHの第2の水溶液中で精製すべきである。典型的には第1の水溶液のpHは4.0〜6.0の範囲であり、第2の水溶液のpHは6.5〜9.0、好ましくは6.5〜8.5又は6.5〜8.0の範囲である。誘導体化反応のpHが低いと、任意の中間鎖部位ではなく、タンパク質のN末端での選択的誘導体化が促進される。
【0076】
さらに、本発明者らは、或る特定の製剤添加物を使用すると、選択的で安定的な多糖EPO誘導体の形成が促進されることを見出した。製剤添加物は、1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸から選択され得る。これらは反応媒体に添加してもよく、又は代替的には安定化剤として最終製品組成物に添加してもよい。
【0077】
本発明の一実施形態において、製剤添加物は、ソルビトール、トレハロース又はスクロースである。異なる実施形態において、製剤添加物は、非イオン性界面活性剤である。製剤添加物は代替的にはPSA、PEG又はヒドロキシ−β−シクロデキストリンから選択されるポリマーであり得る。異なる実施形態において、製剤添加物は、二価金属イオンである。好ましい二価金属イオンとしては、Zn2+、Ni2+、Co2+、Sr2+、F
e2+、Mg2+又はCa2+が挙げられる。
【0078】
製剤添加物はバッファーであり得る。好ましくは、製剤添加物がバッファーの場合、それはリン酸ナトリウムである。
【0079】
本発明の方法における多糖誘導体の精製は、当該技術分野において既知の種々の方法を使用して実行され得る。好適な精製方法の例としては、HIC(疎水性相互作用クロマトグラフィ)、SEC(サイズ排除クロマトグラフィ)、HPLC(高速液体クロマトグラフィ)、AEC(陰イオン交換クロマトグラフィ)及び金属アフィニティクロマトグラフィが挙げられる。
【0080】
分子量分布の広いポリシアル酸の集団を、多分散度が低めの画分、すなわち、異なる平均分子量の画分に分画し得る。分画は、好ましくは本発明者らの先の国際特許出願第WO2005/016794号及び同第WO2005/03149号に記載されているように、好適な塩基性バッファーを溶出に使用する陰イオン交換クロマトグラフィによって実施される。この分画方法は、多糖出発原料及び誘導体に好適である。したがって、この技法は、本発明の必須の工程段階の前後に適用され得る。好ましくは、得られるEPOの多糖誘導体の多分散度は1.3未満、より好ましくは1.2未満、最も好ましくは1.1未満である。
【0081】
本発明によるEPOの誘導体化の結果、タンパク質の半減期が増大し、安定性が改善し、免疫原性が低減し、且つ/又は溶解度が制御される。それゆえ、EPOのバイオアベイラビリティ及び薬物動態特性が改善する。モノポリシアル化EPO複合体を作製する新規な方法には特別な価値がある。
【0082】
本発明を、実施例1〜実施例3.12によって、また添付の図面を参照して説明する。
【図面の簡単な説明】
【0083】
【図1】従来技術の非還元シアル酸末端ユニットの活性化を示す反応スキーム図である。
【図2】タンパク質のN末端又はランダム誘導体化を示す反応スキーム図である。
【図3a】Triple Detection GPC(Viscotek:RI+RALS+Viscosiometer)を使用する、異なるpHでの24kDaのコロミン酸(CA)の分解を示す図である。
【図3b】CAポリマーのゲル浸透クロマトグラフィを示す図である。
【図4】SDS−PAGEによる、PEG化EPO及びポリシアル化EPOの特徴付けを示す図である。
【図5】SDS−PAGE(右手側)及びSE−HPLC(左手側)による、ポリシアル化EPOの特徴付けを示す図である。
【図6】SE−HPLCによる、EPO、ポリシアル化EPO及びPEG化EPOの特徴付けを示す図である。
【図7】EPOに関するFACSデータ(網状赤血球数)を示す図である。
【図8】EPO製剤のin vivoでのクリアランスを示す図である。
【図9】EPO製剤のin vivoでのクリアランスを示す図である。
【図10】SE−HPLCによる、EPO−CA複合体の特徴付けを示す図である。
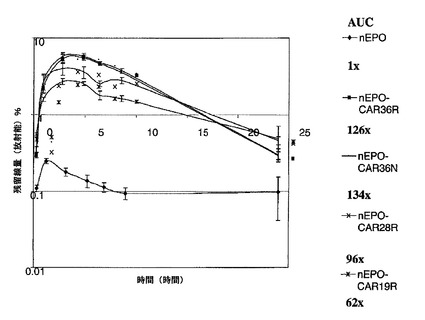
【図11】非グリコシル化EPO対ポリシアル化非グリコシル化EPOのin vivoでのクリアランスを示す図である。
【図12】非グリコシル化EPO対ポリシアル化非グリコシル化EPOのin vivoでのクリアランスを示す図である。
【図13】SE−HPLC及びSDS−PAGEによる、NG−EPO−CA複合体の特徴付けを示す図である。
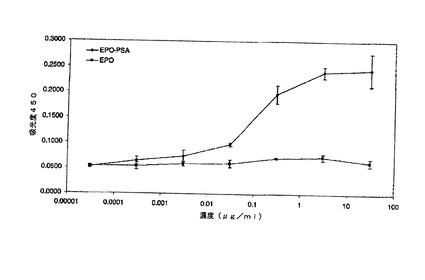
【図14】サンドイッチELISAによるEPO−PSAの検出を示す図である。
【図15】試薬希釈液中のEPO−PSAに対するELISAの感度を示す図である。
【図16】サイズ排除HPLCによるEPO複合体の安定性を示す図である。
【図17】ポリシアル化EPOのSDS−PAGEを示す図である。
【図18】EPO製剤のin vivoでの効力を示す図である(n=3〜4±SE)(非近交系雌性マウス;12週齢;SC(アルデヒドケミストリー))。
【図19】EPO製剤のin vivoでの効力を示す図である(雌性ウィスターラット;8週齢〜9週齢;n=5±SEM)。
【図20】SE−HPLCによる、NG EPOのPEG化を示す図である。
【実施例】
【0084】
材料
炭酸アンモニウム、エチレングリコール、ポリエチレングリコール(8KDa)、シアノ水素化ホウ素ナトリウム(sodium cyanoborohydride)(純度98%超)、メタ過ヨウ素
酸ナトリウム及び分子量マーカー、硫酸アンモニウム、塩化ナトリウム、リン酸ナトリウム、ソルビトール、Tween 20並びにびTrisは、Sigma Chemical Laboratory
、英国から入手した。酢酸ナトリウム及びリン酸ナトリウムは、BDH、英国から入手した
。使用するコロミン酸、直鎖α−(2,8)−結合大腸菌K1ポリシアル酸(平均22.7kDa、高多分散度1.34、39kDa、多分散度1.4;11kDa、多分散度1.27)は、Camida、アイルランド及びS.I.I.L. India Ltd.から入手した。他の材料と
しては、2,4−ジニトロフェニルヒドラジン(Aldrich Chemical Company、英国)、透析管(3.5KDa及び10KDaカットオフ限界;Medicell International Limited、英国)、Sepharose SP HiTrap、PD−10カラム、Q FF[カラム1ml又は5ml];Hitrap Butyl HPカラム[1ml又は5ml](Pharmacia、英国)、Tris−グリシンポリアクリルアミドゲル(4%〜20%及び8
%〜16%)、Tris−グリシンドデシル硫酸ナトリウムランニングバッファー及びローディングバッファー(Novex、英国)が含まれた。脱イオン水は、Elgastat
Option 4水精製ユニット(Elga Limited、英国)から入手した。使用した全ての試薬は分析用(analytical grade)である。プレートリーダー(Dynex Technologies、英国)をタンパク質アッセイ及びCAアッセイにおける分光光度定量に使用した。マウス及びラットは、Harlan、英国から購入し、使用前の少なくとも1週間馴化させた。EPOはSIIL、インドから入手した。
1.タンパク質及びコロミン酸の定量
他で記載されているように[Gregoriadis他、1993;Fernandes及びGregoriadis、1996
、1997]、レゾルシノール法[Svennerholm、1957]によって、レゾルシノール試薬を用
いた(シアル酸としての)ポリシアル酸の定量的評価を実行した。タンパク質を、BCA比色法又は280nmでのUV吸光度によって測定した。
2.1.コロミン酸の活性化
新たに調製した0.02Mメタ過ヨウ素酸ナトリウム(NaIO4)溶液(8倍モル過剰)を20℃でCAと混合し、反応混合物を暗所で15分間、磁気的に撹拌した。次に、2倍容量のエチレングリコールを反応混合物に加えて、過剰のNaIO4を消費し、混合物をそのまま20℃でさらに30分間撹拌した。酸化したコロミン酸を、4℃の0.01%炭酸アンモニウムバッファー(pH7.4)に対して長時間(extensively)(24時
間)透析した(3.5KDa分子量カットオフ透析管)。限外濾過(分子量カットオフ3.5kDaに対する)を使用して、透析管からCAO溶液を濃縮した。必要容量まで濃縮した後、濾液を凍結乾燥し、さらに使用するまで−40℃で保存した。代替的には、CAOを、エタノールによる沈殿(2回)によって反応混合物から回収した。
2.2.CA及び誘導体の酸化状態の定量
カルボニル化合物との相互作用で難溶性の2,4−ジニトロフェニルヒドラゾンを生じる、2,4−ジニトロフェニルヒドラジン(2,4−DNPH)を用いて、コロミン酸酸化度の定性的評価を実行した。非酸化コロミン酸(CA)/酸化コロミン酸(CAO)を2,4−DNPH試薬(1.0ml)に加え、溶液を振盪した後、結晶性の沈殿物が観察されるまで37℃で放置した[Shriner他、1980]。CAの酸化度(定量的)を、アルカ
リ性溶液中でのフェリシアン化物イオンのフェロシアン化第二鉄(プルシアンブルー(Persian blue))への還元に基づく方法[Park及びJohnson、1949]を用いて測定した(こ
れは次に630nmで測定される)。この場合、グルコースを標準として使用した。
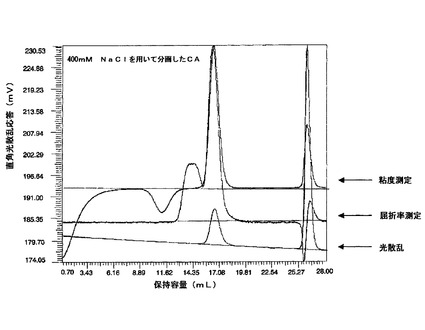
2.3.ゲル浸透クロマトグラフィ
コロミン酸試料(CA及びCAO)をNaNO3(0.2M)、CH3CN(10%;5mg/ml)に溶解し、2×GMPWXLカラムでクロマトグラフィにかけ、屈折率により検出した(GPC系:VE1121 GPC溶媒ポンプ、VE3580 RI検出器及びTrisec 3ソフトウェアによる照合、Viscotek Europe Ltd)。試料(5mg
/ml)を0.45μmのナイロン膜で濾過し、移動相として0.2M NaNO3 及びCH3CN(10%)を用いて0.7cm/分で流した。
【0085】
結果を図3b並びに第5表及び第6表に示す。
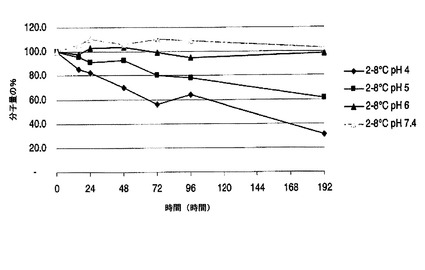
2.4.コロミン酸の安定性
PEG化の化学反応に関するルールをそのままポリシアル化に応用することができないのは、これらの分子の生理化学的特性が異なるからである。PSAは、酸に不安定なポリマーであると共に、中性付近のpHでは何週間も安定である(図3a)。図3aの結果から、pH6.0及び7.4では、CAは8日間安定であり、pH5.0では緩徐に分解し(48時間後、初期分子量の92%)、pH4.0では緩徐に分解する(48時間後、初期分子量の70%)ことが示される。ポリシアル酸は親水性が高いが、PEGは両親媒性である。PEG化に使用される条件を使用してポリシアル化を実行すると、多くの場合、タンパク質の凝集及び沈殿が観察される。
3.製剤添加物を含むN末端タンパク質−CA複合体の調製
3.1.EPO−CA複合体(N末端法)の調製
EPOを溶液(10mMリン酸ナトリウムバッファー、130mM NaCl、pH7.0中、0.34mg/ml;比活性:110,000U/mg、分子量:30600)
として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を、2mlのエッペンドルフチューブに採取した。必要量(25倍モル過剰)のコロミン酸を取り、タンパク質溶液を固体CAに加え、緩やかに混合した。反応混合物中で50mM又は3.17mg/mlとなるように、必要容量のシアノ水素化ホウ素ナトリウム溶液を加え、ボルテックスし、最終反応混合物のpHをチェックした(必要に応じて、pHを6.0に調整する)。チューブをシールし、所望の温度で24時間撹拌するか、又は反応混合物を最初に室温(22℃)で8時間インキュベートした後、4℃±1℃で一晩(14時間)保持した。インキュベーション後、必要な試料を採取した(例えば、活性アッセイ、SDS−PAGE、SE−HPLC用)。
3.1.1.EPO−CA複合体の精製及び特徴付け(N末端法)
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で希釈し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中、試料を氷上(4℃±1℃)に保持した。
【0086】
タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光
度は約0.743である)。SDS−PAGE用の試料を採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を陰イオン交換クロマトグラフィ(AEC)を使用して実施した。AECでは、タンパク質を含有するHIC画分をAEC バッファーA(50mM
Tris、150mM NaCl、pH8.0)(試料1ml+AECバッファーA 5ml)で希釈し、1.0ml/分でAECバッファーAを用いて事前に平衡化したECカラムにローディングした。ローディング画分を回収及び標識した。カラムをAECバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファーB(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0087】
代替的には、精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaの場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.
743である)。SDS−PAGE用の試料を採取した。
【0088】
一定分量をタンパク質アッセイ及びCAアッセイ用に取り出した。残りの溶液は、使用するまで−20℃で保存した。生成物をSDS−PAGEによって特徴付けした。フェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしたマウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料及びNG EPO試料の活性の定量を使用した。プロトコルは、Krystal(1972)により報告されている方法
に基づき改作した。アッセイは、赤血球前駆細胞へのEPOの添加及び3H−チミジン取り込み速度の定量によるDNA複製速度の測定に基づく。B6D2F1マウスにおいて、in vivoでの薬物動態(PK)試験及び薬力学(PD)試験を行った。
3.2.EPO−CA複合体(ランダム)の調製
EPOを溶液(10mMリン酸ナトリウムバッファー、130mM NaCl、pH7.0中、0.34mg/ml;比活性:110000U/mg、分子量:30600)として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を2mlのエッペンドルフチューブに採取した。必要量のコロミン酸を取り、タンパク質溶液を固体CAに加え、緩やかに混合した。反応混合物中で50mM又は3.17mg/mlとなるよ
うに、必要容量のシアノ水素化ホウ素ナトリウム溶液を加え、ボルテックスし、最終反応混合物のpHをチェックした(必要に応じて、pHを7.4に調整する)。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。インキュベーション後、必要な試料を採取した(例えば、活性アッセイ、SDS−PAGE、SE−HPLC用)。
3.2.1.EPO−CA複合体(ランダム)の精製及び特徴付け
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で溶解し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、洗浄画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中、試料を氷上(4℃±1℃)に保持した。
【0089】
タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を陰イオン交換クロマトグラフィ(AXC)を使用して実施した。AXCでは、タンパク質を含有するHIC画分をAXCバッファーA(50mM
Tris、150mM NaCl、pH8.0)(試料1ml+AXCバッファーA 5ml)で希釈し、1.0ml/分でAXCバッファーAで事前に平衡化したAXCカラムにローディングした。ローディング画分を回収及び標識した。カラムをAXCバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファー(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0090】
代替的な精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaの場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.74
3である)。SDS−PAGE用の試料を採取した。
3.3.グリコンケミストリー
ヒドラジドコロミン酸を、最終CA濃度が10mMとなるようにEPO溶液に溶解した。溶液のpHを5.5に調整した。必要容量のNaIO4溶液のNaOAc溶液を、最終濃度が5mM NaIO4となるように加えた。NaHSO3(NaHSO3の最終濃度20mM)を用いて反応を停止した。反応混合物を室温でインキュベートした。最後に、必要容量のNaCNBH3溶液のNaOAc溶液を、最終濃度が50mM NaCNBH3となるように加えた。振盪器上で反応を4℃±1℃で1時間継続した。インキュベーション後、SDS、SE−HPLC、活性アッセイに必要な試料を採取した。
3.3.1.EPO−CA複合体の精製及び特徴付け(グリコンケミストリー)
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で希釈し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した(L1〜Lx)。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中は、試料を氷上(4℃±1℃)に保持した。
【0091】
タンパク質濃度をUV(276nm)によって分析した(1mg/mlのEPOの吸光度は約0.743であった)。試料をSDS−PAGE用に採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を、陰イオン交換クロマトグラフィ陰イオン交換(AXC)を使用して実施した。AXCでは、タンパク質を含有するHIC画分をAXCバッファーA(50mM Tris、150mM NaCl、pH8.0)で希釈し(試料1ml+AXCバッファーA 5ml)、1.0ml/分でAXCバッファーAで事前に平衡化したAXCカラムにローディングした。ローディング画分を回収及び標識した。カラムをAXCバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファー(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0092】
代替的な精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaである場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。
3.4.EPOのPEG化:
EPO(30.6kDa)を溶液(5%ソルビトール、0.025mg/mlポリソルベート80を含有する10mM酢酸ナトリウムバッファー、pH4.0中、0.954m
g/ml)として供給し、2℃〜8℃で保存した。EPO溶液を濃縮して、約1.0mg/mlの溶液とした。必要量のEPOをエッペンドルフチューブに取り、氷上に載置した
。複合体形成用に添加したPEGの量は、式:
PEGの重量={タンパク質の量(g)/(タンパク質の分子量)}×(PEGの分子量)×(PEGのモル過剰)
に基づいて計算した。
【0093】
必要量のPEG 20Kを秤量した。10mM NaOAc、5%ソルビトール、pH5.5(ここで使用するのは、最終反応容量の20%容量)中に可溶化し、混合物を、全てのPEGが溶解するまで緩やかにボルテックスした後、濾過して新しいエッペンドルフに入れるか、又は4000rpmで5分間遠心し、上清を新しいエッペンドルフに移すことにより、いかなる凝集/沈殿物質も除去した。PEGが25倍モル過剰となるように、必要量のEPOをタンパク質溶液をPEG溶液に加え、4℃±1℃で緩速振盪器上に反応混合物を保持することによって緩やかに混合した。最終反応混合物中で50mM又は3.17mg/mlとなるように、必要容量の100mg/ml NaCNBH3溶液を加え、緩やかに混合し、最終反応混合物のpHをチェックし、必要に応じて、4℃±1℃の1M NaOH/HClを用いて5.5に調整した。最後に、反応の容量を、反応混合物中のタンパク質濃度が1mg/mlとなるように、20mM NaOAC、5%ソルビトー
ル、pH5.5を使用して調整した。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。反応を適切な方法によって停止し、in vitro活性、SDS−PAGE(4%〜20%Tris−グリシンゲルを使用)、SE−HPLC(superose 6カラム)用の試料を採取し、反応混合物のpHをチェックした。いかなる沈殿物も除去するために、SE−HPLC分析及び精製前に反応混合物を13000rpmで5分間遠心した。好ましいSE−HPLC用バッファーは0.1Mリン酸ナトリウム(pH6.9)であった。結果を図5に示す。
3.5.N末端非グリコシル化エリスロポエチン(NG EPO−CA)複合体の調製
NG EPOを溶液(20mMリン酸ナトリウムバッファー、300mM NaCl、pH6.65中、0.18mg/ml;比活性:100000U/mg;分子量:19000)として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を2mlのエッペンドルフに採取した。複合体形成に必要なコロミン酸(例えば、酸化コロミン酸又は非酸化コロミン酸)の量を計算した。必要量のコロミン酸を秤量した。タンパク質溶液を固体CAに加え、穏やかに混合した。反応混合物中のシアノ水素化ホウ素ナトリウムの最終濃度が50mM又は3.17mg/mlとなるように、必要容量のシアノ水素化ホウ素ナトリウム溶液を、反応混合物に加えた。最終反応混合物をボルテックスし、pHをチェックした(必要に応じて、pHを7.4に調整した)。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。インキュベーション後、活性アッセイ、SDS−PAGE、SE−HPLC等に必要な試料を採取した。
3.5.1.NG EPO−CA複合体の精製及び特徴付け
残りの反応混合物試料をHICバッファーA(1.2M硫酸アンモニウム、pH6.3)で希釈し(試料1ml+バッファーA 4ml)、HICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA(少なくとも10ml)で洗浄した。洗浄画分を回収し標識した。生成物をHICバッファーBで溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのnEPOの吸光度は約0.743であった)。試料をSDS−PAGE用に採取した。反応条件により、反応混合物中に有意な遊離のNG EPOが存在しなかったため、さらなる精製は不必要であった。NG
EPOが反応混合物中に存在した場合には、タンパク質を含有するHIC画分をVivaspin 6(5000 MWCO)を使用して濃縮し、SE−HPLCによって精製を行った。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのNG
EPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。
【0094】
一定分量をタンパク質アッセイ及びCAアッセイ用に取り出した。残りは、使用するまで−20℃で保存した。生成物をSDS−PAGEによって特徴付けした。
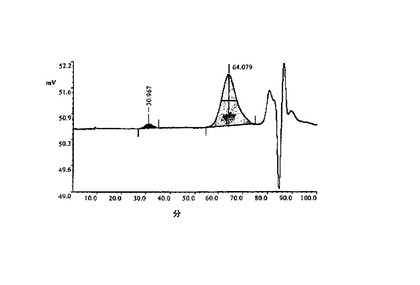
3.6.EPO製剤のSE−HPLC
4℃で冷却したJascoのAS−2057 plusオートサンプラー及びJascoのUV−975 UV/VIS検出器を備えたLiquid Chromatograph(Jasco)でHPLCを実施した。データをIBM/PC上のEZchrom Eliteソフ
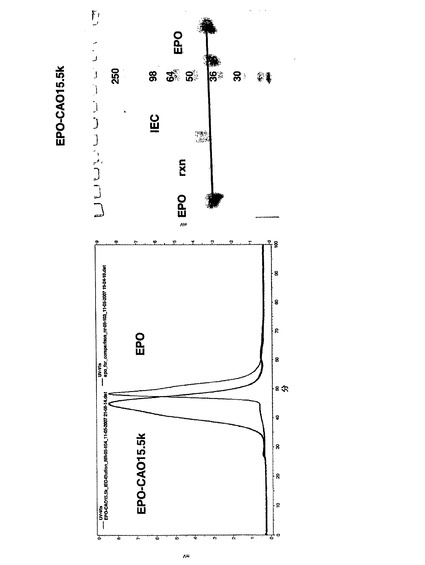
トウェアによって記録した。SEC試料を、Superose 6カラムで、0.1Mリン酸Na、pH6.9の定組成移動相を用いて分析した(図5)。図6は室温での唯一のピーク=76.408を示し、これはEPOに帰属される。
【0095】
図5の左手側に示すSECのピーク表は以下の通りである:
【0096】
【表1】
【0097】
3.7.SDSポリアクリルアミドゲル電気泳動、ウエスタンブロッティング&ELISA
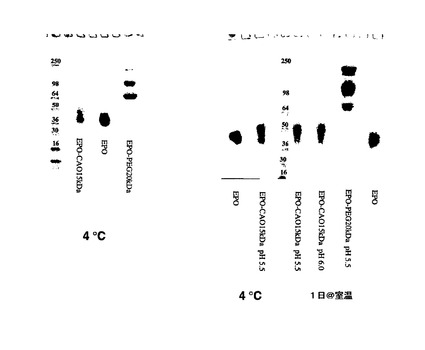
4%〜20%のTris−グリシンゲルを使用して、SDS−PAGEを実施した。試料を、還元バッファー又は非還元バッファーのいずれかで希釈し、タンパク質5.0μgを各ウェルにローディングした。ゲルをトリグリセリンバッファー系で試験し、Coomassie Blueで染色した。抗PSA抗体を使用してウエスタンブロッティングを実施した(図4)。図4はEPO製剤のSDS−PAGEを示す(部位特異的、N末端)。
3.8.in vitroでの活性
フェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしたマウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料の活性の定量を使用した。プロトコルは、Krystal[1972]により報告されている方法に基づき改作
した。アッセイは、赤血球前駆細胞へのEPOの添加及び3H−チミジン取り込み速度の定量によるDNA複製速度の測定に基づく。
3.9.安定性試験
無菌のEPO複合体を20mMリン酸ナトリウム、pH7.4;5%ソルビトール及び0.025mg/ml Tween 20中で、4℃で6週間保存した。下記条件の下、SECカラムを使用して試料のSE−HPLCを実施した:注入容量:100μl、流速:0.250ml/分、ランニングバッファー:0.1Mリン酸ナトリウム、pH:6.9。
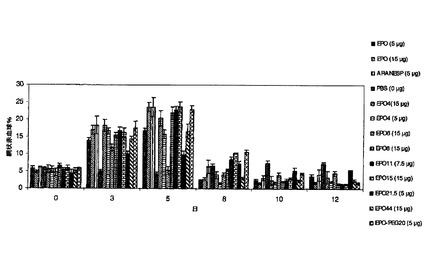
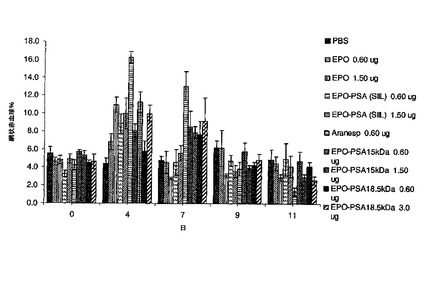
3.10.EPO製剤のin vivoでの効力
EPO製剤のin vivoでの効力を、雌性マウスB6D2F1(7週齢〜8週齢)で試験し、5μg〜15μgのタンパク質用量(同一活性)をマウスに皮下注射した。これらの動物は、4匹ずつ7つの群に分けた。以下のようにEPO製剤を各群の各動物に与えた:EPO、EPO−PSA複合体、PBS、Aranesp(5μg)。各動物から血液50μlを採取し、retic count染料で染色した後、FACSによって分
析した(図8及び図9)。
【0098】
十分に混合した全血5μlを、Retic−Count試薬1mlと混合し、暗所で室温で30分間インキュベートした。その後、試料をFACS機器の助けを借りて網状赤血球を計数することによって分析した。
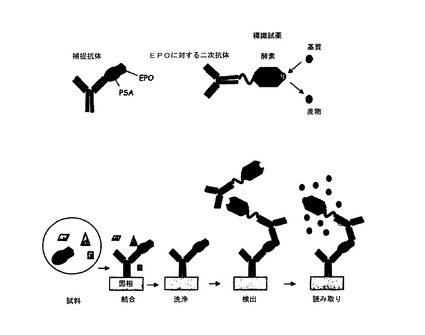
3.11.Elisa
EPO−PSAをプレート全体にコーティングされた抗PSA抗体によって捕捉した。捕捉されたEPO−PSAを抗EPO抗体を用いて検出し、EPOとPSAとの複合体のみを検出した。
3.12.in vivoでのクリアランス
EPO製剤のin vivoでのクリアランスをマウスで試験した。適切量のタンパク質用量をマウスに皮下注射及び静脈内注射した。EPO製剤を125Iで放射標識し、血液試料の放射能を頻繁に測定した。
結果
CAの活性化及び酸化度の定量
N−アセチルノイラミン酸(Neu5Ac)残基の直鎖α−2,8−結合ホモポリマーである、コロミン酸(CA)を使用した。コロミン酸の酸化への曝露を室温で20mM過ヨウ素酸塩を使用して15分間実行した。過ヨウ素酸塩処理後の内部α−2,8結合Neu5Ac残基の完全性をゲル浸透クロマトグラフィによって分析し、酸化コロミン酸(CAO)に対するクロマトグラフが得られ、物質を天然のCAの物質と比較した。酸化CA及び天然CAがほぼ同一の溶出プロファイルを示すことが分かり、逐次酸化段階がポリマー鎖の有意な断片化を引き起こすという証拠は無かった。
【0099】
CAの酸化状態の定量測定を、標準としてグルコースを使用して、フェロシアン化物(プルシアンブルー)へのアルカリ性溶液中でのフェリシアン化物イオンの還元[Park及びJohnson、1949]によって実施した。第1表は、酸化コロミン酸が、化学量論量を上回る
(100%超の)量の還元物質、すなわち、112mol%の見かけ上のアルデヒド含量(還元末端のヘミケタール及び(他の末端、非還元末端に)導入されたアルデヒドの還元力の組合せを含む)を有することが分かった。
【0100】
【表2】
【0101】
EPO複合体の調製、精製及び特徴付け
低pH(pH5.5)及びランダムpH(7.4)並びに4℃±1℃で反応を行うことによってN末端選択的にエリスロポエチン(EPO)のコロミン酸(CA)複合体を調製
及び精製する手順は、上記に詳述されている。これには、シアノ水素化ホウ素ナトリウムの存在下での複合体形成、続く遊離のEPOを除去するためのイオン交換クロマトグラフィ(AEX)を使用する精製、その後の疎水性相互作用クロマトグラフィ(HIC)によるCAの除去が包含される。N末端のα−アミノ基の選択的誘導体化が優勢となるように、また反応中のEPOの凝集を最小限にするために低pHを使用した。最終反応バッファーの組成は、10mM NaOAc中、5%ソルビトール、0.5mg/ml Tween 20(pH5.5)であった。
【0102】
EPO−CA複合体の形成をSE−HPLC(EPOと比較しての、EPO−CAの保持時間の変化;両方の部分が同時に溶出もする);イオン交換クロマトグラフィ(AECカラムへの複合体の結合)及びポリアクリルアミドゲル電気泳動(SDS−PAGE;高分子量種へ向かってのバンドの広がり及びシフト)によって確認した(図4)。ポリシアル化試料は、in vitroで活性であり、未処理のEPOよりも著しく優れたプロファイル(PK及びPD)を示した。図11及び図12はin vivoでの結果を示す。
【0103】
図5の左手側は、24時間後のEPO−CA 39kDa複合体形成のSE−HPLCを示す。第3表はピーク解析表である。特徴付け条件:カラム:Superdex 200、バッファー:炭酸水素アンモニウム0.15M、pH:7.8。
【0104】
【表3】
【0105】
誘導体化の程度は、ポリシアル化EPOよりもPEG化EPOの方が大きいことが分かった(図6)。これは、PEGの不活性な性質及びシアル酸の帯電した性質に起因する可能性がある。FACSデータからの網状赤血球数は、EPOよりもPSA−EPO複合体の方が多かった(図7)。EPO−CAO複合体の精製後、SEC HPLCからのクロマトグラム上に有意なEPOは観察されず、誘導体化は、SE−HPLCでの保持時間の変化並びにSDS−PAGE上の高分子量バンドの広がり及びシフトによって示された。EPOのポリシアル化は、マウス抗PSA抗体を使用するウエスタンブロッティングによっても示された。静脈内投与では、PSA−EPO複合体のin vivoでのクリアランスプロファイルは、EPOと比べて優れていることも分かり(図8)、曲線下面積は7.1倍増大した。同様にEPO−PSAの皮下投与もEPOと比較してより大きな保持を示し、曲線下面積は2.5倍増大した。EPOのポリシアル化が反応混合物のインキュベーション時間、モル過剰及びpHに比例することも分かった。ポリシアル化NG EPOのin vivoでのクリアランスプロファイル(静脈内及び皮下)は、NG EPOよりも良好であることが分かった(図11及び図12)。幾つかのSDSゲルにおいて、EPOのジポリシアル化も観察された。EPO−PSA複合体は、ELISA法によっても確認した(図14及び図15)。赤血球造血現象は、EPOと比較してEPO−PSA複合体の方が大きいことが分かり、6KDa〜15KDaではポリマーの分子量に比例することが分かった(図18)。シアル酸の鎖が大きくなるにつれて赤血球造血の現象は低減するため、15KDaがEPOに最適な鎖長であることが分かった。これは、受容体から
の反発で生じるポリマーの負に帯電した性質に起因する可能性がある。この試験は、図18の助けを借りて確認した。AranespのEmaxは、臨床的に良好ではなく、血栓症をもたらし、骨髄枯渇の原因となり得るEPO−PSAよりも遥かに大きいことが分かり、処置後、ベースライン未満まで網状赤血球を低下させることも分かった。EPO−PSAは、EPOよりも著しく優れていることが分かり、EPO−PSAは、EPO−PEGと同様に優れていることが分かり、EPO−PSAも一定の赤血球造血をもたらす。
【0106】
PSA複合体が、in vitro活性アッセイにおいて活性であることが分かった。PSA−EPO複合体がPEG複合体と同様に良好であり、EPOよりもはるかに優れていることがin vivoでの効力試験から示される(図8及び図9)。
【0107】
NG EPO−CA複合体の形成をSE−HPLC(NGEPOと比較しての、NG EPO−CAの保持時間の変化;両方の部分が同時に溶出もする);イオン交換クロマトグラフィ(AECカラムへの複合体の結合)及びポリアクリルアミドゲル電気泳動(SDS−PAGE;高分子量種のバンドのシフト)によって確認した。図は、24時間後のEPO−CA 39kDa複合体形成を示す。ポリシアル化試料は、in vitroで活性であり、未処理のNGepoよりも著しく優れたプロファイル(PK及びPD)を示した。
【0108】
図10は、SE−HPLCの結果を示す。下記第4表にピーク解析を示す。特徴付け条件−カラム:Superdex 200、バッファー:炭酸水素アンモニウム0.15M、pH:7.8。
【0109】
【表4】
【0110】
図12は、in vivoでのクリアランスの結果を示す。PSA−NG EPOは、NG EPOと比べて、著しく優れたプロファイルを示した。
【0111】
第5表は、使用した各種パラメータの値を示し、第6表は、CA画分の分子量及び多分散度を示す。
【0112】
【表5】
【0113】
【表6】
【0114】
参照文献
【0115】
【表7】
【技術分野】
【0001】
本発明は、新規なEPOの多糖誘導体及びかかる誘導体の製造方法に関する。誘導体は、EPOの安定性、薬物動態(pharmacokinetics)及び薬力学(pharmacodynamics)の改善に有用である。
【背景技術】
【0002】
エリスロポエチンは糖タンパク質ホルモンであると共に、骨髄の赤血球(erythrocyte
(red blood cell))前駆細胞に対するサイトカインである。ヘマトポエチン又はヘモポエチンとも呼ばれ、腎臓によって産生され、赤血球産生を調節する。
【0003】
エリスロポエチンは、身体における赤血球造血(erythropoiesis:赤血球形成)の主要な調節因子である(Martindale、1996)。組換えヒトエリスロポエチンは、エポエチンα及びエポエチンβとして知られる形態で市販されている。これらは、慢性腎不全患者における貧血の管理で使用される。エポエチンγも利用可能である。全て、同一の165アミノ酸配列を有するが、そのグリコシル化パターンが異なる。
【0004】
組換えヒトエリスロポエチンは、高血圧患者、発作歴のある患者、血小板増加症患者、慢性肝不全患者、虚血性血管疾患患者又は悪性腫瘍患者においては、慎重に使用するべきである。エポエチンα及びエポエチンβは、場合によってはグリコシル化及び市販製剤の配合の違いに起因して、その薬物動態が幾分異なる。エポエチンαは、皮下注射後の吸収が緩徐且つ不完全であり、静脈内投与に対する相対的バイオアベイラビリティは約10%〜50%と報告されている。エポエチンβも吸収が緩徐且つ不完全であり、その絶対的バイオアベイラビリティはおよそ40%と報告されている。
【0005】
EPOを誘導体化してその薬物動態特性を改善させる試みが為されている。EPOのポリエチレングリコール誘導体である、CERA(持続性エリスロポエチン受容体活性化剤(Constant Erythropoiesis Receptor Activator))として知られる、Rocheにより開発
中の製品がある。これは、EPOよりも長い半減期を有することが示されており、頻繁な注射の必要性を低減させる。さらなる新規赤血球造血刺激剤は、慢性腎疾患及び癌に随伴する貧血を治療するための新規PEG化合成ペプチド、Hematideである。これはFan他(2006)によってさらに記載されている。
【0006】
静脈内投与後の最終相半減期が組換えヒトEPO及び天然ホルモンよりもおよそ3倍長い、組換えヒトエリスロポエチンの高度にグリコシル化されたアナログである、Darbepoetin等、EPOの他の形態も開発されている。
【0007】
特許文献1には、循環における血漿中半減期が長期化した、微生物から製造されたEPOの修飾変異タンパク質が記載されている。無細胞タンパク質合成技法を使用することにより、PEG又は多糖等の修飾因子と反応し得る非天然アミノ酸を有する、EPOの変異タンパク質が製造される。概して、PEGは、EPOの変異タンパク質中の遊離のスルフヒドリル基と結合する。
【0008】
特許文献2は、EPOとPEGとのN末端複合体を対象とする。複合体は、長い循環半減期及び血漿中滞留時間を有する。
【0009】
特許文献3には、PEGと複合体を形成したEPOの製造方法の改善が記載されている。本発明者らは、rhEPO1分子当たり1個〜3個の直鎖PEG分子を有する複合体の
組成物が最も持続的な効力を示すことを見出した。
【0010】
特許文献4も、生物学的に活性なEPO複合体組成物を提供するという課題に取り組んでいる。EPOは、組成物の循環血清中半減期を増大させる有機分子と共有結合した非抗原性の親水性ポリマーと共有結合で複合体を形成する。水溶性ポリマーは、とりわけ、ポリアルキレンオキシド、ポリアミド又は糖質であり得る。
【0011】
しかしながら、ポリシアル酸(PSA)等のアニオン性多糖を用いたEPOの誘導体化について記載する公表された著作は今日まで全く存在しない。
【0012】
ポリシアル酸(PSA)は、或る特定の菌株により、また哺乳類の或る特定の細胞で産生されるシアル酸の天然非分岐ポリマーである。ポリシアル酸は、天然の細菌由来型ポリマーの、限定(limited)酸加水分解若しくはノイラミニダーゼによる消化、又は分画に
よって、n=約80以上のシアル酸残基から、最小でn=2のシアル酸残基までの各種重合度で製造することができる。
【0013】
近年、ポリシアル酸の生物学的特性、特にα−2,8結合ポリシアル酸ホモポリマーの生物学的特性が、タンパク質及び低分子量薬物分子の薬物動態特性を修飾するために活用されている。ポリシアル酸誘導体化により、多数の治療用タンパク質、例えば、カタラーゼ及びアスパラギナーゼの循環半減期が劇的に改善し、治療用タンパク質に対する先の曝露の望ましくない(且つ時には不可避の)結果として、既存抗体が増加するにもかかわらず、かかるタンパク質を使用することが可能である。α−2,8結合ポリシアル酸はPEGの魅力的な代替物であり、元々人体の一部であり、且つ組織ノイラミニダーゼにより、無毒性の糖であるシアル酸に分解する、免疫学的には不可視の生分解性ポリマーである。
【0014】
本発明者らは以前に、多糖(特にPSA)をタンパク質等の治療薬に結合させる方法を記載している[特許文献5、特許文献6]。これらの方法のうちの幾つかは、ポリマーの「非還元」末端を化学的に誘導体化して、第1級アミン基で反応するタンパク質反応性のアルデヒド部分を作り出すことに基づいている。非還元シアル酸末端ユニットは、隣接ジオールを含有するため、過ヨウ素酸塩を用いて容易に(且つ選択的に)酸化することにより、タンパク質に対する反応性が遥かに高く、且つ還元的アミノ化及び他の化学反応(chemistries:ケミストリー)によるタンパク質の結合に好適な反応要素を含む、モノアル
デヒド体を得ることができる。この反応を図1及び図2に示す。ここで、
図1は、過ヨウ素酸ナトリウムを用いたコロミン酸(大腸菌由来のα−2,8結合ポリシアル酸)の酸化により、非還元末端でタンパク質反応性アルデヒドが形成されることを示し、
図2は、シアノ水素化ホウ素ナトリウムを用いたシッフ塩基の選択的還元により、タンパク質のアミノ基との安定で不可逆的な共有結合が形成されることを示す。
【0015】
上記従来の複合体形成反応中に、例えば、コロミン酸がアミノ酸の側鎖と反応することによって、意図しない副生成物が生成する可能性がある。これは、ヒト及び動物の治療用途で監督機関によって要求される既知組成の複合体の製造において十分に厄介なものとなる可能性がある。
【0016】
反応生成物の殆どの物理化学的特性が類似しているため、意図する反応生成物(例えば、モノポリシアル化体)を各種の意図しない生成物と切り離して精製することは容易ではない。このことは、イオン交換クロマトグラフィ及びゲル浸透クロマトグラフィ(それぞれ電荷及びサイズに基づいて分離する)等の技法によっては、好ましい精製プロファイルが得られないことを意味する。この問題は、複合体形成反応における生成物の複雑度を低減させることによって克服することができる。本発明者らは、多糖とタンパク質との複合
体を形成する新規な方法であって、それによりタンパク質のN末端の高い反応性を利用することができ、且つタンパク質と過ヨウ素酸塩酸化した天然のコロミン酸との還元的アミノ化に関する確立された方法(図1及び図2)を使用して得られる生成物の複雑度を回避する、新規な方法を開発した。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】欧州特許第1219636号
【特許文献2】米国特許第7,128,913号
【特許文献3】米国特許出願公開第2004/0082765号
【特許文献4】米国特許第7,074,755号
【特許文献5】米国特許第5,846,951号
【特許文献6】国際公開第WO−A−0187922号
【発明の概要】
【発明が解決しようとする課題】
【0018】
従来技術を鑑みて、ヒト及び動物の治療で使用することができ、且つ安定性、半減期が最適化され、毒性の低い、改善されたEPO誘導体の提供が必要とされている。本発明者らは、PSA等の多糖をEPOと結合させることで、かかる性質が付与されることを見出し、それにより本発明に到達した。アニオン性多糖と結合したEPOを記載したのはこれが初めてである。
【課題を解決するための手段】
【0019】
本発明の第1の態様により、本発明者らは、EPO又はEPO様タンパク質の多糖誘導体である化合物であって、多糖がアニオン性であり、且つ2個〜200個の糖ユニットを含む、化合物を提供する。
【0020】
以下、「EPO」という用語を使用する場合、本発明者らは、EPO様タンパク質を包含することも意図している。EPO様タンパク質とは、EPOの活性と同等の活性を有するタンパク質を意味する。EPOは、上記で詳述したように赤血球産生を調節する。EPO又はEPO様タンパク質の活性は、Krystal(1983)に記載の標準的なアッセイを使用
して測定することができる。マウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料の活性を測定する。マウスは、事前にフェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしている。アッセイにおいて、EPOを赤血球前駆細胞に加え、DNA複製速度を、3H−チミジン取り込み速度を求めることによって測定する。タンパク質は、NIBSC由来の標準EPOと比較して、10%〜200%の複製速度を誘導する場合、「EPO様」として分類される。典型的には、EPO様タンパク質は、標準EPOの活性の少なくとも35%、好ましくは、標準EPOの活性の少なくとも50%を有する。
【0021】
上記で詳述した、必要な活性を有するEPOの変異体も使用し得る。「EPO様」タンパク質は、「EPOホモログ(homologue)」とも称され得る。2個の配列が相同である
かどうかは、当該技術分野において既知の用語である、%類似性又は%同一性を使用してルーチン的に計算される。配列は、スイスプロット(swissprot)アクセッション番号P
O1588のヒトEPO前駆体である、配列番号1と比較するものとする。活性EPOは、この配列の28残基〜193残基である。EPOホモログ配列は、配列番号1の全体と比較してもよく、又はその28残基〜193残基と比較してもよい。好ましくは、EPOホモログ配列は、活性EPO、すなわち、28残基〜193残基と比較される。
【0022】
本発明において、ホモログは、50%以上の類似性又は同一性(核酸レベル又はアミノ
酸レベル)、好ましくは60%、70%、80%以上、より好ましくは90%以上、例えば、95%又は99%の同一性又は類似性(アミノ酸レベル)を有する。類似性又は同一性の計算には多数のプログラムが利用可能であるが、好ましいプログラムは、BLASTnプログラム、BLASTpプログラム及びBLASTxプログラムであり、初期設定パラメータで実行され、www.ncbi.nlm.nih.govで利用可能である。例えば、2個のアミノ酸配列は、初期設定パラメータ(スコア=100、ワード長=11、期待値=11、低複雑度フィルタリング=オン)でBLASTnプログラムを使用して比較し得る。上記の相同性のレベルは、これらの初期設定パラメータを使用して計算し得る。
【0023】
EPOは、グリコシル化されてもよく、又はグリコシル化されなくてもよい。EPOがグリコシル化される場合、化合物は、典型的には2個〜100個の糖ユニットを含む。より典型的には、化合物は、10個〜80個の糖ユニット、好ましくは20個〜60個の糖ユニット、最も好ましくは40個〜50個の糖ユニットを含む。
【0024】
EPOがグリコシル化されない場合、化合物は、典型的には80個〜180個の糖ユニット、好ましくは100個〜150個の糖ユニット、より好ましくは120個〜145個の糖ユニット、最も好ましくは130個〜140個の糖ユニットを含む。
【0025】
好ましくは、アニオン性多糖は、少なくとも2個、より好ましくは少なくとも5個、最も好ましくは少なくとも10個、例えば少なくとも50個の糖ユニットを有する。
【0026】
アニオン性多糖は、好ましくは、ポリシアル酸、ヘパリン、ヒアルロン酸及びコンドロイチン硫酸から選択される。好ましくは、多糖はポリシアル酸であり、実質的にシアル酸ユニットのみから成る。しかしながら、多糖は、分子中にシアル酸以外のユニットを有し得る。例えば、シアル酸ユニットを他の糖ユニットと交互にしてもよい。しかしながら、好ましくは、多糖は、実質的にシアル酸のユニットから成る。
【0027】
好ましくは、化合物は、EPO又はEPO様タンパク質のN末端誘導体である。すなわち、多糖は、そのN末端でEPOと結合する。本明細書中において、N末端での誘導体化とは、EPOのN末端アミン基での誘導体化を意味する。しかしながら、代替的には、多糖は、中間鎖(mid-chain)アミノ酸、例えば、リジン、システイン、アスパラギン酸、
アルギニン、グルタミン、チロシン、グルタミン酸又はヒスチジンの側鎖で、EPO又はEPO様タンパク質と結合し得る。典型的には、側鎖は、リジンアミノ酸又はシステインアミノ酸の側鎖である。
【0028】
好ましくは、多糖は末端シアル酸基を有し、上記で詳述したように、より好ましくは、α−2,8結合又はα−2,9結合を通じて相互に結合した少なくとも2個のシアル酸ユニットを含む多糖である、ポリシアル酸である。好適なポリシアル酸の重量平均分子量は2kDa〜50kDaの範囲、好ましくは5kDa〜50kDaの範囲である。最も好ましくは、ポリシアル酸は、細菌源、例えば、大腸菌KI、N.meningitidis、Maraxella liquefaciens若しくはPasteurella aeruginosaの多糖B、又は大腸菌K92株由来のK92多糖に由来する。最も好ましくは、大腸菌K1に由来のコロミン酸である。
【0029】
ポリシアル酸は、塩又は遊離酸の形態であり得る。細菌源からの回収後に分子量が低下した、加水分解形態であってもよい。
【0030】
好ましくは、ポリシアル酸である多糖は、分子量が広範な(例えば多分散度が1.3超、例えば2以上にもなる)物質であり得る。好ましくは分子量の多分散度は1.3未満、より好ましくは1.2未満、例えば1.1未満である。多分散度は低いと1.01にもな
り得る。
【0031】
EPOは、2個以上のアニオン性多糖を用いて誘導体化され得る。例えば、EPOは、そのN末端及び内部のアミノ酸側鎖の両方で誘導体化され得る。例えば、リジン、システイン、アスパラギン酸、アルギニン、グルタミン、チロシン、グルタミン酸、セリン及びヒスチジンの側鎖が、アニオン性多糖によって誘導体化され得る。EPOはグリコン(glycon)ユニットでも誘導体化され得る。しかしながら、本発明の好ましい実施形態において、EPOはそのN末端のみで誘導体化される。
【0032】
本明細書中において、N末端での誘導体化とは、EPOのN末端アミン基での誘導体化を意味する。
【0033】
本発明の第1の態様による化合物は、EPOとアニオン性多糖とが共有結合した複合体であり得る。多糖とEPOとの間を結び付ける他の手段としては静電引力が挙げられる。しかしながら、共有結合が好ましい。EPOは、そのN末端アミノ酸で多糖と共有結合し得る。共有結合は、カルボキシル基とアミン基との間のアミド結合であり得る。EPOが多糖と共有結合することができる別の結合は、シッフ塩基を介する。アミンとの複合体形成に好適な基は、国際公開第WO2006/016168号にさらに記載されている。
【0034】
本発明において、多糖は、天然多糖又は天然多糖の誘導体、例えば、糖残基上での1個又は複数の活性基の反応によって誘導体化されているか、又は多糖鎖の末端で誘導体化基と共有結合している多糖であり得る。
【0035】
多糖は、その還元末端ユニット又は非還元末端ユニットのいずれかを介してEPOと結合し得る。このことは、1個の多糖鎖が、2個のEPOタンパク質と結合し得る、すなわち、その還元末端及び非還元末端の両方で誘導体化され得ることを意味する。
【0036】
多糖をタンパク質と結合させる方法は、当該技術分野において既知であり、国際公開第WO92/22331号及び特許文献6において、より詳細に記載されている。本発明における好ましい方法を以下でより詳細に説明する。方法は、本出願の図1及び図2にも記載されている。
【0037】
多糖は、EPO又はEPO様タンパク質と直接、すなわち、図1及び図2に示すように、又はリンカーを介して結合し得る。好適なリンカーは、N−マレイミド、ビニルスルホン、N−ヨードアセトアミド、オルトピリジル又はN−ヒドロキシコハク酸イミドを含有する試薬に由来する。リンカーはまた、生物学的に安定である(biostable)か、又は生
分解性であり、例えば、ポリペプチド又は合成オリゴマーを含み得る。リンカーは、国際公開第WO2005/016973号でさらに記載されるように、二官能性部分に由来し得る。好適な二官能性試薬は、例えば、Bis−NHSである。試薬は、一般式Z−R1−Z(式中、各Zは官能基であり、同一であってもよく、又は異なっていてもよく、且つR1は二官能性有機基である)を有し得る。好ましくは、R1は、アルカンジイル、アリーレン、アルカリーレン、ヘテロアリーレン及びアルキルヘテロアリーレン(これらは任意でカルボニル結合、エステル結合、スルフィド結合、エーテル結合、アミド結合及び/又はアミン結合によって置換及び/又は介在され得る)から成る群より選択される。特に好ましくは、C3〜C6アルカンジイルである。最も好ましくは、R1は、好適な二官能性試薬の適切な部分に相当する。
【0038】
本発明の好ましい化合物は、一般式(I):
【0039】
【化1】
【0040】
(式中、mは、少なくとも1であり、
XBは、EPO又はEPO様タンパク質であるB−XHに由来し(ここで、XHは、NH2又はSHである)、
Lは、結合、連結基であるか、又はポリペプチド若しくは合成オリゴマーを含み、
GlyOは、アニオン性糖ユニットであり、
結合基は、存在すれば、一般式−Y−C(O)−R1−C(O)−であり、Yは、NR2又はNR2−NR2であり、且つR1は、上記で規定した二官能性有機基であり、且つR2は、H又はC1−6アルキルである)
のものである。
【0041】
本発明の本態様において、EPOは、多糖の非還元末端と結合する。末端多糖ユニットはシアル酸ユニットである。多糖中の他の糖ユニットは、GlyOで表され、同一であってもよく、又は異なっていてもよい。好適な糖ユニットとしては、ヘパリン、ヒアルロン酸及びコンドロイチン硫酸が挙げられる。
【0042】
EPOが多糖と直接結合する場合、L基は結合である。しかしながら、L基は代替的には、N−マレイミド、ビニルスルホン、N−ヨードアセトアミド、オルトピリジル又はN−ヒドロキシコハク酸イミドを含有する試薬に由来し得る。試薬は、上記で規定したように、一般式Z−R1−Zを有し得る。本実施形態において、Lは典型的には、基
【0043】
【化2】
【0044】
である。
【0045】
好ましくは、XHはNH2であり、且つEPO又はEPO様タンパク質のN末端である。代替的には、NH2はリジンアミノ酸側鎖の第1級アミンである。異なる実施形態において、XHはシステインアミノ酸の側鎖のチオール基、SHである。
【0046】
本発明の別の態様は、上記で規定したような新規化合物及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物である。
【0047】
医薬組成物は、水性懸濁液の形態であってもよい。水性懸濁液は、水性懸濁液の製造に好適な賦形剤と混合した新規化合物を含有する。医薬組成物は、無菌の注射可能な水性懸濁液又は均質懸濁液の形態であってもよい。懸濁液は、好適な分散剤又は湿潤剤及び懸濁剤を使用する既知の技術に従って製剤化され得る。
【0048】
医薬組成物は、ヒト用途又は家畜(veterinary)用途で、経口投与、静脈内投与、腹腔内投与、筋肉内投与、皮下投与、鼻腔内投与、皮内投与、局所投与又は気管内投与され得る。
【0049】
組成物は製剤添加物をさらに含み得る。製剤添加物とは、Wang他(1999)に記載されている、内的に又は外的にEPOを安定化させることが可能な賦形剤を意味する。賦形剤は、安定化剤、可溶化剤又は金属イオンであり得る。製剤添加物の好適例としては、1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸が挙げられる。これらは単独で使用してもよく、又は組み合わせて使用してもよい。
【0050】
安定化剤は、典型的には、タンパク質のアンフォールディングに対するギブス自由エネルギー変化の増大を引き起こすタンパク質の変性状態の不安定化により作用する。安定化剤は、好ましくは糖又はポリオール、例えばスクロース、ソルビトール、トレハロース、グリセロール、マンニトール、ラクトース及びエチレングリコールである。安定化バッファーはリン酸ナトリウムである。
【0051】
可溶化剤は、好ましくは界面活性剤、好ましくは非イオン性界面活性剤である。好適例としては、Tween 80、Tween 20、Tween 40、Pluoronic F68、Brij 35及びTriton X100が挙げられる。
【0052】
金属イオンは、好ましくは二価である。好適な金属イオンとしては、Zn2+、Ni2+、Co2+、Sr2+、Cu2+及びFe2+が挙げられる。
【0053】
製剤添加物はまた、ヒト血清アルブミン、PSA、PEG又はヒドロキシ−β−シクロデキストリンから選択されるポリマーであり得る。
【0054】
製剤添加物としての使用に好適なアミノ酸及びアミノ酸誘導体としては、ヒスチジン、グリシン、他の類似のアミノ酸及びアスパラギン酸ナトリウムが挙げられる。
【0055】
本発明の別の態様は、EPO又はEPO様タンパク質のアニオン性多糖誘導体の集団を含む、組成物であって、誘導体が2個〜125個の糖ユニットを含み、且つ集団が実質的にタンパク質のN末端誘導体のみから成る、組成物である。「集団」とは、組成物中に2個以上の多糖誘導体が存在することを意味する。誘導体は、同一であるか又は異なる数の糖ユニットを含み得る。好ましくは、組成物中の多糖の多分散度は、1.3未満、より好ましくは1.1未満である。好ましい多糖は、上記本発明の他の態様に関して詳述した通りである。
【0056】
集団において、EPOの実質的に全てがN末端のみで誘導体化される。このことは、集団内のタンパク質の85%、好ましくは少なくとも90%、最も好ましくは少なくとも95%がPSAによりN末端のみで誘導体化されることを意味する。
【0057】
N末端での誘導体化の程度は、当該技術分野において既知の技法、例えばペプチドマッピング又はエドマン分解によって求められ得る。
【0058】
本発明のさらなる態様は、治療で使用するための上記化合物である。
【0059】
本発明の最後の態様により、本発明者らは、EPO又はEPO様タンパク質の多糖誘導体の製造方法であって、2個〜200個の糖ユニットを含むアニオン性多糖が、EPO又はEPO様タンパク質と化学的に反応する、製造方法を提供する。
【0060】
なお、本発明の本態様において、多糖は、EPO又はEPO様タンパク質上の任意の基で反応し得る。例えば、多糖は、アミン基、ヒドロキシル基、カルボキシル基又はスルフ
ヒドリル基と反応し得る。好ましくは、基は、アミン基、より好ましくは末端アミン基である。アミンは、代替的には、アミノ酸、例えばリジンアミノ酸のアミン側鎖であり得る。多糖はまた、EPO上の任意の糖質残基、例えばペンダントグリコン基で反応し得る。
【0061】
多糖は、当該技術分野で既知の方法によってアミノ酸側鎖と結合し得る。例えば、多糖は、in vitroでのカップリングによってAsp又はGluのC末端、−COOH又はカルボキシル側鎖とカップリングし得る。システインアミノ酸のチオール基もin vitroでのカップリングによって多糖と結合し得る。これらの方法は、国際公開第WO03/055526号、特に第6頁及び第7頁の表にさらに記載されている。この参考文献において、in vitroでのカップリングは、オリゴ糖部分とGlnの側鎖上のアミド基との結合にも使用されている。in vitroでのArg残基及びHis残基のイミダゾール基もそれぞれ記載されている。これらの方法をそれぞれ使用して、本発明のEPOを誘導体化し得る。
【0062】
多糖はEPOの修飾形態とも反応し得る。例えば、EPO上の1個又は複数の基は、例えば、還元又は酸化による化学変換を受けた可能性がある。例えば酸化条件を使用して、EPOの末端アミノ基の代わりに反応性カルボニルを生成することができる。
【0063】
本発明の方法での使用に好適な多糖は、新規化合物について先に記載した通りである。
【0064】
本発明の化合物は、従来技術で記載した好適な方法のいずれかによって製造し得る。例えば、典型的な方法は、本発明者らの先の国際特許出願第WO92/22331号に記載されている。
【0065】
典型的には、アニオン性多糖は、EPOに対する誘導体化の前に活性化されている。例えば、反応性アルデヒド基を有する可能性があり、誘導体化反応が還元条件下で実行され得る。反応性アルデヒド基は、多糖のヒドロキシル基の制御酸化によって生成し得る。最も好ましくは、この反応性アルデヒドは予備段階(ここで多糖を、例えば、水溶液中で過ヨウ素酸ナトリウムを使用する制御酸化条件下で反応させる)で生じる。好ましくは、酸化は化学酸化であるが、この段階を実行することが可能な酵素も使用され得る。反応性アルデヒド基は、多糖の非還元末端又は還元末端に存在し得る。その後、EPO、典型的にはN末端が反応性アルデヒド基と反応することにより、還元時にEPOのN末端誘導体を生成する付加体を得ることができる。
【0066】
多糖の活性化は、好ましくは多糖の中間鎖切断が実質的に無い、すなわち、実質的に分子量が低減しない条件下で実行されるべきである。酸化剤は、好適には過ルテニウム酸塩、又は好ましくは過ヨウ素酸塩である。酸化は、1mM〜1Mの範囲の濃度の過ヨウ素酸塩を用い、3〜10の範囲のpH、0℃〜60℃の範囲の温度で、1分〜48時間の範囲の時間、実行され得る。
【0067】
誘導体化反応に好適な還元条件は、水素と触媒、又は好ましくはホウ水素化物(borohydride)等の水素化物を利用し得る。これらは固定化してもよい(例えば、Amberli
te(商標)−担持型ホウ水素化物)。好ましくは、水素化ホウ素ナトリウム等のアルカリ金属水素化物が、1μM〜0.1Mの範囲の濃度、5.0〜10の範囲のpH、0℃〜60℃の範囲の温度、及び1分〜48時間の範囲の期間で、還元剤として使用される。出発原料上のペンダントカルボキシル基が還元されない反応条件が選択される。他の好適な還元剤は、酸性条件下でのシアノホウ水素化物(cyanoborohydride)、例えば、ポリマー担持型シアノホウ水素化物又はアルカリ金属シアノホウ水素化物、L−アスコルビン酸、メタ重亜硫酸ナトリウム、L−セレクトリド、トリアセトキシホウ水素化物等である。
【0068】
多糖の他の活性化誘導体、例えば、本発明者らの先の国際特許出願第WO06/00540号に記載されている、NHS等のペンダント官能基を有するものは、本発明において有用であり得る。
【0069】
一実施形態において、反応性アルデヒドは、多糖の還元末端に存在し、非還元末端は、EPO上のペンダント基と反応しないように不動態化(passivated)されている。
【0070】
コロミン酸の還元末端の反応性は、タンパク質標的に対しては弱いものの、化学的に規定された複合体の製造においては十分に厄介である。
【0071】
多糖の還元末端に反応性アルデヒドを有する多糖を調製するのに好適な化学反応は、本発明者らの先の国際特許出願第WO05/016974号に記載されている。この工程は、予備的な選択的酸化段階、続いての還元、及びその後のさらなる酸化により、還元末端にアルデヒドを有し、且つ非還元末端が不動態化された化合物を得ることを包含する。
【0072】
国際公開第WO2005/016973号には、タンパク質との複合体形成に有用なポリシアル酸誘導体、特に遊離のスルフヒドリル薬物を有するものが記載されている。ポ0リシアル酸化合物をヘテロ二官能性試薬と反応させることにより、スルフヒドリル基との部位特異的複合体形成のためのペンダント官能基を導入する。本発明で使用するアニオン性多糖も、このようにヘテロ二官能性試薬を用いて誘導体化され得る。
【0073】
多糖は、EPOと反応する前に誘導体化され得る。例えば、多糖は二官能性試薬と反応し得る。
【0074】
多糖は、国際公開第WO2006/016168号でさらに記載されているように、予備反応段階(ここで、第1級アミン基、第2級アミン基及びヒドラジンから選択される基が、好ましくはシアル酸である末端糖上に形成される)と、続く反応段階(ここで、これが二官能性試薬と反応することにより、反応中間体が形成される)に付され得る。次いで、中間体はEPOと反応し得る。二官能性試薬は、先に規定したように、一般式Z−R1−Zを有し得る。
【0075】
本発明者らは、或る特定の反応条件が、EPOのN末端での選択的誘導体化を促進することを見出した。N末端での選択的反応を促進するために、誘導体化反応は、酸性pHの第1の水溶液中で実行すべきであり、次いで得られた多糖誘導体は、第1の水溶液よりも高いpHの第2の水溶液中で精製すべきである。典型的には第1の水溶液のpHは4.0〜6.0の範囲であり、第2の水溶液のpHは6.5〜9.0、好ましくは6.5〜8.5又は6.5〜8.0の範囲である。誘導体化反応のpHが低いと、任意の中間鎖部位ではなく、タンパク質のN末端での選択的誘導体化が促進される。
【0076】
さらに、本発明者らは、或る特定の製剤添加物を使用すると、選択的で安定的な多糖EPO誘導体の形成が促進されることを見出した。製剤添加物は、1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸から選択され得る。これらは反応媒体に添加してもよく、又は代替的には安定化剤として最終製品組成物に添加してもよい。
【0077】
本発明の一実施形態において、製剤添加物は、ソルビトール、トレハロース又はスクロースである。異なる実施形態において、製剤添加物は、非イオン性界面活性剤である。製剤添加物は代替的にはPSA、PEG又はヒドロキシ−β−シクロデキストリンから選択されるポリマーであり得る。異なる実施形態において、製剤添加物は、二価金属イオンである。好ましい二価金属イオンとしては、Zn2+、Ni2+、Co2+、Sr2+、F
e2+、Mg2+又はCa2+が挙げられる。
【0078】
製剤添加物はバッファーであり得る。好ましくは、製剤添加物がバッファーの場合、それはリン酸ナトリウムである。
【0079】
本発明の方法における多糖誘導体の精製は、当該技術分野において既知の種々の方法を使用して実行され得る。好適な精製方法の例としては、HIC(疎水性相互作用クロマトグラフィ)、SEC(サイズ排除クロマトグラフィ)、HPLC(高速液体クロマトグラフィ)、AEC(陰イオン交換クロマトグラフィ)及び金属アフィニティクロマトグラフィが挙げられる。
【0080】
分子量分布の広いポリシアル酸の集団を、多分散度が低めの画分、すなわち、異なる平均分子量の画分に分画し得る。分画は、好ましくは本発明者らの先の国際特許出願第WO2005/016794号及び同第WO2005/03149号に記載されているように、好適な塩基性バッファーを溶出に使用する陰イオン交換クロマトグラフィによって実施される。この分画方法は、多糖出発原料及び誘導体に好適である。したがって、この技法は、本発明の必須の工程段階の前後に適用され得る。好ましくは、得られるEPOの多糖誘導体の多分散度は1.3未満、より好ましくは1.2未満、最も好ましくは1.1未満である。
【0081】
本発明によるEPOの誘導体化の結果、タンパク質の半減期が増大し、安定性が改善し、免疫原性が低減し、且つ/又は溶解度が制御される。それゆえ、EPOのバイオアベイラビリティ及び薬物動態特性が改善する。モノポリシアル化EPO複合体を作製する新規な方法には特別な価値がある。
【0082】
本発明を、実施例1〜実施例3.12によって、また添付の図面を参照して説明する。
【図面の簡単な説明】
【0083】
【図1】従来技術の非還元シアル酸末端ユニットの活性化を示す反応スキーム図である。
【図2】タンパク質のN末端又はランダム誘導体化を示す反応スキーム図である。
【図3a】Triple Detection GPC(Viscotek:RI+RALS+Viscosiometer)を使用する、異なるpHでの24kDaのコロミン酸(CA)の分解を示す図である。
【図3b】CAポリマーのゲル浸透クロマトグラフィを示す図である。
【図4】SDS−PAGEによる、PEG化EPO及びポリシアル化EPOの特徴付けを示す図である。
【図5】SDS−PAGE(右手側)及びSE−HPLC(左手側)による、ポリシアル化EPOの特徴付けを示す図である。
【図6】SE−HPLCによる、EPO、ポリシアル化EPO及びPEG化EPOの特徴付けを示す図である。
【図7】EPOに関するFACSデータ(網状赤血球数)を示す図である。
【図8】EPO製剤のin vivoでのクリアランスを示す図である。
【図9】EPO製剤のin vivoでのクリアランスを示す図である。
【図10】SE−HPLCによる、EPO−CA複合体の特徴付けを示す図である。
【図11】非グリコシル化EPO対ポリシアル化非グリコシル化EPOのin vivoでのクリアランスを示す図である。
【図12】非グリコシル化EPO対ポリシアル化非グリコシル化EPOのin vivoでのクリアランスを示す図である。
【図13】SE−HPLC及びSDS−PAGEによる、NG−EPO−CA複合体の特徴付けを示す図である。
【図14】サンドイッチELISAによるEPO−PSAの検出を示す図である。
【図15】試薬希釈液中のEPO−PSAに対するELISAの感度を示す図である。
【図16】サイズ排除HPLCによるEPO複合体の安定性を示す図である。
【図17】ポリシアル化EPOのSDS−PAGEを示す図である。
【図18】EPO製剤のin vivoでの効力を示す図である(n=3〜4±SE)(非近交系雌性マウス;12週齢;SC(アルデヒドケミストリー))。
【図19】EPO製剤のin vivoでの効力を示す図である(雌性ウィスターラット;8週齢〜9週齢;n=5±SEM)。
【図20】SE−HPLCによる、NG EPOのPEG化を示す図である。
【実施例】
【0084】
材料
炭酸アンモニウム、エチレングリコール、ポリエチレングリコール(8KDa)、シアノ水素化ホウ素ナトリウム(sodium cyanoborohydride)(純度98%超)、メタ過ヨウ素
酸ナトリウム及び分子量マーカー、硫酸アンモニウム、塩化ナトリウム、リン酸ナトリウム、ソルビトール、Tween 20並びにびTrisは、Sigma Chemical Laboratory
、英国から入手した。酢酸ナトリウム及びリン酸ナトリウムは、BDH、英国から入手した
。使用するコロミン酸、直鎖α−(2,8)−結合大腸菌K1ポリシアル酸(平均22.7kDa、高多分散度1.34、39kDa、多分散度1.4;11kDa、多分散度1.27)は、Camida、アイルランド及びS.I.I.L. India Ltd.から入手した。他の材料と
しては、2,4−ジニトロフェニルヒドラジン(Aldrich Chemical Company、英国)、透析管(3.5KDa及び10KDaカットオフ限界;Medicell International Limited、英国)、Sepharose SP HiTrap、PD−10カラム、Q FF[カラム1ml又は5ml];Hitrap Butyl HPカラム[1ml又は5ml](Pharmacia、英国)、Tris−グリシンポリアクリルアミドゲル(4%〜20%及び8
%〜16%)、Tris−グリシンドデシル硫酸ナトリウムランニングバッファー及びローディングバッファー(Novex、英国)が含まれた。脱イオン水は、Elgastat
Option 4水精製ユニット(Elga Limited、英国)から入手した。使用した全ての試薬は分析用(analytical grade)である。プレートリーダー(Dynex Technologies、英国)をタンパク質アッセイ及びCAアッセイにおける分光光度定量に使用した。マウス及びラットは、Harlan、英国から購入し、使用前の少なくとも1週間馴化させた。EPOはSIIL、インドから入手した。
1.タンパク質及びコロミン酸の定量
他で記載されているように[Gregoriadis他、1993;Fernandes及びGregoriadis、1996
、1997]、レゾルシノール法[Svennerholm、1957]によって、レゾルシノール試薬を用
いた(シアル酸としての)ポリシアル酸の定量的評価を実行した。タンパク質を、BCA比色法又は280nmでのUV吸光度によって測定した。
2.1.コロミン酸の活性化
新たに調製した0.02Mメタ過ヨウ素酸ナトリウム(NaIO4)溶液(8倍モル過剰)を20℃でCAと混合し、反応混合物を暗所で15分間、磁気的に撹拌した。次に、2倍容量のエチレングリコールを反応混合物に加えて、過剰のNaIO4を消費し、混合物をそのまま20℃でさらに30分間撹拌した。酸化したコロミン酸を、4℃の0.01%炭酸アンモニウムバッファー(pH7.4)に対して長時間(extensively)(24時
間)透析した(3.5KDa分子量カットオフ透析管)。限外濾過(分子量カットオフ3.5kDaに対する)を使用して、透析管からCAO溶液を濃縮した。必要容量まで濃縮した後、濾液を凍結乾燥し、さらに使用するまで−40℃で保存した。代替的には、CAOを、エタノールによる沈殿(2回)によって反応混合物から回収した。
2.2.CA及び誘導体の酸化状態の定量
カルボニル化合物との相互作用で難溶性の2,4−ジニトロフェニルヒドラゾンを生じる、2,4−ジニトロフェニルヒドラジン(2,4−DNPH)を用いて、コロミン酸酸化度の定性的評価を実行した。非酸化コロミン酸(CA)/酸化コロミン酸(CAO)を2,4−DNPH試薬(1.0ml)に加え、溶液を振盪した後、結晶性の沈殿物が観察されるまで37℃で放置した[Shriner他、1980]。CAの酸化度(定量的)を、アルカ
リ性溶液中でのフェリシアン化物イオンのフェロシアン化第二鉄(プルシアンブルー(Persian blue))への還元に基づく方法[Park及びJohnson、1949]を用いて測定した(こ
れは次に630nmで測定される)。この場合、グルコースを標準として使用した。
2.3.ゲル浸透クロマトグラフィ
コロミン酸試料(CA及びCAO)をNaNO3(0.2M)、CH3CN(10%;5mg/ml)に溶解し、2×GMPWXLカラムでクロマトグラフィにかけ、屈折率により検出した(GPC系:VE1121 GPC溶媒ポンプ、VE3580 RI検出器及びTrisec 3ソフトウェアによる照合、Viscotek Europe Ltd)。試料(5mg
/ml)を0.45μmのナイロン膜で濾過し、移動相として0.2M NaNO3 及びCH3CN(10%)を用いて0.7cm/分で流した。
【0085】
結果を図3b並びに第5表及び第6表に示す。
2.4.コロミン酸の安定性
PEG化の化学反応に関するルールをそのままポリシアル化に応用することができないのは、これらの分子の生理化学的特性が異なるからである。PSAは、酸に不安定なポリマーであると共に、中性付近のpHでは何週間も安定である(図3a)。図3aの結果から、pH6.0及び7.4では、CAは8日間安定であり、pH5.0では緩徐に分解し(48時間後、初期分子量の92%)、pH4.0では緩徐に分解する(48時間後、初期分子量の70%)ことが示される。ポリシアル酸は親水性が高いが、PEGは両親媒性である。PEG化に使用される条件を使用してポリシアル化を実行すると、多くの場合、タンパク質の凝集及び沈殿が観察される。
3.製剤添加物を含むN末端タンパク質−CA複合体の調製
3.1.EPO−CA複合体(N末端法)の調製
EPOを溶液(10mMリン酸ナトリウムバッファー、130mM NaCl、pH7.0中、0.34mg/ml;比活性:110,000U/mg、分子量:30600)
として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を、2mlのエッペンドルフチューブに採取した。必要量(25倍モル過剰)のコロミン酸を取り、タンパク質溶液を固体CAに加え、緩やかに混合した。反応混合物中で50mM又は3.17mg/mlとなるように、必要容量のシアノ水素化ホウ素ナトリウム溶液を加え、ボルテックスし、最終反応混合物のpHをチェックした(必要に応じて、pHを6.0に調整する)。チューブをシールし、所望の温度で24時間撹拌するか、又は反応混合物を最初に室温(22℃)で8時間インキュベートした後、4℃±1℃で一晩(14時間)保持した。インキュベーション後、必要な試料を採取した(例えば、活性アッセイ、SDS−PAGE、SE−HPLC用)。
3.1.1.EPO−CA複合体の精製及び特徴付け(N末端法)
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で希釈し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中、試料を氷上(4℃±1℃)に保持した。
【0086】
タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光
度は約0.743である)。SDS−PAGE用の試料を採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を陰イオン交換クロマトグラフィ(AEC)を使用して実施した。AECでは、タンパク質を含有するHIC画分をAEC バッファーA(50mM
Tris、150mM NaCl、pH8.0)(試料1ml+AECバッファーA 5ml)で希釈し、1.0ml/分でAECバッファーAを用いて事前に平衡化したECカラムにローディングした。ローディング画分を回収及び標識した。カラムをAECバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファーB(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0087】
代替的には、精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaの場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.
743である)。SDS−PAGE用の試料を採取した。
【0088】
一定分量をタンパク質アッセイ及びCAアッセイ用に取り出した。残りの溶液は、使用するまで−20℃で保存した。生成物をSDS−PAGEによって特徴付けした。フェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしたマウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料及びNG EPO試料の活性の定量を使用した。プロトコルは、Krystal(1972)により報告されている方法
に基づき改作した。アッセイは、赤血球前駆細胞へのEPOの添加及び3H−チミジン取り込み速度の定量によるDNA複製速度の測定に基づく。B6D2F1マウスにおいて、in vivoでの薬物動態(PK)試験及び薬力学(PD)試験を行った。
3.2.EPO−CA複合体(ランダム)の調製
EPOを溶液(10mMリン酸ナトリウムバッファー、130mM NaCl、pH7.0中、0.34mg/ml;比活性:110000U/mg、分子量:30600)として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を2mlのエッペンドルフチューブに採取した。必要量のコロミン酸を取り、タンパク質溶液を固体CAに加え、緩やかに混合した。反応混合物中で50mM又は3.17mg/mlとなるよ
うに、必要容量のシアノ水素化ホウ素ナトリウム溶液を加え、ボルテックスし、最終反応混合物のpHをチェックした(必要に応じて、pHを7.4に調整する)。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。インキュベーション後、必要な試料を採取した(例えば、活性アッセイ、SDS−PAGE、SE−HPLC用)。
3.2.1.EPO−CA複合体(ランダム)の精製及び特徴付け
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で溶解し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、洗浄画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中、試料を氷上(4℃±1℃)に保持した。
【0089】
タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を陰イオン交換クロマトグラフィ(AXC)を使用して実施した。AXCでは、タンパク質を含有するHIC画分をAXCバッファーA(50mM
Tris、150mM NaCl、pH8.0)(試料1ml+AXCバッファーA 5ml)で希釈し、1.0ml/分でAXCバッファーAで事前に平衡化したAXCカラムにローディングした。ローディング画分を回収及び標識した。カラムをAXCバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファー(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0090】
代替的な精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaの場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.74
3である)。SDS−PAGE用の試料を採取した。
3.3.グリコンケミストリー
ヒドラジドコロミン酸を、最終CA濃度が10mMとなるようにEPO溶液に溶解した。溶液のpHを5.5に調整した。必要容量のNaIO4溶液のNaOAc溶液を、最終濃度が5mM NaIO4となるように加えた。NaHSO3(NaHSO3の最終濃度20mM)を用いて反応を停止した。反応混合物を室温でインキュベートした。最後に、必要容量のNaCNBH3溶液のNaOAc溶液を、最終濃度が50mM NaCNBH3となるように加えた。振盪器上で反応を4℃±1℃で1時間継続した。インキュベーション後、SDS、SE−HPLC、活性アッセイに必要な試料を採取した。
3.3.1.EPO−CA複合体の精製及び特徴付け(グリコンケミストリー)
残りの反応混合物試料を、最終濃度が2Mとなるように、HICバッファーA1(3M硫酸アンモニウム、pH6.3)で希釈し、室温で1.5ml/分の速度でHICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した(L1〜Lx)。カラムをHICバッファーA2(2M硫酸アンモニウム、pH6.3)(少なくとも6カラム容量)で洗浄し、画分を回収及び標識した。生成物をHICバッファーB(50mM Na2HPO4、pH7.4)で溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)(6CV)を回収及び標識した。精製中は、試料を氷上(4℃±1℃)に保持した。
【0091】
タンパク質濃度をUV(276nm)によって分析した(1mg/mlのEPOの吸光度は約0.743であった)。試料をSDS−PAGE用に採取した。CAの分子量が、SE−HPLCによる複合体及びEPOの分離には小さ過ぎる場合(例えば、22kDa)、非複合体形成EPOの分離を、陰イオン交換クロマトグラフィ陰イオン交換(AXC)を使用して実施した。AXCでは、タンパク質を含有するHIC画分をAXCバッファーA(50mM Tris、150mM NaCl、pH8.0)で希釈し(試料1ml+AXCバッファーA 5ml)、1.0ml/分でAXCバッファーAで事前に平衡化したAXCカラムにローディングした。ローディング画分を回収及び標識した。カラムをAXCバッファーA(少なくとも10ml)で洗浄し、画分を回収及び標識した。生成物を溶出バッファー(50mM Tris、600mM NaCl、pH8.0)で溶出し、2.0ml/分での第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。
【0092】
代替的な精製は、SE−HPLCによって行うことができる(例えば、使用するCAが高分子量、例えば、39kDaである場合に、EPOから複合体を分離する)。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのEPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。
3.4.EPOのPEG化:
EPO(30.6kDa)を溶液(5%ソルビトール、0.025mg/mlポリソルベート80を含有する10mM酢酸ナトリウムバッファー、pH4.0中、0.954m
g/ml)として供給し、2℃〜8℃で保存した。EPO溶液を濃縮して、約1.0mg/mlの溶液とした。必要量のEPOをエッペンドルフチューブに取り、氷上に載置した
。複合体形成用に添加したPEGの量は、式:
PEGの重量={タンパク質の量(g)/(タンパク質の分子量)}×(PEGの分子量)×(PEGのモル過剰)
に基づいて計算した。
【0093】
必要量のPEG 20Kを秤量した。10mM NaOAc、5%ソルビトール、pH5.5(ここで使用するのは、最終反応容量の20%容量)中に可溶化し、混合物を、全てのPEGが溶解するまで緩やかにボルテックスした後、濾過して新しいエッペンドルフに入れるか、又は4000rpmで5分間遠心し、上清を新しいエッペンドルフに移すことにより、いかなる凝集/沈殿物質も除去した。PEGが25倍モル過剰となるように、必要量のEPOをタンパク質溶液をPEG溶液に加え、4℃±1℃で緩速振盪器上に反応混合物を保持することによって緩やかに混合した。最終反応混合物中で50mM又は3.17mg/mlとなるように、必要容量の100mg/ml NaCNBH3溶液を加え、緩やかに混合し、最終反応混合物のpHをチェックし、必要に応じて、4℃±1℃の1M NaOH/HClを用いて5.5に調整した。最後に、反応の容量を、反応混合物中のタンパク質濃度が1mg/mlとなるように、20mM NaOAC、5%ソルビトー
ル、pH5.5を使用して調整した。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。反応を適切な方法によって停止し、in vitro活性、SDS−PAGE(4%〜20%Tris−グリシンゲルを使用)、SE−HPLC(superose 6カラム)用の試料を採取し、反応混合物のpHをチェックした。いかなる沈殿物も除去するために、SE−HPLC分析及び精製前に反応混合物を13000rpmで5分間遠心した。好ましいSE−HPLC用バッファーは0.1Mリン酸ナトリウム(pH6.9)であった。結果を図5に示す。
3.5.N末端非グリコシル化エリスロポエチン(NG EPO−CA)複合体の調製
NG EPOを溶液(20mMリン酸ナトリウムバッファー、300mM NaCl、pH6.65中、0.18mg/ml;比活性:100000U/mg;分子量:19000)として供給し、−32℃で保存し、タンパク質を2℃〜8℃で解凍し、必要量を2mlのエッペンドルフに採取した。複合体形成に必要なコロミン酸(例えば、酸化コロミン酸又は非酸化コロミン酸)の量を計算した。必要量のコロミン酸を秤量した。タンパク質溶液を固体CAに加え、穏やかに混合した。反応混合物中のシアノ水素化ホウ素ナトリウムの最終濃度が50mM又は3.17mg/mlとなるように、必要容量のシアノ水素化ホウ素ナトリウム溶液を、反応混合物に加えた。最終反応混合物をボルテックスし、pHをチェックした(必要に応じて、pHを7.4に調整した)。チューブをシールし、所望の温度(4℃±1℃)で24時間撹拌した。インキュベーション後、活性アッセイ、SDS−PAGE、SE−HPLC等に必要な試料を採取した。
3.5.1.NG EPO−CA複合体の精製及び特徴付け
残りの反応混合物試料をHICバッファーA(1.2M硫酸アンモニウム、pH6.3)で希釈し(試料1ml+バッファーA 4ml)、HICバッファーAで事前に平衡化したHICカラムにローディングした。ローディング画分を回収及び標識した。カラムをHICバッファーA(少なくとも10ml)で洗浄した。洗浄画分を回収し標識した。生成物をHICバッファーBで溶出し、第1の画分(0.5ml)及びその後の画分(0.5ml〜1ml)を回収及び標識した。精製中、試料を氷上に保持した。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのnEPOの吸光度は約0.743であった)。試料をSDS−PAGE用に採取した。反応条件により、反応混合物中に有意な遊離のNG EPOが存在しなかったため、さらなる精製は不必要であった。NG
EPOが反応混合物中に存在した場合には、タンパク質を含有するHIC画分をVivaspin 6(5000 MWCO)を使用して濃縮し、SE−HPLCによって精製を行った。タンパク質濃度をUV(280nm)によって分析した(1mg/mlのNG
EPOの吸光度は約0.743である)。試料をSDS−PAGE用に採取した。
【0094】
一定分量をタンパク質アッセイ及びCAアッセイ用に取り出した。残りは、使用するまで−20℃で保存した。生成物をSDS−PAGEによって特徴付けした。
3.6.EPO製剤のSE−HPLC
4℃で冷却したJascoのAS−2057 plusオートサンプラー及びJascoのUV−975 UV/VIS検出器を備えたLiquid Chromatograph(Jasco)でHPLCを実施した。データをIBM/PC上のEZchrom Eliteソフ
トウェアによって記録した。SEC試料を、Superose 6カラムで、0.1Mリン酸Na、pH6.9の定組成移動相を用いて分析した(図5)。図6は室温での唯一のピーク=76.408を示し、これはEPOに帰属される。
【0095】
図5の左手側に示すSECのピーク表は以下の通りである:
【0096】
【表1】
【0097】
3.7.SDSポリアクリルアミドゲル電気泳動、ウエスタンブロッティング&ELISA
4%〜20%のTris−グリシンゲルを使用して、SDS−PAGEを実施した。試料を、還元バッファー又は非還元バッファーのいずれかで希釈し、タンパク質5.0μgを各ウェルにローディングした。ゲルをトリグリセリンバッファー系で試験し、Coomassie Blueで染色した。抗PSA抗体を使用してウエスタンブロッティングを実施した(図4)。図4はEPO製剤のSDS−PAGEを示す(部位特異的、N末端)。
3.8.in vitroでの活性
フェニルヒドラジンの腹腔内注射を通じて人工的に貧血状態にしたマウスの脾臓から単離した赤血球前駆細胞のin vitroでの増殖を誘導する際のEPO試料の活性の定量を使用した。プロトコルは、Krystal[1972]により報告されている方法に基づき改作
した。アッセイは、赤血球前駆細胞へのEPOの添加及び3H−チミジン取り込み速度の定量によるDNA複製速度の測定に基づく。
3.9.安定性試験
無菌のEPO複合体を20mMリン酸ナトリウム、pH7.4;5%ソルビトール及び0.025mg/ml Tween 20中で、4℃で6週間保存した。下記条件の下、SECカラムを使用して試料のSE−HPLCを実施した:注入容量:100μl、流速:0.250ml/分、ランニングバッファー:0.1Mリン酸ナトリウム、pH:6.9。
3.10.EPO製剤のin vivoでの効力
EPO製剤のin vivoでの効力を、雌性マウスB6D2F1(7週齢〜8週齢)で試験し、5μg〜15μgのタンパク質用量(同一活性)をマウスに皮下注射した。これらの動物は、4匹ずつ7つの群に分けた。以下のようにEPO製剤を各群の各動物に与えた:EPO、EPO−PSA複合体、PBS、Aranesp(5μg)。各動物から血液50μlを採取し、retic count染料で染色した後、FACSによって分
析した(図8及び図9)。
【0098】
十分に混合した全血5μlを、Retic−Count試薬1mlと混合し、暗所で室温で30分間インキュベートした。その後、試料をFACS機器の助けを借りて網状赤血球を計数することによって分析した。
3.11.Elisa
EPO−PSAをプレート全体にコーティングされた抗PSA抗体によって捕捉した。捕捉されたEPO−PSAを抗EPO抗体を用いて検出し、EPOとPSAとの複合体のみを検出した。
3.12.in vivoでのクリアランス
EPO製剤のin vivoでのクリアランスをマウスで試験した。適切量のタンパク質用量をマウスに皮下注射及び静脈内注射した。EPO製剤を125Iで放射標識し、血液試料の放射能を頻繁に測定した。
結果
CAの活性化及び酸化度の定量
N−アセチルノイラミン酸(Neu5Ac)残基の直鎖α−2,8−結合ホモポリマーである、コロミン酸(CA)を使用した。コロミン酸の酸化への曝露を室温で20mM過ヨウ素酸塩を使用して15分間実行した。過ヨウ素酸塩処理後の内部α−2,8結合Neu5Ac残基の完全性をゲル浸透クロマトグラフィによって分析し、酸化コロミン酸(CAO)に対するクロマトグラフが得られ、物質を天然のCAの物質と比較した。酸化CA及び天然CAがほぼ同一の溶出プロファイルを示すことが分かり、逐次酸化段階がポリマー鎖の有意な断片化を引き起こすという証拠は無かった。
【0099】
CAの酸化状態の定量測定を、標準としてグルコースを使用して、フェロシアン化物(プルシアンブルー)へのアルカリ性溶液中でのフェリシアン化物イオンの還元[Park及びJohnson、1949]によって実施した。第1表は、酸化コロミン酸が、化学量論量を上回る
(100%超の)量の還元物質、すなわち、112mol%の見かけ上のアルデヒド含量(還元末端のヘミケタール及び(他の末端、非還元末端に)導入されたアルデヒドの還元力の組合せを含む)を有することが分かった。
【0100】
【表2】
【0101】
EPO複合体の調製、精製及び特徴付け
低pH(pH5.5)及びランダムpH(7.4)並びに4℃±1℃で反応を行うことによってN末端選択的にエリスロポエチン(EPO)のコロミン酸(CA)複合体を調製
及び精製する手順は、上記に詳述されている。これには、シアノ水素化ホウ素ナトリウムの存在下での複合体形成、続く遊離のEPOを除去するためのイオン交換クロマトグラフィ(AEX)を使用する精製、その後の疎水性相互作用クロマトグラフィ(HIC)によるCAの除去が包含される。N末端のα−アミノ基の選択的誘導体化が優勢となるように、また反応中のEPOの凝集を最小限にするために低pHを使用した。最終反応バッファーの組成は、10mM NaOAc中、5%ソルビトール、0.5mg/ml Tween 20(pH5.5)であった。
【0102】
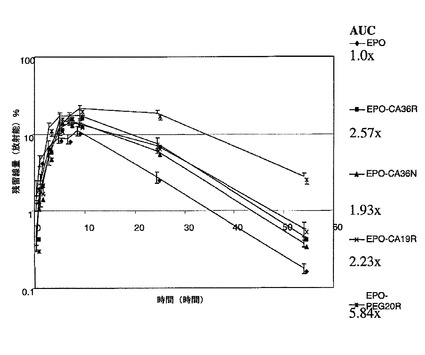
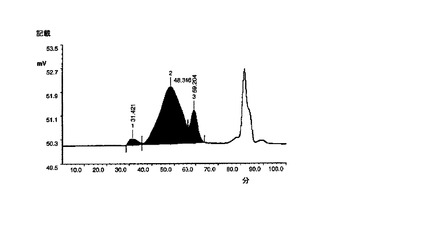
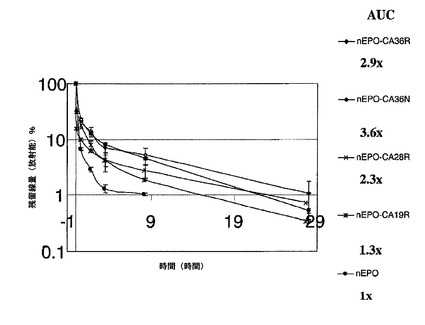
EPO−CA複合体の形成をSE−HPLC(EPOと比較しての、EPO−CAの保持時間の変化;両方の部分が同時に溶出もする);イオン交換クロマトグラフィ(AECカラムへの複合体の結合)及びポリアクリルアミドゲル電気泳動(SDS−PAGE;高分子量種へ向かってのバンドの広がり及びシフト)によって確認した(図4)。ポリシアル化試料は、in vitroで活性であり、未処理のEPOよりも著しく優れたプロファイル(PK及びPD)を示した。図11及び図12はin vivoでの結果を示す。
【0103】
図5の左手側は、24時間後のEPO−CA 39kDa複合体形成のSE−HPLCを示す。第3表はピーク解析表である。特徴付け条件:カラム:Superdex 200、バッファー:炭酸水素アンモニウム0.15M、pH:7.8。
【0104】
【表3】
【0105】
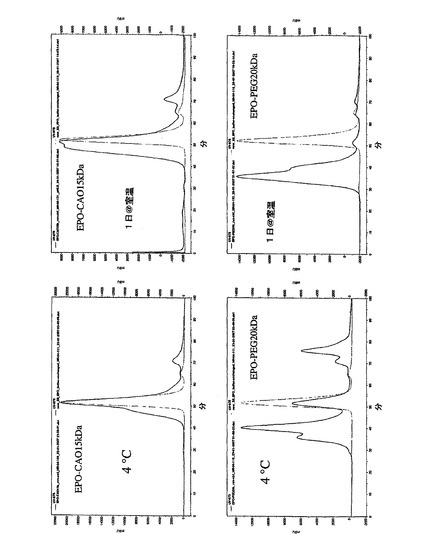
誘導体化の程度は、ポリシアル化EPOよりもPEG化EPOの方が大きいことが分かった(図6)。これは、PEGの不活性な性質及びシアル酸の帯電した性質に起因する可能性がある。FACSデータからの網状赤血球数は、EPOよりもPSA−EPO複合体の方が多かった(図7)。EPO−CAO複合体の精製後、SEC HPLCからのクロマトグラム上に有意なEPOは観察されず、誘導体化は、SE−HPLCでの保持時間の変化並びにSDS−PAGE上の高分子量バンドの広がり及びシフトによって示された。EPOのポリシアル化は、マウス抗PSA抗体を使用するウエスタンブロッティングによっても示された。静脈内投与では、PSA−EPO複合体のin vivoでのクリアランスプロファイルは、EPOと比べて優れていることも分かり(図8)、曲線下面積は7.1倍増大した。同様にEPO−PSAの皮下投与もEPOと比較してより大きな保持を示し、曲線下面積は2.5倍増大した。EPOのポリシアル化が反応混合物のインキュベーション時間、モル過剰及びpHに比例することも分かった。ポリシアル化NG EPOのin vivoでのクリアランスプロファイル(静脈内及び皮下)は、NG EPOよりも良好であることが分かった(図11及び図12)。幾つかのSDSゲルにおいて、EPOのジポリシアル化も観察された。EPO−PSA複合体は、ELISA法によっても確認した(図14及び図15)。赤血球造血現象は、EPOと比較してEPO−PSA複合体の方が大きいことが分かり、6KDa〜15KDaではポリマーの分子量に比例することが分かった(図18)。シアル酸の鎖が大きくなるにつれて赤血球造血の現象は低減するため、15KDaがEPOに最適な鎖長であることが分かった。これは、受容体から
の反発で生じるポリマーの負に帯電した性質に起因する可能性がある。この試験は、図18の助けを借りて確認した。AranespのEmaxは、臨床的に良好ではなく、血栓症をもたらし、骨髄枯渇の原因となり得るEPO−PSAよりも遥かに大きいことが分かり、処置後、ベースライン未満まで網状赤血球を低下させることも分かった。EPO−PSAは、EPOよりも著しく優れていることが分かり、EPO−PSAは、EPO−PEGと同様に優れていることが分かり、EPO−PSAも一定の赤血球造血をもたらす。
【0106】
PSA複合体が、in vitro活性アッセイにおいて活性であることが分かった。PSA−EPO複合体がPEG複合体と同様に良好であり、EPOよりもはるかに優れていることがin vivoでの効力試験から示される(図8及び図9)。
【0107】
NG EPO−CA複合体の形成をSE−HPLC(NGEPOと比較しての、NG EPO−CAの保持時間の変化;両方の部分が同時に溶出もする);イオン交換クロマトグラフィ(AECカラムへの複合体の結合)及びポリアクリルアミドゲル電気泳動(SDS−PAGE;高分子量種のバンドのシフト)によって確認した。図は、24時間後のEPO−CA 39kDa複合体形成を示す。ポリシアル化試料は、in vitroで活性であり、未処理のNGepoよりも著しく優れたプロファイル(PK及びPD)を示した。
【0108】
図10は、SE−HPLCの結果を示す。下記第4表にピーク解析を示す。特徴付け条件−カラム:Superdex 200、バッファー:炭酸水素アンモニウム0.15M、pH:7.8。
【0109】
【表4】
【0110】
図12は、in vivoでのクリアランスの結果を示す。PSA−NG EPOは、NG EPOと比べて、著しく優れたプロファイルを示した。
【0111】
第5表は、使用した各種パラメータの値を示し、第6表は、CA画分の分子量及び多分散度を示す。
【0112】
【表5】
【0113】
【表6】
【0114】
参照文献
【0115】
【表7】
【特許請求の範囲】
【請求項1】
EPO又はEPOホモログのN末端多糖誘導体である化合物であって、該多糖がアニオン性であり、且つ2個〜125個の糖ユニットを含み、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、化合物。
【請求項2】
前記多糖が、ポリシアル酸である、請求項1に記載の化合物。
【請求項3】
前記多糖が、シアル酸ユニットのみから成る、請求項2に記載の化合物。
【請求項4】
前記EPO又は前記EPOホモログが、前記多糖の還元末端ユニットで誘導体化される、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
一般式(I):
【化1】
(式中、mは、少なくとも1であり、
XBは、EPO又はEPOホモログであるB−XHに由来し(ここで、XHは、NH2であり、且つ前記EPO又は前記EPO様タンパク質のN末端である、又は、NH2であり、且つリジンアミノ酸側鎖のアミン基である)、
Lは、結合、結合基であるか、又はポリペプチドを含み、
GlyOは、アニオン性糖ユニットであり、
前記結合基は、存在すれば、一般式−Y−C(O)−R1−C(O)−であり、
Yは、NR2又はNR2−NR2であり、R1は、アルカンジイル、アリーレン、アルカリーレン、ヘテロアリーレン及びアルキルヘテロアリーレン(これらのいずれかはカルボニル結合、エステル結合、スルフィド結合、エーテル結合、アミド結合及び/又はアミン結合によって置換及び/又は介在されていてもよい)から成る群より選択される二官能性有機基であり、
且つR2は、H又はC1−6アルキルであり、
該EPOホモログは、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する)の化合物。
【請求項6】
Lが結合であるか、又は基
【化2】
である、請求項5に記載の化合物。
【請求項7】
前記EPOがグリコシル化され、且つ2個〜100個の糖ユニットを含む、請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
前記EPOがグリコシル化されず、且つ80個〜180個の糖ユニットを含む、請求項1〜6のいずれか一項に記載の化合物。
【請求項9】
請求項1〜8のいずれか一項に記載の化合物、及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物。
【請求項10】
治療で使用するための、請求項1〜8のいずれか一項に記載の化合物。
【請求項11】
EPO又はEPOホモログのN末端多糖誘導体の製造方法であって、2個〜200個の糖ユニットを含むアニオン性多糖を、前記EPO又は前記EPOホモログと化学的に反応させ、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、製造方法。
【請求項12】
前記アニオン性多糖が、前記EPO又は前記EPOホモログと反応する反応性アルデヒド基を有し、且つ前記誘導体が還元条件下で生成される、請求項11に記載の方法。
【請求項13】
前記反応性アルデヒド基が前記多糖の非還元末端に存在する、請求項12に記載の方法。
【請求項14】
前記多糖が、前記EPO又は前記EPOホモログのアミン基と反応する、請求項11〜13のいずれか一項に記載の方法。
【請求項15】
前記アミンが末端アミン基である、又は、
前記EPO又は前記EPOホモログのリジンアミノ酸に由来する、
請求項14に記載の方法。
【請求項16】
前記アニオン性多糖又は反応中間体が、酸性pHの第1の水溶液中で、前記EPO又は前記EPOホモログの末端アミン基と反応し、且つ得られた多糖誘導体を、前記第1の水溶液よりも高いpHの第2の水溶液中で精製する、請求項11〜15のいずれか一項に記載の方法。
【請求項17】
前記第1の水溶液の前記pHが4.0〜6.0の範囲であり、前記第2の水溶液の前記pHが6.5〜9.0の範囲である、請求項16に記載の方法。
【請求項18】
前記多糖が、前記EPO又は前記EPOホモログ上の糖質残基で反応する、請求項11〜13のいずれか一項に記載の方法。
【請求項19】
前記糖質残基が、前記EPO又は前記EPOホモログ上のペンダントグリコン基に位置する、請求項18に記載の方法。
【請求項20】
1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸から選択される製剤添加物の存在下で実行される、請求項11〜17のいずれか一項に記載の方法。
【請求項21】
前記製剤添加物が、バッファーであり、且つ該バッファーがリン酸ナトリウム/酢酸ナトリウムである、請求項20に記載の方法。
【請求項22】
EPO又はEPOホモログの多糖誘導体の化合物であって、
該多糖がアニオン性であり、且つ20個〜60個の糖ユニットを含み、
更に、該多糖が、前記EPO又は前記EPOホモログ上の糖質残基で化学的に反応し、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、化合物。
【請求項23】
前記多糖が、ポリシアル酸である、請求項22に記載の化合物。
【請求項24】
請求項18又は19に記載の方法により得られる、EPO又はEPOホモログの多糖誘導体の化合物。
【請求項25】
請求項22〜24のいずれか一項に記載の化合物、及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物。
【請求項26】
治療で使用するための、請求項22〜24のいずれか一項に記載のEPO又はEPOホモログの多糖誘導体の化合物。
【請求項1】
EPO又はEPOホモログのN末端多糖誘導体である化合物であって、該多糖がアニオン性であり、且つ2個〜125個の糖ユニットを含み、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、化合物。
【請求項2】
前記多糖が、ポリシアル酸である、請求項1に記載の化合物。
【請求項3】
前記多糖が、シアル酸ユニットのみから成る、請求項2に記載の化合物。
【請求項4】
前記EPO又は前記EPOホモログが、前記多糖の還元末端ユニットで誘導体化される、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
一般式(I):
【化1】
(式中、mは、少なくとも1であり、
XBは、EPO又はEPOホモログであるB−XHに由来し(ここで、XHは、NH2であり、且つ前記EPO又は前記EPO様タンパク質のN末端である、又は、NH2であり、且つリジンアミノ酸側鎖のアミン基である)、
Lは、結合、結合基であるか、又はポリペプチドを含み、
GlyOは、アニオン性糖ユニットであり、
前記結合基は、存在すれば、一般式−Y−C(O)−R1−C(O)−であり、
Yは、NR2又はNR2−NR2であり、R1は、アルカンジイル、アリーレン、アルカリーレン、ヘテロアリーレン及びアルキルヘテロアリーレン(これらのいずれかはカルボニル結合、エステル結合、スルフィド結合、エーテル結合、アミド結合及び/又はアミン結合によって置換及び/又は介在されていてもよい)から成る群より選択される二官能性有機基であり、
且つR2は、H又はC1−6アルキルであり、
該EPOホモログは、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する)の化合物。
【請求項6】
Lが結合であるか、又は基
【化2】
である、請求項5に記載の化合物。
【請求項7】
前記EPOがグリコシル化され、且つ2個〜100個の糖ユニットを含む、請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
前記EPOがグリコシル化されず、且つ80個〜180個の糖ユニットを含む、請求項1〜6のいずれか一項に記載の化合物。
【請求項9】
請求項1〜8のいずれか一項に記載の化合物、及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物。
【請求項10】
治療で使用するための、請求項1〜8のいずれか一項に記載の化合物。
【請求項11】
EPO又はEPOホモログのN末端多糖誘導体の製造方法であって、2個〜200個の糖ユニットを含むアニオン性多糖を、前記EPO又は前記EPOホモログと化学的に反応させ、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、製造方法。
【請求項12】
前記アニオン性多糖が、前記EPO又は前記EPOホモログと反応する反応性アルデヒド基を有し、且つ前記誘導体が還元条件下で生成される、請求項11に記載の方法。
【請求項13】
前記反応性アルデヒド基が前記多糖の非還元末端に存在する、請求項12に記載の方法。
【請求項14】
前記多糖が、前記EPO又は前記EPOホモログのアミン基と反応する、請求項11〜13のいずれか一項に記載の方法。
【請求項15】
前記アミンが末端アミン基である、又は、
前記EPO又は前記EPOホモログのリジンアミノ酸に由来する、
請求項14に記載の方法。
【請求項16】
前記アニオン性多糖又は反応中間体が、酸性pHの第1の水溶液中で、前記EPO又は前記EPOホモログの末端アミン基と反応し、且つ得られた多糖誘導体を、前記第1の水溶液よりも高いpHの第2の水溶液中で精製する、請求項11〜15のいずれか一項に記載の方法。
【請求項17】
前記第1の水溶液の前記pHが4.0〜6.0の範囲であり、前記第2の水溶液の前記pHが6.5〜9.0の範囲である、請求項16に記載の方法。
【請求項18】
前記多糖が、前記EPO又は前記EPOホモログ上の糖質残基で反応する、請求項11〜13のいずれか一項に記載の方法。
【請求項19】
前記糖質残基が、前記EPO又は前記EPOホモログ上のペンダントグリコン基に位置する、請求項18に記載の方法。
【請求項20】
1個又は複数のバッファー、安定化剤、界面活性剤、塩、ポリマー、金属イオン、糖、ポリオール又はアミノ酸から選択される製剤添加物の存在下で実行される、請求項11〜17のいずれか一項に記載の方法。
【請求項21】
前記製剤添加物が、バッファーであり、且つ該バッファーがリン酸ナトリウム/酢酸ナトリウムである、請求項20に記載の方法。
【請求項22】
EPO又はEPOホモログの多糖誘導体の化合物であって、
該多糖がアニオン性であり、且つ20個〜60個の糖ユニットを含み、
更に、該多糖が、前記EPO又は前記EPOホモログ上の糖質残基で化学的に反応し、該EPOホモログが、配列番号1に示すアミノ酸配列と50%以上の類似性又は相同性を有する、化合物。
【請求項23】
前記多糖が、ポリシアル酸である、請求項22に記載の化合物。
【請求項24】
請求項18又は19に記載の方法により得られる、EPO又はEPOホモログの多糖誘導体の化合物。
【請求項25】
請求項22〜24のいずれか一項に記載の化合物、及び1個又は複数の薬学的に許容される賦形剤を含む、医薬組成物。
【請求項26】
治療で使用するための、請求項22〜24のいずれか一項に記載のEPO又はEPOホモログの多糖誘導体の化合物。
【図1】


【図2】
【図3a】
【図3b】
【図4】
【図5】
【図6】
【図7】

【図8】

【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】

【図18】
【図19】
【図20】
【図2】
【図3a】
【図3b】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2013−100309(P2013−100309A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2012−287324(P2012−287324)
【出願日】平成24年12月28日(2012.12.28)
【分割の表示】特願2009−521343(P2009−521343)の分割
【原出願日】平成19年7月25日(2007.7.25)
【出願人】(507042545)リポクセン テクノロジーズ リミテッド (15)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成24年12月28日(2012.12.28)
【分割の表示】特願2009−521343(P2009−521343)の分割
【原出願日】平成19年7月25日(2007.7.25)
【出願人】(507042545)リポクセン テクノロジーズ リミテッド (15)
【Fターム(参考)】
[ Back to top ]