
オキサゾリジノン誘導体および使用方法
本発明は、新規なN−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミド誘導体、それらの許容できる酸付加塩、溶媒和物および水和物に関する。本発明はまた、本発明の化合物を含む組成物、ならびに抗菌剤によって有益に治療される疾患および病状を治療する方法におけるかかる組成物の使用も提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本願は、2006年10月23日出願の米国仮特許出願第60/853,890号、および2007年9月24日出願の同第60/974,637号の利益を主張する。これらの出願の内容を、参照によりその全体を本願明細書に援用する。
【0002】
本発明は、新規なN−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミド誘導体、それらの許容できる酸付加塩、溶媒和物、および水和物に関する。本発明はまた、本発明の化合物を含む組成物、および抗菌剤によって有益に治療される疾患および病状の治療方法におけるかかる組成物の使用をも提供する。
【背景技術】
【0003】
リネゾリドは、(S)−N−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミドについての一般名である。それは、多くの動物モデルにおいて抗菌剤として有効であることが示されている。マウス大腿症感染モデルで確立されたPK/PD関係から、有効性を決定する主要パラメータはMICを超える時間であることが示された。リネゾリドは、グラム陽性菌ならびに一部のグラム陰性菌および嫌気性菌を含めた多くのヒトの病原体および獣医学的病原体に対して有効である有用な抗菌剤であることが公知である。特許文献1:米国特許第5,688,792号および特許文献2:国際公開第95/07271号を参照のこと。
【0004】
臨床試験において、リネゾリドは、以下の感染症の治療において有効であることが示されている:バンコマイシン耐性フェシウム菌;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;および肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎(Barbachyn,MRら, Pharmacia & Upjohn Co.に対する特許文献3:米国特許第5,688,792号;2006年7月に改訂されたZYVOXラベル)。
【0005】
推奨されるヒトの用量は、バンコマイシン耐性フェシウム菌(菌血症を含む)、院内肺炎;併発性皮膚・皮膚組織感染症;および市中肺炎(菌血症を含む)に対しては、12時間ごとに600mgである。400mgのBIDの用量は、無併発性皮膚・皮膚組織感染症に対して推奨される。臨床試験において、この用量は、トラフ値における黄色ブドウ球菌に対するMIC90を超えることが示された。ヒトにおけるPK/PD関係は明確には確立されていない。1つの研究では、AUC/MICが有効性予測判断材料であることが見出されたが、しかしこのPK/PD予測判断材料は信頼性がないとみなされた。リネゾリドは、高用量では非線形の動力学を示す。1日3回の725mgの用量は、血清クレアチニンの増加によって認容性を認めることができた。骨髄抑制がリネゾリドを受けた患者で報告されている。この骨髄抑制は可逆的であり、リネゾリドを受けた患者は、毎週モニターされるべきである。ヒトにおけるリネゾリドについてのPK/PDはまだ十分に確立されていないが、同等またはより低い用量でMICを超える曝露レベルを維持できる、より長い血清半減期を有する化合物を同定することは明らかに有利であろう。これによって、必要とされるMICを維持しながらのより低いBID用量、またはAUCを減少しつつ必要とされるMICを維持するであろうより高い投与量QDの投与が可能になるであろう。
【0006】
リネゾリドの代謝はマウス、ラット、イヌおよびヒトですでに研究されており、2つの主な代謝経路が同定されている。排出される主な代謝産物は、それぞれ、モルホリン基の酸化によって形成されるラクトン環およびラクタム環の加水分解に由来するM4およびM6として公知のカルボン酸である。これらの代謝産物は不活性である。ヒトでは、主要な代謝経路はラクトン経路である。非特許文献1:Slatter,JGら, Xenobiotica 2002,32,907頁および非特許文献2:Drug Metab Dispos 2001,29,1136頁を参照。ヒトで投与された用量のおよそ35%は、尿中で親化合物として見出されるのに対し、その用量の50%は上記2つの代謝産物が占める。モルホリン環の酸化はCyp酵素によるものではない。生体外研究によって、リネゾリドは臨床的に関連するCypアイソフォーム(1A2;2C9;2C19;2D6;2E1;3A4)の基質でもなく、阻害薬でもなく、誘導因子でもないことが示された。非特許文献3:US NDA 第02130号を参照。
【0007】
リネゾリドのN−オキシドも、前臨床試験において抗菌剤として検討されている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第5,688,792号明細書
【特許文献2】国際公開第95/07271号パンフレット
【特許文献3】米国特許第5,688,792号明細書
【非特許文献】
【0009】
【非特許文献1】Slatter,JGら, Xenobiotica 2002,32,907頁
【非特許文献2】Drug Metab Dispos 2001,29,1136頁
【非特許文献3】US NDA 第02130号
【発明の概要】
【発明が解決しようとする課題】
【0010】
それゆえ、リネゾリドの有益な活性を示し、他の恩恵、例えば、代謝での問題の軽減を伴う有害な副作用の減少、も有する可能性がある化合物を生み出すこと、その薬理学的有効寿命をさらに延ばし、患者の服薬を高めること、そして潜在的には、個体群での薬物動態のばらつきを減少させ、および/または危険な薬物−薬物相互作用についてのその可能性を減少させることが望ましい。
【課題を解決するための手段】
【0011】
上記の課題は、本発明の新規なN−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミド誘導体、それらの許容できる酸付加塩、溶媒和物、および水和物によって解決される。
【図面の簡単な説明】
【0012】
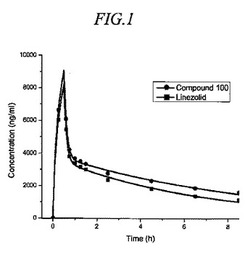
【図1】雌のチンパンジーへの点滴静注後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図2】雄のチンパンジーへの点滴静注後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図3】雌のチンパンジーへの経口投与後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図4】雄のチンパンジーへの経口投与後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【発明を実施するための形態】
【0013】
(定義)
「改善する」および「治療する」という用語は同じ意味で用いられ、両用語は、疾患(例えば、感染症、微生物)の発症または進行を減少、抑圧、減衰、縮小、停止、または安定させることを意味する。
【0014】
「疾患」は、細胞、組織、または臓器の正常機能を損傷するか、または妨げるあらゆる病状または障害を意味する。
【0015】
ある変動の天然の同位体存在量は、合成化合物において、合成に使用される化学物質の原材料に応じて発生することが分かる。従って、リネゾリドの調製は、少量の重水素化された同位体的同族体(isotopologues)および/または13Cを含有する同位体的同族体を本質的に含有する。この変動があったとしても、天然で存在する安定水素同位体および炭素同位体の濃度は、本発明の化合物の安定した同位体置換の度合いと比較して小さく、重要ではない。例えば、Wada EおよびHanba Y,Seikagaku 1994,66:15、Ganes LZら,Comp.Biochem.Physiol.Mol.Integr.Physiol.1998,119:725参照。本発明の化合物において、特定の位置が重水素を有すると示される場合、その位置の重水素の存在量は、0.015%である天然の重水素の存在量よりも実質的に大きいと理解される。重水素を有すると示される位置は、通常、当該化合物における重水素として指定された各原子で、少なくとも3000(45%の重水素導入)の最小限の同位体濃縮係数を有する。
【0016】
本願明細書で使用する「同位体濃縮係数」という用語は、特定の同位体の同位体存在量と天然存在量との比率を意味する。
【0017】
他の実施形態では、本発明の化合物は、式Iまたは式Iaで重水素として指定された各原子について、少なくとも3500(重水素として指定された各原子で、52.5%の重水素導入)、少なくとも4000(60%の重水素導入)、少なくとも4500(67.5%の重水素導入)、少なくとも5000(75%の重水素導入)、少なくとも5500(82.5%の重水素導入)、少なくとも6000(90%の重水素導入)、少なくとも6333.3(95%の重水素導入)、少なくとも6466.7(97%の重水素導入合)、少なくとも6600(99%の重水素導入)、または少なくとも6633.3(99.5%の重水素導入)の同位体濃縮係数を有する。
【0018】
本発明の化合物において、特定の同位体として特に指定されない任意の原子は、その原子の任意の安定した同位体を表すことを意味する。別段の記載がない限り、位置が具体的に「H」または「水素」と指定される場合、その位置は、その天然存在量同位体組成において水素を有すると理解される。
【0019】
別の実施形態では、本願明細書で使用する「化合物」は、10%未満、好ましくは6%未満、より好ましくは3%未満の合わせたすべての他の同位体的同族体(いずれの重水素または13Cを除く種類を含む)を含有する。特定の態様では、この化合物は「X」%未満の合わせたすべての他の同位体的同族体(いずれの重水素または13Cを除く種類を含む)を含有し、このXは0および10を含んで0〜10の任意の数字(例えば、1、0.5、0.001)である。10%を超える、合わせたすべての他の同位体的同族体を含有する組成物は、本願明細書で「混合物」と呼ばれ、それらは以下に示されるパラメータを満たさなければならない。本願明細書における同位体組成のこれらの限界および同位体組成へのすべての言及は、単に、式I/Iaの化合物のその活性な遊離の塩基形態中に存在する重水素/水素および13C/12Cの相対量を指し、対イオンの加水分解性部分の同位体組成を含まない。
【0020】
「同位体的同族体」という用語は、その分子またはイオンの同位体の組成だけにおいて、本発明の特定の化合物とは異なる種を指す。
【0021】
本願明細書において使用する「化合物」という用語は、その塩、溶媒和物、またはその水和物を含むことも意図する。本願で記載される本発明の特定の態様における「塩」、「溶媒和物」、または「水和物」という具体的な記述は、「化合物」という用語がこれらの他の形態の記述なく使用される本発明の他の態様では、これらの形態が意図的に省かれていると解釈されるべきではない。
【0022】
本発明の化合物の塩は、酸とアミノ官能基などのその化合物の塩基性基の間で、または、塩基とカルボキシル官能基などのその化合物の酸性基の間で形成される。別の好ましい実施形態によれば、化合物は、薬理学的に許容できる酸付加塩である。
【0023】
本願明細書において使用する「薬理学的に許容できる」という用語は、適切な医学的判断の範囲で、ヒトおよび他の哺乳動物の組織に接触させる使用において、過度の毒性、刺激、アレルギー反応などがなく好適で、適度の利益/危険性の比でつり合いの取れている、成分を指す。「薬理学的に許容できる塩」とは、服用者への投与において、直接的にまたは間接的に本発明の化合物を提供できる非毒性の塩を意味する。「薬理学的に許容できる対イオン」は、服用者への投与において塩から放出される場合に非毒性である、その塩のイオン部分である。
【0024】
薬理学的に許容できる塩を形成するために一般的に使用される酸としては、二硫化水素、塩化水素酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸などの無機酸、ならびに、p−トルエンスルホン酸、サリチル酸、酒石酸、酸性酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、p−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸などの有機酸、ならびに関連した無機酸および有機酸が挙げられる。かかる薬理学的に許容できる塩としては、従って、硫酸塩、ピロ硫酸塩、硫酸水素塩、亜硫酸塩、亜硫酸水素塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−二酸塩、ヘキシン−1,6−二酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スフホン酸塩、マンデル酸塩、および他の塩が挙げられる。好ましい薬理学的に許容できる酸付加塩としては、塩化水素酸および臭化水素酸などの鉱酸から形成されるもの、特にマレイン酸などの有機酸から形成されるものが挙げられる。
【0025】
本願明細書において使用する「水和物」という用語は、非共有結合性の分子間力によって結合された、化学量論量または非化学量論量の水を更に含む化合物を意味する。
【0026】
本願明細書において使用する「溶媒和物」という用語は、非共有結合性の分子間力によって結合された、化学量論量または非化学量論量の溶媒(水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノールなど)を更に含む化合物を意味する。
【0027】
本発明の化合物は、1つ以上の不斉炭素原子を含有してよい。従って、本発明の化合物は、個々の立体異性体(鏡像異性体もしくはジアステレオマー)、および立体異性体の混合物として存在することができる。従って、本発明の化合物としては、立体異性体混合物だけではなく、他の立体異性体が実質的に含まれない個々のそれぞれの立体異性体も挙げることができる。本願明細書において使用する「他の立体異性体が実質的に含まれない」という用語は、25%より少ない他の立体異性体が存在すること、好ましくは10%より少ない他の立体異性体が存在すること、より好ましくは5%より少ない他の立体異性体が存在すること、最も好ましくは2%より少ない他の立体異性体が存在すること、または「X」%より少ない他の立体異性体が存在すること(ここで、Xは0と100を含み、0〜100間の数字である)を意味する。ジアステレオマーを得る方法、または合成する方法は、当該技術分野で周知であり、最終化合物、または出発物質、または中間物に実施できるものとして、適用してよい。他の実施形態は、上記化合物が単離された化合物である実施形態である。本願明細書において使用する「少なくともX%鏡像異性体に富む」という用語は、その化合物の少なくともX%が単一の鏡像異性体形態にあることを意味する(ここで、Xは0と100を含み、0〜100間の数字である)。
【0028】
本願明細書において使用する「安定化合物」という用語は、その製造を可能にするのに十分な安定性を有し、本願明細書に記載した目的(例えば、治療薬にする製剤化、治療用の化合物の生産で使用するための中間体、単離可能な中間体または保存可能な中間体化合物、非定型抗精神病薬に応答する疾患または症状を治療すること)に有用となるように、十分な時間にわたって化合物の完全性を保持する化合物を指す。
【0029】
「2H」および「D」はともに重水素を指す。「立体異性体」は、鏡像異性体およびジアステレオマーの両方を指す。「tert」は、各々第三級を指す。「CDI」は1,1’−カルボニルジイミダゾールを指す。
【0030】
本願明細書中の変動要素のいずれかの定義において化学基の一覧を記述する際は、いずれかの単独の基または列挙された基の組合せとしてのその変動要素の定義を含む。本願明細書中の変動要素についてのある実施形態を記述する際は、いずれかの単独の実施形態または任意の他の実施形態もしくはその一部分との組合せとしてのその実施形態を含む。
【0031】
本願明細書全体を通して、「各Y」と言及することは、独立に、当てはまる場合にはすべての「Y」基(例えば、Y1、Y2、Y3、およびY4)を含み、「各W」は独立に、当てはまる場合にはすべての「W」基(例えば、W1、W2、W3、W4、およびW5)を含み、「各Z」は独立に、当てはまる場合にはすべての「Z」基(例えば、Z1、Z2、Z3、およびZ4)を含む。
【0032】
(治療用化合物)
本発明は、式Iもしくは式Iaの化合物:
【化1】

または式Iの塩;または式Iもしくは式Iaの水和物もしくは溶媒和物
(式中、
各Wは独立に、水素または重水素であり、
各Yは独立に、水素または重水素であり、
各Zは独立に、水素、重水素、またはフッ素であり、かつ
W、YまたはZの少なくとも1つは重水素である)
を提供する。
【0033】
一実施形態では、少なくとも1つのWは、重水素であり、少なくとも2つのY部分は重水素であり、かつ少なくとも2つのZ部分は重水素またはフッ素である。
【0034】
一実施形態では、W1およびW2は同時に重水素である。
【0035】
一実施形態では、Y1、Y2、Y3およびY4は同時に重水素である。
【0036】
一実施形態では、Z1、Z2、Z3およびZ4の各々は独立に、重水素およびフッ素から選択される。より具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素である。
【0037】
特定の実施形態では、式Iまたは式Iaの化合物の配置は(S)である。
【0038】
より具体的な実施形態では、Y1、Y2、Y3、Y4、W1およびW2は同時に重水素である。
【0039】
別の具体的な実施形態では、Z1、Z2、Z3およびZ4の各々は独立に、重水素およびフッ素から選択され、W1およびW2は同時に重水素である。
【0040】
別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3およびZ4は同時に重水素である。別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3およびZ4は同時に重水素であり、かつW3、W4およびW5は同時に水素である。
【0041】
さらに別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時にフッ素であり、かつY1、Y2、Y3およびY4は同時に重水素である。
【0042】
なお別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3、Z4、W1およびW2は同時に重水素である。
【0043】
別の実施形態では、Z1、Z2、Z3およびZ4は同時にフッ素であり、かつY1、Y2、Y3、Y4、W1、およびW2は同時に重水素である。
【0044】
別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素であり、かつY1、Y2、Y3、Y4、W1およびW2は同時に水素である。別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素であり、かつW3、W4およびW5は同時に水素である。
【0045】
さらに別の具体的な実施形態では、Z1、Z2、Z3Z4、Y1、Y2、Y3およびY4は同時に重水素であり、かつW1およびW2は同時に水素である。
【0046】
一実施形態では、式Iまたは式Iaの化合物は少なくとも3個の重水素原子を含有する。
【0047】
一実施形態では、式Iまたは式Iaの化合物は少なくとも4個の重水素原子を含有する。
【0048】
一実施形態では、式Iまたは式Iaの化合物は少なくとも5個の重水素原子を含有する。
【0049】
本発明の具体的な化合物の例としては、
【化2】
が挙げられる。
【0050】
本発明の具体的な化合物の他の例としては、以下の
【化3】
が挙げられる。
【0051】
別の一連の実施形態では、上述の実施形態のいずれかにおいて重水素として指定されない任意の原子は、その天然の同位体存在量で存在する。
【0052】
類似の化合物の中に重水素を導入する一般的方法が、広範囲にわたって文書化されている。例えば、そのほとんどの号が、生物活性有機低分子への重水素の特異的導入についての詳細な実験的記述を含むThe Journal of Labelled Compounds and Radiopharmaceuticals(John Wiley & Sons)を参照。また、例えば、Leis HJ,Curr Org Chem,1998,2:131およびその中の参考文献、ならびにMoebius G,ZfI−Mitteilungen 1989,150:297も参照。重水素標識した試薬の好適な商用供給業者としては、とりわけ、Isotec,Inc.(オハイオ州、マイアミズバーグ)、Cambridge Isotope Laboratories(マサチューセッツ州、アンドーバー)、ICON Services Inc.(ニュージャージー州、サミット)、およびC/D/N Isotopes,Inc.(カナダ、ケベック州、ポアントクレール)が挙げられる。
【0053】
式I/Iaの化合物の合成は、通常の技能を有する合成化学者により、有機合成の分野で公知の手段によって容易に行うことができる。かかる方法は、本願明細書に記載される化合物を合成するための対応する重水素化されかつ任意に他の同位体を含有する試薬および/または中間体を利用して、または同位体原子を化学構造に導入するための当該技術分野で公知の標準的合成手順を活用して実施することができる。関連する手順および中間体は、例えば、PCT公開公報WO1997010223、WO2005099353、WO1995007271、WO2006008754;Lizondo,Jら, Drugs Fut 1996,21(11):1116;Brickner,SJら, J Med Chem 1996,39(3):673;およびMallesham,Bら, Org Lett 2003,5(7):963に記載されている。以下のスキームは、式Iまたは式Iaの化合物がどうやって調製され得るかを図示する。
【0054】
スキーム1.式Iの化合物を調製するための一般的経路
【化4】
【0055】
上記のスキーム1は、式Iの化合物を調製するための一般的経路を示す。式Iaの化合物は、トリフルオロ過酢酸またはm−クロロ過安息香酸などの適切な酸化剤を使用して式Iの化合物から作製することができる。国際公開第1997010223号を参照。式I/Iaの化合物を合成するための他のアプローチは、実施例に示されているか、または本願明細書に引用された引用文献から容易に適合させることができる。これらの手順の変法およびそれらの最適化は、当業者の技能の範囲内である。
【0056】
上に示した特定のアプローチおよび化合物は、限定を意図したものではない。本願明細書のスキームにおいて明確に示されない経路の範囲内にあるものを含む、式I/Iaの化合物およびその合成前駆体のさらなる合成方法は、当業者の化学者らの手法の範囲にある。必要に応じて競合する副生成物を最小化して反応条件を最適化するための方法は当該分野で公知である。反応最適化およびスケールアップでは、高速パラレル合成装置およびコンピュータ制御マイクロリアクタを有利に利用してよい(例えば、Design And Optimization in Organic Synthesis,第2版,Carlson R編,2005;Elsevier Science Ltd.;Jaehnisch,Kら, Angew.Chem.Int.Ed.Engl.2004 43:406;およびこれらで引用される引用文献)。本願明細書で引用した合成の引用文献に加えて、反応スキームおよび手順は、例えば、SciFinder(登録商標)(米国化学会のCAS部門)、STN(登録商標)(米国化学会のCAS部門)、CrossFire Beilstein(登録商標)(Elsevier MDL)などの市販の構造検索可能なデータベースソフトウェア、あるいはグーグル(登録商標)などのインターネット検索エンジンまたは米国特許商標庁のテキストデータベースなどのキーワードデータベースを用いて、当業者によって決定されてよい。本願明細書に記載された方法は、本願明細書に具体的に記載された工程の前か後に、本願明細書の化合物の合成を最終的に可能にするために、好適な保護基を付加または除去する工程をさらに含んでもよい。さらに、様々な合成工程は、所望の化合物を得るために、別の順序または順番で実施されてもよい。適用できる化合物を合成するのに有用な合成化学変換および保護基方法論(保護および脱保護)は、当該分野で公知であり、例えば、R.Larock, Comprehensive Organic Transformations,VCH Publishers(1989);T.W.GreeneおよびP.G.M.Wuts, Protective Groups in Organic Synthesis,第3版、John Wiley and Sons(1999);L.FieserおよびM.Fieser, Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994);およびL.Paquette編集, Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、ならびにこれらのこれらに続く版に記載されているものが挙げられる。
【0057】
本願明細書に記載される合成方法は、さらに、前述のスキームで記載された工程のいずれかの前または後に、本願明細書に記載される式の化合物の合成を最終的に可能にするために、適切な保護基を付加もしくは除去する工程をも含んでいてよい。本願明細書に記載される方法は、1つの式の化合物を別の式の化合物に変換することを企図する。変換プロセスは、その場で実施してもよいし、または中間体化合物を単離して実施してもよい1つ以上の化学変換を指す。この変換は、本願明細書で引用された参考文献の中のものを含めて当該技術分野で公知の技術および手順を用いて、出発化合物または中間体を付加的な試薬と反応させることを含んでいてもよい。ある中間体は、精製(例えば、濾過、蒸留、昇華、結晶化、粉末化、固相抽出、およびクロマトグラフィー)して、または精製せずに使用することができる。
【0058】
本発明によって想定される置換基および変動要素の組み合わせは、安定化合物の形成の結果によるものだけである。
【0059】
(組成物)
本発明は、有効量の式I/Ia(例えば、本願明細書中の式のいずれも含む)の化合物、あるいは式Iの化合物の薬理学的に許容できる塩、式Iまたは式Iaの水和物または溶媒和物と、許容できる担体とを含む組成物も提供する。一実施形態では、この組成物は発熱物質を含まない。好ましくは、本発明の組成物は、担体が薬理学的に許容できる担体である薬理学的な用途(医薬組成物)のために処方される。この担体は、製剤の他の成分と適合し、薬理学的に許容できる担体の場合は、薬剤に通常使用する量においては、その服用者に無害であるという意味で「許容できる」ものでなければならない。
【0060】
本発明の医薬組成物に使用してよい薬理学的に許容できる担体、アジュバント、およびビヒクルとしては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミンなどの血清タンパク質、リン酸塩などの緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、硫酸プロタミン、リン酸一水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩などの塩または電解質、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースをベースとした物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリル酸塩、ワックス、ポリエチレン−ポリオキシプロピレンブロック重合体、ポリエチレングリコール、および羊毛脂を挙げることができるが、これらに限定されない。
【0061】
必要に応じて、上記医薬組成物中の本発明の化合物の溶解性およびバイオアベイラビリティは、当該技術分野で周知の方法により高めることができる。1つの方法としては、その製剤で脂質賦形剤を使用することが挙げられる。「Oral Lipid−Based Formulations:Enhancing the Bioavailability of Poorly Water−Soluble Drugs(Drugs and the Pharmaceutical Sciences)」,David J.Hauss編,Informa Healthcare,2007;および「Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery:Basic Principles and Biological Examples」,Kishor M.Wasan編,Wiley−Interscience,2006を参照。
【0062】
バイオアベイラビリティを高める別の公知の方法は、必要に応じてポリキサマー(LUTROL(商標)およびPLURONIC(商標)(BASF Corporation)など)、またはエチレンオキシドおよびプロピレンオキシドのブロックコポリマーを配合した本発明の化合物の非晶質形を使用することである。米国特許第7,014,866号および米国特許出願公開第20060094744号および同第20060079502号を参照。
【0063】
本発明の医薬組成物としては、経口投与、経直腸投与、経鼻投与、局所投与(口腔投与および舌下投与を含む)、膣内投与、または非経口投与(例えば、皮下投与、筋肉投与、静脈内投与および皮内投与を含む)に好適なものが挙げられる。一部の実施形態では、本願明細書の式の化合物は、(例えば、経皮パッチ、またはイオン導入法の技術を用いて)経皮投与される。他の製剤は、例えば、錠剤、持続放出性カプセルなどの単位量分の剤形として、およびリポソームとして、提供されることが好都合であるかも知れず、薬学分野で周知の任意の方法で調製してよい。例えば、Remington’s Pharmaceutical Sciences,Mack Publishing Company,ペンシルベニア州、フィラデルフィア(第17版、1985)を参照。
【0064】
かかる調製方法は、1つ以上の副成分を構成する担体のような成分を、投与されるべき分子と一緒にする工程を含む。通常、組成物は、活性成分を、液体担体、リポソーム、または微粉化した固体担体あるいはその両方と、均一におよび密に一緒にすることによって、および必要に応じて生成物を成形することによって調製される。
【0065】
一部の実施形態では、この化合物は経口投与される。経口投与に好適な本発明の組成物は、各々が所定量の活性成分を含有するカプセル剤、サシェ剤、または錠剤などの個別の単位として、粉剤または顆粒剤として、水性液体または非水性液体中の液剤または懸濁剤として、水中油型乳濁液として、油中水型乳濁液として、リポソームに詰められて、またはボーラスとしてなどで、提供されてもよい。軟ゼラチンカプセルは、化合物の吸収速度を有益に速くできる場合がある懸濁剤を含有するのに有用であり得る。
【0066】
経口用途の錠剤の場合、一般に使用される担体としては、乳糖およびトウモロコシでんぷんが挙げられる。ステアリン酸マグネシウムなどの滑沢剤も、通常添加される。カプセル剤形の経口投与に有用な希釈剤としては、乳糖および乾燥トウモロコシでんぷんが挙げられる。水系懸濁剤が経口投与される場合、活性成分は、乳化剤および懸濁化剤と混合される。必要に応じて、一部の甘味料、および/または矯味矯臭剤、および/または着色料を添加してもよい。
【0067】
経口投与に好適な組成物としては、風味ベースの成分、一般的にはショ糖およびアラビアゴムまたはトラガカントを含む薬用ドロップ、ならびにゼラチンおよびグリセリンまたはショ糖およびアラビアゴムなどの不活性ベース中に活性成分を含むトローチが挙げられる。
【0068】
非経口投与に好適な組成物としては、酸化防止剤、緩衝剤、細菌発育抑制剤、および意図する服用者の血液に対して製剤を等張にする溶質を含有してもよい水系無菌注入液および非水系無菌注入液、ならびに、懸濁化剤および増粘剤を含んでもよい水系無菌注入液および非水系無菌懸濁液が挙げられる。この製剤は、例えば、密封されたアンプルおよびバイアルなどの、1回の用量または複数回の用量の容器で提供されてもよく、使用する直前に、例えば注入用の水などの無菌液体担体の添加だけを必要とする凍結乾燥(冷凍乾燥)状態で保管されてもよい。即席注入液および即席注入懸濁液は、無菌粉剤、無菌顆粒剤、および無菌錠剤から調製されてもよい。
【0069】
かかる注入液は、例えば、無菌の注入可能な水系懸濁液または油性懸濁液の剤形であってもよい。この懸濁液は、好適な分散剤または湿潤剤(例えばTween80など)および懸濁化剤を使用して、当該分野で公知の技術に従って調製されてもよい。無菌の注入可能な調剤は、例えば1,3−ブタンジオール中の溶液のように、非毒性の非経口で許容できる希釈剤または溶媒中の無菌の注入可能な液剤または懸濁剤であってもよい。許容できるビヒクルおよび溶媒のうち、使用可能なものは、マンニトール、水、リンガー液、および生理食塩液である。加えて、無菌の不揮発性油は、溶媒または懸濁媒体として、従来から使用されている。この目的で、合成モノグリセリドまたはジグリセリドを含む、任意の無刺激不揮発性油を使用してもよい。オレイン酸およびそのグリセリド誘導体などの脂肪酸は、オリーブ油またはヒマシ油などの天然の薬理学的に許容できる油がそうであるように、注入液の調製に有用であり、特にそのポリオキシエチレン化したそのバージョンが有用である。これらの油液剤または油懸濁剤は、長鎖アルコール希釈剤または分散剤を含有してもよい。
【0070】
本発明の医薬組成物は、経直腸投与用の坐薬という剤形で投与されてもよい。これらの組成物は、本発明の化合物と、室温で固体であるが直腸温では液体であり、従って直腸で溶け活性成分を放出する好適な非刺激性の賦形剤とを混合することによって調製できる。かかる物質としては、ココアバター、蜜ロウ、およびポリエチレングリコールが挙げられるが、これらに限定されない。
【0071】
本発明の医薬組成物は、鼻エアロゾルまたは鼻孔吸入によって投与されてもよい。かかる組成物は、医薬製剤分野において周知の技術に従って調製され、ベンジルアルコールまたは他の好適な保存料、バイオアベイラビリティを高める吸収促進剤、フッ化炭素、および/あるいは当該分野で公知の他の可溶化剤または分散剤を使用して、生理食塩水中の溶液として調製されてもよい。かかる投与は、勃起不全に対する薬物について有効であることが公知である。例えば、Alexza Molecular Delivery Corporationに譲渡されたRabinowitz JDおよびZaffaroni ACの米国特許第6,803,031号明細書を参照。
【0072】
本発明の医薬組成物の局所投与は、所望の治療が、局所的付与によって容易に到達できる範囲または器官に関係する場合に特に有用である。皮膚への局所投与のために、この医薬組成物は、担体に懸濁または溶解された活性成分を含有する好適な軟膏剤を用いて処方されるべきである。本発明の化合物の局所投与のための担体としては、鉱油(mineral oil)、鉱油(liquid petroleum)、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ロウ、および水が挙げられるが、これらに限定されない。あるいは、この医薬組成物は、担体に懸濁または溶解された活性化合物を含有する好適なローション剤またはクリームを用いて処方できる。好適な担体としては、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられるが、これらに限定されない。本発明の医薬組成物は、直腸の坐薬製剤によって、または好適なかん腸製剤として、下部消化管に局所的に付与されてもよい。局所的な経皮パッチおよびイオン導入法による投与も、本発明に含まれる。
【0073】
主題の治療成分の投与は、関心部位に投与するように、局所的であってよい。関心部位に主題の組成物を提供するためには、注射、カテーテル、トロカール、噴出装置(projectile)、プルロニックゲル(pluronic gel)、ステント、持続した薬物放出をもたらすポリマー、または内部到達をもたらす他の装置の使用などのさまざまな技法を使用することができる。
【0074】
従って、また別の実施形態によれば、本発明の化合物は、人工器官、人工弁、代用血管、ステント、またはカテーテルなどの、移植可能な医療装置を被覆するための組成物に導入されてもよい。好適な被覆物および被覆された移植可能な装置の一般的な調製物は、当該分野において公知であり、米国特許第6,099,562号明細書、同第5,886,026号明細書、および同第5,304,121号明細書に例示されている。この被覆物は、一般的に、ヒドロゲルポリマー、ポリメチルジシロキサン、ポリカプロラクトン、ポリエチレングリコール、ポリ乳酸、エチレン酢酸ビニル、およびこれらの混合物などの生体適合性のポリマー材料である。この被覆物は、必要に応じて、フルオロシリコーン、多糖類、ポリエチレングリコール、リン脂質、またはこれらの組み合わせの好適な上塗りによってさらに被覆し、組成物に制御放出特性を与えてもよい。侵襲装置の被覆物は、本願明細書で使用される薬理学的に許容できる担体、アジュバント、またはビヒクルの定義の範囲内に含まれる。
【0075】
別の実施形態によれば、本発明は、移植可能な医療装置を被覆する方法であって、この装置を上に記載した被覆組成物に接触させる工程を含む方法を提供する。この装置の被覆が哺乳動物への移植の前に実施されることは、当業者には明らかである。
【0076】
別の実施形態によれば、本発明は、移植可能な薬物放出装置に含侵させる方法であって、この薬物放出装置を本発明の化合物または組成物に接触させる工程を含む方法を提供する。移植可能な薬物放出装置としては、生分解性ポリマーカプセルまたはカプセル(bullet)、非分解性で拡散性のポリマーカプセルおよび生分解性ポリマーウエハが挙げられるが、これらに限定されない。
【0077】
別の実施形態によれば、本発明は、本発明の化合物または本発明の化合物を含む組成物で被覆され、従って、この化合物が薬理学的に活性である、移植可能な医療装置を提供する。
【0078】
別の実施形態によれば、本発明は、本発明の化合物または本発明の化合物を含む組成物を含侵させているか、あるいはこの化合物または組成物を含有し、従って、この化合物がこの装置から放出され、この化合物が薬理学的に活性である、移植可能な薬物放出装置を提供する。
【0079】
患者から取り出すことによって器官または組織に到達可能な場合、かかる器官または組織は本発明の組成物を含有する媒体に侵漬されてもよいし、本発明の組成物が器官に塗布されてもよいし、本発明の組成物が他の任意の好都合な方法で付与されてもよい。
【0080】
別の実施形態では、本発明の組成物は、第2の治療剤をさらに含む。この第2の治療剤は、特に抗菌治療において、抗菌性化合物とともに投与した場合に有利な特性を有することが知られているか、または有利な特性を示す任意の化合物または治療剤が挙げられ、他の抗菌剤および/または抗炎症剤との併用療法が想起される。本発明に係る併用療法は、従って、少なくとも1種の式Iまたは式Iaの化合物の投与、および他の抗菌剤の任意の使用、ならびにシクロオキシゲナーゼ阻害薬、特にシクロオキシゲナーゼ−2の選択的阻害薬の任意の使用を含む。他の抗菌療法および抗炎症薬は、例えば、国際公開第01/34128号パンフレットおよび同第03/061704号パンフレットに開示されており、これらの出願を、それらが抗菌療法および抗炎症薬療法の組合せを開示する程度まで、参照によって本願明細書に援用する。
【0081】
本発明の化合物と配合することができる第2の治療剤の例としては、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブが挙げられるが、これらに限定されない。
【0082】
別の実施形態では、本発明は、本発明の化合物および第2の治療剤が互いに関与する、本発明の化合物および第2の治療剤の個別の剤形を提供する。本願明細書で使用する「互いに関与する」という用語は、個別の剤形が一緒に包装されているか、または別の形で互いにくっついており、従って、個別の剤形は一緒に市販され(互いに24時間より短い時間内に順次または同時に)投与されるという意図が容易に明らであることを意味する。
【0083】
本発明の化合物は、より長い半減期を示し、モル基準で同じ量のリネゾリドと比べて、投薬後24時間のより高い血清濃度レベルをもたらす。従って、一実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドの血清最終排泄半減期よりも大きい、上記化合物の血清最終排泄半減期をもたらす医薬組成物を提供する。他の実施形態では、式Iの化合物の血清最終排泄半減期は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるリネゾリドの血清最終排泄半減期の少なくとも125%、130%、135%、140%またはこれより大きい。
【0084】
関連する実施形態では、本発明は、有効量の式Iの化合物、またはその薬理学的に許容できる塩を含み、第1の組成物の単回用量が試験被験者に投与された後の化合物の血清最終排泄半減期が7時間を超える医薬組成物を提供する。
【0085】
別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合の投与後24時間のリネゾリドの血清濃度よりも大きい、投与後24時間の上記化合物の血清濃度をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物の投与後24時間でもたらされる式Iの化合物の血清濃度は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるリネゾリドの血清濃度の少なくとも150%、175%、200%、225%、250%、275%、300%またはこれより大きい。
【0086】
一実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドのAUC0−24よりも大きい、上記化合物のAUC0−24をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物によってもたらされるAUC0−24は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるAUC0−24の少なくとも125%、130%、135%、140%、145%またはこれより大きい。
【0087】
本発明の化合物はまた、リネゾリドと比べて特定の代謝に対してより大きい耐性を示す。従って、別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者への投与後24時間において無瑕で排出される化合物の量が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された後24時間において無瑕で排出されるリネゾリドの量よりも大きい医薬組成物を提供する。他の実施形態では、本発明の組成物の投与後24時間において無瑕で排出される式Iの化合物の量は、同じ投薬治療方式で投与された対応するリネゾリド組成物の投与後24時間において無瑕で排出されるリネゾリドの量の少なくとも150%、160%、170%、180%、190%、200%、210%、またはこれより大きい。
【0088】
関連する実施形態では、本発明は、有効量の式Iの化合物、またはその薬理学的に許容できる塩を含み、かつ被験者へのこの組成物の投与後24時間において、この化合物の有効量の少なくとも45%が被験者によって無疵で排出される医薬組成物を提供する。
【0089】
さらに別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者への投与が、活性成分のモル基準で式Iの化合物の量よりも多くかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドと、a)同様のAUC0−24、b)同様のCmaxまたはc)同様のCmin、のうちの1つ以上をもたらす医薬組成物を提供する。他の実施形態では、式Iの化合物の有効量は、式Iの化合物と同じ投薬治療方式で投与された場合にa)同様のAUC0−24、b)同様のCmax、またはc)同様のCmin、のうちの1つ以上をもたらすために必要とされるリネゾリドの量の80%、70%、60%、50%、40%、33%未満,であるかまたはこれより少ない。
【0090】
さらに別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその1つ以上の投与量の投与が、a)第1の投与量の投与後24時間の間6mg/Lを超える上記化合物の血清濃度の維持、およびb)投与後24時間の間6mg/Lを超えるリネゾリドの血清レベルを維持するために必要とされるリネゾリドの量を含む医薬組成物で等価な試験被験者にリネゾリドが投与された場合のリネゾリドのAUC0−24未満である、その化合物のAUC0−24をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物によってもたらされるAUC0−24は、リネゾリド組成物の必要とされる投与量によってもたらされるAUC0−24の85%、80%、75%、70%、65%未満,であるかまたはこれより少ない。
【0091】
上記の実施形態の各々において、式Iの化合物の薬理学的に許容できる塩、および/またはリネゾリドを、遊離塩基形態の代わりに使用してもよい。
【0092】
より具体的な実施形態では、上で示した組成物の各々において、上記化合物は化合物100、化合物101、化合物102または化合物103から選択される。
【0093】
「試験被験者」は、任意の哺乳動物、好ましくはヒトである。
【0094】
「等価な試験被験者」は、本願明細書では、試験被験者と同じ種および性別でありかつ試験被験者および等価な被験者の両方に対して等量のリネゾリドを投与した後に試験される薬物動態パラメータにおいて試験被験者と比べて10%以下のばらつきを示すものと定義される。
【0095】
本発明の医薬組成物において、本発明の化合物は有効量で存在する。本願明細書で使用する「有効量」という用語は、適切な投薬治療方式で投与された場合に、治療される障害の重症度、期間もしくは進行を縮小または改善するのに十分な量、治療される障害の進行を防ぐのに十分な量、治療される障害の退縮をもたらすのに十分な量、あるいは別の治療の予防効果または治療効果を高めるかもしくは向上させるのに十分な量を指す。
【0096】
動物とヒトに対する(体表面の1平方メートル当たりのミリグラムに基づいた)投与量の相関関係は、Freireichら、(1966)Cancer Chemother Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定されてよい。例えば、Scientific Tables,Geigy Pharmaceuticals,ニューヨーク州,アーズリー,1970,537を参照。
【0097】
本発明の化合物は等しい投与量でリネゾリドより長い血清半減期を示すため、それらを、最小阻止濃度(「MIC」)を超える必要とされる時間を依然維持しつつ、リネゾリドよりもより低い用量および/またはより低い頻度間隔で投与することができる。リネゾリドと比べて、本発明の化合物の投与のより低い頻度間隔は、各投薬に伴う血清濃度の急上昇の数を減少させるであろう。これは、ひいては、その患者の経時的な本発明の化合物への暴露総量(累積的AUC暴露(cumulative AUC exposure))を減少させるであろう。リネゾリドがミトコンドリアのタンパク質合成を阻害することによって引き起こされると考えられる、リネゾリドの累加性毒性に関連付けられてきたのはこの累積的AUC暴露である(Devriese ASら, Clin Infect Dis 2006,42:1111)。リネゾリドの累加性毒性は、患者がその薬物を摂取できる時間量を制限する。
【0098】
リネゾリドと比べた場合の累積的AUC暴露の減少は、本発明の化合物を含む制御放出製剤の使用によって、さらに高めることができる。かかる制御放出製剤は、当該技術分野で周知の方法を使用して調製される。例えば、Remmington:The Science and Practice of Pharmacy, 第21版(Lippincott Williams & Wilkins 2005); およびModern Pharmaceutics 第4版(Drugs and the Pharmaceutical Sciences 第121巻),Banker GSおよびRhodes CT編(Informa Healthcare 2002)を参照。
【0099】
動物とヒトに対する(体表面の1平方メートル当たりのミリグラムに基づいた)投与量の相関関係は、Freireichら、(1966)Cancer Chemother Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定されてよい。例えば、Scientific Tables,Geigy Pharmaceuticals,ニューヨーク州,アーズリー,1970,537を参照。本発明の化合物の有効量は、適切な場合にはいくつかの個々の用量の形態で、24時間ごとに約50mg〜約2000mgの範囲であってよい。一実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約250mg〜約1250mg、または各々12時間ごとに与えられる約125mg〜約625mgの2回の別々の投与量の範囲である。別の実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約750mg〜約1250mg、または各々12時間ごとに与えられる約375mg〜約625mgの2回の別々の投与量の範囲である。さらに別の実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約450mg〜約1200mg、または各々12時間ごとに与えられる約225mg〜約625mgの2回の別々の投与量の範囲である。より具体的な実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約450mg〜約750mg、または各々12時間ごとに与えられる約225mg〜約375mgの2回の別々の投与量の範囲である。上に記載した範囲の中またはその範囲の間に入る本発明の化合物の他の範囲もまた、本発明の範囲内である。有効用量は、当業者により認識されるように、治療される疾患、疾患の重症度、投与経路、患者の性別、年齢、および全体的な健康状態、賦形剤の使用法、他の薬剤の使用などの他の治療との併用の可能性、および主治医の判断によっても変化する。
【0100】
本発明の医薬組成物に存在し本発明の方法で使用されるミリグラム量の化合物は、遊離塩基の化合物の量を表す。本発明の化合物の薬理学的塩の使用では、等価なモル量の遊離塩基化合物が使用されるように明記された量を増やすことが必要であることを理解されたい。
【0101】
第2の治療剤を含む医薬組成物についての第2の治療剤の有効量は、単剤治療法において、その薬剤だけを使用する場合に通常使用される投与量の約20%〜100%の間である。有効量は、通常の単剤治療用量の約70%〜100%の間であることが好ましい。これらの第2の治療剤の通常の単剤治療投与量は、当該分野において周知である。例えば、Wellsら編集、Pharmacotherapy Handbook,第2版,Appleton and Lange,コネティカット州,スタンフォード(2000);PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000,デラックス版,Tarascon Publishing,カリフォルニア州,ロマリンダ(2000)を参照。これら各々の参考文献の内容全体を、参照によって、本願明細書に援用する。
【0102】
上で言及した第2の治療剤の一部は、本発明の化合物と相乗的に作用することが予想される。これが生じる場合、第2の治療剤および/または本発明の化合物の有効用量は、単剤治療において必要とされる量よりも減らされる。これは、第2の治療剤または本発明の化合物のどちらかの中毒性副作用の最小化、有効性の相乗的向上、投与または使用の容易性の向上、および/あるいは化合物の調製または処方の総合的な費用の削減に優位性を有する。
【0103】
(治療方法)
別の実施形態によれば、本発明は、リネゾリドによって有益に治療される疾患を患っているか、またはそれらにかかり易い被験者を治療する方法であって、上記被験者に有効量の本発明の化合物もしくは組成物を投与する工程を含む方法を提供する。かかる疾患は当該技術分野で周知であり、例えば、典型的には抗菌療法によって治療される種々の疾患状態(例えば、感染症、真菌性障害)の治療または予防を含む。それゆえ、式I/Iaの化合物は、グラム陽性菌および一部のグラム陰性菌および嫌気性菌によって媒介される障害を含めた障害の治療において有用性を有する。
【0104】
一実施形態では、本発明は、フェシウム菌、黄色ブドウ球菌、ストレプトコッカス・アガラクチア、肺炎球菌、化膿性連鎖球菌、フェカリス菌、表皮ブドウ球菌、スタフィロコッカス・ヘモリティカス、およびパスツレラ・マルトシダから選択される細菌によって引き起こされる感染症を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0105】
別の実施形態では、本発明は、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎;および結核から選択される疾患もしくは障害(またはそれらの症状)を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0106】
別の実施形態では、本発明は、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;および肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎から選択される疾患もしくは障害(またはそれらの症状)を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0107】
本発明の方法は、細菌感染治療の他の治療方法とともに用いることができる。特に、抗菌療法では、他の抗菌剤および/または抗炎症薬を用いた併用療法が想起される。少なくとも1種の式Iもしくは式Iaの化合物の投与、ならびに他の抗菌剤の任意の使用、およびシクロオキシゲナーゼ阻害薬、特にシクロオキシゲナーゼ−2の選択的阻害薬の任意の使用。かかる薬剤の組合せは、一緒に投与されてもよいし、または別々に投与されてもよく、別々に投与されるときは、投与は同時に行ってもよいし、または(時間的に近接する場合および離れている場合の両方とも)任意の順番で逐次的に行ってもよい。他の抗菌療法および抗炎症薬は、例えば、国際公開第01/34128号および同第03/061704号に記載されており、これらの出願を、それらが抗菌療法および抗炎症療法の組合せを開示する程度まで、参照により本願明細書に援用する。
【0108】
本願明細書に記載される方法は、被験者が特に明記された治療を必要とすると同定される方法を含む。かかる治療を必要とする被験者を同定することは、被験者または医療専門家の判断よるものであってよく、主観的(例えば、所見)であってもよいし、客観的(例えば、試験または診断方法によって測定できる)であってもよい。
【0109】
別の実施形態では、本発明は、細胞の活性を調節する方法であって、細胞を本願明細書中の式のいずれかの化合物の1種以上と接触させることを含む方法を提供する。
【0110】
別の実施形態では、本発明は、細菌感染症を患っているか、またはそれらにかかり易い患者の治療方法であって、それを必要とする患者に24時間の期間にわたって約450mg〜約750mgの式Iまたは式Iaの化合物を投与する工程を含む方法を提供する。別の実施形態では、患者は、450mg〜700mgの式Iまたは式Iaの化合物を投与される。
【0111】
別の実施形態では、上記の方法は、約3mg/Lを超える定常状態Cminをもたらす。別の実施形態では、上記の方法は、約4mg/Lを超える定常状態Cminをもたらす。別の実施形態では、上記の方法は、約6mg/Lを超える定常状態Cminをもたらす。さらに別の実施形態では、上記の方法は、約18mg/L未満の定常状態Cmaxをもたらす。別の実施形態では、上記の方法は、約16mg/L未満の定常状態Cmaxをもたらす。別の実施形態では、上記の方法は、約13mg/L未満の定常状態Cmaxをもたらす。さらに別の実施形態では、上記の方法は、約11.5mg/L未満の定常状態Cmaxをもたらす。
【0112】
別の実施形態では、上記の治療方法は、患者に1種以上の第2の治療剤を併用投与するさらなる工程を含む。第2の治療剤の選択は、リネゾリドとの併用投与に有用であることが公知の任意の第2の治療剤から行ってよい。
【0113】
具体的な実施形態では、本発明の併用療法は、式Iの化合物と、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される第2の治療剤とを投与することを含む。
【0114】
本願明細書で使用する「併用投与」という用語は、第2の治療剤が、1つの剤形(上述したように、本発明の化合物および第2の治療剤を含む本発明の組成物など)の一部として、または複数の個別の剤形として、本発明の化合物と一緒に投与されてもよいという意味である。あるいは、さらなる薬剤が、本発明の化合物の投与の前に、本発明の化合物の投与に続いて、または本発明の化合物の投与の後に投与されてもよい。かかる併用療法において、本発明の化合物および第2の治療剤はともに、従来の方法で投与される。本発明の化合物と第2の治療剤の両方を含む本発明の組成物を患者に投与することは、同じ治療剤、他の任意の第2の治療剤、または本発明の任意の化合物を、当該患者に、治療期間の別の時間に、別途投与することを除外しない。
【0115】
これらの第2の治療剤の有効量は当業者に周知であり、投与の案内は、本願明細書で引用した特許公報および特許公開公報、ならびに、Wellsら編集、Pharmacotherapy Handbook,第2版,Appleton and Lange,コネティカット州,スタンフォード(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,デラックス版,Tarascon Publishing,カリフォルニア州,ロマリンダ(2000)ならびに他の医療文献において見出してよい。しかしながら、第2の治療剤の最適な有効量の範囲を決定することは、十分に当業者の視野の範囲内である。
【0116】
本発明の一実施形態では、第2の治療剤が被験者に投与される場合、本発明の化合物の有効量は、第2の治療剤が投与されない場合の本発明の化合物の有効量よりも少なくなる。他の実施形態では、第2の治療剤の有効量は、本発明の化合物が投与されない場合の第2の治療剤の有効量よりも少なくなる。このようにして、どちらか一方の薬剤の多用量に関連した好ましくない副作用は、最小化される場合がある。他の潜在的な利点(投薬治療方式の改善および/または薬物費用の削減が挙げられるが、これらに限定されない)は、当業者に明らかである。
【0117】
さらに別の態様では、本発明は、被験者における上に記載の疾患、障害またはその症状の治療または予防のための薬剤の、単一組成物または個別の剤形としての製造において、式Iまたは式Iaの化合物の単独の使用または上述の第2の治療剤のうちの1つ以上との併用を提供する。本発明の他の態様は、被験者における本願明細書において記載したその疾患、障害、または症状の治療または予防に使用する本願明細書中の式の化合物である。
【0118】
(診断法およびキット)
本発明の化合物および組成物は、溶液中または血漿などの生体試料中のリネゾリドの濃度の測定方法、リネゾリドの代謝の検査方法、および他の分析的研究方法における試薬としても有用である。
【0119】
一実施形態によれば、本発明は、溶液中または生体試料中のリネゾリドの濃度の測定方法を提供し、この測定方法は、
a)既知濃度の式Iまたは式Iaの化合物を、生体試料の溶液に添加する工程と、
b)式Iまたは式Iaの化合物からリネゾリドを識別する測定装置にその溶液または生体試料を曝露する工程と、
c)検出された量の式Iまたは式Iaの化合物を、その生体試料または溶液に添加された既知濃度の式Iまたは式Iaの化合物に関連付けるために、測定装置を較正する工程と、
d)この較正された測定装置で、生体試料中のリネゾリドの量を測定する工程と、
e)式Iまたは式Iaの化合物について得られた検出量と濃度との間の相関を用いて、試料の溶液中のリネゾリドの濃度を決定する工程と
を含む。
【0120】
式Iまたは式Iaの対応する化合物からリネゾリドを識別できる測定装置としては、同位体存在量だけが互いに異なる2つの化合物を識別できる任意の測定装置が挙げられる。測定装置の例としては、質量分析計、NMR分光計、またはIR分光計が挙げられる。
【0121】
他の実施形態では、本発明は、式Iまたは式Iaの化合物の代謝安定性の評価方法を提供し、この評価方法は、ある時間にわたって、この式Iまたは式Iaの化合物を代謝酵素源に接触させる工程と、その時間の後に、式Iまたは式Iaの化合物の量を、式Iまたは式Iaの化合物の代謝産物と比較する工程と、を含む。
【0122】
関連する実施形態では、本発明は、この式Iまたは式Iaの化合物を投与した後、患者における式Iまたは式Iaの化合物の代謝安定性を評価する方法を提供する。この方法は、被験者へ式Iまたは式Iaの化合物を投与した後のある時間において、患者からの血清、尿または糞便試料を入手する工程と、式Iまたは式Iaの化合物の量を、血清、尿または糞便試料における式Iまたは式Iaの化合物の代謝産物と比較する工程とを含む。
【0123】
本発明はまた、本願明細書に記載されたものを含めた感染性疾患または感染性障害を治療するために使用するキットも提供する。これらのキットは、(a)式I/Iaの化合物、または式Iの塩、または式Iもしくは式Iaの水和物もしくは溶媒和物を含み、容器に入っている医薬組成物と、(b)本願明細書に記載されたものを含めた感染性疾患または感染性障害を治療するための医薬組成物の使用方法を説明する説明書とを含む。
【0124】
容器は、この医薬組成物を保持できる任意の容器あるいは他の密封されたまたは密封可能な器具であってよい。例としては、瓶、アンプル、各区画もしくはチャンバがこの組成物の1回の用量を含む、区画されたホルダーを有するボトルすなわち多室型のホルダーを有するボトル、各区画がこの組成物の1回の用量を含む分けられた薄片の包み、またはこの組成物の1回の用量を分配するディスペンサが挙げられる。容器は、当該分野で公知の任意の従来の形状または形態で、例えば、紙もしくはダンボール箱、ガラスもしくはプラスチックの瓶またはジャー、再封可能な袋(例えば、異なる容器に入れるために錠剤の「詰め替え」を保管するため)、または治療のスケジュールに従ってパックの外に押し出す個々の用量を入れたブリスターパックなどの薬理学的に許容できる材料で作られた容器であってよい。使用する容器は、関連する具体的な剤形に応じることができ、例えば従来のダンボール箱は、一般に液体懸濁液を保持するためには使用されない。1回の用量の剤形を市場に出すために、単一のパッケージにおいて、複数の容器を一緒に使うことができることは、実用的である。例えば、錠剤は瓶に入れられ、次にこの瓶が箱の中に入れられてもよい。容器はブリスターパックであることが好ましい。
【0125】
このキットはさらに、情報および/または医師、薬剤師もしくは被験者に対する指示を含むタイプの記憶を助けるものを含んでよい。かかる記憶を助けるものとしては、そのように特定された錠剤またはカプセルが摂取されるべき投与計画の日に対応する、投与量が入っている各チャンバもしくは区画の上に印刷された数字、または各チャンバもしくは区画の上に印刷されたその週の曜日、または何らかのタイプの情報を含むカードが挙げられる。1回用量のディスペンサについては、記憶を助けるものとしてはさらに、すでに分注された1日の用量の数を示す機械式計数器、ならびに、例えば最後の1日の用量が摂取された日付を読み出し、かつ/またはいつ次の用量を摂取するべきかを思い出させる液晶表示および/または可聴式の備忘録信号に接続された電池式のマイクロチップメモリが挙げられる。かかるキットで有用な他の記憶を助けるものは、カードに印刷されたカレンダー、および容易に分かる他のバリエーションである。
【実施例】
【0126】
(実施例1.中間体12の合成)
【化5】
氷浴で冷却した、酢酸エチル(「EtOAc」)(140ml)中のd8−モルホリン(10;23.5g、0.25mol)およびiPr2EtN(44ml、0.25mol)の撹拌した溶液に、3,4−ジフルオロニトロベンゼン(11、27.4ml、0.25mol)を10分間かけて滴下した。この反応混合物を室温で48時間撹拌した。この反応混合物をEtOAc(300ml)およびCH2Cl2(50ml)で希釈し、次いで水(350ml)を加えた。層を分離させ、水層をEtOAc(3×300ml)で洗浄した。合わせた有機物を乾燥し(MgSO4)、濾過し、真空中で濃縮した。20% EtOAc/ヘキサンで溶出するカラムクロマトグラフィ(1kgシリカ)を使用して粗生成物を生成し、中間体12を87%収率で得た。
【0127】
(実施例2.中間体13の合成)
【化6】
N2下の変性EtOH(875ml)中の12(50g、0.21mol)の撹拌した懸濁液に10% Pd/C(50%湿体、17.5g)を加えた。この反応容器にN2を10分間、H2を10分間パージし、大気圧のH2下で一晩撹拌した。水素化を停止し、この容器にN2を15分間パージした。この混合物をセライトを通して濾過し、変性EtOH(500ml)およびDCM(3×700ml)で洗浄した。合わせた濾液を真空中で濃縮し、所望のアニリン13(38.5g、90%収率)を桃色固体として得た。
【0128】
(実施例3.(R)−d2−エピクロロヒドリン14Bの合成)
【化7】
【0129】
Et2O(1000ml)中のメチル−α,β−イソプロピリデン−D−グリセレート(20;175g、1.09mol、1当量)の氷冷した溶液に、LiAlD4(34.43g、0.82mol、0.75当量)をEt2O(1000ml)中の懸濁液として3時間かけて加えた。この反応混合物を5時間還流させた。この混合物を室温まで冷却し、Et2O(1000ml)で希釈し、水(40ml)でクエンチした。この混合物を15分間撹拌し、濾過し、固体をEt2O(1000ml)で洗浄した。濾液を真空中で濃縮し、対応するアルコール21を高純度で得た(121.3g、83%収率)。
【0130】
アルコール(21;60.65g、0.45mol、1当量)を、PPh3(124g、0.47mol、1.05当量)およびDBU(34ml、0.225mol、0.5当量)とともにベンゼン(110ml)に溶解させた。この混合物を、DBU(17ml、0.11mol、0.25当量)を含むCCl4(110ml)の還流溶液に30分間かけて滴下した。この反応混合物を還流状態で一晩撹拌した。この反応混合物を室温まで冷却し、真空中で濃縮し、粗製混合物を得た。この粗製混合物をシリカ(60g)上に吸収させ、EtOAc:ヘキサン 1:1で溶出するカラムクロマトグラフィ(シリカ:1200g)を使用して精製し、所望のクロリド22を54%収率で得た。
【0131】
クロリド22(37.6g、0.25mol)を、アセトン(60ml)および1M HCl(150ml)の溶液に加えた。この混合物を55℃で30分間加熱した。この反応混合物を室温まで冷却し、アセトンを真空中で除去した。この混合物をNaCl(43g)で飽和させ、EtOAc(2×250ml)で抽出した。合わせたEtOAc層をMgSO4で乾燥し、濃縮し、ジオール、(R)−d2−クロロヒドリン23(21.8g、79%収率)を得た。
【0132】
ピリジン(210ml)中の(R)−d2−クロロヒドリン23(21.0g、0.19mol、1当量)の氷冷した溶液に、トルエンスルホニルクロリド(35.5g、0.19mol、1当量)を少しずつ加えた。このスルホニルクロリドの添加終了後、この混合物を室温まで加温し、1時間撹拌した。この混合物にEt2O(300ml)を加え、水性洗浄液が酸性になるまでこの混合物を1M HCl(3×500ml)で洗浄した。この有機抽出液を飽和NaHCO3水溶液(300ml)で洗浄し、MgSO4で乾燥し、濃縮し、対応するトシレート24(31.3g、63%収率)を得た。
【0133】
ナトリウム金属(5g、37.48mmol、2当量)をエチレングリコール(40ml)に加え、この混合物を20℃で一晩撹拌し、ナトリウムエチレングリコレートのエチレングリコール溶液を生成した。エチレングリコール(5ml)中のトシレート24(5g、18.74mmol、1当量)の溶液を加え、この混合物を20℃で15分間撹拌した。生成物を減圧下でこの混合物から除去し、ドライアイス/IPAコールドフィンガー中で透明な液体として集め、鏡像異性体に富む(R)−d2−エピクロロヒドリン25(1.02g、58%収率)を得た。キラルGCは、79.4%の鏡像体過剰率を示した。
【0134】
(S,S)−コバルト(II)触媒(43.2mg、0.0716mmol)をトルエン(8μl)に溶解させた。酢酸(8.6μl、0.143mmol、2当量)を加え、生成した混合物を開放系で室温で30分間撹拌し、この間にこの混合物の色は橙色から暗褐色に変化した。すべての揮発性物質を真空中で除去し、コバルト(III)触媒のアセテート錯体を褐色残渣として得た。この調製した触媒に、80%鏡像異性体に富む(R)−d2−エピクロロヒドリン25(2.5g)およびTHF(2.5ml)を加えた。この反応フラスコを0℃に冷却し、H2O(0.05ml)を15分間かけて滴下した。この反応液を室温まで加温し、18時間撹拌した。この反応混合物に、少量のMgSO4を加え、室温での蒸留により(R)−d2−エピクロロヒドリン26を単離し、エピクロロヒドリンおよびTHFの1:1混合物を得た。キラルGC分析は99.1%eeを示した。
【0135】
(実施例4.中間体15Aおよび15Bの合成)
【化8】
アニリン13(15g、74mmol、1.0当量)をN2下でイソプロパノール(75ml)に溶解させ、(R)−エピクロロヒドリン(14A;7.0g、81.4mmol、1.1当量)を加えた。この混合物を還流状態で一晩撹拌した。溶媒を真空中で除去し、残渣をカラムクロマトグラフィ(750gシリカ、CH2Cl2、次いで1% MeOH、CH2Cl2)によって精製し、15Aを淡褐色油状物として得た(12.8g、58%収率)。
【0136】
アニリン(4.1g、20.1mmol、1.0当量)をN2下でイソプロパノール(20ml)に溶解させ、(R)−d2−エピクロロヒドリン(14B;2.0g、22.1mmol、1.1当量)を加えた。この混合物を還流状態で一晩撹拌した。溶媒を真空中で除去し、残渣をカラムクロマトグラフィ(300gシリカ、CH2Cl2、次いで1% MeOH、CH2Cl2)によって精製し、15Bを淡褐色油状物として得た(3.0g、50%収率)。LCは純度97%を示した。キラルLCは、95.7%の鏡像体過剰率を示した。
【0137】
(実施例5.中間体17Aおよび17Bの合成)
【化9】
中間体15A(12.8g、0.043mol、1当量)、フタルイミドカリウム(16;10.4g、0.056mol、1.3当量)およびDMF(100ml)を100℃で5時間加熱した。LC分析は反応の完結を示した。この反応混合物を室温まで冷却し、水(450ml)の中へ注ぎ込んだ。この混合物を2時間撹拌し、濾過し、固体ケーキを真空オーブン中、40℃で一晩乾燥し、17Bを得た(9g、51%収率)。LCMSは純度95.0%を示した。
【0138】
中間体15B(3.2g、10.7mmol、1当量)、フタルイミドカリウム(16;2.58g、13.9mmol、1.3当量)およびDMF(23ml)を100℃で5時間加熱した。この反応混合物を室温まで冷却し、水(100ml)の中へ注ぎ込んだ。この混合物を2時間撹拌し、濾過し、固体ケーキを真空オーブン中、40℃で一晩乾燥し、17Bを得た(2.27g、52%収率)。LCは純度98.2%を示した。キラルLCは98.6%の鏡像体過剰率を示した。
【0139】
(実施例6.中間体18Aおよび18Bの合成)
【化10】
中間体17A(9.0g、22mmol、1当量)をDCM(100ml)に溶解させ、カルボニルジイミダゾール(5.0g、31mmol、1.4当量)を室温で加え、この混合物を窒素下で一晩撹拌した。LC分析は反応の完結を示した。水(300ml)をこの混合物に加え、水層をDCM(300ml)で抽出した。合わせたDCM層をMgSO4で乾燥し、真空中で濃縮し、18Aを得た。LCMSは純度94.4%を示した。
【0140】
中間体17B(2.08g、5.08mmol、1当量)をDCM(25ml)に溶解させ、カルボニルジイミダゾール(1.15g、7.11mmol、1.4当量)を室温で加え、この混合物を窒素下で一晩撹拌した。LC分析は反応の完結を示した。水(70ml)をこの混合物に加え、水層をDCM(70ml)で抽出した。合わせたDCM層をMgSO4で乾燥し、真空中で濃縮し、18Bを得た(2.1g、95%収率)。キラルLCは99.0%の鏡像体過剰率を示した。
【0141】
(実施例7.中間体19Aおよび19Bの合成)
【化11】
MeOH(100ml)およびヒドラジン水和物(6.1ml、0.125mol、5.5当量)を18A(9.9g、23mmol、1当量)が入っているフラスコに加えた。この混合物を還流温度で1時間撹拌した。この反応混合物を室温まで冷却し、水(200ml)を加え、この混合物をDCM(2×200ml)で抽出した。合わせたDCM抽出液を水(100ml)で洗浄し、MgSO4で乾燥し、真空中で濃縮し、19Aを得た(6.0g、87%収率)。LCMSは純度96.8%を示した。
【0142】
MeOH(20ml)およびヒドラジン水和物(1.4ml、26.5mmol、5.5当量)を18B(2.1g、4.8mmol、1当量)が入っているフラスコに加えた。この混合物を還流温度で1時間撹拌した。この反応混合物を室温まで冷却し、水(40ml)を加え、この混合物をDCM(2×40ml)で抽出した。合わせたDCM抽出液を水(100ml)で洗浄し、MgSO4で乾燥し、真空中で濃縮し、19Bを得た(1.26g、86%収率)。キラルLCは99.3%の鏡像体過剰率を示した。
【0143】
(実施例8.中間体20Aおよび20Bの合成)
【化12】
中間体19A(6.0g、0.02mol、1当量)をトルエン(90ml)中で15分間撹拌した。無水酢酸(5.4ml、0.057mol、2.9当量)を室温で滴下した。水浴を5分間用いてこの混合物を35℃に加温し、19Aの溶解度を高めた。この反応混合物を室温で1時間撹拌し、0℃に冷却し、濾過し、化合物101を得た(5.3g、78%収率)。LCは純度99.1%を示した。キラルLCは99.4%の鏡像体過剰率を示した。1H−NMR(300MHz,CDCl3):δ 2.03(s,3H),3.61−3.66(m,2H),3.71−3.77(m,1H),4.00(見かけ上t,J=8.9,1H),4.71−4.79(m,1H),6.50−6.54(m,1H),6.88(見かけ上t,J=8.9,1H),7.04(dd,J1=10.0,J2=1.6,1H),7.41(dd,J1=14.6,J2=2.7,1H)。HPLC(方法:RP80A、150mm×4.6mmカラム−勾配方法 5−95%ACN+0.1%ギ酸、勾配の前に5%ACNで5分間保持、10分間にわたる勾配、次いで95%ACNで10分間保持;T=30℃;波長:258nm):保持時間:11.45分。キラルHPLC(方法:Chiralpak AD−H;250×4.6mmカラム;5μm粒径−ヘキサン/IPA/DEA(80:20:01);T=40℃;波長:258nm):所望の鏡像異性体について11.80分、少量の鏡像異性体について14.21分;ee=99.8%。MS(M+H+):346.5。中間体19B(1.25g、4.09mmol、1当量)をトルエン(20ml)中で15分間撹拌した。無水酢酸(1.12ml、11.86mol、2.9当量)を室温で滴下した。水浴を5分間用いてこの混合物を35℃に加温し、19Bの溶解度を高めた。この反応混合物を室温で1時間撹拌し、0℃に冷却し、濾過し、化合物100を得た(1.05g、74%収率)。LCは純度99.4%を示した。キラルLCは98.9%の鏡像体過剰率を示した。1H−NMR(300MHz,CDCl3):δ 2.00(s,3H),3.74(dd,J1=9.1,J2=6.8,1H),4.00(t,J=9.1,1H),4.75(dd,J1=8.8,J2=6.8,1H),6.52(bs,1H),6.88(t,J=8.8,1H),7.02−7.06(m,1H),7.41(dd,J1=4.5,J2=2.6,1H)。HPLC(方法:RP80A、150mm×4.6mmカラム−勾配方法 5−95%ACN+0.1%ギ酸、勾配の前に5%ACNで5分間保持、10分間にわたる勾配、次いで95%ACNで10分間保持;T=30℃;波長:258nm):保持時間:11.55分間。キラルHPLC(方法:Chiralpak AD−H;250×4.6mmカラム;5μm粒径−ヘキサン/IPA/DEA(80:20:01);T=40℃;波長:258nm):所望の鏡像異性体について10.63分、少量の鏡像異性体について13.18分;ee=98.9%.MS(M+H+):348.3。
【0144】
(実施例9.2,2,6,6−d4−モルホリン(33)および全重水素化モルホリン(10)の合成)
【化13】
ジグルコール酸(30)を水酸化ナトリウムのD2O溶液で処理し、対応する重水素化二ナトリウム化合物31を生成した。化合物31を全重水素化塩化アンモニウムの存在下で加熱して、d4−ジオキソモルホリン32を生成し、これをTHF中で水素化ホウ素によって還元し、所望の2,2,6,6−d4−モルホリン(33)を生成した。テトラジュウテロモルホリン33を上記の実施例1の全重水素化モルホリン10の代わりに使用して、各Zが重水素であり、各Yが水素である式Iおよび式Iaの化合物、例えば化合物102および化合物103を生成することができる。
【0145】
(実施例10)
抗菌活性を、マウスアッセイ手順を使用して生体内で試験した。雌のマウス(各々18〜20gの6匹)の群に、使用直前に解凍し4% ビール酵母を含む脳心臓浸出物(黄色ブドウ球菌)または脳心臓浸出物(連鎖球菌種)に懸濁させた黄色ブドウ球菌を腹腔内に注射した。1つの薬物あたり6回用量レベルで抗生物質処置を、経口挿管または皮下経路のいずれかによる感染の1.5時間後に投与した。6日間、毎日生存を観察した。死亡率に基づくED50値をプロビット分析を使用して算出した。主題の化合物を対照(例えば、バンコマイシン)と比較した。
【0146】
(実施例11)
生体外活性実験を、当業者に公知の標準的な希釈方法によって行った。手短に言えば、抗生物質の系列2倍希釈液を希釈剤中で調製し,標準的な量のミコバクテリア増殖培地を薬物のアリコートに加えた。この培地に標準化ミコバクテリア細胞懸濁液を植え付け、適切な条件下でインキュベートした。インキュベーション後、最小阻止濃度(MIC)を目視の観察によって測定した。MICは、ミコバクテリア増殖を阻止するために必要とする最小薬物濃度(μg/ml単位)と定義される。
【0147】
(実施例12)
生体内データを、1×107個の生きた結核菌(Erdman株)を用いて静脈内感染させたCD−1マウスから得た。24時間後、薬物処置を開始した。すべての薬物を、1日2回、4週間、経口摂食によって与えた。治療の最後に、生きた細胞数を脾臓および肺のホモジネートから測定した。
【0148】
(実施例13)
チンパンジーでの薬物動態
リネゾリドとの50:50 混合物としてチンパンジーに経口投与または静脈内投与した場合の化合物100の薬物動態を研究した。経口投与および静脈内投与の両方のための溶液を、撹拌しながら、65℃で900mlの注射用の滅菌水中でリネゾリド(200mg)、化合物100(200mg)、クエン酸ナトリウム二水和物(164mg)、無水クエン酸(85mg)、およびデキストロース一水和物(5.024g)を混合することにより調製した。この混合物を25℃に冷却し、生成した溶液のpHを必要に応じて10% HClまたは10% NaOHのいずれかを用いて4.8に調整した。この溶液の最終体積を、滅菌水を用いて1lにした。この投薬溶液を、投薬前に0.22μmフィルターを通して濾過した。
【0149】
4匹のチンパンジー(2匹の雄および2匹の雌)をこの研究で使用した。1匹の雄および1匹の雌を静脈内研究のために使用し、1匹の雄および1匹の雌を経口投薬研究のために使用した。すべての動物を、投薬前に一晩絶食させた。すべての研究について、動物を、投薬前にケタミン(およそ10mg/ml)またはテラゾール(telazol)(およそ5mg/ml)で鎮静させた。各研究について、動物に300mgの併用薬物(各々150mgのリネゾリドおよび化合物100)を投薬した。静脈内用量は、30分間にわたる点滴によって投与した(2mg/mlで150mlの併用薬物)。経口用量は、2mg/mlで150mlの体積の併用薬物を投与した。
【0150】
静脈内研究については、4.5mlのアリコートの血液を、点滴の開始前、点滴開始15分後、および点滴の最後の直前に各動物から採取した。さらなる試料を、点滴後6分、15分、30分、および60分および1.5時間、2時間、4時間、6時間、8時間および24時間に採取した。経口研究については、4.5mlのアリコートの血液を、投薬前、および投薬後15分、20分および60分および1.5時間、2時間、4時間、6時間、8時間および24時間に各動物から採取した。すべての血液試料を抗凝血剤としてのヘパリンナトリウムが入っているバキュテナーチューブに集め、十分に混合し、氷上で保存した。試料を、回収から1時間以内に遠心分離にかけ、血漿を集め、分析まで−70℃で凍結させた。尿も、投薬後24時間にわたって各動物から集めた。
【0151】
各試料を、リネゾリドおよび化合物100の両方の存在について、LC−MS/MSによって以下のようにして分析した。チンパンジーの血漿試料(100μL)を、LC−MS/MS分析の前に300μLの内部標準溶液と混合した。内部標準は、アセトニトリル/水(90/10、体積/体積)中の250ng/mL ハロペリドールであった。タンパク質の沈殿後、10μLの上清をZorbax SB−C8(高速分解)カラム(2.1×30mm、3.5μm)に注入した。最初の移動相条件は、0.5mL/分の流量での、100% A(0.1% ギ酸を含む水)および0% B(0.1% ギ酸を含むアセトニトリル)であった。移動相Bを、2分以内に90%に到達させ、1分間保持してから、3.2分に再び0%に減少させた。全体の分析時間は6分であった。前駆体/生成物イオン対を、リネゾリド、化合物100およびハロペリドールの検出について、それぞれm/z 338/296、m/z 348/306およびm/z 376/165に設定した。
【0152】
尿試料を同様に分析した。チンパンジーの尿試料(10μL)を、独立に、Zorbax SB−C8(高速分解)カラム(2.1×30mm、3.5μm)に注入した。最初の移動相条件は、0.4mL/分の流量での100% A(0.1% ギ酸を含む水)および0% B(0.1% ギ酸を含むアセトニトリル)であった。移動相Bを、42分以内に25%に到達させ、次いで2分間かけて25%から90%に到達させてから、4分かけて再び0%に減少させた。全体の分析時間は48分であった。質量分析計を陽イオンモードに設定し、イオンをm/z 100〜1000で走査した。代謝産物の特定の分子イオンを同定すると、MS/MS実験を実施して生成物イオンを生成した。
【0153】
図1および図2は、静脈内投薬研究の結果を示す。雌(図1)および雄のチンパンジー(図2)の両方が、リネゾリドと比べて、化合物100についての半減期およびAUCの増加を示した。静脈内投薬についての半減期の算出値を表1に示す。
【0154】
表1:静脈内投薬後の化合物100およびリネゾリドの半減期
【表1】
【0155】
図3および図4は、経口投薬研究の結果を示す。雌(図3)および雄のチンパンジー(図4)の両方が、リネゾリドと比べて、化合物100についての半減期およびAUCの増加を示した。8時間および24時間におけるリネゾリドに対する化合物100の血清濃度の比を表2に示す。各化合物についてのAUCの平均算出値を表3に示す。
【0156】
表2:経口投薬後のリネゾリドに対する化合物100の血清濃度の比
【表2】
【0157】
表3.経口投薬後の化合物100およびリネゾリドの平均AUC0−24時間
【表3】
【0158】
リネゾリドと比べたときの化合物100の代謝の最終結果を、静脈内投薬または経口投薬後の尿中の各化合物の排出を追跡することにより、分析した。この分析の結果を表4に示す。
【0159】
表4.尿における無疵のリネゾリドおよび化合物100の排出
【表4】
表4に示す結果は、投与経路にも被験体の性別にも依らず、リネゾリドのおよそ2倍も多い化合物100が無疵で尿中に排出されたことを明らかにする。加えて、さらなる分析から、M6代謝産物およびその重水素化等価体の量は本質的に同じであるが、重水素化M4代謝産物の量はリネゾリドM4代謝産物よりも有意に低いことが明らかとなった。
【0160】
これらのチンパンジーでの研究は、化合物100はリネゾリドよりもゆっくりと代謝され、そしてその代謝の最終結果は、リネゾリドと比べて、M4代謝産物から無疵の排出にシフトすることを示す。
【0161】
さらなる記載がなくとも、当業者は、前述の記載および例示となる実施例を使用して、本発明の化合物を作製して利用し、請求項に係る方法を実施することができると考えられる。前述の説明および実施例は単に特定の好ましい実施形態の詳細な説明を提供するだけであることを理解されたい。種々の変更態様および均等物は、本発明の趣旨および範囲から逸脱することなく作製され得る。
【技術分野】
【0001】
(関連出願)
本願は、2006年10月23日出願の米国仮特許出願第60/853,890号、および2007年9月24日出願の同第60/974,637号の利益を主張する。これらの出願の内容を、参照によりその全体を本願明細書に援用する。
【0002】
本発明は、新規なN−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミド誘導体、それらの許容できる酸付加塩、溶媒和物、および水和物に関する。本発明はまた、本発明の化合物を含む組成物、および抗菌剤によって有益に治療される疾患および病状の治療方法におけるかかる組成物の使用をも提供する。
【背景技術】
【0003】
リネゾリドは、(S)−N−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミドについての一般名である。それは、多くの動物モデルにおいて抗菌剤として有効であることが示されている。マウス大腿症感染モデルで確立されたPK/PD関係から、有効性を決定する主要パラメータはMICを超える時間であることが示された。リネゾリドは、グラム陽性菌ならびに一部のグラム陰性菌および嫌気性菌を含めた多くのヒトの病原体および獣医学的病原体に対して有効である有用な抗菌剤であることが公知である。特許文献1:米国特許第5,688,792号および特許文献2:国際公開第95/07271号を参照のこと。
【0004】
臨床試験において、リネゾリドは、以下の感染症の治療において有効であることが示されている:バンコマイシン耐性フェシウム菌;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;および肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎(Barbachyn,MRら, Pharmacia & Upjohn Co.に対する特許文献3:米国特許第5,688,792号;2006年7月に改訂されたZYVOXラベル)。
【0005】
推奨されるヒトの用量は、バンコマイシン耐性フェシウム菌(菌血症を含む)、院内肺炎;併発性皮膚・皮膚組織感染症;および市中肺炎(菌血症を含む)に対しては、12時間ごとに600mgである。400mgのBIDの用量は、無併発性皮膚・皮膚組織感染症に対して推奨される。臨床試験において、この用量は、トラフ値における黄色ブドウ球菌に対するMIC90を超えることが示された。ヒトにおけるPK/PD関係は明確には確立されていない。1つの研究では、AUC/MICが有効性予測判断材料であることが見出されたが、しかしこのPK/PD予測判断材料は信頼性がないとみなされた。リネゾリドは、高用量では非線形の動力学を示す。1日3回の725mgの用量は、血清クレアチニンの増加によって認容性を認めることができた。骨髄抑制がリネゾリドを受けた患者で報告されている。この骨髄抑制は可逆的であり、リネゾリドを受けた患者は、毎週モニターされるべきである。ヒトにおけるリネゾリドについてのPK/PDはまだ十分に確立されていないが、同等またはより低い用量でMICを超える曝露レベルを維持できる、より長い血清半減期を有する化合物を同定することは明らかに有利であろう。これによって、必要とされるMICを維持しながらのより低いBID用量、またはAUCを減少しつつ必要とされるMICを維持するであろうより高い投与量QDの投与が可能になるであろう。
【0006】
リネゾリドの代謝はマウス、ラット、イヌおよびヒトですでに研究されており、2つの主な代謝経路が同定されている。排出される主な代謝産物は、それぞれ、モルホリン基の酸化によって形成されるラクトン環およびラクタム環の加水分解に由来するM4およびM6として公知のカルボン酸である。これらの代謝産物は不活性である。ヒトでは、主要な代謝経路はラクトン経路である。非特許文献1:Slatter,JGら, Xenobiotica 2002,32,907頁および非特許文献2:Drug Metab Dispos 2001,29,1136頁を参照。ヒトで投与された用量のおよそ35%は、尿中で親化合物として見出されるのに対し、その用量の50%は上記2つの代謝産物が占める。モルホリン環の酸化はCyp酵素によるものではない。生体外研究によって、リネゾリドは臨床的に関連するCypアイソフォーム(1A2;2C9;2C19;2D6;2E1;3A4)の基質でもなく、阻害薬でもなく、誘導因子でもないことが示された。非特許文献3:US NDA 第02130号を参照。
【0007】
リネゾリドのN−オキシドも、前臨床試験において抗菌剤として検討されている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第5,688,792号明細書
【特許文献2】国際公開第95/07271号パンフレット
【特許文献3】米国特許第5,688,792号明細書
【非特許文献】
【0009】
【非特許文献1】Slatter,JGら, Xenobiotica 2002,32,907頁
【非特許文献2】Drug Metab Dispos 2001,29,1136頁
【非特許文献3】US NDA 第02130号
【発明の概要】
【発明が解決しようとする課題】
【0010】
それゆえ、リネゾリドの有益な活性を示し、他の恩恵、例えば、代謝での問題の軽減を伴う有害な副作用の減少、も有する可能性がある化合物を生み出すこと、その薬理学的有効寿命をさらに延ばし、患者の服薬を高めること、そして潜在的には、個体群での薬物動態のばらつきを減少させ、および/または危険な薬物−薬物相互作用についてのその可能性を減少させることが望ましい。
【課題を解決するための手段】
【0011】
上記の課題は、本発明の新規なN−[[3−[3−フルオロ−4−(4−モルホリニル)フェニル]−2−オキソ−5−オキサゾリジニル]メチル]−アセトアミド誘導体、それらの許容できる酸付加塩、溶媒和物、および水和物によって解決される。
【図面の簡単な説明】
【0012】
【図1】雌のチンパンジーへの点滴静注後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図2】雄のチンパンジーへの点滴静注後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図3】雌のチンパンジーへの経口投与後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【図4】雄のチンパンジーへの経口投与後の、リネゾリドおよび化合物100の組合せの血清薬物動態を示す図である。
【発明を実施するための形態】
【0013】
(定義)
「改善する」および「治療する」という用語は同じ意味で用いられ、両用語は、疾患(例えば、感染症、微生物)の発症または進行を減少、抑圧、減衰、縮小、停止、または安定させることを意味する。
【0014】
「疾患」は、細胞、組織、または臓器の正常機能を損傷するか、または妨げるあらゆる病状または障害を意味する。
【0015】
ある変動の天然の同位体存在量は、合成化合物において、合成に使用される化学物質の原材料に応じて発生することが分かる。従って、リネゾリドの調製は、少量の重水素化された同位体的同族体(isotopologues)および/または13Cを含有する同位体的同族体を本質的に含有する。この変動があったとしても、天然で存在する安定水素同位体および炭素同位体の濃度は、本発明の化合物の安定した同位体置換の度合いと比較して小さく、重要ではない。例えば、Wada EおよびHanba Y,Seikagaku 1994,66:15、Ganes LZら,Comp.Biochem.Physiol.Mol.Integr.Physiol.1998,119:725参照。本発明の化合物において、特定の位置が重水素を有すると示される場合、その位置の重水素の存在量は、0.015%である天然の重水素の存在量よりも実質的に大きいと理解される。重水素を有すると示される位置は、通常、当該化合物における重水素として指定された各原子で、少なくとも3000(45%の重水素導入)の最小限の同位体濃縮係数を有する。
【0016】
本願明細書で使用する「同位体濃縮係数」という用語は、特定の同位体の同位体存在量と天然存在量との比率を意味する。
【0017】
他の実施形態では、本発明の化合物は、式Iまたは式Iaで重水素として指定された各原子について、少なくとも3500(重水素として指定された各原子で、52.5%の重水素導入)、少なくとも4000(60%の重水素導入)、少なくとも4500(67.5%の重水素導入)、少なくとも5000(75%の重水素導入)、少なくとも5500(82.5%の重水素導入)、少なくとも6000(90%の重水素導入)、少なくとも6333.3(95%の重水素導入)、少なくとも6466.7(97%の重水素導入合)、少なくとも6600(99%の重水素導入)、または少なくとも6633.3(99.5%の重水素導入)の同位体濃縮係数を有する。
【0018】
本発明の化合物において、特定の同位体として特に指定されない任意の原子は、その原子の任意の安定した同位体を表すことを意味する。別段の記載がない限り、位置が具体的に「H」または「水素」と指定される場合、その位置は、その天然存在量同位体組成において水素を有すると理解される。
【0019】
別の実施形態では、本願明細書で使用する「化合物」は、10%未満、好ましくは6%未満、より好ましくは3%未満の合わせたすべての他の同位体的同族体(いずれの重水素または13Cを除く種類を含む)を含有する。特定の態様では、この化合物は「X」%未満の合わせたすべての他の同位体的同族体(いずれの重水素または13Cを除く種類を含む)を含有し、このXは0および10を含んで0〜10の任意の数字(例えば、1、0.5、0.001)である。10%を超える、合わせたすべての他の同位体的同族体を含有する組成物は、本願明細書で「混合物」と呼ばれ、それらは以下に示されるパラメータを満たさなければならない。本願明細書における同位体組成のこれらの限界および同位体組成へのすべての言及は、単に、式I/Iaの化合物のその活性な遊離の塩基形態中に存在する重水素/水素および13C/12Cの相対量を指し、対イオンの加水分解性部分の同位体組成を含まない。
【0020】
「同位体的同族体」という用語は、その分子またはイオンの同位体の組成だけにおいて、本発明の特定の化合物とは異なる種を指す。
【0021】
本願明細書において使用する「化合物」という用語は、その塩、溶媒和物、またはその水和物を含むことも意図する。本願で記載される本発明の特定の態様における「塩」、「溶媒和物」、または「水和物」という具体的な記述は、「化合物」という用語がこれらの他の形態の記述なく使用される本発明の他の態様では、これらの形態が意図的に省かれていると解釈されるべきではない。
【0022】
本発明の化合物の塩は、酸とアミノ官能基などのその化合物の塩基性基の間で、または、塩基とカルボキシル官能基などのその化合物の酸性基の間で形成される。別の好ましい実施形態によれば、化合物は、薬理学的に許容できる酸付加塩である。
【0023】
本願明細書において使用する「薬理学的に許容できる」という用語は、適切な医学的判断の範囲で、ヒトおよび他の哺乳動物の組織に接触させる使用において、過度の毒性、刺激、アレルギー反応などがなく好適で、適度の利益/危険性の比でつり合いの取れている、成分を指す。「薬理学的に許容できる塩」とは、服用者への投与において、直接的にまたは間接的に本発明の化合物を提供できる非毒性の塩を意味する。「薬理学的に許容できる対イオン」は、服用者への投与において塩から放出される場合に非毒性である、その塩のイオン部分である。
【0024】
薬理学的に許容できる塩を形成するために一般的に使用される酸としては、二硫化水素、塩化水素酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸などの無機酸、ならびに、p−トルエンスルホン酸、サリチル酸、酒石酸、酸性酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、p−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸などの有機酸、ならびに関連した無機酸および有機酸が挙げられる。かかる薬理学的に許容できる塩としては、従って、硫酸塩、ピロ硫酸塩、硫酸水素塩、亜硫酸塩、亜硫酸水素塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−二酸塩、ヘキシン−1,6−二酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スフホン酸塩、マンデル酸塩、および他の塩が挙げられる。好ましい薬理学的に許容できる酸付加塩としては、塩化水素酸および臭化水素酸などの鉱酸から形成されるもの、特にマレイン酸などの有機酸から形成されるものが挙げられる。
【0025】
本願明細書において使用する「水和物」という用語は、非共有結合性の分子間力によって結合された、化学量論量または非化学量論量の水を更に含む化合物を意味する。
【0026】
本願明細書において使用する「溶媒和物」という用語は、非共有結合性の分子間力によって結合された、化学量論量または非化学量論量の溶媒(水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノールなど)を更に含む化合物を意味する。
【0027】
本発明の化合物は、1つ以上の不斉炭素原子を含有してよい。従って、本発明の化合物は、個々の立体異性体(鏡像異性体もしくはジアステレオマー)、および立体異性体の混合物として存在することができる。従って、本発明の化合物としては、立体異性体混合物だけではなく、他の立体異性体が実質的に含まれない個々のそれぞれの立体異性体も挙げることができる。本願明細書において使用する「他の立体異性体が実質的に含まれない」という用語は、25%より少ない他の立体異性体が存在すること、好ましくは10%より少ない他の立体異性体が存在すること、より好ましくは5%より少ない他の立体異性体が存在すること、最も好ましくは2%より少ない他の立体異性体が存在すること、または「X」%より少ない他の立体異性体が存在すること(ここで、Xは0と100を含み、0〜100間の数字である)を意味する。ジアステレオマーを得る方法、または合成する方法は、当該技術分野で周知であり、最終化合物、または出発物質、または中間物に実施できるものとして、適用してよい。他の実施形態は、上記化合物が単離された化合物である実施形態である。本願明細書において使用する「少なくともX%鏡像異性体に富む」という用語は、その化合物の少なくともX%が単一の鏡像異性体形態にあることを意味する(ここで、Xは0と100を含み、0〜100間の数字である)。
【0028】
本願明細書において使用する「安定化合物」という用語は、その製造を可能にするのに十分な安定性を有し、本願明細書に記載した目的(例えば、治療薬にする製剤化、治療用の化合物の生産で使用するための中間体、単離可能な中間体または保存可能な中間体化合物、非定型抗精神病薬に応答する疾患または症状を治療すること)に有用となるように、十分な時間にわたって化合物の完全性を保持する化合物を指す。
【0029】
「2H」および「D」はともに重水素を指す。「立体異性体」は、鏡像異性体およびジアステレオマーの両方を指す。「tert」は、各々第三級を指す。「CDI」は1,1’−カルボニルジイミダゾールを指す。
【0030】
本願明細書中の変動要素のいずれかの定義において化学基の一覧を記述する際は、いずれかの単独の基または列挙された基の組合せとしてのその変動要素の定義を含む。本願明細書中の変動要素についてのある実施形態を記述する際は、いずれかの単独の実施形態または任意の他の実施形態もしくはその一部分との組合せとしてのその実施形態を含む。
【0031】
本願明細書全体を通して、「各Y」と言及することは、独立に、当てはまる場合にはすべての「Y」基(例えば、Y1、Y2、Y3、およびY4)を含み、「各W」は独立に、当てはまる場合にはすべての「W」基(例えば、W1、W2、W3、W4、およびW5)を含み、「各Z」は独立に、当てはまる場合にはすべての「Z」基(例えば、Z1、Z2、Z3、およびZ4)を含む。
【0032】
(治療用化合物)
本発明は、式Iもしくは式Iaの化合物:
【化1】
または式Iの塩;または式Iもしくは式Iaの水和物もしくは溶媒和物
(式中、
各Wは独立に、水素または重水素であり、
各Yは独立に、水素または重水素であり、
各Zは独立に、水素、重水素、またはフッ素であり、かつ
W、YまたはZの少なくとも1つは重水素である)
を提供する。
【0033】
一実施形態では、少なくとも1つのWは、重水素であり、少なくとも2つのY部分は重水素であり、かつ少なくとも2つのZ部分は重水素またはフッ素である。
【0034】
一実施形態では、W1およびW2は同時に重水素である。
【0035】
一実施形態では、Y1、Y2、Y3およびY4は同時に重水素である。
【0036】
一実施形態では、Z1、Z2、Z3およびZ4の各々は独立に、重水素およびフッ素から選択される。より具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素である。
【0037】
特定の実施形態では、式Iまたは式Iaの化合物の配置は(S)である。
【0038】
より具体的な実施形態では、Y1、Y2、Y3、Y4、W1およびW2は同時に重水素である。
【0039】
別の具体的な実施形態では、Z1、Z2、Z3およびZ4の各々は独立に、重水素およびフッ素から選択され、W1およびW2は同時に重水素である。
【0040】
別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3およびZ4は同時に重水素である。別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3およびZ4は同時に重水素であり、かつW3、W4およびW5は同時に水素である。
【0041】
さらに別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時にフッ素であり、かつY1、Y2、Y3およびY4は同時に重水素である。
【0042】
なお別の具体的な実施形態では、Y1、Y2、Y3、Y4、Z1、Z2、Z3、Z4、W1およびW2は同時に重水素である。
【0043】
別の実施形態では、Z1、Z2、Z3およびZ4は同時にフッ素であり、かつY1、Y2、Y3、Y4、W1、およびW2は同時に重水素である。
【0044】
別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素であり、かつY1、Y2、Y3、Y4、W1およびW2は同時に水素である。別の具体的な実施形態では、Z1、Z2、Z3およびZ4は同時に重水素であり、かつW3、W4およびW5は同時に水素である。
【0045】
さらに別の具体的な実施形態では、Z1、Z2、Z3Z4、Y1、Y2、Y3およびY4は同時に重水素であり、かつW1およびW2は同時に水素である。
【0046】
一実施形態では、式Iまたは式Iaの化合物は少なくとも3個の重水素原子を含有する。
【0047】
一実施形態では、式Iまたは式Iaの化合物は少なくとも4個の重水素原子を含有する。
【0048】
一実施形態では、式Iまたは式Iaの化合物は少なくとも5個の重水素原子を含有する。
【0049】
本発明の具体的な化合物の例としては、
【化2】
が挙げられる。
【0050】
本発明の具体的な化合物の他の例としては、以下の
【化3】
が挙げられる。
【0051】
別の一連の実施形態では、上述の実施形態のいずれかにおいて重水素として指定されない任意の原子は、その天然の同位体存在量で存在する。
【0052】
類似の化合物の中に重水素を導入する一般的方法が、広範囲にわたって文書化されている。例えば、そのほとんどの号が、生物活性有機低分子への重水素の特異的導入についての詳細な実験的記述を含むThe Journal of Labelled Compounds and Radiopharmaceuticals(John Wiley & Sons)を参照。また、例えば、Leis HJ,Curr Org Chem,1998,2:131およびその中の参考文献、ならびにMoebius G,ZfI−Mitteilungen 1989,150:297も参照。重水素標識した試薬の好適な商用供給業者としては、とりわけ、Isotec,Inc.(オハイオ州、マイアミズバーグ)、Cambridge Isotope Laboratories(マサチューセッツ州、アンドーバー)、ICON Services Inc.(ニュージャージー州、サミット)、およびC/D/N Isotopes,Inc.(カナダ、ケベック州、ポアントクレール)が挙げられる。
【0053】
式I/Iaの化合物の合成は、通常の技能を有する合成化学者により、有機合成の分野で公知の手段によって容易に行うことができる。かかる方法は、本願明細書に記載される化合物を合成するための対応する重水素化されかつ任意に他の同位体を含有する試薬および/または中間体を利用して、または同位体原子を化学構造に導入するための当該技術分野で公知の標準的合成手順を活用して実施することができる。関連する手順および中間体は、例えば、PCT公開公報WO1997010223、WO2005099353、WO1995007271、WO2006008754;Lizondo,Jら, Drugs Fut 1996,21(11):1116;Brickner,SJら, J Med Chem 1996,39(3):673;およびMallesham,Bら, Org Lett 2003,5(7):963に記載されている。以下のスキームは、式Iまたは式Iaの化合物がどうやって調製され得るかを図示する。
【0054】
スキーム1.式Iの化合物を調製するための一般的経路
【化4】
【0055】
上記のスキーム1は、式Iの化合物を調製するための一般的経路を示す。式Iaの化合物は、トリフルオロ過酢酸またはm−クロロ過安息香酸などの適切な酸化剤を使用して式Iの化合物から作製することができる。国際公開第1997010223号を参照。式I/Iaの化合物を合成するための他のアプローチは、実施例に示されているか、または本願明細書に引用された引用文献から容易に適合させることができる。これらの手順の変法およびそれらの最適化は、当業者の技能の範囲内である。
【0056】
上に示した特定のアプローチおよび化合物は、限定を意図したものではない。本願明細書のスキームにおいて明確に示されない経路の範囲内にあるものを含む、式I/Iaの化合物およびその合成前駆体のさらなる合成方法は、当業者の化学者らの手法の範囲にある。必要に応じて競合する副生成物を最小化して反応条件を最適化するための方法は当該分野で公知である。反応最適化およびスケールアップでは、高速パラレル合成装置およびコンピュータ制御マイクロリアクタを有利に利用してよい(例えば、Design And Optimization in Organic Synthesis,第2版,Carlson R編,2005;Elsevier Science Ltd.;Jaehnisch,Kら, Angew.Chem.Int.Ed.Engl.2004 43:406;およびこれらで引用される引用文献)。本願明細書で引用した合成の引用文献に加えて、反応スキームおよび手順は、例えば、SciFinder(登録商標)(米国化学会のCAS部門)、STN(登録商標)(米国化学会のCAS部門)、CrossFire Beilstein(登録商標)(Elsevier MDL)などの市販の構造検索可能なデータベースソフトウェア、あるいはグーグル(登録商標)などのインターネット検索エンジンまたは米国特許商標庁のテキストデータベースなどのキーワードデータベースを用いて、当業者によって決定されてよい。本願明細書に記載された方法は、本願明細書に具体的に記載された工程の前か後に、本願明細書の化合物の合成を最終的に可能にするために、好適な保護基を付加または除去する工程をさらに含んでもよい。さらに、様々な合成工程は、所望の化合物を得るために、別の順序または順番で実施されてもよい。適用できる化合物を合成するのに有用な合成化学変換および保護基方法論(保護および脱保護)は、当該分野で公知であり、例えば、R.Larock, Comprehensive Organic Transformations,VCH Publishers(1989);T.W.GreeneおよびP.G.M.Wuts, Protective Groups in Organic Synthesis,第3版、John Wiley and Sons(1999);L.FieserおよびM.Fieser, Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994);およびL.Paquette編集, Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、ならびにこれらのこれらに続く版に記載されているものが挙げられる。
【0057】
本願明細書に記載される合成方法は、さらに、前述のスキームで記載された工程のいずれかの前または後に、本願明細書に記載される式の化合物の合成を最終的に可能にするために、適切な保護基を付加もしくは除去する工程をも含んでいてよい。本願明細書に記載される方法は、1つの式の化合物を別の式の化合物に変換することを企図する。変換プロセスは、その場で実施してもよいし、または中間体化合物を単離して実施してもよい1つ以上の化学変換を指す。この変換は、本願明細書で引用された参考文献の中のものを含めて当該技術分野で公知の技術および手順を用いて、出発化合物または中間体を付加的な試薬と反応させることを含んでいてもよい。ある中間体は、精製(例えば、濾過、蒸留、昇華、結晶化、粉末化、固相抽出、およびクロマトグラフィー)して、または精製せずに使用することができる。
【0058】
本発明によって想定される置換基および変動要素の組み合わせは、安定化合物の形成の結果によるものだけである。
【0059】
(組成物)
本発明は、有効量の式I/Ia(例えば、本願明細書中の式のいずれも含む)の化合物、あるいは式Iの化合物の薬理学的に許容できる塩、式Iまたは式Iaの水和物または溶媒和物と、許容できる担体とを含む組成物も提供する。一実施形態では、この組成物は発熱物質を含まない。好ましくは、本発明の組成物は、担体が薬理学的に許容できる担体である薬理学的な用途(医薬組成物)のために処方される。この担体は、製剤の他の成分と適合し、薬理学的に許容できる担体の場合は、薬剤に通常使用する量においては、その服用者に無害であるという意味で「許容できる」ものでなければならない。
【0060】
本発明の医薬組成物に使用してよい薬理学的に許容できる担体、アジュバント、およびビヒクルとしては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミンなどの血清タンパク質、リン酸塩などの緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、硫酸プロタミン、リン酸一水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩などの塩または電解質、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースをベースとした物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリル酸塩、ワックス、ポリエチレン−ポリオキシプロピレンブロック重合体、ポリエチレングリコール、および羊毛脂を挙げることができるが、これらに限定されない。
【0061】
必要に応じて、上記医薬組成物中の本発明の化合物の溶解性およびバイオアベイラビリティは、当該技術分野で周知の方法により高めることができる。1つの方法としては、その製剤で脂質賦形剤を使用することが挙げられる。「Oral Lipid−Based Formulations:Enhancing the Bioavailability of Poorly Water−Soluble Drugs(Drugs and the Pharmaceutical Sciences)」,David J.Hauss編,Informa Healthcare,2007;および「Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery:Basic Principles and Biological Examples」,Kishor M.Wasan編,Wiley−Interscience,2006を参照。
【0062】
バイオアベイラビリティを高める別の公知の方法は、必要に応じてポリキサマー(LUTROL(商標)およびPLURONIC(商標)(BASF Corporation)など)、またはエチレンオキシドおよびプロピレンオキシドのブロックコポリマーを配合した本発明の化合物の非晶質形を使用することである。米国特許第7,014,866号および米国特許出願公開第20060094744号および同第20060079502号を参照。
【0063】
本発明の医薬組成物としては、経口投与、経直腸投与、経鼻投与、局所投与(口腔投与および舌下投与を含む)、膣内投与、または非経口投与(例えば、皮下投与、筋肉投与、静脈内投与および皮内投与を含む)に好適なものが挙げられる。一部の実施形態では、本願明細書の式の化合物は、(例えば、経皮パッチ、またはイオン導入法の技術を用いて)経皮投与される。他の製剤は、例えば、錠剤、持続放出性カプセルなどの単位量分の剤形として、およびリポソームとして、提供されることが好都合であるかも知れず、薬学分野で周知の任意の方法で調製してよい。例えば、Remington’s Pharmaceutical Sciences,Mack Publishing Company,ペンシルベニア州、フィラデルフィア(第17版、1985)を参照。
【0064】
かかる調製方法は、1つ以上の副成分を構成する担体のような成分を、投与されるべき分子と一緒にする工程を含む。通常、組成物は、活性成分を、液体担体、リポソーム、または微粉化した固体担体あるいはその両方と、均一におよび密に一緒にすることによって、および必要に応じて生成物を成形することによって調製される。
【0065】
一部の実施形態では、この化合物は経口投与される。経口投与に好適な本発明の組成物は、各々が所定量の活性成分を含有するカプセル剤、サシェ剤、または錠剤などの個別の単位として、粉剤または顆粒剤として、水性液体または非水性液体中の液剤または懸濁剤として、水中油型乳濁液として、油中水型乳濁液として、リポソームに詰められて、またはボーラスとしてなどで、提供されてもよい。軟ゼラチンカプセルは、化合物の吸収速度を有益に速くできる場合がある懸濁剤を含有するのに有用であり得る。
【0066】
経口用途の錠剤の場合、一般に使用される担体としては、乳糖およびトウモロコシでんぷんが挙げられる。ステアリン酸マグネシウムなどの滑沢剤も、通常添加される。カプセル剤形の経口投与に有用な希釈剤としては、乳糖および乾燥トウモロコシでんぷんが挙げられる。水系懸濁剤が経口投与される場合、活性成分は、乳化剤および懸濁化剤と混合される。必要に応じて、一部の甘味料、および/または矯味矯臭剤、および/または着色料を添加してもよい。
【0067】
経口投与に好適な組成物としては、風味ベースの成分、一般的にはショ糖およびアラビアゴムまたはトラガカントを含む薬用ドロップ、ならびにゼラチンおよびグリセリンまたはショ糖およびアラビアゴムなどの不活性ベース中に活性成分を含むトローチが挙げられる。
【0068】
非経口投与に好適な組成物としては、酸化防止剤、緩衝剤、細菌発育抑制剤、および意図する服用者の血液に対して製剤を等張にする溶質を含有してもよい水系無菌注入液および非水系無菌注入液、ならびに、懸濁化剤および増粘剤を含んでもよい水系無菌注入液および非水系無菌懸濁液が挙げられる。この製剤は、例えば、密封されたアンプルおよびバイアルなどの、1回の用量または複数回の用量の容器で提供されてもよく、使用する直前に、例えば注入用の水などの無菌液体担体の添加だけを必要とする凍結乾燥(冷凍乾燥)状態で保管されてもよい。即席注入液および即席注入懸濁液は、無菌粉剤、無菌顆粒剤、および無菌錠剤から調製されてもよい。
【0069】
かかる注入液は、例えば、無菌の注入可能な水系懸濁液または油性懸濁液の剤形であってもよい。この懸濁液は、好適な分散剤または湿潤剤(例えばTween80など)および懸濁化剤を使用して、当該分野で公知の技術に従って調製されてもよい。無菌の注入可能な調剤は、例えば1,3−ブタンジオール中の溶液のように、非毒性の非経口で許容できる希釈剤または溶媒中の無菌の注入可能な液剤または懸濁剤であってもよい。許容できるビヒクルおよび溶媒のうち、使用可能なものは、マンニトール、水、リンガー液、および生理食塩液である。加えて、無菌の不揮発性油は、溶媒または懸濁媒体として、従来から使用されている。この目的で、合成モノグリセリドまたはジグリセリドを含む、任意の無刺激不揮発性油を使用してもよい。オレイン酸およびそのグリセリド誘導体などの脂肪酸は、オリーブ油またはヒマシ油などの天然の薬理学的に許容できる油がそうであるように、注入液の調製に有用であり、特にそのポリオキシエチレン化したそのバージョンが有用である。これらの油液剤または油懸濁剤は、長鎖アルコール希釈剤または分散剤を含有してもよい。
【0070】
本発明の医薬組成物は、経直腸投与用の坐薬という剤形で投与されてもよい。これらの組成物は、本発明の化合物と、室温で固体であるが直腸温では液体であり、従って直腸で溶け活性成分を放出する好適な非刺激性の賦形剤とを混合することによって調製できる。かかる物質としては、ココアバター、蜜ロウ、およびポリエチレングリコールが挙げられるが、これらに限定されない。
【0071】
本発明の医薬組成物は、鼻エアロゾルまたは鼻孔吸入によって投与されてもよい。かかる組成物は、医薬製剤分野において周知の技術に従って調製され、ベンジルアルコールまたは他の好適な保存料、バイオアベイラビリティを高める吸収促進剤、フッ化炭素、および/あるいは当該分野で公知の他の可溶化剤または分散剤を使用して、生理食塩水中の溶液として調製されてもよい。かかる投与は、勃起不全に対する薬物について有効であることが公知である。例えば、Alexza Molecular Delivery Corporationに譲渡されたRabinowitz JDおよびZaffaroni ACの米国特許第6,803,031号明細書を参照。
【0072】
本発明の医薬組成物の局所投与は、所望の治療が、局所的付与によって容易に到達できる範囲または器官に関係する場合に特に有用である。皮膚への局所投与のために、この医薬組成物は、担体に懸濁または溶解された活性成分を含有する好適な軟膏剤を用いて処方されるべきである。本発明の化合物の局所投与のための担体としては、鉱油(mineral oil)、鉱油(liquid petroleum)、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ロウ、および水が挙げられるが、これらに限定されない。あるいは、この医薬組成物は、担体に懸濁または溶解された活性化合物を含有する好適なローション剤またはクリームを用いて処方できる。好適な担体としては、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられるが、これらに限定されない。本発明の医薬組成物は、直腸の坐薬製剤によって、または好適なかん腸製剤として、下部消化管に局所的に付与されてもよい。局所的な経皮パッチおよびイオン導入法による投与も、本発明に含まれる。
【0073】
主題の治療成分の投与は、関心部位に投与するように、局所的であってよい。関心部位に主題の組成物を提供するためには、注射、カテーテル、トロカール、噴出装置(projectile)、プルロニックゲル(pluronic gel)、ステント、持続した薬物放出をもたらすポリマー、または内部到達をもたらす他の装置の使用などのさまざまな技法を使用することができる。
【0074】
従って、また別の実施形態によれば、本発明の化合物は、人工器官、人工弁、代用血管、ステント、またはカテーテルなどの、移植可能な医療装置を被覆するための組成物に導入されてもよい。好適な被覆物および被覆された移植可能な装置の一般的な調製物は、当該分野において公知であり、米国特許第6,099,562号明細書、同第5,886,026号明細書、および同第5,304,121号明細書に例示されている。この被覆物は、一般的に、ヒドロゲルポリマー、ポリメチルジシロキサン、ポリカプロラクトン、ポリエチレングリコール、ポリ乳酸、エチレン酢酸ビニル、およびこれらの混合物などの生体適合性のポリマー材料である。この被覆物は、必要に応じて、フルオロシリコーン、多糖類、ポリエチレングリコール、リン脂質、またはこれらの組み合わせの好適な上塗りによってさらに被覆し、組成物に制御放出特性を与えてもよい。侵襲装置の被覆物は、本願明細書で使用される薬理学的に許容できる担体、アジュバント、またはビヒクルの定義の範囲内に含まれる。
【0075】
別の実施形態によれば、本発明は、移植可能な医療装置を被覆する方法であって、この装置を上に記載した被覆組成物に接触させる工程を含む方法を提供する。この装置の被覆が哺乳動物への移植の前に実施されることは、当業者には明らかである。
【0076】
別の実施形態によれば、本発明は、移植可能な薬物放出装置に含侵させる方法であって、この薬物放出装置を本発明の化合物または組成物に接触させる工程を含む方法を提供する。移植可能な薬物放出装置としては、生分解性ポリマーカプセルまたはカプセル(bullet)、非分解性で拡散性のポリマーカプセルおよび生分解性ポリマーウエハが挙げられるが、これらに限定されない。
【0077】
別の実施形態によれば、本発明は、本発明の化合物または本発明の化合物を含む組成物で被覆され、従って、この化合物が薬理学的に活性である、移植可能な医療装置を提供する。
【0078】
別の実施形態によれば、本発明は、本発明の化合物または本発明の化合物を含む組成物を含侵させているか、あるいはこの化合物または組成物を含有し、従って、この化合物がこの装置から放出され、この化合物が薬理学的に活性である、移植可能な薬物放出装置を提供する。
【0079】
患者から取り出すことによって器官または組織に到達可能な場合、かかる器官または組織は本発明の組成物を含有する媒体に侵漬されてもよいし、本発明の組成物が器官に塗布されてもよいし、本発明の組成物が他の任意の好都合な方法で付与されてもよい。
【0080】
別の実施形態では、本発明の組成物は、第2の治療剤をさらに含む。この第2の治療剤は、特に抗菌治療において、抗菌性化合物とともに投与した場合に有利な特性を有することが知られているか、または有利な特性を示す任意の化合物または治療剤が挙げられ、他の抗菌剤および/または抗炎症剤との併用療法が想起される。本発明に係る併用療法は、従って、少なくとも1種の式Iまたは式Iaの化合物の投与、および他の抗菌剤の任意の使用、ならびにシクロオキシゲナーゼ阻害薬、特にシクロオキシゲナーゼ−2の選択的阻害薬の任意の使用を含む。他の抗菌療法および抗炎症薬は、例えば、国際公開第01/34128号パンフレットおよび同第03/061704号パンフレットに開示されており、これらの出願を、それらが抗菌療法および抗炎症薬療法の組合せを開示する程度まで、参照によって本願明細書に援用する。
【0081】
本発明の化合物と配合することができる第2の治療剤の例としては、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブが挙げられるが、これらに限定されない。
【0082】
別の実施形態では、本発明は、本発明の化合物および第2の治療剤が互いに関与する、本発明の化合物および第2の治療剤の個別の剤形を提供する。本願明細書で使用する「互いに関与する」という用語は、個別の剤形が一緒に包装されているか、または別の形で互いにくっついており、従って、個別の剤形は一緒に市販され(互いに24時間より短い時間内に順次または同時に)投与されるという意図が容易に明らであることを意味する。
【0083】
本発明の化合物は、より長い半減期を示し、モル基準で同じ量のリネゾリドと比べて、投薬後24時間のより高い血清濃度レベルをもたらす。従って、一実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドの血清最終排泄半減期よりも大きい、上記化合物の血清最終排泄半減期をもたらす医薬組成物を提供する。他の実施形態では、式Iの化合物の血清最終排泄半減期は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるリネゾリドの血清最終排泄半減期の少なくとも125%、130%、135%、140%またはこれより大きい。
【0084】
関連する実施形態では、本発明は、有効量の式Iの化合物、またはその薬理学的に許容できる塩を含み、第1の組成物の単回用量が試験被験者に投与された後の化合物の血清最終排泄半減期が7時間を超える医薬組成物を提供する。
【0085】
別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合の投与後24時間のリネゾリドの血清濃度よりも大きい、投与後24時間の上記化合物の血清濃度をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物の投与後24時間でもたらされる式Iの化合物の血清濃度は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるリネゾリドの血清濃度の少なくとも150%、175%、200%、225%、250%、275%、300%またはこれより大きい。
【0086】
一実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその投与が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドのAUC0−24よりも大きい、上記化合物のAUC0−24をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物によってもたらされるAUC0−24は、同じ投薬治療方式で投与された対応するリネゾリド組成物によってもたらされるAUC0−24の少なくとも125%、130%、135%、140%、145%またはこれより大きい。
【0087】
本発明の化合物はまた、リネゾリドと比べて特定の代謝に対してより大きい耐性を示す。従って、別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者への投与後24時間において無瑕で排出される化合物の量が、活性成分のモル基準で式Iの化合物の量と同じでありかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された後24時間において無瑕で排出されるリネゾリドの量よりも大きい医薬組成物を提供する。他の実施形態では、本発明の組成物の投与後24時間において無瑕で排出される式Iの化合物の量は、同じ投薬治療方式で投与された対応するリネゾリド組成物の投与後24時間において無瑕で排出されるリネゾリドの量の少なくとも150%、160%、170%、180%、190%、200%、210%、またはこれより大きい。
【0088】
関連する実施形態では、本発明は、有効量の式Iの化合物、またはその薬理学的に許容できる塩を含み、かつ被験者へのこの組成物の投与後24時間において、この化合物の有効量の少なくとも45%が被験者によって無疵で排出される医薬組成物を提供する。
【0089】
さらに別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者への投与が、活性成分のモル基準で式Iの化合物の量よりも多くかつ式Iの化合物と同じ投薬治療方式で投与されたある量のリネゾリドを含む医薬組成物でリネゾリドが等価な試験被験者に投与された場合のリネゾリドと、a)同様のAUC0−24、b)同様のCmaxまたはc)同様のCmin、のうちの1つ以上をもたらす医薬組成物を提供する。他の実施形態では、式Iの化合物の有効量は、式Iの化合物と同じ投薬治療方式で投与された場合にa)同様のAUC0−24、b)同様のCmax、またはc)同様のCmin、のうちの1つ以上をもたらすために必要とされるリネゾリドの量の80%、70%、60%、50%、40%、33%未満,であるかまたはこれより少ない。
【0090】
さらに別の実施形態では、本発明は、有効量の式Iの化合物を含み、かつ試験被験者へのその1つ以上の投与量の投与が、a)第1の投与量の投与後24時間の間6mg/Lを超える上記化合物の血清濃度の維持、およびb)投与後24時間の間6mg/Lを超えるリネゾリドの血清レベルを維持するために必要とされるリネゾリドの量を含む医薬組成物で等価な試験被験者にリネゾリドが投与された場合のリネゾリドのAUC0−24未満である、その化合物のAUC0−24をもたらす医薬組成物を提供する。他の実施形態では、本発明の組成物によってもたらされるAUC0−24は、リネゾリド組成物の必要とされる投与量によってもたらされるAUC0−24の85%、80%、75%、70%、65%未満,であるかまたはこれより少ない。
【0091】
上記の実施形態の各々において、式Iの化合物の薬理学的に許容できる塩、および/またはリネゾリドを、遊離塩基形態の代わりに使用してもよい。
【0092】
より具体的な実施形態では、上で示した組成物の各々において、上記化合物は化合物100、化合物101、化合物102または化合物103から選択される。
【0093】
「試験被験者」は、任意の哺乳動物、好ましくはヒトである。
【0094】
「等価な試験被験者」は、本願明細書では、試験被験者と同じ種および性別でありかつ試験被験者および等価な被験者の両方に対して等量のリネゾリドを投与した後に試験される薬物動態パラメータにおいて試験被験者と比べて10%以下のばらつきを示すものと定義される。
【0095】
本発明の医薬組成物において、本発明の化合物は有効量で存在する。本願明細書で使用する「有効量」という用語は、適切な投薬治療方式で投与された場合に、治療される障害の重症度、期間もしくは進行を縮小または改善するのに十分な量、治療される障害の進行を防ぐのに十分な量、治療される障害の退縮をもたらすのに十分な量、あるいは別の治療の予防効果または治療効果を高めるかもしくは向上させるのに十分な量を指す。
【0096】
動物とヒトに対する(体表面の1平方メートル当たりのミリグラムに基づいた)投与量の相関関係は、Freireichら、(1966)Cancer Chemother Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定されてよい。例えば、Scientific Tables,Geigy Pharmaceuticals,ニューヨーク州,アーズリー,1970,537を参照。
【0097】
本発明の化合物は等しい投与量でリネゾリドより長い血清半減期を示すため、それらを、最小阻止濃度(「MIC」)を超える必要とされる時間を依然維持しつつ、リネゾリドよりもより低い用量および/またはより低い頻度間隔で投与することができる。リネゾリドと比べて、本発明の化合物の投与のより低い頻度間隔は、各投薬に伴う血清濃度の急上昇の数を減少させるであろう。これは、ひいては、その患者の経時的な本発明の化合物への暴露総量(累積的AUC暴露(cumulative AUC exposure))を減少させるであろう。リネゾリドがミトコンドリアのタンパク質合成を阻害することによって引き起こされると考えられる、リネゾリドの累加性毒性に関連付けられてきたのはこの累積的AUC暴露である(Devriese ASら, Clin Infect Dis 2006,42:1111)。リネゾリドの累加性毒性は、患者がその薬物を摂取できる時間量を制限する。
【0098】
リネゾリドと比べた場合の累積的AUC暴露の減少は、本発明の化合物を含む制御放出製剤の使用によって、さらに高めることができる。かかる制御放出製剤は、当該技術分野で周知の方法を使用して調製される。例えば、Remmington:The Science and Practice of Pharmacy, 第21版(Lippincott Williams & Wilkins 2005); およびModern Pharmaceutics 第4版(Drugs and the Pharmaceutical Sciences 第121巻),Banker GSおよびRhodes CT編(Informa Healthcare 2002)を参照。
【0099】
動物とヒトに対する(体表面の1平方メートル当たりのミリグラムに基づいた)投与量の相関関係は、Freireichら、(1966)Cancer Chemother Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定されてよい。例えば、Scientific Tables,Geigy Pharmaceuticals,ニューヨーク州,アーズリー,1970,537を参照。本発明の化合物の有効量は、適切な場合にはいくつかの個々の用量の形態で、24時間ごとに約50mg〜約2000mgの範囲であってよい。一実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約250mg〜約1250mg、または各々12時間ごとに与えられる約125mg〜約625mgの2回の別々の投与量の範囲である。別の実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約750mg〜約1250mg、または各々12時間ごとに与えられる約375mg〜約625mgの2回の別々の投与量の範囲である。さらに別の実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約450mg〜約1200mg、または各々12時間ごとに与えられる約225mg〜約625mgの2回の別々の投与量の範囲である。より具体的な実施形態では、本発明の化合物の有効量は、24時間ごとの単回投与量の形態での約450mg〜約750mg、または各々12時間ごとに与えられる約225mg〜約375mgの2回の別々の投与量の範囲である。上に記載した範囲の中またはその範囲の間に入る本発明の化合物の他の範囲もまた、本発明の範囲内である。有効用量は、当業者により認識されるように、治療される疾患、疾患の重症度、投与経路、患者の性別、年齢、および全体的な健康状態、賦形剤の使用法、他の薬剤の使用などの他の治療との併用の可能性、および主治医の判断によっても変化する。
【0100】
本発明の医薬組成物に存在し本発明の方法で使用されるミリグラム量の化合物は、遊離塩基の化合物の量を表す。本発明の化合物の薬理学的塩の使用では、等価なモル量の遊離塩基化合物が使用されるように明記された量を増やすことが必要であることを理解されたい。
【0101】
第2の治療剤を含む医薬組成物についての第2の治療剤の有効量は、単剤治療法において、その薬剤だけを使用する場合に通常使用される投与量の約20%〜100%の間である。有効量は、通常の単剤治療用量の約70%〜100%の間であることが好ましい。これらの第2の治療剤の通常の単剤治療投与量は、当該分野において周知である。例えば、Wellsら編集、Pharmacotherapy Handbook,第2版,Appleton and Lange,コネティカット州,スタンフォード(2000);PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000,デラックス版,Tarascon Publishing,カリフォルニア州,ロマリンダ(2000)を参照。これら各々の参考文献の内容全体を、参照によって、本願明細書に援用する。
【0102】
上で言及した第2の治療剤の一部は、本発明の化合物と相乗的に作用することが予想される。これが生じる場合、第2の治療剤および/または本発明の化合物の有効用量は、単剤治療において必要とされる量よりも減らされる。これは、第2の治療剤または本発明の化合物のどちらかの中毒性副作用の最小化、有効性の相乗的向上、投与または使用の容易性の向上、および/あるいは化合物の調製または処方の総合的な費用の削減に優位性を有する。
【0103】
(治療方法)
別の実施形態によれば、本発明は、リネゾリドによって有益に治療される疾患を患っているか、またはそれらにかかり易い被験者を治療する方法であって、上記被験者に有効量の本発明の化合物もしくは組成物を投与する工程を含む方法を提供する。かかる疾患は当該技術分野で周知であり、例えば、典型的には抗菌療法によって治療される種々の疾患状態(例えば、感染症、真菌性障害)の治療または予防を含む。それゆえ、式I/Iaの化合物は、グラム陽性菌および一部のグラム陰性菌および嫌気性菌によって媒介される障害を含めた障害の治療において有用性を有する。
【0104】
一実施形態では、本発明は、フェシウム菌、黄色ブドウ球菌、ストレプトコッカス・アガラクチア、肺炎球菌、化膿性連鎖球菌、フェカリス菌、表皮ブドウ球菌、スタフィロコッカス・ヘモリティカス、およびパスツレラ・マルトシダから選択される細菌によって引き起こされる感染症を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0105】
別の実施形態では、本発明は、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎;および結核から選択される疾患もしくは障害(またはそれらの症状)を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0106】
別の実施形態では、本発明は、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;および肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎から選択される疾患もしくは障害(またはそれらの症状)を患っているか、またはそれらにかかり易い被験者を治療する方法を提供する。
【0107】
本発明の方法は、細菌感染治療の他の治療方法とともに用いることができる。特に、抗菌療法では、他の抗菌剤および/または抗炎症薬を用いた併用療法が想起される。少なくとも1種の式Iもしくは式Iaの化合物の投与、ならびに他の抗菌剤の任意の使用、およびシクロオキシゲナーゼ阻害薬、特にシクロオキシゲナーゼ−2の選択的阻害薬の任意の使用。かかる薬剤の組合せは、一緒に投与されてもよいし、または別々に投与されてもよく、別々に投与されるときは、投与は同時に行ってもよいし、または(時間的に近接する場合および離れている場合の両方とも)任意の順番で逐次的に行ってもよい。他の抗菌療法および抗炎症薬は、例えば、国際公開第01/34128号および同第03/061704号に記載されており、これらの出願を、それらが抗菌療法および抗炎症療法の組合せを開示する程度まで、参照により本願明細書に援用する。
【0108】
本願明細書に記載される方法は、被験者が特に明記された治療を必要とすると同定される方法を含む。かかる治療を必要とする被験者を同定することは、被験者または医療専門家の判断よるものであってよく、主観的(例えば、所見)であってもよいし、客観的(例えば、試験または診断方法によって測定できる)であってもよい。
【0109】
別の実施形態では、本発明は、細胞の活性を調節する方法であって、細胞を本願明細書中の式のいずれかの化合物の1種以上と接触させることを含む方法を提供する。
【0110】
別の実施形態では、本発明は、細菌感染症を患っているか、またはそれらにかかり易い患者の治療方法であって、それを必要とする患者に24時間の期間にわたって約450mg〜約750mgの式Iまたは式Iaの化合物を投与する工程を含む方法を提供する。別の実施形態では、患者は、450mg〜700mgの式Iまたは式Iaの化合物を投与される。
【0111】
別の実施形態では、上記の方法は、約3mg/Lを超える定常状態Cminをもたらす。別の実施形態では、上記の方法は、約4mg/Lを超える定常状態Cminをもたらす。別の実施形態では、上記の方法は、約6mg/Lを超える定常状態Cminをもたらす。さらに別の実施形態では、上記の方法は、約18mg/L未満の定常状態Cmaxをもたらす。別の実施形態では、上記の方法は、約16mg/L未満の定常状態Cmaxをもたらす。別の実施形態では、上記の方法は、約13mg/L未満の定常状態Cmaxをもたらす。さらに別の実施形態では、上記の方法は、約11.5mg/L未満の定常状態Cmaxをもたらす。
【0112】
別の実施形態では、上記の治療方法は、患者に1種以上の第2の治療剤を併用投与するさらなる工程を含む。第2の治療剤の選択は、リネゾリドとの併用投与に有用であることが公知の任意の第2の治療剤から行ってよい。
【0113】
具体的な実施形態では、本発明の併用療法は、式Iの化合物と、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される第2の治療剤とを投与することを含む。
【0114】
本願明細書で使用する「併用投与」という用語は、第2の治療剤が、1つの剤形(上述したように、本発明の化合物および第2の治療剤を含む本発明の組成物など)の一部として、または複数の個別の剤形として、本発明の化合物と一緒に投与されてもよいという意味である。あるいは、さらなる薬剤が、本発明の化合物の投与の前に、本発明の化合物の投与に続いて、または本発明の化合物の投与の後に投与されてもよい。かかる併用療法において、本発明の化合物および第2の治療剤はともに、従来の方法で投与される。本発明の化合物と第2の治療剤の両方を含む本発明の組成物を患者に投与することは、同じ治療剤、他の任意の第2の治療剤、または本発明の任意の化合物を、当該患者に、治療期間の別の時間に、別途投与することを除外しない。
【0115】
これらの第2の治療剤の有効量は当業者に周知であり、投与の案内は、本願明細書で引用した特許公報および特許公開公報、ならびに、Wellsら編集、Pharmacotherapy Handbook,第2版,Appleton and Lange,コネティカット州,スタンフォード(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,デラックス版,Tarascon Publishing,カリフォルニア州,ロマリンダ(2000)ならびに他の医療文献において見出してよい。しかしながら、第2の治療剤の最適な有効量の範囲を決定することは、十分に当業者の視野の範囲内である。
【0116】
本発明の一実施形態では、第2の治療剤が被験者に投与される場合、本発明の化合物の有効量は、第2の治療剤が投与されない場合の本発明の化合物の有効量よりも少なくなる。他の実施形態では、第2の治療剤の有効量は、本発明の化合物が投与されない場合の第2の治療剤の有効量よりも少なくなる。このようにして、どちらか一方の薬剤の多用量に関連した好ましくない副作用は、最小化される場合がある。他の潜在的な利点(投薬治療方式の改善および/または薬物費用の削減が挙げられるが、これらに限定されない)は、当業者に明らかである。
【0117】
さらに別の態様では、本発明は、被験者における上に記載の疾患、障害またはその症状の治療または予防のための薬剤の、単一組成物または個別の剤形としての製造において、式Iまたは式Iaの化合物の単独の使用または上述の第2の治療剤のうちの1つ以上との併用を提供する。本発明の他の態様は、被験者における本願明細書において記載したその疾患、障害、または症状の治療または予防に使用する本願明細書中の式の化合物である。
【0118】
(診断法およびキット)
本発明の化合物および組成物は、溶液中または血漿などの生体試料中のリネゾリドの濃度の測定方法、リネゾリドの代謝の検査方法、および他の分析的研究方法における試薬としても有用である。
【0119】
一実施形態によれば、本発明は、溶液中または生体試料中のリネゾリドの濃度の測定方法を提供し、この測定方法は、
a)既知濃度の式Iまたは式Iaの化合物を、生体試料の溶液に添加する工程と、
b)式Iまたは式Iaの化合物からリネゾリドを識別する測定装置にその溶液または生体試料を曝露する工程と、
c)検出された量の式Iまたは式Iaの化合物を、その生体試料または溶液に添加された既知濃度の式Iまたは式Iaの化合物に関連付けるために、測定装置を較正する工程と、
d)この較正された測定装置で、生体試料中のリネゾリドの量を測定する工程と、
e)式Iまたは式Iaの化合物について得られた検出量と濃度との間の相関を用いて、試料の溶液中のリネゾリドの濃度を決定する工程と
を含む。
【0120】
式Iまたは式Iaの対応する化合物からリネゾリドを識別できる測定装置としては、同位体存在量だけが互いに異なる2つの化合物を識別できる任意の測定装置が挙げられる。測定装置の例としては、質量分析計、NMR分光計、またはIR分光計が挙げられる。
【0121】
他の実施形態では、本発明は、式Iまたは式Iaの化合物の代謝安定性の評価方法を提供し、この評価方法は、ある時間にわたって、この式Iまたは式Iaの化合物を代謝酵素源に接触させる工程と、その時間の後に、式Iまたは式Iaの化合物の量を、式Iまたは式Iaの化合物の代謝産物と比較する工程と、を含む。
【0122】
関連する実施形態では、本発明は、この式Iまたは式Iaの化合物を投与した後、患者における式Iまたは式Iaの化合物の代謝安定性を評価する方法を提供する。この方法は、被験者へ式Iまたは式Iaの化合物を投与した後のある時間において、患者からの血清、尿または糞便試料を入手する工程と、式Iまたは式Iaの化合物の量を、血清、尿または糞便試料における式Iまたは式Iaの化合物の代謝産物と比較する工程とを含む。
【0123】
本発明はまた、本願明細書に記載されたものを含めた感染性疾患または感染性障害を治療するために使用するキットも提供する。これらのキットは、(a)式I/Iaの化合物、または式Iの塩、または式Iもしくは式Iaの水和物もしくは溶媒和物を含み、容器に入っている医薬組成物と、(b)本願明細書に記載されたものを含めた感染性疾患または感染性障害を治療するための医薬組成物の使用方法を説明する説明書とを含む。
【0124】
容器は、この医薬組成物を保持できる任意の容器あるいは他の密封されたまたは密封可能な器具であってよい。例としては、瓶、アンプル、各区画もしくはチャンバがこの組成物の1回の用量を含む、区画されたホルダーを有するボトルすなわち多室型のホルダーを有するボトル、各区画がこの組成物の1回の用量を含む分けられた薄片の包み、またはこの組成物の1回の用量を分配するディスペンサが挙げられる。容器は、当該分野で公知の任意の従来の形状または形態で、例えば、紙もしくはダンボール箱、ガラスもしくはプラスチックの瓶またはジャー、再封可能な袋(例えば、異なる容器に入れるために錠剤の「詰め替え」を保管するため)、または治療のスケジュールに従ってパックの外に押し出す個々の用量を入れたブリスターパックなどの薬理学的に許容できる材料で作られた容器であってよい。使用する容器は、関連する具体的な剤形に応じることができ、例えば従来のダンボール箱は、一般に液体懸濁液を保持するためには使用されない。1回の用量の剤形を市場に出すために、単一のパッケージにおいて、複数の容器を一緒に使うことができることは、実用的である。例えば、錠剤は瓶に入れられ、次にこの瓶が箱の中に入れられてもよい。容器はブリスターパックであることが好ましい。
【0125】
このキットはさらに、情報および/または医師、薬剤師もしくは被験者に対する指示を含むタイプの記憶を助けるものを含んでよい。かかる記憶を助けるものとしては、そのように特定された錠剤またはカプセルが摂取されるべき投与計画の日に対応する、投与量が入っている各チャンバもしくは区画の上に印刷された数字、または各チャンバもしくは区画の上に印刷されたその週の曜日、または何らかのタイプの情報を含むカードが挙げられる。1回用量のディスペンサについては、記憶を助けるものとしてはさらに、すでに分注された1日の用量の数を示す機械式計数器、ならびに、例えば最後の1日の用量が摂取された日付を読み出し、かつ/またはいつ次の用量を摂取するべきかを思い出させる液晶表示および/または可聴式の備忘録信号に接続された電池式のマイクロチップメモリが挙げられる。かかるキットで有用な他の記憶を助けるものは、カードに印刷されたカレンダー、および容易に分かる他のバリエーションである。
【実施例】
【0126】
(実施例1.中間体12の合成)
【化5】
氷浴で冷却した、酢酸エチル(「EtOAc」)(140ml)中のd8−モルホリン(10;23.5g、0.25mol)およびiPr2EtN(44ml、0.25mol)の撹拌した溶液に、3,4−ジフルオロニトロベンゼン(11、27.4ml、0.25mol)を10分間かけて滴下した。この反応混合物を室温で48時間撹拌した。この反応混合物をEtOAc(300ml)およびCH2Cl2(50ml)で希釈し、次いで水(350ml)を加えた。層を分離させ、水層をEtOAc(3×300ml)で洗浄した。合わせた有機物を乾燥し(MgSO4)、濾過し、真空中で濃縮した。20% EtOAc/ヘキサンで溶出するカラムクロマトグラフィ(1kgシリカ)を使用して粗生成物を生成し、中間体12を87%収率で得た。
【0127】
(実施例2.中間体13の合成)
【化6】
N2下の変性EtOH(875ml)中の12(50g、0.21mol)の撹拌した懸濁液に10% Pd/C(50%湿体、17.5g)を加えた。この反応容器にN2を10分間、H2を10分間パージし、大気圧のH2下で一晩撹拌した。水素化を停止し、この容器にN2を15分間パージした。この混合物をセライトを通して濾過し、変性EtOH(500ml)およびDCM(3×700ml)で洗浄した。合わせた濾液を真空中で濃縮し、所望のアニリン13(38.5g、90%収率)を桃色固体として得た。
【0128】
(実施例3.(R)−d2−エピクロロヒドリン14Bの合成)
【化7】
【0129】
Et2O(1000ml)中のメチル−α,β−イソプロピリデン−D−グリセレート(20;175g、1.09mol、1当量)の氷冷した溶液に、LiAlD4(34.43g、0.82mol、0.75当量)をEt2O(1000ml)中の懸濁液として3時間かけて加えた。この反応混合物を5時間還流させた。この混合物を室温まで冷却し、Et2O(1000ml)で希釈し、水(40ml)でクエンチした。この混合物を15分間撹拌し、濾過し、固体をEt2O(1000ml)で洗浄した。濾液を真空中で濃縮し、対応するアルコール21を高純度で得た(121.3g、83%収率)。
【0130】
アルコール(21;60.65g、0.45mol、1当量)を、PPh3(124g、0.47mol、1.05当量)およびDBU(34ml、0.225mol、0.5当量)とともにベンゼン(110ml)に溶解させた。この混合物を、DBU(17ml、0.11mol、0.25当量)を含むCCl4(110ml)の還流溶液に30分間かけて滴下した。この反応混合物を還流状態で一晩撹拌した。この反応混合物を室温まで冷却し、真空中で濃縮し、粗製混合物を得た。この粗製混合物をシリカ(60g)上に吸収させ、EtOAc:ヘキサン 1:1で溶出するカラムクロマトグラフィ(シリカ:1200g)を使用して精製し、所望のクロリド22を54%収率で得た。
【0131】
クロリド22(37.6g、0.25mol)を、アセトン(60ml)および1M HCl(150ml)の溶液に加えた。この混合物を55℃で30分間加熱した。この反応混合物を室温まで冷却し、アセトンを真空中で除去した。この混合物をNaCl(43g)で飽和させ、EtOAc(2×250ml)で抽出した。合わせたEtOAc層をMgSO4で乾燥し、濃縮し、ジオール、(R)−d2−クロロヒドリン23(21.8g、79%収率)を得た。
【0132】
ピリジン(210ml)中の(R)−d2−クロロヒドリン23(21.0g、0.19mol、1当量)の氷冷した溶液に、トルエンスルホニルクロリド(35.5g、0.19mol、1当量)を少しずつ加えた。このスルホニルクロリドの添加終了後、この混合物を室温まで加温し、1時間撹拌した。この混合物にEt2O(300ml)を加え、水性洗浄液が酸性になるまでこの混合物を1M HCl(3×500ml)で洗浄した。この有機抽出液を飽和NaHCO3水溶液(300ml)で洗浄し、MgSO4で乾燥し、濃縮し、対応するトシレート24(31.3g、63%収率)を得た。
【0133】
ナトリウム金属(5g、37.48mmol、2当量)をエチレングリコール(40ml)に加え、この混合物を20℃で一晩撹拌し、ナトリウムエチレングリコレートのエチレングリコール溶液を生成した。エチレングリコール(5ml)中のトシレート24(5g、18.74mmol、1当量)の溶液を加え、この混合物を20℃で15分間撹拌した。生成物を減圧下でこの混合物から除去し、ドライアイス/IPAコールドフィンガー中で透明な液体として集め、鏡像異性体に富む(R)−d2−エピクロロヒドリン25(1.02g、58%収率)を得た。キラルGCは、79.4%の鏡像体過剰率を示した。
【0134】
(S,S)−コバルト(II)触媒(43.2mg、0.0716mmol)をトルエン(8μl)に溶解させた。酢酸(8.6μl、0.143mmol、2当量)を加え、生成した混合物を開放系で室温で30分間撹拌し、この間にこの混合物の色は橙色から暗褐色に変化した。すべての揮発性物質を真空中で除去し、コバルト(III)触媒のアセテート錯体を褐色残渣として得た。この調製した触媒に、80%鏡像異性体に富む(R)−d2−エピクロロヒドリン25(2.5g)およびTHF(2.5ml)を加えた。この反応フラスコを0℃に冷却し、H2O(0.05ml)を15分間かけて滴下した。この反応液を室温まで加温し、18時間撹拌した。この反応混合物に、少量のMgSO4を加え、室温での蒸留により(R)−d2−エピクロロヒドリン26を単離し、エピクロロヒドリンおよびTHFの1:1混合物を得た。キラルGC分析は99.1%eeを示した。
【0135】
(実施例4.中間体15Aおよび15Bの合成)
【化8】
アニリン13(15g、74mmol、1.0当量)をN2下でイソプロパノール(75ml)に溶解させ、(R)−エピクロロヒドリン(14A;7.0g、81.4mmol、1.1当量)を加えた。この混合物を還流状態で一晩撹拌した。溶媒を真空中で除去し、残渣をカラムクロマトグラフィ(750gシリカ、CH2Cl2、次いで1% MeOH、CH2Cl2)によって精製し、15Aを淡褐色油状物として得た(12.8g、58%収率)。
【0136】
アニリン(4.1g、20.1mmol、1.0当量)をN2下でイソプロパノール(20ml)に溶解させ、(R)−d2−エピクロロヒドリン(14B;2.0g、22.1mmol、1.1当量)を加えた。この混合物を還流状態で一晩撹拌した。溶媒を真空中で除去し、残渣をカラムクロマトグラフィ(300gシリカ、CH2Cl2、次いで1% MeOH、CH2Cl2)によって精製し、15Bを淡褐色油状物として得た(3.0g、50%収率)。LCは純度97%を示した。キラルLCは、95.7%の鏡像体過剰率を示した。
【0137】
(実施例5.中間体17Aおよび17Bの合成)
【化9】
中間体15A(12.8g、0.043mol、1当量)、フタルイミドカリウム(16;10.4g、0.056mol、1.3当量)およびDMF(100ml)を100℃で5時間加熱した。LC分析は反応の完結を示した。この反応混合物を室温まで冷却し、水(450ml)の中へ注ぎ込んだ。この混合物を2時間撹拌し、濾過し、固体ケーキを真空オーブン中、40℃で一晩乾燥し、17Bを得た(9g、51%収率)。LCMSは純度95.0%を示した。
【0138】
中間体15B(3.2g、10.7mmol、1当量)、フタルイミドカリウム(16;2.58g、13.9mmol、1.3当量)およびDMF(23ml)を100℃で5時間加熱した。この反応混合物を室温まで冷却し、水(100ml)の中へ注ぎ込んだ。この混合物を2時間撹拌し、濾過し、固体ケーキを真空オーブン中、40℃で一晩乾燥し、17Bを得た(2.27g、52%収率)。LCは純度98.2%を示した。キラルLCは98.6%の鏡像体過剰率を示した。
【0139】
(実施例6.中間体18Aおよび18Bの合成)
【化10】
中間体17A(9.0g、22mmol、1当量)をDCM(100ml)に溶解させ、カルボニルジイミダゾール(5.0g、31mmol、1.4当量)を室温で加え、この混合物を窒素下で一晩撹拌した。LC分析は反応の完結を示した。水(300ml)をこの混合物に加え、水層をDCM(300ml)で抽出した。合わせたDCM層をMgSO4で乾燥し、真空中で濃縮し、18Aを得た。LCMSは純度94.4%を示した。
【0140】
中間体17B(2.08g、5.08mmol、1当量)をDCM(25ml)に溶解させ、カルボニルジイミダゾール(1.15g、7.11mmol、1.4当量)を室温で加え、この混合物を窒素下で一晩撹拌した。LC分析は反応の完結を示した。水(70ml)をこの混合物に加え、水層をDCM(70ml)で抽出した。合わせたDCM層をMgSO4で乾燥し、真空中で濃縮し、18Bを得た(2.1g、95%収率)。キラルLCは99.0%の鏡像体過剰率を示した。
【0141】
(実施例7.中間体19Aおよび19Bの合成)
【化11】
MeOH(100ml)およびヒドラジン水和物(6.1ml、0.125mol、5.5当量)を18A(9.9g、23mmol、1当量)が入っているフラスコに加えた。この混合物を還流温度で1時間撹拌した。この反応混合物を室温まで冷却し、水(200ml)を加え、この混合物をDCM(2×200ml)で抽出した。合わせたDCM抽出液を水(100ml)で洗浄し、MgSO4で乾燥し、真空中で濃縮し、19Aを得た(6.0g、87%収率)。LCMSは純度96.8%を示した。
【0142】
MeOH(20ml)およびヒドラジン水和物(1.4ml、26.5mmol、5.5当量)を18B(2.1g、4.8mmol、1当量)が入っているフラスコに加えた。この混合物を還流温度で1時間撹拌した。この反応混合物を室温まで冷却し、水(40ml)を加え、この混合物をDCM(2×40ml)で抽出した。合わせたDCM抽出液を水(100ml)で洗浄し、MgSO4で乾燥し、真空中で濃縮し、19Bを得た(1.26g、86%収率)。キラルLCは99.3%の鏡像体過剰率を示した。
【0143】
(実施例8.中間体20Aおよび20Bの合成)
【化12】
中間体19A(6.0g、0.02mol、1当量)をトルエン(90ml)中で15分間撹拌した。無水酢酸(5.4ml、0.057mol、2.9当量)を室温で滴下した。水浴を5分間用いてこの混合物を35℃に加温し、19Aの溶解度を高めた。この反応混合物を室温で1時間撹拌し、0℃に冷却し、濾過し、化合物101を得た(5.3g、78%収率)。LCは純度99.1%を示した。キラルLCは99.4%の鏡像体過剰率を示した。1H−NMR(300MHz,CDCl3):δ 2.03(s,3H),3.61−3.66(m,2H),3.71−3.77(m,1H),4.00(見かけ上t,J=8.9,1H),4.71−4.79(m,1H),6.50−6.54(m,1H),6.88(見かけ上t,J=8.9,1H),7.04(dd,J1=10.0,J2=1.6,1H),7.41(dd,J1=14.6,J2=2.7,1H)。HPLC(方法:RP80A、150mm×4.6mmカラム−勾配方法 5−95%ACN+0.1%ギ酸、勾配の前に5%ACNで5分間保持、10分間にわたる勾配、次いで95%ACNで10分間保持;T=30℃;波長:258nm):保持時間:11.45分。キラルHPLC(方法:Chiralpak AD−H;250×4.6mmカラム;5μm粒径−ヘキサン/IPA/DEA(80:20:01);T=40℃;波長:258nm):所望の鏡像異性体について11.80分、少量の鏡像異性体について14.21分;ee=99.8%。MS(M+H+):346.5。中間体19B(1.25g、4.09mmol、1当量)をトルエン(20ml)中で15分間撹拌した。無水酢酸(1.12ml、11.86mol、2.9当量)を室温で滴下した。水浴を5分間用いてこの混合物を35℃に加温し、19Bの溶解度を高めた。この反応混合物を室温で1時間撹拌し、0℃に冷却し、濾過し、化合物100を得た(1.05g、74%収率)。LCは純度99.4%を示した。キラルLCは98.9%の鏡像体過剰率を示した。1H−NMR(300MHz,CDCl3):δ 2.00(s,3H),3.74(dd,J1=9.1,J2=6.8,1H),4.00(t,J=9.1,1H),4.75(dd,J1=8.8,J2=6.8,1H),6.52(bs,1H),6.88(t,J=8.8,1H),7.02−7.06(m,1H),7.41(dd,J1=4.5,J2=2.6,1H)。HPLC(方法:RP80A、150mm×4.6mmカラム−勾配方法 5−95%ACN+0.1%ギ酸、勾配の前に5%ACNで5分間保持、10分間にわたる勾配、次いで95%ACNで10分間保持;T=30℃;波長:258nm):保持時間:11.55分間。キラルHPLC(方法:Chiralpak AD−H;250×4.6mmカラム;5μm粒径−ヘキサン/IPA/DEA(80:20:01);T=40℃;波長:258nm):所望の鏡像異性体について10.63分、少量の鏡像異性体について13.18分;ee=98.9%.MS(M+H+):348.3。
【0144】
(実施例9.2,2,6,6−d4−モルホリン(33)および全重水素化モルホリン(10)の合成)
【化13】
ジグルコール酸(30)を水酸化ナトリウムのD2O溶液で処理し、対応する重水素化二ナトリウム化合物31を生成した。化合物31を全重水素化塩化アンモニウムの存在下で加熱して、d4−ジオキソモルホリン32を生成し、これをTHF中で水素化ホウ素によって還元し、所望の2,2,6,6−d4−モルホリン(33)を生成した。テトラジュウテロモルホリン33を上記の実施例1の全重水素化モルホリン10の代わりに使用して、各Zが重水素であり、各Yが水素である式Iおよび式Iaの化合物、例えば化合物102および化合物103を生成することができる。
【0145】
(実施例10)
抗菌活性を、マウスアッセイ手順を使用して生体内で試験した。雌のマウス(各々18〜20gの6匹)の群に、使用直前に解凍し4% ビール酵母を含む脳心臓浸出物(黄色ブドウ球菌)または脳心臓浸出物(連鎖球菌種)に懸濁させた黄色ブドウ球菌を腹腔内に注射した。1つの薬物あたり6回用量レベルで抗生物質処置を、経口挿管または皮下経路のいずれかによる感染の1.5時間後に投与した。6日間、毎日生存を観察した。死亡率に基づくED50値をプロビット分析を使用して算出した。主題の化合物を対照(例えば、バンコマイシン)と比較した。
【0146】
(実施例11)
生体外活性実験を、当業者に公知の標準的な希釈方法によって行った。手短に言えば、抗生物質の系列2倍希釈液を希釈剤中で調製し,標準的な量のミコバクテリア増殖培地を薬物のアリコートに加えた。この培地に標準化ミコバクテリア細胞懸濁液を植え付け、適切な条件下でインキュベートした。インキュベーション後、最小阻止濃度(MIC)を目視の観察によって測定した。MICは、ミコバクテリア増殖を阻止するために必要とする最小薬物濃度(μg/ml単位)と定義される。
【0147】
(実施例12)
生体内データを、1×107個の生きた結核菌(Erdman株)を用いて静脈内感染させたCD−1マウスから得た。24時間後、薬物処置を開始した。すべての薬物を、1日2回、4週間、経口摂食によって与えた。治療の最後に、生きた細胞数を脾臓および肺のホモジネートから測定した。
【0148】
(実施例13)
チンパンジーでの薬物動態
リネゾリドとの50:50 混合物としてチンパンジーに経口投与または静脈内投与した場合の化合物100の薬物動態を研究した。経口投与および静脈内投与の両方のための溶液を、撹拌しながら、65℃で900mlの注射用の滅菌水中でリネゾリド(200mg)、化合物100(200mg)、クエン酸ナトリウム二水和物(164mg)、無水クエン酸(85mg)、およびデキストロース一水和物(5.024g)を混合することにより調製した。この混合物を25℃に冷却し、生成した溶液のpHを必要に応じて10% HClまたは10% NaOHのいずれかを用いて4.8に調整した。この溶液の最終体積を、滅菌水を用いて1lにした。この投薬溶液を、投薬前に0.22μmフィルターを通して濾過した。
【0149】
4匹のチンパンジー(2匹の雄および2匹の雌)をこの研究で使用した。1匹の雄および1匹の雌を静脈内研究のために使用し、1匹の雄および1匹の雌を経口投薬研究のために使用した。すべての動物を、投薬前に一晩絶食させた。すべての研究について、動物を、投薬前にケタミン(およそ10mg/ml)またはテラゾール(telazol)(およそ5mg/ml)で鎮静させた。各研究について、動物に300mgの併用薬物(各々150mgのリネゾリドおよび化合物100)を投薬した。静脈内用量は、30分間にわたる点滴によって投与した(2mg/mlで150mlの併用薬物)。経口用量は、2mg/mlで150mlの体積の併用薬物を投与した。
【0150】
静脈内研究については、4.5mlのアリコートの血液を、点滴の開始前、点滴開始15分後、および点滴の最後の直前に各動物から採取した。さらなる試料を、点滴後6分、15分、30分、および60分および1.5時間、2時間、4時間、6時間、8時間および24時間に採取した。経口研究については、4.5mlのアリコートの血液を、投薬前、および投薬後15分、20分および60分および1.5時間、2時間、4時間、6時間、8時間および24時間に各動物から採取した。すべての血液試料を抗凝血剤としてのヘパリンナトリウムが入っているバキュテナーチューブに集め、十分に混合し、氷上で保存した。試料を、回収から1時間以内に遠心分離にかけ、血漿を集め、分析まで−70℃で凍結させた。尿も、投薬後24時間にわたって各動物から集めた。
【0151】
各試料を、リネゾリドおよび化合物100の両方の存在について、LC−MS/MSによって以下のようにして分析した。チンパンジーの血漿試料(100μL)を、LC−MS/MS分析の前に300μLの内部標準溶液と混合した。内部標準は、アセトニトリル/水(90/10、体積/体積)中の250ng/mL ハロペリドールであった。タンパク質の沈殿後、10μLの上清をZorbax SB−C8(高速分解)カラム(2.1×30mm、3.5μm)に注入した。最初の移動相条件は、0.5mL/分の流量での、100% A(0.1% ギ酸を含む水)および0% B(0.1% ギ酸を含むアセトニトリル)であった。移動相Bを、2分以内に90%に到達させ、1分間保持してから、3.2分に再び0%に減少させた。全体の分析時間は6分であった。前駆体/生成物イオン対を、リネゾリド、化合物100およびハロペリドールの検出について、それぞれm/z 338/296、m/z 348/306およびm/z 376/165に設定した。
【0152】
尿試料を同様に分析した。チンパンジーの尿試料(10μL)を、独立に、Zorbax SB−C8(高速分解)カラム(2.1×30mm、3.5μm)に注入した。最初の移動相条件は、0.4mL/分の流量での100% A(0.1% ギ酸を含む水)および0% B(0.1% ギ酸を含むアセトニトリル)であった。移動相Bを、42分以内に25%に到達させ、次いで2分間かけて25%から90%に到達させてから、4分かけて再び0%に減少させた。全体の分析時間は48分であった。質量分析計を陽イオンモードに設定し、イオンをm/z 100〜1000で走査した。代謝産物の特定の分子イオンを同定すると、MS/MS実験を実施して生成物イオンを生成した。
【0153】
図1および図2は、静脈内投薬研究の結果を示す。雌(図1)および雄のチンパンジー(図2)の両方が、リネゾリドと比べて、化合物100についての半減期およびAUCの増加を示した。静脈内投薬についての半減期の算出値を表1に示す。
【0154】
表1:静脈内投薬後の化合物100およびリネゾリドの半減期
【表1】
【0155】
図3および図4は、経口投薬研究の結果を示す。雌(図3)および雄のチンパンジー(図4)の両方が、リネゾリドと比べて、化合物100についての半減期およびAUCの増加を示した。8時間および24時間におけるリネゾリドに対する化合物100の血清濃度の比を表2に示す。各化合物についてのAUCの平均算出値を表3に示す。
【0156】
表2:経口投薬後のリネゾリドに対する化合物100の血清濃度の比
【表2】
【0157】
表3.経口投薬後の化合物100およびリネゾリドの平均AUC0−24時間
【表3】
【0158】
リネゾリドと比べたときの化合物100の代謝の最終結果を、静脈内投薬または経口投薬後の尿中の各化合物の排出を追跡することにより、分析した。この分析の結果を表4に示す。
【0159】
表4.尿における無疵のリネゾリドおよび化合物100の排出
【表4】
表4に示す結果は、投与経路にも被験体の性別にも依らず、リネゾリドのおよそ2倍も多い化合物100が無疵で尿中に排出されたことを明らかにする。加えて、さらなる分析から、M6代謝産物およびその重水素化等価体の量は本質的に同じであるが、重水素化M4代謝産物の量はリネゾリドM4代謝産物よりも有意に低いことが明らかとなった。
【0160】
これらのチンパンジーでの研究は、化合物100はリネゾリドよりもゆっくりと代謝され、そしてその代謝の最終結果は、リネゾリドと比べて、M4代謝産物から無疵の排出にシフトすることを示す。
【0161】
さらなる記載がなくとも、当業者は、前述の記載および例示となる実施例を使用して、本発明の化合物を作製して利用し、請求項に係る方法を実施することができると考えられる。前述の説明および実施例は単に特定の好ましい実施形態の詳細な説明を提供するだけであることを理解されたい。種々の変更態様および均等物は、本発明の趣旨および範囲から逸脱することなく作製され得る。
【特許請求の範囲】
【請求項1】
式Iまたは式Iaの化合物:
【化1】
(式中、
各Wは独立に、水素または重水素であり、
各Yは独立に、水素または重水素であり、
各Zは独立に、水素、重水素、もしくはフッ素であり、かつ
少なくとも1つのW、YまたはZは重水素である)
またはその薬理学的に許容できる塩。
【請求項2】
少なくとも1つのWが重水素であり、少なくとも2つのY部分が重水素であり、かつ少なくとも2つのZ部分が重水素またはフッ素である、請求項1に記載の化合物。
【請求項3】
W1およびW2が同時に重水素である、請求項1または請求項2に記載の化合物。
【請求項4】
W1およびW2が同時に水素である、請求項1または請求項2に記載の化合物。
【請求項5】
Y1、Y2、Y3およびY4が同時に重水素である、請求項1から請求項4のいずれか1項に記載の化合物。
【請求項6】
Y1、Y2、Y3およびY4が同時に水素である、請求項1から請求項4のいずれか1項に記載の化合物。
【請求項7】
Z1、Z2、Z3およびZ4の各々が独立に、重水素およびフッ素から選択される、請求項1から請求項6のいずれか1項に記載の化合物。
【請求項8】
Z1、Z2、Z3およびZ4が同時に重水素である、請求項7に記載の化合物。
【請求項9】
前記式Iまたは式Iaの化合物の配置が(S)である、請求項1から請求項8のいずれか1項に記載の化合物。
【請求項10】
以下の
【化2】
の群から選択される、請求項1に記載の化合物。
【請求項11】
重水素と指定されない任意の原子が天然の同位体存在量で存在する、請求項1から請求項10のいずれか1項に記載の化合物。
【請求項12】
請求項1に記載の化合物と、許容できる担体とを含む、発熱物質不含組成物。
【請求項13】
前記担体が薬理学的に許容できる担体である、薬理学的投与用に処方された請求項12に記載の組成物。
【請求項14】
さらに、抗菌剤およびシクロオキシゲナーゼ阻害薬から選択される第2の治療剤を含む、請求項13に記載の組成物。
【請求項15】
前記第2の治療剤が、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される、請求項14に記載の組成物。
【請求項16】
細菌感染症または真菌障害を患っているか、またはそれらにかかり易い被験者を治療する方法であって、それを必要とする前記被験者に請求項11に記載の組成物を投与する工程を含む方法。
【請求項17】
前記被験者が、フェシウム菌、黄色ブドウ球菌、ストレプトコッカス・アガラクチア、肺炎球菌、化膿性連鎖球菌、フェカリス菌、表皮ブドウ球菌、スタフィロコッカス・ヘモリティカス、およびパスツレラ・マルトシダから選択される細菌によって引き起こされる感染症を患っているか、またはそれにかかり易い、請求項16に記載の方法。
【請求項18】
前記被験者が、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎;ならびに結核から選択される疾患もしくは障害を患っているか、またはそれらにかかり易い、請求項16に記載の方法。
【請求項19】
前記被験者が、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;ならびに肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎から選択される疾患もしくは障害を患っているか、またはそれらにかかり易い、請求項18に記載の方法。
【請求項20】
ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される第2の治療剤を投与することを必要とする前記被験者に、かかる投与をするさらなる工程を含む、請求項16から請求項19のいずれか1項に記載の方法。
【請求項1】
式Iまたは式Iaの化合物:
【化1】
(式中、
各Wは独立に、水素または重水素であり、
各Yは独立に、水素または重水素であり、
各Zは独立に、水素、重水素、もしくはフッ素であり、かつ
少なくとも1つのW、YまたはZは重水素である)
またはその薬理学的に許容できる塩。
【請求項2】
少なくとも1つのWが重水素であり、少なくとも2つのY部分が重水素であり、かつ少なくとも2つのZ部分が重水素またはフッ素である、請求項1に記載の化合物。
【請求項3】
W1およびW2が同時に重水素である、請求項1または請求項2に記載の化合物。
【請求項4】
W1およびW2が同時に水素である、請求項1または請求項2に記載の化合物。
【請求項5】
Y1、Y2、Y3およびY4が同時に重水素である、請求項1から請求項4のいずれか1項に記載の化合物。
【請求項6】
Y1、Y2、Y3およびY4が同時に水素である、請求項1から請求項4のいずれか1項に記載の化合物。
【請求項7】
Z1、Z2、Z3およびZ4の各々が独立に、重水素およびフッ素から選択される、請求項1から請求項6のいずれか1項に記載の化合物。
【請求項8】
Z1、Z2、Z3およびZ4が同時に重水素である、請求項7に記載の化合物。
【請求項9】
前記式Iまたは式Iaの化合物の配置が(S)である、請求項1から請求項8のいずれか1項に記載の化合物。
【請求項10】
以下の
【化2】
の群から選択される、請求項1に記載の化合物。
【請求項11】
重水素と指定されない任意の原子が天然の同位体存在量で存在する、請求項1から請求項10のいずれか1項に記載の化合物。
【請求項12】
請求項1に記載の化合物と、許容できる担体とを含む、発熱物質不含組成物。
【請求項13】
前記担体が薬理学的に許容できる担体である、薬理学的投与用に処方された請求項12に記載の組成物。
【請求項14】
さらに、抗菌剤およびシクロオキシゲナーゼ阻害薬から選択される第2の治療剤を含む、請求項13に記載の組成物。
【請求項15】
前記第2の治療剤が、ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される、請求項14に記載の組成物。
【請求項16】
細菌感染症または真菌障害を患っているか、またはそれらにかかり易い被験者を治療する方法であって、それを必要とする前記被験者に請求項11に記載の組成物を投与する工程を含む方法。
【請求項17】
前記被験者が、フェシウム菌、黄色ブドウ球菌、ストレプトコッカス・アガラクチア、肺炎球菌、化膿性連鎖球菌、フェカリス菌、表皮ブドウ球菌、スタフィロコッカス・ヘモリティカス、およびパスツレラ・マルトシダから選択される細菌によって引き起こされる感染症を患っているか、またはそれにかかり易い、請求項16に記載の方法。
【請求項18】
前記被験者が、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎;ならびに結核から選択される疾患もしくは障害を患っているか、またはそれらにかかり易い、請求項16に記載の方法。
【請求項19】
前記被験者が、グラム陽性菌感染症、バンコマイシン耐性フェシウム菌感染症;黄色ブドウ球菌および肺炎球菌による院内肺炎;黄色ブドウ球菌、化膿性連鎖球菌、またはストレプトコッカス・アガラクチアによって引き起こされる併発性皮膚・皮膚組織感染症;黄色ブドウ球菌または化膿性連鎖球菌によって引き起こされる無併発性皮膚・皮膚組織感染症;ならびに肺炎球菌または黄色ブドウ球菌によって引き起こされる市中肺炎から選択される疾患もしくは障害を患っているか、またはそれらにかかり易い、請求項18に記載の方法。
【請求項20】
ゲンタマイシン、トブラマイシン、アズトレオナム、セファゾリン、セフタジジム、ピペラシリン、シプロフロキサシン、オフロキサシン、レボフロキサシン、セレコキシブ、およびロフェコキシブから選択される第2の治療剤を投与することを必要とする前記被験者に、かかる投与をするさらなる工程を含む、請求項16から請求項19のいずれか1項に記載の方法。
【図1】

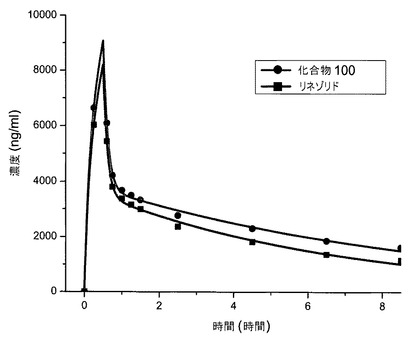
【図2】
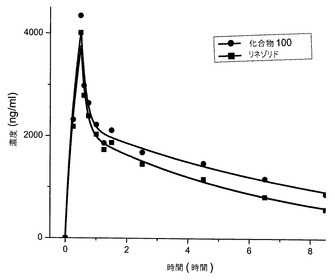
【図3】
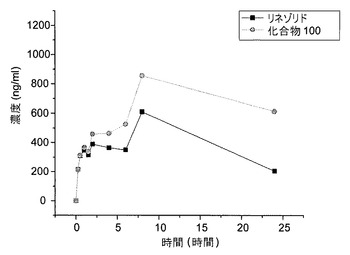
【図4】
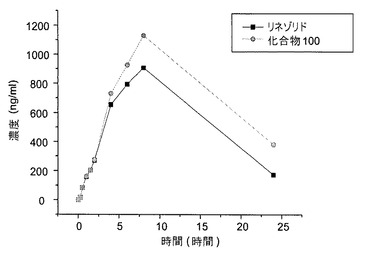
【図2】
【図3】
【図4】
【公表番号】特表2010−507576(P2010−507576A)
【公表日】平成22年3月11日(2010.3.11)
【国際特許分類】
【出願番号】特願2009−533406(P2009−533406)
【出願日】平成19年10月23日(2007.10.23)
【国際出願番号】PCT/US2007/022516
【国際公開番号】WO2008/127300
【国際公開日】平成20年10月23日(2008.10.23)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
【公表日】平成22年3月11日(2010.3.11)
【国際特許分類】
【出願日】平成19年10月23日(2007.10.23)
【国際出願番号】PCT/US2007/022516
【国際公開番号】WO2008/127300
【国際公開日】平成20年10月23日(2008.10.23)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
[ Back to top ]