
オキシコドンおよびナロキソンを含む即時放出医薬組成物
本発明は、オキシコドンおよびナロキソンまたはそれらの薬学的に許容される塩を含む、疼痛に苦しむ患者を治療するのに適した経口即時放出医薬組成物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、疼痛に苦しむ患者を治療するのに適した医薬組成物に関する。
【背景技術】
【0002】
疼痛に苦しむ患者を適切に治療することの重要性は、過去数十年にわたって、ますます認識されつつある。
【0003】
がん患者において起こるような慢性の中等度から高度のおよびさらには重度の疼痛の治療のために、オピオイド系鎮痛剤が、最近の数十年にわたってますます普及しつつある。この発展に寄与している要因の中には、以前に利用可能であったそれらの薬剤の即時放出製剤に比較して、患者が、低減された頻度で服用することができる、モルフィン、ヒドロモルホンおよびオキシコドンなどのオピオイドの制御放出製剤の導入があった。
【0004】
活性薬剤としてオキシコドン塩酸塩を含む製品Oxygesic(登録商標)錠剤、およびオキシコドン塩酸塩とオピオイド拮抗薬ナロキソン塩酸塩との組合せを含むTargin(登録商標)錠剤などのオピオイドオキシコドン制御放出製剤は、商業的展望から、ならびに患者および医師による受容性の観点でどちらも成功した。
【0005】
しかし、オピオイドの制御放出製剤は、疼痛に苦しむ患者を治療する場合に、必ずしも第1選択の薬剤であるとは限らない可能性がある。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の1つの目的は、疼痛を治療するのに、とりわけ慢性の中等度〜高度のおよびさらには重度の疼痛を治療するのに適し、標準的な疼痛療法中に発生する可能性のあるような望ましくない副作用を同時に回避しながら、疼痛に苦しむ患者の用量設定するのにおよび/または疼痛に苦しむ患者における突出痛を治療するのに使用することができる医薬組成物を提供することである。
【0007】
後に続く説明から明らかになるようなこれらのおよびその他の目的は、独立請求項の主題によって達成される。その後に示される従属請求項は、本発明の好ましい実施形態の一部を示す。
【課題を解決するための手段】
【0008】
本発明は、一実施形態において、少なくともオキシコドンまたはその薬学上許容される塩およびナロキソンまたはその薬学上許容される塩を約2:1の重量比(オキシコドンまたはその薬学上許容される塩:ナロキソンまたはその薬学上許容される塩)で含む経口即時放出医薬組成物に関する。
【0009】
一般に、本発明による経口即時放出医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩に加えて、その他の薬学的に活性な薬剤を含むことができる。しかし、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が、本発明による医薬組成物の唯一の薬学的に活性な薬剤であることが本発明の好ましい実施形態である場合もある。
【0010】
一般に、本発明による即時放出医薬組成物は、オキシコドンをその遊離塩基またはその塩の形態で、およびナロキソンをその遊離塩基またはその塩の形態で含むことができる。しかし、本発明による経口即時放出医薬組成物が、オキシコドンをオキシコドン塩酸塩の形態で、およびナロキソンをナロキソン塩酸塩の形態で含むことが好ましい場合もある。オキシコドン塩酸塩およびナロキソン塩酸塩が、医薬組成物の唯一の薬学的に活性な薬剤を構成することがさらにより好ましい可能性もある。
【0011】
第1態様において、本発明による経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。1つの好ましい実施形態において、本発明による経口即時放出医薬組成物は、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。これらの医薬組成物は、該薬学的に活性な薬剤を約2:1の重量比で含むことができる。
【0012】
この第1態様の好ましい実施形態は、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を約2:1の比率(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み、オキシコドン塩酸塩が約2.5mg〜約40mgのオキシコドン塩酸塩の量で存在し、ナロキソン塩酸塩が約1.25mg〜約20mgのナロキソン塩酸塩の量で存在する、経口即時放出医薬組成物に関し得る。
【0013】
この第1態様のさらにより好ましい実施形態は、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を約2:1の比率(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み、オキシコドン塩酸塩が約2.5mg〜約20mgのオキシコドン塩酸塩の量で存在し、ナロキソン塩酸塩が約1.25mg〜約10mgのナロキソン塩酸塩の量で存在する、経口即時放出医薬組成物に関し得る。
【0014】
本発明の第2態様は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、かつここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する、経口即時放出医薬組成物に関する。
【0015】
この第2態様の好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0016】
この第2態様のより好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0017】
この第2態様のさらにより好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0018】
第2態様のすべての実施形態において、経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。
【0019】
約1mg〜約80mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約40mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約1mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、あるいは約1mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩が、さらにより好ましい可能性がある。
【0020】
本発明の第3態様において、経口即時放出医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含むことができ、ここで、該組成物は、固体形態であり、かつ該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する。
【0021】
この第3態様の好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0022】
この第3態様のより好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0023】
この第3態様のさらにより好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0024】
第3態様のすべての実施形態において、経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。
【0025】
約1mg〜約80mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約40mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約1mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、あるいは約1mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩が、さらにより好ましい可能性がある。
【0026】
第3態様のすべての実施形態において、医薬組成物は、崩壊剤、および任意選択で充填剤、および任意選択でその他の薬学的に許容される添加剤を含むことができる。好ましくは、崩壊剤として、例えば、デンプンと乳糖の組合せを使用することができる。乳糖は、単独で、同時に充填剤として機能することができる。とりわけ好ましい実施形態は、崩壊剤および充填剤の双方として機能することができる、乳糖85%とデンプン15%との組合せである製品Starlac(登録商標)に依拠する。異なる種類の崩壊剤、充填剤またはその他の種類の薬学的に許容される添加剤を後に示す。組み合わせた充填剤/崩壊剤は、医薬組成物内に、該組成物の重量を基準にて約40重量%〜約90重量%の量で、好ましくは約50重量%〜約85重量%の量で、さらにより好ましくは約60重量%〜約80重量%の量で含むことができる。StarLac(登録商標)のように崩壊剤および充填剤の双方として二重の機能を有する添加剤を使用する場合、これらの数字がとりわけ適用される。
【0027】
第3態様のすべての実施形態において、医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含むことができる。
【0028】
本発明の第3態様のすべての実施形態において、医薬組成物は、錠剤、カプセル剤、顆粒剤、多粒子剤などの形態で提供することができる。錠剤は、とりわけ好ましい可能性がある。
【0029】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、ヒト健常志願者での単回投与治験で投与された場合に、約15ng.h/mL〜約500ng.h/mLの範囲の、好ましくは約20ng.h/mL〜約400ng.h/mLの範囲の、より好ましくは約25ng.h/mL〜約300ng.h/mLの範囲の、さらにより好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0030】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、ヒト健常志願者での単回投与治験で投与された場合に、約1ng/mL〜約300ng/mLの範囲の、好ましくは約2ng/mL〜約200ng/mLの範囲の、より好ましくは約3ng/mL〜約100ng/mLの範囲の、さらにより好ましくは約4ng/mL〜約75ng/mLの範囲の、最も好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0031】
本発明による経口即時放出医薬組成物は、限定はされないが、錠剤、カプセル剤、多粒子剤(例えば、顆粒、長球またはビーズ)、および液剤(例えば溶液、懸濁液または乳液)の形態であり得る。
【0032】
本発明による経口即時放出医薬組成物は、薬学的に許容される添加剤として、少なくとも賦形剤および任意選択で崩壊剤を含むことができる。さらに、それらの組成物は、滑沢剤、充填剤、着色剤、着香剤、pH調整剤、可塑剤、抗粘着剤、結合剤などのその他の薬学的に許容される添加剤を含むことができる。本発明による経口即時放出医薬組成物は、また、薬学的に許容される添加剤として、少なくとも崩壊剤および任意選択で賦形剤を含むことができる。さらに、それらの組成物は、滑沢剤、充填剤、着色剤、着香剤、pH調整剤、可塑剤、抗粘着剤、結合剤などのその他の薬学的に許容される添加剤を含むことができる。Starlac(登録商標)製品などの崩壊剤および賦形剤の双方としての二重機能を有する添加剤を好ましくは使用することができることを理解されたい。
【0033】
本発明のとりわけ好ましい実施形態において、経口即時放出医薬組成物は、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1(オキシコドン塩酸塩:ナロキソン塩酸塩)の重量比含み、ここで、該組成物は、唯一の薬学的に許容される活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含み、組成物は錠剤の形態であり、製剤は、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含み、かつ製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する。本発明のこれらのとりわけ好ましい実施形態のさらなる好ましい態様において、該医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の95重量%以上を、およびナロキソン塩酸塩の95重量%以上をin vitroで放出する。
【0034】
本発明のこれらのとりわけ好ましい実施形態の別の態様は、ヒト健常志願者での単回投与研究で投与された場合に、約25ng.h/mL〜約300ng.h/mLの範囲の、より好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供する医薬組成物に関する。このような経口即時放出医薬組成物は、オキシコドンおよびナロキソンの塩酸塩を好ましくは含むことができる。
【0035】
本発明のこれらのとりわけ好ましい実施形態の別の態様は、ヒト健常志願者での単回投与研究で投与された場合に、約4ng/mL〜約75ng/mLの範囲の、より好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供する医薬組成物に関する。このような経口即時放出医薬組成物は、オキシコドンおよびナロキソンの塩酸塩を好ましくは含むことができる。加えて先の段落のAUC値を提供する医薬組成物が、とりわけ好ましい。
【0036】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者の用量設定するのに使用することができる。
【0037】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を治療するのに使用することができる。
【0038】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者における、疼痛、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛を治療するのに使用することができる。
【0039】
本発明は、また、前記のような経口即時放出医薬組成物の使用、および、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者の用量設定するための薬剤の製造に関する。
【0040】
さらに、本発明は、前記のような経口即時放出医薬組成物の使用、および疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を治療するための薬剤の製造に関する。
【0041】
さらに、本発明は、前記のような経口即時放出医薬組成物の使用、および疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における疼痛を治療するための薬剤の製造に関する。
【0042】
本発明は、別の態様において、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者を、前記のような経口即時放出医薬組成物を投与することによって患者の用量設定する方法に関する。
【0043】
本発明のさらに別の態様は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を、前記のような経口即時放出医薬組成物を投与することによって治療する方法に関する。
【0044】
本発明のさらに別の態様は、疼痛に苦しむ患者における、疼痛、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛を、前記のような経口即時放出医薬組成物を投与することによって治療する方法に関する。
【0045】
さらに、本発明は、前記のような経口即時放出医薬組成物の製造方法に関する。
【図面の簡単な説明】
【0046】
【図1】本発明の医薬組成物の製造方法に関する流れ図を描いた図である。
【図2】実験2に記載の臨床試験に関する治療スケジュールを描いた図である。
【図3】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、オキシコドンの平均血漿中濃度を描いた図である。
【図4】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、ナロキソンの平均血漿中濃度を描いた図である。
【図5】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、ナロキソン−3−グルクロニドの平均血漿中濃度を描いた図である。
【図6】実験3に記載の臨床試験に関する治療スケジュールを描いた図である。
【図7】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、オキシコドンの平均血漿中濃度を描いた図である。
【図8】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、ナロキソンの平均血漿中濃度を描いた図である。
【図9】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、ナロキソン−3−グルクロニドの平均血漿中濃度を描いた図である。
【発明を実施するための形態】
【0047】
以下で例示的に説明するような本発明は、本明細書中で具体的に開示されない任意の要素または要素群、限定または限定群の不在下で適切に実施される可能性がある。
【0048】
本発明は、特定の実施形態に関して、かついくつかの図を参照して説明されるが、本発明はそれらに限定されるものではなく、特許請求の範囲によってのみ限定される。記載の図面は、単なる略図であって、それに限定するものではない。以下で示される用語は、そうでないことを指摘しない限り、一般にはそれらの通常的な意味で解釈されるものとする。
【0049】
用語「含む」が本明細書および特許請求の範囲中で使用される場合、該用語は、その他の要素を排除するものではない。本発明の目的に関して、用語「〜からなる」は、用語「〜から成り立つ」のある好ましい実施形態であると考えられる。以下で、ある群が、少なくともある数の実施形態を含むと定義される場合、このことは、また、好ましくは、これらの実施形態からのみなる群を開示していると解釈されるものとする。
【0050】
不定冠詞または定冠詞、例えば「a」、「an」、「the」が単数名詞を指す際に使用される場合、これは、特記しない限り、その名詞の複数形を含む。本発明の文脈中の用語「約」または「ほぼ」は、当業者が当該特徴の技術的効果をなお保証すると解釈する、正確さの隔たりを意味する。該用語は、典型的には、指摘した数値からの±10%、好ましくは±5%の偏差の存在を示す。
【0051】
以下でオキシコドンまたはナロキソンについて言及する場合、用語「オキシコドン」または「ナロキソン」が遊離塩基のみを指すのが当然であることを具体的に指摘しない限り、その言及は、常に、オキシコドン遊離塩基の薬学的に許容される塩またはナロキソン遊離塩基の薬学的に許容される塩あるいはそれらの誘導体についての言及をも含む。
【0052】
用語「即時放出」は、活性成分、すなわち、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が医薬組成物から放出される放出速度に関連している。一般的理解との関連で、用語「即時放出」は、特殊な製剤設計および/または製造方法によって故意に変更されていない、活性物質(群)の放出を示す医薬組成物に関連している。
【0053】
用語「即時放出」は、とりわけ、本発明による医薬組成物が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する特性を指す。
【0054】
in vitro放出速度は、欧州薬局方2.9.3(第6版)中に記載の通りの欧州薬局方(Ph.Eur.)パドル法により測定される。パドル速度は、模擬胃液(ペプシン不含USP(米国薬局方))溶出媒質中で100rpmに設定される。溶出媒質のアリコートを、15分および45分の時点で抜き取り、60℃で維持された逆相Merck LiChrospher 60RP Select Bカラムを使用するHPLCで分析する。移動相は、85:15v/v、pH2.0の塩化カリウム:メタノールからなる。紫外検出は230nmで行われる。オキシコドンおよびナロキソンは、外部標準アッセイによって定量される。
【0055】
経口即時放出医薬剤形が、5mgまでの量のオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および2.5mgまでの量のナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む場合、欧州薬局方パドル試験は、500mLの0.1N塩酸中で実施される。
【0056】
経口即時放出医薬剤形が、5mgを超える量のオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および2.5mgを超える量のナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む場合、欧州薬局方パドル試験は、900mLの0.1N塩酸中で実施される。
【0057】
以下で言及されるような用語「用量設定(titration)」およびその文法上の変形体は、医師が、疼痛、とりわけ慢性の中等度から高度およびさらには重度の疼痛に苦しむ患者のために、オキシコドン塩酸塩などの具体的なオピオイドについて適切な投与量を確認する過程を指す。疼痛に苦しむ患者は、彼らの異なる代謝特性のため、投与すべきオピオイド系鎮痛剤の量に関して異なる要求を有するので、典型的には、医師は、許容される疼痛コントロールを提供するため、先ず、オピオイド製剤の即時放出製剤の投与量を増大させることを試み、患者が無疼痛であるか、あるいは少なくとも有意な疼痛低減を有するのに十分であるオピオイドの投与量を確認する。
【0058】
投与量が、これらの患者によって経験される(慢性)疼痛を永続的にコントロールするのに適している場合、患者は、典型的には、薬剤を低減された頻度で時刻を決めた規則正しいレジメンで、例えば12時間毎または1日1回のみで摂取できるような対応投与量を含む制御放出製剤に変更される。疼痛治療なしで慢性疼痛感覚に苦しむような患者は、典型的には、制御された背景痛(background pain)を伴う患者と呼ばれる。
【0059】
本発明の文脈で、用語「突出痛」およびその文法上の変形体は、オピオイドの時刻を決めた規則正しいレジメンで治療されている患者によって経験される持続痛または背景痛を超える疼痛の一時的増大を指す。
【0060】
用語「持続痛」は、以下で使用する場合、典型的には、疼痛治療用のオピオイドレジメンを受けている患者が例えば12時間以上の間の平均疼痛強度の体験と報告する疼痛である。
【0061】
持続痛の疼痛強度は、典型的には、数値アナログ尺度試験(NAS)などの通常の方法を使用して測定される。突出痛発作および持続痛の測定は、例えば、Portenoyら(1999)The Journal of Pain 7(8):583〜591頁;およびPortenoyら(1999)Pain 81:129〜134頁に記載されている。これらの刊行物中で与えられているような突出痛および制御された持続痛の定義は、参照により本明細書に組み込まれる。したがって、制御された持続痛の名称は、2つの判断基準が合致することを典型的には要求する。第1に、患者は、質問[あなたの疼痛は、最近、あなたが「一定」または「ほとんど一定」と評する、あるいは肯定的返答の中で受けている治療のためではないにしても「一定またはほとんど一定である」要素を持っていますか]に返答しなければならない。第2に、患者は、比較的良好な疼痛コントロールと矛盾しないオピオイドレジメンによって治療されることを要求されなければならない。当業者は、2つの参考文献中で提供される情報に基づいて制御された持続痛を測定する方法を承知しているであろう。
【0062】
突出痛は、次いで、制御された持続痛のレベルを超える患者によって経験される疼痛の再燃として確認される。疼痛強度は、例えば、項目「なし」、「軽度」、「中等度」、「重度」および「極度に痛い」を有する5点分類尺度を使用して評価することができる。この発作が患者によって重度または極度に痛いと評価された場合、患者は、典型的には、突出痛発作を経験する。
【0063】
以下で言及するような用語「生物学的同等性」およびその文法上の変形体は、それらの通常の意味で使用される。とりわけ、試験医薬組成物の血漿中レベルの曲線下面積(AUC)(AUCtまたはAUCinf)に関する平均値の90%信頼区間が、参照医薬組成物の対応する平均値の80%〜125%の範囲内にあり、かつ試験医薬組成物の血漿中レベルの最大濃度(Cmax)に関する平均値の90%信頼区間が、参照医薬組成物の対応する平均値の80%〜125%の範囲内にある場合、医薬組成物は、参照医薬組成物に対して生物学的に同等であると言われる。
【0064】
Cmax値は、活性薬剤、すなわち、オキシコドンおよび/またはナロキソン(または塩酸塩などのそれらの塩)の最大血漿中濃度を示す。
【0065】
tmax値は、Cmax値に到達する時点を示す。換言すれば、tmaxは、最大血漿中濃度が観察される時点である。
【0066】
AUC(曲線下面積)値は、濃度曲線の面積に相当する。AUC値は、血液循環中に吸収された活性薬剤、すなわち、オキシコドンおよびナロキソン(または塩酸塩などのそれらの塩)の全体量に比例し、それゆえバイオアベイラビリティーの尺度である。
【0067】
AUCt値は、投与時点から測定可能最終濃度までの血漿中濃度−時間曲線下の面積に関する値である。AUCt値は、通常、台形法を使用して計算される。可能な場合、終末相速度定数であるλZを、終末対数−線形相中にあると判定されるそれらの箇所を使用して推定する。見かけの終末相半減期であるt1/2Zは、通常的には、Ln2のλZに対する比率から決定される。最終測定点と無限大との間の血漿中濃度−時間曲線下面積は、観察される最終血漿中濃度(Clast)のλZに対する比率から計算することができる。次いで、これをAUCtに加算して、投与時から無限大までの血漿中濃度−曲線下面積であるAUCinfを得る。
【0068】
用語「バイオアベイラビリティー」は、本発明の目的の場合、オキシコドンおよびナロキソン(または塩酸塩などのそれらの塩)などの活性薬剤が、該医薬組成物の経口投与後に吸収される度合いと定義される。
【0069】
用語「定常状態」は、次のように説明することができる:最初の用量が投与される時点、t=0で、濃度C=0である。次いで、濃度は、最初の極大点を通り、次いで、最初の極小点まで降下する。濃度が0まで降下する前に、別の用量が、投与され、その結果、第2の濃度増加は0から始まるのではない。
この最初の濃度極小値上に積み上げると、二回目の用量を投与した後に、曲線は、最初の極大値を超える二番目の極大値を通過し、かつ最初の最小値を超える二番目の極小値まで降下する。したがって、反復投与および付随する活性薬剤の段階的な蓄積のため、血漿中濃度は、それが、吸収および排出が平衡状態にある点まで安定化するまで、上昇する。吸収および排出が平衡状態にあり、濃度が、規定された極小値と規定された極大値との間を絶えず振動する状態は、定常状態と呼ばれる。
【0070】
用語「維持療法」および「長期療法」は、本発明の目的の場合、患者が、オピオイド鎮痛剤を用いて上で定義したような定常状態に用量設定された後に患者に投与される薬物療法と定義される。前述の持続痛は、維持/長期療法中の疼痛感覚を指す。
【0071】
血漿中曲線を記述するパラメーターは、臨床試験中に、先ず、ある数の被験者にオキシコドンおよびナロキソン(またはそれらの塩酸塩などの塩)などの活性薬剤を1回だけ投与することによって得ることができる。個々の被験者の血漿中値は、次いで平均化され、例えば、平均のAUC、Cmaxおよびtmax値が得られる。
【0072】
本発明の文脈で、AUC、Cmaxおよびtmaxなどの薬物動態パラメーターは、そうでないことを指摘しない限り、平均値を指す。さらに、本発明の文脈で、AUC、Cmax、tmaxまたは鎮痛効力に関する値などのin vivoパラメーターは、投与後に定常状態で得られる、あるいはヒト患者および/または健常ヒト対象への単回投与のパラメーターまたは値を指す。
【0073】
平均tmax、CmaxおよびAUCなどの薬物動態パラメーターが、健常ヒト対象について測定される場合、それらのパラメーターは、典型的には、ほぼ16〜24名の健常ヒト対象からなる試験集団中で長時間にわたって血漿中値の進展を測定することによって得られる。
欧州医薬品審査庁(EMEA)または米国食品医薬品庁(FDA)などの規制組織は、通常、例えば20または24名の被験者から得られたデータを受理する。しかし、5名の被験者などのより少ない参加者を含む初期試験も受理される可能性がある。
【0074】
この文脈における用語「健常」ヒト対象は、通常的には、身長、体重、および血圧などの生理学的パラメーターに関して平均的な値を有する血統が白色人種の典型的な男性または女性を指す。本発明の目的のための健常ヒト対象は、臨床試験の調和に関する国際会議(ICH)の推奨に基づき、かつそれと一致した組入れおよび除外基準により選択される。本発明の目的の場合、健常対象は、実施例2および実施例3中で説明されるような組入および除外基準により識別することができる。
【0075】
したがって、組入れ基準は、例えば18歳以上60歳以下の年齢、例えば19〜29kg/m2の範囲内のBMI、および例えば、男性では60〜100kg、女性では55〜90kgの範囲内の体重を含み;女性は、非授乳、非妊娠であり、かつ治験薬剤を受入れる前の24時間以内に陰性の尿β−hCG妊娠検査;病歴、身体的検査、臨床検査、バイタルサイン、およびECGなどに関して有意に異常な所見の欠如によって証明される一般的に良好な健康状態を示す必要がある。
【0076】
除外基準は、例えば、治験薬剤の初回投与の3ヶ月以内での任意の治験物またはプラセボへの暴露;治験薬剤の初回投与前30日以内での任意の重大な疾患;病歴、身体的検査または試験室での分析に関する治験前スクリーニングの時点で確認された臨床的に重大な任意の異常;治験薬剤の初回投与前の21日間における任意の処方薬剤(閉経女性に対するHRTおよび避妊薬剤を除く)または治験薬剤の初回最初前の7日間における制酸剤、ビタミン、生薬および/またはミネラル栄養補助食品を含むOTC薬の使用;消化管薬物吸収(例えば、胃内容物排出遅延、吸収不良症候群)、分布(例えば、肥満)、代謝もしくは排泄(例えば、肝炎、糸球体腎炎)を妨害することが知られている並存的な医学的状態;治験責任医師の意見の中で、対象が治験を安全に完了する能力を危うくする並存的な医学的状態の既往歴;対象が薬理学的治療を必要とした発作性疾患の既往歴;1日に5本を超える現在の喫煙歴;DSM−IV基準による薬物もしくはアルコール乱用の活動歴もしくは既往歴の証拠を有する対象;1日に2本以上のアルコール飲料の規則的消費を報告した、またはスクリーニング時点で0.5%以上の血中アルコールレベルを有する対象;治験薬剤の初回投与前の3ヶ月間における500mLを超える血液もしくは血液産物の献血またはその他の重大な血液喪失;治験前スクリーニングにおいてスクリーニング時点で採集した尿検体中のエタノール、アヘン剤、バルビツール剤、アンフェタミン剤、コカイン代謝産物、メタドン、プロポキシフェン、フェニルシクリジン、ベンゾジアゼピン、およびカンナビノイドに関する任意の陽性結果;オキシコドン、ナロキソン、または関連化合物などに対する既知の敏感性などを含む。
【0077】
平均のtmax、CmaxおよびAUCなどの薬物動態パラメーターを患者で得る場合、該患者群は、典型的には、10〜200名の患者から構成される。妥当な患者数は、例えば、10、20、30、40、50、75、100、125または150である。患者は、治療予定の状態の症状により選択される。患者は、例えば、18歳以上でよく、腫瘍および非腫瘍起源の中等度から高度およびさらには重度の慢性痛に苦しみ、WHOステップIIまたはIIIの鎮痛剤などで不十分な効力および/または忍容性を示す。患者は、最近のアルコールもしくは薬物乱用の、最近の重度の心血管および呼吸器疾患の、重度の肝および腎不全などの徴候が存在する場合、例えば、薬物動態パラメーターの測定について考慮に入れない可能性がある。
【0078】
前におよび後に示すような薬物動態パラメーターの値は、実施例2および3で得られたデータに基づいて導き出され、そのすべては、健常ヒト対象での単回投与治験に関連することを理解されたい。しかし、類似の結果が、健常ヒト対象における定常状態での投与で、またはヒト患者における単回および定常状態投与で得られると想定される。当業者は、もちろん、定常状態のCmaxが単回投与後のCmaxよりも高いことを承知している。
【0079】
薬物動態パラメーターの計算は、WinNonlin Enterprise Edition、Version 4.1を用いて実施することができる。
【0080】
前に言及したように、本発明は、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物に関する。
【0081】
以下で示すように、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含むこのような経口即時放出医薬組成物は、オキシコドン塩酸塩およびナロキソン塩酸塩を2:1の重量比で(オキシコドン塩酸塩:ナロキソン塩酸塩)含む参照の制御放出医薬組成物と対照して試験した場合、生物学的に同等である。オキシコドン塩酸塩およびナロキソン塩酸塩を2:1の重量比で含む実施例3で使用される制御放出参照医薬組成物は、Targin(登録商標)錠の名称下にドイツ市場で入手可能である。Targin(登録商標)錠について、この制御放出製剤は、例えば高度〜重度の疼痛に苦しむ患者において優れた疼痛コントロールを提供するだけでなく、極めて優れた副作用プロファイルを提供することが示されている。Targin(登録商標)錠の使用による副作用の改善は、腸機能の改善として、オピオイド誘発性便秘の低減として、オピオイド誘発性尿滞留の低減として、かつ疼痛に苦しむ患者による薬物に対する忍容性および好みの増大として評価することができる(例えば、Vondrackovaら、J.Pain(2008),9(12):1144〜1154頁;Nadstawekら、Int.J.Clin.Pract.(2008),62(8):1159〜1167頁;およびMeissnerら、Eur.J.Pain(2008)(印刷中の論文)を参照されたい)。
【0082】
さらに、実験2から明らかになるように、本発明の経口即時放出医薬組成物は、とりわけ、それらが、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む場合に、用量比例性である。
【0083】
この場合、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む即時放出医薬組成物の制御放出製剤Targin(登録商標)錠に対する送達程度の同等性である生物学的同等性を考慮すれば(特に予想外のバイオアベイラビリティーの程度、およびナロキソンの初回通過効果が即時放出製剤についても維持されるという事実を考慮すれば)、本発明による即時放出医薬組成物が、疼痛、とりわけ慢性の中等度から高度およびさらには重度の疼痛に苦しむ患者における疼痛緩和を十分に提供するのに有効であるのみならず、これらの組成物で治療された患者は、とりわけオピオイド作用薬であるオキシコドン塩酸塩のみを含む先行技術から公知の経口即時放出製剤と比較して、Targin(登録商標)錠について観察される副作用プロファイルに関して同一ではないが類似の改善を経験することを想定することは正当化されると思われる。
【0084】
したがって、本発明による経口即時放出医薬組成物は、疼痛治療において有効であり、かつ、オピオイド作用薬であるオキシコドン塩酸塩のみを含む先行技術の即時放出医薬組成物と比較した場合の、オピオイド誘発性便秘の低減、オピオイド誘発性尿滞留の低減などの副作用に関する改善、ならびに患者による薬剤に対する忍容性および好みの改善を示すことが想定される。
【0085】
本発明の経口即時放出医薬組成物の用量比例性と組み合わせたこれらの特性は、過去において、制御放出製剤の改善された副作用プロファイルを考慮して、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む制御放出医薬組成物が、オピオイド作用薬であるオキシコドンのみを含む即時放出製剤に優先して選択されたであろう状況において、医師が本発明による医薬組成物を使用することを可能にする。
【0086】
本発明の経口即時放出医薬組成物は、疼痛に苦しむ患者の用量設定に、および/または疼痛に苦しむ患者における突出痛の治療にとりわけ適している。
【0087】
とりわけ、本発明の経口即時放出医薬組成物は、疼痛、とりわけ高度〜重度(慢性)疼痛に苦しむ患者の用量設定するのに使用することができ、過去における、オキシコドンおよびナロキソンの制御放出製剤を用いる患者のための用量設定は、受容できない疼痛の経験ではないにせよ、制御放出製剤の遅延された作用による不快な経験を意味した。さらに、本発明による経口即時放出製剤は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の制御放出製剤で既に治療されているが、それにもかかわらず突然の疼痛発作を経験し、それゆえオピオイドであるオキシコドン塩酸塩のみを含む即時放出製剤を必要とした患者における、例えば突出痛の発作を治療するのに使用することができる。
【0088】
言及したように、本発明の医薬剤形は、また、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物に関し、ここで、該組成物は、錠剤の形態で提供され、かつ該組成物は、例えば、15分後に活性物の90%超またはさらには95%超が放出されるような急速なin vitro溶出プロファイルを提供する。これらの組み合わされた特性は、組成物を、固体形態で、例えば、嚥下可能な錠剤として投与することができる利点を有する。同時に、嚥下に伴う問題を有する可能性のある子供または高齢者などの患者群は、投与に先立って錠剤を最初に液体中に急速に溶解することによって投与することでき、あるいは唾液中での急速溶解のために口中に直接的に入れることができる。これは、疼痛発作が突然に発生し、その結果、迅速な疼痛緩和が要求される場合に、重要な利便性である可能性がある。
【0089】
既に言及したように、本発明は、本質的には、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、好ましくは約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物に関する。
【0090】
原則として、経口即時放出医薬組成物は、オキシコドンおよびナロキソンに加えて薬学的に許容される活性薬剤を含むことができる。しかし、本発明のすべての態様の好ましい実施形態において、本発明による経口即時放出医薬組成物は、唯一の薬学的に活性な薬剤としてオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む。
【0091】
オキシコドンおよびナロキソンは、本発明の経口即時放出医薬剤形中に遊離塩基として存在することができる。
【0092】
しかし、オキシコドンまたはナロキソンは、また、それらの薬学的に許容される塩の形態で存在することができる。このような塩としては、例えば、塩酸塩、硫酸塩、重亜硫酸塩、酒石酸塩、硝酸塩、クエン酸塩、バルビツール酸塩、リン酸塩、リンゴ酸塩、マレイン酸塩、臭化水素酸塩、ヨウ化水素酸塩、フマル酸塩、コハク酸塩などが挙げられる。
【0093】
以下で言及するようなオキシコドンおよびナロキソンは、また、リチウム、ナトリウムおよびカリウムを含むアルカリ金属の金属塩などの塩基付加塩として存在することができる。それらは、また、遊離塩基の誘導体の形態で存在することができる。このような誘導体としては、例えばエステルが挙げられる。
【0094】
好ましい実施形態において、本発明は、オキシコドン塩酸塩およびナロキソン塩酸塩を使用する。本発明のさらに好ましい実施形態において、経口即時放出医薬組成物は、唯一の薬学的に活性な薬剤として、オキシコドン塩酸塩およびナロキソン塩酸塩を好ましくは約2:1の重量比で含む。
【0095】
本発明の経口即時放出医薬組成物は、患者における疼痛、とりわけ高度〜重度(慢性)の疼痛を治療するのに十分である量のオキシコドンを含む。典型的には、本発明の医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩を含む。
【0096】
本発明の医薬組成物中のナロキソンの量は、投与されるナロキソンの量が、オキシコドンで仲介される疼痛緩和に実質上マイナスに影響しないように選択される。典型的には、本発明の医薬組成物は、約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。
【0097】
オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の量は、オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩の比率が、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の重量を基準にして、重量で約2:1であるように選択することができる。
【0098】
したがって、本発明の医薬組成物は、典型的には、約1mg〜約160mgのオキシコドン塩酸塩に等価な、約1mg〜約80mgのオキシコドン塩酸塩に等価な、約1mg〜約40mgのオキシコドン塩酸塩に等価な、約1mg〜約20mgのオキシコドン塩酸塩に等価な、約1mg〜約10mgのオキシコドン塩酸塩に等価な、約1mg〜約5mgのオキシコドン塩酸塩に等価な、および約1mg〜約2.5mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩を含む。
【0099】
さらに、本発明の医薬組成物は、典型的には、約0.5mg〜約80mgのナロキソンに等価な、約0.5mg〜約40mgのナロキソン塩酸塩に等価な、約0.5mg〜約20mgのナロキソン塩酸塩に等価な、約0.5mg〜約10mgのナロキソン塩酸塩に等価な、約0.5mg〜約5mgのナロキソン塩酸塩に等価な、約0.5mg〜約2.5mgのナロキソン塩酸塩に等価な、および約0.5mg〜約1.25mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。
【0100】
本発明の好ましい実施形態は、約2.5mg〜約40mgのオキシコドン塩酸塩、好ましくは約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約1.25mg〜約20mgのナロキソン塩酸塩、好ましくは約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む医薬組成物に関する。これらの組成物は、とりわけ、オキシコドンおよびナロキソンの塩酸塩を使用することができる。
【0101】
本発明によるとりわけ好ましい投与強度の経口即時放出医薬組成物は、約2.5mに等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mgに等価な量のナロキソンまたはその薬学的に許容される塩、約5mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約2.5mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約10mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約5mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、ならびに約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。これらの組成物は、とりわけ、オキシコドンおよびナロキソンの塩酸塩を使用することができる。
【0102】
本発明のとりわけ好ましい実施形態は、2.5mgのオキシコドン塩酸塩および1.25mgのナロキソン塩酸塩、5mgのオキシコドン塩酸塩および2.5mgのナロキソン塩酸塩、10mgのオキシコドン塩酸塩および5mgのナロキソン塩酸塩、ならびに20mgのオキシコドン塩酸塩および10mgのナロキソン塩酸塩を含む経口即時放出医薬組成物に関する。
【0103】
オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明による経口即時放出医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、典型的には、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する。より好ましくは、本発明による医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する。
【0104】
さらにより好ましくは、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明による医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80%以上、好ましくは90%以上、さらにより好ましくは95%以上を、およびナロキソンまたはその薬学的に許容される塩の80%以上、好ましくは90%以上、さらにより好ましくは95重量%以上をin vitroで放出する。
【0105】
本発明による経口即時放出医薬組成物は、即時放出製剤にとって一般的な剤形で提供することができる。したがって、本発明による経口即時放出医薬組成物は、錠剤、カプセル剤、多粒子剤(例えば、顆粒、長円またはビーズ)などの固体形態で、あるいは液剤(例えば、溶液、懸濁液または乳液)で存在することができる。本発明の経口即時放出医薬組成物にとって好ましい剤形は錠剤である。
【0106】
本発明による経口即時放出医薬組成物を製造する場合、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩に加えて、製剤技術の分野で一般的である薬学的に許容される添加剤を使用することができる。典型的な薬学的に許容される添加剤は、崩壊剤、賦形剤、滑沢剤、流動促進剤、抗粘着剤、可塑剤、着色剤、着香剤、結合剤、pH調整剤などである。これらの添加剤(崩壊剤を除いて)は、それらが、前に記載したような即時放出のin vitro放出速度を実質上変更しないように選択すべきである。
【0107】
とりわけ本発明の医薬組成物を錠剤として提供する場合、本発明の医薬組成物が、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含むことが好ましい場合がある。また、とりわけ本発明の医薬組成物を錠剤として提供する場合、本発明の医薬組成物が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含むことが好ましい場合がある。さらに、崩壊剤および賦形剤の双方として機能を発揮する添加剤を使用することが好ましい場合がある。
【0108】
崩壊剤は、例えば、投与後の錠剤が急速に崩壊し、その結果、活性成分であるオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が吸収のために容易に利用可能になることを確実にする。
【0109】
賦形剤は、限定はされないが、乳糖(乳糖一水和物、無水乳糖など)、デンプン(トウモロコシデンプン、α化デンプンなど)、微結晶セルロース、ブドウ糖、マンニトール、マルチトール、StarLac(登録商標)(噴霧乾燥乳糖85%、トウモロコシデンプン15%)、ショ糖、カルシウム塩(リン酸水素カルシウムなど)、またはこれらの任意の組合せから選択することができる。
【0110】
崩壊剤は、限定はされないが、とりわけ、StarLac(登録商標)(噴霧乾燥乳糖85%、トウモロコシデンプン15%)、クロスカルメロース(クロスカルメロースナトリウムなど)、デンプングリコール酸ナトリウム、クロスポビドン、アルギン酸、または低置換ヒドロキシプロピルセルロースから選択することができる。
【0111】
StarLac(登録商標)製品のような乳糖とデンプンとの組合せは、それが充填剤および崩壊剤の特性を兼ね備えているのでとりわけ好ましい可能性がある。
【0112】
流動促進剤および滑沢剤は、限定はされないが、とりわけ、高分散性シリカ、タルク、酸化マグネシウム、ステアリン酸マグネシウム、フマル酸ステアリルナトリウムなどから選択することができる。
【0113】
流動化剤および滑沢剤は、とりわけ、高分散性シリカ、タルク、酸化マグネシウム、ステアリン酸マグネシウム、フマル酸ステアリルナトリウムなどを含む。
【0114】
本発明の医薬組成物を錠剤として提供する場合、それらの錠剤を、区別の目的で、化粧コーティングを用いて被覆することができる。このようなコーティングは、本発明による医薬組成物の即時放出特性に実質的な影響を有さない。
【0115】
好ましい実施形態において、本発明は、約2.5mg〜約160mgのオキシコドン塩酸塩および約1.25mg〜約80mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0116】
別の好ましい実施形態において、本発明は、約2.5mg〜約80mgのオキシコドン塩酸塩および約1.25mg〜約40mgのナロキソン塩酸塩を2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0117】
別の好ましい実施形態において、本発明は、約2.5mg〜約40mgのオキシコドン塩酸塩および約1.25mg〜約20mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0118】
別の好ましい実施形態において、本発明は、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0119】
さらに別の好ましい実施形態において、本発明は、約2.5mg〜約10mgのオキシコドン塩酸塩および約1.25mg〜約5mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0120】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、健常ヒト志願者において単回投与で投与された場合に、約15ng.h/mL〜約500ng.h/mLの範囲の、好ましくは約20ng.h/mL〜約400ng.h/mLの範囲の、より好ましくは約25ng.h/mL〜約300ng.h/mLの範囲の、さらにより好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0121】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、健常ヒト志願者において単回投与治験で投与された場合に、約1ng/mL〜約300ng/mLの範囲の、好ましくは約2ng/mL〜約200ng/mLの範囲の、より好ましくは約3ng/mL〜約100ng/mLの範囲の、さらにより好ましくは約4ng/mL〜約75ng/mLの範囲の、最も好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0122】
本発明の実施形態は、また、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物であって、ストレス条件下で貯蔵した後の該医薬組成物が、薬学的に活性な薬剤を、該医薬組成物をストレス条件にさらす前と実質的に同じ放出速度で放出する経口即時放出医薬組成物に関する。
【0123】
本発明の他の実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物であって、ストレス条件下で貯蔵した後の該医薬組成物が、オキシコドンまたはその薬学的に許容される塩に関連するおよび/またはナロキソンまたはその薬学的に許容される塩に関連する全部で3.0%未満の物質を有する経口即時放出医薬組成物に関する。
【0124】
本発明の文脈中のストレス条件下での貯蔵は、医薬組成物を、高められた温度および/または相対湿度(RH)に長時間さらすことを意味する。例えば、典型的なストレス条件は、25℃および60%RHでの少なくとも1、2、3、4、5、6、9または12ヶ月間にわたる貯蔵を指す。別のストレス条件は、40℃および75%RHでの少なくとも1、2、3、4、または5ヶ月間にわたる貯蔵を指す。
【0125】
このようなストレス貯蔵条件は、医薬組成物が、その安全性および有効性に対するマイナスの影響なしに患者の家庭内で一般的である条件下で長期間十分な貯蔵寿命を有するかどうかを判定するために使用される。このようなマイナスの影響としては、in vitroでの放出速度が、長い間に変化し、その結果、投与後に異なる量の活性物が放出されるので、組成物の有効性が影響を受けることが挙げられる。同様に、マイナスの影響は、また、機能を果たす薬学的に活性な薬剤の全体量を減少させるか、または毒性副生物の形成につながる可能性のある、薬学的に活性な薬剤の分解に由来する可能性がある。
【0126】
in vitro放出プロファイルにおける、または医薬組成物中の活性薬剤(群)の量に関する変化がストレス条件下での貯蔵後に観察される場合、このことは、安定性の問題を暗示している可能性がある。このような変化が観察されない場合、このことは、逆に、医薬組成物が、貯蔵上安定であることを意味する。
【0127】
用語「実質上同じ放出速度」は、ストレス条件にさらされた医薬組成物についてのin vitro放出速度が、参照組成物と同等である状況を指す。参照組成物は、全く同じ医薬組成物ではあるが、ストレス条件にさらされていない。ストレス条件にさらされた組成物のin vitro放出プロファイルが、参照組成物のin vitro放出プロファイルから、約20%超、好ましくは約15%以下、より好ましくは約10%以下、さらにより好ましくは約5%以下の逸脱をしない場合、そのin vitro放出速度は、実質的に同一であると見なされる。
【0128】
用語「すべてのオキシコドンおよびナロキソン類縁物質」は、これらの薬学的に活性な薬剤の化学反応から生じる物質を指す。これらには、例えば、ナロキソンn−オキシドなどが含まれる。
【0129】
安定性を評価するために、医薬組成物を前述のようなストレス条件にさらし、オキシコドンおよび/またはナロキソン類縁物質の量を測定することができる。次いで、ストレス条件にさらされなかった全く同一の医薬組成物についてオキシコドンおよび/またはナロキソン類縁物質の量を測定する。この組成物は、参照組成物であると見なされる。「オキシコドン類縁物質」および「ナロキソン類縁物質」の検出は、典型的には、HPLC分析で実施される。物質の同定は、既知の純粋な参照物質を用いて同様の分析を行うことによって判定することができる。
【0130】
医薬組成物は、それをストレス条件にさらした後に、それが、組成物内に存在するヒドロモルホンまたはナロキソンの量と比較して、約3%以下の、好ましくは約2%以下の、より好ましくは約1%以下の、さらにより好ましくは0.5%以下のオキシコドンおよび/またはナロキソン類縁物質を有する場合、安定であると見なされる。
【0131】
ストレス条件下での貯蔵後に実質上同一の放出速度を有する、および/またはストレス条件下での貯蔵後に3%未満のオキシコドンおよびナロキソン類縁物質を有する組成物の特性は、とりわけ組成物が錠剤などの固体形態をとる場合、2:1の重量比を有する組成物、前述の量を有する組成物および/または前述のin vitro放出プロファイルを有する組成物などの、前に述べた実施形態にも関係すると理解されたい。
【0132】
固体形態で、例えば錠剤の形態で提供される本発明による医薬組成物は、例えば、次のステップ:
a)双方とも適切な粒径のオキシコドンまたはその薬学的に許容される塩、ナロキソンまたはその薬学的に許容される塩、賦形剤、および任意選択で崩壊剤をブレンドするステップ、
b)任意選択で、前記ブレンド物を滑沢化するステップ、
c)前記ブレンド物を直接的に圧縮して錠剤を得るステップ、
を含む方法によって製造することができる。
【0133】
固体形態で、例えば錠剤の形態で提供される本発明による医薬組成物は、例えば、次のステップ:
a)双方とも適切な粒径のオキシコドンまたはその薬学的に許容される塩、ナロキソンまたはその薬学的に許容される塩、崩壊剤、および任意選択で賦形剤をブレンドするステップ、
b)任意選択で、前記ブレンド物を滑沢化するステップ、
c)前記ブレンド物を直接的に圧縮して錠剤を得るステップ、
を含む方法によって製造することができる。
【0134】
必要と考えられる場合、上記方法は、均一なブレンド化のため塊のない成分を確保にするために、種々の機会での篩い分けステップを含むことができる。
【0135】
また、好ましくは、すべての添加剤および任意選択で活性成分は、同一の大きさの範囲内にある。典型的な大きさの範囲は、約100μm〜約300μmである。
【0136】
このような手順によって得られる錠剤は、急速に崩壊し、前に概略を述べたような欧州薬局方パドル法で測定した場合、15分の時点でオキシコドンまたはその薬学的に許容される塩の90%以上、好ましくは95%以上を、およびナロキソンまたはその薬学的に許容される塩の90%以上、好ましくは95%以上をin vitroで放出することが見出された。実施例4から理解できるように、このような手順により製造された錠剤は、貯蔵安定性があり、それらは、ストレス条件下での長期貯蔵後にin vitroでのそれらの放出挙動を実質的に変化させないことを意味する。
【0137】
前記のようなおよび前に製造されたような即時放出医薬組成物は、疼痛、とりわけ中等度から重度(慢性)疼痛に苦しむ患者の用量設定するのに使用することができる。このような製剤は、また、疼痛、とりわけ中等度から重度(慢性)疼痛に苦しむ患者における突出痛を治療するのに使用することができる。
【0138】
患者の用量設定のための、および患者における突出痛を治療するための経口即時放出医薬組成物の使用は、便秘および尿滞留などの典型的なオピオイド誘発性副作用に苦しむことなく、即時放出特性の結果として患者における迅速な疼痛緩和を確実にする。
【0139】
本発明を、一部の具体的実施例に関して以下で説明する。しかし、これらの実施例を限定と解釈すべきでない。
(実施例)
【実施例1】
【0140】
オキシコドン塩酸塩およびナロキソン塩酸塩を含む即時放出医薬組成物の調製
オキシコドン塩酸塩20mg/ナロキソン塩酸塩10mg(IR−OXN20/10)、オキシコドン塩酸塩10mg/ナロキソン塩酸塩5mg(IR−OXN10/5)、オキシコドン塩酸塩5mg/ナロキソン塩酸塩2.5mg(IR−OXN5/2.5)、およびオキシコドン塩酸塩2.5mg/ナロキソン塩酸塩1.25mg(IR−OX2.5/1.25)を含む即時放出医薬組成物を、下記の通り製造した。それらの組成を表1に詳述する。
【0141】
【表1】

【0142】
【表2】
【0143】
【表3】
【0144】
【表4】
【0145】
【表5】
【0146】
表1〜5の成分を、図1の流れ図に記載のように処理した。
【0147】
詳細には、微結晶セルロース、クロスカルメロースナトリウム、および活性成分を、500μmの篩いを通して篩い分けて任意の凝集物を除去し、StarLac(登録商標)と共にブレンダーに仕込み、均一なブレンドが達成されるまでブレンドした。StarLac(登録商標)は自由流動性であるので、その篩い分けは必要なかった。滑沢剤であるフマル酸ステアリルナトリウムを、500μmの篩いを通して篩い分け、ブレンド物に添加し、さらにブレンドした。次いで、ブレンド物を、直接圧縮によって圧縮して錠剤とした。許容されるブレンド物の均一性を確保するために、粉砕級のナロキソン塩酸塩を利用してオキシコドン塩酸塩およびその他の添加剤と類似の粒径範囲を得た。
【0148】
異なる強度の製品間の区別を提供するために、着色された化粧フィルムコートを塗布した。フィルムコーティングの条件は、適切な審美的品質の被覆錠剤を一貫して生じるように最適化した。
【0149】
次いで、IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5、およびIR−OXN2.5/1.25錠剤を、欧州薬局方のパドル試験によって試験した。
【0150】
すべての場合において、15分の時点でオキシコドン塩酸塩およびナロキソン塩酸塩の95%超が放出された。
【実施例2】
【0151】
健常対象におけるIR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5、およびIR−OXN2.5/1.25錠剤の用量比例性を比較するための単回投与治験
1.目的
目的は、2.5/1.25mg、5/2.5mg、10/5mg、および20/10mgの強度のIR−OXN錠剤から、オキシコドンおよびナロキソン(または、代用物であるナロキソン−3−グルクロニド)の用量比例性を評価することとした。
【0152】
2.試験集団
18名の対象が治験を完了し、有効な薬物動態データを提供することを目標に、治験薬剤を投与される全部で21名の健常成人、男性および女性対象を無作為化するように計画した。全部で21名の対象を、実際に登録し、無作為化し、20名の対象が治験を完了した。
【0153】
<組入れ基準>
治験に組み入れ予定の対象は、次の基準のすべてに合致する者とした:
1.年齢が18〜55歳(両端を含む)の男性または女性対象。
2.治験中に性的に活発であった、または性的に活発になった女性対象は、治験中を通して高度に有効な避妊法を進んで使用したにちがいない者。高度に有効な受胎調節法は、不妊手術、インプラント、注射剤、併用経口避妊薬、一部の子宮内デバイス、または配偶者の精管切除などの、一貫してかつ正しく使用された場合に低い失敗率(すなわち、1年に1%未満)をもたらすものと定義した。
3.閉経1年未満の女性対象は、血清妊娠検査で陰性であり、かつ非授乳であったにちがいない者。
4.閉経後1年間を超え、かつ高めたれた血清卵胞刺激ホルモン(FSH)を有するか、またはホルモン補充療法(HRT)で治療された女性対象。
5.男性対象は、彼らの配偶者との避妊を治験中および治験完了後の30日間進んで使用したにちがいなく、かつ彼らの配偶者がこの期間に妊娠した場合治験責任医師に伝えることに同意した者。
6.55〜100kgの範囲の体重、および18以上かつ29以下の体型指数(BMI)。
7.健康で、かつ病歴、身体的検査、バイタルサイン、臨床検査、および心電図(ECG)によって判定されるような有意な異常所見がない。
8.治験中に供給されるすべての食物を進んで食すること。
9.対象のプライマリーケア医師が、事前の12ヶ月以内に、対象の病歴中に彼らの臨床試験への登録の除外となるものが存在しないことを確認した者。
【0154】
<除外基準>
治験から除外すべき対象は、次の基準のいずれかに合致する者とした:
1.薬物またはアルコール乱用の任意の既往歴。
2.薬物の吸収、分布、代謝または排泄を妨害する可能性のある状態の任意の既往歴。
3.事前の30日間でのオピオイドまたはオピオイド拮抗薬を含有する薬剤の使用。
4.病因にかかわらず、頻回の悪心または嘔吐の任意の既往歴。
5.発作または症候性頭部外傷の任意の既往歴。
6.この治験の初回投与に先立つ90日間中の臨床薬物治験への参加。
7.この治験への登録に先立つ4週間中の任意の重大な疾患。
8.初回投与に先立つ7日間中、またはこの治験過程中のビタミン、生薬および/またはミネラル補助食品を含む任意の薬剤の使用(HRTおよび避妊薬の連続的使用を除いて)。
9.治験薬物の投与に先立つ8時間中、または投与に続く4時間中に食物を控えること、および各拘束期間中完全にカフェインまたはキサンチン含有飲料を控えることの拒絶。
10.女性では14単位/週、男性では21単位/週の等価量を超える毎週のアルコール摂取。
11.治験薬物投与前48時間以内のアルコール飲料の消費、および治験薬物投与後の少なくとも48時間アルコールを控えることの拒絶。
12.治験薬物投与の45日以内の喫煙暦、および治験中の禁煙の拒絶。
13.プロトコールによって要求された場合を除く、治験薬物投与に先立つ30日以内のまたは治験中のいずれかに時点での血液または血液産物の提供。
14.尿中薬物スクリーニング、アルコール検査、B型肝炎表面抗原(HBsAg)、C型肝炎抗体、またはヒト免疫不全ウイルス(HIV)検査の陽性結果。
15.オキシコドン、ナロキソン、ナルトレキソンまたは関連化合物に対する既知の感受性。
16.対象のプライマリーケア医師に報告することを承認することへの拒絶。
【0155】
対象集団の人口統計学的データを表6に示す。治験集団は、男性14名および女性7名の平均年齢が31歳(範囲:21〜53歳)の対象から構成した。20名の対象は白人であり、1名の対象は黒人であった。
【0156】
【表6】
【0157】
3.治験デザイン
治験は、単一治験センターで実施される健常な男性および女性対象における、オープンラベル、単回投与、4治療薬、4期間、無作為化クロスオーバー治験とした。各対象は、2.5/1.25mg、5/2.5mg、10/5mg、および20/10mgの強度のIR−OXN錠剤の単回投与を受けた。対象は、無作為割付スケジュール(RAS)により、各投与の間に少なくとも7日間のウォッシュアウト期間を伴う4治療薬のそれぞれを投与された。
【0158】
対象は、治験期間1の初回投与日の21日以内にスクリーニングのために来院した。次いで、適格対象は、各治験期間における投与の前日に治験施設にチェックインした。翌朝、対象に、絶食状態で適切な治験薬物を投与した。
【0159】
薬物動態用血液検体を、各治験期間における治験薬物投与後の36時間の間に採取し、36時間の時点の血液検体採取後に対象を退院させた。
【0160】
各治験期間中、バイタルサイン(脈拍、血圧、および呼吸数)を、投与前、次いで投与後の1、2、4、6、8、12、24および36時間の時点で測定した。口腔温を、投与前、ならびに投与後の24および36時間の時点で測定した。治験中、有害事象を記録した。対象は、治験の完了/中止の場合、彼らの治験薬剤の最終投与の7〜10日後に治験後来院した。
【0161】
治験デザインを図2に示す。図2中の略語は次の通りである:
R=無作為化
P1、P2、P3、P4=それぞれ、RASによる治験薬物の単回投与、それに続く血液サンプリング、および投与後36時間までの安全性評価からなる期間1〜4。各治験期間の間には、治験薬物の投与の間に少なくとも7日間のウォッシュアウトを存在させた。
PS med=治験の完了/中止の場合における、治験薬物の最終投与の7〜10日後の治験後健診
V1〜V6=来院
【0162】
4.薬物動態用検体の採集
1、8、15および22日目に開始して、一連の血液採集物(それぞれ6mL)を次の時点で得た:
投与前、投与後0.25、0.5、0.75、1、1.25、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1つの投与期間につき22の血液検体)。
指定の血液サンプリング時間からの許容される時間枠は±5分とした。
薬物動態測定のため、ほぼ528mLの血液(6mLの検体22個を4件)を各対象から採取した。
【0163】
5.投与治療薬
治療薬は、絶食状態において次の通り経口投与した:
A:IR OXN2.5/1.25mg錠剤を1錠
B:IR OXN5/2.5mg錠剤を1錠
C:IR OXN10/5mg錠剤を1錠
D:IR OXN20/10mg錠剤を1錠。
【0164】
オピオイド関連有害事象を低減するため、ナルトレキソンHClを、すべての治療薬(A〜D)と共に経口投与した。ナルトレキソンHCl50mg錠剤(Nalorex(登録商標)錠、Bristol−Myers Squibb Pharmaceuticals Ltd)。各治験期間において、治験薬物投与の−13時間、−1時間、および+11時間の時点で1錠のナルトレキソン錠剤を投与した。
【0165】
6.薬物動態パラメーター
次の薬物動態パラメーターを、オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、ナロキソン、6β−ナロキソール、ナロキソン−3−グルクロニド、および6β−ナロキソール−3−グルクロニドの血漿中濃度から計算した:
・投与時から測定可能な最終濃度まで測定された血漿中濃度−時間曲線下面積(AUCt)
・投与時から測定され無限大まで外挿された血漿中濃度−時間曲線下面積(AUCinf)
・観察される最大血漿中濃度(Cmax)
・観察される最大血漿中濃度到達時間(tmax)
・終末相速度定数(λZ)
・終末相半減期(t1/2Z)。
【0166】
オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、およびナロキソン−3−グルクロニドに関するAUC値はng.h/mLで、Cmax値はng/mLで報告した。ナロキソン、6β−ナロキソール、および6β−ナロキソール−3−グルクロニドに関して、AUC値はpg.h/mLで、Cmax値はpg/mLで報告した。
【0167】
AUCt値は、線形台形法を使用して計算した。可能な場合、λZを、終末対数−線形相にあると判定されるそれらの箇所を使用して推定した。t1/2Zは、In2のλZに対する比率から決定した。最終測定点と無限大との間の血漿中濃度−時間曲線下面積は、観察される最終血漿中濃度(Clast)のλZに対する比率から計算した。次いで、これをAUCtに加算してAUCINFを得た。
【0168】
すべての薬物動態計算は、WinNonlin Enterprise Edition、version 4.1を用いて実施した。
【0169】
とりわけ低用量で、ナロキソンの血漿中濃度は極度に低く、薬物動態の完全な特徴づけはできなかった。後の結果および考察の部では、その主要代謝産物であるナロキソン−3−グルクロニドにより大きな強調が置かれる。
【0170】
治験の主要目的は、2.1/1.25mg、5/2.5mg、10/5mgおよび20/10mgの強度のIR OXN錠剤からオキシコドンおよびナロキソン(または代用物ナロキソン−3−グルクロニド)の用量比例性を判定することとした。
【0171】
用量調整された相対的な全身性バイオアベイラビリティーを、AUCt、および可能な場合AUCINFの比率から計算した。オキシコドン、ナロキソンおよびそれらの代謝産物に関する重要な比較は、
・OXN IR5/2.5mg錠剤対OXN IR2.5/1.25mg錠剤
・OXN IR10/5mg錠剤対OXN IR2.5/1.25mg錠剤
・OXN IR20/10mg錠剤対OXN IR2.5/1.25mg錠剤、とした。
【0172】
観察される最大血漿中濃度(Cmax)およびCmaxが生じる時刻(tmax)は、報告された血漿中濃度−時間データから直接に得られた。用量調整されたCmax比を、上に概略を示した比較を行って計算した。
【0173】
7.結果
オキシコドン、ナロキソンおよびナロキソン−3−グルクロニドの薬物動態パラメーターに関する結果をそれぞれ図3〜5に、解析された21名の対象について示す。
【0174】
<オキシコドン>
オキシコドンに関する要約統計量を表7に示す。
【0175】
【表7】
【0176】
すべてのIR OXN錠剤強度で、tmaxおよびt1/2Zパラメーターについて類似の値が記録された。
【0177】
IR OXN錠剤強度のそれぞれは、AUCt、AUCINFおよびCmaxに関して、オキシコドンのIR OXN2.5/1.25mg錠剤に対して同等の用量調整バイオアベイラビリティーを提供し、比較比率のそれぞれに関する90%信頼区間は、生物学的同等性に対する80〜125%の許容性限界内に含まれた。オキシコドンの薬物動態パラメーターに関する統計解析の結果を表8に示す。
【0178】
【表8】
【0179】
<ナロキソン−3−グルクロニド>
ナロキソン−3−グルクロニドの薬物動態パラメーターを、ナロキソンの代用物として分析した。
【0180】
ナロキソン−3−グルクロニドに関する要約統計量を表9に示す。
【0181】
【表9】
【0182】
すべてのIR OXN錠剤強度で、tmaxについては類似の値が記録された。平均のt1/2Z値は、IR OXN5/2.5mg錠剤強度の8.88時間からIR OXN20/10mg錠剤強度の15.68時間までの範囲にわたった。
【0183】
IR OXN錠剤強度のそれぞれは、AUCt、AUCINFおよびCmaxに関してナロキソン−3−グルクロニドのIR OXN2.5/1.25mg錠剤に対して同等の用量調整バイオアベイラビリティーを提供し、比較比率のそれぞれに関する90%信頼区間は、131.9%であるIR OXN5/2.5mg対IR OXN2.5/1.25mgに関するCmax比率に関する上側90%信頼区間を除いて、生物学的同等性に対する80〜125%の許容性限界内に含まれた。ナロキソン−3−グルクロニドの薬物動態パラメーターに関する統計解析の結果を表10に示す。
【0184】
【表10】
【0185】
8.結論
用量調整した生物学的同等性が、オキシコドンおよびナロキソン−3−グルクロニドの主要分析対象物に関して、IROXN2.5/1.25mg錠剤に対してそれぞれのOXN IR錠剤強度について達成された。用量比例性は、2.5/1.25mg〜20/10mgの範囲の強度のIR OXN錠剤強度について確認された。ナロキソンの血漿中濃度は、とりわけ低用量で極度に低く、前の治験結果を追認し、代用物ナロキソン−3−グルクロニドの分析を支持した。
【実施例3】
【0186】
IR−OXN20/10錠剤のバイオアベイラビリティーをオキシコドン/ナロキソン20/10mg長期放出錠剤(Targin(登録商標)と比較するための、健常対象における単回投与治験
1.目的
この治験の目的は、絶食状態で健常対象に投与した場合の、OXN IR20/10mg錠剤、OXN PR20/10mg錠剤、およびオキシコドンIR20mgカプセル剤の薬物動態、バイオアベイラビリティー、および安全性を評価することとした。
【0187】
2.試験集団
18名の対象が治験を完了し、有効な薬物動態データを提供することを目標に、治験薬剤を投与される全部で21名の健常成人、男性および女性対象を無作為化するように計画した。全部で22名の対象を、実際に登録し、無作為化し、21名の対象が治験を完了した。1名の対象は、治験薬剤(すなわち、OXNまたはオキシコドン)の投与前に取りやめた。
【0188】
<組入れ基準>
治験に組み入れ予定の対象は、次の基準のすべてに合致する者とした:
1.年齢が18〜55歳(両端を含む)の男性または女性対象。
2.治験中に性的に活発であった、または性的に活発になった女性対象は、治験中を通して高度に有効な避妊法を進んで使用したにちがいない者。高度に有効な受胎調節法は、不妊手術、インプラント、注射剤、併用経口避妊薬、一部の子宮内デバイス、または配偶者の精管切除などの、一貫してかつ正しく使用された場合に低い失敗率(すなわち、1年に1%未満)をもたらすものと定義した。
3.閉経1年未満の女性対象は、血清妊娠検査で陰性であり、かつ非授乳であったにちがいない者。
4.閉経後1年間を超え、かつ高めたれた血清卵胞刺激ホルモン(FSH)を有するか、またはホルモン補充療法(HRT)で治療された女性対象。
5.55〜100kgの範囲の体重、および18以上かつ29以下の体型指数(BMI)。
6.健康で、かつ病歴、身体的検査、バイタルサイン、臨床検査、および心電図(ECG)によって判定されるような有意な異常所見がない。
7.治験中に供給されるすべての食物を進んで食すること。
8.対象のプライマリーケア医師が、事前の12ヶ月以内に、対象が臨床試験へ参加するのに適していることを確認した者。
【0189】
<除外基準>
治験から除外すべき対象は、次の基準のいずれかに合致する者とした:
1.薬物またはアルコール乱用の任意の既往歴。
2.薬物の吸収、分布、代謝または排泄を妨害する可能性のある状態の任意の既往歴。
3.事前の30日間でのオピオイドまたはオピオイド拮抗薬を含有する薬剤の使用。
4.病因にかかわらず、頻回の悪心または嘔吐の任意の既往歴。
5.発作または症候性頭部外傷の任意の既往歴。
6.この治験の初回投与に先立つ90日間中の臨床薬物治験への参加。
7.この治験への登録に先立つ4週間中の任意の重大な疾患。
8.初回投与に先立つ7日間中、またはこの治験過程中のビタミン、生薬および/またはミネラル補助食品を含む任意の薬剤の使用(HRTおよび避妊薬の連続的使用を除いて)。
9.治験薬物の投与に先立つ8時間中、または投与に続く4時間中に食物を控えること、および各拘束期間中完全にカフェインまたはキサンチン含有飲料を控えることの拒絶。
10.女性では14単位/週、男性では21単位/週の等価量を超える毎週のアルコール摂取。
11.治験薬物投与前48時間以内のアルコール飲料の消費、および治験薬物投与後の少なくとも48時間アルコールを控えることの拒絶。
12.治験薬物投与の45日以内の喫煙暦、および治験中の禁煙の拒絶。
13.プロトコールによって要求された場合を除く、治験薬物投与に先立つ30日以内または治験中の任意いずれかの時点での血液または血液産物の提供。
14.尿中薬物スクリーニング、アルコール検査、B型肝炎表面抗原(HBsAg)、C型肝炎抗体、またはヒト免疫不全ウイルス(HIV)検査の陽性結果。
15.オキシコドン、ナロキソン、ナルトレキソンまたは関連化合物に対する既知の感受性。
16.対象のプライマリーケア医師に報告することを承認することへの拒絶。
【0190】
対象集団の人口統計学的データを表11に示す。治験集団は、男性11名および女性10名の平均年齢が33歳(範囲:20〜52歳)の対象から構成した。すべての対象が白人であった。
【0191】
【表11】
【0192】
3.治験デザイン
治験は、オープンラベル、単回投与、3治療薬、3期間、無作為化クロスオーバーとした。各対象は、IROXN20/10mg錠剤、オキシコIR20mgカプセル剤、およびOXN PR20/10mg(Targin(登録商標))の単回投与を受けた。対象は、無作為割付スケジュール(RAS)により、各投与の間に少なくとも7日間のウォッシュアウト期間を伴う3治療薬のそれぞれを投与された。
【0193】
対象は、治療期間1の初回投与日の21日間以内にスクリーニングのために来院した。次いで、適格対象は、各治験期間における投与の前日に治験施設にチェックインした。翌朝、対象に絶食状態で適切な治験薬物を投与した。
【0194】
薬物動態用血液検体を、各治験期間における治験薬物投与後の36時間の間に採取し、36時間の時点の血液検体採取後に対象を退院させた。
【0195】
各治験期間中、バイタルサイン(脈拍、血圧、および呼吸数)を、投与前、次いで投与後の1、2、4、6、8、12、24および36時間の時点で測定した。口腔温を、投与前、ならびに投与後の24および36時間の時点で測定した。治験中、有害事象を通して記録した。
【0196】
対象は、治験の完了/中止の場合、彼らの治験薬剤の最終投与の4〜7日後に治験後来院した。
【0197】
治験デザインを6に示す。図6中の略語は次の通りである:
R=無作為化
P1、P2、P3=それぞれ、治験薬物の単回投与、それに続く血液サンプリング、および36時間までの安全性評価からなる期間1〜3。各治験期間の治験薬物投与の間に少なくとも7日間のウォッシュアウトを存在させた。
PS med=治験の完了/中止の場合、最終治験薬物投与の7〜10日後の治験後健診。治験中止以前だけナルトレキソンを投与された対象は、施設からの退院に先立って治験後健診を受けた。
V1〜V5=来院
【0198】
4.薬物動態用検体の採集
1、8および15日目に開始して、次の時点での一連の血液採集物(それぞれ6mL)を得た:
治療薬AおよびB:
投与前、投与後0.25、0.5、0.75、1、1.25、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1投与期間につき22個の血液検体)。
治療薬C:
投与前、投与後0.5、1、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1投与期間につき19個の血液検体)。
対象が、OXN PR20/10mg錠剤の投与後12時間以内に、あるいはOXN IR20/10mgまたはオキシコドンIR20mgカプセル剤の投与後6時間以内に嘔吐を経験した場合、その対象の残りの治験期間について、さらなる薬学動態用血液検体を採取しなかった。
指定の血液サンプリング時間からの許容される時間枠は±5分とした。
薬物動態測定のため、ほぼ378mLの血液(6mLの検体22個を2件および6mLの検体19個を1件)を各対象から採取した。
【0199】
5.投与治療薬
この治験で投与される治療薬を以下に示す:
試験治療薬:
IR OXN20/10mg錠剤。該治療薬は、絶食状態において次の通り経口投与した:
治療薬A:OXN IR20/10mg錠剤 1錠。
参照治療薬:
IRオキシコドン20mgカプセル剤(OxyNorm(登録商標)Napp Pharmaceuticals Ltd、英国)。該治療薬は、絶食状態において次の通り経口投与した:
治療薬B:オキシコドンIR20mgカプセル剤 1カプセル
OXN PR20/10mg錠剤(Targin(登録商標))。英国Bard Pharmaceuticals Ltd製造。該治療薬は、絶食状態において次の通り経口投与した:
治療薬C:OXN PR20/10mg錠剤 1錠。
【0200】
治験薬物の投与は、各治験期間においてオピオイド関連有害事象の危険を低減するために、ナルトレキソンでの援護下に行われた。ナルトレキソン塩酸塩50mg錠剤(Nalorex(登録商標)錠、Bristol−Myers Squibb Pharmaceuticals Ltd)を次の通り経口投与した:
ナルトレキソン50mg錠剤を1錠、治験薬物投与の13時間前(−1、7、14日目)、1時間前および11時間後(1、8および15日目)の時点で、すべて100mLの水で嚥下した(全部で3回投与)。
【0201】
6.薬物動態パラメーター
血漿中濃度データおよび薬物動態パラメーターを、登録された集団中の対象について列挙した。
【0202】
各分析対象物についての血漿中濃度データを、登録された集団中の対象について時点および治療薬によって記述的に要約した。また、各分析対象物についての個別および平均の血漿中濃度を、各治療薬について時間とともにプロットした。
【0203】
各分析対象物についての薬物動態パラメーター(AUCt、AUCINF、Cmax、tmax、λZ、およびt1/2Z)を、薬物動態パラメーターのための完全分析集団中のすべての対象について治療薬によって記述的に要約した。有効な薬物動態パラメーターを得るために、対象は、OXN PR20/10錠剤の投与後12時間以内の、またはOXN IR20/10錠剤もしくはオキシコドンIR20mgカプセル剤の投与後6時間以内の嘔吐を経験してはならない。
【0204】
分析対象物オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、ナロキソン、6β−ナロキソール、ナロキソン−3−グルクロニド、および6β−ナロキソール−3−グルクロニドに関する対数変換データ、ならびに薬物動態パラメーターAUCt、AUCINF、およびCmaxを、治療薬および期間については固定項、対象については無作為効果で、混合効果線形モデル18を使用して解析した。
【0205】
治療薬の比率/差異およびそれらに付随する90%信頼区間を、最小二乗平均から計算した。
【0206】
比較予定の治療薬は次の通りである:
・IR OXN20/10mg対オキシコドンIR20mg
・IR OXN20/10mg対OXN PR20/10mg
【0207】
代謝産物:親薬物の比率は、AUCt値、および可能な場合AUCINF値を使用して各治療薬に関して計算した。
【0208】
すべての薬物動態の計算は、WinNonlin Enterprise Edition、version 4.1.を使用して行った。
【0209】
7.結果:
オキシコドン、ナロキソン、およびナロキソン−3−グルクロニドの薬物動態パラメーターに関する結果を、それぞれ図7〜9に分析された対象について示す。
【0210】
<オキシコドン>
オキシコドンに関する要約統計量を表12に示す。
【0211】
【表12】
【0212】
OXN PR20/10mg錠剤と比較して、IR OXN IR20/10mg錠剤は、90%信頼区間で、生物学的同等性の基準に合致する96.3%のオキシコドンの平均経口アベイラビリティーを有した。即時放出および長期放出製剤を比較すると、OXN IR20/10mg錠剤のCmaxは、OXN PR20/10mg錠剤に比べて有意により高かった。
【0213】
オキシコドンIRカプセル剤と比較して、IR OXN20/10は、99.2%のオキシコドンの平均経口アベイラビリティー、および100%の平均Cmax比率を有した。これらの比較に関する生物学的同等性の評価は、すべて、生物学的同等性に関する判定基準に合致した90%信頼区間を有した。
【0214】
オキシコドンに関する平均半減期の値は、すべての治療薬にわたって類似し、4.2〜4.3時間の範囲にあった。
【0215】
IR OXN20/10およびオキシコドンIR20mgカプセル剤は、類似のtmax中央値を有した。OXN PR20/10は、他の治療薬に比べてより遅いtmaxを有した。
【0216】
オキシコドンの薬物動態パラメーターの統計解析結果を表13に示す。
【0217】
【表13】
【0218】
<ナロキソン−3−グルクロニド>
ナロキソン−3−グルクロニドに関する要約統計量を表14に示す。
【0219】
【表14】
【0220】
OXN PR20/10錠剤と比較して、IR OXN20/10は、90%信頼区間で、生物学的同等性の基準に合致する90.4%のナロキソン−3−グルクロニドの平均経口アベイラビリティーを有した。即時放出および長期放出製剤を比較すると、IR OXN20/10に関するCmaxは、OXN PR20/10錠剤に比べて有意により高かった。
【0221】
平均半減期の値は、IR OXN(8.7時間)およびOXN PR(9.1時間)錠剤の間で類似していた。
【0222】
OXN PR20/10錠剤に関するtmax値は、IR OXN20/10に比べてより遅かった。
【0223】
ナロキソン−3−グルクロニドの薬物動態パラメーターに関する統計解析結果を表15に示す。
【0224】
【表15】
【0225】
8.結論:
薬物動態パラメーターに関する結果は、IR OXN20/10mgおよびOXN PR20/10mg錠剤から、オキシコドンおよびナロキソン(または代用物ナロキソン−3−グルクロニド)の十分に同等なバイオアベイラビリティーを、かつオキシコドンに関するIR OXN20/10mgおよびオキシコドンIR20mgカプセル剤の生物学的同等性を示した。ナロキソンは、予想したように、極めて低いバイオアベイラビリティーを有し、以前の治験結果を確証し、代用物ナロキソン−3−グルクロニドの分析を支持していた。
実験4
【0226】
IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤の貯蔵安定性
IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤を、PVC/PVdCアルミニウムホイルのブリスターパック中に25℃/60%RTおよび40℃/75%RHで貯蔵した。
【0227】
安定性データは、25℃/60%RTおよび40℃/75%RHで3ヶ月間貯蔵した後のIR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤に関して準備する。
【0228】
下記に示すデータは、物理的特性および溶出に関するオキシコドン塩酸塩/ナロキソン塩酸塩の即時放出錠剤の安定性を確認する。
【0229】
【表16】
【0230】
【表17】
【0231】
【表18】
【0232】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ月間の貯蔵後に最大で約0.5%まで変化した。
【0233】
【表19】
【0234】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で約2.3%まで変化した。
【0235】
【表20】
【0236】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ月の貯蔵期間にわたって一定のままであった。
【0237】
【表21】
【0238】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で1.7%まで変化した。
【0239】
【表22】
【0240】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ間の貯蔵後に最大で0.4%まで変化した。
【0241】
【表23】
【0242】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で0.7%まで変化した。
【技術分野】
【0001】
本発明は、疼痛に苦しむ患者を治療するのに適した医薬組成物に関する。
【背景技術】
【0002】
疼痛に苦しむ患者を適切に治療することの重要性は、過去数十年にわたって、ますます認識されつつある。
【0003】
がん患者において起こるような慢性の中等度から高度のおよびさらには重度の疼痛の治療のために、オピオイド系鎮痛剤が、最近の数十年にわたってますます普及しつつある。この発展に寄与している要因の中には、以前に利用可能であったそれらの薬剤の即時放出製剤に比較して、患者が、低減された頻度で服用することができる、モルフィン、ヒドロモルホンおよびオキシコドンなどのオピオイドの制御放出製剤の導入があった。
【0004】
活性薬剤としてオキシコドン塩酸塩を含む製品Oxygesic(登録商標)錠剤、およびオキシコドン塩酸塩とオピオイド拮抗薬ナロキソン塩酸塩との組合せを含むTargin(登録商標)錠剤などのオピオイドオキシコドン制御放出製剤は、商業的展望から、ならびに患者および医師による受容性の観点でどちらも成功した。
【0005】
しかし、オピオイドの制御放出製剤は、疼痛に苦しむ患者を治療する場合に、必ずしも第1選択の薬剤であるとは限らない可能性がある。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の1つの目的は、疼痛を治療するのに、とりわけ慢性の中等度〜高度のおよびさらには重度の疼痛を治療するのに適し、標準的な疼痛療法中に発生する可能性のあるような望ましくない副作用を同時に回避しながら、疼痛に苦しむ患者の用量設定するのにおよび/または疼痛に苦しむ患者における突出痛を治療するのに使用することができる医薬組成物を提供することである。
【0007】
後に続く説明から明らかになるようなこれらのおよびその他の目的は、独立請求項の主題によって達成される。その後に示される従属請求項は、本発明の好ましい実施形態の一部を示す。
【課題を解決するための手段】
【0008】
本発明は、一実施形態において、少なくともオキシコドンまたはその薬学上許容される塩およびナロキソンまたはその薬学上許容される塩を約2:1の重量比(オキシコドンまたはその薬学上許容される塩:ナロキソンまたはその薬学上許容される塩)で含む経口即時放出医薬組成物に関する。
【0009】
一般に、本発明による経口即時放出医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩に加えて、その他の薬学的に活性な薬剤を含むことができる。しかし、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が、本発明による医薬組成物の唯一の薬学的に活性な薬剤であることが本発明の好ましい実施形態である場合もある。
【0010】
一般に、本発明による即時放出医薬組成物は、オキシコドンをその遊離塩基またはその塩の形態で、およびナロキソンをその遊離塩基またはその塩の形態で含むことができる。しかし、本発明による経口即時放出医薬組成物が、オキシコドンをオキシコドン塩酸塩の形態で、およびナロキソンをナロキソン塩酸塩の形態で含むことが好ましい場合もある。オキシコドン塩酸塩およびナロキソン塩酸塩が、医薬組成物の唯一の薬学的に活性な薬剤を構成することがさらにより好ましい可能性もある。
【0011】
第1態様において、本発明による経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。1つの好ましい実施形態において、本発明による経口即時放出医薬組成物は、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。これらの医薬組成物は、該薬学的に活性な薬剤を約2:1の重量比で含むことができる。
【0012】
この第1態様の好ましい実施形態は、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を約2:1の比率(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み、オキシコドン塩酸塩が約2.5mg〜約40mgのオキシコドン塩酸塩の量で存在し、ナロキソン塩酸塩が約1.25mg〜約20mgのナロキソン塩酸塩の量で存在する、経口即時放出医薬組成物に関し得る。
【0013】
この第1態様のさらにより好ましい実施形態は、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を約2:1の比率(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み、オキシコドン塩酸塩が約2.5mg〜約20mgのオキシコドン塩酸塩の量で存在し、ナロキソン塩酸塩が約1.25mg〜約10mgのナロキソン塩酸塩の量で存在する、経口即時放出医薬組成物に関し得る。
【0014】
本発明の第2態様は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、かつここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する、経口即時放出医薬組成物に関する。
【0015】
この第2態様の好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0016】
この第2態様のより好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0017】
この第2態様のさらにより好ましい実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含み、ここで、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、経口即時放出医薬組成物に関し得る。
【0018】
第2態様のすべての実施形態において、経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。
【0019】
約1mg〜約80mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約40mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約1mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、あるいは約1mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩が、さらにより好ましい可能性がある。
【0020】
本発明の第3態様において、経口即時放出医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含むことができ、ここで、該組成物は、固体形態であり、かつ該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する。
【0021】
この第3態様の好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0022】
この第3態様のより好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0023】
この第3態様のさらにより好ましい実施形態は、該製剤が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する経口即時放出医薬組成物に関し得る。
【0024】
第3態様のすべての実施形態において、経口即時放出医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含むことができる。
【0025】
約1mg〜約80mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約40mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約1mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、あるいは約1mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩が、さらにより好ましい可能性がある。
【0026】
第3態様のすべての実施形態において、医薬組成物は、崩壊剤、および任意選択で充填剤、および任意選択でその他の薬学的に許容される添加剤を含むことができる。好ましくは、崩壊剤として、例えば、デンプンと乳糖の組合せを使用することができる。乳糖は、単独で、同時に充填剤として機能することができる。とりわけ好ましい実施形態は、崩壊剤および充填剤の双方として機能することができる、乳糖85%とデンプン15%との組合せである製品Starlac(登録商標)に依拠する。異なる種類の崩壊剤、充填剤またはその他の種類の薬学的に許容される添加剤を後に示す。組み合わせた充填剤/崩壊剤は、医薬組成物内に、該組成物の重量を基準にて約40重量%〜約90重量%の量で、好ましくは約50重量%〜約85重量%の量で、さらにより好ましくは約60重量%〜約80重量%の量で含むことができる。StarLac(登録商標)のように崩壊剤および充填剤の双方として二重の機能を有する添加剤を使用する場合、これらの数字がとりわけ適用される。
【0027】
第3態様のすべての実施形態において、医薬組成物は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含むことができる。
【0028】
本発明の第3態様のすべての実施形態において、医薬組成物は、錠剤、カプセル剤、顆粒剤、多粒子剤などの形態で提供することができる。錠剤は、とりわけ好ましい可能性がある。
【0029】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、ヒト健常志願者での単回投与治験で投与された場合に、約15ng.h/mL〜約500ng.h/mLの範囲の、好ましくは約20ng.h/mL〜約400ng.h/mLの範囲の、より好ましくは約25ng.h/mL〜約300ng.h/mLの範囲の、さらにより好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0030】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、ヒト健常志願者での単回投与治験で投与された場合に、約1ng/mL〜約300ng/mLの範囲の、好ましくは約2ng/mL〜約200ng/mLの範囲の、より好ましくは約3ng/mL〜約100ng/mLの範囲の、さらにより好ましくは約4ng/mL〜約75ng/mLの範囲の、最も好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0031】
本発明による経口即時放出医薬組成物は、限定はされないが、錠剤、カプセル剤、多粒子剤(例えば、顆粒、長球またはビーズ)、および液剤(例えば溶液、懸濁液または乳液)の形態であり得る。
【0032】
本発明による経口即時放出医薬組成物は、薬学的に許容される添加剤として、少なくとも賦形剤および任意選択で崩壊剤を含むことができる。さらに、それらの組成物は、滑沢剤、充填剤、着色剤、着香剤、pH調整剤、可塑剤、抗粘着剤、結合剤などのその他の薬学的に許容される添加剤を含むことができる。本発明による経口即時放出医薬組成物は、また、薬学的に許容される添加剤として、少なくとも崩壊剤および任意選択で賦形剤を含むことができる。さらに、それらの組成物は、滑沢剤、充填剤、着色剤、着香剤、pH調整剤、可塑剤、抗粘着剤、結合剤などのその他の薬学的に許容される添加剤を含むことができる。Starlac(登録商標)製品などの崩壊剤および賦形剤の双方としての二重機能を有する添加剤を好ましくは使用することができることを理解されたい。
【0033】
本発明のとりわけ好ましい実施形態において、経口即時放出医薬組成物は、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1(オキシコドン塩酸塩:ナロキソン塩酸塩)の重量比含み、ここで、該組成物は、唯一の薬学的に許容される活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含み、組成物は錠剤の形態であり、製剤は、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含み、かつ製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する。本発明のこれらのとりわけ好ましい実施形態のさらなる好ましい態様において、該医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の95重量%以上を、およびナロキソン塩酸塩の95重量%以上をin vitroで放出する。
【0034】
本発明のこれらのとりわけ好ましい実施形態の別の態様は、ヒト健常志願者での単回投与研究で投与された場合に、約25ng.h/mL〜約300ng.h/mLの範囲の、より好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供する医薬組成物に関する。このような経口即時放出医薬組成物は、オキシコドンおよびナロキソンの塩酸塩を好ましくは含むことができる。
【0035】
本発明のこれらのとりわけ好ましい実施形態の別の態様は、ヒト健常志願者での単回投与研究で投与された場合に、約4ng/mL〜約75ng/mLの範囲の、より好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供する医薬組成物に関する。このような経口即時放出医薬組成物は、オキシコドンおよびナロキソンの塩酸塩を好ましくは含むことができる。加えて先の段落のAUC値を提供する医薬組成物が、とりわけ好ましい。
【0036】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者の用量設定するのに使用することができる。
【0037】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を治療するのに使用することができる。
【0038】
本発明による経口即時放出医薬組成物は、疼痛に苦しむ患者における、疼痛、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛を治療するのに使用することができる。
【0039】
本発明は、また、前記のような経口即時放出医薬組成物の使用、および、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者の用量設定するための薬剤の製造に関する。
【0040】
さらに、本発明は、前記のような経口即時放出医薬組成物の使用、および疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を治療するための薬剤の製造に関する。
【0041】
さらに、本発明は、前記のような経口即時放出医薬組成物の使用、および疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における疼痛を治療するための薬剤の製造に関する。
【0042】
本発明は、別の態様において、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者を、前記のような経口即時放出医薬組成物を投与することによって患者の用量設定する方法に関する。
【0043】
本発明のさらに別の態様は、疼痛に苦しむ患者、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛に苦しむ患者における突出痛を、前記のような経口即時放出医薬組成物を投与することによって治療する方法に関する。
【0044】
本発明のさらに別の態様は、疼痛に苦しむ患者における、疼痛、とりわけ慢性の中等度から高度のおよびさらには重度の疼痛を、前記のような経口即時放出医薬組成物を投与することによって治療する方法に関する。
【0045】
さらに、本発明は、前記のような経口即時放出医薬組成物の製造方法に関する。
【図面の簡単な説明】
【0046】
【図1】本発明の医薬組成物の製造方法に関する流れ図を描いた図である。
【図2】実験2に記載の臨床試験に関する治療スケジュールを描いた図である。
【図3】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、オキシコドンの平均血漿中濃度を描いた図である。
【図4】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、ナロキソンの平均血漿中濃度を描いた図である。
【図5】対象が21名の実験2において、IR OXN2.5/1.25、IR OXN5/2.5、IR OXN10/5、およびIR OXN20/10に関して測定した場合の、ナロキソン−3−グルクロニドの平均血漿中濃度を描いた図である。
【図6】実験3に記載の臨床試験に関する治療スケジュールを描いた図である。
【図7】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、オキシコドンの平均血漿中濃度を描いた図である。
【図8】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、ナロキソンの平均血漿中濃度を描いた図である。
【図9】対象が21名の実験3において、IR OXN20/10、IRオキシコドン20mg、およびPRオキシコドン20/10(Targin(登録商標))に関して測定した場合の、ナロキソン−3−グルクロニドの平均血漿中濃度を描いた図である。
【発明を実施するための形態】
【0047】
以下で例示的に説明するような本発明は、本明細書中で具体的に開示されない任意の要素または要素群、限定または限定群の不在下で適切に実施される可能性がある。
【0048】
本発明は、特定の実施形態に関して、かついくつかの図を参照して説明されるが、本発明はそれらに限定されるものではなく、特許請求の範囲によってのみ限定される。記載の図面は、単なる略図であって、それに限定するものではない。以下で示される用語は、そうでないことを指摘しない限り、一般にはそれらの通常的な意味で解釈されるものとする。
【0049】
用語「含む」が本明細書および特許請求の範囲中で使用される場合、該用語は、その他の要素を排除するものではない。本発明の目的に関して、用語「〜からなる」は、用語「〜から成り立つ」のある好ましい実施形態であると考えられる。以下で、ある群が、少なくともある数の実施形態を含むと定義される場合、このことは、また、好ましくは、これらの実施形態からのみなる群を開示していると解釈されるものとする。
【0050】
不定冠詞または定冠詞、例えば「a」、「an」、「the」が単数名詞を指す際に使用される場合、これは、特記しない限り、その名詞の複数形を含む。本発明の文脈中の用語「約」または「ほぼ」は、当業者が当該特徴の技術的効果をなお保証すると解釈する、正確さの隔たりを意味する。該用語は、典型的には、指摘した数値からの±10%、好ましくは±5%の偏差の存在を示す。
【0051】
以下でオキシコドンまたはナロキソンについて言及する場合、用語「オキシコドン」または「ナロキソン」が遊離塩基のみを指すのが当然であることを具体的に指摘しない限り、その言及は、常に、オキシコドン遊離塩基の薬学的に許容される塩またはナロキソン遊離塩基の薬学的に許容される塩あるいはそれらの誘導体についての言及をも含む。
【0052】
用語「即時放出」は、活性成分、すなわち、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が医薬組成物から放出される放出速度に関連している。一般的理解との関連で、用語「即時放出」は、特殊な製剤設計および/または製造方法によって故意に変更されていない、活性物質(群)の放出を示す医薬組成物に関連している。
【0053】
用語「即時放出」は、とりわけ、本発明による医薬組成物が、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する特性を指す。
【0054】
in vitro放出速度は、欧州薬局方2.9.3(第6版)中に記載の通りの欧州薬局方(Ph.Eur.)パドル法により測定される。パドル速度は、模擬胃液(ペプシン不含USP(米国薬局方))溶出媒質中で100rpmに設定される。溶出媒質のアリコートを、15分および45分の時点で抜き取り、60℃で維持された逆相Merck LiChrospher 60RP Select Bカラムを使用するHPLCで分析する。移動相は、85:15v/v、pH2.0の塩化カリウム:メタノールからなる。紫外検出は230nmで行われる。オキシコドンおよびナロキソンは、外部標準アッセイによって定量される。
【0055】
経口即時放出医薬剤形が、5mgまでの量のオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および2.5mgまでの量のナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む場合、欧州薬局方パドル試験は、500mLの0.1N塩酸中で実施される。
【0056】
経口即時放出医薬剤形が、5mgを超える量のオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および2.5mgを超える量のナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む場合、欧州薬局方パドル試験は、900mLの0.1N塩酸中で実施される。
【0057】
以下で言及されるような用語「用量設定(titration)」およびその文法上の変形体は、医師が、疼痛、とりわけ慢性の中等度から高度およびさらには重度の疼痛に苦しむ患者のために、オキシコドン塩酸塩などの具体的なオピオイドについて適切な投与量を確認する過程を指す。疼痛に苦しむ患者は、彼らの異なる代謝特性のため、投与すべきオピオイド系鎮痛剤の量に関して異なる要求を有するので、典型的には、医師は、許容される疼痛コントロールを提供するため、先ず、オピオイド製剤の即時放出製剤の投与量を増大させることを試み、患者が無疼痛であるか、あるいは少なくとも有意な疼痛低減を有するのに十分であるオピオイドの投与量を確認する。
【0058】
投与量が、これらの患者によって経験される(慢性)疼痛を永続的にコントロールするのに適している場合、患者は、典型的には、薬剤を低減された頻度で時刻を決めた規則正しいレジメンで、例えば12時間毎または1日1回のみで摂取できるような対応投与量を含む制御放出製剤に変更される。疼痛治療なしで慢性疼痛感覚に苦しむような患者は、典型的には、制御された背景痛(background pain)を伴う患者と呼ばれる。
【0059】
本発明の文脈で、用語「突出痛」およびその文法上の変形体は、オピオイドの時刻を決めた規則正しいレジメンで治療されている患者によって経験される持続痛または背景痛を超える疼痛の一時的増大を指す。
【0060】
用語「持続痛」は、以下で使用する場合、典型的には、疼痛治療用のオピオイドレジメンを受けている患者が例えば12時間以上の間の平均疼痛強度の体験と報告する疼痛である。
【0061】
持続痛の疼痛強度は、典型的には、数値アナログ尺度試験(NAS)などの通常の方法を使用して測定される。突出痛発作および持続痛の測定は、例えば、Portenoyら(1999)The Journal of Pain 7(8):583〜591頁;およびPortenoyら(1999)Pain 81:129〜134頁に記載されている。これらの刊行物中で与えられているような突出痛および制御された持続痛の定義は、参照により本明細書に組み込まれる。したがって、制御された持続痛の名称は、2つの判断基準が合致することを典型的には要求する。第1に、患者は、質問[あなたの疼痛は、最近、あなたが「一定」または「ほとんど一定」と評する、あるいは肯定的返答の中で受けている治療のためではないにしても「一定またはほとんど一定である」要素を持っていますか]に返答しなければならない。第2に、患者は、比較的良好な疼痛コントロールと矛盾しないオピオイドレジメンによって治療されることを要求されなければならない。当業者は、2つの参考文献中で提供される情報に基づいて制御された持続痛を測定する方法を承知しているであろう。
【0062】
突出痛は、次いで、制御された持続痛のレベルを超える患者によって経験される疼痛の再燃として確認される。疼痛強度は、例えば、項目「なし」、「軽度」、「中等度」、「重度」および「極度に痛い」を有する5点分類尺度を使用して評価することができる。この発作が患者によって重度または極度に痛いと評価された場合、患者は、典型的には、突出痛発作を経験する。
【0063】
以下で言及するような用語「生物学的同等性」およびその文法上の変形体は、それらの通常の意味で使用される。とりわけ、試験医薬組成物の血漿中レベルの曲線下面積(AUC)(AUCtまたはAUCinf)に関する平均値の90%信頼区間が、参照医薬組成物の対応する平均値の80%〜125%の範囲内にあり、かつ試験医薬組成物の血漿中レベルの最大濃度(Cmax)に関する平均値の90%信頼区間が、参照医薬組成物の対応する平均値の80%〜125%の範囲内にある場合、医薬組成物は、参照医薬組成物に対して生物学的に同等であると言われる。
【0064】
Cmax値は、活性薬剤、すなわち、オキシコドンおよび/またはナロキソン(または塩酸塩などのそれらの塩)の最大血漿中濃度を示す。
【0065】
tmax値は、Cmax値に到達する時点を示す。換言すれば、tmaxは、最大血漿中濃度が観察される時点である。
【0066】
AUC(曲線下面積)値は、濃度曲線の面積に相当する。AUC値は、血液循環中に吸収された活性薬剤、すなわち、オキシコドンおよびナロキソン(または塩酸塩などのそれらの塩)の全体量に比例し、それゆえバイオアベイラビリティーの尺度である。
【0067】
AUCt値は、投与時点から測定可能最終濃度までの血漿中濃度−時間曲線下の面積に関する値である。AUCt値は、通常、台形法を使用して計算される。可能な場合、終末相速度定数であるλZを、終末対数−線形相中にあると判定されるそれらの箇所を使用して推定する。見かけの終末相半減期であるt1/2Zは、通常的には、Ln2のλZに対する比率から決定される。最終測定点と無限大との間の血漿中濃度−時間曲線下面積は、観察される最終血漿中濃度(Clast)のλZに対する比率から計算することができる。次いで、これをAUCtに加算して、投与時から無限大までの血漿中濃度−曲線下面積であるAUCinfを得る。
【0068】
用語「バイオアベイラビリティー」は、本発明の目的の場合、オキシコドンおよびナロキソン(または塩酸塩などのそれらの塩)などの活性薬剤が、該医薬組成物の経口投与後に吸収される度合いと定義される。
【0069】
用語「定常状態」は、次のように説明することができる:最初の用量が投与される時点、t=0で、濃度C=0である。次いで、濃度は、最初の極大点を通り、次いで、最初の極小点まで降下する。濃度が0まで降下する前に、別の用量が、投与され、その結果、第2の濃度増加は0から始まるのではない。
この最初の濃度極小値上に積み上げると、二回目の用量を投与した後に、曲線は、最初の極大値を超える二番目の極大値を通過し、かつ最初の最小値を超える二番目の極小値まで降下する。したがって、反復投与および付随する活性薬剤の段階的な蓄積のため、血漿中濃度は、それが、吸収および排出が平衡状態にある点まで安定化するまで、上昇する。吸収および排出が平衡状態にあり、濃度が、規定された極小値と規定された極大値との間を絶えず振動する状態は、定常状態と呼ばれる。
【0070】
用語「維持療法」および「長期療法」は、本発明の目的の場合、患者が、オピオイド鎮痛剤を用いて上で定義したような定常状態に用量設定された後に患者に投与される薬物療法と定義される。前述の持続痛は、維持/長期療法中の疼痛感覚を指す。
【0071】
血漿中曲線を記述するパラメーターは、臨床試験中に、先ず、ある数の被験者にオキシコドンおよびナロキソン(またはそれらの塩酸塩などの塩)などの活性薬剤を1回だけ投与することによって得ることができる。個々の被験者の血漿中値は、次いで平均化され、例えば、平均のAUC、Cmaxおよびtmax値が得られる。
【0072】
本発明の文脈で、AUC、Cmaxおよびtmaxなどの薬物動態パラメーターは、そうでないことを指摘しない限り、平均値を指す。さらに、本発明の文脈で、AUC、Cmax、tmaxまたは鎮痛効力に関する値などのin vivoパラメーターは、投与後に定常状態で得られる、あるいはヒト患者および/または健常ヒト対象への単回投与のパラメーターまたは値を指す。
【0073】
平均tmax、CmaxおよびAUCなどの薬物動態パラメーターが、健常ヒト対象について測定される場合、それらのパラメーターは、典型的には、ほぼ16〜24名の健常ヒト対象からなる試験集団中で長時間にわたって血漿中値の進展を測定することによって得られる。
欧州医薬品審査庁(EMEA)または米国食品医薬品庁(FDA)などの規制組織は、通常、例えば20または24名の被験者から得られたデータを受理する。しかし、5名の被験者などのより少ない参加者を含む初期試験も受理される可能性がある。
【0074】
この文脈における用語「健常」ヒト対象は、通常的には、身長、体重、および血圧などの生理学的パラメーターに関して平均的な値を有する血統が白色人種の典型的な男性または女性を指す。本発明の目的のための健常ヒト対象は、臨床試験の調和に関する国際会議(ICH)の推奨に基づき、かつそれと一致した組入れおよび除外基準により選択される。本発明の目的の場合、健常対象は、実施例2および実施例3中で説明されるような組入および除外基準により識別することができる。
【0075】
したがって、組入れ基準は、例えば18歳以上60歳以下の年齢、例えば19〜29kg/m2の範囲内のBMI、および例えば、男性では60〜100kg、女性では55〜90kgの範囲内の体重を含み;女性は、非授乳、非妊娠であり、かつ治験薬剤を受入れる前の24時間以内に陰性の尿β−hCG妊娠検査;病歴、身体的検査、臨床検査、バイタルサイン、およびECGなどに関して有意に異常な所見の欠如によって証明される一般的に良好な健康状態を示す必要がある。
【0076】
除外基準は、例えば、治験薬剤の初回投与の3ヶ月以内での任意の治験物またはプラセボへの暴露;治験薬剤の初回投与前30日以内での任意の重大な疾患;病歴、身体的検査または試験室での分析に関する治験前スクリーニングの時点で確認された臨床的に重大な任意の異常;治験薬剤の初回投与前の21日間における任意の処方薬剤(閉経女性に対するHRTおよび避妊薬剤を除く)または治験薬剤の初回最初前の7日間における制酸剤、ビタミン、生薬および/またはミネラル栄養補助食品を含むOTC薬の使用;消化管薬物吸収(例えば、胃内容物排出遅延、吸収不良症候群)、分布(例えば、肥満)、代謝もしくは排泄(例えば、肝炎、糸球体腎炎)を妨害することが知られている並存的な医学的状態;治験責任医師の意見の中で、対象が治験を安全に完了する能力を危うくする並存的な医学的状態の既往歴;対象が薬理学的治療を必要とした発作性疾患の既往歴;1日に5本を超える現在の喫煙歴;DSM−IV基準による薬物もしくはアルコール乱用の活動歴もしくは既往歴の証拠を有する対象;1日に2本以上のアルコール飲料の規則的消費を報告した、またはスクリーニング時点で0.5%以上の血中アルコールレベルを有する対象;治験薬剤の初回投与前の3ヶ月間における500mLを超える血液もしくは血液産物の献血またはその他の重大な血液喪失;治験前スクリーニングにおいてスクリーニング時点で採集した尿検体中のエタノール、アヘン剤、バルビツール剤、アンフェタミン剤、コカイン代謝産物、メタドン、プロポキシフェン、フェニルシクリジン、ベンゾジアゼピン、およびカンナビノイドに関する任意の陽性結果;オキシコドン、ナロキソン、または関連化合物などに対する既知の敏感性などを含む。
【0077】
平均のtmax、CmaxおよびAUCなどの薬物動態パラメーターを患者で得る場合、該患者群は、典型的には、10〜200名の患者から構成される。妥当な患者数は、例えば、10、20、30、40、50、75、100、125または150である。患者は、治療予定の状態の症状により選択される。患者は、例えば、18歳以上でよく、腫瘍および非腫瘍起源の中等度から高度およびさらには重度の慢性痛に苦しみ、WHOステップIIまたはIIIの鎮痛剤などで不十分な効力および/または忍容性を示す。患者は、最近のアルコールもしくは薬物乱用の、最近の重度の心血管および呼吸器疾患の、重度の肝および腎不全などの徴候が存在する場合、例えば、薬物動態パラメーターの測定について考慮に入れない可能性がある。
【0078】
前におよび後に示すような薬物動態パラメーターの値は、実施例2および3で得られたデータに基づいて導き出され、そのすべては、健常ヒト対象での単回投与治験に関連することを理解されたい。しかし、類似の結果が、健常ヒト対象における定常状態での投与で、またはヒト患者における単回および定常状態投与で得られると想定される。当業者は、もちろん、定常状態のCmaxが単回投与後のCmaxよりも高いことを承知している。
【0079】
薬物動態パラメーターの計算は、WinNonlin Enterprise Edition、Version 4.1を用いて実施することができる。
【0080】
前に言及したように、本発明は、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物に関する。
【0081】
以下で示すように、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含むこのような経口即時放出医薬組成物は、オキシコドン塩酸塩およびナロキソン塩酸塩を2:1の重量比で(オキシコドン塩酸塩:ナロキソン塩酸塩)含む参照の制御放出医薬組成物と対照して試験した場合、生物学的に同等である。オキシコドン塩酸塩およびナロキソン塩酸塩を2:1の重量比で含む実施例3で使用される制御放出参照医薬組成物は、Targin(登録商標)錠の名称下にドイツ市場で入手可能である。Targin(登録商標)錠について、この制御放出製剤は、例えば高度〜重度の疼痛に苦しむ患者において優れた疼痛コントロールを提供するだけでなく、極めて優れた副作用プロファイルを提供することが示されている。Targin(登録商標)錠の使用による副作用の改善は、腸機能の改善として、オピオイド誘発性便秘の低減として、オピオイド誘発性尿滞留の低減として、かつ疼痛に苦しむ患者による薬物に対する忍容性および好みの増大として評価することができる(例えば、Vondrackovaら、J.Pain(2008),9(12):1144〜1154頁;Nadstawekら、Int.J.Clin.Pract.(2008),62(8):1159〜1167頁;およびMeissnerら、Eur.J.Pain(2008)(印刷中の論文)を参照されたい)。
【0082】
さらに、実験2から明らかになるように、本発明の経口即時放出医薬組成物は、とりわけ、それらが、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む場合に、用量比例性である。
【0083】
この場合、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む即時放出医薬組成物の制御放出製剤Targin(登録商標)錠に対する送達程度の同等性である生物学的同等性を考慮すれば(特に予想外のバイオアベイラビリティーの程度、およびナロキソンの初回通過効果が即時放出製剤についても維持されるという事実を考慮すれば)、本発明による即時放出医薬組成物が、疼痛、とりわけ慢性の中等度から高度およびさらには重度の疼痛に苦しむ患者における疼痛緩和を十分に提供するのに有効であるのみならず、これらの組成物で治療された患者は、とりわけオピオイド作用薬であるオキシコドン塩酸塩のみを含む先行技術から公知の経口即時放出製剤と比較して、Targin(登録商標)錠について観察される副作用プロファイルに関して同一ではないが類似の改善を経験することを想定することは正当化されると思われる。
【0084】
したがって、本発明による経口即時放出医薬組成物は、疼痛治療において有効であり、かつ、オピオイド作用薬であるオキシコドン塩酸塩のみを含む先行技術の即時放出医薬組成物と比較した場合の、オピオイド誘発性便秘の低減、オピオイド誘発性尿滞留の低減などの副作用に関する改善、ならびに患者による薬剤に対する忍容性および好みの改善を示すことが想定される。
【0085】
本発明の経口即時放出医薬組成物の用量比例性と組み合わせたこれらの特性は、過去において、制御放出製剤の改善された副作用プロファイルを考慮して、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む制御放出医薬組成物が、オピオイド作用薬であるオキシコドンのみを含む即時放出製剤に優先して選択されたであろう状況において、医師が本発明による医薬組成物を使用することを可能にする。
【0086】
本発明の経口即時放出医薬組成物は、疼痛に苦しむ患者の用量設定に、および/または疼痛に苦しむ患者における突出痛の治療にとりわけ適している。
【0087】
とりわけ、本発明の経口即時放出医薬組成物は、疼痛、とりわけ高度〜重度(慢性)疼痛に苦しむ患者の用量設定するのに使用することができ、過去における、オキシコドンおよびナロキソンの制御放出製剤を用いる患者のための用量設定は、受容できない疼痛の経験ではないにせよ、制御放出製剤の遅延された作用による不快な経験を意味した。さらに、本発明による経口即時放出製剤は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の制御放出製剤で既に治療されているが、それにもかかわらず突然の疼痛発作を経験し、それゆえオピオイドであるオキシコドン塩酸塩のみを含む即時放出製剤を必要とした患者における、例えば突出痛の発作を治療するのに使用することができる。
【0088】
言及したように、本発明の医薬剤形は、また、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物に関し、ここで、該組成物は、錠剤の形態で提供され、かつ該組成物は、例えば、15分後に活性物の90%超またはさらには95%超が放出されるような急速なin vitro溶出プロファイルを提供する。これらの組み合わされた特性は、組成物を、固体形態で、例えば、嚥下可能な錠剤として投与することができる利点を有する。同時に、嚥下に伴う問題を有する可能性のある子供または高齢者などの患者群は、投与に先立って錠剤を最初に液体中に急速に溶解することによって投与することでき、あるいは唾液中での急速溶解のために口中に直接的に入れることができる。これは、疼痛発作が突然に発生し、その結果、迅速な疼痛緩和が要求される場合に、重要な利便性である可能性がある。
【0089】
既に言及したように、本発明は、本質的には、少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を、好ましくは約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物に関する。
【0090】
原則として、経口即時放出医薬組成物は、オキシコドンおよびナロキソンに加えて薬学的に許容される活性薬剤を含むことができる。しかし、本発明のすべての態様の好ましい実施形態において、本発明による経口即時放出医薬組成物は、唯一の薬学的に活性な薬剤としてオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む。
【0091】
オキシコドンおよびナロキソンは、本発明の経口即時放出医薬剤形中に遊離塩基として存在することができる。
【0092】
しかし、オキシコドンまたはナロキソンは、また、それらの薬学的に許容される塩の形態で存在することができる。このような塩としては、例えば、塩酸塩、硫酸塩、重亜硫酸塩、酒石酸塩、硝酸塩、クエン酸塩、バルビツール酸塩、リン酸塩、リンゴ酸塩、マレイン酸塩、臭化水素酸塩、ヨウ化水素酸塩、フマル酸塩、コハク酸塩などが挙げられる。
【0093】
以下で言及するようなオキシコドンおよびナロキソンは、また、リチウム、ナトリウムおよびカリウムを含むアルカリ金属の金属塩などの塩基付加塩として存在することができる。それらは、また、遊離塩基の誘導体の形態で存在することができる。このような誘導体としては、例えばエステルが挙げられる。
【0094】
好ましい実施形態において、本発明は、オキシコドン塩酸塩およびナロキソン塩酸塩を使用する。本発明のさらに好ましい実施形態において、経口即時放出医薬組成物は、唯一の薬学的に活性な薬剤として、オキシコドン塩酸塩およびナロキソン塩酸塩を好ましくは約2:1の重量比で含む。
【0095】
本発明の経口即時放出医薬組成物は、患者における疼痛、とりわけ高度〜重度(慢性)の疼痛を治療するのに十分である量のオキシコドンを含む。典型的には、本発明の医薬組成物は、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩を含む。
【0096】
本発明の医薬組成物中のナロキソンの量は、投与されるナロキソンの量が、オキシコドンで仲介される疼痛緩和に実質上マイナスに影響しないように選択される。典型的には、本発明の医薬組成物は、約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。
【0097】
オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の量は、オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩の比率が、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩の重量を基準にして、重量で約2:1であるように選択することができる。
【0098】
したがって、本発明の医薬組成物は、典型的には、約1mg〜約160mgのオキシコドン塩酸塩に等価な、約1mg〜約80mgのオキシコドン塩酸塩に等価な、約1mg〜約40mgのオキシコドン塩酸塩に等価な、約1mg〜約20mgのオキシコドン塩酸塩に等価な、約1mg〜約10mgのオキシコドン塩酸塩に等価な、約1mg〜約5mgのオキシコドン塩酸塩に等価な、および約1mg〜約2.5mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩を含む。
【0099】
さらに、本発明の医薬組成物は、典型的には、約0.5mg〜約80mgのナロキソンに等価な、約0.5mg〜約40mgのナロキソン塩酸塩に等価な、約0.5mg〜約20mgのナロキソン塩酸塩に等価な、約0.5mg〜約10mgのナロキソン塩酸塩に等価な、約0.5mg〜約5mgのナロキソン塩酸塩に等価な、約0.5mg〜約2.5mgのナロキソン塩酸塩に等価な、および約0.5mg〜約1.25mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。
【0100】
本発明の好ましい実施形態は、約2.5mg〜約40mgのオキシコドン塩酸塩、好ましくは約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩、および約1.25mg〜約20mgのナロキソン塩酸塩、好ましくは約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む医薬組成物に関する。これらの組成物は、とりわけ、オキシコドンおよびナロキソンの塩酸塩を使用することができる。
【0101】
本発明によるとりわけ好ましい投与強度の経口即時放出医薬組成物は、約2.5mに等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mgに等価な量のナロキソンまたはその薬学的に許容される塩、約5mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約2.5mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、約10mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約5mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩、ならびに約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む。これらの組成物は、とりわけ、オキシコドンおよびナロキソンの塩酸塩を使用することができる。
【0102】
本発明のとりわけ好ましい実施形態は、2.5mgのオキシコドン塩酸塩および1.25mgのナロキソン塩酸塩、5mgのオキシコドン塩酸塩および2.5mgのナロキソン塩酸塩、10mgのオキシコドン塩酸塩および5mgのナロキソン塩酸塩、ならびに20mgのオキシコドン塩酸塩および10mgのナロキソン塩酸塩を含む経口即時放出医薬組成物に関する。
【0103】
オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明による経口即時放出医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、典型的には、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する。より好ましくは、本発明による医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する。
【0104】
さらにより好ましくは、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明による医薬組成物は、欧州薬局方パドル法を37℃の0.1N塩酸、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80%以上、好ましくは90%以上、さらにより好ましくは95%以上を、およびナロキソンまたはその薬学的に許容される塩の80%以上、好ましくは90%以上、さらにより好ましくは95重量%以上をin vitroで放出する。
【0105】
本発明による経口即時放出医薬組成物は、即時放出製剤にとって一般的な剤形で提供することができる。したがって、本発明による経口即時放出医薬組成物は、錠剤、カプセル剤、多粒子剤(例えば、顆粒、長円またはビーズ)などの固体形態で、あるいは液剤(例えば、溶液、懸濁液または乳液)で存在することができる。本発明の経口即時放出医薬組成物にとって好ましい剤形は錠剤である。
【0106】
本発明による経口即時放出医薬組成物を製造する場合、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩に加えて、製剤技術の分野で一般的である薬学的に許容される添加剤を使用することができる。典型的な薬学的に許容される添加剤は、崩壊剤、賦形剤、滑沢剤、流動促進剤、抗粘着剤、可塑剤、着色剤、着香剤、結合剤、pH調整剤などである。これらの添加剤(崩壊剤を除いて)は、それらが、前に記載したような即時放出のin vitro放出速度を実質上変更しないように選択すべきである。
【0107】
とりわけ本発明の医薬組成物を錠剤として提供する場合、本発明の医薬組成物が、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含むことが好ましい場合がある。また、とりわけ本発明の医薬組成物を錠剤として提供する場合、本発明の医薬組成物が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含むことが好ましい場合がある。さらに、崩壊剤および賦形剤の双方として機能を発揮する添加剤を使用することが好ましい場合がある。
【0108】
崩壊剤は、例えば、投与後の錠剤が急速に崩壊し、その結果、活性成分であるオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩が吸収のために容易に利用可能になることを確実にする。
【0109】
賦形剤は、限定はされないが、乳糖(乳糖一水和物、無水乳糖など)、デンプン(トウモロコシデンプン、α化デンプンなど)、微結晶セルロース、ブドウ糖、マンニトール、マルチトール、StarLac(登録商標)(噴霧乾燥乳糖85%、トウモロコシデンプン15%)、ショ糖、カルシウム塩(リン酸水素カルシウムなど)、またはこれらの任意の組合せから選択することができる。
【0110】
崩壊剤は、限定はされないが、とりわけ、StarLac(登録商標)(噴霧乾燥乳糖85%、トウモロコシデンプン15%)、クロスカルメロース(クロスカルメロースナトリウムなど)、デンプングリコール酸ナトリウム、クロスポビドン、アルギン酸、または低置換ヒドロキシプロピルセルロースから選択することができる。
【0111】
StarLac(登録商標)製品のような乳糖とデンプンとの組合せは、それが充填剤および崩壊剤の特性を兼ね備えているのでとりわけ好ましい可能性がある。
【0112】
流動促進剤および滑沢剤は、限定はされないが、とりわけ、高分散性シリカ、タルク、酸化マグネシウム、ステアリン酸マグネシウム、フマル酸ステアリルナトリウムなどから選択することができる。
【0113】
流動化剤および滑沢剤は、とりわけ、高分散性シリカ、タルク、酸化マグネシウム、ステアリン酸マグネシウム、フマル酸ステアリルナトリウムなどを含む。
【0114】
本発明の医薬組成物を錠剤として提供する場合、それらの錠剤を、区別の目的で、化粧コーティングを用いて被覆することができる。このようなコーティングは、本発明による医薬組成物の即時放出特性に実質的な影響を有さない。
【0115】
好ましい実施形態において、本発明は、約2.5mg〜約160mgのオキシコドン塩酸塩および約1.25mg〜約80mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0116】
別の好ましい実施形態において、本発明は、約2.5mg〜約80mgのオキシコドン塩酸塩および約1.25mg〜約40mgのナロキソン塩酸塩を2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0117】
別の好ましい実施形態において、本発明は、約2.5mg〜約40mgのオキシコドン塩酸塩および約1.25mg〜約20mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0118】
別の好ましい実施形態において、本発明は、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0119】
さらに別の好ましい実施形態において、本発明は、約2.5mg〜約10mgのオキシコドン塩酸塩および約1.25mg〜約5mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含む経口即時放出医薬組成物に関し、ここで、該製剤は、欧州薬局方パドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の90重量%以上、好ましくは95重量%以上を、およびナロキソン塩酸塩の90重量%以上、好ましくは95重量%以上をin vitroで放出する。好ましくは、このような組成物は、崩壊剤を含み、かつ錠剤などの固体形態をとる。
【0120】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、健常ヒト志願者において単回投与で投与された場合に、約15ng.h/mL〜約500ng.h/mLの範囲の、好ましくは約20ng.h/mL〜約400ng.h/mLの範囲の、より好ましくは約25ng.h/mL〜約300ng.h/mLの範囲の、さらにより好ましくは約30ng.h/mL〜約250ng.h/mLの範囲のオキシコドンのAUCtを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0121】
約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を約2:1の重量比で含む本発明の経口即時放出医薬組成物の中で、健常ヒト志願者において単回投与治験で投与された場合に、約1ng/mL〜約300ng/mLの範囲の、好ましくは約2ng/mL〜約200ng/mLの範囲の、より好ましくは約3ng/mL〜約100ng/mLの範囲の、さらにより好ましくは約4ng/mL〜約75ng/mLの範囲の、最も好ましくは約6ng/mL〜約50ng/mLの範囲のオキシコドンのCmaxを提供するものが好ましい可能性がある。このような経口即時放出医薬組成物は、好ましくは、オキシコドンおよびナロキソンの塩酸塩を含むことができる。
【0122】
本発明の実施形態は、また、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物であって、ストレス条件下で貯蔵した後の該医薬組成物が、薬学的に活性な薬剤を、該医薬組成物をストレス条件にさらす前と実質的に同じ放出速度で放出する経口即時放出医薬組成物に関する。
【0123】
本発明の他の実施形態は、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む経口即時放出医薬組成物であって、ストレス条件下で貯蔵した後の該医薬組成物が、オキシコドンまたはその薬学的に許容される塩に関連するおよび/またはナロキソンまたはその薬学的に許容される塩に関連する全部で3.0%未満の物質を有する経口即時放出医薬組成物に関する。
【0124】
本発明の文脈中のストレス条件下での貯蔵は、医薬組成物を、高められた温度および/または相対湿度(RH)に長時間さらすことを意味する。例えば、典型的なストレス条件は、25℃および60%RHでの少なくとも1、2、3、4、5、6、9または12ヶ月間にわたる貯蔵を指す。別のストレス条件は、40℃および75%RHでの少なくとも1、2、3、4、または5ヶ月間にわたる貯蔵を指す。
【0125】
このようなストレス貯蔵条件は、医薬組成物が、その安全性および有効性に対するマイナスの影響なしに患者の家庭内で一般的である条件下で長期間十分な貯蔵寿命を有するかどうかを判定するために使用される。このようなマイナスの影響としては、in vitroでの放出速度が、長い間に変化し、その結果、投与後に異なる量の活性物が放出されるので、組成物の有効性が影響を受けることが挙げられる。同様に、マイナスの影響は、また、機能を果たす薬学的に活性な薬剤の全体量を減少させるか、または毒性副生物の形成につながる可能性のある、薬学的に活性な薬剤の分解に由来する可能性がある。
【0126】
in vitro放出プロファイルにおける、または医薬組成物中の活性薬剤(群)の量に関する変化がストレス条件下での貯蔵後に観察される場合、このことは、安定性の問題を暗示している可能性がある。このような変化が観察されない場合、このことは、逆に、医薬組成物が、貯蔵上安定であることを意味する。
【0127】
用語「実質上同じ放出速度」は、ストレス条件にさらされた医薬組成物についてのin vitro放出速度が、参照組成物と同等である状況を指す。参照組成物は、全く同じ医薬組成物ではあるが、ストレス条件にさらされていない。ストレス条件にさらされた組成物のin vitro放出プロファイルが、参照組成物のin vitro放出プロファイルから、約20%超、好ましくは約15%以下、より好ましくは約10%以下、さらにより好ましくは約5%以下の逸脱をしない場合、そのin vitro放出速度は、実質的に同一であると見なされる。
【0128】
用語「すべてのオキシコドンおよびナロキソン類縁物質」は、これらの薬学的に活性な薬剤の化学反応から生じる物質を指す。これらには、例えば、ナロキソンn−オキシドなどが含まれる。
【0129】
安定性を評価するために、医薬組成物を前述のようなストレス条件にさらし、オキシコドンおよび/またはナロキソン類縁物質の量を測定することができる。次いで、ストレス条件にさらされなかった全く同一の医薬組成物についてオキシコドンおよび/またはナロキソン類縁物質の量を測定する。この組成物は、参照組成物であると見なされる。「オキシコドン類縁物質」および「ナロキソン類縁物質」の検出は、典型的には、HPLC分析で実施される。物質の同定は、既知の純粋な参照物質を用いて同様の分析を行うことによって判定することができる。
【0130】
医薬組成物は、それをストレス条件にさらした後に、それが、組成物内に存在するヒドロモルホンまたはナロキソンの量と比較して、約3%以下の、好ましくは約2%以下の、より好ましくは約1%以下の、さらにより好ましくは0.5%以下のオキシコドンおよび/またはナロキソン類縁物質を有する場合、安定であると見なされる。
【0131】
ストレス条件下での貯蔵後に実質上同一の放出速度を有する、および/またはストレス条件下での貯蔵後に3%未満のオキシコドンおよびナロキソン類縁物質を有する組成物の特性は、とりわけ組成物が錠剤などの固体形態をとる場合、2:1の重量比を有する組成物、前述の量を有する組成物および/または前述のin vitro放出プロファイルを有する組成物などの、前に述べた実施形態にも関係すると理解されたい。
【0132】
固体形態で、例えば錠剤の形態で提供される本発明による医薬組成物は、例えば、次のステップ:
a)双方とも適切な粒径のオキシコドンまたはその薬学的に許容される塩、ナロキソンまたはその薬学的に許容される塩、賦形剤、および任意選択で崩壊剤をブレンドするステップ、
b)任意選択で、前記ブレンド物を滑沢化するステップ、
c)前記ブレンド物を直接的に圧縮して錠剤を得るステップ、
を含む方法によって製造することができる。
【0133】
固体形態で、例えば錠剤の形態で提供される本発明による医薬組成物は、例えば、次のステップ:
a)双方とも適切な粒径のオキシコドンまたはその薬学的に許容される塩、ナロキソンまたはその薬学的に許容される塩、崩壊剤、および任意選択で賦形剤をブレンドするステップ、
b)任意選択で、前記ブレンド物を滑沢化するステップ、
c)前記ブレンド物を直接的に圧縮して錠剤を得るステップ、
を含む方法によって製造することができる。
【0134】
必要と考えられる場合、上記方法は、均一なブレンド化のため塊のない成分を確保にするために、種々の機会での篩い分けステップを含むことができる。
【0135】
また、好ましくは、すべての添加剤および任意選択で活性成分は、同一の大きさの範囲内にある。典型的な大きさの範囲は、約100μm〜約300μmである。
【0136】
このような手順によって得られる錠剤は、急速に崩壊し、前に概略を述べたような欧州薬局方パドル法で測定した場合、15分の時点でオキシコドンまたはその薬学的に許容される塩の90%以上、好ましくは95%以上を、およびナロキソンまたはその薬学的に許容される塩の90%以上、好ましくは95%以上をin vitroで放出することが見出された。実施例4から理解できるように、このような手順により製造された錠剤は、貯蔵安定性があり、それらは、ストレス条件下での長期貯蔵後にin vitroでのそれらの放出挙動を実質的に変化させないことを意味する。
【0137】
前記のようなおよび前に製造されたような即時放出医薬組成物は、疼痛、とりわけ中等度から重度(慢性)疼痛に苦しむ患者の用量設定するのに使用することができる。このような製剤は、また、疼痛、とりわけ中等度から重度(慢性)疼痛に苦しむ患者における突出痛を治療するのに使用することができる。
【0138】
患者の用量設定のための、および患者における突出痛を治療するための経口即時放出医薬組成物の使用は、便秘および尿滞留などの典型的なオピオイド誘発性副作用に苦しむことなく、即時放出特性の結果として患者における迅速な疼痛緩和を確実にする。
【0139】
本発明を、一部の具体的実施例に関して以下で説明する。しかし、これらの実施例を限定と解釈すべきでない。
(実施例)
【実施例1】
【0140】
オキシコドン塩酸塩およびナロキソン塩酸塩を含む即時放出医薬組成物の調製
オキシコドン塩酸塩20mg/ナロキソン塩酸塩10mg(IR−OXN20/10)、オキシコドン塩酸塩10mg/ナロキソン塩酸塩5mg(IR−OXN10/5)、オキシコドン塩酸塩5mg/ナロキソン塩酸塩2.5mg(IR−OXN5/2.5)、およびオキシコドン塩酸塩2.5mg/ナロキソン塩酸塩1.25mg(IR−OX2.5/1.25)を含む即時放出医薬組成物を、下記の通り製造した。それらの組成を表1に詳述する。
【0141】
【表1】
【0142】
【表2】
【0143】
【表3】
【0144】
【表4】
【0145】
【表5】
【0146】
表1〜5の成分を、図1の流れ図に記載のように処理した。
【0147】
詳細には、微結晶セルロース、クロスカルメロースナトリウム、および活性成分を、500μmの篩いを通して篩い分けて任意の凝集物を除去し、StarLac(登録商標)と共にブレンダーに仕込み、均一なブレンドが達成されるまでブレンドした。StarLac(登録商標)は自由流動性であるので、その篩い分けは必要なかった。滑沢剤であるフマル酸ステアリルナトリウムを、500μmの篩いを通して篩い分け、ブレンド物に添加し、さらにブレンドした。次いで、ブレンド物を、直接圧縮によって圧縮して錠剤とした。許容されるブレンド物の均一性を確保するために、粉砕級のナロキソン塩酸塩を利用してオキシコドン塩酸塩およびその他の添加剤と類似の粒径範囲を得た。
【0148】
異なる強度の製品間の区別を提供するために、着色された化粧フィルムコートを塗布した。フィルムコーティングの条件は、適切な審美的品質の被覆錠剤を一貫して生じるように最適化した。
【0149】
次いで、IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5、およびIR−OXN2.5/1.25錠剤を、欧州薬局方のパドル試験によって試験した。
【0150】
すべての場合において、15分の時点でオキシコドン塩酸塩およびナロキソン塩酸塩の95%超が放出された。
【実施例2】
【0151】
健常対象におけるIR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5、およびIR−OXN2.5/1.25錠剤の用量比例性を比較するための単回投与治験
1.目的
目的は、2.5/1.25mg、5/2.5mg、10/5mg、および20/10mgの強度のIR−OXN錠剤から、オキシコドンおよびナロキソン(または、代用物であるナロキソン−3−グルクロニド)の用量比例性を評価することとした。
【0152】
2.試験集団
18名の対象が治験を完了し、有効な薬物動態データを提供することを目標に、治験薬剤を投与される全部で21名の健常成人、男性および女性対象を無作為化するように計画した。全部で21名の対象を、実際に登録し、無作為化し、20名の対象が治験を完了した。
【0153】
<組入れ基準>
治験に組み入れ予定の対象は、次の基準のすべてに合致する者とした:
1.年齢が18〜55歳(両端を含む)の男性または女性対象。
2.治験中に性的に活発であった、または性的に活発になった女性対象は、治験中を通して高度に有効な避妊法を進んで使用したにちがいない者。高度に有効な受胎調節法は、不妊手術、インプラント、注射剤、併用経口避妊薬、一部の子宮内デバイス、または配偶者の精管切除などの、一貫してかつ正しく使用された場合に低い失敗率(すなわち、1年に1%未満)をもたらすものと定義した。
3.閉経1年未満の女性対象は、血清妊娠検査で陰性であり、かつ非授乳であったにちがいない者。
4.閉経後1年間を超え、かつ高めたれた血清卵胞刺激ホルモン(FSH)を有するか、またはホルモン補充療法(HRT)で治療された女性対象。
5.男性対象は、彼らの配偶者との避妊を治験中および治験完了後の30日間進んで使用したにちがいなく、かつ彼らの配偶者がこの期間に妊娠した場合治験責任医師に伝えることに同意した者。
6.55〜100kgの範囲の体重、および18以上かつ29以下の体型指数(BMI)。
7.健康で、かつ病歴、身体的検査、バイタルサイン、臨床検査、および心電図(ECG)によって判定されるような有意な異常所見がない。
8.治験中に供給されるすべての食物を進んで食すること。
9.対象のプライマリーケア医師が、事前の12ヶ月以内に、対象の病歴中に彼らの臨床試験への登録の除外となるものが存在しないことを確認した者。
【0154】
<除外基準>
治験から除外すべき対象は、次の基準のいずれかに合致する者とした:
1.薬物またはアルコール乱用の任意の既往歴。
2.薬物の吸収、分布、代謝または排泄を妨害する可能性のある状態の任意の既往歴。
3.事前の30日間でのオピオイドまたはオピオイド拮抗薬を含有する薬剤の使用。
4.病因にかかわらず、頻回の悪心または嘔吐の任意の既往歴。
5.発作または症候性頭部外傷の任意の既往歴。
6.この治験の初回投与に先立つ90日間中の臨床薬物治験への参加。
7.この治験への登録に先立つ4週間中の任意の重大な疾患。
8.初回投与に先立つ7日間中、またはこの治験過程中のビタミン、生薬および/またはミネラル補助食品を含む任意の薬剤の使用(HRTおよび避妊薬の連続的使用を除いて)。
9.治験薬物の投与に先立つ8時間中、または投与に続く4時間中に食物を控えること、および各拘束期間中完全にカフェインまたはキサンチン含有飲料を控えることの拒絶。
10.女性では14単位/週、男性では21単位/週の等価量を超える毎週のアルコール摂取。
11.治験薬物投与前48時間以内のアルコール飲料の消費、および治験薬物投与後の少なくとも48時間アルコールを控えることの拒絶。
12.治験薬物投与の45日以内の喫煙暦、および治験中の禁煙の拒絶。
13.プロトコールによって要求された場合を除く、治験薬物投与に先立つ30日以内のまたは治験中のいずれかに時点での血液または血液産物の提供。
14.尿中薬物スクリーニング、アルコール検査、B型肝炎表面抗原(HBsAg)、C型肝炎抗体、またはヒト免疫不全ウイルス(HIV)検査の陽性結果。
15.オキシコドン、ナロキソン、ナルトレキソンまたは関連化合物に対する既知の感受性。
16.対象のプライマリーケア医師に報告することを承認することへの拒絶。
【0155】
対象集団の人口統計学的データを表6に示す。治験集団は、男性14名および女性7名の平均年齢が31歳(範囲:21〜53歳)の対象から構成した。20名の対象は白人であり、1名の対象は黒人であった。
【0156】
【表6】
【0157】
3.治験デザイン
治験は、単一治験センターで実施される健常な男性および女性対象における、オープンラベル、単回投与、4治療薬、4期間、無作為化クロスオーバー治験とした。各対象は、2.5/1.25mg、5/2.5mg、10/5mg、および20/10mgの強度のIR−OXN錠剤の単回投与を受けた。対象は、無作為割付スケジュール(RAS)により、各投与の間に少なくとも7日間のウォッシュアウト期間を伴う4治療薬のそれぞれを投与された。
【0158】
対象は、治験期間1の初回投与日の21日以内にスクリーニングのために来院した。次いで、適格対象は、各治験期間における投与の前日に治験施設にチェックインした。翌朝、対象に、絶食状態で適切な治験薬物を投与した。
【0159】
薬物動態用血液検体を、各治験期間における治験薬物投与後の36時間の間に採取し、36時間の時点の血液検体採取後に対象を退院させた。
【0160】
各治験期間中、バイタルサイン(脈拍、血圧、および呼吸数)を、投与前、次いで投与後の1、2、4、6、8、12、24および36時間の時点で測定した。口腔温を、投与前、ならびに投与後の24および36時間の時点で測定した。治験中、有害事象を記録した。対象は、治験の完了/中止の場合、彼らの治験薬剤の最終投与の7〜10日後に治験後来院した。
【0161】
治験デザインを図2に示す。図2中の略語は次の通りである:
R=無作為化
P1、P2、P3、P4=それぞれ、RASによる治験薬物の単回投与、それに続く血液サンプリング、および投与後36時間までの安全性評価からなる期間1〜4。各治験期間の間には、治験薬物の投与の間に少なくとも7日間のウォッシュアウトを存在させた。
PS med=治験の完了/中止の場合における、治験薬物の最終投与の7〜10日後の治験後健診
V1〜V6=来院
【0162】
4.薬物動態用検体の採集
1、8、15および22日目に開始して、一連の血液採集物(それぞれ6mL)を次の時点で得た:
投与前、投与後0.25、0.5、0.75、1、1.25、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1つの投与期間につき22の血液検体)。
指定の血液サンプリング時間からの許容される時間枠は±5分とした。
薬物動態測定のため、ほぼ528mLの血液(6mLの検体22個を4件)を各対象から採取した。
【0163】
5.投与治療薬
治療薬は、絶食状態において次の通り経口投与した:
A:IR OXN2.5/1.25mg錠剤を1錠
B:IR OXN5/2.5mg錠剤を1錠
C:IR OXN10/5mg錠剤を1錠
D:IR OXN20/10mg錠剤を1錠。
【0164】
オピオイド関連有害事象を低減するため、ナルトレキソンHClを、すべての治療薬(A〜D)と共に経口投与した。ナルトレキソンHCl50mg錠剤(Nalorex(登録商標)錠、Bristol−Myers Squibb Pharmaceuticals Ltd)。各治験期間において、治験薬物投与の−13時間、−1時間、および+11時間の時点で1錠のナルトレキソン錠剤を投与した。
【0165】
6.薬物動態パラメーター
次の薬物動態パラメーターを、オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、ナロキソン、6β−ナロキソール、ナロキソン−3−グルクロニド、および6β−ナロキソール−3−グルクロニドの血漿中濃度から計算した:
・投与時から測定可能な最終濃度まで測定された血漿中濃度−時間曲線下面積(AUCt)
・投与時から測定され無限大まで外挿された血漿中濃度−時間曲線下面積(AUCinf)
・観察される最大血漿中濃度(Cmax)
・観察される最大血漿中濃度到達時間(tmax)
・終末相速度定数(λZ)
・終末相半減期(t1/2Z)。
【0166】
オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、およびナロキソン−3−グルクロニドに関するAUC値はng.h/mLで、Cmax値はng/mLで報告した。ナロキソン、6β−ナロキソール、および6β−ナロキソール−3−グルクロニドに関して、AUC値はpg.h/mLで、Cmax値はpg/mLで報告した。
【0167】
AUCt値は、線形台形法を使用して計算した。可能な場合、λZを、終末対数−線形相にあると判定されるそれらの箇所を使用して推定した。t1/2Zは、In2のλZに対する比率から決定した。最終測定点と無限大との間の血漿中濃度−時間曲線下面積は、観察される最終血漿中濃度(Clast)のλZに対する比率から計算した。次いで、これをAUCtに加算してAUCINFを得た。
【0168】
すべての薬物動態計算は、WinNonlin Enterprise Edition、version 4.1を用いて実施した。
【0169】
とりわけ低用量で、ナロキソンの血漿中濃度は極度に低く、薬物動態の完全な特徴づけはできなかった。後の結果および考察の部では、その主要代謝産物であるナロキソン−3−グルクロニドにより大きな強調が置かれる。
【0170】
治験の主要目的は、2.1/1.25mg、5/2.5mg、10/5mgおよび20/10mgの強度のIR OXN錠剤からオキシコドンおよびナロキソン(または代用物ナロキソン−3−グルクロニド)の用量比例性を判定することとした。
【0171】
用量調整された相対的な全身性バイオアベイラビリティーを、AUCt、および可能な場合AUCINFの比率から計算した。オキシコドン、ナロキソンおよびそれらの代謝産物に関する重要な比較は、
・OXN IR5/2.5mg錠剤対OXN IR2.5/1.25mg錠剤
・OXN IR10/5mg錠剤対OXN IR2.5/1.25mg錠剤
・OXN IR20/10mg錠剤対OXN IR2.5/1.25mg錠剤、とした。
【0172】
観察される最大血漿中濃度(Cmax)およびCmaxが生じる時刻(tmax)は、報告された血漿中濃度−時間データから直接に得られた。用量調整されたCmax比を、上に概略を示した比較を行って計算した。
【0173】
7.結果
オキシコドン、ナロキソンおよびナロキソン−3−グルクロニドの薬物動態パラメーターに関する結果をそれぞれ図3〜5に、解析された21名の対象について示す。
【0174】
<オキシコドン>
オキシコドンに関する要約統計量を表7に示す。
【0175】
【表7】
【0176】
すべてのIR OXN錠剤強度で、tmaxおよびt1/2Zパラメーターについて類似の値が記録された。
【0177】
IR OXN錠剤強度のそれぞれは、AUCt、AUCINFおよびCmaxに関して、オキシコドンのIR OXN2.5/1.25mg錠剤に対して同等の用量調整バイオアベイラビリティーを提供し、比較比率のそれぞれに関する90%信頼区間は、生物学的同等性に対する80〜125%の許容性限界内に含まれた。オキシコドンの薬物動態パラメーターに関する統計解析の結果を表8に示す。
【0178】
【表8】
【0179】
<ナロキソン−3−グルクロニド>
ナロキソン−3−グルクロニドの薬物動態パラメーターを、ナロキソンの代用物として分析した。
【0180】
ナロキソン−3−グルクロニドに関する要約統計量を表9に示す。
【0181】
【表9】
【0182】
すべてのIR OXN錠剤強度で、tmaxについては類似の値が記録された。平均のt1/2Z値は、IR OXN5/2.5mg錠剤強度の8.88時間からIR OXN20/10mg錠剤強度の15.68時間までの範囲にわたった。
【0183】
IR OXN錠剤強度のそれぞれは、AUCt、AUCINFおよびCmaxに関してナロキソン−3−グルクロニドのIR OXN2.5/1.25mg錠剤に対して同等の用量調整バイオアベイラビリティーを提供し、比較比率のそれぞれに関する90%信頼区間は、131.9%であるIR OXN5/2.5mg対IR OXN2.5/1.25mgに関するCmax比率に関する上側90%信頼区間を除いて、生物学的同等性に対する80〜125%の許容性限界内に含まれた。ナロキソン−3−グルクロニドの薬物動態パラメーターに関する統計解析の結果を表10に示す。
【0184】
【表10】
【0185】
8.結論
用量調整した生物学的同等性が、オキシコドンおよびナロキソン−3−グルクロニドの主要分析対象物に関して、IROXN2.5/1.25mg錠剤に対してそれぞれのOXN IR錠剤強度について達成された。用量比例性は、2.5/1.25mg〜20/10mgの範囲の強度のIR OXN錠剤強度について確認された。ナロキソンの血漿中濃度は、とりわけ低用量で極度に低く、前の治験結果を追認し、代用物ナロキソン−3−グルクロニドの分析を支持した。
【実施例3】
【0186】
IR−OXN20/10錠剤のバイオアベイラビリティーをオキシコドン/ナロキソン20/10mg長期放出錠剤(Targin(登録商標)と比較するための、健常対象における単回投与治験
1.目的
この治験の目的は、絶食状態で健常対象に投与した場合の、OXN IR20/10mg錠剤、OXN PR20/10mg錠剤、およびオキシコドンIR20mgカプセル剤の薬物動態、バイオアベイラビリティー、および安全性を評価することとした。
【0187】
2.試験集団
18名の対象が治験を完了し、有効な薬物動態データを提供することを目標に、治験薬剤を投与される全部で21名の健常成人、男性および女性対象を無作為化するように計画した。全部で22名の対象を、実際に登録し、無作為化し、21名の対象が治験を完了した。1名の対象は、治験薬剤(すなわち、OXNまたはオキシコドン)の投与前に取りやめた。
【0188】
<組入れ基準>
治験に組み入れ予定の対象は、次の基準のすべてに合致する者とした:
1.年齢が18〜55歳(両端を含む)の男性または女性対象。
2.治験中に性的に活発であった、または性的に活発になった女性対象は、治験中を通して高度に有効な避妊法を進んで使用したにちがいない者。高度に有効な受胎調節法は、不妊手術、インプラント、注射剤、併用経口避妊薬、一部の子宮内デバイス、または配偶者の精管切除などの、一貫してかつ正しく使用された場合に低い失敗率(すなわち、1年に1%未満)をもたらすものと定義した。
3.閉経1年未満の女性対象は、血清妊娠検査で陰性であり、かつ非授乳であったにちがいない者。
4.閉経後1年間を超え、かつ高めたれた血清卵胞刺激ホルモン(FSH)を有するか、またはホルモン補充療法(HRT)で治療された女性対象。
5.55〜100kgの範囲の体重、および18以上かつ29以下の体型指数(BMI)。
6.健康で、かつ病歴、身体的検査、バイタルサイン、臨床検査、および心電図(ECG)によって判定されるような有意な異常所見がない。
7.治験中に供給されるすべての食物を進んで食すること。
8.対象のプライマリーケア医師が、事前の12ヶ月以内に、対象が臨床試験へ参加するのに適していることを確認した者。
【0189】
<除外基準>
治験から除外すべき対象は、次の基準のいずれかに合致する者とした:
1.薬物またはアルコール乱用の任意の既往歴。
2.薬物の吸収、分布、代謝または排泄を妨害する可能性のある状態の任意の既往歴。
3.事前の30日間でのオピオイドまたはオピオイド拮抗薬を含有する薬剤の使用。
4.病因にかかわらず、頻回の悪心または嘔吐の任意の既往歴。
5.発作または症候性頭部外傷の任意の既往歴。
6.この治験の初回投与に先立つ90日間中の臨床薬物治験への参加。
7.この治験への登録に先立つ4週間中の任意の重大な疾患。
8.初回投与に先立つ7日間中、またはこの治験過程中のビタミン、生薬および/またはミネラル補助食品を含む任意の薬剤の使用(HRTおよび避妊薬の連続的使用を除いて)。
9.治験薬物の投与に先立つ8時間中、または投与に続く4時間中に食物を控えること、および各拘束期間中完全にカフェインまたはキサンチン含有飲料を控えることの拒絶。
10.女性では14単位/週、男性では21単位/週の等価量を超える毎週のアルコール摂取。
11.治験薬物投与前48時間以内のアルコール飲料の消費、および治験薬物投与後の少なくとも48時間アルコールを控えることの拒絶。
12.治験薬物投与の45日以内の喫煙暦、および治験中の禁煙の拒絶。
13.プロトコールによって要求された場合を除く、治験薬物投与に先立つ30日以内または治験中の任意いずれかの時点での血液または血液産物の提供。
14.尿中薬物スクリーニング、アルコール検査、B型肝炎表面抗原(HBsAg)、C型肝炎抗体、またはヒト免疫不全ウイルス(HIV)検査の陽性結果。
15.オキシコドン、ナロキソン、ナルトレキソンまたは関連化合物に対する既知の感受性。
16.対象のプライマリーケア医師に報告することを承認することへの拒絶。
【0190】
対象集団の人口統計学的データを表11に示す。治験集団は、男性11名および女性10名の平均年齢が33歳(範囲:20〜52歳)の対象から構成した。すべての対象が白人であった。
【0191】
【表11】
【0192】
3.治験デザイン
治験は、オープンラベル、単回投与、3治療薬、3期間、無作為化クロスオーバーとした。各対象は、IROXN20/10mg錠剤、オキシコIR20mgカプセル剤、およびOXN PR20/10mg(Targin(登録商標))の単回投与を受けた。対象は、無作為割付スケジュール(RAS)により、各投与の間に少なくとも7日間のウォッシュアウト期間を伴う3治療薬のそれぞれを投与された。
【0193】
対象は、治療期間1の初回投与日の21日間以内にスクリーニングのために来院した。次いで、適格対象は、各治験期間における投与の前日に治験施設にチェックインした。翌朝、対象に絶食状態で適切な治験薬物を投与した。
【0194】
薬物動態用血液検体を、各治験期間における治験薬物投与後の36時間の間に採取し、36時間の時点の血液検体採取後に対象を退院させた。
【0195】
各治験期間中、バイタルサイン(脈拍、血圧、および呼吸数)を、投与前、次いで投与後の1、2、4、6、8、12、24および36時間の時点で測定した。口腔温を、投与前、ならびに投与後の24および36時間の時点で測定した。治験中、有害事象を通して記録した。
【0196】
対象は、治験の完了/中止の場合、彼らの治験薬剤の最終投与の4〜7日後に治験後来院した。
【0197】
治験デザインを6に示す。図6中の略語は次の通りである:
R=無作為化
P1、P2、P3=それぞれ、治験薬物の単回投与、それに続く血液サンプリング、および36時間までの安全性評価からなる期間1〜3。各治験期間の治験薬物投与の間に少なくとも7日間のウォッシュアウトを存在させた。
PS med=治験の完了/中止の場合、最終治験薬物投与の7〜10日後の治験後健診。治験中止以前だけナルトレキソンを投与された対象は、施設からの退院に先立って治験後健診を受けた。
V1〜V5=来院
【0198】
4.薬物動態用検体の採集
1、8および15日目に開始して、次の時点での一連の血液採集物(それぞれ6mL)を得た:
治療薬AおよびB:
投与前、投与後0.25、0.5、0.75、1、1.25、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1投与期間につき22個の血液検体)。
治療薬C:
投与前、投与後0.5、1、1.5、2、2.5、3、3.5、4、5、6、8、10、12、16、24、28、32および36時間(1投与期間につき19個の血液検体)。
対象が、OXN PR20/10mg錠剤の投与後12時間以内に、あるいはOXN IR20/10mgまたはオキシコドンIR20mgカプセル剤の投与後6時間以内に嘔吐を経験した場合、その対象の残りの治験期間について、さらなる薬学動態用血液検体を採取しなかった。
指定の血液サンプリング時間からの許容される時間枠は±5分とした。
薬物動態測定のため、ほぼ378mLの血液(6mLの検体22個を2件および6mLの検体19個を1件)を各対象から採取した。
【0199】
5.投与治療薬
この治験で投与される治療薬を以下に示す:
試験治療薬:
IR OXN20/10mg錠剤。該治療薬は、絶食状態において次の通り経口投与した:
治療薬A:OXN IR20/10mg錠剤 1錠。
参照治療薬:
IRオキシコドン20mgカプセル剤(OxyNorm(登録商標)Napp Pharmaceuticals Ltd、英国)。該治療薬は、絶食状態において次の通り経口投与した:
治療薬B:オキシコドンIR20mgカプセル剤 1カプセル
OXN PR20/10mg錠剤(Targin(登録商標))。英国Bard Pharmaceuticals Ltd製造。該治療薬は、絶食状態において次の通り経口投与した:
治療薬C:OXN PR20/10mg錠剤 1錠。
【0200】
治験薬物の投与は、各治験期間においてオピオイド関連有害事象の危険を低減するために、ナルトレキソンでの援護下に行われた。ナルトレキソン塩酸塩50mg錠剤(Nalorex(登録商標)錠、Bristol−Myers Squibb Pharmaceuticals Ltd)を次の通り経口投与した:
ナルトレキソン50mg錠剤を1錠、治験薬物投与の13時間前(−1、7、14日目)、1時間前および11時間後(1、8および15日目)の時点で、すべて100mLの水で嚥下した(全部で3回投与)。
【0201】
6.薬物動態パラメーター
血漿中濃度データおよび薬物動態パラメーターを、登録された集団中の対象について列挙した。
【0202】
各分析対象物についての血漿中濃度データを、登録された集団中の対象について時点および治療薬によって記述的に要約した。また、各分析対象物についての個別および平均の血漿中濃度を、各治療薬について時間とともにプロットした。
【0203】
各分析対象物についての薬物動態パラメーター(AUCt、AUCINF、Cmax、tmax、λZ、およびt1/2Z)を、薬物動態パラメーターのための完全分析集団中のすべての対象について治療薬によって記述的に要約した。有効な薬物動態パラメーターを得るために、対象は、OXN PR20/10錠剤の投与後12時間以内の、またはOXN IR20/10錠剤もしくはオキシコドンIR20mgカプセル剤の投与後6時間以内の嘔吐を経験してはならない。
【0204】
分析対象物オキシコドン、ノルオキシコドン、オキシモルホン、ノルオキシモルホン、ナロキソン、6β−ナロキソール、ナロキソン−3−グルクロニド、および6β−ナロキソール−3−グルクロニドに関する対数変換データ、ならびに薬物動態パラメーターAUCt、AUCINF、およびCmaxを、治療薬および期間については固定項、対象については無作為効果で、混合効果線形モデル18を使用して解析した。
【0205】
治療薬の比率/差異およびそれらに付随する90%信頼区間を、最小二乗平均から計算した。
【0206】
比較予定の治療薬は次の通りである:
・IR OXN20/10mg対オキシコドンIR20mg
・IR OXN20/10mg対OXN PR20/10mg
【0207】
代謝産物:親薬物の比率は、AUCt値、および可能な場合AUCINF値を使用して各治療薬に関して計算した。
【0208】
すべての薬物動態の計算は、WinNonlin Enterprise Edition、version 4.1.を使用して行った。
【0209】
7.結果:
オキシコドン、ナロキソン、およびナロキソン−3−グルクロニドの薬物動態パラメーターに関する結果を、それぞれ図7〜9に分析された対象について示す。
【0210】
<オキシコドン>
オキシコドンに関する要約統計量を表12に示す。
【0211】
【表12】
【0212】
OXN PR20/10mg錠剤と比較して、IR OXN IR20/10mg錠剤は、90%信頼区間で、生物学的同等性の基準に合致する96.3%のオキシコドンの平均経口アベイラビリティーを有した。即時放出および長期放出製剤を比較すると、OXN IR20/10mg錠剤のCmaxは、OXN PR20/10mg錠剤に比べて有意により高かった。
【0213】
オキシコドンIRカプセル剤と比較して、IR OXN20/10は、99.2%のオキシコドンの平均経口アベイラビリティー、および100%の平均Cmax比率を有した。これらの比較に関する生物学的同等性の評価は、すべて、生物学的同等性に関する判定基準に合致した90%信頼区間を有した。
【0214】
オキシコドンに関する平均半減期の値は、すべての治療薬にわたって類似し、4.2〜4.3時間の範囲にあった。
【0215】
IR OXN20/10およびオキシコドンIR20mgカプセル剤は、類似のtmax中央値を有した。OXN PR20/10は、他の治療薬に比べてより遅いtmaxを有した。
【0216】
オキシコドンの薬物動態パラメーターの統計解析結果を表13に示す。
【0217】
【表13】
【0218】
<ナロキソン−3−グルクロニド>
ナロキソン−3−グルクロニドに関する要約統計量を表14に示す。
【0219】
【表14】
【0220】
OXN PR20/10錠剤と比較して、IR OXN20/10は、90%信頼区間で、生物学的同等性の基準に合致する90.4%のナロキソン−3−グルクロニドの平均経口アベイラビリティーを有した。即時放出および長期放出製剤を比較すると、IR OXN20/10に関するCmaxは、OXN PR20/10錠剤に比べて有意により高かった。
【0221】
平均半減期の値は、IR OXN(8.7時間)およびOXN PR(9.1時間)錠剤の間で類似していた。
【0222】
OXN PR20/10錠剤に関するtmax値は、IR OXN20/10に比べてより遅かった。
【0223】
ナロキソン−3−グルクロニドの薬物動態パラメーターに関する統計解析結果を表15に示す。
【0224】
【表15】
【0225】
8.結論:
薬物動態パラメーターに関する結果は、IR OXN20/10mgおよびOXN PR20/10mg錠剤から、オキシコドンおよびナロキソン(または代用物ナロキソン−3−グルクロニド)の十分に同等なバイオアベイラビリティーを、かつオキシコドンに関するIR OXN20/10mgおよびオキシコドンIR20mgカプセル剤の生物学的同等性を示した。ナロキソンは、予想したように、極めて低いバイオアベイラビリティーを有し、以前の治験結果を確証し、代用物ナロキソン−3−グルクロニドの分析を支持していた。
実験4
【0226】
IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤の貯蔵安定性
IR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤を、PVC/PVdCアルミニウムホイルのブリスターパック中に25℃/60%RTおよび40℃/75%RHで貯蔵した。
【0227】
安定性データは、25℃/60%RTおよび40℃/75%RHで3ヶ月間貯蔵した後のIR−OXN20/10、IR−OXN10/5、IR−OXN5/2.5およびIR−OXN2.5/1.25錠剤に関して準備する。
【0228】
下記に示すデータは、物理的特性および溶出に関するオキシコドン塩酸塩/ナロキソン塩酸塩の即時放出錠剤の安定性を確認する。
【0229】
【表16】
【0230】
【表17】
【0231】
【表18】
【0232】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ月間の貯蔵後に最大で約0.5%まで変化した。
【0233】
【表19】
【0234】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で約2.3%まで変化した。
【0235】
【表20】
【0236】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ月の貯蔵期間にわたって一定のままであった。
【0237】
【表21】
【0238】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で1.7%まで変化した。
【0239】
【表22】
【0240】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、12ヶ間の貯蔵後に最大で0.4%まで変化した。
【0241】
【表23】
【0242】
初回貯蔵後の全類縁物質は、約0.3%であった。これは、6ヶ月間の貯蔵後に最大で0.7%まで変化した。
【特許請求の範囲】
【請求項1】
少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物。
【請求項2】
前記オキシコドンの薬学的に許容される塩およびナロキソンの薬学的に許容される塩が、オキシコドンおよびナロキソンの塩酸塩から選択される、請求項1に記載の経口即時放出医薬組成物。
【請求項3】
前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項2に記載の経口即時放出医薬組成物。
【請求項4】
前記製剤が、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項1、2または3のいずれか一項に記載の経口即時放出医薬組成物。
【請求項5】
前記製剤が、約2.5mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項4に記載の経口即時放出医薬組成物。
【請求項6】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項5に記載の経口即時放出医薬組成物。
【請求項7】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を含む、請求項6に記載の経口即時放出医薬組成物。
【請求項8】
前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項7に記載の経口即時放出医薬組成物。
【請求項9】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する、請求項1、2、3、4、5、6、7または8のいずれか一項に記載の経口即時放出医薬組成物。
【請求項10】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、請求項9に記載の経口即時放出医薬組成物。
【請求項11】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、請求項9または10に記載の経口即時放出医薬組成物。
【請求項12】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、請求項11に記載の経口即時放出医薬組成物。
【請求項13】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、請求項12に記載の経口即時放出医薬組成物。
【請求項14】
前記組成物が、錠剤、カプセル剤、多粒子剤、ペレット剤または液剤の形態である、請求項1、2、3、4、5、6、7、8、9、10、11、12、または13のいずれか一項に記載の経口即時放出医薬組成物。
【請求項15】
前記製剤が、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含む、請求項1、2、3、4、5、6、7、8、9、10、11、12、13または14のいずれか一項に記載の経口即時放出医薬組成物。
【請求項16】
前記製剤が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含む、請求項1、2、3、4、5、6、7、8、9、10、11、12、13または14のいずれか一項に記載の経口即時放出医薬組成物。
【請求項17】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み;前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含み;前記組成物が、固体形態で、任意選択で錠剤の形態であり;前記製剤が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含み;かつ前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドン塩酸塩の80重量%以上を、およびナロキソン塩酸塩の80重量%以上をin vitroで放出する、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15または16のいずれか一項に記載の経口即時放出医薬組成物。
【請求項18】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の95重量%以上を、およびナロキソン塩酸塩の95重量%以上をin vitroで放出する、請求項17に記載の経口即時放出医薬組成物。
【請求項19】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む、経口即時放出医薬組成物。
【請求項20】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、請求項19に記載の経口即時放出医薬組成物。
【請求項21】
前記医薬組成物が、約1mg〜160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項19または20に記載の経口即時放出医薬組成物。
【請求項22】
前記医薬組成物が、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む、請求項19、20または21に記載の経口即時放出医薬組成物。
【請求項23】
前記医薬組成物が、固体形態である、請求項19、20、21または22に記載の経口即時放出医薬組成物。
【請求項24】
前記医薬組成物が、少なくとも崩壊剤を付加的に含む、請求項19、20、21、22または23に記載の経口即時放出医薬組成物。
【請求項25】
前記医薬組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項19、20、21、22、23または24に記載の経口即時放出医薬組成物。
【請求項26】
疼痛に苦しむ患者の用量設定するのに使用するための、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物。
【請求項27】
疼痛に苦しむ患者の突出痛を治療するための、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物。
【請求項28】
疼痛に苦しむ患者の用量設定するための薬剤の製造における、請求項、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物の使用。
【請求項29】
疼痛に苦しむ患者の突出痛を治療する薬剤の製造における、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物の使用。
【請求項30】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物を投与することによって、疼痛に苦しむ患者の用量設定する方法。
【請求項31】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物を投与することによって、疼痛に苦しむ患者の突出痛を治療する方法。
【請求項1】
少なくともオキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む経口即時放出医薬組成物。
【請求項2】
前記オキシコドンの薬学的に許容される塩およびナロキソンの薬学的に許容される塩が、オキシコドンおよびナロキソンの塩酸塩から選択される、請求項1に記載の経口即時放出医薬組成物。
【請求項3】
前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項2に記載の経口即時放出医薬組成物。
【請求項4】
前記製剤が、約1mg〜約160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項1、2または3のいずれか一項に記載の経口即時放出医薬組成物。
【請求項5】
前記製剤が、約2.5mg〜約40mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約20mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項4に記載の経口即時放出医薬組成物。
【請求項6】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約1.25mg〜約10mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項5に記載の経口即時放出医薬組成物。
【請求項7】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を含む、請求項6に記載の経口即時放出医薬組成物。
【請求項8】
前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項7に記載の経口即時放出医薬組成物。
【請求項9】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の75重量%以上を、およびナロキソンまたはその薬学的に許容される塩の75重量%以上をin vitroで放出する、請求項1、2、3、4、5、6、7または8のいずれか一項に記載の経口即時放出医薬組成物。
【請求項10】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、請求項9に記載の経口即時放出医薬組成物。
【請求項11】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の80重量%以上を、およびナロキソンまたはその薬学的に許容される塩の80重量%以上をin vitroで放出する、請求項9または10に記載の経口即時放出医薬組成物。
【請求項12】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、請求項11に記載の経口即時放出医薬組成物。
【請求項13】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、請求項12に記載の経口即時放出医薬組成物。
【請求項14】
前記組成物が、錠剤、カプセル剤、多粒子剤、ペレット剤または液剤の形態である、請求項1、2、3、4、5、6、7、8、9、10、11、12、または13のいずれか一項に記載の経口即時放出医薬組成物。
【請求項15】
前記製剤が、薬学的に許容される添加剤として少なくとも賦形剤および任意選択で崩壊剤を含む、請求項1、2、3、4、5、6、7、8、9、10、11、12、13または14のいずれか一項に記載の経口即時放出医薬組成物。
【請求項16】
前記製剤が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含む、請求項1、2、3、4、5、6、7、8、9、10、11、12、13または14のいずれか一項に記載の経口即時放出医薬組成物。
【請求項17】
前記製剤が、約2.5mg〜約20mgのオキシコドン塩酸塩および約1.25mg〜約10mgのナロキソン塩酸塩を約2:1の重量比(オキシコドン塩酸塩:ナロキソン塩酸塩)で含み;前記組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含み;前記組成物が、固体形態で、任意選択で錠剤の形態であり;前記製剤が、薬学的に許容される添加剤として少なくとも崩壊剤および任意選択で賦形剤を含み;かつ前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、45分の時点でオキシコドン塩酸塩の80重量%以上を、およびナロキソン塩酸塩の80重量%以上をin vitroで放出する、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15または16のいずれか一項に記載の経口即時放出医薬組成物。
【請求項18】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドン塩酸塩の95重量%以上を、およびナロキソン塩酸塩の95重量%以上をin vitroで放出する、請求項17に記載の経口即時放出医薬組成物。
【請求項19】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の90重量%以上を、およびナロキソンまたはその薬学的に許容される塩の90重量%以上をin vitroで放出する、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を含む、経口即時放出医薬組成物。
【請求項20】
前記製剤が、欧州薬局方のパドル法を37℃の0.1N塩酸中、100rpmで使用し、かつ230nmのUV検出を使用して測定された場合に、15分の時点でオキシコドンまたはその薬学的に許容される塩の95重量%以上を、およびナロキソンまたはその薬学的に許容される塩の95重量%以上をin vitroで放出する、請求項19に記載の経口即時放出医薬組成物。
【請求項21】
前記医薬組成物が、約1mg〜160mgのオキシコドン塩酸塩に等価な量のオキシコドンまたはその薬学的に許容される塩および約0.5mg〜約80mgのナロキソン塩酸塩に等価な量のナロキソンまたはその薬学的に許容される塩を含む、請求項19または20に記載の経口即時放出医薬組成物。
【請求項22】
前記医薬組成物が、オキシコドンまたはその薬学的に許容される塩およびナロキソンまたはその薬学的に許容される塩を約2:1の重量比(オキシコドンまたはその薬学的に許容される塩:ナロキソンまたはその薬学的に許容される塩)で含む、請求項19、20または21に記載の経口即時放出医薬組成物。
【請求項23】
前記医薬組成物が、固体形態である、請求項19、20、21または22に記載の経口即時放出医薬組成物。
【請求項24】
前記医薬組成物が、少なくとも崩壊剤を付加的に含む、請求項19、20、21、22または23に記載の経口即時放出医薬組成物。
【請求項25】
前記医薬組成物が、唯一の薬学的に活性な薬剤としてオキシコドン塩酸塩およびナロキソン塩酸塩を含む、請求項19、20、21、22、23または24に記載の経口即時放出医薬組成物。
【請求項26】
疼痛に苦しむ患者の用量設定するのに使用するための、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物。
【請求項27】
疼痛に苦しむ患者の突出痛を治療するための、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物。
【請求項28】
疼痛に苦しむ患者の用量設定するための薬剤の製造における、請求項、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物の使用。
【請求項29】
疼痛に苦しむ患者の突出痛を治療する薬剤の製造における、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物の使用。
【請求項30】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物を投与することによって、疼痛に苦しむ患者の用量設定する方法。
【請求項31】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25または26のいずれか一項に記載の経口即時放出医薬組成物を投与することによって、疼痛に苦しむ患者の突出痛を治療する方法。
【図1】

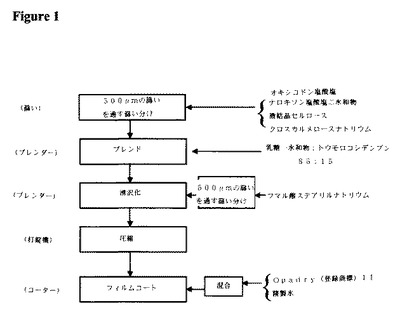
【図2】
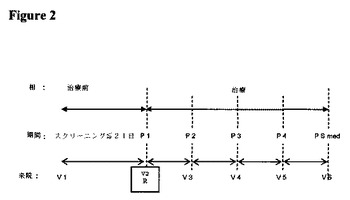
【図3】
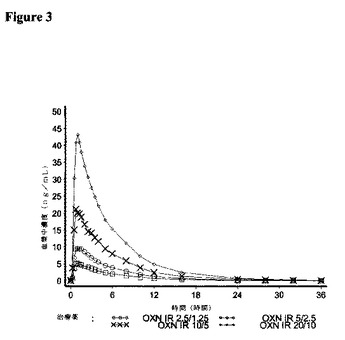
【図4】
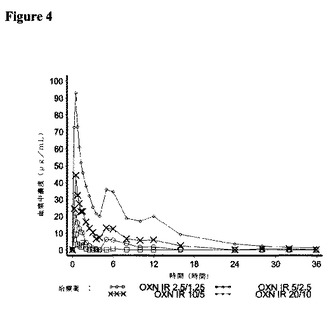
【図5】
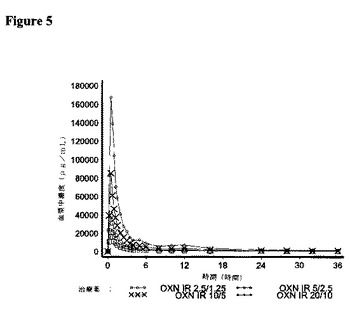
【図6】
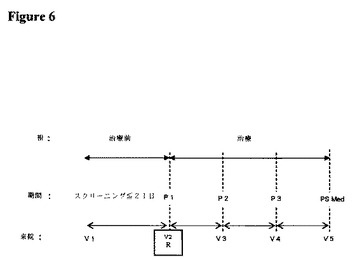
【図7】
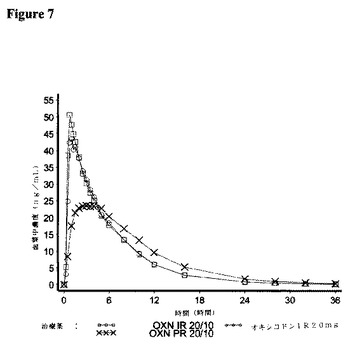
【図8】
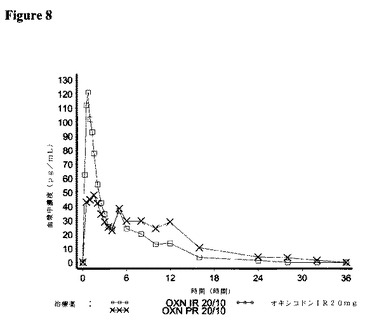
【図9】
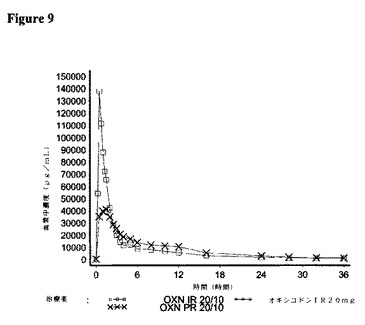
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2012−520261(P2012−520261A)
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2011−553441(P2011−553441)
【出願日】平成22年3月10日(2010.3.10)
【国際出願番号】PCT/EP2010/053028
【国際公開番号】WO2010/103039
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成22年3月10日(2010.3.10)
【国際出願番号】PCT/EP2010/053028
【国際公開番号】WO2010/103039
【国際公開日】平成22年9月16日(2010.9.16)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]