
オキシコドン及びナロキソンの医薬配合物
【課題】オキシコドンまたはそれの塩を含む、他の剤形よりも非経口的に乱用されにくい、他の剤形よりも流用されにくい、オキシコドン医薬組成物の提供。
【解決手段】特定の実施態様では、10mgから40mgまでのオキシコドンハイドロクロライドまたはそれの薬学的に許容可能な塩と、0.65mgから0.90mgまでのナロキソンハイドロクロライドまたはそれの薬学的に許容可能な塩とを含む医薬組成物。好ましくは、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、該オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、該医薬組成物。
【解決手段】特定の実施態様では、10mgから40mgまでのオキシコドンハイドロクロライドまたはそれの薬学的に許容可能な塩と、0.65mgから0.90mgまでのナロキソンハイドロクロライドまたはそれの薬学的に許容可能な塩とを含む医薬組成物。好ましくは、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、該オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、該医薬組成物。
【発明の詳細な説明】
【技術分野】
【0001】
オキシコドン製剤は、時として、乱用の対象である。特定の用量のオキシコドンは、非
経口的に投与されると、同じ用量を経口的に投与した場合に比べ、一層効力が高くなり得
る。経口用オキシコドン製剤を乱用する一つの方法は、その活性物質を溶液に為し、それ
を注射する操作を含む。これらの薬剤の非経口的な乱用を防止するため、オピオイド拮抗
薬が特定のオピオイド作動薬と配合されている。
【背景技術】
【0002】
先行技術においては、即時放出性(immediate release)のペンタゾ
シンとナロキソンの配合物が、米国において入手可能な錠剤の形態で利用されており、S
anofi−WinthropからTalwin(登録商標)Nxとして市販されている
。Talwin(登録商標)Nxは、50mgの塩基に相当する即時放出性のペンタゾシ
ンハイドロクロライドと0.5mgの塩基に相当するナロキソンハイドロクロライドを含
んでいる。チリジン(50mg)とナロキソン(4mg)を含む固定の配合物療法が、1
978年以来、疼痛を管理するため、ドイツで利用されている(Valoron(登録商
標)N、Goedecke)。ブプレノルフィンとナロキソンの固定の配合物が、疼痛を
治療するため、ニュージーランドで1991年に導入された(Temgesic(登録商
標)Nx、Reckitt & Colman)。
【0003】
Purdue Pharma L.Pは、現在、OxyContin(登録商標)とい
う商品名で、10、20、40、80、及び160mgのオキシコドンハイドロクロライ
ドを含む剤形の徐放出性(sustained−release)のオキシコドンを市販
している。
【0004】
米国特許第5,266,331号;第5,508,042号;第5,549,912号
及び第5,656,295号は、徐放出性オキシコドン製剤を開示している。
【0005】
Kreekに付与された米国特許第4,769,372号及び第4,785,000号
は、1日に1回から5回まで、約1.5mgから約100mgまでのオピオイド鎮痛薬、
又は経口的に投与されたときに殆どもしくは全く系統的な拮抗活性を持たない約1mgか
ら約18mgまでのオピオイド拮抗薬を含む1用量単位から2用量単位までを投与するこ
とにより、腸管の自動能不全を誘発することなく、慢性の疼痛または慢性の咳を被ってい
る患者を治療する方法を開示することを主旨としている。
【0006】
Crainらに付与された米国特許第5,472,943号は、二つのモードで作用す
るオピオイド作動薬をオピオイド拮抗薬と共に投与することにより、その作動薬の鎮痛効
力を高める方法を開示することを主旨としている。
【0007】
米国特許第3,773,955号は、1mgから2.5mgまでのナロキソンとの配合
においてオピオイド作動薬を含む即時放出性の製剤を開示することを主旨としている。
【0008】
先述のものを含め、ここで引用されるすべての文書は、あらゆる目的で、それらの全体
が参照により本明細書に組み入れられる。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の一つの目的は、オキシコドンまたはそれの塩を含む経口用剤形を提供すること
である。
【0010】
本発明の特定の実施態様における一つの目的は、他の剤形よりも非経口的に乱用されに
くい、オキシコドンまたはそれの塩の経口用剤形を提供することである。
【0011】
本発明の特定の実施態様における一つの目的は、他の剤形よりも常習者に興味を持たれ
にくい、オキシコドンまたはそれの塩の経口用剤形を提供することである。
【0012】
本発明の特定の実施態様における一つの目的は、他の剤形よりも流用されにくい、オキ
シコドンまたはそれの塩の経口用剤形を提供することである。
【0013】
本発明の特定の実施態様における一つの目的は、オキシコドンまたはそれの塩の経口用
剤形を用いてヒト患者における疼痛を治療し、その一方で、その剤形の乱用の可能性を低
減する方法を提供することである。
【0014】
本発明の特定の実施態様における一つの目的は、乱用の可能性が比較的少なくなるよう
な仕方で、オキシコドンまたはそれの塩の経口用剤形を製造する方法を提供することであ
る。
【0015】
これらの目的及び他の目的は、10mgから40mgまでのオキシコドンまたはそれの
薬学的に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの
薬学的に許容可能な塩とを含む医薬組成物に向けられた本発明により達成される。
【課題を解決するための手段】
【0016】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において、約10mgのオキシコドンハイドロクロライドと、0.80m
gから0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられ
ている。
【0017】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられて
いる。
【0018】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられて
いる。
【0019】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約10mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0020】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0021】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0022】
ここに開示されている本発明の特定の実施態様では、その剤形は、ナロキソンまたはそ
れの薬学的に許容可能な塩の徐放出をもたらす。
【0023】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす特定
の実施態様では、その剤形は、37℃における900mlの模擬胃液(SGF)中におい
て100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法
によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、オキシ
コドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±30%の範囲内にあるナ
ロキソンまたはそれの薬学的に許容可能な塩の溶解をもたらす。例えば、もしオキシコド
ンまたはそれの薬学的に許容可能な塩の溶解速度が1時間で40%の場合、ナロキソンま
たはそれの薬学的に許容可能な塩の溶解速度は28%から52%までの範囲であろう。
【0024】
上述の実施態様における特定の実施態様では、ナロキソンまたはそれの薬学的に許容可
能な塩は、1時間、4時間、及び12時間経過時点において、オキシコドンまたはそれの
薬学的に許容可能な塩の溶解速度に対して±20%の範囲内にあり;代替的に、オキシコ
ドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±10%の範囲内にあり;あ
るいは、代替的に、オキシコドンまたはそれの薬学的に許容可能な塩の溶解速度に対して
±5%の範囲内にあるイン・ビトロ溶解速度を有している。
【0025】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす別の
実施態様では、オキシコドンまたはそれの薬学的に許容可能な塩並びにナロキソンまたは
それの薬学的に許容可能な塩のイン・ビトロ溶解速度は、37℃における900mlの模
擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUS
P装置I(バスケット)法により測定したときに、1時間では、約20重量%から約60
重量%までのオキシコドンまたはそれの薬学的に許容可能な塩と、約20重量%から約6
0重量%までのナロキソンまたはそれの薬学的に許容可能な塩とが放出され;2時間では
、約30重量%から約75重量%までのオキシコドンまたはそれの薬学的に許容可能な塩
と、約30重量%から約75重量%までのナロキソンまたはそれの薬学的に許容可能な塩
とが放出され;4時間では、約40重量%から約90重量%までのオキシコドンまたはそ
れの薬学的に許容可能な塩と、約40重量%から約90重量%までのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;8時間では、約60重量%より多くのオキシコ
ドンまたはそれの薬学的に許容可能な塩と、約60重量%より多くのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;そして、12時間では、約70重量%より多く
のオキシコドンまたはそれの薬学的に許容可能な塩と、約70重量%より多くのナロキソ
ンまたはそれの薬学的に許容可能な塩とが放出されるような溶解速度である。
【0026】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす別の
実施態様では、オキシコドンまたはそれの薬学的に許容可能な塩並びにナロキソンまたは
それの薬学的に許容可能な塩のイン・ビトロ溶解速度は、37℃における900mlの模
擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUS
P装置I(バスケット)法により測定したときに、1時間では、約30重量%から約60
重量%までのオキシコドンまたはそれの薬学的に許容可能な塩と、約30重量%から約6
0重量%までのナロキソンまたはそれの薬学的に許容可能な塩とが放出され;2時間では
、約40重量%から約70重量%までのオキシコドンまたはそれの薬学的に許容可能な塩
と、約40重量%から約70重量%までのナロキソンまたはそれの薬学的に許容可能な塩
とが放出され;4時間では、約55重量%から約90重量%までのオキシコドンまたはそ
れの薬学的に許容可能な塩と、約55重量%から約90重量%までのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;8時間では、約70重量%より多くのオキシコ
ドンまたはそれの薬学的に許容可能な塩と、約70重量%より多くのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;そして、12時でには、約80重量%より多く
のオキシコドンまたはそれの薬学的に許容可能な塩と、約80重量%より多くのナロキソ
ンまたはそれの薬学的に許容可能な塩とが放出されるような溶解速度である。
【0027】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに、制御放出性(controlled release)対照製品(Ph
ysician’s Desk Reference 2002年版に記載されている通
りのOxyContin(登録商標))のオキシコドン塩基同等量によりもたらされる血
漿中オキシコドンの平均AUCの80%から125%までの範囲内における血漿中オキシ
コドンの平均AUCをもたらす。別の実施態様では、血漿中オキシコドンの平均AUCは
、ヒト被験者の母集団に対して単回用量投与をしたときに制御放出性対照製品のオキシコ
ドン塩基同等量によりもたらされる血漿中オキシコドンの平均AUCの90%から110
%までの範囲内、あるいは95%から105%までの範囲内である。
【0028】
特定の実施態様では、本発明の医薬組成物は、ナロキソンハイドロクロライドを含有す
る即時放出性対照製品、即ち、Physician’s Desk Reference
2002年版に記載されている通りのTalwin(登録商標)Nxのナロキソン塩基
同等量によりもたらされる血漿中ナロキソンの平均Cmaxよりも少なくとも50%下回
った血漿中ナロキソンの平均Cmaxをもたらす。別の実施態様では、この血漿中ナロキ
ソンの平均Cmaxは、ナロキソンハイドロクロライドを含有する即時放出性対照製品、
即ち、Physician’s Desk Reference 2002年版に記載さ
れている通りのTalwin(登録商標)Nxのナロキソン塩基同等量によりもたらされ
る血漿中ナロキソンの平均Cmaxよりも少なくとも65%下回っており、あるいは少な
くとも80%下回っている。
【0029】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに、180pg/ml未満、150pg/ml未満、または100pg/m
l未満の血漿中ナロキソンの平均Cmaxをもたらす。より好適な実施態様では、この血
漿中ナロキソンの平均Cmaxは、ヒト被験者の母集団に対して単回用量投与をしたとき
に、50pg/ml未満、10pg/ml未満、または5pg/ml未満である。
【0030】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに5pg/ml未満の血漿中ナロキソンの平均Cmaxをもたらすような量の
ナロキソンまたはそれの薬学的に許容可能な塩を含んでいる。
【0031】
ここに開示されている本発明の特定の実施態様では、その剤形は、ヒト患者に対する定
常状態での経口投与後、少なくとも12時間は疼痛の効果的な軽減をもたらす。
【0032】
ここに開示されている本発明の特定の実施態様では、その剤形は、ヒト患者に対する定
常状態での経口投与後、少なくとも24時間は疼痛の効果的な軽減をもたらす。
【0033】
ここに開示されている本発明の特定の実施態様では、その剤形は、徐放出性賦形剤に相
互分散された(interdispersed)オキシコドンハイドロクロライドとナロキソンハイドロク
ロライドを包含したマトリックスを含んでいる。
【0034】
特定の実施態様では、本発明は、ここに開示されている組成物を調製することを含有す
る、オキシコドン製剤の非経口的な乱用の可能性を低減する方法に向けられている。
【0035】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を経
口的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0036】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を、
少なくとも定常状態が達成されるまで12時間毎に経口的に投与することを含む、ヒト患
者における疼痛を治療する方法に向けられている。
【0037】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を、
少なくとも定常状態が達成されるまで24時間毎に経口的に投与することを含む、ヒト患
者における疼痛を治療する方法に向けられている。
【0038】
特定の実施態様では、本発明は、患者に対する定常状態での経口投与後、少なくとも1
2時間は疼痛の効果的な軽減をもたらす、ここに開示されている通りの医薬組成物を経口
的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0039】
特定の実施態様では、本発明は、患者に対する定常状態での経口投与後、少なくとも2
4時間は疼痛の効果的な軽減をもたらす、ここに開示されている通りの医薬組成物を経口
的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0040】
特定の実施態様では、本発明は、オキシコドンまたはそれの薬学的に許容可能な塩を徐
放出性の賦形剤と配合してオキシコドン/賦形剤配合物を形成すること;そのオキシコド
ン/賦形剤配合物にナロキソンまたはそれの薬学的に許容可能な塩を加えること;並びに
、オキシコドンまたはそれの薬学的に許容可能な塩と、ナロキソンまたはそれの薬学的に
許容可能な塩との徐放出性経口用剤形を形成することを含む、徐放出性経口用剤形を製造
する方法に向けられている。代替的に、最初にナロキソンを賦形剤と配合し、オキシコド
ンをそのナロキソン/賦形剤混合物に加えてもよい。別の実施態様では、オキシコドンと
ナロキソンを、賦形剤と配合するのと同時的に、もしくは、賦形剤と配合する前に配合す
ることができる。特定の実施態様では、一つの物質が、他の物質との配合に先だって、徐
放出性形態となるように前処理されない。
【0041】
特定の実施態様では、本発明は、約10mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約20
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療す
る方法に向けられている。
【0042】
特定の実施態様では、本発明は、約20mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約40
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療す
る方法に向けられている。
【0043】
特定の実施態様では、本発明は、約10mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約20
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することによりその用量を増量し、更にその後、約40mgの
オキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的に許容
可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的に許容
可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐放出を
もたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療する方法
に向けられている。
【0044】
特定の実施態様では、本発明は、約10mgから約40mgまでのオキシコドンまたは
それの薬学的に許容可能な塩と、ナロキソンまたはそれの薬学的に許容可能な塩とを含む
少なくとも1つの製剤を含有した容器を包含する、疼痛を治療するためのキットに向けら
れており;そのキットは、更に、上述の製剤の使用を指示する証印(indicia)を
含んでいる。特定の実施態様では、そのキットの製剤は、0.65mgから0.9mgま
でのナロキソンまたはそれの薬学的に許容可能な塩を含んでおり、そして、特定の実施態
様では、上述の証印は、その使用が製剤の非経口的な乱用を低減するためのものであるこ
とを指示している。
【0045】
「徐放出性」という用語は、本発明の目的上、即時放出性製品と比べて長い期間、例え
ば約12時間から約24時間までの期間にわたり、血液(例えば血漿)中の濃度を治療的
範囲内であるが毒性レベル以下に維持するような仕方でオキシコドンまたはそれの塩を放
出することとして定義される。都合のよいことに、この徐放出により、十分に1日に2回
または1日に1回製剤が提供される。特定の実施態様では、1時間、4時間、及び12時
間におけるナロキソンまたはそれの塩の放出速度は、オキシコドンまたはそれの薬学的に
許容可能な塩の溶解速度に対して±30%の範囲内である。
【0046】
本明細書で使用される場合、「非経口的に」という用語は、皮下注射、静脈内、筋肉内
、胸骨内への注射、注入技術、あるいは、当技術分野において知られている他の注入方法
を含む。
【0047】
別な具合に特記されていない限り、「オキシコドン」という用語はオキシコドン塩基を
意味している。別な具合に特記されていない限り、「ナロキソン」という用語はナロキソ
ン塩基を意味している。塩という用語は、薬学的に許容可能な塩を意味している。
【0048】
「定常状態」という用語は、その系に到達する薬剤の量が、その系を去る薬剤の量とほ
ぼ同じであることを意味している。従って、「定常状態」の場合、患者の身体は、薬剤が
血流中への吸収を通じてその患者の系で利用できるようになるのとほぼ同じ速度でその薬
剤を排出する。
【0049】
「非経口的に有効な量のナロキソンまたはそれの薬学的に許容可能な塩」という用語は
、ナロキソンまたはそれの薬学的に許容可能な塩が、この剤形を非経口的に投与したとき
のオキシコドンの作用を無効にするのに充分な量、もしくは、その作用を部分的に無効に
する量であり、且つ、この剤形を経口投与したときのオキシコドンの作用を無効もしくは
部分的に無効にする量以下の量であることを意味している。
【0050】
「平均」という用語は、別な具合に特定されていない限り、算術平均を表している。
【0051】
「ヒト被験者」という用語は、当業者により理解されるように、正常な代謝機能を有す
る健常なヒト被験者を意味している。
【発明を実施するための最良の形態】
【0052】
本発明の剤形は、約10mgから約40mgまでのオキシコドンハイドロクロライドに
相当する量のオキシコドンまたはそれの薬学的に許容可能な塩を含んでいる。オキシコド
ンハイドロクロライドの特に好適な用量は、約10mg、約20mg、約30mg、また
は約40mgである。オキシコドンまたはそれの塩は、そのオキシコドンの徐放出をもた
らすべく、薬学的に許容可能な適切な賦形剤と調合される。
【0053】
本発明の剤形は、約0.65mgから約0.90mgまでのナロキソンまたはそれの薬
学的に許容可能な塩を含んでいる。ナロキソン塩の特に好適な用量範囲は、約0.82m
gから約0.88mgまで、及び約0.84mgから約0.86mgまでである。特に好
適な用量は、約0.85mgのナロキソン塩を有している。ナロキソン塩基の特に好適な
用量範囲は、約0.65mgから約0.75mgまで、及び約0.67mgから約0.7
3mgまでである。特に好適な用量は、約0.70mgのナロキソン塩基を有している。
【0054】
本発明の剤形は、約0.65mgから約0.90mgまでのナロキソンハイドロクロラ
イドジハイドレートに相当する量のナロキソンまたはそれの薬学的に許容可能な塩を含ん
でいる。ナロキソンハイドロクロライドジハイドレートの特に好適な用量範囲は、約0.
82mgから約0.88mgまで、または約0.84mgから約0.86mgまでである
。特に好適な用量は、約0.85mgのナロキソンハイドロクロライドジハイドレートを
有している。ナロキソン塩基の特に好適な用量範囲は、約0.65mgから約0.75m
gまで、または同等量のそれの塩、及び約0.67mgから約0.73mgまで、または
同等量のそれの塩である。特に好適な用量は、約0.70mgのナロキソン塩基、または
同等量のそれの塩を有している。
【0055】
ナロキソンまたはそれの塩は、そのナロキソンまたはそれの塩の即時放出をもたらすべ
く調合され、薬学的に許容可能な適切な賦形剤と配合されてよい。ナロキソンまたはそれ
の塩の徐放出速度は、オキシコドンまたはそれの塩の徐放出速度と同じであってもよいし
、あるいは異なっていてもよい。本発明の特に好適な実施態様は、10mgのオキシコド
ンハイドロクロライドと0.85mgのナロキソンハイドロクロライドを含有する剤形;
20mgのオキシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロラ
イドを含有する剤形;30mgのオキシコドンハイドロクロライドと0.85mgのナロ
キソンハイドロクロライドを含有する剤形;40mgのオキシコドンハイドロクロライド
と0.85mgのナロキソンハイドロクロライドを含有する剤形;あるいは、同等量のオ
キシコドン塩基、ナロキソン塩基、または他の薬学的に許容可能なそれの塩を含有する剤
形である。オキシコドン及びナロキソンのハイドロクロライド塩は特に好適である。
【0056】
本発明の他の特に好適な実施態様は、10mgのオキシコドンハイドロクロライドと0
.85mgのナロキソンハイドロクロライドジハイドレートを含有する剤形;20mgの
オキシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロライドジハイ
ドレートを含有する剤形;30mgのオキシコドンハイドロクロライドと0.85mgの
ナロキソンハイドロクロライドジハイドレートを含有する剤形;あるいは、40mgのオ
キシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロライドジハイド
レートを含有する剤形である。
【0057】
それらの物質が共に徐放出性の形態を為している実施態様では、都合のよいことに、そ
の剤形は、37℃における900mlの模擬胃液(SGF)中において100rpmで、
米国薬局方XXIV(2000年)のUSP装置I(バスケット)法により測定したとき
に、本発明の目的に適合したイン・ビトロ溶解速度をもたらすような仕方でオキシコドン
またはそれの薬学的に許容可能な塩とナロキソンまたはそれの薬学的に許容可能な塩を放
出する。
【0058】
本発明において開示されているナロキソンまたはそれの塩の範囲は、もしその製剤が溶
解されて非経口的に投与された場合に、オキシコドンのオピオイド効果を少なくとも部分
的に阻止することによって静脈内乱用を防止するのに非経口的に有効な量である。好適に
は、その量は、物理的に最も依存性の高い個人が非経口投与をしたときに、オピオイドの
使用中止後に見られる症状と非常によく似た中等度から重度の禁断症候群を突如として引
き起こすのに充分な量でもある。この禁断症候群のうちで最も一般的な症状は、食欲不振
、体重減少、瞳孔拡大、過剰な発汗と交互に生じる悪寒、異常な痙攣、悪心、嘔吐、筋肉
痙攣、過剰刺激感受性、流涙、鼻漏(rinorrhea)、鳥肌、及び心拍数上昇を含
む。ナロキソンの量は、疼痛を被っている患者に経口投与したときに、有害効果を引き起
こしたり、あるいは、鎮痛効能を低減させるような量であるべきではない。
【0059】
(徐放出性剤形)
オキシコドン(またはオキシコドン塩)、及び、場合によってはナロキソン(またはナ
ロキソン塩)は、当業者にとって既知の何らかの適した錠剤、被覆錠剤、または多重粒状
体(multiparticulate)製剤の形態における徐放出性経口用製剤として
調合される。その徐放出性剤形は、ナロキソンまたはそれの塩を伴って、もしくはそれら
を伴わずに、オキシコドンまたはそれの塩と共にマトリックスに組み込まれた徐放出性材
料を含んでいてよい。例えば、オキシコドン塩が徐放出性マトリックスに組み込まれてい
てよく、そして、ナロキソン塩が、そのマトリックスとは別になっていてもよいし、ある
いは、そのマトリックスに組み込まれていてもよい。
【0060】
この徐放出性剤形は、場合によっては、ナロキソンまたはそれの塩を伴って、もしくは
それらを伴わずに、オキシコドンまたはそれの塩を含有する粒子を含んでいてよい。特定
の実施態様では、それらの粒子は、約0.1mmから約2.5mmまでの直径、好適には
約0.5mmから約2mmまでの直径を有している。ナロキソンまたはナロキソン塩は、
オキシコドンまたはオキシコドン塩を含有する粒子に組み込まれていてよく、あるいは、
オキシコドンまたはオキシコドン塩粒子を含有する錠剤またはカプセル剤に組み込まれて
いてよい。好適には、それらの粒子は、水性媒質中における持続的な速度での(1つもし
くは複数の)本活性物質の放出を可能にする材料でフィルムコーティングされる。そのフ
ィルムコーティングは、ここで述べられている他の特性と組み合わせて、望ましいイン・
ビトロ放出速度を達成できるように選択される。本発明の徐放出性コーティング製剤は、
滑らか且つエレガントで、顔料及び他のコーティング用添加剤を支持することができ、無
毒且つ不活性で粘着性のない強い連続的なフィルムを作製できるべきである。
【0061】
(被覆ビーズ)
本発明の特定の実施態様では、不活性な製薬用ビーズ、例えばnu pariel 1
8/20ビーズ等をコーティングするために疎水性材料が使用され、その後、結果として
得られた複数の固体の徐放出性ビーズを、経口摂取し環境流体、例えば胃液または溶解媒
質と接触したときに効果的な徐放出性の用量を供給するのに充分な量で、ゼラチンカプセ
ルに入れることができる。特定の実施態様では、ナロキソンまたはナロキソン塩は、オキ
シコドンまたはオキシコドン塩を含有する徐放出性ビーズにコーティングされてよく、あ
るいは、徐放出性オキシコドンまたはオキシコドン塩ビーズと共にカプセルに入れられて
もよい。
【0062】
本発明の徐放出性ビーズ製剤は、例えば、経口摂取されて胃液次いで腸液に晒されたと
きに、本発明の(1つもしくは複数の)物質をゆっくりと放出する。本発明の製剤の徐放
出プロフィールは、例えば、疎水性材料を用いるオーバーコーティングの量を変更するこ
とにより、疎水性材料に可塑剤を加える仕方を変更することにより、疎水性材料に対する
可塑剤の量を変更することにより、付加的な成分または賦形剤を含めることにより、ある
いは製造方法を変更することなどにより変えることができる。また、最終製品の溶解プロ
フィールも、例えばその遅延剤コーティングの厚みを増加または減少させることにより変
えることができる。
【0063】
本発明の(1つもしくは複数の)物質でコーティングされたスフェロイド(spher
oids)またはビーズは、例えば、(1つもしくは複数の)物質を水に溶かし、次いで
、Wusterインサートを用いて、その溶液を基材、例えばnu pariel 18
/20ビーズに噴霧することにより調製される。場合によっては、ビーズへの(1つもし
くは複数の)本物質の結合を補助するため、及び/又は、その溶液を着色するため等、ビ
ーズをコーティングする前に付加的な成分も加えられる。例えば、着色剤を伴って、もし
くは、着色剤を伴わずに、ヒドロキシプロピルメチルセルロース等を含む製品(例えば、
Opadry(登録商標)、Colorcon,Inc.から商業的に入手可能)をその
溶液に加えてよく、そして、その溶液は、ビーズに同じく塗布する前に混合(例えば約1
時間)される。その後、結果として生じる被覆された基材、この例ではビーズは、場合に
よっては、疎水性の徐放出性コーティングから(1つもしくは複数の)本活性物質を分離
するため、バリヤー剤でオーバーコーティングされる。適切なバリヤー剤の一つの例は、
ヒドロキシプロピルメチルセルロースを含むバリヤー剤である。しかし、当技術分野にお
いて既知のあらゆるフィルム形成剤が使用されてよい。そのバリヤー剤は、最終製品の溶
解速度に影響を及ぼさないことが好適である。
【0064】
それらのビーズは、疎水性材料の水性分散系でオーバーコーティングされてよい。この
疎水性材料の水性分散系は、好適には、更に、有効量の可塑剤、例えばトリエチルシトレ
ートを含む。予め調合済みのエチルセルロースの水性分散系、例えばAquacoat(
登録商標)またはSurelease(登録商標)を使用することができる。Surel
ease(登録商標)を使用する場合には、可塑剤を別個に加える必要はない。代替的に
、予め調合済みのアクリルポリマーの水性分散系、例えばEudragit(登録商標)
を使用することもできる。
【0065】
本発明のコーティング溶液は、好適には、フィルム形成剤、可塑剤、及び溶媒系(即ち
、水)のほかに、エレガンスさと製品識別能力を与えるための着色剤を含んでいる。疎水
性材料の水性分散系の代わりに、あるいは、疎水性材料の水性分散系に加えて、その治療
的に活性な物質の溶液に色を付けてもよい。例えば、アルコールまたはプロピレングリコ
ールをベースとしたカラー分散系、破砕アルミニウムレーキ、及び乳白剤、例えば二酸化
チタン等の使用を介し、剪断力(shear)を利用して水溶性ポリマー溶液に、次いで
、低剪断力を利用してその可塑化されたAquacoat(登録商標)に色を付けること
により、Aquacoat(登録商標)にも色を付けることができる。代替的に、本発明
の製剤に色をもたらす何らかの適切な方法を用いてもよい。アクリルポリマーの水性分散
系が使用される場合、本製剤に色をもたらすのに適した成分は、二酸化チタン、及び着色
顔料、例えば酸化鉄顔料等を含む。しかし、顔料を組み入れると、そのコーティングの遅
延効果(retard effect)を高める可能性がある。
【0066】
可塑化された疎水性材料は、当技術分野において既知の何らかの適切な噴霧装置を用い
て噴霧することにより、(1つもしくは複数の)本物質を含有する基材上に塗布されてよ
い。一つの好適な方法では、Wurster流動床システムが使用され、そこでは、下側
から注入されたエアージェットがそのコア材料を流動化し、そして、アクリルポリマーコ
ーティングが噴霧されている間に乾燥機能を果たす。その被覆基材が水性溶液、例えば胃
液等に晒されたときに、(1つもしくは複数の)本物質の予め決定された徐放出性を得る
のに充分な量の疎水性材料が塗布されてよい。疎水性材料でコーティングした後、場合に
よっては、それらのビーズに、フィルム形成剤、例えばOpadry(登録商標)等の更
なるオーバーコーティングが施される。適用される場合、このオーバーコーティングは、
それらのビーズのアグロメレーションを実質的に低減するために施される。
【0067】
本発明の徐放出性製剤からの(1つもしくは複数の)本物質の放出は、1つもしくはそ
れ以上の放出変性剤(release−modifying agents)を加えるこ
とにより、あるいは、そのコーティングを通じる1つもしくはそれ以上の通路を設けるこ
とにより、更なる影響を受け、即ち、所望の速度に調節することができる。疎水性材料と
水溶性材料との比は、他の数あるファクターの中でもとりわけ、必要な放出速度、及び、
選定された材料の溶解度特性によって決定される。
【0068】
細孔形成剤として機能する放出変性剤は、有機または無機であってよく、そして、使用
環境において、そのコーティングから溶解、抽出、または浸出され得る材料を含む。この
細孔形成剤は、ヒドロキシプロピルメチルセルロース等の1つもしくはそれ以上の親水性
材料を含んでいてよい。
【0069】
本発明の徐放出性コーティングは、浸食促進剤、例えばデンプン及びゴム等を含むこと
もできる。
【0070】
また、本発明の徐放出性コーティングは、使用環境において微孔性薄膜を作製するのに
有用な材料、例えば、炭酸の線状ポリエステルを含むポリカーボネート(そこでは、ポリ
マー鎖中にカーボネート基が繰返し現れる)も含むことができる。
【0071】
放出変性剤は半透水性ポリマーも含んでいてよい。
【0072】
特定の好適な実施態様では、その放出変性剤は、ヒドロキシプロピルメチルセルロース
、ラクトース、金属ステアレート、及び前述の物質の何らかの混合物から選択される。
【0073】
本発明の徐放出性コーティングは、少なくとも1つの通路、オリフィス等を含む出口手
段(exit means)も含むことができる。その通路は、米国特許第3,845,
770号;第3,916,899号;第4,063,064号;及び第4,088,86
4号に開示されているような方法により形成されてよい。また、その通路は、あらゆる形
状を有していてよく、例えば丸形、三角形、矩形、楕円形、不規則な形等であってよい。
【0074】
(マトリックス製剤)
本発明の別の実施態様では、徐放出性製剤は、場合によってはここで説明されている通
りの徐放出性コーティングを有するマトリックスにより達成される。徐放出性マトリック
スに包含させるのに適した材料は、そのマトリックスを形成するために使用する方法に依
存するであろう。
【0075】
例えば、マトリックスは、オキシコドン(またはオキシコドン塩)、及び、場合によっ
てはナロキソン(またはナロキソン塩)に加え、以下のものを含んでいてよい:
親水性及び/又は疎水性の材料、例えばゴム、セルロースエーテル、アクリル樹脂、タ
ンパク質誘導材料等;このリストは、排他的であることを意図したものではなく、(1つ
もしくは複数の)本物質の徐放出性を付与することができ、且つ、溶融する(もしくは、
押し出されるのに必要な程度にまで軟化する)、薬学的に許容可能なあらゆる疎水性材料
または親水性材料を本発明に従って使用することができる。
【0076】
消化可能な長鎖(C8−C50、特にはC12−C40)の置換または非置換炭化水素、例え
ば脂肪酸、脂肪族アルコール、脂肪酸のグリセリルエステル、鉱油及び植物油及びロウ、
及びステアリルアルコール;並びにポリアルキレングリコール等。
【0077】
これらのポリマーのうち、アクリルポリマー、特にはEudragit(登録商標)R
SPO−セルロースエーテル、特にはヒドロキシアルキルセルロース及びカルボキシアル
キルセルロースが好適である。本経口用剤形は、1重量%から80重量%までの間の少な
くとも1つの親水性または疎水性材料を含んでいてよい。
【0078】
疎水性材料が炭化水素の場合、その炭化水素は、好適には、25℃から90℃までの間
の融点を有している。長鎖炭化水素材料のうち、脂肪(脂肪族)アルコールが好適である
。本経口用剤形は、60重量%までの少なくとも1つの消化可能な長鎖炭化水素を含んで
いてよい。
【0079】
好適には、本経口用剤形は、60重量%までの少なくとも1つのポリアルキレングリコ
ールを含んでいる。
【0080】
疎水性材料は、好適には、アルキルセルロース、アクリル酸及びメタクリル酸のポリマ
ー及びコポリマー、セラック、ゼイン、硬化(hydrogenated)ヒマシ油、硬
化植物油、またはそれらの混合物からなるグループから選択される。本発明の特定の好適
な実施態様では、疎水性材料は薬学的に許容可能なアクリルポリマーであり、それらのア
クリルポリマーは、これらに限定するものではないが、アクリル酸とメタクリル酸のコポ
リマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキシエチルメタ
クリレート、シアノエチルメタクリレート、アミノアルキルメタクリレートコポリマー、
ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、
ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレート
、ポリアクリルアミド、ポリ(無水メタクリル酸)、及びグリシジルメタクリレートコポ
リマーを含む。別の実施態様では、疎水性材料は、ヒドロキシプロピルメチルセルロース
等のヒドロキシアルキルセルロース、及び前述の物質の混合物等の材料から選択される。
【0081】
好適な疎水性材料は、多少なりとも明白な親水性及び/又は疎水性の傾向を伴って、水
に不溶性である。好適には、本発明に有用な疎水性材料は、約25℃から約200℃まで
、好適には約45℃から約90℃までの融点を有している。具体的には、疎水性材料は、
天然または合成のロウ、脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアル
コール、ステアリルアルコール、セチルアルコール、または、好適にはセトステアリルア
ルコール等)、これらに限定するものではないが、脂肪酸エステル、脂肪酸グリセリド(
モノ−、ジ−、及びトリ−グリセリド)を含めた脂肪酸、硬化脂肪、炭化水素、通常のロ
ウ(normal waxes)、ステアリン酸、ステアリルアルコール、及び、炭化水
素骨格を有する疎水性及び親水性材料を含んでいてよい。適切なロウは、例えば、蜜ロウ
、糖ロウ、ヒマシロウ(castor wax)、及びカルナウバロウを含む。本発明の目的上、ロウ
様物質は、室温では通常固体であって、約25℃から約100℃までの融点を有するあら
ゆる材料として定義される。
【0082】
本発明に従って使用され得る適切な疎水性材料は、消化可能な長鎖(C8−C50、特に
はC12−C40)の置換または非置換炭化水素、例えば脂肪酸、脂肪族アルコール、脂肪酸
のグリセリルエステル、鉱油及び植物油、及び、天然及び合成のロウ等を含む。25℃か
ら90℃までの間の融点を有する炭化水素が好適である。特定の実施態様では、長鎖炭化
水素材料のうち、脂肪(脂肪族)アルコールが好適である。本経口用剤形は、60重量%
までの少なくとも1つの消化可能な長鎖炭化水素を含んでいてよい。
【0083】
好適には、2つもしくはそれ以上の疎水性材料の組み合わせが本マトリックス製剤に含
まれている。付加的な疎水性材料が含まれている場合、その材料は、好適には、天然及び
合成のロウ、脂肪酸、脂肪族アルコール、及びそれらの混合物から選択される。それらの
例は、蜜ロウ、カルナウバロウ、ステアリン酸、及びステアリルアルコールを含む。この
リストは、排他的であることを意図したものではない。
【0084】
一つの特に好適なマトリックスは、少なくとも1つの水溶性ヒドロキシアルキルセルロ
ース、少なくとも1つのC12−C36、好適にはC14−C22の脂肪族アルコール、及び、場
合によっては、少なくとも1つのポリアルキレングリコールを含む。前述の少なくとも1
つのヒドロキシアルキルセルロースは、好適には、ヒドロキシ(C1からC6までの)アル
キルセルロース、例えばヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセル
ロース、及び、特にはヒドロキシエチルセルロース等である。本経口用剤形における前述
の少なくとも1つのヒドロキシアルキルセルロースの量は、とりわけ、必要とされるオキ
シコドンの精確な放出速度によって決まるであろう。前述の少なくとも1つの脂肪族アル
コールは、例えば、ラウリルアルコール、ミリスチルアルコール、またはステアリルアル
コールであってよい。しかし、本経口用剤形の特に好適な実施態様では、その少なくとも
1つの脂肪族アルコールはセチルアルコールまたはセトステアリルアルコールである。本
経口用剤形における前述の少なくとも1つの脂肪族アルコールの量は、上述の如く、必要
とされるオキシコドンの精確な放出速度によって決まるであろう。また、その量は、本経
口用剤形に少なくとも1つのポリアルキレングリコールが存在しているか存在していない
かにも依存するであろう。少なくとも1つのポリアルキレングリコールが存在していない
場合、本経口用剤形は、好適には、20重量%から50重量%までの間の少なくとも1つ
の脂肪族アルコールを含む。本経口用剤形に少なくとも1つのポリアルキレングリコール
が存在している場合には、その少なくとも1つの脂肪族アルコールとその少なくとも1つ
のポリアルキレングリコールを合わせた重量が、好適には、合計用量のうちの20重量%
から50重量%までを構成する。
【0085】
一つの実施態様では、例えば前述の少なくとも1つのヒドロキシアルキルセルロースま
たはアクリル樹脂と前述の少なくとも1つの脂肪族アルコール/ポリアルキレングリコー
ルとの比は、その少なくとも1つのヒドロキシアルキルセルロースとその少なくとも1つ
の脂肪族アルコール/ポリアルキレングリコールの(w/w)が1:2から1:4までの
間であることが好適であり、1:3から1:4までの間の比が特に好適であることから決
定される。
【0086】
前述の少なくとも1つのポリアルキレングリコールは、例えば、ポリプロピレングリコ
ールであってよく、好適には、ポリエチレングリコールであってよい。この少なくとも1
つのポリアルキレングリコールの数平均分子量は、好適には1,000から15,000
までの間であり、特には1,500から12,000までの間である。
【0087】
別の適切な徐放出性マトリックスは、アルキルセルロース(特にはエチルセルロース)
、C12からC36までの脂肪族アルコール、及び、場合によっては、ポリアルキレングリコ
ールを含むであろう。
【0088】
別の好適な実施態様では、本マトリックスは、少なくとも2つの疎水性材料からなる薬
学的に許容可能な組み合わせを含む。
【0089】
上述の成分に加え、徐放出性マトリックスは、適切な量の他の材料、例えば製薬業界に
おいて普通に用いられている希釈剤、潤滑剤、結合剤、粒状化助剤(granulating aids)、
着色剤、風味剤(flavorants)、及びグリダント(glidants)等も含んでいてよい
。
【0090】
(マトリックス−粒状体)
本発明による固体の徐放出性経口用剤形の調製を容易化するため、当業者に知られたマ
トリックス製剤のあらゆる調製方法を使用することができる。例えばマトリックスへの組
み込みは、例えば(a)少なくとも1つの水溶性ヒドロキシアルキルセルロースと、オキ
シコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(またはナロキ
ソン塩)を含有する顆粒を形成し;(b)それらのヒドロキシアルキルセルロース含有顆
粒を少なくとも1つのC12−C36脂肪族アルコールと混合し;そして、(c)場合によっ
ては、それらの顆粒を圧縮及び成形することにより果たすことができる。好適には、それ
らの顆粒は、水を用いて上述のヒドロキシアルキルセルロース顆粒を湿式造粒することに
より形成される。
【0091】
更に別の代替的な実施態様では、スフェロイドを形成すべく、球状化剤(spheronizing
agent)をオキシコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(
またはナロキソン塩)と共に球状化することができる。微結晶セルロースは好適な球状化
剤である。適切な微結晶セルロースは、例えば、Avicel PH 101(商標、F
MC Corporation)として販売されている材料である。そのような実施態様
では、本活性成分及び球状化剤に加え、それらのスフェロイドは結合剤も含んでいてよい
。適切な結合剤、例えば低粘度の水溶性ポリマー等は、製薬業界における当業者に広く知
られている。しかし、水溶性のヒドロキシ低級アルキルセルロース、例えばヒドロキシプ
ロピルセルロース等が好適である。付加的に(あるいは代替的に)、それらのスフェロイ
ドは、水に不溶性のポリマー、特にはアクリルポリマー、アクリルコポリマー、例えばメ
タクリル酸−エチルアクリレートコポリマー等、あるいはエチルセルロースを含んでいて
よい。そのような実施態様では、本徐放出性コーティングは、一般的に、疎水性材料、例
えば(a)単独もしくは脂肪族アルコールとの混和物のいずれかの形態におけるロウ;ま
たは(b)セラックあるいはゼイン等を含むであろう。
【0092】
(溶融押出しマトリックス)
また、徐放出性マトリックスは、溶融造粒法または溶融押出し法によって調製すること
もできる。一般的に、溶融造粒法は、常態では固体の疎水性材料、例えばロウ等を溶融し
、そこへ粉末化された薬剤を組み入れる操作を含む。徐放出性剤形を得るためには、その
溶融ロウ疎水性材料に付加的な疎水性物質、例えばエチルセルロース、または水に不溶性
のアクリルポリマー等を組み入れることが必要になり得る。溶融造粒法により調製された
徐放出性製剤の例は、米国特許第4,861,598号で見ることができる。
【0093】
上述の付加的な疎水性材料は、1つもしくはそれ以上の水に不溶性のロウ様熱可塑性物
質を含んでいてよく、そして、その熱可塑性物質は、恐らく、この1つもしくはそれ以上
の水に不溶性のロウ様物質よりも疎水性に劣る1つもしくはそれ以上ロウ様熱可塑性物質
と混合されていよう。一定の放出を達成するため、本製剤におけるそれら個々のロウ様物
質は、初期放出段階の間、胃腸液中において実質的に非分解性であって且つ不溶性である
べきである。水に不溶性の有用なロウ様物質は、約1:5,000(w/w)より低い水
溶解度を有する物質であり得る。
【0094】
上述の成分に加え、徐放出性マトリックスは、適切な量の他の材料、例えば製薬業界に
おいて普通に用いられている希釈剤、潤滑剤、結合剤、粒状化助剤、着色剤、風味剤、及
びグリダント等も含んでいてよい。これらの付加的な材料の量は、所望の製剤に望ましい
効果をもたらすのに充分な量であろう。
【0095】
上述の成分に加え、溶融押出し多重粒状体を組み入れた徐放出性マトリックスは、適切
な量の他の材料、例えば製薬業界において普通に用いられている希釈剤、潤滑剤、結合剤
、粒状化助剤、着色剤、風味剤、及びグリダント等も、望ましい場合には、重量で粒状体
の約50%の量まで含んでいてよい。
【0096】
経口用剤形を調合するのに使用され得る、薬学的に許容可能な担体及び賦形剤の具体的
な例が、Handbook of Pharmaceutical Excipients、American Pharmaceutical Associati
on(1986年)に記載されている。
【0097】
(溶融押出し多重粒状体)
本発明による適切な溶融押出しマトリックスの調製は、例えば、オキシコドン(または
オキシコドン塩)及び/又はナロキソン(またはナロキソン塩)を、少なくとも1つの疎
水性材料、及び、好適には、均一な混合物を得るための付加的な疎水性材料と共に混合す
るステップを含んでいてよい。次いで、その均一な混合物は、少なくともその混合物を充
分に押し出せるほどに軟化させるのに充分な温度にまで加熱される。その後、結果として
得られたその均一な混合物は、ストランドを形成すべく押し出される。その押出し物は、
好適には、冷却され、そして、当技術分野において既知の何らかの手段により多重粒状体
の形態に切断される。それらのストランドは、冷却され、そして、多重粒状体の形態に切
断される。その後、それらの多重粒状体は単位量に分けられる。この押出し物は、好適に
は、約0.1mmから約5mmまでの範囲の直径を有しており、そして、約8時間から約
24時間までの期間にわたる治療的に活性な物質の徐放出性をもたらす。
【0098】
本発明の溶融押出し品を調製するための一つの随意的なプロセスは、疎水性材料、オキ
シコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(またはナロキ
ソン塩)、及び、随意的な結合剤を計量して押出機に直接的に供給するステップ;その均
一な混合物を加熱するステップ;均一な混合物を押し出し、これにより、ストランドを形
成するステップ;均一な混合物を含有するそれらのストランドを冷却するステップ;それ
らのストランドを切断して、約0.1mmから約12mmまでのサイズを有する粒子に為
すステップ;及び、それらの粒子を単位量に分けるステップを含む。本発明のこの態様で
は、比較的連続的な製造手順が実現される。
【0099】
また、押出機の開口または出口ポートの直径も、押し出されるストランドの太さを変え
るべく調節することができる。更に、押出機の出口部分は丸形である必要はない;長円形
や矩形等であってよい。出てくるストランドは、熱線切断機や断裁機等を用いて粒子に為
すことができる。
【0100】
溶融押出し多重粒状体系は、押出機の出口オリフィスに応じて、例えば顆粒、スフェロ
イド、またはペレットの形態であってよい。本発明の目的上、「(1つもしくは複数の)
溶融押出し多重粒状体」及び「(1つもしくは複数の)溶融押出し多重粒状体系」及び「
溶融押出し粒子」という用語は、好適には同様なサイズ及び/又は形の範囲内にあり、そ
して、好適には本明細書で説明されている通りの疎水性材料を含め、1つもしくはそれ以
上の活性物質と1つもしくはそれ以上の賦形剤を含有する複数の単位を表すものとする。
この点について、本溶融押出し多重粒状体は、長さが約0.1mmから約12mmまでの
範囲であり、そして、約0.1mmから約5mmまでの直径を有するであろう。更に、本
溶融押出し多重粒状体は、このサイズ範囲内において、あらゆる幾何学的形状であってよ
いことを理解すべきである。代替的に、その押出し物を単に所望の長さに切断し、そして
、球状化ステップを必要とせずに、治療的に活性な物質の単位量に分けることもできる。
【0101】
一つの好適な実施態様では、経口用剤形は、有効量の溶融押出し多重粒状体をカプセル
内に包含させるべく調製される。例えば、複数の本溶融押出し多重粒状体を、経口摂取さ
れて胃液と接触したときに効果的な徐放出性の用量をもたらすのに充分な量でゼラチンカ
プセルに入れることができる。
【0102】
別の好適な実施態様では、適切な量の多重粒状体押出し物が、標準的な技術を利用した
通常の錠剤機を用いて経口用錠剤の形態に圧縮される。(圧縮及び成形される)錠剤、(
硬質及び軟質のゼラチン)カプセル剤、及び丸剤を製造するための技術及び組成物につい
ても、Remington’s Pharmaceutical Sciences(編集者、Arthur Osol)、1
553−1593頁(1980年)に記載されている。
【0103】
更に別の好適な実施態様では、上でもっと詳しく記載されている米国特許第4,957
,681号(Klimeschら)で説明されているようにして、その押出し物を錠剤の
形態に成形することができる。
【0104】
場合によっては、それらの徐放出性溶融押出し多重粒状体系または錠剤は、徐放出性コ
ーティング、例えば上で説明されている徐放出性コーティング等でコーティングされてよ
く、あるいは、ゼラチンカプセルを例えば上で説明されている徐放出性コーティング等で
更に被覆することができる。そのオーバーコーティングは、他のものの中でもとりわけ、
所望の放出速度に大きく依存するが、そのようなコーティングは、好適には、約2パーセ
ントから約30パーセントまでの重量増加レベルを得るのに充分な量の疎水性材料を含む
。
【0105】
本発明の溶融押出し単位剤形は、更に、カプセルに入れられる前の溶融押出し粒子(例
えば、オキシコドン(またはオキシコドン塩)を伴う一群の粒子とナロキソン(またはナ
ロキソン塩)を伴う一群の粒子)の組み合わせを含んでいてよい。更に、それらの単位剤
形は、即座に放出するためのある量の即時放出性物質も含むことができる。その即時放出
性物質は、例えばゼラチンカプセル内における分離したペレットとして組み入れられてよ
く、あるいは、それらの剤形(例えば、徐放出性コーティング剤形またはマトリックスを
ベースとした剤形)の調製後、多重粒状体の表面にコーティングされてもよい。また、本
発明の単位剤形は、所望の効果を達成するため、徐放出性のビーズとマトリックス多重粒
状体の組み合わせも含んでいてよい。
【0106】
好適には、本発明の徐放出性製剤は、例えば経口摂取されて胃液次いで腸液に晒された
ときに、(1つもしくは複数の)本物質をゆっくりと放出する。本発明の溶融押出し製剤
の徐放出プロフィールは、例えば遅延剤の量、即ち、疎水性材料の量を変更することによ
り、疎水性材料に対する可塑剤の量を変更することにより、付加的な成分または賦形剤を
含めることにより、製造方法を変更することなどにより変えることができる。
【0107】
本発明の別の実施態様では、溶融押出し材料は、オキシコドン(またはオキシコドン塩
)及びナロキソン(またはナロキソン塩)の包含を伴うことなく調製され、そして、その
後、オキシコドン(またはオキシコドン塩)及びナロキソン(またはナロキソン塩)をそ
の押出し物に加えることができる。そのような製剤は、典型的には、押出しマトリックス
材料と共に混合された本物質を有しており、その後、ゆっくりと放出する製剤をもたらす
ため、その混合物が錠剤化されよう。
【0108】
(コーティング)
本発明の剤形は、場合によっては、1つもしくはそれ以上の放出を調節するのに適した
材料もしくは本製剤を保護するのに適した材料でコーティングされてよい。一つの実施態
様では、コーティングは、pH−依存性の放出もしくはpH−非依存性の放出のいずれか
を可能にするために施される。pH−依存性のコーティングは、患者に対して少なくとも
約8時間、好適には約12時間から約24時間までにわたる鎮痛効果を与えることができ
る吸収プロフィールがもたらされるように、オキシコドンを胃腸(GI)管の望ましい領
域で、例えば胃または小腸で放出するように機能する。pH−非依存性コーティングが所
望の場合、そのコーティングは、例えばGI管等の環境流体におけるpH−変化に関係な
く最適な放出が達成されるように設計される。また、GI管の一つの望ましい領域、例え
ば胃等で用量の一部を放出し、そして、GI管の別の領域、例えば小腸で残りの用量を放
出する組成物を調合することもできる。
【0109】
製剤を得るためにpH−依存性コーティングを利用する本発明による製剤は、反復−作
用効果(repeat−action effect)を与えることもでき、そこでは、
非保護薬剤は、腸溶コーティング上にコーティングされて、胃で放出され、一方、腸溶コ
ーティングで保護されている残りの薬剤は、胃腸管を更に下って放出される。本発明に従
って使用され得るpH−依存性のコーティングは、セラック、セルロースアセテートフタ
レート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピル
メチルセルロースフタレート、及びメタクリル酸エステルコポリマー、ゼイン等を含む。
【0110】
特定の好適な実施態様では、オキシコドンまたはそれの塩、及び、場合によっては、ナ
ロキソンまたはそれの塩を含有する基材(例えば、錠剤のコアビーズ、マトリックス粒子
)は、(i)アルキルセルロース;(ii)アクリルポリマー;または(iii)それら
の混合物から選択される疎水性材料でコーティングされる。そのコーティングは、有機ま
たは水性の溶液または分散系の形態で適用されてよい。そのコーティングは、所望の徐放
出プロフィールを得るため、基材の約2%から約25%までの重量増加が得られるように
適用されてよい。水性分散系から誘導されるコーティングについては、例えば米国特許第
5,273,760号及び第5,286,493号で詳しく説明されている。
【0111】
本発明に従って使用され得る徐放出性製剤またはコーティングの他の例は、米国特許第
5,324,351号;第5,356,467号、及び第5,472,712号を含む。
【0112】
(アルキルセルロースポリマー)
アルキルセルロースを含め、セルロース性の材料及びポリマーは、本発明によるビーズ
をコーティングするのに非常に適した疎水性材料を提供する。単なる例として挙げると、
一つの好適なアルキルセルロースポリマーはエチルセルロースであるが、当業者であれば
、本発明による疎水性コーティングの全体または一部として、他のセルロース及び/又は
アルキルセルロースポリマーも、単独もしくは何らかの組み合わせにおいて、容易に使用
され得ることが認識されよう。
【0113】
一つの商業的に入手可能なエチルセルロースの水性分散系はAquacoat(登録商
標)(FMC Corp.、フィラデルフィア、ペンシルバニア州、U.S.A.)であ
る。Aquacoat(登録商標)は、エチルセルロースを水に不混和性の有機溶媒中に
溶解し、その後、界面活性剤と安定剤の存在下において、それを水中に乳化させることに
より調製される。均質化してサブミクロンオーダーの液滴を発生させた後、真空下でその
有機溶媒を蒸発させることにより、シュードラテックス(pseudolatex)を形成する。可塑
剤は、製造段階中には、このシュードラテックスに組み入れられない。従って、それをコ
ーティングとして使用するのに先立って、Aquacoat(登録商標)を適切な可塑剤
と使用前に直接的に混合する必要がある。エチルセルロースの別の水性分散系は、Sur
elease(登録商標)(Colorcon,Inc.、ウェストポイント、ペンシル
バニア州、U.S.A.)として商業的に入手することができる。この製品は、可塑剤を
製造段階中に分散系に組み入れることにより調製される。ポリマー、可塑剤(セバシン酸
ジブチル(dibutyl sebacate))、及び安定剤(オレイン酸)のホットメルトが均一な混合
物として調製され、その後、基材に直接的に適用できる水性分散系を得るべく、この混合
物がアルカリ性溶液で希釈される。
【0114】
(アクリルポリマー)
本発明の他の好適な実施態様では、徐放出性コーティングを含む疎水性材料は、薬学的
に許容可能なアクリルポリマーであり、それらのアクリルポリマーは、これらに限定する
ものではないが、アクリル酸とメタクリル酸のコポリマー、メチルメタクリレートコポリ
マー、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸
)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メチルメタ
クリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)コポリマー、ポリア
クリルアミド、アミノアルキルメタクリレートコポリマー、ポリ(無水メタクリル酸)、
及びグリシジルメタクリレートコポリマーを含む。
【0115】
特定の好適な実施態様では、そのアクリルポリマーは1つもしくはそれ以上のアンモニ
オメタクリレートコポリマーを含む。アンモニオメタクリレートコポリマーは、当技術分
野において広く知られており、低含量の第四級アンモニウム基を伴うアクリル酸及びメタ
クリル酸エステルの充分に重合されたコポリマーとしてNF XVIIに記載されている
。
【0116】
望ましい溶解プロフィールを得るため、例えば中性(メタ)アクリルエステルに対する
第四級アンモニウム基のモル比が異なっている等の異なる物理的特性を有する2つもしく
はそれ以上のアンモニオメタクリレートコポリマーを組み入れることが必要になり得る。
【0117】
特定のメタクリル酸エステル型ポリマーは、本発明に従って使用され得るpH−依存性
コーティングを調製するのに有用である。例えば、メタクリル酸コポリマーまたは高分子
メタクリレートとしても知られているジエチルアミノエチルメタクリレートと他の中性メ
タクリルエステルから合成される一群のコポリマーがあり、Rohm Tech,Inc
.からEudragit(登録商標)として商業的に入手可能である。幾つかの異なるタ
イプのEudragit(登録商標)がある。例えば、Eudragit(登録商標)E
は、酸性媒質中において膨潤及び溶解するメタクリル酸コポリマーの一例である。Eud
ragit(登録商標)Lは、約pH<5.7では膨潤せず、約pH>6で溶けるメタク
リル酸コポリマーである。Eudragit(登録商標)Sは、約pH<6.5では膨潤
せず、約pH>7で溶ける。Eudragit(登録商標)RL及びEudragit(
登録商標)RSは水に対して膨潤性であり、これらのポリマーによって吸収される水の量
はpH−依存性であるが、Eudragit(登録商標)RL及びRSでコーティングさ
れた剤形はpH−非依存性である。
【0118】
特定の好適な実施態様では、そのアクリルコーティングは、それぞれEudragit
(登録商標)RL30D及びEudragit(登録商標)RS30Dという商品名でR
ohm Pharmaから商業的に入手可能な2つのアクリル樹脂ラッカーの混合物を含
んでいる。Eudragit(登録商標)RL30D及びEudragit(登録商標)
RS30Dは、低含量の第四級アンモニウム基を伴うアクリル及びメタクリルエステルの
コポリマーであり、残りの中性(メタ)アクリルエステルに対するアンモニウム基のモル
比は、Eudragit(登録商標)RL30Dの場合は1:20であり、Eudrag
it(登録商標)RS30Dの場合は1:40である。その平均分子量は約150,00
0である。RL(高浸透性)及びRS(低浸透性)というコードの意味は、これらの物質
の浸透特性を表している。Eudragit(登録商標)RL/RS混合物は、水及び消
化液中において不溶性である。しかし、これらから形成されるコーティングは、水性溶液
及び消化液中において膨潤性であり、且つ、浸透性である。
【0119】
本発明のEudragit(登録商標)RL/RS分散系は、望ましい溶解プロフィー
ルを有する徐放出性製剤を最終的に得るため、あらゆる望ましい比で相互に混合されてよ
い。望ましい徐放出性製剤は、例えば、100%のEudragit(登録商標)RL、
50%のEudragit(登録商標)RLと50%のEudragit(登録商標)R
S、及び10%のEudragit(登録商標)RL:90%のEudragit(登録
商標)RSから誘導される遅延剤コーティングから得ることができる。当業者であれば、
勿論、例えばEudragit(登録商標)L等の他のアクリルポリマーも使用し得るこ
とが認識されよう。
【0120】
(可塑剤)
コーティングが疎水性材料の水性分散系を含む本発明の実施態様では、その疎水性材料
の水性分散系に有効量の可塑剤を含めることにより、その徐放出性コーティングの物理的
特性が一層改善される。例えば、エチルセルロースは、比較的高いガラス転移温度を有し
ており、通常のコーティング条件下では可撓性のフィルムを形成しないため、それをコー
ティング材料として使用する前に、エチルセルロースコーティングを含有する徐放出性コ
ーティングに可塑剤を組み入れることが好適である。一般的に、コーティング溶液に組み
込まれる可塑剤の量は、そのフィルム形成剤の濃度に基づいており、例えば、フィルム形
成剤の重量で約1パーセントから約50パーセントまでであることが最も多い。しかし、
可塑剤の濃度は、個々のコーティング溶液と適用方法を用いて慎重な実験を行った後にの
み、適切に決定することができる。
【0121】
エチルセルロースに対する適切な可塑剤の例は、水に不溶性の可塑剤、例えばセバシン
酸ジブチル、ジエチルフタレート、トリエチルシトレート、トリブチルシトレート、及び
トリアセチンを含むが、但し、水に不溶性な他の可塑剤(例えばアセチル化モノグリセリ
ド、フタル酸エステル、ヒマシ油、等)も使用することができる。トリエチルシトレート
は、本発明のエチルセルロースの水性分散系に対する特に好適な可塑剤である。
【0122】
本発明のアクリルポリマーに対する適切な可塑剤の例は、これらに限定するものではな
いが、トリエチルシトレートNF XVI等のクエン酸エステル、トリブチルシトレート
、ジブチルフタレート、及び、恐らくは1,2−プロピレングリコールを含む。Eudr
agit(登録商標)RL/RSラッカー溶液等のアクリルフィルムから形成されるフィ
ルムの弾性を高めるのに適していることが判明している他の可塑剤は、ポリエチレングリ
コール、プロピレングリコール、ジエチルフタレート、ヒマシ油、及びトリアセチンを含
む。トリエチルシトレートは、本発明のエチルセルロースの水性分散系に対する特に好適
な可塑剤である。
【0123】
更に、少量のタルクの付加は、処理中にこの水性分散系がくっつく傾向を低減し、且つ
、つや出し剤として作用することが判明している。
【0124】
(徐放出性浸透圧性剤形)
また、本発明による徐放出性剤形は、浸透圧性投与製剤として調製されてもよい。この
浸透圧性剤形は、好適には、薬剤層(オキシコドン(またはオキシコドン塩)、及び、場
合によっては、ナロキソン(またはナロキソン塩)を含む)と送給または押し出し層(ナ
ロキソン(またはナロキソン塩)を含んでいてよい)を含有する二層のコアを含んでおり
、そして、その二層のコアは、場合によっては少なくとも1つの通路が配置された、半透
性の壁で覆われている。
【0125】
本発明の目的で使用される「通路」という表現は、そこを通ってオキシコドンまたはオ
キシコドン塩が(ナロキソンまたはナロキソン塩と共に、もしくはそれらを伴わずに)繊
維、毛細管、多孔性オーバーレイ、多孔質インサート、微孔性メンバー、または多孔性組
成物を通じてポンピング、拡散、または移動され得る開口、オリフィス、穴、細孔、多孔
質エレメントを含む。また、この通路は、使用環境流体中において壁から浸食または浸出
されて少なくとも1つの通路を生成する化合物を含んでいてもよい。通路を形成するため
の代表的な化合物は、上述の壁中において浸食可能なポリ(グリコール)酸またはポリ(
乳)酸;ゼラチン状フィラメント;水により除去可能なポリ(ビニルアルコール);浸出
可能な化合物、例えば流体により除去可能な細孔形成ポリサッカリド、酸、塩、または酸
化物を含む。通路は、徐放出寸法の細孔−通路を形成すべく、上述の壁からある化合物、
例えばソルビトール、スクロース、ラクトース、マルトース、またはフルクトース等を浸
出させることにより形成することができる。その通路は、本剤形からのオキシコドンまた
はオキシコドン塩の持続的な計量的放出に役立つあらゆる形状、例えば丸形、三角形、四
角形、及び長円形等を為していてよい。この剤形は、本剤形の1つもしくはそれ以上の表
面に離間した関係において1つもしくはそれ以上の通路を有する状態で製造することがで
きる。通路及び通路を形成するための装置が米国特許第3,845,770号;第3,9
16,899号;第4,063,064号及び第4,088,864号に開示されている
。ある徐放出速度の放出用細孔をもたらすべく水性浸出により形成された放出用細孔とし
てサイジング、成形、及び適合化された徐放出寸法を含む通路が米国特許第4,200,
098号及び第4,285,987号に開示されている。
【0126】
特定の実施態様では、上述の二層のコアは、オキシコドンまたはそれの塩を有する薬剤
層と、ナロキソンまたはそれの塩を含有する変位または押し出し層を含んでいる。特定の
実施態様では、その薬剤層は、少なくとも1つのポリマーヒドロゲルも含んでいてよい。
そのポリマーヒドロゲルは、約500から約6,000,000までの間の平均分子量を
有していてよい。ポリマーヒドロゲルの例は、これらに限定されるものではないが、化学
式(C6H12O5)n・H2O[式中、nは3から7,500までである]を含み、500か
ら1,250,000までの数平均分子量を含むマルトデキストリンポリマー;例えば5
0,000から750,000までの重量平均分子量を有するポリ(エチレンオキシド)
及びポリ(プロピレンオキシド)で代表され、より特定的には、少なくとも100,00
0、200,000、300,000、または400,000の重量平均分子量のうちの
1つからなるポリ(エチレンオキシド)で代表されるポリ(アルキレンオキシド);10
,000から175,000までの重量平均分子量からなるアルカリカルボキシアルキル
セルロース[ここで、アルカリはナトリウムまたはカリウムであり、アルキルはメチル、
エチル、プロピル、またはブチルである];及び10,000から500,000までの
数平均分子量からなるエチレン−アクリル酸(メタクリル及びエタクリル酸を含む)のコ
ポリマーを含む。
【0127】
本発明の特定の実施態様では、上述の送給または押し出し層はオスモポリマー(osm
opolymer)を含んでいる。オスモポリマーの例は、これらに限定されるものでは
ないが、ポリアルキレンオキシド及びカルボキシアルキルセルロースからなるグループか
ら選択されるメンバーを含む。このポリアルキレンオキシドは、1,000,000から
10,000,000までの重量平均分子量を有している。そのポリアルキレンオキシド
は、ポリメチレンオキシド、ポリエチレンオキシド、ポリプロピレンオキシド、1,00
0,000の平均分子量を有するポリエチレンオキシド、5,000,000の平均分子
量を含むポリエチレンオキシド、7,000,000の平均分子量を含むポリエチレンオ
キシド、1,000,000の平均分子量を有する架橋ポリメチレンオキシド、及び平均
分子量が1,200,000のポリプロピレンオキシドからなるグループから選択される
メンバーであってよい。典型的なオスモポリマーカルボキシアルキルセルロースは、アル
カリカルボキシアルキルセルロース、ナトリウムカルボキシメチルセルロース、カリウム
カルボキシメチルセルロース、ナトリウムカルボキシエチルセルロース、リチウムカルボ
キシメチルセルロース、ナトリウムカルボキシエチルセルロース、カルボキシアルキルヒ
ドロキシアルキルセルロース、カルボキシメチルヒドロキシエチルセルロース、カルボキ
シエチルヒドロキシエチルセルロース、及びカルボキシメチルヒドロキシプロピルセルロ
ースからなるグループから選択されるメンバーを含む。この変位層で使用されるこれらの
オスモポリマーは、上述の半透壁を横断する浸透圧勾配を提示する。オスモポリマーは、
剤形内へ流体を吸い込むことによって浸透圧性ヒドロゲル(オスモゲルとしても知られて
いる)として膨潤及び膨張し、これにより、それらは、この浸透圧性剤形からオキシコド
ンまたはそれの薬学的に許容可能な塩を押し出す。
【0128】
また、上述の押し出し層は、オスマジェント(osmagents)及び浸透圧的に有
効な溶質としても知られている1つもしくはそれ以上の浸透圧的に有効な化合物も含んで
いてよい。それらは、例えば胃腸管からの環境液を剤形内へ吸い込み、上述の変位層の送
給運動力学に寄与する。浸透圧的に活性な化合物の例は、浸透圧性の塩及び浸透圧性の炭
水化物からなるグループから選択されるメンバーを含む。特定のオスマジェントの例は、
これらに限定されるものではないが、ナトリウムクロライド、カリウムクロライド、マグ
ネシウムサルフェート、リチウムホスフェート、リチウムクロライド、ナトリウムホスフ
ェート、カリウムサルフェート、ナトリウムサルフェート、カリウムホスフェート、グル
コース、フルクトース、及びマルトースを含む。
【0129】
上述の押し出し層は、場合によっては、9,000から450,000までの数平均分
子量を有するヒドロキシプロピルアルキルセルロースを含んでいてよい。そのヒドロキシ
プロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロ
ピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピ
ルブチルセルロース、及びヒドロキシプロピルペンチルセルロースからなるグループから
選択されるメンバーで代表される。
【0130】
上述の押し出し層は、場合によっては、無毒の着色剤または染料を含んでいてよい。着
色剤または染料の例は、これらに限定されるものではないが、食品医薬品局着色剤(FD
&C)、例えばFD&C第1号青色染料、FD&C第4号赤色染料等や、赤色酸化第二鉄
、黄色酸化第二鉄、チタンジオキシド、カーボンブラック、及びインジゴを含む。
【0131】
また、上述の押し出し層は、場合によっては、成分の酸化を抑制するための酸化防止剤
も含んでいてよい。酸化防止剤の幾つかの例は、これらに限定されるものではないが、ア
スコルビン酸、アスコルビルパルミテート、ブチル化ヒドロキシアニソール、2及び3t
ert−ブチル−4−ヒドロキシアニソールの混合物、ブチル化ヒドロキシトルエン、ナ
トリウムイソアスコルベート、ジヒドログアレチン酸(dihydroguaretic
acid)、カリウムソルベート、ナトリウムバイサルフェート、ナトリウムメタバイ
サルフェート、ソルビン酸、カリウムアスコルベート、ビタミンE、4−クロロ−2,6
−ジtertブチルフェノール、アルファトコフェロール、及びプロピルガレートからな
るグループから選択されるメンバーを含む。
【0132】
特定の代替的な実施態様では、本剤形は、オキシコドンまたはそれの薬学的に許容可能
な塩、ナロキソンまたはそれの薬学的に許容可能な塩、薬学的に許容可能なポリマー(例
えばポリエチレンオキシド)、場合によっては崩壊剤(例えばポリビニルピロリドン)、
場合によっては吸収増強剤(例えば脂肪酸、界面活性剤、キレート化剤、胆汁酸塩等)を
含有する均質なコアを含む。この均質なコアは、オキシコドンまたはそれの薬学的に許容
可能な塩を放出するための(上で定義されている通りの)通路を有する半透性の壁で覆わ
れている。
【0133】
特定の実施態様では、上述の半透性の壁は、セルロースエステルポリマー、セルロース
エーテルポリマー、及びセルロースエステル−エーテルポリマーからなるグループから選
択されるメンバーを含む。代表的な壁のポリマーは、セルロースアシレート、セルロース
ジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテ
ート、セルローストリアセテート、モノ−、ジ−、及びトリセルロースアルケニレート、
及び、モノ−、ジ−、及びトリセルロースアルキニレートからなるグループから選択され
るメンバーを含む。本発明で使用されるポリ(セルロース)は、20,000から7,5
00,000までの数平均分子量を有している。
【0134】
本発明の目的にかなった付加的な半透性ポリマーは、アセトアルデヒドジメチルセルロ
ースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチ
ルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテー
トジエチルアミノアセテート;半透性ポリアミド;半透性ポリウレタン;半透性スルホン
化ポリスチレン;米国特許第3,173,876号;第3,276,586号;第3,5
41,005号;第3,541,006号及び第3,546,876号に開示されている
ようなポリアニオンとポリカチオンの共沈により形成される半透性架橋ポリマー;米国特
許第3,133,132号でLoebとSourirajanが開示しているような半透
性ポリマー;半透性架橋ポリスチレン;半透性架橋ポリ(ナトリウムスチレンスルホネー
ト);半透性架橋ポリ(ビニルベンジルトリメチルアンモニウムクロライド);及び、半
透性の壁を横断する静水圧差または浸透圧差を大気圧で表したときに2.5×10-8(c
m2/hr・atm)から2.5×10-2(cm2/hr・atm)までの流体透過性を有
する半透性ポリマーを含む。本発明に有用な他のポリマーは、米国特許第3,845,7
70号;第3.916,899号及び第4,160,020号により;及び、Handbook o
f Common Polymers(スコット(Scott),J.R.及びW.J.Roff、197
1年、CRC Press、クリーブランド、オハイオ州)により、当技術分野において
既知である。
【0135】
特定の実施態様では、好適には、上述の半透性の壁は、無毒で不活性であり、そして、
その薬剤の調剤寿命(dispensing life)の間、それの物理的及び化学的
な完全性を維持する。特定の実施態様では、本剤形は結合剤を含む。結合剤の例は、これ
らに限定されるものではないが、5,000から350,000までの粘度平均分子量を
有する治療上許容可能なビニルポリマーを含み、そのようなビニルポリマーは、ポリ−n
−ビニルアミド、ポリ−n−ビニルアセトアミド、ポリ−n−ビニルピロリドンとしても
知られているポリ(ビニルピロリドン)、ポリ−n−ビニルカプロラクトン、ポリ−n−
ビニル−5−メチル−2−ピロリドン、及び、ポリ−n−ビニルピロリドンとビニルアセ
テート、ビニルアルコール、ビニルクロライド、ビニルフルオリド、ビニルブチレート、
ビニルラウレエート(laureate)、及びビニルステアレートからなるグループから選択され
るメンバーとのコポリマーからなるグループから選択されるメンバーによって代表される
。他の結合剤は、例えば、アカシア、デンプン、ゼラチン、及び平均分子量が9,200
から250,000までのヒドロキシプロピルアルキルセルロースを含む。
【0136】
特定の実施態様では、本剤形は潤滑剤を含み、そして、その潤滑剤は、金型の壁やパン
チの表面にくっつくのを防ぐため、その剤形の製造中に使用されてよい。潤滑剤の例は、
これらに限定されるものではないが、マグネシウムステアレート、ナトリウムステアレー
ト、ステアリン酸、カルシウムステアレート、マグネシウムオレエート、オレイン酸、カ
リウムオレエート、カプリル酸、ナトリウムステアリルフマレート、及びマグネシウムパ
ルミテートを含む。
【0137】
特定の好適な実施態様では、本発明は、10mgから40mgまでのオキシコドンまた
はそれの薬学的に許容可能な塩と、150,000から500,000までの平均分子量
を有する25mgから500mgまでのポリ(アルキレンオキシド)と、40,000の
平均分子量を有する1mgから50mgまでのポリビニルピロリドンと、0mgから約7
.5mgまでの潤滑剤、及び0.80mgから0.90mgまでのナロキソンまたはそれ
の塩とを含有する治療用組成物を含む。この0.80mgから0.90mgまでのナロキ
ソンまたはそれの塩は、好適には、上述の薬剤層にある。
【0138】
(坐剤)
本発明の徐放出性製剤は、適切な坐薬用基剤と本明細書に開示されている用量のオキシ
コドン(またはオキシコドン塩)及びナロキソン(またはナロキソン塩)とを含有する直
腸投与用の薬学的な坐薬として調合されてよい。徐放出性坐薬製剤の調製は、例えば米国
特許第5,215,758号に記載されている。
【0139】
吸収に先立って、薬剤は溶液の形態を為していなければならない。坐薬の場合には、溶
液となる前に、その坐薬基剤の溶解もしくは基剤の溶融が先んじて生じなければならず、
その後、その坐薬基剤からの薬剤が直腸液に分配される。身体内への薬剤の吸収は、坐薬
基剤によって変わり得る。従って、特定の薬剤と組み合わせて使用されるべき特定の坐薬
基剤は、その薬剤の物理的な特性を考慮に入れて選択されねばならない。例えば、脂質−
可溶性の薬剤は、直腸液内へ容易に分配されないが、脂質基剤に僅かにしか溶けない薬剤
は、直腸液内へ容易に分配されるであろう。
【0140】
薬剤の溶解時間(または放出速度)に影響を及ぼす種々の異なるファクターの中でもと
りわけ重要なファクターは、溶解溶媒媒質に供される薬剤物質の表面積、その溶液のpH
、その特定の溶媒媒質中におけるその物質の溶解度、及び、その溶媒媒質中における溶解
材料の飽和濃度の駆動力である。一般的に、直腸投与された坐剤からの薬物の吸収に影響
を及ぼすファクターは、坐剤のビヒクル、吸収部位のpH、薬剤のpKa、イオン化の程
度、及び脂質溶解度を含む。
【0141】
選択される坐薬基剤は、本発明の(1つもしくは複数の)物質に対して適合性を有して
いるべきである。更に、その坐薬基剤は、無毒で粘膜を刺激せず、直腸液中において溶融
または溶解し、そして、保管中は安定していることが好適である。
【0142】
水溶性の薬剤と水に不溶性の薬剤の両方に対する本発明の特定の好適な実施態様では、
その坐薬基剤は、鎖長がC12からC18までの飽和天然脂肪酸のモノ−、ジ−、及びトリグ
リセリドからなるグループから選択される脂肪酸ロウを含んでいる。
【0143】
本発明の坐剤の調製では、他の賦形剤を使用することもできる。例えば、直腸ルートを
介して投与するのに適した形を形成するため、ロウが使用されてよい。この系は、ロウを
伴わずに使用することもできるが、直腸投与及び経口投与のどちらの場合にも、ゼラチン
カプセルに充填される希釈剤の付加を伴う。
【0144】
商業的に入手可能な適切なモノ−、ジ−、及びトリグリセリドの例は、Novata
TM(タイプAB、AB、B、BC、BD、BBC、E、BCF、C、D、及び299)
(Henkel社製)、及びWitepsol TM(タイプH5、H12、H15、H
175、H185、H19、H32、H35、H39、H42、W25、W31、W35
、W45、S55、S58、E75、E76、及びE85)(Dynamit Nobe
l社製)という商品名の下で販売されている、12−18炭素原子鎖の飽和天然脂肪酸を
含む。
【0145】
他の薬学的に許容可能な坐薬基剤は、上述のモノ−、ジ−、及びトリグリセリドに対し
て全体的または部分的に置換されていてよい。この坐薬における基剤の量は、本剤形のサ
イズ(即ち、実際の重量)、使用される基剤(例えばアルギネート)及び薬剤の量によっ
て決定される。一般的には、坐薬基剤の量は、その坐剤の全重量のうち、重量で約20%
から約90%までである。好適には、坐薬中における坐薬基剤の量は、その坐剤の全重量
のうち、重量で約65%から約80%までである。
【0146】
(他の形態)
ここで開示されている本発明は、オキシコドン及びナロキソンの薬学的に許容可能なあ
らゆる塩を包含すべく意図されている。それらの薬学的に許容可能な塩は、これらに限定
されるものではないが、金属塩、例えばナトリウム塩、カリウム塩、セシウム塩等;アル
カリ土類金属、例えばカルシウム塩、マグネシウム塩等;有機アミン塩、例えばトリエチ
ルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、トリエタノールアミン塩、
ジシクロヘキシルアミン塩、N,N’−ジベンジルエチレンジアミン塩等;無機酸塩、例
えばハイドロクロライド、ハイドロブロミド、サルフェート、ホスフェート、テレフタレ
ート等;有機酸塩、例えばホルメート、アセテート、トリフルオロアセテート、マレエー
ト、酒石酸塩等;スルホネート、例えばメタンスルホネート、ベンゼンスルホネート、p
−トルエンスルホネート等;アミノ酸塩、例えばアルギネート(arginate)、ア
スパルギネート、グルタメート等を含む。
【0147】
オキシコドン(またはオキシコドン塩)とナロキソン(またはナロキソン塩)の組み合
わせは、通常の賦形剤、即ち、少なくともオキシコドンまたはそれの塩の徐放出性をもた
らすために当技術分野において知られている経口投与に適した薬学的に許容可能な有機ま
たは無機の担体物質と混ぜて使用することができる。薬学的に許容可能な適切な担体は、
これらに限定されるものではないが、アルコール、アラビアゴム、植物油、ベンジルアル
コール、ポリエチレングリコール、ゲレート(gelate)、炭水化物、例えばラクト
ース、アミロース、またはデンプン等、マグネシウムステアレート、タルク、ケイ酸、粘
性パラフィン、香油、脂肪酸モノグリセリド及びジグリセリド、ペンタエリトリトール(p
entaerythritol)脂肪酸エステル、ヒドロキシメチルセルロース、ポリビニルピロリドン
等を含む。これらの薬学的調製物は、滅菌されてよく、そして、望ましい場合には、補助
的な物質、例えば潤滑剤、崩壊剤、保存剤、安定剤、湿潤剤、乳化剤、浸透圧緩衝剤に影
響を及ぼすための塩、着色剤、矯味・矯臭(flavoring)及び/又は芳香剤等と混合されて
よい。経口的な使用が意図された本組成物は、当技術分野において既知のどんな方法によ
って調製されてもよく、そして、そのような組成物は、錠剤を製造するのに適した不活性
で無毒の薬学的に許容可能な賦形剤からなるグループから選択される1つもしくはそれ以
上の物質を含んでいてよい。そのような賦形剤は、例えば、ラクトース等の不活性な希釈
剤;コーンスターチ等の顆粒化及び崩壊剤;デンプン等の結合剤;及び、マグネシウムス
テアレート等の潤滑剤を含む。それらの錠剤は、コーティングされなくてもよいし、ある
いは、エレガンスさを得るため、または、活性成分の放出を遅らせるため、既知の技術に
よりコーティングされてもよい。また、経口的に使用するための製剤は、活性成分が不活
性な希釈剤と混合された硬質のゼラチンカプセル剤として提供されてもよい。
【0148】
本発明の経口用剤形は、錠剤、トローチ剤、甘味入り錠剤、粉末剤または顆粒剤、硬質
または軟質のカプセル剤、微小粒子(例えばマイクロカプセル、極小球体(microspheres)
等)、バッカル錠剤、坐剤等の形態であってよい。オキシコドン(またはオキシコドン塩
)とナロキソン(またはナロキソン塩)は、実質的に互いに相互分散(interdespersed)さ
れていてよい。
【0149】
特定の実施態様では、本発明は、上で説明されている通りの剤形を調製することにより
、経口用オキシコドン剤形(またはオキシコドン塩)の非経口的な乱用を防止する方法を
提供する。
【0150】
特定の実施態様では、本発明は、上で説明されている通りの剤形を調製することを含む
、経口用オキシコドン剤形の流用を防止する方法を提供する。
【0151】
特定の実施態様では、本発明は、上で説明されている剤形をヒト患者に投与することに
より疼痛を治療する方法を提供する。
【0152】
以下の実施例は、本発明の様々な態様を例証するものである。それらの実施例は、どの
ような仕方においても決して特許請求の範囲を限定するものと解釈すべきではない。
【実施例1】
【0153】
ナロキソンハイドロクロライドを含有する徐放出性オキシコドンハイドロクロライド製
剤を、以下の表1における調合により調製する:
【表1】

【0154】
この実施例では、ナロキソンハイドロクロライドジハイドレートが、顆粒化プロセス中
に本製剤に加えられる。そのプロセスを以下に説明する:
1.分散:ナロキソンHClを水に溶解し、その溶液をEudragit/トリアセチン
分散系に加える。
2.顆粒化:流動床グラニュレーターを用いて、そのEudragit/トリアセチン分
散系を、オキシコドンHCl、噴霧乾燥ラクトース、及びポビドンの混合物に噴霧する。
3.粉砕:その顆粒化物を取り出し、約1mmの開口(18メッシュのスクリーン)を有
するミルに通す。
4.ロウ引き:ステアリルアルコールを約50℃で融かし、高剪断混合機を用いて、上述
の粉砕顆粒化物に加える。トレーまたは流動床上で室温にまで冷却する。
5.粉砕:その冷却された顆粒化物を、約18メッシュのスクリーンを有するミルに通す
。
6.潤滑:混合機を用いて、その顆粒化物をタルク及びマグネシウムステアレートで潤滑
する。
7.圧縮:Kilian(登録商標)タブレットプレスを用いてその顆粒化物を圧縮し、
錠剤にする。
8.フィルムコーティング:ロータリーパンを用いて、その錠剤に水性のフィルムコーテ
ィングを施す。
【実施例2】
【0155】
以下の表2に示されている調合により、オキシコドン塩/ナロキソン塩の徐放出性浸透
圧性錠剤を製造する:
【表2】
【0156】
上述の調合を有する剤形を以下の手順により調製する:
先ず、オキシコドンハイドロクロライド、ナロキソンハイドロクロライドジハイドレー
ト、200,000の平均分子量を有するポリ(エチレンオキシド)、及び40,000
の平均分子量を有するポリビニルピロリドンを混合機に加え、10分間混合する。次いで
、その混合された材料に変性無水アルコールを10分間連続的に混合しながら加える。次
いで、その湿性顆粒化物を20メッシュのスクリーンに通し、室温で20時間乾燥させた
後、16メッシュのスクリーンに通す。次に、その顆粒化物を混合機へ移して混合し、マ
グネシウムステアレートで潤滑する。
【0157】
次いで、本剤形からオキシコドンHCl/ナロキソンHCl混合物を押し出すための変
位または押し出し用組成物を以下のようにして調製する:先ず、11,200の平均分子
量を有する3910gのヒドロキシプロピルメチルセルロースを45,339gの水に溶
解する。次いで、101gのブチル化ヒドロキシトルエンを650gの変性無水アルコー
ルに溶解する。次に、2.5kgのヒドロキシプロピルメチルセルロース水溶液をブチル
化ヒドロキシトルエンアルコール溶液に連続的に混合しながら加える。次いで、残りのヒ
ドロキシプロピルメチルセルロース水溶液をブチル化ヒドロキシトルエンアルコール溶液
に連続的に混合しながら加えることにより、結合剤溶液の調製を果たす。
【0158】
次に、21メッシュのスクリーンを備えたQuadro Comil(登録商標)ミル
を用いて、36,000gのナトリウムクロライドをサイジングする。次いで、1200
gの酸化第二鉄を40メッシュのスクリーンに通す。次いで、そのスクリーニングされた
材料、7,500,000の平均分子量を有する76,400gの薬学的に許容可能なポ
リ(エチレンオキシド)、11,200の平均分子量を有する2500gのヒドロキシプ
ロピルメチルセルロースを、Glatt(登録商標)流動床造粒機のボウルに加える。ボ
ウルを造粒機に取り付け、造粒プロセスを開始し、顆粒化を果たす。次に、その乾燥粉末
を空気懸濁させ、10分間混合する。次に、この粉末に上述の結合剤溶液を3本のノズル
から噴霧する。このプロセスの間、以下の如くに造粒を監視する:800g/分の合計溶
液噴霧速度;43℃のインレット温度、及び4300m3/時の空気流量。45,033
gの溶液噴霧終了後、結果として得られた被覆顆粒化粒子を35分間乾燥プロセスに掛け
る。
【0159】
8メッシュのスクリーンを有するQuadro Comil(登録商標)ミルを用いて
、それらのコーティングされた顆粒をサイジングする。その顆粒化物をTote(登録商
標)タンブラーへ移して混合し、281.7gのマグネシウムステアレートで潤滑する。
【0160】
次に、オキシコドンハイドロクロライド/ナロキソンハイドロクロライドを含有する医
薬組成物と押し出し用組成物を、Kilian(登録商標)タブレットプレスで二層錠剤
の形態に圧縮する。先ず、医薬組成物を金型のキャビティーに加えて予備圧縮した後、1
35mgの押し出し用組成物を加え、それらの層を3メートルトンの圧力ヘッド下におい
てプレスし、直径が11/32インチ(0.873cm)の接触層配列にする。
【0161】
この二層化された配列を半透性の壁でコーティングする。この壁形成組成物は、39.
8%のアセチル含量を有する100%セルロースアセテートを含む。その壁形成組成物を
アセトン:水(95:5の重量:重量)共溶媒に溶解して4%の固溶体を作製する。その
壁形成組成物を、24インチ(60cm)のVector(登録商標)ハイ−コーター内
において、上述の二層上及び二層の周りに噴霧する。次に、薬剤オキシコドン層を本剤形
の外面とつなぐため、その半透性の壁を通じて20ミル(0.508mm)の1つの出口
通路を穿孔する。45℃で45%の湿度において72時間乾燥させることにより、残存す
る溶媒を取り除く。次に、その浸透圧性投薬系を45℃で4時間乾燥させ、過剰な水分を
取り除く。
【実施例3】
【0162】
以下の表4に示されている調合により、オキシコドン10mg/ナロキソン0.85m
gの徐放出性カプセル剤を調製する:
【表3】
【0163】
上述の製剤を以下の手順により調製する:
1.ステアリルアルコールのフレークをインパクトミルに通す。
2.オキシコドンHCl、ナロキソンHCl、ステアリン酸、ステアリルアルコール、
及びEudragit RSPOを適切なブレンダー/ミキサー内で混合する。
3.その混合された材料を高温で双軸スクリュー押出し機に連続的に供給し、結果とし
て生じたストランドをコンベヤー上に収集する。
4.それらのストランドをコンベヤー上で冷却させる。
5.ペレタイザーを用いて、それらのストランドを1mmのペレットに切断する。
6.それらのペレットを、微細なペレットや大きすぎるペレットに関してスクリーニン
グし、サイズを約0.8mm−1.4mmの許容可能な範囲にする。
7.120mg/カプセルの充填重量でカプセルに充填する(サイズ2のカプセルに充
填する)。
【実施例4】
【0164】
以下の手順により、オキシコドン10mg/ナロキソン0.85mgの徐放出性カプセ
ル剤を調製する:
最初に、以下の表5に示されている調合により、即時放出性のオキシコドンビーズを調
製する:
【表4】
【0165】
(プロセス)
1.薬剤層形成溶液:オキシコドンHClとOpadryクリアーを水に溶解する。
2.薬剤のローディング:流動床乾燥機内において、薬剤溶液をNuPareilビー
ズに噴霧する。
3.コーティング:Opadryバタースコッチを水に分散させる。薬剤がローディン
グされたビーズに噴霧する。
【0166】
その後、以下の表6に示されている調合により徐放出性のビーズを調製する:
【表5】
【0167】
(プロセス)
1.制御放出性コーティング溶液:トリエチルシトレートを水中において均一化する。
その分散系をEudragit(登録商標)RS30D及びEudragit(登録商標
)RL30Dに加えた後、Cab−O−Sil(登録商標)をその混合物に加える。
2.シールコーティング溶液:Opadry(登録商標)クリアーを水に溶解する。
3.コーティング:流動床底面−噴霧法を用いて、オキシコドンHCl IRビーズに
、制御放出性コーティング溶液、続いて、シールコーティング溶液を適用する。
4.硬化:コーティングされたビーズをトレーに載せ、オーブン内において45℃で2
4時間硬化させる。
【0168】
オキシコドン/ナロキソンの徐放出性ビーズをもたらすため、上述の調合に、単位当た
り0.85mgのナロキソンを含めることができる。そのナロキソンは、NuParei
lビーズに噴霧する前に、オキシコドンHClと共に精製水中に溶解されてよい。
【実施例5】
【0169】
以下の表7における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の10mgオキシコドンハイドロクロライド製剤を調製した:
【表6】
【0170】
表7の製剤を以下の手順により調製した:
1.Niro MP−6流動床プロセッサー(Glatt GPCG−60/120で
置き換えることもできる)内において、オキシコドンHCl、ラクトース、及びポビドン
の一部を混合する。
2.Lightin’エアーミキサを用いて、Eudragit(登録商標)RS30
Dとトリアセチンをステンレス鋼製の容器内で混合する。
3.ステップ2から得られるEudragit(登録商標)の分散系を噴霧することに
より、Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、ステップ1から得られる混合物を顆粒化する。
4.Lightin’エアーミキサを用い、ステンレス鋼製の容器内において、残りの
ポビドン、ナロキソンHCl、及び水を混合し、透明な溶液を形成する。
5.Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、ステップ4から得られるナロキソンHCl溶液をステップ3から得
られる顆粒化物に噴霧する。
6.Quadro Comilを用いて、ステップ5から得られる顆粒化物をスクリー
ニングする。
7.Colletteグラニュレーター/ミキサー内において、ステアリルアルコール
(スチームジャケット付きのケトル内で前もって融かしておく)をステップ6から得られ
るスクリーニングされた顆粒化物と混合する。
8.Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、もしくは代替的に、ステンレス鋼製のトレー上において、ステップ
7から得られるロウ引きされた顆粒化物を冷却する。
9.Quadro Comilを用いて、ステップ8から得られる冷却された顆粒化物
の塊を除去する。
10.Colletteグラニュレーター/ミキサー内において、ステップ8から得ら
れる顆粒化物をタルク及びマグネシウムステアレートと混合する。
11.Kilian S−250タブレットプレスを用いて、顆粒化物を錠剤の形態に
圧縮する。
12.48″のAccela−Cotaコーティングパン内において、錠剤をOpad
ryでコーティングする。
【0171】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0172】
その溶解の結果が以下の表7Aに示されている:
【表7】
【実施例6】
【0173】
以下の表8における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の20mgオキシコドンハイドロクロライド製剤を調製した:
【表8】
【0174】
表8の製剤を実施例5と同じ手順により調製した。
【0175】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0176】
その溶解の結果が以下の表8Aに示されている:
【表9】
【実施例7】
【0177】
以下の表9における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の40mgオキシコドンハイドロクロライド製剤を調製した:
【表10】
【0178】
表9の製剤を実施例5と同じ手順により調製した。
【0179】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0180】
その溶解の結果が以下の表9Aに示されている:
【表11】
【実施例8】
【0181】
実施例8では、絶食状態下にある18人の正常で健常な若い成人の男性及び女性被験者
を対象として、単一施設におけるオープン−ラベル方式の無作為、4−期間、4−シーケ
ンス、4−治療様式交差試験を実施した。この試験は、Talwin(登録商標)Nx(
〜0.55mgのナロキソンハイドロクロライドを含有)の単回錠剤投与、単回投与にお
ける1.5mgの経口的なナロキソンハイドロクロライド、分割投与(0時間、3時間、
6時間、及び9時間において、それぞれ、0.75mg、〜0.25mg、〜0.25m
g、及び〜0.25mg)としての1.5mgの経口的なナロキソンハイドロクロライド
、及び0.5mgの0.4mg/mlの静注によるナロキソンハイドロクロライドにおけ
るナロキソンの薬物動力学的パラメーターを比較するために行われた。被験者は、以下の
ものからなる期間と絡めた4種類のシーケンスに無作為に割り当てられた:
期間1は、1−8日目(一回目の投与時点から二回目の投与時点まで)であり、
期間2は、8−15日目(二回目の投与時点から三回目の投与時点まで)であり、
期間3は、15−22日目(三回目の投与時点から四回目の投与時点まで)であり、そ
して
期間4は22日目(最後の投与時点から本試験の終了まで)であった。
【0182】
投与様式の無作為化シーケンスが以下の表10に列挙されている:
【表12】
【0183】
被験者は、0日目、7日目、14日目、及び21日目の晩に、少なくとも投与の12時
間前に試験施設に来院し、そして、投与してから少なくとも6時間後まで(ナロキソンH
Clを経口的に分割用量で服用するグループの場合には、投与してから少なくとも12時
間後まで)は施設内に留め置かれた。投与は、1日目、8日目、15日目、及び22日目
に行われた。被験者は、試験期間中、激しい運動をしないように指示された。
【0184】
本試験で使用したストック溶液及び薬剤は、Sabex Inc.から購入した2ml
バイアルの1mg/mlナロキソンHCl注射剤USPでSabex Inc.から購入
したもの、10mlバイアルの0.4mg/mlナロキソンHCl注射剤USPでSab
ex Inc.から購入したもの、及びTalwin(登録商標)Nx(それぞれ50m
gの塩基及び0.5mgの塩基に相当するペンタゾシン及びナロキソンハイドロクロライ
ドの錠剤(USP))からなる。スクリーニングでの静注によるナロキソンチャレンジ試
験では1mg/mlのナロキソン濃度を使用し、そして、試験期間中の静脈による投与で
は0.4mg/mlの濃度を使用した。経口的な投与では、1mg/mlのストック溶液
を水で0.02mg/mlまで希釈した。用量は以下の通りであった:1錠のTalwi
n(登録商標)Nx、0.5mgの静注によるナロキソンHCl(1.25mlの0.4
mg/ml)、75mlの単回経口用量(濃度0.02mg/ml)としての1.5mg
の経口的なナロキソンHCl、及び、0時間、3時間、6時間、及び9時間において投与
される、それぞれ、50%(0.75mg)、17%(〜0/25mg)、17%(〜0
.25mg)、及び16%(〜0.25mg)の用量における分割経口用量としての1.
5mgのナロキソンHCl。
【0185】
Talwin(登録商標)Nxは8オンスの水と共に投与され、被験者はこれを完全に
飲み干した。その後、口腔を検査し、各被験者がこの錠剤を飲み込んだことを確認した。
【0186】
ナロキソンの単回経口投与後、被験者は、2回の付加的な75mlのすすぎ水を飲み、
合計の容量で225mlを飲んだ。分割投与(同じく0.02mg/ml)の場合は、最
初の投与後、2回の94mlのすすぎ水を飲み、それ以降の投与後には2回の75mlの
すすぎ水を飲んだ。
【0187】
静注によるナロキソンは、IVプッシュにより投与された。IVナロキソン後、スタッ
フのメンバーが少なくとも20分間連続的に被験者を観察した。
【0188】
各被験者は、それぞれの治療様式を1回受けた。それぞれの治療は、少なくとも10時
間の夜通しの絶食(水を除く)後に行われ、そして、被験者は、投与後に更に4時間絶食
させられた。投与する日を除き、食物に制限はなかった。被験者が臨床室に留め置かれて
いる間、すべての食事が標準化された時間に出され、そして、食事の内容も、本試験期間
全体を通じ、且つ、被験者間で標準化された。その内容を含め、食事の記録を維持した。
被験者が本臨床施設にいる間、カフェインやキサンチンを含有する飲み物は出されなかっ
た。
【0189】
本試験では、18人の無作為化された被験者のうち17人が4種類すべての治療を全う
した。シーケンス4に無作為に割り当てられた1人の被験者は、最初の治療段階(単回投
与の経口ナロキソン)完了後、同意を撤回した。
【0190】
(薬物動力学の結果)
本試験での薬物動力学データ(AUCt、AUC0-∞、Cmax、t1/2、tmax、Cl、及
びVssを得た)の概要が以下の表11に列挙されている:
【表13】
【0191】
以下の表12は、用量を0.5mgのIVナロキソンHCl用量に規格化した薬物動力
学データの概要を列挙したものである。
【表14】
【0192】
4種類の治療様式でのナロキソンに対する算術平均観測血漿濃度−時間プロフィールが
図2A−Dに示されている。4種類の治療様式でのナロキソンに対する個々の観測血漿濃
度−時間プロフィールのスパゲッティープロットが図3A−Dに示されている。試験した
4種類すべての治療様式で、ナロキソンのピーク血漿濃度へ向けた急速な増大が観測され
た。予想していた通り、ピーク血漿濃度に達するまでの時間は、IV製剤(0.14時間
、〜9分)の場合が最も短く、続いて、Talwin(登録商標)Nx(0.71時間)
、ナロキソンの単回経口溶液投与(0.78時間)であり、最後がナロキソンの分割経口
投与(3.18時間)であった。0.5mgのIVナロキソンHCl、1.5mgのナロ
キソンHClの単回経口投与、1.5mgのナロキソンHClの分割経口投与、及びTa
lwin(登録商標)Nx(0.55mgのナロキソンHClを含有)で、それぞれ、6
252pg/ml、23.0pg/ml、13.4pg/ml、及び8.00pg/ml
の平均ピーク血漿露出(Cmax)が観測された。
【0193】
IV治療様式に対する平均見掛け終末(terminal)消失半減期(t1/2)は0
.92時間であった。血漿濃度時間曲線9及び15の終末部分は、この半減期の値を決定
するために使用することができなかった。3種類の経口治療様式に対しては、終末消失半
減期を見積もりしなかった(見掛け終末消失速度定数λを、大多数の被験者の「鋸歯」濃
度時間プロフィールに基づいて精確に決定することができなかったため)。
【0194】
1.5mgの単回経口投与(47.4pg・h/ml)の平均合計ナロキソン露出(A
UCt)は、1.5mgのナロキソンの分割経口投与(64.9pg・h/ml)の約7
3%であった。予想していた通り、0.5mgのナロキソンのIV投与は3199pg・
h/mlの最も高い平均合計露出をもたらし、一方、Talwin(登録商標)Nxによ
る治療様式は5.92pg・h/mlの平均合計露出値をもたらした。0.5mgのIV
治療様式の場合、無限大に外挿された平均合計露出(AUC0-∞)は3051pg・h/
mlであった。一方、3種類の経口治療様式に対しては、17人の被験者のうち15人で
終末消失半減期を見積もることができなかったため、AUC0-∞は見積もり不能であった
。
【0195】
IV治療様式の場合、定常状態における平均血漿クリアランス及び分配容量は、それぞ
れ、2854ml/分及び217Lと見積もられた。
【0196】
1.5mgのナロキソンHClの単回経口投与における平均絶対バイオアベイラビリテ
ィー(F)(AUCtに基づく)は0.47%(範囲:0.15−1.11%)であり、
1.5mgのナロキソンHClの分割経口投与の場合は0.66%(範囲:0.01−2
.08%)であり、そして、Talwin(登録商標)Nx(0.55mgのナロキソン
HClを含有)の単回錠剤投与の場合は0.17%(範囲:0.0−0.79%)であっ
た。これらの低いバイオアベイラビリティー値は、ナロキソンが経口的に投与されたとき
に生じる有意な第一径路(first pass)代謝を反映するものである。
【0197】
全体的に、すべての治療様式にわたり、ナロキソンCmax値に関連して、%CVの観点
における有意な変動性が見られた(範囲:52−72%)。IV治療様式に対するAUC
t値は比較的変動性が小さかった(%CV−26%)が、一方、経口的な治療様式は変動
性が大きかった(%CV範囲:68−122%)。
【0198】
(有害事象)
死亡または重篤な有害事象は無かった。有害事象のために中止した被験者はいなかった
。薬剤に関係する有害事象の大多数は、被験者がTalwin(登録商標)Nxを投与さ
れたときに起こった。これらの有害事象は、悪心、嘔吐、めまい、及び頭痛を含んだ。1
人の被験者は、スケジュールが合わなかったため、同意を撤回した。
【0199】
18人の被験者のうち11人が合計25例の有害事象を経験したが、どれも重篤なもの
ではなかった。これらの被験者のうち7人は、本試験の薬物適用に多分、恐らく、もしく
は確実に関係していると記述された有害事象を経験した。全体的に、有害事象は、ペンタ
ゾシンに関係した予想される有害事象と矛盾無く、Talwin(登録商標)Nxの投与
を受けた被験者で一層頻繁に報告された。すべての被験者がすべての有害事象から回復し
た。本試験薬剤に関しては、措置(例えば、中止)を何も講じなかった。
【0200】
Talwin(登録商標)Nxの投与後、6人の被験者が有害事象を報告し、それらの
有害事象は、すべて、本試験の薬物適用に恐らくまたは確実に関係していると記述され、
めまい(N=5)、悪心(N=2)、嘔吐(N=1)、及び疲労(N=1)を含んだ。T
alwin(登録商標)Nxの投与を受けた被験者の中でもとりわけ重度な有害事象は、
めまい(N=1)及び悪心(N=2)を含んだ。
【0201】
他の3種類の治療様式の期間中には、本試験の薬物適用と多分、恐らく、または確実に
関係している重度な有害事象を報告した被験者は誰もおらず、また、めまいまたは何らか
の重大性を報告した被験者も誰もいなかった。また、他の3つの治療グループの期間中に
は、2例の有害事象のみが、試験薬剤に多分、恐らく、または確実に関係していると考え
られた:分割投与における経口ナロキソンに多分関係した頭痛、及び、静注によるナロキ
ソンに多分関係した悪心。ナロキソンのチャレンジ試験期間中には、無作為に振り分けら
れた18人の被験者で有害事象は観察されなかった。
【0202】
最も一般的に報告された有害事象はめまいと頭痛であった。頭痛は5人の被験者で報告
され、それらの被験者は誰もTalwin(登録商標)Nx投与期間中の被験者ではなか
った。そのうちの1人の被験者の場合、無作為化の前に頭痛が起こり、3人の被験者では
、僅か1回の治療期間の間に頭痛が起こり、そして、1人の被験者では、2回の治療期間
の間に頭痛が起こった。分割投与における経口ナロキソン後に1人の被験者でのみ、その
頭痛が試験薬剤に多分、恐らく、または確実に関係していると考えられ、そして、その重
傷度は軽度であった。それ以外の被験者では誰も、試験薬剤の投与後における投与日に頭
痛の発現を報告しなかった。
【0203】
(本試験からの結論)
ナロキソンの絶対バイオアベイラビリティーは、試験した3種類すべての経口的な治療
様式で<1%であった。血漿ナロキソン濃度は、すべての治療様式で、変動性が大きかっ
た。平均ナロキソンピーク露出は、8pg/mlで、範囲が0−16.6pg/mlであ
り、ナロキソンのi.v.投与後の場合の約0.1%であった。ナロキソンの終末半減期
は短く、約1hであった。
【0204】
単独で与えられるナロキソン、HClは、0.5mgIVの合計用量で充分に許容され
、そして、経口的には1.5mgまで充分に許容される。
【実施例9】
【0205】
実施例9では、OxyContin(登録商標)40mgと実施例7のオキシコドン/
ナロキソン制御放出性剤形(40mg/0.85mg)との生物学的同等性を評価すべく
管理された正常且つ健常な成人の男性及び女性被験者において、単回投与によるオープン
−ラベル方式の無作為、2−期間、2−治療様式交差試験を実施した。薬物動力学を評価
可能な被験者を少なくとも48人そろえるため、96人までの被験者を登録することとし
た。スクリーニング要件を満たした被験者は、1日目に本試験施設に入院し、1日目にN
arcan(登録商標)(ナロキソンハイドロクロライド溶液)チャレンジ試験を受けた
。Narcan(登録商標)チャレンジを成功裏に完了した被験者は、投与を2つの期間
に分離する5日間のウォッシュアウト期間を伴う2つの交差治療様式シーケンス(対照治
療様式から試験治療様式へ、あるいは、試験治療様式から対照治療様式へ)のうちの一方
に無作為に振り分けられた。被験者は、1日目から10日目まで連続的に本試験施設に留
め置かれた。被験者は、無作為化コードに従って、2日目と7日目の8:00AM頃に、
それらの治療様式のうちの一方による投与を受けた。すべての用量は、夜通しの絶食(水
を除く)後、8オンスの水と共に投与された。2日目と7日目の投与後、投与前から投与
の72時間後までの期間にわたり、血液サンプルを連続的に採取した。オキシコドンとナ
ロキソン(該当する場合)の血漿濃度を決定し、薬物動力学的な測定基準を算出して解析
した。安全性の測定は、有害事象の報告、生命徴候の測定、パルスオキシメトリー、臨床
検査室パラメーター、心電図(ECG)、及び理学的検査から構成された。統計分析の目
的上、期間1は、最初の用量の投与(2日目)から2回目の用量の投与(7日目)の直前
までの時間と定義された。期間2は、2回目の用量の投与(7日目)から10日目に行わ
れた投与の72時間後における血液採取までの時間と定義された。(生物学的同等性の評
価の観点における)評価可能な被験者は、これら2つの試験治療様式のうちのいずれかの
後、少なくとも12時間は嘔吐の発生があってはならない。
【0206】
ナロキソンを伴うオキシコドンの制御放出とOxyContin(登録商標)とに対す
る薬物動力学的測定基準(平均値±SD)が以下の表13にまとめられている。決定及び
算出された各薬物動力学的計量測定基準に対し、試験及び対照治療様式による平均値は同
様であり、すべてのパラメーターの算術平均(±SD)は、相互の約10%以内にあった
。
【表15】
【0207】
ナロキソンに対する薬物動力学的計量パラメーター(平均値±SD)が以下の表14に
まとめられている。
【表16】
【0208】
ナロキソンを伴うオキシコドンの制御放出(試験)対OxyContin(登録商標)
(対照)の生物学的同等性は、以下の表15に列挙されている各治療様式でのLS(最小
二乗)平均AUC及びCmax値に基づく生物学的同等性判定基準を用いて評価された。以
下の表15には、Cmax及びAUC値に対する指数化LS平均の比についての信頼区間(
90%)も示されている。
【表17】
上述の比及び90%CIの算出では、対数変換パラメーターを使用し、その後、指数化
し直した。また、上述の比及び90%CIは、シーケンス、被験者(シーケンス)、期間
、及びANOVAモデルにおける治療に対する影響を伴う指数化LS平均に基づいて決定
された。
【0209】
上の表から分かるように、AUCt、AUC∞、及びCmaxに対して、それらの比の90
%信頼区間の上限は125%未満(AUCt、AUC∞、及びCmaxに対して、それぞれ、
100%、100%、及び108%)であり、一方、それらの比の90%信頼区間の下限
は80%以上(AUCt、AUC∞、及びCmaxに対して、それぞれ、94%、93%、及
び100%)であった。これらの結果は、ナロキソンを伴う40mgのオキシコドン制御
放出が40mgのOxyContin(登録商標)と生物学的同等性を有していることを
示している。
【0210】
(安全性分析)
この試験における安全性パラメーターは、オピオイドのフェーズ1試験における安全性
を評価するために利用したパラメーターと同じものであった。全体的に、ナロキソンを伴
うオキシコドンの制御放出は、健常な被験者により広く許容された。死亡または重篤な有
害事象の報告はなかった。2人の被験者が、有害事象のため、本試験を中止した。最も一
般的な有害事象は、かゆみ、悪心、めまい、傾眠、及び嘔吐であった。すべての有害事象
は、軽度ないし中等度の強度であった。1人の被験者での一つの有害事象(この被験者の
咳の増加は、試験薬剤に関係しているとは考えられず、本試験の終了後も続いた)を例外
として、すべての有害事象は本試験を完了するまでに消散した。すべての試験薬剤関連有
害事象が、オキシコドンに関する既知の安全性プロフィールと合致した。臨床検査室値に
おける変化は散発性のものであり、本試験の薬物適用には関係していないものと考えられ
た。理学的検査、生命徴候、または心電図(ECGs)には、臨床的に意味のある薬剤関
連の変化はなかった。
【0211】
全体的な有害事象(発生率10%以上)の発生が以下の表16に示されている。
【表18】
【0212】
各治療後、85人の被験者が有害事象を報告した。有害事象の発生率は両治療様式間で
同様であった(OxyContin(登録商標)による治療後には85人[90.4%]
であったのに比べ、ナロキソンを伴うオキシコドンの制御放出による治療後には85人[
91.4%]であった)。OxyContin(登録商標)(対照)による治療後の場合
には、85人の被験者により415例の有害事象が報告されたのに比べ、ナロキソンを伴
うオキシコドンの制御放出(試験)による治療後の場合には、85人の被験者により41
3例の有害事象が報告された。かゆみ、悪心、めまい、傾眠、及び嘔吐が、どちらかの治
療用式後に最も頻繁に報告された有害事象であった。これらの有害事象は、ナロキソンを
伴うオキシコドンの制御放出とOxyContin(登録商標)により代表されるオピオ
イド鎮痛薬クラスの薬剤から予想される有害事象プロフィールと合致する。
【0213】
いずれかの治療用式後に報告された他の一般的な有害事象は、陶酔、頭痛、血管拡張、
及び発汗を含んだ。ナロキソンを伴うオキシコドンの制御放出による治療後の場合(23
/93、24.7%)には、OxyContin(登録商標)の場合(14/94、14
.9%)に比べ、もっと多数の被験者が血管拡張を報告した。ナロキソンを伴うオキシコ
ドンの制御放出による治療後及びOxyContin(登録商標)による治療後には、そ
れぞれ、以下の有害事象が同様な割合で報告された:頭痛(23.7%、22.3%)、
陶酔(31.2%、26.6%)、及び発汗(10.8%、8.5%)。
【0214】
全体的に、期間1及び2で報告されたあらゆる特定の有害事象の発生率間に顕著な差異
は観測されなかった。
【0215】
治療様式に少なくとも多分関係していると考えられた事象のうち、ナロキソンを伴うオ
キシコドンの制御放出による治療後及びOxyContin(登録商標)による治療後に
最も高い発生率で報告された事象は、それぞれ:かゆみ(52.7%、50.0%);悪
心(47.3%、36.2%);めまい(36.6%、45.7%);傾眠(40.9%
、39.4%);嘔吐(33.3%、30.9%);陶酔(31.2%、26.6%);
血管拡張(23.7%、14.9%)、及び頭痛(19.4%、19.1%)であった。
【0216】
全体的に、期間1及び2で報告された、治療様式に少なくとも多分関係していると考え
られた有害事象の発生率間に顕著な差異は観測されなかった。
【0217】
(血漿濃度−時間曲線)
図4は、両治療用式後の時間経過に伴うオキシコドンの平均血漿濃度を示している。
【0218】
図5及び6は、安全性について評価可能なすべての被験者でのナロキソンに対する平均
観測血漿濃度−時間曲線を示している。
【0219】
評価した被験者の約3分の2が、大多数の時点において、定量限界(BLQ[5pg/
mL])以下の血漿ナロキソン濃度を有していた。一般的に、平均血漿ナロキソン濃度は
低く、非常に変動性であった。時間経過に伴う平均ナロキソン濃度は、すべての時点にお
いて<1pg/mLであった。しかし、個別的に見れば、26pg/mLもの高い血漿濃
度を有した被験者もいた。
【0220】
(本試験からの結論)
この試験治療様式(0.85mgのナロキソンを伴う40mgのオキシコドンの制御放
出性錠剤)は、オキシコドンに対するこの対照治療様式(OxyContin(登録商標
)40mg錠)に対して生物学的同等性を有している。
【0221】
試験及び対照治療様式後における平均オキシコドンAUCt、AUC∞、Cmax、tmax
、及びびt1/2の値は同様であり、すべての値が相互の約10%以内にあった。
【0222】
平均血漿ナロキソン濃度は一般的に低く(<1pg/ml)、且つ、非常に変動性であ
り、数人の被験者では約0pg/mLから26pg/mLまでの範囲に及んだ。評価した
被験者の約3分の2がBLQ(5pg/mL)の血漿ナロキソン濃度を有していた。男性
患者と女性患者間の薬物動力学的データには臨床的に有意差がなかった。
【0223】
0.85mgのナロキソンを伴う40mgのオキシコドンの制御放出と40mgのOx
yContin(登録商標)は、同様な安全性プロフィールを呈した。両方とも、健常な
男性及び女性のボランティアに広く許容された。
【0224】
本発明の多くの他の変形態様が当業者にとって明らかであり、それらの変形態様も、添
付の特許請求の範囲に包含されるべく意図されている。
【図面の簡単な説明】
【0225】
【図1】図1は、本発明の一つの実施態様を実現する方法の作業系統図である。
【図2】図2A、2B、2C、及び2Dは、実施例8における4種類の治療様式で観測されたナロキソンに対する血漿濃度−時間プロフィールの平均値をグラフで表したものである。
【図3】図3A、3B、3C、及び3Dは、4種類の治療様式で観測されたナロキソンに対する個々の血漿濃度−時間プロフィールをグラフで表したものである。
【図4】図4は、実施例9の両様式による治療後における、時間の経過に伴うオキシコドンの平均血漿濃度をグラフで表したものである。
【図5】図5は、安全性について評価可能なすべての被験者での、時間の経過に伴うナロキソンに対する平均観測血漿濃度をグラフで表したものである。
【図6】図6は、安全性について評価可能なすべての被験者を性別に見た、時間の経過に伴うナロキソンに対する平均観測血漿濃度をグラフで表したものである。
【技術分野】
【0001】
オキシコドン製剤は、時として、乱用の対象である。特定の用量のオキシコドンは、非
経口的に投与されると、同じ用量を経口的に投与した場合に比べ、一層効力が高くなり得
る。経口用オキシコドン製剤を乱用する一つの方法は、その活性物質を溶液に為し、それ
を注射する操作を含む。これらの薬剤の非経口的な乱用を防止するため、オピオイド拮抗
薬が特定のオピオイド作動薬と配合されている。
【背景技術】
【0002】
先行技術においては、即時放出性(immediate release)のペンタゾ
シンとナロキソンの配合物が、米国において入手可能な錠剤の形態で利用されており、S
anofi−WinthropからTalwin(登録商標)Nxとして市販されている
。Talwin(登録商標)Nxは、50mgの塩基に相当する即時放出性のペンタゾシ
ンハイドロクロライドと0.5mgの塩基に相当するナロキソンハイドロクロライドを含
んでいる。チリジン(50mg)とナロキソン(4mg)を含む固定の配合物療法が、1
978年以来、疼痛を管理するため、ドイツで利用されている(Valoron(登録商
標)N、Goedecke)。ブプレノルフィンとナロキソンの固定の配合物が、疼痛を
治療するため、ニュージーランドで1991年に導入された(Temgesic(登録商
標)Nx、Reckitt & Colman)。
【0003】
Purdue Pharma L.Pは、現在、OxyContin(登録商標)とい
う商品名で、10、20、40、80、及び160mgのオキシコドンハイドロクロライ
ドを含む剤形の徐放出性(sustained−release)のオキシコドンを市販
している。
【0004】
米国特許第5,266,331号;第5,508,042号;第5,549,912号
及び第5,656,295号は、徐放出性オキシコドン製剤を開示している。
【0005】
Kreekに付与された米国特許第4,769,372号及び第4,785,000号
は、1日に1回から5回まで、約1.5mgから約100mgまでのオピオイド鎮痛薬、
又は経口的に投与されたときに殆どもしくは全く系統的な拮抗活性を持たない約1mgか
ら約18mgまでのオピオイド拮抗薬を含む1用量単位から2用量単位までを投与するこ
とにより、腸管の自動能不全を誘発することなく、慢性の疼痛または慢性の咳を被ってい
る患者を治療する方法を開示することを主旨としている。
【0006】
Crainらに付与された米国特許第5,472,943号は、二つのモードで作用す
るオピオイド作動薬をオピオイド拮抗薬と共に投与することにより、その作動薬の鎮痛効
力を高める方法を開示することを主旨としている。
【0007】
米国特許第3,773,955号は、1mgから2.5mgまでのナロキソンとの配合
においてオピオイド作動薬を含む即時放出性の製剤を開示することを主旨としている。
【0008】
先述のものを含め、ここで引用されるすべての文書は、あらゆる目的で、それらの全体
が参照により本明細書に組み入れられる。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の一つの目的は、オキシコドンまたはそれの塩を含む経口用剤形を提供すること
である。
【0010】
本発明の特定の実施態様における一つの目的は、他の剤形よりも非経口的に乱用されに
くい、オキシコドンまたはそれの塩の経口用剤形を提供することである。
【0011】
本発明の特定の実施態様における一つの目的は、他の剤形よりも常習者に興味を持たれ
にくい、オキシコドンまたはそれの塩の経口用剤形を提供することである。
【0012】
本発明の特定の実施態様における一つの目的は、他の剤形よりも流用されにくい、オキ
シコドンまたはそれの塩の経口用剤形を提供することである。
【0013】
本発明の特定の実施態様における一つの目的は、オキシコドンまたはそれの塩の経口用
剤形を用いてヒト患者における疼痛を治療し、その一方で、その剤形の乱用の可能性を低
減する方法を提供することである。
【0014】
本発明の特定の実施態様における一つの目的は、乱用の可能性が比較的少なくなるよう
な仕方で、オキシコドンまたはそれの塩の経口用剤形を製造する方法を提供することであ
る。
【0015】
これらの目的及び他の目的は、10mgから40mgまでのオキシコドンまたはそれの
薬学的に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの
薬学的に許容可能な塩とを含む医薬組成物に向けられた本発明により達成される。
【課題を解決するための手段】
【0016】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において、約10mgのオキシコドンハイドロクロライドと、0.80m
gから0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられ
ている。
【0017】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられて
いる。
【0018】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物に向けられて
いる。
【0019】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約10mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0020】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0021】
特定の実施態様では、本発明は、少なくともオキシコドンハイドロクロライドの徐放出
をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80mg
から0.90mgまでのナロキソンハイドロクロライドジハイドレートとを含む医薬組成
物に向けられている。
【0022】
ここに開示されている本発明の特定の実施態様では、その剤形は、ナロキソンまたはそ
れの薬学的に許容可能な塩の徐放出をもたらす。
【0023】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす特定
の実施態様では、その剤形は、37℃における900mlの模擬胃液(SGF)中におい
て100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法
によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、オキシ
コドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±30%の範囲内にあるナ
ロキソンまたはそれの薬学的に許容可能な塩の溶解をもたらす。例えば、もしオキシコド
ンまたはそれの薬学的に許容可能な塩の溶解速度が1時間で40%の場合、ナロキソンま
たはそれの薬学的に許容可能な塩の溶解速度は28%から52%までの範囲であろう。
【0024】
上述の実施態様における特定の実施態様では、ナロキソンまたはそれの薬学的に許容可
能な塩は、1時間、4時間、及び12時間経過時点において、オキシコドンまたはそれの
薬学的に許容可能な塩の溶解速度に対して±20%の範囲内にあり;代替的に、オキシコ
ドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±10%の範囲内にあり;あ
るいは、代替的に、オキシコドンまたはそれの薬学的に許容可能な塩の溶解速度に対して
±5%の範囲内にあるイン・ビトロ溶解速度を有している。
【0025】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす別の
実施態様では、オキシコドンまたはそれの薬学的に許容可能な塩並びにナロキソンまたは
それの薬学的に許容可能な塩のイン・ビトロ溶解速度は、37℃における900mlの模
擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUS
P装置I(バスケット)法により測定したときに、1時間では、約20重量%から約60
重量%までのオキシコドンまたはそれの薬学的に許容可能な塩と、約20重量%から約6
0重量%までのナロキソンまたはそれの薬学的に許容可能な塩とが放出され;2時間では
、約30重量%から約75重量%までのオキシコドンまたはそれの薬学的に許容可能な塩
と、約30重量%から約75重量%までのナロキソンまたはそれの薬学的に許容可能な塩
とが放出され;4時間では、約40重量%から約90重量%までのオキシコドンまたはそ
れの薬学的に許容可能な塩と、約40重量%から約90重量%までのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;8時間では、約60重量%より多くのオキシコ
ドンまたはそれの薬学的に許容可能な塩と、約60重量%より多くのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;そして、12時間では、約70重量%より多く
のオキシコドンまたはそれの薬学的に許容可能な塩と、約70重量%より多くのナロキソ
ンまたはそれの薬学的に許容可能な塩とが放出されるような溶解速度である。
【0026】
オキシコドンまたはそれの塩並びにナロキソンまたはそれの塩の徐放出をもたらす別の
実施態様では、オキシコドンまたはそれの薬学的に許容可能な塩並びにナロキソンまたは
それの薬学的に許容可能な塩のイン・ビトロ溶解速度は、37℃における900mlの模
擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUS
P装置I(バスケット)法により測定したときに、1時間では、約30重量%から約60
重量%までのオキシコドンまたはそれの薬学的に許容可能な塩と、約30重量%から約6
0重量%までのナロキソンまたはそれの薬学的に許容可能な塩とが放出され;2時間では
、約40重量%から約70重量%までのオキシコドンまたはそれの薬学的に許容可能な塩
と、約40重量%から約70重量%までのナロキソンまたはそれの薬学的に許容可能な塩
とが放出され;4時間では、約55重量%から約90重量%までのオキシコドンまたはそ
れの薬学的に許容可能な塩と、約55重量%から約90重量%までのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;8時間では、約70重量%より多くのオキシコ
ドンまたはそれの薬学的に許容可能な塩と、約70重量%より多くのナロキソンまたはそ
れの薬学的に許容可能な塩とが放出され;そして、12時でには、約80重量%より多く
のオキシコドンまたはそれの薬学的に許容可能な塩と、約80重量%より多くのナロキソ
ンまたはそれの薬学的に許容可能な塩とが放出されるような溶解速度である。
【0027】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに、制御放出性(controlled release)対照製品(Ph
ysician’s Desk Reference 2002年版に記載されている通
りのOxyContin(登録商標))のオキシコドン塩基同等量によりもたらされる血
漿中オキシコドンの平均AUCの80%から125%までの範囲内における血漿中オキシ
コドンの平均AUCをもたらす。別の実施態様では、血漿中オキシコドンの平均AUCは
、ヒト被験者の母集団に対して単回用量投与をしたときに制御放出性対照製品のオキシコ
ドン塩基同等量によりもたらされる血漿中オキシコドンの平均AUCの90%から110
%までの範囲内、あるいは95%から105%までの範囲内である。
【0028】
特定の実施態様では、本発明の医薬組成物は、ナロキソンハイドロクロライドを含有す
る即時放出性対照製品、即ち、Physician’s Desk Reference
2002年版に記載されている通りのTalwin(登録商標)Nxのナロキソン塩基
同等量によりもたらされる血漿中ナロキソンの平均Cmaxよりも少なくとも50%下回
った血漿中ナロキソンの平均Cmaxをもたらす。別の実施態様では、この血漿中ナロキ
ソンの平均Cmaxは、ナロキソンハイドロクロライドを含有する即時放出性対照製品、
即ち、Physician’s Desk Reference 2002年版に記載さ
れている通りのTalwin(登録商標)Nxのナロキソン塩基同等量によりもたらされ
る血漿中ナロキソンの平均Cmaxよりも少なくとも65%下回っており、あるいは少な
くとも80%下回っている。
【0029】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに、180pg/ml未満、150pg/ml未満、または100pg/m
l未満の血漿中ナロキソンの平均Cmaxをもたらす。より好適な実施態様では、この血
漿中ナロキソンの平均Cmaxは、ヒト被験者の母集団に対して単回用量投与をしたとき
に、50pg/ml未満、10pg/ml未満、または5pg/ml未満である。
【0030】
特定の実施態様では、本発明の医薬組成物は、ヒト被験者の母集団に対して単回用量投
与をしたときに5pg/ml未満の血漿中ナロキソンの平均Cmaxをもたらすような量の
ナロキソンまたはそれの薬学的に許容可能な塩を含んでいる。
【0031】
ここに開示されている本発明の特定の実施態様では、その剤形は、ヒト患者に対する定
常状態での経口投与後、少なくとも12時間は疼痛の効果的な軽減をもたらす。
【0032】
ここに開示されている本発明の特定の実施態様では、その剤形は、ヒト患者に対する定
常状態での経口投与後、少なくとも24時間は疼痛の効果的な軽減をもたらす。
【0033】
ここに開示されている本発明の特定の実施態様では、その剤形は、徐放出性賦形剤に相
互分散された(interdispersed)オキシコドンハイドロクロライドとナロキソンハイドロク
ロライドを包含したマトリックスを含んでいる。
【0034】
特定の実施態様では、本発明は、ここに開示されている組成物を調製することを含有す
る、オキシコドン製剤の非経口的な乱用の可能性を低減する方法に向けられている。
【0035】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を経
口的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0036】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を、
少なくとも定常状態が達成されるまで12時間毎に経口的に投与することを含む、ヒト患
者における疼痛を治療する方法に向けられている。
【0037】
特定の実施態様では、本発明は、ここで説明されている通りの徐放出性経口用剤形を、
少なくとも定常状態が達成されるまで24時間毎に経口的に投与することを含む、ヒト患
者における疼痛を治療する方法に向けられている。
【0038】
特定の実施態様では、本発明は、患者に対する定常状態での経口投与後、少なくとも1
2時間は疼痛の効果的な軽減をもたらす、ここに開示されている通りの医薬組成物を経口
的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0039】
特定の実施態様では、本発明は、患者に対する定常状態での経口投与後、少なくとも2
4時間は疼痛の効果的な軽減をもたらす、ここに開示されている通りの医薬組成物を経口
的に投与することを含む、ヒト患者における疼痛を治療する方法に向けられている。
【0040】
特定の実施態様では、本発明は、オキシコドンまたはそれの薬学的に許容可能な塩を徐
放出性の賦形剤と配合してオキシコドン/賦形剤配合物を形成すること;そのオキシコド
ン/賦形剤配合物にナロキソンまたはそれの薬学的に許容可能な塩を加えること;並びに
、オキシコドンまたはそれの薬学的に許容可能な塩と、ナロキソンまたはそれの薬学的に
許容可能な塩との徐放出性経口用剤形を形成することを含む、徐放出性経口用剤形を製造
する方法に向けられている。代替的に、最初にナロキソンを賦形剤と配合し、オキシコド
ンをそのナロキソン/賦形剤混合物に加えてもよい。別の実施態様では、オキシコドンと
ナロキソンを、賦形剤と配合するのと同時的に、もしくは、賦形剤と配合する前に配合す
ることができる。特定の実施態様では、一つの物質が、他の物質との配合に先だって、徐
放出性形態となるように前処理されない。
【0041】
特定の実施態様では、本発明は、約10mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約20
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療す
る方法に向けられている。
【0042】
特定の実施態様では、本発明は、約20mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約40
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療す
る方法に向けられている。
【0043】
特定の実施態様では、本発明は、約10mgのオキシコドンハイドロクロライドに相当
する量のオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.90
mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物を、少なく
ともオキシコドンハイドロクロライドの徐放出をもたらす剤形で投与し、その後、約20
mgのオキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的
に許容可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的
に許容可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐
放出をもたらす剤形で投与することによりその用量を増量し、更にその後、約40mgの
オキシコドンハイドロクロライドに相当する量のオキシコドンまたはそれの薬学的に許容
可能な塩と、0.65mgから0.90mgまでのナロキソンまたはそれの薬学的に許容
可能な塩とを含む医薬組成物を、少なくともオキシコドンハイドロクロライドの徐放出を
もたらす剤形で投与することにより、その用量を増量することを含む疼痛を治療する方法
に向けられている。
【0044】
特定の実施態様では、本発明は、約10mgから約40mgまでのオキシコドンまたは
それの薬学的に許容可能な塩と、ナロキソンまたはそれの薬学的に許容可能な塩とを含む
少なくとも1つの製剤を含有した容器を包含する、疼痛を治療するためのキットに向けら
れており;そのキットは、更に、上述の製剤の使用を指示する証印(indicia)を
含んでいる。特定の実施態様では、そのキットの製剤は、0.65mgから0.9mgま
でのナロキソンまたはそれの薬学的に許容可能な塩を含んでおり、そして、特定の実施態
様では、上述の証印は、その使用が製剤の非経口的な乱用を低減するためのものであるこ
とを指示している。
【0045】
「徐放出性」という用語は、本発明の目的上、即時放出性製品と比べて長い期間、例え
ば約12時間から約24時間までの期間にわたり、血液(例えば血漿)中の濃度を治療的
範囲内であるが毒性レベル以下に維持するような仕方でオキシコドンまたはそれの塩を放
出することとして定義される。都合のよいことに、この徐放出により、十分に1日に2回
または1日に1回製剤が提供される。特定の実施態様では、1時間、4時間、及び12時
間におけるナロキソンまたはそれの塩の放出速度は、オキシコドンまたはそれの薬学的に
許容可能な塩の溶解速度に対して±30%の範囲内である。
【0046】
本明細書で使用される場合、「非経口的に」という用語は、皮下注射、静脈内、筋肉内
、胸骨内への注射、注入技術、あるいは、当技術分野において知られている他の注入方法
を含む。
【0047】
別な具合に特記されていない限り、「オキシコドン」という用語はオキシコドン塩基を
意味している。別な具合に特記されていない限り、「ナロキソン」という用語はナロキソ
ン塩基を意味している。塩という用語は、薬学的に許容可能な塩を意味している。
【0048】
「定常状態」という用語は、その系に到達する薬剤の量が、その系を去る薬剤の量とほ
ぼ同じであることを意味している。従って、「定常状態」の場合、患者の身体は、薬剤が
血流中への吸収を通じてその患者の系で利用できるようになるのとほぼ同じ速度でその薬
剤を排出する。
【0049】
「非経口的に有効な量のナロキソンまたはそれの薬学的に許容可能な塩」という用語は
、ナロキソンまたはそれの薬学的に許容可能な塩が、この剤形を非経口的に投与したとき
のオキシコドンの作用を無効にするのに充分な量、もしくは、その作用を部分的に無効に
する量であり、且つ、この剤形を経口投与したときのオキシコドンの作用を無効もしくは
部分的に無効にする量以下の量であることを意味している。
【0050】
「平均」という用語は、別な具合に特定されていない限り、算術平均を表している。
【0051】
「ヒト被験者」という用語は、当業者により理解されるように、正常な代謝機能を有す
る健常なヒト被験者を意味している。
【発明を実施するための最良の形態】
【0052】
本発明の剤形は、約10mgから約40mgまでのオキシコドンハイドロクロライドに
相当する量のオキシコドンまたはそれの薬学的に許容可能な塩を含んでいる。オキシコド
ンハイドロクロライドの特に好適な用量は、約10mg、約20mg、約30mg、また
は約40mgである。オキシコドンまたはそれの塩は、そのオキシコドンの徐放出をもた
らすべく、薬学的に許容可能な適切な賦形剤と調合される。
【0053】
本発明の剤形は、約0.65mgから約0.90mgまでのナロキソンまたはそれの薬
学的に許容可能な塩を含んでいる。ナロキソン塩の特に好適な用量範囲は、約0.82m
gから約0.88mgまで、及び約0.84mgから約0.86mgまでである。特に好
適な用量は、約0.85mgのナロキソン塩を有している。ナロキソン塩基の特に好適な
用量範囲は、約0.65mgから約0.75mgまで、及び約0.67mgから約0.7
3mgまでである。特に好適な用量は、約0.70mgのナロキソン塩基を有している。
【0054】
本発明の剤形は、約0.65mgから約0.90mgまでのナロキソンハイドロクロラ
イドジハイドレートに相当する量のナロキソンまたはそれの薬学的に許容可能な塩を含ん
でいる。ナロキソンハイドロクロライドジハイドレートの特に好適な用量範囲は、約0.
82mgから約0.88mgまで、または約0.84mgから約0.86mgまでである
。特に好適な用量は、約0.85mgのナロキソンハイドロクロライドジハイドレートを
有している。ナロキソン塩基の特に好適な用量範囲は、約0.65mgから約0.75m
gまで、または同等量のそれの塩、及び約0.67mgから約0.73mgまで、または
同等量のそれの塩である。特に好適な用量は、約0.70mgのナロキソン塩基、または
同等量のそれの塩を有している。
【0055】
ナロキソンまたはそれの塩は、そのナロキソンまたはそれの塩の即時放出をもたらすべ
く調合され、薬学的に許容可能な適切な賦形剤と配合されてよい。ナロキソンまたはそれ
の塩の徐放出速度は、オキシコドンまたはそれの塩の徐放出速度と同じであってもよいし
、あるいは異なっていてもよい。本発明の特に好適な実施態様は、10mgのオキシコド
ンハイドロクロライドと0.85mgのナロキソンハイドロクロライドを含有する剤形;
20mgのオキシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロラ
イドを含有する剤形;30mgのオキシコドンハイドロクロライドと0.85mgのナロ
キソンハイドロクロライドを含有する剤形;40mgのオキシコドンハイドロクロライド
と0.85mgのナロキソンハイドロクロライドを含有する剤形;あるいは、同等量のオ
キシコドン塩基、ナロキソン塩基、または他の薬学的に許容可能なそれの塩を含有する剤
形である。オキシコドン及びナロキソンのハイドロクロライド塩は特に好適である。
【0056】
本発明の他の特に好適な実施態様は、10mgのオキシコドンハイドロクロライドと0
.85mgのナロキソンハイドロクロライドジハイドレートを含有する剤形;20mgの
オキシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロライドジハイ
ドレートを含有する剤形;30mgのオキシコドンハイドロクロライドと0.85mgの
ナロキソンハイドロクロライドジハイドレートを含有する剤形;あるいは、40mgのオ
キシコドンハイドロクロライドと0.85mgのナロキソンハイドロクロライドジハイド
レートを含有する剤形である。
【0057】
それらの物質が共に徐放出性の形態を為している実施態様では、都合のよいことに、そ
の剤形は、37℃における900mlの模擬胃液(SGF)中において100rpmで、
米国薬局方XXIV(2000年)のUSP装置I(バスケット)法により測定したとき
に、本発明の目的に適合したイン・ビトロ溶解速度をもたらすような仕方でオキシコドン
またはそれの薬学的に許容可能な塩とナロキソンまたはそれの薬学的に許容可能な塩を放
出する。
【0058】
本発明において開示されているナロキソンまたはそれの塩の範囲は、もしその製剤が溶
解されて非経口的に投与された場合に、オキシコドンのオピオイド効果を少なくとも部分
的に阻止することによって静脈内乱用を防止するのに非経口的に有効な量である。好適に
は、その量は、物理的に最も依存性の高い個人が非経口投与をしたときに、オピオイドの
使用中止後に見られる症状と非常によく似た中等度から重度の禁断症候群を突如として引
き起こすのに充分な量でもある。この禁断症候群のうちで最も一般的な症状は、食欲不振
、体重減少、瞳孔拡大、過剰な発汗と交互に生じる悪寒、異常な痙攣、悪心、嘔吐、筋肉
痙攣、過剰刺激感受性、流涙、鼻漏(rinorrhea)、鳥肌、及び心拍数上昇を含
む。ナロキソンの量は、疼痛を被っている患者に経口投与したときに、有害効果を引き起
こしたり、あるいは、鎮痛効能を低減させるような量であるべきではない。
【0059】
(徐放出性剤形)
オキシコドン(またはオキシコドン塩)、及び、場合によってはナロキソン(またはナ
ロキソン塩)は、当業者にとって既知の何らかの適した錠剤、被覆錠剤、または多重粒状
体(multiparticulate)製剤の形態における徐放出性経口用製剤として
調合される。その徐放出性剤形は、ナロキソンまたはそれの塩を伴って、もしくはそれら
を伴わずに、オキシコドンまたはそれの塩と共にマトリックスに組み込まれた徐放出性材
料を含んでいてよい。例えば、オキシコドン塩が徐放出性マトリックスに組み込まれてい
てよく、そして、ナロキソン塩が、そのマトリックスとは別になっていてもよいし、ある
いは、そのマトリックスに組み込まれていてもよい。
【0060】
この徐放出性剤形は、場合によっては、ナロキソンまたはそれの塩を伴って、もしくは
それらを伴わずに、オキシコドンまたはそれの塩を含有する粒子を含んでいてよい。特定
の実施態様では、それらの粒子は、約0.1mmから約2.5mmまでの直径、好適には
約0.5mmから約2mmまでの直径を有している。ナロキソンまたはナロキソン塩は、
オキシコドンまたはオキシコドン塩を含有する粒子に組み込まれていてよく、あるいは、
オキシコドンまたはオキシコドン塩粒子を含有する錠剤またはカプセル剤に組み込まれて
いてよい。好適には、それらの粒子は、水性媒質中における持続的な速度での(1つもし
くは複数の)本活性物質の放出を可能にする材料でフィルムコーティングされる。そのフ
ィルムコーティングは、ここで述べられている他の特性と組み合わせて、望ましいイン・
ビトロ放出速度を達成できるように選択される。本発明の徐放出性コーティング製剤は、
滑らか且つエレガントで、顔料及び他のコーティング用添加剤を支持することができ、無
毒且つ不活性で粘着性のない強い連続的なフィルムを作製できるべきである。
【0061】
(被覆ビーズ)
本発明の特定の実施態様では、不活性な製薬用ビーズ、例えばnu pariel 1
8/20ビーズ等をコーティングするために疎水性材料が使用され、その後、結果として
得られた複数の固体の徐放出性ビーズを、経口摂取し環境流体、例えば胃液または溶解媒
質と接触したときに効果的な徐放出性の用量を供給するのに充分な量で、ゼラチンカプセ
ルに入れることができる。特定の実施態様では、ナロキソンまたはナロキソン塩は、オキ
シコドンまたはオキシコドン塩を含有する徐放出性ビーズにコーティングされてよく、あ
るいは、徐放出性オキシコドンまたはオキシコドン塩ビーズと共にカプセルに入れられて
もよい。
【0062】
本発明の徐放出性ビーズ製剤は、例えば、経口摂取されて胃液次いで腸液に晒されたと
きに、本発明の(1つもしくは複数の)物質をゆっくりと放出する。本発明の製剤の徐放
出プロフィールは、例えば、疎水性材料を用いるオーバーコーティングの量を変更するこ
とにより、疎水性材料に可塑剤を加える仕方を変更することにより、疎水性材料に対する
可塑剤の量を変更することにより、付加的な成分または賦形剤を含めることにより、ある
いは製造方法を変更することなどにより変えることができる。また、最終製品の溶解プロ
フィールも、例えばその遅延剤コーティングの厚みを増加または減少させることにより変
えることができる。
【0063】
本発明の(1つもしくは複数の)物質でコーティングされたスフェロイド(spher
oids)またはビーズは、例えば、(1つもしくは複数の)物質を水に溶かし、次いで
、Wusterインサートを用いて、その溶液を基材、例えばnu pariel 18
/20ビーズに噴霧することにより調製される。場合によっては、ビーズへの(1つもし
くは複数の)本物質の結合を補助するため、及び/又は、その溶液を着色するため等、ビ
ーズをコーティングする前に付加的な成分も加えられる。例えば、着色剤を伴って、もし
くは、着色剤を伴わずに、ヒドロキシプロピルメチルセルロース等を含む製品(例えば、
Opadry(登録商標)、Colorcon,Inc.から商業的に入手可能)をその
溶液に加えてよく、そして、その溶液は、ビーズに同じく塗布する前に混合(例えば約1
時間)される。その後、結果として生じる被覆された基材、この例ではビーズは、場合に
よっては、疎水性の徐放出性コーティングから(1つもしくは複数の)本活性物質を分離
するため、バリヤー剤でオーバーコーティングされる。適切なバリヤー剤の一つの例は、
ヒドロキシプロピルメチルセルロースを含むバリヤー剤である。しかし、当技術分野にお
いて既知のあらゆるフィルム形成剤が使用されてよい。そのバリヤー剤は、最終製品の溶
解速度に影響を及ぼさないことが好適である。
【0064】
それらのビーズは、疎水性材料の水性分散系でオーバーコーティングされてよい。この
疎水性材料の水性分散系は、好適には、更に、有効量の可塑剤、例えばトリエチルシトレ
ートを含む。予め調合済みのエチルセルロースの水性分散系、例えばAquacoat(
登録商標)またはSurelease(登録商標)を使用することができる。Surel
ease(登録商標)を使用する場合には、可塑剤を別個に加える必要はない。代替的に
、予め調合済みのアクリルポリマーの水性分散系、例えばEudragit(登録商標)
を使用することもできる。
【0065】
本発明のコーティング溶液は、好適には、フィルム形成剤、可塑剤、及び溶媒系(即ち
、水)のほかに、エレガンスさと製品識別能力を与えるための着色剤を含んでいる。疎水
性材料の水性分散系の代わりに、あるいは、疎水性材料の水性分散系に加えて、その治療
的に活性な物質の溶液に色を付けてもよい。例えば、アルコールまたはプロピレングリコ
ールをベースとしたカラー分散系、破砕アルミニウムレーキ、及び乳白剤、例えば二酸化
チタン等の使用を介し、剪断力(shear)を利用して水溶性ポリマー溶液に、次いで
、低剪断力を利用してその可塑化されたAquacoat(登録商標)に色を付けること
により、Aquacoat(登録商標)にも色を付けることができる。代替的に、本発明
の製剤に色をもたらす何らかの適切な方法を用いてもよい。アクリルポリマーの水性分散
系が使用される場合、本製剤に色をもたらすのに適した成分は、二酸化チタン、及び着色
顔料、例えば酸化鉄顔料等を含む。しかし、顔料を組み入れると、そのコーティングの遅
延効果(retard effect)を高める可能性がある。
【0066】
可塑化された疎水性材料は、当技術分野において既知の何らかの適切な噴霧装置を用い
て噴霧することにより、(1つもしくは複数の)本物質を含有する基材上に塗布されてよ
い。一つの好適な方法では、Wurster流動床システムが使用され、そこでは、下側
から注入されたエアージェットがそのコア材料を流動化し、そして、アクリルポリマーコ
ーティングが噴霧されている間に乾燥機能を果たす。その被覆基材が水性溶液、例えば胃
液等に晒されたときに、(1つもしくは複数の)本物質の予め決定された徐放出性を得る
のに充分な量の疎水性材料が塗布されてよい。疎水性材料でコーティングした後、場合に
よっては、それらのビーズに、フィルム形成剤、例えばOpadry(登録商標)等の更
なるオーバーコーティングが施される。適用される場合、このオーバーコーティングは、
それらのビーズのアグロメレーションを実質的に低減するために施される。
【0067】
本発明の徐放出性製剤からの(1つもしくは複数の)本物質の放出は、1つもしくはそ
れ以上の放出変性剤(release−modifying agents)を加えるこ
とにより、あるいは、そのコーティングを通じる1つもしくはそれ以上の通路を設けるこ
とにより、更なる影響を受け、即ち、所望の速度に調節することができる。疎水性材料と
水溶性材料との比は、他の数あるファクターの中でもとりわけ、必要な放出速度、及び、
選定された材料の溶解度特性によって決定される。
【0068】
細孔形成剤として機能する放出変性剤は、有機または無機であってよく、そして、使用
環境において、そのコーティングから溶解、抽出、または浸出され得る材料を含む。この
細孔形成剤は、ヒドロキシプロピルメチルセルロース等の1つもしくはそれ以上の親水性
材料を含んでいてよい。
【0069】
本発明の徐放出性コーティングは、浸食促進剤、例えばデンプン及びゴム等を含むこと
もできる。
【0070】
また、本発明の徐放出性コーティングは、使用環境において微孔性薄膜を作製するのに
有用な材料、例えば、炭酸の線状ポリエステルを含むポリカーボネート(そこでは、ポリ
マー鎖中にカーボネート基が繰返し現れる)も含むことができる。
【0071】
放出変性剤は半透水性ポリマーも含んでいてよい。
【0072】
特定の好適な実施態様では、その放出変性剤は、ヒドロキシプロピルメチルセルロース
、ラクトース、金属ステアレート、及び前述の物質の何らかの混合物から選択される。
【0073】
本発明の徐放出性コーティングは、少なくとも1つの通路、オリフィス等を含む出口手
段(exit means)も含むことができる。その通路は、米国特許第3,845,
770号;第3,916,899号;第4,063,064号;及び第4,088,86
4号に開示されているような方法により形成されてよい。また、その通路は、あらゆる形
状を有していてよく、例えば丸形、三角形、矩形、楕円形、不規則な形等であってよい。
【0074】
(マトリックス製剤)
本発明の別の実施態様では、徐放出性製剤は、場合によってはここで説明されている通
りの徐放出性コーティングを有するマトリックスにより達成される。徐放出性マトリック
スに包含させるのに適した材料は、そのマトリックスを形成するために使用する方法に依
存するであろう。
【0075】
例えば、マトリックスは、オキシコドン(またはオキシコドン塩)、及び、場合によっ
てはナロキソン(またはナロキソン塩)に加え、以下のものを含んでいてよい:
親水性及び/又は疎水性の材料、例えばゴム、セルロースエーテル、アクリル樹脂、タ
ンパク質誘導材料等;このリストは、排他的であることを意図したものではなく、(1つ
もしくは複数の)本物質の徐放出性を付与することができ、且つ、溶融する(もしくは、
押し出されるのに必要な程度にまで軟化する)、薬学的に許容可能なあらゆる疎水性材料
または親水性材料を本発明に従って使用することができる。
【0076】
消化可能な長鎖(C8−C50、特にはC12−C40)の置換または非置換炭化水素、例え
ば脂肪酸、脂肪族アルコール、脂肪酸のグリセリルエステル、鉱油及び植物油及びロウ、
及びステアリルアルコール;並びにポリアルキレングリコール等。
【0077】
これらのポリマーのうち、アクリルポリマー、特にはEudragit(登録商標)R
SPO−セルロースエーテル、特にはヒドロキシアルキルセルロース及びカルボキシアル
キルセルロースが好適である。本経口用剤形は、1重量%から80重量%までの間の少な
くとも1つの親水性または疎水性材料を含んでいてよい。
【0078】
疎水性材料が炭化水素の場合、その炭化水素は、好適には、25℃から90℃までの間
の融点を有している。長鎖炭化水素材料のうち、脂肪(脂肪族)アルコールが好適である
。本経口用剤形は、60重量%までの少なくとも1つの消化可能な長鎖炭化水素を含んで
いてよい。
【0079】
好適には、本経口用剤形は、60重量%までの少なくとも1つのポリアルキレングリコ
ールを含んでいる。
【0080】
疎水性材料は、好適には、アルキルセルロース、アクリル酸及びメタクリル酸のポリマ
ー及びコポリマー、セラック、ゼイン、硬化(hydrogenated)ヒマシ油、硬
化植物油、またはそれらの混合物からなるグループから選択される。本発明の特定の好適
な実施態様では、疎水性材料は薬学的に許容可能なアクリルポリマーであり、それらのア
クリルポリマーは、これらに限定するものではないが、アクリル酸とメタクリル酸のコポ
リマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキシエチルメタ
クリレート、シアノエチルメタクリレート、アミノアルキルメタクリレートコポリマー、
ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、
ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレート
、ポリアクリルアミド、ポリ(無水メタクリル酸)、及びグリシジルメタクリレートコポ
リマーを含む。別の実施態様では、疎水性材料は、ヒドロキシプロピルメチルセルロース
等のヒドロキシアルキルセルロース、及び前述の物質の混合物等の材料から選択される。
【0081】
好適な疎水性材料は、多少なりとも明白な親水性及び/又は疎水性の傾向を伴って、水
に不溶性である。好適には、本発明に有用な疎水性材料は、約25℃から約200℃まで
、好適には約45℃から約90℃までの融点を有している。具体的には、疎水性材料は、
天然または合成のロウ、脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアル
コール、ステアリルアルコール、セチルアルコール、または、好適にはセトステアリルア
ルコール等)、これらに限定するものではないが、脂肪酸エステル、脂肪酸グリセリド(
モノ−、ジ−、及びトリ−グリセリド)を含めた脂肪酸、硬化脂肪、炭化水素、通常のロ
ウ(normal waxes)、ステアリン酸、ステアリルアルコール、及び、炭化水
素骨格を有する疎水性及び親水性材料を含んでいてよい。適切なロウは、例えば、蜜ロウ
、糖ロウ、ヒマシロウ(castor wax)、及びカルナウバロウを含む。本発明の目的上、ロウ
様物質は、室温では通常固体であって、約25℃から約100℃までの融点を有するあら
ゆる材料として定義される。
【0082】
本発明に従って使用され得る適切な疎水性材料は、消化可能な長鎖(C8−C50、特に
はC12−C40)の置換または非置換炭化水素、例えば脂肪酸、脂肪族アルコール、脂肪酸
のグリセリルエステル、鉱油及び植物油、及び、天然及び合成のロウ等を含む。25℃か
ら90℃までの間の融点を有する炭化水素が好適である。特定の実施態様では、長鎖炭化
水素材料のうち、脂肪(脂肪族)アルコールが好適である。本経口用剤形は、60重量%
までの少なくとも1つの消化可能な長鎖炭化水素を含んでいてよい。
【0083】
好適には、2つもしくはそれ以上の疎水性材料の組み合わせが本マトリックス製剤に含
まれている。付加的な疎水性材料が含まれている場合、その材料は、好適には、天然及び
合成のロウ、脂肪酸、脂肪族アルコール、及びそれらの混合物から選択される。それらの
例は、蜜ロウ、カルナウバロウ、ステアリン酸、及びステアリルアルコールを含む。この
リストは、排他的であることを意図したものではない。
【0084】
一つの特に好適なマトリックスは、少なくとも1つの水溶性ヒドロキシアルキルセルロ
ース、少なくとも1つのC12−C36、好適にはC14−C22の脂肪族アルコール、及び、場
合によっては、少なくとも1つのポリアルキレングリコールを含む。前述の少なくとも1
つのヒドロキシアルキルセルロースは、好適には、ヒドロキシ(C1からC6までの)アル
キルセルロース、例えばヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセル
ロース、及び、特にはヒドロキシエチルセルロース等である。本経口用剤形における前述
の少なくとも1つのヒドロキシアルキルセルロースの量は、とりわけ、必要とされるオキ
シコドンの精確な放出速度によって決まるであろう。前述の少なくとも1つの脂肪族アル
コールは、例えば、ラウリルアルコール、ミリスチルアルコール、またはステアリルアル
コールであってよい。しかし、本経口用剤形の特に好適な実施態様では、その少なくとも
1つの脂肪族アルコールはセチルアルコールまたはセトステアリルアルコールである。本
経口用剤形における前述の少なくとも1つの脂肪族アルコールの量は、上述の如く、必要
とされるオキシコドンの精確な放出速度によって決まるであろう。また、その量は、本経
口用剤形に少なくとも1つのポリアルキレングリコールが存在しているか存在していない
かにも依存するであろう。少なくとも1つのポリアルキレングリコールが存在していない
場合、本経口用剤形は、好適には、20重量%から50重量%までの間の少なくとも1つ
の脂肪族アルコールを含む。本経口用剤形に少なくとも1つのポリアルキレングリコール
が存在している場合には、その少なくとも1つの脂肪族アルコールとその少なくとも1つ
のポリアルキレングリコールを合わせた重量が、好適には、合計用量のうちの20重量%
から50重量%までを構成する。
【0085】
一つの実施態様では、例えば前述の少なくとも1つのヒドロキシアルキルセルロースま
たはアクリル樹脂と前述の少なくとも1つの脂肪族アルコール/ポリアルキレングリコー
ルとの比は、その少なくとも1つのヒドロキシアルキルセルロースとその少なくとも1つ
の脂肪族アルコール/ポリアルキレングリコールの(w/w)が1:2から1:4までの
間であることが好適であり、1:3から1:4までの間の比が特に好適であることから決
定される。
【0086】
前述の少なくとも1つのポリアルキレングリコールは、例えば、ポリプロピレングリコ
ールであってよく、好適には、ポリエチレングリコールであってよい。この少なくとも1
つのポリアルキレングリコールの数平均分子量は、好適には1,000から15,000
までの間であり、特には1,500から12,000までの間である。
【0087】
別の適切な徐放出性マトリックスは、アルキルセルロース(特にはエチルセルロース)
、C12からC36までの脂肪族アルコール、及び、場合によっては、ポリアルキレングリコ
ールを含むであろう。
【0088】
別の好適な実施態様では、本マトリックスは、少なくとも2つの疎水性材料からなる薬
学的に許容可能な組み合わせを含む。
【0089】
上述の成分に加え、徐放出性マトリックスは、適切な量の他の材料、例えば製薬業界に
おいて普通に用いられている希釈剤、潤滑剤、結合剤、粒状化助剤(granulating aids)、
着色剤、風味剤(flavorants)、及びグリダント(glidants)等も含んでいてよい
。
【0090】
(マトリックス−粒状体)
本発明による固体の徐放出性経口用剤形の調製を容易化するため、当業者に知られたマ
トリックス製剤のあらゆる調製方法を使用することができる。例えばマトリックスへの組
み込みは、例えば(a)少なくとも1つの水溶性ヒドロキシアルキルセルロースと、オキ
シコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(またはナロキ
ソン塩)を含有する顆粒を形成し;(b)それらのヒドロキシアルキルセルロース含有顆
粒を少なくとも1つのC12−C36脂肪族アルコールと混合し;そして、(c)場合によっ
ては、それらの顆粒を圧縮及び成形することにより果たすことができる。好適には、それ
らの顆粒は、水を用いて上述のヒドロキシアルキルセルロース顆粒を湿式造粒することに
より形成される。
【0091】
更に別の代替的な実施態様では、スフェロイドを形成すべく、球状化剤(spheronizing
agent)をオキシコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(
またはナロキソン塩)と共に球状化することができる。微結晶セルロースは好適な球状化
剤である。適切な微結晶セルロースは、例えば、Avicel PH 101(商標、F
MC Corporation)として販売されている材料である。そのような実施態様
では、本活性成分及び球状化剤に加え、それらのスフェロイドは結合剤も含んでいてよい
。適切な結合剤、例えば低粘度の水溶性ポリマー等は、製薬業界における当業者に広く知
られている。しかし、水溶性のヒドロキシ低級アルキルセルロース、例えばヒドロキシプ
ロピルセルロース等が好適である。付加的に(あるいは代替的に)、それらのスフェロイ
ドは、水に不溶性のポリマー、特にはアクリルポリマー、アクリルコポリマー、例えばメ
タクリル酸−エチルアクリレートコポリマー等、あるいはエチルセルロースを含んでいて
よい。そのような実施態様では、本徐放出性コーティングは、一般的に、疎水性材料、例
えば(a)単独もしくは脂肪族アルコールとの混和物のいずれかの形態におけるロウ;ま
たは(b)セラックあるいはゼイン等を含むであろう。
【0092】
(溶融押出しマトリックス)
また、徐放出性マトリックスは、溶融造粒法または溶融押出し法によって調製すること
もできる。一般的に、溶融造粒法は、常態では固体の疎水性材料、例えばロウ等を溶融し
、そこへ粉末化された薬剤を組み入れる操作を含む。徐放出性剤形を得るためには、その
溶融ロウ疎水性材料に付加的な疎水性物質、例えばエチルセルロース、または水に不溶性
のアクリルポリマー等を組み入れることが必要になり得る。溶融造粒法により調製された
徐放出性製剤の例は、米国特許第4,861,598号で見ることができる。
【0093】
上述の付加的な疎水性材料は、1つもしくはそれ以上の水に不溶性のロウ様熱可塑性物
質を含んでいてよく、そして、その熱可塑性物質は、恐らく、この1つもしくはそれ以上
の水に不溶性のロウ様物質よりも疎水性に劣る1つもしくはそれ以上ロウ様熱可塑性物質
と混合されていよう。一定の放出を達成するため、本製剤におけるそれら個々のロウ様物
質は、初期放出段階の間、胃腸液中において実質的に非分解性であって且つ不溶性である
べきである。水に不溶性の有用なロウ様物質は、約1:5,000(w/w)より低い水
溶解度を有する物質であり得る。
【0094】
上述の成分に加え、徐放出性マトリックスは、適切な量の他の材料、例えば製薬業界に
おいて普通に用いられている希釈剤、潤滑剤、結合剤、粒状化助剤、着色剤、風味剤、及
びグリダント等も含んでいてよい。これらの付加的な材料の量は、所望の製剤に望ましい
効果をもたらすのに充分な量であろう。
【0095】
上述の成分に加え、溶融押出し多重粒状体を組み入れた徐放出性マトリックスは、適切
な量の他の材料、例えば製薬業界において普通に用いられている希釈剤、潤滑剤、結合剤
、粒状化助剤、着色剤、風味剤、及びグリダント等も、望ましい場合には、重量で粒状体
の約50%の量まで含んでいてよい。
【0096】
経口用剤形を調合するのに使用され得る、薬学的に許容可能な担体及び賦形剤の具体的
な例が、Handbook of Pharmaceutical Excipients、American Pharmaceutical Associati
on(1986年)に記載されている。
【0097】
(溶融押出し多重粒状体)
本発明による適切な溶融押出しマトリックスの調製は、例えば、オキシコドン(または
オキシコドン塩)及び/又はナロキソン(またはナロキソン塩)を、少なくとも1つの疎
水性材料、及び、好適には、均一な混合物を得るための付加的な疎水性材料と共に混合す
るステップを含んでいてよい。次いで、その均一な混合物は、少なくともその混合物を充
分に押し出せるほどに軟化させるのに充分な温度にまで加熱される。その後、結果として
得られたその均一な混合物は、ストランドを形成すべく押し出される。その押出し物は、
好適には、冷却され、そして、当技術分野において既知の何らかの手段により多重粒状体
の形態に切断される。それらのストランドは、冷却され、そして、多重粒状体の形態に切
断される。その後、それらの多重粒状体は単位量に分けられる。この押出し物は、好適に
は、約0.1mmから約5mmまでの範囲の直径を有しており、そして、約8時間から約
24時間までの期間にわたる治療的に活性な物質の徐放出性をもたらす。
【0098】
本発明の溶融押出し品を調製するための一つの随意的なプロセスは、疎水性材料、オキ
シコドン(またはオキシコドン塩)、及び、場合によっては、ナロキソン(またはナロキ
ソン塩)、及び、随意的な結合剤を計量して押出機に直接的に供給するステップ;その均
一な混合物を加熱するステップ;均一な混合物を押し出し、これにより、ストランドを形
成するステップ;均一な混合物を含有するそれらのストランドを冷却するステップ;それ
らのストランドを切断して、約0.1mmから約12mmまでのサイズを有する粒子に為
すステップ;及び、それらの粒子を単位量に分けるステップを含む。本発明のこの態様で
は、比較的連続的な製造手順が実現される。
【0099】
また、押出機の開口または出口ポートの直径も、押し出されるストランドの太さを変え
るべく調節することができる。更に、押出機の出口部分は丸形である必要はない;長円形
や矩形等であってよい。出てくるストランドは、熱線切断機や断裁機等を用いて粒子に為
すことができる。
【0100】
溶融押出し多重粒状体系は、押出機の出口オリフィスに応じて、例えば顆粒、スフェロ
イド、またはペレットの形態であってよい。本発明の目的上、「(1つもしくは複数の)
溶融押出し多重粒状体」及び「(1つもしくは複数の)溶融押出し多重粒状体系」及び「
溶融押出し粒子」という用語は、好適には同様なサイズ及び/又は形の範囲内にあり、そ
して、好適には本明細書で説明されている通りの疎水性材料を含め、1つもしくはそれ以
上の活性物質と1つもしくはそれ以上の賦形剤を含有する複数の単位を表すものとする。
この点について、本溶融押出し多重粒状体は、長さが約0.1mmから約12mmまでの
範囲であり、そして、約0.1mmから約5mmまでの直径を有するであろう。更に、本
溶融押出し多重粒状体は、このサイズ範囲内において、あらゆる幾何学的形状であってよ
いことを理解すべきである。代替的に、その押出し物を単に所望の長さに切断し、そして
、球状化ステップを必要とせずに、治療的に活性な物質の単位量に分けることもできる。
【0101】
一つの好適な実施態様では、経口用剤形は、有効量の溶融押出し多重粒状体をカプセル
内に包含させるべく調製される。例えば、複数の本溶融押出し多重粒状体を、経口摂取さ
れて胃液と接触したときに効果的な徐放出性の用量をもたらすのに充分な量でゼラチンカ
プセルに入れることができる。
【0102】
別の好適な実施態様では、適切な量の多重粒状体押出し物が、標準的な技術を利用した
通常の錠剤機を用いて経口用錠剤の形態に圧縮される。(圧縮及び成形される)錠剤、(
硬質及び軟質のゼラチン)カプセル剤、及び丸剤を製造するための技術及び組成物につい
ても、Remington’s Pharmaceutical Sciences(編集者、Arthur Osol)、1
553−1593頁(1980年)に記載されている。
【0103】
更に別の好適な実施態様では、上でもっと詳しく記載されている米国特許第4,957
,681号(Klimeschら)で説明されているようにして、その押出し物を錠剤の
形態に成形することができる。
【0104】
場合によっては、それらの徐放出性溶融押出し多重粒状体系または錠剤は、徐放出性コ
ーティング、例えば上で説明されている徐放出性コーティング等でコーティングされてよ
く、あるいは、ゼラチンカプセルを例えば上で説明されている徐放出性コーティング等で
更に被覆することができる。そのオーバーコーティングは、他のものの中でもとりわけ、
所望の放出速度に大きく依存するが、そのようなコーティングは、好適には、約2パーセ
ントから約30パーセントまでの重量増加レベルを得るのに充分な量の疎水性材料を含む
。
【0105】
本発明の溶融押出し単位剤形は、更に、カプセルに入れられる前の溶融押出し粒子(例
えば、オキシコドン(またはオキシコドン塩)を伴う一群の粒子とナロキソン(またはナ
ロキソン塩)を伴う一群の粒子)の組み合わせを含んでいてよい。更に、それらの単位剤
形は、即座に放出するためのある量の即時放出性物質も含むことができる。その即時放出
性物質は、例えばゼラチンカプセル内における分離したペレットとして組み入れられてよ
く、あるいは、それらの剤形(例えば、徐放出性コーティング剤形またはマトリックスを
ベースとした剤形)の調製後、多重粒状体の表面にコーティングされてもよい。また、本
発明の単位剤形は、所望の効果を達成するため、徐放出性のビーズとマトリックス多重粒
状体の組み合わせも含んでいてよい。
【0106】
好適には、本発明の徐放出性製剤は、例えば経口摂取されて胃液次いで腸液に晒された
ときに、(1つもしくは複数の)本物質をゆっくりと放出する。本発明の溶融押出し製剤
の徐放出プロフィールは、例えば遅延剤の量、即ち、疎水性材料の量を変更することによ
り、疎水性材料に対する可塑剤の量を変更することにより、付加的な成分または賦形剤を
含めることにより、製造方法を変更することなどにより変えることができる。
【0107】
本発明の別の実施態様では、溶融押出し材料は、オキシコドン(またはオキシコドン塩
)及びナロキソン(またはナロキソン塩)の包含を伴うことなく調製され、そして、その
後、オキシコドン(またはオキシコドン塩)及びナロキソン(またはナロキソン塩)をそ
の押出し物に加えることができる。そのような製剤は、典型的には、押出しマトリックス
材料と共に混合された本物質を有しており、その後、ゆっくりと放出する製剤をもたらす
ため、その混合物が錠剤化されよう。
【0108】
(コーティング)
本発明の剤形は、場合によっては、1つもしくはそれ以上の放出を調節するのに適した
材料もしくは本製剤を保護するのに適した材料でコーティングされてよい。一つの実施態
様では、コーティングは、pH−依存性の放出もしくはpH−非依存性の放出のいずれか
を可能にするために施される。pH−依存性のコーティングは、患者に対して少なくとも
約8時間、好適には約12時間から約24時間までにわたる鎮痛効果を与えることができ
る吸収プロフィールがもたらされるように、オキシコドンを胃腸(GI)管の望ましい領
域で、例えば胃または小腸で放出するように機能する。pH−非依存性コーティングが所
望の場合、そのコーティングは、例えばGI管等の環境流体におけるpH−変化に関係な
く最適な放出が達成されるように設計される。また、GI管の一つの望ましい領域、例え
ば胃等で用量の一部を放出し、そして、GI管の別の領域、例えば小腸で残りの用量を放
出する組成物を調合することもできる。
【0109】
製剤を得るためにpH−依存性コーティングを利用する本発明による製剤は、反復−作
用効果(repeat−action effect)を与えることもでき、そこでは、
非保護薬剤は、腸溶コーティング上にコーティングされて、胃で放出され、一方、腸溶コ
ーティングで保護されている残りの薬剤は、胃腸管を更に下って放出される。本発明に従
って使用され得るpH−依存性のコーティングは、セラック、セルロースアセテートフタ
レート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピル
メチルセルロースフタレート、及びメタクリル酸エステルコポリマー、ゼイン等を含む。
【0110】
特定の好適な実施態様では、オキシコドンまたはそれの塩、及び、場合によっては、ナ
ロキソンまたはそれの塩を含有する基材(例えば、錠剤のコアビーズ、マトリックス粒子
)は、(i)アルキルセルロース;(ii)アクリルポリマー;または(iii)それら
の混合物から選択される疎水性材料でコーティングされる。そのコーティングは、有機ま
たは水性の溶液または分散系の形態で適用されてよい。そのコーティングは、所望の徐放
出プロフィールを得るため、基材の約2%から約25%までの重量増加が得られるように
適用されてよい。水性分散系から誘導されるコーティングについては、例えば米国特許第
5,273,760号及び第5,286,493号で詳しく説明されている。
【0111】
本発明に従って使用され得る徐放出性製剤またはコーティングの他の例は、米国特許第
5,324,351号;第5,356,467号、及び第5,472,712号を含む。
【0112】
(アルキルセルロースポリマー)
アルキルセルロースを含め、セルロース性の材料及びポリマーは、本発明によるビーズ
をコーティングするのに非常に適した疎水性材料を提供する。単なる例として挙げると、
一つの好適なアルキルセルロースポリマーはエチルセルロースであるが、当業者であれば
、本発明による疎水性コーティングの全体または一部として、他のセルロース及び/又は
アルキルセルロースポリマーも、単独もしくは何らかの組み合わせにおいて、容易に使用
され得ることが認識されよう。
【0113】
一つの商業的に入手可能なエチルセルロースの水性分散系はAquacoat(登録商
標)(FMC Corp.、フィラデルフィア、ペンシルバニア州、U.S.A.)であ
る。Aquacoat(登録商標)は、エチルセルロースを水に不混和性の有機溶媒中に
溶解し、その後、界面活性剤と安定剤の存在下において、それを水中に乳化させることに
より調製される。均質化してサブミクロンオーダーの液滴を発生させた後、真空下でその
有機溶媒を蒸発させることにより、シュードラテックス(pseudolatex)を形成する。可塑
剤は、製造段階中には、このシュードラテックスに組み入れられない。従って、それをコ
ーティングとして使用するのに先立って、Aquacoat(登録商標)を適切な可塑剤
と使用前に直接的に混合する必要がある。エチルセルロースの別の水性分散系は、Sur
elease(登録商標)(Colorcon,Inc.、ウェストポイント、ペンシル
バニア州、U.S.A.)として商業的に入手することができる。この製品は、可塑剤を
製造段階中に分散系に組み入れることにより調製される。ポリマー、可塑剤(セバシン酸
ジブチル(dibutyl sebacate))、及び安定剤(オレイン酸)のホットメルトが均一な混合
物として調製され、その後、基材に直接的に適用できる水性分散系を得るべく、この混合
物がアルカリ性溶液で希釈される。
【0114】
(アクリルポリマー)
本発明の他の好適な実施態様では、徐放出性コーティングを含む疎水性材料は、薬学的
に許容可能なアクリルポリマーであり、それらのアクリルポリマーは、これらに限定する
ものではないが、アクリル酸とメタクリル酸のコポリマー、メチルメタクリレートコポリ
マー、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸
)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メチルメタ
クリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)コポリマー、ポリア
クリルアミド、アミノアルキルメタクリレートコポリマー、ポリ(無水メタクリル酸)、
及びグリシジルメタクリレートコポリマーを含む。
【0115】
特定の好適な実施態様では、そのアクリルポリマーは1つもしくはそれ以上のアンモニ
オメタクリレートコポリマーを含む。アンモニオメタクリレートコポリマーは、当技術分
野において広く知られており、低含量の第四級アンモニウム基を伴うアクリル酸及びメタ
クリル酸エステルの充分に重合されたコポリマーとしてNF XVIIに記載されている
。
【0116】
望ましい溶解プロフィールを得るため、例えば中性(メタ)アクリルエステルに対する
第四級アンモニウム基のモル比が異なっている等の異なる物理的特性を有する2つもしく
はそれ以上のアンモニオメタクリレートコポリマーを組み入れることが必要になり得る。
【0117】
特定のメタクリル酸エステル型ポリマーは、本発明に従って使用され得るpH−依存性
コーティングを調製するのに有用である。例えば、メタクリル酸コポリマーまたは高分子
メタクリレートとしても知られているジエチルアミノエチルメタクリレートと他の中性メ
タクリルエステルから合成される一群のコポリマーがあり、Rohm Tech,Inc
.からEudragit(登録商標)として商業的に入手可能である。幾つかの異なるタ
イプのEudragit(登録商標)がある。例えば、Eudragit(登録商標)E
は、酸性媒質中において膨潤及び溶解するメタクリル酸コポリマーの一例である。Eud
ragit(登録商標)Lは、約pH<5.7では膨潤せず、約pH>6で溶けるメタク
リル酸コポリマーである。Eudragit(登録商標)Sは、約pH<6.5では膨潤
せず、約pH>7で溶ける。Eudragit(登録商標)RL及びEudragit(
登録商標)RSは水に対して膨潤性であり、これらのポリマーによって吸収される水の量
はpH−依存性であるが、Eudragit(登録商標)RL及びRSでコーティングさ
れた剤形はpH−非依存性である。
【0118】
特定の好適な実施態様では、そのアクリルコーティングは、それぞれEudragit
(登録商標)RL30D及びEudragit(登録商標)RS30Dという商品名でR
ohm Pharmaから商業的に入手可能な2つのアクリル樹脂ラッカーの混合物を含
んでいる。Eudragit(登録商標)RL30D及びEudragit(登録商標)
RS30Dは、低含量の第四級アンモニウム基を伴うアクリル及びメタクリルエステルの
コポリマーであり、残りの中性(メタ)アクリルエステルに対するアンモニウム基のモル
比は、Eudragit(登録商標)RL30Dの場合は1:20であり、Eudrag
it(登録商標)RS30Dの場合は1:40である。その平均分子量は約150,00
0である。RL(高浸透性)及びRS(低浸透性)というコードの意味は、これらの物質
の浸透特性を表している。Eudragit(登録商標)RL/RS混合物は、水及び消
化液中において不溶性である。しかし、これらから形成されるコーティングは、水性溶液
及び消化液中において膨潤性であり、且つ、浸透性である。
【0119】
本発明のEudragit(登録商標)RL/RS分散系は、望ましい溶解プロフィー
ルを有する徐放出性製剤を最終的に得るため、あらゆる望ましい比で相互に混合されてよ
い。望ましい徐放出性製剤は、例えば、100%のEudragit(登録商標)RL、
50%のEudragit(登録商標)RLと50%のEudragit(登録商標)R
S、及び10%のEudragit(登録商標)RL:90%のEudragit(登録
商標)RSから誘導される遅延剤コーティングから得ることができる。当業者であれば、
勿論、例えばEudragit(登録商標)L等の他のアクリルポリマーも使用し得るこ
とが認識されよう。
【0120】
(可塑剤)
コーティングが疎水性材料の水性分散系を含む本発明の実施態様では、その疎水性材料
の水性分散系に有効量の可塑剤を含めることにより、その徐放出性コーティングの物理的
特性が一層改善される。例えば、エチルセルロースは、比較的高いガラス転移温度を有し
ており、通常のコーティング条件下では可撓性のフィルムを形成しないため、それをコー
ティング材料として使用する前に、エチルセルロースコーティングを含有する徐放出性コ
ーティングに可塑剤を組み入れることが好適である。一般的に、コーティング溶液に組み
込まれる可塑剤の量は、そのフィルム形成剤の濃度に基づいており、例えば、フィルム形
成剤の重量で約1パーセントから約50パーセントまでであることが最も多い。しかし、
可塑剤の濃度は、個々のコーティング溶液と適用方法を用いて慎重な実験を行った後にの
み、適切に決定することができる。
【0121】
エチルセルロースに対する適切な可塑剤の例は、水に不溶性の可塑剤、例えばセバシン
酸ジブチル、ジエチルフタレート、トリエチルシトレート、トリブチルシトレート、及び
トリアセチンを含むが、但し、水に不溶性な他の可塑剤(例えばアセチル化モノグリセリ
ド、フタル酸エステル、ヒマシ油、等)も使用することができる。トリエチルシトレート
は、本発明のエチルセルロースの水性分散系に対する特に好適な可塑剤である。
【0122】
本発明のアクリルポリマーに対する適切な可塑剤の例は、これらに限定するものではな
いが、トリエチルシトレートNF XVI等のクエン酸エステル、トリブチルシトレート
、ジブチルフタレート、及び、恐らくは1,2−プロピレングリコールを含む。Eudr
agit(登録商標)RL/RSラッカー溶液等のアクリルフィルムから形成されるフィ
ルムの弾性を高めるのに適していることが判明している他の可塑剤は、ポリエチレングリ
コール、プロピレングリコール、ジエチルフタレート、ヒマシ油、及びトリアセチンを含
む。トリエチルシトレートは、本発明のエチルセルロースの水性分散系に対する特に好適
な可塑剤である。
【0123】
更に、少量のタルクの付加は、処理中にこの水性分散系がくっつく傾向を低減し、且つ
、つや出し剤として作用することが判明している。
【0124】
(徐放出性浸透圧性剤形)
また、本発明による徐放出性剤形は、浸透圧性投与製剤として調製されてもよい。この
浸透圧性剤形は、好適には、薬剤層(オキシコドン(またはオキシコドン塩)、及び、場
合によっては、ナロキソン(またはナロキソン塩)を含む)と送給または押し出し層(ナ
ロキソン(またはナロキソン塩)を含んでいてよい)を含有する二層のコアを含んでおり
、そして、その二層のコアは、場合によっては少なくとも1つの通路が配置された、半透
性の壁で覆われている。
【0125】
本発明の目的で使用される「通路」という表現は、そこを通ってオキシコドンまたはオ
キシコドン塩が(ナロキソンまたはナロキソン塩と共に、もしくはそれらを伴わずに)繊
維、毛細管、多孔性オーバーレイ、多孔質インサート、微孔性メンバー、または多孔性組
成物を通じてポンピング、拡散、または移動され得る開口、オリフィス、穴、細孔、多孔
質エレメントを含む。また、この通路は、使用環境流体中において壁から浸食または浸出
されて少なくとも1つの通路を生成する化合物を含んでいてもよい。通路を形成するため
の代表的な化合物は、上述の壁中において浸食可能なポリ(グリコール)酸またはポリ(
乳)酸;ゼラチン状フィラメント;水により除去可能なポリ(ビニルアルコール);浸出
可能な化合物、例えば流体により除去可能な細孔形成ポリサッカリド、酸、塩、または酸
化物を含む。通路は、徐放出寸法の細孔−通路を形成すべく、上述の壁からある化合物、
例えばソルビトール、スクロース、ラクトース、マルトース、またはフルクトース等を浸
出させることにより形成することができる。その通路は、本剤形からのオキシコドンまた
はオキシコドン塩の持続的な計量的放出に役立つあらゆる形状、例えば丸形、三角形、四
角形、及び長円形等を為していてよい。この剤形は、本剤形の1つもしくはそれ以上の表
面に離間した関係において1つもしくはそれ以上の通路を有する状態で製造することがで
きる。通路及び通路を形成するための装置が米国特許第3,845,770号;第3,9
16,899号;第4,063,064号及び第4,088,864号に開示されている
。ある徐放出速度の放出用細孔をもたらすべく水性浸出により形成された放出用細孔とし
てサイジング、成形、及び適合化された徐放出寸法を含む通路が米国特許第4,200,
098号及び第4,285,987号に開示されている。
【0126】
特定の実施態様では、上述の二層のコアは、オキシコドンまたはそれの塩を有する薬剤
層と、ナロキソンまたはそれの塩を含有する変位または押し出し層を含んでいる。特定の
実施態様では、その薬剤層は、少なくとも1つのポリマーヒドロゲルも含んでいてよい。
そのポリマーヒドロゲルは、約500から約6,000,000までの間の平均分子量を
有していてよい。ポリマーヒドロゲルの例は、これらに限定されるものではないが、化学
式(C6H12O5)n・H2O[式中、nは3から7,500までである]を含み、500か
ら1,250,000までの数平均分子量を含むマルトデキストリンポリマー;例えば5
0,000から750,000までの重量平均分子量を有するポリ(エチレンオキシド)
及びポリ(プロピレンオキシド)で代表され、より特定的には、少なくとも100,00
0、200,000、300,000、または400,000の重量平均分子量のうちの
1つからなるポリ(エチレンオキシド)で代表されるポリ(アルキレンオキシド);10
,000から175,000までの重量平均分子量からなるアルカリカルボキシアルキル
セルロース[ここで、アルカリはナトリウムまたはカリウムであり、アルキルはメチル、
エチル、プロピル、またはブチルである];及び10,000から500,000までの
数平均分子量からなるエチレン−アクリル酸(メタクリル及びエタクリル酸を含む)のコ
ポリマーを含む。
【0127】
本発明の特定の実施態様では、上述の送給または押し出し層はオスモポリマー(osm
opolymer)を含んでいる。オスモポリマーの例は、これらに限定されるものでは
ないが、ポリアルキレンオキシド及びカルボキシアルキルセルロースからなるグループか
ら選択されるメンバーを含む。このポリアルキレンオキシドは、1,000,000から
10,000,000までの重量平均分子量を有している。そのポリアルキレンオキシド
は、ポリメチレンオキシド、ポリエチレンオキシド、ポリプロピレンオキシド、1,00
0,000の平均分子量を有するポリエチレンオキシド、5,000,000の平均分子
量を含むポリエチレンオキシド、7,000,000の平均分子量を含むポリエチレンオ
キシド、1,000,000の平均分子量を有する架橋ポリメチレンオキシド、及び平均
分子量が1,200,000のポリプロピレンオキシドからなるグループから選択される
メンバーであってよい。典型的なオスモポリマーカルボキシアルキルセルロースは、アル
カリカルボキシアルキルセルロース、ナトリウムカルボキシメチルセルロース、カリウム
カルボキシメチルセルロース、ナトリウムカルボキシエチルセルロース、リチウムカルボ
キシメチルセルロース、ナトリウムカルボキシエチルセルロース、カルボキシアルキルヒ
ドロキシアルキルセルロース、カルボキシメチルヒドロキシエチルセルロース、カルボキ
シエチルヒドロキシエチルセルロース、及びカルボキシメチルヒドロキシプロピルセルロ
ースからなるグループから選択されるメンバーを含む。この変位層で使用されるこれらの
オスモポリマーは、上述の半透壁を横断する浸透圧勾配を提示する。オスモポリマーは、
剤形内へ流体を吸い込むことによって浸透圧性ヒドロゲル(オスモゲルとしても知られて
いる)として膨潤及び膨張し、これにより、それらは、この浸透圧性剤形からオキシコド
ンまたはそれの薬学的に許容可能な塩を押し出す。
【0128】
また、上述の押し出し層は、オスマジェント(osmagents)及び浸透圧的に有
効な溶質としても知られている1つもしくはそれ以上の浸透圧的に有効な化合物も含んで
いてよい。それらは、例えば胃腸管からの環境液を剤形内へ吸い込み、上述の変位層の送
給運動力学に寄与する。浸透圧的に活性な化合物の例は、浸透圧性の塩及び浸透圧性の炭
水化物からなるグループから選択されるメンバーを含む。特定のオスマジェントの例は、
これらに限定されるものではないが、ナトリウムクロライド、カリウムクロライド、マグ
ネシウムサルフェート、リチウムホスフェート、リチウムクロライド、ナトリウムホスフ
ェート、カリウムサルフェート、ナトリウムサルフェート、カリウムホスフェート、グル
コース、フルクトース、及びマルトースを含む。
【0129】
上述の押し出し層は、場合によっては、9,000から450,000までの数平均分
子量を有するヒドロキシプロピルアルキルセルロースを含んでいてよい。そのヒドロキシ
プロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロ
ピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピ
ルブチルセルロース、及びヒドロキシプロピルペンチルセルロースからなるグループから
選択されるメンバーで代表される。
【0130】
上述の押し出し層は、場合によっては、無毒の着色剤または染料を含んでいてよい。着
色剤または染料の例は、これらに限定されるものではないが、食品医薬品局着色剤(FD
&C)、例えばFD&C第1号青色染料、FD&C第4号赤色染料等や、赤色酸化第二鉄
、黄色酸化第二鉄、チタンジオキシド、カーボンブラック、及びインジゴを含む。
【0131】
また、上述の押し出し層は、場合によっては、成分の酸化を抑制するための酸化防止剤
も含んでいてよい。酸化防止剤の幾つかの例は、これらに限定されるものではないが、ア
スコルビン酸、アスコルビルパルミテート、ブチル化ヒドロキシアニソール、2及び3t
ert−ブチル−4−ヒドロキシアニソールの混合物、ブチル化ヒドロキシトルエン、ナ
トリウムイソアスコルベート、ジヒドログアレチン酸(dihydroguaretic
acid)、カリウムソルベート、ナトリウムバイサルフェート、ナトリウムメタバイ
サルフェート、ソルビン酸、カリウムアスコルベート、ビタミンE、4−クロロ−2,6
−ジtertブチルフェノール、アルファトコフェロール、及びプロピルガレートからな
るグループから選択されるメンバーを含む。
【0132】
特定の代替的な実施態様では、本剤形は、オキシコドンまたはそれの薬学的に許容可能
な塩、ナロキソンまたはそれの薬学的に許容可能な塩、薬学的に許容可能なポリマー(例
えばポリエチレンオキシド)、場合によっては崩壊剤(例えばポリビニルピロリドン)、
場合によっては吸収増強剤(例えば脂肪酸、界面活性剤、キレート化剤、胆汁酸塩等)を
含有する均質なコアを含む。この均質なコアは、オキシコドンまたはそれの薬学的に許容
可能な塩を放出するための(上で定義されている通りの)通路を有する半透性の壁で覆わ
れている。
【0133】
特定の実施態様では、上述の半透性の壁は、セルロースエステルポリマー、セルロース
エーテルポリマー、及びセルロースエステル−エーテルポリマーからなるグループから選
択されるメンバーを含む。代表的な壁のポリマーは、セルロースアシレート、セルロース
ジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテ
ート、セルローストリアセテート、モノ−、ジ−、及びトリセルロースアルケニレート、
及び、モノ−、ジ−、及びトリセルロースアルキニレートからなるグループから選択され
るメンバーを含む。本発明で使用されるポリ(セルロース)は、20,000から7,5
00,000までの数平均分子量を有している。
【0134】
本発明の目的にかなった付加的な半透性ポリマーは、アセトアルデヒドジメチルセルロ
ースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチ
ルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテー
トジエチルアミノアセテート;半透性ポリアミド;半透性ポリウレタン;半透性スルホン
化ポリスチレン;米国特許第3,173,876号;第3,276,586号;第3,5
41,005号;第3,541,006号及び第3,546,876号に開示されている
ようなポリアニオンとポリカチオンの共沈により形成される半透性架橋ポリマー;米国特
許第3,133,132号でLoebとSourirajanが開示しているような半透
性ポリマー;半透性架橋ポリスチレン;半透性架橋ポリ(ナトリウムスチレンスルホネー
ト);半透性架橋ポリ(ビニルベンジルトリメチルアンモニウムクロライド);及び、半
透性の壁を横断する静水圧差または浸透圧差を大気圧で表したときに2.5×10-8(c
m2/hr・atm)から2.5×10-2(cm2/hr・atm)までの流体透過性を有
する半透性ポリマーを含む。本発明に有用な他のポリマーは、米国特許第3,845,7
70号;第3.916,899号及び第4,160,020号により;及び、Handbook o
f Common Polymers(スコット(Scott),J.R.及びW.J.Roff、197
1年、CRC Press、クリーブランド、オハイオ州)により、当技術分野において
既知である。
【0135】
特定の実施態様では、好適には、上述の半透性の壁は、無毒で不活性であり、そして、
その薬剤の調剤寿命(dispensing life)の間、それの物理的及び化学的
な完全性を維持する。特定の実施態様では、本剤形は結合剤を含む。結合剤の例は、これ
らに限定されるものではないが、5,000から350,000までの粘度平均分子量を
有する治療上許容可能なビニルポリマーを含み、そのようなビニルポリマーは、ポリ−n
−ビニルアミド、ポリ−n−ビニルアセトアミド、ポリ−n−ビニルピロリドンとしても
知られているポリ(ビニルピロリドン)、ポリ−n−ビニルカプロラクトン、ポリ−n−
ビニル−5−メチル−2−ピロリドン、及び、ポリ−n−ビニルピロリドンとビニルアセ
テート、ビニルアルコール、ビニルクロライド、ビニルフルオリド、ビニルブチレート、
ビニルラウレエート(laureate)、及びビニルステアレートからなるグループから選択され
るメンバーとのコポリマーからなるグループから選択されるメンバーによって代表される
。他の結合剤は、例えば、アカシア、デンプン、ゼラチン、及び平均分子量が9,200
から250,000までのヒドロキシプロピルアルキルセルロースを含む。
【0136】
特定の実施態様では、本剤形は潤滑剤を含み、そして、その潤滑剤は、金型の壁やパン
チの表面にくっつくのを防ぐため、その剤形の製造中に使用されてよい。潤滑剤の例は、
これらに限定されるものではないが、マグネシウムステアレート、ナトリウムステアレー
ト、ステアリン酸、カルシウムステアレート、マグネシウムオレエート、オレイン酸、カ
リウムオレエート、カプリル酸、ナトリウムステアリルフマレート、及びマグネシウムパ
ルミテートを含む。
【0137】
特定の好適な実施態様では、本発明は、10mgから40mgまでのオキシコドンまた
はそれの薬学的に許容可能な塩と、150,000から500,000までの平均分子量
を有する25mgから500mgまでのポリ(アルキレンオキシド)と、40,000の
平均分子量を有する1mgから50mgまでのポリビニルピロリドンと、0mgから約7
.5mgまでの潤滑剤、及び0.80mgから0.90mgまでのナロキソンまたはそれ
の塩とを含有する治療用組成物を含む。この0.80mgから0.90mgまでのナロキ
ソンまたはそれの塩は、好適には、上述の薬剤層にある。
【0138】
(坐剤)
本発明の徐放出性製剤は、適切な坐薬用基剤と本明細書に開示されている用量のオキシ
コドン(またはオキシコドン塩)及びナロキソン(またはナロキソン塩)とを含有する直
腸投与用の薬学的な坐薬として調合されてよい。徐放出性坐薬製剤の調製は、例えば米国
特許第5,215,758号に記載されている。
【0139】
吸収に先立って、薬剤は溶液の形態を為していなければならない。坐薬の場合には、溶
液となる前に、その坐薬基剤の溶解もしくは基剤の溶融が先んじて生じなければならず、
その後、その坐薬基剤からの薬剤が直腸液に分配される。身体内への薬剤の吸収は、坐薬
基剤によって変わり得る。従って、特定の薬剤と組み合わせて使用されるべき特定の坐薬
基剤は、その薬剤の物理的な特性を考慮に入れて選択されねばならない。例えば、脂質−
可溶性の薬剤は、直腸液内へ容易に分配されないが、脂質基剤に僅かにしか溶けない薬剤
は、直腸液内へ容易に分配されるであろう。
【0140】
薬剤の溶解時間(または放出速度)に影響を及ぼす種々の異なるファクターの中でもと
りわけ重要なファクターは、溶解溶媒媒質に供される薬剤物質の表面積、その溶液のpH
、その特定の溶媒媒質中におけるその物質の溶解度、及び、その溶媒媒質中における溶解
材料の飽和濃度の駆動力である。一般的に、直腸投与された坐剤からの薬物の吸収に影響
を及ぼすファクターは、坐剤のビヒクル、吸収部位のpH、薬剤のpKa、イオン化の程
度、及び脂質溶解度を含む。
【0141】
選択される坐薬基剤は、本発明の(1つもしくは複数の)物質に対して適合性を有して
いるべきである。更に、その坐薬基剤は、無毒で粘膜を刺激せず、直腸液中において溶融
または溶解し、そして、保管中は安定していることが好適である。
【0142】
水溶性の薬剤と水に不溶性の薬剤の両方に対する本発明の特定の好適な実施態様では、
その坐薬基剤は、鎖長がC12からC18までの飽和天然脂肪酸のモノ−、ジ−、及びトリグ
リセリドからなるグループから選択される脂肪酸ロウを含んでいる。
【0143】
本発明の坐剤の調製では、他の賦形剤を使用することもできる。例えば、直腸ルートを
介して投与するのに適した形を形成するため、ロウが使用されてよい。この系は、ロウを
伴わずに使用することもできるが、直腸投与及び経口投与のどちらの場合にも、ゼラチン
カプセルに充填される希釈剤の付加を伴う。
【0144】
商業的に入手可能な適切なモノ−、ジ−、及びトリグリセリドの例は、Novata
TM(タイプAB、AB、B、BC、BD、BBC、E、BCF、C、D、及び299)
(Henkel社製)、及びWitepsol TM(タイプH5、H12、H15、H
175、H185、H19、H32、H35、H39、H42、W25、W31、W35
、W45、S55、S58、E75、E76、及びE85)(Dynamit Nobe
l社製)という商品名の下で販売されている、12−18炭素原子鎖の飽和天然脂肪酸を
含む。
【0145】
他の薬学的に許容可能な坐薬基剤は、上述のモノ−、ジ−、及びトリグリセリドに対し
て全体的または部分的に置換されていてよい。この坐薬における基剤の量は、本剤形のサ
イズ(即ち、実際の重量)、使用される基剤(例えばアルギネート)及び薬剤の量によっ
て決定される。一般的には、坐薬基剤の量は、その坐剤の全重量のうち、重量で約20%
から約90%までである。好適には、坐薬中における坐薬基剤の量は、その坐剤の全重量
のうち、重量で約65%から約80%までである。
【0146】
(他の形態)
ここで開示されている本発明は、オキシコドン及びナロキソンの薬学的に許容可能なあ
らゆる塩を包含すべく意図されている。それらの薬学的に許容可能な塩は、これらに限定
されるものではないが、金属塩、例えばナトリウム塩、カリウム塩、セシウム塩等;アル
カリ土類金属、例えばカルシウム塩、マグネシウム塩等;有機アミン塩、例えばトリエチ
ルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、トリエタノールアミン塩、
ジシクロヘキシルアミン塩、N,N’−ジベンジルエチレンジアミン塩等;無機酸塩、例
えばハイドロクロライド、ハイドロブロミド、サルフェート、ホスフェート、テレフタレ
ート等;有機酸塩、例えばホルメート、アセテート、トリフルオロアセテート、マレエー
ト、酒石酸塩等;スルホネート、例えばメタンスルホネート、ベンゼンスルホネート、p
−トルエンスルホネート等;アミノ酸塩、例えばアルギネート(arginate)、ア
スパルギネート、グルタメート等を含む。
【0147】
オキシコドン(またはオキシコドン塩)とナロキソン(またはナロキソン塩)の組み合
わせは、通常の賦形剤、即ち、少なくともオキシコドンまたはそれの塩の徐放出性をもた
らすために当技術分野において知られている経口投与に適した薬学的に許容可能な有機ま
たは無機の担体物質と混ぜて使用することができる。薬学的に許容可能な適切な担体は、
これらに限定されるものではないが、アルコール、アラビアゴム、植物油、ベンジルアル
コール、ポリエチレングリコール、ゲレート(gelate)、炭水化物、例えばラクト
ース、アミロース、またはデンプン等、マグネシウムステアレート、タルク、ケイ酸、粘
性パラフィン、香油、脂肪酸モノグリセリド及びジグリセリド、ペンタエリトリトール(p
entaerythritol)脂肪酸エステル、ヒドロキシメチルセルロース、ポリビニルピロリドン
等を含む。これらの薬学的調製物は、滅菌されてよく、そして、望ましい場合には、補助
的な物質、例えば潤滑剤、崩壊剤、保存剤、安定剤、湿潤剤、乳化剤、浸透圧緩衝剤に影
響を及ぼすための塩、着色剤、矯味・矯臭(flavoring)及び/又は芳香剤等と混合されて
よい。経口的な使用が意図された本組成物は、当技術分野において既知のどんな方法によ
って調製されてもよく、そして、そのような組成物は、錠剤を製造するのに適した不活性
で無毒の薬学的に許容可能な賦形剤からなるグループから選択される1つもしくはそれ以
上の物質を含んでいてよい。そのような賦形剤は、例えば、ラクトース等の不活性な希釈
剤;コーンスターチ等の顆粒化及び崩壊剤;デンプン等の結合剤;及び、マグネシウムス
テアレート等の潤滑剤を含む。それらの錠剤は、コーティングされなくてもよいし、ある
いは、エレガンスさを得るため、または、活性成分の放出を遅らせるため、既知の技術に
よりコーティングされてもよい。また、経口的に使用するための製剤は、活性成分が不活
性な希釈剤と混合された硬質のゼラチンカプセル剤として提供されてもよい。
【0148】
本発明の経口用剤形は、錠剤、トローチ剤、甘味入り錠剤、粉末剤または顆粒剤、硬質
または軟質のカプセル剤、微小粒子(例えばマイクロカプセル、極小球体(microspheres)
等)、バッカル錠剤、坐剤等の形態であってよい。オキシコドン(またはオキシコドン塩
)とナロキソン(またはナロキソン塩)は、実質的に互いに相互分散(interdespersed)さ
れていてよい。
【0149】
特定の実施態様では、本発明は、上で説明されている通りの剤形を調製することにより
、経口用オキシコドン剤形(またはオキシコドン塩)の非経口的な乱用を防止する方法を
提供する。
【0150】
特定の実施態様では、本発明は、上で説明されている通りの剤形を調製することを含む
、経口用オキシコドン剤形の流用を防止する方法を提供する。
【0151】
特定の実施態様では、本発明は、上で説明されている剤形をヒト患者に投与することに
より疼痛を治療する方法を提供する。
【0152】
以下の実施例は、本発明の様々な態様を例証するものである。それらの実施例は、どの
ような仕方においても決して特許請求の範囲を限定するものと解釈すべきではない。
【実施例1】
【0153】
ナロキソンハイドロクロライドを含有する徐放出性オキシコドンハイドロクロライド製
剤を、以下の表1における調合により調製する:
【表1】
【0154】
この実施例では、ナロキソンハイドロクロライドジハイドレートが、顆粒化プロセス中
に本製剤に加えられる。そのプロセスを以下に説明する:
1.分散:ナロキソンHClを水に溶解し、その溶液をEudragit/トリアセチン
分散系に加える。
2.顆粒化:流動床グラニュレーターを用いて、そのEudragit/トリアセチン分
散系を、オキシコドンHCl、噴霧乾燥ラクトース、及びポビドンの混合物に噴霧する。
3.粉砕:その顆粒化物を取り出し、約1mmの開口(18メッシュのスクリーン)を有
するミルに通す。
4.ロウ引き:ステアリルアルコールを約50℃で融かし、高剪断混合機を用いて、上述
の粉砕顆粒化物に加える。トレーまたは流動床上で室温にまで冷却する。
5.粉砕:その冷却された顆粒化物を、約18メッシュのスクリーンを有するミルに通す
。
6.潤滑:混合機を用いて、その顆粒化物をタルク及びマグネシウムステアレートで潤滑
する。
7.圧縮:Kilian(登録商標)タブレットプレスを用いてその顆粒化物を圧縮し、
錠剤にする。
8.フィルムコーティング:ロータリーパンを用いて、その錠剤に水性のフィルムコーテ
ィングを施す。
【実施例2】
【0155】
以下の表2に示されている調合により、オキシコドン塩/ナロキソン塩の徐放出性浸透
圧性錠剤を製造する:
【表2】
【0156】
上述の調合を有する剤形を以下の手順により調製する:
先ず、オキシコドンハイドロクロライド、ナロキソンハイドロクロライドジハイドレー
ト、200,000の平均分子量を有するポリ(エチレンオキシド)、及び40,000
の平均分子量を有するポリビニルピロリドンを混合機に加え、10分間混合する。次いで
、その混合された材料に変性無水アルコールを10分間連続的に混合しながら加える。次
いで、その湿性顆粒化物を20メッシュのスクリーンに通し、室温で20時間乾燥させた
後、16メッシュのスクリーンに通す。次に、その顆粒化物を混合機へ移して混合し、マ
グネシウムステアレートで潤滑する。
【0157】
次いで、本剤形からオキシコドンHCl/ナロキソンHCl混合物を押し出すための変
位または押し出し用組成物を以下のようにして調製する:先ず、11,200の平均分子
量を有する3910gのヒドロキシプロピルメチルセルロースを45,339gの水に溶
解する。次いで、101gのブチル化ヒドロキシトルエンを650gの変性無水アルコー
ルに溶解する。次に、2.5kgのヒドロキシプロピルメチルセルロース水溶液をブチル
化ヒドロキシトルエンアルコール溶液に連続的に混合しながら加える。次いで、残りのヒ
ドロキシプロピルメチルセルロース水溶液をブチル化ヒドロキシトルエンアルコール溶液
に連続的に混合しながら加えることにより、結合剤溶液の調製を果たす。
【0158】
次に、21メッシュのスクリーンを備えたQuadro Comil(登録商標)ミル
を用いて、36,000gのナトリウムクロライドをサイジングする。次いで、1200
gの酸化第二鉄を40メッシュのスクリーンに通す。次いで、そのスクリーニングされた
材料、7,500,000の平均分子量を有する76,400gの薬学的に許容可能なポ
リ(エチレンオキシド)、11,200の平均分子量を有する2500gのヒドロキシプ
ロピルメチルセルロースを、Glatt(登録商標)流動床造粒機のボウルに加える。ボ
ウルを造粒機に取り付け、造粒プロセスを開始し、顆粒化を果たす。次に、その乾燥粉末
を空気懸濁させ、10分間混合する。次に、この粉末に上述の結合剤溶液を3本のノズル
から噴霧する。このプロセスの間、以下の如くに造粒を監視する:800g/分の合計溶
液噴霧速度;43℃のインレット温度、及び4300m3/時の空気流量。45,033
gの溶液噴霧終了後、結果として得られた被覆顆粒化粒子を35分間乾燥プロセスに掛け
る。
【0159】
8メッシュのスクリーンを有するQuadro Comil(登録商標)ミルを用いて
、それらのコーティングされた顆粒をサイジングする。その顆粒化物をTote(登録商
標)タンブラーへ移して混合し、281.7gのマグネシウムステアレートで潤滑する。
【0160】
次に、オキシコドンハイドロクロライド/ナロキソンハイドロクロライドを含有する医
薬組成物と押し出し用組成物を、Kilian(登録商標)タブレットプレスで二層錠剤
の形態に圧縮する。先ず、医薬組成物を金型のキャビティーに加えて予備圧縮した後、1
35mgの押し出し用組成物を加え、それらの層を3メートルトンの圧力ヘッド下におい
てプレスし、直径が11/32インチ(0.873cm)の接触層配列にする。
【0161】
この二層化された配列を半透性の壁でコーティングする。この壁形成組成物は、39.
8%のアセチル含量を有する100%セルロースアセテートを含む。その壁形成組成物を
アセトン:水(95:5の重量:重量)共溶媒に溶解して4%の固溶体を作製する。その
壁形成組成物を、24インチ(60cm)のVector(登録商標)ハイ−コーター内
において、上述の二層上及び二層の周りに噴霧する。次に、薬剤オキシコドン層を本剤形
の外面とつなぐため、その半透性の壁を通じて20ミル(0.508mm)の1つの出口
通路を穿孔する。45℃で45%の湿度において72時間乾燥させることにより、残存す
る溶媒を取り除く。次に、その浸透圧性投薬系を45℃で4時間乾燥させ、過剰な水分を
取り除く。
【実施例3】
【0162】
以下の表4に示されている調合により、オキシコドン10mg/ナロキソン0.85m
gの徐放出性カプセル剤を調製する:
【表3】
【0163】
上述の製剤を以下の手順により調製する:
1.ステアリルアルコールのフレークをインパクトミルに通す。
2.オキシコドンHCl、ナロキソンHCl、ステアリン酸、ステアリルアルコール、
及びEudragit RSPOを適切なブレンダー/ミキサー内で混合する。
3.その混合された材料を高温で双軸スクリュー押出し機に連続的に供給し、結果とし
て生じたストランドをコンベヤー上に収集する。
4.それらのストランドをコンベヤー上で冷却させる。
5.ペレタイザーを用いて、それらのストランドを1mmのペレットに切断する。
6.それらのペレットを、微細なペレットや大きすぎるペレットに関してスクリーニン
グし、サイズを約0.8mm−1.4mmの許容可能な範囲にする。
7.120mg/カプセルの充填重量でカプセルに充填する(サイズ2のカプセルに充
填する)。
【実施例4】
【0164】
以下の手順により、オキシコドン10mg/ナロキソン0.85mgの徐放出性カプセ
ル剤を調製する:
最初に、以下の表5に示されている調合により、即時放出性のオキシコドンビーズを調
製する:
【表4】
【0165】
(プロセス)
1.薬剤層形成溶液:オキシコドンHClとOpadryクリアーを水に溶解する。
2.薬剤のローディング:流動床乾燥機内において、薬剤溶液をNuPareilビー
ズに噴霧する。
3.コーティング:Opadryバタースコッチを水に分散させる。薬剤がローディン
グされたビーズに噴霧する。
【0166】
その後、以下の表6に示されている調合により徐放出性のビーズを調製する:
【表5】
【0167】
(プロセス)
1.制御放出性コーティング溶液:トリエチルシトレートを水中において均一化する。
その分散系をEudragit(登録商標)RS30D及びEudragit(登録商標
)RL30Dに加えた後、Cab−O−Sil(登録商標)をその混合物に加える。
2.シールコーティング溶液:Opadry(登録商標)クリアーを水に溶解する。
3.コーティング:流動床底面−噴霧法を用いて、オキシコドンHCl IRビーズに
、制御放出性コーティング溶液、続いて、シールコーティング溶液を適用する。
4.硬化:コーティングされたビーズをトレーに載せ、オーブン内において45℃で2
4時間硬化させる。
【0168】
オキシコドン/ナロキソンの徐放出性ビーズをもたらすため、上述の調合に、単位当た
り0.85mgのナロキソンを含めることができる。そのナロキソンは、NuParei
lビーズに噴霧する前に、オキシコドンHClと共に精製水中に溶解されてよい。
【実施例5】
【0169】
以下の表7における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の10mgオキシコドンハイドロクロライド製剤を調製した:
【表6】
【0170】
表7の製剤を以下の手順により調製した:
1.Niro MP−6流動床プロセッサー(Glatt GPCG−60/120で
置き換えることもできる)内において、オキシコドンHCl、ラクトース、及びポビドン
の一部を混合する。
2.Lightin’エアーミキサを用いて、Eudragit(登録商標)RS30
Dとトリアセチンをステンレス鋼製の容器内で混合する。
3.ステップ2から得られるEudragit(登録商標)の分散系を噴霧することに
より、Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、ステップ1から得られる混合物を顆粒化する。
4.Lightin’エアーミキサを用い、ステンレス鋼製の容器内において、残りの
ポビドン、ナロキソンHCl、及び水を混合し、透明な溶液を形成する。
5.Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、ステップ4から得られるナロキソンHCl溶液をステップ3から得
られる顆粒化物に噴霧する。
6.Quadro Comilを用いて、ステップ5から得られる顆粒化物をスクリー
ニングする。
7.Colletteグラニュレーター/ミキサー内において、ステアリルアルコール
(スチームジャケット付きのケトル内で前もって融かしておく)をステップ6から得られ
るスクリーニングされた顆粒化物と混合する。
8.Niro MP−6(または、Glatt GPCG−60/120)流動床プロ
セッサー内において、もしくは代替的に、ステンレス鋼製のトレー上において、ステップ
7から得られるロウ引きされた顆粒化物を冷却する。
9.Quadro Comilを用いて、ステップ8から得られる冷却された顆粒化物
の塊を除去する。
10.Colletteグラニュレーター/ミキサー内において、ステップ8から得ら
れる顆粒化物をタルク及びマグネシウムステアレートと混合する。
11.Kilian S−250タブレットプレスを用いて、顆粒化物を錠剤の形態に
圧縮する。
12.48″のAccela−Cotaコーティングパン内において、錠剤をOpad
ryでコーティングする。
【0171】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0172】
その溶解の結果が以下の表7Aに示されている:
【表7】
【実施例6】
【0173】
以下の表8における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の20mgオキシコドンハイドロクロライド製剤を調製した:
【表8】
【0174】
表8の製剤を実施例5と同じ手順により調製した。
【0175】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0176】
その溶解の結果が以下の表8Aに示されている:
【表9】
【実施例7】
【0177】
以下の表9における調合により、ナロキソンハイドロクロライドを含有する、徐放出性
の40mgオキシコドンハイドロクロライド製剤を調製した:
【表10】
【0178】
表9の製剤を実施例5と同じ手順により調製した。
【0179】
その後、それらの錠剤を、37℃における900mlの模擬胃液(SGF)中において
100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法を
用いて、溶解について試験した。
【0180】
その溶解の結果が以下の表9Aに示されている:
【表11】
【実施例8】
【0181】
実施例8では、絶食状態下にある18人の正常で健常な若い成人の男性及び女性被験者
を対象として、単一施設におけるオープン−ラベル方式の無作為、4−期間、4−シーケ
ンス、4−治療様式交差試験を実施した。この試験は、Talwin(登録商標)Nx(
〜0.55mgのナロキソンハイドロクロライドを含有)の単回錠剤投与、単回投与にお
ける1.5mgの経口的なナロキソンハイドロクロライド、分割投与(0時間、3時間、
6時間、及び9時間において、それぞれ、0.75mg、〜0.25mg、〜0.25m
g、及び〜0.25mg)としての1.5mgの経口的なナロキソンハイドロクロライド
、及び0.5mgの0.4mg/mlの静注によるナロキソンハイドロクロライドにおけ
るナロキソンの薬物動力学的パラメーターを比較するために行われた。被験者は、以下の
ものからなる期間と絡めた4種類のシーケンスに無作為に割り当てられた:
期間1は、1−8日目(一回目の投与時点から二回目の投与時点まで)であり、
期間2は、8−15日目(二回目の投与時点から三回目の投与時点まで)であり、
期間3は、15−22日目(三回目の投与時点から四回目の投与時点まで)であり、そ
して
期間4は22日目(最後の投与時点から本試験の終了まで)であった。
【0182】
投与様式の無作為化シーケンスが以下の表10に列挙されている:
【表12】
【0183】
被験者は、0日目、7日目、14日目、及び21日目の晩に、少なくとも投与の12時
間前に試験施設に来院し、そして、投与してから少なくとも6時間後まで(ナロキソンH
Clを経口的に分割用量で服用するグループの場合には、投与してから少なくとも12時
間後まで)は施設内に留め置かれた。投与は、1日目、8日目、15日目、及び22日目
に行われた。被験者は、試験期間中、激しい運動をしないように指示された。
【0184】
本試験で使用したストック溶液及び薬剤は、Sabex Inc.から購入した2ml
バイアルの1mg/mlナロキソンHCl注射剤USPでSabex Inc.から購入
したもの、10mlバイアルの0.4mg/mlナロキソンHCl注射剤USPでSab
ex Inc.から購入したもの、及びTalwin(登録商標)Nx(それぞれ50m
gの塩基及び0.5mgの塩基に相当するペンタゾシン及びナロキソンハイドロクロライ
ドの錠剤(USP))からなる。スクリーニングでの静注によるナロキソンチャレンジ試
験では1mg/mlのナロキソン濃度を使用し、そして、試験期間中の静脈による投与で
は0.4mg/mlの濃度を使用した。経口的な投与では、1mg/mlのストック溶液
を水で0.02mg/mlまで希釈した。用量は以下の通りであった:1錠のTalwi
n(登録商標)Nx、0.5mgの静注によるナロキソンHCl(1.25mlの0.4
mg/ml)、75mlの単回経口用量(濃度0.02mg/ml)としての1.5mg
の経口的なナロキソンHCl、及び、0時間、3時間、6時間、及び9時間において投与
される、それぞれ、50%(0.75mg)、17%(〜0/25mg)、17%(〜0
.25mg)、及び16%(〜0.25mg)の用量における分割経口用量としての1.
5mgのナロキソンHCl。
【0185】
Talwin(登録商標)Nxは8オンスの水と共に投与され、被験者はこれを完全に
飲み干した。その後、口腔を検査し、各被験者がこの錠剤を飲み込んだことを確認した。
【0186】
ナロキソンの単回経口投与後、被験者は、2回の付加的な75mlのすすぎ水を飲み、
合計の容量で225mlを飲んだ。分割投与(同じく0.02mg/ml)の場合は、最
初の投与後、2回の94mlのすすぎ水を飲み、それ以降の投与後には2回の75mlの
すすぎ水を飲んだ。
【0187】
静注によるナロキソンは、IVプッシュにより投与された。IVナロキソン後、スタッ
フのメンバーが少なくとも20分間連続的に被験者を観察した。
【0188】
各被験者は、それぞれの治療様式を1回受けた。それぞれの治療は、少なくとも10時
間の夜通しの絶食(水を除く)後に行われ、そして、被験者は、投与後に更に4時間絶食
させられた。投与する日を除き、食物に制限はなかった。被験者が臨床室に留め置かれて
いる間、すべての食事が標準化された時間に出され、そして、食事の内容も、本試験期間
全体を通じ、且つ、被験者間で標準化された。その内容を含め、食事の記録を維持した。
被験者が本臨床施設にいる間、カフェインやキサンチンを含有する飲み物は出されなかっ
た。
【0189】
本試験では、18人の無作為化された被験者のうち17人が4種類すべての治療を全う
した。シーケンス4に無作為に割り当てられた1人の被験者は、最初の治療段階(単回投
与の経口ナロキソン)完了後、同意を撤回した。
【0190】
(薬物動力学の結果)
本試験での薬物動力学データ(AUCt、AUC0-∞、Cmax、t1/2、tmax、Cl、及
びVssを得た)の概要が以下の表11に列挙されている:
【表13】
【0191】
以下の表12は、用量を0.5mgのIVナロキソンHCl用量に規格化した薬物動力
学データの概要を列挙したものである。
【表14】
【0192】
4種類の治療様式でのナロキソンに対する算術平均観測血漿濃度−時間プロフィールが
図2A−Dに示されている。4種類の治療様式でのナロキソンに対する個々の観測血漿濃
度−時間プロフィールのスパゲッティープロットが図3A−Dに示されている。試験した
4種類すべての治療様式で、ナロキソンのピーク血漿濃度へ向けた急速な増大が観測され
た。予想していた通り、ピーク血漿濃度に達するまでの時間は、IV製剤(0.14時間
、〜9分)の場合が最も短く、続いて、Talwin(登録商標)Nx(0.71時間)
、ナロキソンの単回経口溶液投与(0.78時間)であり、最後がナロキソンの分割経口
投与(3.18時間)であった。0.5mgのIVナロキソンHCl、1.5mgのナロ
キソンHClの単回経口投与、1.5mgのナロキソンHClの分割経口投与、及びTa
lwin(登録商標)Nx(0.55mgのナロキソンHClを含有)で、それぞれ、6
252pg/ml、23.0pg/ml、13.4pg/ml、及び8.00pg/ml
の平均ピーク血漿露出(Cmax)が観測された。
【0193】
IV治療様式に対する平均見掛け終末(terminal)消失半減期(t1/2)は0
.92時間であった。血漿濃度時間曲線9及び15の終末部分は、この半減期の値を決定
するために使用することができなかった。3種類の経口治療様式に対しては、終末消失半
減期を見積もりしなかった(見掛け終末消失速度定数λを、大多数の被験者の「鋸歯」濃
度時間プロフィールに基づいて精確に決定することができなかったため)。
【0194】
1.5mgの単回経口投与(47.4pg・h/ml)の平均合計ナロキソン露出(A
UCt)は、1.5mgのナロキソンの分割経口投与(64.9pg・h/ml)の約7
3%であった。予想していた通り、0.5mgのナロキソンのIV投与は3199pg・
h/mlの最も高い平均合計露出をもたらし、一方、Talwin(登録商標)Nxによ
る治療様式は5.92pg・h/mlの平均合計露出値をもたらした。0.5mgのIV
治療様式の場合、無限大に外挿された平均合計露出(AUC0-∞)は3051pg・h/
mlであった。一方、3種類の経口治療様式に対しては、17人の被験者のうち15人で
終末消失半減期を見積もることができなかったため、AUC0-∞は見積もり不能であった
。
【0195】
IV治療様式の場合、定常状態における平均血漿クリアランス及び分配容量は、それぞ
れ、2854ml/分及び217Lと見積もられた。
【0196】
1.5mgのナロキソンHClの単回経口投与における平均絶対バイオアベイラビリテ
ィー(F)(AUCtに基づく)は0.47%(範囲:0.15−1.11%)であり、
1.5mgのナロキソンHClの分割経口投与の場合は0.66%(範囲:0.01−2
.08%)であり、そして、Talwin(登録商標)Nx(0.55mgのナロキソン
HClを含有)の単回錠剤投与の場合は0.17%(範囲:0.0−0.79%)であっ
た。これらの低いバイオアベイラビリティー値は、ナロキソンが経口的に投与されたとき
に生じる有意な第一径路(first pass)代謝を反映するものである。
【0197】
全体的に、すべての治療様式にわたり、ナロキソンCmax値に関連して、%CVの観点
における有意な変動性が見られた(範囲:52−72%)。IV治療様式に対するAUC
t値は比較的変動性が小さかった(%CV−26%)が、一方、経口的な治療様式は変動
性が大きかった(%CV範囲:68−122%)。
【0198】
(有害事象)
死亡または重篤な有害事象は無かった。有害事象のために中止した被験者はいなかった
。薬剤に関係する有害事象の大多数は、被験者がTalwin(登録商標)Nxを投与さ
れたときに起こった。これらの有害事象は、悪心、嘔吐、めまい、及び頭痛を含んだ。1
人の被験者は、スケジュールが合わなかったため、同意を撤回した。
【0199】
18人の被験者のうち11人が合計25例の有害事象を経験したが、どれも重篤なもの
ではなかった。これらの被験者のうち7人は、本試験の薬物適用に多分、恐らく、もしく
は確実に関係していると記述された有害事象を経験した。全体的に、有害事象は、ペンタ
ゾシンに関係した予想される有害事象と矛盾無く、Talwin(登録商標)Nxの投与
を受けた被験者で一層頻繁に報告された。すべての被験者がすべての有害事象から回復し
た。本試験薬剤に関しては、措置(例えば、中止)を何も講じなかった。
【0200】
Talwin(登録商標)Nxの投与後、6人の被験者が有害事象を報告し、それらの
有害事象は、すべて、本試験の薬物適用に恐らくまたは確実に関係していると記述され、
めまい(N=5)、悪心(N=2)、嘔吐(N=1)、及び疲労(N=1)を含んだ。T
alwin(登録商標)Nxの投与を受けた被験者の中でもとりわけ重度な有害事象は、
めまい(N=1)及び悪心(N=2)を含んだ。
【0201】
他の3種類の治療様式の期間中には、本試験の薬物適用と多分、恐らく、または確実に
関係している重度な有害事象を報告した被験者は誰もおらず、また、めまいまたは何らか
の重大性を報告した被験者も誰もいなかった。また、他の3つの治療グループの期間中に
は、2例の有害事象のみが、試験薬剤に多分、恐らく、または確実に関係していると考え
られた:分割投与における経口ナロキソンに多分関係した頭痛、及び、静注によるナロキ
ソンに多分関係した悪心。ナロキソンのチャレンジ試験期間中には、無作為に振り分けら
れた18人の被験者で有害事象は観察されなかった。
【0202】
最も一般的に報告された有害事象はめまいと頭痛であった。頭痛は5人の被験者で報告
され、それらの被験者は誰もTalwin(登録商標)Nx投与期間中の被験者ではなか
った。そのうちの1人の被験者の場合、無作為化の前に頭痛が起こり、3人の被験者では
、僅か1回の治療期間の間に頭痛が起こり、そして、1人の被験者では、2回の治療期間
の間に頭痛が起こった。分割投与における経口ナロキソン後に1人の被験者でのみ、その
頭痛が試験薬剤に多分、恐らく、または確実に関係していると考えられ、そして、その重
傷度は軽度であった。それ以外の被験者では誰も、試験薬剤の投与後における投与日に頭
痛の発現を報告しなかった。
【0203】
(本試験からの結論)
ナロキソンの絶対バイオアベイラビリティーは、試験した3種類すべての経口的な治療
様式で<1%であった。血漿ナロキソン濃度は、すべての治療様式で、変動性が大きかっ
た。平均ナロキソンピーク露出は、8pg/mlで、範囲が0−16.6pg/mlであ
り、ナロキソンのi.v.投与後の場合の約0.1%であった。ナロキソンの終末半減期
は短く、約1hであった。
【0204】
単独で与えられるナロキソン、HClは、0.5mgIVの合計用量で充分に許容され
、そして、経口的には1.5mgまで充分に許容される。
【実施例9】
【0205】
実施例9では、OxyContin(登録商標)40mgと実施例7のオキシコドン/
ナロキソン制御放出性剤形(40mg/0.85mg)との生物学的同等性を評価すべく
管理された正常且つ健常な成人の男性及び女性被験者において、単回投与によるオープン
−ラベル方式の無作為、2−期間、2−治療様式交差試験を実施した。薬物動力学を評価
可能な被験者を少なくとも48人そろえるため、96人までの被験者を登録することとし
た。スクリーニング要件を満たした被験者は、1日目に本試験施設に入院し、1日目にN
arcan(登録商標)(ナロキソンハイドロクロライド溶液)チャレンジ試験を受けた
。Narcan(登録商標)チャレンジを成功裏に完了した被験者は、投与を2つの期間
に分離する5日間のウォッシュアウト期間を伴う2つの交差治療様式シーケンス(対照治
療様式から試験治療様式へ、あるいは、試験治療様式から対照治療様式へ)のうちの一方
に無作為に振り分けられた。被験者は、1日目から10日目まで連続的に本試験施設に留
め置かれた。被験者は、無作為化コードに従って、2日目と7日目の8:00AM頃に、
それらの治療様式のうちの一方による投与を受けた。すべての用量は、夜通しの絶食(水
を除く)後、8オンスの水と共に投与された。2日目と7日目の投与後、投与前から投与
の72時間後までの期間にわたり、血液サンプルを連続的に採取した。オキシコドンとナ
ロキソン(該当する場合)の血漿濃度を決定し、薬物動力学的な測定基準を算出して解析
した。安全性の測定は、有害事象の報告、生命徴候の測定、パルスオキシメトリー、臨床
検査室パラメーター、心電図(ECG)、及び理学的検査から構成された。統計分析の目
的上、期間1は、最初の用量の投与(2日目)から2回目の用量の投与(7日目)の直前
までの時間と定義された。期間2は、2回目の用量の投与(7日目)から10日目に行わ
れた投与の72時間後における血液採取までの時間と定義された。(生物学的同等性の評
価の観点における)評価可能な被験者は、これら2つの試験治療様式のうちのいずれかの
後、少なくとも12時間は嘔吐の発生があってはならない。
【0206】
ナロキソンを伴うオキシコドンの制御放出とOxyContin(登録商標)とに対す
る薬物動力学的測定基準(平均値±SD)が以下の表13にまとめられている。決定及び
算出された各薬物動力学的計量測定基準に対し、試験及び対照治療様式による平均値は同
様であり、すべてのパラメーターの算術平均(±SD)は、相互の約10%以内にあった
。
【表15】
【0207】
ナロキソンに対する薬物動力学的計量パラメーター(平均値±SD)が以下の表14に
まとめられている。
【表16】
【0208】
ナロキソンを伴うオキシコドンの制御放出(試験)対OxyContin(登録商標)
(対照)の生物学的同等性は、以下の表15に列挙されている各治療様式でのLS(最小
二乗)平均AUC及びCmax値に基づく生物学的同等性判定基準を用いて評価された。以
下の表15には、Cmax及びAUC値に対する指数化LS平均の比についての信頼区間(
90%)も示されている。
【表17】
上述の比及び90%CIの算出では、対数変換パラメーターを使用し、その後、指数化
し直した。また、上述の比及び90%CIは、シーケンス、被験者(シーケンス)、期間
、及びANOVAモデルにおける治療に対する影響を伴う指数化LS平均に基づいて決定
された。
【0209】
上の表から分かるように、AUCt、AUC∞、及びCmaxに対して、それらの比の90
%信頼区間の上限は125%未満(AUCt、AUC∞、及びCmaxに対して、それぞれ、
100%、100%、及び108%)であり、一方、それらの比の90%信頼区間の下限
は80%以上(AUCt、AUC∞、及びCmaxに対して、それぞれ、94%、93%、及
び100%)であった。これらの結果は、ナロキソンを伴う40mgのオキシコドン制御
放出が40mgのOxyContin(登録商標)と生物学的同等性を有していることを
示している。
【0210】
(安全性分析)
この試験における安全性パラメーターは、オピオイドのフェーズ1試験における安全性
を評価するために利用したパラメーターと同じものであった。全体的に、ナロキソンを伴
うオキシコドンの制御放出は、健常な被験者により広く許容された。死亡または重篤な有
害事象の報告はなかった。2人の被験者が、有害事象のため、本試験を中止した。最も一
般的な有害事象は、かゆみ、悪心、めまい、傾眠、及び嘔吐であった。すべての有害事象
は、軽度ないし中等度の強度であった。1人の被験者での一つの有害事象(この被験者の
咳の増加は、試験薬剤に関係しているとは考えられず、本試験の終了後も続いた)を例外
として、すべての有害事象は本試験を完了するまでに消散した。すべての試験薬剤関連有
害事象が、オキシコドンに関する既知の安全性プロフィールと合致した。臨床検査室値に
おける変化は散発性のものであり、本試験の薬物適用には関係していないものと考えられ
た。理学的検査、生命徴候、または心電図(ECGs)には、臨床的に意味のある薬剤関
連の変化はなかった。
【0211】
全体的な有害事象(発生率10%以上)の発生が以下の表16に示されている。
【表18】
【0212】
各治療後、85人の被験者が有害事象を報告した。有害事象の発生率は両治療様式間で
同様であった(OxyContin(登録商標)による治療後には85人[90.4%]
であったのに比べ、ナロキソンを伴うオキシコドンの制御放出による治療後には85人[
91.4%]であった)。OxyContin(登録商標)(対照)による治療後の場合
には、85人の被験者により415例の有害事象が報告されたのに比べ、ナロキソンを伴
うオキシコドンの制御放出(試験)による治療後の場合には、85人の被験者により41
3例の有害事象が報告された。かゆみ、悪心、めまい、傾眠、及び嘔吐が、どちらかの治
療用式後に最も頻繁に報告された有害事象であった。これらの有害事象は、ナロキソンを
伴うオキシコドンの制御放出とOxyContin(登録商標)により代表されるオピオ
イド鎮痛薬クラスの薬剤から予想される有害事象プロフィールと合致する。
【0213】
いずれかの治療用式後に報告された他の一般的な有害事象は、陶酔、頭痛、血管拡張、
及び発汗を含んだ。ナロキソンを伴うオキシコドンの制御放出による治療後の場合(23
/93、24.7%)には、OxyContin(登録商標)の場合(14/94、14
.9%)に比べ、もっと多数の被験者が血管拡張を報告した。ナロキソンを伴うオキシコ
ドンの制御放出による治療後及びOxyContin(登録商標)による治療後には、そ
れぞれ、以下の有害事象が同様な割合で報告された:頭痛(23.7%、22.3%)、
陶酔(31.2%、26.6%)、及び発汗(10.8%、8.5%)。
【0214】
全体的に、期間1及び2で報告されたあらゆる特定の有害事象の発生率間に顕著な差異
は観測されなかった。
【0215】
治療様式に少なくとも多分関係していると考えられた事象のうち、ナロキソンを伴うオ
キシコドンの制御放出による治療後及びOxyContin(登録商標)による治療後に
最も高い発生率で報告された事象は、それぞれ:かゆみ(52.7%、50.0%);悪
心(47.3%、36.2%);めまい(36.6%、45.7%);傾眠(40.9%
、39.4%);嘔吐(33.3%、30.9%);陶酔(31.2%、26.6%);
血管拡張(23.7%、14.9%)、及び頭痛(19.4%、19.1%)であった。
【0216】
全体的に、期間1及び2で報告された、治療様式に少なくとも多分関係していると考え
られた有害事象の発生率間に顕著な差異は観測されなかった。
【0217】
(血漿濃度−時間曲線)
図4は、両治療用式後の時間経過に伴うオキシコドンの平均血漿濃度を示している。
【0218】
図5及び6は、安全性について評価可能なすべての被験者でのナロキソンに対する平均
観測血漿濃度−時間曲線を示している。
【0219】
評価した被験者の約3分の2が、大多数の時点において、定量限界(BLQ[5pg/
mL])以下の血漿ナロキソン濃度を有していた。一般的に、平均血漿ナロキソン濃度は
低く、非常に変動性であった。時間経過に伴う平均ナロキソン濃度は、すべての時点にお
いて<1pg/mLであった。しかし、個別的に見れば、26pg/mLもの高い血漿濃
度を有した被験者もいた。
【0220】
(本試験からの結論)
この試験治療様式(0.85mgのナロキソンを伴う40mgのオキシコドンの制御放
出性錠剤)は、オキシコドンに対するこの対照治療様式(OxyContin(登録商標
)40mg錠)に対して生物学的同等性を有している。
【0221】
試験及び対照治療様式後における平均オキシコドンAUCt、AUC∞、Cmax、tmax
、及びびt1/2の値は同様であり、すべての値が相互の約10%以内にあった。
【0222】
平均血漿ナロキソン濃度は一般的に低く(<1pg/ml)、且つ、非常に変動性であ
り、数人の被験者では約0pg/mLから26pg/mLまでの範囲に及んだ。評価した
被験者の約3分の2がBLQ(5pg/mL)の血漿ナロキソン濃度を有していた。男性
患者と女性患者間の薬物動力学的データには臨床的に有意差がなかった。
【0223】
0.85mgのナロキソンを伴う40mgのオキシコドンの制御放出と40mgのOx
yContin(登録商標)は、同様な安全性プロフィールを呈した。両方とも、健常な
男性及び女性のボランティアに広く許容された。
【0224】
本発明の多くの他の変形態様が当業者にとって明らかであり、それらの変形態様も、添
付の特許請求の範囲に包含されるべく意図されている。
【図面の簡単な説明】
【0225】
【図1】図1は、本発明の一つの実施態様を実現する方法の作業系統図である。
【図2】図2A、2B、2C、及び2Dは、実施例8における4種類の治療様式で観測されたナロキソンに対する血漿濃度−時間プロフィールの平均値をグラフで表したものである。
【図3】図3A、3B、3C、及び3Dは、4種類の治療様式で観測されたナロキソンに対する個々の血漿濃度−時間プロフィールをグラフで表したものである。
【図4】図4は、実施例9の両様式による治療後における、時間の経過に伴うオキシコドンの平均血漿濃度をグラフで表したものである。
【図5】図5は、安全性について評価可能なすべての被験者での、時間の経過に伴うナロキソンに対する平均観測血漿濃度をグラフで表したものである。
【図6】図6は、安全性について評価可能なすべての被験者を性別に見た、時間の経過に伴うナロキソンに対する平均観測血漿濃度をグラフで表したものである。
【特許請求の範囲】
【請求項1】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約10mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項2】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項3】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項4】
上記組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項5】
剤形が、ヒト患者に対する定常状態での経口投与後、12時間の間、効果的な疼痛の軽減をもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項6】
オキシコドンハイドロクロライド及びナロキソンハイドロクロライドが、徐放出性の賦形剤を更に含有するマトリックス内において実質的に相互分散されている(interdispersed)、請求項1から3のいずれかに記載の医薬組成物。
【請求項7】
請求項1から3のいずれかに記載の医薬組成物であって、オキシコドンハイドロクロライド及びナロキソンハイドロクロライドのイン・ビトロ溶解速度が、1時間では、約30から約60重量%までのオキシコドンハイドロクロライド及び約30から約60重量%までのナロキソンハイドロクロライドが放出され;2時間では、約40から約70重量%までのオキシコドンハイドロクロライド及び約40%から約70重量%までのナロキソンハイドロクロライドが放出され;4時間では、約55から約90重量%までのオキシコドンハイドロクロライド及び約55から約90重量%までのナロキソンハイドロクロライドが放出され;8時間では、約70重量%より多くのオキシコドンハイドロクロライド及び約70重量%より多くのナロキソンハイドロクロライドが放出され;そして、12時間では、約80重量%より多くのオキシコドンハイドロクロライド及び約80重量%より多くのナロキソンハイドロクロライドが放出されるイン・ビトロ溶解速度である医薬組成物。
【請求項8】
ある剤形において約10mgから約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩と、0.65から0.90mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物であって、該組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±10%の範囲内にある前記ナロキソンまたはそれの薬学的に許容可能な塩の溶解速度をもたらす、医薬組成物。
【請求項9】
ヒト被験者の母集団に単回用量投与したときに、OxyContin(登録商標)(オキシコドンハイドロクロライド制御放出性)の量に相当するオキシコドン塩基によりもたらされる血漿オキシコドンの平均AUCの95%から105%までの範囲内にある血漿オキシコドンの平均AUCをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項10】
ヒト被験者の母集団に単回用量投与したときに、50pg/ml未満の血漿ナロキソンの平均Cmaxをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項11】
ヒト被験者の母集団に単回用量投与したときに、10pg/ml未満の血漿ナロキソンの平均Cmaxをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項12】
少なくともオキシコドンまたはそれの薬学的に許容可能な塩の徐放出をもたらす剤形において、約10−40mgのオキシコドンハイドロクロライドに相当するオキシコドン塩基の量におけるオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.75mgまでのナロキソン塩基に相当する量におけるナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物。
【請求項13】
10mgのオキシコドンハイドロクロライドを含む、請求項1または12に記載の医薬組成物。
【請求項14】
20mgのオキシコドンハイドロクロライドを含む、請求項2または12に記載の医薬組成物。
【請求項15】
40mgのオキシコドンハイドロクロライドを含む、請求項3または12に記載の医薬組成物。
【請求項16】
約10から約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩を含有する医薬組成物に0.65から0.90mgまでのナロキソンまたはそれの薬学的に許容可能な塩を組み入れることを含む徐放出性剤形の調製方法であって、前記組成物が少なくともオキシコドンまたはそれの薬学的に許容可能な塩の徐放出をもたらす、前記方法。
【請求項17】
疼痛を治療するためのキットであって:約10から約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩と、0.65から0.9mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む少なくとも1つの製剤を含有する容器を含み;前記キットが更に該製剤の使用を指示する証印を含む、疼痛治療用のキット。
【請求項18】
上記証印は、該使用が前記製剤の非経口的な乱用を低減するためのものであることを指示している、請求項17に記載のキット。
【請求項19】
上記組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、請求項7に記載の医薬組成物。
【請求項1】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約10mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項2】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約20mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項3】
少なくともオキシコドンハイドロクロライドの徐放出をもたらす剤形において約40mgのオキシコドンハイドロクロライドと、0.80から0.90mgまでのナロキソンハイドロクロライドとを含む医薬組成物。
【請求項4】
上記組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項5】
剤形が、ヒト患者に対する定常状態での経口投与後、12時間の間、効果的な疼痛の軽減をもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項6】
オキシコドンハイドロクロライド及びナロキソンハイドロクロライドが、徐放出性の賦形剤を更に含有するマトリックス内において実質的に相互分散されている(interdispersed)、請求項1から3のいずれかに記載の医薬組成物。
【請求項7】
請求項1から3のいずれかに記載の医薬組成物であって、オキシコドンハイドロクロライド及びナロキソンハイドロクロライドのイン・ビトロ溶解速度が、1時間では、約30から約60重量%までのオキシコドンハイドロクロライド及び約30から約60重量%までのナロキソンハイドロクロライドが放出され;2時間では、約40から約70重量%までのオキシコドンハイドロクロライド及び約40%から約70重量%までのナロキソンハイドロクロライドが放出され;4時間では、約55から約90重量%までのオキシコドンハイドロクロライド及び約55から約90重量%までのナロキソンハイドロクロライドが放出され;8時間では、約70重量%より多くのオキシコドンハイドロクロライド及び約70重量%より多くのナロキソンハイドロクロライドが放出され;そして、12時間では、約80重量%より多くのオキシコドンハイドロクロライド及び約80重量%より多くのナロキソンハイドロクロライドが放出されるイン・ビトロ溶解速度である医薬組成物。
【請求項8】
ある剤形において約10mgから約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩と、0.65から0.90mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物であって、該組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンまたはそれの薬学的に許容可能な塩の溶解速度に対して±10%の範囲内にある前記ナロキソンまたはそれの薬学的に許容可能な塩の溶解速度をもたらす、医薬組成物。
【請求項9】
ヒト被験者の母集団に単回用量投与したときに、OxyContin(登録商標)(オキシコドンハイドロクロライド制御放出性)の量に相当するオキシコドン塩基によりもたらされる血漿オキシコドンの平均AUCの95%から105%までの範囲内にある血漿オキシコドンの平均AUCをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項10】
ヒト被験者の母集団に単回用量投与したときに、50pg/ml未満の血漿ナロキソンの平均Cmaxをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項11】
ヒト被験者の母集団に単回用量投与したときに、10pg/ml未満の血漿ナロキソンの平均Cmaxをもたらす、請求項1から3のいずれかに記載の医薬組成物。
【請求項12】
少なくともオキシコドンまたはそれの薬学的に許容可能な塩の徐放出をもたらす剤形において、約10−40mgのオキシコドンハイドロクロライドに相当するオキシコドン塩基の量におけるオキシコドンまたはそれの薬学的に許容可能な塩と、0.65mgから0.75mgまでのナロキソン塩基に相当する量におけるナロキソンまたはそれの薬学的に許容可能な塩とを含む医薬組成物。
【請求項13】
10mgのオキシコドンハイドロクロライドを含む、請求項1または12に記載の医薬組成物。
【請求項14】
20mgのオキシコドンハイドロクロライドを含む、請求項2または12に記載の医薬組成物。
【請求項15】
40mgのオキシコドンハイドロクロライドを含む、請求項3または12に記載の医薬組成物。
【請求項16】
約10から約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩を含有する医薬組成物に0.65から0.90mgまでのナロキソンまたはそれの薬学的に許容可能な塩を組み入れることを含む徐放出性剤形の調製方法であって、前記組成物が少なくともオキシコドンまたはそれの薬学的に許容可能な塩の徐放出をもたらす、前記方法。
【請求項17】
疼痛を治療するためのキットであって:約10から約40mgまでのオキシコドンまたはそれの薬学的に許容可能な塩と、0.65から0.9mgまでのナロキソンまたはそれの薬学的に許容可能な塩とを含む少なくとも1つの製剤を含有する容器を含み;前記キットが更に該製剤の使用を指示する証印を含む、疼痛治療用のキット。
【請求項18】
上記証印は、該使用が前記製剤の非経口的な乱用を低減するためのものであることを指示している、請求項17に記載のキット。
【請求項19】
上記組成物が、37℃における900mlの模擬胃液(SGF)中において100rpmで、米国薬局方XXIV(2000年)のUSP装置I(バスケット)法によりイン・ビトロで試験したときに、1時間、4時間、及び12時間において、前記オキシコドンハイドロクロライドの溶解速度に対して±10%の範囲内にある前記ナロキソンハイドロクロライドの溶解速度をもたらす、請求項7に記載の医薬組成物。
【図1】

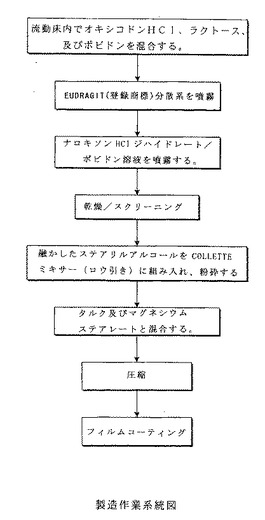
【図2】
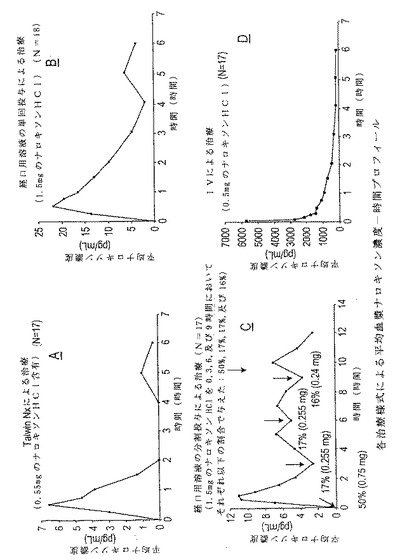
【図3】
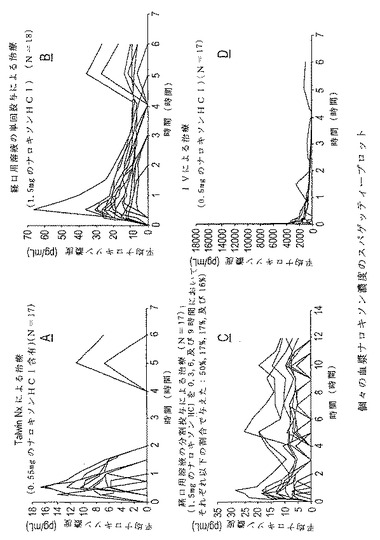
【図4】
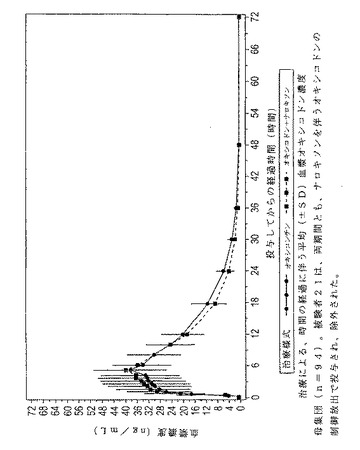
【図5】
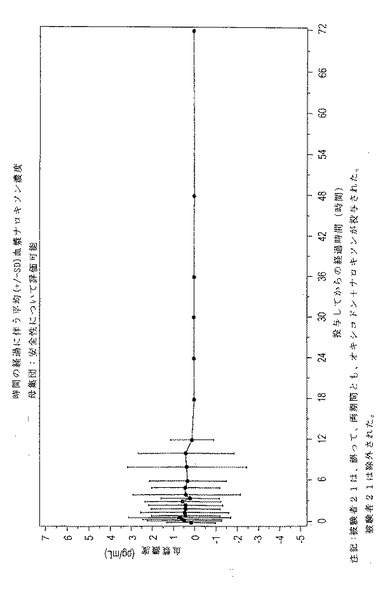
【図6】
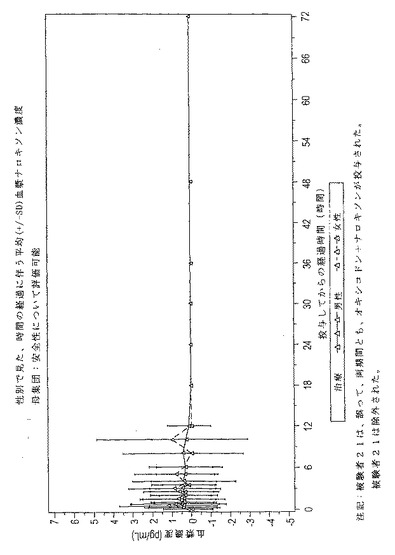
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−79055(P2009−79055A)
【公開日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願番号】特願2008−259189(P2008−259189)
【出願日】平成20年10月6日(2008.10.6)
【分割の表示】特願2003−513416(P2003−513416)の分割
【原出願日】平成14年7月18日(2002.7.18)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公開日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願日】平成20年10月6日(2008.10.6)
【分割の表示】特願2003−513416(P2003−513416)の分割
【原出願日】平成14年7月18日(2002.7.18)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]