
オストワルド熟成を減少させた水不溶性の医薬品物質の固体ナノ粒子処方物
本発明は、薬理学、医薬および医薬品化学の分野に属する。本発明は、オストワルド熟成を減少させた、実質的に水不溶性の医薬品物質(ドセタキセルなど)の、水系媒体中に分散した固体ナノ粒子から構成される新規な医薬組成物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2006年7月24日出願の、本発明者らによる米国特許仮出願第60/832587号の利益を主張する。
【0002】
本発明は、ナノ粒子を作製するためのプロセスに関する。より具体的には、本発明は、水不溶性の物質が水に溶解し得るようにその水不溶性の物質のナノ粒子を作製するためのプロセスに関する。さらにより具体的には、本発明は、水不溶性医薬品を水系媒体で送達することができるように、その水不溶性の医薬品のナノ粒子を作製するためのプロセスに関する。
【背景技術】
【0003】
ほとんどの抗癌剤の治療効力は、腫瘍部位への十分な局所的送達を実現することに基礎を置いている。多くの癌用化学療法薬は生体外で非常に有効であるが、生体内ではそれほど有効でないことが示されている。この不一致は、薬物を腫瘍部位に治療レベルで送達することの難しさ、および回復するためにはほとんど100%の細胞殺滅が必要であることに一部は起因すると考えられている(Jain 1994;Tannock 1998)。治療用分子であるサイトカイン、抗体、およびウイルスベクターは、血管壁を横断することが困難であるため、腫瘍に作用する能力には限界があることが多い(Yuan 1998)。不十分な特異的送達は、現在の癌用化学療法薬に関して認められるしばしば低い治療指数につながる可能性がある。これは、これらの化合物の広範な拡散および非特異的作用に起因する重大な全身毒性に姿を変える。
【0004】
別の問題は、投与に適した薬理学的に許容できるビヒクル中のいくつかの強力な化学療法薬の溶解性である。化学療法で広く使用されている2種類の分子は、微小管阻害剤(タキサン誘導体など)およびトポイソメラーゼI阻害剤(カンプトテシン(campthothecin)誘導体など)である。しかしながら、これらの重要な2種類の薬物がヒトにとって非常に有毒なビヒクル中で処方されてきたことが、実際、現在公知である。本発明は、水不溶性の医薬品物質の溶解性およびビヒクル毒性の問題を克服するための新規な医薬組成物を開示するためになされた。
【0005】
(治療薬としての微小管阻害剤)
パクリタキセル(タキソール、図1)は、Waniら(1971)によってタイヘイヨウイチイの木(Taxus brevifolia)から単離された天然のジテルペン産物である。タキサン、パクリタキセルおよびドセタキセル(特許文献1)は、微小管を安定化し腫瘍細胞死に導く新規な種類の抗癌剤に属する。同定された最初の微小管安定剤であるパクリタキセル(タキソール(登録商標)、Bristol−Myers Squibb Co.、米国、ニュージャージー州)が多くの種類の癌の治療にとって大きな価値があることは明らかになっている(Rowinsky 2001、Holton 1995)。パクリタキセルの臨床的成功によって、第二世代のタキサン、ドセタキセル(タキソテール(登録商標)、Sanofi−Aventis Pharmaceuticals、米国、ニュージャージー州)が開発され、同様の作用機構をもつ他の化合物の集中的な探索が開始された。数種類の構造的に多様な微小管安定化化合物が同定されている。同定された非タキサン安定剤である、エポチロン(Bollag 1995)、Taccalonolide(Tinley 2003)およびディスコデルモライド(discodermolide)(Mooberry 2004および1999;Martello 2000)は優れた前臨床活性を有しており、抗癌剤として臨床試験で評価されている。
【0006】
微小管は、多くの細胞機能に関与するチューブリンポリマーであり(Dustin 1984)、その多くの細胞機能のうちの1つが、細胞分裂の間に新しく形成する細胞の極に移動する染色体にとって必要な紡錘体の形成である(Avila 1990)。細胞機能にとって微小管が重要であることから、微小管は生物学的な微小管の毒に対する鋭敏な標的となる。微小管の安定化または破壊という点で微小管と相互作用するすべての化合物は微小管阻害剤と呼ばれる。それらは、細胞毒性効果を有し、そして細胞を殺すこともある。微小管は細胞増殖において有糸分裂を行うために必要とされるので、微小管阻害剤は、正常細胞よりも頻繁に分裂する癌細胞を主に攻撃するであろう。それゆえ、それらの多くは非常に重要な抗腫瘍化合物である。
【0007】
チューブリンは、四次構造が2つのポリペプチドサブユニット、α−チューブリンおよびβ−チューブリンから構成されているタンパク質である。高等真核生物における各サブユニットについていくつかのイソタイプが記載されている。微小管の機能は、その重合および解重合する能力に基づいている。このプロセスは非常に動的であり、この細胞構造の急激な短縮または伸長を伴う。チューブリンはGTP結合タンパク質であり、このヌクレオチドがタンパク質に結合することは、微小管重合のために必要とされるが、他方、重合したチューブリンに結合したGTPの加水分解は、微小管の解重合のために必要とされる。正常細胞における微小管安定性は、微小管安定化を促す微小管結合タンパク質(MAP)と呼ばれるいくつかのタンパク質の存在下で調節される。微小管重合を調節する細胞機構は、Ca2+の濃度に非常に鋭敏である。ほとんどの真核細胞の休止状態に特徴的な低い細胞質Ca2+濃度は微小管重合を促し、Ca2+の局所的増加は微小管解離(disassembly)を引き起こす(Gelford 1991)。微小管は、各々がα−チューブリンおよびβ−チューブリンを有する1つの分子からなるタンパク質二量体の重合を介して形成される。二量体およびポリマーは動的平衡の状態にあり、そのネットワークは機能上の要求に柔軟かつ迅速に反応することができる。このポリマーは通常は内径および直径がそれぞれ14nmおよび28nmの微細な無分岐の円筒、いわゆる微小管を形成する(図2;Kingston 2001)。重合は、α,β−二量体を一緒に結合して短いプロトフィラメントを形成することによって開始され、続いてそのプロトフィラメントの13本が並んで配列し微小管を形成する。引き続くこの微小管の成長は有極性であり、主に、プロトフィラメントのいわゆるプラス端でさらなる二量体の付加によって起こる。付加にはGTPが関与する。このGTPは二量体に結合され、GDPに切断され、このGDPはチューブリンに結合したままで残る。GTPに対する結合部位は、b−サブユニット上にある。細胞がGTP−チューブリン二量体に富むようになると、GDP−チューブリンへの加水分解が重合速度に遅れ、α,β−チューブリン−GTPキャップがプロトフィラメントのプラス端で生成し、微小管のさらなる成長を遮断する。
【0008】
微小管阻害剤は、様々な生物学的供給源に由来して細胞骨格機能への強い影響力および強い毒性を有する化学的に非常に変化に富んだ一群の化合物を表す。細胞における微小管機能は、チューブリンが重合する能力または微小管が解重合する能力に依存する。これらのプロセスに影響を及ぼすことができる化合物、すなわち微小管阻害剤(抗チューブリン薬、抗有糸分裂薬などとも呼ばれる)は、その作用機構に従って4つの群に分けることができる。1.GTP部位に結合する化合物;2.コルヒチン部位に結合する化合物;3.微小管安定剤として影響を及ぼす化合物;および4.微小管ネットワーク破壊を行う化合物。
【0009】
タキソールの構造には、2つの芳香環およびこの薬物の活性に必要なオキセタン環を含む四環式構造が存在する。この化合物の一次作用は、微小管を安定化し、その解重合を防止することである(図2)。このように、タキソールは細胞周期の中のG2と有糸分裂との間で細胞を増殖させることを遮断するはずである。タキソールの結合は、β−チューブリンのアミノ末端の異なる場所で発生するようであるが、α−チューブリンの中間領域への結合もまた報告されている(Loeb 1997)。
【0010】
新しい種類の微小管安定化化合物が細菌Sorangium cellulosumから単離された。これらのマクロライド化合物は、それらの典型的な構造単位がエポキシド、チアゾール、およびケトンであるため、エポチロン(図3)と呼ばれた(Kowalski 1997;Schinzer 1996)。エポチロンは、2つの構造的に異なるもの、エポチロンAおよびエポチロンBとして存在する。エポチロンBは付加的なメチル基を含む(Hyfle 1996)。エポチロンAは、細菌代謝の主要産物であり、エポチロンBの収率はエポチロンAの収率の20〜30%に達する。化学構造の小さな違いにもかかわらず、多くの試験系においてエポチロンBは、約10倍より有効である。これらの化合物は、微小管の重合を安定化するという顕著な効果を示し、それらは発酵プロセスによって大規模に容易に得られる(Gerth 1994)。両方のエポチロンは、非常に狭い範囲の活性を示し、タキソールがするように細胞をG2−M相で休止させる。エポチロンの全合成は、多くの研究室から報告された(Balog 1996;Su 1997;Yang 1997;Schinzer 1997)。
【0011】
臨床用途を有するタキソールの興味深い半合成アナログは、C−4位およびC−5位でオキセタン環に、そしてC−13でエステル側鎖に連結されたタキサン環を含む化合物であるドセタキセル(タキソテール;図2)である。
【0012】
その幅広い臨床的有用性にもかかわらず、パクリタキセルおよびドセタキセルが水に不要であることに起因して、それらを処方する上での困難さが存在する。パクリタキセルの水溶性は、約12mg/リットルにすぎない。パクリタキセルおよびドセタキセルは、ほとんどの薬理学的に許容できる溶媒にも不溶であり、しかもより溶解しやすい塩を形成するための適切な化学官能性を欠く。その結果、パクリタキセルおよびドセタキセルの非経口投与のために特別の製剤が必要になる。パクリタキセルおよびドセタキセルは、経口投与された場合、ほとんど吸収されない(1%未満)。患者への投与について規制当局の許可を受けたパクリタキセルの経口製剤は得られていない。
【0013】
パクリタキセルは、現在、1mLあたり527mgのポリオキシエチル化ヒマシ油(Cremophor(登録商標)EL)と49.7%(v/v)無水エチルアルコール(米国薬局方)からなるビヒクル中に1mLあたり6mgパクリタキセルを含有する高濃度の非水系液剤である、タキソール(登録商標)として処方されている(Bristol−Myers Squibb Co.、米国、ニュージャージー州から入手可能)。Cremophor ELはその液剤の物理的安定性を改善し、エチルアルコールはパクリタキセルを可溶化する。この液剤は冷蔵して保存され、使用直前に5%デキストロースまたは0.9%生理食塩水中で希釈される。パクリタキセルの静脈内注射は、一般に0.3〜1.2mg/mLの濃度範囲内で患者に投与するように調製される。パクリタキセルに加えて、投与用の希釈溶液は、10%以下のエタノール、10%以下のCremophor ELおよび80%以下の水溶液からなる。しかしながら、所定の濃度に希釈すると、沈殿を生じる可能性がある過飽和溶液を生成する可能性がある。命を脅かす可能性がある微粒子の注入を避けるために、タキソテール(登録商標)の投与の際にはインラインの0.22ミクロンフィルターが使用される。
【0014】
ドセタキセルは、現在、1040mgのポリソルベート80からなるビヒクル中に1mLあたり40mgのドセタキセルを含有する高濃度の非水系液剤であるタキソテール(登録商標)として処方されており、注射のために13%(v/v)無水エチルアルコールを含む水で希釈される(Sanofi−Aventis Pharmaceuticals Inc.、米国、ニュージャージー州から入手可能)。第一段階の希釈溶液は、使用直前に、5%デキストロースまたは0.9%生理食塩水中にさらに希釈される。ドセタキセルの静脈内注射は、一般に0.3〜0.74mg/mLの濃度範囲内で患者に投与するように調製される。しかしながら、所定の濃度に希釈すると、結晶化して沈殿する可能性がある過飽和溶液を生成する可能性がある。命を脅かす可能性がある粒子の注入を避けるために、タキソテール(登録商標)の投与の際にはインラインの0.22ミクロンフィルターが使用される。
【0015】
いくつかの有毒な副作用が、タキソテール(登録商標)製剤中のドセタキセルの投与から生じる。この副作用としては、アナフィラキシー反応、低血圧、血管性浮腫、じんましん、末梢神経障害、関節痛、粘膜炎、吐き気、嘔吐、脱毛症、アルコール中毒、呼吸窮迫症(例えば呼吸困難)、心血管異常、流感のような症状(例えば筋肉痛)、胃腸障害、血液系の合併症(例えば好中球減少)、尿生殖器への影響および皮膚発疹が挙げられる。これらの望ましくない副作用のいくつかは臨床試験において経験されており、ある場合にはその反応は致命的である。これらの反応の発生率および重篤性を低下させるために、患者は、コルチコステロイド、ジフェンヒドラミン、H2アンタゴニスト、抗ヒスタミン薬、または顆粒球コロニー刺激因子(G−CSF)で予め投薬治療され、輸液の継続時間は長時間に設定されてきた。かかる予めの投薬治療は、重篤な超過敏反応の発生率を5%未満にまで低下させたが、軽度の反応は未だ約30%の患者において報告されている。タキソテール(登録商標)での治療を受けたすべての患者は、体液貯留の発生率および重篤性ならびに超過敏反応の重篤性を低下させるために、経口によるコルチコステロイドで予めの投薬治療(タキソテール(登録商標)投与の1日前に始めて3日間、1日あたりデキサメタゾン16mg)を受ける必要がある。
【0016】
現在のタキサン組成物よりも安全で耐性が高いタキサン組成物を生成するために、様々な戦略が進められている。Cremophorおよびポリソルベート80の使用を回避するパクリタキセルおよびドセタキセルの代替的な製剤が提案されている。
【0017】
パクリタキセル、ドセタキセルおよび他の活性なタキサンに対するリン脂質ベースのリポソーム製剤が開発されており(Sharmaら 1993;SharmaおよびStraubinger 1994),これらのタキサン製剤および他のタキサン製剤の物理特性が研究されている(SharmaおよびStraubinger 1994;Balasubramanian 1994;Balasubramanian 1994)。これらの製剤の主な効用は、いくつかの動物腫瘍モデルにおいて実証されているとおり、Cremophor EL賦形剤に関する毒性の排除であり、タキサン自体の毒性の低下である(Sharma 1993;A.Sharma 1995;Sharma 1996)。この観察は、パクリタキセル(Sharma 1995)に加えていくつかのタキサンについても当てはまる。ある場合には、この薬物の抗腫瘍作用は、リポソームベースの製剤よりもわずかに大きいようである(Sharma 1993)。
【0018】
特許文献2は、分散系としてタキサンを安定化させる方法を開示する。この方法は、分散系におけるタキサンの物理的安定性を改善する分子にタキサンを接触させることを含む。分散系のタキサンの物理的安定性を改善することによって、より高いタキサン含有量を実現することができる。この特許は、リポソームに対して20mol%未満の全タキサンの量でリポソーム中に存在する1種以上のタキサンを含有するリポソームを含み、そのリポソームが少なくとも30%グリセロールを有するグリセロール:水 組成物に懸濁している、安定なタキサン含有リポソーム調製物を提供する。
【0019】
特許文献3、特許文献4および特許文献5は、実質的に水不溶性の薬理学的に活性な物質(抗癌剤であるタキソールなど)の体内送達のための組成物を開示する。この薬理学的に活性な薬剤は、可溶性の形態または懸濁した粒子の形態で送達される。特に、この可溶性の形態は、タンパク質の壁のシェル内に収容された生体適合性の分散剤中の薬理学的に活性な薬剤の溶液を含んでいてもよい。あるいは、このタンパク質の壁のシェルは、タキソールの粒子を含有していてもよい。このポリマーシェルは、ジスルフィド結合の存在によって架橋された生体適合性ポリマー(アルブミンなど)である。中に実質的に水不溶性の薬理学的に活性な物質を含有するポリマーシェルは、次いで投与のために生体適合性の水性液体に懸濁される。かかるポリマーシェルを作製するためのプロセスは、非極性溶媒(クロロホルムなど)に溶解した薬物のみおよびアルブミンの水溶液を乳化させ、そのエマルジョンを約50℃で急速にエバポレーションすることによる。これらの特許によれば、このプロセスは、アルブミン分子間でジスルフィド結合を形成することによるアルブミンの架橋されたポリマータンパク質シェルを生成し、薬物は、容器としてのこのポリマーシェルの内部に存在する。さらに、これらの特許は、その発明を、タンパク質の最少の変性を伴う特異的ジスルフィド結合の形成による化学的架橋および熱変性方法により形成されるタンパク質ミクロスフェアとは区別する。加えて、ポリマーシェル内に収容される実質的に水不溶性の薬理学的に活性な物質の粒子は、先行技術の架橋されかまたは熱変性されたタンパク質ミクロスフェアとは異なる。なぜなら、そのプロセスにより生成されるポリマーシェルは、被覆された粒子の直径に比べて相対的に薄いからである。
【0020】
しかしながら、タンパク質を乳化剤として使用する水中油型エマルジョンにおいて、ある量のそのタンパク質が、そのタンパク質と油−水間の界面領域との相互作用に起因して変性するかも知れず、そして、天然のタンパク質に比べて変性したタンパク質の溶解度が低いため、その変性したタンパク質は凝集してより大きい粒径を形成する可能性があるということが先行技術において知られている(Hegg 1982)。そのタンパク質の残りは、水相に単量体として残るであろう。これは、2〜5%アルブミン溶液中のクロロホルムの均質化によって形成される水中油型マイクロエマルジョンの急速なエバポレーションによって約50℃でのエバポレーション後に濁ったタンパク質溶液が生成し、粒径分析器によって測定すると95%を超えるタンパク質が単量体または二量体のいずれかとして溶液中に存在するという事実によって実証することができる。換言すれば、タンパク質は、何らの認められる架橋を伴わずに可溶性の形態で回収することができる。さらに、ジスルフィド架橋は球状タンパク質のゲル生成における決定要因ではなく(Hegg 1982)、そして界面における分子の凝集が、エマルジョン安定性にとって重要である(Dimitrova 2001)ことが示されている。このように、特許文献3は、表面上のアルブミン分子によって囲まれた非晶質タキソールナノ粒子の形成を、タンパク質中の−SH基の架橋により形成されるタンパク質ポリマーシェル中に封入されたタキソールと呼んでいるのかもしれない。
【0021】
さらに特許文献3および特許文献5によれば、ナノ粒子形成の従来の方法とは異なり、ポリマー(例えば、ポリ乳酸)は油相に溶解されない。開示された組成物の調製において用いられる油相は、溶媒に溶解した薬理学的に活性な薬剤しか含有しない。特許文献3および特許文献5は、もっぱら薬理学的に活性な薬剤のみを油相に溶解することに注目しており、他の何かを溶解することには注目していないため、これは重要である。
【0022】
特許文献3に開示された技術を使用して、商業的に実行可能なパクリタキセル製剤が製造され、2005年にヒトへの使用についてFDAに承認された。それは、ABRAXANE(登録商標)(American Pharmaceuticals Partners Inc.、米国、イリノイ州)として上市された。この製品の説明書は、注射用懸濁剤のためのABRAXANE(登録商標)(注射用懸濁剤のためのパクリタキセルのタンパク質結合粒子)は、約130nmの平均粒径を有するパクリタキセルのアルブミン結合形態であると主張する。ABRAXANE(登録商標)は、静脈注射に先立って20mLの0.9%塩化ナトリウム注射液(米国薬局方)を用いて再構成するための白色〜黄色の、無菌の、凍結乾燥された粉末として供給される。各使い捨てのバイアルは100mgのパクリタキセルおよび約900mgのヒトアルブミンを含有する。再構成された懸濁液の1ミリリットル(mL)は5mgのパクリタキセルを含有する。ABRAXANE(登録商標)は、溶媒を含まない。
【0023】
特許文献3に開示される技術は薬物送達にとって非常に有用であるが、それは、タンパク質溶液に懸濁された実質的に水不溶性の医薬品のみの非晶質のナノ粒子を生成する。非晶質の粒子の状態において、粒子間の弱いファンデルワールス相互作用を除いて実質的に水不溶性薬剤の分子間には他の安定化する力は存在しないため、それらは不安定(例えば、オストワルド熟成)になりがちである。なぜなら、非晶質粒子の溶解は、主として、所定の媒体に対する非晶質の粒子中の化合物の溶解性によって決定されるからである。
【0024】
実際、ナノ粒子懸濁液を生成するための特許文献3に記載される方法は、ドセタキセルナノ粒子分散液を生成するために適用した場合、粒子は、オストワルド熟成に起因して調製後1時間以内に沈殿しはじめた。このように、ナノ粒子分散液を生成するための特許文献3および特許文献5に開示される方法は、水系媒体に懸濁された特定の実質的に水不溶性の医薬品(ドセタキセルナノ粒子など)の調製には有用ではなく、水溶液において実質的に水不溶性の医薬品の安定なナノ粒子分散液を製造するための新規なプロセスに対するニーズが存在する。
【0025】
特許文献6は、タンパク質で安定化されたリポソームについての組成物および方法、タンパク質で安定化されたリポソームの作製、ならびにタンパク質で安定化されたリポソームの投与を開示する。このプロセスは、溶媒エバポレーション技法を使用するリポソームの調製のための安定剤としてタンパク質を使用する水中油型エマルジョンの使用を含み、本発明で開示される固体非晶質ナノ粒子とは異なる物理特性を備えたリポソームを生成する。
【0026】
特許文献7は、水系媒体中の固体粒子の安定な分散液を調製するためのプロセスを開示する。この方法は、(a)実質的に水不溶性の物質、水混和性有機溶媒および阻害剤を含む第1の溶液を、(b)水および必要に応じて安定剤を含む水相とを混合し、これによって阻害剤および実質的に水不溶性の物質を含む固体粒子を沈殿させるステップと、必要に応じて、水混和性有機溶媒を除去するステップとを含み、この阻害剤はその記載で定義されるような非ポリマー性の疎水性有機化合物である。このプロセスは、水系媒体中の固体粒子分散液を提供し、その粒子はオストワルド熟成によって媒介される粒子成長が低下している。この出願は、水混和性有機溶媒を使用することによる、沈殿技法によるナノ粒子の調製を記載している。沈殿技法によって粒径を制御することが困難であるため、この方法についての問題は粒子のサイズを制御することである。この方法は、水非混和性の有機溶媒を使用して細かい水中油型エマルジョンを形成し、引き続いて水非混和性有機溶媒をエバポレーションしてナノ粒子を形成する本発明とは全体的に異なる。
【0027】
特許文献8は、水系媒体中の固体粒子の安定な分散液の調製を開示する。この調製法は、(a)チアゾール化合物である実質的に水不溶性の物質、水混和性有機溶媒および阻害剤を含む第1の溶液と、(b)水および必要に応じて安定剤を含む水相とを混合し、これにより阻害剤および実質的に水不溶性の物質を含む固体粒子を沈殿させるステップと、必要に応じて水混和性有機溶媒を除去するステップとを含み、この阻害剤は非ポリマー性疎水性有機化合物である。
【0028】
(ナノ粒子調製のための方法)
固体ナノ粒子を調製するための方法が、文献にいくつか開示されている。例えば、固体脂質ナノ粒子(SLN)は、固体脂質から構成されるマトリックスを備えるナノ粒子である。すなわちその脂質は、室温でも体温でも固体である(Mullerら、1995;LucksおよびMuller、1996;Mullerら、2000;MehnertおよびMader、2001)。この脂質は、その融点の約5℃上で融解し、薬物は融解した脂質に溶解または分散される。引き続いて、この融液は、高速撹拌によって熱界面活性剤溶液中に分散される。得られた粗エマルジョンは高圧ユニット中で、通常、500barおよび3回の均質化サイクルで均質化される。熱水中油型ナノエマルジョンが得られ、冷却され、脂質が再結晶し、固体脂質ナノ粒子を形成する。薬物ナノ結晶と同じく、SLNは、接着特性を有する。それらは、腸壁に付着し、薬物が吸収されるべき場所で正確にその薬物を放出する。加えて、脂質は、脂溶性薬物(ビタミンEなど)に対してのみならず薬物一般に対しても吸収促進特性を有することが知られている(PorterおよびCharman、2001;Sekら、2001;Charman、2000)。それでも脂質の構造に依存して、脂質吸収の増大には差は存在する(Sekら、2002)。基本的には、身体はこの脂質および可溶化された薬物を同時に摂取している。
【0029】
その一方で、固体マトリックスを用いた第二世代の脂質ナノ粒子、いわゆるナノ構造の脂質担体が開発されている(Mullerら、2000b)。NLC(登録商標)は、固体脂質と液体脂質(油)のブレンドから脂質マトリックスを調製することによって、あるナノ構造が粒子マトリックスに付与されていることを特徴とする。この混合物は、40℃ではまだ固体である。これらの粒子は、薬物の最大含有量に関して改良された特性を有し、薬物放出プロフィールを調節することにおいてより柔軟性があり、そして薬物放出を開始させるのに適してもいる(Mullerら、2002;RadtkeおよびMuller、2001b)。それらはまた、SLNと同様、経口薬物投与および非経口薬物投与のためにも使用することができるが、いくつかの付加的な興味深い特徴を有する。
【0030】
LDC(登録商標)ナノ粒子技術(MullerおよびOlbrich、1999b、2000b、2000c)において、「結合体」(この用語はその最も広い意味で使用される)は、(例えば、アミノ基含有分子と脂肪酸との)塩形成あるいは共有結合による連結(例えばエーテル、エステル、例えばトリブチリン)のいずれかによって調製された。脂質結合体のほとんどは約50〜100℃のどこかで融解する。結合体は融解され、熱界面活性剤溶液中に分散される。さらなる処理は、SLNおよびNLCと同様に行われる。得られたエマルジョン系は高圧均質化によって均質化され、得られたナノ分散液は冷却され、結合体が再結晶化し、LDCナノ粒子を形成する。この懸濁液をプロドラッグのナノ懸濁液であると考えることもできよう。
【0031】
特許文献9は、脂質マトリックスが安定な同質異像の状態にある主に異方性形状のコロイド状固体脂質粒子(SLP)の懸濁液の調製、およびミクロン粒子およびサブミクロンの生物活性剤粒子(PBA)の懸濁液の調製を開示する。生物活性剤粒子(PBA)の少なくとも約12ヶ月安定な懸濁液は、以下の(a)少なくとも1種の固体生物活性剤を融解させるステップと、(b)分散液媒体をステップ(a)で形成された少なくとも1種の融解した固体生物活性剤とほぼ同じ温度まで加熱するステップと、(c)分散液媒体中で別々の相を形成しない少なくとも1種の非常に流動的な水溶性または水分散性の安定剤を、再結晶の間に新しく作製される表面を安定化させるために乳化後有効な量で分散液媒体に加え、必要に応じて少なくとも1種の脂溶性または脂質分散性の安定剤を上記少なくとも1種の融解した生物活性剤に加えるステップと、(d)上記少なくとも1種の融解した生物活性剤および分散液媒体を予め混合し、引き続いてこの混合物を高圧均質化、微小流体化および/もしくは超音波処理によって均質化するステップと、(e)分散された生物活性剤の再結晶によって固体粒子が形成されるまで、この均質化された分散液を冷却するステップを有する乳化プロセスによって製造される。
【0032】
タキサンを含有する固体ナノ粒子の調製のためにこれらの技法を適用する際の問題点の1つは、ドセタキセルなどのいくつかのタキサンがこれらの技法で使用される高温で分解する傾向があることである。別の不都合は、封入された薬物の安定性および放出特性に影響を及ぼす可能性がある結晶性ナノ粒子の形成である。
【0033】
固体ナノ粒子を調製するための別の一般的な方法は、水中油型エマルジョンの溶媒エバポレーションによる。この油相は、1つ以上の医薬品物質を含有し、水相は緩衝剤または乳化剤のみを含有する。エマルジョンは2つの非混和性液体(通常は油および水)からなり、その液体のうちの一方は他方の中に小さい球状の液滴として分散している。ほとんどの食品において、例えば、液滴の直径は、通常、0.1〜100μmの間のいずれかにある(DickinsonおよびStainsby 1982、Dickinson 1992)。エマルジョンは、油相および水相の分布に従って便宜的に分類することができる。水相に分散した油液滴からなる系は水中油型エマルジョンまたはO/Wエマルジョンと呼ばれる(例えば、マヨネーズ、牛乳、クリームなど)。油相に分散した水液滴からなる系は油中水型またはW/Oエマルジョンと呼ばれる(例えばマーガリン、バターおよびスプレッド)。2つの別々の非混和性液体をエマルジョンに変換するプロセス、または,既存のエマルジョンの液滴のサイズを小さくするプロセスは、均質化として知られている。
【0034】
純粋な油および純水を一緒に均質化することによってエマルジョンを形成することはできるが、その2つの相は、水の層(高密度)の上に油の層(低密度)が存在する系にすぐに分離する。これは、液滴はその隣あう液滴と合体する傾向があり、最終的に完全な相分離に到るからである。エマルジョンは、通常、熱力学的に不安定な系である。均質化に先立って乳化剤および/または増粘剤として公知の物質を含めることによって、かなりの時間(数分、数時間、数日、数週間、数ヶ月または数年)速度論的に安定な(準安定な)エマルジョンを形成することが可能である。
【0035】
乳化剤は、均質化の中で新たに形成した液滴の表面に吸着して、液滴が凝集するのに十分一緒に近づくのを防止する保護膜を形成する界面活性な分子である。ほとんどの乳化剤は、同じ分子中に極性領域および非極性領域を有する分子である。食品産業で最も一般的に使用される乳化剤は、両親媒性タンパク質、低分子界面活性剤、およびモノグリセリド(脂肪酸のスクロースエステル、モノグリセリドのクエン酸エステル)、脂肪酸の塩などである(Krog、1990)。
【0036】
増粘剤は、エマルジョンの連続相の粘度を高めるために使用される成分であり、それらは液滴の動きを遅延させることによりエマルジョン安定性を高める。安定剤は、エマルジョンの安定性を高めるために使用することができる任意の成分であり、それゆえ、乳化剤または増粘剤のいずれであってもよい。
【0037】
「エマルジョン安定性」との用語は、エマルジョンが経時的なその特性の変化に抵抗することができる能力を記述するために広く使用される。エマルジョンは、クリーム状化、沈降、軟凝集、合体、および相反転が挙げられる種々の物理的プロセスによって不安定になる可能性がある。クリーム状化および沈降はともに重力分離の形態である。クリーム状化は、液滴が周りを取り囲む液体よりも低い密度を有することに由来する液滴の上方向の動きを表し、一方、沈降は、液滴が周りを取り囲む液体よりも高い密度を有することに由来する液滴の下方向の動きを表す。軟凝集および合体はともに液滴の凝集の一種である。軟凝集は、2つ以上の液滴が一緒になって液滴が個々の全体性を保持した凝集体を形成するときに起こり、他方、合体は、2つ以上の液滴が一緒に合体して1つのより大きい液滴を形成するプロセスである。大々的に液滴の合体が起こると、最終的にサンプルの上に分離した油相が形成された状態になり得る。これは「オイルオフ」として公知である。
【0038】
ほとんどのエマルジョンは、便宜的に、異なる物理化学的特性を有する3つの領域、つまり液滴の内部、連続相、および界面からなると考えることができる。エマルジョン中の分子は、その濃度および極性に従ってこれら3つの領域の中に分散する(Wedzicha 1988)。非極性分子は主に油相に局在する傾向があり、極性分子は水相に局在する傾向があり、親媒性分子は界面に局在する傾向がある。平衡状態においてさえ異なる領域間で分子の連続的な交換が起こり、この交換は系全体にわたる分子の物質移行に依存する速度で起こることに留意するべきである。分子は、エマルジョンの環境条件の何らかの変化(例えば、口内での温度変化または希釈)があると、1つの領域から別の領域に動くかも知れない。エマルジョン内の分子の位置および物質移行は、食品の芳香、フレーバー放出、質感、および物理化学的安定性に重大な影響を及ぼす(Dickinson 1982;Wedzicha 1991;Coupland 1996)。
【0039】
エマルジョンの多くの特性は、その動的性質を参照してのみ理解することができる。均質化によるエマルジョンの形成は、液滴の激しい崩壊およびバルク液体から界面領域への界面活性分子の迅速な動きを含む非常に動的なプロセスである。エマルジョンが形成された後でさえ、エマルジョン中の液滴は、ブラウン運動、重力、または印加された機械的な力によって絶え間なく動いており、頻繁に互いに衝突している(Melik 1988;Dukhin 1996)。液滴の絶え間ない動きおよび相互作用のために、エマルジョンの特性は種々の不安定化プロセス(温度の変化または時間の変化)に起因して経時的に変化する。
【0040】
エマルジョンの最も重要な特性は、それが含む液滴のサイズによって決定される(Dickinson 1982;1992)。その結果、エマルジョン中の液滴のサイズを制御、予測および測定することが重要である。エマルジョン中のすべての液滴が同じサイズであれば、そのエマルジョンは単分散性と呼ばれるが、存在するサイズに範囲があれば、そのエマルジョンは多分散性であると呼ばれる。単分散性エマルジョン中の液滴のサイズは、1つの数(例えば、液滴直径(d)または半径(r))によって完全に特徴付けることができる。単分散性エマルジョンは、実験による測定値の解釈が多分散性エマルジョンの場合よりもはるかに単純であるため、それらは基礎的研究のために使用されることがある。しかしながら、均質化によるエマルジョンは常に液滴サイズの分布を含み、その液滴サイズの指定は単分散性系の場合よりも複雑である。理想を言えば、エマルジョンの全粒径分布についての情報(すなわち、その系の液滴の各々のサイズ)を持ちたいところである。多くの場合、液滴の平均サイズおよび分布の幅がわかれば十分である(Hunter 1986)。
【0041】
効率よい乳化剤は、経時的に油相と水相との目に見える分離が存在しないエマルジョンを生成する。何らかのエマルジョンの破壊が起こったとしても、相分離は、長期間人の目には見えないかも知れない。乳化剤の効率を決定するより定量的な方法は、エマルジョンの粒径分布の経時変化を測定することである。効率よい乳化剤は粒径分布が経時的に変化しないエマルジョンを生成するが、他方、低質の乳化剤は合体および/または軟凝集に起因して粒径が大きくなるエマルジョンを生成する。エマルジョン安定性の動力学は、粒径が経時的に大きくなる速度を測定することにより確立することができる。
【0042】
(乳化剤としてのタンパク質)
水中油型エマルジョンにおいて、タンパク質はほとんど界面活性剤および乳化剤として使用される。o/wエマルジョンで使用される食用タンパク質のうちの1つは、乳漿タンパク質である。乳漿タンパク質としては4つのタンパク質が挙げられる:β−ラクトグロブリン、α−ラクトアルブミン、ウシ血清アルブミンおよびイムノグロブリン(Tornberg 1990)。工業的には、等電点〜5の乳漿タンパク質分離物(WPI)(Tornberg 1990)がo/wエマルジョン調製に使用されている。Hunt(1995)によれば、8%の乳漿タンパク質濃度が自己支持性ゲルを生成するために使用されてきた。後になって、自己支持性ゲルを生成するための乳漿タンパク質の限界濃度が4〜5%に低下したことが公知になった。90℃または121℃の温度での熱処理および50mMを超えるイオン強度を使用することにより、2%w/wという低い乳漿タンパク質濃度でゲルを生成することが可能である(Hunt 1995)。
【0043】
特許文献10は、油、水および不溶性タンパク質を高せん断で混合することにより安定な水中油型エマルジョンを調製する方法を開示する。不溶性タンパク質の量を変えることにより、このエマルジョンは液体、半固体または固体になり得る。好ましい不溶性タンパク質は、不溶性線維性タンパク質(コラーゲンなど)である。このエマルジョンは、親水性もしくは疎水性の薬理学的に活性な薬剤とともに投薬することができ、創傷包帯または軟膏としてまたはそれらの中において有用である。
【0044】
特許文献11は、水相、脂肪相および界面活性剤を含み、この脂肪相が少なくとも1つの化粧用油および少なくとも1つの揮発性フルオロ化合物の混和性混合物を含有し、少なくとも1つの揮発性フルオロ化合物が脂肪相の屈折率が水相の屈折率の±0.05に等しくなるような割合で存在する、透明または半透明な化粧品エマルジョンに関する発明を開示する。この発明はまた、上記エマルジョンを調製するためのプロセス、ならびにスキンケア、毛髪のコンディショニングおよび日焼け止めおよび/または人為的な日焼けにおける上記エマルジョンの使用にも関する。
【0045】
乳漿から誘導されたタンパク質は、乳化剤として広く使用されている(Phillips 1994;Dalgleish 1996)。それらは、均質化の間に油液滴の表面に吸収され、液滴が合体するのを防止する保護膜を形成する(Dickinson 1998)。乳漿タンパク質分離物(WPI)によって安定化されたエマルジョンの物理化学的特性は、水相の組成(例えば、イオン強度およびpH)ならびに製品の加工条件および保存条件(例えば、加熱、冷却、および機械的な撹拌)に関連する。エマルジョンはWPIの等電点の周辺で軟凝集しやすいが、高pHまたは低pHでは安定である(Philips 1994)。軟凝集に対する安定性は、液滴間のコロイド相互作用、すなわち、ファンデルワールス力、静電的反発力および立体的な力によって解釈することができた(Philips 1994;Dalgleish 1996)。ファンデルワールス相互作用は、距離の6乗の逆数に依存するため、かなり短距離でしか効果がない。同じ電荷を帯びた液滴間の静電的相互作用は反発的であり、その大きさおよび範囲はイオン強度が大きくなるにつれて減少する。短距離相互作用は、界面層の厚み以下のオーダーの液滴分離で重要になる(例えば、立体的な力、熱変動による力および水和力)(Israelachvili 1992)。かかる相互作用は、界面層の厚みより大きい距離では無視できるが、層が重なる場合には強く反発する力となり、液滴がより近づくのを防止する。タンパク質乳化剤についての基準は、油/水界面で素早く吸収される能力であり、表面疎水性は二次的な重要性しか有しないことが示されている(Lockhead 1999)。
【0046】
このように、溶媒エバポレーション技法を使用するナノ粒子の調製において、タンパク質は微細な水中油型エマルジョンを形成するための乳化剤として使用することができ、その後、エマルジョン中の有機溶媒をエバポレーションしてナノ粒子を形成することができる。ヒト血清アルブミンは、ヒトにおいて免疫原性ではなく、乳化剤としての所望の特性を有し、かつ腫瘍部位に対する選択的な標的特性を有するため、かかる調製物にとっては理想的なものとなり得る。りん光偏光解消法を用いた測定は、BSAの中性溶液中における、ヒト血清アルブミンの結晶構造におけるもののようなアルブミンのかなり剛直なハート型をした構造(8nm×8nm×3.2nm)を支持し(Ferrer 2001)、血清アルブミンは良好なゲル化特性を有することが示されている(Hegg 1982)。
【0047】
(乳化剤としてのポリマー)
乳化剤としてのタンパク質とは別に、いくつかの天然ポリマー、半天然ポリマー、および合成ポリマーを乳化剤として使用することができる。ポリマー乳化剤としては、天然に存在する乳化剤、例えば、寒天、カラギーナン、ファーセレラン(furcellaran)、タマリンド種子多糖体、タラガム(gum tare)、カラヤガム、ペクチン、キサンタンガム、アルギン酸ナトリウム、トラガカントガム、グアーガム、ローカストビーンガム、プルラン、ジェランガム、アラビアガムおよび種々のデンプン類が挙げられる。半合成乳化剤としては、カルボキシメチルセルロース(CMC)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、アルギン酸 プロピレングリコールエステル、化学変性デンプン(可溶性デンプンを含む)が挙げられ、合成ポリマーとしては、ポリビニルアルコール、ポリエチレングリコールおよびポリアクリル酸ナトリウムが挙げられる。これらのポリマー乳化剤は、エマルジョン組成物(エマルジョンフレーバーなど)または粉末組成物(脂肪粉末および油および粉末フレーバーなど)の製造に使用される。粉末組成物は、油、脂溶性フレーバーなどおよび水系成分をポリマー乳化剤を用いて乳化させ、次いでこのエマルジョンを噴霧乾燥などにかけることによって製造される。この場合、粉末組成物は、マイクロカプセルの形態にあることが多い。
【0048】
(オストワルド熟成)
一般に、広い範囲のサイズを有する粒子が媒体に分散される場合、媒体への粒子の溶解速度は異なるであろう。溶解速度が異なると、より大きい粒子と比べてより小さい粒子が熱力学的に不安定になり、より小さい粒子からより大きい粒子への物質の流束が生じる。この効果は、より小さい粒子が媒体に溶解し、その溶解した物質がより大きい粒子の上に堆積し、これにより粒径の増大が生じることである。粒子成長に関する1つのかかるメカニズムは、オストワルド熟成として公知である(Ostwald 1896および1897)。オストワルド熟成は、材料科学および医薬品科学におけるその重要性から精力的に研究されてきた(Baldan 2002;Voorhees 1992;Madras 2001および2002;Lifshitz 1961およびDavies 1981)。
【0049】
分散液中の粒子の成長は、保存中の分散液の不安定性をもたらすことがあり、分散液からの粒子の沈降を引き起こすことがある。薬理学的に活性な化合物の分散液の粒径が一定で留まることは特に重要である。なぜなら、粒径の変化は生物学的利用能、毒性、それゆえその化合物の効力に影響を及ぼすことが予想されるからである。さらに、分散液が静脈内投与のために必要とされる場合、分散液中での粒子の成長が起こると、その分散液はこの目的のために適さないものとなり、恐らくは不都合なまたは危険な副作用につながる。
【0050】
理論上は、分散液中のすべての粒子が同じサイズであれば、オストワルド熟成に由来する粒子の成長は、排除される。しかしながら、現実には完全に均一な粒径を実現することは不可能であり、粒径の小さい差でさえ粒子の成長を引き起し得る。
【0051】
特許文献12は、温度および注入速度を制御した条件下で水系沈殿液を有機液体中の固体の溶液に注ぎ、これによって粒径を制御することによる、固体の均一なサイズの粒子を調製するためのプロセスを記載する。特許文献13は、沈殿液が非水系である類似のプロセスを記載する。しかしながら、その粒子が沈殿媒体に対して小さいが有限の溶解度を有する場合、粒子が沈殿した後に粒径の成長が観察される。これらのプロセスを使用して特定の粒径を維持するためには、粒子が沈殿するや否や粒子その粒子を単離して粒子の成長を最少にすることが必要である。それゆえ、これらのプロセスによって調製された粒子は、分散液として液体媒体中で保存することができない。さらに、ある物質については、オストワルド熟成の速度は非常に大きいので、懸濁液から小さい粒子(特にナノ粒子)を単離することは現実的ではない。
【0052】
HiguchiおよびMisra(非特許文献1)は、疎水性化合物(ヘキサデカンなど)をエマルジョンの油相に加えることによる、水中油型エマルジョン中の油液滴の成長を阻害する方法を記載する。特許文献14は、10,000以下の分子量を有するポリマー物質を水中油型エマルジョンの分散した油相に添加してオストワルド熟成を阻害することを記載する。Welin−Bergerら(非特許文献2)は、疎水性物質を水中油型エマルジョンの油相に添加してエマルジョン中の油液滴のオストワルド熟成を阻害することを記載する。これら後者の3つの先行技術文献においては、油相に添加される物質は油相に溶解され、水系連続媒体に分散された単一の相油を与える。
【0053】
特許文献15は、分散相が疎水性溶媒に溶解された疎水性農薬である水中油型エマルジョンを安定化するためにポリマー安定剤を添加することを記載する。
【0054】
特許文献16は、オストワルド熟成を制御するためにイオン性分散剤が添加された有機溶媒中の固体農薬の分散液を開示する。
【0055】
特許文献17は、分散した油相がナノカプセル外膜を形成するように設計された物質と、有機溶媒と、必要に応じて活性成分とを含む水中油型エマルジョンを形成することによる、小嚢状のナノカプセルを調製するためのプロセスを記載する。安定なエマルジョンの形成後、溶媒は抽出され、ナノカプセルの分散液が残される。
【0056】
特許文献18は、水不溶性の物質とリン脂質との間に複合体を含む粒子がその物質およびリン脂質を水系媒体の中へと共沈殿することにより調製されるプロセスを記載する。一般に、複合体の形成を確実にするためのリン脂質と物質とのモル比は1:1である。
【0057】
特許文献19は、物質の粒子が脂質マトリックスに分散されている脂質マトリックス担体を記載する。脂質マトリックス担体の主要相は、疎水性脂質物質(リン脂質など)を含む。
【先行技術文献】
【特許文献】
【0058】
【特許文献1】米国特許第4,814,470号明細書
【特許文献2】米国特許第6,348,215号明細書
【特許文献3】米国特許第5,439,686号明細書
【特許文献4】米国特許第5,560,933号明細書
【特許文献5】米国特許第5,916,596号明細書
【特許文献6】米国特許出願公開第20040247660号明細書
【特許文献7】米国特許出願公開第20050009908号明細書
【特許文献8】米国特許出願公開第20060141043A号明細書
【特許文献9】米国特許第6,207,178号明細書
【特許文献10】米国特許第6,106,855号明細書
【特許文献11】米国特許第6,616,917号明細書
【特許文献12】米国特許第4,826,689号明細書
【特許文献13】米国特許第4,997,454号明細書
【特許文献14】米国特許第6,074,986号明細書(国際公開第95/07614号パンフレット)
【特許文献15】欧州特許第589838号明細書
【特許文献16】米国特許第4,348,385号明細書
【特許文献17】国際公開第99/04766号パンフレット
【特許文献18】米国特許第5,100,591号明細書
【特許文献19】米国特許第4,610,868号明細書
【非特許文献】
【0059】
【非特許文献1】J.Pharm.Sci.,51(1962)459
【非特許文献2】Int.Jour.of Pharmaceutics 200(2000)249−260頁
【発明の概要】
【発明が解決しようとする課題】
【0060】
オストワルド熟成を阻害することによる実質的に安定なナノ粒子が、タンパク質(血清アルブミンなど)またはポリマー(ポリビニルアルコールなど)を乳化剤として使用して、水中油型エマルジョンの溶媒エバポレーションによって形成できることは、本願に先立つ当該技術分野において認識されておらず、理解もされていない。本発明は、ヒトの疾患に対する毒性が低下した治療のための、感知できるほどのオストワルド熟成効果を伴わない、ナノ粒子として選択された実質的に水不溶性の医薬品物質の送達のための新規な薬物送達システムを開示する。
【課題を解決するための手段】
【0061】
本発明者らは、タンパク質または他のポリマーを界面活性剤として使用する水中油型エマルジョンプロセスを使用して、水系媒体中のドセタキセルの固体粒子の実質的に安定な分散液が調製できることを、驚きをもって見出した。本発明によって調製される分散液は、オストワルド熟成によって媒介される形成後の粒子の成長がほとんどないかまたはまったくない。
【0062】
本発明の第1の態様によれば、水系媒体中の固体粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて水混和性有機溶媒、およびオストワルド熟成阻害剤を含む第1の溶液と、
(b)水および乳化剤(タンパク質が好ましい)を含む水相と、
を混合するステップと、
高圧均質化条件下で水中油型エマルジョンを形成するステップと、
前記水非混和性有機溶媒を真空下で急速にエバポレーションするステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を製造し、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
ことを特徴とするプロセスが提供される。
【0063】
本発明にかかるプロセスにより、非常に小さい粒子(特にナノ粒子)の実質的に安定な分散液が粒子の成長なく高濃度で調製できるようになる。
【0064】
本発明にかかる分散液は実質的に安定である。これは、前記分散液中の前記固体粒子において、オストワルド熟成によって媒介される粒子の成長が減少したかまたは実質的にまったく起こらないことを意味する。「粒子の成長が減少した」とは、オストワルド熟成阻害剤を使用することなく調製した粒子と比べて、オストワルド熟成によって媒介される粒子の成長の速度が低下していることを意味する。「粒子の成長が実質的にまったく起こらない」との用語は、前記水系媒体中の前記粒子の平均粒径が、本発明のプロセスで水相に分散した後20℃で12〜120時間にわたって10%を超えて増加しない(より好ましくは5%以下だけ)ことを意味する。「実質的に安定な粒子またはナノ粒子」との用語は、前記水系媒体中の前記粒子の平均粒径が20℃で12〜120時間にわたって10%を超えて増加しない(より好ましくは5%以下だけ)ことを意味する。前記粒子には、好ましくは12〜120時間にわたって、より好ましくは24〜120時間にわたって、そしてより好ましくは48〜120時間にわたって粒子の成長が実質的にまったくない。
【0065】
固体粒子が非晶質形態で調製される場合、生成した粒子は一般に、水系分散液として保存する間にいずれは熱力学的により安定な結晶形態に戻ることを理解するべきである。かかる分散液が再結晶するために要する時間は物質に依存し、数時間から数日の間で変わり得る。一般に、かかる再結晶は、粒子の成長、および分散液から沈降しがちな大きい結晶性粒子の形成をもたらすであろう。本発明は前記懸濁液中の非晶質の粒子が結晶状態へと変換するのを防止するものではないことは理解するべきである。本発明にかかる粒子中にオストワルド熟成阻害剤が存在することで、本願明細書においてすでに記載したように、オストワルド熟成によって媒介される粒子の成長は有意に軽減または解消される。それゆえ、前記粒子はオストワルド熟成に対して安定であり、本願明細書において使用される「安定な」との用語はこのような内容として解釈されるべきである。
【0066】
前記分散液中の前記固体粒子は、好ましくは10μm未満、より好ましくは5μm未満、さらにより好ましくは1μm未満、特に500nm未満の平均粒径を有する。前記分散液中の前記粒子が10〜500nm、より特に50〜300nm、そしてさらにより特に50〜200nmの平均粒径を有することが特に好ましい。前記分散液中の前記粒子の平均サイズは、従来の技法を使用して、例えば強度平均粒径を測定するための動的光散乱によって測定することができる。
【0067】
一般に、本発明によって調製される分散液中の固体粒子は、狭い単峰性の粒径分布を示す。
【0068】
前記固体粒子は結晶性であっても、半結晶性であってもまたは非晶質であってもよい。一実施形態では、前記固体粒子は、実質的に非晶質形態の薬理学的に活性な物質を含む。多くの薬理学的化合物が、結晶形態または半結晶形態よりも非晶質形態でより高い生物学的利用能を示すため、これは好都合であり得る。得られる粒子の正確な形態は、前記プロセスのエバポレーションステップで使用される条件に依存する。一般に、本発明のプロセスは、前記エマルジョンの急速なエバポレーションを行い、実質的に非晶質の粒子を形成する。
【0069】
本発明は、220nm未満の平均直径サイズ、より好ましくは約20〜200nmの平均直径サイズ、最も好ましくは約50〜180nmの平均直径サイズを有する固体ナノ粒子を製造する方法を提供する。これらの固体ナノ粒子懸濁液は、0.22μmのフィルターを通すことにより濾過滅菌することができ、凍結乾燥することができる。この無菌懸濁液は、抗凍結剤(スクロース、マンニトール、トレハロースなど)を用いて、またはこれを用いずにバイアル中でケーキの形態で凍結乾燥することができる。凍結乾燥されたケーキは、ナノ粒子サイズ、安定性および薬物効力を変えることなくもとの固体ナノ粒子懸濁液に再構成することができ、このケーキは24ヶ月を超えて安定である。
【0070】
別の実施形態では、濾過滅菌された固体ナノ粒子は、抗凍結剤(スクロース、マンニトール、トレハロースなど)を使用して、バイアル中でケーキの形態で凍結乾燥することができる。凍結乾燥されたケーキは、固体ナノ粒子の粒径を変えることなくもとのリポソームに再構成することができる。これらのナノ粒子は、種々の経路、好ましくは静脈内経路、非経口経路、腫瘍内経路および経口経路によって投与される。
【0071】
図面では、同じ要素は同じ参照番号によって描かれている。図面は、手短に以下に説明される。本願明細書に組み込まれ本願明細書の一部を形成する添付の図面は、本発明の実施形態を例証し、詳細な説明と一緒になって本発明の原理を説明する役割を果たす。
【図面の簡単な説明】
【0072】
【図1】タキサンの化学構造の図である。
【図2】チューブリン−微小管の動態の図(Kingston 2001)である。
【図3】エポチロンの化学構造の図である。
【図4】クロロホルムおよびエタノールを用いた均質化後の4%アルブミンの粒径分析のグラフである。
【図5】4%アルブミンの粒径分析のグラフである。
【図6A】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6B】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6C】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6D】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6E】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6F】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7A】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7B】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7C】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7D】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7E】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8A】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8B】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8C】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8D】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8E】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8F】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8G】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8H】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9A】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9B】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9C】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9D】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9E】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9F】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9G】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9H】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図10A】阻害剤をまったく含まないドセタキセルの粒径分析のグラフである。
【図10B】阻害剤をまったく含まないドセタキセルの粒径分析のグラフである。
【図11A】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11B】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11C】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11D】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11E】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11F】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11G】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11H】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11I】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【発明を実施するための形態】
【0073】
微小管動態または安定性を妨害して細胞分裂を阻害し細胞死へと導く能力が「微小管阻害剤」として理解される。かかる作用は、いくつかの天然化合物、半合成化合物および合成化合物によって行われる。それらは、チューブリン上のそれらの結合部位によって分類される。チューブリン上の薬物結合部位には3つの一般的種類、つまりコルヒチン結合部位、タキソール部位およびビンカアルカロイド部位が存在する。ほとんどの他の薬物は、これらの薬物の少なくとも1つと競合してまたは競合しないで結合するようである。これは、それらが重なる結合モチーフを共有することを示唆する。また、3つの一般的な相互作用モード、つまりチューブリン捕捉薬物(コルヒチンなど)、別のポリマーを誘発する薬物(ビンカアルカロイドなど)、および微小管を安定化する薬物(タキソールなど)が存在する。「微小管阻害剤」との用語は、しばしば、チューブリンに結合し微小管動態を妨害するすべての化合物についての一般用語として使用され、同様に、これらの化合物に対する受容体は、「チューブリン」として一般に公知である。微小管阻害剤は、チューブリン阻害剤、抗チューブリン薬、有糸分裂阻害剤、抗微小管薬および抗有糸分裂薬とも呼ばれる。
【0074】
本願明細書において使用する場合、「μm」との用語、または「マイクロメートルまたはミクロン」との用語は、1メートルの百万分の1の測定単位を指す。
【0075】
本願明細書において使用する場合、「nm」との用語または「ナノメートル」との用語は、1メートルの10億分の1の測定単位を指す。
【0076】
本願明細書において使用する場合、「μg」との用語または「マイクログラム」との用語は、1グラムの百万分の1の測定単位を指す。
【0077】
本願明細書において使用する場合、「ng」との用語または「ナノグラム」との用語は、1グラムの10億分の1の測定単位を指す。
【0078】
本願明細書において使用する場合、「mL」との用語は1リットルの千分の1の測定単位を指す。
【0079】
本願明細書において使用する場合、「mM」との用語は、1モルの千分の1の測定単位を指す。
【0080】
本願明細書において使用する場合、「生体適合性」との用語は、ある物質が生物系に導入されたときに、その生物系を感知できるほどには変えないかまたはまったく不都合な影響を及ぼさない物質を表す。
【0081】
本願明細書において使用する場合、「実質的に水不溶性の医薬品物質または薬剤」との用語は、水にあまり溶解しないかまたは水にほとんど溶解しない生物学的に活性な化合物を意味する。かかる化合物の例は、パクリタキセル、ドセタキセル、SN−38、オレアンドリン、シクロスポリン、ジギトキシンなどである。
【0082】
「粒子の成長が低下した」との用語は、オストワルド熟成に媒介される粒子の成長の速度が、オストワルド熟成阻害剤を使用せずに調製した粒子と比べて低下していることを意味する。
【0083】
「実質的に粒子の成長がない」との用語は、本発明のプロセスにおいて水相に分散させた後、20℃で12〜120時間の間に、水系媒体中の粒子の平均粒径が10%を超えて増大しない(より好ましくは5%以内だけしか増大しない)ことを意味する。
【0084】
「実質的に安定な粒子またはナノ粒子」との用語は、20℃で12〜120時間の間に、水系媒体中の粒子の平均粒径が10%を超えて増大しない(より好ましくは5%以内だけしか増大しない)ことを意味する。粒子は、好ましくは12〜120時間の間、より好ましくは24〜120時間、より好ましくは48〜120時間の間、実質的に粒子の成長がない。
【0085】
「細胞増殖性疾患」との用語は、本願明細書においては、形態学的に周囲の組織から異なっているように見えることが多い悪性細胞および良性細胞の集団を意味することが意図されている。
【0086】
「タキサン」との用語は、本願明細書において使用する場合、微小管作用機構を有し、かつ独特のタキサン環系(図1を参照)および細胞増殖抑制作用のために必要な立体特異的側鎖を含む構造を有する種類の抗腫瘍薬または抗有糸分裂薬を指す。パクリタキセル(タキソールとしても公知)は臨床的に使用された最初のタキサンである。活性なアナログであるドセタキセルも臨床的に使用されており、10−DAB IIIから合成される(Colinらに対して1989年3月21日に発行された米国特許第4,814,470号)。SB−T−1011と呼ばれるタキサンは、同様にイチイの針葉から得られる14β−ヒドロキシ−10−DAB IIIから合成された(Ojimaら 1994)。パクリタキセルの側鎖には、アミド結合によって連結された2つの芳香環があるが(図1)、他の活性なアナログ(ドセタキセルなど)(Gueritte−Voegeleinら 1990;Gueritte−Voegeleinら 1991)が存在することは、基本的なパクリタキセル側鎖モチーフに対するある構造上の改変が許容され得ることを実証する。
【0087】
使用することができるタキサンの例としては、タキソール(パクリタキセル);タキソテール(ドセタキセル);MAC−321;TL−909;TL−310;スピカチン(spicatin);タキサン−2,13−ジオン,5β,9β,10β−トリヒドロキシ−,アセトンとの環状9,10−アセタール、アセテート;タキサン−2,13−ジオン,5β,9β,10β−トリヒドロキシ−,アセトンとの環状9,10−アセタール;タキサン−2β,5β,9β,10β−テトロール,アセトンとの環状9,10−アセタール;タキサン;セファロマンニン−7−キシロシド;7−エピ−10−デアセチルセファロマンニン;10−デアセチルセファロマンニン;セファロマンニン(cephalomannine);タキソールB;13−(2’,3’−ジヒドロキシ−3’−フェニルプロピオニル)バッカチン III;ユンナンキソール(yunnanxol);7−(4−アジドベンゾイル)バッカチン III;N−デベンゾイルタキソールA;O−アセチルバッカチン IV;7−(トリエチルシリル)バッカチン III;7,10−ジ−O−[(2,2,2−トリクロロエトキシ)カルボニル]バッカチン III;バッカチン III 13−O−アセテート;バッカチンジアセテート;バッカチン;バッカチンVII;バッカチンVI;バッカチンIV;7−エピ−バッカチンIII;バッカチンV;バッカチンI;バッカチンIII;バッカチンA;10−デアセチル−7−エピタキソール;エピタキソール;10−デアセチルタキソールC;7−キシロシル−10−デアセチルタキソール;10−デアセチルタキソール−7−キシロシド;7−エピ−10−デアセチルダキソール;10−デアセチルタキソール;および10−デアセチルタキソールBが挙げられるが、これらに限定されない。
【0088】
「ドセタキセル」との用語は、タキソテール(登録商標)の活性成分または他にタキソテール(登録商標)自体を指す。
【0089】
「オストワルド熟成」との用語は、沈殿物または媒体に分散した固体粒子の粗大化を指し、これは、溶液における相分離の最終段階にあり、この間により小さい粒子を犠牲にして沈殿物または固体粒子のより大きい粒子が成長し、小さい粒子は消失する。Ostwald(1896および1897)が認識したように、今では彼の名前を冠しているこのプロセスの駆動力は、沈殿物または固体粒子と溶質との間の表面張力に起因するより小さい粒子の溶解性の増大である。溶質が沈殿物または固体粒子と局所的な平衡にあると仮定すると、この溶解性の差は溶質濃度の勾配を誘発し、より小さい粒子からより大きい粒子への拡散する流束を導く。(粒子表面における溶質原子の緩慢な堆積が律速になる成長とは異なり)拡散律速成長とも呼ばれる。
【0090】
「阻害剤」との用語は、一般に、オストワルド熟成に起因する水系媒体に分散した固体ナノ粒子の不安定性を軽減するために実質的に水不溶性の物質に加えられる有機物質を指す。
【0091】
好ましい実施形態では、本発明は、微小管阻害剤から選択される実質的に水不溶性の医薬品物質の、オストワルド熟成に起因する粒子の成長がない固体ナノ粒子処方物、ならびにかかる処方物を調製および使用する方法を提供する。
【0092】
これらのナノ粒子処方物の利点は、実質的に水不溶性の医薬品物質がオストワルド熟成の阻害剤と共沈殿されることである。これらの組成物は、ゆっくりとした輸液によって、またはボーラス注入法によって、または他の非経口もしくは経口送達経路によってナノ粒子または懸濁液の形態で送達することができる薬理学的に活性な薬剤の非常に低い毒性形態を提供することが観察されている。これらのナノ粒子は、400nm未満、好ましくは200nm未満、そしてより好ましくは140nm未満のサイズを有し、親水性タンパク質がそのナノ粒子の表面に吸着されている。これらのナノ粒子は、異なるモルホロジーをとることができ、それらは非晶質の粒子または結晶性粒子として存在することができる。
【0093】
実質的に不溶性とは、25℃の水に対して0.5mg/ml未満、好ましくは0.1mg/ml未満、そして特には0.05mg/ml未満の溶解度を有する物質を意味する。
【0094】
粒子の成長の阻害に対する最大の影響は、その物質が25℃の水に対して0.05μg/mlを超える溶解度を有するときに観察される。好ましい実施形態では、その物質は、0.05μg/ml〜0.5mg/mlの範囲、例えば0.05μg/ml〜0.05mg/mlの範囲の溶解度を有する。
【0095】
水に対するその物質の溶解度は、従来の技法を使用して測定することができる。例えば、25℃で過剰量の物質を水に加え、その溶液を48時間平衡化させることによりその物質の飽和溶液が調製される。過剰の固体は遠心分離または濾過によって取り除かれ、水に対するその物質の濃度は適切な分析技法(HPLCなど)によって測定される。
【0096】
本発明にかかるプロセスは、広範囲の実質的に水不溶性の物質の安定な水系分散液を調製するために使用することができる。適切な物質としては、顔料、農薬、除草剤、殺菌剤、工業的な殺生物剤、化粧品、薬理学的に活性な化合物および薬理学的に不活性な物質(薬理学的に許容できる担体および希釈剤など)が挙げられるが、これらに限定されない。
【0097】
好ましい実施形態では、実質的に水不溶性の物質は、実質的に水不溶性の薬理学的に活性な物質である。多くの種類の薬理学的に活性な化合物が本発明で使用するのに適しており、その例としては、実質的に水不溶性の抗癌剤(例えばビカルタミド)、ステロイド、好ましくはグルココルチコステロイド(特に、抗炎症性グルココルチコステロイド、例えばブデソニド)、血圧降下薬(例えばフェロジピンまたはプラゾシン)、β遮断薬(例えばピンドロールまたはプロプラノロール)、脂質低下薬、抗凝固剤、抗血栓剤、抗真菌薬(例えばグリセオフルビン)、抗ウイルス薬、抗生物質、抗菌剤(例えばシプロフロキサシン)、抗精神病薬、抗うつ剤、鎮痛剤、麻酔薬、抗炎症剤(胃腸の炎症性疾患の治療のための化合物、例えば国際公開第99/55706号パンフレットに記載される化合物および他の抗炎症性化合物、例えばケトプロフェンを含む)、抗ヒスタミン薬、ホルモン(例えばテストステロン)、免疫調整剤または避妊薬が挙げられるが、これらに限定されない。これらの物質は、単一の実質的に水不溶性の物質を含んでいてもよく、または2つ以上のかかる物質の組合せを含んでいてもよい。
【0098】
本発明によって製造されるナノ粒子は約60〜190nmの直径を有するため、それらはRESによる摂取量が減少し、その結果、より長い循環時間、生物学的安定性および化学的安定性の向上、および腫瘍部位での蓄積量の増大を示す。もっとも重要なことは、このナノ粒子が薬物動態および生体内分布を変えることができるため、このナノ粒子処方物が、毒性が実質的に低下しマウスに対して抗腫瘍活性の著しい増加をもたらすことができることである。これは有毒な副作用を軽減することができ、その療法の効力を高めることができる。
【0099】
(オストワルド熟成阻害剤)
オストワルド熟成阻害剤は、水非混和性有機相に存在する実質的に水不溶性の物質よりも水に対する溶解性が低い非ポリマー性疎水性有機化合物であるが、このオストワルド熟成阻害剤はリン脂質ではない。適切なオストワルド熟成阻害剤は、25℃において0.1mg/l未満、より好ましくは0.01mg/l未満の水に対する溶解度を有する。本発明の実施形態にでは、オストワルド熟成阻害剤は、25℃において0.05μg/ml未満、例えば0.1ng/ml〜0.05μg/mlの水に対する溶解度を有する。
【0100】
本発明の実施形態では、オストワルド熟成阻害剤は、2000未満、例えば500未満、例えば400未満の分子量を有する。本発明の別の実施形態では、オストワルド熟成阻害剤は、1000未満、例えば600未満の分子量を有する。例えば、オストワルド熟成阻害剤は、200〜2000の範囲の分子量を有していてよく、好ましくは400〜1000の範囲、より好ましくは200〜600の範囲の分子量を有していてよい。
【0101】
適切なオストワルド熟成阻害剤としては、分類(i)〜(vii)から選択された阻害剤またはかかる阻害剤の2つ以上の組合せが挙げられる。
(i)脂肪酸のモノグリセリド、ジグリセリドまたは(より好ましくは)トリグリセリド。適切な脂肪酸としては、8〜12個、より好ましくは8〜10個の炭素原子を含む中鎖脂肪酸、または12個より多い炭素原子、例えば14〜20個の炭素原子、より好ましくは14〜18個の炭素原子を含む長鎖脂肪酸が挙げられる。この脂肪酸は、飽和脂肪酸であってもよく、不飽和脂肪酸であってもよく、または飽和脂肪酸と不飽和脂肪酸との混合物であってもよい。脂肪酸は任意に1つ以上のヒドロキシル基を含んでいてもよい(例えばリシノール酸)。グリセリドは、周知の技法、例えば、グリセロールを1つ以上の長鎖脂肪酸または中鎖脂肪酸とエステル化することにより、調製することができる。好ましい実施形態では、オストワルド熟成阻害剤は、グリセロールを長鎖脂肪酸、または好ましくは中鎖脂肪酸の混合物とエステル化することにより得られるトリグリセリドの混合物である。脂肪酸の混合物は、天然物から、例えば天然油(ヤシ油など)からの抽出によって得ることができる。ヤシ油から抽出された脂肪酸は、約50〜80重量%のデカン酸および20〜50重量%のオクタン酸を含有する。グリセロールをエステル化するために脂肪酸の混合物を使用すると、異なるアシル鎖長の混合物を含むグリセリドの混合物が得られる。長鎖トリグリセリドおよび中鎖トリグリセリドは市販されている。例えば、8〜12個、より好ましくは8〜10個の炭素原子を有するアシル基を含む中鎖トリグリセリド(MCT)はグリセロールとヤシ油から抽出された脂肪酸とのエステル化により調製され、8〜12個、より好ましくは8〜10個の炭素原子を有するアシル基を含むトリグリセリドの混合物が得られる。このMCTは、Miglyol 812N(Huls、ドイツ)として市販されている。他の市販されているMCTとしては、Miglyol 810およびMiglyol 818(Huls、ドイツ)が挙げられる。さらなる適切な中鎖トリグリセリドは、トリラウリン(グリセロールトリラウレート)である。市販されている長鎖トリグリセリドとしては、グリセリルトリステアレート、グリセリルトリパルミテート、大豆油、ゴマ油、ヒマワリ油、ヒマシ油または菜種油が挙げられる。
モノグリセリドおよびジグリセリドは、グリセロールと適切な脂肪酸、または脂肪酸の混合物との部分エステル化によって得ることができる。必要に応じて、モノグリセリドおよびジグリセリドは、従来の技法を使用して、例えば反応混合物からの抽出および引き続くエステル化によって、分離し生成することができる。モノグリセリドが使用される場合、それは長鎖モノグリセリド、例えばグリセロールと18個の炭素原子を含む脂肪酸とのエステル化により形成されるモノグリセリドであることが好ましい。
(ii)C2−10ジオールの脂肪酸モノエステルまたは(好ましくは)ジエステル。好ましくは、このジオールは、飽和であってもよくまたは不飽和であってもよい脂肪族ジオール、例えば直鎖ジオールであっても分枝鎖ジオールであってもよいC2−10アルカンジオールである。より好ましくは、このジオールは、直鎖ジオールであっても分枝鎖ジオールであってもよいC2−6アルカンジオール、例えばエチレングリコールまたはプロピレングリコールである。適切な脂肪酸としては、グリセリドに関連して上に記載した中鎖脂肪酸および長鎖脂肪酸が挙げられる。好ましいエステルは、プロピレングリコールと10〜18個の炭素原子を含む1つ以上の脂肪酸とのジエステル、例えばMiglyol 840(Huls、ドイツ)である。
(iii)アルカノールまたはシクロアルカノールの脂肪酸エステル。適切なアルカノールとしては、直鎖であっても分枝鎖であってもよいC1−20アルカノール、例えばエタノール、プロパノール、イソプロパノール、n−ブタノール、sec−ブタノールまたはtert−ブタノールが挙げられる。適切なシクロアルカノールとしては、C3−6シクロアルカノール、例えばシクロヘキサノールが挙げられる。適切な脂肪酸としては、グリセリドに関連して上に記載した中鎖脂肪酸および長鎖脂肪酸が挙げられる。好ましいエステルは、C2−6アルカノールと8〜10個の炭素原子、より好ましくは12〜29個の炭素原子を含む1つ以上の脂肪酸とのエステルである。この脂肪酸は飽和であっても不飽和であってもよい。適切なエステルとしては、例えばドデカン酸ドデシルまたはオレイン酸エチルが挙げられる。
(iv)ワックス。適切なワックスとしては、長鎖脂肪酸と少なくとも12個の炭素原子を含むアルコールとのエステルが挙げられる。このアルコールは、脂肪族アルコールであってもよく、芳香族アルコールであってもよく、脂肪族基および芳香族基を含むアルコールであってもよく、2つ以上のかかるアルコールの混合物であってもよい。アルコールが脂肪族アルコールである場合、それは飽和であっても不飽和であってもよい。この脂肪族アルコールは、直鎖であってもよく、分枝鎖であってもよくまたは環状であってもよい。適切な脂肪族アルコールとしては、12個より多い炭素原子、好ましくは14個より多い炭素原子、特に18個より多い炭素原子、例えば12〜40個、より好ましくは14〜36個および特に18〜34個の炭素原子を含む脂肪族アルコールが挙げられる。適切な長鎖脂肪酸としては、グリセリドに関連して上に記載した長鎖脂肪酸、好ましくは14個より多い炭素原子、特に18個より多い炭素原子、例えば14〜40個、より好ましくは14〜36個、特に18〜34個の炭素原子を含む長鎖脂肪酸が挙げられる。このワックスは、天然ワックス(例えば蜜ろう)であってもよく、植物性素材に由来するワックスでもよく、脂肪酸と長鎖アルコールとのエステル化によって調製される合成ワックスであってもよい。他の適切なワックスとしては、石油ろう(パラフィンワックスなど)が挙げられる。
(v)長鎖脂肪族アルコール。適切なアルコールとしては、6個以上の炭素原子、より好ましくは8個以上の炭素原子、例えば12個以上の炭素原子、例えば12〜30個、例えば14〜28個の炭素原子を有するアルコールが挙げられる。長鎖脂肪族アルコールが10〜28個、特に14〜22個の炭素原子、例えば14〜22個の炭素原子を有することが特に好ましい。このアルコールは、直鎖であってもよく、分枝鎖であってもよく、飽和であってもよく不飽和であってもよい。適切な長鎖アルコールの例としては、1−ヘキサデカノール、1−オクタデカノールまたは1−ヘプタデカノールが挙げられる。または、
(vi)硬化植物油、例えば硬化ヒマシ油。
(vii)コレステロールおよびコレステロールの脂肪酸エステル。
【0102】
本発明の一実施形態では、オストワルド熟成阻害剤は、6〜22個、好ましくは10〜20個の炭素原子を含む長鎖トリグリセリドおよび長鎖脂肪族アルコールから選択される。好ましい長鎖トリグリセリドおよび長鎖脂肪族アルコールは、上に定義されている。好ましい実施形態では、オストワルド熟成阻害剤は、12〜18個の炭素原子を有するアシル基を含む長鎖トリグリセリドまたはかかるトリグリセリドの混合物、および10〜22個の炭素原子を含む脂肪族アルコール(好ましくは1−ヘキサデカノール)のエステルまたはこれらの混合物(例えばヘキサデシルヘキサデカノエート)から選択される。
【0103】
本発明の別の実施形態では、オストワルド熟成阻害剤は、コレステロールのエステルおよびコレステロールから選択される。好ましいコレステリルエステルは、パルミチン酸コレステリルまたはステアリン酸コレステリルである。
【0104】
実質的に水不溶性の物質が薬理学的に活性な化合物である場合、オストワルド熟成阻害剤は薬理学的に不活性な物質であることが好ましい。
【0105】
オストワルド熟成阻害剤は、粒子中に、懸濁液中の粒子のオストワルド熟成を防止するのに十分な量で存在する。オストワルド熟成阻害剤は、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む、本発明のプロセスにおいて形成される固体粒子中の少量成分であることが好ましい。それゆえ、オストワルド熟成阻害剤が分散液中の粒子のオストワルド熟成を防止するのにちょうど十分な量で存在し、これにより粒子中に存在するオストワルド熟成阻害剤の量を最少にすることが好ましい。
【0106】
本発明の実施形態では、オストワルド熟成阻害剤および実質的に水不溶性の物質の総重量に対するオストワルド熟成阻害剤の重量分率(すなわち、オストワルド熟成阻害剤の重量/(オストワルド熟成阻害剤の重量+実質的に水不溶性の物質の重量))が0.01〜0.99であり、好ましくは0.05〜0.95であり、特に0.2〜0.95であり、とりわけ0.3〜0.95である。好ましい実施形態では、オストワルド熟成阻害剤および実質的に水不溶性の物質の総重量に対するオストワルド熟成阻害剤の重量分率は、0.95未満であり、より好ましくは0.9以下であり、例えば0.2〜0.9、例えば0.3〜0.9、例えば約0.8である。この実質的に水不溶性の物質が薬理学的に活性な物質であり、オストワルド熟成阻害剤が比較的無毒であり(例えば0.8を超える重量分率)かつ生体内に投与されたときに望ましくない副作用を生じず、そして/または薬理学的に活性な物質の溶解速度/生物学的利用能に影響を及ぼさないかも知れない場合には、これは特に好ましい。
【0107】
さらに、本発明者らは、オストワルド熟成による粒子の成長を防止し、これにより小さい(好ましくは1000nm未満、好ましくは500nm未満)安定な粒子が調製できるようにするためには、オストワルド熟成阻害剤および実質的に水不溶性の物質に対するオストワルド熟成阻害剤の低い重量比(すなわち0.5未満)で一般に十分であることを見出した。小さく一定の粒径が望ましい場合が多く、実質的に水不溶性の物質が例えば静脈内投与に使用される薬理学的に活性な物質である場合には特にそうである。
【0108】
本発明にかかるプロセスによって調製される分散液の1つの応用は、薬理学的に活性な化合物の毒物学の研究である。本発明のプロセスによって調製される分散液は、特に物質の粒径が500nm未満である場合には、別のプロセスを使用して調製される分散液と比べて改善された生物学的利用能を示すことができる。この応用において、オストワルド熟成阻害剤の存在に関連する毒物学に対するいかなる効果も最少になるように、活性な化合物に対するオストワルド熟成阻害剤の量を最少にすることが好都合である。
【0109】
実質的に水不溶性の物質がオストワルド熟成阻害剤に対してかなりの溶解度を有する場合、実質的に水不溶性の物質に対するオストワルド熟成阻害剤の重量比は、実質的に水不溶性の物質の量がオストワルド熟成阻害剤中の実質的に水不溶性の物質の飽和溶液を形成するのに必要とされる量を超えることを確実にするたように選択されるべきである。これにより、実質的に水不溶性の物質の固体粒子が分散液中で形成されることが確実になる。オストワルド熟成阻害剤中の実質的に水不溶性の物質の溶液を含む液体液滴、あるいは固体物質および大領域の液体オストワルド熟成阻害剤を含む2相系がこのプロセスで形成されないことを確実にするために、分散液が調製される温度(例えば周囲温度)でオストワルド熟成阻害剤が液体である場合に、これは重要である。
【0110】
理論に拘束されることは望まないが、本発明者らは、粒子中で物質とオストワルド熟成阻害剤との間で相分離が起こる系は、固体粒子が実質的に単一相系を形成する系よりもオストワルド熟成を起こしやすいと考える。従って、好ましい実施形態では、オストワルド熟成阻害剤は、実質的に水不溶性の物質およびオストワルド熟成阻害剤の実質的に単一相の混合物を含む分散液中の固体粒子を形成するのに十分その物質と混和性である。本発明によって形成される粒子の組成は、従来の技法、例えばオストワルド熟成阻害剤に対する実質的に水不溶性の物質の(熱力学的)溶解度、固体粒子中の相分離を検出するための慣用的な示差走査熱量測定(DSC)法を使用して得られる融解エントロピーおよび融点の分析を用いて分析することができる。さらに、核磁気共鳴(NMR)を用いたナノ懸濁液の研究(例えば、粒子中のいずれかの成分の線幅拡大)を使用して粒子中の相分離を検出することができる。
【0111】
一般に、オストワルド熟成阻害剤は、実質的に単一相粒子を形成するのに十分な上記物質との混和性を有するべきである。これは、オストワルド熟成阻害剤が固体粒子中に分子レベルで分散しているか、または固体粒子全体にわたって分散しているオストワルド熟成阻害剤の小さい領域中に存在することを意味する。多くの物質について、物質/オストワルド熟成阻害剤混合物は非理想的混合物であると考えられる。これは、その2つの成分の混合が非ゼロエンタルピー変化を伴うことを意味する。
【0112】
(本発明のナノ粒子の調製)
水系媒体に分散した固体ナノ粒子を形成するために、実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)は、適切な溶媒(例えば、クロロホルム、塩化メチレン、酢酸エチル、エタノール、テトラヒドロフラン、ジオキサン、アセトニトリル、アセトン、ジメチルスルホキシド、ジメチルホルムアミド、メチルピロリジノンなど、およびこれらのうちの任意の2つ以上の混合物)中に溶解される。本発明を実施するために使用することが想起されるさらなる溶媒としては、大豆油、ココナッツオイル、オリーブ油、サフラワー油、綿実油、ゴマ油、オレンジ油、リモネン油、C1−C20アルコール、C2−C20エステル、C3−C20ケトン、ポリエチレングリコール、脂肪族炭化水素、芳香族炭化水素、ハロゲン化炭化水素およびこれらの組合せが挙げられる。
【0113】
次の段階では、固体ナノ粒子を作製するために、タンパク質(例えば、ヒト血清アルブミン)を(水相に)加えて、安定なナノ液滴の形成のための安定剤または乳化剤として作用させる。タンパク質は、約0.05〜25%(w/v)の範囲、より好ましくは約0.5%〜10%(w/v)の範囲の濃度で加えられる。
【0114】
次の段階では、固体ナノ粒子を作製するために、高圧下および高せん断力下での均質化によってエマルジョンが形成される。かかる均質化は、約3,000〜30,000psi(約21〜約207MPa)の範囲の圧力で通常運転される高圧ホモジナイザー中で実施されることが好都合である。かかるプロセスは、約6,000〜25,000psi(約41〜約172MPa)の範囲の圧力で実施されることが好ましい。生成したエマルジョンは、実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤を含有する非水系溶媒の非常に小さいナノ液滴を含む。許容できる均質化方法としては、高せん断およびキャビテーションを付与するプロセス(高圧均質化、高せん断ミキサー、超音波処理、高せん断インペラなど)が挙げられる。
【0115】
最後に、固体ナノ粒子を作製するために、溶媒が減圧下でエバポレーションされ、固体形態の実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)ならびにタンパク質の固体ナノ粒子から構成されるコロイド系が得られる。許容できるエバポレーション方法としては、ロータリーエバポレーター、流下膜式エバポレーター、噴霧乾燥機、凍結乾燥機などの使用が挙げられる。溶媒のエバポレーション後、液体懸濁液は乾燥され、薬理学的に活性な薬剤およびタンパク質を含有する粉末が得られ得る。生成した粉末は、任意の好都合なときに適切な水系媒体(生理食塩水、緩衝化生理食塩水、水、緩衝化水系媒体、アミノ酸の溶液、ビタミンの溶液、炭水化物の溶液など、ならびにこれらのうちの任意の2つ以上の組合せなど)の中に再懸濁することができ、哺乳動物に投与することができる懸濁液が得られる。この粉末を得るために想起される方法としては、フリーズドライ法(freeze−drying)、噴霧乾燥などが挙げられる。
【0116】
本発明の具体的な実施形態によれば、実質的に水不溶性の医薬品物質およびオストワルド成長に対するオストワルド熟成阻害剤を含有する異常に小さいサブミクロン固体粒子、すなわち直径200ナノメートル未満である粒子を形成するための方法が提供される。かかる粒子は、使用前に液体懸濁液の形態で濾過滅菌することができる。本発明の処方プロセスの最終生成物(すなわち、実質的に水不溶性の医薬品物質の粒子)を濾過滅菌することができるということは、非常に重要である。なぜなら、高濃度のタンパク質(例えば、血清アルブミン)を含有する分散液を従来の手段(高圧蒸気殺菌法など)によって滅菌することができないからである。
【0117】
実質的に水不溶性の医薬品物質の濾過滅菌できる固体ナノ粒子(すなわち、200nm未満の粒子)を得るために、実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)は最初に実質的に水非混和性の有機溶媒(例えば、水に対して約5%未満の溶解度を有する溶媒。例えば、クロロホルム)に高濃度で溶解され、これにより実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤を含有する油相が形成される。適切な溶媒はこれまでに示されている。次に、水混和性の有機溶媒(例えば、水に対して約10%を超える溶解度を有する溶媒。例えば、エタノール)が全有機相の約1%〜99%v/vの範囲、より好ましくは約5%〜25%v/vの範囲の最終濃度で油相に加えられる。この水混和性有機溶媒は、酢酸エチル、エタノール、テトラヒドロフラン、ジオキサン、アセトニトリル、アセトン、ジメチルスルホキシド、ジメチルホルムアミド、メチルピロリジノンなどの溶媒から選択することができる。あるいは、水非混和性溶媒と水混和性溶媒との混合物が最初に調製され、次いで実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤がその混合物に溶解される。有機相中の水混和性溶媒は有機相と水相との間の界面の潤滑剤として作用し、均質化の間に微細な水中油型エマルジョンの形成をもたらすと考えられる。
【0118】
次の段階では、オストワルド成長が減少した実質的に水不溶性の医薬品物質の固体ナノ粒子の形成のために、ヒト血清アルブミンまたはこれまでに記載した任意の他の適切な安定剤が水系媒体に溶解される。この成分は、安定なナノ液滴の形成のための乳化剤として作用する。必要に応じて、十分な量の第1の有機溶媒(例えばクロロホルム)が水相に溶解され、その水系媒体は飽和濃度に近づけられる。別個の測定された量の有機相が飽和水相に加えられ(この時点で、これは実質的に水不溶性の医薬品物質、第1の有機溶媒および第2の有機溶媒を含有する)、有機相の相分率が約0.5%〜15%v/v、より好ましくは1%〜8%v/vであるようにされる。次に、低せん断力での均質化によってマイクロ液滴およびナノ液滴から構成される混合物が形成される。これは、例えば、約2,000〜約15,000rpmの範囲で運転される従来の実験室用のホモジナイザーを用いて、当業者が容易に認識できるような種々の方法で実現することができる。これは、続いて高圧(すなわち、約3,000〜30,000psi(約21〜約207MPa)の範囲)で均質化される。生成した混合物は、タンパク質水溶液(例えば、ヒト血清アルブミン)、実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤(複数種であってもよい)、他の薬剤、第1の溶媒および第2の溶媒を含む。最後に、溶媒は真空下で急速にエバポレーションされ、非常に小さいナノ粒子の形態(すなわち,直径が約50nm〜200nmの範囲の粒子)のコロイド状分散系(実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤ならびにタンパク質の固体)が得られ、このように濾過滅菌することができる。粒子の好ましいサイズ範囲は、処方および操作パラメータに依存して約50nm〜170nmである。
【0119】
本発明によって調製される固体ナノ粒子は、例えば、適切な温度−時間プロフィールで凍結乾燥することにより、そこから水を除去することによって粉末形態へとさらに変換されてもよい。タンパク質(例えば、ヒト血清アルブミン)自体が抗凍結剤として作用し、マンニトール、スクロース、トレハロース、グリシンなどの従来の抗凍結剤を使用する必要なく、粉末は、水、生理食塩水または緩衝液を加えることにより容易に再構成される。必要でないが、所望される場合には従来の抗凍結剤が本発明の処方物に添加されてもよいことは、当然理解される。実質的に水不溶性の医薬品物質を含有する固体ナノ粒子によって、比較的小さい体積で高用量の薬理学的に活性な薬剤の送達が可能になる。
【0120】
本発明の実施形態によれば、実質的に水不溶性の医薬品物質を含有する固体ナノ粒子は約2ミクロンより大きい断面直径を有する。1ミクロン未満の断面直径がより好ましく、静脈内投与経路にとっては0.22ミクロン未満の断面直径が現在最も好ましい。
【0121】
本発明によって安定剤として使用することが想起されるタンパク質としては、アルブミン(35個のシステイン残基を含む)、イムノグロブリン、カゼイン、インスリン(6個のシステインを含む)、ヘモグロビン(1つのα2β2ユニットあたり6個のシステイン残基を含む)、リゾチーム(8個のシステイン残基を含む)、イムノグロブリン、α−2−マクログロブリン、フィブロネクチン、ビトロネクチン、フィブリノゲン、リパーゼなどが挙げられる。タンパク質、ペプチド、酵素、抗体およびこれらの組合せは、本発明で使用することが想起される一般的な種類の安定剤である。
【0122】
使用するために現在好ましいタンパク質はアルブミンである。ヒト血清アルブミン(HSA)は、最も豊富にある血漿タンパク質(〜640
【化1】

)であり、ヒトに対しては免疫原性ではない。タンパク質は、主に、広範な範囲の疎水性低分子リガンド(脂肪酸、ビリルビン、チロキシン、胆汁酸およびステロイドが挙げられる)に結合するその顕著な能力によって特徴付けられる。このタンパク質は、これらの化合物についての可溶化剤および輸送体としての役割を果たし、ある場合には、遊離濃度の重要な緩衝作用をもたらす。HSAはまた、内在性リガンドの結合場所と重なる2つの主要な部位で実に様々な薬物を結合する。このタンパク質は、3つの相同ドメイン(I〜III)を含む66kDのらせん状の単量体であり、その相同ドメインの各々はAサブドメインおよびBサブドメインから構成される。りん光偏光解消法を用いて中性溶液においてエリトロシン−ウシ血清アルブミン複合体ついて行った測定結果は、ナノ秒〜数ミリ秒の時間範囲ではBSAの溶液中の大きいタンパク質セグメントの独立した動きは存在しないことと整合する。これらの測定結果は、BSAの中性溶液中で、ヒト血清アルブミンの結晶構造におけるようなアルブミンのハート型をした構造(8nm×8nm×8nm×3.2nm)を有していることを支持する(Ferrer 2001)。アルブミンの別の利点は、薬物を腫瘍部位へと輸送する能力である。特異的抗体も、特異的場所へナノ粒子を標的化するために利用することができる。
【0123】
本発明の組成物の調製において、実質的に水不溶性実質的に水不溶性の医薬品物質を懸濁または溶解するために実に様々な有機媒体を用いることができる。本発明の実施のために使用することが想起される有機媒体としては、薬理学的に活性な薬剤を懸濁または溶解することはできるが、乳化剤として用いたポリマーとも、薬理学的に活性な薬剤自体とも化学反応しない任意の非水性液体が挙げられる。例としては、植物油(例えば、大豆油、オリーブ油など)、ココナッツオイル、サフラワー油、綿実油、ゴマ油、オレンジ油、リモネン油、4〜30個の炭素原子を有する脂肪族、脂環式または芳香族の炭化水素(例えば、n−ドデカン、n−デカン、n−ヘキサン、シクロヘキサン、トルエン、ベンゼンなど)、2〜30個の炭素原子を有する脂肪族または芳香族のアルコール(例えば、オクタノールなど)、2〜30個の炭素原子を有する脂肪族または芳香族のエステル(例えば、カプリル酸エチル(オクタン酸エチル)など)、2〜30個の炭素原子を有するアルキルエーテル、アリールエーテルまたは環状エーテル(例えば、ジエチルエーテル、テトラヒドロフランなど)、1〜30個の炭素原子(および必要に応じて1個より多いハロゲン置換基、例えば、CH3Cl、CH2Cl2、CH2Cl−CH2Clなど)を有するアルキルハライドまたはアリールハライド、3〜30個の炭素原子を有するケトン(例えば、アセトン、メチルエチルケトンなど)、ポリアルキレングリコール(例えば、ポリエチレングリコールなど)、あるいはこれらのうちの任意の2つ以上の組合せが挙げられる。
【0124】
本発明の実施のために使用することが想起される有機媒体の特に好ましい組合せは、典型的には約200℃以下の沸点を有し、そしてより高分子量の(あまり揮発性でない)有機媒体を伴った、例えばジクロロメタン、クロロホルム、酢酸エチル、ベンゼンなど(すなわち、薬理学的に活性な薬剤に対して高い溶解度を有し、用いる他の有機媒体に可溶である溶媒)のような揮発性液体を含む。他の有機媒体に加えられる場合、これらの揮発性添加剤は、薬理学的に活性な薬剤の有機媒体中への溶解度を駆動することを補助する。このステップは通常時間がかかるため、これは望ましい。溶解後、この揮発性成分は、(必要に応じて真空下での)エバポレーションにより除去されてよい。
【0125】
液滴サイズを小さくするために、本発明によって調製される固体ナノ粒子処方物は、均質化のあいだエマルジョンをさらに安定化するためのある量の生体適合性界面活性剤をさらに含んでいてもよい。これらの生体適合性界面活性剤は、天然のレシチン(卵レシチン、大豆レシチンなど);植物性モノガラクトシルジグリセリド(硬化物)または植物性ジガラクトシルジグリセリド(硬化物);合成レシチン(ジヘキサノイル−L−□−レシチン、ジオクタノイル−L−□−レシチン、ジデカノイル−L−□−レシチン、ジドデカノイル−L−□−レシチン、ジテトラデカノイル−L−□−レシチン、ジヘキサデカノイル−L−□−レシチン、ジオクタデカノイル−L−□−レシチン、ジオレイル−L−□−レシチン、ジリノレオイル−L−□−レシチン、□−パルミト−□−オレオイル−L−□−レシチン、L−□−グリセロホスホリルコリンなど);ポリオキシエチル化炭化水素または植物油(Cremaphor(登録商標) ELもしくはRH40、Emulphor(登録商標) EL−620PもしくはEL−719、Arlacel(登録商標)−186、Pluronic(登録商標) F−68など);ソルビタンエステル(ソルビタンモノラウレート、ソルビタンモノステアレート、ソルビタンモノパルミテート、ソルビタントリステアレート、ソルビタンモノオレエートなど);PEG脂肪酸エステル(PEG200 ジココエート、PEG300 ジステアレート、PEG400 セスキオエレート、PEG400 ジオレエートなど);エトキシ化グリセリンエステル(POE(20) グリセロールモノステアレート、POE(20) グリセロールモノオレエートなど);エトキシ化脂肪族アミン(POE(15) ココリルアミン、POE(25) ココリルアミン、POE(80) オレイルアミンなど);エトキシ化ソルビタンエステル(POE(20) ソルビタンモノラウレート、POE(20) ソルビタンモノステアレート、POE(20) ソルビタントリステアレート、POE(20) ソルビタントリオレエートなど);エトキシ化脂肪酸(POE(5) オレイン酸、POE(5) ヤシ油脂肪酸、POE(14) ヤシ油脂肪酸、POE(9) ステアリン酸、POE(40) ステアリン酸など);アルコール−脂肪酸エステル(パルミチン酸2−エチルヘキシル、オレイン酸イソブチル、アジピン酸ジトリデシルなど);エトキシ化アルコール(POE(2)−2−エチルヘキシルアルコール、POE(10) セチルアルコール、POE(4) デシルアルコール、POE(6) ラウリルアルコールなど);アルコキシ化ヒマシ油(POE(5) ヒマシ油、POE(25) ヒマシ油、POE(25) 硬化ヒマシ油など);グリセリンエステル(グリセロールモノステアレート、グリセロールトリカプリレートなど);ポリエチレングリコール(ポリエチレングリコール−200、ポリエチレングリコール−300、ポリエチレングリコール−400など);糖エステル(スクロース脂肪酸エステルなど)から選択することができる。処方物中の生体適合性界面活性剤の割合(%)は、0.002重量%〜1重量%で変わり得る。
【0126】
本発明によって調製される固体ナノ粒子処方物は、さらに、所定のキレート剤を含んでいてよい。処方物に加えられる生体適合性キレート剤は、エチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコール−ビス(□−アミノエチルエーテル)−四酢酸(EGTA)、N−(ヒドロキシエチル)−エチレンジアミン三酢酸(HEDTA)、ニトリロ三酢酸(NTA)、トリエタノールアミン、8−ヒドロキシキノリン、クエン酸、酒石酸、リン酸、グルコン酸、糖酸、チオジプロピオン酸、アセトンジカルボン酸、ジ(ヒドロキシエチル)グリシン、フェニルアラニン、トリプトファン、グリセリン、ソルビトール、ジグライムおよびこれらの薬理学的に許容できる塩から選択することができる。
【0127】
本発明によって調製されるナノ粒子処方物は、さらに、アスコルビン酸誘導体(アスコルビン酸、エリソルビン酸、アスコルビン酸ナトリウムなど);チオール誘導体(チオグリセロール、システイン、アセチルシステイン、シスチン、ジチオエリスリトール(dithioerythreitol)、ジチオトレイトール、グルタチオンなど);トコフェロール;ブチル化ヒドロキシアニソール;ブチル化ヒドロキシトルエン;亜硫酸塩(硫酸ナトリウム、亜硫酸水素ナトリウム、アセトン亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウム、ホルムアルデヒドナトリウムなど)から選択することができる抗酸化剤を含んでいてもよい。
【0128】
本発明によって調製されるナノ粒子処方物は、さらに、必要に応じてある防腐剤を含んでいてもよい。本発明の処方物に加えるための防腐剤は、フェノール、クロロブタノール、ベンジルアルコール、メチルパラベン、プロピルパラベン、塩化ベンザルコニウムおよび塩化セチルピリジニウムから選択することができる。
【0129】
上で説明したようにして調製される、タンパク質とともに実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤を含有する固体ナノ粒子は、生体適合性水性液体中の懸濁液として送達される。この液体は、水、生理食塩水、適切な緩衝液を含む溶液、栄養剤(アミノ酸、糖類、タンパク質、炭水化物、ビタミンまたは脂肪など)を含む溶液から選択することができる。
【0130】
長期保存安定性を向上させるために、固体ナノ粒子処方は、1つ以上の保護剤(スクロース、マンニトール、トレハロースなど)の存在下で凍結および凍結乾燥されてもよい。凍結乾燥された固体ナノ粒子処方物の再水和によって、懸濁液は、本質的にすべてのそれ以前に加えられた実質的に水不溶性の医薬品物質および粒径を保持する。再水和は、単に純水または滅菌水または0.9%塩化ナトリウム注射液または5%デキストロース溶液を加え、次いでその懸濁液を緩やかにかき混ぜることにより実現される。固体ナノ粒子処方中の実質的に水不溶性の医薬品物質の効力は、凍結乾燥および再構成の後も失われることはない。
【0131】
本発明の固体ナノ粒子処方物は、オストワルド熟成阻害剤の存在に起因してオストワルド熟成を起こしにくいことが示されており、先行技術で開示されている処方物よりも溶液中で安定である。本発明において、様々なオストワルド熟成阻害剤組成物、粒径、および実質的に水不溶性の医薬品物質とタンパク質との比を有する本発明の固体ナノ粒子処方効力は、種々の系(ヒト細胞株および細胞増殖活性についての動物モデルなど)に対して研究された。
【0132】
本発明の固体ナノ粒子処方物は、遊離形態で投与された実質的に水不溶性の医薬品物質ほどには有毒でないことが示された。さらに、様々な肉腫をもつマウスおよび腫瘍をもたない正常なマウスの体重に対する、固体ナノ粒子処方物および遊離形態の種々の実質的に水不溶性の医薬品物質の効果が研究されている。しかしながら、本願明細書に提供された実施例は、決して、本発明の範囲を限定することも制限することも意図されておらず、本発明の技術を実施するためにもっぱら利用しなければならない条件、パラメータ、試薬、または出発物質を提供していると解釈されるべきではない。
【実施例】
【0133】
(実施例1)
(阻害剤としてのコレステロールを有する不安定な固体ナノ粒子の調製)
96mgのコレステロールおよび100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。1.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水(Type I water)に溶解することにより、3%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって0.5〜4mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0134】
この懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8時間にわたってモニタリングした。両方のサンプルの粒子は、1〜2時間後に変化し始め、4時間後にオストワルド熟成によって沈殿し始めた(図6)。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【0135】
(実施例2)
(阻害剤としてのコレステロールおよびステアリン酸コレステリルを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、500mgのステアリン酸コレステリル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜4mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0136】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図7)。粒径は48時間後も変化せず、4日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0137】
(実施例3)
(阻害剤としてのコレステロールおよびステアリン酸コレステリルを有する安定な固体ナノ粒子の調製)
50mgのコレステロール(Northern Lipids、カナダ)、250mgのステアリン酸コレステリル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0138】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした。粒径は48時間後も変化せず、3日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0139】
(実施例4)
(阻害剤としてのコレステロールおよびヘキサデカン酸ヘキサデシルを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、500mgのヘキサデカン酸ヘキサデシル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.0mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0140】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図8)。粒径は48時間後も変化せず、5日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0141】
(実施例5)
(阻害剤としてのコレステロールおよびグリセリルトリステアレートを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、200mgのグリセリルトリステアレート(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、3.0mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0142】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって、測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図9)。粒径は48時間後も変化せず、5日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0143】
(実施例6)
(ヒト血清アルブミンに対する乳化の効果)
3.5mLのクロロホルムと0.6mLの無水エタノールとを混合することにより、有機相を調製した。2gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、4%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.7に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて6000〜10000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を20,000〜30,000psi(約138〜約207MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。次いでこのエマルジョンをロータリーエバポレーター(Buchi、スイス)に移し、急速にエバポレーションし、高圧均質化にかけたアルブミン溶液を得た。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、1〜5mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0144】
アルブミン溶液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。約5〜8nmに1つ、そして約120〜140nmに1つの2つのピークが存在することが観察された、約5〜8nmのピークは約99体積%を占め、約120〜140nmのピークは、1体積%未満を占めていた(図4)。対照として、4%ヒト血清溶液中の粒径分布を測定した。それは約5〜8nmに1つのピークのみを有していた(図5)。これらの研究は、アルブミン溶液を水中油型エマルジョン中で均質化すると、2〜3%未満のアルブミン分子が変性により凝集することになるということを示す。
【0145】
(実施例7)
(阻害剤をまったく含まない不安定な固体ナノ粒子の調製)
100mgのドセタキセル(Guiyuanchempharm、中国)を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのウシ血清アルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ウシ血清アルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを8〜12回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜3mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0146】
エバポレーション後、この溶液が他の処方物よりも濁っていることが判明した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.45μmフィルターを通して濾過し、この溶液を濾過するのに270mm超のフィルターが必要であった。この溶液を0.22μmフィルター(Nalgene、米国)を通してもう一度濾過した。これらのフィルターを各々25mLのアセトニトリルで洗浄し、このフィルター上の沈殿した薬物を回収した。懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。0.22μmフィルターで濾過した懸濁液の粒径は、240〜390nmであった(図10)。0.22μmフィルターで濾過した懸濁液およびフィルターから回収したドセタキセルについて、HPLC法によってドセタキセル濃度を測定した。ドセタキセルの約50%がフィルターから回収され、残りは懸濁液中に存在することが判明した。この懸濁液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。両方のサンプルの粒子は、1〜3時間後に変化し始め、8時間後にオストワルド熟成によって、沈殿し始めた。
【0147】
この処方物を再度調製したが、上記と同じ沈殿の問題を観察した。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【0148】
(実施例8)
(阻害剤をまったく含まない不安定な固体ナノ粒子の調製)
160mgのドセタキセル(Guiyuanchempharm、中国)を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒト血清アルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒト血清アルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このアルブミン溶液のpHを6.2〜6.5に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜24,000psi(約103〜約165MPa)の間で変え、この乳化プロセスを8〜12回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって0.5〜3mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0149】
エバポレーション後、この溶液が他の処方物よりも濁っていることが判明した。懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。濾過していない懸濁液の粒径は200〜1000nmであった(図11)。懸濁液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。両方のサンプルの粒子は、1〜3時間後に変化し始め、8時間後にオストワルド熟成によって沈殿し始めた。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【産業上の利用可能性】
【0150】
本発明は、タンパク質または他のポリマーを界面活性剤として使用する水中油型エマルジョンプロセスを使用して調製される、水系媒体中の非水溶性物質の固体粒子の実質的に安定な分散液の作製に関連するあらゆる目的のために使用することができ、本発明によって調製される分散液は、オストワルド熟成によって媒介される、形成後の粒子の成長がほとんどないかまたはまったくない。本発明は、癌治療として使用する水溶性組成物を作製するために使用することができるが、当業者は他の応用も同様に思いつくであろう。
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2006年7月24日出願の、本発明者らによる米国特許仮出願第60/832587号の利益を主張する。
【0002】
本発明は、ナノ粒子を作製するためのプロセスに関する。より具体的には、本発明は、水不溶性の物質が水に溶解し得るようにその水不溶性の物質のナノ粒子を作製するためのプロセスに関する。さらにより具体的には、本発明は、水不溶性医薬品を水系媒体で送達することができるように、その水不溶性の医薬品のナノ粒子を作製するためのプロセスに関する。
【背景技術】
【0003】
ほとんどの抗癌剤の治療効力は、腫瘍部位への十分な局所的送達を実現することに基礎を置いている。多くの癌用化学療法薬は生体外で非常に有効であるが、生体内ではそれほど有効でないことが示されている。この不一致は、薬物を腫瘍部位に治療レベルで送達することの難しさ、および回復するためにはほとんど100%の細胞殺滅が必要であることに一部は起因すると考えられている(Jain 1994;Tannock 1998)。治療用分子であるサイトカイン、抗体、およびウイルスベクターは、血管壁を横断することが困難であるため、腫瘍に作用する能力には限界があることが多い(Yuan 1998)。不十分な特異的送達は、現在の癌用化学療法薬に関して認められるしばしば低い治療指数につながる可能性がある。これは、これらの化合物の広範な拡散および非特異的作用に起因する重大な全身毒性に姿を変える。
【0004】
別の問題は、投与に適した薬理学的に許容できるビヒクル中のいくつかの強力な化学療法薬の溶解性である。化学療法で広く使用されている2種類の分子は、微小管阻害剤(タキサン誘導体など)およびトポイソメラーゼI阻害剤(カンプトテシン(campthothecin)誘導体など)である。しかしながら、これらの重要な2種類の薬物がヒトにとって非常に有毒なビヒクル中で処方されてきたことが、実際、現在公知である。本発明は、水不溶性の医薬品物質の溶解性およびビヒクル毒性の問題を克服するための新規な医薬組成物を開示するためになされた。
【0005】
(治療薬としての微小管阻害剤)
パクリタキセル(タキソール、図1)は、Waniら(1971)によってタイヘイヨウイチイの木(Taxus brevifolia)から単離された天然のジテルペン産物である。タキサン、パクリタキセルおよびドセタキセル(特許文献1)は、微小管を安定化し腫瘍細胞死に導く新規な種類の抗癌剤に属する。同定された最初の微小管安定剤であるパクリタキセル(タキソール(登録商標)、Bristol−Myers Squibb Co.、米国、ニュージャージー州)が多くの種類の癌の治療にとって大きな価値があることは明らかになっている(Rowinsky 2001、Holton 1995)。パクリタキセルの臨床的成功によって、第二世代のタキサン、ドセタキセル(タキソテール(登録商標)、Sanofi−Aventis Pharmaceuticals、米国、ニュージャージー州)が開発され、同様の作用機構をもつ他の化合物の集中的な探索が開始された。数種類の構造的に多様な微小管安定化化合物が同定されている。同定された非タキサン安定剤である、エポチロン(Bollag 1995)、Taccalonolide(Tinley 2003)およびディスコデルモライド(discodermolide)(Mooberry 2004および1999;Martello 2000)は優れた前臨床活性を有しており、抗癌剤として臨床試験で評価されている。
【0006】
微小管は、多くの細胞機能に関与するチューブリンポリマーであり(Dustin 1984)、その多くの細胞機能のうちの1つが、細胞分裂の間に新しく形成する細胞の極に移動する染色体にとって必要な紡錘体の形成である(Avila 1990)。細胞機能にとって微小管が重要であることから、微小管は生物学的な微小管の毒に対する鋭敏な標的となる。微小管の安定化または破壊という点で微小管と相互作用するすべての化合物は微小管阻害剤と呼ばれる。それらは、細胞毒性効果を有し、そして細胞を殺すこともある。微小管は細胞増殖において有糸分裂を行うために必要とされるので、微小管阻害剤は、正常細胞よりも頻繁に分裂する癌細胞を主に攻撃するであろう。それゆえ、それらの多くは非常に重要な抗腫瘍化合物である。
【0007】
チューブリンは、四次構造が2つのポリペプチドサブユニット、α−チューブリンおよびβ−チューブリンから構成されているタンパク質である。高等真核生物における各サブユニットについていくつかのイソタイプが記載されている。微小管の機能は、その重合および解重合する能力に基づいている。このプロセスは非常に動的であり、この細胞構造の急激な短縮または伸長を伴う。チューブリンはGTP結合タンパク質であり、このヌクレオチドがタンパク質に結合することは、微小管重合のために必要とされるが、他方、重合したチューブリンに結合したGTPの加水分解は、微小管の解重合のために必要とされる。正常細胞における微小管安定性は、微小管安定化を促す微小管結合タンパク質(MAP)と呼ばれるいくつかのタンパク質の存在下で調節される。微小管重合を調節する細胞機構は、Ca2+の濃度に非常に鋭敏である。ほとんどの真核細胞の休止状態に特徴的な低い細胞質Ca2+濃度は微小管重合を促し、Ca2+の局所的増加は微小管解離(disassembly)を引き起こす(Gelford 1991)。微小管は、各々がα−チューブリンおよびβ−チューブリンを有する1つの分子からなるタンパク質二量体の重合を介して形成される。二量体およびポリマーは動的平衡の状態にあり、そのネットワークは機能上の要求に柔軟かつ迅速に反応することができる。このポリマーは通常は内径および直径がそれぞれ14nmおよび28nmの微細な無分岐の円筒、いわゆる微小管を形成する(図2;Kingston 2001)。重合は、α,β−二量体を一緒に結合して短いプロトフィラメントを形成することによって開始され、続いてそのプロトフィラメントの13本が並んで配列し微小管を形成する。引き続くこの微小管の成長は有極性であり、主に、プロトフィラメントのいわゆるプラス端でさらなる二量体の付加によって起こる。付加にはGTPが関与する。このGTPは二量体に結合され、GDPに切断され、このGDPはチューブリンに結合したままで残る。GTPに対する結合部位は、b−サブユニット上にある。細胞がGTP−チューブリン二量体に富むようになると、GDP−チューブリンへの加水分解が重合速度に遅れ、α,β−チューブリン−GTPキャップがプロトフィラメントのプラス端で生成し、微小管のさらなる成長を遮断する。
【0008】
微小管阻害剤は、様々な生物学的供給源に由来して細胞骨格機能への強い影響力および強い毒性を有する化学的に非常に変化に富んだ一群の化合物を表す。細胞における微小管機能は、チューブリンが重合する能力または微小管が解重合する能力に依存する。これらのプロセスに影響を及ぼすことができる化合物、すなわち微小管阻害剤(抗チューブリン薬、抗有糸分裂薬などとも呼ばれる)は、その作用機構に従って4つの群に分けることができる。1.GTP部位に結合する化合物;2.コルヒチン部位に結合する化合物;3.微小管安定剤として影響を及ぼす化合物;および4.微小管ネットワーク破壊を行う化合物。
【0009】
タキソールの構造には、2つの芳香環およびこの薬物の活性に必要なオキセタン環を含む四環式構造が存在する。この化合物の一次作用は、微小管を安定化し、その解重合を防止することである(図2)。このように、タキソールは細胞周期の中のG2と有糸分裂との間で細胞を増殖させることを遮断するはずである。タキソールの結合は、β−チューブリンのアミノ末端の異なる場所で発生するようであるが、α−チューブリンの中間領域への結合もまた報告されている(Loeb 1997)。
【0010】
新しい種類の微小管安定化化合物が細菌Sorangium cellulosumから単離された。これらのマクロライド化合物は、それらの典型的な構造単位がエポキシド、チアゾール、およびケトンであるため、エポチロン(図3)と呼ばれた(Kowalski 1997;Schinzer 1996)。エポチロンは、2つの構造的に異なるもの、エポチロンAおよびエポチロンBとして存在する。エポチロンBは付加的なメチル基を含む(Hyfle 1996)。エポチロンAは、細菌代謝の主要産物であり、エポチロンBの収率はエポチロンAの収率の20〜30%に達する。化学構造の小さな違いにもかかわらず、多くの試験系においてエポチロンBは、約10倍より有効である。これらの化合物は、微小管の重合を安定化するという顕著な効果を示し、それらは発酵プロセスによって大規模に容易に得られる(Gerth 1994)。両方のエポチロンは、非常に狭い範囲の活性を示し、タキソールがするように細胞をG2−M相で休止させる。エポチロンの全合成は、多くの研究室から報告された(Balog 1996;Su 1997;Yang 1997;Schinzer 1997)。
【0011】
臨床用途を有するタキソールの興味深い半合成アナログは、C−4位およびC−5位でオキセタン環に、そしてC−13でエステル側鎖に連結されたタキサン環を含む化合物であるドセタキセル(タキソテール;図2)である。
【0012】
その幅広い臨床的有用性にもかかわらず、パクリタキセルおよびドセタキセルが水に不要であることに起因して、それらを処方する上での困難さが存在する。パクリタキセルの水溶性は、約12mg/リットルにすぎない。パクリタキセルおよびドセタキセルは、ほとんどの薬理学的に許容できる溶媒にも不溶であり、しかもより溶解しやすい塩を形成するための適切な化学官能性を欠く。その結果、パクリタキセルおよびドセタキセルの非経口投与のために特別の製剤が必要になる。パクリタキセルおよびドセタキセルは、経口投与された場合、ほとんど吸収されない(1%未満)。患者への投与について規制当局の許可を受けたパクリタキセルの経口製剤は得られていない。
【0013】
パクリタキセルは、現在、1mLあたり527mgのポリオキシエチル化ヒマシ油(Cremophor(登録商標)EL)と49.7%(v/v)無水エチルアルコール(米国薬局方)からなるビヒクル中に1mLあたり6mgパクリタキセルを含有する高濃度の非水系液剤である、タキソール(登録商標)として処方されている(Bristol−Myers Squibb Co.、米国、ニュージャージー州から入手可能)。Cremophor ELはその液剤の物理的安定性を改善し、エチルアルコールはパクリタキセルを可溶化する。この液剤は冷蔵して保存され、使用直前に5%デキストロースまたは0.9%生理食塩水中で希釈される。パクリタキセルの静脈内注射は、一般に0.3〜1.2mg/mLの濃度範囲内で患者に投与するように調製される。パクリタキセルに加えて、投与用の希釈溶液は、10%以下のエタノール、10%以下のCremophor ELおよび80%以下の水溶液からなる。しかしながら、所定の濃度に希釈すると、沈殿を生じる可能性がある過飽和溶液を生成する可能性がある。命を脅かす可能性がある微粒子の注入を避けるために、タキソテール(登録商標)の投与の際にはインラインの0.22ミクロンフィルターが使用される。
【0014】
ドセタキセルは、現在、1040mgのポリソルベート80からなるビヒクル中に1mLあたり40mgのドセタキセルを含有する高濃度の非水系液剤であるタキソテール(登録商標)として処方されており、注射のために13%(v/v)無水エチルアルコールを含む水で希釈される(Sanofi−Aventis Pharmaceuticals Inc.、米国、ニュージャージー州から入手可能)。第一段階の希釈溶液は、使用直前に、5%デキストロースまたは0.9%生理食塩水中にさらに希釈される。ドセタキセルの静脈内注射は、一般に0.3〜0.74mg/mLの濃度範囲内で患者に投与するように調製される。しかしながら、所定の濃度に希釈すると、結晶化して沈殿する可能性がある過飽和溶液を生成する可能性がある。命を脅かす可能性がある粒子の注入を避けるために、タキソテール(登録商標)の投与の際にはインラインの0.22ミクロンフィルターが使用される。
【0015】
いくつかの有毒な副作用が、タキソテール(登録商標)製剤中のドセタキセルの投与から生じる。この副作用としては、アナフィラキシー反応、低血圧、血管性浮腫、じんましん、末梢神経障害、関節痛、粘膜炎、吐き気、嘔吐、脱毛症、アルコール中毒、呼吸窮迫症(例えば呼吸困難)、心血管異常、流感のような症状(例えば筋肉痛)、胃腸障害、血液系の合併症(例えば好中球減少)、尿生殖器への影響および皮膚発疹が挙げられる。これらの望ましくない副作用のいくつかは臨床試験において経験されており、ある場合にはその反応は致命的である。これらの反応の発生率および重篤性を低下させるために、患者は、コルチコステロイド、ジフェンヒドラミン、H2アンタゴニスト、抗ヒスタミン薬、または顆粒球コロニー刺激因子(G−CSF)で予め投薬治療され、輸液の継続時間は長時間に設定されてきた。かかる予めの投薬治療は、重篤な超過敏反応の発生率を5%未満にまで低下させたが、軽度の反応は未だ約30%の患者において報告されている。タキソテール(登録商標)での治療を受けたすべての患者は、体液貯留の発生率および重篤性ならびに超過敏反応の重篤性を低下させるために、経口によるコルチコステロイドで予めの投薬治療(タキソテール(登録商標)投与の1日前に始めて3日間、1日あたりデキサメタゾン16mg)を受ける必要がある。
【0016】
現在のタキサン組成物よりも安全で耐性が高いタキサン組成物を生成するために、様々な戦略が進められている。Cremophorおよびポリソルベート80の使用を回避するパクリタキセルおよびドセタキセルの代替的な製剤が提案されている。
【0017】
パクリタキセル、ドセタキセルおよび他の活性なタキサンに対するリン脂質ベースのリポソーム製剤が開発されており(Sharmaら 1993;SharmaおよびStraubinger 1994),これらのタキサン製剤および他のタキサン製剤の物理特性が研究されている(SharmaおよびStraubinger 1994;Balasubramanian 1994;Balasubramanian 1994)。これらの製剤の主な効用は、いくつかの動物腫瘍モデルにおいて実証されているとおり、Cremophor EL賦形剤に関する毒性の排除であり、タキサン自体の毒性の低下である(Sharma 1993;A.Sharma 1995;Sharma 1996)。この観察は、パクリタキセル(Sharma 1995)に加えていくつかのタキサンについても当てはまる。ある場合には、この薬物の抗腫瘍作用は、リポソームベースの製剤よりもわずかに大きいようである(Sharma 1993)。
【0018】
特許文献2は、分散系としてタキサンを安定化させる方法を開示する。この方法は、分散系におけるタキサンの物理的安定性を改善する分子にタキサンを接触させることを含む。分散系のタキサンの物理的安定性を改善することによって、より高いタキサン含有量を実現することができる。この特許は、リポソームに対して20mol%未満の全タキサンの量でリポソーム中に存在する1種以上のタキサンを含有するリポソームを含み、そのリポソームが少なくとも30%グリセロールを有するグリセロール:水 組成物に懸濁している、安定なタキサン含有リポソーム調製物を提供する。
【0019】
特許文献3、特許文献4および特許文献5は、実質的に水不溶性の薬理学的に活性な物質(抗癌剤であるタキソールなど)の体内送達のための組成物を開示する。この薬理学的に活性な薬剤は、可溶性の形態または懸濁した粒子の形態で送達される。特に、この可溶性の形態は、タンパク質の壁のシェル内に収容された生体適合性の分散剤中の薬理学的に活性な薬剤の溶液を含んでいてもよい。あるいは、このタンパク質の壁のシェルは、タキソールの粒子を含有していてもよい。このポリマーシェルは、ジスルフィド結合の存在によって架橋された生体適合性ポリマー(アルブミンなど)である。中に実質的に水不溶性の薬理学的に活性な物質を含有するポリマーシェルは、次いで投与のために生体適合性の水性液体に懸濁される。かかるポリマーシェルを作製するためのプロセスは、非極性溶媒(クロロホルムなど)に溶解した薬物のみおよびアルブミンの水溶液を乳化させ、そのエマルジョンを約50℃で急速にエバポレーションすることによる。これらの特許によれば、このプロセスは、アルブミン分子間でジスルフィド結合を形成することによるアルブミンの架橋されたポリマータンパク質シェルを生成し、薬物は、容器としてのこのポリマーシェルの内部に存在する。さらに、これらの特許は、その発明を、タンパク質の最少の変性を伴う特異的ジスルフィド結合の形成による化学的架橋および熱変性方法により形成されるタンパク質ミクロスフェアとは区別する。加えて、ポリマーシェル内に収容される実質的に水不溶性の薬理学的に活性な物質の粒子は、先行技術の架橋されかまたは熱変性されたタンパク質ミクロスフェアとは異なる。なぜなら、そのプロセスにより生成されるポリマーシェルは、被覆された粒子の直径に比べて相対的に薄いからである。
【0020】
しかしながら、タンパク質を乳化剤として使用する水中油型エマルジョンにおいて、ある量のそのタンパク質が、そのタンパク質と油−水間の界面領域との相互作用に起因して変性するかも知れず、そして、天然のタンパク質に比べて変性したタンパク質の溶解度が低いため、その変性したタンパク質は凝集してより大きい粒径を形成する可能性があるということが先行技術において知られている(Hegg 1982)。そのタンパク質の残りは、水相に単量体として残るであろう。これは、2〜5%アルブミン溶液中のクロロホルムの均質化によって形成される水中油型マイクロエマルジョンの急速なエバポレーションによって約50℃でのエバポレーション後に濁ったタンパク質溶液が生成し、粒径分析器によって測定すると95%を超えるタンパク質が単量体または二量体のいずれかとして溶液中に存在するという事実によって実証することができる。換言すれば、タンパク質は、何らの認められる架橋を伴わずに可溶性の形態で回収することができる。さらに、ジスルフィド架橋は球状タンパク質のゲル生成における決定要因ではなく(Hegg 1982)、そして界面における分子の凝集が、エマルジョン安定性にとって重要である(Dimitrova 2001)ことが示されている。このように、特許文献3は、表面上のアルブミン分子によって囲まれた非晶質タキソールナノ粒子の形成を、タンパク質中の−SH基の架橋により形成されるタンパク質ポリマーシェル中に封入されたタキソールと呼んでいるのかもしれない。
【0021】
さらに特許文献3および特許文献5によれば、ナノ粒子形成の従来の方法とは異なり、ポリマー(例えば、ポリ乳酸)は油相に溶解されない。開示された組成物の調製において用いられる油相は、溶媒に溶解した薬理学的に活性な薬剤しか含有しない。特許文献3および特許文献5は、もっぱら薬理学的に活性な薬剤のみを油相に溶解することに注目しており、他の何かを溶解することには注目していないため、これは重要である。
【0022】
特許文献3に開示された技術を使用して、商業的に実行可能なパクリタキセル製剤が製造され、2005年にヒトへの使用についてFDAに承認された。それは、ABRAXANE(登録商標)(American Pharmaceuticals Partners Inc.、米国、イリノイ州)として上市された。この製品の説明書は、注射用懸濁剤のためのABRAXANE(登録商標)(注射用懸濁剤のためのパクリタキセルのタンパク質結合粒子)は、約130nmの平均粒径を有するパクリタキセルのアルブミン結合形態であると主張する。ABRAXANE(登録商標)は、静脈注射に先立って20mLの0.9%塩化ナトリウム注射液(米国薬局方)を用いて再構成するための白色〜黄色の、無菌の、凍結乾燥された粉末として供給される。各使い捨てのバイアルは100mgのパクリタキセルおよび約900mgのヒトアルブミンを含有する。再構成された懸濁液の1ミリリットル(mL)は5mgのパクリタキセルを含有する。ABRAXANE(登録商標)は、溶媒を含まない。
【0023】
特許文献3に開示される技術は薬物送達にとって非常に有用であるが、それは、タンパク質溶液に懸濁された実質的に水不溶性の医薬品のみの非晶質のナノ粒子を生成する。非晶質の粒子の状態において、粒子間の弱いファンデルワールス相互作用を除いて実質的に水不溶性薬剤の分子間には他の安定化する力は存在しないため、それらは不安定(例えば、オストワルド熟成)になりがちである。なぜなら、非晶質粒子の溶解は、主として、所定の媒体に対する非晶質の粒子中の化合物の溶解性によって決定されるからである。
【0024】
実際、ナノ粒子懸濁液を生成するための特許文献3に記載される方法は、ドセタキセルナノ粒子分散液を生成するために適用した場合、粒子は、オストワルド熟成に起因して調製後1時間以内に沈殿しはじめた。このように、ナノ粒子分散液を生成するための特許文献3および特許文献5に開示される方法は、水系媒体に懸濁された特定の実質的に水不溶性の医薬品(ドセタキセルナノ粒子など)の調製には有用ではなく、水溶液において実質的に水不溶性の医薬品の安定なナノ粒子分散液を製造するための新規なプロセスに対するニーズが存在する。
【0025】
特許文献6は、タンパク質で安定化されたリポソームについての組成物および方法、タンパク質で安定化されたリポソームの作製、ならびにタンパク質で安定化されたリポソームの投与を開示する。このプロセスは、溶媒エバポレーション技法を使用するリポソームの調製のための安定剤としてタンパク質を使用する水中油型エマルジョンの使用を含み、本発明で開示される固体非晶質ナノ粒子とは異なる物理特性を備えたリポソームを生成する。
【0026】
特許文献7は、水系媒体中の固体粒子の安定な分散液を調製するためのプロセスを開示する。この方法は、(a)実質的に水不溶性の物質、水混和性有機溶媒および阻害剤を含む第1の溶液を、(b)水および必要に応じて安定剤を含む水相とを混合し、これによって阻害剤および実質的に水不溶性の物質を含む固体粒子を沈殿させるステップと、必要に応じて、水混和性有機溶媒を除去するステップとを含み、この阻害剤はその記載で定義されるような非ポリマー性の疎水性有機化合物である。このプロセスは、水系媒体中の固体粒子分散液を提供し、その粒子はオストワルド熟成によって媒介される粒子成長が低下している。この出願は、水混和性有機溶媒を使用することによる、沈殿技法によるナノ粒子の調製を記載している。沈殿技法によって粒径を制御することが困難であるため、この方法についての問題は粒子のサイズを制御することである。この方法は、水非混和性の有機溶媒を使用して細かい水中油型エマルジョンを形成し、引き続いて水非混和性有機溶媒をエバポレーションしてナノ粒子を形成する本発明とは全体的に異なる。
【0027】
特許文献8は、水系媒体中の固体粒子の安定な分散液の調製を開示する。この調製法は、(a)チアゾール化合物である実質的に水不溶性の物質、水混和性有機溶媒および阻害剤を含む第1の溶液と、(b)水および必要に応じて安定剤を含む水相とを混合し、これにより阻害剤および実質的に水不溶性の物質を含む固体粒子を沈殿させるステップと、必要に応じて水混和性有機溶媒を除去するステップとを含み、この阻害剤は非ポリマー性疎水性有機化合物である。
【0028】
(ナノ粒子調製のための方法)
固体ナノ粒子を調製するための方法が、文献にいくつか開示されている。例えば、固体脂質ナノ粒子(SLN)は、固体脂質から構成されるマトリックスを備えるナノ粒子である。すなわちその脂質は、室温でも体温でも固体である(Mullerら、1995;LucksおよびMuller、1996;Mullerら、2000;MehnertおよびMader、2001)。この脂質は、その融点の約5℃上で融解し、薬物は融解した脂質に溶解または分散される。引き続いて、この融液は、高速撹拌によって熱界面活性剤溶液中に分散される。得られた粗エマルジョンは高圧ユニット中で、通常、500barおよび3回の均質化サイクルで均質化される。熱水中油型ナノエマルジョンが得られ、冷却され、脂質が再結晶し、固体脂質ナノ粒子を形成する。薬物ナノ結晶と同じく、SLNは、接着特性を有する。それらは、腸壁に付着し、薬物が吸収されるべき場所で正確にその薬物を放出する。加えて、脂質は、脂溶性薬物(ビタミンEなど)に対してのみならず薬物一般に対しても吸収促進特性を有することが知られている(PorterおよびCharman、2001;Sekら、2001;Charman、2000)。それでも脂質の構造に依存して、脂質吸収の増大には差は存在する(Sekら、2002)。基本的には、身体はこの脂質および可溶化された薬物を同時に摂取している。
【0029】
その一方で、固体マトリックスを用いた第二世代の脂質ナノ粒子、いわゆるナノ構造の脂質担体が開発されている(Mullerら、2000b)。NLC(登録商標)は、固体脂質と液体脂質(油)のブレンドから脂質マトリックスを調製することによって、あるナノ構造が粒子マトリックスに付与されていることを特徴とする。この混合物は、40℃ではまだ固体である。これらの粒子は、薬物の最大含有量に関して改良された特性を有し、薬物放出プロフィールを調節することにおいてより柔軟性があり、そして薬物放出を開始させるのに適してもいる(Mullerら、2002;RadtkeおよびMuller、2001b)。それらはまた、SLNと同様、経口薬物投与および非経口薬物投与のためにも使用することができるが、いくつかの付加的な興味深い特徴を有する。
【0030】
LDC(登録商標)ナノ粒子技術(MullerおよびOlbrich、1999b、2000b、2000c)において、「結合体」(この用語はその最も広い意味で使用される)は、(例えば、アミノ基含有分子と脂肪酸との)塩形成あるいは共有結合による連結(例えばエーテル、エステル、例えばトリブチリン)のいずれかによって調製された。脂質結合体のほとんどは約50〜100℃のどこかで融解する。結合体は融解され、熱界面活性剤溶液中に分散される。さらなる処理は、SLNおよびNLCと同様に行われる。得られたエマルジョン系は高圧均質化によって均質化され、得られたナノ分散液は冷却され、結合体が再結晶化し、LDCナノ粒子を形成する。この懸濁液をプロドラッグのナノ懸濁液であると考えることもできよう。
【0031】
特許文献9は、脂質マトリックスが安定な同質異像の状態にある主に異方性形状のコロイド状固体脂質粒子(SLP)の懸濁液の調製、およびミクロン粒子およびサブミクロンの生物活性剤粒子(PBA)の懸濁液の調製を開示する。生物活性剤粒子(PBA)の少なくとも約12ヶ月安定な懸濁液は、以下の(a)少なくとも1種の固体生物活性剤を融解させるステップと、(b)分散液媒体をステップ(a)で形成された少なくとも1種の融解した固体生物活性剤とほぼ同じ温度まで加熱するステップと、(c)分散液媒体中で別々の相を形成しない少なくとも1種の非常に流動的な水溶性または水分散性の安定剤を、再結晶の間に新しく作製される表面を安定化させるために乳化後有効な量で分散液媒体に加え、必要に応じて少なくとも1種の脂溶性または脂質分散性の安定剤を上記少なくとも1種の融解した生物活性剤に加えるステップと、(d)上記少なくとも1種の融解した生物活性剤および分散液媒体を予め混合し、引き続いてこの混合物を高圧均質化、微小流体化および/もしくは超音波処理によって均質化するステップと、(e)分散された生物活性剤の再結晶によって固体粒子が形成されるまで、この均質化された分散液を冷却するステップを有する乳化プロセスによって製造される。
【0032】
タキサンを含有する固体ナノ粒子の調製のためにこれらの技法を適用する際の問題点の1つは、ドセタキセルなどのいくつかのタキサンがこれらの技法で使用される高温で分解する傾向があることである。別の不都合は、封入された薬物の安定性および放出特性に影響を及ぼす可能性がある結晶性ナノ粒子の形成である。
【0033】
固体ナノ粒子を調製するための別の一般的な方法は、水中油型エマルジョンの溶媒エバポレーションによる。この油相は、1つ以上の医薬品物質を含有し、水相は緩衝剤または乳化剤のみを含有する。エマルジョンは2つの非混和性液体(通常は油および水)からなり、その液体のうちの一方は他方の中に小さい球状の液滴として分散している。ほとんどの食品において、例えば、液滴の直径は、通常、0.1〜100μmの間のいずれかにある(DickinsonおよびStainsby 1982、Dickinson 1992)。エマルジョンは、油相および水相の分布に従って便宜的に分類することができる。水相に分散した油液滴からなる系は水中油型エマルジョンまたはO/Wエマルジョンと呼ばれる(例えば、マヨネーズ、牛乳、クリームなど)。油相に分散した水液滴からなる系は油中水型またはW/Oエマルジョンと呼ばれる(例えばマーガリン、バターおよびスプレッド)。2つの別々の非混和性液体をエマルジョンに変換するプロセス、または,既存のエマルジョンの液滴のサイズを小さくするプロセスは、均質化として知られている。
【0034】
純粋な油および純水を一緒に均質化することによってエマルジョンを形成することはできるが、その2つの相は、水の層(高密度)の上に油の層(低密度)が存在する系にすぐに分離する。これは、液滴はその隣あう液滴と合体する傾向があり、最終的に完全な相分離に到るからである。エマルジョンは、通常、熱力学的に不安定な系である。均質化に先立って乳化剤および/または増粘剤として公知の物質を含めることによって、かなりの時間(数分、数時間、数日、数週間、数ヶ月または数年)速度論的に安定な(準安定な)エマルジョンを形成することが可能である。
【0035】
乳化剤は、均質化の中で新たに形成した液滴の表面に吸着して、液滴が凝集するのに十分一緒に近づくのを防止する保護膜を形成する界面活性な分子である。ほとんどの乳化剤は、同じ分子中に極性領域および非極性領域を有する分子である。食品産業で最も一般的に使用される乳化剤は、両親媒性タンパク質、低分子界面活性剤、およびモノグリセリド(脂肪酸のスクロースエステル、モノグリセリドのクエン酸エステル)、脂肪酸の塩などである(Krog、1990)。
【0036】
増粘剤は、エマルジョンの連続相の粘度を高めるために使用される成分であり、それらは液滴の動きを遅延させることによりエマルジョン安定性を高める。安定剤は、エマルジョンの安定性を高めるために使用することができる任意の成分であり、それゆえ、乳化剤または増粘剤のいずれであってもよい。
【0037】
「エマルジョン安定性」との用語は、エマルジョンが経時的なその特性の変化に抵抗することができる能力を記述するために広く使用される。エマルジョンは、クリーム状化、沈降、軟凝集、合体、および相反転が挙げられる種々の物理的プロセスによって不安定になる可能性がある。クリーム状化および沈降はともに重力分離の形態である。クリーム状化は、液滴が周りを取り囲む液体よりも低い密度を有することに由来する液滴の上方向の動きを表し、一方、沈降は、液滴が周りを取り囲む液体よりも高い密度を有することに由来する液滴の下方向の動きを表す。軟凝集および合体はともに液滴の凝集の一種である。軟凝集は、2つ以上の液滴が一緒になって液滴が個々の全体性を保持した凝集体を形成するときに起こり、他方、合体は、2つ以上の液滴が一緒に合体して1つのより大きい液滴を形成するプロセスである。大々的に液滴の合体が起こると、最終的にサンプルの上に分離した油相が形成された状態になり得る。これは「オイルオフ」として公知である。
【0038】
ほとんどのエマルジョンは、便宜的に、異なる物理化学的特性を有する3つの領域、つまり液滴の内部、連続相、および界面からなると考えることができる。エマルジョン中の分子は、その濃度および極性に従ってこれら3つの領域の中に分散する(Wedzicha 1988)。非極性分子は主に油相に局在する傾向があり、極性分子は水相に局在する傾向があり、親媒性分子は界面に局在する傾向がある。平衡状態においてさえ異なる領域間で分子の連続的な交換が起こり、この交換は系全体にわたる分子の物質移行に依存する速度で起こることに留意するべきである。分子は、エマルジョンの環境条件の何らかの変化(例えば、口内での温度変化または希釈)があると、1つの領域から別の領域に動くかも知れない。エマルジョン内の分子の位置および物質移行は、食品の芳香、フレーバー放出、質感、および物理化学的安定性に重大な影響を及ぼす(Dickinson 1982;Wedzicha 1991;Coupland 1996)。
【0039】
エマルジョンの多くの特性は、その動的性質を参照してのみ理解することができる。均質化によるエマルジョンの形成は、液滴の激しい崩壊およびバルク液体から界面領域への界面活性分子の迅速な動きを含む非常に動的なプロセスである。エマルジョンが形成された後でさえ、エマルジョン中の液滴は、ブラウン運動、重力、または印加された機械的な力によって絶え間なく動いており、頻繁に互いに衝突している(Melik 1988;Dukhin 1996)。液滴の絶え間ない動きおよび相互作用のために、エマルジョンの特性は種々の不安定化プロセス(温度の変化または時間の変化)に起因して経時的に変化する。
【0040】
エマルジョンの最も重要な特性は、それが含む液滴のサイズによって決定される(Dickinson 1982;1992)。その結果、エマルジョン中の液滴のサイズを制御、予測および測定することが重要である。エマルジョン中のすべての液滴が同じサイズであれば、そのエマルジョンは単分散性と呼ばれるが、存在するサイズに範囲があれば、そのエマルジョンは多分散性であると呼ばれる。単分散性エマルジョン中の液滴のサイズは、1つの数(例えば、液滴直径(d)または半径(r))によって完全に特徴付けることができる。単分散性エマルジョンは、実験による測定値の解釈が多分散性エマルジョンの場合よりもはるかに単純であるため、それらは基礎的研究のために使用されることがある。しかしながら、均質化によるエマルジョンは常に液滴サイズの分布を含み、その液滴サイズの指定は単分散性系の場合よりも複雑である。理想を言えば、エマルジョンの全粒径分布についての情報(すなわち、その系の液滴の各々のサイズ)を持ちたいところである。多くの場合、液滴の平均サイズおよび分布の幅がわかれば十分である(Hunter 1986)。
【0041】
効率よい乳化剤は、経時的に油相と水相との目に見える分離が存在しないエマルジョンを生成する。何らかのエマルジョンの破壊が起こったとしても、相分離は、長期間人の目には見えないかも知れない。乳化剤の効率を決定するより定量的な方法は、エマルジョンの粒径分布の経時変化を測定することである。効率よい乳化剤は粒径分布が経時的に変化しないエマルジョンを生成するが、他方、低質の乳化剤は合体および/または軟凝集に起因して粒径が大きくなるエマルジョンを生成する。エマルジョン安定性の動力学は、粒径が経時的に大きくなる速度を測定することにより確立することができる。
【0042】
(乳化剤としてのタンパク質)
水中油型エマルジョンにおいて、タンパク質はほとんど界面活性剤および乳化剤として使用される。o/wエマルジョンで使用される食用タンパク質のうちの1つは、乳漿タンパク質である。乳漿タンパク質としては4つのタンパク質が挙げられる:β−ラクトグロブリン、α−ラクトアルブミン、ウシ血清アルブミンおよびイムノグロブリン(Tornberg 1990)。工業的には、等電点〜5の乳漿タンパク質分離物(WPI)(Tornberg 1990)がo/wエマルジョン調製に使用されている。Hunt(1995)によれば、8%の乳漿タンパク質濃度が自己支持性ゲルを生成するために使用されてきた。後になって、自己支持性ゲルを生成するための乳漿タンパク質の限界濃度が4〜5%に低下したことが公知になった。90℃または121℃の温度での熱処理および50mMを超えるイオン強度を使用することにより、2%w/wという低い乳漿タンパク質濃度でゲルを生成することが可能である(Hunt 1995)。
【0043】
特許文献10は、油、水および不溶性タンパク質を高せん断で混合することにより安定な水中油型エマルジョンを調製する方法を開示する。不溶性タンパク質の量を変えることにより、このエマルジョンは液体、半固体または固体になり得る。好ましい不溶性タンパク質は、不溶性線維性タンパク質(コラーゲンなど)である。このエマルジョンは、親水性もしくは疎水性の薬理学的に活性な薬剤とともに投薬することができ、創傷包帯または軟膏としてまたはそれらの中において有用である。
【0044】
特許文献11は、水相、脂肪相および界面活性剤を含み、この脂肪相が少なくとも1つの化粧用油および少なくとも1つの揮発性フルオロ化合物の混和性混合物を含有し、少なくとも1つの揮発性フルオロ化合物が脂肪相の屈折率が水相の屈折率の±0.05に等しくなるような割合で存在する、透明または半透明な化粧品エマルジョンに関する発明を開示する。この発明はまた、上記エマルジョンを調製するためのプロセス、ならびにスキンケア、毛髪のコンディショニングおよび日焼け止めおよび/または人為的な日焼けにおける上記エマルジョンの使用にも関する。
【0045】
乳漿から誘導されたタンパク質は、乳化剤として広く使用されている(Phillips 1994;Dalgleish 1996)。それらは、均質化の間に油液滴の表面に吸収され、液滴が合体するのを防止する保護膜を形成する(Dickinson 1998)。乳漿タンパク質分離物(WPI)によって安定化されたエマルジョンの物理化学的特性は、水相の組成(例えば、イオン強度およびpH)ならびに製品の加工条件および保存条件(例えば、加熱、冷却、および機械的な撹拌)に関連する。エマルジョンはWPIの等電点の周辺で軟凝集しやすいが、高pHまたは低pHでは安定である(Philips 1994)。軟凝集に対する安定性は、液滴間のコロイド相互作用、すなわち、ファンデルワールス力、静電的反発力および立体的な力によって解釈することができた(Philips 1994;Dalgleish 1996)。ファンデルワールス相互作用は、距離の6乗の逆数に依存するため、かなり短距離でしか効果がない。同じ電荷を帯びた液滴間の静電的相互作用は反発的であり、その大きさおよび範囲はイオン強度が大きくなるにつれて減少する。短距離相互作用は、界面層の厚み以下のオーダーの液滴分離で重要になる(例えば、立体的な力、熱変動による力および水和力)(Israelachvili 1992)。かかる相互作用は、界面層の厚みより大きい距離では無視できるが、層が重なる場合には強く反発する力となり、液滴がより近づくのを防止する。タンパク質乳化剤についての基準は、油/水界面で素早く吸収される能力であり、表面疎水性は二次的な重要性しか有しないことが示されている(Lockhead 1999)。
【0046】
このように、溶媒エバポレーション技法を使用するナノ粒子の調製において、タンパク質は微細な水中油型エマルジョンを形成するための乳化剤として使用することができ、その後、エマルジョン中の有機溶媒をエバポレーションしてナノ粒子を形成することができる。ヒト血清アルブミンは、ヒトにおいて免疫原性ではなく、乳化剤としての所望の特性を有し、かつ腫瘍部位に対する選択的な標的特性を有するため、かかる調製物にとっては理想的なものとなり得る。りん光偏光解消法を用いた測定は、BSAの中性溶液中における、ヒト血清アルブミンの結晶構造におけるもののようなアルブミンのかなり剛直なハート型をした構造(8nm×8nm×3.2nm)を支持し(Ferrer 2001)、血清アルブミンは良好なゲル化特性を有することが示されている(Hegg 1982)。
【0047】
(乳化剤としてのポリマー)
乳化剤としてのタンパク質とは別に、いくつかの天然ポリマー、半天然ポリマー、および合成ポリマーを乳化剤として使用することができる。ポリマー乳化剤としては、天然に存在する乳化剤、例えば、寒天、カラギーナン、ファーセレラン(furcellaran)、タマリンド種子多糖体、タラガム(gum tare)、カラヤガム、ペクチン、キサンタンガム、アルギン酸ナトリウム、トラガカントガム、グアーガム、ローカストビーンガム、プルラン、ジェランガム、アラビアガムおよび種々のデンプン類が挙げられる。半合成乳化剤としては、カルボキシメチルセルロース(CMC)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、アルギン酸 プロピレングリコールエステル、化学変性デンプン(可溶性デンプンを含む)が挙げられ、合成ポリマーとしては、ポリビニルアルコール、ポリエチレングリコールおよびポリアクリル酸ナトリウムが挙げられる。これらのポリマー乳化剤は、エマルジョン組成物(エマルジョンフレーバーなど)または粉末組成物(脂肪粉末および油および粉末フレーバーなど)の製造に使用される。粉末組成物は、油、脂溶性フレーバーなどおよび水系成分をポリマー乳化剤を用いて乳化させ、次いでこのエマルジョンを噴霧乾燥などにかけることによって製造される。この場合、粉末組成物は、マイクロカプセルの形態にあることが多い。
【0048】
(オストワルド熟成)
一般に、広い範囲のサイズを有する粒子が媒体に分散される場合、媒体への粒子の溶解速度は異なるであろう。溶解速度が異なると、より大きい粒子と比べてより小さい粒子が熱力学的に不安定になり、より小さい粒子からより大きい粒子への物質の流束が生じる。この効果は、より小さい粒子が媒体に溶解し、その溶解した物質がより大きい粒子の上に堆積し、これにより粒径の増大が生じることである。粒子成長に関する1つのかかるメカニズムは、オストワルド熟成として公知である(Ostwald 1896および1897)。オストワルド熟成は、材料科学および医薬品科学におけるその重要性から精力的に研究されてきた(Baldan 2002;Voorhees 1992;Madras 2001および2002;Lifshitz 1961およびDavies 1981)。
【0049】
分散液中の粒子の成長は、保存中の分散液の不安定性をもたらすことがあり、分散液からの粒子の沈降を引き起こすことがある。薬理学的に活性な化合物の分散液の粒径が一定で留まることは特に重要である。なぜなら、粒径の変化は生物学的利用能、毒性、それゆえその化合物の効力に影響を及ぼすことが予想されるからである。さらに、分散液が静脈内投与のために必要とされる場合、分散液中での粒子の成長が起こると、その分散液はこの目的のために適さないものとなり、恐らくは不都合なまたは危険な副作用につながる。
【0050】
理論上は、分散液中のすべての粒子が同じサイズであれば、オストワルド熟成に由来する粒子の成長は、排除される。しかしながら、現実には完全に均一な粒径を実現することは不可能であり、粒径の小さい差でさえ粒子の成長を引き起し得る。
【0051】
特許文献12は、温度および注入速度を制御した条件下で水系沈殿液を有機液体中の固体の溶液に注ぎ、これによって粒径を制御することによる、固体の均一なサイズの粒子を調製するためのプロセスを記載する。特許文献13は、沈殿液が非水系である類似のプロセスを記載する。しかしながら、その粒子が沈殿媒体に対して小さいが有限の溶解度を有する場合、粒子が沈殿した後に粒径の成長が観察される。これらのプロセスを使用して特定の粒径を維持するためには、粒子が沈殿するや否や粒子その粒子を単離して粒子の成長を最少にすることが必要である。それゆえ、これらのプロセスによって調製された粒子は、分散液として液体媒体中で保存することができない。さらに、ある物質については、オストワルド熟成の速度は非常に大きいので、懸濁液から小さい粒子(特にナノ粒子)を単離することは現実的ではない。
【0052】
HiguchiおよびMisra(非特許文献1)は、疎水性化合物(ヘキサデカンなど)をエマルジョンの油相に加えることによる、水中油型エマルジョン中の油液滴の成長を阻害する方法を記載する。特許文献14は、10,000以下の分子量を有するポリマー物質を水中油型エマルジョンの分散した油相に添加してオストワルド熟成を阻害することを記載する。Welin−Bergerら(非特許文献2)は、疎水性物質を水中油型エマルジョンの油相に添加してエマルジョン中の油液滴のオストワルド熟成を阻害することを記載する。これら後者の3つの先行技術文献においては、油相に添加される物質は油相に溶解され、水系連続媒体に分散された単一の相油を与える。
【0053】
特許文献15は、分散相が疎水性溶媒に溶解された疎水性農薬である水中油型エマルジョンを安定化するためにポリマー安定剤を添加することを記載する。
【0054】
特許文献16は、オストワルド熟成を制御するためにイオン性分散剤が添加された有機溶媒中の固体農薬の分散液を開示する。
【0055】
特許文献17は、分散した油相がナノカプセル外膜を形成するように設計された物質と、有機溶媒と、必要に応じて活性成分とを含む水中油型エマルジョンを形成することによる、小嚢状のナノカプセルを調製するためのプロセスを記載する。安定なエマルジョンの形成後、溶媒は抽出され、ナノカプセルの分散液が残される。
【0056】
特許文献18は、水不溶性の物質とリン脂質との間に複合体を含む粒子がその物質およびリン脂質を水系媒体の中へと共沈殿することにより調製されるプロセスを記載する。一般に、複合体の形成を確実にするためのリン脂質と物質とのモル比は1:1である。
【0057】
特許文献19は、物質の粒子が脂質マトリックスに分散されている脂質マトリックス担体を記載する。脂質マトリックス担体の主要相は、疎水性脂質物質(リン脂質など)を含む。
【先行技術文献】
【特許文献】
【0058】
【特許文献1】米国特許第4,814,470号明細書
【特許文献2】米国特許第6,348,215号明細書
【特許文献3】米国特許第5,439,686号明細書
【特許文献4】米国特許第5,560,933号明細書
【特許文献5】米国特許第5,916,596号明細書
【特許文献6】米国特許出願公開第20040247660号明細書
【特許文献7】米国特許出願公開第20050009908号明細書
【特許文献8】米国特許出願公開第20060141043A号明細書
【特許文献9】米国特許第6,207,178号明細書
【特許文献10】米国特許第6,106,855号明細書
【特許文献11】米国特許第6,616,917号明細書
【特許文献12】米国特許第4,826,689号明細書
【特許文献13】米国特許第4,997,454号明細書
【特許文献14】米国特許第6,074,986号明細書(国際公開第95/07614号パンフレット)
【特許文献15】欧州特許第589838号明細書
【特許文献16】米国特許第4,348,385号明細書
【特許文献17】国際公開第99/04766号パンフレット
【特許文献18】米国特許第5,100,591号明細書
【特許文献19】米国特許第4,610,868号明細書
【非特許文献】
【0059】
【非特許文献1】J.Pharm.Sci.,51(1962)459
【非特許文献2】Int.Jour.of Pharmaceutics 200(2000)249−260頁
【発明の概要】
【発明が解決しようとする課題】
【0060】
オストワルド熟成を阻害することによる実質的に安定なナノ粒子が、タンパク質(血清アルブミンなど)またはポリマー(ポリビニルアルコールなど)を乳化剤として使用して、水中油型エマルジョンの溶媒エバポレーションによって形成できることは、本願に先立つ当該技術分野において認識されておらず、理解もされていない。本発明は、ヒトの疾患に対する毒性が低下した治療のための、感知できるほどのオストワルド熟成効果を伴わない、ナノ粒子として選択された実質的に水不溶性の医薬品物質の送達のための新規な薬物送達システムを開示する。
【課題を解決するための手段】
【0061】
本発明者らは、タンパク質または他のポリマーを界面活性剤として使用する水中油型エマルジョンプロセスを使用して、水系媒体中のドセタキセルの固体粒子の実質的に安定な分散液が調製できることを、驚きをもって見出した。本発明によって調製される分散液は、オストワルド熟成によって媒介される形成後の粒子の成長がほとんどないかまたはまったくない。
【0062】
本発明の第1の態様によれば、水系媒体中の固体粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて水混和性有機溶媒、およびオストワルド熟成阻害剤を含む第1の溶液と、
(b)水および乳化剤(タンパク質が好ましい)を含む水相と、
を混合するステップと、
高圧均質化条件下で水中油型エマルジョンを形成するステップと、
前記水非混和性有機溶媒を真空下で急速にエバポレーションするステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を製造し、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
ことを特徴とするプロセスが提供される。
【0063】
本発明にかかるプロセスにより、非常に小さい粒子(特にナノ粒子)の実質的に安定な分散液が粒子の成長なく高濃度で調製できるようになる。
【0064】
本発明にかかる分散液は実質的に安定である。これは、前記分散液中の前記固体粒子において、オストワルド熟成によって媒介される粒子の成長が減少したかまたは実質的にまったく起こらないことを意味する。「粒子の成長が減少した」とは、オストワルド熟成阻害剤を使用することなく調製した粒子と比べて、オストワルド熟成によって媒介される粒子の成長の速度が低下していることを意味する。「粒子の成長が実質的にまったく起こらない」との用語は、前記水系媒体中の前記粒子の平均粒径が、本発明のプロセスで水相に分散した後20℃で12〜120時間にわたって10%を超えて増加しない(より好ましくは5%以下だけ)ことを意味する。「実質的に安定な粒子またはナノ粒子」との用語は、前記水系媒体中の前記粒子の平均粒径が20℃で12〜120時間にわたって10%を超えて増加しない(より好ましくは5%以下だけ)ことを意味する。前記粒子には、好ましくは12〜120時間にわたって、より好ましくは24〜120時間にわたって、そしてより好ましくは48〜120時間にわたって粒子の成長が実質的にまったくない。
【0065】
固体粒子が非晶質形態で調製される場合、生成した粒子は一般に、水系分散液として保存する間にいずれは熱力学的により安定な結晶形態に戻ることを理解するべきである。かかる分散液が再結晶するために要する時間は物質に依存し、数時間から数日の間で変わり得る。一般に、かかる再結晶は、粒子の成長、および分散液から沈降しがちな大きい結晶性粒子の形成をもたらすであろう。本発明は前記懸濁液中の非晶質の粒子が結晶状態へと変換するのを防止するものではないことは理解するべきである。本発明にかかる粒子中にオストワルド熟成阻害剤が存在することで、本願明細書においてすでに記載したように、オストワルド熟成によって媒介される粒子の成長は有意に軽減または解消される。それゆえ、前記粒子はオストワルド熟成に対して安定であり、本願明細書において使用される「安定な」との用語はこのような内容として解釈されるべきである。
【0066】
前記分散液中の前記固体粒子は、好ましくは10μm未満、より好ましくは5μm未満、さらにより好ましくは1μm未満、特に500nm未満の平均粒径を有する。前記分散液中の前記粒子が10〜500nm、より特に50〜300nm、そしてさらにより特に50〜200nmの平均粒径を有することが特に好ましい。前記分散液中の前記粒子の平均サイズは、従来の技法を使用して、例えば強度平均粒径を測定するための動的光散乱によって測定することができる。
【0067】
一般に、本発明によって調製される分散液中の固体粒子は、狭い単峰性の粒径分布を示す。
【0068】
前記固体粒子は結晶性であっても、半結晶性であってもまたは非晶質であってもよい。一実施形態では、前記固体粒子は、実質的に非晶質形態の薬理学的に活性な物質を含む。多くの薬理学的化合物が、結晶形態または半結晶形態よりも非晶質形態でより高い生物学的利用能を示すため、これは好都合であり得る。得られる粒子の正確な形態は、前記プロセスのエバポレーションステップで使用される条件に依存する。一般に、本発明のプロセスは、前記エマルジョンの急速なエバポレーションを行い、実質的に非晶質の粒子を形成する。
【0069】
本発明は、220nm未満の平均直径サイズ、より好ましくは約20〜200nmの平均直径サイズ、最も好ましくは約50〜180nmの平均直径サイズを有する固体ナノ粒子を製造する方法を提供する。これらの固体ナノ粒子懸濁液は、0.22μmのフィルターを通すことにより濾過滅菌することができ、凍結乾燥することができる。この無菌懸濁液は、抗凍結剤(スクロース、マンニトール、トレハロースなど)を用いて、またはこれを用いずにバイアル中でケーキの形態で凍結乾燥することができる。凍結乾燥されたケーキは、ナノ粒子サイズ、安定性および薬物効力を変えることなくもとの固体ナノ粒子懸濁液に再構成することができ、このケーキは24ヶ月を超えて安定である。
【0070】
別の実施形態では、濾過滅菌された固体ナノ粒子は、抗凍結剤(スクロース、マンニトール、トレハロースなど)を使用して、バイアル中でケーキの形態で凍結乾燥することができる。凍結乾燥されたケーキは、固体ナノ粒子の粒径を変えることなくもとのリポソームに再構成することができる。これらのナノ粒子は、種々の経路、好ましくは静脈内経路、非経口経路、腫瘍内経路および経口経路によって投与される。
【0071】
図面では、同じ要素は同じ参照番号によって描かれている。図面は、手短に以下に説明される。本願明細書に組み込まれ本願明細書の一部を形成する添付の図面は、本発明の実施形態を例証し、詳細な説明と一緒になって本発明の原理を説明する役割を果たす。
【図面の簡単な説明】
【0072】
【図1】タキサンの化学構造の図である。
【図2】チューブリン−微小管の動態の図(Kingston 2001)である。
【図3】エポチロンの化学構造の図である。
【図4】クロロホルムおよびエタノールを用いた均質化後の4%アルブミンの粒径分析のグラフである。
【図5】4%アルブミンの粒径分析のグラフである。
【図6A】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6B】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6C】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6D】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6E】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図6F】阻害剤としてコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7A】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7B】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7C】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7D】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図7E】阻害剤としてステアリン酸コレステリルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8A】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8B】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8C】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8D】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8E】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8F】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8G】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図8H】阻害剤としてヘキサデカン酸ヘキサデシルおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9A】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9B】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9C】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9D】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9E】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9F】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9G】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図9H】阻害剤としてグリセリルトリステアレートおよびコレステロールを含有するドセタキセルの粒径分析のグラフである。
【図10A】阻害剤をまったく含まないドセタキセルの粒径分析のグラフである。
【図10B】阻害剤をまったく含まないドセタキセルの粒径分析のグラフである。
【図11A】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11B】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11C】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11D】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11E】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11F】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11G】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11H】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【図11I】有機溶媒のエバポレーションのプロセスの間で比較した粒径の図である。
【発明を実施するための形態】
【0073】
微小管動態または安定性を妨害して細胞分裂を阻害し細胞死へと導く能力が「微小管阻害剤」として理解される。かかる作用は、いくつかの天然化合物、半合成化合物および合成化合物によって行われる。それらは、チューブリン上のそれらの結合部位によって分類される。チューブリン上の薬物結合部位には3つの一般的種類、つまりコルヒチン結合部位、タキソール部位およびビンカアルカロイド部位が存在する。ほとんどの他の薬物は、これらの薬物の少なくとも1つと競合してまたは競合しないで結合するようである。これは、それらが重なる結合モチーフを共有することを示唆する。また、3つの一般的な相互作用モード、つまりチューブリン捕捉薬物(コルヒチンなど)、別のポリマーを誘発する薬物(ビンカアルカロイドなど)、および微小管を安定化する薬物(タキソールなど)が存在する。「微小管阻害剤」との用語は、しばしば、チューブリンに結合し微小管動態を妨害するすべての化合物についての一般用語として使用され、同様に、これらの化合物に対する受容体は、「チューブリン」として一般に公知である。微小管阻害剤は、チューブリン阻害剤、抗チューブリン薬、有糸分裂阻害剤、抗微小管薬および抗有糸分裂薬とも呼ばれる。
【0074】
本願明細書において使用する場合、「μm」との用語、または「マイクロメートルまたはミクロン」との用語は、1メートルの百万分の1の測定単位を指す。
【0075】
本願明細書において使用する場合、「nm」との用語または「ナノメートル」との用語は、1メートルの10億分の1の測定単位を指す。
【0076】
本願明細書において使用する場合、「μg」との用語または「マイクログラム」との用語は、1グラムの百万分の1の測定単位を指す。
【0077】
本願明細書において使用する場合、「ng」との用語または「ナノグラム」との用語は、1グラムの10億分の1の測定単位を指す。
【0078】
本願明細書において使用する場合、「mL」との用語は1リットルの千分の1の測定単位を指す。
【0079】
本願明細書において使用する場合、「mM」との用語は、1モルの千分の1の測定単位を指す。
【0080】
本願明細書において使用する場合、「生体適合性」との用語は、ある物質が生物系に導入されたときに、その生物系を感知できるほどには変えないかまたはまったく不都合な影響を及ぼさない物質を表す。
【0081】
本願明細書において使用する場合、「実質的に水不溶性の医薬品物質または薬剤」との用語は、水にあまり溶解しないかまたは水にほとんど溶解しない生物学的に活性な化合物を意味する。かかる化合物の例は、パクリタキセル、ドセタキセル、SN−38、オレアンドリン、シクロスポリン、ジギトキシンなどである。
【0082】
「粒子の成長が低下した」との用語は、オストワルド熟成に媒介される粒子の成長の速度が、オストワルド熟成阻害剤を使用せずに調製した粒子と比べて低下していることを意味する。
【0083】
「実質的に粒子の成長がない」との用語は、本発明のプロセスにおいて水相に分散させた後、20℃で12〜120時間の間に、水系媒体中の粒子の平均粒径が10%を超えて増大しない(より好ましくは5%以内だけしか増大しない)ことを意味する。
【0084】
「実質的に安定な粒子またはナノ粒子」との用語は、20℃で12〜120時間の間に、水系媒体中の粒子の平均粒径が10%を超えて増大しない(より好ましくは5%以内だけしか増大しない)ことを意味する。粒子は、好ましくは12〜120時間の間、より好ましくは24〜120時間、より好ましくは48〜120時間の間、実質的に粒子の成長がない。
【0085】
「細胞増殖性疾患」との用語は、本願明細書においては、形態学的に周囲の組織から異なっているように見えることが多い悪性細胞および良性細胞の集団を意味することが意図されている。
【0086】
「タキサン」との用語は、本願明細書において使用する場合、微小管作用機構を有し、かつ独特のタキサン環系(図1を参照)および細胞増殖抑制作用のために必要な立体特異的側鎖を含む構造を有する種類の抗腫瘍薬または抗有糸分裂薬を指す。パクリタキセル(タキソールとしても公知)は臨床的に使用された最初のタキサンである。活性なアナログであるドセタキセルも臨床的に使用されており、10−DAB IIIから合成される(Colinらに対して1989年3月21日に発行された米国特許第4,814,470号)。SB−T−1011と呼ばれるタキサンは、同様にイチイの針葉から得られる14β−ヒドロキシ−10−DAB IIIから合成された(Ojimaら 1994)。パクリタキセルの側鎖には、アミド結合によって連結された2つの芳香環があるが(図1)、他の活性なアナログ(ドセタキセルなど)(Gueritte−Voegeleinら 1990;Gueritte−Voegeleinら 1991)が存在することは、基本的なパクリタキセル側鎖モチーフに対するある構造上の改変が許容され得ることを実証する。
【0087】
使用することができるタキサンの例としては、タキソール(パクリタキセル);タキソテール(ドセタキセル);MAC−321;TL−909;TL−310;スピカチン(spicatin);タキサン−2,13−ジオン,5β,9β,10β−トリヒドロキシ−,アセトンとの環状9,10−アセタール、アセテート;タキサン−2,13−ジオン,5β,9β,10β−トリヒドロキシ−,アセトンとの環状9,10−アセタール;タキサン−2β,5β,9β,10β−テトロール,アセトンとの環状9,10−アセタール;タキサン;セファロマンニン−7−キシロシド;7−エピ−10−デアセチルセファロマンニン;10−デアセチルセファロマンニン;セファロマンニン(cephalomannine);タキソールB;13−(2’,3’−ジヒドロキシ−3’−フェニルプロピオニル)バッカチン III;ユンナンキソール(yunnanxol);7−(4−アジドベンゾイル)バッカチン III;N−デベンゾイルタキソールA;O−アセチルバッカチン IV;7−(トリエチルシリル)バッカチン III;7,10−ジ−O−[(2,2,2−トリクロロエトキシ)カルボニル]バッカチン III;バッカチン III 13−O−アセテート;バッカチンジアセテート;バッカチン;バッカチンVII;バッカチンVI;バッカチンIV;7−エピ−バッカチンIII;バッカチンV;バッカチンI;バッカチンIII;バッカチンA;10−デアセチル−7−エピタキソール;エピタキソール;10−デアセチルタキソールC;7−キシロシル−10−デアセチルタキソール;10−デアセチルタキソール−7−キシロシド;7−エピ−10−デアセチルダキソール;10−デアセチルタキソール;および10−デアセチルタキソールBが挙げられるが、これらに限定されない。
【0088】
「ドセタキセル」との用語は、タキソテール(登録商標)の活性成分または他にタキソテール(登録商標)自体を指す。
【0089】
「オストワルド熟成」との用語は、沈殿物または媒体に分散した固体粒子の粗大化を指し、これは、溶液における相分離の最終段階にあり、この間により小さい粒子を犠牲にして沈殿物または固体粒子のより大きい粒子が成長し、小さい粒子は消失する。Ostwald(1896および1897)が認識したように、今では彼の名前を冠しているこのプロセスの駆動力は、沈殿物または固体粒子と溶質との間の表面張力に起因するより小さい粒子の溶解性の増大である。溶質が沈殿物または固体粒子と局所的な平衡にあると仮定すると、この溶解性の差は溶質濃度の勾配を誘発し、より小さい粒子からより大きい粒子への拡散する流束を導く。(粒子表面における溶質原子の緩慢な堆積が律速になる成長とは異なり)拡散律速成長とも呼ばれる。
【0090】
「阻害剤」との用語は、一般に、オストワルド熟成に起因する水系媒体に分散した固体ナノ粒子の不安定性を軽減するために実質的に水不溶性の物質に加えられる有機物質を指す。
【0091】
好ましい実施形態では、本発明は、微小管阻害剤から選択される実質的に水不溶性の医薬品物質の、オストワルド熟成に起因する粒子の成長がない固体ナノ粒子処方物、ならびにかかる処方物を調製および使用する方法を提供する。
【0092】
これらのナノ粒子処方物の利点は、実質的に水不溶性の医薬品物質がオストワルド熟成の阻害剤と共沈殿されることである。これらの組成物は、ゆっくりとした輸液によって、またはボーラス注入法によって、または他の非経口もしくは経口送達経路によってナノ粒子または懸濁液の形態で送達することができる薬理学的に活性な薬剤の非常に低い毒性形態を提供することが観察されている。これらのナノ粒子は、400nm未満、好ましくは200nm未満、そしてより好ましくは140nm未満のサイズを有し、親水性タンパク質がそのナノ粒子の表面に吸着されている。これらのナノ粒子は、異なるモルホロジーをとることができ、それらは非晶質の粒子または結晶性粒子として存在することができる。
【0093】
実質的に不溶性とは、25℃の水に対して0.5mg/ml未満、好ましくは0.1mg/ml未満、そして特には0.05mg/ml未満の溶解度を有する物質を意味する。
【0094】
粒子の成長の阻害に対する最大の影響は、その物質が25℃の水に対して0.05μg/mlを超える溶解度を有するときに観察される。好ましい実施形態では、その物質は、0.05μg/ml〜0.5mg/mlの範囲、例えば0.05μg/ml〜0.05mg/mlの範囲の溶解度を有する。
【0095】
水に対するその物質の溶解度は、従来の技法を使用して測定することができる。例えば、25℃で過剰量の物質を水に加え、その溶液を48時間平衡化させることによりその物質の飽和溶液が調製される。過剰の固体は遠心分離または濾過によって取り除かれ、水に対するその物質の濃度は適切な分析技法(HPLCなど)によって測定される。
【0096】
本発明にかかるプロセスは、広範囲の実質的に水不溶性の物質の安定な水系分散液を調製するために使用することができる。適切な物質としては、顔料、農薬、除草剤、殺菌剤、工業的な殺生物剤、化粧品、薬理学的に活性な化合物および薬理学的に不活性な物質(薬理学的に許容できる担体および希釈剤など)が挙げられるが、これらに限定されない。
【0097】
好ましい実施形態では、実質的に水不溶性の物質は、実質的に水不溶性の薬理学的に活性な物質である。多くの種類の薬理学的に活性な化合物が本発明で使用するのに適しており、その例としては、実質的に水不溶性の抗癌剤(例えばビカルタミド)、ステロイド、好ましくはグルココルチコステロイド(特に、抗炎症性グルココルチコステロイド、例えばブデソニド)、血圧降下薬(例えばフェロジピンまたはプラゾシン)、β遮断薬(例えばピンドロールまたはプロプラノロール)、脂質低下薬、抗凝固剤、抗血栓剤、抗真菌薬(例えばグリセオフルビン)、抗ウイルス薬、抗生物質、抗菌剤(例えばシプロフロキサシン)、抗精神病薬、抗うつ剤、鎮痛剤、麻酔薬、抗炎症剤(胃腸の炎症性疾患の治療のための化合物、例えば国際公開第99/55706号パンフレットに記載される化合物および他の抗炎症性化合物、例えばケトプロフェンを含む)、抗ヒスタミン薬、ホルモン(例えばテストステロン)、免疫調整剤または避妊薬が挙げられるが、これらに限定されない。これらの物質は、単一の実質的に水不溶性の物質を含んでいてもよく、または2つ以上のかかる物質の組合せを含んでいてもよい。
【0098】
本発明によって製造されるナノ粒子は約60〜190nmの直径を有するため、それらはRESによる摂取量が減少し、その結果、より長い循環時間、生物学的安定性および化学的安定性の向上、および腫瘍部位での蓄積量の増大を示す。もっとも重要なことは、このナノ粒子が薬物動態および生体内分布を変えることができるため、このナノ粒子処方物が、毒性が実質的に低下しマウスに対して抗腫瘍活性の著しい増加をもたらすことができることである。これは有毒な副作用を軽減することができ、その療法の効力を高めることができる。
【0099】
(オストワルド熟成阻害剤)
オストワルド熟成阻害剤は、水非混和性有機相に存在する実質的に水不溶性の物質よりも水に対する溶解性が低い非ポリマー性疎水性有機化合物であるが、このオストワルド熟成阻害剤はリン脂質ではない。適切なオストワルド熟成阻害剤は、25℃において0.1mg/l未満、より好ましくは0.01mg/l未満の水に対する溶解度を有する。本発明の実施形態にでは、オストワルド熟成阻害剤は、25℃において0.05μg/ml未満、例えば0.1ng/ml〜0.05μg/mlの水に対する溶解度を有する。
【0100】
本発明の実施形態では、オストワルド熟成阻害剤は、2000未満、例えば500未満、例えば400未満の分子量を有する。本発明の別の実施形態では、オストワルド熟成阻害剤は、1000未満、例えば600未満の分子量を有する。例えば、オストワルド熟成阻害剤は、200〜2000の範囲の分子量を有していてよく、好ましくは400〜1000の範囲、より好ましくは200〜600の範囲の分子量を有していてよい。
【0101】
適切なオストワルド熟成阻害剤としては、分類(i)〜(vii)から選択された阻害剤またはかかる阻害剤の2つ以上の組合せが挙げられる。
(i)脂肪酸のモノグリセリド、ジグリセリドまたは(より好ましくは)トリグリセリド。適切な脂肪酸としては、8〜12個、より好ましくは8〜10個の炭素原子を含む中鎖脂肪酸、または12個より多い炭素原子、例えば14〜20個の炭素原子、より好ましくは14〜18個の炭素原子を含む長鎖脂肪酸が挙げられる。この脂肪酸は、飽和脂肪酸であってもよく、不飽和脂肪酸であってもよく、または飽和脂肪酸と不飽和脂肪酸との混合物であってもよい。脂肪酸は任意に1つ以上のヒドロキシル基を含んでいてもよい(例えばリシノール酸)。グリセリドは、周知の技法、例えば、グリセロールを1つ以上の長鎖脂肪酸または中鎖脂肪酸とエステル化することにより、調製することができる。好ましい実施形態では、オストワルド熟成阻害剤は、グリセロールを長鎖脂肪酸、または好ましくは中鎖脂肪酸の混合物とエステル化することにより得られるトリグリセリドの混合物である。脂肪酸の混合物は、天然物から、例えば天然油(ヤシ油など)からの抽出によって得ることができる。ヤシ油から抽出された脂肪酸は、約50〜80重量%のデカン酸および20〜50重量%のオクタン酸を含有する。グリセロールをエステル化するために脂肪酸の混合物を使用すると、異なるアシル鎖長の混合物を含むグリセリドの混合物が得られる。長鎖トリグリセリドおよび中鎖トリグリセリドは市販されている。例えば、8〜12個、より好ましくは8〜10個の炭素原子を有するアシル基を含む中鎖トリグリセリド(MCT)はグリセロールとヤシ油から抽出された脂肪酸とのエステル化により調製され、8〜12個、より好ましくは8〜10個の炭素原子を有するアシル基を含むトリグリセリドの混合物が得られる。このMCTは、Miglyol 812N(Huls、ドイツ)として市販されている。他の市販されているMCTとしては、Miglyol 810およびMiglyol 818(Huls、ドイツ)が挙げられる。さらなる適切な中鎖トリグリセリドは、トリラウリン(グリセロールトリラウレート)である。市販されている長鎖トリグリセリドとしては、グリセリルトリステアレート、グリセリルトリパルミテート、大豆油、ゴマ油、ヒマワリ油、ヒマシ油または菜種油が挙げられる。
モノグリセリドおよびジグリセリドは、グリセロールと適切な脂肪酸、または脂肪酸の混合物との部分エステル化によって得ることができる。必要に応じて、モノグリセリドおよびジグリセリドは、従来の技法を使用して、例えば反応混合物からの抽出および引き続くエステル化によって、分離し生成することができる。モノグリセリドが使用される場合、それは長鎖モノグリセリド、例えばグリセロールと18個の炭素原子を含む脂肪酸とのエステル化により形成されるモノグリセリドであることが好ましい。
(ii)C2−10ジオールの脂肪酸モノエステルまたは(好ましくは)ジエステル。好ましくは、このジオールは、飽和であってもよくまたは不飽和であってもよい脂肪族ジオール、例えば直鎖ジオールであっても分枝鎖ジオールであってもよいC2−10アルカンジオールである。より好ましくは、このジオールは、直鎖ジオールであっても分枝鎖ジオールであってもよいC2−6アルカンジオール、例えばエチレングリコールまたはプロピレングリコールである。適切な脂肪酸としては、グリセリドに関連して上に記載した中鎖脂肪酸および長鎖脂肪酸が挙げられる。好ましいエステルは、プロピレングリコールと10〜18個の炭素原子を含む1つ以上の脂肪酸とのジエステル、例えばMiglyol 840(Huls、ドイツ)である。
(iii)アルカノールまたはシクロアルカノールの脂肪酸エステル。適切なアルカノールとしては、直鎖であっても分枝鎖であってもよいC1−20アルカノール、例えばエタノール、プロパノール、イソプロパノール、n−ブタノール、sec−ブタノールまたはtert−ブタノールが挙げられる。適切なシクロアルカノールとしては、C3−6シクロアルカノール、例えばシクロヘキサノールが挙げられる。適切な脂肪酸としては、グリセリドに関連して上に記載した中鎖脂肪酸および長鎖脂肪酸が挙げられる。好ましいエステルは、C2−6アルカノールと8〜10個の炭素原子、より好ましくは12〜29個の炭素原子を含む1つ以上の脂肪酸とのエステルである。この脂肪酸は飽和であっても不飽和であってもよい。適切なエステルとしては、例えばドデカン酸ドデシルまたはオレイン酸エチルが挙げられる。
(iv)ワックス。適切なワックスとしては、長鎖脂肪酸と少なくとも12個の炭素原子を含むアルコールとのエステルが挙げられる。このアルコールは、脂肪族アルコールであってもよく、芳香族アルコールであってもよく、脂肪族基および芳香族基を含むアルコールであってもよく、2つ以上のかかるアルコールの混合物であってもよい。アルコールが脂肪族アルコールである場合、それは飽和であっても不飽和であってもよい。この脂肪族アルコールは、直鎖であってもよく、分枝鎖であってもよくまたは環状であってもよい。適切な脂肪族アルコールとしては、12個より多い炭素原子、好ましくは14個より多い炭素原子、特に18個より多い炭素原子、例えば12〜40個、より好ましくは14〜36個および特に18〜34個の炭素原子を含む脂肪族アルコールが挙げられる。適切な長鎖脂肪酸としては、グリセリドに関連して上に記載した長鎖脂肪酸、好ましくは14個より多い炭素原子、特に18個より多い炭素原子、例えば14〜40個、より好ましくは14〜36個、特に18〜34個の炭素原子を含む長鎖脂肪酸が挙げられる。このワックスは、天然ワックス(例えば蜜ろう)であってもよく、植物性素材に由来するワックスでもよく、脂肪酸と長鎖アルコールとのエステル化によって調製される合成ワックスであってもよい。他の適切なワックスとしては、石油ろう(パラフィンワックスなど)が挙げられる。
(v)長鎖脂肪族アルコール。適切なアルコールとしては、6個以上の炭素原子、より好ましくは8個以上の炭素原子、例えば12個以上の炭素原子、例えば12〜30個、例えば14〜28個の炭素原子を有するアルコールが挙げられる。長鎖脂肪族アルコールが10〜28個、特に14〜22個の炭素原子、例えば14〜22個の炭素原子を有することが特に好ましい。このアルコールは、直鎖であってもよく、分枝鎖であってもよく、飽和であってもよく不飽和であってもよい。適切な長鎖アルコールの例としては、1−ヘキサデカノール、1−オクタデカノールまたは1−ヘプタデカノールが挙げられる。または、
(vi)硬化植物油、例えば硬化ヒマシ油。
(vii)コレステロールおよびコレステロールの脂肪酸エステル。
【0102】
本発明の一実施形態では、オストワルド熟成阻害剤は、6〜22個、好ましくは10〜20個の炭素原子を含む長鎖トリグリセリドおよび長鎖脂肪族アルコールから選択される。好ましい長鎖トリグリセリドおよび長鎖脂肪族アルコールは、上に定義されている。好ましい実施形態では、オストワルド熟成阻害剤は、12〜18個の炭素原子を有するアシル基を含む長鎖トリグリセリドまたはかかるトリグリセリドの混合物、および10〜22個の炭素原子を含む脂肪族アルコール(好ましくは1−ヘキサデカノール)のエステルまたはこれらの混合物(例えばヘキサデシルヘキサデカノエート)から選択される。
【0103】
本発明の別の実施形態では、オストワルド熟成阻害剤は、コレステロールのエステルおよびコレステロールから選択される。好ましいコレステリルエステルは、パルミチン酸コレステリルまたはステアリン酸コレステリルである。
【0104】
実質的に水不溶性の物質が薬理学的に活性な化合物である場合、オストワルド熟成阻害剤は薬理学的に不活性な物質であることが好ましい。
【0105】
オストワルド熟成阻害剤は、粒子中に、懸濁液中の粒子のオストワルド熟成を防止するのに十分な量で存在する。オストワルド熟成阻害剤は、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む、本発明のプロセスにおいて形成される固体粒子中の少量成分であることが好ましい。それゆえ、オストワルド熟成阻害剤が分散液中の粒子のオストワルド熟成を防止するのにちょうど十分な量で存在し、これにより粒子中に存在するオストワルド熟成阻害剤の量を最少にすることが好ましい。
【0106】
本発明の実施形態では、オストワルド熟成阻害剤および実質的に水不溶性の物質の総重量に対するオストワルド熟成阻害剤の重量分率(すなわち、オストワルド熟成阻害剤の重量/(オストワルド熟成阻害剤の重量+実質的に水不溶性の物質の重量))が0.01〜0.99であり、好ましくは0.05〜0.95であり、特に0.2〜0.95であり、とりわけ0.3〜0.95である。好ましい実施形態では、オストワルド熟成阻害剤および実質的に水不溶性の物質の総重量に対するオストワルド熟成阻害剤の重量分率は、0.95未満であり、より好ましくは0.9以下であり、例えば0.2〜0.9、例えば0.3〜0.9、例えば約0.8である。この実質的に水不溶性の物質が薬理学的に活性な物質であり、オストワルド熟成阻害剤が比較的無毒であり(例えば0.8を超える重量分率)かつ生体内に投与されたときに望ましくない副作用を生じず、そして/または薬理学的に活性な物質の溶解速度/生物学的利用能に影響を及ぼさないかも知れない場合には、これは特に好ましい。
【0107】
さらに、本発明者らは、オストワルド熟成による粒子の成長を防止し、これにより小さい(好ましくは1000nm未満、好ましくは500nm未満)安定な粒子が調製できるようにするためには、オストワルド熟成阻害剤および実質的に水不溶性の物質に対するオストワルド熟成阻害剤の低い重量比(すなわち0.5未満)で一般に十分であることを見出した。小さく一定の粒径が望ましい場合が多く、実質的に水不溶性の物質が例えば静脈内投与に使用される薬理学的に活性な物質である場合には特にそうである。
【0108】
本発明にかかるプロセスによって調製される分散液の1つの応用は、薬理学的に活性な化合物の毒物学の研究である。本発明のプロセスによって調製される分散液は、特に物質の粒径が500nm未満である場合には、別のプロセスを使用して調製される分散液と比べて改善された生物学的利用能を示すことができる。この応用において、オストワルド熟成阻害剤の存在に関連する毒物学に対するいかなる効果も最少になるように、活性な化合物に対するオストワルド熟成阻害剤の量を最少にすることが好都合である。
【0109】
実質的に水不溶性の物質がオストワルド熟成阻害剤に対してかなりの溶解度を有する場合、実質的に水不溶性の物質に対するオストワルド熟成阻害剤の重量比は、実質的に水不溶性の物質の量がオストワルド熟成阻害剤中の実質的に水不溶性の物質の飽和溶液を形成するのに必要とされる量を超えることを確実にするたように選択されるべきである。これにより、実質的に水不溶性の物質の固体粒子が分散液中で形成されることが確実になる。オストワルド熟成阻害剤中の実質的に水不溶性の物質の溶液を含む液体液滴、あるいは固体物質および大領域の液体オストワルド熟成阻害剤を含む2相系がこのプロセスで形成されないことを確実にするために、分散液が調製される温度(例えば周囲温度)でオストワルド熟成阻害剤が液体である場合に、これは重要である。
【0110】
理論に拘束されることは望まないが、本発明者らは、粒子中で物質とオストワルド熟成阻害剤との間で相分離が起こる系は、固体粒子が実質的に単一相系を形成する系よりもオストワルド熟成を起こしやすいと考える。従って、好ましい実施形態では、オストワルド熟成阻害剤は、実質的に水不溶性の物質およびオストワルド熟成阻害剤の実質的に単一相の混合物を含む分散液中の固体粒子を形成するのに十分その物質と混和性である。本発明によって形成される粒子の組成は、従来の技法、例えばオストワルド熟成阻害剤に対する実質的に水不溶性の物質の(熱力学的)溶解度、固体粒子中の相分離を検出するための慣用的な示差走査熱量測定(DSC)法を使用して得られる融解エントロピーおよび融点の分析を用いて分析することができる。さらに、核磁気共鳴(NMR)を用いたナノ懸濁液の研究(例えば、粒子中のいずれかの成分の線幅拡大)を使用して粒子中の相分離を検出することができる。
【0111】
一般に、オストワルド熟成阻害剤は、実質的に単一相粒子を形成するのに十分な上記物質との混和性を有するべきである。これは、オストワルド熟成阻害剤が固体粒子中に分子レベルで分散しているか、または固体粒子全体にわたって分散しているオストワルド熟成阻害剤の小さい領域中に存在することを意味する。多くの物質について、物質/オストワルド熟成阻害剤混合物は非理想的混合物であると考えられる。これは、その2つの成分の混合が非ゼロエンタルピー変化を伴うことを意味する。
【0112】
(本発明のナノ粒子の調製)
水系媒体に分散した固体ナノ粒子を形成するために、実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)は、適切な溶媒(例えば、クロロホルム、塩化メチレン、酢酸エチル、エタノール、テトラヒドロフラン、ジオキサン、アセトニトリル、アセトン、ジメチルスルホキシド、ジメチルホルムアミド、メチルピロリジノンなど、およびこれらのうちの任意の2つ以上の混合物)中に溶解される。本発明を実施するために使用することが想起されるさらなる溶媒としては、大豆油、ココナッツオイル、オリーブ油、サフラワー油、綿実油、ゴマ油、オレンジ油、リモネン油、C1−C20アルコール、C2−C20エステル、C3−C20ケトン、ポリエチレングリコール、脂肪族炭化水素、芳香族炭化水素、ハロゲン化炭化水素およびこれらの組合せが挙げられる。
【0113】
次の段階では、固体ナノ粒子を作製するために、タンパク質(例えば、ヒト血清アルブミン)を(水相に)加えて、安定なナノ液滴の形成のための安定剤または乳化剤として作用させる。タンパク質は、約0.05〜25%(w/v)の範囲、より好ましくは約0.5%〜10%(w/v)の範囲の濃度で加えられる。
【0114】
次の段階では、固体ナノ粒子を作製するために、高圧下および高せん断力下での均質化によってエマルジョンが形成される。かかる均質化は、約3,000〜30,000psi(約21〜約207MPa)の範囲の圧力で通常運転される高圧ホモジナイザー中で実施されることが好都合である。かかるプロセスは、約6,000〜25,000psi(約41〜約172MPa)の範囲の圧力で実施されることが好ましい。生成したエマルジョンは、実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤を含有する非水系溶媒の非常に小さいナノ液滴を含む。許容できる均質化方法としては、高せん断およびキャビテーションを付与するプロセス(高圧均質化、高せん断ミキサー、超音波処理、高せん断インペラなど)が挙げられる。
【0115】
最後に、固体ナノ粒子を作製するために、溶媒が減圧下でエバポレーションされ、固体形態の実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)ならびにタンパク質の固体ナノ粒子から構成されるコロイド系が得られる。許容できるエバポレーション方法としては、ロータリーエバポレーター、流下膜式エバポレーター、噴霧乾燥機、凍結乾燥機などの使用が挙げられる。溶媒のエバポレーション後、液体懸濁液は乾燥され、薬理学的に活性な薬剤およびタンパク質を含有する粉末が得られ得る。生成した粉末は、任意の好都合なときに適切な水系媒体(生理食塩水、緩衝化生理食塩水、水、緩衝化水系媒体、アミノ酸の溶液、ビタミンの溶液、炭水化物の溶液など、ならびにこれらのうちの任意の2つ以上の組合せなど)の中に再懸濁することができ、哺乳動物に投与することができる懸濁液が得られる。この粉末を得るために想起される方法としては、フリーズドライ法(freeze−drying)、噴霧乾燥などが挙げられる。
【0116】
本発明の具体的な実施形態によれば、実質的に水不溶性の医薬品物質およびオストワルド成長に対するオストワルド熟成阻害剤を含有する異常に小さいサブミクロン固体粒子、すなわち直径200ナノメートル未満である粒子を形成するための方法が提供される。かかる粒子は、使用前に液体懸濁液の形態で濾過滅菌することができる。本発明の処方プロセスの最終生成物(すなわち、実質的に水不溶性の医薬品物質の粒子)を濾過滅菌することができるということは、非常に重要である。なぜなら、高濃度のタンパク質(例えば、血清アルブミン)を含有する分散液を従来の手段(高圧蒸気殺菌法など)によって滅菌することができないからである。
【0117】
実質的に水不溶性の医薬品物質の濾過滅菌できる固体ナノ粒子(すなわち、200nm未満の粒子)を得るために、実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤(複数種であってもよい)は最初に実質的に水非混和性の有機溶媒(例えば、水に対して約5%未満の溶解度を有する溶媒。例えば、クロロホルム)に高濃度で溶解され、これにより実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤を含有する油相が形成される。適切な溶媒はこれまでに示されている。次に、水混和性の有機溶媒(例えば、水に対して約10%を超える溶解度を有する溶媒。例えば、エタノール)が全有機相の約1%〜99%v/vの範囲、より好ましくは約5%〜25%v/vの範囲の最終濃度で油相に加えられる。この水混和性有機溶媒は、酢酸エチル、エタノール、テトラヒドロフラン、ジオキサン、アセトニトリル、アセトン、ジメチルスルホキシド、ジメチルホルムアミド、メチルピロリジノンなどの溶媒から選択することができる。あるいは、水非混和性溶媒と水混和性溶媒との混合物が最初に調製され、次いで実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤がその混合物に溶解される。有機相中の水混和性溶媒は有機相と水相との間の界面の潤滑剤として作用し、均質化の間に微細な水中油型エマルジョンの形成をもたらすと考えられる。
【0118】
次の段階では、オストワルド成長が減少した実質的に水不溶性の医薬品物質の固体ナノ粒子の形成のために、ヒト血清アルブミンまたはこれまでに記載した任意の他の適切な安定剤が水系媒体に溶解される。この成分は、安定なナノ液滴の形成のための乳化剤として作用する。必要に応じて、十分な量の第1の有機溶媒(例えばクロロホルム)が水相に溶解され、その水系媒体は飽和濃度に近づけられる。別個の測定された量の有機相が飽和水相に加えられ(この時点で、これは実質的に水不溶性の医薬品物質、第1の有機溶媒および第2の有機溶媒を含有する)、有機相の相分率が約0.5%〜15%v/v、より好ましくは1%〜8%v/vであるようにされる。次に、低せん断力での均質化によってマイクロ液滴およびナノ液滴から構成される混合物が形成される。これは、例えば、約2,000〜約15,000rpmの範囲で運転される従来の実験室用のホモジナイザーを用いて、当業者が容易に認識できるような種々の方法で実現することができる。これは、続いて高圧(すなわち、約3,000〜30,000psi(約21〜約207MPa)の範囲)で均質化される。生成した混合物は、タンパク質水溶液(例えば、ヒト血清アルブミン)、実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤(複数種であってもよい)、他の薬剤、第1の溶媒および第2の溶媒を含む。最後に、溶媒は真空下で急速にエバポレーションされ、非常に小さいナノ粒子の形態(すなわち,直径が約50nm〜200nmの範囲の粒子)のコロイド状分散系(実質的に水不溶性の医薬品物質、オストワルド熟成阻害剤および他の薬剤ならびにタンパク質の固体)が得られ、このように濾過滅菌することができる。粒子の好ましいサイズ範囲は、処方および操作パラメータに依存して約50nm〜170nmである。
【0119】
本発明によって調製される固体ナノ粒子は、例えば、適切な温度−時間プロフィールで凍結乾燥することにより、そこから水を除去することによって粉末形態へとさらに変換されてもよい。タンパク質(例えば、ヒト血清アルブミン)自体が抗凍結剤として作用し、マンニトール、スクロース、トレハロース、グリシンなどの従来の抗凍結剤を使用する必要なく、粉末は、水、生理食塩水または緩衝液を加えることにより容易に再構成される。必要でないが、所望される場合には従来の抗凍結剤が本発明の処方物に添加されてもよいことは、当然理解される。実質的に水不溶性の医薬品物質を含有する固体ナノ粒子によって、比較的小さい体積で高用量の薬理学的に活性な薬剤の送達が可能になる。
【0120】
本発明の実施形態によれば、実質的に水不溶性の医薬品物質を含有する固体ナノ粒子は約2ミクロンより大きい断面直径を有する。1ミクロン未満の断面直径がより好ましく、静脈内投与経路にとっては0.22ミクロン未満の断面直径が現在最も好ましい。
【0121】
本発明によって安定剤として使用することが想起されるタンパク質としては、アルブミン(35個のシステイン残基を含む)、イムノグロブリン、カゼイン、インスリン(6個のシステインを含む)、ヘモグロビン(1つのα2β2ユニットあたり6個のシステイン残基を含む)、リゾチーム(8個のシステイン残基を含む)、イムノグロブリン、α−2−マクログロブリン、フィブロネクチン、ビトロネクチン、フィブリノゲン、リパーゼなどが挙げられる。タンパク質、ペプチド、酵素、抗体およびこれらの組合せは、本発明で使用することが想起される一般的な種類の安定剤である。
【0122】
使用するために現在好ましいタンパク質はアルブミンである。ヒト血清アルブミン(HSA)は、最も豊富にある血漿タンパク質(〜640
【化1】
)であり、ヒトに対しては免疫原性ではない。タンパク質は、主に、広範な範囲の疎水性低分子リガンド(脂肪酸、ビリルビン、チロキシン、胆汁酸およびステロイドが挙げられる)に結合するその顕著な能力によって特徴付けられる。このタンパク質は、これらの化合物についての可溶化剤および輸送体としての役割を果たし、ある場合には、遊離濃度の重要な緩衝作用をもたらす。HSAはまた、内在性リガンドの結合場所と重なる2つの主要な部位で実に様々な薬物を結合する。このタンパク質は、3つの相同ドメイン(I〜III)を含む66kDのらせん状の単量体であり、その相同ドメインの各々はAサブドメインおよびBサブドメインから構成される。りん光偏光解消法を用いて中性溶液においてエリトロシン−ウシ血清アルブミン複合体ついて行った測定結果は、ナノ秒〜数ミリ秒の時間範囲ではBSAの溶液中の大きいタンパク質セグメントの独立した動きは存在しないことと整合する。これらの測定結果は、BSAの中性溶液中で、ヒト血清アルブミンの結晶構造におけるようなアルブミンのハート型をした構造(8nm×8nm×8nm×3.2nm)を有していることを支持する(Ferrer 2001)。アルブミンの別の利点は、薬物を腫瘍部位へと輸送する能力である。特異的抗体も、特異的場所へナノ粒子を標的化するために利用することができる。
【0123】
本発明の組成物の調製において、実質的に水不溶性実質的に水不溶性の医薬品物質を懸濁または溶解するために実に様々な有機媒体を用いることができる。本発明の実施のために使用することが想起される有機媒体としては、薬理学的に活性な薬剤を懸濁または溶解することはできるが、乳化剤として用いたポリマーとも、薬理学的に活性な薬剤自体とも化学反応しない任意の非水性液体が挙げられる。例としては、植物油(例えば、大豆油、オリーブ油など)、ココナッツオイル、サフラワー油、綿実油、ゴマ油、オレンジ油、リモネン油、4〜30個の炭素原子を有する脂肪族、脂環式または芳香族の炭化水素(例えば、n−ドデカン、n−デカン、n−ヘキサン、シクロヘキサン、トルエン、ベンゼンなど)、2〜30個の炭素原子を有する脂肪族または芳香族のアルコール(例えば、オクタノールなど)、2〜30個の炭素原子を有する脂肪族または芳香族のエステル(例えば、カプリル酸エチル(オクタン酸エチル)など)、2〜30個の炭素原子を有するアルキルエーテル、アリールエーテルまたは環状エーテル(例えば、ジエチルエーテル、テトラヒドロフランなど)、1〜30個の炭素原子(および必要に応じて1個より多いハロゲン置換基、例えば、CH3Cl、CH2Cl2、CH2Cl−CH2Clなど)を有するアルキルハライドまたはアリールハライド、3〜30個の炭素原子を有するケトン(例えば、アセトン、メチルエチルケトンなど)、ポリアルキレングリコール(例えば、ポリエチレングリコールなど)、あるいはこれらのうちの任意の2つ以上の組合せが挙げられる。
【0124】
本発明の実施のために使用することが想起される有機媒体の特に好ましい組合せは、典型的には約200℃以下の沸点を有し、そしてより高分子量の(あまり揮発性でない)有機媒体を伴った、例えばジクロロメタン、クロロホルム、酢酸エチル、ベンゼンなど(すなわち、薬理学的に活性な薬剤に対して高い溶解度を有し、用いる他の有機媒体に可溶である溶媒)のような揮発性液体を含む。他の有機媒体に加えられる場合、これらの揮発性添加剤は、薬理学的に活性な薬剤の有機媒体中への溶解度を駆動することを補助する。このステップは通常時間がかかるため、これは望ましい。溶解後、この揮発性成分は、(必要に応じて真空下での)エバポレーションにより除去されてよい。
【0125】
液滴サイズを小さくするために、本発明によって調製される固体ナノ粒子処方物は、均質化のあいだエマルジョンをさらに安定化するためのある量の生体適合性界面活性剤をさらに含んでいてもよい。これらの生体適合性界面活性剤は、天然のレシチン(卵レシチン、大豆レシチンなど);植物性モノガラクトシルジグリセリド(硬化物)または植物性ジガラクトシルジグリセリド(硬化物);合成レシチン(ジヘキサノイル−L−□−レシチン、ジオクタノイル−L−□−レシチン、ジデカノイル−L−□−レシチン、ジドデカノイル−L−□−レシチン、ジテトラデカノイル−L−□−レシチン、ジヘキサデカノイル−L−□−レシチン、ジオクタデカノイル−L−□−レシチン、ジオレイル−L−□−レシチン、ジリノレオイル−L−□−レシチン、□−パルミト−□−オレオイル−L−□−レシチン、L−□−グリセロホスホリルコリンなど);ポリオキシエチル化炭化水素または植物油(Cremaphor(登録商標) ELもしくはRH40、Emulphor(登録商標) EL−620PもしくはEL−719、Arlacel(登録商標)−186、Pluronic(登録商標) F−68など);ソルビタンエステル(ソルビタンモノラウレート、ソルビタンモノステアレート、ソルビタンモノパルミテート、ソルビタントリステアレート、ソルビタンモノオレエートなど);PEG脂肪酸エステル(PEG200 ジココエート、PEG300 ジステアレート、PEG400 セスキオエレート、PEG400 ジオレエートなど);エトキシ化グリセリンエステル(POE(20) グリセロールモノステアレート、POE(20) グリセロールモノオレエートなど);エトキシ化脂肪族アミン(POE(15) ココリルアミン、POE(25) ココリルアミン、POE(80) オレイルアミンなど);エトキシ化ソルビタンエステル(POE(20) ソルビタンモノラウレート、POE(20) ソルビタンモノステアレート、POE(20) ソルビタントリステアレート、POE(20) ソルビタントリオレエートなど);エトキシ化脂肪酸(POE(5) オレイン酸、POE(5) ヤシ油脂肪酸、POE(14) ヤシ油脂肪酸、POE(9) ステアリン酸、POE(40) ステアリン酸など);アルコール−脂肪酸エステル(パルミチン酸2−エチルヘキシル、オレイン酸イソブチル、アジピン酸ジトリデシルなど);エトキシ化アルコール(POE(2)−2−エチルヘキシルアルコール、POE(10) セチルアルコール、POE(4) デシルアルコール、POE(6) ラウリルアルコールなど);アルコキシ化ヒマシ油(POE(5) ヒマシ油、POE(25) ヒマシ油、POE(25) 硬化ヒマシ油など);グリセリンエステル(グリセロールモノステアレート、グリセロールトリカプリレートなど);ポリエチレングリコール(ポリエチレングリコール−200、ポリエチレングリコール−300、ポリエチレングリコール−400など);糖エステル(スクロース脂肪酸エステルなど)から選択することができる。処方物中の生体適合性界面活性剤の割合(%)は、0.002重量%〜1重量%で変わり得る。
【0126】
本発明によって調製される固体ナノ粒子処方物は、さらに、所定のキレート剤を含んでいてよい。処方物に加えられる生体適合性キレート剤は、エチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコール−ビス(□−アミノエチルエーテル)−四酢酸(EGTA)、N−(ヒドロキシエチル)−エチレンジアミン三酢酸(HEDTA)、ニトリロ三酢酸(NTA)、トリエタノールアミン、8−ヒドロキシキノリン、クエン酸、酒石酸、リン酸、グルコン酸、糖酸、チオジプロピオン酸、アセトンジカルボン酸、ジ(ヒドロキシエチル)グリシン、フェニルアラニン、トリプトファン、グリセリン、ソルビトール、ジグライムおよびこれらの薬理学的に許容できる塩から選択することができる。
【0127】
本発明によって調製されるナノ粒子処方物は、さらに、アスコルビン酸誘導体(アスコルビン酸、エリソルビン酸、アスコルビン酸ナトリウムなど);チオール誘導体(チオグリセロール、システイン、アセチルシステイン、シスチン、ジチオエリスリトール(dithioerythreitol)、ジチオトレイトール、グルタチオンなど);トコフェロール;ブチル化ヒドロキシアニソール;ブチル化ヒドロキシトルエン;亜硫酸塩(硫酸ナトリウム、亜硫酸水素ナトリウム、アセトン亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウム、ホルムアルデヒドナトリウムなど)から選択することができる抗酸化剤を含んでいてもよい。
【0128】
本発明によって調製されるナノ粒子処方物は、さらに、必要に応じてある防腐剤を含んでいてもよい。本発明の処方物に加えるための防腐剤は、フェノール、クロロブタノール、ベンジルアルコール、メチルパラベン、プロピルパラベン、塩化ベンザルコニウムおよび塩化セチルピリジニウムから選択することができる。
【0129】
上で説明したようにして調製される、タンパク質とともに実質的に水不溶性の医薬品物質およびオストワルド熟成阻害剤を含有する固体ナノ粒子は、生体適合性水性液体中の懸濁液として送達される。この液体は、水、生理食塩水、適切な緩衝液を含む溶液、栄養剤(アミノ酸、糖類、タンパク質、炭水化物、ビタミンまたは脂肪など)を含む溶液から選択することができる。
【0130】
長期保存安定性を向上させるために、固体ナノ粒子処方は、1つ以上の保護剤(スクロース、マンニトール、トレハロースなど)の存在下で凍結および凍結乾燥されてもよい。凍結乾燥された固体ナノ粒子処方物の再水和によって、懸濁液は、本質的にすべてのそれ以前に加えられた実質的に水不溶性の医薬品物質および粒径を保持する。再水和は、単に純水または滅菌水または0.9%塩化ナトリウム注射液または5%デキストロース溶液を加え、次いでその懸濁液を緩やかにかき混ぜることにより実現される。固体ナノ粒子処方中の実質的に水不溶性の医薬品物質の効力は、凍結乾燥および再構成の後も失われることはない。
【0131】
本発明の固体ナノ粒子処方物は、オストワルド熟成阻害剤の存在に起因してオストワルド熟成を起こしにくいことが示されており、先行技術で開示されている処方物よりも溶液中で安定である。本発明において、様々なオストワルド熟成阻害剤組成物、粒径、および実質的に水不溶性の医薬品物質とタンパク質との比を有する本発明の固体ナノ粒子処方効力は、種々の系(ヒト細胞株および細胞増殖活性についての動物モデルなど)に対して研究された。
【0132】
本発明の固体ナノ粒子処方物は、遊離形態で投与された実質的に水不溶性の医薬品物質ほどには有毒でないことが示された。さらに、様々な肉腫をもつマウスおよび腫瘍をもたない正常なマウスの体重に対する、固体ナノ粒子処方物および遊離形態の種々の実質的に水不溶性の医薬品物質の効果が研究されている。しかしながら、本願明細書に提供された実施例は、決して、本発明の範囲を限定することも制限することも意図されておらず、本発明の技術を実施するためにもっぱら利用しなければならない条件、パラメータ、試薬、または出発物質を提供していると解釈されるべきではない。
【実施例】
【0133】
(実施例1)
(阻害剤としてのコレステロールを有する不安定な固体ナノ粒子の調製)
96mgのコレステロールおよび100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。1.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水(Type I water)に溶解することにより、3%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって0.5〜4mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0134】
この懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8時間にわたってモニタリングした。両方のサンプルの粒子は、1〜2時間後に変化し始め、4時間後にオストワルド熟成によって沈殿し始めた(図6)。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【0135】
(実施例2)
(阻害剤としてのコレステロールおよびステアリン酸コレステリルを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、500mgのステアリン酸コレステリル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜4mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0136】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図7)。粒径は48時間後も変化せず、4日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0137】
(実施例3)
(阻害剤としてのコレステロールおよびステアリン酸コレステリルを有する安定な固体ナノ粒子の調製)
50mgのコレステロール(Northern Lipids、カナダ)、250mgのステアリン酸コレステリル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0138】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした。粒径は48時間後も変化せず、3日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0139】
(実施例4)
(阻害剤としてのコレステロールおよびヘキサデカン酸ヘキサデシルを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、500mgのヘキサデカン酸ヘキサデシル(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、2.0mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0140】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図8)。粒径は48時間後も変化せず、5日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0141】
(実施例5)
(阻害剤としてのコレステロールおよびグリセリルトリステアレートを有する安定な固体ナノ粒子の調製)
100mgのコレステロール(Northern Lipids、カナダ)、200mgのグリセリルトリステアレート(Sigma Aldrich、ミズーリ州)および100mgのドセタキセル(Guiyuanchempharm、中国)の混合物を、3.0mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜2mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0142】
懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって、測定した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.22μmフィルター(Nalgene、米国)を通して濾過滅菌した。この懸濁液の粒径は、30〜220nmであった。この懸濁液を−40℃よりも低い温度で凍結し、凍結乾燥した。凍結乾燥したケーキを、さらなる使用の前に再構成した。再構成した溶液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。これら2つのアリコートの粒径を、24℃で8日間にわたってモニタリングした(図9)。粒径は48時間後も変化せず、5日間安定であった。上記の組成を含有する処方物を、オストワルド熟成によって安定なものである、と分類した。
【0143】
(実施例6)
(ヒト血清アルブミンに対する乳化の効果)
3.5mLのクロロホルムと0.6mLの無水エタノールとを混合することにより、有機相を調製した。2gのヒトアルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、4%ヒトアルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.7に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて6000〜10000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を20,000〜30,000psi(約138〜約207MPa)の間で変え、この乳化プロセスを5〜8回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。次いでこのエマルジョンをロータリーエバポレーター(Buchi、スイス)に移し、急速にエバポレーションし、高圧均質化にかけたアルブミン溶液を得た。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、1〜5mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0144】
アルブミン溶液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。約5〜8nmに1つ、そして約120〜140nmに1つの2つのピークが存在することが観察された、約5〜8nmのピークは約99体積%を占め、約120〜140nmのピークは、1体積%未満を占めていた(図4)。対照として、4%ヒト血清溶液中の粒径分布を測定した。それは約5〜8nmに1つのピークのみを有していた(図5)。これらの研究は、アルブミン溶液を水中油型エマルジョン中で均質化すると、2〜3%未満のアルブミン分子が変性により凝集することになるということを示す。
【0145】
(実施例7)
(阻害剤をまったく含まない不安定な固体ナノ粒子の調製)
100mgのドセタキセル(Guiyuanchempharm、中国)を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのウシ血清アルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ウシ血清アルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このヒトアルブミン溶液のpHを6.0〜6.8に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜20,000psi(約103〜約138MPa)の間で変え、この乳化プロセスを8〜12回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって、0.5〜3mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0146】
エバポレーション後、この溶液が他の処方物よりも濁っていることが判明した。2.5gの抗凍結剤トレハロース二水和物(Sigma−Aldrich Co、米国)を10mLの無菌のI型水に溶解し、懸濁液中のトレハロース二水和物の濃度が4〜9重量%の範囲にあるようにこの溶液を上記懸濁液に加えた。この懸濁液を0.45μmフィルターを通して濾過し、この溶液を濾過するのに270mm超のフィルターが必要であった。この溶液を0.22μmフィルター(Nalgene、米国)を通してもう一度濾過した。これらのフィルターを各々25mLのアセトニトリルで洗浄し、このフィルター上の沈殿した薬物を回収した。懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。0.22μmフィルターで濾過した懸濁液の粒径は、240〜390nmであった(図10)。0.22μmフィルターで濾過した懸濁液およびフィルターから回収したドセタキセルについて、HPLC法によってドセタキセル濃度を測定した。ドセタキセルの約50%がフィルターから回収され、残りは懸濁液中に存在することが判明した。この懸濁液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。両方のサンプルの粒子は、1〜3時間後に変化し始め、8時間後にオストワルド熟成によって、沈殿し始めた。
【0147】
この処方物を再度調製したが、上記と同じ沈殿の問題を観察した。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【0148】
(実施例8)
(阻害剤をまったく含まない不安定な固体ナノ粒子の調製)
160mgのドセタキセル(Guiyuanchempharm、中国)を、2.5mLのクロロホルムと0.5mLのエタノールの混合物に溶解した。2.5gのヒト血清アルブミン(Sigma−Aldrich Co、米国)を50mLの無菌のI型水に溶解することにより、5%ヒト血清アルブミン溶液を調製した。滅菌水中の1N塩酸または1N水酸化ナトリウム溶液のいずれかを加えることにより、このアルブミン溶液のpHを6.2〜6.5に調整した。上記の有機溶液を上記アルブミン相に加え、混合物をIKAホモジナイザー(IKA Works、ドイツ)を用いて4000〜6000RPMで予均一化した。生成したエマルジョンを高圧均質化(Avestin Inc、米国)にかけた。圧力を15,000〜24,000psi(約103〜約165MPa)の間で変え、この乳化プロセスを8〜12回続けた。均質化の間、温度制御した熱交換器(Julabo、米国)からホモジナイザーを通して冷媒を循環させることにより、エマルジョンを5℃〜10℃で冷却した。これにより、均質で非常に微細な水中油型エマルジョンが得られた。このエマルジョンをロータリーエバポレータ(Buchi、スイス)に移し、ナノ粒子懸濁液へと急速にエバポレーションした。エバポレータの圧力は、エバポレーションの間、真空ポンプ(Welch)によって0.5〜3mmHgに設定し、エバポレーションの間の浴槽温度は35℃に設定した。
【0149】
エバポレーション後、この溶液が他の処方物よりも濁っていることが判明した。懸濁液の粒径は、Malvern Zetasizerを用いて光子相関分光法によって測定した。濾過していない懸濁液の粒径は200〜1000nmであった(図11)。懸濁液の1つのアリコートを25℃で保存し、他は2〜6℃で保存した。両方のサンプルの粒子は、1〜3時間後に変化し始め、8時間後にオストワルド熟成によって沈殿し始めた。上記の組成を含有する処方物を、オストワルド熟成によって不安定なものである、と分類した。
【産業上の利用可能性】
【0150】
本発明は、タンパク質または他のポリマーを界面活性剤として使用する水中油型エマルジョンプロセスを使用して調製される、水系媒体中の非水溶性物質の固体粒子の実質的に安定な分散液の作製に関連するあらゆる目的のために使用することができ、本発明によって調製される分散液は、オストワルド熟成によって媒介される、形成後の粒子の成長がほとんどないかまたはまったくない。本発明は、癌治療として使用する水溶性組成物を作製するために使用することができるが、当業者は他の応用も同様に思いつくであろう。
【特許請求の範囲】
【請求項1】
水系媒体中の240nm未満の平均粒径を有する固体ナノ粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記媒体から前記水非混和性有機溶媒、および界面潤滑剤として使用した前記任意の有機溶媒を真空下で除去するステップと、
を含み、これによって前記阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成し、必要に応じて
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
ことを特徴とするプロセス。
【請求項2】
前記実質的に水不溶性の物質が実質的に水不溶性の薬理学的に活性な化合物である、請求項1に記載のプロセス。
【請求項3】
前記実質的に水不溶性の薬理学的に活性な化合物が微小管阻害剤からなる群から選択される、請求項2に記載のプロセス。
【請求項4】
前記微小管阻害剤が、タキサン、エポチロン、エポチロン誘導体、ビンカアルカロイド、ポドフィロトキシンおよびその誘導体、コルヒチンおよびその誘導体、ドラスタチンおよびその誘導体、リゾキシンおよびその誘導体、エクチナサイジン、コンブレタスタチンおよびその誘導体、インドール誘導体、ディスコデルモライドおよびその誘導体、スポンジスタチンおよびその誘導体、クリプトフィシンおよびその誘導体、ジデムニンおよびその誘導体、グリセオフルビンおよびその誘導体、メイタンシンおよびその誘導体、ハリコンドリンおよびその誘導体、ヘミアスタリンおよびその誘導体ならびにラメラリンおよびその誘導体である、請求項3に記載のプロセス。
【請求項5】
前記微小管阻害剤がドセタキセルおよびパクリタキセルである、請求項3に記載のプロセス。
【請求項6】
前記微小管阻害剤がエポチロン−Aおよびエポチロン−Bである、請求項3に記載のプロセス。
【請求項7】
前記微小管阻害剤がMAC−321、TL−909およびTL−310である、請求項3に記載のプロセス。
【請求項8】
前記オストワルド熟成阻害剤が、グリセロールと中鎖脂肪酸および長鎖脂肪酸の混合物とをエステル化することにより得られるトリグリセリドの混合物である、請求項1に記載のプロセス。
【請求項9】
前記オストワルド熟成阻害剤が、8〜18個の炭素原子を有するアシル基を含むトリグリセリドの混合物である、請求項8に記載のプロセス。
【請求項10】
前記オストワルド熟成阻害剤が、コレステロールと中鎖脂肪酸および長鎖脂肪酸の混合物とをエステル化することにより得られるコレステリルエステルの混合物である、請求項1に記載のプロセス。
【請求項11】
前記有機相がさらに、6個以上の炭素原子を含む長鎖脂肪族アルコールである共阻害剤を含む、請求項1に記載のプロセス。
【請求項12】
前記有機相がさらに、コレステロールである共阻害剤を含む、請求項1に記載のプロセス。
【請求項13】
前記オストワルド熟成阻害剤が前記分散液中の固体粒子を形成するのに充分、前記実質的に水不溶性の物質と混和性であり、前記粒子が前記物質および前記オストワルド熟成阻害剤の実質的に単一相の混合物を含む、請求項1に記載のプロセス。
【請求項14】
前記乳化剤がタンパク質であり、前記タンパク質が、アルブミン、イムノグロブリン、カゼイン、インスリン、ヘモグロビン、リゾチーム、イムノグロブリン、α−2−マクログロブリン、フィブロネクチン、ビトロネクチン、フィブリノゲン、リパーゼ、酵素および抗体から選択される、請求項1に記載のプロセス。
【請求項15】
前記タンパク質がヒト血清アルブミンである、請求項1に記載のプロセス。
【請求項16】
前記乳化剤がポリマー分散剤および界面活性剤を含む、請求項1に記載のプロセス。
【請求項17】
水系媒体中の500nm未満の平均粒径を有する実質的に水不溶性の薬理学的に活性な物質の固体ナノ粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記分散液から水非混和性有機溶媒、および界面潤滑剤として使用した前記任意の水混和性有機溶媒を真空下で除去するステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成し、
前記オストワルド熟成阻害剤は前記薬理学的に活性な物質よりも水溶性が小さく、前記オストワルド熟成阻害剤は、
(i)脂肪酸のモノグリセリド、ジグリセリドまたはトリグリセリド、
(ii)C2−12ジオールの脂肪酸モノエステルまたはジエステル、
(iii)アルカノールまたはシクロアルカノールの脂肪酸エステル、
(iv)ワックス、
(v)長鎖脂肪族アルコールおよびコレステロール、
(vi)コレステロールの脂肪酸エステル、ならびに
(vii)硬化植物油、
からなる群から選択される1つ以上の物質である、プロセス。
【請求項18】
前記固体粒子の平均粒径が500nm未満であることを特徴とする、請求項17に記載のプロセス。
【請求項19】
請求項1から請求項18のいずれか1項に記載のプロセスによって調製される実質的に安定な水系分散液であって、前記分散液は、連続する水相および前記連続する水相に分散した固体粒子を含み、前記固体粒子はオストワルド熟成阻害剤および実質的に水不溶性の物質を含み、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性である非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
実質的に安定な水系分散液。
【請求項20】
請求項1から請求項19のいずれか1項に記載のプロセスによって調製される、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む固体粒子。
【請求項21】
前記実質的に水不溶性の物質が実質的に水不溶性の薬理学的に活性な物質である、請求項17に記載の固体粒子。
【請求項22】
薬理学的に許容できる担体または希釈剤と合わせて、請求項1から請求項21のいずれか1項に記載の固体粒子を含む医薬製剤。
【請求項23】
水系媒体中の実質的に水不溶性の物質の固体ナノ粒子の分散液におけるオストワルド熟成を阻害する方法であって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記分散液から前記水非混和性有機溶媒、および界面潤滑剤として使用した任意の水混和性有機溶媒を真空下で除去するステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成して、水系媒体中の前記実質的に水不溶性の固体粒子の分散液をもたらし、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さくかつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではないことを特徴とする、方法。
【請求項24】
前記固体粒子の平均粒径が1μm未満である、請求項23に記載の方法。
【請求項25】
前記第1の溶液が1つ以上の阻害剤を含む、請求項1に記載のプロセス。
【請求項26】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項1に記載のプロセス。
【請求項27】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項28】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性のイオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項29】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤とを含有する、固体ナノ粒子組成物。
【請求項30】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項31】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項32】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項33】
前記薬理学的に許容できる防腐剤が、フェノール、クロロブタノール、ベンジルアルコール、メチルパラベン、プロピルパラベン、メチルパラベンナトリウム、プロピルパラベンナトリウム、塩化ベンザルコニウムおよび塩化セチルピリジニウムから選択される、請求項27に記載のナノ粒子組成物。
【請求項34】
前記生体適合性キレート剤が、エチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコール−ビス(□−アミノエチルエーテル)−四酢酸(EGTA)、N−(ヒドロキシエチル)−エチレンジアミン三酢酸(HEDTA)、ニトリロ三酢酸(NTA)、トリエタノールアミン、8−ヒドロキシキノリン、クエン酸、酒石酸、リン酸、グルコン酸、糖酸、チオジプロピオン酸、アセトンジカルボン酸、ジ(ヒドロキシエチル)グリシン、フェニルアラニン、トリプトファン、グリセリン、ソルビトール、ジグライムおよびこれらの薬理学的に許容できる塩から選択される、請求項29に記載の固体ナノ粒子組成物。
【請求項35】
前記生体適合性抗酸化剤が、アスコルビン酸誘導体(アスコルビン酸、エリソルビン酸、アスコルビン酸ナトリウムなど);チオール誘導体(チオグリセロール、システイン、アセチルシステイン、シスチン、ジチオエリスリトール、ジチオトレイトール、グルタチオンなど);トコフェロール;ブチル化ヒドロキシアニソール;ブチル化ヒドロキシトルエン;亜硫酸塩(硫酸ナトリウム、亜硫酸水素ナトリウム、アセトン亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウム、ホルムアルデヒドスルホキシル酸ナトリウム、チオ硫酸ナトリウムなど)およびノルジヒドログアヤレト酸から選択される、請求項30に記載の固体ナノ粒子脂質組成物。
【請求項36】
前記緩衝剤が、第1リン酸ナトリウムおよび第二リン酸ナトリウム、安息香酸ナトリウム、安息香酸カリウム、クエン酸ナトリウム、酢酸ナトリウム、酒石酸ナトリウムおよび他の薬剤である、請求項31に記載の固体ナノ粒子組成物。
【請求項37】
前記賦形剤が、マンニトール、ソルビトール、フルクトース、グルコース、ラクトース、スクロース、トレハロースおよび任意の他の水溶性糖類である、請求項32に記載の固体ナノ粒子組成物。
【請求項38】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項1に記載の固体ナノ粒子医薬組成物。
【請求項39】
請求項1に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項40】
前記医薬組成物が種々のサイズのバイアルの中でフリーズドライ法にかけられるかまたは凍結乾燥され、後の哺乳動物に対しての使用に適した真空条件下で栓で塞がれる、請求項39に記載の固体ナノ粒子医薬組成物を調製するためのプロセス。
【請求項41】
前記哺乳動物がヒトである、請求項40に記載のプロセス。
【請求項42】
請求項39に記載の方法により製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.01mg/mL〜約100mg/mLである、固体ナノ粒子組成物。
【請求項43】
細胞増殖性疾患を治療する方法であって、かかる治療を必要とする患者に、治療上有効量の請求項42に記載の組成物を投与することを含む、方法。
【請求項44】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項1に記載のプロセス。
【請求項45】
前記合成ポリマーが、合成ポリアミノ酸、ポリビニルアルコール、ポリメタクリル酸ヒドロキシエチル、ポリアクリル酸、ポリエチルオキサゾリン、ポリアクリルアミド、ポリビニルピロリジノン、ポリアルキレングリコール、ポリラクチド、ポリグリコリド、ポリカプロラクトン、またはこれらのコポリマー、ならびにこれらのうちの任意の2つ以上の混合物から選択される、請求項44に記載のプロセス。
【請求項46】
請求項21から請求項29のいずれか1項に記載のプロセスによって調製される、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む固体粒子。
【請求項47】
前記第1の溶液が1つ以上の阻害剤を含む、請求項17に記載のプロセス。
【請求項48】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項17に記載のプロセス。
【請求項49】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項50】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性イオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項51】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤を含有する、固体ナノ粒子組成物。
【請求項52】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項53】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項54】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項55】
前記第1の溶液が1つ以上の阻害剤を含む、請求項23に記載のプロセス。
【請求項56】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項23に記載のプロセス。
【請求項57】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項58】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性イオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項59】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤とを含有する、固体ナノ粒子組成物。
【請求項60】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項61】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項62】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項63】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項17に記載の固体ナノ粒子医薬組成物。
【請求項64】
請求項17に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項65】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項23に記載の固体ナノ粒子医薬組成物。
【請求項66】
請求項23に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項67】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項17に記載のプロセス。
【請求項68】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項23に記載のプロセス。
【請求項69】
前記固体粒子の平均粒径が300nm未満である、請求項17に記載のプロセス。
【請求項70】
前記固体粒子の平均粒径が220nm未満である、請求項17に記載のプロセス。
【請求項71】
前記固体粒子の平均粒径が500nm未満である、請求項23に記載のプロセス。
【請求項72】
前記固体粒子の平均粒径が220nm未満である、請求項23に記載のプロセス。
【請求項73】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項1に記載のプロセス。
【請求項74】
請求項39に記載の方法によって製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.1mg/mL〜約50mg/mLである、固体ナノ粒子組成物。
【請求項75】
請求項39に記載の方法によって製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.5mg/mL〜約10mg/mLである、固体ナノ粒子組成物。
【請求項76】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項17に記載のプロセス。
【請求項77】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項23に記載のプロセス。
【請求項1】
水系媒体中の240nm未満の平均粒径を有する固体ナノ粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記媒体から前記水非混和性有機溶媒、および界面潤滑剤として使用した前記任意の有機溶媒を真空下で除去するステップと、
を含み、これによって前記阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成し、必要に応じて
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
ことを特徴とするプロセス。
【請求項2】
前記実質的に水不溶性の物質が実質的に水不溶性の薬理学的に活性な化合物である、請求項1に記載のプロセス。
【請求項3】
前記実質的に水不溶性の薬理学的に活性な化合物が微小管阻害剤からなる群から選択される、請求項2に記載のプロセス。
【請求項4】
前記微小管阻害剤が、タキサン、エポチロン、エポチロン誘導体、ビンカアルカロイド、ポドフィロトキシンおよびその誘導体、コルヒチンおよびその誘導体、ドラスタチンおよびその誘導体、リゾキシンおよびその誘導体、エクチナサイジン、コンブレタスタチンおよびその誘導体、インドール誘導体、ディスコデルモライドおよびその誘導体、スポンジスタチンおよびその誘導体、クリプトフィシンおよびその誘導体、ジデムニンおよびその誘導体、グリセオフルビンおよびその誘導体、メイタンシンおよびその誘導体、ハリコンドリンおよびその誘導体、ヘミアスタリンおよびその誘導体ならびにラメラリンおよびその誘導体である、請求項3に記載のプロセス。
【請求項5】
前記微小管阻害剤がドセタキセルおよびパクリタキセルである、請求項3に記載のプロセス。
【請求項6】
前記微小管阻害剤がエポチロン−Aおよびエポチロン−Bである、請求項3に記載のプロセス。
【請求項7】
前記微小管阻害剤がMAC−321、TL−909およびTL−310である、請求項3に記載のプロセス。
【請求項8】
前記オストワルド熟成阻害剤が、グリセロールと中鎖脂肪酸および長鎖脂肪酸の混合物とをエステル化することにより得られるトリグリセリドの混合物である、請求項1に記載のプロセス。
【請求項9】
前記オストワルド熟成阻害剤が、8〜18個の炭素原子を有するアシル基を含むトリグリセリドの混合物である、請求項8に記載のプロセス。
【請求項10】
前記オストワルド熟成阻害剤が、コレステロールと中鎖脂肪酸および長鎖脂肪酸の混合物とをエステル化することにより得られるコレステリルエステルの混合物である、請求項1に記載のプロセス。
【請求項11】
前記有機相がさらに、6個以上の炭素原子を含む長鎖脂肪族アルコールである共阻害剤を含む、請求項1に記載のプロセス。
【請求項12】
前記有機相がさらに、コレステロールである共阻害剤を含む、請求項1に記載のプロセス。
【請求項13】
前記オストワルド熟成阻害剤が前記分散液中の固体粒子を形成するのに充分、前記実質的に水不溶性の物質と混和性であり、前記粒子が前記物質および前記オストワルド熟成阻害剤の実質的に単一相の混合物を含む、請求項1に記載のプロセス。
【請求項14】
前記乳化剤がタンパク質であり、前記タンパク質が、アルブミン、イムノグロブリン、カゼイン、インスリン、ヘモグロビン、リゾチーム、イムノグロブリン、α−2−マクログロブリン、フィブロネクチン、ビトロネクチン、フィブリノゲン、リパーゼ、酵素および抗体から選択される、請求項1に記載のプロセス。
【請求項15】
前記タンパク質がヒト血清アルブミンである、請求項1に記載のプロセス。
【請求項16】
前記乳化剤がポリマー分散剤および界面活性剤を含む、請求項1に記載のプロセス。
【請求項17】
水系媒体中の500nm未満の平均粒径を有する実質的に水不溶性の薬理学的に活性な物質の固体ナノ粒子の実質的に安定な分散液を調製するためのプロセスであって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記分散液から水非混和性有機溶媒、および界面潤滑剤として使用した前記任意の水混和性有機溶媒を真空下で除去するステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成し、
前記オストワルド熟成阻害剤は前記薬理学的に活性な物質よりも水溶性が小さく、前記オストワルド熟成阻害剤は、
(i)脂肪酸のモノグリセリド、ジグリセリドまたはトリグリセリド、
(ii)C2−12ジオールの脂肪酸モノエステルまたはジエステル、
(iii)アルカノールまたはシクロアルカノールの脂肪酸エステル、
(iv)ワックス、
(v)長鎖脂肪族アルコールおよびコレステロール、
(vi)コレステロールの脂肪酸エステル、ならびに
(vii)硬化植物油、
からなる群から選択される1つ以上の物質である、プロセス。
【請求項18】
前記固体粒子の平均粒径が500nm未満であることを特徴とする、請求項17に記載のプロセス。
【請求項19】
請求項1から請求項18のいずれか1項に記載のプロセスによって調製される実質的に安定な水系分散液であって、前記分散液は、連続する水相および前記連続する水相に分散した固体粒子を含み、前記固体粒子はオストワルド熟成阻害剤および実質的に水不溶性の物質を含み、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性である非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さく、かつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではない、
実質的に安定な水系分散液。
【請求項20】
請求項1から請求項19のいずれか1項に記載のプロセスによって調製される、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む固体粒子。
【請求項21】
前記実質的に水不溶性の物質が実質的に水不溶性の薬理学的に活性な物質である、請求項17に記載の固体粒子。
【請求項22】
薬理学的に許容できる担体または希釈剤と合わせて、請求項1から請求項21のいずれか1項に記載の固体粒子を含む医薬製剤。
【請求項23】
水系媒体中の実質的に水不溶性の物質の固体ナノ粒子の分散液におけるオストワルド熟成を阻害する方法であって、
(a)水および乳化剤を含む水相と、
(b)実質的に水不溶性の物質、水非混和性有機溶媒、必要に応じて界面潤滑剤としての水混和性有機溶媒、およびオストワルド熟成阻害剤を含む有機相と、
を混合するステップと、
(c)高圧ホモジナイザーを使用して水中油型エマルジョンを形成するステップと、
(d)前記分散液から前記水非混和性有機溶媒、および界面潤滑剤として使用した任意の水混和性有機溶媒を真空下で除去するステップと、
を含み、これによって前記オストワルド熟成阻害剤および前記実質的に水不溶性の物質を含む固体粒子を形成して、水系媒体中の前記実質的に水不溶性の固体粒子の分散液をもたらし、
(i)前記オストワルド熟成阻害剤は実質的に水に不溶性の非ポリマー性疎水性有機化合物であり、
(ii)前記オストワルド熟成阻害剤は前記実質的に水不溶性の物質よりも水溶性が小さくかつ
(iii)前記オストワルド熟成阻害剤はリン脂質ではないことを特徴とする、方法。
【請求項24】
前記固体粒子の平均粒径が1μm未満である、請求項23に記載の方法。
【請求項25】
前記第1の溶液が1つ以上の阻害剤を含む、請求項1に記載のプロセス。
【請求項26】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項1に記載のプロセス。
【請求項27】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項28】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性のイオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項29】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤とを含有する、固体ナノ粒子組成物。
【請求項30】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項31】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項32】
請求項1に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項33】
前記薬理学的に許容できる防腐剤が、フェノール、クロロブタノール、ベンジルアルコール、メチルパラベン、プロピルパラベン、メチルパラベンナトリウム、プロピルパラベンナトリウム、塩化ベンザルコニウムおよび塩化セチルピリジニウムから選択される、請求項27に記載のナノ粒子組成物。
【請求項34】
前記生体適合性キレート剤が、エチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコール−ビス(□−アミノエチルエーテル)−四酢酸(EGTA)、N−(ヒドロキシエチル)−エチレンジアミン三酢酸(HEDTA)、ニトリロ三酢酸(NTA)、トリエタノールアミン、8−ヒドロキシキノリン、クエン酸、酒石酸、リン酸、グルコン酸、糖酸、チオジプロピオン酸、アセトンジカルボン酸、ジ(ヒドロキシエチル)グリシン、フェニルアラニン、トリプトファン、グリセリン、ソルビトール、ジグライムおよびこれらの薬理学的に許容できる塩から選択される、請求項29に記載の固体ナノ粒子組成物。
【請求項35】
前記生体適合性抗酸化剤が、アスコルビン酸誘導体(アスコルビン酸、エリソルビン酸、アスコルビン酸ナトリウムなど);チオール誘導体(チオグリセロール、システイン、アセチルシステイン、シスチン、ジチオエリスリトール、ジチオトレイトール、グルタチオンなど);トコフェロール;ブチル化ヒドロキシアニソール;ブチル化ヒドロキシトルエン;亜硫酸塩(硫酸ナトリウム、亜硫酸水素ナトリウム、アセトン亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウム、ホルムアルデヒドスルホキシル酸ナトリウム、チオ硫酸ナトリウムなど)およびノルジヒドログアヤレト酸から選択される、請求項30に記載の固体ナノ粒子脂質組成物。
【請求項36】
前記緩衝剤が、第1リン酸ナトリウムおよび第二リン酸ナトリウム、安息香酸ナトリウム、安息香酸カリウム、クエン酸ナトリウム、酢酸ナトリウム、酒石酸ナトリウムおよび他の薬剤である、請求項31に記載の固体ナノ粒子組成物。
【請求項37】
前記賦形剤が、マンニトール、ソルビトール、フルクトース、グルコース、ラクトース、スクロース、トレハロースおよび任意の他の水溶性糖類である、請求項32に記載の固体ナノ粒子組成物。
【請求項38】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項1に記載の固体ナノ粒子医薬組成物。
【請求項39】
請求項1に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項40】
前記医薬組成物が種々のサイズのバイアルの中でフリーズドライ法にかけられるかまたは凍結乾燥され、後の哺乳動物に対しての使用に適した真空条件下で栓で塞がれる、請求項39に記載の固体ナノ粒子医薬組成物を調製するためのプロセス。
【請求項41】
前記哺乳動物がヒトである、請求項40に記載のプロセス。
【請求項42】
請求項39に記載の方法により製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.01mg/mL〜約100mg/mLである、固体ナノ粒子組成物。
【請求項43】
細胞増殖性疾患を治療する方法であって、かかる治療を必要とする患者に、治療上有効量の請求項42に記載の組成物を投与することを含む、方法。
【請求項44】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項1に記載のプロセス。
【請求項45】
前記合成ポリマーが、合成ポリアミノ酸、ポリビニルアルコール、ポリメタクリル酸ヒドロキシエチル、ポリアクリル酸、ポリエチルオキサゾリン、ポリアクリルアミド、ポリビニルピロリジノン、ポリアルキレングリコール、ポリラクチド、ポリグリコリド、ポリカプロラクトン、またはこれらのコポリマー、ならびにこれらのうちの任意の2つ以上の混合物から選択される、請求項44に記載のプロセス。
【請求項46】
請求項21から請求項29のいずれか1項に記載のプロセスによって調製される、オストワルド熟成阻害剤および実質的に水不溶性の物質を含む固体粒子。
【請求項47】
前記第1の溶液が1つ以上の阻害剤を含む、請求項17に記載のプロセス。
【請求項48】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項17に記載のプロセス。
【請求項49】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項50】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性イオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項51】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤を含有する、固体ナノ粒子組成物。
【請求項52】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項53】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項54】
請求項17に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項55】
前記第1の溶液が1つ以上の阻害剤を含む、請求項23に記載のプロセス。
【請求項56】
前記タンパク質を含有する前記水相のpHが3〜10の間に調整される、請求項23に記載のプロセス。
【請求項57】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに薬理学的に許容できる防腐剤とを含有する、ナノ粒子組成物。
【請求項58】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性イオン性界面活性剤および非イオン性界面活性剤とを含有する、固体ナノ粒子組成物。
【請求項59】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性キレート剤とを含有する、固体ナノ粒子組成物。
【請求項60】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性抗酸化剤とを含有する、固体ナノ粒子脂質組成物。
【請求項61】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性緩衝剤とを含有する、固体ナノ粒子脂質組成物。
【請求項62】
請求項23に記載の方法によって製造される実質的に水不溶性の医薬品物質と、さらに生体適合性賦形剤とを含有する、固体ナノ粒子脂質組成物。
【請求項63】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項17に記載の固体ナノ粒子医薬組成物。
【請求項64】
請求項17に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項65】
前記阻害剤に対する前記実質的に水不溶性の物質の重量比が約99:1〜1:99である、請求項23に記載の固体ナノ粒子医薬組成物。
【請求項66】
請求項23に記載の無菌の固体ナノ粒子医薬組成物を調製するためのプロセスであって、前記実質的に水不溶性の物質を含有する水溶液が、無菌環境で0.22ミクロンのフィルターを通して濾過することにより滅菌される、プロセス。
【請求項67】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項17に記載のプロセス。
【請求項68】
前記生体適合性ポリマーが天然に存在するポリマー、半合成ポリマー、合成ポリマー、またはこれらの組合せである、請求項23に記載のプロセス。
【請求項69】
前記固体粒子の平均粒径が300nm未満である、請求項17に記載のプロセス。
【請求項70】
前記固体粒子の平均粒径が220nm未満である、請求項17に記載のプロセス。
【請求項71】
前記固体粒子の平均粒径が500nm未満である、請求項23に記載のプロセス。
【請求項72】
前記固体粒子の平均粒径が220nm未満である、請求項23に記載のプロセス。
【請求項73】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項1に記載のプロセス。
【請求項74】
請求項39に記載の方法によって製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.1mg/mL〜約50mg/mLである、固体ナノ粒子組成物。
【請求項75】
請求項39に記載の方法によって製造される実質的に水不溶性の医薬品物質を含み、前記実質的に水不溶性の医薬品物質の量が0.5mg/mL〜約10mg/mLである、固体ナノ粒子組成物。
【請求項76】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項17に記載のプロセス。
【請求項77】
前記タンパク質を含有する前記水相のpHが5〜8の間に調整される、請求項23に記載のプロセス。
【図1】

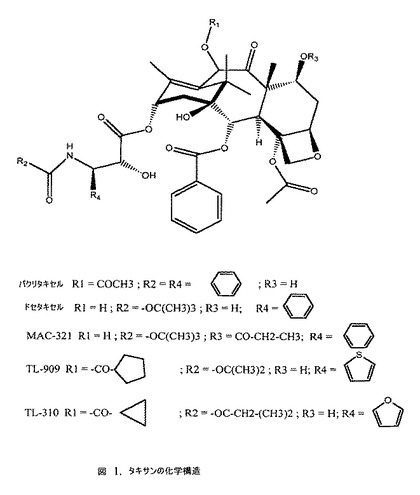
【図2】
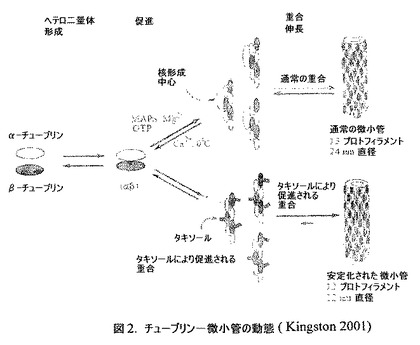
【図3】
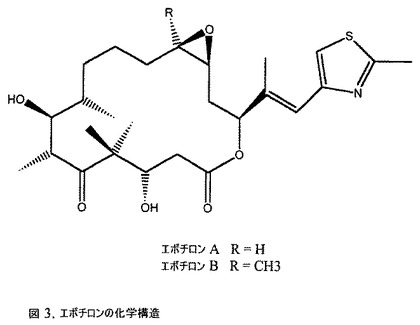
【図4】
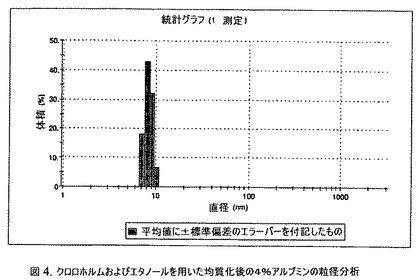
【図5】
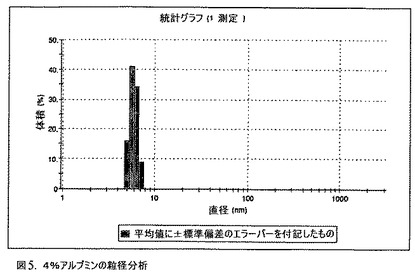
【図6A】
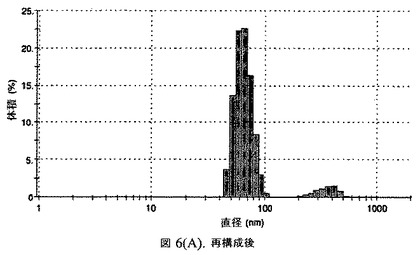
【図6B】
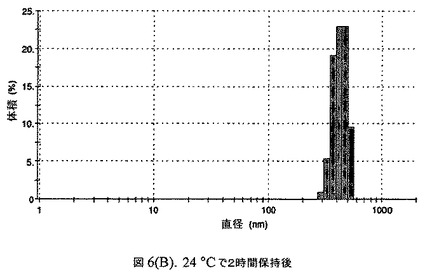
【図6C】
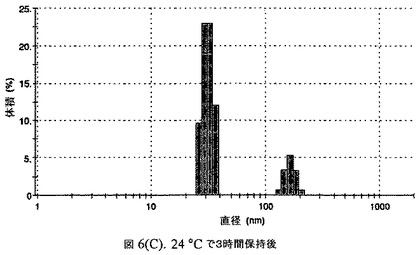
【図6D】
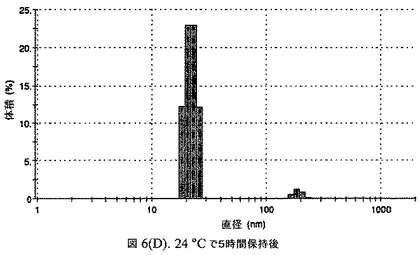
【図6E】
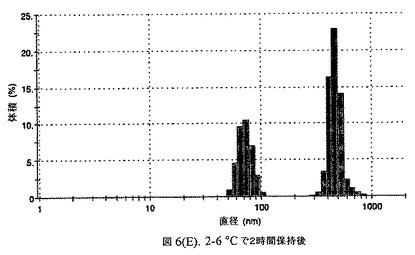
【図6F】
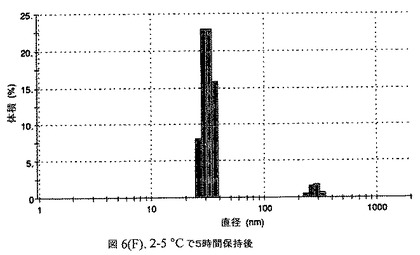
【図7A】
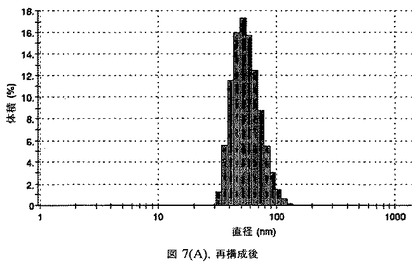
【図7B】
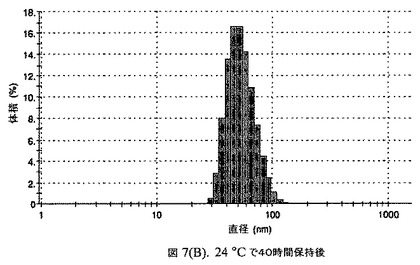
【図7C】
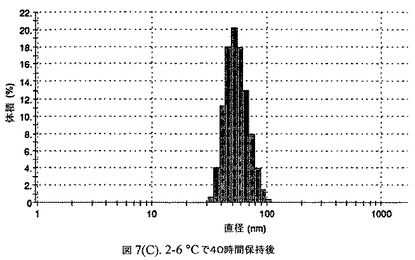
【図7D】
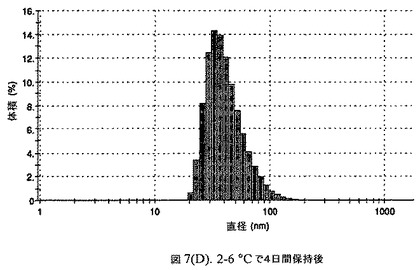
【図7E】
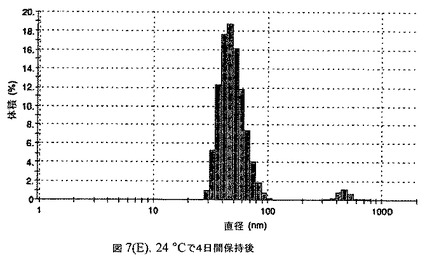
【図8A】
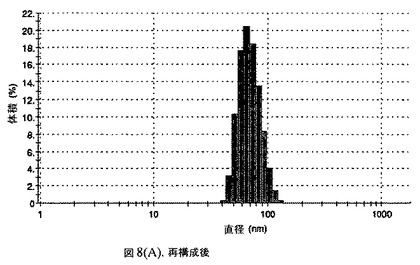
【図8B】
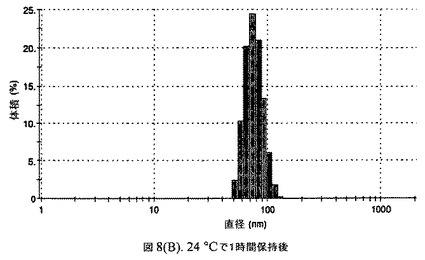
【図8C】
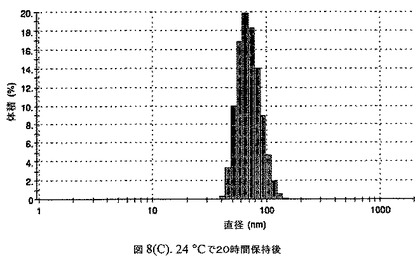
【図8D】
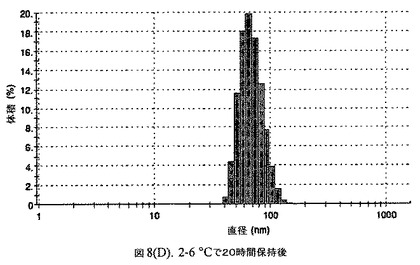
【図8E】
【図8F】
【図8G】
【図8H】
【図9A】
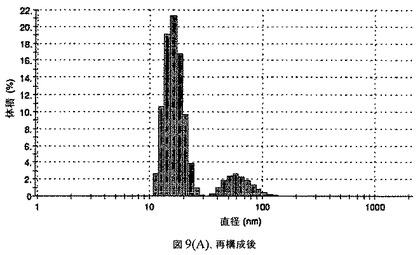
【図9B】
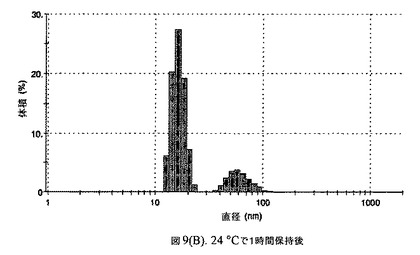
【図9C】
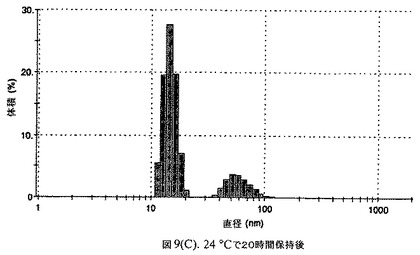
【図9D】
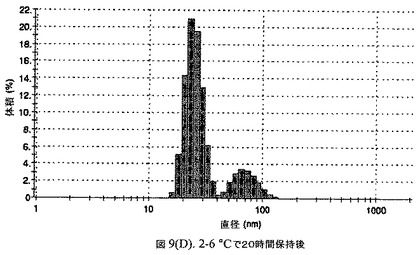
【図9E】
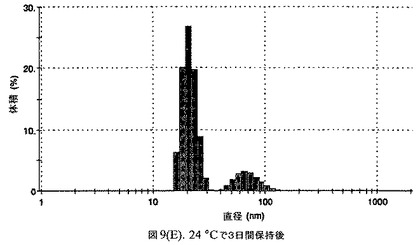
【図9F】
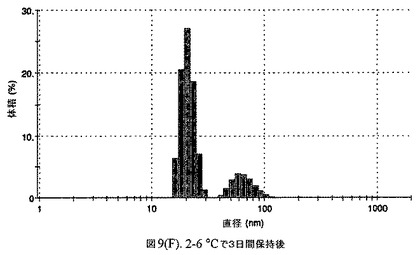
【図9G】
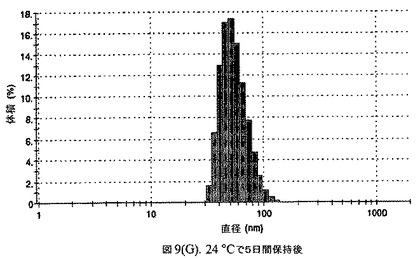
【図9H】
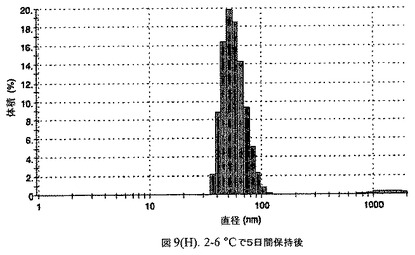
【図10A】
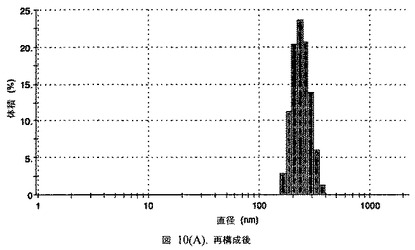
【図10B】
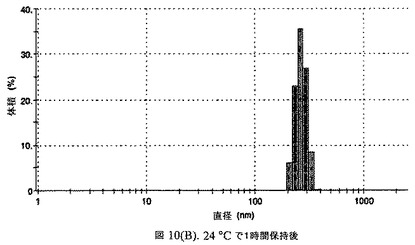
【図11A】
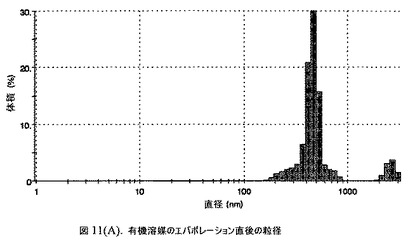
【図11B】
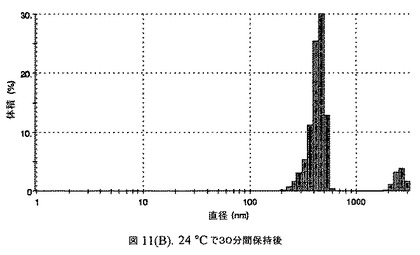
【図11C】
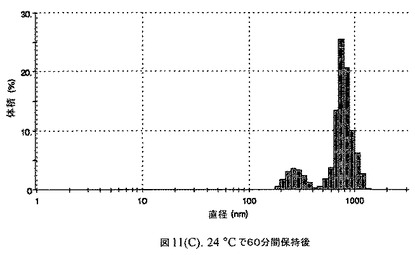
【図11D】
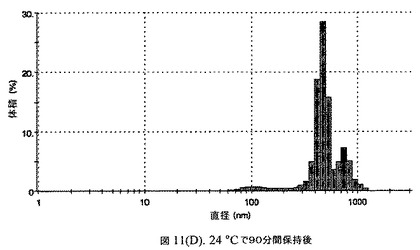
【図11E】
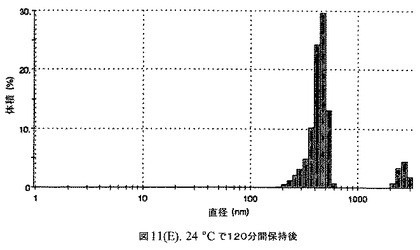
【図11F】
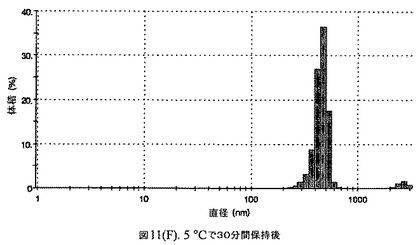
【図11G】
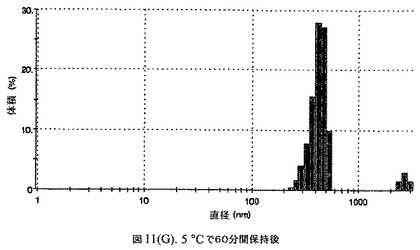
【図11H】
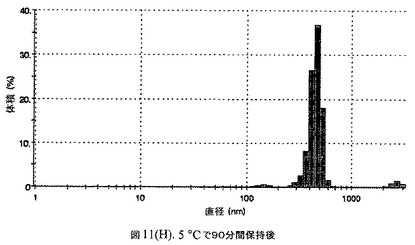
【図11I】
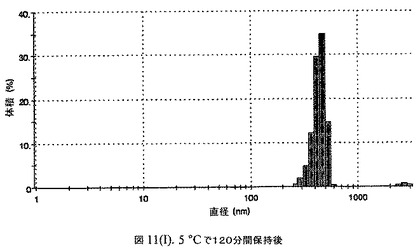
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図8A】
【図8B】
【図8C】
【図8D】
【図8E】
【図8F】
【図8G】
【図8H】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図9F】
【図9G】
【図9H】
【図10A】
【図10B】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図11I】
【公表番号】特表2010−505748(P2010−505748A)
【公表日】平成22年2月25日(2010.2.25)
【国際特許分類】
【出願番号】特願2009−521797(P2009−521797)
【出願日】平成19年7月24日(2007.7.24)
【国際出願番号】PCT/US2007/016599
【国際公開番号】WO2008/013785
【国際公開日】平成20年1月31日(2008.1.31)
【出願人】(509022587)シング−ブルーマー アンド カンパニー, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成22年2月25日(2010.2.25)
【国際特許分類】
【出願日】平成19年7月24日(2007.7.24)
【国際出願番号】PCT/US2007/016599
【国際公開番号】WO2008/013785
【国際公開日】平成20年1月31日(2008.1.31)
【出願人】(509022587)シング−ブルーマー アンド カンパニー, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]