
オピオイド作動薬/拮抗薬の併用
【課題】市販されている従来の投薬剤形よりも経口投与によって乱用の可能性が低くなるオピオイド鎮痛薬の経口投薬剤形の提供。
【解決手段】経口的に鎮痛薬として有効な量のオピオイド作動薬と経口的に活性なオピオイド拮抗薬との組合せを含有する経口投薬剤形に関し、該オピオイド拮抗薬は、該組合せを経口投与するときには鎮痛薬として有効であるが、身体依存性被験者には嫌われる組合せ製剤を提供するような、オピオイド作動薬に対する比率で配合される。好ましくは、前記組合せ製剤中に配合されるオピオイド拮抗薬の量は、身体依存性麻薬中毒者に少なくとも穏やかに拒否的な「嫌悪」の経験(例えば、性急型禁断症状)を与えるものである。
【解決手段】経口的に鎮痛薬として有効な量のオピオイド作動薬と経口的に活性なオピオイド拮抗薬との組合せを含有する経口投薬剤形に関し、該オピオイド拮抗薬は、該組合せを経口投与するときには鎮痛薬として有効であるが、身体依存性被験者には嫌われる組合せ製剤を提供するような、オピオイド作動薬に対する比率で配合される。好ましくは、前記組合せ製剤中に配合されるオピオイド拮抗薬の量は、身体依存性麻薬中毒者に少なくとも穏やかに拒否的な「嫌悪」の経験(例えば、性急型禁断症状)を与えるものである。
【発明の詳細な説明】
【技術分野】
【0001】
オピオイドは、オピオイド作動薬としても知られているもので、アヘンやモルヒネのよ
うな性質を示す薬物の1グループである。オピオイドは主に中程度ないし強力な鎮痛薬と
して使用されるが、他にも多くの薬理作用を有しており、例えば、睡眠作用、呼吸抑制作
用、気分の変化、意識の低下を起こさない精神混濁などを示す。オピオイドは脳や他の組
織にある立体特異的で飽和性の結合部位と相互作用して、作動薬として機能する。内在性
のオピオイド様ペプチドが特に中枢神経系の領域に存在しており、痛みの認識;運動、気
分および行動;神経内分泌学的機能の調節に関係していると推測される。アヘンは20種類
を上回る種々のアルカロイドを含有する。モルヒネ、コデインおよびパパベリンはこのグ
ループに属している。
【背景技術】
【0002】
19世紀の中頃までに、粗製のアヘン製剤ではなく純粋なアルカロイド(例:モルヒネ)
の使用が医療分野全体に広まり始めた。モルヒネの非経口使用はより重症のさまざまな強
迫的薬物使用を生じる傾向があった。オピオイドに関する耽溺性の問題から、耽溺性を生
じる可能性のない鎮痛薬の研究が促進されるようになった。1967年頃までに、モルヒネ様
薬物と、拮抗薬と、その後「混合作動薬−拮抗薬」と呼ばれたものの複雑な相互作用は、
オピオイドおよびその関連薬物の2以上のタイプの受容体の存在を仮定することで最もよ
く説明できると、研究者らにより結論づけられた。モルヒネ様作用を有する新しい完全合
成物質の出現により、「オピオイド」という用語は一般的に、オピオイド受容体の亜種の
いずれかと立体特異的に結合してアゴニスト作用を生じる全ての外因性物質の総称として
残されたものである。
【0003】
オピオイドの反復使用に伴う耐薬性と身体的依存性の発現の可能性はあらゆるオピオイ
ド薬物に特徴的な性質であり、精神的依存性(すなわち、耽溺性)を生じる可能性は、た
とえ医原性耽溺が稀であっても、オピオイドによる疼痛治療をおこなううえで重大な関心
事の一つとなっている。オピオイドの使用に関連したもう一つの重大な問題は、気晴らし
を目的として疼痛患者から(患者ではない)別人、例えば麻薬中毒者にこれらの薬物が渡
ることである。
【0004】
オピオイドの総合的な乱用可能性はただ一つの要因によって確立されるものではない。
実際、各種の要因が存在し、例えば、薬物の中止が薬物を見つけようとする行動をとらせ
るのに十分な苦痛を引き起こす種類の身体的依存性を生じる薬物の能力、他の麻薬の中止
により生じる禁断症状を抑制する能力、モルヒネや他のオピオイドにより生じる多幸感と
同様の多幸感を誘発する程度、薬物を通常の治療範囲を超えて投与したときに起こる毒性
のパターン、水溶性のような薬物の物理的性質などがある。そのような物理的性質により
、その薬物が腸管外経路によって乱用されそうであるか否かを判定することができる。
【0005】
米国では、強迫的薬物常用者を規制する努力として、強迫的薬物常用者の疼痛治療にオ
ピオイドを使用することに制限を設けることで、薬物の入手可能性を抑えることが含まれ
ている。実際、医師は、このような薬物に対して精神的依存性(すなわち、耽溺性)を発
現する傾向があると思われる人達にさえ強力なオピオイド鎮痛薬を投与するという選択に
直面する場合がしばしばある。この問題のため、乱用の可能性がない別の薬物で十分であ
る場合はこれらの患者にオピオイドを投与すべきでないこと、さらにこれらの患者には薬
物を非経口的に自己投与させないようにすべきで、一度に二三日分しか処方しないように
すべきことが勧告された。
【0006】
オピオイドの常用および依存性が生ずるには少なくとも3つの基本的なパターンが確認
されている。第1のパターンは、個人の薬物使用が医学的治療に関係して始まり、薬物が
例えば医師から供給されることによるものである。第2のパターンは、経験的または「気
晴らし」的薬物使用から始まって、使用回数が次第に増えていくというものである。第3
のパターンは、薬物使用が前記の方法のいずれかで始まるが、その後、体制化された耽溺
治療プログラムから得られるメタドンのような経口オピオイドに移るというものである。
【0007】
耐薬性とは、同じレベルの鎮痛または多幸感を達成するために、ある期間にわたってオ
ピオイドの用量を増加する必要があること、または同量の反復投与によって鎮痛、多幸感
または他のオピオイド効果の低下が観察されることを意味する。オピオイドの呼吸抑制、
鎮痛、鎮静、催吐、多幸感作用に対して顕著な耐薬性が生じることはわかっている。しか
しながら、この耐薬性が常用者または疼痛治療の必要な患者において発生する速度は使用
パターンに依存している。オピオイドを頻繁に使用する場合は、用量の増加が必要であり
うる。耐薬性はオピオイドのすべての効果に対して同等にまたは同一速度で発生するもの
ではない。呼吸抑制作用に対して高い耐性を示す使用者でさえ縮瞳や便秘症を呈し続ける
。オピオイドに対する耐性はたいてい禁断症状がなくなったときに消失する。
【0008】
身体的依存性はオピオイドの反復投与または長期間の使用により発生しうる。身体的依
存性はオピオイド使用の中止後に徐々に現れるか、または麻薬拮抗薬の投与後すぐ(例え
ば20分以内)に現れる(「性急型禁断症状」という)。依存性が確立されている薬物、そ
の使用期間および投与量に応じて、禁断症状の回数、種類、持続期間および重症度はさま
ざまに変化する。最も普通に見られる禁断症状としては、食欲不振、体重減少、瞳孔拡大
、異常な発汗と交互に起こる悪寒、腹部の痙攣、悪心、嘔吐、筋痙縮、高興奮性、流涙、
鼻漏、鳥肌、および心拍数の増加が挙げられる。典型的には、禁断症状は最後に投薬して
から24〜28時間後に発生し、3日目ごろに最大強度に達し、3週目まで軽減しない可能性
がある。
【0009】
オピオイドに対する精神的依存性(すなわち、耽溺性)は、多幸感や、例えば心理社会
経済的圧力からの逃避を達成する方向に向かう、薬物を見つけようとする行動により特徴
づけられる。中毒者は非医療目的で、また、自己危害に直面して、オピオイドを投与し続
けるであろう。
【0010】
薬理学的には、オピオイド拮抗薬は一般にオピオイド作動薬のあらゆる作用効果をブロ
ックするか逆転させるものである。オピオイド拮抗薬の一つの用途は、オピオイドを中毒
者に投与した際に現れる可能性のある多幸感作用をブロックするために、ナルトレキソン
(naltrexone)で1日1回処置するものである。低用量のオピオイド拮抗薬は、個体がオピ
オイドに対して身体的依存性を示すか否かを判定するために使用されている。最も一般的
には、オピオイド作動薬を過剰投与された個体に対してオピオイド効果を逆転させるため
にオピオイド拮抗薬が使用される。
【0011】
以前に当技術分野では、オピオイド鎮痛薬に伴う乱用性を防止しようとする試みがなさ
れた。典型的には、一定量のオピオイド鎮痛薬は、同量を経口投与したときと比べて非経
口投与したときの方が効力が強い。したがって、流行している経口投薬の乱用様式の一つ
は、その剤形からオピオイドを抽出し、その後「恍惚感」を得るために(「適当な」注射
用ビヒクルを用いて)オピオイドを注射するというものである。したがって、乱用を抑え
る試みは次のことに集中していた。すなわち、経口的には活性でないが、ある人がオピオ
イドを溶解して非経口的にそれを投与しようとする場合にオピオイドの鎮痛効果を実質的
にブロックしうるオピオイド拮抗薬を経口投薬剤形中に配合することである。
【0012】
例えば、Sanofi-WinthropからTalwin(登録商標)Nxとして市販されて米国で入手できる
錠剤には、ペンタゾシンとナロキソンの組合せが利用されている。Talwin(登録商標)Nxに
は50mg塩基に等しい塩酸ペンタゾシンおよび0.5mg塩基に等しい塩酸ナロキソンが含まれ
ている。Talwin(登録商標)Nxは中程度から重症の疼痛の緩和に効果があると表示されてい
る。この組合せ物中に存在するナロキソンの量は経口摂取したときには何の作用も示さず
、ペンタゾシンの薬理作用を妨害しない。ところが、この量のナロキソンを注射により投
与すると、麻薬性鎮痛薬に対する強い拮抗作用が認められた。こうして、ナロキソンの配
合は、経口投薬剤形を可溶化して注射したときに起こる経口ペンタゾシンの不正な使用形
態を制限することを意図したものである。したがって、この投薬剤形は以前の経口ペンタ
ゾシン製剤よりも不正な非経口使用の可能性が少ない。しかしながら、それは依然として
経口による患者の不正な使用および乱用を受けやすく、例えば、患者が一度に複数回分の
用量を摂取する場合である。
【0013】
Sunshineらは、Clin. J. Pain, 1988, 4:35-40(「経口投与後のペンタゾシン対ペンタ
ゾシン−ナロキソン併用の鎮痛効果」)において、ペンタゾシン50mgの鎮痛効果に及ぼす
0.5mgナロキソンの添加の影響を報告した。この併用は、疼痛強度差の合計(SPID)に関し
て、また、4時間目の緩和および疼痛強度差(PID)に関してペンタゾシンより著しく効果
のないことが見出された。中程度の基線疼痛を有する患者の場合、この併用はSPIDに関し
てと3および4時間目の緩和およびPIDに関してペンタゾシンより疼痛緩和が有意に低か
った。重度の基線疼痛を有する患者においては、ペンタゾシンとペンタゾシン−ナロキソ
ン併用との間に有意差はなかった。
【0014】
Wangらは、J. Clin. Pharmacol., 1981, 21:162-8(「経口鎮痛薬の交差および平行研
究」)において、慢性疼痛患者の交差研究でのパーコダン(Percodan:登録商標)(オキシコ
ドンHCl 4.5mg、オキシコドンテレフタレート0.28mg、アスピリン224mg、フェナセチン16
0mgおよびカフェイン32mgを含む)単独およびプラシーボと比較したときのナロキソン0.25
mgとパーコダンとの併用を報告した。この併用は試験の後半時間では鎮痛毎時パラメータ
ーのほとんどに関してパーコダン単独よりも低い平均スコアを有していた。しかし、概略
変数に関しては、この併用はプラシーボまたはパーコダン単独のいずれとも有意差を示さ
なかった。
【0015】
疼痛治療のためにブプレノルフィンとナロキソンを一定割合で組合せたもの(Temgesic(
登録商標)Nx, Reckitt & Colman)がニュージーランドで1991年に導入された。
【0016】
ドイツでは重度の疼痛の管理のため1978年以来チリジン(50mg)とナロキソン(4mg)から
なる一定の併用療法が利用可能になっている(Valoron(登録商標)N, Goedecke)。これらの
薬物を併用することの論理的根拠は、有効な疼痛緩和と、モルヒネ受容体でのナロキソン
誘導拮抗作用によるチリジン中毒の予防にある。
【0017】
米国特許第3,773,955号(Pachterら)には、非経口投与時には鎮痛、多幸感、または身体
的依存性を起こさず、それにより鎮痛薬の非経口乱用を防止できる、経口的に有効な鎮痛
組成物が記載されている。この種の組成物は鎮痛薬の経口量につき約0.1〜10mgのナロキ
ソンを含んでいた。この文献はオピオイドの経口乱用に関心をもっていなかった。
【0018】
米国特許第3,493,657号(Lewensteinら)には、ナロキソンとモルヒネまたはオキシモル
フォンを含む組成物が記載されており、この組成物は幻覚のような副作用を発生させずに
強力な鎮痛効果を発揮するとされている。
【0019】
米国特許第4,457,933号(Gordonら)には、鎮痛量のオピオイドを比較的狭い特定の範囲
でナロキソンと組み合わせることにより、オキシコドン、プロポキシフェンおよびペンタ
ゾシンなどの強力な鎮痛薬の経口および非経口の両方の乱用可能性を減少させる方法が記
載されている。2.5〜5:1(重量部)の比を有するオキシコドン−ナロキソン組成物および16
〜50:1(重量部)の比を有するペンタゾシン−ナロキソン組成物が好ましいものであった。
オピオイドと併用されるべき用量のナロキソンは、オピオイドの経口鎮痛活性に実質的に
影響を及ぼさずに、オピオイドの経口または非経口乱用の可能性を実質的に排除すると述
べられている。
【0020】
米国特許第4,582,835号(Lewis)には、舌下投与に有効な量のブプレノルフィンをナロキ
ソンと共に投与することによる疼痛の治療方法が記載されている。Lewisによれば、ナロ
キソンとブプレノルフィンの投与量比は、非経口投与の場合が1:3〜1:1、舌下投与の場合
が1:2〜2:1である。
【0021】
当分野においては、経口オピオイド製剤は、非経口投与のみならず、患者や中毒者が投
薬間隔の間に処方量より多い経口量を経口的に自己投与する場合には経口によっても乱用
されることが次第に認識されてきた。したがって、経口投与可能で、経口乱用の可能性が
低い疼痛治療用の製剤を開発する必要がある。
【0022】
本発明らの知るかぎりでは、経口的に併用した場合に鎮痛薬として有効であるが、身体
依存性被験者には忌み嫌われる、ある比率のオピオイド作動薬とオピオイド拮抗薬はこれ
まで認識されていない。
【発明の開示】
【発明が解決しようとする課題】
【0023】
本発明の目的および概要 本発明の目的は、市販されている従来の投薬剤形よりも経口
投与によって乱用の可能性が低くなるオピオイド鎮痛薬の経口投薬剤形を提供することで
ある。
【0024】
本発明の更なる目的は、治療的痛覚消失を付与し、さらに身体依存性被験者が多量(例
えば、通常の処方量の2〜3倍ほど)のオピオイドを摂取したり投与されたときには拒否
的な「嫌悪」体験を与える方法ならびにオピオイド鎮痛薬の経口投薬剤形を提供すること
である。
【0025】
本発明の更なる目的は、通常の処方量より多い量(例えば、通常の処方量の約2〜3倍
)のオピオイドを摂取する非身体依存性被験者において、拮抗薬不在下での同量のオピオ
イドと比べたときほど正の強化(positively reinforcing)を与えないやり方で治療的痛覚
消失を付与する方法ならびにオピオイド鎮痛薬の経口投薬剤形を提供することである。
【0026】
本発明の更なる目的は、オピオイド鎮痛薬の経口投薬剤形を用いて、該投薬剤形の経口
乱用可能性を低下させながらヒト患者の疼痛を治療する方法を提供することである。
【0027】
本発明の更なる目的は、経口乱用可能性が低くなるようなオピオイド鎮痛薬の経口投薬
剤形を製造する方法を提供することである。
【課題を解決するための手段】
【0028】
前記および他の目的は本発明によって達成される。本発明は、一部には、経口的に併用
した場合に鎮痛薬として有効であるが、身体依存性被験者には嫌われる、オピオイド拮抗
薬とオピオイド作動薬(鎮痛薬)のある比率が存在する、という驚くべき知見に基づいて
いる。本発明者らの知るかぎりでは、このことは当業者、例えば、麻薬中毒学者、鎮痛学
者、臨床薬理学者によってさえ考慮されたことがない。ある組合せ製剤(拮抗薬/作動薬
の組合せ)が、ある集団(疼痛患者)には本質的に治療的であるのに、通常の処方量と同
量またはそれより多量(例えば、通常の処方量の約2〜3倍)のオピオイドを投与した場
合に別の集団(例えば、身体依存性被験者)には受け入れられない(嫌われる)というこ
とは驚くべきことである。
【0029】
本発明は、一部には、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者
における直接測定により、またはヒト被験者におけるオピオイド効力(痛覚消失)の1以
上の代理指標の使用により評価したとき、痛覚消失をやや低下させうる比率で、経口的に
鎮痛有効量のオピオイド作動薬とオピオイド拮抗薬を含有する経口投薬剤形に関する。オ
ピオイド効力(痛覚消失)の代理指標としては、鎮静、呼吸速度および/または瞳孔の大
きさ(瞳孔測定による)、および「薬効」のための視覚アナログスケール(visual analog
ue scale: VAS)が挙げられる。このような代理指標は、共存量のオピオイド拮抗薬を用い
ないでオピオイドを同量使用したときに比べて、オピオイド効果の低下を示す方向での影
響を受ける。
【0030】
オピオイドがヒドロコドンで、拮抗薬がナルトレキソンである好ましい実施形態におい
ては、経口投薬剤形が二酒石酸塩の形のヒドロコドンと塩酸塩の形のナルトレキソンを含
む。
【0031】
オピオイドがヒドロコドンで、拮抗薬がナルトレキソンである好ましい実施形態におい
ては、ナルトレキソン対ヒドロコドンの重量比を約0.03〜0.27:1とすることが好ましく、
約0.05〜0.20:1とすることがより好ましい。
【0032】
本発明は、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者における直
接測定により、またはヒト被験者におけるオピオイド効果の1以上の代理指標の使用によ
り評価したとき、痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピオイド
作動薬とオピオイド拮抗薬を含有する経口投薬剤形を調製することを含んでなる、被験者
による経口オピオイド製剤の経口乱用を防止する方法に関する。前記経口投薬剤形が身体
依存性被験者によって比較的多量に(例えば、通常の処方量の約2〜3倍)摂取される場
合には、その使用は身体依存性被験者に嫌われ、かつ非身体依存性ヒト被験者においては
(単独で摂取された)オピオイドほどに正の強化を与えないことが好ましい。
【0033】
本発明はまた、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者におけ
る直接測定によって、またはヒト被験者におけるオピオイド効果の1以上の代理指標の使
用によって痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピオイド作動薬
をオピオイド拮抗薬と共に経口投与することを含んでなる治療方法に関する。
【0034】
本発明はさらに、一部には、経口的に鎮痛有効量のオピオイド作動薬と経口的に活性な
オピオイド拮抗薬の組合せを含む経口投薬剤形に関し、オピオイド拮抗薬は、(i)経口投
与した際に該投薬剤形から引き出される痛覚消失のレベルを非治療レベルにまで低減させ
ない量で、かつ(ii)身体依存性被験者が通常の処方量の少なくとも2倍(しばしば処方量
の2〜3倍またはそれ以上)の量を一度に摂取しようとするとき、オピオイド拮抗薬の存
在しない同量のオピオイドに比べて、身体依存性被験者に少なくとも穏やかに拒否的な「
嫌悪」の経験(例えば、性急型禁断症状)を与える量で含まれる。好ましくは、前記経口
投薬剤形に含まれるナルトレキソンの量は、非身体依存性オピオイド中毒者に、拮抗薬の
存在しない同様の経口投薬剤形よりも少ない正の強化(例えば、より少ない「嗜好」)を
与える量である。好ましくは、前記製剤は経口投与したときに効果的な痛覚消失をもたら
すものである。
【0035】
本発明にとって、「患者における直接測定により、またはヒト被験者におけるオピオイ
ド鎮痛効力の1以上の代理指標の使用により評価したとき、痛覚消失をやや低下させうる
」という表現は、疼痛患者が、本発明に従って投与された製剤(すなわち、オピオイド作
動薬/拮抗薬の組合せ)と、同量のオピオイド作動薬を含むがオピオイド拮抗薬を含まな
い同様の製剤と、の間の差異に気づくかまたは容易には気づかないが、その組合せから鎮
痛効果を得ていることを意味する。本発明に従って投与される製剤の薬理学的効果(痛覚
消失)は、例えば、該投薬剤形を投与してから連続した時間後に患者によって報告された
鎮痛薬アンケートからのスコアによって説明することができる。痛覚消失の概略指標とし
ては、疼痛強度差の合計(SPID)および全疼痛緩和(TOTPAR)が含まれる。
【0036】
好ましい実施形態において、前記投薬剤形に含まれるオピオイド拮抗薬の量は、「薬効
」のための視覚アナログスケール(VAS)のような代理指標によって測定したとき、経口投
与した際に該投薬剤形から引き出される痛覚消失のレベルを臨床上有意に低下させうる量
である。他の実施形態において、経口投薬剤形に含まれるオピオイド拮抗薬の量は、経口
投与した際に該投薬剤形から引き出される痛覚消失のレベルをそれとわかるほどに低下さ
せうるが、痛覚消失のレベルを治療レベル以下にまで低下させないものである。
【0037】
好ましくは、経口投薬剤形に含まれる拮抗薬の量は、非身体依存性オピオイド被験者に
、拮抗薬を含まない同様の経口投薬剤形よりも少ない正の強化(例えば、より少ない「嗜
好」)を与えるものである。
【0038】
本発明はまた、ヒト患者の疼痛の治療を意図したオピオイド鎮痛薬の経口投薬剤形の調
製方法に関する。この方法は、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが
、患者における直接測定によりまたはヒト被験者における痛覚消失の1以上の代理指標の
使用により評価したとき痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピ
オイド作動薬をオピオイド拮抗薬と組み合わせて、経口投薬剤形の経口乱用の可能性を最
小限にする様式で行われる。特定の実施形態において、この組合せは、経口投与した場合
に、(同量のオピオイド単独と比べて)該投薬剤形から引き出される痛覚消失のレベルの
臨床的に有意な低下をもたらし、また、身体依存性被験者が通常の処方量つまり常用量よ
り多いオピオイドを摂取するときには該被験者に少なくとも穏やかに拒否的な「嫌悪」の
経験(例えば、性急型禁断症状)を与える。この被験者は例えば通常の処方量より多量(
例えば、少なくとも2〜3倍)を一度に摂取することによって多幸感(「恍惚感」)を得
ようとする麻薬中毒者でありうる。前記投薬剤形に含まれる量のオピオイド拮抗薬は、薬
効のための視覚アナログスケール(VAS)のような薬理学的パラメーターによって測定した
とき、経口投与時に該投薬剤形から引き出される痛覚消失のレベルを低下させうるか、ま
たはそれとわかるほどに低下させうるが、それにもかかわらず効果的な痛覚消失を該投薬
剤形に与えることが好ましい。前記方法の好ましい実施形態において、オピオイド拮抗薬
の用量はオピオイドの鎮痛効果の代理指標に認めうる程度に影響を与える量である。好ま
しい実施形態において、経口投薬剤形に含まれる量の拮抗薬は、非身体依存性オピオイド
被験者に、拮抗薬を含まない同様の経口投薬剤形よりも少ない正の強化(例えば、より少
ない「嗜好」)を与えるものである。
【0039】
ここに記載する本発明の薬物組合せを含む経口医薬組成物は錠剤、液剤、トローチ剤、
ロゼンジ剤、水性または油性懸濁剤、分散性の粉末、顆粒、マトリックス球体または被覆
不活性ビーズを含む多粒子製剤、乳濁剤、硬質または軟質カプセル剤、シロップ剤または
エリキシル剤、微粒子(例:マイクロカプセル、マイクロスフェアなど)、バッカル錠剤
などでありうる。本発明の投薬剤形は当業者に公知のどのような望ましい製薬上許容され
る賦形剤を含んでいてもよい。前記投薬剤形はさらに、オピオイド作動薬とオピオイド拮
抗薬の即時放出を提供することができる。好ましい実施形態では、前記投薬剤形がオピオ
イド作動薬の持続放出を提供し、かつオピオイド拮抗薬の用量の一部または全部を(i)即
時放出形態、(ii)持続放出形態、または(iii)即時放出および持続放出の両形態で提供す
る。かかる実施形態はさらにオピオイド作動薬の一部を即時放出形態で含んでいてもよい
。持続放出は医薬製剤の分野の当業者に知られた製剤化/製造法に従って、例えば、オピ
オイド作動薬とオピオイド拮抗薬を含むマトリックス中に持続放出性担体を配合すること
により、またはオピオイド作動薬とオピオイド拮抗薬を含むマトリックスに持続放出性コ
ーティングを施すことによって行うことができる。
【0040】
本発明はより安全な製剤(例えば、より少ない呼吸抑制)ならびにオピオイド耐性およ
び身体依存性発現の速度が比較的遅い製剤を提供する。
【0041】
他の好ましい実施形態において、前記投薬剤形に含まれるオピオイドは、経口的に活性
な、ヒドロコドンと異なるオピオイド作動薬である。このような製剤に含まれるナルトレ
キソンの比率は、ヒドロコドンと比較したときの各種オピオイド鎮痛薬の公知の等価鎮痛
薬用量を考慮に入れて、単純な計算から容易に決定することができる。各種オピオイド鎮
痛薬の等価鎮痛薬用量は以下に提示されるか、さもなくば例えばFoley, K., N. Engl. J.
Med., 1985, 313:84-95「癌による疼痛の治療」(参照により本明細書に含めるものとす
る)から当業者には公知である。この実施形態の別の面では、ナルトレキソンの代わりに
別のオピオイド拮抗薬が、その等価拮抗薬用量で用いられる。
【0042】
特定の実施形態においては、前記製剤中に2種類のオピオイド鎮痛薬の組合せを含める
。別の実施形態では、1種類以上のオピオイド鎮痛薬を含め、さらにオピオイド拮抗薬に
加えて別の非オピオイド薬物を含める。このような非オピオイド薬物は好ましくは更なる
痛覚消失を与えるものであり、例えば、アスピリン、アセトアミノフェン、非ステロイド
系抗炎症薬(NSAIDS)、NMDA拮抗薬、およびサイコオキシゲナーゼ-II阻害剤(COX-II阻害剤
)などである。更なる実施形態において、痛覚消失以外の所望の効果を与える非オピオイ
ド薬物、例えば、鎮咳薬、去痰薬、うっ血除去剤、抗ヒスタミン剤などを含めてもよい。
【0043】
本明細書中で用いる「非経口」とは、皮下注射、静脈内、筋肉内、胸骨内への注入また
は輸液の技術を含む。
【0044】
「効果的な痛覚消失」とは、本発明においては、ヒト患者で測定したときの、許容レベ
ルの副作用と共に、疼痛の満足のゆく減少または消失として定義される。
【0045】
「持続放出」とは、本発明においては、血液(例:血漿)濃度(レベル)が、1日2回
または1日1回製剤を表示する時間にわたって、治療範囲内(最少有効鎮痛薬濃度つまり
「MEAC」以上)に、しかし毒性レベル以下に維持されるような速度での経口製剤からの薬
物(オピオイド鎮痛薬)の放出として定義される。
【0046】
「定常状態」とは、薬物の除去速度が体内への該薬物の吸収速度と同じになる時期をさ
す。
【0047】
本発明において、「オピオイド作動薬」は「オピオイド」または「オピオイド鎮痛薬」
と相互交換可能であり、オピオイドの塩基、混合作動薬−拮抗薬、部分的作動薬、その製
薬上許容される塩、その立体異性体、そのエーテルおよびエステル、それらの混合物を含
むものである。
【発明を実施するための最良の形態】
【0048】
発明の詳細な説明 オピオイド受容体にはμ、κ、およびδと呼ばれる少なくとも3つの
亜型が存在するとみなされてきた。この枠内で、μ受容体は、上部脊椎麻酔(superspinal
analgesia)、呼吸抑制、多幸感、および身体依存性の生成に関与すると考えられる。κ
受容体は、脊椎麻酔、縮瞳、および鎮静の誘発に関与すると考えられる。γ受容体を活性
化すると、不快気分および幻覚、更に呼吸促進作用および血管中枢興奮作用が誘発される
。Lord, et al. Nature, 1977, 267, 495-99には、μ受容体と異なるγと呼ばれる受容体
がマウス精管中に存在すると記載されている。オピオイド作動薬は主にμ受容体でその作
動薬作用を示し、それより程度は少ないがκ受容体でもそうした作用を示すと考えられる
。1つのまたはもう1つの受容体タイプで半作動薬として作用するようにみえる薬剤が数
種存在する。このような薬剤としては、ナロルフィン、プロピラム、およびブプレノルフ
ィンが挙げられる。更に他の薬剤は、μ受容体で競合的拮抗薬として機能し、κおよびω
受容体で作動薬作用を示すことによりモルヒネ様薬剤の作用をブロックする。そのような
作用機構を記述すべく用語「作動薬-拮抗薬」が使用されるようになった。オピオイドの
働きに対する拮抗作用の考え方は複雑であると思われる。
【0049】
オピオイド作動薬-拮抗薬および半作動薬を投与した場合、薬剤の作動薬作用に対する
耐性は現れるが、薬剤の拮抗薬作用に対する耐性は現れないことが判明した。長期間にわ
たり高用量の投与を行った後でさえも、ナロキソンの投与中止は、いかなる識別可能な禁
断症状によっても特性付けられない。また、もう1つの比較的純粋なオピオイド拮抗薬で
あるナルトレキソンの投与を停止した場合、非常にわずかな徴候および症状が現れる。し
かしながら、長期間にわたり高用量の投与を行った後、オピオイド作動薬-拮抗薬のナロ
ルフィンまたはシクラゾシンの投与を急激に停止すると、両方の薬剤で類似した特徴的な
禁断症状が現れる。
【0050】
ナロキソンは、作動薬作用をほとんど示さないオピオイド拮抗薬である。12mgまでの用
量でナロキソンを皮下投与した場合、識別可能な本質的な影響は現れない。また、24mgの
ナロキソンの場合、わずかな眠気が誘発されるにすぎない。ヒトにおいて小用量(0.4-0.8
mg)のナロキソンを筋肉内投与または静脈内投与した場合、モルヒネ様オピオイド作動薬
の作用が阻害されるかまたは急速に逆方向に向かう。1mgのナロキソンを静脈内投与する
と25mgのヘロインの作用が完全にブロックされると報告されている。ナロキソンの作用は
、ほぼ静脈内投与の直後に現れる。この薬剤は経口投与の後で吸収されるが、肝臓を1回
通ると代謝されて急速に不活性な形態になると報告されている。その結果、その作用は非
経口投与を行った場合の1/15にすぎないと報告されている。1gよりも多く経口投与し
た場合、24時間未満でほとんど完全に代謝されると報告されている。
【0051】
他のオピオイド拮抗薬、例えば、いずれも窒素上にシクロプロピルメチル基が置換した
シクラゾシンおよびナルトレキソンは、経口投与の場合、それらの効力の大部分を保持し
、また、その作用の持続時間はかなり長く、経口投与後24時間に達する。最も好ましい
オピオイド拮抗薬は、ナルトレキソンである。しかしながら、他のオピオイド拮抗薬、例
えば、限定されるものではないが、ナロキソン、ナルメフェン、シクラゾシン、およびレ
バロルファンを同等な拮抗薬として経口投与することも、本発明に従って利用可能である
。このような他の拮抗薬対特定のオピオイド作動薬の比は、ナルトレキソン以外の様々な
オピオイド拮抗薬の利用を望む当業者には、過度の実験を行うことなく容易に決定可能で
ある。オピオイド作動薬に対するそれらの比について、本明細書中で具体的に示し、詳細
に説明する。当業者は、例えば、本明細書中に記載のものと同じかまたは類似の臨床研究
を行うことによって、こうした他の拮抗薬対オピオイド作動薬の比を決定してもよい。こ
の場合、例えば、本明細書中に記載のナルトレキソン対ヒドロコドンの比と同等な比で経
口投与されるオピオイド拮抗薬/オピオイド作動薬の組合せ製剤は、本発明の範囲および
特許請求の範囲に含まれるものとみなされる。例えば、本発明の特定の実施形態では、ナ
ロキソンをオピオイド拮抗薬として利用し、投薬剤形中に含まれるナロキソンの量は、こ
の組合せ製剤中にナルトレキソンが含まれる場合と同等な拮抗薬作用を呈するのに十分な
量とする。
【0052】
既にオピオイド常用癖のある患者を治療する場合、オピオイド作動薬の多幸化作用を防
止するために、ナルトレキソンが高用量(100mgを超える量)で経口投与されてきた。ナル
トレキソンはδ部位よりもμ部位に対してかなり優先的なブロッキング作用を示すことが
報告されている。ナルトレキソンは、オピオイド作動薬の性質をもたないオキシモルホン
の合成同族体として知られており、オキシモルホンの窒素原子上に位置するメチル基がシ
クロプロピルメチル基で置換されている点がオキシモルホンと構造上異なっている。ナル
トレキソンの塩酸塩は、約100mg/ccまで水に可溶である。ナルトレキソンの薬理学的およ
び薬物動態学的性質は、多数の動物および臨床実験で評価された。例えば、参照により本
明細書に組み入れるGonzalez JP, et al. Naltrexone: A review of its Pharmacodynami
c and Pharmacokinetic Properties and Therapeutic Effficacy in the Management of
Opioid Dependence. Drugs 1988; 35:192-213を参照されたい。経口投与後、ナルトレキ
ソンは急速に吸収され(1時間以内)、その経口投与時の生物学的利用率は5〜40%の範囲で
ある。ナルトレキソンのタンパク質結合率は約21%であり、1回投与後の分布容積は16.1 L
/kgである。
【0053】
ナルトレキソンは、アルコール依存症を治療するために、また外因的に投与されたオピ
オイドをブロックするために、錠剤の形態で市販されている(Revia(登録商標)、DuPont)
。例えば、Revia(塩酸ナルトレキソン錠剤). Physician's Desk Reference 51st ed., Mo
ntvale, NJ. "Medical Economics" 1997; 51:957-959を参照されたい。50mgのReVia(登録
商標)を投与すると、25mgのヘロインを静脈内に投与したときの薬理学的作用が24時間後
までブロックされる。
【0054】
モルヒネ、ヘロイン、または他のオピオイドとの同時投与を連用した場合、ナルトレキ
ソンはオピオイドに対する身体依存性の発生をブロックすることが知られている。ナルト
レキソンによりヘロインの作用をブロックする方法はオピオイド受容体での競合的な結合
に基づくものと考えられている。ナルトレキソンは、オピオイドの作用を完全にブロック
することによって麻薬中毒を治療すべく使用されてきた。麻薬中毒に対してナルトレキソ
ンが最もうまく使用できるのは、予後の良好な麻薬常用者を対象にして、行動コントロー
ルまたは他の指示遵守性向上法(compliance enhancing method)を含む総合的な作業また
はリハビリプログラムの一部分として使用する場合である。ナルトレキソンを用いて麻薬
依存症を治療する場合、少なくとも7〜10日間にわたり患者にオピオイドを与えないこと
が好ましい。このような目的では、ナルトレキソンの初期用量は、典型的には約25mgであ
り、禁断症状が現れないときは1日あたり50mgに増量してもよい。1日あたりの用量を50mg
にすれば、非経口的に投与されたオピオイドの作用は臨床的に適切にブロックされると考
えられる。また、ナルトレキソンは、社会的および心理療法的手法の補助としてアルコー
ル中毒の治療に使用されてきた。
【0055】
本発明の投薬剤形および方法では、含まれるナルトレキソンの量は、これまでの市販品
で利用されてきた投与量よりもかなり少ない。この原因の一部分として、本発明における
ナルトレキソンの使用法が異なっている点が挙げられる。すなわち、本発明の目標は、オ
ピオイドの作用をブロックすることではなく、身体依存性被験者が大量の組合せ製剤、例
えば、通常処方される用量の約2〜3倍の組合せ製剤を摂取した場合または投与された場合
に否定的な「嫌悪」体験が得られるようにすることである。
【0056】
従って、例えば、オピオイドが酒石酸水素ヒドロコドン15mgである本発明の製剤の場合
、製剤中に含まれる塩酸ナルトレキソンの量は、ヒドロコドン15mgあたりナルトレキソン
約0.5mg〜約4mg、好ましくは約0.75mg〜約3mgである。
【0057】
本発明に有用なオピオイド鎮痛薬としては、すべてのオピオイド作動薬または作動薬-
拮抗薬混合体、半作動薬、例えば、限定されるものではないが、アルフェンタニル、アリ
ルプロジン、アルファプロジン、アニレリジン、ベルジルモルヒネ、ベジトラミド、ブプ
レノルフィン、ブトルファノール、クロニタゼン、コデイン、デソモルヒネ、デキストロ
モラミド、デゾシン、ジアムプロミド、ジアモルホン、ジヒドロコデイン、ジヒドロモル
ヒネ、ジメノキサドール、ジメフェプタノール、ジメチルチアムブテン、ジオキサフェチ
ルブチレート、ジピパノン、エプタゾシン、エトヘプタジン、エチルメチルチアムブテン
、エチルモルヒネ、エトニタゼン、フェンタニール、ヘロイン、ヒドロコドン、ヒドロモ
ルホン、ヒドロキシペチジン、イソメサドン、ケトベミドン、レボルファノール、レボフ
ェナシルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタゾシン、メタ
ドン、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノルレボルファノ
ール、ノルメタドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピパノン、アヘ
ン、オキシコドン、オキシモルホン、パパベレツム、ペンタゾシン、フェナドキソン、フ
ェノモルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミド、プロフェ
プタジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニル、チリジン
、トラマドール、それらの任意の組合せ、それらの任意の塩などが挙げられる。
【0058】
特定の好ましい実施形態では、オピオイド作動薬または鎮痛薬は、ヒドロコドン、モル
ヒネ、ヒドロモルホン、オキシコドン、コデイン、レボルファノール、メペリジン、メタ
ドン、もしくはそれらの塩、またはそれの混合物からなる群より選ばれる。特定の好まし
い実施形態では、オピオイド作動薬はヒドロコドンである。ヒドロコドンの用量15mgと比
較して、これらのオピオイドで同等な鎮痛作用の得られる用量が以下の表1に示されてい
る。
【0059】
【表1】

@0012
【0060】
ナルトレキソンの好ましい比がヒドロコドン15mgあたり約0.5〜約4mgの量であることに
基づいて、各オピオイド1mgに対するナルトレキソンのおよその比が表2に示されている。
【0061】
【表2】
@0013
【0062】
ナルトレキソンのより好ましい比がヒドロコドン15mgあたりナルトレキソン約0.75mg〜
約3mgであることに基づいて、各オピオイド1mgに対するナルトレキソンのおよその比が表
3に示されている。
【0063】
【表3】
@0014
【0064】
ヒドロコドンは疼痛に対処するのに有効であるが、心理的にオピオイドに依存している
者または治療以外の理由でオピオイドを誤用している者によるオピオイドの乱用が増大し
ている。既に他のオピオイドを経験している場合、オピオイドを麻薬拮抗薬と組み合わせ
て投与すると、特に、以前常用者であった患者では、乱用の可能性が減少することが示さ
れた。Weinhold LL, et al. Bpurenorphine Alone and in Combination with Naltrexone
in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30:263-274; Mendelso
n J., et. al., Buprenorphine and Naloxone Interactions in Opiate-Dependent Volun
teers, Clin Pharm Ther 1996; 60:105-114。いずれも参照により本明細書に組み入れる
。
【0065】
ヒドロコドンは、多面的に中枢神経系および胃腸に作用する半合成麻薬性鎮痛薬および
鎮咳薬である。化学的には、ヒドロコドンは、4、5-エポキシ-3-メトキシ-17-メチルモル
フィナン-6-オンであり、ジヒドロコデイノンとしても知られている。他のオピオイドと
同様に、ヒドロコドンは習慣性になることもあり、モルヒネタイプの薬物依存性を生じる
可能性がある。他のアヘン誘導体と同様に、ヒドロコドンは、過剰に投与した場合、呼吸
を抑制する。
【0066】
また、経口用ヒドロコドンは、鎮咳薬としてヨーロッパ(ベルギー、ドイツ、ギリシア
、イタリア、ルクセンブルク、ノルウェー、およびスイス)で利用可能である。また、ド
イツでは、鎮咳薬として非経口製剤が利用可能である。米国では、鎮痛薬としての使用の
ために、中程度または多少重度の疼痛を和らげるための非アヘン製剤(すなわち、イブプ
ロフェン、アセトアミノフェン、アスピリンなど)との一定の組合せ製剤としてのみ、酒
石酸水素ヒドロコドンが市販されている。
【0067】
通常のヒドロコドン投薬剤形では、アセトアミノフェンが併用され、例えば、米国では
UCB Pharma, Inc.からLortab (登録商標)として、2.5/500mg、5/500mg、7.5/500mg、およ
び10/500mgのヒドロコドン/アセトアミノフェン錠剤が市販されている。酒石酸水素ヒド
ロコドン7.5mg+アセトアミノフェン650mgおよび酒石酸水素ヒドロコドン7.5mg+アセト
アミノフェン750mgの割合の錠剤としても利用可能である。疼痛を軽減するために必要な
場合、ヒドロコドンをアスピリンと組み合わせ、成人に対して一般に4〜6時間ごとに1〜2
個の錠剤が経口投薬剤形として投与される。錠剤の形態の場合、酒石酸水素ヒドロコドン
5mgおよびアスピリン224mg更にカフェイン32mgが含まれるか、または、酒石酸水素ヒドロ
コドン5mgおよびアスピリン500mgが含まれる。比較的新しい製剤には、酒石酸水素ヒドロ
コドンおよびイブプロフェンが含まれる。米国でKnoll Laboratoriesから市販されている
Vicoprofen(登録商標)は、酒石酸水素ヒドロコドン7.5mgとイブプロフェン200mgを含む錠
剤である。本発明には、このような製剤すべてが含まれるとともに、本明細書中に記載さ
れている本発明の用量範囲内の経口活性オピオイド拮抗薬も含まれるものとする。
【0068】
ヒドロコドンなどのオピオイド鎮痛薬が乱用される可能性は、本発明の組合せによって
驚くほど低減される。より詳細には、単一の経口投薬剤形中でオピオイド鎮痛薬を少量の
オピオイド拮抗薬と組み合わせた場合、依然として痛覚消失を呈する製剤が得られるが、
この製剤を用いると、1回に1錠より多く、例えば、通常処方される用量の2〜3倍の用量で
投与することによって、身体依存性ヒト被験者が薬剤を乱用し続ける可能性が実質的にな
くなることを見出した。
【0069】
本発明の経口投薬剤形には、経口的に治療上有効な量のオピオイド作動薬が含まれると
ともに、ナルトレキソンなどのオピオイド拮抗薬が、次の条件、すなわち(i)経口投与時
に投薬剤形により誘発される痛覚消失のレベルが非治療レベルまで低減せず、かつ(ii)通
常処方された用量より多くの用量を1度に摂取した場合、身体依存性ヒト被験者、例えば
、身体依存性常用者(具体的には、急発禁断症候群の常用者)が少なくとも穏やかな否定的
「嫌悪」体験をするという条件を満たす量で含まれる。好ましくは、経口投薬剤形中に含
まれる拮抗薬の量は、(iii)拮抗薬の含まれない同等な経口投薬剤形を使用した場合より
も、非身体依存性ヒト被験者、例えば、オピオイド常用者による増量の要求が低減する(
例えば、「嗜好度」が減少する)量である。
【0070】
上のパラグラフに記載の要件(i)〜(iii)を達成するのに有用な拮抗薬の量は、少なくと
も部分的には、例えば、VASスケール(この場合、投薬剤形の作用に対する自覚症状を被験
者が等級化する)などの「代用」試験および/または瞳孔サイズ(瞳孔測定)などの測定を
用いて決定してもよい。こうした測定を行うことにより、当業者は、作動薬のオピエート
効果を低減させる拮抗薬の用量を作動薬の用量を基準に決定することができる。その後、
当業者は、身体依存性被験者に対して嫌悪作用を誘発するオピオイド拮抗薬のレベルおよ
び非身体依存性常用者の「嗜好度」またはオピオイド増量要求を最小限に抑えるオピオイ
ド拮抗薬のレベルを決定することができる。オピオイド拮抗薬のこれらのレベルが決まれ
ば、上のパラグラフに記載の要件(i)〜(iii)を達成するのに有用な拮抗薬のこうしたレベ
ルにまたはこうしたレベル未満に拮抗薬の用量範囲を設定することができる。
【0071】
オピオイド作動薬とオピオイド拮抗薬との組合せ製剤は、従来の賦形剤、すなわち、当
技術分野で周知であり経口投与に適した製薬上許容される有機または無機の担体物質との
混合物の形態で利用することができる。好適な製薬上許容される担体としては、水、塩類
溶液、アルコール、アラビアゴム、植物油、ベンジルアルコール、ポリエチレングリコー
ル、ゲル化剤、炭水化物、例えば、ラクトース、アミロース、またはデンプン、ステアリ
ン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、香油、脂肪酸モノグリセリドおよ
びジグリセリド、ペンタエリトリトール脂肪酸エステル、ヒドロキシメチルセルロース、
ポリビニルピロリドンなどが挙げられるが、これらに限定されるものではない。製剤は、
殺菌が可能であり、更に、所望により、補助剤、例えば、滑沢剤、保存剤、安定剤、湿潤
剤、乳化剤、浸透圧を変化させるための塩、緩衝剤、着色剤、着香剤、および/または芳
香剤との混合が可能である。また、製剤は、所望により、他の活性剤、例えば、他の鎮痛
薬と併用することも可能である。経口投与のために、特に好適なのは、錠剤、糖衣錠、液
剤、滴剤、座剤、カプセル剤、カプレット剤、およびゲルキャップ剤である。経口用に使
用される組成物は、当技術分野で周知のいずれの方法により調製してもよく、このような
組成物には、錠剤の製造に好適である不活性で無毒な医薬用賦形剤からなる群より選ばれ
る1種以上の物質が含まれていてもよい。このような賦形剤としては、例えば、ラクトー
スなどの不活性な希釈剤、トウモロコシデンプンなどの造粒剤および崩壊剤、デンプンな
どの結合剤、ステアリン酸マグネシウムなどの滑沢剤が挙げられる。錠剤は、コーティン
グされていなくてもよいし、外観をよくするために、または有効成分の遅延放出を行うた
めに、周知の手法によりコーティングしてもよい。また、経口用製剤は、有効成分が不活
性希釈剤と混合されている硬ゼラチンカプセル剤として提供してもよい。
【0072】
水性懸濁剤には、上記の組合せの薬剤が含まれ、この混合物には、懸濁化剤、例えば、
ヒドロキシプロピルメチルセルロースのような製薬上許容される合成ガムまたは天然ガム
が一種以上含まれる。油性懸濁剤は、上記の組合せの薬剤を植物油または鉱油中に懸濁さ
せることによって調合可能である。油性懸濁剤には、蜜蝋やセチルアルコールなどの粘稠
化剤か含まれていてもよい。甘味化担体を利用する場合、シロップ剤、エリキシル剤など
を使用することができる。また、注射可能な懸濁剤を調製することも可能であり、この場
合には、適切な液状担体、懸濁化剤などを利用してもよい。
【0073】
本発明の治療方法および製剤には、オピオイド鎮痛薬およびオピオイド拮抗薬のほかに
1種以上の薬剤が含まれていてもよい。こうして追加される1種または複数種の薬剤は、相
乗的に作用するものであってもよいし、そうでないものであってもよい。従って、特定の
実施形態では、製剤中でオピオイド拮抗薬のほかに2種のオピオイド鎮痛薬を組み合わせ
てもよい。例えば、投薬剤形には、半減期、溶解性、効力などの性質やこれらの複数の性
質が異なる2種のオピオイド鎮痛薬が含まれていてもよい。更なる実施形態では、1種以上
のオピオイド鎮痛薬が含まれ、更に、オピオイド拮抗薬のほかに非オピオイド薬剤が含ま
れる。このような非オピオイド薬剤は、好ましくは、更なる痛覚消失を呈するものであり
、具体的は、アスピリン;アセトアミノフェン;非ステロイド系抗炎症薬(「NSAIDS」)、例
えば、イブプロフェン、ケトプロフェンなど;N-メチル-D-アスパルテート(NMDA)受容体拮
抗薬、例えば、デキストロメトルファン、デキストロルファン、またはケタミンのような
モルフィナン;シクロオキシゲナーゼーII阻害剤(「COX-II阻害剤」);および/またはグリ
シン受容体拮抗薬が挙げられる。
【0074】
本発明の特定の好ましい実施形態では、本発明によりNSAIDやCOX-2阻害剤のような追加
の非オピオイド作動薬を導入できるようになるため、オピオイド鎮痛薬をより低用量で使
用することができる。薬剤の一方または両方をより少ない量で使用することにより、ヒト
において、有効な疼痛治療に伴う副作用が減少する。
【0075】
好適な非ステロイド系抗炎症薬としては、イブプロフェン、ジクロフェナク、ナプロキ
セン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン、ケ
トプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、
プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロ
フェン、チアプロフェン酸、フルプロフェン、ブクロキシ酸、インドメタシン、スリンダ
ク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザ
ク、クリダナク、オキスピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニ
フルム酸、トルフェナム酸、ジフルリサル、フルフェニサル、ピロキシカム、スドキシカ
ム、またはイソキシカムなどが挙げられる。これらの薬剤の有用な用量は、当業者には周
知である。
【0076】
N-メチル-D-アスパルテート(NMDA)受容体拮抗薬は、当技術分野で周知であり、具体的
には、モルフィナン、例えば、デキストロメトルファンもしくはデキストロルファン、ケ
タミン、d-メタドン、または製薬上許容されるそれらの塩が挙げられる。本発明の目的に
対して、「NMDA拮抗薬」という用語には、NMDA-受容体活性化の結果として細胞内で生じ
る主要な作用をブロックする薬剤、例えば、GM1やGT1bなどのガングリオシド、トリフル
オペラジンなどのフェノチアジン、またはN-(6-アミノヘキシル)-5-クロロ-1-ナフタレン
スルホンアミドなどのナフタレンスルホンアミドも含まれるものとみなされる。米国特許
第5,321,012号および同第5,556,838号(いずれもMayerらに付与されている)には、これら
の薬剤により、嗜癖性薬物、例えば、モルヒネ、コデインなどの麻薬性鎮痛薬に対する耐
性および/または依存性の出現が抑制されると記載されており、また、米国特許第5,502,
058号(Mayerらに付与されている)には、慢性的な疼痛の治療ができると記載されている。
これらの特許はいずれも、参照により本明細書に組み入れる。Mayerらの特許に記載され
ているように、NMDA拮抗薬は、単独で含まれていてもよいし、リドカインのような局所麻
酔薬と一緒に含まれていてもよい。
【0077】
グリシン受容体拮抗薬を用いる慢性的疼痛の治療およびこのような薬剤の同定について
は、参照により本明細書に組み入れる米国特許第5,514,680号(Weberら)に記載されている
。
【0078】
COX-2阻害剤について当技術分野で報告がなされており、多くの化学構造がシクロオキ
シゲナーゼ-2を阻害することが知られている。COX-2阻害剤については、例えば、米国特
許第5,616,601号、同第5,604,260号、同第5,593,994号、同第5,550,142号、同第5,536,75
2号、同第5,521,213号、同第5,475,995号、同第5,639,780号、同第5,604,253号、同第5,5
52,422号、同第5,510,368号、同第5,436,265号、同第5,409,944号、および同第5,130,311
号に記載されている。これらの特許はいずれも、参照により本明細書に組み入れる。特定
の好ましいCOX-2阻害剤としては、セレコキシブ(SC-58635)、DUP-697、フロスリド(CGP-2
8238)、メロキシカム、6-メトキシ-2-ナフチル酢酸(6-MNA)、MIK-966、ナブメトン(6-MNA
用のプロドラッグ)、ニメスリド、NS-398、SC-5766、SC-58215、T-614、またはそれの組
合せが挙げられる。1日あたり体重1キログラムにつき約0.005mg〜約140mg程度のCOX-2阻
害剤の用量レベルが、オピオイド鎮痛薬と組み合わせる場合、治療上有効である。また、
1日あたり患者1人につき約0.25mg〜約7gのCOX-2阻害剤が、オピオイド鎮痛薬と組み合わ
せて投与される。
【0079】
更なる実施形態では、痛覚消失以外の所望の作用を呈する非オピオイド薬剤、例えば、
鎮咳薬、去痰薬、充血除去薬、抗ヒスタミン薬、局所麻酔薬などを含めることができる。
【0080】
本発明に係る経口投薬剤形は、例えば、顆粒剤、スフェロイド剤、ビーズ剤、ペレット
剤(これ以降では、まとめて「多粒子剤」と記す)として提供してもよい。所定の時間にわ
たり所望の用量のオピオイドを提供するのに有効な量の多粒子剤をカプセル剤中に入れて
もよいし、任意の他の効果的な形態の経口固形剤中に導入してもよい。このほか、経口投
薬剤形は、錠剤の形態であってもよい。
【0081】
制御放出性投薬剤形 オピオイド作動薬/オピオイド拮抗薬組合せ剤は、当業者に公知の
任意の好適な錠剤、コーティング錠、または多粒子製剤の形態の制御放出性または持続放
出性経口製剤として調合可能である。持続放出性投薬剤形には、場合により、オピオイド
作動薬およびオピオイド拮抗薬と一緒にマトリックス中に導入される持続放出性担体が含
まれていてもよく、あるいは持続放出性投薬剤形を持続放出性剤皮として適用してもよい
。
【0082】
オピオイド鎮痛薬がヒドロコドンを含む実施形態では、持続放出性経口投薬剤形には、
投薬単位あたりヒドロコドン約8mg〜約50mgの鎮痛薬が含まれていてもよい。ヒドロモル
ホンが治療上活性なオピオイドである持続放出性経口投薬剤形では、塩酸ヒドロモルホン
が約2mg〜約64mgの量で含まれている。別の実施形態では、オピオイド鎮痛薬にはモルヒ
ネが含まれ、本発明の持続放出性経口投薬剤形には、重量基準で約2.5mg〜約800mgのモル
ヒネが含まれる。さらにもう1つの実施形態では、オピオイド鎮痛薬にはオキシコドンが
含まれ、持続放出性経口投薬剤形には、約2.5mg〜約800mgのオキシコドンが含まれる。オ
ピオイド鎮痛薬にはトラマドールが含まれていてもよく、持続放出性経口投薬剤形には、
投薬単位あたり約25mg〜800mgのトラマドールが含まれていてもよい。投薬剤形には、実
質的に等価な治療効果が得られるように1種以上のオピオイド鎮痛薬が含まれていてもよ
い。このほか、投薬剤形には、本発明に有用なオピオイドの他の塩が等モル量で含まれて
いてもよい。
【0083】
本発明の好ましい1実施形態では、持続放出性投薬剤形には、有効成分を含有または包
含し、約0.1mm〜約2.5mm、好ましくは約0.5mm〜約2mmの直径を有する粒子が含まれていて
もよい。
【0084】
該粒子は、好ましくは、水性媒質中にオピオイド作動薬/拮抗薬組合せ剤を持続速度で
放出させることのできる物質で皮膜されている。皮膜は、他の所定の性質と所望のin-vit
ro放出速度とが同時に得られるように選択される。本発明の持続放出性コーティング製剤
は、強力な連続膜を形成可能なものでなければならず、しかもこの膜は、平滑で外観がよ
く、顔料および他のコーティング添加剤を保持することができ、無毒で、不活性で、不粘
着性でなければならない。
【0085】
特定の実施形態では、粒子には、オピオイド拮抗薬と共にオピオイド鎮痛薬を含んでな
る通常の放出性マトリックスが含まれる。
【0086】
剤皮 本発明の投薬剤形は、場合により、製剤の放出調節または保護に好適な1種以上の
物質でコーティングされていてもよい。1実施形態では、例えば、胃腸液に触れたときに
pH依存的放出またはpH非依存的放出を可能にする剤皮(コーティング)が施される。pH依
存性剤皮は、患者に対して少なくとも約8時間、好ましくは約12時間から約24時間までの
痛覚消失を引き起こすことのできる吸収プロフィルが得られるように、胃腸(GI)管、例え
ば、胃または小腸の所望の領域でオピオイドを放出する働きをする。pH非依存性剤皮が望
まれる場合、周囲の流体中で、例えば、GI管中で、pH変化に関係なく最適な放出が達成さ
れるように剤皮をデザインする。また、GI管の所望の1領域、例えば、胃において用量の
一部分を放出し、GI管の他の領域、例えば、小腸において用量の残りの部分を放出する組
成物を調合することも可能である。
【0087】
また、腸溶剤皮上にコーティングされた未保護薬剤を胃で放出し、腸溶剤皮で保護され
た残りの薬剤を胃腸管の下流で更に放出させるようにすべく、pH依存性剤皮を利用して製
剤を作製する本発明に係る製剤に対して反復作用効果を付与してもよい。セラック、酢酸
セルロースフタレート(CAP)、ポリ酢酸ビニルフタレート(PVAP)、ヒドロキシプロピルメ
チルセルロースフタレート、メタクリル酸エステルコポリマー、ゼインなどを含んでなる
pH依存性の剤皮を本発明に従って使用してもよい。
【0088】
特定の好ましい実施形態では、オピオイド鎮痛薬(COX-2インヒビターの併用されたまた
は併用されていないオピオイド鎮痛薬)を含む基体(例えば、錠剤コアビーズ、マトリック
ス粒子)は、(i)アルキルセルロース、(ii)アクリルポリマー、または (iii)それらの混合
物から選択される疎水性材料でコーティングされる。有機性または水性の溶液または分散
体の形態で剤皮を適用することができる。所望の持続放出プロフィルを得るべく、基体に
対して約2〜約25%の重量増となるように剤皮を適用してもよい。水性分散物から得られる
剤皮については、米国特許第5,273,760号および同第5,286,493号に詳細が記載されている
。これらの特許は、本発明の譲受人に譲渡されており、参照により本明細書に組み入れる
。
【0089】
本発明により使用可能な持続放出性製剤および剤皮の他の例は、本発明の譲受人に譲渡
されている米国特許第5,324,351号、同第5,356,467号、および同第5,472,712号に記載さ
れているものを含む。参照により、これらの特許の全内容を本明細書に組み入れる。
【0090】
アルキルセルロースポリマー アルキルセルロースなどのセルロース系の材料およびポ
リマーは、本発明においてビーズ剤をコーティングするのに好適な疎水性材料である。簡
単に例を示すと、好ましいアルキルセルロースポリマーの1つは、エチルセルロースであ
るが、他のセルロースおよび/またはアルキルセルロースポリマーを単独でまたは任意に
組み合わせて本発明に係る疎水性剤皮の全体または一部として容易に利用できることは、
当業者には分かるであろう。
【0091】
市販されているエチルセルロースの水性分散体の1つは、Aquacoat(登録商標)(FMC Corp
., Philadelphia, Pennsylvania, U.S.A.)である。Aquacoat(登録商標)の調製は次のよう
に行われる。まず、水不混和性有機溶剤中にエチルセルロースを溶解し、次に、界面活性
剤および安定剤の存在下でこれを水中に乳化する。均質化処理を行ってサブミクロンの液
滴を形成した後、減圧下で有機溶剤を蒸発させて擬似ラテックスを形成する。製造段階で
は、擬似ラテックス中に可塑剤を導入しない。従って、剤皮としてこれを使用する前に、
予めAquacoat(登録商標)を好適な可塑剤と均質混合する必要がある。
【0092】
エチルセルロースのもう1つの水性分散体が、Surelease(登録商標)(Colorcon, inc.,
West Point, Pennsylvania, U.S.A.)として市販されている。この製品は、製造プロセス
時に分散体中に可塑剤を導入することにより調製される。ポリマー、可塑剤(ジブチルセ
バケート)と安定剤(オレイン酸)の高温溶融体を均質混合物として調製し、次に、アルカ
リ溶液で希釈することにより、基体上に直接適用可能な水性分散体が得られる。
【0093】
アクリルポリマー 本発明の他の好ましい実施形態では、制御放出性剤皮を構成する疎
水性材料は製薬上許容されるアクリルポリマーであり、具体的には、アクリル酸およびメ
タクリル酸のコポリマー、メチルメタクリレートコポリマー、エトキシエチルメタクリレ
ート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリ
ル酸アルキルアミドコポリマー、ポリ(メチルメタクリレート)、ポリメタクリレート、ポ
リ(メチルメタクリレート)コポリマー、ポリアクリルアミド、アミノアルキルメタクリレ
ートコポリマー、ポリ(メタクリル酸無水物)、およびグリシジルメタクリレートコポリマ
ーが挙げられるが、これらに限定されるものではない。
【0094】
特定の好ましい実施形態では、アクリルポリマーには、1種以上のアンモニオメタクリ
レートコポリマーが含まれる。アンモニオメタクリレートコポリマーは当技術分野で公知
であり、低含有量の第4級アンモニウム基を含むアクリル酸エステルとメタクリル酸エス
テルとの完全重合コポリマーとしてNF XVII中に記載されている。
【0095】
望ましい溶解プロフィルを得るために、異なる物理的性質を有する2種以上のアンモニ
オメタクリレートコポリマーを導入する必要がある。例えば、中性の(メタ)アクリル酸エ
ステルに第4級アンモニウム基を異なるモル比で導入する。
【0096】
特定のメタクリル酸エステル型ポリマーは、本発明に従って使用可能なpH依存性剤皮を
調製するのに有用である。具体的には、ジエチルアミノエチルメタクリレートと他の中性
のメタクリル酸エステルとから合成されるコポリマーのファミリーが挙げられ、これはメ
タクリル酸コポリマーまたはメタクリレートポリマーとも呼ばれ、Rohm Tech, Inc.からE
udragit(登録商標)として市販されている。
いくつかの異なるタイプのEudragit(登録商標)が存在する。例えば、Eudragit(登録商
標) Eは、酸性媒質中で膨潤および溶解するメタクリル酸コポリマーの例である。Eudragi
t(登録商標) Lは、pH<約5.7では膨潤せず、pH>約6で溶解するメタクリル酸コポリマーで
ある。Eudragit(登録商標) Sは、pH<約6.5では膨潤せず、pH>約7で溶解する。Eudragit(
登録商標) RLおよびEudragit(登録商標) RSは水で膨潤可能であり、これらのポリマーに
より吸収される水の量はpH依存性であるが、Eudragit(登録商標) RLおよびRSでコーティ
ングされた投薬剤形はpH非依存性である。
【0097】
特定の好ましい実施形態では、アクリル剤皮には、それぞれ商品名Eudragit(登録商標)
RL3ODおよびEudragit(登録商標)RS3ODとしてRohm Pharmaから市販されている2種のアクリ
ル樹脂ラッカーの混合物が含まれる。Eudragit(登録商標)RL3ODおよびEudragit(登録商標
)RS3ODは、低含有量の第4級アンモニウム基を含むアクリル酸エステルとメタクリル酸エ
ステルとのコポリマーであり、アンモニウム基対残りの中性(メタ)アクリル酸エステルの
モル比は、Eudragit(登録商標)RL3ODでは1:20であり、Eudragit(登録商標)RS3ODでは1:40
である。平均分子量は、約150,000である。コード表示RL(高浸透性)およびRS(低浸透性)
は、これらの薬剤の浸透性を表している。Eudragit(登録商標)RL/RS混合物は、水および
消化液に不溶である。しかしながら、これらの物質から形成される剤皮は、水溶液および
消化液中で膨潤および浸透が可能である。
【0098】
本発明のEudragit(登録商標) RL/RS分散体は、望ましい溶解プロフィルを有する持続放
出性製剤が最終的に得られるように、任意の所望の比で混合可能である。例えば、Eudrag
it(登録商標) RL 100%、Eudragit(登録商標) RL 50% + Eudragit(登録商標) RS 50%、お
よびEudragit(登録商標) RL 10% + Eudragit(登録商標) RS 90%から誘導される遅延性剤
皮を用いて、望ましい持続放出性製剤を得ることができる。もちろん、他のアクリルポリ
マー、例えば、Eudragit(登録商標) Lなども使用可能であることは、当業者には分かるで
あろう。
【0099】
可塑剤 剤皮に疎水性材料の水性分散体が含まれる本発明の実施形態では、疎水性材料
の水性分散体中に有効量の可塑剤を導入すると、持続放出性剤皮の物理的性質が改良され
るであろう。例えば、エチルセルロースは、比較的高いガラス転移温度を有し、通常のコ
ーティング条件下では可撓性の膜を形成しないため、それを剤皮材料として使用する前に
、エチルセルロース剤皮を含有する持続放出性剤皮中に可塑剤を導入することが望ましい
。一般的には、剤皮溶液中に含まれる可塑剤の量は、被膜形成剤の濃度を基準にして、例
えば、ほとんどの場合、被膜形成剤の重量の約1〜約50%である。しかしながら、可塑剤の
濃度は、特定のコーティング溶液および適用方法を用いて注意深く実験を行った後で適切
に決めなければならない。
【0100】
エチルセルロース用の好適な可塑剤としては、例えば、ジブチルセバケート、ジエチル
フタレート、トリエチルシトレート、トリブチルシトレート、およびトリアセチンのよう
な水不溶性の可塑剤が挙げられるが、他の水不溶性可塑剤(例えば、アセチル化モノグリ
セリド、フタル酸エステル、ヒマシ油など)を使用することも可能である。トリエチルシ
トレートは、本発明のエチルセルロース水性分散体には特に好ましい可塑剤である。
【0101】
本発明のアクリルポリマー用の好適な可塑剤としては、例えば、トリエチルシトレート
NF XVIやトリブチルシトレートのようなクエン酸エステル、ジブチルフタレート、および
場合により1,2-プロピレングリコールが挙げられるが、これらに限定されるものではない
。アクリル皮膜から形成される膜、例えば、Eudragit(登録商標) RL/RSラッカー溶液から
形成される膜の弾性を増強するのに好適であることが判明している他の可塑剤としては、
ポリエチレングリコール、プロピレングリコール、ジエチルフタレート、ヒマシ油、およ
びトリアセチンが挙げられる。トリエチルシトレートは、本発明のエチルセルロースの水
性分散体には特に好ましい可塑剤である。
【0102】
このほか、少量のタルクを添加すると、加工時における水性分散体の付着傾向が減少し
、艶出剤としての効能が現れることが判明した。
【0103】
コーティングされたビーズ剤の調製方法 疎水性材料を用いて不活性な医薬用ビーズ剤
、例えば、nu pariel 18/20ビーズ剤をコーティングする場合、コーティング後得られた
複数の固体制御放出性ビーズ剤を、周囲の流体、例えば、胃液または溶解媒質による消化
を受けたりそれと接触したときに有効な制御放出用量を確保するのに十分な量で、ゼラチ
ンカプセル剤中に配置してもよい。
【0104】
本発明の制御放出性ビーズ製剤は、例えば、摂取後、胃液や腸液に触れると、治療上有
効な薬剤を徐々に放出する。本発明の製剤の制御放出プロフィルは、例えば、疎水性材料
を含む被覆膜の量の変更、疎水性材料中に可塑剤を添加する方法の変更、疎水性材料に関
連する可塑剤の量の変更、他の成分または賦形剤の導入、製造方法の変更などによって、
調節可能である。最終的な製品の溶解プロフィルに関しても、例えば、遅延性剤皮の厚さ
の増大または減少によって、調節可能である。
【0105】
治療上有効な薬剤でコーティングされたスフェロイド剤またはビーズ剤は、例えば、治
療上有効な薬剤を水中に溶解させ、次に、Wusterインサートを用いてnu pariel 18/20ビ
ーズ剤などの基体上に溶液をスプレーすることによって、調製される。場合により、ビー
ズ剤へのオピオイドの結合および/または溶液の着色などを促進するために、ビーズ剤に
コーティングする前に他の成分を添加する。例えば、着色剤(例えば、Colorcon, Inc.か
ら市販されているOpadry(登録商標))を併用してまたは併用せずに、ヒドロキシプロピル
メチルセルロースを含む物質を溶液に添加し、この溶液を混合した後(例えば、約1時間混
合した後)、これをビーズ剤上に適用してもよい。次に、こうしてコーティングされた基
体(この例ではビーズ剤)を、場合により、障壁剤でオーバーコーティングすることにより
、治療上有効な薬剤を疎水性制御放出性剤皮から分離させてもよい。好適な障壁剤の例は
、ヒドロキシプロピルメチルセルロースが含まれる障壁剤である。しかしながら、当技術
分野で公知の被膜形成剤はいずれも使用可能である。完成品の溶解速度に影響を及ぼさな
い障壁剤が好ましい。
【0106】
次に、疎水性材料の水性分散体でビーズ剤をオーバーコーティングしてもよい。疎水性
材料の水性分散体には、好ましくは、有効量の可塑剤、例えば、トリエチルシトレートが
更に含まれる。既に調合済みであるエチルセルロースの水性分散体、例えば、Aquacoat(
登録商標)やSurelease(登録商標)を使用してもよい。Surelease(登録商標)を使用する場
合には可塑剤を別に添加する必要はない。このほか、既に調合済みであるアクリルポリマ
ーの水性分散体、例えば、Eudragit(登録商標)を使用することもできる。
【0107】
本発明のコーティング溶液には、被膜形成剤、可塑剤、および溶剤系(すなわち、水)の
ほかに、好ましくは、外観の改良および製品の識別を行うための着色剤が含まれる。疎水
性材料の水性分散体を着色する代わりに、またはそれを着色するとともに、治療上有効な
薬剤の溶液を着色してもよい。例えば、アルコールまたはプロピレングリコールベースの
着色分散体、微粉砕アンモニウムレーキ、および二酸化チタンのような不透明剤を用いて
Aquacoat(登録商標)を着色してもよい。この場合、水溶性ポリマー溶液を剪断力下で着色
し、次に、可塑化Aquacoat(登録商標)を低剪断力下で着色する。このほか、本発明の製剤
を着色するための任意の好適な方法が使用可能である。アクリルポリマーの水性分散体を
使用する場合、製剤を着色するのに好適な成分としては、二酸化チタンおよび有色顔料、
例えば、酸化鉄顔料が挙げられる。しかしながら、顔料を導入すると、剤皮の遅延効果が
増大する。
【0108】
当技術分野で公知の任意の好適なスプレー装置を用いてスプレーすることにより、治療
上有効な薬剤を含む基体上に可塑化疎水性材料を適用することが可能である。好ましい方
法では、Wurster流動床系を使用する。この系では、下方から噴射されたエアジェットに
よりコア材料を流動させ、アクリルポリマー剤皮をコア上にスプレーしながら乾燥を行う
。好ましくは、治療上有効な薬剤の物理的な特性や可塑剤の導入方法などを考慮に入れて
、コーティングされた基体が胃液などの水溶液に触れたときに治療上有効な該薬剤を予め
決められたように制御放出させるのに十分な量で疎水性材料を適用する。疎水性材料でコ
ーティングした後、場合により、Opadry(登録商標)のような被膜形成剤の被覆膜をさらに
ビーズ剤に適用する。こうした被覆膜を設けると、多少なりとも、ビーズ剤の凝集は実質
的に低減する。
【0109】
1種以上の放出調節剤を添加することによって、または剤皮を貫通する1つ以上の通路を
提供することによって、本発明の制御放出性製剤からの治療上有効な薬剤の放出を更に変
化させることができる。すなわち、所望の速度に調節することができる。疎水性材料対水
溶性材料の比は、いくつかある因子の中で特に、所要の放出速度および選択される材料の
溶解特性によって決定される。
【0110】
細孔形成剤として機能する放出調節剤は、有機物であっても無機物であってもよく、こ
うした放出調節剤には、使用環境下で剤皮から溶解、抽出、または浸出させることのでき
る物質が含まれる。細孔形成剤には、1種以上の親水性材料、例えば、ヒドロキシプロピ
ルメチルセルロースが含まれていてもよい。
【0111】
また、デンプンやガムなどの浸食促進剤を本発明の持続放出性剤皮に含有させることも
できる。
【0112】
また、使用環境下でミクロ細孔性単層を形成するのに有用な材料、例えば、カーボネー
ト基がポリマー鎖中に複数回出現する炭酸の直鎖状ポリエステルを含んでなるポリカーボ
ネートを本発明の持続放出性剤皮に含有させることができる。
【0113】
また、放出調節剤には、半透過性ポリマーが含まれていてもよい。
【0114】
特定の好ましい実施形態では、放出調節剤は、ヒドロキシプロピルメチルセルロース、
ラクトース、金属ステアリン酸塩、およびそれらの任意の混合物から選択される。
【0115】
また、本発明の持続放出性剤皮には、少なくとも1つの通路、オリフィスなどを含んで
なる送出手段が含まれていてもよい。米国特許第3,845,770号、同第3,916,889号、同第4,
063,064号、および同第4,088,864号に開示されているような方法によって経路を形成する
ことが可能である(これらの特許はいずれも、参照により本明細書に組み入れる)。経路は
、円形、三角形、四角形、楕円形、不規則形など、任意の形状をとることができる。
【0116】
マトリックスビーズ製剤 本発明の他の実施形態では、先に記載したような制御放出性
剤皮を有するマトリックスを用いることによって制御放出性製剤が得られる。また、本発
明においては、オピオイドのin vitro溶解速度が好ましい範囲内に設定されかつpH依存的
またはpH非依存的にオピオイドが放出される制御放出性マトリックスも利用可能である。
制御放出性マトリックス中へ導入するのに好適な物質は、マトリックス形成に使用される
方法に依存する。
【0117】
例えば、オピオイド鎮痛薬および(場合により)COX-2のほかに、以下の物質がマトリッ
クス中に含まれていてもよい。
【0118】
親水性および/または疎水性材料、例えば、ガム、セルロースエーテル、アクリル樹脂
、タンパク質誘導物質。このリストは、限定的なものではなく、活性剤の制御放出を行う
ことができかつ融解する(または押出に必要な程度まで軟化する)任意の製薬上許容される
疎水性材料または親水性材料を、本発明に従って使用することが可能である。
【0119】
消化可能な長鎖状の(C8〜C50、特に、C12〜C40)置換もしくは無置換炭化水素、例えば
、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、蝋、ステアリ
ルアルコール、ポリアルキレングリコール。
【0120】
これらのポリマーのうちで、アクリルポリマー、特に、Eudragit(登録商標) RSPO、セ
ルロースエーテル、特に、ヒドロキシアルキルセルロースおよびカルボキシアルキルセル
ロースが好ましい。経口投薬剤形には、少なくとも1種の親水性または疎水性の材料が1%
〜80%(重量基準)の量で含まれていてもよい。
【0121】
疎水性材料が炭化水素である場合、好ましくは、炭化水素の融点は、25〜90℃である。
長鎖状炭化水素材料のうちで、脂肪(脂肪族)アルコールが好ましい。経口投薬剤形には、
少なくとも1種の消化可能な長鎖状炭化水素が60%(重量基準)までの量で含まれていてもよ
い。
【0122】
好ましくは、経口投薬剤形には、少なくとも1種のポリアルキレングリコールが60%(重
量基準)までの量で含まれていてもよい。
【0123】
疎水性材料は、好ましくは、アルキルセルロース、アクリル酸およびメタクリル酸のポ
リマーおよびコポリマー、セラック、ゼイン、水素化ヒマシ油、水素化植物油、またはそ
れらの混合物からなる群より選ばれる。本発明の特定の好ましい実施形態では、疎水性材
料は、製薬上許容されるアクリルポリマーであり、具体的には、アクリル酸およびメタク
リル酸のコポリマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキ
シエチルメタクリレート、シアノエチルメタクリレート、アミノアルキルメタクリレート
コポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポ
リマー、ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレー
ト、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびグリシジルメタクリレート
コポリマーが挙げられるが、これらに限定されるものではない。他の実施形態では、疎水
性材料は、ヒドロキシアルキルセルロースのような材料、例えば、ヒドロキシプロピルメ
チルセルロース、およびそれらの混合物から選択される。
【0124】
好ましい疎水性材料は水不溶性であり、多かれ少なかれ、はっきりと親水性および/ま
たは疎水性の傾向を有する。好ましくは、本発明に有用な疎水性材料の融点は、約30〜約
200℃、好ましくは約45〜約90℃である。特に、疎水性材料には、天然蝋または合成蝋、
脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアルコール、ステアリルアル
コール、セチルアルコール、または好ましくはセトステアリルアルコール)、脂肪酸類、
例えば、限定されるものではないが、脂肪酸エステル、脂肪酸グリセリド(モノ-、ジ-、
およびトリ-グリセリド)、水素化脂肪、炭化水素、ノルマルワックス、ステアリン酸、ス
テアリルアルコール、ならびに炭化水素主鎖を有する疎水性および親水性材料が含まれて
いてもよい。好適な蝋としては、例えば、蜜蝋、グリコワックス(glycowax)、カストール
ワックス、およびカルナウバワックスが挙げられる。本発明の目的では、蝋様物質とは、
室温で通常固体であり約30〜約100℃の融点を有する任意の物質として定義付けられる。
【0125】
本発明に従って使用しうる好適な疎水性材料としては、消化可能な長鎖状の(C8〜C50、
特に、C12〜C40)置換もしくは無置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪
酸のグリセリルエステル、鉱油、植物油、天然蝋、および合成蝋が挙げられる。25〜90℃
の融点を有する炭化水素が好ましい。特定の実施形態では、長鎖状炭化水素材料のうちで
、脂肪(脂肪族)アルコールが好ましい。経口投薬剤形には、少なくとも1種の消化可能な
長鎖状炭化水素が60%(重量基準)までの量で含まれていてもよい。
【0126】
好ましくは、マトリックス製剤中に2種以上の疎水性材料が一緒に含まれる。
他の疎水性材料を含める場合、こうした材料は、好ましくは、天然蝋、合成蝋、脂肪酸
、脂肪アルコール、およびそれらの混合物から選択される。具体的には、蜜蝋、カルナウ
バワックス、ステアリン酸、およびステアリルアルコールが挙げられる。このリストは、
限定的なものではない。
【0127】
特定の好適なマトリックスの1つには、少なくとも1種の水溶性ヒドロキシアルキルセル
ロース、少なくとも1種のC12〜C36、好ましくはC14〜C22脂肪族アルコール、および場合
により少なくとも1種のポリアルキレングリコールが含まれる。上記の少なくとも1種のヒ
ドロキシアルキルセルロースは、好ましくは、ヒドロキシ(C1〜C6)アルキルセルロース、
例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、および
特にヒドロキシエチルセルロースである。本発明の経口投薬剤形中の少なくとも1種のヒ
ドロキシアルキルセルロースの量は、特に、所要の正確なオピオイド放出速度によって決
定されるであろう。上記の少なくとも1種の脂肪族アルコールは、例えば、ラウリルアル
コール、ミリスチルアルコール、またはステアリルアルコールであってよい。しかしなが
ら、本発明の経口投薬剤形の特に好ましい実施形態では、少なくとも1種の脂肪族アルコ
ールは、セチルアルコールまたはセトステアリルアルコールである。本発明の経口投薬剤
形中の少なくとも1種の脂肪族アルコールの量は、先に述べたように、所要の正確なオピ
オイド放出速度によって決定されるであろう。この量はまた、少なくとも1種のポリアル
キレングリコールが経口投薬剤形中に存在するかしないかにも依存する。少なくとも1種
のポリアルキレングリコールの不在下では、経口投薬剤形には、好ましくは、少なくとも
1種の脂肪族アルコールが20%〜50%(重量基準)の量で含まれる。少なくとも1種のポリアル
キレングリコールが経口投薬剤形中に存在する場合、少なくとも1種の脂肪族アルコール
と少なくとも1種のポリアルキレングリコールとの合計重量は、好ましくは、全用量の20%
〜50%(重量基準)を占める。
【0128】
1実施形態では、例えば、少なくとも1種のヒドロキシアルキルセルロースまたはアクリ
ル樹脂対少なくとも1種の脂肪族アルコール/ポリアルキレングリコールの比によって、製
剤からのオピオイドの放出速度はかなりの影響を受ける。少なくとも1種のヒドロキシア
ルキルセルロース対少なくとも1種の脂肪族アルコール/ポリアルキレングリコールの比は
1:2〜1:4が好ましく、1:3〜1:4が特に好ましい。
【0129】
上記の少なくとも1種のポリアルキレングリコールは、例えば、ポリプロピレングリコ
ールであってよく、好ましくは、ポリエチレングリコールである。少なくとも1種のポリ
アルキレングリコールの数平均分子量は1,000〜15,000が好ましく、特に、1,500〜12,000
が好ましい。
【0130】
別の好適な制御放出性マトリックスには、アルキルセルロース(特にエチルセルロース)
、C12〜C36脂肪族アルコール、および場合によりポリアルキレングリコールが含まれるで
あろう。
【0131】
別の好ましい実施形態では、マトリックスには、少なくとも2種の疎水性材料の製薬上
許容される組合せが含まれる。
【0132】
上の成分のほかに、制御放出性マトリックスには、好適な量の他の物質、例えば医薬分
野で以前より知られている、希釈剤、滑沢剤、結合剤、造粒助剤、着色剤、着香剤、およ
び滑走剤(glidant)が含まれていてもよい。
【0133】
マトリックスベースのビーズ剤を調製するための方法 本発明に係る固形制御放出性経
口投薬剤形の調製を容易にするために、当業者に周知であるマトリックス製剤の調製方法
のいずれを使用してもよい。具体例として、マトリックス中への導入は、例えば、次のよ
うに行うことができる。(a)少なくとも1種の水溶性ヒドロキシアルキルセルロースとオピ
オイドまたはオピオイド塩とを含む顆粒を形成、(b)ヒドロキシアルキルセルロースを含
有する顆粒を少なくとも1種のC12〜C36脂肪族アルコールと混合し、(c)場合により、顆粒
を圧縮および造形する。好ましくは、水を用いてヒドロキシアルキルセルロース/オピオ
イドを湿式造粒することによって、顆粒を形成する。この方法の特に好ましい実施形態で
は、湿式造粒ステップで添加される水の量は、好ましくは、オピオイドの乾燥重量の1.5
〜5倍、特に、1.75〜3.5倍である。
【0134】
更に他の実施形態では、有効成分と一緒に球状化剤を用いて球状化処理することにより
、スフェロイド剤を形成することができる。微晶質セルロースが好ましい。好適な微晶質
セルロースは、例えば、Avicel PH 101(商標、FMC Corporation)として販売される物質で
ある。このような実施形態では、有効成分および球状化剤のほかに、スフェロイド剤には
結合剤が含まれていてもよい。好適な結合剤、例えば、低粘度の水溶性ポリマーは、医薬
分野の当業者には周知であろう。しかしながら、ヒドロキシプロピルセルロースのような
水溶性ヒドロキシ低級アルキルセルロースが好ましい。このほかに(またはこの代わりに)
、スフェロイド剤には、水不溶性ポリマー、特に、アクリルポリマー、アクリルコポリマ
ー、例えば、メタクリル酸-エチルアクリル酸コポリマー、またはエチルセルロースが含
まれていてもよい。このような実施形態では、持続放出性剤皮には、一般に、(a)蝋(単独
でもしくは脂肪アルコールとの混合物として)、または(b)セラックもしくはゼインのよう
な疎水性材料が含まれるであろう。
【0135】
溶融押出マトリックス 溶融造粒法または溶融押出法を用いて、持続放出性マトリクス
を調製することも可能である。一般的には、溶融造粒法では、通常は固体の疎水性材料、
例えば、蝋を溶融させ、その中に粉末薬剤を導入する。持続放出性投薬剤形を得るために
、追加の疎水性物質、例えば、エチルセルロースまたは水不溶性アクリルポリマーを、溶
融蝋疎水性材料中に導入する必要がある。溶融造粒法を用いて調製される持続放出性製剤
の例が、米国特許第4,861,598号に記載されている。この特許は、本発明の譲受人に譲渡
されており、その全内容を、参照により本明細書に組み入れる。
【0136】
追加の疎水性材料には、1種以上の水不溶性蝋様可塑性物質が、1種以上の該水不溶性蝋
様物質よりも疎水性の弱い1種以上の蝋様熱可塑性物質と混合されて含まれていてもよい
。一定した放出が行われるようにすべく、製剤中の各蝋様物質は、初期放出段階において
、実質的に胃腸液中で分解されず、かつ不溶でなければならない。有用な水不溶性蝋様物
質は、水への溶解度が約1:5,000(w/w)よりも低い物質であってよい。
【0137】
また、持続放出性マトリックスには、上記の成分のほかに、適量の他の材料、例えば医
薬分野で以前より知られている、希釈剤、滑沢剤、結合剤、造粒助剤、着色剤、着香剤、
および滑走剤(glidant)が含まれていてもよい。これらの追加材料の量は、所望の作用
を所望の製剤に付与するのに十分な量であろう。
【0138】
また、溶融押出された多粒子剤を含んでなる持続放出性マトリックスには、上記の成分
のほかに、適量の他の材料、例えば医薬分野で以前より知られている、希釈剤、滑沢剤、
結合剤、造粒助剤、着色剤、着香剤、および滑走剤(glidant)が、所望により顆粒の約50
重量%までの量で含まれていてもよい。
【0139】
経口投薬剤形の調合に使用しうる製薬上許容される担体および賦形剤の特定の具体例が
、参照により本明細書に組み入れるHandbook of Pharmaceutical Excipients, American
Pharmaceutical Association (1986)に記載されている。
【0140】
溶融押出多粒子剤 本発明に係る好適な溶融押出マトリックスの調製には、例えば、オ
ピオイド鎮痛薬を、少なくとも1種の疎水性材料および好ましくは追加の疎水性材料とブ
レンドして均質混合物を得るステップが含まれていてもよい。次に、十分な温度まで均質
混合物を加熱することにより、押出するのに十分な程度まで均質混合物を少なくとも軟化
させる。その後、得られた均質混合物を押出してストランドを形成する。好ましくは、当
技術分野で公知の任意の手段により押出物の冷却および切削を行って多粒子剤にする。ス
トランドの冷却および切削を行って多粒子剤にする。次に、多粒子剤を単位用量に分割す
る。押出物は、好ましくは、約0.1〜約5mmの直径を有し、治療上有効な薬剤を約8〜約24
時間にわたり持続放出させる。
【0141】
本発明の溶融押出物を調製するために場合により使用される方法では、疎水性材料、治
療上有効な薬剤、および必要に応じて使用される結合剤を計量して押出機に直接供給し、
均質混合物を熱し、均質混合物を押出すことによりストランドを形成し、均質混合物を含
有するストランドを冷却し、ストランドを切断して約0.1mm〜約12mmのサイズの粒子にし
、更に、この粒子を単位用量まで分割する。
本発明のこの態様では、比較的連続した製造工程が実現される。
【0142】
また、押出機のアパーチャーすなわち送出口の直径を調節することにより、押出される
ストランドの厚さを変えることができる。更に、押出機の送出部分は、円形である必要は
なく、楕円形、矩形などにすることも可能である。熱線カッター、裁断機などを用いて、
送出ストランドを分割して粒子にすることもできる。
【0143】
溶融押出多粒子系は、押出機の送出オリフィスに応じて、例えば、顆粒剤、スフェロイ
ド剤、またはペレット剤の形態をとることができる。本発明の目的では、用語「溶融押出
多粒子剤」、「溶融押出多粒子系」、および「溶融押出粒子」とは、好ましくは、所定の
範囲内の類似したサイズおよび/または形状を有し、1種以上の有効な薬剤と、1種以上の
賦形剤と、好ましくは、本明細書中に記載の疎水性材料とを含む複数のユニットを意味す
る。この場合、溶融押出多粒子剤の長さは、約0.1〜約12mmであり、その直径は約0.1〜約
5mmである。また、溶融押出多粒子剤は、このサイズの範囲内で任意の幾何学形状をとる
ことができると考えられる。このほか、球状化ステップを用いずに、押出物を単に所望の
長さに切断し、治療上有効な薬剤の単位用量まで分割してもよい。
【0144】
好ましい1実施形態では、有効量の溶融押出多粒子剤がカプセル剤中に含まれるように
経口投薬剤を調製する。例えば、胃液による消化および胃液との接触の際に有効な持続放
出を行うのに十分な量で複数の溶融押出多粒子剤をゼラチンカプセル剤中に配置してもよ
い。
【0145】
もう1つの好ましい実施形態では、従来型の錠剤機を用いて標準的な方法により適量の
多粒子剤押出物を圧縮して経口錠剤にする。また、錠剤(圧縮錠剤、すりこみ錠剤)、カプ
セル剤(硬質ゼラチンカプセル剤および軟質ゼラチンカプセル剤)、および丸剤を作製する
ための方法および組成については、参照により本明細書に組み入れるRemington's Pharma
ceutical Sciences, (Arthur Osol, editor), 1553-1593 (1980)に記載されている。
【0146】
さらに別の好ましい実施形態では、前記でさらに詳細に記載され、そのため参照により
本明細書中に組み入れる米国特許第4,957,681号(Klimeschら)に記載されているようにし
て押出物を錠剤に成形することができる。
【0147】
場合により、持続放出性剤皮(例えば前記の持続放出性剤皮)を用いて、持続放出性溶融
押出多粒子系または錠剤にコーティングを施すことが可能であり、あるいはゼラチンカプ
セル剤に更にコーティングを施すことも可能である。このような剤皮には、好ましくは、
重量を約2〜約30%増加させるのに十分な量の疎水性材料が含まれる。しかしながら、特に
、利用する特定のオピオイド鎮痛薬化合物の物理的性質および所望の放出速度にもよるが
、被覆膜を更に増大させることも可能である。
【0148】
本発明の溶融押出単位投薬剤には、更に、先に開示した治療上有効な薬剤を1種以上含
有する溶融押出多粒子剤がカプセル化前に組み合わせされて含まれていてもよい。このほ
か、単位投薬剤には、直ぐに治療効果を得るべく、即時放出性の治療上有効な薬剤を所定
量含有させることができる。即時放出性の治療上有効な薬剤は、例えば、別のペレット剤
としてゼラチンカプセル剤中に導入してもよいし、あるいは投薬剤を調製した後、多粒子
剤の表面上にコーティングしてもよい(例えば、制御放出性の剤皮またはマトリックスベ
ースの剤皮)。また、本発明の単位投薬剤には、所望の効果を得るべく、制御放出性ビー
ズ剤およびマトリックス多粒子剤が組み合わされて含まれていてもよい。
【0149】
本発明の持続放出性製剤は、好ましくは、例えば、摂取後、胃液や腸液に触れると、治
療上有効な薬剤を徐々に放出する。発明の溶融押出製剤の持続放出プロフィルは、例えば
、遅延剤すなわち疎水性材料の量の変更、疎水性材料に対する可塑剤の量の変更、他の成
分または賦形剤の導入、製造方法の変更などによって、調節可能である。
【0150】
本発明の他の実施形態では、溶融押出される材料を、治療上有効な薬剤を含有させずに
調製し、その後、該薬剤を押出物に添加する。このような製剤に含まれる治療上有効な薬
剤は、典型的には、押出マトリックス材料と一緒にブレンドされ、次に、該混合物の錠剤
化を行うことにより、徐放性製剤を形成することになるであろう。このような製剤は、例
えば、製剤中に含まれる治療上有効な薬剤が疎水性材料および/または遅延剤材料の軟化
に必要な温度の影響を受け易い場合に有利である。
【0151】
好ましい実施形態の詳細な説明 以下の実施例により本発明の種々の態様について具体
的に説明する。これらの実施例により特許請求の範囲はなんら制限されるものではない。
【0152】
本発明者らの知るかぎり、ナルトレキソンを種々のオピオイドアゴニストと一緒に同時
投与した後でその競合的アゴニストの性質を直接比較することはこれまでに行われていな
い。しかしながら、ヘロインまたはモルヒネのいずれかのチャレンジを受けた被験者にお
いて、用量変化の研究が行われ、オピオイドアンタゴニストの性質が評価された。一般的
には、ナルトレキソン50mgを予め投与してからその24時間後にヘロイン25mgを静脈内投与
した場合、オピオイドアゴニストの作用は完全にブロックされるかまたは低減した。Gonz
alez JP, Brogden RN. 「ナルトレキソン:オピオイド依存症の治療におけるその薬力学的
および薬動学的性質ならびに治療効果に関するレビュー」Drugs 1988; 35:192-213; Resn
ick RR, Valavka J, Freedman AM, Thomas M. 「EN-1639A(ナルトレキソン):新しい麻薬
アンタゴニストに関する研究」Am. J. Psychiatry 1974; 131:646-650を参照されたい。
これらはいずれも参照により本明細書に組み入れる。
【実施例】
【0153】
実施例1 実施例1では、ランダム化、単純盲検、プラシーボ対照、および単回投与によ
る4法交差試験を行った。これにより、ナルトレキソン経口液剤6.4mgが6人の正常な健常
女性ボランティアにおいてヒドロコドン15mgのオピオイドアゴニスト作用をブロックする
かを評価した。実験個体群には女性だけが含まれていた。なぜなら、以前の観察において
、女性の方が男性よりもオピオイドアゴニスト作用に対する感受性が高いことが示唆され
たためである。4種の治療薬は、HYIR/APAP(ヒドロコドン7.5およびアセトアミノフェン75
0mgの2つの錠剤、Vicodin ES(登録商標)) + ナルトレキソン経口液剤3.2mg、HYIR/APAP(2
×7.5mg) + ナルトレキソン経口液剤6.4mg、HYIR比較(comparator)錠剤(2×750mg Trilis
ate(登録商標)錠剤) + ナルトレキソン経口液剤(プラシーボ)、HYIR/APAP(Vicodin ES(登
録商標)の2つの錠剤) + ナルトレキソン経口液剤(プラシーボ)であった。いずれの治療薬
についても、絶食状態で投与した。それぞれの投与の間で48時間のウォッシュアウト期間
を設けた。被験者を、これらの4種の治療薬グループの4つの治療薬投与順にランダムに割
り当てた。最初の投与を行う前の夕方に被験者は試験施設に集まり、最後の投与を行って
から24時間後の投与評価が終了するまで拘束状態にあった。安全性の測定は、副作用の報
告、生命徴候、異常な実験値、異常な外診結果、およびECGの結果からなっていた。また
、薬力学的パラメーター(瞳孔サイズおよび改良型特異的薬効アンケート調査)についても
評価した。
【0154】
試験治療薬 4種の治療薬は以下の通りであった。
【0155】
ヒドロコドン即時放出性錠剤(2×7.5mg) + ナルトレキソン経口液剤3.2mg。
【0156】
ヒドロコドン即時放出性錠剤(2×7.5mg) + ナルトレキソン経口液剤6.4mg。
【0157】
ヒドロコドン即時放出性比較錠剤 + プラシーボナルトレキソン経口液剤。
【0158】
プラシーボヒドロコドン即時放出性錠剤(2×7.5mg) + プラシーボナルトレキソン経口液
剤。
【0159】
試験製品 この試験で評価した製品には、Vicodin ES(登録商標)(酒石酸水素ヒドロコド
ン7.5mgおよびアセトアミノフェン750mg、Knoll Pharmaceuticals)、比較物質(comparato
r)として利用したTrilisate(登録商標) (コリンマグネシウムトリサリチレート750mg、Pu
rdue Frederick)、およびナルトレキソン粉末薬が含まれていた。Vicodin ES(登録商標)
を活性治療薬として選択した。なぜなら、この製品中のアセトアミノフェン部分は、中枢
神経系にも瞳孔測定にも影響を及ぼさないと考えられるからである。「比較物質」として
使用するためにTrilisateを選択した。なぜなら、その物理的外観がVicodin ES(登録商標
)と類似しているうえに、中枢神経系にも瞳孔測定にも影響を及ぼさないからである。市
販の錠剤(Revia(登録商標) 50mg錠剤、DuPont)を用いずにナルトレキソン散剤を選択する
ことにより、経口液剤を調製する際の全体的な精度を向上させた。現場の研究薬剤師が、
適切な医薬品調合法を利用して滅菌環境下でナルトレキソン散剤から経口液剤を再調製し
た。ナルトレキソン散剤(Mallinckrodt Chemical)を使用してナルトレキソン液剤を調合
した。ナルトレキソンの個々のストック溶液は、TsangおよびHoltsmanにより提案された
方法に変更を加えて調製した。Tsang BK, Holtsman R. 「液状ナルトレキソンの室温安定
性」Anesthesiology 1995:83:A864。この文献は参照により本明細書に組み入れる。ナル
トレキソンストック溶液の調製は、次のように行った。各投与期間の直前(60分以内)に、
ナルトレキソン散剤32mgおよび64mgを秤量した。これらの部分をそれぞれ、蒸留水50mL +
単純シロップ50mL(NF:米国医薬品集)中に溶解し、最終体積を100mLとした。最終溶液の
濃度はそれぞれ、0.32mg /mL(32mg/100mL)および0.64mg/mL(64mg/100mL)であった。これ
らの濃度を用いることにより、各投与期間中に同じ体積(10mL)のナルトレキソン経口液剤
を投与できるようにした。ナルトレキソン経口液剤プラシーボは、活性溶液と同じビヒク
ルで調製した。活性溶液と同じような味を呈するように、苦味剤Bitterguard(デナトニウ
ムベンゾエート、NF)の添加を行った。
【0160】
薬力学的測定a. 瞳孔サイズ - 瞳孔測定により調べる。
【0161】
75mmレンズおよび内蔵エレクトロニックリングフラッシュを備えたPolaroid CU-5カメ
ラにPolacolor ER 669インスタントパックフィルム12を装填して、瞳孔直径の測定を行っ
た。この方法は、瞳孔を調べるための安全で正確な方法として受け入れられるようになっ
ており、一般に、赤外線テレビ瞳孔測定法(より汎用性がありかつ高性能であるが、かな
り高価で取扱いの難しい方法)に次ぐ方法であるとみなされている。Polaroid CU-5法の精
度は0.1mm以内であると言われている。Czarnecki JS, Pilley SF, Thompson HS. “The u
se of Photography in the Clinical Evaluation of Unequal Pupils”, Canad. J. Opht
hal. 1979;14:297-302を参照されたい。この文献は、参照により本明細書に組み入れる。
【0162】
次のように瞳孔直径を測定した。フラッシュの角膜反射によって水平方向の瞳孔周辺が
不明瞭になるのを避けるために、リングフラッシュの3時および9時の方向を2つの小片で
カバーするようにカメラを改造した。被験者の顔の前面で3インチフレームが側方眼窩縁
に当接するようにカメラを配置し、視線がそのカメラ領域の上部すれすれを向くようにし
た(上方すれすれ入射(upgaze)を最小限に抑えるため)。カメラ本体の上部越しに遠方の非
調節性標的に視線を固定するように被験者に指示し、調節性瞳孔反射を最小限に抑えるよ
うにした。被験者が遠方を凝視している状態で写真を撮影した。写真はいずれも、一定の
周囲光の下で撮影した。瞳孔潜伏時間は、フラッシュが瞳孔直径に影響を及ぼさない時間
であった。フラッシュの後、瞳孔の持続性収縮は起こらないが、短時間の収縮は観測され
る。従って、フラッシュは、この試験に必要な測定には影響を及ぼさなかった。Smith SA
, Dewhist RR. “A Single Diagnostic Test for Pupillary Abnormality in Diabetic A
utonomic Neuropathy”, Diabetic Medicine 1988;3:38-41を参照されたい。この文献は
、参照により本明細書に組み入れる。所定の時間(周囲温度にもよるが、約1分間)でプリ
ントの現像を行うと、被験者の顔中部の1対1の写真が得られ、瞳孔はプリントの上部に現
れる。次に、0.1mmまで校正された網線の内蔵された単純なルーペを用いて瞳孔直径を測
定する。プロトコルに規定された各時間で左眼だけを対象に瞳孔効果を調べた。
【0163】
b. 改良型特異的薬効アンケート調査 アンケート調査は、JasinskiおよびPrestonが使
用した22の質問事項に改良を加えたものである。Jasinski DR., “Assessment of the Ab
use Potential of Morphine-Like Drugs (男性に利用される方法)”, Drug Addiction I
(Martin, W.R., ed.), 1997:197-258. Springer-Verlag, New York; Preson KL, Jasinsk
i DR. Testa M., “Abuse Potential and Pharmacological Comparison of Tramadol and
Morphine”, Drug and Alcohol Dependence 1991;27:7-17を参照されたい。これらの文
献はいずれも、参照により本明細書に組み入れる。このアンケートは10項目からなり、血
液を採取する10分前に被験者が評価した。この項目は、アヘン作動薬薬剤の徴候に関連付
けられたものであり、次の通りであった。
【0164】
被験者への質問内容: 1)あなたは薬剤の効果を感じますか? 2)あなたは皮膚にかゆみを
感じますか? 3)あなたは楽な気持ちを感じますか? 4)あなたは眠気を感じますか? 5)あな
たは酔いを感じますか? 6)あなたは神経のいらだちを感じますか? 7)あなたは元気いっぱ
いであると感じますか? 8)あなたは話をする必要性を感じますか? 9)あなたは胃のむかつ
きを感じますか? 10)あなたはめまいを感じますか? この際、被験者は、100mmの視覚的ア
ナログスケール(VAS)に沿って縦線を引くことにより、項目の評価を行った。このスケー
ルの一端には「まったく感じない」と記され、他端には「かなり感じる」と記されていた
。
【0165】
左眼の瞳孔サイズは、基準時間(投与前30分以内)および投与後0.5、1、2、4、6、9、お
よび12時間で測定した。また、被験者は、基準時間および投与後0.5、1、2、4、6、9、お
よび12時間で薬効スコアを評価し、改良型特異的薬効アンケート調査(“MSDEQ")用の視覚
的アナログスケール上に記入した。
【0166】
ナルトレキソン用量に対する11種のレスポンス(MSDEQの質問および瞳孔直径の測定)を
示すそれぞれのグラフを、視覚的および統計的に調べ、この試験に使用したヒドロコドン
の用量と組み合わせて、ナルトレキソンの公称有効用量を決定した。
【0167】
報告した副作用は、一般にオピオイド鎮痛薬の投与に関連付けられるものであり、その
ほとんどは「中程度」のものとして分類された。重度の副作用や死亡はまったく起こらな
かった。また、副作用が原因で試験を中断した患者はいなかった。
【0168】
結果は図1および2に示されている。
【0169】
図1は、ヒドロコドンにより誘発されたVAS(視覚的アナログスケール)「薬効」に対する
ナルトレキソンの拮抗作用を示している。この図は、改良型特異的薬効アンケート調査の
最初の質問に関するものであり、被験者への質問内容は、「あなたは薬剤の効果を感じま
すか?」である。これらの結果から示唆されるように、ナルトレキソンに関して用量‐応
答効果が存在し、ナルトレキソン用量の増大に伴ってヒドロコドンのVAS「薬効」は減少
した。ナルトレキソン用量6.4mgの場合、ナルトレキソン用量3.2mgの場合よりも、ヒドロ
コドン用量15mgの薬効に対してより大きな拮抗作用を示した。ヒドロコドンのオピオイド
作用は、ナルトレキソン用量6.4mgによって完全にはブロックされなかった。図2は、ヒド
ロコドンにより誘発された瞳孔収縮に対するナルトレキソンの拮抗作用を示している。こ
れらの結果からもナルトレキソンに関して用量‐応答効果が示唆され、ヒドロコドン15mg
を投与された被験者の瞳孔収縮は、ナルトレキソンの用量を増大させると減少した。ナル
トレキソン用量6.4mgの場合、ナルトレキソン用量3.2mgの場合よりも、ヒドロコドンによ
り誘発された瞳孔収縮に対してより大きな拮抗作用を示した。ヒドロコドンによる瞳孔収
縮は、ナルトレキソン用量6.4mgによって完全にはブロックされなかった。偽薬グループ
で、最小量の瞳孔収縮を生じた。ヒドロコドン+ナルトレキソン偽薬グループは、最大の
瞳孔収縮を示し、従って、瞳孔直径の測定値は最小であった。
【0170】
実施例2 実施例2では、正常な健常女性被験者において、10期、ランダム化、交差、お
よび単純盲検による試験を行って、経口ナルトレキソン対経口ヒドロコドンの比を評価し
た。これにより、名目上、オピオイド作動薬作用は最小限に抑えられた。21人の被験者が
この試験に登録し、16人が試験を完了した。10種の治療薬には、HYIR/APAP(錠剤1個あた
りヒドロコドン7.5およびアセトアミノフェン750mgの2つの錠剤、Vicodin ES(登録商標))
+次の用量:0.4mg/10mL、0.8mg/10mL、1.6mg/10mL、3.2mg/10mL、4.8mg/10mL、6.4mg/10
mL、9.6mg/10mL、12.8mg/10mLのナルトレキソン経口液剤および偽薬ナルトレキソン経口
液剤、更に、ヒドロコドン即時放出性コンパラトール錠剤(2×750mg Trilisate(登録商標
)錠剤)+偽薬ナルトレキソン経口液剤が含まれていた。いずれの治療薬についても、絶食
状態で投与した。それぞれの投与の間で48時間のウォッシャアウト期間を設けた。被験者
を、これらの10種の治療薬グループの10期の治療シーケンスにランダムに割り当てた。最
初の投与を行う前の夕方に被験者は試験施設に集まり、最後の投与を行ってから24時間後
の投与評価が終了するまで拘束状態にあった。安全性の測定は、副作用の報告、生命徴候
、異常な実験値、異常な外診結果、およびECGの結果からなっていた。血漿中のヒドロコ
ドン、ナルトレキソン、および6-β-ナルトレキソールレベルを調べた。これから薬物動
態学的値が計算および分析されるであろう。また、薬力学的パラメーター(瞳孔サイズお
よび改良型特異的薬効アンケート調査)についても評価した。
【0171】
投与レジメ 投与レジメは次の通りであった。
【0172】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(偽薬)と一緒にヒドロコドン即時放出性コンパラトール(偽薬)錠剤を投与した。投
与後さらに4時間絶食を続けた。
【0173】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(偽薬)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さらに
4時間絶食を続けた。
【0174】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(0.4mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0175】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(0.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0176】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(1.6mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0177】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(3.2mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0178】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(4.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0179】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(6.4mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0180】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(9.6mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0181】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(12.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0182】
各投与日に所定の薬剤を各用量で投与したが、被験者はその投与前8時間にわたり絶食
し、投与後さらに4時間絶食を続けた。最初の投与(0時間)を行う前に(投与前30分以内に
)ベースライン用血液サンプル(血漿中のヒドロコドン、ナルトレキソン、および6-β-ナ
ルトレキソールを調べるため)を採取し、更に、投与後0.5、1、2、4、6および9時間で採
取した。サンプルはすべて、予定時間の±2分以内に採取した。基準時間(投与前30分以内
)ならびに投与後0.5、1、2、4、6および9時間で血液サンプルの採取を行う直前に以下の
薬力学的パラメーターの測定を行った。
【0183】
各投与期間の直前にナルトレキソン散剤4、8、16、32、48、64、96、および128mgを秤
量し、8種のナルトレキソンストック溶液を調製した。これらの散剤アリコートはそれぞ
れ、蒸留水50ml+単シロップ50ml中に溶解した。最終溶液は100mLであり、その濃度は0.0
4、0.08、0.16、0.32、0.48、0.96、および1.28mg/mLであった。これらの濃度を用いるこ
とにより、各投与期間中に同じ体積(10mL)のナルトレキソン経口液剤を投与できるように
した。ナルトレキソン偽薬液剤は、活性溶液と同じビヒクルで調製した。活性溶液と同じ
ような味を呈するように、苦味剤Bitterguard Powder(デナトニウムベンゾエート)の添加
を行った。
【0184】
薬力学的測定 実施例2の薬力学的測定値は、上記の実施例1で述べた手順に従って得た
。
【0185】
所定の時間における各処置に関する平均「薬効」VASスコアおよび瞳孔直径を図3および
4にそれぞれ示す。一般的には、ナルトレキソンの用量を増大させて(0mg〜12.8mgの範囲)
ヒドロコドン即時放出剤/アセトアミノフェン(「HYIR/APAP」)を1回投与すると、「薬効
」VASスコアが全体的に減少するとともに瞳孔収縮も減少した。図5および6は、各ナルト
レキソン用量の対数値に対して、対応する平均最大「薬効」VASスコア(±95% CI)および
平均最小瞳孔直径(±95% CI)を表している。これらの図から、瞳孔効果に関する用量‐応
答関係の方が「薬効」VAS応答の場合よりも大きな用量‐応答関係を示すことが分かる。
【0186】
これらの結果から分かるように、ナルトレキソン0.4mgを投与した場合でさえも、ヒド
ロコドン投与の薬理学的効果は低減した。ナルトレキソン約0.4mgのときにヒドロコドン
用量15mgに対して最小の拮抗作用を示した。ナルトレキソン用量が0.4mgを超えると、ヒ
ドロコドン投与の効果が減少し始めた。
【0187】
報告した副作用は、一般にオピオイド鎮痛薬の投与に関連付けられるものであり、その
ほとんどは「マイルドな」ものとして分類された。全体で5人の被験者(5/21)は、試験を
中断した。3人の被験者は副作用が原因で中断した。これらの被験者のうちの2人は、重度
でないと分類された副作用を伴った。1人の被験者は、重度であると分類された貧血を起
こし、鉄治療を必要とした。他の2人の被験者は、試験に参加できなくする情報が病歴中
に存在すると担当医師が判断したために中断した。この試験で死亡は起こらなかった。
【0188】
一般的には、ナルトレキソン経口液剤の用量を増大させて(0mg〜12.8mgの範囲)ヒドロ
コドン即時放出性錠剤15mgを1回投与すると、「薬効」VASスコアが全体的に減少し瞳孔直
径は増加した。
【0189】
実施例3 実施例3では、ヒドロコドン即時放出性錠剤およびナルトレキソン経口液剤を
投与したモルヒネ依存性ボランティアにおいて性急型禁断症状を評価した試験の結果を提
示する。この試験は、単純盲検、1回投与、およびプラシーボ対照によるナルトレキソン
用量増大試験であり、オピオイドに対して身体的依存のある被験者を対象とした。実験対
象被験者(5人)は、Narcanチャレンジ、嗜癖重度指数スコア、身体検査、観察、および尿
薬剤スクリーニングの結果から判断して、オピオイド依存性であり、その時点で嗜癖に対
する治療を望んではいなかった。ヒドロコドン即時放出剤およびナルトレキソンの同時投
与を行った後で性急型禁断症状を評価するために、ヒドロコドン乱用患者の使用する用量
レベルをシミュレートすべく、ヒドロコドン即時放出剤の用量30mgを選択した。これは、
オピオイドの投与されたことのない被験者において、他の一般に使用されるオピオイドと
等価な鎮痛作用を示すと考えられる用量である。ヒドロコドンの相対的鎮痛効果は、オキ
シコドンの鎮痛効果と同等であり、経口モルヒネの鎮痛効果の約2倍であると考えられて
いる。
【0190】
試験処置 処置は次の通りであった。
【0191】
ヒドロコドン/アセトアミノフェン即時放出性(HYIR/APAP)錠剤30mg(Lortab(登録商標)
3×10mg) + 用量を増大させたナルトレキソン経口液剤0、0.25mg、0.5mg、1.0mg、および
2.0mg。ヒドロコドン/アセトアミノフェン即時放出性(HYIR/APAP)錠剤30mg(Lortab(登録
商標) 3×10mg) + ナルトレキソンプラシーボ経口液剤。ナルトレキソン経口液剤および
プラシーボ液剤は、実施例1〜2に従って調製した。
【0192】
規則的な時間間隔で、すなわち毎日午前6時および10時ならびに午後4時および10時に硫
酸モルヒネ15mgを筋肉内投与することによって、5日間かけて被験者を安定化させた。硫
酸モルヒネ15mgの筋肉内投与は、ヒドロコドン30mgの経口投与と同等である。安定化を行
った後、試験治療薬の投与日の午前10時に試験治療薬を投与、それから6時間にわたり観
察を行った。6時間後に性急型禁断症状が現れない場合、午後4時に硫酸モルヒネ15mgを再
び筋肉内投与した。次の試験薬剤の投与を行う前に48時間かけて被験者を安定化させた。
各処置(1〜4)後、性急型禁断症状が現れない場合、以下の処置の中から次の番号順に試験
治療薬を被験者に投与した。
【0193】
処置番号1: 8時間の絶食を行った後、投与日のほぼ10:00にプラシーボナルトレキソン(
10mL)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与
後、引き続き(4)時間絶食を行う。
【0194】
処置番号2: 8時間の絶食を行った後、投与日のほぼ10:00に0.25mgナルトレキソン(10m
L)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後
、引き続き(4)時間絶食を行う。
【0195】
処置番号3: 8時間の絶食を行った後、投与日のほぼ10:00に0.5mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0196】
処置番号4: 8時間の絶食を行った後、投与日のほぼ10:00に1.0mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0197】
処置番号5: 8時間の絶食を行った後、投与日のほぼ10:00に2.0mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0198】
投与の0.5時間前および投与の0.5、1、2、4、および6時間後に、血液サンプルを採取し
た。Pupilscan瞳孔計を用いて瞳孔直径の測定値を求め、ミリメートルの少数以下を四捨
五入してミリメートル単位で記録した。各試験期間の後、48時間にわたるウォッシュアウ
ト期間を設けた。4人の被験者が試験を終了し、1人の被験者はとりやめた。ナルトレキソ
ンの効果として、1および2mgの場合、わずかな禁断(禁断の徴候)が得られた。
【0199】
プロトコルを修正し、12人の実験対象被験者をこのプロトコルに従って試験した。この
プロトコルは、ナルトレキソンの比を増大させたこと以外は先に述べた試験と同じであっ
た。修正プロトコルのナルトレキソン用量は、0、1、2、4、および8mgであった。実験対
象被験者のうちの8人が試験を終了し、4人はとりやめた。
【0200】
各被験者に対して生命徴候を監視し、更に、オピオイド禁断の徴候および症状を監視し
た。禁断徴候としては、鼻づまりもしくは鼻水、流涙、あくび、発汗、震え、嘔吐、起毛
、瞳孔散大、被刺激性、および不眠が挙げられる。禁断症状としては、温度変化の感覚、
関節、骨、もしくは筋肉の痛み、胃の痙攣、皮膚のかゆみ、および吐き気が挙げられ、被
験者は、これらの症状の主観的な経験を報告する。
【0201】
薬剤併用の主観的経験の尺度を示すために、被験者は、試験期間にわたりアンケートの
質問に答えた。実施例1に記載したように、質問に対する答えを視覚的アナログスケール
上で等級分けした。評価した主観的経験は、次の通りであった:薬剤の好き嫌い、薬効の
知覚能力、発汗、不眠、震え、流涙、鳥肌、胃の不調、鼻血、眠気、寒気、発熱、筋肉痛
、緊張もしくは弛緩、錯乱、恐怖感、被刺激性、多弁性、禁断の感覚、疾患の感覚。また
、被験者の観察を次の観点から行った:あくび、ひっかき、弛緩、鼻血、被刺激性、禁断
。このほか、血圧、脈拍数、呼吸数、瞳孔サイズ、および体温の監視も行った。
【0202】
被験者のうちの5人に対するデータを以下に示す。図7A〜Cは、アンケートの質問から得
られたヒドロコドンの主観的知覚の平均スコアを表しており、投与後の時間の関数として
、またナルトレキソンの用量の関数としてプロットしたものである。図7Aは、種々の量の
ナルトレキソンの存在下で被験者がヒドロコドンの効果を感じる能力を示している。図7B
および7Cは、それぞれ、種々の量のナルトレキソンの存在下におけるヒドロコドンに対す
る被験者の好ましいと感じるまたは好ましくないと感じる主観的経験を示している。
【0203】
図8AおよびBは、ヒドロコドンの効果についての主観的知覚の平均スコアを表しており
、投与後の時間の関数として、またナルトレキソンの用量の関数としてプロットしたもの
である。図8Aは、種々の量のナルトレキソンの存在下におけるヒドロコドンの効果により
禁断症状の主観的知覚を示している。図8Bは、種々の量のナルトレキソンの存在下におけ
る疾患についての主観的経験を示している。図9Aは、種々の量のナルトレキソンの存在下
においてヒドロコドンが瞳孔サイズに及ぼす影響を示している。図9Bは、種々の量のナル
トレキソンの存在下におけるヒドロコドンの効果による明らかな禁断症状を観測者の立場
から示したものである。
【0204】
図10A〜Cは、図7A〜Cに示されている曲線の下側面積を6時間の観察時間にわたり積分し
てナルトレキソンの用量の関数として示したものであり、更に、ナルトレキソンのプラシ
ーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルも示されている
。図10Aから分かるように、8mgまでのナルトレキソンを用いた場合、被験者がヒドロコド
ンの効果を知覚する能力はなくならない。すなわち、各ナルトレキソン用量に対して観測
された実測値から得られるAUC(0〜6時間)は、ナルトレキソンプラシーボ応答に対する95%
信頼限界内に完全に含まれる。図10Bは、ヒドロコドンに対する被験者の好ましいと感じ
る主観的経験のAUC(0〜6時間)をナルトレキソンの用量の関数として示している。図10Bか
ら分かるように、ナルトレキソンの用量が1mgを超えると、被験者の好ましいと感じる経
験は低減する。すなわち、AUCの実測値(0〜6時間)は、ナルトレキソン約1mgのところでナ
ルトレキソンプラシーボに対する95%信頼限界よりも小さくなる。図10Cから分かるように
、ナルトレキソンの用量が1mgを超えると、被験者の好ましくないと感じる経験は増大す
る。すなわち、AUCの実測値(0〜6時間)は、ナルトレキソン約1mgのところでナルトレキソ
ンプラシーボに対する95%信頼限界よりも大きくなる。
【0205】
図11A〜Cは、図8A〜Bおよび図9Aに示されている曲線の下側面積を6時間の観察時間にわ
たり積分してナルトレキソンの用量の関数として示したものであり、更に、ナルトレキソ
ンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルも示
されている。図11Aは、種々の量のナルトレキソンの存在下で禁断の主観的経験のAUC(0〜
6時間)を示している。図11Aから分かるように、ナルトレキソンの用量が約0.75mgを超え
ると、疾患の主観的経験が現れる。すなわち、各ナルトレキソン用量に対して図8Aで観測
されたAUCの実測値(0〜6時間)は、ナルトレキソン約0.75mgのところでナルトレキソンプ
ラシーボ応答に対する95%信頼限界より大きくなる。図11Bは、種々の量のナルトレキソン
の存在下で疾患の主観的知覚のAUC(0〜6時間)を示している。図11Bから分かるように、ナ
ルトレキソンの用量が約0.75mgを超えると、疾患の主観的経験が現れる。すなわち、各ナ
ルトレキソン用量に対して図8Bで観測されたAUCの実測値(0〜6時間)は、ナルトレキソン
約0.75mgのところでナルトレキソンプラシーボ応答に対する95%信頼限界より大きくなる
。図11Cは、瞳孔サイズ変化の実測値のAUC(0〜6時間)をナルトレキソンの用量の関数とし
て示している。図11Cから分かるように、8mgまでのナルトレキソンを用いた場合、ヒドロ
コドンの縮瞳効果はなくならない。すなわち、各ナルトレキソン用量に対して図9Aで観測
された実測値から得られるAUC(0〜6時間)は、ナルトレキソンプラシーボ応答に対する95%
信頼限界内に完全に含まれる。
【0206】
臨床試験の結果、ヒドロコドンは、ナルトレキソンと併用した場合、0.5時間未満に出
現し、0.5〜1時間以内にピークに達し、3〜4時間以内に著しく減少することが分かった。
弱い用量‐応答曲線が得られた。ナルトレキソンを添加すると、ヒドロコドンに対する好
ましいと感じる主観的経験は低減し、ヒドロコドンに対する嫌悪の主観的経験は増大し、
疾患についての主観的経験およびヒドロコドン効果により禁断症状の主観的経験は増大し
た。これらの経験は、明らかに嫌悪感を生じるものであった。
【0207】
本発明の特定の好ましい実施例を参照しながら本発明について説明および例示を行って
きたが、本発明の精神および範囲から逸脱することなく、本発明の範囲内で容易に変更を
加えることができるは、当業者には分かるであろう。このような変更は、添付の特許請求
の範囲に含めるものとする。
【図面の簡単な説明】
【0208】
以下の図面は本発明の実施形態を例示するものであり、特許請求の範囲により包含され
る本発明の範囲を制限するものではない。
【図1】図1は、実施例1についてのヒドロコドン誘導VAS(視覚アナログスケール)「薬効」のナルトレキソン拮抗作用を示す。
【図2】図2は、実施例1についてのヒドロコドン誘導瞳孔縮小のナルトレキソン拮抗作用を示す。
【図3】図3は、実施例2のそれぞれの治療についての時間経過に対する平均「薬効」VASスコアを示す。
【図4】図4は、実施例2のそれぞれの治療についての時間経過に対する平均「薬効」瞳孔直径を示す。
【図5】図5は、実施例2のそれぞれのナルトレキソン用量からの対数に対する平均最大「薬効」VASスコア(±95% CI)を示す。
【図6】図6は、実施例2のそれぞれのナルトレキソン用量からの対数に対する平均最少瞳孔直径(±95% CI)を示す。
【図7】A:実施例3において可変量のナルトレキソン存在下でヒドロコドン効果を感じる被験者の能力を示す。 BおよびC:実施例3において可変量のナルトレキソン存在下でヒドロコドンを被験者が好ましいまたは嫌だとそれぞれ感じる主観的経験を示す。
【図8】A:実施例3における可変量のナルトレキソン存在下でのヒドロコドン効果からの禁断症状の被験者の認識を示す。 B:実施例3における可変量のナルトレキソン存在下での疾病の主観的経験を示す。
【図9】A:実施例3において可変量のナルトレキソン存在下でヒドロコドンが瞳孔サイズに及ぼす効果を示す。 B:実施例3において可変量のナルトレキソン存在下でヒドロコドン効果からの禁断症状の、観測者から見たときの見かけの程度を示す。
【図10】A〜C:ナルトレキソン用量の関数としての、6時間の観察期間にわたって積分した図7A〜Cに示される曲線下面積、およびナルトレキソンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルを示す。
【図11】A〜C:ナルトレキソン用量の関数としての、6時間の観察期間にわたって積分した図8A〜Bおよび図9Aに示される曲線下面積、およびナルトレキソンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルを示す。
【技術分野】
【0001】
オピオイドは、オピオイド作動薬としても知られているもので、アヘンやモルヒネのよ
うな性質を示す薬物の1グループである。オピオイドは主に中程度ないし強力な鎮痛薬と
して使用されるが、他にも多くの薬理作用を有しており、例えば、睡眠作用、呼吸抑制作
用、気分の変化、意識の低下を起こさない精神混濁などを示す。オピオイドは脳や他の組
織にある立体特異的で飽和性の結合部位と相互作用して、作動薬として機能する。内在性
のオピオイド様ペプチドが特に中枢神経系の領域に存在しており、痛みの認識;運動、気
分および行動;神経内分泌学的機能の調節に関係していると推測される。アヘンは20種類
を上回る種々のアルカロイドを含有する。モルヒネ、コデインおよびパパベリンはこのグ
ループに属している。
【背景技術】
【0002】
19世紀の中頃までに、粗製のアヘン製剤ではなく純粋なアルカロイド(例:モルヒネ)
の使用が医療分野全体に広まり始めた。モルヒネの非経口使用はより重症のさまざまな強
迫的薬物使用を生じる傾向があった。オピオイドに関する耽溺性の問題から、耽溺性を生
じる可能性のない鎮痛薬の研究が促進されるようになった。1967年頃までに、モルヒネ様
薬物と、拮抗薬と、その後「混合作動薬−拮抗薬」と呼ばれたものの複雑な相互作用は、
オピオイドおよびその関連薬物の2以上のタイプの受容体の存在を仮定することで最もよ
く説明できると、研究者らにより結論づけられた。モルヒネ様作用を有する新しい完全合
成物質の出現により、「オピオイド」という用語は一般的に、オピオイド受容体の亜種の
いずれかと立体特異的に結合してアゴニスト作用を生じる全ての外因性物質の総称として
残されたものである。
【0003】
オピオイドの反復使用に伴う耐薬性と身体的依存性の発現の可能性はあらゆるオピオイ
ド薬物に特徴的な性質であり、精神的依存性(すなわち、耽溺性)を生じる可能性は、た
とえ医原性耽溺が稀であっても、オピオイドによる疼痛治療をおこなううえで重大な関心
事の一つとなっている。オピオイドの使用に関連したもう一つの重大な問題は、気晴らし
を目的として疼痛患者から(患者ではない)別人、例えば麻薬中毒者にこれらの薬物が渡
ることである。
【0004】
オピオイドの総合的な乱用可能性はただ一つの要因によって確立されるものではない。
実際、各種の要因が存在し、例えば、薬物の中止が薬物を見つけようとする行動をとらせ
るのに十分な苦痛を引き起こす種類の身体的依存性を生じる薬物の能力、他の麻薬の中止
により生じる禁断症状を抑制する能力、モルヒネや他のオピオイドにより生じる多幸感と
同様の多幸感を誘発する程度、薬物を通常の治療範囲を超えて投与したときに起こる毒性
のパターン、水溶性のような薬物の物理的性質などがある。そのような物理的性質により
、その薬物が腸管外経路によって乱用されそうであるか否かを判定することができる。
【0005】
米国では、強迫的薬物常用者を規制する努力として、強迫的薬物常用者の疼痛治療にオ
ピオイドを使用することに制限を設けることで、薬物の入手可能性を抑えることが含まれ
ている。実際、医師は、このような薬物に対して精神的依存性(すなわち、耽溺性)を発
現する傾向があると思われる人達にさえ強力なオピオイド鎮痛薬を投与するという選択に
直面する場合がしばしばある。この問題のため、乱用の可能性がない別の薬物で十分であ
る場合はこれらの患者にオピオイドを投与すべきでないこと、さらにこれらの患者には薬
物を非経口的に自己投与させないようにすべきで、一度に二三日分しか処方しないように
すべきことが勧告された。
【0006】
オピオイドの常用および依存性が生ずるには少なくとも3つの基本的なパターンが確認
されている。第1のパターンは、個人の薬物使用が医学的治療に関係して始まり、薬物が
例えば医師から供給されることによるものである。第2のパターンは、経験的または「気
晴らし」的薬物使用から始まって、使用回数が次第に増えていくというものである。第3
のパターンは、薬物使用が前記の方法のいずれかで始まるが、その後、体制化された耽溺
治療プログラムから得られるメタドンのような経口オピオイドに移るというものである。
【0007】
耐薬性とは、同じレベルの鎮痛または多幸感を達成するために、ある期間にわたってオ
ピオイドの用量を増加する必要があること、または同量の反復投与によって鎮痛、多幸感
または他のオピオイド効果の低下が観察されることを意味する。オピオイドの呼吸抑制、
鎮痛、鎮静、催吐、多幸感作用に対して顕著な耐薬性が生じることはわかっている。しか
しながら、この耐薬性が常用者または疼痛治療の必要な患者において発生する速度は使用
パターンに依存している。オピオイドを頻繁に使用する場合は、用量の増加が必要であり
うる。耐薬性はオピオイドのすべての効果に対して同等にまたは同一速度で発生するもの
ではない。呼吸抑制作用に対して高い耐性を示す使用者でさえ縮瞳や便秘症を呈し続ける
。オピオイドに対する耐性はたいてい禁断症状がなくなったときに消失する。
【0008】
身体的依存性はオピオイドの反復投与または長期間の使用により発生しうる。身体的依
存性はオピオイド使用の中止後に徐々に現れるか、または麻薬拮抗薬の投与後すぐ(例え
ば20分以内)に現れる(「性急型禁断症状」という)。依存性が確立されている薬物、そ
の使用期間および投与量に応じて、禁断症状の回数、種類、持続期間および重症度はさま
ざまに変化する。最も普通に見られる禁断症状としては、食欲不振、体重減少、瞳孔拡大
、異常な発汗と交互に起こる悪寒、腹部の痙攣、悪心、嘔吐、筋痙縮、高興奮性、流涙、
鼻漏、鳥肌、および心拍数の増加が挙げられる。典型的には、禁断症状は最後に投薬して
から24〜28時間後に発生し、3日目ごろに最大強度に達し、3週目まで軽減しない可能性
がある。
【0009】
オピオイドに対する精神的依存性(すなわち、耽溺性)は、多幸感や、例えば心理社会
経済的圧力からの逃避を達成する方向に向かう、薬物を見つけようとする行動により特徴
づけられる。中毒者は非医療目的で、また、自己危害に直面して、オピオイドを投与し続
けるであろう。
【0010】
薬理学的には、オピオイド拮抗薬は一般にオピオイド作動薬のあらゆる作用効果をブロ
ックするか逆転させるものである。オピオイド拮抗薬の一つの用途は、オピオイドを中毒
者に投与した際に現れる可能性のある多幸感作用をブロックするために、ナルトレキソン
(naltrexone)で1日1回処置するものである。低用量のオピオイド拮抗薬は、個体がオピ
オイドに対して身体的依存性を示すか否かを判定するために使用されている。最も一般的
には、オピオイド作動薬を過剰投与された個体に対してオピオイド効果を逆転させるため
にオピオイド拮抗薬が使用される。
【0011】
以前に当技術分野では、オピオイド鎮痛薬に伴う乱用性を防止しようとする試みがなさ
れた。典型的には、一定量のオピオイド鎮痛薬は、同量を経口投与したときと比べて非経
口投与したときの方が効力が強い。したがって、流行している経口投薬の乱用様式の一つ
は、その剤形からオピオイドを抽出し、その後「恍惚感」を得るために(「適当な」注射
用ビヒクルを用いて)オピオイドを注射するというものである。したがって、乱用を抑え
る試みは次のことに集中していた。すなわち、経口的には活性でないが、ある人がオピオ
イドを溶解して非経口的にそれを投与しようとする場合にオピオイドの鎮痛効果を実質的
にブロックしうるオピオイド拮抗薬を経口投薬剤形中に配合することである。
【0012】
例えば、Sanofi-WinthropからTalwin(登録商標)Nxとして市販されて米国で入手できる
錠剤には、ペンタゾシンとナロキソンの組合せが利用されている。Talwin(登録商標)Nxに
は50mg塩基に等しい塩酸ペンタゾシンおよび0.5mg塩基に等しい塩酸ナロキソンが含まれ
ている。Talwin(登録商標)Nxは中程度から重症の疼痛の緩和に効果があると表示されてい
る。この組合せ物中に存在するナロキソンの量は経口摂取したときには何の作用も示さず
、ペンタゾシンの薬理作用を妨害しない。ところが、この量のナロキソンを注射により投
与すると、麻薬性鎮痛薬に対する強い拮抗作用が認められた。こうして、ナロキソンの配
合は、経口投薬剤形を可溶化して注射したときに起こる経口ペンタゾシンの不正な使用形
態を制限することを意図したものである。したがって、この投薬剤形は以前の経口ペンタ
ゾシン製剤よりも不正な非経口使用の可能性が少ない。しかしながら、それは依然として
経口による患者の不正な使用および乱用を受けやすく、例えば、患者が一度に複数回分の
用量を摂取する場合である。
【0013】
Sunshineらは、Clin. J. Pain, 1988, 4:35-40(「経口投与後のペンタゾシン対ペンタ
ゾシン−ナロキソン併用の鎮痛効果」)において、ペンタゾシン50mgの鎮痛効果に及ぼす
0.5mgナロキソンの添加の影響を報告した。この併用は、疼痛強度差の合計(SPID)に関し
て、また、4時間目の緩和および疼痛強度差(PID)に関してペンタゾシンより著しく効果
のないことが見出された。中程度の基線疼痛を有する患者の場合、この併用はSPIDに関し
てと3および4時間目の緩和およびPIDに関してペンタゾシンより疼痛緩和が有意に低か
った。重度の基線疼痛を有する患者においては、ペンタゾシンとペンタゾシン−ナロキソ
ン併用との間に有意差はなかった。
【0014】
Wangらは、J. Clin. Pharmacol., 1981, 21:162-8(「経口鎮痛薬の交差および平行研
究」)において、慢性疼痛患者の交差研究でのパーコダン(Percodan:登録商標)(オキシコ
ドンHCl 4.5mg、オキシコドンテレフタレート0.28mg、アスピリン224mg、フェナセチン16
0mgおよびカフェイン32mgを含む)単独およびプラシーボと比較したときのナロキソン0.25
mgとパーコダンとの併用を報告した。この併用は試験の後半時間では鎮痛毎時パラメータ
ーのほとんどに関してパーコダン単独よりも低い平均スコアを有していた。しかし、概略
変数に関しては、この併用はプラシーボまたはパーコダン単独のいずれとも有意差を示さ
なかった。
【0015】
疼痛治療のためにブプレノルフィンとナロキソンを一定割合で組合せたもの(Temgesic(
登録商標)Nx, Reckitt & Colman)がニュージーランドで1991年に導入された。
【0016】
ドイツでは重度の疼痛の管理のため1978年以来チリジン(50mg)とナロキソン(4mg)から
なる一定の併用療法が利用可能になっている(Valoron(登録商標)N, Goedecke)。これらの
薬物を併用することの論理的根拠は、有効な疼痛緩和と、モルヒネ受容体でのナロキソン
誘導拮抗作用によるチリジン中毒の予防にある。
【0017】
米国特許第3,773,955号(Pachterら)には、非経口投与時には鎮痛、多幸感、または身体
的依存性を起こさず、それにより鎮痛薬の非経口乱用を防止できる、経口的に有効な鎮痛
組成物が記載されている。この種の組成物は鎮痛薬の経口量につき約0.1〜10mgのナロキ
ソンを含んでいた。この文献はオピオイドの経口乱用に関心をもっていなかった。
【0018】
米国特許第3,493,657号(Lewensteinら)には、ナロキソンとモルヒネまたはオキシモル
フォンを含む組成物が記載されており、この組成物は幻覚のような副作用を発生させずに
強力な鎮痛効果を発揮するとされている。
【0019】
米国特許第4,457,933号(Gordonら)には、鎮痛量のオピオイドを比較的狭い特定の範囲
でナロキソンと組み合わせることにより、オキシコドン、プロポキシフェンおよびペンタ
ゾシンなどの強力な鎮痛薬の経口および非経口の両方の乱用可能性を減少させる方法が記
載されている。2.5〜5:1(重量部)の比を有するオキシコドン−ナロキソン組成物および16
〜50:1(重量部)の比を有するペンタゾシン−ナロキソン組成物が好ましいものであった。
オピオイドと併用されるべき用量のナロキソンは、オピオイドの経口鎮痛活性に実質的に
影響を及ぼさずに、オピオイドの経口または非経口乱用の可能性を実質的に排除すると述
べられている。
【0020】
米国特許第4,582,835号(Lewis)には、舌下投与に有効な量のブプレノルフィンをナロキ
ソンと共に投与することによる疼痛の治療方法が記載されている。Lewisによれば、ナロ
キソンとブプレノルフィンの投与量比は、非経口投与の場合が1:3〜1:1、舌下投与の場合
が1:2〜2:1である。
【0021】
当分野においては、経口オピオイド製剤は、非経口投与のみならず、患者や中毒者が投
薬間隔の間に処方量より多い経口量を経口的に自己投与する場合には経口によっても乱用
されることが次第に認識されてきた。したがって、経口投与可能で、経口乱用の可能性が
低い疼痛治療用の製剤を開発する必要がある。
【0022】
本発明らの知るかぎりでは、経口的に併用した場合に鎮痛薬として有効であるが、身体
依存性被験者には忌み嫌われる、ある比率のオピオイド作動薬とオピオイド拮抗薬はこれ
まで認識されていない。
【発明の開示】
【発明が解決しようとする課題】
【0023】
本発明の目的および概要 本発明の目的は、市販されている従来の投薬剤形よりも経口
投与によって乱用の可能性が低くなるオピオイド鎮痛薬の経口投薬剤形を提供することで
ある。
【0024】
本発明の更なる目的は、治療的痛覚消失を付与し、さらに身体依存性被験者が多量(例
えば、通常の処方量の2〜3倍ほど)のオピオイドを摂取したり投与されたときには拒否
的な「嫌悪」体験を与える方法ならびにオピオイド鎮痛薬の経口投薬剤形を提供すること
である。
【0025】
本発明の更なる目的は、通常の処方量より多い量(例えば、通常の処方量の約2〜3倍
)のオピオイドを摂取する非身体依存性被験者において、拮抗薬不在下での同量のオピオ
イドと比べたときほど正の強化(positively reinforcing)を与えないやり方で治療的痛覚
消失を付与する方法ならびにオピオイド鎮痛薬の経口投薬剤形を提供することである。
【0026】
本発明の更なる目的は、オピオイド鎮痛薬の経口投薬剤形を用いて、該投薬剤形の経口
乱用可能性を低下させながらヒト患者の疼痛を治療する方法を提供することである。
【0027】
本発明の更なる目的は、経口乱用可能性が低くなるようなオピオイド鎮痛薬の経口投薬
剤形を製造する方法を提供することである。
【課題を解決するための手段】
【0028】
前記および他の目的は本発明によって達成される。本発明は、一部には、経口的に併用
した場合に鎮痛薬として有効であるが、身体依存性被験者には嫌われる、オピオイド拮抗
薬とオピオイド作動薬(鎮痛薬)のある比率が存在する、という驚くべき知見に基づいて
いる。本発明者らの知るかぎりでは、このことは当業者、例えば、麻薬中毒学者、鎮痛学
者、臨床薬理学者によってさえ考慮されたことがない。ある組合せ製剤(拮抗薬/作動薬
の組合せ)が、ある集団(疼痛患者)には本質的に治療的であるのに、通常の処方量と同
量またはそれより多量(例えば、通常の処方量の約2〜3倍)のオピオイドを投与した場
合に別の集団(例えば、身体依存性被験者)には受け入れられない(嫌われる)というこ
とは驚くべきことである。
【0029】
本発明は、一部には、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者
における直接測定により、またはヒト被験者におけるオピオイド効力(痛覚消失)の1以
上の代理指標の使用により評価したとき、痛覚消失をやや低下させうる比率で、経口的に
鎮痛有効量のオピオイド作動薬とオピオイド拮抗薬を含有する経口投薬剤形に関する。オ
ピオイド効力(痛覚消失)の代理指標としては、鎮静、呼吸速度および/または瞳孔の大
きさ(瞳孔測定による)、および「薬効」のための視覚アナログスケール(visual analog
ue scale: VAS)が挙げられる。このような代理指標は、共存量のオピオイド拮抗薬を用い
ないでオピオイドを同量使用したときに比べて、オピオイド効果の低下を示す方向での影
響を受ける。
【0030】
オピオイドがヒドロコドンで、拮抗薬がナルトレキソンである好ましい実施形態におい
ては、経口投薬剤形が二酒石酸塩の形のヒドロコドンと塩酸塩の形のナルトレキソンを含
む。
【0031】
オピオイドがヒドロコドンで、拮抗薬がナルトレキソンである好ましい実施形態におい
ては、ナルトレキソン対ヒドロコドンの重量比を約0.03〜0.27:1とすることが好ましく、
約0.05〜0.20:1とすることがより好ましい。
【0032】
本発明は、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者における直
接測定により、またはヒト被験者におけるオピオイド効果の1以上の代理指標の使用によ
り評価したとき、痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピオイド
作動薬とオピオイド拮抗薬を含有する経口投薬剤形を調製することを含んでなる、被験者
による経口オピオイド製剤の経口乱用を防止する方法に関する。前記経口投薬剤形が身体
依存性被験者によって比較的多量に(例えば、通常の処方量の約2〜3倍)摂取される場
合には、その使用は身体依存性被験者に嫌われ、かつ非身体依存性ヒト被験者においては
(単独で摂取された)オピオイドほどに正の強化を与えないことが好ましい。
【0033】
本発明はまた、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが、患者におけ
る直接測定によって、またはヒト被験者におけるオピオイド効果の1以上の代理指標の使
用によって痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピオイド作動薬
をオピオイド拮抗薬と共に経口投与することを含んでなる治療方法に関する。
【0034】
本発明はさらに、一部には、経口的に鎮痛有効量のオピオイド作動薬と経口的に活性な
オピオイド拮抗薬の組合せを含む経口投薬剤形に関し、オピオイド拮抗薬は、(i)経口投
与した際に該投薬剤形から引き出される痛覚消失のレベルを非治療レベルにまで低減させ
ない量で、かつ(ii)身体依存性被験者が通常の処方量の少なくとも2倍(しばしば処方量
の2〜3倍またはそれ以上)の量を一度に摂取しようとするとき、オピオイド拮抗薬の存
在しない同量のオピオイドに比べて、身体依存性被験者に少なくとも穏やかに拒否的な「
嫌悪」の経験(例えば、性急型禁断症状)を与える量で含まれる。好ましくは、前記経口
投薬剤形に含まれるナルトレキソンの量は、非身体依存性オピオイド中毒者に、拮抗薬の
存在しない同様の経口投薬剤形よりも少ない正の強化(例えば、より少ない「嗜好」)を
与える量である。好ましくは、前記製剤は経口投与したときに効果的な痛覚消失をもたら
すものである。
【0035】
本発明にとって、「患者における直接測定により、またはヒト被験者におけるオピオイ
ド鎮痛効力の1以上の代理指標の使用により評価したとき、痛覚消失をやや低下させうる
」という表現は、疼痛患者が、本発明に従って投与された製剤(すなわち、オピオイド作
動薬/拮抗薬の組合せ)と、同量のオピオイド作動薬を含むがオピオイド拮抗薬を含まな
い同様の製剤と、の間の差異に気づくかまたは容易には気づかないが、その組合せから鎮
痛効果を得ていることを意味する。本発明に従って投与される製剤の薬理学的効果(痛覚
消失)は、例えば、該投薬剤形を投与してから連続した時間後に患者によって報告された
鎮痛薬アンケートからのスコアによって説明することができる。痛覚消失の概略指標とし
ては、疼痛強度差の合計(SPID)および全疼痛緩和(TOTPAR)が含まれる。
【0036】
好ましい実施形態において、前記投薬剤形に含まれるオピオイド拮抗薬の量は、「薬効
」のための視覚アナログスケール(VAS)のような代理指標によって測定したとき、経口投
与した際に該投薬剤形から引き出される痛覚消失のレベルを臨床上有意に低下させうる量
である。他の実施形態において、経口投薬剤形に含まれるオピオイド拮抗薬の量は、経口
投与した際に該投薬剤形から引き出される痛覚消失のレベルをそれとわかるほどに低下さ
せうるが、痛覚消失のレベルを治療レベル以下にまで低下させないものである。
【0037】
好ましくは、経口投薬剤形に含まれる拮抗薬の量は、非身体依存性オピオイド被験者に
、拮抗薬を含まない同様の経口投薬剤形よりも少ない正の強化(例えば、より少ない「嗜
好」)を与えるものである。
【0038】
本発明はまた、ヒト患者の疼痛の治療を意図したオピオイド鎮痛薬の経口投薬剤形の調
製方法に関する。この方法は、オピオイド鎮痛薬による鎮痛効力を維持する比率であるが
、患者における直接測定によりまたはヒト被験者における痛覚消失の1以上の代理指標の
使用により評価したとき痛覚消失をやや低下させうる比率で、経口的に鎮痛有効量のオピ
オイド作動薬をオピオイド拮抗薬と組み合わせて、経口投薬剤形の経口乱用の可能性を最
小限にする様式で行われる。特定の実施形態において、この組合せは、経口投与した場合
に、(同量のオピオイド単独と比べて)該投薬剤形から引き出される痛覚消失のレベルの
臨床的に有意な低下をもたらし、また、身体依存性被験者が通常の処方量つまり常用量よ
り多いオピオイドを摂取するときには該被験者に少なくとも穏やかに拒否的な「嫌悪」の
経験(例えば、性急型禁断症状)を与える。この被験者は例えば通常の処方量より多量(
例えば、少なくとも2〜3倍)を一度に摂取することによって多幸感(「恍惚感」)を得
ようとする麻薬中毒者でありうる。前記投薬剤形に含まれる量のオピオイド拮抗薬は、薬
効のための視覚アナログスケール(VAS)のような薬理学的パラメーターによって測定した
とき、経口投与時に該投薬剤形から引き出される痛覚消失のレベルを低下させうるか、ま
たはそれとわかるほどに低下させうるが、それにもかかわらず効果的な痛覚消失を該投薬
剤形に与えることが好ましい。前記方法の好ましい実施形態において、オピオイド拮抗薬
の用量はオピオイドの鎮痛効果の代理指標に認めうる程度に影響を与える量である。好ま
しい実施形態において、経口投薬剤形に含まれる量の拮抗薬は、非身体依存性オピオイド
被験者に、拮抗薬を含まない同様の経口投薬剤形よりも少ない正の強化(例えば、より少
ない「嗜好」)を与えるものである。
【0039】
ここに記載する本発明の薬物組合せを含む経口医薬組成物は錠剤、液剤、トローチ剤、
ロゼンジ剤、水性または油性懸濁剤、分散性の粉末、顆粒、マトリックス球体または被覆
不活性ビーズを含む多粒子製剤、乳濁剤、硬質または軟質カプセル剤、シロップ剤または
エリキシル剤、微粒子(例:マイクロカプセル、マイクロスフェアなど)、バッカル錠剤
などでありうる。本発明の投薬剤形は当業者に公知のどのような望ましい製薬上許容され
る賦形剤を含んでいてもよい。前記投薬剤形はさらに、オピオイド作動薬とオピオイド拮
抗薬の即時放出を提供することができる。好ましい実施形態では、前記投薬剤形がオピオ
イド作動薬の持続放出を提供し、かつオピオイド拮抗薬の用量の一部または全部を(i)即
時放出形態、(ii)持続放出形態、または(iii)即時放出および持続放出の両形態で提供す
る。かかる実施形態はさらにオピオイド作動薬の一部を即時放出形態で含んでいてもよい
。持続放出は医薬製剤の分野の当業者に知られた製剤化/製造法に従って、例えば、オピ
オイド作動薬とオピオイド拮抗薬を含むマトリックス中に持続放出性担体を配合すること
により、またはオピオイド作動薬とオピオイド拮抗薬を含むマトリックスに持続放出性コ
ーティングを施すことによって行うことができる。
【0040】
本発明はより安全な製剤(例えば、より少ない呼吸抑制)ならびにオピオイド耐性およ
び身体依存性発現の速度が比較的遅い製剤を提供する。
【0041】
他の好ましい実施形態において、前記投薬剤形に含まれるオピオイドは、経口的に活性
な、ヒドロコドンと異なるオピオイド作動薬である。このような製剤に含まれるナルトレ
キソンの比率は、ヒドロコドンと比較したときの各種オピオイド鎮痛薬の公知の等価鎮痛
薬用量を考慮に入れて、単純な計算から容易に決定することができる。各種オピオイド鎮
痛薬の等価鎮痛薬用量は以下に提示されるか、さもなくば例えばFoley, K., N. Engl. J.
Med., 1985, 313:84-95「癌による疼痛の治療」(参照により本明細書に含めるものとす
る)から当業者には公知である。この実施形態の別の面では、ナルトレキソンの代わりに
別のオピオイド拮抗薬が、その等価拮抗薬用量で用いられる。
【0042】
特定の実施形態においては、前記製剤中に2種類のオピオイド鎮痛薬の組合せを含める
。別の実施形態では、1種類以上のオピオイド鎮痛薬を含め、さらにオピオイド拮抗薬に
加えて別の非オピオイド薬物を含める。このような非オピオイド薬物は好ましくは更なる
痛覚消失を与えるものであり、例えば、アスピリン、アセトアミノフェン、非ステロイド
系抗炎症薬(NSAIDS)、NMDA拮抗薬、およびサイコオキシゲナーゼ-II阻害剤(COX-II阻害剤
)などである。更なる実施形態において、痛覚消失以外の所望の効果を与える非オピオイ
ド薬物、例えば、鎮咳薬、去痰薬、うっ血除去剤、抗ヒスタミン剤などを含めてもよい。
【0043】
本明細書中で用いる「非経口」とは、皮下注射、静脈内、筋肉内、胸骨内への注入また
は輸液の技術を含む。
【0044】
「効果的な痛覚消失」とは、本発明においては、ヒト患者で測定したときの、許容レベ
ルの副作用と共に、疼痛の満足のゆく減少または消失として定義される。
【0045】
「持続放出」とは、本発明においては、血液(例:血漿)濃度(レベル)が、1日2回
または1日1回製剤を表示する時間にわたって、治療範囲内(最少有効鎮痛薬濃度つまり
「MEAC」以上)に、しかし毒性レベル以下に維持されるような速度での経口製剤からの薬
物(オピオイド鎮痛薬)の放出として定義される。
【0046】
「定常状態」とは、薬物の除去速度が体内への該薬物の吸収速度と同じになる時期をさ
す。
【0047】
本発明において、「オピオイド作動薬」は「オピオイド」または「オピオイド鎮痛薬」
と相互交換可能であり、オピオイドの塩基、混合作動薬−拮抗薬、部分的作動薬、その製
薬上許容される塩、その立体異性体、そのエーテルおよびエステル、それらの混合物を含
むものである。
【発明を実施するための最良の形態】
【0048】
発明の詳細な説明 オピオイド受容体にはμ、κ、およびδと呼ばれる少なくとも3つの
亜型が存在するとみなされてきた。この枠内で、μ受容体は、上部脊椎麻酔(superspinal
analgesia)、呼吸抑制、多幸感、および身体依存性の生成に関与すると考えられる。κ
受容体は、脊椎麻酔、縮瞳、および鎮静の誘発に関与すると考えられる。γ受容体を活性
化すると、不快気分および幻覚、更に呼吸促進作用および血管中枢興奮作用が誘発される
。Lord, et al. Nature, 1977, 267, 495-99には、μ受容体と異なるγと呼ばれる受容体
がマウス精管中に存在すると記載されている。オピオイド作動薬は主にμ受容体でその作
動薬作用を示し、それより程度は少ないがκ受容体でもそうした作用を示すと考えられる
。1つのまたはもう1つの受容体タイプで半作動薬として作用するようにみえる薬剤が数
種存在する。このような薬剤としては、ナロルフィン、プロピラム、およびブプレノルフ
ィンが挙げられる。更に他の薬剤は、μ受容体で競合的拮抗薬として機能し、κおよびω
受容体で作動薬作用を示すことによりモルヒネ様薬剤の作用をブロックする。そのような
作用機構を記述すべく用語「作動薬-拮抗薬」が使用されるようになった。オピオイドの
働きに対する拮抗作用の考え方は複雑であると思われる。
【0049】
オピオイド作動薬-拮抗薬および半作動薬を投与した場合、薬剤の作動薬作用に対する
耐性は現れるが、薬剤の拮抗薬作用に対する耐性は現れないことが判明した。長期間にわ
たり高用量の投与を行った後でさえも、ナロキソンの投与中止は、いかなる識別可能な禁
断症状によっても特性付けられない。また、もう1つの比較的純粋なオピオイド拮抗薬で
あるナルトレキソンの投与を停止した場合、非常にわずかな徴候および症状が現れる。し
かしながら、長期間にわたり高用量の投与を行った後、オピオイド作動薬-拮抗薬のナロ
ルフィンまたはシクラゾシンの投与を急激に停止すると、両方の薬剤で類似した特徴的な
禁断症状が現れる。
【0050】
ナロキソンは、作動薬作用をほとんど示さないオピオイド拮抗薬である。12mgまでの用
量でナロキソンを皮下投与した場合、識別可能な本質的な影響は現れない。また、24mgの
ナロキソンの場合、わずかな眠気が誘発されるにすぎない。ヒトにおいて小用量(0.4-0.8
mg)のナロキソンを筋肉内投与または静脈内投与した場合、モルヒネ様オピオイド作動薬
の作用が阻害されるかまたは急速に逆方向に向かう。1mgのナロキソンを静脈内投与する
と25mgのヘロインの作用が完全にブロックされると報告されている。ナロキソンの作用は
、ほぼ静脈内投与の直後に現れる。この薬剤は経口投与の後で吸収されるが、肝臓を1回
通ると代謝されて急速に不活性な形態になると報告されている。その結果、その作用は非
経口投与を行った場合の1/15にすぎないと報告されている。1gよりも多く経口投与し
た場合、24時間未満でほとんど完全に代謝されると報告されている。
【0051】
他のオピオイド拮抗薬、例えば、いずれも窒素上にシクロプロピルメチル基が置換した
シクラゾシンおよびナルトレキソンは、経口投与の場合、それらの効力の大部分を保持し
、また、その作用の持続時間はかなり長く、経口投与後24時間に達する。最も好ましい
オピオイド拮抗薬は、ナルトレキソンである。しかしながら、他のオピオイド拮抗薬、例
えば、限定されるものではないが、ナロキソン、ナルメフェン、シクラゾシン、およびレ
バロルファンを同等な拮抗薬として経口投与することも、本発明に従って利用可能である
。このような他の拮抗薬対特定のオピオイド作動薬の比は、ナルトレキソン以外の様々な
オピオイド拮抗薬の利用を望む当業者には、過度の実験を行うことなく容易に決定可能で
ある。オピオイド作動薬に対するそれらの比について、本明細書中で具体的に示し、詳細
に説明する。当業者は、例えば、本明細書中に記載のものと同じかまたは類似の臨床研究
を行うことによって、こうした他の拮抗薬対オピオイド作動薬の比を決定してもよい。こ
の場合、例えば、本明細書中に記載のナルトレキソン対ヒドロコドンの比と同等な比で経
口投与されるオピオイド拮抗薬/オピオイド作動薬の組合せ製剤は、本発明の範囲および
特許請求の範囲に含まれるものとみなされる。例えば、本発明の特定の実施形態では、ナ
ロキソンをオピオイド拮抗薬として利用し、投薬剤形中に含まれるナロキソンの量は、こ
の組合せ製剤中にナルトレキソンが含まれる場合と同等な拮抗薬作用を呈するのに十分な
量とする。
【0052】
既にオピオイド常用癖のある患者を治療する場合、オピオイド作動薬の多幸化作用を防
止するために、ナルトレキソンが高用量(100mgを超える量)で経口投与されてきた。ナル
トレキソンはδ部位よりもμ部位に対してかなり優先的なブロッキング作用を示すことが
報告されている。ナルトレキソンは、オピオイド作動薬の性質をもたないオキシモルホン
の合成同族体として知られており、オキシモルホンの窒素原子上に位置するメチル基がシ
クロプロピルメチル基で置換されている点がオキシモルホンと構造上異なっている。ナル
トレキソンの塩酸塩は、約100mg/ccまで水に可溶である。ナルトレキソンの薬理学的およ
び薬物動態学的性質は、多数の動物および臨床実験で評価された。例えば、参照により本
明細書に組み入れるGonzalez JP, et al. Naltrexone: A review of its Pharmacodynami
c and Pharmacokinetic Properties and Therapeutic Effficacy in the Management of
Opioid Dependence. Drugs 1988; 35:192-213を参照されたい。経口投与後、ナルトレキ
ソンは急速に吸収され(1時間以内)、その経口投与時の生物学的利用率は5〜40%の範囲で
ある。ナルトレキソンのタンパク質結合率は約21%であり、1回投与後の分布容積は16.1 L
/kgである。
【0053】
ナルトレキソンは、アルコール依存症を治療するために、また外因的に投与されたオピ
オイドをブロックするために、錠剤の形態で市販されている(Revia(登録商標)、DuPont)
。例えば、Revia(塩酸ナルトレキソン錠剤). Physician's Desk Reference 51st ed., Mo
ntvale, NJ. "Medical Economics" 1997; 51:957-959を参照されたい。50mgのReVia(登録
商標)を投与すると、25mgのヘロインを静脈内に投与したときの薬理学的作用が24時間後
までブロックされる。
【0054】
モルヒネ、ヘロイン、または他のオピオイドとの同時投与を連用した場合、ナルトレキ
ソンはオピオイドに対する身体依存性の発生をブロックすることが知られている。ナルト
レキソンによりヘロインの作用をブロックする方法はオピオイド受容体での競合的な結合
に基づくものと考えられている。ナルトレキソンは、オピオイドの作用を完全にブロック
することによって麻薬中毒を治療すべく使用されてきた。麻薬中毒に対してナルトレキソ
ンが最もうまく使用できるのは、予後の良好な麻薬常用者を対象にして、行動コントロー
ルまたは他の指示遵守性向上法(compliance enhancing method)を含む総合的な作業また
はリハビリプログラムの一部分として使用する場合である。ナルトレキソンを用いて麻薬
依存症を治療する場合、少なくとも7〜10日間にわたり患者にオピオイドを与えないこと
が好ましい。このような目的では、ナルトレキソンの初期用量は、典型的には約25mgであ
り、禁断症状が現れないときは1日あたり50mgに増量してもよい。1日あたりの用量を50mg
にすれば、非経口的に投与されたオピオイドの作用は臨床的に適切にブロックされると考
えられる。また、ナルトレキソンは、社会的および心理療法的手法の補助としてアルコー
ル中毒の治療に使用されてきた。
【0055】
本発明の投薬剤形および方法では、含まれるナルトレキソンの量は、これまでの市販品
で利用されてきた投与量よりもかなり少ない。この原因の一部分として、本発明における
ナルトレキソンの使用法が異なっている点が挙げられる。すなわち、本発明の目標は、オ
ピオイドの作用をブロックすることではなく、身体依存性被験者が大量の組合せ製剤、例
えば、通常処方される用量の約2〜3倍の組合せ製剤を摂取した場合または投与された場合
に否定的な「嫌悪」体験が得られるようにすることである。
【0056】
従って、例えば、オピオイドが酒石酸水素ヒドロコドン15mgである本発明の製剤の場合
、製剤中に含まれる塩酸ナルトレキソンの量は、ヒドロコドン15mgあたりナルトレキソン
約0.5mg〜約4mg、好ましくは約0.75mg〜約3mgである。
【0057】
本発明に有用なオピオイド鎮痛薬としては、すべてのオピオイド作動薬または作動薬-
拮抗薬混合体、半作動薬、例えば、限定されるものではないが、アルフェンタニル、アリ
ルプロジン、アルファプロジン、アニレリジン、ベルジルモルヒネ、ベジトラミド、ブプ
レノルフィン、ブトルファノール、クロニタゼン、コデイン、デソモルヒネ、デキストロ
モラミド、デゾシン、ジアムプロミド、ジアモルホン、ジヒドロコデイン、ジヒドロモル
ヒネ、ジメノキサドール、ジメフェプタノール、ジメチルチアムブテン、ジオキサフェチ
ルブチレート、ジピパノン、エプタゾシン、エトヘプタジン、エチルメチルチアムブテン
、エチルモルヒネ、エトニタゼン、フェンタニール、ヘロイン、ヒドロコドン、ヒドロモ
ルホン、ヒドロキシペチジン、イソメサドン、ケトベミドン、レボルファノール、レボフ
ェナシルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタゾシン、メタ
ドン、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノルレボルファノ
ール、ノルメタドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピパノン、アヘ
ン、オキシコドン、オキシモルホン、パパベレツム、ペンタゾシン、フェナドキソン、フ
ェノモルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミド、プロフェ
プタジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニル、チリジン
、トラマドール、それらの任意の組合せ、それらの任意の塩などが挙げられる。
【0058】
特定の好ましい実施形態では、オピオイド作動薬または鎮痛薬は、ヒドロコドン、モル
ヒネ、ヒドロモルホン、オキシコドン、コデイン、レボルファノール、メペリジン、メタ
ドン、もしくはそれらの塩、またはそれの混合物からなる群より選ばれる。特定の好まし
い実施形態では、オピオイド作動薬はヒドロコドンである。ヒドロコドンの用量15mgと比
較して、これらのオピオイドで同等な鎮痛作用の得られる用量が以下の表1に示されてい
る。
【0059】
【表1】
@0012
【0060】
ナルトレキソンの好ましい比がヒドロコドン15mgあたり約0.5〜約4mgの量であることに
基づいて、各オピオイド1mgに対するナルトレキソンのおよその比が表2に示されている。
【0061】
【表2】
@0013
【0062】
ナルトレキソンのより好ましい比がヒドロコドン15mgあたりナルトレキソン約0.75mg〜
約3mgであることに基づいて、各オピオイド1mgに対するナルトレキソンのおよその比が表
3に示されている。
【0063】
【表3】
@0014
【0064】
ヒドロコドンは疼痛に対処するのに有効であるが、心理的にオピオイドに依存している
者または治療以外の理由でオピオイドを誤用している者によるオピオイドの乱用が増大し
ている。既に他のオピオイドを経験している場合、オピオイドを麻薬拮抗薬と組み合わせ
て投与すると、特に、以前常用者であった患者では、乱用の可能性が減少することが示さ
れた。Weinhold LL, et al. Bpurenorphine Alone and in Combination with Naltrexone
in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30:263-274; Mendelso
n J., et. al., Buprenorphine and Naloxone Interactions in Opiate-Dependent Volun
teers, Clin Pharm Ther 1996; 60:105-114。いずれも参照により本明細書に組み入れる
。
【0065】
ヒドロコドンは、多面的に中枢神経系および胃腸に作用する半合成麻薬性鎮痛薬および
鎮咳薬である。化学的には、ヒドロコドンは、4、5-エポキシ-3-メトキシ-17-メチルモル
フィナン-6-オンであり、ジヒドロコデイノンとしても知られている。他のオピオイドと
同様に、ヒドロコドンは習慣性になることもあり、モルヒネタイプの薬物依存性を生じる
可能性がある。他のアヘン誘導体と同様に、ヒドロコドンは、過剰に投与した場合、呼吸
を抑制する。
【0066】
また、経口用ヒドロコドンは、鎮咳薬としてヨーロッパ(ベルギー、ドイツ、ギリシア
、イタリア、ルクセンブルク、ノルウェー、およびスイス)で利用可能である。また、ド
イツでは、鎮咳薬として非経口製剤が利用可能である。米国では、鎮痛薬としての使用の
ために、中程度または多少重度の疼痛を和らげるための非アヘン製剤(すなわち、イブプ
ロフェン、アセトアミノフェン、アスピリンなど)との一定の組合せ製剤としてのみ、酒
石酸水素ヒドロコドンが市販されている。
【0067】
通常のヒドロコドン投薬剤形では、アセトアミノフェンが併用され、例えば、米国では
UCB Pharma, Inc.からLortab (登録商標)として、2.5/500mg、5/500mg、7.5/500mg、およ
び10/500mgのヒドロコドン/アセトアミノフェン錠剤が市販されている。酒石酸水素ヒド
ロコドン7.5mg+アセトアミノフェン650mgおよび酒石酸水素ヒドロコドン7.5mg+アセト
アミノフェン750mgの割合の錠剤としても利用可能である。疼痛を軽減するために必要な
場合、ヒドロコドンをアスピリンと組み合わせ、成人に対して一般に4〜6時間ごとに1〜2
個の錠剤が経口投薬剤形として投与される。錠剤の形態の場合、酒石酸水素ヒドロコドン
5mgおよびアスピリン224mg更にカフェイン32mgが含まれるか、または、酒石酸水素ヒドロ
コドン5mgおよびアスピリン500mgが含まれる。比較的新しい製剤には、酒石酸水素ヒドロ
コドンおよびイブプロフェンが含まれる。米国でKnoll Laboratoriesから市販されている
Vicoprofen(登録商標)は、酒石酸水素ヒドロコドン7.5mgとイブプロフェン200mgを含む錠
剤である。本発明には、このような製剤すべてが含まれるとともに、本明細書中に記載さ
れている本発明の用量範囲内の経口活性オピオイド拮抗薬も含まれるものとする。
【0068】
ヒドロコドンなどのオピオイド鎮痛薬が乱用される可能性は、本発明の組合せによって
驚くほど低減される。より詳細には、単一の経口投薬剤形中でオピオイド鎮痛薬を少量の
オピオイド拮抗薬と組み合わせた場合、依然として痛覚消失を呈する製剤が得られるが、
この製剤を用いると、1回に1錠より多く、例えば、通常処方される用量の2〜3倍の用量で
投与することによって、身体依存性ヒト被験者が薬剤を乱用し続ける可能性が実質的にな
くなることを見出した。
【0069】
本発明の経口投薬剤形には、経口的に治療上有効な量のオピオイド作動薬が含まれると
ともに、ナルトレキソンなどのオピオイド拮抗薬が、次の条件、すなわち(i)経口投与時
に投薬剤形により誘発される痛覚消失のレベルが非治療レベルまで低減せず、かつ(ii)通
常処方された用量より多くの用量を1度に摂取した場合、身体依存性ヒト被験者、例えば
、身体依存性常用者(具体的には、急発禁断症候群の常用者)が少なくとも穏やかな否定的
「嫌悪」体験をするという条件を満たす量で含まれる。好ましくは、経口投薬剤形中に含
まれる拮抗薬の量は、(iii)拮抗薬の含まれない同等な経口投薬剤形を使用した場合より
も、非身体依存性ヒト被験者、例えば、オピオイド常用者による増量の要求が低減する(
例えば、「嗜好度」が減少する)量である。
【0070】
上のパラグラフに記載の要件(i)〜(iii)を達成するのに有用な拮抗薬の量は、少なくと
も部分的には、例えば、VASスケール(この場合、投薬剤形の作用に対する自覚症状を被験
者が等級化する)などの「代用」試験および/または瞳孔サイズ(瞳孔測定)などの測定を
用いて決定してもよい。こうした測定を行うことにより、当業者は、作動薬のオピエート
効果を低減させる拮抗薬の用量を作動薬の用量を基準に決定することができる。その後、
当業者は、身体依存性被験者に対して嫌悪作用を誘発するオピオイド拮抗薬のレベルおよ
び非身体依存性常用者の「嗜好度」またはオピオイド増量要求を最小限に抑えるオピオイ
ド拮抗薬のレベルを決定することができる。オピオイド拮抗薬のこれらのレベルが決まれ
ば、上のパラグラフに記載の要件(i)〜(iii)を達成するのに有用な拮抗薬のこうしたレベ
ルにまたはこうしたレベル未満に拮抗薬の用量範囲を設定することができる。
【0071】
オピオイド作動薬とオピオイド拮抗薬との組合せ製剤は、従来の賦形剤、すなわち、当
技術分野で周知であり経口投与に適した製薬上許容される有機または無機の担体物質との
混合物の形態で利用することができる。好適な製薬上許容される担体としては、水、塩類
溶液、アルコール、アラビアゴム、植物油、ベンジルアルコール、ポリエチレングリコー
ル、ゲル化剤、炭水化物、例えば、ラクトース、アミロース、またはデンプン、ステアリ
ン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、香油、脂肪酸モノグリセリドおよ
びジグリセリド、ペンタエリトリトール脂肪酸エステル、ヒドロキシメチルセルロース、
ポリビニルピロリドンなどが挙げられるが、これらに限定されるものではない。製剤は、
殺菌が可能であり、更に、所望により、補助剤、例えば、滑沢剤、保存剤、安定剤、湿潤
剤、乳化剤、浸透圧を変化させるための塩、緩衝剤、着色剤、着香剤、および/または芳
香剤との混合が可能である。また、製剤は、所望により、他の活性剤、例えば、他の鎮痛
薬と併用することも可能である。経口投与のために、特に好適なのは、錠剤、糖衣錠、液
剤、滴剤、座剤、カプセル剤、カプレット剤、およびゲルキャップ剤である。経口用に使
用される組成物は、当技術分野で周知のいずれの方法により調製してもよく、このような
組成物には、錠剤の製造に好適である不活性で無毒な医薬用賦形剤からなる群より選ばれ
る1種以上の物質が含まれていてもよい。このような賦形剤としては、例えば、ラクトー
スなどの不活性な希釈剤、トウモロコシデンプンなどの造粒剤および崩壊剤、デンプンな
どの結合剤、ステアリン酸マグネシウムなどの滑沢剤が挙げられる。錠剤は、コーティン
グされていなくてもよいし、外観をよくするために、または有効成分の遅延放出を行うた
めに、周知の手法によりコーティングしてもよい。また、経口用製剤は、有効成分が不活
性希釈剤と混合されている硬ゼラチンカプセル剤として提供してもよい。
【0072】
水性懸濁剤には、上記の組合せの薬剤が含まれ、この混合物には、懸濁化剤、例えば、
ヒドロキシプロピルメチルセルロースのような製薬上許容される合成ガムまたは天然ガム
が一種以上含まれる。油性懸濁剤は、上記の組合せの薬剤を植物油または鉱油中に懸濁さ
せることによって調合可能である。油性懸濁剤には、蜜蝋やセチルアルコールなどの粘稠
化剤か含まれていてもよい。甘味化担体を利用する場合、シロップ剤、エリキシル剤など
を使用することができる。また、注射可能な懸濁剤を調製することも可能であり、この場
合には、適切な液状担体、懸濁化剤などを利用してもよい。
【0073】
本発明の治療方法および製剤には、オピオイド鎮痛薬およびオピオイド拮抗薬のほかに
1種以上の薬剤が含まれていてもよい。こうして追加される1種または複数種の薬剤は、相
乗的に作用するものであってもよいし、そうでないものであってもよい。従って、特定の
実施形態では、製剤中でオピオイド拮抗薬のほかに2種のオピオイド鎮痛薬を組み合わせ
てもよい。例えば、投薬剤形には、半減期、溶解性、効力などの性質やこれらの複数の性
質が異なる2種のオピオイド鎮痛薬が含まれていてもよい。更なる実施形態では、1種以上
のオピオイド鎮痛薬が含まれ、更に、オピオイド拮抗薬のほかに非オピオイド薬剤が含ま
れる。このような非オピオイド薬剤は、好ましくは、更なる痛覚消失を呈するものであり
、具体的は、アスピリン;アセトアミノフェン;非ステロイド系抗炎症薬(「NSAIDS」)、例
えば、イブプロフェン、ケトプロフェンなど;N-メチル-D-アスパルテート(NMDA)受容体拮
抗薬、例えば、デキストロメトルファン、デキストロルファン、またはケタミンのような
モルフィナン;シクロオキシゲナーゼーII阻害剤(「COX-II阻害剤」);および/またはグリ
シン受容体拮抗薬が挙げられる。
【0074】
本発明の特定の好ましい実施形態では、本発明によりNSAIDやCOX-2阻害剤のような追加
の非オピオイド作動薬を導入できるようになるため、オピオイド鎮痛薬をより低用量で使
用することができる。薬剤の一方または両方をより少ない量で使用することにより、ヒト
において、有効な疼痛治療に伴う副作用が減少する。
【0075】
好適な非ステロイド系抗炎症薬としては、イブプロフェン、ジクロフェナク、ナプロキ
セン、ベノキサプロフェン、フルルビプロフェン、フェノプロフェン、フルブフェン、ケ
トプロフェン、インドプロフェン、ピロプロフェン、カルプロフェン、オキサプロジン、
プラモプロフェン、ムロプロフェン、トリオキサプロフェン、スプロフェン、アミノプロ
フェン、チアプロフェン酸、フルプロフェン、ブクロキシ酸、インドメタシン、スリンダ
ク、トルメチン、ゾメピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザ
ク、クリダナク、オキスピナク、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニ
フルム酸、トルフェナム酸、ジフルリサル、フルフェニサル、ピロキシカム、スドキシカ
ム、またはイソキシカムなどが挙げられる。これらの薬剤の有用な用量は、当業者には周
知である。
【0076】
N-メチル-D-アスパルテート(NMDA)受容体拮抗薬は、当技術分野で周知であり、具体的
には、モルフィナン、例えば、デキストロメトルファンもしくはデキストロルファン、ケ
タミン、d-メタドン、または製薬上許容されるそれらの塩が挙げられる。本発明の目的に
対して、「NMDA拮抗薬」という用語には、NMDA-受容体活性化の結果として細胞内で生じ
る主要な作用をブロックする薬剤、例えば、GM1やGT1bなどのガングリオシド、トリフル
オペラジンなどのフェノチアジン、またはN-(6-アミノヘキシル)-5-クロロ-1-ナフタレン
スルホンアミドなどのナフタレンスルホンアミドも含まれるものとみなされる。米国特許
第5,321,012号および同第5,556,838号(いずれもMayerらに付与されている)には、これら
の薬剤により、嗜癖性薬物、例えば、モルヒネ、コデインなどの麻薬性鎮痛薬に対する耐
性および/または依存性の出現が抑制されると記載されており、また、米国特許第5,502,
058号(Mayerらに付与されている)には、慢性的な疼痛の治療ができると記載されている。
これらの特許はいずれも、参照により本明細書に組み入れる。Mayerらの特許に記載され
ているように、NMDA拮抗薬は、単独で含まれていてもよいし、リドカインのような局所麻
酔薬と一緒に含まれていてもよい。
【0077】
グリシン受容体拮抗薬を用いる慢性的疼痛の治療およびこのような薬剤の同定について
は、参照により本明細書に組み入れる米国特許第5,514,680号(Weberら)に記載されている
。
【0078】
COX-2阻害剤について当技術分野で報告がなされており、多くの化学構造がシクロオキ
シゲナーゼ-2を阻害することが知られている。COX-2阻害剤については、例えば、米国特
許第5,616,601号、同第5,604,260号、同第5,593,994号、同第5,550,142号、同第5,536,75
2号、同第5,521,213号、同第5,475,995号、同第5,639,780号、同第5,604,253号、同第5,5
52,422号、同第5,510,368号、同第5,436,265号、同第5,409,944号、および同第5,130,311
号に記載されている。これらの特許はいずれも、参照により本明細書に組み入れる。特定
の好ましいCOX-2阻害剤としては、セレコキシブ(SC-58635)、DUP-697、フロスリド(CGP-2
8238)、メロキシカム、6-メトキシ-2-ナフチル酢酸(6-MNA)、MIK-966、ナブメトン(6-MNA
用のプロドラッグ)、ニメスリド、NS-398、SC-5766、SC-58215、T-614、またはそれの組
合せが挙げられる。1日あたり体重1キログラムにつき約0.005mg〜約140mg程度のCOX-2阻
害剤の用量レベルが、オピオイド鎮痛薬と組み合わせる場合、治療上有効である。また、
1日あたり患者1人につき約0.25mg〜約7gのCOX-2阻害剤が、オピオイド鎮痛薬と組み合わ
せて投与される。
【0079】
更なる実施形態では、痛覚消失以外の所望の作用を呈する非オピオイド薬剤、例えば、
鎮咳薬、去痰薬、充血除去薬、抗ヒスタミン薬、局所麻酔薬などを含めることができる。
【0080】
本発明に係る経口投薬剤形は、例えば、顆粒剤、スフェロイド剤、ビーズ剤、ペレット
剤(これ以降では、まとめて「多粒子剤」と記す)として提供してもよい。所定の時間にわ
たり所望の用量のオピオイドを提供するのに有効な量の多粒子剤をカプセル剤中に入れて
もよいし、任意の他の効果的な形態の経口固形剤中に導入してもよい。このほか、経口投
薬剤形は、錠剤の形態であってもよい。
【0081】
制御放出性投薬剤形 オピオイド作動薬/オピオイド拮抗薬組合せ剤は、当業者に公知の
任意の好適な錠剤、コーティング錠、または多粒子製剤の形態の制御放出性または持続放
出性経口製剤として調合可能である。持続放出性投薬剤形には、場合により、オピオイド
作動薬およびオピオイド拮抗薬と一緒にマトリックス中に導入される持続放出性担体が含
まれていてもよく、あるいは持続放出性投薬剤形を持続放出性剤皮として適用してもよい
。
【0082】
オピオイド鎮痛薬がヒドロコドンを含む実施形態では、持続放出性経口投薬剤形には、
投薬単位あたりヒドロコドン約8mg〜約50mgの鎮痛薬が含まれていてもよい。ヒドロモル
ホンが治療上活性なオピオイドである持続放出性経口投薬剤形では、塩酸ヒドロモルホン
が約2mg〜約64mgの量で含まれている。別の実施形態では、オピオイド鎮痛薬にはモルヒ
ネが含まれ、本発明の持続放出性経口投薬剤形には、重量基準で約2.5mg〜約800mgのモル
ヒネが含まれる。さらにもう1つの実施形態では、オピオイド鎮痛薬にはオキシコドンが
含まれ、持続放出性経口投薬剤形には、約2.5mg〜約800mgのオキシコドンが含まれる。オ
ピオイド鎮痛薬にはトラマドールが含まれていてもよく、持続放出性経口投薬剤形には、
投薬単位あたり約25mg〜800mgのトラマドールが含まれていてもよい。投薬剤形には、実
質的に等価な治療効果が得られるように1種以上のオピオイド鎮痛薬が含まれていてもよ
い。このほか、投薬剤形には、本発明に有用なオピオイドの他の塩が等モル量で含まれて
いてもよい。
【0083】
本発明の好ましい1実施形態では、持続放出性投薬剤形には、有効成分を含有または包
含し、約0.1mm〜約2.5mm、好ましくは約0.5mm〜約2mmの直径を有する粒子が含まれていて
もよい。
【0084】
該粒子は、好ましくは、水性媒質中にオピオイド作動薬/拮抗薬組合せ剤を持続速度で
放出させることのできる物質で皮膜されている。皮膜は、他の所定の性質と所望のin-vit
ro放出速度とが同時に得られるように選択される。本発明の持続放出性コーティング製剤
は、強力な連続膜を形成可能なものでなければならず、しかもこの膜は、平滑で外観がよ
く、顔料および他のコーティング添加剤を保持することができ、無毒で、不活性で、不粘
着性でなければならない。
【0085】
特定の実施形態では、粒子には、オピオイド拮抗薬と共にオピオイド鎮痛薬を含んでな
る通常の放出性マトリックスが含まれる。
【0086】
剤皮 本発明の投薬剤形は、場合により、製剤の放出調節または保護に好適な1種以上の
物質でコーティングされていてもよい。1実施形態では、例えば、胃腸液に触れたときに
pH依存的放出またはpH非依存的放出を可能にする剤皮(コーティング)が施される。pH依
存性剤皮は、患者に対して少なくとも約8時間、好ましくは約12時間から約24時間までの
痛覚消失を引き起こすことのできる吸収プロフィルが得られるように、胃腸(GI)管、例え
ば、胃または小腸の所望の領域でオピオイドを放出する働きをする。pH非依存性剤皮が望
まれる場合、周囲の流体中で、例えば、GI管中で、pH変化に関係なく最適な放出が達成さ
れるように剤皮をデザインする。また、GI管の所望の1領域、例えば、胃において用量の
一部分を放出し、GI管の他の領域、例えば、小腸において用量の残りの部分を放出する組
成物を調合することも可能である。
【0087】
また、腸溶剤皮上にコーティングされた未保護薬剤を胃で放出し、腸溶剤皮で保護され
た残りの薬剤を胃腸管の下流で更に放出させるようにすべく、pH依存性剤皮を利用して製
剤を作製する本発明に係る製剤に対して反復作用効果を付与してもよい。セラック、酢酸
セルロースフタレート(CAP)、ポリ酢酸ビニルフタレート(PVAP)、ヒドロキシプロピルメ
チルセルロースフタレート、メタクリル酸エステルコポリマー、ゼインなどを含んでなる
pH依存性の剤皮を本発明に従って使用してもよい。
【0088】
特定の好ましい実施形態では、オピオイド鎮痛薬(COX-2インヒビターの併用されたまた
は併用されていないオピオイド鎮痛薬)を含む基体(例えば、錠剤コアビーズ、マトリック
ス粒子)は、(i)アルキルセルロース、(ii)アクリルポリマー、または (iii)それらの混合
物から選択される疎水性材料でコーティングされる。有機性または水性の溶液または分散
体の形態で剤皮を適用することができる。所望の持続放出プロフィルを得るべく、基体に
対して約2〜約25%の重量増となるように剤皮を適用してもよい。水性分散物から得られる
剤皮については、米国特許第5,273,760号および同第5,286,493号に詳細が記載されている
。これらの特許は、本発明の譲受人に譲渡されており、参照により本明細書に組み入れる
。
【0089】
本発明により使用可能な持続放出性製剤および剤皮の他の例は、本発明の譲受人に譲渡
されている米国特許第5,324,351号、同第5,356,467号、および同第5,472,712号に記載さ
れているものを含む。参照により、これらの特許の全内容を本明細書に組み入れる。
【0090】
アルキルセルロースポリマー アルキルセルロースなどのセルロース系の材料およびポ
リマーは、本発明においてビーズ剤をコーティングするのに好適な疎水性材料である。簡
単に例を示すと、好ましいアルキルセルロースポリマーの1つは、エチルセルロースであ
るが、他のセルロースおよび/またはアルキルセルロースポリマーを単独でまたは任意に
組み合わせて本発明に係る疎水性剤皮の全体または一部として容易に利用できることは、
当業者には分かるであろう。
【0091】
市販されているエチルセルロースの水性分散体の1つは、Aquacoat(登録商標)(FMC Corp
., Philadelphia, Pennsylvania, U.S.A.)である。Aquacoat(登録商標)の調製は次のよう
に行われる。まず、水不混和性有機溶剤中にエチルセルロースを溶解し、次に、界面活性
剤および安定剤の存在下でこれを水中に乳化する。均質化処理を行ってサブミクロンの液
滴を形成した後、減圧下で有機溶剤を蒸発させて擬似ラテックスを形成する。製造段階で
は、擬似ラテックス中に可塑剤を導入しない。従って、剤皮としてこれを使用する前に、
予めAquacoat(登録商標)を好適な可塑剤と均質混合する必要がある。
【0092】
エチルセルロースのもう1つの水性分散体が、Surelease(登録商標)(Colorcon, inc.,
West Point, Pennsylvania, U.S.A.)として市販されている。この製品は、製造プロセス
時に分散体中に可塑剤を導入することにより調製される。ポリマー、可塑剤(ジブチルセ
バケート)と安定剤(オレイン酸)の高温溶融体を均質混合物として調製し、次に、アルカ
リ溶液で希釈することにより、基体上に直接適用可能な水性分散体が得られる。
【0093】
アクリルポリマー 本発明の他の好ましい実施形態では、制御放出性剤皮を構成する疎
水性材料は製薬上許容されるアクリルポリマーであり、具体的には、アクリル酸およびメ
タクリル酸のコポリマー、メチルメタクリレートコポリマー、エトキシエチルメタクリレ
ート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリ
ル酸アルキルアミドコポリマー、ポリ(メチルメタクリレート)、ポリメタクリレート、ポ
リ(メチルメタクリレート)コポリマー、ポリアクリルアミド、アミノアルキルメタクリレ
ートコポリマー、ポリ(メタクリル酸無水物)、およびグリシジルメタクリレートコポリマ
ーが挙げられるが、これらに限定されるものではない。
【0094】
特定の好ましい実施形態では、アクリルポリマーには、1種以上のアンモニオメタクリ
レートコポリマーが含まれる。アンモニオメタクリレートコポリマーは当技術分野で公知
であり、低含有量の第4級アンモニウム基を含むアクリル酸エステルとメタクリル酸エス
テルとの完全重合コポリマーとしてNF XVII中に記載されている。
【0095】
望ましい溶解プロフィルを得るために、異なる物理的性質を有する2種以上のアンモニ
オメタクリレートコポリマーを導入する必要がある。例えば、中性の(メタ)アクリル酸エ
ステルに第4級アンモニウム基を異なるモル比で導入する。
【0096】
特定のメタクリル酸エステル型ポリマーは、本発明に従って使用可能なpH依存性剤皮を
調製するのに有用である。具体的には、ジエチルアミノエチルメタクリレートと他の中性
のメタクリル酸エステルとから合成されるコポリマーのファミリーが挙げられ、これはメ
タクリル酸コポリマーまたはメタクリレートポリマーとも呼ばれ、Rohm Tech, Inc.からE
udragit(登録商標)として市販されている。
いくつかの異なるタイプのEudragit(登録商標)が存在する。例えば、Eudragit(登録商
標) Eは、酸性媒質中で膨潤および溶解するメタクリル酸コポリマーの例である。Eudragi
t(登録商標) Lは、pH<約5.7では膨潤せず、pH>約6で溶解するメタクリル酸コポリマーで
ある。Eudragit(登録商標) Sは、pH<約6.5では膨潤せず、pH>約7で溶解する。Eudragit(
登録商標) RLおよびEudragit(登録商標) RSは水で膨潤可能であり、これらのポリマーに
より吸収される水の量はpH依存性であるが、Eudragit(登録商標) RLおよびRSでコーティ
ングされた投薬剤形はpH非依存性である。
【0097】
特定の好ましい実施形態では、アクリル剤皮には、それぞれ商品名Eudragit(登録商標)
RL3ODおよびEudragit(登録商標)RS3ODとしてRohm Pharmaから市販されている2種のアクリ
ル樹脂ラッカーの混合物が含まれる。Eudragit(登録商標)RL3ODおよびEudragit(登録商標
)RS3ODは、低含有量の第4級アンモニウム基を含むアクリル酸エステルとメタクリル酸エ
ステルとのコポリマーであり、アンモニウム基対残りの中性(メタ)アクリル酸エステルの
モル比は、Eudragit(登録商標)RL3ODでは1:20であり、Eudragit(登録商標)RS3ODでは1:40
である。平均分子量は、約150,000である。コード表示RL(高浸透性)およびRS(低浸透性)
は、これらの薬剤の浸透性を表している。Eudragit(登録商標)RL/RS混合物は、水および
消化液に不溶である。しかしながら、これらの物質から形成される剤皮は、水溶液および
消化液中で膨潤および浸透が可能である。
【0098】
本発明のEudragit(登録商標) RL/RS分散体は、望ましい溶解プロフィルを有する持続放
出性製剤が最終的に得られるように、任意の所望の比で混合可能である。例えば、Eudrag
it(登録商標) RL 100%、Eudragit(登録商標) RL 50% + Eudragit(登録商標) RS 50%、お
よびEudragit(登録商標) RL 10% + Eudragit(登録商標) RS 90%から誘導される遅延性剤
皮を用いて、望ましい持続放出性製剤を得ることができる。もちろん、他のアクリルポリ
マー、例えば、Eudragit(登録商標) Lなども使用可能であることは、当業者には分かるで
あろう。
【0099】
可塑剤 剤皮に疎水性材料の水性分散体が含まれる本発明の実施形態では、疎水性材料
の水性分散体中に有効量の可塑剤を導入すると、持続放出性剤皮の物理的性質が改良され
るであろう。例えば、エチルセルロースは、比較的高いガラス転移温度を有し、通常のコ
ーティング条件下では可撓性の膜を形成しないため、それを剤皮材料として使用する前に
、エチルセルロース剤皮を含有する持続放出性剤皮中に可塑剤を導入することが望ましい
。一般的には、剤皮溶液中に含まれる可塑剤の量は、被膜形成剤の濃度を基準にして、例
えば、ほとんどの場合、被膜形成剤の重量の約1〜約50%である。しかしながら、可塑剤の
濃度は、特定のコーティング溶液および適用方法を用いて注意深く実験を行った後で適切
に決めなければならない。
【0100】
エチルセルロース用の好適な可塑剤としては、例えば、ジブチルセバケート、ジエチル
フタレート、トリエチルシトレート、トリブチルシトレート、およびトリアセチンのよう
な水不溶性の可塑剤が挙げられるが、他の水不溶性可塑剤(例えば、アセチル化モノグリ
セリド、フタル酸エステル、ヒマシ油など)を使用することも可能である。トリエチルシ
トレートは、本発明のエチルセルロース水性分散体には特に好ましい可塑剤である。
【0101】
本発明のアクリルポリマー用の好適な可塑剤としては、例えば、トリエチルシトレート
NF XVIやトリブチルシトレートのようなクエン酸エステル、ジブチルフタレート、および
場合により1,2-プロピレングリコールが挙げられるが、これらに限定されるものではない
。アクリル皮膜から形成される膜、例えば、Eudragit(登録商標) RL/RSラッカー溶液から
形成される膜の弾性を増強するのに好適であることが判明している他の可塑剤としては、
ポリエチレングリコール、プロピレングリコール、ジエチルフタレート、ヒマシ油、およ
びトリアセチンが挙げられる。トリエチルシトレートは、本発明のエチルセルロースの水
性分散体には特に好ましい可塑剤である。
【0102】
このほか、少量のタルクを添加すると、加工時における水性分散体の付着傾向が減少し
、艶出剤としての効能が現れることが判明した。
【0103】
コーティングされたビーズ剤の調製方法 疎水性材料を用いて不活性な医薬用ビーズ剤
、例えば、nu pariel 18/20ビーズ剤をコーティングする場合、コーティング後得られた
複数の固体制御放出性ビーズ剤を、周囲の流体、例えば、胃液または溶解媒質による消化
を受けたりそれと接触したときに有効な制御放出用量を確保するのに十分な量で、ゼラチ
ンカプセル剤中に配置してもよい。
【0104】
本発明の制御放出性ビーズ製剤は、例えば、摂取後、胃液や腸液に触れると、治療上有
効な薬剤を徐々に放出する。本発明の製剤の制御放出プロフィルは、例えば、疎水性材料
を含む被覆膜の量の変更、疎水性材料中に可塑剤を添加する方法の変更、疎水性材料に関
連する可塑剤の量の変更、他の成分または賦形剤の導入、製造方法の変更などによって、
調節可能である。最終的な製品の溶解プロフィルに関しても、例えば、遅延性剤皮の厚さ
の増大または減少によって、調節可能である。
【0105】
治療上有効な薬剤でコーティングされたスフェロイド剤またはビーズ剤は、例えば、治
療上有効な薬剤を水中に溶解させ、次に、Wusterインサートを用いてnu pariel 18/20ビ
ーズ剤などの基体上に溶液をスプレーすることによって、調製される。場合により、ビー
ズ剤へのオピオイドの結合および/または溶液の着色などを促進するために、ビーズ剤に
コーティングする前に他の成分を添加する。例えば、着色剤(例えば、Colorcon, Inc.か
ら市販されているOpadry(登録商標))を併用してまたは併用せずに、ヒドロキシプロピル
メチルセルロースを含む物質を溶液に添加し、この溶液を混合した後(例えば、約1時間混
合した後)、これをビーズ剤上に適用してもよい。次に、こうしてコーティングされた基
体(この例ではビーズ剤)を、場合により、障壁剤でオーバーコーティングすることにより
、治療上有効な薬剤を疎水性制御放出性剤皮から分離させてもよい。好適な障壁剤の例は
、ヒドロキシプロピルメチルセルロースが含まれる障壁剤である。しかしながら、当技術
分野で公知の被膜形成剤はいずれも使用可能である。完成品の溶解速度に影響を及ぼさな
い障壁剤が好ましい。
【0106】
次に、疎水性材料の水性分散体でビーズ剤をオーバーコーティングしてもよい。疎水性
材料の水性分散体には、好ましくは、有効量の可塑剤、例えば、トリエチルシトレートが
更に含まれる。既に調合済みであるエチルセルロースの水性分散体、例えば、Aquacoat(
登録商標)やSurelease(登録商標)を使用してもよい。Surelease(登録商標)を使用する場
合には可塑剤を別に添加する必要はない。このほか、既に調合済みであるアクリルポリマ
ーの水性分散体、例えば、Eudragit(登録商標)を使用することもできる。
【0107】
本発明のコーティング溶液には、被膜形成剤、可塑剤、および溶剤系(すなわち、水)の
ほかに、好ましくは、外観の改良および製品の識別を行うための着色剤が含まれる。疎水
性材料の水性分散体を着色する代わりに、またはそれを着色するとともに、治療上有効な
薬剤の溶液を着色してもよい。例えば、アルコールまたはプロピレングリコールベースの
着色分散体、微粉砕アンモニウムレーキ、および二酸化チタンのような不透明剤を用いて
Aquacoat(登録商標)を着色してもよい。この場合、水溶性ポリマー溶液を剪断力下で着色
し、次に、可塑化Aquacoat(登録商標)を低剪断力下で着色する。このほか、本発明の製剤
を着色するための任意の好適な方法が使用可能である。アクリルポリマーの水性分散体を
使用する場合、製剤を着色するのに好適な成分としては、二酸化チタンおよび有色顔料、
例えば、酸化鉄顔料が挙げられる。しかしながら、顔料を導入すると、剤皮の遅延効果が
増大する。
【0108】
当技術分野で公知の任意の好適なスプレー装置を用いてスプレーすることにより、治療
上有効な薬剤を含む基体上に可塑化疎水性材料を適用することが可能である。好ましい方
法では、Wurster流動床系を使用する。この系では、下方から噴射されたエアジェットに
よりコア材料を流動させ、アクリルポリマー剤皮をコア上にスプレーしながら乾燥を行う
。好ましくは、治療上有効な薬剤の物理的な特性や可塑剤の導入方法などを考慮に入れて
、コーティングされた基体が胃液などの水溶液に触れたときに治療上有効な該薬剤を予め
決められたように制御放出させるのに十分な量で疎水性材料を適用する。疎水性材料でコ
ーティングした後、場合により、Opadry(登録商標)のような被膜形成剤の被覆膜をさらに
ビーズ剤に適用する。こうした被覆膜を設けると、多少なりとも、ビーズ剤の凝集は実質
的に低減する。
【0109】
1種以上の放出調節剤を添加することによって、または剤皮を貫通する1つ以上の通路を
提供することによって、本発明の制御放出性製剤からの治療上有効な薬剤の放出を更に変
化させることができる。すなわち、所望の速度に調節することができる。疎水性材料対水
溶性材料の比は、いくつかある因子の中で特に、所要の放出速度および選択される材料の
溶解特性によって決定される。
【0110】
細孔形成剤として機能する放出調節剤は、有機物であっても無機物であってもよく、こ
うした放出調節剤には、使用環境下で剤皮から溶解、抽出、または浸出させることのでき
る物質が含まれる。細孔形成剤には、1種以上の親水性材料、例えば、ヒドロキシプロピ
ルメチルセルロースが含まれていてもよい。
【0111】
また、デンプンやガムなどの浸食促進剤を本発明の持続放出性剤皮に含有させることも
できる。
【0112】
また、使用環境下でミクロ細孔性単層を形成するのに有用な材料、例えば、カーボネー
ト基がポリマー鎖中に複数回出現する炭酸の直鎖状ポリエステルを含んでなるポリカーボ
ネートを本発明の持続放出性剤皮に含有させることができる。
【0113】
また、放出調節剤には、半透過性ポリマーが含まれていてもよい。
【0114】
特定の好ましい実施形態では、放出調節剤は、ヒドロキシプロピルメチルセルロース、
ラクトース、金属ステアリン酸塩、およびそれらの任意の混合物から選択される。
【0115】
また、本発明の持続放出性剤皮には、少なくとも1つの通路、オリフィスなどを含んで
なる送出手段が含まれていてもよい。米国特許第3,845,770号、同第3,916,889号、同第4,
063,064号、および同第4,088,864号に開示されているような方法によって経路を形成する
ことが可能である(これらの特許はいずれも、参照により本明細書に組み入れる)。経路は
、円形、三角形、四角形、楕円形、不規則形など、任意の形状をとることができる。
【0116】
マトリックスビーズ製剤 本発明の他の実施形態では、先に記載したような制御放出性
剤皮を有するマトリックスを用いることによって制御放出性製剤が得られる。また、本発
明においては、オピオイドのin vitro溶解速度が好ましい範囲内に設定されかつpH依存的
またはpH非依存的にオピオイドが放出される制御放出性マトリックスも利用可能である。
制御放出性マトリックス中へ導入するのに好適な物質は、マトリックス形成に使用される
方法に依存する。
【0117】
例えば、オピオイド鎮痛薬および(場合により)COX-2のほかに、以下の物質がマトリッ
クス中に含まれていてもよい。
【0118】
親水性および/または疎水性材料、例えば、ガム、セルロースエーテル、アクリル樹脂
、タンパク質誘導物質。このリストは、限定的なものではなく、活性剤の制御放出を行う
ことができかつ融解する(または押出に必要な程度まで軟化する)任意の製薬上許容される
疎水性材料または親水性材料を、本発明に従って使用することが可能である。
【0119】
消化可能な長鎖状の(C8〜C50、特に、C12〜C40)置換もしくは無置換炭化水素、例えば
、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、蝋、ステアリ
ルアルコール、ポリアルキレングリコール。
【0120】
これらのポリマーのうちで、アクリルポリマー、特に、Eudragit(登録商標) RSPO、セ
ルロースエーテル、特に、ヒドロキシアルキルセルロースおよびカルボキシアルキルセル
ロースが好ましい。経口投薬剤形には、少なくとも1種の親水性または疎水性の材料が1%
〜80%(重量基準)の量で含まれていてもよい。
【0121】
疎水性材料が炭化水素である場合、好ましくは、炭化水素の融点は、25〜90℃である。
長鎖状炭化水素材料のうちで、脂肪(脂肪族)アルコールが好ましい。経口投薬剤形には、
少なくとも1種の消化可能な長鎖状炭化水素が60%(重量基準)までの量で含まれていてもよ
い。
【0122】
好ましくは、経口投薬剤形には、少なくとも1種のポリアルキレングリコールが60%(重
量基準)までの量で含まれていてもよい。
【0123】
疎水性材料は、好ましくは、アルキルセルロース、アクリル酸およびメタクリル酸のポ
リマーおよびコポリマー、セラック、ゼイン、水素化ヒマシ油、水素化植物油、またはそ
れらの混合物からなる群より選ばれる。本発明の特定の好ましい実施形態では、疎水性材
料は、製薬上許容されるアクリルポリマーであり、具体的には、アクリル酸およびメタク
リル酸のコポリマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキ
シエチルメタクリレート、シアノエチルメタクリレート、アミノアルキルメタクリレート
コポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポ
リマー、ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレー
ト、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびグリシジルメタクリレート
コポリマーが挙げられるが、これらに限定されるものではない。他の実施形態では、疎水
性材料は、ヒドロキシアルキルセルロースのような材料、例えば、ヒドロキシプロピルメ
チルセルロース、およびそれらの混合物から選択される。
【0124】
好ましい疎水性材料は水不溶性であり、多かれ少なかれ、はっきりと親水性および/ま
たは疎水性の傾向を有する。好ましくは、本発明に有用な疎水性材料の融点は、約30〜約
200℃、好ましくは約45〜約90℃である。特に、疎水性材料には、天然蝋または合成蝋、
脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアルコール、ステアリルアル
コール、セチルアルコール、または好ましくはセトステアリルアルコール)、脂肪酸類、
例えば、限定されるものではないが、脂肪酸エステル、脂肪酸グリセリド(モノ-、ジ-、
およびトリ-グリセリド)、水素化脂肪、炭化水素、ノルマルワックス、ステアリン酸、ス
テアリルアルコール、ならびに炭化水素主鎖を有する疎水性および親水性材料が含まれて
いてもよい。好適な蝋としては、例えば、蜜蝋、グリコワックス(glycowax)、カストール
ワックス、およびカルナウバワックスが挙げられる。本発明の目的では、蝋様物質とは、
室温で通常固体であり約30〜約100℃の融点を有する任意の物質として定義付けられる。
【0125】
本発明に従って使用しうる好適な疎水性材料としては、消化可能な長鎖状の(C8〜C50、
特に、C12〜C40)置換もしくは無置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪
酸のグリセリルエステル、鉱油、植物油、天然蝋、および合成蝋が挙げられる。25〜90℃
の融点を有する炭化水素が好ましい。特定の実施形態では、長鎖状炭化水素材料のうちで
、脂肪(脂肪族)アルコールが好ましい。経口投薬剤形には、少なくとも1種の消化可能な
長鎖状炭化水素が60%(重量基準)までの量で含まれていてもよい。
【0126】
好ましくは、マトリックス製剤中に2種以上の疎水性材料が一緒に含まれる。
他の疎水性材料を含める場合、こうした材料は、好ましくは、天然蝋、合成蝋、脂肪酸
、脂肪アルコール、およびそれらの混合物から選択される。具体的には、蜜蝋、カルナウ
バワックス、ステアリン酸、およびステアリルアルコールが挙げられる。このリストは、
限定的なものではない。
【0127】
特定の好適なマトリックスの1つには、少なくとも1種の水溶性ヒドロキシアルキルセル
ロース、少なくとも1種のC12〜C36、好ましくはC14〜C22脂肪族アルコール、および場合
により少なくとも1種のポリアルキレングリコールが含まれる。上記の少なくとも1種のヒ
ドロキシアルキルセルロースは、好ましくは、ヒドロキシ(C1〜C6)アルキルセルロース、
例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、および
特にヒドロキシエチルセルロースである。本発明の経口投薬剤形中の少なくとも1種のヒ
ドロキシアルキルセルロースの量は、特に、所要の正確なオピオイド放出速度によって決
定されるであろう。上記の少なくとも1種の脂肪族アルコールは、例えば、ラウリルアル
コール、ミリスチルアルコール、またはステアリルアルコールであってよい。しかしなが
ら、本発明の経口投薬剤形の特に好ましい実施形態では、少なくとも1種の脂肪族アルコ
ールは、セチルアルコールまたはセトステアリルアルコールである。本発明の経口投薬剤
形中の少なくとも1種の脂肪族アルコールの量は、先に述べたように、所要の正確なオピ
オイド放出速度によって決定されるであろう。この量はまた、少なくとも1種のポリアル
キレングリコールが経口投薬剤形中に存在するかしないかにも依存する。少なくとも1種
のポリアルキレングリコールの不在下では、経口投薬剤形には、好ましくは、少なくとも
1種の脂肪族アルコールが20%〜50%(重量基準)の量で含まれる。少なくとも1種のポリアル
キレングリコールが経口投薬剤形中に存在する場合、少なくとも1種の脂肪族アルコール
と少なくとも1種のポリアルキレングリコールとの合計重量は、好ましくは、全用量の20%
〜50%(重量基準)を占める。
【0128】
1実施形態では、例えば、少なくとも1種のヒドロキシアルキルセルロースまたはアクリ
ル樹脂対少なくとも1種の脂肪族アルコール/ポリアルキレングリコールの比によって、製
剤からのオピオイドの放出速度はかなりの影響を受ける。少なくとも1種のヒドロキシア
ルキルセルロース対少なくとも1種の脂肪族アルコール/ポリアルキレングリコールの比は
1:2〜1:4が好ましく、1:3〜1:4が特に好ましい。
【0129】
上記の少なくとも1種のポリアルキレングリコールは、例えば、ポリプロピレングリコ
ールであってよく、好ましくは、ポリエチレングリコールである。少なくとも1種のポリ
アルキレングリコールの数平均分子量は1,000〜15,000が好ましく、特に、1,500〜12,000
が好ましい。
【0130】
別の好適な制御放出性マトリックスには、アルキルセルロース(特にエチルセルロース)
、C12〜C36脂肪族アルコール、および場合によりポリアルキレングリコールが含まれるで
あろう。
【0131】
別の好ましい実施形態では、マトリックスには、少なくとも2種の疎水性材料の製薬上
許容される組合せが含まれる。
【0132】
上の成分のほかに、制御放出性マトリックスには、好適な量の他の物質、例えば医薬分
野で以前より知られている、希釈剤、滑沢剤、結合剤、造粒助剤、着色剤、着香剤、およ
び滑走剤(glidant)が含まれていてもよい。
【0133】
マトリックスベースのビーズ剤を調製するための方法 本発明に係る固形制御放出性経
口投薬剤形の調製を容易にするために、当業者に周知であるマトリックス製剤の調製方法
のいずれを使用してもよい。具体例として、マトリックス中への導入は、例えば、次のよ
うに行うことができる。(a)少なくとも1種の水溶性ヒドロキシアルキルセルロースとオピ
オイドまたはオピオイド塩とを含む顆粒を形成、(b)ヒドロキシアルキルセルロースを含
有する顆粒を少なくとも1種のC12〜C36脂肪族アルコールと混合し、(c)場合により、顆粒
を圧縮および造形する。好ましくは、水を用いてヒドロキシアルキルセルロース/オピオ
イドを湿式造粒することによって、顆粒を形成する。この方法の特に好ましい実施形態で
は、湿式造粒ステップで添加される水の量は、好ましくは、オピオイドの乾燥重量の1.5
〜5倍、特に、1.75〜3.5倍である。
【0134】
更に他の実施形態では、有効成分と一緒に球状化剤を用いて球状化処理することにより
、スフェロイド剤を形成することができる。微晶質セルロースが好ましい。好適な微晶質
セルロースは、例えば、Avicel PH 101(商標、FMC Corporation)として販売される物質で
ある。このような実施形態では、有効成分および球状化剤のほかに、スフェロイド剤には
結合剤が含まれていてもよい。好適な結合剤、例えば、低粘度の水溶性ポリマーは、医薬
分野の当業者には周知であろう。しかしながら、ヒドロキシプロピルセルロースのような
水溶性ヒドロキシ低級アルキルセルロースが好ましい。このほかに(またはこの代わりに)
、スフェロイド剤には、水不溶性ポリマー、特に、アクリルポリマー、アクリルコポリマ
ー、例えば、メタクリル酸-エチルアクリル酸コポリマー、またはエチルセルロースが含
まれていてもよい。このような実施形態では、持続放出性剤皮には、一般に、(a)蝋(単独
でもしくは脂肪アルコールとの混合物として)、または(b)セラックもしくはゼインのよう
な疎水性材料が含まれるであろう。
【0135】
溶融押出マトリックス 溶融造粒法または溶融押出法を用いて、持続放出性マトリクス
を調製することも可能である。一般的には、溶融造粒法では、通常は固体の疎水性材料、
例えば、蝋を溶融させ、その中に粉末薬剤を導入する。持続放出性投薬剤形を得るために
、追加の疎水性物質、例えば、エチルセルロースまたは水不溶性アクリルポリマーを、溶
融蝋疎水性材料中に導入する必要がある。溶融造粒法を用いて調製される持続放出性製剤
の例が、米国特許第4,861,598号に記載されている。この特許は、本発明の譲受人に譲渡
されており、その全内容を、参照により本明細書に組み入れる。
【0136】
追加の疎水性材料には、1種以上の水不溶性蝋様可塑性物質が、1種以上の該水不溶性蝋
様物質よりも疎水性の弱い1種以上の蝋様熱可塑性物質と混合されて含まれていてもよい
。一定した放出が行われるようにすべく、製剤中の各蝋様物質は、初期放出段階において
、実質的に胃腸液中で分解されず、かつ不溶でなければならない。有用な水不溶性蝋様物
質は、水への溶解度が約1:5,000(w/w)よりも低い物質であってよい。
【0137】
また、持続放出性マトリックスには、上記の成分のほかに、適量の他の材料、例えば医
薬分野で以前より知られている、希釈剤、滑沢剤、結合剤、造粒助剤、着色剤、着香剤、
および滑走剤(glidant)が含まれていてもよい。これらの追加材料の量は、所望の作用
を所望の製剤に付与するのに十分な量であろう。
【0138】
また、溶融押出された多粒子剤を含んでなる持続放出性マトリックスには、上記の成分
のほかに、適量の他の材料、例えば医薬分野で以前より知られている、希釈剤、滑沢剤、
結合剤、造粒助剤、着色剤、着香剤、および滑走剤(glidant)が、所望により顆粒の約50
重量%までの量で含まれていてもよい。
【0139】
経口投薬剤形の調合に使用しうる製薬上許容される担体および賦形剤の特定の具体例が
、参照により本明細書に組み入れるHandbook of Pharmaceutical Excipients, American
Pharmaceutical Association (1986)に記載されている。
【0140】
溶融押出多粒子剤 本発明に係る好適な溶融押出マトリックスの調製には、例えば、オ
ピオイド鎮痛薬を、少なくとも1種の疎水性材料および好ましくは追加の疎水性材料とブ
レンドして均質混合物を得るステップが含まれていてもよい。次に、十分な温度まで均質
混合物を加熱することにより、押出するのに十分な程度まで均質混合物を少なくとも軟化
させる。その後、得られた均質混合物を押出してストランドを形成する。好ましくは、当
技術分野で公知の任意の手段により押出物の冷却および切削を行って多粒子剤にする。ス
トランドの冷却および切削を行って多粒子剤にする。次に、多粒子剤を単位用量に分割す
る。押出物は、好ましくは、約0.1〜約5mmの直径を有し、治療上有効な薬剤を約8〜約24
時間にわたり持続放出させる。
【0141】
本発明の溶融押出物を調製するために場合により使用される方法では、疎水性材料、治
療上有効な薬剤、および必要に応じて使用される結合剤を計量して押出機に直接供給し、
均質混合物を熱し、均質混合物を押出すことによりストランドを形成し、均質混合物を含
有するストランドを冷却し、ストランドを切断して約0.1mm〜約12mmのサイズの粒子にし
、更に、この粒子を単位用量まで分割する。
本発明のこの態様では、比較的連続した製造工程が実現される。
【0142】
また、押出機のアパーチャーすなわち送出口の直径を調節することにより、押出される
ストランドの厚さを変えることができる。更に、押出機の送出部分は、円形である必要は
なく、楕円形、矩形などにすることも可能である。熱線カッター、裁断機などを用いて、
送出ストランドを分割して粒子にすることもできる。
【0143】
溶融押出多粒子系は、押出機の送出オリフィスに応じて、例えば、顆粒剤、スフェロイ
ド剤、またはペレット剤の形態をとることができる。本発明の目的では、用語「溶融押出
多粒子剤」、「溶融押出多粒子系」、および「溶融押出粒子」とは、好ましくは、所定の
範囲内の類似したサイズおよび/または形状を有し、1種以上の有効な薬剤と、1種以上の
賦形剤と、好ましくは、本明細書中に記載の疎水性材料とを含む複数のユニットを意味す
る。この場合、溶融押出多粒子剤の長さは、約0.1〜約12mmであり、その直径は約0.1〜約
5mmである。また、溶融押出多粒子剤は、このサイズの範囲内で任意の幾何学形状をとる
ことができると考えられる。このほか、球状化ステップを用いずに、押出物を単に所望の
長さに切断し、治療上有効な薬剤の単位用量まで分割してもよい。
【0144】
好ましい1実施形態では、有効量の溶融押出多粒子剤がカプセル剤中に含まれるように
経口投薬剤を調製する。例えば、胃液による消化および胃液との接触の際に有効な持続放
出を行うのに十分な量で複数の溶融押出多粒子剤をゼラチンカプセル剤中に配置してもよ
い。
【0145】
もう1つの好ましい実施形態では、従来型の錠剤機を用いて標準的な方法により適量の
多粒子剤押出物を圧縮して経口錠剤にする。また、錠剤(圧縮錠剤、すりこみ錠剤)、カプ
セル剤(硬質ゼラチンカプセル剤および軟質ゼラチンカプセル剤)、および丸剤を作製する
ための方法および組成については、参照により本明細書に組み入れるRemington's Pharma
ceutical Sciences, (Arthur Osol, editor), 1553-1593 (1980)に記載されている。
【0146】
さらに別の好ましい実施形態では、前記でさらに詳細に記載され、そのため参照により
本明細書中に組み入れる米国特許第4,957,681号(Klimeschら)に記載されているようにし
て押出物を錠剤に成形することができる。
【0147】
場合により、持続放出性剤皮(例えば前記の持続放出性剤皮)を用いて、持続放出性溶融
押出多粒子系または錠剤にコーティングを施すことが可能であり、あるいはゼラチンカプ
セル剤に更にコーティングを施すことも可能である。このような剤皮には、好ましくは、
重量を約2〜約30%増加させるのに十分な量の疎水性材料が含まれる。しかしながら、特に
、利用する特定のオピオイド鎮痛薬化合物の物理的性質および所望の放出速度にもよるが
、被覆膜を更に増大させることも可能である。
【0148】
本発明の溶融押出単位投薬剤には、更に、先に開示した治療上有効な薬剤を1種以上含
有する溶融押出多粒子剤がカプセル化前に組み合わせされて含まれていてもよい。このほ
か、単位投薬剤には、直ぐに治療効果を得るべく、即時放出性の治療上有効な薬剤を所定
量含有させることができる。即時放出性の治療上有効な薬剤は、例えば、別のペレット剤
としてゼラチンカプセル剤中に導入してもよいし、あるいは投薬剤を調製した後、多粒子
剤の表面上にコーティングしてもよい(例えば、制御放出性の剤皮またはマトリックスベ
ースの剤皮)。また、本発明の単位投薬剤には、所望の効果を得るべく、制御放出性ビー
ズ剤およびマトリックス多粒子剤が組み合わされて含まれていてもよい。
【0149】
本発明の持続放出性製剤は、好ましくは、例えば、摂取後、胃液や腸液に触れると、治
療上有効な薬剤を徐々に放出する。発明の溶融押出製剤の持続放出プロフィルは、例えば
、遅延剤すなわち疎水性材料の量の変更、疎水性材料に対する可塑剤の量の変更、他の成
分または賦形剤の導入、製造方法の変更などによって、調節可能である。
【0150】
本発明の他の実施形態では、溶融押出される材料を、治療上有効な薬剤を含有させずに
調製し、その後、該薬剤を押出物に添加する。このような製剤に含まれる治療上有効な薬
剤は、典型的には、押出マトリックス材料と一緒にブレンドされ、次に、該混合物の錠剤
化を行うことにより、徐放性製剤を形成することになるであろう。このような製剤は、例
えば、製剤中に含まれる治療上有効な薬剤が疎水性材料および/または遅延剤材料の軟化
に必要な温度の影響を受け易い場合に有利である。
【0151】
好ましい実施形態の詳細な説明 以下の実施例により本発明の種々の態様について具体
的に説明する。これらの実施例により特許請求の範囲はなんら制限されるものではない。
【0152】
本発明者らの知るかぎり、ナルトレキソンを種々のオピオイドアゴニストと一緒に同時
投与した後でその競合的アゴニストの性質を直接比較することはこれまでに行われていな
い。しかしながら、ヘロインまたはモルヒネのいずれかのチャレンジを受けた被験者にお
いて、用量変化の研究が行われ、オピオイドアンタゴニストの性質が評価された。一般的
には、ナルトレキソン50mgを予め投与してからその24時間後にヘロイン25mgを静脈内投与
した場合、オピオイドアゴニストの作用は完全にブロックされるかまたは低減した。Gonz
alez JP, Brogden RN. 「ナルトレキソン:オピオイド依存症の治療におけるその薬力学的
および薬動学的性質ならびに治療効果に関するレビュー」Drugs 1988; 35:192-213; Resn
ick RR, Valavka J, Freedman AM, Thomas M. 「EN-1639A(ナルトレキソン):新しい麻薬
アンタゴニストに関する研究」Am. J. Psychiatry 1974; 131:646-650を参照されたい。
これらはいずれも参照により本明細書に組み入れる。
【実施例】
【0153】
実施例1 実施例1では、ランダム化、単純盲検、プラシーボ対照、および単回投与によ
る4法交差試験を行った。これにより、ナルトレキソン経口液剤6.4mgが6人の正常な健常
女性ボランティアにおいてヒドロコドン15mgのオピオイドアゴニスト作用をブロックする
かを評価した。実験個体群には女性だけが含まれていた。なぜなら、以前の観察において
、女性の方が男性よりもオピオイドアゴニスト作用に対する感受性が高いことが示唆され
たためである。4種の治療薬は、HYIR/APAP(ヒドロコドン7.5およびアセトアミノフェン75
0mgの2つの錠剤、Vicodin ES(登録商標)) + ナルトレキソン経口液剤3.2mg、HYIR/APAP(2
×7.5mg) + ナルトレキソン経口液剤6.4mg、HYIR比較(comparator)錠剤(2×750mg Trilis
ate(登録商標)錠剤) + ナルトレキソン経口液剤(プラシーボ)、HYIR/APAP(Vicodin ES(登
録商標)の2つの錠剤) + ナルトレキソン経口液剤(プラシーボ)であった。いずれの治療薬
についても、絶食状態で投与した。それぞれの投与の間で48時間のウォッシュアウト期間
を設けた。被験者を、これらの4種の治療薬グループの4つの治療薬投与順にランダムに割
り当てた。最初の投与を行う前の夕方に被験者は試験施設に集まり、最後の投与を行って
から24時間後の投与評価が終了するまで拘束状態にあった。安全性の測定は、副作用の報
告、生命徴候、異常な実験値、異常な外診結果、およびECGの結果からなっていた。また
、薬力学的パラメーター(瞳孔サイズおよび改良型特異的薬効アンケート調査)についても
評価した。
【0154】
試験治療薬 4種の治療薬は以下の通りであった。
【0155】
ヒドロコドン即時放出性錠剤(2×7.5mg) + ナルトレキソン経口液剤3.2mg。
【0156】
ヒドロコドン即時放出性錠剤(2×7.5mg) + ナルトレキソン経口液剤6.4mg。
【0157】
ヒドロコドン即時放出性比較錠剤 + プラシーボナルトレキソン経口液剤。
【0158】
プラシーボヒドロコドン即時放出性錠剤(2×7.5mg) + プラシーボナルトレキソン経口液
剤。
【0159】
試験製品 この試験で評価した製品には、Vicodin ES(登録商標)(酒石酸水素ヒドロコド
ン7.5mgおよびアセトアミノフェン750mg、Knoll Pharmaceuticals)、比較物質(comparato
r)として利用したTrilisate(登録商標) (コリンマグネシウムトリサリチレート750mg、Pu
rdue Frederick)、およびナルトレキソン粉末薬が含まれていた。Vicodin ES(登録商標)
を活性治療薬として選択した。なぜなら、この製品中のアセトアミノフェン部分は、中枢
神経系にも瞳孔測定にも影響を及ぼさないと考えられるからである。「比較物質」として
使用するためにTrilisateを選択した。なぜなら、その物理的外観がVicodin ES(登録商標
)と類似しているうえに、中枢神経系にも瞳孔測定にも影響を及ぼさないからである。市
販の錠剤(Revia(登録商標) 50mg錠剤、DuPont)を用いずにナルトレキソン散剤を選択する
ことにより、経口液剤を調製する際の全体的な精度を向上させた。現場の研究薬剤師が、
適切な医薬品調合法を利用して滅菌環境下でナルトレキソン散剤から経口液剤を再調製し
た。ナルトレキソン散剤(Mallinckrodt Chemical)を使用してナルトレキソン液剤を調合
した。ナルトレキソンの個々のストック溶液は、TsangおよびHoltsmanにより提案された
方法に変更を加えて調製した。Tsang BK, Holtsman R. 「液状ナルトレキソンの室温安定
性」Anesthesiology 1995:83:A864。この文献は参照により本明細書に組み入れる。ナル
トレキソンストック溶液の調製は、次のように行った。各投与期間の直前(60分以内)に、
ナルトレキソン散剤32mgおよび64mgを秤量した。これらの部分をそれぞれ、蒸留水50mL +
単純シロップ50mL(NF:米国医薬品集)中に溶解し、最終体積を100mLとした。最終溶液の
濃度はそれぞれ、0.32mg /mL(32mg/100mL)および0.64mg/mL(64mg/100mL)であった。これ
らの濃度を用いることにより、各投与期間中に同じ体積(10mL)のナルトレキソン経口液剤
を投与できるようにした。ナルトレキソン経口液剤プラシーボは、活性溶液と同じビヒク
ルで調製した。活性溶液と同じような味を呈するように、苦味剤Bitterguard(デナトニウ
ムベンゾエート、NF)の添加を行った。
【0160】
薬力学的測定a. 瞳孔サイズ - 瞳孔測定により調べる。
【0161】
75mmレンズおよび内蔵エレクトロニックリングフラッシュを備えたPolaroid CU-5カメ
ラにPolacolor ER 669インスタントパックフィルム12を装填して、瞳孔直径の測定を行っ
た。この方法は、瞳孔を調べるための安全で正確な方法として受け入れられるようになっ
ており、一般に、赤外線テレビ瞳孔測定法(より汎用性がありかつ高性能であるが、かな
り高価で取扱いの難しい方法)に次ぐ方法であるとみなされている。Polaroid CU-5法の精
度は0.1mm以内であると言われている。Czarnecki JS, Pilley SF, Thompson HS. “The u
se of Photography in the Clinical Evaluation of Unequal Pupils”, Canad. J. Opht
hal. 1979;14:297-302を参照されたい。この文献は、参照により本明細書に組み入れる。
【0162】
次のように瞳孔直径を測定した。フラッシュの角膜反射によって水平方向の瞳孔周辺が
不明瞭になるのを避けるために、リングフラッシュの3時および9時の方向を2つの小片で
カバーするようにカメラを改造した。被験者の顔の前面で3インチフレームが側方眼窩縁
に当接するようにカメラを配置し、視線がそのカメラ領域の上部すれすれを向くようにし
た(上方すれすれ入射(upgaze)を最小限に抑えるため)。カメラ本体の上部越しに遠方の非
調節性標的に視線を固定するように被験者に指示し、調節性瞳孔反射を最小限に抑えるよ
うにした。被験者が遠方を凝視している状態で写真を撮影した。写真はいずれも、一定の
周囲光の下で撮影した。瞳孔潜伏時間は、フラッシュが瞳孔直径に影響を及ぼさない時間
であった。フラッシュの後、瞳孔の持続性収縮は起こらないが、短時間の収縮は観測され
る。従って、フラッシュは、この試験に必要な測定には影響を及ぼさなかった。Smith SA
, Dewhist RR. “A Single Diagnostic Test for Pupillary Abnormality in Diabetic A
utonomic Neuropathy”, Diabetic Medicine 1988;3:38-41を参照されたい。この文献は
、参照により本明細書に組み入れる。所定の時間(周囲温度にもよるが、約1分間)でプリ
ントの現像を行うと、被験者の顔中部の1対1の写真が得られ、瞳孔はプリントの上部に現
れる。次に、0.1mmまで校正された網線の内蔵された単純なルーペを用いて瞳孔直径を測
定する。プロトコルに規定された各時間で左眼だけを対象に瞳孔効果を調べた。
【0163】
b. 改良型特異的薬効アンケート調査 アンケート調査は、JasinskiおよびPrestonが使
用した22の質問事項に改良を加えたものである。Jasinski DR., “Assessment of the Ab
use Potential of Morphine-Like Drugs (男性に利用される方法)”, Drug Addiction I
(Martin, W.R., ed.), 1997:197-258. Springer-Verlag, New York; Preson KL, Jasinsk
i DR. Testa M., “Abuse Potential and Pharmacological Comparison of Tramadol and
Morphine”, Drug and Alcohol Dependence 1991;27:7-17を参照されたい。これらの文
献はいずれも、参照により本明細書に組み入れる。このアンケートは10項目からなり、血
液を採取する10分前に被験者が評価した。この項目は、アヘン作動薬薬剤の徴候に関連付
けられたものであり、次の通りであった。
【0164】
被験者への質問内容: 1)あなたは薬剤の効果を感じますか? 2)あなたは皮膚にかゆみを
感じますか? 3)あなたは楽な気持ちを感じますか? 4)あなたは眠気を感じますか? 5)あな
たは酔いを感じますか? 6)あなたは神経のいらだちを感じますか? 7)あなたは元気いっぱ
いであると感じますか? 8)あなたは話をする必要性を感じますか? 9)あなたは胃のむかつ
きを感じますか? 10)あなたはめまいを感じますか? この際、被験者は、100mmの視覚的ア
ナログスケール(VAS)に沿って縦線を引くことにより、項目の評価を行った。このスケー
ルの一端には「まったく感じない」と記され、他端には「かなり感じる」と記されていた
。
【0165】
左眼の瞳孔サイズは、基準時間(投与前30分以内)および投与後0.5、1、2、4、6、9、お
よび12時間で測定した。また、被験者は、基準時間および投与後0.5、1、2、4、6、9、お
よび12時間で薬効スコアを評価し、改良型特異的薬効アンケート調査(“MSDEQ")用の視覚
的アナログスケール上に記入した。
【0166】
ナルトレキソン用量に対する11種のレスポンス(MSDEQの質問および瞳孔直径の測定)を
示すそれぞれのグラフを、視覚的および統計的に調べ、この試験に使用したヒドロコドン
の用量と組み合わせて、ナルトレキソンの公称有効用量を決定した。
【0167】
報告した副作用は、一般にオピオイド鎮痛薬の投与に関連付けられるものであり、その
ほとんどは「中程度」のものとして分類された。重度の副作用や死亡はまったく起こらな
かった。また、副作用が原因で試験を中断した患者はいなかった。
【0168】
結果は図1および2に示されている。
【0169】
図1は、ヒドロコドンにより誘発されたVAS(視覚的アナログスケール)「薬効」に対する
ナルトレキソンの拮抗作用を示している。この図は、改良型特異的薬効アンケート調査の
最初の質問に関するものであり、被験者への質問内容は、「あなたは薬剤の効果を感じま
すか?」である。これらの結果から示唆されるように、ナルトレキソンに関して用量‐応
答効果が存在し、ナルトレキソン用量の増大に伴ってヒドロコドンのVAS「薬効」は減少
した。ナルトレキソン用量6.4mgの場合、ナルトレキソン用量3.2mgの場合よりも、ヒドロ
コドン用量15mgの薬効に対してより大きな拮抗作用を示した。ヒドロコドンのオピオイド
作用は、ナルトレキソン用量6.4mgによって完全にはブロックされなかった。図2は、ヒド
ロコドンにより誘発された瞳孔収縮に対するナルトレキソンの拮抗作用を示している。こ
れらの結果からもナルトレキソンに関して用量‐応答効果が示唆され、ヒドロコドン15mg
を投与された被験者の瞳孔収縮は、ナルトレキソンの用量を増大させると減少した。ナル
トレキソン用量6.4mgの場合、ナルトレキソン用量3.2mgの場合よりも、ヒドロコドンによ
り誘発された瞳孔収縮に対してより大きな拮抗作用を示した。ヒドロコドンによる瞳孔収
縮は、ナルトレキソン用量6.4mgによって完全にはブロックされなかった。偽薬グループ
で、最小量の瞳孔収縮を生じた。ヒドロコドン+ナルトレキソン偽薬グループは、最大の
瞳孔収縮を示し、従って、瞳孔直径の測定値は最小であった。
【0170】
実施例2 実施例2では、正常な健常女性被験者において、10期、ランダム化、交差、お
よび単純盲検による試験を行って、経口ナルトレキソン対経口ヒドロコドンの比を評価し
た。これにより、名目上、オピオイド作動薬作用は最小限に抑えられた。21人の被験者が
この試験に登録し、16人が試験を完了した。10種の治療薬には、HYIR/APAP(錠剤1個あた
りヒドロコドン7.5およびアセトアミノフェン750mgの2つの錠剤、Vicodin ES(登録商標))
+次の用量:0.4mg/10mL、0.8mg/10mL、1.6mg/10mL、3.2mg/10mL、4.8mg/10mL、6.4mg/10
mL、9.6mg/10mL、12.8mg/10mLのナルトレキソン経口液剤および偽薬ナルトレキソン経口
液剤、更に、ヒドロコドン即時放出性コンパラトール錠剤(2×750mg Trilisate(登録商標
)錠剤)+偽薬ナルトレキソン経口液剤が含まれていた。いずれの治療薬についても、絶食
状態で投与した。それぞれの投与の間で48時間のウォッシャアウト期間を設けた。被験者
を、これらの10種の治療薬グループの10期の治療シーケンスにランダムに割り当てた。最
初の投与を行う前の夕方に被験者は試験施設に集まり、最後の投与を行ってから24時間後
の投与評価が終了するまで拘束状態にあった。安全性の測定は、副作用の報告、生命徴候
、異常な実験値、異常な外診結果、およびECGの結果からなっていた。血漿中のヒドロコ
ドン、ナルトレキソン、および6-β-ナルトレキソールレベルを調べた。これから薬物動
態学的値が計算および分析されるであろう。また、薬力学的パラメーター(瞳孔サイズお
よび改良型特異的薬効アンケート調査)についても評価した。
【0171】
投与レジメ 投与レジメは次の通りであった。
【0172】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(偽薬)と一緒にヒドロコドン即時放出性コンパラトール(偽薬)錠剤を投与した。投
与後さらに4時間絶食を続けた。
【0173】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(偽薬)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さらに
4時間絶食を続けた。
【0174】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(0.4mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0175】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(0.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0176】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(1.6mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0177】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(3.2mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0178】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(4.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0179】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(6.4mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0180】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(9.6mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0181】
8時間の絶食を行ってから第1期〜第10期の投与日のほぼ08:00にナルトレキソン経口液
剤10ml(12.8mg)と一緒にヒドロコドン即時放出性錠剤(2×7.5mg)を投与した。投与後さら
に4時間絶食を続けた。
【0182】
各投与日に所定の薬剤を各用量で投与したが、被験者はその投与前8時間にわたり絶食
し、投与後さらに4時間絶食を続けた。最初の投与(0時間)を行う前に(投与前30分以内に
)ベースライン用血液サンプル(血漿中のヒドロコドン、ナルトレキソン、および6-β-ナ
ルトレキソールを調べるため)を採取し、更に、投与後0.5、1、2、4、6および9時間で採
取した。サンプルはすべて、予定時間の±2分以内に採取した。基準時間(投与前30分以内
)ならびに投与後0.5、1、2、4、6および9時間で血液サンプルの採取を行う直前に以下の
薬力学的パラメーターの測定を行った。
【0183】
各投与期間の直前にナルトレキソン散剤4、8、16、32、48、64、96、および128mgを秤
量し、8種のナルトレキソンストック溶液を調製した。これらの散剤アリコートはそれぞ
れ、蒸留水50ml+単シロップ50ml中に溶解した。最終溶液は100mLであり、その濃度は0.0
4、0.08、0.16、0.32、0.48、0.96、および1.28mg/mLであった。これらの濃度を用いるこ
とにより、各投与期間中に同じ体積(10mL)のナルトレキソン経口液剤を投与できるように
した。ナルトレキソン偽薬液剤は、活性溶液と同じビヒクルで調製した。活性溶液と同じ
ような味を呈するように、苦味剤Bitterguard Powder(デナトニウムベンゾエート)の添加
を行った。
【0184】
薬力学的測定 実施例2の薬力学的測定値は、上記の実施例1で述べた手順に従って得た
。
【0185】
所定の時間における各処置に関する平均「薬効」VASスコアおよび瞳孔直径を図3および
4にそれぞれ示す。一般的には、ナルトレキソンの用量を増大させて(0mg〜12.8mgの範囲)
ヒドロコドン即時放出剤/アセトアミノフェン(「HYIR/APAP」)を1回投与すると、「薬効
」VASスコアが全体的に減少するとともに瞳孔収縮も減少した。図5および6は、各ナルト
レキソン用量の対数値に対して、対応する平均最大「薬効」VASスコア(±95% CI)および
平均最小瞳孔直径(±95% CI)を表している。これらの図から、瞳孔効果に関する用量‐応
答関係の方が「薬効」VAS応答の場合よりも大きな用量‐応答関係を示すことが分かる。
【0186】
これらの結果から分かるように、ナルトレキソン0.4mgを投与した場合でさえも、ヒド
ロコドン投与の薬理学的効果は低減した。ナルトレキソン約0.4mgのときにヒドロコドン
用量15mgに対して最小の拮抗作用を示した。ナルトレキソン用量が0.4mgを超えると、ヒ
ドロコドン投与の効果が減少し始めた。
【0187】
報告した副作用は、一般にオピオイド鎮痛薬の投与に関連付けられるものであり、その
ほとんどは「マイルドな」ものとして分類された。全体で5人の被験者(5/21)は、試験を
中断した。3人の被験者は副作用が原因で中断した。これらの被験者のうちの2人は、重度
でないと分類された副作用を伴った。1人の被験者は、重度であると分類された貧血を起
こし、鉄治療を必要とした。他の2人の被験者は、試験に参加できなくする情報が病歴中
に存在すると担当医師が判断したために中断した。この試験で死亡は起こらなかった。
【0188】
一般的には、ナルトレキソン経口液剤の用量を増大させて(0mg〜12.8mgの範囲)ヒドロ
コドン即時放出性錠剤15mgを1回投与すると、「薬効」VASスコアが全体的に減少し瞳孔直
径は増加した。
【0189】
実施例3 実施例3では、ヒドロコドン即時放出性錠剤およびナルトレキソン経口液剤を
投与したモルヒネ依存性ボランティアにおいて性急型禁断症状を評価した試験の結果を提
示する。この試験は、単純盲検、1回投与、およびプラシーボ対照によるナルトレキソン
用量増大試験であり、オピオイドに対して身体的依存のある被験者を対象とした。実験対
象被験者(5人)は、Narcanチャレンジ、嗜癖重度指数スコア、身体検査、観察、および尿
薬剤スクリーニングの結果から判断して、オピオイド依存性であり、その時点で嗜癖に対
する治療を望んではいなかった。ヒドロコドン即時放出剤およびナルトレキソンの同時投
与を行った後で性急型禁断症状を評価するために、ヒドロコドン乱用患者の使用する用量
レベルをシミュレートすべく、ヒドロコドン即時放出剤の用量30mgを選択した。これは、
オピオイドの投与されたことのない被験者において、他の一般に使用されるオピオイドと
等価な鎮痛作用を示すと考えられる用量である。ヒドロコドンの相対的鎮痛効果は、オキ
シコドンの鎮痛効果と同等であり、経口モルヒネの鎮痛効果の約2倍であると考えられて
いる。
【0190】
試験処置 処置は次の通りであった。
【0191】
ヒドロコドン/アセトアミノフェン即時放出性(HYIR/APAP)錠剤30mg(Lortab(登録商標)
3×10mg) + 用量を増大させたナルトレキソン経口液剤0、0.25mg、0.5mg、1.0mg、および
2.0mg。ヒドロコドン/アセトアミノフェン即時放出性(HYIR/APAP)錠剤30mg(Lortab(登録
商標) 3×10mg) + ナルトレキソンプラシーボ経口液剤。ナルトレキソン経口液剤および
プラシーボ液剤は、実施例1〜2に従って調製した。
【0192】
規則的な時間間隔で、すなわち毎日午前6時および10時ならびに午後4時および10時に硫
酸モルヒネ15mgを筋肉内投与することによって、5日間かけて被験者を安定化させた。硫
酸モルヒネ15mgの筋肉内投与は、ヒドロコドン30mgの経口投与と同等である。安定化を行
った後、試験治療薬の投与日の午前10時に試験治療薬を投与、それから6時間にわたり観
察を行った。6時間後に性急型禁断症状が現れない場合、午後4時に硫酸モルヒネ15mgを再
び筋肉内投与した。次の試験薬剤の投与を行う前に48時間かけて被験者を安定化させた。
各処置(1〜4)後、性急型禁断症状が現れない場合、以下の処置の中から次の番号順に試験
治療薬を被験者に投与した。
【0193】
処置番号1: 8時間の絶食を行った後、投与日のほぼ10:00にプラシーボナルトレキソン(
10mL)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与
後、引き続き(4)時間絶食を行う。
【0194】
処置番号2: 8時間の絶食を行った後、投与日のほぼ10:00に0.25mgナルトレキソン(10m
L)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後
、引き続き(4)時間絶食を行う。
【0195】
処置番号3: 8時間の絶食を行った後、投与日のほぼ10:00に0.5mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0196】
処置番号4: 8時間の絶食を行った後、投与日のほぼ10:00に1.0mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0197】
処置番号5: 8時間の絶食を行った後、投与日のほぼ10:00に2.0mgナルトレキソン(10mL
)経口液剤と一緒にHYIR/APAP錠剤30mg(Lortab(登録商標) 3×10mg)を投与する。投与後、
引き続き(4)時間絶食を行う。
【0198】
投与の0.5時間前および投与の0.5、1、2、4、および6時間後に、血液サンプルを採取し
た。Pupilscan瞳孔計を用いて瞳孔直径の測定値を求め、ミリメートルの少数以下を四捨
五入してミリメートル単位で記録した。各試験期間の後、48時間にわたるウォッシュアウ
ト期間を設けた。4人の被験者が試験を終了し、1人の被験者はとりやめた。ナルトレキソ
ンの効果として、1および2mgの場合、わずかな禁断(禁断の徴候)が得られた。
【0199】
プロトコルを修正し、12人の実験対象被験者をこのプロトコルに従って試験した。この
プロトコルは、ナルトレキソンの比を増大させたこと以外は先に述べた試験と同じであっ
た。修正プロトコルのナルトレキソン用量は、0、1、2、4、および8mgであった。実験対
象被験者のうちの8人が試験を終了し、4人はとりやめた。
【0200】
各被験者に対して生命徴候を監視し、更に、オピオイド禁断の徴候および症状を監視し
た。禁断徴候としては、鼻づまりもしくは鼻水、流涙、あくび、発汗、震え、嘔吐、起毛
、瞳孔散大、被刺激性、および不眠が挙げられる。禁断症状としては、温度変化の感覚、
関節、骨、もしくは筋肉の痛み、胃の痙攣、皮膚のかゆみ、および吐き気が挙げられ、被
験者は、これらの症状の主観的な経験を報告する。
【0201】
薬剤併用の主観的経験の尺度を示すために、被験者は、試験期間にわたりアンケートの
質問に答えた。実施例1に記載したように、質問に対する答えを視覚的アナログスケール
上で等級分けした。評価した主観的経験は、次の通りであった:薬剤の好き嫌い、薬効の
知覚能力、発汗、不眠、震え、流涙、鳥肌、胃の不調、鼻血、眠気、寒気、発熱、筋肉痛
、緊張もしくは弛緩、錯乱、恐怖感、被刺激性、多弁性、禁断の感覚、疾患の感覚。また
、被験者の観察を次の観点から行った:あくび、ひっかき、弛緩、鼻血、被刺激性、禁断
。このほか、血圧、脈拍数、呼吸数、瞳孔サイズ、および体温の監視も行った。
【0202】
被験者のうちの5人に対するデータを以下に示す。図7A〜Cは、アンケートの質問から得
られたヒドロコドンの主観的知覚の平均スコアを表しており、投与後の時間の関数として
、またナルトレキソンの用量の関数としてプロットしたものである。図7Aは、種々の量の
ナルトレキソンの存在下で被験者がヒドロコドンの効果を感じる能力を示している。図7B
および7Cは、それぞれ、種々の量のナルトレキソンの存在下におけるヒドロコドンに対す
る被験者の好ましいと感じるまたは好ましくないと感じる主観的経験を示している。
【0203】
図8AおよびBは、ヒドロコドンの効果についての主観的知覚の平均スコアを表しており
、投与後の時間の関数として、またナルトレキソンの用量の関数としてプロットしたもの
である。図8Aは、種々の量のナルトレキソンの存在下におけるヒドロコドンの効果により
禁断症状の主観的知覚を示している。図8Bは、種々の量のナルトレキソンの存在下におけ
る疾患についての主観的経験を示している。図9Aは、種々の量のナルトレキソンの存在下
においてヒドロコドンが瞳孔サイズに及ぼす影響を示している。図9Bは、種々の量のナル
トレキソンの存在下におけるヒドロコドンの効果による明らかな禁断症状を観測者の立場
から示したものである。
【0204】
図10A〜Cは、図7A〜Cに示されている曲線の下側面積を6時間の観察時間にわたり積分し
てナルトレキソンの用量の関数として示したものであり、更に、ナルトレキソンのプラシ
ーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルも示されている
。図10Aから分かるように、8mgまでのナルトレキソンを用いた場合、被験者がヒドロコド
ンの効果を知覚する能力はなくならない。すなわち、各ナルトレキソン用量に対して観測
された実測値から得られるAUC(0〜6時間)は、ナルトレキソンプラシーボ応答に対する95%
信頼限界内に完全に含まれる。図10Bは、ヒドロコドンに対する被験者の好ましいと感じ
る主観的経験のAUC(0〜6時間)をナルトレキソンの用量の関数として示している。図10Bか
ら分かるように、ナルトレキソンの用量が1mgを超えると、被験者の好ましいと感じる経
験は低減する。すなわち、AUCの実測値(0〜6時間)は、ナルトレキソン約1mgのところでナ
ルトレキソンプラシーボに対する95%信頼限界よりも小さくなる。図10Cから分かるように
、ナルトレキソンの用量が1mgを超えると、被験者の好ましくないと感じる経験は増大す
る。すなわち、AUCの実測値(0〜6時間)は、ナルトレキソン約1mgのところでナルトレキソ
ンプラシーボに対する95%信頼限界よりも大きくなる。
【0205】
図11A〜Cは、図8A〜Bおよび図9Aに示されている曲線の下側面積を6時間の観察時間にわ
たり積分してナルトレキソンの用量の関数として示したものであり、更に、ナルトレキソ
ンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルも示
されている。図11Aは、種々の量のナルトレキソンの存在下で禁断の主観的経験のAUC(0〜
6時間)を示している。図11Aから分かるように、ナルトレキソンの用量が約0.75mgを超え
ると、疾患の主観的経験が現れる。すなわち、各ナルトレキソン用量に対して図8Aで観測
されたAUCの実測値(0〜6時間)は、ナルトレキソン約0.75mgのところでナルトレキソンプ
ラシーボ応答に対する95%信頼限界より大きくなる。図11Bは、種々の量のナルトレキソン
の存在下で疾患の主観的知覚のAUC(0〜6時間)を示している。図11Bから分かるように、ナ
ルトレキソンの用量が約0.75mgを超えると、疾患の主観的経験が現れる。すなわち、各ナ
ルトレキソン用量に対して図8Bで観測されたAUCの実測値(0〜6時間)は、ナルトレキソン
約0.75mgのところでナルトレキソンプラシーボ応答に対する95%信頼限界より大きくなる
。図11Cは、瞳孔サイズ変化の実測値のAUC(0〜6時間)をナルトレキソンの用量の関数とし
て示している。図11Cから分かるように、8mgまでのナルトレキソンを用いた場合、ヒドロ
コドンの縮瞳効果はなくならない。すなわち、各ナルトレキソン用量に対して図9Aで観測
された実測値から得られるAUC(0〜6時間)は、ナルトレキソンプラシーボ応答に対する95%
信頼限界内に完全に含まれる。
【0206】
臨床試験の結果、ヒドロコドンは、ナルトレキソンと併用した場合、0.5時間未満に出
現し、0.5〜1時間以内にピークに達し、3〜4時間以内に著しく減少することが分かった。
弱い用量‐応答曲線が得られた。ナルトレキソンを添加すると、ヒドロコドンに対する好
ましいと感じる主観的経験は低減し、ヒドロコドンに対する嫌悪の主観的経験は増大し、
疾患についての主観的経験およびヒドロコドン効果により禁断症状の主観的経験は増大し
た。これらの経験は、明らかに嫌悪感を生じるものであった。
【0207】
本発明の特定の好ましい実施例を参照しながら本発明について説明および例示を行って
きたが、本発明の精神および範囲から逸脱することなく、本発明の範囲内で容易に変更を
加えることができるは、当業者には分かるであろう。このような変更は、添付の特許請求
の範囲に含めるものとする。
【図面の簡単な説明】
【0208】
以下の図面は本発明の実施形態を例示するものであり、特許請求の範囲により包含され
る本発明の範囲を制限するものではない。
【図1】図1は、実施例1についてのヒドロコドン誘導VAS(視覚アナログスケール)「薬効」のナルトレキソン拮抗作用を示す。
【図2】図2は、実施例1についてのヒドロコドン誘導瞳孔縮小のナルトレキソン拮抗作用を示す。
【図3】図3は、実施例2のそれぞれの治療についての時間経過に対する平均「薬効」VASスコアを示す。
【図4】図4は、実施例2のそれぞれの治療についての時間経過に対する平均「薬効」瞳孔直径を示す。
【図5】図5は、実施例2のそれぞれのナルトレキソン用量からの対数に対する平均最大「薬効」VASスコア(±95% CI)を示す。
【図6】図6は、実施例2のそれぞれのナルトレキソン用量からの対数に対する平均最少瞳孔直径(±95% CI)を示す。
【図7】A:実施例3において可変量のナルトレキソン存在下でヒドロコドン効果を感じる被験者の能力を示す。 BおよびC:実施例3において可変量のナルトレキソン存在下でヒドロコドンを被験者が好ましいまたは嫌だとそれぞれ感じる主観的経験を示す。
【図8】A:実施例3における可変量のナルトレキソン存在下でのヒドロコドン効果からの禁断症状の被験者の認識を示す。 B:実施例3における可変量のナルトレキソン存在下での疾病の主観的経験を示す。
【図9】A:実施例3において可変量のナルトレキソン存在下でヒドロコドンが瞳孔サイズに及ぼす効果を示す。 B:実施例3において可変量のナルトレキソン存在下でヒドロコドン効果からの禁断症状の、観測者から見たときの見かけの程度を示す。
【図10】A〜C:ナルトレキソン用量の関数としての、6時間の観察期間にわたって積分した図7A〜Cに示される曲線下面積、およびナルトレキソンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルを示す。
【図11】A〜C:ナルトレキソン用量の関数としての、6時間の観察期間にわたって積分した図8A〜Bおよび図9Aに示される曲線下面積、およびナルトレキソンのプラシーボ応答(ヒドロコドン30mg、ナルトレキソン0mg)に対する95%信頼レベルを示す。
【特許請求の範囲】
【請求項1】
経口的に治療上有効な量のオピオイド作動薬と、オピオイド拮抗薬とを含有する経口投薬剤形であって、
前記オピオイド拮抗薬は、即時放出形態であり、
前記投薬剤形は、前記作動薬と拮抗薬の組合せを経口的に投与したときには鎮痛薬として有効であるが、(i)前記治療有効量と同量でまたはそれより多量に投与したときには身体依存性ヒト被験者に嫌悪作用を生じさせ、かつ、(ii)前記オピオイド拮抗薬なしでヒト患者に投与したときには、鎮痛効果を維持するが、同じ治療量のオピオイド鎮痛薬と比較して前記オピオイド作動薬の鎮痛効果を増加させない、組合せ製剤を提供するオピオイド拮抗薬対オピオイド作動薬の比を有し、
前記オピオイド拮抗薬は、ナルトレキソン、ナロキソン、ナルメフェン、シクラゾシン、レバロルファン、それらの薬学的に許容しうる塩、及びそれらの混合物からなる群より選択され、
前記オピオイド拮抗薬対前記オピオイド作動薬の比は、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩とに基づく0.03:1〜0.27:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、オキシコドンまたはその薬学的に許容しうる塩とに基づく0.037:1〜0.296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、コデインまたはその薬学的に許容しうる塩とに基づく0.005:1〜0.044:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロモルホンまたはその薬学的に許容しうる塩とに基づく0.148:1〜1.185:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、レボルファノールまたはその薬学的に許容しうる塩とに基づく0.278:1〜2.222:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メペリジンまたはその薬学的に許容しうる塩とに基づく0.0037:1〜0.0296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メタドンまたはその薬学的に許容しうる塩とに基づく0.056:1〜0.444:1の重量比、および
ナルトレキソンまたはその薬学的に許容しうる塩と、モルヒネまたはその薬学的に許容しうる塩とに基づく0.018:1〜0.148:1の重量比、からなる群より選択される、前記経口投薬剤形。
【請求項2】
前記経口投薬剤形に含まれる拮抗薬の量が、前記治療有効量の2〜3倍のオピオイドを摂取する身体依存性中毒者に嫌悪の経験を生じさせる、請求項1に記載の経口投薬剤形。
【請求項3】
前記オピオイド作動薬がヒドロコドンまたはその薬学的に許容しうる塩であり、前記オピオイド拮抗薬がナルトレキソンまたはその薬学的に許容しうる塩である、請求項1に記載の経口投薬剤形。
【請求項4】
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩との比が、0.03:1〜0.27:1である、請求項3に記載の経口投薬剤形。
【請求項5】
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩との比が、0.05:1〜0.20:1である、請求項3に記載の経口投薬剤形。
【請求項6】
前記オピオイド作動薬がモルヒネ、ヒドロモルフォン、ヒドロコドン、オキシコドン、コデイン、レボルファノール、メペリジン、メタドン、これらの薬学的に許容しうる塩およびこれらの混合物からなる群より選択される、請求項1に記載の経口投薬剤形。
【請求項7】
NSAID、COX-2阻害剤、アセトアミノフェン、アスピリン、NMDA受容体拮抗薬、NMDA受容体活性化の主要な細胞内結果をブロックする薬物、鎮咳薬、去痰薬、うっ血除去薬、抗ヒスタミン薬、およびこれらの混合物からなる群より選択される非オピオイド薬物をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項8】
1種以上の製薬上許容される不活性賦形剤をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項9】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩である、請求項6に記載の経口投薬剤形。
【請求項10】
前記オピオイド作動薬に対して持続放出性を与える持続放出性担体をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項11】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、オキシコドンまたはその薬学的に許容しうる塩であり、それらの比が0.037:1〜0.296:1である、請求項1に記載の経口投薬剤形。
【請求項12】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、コデインまたはその薬学的に許容しうる塩であり、それらの比が0.005:1〜0.044:1である、請求項1に記載の経口投薬剤形。
【請求項13】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、ヒドロモルホンまたはその薬学的に許容しうる塩であり、それらの比が0.148:1〜1.185:1である、請求項1に記載の経口投薬剤形。
【請求項14】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、レボルファノールまたはその薬学的に許容しうる塩であり、それらの比が0.278:1〜2.222:1である、請求項1に記載の経口投薬剤形。
【請求項15】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メペリジンまたはその薬学的に許容しうる塩であり、それらの比が0.0037:1〜0.0296:1である、請求項1に記載の経口投薬剤形。
【請求項16】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メタドンまたはその薬学的に許容しうる塩であり、それらの比が0.056:1〜0.444:1である、請求項1に記載の経口投薬剤形。
【請求項17】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、モルヒネまたはその薬学的に許容しうる塩であり、それらの比が0.018:1〜0.148:1である、請求項1に記載の経口投薬剤形。
【請求項18】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、オキシコドンまたはその薬学的に許容しうる塩であり、それらの比が0.056:1〜0.222:1である、請求項1に記載の経口投薬剤形。
【請求項19】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、コデインまたはその薬学的に許容しうる塩であり、それらの比が0.0083:1〜0.033:1である、請求項1に記載の経口投薬剤形。
【請求項20】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、ヒドロモルホンまたはその薬学的に許容しうる塩であり、それらの比が0.222:1〜0.889:1である、請求項1に記載の経口投薬剤形。
【請求項21】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、レボルファノールまたはその薬学的に許容しうる塩であり、それらの比が0.417:1〜1.667:1である、請求項1に記載の経口投薬剤形。
【請求項22】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メペリジンまたはその薬学的に許容しうる塩であり、それらの比が0.0056:1〜0.022:1である、請求項1に記載の経口投薬剤形。
【請求項23】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メタドンまたはその薬学的に許容しうる塩であり、それらの比が0.083:1〜0.333:1である、請求項1に記載の経口投薬剤形。
【請求項24】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、モルヒネまたはその薬学的に許容しうる塩であり、それらの比が0.028:1〜0.111:1である、請求項1に記載の経口投薬剤形。
【請求項25】
前記即時放出形態のオピオイド拮抗薬が、ゼラチンカプセル内に別のペレットとして導入されるか、前記オピオイド作動薬、オピオイド拮抗薬、またはそれらの両方を含有する溶融押出多粒子剤の表面上にコーティングされる、請求項1に記載の経口投薬剤形。
【請求項26】
経口的に治療上有効な量のオピオイド作動薬と、オピオイド拮抗薬とを含有する経口投薬剤形であって、
前記オピオイド拮抗薬は、即時放出形態であり、
前記投薬剤形は、前記作動薬と拮抗薬の組合せを経口的に投与したときには鎮痛薬として有効であるが、(i)前記治療有効量と同量でまたはそれより多量に投与したときには身体依存性ヒト被験者に嫌悪作用を生じさせ、かつ、(ii)前記オピオイド拮抗薬なしでヒト患者に投与したときには、同じ治療量のオピオイド鎮痛薬と比較して前記オピオイド作動薬の鎮痛効果を維持するか減少させる、組合せ製剤を提供するオピオイド拮抗薬対オピオイド作動薬の比を有し、
前記オピオイド拮抗薬は、ナルトレキソン、ナロキソン、ナルメフェン、シクラゾシン、レバロルファン、それらの薬学的に許容しうる塩、及びそれらの混合物からなる群より選択され、
前記オピオイド拮抗薬対前記オピオイド作動薬の比は、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩とに基づく0.03:1〜0.27:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、オキシコドンまたはその薬学的に許容しうる塩とに基づく0.037:1〜0.296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、コデインまたはその薬学的に許容しうる塩とに基づく0.005:1〜0.044:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロモルホンまたはその薬学的に許容しうる塩とに基づく0.148:1〜1.185:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、レボルファノールまたはその薬学的に許容しうる塩とに基づく0.278:1〜2.222:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メペリジンまたはその薬学的に許容しうる塩とに基づく0.0037:1〜0.0296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メタドンまたはその薬学的に許容しうる塩とに基づく0.056:1〜0.444:1の重量比、および
ナルトレキソンまたはその薬学的に許容しうる塩と、モルヒネまたはその薬学的に許容しうる塩とに基づく0.018:1〜0.148:1の重量比、からなる群より選択される、前記経口投薬剤形。
【請求項27】
前記即時放出形態のオピオイド拮抗薬が、ゼラチンカプセル内に別のペレットとして導入されるか、前記オピオイド作動薬、オピオイド拮抗薬、またはそれらの両方を含有する溶融押出多粒子剤の表面上にコーティングされる、請求項26に記載の経口投薬剤形。
【請求項1】
経口的に治療上有効な量のオピオイド作動薬と、オピオイド拮抗薬とを含有する経口投薬剤形であって、
前記オピオイド拮抗薬は、即時放出形態であり、
前記投薬剤形は、前記作動薬と拮抗薬の組合せを経口的に投与したときには鎮痛薬として有効であるが、(i)前記治療有効量と同量でまたはそれより多量に投与したときには身体依存性ヒト被験者に嫌悪作用を生じさせ、かつ、(ii)前記オピオイド拮抗薬なしでヒト患者に投与したときには、鎮痛効果を維持するが、同じ治療量のオピオイド鎮痛薬と比較して前記オピオイド作動薬の鎮痛効果を増加させない、組合せ製剤を提供するオピオイド拮抗薬対オピオイド作動薬の比を有し、
前記オピオイド拮抗薬は、ナルトレキソン、ナロキソン、ナルメフェン、シクラゾシン、レバロルファン、それらの薬学的に許容しうる塩、及びそれらの混合物からなる群より選択され、
前記オピオイド拮抗薬対前記オピオイド作動薬の比は、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩とに基づく0.03:1〜0.27:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、オキシコドンまたはその薬学的に許容しうる塩とに基づく0.037:1〜0.296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、コデインまたはその薬学的に許容しうる塩とに基づく0.005:1〜0.044:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロモルホンまたはその薬学的に許容しうる塩とに基づく0.148:1〜1.185:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、レボルファノールまたはその薬学的に許容しうる塩とに基づく0.278:1〜2.222:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メペリジンまたはその薬学的に許容しうる塩とに基づく0.0037:1〜0.0296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メタドンまたはその薬学的に許容しうる塩とに基づく0.056:1〜0.444:1の重量比、および
ナルトレキソンまたはその薬学的に許容しうる塩と、モルヒネまたはその薬学的に許容しうる塩とに基づく0.018:1〜0.148:1の重量比、からなる群より選択される、前記経口投薬剤形。
【請求項2】
前記経口投薬剤形に含まれる拮抗薬の量が、前記治療有効量の2〜3倍のオピオイドを摂取する身体依存性中毒者に嫌悪の経験を生じさせる、請求項1に記載の経口投薬剤形。
【請求項3】
前記オピオイド作動薬がヒドロコドンまたはその薬学的に許容しうる塩であり、前記オピオイド拮抗薬がナルトレキソンまたはその薬学的に許容しうる塩である、請求項1に記載の経口投薬剤形。
【請求項4】
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩との比が、0.03:1〜0.27:1である、請求項3に記載の経口投薬剤形。
【請求項5】
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩との比が、0.05:1〜0.20:1である、請求項3に記載の経口投薬剤形。
【請求項6】
前記オピオイド作動薬がモルヒネ、ヒドロモルフォン、ヒドロコドン、オキシコドン、コデイン、レボルファノール、メペリジン、メタドン、これらの薬学的に許容しうる塩およびこれらの混合物からなる群より選択される、請求項1に記載の経口投薬剤形。
【請求項7】
NSAID、COX-2阻害剤、アセトアミノフェン、アスピリン、NMDA受容体拮抗薬、NMDA受容体活性化の主要な細胞内結果をブロックする薬物、鎮咳薬、去痰薬、うっ血除去薬、抗ヒスタミン薬、およびこれらの混合物からなる群より選択される非オピオイド薬物をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項8】
1種以上の製薬上許容される不活性賦形剤をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項9】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩である、請求項6に記載の経口投薬剤形。
【請求項10】
前記オピオイド作動薬に対して持続放出性を与える持続放出性担体をさらに含有する、請求項1に記載の経口投薬剤形。
【請求項11】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、オキシコドンまたはその薬学的に許容しうる塩であり、それらの比が0.037:1〜0.296:1である、請求項1に記載の経口投薬剤形。
【請求項12】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、コデインまたはその薬学的に許容しうる塩であり、それらの比が0.005:1〜0.044:1である、請求項1に記載の経口投薬剤形。
【請求項13】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、ヒドロモルホンまたはその薬学的に許容しうる塩であり、それらの比が0.148:1〜1.185:1である、請求項1に記載の経口投薬剤形。
【請求項14】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、レボルファノールまたはその薬学的に許容しうる塩であり、それらの比が0.278:1〜2.222:1である、請求項1に記載の経口投薬剤形。
【請求項15】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メペリジンまたはその薬学的に許容しうる塩であり、それらの比が0.0037:1〜0.0296:1である、請求項1に記載の経口投薬剤形。
【請求項16】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メタドンまたはその薬学的に許容しうる塩であり、それらの比が0.056:1〜0.444:1である、請求項1に記載の経口投薬剤形。
【請求項17】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、モルヒネまたはその薬学的に許容しうる塩であり、それらの比が0.018:1〜0.148:1である、請求項1に記載の経口投薬剤形。
【請求項18】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、オキシコドンまたはその薬学的に許容しうる塩であり、それらの比が0.056:1〜0.222:1である、請求項1に記載の経口投薬剤形。
【請求項19】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、コデインまたはその薬学的に許容しうる塩であり、それらの比が0.0083:1〜0.033:1である、請求項1に記載の経口投薬剤形。
【請求項20】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、ヒドロモルホンまたはその薬学的に許容しうる塩であり、それらの比が0.222:1〜0.889:1である、請求項1に記載の経口投薬剤形。
【請求項21】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、レボルファノールまたはその薬学的に許容しうる塩であり、それらの比が0.417:1〜1.667:1である、請求項1に記載の経口投薬剤形。
【請求項22】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メペリジンまたはその薬学的に許容しうる塩であり、それらの比が0.0056:1〜0.022:1である、請求項1に記載の経口投薬剤形。
【請求項23】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、メタドンまたはその薬学的に許容しうる塩であり、それらの比が0.083:1〜0.333:1である、請求項1に記載の経口投薬剤形。
【請求項24】
前記オピオイド拮抗薬が、ナルトレキソンまたはその薬学的に許容しうる塩であり、前記オピオイド作動薬が、モルヒネまたはその薬学的に許容しうる塩であり、それらの比が0.028:1〜0.111:1である、請求項1に記載の経口投薬剤形。
【請求項25】
前記即時放出形態のオピオイド拮抗薬が、ゼラチンカプセル内に別のペレットとして導入されるか、前記オピオイド作動薬、オピオイド拮抗薬、またはそれらの両方を含有する溶融押出多粒子剤の表面上にコーティングされる、請求項1に記載の経口投薬剤形。
【請求項26】
経口的に治療上有効な量のオピオイド作動薬と、オピオイド拮抗薬とを含有する経口投薬剤形であって、
前記オピオイド拮抗薬は、即時放出形態であり、
前記投薬剤形は、前記作動薬と拮抗薬の組合せを経口的に投与したときには鎮痛薬として有効であるが、(i)前記治療有効量と同量でまたはそれより多量に投与したときには身体依存性ヒト被験者に嫌悪作用を生じさせ、かつ、(ii)前記オピオイド拮抗薬なしでヒト患者に投与したときには、同じ治療量のオピオイド鎮痛薬と比較して前記オピオイド作動薬の鎮痛効果を維持するか減少させる、組合せ製剤を提供するオピオイド拮抗薬対オピオイド作動薬の比を有し、
前記オピオイド拮抗薬は、ナルトレキソン、ナロキソン、ナルメフェン、シクラゾシン、レバロルファン、それらの薬学的に許容しうる塩、及びそれらの混合物からなる群より選択され、
前記オピオイド拮抗薬対前記オピオイド作動薬の比は、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロコドンまたはその薬学的に許容しうる塩とに基づく0.03:1〜0.27:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、オキシコドンまたはその薬学的に許容しうる塩とに基づく0.037:1〜0.296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、コデインまたはその薬学的に許容しうる塩とに基づく0.005:1〜0.044:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、ヒドロモルホンまたはその薬学的に許容しうる塩とに基づく0.148:1〜1.185:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、レボルファノールまたはその薬学的に許容しうる塩とに基づく0.278:1〜2.222:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メペリジンまたはその薬学的に許容しうる塩とに基づく0.0037:1〜0.0296:1の重量比、
ナルトレキソンまたはその薬学的に許容しうる塩と、メタドンまたはその薬学的に許容しうる塩とに基づく0.056:1〜0.444:1の重量比、および
ナルトレキソンまたはその薬学的に許容しうる塩と、モルヒネまたはその薬学的に許容しうる塩とに基づく0.018:1〜0.148:1の重量比、からなる群より選択される、前記経口投薬剤形。
【請求項27】
前記即時放出形態のオピオイド拮抗薬が、ゼラチンカプセル内に別のペレットとして導入されるか、前記オピオイド作動薬、オピオイド拮抗薬、またはそれらの両方を含有する溶融押出多粒子剤の表面上にコーティングされる、請求項26に記載の経口投薬剤形。
【図1】

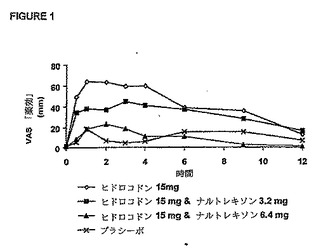
【図2】
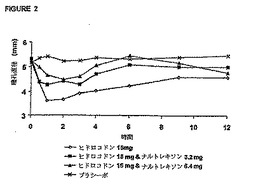
【図3】
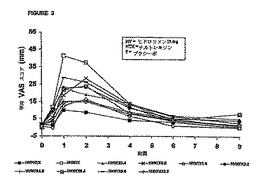
【図4】
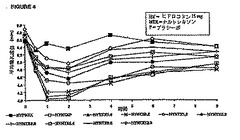
【図5】
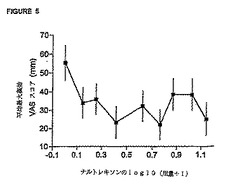
【図6】
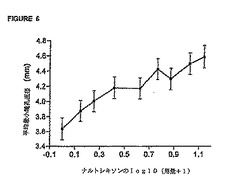
【図7】
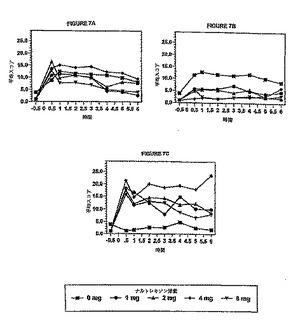
【図8】
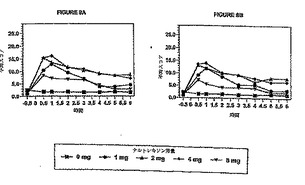
【図9】
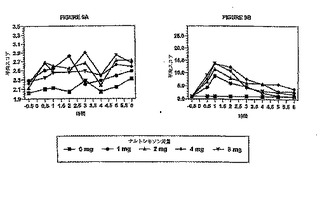
【図10】
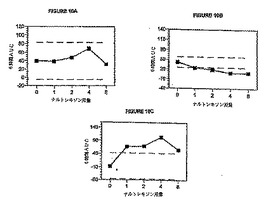
【図11】
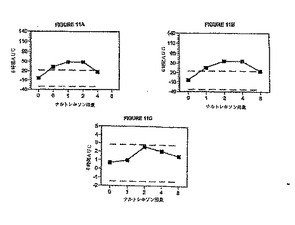
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2010−280681(P2010−280681A)
【公開日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願番号】特願2010−166429(P2010−166429)
【出願日】平成22年7月23日(2010.7.23)
【分割の表示】特願2005−97816(P2005−97816)の分割
【原出願日】平成10年12月22日(1998.12.22)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.POLAROID
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公開日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願日】平成22年7月23日(2010.7.23)
【分割の表示】特願2005−97816(P2005−97816)の分割
【原出願日】平成10年12月22日(1998.12.22)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.POLAROID
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]