
オピオイド拮抗薬とmTOR阻害薬を用いた治療
本発明の実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与することによって、細胞の増殖及び遊走を特徴とする障害又は疾患を治療する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
この出願は、2008年3月21日提出の全開示が参照により本明細書に援用される米国仮出願第61/038,577号の優先権を35 U.S.C.§119(e)の下で主張する。
(連邦政府による資金提供を受けた研究開発の記載)
適用なし。
【背景技術】
【0002】
細胞の成長及び増殖は、全ての生体における正常な進行中プロセスであり、規則的な細胞周期を維持するため微妙に均衡が保たれている多くの因子及びシグナルに関与する。哺乳動物細胞が成長及び分割するか否かは、例えば細胞が成長できる空間の利用能、及び現在置かれている環境内における特有の刺激因子や阻害因子の分泌などの種々のフィードバック制御機構によって決まる。
mTORは、ホスファチジルイノシトール3−キナーゼ(PI3K)−関連キナーゼ(PIKK)ファミリーの大型ポリペプチドセリン/スレオニンキナーゼである。mTORは、PI3K径路から下流にあり、細胞の成長及び増殖を調節するための種々の細胞シグナル伝達事象における中間体として機能する。mTOR活性はセリン/スレオニンキナーゼAktによって調節され、最近の証拠は、これらのキナーゼが複雑なフィードバック阻害径路を介して相互作用することを示している。mTORは、G1期からS期を通して細胞周期の進行に必要な主要タンパク質の翻訳を制御することによって、細胞の複製を調節する。すなわち、mTORは、mRNA、主に4E−PB1、P7056K及びeEFZの翻訳に関与するいくつかのタンパク質のリン酸化状態の調節によって特有のmRNAの翻訳を制御する。
mTOR径路は、そのPI3K及びAkt成分と共に、栄養素、ホルモン及び成長因子、例えばVEGFに応答する細胞の増殖の重要なレギュレーターである。成長因子は、同族細胞表面受容体に結合することによって、PI3Kシグナル伝達を活性化し、それによってAktを介したシグナル伝達カスケードを惹起し、mTORの活性化をもたらすことができる。最近の研究は、mTOR阻害薬が、成長因子媒介シグナル伝達と成長因子の翻訳を両方とも阻害することによって、抗増殖作用及び血管新生抑制作用を有することを実証した。
【0003】
癌細胞では、mTORの上流のPI3K径路内の複数の調節不全機構がmTOR活性を上昇させ、結果として腫瘍の成長を増やすとして実証されている。このように、mTORシグナル伝達経路の調節不全が癌の進行に関係しており、現在、癌の治療薬としてmTORの阻害薬が調査されている。mTOR阻害薬は強力な免疫抑制薬でもある。1つの該薬剤であるシロリムスは、現在、臓器拒絶反応の予防用に使われている。現在市販されているか又は開発中である他のmTOR阻害薬として、テムシロリムス(ToriselTM;Wyeth)、RAD001(エベロリムス;Novartis)、MK−8669(デフォロリムス(deforolimus);Merck & Ariad pharma)、FK506(タクロリムス;Astellas)、TOP216(Toptarget A/S)、OSI−027(OSI Pharma)、TAFA93(Isotechnika)、及びnab−ラパマイシン(APP Pharma)が挙げられる。これらのmTOR阻害薬の多くは、ラパマイシン又はラパマイシン誘発体である。
臓器移植拒絶反応予防薬及び抗癌薬としての開発に加え、mTOR阻害薬は、関節リウマチ、自己免疫障害、乾癬、多発性硬化症、パーキンソン病、脳卒中及び末梢神経障害の治療のためにも開発されている。例えば、mTOR阻害薬ラパマイシンは、例えば実験的アレルギー性脳脊髄炎、インスリン依存性糖尿病、マウスループス及びアジュバント関節炎などの自己免疫のいくつかの動物モデルにおいて効力が示され、かつリウマチ様疾患の治療に可能性があると提案されている。リウマチ様疾患の治療については、リウマチ様疾患又はグレーブス病の個体由来の抗体はmTOR径路を介して線維芽細胞を活性化するが、ラパマイシンはこれを阻害することができ、mTORの遮断がこれらの疾患における炎症を制限するのに価値があること示唆している。ラパマイシンは、ヒト線維芽細胞内のコラーゲンmRNAレベルを下げることも分かっており、mTORがI型コラーゲンの合成をポジティブに調節することを示唆している。従って、mTORの遮断は、線維性病変が正常な組織構造を破壊し、臓器不全に寄与する強皮症のような線維性疾患にも有益でありうる。HIV−1がCD4T細胞及びマクロファージに侵入するために利用するCCR5ケモカイン受容体のタンパク質発現もラパマイシンによって阻害され、免疫細胞内におけるCCR5媒介ウイルス侵入の遮断の臨床用途を示唆している。最近、ラパマイシン治療が、筋肉及び皮膚に影響を及ぼす自己免疫状態である皮膚筋炎の患者において臨床疾患を有意に軽減することが分かった。mTOR阻害薬は、培養及び動物モデルにおいて強力な神経保護特性及び神経再生特性をも有する。
しかしながら、臨床研究においていくつかの副作用が観察されている。副作用としては、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。これらの副作用の少なくとも一部は用量依存性であると思われる。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の実施形態の原理を具体化する方法及び医薬の併用は、mTOR阻害薬を、一般的にμ−オピオイド受容体拮抗薬と記述される分類の化合物と共投与することを包含する。発明者らは、メチルナルトレキソン等のμ−オピオイド受容体拮抗薬をmTOR阻害薬と相乗様式で共投与して、mTOR阻害薬の治療的に有効な投与量を低減できることを見出した。上述したように、mTOR阻害薬に伴う副作用の少なくとも一部は用量依存性であると思われるので、μ−オピオイド受容体拮抗薬の共投与は、低用量でこれらのmTOR阻害薬を使用できるようにし、同時に副作用の発生及び/又は重症度を低減しうる。発明者らは、メチルナルトレキソンが、mTORの活性化の上流事象であるキナーゼAktの活性化を阻害することを実証した。Akt及びmTORは、複雑な制御フィードバックループに関与しており、本発明の実施形態のよる両Akt及びmTORの同時ターゲティングは相乗効果を有する。
【課題を解決するための手段】
【0005】
mTOR阻害薬と組み合わせたμ−オピオイド受容体拮抗薬の使用は、これらの阻害薬の抗癌及び免疫抑制効力を大いに高め、それらの低用量での使用を可能にし、ひいては副作用の発生/重症度を低減しうる。さらに、メチルナルトレキソン等のμ−オピオイド受容体拮抗薬をこれらのmTOR阻害薬の効力を高めるために使用すると、結果として、治療作用に必要な用量が減少するので、mTOR阻害薬に伴う治療の高いコストをも大いに下げるであろう。さらに、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与することによって、mTOR阻害薬の治療指数又は有用性を改善する方法を企図する。
本発明の実施形態の原理を具体化する方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬の組合せを用いて、細胞の増殖及び遊走、特に内皮細胞の増殖及び遊走を減弱、例えば阻害又は低減することを含む。前記μ−オピオイド受容体拮抗薬としては、限定するものではないが、末梢限定拮抗薬が挙げられる。本発明の一態様により、細胞の不必要な遊走及び/又は増殖を特徴とする障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む治療方法を提供する。この治療は、細胞の遊走と増殖の一方又は両方を阻害し、細胞は適宜、内皮細胞であってよく、血管内皮細胞が特に興味深い。従って、別の実施形態では、血管内皮細胞の不必要な遊走又は増殖を特徴とする前記障害は、不必要な血管新生である。言い換えれば、不必要な血管新生を治療する方法を企図する。
【0006】
別の態様により、腫瘍細胞若しくは癌細胞の遊走及び/又は増殖を減弱する方法であって、前記細胞を抗遊走又は抗増殖量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。異常な細胞増殖を減弱する工程では、治療的に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を哺乳動物に投与することによって、哺乳動物内でmTOR/Aktシグナル伝達経路の活性化を阻害する。従って、本発明の実施形態により、細胞、例えば内皮細胞においてシグナル伝達するmTOR/Akt径路を阻害する方法であって、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。本発明の別の実施形態はさらに、対象の癌組織を治療する方法であって、前記癌組織内におけるmTOR/Akt径路媒介作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、mTOR/Akt径路誘発細胞増殖及び遊走を相乗的に阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法を包含する。
さらなる態様では、細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明のさらに別の実施形態は、癌の治療方法、例えば、治療が必要な対象の腫瘍の成長を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を包含する。
【0007】
本発明のさらなる実施形態は、哺乳動物内における成長因子受容体を発現する細胞の異常な増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与することによって治療する方法を提供する。特定の実施形態では、前記成長因子受容体が、血管内皮成長因子受容体(VEGF−R)、上皮成長因子受容体(EGF−R)又はインスリン様成長因子受容体(IGF−R)である。従って、本発明の他の実施形態により、内皮細胞内の成長因子シグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記成長因子がVEGF、EGF又はIGFである。
本発明の実施形態に従い、mTOR阻害薬とμ−オピオイド拮抗薬の相乗的併用剤を投与することによる自己免疫疾患、乾癬、神経変性疾患、免疫細胞内へのCCRS媒介ウイルス侵入、並びに吐き気及び嘔吐の治療方法をも企図する。
本発明の実施形態により、mTOR阻害薬及びμ−オピオイド受容体拮抗薬のみならず、共投与についての使用説明書を伴う医薬併用剤及びパッケージをも提供する。
【0008】
本発明の実施形態の方法で使うmTOR阻害薬は、一般的にmTORによるp70S6キナーゼのセリン389のリン酸化を阻害することによってG1期からS期への細胞周期の進行を遮断することによって細胞の複製を阻害する化合物を含む。既知のmTOR阻害薬としては、ラパマイシン及びラパマイシン誘発体が挙げられる。mTOR活性のいくつかの他の小分子阻害薬も同定されている。本発明の実施形態の併用療法では、1種より多くのmTOR阻害薬の使用も企図する。
一部の実施形態では、μ−オピオイド受容体拮抗薬が末梢性μ−オピオイド受容体拮抗薬である。末梢限定μ−オピオイド受容体拮抗薬は一般的にヘテロ環式アミン化合物であり、化合物のいくつかの異なる分類にも属する。例えば、1つの分類は、モルフィナンの四級誘発体、特にノルオキシモルフォン(noroxymorphone)の四級誘発体である。一実施形態では、ノルオキシモルフォンの四級誘発体は適宜、例えば、N−メチルナルトレキソン(又は単にメチルナルトレキソン)、N−メチルナロキソン、N−メチルナロルフィン、N−ジアリルノルモルフィン、N−アリルレバロルファン、又はN−メチルナルメフェンである。別分類の末梢限定拮抗薬は、N−置換ピペリジンである。一実施形態では、N−ピペリジンがピペリジン−N−アルキルカルボナート、例えばアルビモパンである。本発明の実施形態で価値がありうる他分類の化合物は、ベンゾモルファンの四級誘発体、ノルモルファニン(normorphanin)の四級誘発体並びにモルファニンの三級誘発体、ベンゾモルファン及びノルモルファニンのポリマー複合体である。
本発明の実施形態は、併用療法において、1種より多くのμ−オピオイド受容体拮抗薬の投与をも包含する。拮抗薬の組合せとしては、μ−拮抗薬の組合せ並びにμ−及びκ−拮抗薬の組合せ、例えば、メチルナルトレキソンとアルビモパンの組合せ、又はナロキソンとメチルナルトレキソンの組合せが挙げられる。
本発明の実施形態の方法で使用可能な他のμ−オピオイド受容体拮抗薬として、モルフィナンの三級誘発体、特に、ノルオキシモルフォンの三級誘発体、例えばナロキソンが挙げられる。
添付図面と関連して本明細書で提示する詳細な説明を参照することによって、本発明の実施形態をより良く理解かつ認識できるであろう。
【図面の簡単な説明】
【0009】
【図1】ナルトレキソンとメチルナルトレキソンの化学構造、及びナルトレキソンからメチルナルトレキソンへの変換反応を示す。
【図2】ベバシズマブ、5−FU、メチルナルトレキソン、ベバシズマブとメチルナルトレキソンの組合せ及び5−FUとメチルナルトレキソンの組合せの存在下、ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いたセリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)を実証する免疫ブロットである。
【図3】セリン473(A)及びスレオニン308(B)におけるAktのVEGF誘発リン酸化のメチルナルトレキソンによる阻害の用量関連効果を示すグラフである。
【図4】VEGF誘発内皮細胞増殖の阻害に及ぼすラパマイシンの用量関連効果を示すグラフである。
【図5】VEGF誘発内皮細胞遊走の阻害に及ぼすラパマイシンの用量関連効果を示すグラフである。
【図6】メチルナルトレキソンとラパマイシンの組合せを用いたVEGF誘発内皮細胞増殖の相乗的阻害を示すグラフである。
【図7】メチルナルトレキソンとラパマイシンの組合せを用いたVEGF誘発内皮細胞遊走の相乗的阻害を示すグラフである。
【図8】ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いたセリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)に及ぼすメチルナルトレキソンとテムシロリムスの効果を実証する免疫ブロットである。
【図9】ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いた、(A) mTOR複合体1及びmTOR複合体2のVEGF誘発形成に及ぼすメチルナルトレキソン及びテムシロリムスの効果、並びに(B)セリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)に及ぼすPI3キナーゼ阻害、Src枯渇及びRictor枯渇の効果を実証する免疫ブロットである。
【図10】(A)メチルナルトレキソンとテムシロリムスの組合せを用いたVEGF誘発内皮細胞増殖の相乗的阻害及び(B)メチルナルトレキソンとテムシロリムスの組合せを用いたVEGF誘発内皮細胞遊走の相乗的阻害を示すグラスを示す。
【図11】メチルナルトレキソンとテムシロリムスの組合せ用いた内皮細胞の(A)増殖及び(B)遊走の相乗的阻害がチロシンホスファターゼ活性によって制御されることを実証するグラフを示す。
【図12】メチルナルトレキソンとmTOR阻害薬の相乗的活性の分子基盤の略図である。
【発明を実施するための形態】
【0010】
本発明の実施形態で明白な原理により、mTOR阻害薬とμ−オピオイド受容体拮抗薬の組合せを利用する医薬併用剤及び方法を提供する。本発明の実施形態の方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬を共投与することによって、細胞の不必要及び望ましくない増殖及び遊走、特に内皮細胞の不必要及び望ましくない増殖及び/又は遊走を特徴とする障害を治療する工程を含む。以下の実施例で説明するように、mTOR阻害薬とμ−オピオイド受容体拮抗薬(例えばメチルナルトレキソン(MNTX))の組合せは、細胞の不必要な増殖及び遊走、例えば、内皮細胞のVEGF誘発増殖及び遊走の低減において予想外の相乗作用をもたらす。
本発明の少なくとも1つの実施形態を説明する前に、本発明は、下記説明で述べ、また実施例によって例示する詳細に本発明を適用することに限定されないことを理解すべきである。このような説明及び実施例は、添付の特許請求の範囲に記載の本発明の範囲を制限する意図ではない。本発明は、他の実施形態が可能であり、或いは種々のやり方で実施又は実行することができる。以下の詳細な説明及び実施例は、好適な薬物としてラパマイシン、テムシロリムス及びメチルナルトレキソンを利用する実施形態を参照することにより本発明を記述しているが、他のmTOR阻害薬及びμ−オピオイド受容体拮抗薬も本発明の原理に従って使うのに適していることを理解すべきである。
さらに、いずれの特許又は特許文献をも含め、本明細書で引用したいずれの参考文献も先行技術を構成することを認めるものではない。特に、当然のことながら、特に断らない限り、本明細書におけるいずれの文献の参照も、これらの文献のいずれかが米国又は他のいずれかの国の当該技術で共通の一般知識の一部を形成するという承認を構成しない。参考文献のいずれの議論もそれらの著者が主張することを述べており、出願人は、本明細書で引用したいずれの文献の正確さ及び適切性をも検証する権利を保留する。
【0011】
この開示全体を通じて、この発明の種々の態様を範囲形式で提示することがある。範囲形式の説明は、単に便利さ及び簡潔さのためであることを理解すべきであり、かつ本発明の範囲に対する確固たる限定とみなすべきでない。従って、当業者には分かるように、ありとあらゆる目的のため、特に書面による明細を提供するという観点において、本明細書で開示する全ての範囲は、そのありとあらゆる可能な部分的範囲及び部分的範囲の組合せ、並びに当該範囲内の全ての整数及び分数の値をも包含する。例として、20%〜40%という範囲は20%〜32.5%及び32.5%〜40%、20%〜27.5%及び27.5%〜40%などの範囲に分解することができ、その全てがこの明細書で明確に列挙されているものと解釈すべきである。リストアップしたいずれの範囲も、少なくとも2等分、3等分、4等分、5等分、10等分などに分解される同一範囲を十分に記述し、かつ有効にするものと容易に認識することができる。非限定例として、本明細書で論じる各範囲は、容易に下部3分の1、中部3分の1及び上部3分の1等に分解することができる。
また、当業者には分かるように、「まで」、「少なくとも」、「より大きい」、「未満」、「より多い」等の全ての言葉は、列挙した数を包含し、上述したように引き続き部分的範囲に分解できる範囲を指す。同様に、本明細書で開示する全ての比率も、より広い比率に含まれる全ての部分的範囲を包含する。これらは、具体的に意図しているものの例にすぎない。さらに、第1の表示数と第2の表示数の「間の範囲」という表現と、「第1の表示数〜第2の表示数の範囲」という表現は、本明細書では相互交換可能に使用される。
さらに、本明細書において「含む(comprising)」、「含む(including)」、「有する(having)」、及びその変形の使用は、その後に列挙する項目及びその等価項目並びに追加項目、例えば他の工程及び/又は成分を包含することを意味する。これらの用語は、「〜から成る」及び「本質的に〜から成る」という用語を包含する。「本質的に〜から成る」の使用は、該組成物又は方法が追加の成分及び/又は工程を含みうることを意味するが、その追加の成分及び/又は工程が、請求した組成物又は方法の基本的及び新規な特徴を実質的に変えない場合に限る。
【0012】
特に定義しない限り、全ての科学用語及び技術用語は、本明細書では通常の用法に従って使用され、本発明が属する技術の当業者が一般的に解釈するのと同じ意味を有する。しかし、本明細書で使用する場合、以下の定義は、本発明を理解するのに熟練実務家を助けるのに役立つであろう。
「対象」は、哺乳動物、例えばヒト、マウス、イヌ、ネコ、ラットを表す。
「アルキル」は、飽和しており、かつ鎖内に1〜約10個の炭素原子、並びにその中に全ての組合せ及び副組合せの鎖を有する直鎖、分岐鎖、又は環式であってよい、一価の脂肪族炭化水素基を表す。典型的アルキル基として、限定するものではないが、メチル、エチル、n−プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、シクロプロピル、シクロブチル、シクロペンチル、及びシクロヘキシルが挙げられる。
「低級アルキル」は、1〜約6個の炭素原子を有するアルキル基を表す。
「アルケニル」は、鎖内に少なくとも1個の炭素−炭素二重結合を含み、かつ鎖内に2〜約10個の炭素原子、並びにその中に全ての組合せ及び副組合せの鎖を有する一価の脂肪族炭化水素基を表す。典型的アルケニル基として、限定するものではないが、ビニル、プロペニル、ブチニル、ペンテニル、ヘキセニル、及びヘプトニルが挙げられる。
「アルキニル」は、少なくとも1個の炭素−炭素三重結合を含み、かつ鎖内に2〜約10個の炭素原子、並びにその中に組合せ及び副組合せの鎖を有する一価の脂肪族炭化水素基を表す。典型的アルキニル基として、限定するものではないが、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、及びヘプチニルが挙げられる。
「アルキレン」は、その中に1〜約6個の炭素原子、並びに全ての組合せ及び副組合せの鎖を有する二価の脂肪族炭化水素基を表す。アルキレン基は、直鎖、分岐鎖、又は環式であってよい。任意に、アルキレン基に沿って、1個以上の酸素、イオウ、又は任意に置換されていてもよい窒素原子(窒素の置換基は前述したアルキル基である)が挿入されていてもよい。
「アルケニレン」は、少なくとも1個の炭素−炭素二重結合を含み、直鎖、分岐鎖、又は環式であってよい二価のアルキレン基を表す。典型的アルケニレン基として、限定するものではないが、エテニレン(CH=CH)及びプロペニレン(CH=CHCH2)が挙げられる。
【0013】
「シクロアルキル」は、その中に約3〜約10個の炭素、並びに全ての組合せ及び副組合せの環を有する飽和単環式又は二環式炭化水素環を表す。シクロアルキル基は任意に1つ以上のシクロアルキル基置換基で置換されていてもよい。典型的シクロアルキル基として、限定するものではないが、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、及びシクロヘプチルが挙げられる。
「アシル」は、アルキル−CO基(アルキルは前述したとおり)を意味する。典型的アシル基として、限定するものではないが、アセチル、プロパノイル、2−メチルプロパノイル、ブタノイル、及びパルミトイルが挙げられる。
「アリール」は、その中に約6〜約10個の炭素、並びに全ての組合せ及び副組合せの環を有する芳香族炭素環式基を表す。アリール基は、任意に1又は2つ以上のアリール基置換基で置換されていてもよい。典型的アリール基として、限定するものではないが、フェニル及びナフチルが挙げられる。
「アリール置換アルキル」は、末端炭素のところで、任意に置換されていてもよいアリール基、好ましくは任意に置換されていてもよいフェニル環によって置換されている直鎖アルキル基、好ましくは低級アルキル基を表す。典型的アリール置換アルキル基として、例えば、フェニルメチル、フェニルエチル、及び3(4−メチルフェニル)プロピルが挙げられる。
「ヘテロ環式」は、その中に約4〜約10個の環員、並びに全ての組合せ及び副組合せの環を含み、1個以上の環員が炭素以外の元素、例えば、窒素、酸素、又はイオウである、単環式又は多環式環系炭素環式基を表す。ヘテロ環式基は、芳香族又は非芳香族であってよい。典型的ヘテロ環式基として、例えば、ピロール及びピペリジン基が挙げられる。
「ハロ」は、フルオロ、クロロ、ブロモ、又はヨードを表す。
【0014】
「共投与」又は「共投与する」とは、2種以上の薬剤をいずれかの投与径路で患者又は対象に投与する併用療法を表すことを意味する。薬剤の共投与は、併用療法又は併用治療と呼ばれることもある。薬剤は、同一の投与製剤又は別々の製剤中にあってよい。活性薬が別々の投与製剤中にある1種より多くの活性薬による併用治療では、活性薬を同時に投与することができ、或いは活性薬をそれぞれ別々に時間差で投与することができる。併用治療の薬剤は、それら両薬剤が体内で有効濃度を達成させるのに十分な様式で与えられる限り、同時又は逐次投与してよい(例えば、一方の薬剤を他方の投与直後に投与するか、或いは偶発的に、例えば、ある時に一方の薬剤を与えて、その後、例えば1週間以内に他方の薬剤を与えてよい)。薬剤を異なる経路で投与してもよく、例えば、一方の薬剤を静脈内投与し、第2の薬剤を筋肉内、静脈内、又は経口投与してよい。
μ−オピオイド受容体拮抗薬に関する用語「末梢性」、又は「末梢限定」又は「末梢作用性」は、中枢神経系の外側にある生理学的な系及び要素に主として作用するμ−オピオイド受容体拮抗薬を指定する。換言すれば、それらは低減した中枢神経系(CNS)活性を示すか又は該活性が実質的にない。例えば、それらはオピオイドの中枢作用を阻害するのに有効な量で血液脳関門を容易には横断できない。すなわち、それらは末梢投与したとき、オピオイドの鎮痛作用を有効には阻害せず、すなわち、それらはオピオイドの鎮痛作用を低減しない。本発明の実施形態で利用する末梢性μ−オピオイド受容体拮抗薬化合物は、CNS内でその薬理活性の約5〜15%未満を示し、約0%(すなわち、無い)のCNS活性が最も適している。末梢性μ−オピオイド受容体拮抗薬の非中枢作用特性は、分子又は化学種の電荷、極性、及び/又は大きさに関連することが多い。例えば、本明細書に記載の末梢作用性四級アミンμ−オピオイド受容体拮抗薬は正の電荷をもつが、中枢作用性三級アミンμ−オピオイド受容体拮抗薬は中性分子である。
【0015】
本明細書で使用する場合、用語「mTOR阻害薬」は、mTORの活性又は発現を調節することによって、G1期〜S期の細胞周期の進行を遮断することで細胞の複製を阻害する化合物若しくはリガンド、又はその医薬的に許容しうる塩を意味する。この用語は、mTOR活性を阻害する、中性三環式化合物ラパマイシン(シロリムス)及び他のラパマイシン化合物(例えば、ラパマイシン誘発体、ラパマイシン類似体など)、及び他のマクロライド化合物並びに他の構造的に異なる小分子、例えば縮合二環式化合物を包含する。これらには、ラパマイシンに構造的に類似する化合物、例えば、治療利益を高めるために修飾された同様の大環状構造を有する化合物が含まれる。ラパマイシン誘発体などのmTOR阻害薬については本明細書で後述する。
本明細書で使用する用語「治療する」又は「治療」は、医学的又は生理学的状態のいずれの手段の制御、例えば、該状態の予防、ケア、軽減、該状態又は該状態の症状の減弱、緩和、低減、及び該状態の進行の阻害又は静止をも包含する。
本明細書で使用する場合、用語「副作用」は、薬物の目的又は所望の作用以外の作用を表すことを意味する。副作用は有益又は望ましくない、すなわち有害でありうる。本ケースでは、望ましくない作用はmTOR阻害薬の投与後に起こることが多い。このような副作用として、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。
【0016】
用語「溶媒和物」は、さらに、非共有結合性の分子内力で化学量論又は非化学量論量の溶媒が結合している、本明細書で提供する化合物又はその塩を表す。溶媒が水の場合は、溶媒和物は水和物である。
本明細書で使用する場合、用語「効力」は、癌を患っている対象の癌を治療する抗癌薬の性能又は能力を表す。効力を所定強度の特有の効果を生じさせるのに必要な薬物の用量として表すこともある。
特定の実施形態では、細胞の増殖及び遊走に関連する用語「不必要な」、例えば「不必要な増殖」又は「不必要な遊走」は、正常な細胞の代謝回転、代謝、成長又は繁殖から外れている、細胞の「異常又は病的又は調節不全的又は望ましくない又は不適切な」増殖、分割、成長又は遊走を表し、一般的に、正常に機能する細胞で典型的に起こるより急速又は有意に大きい程度で起こり、正常な機能を果たさない。不必要な増殖及び不必要な遊走は、本質的に過剰増殖性である障害で顕著であり、該障害としては、限定するものではないが、癌、例えばメラノーマ、肺癌、乳癌、膵臓癌、前立腺癌、結腸癌又は卵巣癌、乾癬、関節リウマチ、角質増殖(perkeratosis)による表皮溶解、レストラトシス(restratosis)、再狭窄、子宮内膜症及び異常な創傷治癒が挙げられる。
【0017】
光学活性化合物について述べる場合、プレフィックスR及びSを用いて、分子のキラル中心の回りの分子の絶対配置を示す。記号(+)及び(−)を用いて、化合物の旋光性、すなわち、光学活性化合物が偏光面を回転させる方向を表す。(−)プレフィックスは、化合物が左旋性、すなわち、化合物が偏光面を左又は反時計回りに回転させることを示す。(+)プレフィックスは、化合物が右旋性、すなわち、化合物が偏光面を右又は時計回りに回転させることを示す。しかし、旋光性の記号、(+)及び(−)は、分子の絶対配置、R及びSとは無関係である。
本発明の方法の以下の説明では、特に断らない限り、プロセス工程を室温及び大気圧で行なう。
一態様では、本発明の実施形態は、異常又は望ましくない細胞プロセス、例えば、細胞の不必要な遊走及び/又は増殖、特に内皮細胞の不必要な遊走及び/又は増殖を減弱する方法に関する。方法は、1種以上のmTOR阻害薬と1種以上のμ−オピオイド受容体拮抗薬を相乗的有効量で、例えば、患者若しくは対象の組織又は器官の細胞、特に患者の組織又は器官の内皮細胞に投与して、細胞の遊走及び/又は増殖、例えば、内皮細胞の遊走及び/又は増殖を阻害する工程を含む。
μ−オピオイド受容体拮抗薬は、オピオイド、内因性又は外因性、及び成長因子、例えばVEGF、PDGF、S1P等によって誘発される不必要な増殖及び遊走を阻害することが分かった。特に末梢性μ−オピオイド受容体拮抗薬は、同時係属米国特許出願第11/908,058号に開示されているように、内皮細胞のオピオイド及び成長因子誘発増殖及び遊走の阻害において実質的な効力を示した。末梢性μ−オピオイド受容体拮抗薬メチルナルトレキソン(MNTX)は、濃度依存様式で、オピオイド及び成長因子誘発の両方の増殖及び遊走を阻害する。さらに、μ−オピオイド受容体拮抗薬、特に末梢性μ−オピオイド受容体拮抗薬MNTXは、Aktの阻害によって、作用物質誘発内皮細胞(EC)増殖及び遊走を阻害することを発見した。作用物質は、オピオイド、外因性及び/又は内因性、血管新生因子(例えば、VEGF)、並びに他の増殖及び/又は遊走刺激因子(例えば、PDGF、S1P、S1P3受容体、RhoA等)でありうる。
μ−オピオイド受容体拮抗薬は、標的細胞を感染させるウイルスの能力を阻害することも分かった。特に、末梢性μ−オピオイド受容体拮抗薬は、参照により本明細書に援用される同時係属米国特許出願第10/163,482号に開示されているように、HIVのウイルス侵入及びこのウイルスの細胞表面受容体であるCCR5の発現の阻害において実質的な効力を示した。
【0018】
本発明の実施形態は、mTOR阻害薬と共投与するμ−オピオイド受容体拮抗薬が、癌細胞に及ぼす抗増殖作用を有意に高め、ひいては治療作用の増加をもたらすこと、例えば、末梢性μ−オピオイド受容体拮抗薬を特定腫瘍に投与するとmTOR阻害薬への腫瘍の反応を増強できることを実証する。以下の実施例で示すように、薬物を単独で使用する治療計画に比べて低濃度のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を利用する上記共投与併用剤では、有意に高い抗増殖作用及び抗遊走作用が得られる。例えば、本発明の実施形態に従うμ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、mTOR阻害薬の用量を減らすか又は効力若しくは効率又は両方を高めうる。さらに、本発明の実施形態に従うμ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、予防的価値をも有する。
mTOR阻害薬に伴う有害な副作用が、mTOR阻害薬のみをより多い用量で使用する場合に一般的に観察される当該副作用に比べて相当に低減した療法をもたらす可能性もある。これらの副作用としては、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。これらの副作用の少なくとも一部は用量依存性である。本発明の実施形態を利用すれば、より低い用量の可能性が達成される。
さらに、mTOR阻害薬療法に伴う高い費用が、mTOR阻害薬のみをより多い用量で使用する場合に一般的に観察される当該費用に比べて相当に低減した療法をもたらす可能性がある。その上、μ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、mTORのみを用いて一般的に観察される場合に比し、mTOR阻害薬の効率及び効力を相当高めることができる。
【0019】
本発明の実施形態で明白な原理を具体化する方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬(限定するものではないが、末梢限定拮抗薬であるものが挙げられる)を用いて、細胞の不必要な増殖及び遊走、特に内皮細胞の増殖及び遊走を減弱、例えば、阻害又は低減する工程を含む。本発明の一態様により、内皮細胞の不必要な遊走又は増殖を特徴とする障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む治療方法を提供する。本治療は、遊走と増殖の一方又は両方を阻害しうる。さらなる実施形態では、不必要な遊走又は増殖は、血管内皮細胞の遊走又は増殖であり、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を用いて治療される。別の実施形態では、血管内皮細胞の不必要な遊走又は増殖は、不必要な血管新生である。従って、不必要な血管新生を治療する方法を企図する。
【0020】
さらに別の態様により、腫瘍又は癌の細胞の遊走及び/又は増殖を減弱する方法であって、細胞を抗遊走又は抗増殖量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。細胞増殖の減弱においては、哺乳動物内でmTOR/Aktシグナル伝達経路の活性化亢進を示す異常な細胞増殖を治療方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。従って、内皮細胞内でシグナル伝達するmTOR/Akt径路を阻害する方法をも提供する。方法は、細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記対象に、前記癌組織内でmTOR/Akt径路媒介作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、mTOR/Akt径路誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。
【0021】
さらに別の実施形態では、哺乳動物内で成長因子受容体を発現する細胞の異常な増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。特定の実施形態では、前記成長因子受容体が血管内皮成長因子受容体(VEGF−R)、上皮成長因子受容体(EGF−R)又はインスリン様成長因子受容体(IGF−R)である。従って、内皮細胞内で成長因子シグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記成長因子がVEGF、EGF又はIGFである。方法は、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記癌組織内で成長因子誘発作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、成長因子誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。癌組織内におけるこれらの作用がVEGF誘発、EGF誘発又はIGF誘発作用である場合を特に企図する。
【0022】
なおさらなる実施形態では、哺乳動物内でホルモン受容体を発現する細胞の異常な細胞増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。特定の実施形態では、ホルモン受容体がエストロゲン受容体(ER)、プロゲステロン受容体(PR)又はアンドロゲン受容体(AR)である。従って、内皮細胞内でホルモンシグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記ホルモンがエストロゲン、プロゲステロン又はアンドロゲンである。方法は、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記癌組織内でホルモン誘発作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、ホルモン誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。癌組織内におけるこれらの作用がエストロゲン誘発、プロゲステロン誘発又はアンドロゲン誘発作用である場合を特に企図する。
【0023】
なおさらなる態様では、細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明のさらに別の実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、癌の治療が必要な対象の癌を治療する方法である。本発明のさらなる実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、腫瘍増殖の阻害が必要な対象の腫瘍の増殖を阻害する方法を提供する。
さらなる態様では、患者の自己免疫疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明の自己免疫疾患としては、限定するものではないが、アレルギー性脳脊髄炎、インスリン依存性糖尿病、ループス、関節リウマチ、多発性硬化症、皮膚筋炎、グレーブス病及びアジュバント関節炎が挙げられる。
本発明のさらに別の実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、患者の乾癬を治療する方法を提供する。特定の実施形態では、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を局所に適用する。
本発明のさらなる実施形態は、神経変性疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬の共投与を含む方法を提供する。特定の実施形態では、神経変性疾患がパーキンソン病又は多発性硬化症である。
本発明のなおさらなる実施形態は、免疫細胞へのCCR5媒介ウイルス侵入を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬の共投与を含む方法である。特に免疫細胞へのCCR5媒介HIV侵入の阻害を企図する。さらに、HIV/AIDSに苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、HIV/AIDSを治療する方法を提供する。
別の態様では、mTOR阻害薬を用いた癌の治療によって誘発される吐き気及び嘔吐は、末梢性μ−オピオイド受容体拮抗薬メチルナルトレキソン等のμ−オピオイド受容体拮抗薬を共投与することによって軽減されうる。
【0024】
上述したように、mTOR阻害薬には、mTORの活性又は発現の調節を通じてG1期〜S期の細胞周期の進行を遮断することによって、細胞の複製を阻害する、化合物若しくはリガンド、又はその医薬的に許容しうる塩が包含される。現在利用可能であるか又は開発中であるmTOR阻害薬としては、テムシロリムス(ToriselTM;Wyeth)、RAD001(エベロリムス;Novartis)、MK−8669(デフォロリムス;Merck & Ariad pharma)、TOP216(Toptarget A/S)、OSI−027(OSI Pharma)、TAFA93(Isotechnika)、nab−ラパマイシン(APP Phama)及びタクロリムス(FK506; Astellas)が挙げられる。
mTOR阻害薬には、ラパマイシン及び関連化合物が包含される。ラパマイシンはストレプトマイセス(Streptomyces)によって産生されるマクロライドである。ラパマイシンは強力な免疫抑制薬であり、移植された臓器の拒絶反応を防止するために臨床的に使用される。ラパマイシン及び関連化合物は、抗癌治療薬としても現在開発中である。本発明の実施形態で有用なラパマイシンは、ラパマイシン核の誘導体として化学的又は生物学的に修飾されているが、免疫抑制又は抗癌特性を保持したままである化合物を包含する。従って、ラパマイシンには、ラパマイシン自体、並びにラパマイシンのエステル、エーテル、カルボナート、オキシム、ヒドラゾン、 及びヒドロキシルアミン、並びにラパマイシン核上の官能基を例えば還元又は酸化によって修飾したラパマイシンが含まれる。
詳細には、ラパマイシンの構造を以下に示す式(A)として与える。
【0025】
【化1】
【0026】
上で開示した現在使用又は開発中の多くのラパマイシン誘導体は、C−40位に置換基がある基本的なラパマイシン構造を有する。位置40の置換基をRとして表すと、以下の置換基と化合物が対応する:R=−OP(O)(Me)2、AP23573(国際特許公開第WO98/02441号及び第WO2001/14387号);R=−OC(O)C(CH3)(CH2OH)、テムシロリムス(米国特許第5,362,718号);R=−OCH2CH2OH、エベロリムス(米国特許第5,665,772号);R=−OCH2CH2OEt、バイオリムス;R=−テトラゾール、ABT−578((国際特許公開第WO 99/15530号)。全ての特許及び出願は、参照により本明細書に援用される。
多くの他のラパマイシン誘導体は、C−40及び/又はC−16及び/又はC−32位に置換を含む。ラパマイシンのエステル及びエーテルは、全て参照により本明細書に援用される下記特許に記載されている:アルキルエステル(米国特許第4,316,885号);アミノアルキルエステル(米国特許第4,650,803号);フッ素化エステル(米国特許第5,100,883号);アミドエステル(米国特許第5,118,677号);カルバミン酸エステル(米国特許第5,118,678号;第5,411,967号;第5,434,260号;第5,480,988号;第5,480,989号;第5,489,680号);シリルエステル(米国特許第5,120,842号);アミノジエステル(米国特許第5,162,333号);スルホン酸エステル及び硫酸エステル(米国特許第5,177,203号);エステル(米国特許第5,221,670号);アルコキシエステル(米国特許第5,233,036号);O−アリール、−アルキル、−アルケニル、及び−アルキニルエーテル(米国特許第5,258,389号);炭酸エステル(米国特許第5,260,300号);アリールカルボニル及びアルコキシカルボニルカルバミン酸エステル(米国特許第5,262,423号);カルバミン酸エステル(米国特許第5,302,584号);ヒドロキシエステル(米国特許第5,362,718号);ヒンダードエステル(米国特許第5,385,908号);ヘテロ環式エステル(米国特許第5,385,909号);gem−二置換エステル(米国特許第5,385,910号);アミノアルカン酸エステル(米国特許第5,389,639号);ホスホリルカルバミン酸エステル(米国特許第5,391,730号);アミジノカルバミン酸エステル(米国特許第5,463,048号);ヒンダードN−オキシドエステル(米国特許第5,491,231号);ビオチンエステル(米国特許第5,504,091号);O−アルキルエーテル(米国特許第5,665,772号);及びラパマイシンPEGエステル(米国特許第5,780,462号);32−エステル及びエーテル(米国特許第5,256,790号)。これらのエステル及びエーテルの調製は、上掲特許に開示されている。全ての特許及び出願は、参照により本明細書に援用される。
【0027】
米国特許第5,373,014号、第5,378,836号、第5,023,264号、及び第5,563,145号に開示されているように、ラパマイシンのオキシム、ヒドラゾン、及びヒドロキシルアミンも包含される。これらのオキシム、ヒドラゾン、及びヒドロキシルアミンのの調製は、上掲特許に開示されている。40−オキソラパマイシンの調製は米国特許第5,023,263号に開示されてる。全ての特許は、参照により本明細書に援用される。
mTORの他の小分子阻害薬として、縮合二環状化合物(国際特許公開第WO2007/61737号、第WO2007/87395号及び第WO2007/64993号)、ヘテロ芳香族アミン(国際特許公開第WO 2001/19828号)、ピロロピリミジン化合物(国際特許公開第WO2005/47289号)、ジフェニル−ジヒドロ−インドール−2−オン誘導体(国際特許公開第WO2005/97107号)、及びトリメチル−ドデカ−トリエン誘導体(米国特許第特許公開第2007/037887号)が挙げられる。これらの全ての特許は、参照により本明細書に援用される。
【0028】
本発明の実施形態のμ−オピオイド受容体拮抗薬は、中枢作用性及び末梢作用性の両μ−オピオイド受容体拮抗薬を包含する。しかし、特定価値の当該拮抗薬が好適には末梢限定μ−オピオイド受容体拮抗薬である場合を企図する。
μ−オピオイド受容体拮抗薬は、その拮抗特性を維持しながら構造が変化しうる分類の化合物を形成する。これらの化合物として、三級及び四級モルフィナン、特にノルオキシモルフォン誘導体、N−置換ピペリジン、特に、ピペリジン−N−アルキルカルボキシラート、並びに三級及び四級ベンゾモルファン、並びに三級及び四級ノルモルフィナン誘導体、特に6−カルボキシ−ノルモルフィナン誘導体が挙げられる。三級化合物拮抗薬は、かなり脂溶性であり、血液脳関門を容易に横断する。血液脳関門を横断し、中枢的(及び末梢的)に活性なμ−オピオイド受容体拮抗薬の例として、例えば、ナロキソン(Baxter Pharmaceutical Products, Inc.から商業的に入手可能)、及びナルメフェン(例えば、DuPont Pharmaから入手可能)が挙げられる。他方、末梢限定拮抗薬は、典型的に電荷をもち、極性、及び/又は高分子量であり、それぞれが拮抗薬の血液脳関門の横断を妨害する。メチルナルトレキソンは、三級μ−オピオイド受容体拮抗薬ナルトレキソンの四級誘導体である。ナルトレキソンにメチル基を付加すると、さらに極性が高く、脂溶性が低い化合物を形成する。従って、メチルナルトレキソンは血液脳関門を横断せず、末梢に位置する受容体によって典型的に媒介される望ましくない副作用を遮断する可能性を有する。
本発明の実施形態で使う末梢性μ−オピオイド受容体拮抗薬は、四級モルフィナン誘導体である化合物、特に、下記式(I)
【0029】
【化2】
【0030】
の四級ノルオキシモルフォン、その単一のエナンチオマー、エナンチオマー混合物、個々のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物、若しくはプロドラッグであってよく、式中、Rは、アルキル、アルケニル、アルキニル、アリール、シクロアルキル置換アルキル、又はアリール置換アルキルであり、X−は、アニオン、特にクロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである。式(I)のノルオキシモルフォン誘導体は、例えば、参照により本明細書に援用される米国特許第4,176,186号の手順に従って調製され;全て参照により本明細書に援用される米国特許第4,719,215号;第4,861,781号;第5,102,887号;第5,972,954号;及び第6,274,591号;米国特許出願第2002/0028825号及び第2003/0022909号;並びにPCT公開第WO99/22737号及び第WO98/25613をも参照されたい。
【0031】
特に有益な式(I)の化合物は、式中、Rが、下記式(II)に示すように、シクロプロピルメチル
【0032】
【化3】
【0033】
である、N−メチルナルトレキソン(又は単にメチルナルトレキソン)、その単一のエナンチオマー、エナンチオマー混合物、個々のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物、若しくはプロドラッグであり、式中、X−は上述したとおりである。メチルナルトレキソンは、μ−オピオイド受容体拮抗薬ナルトレキソンの四級誘導体である。メチルナルトレキソンは塩(例えば、N−メチルナルトレキソンブロミド)として存在するので、本明細書で使用する場合、用語「メチルナルトレキソン」又は「MNTX」は、該塩を包含する。従って、「メチルナルトレキソン」又は「MNTX」としては具体的に、限定するものではないが、メチルナルトレキソンの臭化水素酸塩、塩酸塩、ヨウ化水素酸塩、炭酸塩、及びメチル硫酸塩が挙げられる。文献でMNTXの臭化水素酸塩として使われる名称として、例えば、メチルナルトレキソンブロミド;N−メチルナルトレキソンブロミド;ナルトレキソンメトブロミド;ナルトレキソンメチルブロミド;SC−37359;MRZ−2663−BR;及びN−シクロプロピルメチルノルオキシ−モルフィン−メトブロミドが挙げられる。従って、本明細書では、用語「メチルナルトレキソン」はメチルナルトレキソンのいずれの適切な形態、例えば、N−メチルナルトレキソン又はその医薬的に許容しうる塩、そのいずれのプロドラッグ、そのエナンチオマー、そのエピマー(エピマーについては後述する)をも意味するものと解釈すべきである。
【0034】
式(I)及び(II)の化合物、例えば、メチルナルトレキソンは、キラル中心を有しうるので、当該キラル中心上の置換基の配置によって立体化学的異性体として存在することがある。このような立体化学的異性体、例えば、エナンチオマー、ジアステレオマーは、本発明の実施形態で使うために企図される化合物の範囲内である。本発明の組成物及び方法において、使用する化合物は個々の立体異性体、及び立体異性体の混合物、例えば、エナンチオマーの混合物、ジアステレオマーの混合物でありうる。一定の態様では、実質的に純粋な立体異性体である化合物を利用する方法を提供する。全ての互変異性体も本発明の組成物に包含されるものとする。
例えば、メチルナルトレキソンのR及びS配置が知られており、一部の実施形態では、製剤及び方法において、メチルナルトレキソンの単離されたR−−N異性体を利用しうる。本明細書で使用する場合、メチルナルトレキソンの「R−−N異性体」という名称は、窒素に関して(R)配置の該化合物を表す。単離された異性体化合物としては、限定するものではないが、参照により本明細書に援用される米国特許出願第11/441,395号、及び特許協力条約公開出願WO2006/127899に記載されたR−−N異性体メチルナルトレキソン化合物が挙げられる。一部の実施形態では、活性化合物がR−−N異性体メチルナルトレキソン、又はその塩である。米国特許出願第11/441,395号に記載されたメチルナルトレキソンのR−−N異性体はオピオイド拮抗薬である。
【0035】
一部の実施形態では、製剤及び方法でメチルナルトレキソンの単離されたS−−N異性体を利用しうる。本明細書で使用する場合、メチルナルトレキソンの「S−−N異性体」という名称は、窒素に関して(S)配置の該化合物を表す。単離された異性体化合物としては、限定するものではないが、参照により本明細書に援用される米国特許出願第11/441,452号、及び特許協力条約公開出願WO2006/127898に記載されたメチルナルトレキソン化合物のS−−N異性体が挙げられる。一部の実施形態では、活性化合物がS−−N異性体メチルナルトレキソン、又はその塩である。米国特許出願第11/441,452号に記載されたメチルナルトレキソンのS−−N異性体はオピオイド作動薬である。
一定の実施形態では、本明細書で述べる製剤又は薬用量調製で利用するメチルナルトレキソンは、全体的なオピオイド拮抗作用を有することを特徴とする立体異性体の混合物である。例えば、メチルナルトレキソンは、混合物自体が拮抗薬として作用し、本明細書に記載の使用方法でオピオイド拮抗薬として有用であるような、R−−N及びS−−Nメチルナルトレキソンの混合物でありうる。一定の実施形態では、実質的にS−−NメチルナルトレキソンのないR−−Nメチルナルトレキソンを使用する。
本発明の一定の実施形態では、少なくとも約99.6%、99.7%、99.8%、99.85%、99.9%、又は99.95%のメチルナルトレキソンが窒素に関して(R)配置である。サンプル中に存在する(R)−−N−異性体の量を当該同一サンプル中に存在する(S)−−N−異性体の量と比較して決定する方法は、全体が参照により本明細書に援用されるWO2006/127899に詳述されている。他の実施形態では、メチルナルトレキソンが0.15%、0.10%、それ未満の(S)−−N−異性体を含む。
【0036】
当業者には当然のことながら、製剤、薬用量調製、又は方法において利用するメチルナルトレキソンの量に言及している場合、当該量は、メチルナルトレキソン(若しくはその塩)の総量を表すか、又は特定目的(例えば、オピオイド拮抗作用)のためには、メチルナルトレキソンの他の形態も存在するにせよ、メチルナルトレキソンの関連性のある活性形態の量を表すことがある。さらに、本明細書で指定する場合、メチルナルトレキソンの特定形態(例えば、N−メチルナルトレキソンブロミド)について薬用量又は量を定義することがある。メチルナルトレキソンの異なる形態又は塩を使用する場合、当業者は、該薬用量又は量を、等価量の活性メチルナルトレキソンがもたらす用量又は量に調整できることを認めるであろう。
メチルナルトレキソンは、例えばMallinckrodt Pharmaceuticals, St. Louis, Mo.から商業的に入手可能である。メチルナルトレキソンは、水に自由に溶ける白色の結晶性粉末として、典型的に臭化水素酸塩として提供される。提供されたままの化合物は、逆相HPLCで99.4%純粋であり、同法によって0.011%未満の非四級化ナルトレキソンを含む。例えば、約5mg/mLの濃度の無菌溶液として、メチルナルトレキソンを調製することができる。
【0037】
他の末梢性μ−オピオイド受容体拮抗薬として、N−置換ピペリジン、特に下記式(III)で表されるピペリジン−N−アルキルカルボキシラートが挙げられる。
【0038】
【化4】
【0039】
式中、R1は、水素又はアルキルであり;R2は、水素、アルキル、又はアルケニルであり;R3は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R4は、水素、アルキル、又はアルケニルであり;Aは、OR5又はNR6R7であり;このときR5は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R6は、水素又はアルキルであり;R7は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル又はアリール−置換アルキル、又はアルキレン置換Bであるか或いはR6及びR7が結合している窒素原子と一緒に、R6とR7が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Bは、下記基であり、
【0040】
【化5】
【0041】
式中、R8は、水素又はアルキルであり;R9は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル又はアリール置換アルキルであるか或いはR8及びR9が結合している窒素原子と一緒に、R8とR9が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Wは、OR10、NR11R12、又はOEであり;このときR10は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルケニル、又はアリール置換アルキルであり;R11は、水素又はアルキルであり;R12は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、アリール置換アルキル、又はアルキレン置換C(=O)Yであるか或いはR11及びR12が結合している窒素原子と一緒に、R11とR12が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Eは、下記基
【0042】
【化6】
【0043】
アルキレン置換(C=O)D、又は−R13OC(=O)R14であり;このときR13は、アルキル置換アルキレンであり;R14は、アルキルであり;Dは、OR15又はNR16R17であり;このときR15は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R16は、水素、アルキル、アルケニル、アリール、アリール置換アルキル、シクロアルキル、シクロアルケニル、シクロアルキル−置換アルキル、又はシクロアルケニル−置換アルキルであり;R17は、水素又はアルキルであるか或いはR16及びR17が結合している窒素原子と一緒に、R16とR17が、ピロール又はピペリジンから成る群より選択されるヘテロ環式環を形成し;
Yは、OR18又はNR19R20であり;このときR18は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル−置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R19は、水素又はアルキルであり;R20は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであるか或いはR19及びR20が結合している窒素原子と一緒に、R19とR20が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;R21は、水素又はアルキルであり;nは0〜4である。
【0044】
価値がありうる特定のピペリジン−N−アルキルカルボナートは、N−アルキルアミノ−3,4,4置換ピペリジン、例えば下記式(IV)で表されるアルビモパンである。
【0045】
【化7】
【0046】
N−置換ピペリジンは、米国特許第5,270,328号;第6,451,806号;第6,469,030号(全て参照により本明細書に援用される)に開示されているように調製される。アルビモパンは、Adolor Corp.、Exton、PAから商業的に入手可能である。該化合物は、適度な高分子量、双性イオン形態を有し、かつ血液脳関門の透過を防止する極性を有する。
さらに他の末梢性μ−オピオイド受容体拮抗薬化合物として、四級ベンゾモルファン化合物が挙げられる。本発明の実施形態で利用しうる四級ベンゾモルファン化合物は、下記式(V)を有する。
【0047】
【化8】
【0048】
式中、R1は、水素、アシル、又はアセトキシであり;R2は、アルキル又はアルケニルであり;Rは、アルキル、アルケニル、又はアルキニルであり、かつX−は、アニオン、特にクロリド、ブロミド、ヨージド、又はメチルスルファートアニオンである。
本発明の実施形態で利用しうるベンゾモルファン化合物の特有の四級誘導体としては、式(V)の下記化合物が挙げられる:2’−ヒドロキシ−5,9−ジメチル−2,2−ジアリル−6,7−ベンゾモルファニウム−ブロミド;2’−ヒドロキシ−5,9−ジメチル−2−n−プロピル−2−アリル−6,7−ベンゾモルファニウム−ブロミド;2’−ヒドロキシ−5,9−ジメチル−2−n−プロピル−2−プロパルギル−6,7−ベンゾモルファニウム−ブロミド;及び2’−アセトキシ−5,9−ジメチル−2−n−プロピル−2−アリル−6,7−ベンゾモルファニウム−ブロミド。
本発明の実施形態で利用しうる他の四級ベンゾモルファン化合物は、例えば、その開示全体が参照により本明細書に援用される米国特許第3,723,440号に記載されている。
他の末梢性オピオイド拮抗薬として、全体が参照により本明細書に援用され、かつ下記式(VI)を有する化合物を含む、発明の名称が「6−カルボキシ−ノルモルフィナン誘導体、その合成及び使用」である米国特許第7,501,434号に記載されている6−カルボキシ−ノルモルフィナン誘導体、特にN−メチル−C−ノルモルフィナン誘導体が挙げられる。
【0049】
【化9】
【0050】
末梢性オピオイド拮抗薬として、参照により本明細書に援用される米国特許出願第11/332,964号に記載されているオピオイド拮抗薬のポリマー複合体も挙げられる。特有のポリマー複合体として、PEG化ナロキソン及びナルトレキソンが挙げられる。
本発明の実施形態は、1種より多くのμ−オピオイド受容体拮抗薬(μ−オピオイド受容体拮抗薬の組合せ、μ及びκ拮抗薬の組合せ、例えば、メチルナルトレキソンとアルビモパンの組合せが挙げられる)の投与をも包含する。
本発明の実施形態で利用する化合物はプロドラッグ形態で存在しうる。本明細書で使用する場合、「プロドラッグ」は、哺乳動物対象に該プロドラッグを投与すると、本発明の実施形態においてin vivoで利用する活性薬又は他の化合物にin vivoで代謝される活性親薬又は化合物を放出するいずれの共有結合した担体をも含むものとする。プロドラッグは、医薬品のいくつかの望ましい品質(例えば、溶解性、生物学的利用能、製造品質など)を高めることが分かっているので、本発明の一部の実施形態で利用する化合物は、所望により、プロドラッグ形態で送達されうる。従って、本発明の実施形態は、プロドラッグを送達する方法を企図する。本発明の実施形態で利用する化合物のプロドラッグは、化合物中に存在する官能基を修飾(該修飾が日常的操作又はin vivoで開裂して親化合物になるように)することによって調製される。
従って、プロドラッグとしては、例えば、ヒドロキシ、アミノ、又はカルボキシ基がいずれかの基に結合しており、該プロドラッグを哺乳動物対象に投与すると、開裂してそれぞれ自由なヒドロキシル、自由なアミノ、又はカルボン酸を形成する、本明細書に記載の化合物が挙げられる。他の例として、限定するものではないが、アルコール及びアミン官能基のアセタート、ホルマート、及びベンゾアート誘導体;並びにアルキルエステル、カルボン酸エステル、アリールエステル、及びアルキルアリールエステル、例えばメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、シクロプロピル、フェニル、ベンジルフェネチルエステル等が挙げられる。
【0051】
上述したように、本発明の実施形態で利用する化合物は、当業者に周知のいくつかの方法で調製されうる。本発明の実施形態で述べる全ての調製は、ミリグラム、グラム、マルチグラム、キログラム、マルチキログラム、又は市販医薬品スケールを含めたいずれのスケールでも実施することを企図している。
メチルナルトレキソンについて上述したように、本発明の実施形態で利用する化合物は、1つ以上の非対称に置換された炭素原子を含むことがあり、光学活性又はラセミ形で単離されうる。従って、特有の立体化学又は異性形が具体的に指定されていない限り、全てのキラル、ジアステレオマー、ラセミ形、エピマー形、及び全ての互変異性体形を意図している。このような光学活性形を調製及び単離する方法は技術上周知である。例えば、限定するものではないが、ラセミ形の分割、順相、逆相、及びキラルクロマトグラフィー、優先晶出法(preferential salt formation)、再結晶法などの標準的技術によって、或いはキラル出発原料からのキラル合成又は標的キラル中心の計画的合成によって、立体異性体の混合物を分離しうる。
【0052】
一般的に言えば、医学的に許容しうるいずれの投与様式、例えば、臨床的に容認できない副作用を引き起こすことなく、活性化合物の有効レベルを生じさせるいずれの投与様式を用いても本発明の実施形態の方法を実施することができる。このような投与様式としては、経口、直腸、局所(散剤、軟膏、液滴、経皮パッチ、又はイオン注入装置によってのうに)、経皮、舌下、筋肉内、注入、静脈内、肺、筋肉内、腔内、エアロゾルとして、耳(例えば、点耳薬によって)、鼻腔内、吸入、眼内、又は皮下投与が挙げられる。
さらに、腸溶コーティング錠剤又はカプセル剤として本発明の実施形態の化合物を投与することができる。一部の実施形態では、遅速注入法又は持続放出若しくは制御放出法によるか又は凍結乾燥散剤としてμ−オピオイド受容体拮抗薬を投与する。
さらに、本発明の実施形態の化合物を局所投与することができる。局所投与用製剤としては、軟膏、ローション、クリーム、ゲル、液滴、座剤、スプレー、液体及び散剤が挙げられる。通常の医薬担体、水、粉末又は油性基剤、増粘剤などが必要であるか又は望ましいことがある。
投与するとき、本発明の実施形態の化合物は、医薬的に許容しうる量及び医薬的に許容しうる組成物若しくは製剤で与えられる。該製剤は、日常的に塩、緩衝剤、保存剤、及び必要に応じて他の治療成分を含有しうる。
医薬中で使用する場合、本発明の実施形態の化合物の医薬的に許容しうる塩を使用しうるが、便宜上医薬的に許容しえない塩を用いてその医薬的に許容しうる塩を調製することがあり、このような医薬的に許容しえない塩は本発明の範囲から除外されない。このような薬理学的及び医薬的に許容しうる塩として、限定するものではないが、以下の酸から調製される当該塩が挙げられる:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、p−トルエンスルホン酸、酒石酸、クエン酸、メタンスルホン酸、ギ酸、コハク酸、ナフタレン−2−スルホン酸、パモ酸、3−ヒドロキシ−2−ナフタレンカルボン酸、及びベンゼンスルホン酸。
【0053】
本発明の実施形態の製剤に緩衝剤及び保存剤を含めてもよい。適切な緩衝剤として、限定するものではないが、酢酸とその塩(1〜2%w/v);クエン酸とその塩(1〜3%w/v);ホウ酸とその塩(0.5〜2.5%w/v);及びリン酸とその塩(0.8〜2%w/v)が挙げられる。適切な保存剤として、限定するものではないが、塩化ベンザルコニウム(0.003〜0.03%w/v);クロロブタノール(0.3〜0.9%w/v);パラベン(0.01〜0.25%w/v);及びチメロサール(0.004〜0.02%w/v)が挙げられる。
投与を容易にするため、本発明の実施形態の医薬組成物は、1種以上の医薬的に許容しうる賦形剤、例えば潤沢剤、希釈剤、結合剤、担体、及び崩壊剤を含んでもよい。他の補助剤として、例えば、安定剤、湿潤剤、乳化剤、浸透圧に影響を与える塩、着色剤、香料、及び/又は芳香族活性化合物が挙げられる。
医薬的に許容しうる担体又は賦形剤は、無毒の固体、半固体若しくは液体フィラー、希釈剤、被包材、又はいずれのタイプの製剤助剤をも表す。例えば、適切な医薬的に許容しうる担体、希釈剤、溶媒、又はビヒクルとして、限定するものではないが、水、塩(緩衝)溶液、アルコール、アラビアゴム、鉱油、植物油、ベンジルアルコール、ポリエチレングリコール、ゼラチン、炭水化物(例えばラクトース、アミロース又はデンプン)、ステアリン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、植物油、脂肪酸モノグリセリド、脂肪酸ジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシルメチルセルロース、ポリビニルピロリドン等が挙げられる。例えば、レシチン等のコーティング材の使用によって、又は分散系の場合には所要の粒径の維持によって、及び界面活性剤の使用によって、適正な流動性を維持することができる。種々の抗菌剤、例えば、抗細菌剤及び抗真菌剤、パラベン、クロロブタノール、フェノール、ソルビン酸などの薬剤を含めることによって、微生物の作用の阻止を確実にすることができる。
【0054】
医薬的に許容しうる固体担体を使用する場合、本発明の実施形態で使うのに適した化合物の剤形は、錠剤、カプセル剤、散剤、座剤、又はロゼンジ剤でありうる。液体担体を使用する場合、剤形は、軟ゼラチンカプセル剤、経皮パッチ、エアロゾルスプレー、局所クリーム、シロップ又は液体座剤、エマルション、又は溶液でありうる。
非経口適用では、特に注射用無菌溶液、好ましくは非水性若しくは水性溶液、並びに分散液、懸濁液、エマルション、又は座剤を含めたインプラントが適している。アンプルは多くの場合便利な単位剤形である。注射用デポー形も好適であり、ポリラクチド−ポリグリコリド、ポリ(オルトエステル)、及びポリ(酸無水物)等の生分解性ポリマー中で薬物のマイクロカプセルマトリクス(microencapsule matrices)を形成することによって製造しうる。ポリマーに対する薬物の比率及び利用する特定ポリマーの性質によっては、薬物放出速度を制御することができる。
経腸適用では、特に錠剤、糖衣錠(dragee)、液体、液滴、座剤、又はカプセル剤、例えば軟ゼラチンカプセル剤が適している。甘味ビヒクルを利用するシロップ剤、エリキシル剤などを使用することができる。
【0055】
上述したように、他の医薬送達システムとして、持続放出、遅延放出、又は徐放送達システムが挙げられる。このようなシステムは、本発明の化合物の反復投与を回避し、患者及び医師の便利さを高め、かつ化合物の持続性血漿レベルを維持することができる。多くのタイプの制御放出送達システムが利用可能であり、当業者には周知である。
例えば、本発明の実施形態の化合物を医薬的に許容しうる徐放マトリックス、例えば生分解性ポリマーと併用して、治療組成物を形成することができる。本明細書で使用する場合、徐放マトリックスは、酵素加水分解若しくは酸塩基加水分解又は溶解によって分解しうる材料、通常はポリマー製のマトリックスである。体内に挿入されると、該マトリックスは酵素及び体液によって作用を受ける。徐放マトリックスは、望ましくは生体適合性材料、例えばリポソーム;ポリマーをベースとしたシステム、例えばポリラクチド(ポリ乳酸)、ポリグリコリド(グリコール酸のポリマー)、ポリラクチド−コ−グリコリド(乳酸とグリコール酸のコポリマー)、ポリ酸無水物、ポリ(オルト)エステル、多糖、ポリアミノ酸、ヒアルロン酸、コラーゲン、コンドロイチン硫酸、ポリヌクレオチド、ポリビニルプロピレン、ポリビニルピロリドン、及びシリコーン;非ポリマーシステム、例えばカルボン酸、脂肪酸、リン脂質、アミノ酸、及び脂質、例えばステロール;ヒドロゲル放出システム;サイラスティック(silastic)システム;ペプチドベースシステム;インプラント等から選択される。具体例として、限定するものではないが、以下のものが挙げられる:(a)米国特許第4,452,775号、第4,675,189号、及び第5,736,152号(全体が参照により本明細書に援用される)で見つかるマトリックス内の形態で多糖が含まれる浸食システム、及び(b)米国特許第3,854,480号、第5,133,974号、及び第5,407,686号(全体が参照により本明細書に援用される)に記載されているようなポリマーから活性成分が制御された速度で浸透する拡散システム。さらに、ポンプをベースとしたハードワイヤード送達システム(pump−basedhard−wired delivery system)を使用でき、移植に適合したシステムもある。好適な腸溶性コーティングは、PCT公開第WO98/25613号及び米国特許第6,274,591号(両方とも参照により本明細書に援用される)に記載されている。マイクロカプセル化、多重コーティング等によってのように差動分解性コーティングを利用して活性化合物を保護する当該組成物として徐放又は制御放出組成物を調製することもできる。
【0056】
特にメチルナルトレキソンに関して、水性製剤は、キレート化剤、緩衝剤、抗酸化剤及び必要に応じて等張剤(好ましくは3.0〜3.5のpHに調整)を含有しうる。オートクレーブ処理及び長期貯蔵に安定した製剤は、その開示内容が参照により本明細書に援用され、発明の名称が「医薬製剤」であり、第2004/0266806号として公開された米国特許出願第10/821811号に記載されている。有効期間を増やしたメチルナルトレキソンの製剤も、参照によって本明細書に援用される、発明の名称が「化合物の非経口送達用製剤及びその使用」である国際特許公開第WO2008/19115号に記載されている。メチルナルトレキソンの凍結乾燥製剤は、米国特許出願第11/899,724号に記載され、メチルナルトレキソンを含有する粒子を含む製剤は、参照により本明細書に援用される米国特許第6,419,959号に記載されている。メチルナルトレキソンの経皮送達に適した製剤は、参照により本明細書に援用される国際特許公開第2007/41544号に記載されている。
【0057】
本発明の実施形態の化合物、mTOR阻害薬及びμ−オピオイド受容体拮抗薬は、相乗的な抗増殖及び抗遊走に有効な量で提供される。しかし、当然のことながら、本発明の化合物及び組成物の総投与量は、ゾンデ医療判断の範囲内で担当医師によって決定される。いずれの特定の患者に特有の治療的に有効な用量レベルも種々の因子によって左右される。該因子としては、治療する障害及び障害の重症度;利用する特有化合物の活性;利用する特有組成物;患者の年齢、体重、一般的健康状態、性別、及び食事制限;投与時間;投与経路;利用する特有化合物の排泄速度;治療の持続期間;利用する特有化合物と併用又は同時に使用する薬物など、医術で周知の因子が挙げられる。例えば、ある技法は、所望の治療効果を達成するのに必要な用量未満のレベルで化合物の用量を開始して、所望効果が達成されるまで投与量を徐々に増やすことである。
所望により、1日の有効な用量を投与目的によって複数の用量に分割してよい。結果として、単一用量組成物は、1日の用量を構成する量又はその約数の量を含有しうる。上述したように、当業者は、良い医療行為及び個々の患者の臨床状態によって決まる有効湯量及び共投与計画(本明細書で述べるように)を容易に最適化することができる。
一般的に、μ−オピオイド受容体拮抗薬、特に末梢性拮抗薬の経口用量は、1日当たり約0.01〜約80mg/kg(体重)の範囲である。1〜20mg/kg(体重)の経口用量が望ましい結果をもたらすと予想される。一般的に、静脈内及び皮下投与などの非経口投与は、約0.001〜5mg/kg(体重)の範囲である。0.05〜0.5mg/kg(体重)の範囲の用量が所望の結果をもたらすと予想される。薬用量を適宜調整して、投与様式によって決まる局所又は全身の所望薬物レベルを達成することができる。例えば、腸溶コーティング製剤中のμ−オピオイド受容体拮抗薬の経口投与用薬用量は、非コーティング経口用量の10〜30%であると予想される。患者の応答が該用量では不十分な場合、患者の耐用性が許す程度まで、さらに高い用量(又はより局所的な異なる送達径路によって30%の用量より効率的に高い)を利用してよい。1日当たりの複数回の用量は、化合物の適切な全身レベルを達成するように考慮される。適切なシステムレベルは、例えば、当業者に周知の日常的なHPLC法を用いて、薬物の患者の血漿レベルを測定することで決定される。
【0058】
本発明の実施形態では、必要に応じて、例えば、本発明の実施形態の化合物について知られている投与量で、いずれの投与経路によっても、例えば、経腸(例えば、経口)、又は非経口又は局所的にmTOR阻害薬化合物を投与しうる。mTOR阻害薬について種々の経口及び非経口剤形が知られている。1日の経口投与量は、例えば分散性錠剤の形態で0.1mg〜25mgの範囲であってよい。1週間の投与量は、70mgまで含んでよく、治療する疾患によって決まる。静脈内投与などの非経口投与では、1日の投与計画(5日間毎日、2週間〜3週間毎日)で投与する場合、最初の静脈内投与量が0.1〜100mg/m2であり、さらに好適には、1週間に1回の投与計画で投与する場合、0.1〜1000mg/m2である。例えば、エベロリムスは、治療する疾患によって、例えば分散性錠剤の形態又は固体分散系の形態で、0.1mg〜25mgまで又は0.1mg〜15mg(例えば0.1mg、0.25mg、0.5mg、0.75mg、1mg、2.5mg、5mg、又は10mgなど)の1日の投与量で経口投与することができる。エベロリムスを70mgまで含みうる、例えば10〜70mg、又は30〜50mg(治療する疾患によって決まる)の1週間の薬用量で投与することができる。さらなる例では、タクロリムス(Protopic)を軟膏基剤中0.03%〜0.1%(w/w)の軟膏として投与することができる。同様に、例えば、同様の投与量範囲で他のmTOR阻害薬を投与することができる。
【0059】
本発明の例示実施形態では、μ−オピオイド受容体拮抗薬をmTOR阻害薬と共投与する。換言すれば、μ−オピオイド受容体拮抗薬化合物とmTOR阻害薬の共投与は、適切にμ−オピオイド受容体拮抗薬とmTOR阻害薬を含む医薬の組合せと考えられ、この組合せを毎日又は間欠ベースの末梢性μ−オピオイド受容体拮抗薬の投与、及び毎日又は間欠ベースのmTOR阻害薬の投与に適合させる。従って、μ−オピオイド受容体拮抗薬をmTOR阻害薬の投与前、投与と同時、又は投与後に投与してよい。典型的投与計画では、患者は、mTOR阻害薬の30分の静脈内注入を受けた直後にμ−オピオイド受容体拮抗薬を受けるか又はμ−オピオイド受容体拮抗薬の投与を先に受ける。1回以上の治療周期後、得られた結果及び観察されたいずれもかの副作用によって、薬用量を上方又は下方調整することができる。例示実施形態では、mTOR阻害薬の投与前のμ−オピオイド受容体拮抗薬の投与が特に好適である。
本発明の実施形態で共投与可能な薬剤を混合物として、例えば、単一製剤又は単一錠剤中の混合物のように調製してもよい。これらの製剤は、非経口又は経口製剤、例えば、米国特許第6,277,384号;第6,261,599号;第5,958,452号及びPCT公開第WO98/25613号(それぞれ参照により本明細書に援用される)に記載されている製剤であってよい。
【0060】
本発明の実施形態の方法を単独で使用してよく、或いは上述した種々の状態と関連する内皮細胞の成長又は遊走を制御するための他の治療と併用してよい。mTOR阻害薬及び末梢性μ−オピオイド受容体拮抗薬を、オピオイド若しくはμ−オピオイド受容体拮抗薬又はmTOR阻害薬でない別の治療薬と共投与してよい。このような適切な治療薬として、他の抗癌薬が挙げられる。
本発明の実施形態は、癌の治療をも包含する。治療しうる癌のタイプは、mTORの関与によってのみ制限される。従って、これらの療法を用いて、脳、肺、肝臓、脾臓、腎臓、リンパ節、膵臓、小腸、血液細胞、結腸、胃、乳房、子宮内膜、前立腺、精巣、卵巣、皮膚、頭頚部、食道、骨髄、血液又は他の組織の癌を含めた種々多様の腫瘍を治療できると考えられる。
多くの文脈で、必ずしも腫瘍細胞が殺されるか又は正常細胞の死若しくは「アポトーシス」を受けるように誘発される必要はない。むしろ、意味ある治療を達成するために必要なのは、腫瘍の成長をある程度まで遅らせることだけである。しかし、もしかしたら腫瘍の成長が完全に遮断されるか、又は一部の腫瘍の退縮が達成されるかもしれない。「寛解」及び「腫瘍量(tumor burden)の減少」などの臨床的用語は、所定のそれらの普通の用法も考えられる。
以下の実施例で本発明の実施形態をさらに説明するが、本発明の範囲を限定する目的で実施例を解釈すべきでない。
【実施例】
【0061】
実施例1:VEGF誘発Akt活性化の阻害
セリン/スレオニンキナーゼAktのVEGF誘発活性化に及ぼすメチルナルトレキソン(MNTX)の作用を評価するため、特徴がはっきりした内皮細胞系であるヒト肺微小血管内皮細胞(HPMVEC)を使用した。HPMVECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX、100ng/mlのベバシズマブ、100μMの5−FU、100nMのMNTX+100ng/mlのベバシズマブ又は100nMのMNTX+100μMの5−FUを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。細胞ライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt、抗Akt又は抗アクチン抗体を用いて免疫ブロットした。結果は、メチルナルトレキソンがセリン及びスレオニンのそれぞれ位置473及び308でAktのVEGF誘発リン酸化を抑止することを示した(図2)。さらに、ベバシズマブ及び5−FUと併用したメチルナルトレキソンは、相乗的にAktの活性化を阻害した。VEGF誘発pSer473Akt(図3A)及びpThr308Akt(図3B)の免疫反応性の阻害に及ぼす種々の濃度のMNTX(1.0、10、100、1000nM)の作用は、この阻害が用量依存性であることを実証した。
【0062】
細胞培養及び試薬−ヒト肺微小血管ECをCambrex(Walkersville, MD)から得、以前に記載されているように(Singleton et al. (2006), Microvasc. Res. 72(1−2):3−11; Singleton et al. (2007), Am. J. Respir. Cell Mol. Biol. 37(2):222−231)、EBM−2完全培地(Cambrex)内で37℃にて5%CO2、95%空気の加湿雰囲気内で培養し、実験用には継代6〜10を用いた。特に断らない限り、Sigma (St. Louis, MO)から試薬を得た。血管内皮成長因子(VEGF)はR&D Systems(Minneapolis, MN)から購入した。メチルナルトレキソンブロミド(MNTX)はMallinckrodt Specialty Chemicals (Phillipsburg, NJ)から購入した。ベバシズマブはGenentech(South San Francisco, CA)から購入した。5−フルオロウラシル(5−FU)はAbraxis Pharmaceutical Products (Schaumburg, IL)から購入した。ナルトレキソン及びラパマイシンはSigma(St. Louis, MO)から購入した。SDS−PAGE電気泳動用の試薬はBio−Rad(Richmond, CA)から購入し、Immobilon−P転写膜はMillipore(Millipore Corp., Bedford, MA)から購入した。ウサギ抗pSer473Akt、ウサギ抗pThr308Akt及びウサギ抗Akt抗体はCell Cignalling Technologies(Danvers, MA)から購入した。マウス抗β−アクチン抗体はSigma(St. Louis, MO)から購入した。二次西洋ワサビペルオキシダーゼ(HRP)標識抗体はAmersham Biosciences(Piscataway, NJ)から購入した。
SDS−PAGE及び免疫ブロット法−処理済み又は未処理HPMVECから得た細胞材料をIP緩衝液(50mM HEPES(pH 7.5)、150mM NaCl、20mM MgCl2、1% Nonidet P−40(NP−40)、0.4mM Na3VO4、40mM NaF、50μM オカダ酸、0.2mM フェニルメチルスルホニルフルオリド、1:250希釈のCalbiochemプロテアーゼ阻害薬混合物3)とインキュベートし、4〜15%のポリアクリルアミドゲル中のSDS−PAGEに供し、ImmobilonTM膜上に移し、特異性一次抗体及び二次抗体を用いて展開した。増強化学発光(Amersham Biosciences)を用いて免疫反応性バンドの可視化を達成した。活性化Aktの相対量を調べるため、pSer473Akt及びpThr308Akt免疫反応性バンド強度を総Akt免疫反応性バンド強度で割った。
【0063】
実施例2:VEGF誘発内皮細胞遊走及び増殖に及ぼすラパマイシンの作用
細胞の遊走及び増殖におけるmTORの役割を調べるため、HPMVECの遊走及び増殖アッセイに及ぼすラパマイシンの作用を定量した。ヒトECをVEGF(100nM)誘発増殖(図4)及び遊走(図5)について0.01、0.1、1.0若しくは10nMのラパマイシンの存在下又は非存在下でアッセイした。結果は、ラパマイシンによるmTORの阻害が内皮細胞の増殖及び遊走の用量依存性阻害をもたらすことを実証する。このことは、mTORがこれらのプロセスに関与することを実証している以前の研究と一致する。
ヒト肺微小血管EC遊走アッセイ−in vitroで細胞の遊走をモニターするために8μMのポアサイズの24のトランスウェル(transwell)ユニットを用いた。HPMVEC(約1×104細胞/ウェル)を種々の処理をして上方チャンバーに蒔き、VEGF(100nM)を下方チャンバーに添加した。細胞を18時間遊走させた。上方及び下方チャンバーから得た細胞をCellTiter96TM MTSアッセイ(Promega, San Luis Obispo, CA)を用いて定量化し、492nmで解読した。上方と下方の両チャンバー内の細胞数で割った下方チャンバー内の細胞数として遊走率を定義した。各アッセイを三通り準備し、少なくとも5回繰り返し、スチューデントt−検定で統計的に解析した(P<0.05で設定される統計的有意性を有する)。
ヒト肺微小血管EC増殖アッセイ−細胞の成長を測定するため、種々の薬剤で前処理したHPMVEC[5×103細胞/ウェル]を、5%CO2/95%空気中37℃にて96−ウェル培養プレート内で100nMのVEGFを含む0.2mlの無血清培地と24時間インキュベートした。96TM MTSアッセイ(Promega, San Luis Obispo, CA)を用いて細胞数の増加を測定することによってin vitro細胞増殖アッセイを分析し、492nmで解読した。各アッセイを三通り準備し、少なくとも5回繰り返し、スチューデントt−検定で統計的に解析した(P<0.05で設定される統計的有意性を有する)。
統計解析−スチューデントt−検定を用いて、2以上の異なる実験群から得たデータの平均値を比較した。結果を平均値±S.E.として表す。
【0064】
実施例3:VEGF誘発内皮細胞遊走及び増殖に及ぼすメチルナルトレキソンとmTOR阻害薬の相乗効果
AktとmTORの間に存在する複雑なフィードバック阻害シグナル伝達ネットワークに鑑み、上述した遊走及び増殖アッセイを用いて、ラパマイシンによるmTOR活性化の阻害とメチルナルトレキソンによるAktの阻害を同時に行なうと、これらのプロセスに対して相乗効果をもたらすかを決定した。ヒト内皮細胞をVEGF(100nM)誘発増殖(図6)及び遊走(図7)について10若しくは100nMのMNTX、10nMのナルトレキソン、0.1nMのラパマイシン、100nMのMNTX+0.1nMのラパマイシン又は10nMのナルトレキソン+0.1nMのラパマイシンの存在下或いは非存在下でアッセイした。メチルナルトレキソンとテムシロリムスを併用して同様の結果を得た(図10)。
結果は、メチルナルトレキソンとmTOR阻害薬が相乗的に内皮細胞の遊走及び増殖を阻害することを実証した。メチルナルトレキソンで観察された結果と異なり、テムシロリムスを用いたヒト内皮細胞の処理は、AktのVEGF誘発活性化を阻害しなかった(図8)。これは、メチルナルトレキソンとmTOR阻害薬の併用で観察された相乗効果が、おそらく2つの別個の点におけるmTORシグナル伝達経路の阻害のためであり、メチルナルトレキソン誘発阻害はAkt活性化の上流で起こり、mTOR阻害薬による阻害はAkt活性化の下流で起こることを示唆している(図12)。この仮説は、メチルナルトレキソンはmTOR複合体IとmTOR複合体IIのVEGF誘発形成を阻害するが、テムシロリムスはmTOR複合体IIの形成だけを阻害するという事実によって支持される(図9A)。
VEGF誘発Akt活性化の機構をさらに解明するため、PI3キナーゼ阻害薬、LY294002、Src siRNA又はRictor(mTOR複合体IIの成分)siRNAで前処理した場合としない場合のVEGFでヒト内皮細胞を処理した。抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いてライセートを免疫ブロットした(図9B)。PI3キナーゼ阻害薬は、VEGF誘発Aktスレオニン308リン酸化を阻害する。mTOR複合体2の形成(Rictor siRNA)の阻害がVEGF誘発Aktセリン473のリン酸化を遮断する。Srcの発現(siRNA)の阻害がAktセリン473及びAktスレオニン308の両リン酸化を遮断する。さらに、メチルナルトレキソンとmTOR阻害薬の併用がヒト内皮細胞の増殖と遊走に及ぼす相乗効果におけるチロシンホスファターゼ(PTP)の中心的役割を、これらのVEGF誘発血管新生促進事象を阻害する強力なPTP阻害薬3,4−デホスタチン(dephostatin)を用いて実証した(図11)。
【0065】
Aktリン酸化のMNTX及びテムシロリムス制御−ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX又は100nMのテムシロリムスを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いて免疫ブロットした。
mTOR複合体形成及びAktリン酸化制御の分析−ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX又は100nMのテムシロリムスを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、抗Raptor(mTOR複合体1の成分)又は抗Rictor(mTOR複合体2の成分)抗体を用いて免疫ブロットした。免疫ブロットした材料でSDS−PAGEを実行し、抗mTOR、抗FKBP12、抗Raptor、抗SIN1又は抗Rictor抗体のどれかを用いて免疫ブロットした。Aktリン酸化の制御を調べるため、ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いはPI3キナーゼ阻害薬、LY294002(10μM、1時間)、Src siRNA又はRictor(mTOR複合体2の成分)siRNAを用いて前処理した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いて免疫ブロットした。
VEGF誘発ヒトEC増殖及び遊走に及ぼすMNTXとテムシロリムスの相乗効果。ヒトECの阻害曲線を、0.1、1.0、10、100若しくは500nMのMNTX、テムシロリムス又は10nMのMNTX+テムシロリムスの存在下或いは非存在下でVEGF(100nM)誘発増殖及び遊走についてアッセイした(24時間)。MNTXは、ECのVEGF誘発増殖を約100nMのIC50で阻害した。ECに10nMのMNTXを添加すると、VEGF誘発増殖のテムシロリムス阻害のIC50が約10nM〜約1nMにシフトした。実験は三通り行なった。エラーバー=標準偏差。
テムシロリムとのMNTXの相乗効果はチロシンホスファターゼの活性によって制御される。ヒトECを、10nMのMNTXがある場合とない場合で10nM又は15nMのテムシロリムスの存在下でVEGF(100nM)誘発増殖及び遊走(24時間)についてアッセイした(それぞれ3,4−デホスタチンの非存在下又は存在下の増殖の阻害についてのIC50濃度)。実験は三通り行なった。エラーバー=標準偏差。
如何なる特定の理論によっても制限されたくないが、図12は、実施例の結果によって実証された相乗効果に可能な機構を示す。
【0066】
実施例4:mTOR阻害薬とメチルナルトレキソンを併用した哺乳動物対象の治療
第1セットの実験では、腫瘍細胞のトランスフォーメーション、近親交配又は移植によってマウスに腫瘍を発生させる。それぞれ少なくとも60mm3の体積を有する腫瘍を保持する48匹のマウスをランダムに4群に分ける。第1群は、オピオイド拮抗薬もmTOR阻害薬をも含まないコントロール物質を受ける。第2群は、治療的に有効な量のメチルナルトレキソンを用いて腫瘍と接するのに容認できる径路を介して投与される末梢性オピオイド拮抗薬メチルナルトレキソン、例えば5mg/kg/日の用量で経口投与を受ける。第3群は、治療的に有効な量のmTOR阻害薬を用いて腫瘍と接するのに容認できる径路を介して投与されるラパマイシン等のmTOR阻害薬、例えば、1mg/kg/日の用量のラパマイシンの注射を受ける。第4群は、メチルナルトレキソンとmTOR阻害薬の組合せを受ける。
各群のマウス間の腫瘍の成長速度、腫瘍サイズ、腫瘍内の血管新生及び死亡率の差異を記録する。mTOR阻害薬、例えばラパマイシンの治療用量を変えて追加実験を行ない、メチルナルトレキソンとの共投与に起因するmTOR阻害薬の治療用量の減少を定量する。
【0067】
実施例5:mTOR阻害薬とアルビモパンを併用した哺乳動物対象の治療
第1セットの実験では、腫瘍細胞のトランスフォーメーション、近親交配又は移植によってマウスに腫瘍を発生させる。それぞれ少なくとも60mm3の体積を有する腫瘍を保持する48匹のマウスをランダムに4群に分ける。第1群は、オピオイド拮抗薬もmTOR阻害薬をも含まないコントロール物質を受ける。第2群は、治療的に有効な量のアルビモパンを用いて腫瘍と接するのに容認できる径路を介して投与される末梢性オピオイド拮抗薬アルビモパンを受ける。第3群は、治療的に有効な量のmTOR阻害薬を用いて腫瘍と接するのに容認できる径路を介して投与されるラパマイシン等のmTOR阻害薬、例えば、1mg/kg/日の用量の注射を受ける。第4群は、アルビモパンとmTOR阻害薬の組合せを受ける。
各群のマウス間の腫瘍の成長速度、腫瘍サイズ、腫瘍内の血管新生及び死亡率の差異を記録する。mTOR阻害薬、例えばラパマイシンの治療用量を変えて追加実験を行ない、アルビモパンとの共投与に起因するmTOR阻害薬の治療用量の減少を定量する。
【0068】
要約すれば、本発明の実施形態の方法は、癌及び他の過剰増殖疾患並びに自己免疫疾患といった細胞の増殖及び遊走に関連する疾患又は障害を治療するために提供され、この方法はmTOR阻害薬とμ−オピオイド受容体拮抗薬の共投与を含む。
以上、種々の特有の実施形態及び手法に関連して本発明について述べた。しかし、本発明の精神及び範囲内にとどまりながら、多くの変更及び修正を行なえることを理解すべきである。
全ての出版物、特許、及び特許出願は、それぞれ個々の出版物、特許、及び特許出願が具体的かつ個々に参照によって指示されたのと同程度に、参照により本明細書に明確に援用される。本開示と、援用された特許、出版物及び参考文献との間で矛盾する場合、本開示が支配するものとする。
【技術分野】
【0001】
(関連出願の相互参照)
この出願は、2008年3月21日提出の全開示が参照により本明細書に援用される米国仮出願第61/038,577号の優先権を35 U.S.C.§119(e)の下で主張する。
(連邦政府による資金提供を受けた研究開発の記載)
適用なし。
【背景技術】
【0002】
細胞の成長及び増殖は、全ての生体における正常な進行中プロセスであり、規則的な細胞周期を維持するため微妙に均衡が保たれている多くの因子及びシグナルに関与する。哺乳動物細胞が成長及び分割するか否かは、例えば細胞が成長できる空間の利用能、及び現在置かれている環境内における特有の刺激因子や阻害因子の分泌などの種々のフィードバック制御機構によって決まる。
mTORは、ホスファチジルイノシトール3−キナーゼ(PI3K)−関連キナーゼ(PIKK)ファミリーの大型ポリペプチドセリン/スレオニンキナーゼである。mTORは、PI3K径路から下流にあり、細胞の成長及び増殖を調節するための種々の細胞シグナル伝達事象における中間体として機能する。mTOR活性はセリン/スレオニンキナーゼAktによって調節され、最近の証拠は、これらのキナーゼが複雑なフィードバック阻害径路を介して相互作用することを示している。mTORは、G1期からS期を通して細胞周期の進行に必要な主要タンパク質の翻訳を制御することによって、細胞の複製を調節する。すなわち、mTORは、mRNA、主に4E−PB1、P7056K及びeEFZの翻訳に関与するいくつかのタンパク質のリン酸化状態の調節によって特有のmRNAの翻訳を制御する。
mTOR径路は、そのPI3K及びAkt成分と共に、栄養素、ホルモン及び成長因子、例えばVEGFに応答する細胞の増殖の重要なレギュレーターである。成長因子は、同族細胞表面受容体に結合することによって、PI3Kシグナル伝達を活性化し、それによってAktを介したシグナル伝達カスケードを惹起し、mTORの活性化をもたらすことができる。最近の研究は、mTOR阻害薬が、成長因子媒介シグナル伝達と成長因子の翻訳を両方とも阻害することによって、抗増殖作用及び血管新生抑制作用を有することを実証した。
【0003】
癌細胞では、mTORの上流のPI3K径路内の複数の調節不全機構がmTOR活性を上昇させ、結果として腫瘍の成長を増やすとして実証されている。このように、mTORシグナル伝達経路の調節不全が癌の進行に関係しており、現在、癌の治療薬としてmTORの阻害薬が調査されている。mTOR阻害薬は強力な免疫抑制薬でもある。1つの該薬剤であるシロリムスは、現在、臓器拒絶反応の予防用に使われている。現在市販されているか又は開発中である他のmTOR阻害薬として、テムシロリムス(ToriselTM;Wyeth)、RAD001(エベロリムス;Novartis)、MK−8669(デフォロリムス(deforolimus);Merck & Ariad pharma)、FK506(タクロリムス;Astellas)、TOP216(Toptarget A/S)、OSI−027(OSI Pharma)、TAFA93(Isotechnika)、及びnab−ラパマイシン(APP Pharma)が挙げられる。これらのmTOR阻害薬の多くは、ラパマイシン又はラパマイシン誘発体である。
臓器移植拒絶反応予防薬及び抗癌薬としての開発に加え、mTOR阻害薬は、関節リウマチ、自己免疫障害、乾癬、多発性硬化症、パーキンソン病、脳卒中及び末梢神経障害の治療のためにも開発されている。例えば、mTOR阻害薬ラパマイシンは、例えば実験的アレルギー性脳脊髄炎、インスリン依存性糖尿病、マウスループス及びアジュバント関節炎などの自己免疫のいくつかの動物モデルにおいて効力が示され、かつリウマチ様疾患の治療に可能性があると提案されている。リウマチ様疾患の治療については、リウマチ様疾患又はグレーブス病の個体由来の抗体はmTOR径路を介して線維芽細胞を活性化するが、ラパマイシンはこれを阻害することができ、mTORの遮断がこれらの疾患における炎症を制限するのに価値があること示唆している。ラパマイシンは、ヒト線維芽細胞内のコラーゲンmRNAレベルを下げることも分かっており、mTORがI型コラーゲンの合成をポジティブに調節することを示唆している。従って、mTORの遮断は、線維性病変が正常な組織構造を破壊し、臓器不全に寄与する強皮症のような線維性疾患にも有益でありうる。HIV−1がCD4T細胞及びマクロファージに侵入するために利用するCCR5ケモカイン受容体のタンパク質発現もラパマイシンによって阻害され、免疫細胞内におけるCCR5媒介ウイルス侵入の遮断の臨床用途を示唆している。最近、ラパマイシン治療が、筋肉及び皮膚に影響を及ぼす自己免疫状態である皮膚筋炎の患者において臨床疾患を有意に軽減することが分かった。mTOR阻害薬は、培養及び動物モデルにおいて強力な神経保護特性及び神経再生特性をも有する。
しかしながら、臨床研究においていくつかの副作用が観察されている。副作用としては、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。これらの副作用の少なくとも一部は用量依存性であると思われる。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の実施形態の原理を具体化する方法及び医薬の併用は、mTOR阻害薬を、一般的にμ−オピオイド受容体拮抗薬と記述される分類の化合物と共投与することを包含する。発明者らは、メチルナルトレキソン等のμ−オピオイド受容体拮抗薬をmTOR阻害薬と相乗様式で共投与して、mTOR阻害薬の治療的に有効な投与量を低減できることを見出した。上述したように、mTOR阻害薬に伴う副作用の少なくとも一部は用量依存性であると思われるので、μ−オピオイド受容体拮抗薬の共投与は、低用量でこれらのmTOR阻害薬を使用できるようにし、同時に副作用の発生及び/又は重症度を低減しうる。発明者らは、メチルナルトレキソンが、mTORの活性化の上流事象であるキナーゼAktの活性化を阻害することを実証した。Akt及びmTORは、複雑な制御フィードバックループに関与しており、本発明の実施形態のよる両Akt及びmTORの同時ターゲティングは相乗効果を有する。
【課題を解決するための手段】
【0005】
mTOR阻害薬と組み合わせたμ−オピオイド受容体拮抗薬の使用は、これらの阻害薬の抗癌及び免疫抑制効力を大いに高め、それらの低用量での使用を可能にし、ひいては副作用の発生/重症度を低減しうる。さらに、メチルナルトレキソン等のμ−オピオイド受容体拮抗薬をこれらのmTOR阻害薬の効力を高めるために使用すると、結果として、治療作用に必要な用量が減少するので、mTOR阻害薬に伴う治療の高いコストをも大いに下げるであろう。さらに、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与することによって、mTOR阻害薬の治療指数又は有用性を改善する方法を企図する。
本発明の実施形態の原理を具体化する方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬の組合せを用いて、細胞の増殖及び遊走、特に内皮細胞の増殖及び遊走を減弱、例えば阻害又は低減することを含む。前記μ−オピオイド受容体拮抗薬としては、限定するものではないが、末梢限定拮抗薬が挙げられる。本発明の一態様により、細胞の不必要な遊走及び/又は増殖を特徴とする障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む治療方法を提供する。この治療は、細胞の遊走と増殖の一方又は両方を阻害し、細胞は適宜、内皮細胞であってよく、血管内皮細胞が特に興味深い。従って、別の実施形態では、血管内皮細胞の不必要な遊走又は増殖を特徴とする前記障害は、不必要な血管新生である。言い換えれば、不必要な血管新生を治療する方法を企図する。
【0006】
別の態様により、腫瘍細胞若しくは癌細胞の遊走及び/又は増殖を減弱する方法であって、前記細胞を抗遊走又は抗増殖量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。異常な細胞増殖を減弱する工程では、治療的に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を哺乳動物に投与することによって、哺乳動物内でmTOR/Aktシグナル伝達経路の活性化を阻害する。従って、本発明の実施形態により、細胞、例えば内皮細胞においてシグナル伝達するmTOR/Akt径路を阻害する方法であって、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。本発明の別の実施形態はさらに、対象の癌組織を治療する方法であって、前記癌組織内におけるmTOR/Akt径路媒介作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、mTOR/Akt径路誘発細胞増殖及び遊走を相乗的に阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法を包含する。
さらなる態様では、細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明のさらに別の実施形態は、癌の治療方法、例えば、治療が必要な対象の腫瘍の成長を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を包含する。
【0007】
本発明のさらなる実施形態は、哺乳動物内における成長因子受容体を発現する細胞の異常な増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与することによって治療する方法を提供する。特定の実施形態では、前記成長因子受容体が、血管内皮成長因子受容体(VEGF−R)、上皮成長因子受容体(EGF−R)又はインスリン様成長因子受容体(IGF−R)である。従って、本発明の他の実施形態により、内皮細胞内の成長因子シグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記成長因子がVEGF、EGF又はIGFである。
本発明の実施形態に従い、mTOR阻害薬とμ−オピオイド拮抗薬の相乗的併用剤を投与することによる自己免疫疾患、乾癬、神経変性疾患、免疫細胞内へのCCRS媒介ウイルス侵入、並びに吐き気及び嘔吐の治療方法をも企図する。
本発明の実施形態により、mTOR阻害薬及びμ−オピオイド受容体拮抗薬のみならず、共投与についての使用説明書を伴う医薬併用剤及びパッケージをも提供する。
【0008】
本発明の実施形態の方法で使うmTOR阻害薬は、一般的にmTORによるp70S6キナーゼのセリン389のリン酸化を阻害することによってG1期からS期への細胞周期の進行を遮断することによって細胞の複製を阻害する化合物を含む。既知のmTOR阻害薬としては、ラパマイシン及びラパマイシン誘発体が挙げられる。mTOR活性のいくつかの他の小分子阻害薬も同定されている。本発明の実施形態の併用療法では、1種より多くのmTOR阻害薬の使用も企図する。
一部の実施形態では、μ−オピオイド受容体拮抗薬が末梢性μ−オピオイド受容体拮抗薬である。末梢限定μ−オピオイド受容体拮抗薬は一般的にヘテロ環式アミン化合物であり、化合物のいくつかの異なる分類にも属する。例えば、1つの分類は、モルフィナンの四級誘発体、特にノルオキシモルフォン(noroxymorphone)の四級誘発体である。一実施形態では、ノルオキシモルフォンの四級誘発体は適宜、例えば、N−メチルナルトレキソン(又は単にメチルナルトレキソン)、N−メチルナロキソン、N−メチルナロルフィン、N−ジアリルノルモルフィン、N−アリルレバロルファン、又はN−メチルナルメフェンである。別分類の末梢限定拮抗薬は、N−置換ピペリジンである。一実施形態では、N−ピペリジンがピペリジン−N−アルキルカルボナート、例えばアルビモパンである。本発明の実施形態で価値がありうる他分類の化合物は、ベンゾモルファンの四級誘発体、ノルモルファニン(normorphanin)の四級誘発体並びにモルファニンの三級誘発体、ベンゾモルファン及びノルモルファニンのポリマー複合体である。
本発明の実施形態は、併用療法において、1種より多くのμ−オピオイド受容体拮抗薬の投与をも包含する。拮抗薬の組合せとしては、μ−拮抗薬の組合せ並びにμ−及びκ−拮抗薬の組合せ、例えば、メチルナルトレキソンとアルビモパンの組合せ、又はナロキソンとメチルナルトレキソンの組合せが挙げられる。
本発明の実施形態の方法で使用可能な他のμ−オピオイド受容体拮抗薬として、モルフィナンの三級誘発体、特に、ノルオキシモルフォンの三級誘発体、例えばナロキソンが挙げられる。
添付図面と関連して本明細書で提示する詳細な説明を参照することによって、本発明の実施形態をより良く理解かつ認識できるであろう。
【図面の簡単な説明】
【0009】
【図1】ナルトレキソンとメチルナルトレキソンの化学構造、及びナルトレキソンからメチルナルトレキソンへの変換反応を示す。
【図2】ベバシズマブ、5−FU、メチルナルトレキソン、ベバシズマブとメチルナルトレキソンの組合せ及び5−FUとメチルナルトレキソンの組合せの存在下、ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いたセリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)を実証する免疫ブロットである。
【図3】セリン473(A)及びスレオニン308(B)におけるAktのVEGF誘発リン酸化のメチルナルトレキソンによる阻害の用量関連効果を示すグラフである。
【図4】VEGF誘発内皮細胞増殖の阻害に及ぼすラパマイシンの用量関連効果を示すグラフである。
【図5】VEGF誘発内皮細胞遊走の阻害に及ぼすラパマイシンの用量関連効果を示すグラフである。
【図6】メチルナルトレキソンとラパマイシンの組合せを用いたVEGF誘発内皮細胞増殖の相乗的阻害を示すグラフである。
【図7】メチルナルトレキソンとラパマイシンの組合せを用いたVEGF誘発内皮細胞遊走の相乗的阻害を示すグラフである。
【図8】ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いたセリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)に及ぼすメチルナルトレキソンとテムシロリムスの効果を実証する免疫ブロットである。
【図9】ヒト内皮細胞内における抗ホスホ−セリン473−Akt、抗ホスホ−スレオニン308−Akt及び抗Akt特異性抗体を用いた、(A) mTOR複合体1及びmTOR複合体2のVEGF誘発形成に及ぼすメチルナルトレキソン及びテムシロリムスの効果、並びに(B)セリン473及びスレオニン308におけるAktのVEGF誘発リン酸化(活性化)に及ぼすPI3キナーゼ阻害、Src枯渇及びRictor枯渇の効果を実証する免疫ブロットである。
【図10】(A)メチルナルトレキソンとテムシロリムスの組合せを用いたVEGF誘発内皮細胞増殖の相乗的阻害及び(B)メチルナルトレキソンとテムシロリムスの組合せを用いたVEGF誘発内皮細胞遊走の相乗的阻害を示すグラスを示す。
【図11】メチルナルトレキソンとテムシロリムスの組合せ用いた内皮細胞の(A)増殖及び(B)遊走の相乗的阻害がチロシンホスファターゼ活性によって制御されることを実証するグラフを示す。
【図12】メチルナルトレキソンとmTOR阻害薬の相乗的活性の分子基盤の略図である。
【発明を実施するための形態】
【0010】
本発明の実施形態で明白な原理により、mTOR阻害薬とμ−オピオイド受容体拮抗薬の組合せを利用する医薬併用剤及び方法を提供する。本発明の実施形態の方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬を共投与することによって、細胞の不必要及び望ましくない増殖及び遊走、特に内皮細胞の不必要及び望ましくない増殖及び/又は遊走を特徴とする障害を治療する工程を含む。以下の実施例で説明するように、mTOR阻害薬とμ−オピオイド受容体拮抗薬(例えばメチルナルトレキソン(MNTX))の組合せは、細胞の不必要な増殖及び遊走、例えば、内皮細胞のVEGF誘発増殖及び遊走の低減において予想外の相乗作用をもたらす。
本発明の少なくとも1つの実施形態を説明する前に、本発明は、下記説明で述べ、また実施例によって例示する詳細に本発明を適用することに限定されないことを理解すべきである。このような説明及び実施例は、添付の特許請求の範囲に記載の本発明の範囲を制限する意図ではない。本発明は、他の実施形態が可能であり、或いは種々のやり方で実施又は実行することができる。以下の詳細な説明及び実施例は、好適な薬物としてラパマイシン、テムシロリムス及びメチルナルトレキソンを利用する実施形態を参照することにより本発明を記述しているが、他のmTOR阻害薬及びμ−オピオイド受容体拮抗薬も本発明の原理に従って使うのに適していることを理解すべきである。
さらに、いずれの特許又は特許文献をも含め、本明細書で引用したいずれの参考文献も先行技術を構成することを認めるものではない。特に、当然のことながら、特に断らない限り、本明細書におけるいずれの文献の参照も、これらの文献のいずれかが米国又は他のいずれかの国の当該技術で共通の一般知識の一部を形成するという承認を構成しない。参考文献のいずれの議論もそれらの著者が主張することを述べており、出願人は、本明細書で引用したいずれの文献の正確さ及び適切性をも検証する権利を保留する。
【0011】
この開示全体を通じて、この発明の種々の態様を範囲形式で提示することがある。範囲形式の説明は、単に便利さ及び簡潔さのためであることを理解すべきであり、かつ本発明の範囲に対する確固たる限定とみなすべきでない。従って、当業者には分かるように、ありとあらゆる目的のため、特に書面による明細を提供するという観点において、本明細書で開示する全ての範囲は、そのありとあらゆる可能な部分的範囲及び部分的範囲の組合せ、並びに当該範囲内の全ての整数及び分数の値をも包含する。例として、20%〜40%という範囲は20%〜32.5%及び32.5%〜40%、20%〜27.5%及び27.5%〜40%などの範囲に分解することができ、その全てがこの明細書で明確に列挙されているものと解釈すべきである。リストアップしたいずれの範囲も、少なくとも2等分、3等分、4等分、5等分、10等分などに分解される同一範囲を十分に記述し、かつ有効にするものと容易に認識することができる。非限定例として、本明細書で論じる各範囲は、容易に下部3分の1、中部3分の1及び上部3分の1等に分解することができる。
また、当業者には分かるように、「まで」、「少なくとも」、「より大きい」、「未満」、「より多い」等の全ての言葉は、列挙した数を包含し、上述したように引き続き部分的範囲に分解できる範囲を指す。同様に、本明細書で開示する全ての比率も、より広い比率に含まれる全ての部分的範囲を包含する。これらは、具体的に意図しているものの例にすぎない。さらに、第1の表示数と第2の表示数の「間の範囲」という表現と、「第1の表示数〜第2の表示数の範囲」という表現は、本明細書では相互交換可能に使用される。
さらに、本明細書において「含む(comprising)」、「含む(including)」、「有する(having)」、及びその変形の使用は、その後に列挙する項目及びその等価項目並びに追加項目、例えば他の工程及び/又は成分を包含することを意味する。これらの用語は、「〜から成る」及び「本質的に〜から成る」という用語を包含する。「本質的に〜から成る」の使用は、該組成物又は方法が追加の成分及び/又は工程を含みうることを意味するが、その追加の成分及び/又は工程が、請求した組成物又は方法の基本的及び新規な特徴を実質的に変えない場合に限る。
【0012】
特に定義しない限り、全ての科学用語及び技術用語は、本明細書では通常の用法に従って使用され、本発明が属する技術の当業者が一般的に解釈するのと同じ意味を有する。しかし、本明細書で使用する場合、以下の定義は、本発明を理解するのに熟練実務家を助けるのに役立つであろう。
「対象」は、哺乳動物、例えばヒト、マウス、イヌ、ネコ、ラットを表す。
「アルキル」は、飽和しており、かつ鎖内に1〜約10個の炭素原子、並びにその中に全ての組合せ及び副組合せの鎖を有する直鎖、分岐鎖、又は環式であってよい、一価の脂肪族炭化水素基を表す。典型的アルキル基として、限定するものではないが、メチル、エチル、n−プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、シクロプロピル、シクロブチル、シクロペンチル、及びシクロヘキシルが挙げられる。
「低級アルキル」は、1〜約6個の炭素原子を有するアルキル基を表す。
「アルケニル」は、鎖内に少なくとも1個の炭素−炭素二重結合を含み、かつ鎖内に2〜約10個の炭素原子、並びにその中に全ての組合せ及び副組合せの鎖を有する一価の脂肪族炭化水素基を表す。典型的アルケニル基として、限定するものではないが、ビニル、プロペニル、ブチニル、ペンテニル、ヘキセニル、及びヘプトニルが挙げられる。
「アルキニル」は、少なくとも1個の炭素−炭素三重結合を含み、かつ鎖内に2〜約10個の炭素原子、並びにその中に組合せ及び副組合せの鎖を有する一価の脂肪族炭化水素基を表す。典型的アルキニル基として、限定するものではないが、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、及びヘプチニルが挙げられる。
「アルキレン」は、その中に1〜約6個の炭素原子、並びに全ての組合せ及び副組合せの鎖を有する二価の脂肪族炭化水素基を表す。アルキレン基は、直鎖、分岐鎖、又は環式であってよい。任意に、アルキレン基に沿って、1個以上の酸素、イオウ、又は任意に置換されていてもよい窒素原子(窒素の置換基は前述したアルキル基である)が挿入されていてもよい。
「アルケニレン」は、少なくとも1個の炭素−炭素二重結合を含み、直鎖、分岐鎖、又は環式であってよい二価のアルキレン基を表す。典型的アルケニレン基として、限定するものではないが、エテニレン(CH=CH)及びプロペニレン(CH=CHCH2)が挙げられる。
【0013】
「シクロアルキル」は、その中に約3〜約10個の炭素、並びに全ての組合せ及び副組合せの環を有する飽和単環式又は二環式炭化水素環を表す。シクロアルキル基は任意に1つ以上のシクロアルキル基置換基で置換されていてもよい。典型的シクロアルキル基として、限定するものではないが、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、及びシクロヘプチルが挙げられる。
「アシル」は、アルキル−CO基(アルキルは前述したとおり)を意味する。典型的アシル基として、限定するものではないが、アセチル、プロパノイル、2−メチルプロパノイル、ブタノイル、及びパルミトイルが挙げられる。
「アリール」は、その中に約6〜約10個の炭素、並びに全ての組合せ及び副組合せの環を有する芳香族炭素環式基を表す。アリール基は、任意に1又は2つ以上のアリール基置換基で置換されていてもよい。典型的アリール基として、限定するものではないが、フェニル及びナフチルが挙げられる。
「アリール置換アルキル」は、末端炭素のところで、任意に置換されていてもよいアリール基、好ましくは任意に置換されていてもよいフェニル環によって置換されている直鎖アルキル基、好ましくは低級アルキル基を表す。典型的アリール置換アルキル基として、例えば、フェニルメチル、フェニルエチル、及び3(4−メチルフェニル)プロピルが挙げられる。
「ヘテロ環式」は、その中に約4〜約10個の環員、並びに全ての組合せ及び副組合せの環を含み、1個以上の環員が炭素以外の元素、例えば、窒素、酸素、又はイオウである、単環式又は多環式環系炭素環式基を表す。ヘテロ環式基は、芳香族又は非芳香族であってよい。典型的ヘテロ環式基として、例えば、ピロール及びピペリジン基が挙げられる。
「ハロ」は、フルオロ、クロロ、ブロモ、又はヨードを表す。
【0014】
「共投与」又は「共投与する」とは、2種以上の薬剤をいずれかの投与径路で患者又は対象に投与する併用療法を表すことを意味する。薬剤の共投与は、併用療法又は併用治療と呼ばれることもある。薬剤は、同一の投与製剤又は別々の製剤中にあってよい。活性薬が別々の投与製剤中にある1種より多くの活性薬による併用治療では、活性薬を同時に投与することができ、或いは活性薬をそれぞれ別々に時間差で投与することができる。併用治療の薬剤は、それら両薬剤が体内で有効濃度を達成させるのに十分な様式で与えられる限り、同時又は逐次投与してよい(例えば、一方の薬剤を他方の投与直後に投与するか、或いは偶発的に、例えば、ある時に一方の薬剤を与えて、その後、例えば1週間以内に他方の薬剤を与えてよい)。薬剤を異なる経路で投与してもよく、例えば、一方の薬剤を静脈内投与し、第2の薬剤を筋肉内、静脈内、又は経口投与してよい。
μ−オピオイド受容体拮抗薬に関する用語「末梢性」、又は「末梢限定」又は「末梢作用性」は、中枢神経系の外側にある生理学的な系及び要素に主として作用するμ−オピオイド受容体拮抗薬を指定する。換言すれば、それらは低減した中枢神経系(CNS)活性を示すか又は該活性が実質的にない。例えば、それらはオピオイドの中枢作用を阻害するのに有効な量で血液脳関門を容易には横断できない。すなわち、それらは末梢投与したとき、オピオイドの鎮痛作用を有効には阻害せず、すなわち、それらはオピオイドの鎮痛作用を低減しない。本発明の実施形態で利用する末梢性μ−オピオイド受容体拮抗薬化合物は、CNS内でその薬理活性の約5〜15%未満を示し、約0%(すなわち、無い)のCNS活性が最も適している。末梢性μ−オピオイド受容体拮抗薬の非中枢作用特性は、分子又は化学種の電荷、極性、及び/又は大きさに関連することが多い。例えば、本明細書に記載の末梢作用性四級アミンμ−オピオイド受容体拮抗薬は正の電荷をもつが、中枢作用性三級アミンμ−オピオイド受容体拮抗薬は中性分子である。
【0015】
本明細書で使用する場合、用語「mTOR阻害薬」は、mTORの活性又は発現を調節することによって、G1期〜S期の細胞周期の進行を遮断することで細胞の複製を阻害する化合物若しくはリガンド、又はその医薬的に許容しうる塩を意味する。この用語は、mTOR活性を阻害する、中性三環式化合物ラパマイシン(シロリムス)及び他のラパマイシン化合物(例えば、ラパマイシン誘発体、ラパマイシン類似体など)、及び他のマクロライド化合物並びに他の構造的に異なる小分子、例えば縮合二環式化合物を包含する。これらには、ラパマイシンに構造的に類似する化合物、例えば、治療利益を高めるために修飾された同様の大環状構造を有する化合物が含まれる。ラパマイシン誘発体などのmTOR阻害薬については本明細書で後述する。
本明細書で使用する用語「治療する」又は「治療」は、医学的又は生理学的状態のいずれの手段の制御、例えば、該状態の予防、ケア、軽減、該状態又は該状態の症状の減弱、緩和、低減、及び該状態の進行の阻害又は静止をも包含する。
本明細書で使用する場合、用語「副作用」は、薬物の目的又は所望の作用以外の作用を表すことを意味する。副作用は有益又は望ましくない、すなわち有害でありうる。本ケースでは、望ましくない作用はmTOR阻害薬の投与後に起こることが多い。このような副作用として、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。
【0016】
用語「溶媒和物」は、さらに、非共有結合性の分子内力で化学量論又は非化学量論量の溶媒が結合している、本明細書で提供する化合物又はその塩を表す。溶媒が水の場合は、溶媒和物は水和物である。
本明細書で使用する場合、用語「効力」は、癌を患っている対象の癌を治療する抗癌薬の性能又は能力を表す。効力を所定強度の特有の効果を生じさせるのに必要な薬物の用量として表すこともある。
特定の実施形態では、細胞の増殖及び遊走に関連する用語「不必要な」、例えば「不必要な増殖」又は「不必要な遊走」は、正常な細胞の代謝回転、代謝、成長又は繁殖から外れている、細胞の「異常又は病的又は調節不全的又は望ましくない又は不適切な」増殖、分割、成長又は遊走を表し、一般的に、正常に機能する細胞で典型的に起こるより急速又は有意に大きい程度で起こり、正常な機能を果たさない。不必要な増殖及び不必要な遊走は、本質的に過剰増殖性である障害で顕著であり、該障害としては、限定するものではないが、癌、例えばメラノーマ、肺癌、乳癌、膵臓癌、前立腺癌、結腸癌又は卵巣癌、乾癬、関節リウマチ、角質増殖(perkeratosis)による表皮溶解、レストラトシス(restratosis)、再狭窄、子宮内膜症及び異常な創傷治癒が挙げられる。
【0017】
光学活性化合物について述べる場合、プレフィックスR及びSを用いて、分子のキラル中心の回りの分子の絶対配置を示す。記号(+)及び(−)を用いて、化合物の旋光性、すなわち、光学活性化合物が偏光面を回転させる方向を表す。(−)プレフィックスは、化合物が左旋性、すなわち、化合物が偏光面を左又は反時計回りに回転させることを示す。(+)プレフィックスは、化合物が右旋性、すなわち、化合物が偏光面を右又は時計回りに回転させることを示す。しかし、旋光性の記号、(+)及び(−)は、分子の絶対配置、R及びSとは無関係である。
本発明の方法の以下の説明では、特に断らない限り、プロセス工程を室温及び大気圧で行なう。
一態様では、本発明の実施形態は、異常又は望ましくない細胞プロセス、例えば、細胞の不必要な遊走及び/又は増殖、特に内皮細胞の不必要な遊走及び/又は増殖を減弱する方法に関する。方法は、1種以上のmTOR阻害薬と1種以上のμ−オピオイド受容体拮抗薬を相乗的有効量で、例えば、患者若しくは対象の組織又は器官の細胞、特に患者の組織又は器官の内皮細胞に投与して、細胞の遊走及び/又は増殖、例えば、内皮細胞の遊走及び/又は増殖を阻害する工程を含む。
μ−オピオイド受容体拮抗薬は、オピオイド、内因性又は外因性、及び成長因子、例えばVEGF、PDGF、S1P等によって誘発される不必要な増殖及び遊走を阻害することが分かった。特に末梢性μ−オピオイド受容体拮抗薬は、同時係属米国特許出願第11/908,058号に開示されているように、内皮細胞のオピオイド及び成長因子誘発増殖及び遊走の阻害において実質的な効力を示した。末梢性μ−オピオイド受容体拮抗薬メチルナルトレキソン(MNTX)は、濃度依存様式で、オピオイド及び成長因子誘発の両方の増殖及び遊走を阻害する。さらに、μ−オピオイド受容体拮抗薬、特に末梢性μ−オピオイド受容体拮抗薬MNTXは、Aktの阻害によって、作用物質誘発内皮細胞(EC)増殖及び遊走を阻害することを発見した。作用物質は、オピオイド、外因性及び/又は内因性、血管新生因子(例えば、VEGF)、並びに他の増殖及び/又は遊走刺激因子(例えば、PDGF、S1P、S1P3受容体、RhoA等)でありうる。
μ−オピオイド受容体拮抗薬は、標的細胞を感染させるウイルスの能力を阻害することも分かった。特に、末梢性μ−オピオイド受容体拮抗薬は、参照により本明細書に援用される同時係属米国特許出願第10/163,482号に開示されているように、HIVのウイルス侵入及びこのウイルスの細胞表面受容体であるCCR5の発現の阻害において実質的な効力を示した。
【0018】
本発明の実施形態は、mTOR阻害薬と共投与するμ−オピオイド受容体拮抗薬が、癌細胞に及ぼす抗増殖作用を有意に高め、ひいては治療作用の増加をもたらすこと、例えば、末梢性μ−オピオイド受容体拮抗薬を特定腫瘍に投与するとmTOR阻害薬への腫瘍の反応を増強できることを実証する。以下の実施例で示すように、薬物を単独で使用する治療計画に比べて低濃度のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を利用する上記共投与併用剤では、有意に高い抗増殖作用及び抗遊走作用が得られる。例えば、本発明の実施形態に従うμ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、mTOR阻害薬の用量を減らすか又は効力若しくは効率又は両方を高めうる。さらに、本発明の実施形態に従うμ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、予防的価値をも有する。
mTOR阻害薬に伴う有害な副作用が、mTOR阻害薬のみをより多い用量で使用する場合に一般的に観察される当該副作用に比べて相当に低減した療法をもたらす可能性もある。これらの副作用としては、発疹、無力症、粘膜炎、食欲不振、末梢性浮腫、高トリグリセリド血症、高血圧症、高コレステロール血症、高クレアチン血症、便秘、腹痛、下痢、頭痛、発熱、尿路感染、貧血、吐き気、関節痛、疼痛、及び血小板減少が挙げられる。これらの副作用の少なくとも一部は用量依存性である。本発明の実施形態を利用すれば、より低い用量の可能性が達成される。
さらに、mTOR阻害薬療法に伴う高い費用が、mTOR阻害薬のみをより多い用量で使用する場合に一般的に観察される当該費用に比べて相当に低減した療法をもたらす可能性がある。その上、μ−オピオイド受容体拮抗薬とmTOR阻害薬の共投与は、mTORのみを用いて一般的に観察される場合に比し、mTOR阻害薬の効率及び効力を相当高めることができる。
【0019】
本発明の実施形態で明白な原理を具体化する方法は、mTOR阻害薬とμ−オピオイド受容体拮抗薬(限定するものではないが、末梢限定拮抗薬であるものが挙げられる)を用いて、細胞の不必要な増殖及び遊走、特に内皮細胞の増殖及び遊走を減弱、例えば、阻害又は低減する工程を含む。本発明の一態様により、内皮細胞の不必要な遊走又は増殖を特徴とする障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む治療方法を提供する。本治療は、遊走と増殖の一方又は両方を阻害しうる。さらなる実施形態では、不必要な遊走又は増殖は、血管内皮細胞の遊走又は増殖であり、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を用いて治療される。別の実施形態では、血管内皮細胞の不必要な遊走又は増殖は、不必要な血管新生である。従って、不必要な血管新生を治療する方法を企図する。
【0020】
さらに別の態様により、腫瘍又は癌の細胞の遊走及び/又は増殖を減弱する方法であって、細胞を抗遊走又は抗増殖量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法を提供する。細胞増殖の減弱においては、哺乳動物内でmTOR/Aktシグナル伝達経路の活性化亢進を示す異常な細胞増殖を治療方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。従って、内皮細胞内でシグナル伝達するmTOR/Akt径路を阻害する方法をも提供する。方法は、細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記対象に、前記癌組織内でmTOR/Akt径路媒介作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、mTOR/Akt径路誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。
【0021】
さらに別の実施形態では、哺乳動物内で成長因子受容体を発現する細胞の異常な増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。特定の実施形態では、前記成長因子受容体が血管内皮成長因子受容体(VEGF−R)、上皮成長因子受容体(EGF−R)又はインスリン様成長因子受容体(IGF−R)である。従って、内皮細胞内で成長因子シグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記成長因子がVEGF、EGF又はIGFである。方法は、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記癌組織内で成長因子誘発作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、成長因子誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。癌組織内におけるこれらの作用がVEGF誘発、EGF誘発又はIGF誘発作用である場合を特に企図する。
【0022】
なおさらなる実施形態では、哺乳動物内でホルモン受容体を発現する細胞の異常な細胞増殖を治療する方法であって、前記哺乳動物に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法を提供する。特定の実施形態では、ホルモン受容体がエストロゲン受容体(ER)、プロゲステロン受容体(PR)又はアンドロゲン受容体(AR)である。従って、内皮細胞内でホルモンシグナル伝達を阻害する方法をも提供する。特有の実施形態では、前記ホルモンがエストロゲン、プロゲステロン又はアンドロゲンである。方法は、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む。対象の癌組織を治療する方法であって、前記癌組織内でホルモン誘発作用を阻害するのに十分な量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法、並びに組織又は細胞集団を、ホルモン誘発増殖及び遊走を阻害するのに有効な条件下の量の少なくとも1種のmTOR阻害薬及び少なくとも1種のμ−オピオイド受容体拮抗薬を含む組成物又は併用剤と接触させる工程を含む方法をも提供する。癌組織内におけるこれらの作用がエストロゲン誘発、プロゲステロン誘発又はアンドロゲン誘発作用である場合を特に企図する。
【0023】
なおさらなる態様では、細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明のさらに別の実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、癌の治療が必要な対象の癌を治療する方法である。本発明のさらなる実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、腫瘍増殖の阻害が必要な対象の腫瘍の増殖を阻害する方法を提供する。
さらなる態様では、患者の自己免疫疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法を提供する。本発明の自己免疫疾患としては、限定するものではないが、アレルギー性脳脊髄炎、インスリン依存性糖尿病、ループス、関節リウマチ、多発性硬化症、皮膚筋炎、グレーブス病及びアジュバント関節炎が挙げられる。
本発明のさらに別の実施形態は、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、患者の乾癬を治療する方法を提供する。特定の実施形態では、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を局所に適用する。
本発明のさらなる実施形態は、神経変性疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬の共投与を含む方法を提供する。特定の実施形態では、神経変性疾患がパーキンソン病又は多発性硬化症である。
本発明のなおさらなる実施形態は、免疫細胞へのCCR5媒介ウイルス侵入を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬の共投与を含む方法である。特に免疫細胞へのCCR5媒介HIV侵入の阻害を企図する。さらに、HIV/AIDSに苦しむ対象に相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む、HIV/AIDSを治療する方法を提供する。
別の態様では、mTOR阻害薬を用いた癌の治療によって誘発される吐き気及び嘔吐は、末梢性μ−オピオイド受容体拮抗薬メチルナルトレキソン等のμ−オピオイド受容体拮抗薬を共投与することによって軽減されうる。
【0024】
上述したように、mTOR阻害薬には、mTORの活性又は発現の調節を通じてG1期〜S期の細胞周期の進行を遮断することによって、細胞の複製を阻害する、化合物若しくはリガンド、又はその医薬的に許容しうる塩が包含される。現在利用可能であるか又は開発中であるmTOR阻害薬としては、テムシロリムス(ToriselTM;Wyeth)、RAD001(エベロリムス;Novartis)、MK−8669(デフォロリムス;Merck & Ariad pharma)、TOP216(Toptarget A/S)、OSI−027(OSI Pharma)、TAFA93(Isotechnika)、nab−ラパマイシン(APP Phama)及びタクロリムス(FK506; Astellas)が挙げられる。
mTOR阻害薬には、ラパマイシン及び関連化合物が包含される。ラパマイシンはストレプトマイセス(Streptomyces)によって産生されるマクロライドである。ラパマイシンは強力な免疫抑制薬であり、移植された臓器の拒絶反応を防止するために臨床的に使用される。ラパマイシン及び関連化合物は、抗癌治療薬としても現在開発中である。本発明の実施形態で有用なラパマイシンは、ラパマイシン核の誘導体として化学的又は生物学的に修飾されているが、免疫抑制又は抗癌特性を保持したままである化合物を包含する。従って、ラパマイシンには、ラパマイシン自体、並びにラパマイシンのエステル、エーテル、カルボナート、オキシム、ヒドラゾン、 及びヒドロキシルアミン、並びにラパマイシン核上の官能基を例えば還元又は酸化によって修飾したラパマイシンが含まれる。
詳細には、ラパマイシンの構造を以下に示す式(A)として与える。
【0025】
【化1】
【0026】
上で開示した現在使用又は開発中の多くのラパマイシン誘導体は、C−40位に置換基がある基本的なラパマイシン構造を有する。位置40の置換基をRとして表すと、以下の置換基と化合物が対応する:R=−OP(O)(Me)2、AP23573(国際特許公開第WO98/02441号及び第WO2001/14387号);R=−OC(O)C(CH3)(CH2OH)、テムシロリムス(米国特許第5,362,718号);R=−OCH2CH2OH、エベロリムス(米国特許第5,665,772号);R=−OCH2CH2OEt、バイオリムス;R=−テトラゾール、ABT−578((国際特許公開第WO 99/15530号)。全ての特許及び出願は、参照により本明細書に援用される。
多くの他のラパマイシン誘導体は、C−40及び/又はC−16及び/又はC−32位に置換を含む。ラパマイシンのエステル及びエーテルは、全て参照により本明細書に援用される下記特許に記載されている:アルキルエステル(米国特許第4,316,885号);アミノアルキルエステル(米国特許第4,650,803号);フッ素化エステル(米国特許第5,100,883号);アミドエステル(米国特許第5,118,677号);カルバミン酸エステル(米国特許第5,118,678号;第5,411,967号;第5,434,260号;第5,480,988号;第5,480,989号;第5,489,680号);シリルエステル(米国特許第5,120,842号);アミノジエステル(米国特許第5,162,333号);スルホン酸エステル及び硫酸エステル(米国特許第5,177,203号);エステル(米国特許第5,221,670号);アルコキシエステル(米国特許第5,233,036号);O−アリール、−アルキル、−アルケニル、及び−アルキニルエーテル(米国特許第5,258,389号);炭酸エステル(米国特許第5,260,300号);アリールカルボニル及びアルコキシカルボニルカルバミン酸エステル(米国特許第5,262,423号);カルバミン酸エステル(米国特許第5,302,584号);ヒドロキシエステル(米国特許第5,362,718号);ヒンダードエステル(米国特許第5,385,908号);ヘテロ環式エステル(米国特許第5,385,909号);gem−二置換エステル(米国特許第5,385,910号);アミノアルカン酸エステル(米国特許第5,389,639号);ホスホリルカルバミン酸エステル(米国特許第5,391,730号);アミジノカルバミン酸エステル(米国特許第5,463,048号);ヒンダードN−オキシドエステル(米国特許第5,491,231号);ビオチンエステル(米国特許第5,504,091号);O−アルキルエーテル(米国特許第5,665,772号);及びラパマイシンPEGエステル(米国特許第5,780,462号);32−エステル及びエーテル(米国特許第5,256,790号)。これらのエステル及びエーテルの調製は、上掲特許に開示されている。全ての特許及び出願は、参照により本明細書に援用される。
【0027】
米国特許第5,373,014号、第5,378,836号、第5,023,264号、及び第5,563,145号に開示されているように、ラパマイシンのオキシム、ヒドラゾン、及びヒドロキシルアミンも包含される。これらのオキシム、ヒドラゾン、及びヒドロキシルアミンのの調製は、上掲特許に開示されている。40−オキソラパマイシンの調製は米国特許第5,023,263号に開示されてる。全ての特許は、参照により本明細書に援用される。
mTORの他の小分子阻害薬として、縮合二環状化合物(国際特許公開第WO2007/61737号、第WO2007/87395号及び第WO2007/64993号)、ヘテロ芳香族アミン(国際特許公開第WO 2001/19828号)、ピロロピリミジン化合物(国際特許公開第WO2005/47289号)、ジフェニル−ジヒドロ−インドール−2−オン誘導体(国際特許公開第WO2005/97107号)、及びトリメチル−ドデカ−トリエン誘導体(米国特許第特許公開第2007/037887号)が挙げられる。これらの全ての特許は、参照により本明細書に援用される。
【0028】
本発明の実施形態のμ−オピオイド受容体拮抗薬は、中枢作用性及び末梢作用性の両μ−オピオイド受容体拮抗薬を包含する。しかし、特定価値の当該拮抗薬が好適には末梢限定μ−オピオイド受容体拮抗薬である場合を企図する。
μ−オピオイド受容体拮抗薬は、その拮抗特性を維持しながら構造が変化しうる分類の化合物を形成する。これらの化合物として、三級及び四級モルフィナン、特にノルオキシモルフォン誘導体、N−置換ピペリジン、特に、ピペリジン−N−アルキルカルボキシラート、並びに三級及び四級ベンゾモルファン、並びに三級及び四級ノルモルフィナン誘導体、特に6−カルボキシ−ノルモルフィナン誘導体が挙げられる。三級化合物拮抗薬は、かなり脂溶性であり、血液脳関門を容易に横断する。血液脳関門を横断し、中枢的(及び末梢的)に活性なμ−オピオイド受容体拮抗薬の例として、例えば、ナロキソン(Baxter Pharmaceutical Products, Inc.から商業的に入手可能)、及びナルメフェン(例えば、DuPont Pharmaから入手可能)が挙げられる。他方、末梢限定拮抗薬は、典型的に電荷をもち、極性、及び/又は高分子量であり、それぞれが拮抗薬の血液脳関門の横断を妨害する。メチルナルトレキソンは、三級μ−オピオイド受容体拮抗薬ナルトレキソンの四級誘導体である。ナルトレキソンにメチル基を付加すると、さらに極性が高く、脂溶性が低い化合物を形成する。従って、メチルナルトレキソンは血液脳関門を横断せず、末梢に位置する受容体によって典型的に媒介される望ましくない副作用を遮断する可能性を有する。
本発明の実施形態で使う末梢性μ−オピオイド受容体拮抗薬は、四級モルフィナン誘導体である化合物、特に、下記式(I)
【0029】
【化2】
【0030】
の四級ノルオキシモルフォン、その単一のエナンチオマー、エナンチオマー混合物、個々のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物、若しくはプロドラッグであってよく、式中、Rは、アルキル、アルケニル、アルキニル、アリール、シクロアルキル置換アルキル、又はアリール置換アルキルであり、X−は、アニオン、特にクロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである。式(I)のノルオキシモルフォン誘導体は、例えば、参照により本明細書に援用される米国特許第4,176,186号の手順に従って調製され;全て参照により本明細書に援用される米国特許第4,719,215号;第4,861,781号;第5,102,887号;第5,972,954号;及び第6,274,591号;米国特許出願第2002/0028825号及び第2003/0022909号;並びにPCT公開第WO99/22737号及び第WO98/25613をも参照されたい。
【0031】
特に有益な式(I)の化合物は、式中、Rが、下記式(II)に示すように、シクロプロピルメチル
【0032】
【化3】
【0033】
である、N−メチルナルトレキソン(又は単にメチルナルトレキソン)、その単一のエナンチオマー、エナンチオマー混合物、個々のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物、若しくはプロドラッグであり、式中、X−は上述したとおりである。メチルナルトレキソンは、μ−オピオイド受容体拮抗薬ナルトレキソンの四級誘導体である。メチルナルトレキソンは塩(例えば、N−メチルナルトレキソンブロミド)として存在するので、本明細書で使用する場合、用語「メチルナルトレキソン」又は「MNTX」は、該塩を包含する。従って、「メチルナルトレキソン」又は「MNTX」としては具体的に、限定するものではないが、メチルナルトレキソンの臭化水素酸塩、塩酸塩、ヨウ化水素酸塩、炭酸塩、及びメチル硫酸塩が挙げられる。文献でMNTXの臭化水素酸塩として使われる名称として、例えば、メチルナルトレキソンブロミド;N−メチルナルトレキソンブロミド;ナルトレキソンメトブロミド;ナルトレキソンメチルブロミド;SC−37359;MRZ−2663−BR;及びN−シクロプロピルメチルノルオキシ−モルフィン−メトブロミドが挙げられる。従って、本明細書では、用語「メチルナルトレキソン」はメチルナルトレキソンのいずれの適切な形態、例えば、N−メチルナルトレキソン又はその医薬的に許容しうる塩、そのいずれのプロドラッグ、そのエナンチオマー、そのエピマー(エピマーについては後述する)をも意味するものと解釈すべきである。
【0034】
式(I)及び(II)の化合物、例えば、メチルナルトレキソンは、キラル中心を有しうるので、当該キラル中心上の置換基の配置によって立体化学的異性体として存在することがある。このような立体化学的異性体、例えば、エナンチオマー、ジアステレオマーは、本発明の実施形態で使うために企図される化合物の範囲内である。本発明の組成物及び方法において、使用する化合物は個々の立体異性体、及び立体異性体の混合物、例えば、エナンチオマーの混合物、ジアステレオマーの混合物でありうる。一定の態様では、実質的に純粋な立体異性体である化合物を利用する方法を提供する。全ての互変異性体も本発明の組成物に包含されるものとする。
例えば、メチルナルトレキソンのR及びS配置が知られており、一部の実施形態では、製剤及び方法において、メチルナルトレキソンの単離されたR−−N異性体を利用しうる。本明細書で使用する場合、メチルナルトレキソンの「R−−N異性体」という名称は、窒素に関して(R)配置の該化合物を表す。単離された異性体化合物としては、限定するものではないが、参照により本明細書に援用される米国特許出願第11/441,395号、及び特許協力条約公開出願WO2006/127899に記載されたR−−N異性体メチルナルトレキソン化合物が挙げられる。一部の実施形態では、活性化合物がR−−N異性体メチルナルトレキソン、又はその塩である。米国特許出願第11/441,395号に記載されたメチルナルトレキソンのR−−N異性体はオピオイド拮抗薬である。
【0035】
一部の実施形態では、製剤及び方法でメチルナルトレキソンの単離されたS−−N異性体を利用しうる。本明細書で使用する場合、メチルナルトレキソンの「S−−N異性体」という名称は、窒素に関して(S)配置の該化合物を表す。単離された異性体化合物としては、限定するものではないが、参照により本明細書に援用される米国特許出願第11/441,452号、及び特許協力条約公開出願WO2006/127898に記載されたメチルナルトレキソン化合物のS−−N異性体が挙げられる。一部の実施形態では、活性化合物がS−−N異性体メチルナルトレキソン、又はその塩である。米国特許出願第11/441,452号に記載されたメチルナルトレキソンのS−−N異性体はオピオイド作動薬である。
一定の実施形態では、本明細書で述べる製剤又は薬用量調製で利用するメチルナルトレキソンは、全体的なオピオイド拮抗作用を有することを特徴とする立体異性体の混合物である。例えば、メチルナルトレキソンは、混合物自体が拮抗薬として作用し、本明細書に記載の使用方法でオピオイド拮抗薬として有用であるような、R−−N及びS−−Nメチルナルトレキソンの混合物でありうる。一定の実施形態では、実質的にS−−NメチルナルトレキソンのないR−−Nメチルナルトレキソンを使用する。
本発明の一定の実施形態では、少なくとも約99.6%、99.7%、99.8%、99.85%、99.9%、又は99.95%のメチルナルトレキソンが窒素に関して(R)配置である。サンプル中に存在する(R)−−N−異性体の量を当該同一サンプル中に存在する(S)−−N−異性体の量と比較して決定する方法は、全体が参照により本明細書に援用されるWO2006/127899に詳述されている。他の実施形態では、メチルナルトレキソンが0.15%、0.10%、それ未満の(S)−−N−異性体を含む。
【0036】
当業者には当然のことながら、製剤、薬用量調製、又は方法において利用するメチルナルトレキソンの量に言及している場合、当該量は、メチルナルトレキソン(若しくはその塩)の総量を表すか、又は特定目的(例えば、オピオイド拮抗作用)のためには、メチルナルトレキソンの他の形態も存在するにせよ、メチルナルトレキソンの関連性のある活性形態の量を表すことがある。さらに、本明細書で指定する場合、メチルナルトレキソンの特定形態(例えば、N−メチルナルトレキソンブロミド)について薬用量又は量を定義することがある。メチルナルトレキソンの異なる形態又は塩を使用する場合、当業者は、該薬用量又は量を、等価量の活性メチルナルトレキソンがもたらす用量又は量に調整できることを認めるであろう。
メチルナルトレキソンは、例えばMallinckrodt Pharmaceuticals, St. Louis, Mo.から商業的に入手可能である。メチルナルトレキソンは、水に自由に溶ける白色の結晶性粉末として、典型的に臭化水素酸塩として提供される。提供されたままの化合物は、逆相HPLCで99.4%純粋であり、同法によって0.011%未満の非四級化ナルトレキソンを含む。例えば、約5mg/mLの濃度の無菌溶液として、メチルナルトレキソンを調製することができる。
【0037】
他の末梢性μ−オピオイド受容体拮抗薬として、N−置換ピペリジン、特に下記式(III)で表されるピペリジン−N−アルキルカルボキシラートが挙げられる。
【0038】
【化4】
【0039】
式中、R1は、水素又はアルキルであり;R2は、水素、アルキル、又はアルケニルであり;R3は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R4は、水素、アルキル、又はアルケニルであり;Aは、OR5又はNR6R7であり;このときR5は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R6は、水素又はアルキルであり;R7は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル又はアリール−置換アルキル、又はアルキレン置換Bであるか或いはR6及びR7が結合している窒素原子と一緒に、R6とR7が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Bは、下記基であり、
【0040】
【化5】
【0041】
式中、R8は、水素又はアルキルであり;R9は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル又はアリール置換アルキルであるか或いはR8及びR9が結合している窒素原子と一緒に、R8とR9が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Wは、OR10、NR11R12、又はOEであり;このときR10は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルケニル、又はアリール置換アルキルであり;R11は、水素又はアルキルであり;R12は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、アリール置換アルキル、又はアルキレン置換C(=O)Yであるか或いはR11及びR12が結合している窒素原子と一緒に、R11とR12が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;Eは、下記基
【0042】
【化6】
【0043】
アルキレン置換(C=O)D、又は−R13OC(=O)R14であり;このときR13は、アルキル置換アルキレンであり;R14は、アルキルであり;Dは、OR15又はNR16R17であり;このときR15は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R16は、水素、アルキル、アルケニル、アリール、アリール置換アルキル、シクロアルキル、シクロアルケニル、シクロアルキル−置換アルキル、又はシクロアルケニル−置換アルキルであり;R17は、水素又はアルキルであるか或いはR16及びR17が結合している窒素原子と一緒に、R16とR17が、ピロール又はピペリジンから成る群より選択されるヘテロ環式環を形成し;
Yは、OR18又はNR19R20であり;このときR18は、水素、アルキル、アルケニル、シクロアルキル、シクロアルケニル、シクロアルキル−置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであり;R19は、水素又はアルキルであり;R20は、水素、アルキル、アルケニル、アリール、シクロアルキル、シクロアルケニル、シクロアルキル置換アルキル、シクロアルケニル置換アルキル、又はアリール置換アルキルであるか或いはR19及びR20が結合している窒素原子と一緒に、R19とR20が、ピロール及びピペリジンから選択されるヘテロ環式環を形成し;R21は、水素又はアルキルであり;nは0〜4である。
【0044】
価値がありうる特定のピペリジン−N−アルキルカルボナートは、N−アルキルアミノ−3,4,4置換ピペリジン、例えば下記式(IV)で表されるアルビモパンである。
【0045】
【化7】
【0046】
N−置換ピペリジンは、米国特許第5,270,328号;第6,451,806号;第6,469,030号(全て参照により本明細書に援用される)に開示されているように調製される。アルビモパンは、Adolor Corp.、Exton、PAから商業的に入手可能である。該化合物は、適度な高分子量、双性イオン形態を有し、かつ血液脳関門の透過を防止する極性を有する。
さらに他の末梢性μ−オピオイド受容体拮抗薬化合物として、四級ベンゾモルファン化合物が挙げられる。本発明の実施形態で利用しうる四級ベンゾモルファン化合物は、下記式(V)を有する。
【0047】
【化8】
【0048】
式中、R1は、水素、アシル、又はアセトキシであり;R2は、アルキル又はアルケニルであり;Rは、アルキル、アルケニル、又はアルキニルであり、かつX−は、アニオン、特にクロリド、ブロミド、ヨージド、又はメチルスルファートアニオンである。
本発明の実施形態で利用しうるベンゾモルファン化合物の特有の四級誘導体としては、式(V)の下記化合物が挙げられる:2’−ヒドロキシ−5,9−ジメチル−2,2−ジアリル−6,7−ベンゾモルファニウム−ブロミド;2’−ヒドロキシ−5,9−ジメチル−2−n−プロピル−2−アリル−6,7−ベンゾモルファニウム−ブロミド;2’−ヒドロキシ−5,9−ジメチル−2−n−プロピル−2−プロパルギル−6,7−ベンゾモルファニウム−ブロミド;及び2’−アセトキシ−5,9−ジメチル−2−n−プロピル−2−アリル−6,7−ベンゾモルファニウム−ブロミド。
本発明の実施形態で利用しうる他の四級ベンゾモルファン化合物は、例えば、その開示全体が参照により本明細書に援用される米国特許第3,723,440号に記載されている。
他の末梢性オピオイド拮抗薬として、全体が参照により本明細書に援用され、かつ下記式(VI)を有する化合物を含む、発明の名称が「6−カルボキシ−ノルモルフィナン誘導体、その合成及び使用」である米国特許第7,501,434号に記載されている6−カルボキシ−ノルモルフィナン誘導体、特にN−メチル−C−ノルモルフィナン誘導体が挙げられる。
【0049】
【化9】
【0050】
末梢性オピオイド拮抗薬として、参照により本明細書に援用される米国特許出願第11/332,964号に記載されているオピオイド拮抗薬のポリマー複合体も挙げられる。特有のポリマー複合体として、PEG化ナロキソン及びナルトレキソンが挙げられる。
本発明の実施形態は、1種より多くのμ−オピオイド受容体拮抗薬(μ−オピオイド受容体拮抗薬の組合せ、μ及びκ拮抗薬の組合せ、例えば、メチルナルトレキソンとアルビモパンの組合せが挙げられる)の投与をも包含する。
本発明の実施形態で利用する化合物はプロドラッグ形態で存在しうる。本明細書で使用する場合、「プロドラッグ」は、哺乳動物対象に該プロドラッグを投与すると、本発明の実施形態においてin vivoで利用する活性薬又は他の化合物にin vivoで代謝される活性親薬又は化合物を放出するいずれの共有結合した担体をも含むものとする。プロドラッグは、医薬品のいくつかの望ましい品質(例えば、溶解性、生物学的利用能、製造品質など)を高めることが分かっているので、本発明の一部の実施形態で利用する化合物は、所望により、プロドラッグ形態で送達されうる。従って、本発明の実施形態は、プロドラッグを送達する方法を企図する。本発明の実施形態で利用する化合物のプロドラッグは、化合物中に存在する官能基を修飾(該修飾が日常的操作又はin vivoで開裂して親化合物になるように)することによって調製される。
従って、プロドラッグとしては、例えば、ヒドロキシ、アミノ、又はカルボキシ基がいずれかの基に結合しており、該プロドラッグを哺乳動物対象に投与すると、開裂してそれぞれ自由なヒドロキシル、自由なアミノ、又はカルボン酸を形成する、本明細書に記載の化合物が挙げられる。他の例として、限定するものではないが、アルコール及びアミン官能基のアセタート、ホルマート、及びベンゾアート誘導体;並びにアルキルエステル、カルボン酸エステル、アリールエステル、及びアルキルアリールエステル、例えばメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、シクロプロピル、フェニル、ベンジルフェネチルエステル等が挙げられる。
【0051】
上述したように、本発明の実施形態で利用する化合物は、当業者に周知のいくつかの方法で調製されうる。本発明の実施形態で述べる全ての調製は、ミリグラム、グラム、マルチグラム、キログラム、マルチキログラム、又は市販医薬品スケールを含めたいずれのスケールでも実施することを企図している。
メチルナルトレキソンについて上述したように、本発明の実施形態で利用する化合物は、1つ以上の非対称に置換された炭素原子を含むことがあり、光学活性又はラセミ形で単離されうる。従って、特有の立体化学又は異性形が具体的に指定されていない限り、全てのキラル、ジアステレオマー、ラセミ形、エピマー形、及び全ての互変異性体形を意図している。このような光学活性形を調製及び単離する方法は技術上周知である。例えば、限定するものではないが、ラセミ形の分割、順相、逆相、及びキラルクロマトグラフィー、優先晶出法(preferential salt formation)、再結晶法などの標準的技術によって、或いはキラル出発原料からのキラル合成又は標的キラル中心の計画的合成によって、立体異性体の混合物を分離しうる。
【0052】
一般的に言えば、医学的に許容しうるいずれの投与様式、例えば、臨床的に容認できない副作用を引き起こすことなく、活性化合物の有効レベルを生じさせるいずれの投与様式を用いても本発明の実施形態の方法を実施することができる。このような投与様式としては、経口、直腸、局所(散剤、軟膏、液滴、経皮パッチ、又はイオン注入装置によってのうに)、経皮、舌下、筋肉内、注入、静脈内、肺、筋肉内、腔内、エアロゾルとして、耳(例えば、点耳薬によって)、鼻腔内、吸入、眼内、又は皮下投与が挙げられる。
さらに、腸溶コーティング錠剤又はカプセル剤として本発明の実施形態の化合物を投与することができる。一部の実施形態では、遅速注入法又は持続放出若しくは制御放出法によるか又は凍結乾燥散剤としてμ−オピオイド受容体拮抗薬を投与する。
さらに、本発明の実施形態の化合物を局所投与することができる。局所投与用製剤としては、軟膏、ローション、クリーム、ゲル、液滴、座剤、スプレー、液体及び散剤が挙げられる。通常の医薬担体、水、粉末又は油性基剤、増粘剤などが必要であるか又は望ましいことがある。
投与するとき、本発明の実施形態の化合物は、医薬的に許容しうる量及び医薬的に許容しうる組成物若しくは製剤で与えられる。該製剤は、日常的に塩、緩衝剤、保存剤、及び必要に応じて他の治療成分を含有しうる。
医薬中で使用する場合、本発明の実施形態の化合物の医薬的に許容しうる塩を使用しうるが、便宜上医薬的に許容しえない塩を用いてその医薬的に許容しうる塩を調製することがあり、このような医薬的に許容しえない塩は本発明の範囲から除外されない。このような薬理学的及び医薬的に許容しうる塩として、限定するものではないが、以下の酸から調製される当該塩が挙げられる:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、p−トルエンスルホン酸、酒石酸、クエン酸、メタンスルホン酸、ギ酸、コハク酸、ナフタレン−2−スルホン酸、パモ酸、3−ヒドロキシ−2−ナフタレンカルボン酸、及びベンゼンスルホン酸。
【0053】
本発明の実施形態の製剤に緩衝剤及び保存剤を含めてもよい。適切な緩衝剤として、限定するものではないが、酢酸とその塩(1〜2%w/v);クエン酸とその塩(1〜3%w/v);ホウ酸とその塩(0.5〜2.5%w/v);及びリン酸とその塩(0.8〜2%w/v)が挙げられる。適切な保存剤として、限定するものではないが、塩化ベンザルコニウム(0.003〜0.03%w/v);クロロブタノール(0.3〜0.9%w/v);パラベン(0.01〜0.25%w/v);及びチメロサール(0.004〜0.02%w/v)が挙げられる。
投与を容易にするため、本発明の実施形態の医薬組成物は、1種以上の医薬的に許容しうる賦形剤、例えば潤沢剤、希釈剤、結合剤、担体、及び崩壊剤を含んでもよい。他の補助剤として、例えば、安定剤、湿潤剤、乳化剤、浸透圧に影響を与える塩、着色剤、香料、及び/又は芳香族活性化合物が挙げられる。
医薬的に許容しうる担体又は賦形剤は、無毒の固体、半固体若しくは液体フィラー、希釈剤、被包材、又はいずれのタイプの製剤助剤をも表す。例えば、適切な医薬的に許容しうる担体、希釈剤、溶媒、又はビヒクルとして、限定するものではないが、水、塩(緩衝)溶液、アルコール、アラビアゴム、鉱油、植物油、ベンジルアルコール、ポリエチレングリコール、ゼラチン、炭水化物(例えばラクトース、アミロース又はデンプン)、ステアリン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、植物油、脂肪酸モノグリセリド、脂肪酸ジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシルメチルセルロース、ポリビニルピロリドン等が挙げられる。例えば、レシチン等のコーティング材の使用によって、又は分散系の場合には所要の粒径の維持によって、及び界面活性剤の使用によって、適正な流動性を維持することができる。種々の抗菌剤、例えば、抗細菌剤及び抗真菌剤、パラベン、クロロブタノール、フェノール、ソルビン酸などの薬剤を含めることによって、微生物の作用の阻止を確実にすることができる。
【0054】
医薬的に許容しうる固体担体を使用する場合、本発明の実施形態で使うのに適した化合物の剤形は、錠剤、カプセル剤、散剤、座剤、又はロゼンジ剤でありうる。液体担体を使用する場合、剤形は、軟ゼラチンカプセル剤、経皮パッチ、エアロゾルスプレー、局所クリーム、シロップ又は液体座剤、エマルション、又は溶液でありうる。
非経口適用では、特に注射用無菌溶液、好ましくは非水性若しくは水性溶液、並びに分散液、懸濁液、エマルション、又は座剤を含めたインプラントが適している。アンプルは多くの場合便利な単位剤形である。注射用デポー形も好適であり、ポリラクチド−ポリグリコリド、ポリ(オルトエステル)、及びポリ(酸無水物)等の生分解性ポリマー中で薬物のマイクロカプセルマトリクス(microencapsule matrices)を形成することによって製造しうる。ポリマーに対する薬物の比率及び利用する特定ポリマーの性質によっては、薬物放出速度を制御することができる。
経腸適用では、特に錠剤、糖衣錠(dragee)、液体、液滴、座剤、又はカプセル剤、例えば軟ゼラチンカプセル剤が適している。甘味ビヒクルを利用するシロップ剤、エリキシル剤などを使用することができる。
【0055】
上述したように、他の医薬送達システムとして、持続放出、遅延放出、又は徐放送達システムが挙げられる。このようなシステムは、本発明の化合物の反復投与を回避し、患者及び医師の便利さを高め、かつ化合物の持続性血漿レベルを維持することができる。多くのタイプの制御放出送達システムが利用可能であり、当業者には周知である。
例えば、本発明の実施形態の化合物を医薬的に許容しうる徐放マトリックス、例えば生分解性ポリマーと併用して、治療組成物を形成することができる。本明細書で使用する場合、徐放マトリックスは、酵素加水分解若しくは酸塩基加水分解又は溶解によって分解しうる材料、通常はポリマー製のマトリックスである。体内に挿入されると、該マトリックスは酵素及び体液によって作用を受ける。徐放マトリックスは、望ましくは生体適合性材料、例えばリポソーム;ポリマーをベースとしたシステム、例えばポリラクチド(ポリ乳酸)、ポリグリコリド(グリコール酸のポリマー)、ポリラクチド−コ−グリコリド(乳酸とグリコール酸のコポリマー)、ポリ酸無水物、ポリ(オルト)エステル、多糖、ポリアミノ酸、ヒアルロン酸、コラーゲン、コンドロイチン硫酸、ポリヌクレオチド、ポリビニルプロピレン、ポリビニルピロリドン、及びシリコーン;非ポリマーシステム、例えばカルボン酸、脂肪酸、リン脂質、アミノ酸、及び脂質、例えばステロール;ヒドロゲル放出システム;サイラスティック(silastic)システム;ペプチドベースシステム;インプラント等から選択される。具体例として、限定するものではないが、以下のものが挙げられる:(a)米国特許第4,452,775号、第4,675,189号、及び第5,736,152号(全体が参照により本明細書に援用される)で見つかるマトリックス内の形態で多糖が含まれる浸食システム、及び(b)米国特許第3,854,480号、第5,133,974号、及び第5,407,686号(全体が参照により本明細書に援用される)に記載されているようなポリマーから活性成分が制御された速度で浸透する拡散システム。さらに、ポンプをベースとしたハードワイヤード送達システム(pump−basedhard−wired delivery system)を使用でき、移植に適合したシステムもある。好適な腸溶性コーティングは、PCT公開第WO98/25613号及び米国特許第6,274,591号(両方とも参照により本明細書に援用される)に記載されている。マイクロカプセル化、多重コーティング等によってのように差動分解性コーティングを利用して活性化合物を保護する当該組成物として徐放又は制御放出組成物を調製することもできる。
【0056】
特にメチルナルトレキソンに関して、水性製剤は、キレート化剤、緩衝剤、抗酸化剤及び必要に応じて等張剤(好ましくは3.0〜3.5のpHに調整)を含有しうる。オートクレーブ処理及び長期貯蔵に安定した製剤は、その開示内容が参照により本明細書に援用され、発明の名称が「医薬製剤」であり、第2004/0266806号として公開された米国特許出願第10/821811号に記載されている。有効期間を増やしたメチルナルトレキソンの製剤も、参照によって本明細書に援用される、発明の名称が「化合物の非経口送達用製剤及びその使用」である国際特許公開第WO2008/19115号に記載されている。メチルナルトレキソンの凍結乾燥製剤は、米国特許出願第11/899,724号に記載され、メチルナルトレキソンを含有する粒子を含む製剤は、参照により本明細書に援用される米国特許第6,419,959号に記載されている。メチルナルトレキソンの経皮送達に適した製剤は、参照により本明細書に援用される国際特許公開第2007/41544号に記載されている。
【0057】
本発明の実施形態の化合物、mTOR阻害薬及びμ−オピオイド受容体拮抗薬は、相乗的な抗増殖及び抗遊走に有効な量で提供される。しかし、当然のことながら、本発明の化合物及び組成物の総投与量は、ゾンデ医療判断の範囲内で担当医師によって決定される。いずれの特定の患者に特有の治療的に有効な用量レベルも種々の因子によって左右される。該因子としては、治療する障害及び障害の重症度;利用する特有化合物の活性;利用する特有組成物;患者の年齢、体重、一般的健康状態、性別、及び食事制限;投与時間;投与経路;利用する特有化合物の排泄速度;治療の持続期間;利用する特有化合物と併用又は同時に使用する薬物など、医術で周知の因子が挙げられる。例えば、ある技法は、所望の治療効果を達成するのに必要な用量未満のレベルで化合物の用量を開始して、所望効果が達成されるまで投与量を徐々に増やすことである。
所望により、1日の有効な用量を投与目的によって複数の用量に分割してよい。結果として、単一用量組成物は、1日の用量を構成する量又はその約数の量を含有しうる。上述したように、当業者は、良い医療行為及び個々の患者の臨床状態によって決まる有効湯量及び共投与計画(本明細書で述べるように)を容易に最適化することができる。
一般的に、μ−オピオイド受容体拮抗薬、特に末梢性拮抗薬の経口用量は、1日当たり約0.01〜約80mg/kg(体重)の範囲である。1〜20mg/kg(体重)の経口用量が望ましい結果をもたらすと予想される。一般的に、静脈内及び皮下投与などの非経口投与は、約0.001〜5mg/kg(体重)の範囲である。0.05〜0.5mg/kg(体重)の範囲の用量が所望の結果をもたらすと予想される。薬用量を適宜調整して、投与様式によって決まる局所又は全身の所望薬物レベルを達成することができる。例えば、腸溶コーティング製剤中のμ−オピオイド受容体拮抗薬の経口投与用薬用量は、非コーティング経口用量の10〜30%であると予想される。患者の応答が該用量では不十分な場合、患者の耐用性が許す程度まで、さらに高い用量(又はより局所的な異なる送達径路によって30%の用量より効率的に高い)を利用してよい。1日当たりの複数回の用量は、化合物の適切な全身レベルを達成するように考慮される。適切なシステムレベルは、例えば、当業者に周知の日常的なHPLC法を用いて、薬物の患者の血漿レベルを測定することで決定される。
【0058】
本発明の実施形態では、必要に応じて、例えば、本発明の実施形態の化合物について知られている投与量で、いずれの投与経路によっても、例えば、経腸(例えば、経口)、又は非経口又は局所的にmTOR阻害薬化合物を投与しうる。mTOR阻害薬について種々の経口及び非経口剤形が知られている。1日の経口投与量は、例えば分散性錠剤の形態で0.1mg〜25mgの範囲であってよい。1週間の投与量は、70mgまで含んでよく、治療する疾患によって決まる。静脈内投与などの非経口投与では、1日の投与計画(5日間毎日、2週間〜3週間毎日)で投与する場合、最初の静脈内投与量が0.1〜100mg/m2であり、さらに好適には、1週間に1回の投与計画で投与する場合、0.1〜1000mg/m2である。例えば、エベロリムスは、治療する疾患によって、例えば分散性錠剤の形態又は固体分散系の形態で、0.1mg〜25mgまで又は0.1mg〜15mg(例えば0.1mg、0.25mg、0.5mg、0.75mg、1mg、2.5mg、5mg、又は10mgなど)の1日の投与量で経口投与することができる。エベロリムスを70mgまで含みうる、例えば10〜70mg、又は30〜50mg(治療する疾患によって決まる)の1週間の薬用量で投与することができる。さらなる例では、タクロリムス(Protopic)を軟膏基剤中0.03%〜0.1%(w/w)の軟膏として投与することができる。同様に、例えば、同様の投与量範囲で他のmTOR阻害薬を投与することができる。
【0059】
本発明の例示実施形態では、μ−オピオイド受容体拮抗薬をmTOR阻害薬と共投与する。換言すれば、μ−オピオイド受容体拮抗薬化合物とmTOR阻害薬の共投与は、適切にμ−オピオイド受容体拮抗薬とmTOR阻害薬を含む医薬の組合せと考えられ、この組合せを毎日又は間欠ベースの末梢性μ−オピオイド受容体拮抗薬の投与、及び毎日又は間欠ベースのmTOR阻害薬の投与に適合させる。従って、μ−オピオイド受容体拮抗薬をmTOR阻害薬の投与前、投与と同時、又は投与後に投与してよい。典型的投与計画では、患者は、mTOR阻害薬の30分の静脈内注入を受けた直後にμ−オピオイド受容体拮抗薬を受けるか又はμ−オピオイド受容体拮抗薬の投与を先に受ける。1回以上の治療周期後、得られた結果及び観察されたいずれもかの副作用によって、薬用量を上方又は下方調整することができる。例示実施形態では、mTOR阻害薬の投与前のμ−オピオイド受容体拮抗薬の投与が特に好適である。
本発明の実施形態で共投与可能な薬剤を混合物として、例えば、単一製剤又は単一錠剤中の混合物のように調製してもよい。これらの製剤は、非経口又は経口製剤、例えば、米国特許第6,277,384号;第6,261,599号;第5,958,452号及びPCT公開第WO98/25613号(それぞれ参照により本明細書に援用される)に記載されている製剤であってよい。
【0060】
本発明の実施形態の方法を単独で使用してよく、或いは上述した種々の状態と関連する内皮細胞の成長又は遊走を制御するための他の治療と併用してよい。mTOR阻害薬及び末梢性μ−オピオイド受容体拮抗薬を、オピオイド若しくはμ−オピオイド受容体拮抗薬又はmTOR阻害薬でない別の治療薬と共投与してよい。このような適切な治療薬として、他の抗癌薬が挙げられる。
本発明の実施形態は、癌の治療をも包含する。治療しうる癌のタイプは、mTORの関与によってのみ制限される。従って、これらの療法を用いて、脳、肺、肝臓、脾臓、腎臓、リンパ節、膵臓、小腸、血液細胞、結腸、胃、乳房、子宮内膜、前立腺、精巣、卵巣、皮膚、頭頚部、食道、骨髄、血液又は他の組織の癌を含めた種々多様の腫瘍を治療できると考えられる。
多くの文脈で、必ずしも腫瘍細胞が殺されるか又は正常細胞の死若しくは「アポトーシス」を受けるように誘発される必要はない。むしろ、意味ある治療を達成するために必要なのは、腫瘍の成長をある程度まで遅らせることだけである。しかし、もしかしたら腫瘍の成長が完全に遮断されるか、又は一部の腫瘍の退縮が達成されるかもしれない。「寛解」及び「腫瘍量(tumor burden)の減少」などの臨床的用語は、所定のそれらの普通の用法も考えられる。
以下の実施例で本発明の実施形態をさらに説明するが、本発明の範囲を限定する目的で実施例を解釈すべきでない。
【実施例】
【0061】
実施例1:VEGF誘発Akt活性化の阻害
セリン/スレオニンキナーゼAktのVEGF誘発活性化に及ぼすメチルナルトレキソン(MNTX)の作用を評価するため、特徴がはっきりした内皮細胞系であるヒト肺微小血管内皮細胞(HPMVEC)を使用した。HPMVECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX、100ng/mlのベバシズマブ、100μMの5−FU、100nMのMNTX+100ng/mlのベバシズマブ又は100nMのMNTX+100μMの5−FUを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。細胞ライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt、抗Akt又は抗アクチン抗体を用いて免疫ブロットした。結果は、メチルナルトレキソンがセリン及びスレオニンのそれぞれ位置473及び308でAktのVEGF誘発リン酸化を抑止することを示した(図2)。さらに、ベバシズマブ及び5−FUと併用したメチルナルトレキソンは、相乗的にAktの活性化を阻害した。VEGF誘発pSer473Akt(図3A)及びpThr308Akt(図3B)の免疫反応性の阻害に及ぼす種々の濃度のMNTX(1.0、10、100、1000nM)の作用は、この阻害が用量依存性であることを実証した。
【0062】
細胞培養及び試薬−ヒト肺微小血管ECをCambrex(Walkersville, MD)から得、以前に記載されているように(Singleton et al. (2006), Microvasc. Res. 72(1−2):3−11; Singleton et al. (2007), Am. J. Respir. Cell Mol. Biol. 37(2):222−231)、EBM−2完全培地(Cambrex)内で37℃にて5%CO2、95%空気の加湿雰囲気内で培養し、実験用には継代6〜10を用いた。特に断らない限り、Sigma (St. Louis, MO)から試薬を得た。血管内皮成長因子(VEGF)はR&D Systems(Minneapolis, MN)から購入した。メチルナルトレキソンブロミド(MNTX)はMallinckrodt Specialty Chemicals (Phillipsburg, NJ)から購入した。ベバシズマブはGenentech(South San Francisco, CA)から購入した。5−フルオロウラシル(5−FU)はAbraxis Pharmaceutical Products (Schaumburg, IL)から購入した。ナルトレキソン及びラパマイシンはSigma(St. Louis, MO)から購入した。SDS−PAGE電気泳動用の試薬はBio−Rad(Richmond, CA)から購入し、Immobilon−P転写膜はMillipore(Millipore Corp., Bedford, MA)から購入した。ウサギ抗pSer473Akt、ウサギ抗pThr308Akt及びウサギ抗Akt抗体はCell Cignalling Technologies(Danvers, MA)から購入した。マウス抗β−アクチン抗体はSigma(St. Louis, MO)から購入した。二次西洋ワサビペルオキシダーゼ(HRP)標識抗体はAmersham Biosciences(Piscataway, NJ)から購入した。
SDS−PAGE及び免疫ブロット法−処理済み又は未処理HPMVECから得た細胞材料をIP緩衝液(50mM HEPES(pH 7.5)、150mM NaCl、20mM MgCl2、1% Nonidet P−40(NP−40)、0.4mM Na3VO4、40mM NaF、50μM オカダ酸、0.2mM フェニルメチルスルホニルフルオリド、1:250希釈のCalbiochemプロテアーゼ阻害薬混合物3)とインキュベートし、4〜15%のポリアクリルアミドゲル中のSDS−PAGEに供し、ImmobilonTM膜上に移し、特異性一次抗体及び二次抗体を用いて展開した。増強化学発光(Amersham Biosciences)を用いて免疫反応性バンドの可視化を達成した。活性化Aktの相対量を調べるため、pSer473Akt及びpThr308Akt免疫反応性バンド強度を総Akt免疫反応性バンド強度で割った。
【0063】
実施例2:VEGF誘発内皮細胞遊走及び増殖に及ぼすラパマイシンの作用
細胞の遊走及び増殖におけるmTORの役割を調べるため、HPMVECの遊走及び増殖アッセイに及ぼすラパマイシンの作用を定量した。ヒトECをVEGF(100nM)誘発増殖(図4)及び遊走(図5)について0.01、0.1、1.0若しくは10nMのラパマイシンの存在下又は非存在下でアッセイした。結果は、ラパマイシンによるmTORの阻害が内皮細胞の増殖及び遊走の用量依存性阻害をもたらすことを実証する。このことは、mTORがこれらのプロセスに関与することを実証している以前の研究と一致する。
ヒト肺微小血管EC遊走アッセイ−in vitroで細胞の遊走をモニターするために8μMのポアサイズの24のトランスウェル(transwell)ユニットを用いた。HPMVEC(約1×104細胞/ウェル)を種々の処理をして上方チャンバーに蒔き、VEGF(100nM)を下方チャンバーに添加した。細胞を18時間遊走させた。上方及び下方チャンバーから得た細胞をCellTiter96TM MTSアッセイ(Promega, San Luis Obispo, CA)を用いて定量化し、492nmで解読した。上方と下方の両チャンバー内の細胞数で割った下方チャンバー内の細胞数として遊走率を定義した。各アッセイを三通り準備し、少なくとも5回繰り返し、スチューデントt−検定で統計的に解析した(P<0.05で設定される統計的有意性を有する)。
ヒト肺微小血管EC増殖アッセイ−細胞の成長を測定するため、種々の薬剤で前処理したHPMVEC[5×103細胞/ウェル]を、5%CO2/95%空気中37℃にて96−ウェル培養プレート内で100nMのVEGFを含む0.2mlの無血清培地と24時間インキュベートした。96TM MTSアッセイ(Promega, San Luis Obispo, CA)を用いて細胞数の増加を測定することによってin vitro細胞増殖アッセイを分析し、492nmで解読した。各アッセイを三通り準備し、少なくとも5回繰り返し、スチューデントt−検定で統計的に解析した(P<0.05で設定される統計的有意性を有する)。
統計解析−スチューデントt−検定を用いて、2以上の異なる実験群から得たデータの平均値を比較した。結果を平均値±S.E.として表す。
【0064】
実施例3:VEGF誘発内皮細胞遊走及び増殖に及ぼすメチルナルトレキソンとmTOR阻害薬の相乗効果
AktとmTORの間に存在する複雑なフィードバック阻害シグナル伝達ネットワークに鑑み、上述した遊走及び増殖アッセイを用いて、ラパマイシンによるmTOR活性化の阻害とメチルナルトレキソンによるAktの阻害を同時に行なうと、これらのプロセスに対して相乗効果をもたらすかを決定した。ヒト内皮細胞をVEGF(100nM)誘発増殖(図6)及び遊走(図7)について10若しくは100nMのMNTX、10nMのナルトレキソン、0.1nMのラパマイシン、100nMのMNTX+0.1nMのラパマイシン又は10nMのナルトレキソン+0.1nMのラパマイシンの存在下或いは非存在下でアッセイした。メチルナルトレキソンとテムシロリムスを併用して同様の結果を得た(図10)。
結果は、メチルナルトレキソンとmTOR阻害薬が相乗的に内皮細胞の遊走及び増殖を阻害することを実証した。メチルナルトレキソンで観察された結果と異なり、テムシロリムスを用いたヒト内皮細胞の処理は、AktのVEGF誘発活性化を阻害しなかった(図8)。これは、メチルナルトレキソンとmTOR阻害薬の併用で観察された相乗効果が、おそらく2つの別個の点におけるmTORシグナル伝達経路の阻害のためであり、メチルナルトレキソン誘発阻害はAkt活性化の上流で起こり、mTOR阻害薬による阻害はAkt活性化の下流で起こることを示唆している(図12)。この仮説は、メチルナルトレキソンはmTOR複合体IとmTOR複合体IIのVEGF誘発形成を阻害するが、テムシロリムスはmTOR複合体IIの形成だけを阻害するという事実によって支持される(図9A)。
VEGF誘発Akt活性化の機構をさらに解明するため、PI3キナーゼ阻害薬、LY294002、Src siRNA又はRictor(mTOR複合体IIの成分)siRNAで前処理した場合としない場合のVEGFでヒト内皮細胞を処理した。抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いてライセートを免疫ブロットした(図9B)。PI3キナーゼ阻害薬は、VEGF誘発Aktスレオニン308リン酸化を阻害する。mTOR複合体2の形成(Rictor siRNA)の阻害がVEGF誘発Aktセリン473のリン酸化を遮断する。Srcの発現(siRNA)の阻害がAktセリン473及びAktスレオニン308の両リン酸化を遮断する。さらに、メチルナルトレキソンとmTOR阻害薬の併用がヒト内皮細胞の増殖と遊走に及ぼす相乗効果におけるチロシンホスファターゼ(PTP)の中心的役割を、これらのVEGF誘発血管新生促進事象を阻害する強力なPTP阻害薬3,4−デホスタチン(dephostatin)を用いて実証した(図11)。
【0065】
Aktリン酸化のMNTX及びテムシロリムス制御−ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX又は100nMのテムシロリムスを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いて免疫ブロットした。
mTOR複合体形成及びAktリン酸化制御の分析−ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いは100nMのMNTX又は100nMのテムシロリムスを用いて前処理(1時間)した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、抗Raptor(mTOR複合体1の成分)又は抗Rictor(mTOR複合体2の成分)抗体を用いて免疫ブロットした。免疫ブロットした材料でSDS−PAGEを実行し、抗mTOR、抗FKBP12、抗Raptor、抗SIN1又は抗Rictor抗体のどれかを用いて免疫ブロットした。Aktリン酸化の制御を調べるため、ヒトECを1時間、血清不足にし、処理をしない(コントロール)か或いはPI3キナーゼ阻害薬、LY294002(10μM、1時間)、Src siRNA又はRictor(mTOR複合体2の成分)siRNAを用いて前処理した場合としない場合のVEGF(100nM、5分)で処理した。ECライセートを得、SDS−PAGEを実行し、かつ抗pSer473Akt、抗pThr308Akt又は抗Akt抗体を用いて免疫ブロットした。
VEGF誘発ヒトEC増殖及び遊走に及ぼすMNTXとテムシロリムスの相乗効果。ヒトECの阻害曲線を、0.1、1.0、10、100若しくは500nMのMNTX、テムシロリムス又は10nMのMNTX+テムシロリムスの存在下或いは非存在下でVEGF(100nM)誘発増殖及び遊走についてアッセイした(24時間)。MNTXは、ECのVEGF誘発増殖を約100nMのIC50で阻害した。ECに10nMのMNTXを添加すると、VEGF誘発増殖のテムシロリムス阻害のIC50が約10nM〜約1nMにシフトした。実験は三通り行なった。エラーバー=標準偏差。
テムシロリムとのMNTXの相乗効果はチロシンホスファターゼの活性によって制御される。ヒトECを、10nMのMNTXがある場合とない場合で10nM又は15nMのテムシロリムスの存在下でVEGF(100nM)誘発増殖及び遊走(24時間)についてアッセイした(それぞれ3,4−デホスタチンの非存在下又は存在下の増殖の阻害についてのIC50濃度)。実験は三通り行なった。エラーバー=標準偏差。
如何なる特定の理論によっても制限されたくないが、図12は、実施例の結果によって実証された相乗効果に可能な機構を示す。
【0066】
実施例4:mTOR阻害薬とメチルナルトレキソンを併用した哺乳動物対象の治療
第1セットの実験では、腫瘍細胞のトランスフォーメーション、近親交配又は移植によってマウスに腫瘍を発生させる。それぞれ少なくとも60mm3の体積を有する腫瘍を保持する48匹のマウスをランダムに4群に分ける。第1群は、オピオイド拮抗薬もmTOR阻害薬をも含まないコントロール物質を受ける。第2群は、治療的に有効な量のメチルナルトレキソンを用いて腫瘍と接するのに容認できる径路を介して投与される末梢性オピオイド拮抗薬メチルナルトレキソン、例えば5mg/kg/日の用量で経口投与を受ける。第3群は、治療的に有効な量のmTOR阻害薬を用いて腫瘍と接するのに容認できる径路を介して投与されるラパマイシン等のmTOR阻害薬、例えば、1mg/kg/日の用量のラパマイシンの注射を受ける。第4群は、メチルナルトレキソンとmTOR阻害薬の組合せを受ける。
各群のマウス間の腫瘍の成長速度、腫瘍サイズ、腫瘍内の血管新生及び死亡率の差異を記録する。mTOR阻害薬、例えばラパマイシンの治療用量を変えて追加実験を行ない、メチルナルトレキソンとの共投与に起因するmTOR阻害薬の治療用量の減少を定量する。
【0067】
実施例5:mTOR阻害薬とアルビモパンを併用した哺乳動物対象の治療
第1セットの実験では、腫瘍細胞のトランスフォーメーション、近親交配又は移植によってマウスに腫瘍を発生させる。それぞれ少なくとも60mm3の体積を有する腫瘍を保持する48匹のマウスをランダムに4群に分ける。第1群は、オピオイド拮抗薬もmTOR阻害薬をも含まないコントロール物質を受ける。第2群は、治療的に有効な量のアルビモパンを用いて腫瘍と接するのに容認できる径路を介して投与される末梢性オピオイド拮抗薬アルビモパンを受ける。第3群は、治療的に有効な量のmTOR阻害薬を用いて腫瘍と接するのに容認できる径路を介して投与されるラパマイシン等のmTOR阻害薬、例えば、1mg/kg/日の用量の注射を受ける。第4群は、アルビモパンとmTOR阻害薬の組合せを受ける。
各群のマウス間の腫瘍の成長速度、腫瘍サイズ、腫瘍内の血管新生及び死亡率の差異を記録する。mTOR阻害薬、例えばラパマイシンの治療用量を変えて追加実験を行ない、アルビモパンとの共投与に起因するmTOR阻害薬の治療用量の減少を定量する。
【0068】
要約すれば、本発明の実施形態の方法は、癌及び他の過剰増殖疾患並びに自己免疫疾患といった細胞の増殖及び遊走に関連する疾患又は障害を治療するために提供され、この方法はmTOR阻害薬とμ−オピオイド受容体拮抗薬の共投与を含む。
以上、種々の特有の実施形態及び手法に関連して本発明について述べた。しかし、本発明の精神及び範囲内にとどまりながら、多くの変更及び修正を行なえることを理解すべきである。
全ての出版物、特許、及び特許出願は、それぞれ個々の出版物、特許、及び特許出願が具体的かつ個々に参照によって指示されたのと同程度に、参照により本明細書に明確に援用される。本開示と、援用された特許、出版物及び参考文献との間で矛盾する場合、本開示が支配するものとする。
【特許請求の範囲】
【請求項1】
対象の細胞の不必要な遊走及び/又は増殖を特徴とする障害を治療する方法であって、前記障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法。
【請求項2】
前記細胞の不必要な遊走及び/又は増殖が内皮細胞の不必要な遊走及び/又は増殖である、請求項1に記載の方法。
【請求項3】
前記内皮細胞が血管内皮細胞である、請求項2に記載の方法。
【請求項4】
前記血管内皮細胞の不必要な遊走及び/又は増殖が不必要な血管新生である、請求項3に記載の方法。
【請求項5】
前記障害が癌である、請求項1に記載の方法。
【請求項6】
前記障害が糖尿病である、請求項1に記載の方法。
【請求項7】
前記障害が鎌状赤血球貧血である、請求項1に記載の方法。
【請求項8】
前記障害が血管創傷である、請求項1に記載の方法。
【請求項9】
前記障害が増殖網膜症である、請求項1に記載の方法。
【請求項10】
前記mTOR阻害薬がラパマイシン誘導体である、請求項1に記載の方法。
【請求項11】
前記ラパマイシン誘導体がテムシロリムス、エベロリムス、デフォロリムス、TAFA93、AP23573、ABT−573、FK506、又はnab−ラパマイシンである、請求項10に記載の方法。
【請求項12】
前記mTOR阻害薬がTOP216又はOSI−027である、請求項1に記載の方法。
【請求項13】
前記μ−オピオイド受容体拮抗薬が末梢性μ−オピオイド受容体拮抗薬である、請求項1に記載の方法。
【請求項14】
前記μ−オピオイド受容体拮抗薬が下記式(I):
【化1】
(式中、Rは、アルキル、アルケニル、アルキニル、アリール、シクロアルキル置換アルキル、又はアリール置換アルキルであり、X−は、クロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである)
の化合物、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー若しくはジアステレオマー混合物、又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項1に記載の方法。
【請求項15】
前記μ−オピオイド受容体拮抗薬が下記式(II):
【化2】
(式中、X−は、クロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである)
の化合物、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー若しくはジアステレオマー混合物、又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項14に記載の方法。
【請求項16】
前記μ−オピオイド受容体拮抗薬が、四級又は三級モルフィナン誘導体、ピペリジン−N−アルキルカルボキシラート、及び四級ベンゾモルファンから成る群より選択される、請求項1に記載の方法。
【請求項17】
前記四級モルフィナン化合物が、N−メチルナルトレキソン、N−メチルナロキソン、N−メチルナロルフィン、N−ジアリルノルモルフィン、N−アリルレバロルファン、及びN−メチルナルメフェンから成る群より選択される化合物の四級塩を含む、請求項16に記載の方法。
【請求項18】
前記末梢性μ−オピオイド受容体拮抗薬がメチルナルトレキソンである、請求項13に記載の方法。
【請求項19】
前記末梢性μ−オピオイド受容体拮抗薬がアルビモパンである、請求項13に記載の方法。
【請求項20】
前記μ−オピオイド受容体拮抗薬及びmTOR阻害薬を同時に投与する、請求項1に記載の方法。
【請求項21】
前記mTOR阻害薬を投与する前に前記μ−オピオイド受容体拮抗薬を投与する、請求項1に記載の方法。
【請求項22】
前記mTOR阻害薬を投与した後に前記μ−オピオイド受容体拮抗薬を投与する、請求項1に記載の方法。
【請求項23】
前記μ−オピオイド受容体拮抗薬の投与が、経口、舌下、筋肉内、皮下、静脈内、局所又は経皮投与である、請求項1に記載の方法。
【請求項24】
前記mTOR阻害薬の投与が、経口、舌下、筋肉内、皮下、静脈内、局所又は経皮投与である、請求項1に記載の方法。
【請求項25】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約0.001mg/kg〜約80mg/kg(体重)である、請求項19に記載の方法。
【請求項26】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約0.05mg/kg〜約50mg/kg(体重)である、請求項25に記載の方法。
【請求項27】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約1mg/kg〜約20mg/kg(体重)である、請求項26に記載の方法。
【請求項28】
哺乳動物細胞内で成長因子シグナル伝達を阻害する方法であって、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法。
【請求項29】
前記細胞が癌細胞である、請求項28に記載の方法。
【請求項30】
前記成長因子シグナル伝達がVEGFシグナル伝達である、請求項28に記載の方法。
【請求項31】
前記μ−オピオイド受容体拮抗薬が、末梢性μ−オピオイド受容体拮抗薬である、請求項28に記載の方法。
【請求項32】
細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項33】
癌の治療が必要な対象の癌を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含み、前記癌には、脳、肺、肝臓、胃、乳房、子宮内膜、前立腺、精巣、卵巣、皮膚、頭頚部、食道、骨髄及び血液の癌が含まれる、方法。
【請求項34】
腫瘍の成長の阻害が必要な対象の腫瘍の成長を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項35】
mTOR阻害薬の治療的有用性を改善する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項36】
自己免疫疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含み、前記自己免疫疾患が、アレルギー性脳脊髄炎、インスリン依存性糖尿病、ループス、関節リウマチ、多発性硬化症、皮膚筋炎、グレーブス病及びアジュバント関節炎である、方法。
【請求項37】
mTOR阻害薬及びμ−オピオイド受容体拮抗薬を相乗的有効量で含んでなる医薬併用剤。
【請求項38】
mTOR阻害薬及びμ−オピオイド受容体拮抗薬、並びに相乗的有効量で併用投与するための使用説明書を含んでなる医薬パッケージ。
【請求項39】
mTOR阻害薬による治療に伴う有害な副作用を低減する方法であって、前記有害な副作用に苦しむ患者に、mTOR阻害薬と共に、有効量のμ−オピオイド受容体拮抗薬を投与する工程を含む方法。
【請求項40】
前記細胞が、エストロゲン受容体、プロゲステロン受容体又はアンドロゲン受容体であるホルモン受容体を発現する細胞である、請求項1に記載の方法。
【請求項41】
前記μ−オピオイド受容体拮抗薬がメチルナルトレキソン、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項28、32〜36のいずれか1項に記載の方法。
【請求項1】
対象の細胞の不必要な遊走及び/又は増殖を特徴とする障害を治療する方法であって、前記障害のある対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を投与する工程を含む方法。
【請求項2】
前記細胞の不必要な遊走及び/又は増殖が内皮細胞の不必要な遊走及び/又は増殖である、請求項1に記載の方法。
【請求項3】
前記内皮細胞が血管内皮細胞である、請求項2に記載の方法。
【請求項4】
前記血管内皮細胞の不必要な遊走及び/又は増殖が不必要な血管新生である、請求項3に記載の方法。
【請求項5】
前記障害が癌である、請求項1に記載の方法。
【請求項6】
前記障害が糖尿病である、請求項1に記載の方法。
【請求項7】
前記障害が鎌状赤血球貧血である、請求項1に記載の方法。
【請求項8】
前記障害が血管創傷である、請求項1に記載の方法。
【請求項9】
前記障害が増殖網膜症である、請求項1に記載の方法。
【請求項10】
前記mTOR阻害薬がラパマイシン誘導体である、請求項1に記載の方法。
【請求項11】
前記ラパマイシン誘導体がテムシロリムス、エベロリムス、デフォロリムス、TAFA93、AP23573、ABT−573、FK506、又はnab−ラパマイシンである、請求項10に記載の方法。
【請求項12】
前記mTOR阻害薬がTOP216又はOSI−027である、請求項1に記載の方法。
【請求項13】
前記μ−オピオイド受容体拮抗薬が末梢性μ−オピオイド受容体拮抗薬である、請求項1に記載の方法。
【請求項14】
前記μ−オピオイド受容体拮抗薬が下記式(I):
【化1】
(式中、Rは、アルキル、アルケニル、アルキニル、アリール、シクロアルキル置換アルキル、又はアリール置換アルキルであり、X−は、クロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである)
の化合物、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー若しくはジアステレオマー混合物、又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項1に記載の方法。
【請求項15】
前記μ−オピオイド受容体拮抗薬が下記式(II):
【化2】
(式中、X−は、クロリド、ブロミド、ヨージド、カルボナート又はメチルスルファートアニオンである)
の化合物、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー若しくはジアステレオマー混合物、又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項14に記載の方法。
【請求項16】
前記μ−オピオイド受容体拮抗薬が、四級又は三級モルフィナン誘導体、ピペリジン−N−アルキルカルボキシラート、及び四級ベンゾモルファンから成る群より選択される、請求項1に記載の方法。
【請求項17】
前記四級モルフィナン化合物が、N−メチルナルトレキソン、N−メチルナロキソン、N−メチルナロルフィン、N−ジアリルノルモルフィン、N−アリルレバロルファン、及びN−メチルナルメフェンから成る群より選択される化合物の四級塩を含む、請求項16に記載の方法。
【請求項18】
前記末梢性μ−オピオイド受容体拮抗薬がメチルナルトレキソンである、請求項13に記載の方法。
【請求項19】
前記末梢性μ−オピオイド受容体拮抗薬がアルビモパンである、請求項13に記載の方法。
【請求項20】
前記μ−オピオイド受容体拮抗薬及びmTOR阻害薬を同時に投与する、請求項1に記載の方法。
【請求項21】
前記mTOR阻害薬を投与する前に前記μ−オピオイド受容体拮抗薬を投与する、請求項1に記載の方法。
【請求項22】
前記mTOR阻害薬を投与した後に前記μ−オピオイド受容体拮抗薬を投与する、請求項1に記載の方法。
【請求項23】
前記μ−オピオイド受容体拮抗薬の投与が、経口、舌下、筋肉内、皮下、静脈内、局所又は経皮投与である、請求項1に記載の方法。
【請求項24】
前記mTOR阻害薬の投与が、経口、舌下、筋肉内、皮下、静脈内、局所又は経皮投与である、請求項1に記載の方法。
【請求項25】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約0.001mg/kg〜約80mg/kg(体重)である、請求項19に記載の方法。
【請求項26】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約0.05mg/kg〜約50mg/kg(体重)である、請求項25に記載の方法。
【請求項27】
前記メチルナルトレキソンの相乗的有効量が、1日当たり約1mg/kg〜約20mg/kg(体重)である、請求項26に記載の方法。
【請求項28】
哺乳動物細胞内で成長因子シグナル伝達を阻害する方法であって、前記細胞を相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬と接触させる工程を含む方法。
【請求項29】
前記細胞が癌細胞である、請求項28に記載の方法。
【請求項30】
前記成長因子シグナル伝達がVEGFシグナル伝達である、請求項28に記載の方法。
【請求項31】
前記μ−オピオイド受容体拮抗薬が、末梢性μ−オピオイド受容体拮抗薬である、請求項28に記載の方法。
【請求項32】
細胞の過剰増殖を特徴とする障害又は疾患を治療する方法であって、前記障害又は疾患に苦しむ対象に、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項33】
癌の治療が必要な対象の癌を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含み、前記癌には、脳、肺、肝臓、胃、乳房、子宮内膜、前立腺、精巣、卵巣、皮膚、頭頚部、食道、骨髄及び血液の癌が含まれる、方法。
【請求項34】
腫瘍の成長の阻害が必要な対象の腫瘍の成長を阻害する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項35】
mTOR阻害薬の治療的有用性を改善する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含む方法。
【請求項36】
自己免疫疾患を治療する方法であって、相乗的有効量のmTOR阻害薬及びμ−オピオイド受容体拮抗薬を共投与する工程を含み、前記自己免疫疾患が、アレルギー性脳脊髄炎、インスリン依存性糖尿病、ループス、関節リウマチ、多発性硬化症、皮膚筋炎、グレーブス病及びアジュバント関節炎である、方法。
【請求項37】
mTOR阻害薬及びμ−オピオイド受容体拮抗薬を相乗的有効量で含んでなる医薬併用剤。
【請求項38】
mTOR阻害薬及びμ−オピオイド受容体拮抗薬、並びに相乗的有効量で併用投与するための使用説明書を含んでなる医薬パッケージ。
【請求項39】
mTOR阻害薬による治療に伴う有害な副作用を低減する方法であって、前記有害な副作用に苦しむ患者に、mTOR阻害薬と共に、有効量のμ−オピオイド受容体拮抗薬を投与する工程を含む方法。
【請求項40】
前記細胞が、エストロゲン受容体、プロゲステロン受容体又はアンドロゲン受容体であるホルモン受容体を発現する細胞である、請求項1に記載の方法。
【請求項41】
前記μ−オピオイド受容体拮抗薬がメチルナルトレキソン、その単一のエナンチオマー、エナンチオマー混合物、単一のジアステレオマー、若しくはジアステレオマー混合物;又はその医薬的に許容しうる塩、溶媒和物若しくはプロドラッグである、請求項28、32〜36のいずれか1項に記載の方法。
【図1】

【図2】
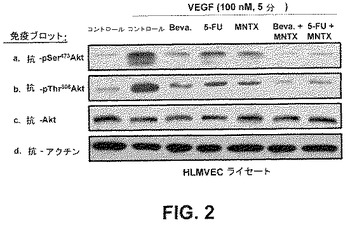
【図3】
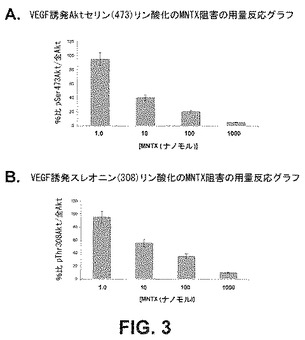
【図4】
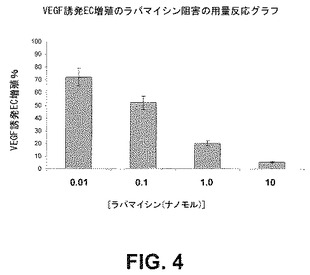
【図5】
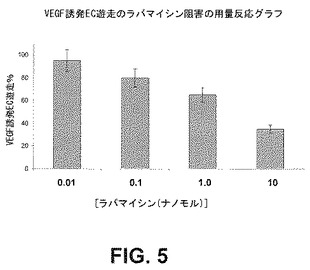
【図6】
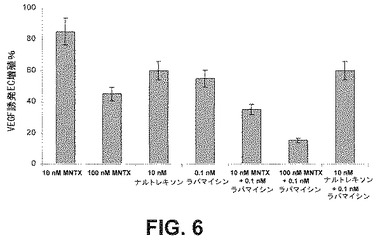
【図7】
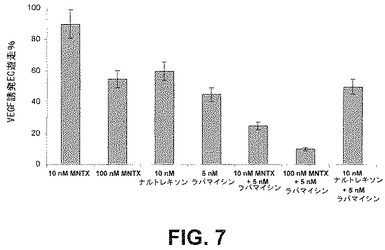
【図8】
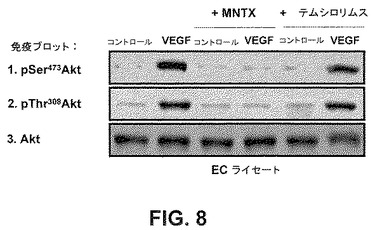
【図9】
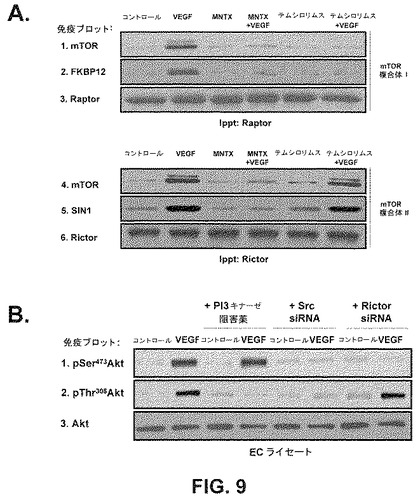
【図10】
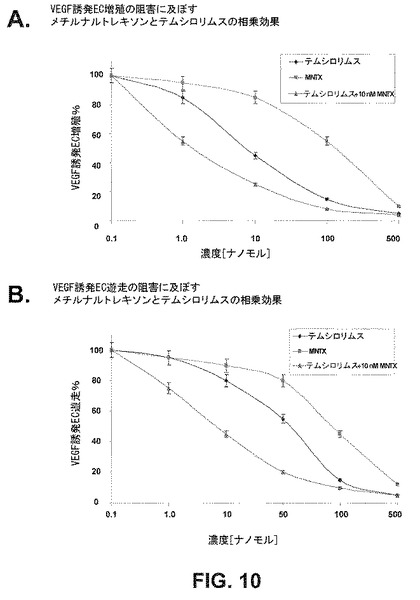
【図11】
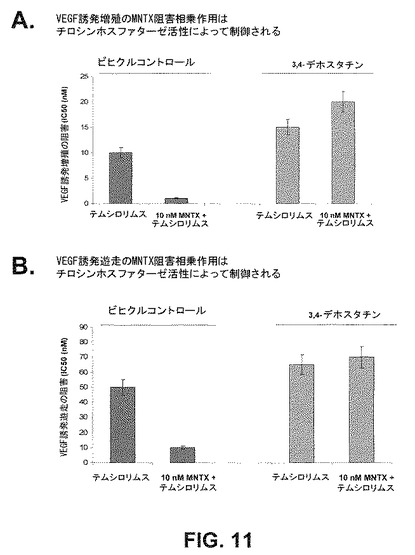
【図12】
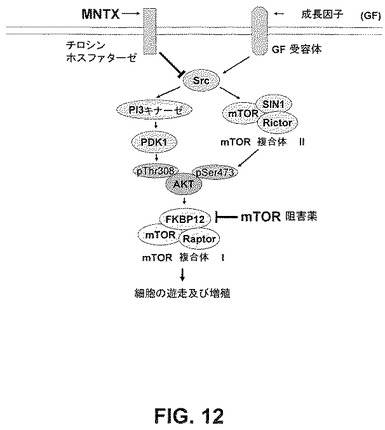
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2011−515415(P2011−515415A)
【公表日】平成23年5月19日(2011.5.19)
【国際特許分類】
【出願番号】特願2011−500985(P2011−500985)
【出願日】平成21年3月20日(2009.3.20)
【国際出願番号】PCT/US2009/037825
【国際公開番号】WO2009/117669
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(501242712)ザ ユニヴァーシティー オヴ シカゴ (19)
【Fターム(参考)】
【公表日】平成23年5月19日(2011.5.19)
【国際特許分類】
【出願日】平成21年3月20日(2009.3.20)
【国際出願番号】PCT/US2009/037825
【国際公開番号】WO2009/117669
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(501242712)ザ ユニヴァーシティー オヴ シカゴ (19)
【Fターム(参考)】
[ Back to top ]