
オメプラゾールナトリウムの結晶性溶媒和物
本発明は、医薬品工業分野に属し、新規結晶性オメプラゾールナトリウム・エタノール溶媒和物およびその調製方法に関し、この新規結晶性オメプラゾールナトリウム・エタノール溶媒和物は、それを種々の結晶形態、とりわけ残留溶媒量の低い、即ち残留溶媒量が0.5重量%未満である公知のオメプラゾールナトリウム形態Aに変換するための方法における中間化合物として作用する。本発明はまた、共に残留溶媒が著しく低濃度である新規な結晶性オメプラゾールナトリウム形態Eおよび結晶性オメプラゾールナトリウム形態F、ならびにそれらの調製方法にも関する。オメプラゾールナトリウム形態Aならびに新規結晶形オメプラゾールナトリウム形態Eおよび形態Fの両者は、胃腸疾患の治療に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬品工業分野に属し、新規のオメプラゾールナトリウム塩の結晶性エタノール溶媒和物に関する。オメプラゾールは、胃腸疾患の治療に使用される(5)6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールの一般名である。更に本発明は、結晶性オメプラゾールナトリウム・エタノール溶媒和物の調製方法、およびオメプラゾールナトリウムの異なる結晶形態への変換方法に関する。とりわけ、該新規オメプラゾールナトリウム・エタノール溶媒和物を、残留溶媒量を少なくして周囲温度で安定な公知の結晶形態のオメプラゾールナトリウム(以降、オメプラゾールナトリウム形態Aと呼ぶ)に変換するための容易に実施できおよび再現性のある方法に関する。
【0002】
本発明はまた、オメプラゾールナトリウムの2つの新規結晶形態にも関し、これらを以降、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fと呼ぶ。更に本発明は、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F(両方とも残留溶媒がかなり低濃度である。)の調製方法に関する。
【0003】
更に本発明は、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fの胃酸分泌過多関連疾患の治療への使用、および有効物質としてオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fを含む医薬組成物に関する。
【0004】
(技術的課題)
有効医薬品成分の化学的安定性、固体状態安定性および「保存寿命」は、薬学的有効化合物に関する重要な性質である。有効医薬品成分の安定性が、特に残留溶媒にも影響されることは一般に公知である。従って、有効医薬品成分の安定で結晶性の固体形態を残留溶媒を低濃度にして、好ましくは工業的に簡便で再現性のある方法で提供することが非常に望ましい。
【0005】
オメプラゾールナトリウムの新規の固体形態およびそれらの調製方法に対する必要性が普遍的にあるが、それは数多くの薬物が特定の固体形態で使用される場合に、望ましい溶出特性、およびいくつかの場合では望ましい生物学的利用率パターンを示すことが認められているからである。更に、多形体の安定性や吸湿性などの特性は種々に異なるであろう。
【背景技術】
【0006】
化学名(5)6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールとして公知であり一般名オメプラゾールを有する化合物は、胃酸分泌を阻害するプロトンポンプ阻害剤として公知である。オメプラゾールは、例えば胃食道逆流、食道炎、胸焼け、胃炎、分泌過多の状態(例えば、ゾリンジャー・エリソン症候群、内分泌腺腫)、十二指腸炎、胃潰瘍および十二指腸潰瘍などの、哺乳類、特にヒトにおける胃酸関連疾患および胃腸炎症性疾患の治療に使用できる。オメプラゾールはまた、例えばヘリコバクター・ピロリにより引き起こされる感染症などの感染症の治療にも有用である。オメプラゾールおよびその薬学的に許容できる塩は、特許EP−B−5129に初めて記載された。
【0007】
例えばナトリウム塩などのオメプラゾールの特定のアルカリ塩は、特許EP−B−124 495に初めて記載された。特許EP−B−124 495の実施例1および2に従って調製したオメプラゾールナトリウム塩は不安定であり、結晶形態と非晶質物質の混合物である。この混合物中に存在する結晶形態の1つは、オメプラゾールナトリウム形態Aであり、および1分子から2分子の水で水和した水和物であり、その水和物の1つの水分子は結晶構造中で強固に結合している一方、もう1つの水分子は乾燥により容易に除去される。1つの強固に結合した水分子を含有する得られる乾燥物質は、非常に吸湿性であり、水分を通常の条件下で迅速に吸収する。
【0008】
明確に定義されたオメプラゾールナトリウム一水和物塩(以降、オメプラゾールナトリウム形態Bと呼ぶ)およびその調製は、米国特許第6,207,188号に開示されている。その記載によるとオメプラゾールナトリウム形態Bは、例えば明確に定義されており、熱力学的に安定であり、非吸湿性でありおよび真に一水和物結晶形態であるなどの有利な特性を示す結晶形態である。形態Bとは対照的に、この特許はオメプラゾールナトリウム形態Aを、一定の保存条件下で完全に、または部分的にオメプラゾールナトリウム形態Bに変換されうる熱力学的に不安定な形態として記述している。米国特許第6,207,188号はまた、そのような不安定なオメプラゾールナトリウム形態Aの調製方法をも示している。記載された方法の欠点は、完結までに3日以上かかるので、時間を浪費することである。
【0009】
米国特許出願公報US 2004/0224987 A1は、類似しているが、オメプラゾールナトリウム形態A調製のための改善された方法を開示しており、この方法は、ナトリウムカチオンを放出する水性塩基Na+B−(Naがナトリウムを表し、Bがヒドロキシドまたはアルコキシド、イオン交換基、樹脂を表す)中のオメプラゾールを、C3からC7の分岐または直鎖炭化水素、C2からC7の分岐または直鎖エーテル、環状エーテル、低級脂肪酸エステル、脂肪族ケトン溶媒、ハロゲン化炭化水素溶媒またはニトリル溶媒からなる、場合により水を含む適切な溶媒中に室温で溶解する段階;生じた溶液を、生成物が溶けにくい適切な貧溶媒で中和する段階;反応混合物を室温で0から24時間撹拌する段階;反応混合物を固体物質が結晶化するまで冷却する段階;単離した固体を従来の方法でろ過する段階;上述の溶媒で洗浄する段階;および単離した化合物を30°から70℃で、好ましくは50°から60℃の温度で乾燥して、オメプラゾールナトリウムの形態Aを得る段階;を含む。得られたオメプラゾールナトリウム形態Aは、熱力学的により安定であり、非吸湿性であり、許容できる残留溶媒限界を有すると述べられている。US 2004/0224987は更に、新規な結晶性オメプラゾールナトリウム形態Cおよび結晶性オメプラゾールナトリウム形態D、およびそれらの調製方法を提供する。
【0010】
ES 2023778には、活性メチレン化合物のアルカリ塩を用いるオメプラゾールの、例えばナトリウム塩などの金属塩の製造方法が開示されている。
【0011】
オメプラゾールの非晶質形塩およびそのスプレードライ法を用いる調製方法がWO 01/87831に開示されている。
【0012】
米国特許第6,262,085号に記載されている発明は、6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールまたはその薬学的に許容できるその塩を提供し、それには、溶液中で塩を製造する結果、溶液中で起こる互変異性に依りメトキシ基をベンゾイミダゾール環上の6位に有するおよび5位に有する両方の化合物が製造されるという説明が付いている。
【0013】
式(I)のスルホキシド化合物またはその薬学的に許容できる塩のアセトン錯体が、ラベプラゾールの実施例を示す特許EP−B−1000943中に開示されている。
【0014】
(実施例を含む発明の説明)
本発明の目的は、周囲温度で安定と思われる残留溶媒量の低いオメプラゾールナトリウム形態Aを調製するための新規方法を見出すことである。更に本発明の目的は、新規な結晶性オメプラゾールナトリウム形態Eおよびその関連調製方法、ならびに新規な結晶性オメプラゾールナトリウム形態Fおよびその関連調製方法に関する。
【0015】
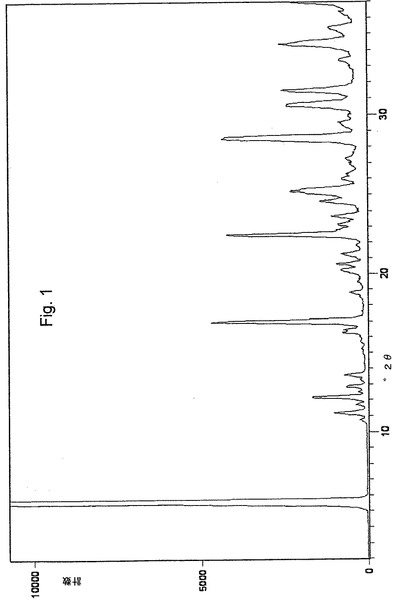
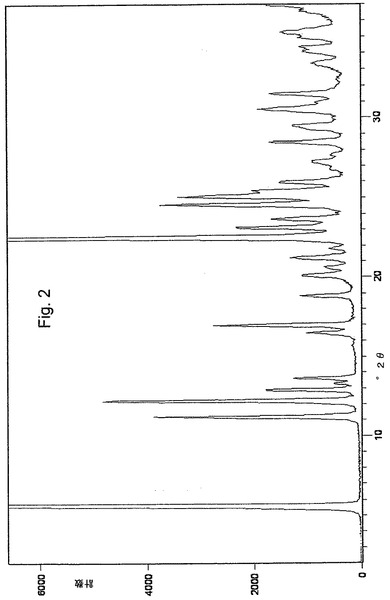
この課題は、安定な結晶性オメプラゾールナトリウム形態A調製のための新規方法、ならびに新規な結晶性オメプラゾールナトリウム形態Eおよび同様に新規な結晶性オメプラゾールナトリウム形態Fの調製方法における中間化合物である、新規の安定な結晶性オメプラゾールナトリウム・エタノール溶媒和物に関する本発明により解決された。粉末X線回折(XRPD)が、安定結晶性オメプラゾールナトリウム形態A、新規な安定結晶性中間体オメプラゾールナトリウム・エタノール溶媒和物、新規な安定結晶性オメプラゾールナトリウム形態E、および新規な安定結晶性オメプラゾールナトリウム形態F間の識別方法として使用される。
【0016】
本発明に従って、オメプラゾールナトリウム・エタノール溶媒和物は、オメプラゾール(原料)を無水エタノール中のNaOH溶液に溶解して調製する。得られた溶液から機械的粒子を、例えばろ過により除去し、更にオメプラゾールナトリウム形態Aの種結晶を加えて結晶化を誘導する。その後、スラリーを周囲温度で数時間、そして収量を上げるために、例えば0°から5℃などの低温で更に数時間撹拌する。沈殿したオメプラゾールナトリウム形態Aを、例えばろ過または遠心分離により分離し、冷無水エタノールで洗浄する。こうして誘導された湿オメプラゾールナトリウム・エタノールを減圧下、40°から50℃で、好ましくは約45℃で乾燥して、安定なエタノール溶媒和物の形態の無水オメプラゾールナトリウムを得る。用語「湿オメプラゾールナトリウム・エタノール」は、減圧下での乾燥の段階前に得られた生成物を意味する。用語「減圧」は一般に、約10mbarから約50mbarの圧力を指す。
【0017】
本発明による無水条件下での方法により調製されるオメプラゾールナトリウム・エタノール溶媒和物は、その結晶格子中に取り込まれた残留エタノールの約8から約11重量%(ガスクロマトグラフィーにより測定して)を含む。この取り込まれたエタノールはオメプラゾールナトリウムのエタノール溶媒和物結晶形態を規定し、更に乾燥しても除去できない。それ自体公知の手法である熱重量分析またはカール・フッシャー法により測定されたオメプラゾールナトリウム・エタノール溶媒和物中の水分定量は、無水生成物に相当する0.5重量%未満であることが見出された。
【0018】
オメプラゾールナトリウム形態Aは水に非常によく溶解し、そのこと自体が非経口製剤に適しており、胃食道逆流症(GERD)を患い、経口治療が受けられない患者を治療する機会を医師に提供する。このように、固体中に取り込まれた残存溶媒を伴う新規オメプラゾールナトリウム・エタノール溶媒和物の非経口製剤は薬学的に許容できなと思われるが、取り込まれた溶媒を実質的に含まないオメプラゾールナトリウムの結晶形態を調製するための重要な中間体として有用である。用語「実質的に含まない」は、残留溶媒(即ち、エタノール)の0.5重量%未満を意味する。
【0019】
従って、オメプラゾールナトリウムの水和物を特定の結晶形態で、即ち公知の形態Aで得るために、オメプラゾールナトリウム・エタノール溶媒和物の結晶格子内に取り込まれたエタノールを水分子と交換することが必要である。我々は、驚くべきことにおよび予期せぬことに、前記交換を新規オメプラゾールナトリウム・エタノール溶媒和物を非溶媒と水との混合物中で温浸することにより好都合に実施できることを見出した。用語「温浸すること」は、生成物をそれが不溶または難溶である溶媒(本明細書中では、非溶媒と称する)中に懸濁させ、次に水の少量を加え、得られた懸濁液を規定された時間の間撹拌する方法として理解される。
【0020】
より具体的には、オメプラゾールナトリウム・エタノール溶媒和物は、オメプラゾールナトリウム・エタノール溶媒和物の結晶を適した非溶媒と水との混合物中で温浸する方法によりオメプラゾールナトリウム形態Aに変換される。用語「適した溶媒」は、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒を意味し、好ましくはジイソプロピルエーテルであり、0°から20℃の、好ましくは5°から10℃の温度範囲で、30分から10時間の、より好ましくは約4時間の時間で用いられる。反応の完結後、沈殿したオメプラゾールナトリウム形態Aを、例えばろ過または遠心分離により優れた収率で回収し、望ましい生成物の劣化を防ぐ条件下で、例えば40°から50℃で減圧下10から24時間乾燥する。
【0021】
本発明のもう1つの態様において、オメプラゾールナトリウム・エタノール溶媒和物の代わりに湿オメプラゾールナトリウム・エタノールもまた、非溶媒と水との混合物中での温浸前に、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒、好ましくはジイソプロピルエーテルの適量であらかじめ洗浄するという条件下ではあるが、温浸の変法で使用してもよい。用語「非溶媒の適量」は、約20重量%から約10重量%の残留エタノールを洗い流してオメプラゾールナトリウム・エタノール溶媒和物を得るための量を意味する。従って、オメプラゾール(原料)を出発物質とし、湿オメプラゾールナトリウム・エタノールの洗浄を用いる結晶性ナトリウム形態Aの調製のための方法全体は、温浸段階前の乾燥を回避することにより、より時間を浪費しなくなる。
【0022】
結晶形態Aのオメプラゾールナトリウムは、オメプラゾールナトリウム1モル当たり水1から2モルで水和された水和物であるため、オメプラゾールナトリウムに関して水の少なくとも約10重量%から約20重量%が、好ましくは水の約10重量%が、残留溶媒量の少ない安定なオメプラゾールナトリウム結晶形態Aを得るために温浸工程において必要である。
【0023】
本発明による温浸工程で使用される混合物中の非溶媒:水の体積比は、40:1から100:1までの範囲内、より好ましくは60:1から80:1までの範囲内である。
【0024】
本発明による方法により調製されたオメプラゾールナトリウム形態A内に存在する残留溶媒濃度は、それ自体公知の方法であるガスクロマトグラフィーにより測定され、0.5重量%限界未満であることが見出された。
【0025】
本発明による方法により調製された結晶性オメプラゾールナトリウム形態A中の水分定量は、それ自体公知の方法である熱重量分析またはカール・フッシャー法により測定され、6から8%までであることが見出されたが、この量は水1モル、即ち4.7%が結晶中に結合する一方、その他の水分子は結晶上に吸収されているだけであることに対応する。
【0026】
従って本発明は、オメプラゾールナトリウム形態A調製のための方法を記述し、その方法は、オメプラゾール(原料)をNaOHの無水エタノール溶液中に溶解すること;オメプラゾールナトリウム形態Aの種結晶を添加して結晶化すること;新規中間体(即ち、無水オメプラゾールナトリウム・エタノール溶媒和物)を単離すること;および、前記新規中間体を非溶媒と水との混合物中での温浸工程により結晶形態Aのオメプラゾールナトリウム水和物に更に変換すること;を含む。従って、オメプラゾールナトリウム形態Aの調製のための本発明は、工業的規模で操作上安全に実施するために好都合な条件を使用する。本方法のもう1つの利点は、簡便であり、経済的であり、迅速であることである。
【0027】
加えて、我々は、本発明による方法により調製されたオメプラゾールナトリウム形態Aは安定であり、オメプラゾールナトリウムのいかなる他の形態をも実質的に含まず(即ち、オメプラゾールナトリウムのいかなる他の形態の検出可能量も含まない)、扱いやすく、例えば化学組成、吸湿性、溶解性および結晶形態などの物理化学的特性における顕著な変化を示さずにかなりの期間にわたって保存可能であることを見出した。
【0028】
本発明の方法により得られるオメプラゾールナトリウム形態Aの安定性は、薬学的有効物質の安定性を特徴付けるための標準手順(EU:CPMP採択、2003年3月、CPMP/ICH/2736/99として発行 − “Committee for proprietary medical products: Note for Guidance on ICH Q1A(R2) Stability testing guidelines: Stability testing of new drug substances and products”)により測定できる。梱包したオメプラゾールナトリウム形態Aが、経年変化加速条件下(医薬品製剤の安定性試験用の標準加速条件である温度40℃、相対湿度75%で)および/または経年変化過酷条件下(薬学的有効物質の安定性試験用の標準過酷条件である温度60℃で)で、規定期間(1ヶ月、3ヶ月、6ヶ月)経年変化させられた。PhEur法(PhEur 3 補足2000)に従って測定された吸光度の定量が、安定性試験中のサンプル品質の評価のための判定基準として使用された。吸光度測定が、例えばクロマトグラフィー法を使用する存在不純物の検出よりも、より感度のよいオメプラゾールナトリウム分解過程のモニター方法であることは以前から示されている(PhEur 3 補足2000)。
【0029】
本発明の簡便で改善された方法に従って調製されたオメプラゾールナトリウム・エタノール溶媒和物およびオメプラゾールナトリウム形態Aは、粉末X線回折(XRPD)パターンにより分析され、その結果図1および図2に示す回折図形を得た。主要なピークは、位置および相対強度と共に回折図形より読み取り、下記表1に示した。ピークの位置(d値)は両化合物とも標準手順により測定した(Kug,H.P. & Aleksander,L.E.,1974)。相対強度はそれほどの信頼性はなく、数値の代わりに以下の定義を使用した。
【0030】
【表1】

【0031】
回折図形中に見いだされる低強度のいくつかの更なるピークは、表1から割愛した。
【0032】
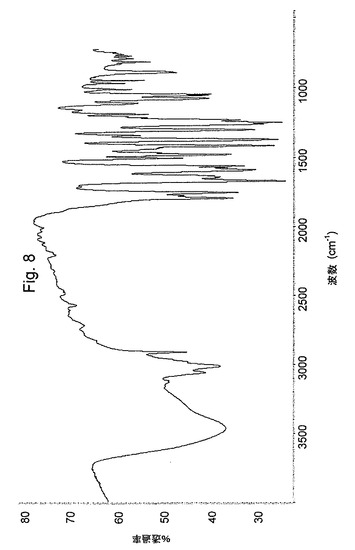
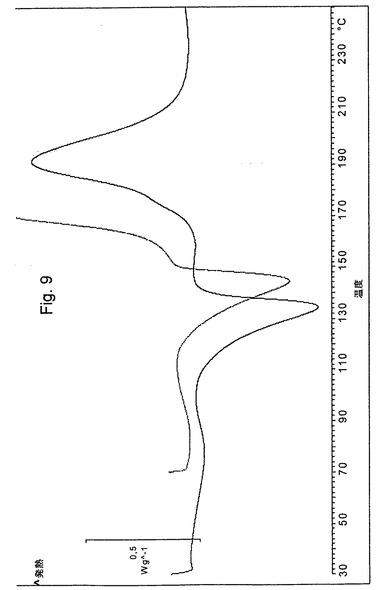
加えて、本発明の新規オメプラゾールナトリウム・エタノール溶媒和物は、図5のIRスペクトルおよび図9の示差走査熱量測定曲線(DSC)(実線)を有する。
【0033】
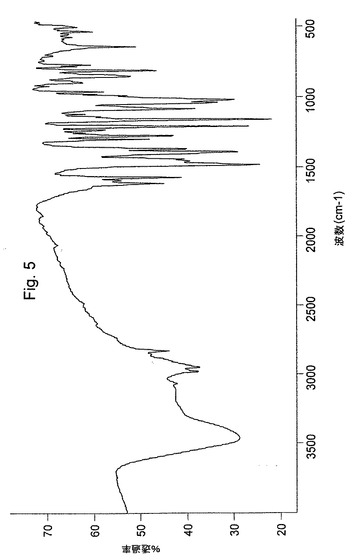
本発明に従って調製された結晶性オメプラゾールナトリウム形態Aは、図6のIRスペクトルおよび図9の示差走査熱量測定曲線(DSC)(点線)を有する。
【0034】
【表2】
【0035】
提示した分析データに基づくと、オメプラゾールナトリウム・エタノール溶媒和物とオメプラゾールナトリウム形態Aとは類似化合物を表すように見えるが、両回折図形を精査すると、一方の回折図形にもう一方には存在しない複数のピークがあり、その逆も同様であり、また配向性によらないピーク強度の違いも大きい。明らかに、オメプラゾールナトリウム・エタノール溶媒和物およびオメプラゾールナトリウム形態Aは、一方が溶媒和物としてもう一方が水和物として異なる結晶構造で結晶化し、従って異なる物理化学的特性(融点、溶解度、吸湿性、安定性を含む)を有する。
【0036】
本発明のもう1つの態様において、我々は、驚くべきことにおよび予期せぬことに、オメプラゾールナトリウムの2つの新規の結晶性多形、即ち本明細書中に開示されるオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fを見出した。
【0037】
オメプラゾールナトリウム・エタノール溶媒和物を、オメプラゾールナトリウム形態Aなどのオメプラゾールのナトリウム塩のいかなる他の形態をも実質的に含まず、および残留溶媒が著しく低濃度であるオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fに変換する簡便な方法も、本明細書中に開示される。
【0038】
上記用語「いかなる他の形態」は、無水物、水和物、溶媒和物、および非晶質物質を、ならびに先行技術で開示された多形を含んで意味する。
【0039】
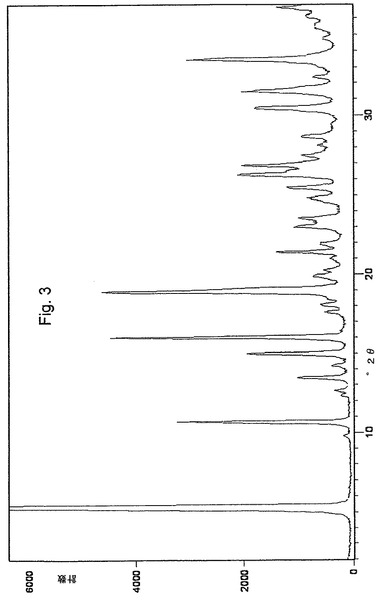
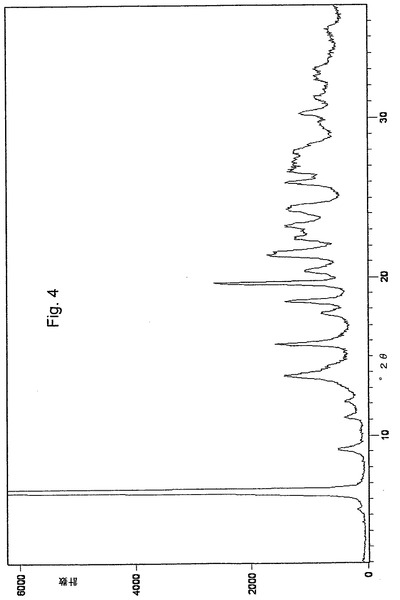
2つの新規な結晶形態、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは、それぞれ図3と図4とに示したそれらの粉末X線回折(XRPD)パターン、および図8と図9とに示したIRスペクトルにより特徴付けられる。これらの特徴は、先行技術で公知のオメプラゾールナトリウムのいかなる他の形態によっても示されない。
【0040】
オメプラゾールナトリウム形態Eは、5.33±0.2°2θ付近の非常に強いX線回折ピークにより特徴付けられる。更に、10.66、16.02、19.01、26.29および33.47±0.2°2θ付近の強い相対強度のピークにより特徴付けられ、13.48、14.98、19.89、21.42、23.02、25.47、30.43および31.47±0.2°2θ付近の中位相対強度のピークを伴う。
【0041】
オメプラゾールナトリウム形態Fは、6.52±0.2°2θ付近の非常に強いX線回折ピークにより特徴付けられる。更に、19.63±0.2°2θ付近の強い相対強度のピークにより特徴付けられることができ、13.79、15.76、18.47、20.38、21.50、22.50、23.22、24.28および25.96±0.2°2θ付近の中位相対強度のピークを伴う。
【0042】
本発明の更なる目的は、2つの新規な結晶形態、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fそれぞれの調製のための簡便な方法である。
【0043】
我々は予期せぬことに、オメプラゾールナトリウム・エタノール溶媒和物の、アセトン、エチルメチルケトン、4−メチル−2−ペンタノンまたはシクロヘキサノンからなる群より選択される適した溶媒からの再結晶により、残留溶媒含有量の低いオメプラゾールナトリウムの新規なオメプラゾール結晶形態が生じうることを見出した。
【0044】
従って、上述のように(実施例1および実施例2も参照)調製された中間化合物としての湿オメプラゾールナトリウム・エタノールまたは乾燥オメプラゾールナトリウム・エタノール溶媒和物は、上記選択された溶媒から好ましくは室温で再結晶することにより、新規オメプラゾールナトリウム形態Eまたは新規オメプラゾールナトリウム形態Fに変換され得る。2つのオメプラゾールナトリウムの新規結晶形態のうちどちらが得られるのかは、中間体の再結晶で使用される上記溶媒の選択に依存する。新規オメプラゾールナトリウムの沈殿結晶は、例えばろ過で収集し、減圧下約45℃で乾燥する。
【0045】
このようにして、オメプラゾールナトリウム・エタノール溶媒和物のアセトンまたはシクロヘキサノンからの再結晶により、オメプラゾール形態Eを0.1重量%未満の有機溶媒量を伴う無水生成物として得る。
【0046】
オメプラゾールナトリウム・エタノール溶媒和物の4−メチル−2−ペンタノンからの再結晶により、残留溶媒濃度がわずかに高め(即ち、約1重量%)の無水物質として、オメプラゾールナトリウム形態Fを得る。
【0047】
オメプラゾールナトリウム・エタノール溶媒和物のエチルメチルケトンからの再結晶により、約0.3重量%の残留溶媒量の無水物質として、両方の形態、即ちオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fの混合物を得る。
【0048】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F中の残存溶媒は、ガスクロマトグラフィーで決定され得る。
【0049】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F中の水分析は、それ自体公知の手法である熱重量分析またはカール・フィッシャー法で決定され得る。
【0050】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは、明確に定義された結晶状態で存在するため容易に特徴付けられる。オメプラゾールナトリウムの前記新規形態の両方は、簡便で再現性のある方法で調製され得る。
【0051】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは特定の条件下(窒素雰囲気下で保存されるなど)で安定な化合物であるが、それは両者とも吸湿性であり、空気の相対湿度に依存して空気からの水分の約7重量%までを吸収するためである。オメプラゾールナトリウムの前記新規形態は両方とも、そういった空気からの水分吸収により公知の結晶性オメプラゾールナトリウム形態Aに逆に変換され得る。
【0052】
EP−B−1 000 943はまた、アセトン中での、またはアセトンと例えばn−ヘキサン、イソプロピルエーテル、トルエンおよび酢酸エチルなどの溶媒との混合物中での再結晶によるスルホキシド化合物ナトリウム塩(例えば、ラベプラゾールナトリウム)のアセトン錯体の調製方法をも記述している。
【0053】
実施したNMR研究、ならびにGC(ガスクロマトグラフィー)分析により、我々は、アセトン中でのオメプラゾールナトリウム・エタノール溶媒和物の再結晶工程中に錯体はまったく生成しなかったことを立証した。
【0054】
オメプラゾールは有用なプロトンポンプ阻害剤であり、哺乳類での、特にヒトでの胃酸分泌調節に使用できることは公知である。特に、オメプラゾールナトリウム形態A、ならびに新規オメプラゾールナトリウム形態Eおよび新規オメプラゾールナトリウム形態Fは、例えば食道逆流、胃炎、十二指腸潰瘍、非潰瘍性消化不良、上部消化管出血、ストレス性潰瘍、ガストリノーマを含む胃酸関連疾患の予防および治療のために、NSAID治療を受ける患者において、および胃酸誤嚥を予防するために術前および術後において使用され得る。更に、オメプラゾールナトリウム形態Aならびに新規オメプラゾールナトリウム形態EおよびFの両方は、乾癬の治療ならびにヘリコバクター感染症およびそれらに関連する疾患の治療で有用でありうる。
【0055】
残留溶媒を実質的に含まないオメプラゾールナトリウム形態Aおよび/または形態Eおよび/または形態Fならびに薬学的に許容される賦形剤を含む医薬組成物の調製も、本明細書中に開示されている。前記医薬組成物は経口投与および非経口投与に適している。本発明によるオメプラゾールナトリウムの最適な投与経路ならびに治療投与量の規模は、いかなる場合においても治療すべき疾患の性質および重症度に依存する。用量および投与頻度はまた、年齢、体重および個々の患者の応答に従って変化させてもよい。一般に、有効成分の適した用量は、1日当たり10mgから80mgの範囲内であり、好ましくは1日総投与量が20から40mgの間である。投与形態は、カプセル剤、錠剤、分散剤、溶液剤、懸濁剤、乳剤、ゲル剤、散剤を含む。
【0056】
方法
粉末X線回折:分解能ステップ0.03°2θで2°から37°2θまでの範囲の反射配置およびCuKα照射を使用するSiemens d−5000粉末回折計。積分時間は1分解能ステップ当たり5秒であり、スリットは20mm(可変開度)および0.6mm(受光側)に設定した。
【0057】
FT−赤外:400から4000cm−1までの走査で16回走査、分解能2cm−1を有する、臭化カリウムペレット法を用いたNicolet Nexus FTIR分光光度計。
【0058】
示差走査熱量測定:Mettler Toledo DSC822e示差走査熱量計。サンプル(4−8g)を密封していない一穴アルミニウム皿中に置き、大気中、70℃から170℃までの温度範囲を3°K/minで加熱して、測定した。
【0059】
ガスクロマトグラフィー:カラムRTX624,30m×0.53mm。Tstarting(開始温度)=40℃、Tgradient(温度勾配)=40℃/minで200℃まで5分。インジェクター:スプリットなし、T=140℃。検出器:FID、T=200℃。移動相:ヘリウム、5psi。Toven(オーブン温度)=80℃。サンプル:115mg/mL DMA(N,N−ジメチルアセトアミド)。
【0060】
本発明は、以下の実施例により説明される。
【実施例1】
【0061】
湿オメプラゾールナトリウム・エタノールのオメプラゾールからの調製
NaOH(36.5g、0.91mol)の無水エタノール(650mL)溶液に、オメプラゾール(300g、0.87mol)を加える。スラリーを周囲温度で20分間撹拌する。得られた溶液を、空隙率B4のブフナーロート上のセライトおよび活性炭の層を通してろ過する。溶液にオメプラゾールナトリウム形態Aの種結晶(1g)を加えて結晶化を開始し、周囲温度で8時間撹拌する。形成したスラリーを0℃から5℃の温度範囲で更に8時間撹拌し、生成物をろ別し、無水エタノール(100mL)で洗浄し、5℃に冷却して湿オメプラゾールナトリウム・エタノール289gを得る。
【実施例2】
【0062】
オメプラゾールナトリウム・エタノール溶媒和物の調製
実施例1に記載されたようにして得られた湿オメプラゾールナトリウム・エタノール289gを減圧下45℃で一晩乾燥してオメプラゾールナトリウム・エタノール溶媒和物245gを得た。
【0063】
含水率%(TgAによるw/w)<0.5%。
【0064】
粉末XRDP、IR(KBr中)およびDSCは、図1、図5、および図9(実線)に示した。
【実施例3】
【0065】
オメプラゾールナトリウム形態Aの調製
実施例1で得られた湿オメプラゾールナトリウム・エタノール289gを、更にジイソプロピルエーテル300mLで3回洗浄し、5L反応容器に移し入れる。ジイソプロピルエーテル(3L)および水(45mL)を加え、スラリーを5℃で4時間、激しく撹拌した。結晶をろ過により分離し、減圧下45℃で一晩乾燥してオメプラゾールナトリウム形態A220gを得る。
【0066】
含水率%(TgAによるw/w)=7.5%。
【0067】
粉末XRDP、IR(KBr中)およびDSCは、図2、図6、および図9(点線)に示した。
【実施例4】
【0068】
オメプラゾールナトリウム形態Eの調製
実施例2で得られた乾燥オメプラゾールナトリウム・エタノール溶媒和物15gをアセトン(150mL)に溶解する。溶液を周囲温度で一晩撹拌後、得られる生成物をろ別し、減圧下45℃で一晩乾燥して表題化合物11.5gを得る。
【0069】
含水率%(TgAによるw/w)<0.5%。
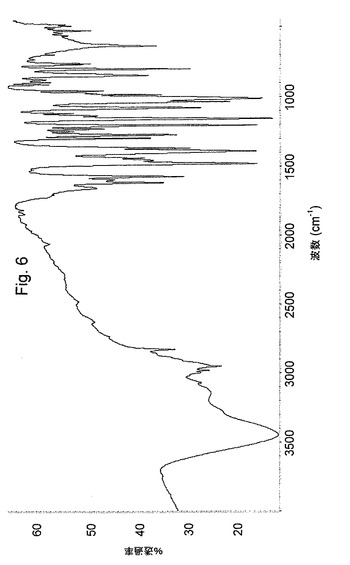
粉末XRDPおよびIR(KBr中)は、図3および図7に示した。
【実施例5】
【0070】
オメプラゾールナトリウム形態Fの調製
実施例2で得られた乾燥オメプラゾールナトリウム・エタノール溶媒和物20gを4−メチル−2−ペンタノン(400mL)に溶解する。溶液を周囲温度で一晩撹拌後、得られた生成物をろ別し、減圧下45℃で一晩乾燥して表題化合物16.4gを得る。
【0071】
含水率%(TgAによるw/w)<0.5%。
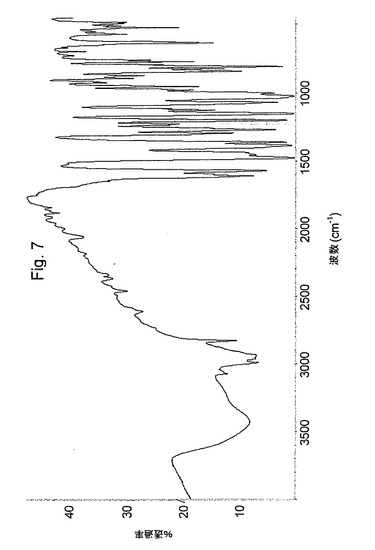
粉末XRDPおよびIR(KBr中)は、図4および図8に示した。
【図面の簡単な説明】
【0072】
【図1】オメプラゾールナトリウム・エタノール溶媒和物の粉末X線回折パターンを示す図である。
【図2】本発明に基づく方法により調製されたオメプラゾールナトリウム形態Aの粉末X線回折パターンを示す図である。
【図3】オメプラゾールナトリウム形態Eの粉末X線回折パターンを示す図である。
【図4】オメプラゾールナトリウム形態Fの粉末X線回折パターンを示す図である。
【図5】オメプラゾールナトリウム・エタノール溶媒和物のIRスペクトルを示す図である。
【図6】本発明による方法により調製されたオメプラゾールナトリウム形態AのIRスペクトルを示す図である。
【図7】オメプラゾールナトリウム形態EのIRスペクトルを示す図である。
【図8】オメプラゾールナトリウム形態FのIRスペクトルを示す図である。
【図9】本発明に従って調製されたオメプラゾールナトリウム・エタノール溶媒和物(実線)およびオメプラゾールナトリウム形態A(点線)の示差走査熱量分析曲線(DSC)を示す図である。
【技術分野】
【0001】
本発明は、医薬品工業分野に属し、新規のオメプラゾールナトリウム塩の結晶性エタノール溶媒和物に関する。オメプラゾールは、胃腸疾患の治療に使用される(5)6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールの一般名である。更に本発明は、結晶性オメプラゾールナトリウム・エタノール溶媒和物の調製方法、およびオメプラゾールナトリウムの異なる結晶形態への変換方法に関する。とりわけ、該新規オメプラゾールナトリウム・エタノール溶媒和物を、残留溶媒量を少なくして周囲温度で安定な公知の結晶形態のオメプラゾールナトリウム(以降、オメプラゾールナトリウム形態Aと呼ぶ)に変換するための容易に実施できおよび再現性のある方法に関する。
【0002】
本発明はまた、オメプラゾールナトリウムの2つの新規結晶形態にも関し、これらを以降、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fと呼ぶ。更に本発明は、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F(両方とも残留溶媒がかなり低濃度である。)の調製方法に関する。
【0003】
更に本発明は、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fの胃酸分泌過多関連疾患の治療への使用、および有効物質としてオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fを含む医薬組成物に関する。
【0004】
(技術的課題)
有効医薬品成分の化学的安定性、固体状態安定性および「保存寿命」は、薬学的有効化合物に関する重要な性質である。有効医薬品成分の安定性が、特に残留溶媒にも影響されることは一般に公知である。従って、有効医薬品成分の安定で結晶性の固体形態を残留溶媒を低濃度にして、好ましくは工業的に簡便で再現性のある方法で提供することが非常に望ましい。
【0005】
オメプラゾールナトリウムの新規の固体形態およびそれらの調製方法に対する必要性が普遍的にあるが、それは数多くの薬物が特定の固体形態で使用される場合に、望ましい溶出特性、およびいくつかの場合では望ましい生物学的利用率パターンを示すことが認められているからである。更に、多形体の安定性や吸湿性などの特性は種々に異なるであろう。
【背景技術】
【0006】
化学名(5)6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールとして公知であり一般名オメプラゾールを有する化合物は、胃酸分泌を阻害するプロトンポンプ阻害剤として公知である。オメプラゾールは、例えば胃食道逆流、食道炎、胸焼け、胃炎、分泌過多の状態(例えば、ゾリンジャー・エリソン症候群、内分泌腺腫)、十二指腸炎、胃潰瘍および十二指腸潰瘍などの、哺乳類、特にヒトにおける胃酸関連疾患および胃腸炎症性疾患の治療に使用できる。オメプラゾールはまた、例えばヘリコバクター・ピロリにより引き起こされる感染症などの感染症の治療にも有用である。オメプラゾールおよびその薬学的に許容できる塩は、特許EP−B−5129に初めて記載された。
【0007】
例えばナトリウム塩などのオメプラゾールの特定のアルカリ塩は、特許EP−B−124 495に初めて記載された。特許EP−B−124 495の実施例1および2に従って調製したオメプラゾールナトリウム塩は不安定であり、結晶形態と非晶質物質の混合物である。この混合物中に存在する結晶形態の1つは、オメプラゾールナトリウム形態Aであり、および1分子から2分子の水で水和した水和物であり、その水和物の1つの水分子は結晶構造中で強固に結合している一方、もう1つの水分子は乾燥により容易に除去される。1つの強固に結合した水分子を含有する得られる乾燥物質は、非常に吸湿性であり、水分を通常の条件下で迅速に吸収する。
【0008】
明確に定義されたオメプラゾールナトリウム一水和物塩(以降、オメプラゾールナトリウム形態Bと呼ぶ)およびその調製は、米国特許第6,207,188号に開示されている。その記載によるとオメプラゾールナトリウム形態Bは、例えば明確に定義されており、熱力学的に安定であり、非吸湿性でありおよび真に一水和物結晶形態であるなどの有利な特性を示す結晶形態である。形態Bとは対照的に、この特許はオメプラゾールナトリウム形態Aを、一定の保存条件下で完全に、または部分的にオメプラゾールナトリウム形態Bに変換されうる熱力学的に不安定な形態として記述している。米国特許第6,207,188号はまた、そのような不安定なオメプラゾールナトリウム形態Aの調製方法をも示している。記載された方法の欠点は、完結までに3日以上かかるので、時間を浪費することである。
【0009】
米国特許出願公報US 2004/0224987 A1は、類似しているが、オメプラゾールナトリウム形態A調製のための改善された方法を開示しており、この方法は、ナトリウムカチオンを放出する水性塩基Na+B−(Naがナトリウムを表し、Bがヒドロキシドまたはアルコキシド、イオン交換基、樹脂を表す)中のオメプラゾールを、C3からC7の分岐または直鎖炭化水素、C2からC7の分岐または直鎖エーテル、環状エーテル、低級脂肪酸エステル、脂肪族ケトン溶媒、ハロゲン化炭化水素溶媒またはニトリル溶媒からなる、場合により水を含む適切な溶媒中に室温で溶解する段階;生じた溶液を、生成物が溶けにくい適切な貧溶媒で中和する段階;反応混合物を室温で0から24時間撹拌する段階;反応混合物を固体物質が結晶化するまで冷却する段階;単離した固体を従来の方法でろ過する段階;上述の溶媒で洗浄する段階;および単離した化合物を30°から70℃で、好ましくは50°から60℃の温度で乾燥して、オメプラゾールナトリウムの形態Aを得る段階;を含む。得られたオメプラゾールナトリウム形態Aは、熱力学的により安定であり、非吸湿性であり、許容できる残留溶媒限界を有すると述べられている。US 2004/0224987は更に、新規な結晶性オメプラゾールナトリウム形態Cおよび結晶性オメプラゾールナトリウム形態D、およびそれらの調製方法を提供する。
【0010】
ES 2023778には、活性メチレン化合物のアルカリ塩を用いるオメプラゾールの、例えばナトリウム塩などの金属塩の製造方法が開示されている。
【0011】
オメプラゾールの非晶質形塩およびそのスプレードライ法を用いる調製方法がWO 01/87831に開示されている。
【0012】
米国特許第6,262,085号に記載されている発明は、6−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾールまたはその薬学的に許容できるその塩を提供し、それには、溶液中で塩を製造する結果、溶液中で起こる互変異性に依りメトキシ基をベンゾイミダゾール環上の6位に有するおよび5位に有する両方の化合物が製造されるという説明が付いている。
【0013】
式(I)のスルホキシド化合物またはその薬学的に許容できる塩のアセトン錯体が、ラベプラゾールの実施例を示す特許EP−B−1000943中に開示されている。
【0014】
(実施例を含む発明の説明)
本発明の目的は、周囲温度で安定と思われる残留溶媒量の低いオメプラゾールナトリウム形態Aを調製するための新規方法を見出すことである。更に本発明の目的は、新規な結晶性オメプラゾールナトリウム形態Eおよびその関連調製方法、ならびに新規な結晶性オメプラゾールナトリウム形態Fおよびその関連調製方法に関する。
【0015】
この課題は、安定な結晶性オメプラゾールナトリウム形態A調製のための新規方法、ならびに新規な結晶性オメプラゾールナトリウム形態Eおよび同様に新規な結晶性オメプラゾールナトリウム形態Fの調製方法における中間化合物である、新規の安定な結晶性オメプラゾールナトリウム・エタノール溶媒和物に関する本発明により解決された。粉末X線回折(XRPD)が、安定結晶性オメプラゾールナトリウム形態A、新規な安定結晶性中間体オメプラゾールナトリウム・エタノール溶媒和物、新規な安定結晶性オメプラゾールナトリウム形態E、および新規な安定結晶性オメプラゾールナトリウム形態F間の識別方法として使用される。
【0016】
本発明に従って、オメプラゾールナトリウム・エタノール溶媒和物は、オメプラゾール(原料)を無水エタノール中のNaOH溶液に溶解して調製する。得られた溶液から機械的粒子を、例えばろ過により除去し、更にオメプラゾールナトリウム形態Aの種結晶を加えて結晶化を誘導する。その後、スラリーを周囲温度で数時間、そして収量を上げるために、例えば0°から5℃などの低温で更に数時間撹拌する。沈殿したオメプラゾールナトリウム形態Aを、例えばろ過または遠心分離により分離し、冷無水エタノールで洗浄する。こうして誘導された湿オメプラゾールナトリウム・エタノールを減圧下、40°から50℃で、好ましくは約45℃で乾燥して、安定なエタノール溶媒和物の形態の無水オメプラゾールナトリウムを得る。用語「湿オメプラゾールナトリウム・エタノール」は、減圧下での乾燥の段階前に得られた生成物を意味する。用語「減圧」は一般に、約10mbarから約50mbarの圧力を指す。
【0017】
本発明による無水条件下での方法により調製されるオメプラゾールナトリウム・エタノール溶媒和物は、その結晶格子中に取り込まれた残留エタノールの約8から約11重量%(ガスクロマトグラフィーにより測定して)を含む。この取り込まれたエタノールはオメプラゾールナトリウムのエタノール溶媒和物結晶形態を規定し、更に乾燥しても除去できない。それ自体公知の手法である熱重量分析またはカール・フッシャー法により測定されたオメプラゾールナトリウム・エタノール溶媒和物中の水分定量は、無水生成物に相当する0.5重量%未満であることが見出された。
【0018】
オメプラゾールナトリウム形態Aは水に非常によく溶解し、そのこと自体が非経口製剤に適しており、胃食道逆流症(GERD)を患い、経口治療が受けられない患者を治療する機会を医師に提供する。このように、固体中に取り込まれた残存溶媒を伴う新規オメプラゾールナトリウム・エタノール溶媒和物の非経口製剤は薬学的に許容できなと思われるが、取り込まれた溶媒を実質的に含まないオメプラゾールナトリウムの結晶形態を調製するための重要な中間体として有用である。用語「実質的に含まない」は、残留溶媒(即ち、エタノール)の0.5重量%未満を意味する。
【0019】
従って、オメプラゾールナトリウムの水和物を特定の結晶形態で、即ち公知の形態Aで得るために、オメプラゾールナトリウム・エタノール溶媒和物の結晶格子内に取り込まれたエタノールを水分子と交換することが必要である。我々は、驚くべきことにおよび予期せぬことに、前記交換を新規オメプラゾールナトリウム・エタノール溶媒和物を非溶媒と水との混合物中で温浸することにより好都合に実施できることを見出した。用語「温浸すること」は、生成物をそれが不溶または難溶である溶媒(本明細書中では、非溶媒と称する)中に懸濁させ、次に水の少量を加え、得られた懸濁液を規定された時間の間撹拌する方法として理解される。
【0020】
より具体的には、オメプラゾールナトリウム・エタノール溶媒和物は、オメプラゾールナトリウム・エタノール溶媒和物の結晶を適した非溶媒と水との混合物中で温浸する方法によりオメプラゾールナトリウム形態Aに変換される。用語「適した溶媒」は、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒を意味し、好ましくはジイソプロピルエーテルであり、0°から20℃の、好ましくは5°から10℃の温度範囲で、30分から10時間の、より好ましくは約4時間の時間で用いられる。反応の完結後、沈殿したオメプラゾールナトリウム形態Aを、例えばろ過または遠心分離により優れた収率で回収し、望ましい生成物の劣化を防ぐ条件下で、例えば40°から50℃で減圧下10から24時間乾燥する。
【0021】
本発明のもう1つの態様において、オメプラゾールナトリウム・エタノール溶媒和物の代わりに湿オメプラゾールナトリウム・エタノールもまた、非溶媒と水との混合物中での温浸前に、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒、好ましくはジイソプロピルエーテルの適量であらかじめ洗浄するという条件下ではあるが、温浸の変法で使用してもよい。用語「非溶媒の適量」は、約20重量%から約10重量%の残留エタノールを洗い流してオメプラゾールナトリウム・エタノール溶媒和物を得るための量を意味する。従って、オメプラゾール(原料)を出発物質とし、湿オメプラゾールナトリウム・エタノールの洗浄を用いる結晶性ナトリウム形態Aの調製のための方法全体は、温浸段階前の乾燥を回避することにより、より時間を浪費しなくなる。
【0022】
結晶形態Aのオメプラゾールナトリウムは、オメプラゾールナトリウム1モル当たり水1から2モルで水和された水和物であるため、オメプラゾールナトリウムに関して水の少なくとも約10重量%から約20重量%が、好ましくは水の約10重量%が、残留溶媒量の少ない安定なオメプラゾールナトリウム結晶形態Aを得るために温浸工程において必要である。
【0023】
本発明による温浸工程で使用される混合物中の非溶媒:水の体積比は、40:1から100:1までの範囲内、より好ましくは60:1から80:1までの範囲内である。
【0024】
本発明による方法により調製されたオメプラゾールナトリウム形態A内に存在する残留溶媒濃度は、それ自体公知の方法であるガスクロマトグラフィーにより測定され、0.5重量%限界未満であることが見出された。
【0025】
本発明による方法により調製された結晶性オメプラゾールナトリウム形態A中の水分定量は、それ自体公知の方法である熱重量分析またはカール・フッシャー法により測定され、6から8%までであることが見出されたが、この量は水1モル、即ち4.7%が結晶中に結合する一方、その他の水分子は結晶上に吸収されているだけであることに対応する。
【0026】
従って本発明は、オメプラゾールナトリウム形態A調製のための方法を記述し、その方法は、オメプラゾール(原料)をNaOHの無水エタノール溶液中に溶解すること;オメプラゾールナトリウム形態Aの種結晶を添加して結晶化すること;新規中間体(即ち、無水オメプラゾールナトリウム・エタノール溶媒和物)を単離すること;および、前記新規中間体を非溶媒と水との混合物中での温浸工程により結晶形態Aのオメプラゾールナトリウム水和物に更に変換すること;を含む。従って、オメプラゾールナトリウム形態Aの調製のための本発明は、工業的規模で操作上安全に実施するために好都合な条件を使用する。本方法のもう1つの利点は、簡便であり、経済的であり、迅速であることである。
【0027】
加えて、我々は、本発明による方法により調製されたオメプラゾールナトリウム形態Aは安定であり、オメプラゾールナトリウムのいかなる他の形態をも実質的に含まず(即ち、オメプラゾールナトリウムのいかなる他の形態の検出可能量も含まない)、扱いやすく、例えば化学組成、吸湿性、溶解性および結晶形態などの物理化学的特性における顕著な変化を示さずにかなりの期間にわたって保存可能であることを見出した。
【0028】
本発明の方法により得られるオメプラゾールナトリウム形態Aの安定性は、薬学的有効物質の安定性を特徴付けるための標準手順(EU:CPMP採択、2003年3月、CPMP/ICH/2736/99として発行 − “Committee for proprietary medical products: Note for Guidance on ICH Q1A(R2) Stability testing guidelines: Stability testing of new drug substances and products”)により測定できる。梱包したオメプラゾールナトリウム形態Aが、経年変化加速条件下(医薬品製剤の安定性試験用の標準加速条件である温度40℃、相対湿度75%で)および/または経年変化過酷条件下(薬学的有効物質の安定性試験用の標準過酷条件である温度60℃で)で、規定期間(1ヶ月、3ヶ月、6ヶ月)経年変化させられた。PhEur法(PhEur 3 補足2000)に従って測定された吸光度の定量が、安定性試験中のサンプル品質の評価のための判定基準として使用された。吸光度測定が、例えばクロマトグラフィー法を使用する存在不純物の検出よりも、より感度のよいオメプラゾールナトリウム分解過程のモニター方法であることは以前から示されている(PhEur 3 補足2000)。
【0029】
本発明の簡便で改善された方法に従って調製されたオメプラゾールナトリウム・エタノール溶媒和物およびオメプラゾールナトリウム形態Aは、粉末X線回折(XRPD)パターンにより分析され、その結果図1および図2に示す回折図形を得た。主要なピークは、位置および相対強度と共に回折図形より読み取り、下記表1に示した。ピークの位置(d値)は両化合物とも標準手順により測定した(Kug,H.P. & Aleksander,L.E.,1974)。相対強度はそれほどの信頼性はなく、数値の代わりに以下の定義を使用した。
【0030】
【表1】
【0031】
回折図形中に見いだされる低強度のいくつかの更なるピークは、表1から割愛した。
【0032】
加えて、本発明の新規オメプラゾールナトリウム・エタノール溶媒和物は、図5のIRスペクトルおよび図9の示差走査熱量測定曲線(DSC)(実線)を有する。
【0033】
本発明に従って調製された結晶性オメプラゾールナトリウム形態Aは、図6のIRスペクトルおよび図9の示差走査熱量測定曲線(DSC)(点線)を有する。
【0034】
【表2】
【0035】
提示した分析データに基づくと、オメプラゾールナトリウム・エタノール溶媒和物とオメプラゾールナトリウム形態Aとは類似化合物を表すように見えるが、両回折図形を精査すると、一方の回折図形にもう一方には存在しない複数のピークがあり、その逆も同様であり、また配向性によらないピーク強度の違いも大きい。明らかに、オメプラゾールナトリウム・エタノール溶媒和物およびオメプラゾールナトリウム形態Aは、一方が溶媒和物としてもう一方が水和物として異なる結晶構造で結晶化し、従って異なる物理化学的特性(融点、溶解度、吸湿性、安定性を含む)を有する。
【0036】
本発明のもう1つの態様において、我々は、驚くべきことにおよび予期せぬことに、オメプラゾールナトリウムの2つの新規の結晶性多形、即ち本明細書中に開示されるオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fを見出した。
【0037】
オメプラゾールナトリウム・エタノール溶媒和物を、オメプラゾールナトリウム形態Aなどのオメプラゾールのナトリウム塩のいかなる他の形態をも実質的に含まず、および残留溶媒が著しく低濃度であるオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fに変換する簡便な方法も、本明細書中に開示される。
【0038】
上記用語「いかなる他の形態」は、無水物、水和物、溶媒和物、および非晶質物質を、ならびに先行技術で開示された多形を含んで意味する。
【0039】
2つの新規な結晶形態、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは、それぞれ図3と図4とに示したそれらの粉末X線回折(XRPD)パターン、および図8と図9とに示したIRスペクトルにより特徴付けられる。これらの特徴は、先行技術で公知のオメプラゾールナトリウムのいかなる他の形態によっても示されない。
【0040】
オメプラゾールナトリウム形態Eは、5.33±0.2°2θ付近の非常に強いX線回折ピークにより特徴付けられる。更に、10.66、16.02、19.01、26.29および33.47±0.2°2θ付近の強い相対強度のピークにより特徴付けられ、13.48、14.98、19.89、21.42、23.02、25.47、30.43および31.47±0.2°2θ付近の中位相対強度のピークを伴う。
【0041】
オメプラゾールナトリウム形態Fは、6.52±0.2°2θ付近の非常に強いX線回折ピークにより特徴付けられる。更に、19.63±0.2°2θ付近の強い相対強度のピークにより特徴付けられることができ、13.79、15.76、18.47、20.38、21.50、22.50、23.22、24.28および25.96±0.2°2θ付近の中位相対強度のピークを伴う。
【0042】
本発明の更なる目的は、2つの新規な結晶形態、オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fそれぞれの調製のための簡便な方法である。
【0043】
我々は予期せぬことに、オメプラゾールナトリウム・エタノール溶媒和物の、アセトン、エチルメチルケトン、4−メチル−2−ペンタノンまたはシクロヘキサノンからなる群より選択される適した溶媒からの再結晶により、残留溶媒含有量の低いオメプラゾールナトリウムの新規なオメプラゾール結晶形態が生じうることを見出した。
【0044】
従って、上述のように(実施例1および実施例2も参照)調製された中間化合物としての湿オメプラゾールナトリウム・エタノールまたは乾燥オメプラゾールナトリウム・エタノール溶媒和物は、上記選択された溶媒から好ましくは室温で再結晶することにより、新規オメプラゾールナトリウム形態Eまたは新規オメプラゾールナトリウム形態Fに変換され得る。2つのオメプラゾールナトリウムの新規結晶形態のうちどちらが得られるのかは、中間体の再結晶で使用される上記溶媒の選択に依存する。新規オメプラゾールナトリウムの沈殿結晶は、例えばろ過で収集し、減圧下約45℃で乾燥する。
【0045】
このようにして、オメプラゾールナトリウム・エタノール溶媒和物のアセトンまたはシクロヘキサノンからの再結晶により、オメプラゾール形態Eを0.1重量%未満の有機溶媒量を伴う無水生成物として得る。
【0046】
オメプラゾールナトリウム・エタノール溶媒和物の4−メチル−2−ペンタノンからの再結晶により、残留溶媒濃度がわずかに高め(即ち、約1重量%)の無水物質として、オメプラゾールナトリウム形態Fを得る。
【0047】
オメプラゾールナトリウム・エタノール溶媒和物のエチルメチルケトンからの再結晶により、約0.3重量%の残留溶媒量の無水物質として、両方の形態、即ちオメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fの混合物を得る。
【0048】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F中の残存溶媒は、ガスクロマトグラフィーで決定され得る。
【0049】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態F中の水分析は、それ自体公知の手法である熱重量分析またはカール・フィッシャー法で決定され得る。
【0050】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは、明確に定義された結晶状態で存在するため容易に特徴付けられる。オメプラゾールナトリウムの前記新規形態の両方は、簡便で再現性のある方法で調製され得る。
【0051】
オメプラゾールナトリウム形態Eおよびオメプラゾールナトリウム形態Fは特定の条件下(窒素雰囲気下で保存されるなど)で安定な化合物であるが、それは両者とも吸湿性であり、空気の相対湿度に依存して空気からの水分の約7重量%までを吸収するためである。オメプラゾールナトリウムの前記新規形態は両方とも、そういった空気からの水分吸収により公知の結晶性オメプラゾールナトリウム形態Aに逆に変換され得る。
【0052】
EP−B−1 000 943はまた、アセトン中での、またはアセトンと例えばn−ヘキサン、イソプロピルエーテル、トルエンおよび酢酸エチルなどの溶媒との混合物中での再結晶によるスルホキシド化合物ナトリウム塩(例えば、ラベプラゾールナトリウム)のアセトン錯体の調製方法をも記述している。
【0053】
実施したNMR研究、ならびにGC(ガスクロマトグラフィー)分析により、我々は、アセトン中でのオメプラゾールナトリウム・エタノール溶媒和物の再結晶工程中に錯体はまったく生成しなかったことを立証した。
【0054】
オメプラゾールは有用なプロトンポンプ阻害剤であり、哺乳類での、特にヒトでの胃酸分泌調節に使用できることは公知である。特に、オメプラゾールナトリウム形態A、ならびに新規オメプラゾールナトリウム形態Eおよび新規オメプラゾールナトリウム形態Fは、例えば食道逆流、胃炎、十二指腸潰瘍、非潰瘍性消化不良、上部消化管出血、ストレス性潰瘍、ガストリノーマを含む胃酸関連疾患の予防および治療のために、NSAID治療を受ける患者において、および胃酸誤嚥を予防するために術前および術後において使用され得る。更に、オメプラゾールナトリウム形態Aならびに新規オメプラゾールナトリウム形態EおよびFの両方は、乾癬の治療ならびにヘリコバクター感染症およびそれらに関連する疾患の治療で有用でありうる。
【0055】
残留溶媒を実質的に含まないオメプラゾールナトリウム形態Aおよび/または形態Eおよび/または形態Fならびに薬学的に許容される賦形剤を含む医薬組成物の調製も、本明細書中に開示されている。前記医薬組成物は経口投与および非経口投与に適している。本発明によるオメプラゾールナトリウムの最適な投与経路ならびに治療投与量の規模は、いかなる場合においても治療すべき疾患の性質および重症度に依存する。用量および投与頻度はまた、年齢、体重および個々の患者の応答に従って変化させてもよい。一般に、有効成分の適した用量は、1日当たり10mgから80mgの範囲内であり、好ましくは1日総投与量が20から40mgの間である。投与形態は、カプセル剤、錠剤、分散剤、溶液剤、懸濁剤、乳剤、ゲル剤、散剤を含む。
【0056】
方法
粉末X線回折:分解能ステップ0.03°2θで2°から37°2θまでの範囲の反射配置およびCuKα照射を使用するSiemens d−5000粉末回折計。積分時間は1分解能ステップ当たり5秒であり、スリットは20mm(可変開度)および0.6mm(受光側)に設定した。
【0057】
FT−赤外:400から4000cm−1までの走査で16回走査、分解能2cm−1を有する、臭化カリウムペレット法を用いたNicolet Nexus FTIR分光光度計。
【0058】
示差走査熱量測定:Mettler Toledo DSC822e示差走査熱量計。サンプル(4−8g)を密封していない一穴アルミニウム皿中に置き、大気中、70℃から170℃までの温度範囲を3°K/minで加熱して、測定した。
【0059】
ガスクロマトグラフィー:カラムRTX624,30m×0.53mm。Tstarting(開始温度)=40℃、Tgradient(温度勾配)=40℃/minで200℃まで5分。インジェクター:スプリットなし、T=140℃。検出器:FID、T=200℃。移動相:ヘリウム、5psi。Toven(オーブン温度)=80℃。サンプル:115mg/mL DMA(N,N−ジメチルアセトアミド)。
【0060】
本発明は、以下の実施例により説明される。
【実施例1】
【0061】
湿オメプラゾールナトリウム・エタノールのオメプラゾールからの調製
NaOH(36.5g、0.91mol)の無水エタノール(650mL)溶液に、オメプラゾール(300g、0.87mol)を加える。スラリーを周囲温度で20分間撹拌する。得られた溶液を、空隙率B4のブフナーロート上のセライトおよび活性炭の層を通してろ過する。溶液にオメプラゾールナトリウム形態Aの種結晶(1g)を加えて結晶化を開始し、周囲温度で8時間撹拌する。形成したスラリーを0℃から5℃の温度範囲で更に8時間撹拌し、生成物をろ別し、無水エタノール(100mL)で洗浄し、5℃に冷却して湿オメプラゾールナトリウム・エタノール289gを得る。
【実施例2】
【0062】
オメプラゾールナトリウム・エタノール溶媒和物の調製
実施例1に記載されたようにして得られた湿オメプラゾールナトリウム・エタノール289gを減圧下45℃で一晩乾燥してオメプラゾールナトリウム・エタノール溶媒和物245gを得た。
【0063】
含水率%(TgAによるw/w)<0.5%。
【0064】
粉末XRDP、IR(KBr中)およびDSCは、図1、図5、および図9(実線)に示した。
【実施例3】
【0065】
オメプラゾールナトリウム形態Aの調製
実施例1で得られた湿オメプラゾールナトリウム・エタノール289gを、更にジイソプロピルエーテル300mLで3回洗浄し、5L反応容器に移し入れる。ジイソプロピルエーテル(3L)および水(45mL)を加え、スラリーを5℃で4時間、激しく撹拌した。結晶をろ過により分離し、減圧下45℃で一晩乾燥してオメプラゾールナトリウム形態A220gを得る。
【0066】
含水率%(TgAによるw/w)=7.5%。
【0067】
粉末XRDP、IR(KBr中)およびDSCは、図2、図6、および図9(点線)に示した。
【実施例4】
【0068】
オメプラゾールナトリウム形態Eの調製
実施例2で得られた乾燥オメプラゾールナトリウム・エタノール溶媒和物15gをアセトン(150mL)に溶解する。溶液を周囲温度で一晩撹拌後、得られる生成物をろ別し、減圧下45℃で一晩乾燥して表題化合物11.5gを得る。
【0069】
含水率%(TgAによるw/w)<0.5%。
粉末XRDPおよびIR(KBr中)は、図3および図7に示した。
【実施例5】
【0070】
オメプラゾールナトリウム形態Fの調製
実施例2で得られた乾燥オメプラゾールナトリウム・エタノール溶媒和物20gを4−メチル−2−ペンタノン(400mL)に溶解する。溶液を周囲温度で一晩撹拌後、得られた生成物をろ別し、減圧下45℃で一晩乾燥して表題化合物16.4gを得る。
【0071】
含水率%(TgAによるw/w)<0.5%。
粉末XRDPおよびIR(KBr中)は、図4および図8に示した。
【図面の簡単な説明】
【0072】
【図1】オメプラゾールナトリウム・エタノール溶媒和物の粉末X線回折パターンを示す図である。
【図2】本発明に基づく方法により調製されたオメプラゾールナトリウム形態Aの粉末X線回折パターンを示す図である。
【図3】オメプラゾールナトリウム形態Eの粉末X線回折パターンを示す図である。
【図4】オメプラゾールナトリウム形態Fの粉末X線回折パターンを示す図である。
【図5】オメプラゾールナトリウム・エタノール溶媒和物のIRスペクトルを示す図である。
【図6】本発明による方法により調製されたオメプラゾールナトリウム形態AのIRスペクトルを示す図である。
【図7】オメプラゾールナトリウム形態EのIRスペクトルを示す図である。
【図8】オメプラゾールナトリウム形態FのIRスペクトルを示す図である。
【図9】本発明に従って調製されたオメプラゾールナトリウム・エタノール溶媒和物(実線)およびオメプラゾールナトリウム形態A(点線)の示差走査熱量分析曲線(DSC)を示す図である。
【特許請求の範囲】
【請求項1】
結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項2】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、表1に実質的に提示されたピークを含む粉末X線回折(XRPD)パターンを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項3】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図1に実質的に一致するピークを含む粉末X線回折パターンを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項4】
残留エタノールの当該化合物中の含水量が、約8重量%から約11重量%の量である、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項5】
当該化合物中の含水量が、0.5重量%未満である、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項6】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図5に実質的に一致する赤外スペクトルを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項7】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図9に実質的に一致する示差走査熱量測定サーモグラムを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項8】
請求項1から7で定義される結晶性オメプラゾール・エタノール溶媒和物の調製のための方法であり、該方法が、
a)オメプラゾールをNaOHの無水エタノール溶液に溶解する段階、
b)前記溶液を結晶化させる段階、
c)得られた結晶をろ過または遠心分離により取り出す段階、
d)こうして誘導された湿オメプラゾールナトリウムの結晶を乾燥する段階、
e)こうして得られた無水オメプラゾールナトリウム・エタノール溶媒和物を単離する段階、
を含む、前記方法。
【請求項9】
オメプラゾールナトリウム形態Aが、結晶化を誘導するために用いられる、請求項8に記載の方法。
【請求項10】
結晶性オメプラゾールナトリウム形態Aの調製のための方法であり、該方法が、
a)無水結晶性オメプラゾール・エタノール溶媒和物を、非溶媒と水との混合物中で温浸する段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Aを単離する段階、
c)こうして得られたオメプラゾールナトリウム形態Aを乾燥する段階、
を含む、前記方法。
【請求項11】
非溶媒が、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される、請求項10a)に記載の方法。
【請求項12】
オメプラゾールナトリウム形態Aの調製のための方法であり、該方法が、
a)湿オメプラゾールナトリウム・エタノールを、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒を用いて洗浄する段階、
b)こうして誘導されたオメプラゾールナトリウム・エタノール溶媒和物を、該非溶媒と水との混合物中で温浸する段階、
c)反応完結後、こうして得られたオメプラゾールナトリウム形態Aを単離する段階、
d)こうして得られたオメプラゾールナトリウム形態Aを乾燥する段階、
を含む、前記方法。
【請求項13】
温浸工程における混合物中の非溶媒:水の体積比が、40:1から100:1の範囲内である、請求項8から12で定義される方法。
【請求項14】
温浸工程における混合物中の非溶媒:水の体積比が、60:1から80:1の範囲内である、請求項8から12で定義される方法。
【請求項15】
オメプラゾールナトリウム形態A中に存在する残留溶媒含有量が、0.5重量%未満である、請求項8から12で定義される方法。
【請求項16】
結晶性オメプラゾールナトリウム形態E。
【請求項17】
結晶性オメプラゾールナトリウム形態Eが、5.33、10.66、13.48、14.98、16.02、19.01、19.89、21.42、23.02、25.47、26.29、30.43、31.47および33.47±0.2°2θのピークを有する粉末X線回折パターンを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項18】
結晶性オメプラゾールナトリウム形態Eが、図3に一致するピークを含むX線回折図パターンを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項19】
結晶性オメプラゾールナトリウム形態Eが、図7に実質的に一致する赤外スペクトルを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項20】
請求項16に記載の結晶性オメプラゾールナトリウム形態Eであり、該化合物中の残留溶媒含有量が0.1重量%未満である、前記結晶性オメプラゾールナトリウム形態E。
【請求項21】
結晶性オメプラゾールナトリウム形態F。
【請求項22】
結晶性オメプラゾールナトリウムFが、6.52、13.79、16.76、18.47、19.63、20.38、21.50、22.50、23.22、24.28、および25.96±2θのピークを有する粉末X線回折パターンを示す、請求項21に記載の結晶性オメプラゾールナトリウムF。
【請求項23】
結晶性オメプラゾールナトリウム形態Fが、図8に一致するピークを含む粉末X線回折パターンを示す、請求項21に記載の結晶性オメプラゾールナトリウム形態F。
【請求項24】
結晶性オメプラゾールナトリウム形態Fが、図8に実質的に一致する赤外スペクトルを示す、請求項21に記載の結晶性オメプラゾールナトリウム形態F。
【請求項25】
請求項21に記載の結晶性オメプラゾールナトリウム形態Fであり、該化合物中の残留溶媒含有量が約1重量%である、前記結晶性オメプラゾールナトリウム形態F。
【請求項26】
請求項16から20で定義される結晶性オメプラゾールナトリウム形態Eの調製のための方法であり、該方法が、
a)無水オメプラゾールナトリウム・エタノール溶媒和物が、アセトンおよびシクロヘキサノンからなる群より選択される溶媒から再結晶される段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Eが、単離される段階、
c)こうして得られたオメプラゾールナトリウム形態Eを乾燥する段階、
を含む、前記方法。
【請求項27】
請求項21から25で定義される結晶性オメプラゾールナトリウム形態Fの調製方法であり、該方法が、
a)無水オメプラゾールナトリウム・エタノール溶媒和物を4−メチル−2−ペンタノンから再結晶する段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Fを単離する段階、
c)こうして得られたオメプラゾールナトリウム形態Fを乾燥する段階、
を含む、前記方法。
【請求項28】
薬学的に許容できる賦形剤との混合物中に請求項16から20で定義されるオメプラゾールナトリウム形態Eを含む、医薬品製剤。
【請求項29】
胃腸疾患治療用の薬物の製造における、有効成分として請求項16から20で定義されるオメプラゾールナトリウム形態Eの使用。
【請求項30】
薬学的に許容できる賦形剤との混合物中に請求項21から25で定義されるオメプラゾールナトリウム形態Fを含む、医薬品製剤。
【請求項31】
胃腸疾患治療用の薬物の製造における、有効成分として請求項21から25で定義されるオメプラゾールナトリウム形態Fの使用。
【請求項1】
結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項2】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、表1に実質的に提示されたピークを含む粉末X線回折(XRPD)パターンを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項3】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図1に実質的に一致するピークを含む粉末X線回折パターンを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項4】
残留エタノールの当該化合物中の含水量が、約8重量%から約11重量%の量である、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項5】
当該化合物中の含水量が、0.5重量%未満である、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項6】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図5に実質的に一致する赤外スペクトルを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項7】
結晶性オメプラゾールナトリウム・エタノール溶媒和物が、図9に実質的に一致する示差走査熱量測定サーモグラムを示す、請求項1に記載の結晶性オメプラゾールナトリウム・エタノール溶媒和物。
【請求項8】
請求項1から7で定義される結晶性オメプラゾール・エタノール溶媒和物の調製のための方法であり、該方法が、
a)オメプラゾールをNaOHの無水エタノール溶液に溶解する段階、
b)前記溶液を結晶化させる段階、
c)得られた結晶をろ過または遠心分離により取り出す段階、
d)こうして誘導された湿オメプラゾールナトリウムの結晶を乾燥する段階、
e)こうして得られた無水オメプラゾールナトリウム・エタノール溶媒和物を単離する段階、
を含む、前記方法。
【請求項9】
オメプラゾールナトリウム形態Aが、結晶化を誘導するために用いられる、請求項8に記載の方法。
【請求項10】
結晶性オメプラゾールナトリウム形態Aの調製のための方法であり、該方法が、
a)無水結晶性オメプラゾール・エタノール溶媒和物を、非溶媒と水との混合物中で温浸する段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Aを単離する段階、
c)こうして得られたオメプラゾールナトリウム形態Aを乾燥する段階、
を含む、前記方法。
【請求項11】
非溶媒が、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される、請求項10a)に記載の方法。
【請求項12】
オメプラゾールナトリウム形態Aの調製のための方法であり、該方法が、
a)湿オメプラゾールナトリウム・エタノールを、ジイソプロピルエーテル、tert−ブチルメチルエーテル、ジエチルエーテル、酢酸エチルおよびアセトニトリルからなる群より選択される非溶媒を用いて洗浄する段階、
b)こうして誘導されたオメプラゾールナトリウム・エタノール溶媒和物を、該非溶媒と水との混合物中で温浸する段階、
c)反応完結後、こうして得られたオメプラゾールナトリウム形態Aを単離する段階、
d)こうして得られたオメプラゾールナトリウム形態Aを乾燥する段階、
を含む、前記方法。
【請求項13】
温浸工程における混合物中の非溶媒:水の体積比が、40:1から100:1の範囲内である、請求項8から12で定義される方法。
【請求項14】
温浸工程における混合物中の非溶媒:水の体積比が、60:1から80:1の範囲内である、請求項8から12で定義される方法。
【請求項15】
オメプラゾールナトリウム形態A中に存在する残留溶媒含有量が、0.5重量%未満である、請求項8から12で定義される方法。
【請求項16】
結晶性オメプラゾールナトリウム形態E。
【請求項17】
結晶性オメプラゾールナトリウム形態Eが、5.33、10.66、13.48、14.98、16.02、19.01、19.89、21.42、23.02、25.47、26.29、30.43、31.47および33.47±0.2°2θのピークを有する粉末X線回折パターンを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項18】
結晶性オメプラゾールナトリウム形態Eが、図3に一致するピークを含むX線回折図パターンを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項19】
結晶性オメプラゾールナトリウム形態Eが、図7に実質的に一致する赤外スペクトルを示す、請求項16に記載の結晶性オメプラゾールナトリウム形態E。
【請求項20】
請求項16に記載の結晶性オメプラゾールナトリウム形態Eであり、該化合物中の残留溶媒含有量が0.1重量%未満である、前記結晶性オメプラゾールナトリウム形態E。
【請求項21】
結晶性オメプラゾールナトリウム形態F。
【請求項22】
結晶性オメプラゾールナトリウムFが、6.52、13.79、16.76、18.47、19.63、20.38、21.50、22.50、23.22、24.28、および25.96±2θのピークを有する粉末X線回折パターンを示す、請求項21に記載の結晶性オメプラゾールナトリウムF。
【請求項23】
結晶性オメプラゾールナトリウム形態Fが、図8に一致するピークを含む粉末X線回折パターンを示す、請求項21に記載の結晶性オメプラゾールナトリウム形態F。
【請求項24】
結晶性オメプラゾールナトリウム形態Fが、図8に実質的に一致する赤外スペクトルを示す、請求項21に記載の結晶性オメプラゾールナトリウム形態F。
【請求項25】
請求項21に記載の結晶性オメプラゾールナトリウム形態Fであり、該化合物中の残留溶媒含有量が約1重量%である、前記結晶性オメプラゾールナトリウム形態F。
【請求項26】
請求項16から20で定義される結晶性オメプラゾールナトリウム形態Eの調製のための方法であり、該方法が、
a)無水オメプラゾールナトリウム・エタノール溶媒和物が、アセトンおよびシクロヘキサノンからなる群より選択される溶媒から再結晶される段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Eが、単離される段階、
c)こうして得られたオメプラゾールナトリウム形態Eを乾燥する段階、
を含む、前記方法。
【請求項27】
請求項21から25で定義される結晶性オメプラゾールナトリウム形態Fの調製方法であり、該方法が、
a)無水オメプラゾールナトリウム・エタノール溶媒和物を4−メチル−2−ペンタノンから再結晶する段階、
b)反応完結後、こうして得られたオメプラゾールナトリウム形態Fを単離する段階、
c)こうして得られたオメプラゾールナトリウム形態Fを乾燥する段階、
を含む、前記方法。
【請求項28】
薬学的に許容できる賦形剤との混合物中に請求項16から20で定義されるオメプラゾールナトリウム形態Eを含む、医薬品製剤。
【請求項29】
胃腸疾患治療用の薬物の製造における、有効成分として請求項16から20で定義されるオメプラゾールナトリウム形態Eの使用。
【請求項30】
薬学的に許容できる賦形剤との混合物中に請求項21から25で定義されるオメプラゾールナトリウム形態Fを含む、医薬品製剤。
【請求項31】
胃腸疾患治療用の薬物の製造における、有効成分として請求項21から25で定義されるオメプラゾールナトリウム形態Fの使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2008−545768(P2008−545768A)
【公表日】平成20年12月18日(2008.12.18)
【国際特許分類】
【出願番号】特願2008−515135(P2008−515135)
【出願日】平成18年6月7日(2006.6.7)
【国際出願番号】PCT/EP2006/005425
【国際公開番号】WO2006/131338
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
【公表日】平成20年12月18日(2008.12.18)
【国際特許分類】
【出願日】平成18年6月7日(2006.6.7)
【国際出願番号】PCT/EP2006/005425
【国際公開番号】WO2006/131338
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
[ Back to top ]