
オランザピンの製法
本発明は、有機溶媒中、式(II)の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンと反応させることによる、式(I)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン(オランザピン)の製法であって、前記反応を、トルエン及び1,3-ジメチル-2-イミダゾリジノンの混合物中で行うことを特徴とする製法に関する。本発明は、式(IB)の新規な2-メチル-4-(4-メチルピペリジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物、その製法及び前記新規化合物を含んでなる医薬組成物にも関する。
式(I)
【化1】

式(II)
【化2】
式(I)
【化1】
式(II)
【化2】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、改善されたオランザピンの製法、新規なオランザピン塩水和物及びその製法に関する。本発明は、前記の新規なオランザピン塩水和物を含有する医薬組成物及び精神病状態の治療におけるその使用にも関する。
【背景技術】
【0002】
式(IA)
【化1】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン(以下、「オランザピン」と称する)は、有用な抗精神病剤であることが知られている。
【0003】
オランザピンは、ヨーロッパ特許第454,436号に初めて記載された。この特許明細書に記載された合成法の最終ステップによれば、窒素雰囲気下、トルエン及びジメチルスルホキシドの1:1混合物中、混合物の沸点において、式(II)
【化2】
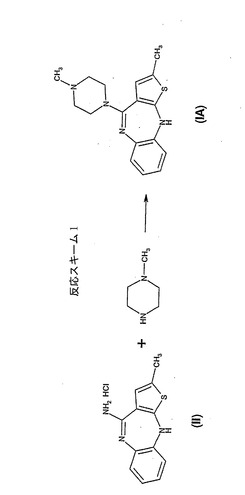
の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、過剰量のN-メチルピペラジンと20時間反応させ、反応混合物を50℃に冷却する。オランザピンは、混合物に少量の水を添加することによって結晶化され、このようにして得られた粗製生成物を、アセトニトリルから再結晶している。所望のオランザピンが収率48%で得られる。反応は図1に示す反応スキーム1で表される。
【0004】
国際特許公開WO 2004/094433号には、オランザピン2塩酸塩の2つの新規な多形相、さらに、新規な多形オランザピン・1塩酸塩が開示されている。
【0005】
ヨーロッパ特許第733,635号明細書には、いわゆる「技術的品質」のオランザピンの製法が開示されている。この製法も、反応スキーム1に従って行われ、6倍量のジメチルスルホキシドが温度120℃で使用される点で異なる。反応は、HPLCによって検知される。反応は、混合物中に残留する式(II)の原料物質の量が5%となるまで行われる。ついで、20℃に冷却し、10倍量のメタノール及び3倍量の水を連続して混合物に添加することによって、オランザピンを温度5℃で結晶化させる。このようにして得られた粗製オランザピンの収率は76.7%である。
【0006】
本発明の発明者らは、従来技術において特定されているように、ジメチルスルホキシド中で又はトルエン及びジメチルスルホキシドの1:1混合物中で行われる上記反応を再現した。再現の間に、反応を不活性ガス(例えば、窒素又はアルゴン)雰囲気下で行う場合にも、式(III)
【化3】
のオランザピンN-オキシドが望ましくない量で生成されるとの知見を得た。これは、利用できる各種の文献によれば、ジメチルスルホキシドは酸化剤として作用するにもかかわらず、発明者らの実験によれば、ジメチルスルホキシドはオランザピンを酸化しないため、極めて驚くべきことである。この事実にもかかわらず、反応スキーム1に示す反応のための溶媒としてジメチルスルホキシドを使用する場合には、より多量の望まれていない式(III)のN-オキシドが生成される。医薬用物質について求められる、より厳格な分析的要件は、式(III)のN-オキシドのような不純物の量の最大限の低減を要求する。発明者らの経験によれば、有効成分としてオランザピンを含有する医薬組成物中に存在する式(III)の不純物の量は、保存の間に増大する。オランザピンとその式(III)のN-オキシドとの間の構造類似性のため、2つの化合物の分離(すなわち、式(III)の不純物の除去)は、一方では、煩雑であり、他方では、不可避的な損失を招く。
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、式(III)のN-オキシドの生成を最少にすることを可能にする、産業上許容されるオランザピンの製法を提供することにある。
【課題を解決するための手段】
【0008】
上記目的は、本発明によって解決される。
【0009】
本発明は、ジメチルスルホキシドの代わりに、反応を、他の二極性の非プロトン性溶媒(すなわち、1,3-ジメチル-2-イミダゾリジノン)及びトルエンの混合物中で行う場合には、式(III)のオランザピンN‐オキシドがかなり少ない量で生成され、長期間の保存においても、医薬組成物について許される制限を越えてほど増大しないため、組成物の安定に関して、N‐オキシドは問題とならないとの知見に基づくものである。
【発明を実施するための最良の形態】
【0010】
本発明の1態様によれば、有機溶媒中、式(II)の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンと反応させることによって、式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン(オランザピン)を製造する方法であって、前記反応を、トルエン及び1,3-ジメチル2-イミダゾリジノンの混合物中で行うことを特徴とする製法が提供される。
【0011】
1,3-ジメチル-2-イミダゾリジノンは、有利な薬学上の特性を有する。この化合物は強力な極性の非プロトン性溶媒であり、酸及びアルカリの存在下において、高温であっても、安定であり、腐食性ではない。1,3-ジメチル-2-イミダゾリジノンの沸点及び発火点は高く(それぞれ、225℃及び120℃)、LD50=2840 mg/kgである。その高誘電率及び溶媒化作用のため、1,3-ジメチル-2-イミダゾリジノンは反応体を活性化する。1,3-ジメチル-2-イミダゾリジノンの適用の他の利点は、ジメチルスルホキシドの酸化性、不快な環境上及び安全上の性質を排除するとの事実にある。
【0012】
反応の間、N-メチルピペラジンは、好ましくは、過剰量で適用される。
【0013】
反応は、トルエン及び1,3-ジメチル-2-イミダゾリジノンの、好ましくは3:1混合物、特に好ましくは1:1混合物、より好ましくは2:1混合物中で行われる。反応は、温度100〜130℃において行われる。反応は、不活性ガス(好ましくは、窒素又はアルゴン)雰囲気下で実施される。
【0014】
本発明による方法の他の利点は、反応が、従来技術によるジメチルスルホキシド中で行われ、20時間を要する方法よりも、かなり短い時間(8〜11時間、好ましくは9時間以内)で行われることにある。反応混合物の蒸発後に得られるオランザピンは高純度である。所望により、生成物をアセトニトリルから再結晶できる。
【0015】
本発明の他の態様によれば、式(IB)
【化4】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を製造する方法であって、式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピンを、水性エタノール媒体中、塩化水素と反応させることを特徴とする製法が提供される。
【0016】
反応は、好ましくは、エタノール及び水の8:2混合物、特に好ましくは9:1混合物中で行われる。
【0017】
式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピンのエタノール懸濁液に、塩化水素水溶液を添加することによって実施される。好ましくは37%塩化水素水溶液が使用される。
【0018】
上記方法によって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、純度99.9%程度(HPLCによる)及び収率83.5%程度で調製できる。
【0019】
本発明のさらに他の態様によれば、新規な式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物が提供される。
【0020】
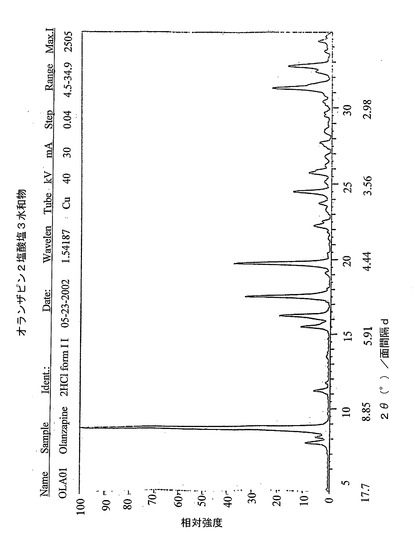
放射線CuKαを使用することによって測定した式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物のX線粉末回折パターンは、図2のチャートに相当し、下記の表1に示すX線粉末回折パターンによって特徴付けられる。
【0021】
【表1】
【0022】
上記データは、下記の条件下で測定されたものである:
装置:PHILIPS 粉末回折装置 XPERT PW 3710
放射線:CuKα(λ:1.54190Å)
モノクロメーター:グラファイト
励起電圧:40kV
アノード電流:30mA
方法:
内標準物質:SRM 675
マイカ粉末(合成フッ素金雲母)Ser. No.:981307
連続測定:θ/2θ スキャン:4.5〜35.00°2θ
ステップサイズ:0.02〜0.04°
サンプル:フラット表面、幅0.5mm、石英サンプルホルダー内、室温で測定及び保存
【0023】
式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物(オランザピン2塩酸塩3水和物)は、高度に有利な結晶型である。生成物は、優秀な濾過、乾燥及び保存特性を有する形態学的に均一な安定した物質であり、その保存に当たって、特殊な環境を必要としない。
【0024】
本発明のさらに他の態様によれば、有効成分として、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体の医薬品キャリヤー及び/又は医薬品添加剤との混合物として含んでなる医薬組成物が提供される。
【0025】
本発明のさらに他の態様によれば、医薬組成物を製造する方法であって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、薬学上許容される固体又は液体のキャリヤー及び/又は添加剤と混合し、混合物を製剤することを特徴とする医薬品の製法が提供される。
【0026】
本発明による医薬組成物は、医薬品工業において一般的に適用されている方法によって調製される。本発明による医薬組成物は、経口(例えば、錠剤、コーチング錠、カプセル、丸剤、溶液、懸濁液又はエマルジョン)、直腸(例えば、座剤)、非経口(例えば、静脈内、腹腔内、等)又は経皮投与される。
【0027】
本発明による医薬組成物は、通常の医薬品キャリヤー及び/又は添加剤を含有できる。キャリヤーとしては、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、ショ糖、乳糖、ペクチン、デキストリン、デンプン、ゼラチン、トラガカント、メチルセルロース、ナトリウムカルボキシメチルセルロース、低融点ワックス、カカオ脂等が使用される。カプセルの場合には、キャリヤーは、一般にカプセルの壁であり、従って、追加のキャリヤーは不要である。経口投与のための剤形として、トローチ剤及び小袋も挙げられる。錠剤、粉末剤、カプセル、丸剤、小袋及びトローチ剤は、経口投与に特に適する固状剤形である。
【0028】
座剤は、キャリヤーとして、低融点ワックス(例えば、脂肪酸トリグリセライドの混合物又はカカオ脂)を含有する。座剤は、ワックスを溶融し、溶融物中に有効成分を均質に分散させ、均質な溶融混合物を好適なサイズ及び形状の型に注加し、冷却下、混合物を固化させることによって調製される。
【0029】
錠剤は、有効成分と好適なキャリヤーとを好適な割合で混合し、混合物を圧縮して、好適なサイズ及び形の錠剤とすることによって調製される。
【0030】
粉末剤は、細かく粉砕した有効成分と、細かく粉砕したキャリヤーとを混合することによって調製される。
【0031】
液体医薬組成物として、徐放性の溶液、懸濁液又はエマルジョンが挙げられる。水溶液及び水性プロピレングリコール溶液が有利である。非経口投与に好適な液体医薬組成物は、好ましくは、水性ポリエチレングリコール溶液の形で調製される。
【0032】
経口投与に適する水溶液は、有効成分を水に溶解し、好適な着色料、芳香剤、安定剤及び粘度調節剤を添加することによって調製される。
【0033】
経口投与に適する水性懸濁液は、有効成分を、粘稠物質(例えば、天然又は合成のゴム、樹脂、メチルセルロース、ナトリウムカルボキシメチルセルロース又は他の公知の懸濁化剤)の存在下、水に懸濁化させることによって調製される。
【0034】
固体医薬組成物の他のタイプは、使用直前に液体組成物に転化され、液体の形でヒトに経口投与されるものである。このような投与の液剤(有効成分に加えて、着色料、芳香剤、保存料、緩衝剤、人工又は天然の甘味料、分散剤、粘度調節剤等を含有する)としては、溶液、懸濁液又はエマルジョンが挙げられる。
【0035】
本発明の医薬組成物は、好ましくは、用量ユニット形で調製される。このような用量ユニットは、所望の量の有効成分を含有する。用量ユニットは、分割した量の組成物を収容するパッケージの形で市販される(例えば、バイアル又はアンプルに詰めた錠剤、カプセル又は粉末剤)。用語「用量ユニット」は、カプセル、錠剤、トローチ剤、小袋自体に関するものであり、好適な数の用量ユニットを収容するパッケージにも関する。
【0036】
有効成分は、本発明による医薬組成物から、直ちに又は徐々に放出される。
【0037】
本発明による医薬組成物は、通常、有効成分約0.1〜100 mg、好ましくは約0.5〜50mgを含有する。
【0038】
本発明のさらに他の態様によれば、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、医薬有効成分として使用が提供される。
【0039】
本発明のさらに他の態様によれば、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、抗精神病活性を有する医薬組成物の調製における使用が提供される。
【0040】
本発明のさらに他の態様によれば、精神病状態を治療する方法であって、このような治療を必要とする患者に、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、薬学上有効な量で投与することを特徴とする精神病状態の治療法が提供される。
【0041】
本発明は下記の利点を包含する:
‐オランザピン塩基の調製の際、望まれていない式(III)のN-オキシド不純物の生成量が少ない。
‐当該方法では、高純度のオランザピンを、優秀な収率で調製できる。
‐オランザピンの調製の際、良好な物理特性を有し、環境及び安全性の点でも好適な溶媒を使用する。
‐オランザピンが、かなり短時間で調製される。
‐本発明による新規なオランザピン2塩酸塩3水和物は、優秀な濾過、乾燥及び保存特性を有する形態学的に均一な結晶形であり、医薬工業において有利に使用される。
【0042】
下記の実施例において、本発明をさらに詳述するが、保護の範囲は、これら実施例に制限されない。
例1
【0043】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩53.77g(0.2モル)、トルエン538 cm3及びDMI(1,3-ジメチル-2-イミダゾリジノン)269 cm3の混合物に、撹拌下、N-メチルピペラジン186 cm3(167.4g, 1.67モル)を添加し、アルゴンストリームを発生させ、油浴を使用して、反応混合物を沸点(126℃)に加熱する。
HPLCによって検知して、原料物質が1%の量となるまで、混合物を9時間沸騰させた。ついで、5〜10ミリバールにて、温度50〜55℃で蒸発させる。このようにして得られた残渣を、氷水で温度約3〜5℃に冷却し、水320 cm3を滴下し、この間に、生成物が反応混合物から連続して分離する。さらに、温度5℃において1時間撹拌し、濾過し、水120 cm3にて洗浄し、赤外線ランプを使用して乾燥させる。このようにして、塩基56g(89.6%)が得られる(融点:189〜193℃;HPLC:98%)。これを14倍量のアセトニトリルから再結晶し、所望の化合物42.6g(76%)を得る(融点:194〜196℃;HPLC:99.87%)。
1HNMR (DMSO, i500):δ:7.59(s, 1H), 6.84(m, 1H), 6.81(m, 1H), 6.79(dd, 1H), 6.69(dd, 1H), 6.33(d, 1H, J=1.1Hz), 3.33(m, 4H), 2.37(t, 4H), 2.27(d, 3H, J=1.1Hz), 2.20(s, 3H)
例2
【0044】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩5g(0.019モル)、トルエン50cm3及びDMI 50cm3の混合物に、撹拌下、N-メチルピペラジン17.5 cm3を添加し、アルゴンストリームを発生させ、油浴を使用して、温度130℃において、反応混合物を11時間沸騰させる。ついで、5〜10ミリバールにて、温度50〜55℃で蒸発させる。
このようにして得られた褐色の残渣を、氷水を使用して、撹拌下、温度約3〜5℃に冷却する。ついで、同じ温度において、水30cm3を滴下する。さらに1時間撹拌し、水で洗浄し、乾燥させる。このようにして、所望の化合物4.5g(77.1%)が得られる(融点:193〜196℃;HPLC:98.8%)。
例3
【0045】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩26.58g(0.1モル)、トルエン266 cm3及びDMI 83cm3の混合物に、撹拌下、N-メチルピペラジン93cm3を添加し、アルゴンストリームを発生させ、油浴内で、温度120℃において、混合物を9時間沸騰させる。ついで、反応混合物を、5〜10ミリバール、温度50〜55℃で蒸発させる。
このようにして得られた褐色の残渣を、撹拌下、氷水で温度約3〜5℃に冷却し、同じ温度において、水160 cm3を滴下する。混合物を、温度約5℃において、さらに1時間撹拌し、濾過し、水60cm3にて洗浄し、乾燥させる。このようにして、所望の化合物23.7g(76%)が得られる(融点:194〜196℃;HPLC:99.18%)。
例4
【0046】
オランザピン2塩酸塩3水和物:(IB)
オランザピン塩基42.6g(0.14モル)を、エタノール680 cm3及び水68cm3の混合物中に懸濁させ、撹拌下、懸濁液に37%塩化水素29.2 cm3を滴下する。塩酸水素の添加の間に、懸濁液が溶液に変化し、ついで、黄色の結晶性生物が連続して分離する。混合物をさらに2時間撹拌する。ついで、懸濁液を濾過し、冷却したエタノール15cm3ずつで2回洗浄し、赤外線ランプ下で乾燥する。このようにして、所望の生成物51.4g(83.5%)が得られる(融点(Koffler):201〜240℃(連続的分解);HPLC:99.9%)。
例5
【0047】
オランザピン2塩酸塩3水和物:(IB)
エタノール80ml中にオランザピン塩基5g(0.016モル)を含有する懸濁液に、激しく撹拌しながら、温度25℃において、2時間で37%塩化水素溶液3.3ml(1.46g, 0.04モル)を滴下する。ついで、混合物を温度25℃においてさらに2時間撹拌し、このようにして得られた懸濁液に、水16mlを滴下し、懸濁液を、温度25℃において24時間撹拌する。このようにして得られた黄色の結晶性物質を濾取し、乾燥する。このようにして、所望の化合物5.8g(82%)が得られる。
例6
【0048】
オランザピン2塩酸塩3水和物:(IB)
エタノール80ml中にオランザピン塩基5g(0.016モル)を含有する懸濁液に、激しく撹拌しながら、温度25℃において、2時間内で37%塩化水素溶液3.3ml(1.46g, 0.04モル)を滴下する。ついで、混合物に水16mlを添加する。水の添加の間に、溶液が形成され、30分で結晶が分離し始める。懸濁液を24時間撹拌し、濾過し、乾燥する。このようにして、所望の化合物4.2g(60%)が得られる。
例7
【0049】
4-{2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン-4-イル}-1-メチルピペラジン1-オキシド(III)(オランザピンN-オキシド)
オランザピン塩基5.0g(0.016モル)をジクロロメタン50cm3中に懸濁させ、懸濁液を0〜5℃に冷却し、これに75% m-クロロ過安息香酸4.05g(3.03g, 0.016モル)を添加する。混合物を、初に、温度0〜5℃において2時間、ついで、10〜12℃において10時間撹拌し、その後、次のように処理する:撹拌下、水100 cm3に注ぎ、ジクロロメタン150cm3ずつで3回抽出する。有機相を、塩化ナトリウム溶液及び水で連続して洗浄する。抽出に続いて、「N-オキシド」が、水相から、白色沈殿の形で分離する。ついで、混合物を濾過する。このようにして、所望の物質4.1g(79.1%)が得られる(融点:204〜206℃;HPLC:99.6%)。
有機相(なお、オランザピン塩基を含有する)の蒸発によって、物質2.78gを得る。これをカラムクロマトグラフィー(溶離液:トルエン及びメタノールの6:4混合物)に供して、物質1.6gを得た後、これをエーテル中に懸濁し、エタノールから再結晶する。このようにして、生成物0.8gがさらに得られる(HPLC:99.7%)。
C17H20N4OS・1.5 H2O(355.44)に関する元素分析:
理論値:C 57.39, H 6.47, N 15.76%
測定値:C 57.25, H 6.5, N 15.8%
IR (KBr): 3213, 3051, 2945, 2915, 1588, 1563, 1466, 1416, 1283, 1220, 1136
1HNMR (DMSO, i500):δ:7.97(s, 1H), 6.85(t, 1H), 6.83(t, 1H), 6.81(dd, 1H), 6.73(dd, 1H), 6.39(s, 1H), 3.78(d, 2H), 3.6(t, 2H), 3.42(t, 2H), 3.11(s, 3H), 2.96(d, 2), 2.27(s, 3H)
13CNMR(DMSO, i500):δ:157.04, 154.2, 144.27, 140.6, 128.33, 127.72, 123.86, 123.66, 122.66, 119.20, 117.78, 64.75, 60.37, 41.47, 15.27
質量分析によれば、M+Hは329であり、断片イオンはm/z値311、285、229において認められる。
【図面の簡単な説明】
【0050】
【図1】4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンを反応させてオランザピンを生成する反応スキームを示す図である。
【図2】式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物のX線粉末回折パターンのチャートを示す図である。
【技術分野】
【0001】
本発明は、改善されたオランザピンの製法、新規なオランザピン塩水和物及びその製法に関する。本発明は、前記の新規なオランザピン塩水和物を含有する医薬組成物及び精神病状態の治療におけるその使用にも関する。
【背景技術】
【0002】
式(IA)
【化1】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン(以下、「オランザピン」と称する)は、有用な抗精神病剤であることが知られている。
【0003】
オランザピンは、ヨーロッパ特許第454,436号に初めて記載された。この特許明細書に記載された合成法の最終ステップによれば、窒素雰囲気下、トルエン及びジメチルスルホキシドの1:1混合物中、混合物の沸点において、式(II)
【化2】
の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、過剰量のN-メチルピペラジンと20時間反応させ、反応混合物を50℃に冷却する。オランザピンは、混合物に少量の水を添加することによって結晶化され、このようにして得られた粗製生成物を、アセトニトリルから再結晶している。所望のオランザピンが収率48%で得られる。反応は図1に示す反応スキーム1で表される。
【0004】
国際特許公開WO 2004/094433号には、オランザピン2塩酸塩の2つの新規な多形相、さらに、新規な多形オランザピン・1塩酸塩が開示されている。
【0005】
ヨーロッパ特許第733,635号明細書には、いわゆる「技術的品質」のオランザピンの製法が開示されている。この製法も、反応スキーム1に従って行われ、6倍量のジメチルスルホキシドが温度120℃で使用される点で異なる。反応は、HPLCによって検知される。反応は、混合物中に残留する式(II)の原料物質の量が5%となるまで行われる。ついで、20℃に冷却し、10倍量のメタノール及び3倍量の水を連続して混合物に添加することによって、オランザピンを温度5℃で結晶化させる。このようにして得られた粗製オランザピンの収率は76.7%である。
【0006】
本発明の発明者らは、従来技術において特定されているように、ジメチルスルホキシド中で又はトルエン及びジメチルスルホキシドの1:1混合物中で行われる上記反応を再現した。再現の間に、反応を不活性ガス(例えば、窒素又はアルゴン)雰囲気下で行う場合にも、式(III)
【化3】
のオランザピンN-オキシドが望ましくない量で生成されるとの知見を得た。これは、利用できる各種の文献によれば、ジメチルスルホキシドは酸化剤として作用するにもかかわらず、発明者らの実験によれば、ジメチルスルホキシドはオランザピンを酸化しないため、極めて驚くべきことである。この事実にもかかわらず、反応スキーム1に示す反応のための溶媒としてジメチルスルホキシドを使用する場合には、より多量の望まれていない式(III)のN-オキシドが生成される。医薬用物質について求められる、より厳格な分析的要件は、式(III)のN-オキシドのような不純物の量の最大限の低減を要求する。発明者らの経験によれば、有効成分としてオランザピンを含有する医薬組成物中に存在する式(III)の不純物の量は、保存の間に増大する。オランザピンとその式(III)のN-オキシドとの間の構造類似性のため、2つの化合物の分離(すなわち、式(III)の不純物の除去)は、一方では、煩雑であり、他方では、不可避的な損失を招く。
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、式(III)のN-オキシドの生成を最少にすることを可能にする、産業上許容されるオランザピンの製法を提供することにある。
【課題を解決するための手段】
【0008】
上記目的は、本発明によって解決される。
【0009】
本発明は、ジメチルスルホキシドの代わりに、反応を、他の二極性の非プロトン性溶媒(すなわち、1,3-ジメチル-2-イミダゾリジノン)及びトルエンの混合物中で行う場合には、式(III)のオランザピンN‐オキシドがかなり少ない量で生成され、長期間の保存においても、医薬組成物について許される制限を越えてほど増大しないため、組成物の安定に関して、N‐オキシドは問題とならないとの知見に基づくものである。
【発明を実施するための最良の形態】
【0010】
本発明の1態様によれば、有機溶媒中、式(II)の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンと反応させることによって、式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン(オランザピン)を製造する方法であって、前記反応を、トルエン及び1,3-ジメチル2-イミダゾリジノンの混合物中で行うことを特徴とする製法が提供される。
【0011】
1,3-ジメチル-2-イミダゾリジノンは、有利な薬学上の特性を有する。この化合物は強力な極性の非プロトン性溶媒であり、酸及びアルカリの存在下において、高温であっても、安定であり、腐食性ではない。1,3-ジメチル-2-イミダゾリジノンの沸点及び発火点は高く(それぞれ、225℃及び120℃)、LD50=2840 mg/kgである。その高誘電率及び溶媒化作用のため、1,3-ジメチル-2-イミダゾリジノンは反応体を活性化する。1,3-ジメチル-2-イミダゾリジノンの適用の他の利点は、ジメチルスルホキシドの酸化性、不快な環境上及び安全上の性質を排除するとの事実にある。
【0012】
反応の間、N-メチルピペラジンは、好ましくは、過剰量で適用される。
【0013】
反応は、トルエン及び1,3-ジメチル-2-イミダゾリジノンの、好ましくは3:1混合物、特に好ましくは1:1混合物、より好ましくは2:1混合物中で行われる。反応は、温度100〜130℃において行われる。反応は、不活性ガス(好ましくは、窒素又はアルゴン)雰囲気下で実施される。
【0014】
本発明による方法の他の利点は、反応が、従来技術によるジメチルスルホキシド中で行われ、20時間を要する方法よりも、かなり短い時間(8〜11時間、好ましくは9時間以内)で行われることにある。反応混合物の蒸発後に得られるオランザピンは高純度である。所望により、生成物をアセトニトリルから再結晶できる。
【0015】
本発明の他の態様によれば、式(IB)
【化4】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を製造する方法であって、式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピンを、水性エタノール媒体中、塩化水素と反応させることを特徴とする製法が提供される。
【0016】
反応は、好ましくは、エタノール及び水の8:2混合物、特に好ましくは9:1混合物中で行われる。
【0017】
式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピンのエタノール懸濁液に、塩化水素水溶液を添加することによって実施される。好ましくは37%塩化水素水溶液が使用される。
【0018】
上記方法によって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、純度99.9%程度(HPLCによる)及び収率83.5%程度で調製できる。
【0019】
本発明のさらに他の態様によれば、新規な式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物が提供される。
【0020】
放射線CuKαを使用することによって測定した式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物のX線粉末回折パターンは、図2のチャートに相当し、下記の表1に示すX線粉末回折パターンによって特徴付けられる。
【0021】
【表1】
【0022】
上記データは、下記の条件下で測定されたものである:
装置:PHILIPS 粉末回折装置 XPERT PW 3710
放射線:CuKα(λ:1.54190Å)
モノクロメーター:グラファイト
励起電圧:40kV
アノード電流:30mA
方法:
内標準物質:SRM 675
マイカ粉末(合成フッ素金雲母)Ser. No.:981307
連続測定:θ/2θ スキャン:4.5〜35.00°2θ
ステップサイズ:0.02〜0.04°
サンプル:フラット表面、幅0.5mm、石英サンプルホルダー内、室温で測定及び保存
【0023】
式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物(オランザピン2塩酸塩3水和物)は、高度に有利な結晶型である。生成物は、優秀な濾過、乾燥及び保存特性を有する形態学的に均一な安定した物質であり、その保存に当たって、特殊な環境を必要としない。
【0024】
本発明のさらに他の態様によれば、有効成分として、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体の医薬品キャリヤー及び/又は医薬品添加剤との混合物として含んでなる医薬組成物が提供される。
【0025】
本発明のさらに他の態様によれば、医薬組成物を製造する方法であって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、薬学上許容される固体又は液体のキャリヤー及び/又は添加剤と混合し、混合物を製剤することを特徴とする医薬品の製法が提供される。
【0026】
本発明による医薬組成物は、医薬品工業において一般的に適用されている方法によって調製される。本発明による医薬組成物は、経口(例えば、錠剤、コーチング錠、カプセル、丸剤、溶液、懸濁液又はエマルジョン)、直腸(例えば、座剤)、非経口(例えば、静脈内、腹腔内、等)又は経皮投与される。
【0027】
本発明による医薬組成物は、通常の医薬品キャリヤー及び/又は添加剤を含有できる。キャリヤーとしては、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、ショ糖、乳糖、ペクチン、デキストリン、デンプン、ゼラチン、トラガカント、メチルセルロース、ナトリウムカルボキシメチルセルロース、低融点ワックス、カカオ脂等が使用される。カプセルの場合には、キャリヤーは、一般にカプセルの壁であり、従って、追加のキャリヤーは不要である。経口投与のための剤形として、トローチ剤及び小袋も挙げられる。錠剤、粉末剤、カプセル、丸剤、小袋及びトローチ剤は、経口投与に特に適する固状剤形である。
【0028】
座剤は、キャリヤーとして、低融点ワックス(例えば、脂肪酸トリグリセライドの混合物又はカカオ脂)を含有する。座剤は、ワックスを溶融し、溶融物中に有効成分を均質に分散させ、均質な溶融混合物を好適なサイズ及び形状の型に注加し、冷却下、混合物を固化させることによって調製される。
【0029】
錠剤は、有効成分と好適なキャリヤーとを好適な割合で混合し、混合物を圧縮して、好適なサイズ及び形の錠剤とすることによって調製される。
【0030】
粉末剤は、細かく粉砕した有効成分と、細かく粉砕したキャリヤーとを混合することによって調製される。
【0031】
液体医薬組成物として、徐放性の溶液、懸濁液又はエマルジョンが挙げられる。水溶液及び水性プロピレングリコール溶液が有利である。非経口投与に好適な液体医薬組成物は、好ましくは、水性ポリエチレングリコール溶液の形で調製される。
【0032】
経口投与に適する水溶液は、有効成分を水に溶解し、好適な着色料、芳香剤、安定剤及び粘度調節剤を添加することによって調製される。
【0033】
経口投与に適する水性懸濁液は、有効成分を、粘稠物質(例えば、天然又は合成のゴム、樹脂、メチルセルロース、ナトリウムカルボキシメチルセルロース又は他の公知の懸濁化剤)の存在下、水に懸濁化させることによって調製される。
【0034】
固体医薬組成物の他のタイプは、使用直前に液体組成物に転化され、液体の形でヒトに経口投与されるものである。このような投与の液剤(有効成分に加えて、着色料、芳香剤、保存料、緩衝剤、人工又は天然の甘味料、分散剤、粘度調節剤等を含有する)としては、溶液、懸濁液又はエマルジョンが挙げられる。
【0035】
本発明の医薬組成物は、好ましくは、用量ユニット形で調製される。このような用量ユニットは、所望の量の有効成分を含有する。用量ユニットは、分割した量の組成物を収容するパッケージの形で市販される(例えば、バイアル又はアンプルに詰めた錠剤、カプセル又は粉末剤)。用語「用量ユニット」は、カプセル、錠剤、トローチ剤、小袋自体に関するものであり、好適な数の用量ユニットを収容するパッケージにも関する。
【0036】
有効成分は、本発明による医薬組成物から、直ちに又は徐々に放出される。
【0037】
本発明による医薬組成物は、通常、有効成分約0.1〜100 mg、好ましくは約0.5〜50mgを含有する。
【0038】
本発明のさらに他の態様によれば、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、医薬有効成分として使用が提供される。
【0039】
本発明のさらに他の態様によれば、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、抗精神病活性を有する医薬組成物の調製における使用が提供される。
【0040】
本発明のさらに他の態様によれば、精神病状態を治療する方法であって、このような治療を必要とする患者に、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、薬学上有効な量で投与することを特徴とする精神病状態の治療法が提供される。
【0041】
本発明は下記の利点を包含する:
‐オランザピン塩基の調製の際、望まれていない式(III)のN-オキシド不純物の生成量が少ない。
‐当該方法では、高純度のオランザピンを、優秀な収率で調製できる。
‐オランザピンの調製の際、良好な物理特性を有し、環境及び安全性の点でも好適な溶媒を使用する。
‐オランザピンが、かなり短時間で調製される。
‐本発明による新規なオランザピン2塩酸塩3水和物は、優秀な濾過、乾燥及び保存特性を有する形態学的に均一な結晶形であり、医薬工業において有利に使用される。
【0042】
下記の実施例において、本発明をさらに詳述するが、保護の範囲は、これら実施例に制限されない。
例1
【0043】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩53.77g(0.2モル)、トルエン538 cm3及びDMI(1,3-ジメチル-2-イミダゾリジノン)269 cm3の混合物に、撹拌下、N-メチルピペラジン186 cm3(167.4g, 1.67モル)を添加し、アルゴンストリームを発生させ、油浴を使用して、反応混合物を沸点(126℃)に加熱する。
HPLCによって検知して、原料物質が1%の量となるまで、混合物を9時間沸騰させた。ついで、5〜10ミリバールにて、温度50〜55℃で蒸発させる。このようにして得られた残渣を、氷水で温度約3〜5℃に冷却し、水320 cm3を滴下し、この間に、生成物が反応混合物から連続して分離する。さらに、温度5℃において1時間撹拌し、濾過し、水120 cm3にて洗浄し、赤外線ランプを使用して乾燥させる。このようにして、塩基56g(89.6%)が得られる(融点:189〜193℃;HPLC:98%)。これを14倍量のアセトニトリルから再結晶し、所望の化合物42.6g(76%)を得る(融点:194〜196℃;HPLC:99.87%)。
1HNMR (DMSO, i500):δ:7.59(s, 1H), 6.84(m, 1H), 6.81(m, 1H), 6.79(dd, 1H), 6.69(dd, 1H), 6.33(d, 1H, J=1.1Hz), 3.33(m, 4H), 2.37(t, 4H), 2.27(d, 3H, J=1.1Hz), 2.20(s, 3H)
例2
【0044】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩5g(0.019モル)、トルエン50cm3及びDMI 50cm3の混合物に、撹拌下、N-メチルピペラジン17.5 cm3を添加し、アルゴンストリームを発生させ、油浴を使用して、温度130℃において、反応混合物を11時間沸騰させる。ついで、5〜10ミリバールにて、温度50〜55℃で蒸発させる。
このようにして得られた褐色の残渣を、氷水を使用して、撹拌下、温度約3〜5℃に冷却する。ついで、同じ温度において、水30cm3を滴下する。さらに1時間撹拌し、水で洗浄し、乾燥させる。このようにして、所望の化合物4.5g(77.1%)が得られる(融点:193〜196℃;HPLC:98.8%)。
例3
【0045】
オランザピン塩基:(IA)
4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩26.58g(0.1モル)、トルエン266 cm3及びDMI 83cm3の混合物に、撹拌下、N-メチルピペラジン93cm3を添加し、アルゴンストリームを発生させ、油浴内で、温度120℃において、混合物を9時間沸騰させる。ついで、反応混合物を、5〜10ミリバール、温度50〜55℃で蒸発させる。
このようにして得られた褐色の残渣を、撹拌下、氷水で温度約3〜5℃に冷却し、同じ温度において、水160 cm3を滴下する。混合物を、温度約5℃において、さらに1時間撹拌し、濾過し、水60cm3にて洗浄し、乾燥させる。このようにして、所望の化合物23.7g(76%)が得られる(融点:194〜196℃;HPLC:99.18%)。
例4
【0046】
オランザピン2塩酸塩3水和物:(IB)
オランザピン塩基42.6g(0.14モル)を、エタノール680 cm3及び水68cm3の混合物中に懸濁させ、撹拌下、懸濁液に37%塩化水素29.2 cm3を滴下する。塩酸水素の添加の間に、懸濁液が溶液に変化し、ついで、黄色の結晶性生物が連続して分離する。混合物をさらに2時間撹拌する。ついで、懸濁液を濾過し、冷却したエタノール15cm3ずつで2回洗浄し、赤外線ランプ下で乾燥する。このようにして、所望の生成物51.4g(83.5%)が得られる(融点(Koffler):201〜240℃(連続的分解);HPLC:99.9%)。
例5
【0047】
オランザピン2塩酸塩3水和物:(IB)
エタノール80ml中にオランザピン塩基5g(0.016モル)を含有する懸濁液に、激しく撹拌しながら、温度25℃において、2時間で37%塩化水素溶液3.3ml(1.46g, 0.04モル)を滴下する。ついで、混合物を温度25℃においてさらに2時間撹拌し、このようにして得られた懸濁液に、水16mlを滴下し、懸濁液を、温度25℃において24時間撹拌する。このようにして得られた黄色の結晶性物質を濾取し、乾燥する。このようにして、所望の化合物5.8g(82%)が得られる。
例6
【0048】
オランザピン2塩酸塩3水和物:(IB)
エタノール80ml中にオランザピン塩基5g(0.016モル)を含有する懸濁液に、激しく撹拌しながら、温度25℃において、2時間内で37%塩化水素溶液3.3ml(1.46g, 0.04モル)を滴下する。ついで、混合物に水16mlを添加する。水の添加の間に、溶液が形成され、30分で結晶が分離し始める。懸濁液を24時間撹拌し、濾過し、乾燥する。このようにして、所望の化合物4.2g(60%)が得られる。
例7
【0049】
4-{2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン-4-イル}-1-メチルピペラジン1-オキシド(III)(オランザピンN-オキシド)
オランザピン塩基5.0g(0.016モル)をジクロロメタン50cm3中に懸濁させ、懸濁液を0〜5℃に冷却し、これに75% m-クロロ過安息香酸4.05g(3.03g, 0.016モル)を添加する。混合物を、初に、温度0〜5℃において2時間、ついで、10〜12℃において10時間撹拌し、その後、次のように処理する:撹拌下、水100 cm3に注ぎ、ジクロロメタン150cm3ずつで3回抽出する。有機相を、塩化ナトリウム溶液及び水で連続して洗浄する。抽出に続いて、「N-オキシド」が、水相から、白色沈殿の形で分離する。ついで、混合物を濾過する。このようにして、所望の物質4.1g(79.1%)が得られる(融点:204〜206℃;HPLC:99.6%)。
有機相(なお、オランザピン塩基を含有する)の蒸発によって、物質2.78gを得る。これをカラムクロマトグラフィー(溶離液:トルエン及びメタノールの6:4混合物)に供して、物質1.6gを得た後、これをエーテル中に懸濁し、エタノールから再結晶する。このようにして、生成物0.8gがさらに得られる(HPLC:99.7%)。
C17H20N4OS・1.5 H2O(355.44)に関する元素分析:
理論値:C 57.39, H 6.47, N 15.76%
測定値:C 57.25, H 6.5, N 15.8%
IR (KBr): 3213, 3051, 2945, 2915, 1588, 1563, 1466, 1416, 1283, 1220, 1136
1HNMR (DMSO, i500):δ:7.97(s, 1H), 6.85(t, 1H), 6.83(t, 1H), 6.81(dd, 1H), 6.73(dd, 1H), 6.39(s, 1H), 3.78(d, 2H), 3.6(t, 2H), 3.42(t, 2H), 3.11(s, 3H), 2.96(d, 2), 2.27(s, 3H)
13CNMR(DMSO, i500):δ:157.04, 154.2, 144.27, 140.6, 128.33, 127.72, 123.86, 123.66, 122.66, 119.20, 117.78, 64.75, 60.37, 41.47, 15.27
質量分析によれば、M+Hは329であり、断片イオンはm/z値311、285、229において認められる。
【図面の簡単な説明】
【0050】
【図1】4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンを反応させてオランザピンを生成する反応スキームを示す図である。
【図2】式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物のX線粉末回折パターンのチャートを示す図である。
【特許請求の範囲】
【請求項1】
有機溶媒中、式(II)
【化1】
の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンと反応させることによる、式(IA)
【化2】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン(オランザピン)を製造する方法であって、前記反応を、トルエン及び1,3-ジメチル-2-イミダゾリジノンの混合物中で行うことを特徴とする、オランザピンの製法。
【請求項2】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの3:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項3】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの1:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項4】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの2:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項5】
反応を温度100〜130℃において行う、請求項1〜4のいずれかに記載の製法。
【請求項6】
反応を8〜11時間で行う、請求項1〜5のいずれかに記載の製法。
【請求項7】
反応を9時間以内で行う、請求項6に記載の製法。
【請求項8】
式(IB)
【化3】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を製造する方法であって、前記反応を、式(IA)
【化4】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピンを、水性エタノール媒体中、塩化水素と反応させることを特徴とする、オランザピン2塩酸塩3水和物の製法。
【請求項9】
反応を、エタノール及び水の8:2(容量比)混合物中で行う、請求項8に記載の製法。
【請求項10】
反応を、エタノール及び水の9:1(容量比)混合物中で行う、請求項8に記載の製法。
【請求項11】
式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピンのエタノール懸濁液に、塩化水素水溶液を添加する、請求項8に記載の製法。
【請求項12】
37%塩化水素水溶液を使用する、請求項11に記載の製法。
【請求項13】
反応を、温度20〜50℃、好ましくは温度25℃において行う、請求項8〜12のいずれかに記載の製法。
【請求項14】
式(IB)
【化5】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物。
【請求項15】
図2に示すチャート及び下記の表に示すX線粉末回折パターン(放射線CuKαを使用して測定)によって特徴付けられるものである、請求項14記載の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物。
表
【表1】
【請求項16】
有効成分として、請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体の医薬品キャリヤー及び/又は添加剤との混合物として含んでなる、医薬組成物。
【請求項17】
請求項16記載の医薬組成物を製造する方法であって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体のキャリヤー及び/又は添加剤との混合し、混合物を製剤することを特徴とする、医薬組成物の製法。
【請求項18】
請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、医薬組成物の調製における使用。
【請求項19】
請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、抗精神病活性を有する医薬組成物の調製における使用。
【請求項20】
抗精神病治療の方法であって、このような治療を必要とする患者に、請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、の薬学上有効な量で投与することを特徴とする、抗精神病治療の方法。
【請求項1】
有機溶媒中、式(II)
【化1】
の4-アミノ-2-メチル-10H-チエノ[2,3-b][1,5]ベンゾジアゼピン塩酸塩を、N-メチルピペラジンと反応させることによる、式(IA)
【化2】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン(オランザピン)を製造する方法であって、前記反応を、トルエン及び1,3-ジメチル-2-イミダゾリジノンの混合物中で行うことを特徴とする、オランザピンの製法。
【請求項2】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの3:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項3】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの1:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項4】
トルエン及び1,3-ジメチル-2-イミダゾリジノンの2:1(容量比)混合物を使用する、請求項1に記載の製法。
【請求項5】
反応を温度100〜130℃において行う、請求項1〜4のいずれかに記載の製法。
【請求項6】
反応を8〜11時間で行う、請求項1〜5のいずれかに記載の製法。
【請求項7】
反応を9時間以内で行う、請求項6に記載の製法。
【請求項8】
式(IB)
【化3】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を製造する方法であって、前記反応を、式(IA)
【化4】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピンを、水性エタノール媒体中、塩化水素と反応させることを特徴とする、オランザピン2塩酸塩3水和物の製法。
【請求項9】
反応を、エタノール及び水の8:2(容量比)混合物中で行う、請求項8に記載の製法。
【請求項10】
反応を、エタノール及び水の9:1(容量比)混合物中で行う、請求項8に記載の製法。
【請求項11】
式(IA)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピンのエタノール懸濁液に、塩化水素水溶液を添加する、請求項8に記載の製法。
【請求項12】
37%塩化水素水溶液を使用する、請求項11に記載の製法。
【請求項13】
反応を、温度20〜50℃、好ましくは温度25℃において行う、請求項8〜12のいずれかに記載の製法。
【請求項14】
式(IB)
【化5】
の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物。
【請求項15】
図2に示すチャート及び下記の表に示すX線粉末回折パターン(放射線CuKαを使用して測定)によって特徴付けられるものである、請求項14記載の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物。
表
【表1】
【請求項16】
有効成分として、請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体の医薬品キャリヤー及び/又は添加剤との混合物として含んでなる、医薬組成物。
【請求項17】
請求項16記載の医薬組成物を製造する方法であって、式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、不活性な固体又は液体のキャリヤー及び/又は添加剤との混合し、混合物を製剤することを特徴とする、医薬組成物の製法。
【請求項18】
請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、医薬組成物の調製における使用。
【請求項19】
請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物の、抗精神病活性を有する医薬組成物の調製における使用。
【請求項20】
抗精神病治療の方法であって、このような治療を必要とする患者に、請求項14記載の式(IB)の2-メチル-4-(4-メチルピペラジン-1-イル)-10H-チエノ-[2,3-b][1,5]ベンゾジアゼピン2塩酸塩3水和物を、の薬学上有効な量で投与することを特徴とする、抗精神病治療の方法。
【図1】

【図2】
【図2】
【公表番号】特表2009−515867(P2009−515867A)
【公表日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願番号】特願2008−539513(P2008−539513)
【出願日】平成18年11月10日(2006.11.10)
【国際出願番号】PCT/HU2006/000096
【国際公開番号】WO2007/054750
【国際公開日】平成19年5月18日(2007.5.18)
【出願人】(507333074)
【Fターム(参考)】
【公表日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願日】平成18年11月10日(2006.11.10)
【国際出願番号】PCT/HU2006/000096
【国際公開番号】WO2007/054750
【国際公開日】平成19年5月18日(2007.5.18)
【出願人】(507333074)
【Fターム(参考)】
[ Back to top ]