
オリゴ糖−タンパク複合体
本明細書において、タンパク質および式I〜VIうちの1つのオリゴ糖を含む、複合体が提供される。かかる複合体を含む医薬組成物がまた、本明細書において提供される。オリゴ糖−糖タンパク複合体を投与することにより、哺乳動物におけるリソソーム蓄積症を治療する方法がさらに提供される。

【発明の詳細な説明】
【技術分野】
【0001】
本願は、米国特許仮出願第61/122,851号の優先権の利益を主張し、その全体を参照により本明細書に組み込む。
【0002】
本発明は、一般に個々のオリゴ糖を含むオリゴ糖−タンパク複合体、およびかかる複合体を含む組成物に関する。本発明はさらに、オリゴ糖−リソソーム酵素複合体を用いた、リソソーム蓄積症の治療方法に関する。
【背景技術】
【0003】
リソソーム蓄積症(LSD)は、リソソーム加水分解酵素の活性欠損に関与する、希少な代謝性疾患の1つであり、40種を超える遺伝子疾患が含まれる。LSDの顕著な特徴は、リソソーム代謝産物の異常蓄積であり、多数の膨張したリソソーム形成を引き起こす。
【0004】
LSDは、欠損酵素の活性化したものを患者に投与することにより治療することができ、当該方法は酵素補充療法(ERT)と呼ばれる。末端にマンノース−6−リン酸(M6P)を有する補充酵素が投与されると、細胞表面結合型カチオン非依存性M6P受容体(CI−MPR)を介したエンドサイト−シスによって標的細胞に取り込まれ、リソソームに作用する。
【0005】
一般に、リン酸化程度が低い補充酵素は、細胞表面のM6P受容体によって効果的に取り込まれず、したがって、機能するリソソームに作用することができない。そのため、マンノースのリン酸化程度が低いと、補充酵素の治療効果に深刻かつ有害な影響が及ぶ可能性がある。
【0006】
補充酵素のM6P含有量を高める方法が開発されている。例えば、米国特許第6,534,300号、第6,670,165号、および第6,861,242号には、末端マンノース残基の酵素的リン酸化が記述されている。別の例として、米国特許第7,001,994号には、M6Pを含むオリゴ糖を、糖タンパク質と結合させる方法が記述されている。当該方法によって製造された、リソソーム酵素である酸性α−グルコシダーゼ(GAA)と、ビスM6Pオリゴ糖との複合体は、ポンペ病のマウスモデルにおいて、組換えヒトGAAより、骨格筋および心筋のグリコーゲンを減少させるのにより効果的であることがわかった。ポンペ病は、GAAの代謝欠陥に起因する常染色体劣性型筋疾患であり、リソソームグリコーゲンの蓄積を特徴とする。同様に、Zhuらにより、合成ビスM6Pオリゴ糖(式A)を、GAAと結合させることが記述されている(Zhuら,Biochem.J.389:619−628(2005))。式Aは、N−結合型グリカン(式B)の天然の三分岐鎖Man9コア構造から、一分岐鎖を取り除き、別の一分岐鎖を短くし、末端のマンノース残基をリン酸化することで作られた。
【0007】
【化1】
【0008】
得られた複合体は、CI−MPRに、より高い親和性で結合し、L6筋芽細胞に関して、より効果的に取り込まれ、ほぼ正常な酵素活性が得られた。しかし、このような成功があるとはいえ、リソソーム酵素と結合した際に、正常またはほぼ正常な酵素活性を有しつつ、CI−MPRとの親和性の改善、および/またはより効果的な細胞吸収を引き起こす、新規のオリゴ糖を同定することは、依然として重要である。しかしながら、取込みを改善するだけでは、必ずしもよりよい治療結果はもたらされない。ある種の結合方法およびオリゴ糖を用いると、得られる複合体の酵素活性は低い。したがって、LSD患者の治療結果を改善することができる、オリゴ糖および複合体を同定することが望まれる。
【0009】
さらに、式AおよびBに示したようなある種のオリゴ糖は、合成が困難かつ高価であることがある。その上、立体選択的にβ−結合型糖鎖を作成することは、糖化学において、困難な問題であった。商用規模での使用に関しては、別のオリゴ糖および合成方法のほうが現実的である可能性がある。オリゴ糖−タンパク複合体を製造するのに使用する方法の最適化がさらに求めらている。特に、治療目的に関しては、複合体製造は非常に不均一であってはならない。これは一貫性のない生物学的機能を生じる場合があるためである。使用するオリゴ糖およびリンカー、結合方法、精製方法、および処方などの複合体の様々な要素が、治療効果に影響を与える得る。
【発明の概要】
【0010】
したがって、本発明のある種の実施形態は、(1)タンパク質、および(2)式I:
【0011】
【化2】
【0012】
[式中、
a=α1,2;α1,3;α1,4;またはα1,6であり;
b=α1,2;α1,3;またはα1,4であり;
c=α1,2;α1,3;α1,4;またはα1,6であり;および
d=α、β、またはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0013】
本発明の他の実施形態は、(1)タンパク質、および(2)式II:
【0014】
【化3】
【0015】
[式中、
e=α1,2;α1,3;α1,4;またはα1,6であり;および
f=α、β、またはαとβとの混合物であり;
ただしe=α1,6のとき、f=αまたはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0016】
本発明のさらに他の実施形態は、(1)タンパク質、および(2)式III:
【0017】
【化4】
【0018】
[式中、
g=α1,2;α1,3;またはα1,4であり;
h=α1,2;α1,3;α1,4;またはα1,6であり;および
i=α、β、またはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0019】
本発明の追加の実施形態は、(1)タンパク質、および(2)式IV:
【0020】
【化5】
【0021】
[式中、
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;ならびに
xが2または3のとき、各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0022】
さらなる実施形態は、(1)タンパク質、および(2)式V:
【0023】
【化6】
【0024】
[式中、
lは、α、β、およびαとβとの混合物から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0025】
追加の実施形態は、(1)タンパク質、および(2)式VI:
【0026】
【化7】
【0027】
[式中、
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、任意にオキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで置換された、且つ任意にN、O、またはSから選択される1または2以上のヘテロ原子で中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4から選択され;
mは、α、β、およびαとβとの混合物から選択され;
yが2、3、または4のとき、各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0028】
追加の実施形態において、本発明は(1)タンパク質、および(2)式Aで表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0029】
ある実施形態において、複合体は、タンパク質1モルにつき、式Aのオリゴ糖を少なくとも2、3、4、または5モル含む。
【0030】
いくつかの実施形態において、本発明のオリゴ糖−タンパク複合体は、この複合体のオリゴ糖およびタンパク質成分間にリンカーを含む。
【0031】
本発明は、式I、II、III、IV、VもしくはVIで表されるオリゴ糖−タンパク複合体、および充填剤、膨張剤、崩壊剤、緩衝剤、安定剤、または賦形剤を含む医薬組成物を提供する。本発明はさらに、式I、II、III、IV、VまたはVIで表されるオリゴ糖−タンパク複合体、あるいはオリゴ糖−タンパク複合体などを含む医薬組成物を用いて、例えば下の表1に開示されるような、リソソーム蓄積症の治療方法を提供する。リソソーム蓄積症は、例えばファブリー病、ポンペ病、ニーマンピックA病、ニーマンピックB病、およびムコ多糖症I型から選択することができる。さらなる実施形態において、本発明は、それを必要とする被験体のリソソーム蓄積症を治療する医薬の製造に関して、(1)タンパク質、および(2)式I、II、III、IV、V、またはVIで表されるオリゴ糖を含む、オリゴ糖−タンパク複合体の使用を提供する。
【0032】
本発明の追加の実施形態は、この出願を通して検討される。本発明の他の目的、特徴、および利点は、以下の詳細な説明により明らかとなる。本発明の1態様に関して検討されたいかなる実施形態も、本発明の別の態様に適用し、逆もまた同様である。実施例の項目の実施形態は、本発明のすべての態様に適用することができる、本発明の実施形態であると理解される。
【0033】
しかしながら、本発明の範囲および趣旨内の様々な変化および修飾は、本願から当業者に明らかになるため、詳細な説明および具体例が、本発明の具体的な実施形態に示されるが、例示のためのみに与えられることが理解されるべきである。
【図面の簡単な説明】
【0034】
【図1】図1は、オリゴ糖82の合成工程を明示する、例示的な逆合成スキームを示す。
【図2】図2は、本明細書に記載されたオリゴ糖合成で使用することができる、単糖のビルディングブロック2aの例示的な合成を示す。
【図3】図3は、本明細書に記載されたオリゴ糖合成で使用することができる、単糖のビルディングブロック1、2、3、および4の例示的な合成を示す。
【図4】図4は、エチレンリンカーの製造に関する、合成スキームを示す。
【図5】図5は、ビルディングブロック2、3、および4を用いる、オリゴ糖82の三糖前駆体の構築に関する合成スキームを示す。
【図6】図6は、図4に記載された三糖前駆体からの、保護された七糖の構築に関する合成スキームを示す。
【図7】図7は、図5に記載された保護された七糖を脱保護して、オリゴ糖82を生成する、合成スキームを示す。
【図8A】図8Aは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8B】図8Bは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8C】図8Cは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8D】図8Dは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8E】図8Eは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図9A】NeoGAAβ SAM6の複合体化能における、酸化レベルの効果を示す。図9Aは、様々なモル比における、GAAと複合体化した、SAM2、SAM3、SAM4、線状SAM4、αSAM6、およびβSAM6オリゴ糖の量を示す。
【図9B】NeoGAAβ SAM6の複合化能における、酸化レベルの効果を示す。図9Bは、異なる量の過ヨウ素酸塩を用いて酸化した、rhGAAと複合体化した六糖(グリカン)の量を示す。
【図10A】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Aは、単糖組成物の分析(定量した単糖量の減少に基づいて推測した酸化)により、測定した酸化を示す。
【図10B】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Bは、ポジティブモードにおける、AA−標識したSAM6オリゴ糖のLTQ MS検出を示す。
【図10C】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Cは、AA−標識した酸化オリゴ糖に対応するMS/MSスペクトルを示す。
【図10D】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Dは、AA−標識した酸化オリゴ糖に対応するMS/MSスペクトルを示す。
【図10E】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す図。図10Eは、異なる量のGAOを用いて滴定したGAM複合体の単糖分析を示す。
【図11A】図11Aは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図11B】図11Bは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図11C】図11Cは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図12】図12は、2および22.5mMの過ヨウ素酸塩で処理した、rhGAAのペプチドマッピングのLC/MS分析を示す。非酸化、単一酸化、および二酸化したトリプシンペプチドT13(メチオニン172および173を含む)の溶出位置を長方形で強調する。
【図13A】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Aは、各サンプルの注入に関する、結合、解離、M6P溶出、および再生段階を示すセンサーグラムである。
【図13B】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Bは、NeoGAAサンプルおよびrhGAAコントロールサンプルに関する、センサーグラムデータの例示的な4パラメータフィットである。
【図13C】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Cは、各製造の異なる結合レベル全体の、2mMvs.7.5mM過ヨウ素酸塩を用いて製造されたNeoGAAサンプルのM6P受容体親和性である。
【図14】図14は、様々なNeoGAA複合体の特異的活性を示す。
【図15A】図15Aは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15B】図15Bは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15C】図15Cは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15D】図15Dは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15E】図15Eは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15F】図15Fは、SAM6複合体の週1回投与を4回行った後のGAAKOマウスにおける、組織グリコーゲンレベルを示す。
【図16】図16は、様々なNeoGAA複合体の吸収を明示する、L6筋芽細胞の取込アッセイの結果を示す。
【図17】図17は、SAM2処理後のGAAノックアウトマウスにおける、心臓、大腿四頭筋、および三頭筋のグリコーゲンクリアランスを示す。
【図18】図18は、SAM4処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図19】図19は、SAM6処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図20】図20は、SAM6およびGAM6処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図20B】図20Bは、rhGAA、GAMおよびSAM複合体の投与後5、15、30、60、120、240、および480分で回収された、マウス血清中のrhGAA濃度を示す。
【発明を実施するための形態】
【0035】
発明の説明
本発明がよく理解されるよう、まず、いくつかの語が定義される。追加の定義は、本願を通して行われる。
【0036】
本明細書および添付の請求項で使用される、単数形「a」、「an」および「the」は、他に文脈で明らかに指示されない限り、複数の対象を含む。したがって、例えば、「1つの化合物(a compound)」を含む方法への引用は、2つまたはそれ以上の化合物の混合物を含む。「または」という語は、他に文脈で明らかに指示されない限り、一般に「および/または」を含む意味で使用される。
【0037】
I.オリゴ糖−タンパク複合体
1つの実施形態において、本発明は、オリゴ糖−タンパク複合体を提供し、これはさらにリンカーを含むことができる。例示的なオリゴ糖、タンパク質、リンカー、複合体化方法、および複合体が開示されている。
【0038】
A.オリゴ糖
オリゴ糖は、上に示したような、式Bまたは式VIで表される二分岐鎖のオリゴ糖誘導体、あるいは式IVおよびVに示されるような直鎖のオリゴ糖誘導体から選択することができる。一般に、二分岐鎖のオリゴ糖は、末端に2つのM6P残基を有し、いくつかの実施形態においては、末端から2番目から1または2以上のM6P残基をさらに含むことができる。直鎖のオリゴ糖は、少なくとも1つのM6P残基を有し、末端のM6P残基を含むことがある。一般に、末端のM6P残基は、α1,2結合で結合し得る(末端のM6Pのα1,2結合は、α1,3またはα1,6結合よりも、CI−MPRおよびカチオン依存性MPR(CD−MPR)に、より結合するということを観察した、Distlerら,J.Biol.Chem.266:21687−21692(1991)を参照のこと)。いくつかの実施形態において、末端のM6P残基は、それぞれ還元末端で、α1,2結合によって隣り合う残基に結合している。いくつかの実施形態において、2つの末端M6P残基の間は、例えばX線結晶学、NMR、および/または分子モデリングにより測定されたように、5、10、15、20、25、30、35、または40Åより離れている。例えば、分子モデリングは、Balajiら,Glycobiology 4:497−515(1994)に記載されたように実施することができる。いくつかの実施形態において、オリゴ糖は、末端のM6P残基が比較的立体障害のないように選択される。いくつかの実施形態において、末端のM6P残基に比較的障害がないオリゴ糖は、末端のM6P残基に障害のあるオリゴ糖より、高い親和性でCI−MPRと結合する。
【0039】
一般に、オリゴ糖はCI−MPRに結合する。例えば、オリゴ糖は、例えば、500、100、50、10、5、1、または0.1nM未満、あるいは例えば、100、50、10、5、2、または1μM未満の解離定数で、CI−MPRに結合することができる。CI−MPRのN−末端領域1〜3の結晶構造は、リガンド結合型および非結合型の両方が知られている(Olsonら,J.Biol.Chem.279:34000−34009(2004)、Olsonら,EMBO J.23:2019−2028(2004))。さらに、構造的に関連のあるCD−MPRも、リガンド結合型および非結合型が知られている(Olsonら,J.Biol.Chem.274:29889−29886(1999)、Olsonら,J.Biol.Chem.277:10156−10161(2002))。したがって、当業者は、その受容体構造情報を使用して、適当なオリゴ糖を選択することができる。
【0040】
オリゴ糖は、例えば、下記で示されるオリゴ糖1〜127を含む、上記で示された式I、II、III、IV、V、またはVIで表されるオリゴ糖のいずれか1つから選択することができる。式I〜IIIで表されるオリゴ糖は、分岐鎖の除去、単糖残基の除去および/または置換、および/または隣接する単糖残基間の結合(α1,2、α1,3、α1,4、もしくはα1,6など)の修飾により、式Bから従来の方法で誘導される。ある種の実施形態において、オリゴ糖は、式I〜VIのいずれか一つに関して、α1,2、α1,3、またはα1,6結合によって結合した、1本または両方の分岐鎖に、例えば1、2、または3つのさらなるマンノース残基を有することがある。
【0041】
オリゴ糖は、例えば1つ、2つまたは3つのM6P残基を有してよい。オリゴ糖は、合計で1、2、3、4、5、6、7、8、9、または10個の単糖残基を有してよい。他の実施形態において、オリゴ糖は、1、2、3、4、5、6、7、8、9、または10個のマンノース残基を有してよく、このうちいずれもリン酸化または非リン酸化されてよい。
【0042】
いくつかの実施形態において、オリゴ糖はオリゴ糖1〜96から選択され、これは式Iの種類である。
【0043】
【化8】
【0044】
【表1】
【0045】
【表2】
【0046】
【表3】
【0047】
いくつかの実施形態において、dはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖1および2、3および4、または95および96の混合物である)。
【0048】
いくつかの実施形態において、オリゴ糖はオリゴ糖97〜103から選択され、これは式IIの種類である。
【0049】
【化9】
【0050】
いくつかの実施形態において、fはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖97および98、99および100、または101および102の混合物である)。
【0051】
いくつかの実施形態において、オリゴ糖はオリゴ糖104〜127から選択され、これは式IIIの種類である。
【0052】
【化10】
【0053】
いくつかの実施形態において、iはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖104および105、106および107、または126および127の混合物である)。
【0054】
いくつかの実施形態において、オリゴ糖は、オリゴ糖128〜133から選択され、これは式IVの種類である。
【0055】
【化11】
【0056】
いくつかの実施形態において、kはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖128および129、130および131、または132および133の混合物である)。
【0057】
いくつかの実施形態において、オリゴ糖はオリゴ糖134および135から選択され、これは式Vの種類である。
【0058】
【化12】
【0059】
いくつかの実施形態において、オリゴ糖はオリゴ糖134および135の混合物である。
【0060】
いくつかの実施形態において、オリゴ糖はオリゴ糖136であり、これは式VIの種類である。
【0061】
【化13】
【0062】
いくつかの実施形態において、オリゴ糖は、天然の供給源から単離することができる。天然の供給源から単離されたオリゴ糖は、関連するオリゴ糖の同種の混合物または異種の混合物であってよい。
【0063】
ある種の実施形態において、オリゴ糖は化学的および/または酵素的合成により製造される。いくつかの実施形態において、オリゴ糖は天然の供給源から単離されたオリゴ糖を、化学的または酵素的に修飾することで製造することができる(「半合成」)。
【0064】
オリゴ糖は、例えば図1〜7、Obsornら,Oligosaccharides:Their Synthesis and Biological Roles,Oxford University Press,2000、Wangら(eds),Synthesis of Carbohydrates through Biotechnology,American Chemical Society,2004、Seeberger,Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries,Wiley−Interscience,2001、Driguezら,Glycoscience:Synthesis of Oligosaccharides and Glycoconjugates,Springer,1999、Duffelsら,Chem.Eur.J.6:1416−1430(2000)、Hojoら,Current Prot.Peptide Sci.1:23−48(2000)、Seebergerら,Nature 446:1046−1051(2007)、Seebergerら,Nature Rev.Drug Discov.4:751−763(2005)、Srivastanaら,Carbohydrate Res.161:195−210(1987)、およびHagiharaら,Chem.Rec.6:290−302(2006)と、米国特許第5,324,663号、第6,156,547号、第6,573,337号、第6,723,843号、第7,019,131号、および第7,160,517号で説明されたように、化学的および/または酵素的に合成することができる。
【0065】
いくつかの実施形態において、オリゴ糖は連続して単糖を追加することで合成することができる。ある種の実施形態において、選択的保護および脱保護により、既存の糖の特定の位置(例えば2−O、3−O、4−O、または6−O)に、単糖を追加することができる。例えば、オリゴ糖82を、図1の逆合成解析および図2〜7に記載の合成スキームに記述されたように、合成することができる。いくつかの実施形態において、ビルディングブロック2aを、図3、5、および6の合成スキームのビルディングブロック2に置換することができる。ビルディングブロック2aが使われていれば、七糖段階でビルディングブロック2のベンジリデン基の除去を避けることができる。
【0066】
マンノース残基を、例えば米国特許第6,905,856号で説明されたように、酵素的にリン酸化することができる。ある種の実施形態において、1、2、または3つのM6P残基を、Berkowitzら,Org.Lett.6:4921−4924(2004)で説明されたように、例えば、マロニルエーテル、マロン酸、およびリン酸など、加水分解酵素耐性M6P模倣体で置き換えることができる。
【0067】
ある種の実施形態において、リンカーは、αまたはβ結合を介して糖に結合することができる。いくつかの実施形態において、β結合は、Crichら,Tetrahedron,54:8321−8348(1998)、Kimら,J.Am.Chem.Soc.,130:8537−8547(2008)、Srivasta et al,Tetrahedron Letters,35:3269−3272(1979)、Hodosiら,J.Am.Chem.Soc.,119:2335−2336(1997)、Nicolaouら,J.Am.Chem.Soc.,119:9057−9058(1997)に記載された方法により形成することができる。1つの実施形態において、不活性の脱離基を含むリンカーと反応させることができる中間体を形成するため、ジブチルスズオキシドを用いて、β結合を形成することができる。
【0068】
1つの実施形態は、式VII:
【0069】
【化14】
【0070】
[式中、
R1は、水素、ヒドロキシル、任意に置換された低級アルキル、ホスフェート、サルフェート、−OR7、保護基、および糖から選択され;
R2、R3、R4およびR5は、それぞれ独立して、水素、サルフェート、ヒドロキシル、−OR8、保護基、および糖から選択され;
R6は、水素、ヒドロキシル、カルボキシル、アルコキシカルボニル、アミノ、アミド、アルキアミノ(alkyamino)、アミノアルキル、アミノキシ、ヒドラジド、ヒドラジン、任意に置換されたアルケニル、および任意に置換されたC2−C6アルキルから選択され;
R7およびR8は、それぞれ独立して、アセチルおよび任意に置換された低級アルキルから選択され;ならびに
nは、1〜10の整数である]
で表される化合物を製造する方法であって、以下のステップ:
a)式VIII:
【0071】
【化15】
【0072】
[式中、
R1からR5は、上に定義された通りであり;ならびに、
R9およびR10は、R9およびR10のうちの1つが、ヒドロキシルのとき、もう一方が水素であるように、水素およびヒドロキシルから選択される]
で表される化合物を、式R11R12(Sn=O)で表される化合物で処理して、式IX:
【0073】
【化16】
【0074】
[式中、
R1からR5は、上で定義された通りであり;ならびに、
R11およびR12はそれぞれ独立に、非置換アルキルから選択され、またはR11およびR12は一緒になって、非置換アルキレンから選択される]
で表される化合物を形成し;
b)式IXの化合物を、任意に金属ハロゲン化物の存在下、式R6−(CH2)n−L:
[式中、
R6およびnは、上で定義された通りであり;ならびに、
Lはハロゲンである]
で表される化合物で処理して式VIIの化合物を形成すること
を含む、前記方法を提供する。
【0075】
「任意の」または「任意に」は、引き続き記述される事象または状況が起こる場合がある、または起こらない場合があること、明細書が事象または状況が起こる例、および起こらない例を含むことを意味する。例えば、「任意に置換されたアルキル」は、下記で定義されるような「アルキル」および「置換アルキル」の両方を含む。1または2以上の置換基を含むどんな基に関しても、かかる基が立体的に現実的でない、合成的に実行不可能である、および/または本質的に不安定な、いかなる置換または置換形式を取り込むことを意図しないことは、当業者により理解される。
【0076】
「アルキル」は、指定された数の炭素原子を有する直鎖および分岐鎖を含み、通常1〜20個の炭素原子を有し、例えば1〜8個の炭素原子を有し、例えば1〜6個の炭素原子を有する。例えば、C1−C6アルキルは、1〜6個の炭素原子の直鎖および分岐鎖の両方のアルキルを含む。アルキル基の例としては、メチル、エチル、プロピル、イソプロピル、n−ブチル、sec−ブチル、tert−ブチル、ペンチル、2−ペンチル、イソペンチル、ネオペンチル、ヘキシル、2−ヘキシル、3−ヘキシル、3−メチルペンチルなどが挙げられる。アルキレンは、アルキルの別の集合であり、アルキルと同じ残基を表すが、2箇所の結合部を有する。アルキレン基は、通常2〜20個の炭素原子、例えば2〜8個の炭素原子、例えば2〜6個の炭素原子を有する。例えば、C0アルキレンは、1つの共有結合を示し、C1アルキレンはメチレン基である。具体的な数の炭素を有するアルキル残基が指定されるとき、その数の炭素を有するすべての幾何異性体が含まれることを意図する。したがって、例えば、「ブチル」はn−ブチル、sec−ブチル、イソブチル、およびt−ブチルを含むことを意味し、「プロピル」は、n−プロピルおよびイソプロピルを含む。「低級アルキル」は1〜4個の炭素を有するアルキル基を示す。
【0077】
「アルケニル」は、親となるアルキルの隣り合う炭素原子から、1分子の水素分子が取り除かれることにより生成される、少なくとも1つの炭素−炭素二重結合を有する、不飽和の分岐または直鎖アルキルを示す。この基は、二重結合がシスまたはトランスのどちらの配置でもよい。例示的なアルキル基には、エテニル;プロプ−1−エン−1−イル、プロプ−1−エン−2−イル、プロプ−2−エン−1−イル(アリル)、プロプ−2−エン−2−イルなどのプロペニル;ブト−1−エン−1−イル、ブト−1−エン−2−イル、2−メチル−プロプ−1−エン−1−イル、ブト−2−エン−1−イル、ブト−2−エン−1−イル、ブト−2−エン−2−イル、ブタ−1,3−ジエン−1−イル、ブタ−1,3−ジエン−2−イルなどのブテニルなどを含むがこれに限定されない。いくつかの実施形態において、アルケニル基は2〜20個の炭素原子を有し、別の実施形態において、2〜6個の炭素原子を有する。
【0078】
本明細書で使用される「置換」という語は、指定された原子または基の1または2以上の水素のいずれかが、指示された基から選ばれたもので置き換わり、ただし指定された原子の通常の原子価を超えないことを意味する。置換基がオキソ(すなわち、=O)であるとき、原子の2つの水素が置き換わる。置換基および/または変数の組合せは、かかる組合せが安定した化合物または有用な合成中間物をもたらす場合に限り、許容される。安定した化合物または安定した構造は、反応混合物からの単離、および少なくとも実際の有用性を有する医薬としての、その後の処方でも存在するほど、十分強固である化合物を示唆するものである。他に断りが無い限り、置換基はそのコア構造に名付けられる。例えば、(シクロアルキル)アルキルが置換基の候補として列挙されるとき、この置換基のコア構造への結合位置は、アルキル部分にあることが、理解されるべきである。
【0079】
他に明らかに定義されない限り、「置換アルキル」という語は、1または2以上の水素原子が、以下から独立に選択される置換基によって置換されるアルキルのことである:
−Ra、−ORb、−O(C1−C2アルキル)O−(例えば、メチレンジオキシ−)、−SRb、−NRbRc、ハロ、シアノ、オキソ、ニトロ、サルフェート、ホスフェート、−CORb、−CO2Rb、−CONRbRc、−OCORb、−OCO2Ra、−OCONRbRc、−NRcCORb、−NRcCO2Ra、−NRcCONRbRc、−SORa、−SO2Ra、−SO2NRbRc、−NRcSO2Ra、エチレングリコール、およびポリエチレングリコール(PEG)。
ここでRaは、任意に置換されたC1−C6アルキル、任意に置換されたアリール、および任意に置換されたヘテロアリールから選択され;
Rbは、水素、−NH2、−NHRc、任意に置換されたC1−C6アルキル、任意に置換されたアリール、および任意に置換されたヘテロアリールから選択され;
Rcは、水素および任意に置換されたC1−C4アルキルから選択され;または
RbおよびRc、並びにそれらが結合する窒素原子は、任意に置換されたヘテロシクロアルキル基を形成し;ならびに
各任意に置換された基は、非置換でよく、あるいは、C1−C4アルキル、アリール、ヘテロアリール、アリール−C1−C4アルキル−、ヘテロアリール−C1−C4アルキル−、C1−C4ハロアルキル−、−OC1−C4アルキル、−OC1−C4アルキルフェニル、−C1−C4アルキル−OH、−OC1−C4ハロアルキル、ハロ、−OH、−NH2、−C1−C4アルキル−NH2、−N(C1−C4アルキル)(C1−C4アルキル)、−NH(C1−C4アルキル)、−N(C1−C4アルキル)(C1−C4アルキルフェニル)、−NH(C1−C4アルキルフェニル)、シアノ、ニトロ、オキソ(ヘテロアリールの置換基として)、−CO2H、−C(O)OC1−C4アルキル、−CON(C1−C4アルキル)(C1−C4アルキル)、−CONH(C1−C4アルキル)、−CONH2、−NHC(O)(C1−C4アルキル)、−NHC(O)(フェニル)、−N(C1−C4アルキル)C(O)(C1−C4アルキル)、−N(C1−C4アルキル)C(O)(フェニル)、−C(O)C1−C4アルキル、−C(O)C1−C4フェニル、−C(O)C1−C4ハロアルキル、−OC(O)C1−C4アルキル、−SO2(C1−C4アルキル)、−SO2(フェニル)、−SO2(C1−C4ハロアルキル)、−SO2NH2、−SO2NH(C1−C4アルキル)、−SO2NH(フェニル)、−NHSO2(C1−C4アルキル)、−NHSO2(フェニル)、および−NHSO2(C1−C4ハロアルキル)から独立して選択される、1または2以上の置換基で独立して置換されてよい。
【0080】
いくつかの実施形態において、R1は、水素、任意に置換された低級アルキル、ホスフェート、サルフェート、−OR7、保護基、および糖から選択される。追加の実施形態において、R2、R3、R4、およびR5は、それぞれ独立に、水素、サルフェート、ヒドロキシル、−OR8、保護基、および糖から選択される。ある種の実施形態において、R7およびR8はそれぞれ独立に、アセチルおよび任意に置換された低級アルキルから選択される。いくつかの実施形態において、R1、R2、R3、R4、およびR5のいずれかまたはすべては、選択的に除去することができる保護基から選択することができ、ベンジル、シリル、およびトリチルエーテル、並びに酢酸以外のエステルが含まれる。いくつかの実施形態において、R1からR5のうちの2つは、一緒になって、保護基を形成する。1つの実施形態において、R1は、−O−ベンジルおよび−OCPh3から選択される。別の実施形態において、R2からR5のうちの少なくとも1つは、−O−ベンジルである。いくつかの実施形態において、R1、R2、およびR4は、それぞれ−O−ベンジルである。さらなる実施形態において、R1は−OCPh3であり、R4は−O−ベンジルである。1つの実施形態において、R11およびR12は、一緒になって、ヘキサメチレンを形成する。別の実施形態において、R11およびR12は、それぞれイソプロピルである。さらに別の実施形態において、R11およびR12は、それぞれヘキシルである。
【0081】
追加の実施形態において、式VIIIの化合物は、任意に保護されたマンノース、ラムノース、イドース、およびアルトロースから選択される。1つの実施形態において、式VIIIの化合物は、任意に保護されたマンノースである。ある状況において、マンノースを、C−3、C−4、またはC−6位の1又は2以上を保護してもよい。いくつかの例において、単一の保護基は、2つの位置に結合することができる。例えば、ベンジリデン基は、C−4およびC−6位の両方を保護するために使用することができる。
【0082】
一般に、式IXの化合物と、強い求電子性または交差反応性でない糖のいかなる保護基も使用することができる。適当な保護基には、任意に置換されたベンジルエーテル、トリチルエーテル、アリルエーテル、またはシリルエーテルなどのエーテル;任意に置換された酢酸、安息香酸、クロロ酢酸、ピバル酸、またはレブリン酸などのエステル;ベンジリデン、イソプロピリデンおよびブタンジアセタールを含むアセタールなどが挙げられる。さらに、保護基を、カルバミン酸およびウレタンから選択することができる。いくつかの実施形態において、例えばベンジル、シリル、およびトリチルエーテル、ならびに酢酸以外のエステルなどの保護基を、選択的に除去することができる。保護基の位置および正体は、所望の最終生成物によって変えることができる。当業者に公知の追加の保護基を、本明細書で記述された実施形態に従って使用することができる。
【0083】
1つの実施形態において、式VIIIの化合物は、式R11R12(Sn=O)で表される化合物で処理され、式R11およびR12は、それぞれ独立に、非置換アルキルから選択され、またはR11およびR12は一緒になって、非置換アルキレンから選択される。いくつかの実施形態において、R11およびR12はブチルである。1つの実施形態において、式R11R12(Sn=O)で表される化合物を、トルエン、ベンゼン、ジメチルホルムアミド、イソプロパノール、メタノール、またはキシレンなどの溶媒中で、式VIIIの化合物と反応させる。いくつかの実施形態において、昇温、任意に還流下で、反応を実施して、式IXの化合物を形成する。いくつかの実施形態において、反応混合物を少なくとも40、50、60、70、または80℃に加熱する。追加の実施形態において、式R11R12(Sn=O)で表される化合物を、少なくとも1、2、5、10、15、または20時間、式VIIIの化合物と反応させる。
【0084】
1つの実施形態において、式IXの化合物を式R6−(CH2)−Lで表される化合物で、任意に金属ハロゲン化物の存在下で処理する。いくつかの実施形態において、R6は水素、ヒドロキシル、カルボキシル、アルコキシカルボニル、アミノ、アミド、アルキアミノ(alkyamino)、アミノアルキル、アミノキシ、ヒドラジド、ヒドラジン、任意に置換されたアルケニル、および任意に置換されたC2−C6アルキルから選択される。追加の実施形態において、nは1〜10の整数である。ある種の実施形態において、nは2、3、4、5、または6であり、R6はC1−C4アルコキシカルボニルである。1つの実施形態において、nは3であり、R6はメトキシカルボニルである。いくつかの実施形態において、Lは活性化されない脱離基である。活性化された脱離基の例としては、トリフレート、スルホネート、トシレート、および他の類似の基が挙げられる。いくつかの実施形態において、低い反応性の脱離基を、アリル基などの隣接基によって活性化することができる。ある種の実施形態において、R6は脱離基を活性化する置換基を含まない。いくつかの実施形態において、Lはブロミド、クロリド、またはヨージドである。1つの実施形態において、Lはブロミドである。追加の実施形態において、式R6−(CH2)n−Lの化合物は、4−ブロモ酪酸メチルである。
【0085】
1つの実施形態において、式VIIIの化合物は、任意に保護されたマンノースであり、式R6−(CH2)n−Lの化合物は、4−ブロモ酪酸メチルである。別の実施形態において、式VIIIの化合物は、3,4,6−トリ−O−ベンジル−D−マンノースおよび3−O−アリル−6−O−トリチル−D−マンノースから選択される。
【0086】
いくつかの実施形態において、式IXの化合物を式R6−(CH2)n−Lで表される化合物で、金属ハロゲン化物の存在下で処理する。金属ハロゲン化物のある種の実施形態は、金属フッ化物を含む。いくつかの実施形態において、金属フッ化物は、フッ化セシウム、フッ化ナトリウム、フッ化カルシウム、フッ化マグネシウム、フッ化リチウム、およびフッ化カリウムから選択される。1つの実施形態において、金属ハロゲン化物はフッ化セシウムである。いくつかの実施形態において、式IXの化合物を式R6−(CH2)n−Lの化合物で処理することは、テトラアルキルアンモニウムハライドの添加をさらに含む。いくつかの例において、テトラアルキルアンモニウムハライドは、テトラブチルアンモニウムヨージドである。追加の実施形態において、金属ハロゲン化物を反応に使用することができる。金属ハロゲン化物の例としては、ヨウ化ナトリウムなどのアルカリ金属ヨウ化物が挙げられる。
【0087】
1つの実施形態において、極性非プロトン性溶媒中で、式IXの化合物を、式R6−(CH2)n−Lの化合物と混合することができる。かかる溶媒には、当業者に公知の中でも、ジメチルホルムアミド、ジメチルアセトアミド、ジメチルスルホキシド、ニトロメタン、ヘキサメチルリン酸アミド、N−メチルピロリドン、アセトン、アセトニトリル、酢酸エチル、およびメチルエチルケトンが挙げられる。
【0088】
ある種の実施形態において、式IXの化合物は、式R6−(CH2)n−Lの化合物と、室温で混合することができる。他の実施形態において、反応物が組み合わされ、少なくとも50、60、70、または80℃に加熱されて、式VIIの化合物を形成する。さらなる実施形態において、混合物は少なくとも1、2、5、10、15、または20時間加熱される。
【0089】
いくつかの実施形態において、本明細書に記載された方法は、少なくとも50、60、70、80、90、95、または99%で立体特異的生成物をもたらす。追加の実施形態において、立体特異的生成物の収率は、最大可能収率の少なくとも50、60、70、75、80、85、90、95、または99%である。ある種の実施形態において、β−結合生成物とα−結合生成物との比率は、少なくとも10:1、20:1、30:1、40:1、50:1、または100:1である。
【0090】
1つの実施形態において、式VIIの化合物を大規模に製造することができる。いくつかの実施形態において、「大規模」は、出発物質、中間体または試薬を少なくとも50、100、500、または1000グラム使用することを示す。追加の実施形態において、「大規模」は、出発物質、中間体または試薬を、少なくとも10、25、50、100、250、または500kg使用することを含む。
【0091】
ジブチルスズオキシド試薬を使用して、式Aの化合物を製造する例示的な合成スキームを図8に示す。
【0092】
B.タンパク質
本明細書で記述されたオリゴ糖−タンパク複合体は、単離されたタンパク質、および組み換えられた、または合成的に生産されたタンパク質を含む、全ての純粋なタンパク質、部分的に精製されたタンパク質、あるいはその断片を含み得る。「純粋な」、「精製された」、および「単離された」という語は、天然の環境中の実質的に遊離の分子を示す。例として、純粋なタンパク質は、由来する細胞または組織の供給源の、実質的に遊離の細胞物質および/または他のタンパク質である。この語は例えば、少なくとも70〜80%、80〜90%、90〜95%、あるいは少なくとも95%、96%、97%、98%、99%、または100%(w/w)純粋である調製物に関する。
【0093】
他の実施形態において、タンパク質は、pH1〜7の範囲、例えば1〜3、2〜5、3〜6、4〜5、5〜6、または4〜6などで、活性アッセイによって測定された、最適な活性を有する酵素であってよい。例えば、酵素はpH4〜6の範囲で、最適pHを有し得る。
【0094】
いくつかの実施形態において、タンパク質は、1〜8の範囲、例えば1〜3、2〜5、3〜8、4〜5、5〜6、4〜6、5〜8、6〜8、または7〜8の範囲の等電点(pI)を有する酵素であり得る。タンパク質のpIを、例えばゲル等電点電気泳動を用いて測定することができる。
【0095】
ある種の実施形態において、タンパク質自体は少なくとも1つのオリゴ糖を有する(すなわち、糖タンパク質である)。個々の実施形態において、タンパク質は、治療用糖タンパク質である。例えば、治療用糖タンパク質は、例えば表1に列挙されたリソソーム加水分解酵素のうちの1つなど、ERT酵素を含むリソソーム酵素であってよい。ある種の実施形態において、リソソーム酵素は、例えばα−グルコシダーゼ(GAA)、α−ガラクトシダーゼA、酸性スフィンゴミエリナーゼ、およびα−L−イズロニダーゼから選択される。個々の実施形態において、リソソーム酵素はGAAである。
【0096】
【表4】
【0097】
いくつかの実施形態において、タンパク質は少なくとも1、2、3、4、5、またはそれ以上のN−結合型またはO−結合型オリゴ糖を有する糖タンパク質であってよい。他の実施形態において、タンパク質は、N−結合型またはO−結合型糖鎖形成のための、1、2、3、4、5、またはそれ以上のコンセンサス部位を有することができ、そしてその少なくとも1つがグリコシル化される。
【0098】
ある種の実施形態において、タンパク質は受容体のリガンドであってよい。例えばいくつかの実施形態において、タンパク質は、例えばマンノースまたはマンノース−6−リン酸などの糖を認識する受容体と結合する糖タンパク質であってよい。個々の実施形態において、糖タンパク質は、例えばアシアロ糖タンパク質受容体、CI−MPR、CD−MPR、またはマンノース受容体と結合することができる。
【0099】
好適なタンパク質配列は当業者に周知である。当業者は、異なる種の配列などを含む関連配列を比較することにより、保存領域および重要な機能的モチーフを容易に同定することができる。保存されたアミノ酸は、活性のためにより重要である可能性が高く、反対に、保存されていないアミノ酸は、変異(variation)を許容する可能性の高いポリペプチド領域を示す。この指針に従って、当業者はただの通常業務によって機能的変異体(functional variant)を容易に同定することができる。さらに、結晶構造が公知である場合に、当業者は結晶構造を分析して、構造および/または機能に重要であり、したがって変異(mutation)に対する許容性が低いと考えられるアミノ酸を同定することができる。当業者はまた、変異(variation)を許容し得るアミノ酸を同定することもできる。さらに当業者は,公知の構造−機能相関の観点で、可能性のある変異(mutation)を評価することができる。
【0100】
例えば、α−ガラクトシダーゼの配列および構造は周知である。例えば、Garmanら,J.Mol.Biol.,337:319−335(2004)、Garmanら,Mol.Genet.Metabol.,77:3−11(2002)、Matsuzawaら,Hum.Genet.117:317−328(2005)を参照のこと。また、GenBank Accession No.X05790も参照のこと。別の例において、GAAの配列は周知である(例えば、Martiniukら,Proc.Natl.Acad.Sci.USA 83:9641−9644(1986)、Hoefslootら,Biochem.J.272:493−497(1990)、Morelandら,J.Biol.Chem.280:6780−6791(2005)を参照のこと)。GenBank Accession No.NM_000152も参照のこと。さらに、E.coliの対応するα−グリコシダーゼの結晶構造は決定されており、構造的見識を他のα−グリコシダーゼに提供することができる。Loveringら,J.Biol.Chem.280:2105−2115(2005)を参照のこと。3つめの例において、酸性スフィンゴミエリナーゼの配列は、酸性スフィンゴミエリナーゼの配列および構造の重要な特徴であるとして周知である(例えば、Lansmannら,Eur.J.Biochem.270:1076−1088(2003)を参照のこと)。例えば、Setoら,Protein Sci.13:3172−3186(2004)、Qiuら,J.Biol.Chem.278:32744−32752(2003)、Takahashiら,Tohoku J.Exp.Med.206:333−340(2005)を参照のこと。GenBank Accession No.AI587087も参照のこと。さらに別の例において、α−L−イズロニダーゼの配列は、α−L−イズロニダーゼの重要な特徴のため、周知である(例えば、Scottら,Proc.Natl.Acad.Sci USA 88:9695−9699(1991)、Scottら,Genomics 13:1311−1313(1992)を参照のこと)。例えば、Scottら,Hum.Mutat.6:288−302(1995)、Rempelら,Mol.Genet.Metab.85:28−37(2005)、Durandら,Glycobiology 7:277−284(1997)、Beesleyら,Hum.Genet.109:503−511(2001)、Brooksら,Glycobiology 11:741−750(2001)、Niemanら,Biochemistry 42:8054−8065(2003)を参照のこと。さらなる例において、イズロン酸−2−スルファターゼの配列は、疾患を引き起こす変異体(mutation)であるとして周知である。例えば、Flomenら,Hum.Mol.Genet.2:5−10(1993)、Robertsら,J.Med.Genet.26:309−313(1989)、Wilsonら,Proc.Natl.Acad.Sci.USA 87:8531−8535(1990)、Wilsonら,Genomics 17:773−775(1993)、Sukegwa−Hayasakaら,J.Inherit.Metab.Dis 29:755−761(2006)およびその中の参考文献を参考のこと。イズロン酸−2−スルファターゼの構造はモデル化されている。例えば、Kimら,Hum.Mutat.21:193−201(2003)を参照のこと。別の例において、N−アセチルガラクトサミン−4−スルファターゼ(アリールスルファターゼB)の配列および構造は、疾患を引き起こす変異体(mutation)であるとして公知である。例えば、Litjensら,Hum.Mut.1:397−402(1992)、Petersら,J.Biol.Chem.265:3374−3381(1990)、Schuchmanら,Genomics 6:149−158(1990)、Bondら,Structure 15:277−289(1997)を参照のこと。
【0101】
C.リンカー
ある種の実施形態において、本発明のオリゴ糖−タンパク複合体は、複合体のオリゴ糖成分およびタンパク質成分との間にリンカーを含む。他の実施形態において、複合体は、リンカーを含まない。リンカーを含む実施形態において、CI−MPRへのオリゴ糖の結合を妨げず、および/またはタンパク質の活性(例えば、酵素的活性を含む)を阻まない限り、当業者に公知の任意の適当なリンカーを使用することができる。例えば、リンカーは、米国特許第4,671,958号、第4,867,973号、第5,691,154号、第5,846,728号、第6,472,506号、第6,541,669号、第7,141,676号、第7,176,185号、第7,232,805号、または米国特許出願公開第2006/0228348号で開示されたリンカーの1つであってよい。いくつかの実施形態において、リンカーは国際公開2008/089403号で開示されたリンカーから選択される。
【0102】
いくつかの実施形態において、リンカーは次式を有することができる。
【0103】
【化17】
【0104】
[式中、
Zは、任意に置換されたアルキル、アルケニル、アルキニル、アリール、エチレングリコール、ポリエチレングリコール(PEG)ヘテロアリール、およびヘテロシクリルから選択され、Pは、水素またはアミノ保護基から選択される]。
【0105】
本明細書で使用される、アミノオキシ化合物(例えば、アルキル、アルケニル、アルキニル、アリール、ヘテロアリール、ヘテロシクリル、アシルオキシ、アルコキシ、アリールオキシ、およびヘテロシクリルオキシなど)のいかなる化学基も、他に述べられない限り、置換または非置換でよく、1または2以上のヘテロ原子、または化学基により中断される場合がある。中断するヘテロ原子には、窒素、酸素、および硫黄が含まれる。置換基または中断する化学基を、例えば、アシル、アシルアミノ、アシルオキシ、アルケニル、アルコキシ、アルキル、アルキニル、アミド、アミノ、アリール、アリールオキシ、アジド、カルバモイル、カルボアルコキシ、カルボキシ、シアノ、シクロアルキル、ホルミル、グアニジノ、ハロ、ヘテロアリール、ヘテロシクリル、ヒドロキシ、イミノアミノ、ニトロ、オキソ、ホスホンアミノ、スルフィニル、スルホンアミノ、スルホナート、スルホニル、チオ、チオアシルアミノ、チオウレイド、およびウレイドから選択することができる。置換基それ自体は、置換または非置換でよく、例えば、窒素、硫黄、および酸素など、1または2以上のヘテロ原子で中断されているかまたは終結している場合がある。
【0106】
1つの実施形態において、リンカーは:
【0107】
【化18】
【0108】
との反応により形成され得る。
【0109】
追加の実施形態において、リンカーは次式を有することができる
【0110】
【化19】
【0111】
[式中、ZおよびPは、上で定義される通りである]。
【0112】
別の実施形態において、リンカーは、ジスルフィド結合を含むことができる。ジスルフィドリンカーを、例えばシステインを介して、タンパク質骨格にオリゴ糖を結合させるのに使用することができる。1つの実施形態において、リンカーは
【0113】
【化20】
【0114】
を含む、またはこれとの反応から形成することができる。
【0115】
一般に、複合体のオリゴ糖成分およびタンパク質成分間の立体障害を避けるよう、CI−MPRへのオリゴ糖の結合、および/またはタンパク質の活性(例えば、酵素的活性を含む)を妨げないようにリンカーを適当な長さにすることができ。例えば、リンカーは1〜100、1〜60、5〜60、5〜40、2〜50、2〜20、5〜10、または5〜20個の直鎖の原子を含むことができ、このリンカーは、エステル、アミド、ヒドラゾン、オキシム、セミカルバゾン、エーテル、チオエーテル、ホスホロチオ酸、ホスホン酸、チオエステル、および/またはジスルフィド結合により、タンパク質およびオリゴ糖に結合する。リンカーの残りの直鎖原子は、例えば、炭素、酸素、窒素、および硫黄から選択され、この原子のうちのいずれも、任意に炭素環、ヘテロ環、アリールまたはヘテロアリール環に含まれていてよい。リンカーの直鎖炭素原子は、任意にハロ、ヒドロキシ、ニトロ、ハロアルキル、アルキル、アルカリル、アリール、アラルキル、アルコキシ、アリールオキシ、アミノ、アシルアミノ、アルキルカルバモイル、アリールカルバモイル、アミノアルキル、アルコキシカルボニル、カルボキシ、ヒドロキシアルキル、アルカンスルホニル、アレンスルホニル、アルカンスルホンアミド、アレンスルホンアミド、アルキルスルホンアミド、アルキルカルボニル、アシルオキシ、シアノおよびウレイドから選択される置換基で置換されていてよい。リンカーの直鎖窒素原子は、任意にアシル、スルホニル、アルキル、アルカリル、アリール、アラルキル、アルコキシカルボニルで置換されていてよい。リンカーの直鎖硫黄原子は、任意に酸化されていてよい。
【0116】
ある種の実施形態において、例えば米国特許出願公開第2006/0228348号および米国特許第4,867,973号、第7,176,185号、第7,232,805号に開示されたように、リンカーを切断することができる。いくつかの実施形態において、リソソーム条件下で、リンカーを切断することができる。
【0117】
D.オリゴ糖−タンパク複合体の製造方法
例えば、式I、II、III、IV、V、またはVIのオリゴ糖を含む複合体など、本発明の複合体は、当業者に公知のいずれかの方法により製造することができる。これら方法のいずれかにおいて、適当なリンカーは、オリゴ糖またはタンパク質のどちらか、あるいはオリゴ糖およびタンパク質の両方に存在することができる。例えば、複合体を、例えばZhuら,Biochem.J.389:619−628(2005)、Zhuら,J.Biol.Chem.279:50336−50341(2004)、米国特許第5,153,312号、第5,212,298号、第5,280,113号、第5,306,492号、第5,521,290号、第7,001,994号、米国特許仮出願第60/885,457号、または第60/885,471号に記述されたように製造することができる。
【0118】
ある種の実施形態において、オリゴ糖はシステインまたはリシンなど、タンパク質のアミノ酸に結合することができる。例えば、糖はスクシンイミジル4−ホルミル安息香酸で、タンパク質のリシン残基を修飾することで、リシンを介して結合することができる。さらに、糖はトラウト試薬またはN−スクシンイミジル−3−(2−ピリジルジチオ)プロピオナート(SPDP)などのジスルフィド、もしくはN−スクシンイミジル−S−アセチルチオ酢酸(SATA)などの保護チオ−ルを含むリンカーで、リシンを修飾することにより、リシンを介して結合することができる。
【0119】
追加の実施形態において、オリゴ糖は糖タンパク質のグリカンに結合することができる。1つの実施形態において、オリゴ糖はグリカンのシアル酸残基に結合することができる。他の実施形態において、オリゴ糖は、グリカンのマンノース、フコース、ガラクトース、および/またはシアル酸残基に結合することができる。ガラクトースを介した結合について、まず、糖タンパク質をシアリダーゼで処理して、シアル酸残基を取り除き、その後オリゴ糖と反応させる前にガラクトースオキシダーゼで処理することができる。
【0120】
例えば、オリゴ糖−タンパク複合体は、(例えば、アミン、チオール、カルボン酸、ヒドロキシルを含む)タンパク質に存在し得るいずれかの官能基、および/または、タンパク質へ導入され得るいずれかの官能基を、オリゴ糖上の好適な第二の官能基と反応させることによって製造され得る。官能基の導入方法は、当技術分野で周知である。例えば、少なくとも1つのカルボニル基を有する糖タンパク質は、例えば、過ヨウ素酸塩(例えば、過ヨウ素酸ナトリウム)またはガラクトースオキシダーゼで、糖タンパク質を酸化することにより得られる。別の例において、カルボニル基は、例えばWangら,Proc.Natl.Acad.Sci.USA 100:56−61(2003)に記載されるように、拡張遺伝暗号を有する発現系を使用して導入され得る。また、例えば、米国特許出願公開第2006/0228348号には、糖タンパク質への反応基の導入について記述されており、参照のこと。
【0121】
いくつかの実施形態において、糖タンパク質は、カルボニル−反応基を含むリンカーで修飾されたオリゴ糖と結合する前に、過ヨウ素酸塩で酸化される。カルボニル−反応基の例としては、アミノオキシ、ヒドラジン、またはヒドラジドなどが挙げられる。ある種の実施形態において、糖タンパク質は、約1、2、3、4、5、7.5、10、または22.5mMの過ヨウ素酸塩で酸化される。ある種の実施形態において、糖タンパク質は、糖タンパク質グリカンのシアル酸残基を酸化し、フコースおよびマンノースの酸化を最小限にするのに十分な条件下で酸化される。例示的実施形態において、過ヨウ素酸塩の濃度は、約2、3、4、または5mM未満とした。1つの実施形態において、過ヨウ素酸塩は過ヨウ素酸ナトリウムである。
【0122】
ある種の実施形態において、結合の間に形成されるタンパク凝集体は、様々なクロマトグラフィー法を用いて、取り除くことができる。1つの実施形態において、疎水性相互作用クロマトグラフィー(HIC)を用いることができる。HICカラムの例としては、Butyl650Cおよび650M、Hexyl650C、Phenyl6FF、Capto OctylおよびCapto Phenylが挙げられる。他の実施形態としては、凝集体を銅、ニッケル、コバルト、または水銀などの金属キレートクロマトグラフィーにより取り除くことができる。1つの実施形態において、銅カラムを、結合・溶出モード(bind−and−elute)またはフロースルーモードで使用することができる。例示的な溶出緩衝液として、グリシンまたはイミダゾールが挙げられる。いくつかの実施形態において、凝集体は10、20、30、40、50、60、70、80、または90%減少する。追加の実施形態において、複合体は0.5、1、1.5、2、2.5、または3%未満の凝集体を含む。
【0123】
E.複合体
複合体のオリゴ糖およびタンパク質成分は、例えば、本明細書で記述されるオリゴ糖およびタンパク質のいずれかであってよい。ある種の実施形態において、オリゴ糖−タンパク複合体は、オリゴ糖−糖タンパク複合体である。いくつかの実施形態において、オリゴ糖−タンパク複合体は、オリゴ糖−リソソーム酵素複合体である。
【0124】
いくつかの実施形態において、複合体は、式I〜VIのオリゴ糖から選択されるオリゴ糖を含む。ある種の実施形態において、複合体は糖タンパク質1につき、オリゴ糖を平均で1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、または1〜2、1〜3、1〜4、1〜6、2〜4、2〜10、2〜12、4〜6、3〜8、5〜6、5〜10、5〜15、5〜20、10〜15、10〜20、12〜15、12〜18、または15〜20分子含む。いくつかの実施形態において、複合体はタンパク質1分子につき、オリゴ糖を少なくとも4、5、6、7、8、9、または10分子含む。追加の実施形態において、複合体はタンパク質1モルにつき、M6Pを少なくとも2、3、4、5、6、7、8、9、または10モル含む。
【0125】
ある種の実施形態において、複合体は複合体化されていないタンパク質と比較して、完全な活性(酵素活性など)を示す。他の実施形態において、複合体は複合体化されていないタンパク質と比較して、少なくとも例えば、50、60、70、75、80、85、90、92、94、95、96、97、98、または99%の活性を示す場合がある。酵素活性を含む活性を測定するアッセイは、当技術分野で周知である。例えば、Eisenthalら,Enzyme Assays:A Practical Approach,Oxford University Press:New York,2002を参照のこと。リソソーム酵素の活性を測定するアッセイは、例えば、Liら,Clin.Chem.50:1785−1796(2004)、Civalleroら,Clin Chim.Acta 372:98−102(2006)に記述されている。GAAの活性を測定する例示的なアッセイは、実施例6に記述されている。van Diggelenら,J.Inherit.Metab.Dis.28:733−741(2005)(酸性スフィンゴミエリナーゼの活性のアッセイを記述)、Downingら,Plant Biotechnol.4:169−181(2006)(α−L−イズロニダーゼの活性のアッセイを記述)、Voznyiら,J.Inherit.Metab.Dis.24:675−80(2001)(イズロン酸−2−スルファターゼ活性のアッセイを記述)、Murrayら,Mol.Genet.Metab.90:307−312(2007)(α−ガラクトシダーゼA活性のアッセイを記述)、Brooksら,J.Inher.Metab.Dis.14:5−12(1991)(N−アセチルガラクトサミン−4−スルファターゼ活性のアッセイを記述)も参照のこと。
【0126】
ある種の実施形態において、複合体は、対応する複合体化されていないタンパク質より、標的細胞(例えば、CI−MPRを介するエンドサイト−シス)によって、より効果的に取り込まれる。例えば、複合体は複合体化されていないタンパク質より、所与の期間に、例えば少なくとも10%、15%、20%、25%、30%、35%、40%、50%、60%、70%、80%、90%、93%、95%、96%、97%、98%、99%、100%、110%、120%、130%、140%、150%、または200%(mol/mol)、より効果的に取り込まれる場合がある。他の実施形態において、所与の期間に、非結合タンパク質と比較して、目的とする細胞型(例えば、L6筋芽細胞またはヒトポンペ病線維芽細胞(NIGMS Human Genetic Cell Repository、Cat.No.GM20005))により、少なくとも2、3、4、5、6、7、8、9、10、20、30、40、50、100、または1000倍(mol/mol)の複合体が取り込まれることがある。基準とされる期間は、例えば、10分、30分、45分、または1、2、3、5、6、12、24、48、または72時間、もしくはそれ以上でよい。L6筋芽細胞のin vitroでの取り込みは、例えば、実施例6およびZhuら,J.Biol.Chem.279:50336−50341(2004)に記述された通りに測定することができる。
【0127】
ある種の実施形態において、複合体化されていないタンパク質と比較して、複合体のCI−MPRへの結合の増加が示される。例えば、複合体化されたタンパク質及び複合体化されていないタンパク質の結合定数または解離定数を比較することにより測定されるとおり、例えば、複合体化されたタンパク質は、複合体化されていないタンパク質と比較して、少なくとも2、3、4、5、10、20、30、40、50、100、1,000、または10,000倍、CI−MPRへの親和性の改善を示し得る。CI−MPRへの結合は、例えば、実施例5およびZhuら,J.Biol.Chem.279:50336−50341(2004)に記述された通りに測定することができる。
【0128】
ある種の実施形態において、複合体は、複合体化されていないタンパク質と比較して、マンノース受容体による取り込みの増加を示さない。あるいは、複合体化されていないタンパク質と比較して、マンノース受容体による取り込みが、5、10、15、20、30、40、または50%未満である。ラット肺胞マクロファージ細胞のマンノース受容体による取り込みは、例えば、Zhuら,Biochem.J.389:619−628(2005)に記述されたように、in vitroで測定することができる。
【0129】
ある種の実施形態において、好適な動物モデルにおける、代謝的に欠損した酵素の蓄積した基質レベルに関して、複合体は、例えば少なくとも2、3、4、5、10、20、30、40、50、100、または1000倍の減少を示す場合がある。例えば、ポンペ病のマウスモデルにおけるグリコーゲンレベルの減少は、例えばZhuら,J.Biol.Chem.279:50336−50341(2004)に記述されたように測定することができる。あるいは、例えばKikuchiら,J.Clin.Invest.101:827−33(1998)に記述された、ポンペ病のウズラモデルを使用することができる。別の例として、肝臓および脾臓中に蓄積されたグリコサミノグリカンの減少を、Kakkisら,Mol.Genet.Metab.72:199−208(2001)に記述されたように、ムコ多糖症I型のネコモデルで測定することができる。さらに、グロボトリアオシルセラミドレベルの減少を、例えば、Ioannouら,Am.J.Hum.Genet.68:14−25(2001)に記述されたように、ファブリー病のマウスモデルで測定することができる。さらに別の例において、スフィンゴミエリンレベルの減少を、例えば、Horinouchiら,Nat.Genet.10:288−293(1995)に記述されたように、ニーマンピック病のA型およびB型のマウスモデルで測定することができる。
【0130】
II.医薬組成物
いくつかの実施形態において、本発明は、それを必要とする被験体のリソソーム蓄積症を治療する医薬の製造における、(1)タンパク質、および(2)式I〜VIのいずれか1つのオリゴ糖を含む、オリゴ糖−タンパク複合体の使用を提供する。
【0131】
本明細書で記述された医薬組成物は、上記で記述されたようなオリゴ糖−タンパク複合体、および充填剤、膨張剤、崩壊剤、緩衝剤、安定剤、または賦形剤など、少なくとも1つの添加剤を含む。いくつかの実施形態において、本発明の医薬組成物は、式I〜VIのいずれか1つののオリゴ糖、およびリソソーム酵素を含む、複合体を含む。
【0132】
標準の医薬処方技術は、当技術者に周知である(例えば、2005Physician’s Desk Reference(登録商標),Thomson Healthcare:Montvale,NJ,2004;Remington:The Science and Practice of Pharmacy,20th ed.,Gennadoら,Eds.Lippincott Williams&Wilkins:Philadelphia,PA,2000を参照のこと)。好適な医薬添加剤には、例えば、マンニトール、デンプン、グルコース、ラクトース、ショ糖、ゼラチン、麦芽、米、小麦粉、石灰、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。組成物はまた、pH緩衝試薬、および湿潤剤または乳化剤を含んでもよい。この組成物は、保存剤を含んでも含まなくてもよい。
【0133】
いくつかの実施形態において、α−ガラクトシダーゼA複合体を含む医薬組成物は、例えば、マンニトール、リン酸一ナトリウム一水和物、および/またはリン酸水素二ナトリウム七水和物など、1または2以上の賦形剤を含むことができる。いくつかの実施形態において、α−グルコシダーゼの複合体を含む医薬組成物は、マンニトール、ポリソルベート80、リン酸水素二ナトリウム七水和物、およびリン酸一ナトリウム一水和物の内、1または2以上のものを含むことができる。別の実施形態において、α−グルコシダーゼの複合体を含む医薬組成物は、グリシン2%まで、マンニトール2%まで、ポリソルベート80を0.01%までを備えた10mMヒスチジンpH6.5を含むことができる。
【0134】
医薬組成物は、単独の活性化合物として、または他の化合物、組成物、もしくは生物学的物質との組合せでも、本明細書に記述された複合体のいずれかを含むことができる。例えば、医薬組成物は、LSDおよび/またはLSDに関連する副作用の治療に有用な、1または2以上の小分子も含むことができる。いくつかの実施形態において、組成物はミグルスタット(miglustat)、および/または例えば米国特許出願公開第2003/0050299号、第2003/0153768号、第2005/0222244号、または第2005/0267094号に記述される1又は2以上の化合物を含むことができる。いくつかの実施形態において、医薬組成物は、1又は2以上の免疫抑制剤も含むことができる。
【0135】
医薬組成物の処方は、目的とする投与方法およびその他の要因に応じて変更してもよい(例えば、Roweら,Handbook of Pharmaceutical Excipients,4th ed.,APhA Publications,2003を参照のこと)。いくつかの実施形態において、組成物を、滅菌注射用蒸留水(米国薬局方)と再構成して、静脈注射による投与のため、滅菌した非発熱性のオフ−ホワイトの凍結乾燥した固形または粉末であってよい。他の実施形態において、組成物を滅菌した非発熱性の溶液であってよい。
【0136】
本明細書で記述された医薬組成物の投与は、いかなる個々の送達システムに限定されず、非経口(皮下、静脈内、頭蓋内、髄内、関節内、筋内、くも膜下、もしくは腹腔内注射を含む)、経皮、または経口(例えば、カプセル、懸濁剤、または錠剤)を挙げることができるがそれには限定されない。個体へは、単回投与または反復投与、様々な生理学的に許容される塩形態のいずれか、および/または医薬組成物の一部として、許容される医薬担体および/または添加剤と共に投与することができる。
【0137】
医薬的に許容される塩には、例えば、酢酸アニオン、ベンゼンスルホン酸アニオン、安息香酸アニオン、重炭酸アニオン、重酒石酸アニオン、ブロミドアニオン、カルシウムエデテート(calcium edetate)アニオン、カンシル酸アニオン、炭酸アニオン、クロリドアニオン、クエン酸アニオン、二塩酸、エデト酸アニオン、エジシル酸アニオン、エストル酸(estolate)アニオン、エシル酸アニオン、フマル酸アニオン、グルセプチン酸アニオン、グルコン酸アニオン、グルタミン酸アニオン、グリコリルアルサニレート(glycollylarsanilate)、ヘキシルレソルシネート(hexylresorcinate)アニオン、ヒドラバミン(hydrabamin)、臭化水素、塩酸、ヒドロキシナフトエート(hydroxynaphthoate)アニオン、ヨージドアニオン、イセチオン酸アニオン、乳酸アニオン、ラクトビオン酸酸アニオン、リンゴ酸アニオン、マレイン酸アニオン、マンデル酸アニオン、メシル酸アニオン、メチル臭化物、メチル硝酸アニオン、メチル硫酸アニオン、ムチン酸アニオン、ナプシル酸アニオン、硝酸アニオン、パモン酸アニオン(エンボネート(embonate))、パントテン酸アニオン、リン酸アニオン/二リン酸アニオン、ポリガラクツロ酸アニオン、サリチル酸アニオン、ステアリン酸アニオン、塩基性酢酸アニオン、コハク酸アニオン、硫酸アニオン、タンニン酸アニオン、酒石酸アニオン、およびテオクル酸アニオン/トリエチオダイドアニオン;ベンザチン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、メグルミン、およびプロカインの(有機)カチオン;並びにアルミニウム、カルシウム、リチウム、マグネシウム、カリウム、ナトリウム、及び亜鉛の(金属)カチオンが挙げられる。医薬的に許容される塩には、例えばBergeら,J.Pharm.Sci.66:1−19(1977)に記述された塩も含まれる。
【0138】
本明細書に記述される複合体は、治療有効量で投与される。一般に治療有効量は、被験体の年齢、全体的な状態、および性別、ならびに被験体の病状の重症度に応じて変更され得る。用量は医師により決定され、治療の実際の効果に合わせて、必要であれば調節することができる。この化合物の毒性および治療効果は、in vitroおよび/またはin vivoで標準的な医薬的手順により決定することができる。毒性および治療効果の用量比率は、治療指数(または治療可能比)であり、比率LD50/ED50として表すことができ、LD50は全個体の50%致死量であり、ED50は全個体の50%に治療効果がある用量である。本発明の複合体は、例えば、少なくとも1、1.5、2、3、4、5、6、7、8、9、10、および20の治療指数を示す場合がある。
【0139】
in vitroアッセイおよび動物試験により得られたデータを、例えば、ヒトにおける用量範囲を決めるのに使用することができる。この化合物の用量は、好ましくは、低い毒性、わずかな毒性、または無毒でED50を含む循環する濃度の範囲内である。使用する剤形および利用する投与経路によって、用量を変えることができる。どの複合体の治療有効量も、in vitroアッセイから、最初に推定することができる。in vitro実験で測定された、IC50値(すなわち、最大数の半数の症状を阻害する試験複合体の濃度)を含む、循環する血漿濃度範囲を実現するため、動物モデルにおいて、用量を決めることができる。血漿中の程度を、例えば、高速液体クロマトグラフィーまたは適当な酵素活性アッセイにより測定することができる。どの特定用量の効果も適当な評価項目のバイオアッセイにより、測定できる。
【0140】
他で指示されない限り、症状の重症度および疾患の進行により、ほぼ1μg/kg〜500mg/kgの用量で、複合体を投与することができる。例えば、外来診療で、例えば1日、2日、3日、4日、5日、6日、7日、8日、9日、10日またはそれ以上の日数おきに、あるいは例えば週1回、週2回、月1回、または1カ月2回の投与でゆっくりした静脈注射により、複合体を投与してよい。化合物の適当な治療有効用量は、治療する臨床医によって選択され、おおよそ1μg/kg〜500mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg/kg〜1mg/kg、および500μg/kg〜5mg/kgの範囲である。いくつかの実施形態において、適当な治療用量は、例えば0.1、0.25、0.5、0.75、1、5、10、15、20、30、40、50、60、70、および100mg/kgから選択される。
【0141】
α−ガラクトシダーゼAを含む複合体を、例えば、2週間に1回または4週間に1回、1.0mg/kg体重の用量で、10、13、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、または33mg/時未満あるいは等しい注入速度で、静脈注射により投与することができる。別の例において、α−グルコシダーゼを含む複合体を、例えば、2週間または4週間に1回、20mg/kgまたは40mg/kgの用量で、おおよそ例えば、1、2、3、4、5、6、7、8、9、または10時間にわたって、静脈注射により投与することができる。いくつかの実施形態において、α−グルコシダーゼの投与速度は、例えば1mg/kg/時で始め、それから例えば30分ごとに2mg/kg/時にし、注入速度に対する被験体の耐性を確立した後、例えば7mg/kg/時である最大まで増やすことができる。N−アセチルガラクトサミン−4−スルファターゼを含む複合体を、例えば、毎1.0mg/kg体重の用量で、毎週、おおよそ例えば1、2、3、4、5、6、7、8、9、または10時間にわたって、静脈注入により投与することができる。さらに具体的な用量の例としては、Physicians’ Desk Reference(登録商標)に記載されている。
【0142】
III.リソソーム蓄積症の治療方法
いくつかの実施形態において、本発明は例えば表1に開示されたリソソーム蓄積症の治療方法を提供する。いくつかの実施形態において、本発明はさらに、マンノース−6−リン酸を含むオリゴ糖と結合することにより、タンパク質をリソソームに向ける方法を提供する。
【0143】
ある種の実施形態において、当該方法は、治療有効量で本発明のオリゴ糖−タンパク複合体を、リソソーム蓄積症である被験体(例えば、ヒト、ネコ、イヌ、マウス、またはラットなどの哺乳動物、あるいは例えばウズラなどの鳥類を含む)に、投与することを含む。オリゴ糖−タンパク複合体は、式I〜VIのいずれかのオリゴ糖など、マンノース−6−リン酸を含むオリゴ糖と、リソソーム酵素(例えば、表1に列挙したリソソーム酵素)などの糖タンパク質との複合体であり得る。1つの実施形態において、当該方法は、本発明の複合体の少なくとも1つを含む医薬組成物を、それを必要とする被験体に投与することを含む。
【0144】
ある種の実施形態において、当該方法は、(1)タンパク質、および(2)式I〜VIのいずれかのオリゴ糖など、マンノース−6−リン酸を含むオリゴ糖を含む複合体を、1または2以上の他の治療剤(therapy)と投与することを含む。1または2以上の他の治療剤は、当該複合体の投与と同時に(混合された製剤としての同時投与を含む)、投与前に、または投与後に投与され得る。
【0145】
いくつかの実施形態において、当該方法は、解熱剤、抗ヒスタミン剤、および/または免疫抑制剤で、(本明細書で記述された複合体での治療前、治療後、または治療中に)被験体を治療することを含む。いくつかの実施形態において、注入に付随する反応を減少、または防止するために、被験体は、オリゴ糖−糖タンパク複合体で治療される前に、解熱剤、抗ヒスタミン剤、および/または免疫抑制剤で治療され得る。例えば、被験体は、アセトアミノフェン、アザチオプリン、シクロホスファミド、シクロスポリンA、ジフェンヒドラミン、メトトレキサート、ミコフェノール酸モフェチル、経口ステロイド、またはラパマイシンのうちの1または2以上で前治療され得る。
【0146】
いくつかの実施形態において、当該方法はアセトアミノフェン、アザチオプリン、シクロホスファミド、シクロスポリンA、ジフェンヒドラミン、メトトレキサート、ミコフェノール酸モフェチル、経口ステロイド、またはラパマイシンのうちの1または2以上で、例えば、t=0(複合体を投与する時間)および/またはt=12、24、36、48、60、72、96、120および144時間で、あるいはおおよそこの時間で、例えば最初の1、2、3、4、5、6、7、8、9、10回またはそれ以上での複合体治療回数で、被験体を治療することを含む。例えば、いくつかの実施例において、ファブリー病またはポンペ病である被験体は、メトトレキサート(例えば、1kgあたり0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2、3、4、5、6、8、10、12、15、25、30、35、40、50、60、70、80mgまたはそれ以上のメトトレキサート)で、例えばt=0、24、および48時間で、またはおおよそこの時間で、例えば最初の1、2、3、4、5、6、7、8週間での複合体の治療で、治療され得る。いくつかの実施形態において、複合体への免疫寛容が、シクロスポリンAおよびアザチオプリンでの治療によって、例えばムコ多糖症I型などのリソソーム蓄積症である被験体に誘導されることがある。例えば、Kakkisら,Proc.Natl.Acad.Sci.U.S.A.101:829−834(2004)に記述されたように、被験体はシクロスポリンAおよびアザチオプリンで治療されてもよい。
【0147】
いくつかの実施形態において、当該方法は、リソソーム蓄積症の治療のために行われる小分子治療および遺伝子治療を含む、小分子治療および/または遺伝子治療で、(複合体での治療前、治療後、または治療中に)被験体を治療することを含む。小分子治療は、ミグルスタットおよび/または例えば、米国特許出願公開第2003/0050299号、第2003/0153768号、第2005/0222244号、および第2005/0267094号に記述された1または2以上の化合物の投与を含んでよい。遺伝子治療は、例えば米国特許第5,952,516号、第6,066,626号、第6,071,890号、第6,287,857号、および米国特許出願公開第2003/0087868号に記述されたように実施することができる。
【0148】
「治療」、「治療方法」という語、およびその同族語は、治療処置および予防/防止対策の両方を示す。したがって、治療を必要とする人として、すでに特定のリソソーム蓄積症を有する個体、および疾患の危険性がある人(すなわち、最終的にその疾患にかかる、または疾患のある種の症状に陥りそうな人)を含んでよい。
【0149】
治療方法は、症状の予防もしくは緩和、または他の所望の生物学的成果をもたらし、臨床兆候の改善もしくは発症遅延、代謝的欠損酵素の活性増加、および/または代謝的欠損酵素の基質の蓄積が減少したレベルにより、評価することができる。
【0150】
いくつかの実施形態において、当該方法は、(1)リソソーム酵素、および(2)式I〜VIのいずれかのオリゴ糖を含む複合体を被験体に投与することを含み、これにより、内在性活性と比べて、例えば少なくとも10%、15%、20%、25%、30%、35%、40%、50%、60%、70%、80%、90%、または100%、被験体内の欠損したリソソーム酵素活性が増加する。いくつかの実施形態において、当該方法は、(1)リソソーム酵素、および(2)式I〜VIのいずれかのオリゴ糖を含む複合体を被験体に投与することを含み、これにより内在性活性と比べて、例えば少なくとも2、3、4、5、6、7、8、9、10、20、30、40、50、100、または1000倍、被験体内の欠損した酵素活性が増加する。増加した酵素活性を、例えば臨床症状の縮小、または適当な臨床的もしくは生物学的アッセイによって、測定することができる。
【0151】
いくつかの実施形態において、当該方法は、GAAを含む本発明の複合体を被験体に投与することを含み、これによって、ポンペ病(酸性α−グルコシダーゼ欠損症、酸性マルターゼ欠損症、グリコーゲン蓄積症II型、糖原病II型、およびリソソーマルα−グルコシダーゼ欠損症としても知られる)を治療する。ある種の実施形態において、組換えヒトGAA(rhGAA)はチャイニーズハムスター卵巣(CHO)細胞を用いて製造される。追加の実施形態において、rhGAAは、オリゴ糖103;103および式Aの混合物;オリゴ糖128、129、130、131、132、133、または136;並びにそれらの任意の組合せから選択されるオリゴ糖と複合体化され得る。1つの実施形態において、オリゴ糖は、オリゴ糖128および136から選択される。追加の実施形態において、GAA1モルにつき、少なくとも5モルの式Aを含む複合体が患者に投与される。
【0152】
ある種の実施形態において、患者は、ポンペ病と診断されている成人である。他の実施形態において、患者は幼児または子供である。
【0153】
GAA活性の増加は、例えば心筋細胞、骨格筋細胞、または皮膚線維芽細胞におけるリソソームグリコーゲンの蓄積の減少を、生化学的(例えば、Zhuら,J.Biol.Chem.279:50336−50341(2004)を参照のこと)または組織学的に観察することにより測定され得る。GAA活性はまた、例えば筋生検サンプル、培養皮膚線維芽細胞、リンパ球、乾燥血斑で測定され得る。乾燥血斑アッセイは、例えば、Umpathysivamら,Clin.Chem.47:1378−1383(2001)およびLiら,Clin.Chem.50:1785−1796(2004)に記述されている。ポンペ病の治療は、例えばクレアチニンキナーゼの血清レベル、運動機能の増幅率(例えば、アルバータ乳幼児運動発達検査法によって評価される)、エコー図で測定される左室心筋重量係数の変化、および心電図で測定される心臓電気活性によって、評価することもできる。GAA複合体の投与はまた、心肥大、心筋症、日中の眠気、労作性呼吸困難、成長障害、摂食困難、「筋緊張低下(floppiness)」、歩行障害、頭痛、低血圧、臓器肥大症(心臓、舌、肝臓の肥大など)、脊柱前弯症、平衡感覚障害、腰痛、朝の頭痛、筋力低下、呼吸不全、翼状肩甲、脊柱側弯症、深部腱反射の低下、睡眠時無呼吸、呼吸器感染症に対する感受性、および嘔吐など、ポンペ病の1または2以上の症状の縮小をもたらし得る。
【0154】
他の実施形態において、当該方法は、α−ガラクトシダーゼAを含む複合体を、被験体に投与することでファブリー病を治療することを含む。ファブリー病、またはアンダーソン−ファブリー病は、α−ガラクトシダーゼAの欠損を特徴とする、希少でX連鎖性のリソソーム蓄積症であり、内臓組織と、内皮、外皮および筋細胞のリソソームに、グロボトリアオシルセラミド(GL3)およびその他の中性スフィンゴ糖脂質の蓄積を引き起こす。血管系への中性スフィンゴ糖脂質の蓄積は、血管の狭窄および拡張、最終的には虚血および梗塞を引き起こす。
【0155】
α−ガラクトシダーゼAを含む本発明の複合体の投与は、例えば、肢端感覚異常、狭心症、角化血管腫、不整脈、歩行失調、四肢の灼熱痛ならびに/ヒリヒリする痛み、白内障、寒冷不耐症、伝導異常、角膜渦濁(corneal whorling)、冠動脈疾患、認知症、うつ病、下痢、心室拡張、めまい、心肥大、心筋症、複視、構音障害、疲労、赤血球沈降速度の上昇を伴う発熱、聴覚問題、心疾患、心臓弁疾患、高熱不耐性、片側運動失調、片側不全麻痺、発汗減少症、発汗不全、梗塞、虚血、関節痛、腎疾患、左室肥大、水晶体異常、レンズ混濁、脂肪尿、筋力低下、心筋梗塞、嘔気、眼振、痛み(例えば、全身に広がる激痛)、多渇症、タンパク尿、食後痛、腎不全、網膜異常、耳鳴り、胃痛、ST−T変化、脳卒中、尿毒症、弁膜症、めまい、嘔吐および虚弱を含む、1または2以上のファブリー病の臨床症状の縮小をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、例えば、血漿、涙液、白血球、生検組織、または培養皮膚線維芽細胞で、α−ガラクトシダーゼA活性の増加をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、複屈折性の脂肪球の増加の減少(例えば、少なくとも10%減少)または消失という、組織学的所見をもたらし得る。それはまた、尿沈渣の脂肪球減少、血清クレアチニンレベルまたはクレアチニンクリアランスによって測定した腎機能の改善、およびタンパク尿の減少をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、腎臓、心臓、および皮膚の毛細血管内皮のGL3含有物の減少をもたらし得る。ファブリー病治療の効果を測定する追加のアッセイは、例えば、MacDermottら,J.Med.Genet.38:750−760(2001)に記載されている。
【0156】
他の実施形態において、当該方法は、酸性スフィンゴミエリナーゼを含む本発明の複合体を、被験体に投与することを含み、これにより、ニーマンピックA病またはニーマンピックB病、あるいは酸性スフィンゴミエリナーゼ欠損症を治療する。酸性スフィンゴミエリナーゼ複合体の投与は、例えば、異常コレステロールレベル、異常脂質レベル、運動失調、血液異常、眼内のさくらんぼ赤色斑、頻繁な肺感染、成長遅延、肝脾腫大症、血小板数低下、リンパ節腫脹症、末梢神経障害、肺機能の問題、息切れ、皮膚色素変化、または黄色腫症を含む、1または2以上のニーマンピックA病またはニーマンピックB病に関する、臨床症状の減少をもたらし得る。いくつかの実施形態において、複合体を頭蓋内に投与することができる。
【0157】
他の実施形態において、当該方法は、α−L−イズロニダーゼを含む、本発明の複合体を、被験体に投与することを含み、これによりムコ多糖症I型(例えば、MPSI型のハーラーおよびハーラー−シャイエ型を含む)を治療する。α−L−イズロニダーゼ複合体の投与は、例えば、大動脈弁逆流、大動脈弁狭窄症、毛根管症候群、慢性鼻炎、伝音難聴、便秘、角膜混濁、発育遅延、下痢、腹部膨張、腰椎後弯症、突背、肝脾腫大症、水頭症、鼠径ヘルニア、脊柱後弯症、精神遅滞、僧帽弁逆流、僧帽弁狭窄症、夜盲症、開放隅角線内症、手の機能低下、進行性関節症、反復呼吸感染、呼吸不全、網膜変性、脊柱側弯症、感音難聴、激しい背中の痛み、鼻漏、睡眠時無呼吸、脊髄圧迫、母指球の萎縮、へそヘルニア、上気道の合併症など、1種または複数のMPSI型の臨床症状の減少をもたらし得る。
【0158】
さらに別の実施形態において、当該方法は、イズロン酸−2−スルファターゼを含む本発明の複合体を、被験体に投与することを含み、これにより、ムコ多糖症II型(ハンター病)を治療する。イズロン酸−2−スルファターゼ複合体の投与は、例えば、腎臓弁膜症、心肺機能不全、毛根管症候群、慢性下痢、慢性の乳頭浮腫、特徴的顔貌、角膜混濁、冠状動脈狭窄、難聴、異形症、異骨症、耳感染、聴覚障害、肝脾腫大症、水頭症、鼠径ヘルニア、関節硬直、後側弯症、精神遅滞、心筋症、心筋肥厚、肺高血圧、網膜機能不全、骨格異常、へそヘルニア、上気道感染、弁機能不全を含む、1または2以上のMPSII型の臨床症状の減少をもたらし得る。
【0159】
さらに他の実施形態において、当該方法はN−アセチルガラクトサミン−4−スルファターゼ(アリールスルファターゼB)を含む、本発明の複合体を被験体に投与することを含み、これによりムコ多糖症VI型(マロトー−ラミー症候群)を治療する。N−アセチルガラクトサミン−4−スルファターゼ複合体の投与は、例えば、失明、心臓異常、心肺疾患、特徴的顔貌、角膜混濁、耳感染、成長遅延、肝腫大、肝脾腫大症、関節変形、神経絞やく症候群、呼吸困難、骨格変形、脊髄圧迫、脾腫、関節の硬直、上気道閉塞を含む、1種または複数のMPSVI型の臨床症状の減少をもたらし得る。
【0160】
前述および以下の説明は、例示および説明のためのみであり、特許請求されている本発明を限定しない。
【実施例】
【0161】
実施例1.オリゴ糖合成の一般的手順
A.グリコシル化
グリコシル化反応を、ドナーである糖およびアクセプターである糖を結合させることによる、標準的な方法を用いて実施した。簡潔に言えば、他に指示されない限り、乾燥窒素下、加熱して活性化したモレキュラーシーブ4Aの存在下で、無水DCMにドナーおよびアクセプター化合物を溶解させた。この溶液を冷却し、0℃で〜30分間保存し、続いてTMSOTf(1eq)をゆっくり添加した。反応をヘキサン/EtOAcを用いて、TLC(シリカゲル)で確認し、TEAまたはヒューニッヒ塩基(1.05eq)を用いて停止した。混合物をろ過し、シロップになるまで濃縮し、指示されない限り、ヘキサン/EtOAcグラジエントを用いたフラッシュカラムクロマトグラフィーによって精製した。
【0162】
B.酸触媒脱アセチル化
ある種の実施形態において、アセチル化化合物は、グリコシル化または他の修飾、例えばリン酸化の前に、脱アセチル化される。以下の実施例において、塩化アセチルを冷(0℃)乾燥メタノールへ加えることによって、塩化水素が生成させた。濃縮した溶液を、DCM/メタノール中のアセチル化化合物の1:3溶液に加えた。溶液のメタノール成分に関して、最終濃度は3%w/vであった。脱アセチル化反応は、1級のアセテートについては、a)約18時間、2級のアセテートについては、b)約48から64時間で行った。反応を、TEAまたはヒューニッヒ塩基で停止し、指示されない限り、続いてDCMまたはEtOAcを水で抽出した。
【0163】
C.リン酸化
いくつかの例において、糖は部位特異的なリン酸化を受ける。室温で乾燥アセトニトリルの糖溶液に、5−メチルテトラゾール(3.4eq)を加え、この混合物を30分間、乾燥窒素下で撹拌した。ジベンジルジイソプロピルホスホルアミダイト(1個のOH基につき1.7eq)を加え、反応が完了するまで(約60分)撹拌した。反応をヘキサン/EtOAcを用いて、TLC(シリカゲル)により確認した。溶液を氷/水で15分冷却し、30%w/v過酸化水素(2eq)を加えた。約60分後、反応が完結し、過剰の飽和チオ硫酸ナトリウムを加えた。混合物をガム状になるまで濃縮し、EtOAcに溶解させ、半飽和食塩水で洗浄し、硫酸ナトリウムで乾燥させた。他で指示されない限り、残渣をヘキサン/EtOAcグラジエントを用いて、フラッシュクロマトグラフィーで精製した。
【0164】
実施例2.二糖アミノオキシアセトアミドプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシドの合成(17)
【0165】
【化21】
【0166】
アリルα−D−マンノシドをPekariら,J.Org.Chem.66:7432(2001)に記述されている方法にしたがって製造した。無水メタノール(100mL)中のアリル−α−D−マンノシド(8.68g、39.4mmol)に、2,3−ブタンジオン(3.63mL、86.1mmol)、トリメチルオルトホルメート(16mL、146mmol)、および10−(+)−カンファースルホン酸(1.37g、5.9mmol)を添加した。混合物を還流下で、9時間、乾燥窒素下で加熱した。反応混合物をTEA(1mL)で停止し、赤いシロップ状になるまで濃縮し、40〜80%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、アリル3−O,4−O−[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド1を白色固体として得た(3.89g、29.4%)。
【0167】
化合物1(2.65g、8mmol)を乾燥ピリジン(20mL)へ溶解し、その溶液を氷/水槽で冷却し、t−ブチルジフェニルシリルクロリド(2.28mL、8.7mmol)を加えた。18時間後、ピリジン(20mL)および無水酢酸(1.6mL、16mmol)を加え、この溶液を50℃まで16時間加熱し、その後、シロップ状になるまで濃縮し、トルエンと共に留去した。残渣をEtOAc(60mL)に溶解させ、1M HCl(2x50mL)、飽和炭酸水素ナトリウム(50mL)で洗浄し、硫酸ナトリウムで乾燥した。混合物をろ過し、真空下で濃縮し、溶液を濃縮し、アリル2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド2をシロップとして得た(5.0g)。
【0168】
塩化パラジウム(II)(0.425g、2.4mmol)を、2(5.0g、8mmol)の無水メタノール(25mL)溶液に撹拌しながら加えた。約3時間後、反応をTEA(0.75mL、4.8mmol)で停止した。メタノールを減圧下で取り除き、残渣を10〜50%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーで精製し、2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド3を、白色泡状物質として得た(2.72g、59.2%)。
【0169】
トリクロロアセトニトリル(1.75mL、17.4mmol)およびDBU(0.05mL,0.35mmol)を3(1.0g、1.74mmol)の乾燥DCM(1mL)溶液に添加した。約75分後、溶液をEtOAc/ヘキサン(0〜30%)を用いて、シリカゲルカラムフラッシュカラムクロマトグラフィーで直接精製し、2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノーストリクロロアセトイミデート4を、白色泡状物質として得た(0.98g、78.2%)。ドナーである4(0.98g、1.36mmol)およびアクセプターである3−N−ベンジルオキシカルボニルアミノプロパノール(0.284g、1.36mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド5を、白色泡状物質として得た(0.66g、64%)。
【0170】
無水メタノール(5mL)中の5(0.66g、0.87mmol)の溶液に、メタノール(0.05mL、0.22mmol)中の25%w/vナトリウムメトキシドの溶液を加えた。約1時間後、反応を氷酢酸(0.025mL)で停止し、シロップ状にまるまで濃縮した。生成物をDCM(10mL)に溶解させ、半飽和食塩水(5mL)で洗浄し、硫酸ナトリウムで乾燥して、白色泡状物質であるN−ベンジルオキシカルボニルアミノプロピル6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド6を得た(0.58g、92%)。
【0171】
【化22】
【0172】
アリルα−D−マンノシド(3.15g、14.3mmol)を乾燥ピリジン(20mL)に溶解させ、氷/水槽で冷却した。t−ブチルジフェニルシリルクロリド(4.03mL、15.7mmol)を加え、この溶液を室温になるまで放置した。18時間撹拌し、ベンゾイルクロリド(5.93mL、51.5mmol)を加え、24時間後、反応を水(3mL)で停止し、30分撹拌した。溶液を真空下で濃縮し、トルエンで分離した(3x50mL)。残渣をEtOAc(100mL)に溶解させ、冷却した1M HCl(50mL)、半飽和食塩水(50mL)、半飽和炭酸水素ナトリウム(50mL)、半飽和食塩水(50mL)で洗浄し、硫酸ナトリウムで乾燥し、ろ過し、シロップ状になるまで濃縮した。0〜50%EtOAc/ヘキサンを用いてシリカゲルフラッシュカラムクロマトグラフィーで精製し、アリル2,3,4−トリ−O−ベンゾイル−6−O−t−ブチルジフェニルシリル−α−D−マンノシド7を、白色泡状物質として得た(8.62g、78.2%)。
【0173】
氷酢酸(11.1mL、18.26mmol)および1M テトラブチルアンモニウムフルオリド(18.26mL、18.26mmol)を7(12.77g、16.6mmol)の乾燥THF(50mL)溶液に加えた。80分で、氷酢酸(0.15mL、25mmol)および1M テトラブチルアンモニウムフルオリド(2.5mL、25mmol)を加え、90分で追加の氷酢酸(0.25mL、4.15mmol)および1M テトラブチルアンモニウムフルオリド(4.15mL、4.15mmol)を加えた。2時間後、溶液を半分の容量まで濃縮し、EtOAc(150mL)で希釈した。溶液を半飽和食塩水(2x150mL)および半飽和重炭酸ナトリウム(200mL)で洗浄し、濃縮し、残渣を10〜50%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーで精製し、アリル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド8を、白色泡状物質として得た(6.75g、76.4%)。化合物8(6.75g、ymmol)を、一般的なリン酸化手順に従って変換し、アリル2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシド9を、白色泡状物質として得た(9.52g、95%)。
【0174】
塩化パラジウム(II)(0.638g、3.6mmol)を9(9.52g、12.1mmol)の乾燥メタノール(50mL)溶液に加えた。約5時間後、さらにパラジウム(II)(0.145g)を加えて、混合物を18時間保存した。この溶液をろ過し、濃縮し、残渣を20〜70%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノース10を、白色泡状物質として得た(3.8g、42,5%)。
【0175】
トリクロロアセトニトリル(5.06mL、5.1mmol)およびDBU(0.15mL、1mmol)を10(3.8g)の乾燥DCM溶液に、乾燥窒素下、0℃で加えた。約90分後、溶液を10〜60%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより直接精製し、2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシドトリクロロアセトイミデート11を、白色泡状物質として得た(3.3g、72.1%)。
【0176】
【化23】
【0177】
ドナーである6(2.62g、2.97mmol)およびアクセプターである11(1.92g、2.7mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2−O−[2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシル]−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド12を、白色泡状物質として得た(1.27g、32.2%)。
【0178】
【化24】
【0179】
TFA/水 19:1 v/v(8.4mL)を、DCM(8mL)中の12(2.1g、1.44mmol)の氷/水槽で冷却した溶液に加えた。約2時間後、出発物質はTLCによって示されたように消費された。エタノール(25mL)をこの溶液に加え、その後濃縮し、エタノール(3x25mL)と共に留去した。残渣を乾燥メタノール(10mL)に溶解させ、氷/水槽で冷却した。塩化アセチル(0.4mL)を加えて、3%w/vのHCl溶液を得た。この溶液を室温まで放置して温めた。約2時間後、出発物質はTLCに示されたように消費された。反応をトリエチルアミン(1mL)で停止し、濃縮し、残渣を30〜100%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、白色泡状物質として13を得た(0.758g、47.9%)。無水メタノール(10mL)中の13(0.758g、0.69mmol)の溶液に、25% w/v ナトリウムメトキシドのメタノール(0.15mL)溶液を加えた。約1時間後、出発物質はtlcに示されたように消費された。反応を1M HClで停止し、14を得た。この溶液に氷酢酸(25μL)、湿10% Pd/C(0.1g)を加え、水素バルーンを取り付けた。6時間反応させたあと、生成物を5%硫酸/EtOHで炭化させたが、UV活性はなかった。混合物をろ過し、油状物になるまで濃縮し、その後水(10mL)に溶解させ、凍結乾燥して、3−アミノプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシド15を得た(0.300g、13から91.3%)。
【0180】
0.1MのNaOH溶液(2mL)を水(8.5mL)中の15(0.1g、0.21mmol)の溶液に加え、その後、THF(8.5mL)中のN−t−ブトキシカルボニル−アミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.14g、0.42mmol)を加えた。18時間後、この溶液を2MのHClでpH4に調節し、この溶液をDCM(3x10mL)で抽出した。水層を凍結乾燥して、N−t−ブトキシカルボニル−アミノオキシアセトアミドプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシド16を得た(0.12g)。化合物16(0.12g)をTFA/DCM1:1(10mL)に溶解し、その溶液を約30分間攪拌し、その後油状物になるまで濃縮した。これを水(5mL)に溶解させ、生成物を凍結乾燥して、固体(0.45g)を得た。この固体をBiogel(登録商標)P2を用いて精製し、水で溶出させて、17を得た(0.063g)。
【0181】
実施例3.三糖(35)の合成
【0182】
【化25】
【0183】
2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノーストリクロロアセトアミデート18を、Yamazakiら,Carb.Res.201:31(1990)に記述されたように製造した。
【0184】
【化26】
【0185】
6−O−アセチル−3,4,6−トリ−O−ベンゾイル−α−D−マンノーストリクロロアセトイミデート19を、Hengら,J.Carb.Chem.20:285(2001)に記述されたように製造した。
【0186】
【化27】
【0187】
ドナー19(5.0g、7.4mmol)およびアクセプターN−9−フルオレニルメチルカルボニルアミノプロパノール(2.41g、8.1mmol)を一般的なグリコシル化手順に従って変換し、白色泡状物質であるN−9−フルオレニルメチルカルボニルアミノプロピル6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド29を得た(4.0g、66.4%)。
【0188】
【化28】
【0189】
化合物29(4.0g、4.9mmol)を、2級アセテートの酸触媒脱アセチル化の一般的な手順(b)に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド30を、白色泡状物質として得た(3.3g、87.3%)。ドナー18(3.27g、5.16mmol)およびアクセプター30(3.3g、4.3mmol)を、一般的なグリコシル化手順に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル6−O−[2−O−アセチル−3,4,6−トリ−O−ベンジルマンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド31を白色泡状物質として得た(4.94g、92.7%)。
【0190】
【化29】
【0191】
化合物31(4.94g、3.96mmol)を、2級アセテートの酸触媒脱アセチル化の一般的な手順(b)に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル6−O−[3,4,6−トリ−O−ベンジルマンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド32を白色泡状物質として得た(1.73g、36%)。化合物11を、実施例2の方法に従って製造した。ドナー11(3.37g、3.74mmol)およびアクセプター32(1.73g、1.44mmol)を、一般的なグリコシル化手順に従って変換し、白色泡状物質として33を得た(1.2g、43.1%)。
【0192】
【化30】
【0193】
無水THF(10mL)中の33(1.2g、0.62mmol)の溶液に、ドデシルチオ−ル(1.48mL、6.2mmol)およびジアザビシクロウンデカ−7−エン(DBU)(0.093mL、0.62mmol)を加えた。約18時間後、出発物質はtlcに示されたように消費された。この溶液をシロップ状になるまで濃縮し、0〜20%メタノール/DCMを用いて、シリカゲルフレッシュカラムクロマトグラフィーで精製した。生成物に、メタノール/水 1:1(20mL)および酢酸(25μL)を加え、Pd/C(0.1g)および水素のバルーンを取り付けた。18時間後、この溶液をセライトでろ過し、白色泡状物質になるまで濃縮した。この泡状物質を乾燥メタノール(10mL)、およびメタノール(0.15mL)の25%w/vナトリウムメトキシドの溶液に溶解させ、6時間後、この溶液を濃縮して、水(10mL)に入れ、DCM(10mL)で洗浄した。水層を凍結乾燥して、アミノプロピル6−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド34を、二ナトリウム塩として得た(0.27g、33から63.5%)。
【0194】
水/DMSO 1:1溶液(10mL)中の34(0.17g、0.3mmol)に、DMSO(2mL)中のN−t−ブトキシカルボニルアミノ−オキシアセチル2,3,5,6−テトラフルオロフェニレート(0.34g、1.14mmol)の溶液およびDMSO(1mL)中の3−ヒドロキシ−1,2,3−ベンゾトリアジン−4(3H)−オン(DHBT)(0.09g、0.6mmol)の溶液を加えた。24時間後、この溶液を,sephadexサイズ排除樹脂を用いて精製した。画分をシリカゲルプレート上で炭化させることで確認し、選択した画分をまとめ、凍結乾燥して固体を得た。TFA/DCM(8mL)に溶解させ、60分撹拌し、その後油状物になるまで濃縮した。水(5mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して35を得た(0.033g、34から16.4%)。
【0195】
実施例4:四糖の合成
A.アミノオキシアセトアミド1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸(28)
【0196】
【化31】
【0197】
化合物19を、実施例3に記述された方法に従って製造した。ドナー19(15.49g、24.3mmol)およびアクセプターである3−N−9−フルオレニルメトキシカルボニルアミノプロパノール(8.7g、29.19mmol)を、一般のグリコシル化手順に従って変換し、N−9−フルオレニルメトキシカルボニルアミノプロピル2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノシド20を、白色泡状物質として得た(10.55g、55%)。
【0198】
【化32】
【0199】
乾燥DCM(50mL)および無水メタノール(100mL)中の20(10.5g、ymmol)の溶液に、約30分の間、塩化アセチル(4.8mL、63mmol)を滴加し、2.3%w/vHClのメタノール溶液を得た。約18時間後、反応をヒューニッヒ塩基(9.81mL、63mmol)で停止し、さらに1.5mL加えた。この溶液をシロップ状になるまで濃縮し、クロロホルム(2x50mL)と共に留去し、DCM(100mL)に溶解させ、半飽和食塩水(100mL)で洗浄し、硫酸ナトリウムで乾燥した。この溶液を濃縮し、0〜100%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、白色泡状物質である、N−9−フルオレニルメトキシカルボニルアミノプロピル3,4,6−トリ−O−ベンジル−α−D−マンノシド21を得た(6.11g、61.2%)。ドナー21(6.11g、13.4mmol)およびアクセプター11(7.1g、7.9mmol)を一般的なグリコシル化手順に従って変換し、N−9−フルオレニルメトキシカルボニルアミノプロピル2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシド22を白色泡状物質として得た(5.8g、50.1%)。
【0200】
無水THF溶液(75mL)中の22(5.8g、3.96mmol)に、ドデシルチオ−ル(9.53mL、40mmol)およびDBU(0.6mL、4mmol)を加えた。約4時間後、反応をメタノール塩酸(5.6mL、8mmol)で停止し、シロップ状になるまで濃縮した。生成物をジエチルエーテル中で粉砕し、3−アミノプロピル2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシド塩酸塩23をゴム状物質(gum)として得た(3.77g、74.5%)。
【0201】
【化33】
【0202】
乾燥アセトニトリル(50mL)中の23(3.77g、2.95mmol)の溶液に、N−ベンジルオキシカルボニルグルタミン酸(0.3661g、1.3mmol)、N−ヒドロキシベンゾトリアゾール(HOBt)(0.4g、2.95mmol)、DBU(4.6mL、4mmol)、および1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド(EDC)(0.75g、8.85mmol)を加えた。16時間後、ヒューニッヒ塩基(0.25mL)を加え、続いてさらにEDC(0.75g、8.85mmol)を加えた。21時間後、この溶液をシロップ状になるまで濃縮し、0〜50%DCMに対して、10%2−プロパノール/DCMを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、N−ベンジルオキシカルボニルアミノ1,5−ジ−[3−アミドプロピル2−O−[2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシル]グルタミン酸24を白色泡状物質として得た(0.97g、11.2%)。
【0203】
無水DCM(20mL)およびメタノール(25mL)中の24(0.912g、0.33mmol)の溶液に、25%w/vナトリウムメトキシドのメタノール(0.09mL、0.41mmol)溶液を加えた。約6時間半後、反応を1M HCl(0.41mL、0.41mmol)で停止し、シロップ状になるまで濃縮し、0〜100%DCMに対して10%2−プロパノール/DCMを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、N−ベンジルオキシカルボニルアミノ1,5−ジ−3−アミノプロピル[2−O−[6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシル]グルタミン酸25を得た(0.306g、44.4%)。
【0204】
THF/水 2:1 v/v(75mL)中の25(0.3g、0.144mmol)の撹拌溶液に、10%湿Pd/C(0.052g)および水素バルーンを取り付けた。16時間後、氷酢酸(25μL)を加え、新鮮な水素バルーンを取り付けた。6時間後、さらに10%Pd/C(0.04g)を追加し、さらに水素を追加した。18時間後、新鮮な5%Pd/C(0.05g)を追加し、さらに水素を追加した。24時間後、生成物を5%硫酸/EtOHで炭化したが、UV活性はなかった。混合物を、セライトを通してろ過し、パッドを洗浄し、約30%まで濃縮して、THFを除去した。その後凍結乾燥して1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸26(0.121g、77.9%)を得た。
【0205】
DMSO(1mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.19g、0.57mmol)の溶液およびDMSO(1mL)中の3,4−ジヒドロ−3−ヒドロキシ−4−オキソ−1,2,3−ベンゾトリアジン(DHBT)(0.052g、0.3mmol)の溶液を、水/DMSO 1:1(7.5mL)中の26(0.16g、0.15mmol)の溶液に加えた。18時間後、この溶液をsephadexサイズ排除樹脂で精製した。シリカゲル上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥してN−t−ブトキシカルボニルアミノオキシアセトアミド1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸27を得た(0.095g、42.1%)。化合物27に、TFA/DCM 1:1(8mL)を加え、混合物を溶解するまで(約60分)撹拌し、その後油状物になるまで濃縮した。水(10mL)を加えて、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して28を得た(0.048g、54.2%)。
【0206】
B.アミノオキシアセトアミドプロピル6−O−([α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド(47)
【0207】
【化34】
【0208】
化合物11を実施例2に記述された通りに製造した。ドナー11(5.0g、7.4mmol)およびアクセプターである3−N−ベンジルオキシカルボニルアミノプロパノール(1.93g、9.25mmol)を実施例1の一般的なグリコシル化手順に従って変換し、白色泡状物質を得た。生成物を、1級アセテートを酸性触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド36を白色泡状物質として得た。
【0209】
【化35】
【0210】
ドナー19(4.47g、6.6mmol)およびアクセプター36(3.6g、5.3mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド37を白色泡状物質として得た(3.8g、63.3%)。化合物37(3.8g、3.17mmol)を、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換して、N−ベンジルオキシカルボニルアミノプロピル6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド38を白色泡状物質として得た(3.5g、95.3%)。
【0211】
化合物18を、Yamazakiら,Carb.Res.201:31(1990)に記述された方法に従って製造した。ドナー18(2.41g、3.75mmol)およびアクセプター38(3.5g、3mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド39が油状物として得られた(5.63g)。化合物39(5.83g)を、2級アセテート(実施例1を参照のこと)を酸触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニル−アミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド40を白色泡状物質として得た(2.0g、38から41.9%)。ドナー19(1.07g、1.63mmol)およびアクセプター41(2.0g、1.3mmol)を、一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド42を白色泡状物質として得た(1.4g、50.8%)。化合物42(1.4g、0.066mmol)を、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニル−アミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド43を白色泡状物質として得た(1.0g、80%)。化合物43(1.0g、0.048mmol)を一般的なリン酸化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド44を白色泡状物質として得た(0.9g、80.7%)。
【0212】
【化36】
【0213】
無水メタノール(20mL)中の44(0.9g、0.039mmol)の溶液に、メタノール(0.09mL、0.4mmol)中の25% w/v ナトリウムメトキシドの溶液を加えた。約7時間後、反応を1M HCl(0.5mL)、10%のPd/C(0.2g)、および水(10mL)で停止した。混合物を24時間、水素バルーン下で保持した。この混合物をセライトでろ過し、EtOAc(20mL)で洗浄した。溶液を凍結乾燥して、白色固体であるN−ベンジルオキシカルボニル−アミノプロピル6−O−([α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジル−ホスホリル−α−D−マンノシル])−α―D−マンノシド45を得た(0.23g、44から74.7%)。
【0214】
DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.375g,1.14mmol)の溶液およびDMSO(1mL)中のDHBT(0.1g、0.6mmol)の溶液を水/DMSO1:1(10mL)中の45(0.23g、0.3mmol)の溶液に加えた。24時間後、溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥した。生成物を、DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.375g、1.14mmol)の溶液およびDMSO(1mL)中のDHBT(0.1g、0.6mmol)の溶液を用いて、水/DMSO 1:1(10mL)中で再アシル化した。24時間後、この溶液をsephadexサイズ排除樹脂で精製し、画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノオキシアセトアミドプロピル6−O−([α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド46を得た(0.11g、37.5%)。化合物46をTFA/DCM(8mL)に溶解させ、追加した。この溶液を60分間撹拌し、その後油状物になるまで濃縮した。水(5mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥して、47を得た(0.07g、70.7%)。
【0215】
実施例5:β−結合型六糖の合成
A.メチルクロトニル3,4,6−トリ−O−ベンジル−β−D−マンノシド(51)
【0216】
【化37】
【0217】
2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノースを、Mayerら,Eur J.Org.Chem.10:2563(1999)に記述の通りに製造した。メタノール(0.5mL)中の25%w/vナトリウムメトキシドを無水メタノール(50mL)中の2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノース(8.0g、23.3mmol)の溶液に加えた。約2時間後、薄層クロマトグラフィー(TLC)で示されたように、出発物質が消費された。この反応を、Amberlite IR120(H+)樹脂で停止し、ろ過し、シロップ状になるまで濃縮して、3,4,6−トリ−O−ベンジル−α−D−マンノース50を得た(6.67g、99%)。化合物50にトルエン(150mL)、続いてジブチルスズオキシド(3.88g、16.28mmol)を加え、Dean−Starkコンデンサを用いながら混合物を還流下で3時間加熱した。得られた溶液を冷却し、シロップ状になるまで濃縮し、乾燥DMF(50mL)に溶解させた。フッ化セシウム(2.28g、12.1mmol)、テトラブチルアンモニウムヨージド(5.47g、14.8mmol)、および4−ブロモクロトン酸メチル(2.46mL、22.2mmol、工業用グレード)を、混合物に加え、約60℃で18時間加熱した。混合物を放冷し、ろ過して固体を取り除いた。イソプロピルエーテル/EtOAc 3.7:1(380mL)で希釈し、半飽和のチオ硫酸ナトリウム(240mL)で洗浄した。水相をイソプロピルエーテル/EtOAc 3.7:1(2x190mL)で抽出し、有機層を合わせて濃縮した。シロップをイソプロパノール(2x25mL)と共に留去し、0〜50%EtOAc/ヘキサンを用いて、フラッシュカラムクロマトグラフィーで精製し、51をシロップ状として得た(4.58g、56.4%)。13C−NMR(100MHz)1J1C,1H(100MHz)、157Hz(β<160Hz、α>170Hz)
【0218】
B.メチルブチリル3,4,6−トリ−O−ベンジル−β−D−マンノシド(52)
化合物50を実施例2に記載の通りに製造した。化合物50(9.4g、21mmol)にトルエン(200mL)、続いてジブチルスズオキシド(5.49g、22mmol)を添加し、Dean−Starkコンデンサを用いながら、混合物を還流下で23時間加熱した。得られた溶液を冷却し、シロップ状になるまで濃縮し、乾燥DMF(100mL)に溶解させた。4−ブロモ酪酸メチル(4.23mL、32mmol)、テトラブチルアンモニウムヨージド(1.94g、5.25mmol)、フッ化セシウム(3.91g、25.5mmol)を添加し、混合物を60℃で2時間加熱し、その後18時間常温とした。混合物を放冷し、セライトでろ過し、EtOAc(50mL)で洗浄した。ゴム状物質になるまで濃縮し、トルエン(3x40mL)と共に留去し、シリカに吸着させ、0〜70%EtOAc/ヘキサンを用いて、フラッシュクロマトグラフィーで精製して、52を油状物として得た(9.11g、78.6%)。13C−NMR(100MHz)1J1C,1H(100MHz)、157.2Hz(β<160Hz、α>170Hz)。α−結合の生成物は観察されなかった。
【0219】
【化38】
【0220】
C.メチルブチリル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド(57)
化合物52を実施例2に記述の通りに製造し、6−O−アセチル−3,4,6−トリ−O−ベンゾイル−α−D−マンノーストリクロロアセトイミデート19を、Hengら,J.Carb.Chem.20:285(2001)の方法に従って製造した。ドナー19(4.29g、6.36mmol)およびアクセプター52(2.9g、5.3mmol)を、実施例1の一般的なグリコシル化手順に従って変換して、メチルブチリル2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド53を白色泡状物質として得た(4.41g、78.5%)。化合物53(4.41g、4.1mmol)を実施例1に記述された、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換して、メチルブチリル2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド54を白色泡状物質として得た(2.25g、53.7%)。化合物54(2.25g、2.2mmol)を実施例1の一般的なリン酸化手順に従って変換して、メチルブチリル2−O−[2,3,4−トリ−O−ベンゾイル−6−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド55を白色泡状物質として得た(2.2g、77.7%)。
【0221】
【化39】
【0222】
55を脱保護するため、メタノール(0.2mL)中の25% w/v ナトリウムメトキシドの溶液を、無水メタノール(20mL)中の55(2.2g、1.7mmol)の溶液に加えた。約24時間後、出発物質が消費されたことをTLCが示した。反応をAmberlite IR120(H+)で停止し、シロップ状になるまで濃縮した。溶液を濃縮し、シリカゲルフラッシュカラムクロマトグラフィーで精製して、メチルブチリル2−O−[6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド56を白色泡状物質として得た(1.10g、67%)。
【0223】
THF/水(20mL)中の56(1.10g、1.15mmol)の撹拌溶液に、氷酢酸(25μL)を加え、10%湿Pd/C(0.1g)および水素バルーンを取り付けた。24時間反応後、生成物を5%硫酸/EtOHで炭化したが、UV下では見えなかった。混合物をセライトでろ過し、パッドを水(20mL)で洗浄した。溶液を濃縮し、真空下で乾燥して、メチルブチリル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド57を得た(0.6g、98%)。
【0224】
【化40】
【0225】
D.アミノオキシアセトアミドヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド(60)
ヒドラジン一水和物(0.44mL、5.75mmol)をメタノール(20mL)中の57(0.6g、1.15mmol)の溶液に撹拌しながら加えた。30分後、水(5mL)を加え、この溶液を18時間撹拌した。さらにヒドラジン(0.44mL、5.75mmol)を加えて、混合物を120時間撹拌した。この溶液を約25%の容積まで濃縮し、水(2x10mL)と共に留去し、生成物を凍結乾燥して、ヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド58を、オフ−ホワイトの固体として得た(0.6g、99%)。
【0226】
DMSO/水 1:1(10mL)中の58(0.2g、0.38mmol)に、DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.49g、1.52mmol)の溶液およびDMSO(2mL)中のDHBT(0.125g、0.76mmol)の溶液を加えた。18時間後、この溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノ−オキシアセトアミドヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド59をオフ−ホワイトの固体として得た(0.1g、37.8%)。化合物59をTFA/DCM1:1(8mL)に溶解させた。この溶液を約60分間撹拌し、その後油状物になるまで濃縮した。水(10mL)を加えて、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、60をオフ−ホワイトの固体として得た(0.045g、52.5%)。
【0227】
【化41】
【0228】
E.メチルブチリル3−O−アリル−6−O−トリチル−β−D−マンノシド(64)の大規模合成
100L CH2Cl2中の50.0kg(128.09mol)d−マンノースペンタアセテートの溶液を26.8kg(243.2mol、1.90eq.)チオフェノールおよび27.3kg(192.3mol、1.50eq.)三フッ化ホウ素ジエチルエーテルで処理し、得られた溶液を22℃で40時間撹拌し、HPLC分析によって、反応が完了したことを判断した。その後115Lの5N NaOH水を、撹拌した反応容器に注意深く入れ、分離した層および有機層を46Lの5N NaOHでもう一度洗浄した。CH2Cl2を蒸留により減圧下で除去し、残渣を60℃で100kgのイソプロパノールに再溶解させた。9℃まで冷却し、生成物F1が結晶化し、それをろ過して単離し、続いてイソプロパノールで洗浄して、35.8kgを得た(63%)。
【0229】
35.80kgのF1(88.28mol)を、143kgのMeOHに22℃で懸濁し、0.73kgの、30%ナトリウムメトキシドのメタノール溶液(4.05mol、0.046eq.)で処理し、透明な溶液を得た。TLC分析により、反応が完了したことを判断されたのち、0.49kgの酢酸(8.14mol、0.09eq.)を加え、溶媒を減圧下で除去した。残渣をトルエンに懸濁し、再び減圧下で濃縮し、最後にアセトンで処理し、生成物であるフェニル−α−D−チオマンノシドを結晶化した。ろ過後、洗浄および乾燥すると、19.65kgの収量(89%)で得られた。
【0230】
ピリジン(43.8kg)中のフェニル−α−D−チオマンノシド(19.25kg、70.69mol)の溶液をトルエン(89kg)中のトリフェニルメチルクロリド(19.7kg、70.66mol)の溶液に40℃で加え、22時間撹拌した。HPLC分析により反応が完了したことが判断された後、溶媒を減圧下で取り除き、残渣をトルエンに溶解させ、再濃縮した。さらにトルエンで希釈した後、溶液を水で1度洗浄した。ヘキサン(840L)およびジイソプロピルエーテル(250L)の混合物にこのトルエン溶液を加えて、生成物を沈殿させ、ろ過および乾燥すると、フェニル6−O−トリチル−1−チオ−α−D−マンノシド61が得られた(32.30kg、89%)。
【0231】
【化42】
【0232】
トルエン(500kg)中の61(30.0kg、58.29mol)およびジブチルスズオキシド(20.3kg、81.5mol)の混合物を還流下で、2時間、濃縮した溶媒の蒸気から水が分離しなくなるまで加熱した。この溶液を40℃に冷却し、DMF(34kg)を加えた。全溶媒のほぼ半分を減圧下で除去し、DMF(216kg)を加え、この溶液をほぼ半分の容量になるまで再び濃縮した。さらにDMF(250kg)を加え、続いて、フッ化セシウム(8.9kg、58.59mol)、DMF(65kg)中のテトラブチルアンモニウムヨージド(23.6kg、63.89mol)の溶液、およびアリルブロミド(21.1kg、174.4mol)を加えた。得られた混合物を50℃で15時間撹拌した。HPLC分析により、反応が完了したことが判断された後、ろ過によって反応混合物から固体を取り除き、ろ液をジイソプロピルエーテル(136kg)および酢酸エチル(30kg)の混合物、続いて、10% w/w チオ硫酸ナトリウム水溶液(300kg)で処理した。相を分離した後、下相を4回ジイソプロピルエーテル(136kg)および酢酸エチル(30kg)の混合物で再抽出し、まとめた上相を水(150kg)で3回洗浄した。上相を減圧下で濃縮し、残渣を75℃でエタノール(160kg)に溶解させた。0℃に冷却すると、生成物は結晶化し、ろ過で単離することができた。フィルターの固形物を洗浄し、乾燥させた後、フェニル3−O−アリル−6−O−トリチル−1−チオ−α−D−マンノシド62を得た(15kg、46%)。
【0233】
【化43】
【0234】
THF(63kg)およびピリジン(18kg、227.5mol)の混合物中の62(12.5kg、22.53mol)の溶液を、水(10.7kg)中のトルエンスルホン酸一水和物(16.7kg、87.79mol)で処理し、続いてN−クロロスクシンイミド(9.6kg、71.89mol)の水(17kg)およびTHF(83kg)の混合物溶液で、15℃で処理した。得られた混合物を22℃に温め、3時間撹拌した。HPLC分析により、反応が完了したことが判断された後、水(15kg)中のチオ硫酸ナトリウム(4.6kg、29.11mol)の溶液を、反応混合物に加えた。相を分離し、上相を減圧下で濃縮し、残渣をトルエンに溶解させ、再び濃縮した。残渣を酢酸エチルに再溶解させ、水、その後16% w/w 塩化ナトリウム水溶液で洗浄した。酢酸エチルを留去後、粗生成物を52kg超のシリカゲルを用いたカラムクロマトグラフィーにより精製し、3〜10% v/v 酢酸エチル/トルエンのグラジエントで溶出して、3−O−アリル−6−トリチル−α−D−マンノース63を得た(7.7kg、73%)。
【0235】
【化44】
【0236】
メタノール(61kg)中の63(7.7kg、16.65mol)およびジブチルスズオキシド(4.56kg、18.32mol)の混合物を、濁った溶液が得られるまで、還流下で加熱し、その後さらに1時間加熱した。この溶液を25℃に冷却し、全溶媒のほぼ半分を減圧下で除去し、DMF(33kg)を加え、この溶液をほぼ半分の容量まで再び濃縮した。さらにDMF(15kg)を加え、続いてフッ化セシウム(2.53kg、16.65mol)、DMF(17kg)中のテトラブチルアンモニウムヨージド(6.15kg、16.65mol)、および4−ブロモ酪酸メチル(4.52kg、24.97mol)を加えた。得られた混合物を80℃で5時間撹拌した。固体を反応混合物からろ過で除去し、ろ液をジイソプロピルエーテル(41kg)、酢酸エチル(14kg)、10% w/w チオ硫酸ナトリウム水溶液(77kg)の混合物で処理した。相の分離後、下相をジイソプロピルエーテル(41kg)および酢酸エチル(51kg)の混合物で再抽出し、まとめた上相を水(39kg)で洗浄した。上相を減圧下で濃縮して、残渣をメタノール(35kg)に溶解させた。
【0237】
反応収率を改善するため、結合反応のプロセスを繰り返した。溶解した残渣を再び減圧下で濃縮し、その後メタノール(122kg)で希釈した。メタノール(60L)を再び留去し、得られた溶液をジブチルスズオキシド(2.28kg、9.16mol)で処理し、混合物を2時間、還流下で加熱した。溶液を29℃に冷却し、全溶媒のほぼ半分を減圧下で留去し、DMF(37kg)を加えて、溶液を再び容量のおよそ半分まで濃縮した。さらにDMF(15kg)を加え、続いてフッ化セシウム(1.26kg、8.29mol)、DMF(17kg)中のテトラブチルアンモニウムヨージド(3.7kg、10.02mol)の溶液、および4−ブロモ酪酸メチル(3.06kg、16.90mol)を加えて、得られた混合物を80℃で2時間撹拌した。HPLC分析により反応が完了したことを判断した後、固体をろ過によって反応混合物から除去し、ろ液をジイソプロピルエーテル(25kg)、酢酸エチル(32kg)、および10% w/w チオ硫酸ナトリウム水溶液(77kg)の混合物で処理した。相の分離後、下相をジイソプロピルエーテル(25kg)および酢酸エチル(32kg)の混合物で再抽出し、まとめた上相を水(39kg)で洗浄した。溶液を減圧下で濃縮し、残渣をトルエン(43kg)に再溶解し、最終的にほぼ30Lの容量まで濃縮した。その後、64の粗メチルエステルを、50kgのシリカゲルを用いたカラムクロマトグラフィーにより精製し、5〜30% v/v 酢酸エチル/トルエンのグラジエントで溶出した。
【0238】
精製したメチルエステルのメタノール溶液(50kg)を、30% w/w 水酸化ナトリウム水溶液(3.29kg)およびメタノール(7.7kg)の混合物で処理し、得られた溶液を14時間撹拌した。HPLC分析により、反応が完了していることを判断し、反応混合物をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物、続いて水(77kg)で処理した。二相混合物を1.2μmのフィルターカートリッジに通し、相を分離した。下相をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物で処理し、下相のpHを、5% w/w クエン酸水溶液(38L)を添加して4.5〜5まで下げた。相を分離し、下相をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物で抽出した。まとめた上相を水(39kg)で洗浄し、その後減圧下で濃縮した。残渣をジイソプロピルエーテル(39kg)と混合し、溶媒を部分的に濃縮して、約20Lの最終容量が得られ、生成物を結晶化し、ろ過により単離することができた。フィルターの固形物を洗浄し、乾燥した後、(3−O−アリル−6−O−トリチル−β−D−マンノシル)−4−ブタン酸64が得られた(4.7kg、51.5%)。
【0239】
【化45】
【0240】
F.四糖中間体(69)の大規模合成
化合物64をパートEの方法に従って製造した。64の保護基をさらなるグリコシル化反応の前に修飾した。THF(14kg)中の64(4.25kg、7.75mol)の溶液を、THF(45kg)中の60%水素化ナトリウム分散体(1.55kg、38.75mol)の撹拌スラリーに注意深く加え、得られた懸濁液を水素が発生しなくなるまで撹拌した。THF(2kg)中のテトラブチルアンモニウムヨージド(0.29g、0.78mol)の懸濁液、続いてベンジルブロミド(9.2kg、53.79mol)を、反応容器に入れた。混合物を22℃で46時間撹拌し、その後30℃、12時間、および35℃、48時間撹拌した。HPLC分析により反応が完了したことを確認した後、混合物を0℃に冷却し、無水メタノール(0.7kg、21.87mol)、続いて30% w/wの、ナトリウムメトキシド(2.1kg、11.66mol)のメタノール溶液を注意深く入れた。その後、酢酸(1.4kg)、続いてトリエチルアミン(9.4kg、92.89mol)を加え、混合物を18時間撹拌した。得られた懸濁液に、水(31kg)を加え、2相に分離した。上相を減圧下で濃縮し、残渣をトルエンに溶かし、再び最終容量が約20Lになるまで濃縮した。粗生成物を42kgのシリカゲルでカラムクロマトグラフィーにより精製し、5〜15%の酢酸エチル/ヘキサンのグラジエントで溶出して、メチルブチリル3−O−アリル−2,4−ジ−O−ベンジル−6−O−トリチル−β−D−マンノシド65を、酢酸エチル溶液として得た(4.4kg、77%)。
【0241】
【化46】
【0242】
37℃のメタノール(19kg)中の65(4.4kg、5.92mol)の溶液に、メタノール(6.3kg)中のトルエンスルホン酸一水和物(0.9kg、4.73mol)の溶液を加え、得られた混合物を1時間撹拌した。HPLC分析により、反応が完了したことを判断した後、トリエチルアミン(1.5kg、14.82mol)を入れ、この溶液を減圧下で濃縮した。その後、トルエン(28kg)を加え、溶液を水(32kg)で洗浄した。相を分離し、上相を減圧下で濃縮した。粗生成物をシリカゲル(32kg)を用いたシリカゲルクロマトグラフィーで、9%、その後17%、その後50% v/v 酢酸エチル/トルエンのグラジエントで溶出させて精製し、メチルブチリル3−O−アリル−2,4−ジ−O−ベンジル−β−D−マンノシド66を、トルエン溶液として得た(2.67kg、90%)。
【0243】
【化47】
【0244】
3.73kg(5.49mol、1.10eq.)の19および2.50kg(4.99mol)の66を34kgの乾燥トルエンに溶解させた。そのうち、約10Lを減圧下留去した。その後、溶液を0℃に冷却し、反応温度を<5℃に維持しつつ、22g(0.099mol、0.02eq.)のTMSOTfを滴加処理した。添加終了後、0℃で1時間撹拌した。HPLC分析により反応が完了したことを判断した後、混合物に30g(0.296mol、0.06eq.)のEt3Nを加えて中和した。ヘキサン(22L)を追加し、得られた懸濁液をろ過し、ろ液を33Lの水で洗浄し、減圧下で濃縮した。この残渣を10Lのトルエンに溶解させ、再度濃縮した。このプロセスを2回以上繰り返した。粗生成物のカラムクロマトグラフィー精製は、50kgシリカゲルで行い、9〜13% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、4.23kg、83%である化合物67を、トルエン溶液として得た。
【0245】
【化48】
【0246】
4.23kg(4.16mol)の67の、5.6LのCH2Cl2および40LのMeOH溶液に、0.72kgの10%Pd/Cを加え、続いて0.12kg(0.63mmol、0.15eq.)のトルエンスルホン酸一水和物溶液を加え、混合物を22℃で24時間撹拌した。HPLC分析により反応が完了したことを判断した後、パラジウム/炭素をろ過により除去し、さらに精製することなく、ろ液を次の工程で使用した。
【0247】
ろ液に4L MeOH中の2.97kg(15.6mol、3.75eq.)トルエンスルホン酸の溶液を加え、得られた混合物を16時間、22℃で撹拌した。HPLC分析により、反応が完了したことを判断した後、混合物を0℃に冷却し、1.64kg(16.2mol、3.89eq.)のトリエチルアミンを加えて中和した。この溶液を減圧下で濃縮し、残渣を82LのMTBEおよび32Lの水に分配した。有機相を減圧下で濃縮し、10Lのトルエンで希釈し、再濃縮した。この手順を2回以上繰り返した。粗生成物のカラムクロマトグラフィー精製を、38kgシリカゲルで行い、23〜26% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、2.59kgの化合物68をトルエン溶液として得た。(67から67%)。
【0248】
【化49】
【0249】
2.90kg(3.10mol)の68および5.24kg(8.23mol、2.65eq.)の18の、30kg乾燥トルエン溶液を、0℃で、0.049kg(0.185mol、0.06eq.)のTBDMSOTfで処理し、0℃で4時間撹拌した。HPLC分析により反応が完了していることを判断した後、0.104kg(1.03mol、0.128eq.)のトリエチルアミン、続いて33Lのヘキサンを加えた。得られた懸濁液をろ過し、ろ液を28Lの水、その後28Lの5%Na2CO3水溶液で洗浄した。トルエン相を減圧下で濃縮し、粗生成物を58kgシリカゲルのカラムクロマトグラフィーで精製し、9〜20% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、4.40kgの四糖中間体69をトルエン溶液として得た(75%)。
【0250】
【化50】
【0251】
G.保護六糖(73)の合成
四糖中間体69を、実施例7に記述された方法に従って製造した。9.7g(5.15mmol)の69の、60ml CH2Cl2溶液に、100mlのMeOHを加え、続いて、混合物の温度を30℃未満に抑えながら、5.7N HClの1,4−ジオキサン溶液を27ml(0.154mol、30eq.)滴加した。その後、反応混合物を22℃で40時間撹拌した。HPLC分析により反応が完了したことを判断した後、混合物の温度が25℃未満に維持されるように、32ml(22.9mmol、44.7eq.)のトリエチルアミンを慎重に加えた。水(250ml)およびトルエン(200ml)を加え、混合物を振とうし、相を分離した。下相を50mlトルエンで再抽出し、まとめた上相を50mlの水で洗浄し、減圧下で濃縮した。粗生成物を100gのシリカゲルを用いてカラムクロマトグラフィー精製し、30〜50% v/v EtOAc/ヘキサンのグラジエントで溶出して、メチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,4−ジ−O−ベンジル−β−D−マンノシド70を得た(6.35g、69%)。
【0252】
【化51】
【0253】
1.0g(0.56mmol)の70および0.95g(1.40mmol、2.5eq.)の19の、9ml乾燥トルエン溶液を、0℃で、0.02g(0.075mmol、0.14eq.)のTBDMSOTfで処理し、0℃で1時間撹拌した。HPLC分析で反応が完了したことを判断した後、22ml(0.158mmol、0.28eq.)のトリエチルアミンを加えた。得られた混合物を、10mlの水で2度洗浄し、減圧下で濃縮した。15gシリカゲルのカラムクロマトグラフィーにより、粗生成物の精製を行い、15〜25% v/v EtOAc/ヘキサン:トルエン 1:1のグラジエントで溶出して、1.75gの化合物メチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド71を、残留トルエンを含む油状物として得た。
【0254】
【化52】
【0255】
10.0g(3.5mmol)の71の、40mL 1,4−ジオキサン溶液を、60mlのMeOHで処理し、その後3.25g(14.0mmol、4eq.)の(+)−カンファースルホン酸で処理し、得られた溶液を100時間、22℃で撹拌した。反応の終了をHPLC分析で判断した後、3ml(21.5mmol、6.2eq.)のトリエチルアミンを加え、溶媒を減圧下で除去した。残渣を200mlのMTBEに溶解させ、200mlのH2Oと共に振とうした。相を分離し、上相を減圧下で濃縮した。粗生成物のクロマトグラフィー精製は、114gのシリカゲルを用い、14〜25% v/v EtOAc/トルエンのグラジエントで溶出して、8.24gのメチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンゾイル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド72を、残留トルエンを含む油状物として得た。
【0256】
【化53】
【0257】
0.965g(0.35mmol)の72の、4g乾燥アセトニトリル溶液に、0.083g(0.70mmol、2eq.)の4,5−ジシアノイミダゾールを加え、続いて0.315g(0.91mmol、2.6eq.)のジベンジルジイソプロピルホスホルアミダイトを加え、混合物を23℃で1時間撹拌した。反応の終了をTLC分析で判断した後、0.5mlの水を加え、この溶液を15分撹拌した。水(9.5ml)および10mlのMTBEを加え、得られた混合物を振とうし、相を分離し、下相を10mlのMTBEと振とうした。上相をまとめて、減圧下で濃縮して、無色の油状物を得た。この残渣をCH2Cl2に溶解させ、−20℃に冷却し、0.247g(1.13mmol、3.2eq.)の70% 3−クロロ過安息香酸で処理した。TLC分析で反応の終了を判断した後、10mlの10%チオ硫酸ナトリウム水溶液を加え、混合物を23℃まで昇温した。下相を分離し、10mlの水と振とうし、減圧下で濃縮した。粗生成物を19gのシリカゲルでカラムクロマトグラフィーにより精製し、25〜50% v/v EtOAc/ヘキサンのグラジエントで溶出して、0.83g(72%)のメチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド73を、無色の油状物として得た。
【0258】
【化54】
【0259】
H. アミノオキシアセトアミドヒドラジド 3−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−(6−O−[α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−β−D−マンノシド(77)の合成
化合物73を、実施例8に記述された方法に従って製造した。氷酢酸(100μL)をメタノール/THF 1:1(600mL)中の73(64g、19.6mmol)に加え、生成物をH−cube(登録商標)を用いて、20%Pd(OH)2/Cを使用し、50℃、H2圧50bar、および流速6mL/分で触媒上を再循環させて水素化した。20時間後に反応の完了をTLCで確認し、この溶液を濃縮して、74を泡状物質として得た(39.88g、93%)。
【0260】
【化55】
【0261】
メタノール(180mL)を74(33.8g、15.5mmol)に、溶解するまで撹拌しながら加え、この溶液を氷/水槽で15分間冷却した。この溶液に64%ヒドラジン一水和物(94ml、1.24mol)を、撹拌しながら加えた。30分後、水(120mL)を加え、この溶液を室温になるまで放置し、18時間保存した。この溶液を約100mLまで濃縮し、水と共に留去し(2x100mL)、最終溶液を水で約180mLに調節した。この溶液をDCM(2x100mL)で抽出し、その後3分割した60mLをsephadexサイズ排除カラムで分離した。もっとも純粋な物質を含む画分をまとめ、凍結乾燥して、75を得た(15.5g、80%)。
【0262】
【化56】
【0263】
DMSO(20mL)を水(30mL)中の75(2.5g、2.0mmol)にゆっくり加え、その後DMSO(6mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(2.58g、7.6mmol)およびDMSO(4mL)中のDHBT(0.65g、4mmol)を加えた。18時間後、溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノオキシアセトアミドヒドラジドブチリル3−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−(6−O−[α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−β−D−マンノシド76(2.57g、90%)を得た。DCM(30mL)、次にTFA(16mL)を化合物76(2.57g、1.8mmol)に加えた。混合物が溶解するまで(約60分)撹拌し、その後油状物になるまで濃縮した。水(20mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、77を得た(1.6g、67.1%)。
【0264】
【化57】
【0265】
実施例6:ジスルフィドリンカーを有する六糖の合成
A.遊離酸形態の六糖の製造
無水MeOHを化合物73に加え、続いてNaOMeを加え、4〜18時間インキュベートした。反応を氷酢酸で停止し、この溶液をシロップ状になるまで濃縮して、78を得た。
【0266】
【化58】
【0267】
化合物78をTHF/メタノール1:1に溶解し、Pd/C−H2で水素化した。溶液を固体になるまで濃縮し、水に溶解させ、NaOH水溶液でけん化した。pHを約4に調整し、Sephadex G−10で精製して、遊離酸81を得た。
【0268】
【化59】
【0269】
B.ジスルフィドリンカーの結合
異なる方法で製造された(Biomiraから入手された)化合物81の粗画分を、過剰なトリエチルアミン(TEA)と混合することで、TEA塩に変換し、続いて、移動相として30%アセトニトリル、0.1%ジカルボン酸TEAを用いて、Superdex Peptide(GEヘルスケア)でクロマトグラフィーを行った。まとめた画分を凍結乾燥し、グリカン:NEA:EDAC:NHS:HOBt:TEA(1:1:1.5:1:1:1 mol:mol)を含む反応中で、NEAと結合させ、軽く撹拌しながら一晩インキュベートした。一部の生成物(0.5mg)を、前回のようにSuperdex Peptideでクロマトグラフィーを行い、凍結乾燥して82を得た(0.28mg)。
【0270】
【化60】
【0271】
実施例7:α−グルコシダーゼ複合体の合成および酸化の最適化
A.複合体化
オリゴ糖を組換えヒト酸性α−グルコシダーゼ(rhGAA)と結合させて、NeoGAAを形成した。主にr^hGAAのシアル酸残基を介して結合したオリゴ糖との複合体を「SAM」とし、ガラクトース残基を介して結合したものを「GAM」とする。
【0272】
NeoGAAβSAM6を、Zhuら,Biochem J,389(Pt3):619−628(2005)に記述されたように、基本的に製造した。この実験で使用するrhGAAのサンプルを、タンパク質1モルにつき、シアル酸を約5.2モル有するように、単糖組成物分析により見出した。簡潔に述べると、5mg/mLのrhGAA(Genzyme Corp.)を100mMの酢酸ナトリウムpH5.6de緩衝液交換し、その後過ヨウ素酸ナトリウム(2、7.5、または22.5mM)と、氷上で、遮光で30分反応させた。反応を2%(vol/vol)になるように、グリセロールを添加することで停止した。酸化反応から小さい分子量の副生物を取り除くため、酸化rhGAAを緩衝液交換し、化合物77(図9に示されるように、タンパク質に対して0〜120倍のモル比)と37℃で6時間複合体化させた。すべての複合体を25mMリン酸ナトリウムpH6.25に緩衝液交換した。これは2%のマンニトールおよび0.005%のTween−80を含む。
【0273】
類似のNeoGAA複合体を、SAM2(化合物17、実施例2)、SAM3(化合物35、実施例3)、SAM4(化合物28、実施例4A)、直鎖SAM4(化合物47、実施例4B)、およびαSAM6(オリゴ糖103)を用いて、7.5mMの過ヨウ素酸塩を使用し、オリゴ糖とrhGAAのモル比を変えることで製造した。
【0274】
別の複合体化方法も実施した。具体的には、アミノキシ、ヒドラジドまたはチオ−ル反応性リンカーのいずれかを有する六糖を、Cys374、リシン、シアル酸、またはガラクトース残基を介して、rhGAAに結合させた。
【0275】
rhGAAのリシン残基を4−ホルミル安息香酸スクシンイミジル(SFB;Solulink Corp.)で修飾し、続いてオリゴ糖と複合体化させることにより、リシンの複合体化を実施した。簡潔に述べれば、rhGAAをまず、150mMの塩化ナトリウムを含む50mMリン酸ナトリウムpH7.2に緩衝液交換した。その後、SFBとGAAとのモル比を20:1として新たに製造された(SFB)で、緩衝化されたrhGAAを処理した。当該混合物を室温で30分インキュベートし、それを100mMの酢酸ナトリウムpH5.5に緩衝液交換し、室温、2時間でヒドラジド六糖へと複合体化させたか、またはSFBで修飾されたGAAを、100mMの酢酸ナトリウムpH5.6に緩衝液交換し、37℃、6時間で、アミノキシ六糖へ複合体化させた。
【0276】
システイン系の複合体化を、チオール反応性NEA−六糖82(実施例6I)と反応させることで実施した。NEA−修飾された六糖82を、水中で再構成し、50mMリン酸ナトリウムおよび50mMヒドロキシアミンpH7.2中で、2時間25℃で、rhGAAと(rhGAAに対するネオグリカンのモル比15:1)インキュベートした。50mMのリン酸ナトリウムpH4.1で、pHを6.2に調整し、インキュベーションを一晩続けた。生成物を25mMリン酸ナトリウムpH6.2に対する遠心ダイアフィルトレーション(centrifugal diafiltration)によって精製した。1molに対して1mol未満のM6Pが導入された。
【0277】
Cys374を介する直接的な複合体化は成功しなかったが、ホモ二官能基性のチオ−ル特異的試薬である、19.9Åのスペーサーアームを有する1,4−ジ−(3’−[2−ピリジルジチオ]−プロピオンアミド)ブタン(DPDPB)が試験され、オリゴ糖との複合体化の前に、溶媒接近がより可能な374位のチオ−ル基を提供した。60倍モル過剰のDPDPBを、共溶媒として10%DMSOまたは10%プロパノールのいずれかの存在下で、rhGAAと反応させた。これは、光散乱によって検出されるように、強い凝集を引き起こした。20%アセトニトリルの存在下での反応はまた、凝集を示したが、反応混合物の限外ろ過液の344nmにおける吸光度は、システインの定量的修飾と一致した。10%までのアセトニトリル濃度の減少は、凝集量を減少させたが、修飾の程度は低かった。
【0278】
別のチオ−ルに基づいた方法を、リシン残基のチオ−ル基の導入により実施した。4時間25℃、リン酸ナトリウムpH6.2中で100倍モル過剰のSATA−dPEG4−NHS(Quanta Biodesign)と酵素反応させることにより、リシン残基に保護チオ−ルが導入された。そしてそれを、同じ緩衝液に対する一晩の透析により精製した。次に、精製された生成物を、システインを基にした複合体に関して、上記で記述された条件下で、NEA−オリゴ糖82と反応させて、リシン−チオ−ル複合体を得た。これは、Man−6P含有量の約10倍の増加を示した(約5個のグリカンが複合体化された)。
【0279】
ヒドラジドとのリシン複合体の安定性は、無傷のタンパク分子量およびM6P含有量を測定することで、37℃、14日まで評価された。50%を超えるネオグリカンが14日間で消失したように、この複合体は安定ではない。リシンを介したアミノキシ複合体を、上記のように、rhGAAに対して0、16.6、25、33および40モル過剰の六糖を用いて製造した。全リシンの約31%だけが複合体化(または5個のネオグリカンが複合体化)されたが、当該複合体化は、16.6倍モル過剰で飽和した。高い凝集レベルが、数種の調製物においても観察された。PEG化されたSFBを試験したが、凝集の減少は見られなかった。
【0280】
2%マンニトールおよび0.005%Tween−80を含む、25mMリン酸ナトリウム、pH6.25中で、37℃で6時間、Clostridium perfringens由来のシアリダーゼ20mU/mgで、rhGAAをまず前処理することにより、ガラクトース複合体化(GAM)を実施した。ジシアリル化後、タンパク質を同一緩衝液中、37℃で一晩、1〜10μg/mgのガラクトースオキシダーゼ(GAO)および2U/mgのカタラーゼ(Sigma)で処理した。その後、Poros50D(陰イオン交換)クロマトグラフィーを用いて再精製して、ノイラミニダーゼおよびカタラーゼを除去した。両方の酵素で処理した生成物を、等量のdH2Oで希釈し、その後、10mMのリン酸ナトリウム緩衝液、pH6.9で事前に平衡状態にあるPoros50Dカラムにアプライした。カラムを10mMの酢酸ナトリウム緩衝液、pH5.0で洗浄し、rhGAAを150mM酢酸ナトリウム緩衝液、pH5.0で溶出させ、そして37℃で6時間、様々なモル比でアミノキシ六糖と複合体化させた。
【0281】
GAM複合体化は、GAAに対して16.6倍モル過剰の六糖で飽和し、約6〜7個のグリカンが複合体化された。凝集レベルは低かった。脱シアリル化の後、シアル酸は検出されなかった。一方、ガラクトースオキシダーゼ処理後にガラクトースもほとんど検出されなかった。いくつかの実例において、20〜30%のガラクトース残基が過剰酸化され、オリゴ糖に結合していないガラクツロン酸を生成した。GAOを滴定すると、1μg/mg超でGAOはグリカン複合体を減少させることを示した。GAOが2μg/mgGAOより多いと、ガラクツロン酸の過剰酸化産物量の明らかな増加がみられた。複合体の最大量は、rhGAA1mgにつき、1〜2μgGAOで実現した(図10E−GAOの滴定後における、GAM複合体の、Man−6P、Gal、GalAを含む単糖の含有量)。GAA 1mgにつき、GAOを0.5〜2μg使用すると、多くの複合体化が、タンパクのMan−6P含有量として観察された。より少ないまたはより多くのGAOを使用すると、より少ないガラクトース、またはより多いGalAのいずれかが生じた。
【0282】
NeoGAAに複合体化したビス−M6P六糖グリカンの量を、M6P含有量分析およびMALDI−TOFにより定量した。M6Pの定量に関して、サンプルをAmicon4、50,000MWCO遠心式フィルターユニットを用いて、5回ろ過を行って、緩衝液交換し、潜在的に過剰のグリカンを除去した。各80マイクログラムのrhGAAまたはNeoGAAサンプルを、6.75MのTFAで1時間半、100℃で加水分解した。サンプルを冷却し、Speed Vacで乾燥させ、200μLの蒸留水で再構成した。再構成したサンプルを再び、Speed Vacで乾燥させ、200μLの50mMクエン酸pH2.0で再構成した。サンプルをS Mini Hカートリッジ(Sartorius)でろ過し、これをクエン酸ナトリウムpH2.0で平衡化して、加水分解物から不純物を除去した。リボース−5−リン酸を、内部標準として、すべてのサンプルおよび標準に追加した。50μLの加水分解物をDionex HPLCに注入し、パルスアンペロメトリック検出機付き高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)によって分析した。加水分解した標準のM6Pで構築した、検量線を用いて定量を行った。その後、複合体化の程度を、グリカン1モルあたり、M6P 2モルの公知のモル比を基に計算した。
【0283】
MALDI−TOF MS分析を、直列モードでVoyagerDE−PRO質量分析計を用いて実施した。0.1%ギ酸水溶液に1:5で希釈し、すべてのサンプルおよび標準で実施し、続いて飽和シナピン酸の50%アセトニトリル/0.1%TFA溶液に1:1で希釈して行った。この混合物の1μLを標的に利用した。サンプル、レファレンス、BSA補正コントロールの3通りで分析した。二点校正を(M+H)+およびBSAの二量体イオンを用いて行った。各NeoGAAサンプルの複合体化の程度は、測定されたグリカン分子量が1323g/モルであることを考慮して、サンプルおよび酸化rhGAAコントロール(グリカンは追加されていない)間の分子量の違いを基に計算した。
【0284】
図9Aは、上記の二糖、三糖、四糖、および六糖複合体を用いた実験の結果を示す。
【0285】
図9Bは、異なる量の過ヨウ素酸塩で製造されたβSAM6複合体の結果を提供する。複合体化反応の飽和を実現するのに必要なオリゴ糖レベルは、酸化工程中に使用される過ヨウ素酸塩の量に比例した(図9B、上のパネル)。2mM過ヨウ素酸塩を用いて、シアル酸約5.2モルを有するrhGAAのサンプルは、タンパク質に対してほぼ25倍モル過剰の六糖で(シアル酸に対して4.8倍モル過剰で)飽和に達した。7.5mMの過ヨウ素酸塩を用いて、グリカンの33倍モル過剰で飽和に達した。グリカンの120倍モル過剰では、飽和に達したが、22.5mMの過ヨウ素酸塩で酸化したrhGAAでは実現しなかった。実現した複合体化の最大レベルは、異なるレベルの過ヨウ素酸塩で製造したサンプルでも異なった。7.5および22.5mMの過ヨウ素酸塩を用いて、タンパク質1モルにつき、それぞれ約8.5および10.5モルのグリカンが組み込まれた。2mM過ヨウ素酸塩での酸化の後、実現可能な複合体化レベルは、タンパク質1モルにつき約5モルのグリカンであり、これは出発物質中のシアル酸残基の数に近い。
【0286】
グリカンの滴定実験を、タンパク質1モルにつき、最初のシアル酸レベルが約7.2モルであるrhGAAを用いて、2mM過ヨウ素酸塩で繰り返した(図9B、下のパネル)。タンパク質1モルにつき、約7モルのグリカンの複合体化レベルは、タンパク質に対して33倍モル過剰以上のグリカンで達成された(シアル酸と比較して、約4.6倍モル過剰)。
【0287】
B.凝集の減少
特定の複合体化方法は、タンパク質の凝集を引き起こす。neoGAAにおいて、凝集を減少させるための2つの方法が開発された。1)様々なHICクロマトグラフィー担体を用いた、疎水性相互作用クロマトグラフィー(HIC)、および2)金属キレート化である。
【0288】
NeoGAAの3gのバッチを調製し、凝集除去に関して、HICおよび銅カラムを評価するために使用した。フロースルーモードで評価したHICカラムは:Butyl650Cおよび650M、Hexyl650C、Phenyl6FF、Capto Octyl、及びCapto Phenylであった。HexylおよびCapto Phenylで、87.5%および90.4%の回収率、並びにそれぞれ3.2%(初期レベル)から1.4%、および3.9%(初期レベル)から1.6%までの凝集減少という同等の結果を示した。表2を参照のこと。
【0289】
【表5】
【0290】
銅キレートカラム(GEまたはTosoh)の操作条件も、フロースルーまたは結合−溶出モードのどちらかで確立した。銅を充填した7mlの金属キレートFFカラム(I.D.、7ml)にまず、10mg/mlの複合体化したGAAをロードして、結合・溶出モードで評価した。室温で、溶出緩衝液として175mMグリシン、100mM酢酸、pH5.5でカラムから溶出されたとき、87%のNeoGAAが回収され、凝集体は1.2%であった。満足な回収率でカラムから溶出されるために、8℃にて175mMより高いグリシンが必要であった。フロースルーモードにおいては(表3)、溶出緩衝液として、150mMグリシン、100mM酢酸塩、pH5.5を用いて、92%の優れた回収率が実現され、凝集体は3.2から1.2%に減少した。
【0291】
【表6】
【0292】
イミダゾール(7.5、8および10mM)も、金属キレート6FFカラムの溶出緩衝液として試された。カラムを溶出するためには、イミダゾールが約8mM必要であった。イミダゾールはカラムから銅を溶出しないため、カラムをコンディショニングする、またはカラムの上方にEDTAで空きスペースを作る必要がなかった。NeoGAA 1mlにつき15mgのカラム容量を達成した。
【0293】
銅を充填したTosoAF−キレート650Mカラムも評価した。結合・溶出モードにおいて、15mg/mlのカラム容量が達成され、8mMのグリシンを用いて、94.1%の溶出および1.2%の凝集体であった。フロースルーモードにおいて、33.6mg/mlの容量を実現した。この溶出緩衝液において、50mMのグリシンで90.6%の回収率、1.2%の凝集体であった。
【0294】
C.オリゴ糖の分析
これらの実験によれば、>2mMの過ヨウ素酸塩の使用で、タンパク質中の出発時レベルのシアル酸レベルを超えたNeoGAAグリカンの取り込みが起こり、これは非シアル酸部分が酸化されたことを示す。過ヨウ素酸塩による、他の炭水化物部位での酸化レベルを測定するため、一連の過ヨウ素酸塩の滴定実験を実施し、他の単糖残基のレベルを観察した。
【0295】
シアル酸含有量を測定するため、サンプルを0.5Mのギ酸を用いて、80℃で1時間、酸加水分解させた。放出されたシアル酸を50〜180mM酢酸ナトリウムグラジエントを用いて、100mM水酸化ナトリウム中で、20分以上、Dionex CarboPac PA1カラムで、パルスアンペロメトリック検出付きの、高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)によって分析した。結果を、rhGAAまたはNeoGAA 1モルにつき、シアル酸(NANAまたはNGNA)のモルとして表し、真正の商用シアル酸標準物の検量線から測定した。
【0296】
フコース、ガラクトース、GlcNAc、およびマンノースを含む、中性単糖のレベルを、110℃で2時間、1MのTFA中で、100μgのrhGAAまたはNeoGAAを加水分解することで測定した。加水分解に続いて、チューブを氷上で冷却し、1分10,000rpmで遠心分離し、Speed Vacにより蒸発乾固した。放出された単糖を250μLの水に再懸濁させ、ボルテックスし、Millipore Ultrafree−MCフィルターチューブを用いてろ過した(10,000MWCO)。放出された単糖を、CarboPacPA1カラムで、パルスアンペロメトリック検出付きの高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)により分析した。定量は、加水分解した単糖の検量線を用いて、同様の方法で行った。
【0297】
上記の実験結果を図10Aに示す。結果から、シアル酸が最も酸化を受けやすい単糖であると示唆された。低レベルのフコース破壊は2mMの過ヨウ素酸塩で検出することができ、その量は5mM以上で測定および再現することができた。マンノースのわずかな酸化は、7.5mM以上の過ヨウ素酸塩でだけ検出することができた。
【0298】
酸化シアル酸、フコース、およびマンノースの存在を確認するため、フラグメンテーション質量分析を、7.5mM過ヨウ素酸塩を用いて酸化したrhGAAから放出されたオリゴ糖に関して実施した。rhGAAおよびNeoGAA中のN−結合型オリゴ糖を、50mMリン酸ナトリウムpH7.0および10mMB−メルカプトエタノール中で、PNGaseFを用いて、一晩かけて分解させて、放出させた。放出されたオリゴ糖を、水を変更しながら、生物的透析(MWCO500Da)により不要なものを取り除いた。透析したサンプルをspeed−vac濃縮機で乾燥し、10mMギ酸アンモニウムpH4.0の50%アセトニトリルおよび50%水の溶液の110μLで再構成した。サンプルをアセトニトリル−水グラジエントおよび10mMギ酸アンモニウムpH4.0で、インラインMS検出器(QStar quadrupole飛行時間型、LCT Premier飛行時間型、およびLTQ直線イオントラップ機)を用いた、TSKGelAmide−80カラム(2x100mmへ100μL注入、5μm粒子サイズ)を使って分析した。
【0299】
オリゴ糖構造を分析するため、オリゴ糖を乾燥し、アントラニル酸(AA)で蛍光標識した。AA標識したオリゴ糖を、アセトニトリル/水グラジエントを用いて、蛍光検出機を使用し、TSKゲルアミド80カラムで、順相HPLCにより分離した。MSフラグメンテーション分析を、陽イオンモードで、LTQ XL直線イオントラップマススペクトロメータを用いて、インラインで行った。スペクトルを400〜2,000m/zでスキャンし、衝突エネルギーは35に(初期設定値、本文に示されない限り)、activationQは0.25に合わせた。
【0300】
シアル酸:シアル酸の完全酸化(C7,8およびC8,9結合の両方で)は、質量で62ダルトンの減少をもたらす。フコースおよびマンノースの酸化は、最初に2ダルトン減少(C2,3またはC3,4結合の酸化)させ、続いて残りのビシナル・ジオールの酸化で、30ダルトン減少する。AAを有する炭酸水素アルデヒドの還元的アミノ化は、酸素の消失を招き、1AAを追加するにたびに、正味121.1ダルトンの分子量の増加が引き起こされる。シアル酸、フコース、およびマンノースを酸化後の、rhGAAオリゴ糖における、理論的な分子量および観察された分子量の変化を表4に示す。
【0301】
【表7】
【0302】
酸化およびAA−誘導型酸化シアル酸、フコース、およびマンノースオリゴ糖種に対応する様々なイオンが観測された。放出されたすべてのオリゴ糖の還元末端のGlcNAcでの誘導化に加えて、AAは存在する反応性アルデヒド種の数と比例して、つまり、酸化シアル酸とでは1:1、酸化マンノースおよびフコースでは2:1で誘導された。
【0303】
図10Bの質量スペクトルは、シアル酸のC7位の酸化およびAA誘導を有するrhGAAオリゴ糖に対応する4つのイオンの検出を示す。シアル酸の酸化に関する2つのイオン(m/z1057および1130)を、さらなるMS/MSフラグメンテーション分析のために選択した。m/z 1057のフラグメーションパターンを図10Cに示し、各イオンは仮定された物質が注釈されている。フラグメンテーションイオンm/z 716、1032、1235、1397、1584、および1747は、MS3分析から選択された。MS3スペクトルは、二分岐鎖グリカンの末端の酸化およびAA標識シアル酸を含む、仮定されたオリゴ糖構造と一致した。特に、m/z 351のフラグメントの放出により、シアル酸のC7型へのAAの結合を確認した。全サンプルの分析において、C7型の酸化されたシアル酸だけが観察された。C8位で酸化されたシアル酸の存在に関する証拠は観察されなかった。
【0304】
フコース酸化:表5にAA−誘導化、酸化A1FおよびA2Fの理論質量および観測質量を列挙する。
【0305】
【表8】
【0306】
m/z1250.4の親イオンのフラグメンテーションパターンを図10Dに示し、酸化シアル酸および酸化フコースを有する、AA標識、一シアル酸、一本鎖、フコシル化コアオリゴ糖で予測されたものと一致する。主なフラグメントイオンはm/z 716、1397、1621、および1783で観測された。これらのイオンは仮定された物質を確認するため、MS3分析のために選択された。m/z 1621および1783のMS3スペクトルにおいて、イオンフラグメンテーションパターンから、親イオンから195ダルトンが消失していることを確認した(それぞれ、親イオン1621および1783から1426および1588まで)。この分子量の変化は、酸化フコースのC1および酸素原子間の切断と一致し、これはC4に結合した誘導体化されたアントラニル酸の消失をもたらす。加えて、さらなるフラグメンテーション、およびC3に結合した別の誘導体化アントラニル酸の消失が、親イオンから386ダルトンの消失として観測した(それぞれ、親イオン1621および1783由来で、1235および1397m/z)。
【0307】
酸化rhGAAオリゴ糖A1FのMSフラグメンテーションパターンは、アントラニル酸が結合したフコースのC3、4の誘導体化を示し、これにより過ヨウ素酸塩による酸化が起こることを確認した。C2−C3結合の酸化の証拠は観察されなかった。ビス−M6P六糖グリカンの酸化rhGAAへの結合は、縮合反応の間に、正味1305.3ダルトンの増加をもたらした。
【0308】
複合体化したオリゴ糖構造の一体性を確認するため、放出された天然オリゴ糖の高精度MS分析を実施した。2および7.5mMの過ヨウ素酸塩で製造したrhGAAおよびNeoGAA SAM6由来のオリゴ糖を、PNGase Fを使用して放出し、10mMのギ酸アンモニウムpH4.0を有するアセトニトリル−水グラジエントで、順相HPLC(TSKゲルアミド−80カラム)により放出した。グリカンの質量分析検出を、インライン、陰イオンモード、QStarまたはLCT飛行時間型質量分析装置を用いて実施した。
【0309】
次の表に、高性能MS/TOF分析を基に、2および7.5mMの過ヨウ素酸塩で酸化したrhGAA由来の天然N−結合型オリゴ糖ピークの同定、並びに2および7.5mMの過ヨウ素酸塩で製造されたSAM6における天然N−結合型オリゴ糖ピークの同定の概要を示す。「Ox」は、酸化の部位数を、「Conj」は複合体化されたビス−M6P六糖グリカンの数を示す。理論質量は理論のオリゴ糖構造のモノアイソトピック分子量から計算され、対応する電荷状態の理論m/zおよび観察m/zを示す。
【0310】
【表9】
【0311】
【表10】
【0312】
【表11】
【0313】
【表12】
【0314】
【表13】
【0315】
20ppm超の質量精度がすべてのオリゴ糖種に関して観測された。MSの結果は、シアル酸の酸化が1mM以上の過ヨウ素酸塩で完了することを示す単糖組成物分析と一致した。マンノースおよびフコースの一部の酸化も明らかであった。酸化マンノースおよび/またはフコースにつき、1および2モルのグリカンの結合に相当するイオンが観測され、これはAAで観測されたように、それぞれの両方のアルデヒド種がグリカンに対して反応性であることを示唆する。
【0316】
高マンノース構造(オリゴマンノース5および6)のいくつかの結合を、2および7.5mMの過ヨウ素酸塩で処理した物質で検出した。2mMの過ヨウ素酸塩で結合した物質において、0または1個複合体化されたグリカンがA1(モノシアル化)種で観測され、0、1、2、および3個複合体化されたグリカンに相当するイオンが、7.5mM SAM6のA1種で観測された。この結果から、7.5mM(2mMではなく)過ヨウ素酸塩を用いて、コア・マンノースおよび/またはガラクトース残基の幾らかの酸化および複合体化が、A1構造で起こることが示唆される。
【0317】
A2およびA2F種(二シアル酸、二分岐鎖、±フコース)について、モノ−およびビ−複合体化種が、2および7.5mMの過ヨウ素酸塩サンプルで観察された。酸化マンノースとのトリ−複合体化の証拠は、7.5mM過ヨウ素酸塩処理サンプルでのみ観察され、2mM処理では観察されず、高い過ヨウ素酸塩濃度のフコースを介した複合体化と一致した。
【0318】
図11Aは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。rhGAAコントロールに関して、N−結合型オリゴ糖種の大部分は、11〜13分に溶出し、これはリン酸化も複合体化もされないオリゴ糖に相当する領域である。NeoGAAオリゴ糖について、ビ−複合体化(bi−conjugated)オリゴ糖種は19〜20分、モノ−複合体化(mono−conjugated)オリゴ糖種は15〜18分、および酸化/無修飾オリゴ糖は10〜13分で溶出した。7.5mM過ヨウ素酸塩で生成されたSAM6サンプルについて、そのオリゴ糖のほぼ半分がビ−複合体化種に相当する領域で溶出し、2mM過ヨウ素酸塩処理したサンプルのオリゴ糖のおおよそ3分の1はビ−複合体であった。これらのサンプルの溶出分析結果は、MALDI−TOFおよびマンノース−6−リン酸含有量分析によって測定された複合体化レベル(SAM6を生成する2および7.5mM過ヨウ素酸塩に関して、NeoGAA1モルあたり、複合体化されたグリカンそれぞれ約7および9モル)に一致した。
【0319】
存在するオリゴ糖構造のより優れた、可視化および定性比較を提供するため、放出されたオリゴ糖を、パルスアンペロメトリック検出付きの高pH陰イオン交換クロマトグラフィーに(HPAEC−PAD)より分析した。サンプルを酢酸ナトリウム/100mM水酸化ナトリウムグラジエントを用いて、Dionex CarboPac PA100カラムで、HPAEC−PADにより分析した。オリゴ糖ピーク物質を、オフラインフラクションコレクション、透析vs.水、およびインラインMS分析を用いた順相HPLCによる分析によって確認した。
【0320】
図11Bは、(MSで測定された)ピーク物質を含む代表的なHPAEC−PADデータを示す。図11Cは、2および7.5mMの過ヨウ素酸塩処理したrhGAAおよびNeoGAASAM6サンプルのHPAEC−PADによるオリゴ糖のプロファイリングを示す。
【0321】
D.過ヨウ素酸塩処理後のrhGAAタンパク質骨格の分析
NeoGAAのタンパク質骨格のタンパク質修飾を調べるため、ペプチドマッピングLC/MSを実施した。0、2、7.5および22.5mMの過ヨウ素酸塩を用いて製造したNeoGAASAM6は、トリプシンを用いて調製され、LCT飛行時間型質量分析器を用いて逆相HPLCにより分析した。システイン、メチオニン、トリプトファン、チロシン、およびヒスチジン残基の酸化、ならびにアスパラギンの脱アミドなど、可能性のあるペプチド修飾を、BioPharmalynxソフトウェアを使用して評価した。検出された有意な修飾は、いくつかの異なる部位のメチオニン酸化のみであった。0、2、および22.5mM過ヨウ素酸塩処理後の、ペプチドT13(メチオニン172および173を含む)の酸化レベルを図12に示す。
【0322】
有意なレベルの酸化が、メチオニン残基122、172および173で見出された。これらのメチオニン残基の酸化を、LC/MS/MS分析により確認した。低レベルの酸化も、メチオニン363で、過ヨウ素酸塩に依存した方法により起こることが観察された。過ヨウ素酸塩の酸化を最小限にする試みにおいて、過ヨウ素酸塩の濃度は、1〜7.5mMのレベルで滴定され、LC/MSによりモニターされた最も感受性の高い部位(ペプチドT13)で酸化した。有意なレベルのメチオニン酸化は、1mMより高い過ヨウ素酸塩濃度で観測され、これによりrhGAAのメチオニンがシアル酸残基と同じくらい酸化に対する感受性が高いことが示された。
【0323】
GAM複合体化の間に、GAOによるガラクトース酸化は、Met172/173の約26%の酸化をもたらした。カタラーゼが2および50ユニット/mgGAAで酸化反応中に含まれた場合、メチオニン酸化は排除された。
【0324】
実施例8:GAA複合体のin vitro特性評価
NeoGAA SAM2を、実施例2の化合物17および7.5mM過ヨウ素酸塩を用いて、実施例7に記述されたように製造した。ガラクトース結合NeoGAA GAM2を二糖17と結合させる前に、ガラクトースオキシダーゼでrhGAAを処理することで製造した。同様に、三糖NeoGAA SAM3(化合物35、実施例3)、四糖NeoGAAs SAM4(化合物28、実施例4A)、および直鎖SAM4(化合物47、実施例4B)も製造した。さらに、六糖NeoGAAβ SAM6を、実施例7に記述されたように、2および7.5mM過ヨウ素酸塩を用いて、化合物77を用いて製造した。追加の六糖複合体αSAM6(α結合、シアル酸残基を介して複合体化されている)およびGAM6(ガラクトース残基を介して複合体化されている)が同様に製造された。
【0325】
A.特異的活性:
活性分析を、rhGAAおよびNeoGAAによって触媒される、合成基質p−ニトロフェニル−D−α−グルコピラノシド(p−NP)の加水分解の比率をモニターすることで実施した。放出される発色団を、アルカリ条件下で、400nmの吸光度で測定した。1単位の活性を、37℃、定義されたアッセイ条件下で、1分あたり、1μmolのp−ニトロフェニル−D−α−グルコピラノシドをp−ニトロフェノールに加水分解するのに必要な酵素量として定義する。
【0326】
NeoGAA SAM2、SAM3、SAM4、直鎖SAM4、αSAM6、およびβSAM6の特異的活性を、図13に示す。さらに、SAM法 vs.GAM法を用いて製造した複合体の特異的活性を評価した。GAAに対するM6Pの数と、小規模および大規模で製造されたNeoGAA複合体の特異的活性との間には逆相関(p<0.001)があり、GAAあたり16.6倍モル過剰のオリゴ糖であった。複合体化(2.5〜33倍モル過剰のオリゴ糖)の間にNeoGAA濃度が滴定される別の実験において、GAA活性の消失がまた、SAMまたはGAM複合体におけるM6P含量の増大と共に観察された。49〜81%のGAA活性を有するSAM(コントロールと比較)について、様々なNeoGAAが1タンパク質につきオリゴ糖を6〜8分子含有した。GAM複合体は67〜92%活性を有し、1タンパク質あたり4〜6個のオリゴ糖を含有した。
【0327】
B.M6P受容体結合:
複合体の機能的効果は、BiacoreおよびM6P受容体アフィニティ−カラムHPLCによって、NeoGAAの可溶性カチオン非依存性マンノース−6−リン酸受容体(sCIMPR)への結合をモニターすること、およびL6筋芽細胞における取り込みによって評価した。ウシ血清から精製した、sCIMPRは、A〜Xの細胞外ドメインを含み、膜透過部分を欠いている。
【0328】
Biacore分析に関して、sCIMPRをCM−5チップにアミン結合させ、NeoGAAサンプルの10μg/mLを表面に固定し、マンノース−6−リン酸の濃度を増加させて溶出した。結合したNeoGAAの50%を置き換えるのに必要なM6Pの濃度として、親和性を定量した(EC50)。当該方法を、受容体親和性の結合および酸化の効果をモニターするのに使用した。
【0329】
図14にBiacore分析の結果を示す。sCIMPRを固定化したBiacoreチップ上に10μg/mLのNeoGAAを注入すると、約250RUのレスポンスの増加を引き起こし、同じ量のrhGAAでは、約100RUの偏差を引き起こした。さらに、rhGAAよりNeoGAAを溶出するのに、ほぼ10倍高いM6P濃度が必要であった(rhGAAおよびNeoGAAについて、それぞれほぼ0.1vs.1.0mMのEC50値)。EC50値と、2および7.5mM過ヨウ素酸塩を用いて製造したNeoGAAの結合レベルには、直線的な関係が見られた(R−2乗>0.95)。調査した結合範囲において(1.6〜4.7モルのグリカン/モル、NeoGAA、2mMの過ヨウ素酸塩で製造、および1.0〜8.5モルのグリカン/モル、7.5mMのNaIO4で製造)、結合親和性に関する複合化レベルの効果は、2および7.5mM過ヨウ素酸塩で製造されたNeoGAAに類似していた。
【0330】
PorosEP樹脂にsCIMPRを固定化し、その後分析用HPLCカラムにパッキングすることで調製された、M6P受容体カラムを使用して、M6P受容体結合をHPLCにより評価した。rhGAAおよびNeoGAAを、0.25、0.85、5、および20mMのM6Pを用いて溶出した(図15AおよびB)。1つの実験において、SAM2およびGAM2を、βSAM6およびGAM6と比較した(図15C)。SAM6およびGAM6では、物質の大部分が溶出される前に(>95%の、カラムに結合した複合体)、20mMのM6Pが必要であった。SAM2およびGAM2は強固には結合せず、5mMのM6Pで大部分が溶出した(>95%がカラムに結合)。
【0331】
さらに、NeoGAA複合体SAM2、SAM3、SAM4、直鎖SAM4、およびαSAM6を、M6P受容体結合に関して評価した(図15D)。SAM2、SAM3、および両方のSAM4複合体の大部分が、5mMのM6Pで溶出したが、両方のSAM6複合体は20mMのM6Pが必要であった。
【0332】
種々の量が複合を有する、NeoGAA複合体を評価した(図15E)。結合画分のNeoGAAの割合は、複合体化されたNeoGAA1モルあたり、>2.0モルのグリカンを有する製造物に関して、一定して>95%だった。複合体化レベルの低いわずかな画分において(1.0〜1.7モルのグリカン)、結合画分は75〜90%であった。NeoGAAに関するM6Pカラム分析データも、rhGAAより高親和性種の量が高いことを示した。具体的には、結合rhGAAの大部分は、0.2mMのM6Pで溶出され、カラムからNeoGAAを溶出するには、20mMのM6Pが必要であった。高親和性種の割合に関して、複合化レベルの全体的な効果は、2.0〜7.5mM過ヨウ化酸塩処理に類似していた。
【0333】
図15Fは、さまざまな量のM6Pの効果を示す。SAM6複合体は、以下のM6P量、SAM6−1(rhGAA 1モルにつき、M6P 4.9モル)、SAM6−2(GAA 1モルにつき、M6P 7.4モル)、SAM6−3(rhGAA 1モルにつき、M6P 10.5モル)、SAM6−4(rhGAA 1モルにつき、M6P 11.2モル)およびSAM6−5(rhGAA 1モルにつき、M6P 16.6モル)を含有した。統計的有意性を^、*および***で示し、それぞれ100mg/kgのrhGAA、SAM6−1、SAM6−3と比較したp<0.05を表す。
【0334】
C.L6筋芽細胞による細胞内移行
L6筋芽細胞の取込みアッセイを、Zhuら,J.Biol.Chem.279:50336−50341(2004)に記述の通りに実施し、カチオン非依存性マンノース−6−リン酸受容体(CIMPR)経路を介して、rhGAAおよびNeoGAAが筋芽細胞を標的とすることが明らかにされた。L6筋芽細胞の取込みアッセイにおいて、rhGAAおよびNeoGAAを、L6筋芽細胞を含むウェル中の+/−5mM M6Pの培地に加え、あらかじめ決定された一晩にてインキュベートした。インキュベート後、細胞を溶解し、4−MUグルコシド基質を用いた活性、およびマイクロBCAアッセイによるタンパク質濃度を分析して、酵素の用量反応曲線を作成した。
【0335】
L6筋芽細胞の取込みアッセイの結果を、図16に示す。SAM2およびGAM2複合体は、無修飾のrhGAAより優位に高い取込みを示したが、二リン酸化されたSAM6またはGAM6複合体ほど高くはなかった(図16、上図)。NeoGAAのSAM2、SAM3、SAM4、直鎖SAM4、およびαSAM6の取込み試験も行った(図16、下図)。同様の実験において、実施例7のリシン−チオ−ル複合体は、約8倍の取込み増加を引き起こした。
【0336】
実施例9:新規GAA複合体のin vivoでの効果
ある種のNeoGAA複合体のin vivoでの効果を、Rabenら,J.Biol.Chem.273(30):19086−92(1998)に記述された、GAAノックアウトマウスモデルを用いて調査した。以下の通り、それぞれ6匹のマウス群を4週間にわたり1週間に1回処置した。
【0337】
【表14】
【0338】
サンプルを心臓、大腿四頭筋、および上腕三頭筋から集め、組織のグリコーゲン含有量を測定した。SAM2、SAM4およびSAM6動物の結果を、それぞれ図17、図18、および図19に示す。SAM6複合体を用いて12匹の動物群で、実験を繰り返し、SAM6複合体が、未修飾のrhGAAより5倍強力であることを確認した。
【0339】
SAMおよびGAM六糖複合体を、溶媒、20、60、または100mg/kgのrhGAA、あるいは4、12、または20mg/kgのSAM6またはGAM6を4週間、1週間に1回摂取した6匹のマウス群を用いて比較した。心臓、大腿四頭筋、上腕三頭筋、隔膜、および腰筋を摘出し、グリコーゲン含有量を分析した。図20に、この試験結果を示す。薬物動態および薬力学試験については、3〜6カ月齢の30匹のGAAノックアウトマウス(オス15匹、メス15匹)を、Charles River Laboratories、ウィルミントン、MAから入手した。各用量群にはオス5匹、メス5匹が含まれる。動物をグループにして飼育し、恒湿25℃、12時間の明暗周期を維持した。すべての動物は、飼料(PicoLab(登録商標)Rodent Diet20)および水を自由に摂取することができるようにした。各用量につき計10匹のマウスとするため、動物を1グループにつきオス5匹、メス5匹の3用量グループに無作為に分けた。各グループには、20mg/kgで、rhGAA、GAMおよびSAM複合体の単回静脈内投与を行った。薬物動態分析用の血液サンプルを、意識のあるマウスの後眼窩静脈叢を介して、投与後、5、15、30、60、120、240、および480分で回収した。血清中のrhGAA濃度を、GAA活性アッセイを用いて測定した。結果を図20Bに示す。
【0340】
実施例10:酸性スフィンゴミエリナーゼ複合体の合成
バキュロウイルス発現系またはチャイニーズハムスター卵巣細胞で発現した組換えヒト酸性スフィンゴミエリナーゼ(rhASM)は、遊離チオ−ル基を有するC−末端システインを有する。Lansmannら,Eur.J.Biochem.270:1076−1088(2003)およびQiuら,J.Biol.Chem.278:32744−32752(2003)を参照のこと。rhASMは、遊離チオ−ル基を介して、オリゴ糖1〜127のいずれかと結合することができ、このオリゴ糖は、米国特許仮出願第60/885,457号、または実施例6に記載された方法によると、リンカーおよびチオ−ル反応性基を含む。
【0341】
実施例11:α−L−イズロニダーゼ複合体の合成
α−L−イズロニダーゼは、オリゴ糖1〜127のいずれかと結合し、このオリゴ糖は、Leeら,Pharm.Res.20:818−825(2003)に記載された方法によると、プロピオンアルデヒド反応性基を含むリンカーを含む。α−L−イズロニダーゼおよびオリゴ糖は、室温、pH5.5、1日間で、還元剤としてシアノ水素化ホウ素ナトリウムの存在下で結合する。その後、小分子が透析または膜分離により、反応混合物から除去される。
【0342】
本明細書で引用されるすべての参考文献は、その全体が参照により本明細書に組み込まれる。参照により組み込まれる参考文献および特許または特許出願が、本明細書に含まれる発明に相反する場合、本明細書があらゆる相反する構成要素に取って代わる。
【0343】
本明細書および請求項で使用される成分、反応条件などの量を表す全数字は、「約」という語により、すべての場合で修飾されるものとして理解されるべきであり、約は例えば±5%を意味する。したがって、反対に指示されない限り、明細書および添付請求項に明記される数値パラメーターは、本明細書によって試みられた所望の特性によって変化する概数である。最低限でも、請求の範囲に相当する原理の応用を限定するいかなる意図は無く、各数値パラメーターは、有効数字の数および概数で表す方法の観点で解釈されるべきである。
【0344】
本発明の範囲および趣旨から逸脱することなく、当業者に明らかな、本発明の多くの修飾および変形を行うことができる。本明細書で記載された具体的な実施形態は、例示のためにのみ記載するものであり、いかなる方法でも限定されるものではない。本明細書および実施例は、例示としてのみの目的であり、本発明の本来の範囲および趣旨は以下の請求項により示される。
【技術分野】
【0001】
本願は、米国特許仮出願第61/122,851号の優先権の利益を主張し、その全体を参照により本明細書に組み込む。
【0002】
本発明は、一般に個々のオリゴ糖を含むオリゴ糖−タンパク複合体、およびかかる複合体を含む組成物に関する。本発明はさらに、オリゴ糖−リソソーム酵素複合体を用いた、リソソーム蓄積症の治療方法に関する。
【背景技術】
【0003】
リソソーム蓄積症(LSD)は、リソソーム加水分解酵素の活性欠損に関与する、希少な代謝性疾患の1つであり、40種を超える遺伝子疾患が含まれる。LSDの顕著な特徴は、リソソーム代謝産物の異常蓄積であり、多数の膨張したリソソーム形成を引き起こす。
【0004】
LSDは、欠損酵素の活性化したものを患者に投与することにより治療することができ、当該方法は酵素補充療法(ERT)と呼ばれる。末端にマンノース−6−リン酸(M6P)を有する補充酵素が投与されると、細胞表面結合型カチオン非依存性M6P受容体(CI−MPR)を介したエンドサイト−シスによって標的細胞に取り込まれ、リソソームに作用する。
【0005】
一般に、リン酸化程度が低い補充酵素は、細胞表面のM6P受容体によって効果的に取り込まれず、したがって、機能するリソソームに作用することができない。そのため、マンノースのリン酸化程度が低いと、補充酵素の治療効果に深刻かつ有害な影響が及ぶ可能性がある。
【0006】
補充酵素のM6P含有量を高める方法が開発されている。例えば、米国特許第6,534,300号、第6,670,165号、および第6,861,242号には、末端マンノース残基の酵素的リン酸化が記述されている。別の例として、米国特許第7,001,994号には、M6Pを含むオリゴ糖を、糖タンパク質と結合させる方法が記述されている。当該方法によって製造された、リソソーム酵素である酸性α−グルコシダーゼ(GAA)と、ビスM6Pオリゴ糖との複合体は、ポンペ病のマウスモデルにおいて、組換えヒトGAAより、骨格筋および心筋のグリコーゲンを減少させるのにより効果的であることがわかった。ポンペ病は、GAAの代謝欠陥に起因する常染色体劣性型筋疾患であり、リソソームグリコーゲンの蓄積を特徴とする。同様に、Zhuらにより、合成ビスM6Pオリゴ糖(式A)を、GAAと結合させることが記述されている(Zhuら,Biochem.J.389:619−628(2005))。式Aは、N−結合型グリカン(式B)の天然の三分岐鎖Man9コア構造から、一分岐鎖を取り除き、別の一分岐鎖を短くし、末端のマンノース残基をリン酸化することで作られた。
【0007】
【化1】
【0008】
得られた複合体は、CI−MPRに、より高い親和性で結合し、L6筋芽細胞に関して、より効果的に取り込まれ、ほぼ正常な酵素活性が得られた。しかし、このような成功があるとはいえ、リソソーム酵素と結合した際に、正常またはほぼ正常な酵素活性を有しつつ、CI−MPRとの親和性の改善、および/またはより効果的な細胞吸収を引き起こす、新規のオリゴ糖を同定することは、依然として重要である。しかしながら、取込みを改善するだけでは、必ずしもよりよい治療結果はもたらされない。ある種の結合方法およびオリゴ糖を用いると、得られる複合体の酵素活性は低い。したがって、LSD患者の治療結果を改善することができる、オリゴ糖および複合体を同定することが望まれる。
【0009】
さらに、式AおよびBに示したようなある種のオリゴ糖は、合成が困難かつ高価であることがある。その上、立体選択的にβ−結合型糖鎖を作成することは、糖化学において、困難な問題であった。商用規模での使用に関しては、別のオリゴ糖および合成方法のほうが現実的である可能性がある。オリゴ糖−タンパク複合体を製造するのに使用する方法の最適化がさらに求めらている。特に、治療目的に関しては、複合体製造は非常に不均一であってはならない。これは一貫性のない生物学的機能を生じる場合があるためである。使用するオリゴ糖およびリンカー、結合方法、精製方法、および処方などの複合体の様々な要素が、治療効果に影響を与える得る。
【発明の概要】
【0010】
したがって、本発明のある種の実施形態は、(1)タンパク質、および(2)式I:
【0011】
【化2】
【0012】
[式中、
a=α1,2;α1,3;α1,4;またはα1,6であり;
b=α1,2;α1,3;またはα1,4であり;
c=α1,2;α1,3;α1,4;またはα1,6であり;および
d=α、β、またはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0013】
本発明の他の実施形態は、(1)タンパク質、および(2)式II:
【0014】
【化3】
【0015】
[式中、
e=α1,2;α1,3;α1,4;またはα1,6であり;および
f=α、β、またはαとβとの混合物であり;
ただしe=α1,6のとき、f=αまたはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0016】
本発明のさらに他の実施形態は、(1)タンパク質、および(2)式III:
【0017】
【化4】
【0018】
[式中、
g=α1,2;α1,3;またはα1,4であり;
h=α1,2;α1,3;α1,4;またはα1,6であり;および
i=α、β、またはαとβとの混合物である]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0019】
本発明の追加の実施形態は、(1)タンパク質、および(2)式IV:
【0020】
【化5】
【0021】
[式中、
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;ならびに
xが2または3のとき、各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0022】
さらなる実施形態は、(1)タンパク質、および(2)式V:
【0023】
【化6】
【0024】
[式中、
lは、α、β、およびαとβとの混合物から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0025】
追加の実施形態は、(1)タンパク質、および(2)式VI:
【0026】
【化7】
【0027】
[式中、
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、任意にオキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで置換された、且つ任意にN、O、またはSから選択される1または2以上のヘテロ原子で中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4から選択され;
mは、α、β、およびαとβとの混合物から選択され;
yが2、3、または4のとき、各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択される]
で表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0028】
追加の実施形態において、本発明は(1)タンパク質、および(2)式Aで表されるオリゴ糖を含む、オリゴ糖−タンパク複合体を提供する。
【0029】
ある実施形態において、複合体は、タンパク質1モルにつき、式Aのオリゴ糖を少なくとも2、3、4、または5モル含む。
【0030】
いくつかの実施形態において、本発明のオリゴ糖−タンパク複合体は、この複合体のオリゴ糖およびタンパク質成分間にリンカーを含む。
【0031】
本発明は、式I、II、III、IV、VもしくはVIで表されるオリゴ糖−タンパク複合体、および充填剤、膨張剤、崩壊剤、緩衝剤、安定剤、または賦形剤を含む医薬組成物を提供する。本発明はさらに、式I、II、III、IV、VまたはVIで表されるオリゴ糖−タンパク複合体、あるいはオリゴ糖−タンパク複合体などを含む医薬組成物を用いて、例えば下の表1に開示されるような、リソソーム蓄積症の治療方法を提供する。リソソーム蓄積症は、例えばファブリー病、ポンペ病、ニーマンピックA病、ニーマンピックB病、およびムコ多糖症I型から選択することができる。さらなる実施形態において、本発明は、それを必要とする被験体のリソソーム蓄積症を治療する医薬の製造に関して、(1)タンパク質、および(2)式I、II、III、IV、V、またはVIで表されるオリゴ糖を含む、オリゴ糖−タンパク複合体の使用を提供する。
【0032】
本発明の追加の実施形態は、この出願を通して検討される。本発明の他の目的、特徴、および利点は、以下の詳細な説明により明らかとなる。本発明の1態様に関して検討されたいかなる実施形態も、本発明の別の態様に適用し、逆もまた同様である。実施例の項目の実施形態は、本発明のすべての態様に適用することができる、本発明の実施形態であると理解される。
【0033】
しかしながら、本発明の範囲および趣旨内の様々な変化および修飾は、本願から当業者に明らかになるため、詳細な説明および具体例が、本発明の具体的な実施形態に示されるが、例示のためのみに与えられることが理解されるべきである。
【図面の簡単な説明】
【0034】
【図1】図1は、オリゴ糖82の合成工程を明示する、例示的な逆合成スキームを示す。
【図2】図2は、本明細書に記載されたオリゴ糖合成で使用することができる、単糖のビルディングブロック2aの例示的な合成を示す。
【図3】図3は、本明細書に記載されたオリゴ糖合成で使用することができる、単糖のビルディングブロック1、2、3、および4の例示的な合成を示す。
【図4】図4は、エチレンリンカーの製造に関する、合成スキームを示す。
【図5】図5は、ビルディングブロック2、3、および4を用いる、オリゴ糖82の三糖前駆体の構築に関する合成スキームを示す。
【図6】図6は、図4に記載された三糖前駆体からの、保護された七糖の構築に関する合成スキームを示す。
【図7】図7は、図5に記載された保護された七糖を脱保護して、オリゴ糖82を生成する、合成スキームを示す。
【図8A】図8Aは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8B】図8Bは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8C】図8Cは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8D】図8Dは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図8E】図8Eは、立体選択的中間体であって且つチオール反応性基の代替形態を形成するため、ジブチルスズを利用し、式Aのβ−結合型六糖を製造する合成スキームを示す。
【図9A】NeoGAAβ SAM6の複合体化能における、酸化レベルの効果を示す。図9Aは、様々なモル比における、GAAと複合体化した、SAM2、SAM3、SAM4、線状SAM4、αSAM6、およびβSAM6オリゴ糖の量を示す。
【図9B】NeoGAAβ SAM6の複合化能における、酸化レベルの効果を示す。図9Bは、異なる量の過ヨウ素酸塩を用いて酸化した、rhGAAと複合体化した六糖(グリカン)の量を示す。
【図10A】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Aは、単糖組成物の分析(定量した単糖量の減少に基づいて推測した酸化)により、測定した酸化を示す。
【図10B】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Bは、ポジティブモードにおける、AA−標識したSAM6オリゴ糖のLTQ MS検出を示す。
【図10C】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Cは、AA−標識した酸化オリゴ糖に対応するMS/MSスペクトルを示す。
【図10D】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す。図10Dは、AA−標識した酸化オリゴ糖に対応するMS/MSスペクトルを示す。
【図10E】様々な量の過ヨウ素酸塩を用いた、シアル酸、フコース、ガラクトース、およびマンノースの酸化を示す図。図10Eは、異なる量のGAOを用いて滴定したGAM複合体の単糖分析を示す。
【図11A】図11Aは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図11B】図11Bは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図11C】図11Cは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。
【図12】図12は、2および22.5mMの過ヨウ素酸塩で処理した、rhGAAのペプチドマッピングのLC/MS分析を示す。非酸化、単一酸化、および二酸化したトリプシンペプチドT13(メチオニン172および173を含む)の溶出位置を長方形で強調する。
【図13A】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Aは、各サンプルの注入に関する、結合、解離、M6P溶出、および再生段階を示すセンサーグラムである。
【図13B】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Bは、NeoGAAサンプルおよびrhGAAコントロールサンプルに関する、センサーグラムデータの例示的な4パラメータフィットである。
【図13C】sCIMPRに対するNeoGAAおよびrhGAAのBiacore結合分析を示す図である。図13Cは、各製造の異なる結合レベル全体の、2mMvs.7.5mM過ヨウ素酸塩を用いて製造されたNeoGAAサンプルのM6P受容体親和性である。
【図14】図14は、様々なNeoGAA複合体の特異的活性を示す。
【図15A】図15Aは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15B】図15Bは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15C】図15Cは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15D】図15Dは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15E】図15Eは、M6P受容体カラムからのNeoGAA複合体の溶出を示す。
【図15F】図15Fは、SAM6複合体の週1回投与を4回行った後のGAAKOマウスにおける、組織グリコーゲンレベルを示す。
【図16】図16は、様々なNeoGAA複合体の吸収を明示する、L6筋芽細胞の取込アッセイの結果を示す。
【図17】図17は、SAM2処理後のGAAノックアウトマウスにおける、心臓、大腿四頭筋、および三頭筋のグリコーゲンクリアランスを示す。
【図18】図18は、SAM4処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図19】図19は、SAM6処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図20】図20は、SAM6およびGAM6処理後のGAAノックアウトマウスにおける、心臓および大腿四頭筋のグリコーゲンクリアランスを示す。
【図20B】図20Bは、rhGAA、GAMおよびSAM複合体の投与後5、15、30、60、120、240、および480分で回収された、マウス血清中のrhGAA濃度を示す。
【発明を実施するための形態】
【0035】
発明の説明
本発明がよく理解されるよう、まず、いくつかの語が定義される。追加の定義は、本願を通して行われる。
【0036】
本明細書および添付の請求項で使用される、単数形「a」、「an」および「the」は、他に文脈で明らかに指示されない限り、複数の対象を含む。したがって、例えば、「1つの化合物(a compound)」を含む方法への引用は、2つまたはそれ以上の化合物の混合物を含む。「または」という語は、他に文脈で明らかに指示されない限り、一般に「および/または」を含む意味で使用される。
【0037】
I.オリゴ糖−タンパク複合体
1つの実施形態において、本発明は、オリゴ糖−タンパク複合体を提供し、これはさらにリンカーを含むことができる。例示的なオリゴ糖、タンパク質、リンカー、複合体化方法、および複合体が開示されている。
【0038】
A.オリゴ糖
オリゴ糖は、上に示したような、式Bまたは式VIで表される二分岐鎖のオリゴ糖誘導体、あるいは式IVおよびVに示されるような直鎖のオリゴ糖誘導体から選択することができる。一般に、二分岐鎖のオリゴ糖は、末端に2つのM6P残基を有し、いくつかの実施形態においては、末端から2番目から1または2以上のM6P残基をさらに含むことができる。直鎖のオリゴ糖は、少なくとも1つのM6P残基を有し、末端のM6P残基を含むことがある。一般に、末端のM6P残基は、α1,2結合で結合し得る(末端のM6Pのα1,2結合は、α1,3またはα1,6結合よりも、CI−MPRおよびカチオン依存性MPR(CD−MPR)に、より結合するということを観察した、Distlerら,J.Biol.Chem.266:21687−21692(1991)を参照のこと)。いくつかの実施形態において、末端のM6P残基は、それぞれ還元末端で、α1,2結合によって隣り合う残基に結合している。いくつかの実施形態において、2つの末端M6P残基の間は、例えばX線結晶学、NMR、および/または分子モデリングにより測定されたように、5、10、15、20、25、30、35、または40Åより離れている。例えば、分子モデリングは、Balajiら,Glycobiology 4:497−515(1994)に記載されたように実施することができる。いくつかの実施形態において、オリゴ糖は、末端のM6P残基が比較的立体障害のないように選択される。いくつかの実施形態において、末端のM6P残基に比較的障害がないオリゴ糖は、末端のM6P残基に障害のあるオリゴ糖より、高い親和性でCI−MPRと結合する。
【0039】
一般に、オリゴ糖はCI−MPRに結合する。例えば、オリゴ糖は、例えば、500、100、50、10、5、1、または0.1nM未満、あるいは例えば、100、50、10、5、2、または1μM未満の解離定数で、CI−MPRに結合することができる。CI−MPRのN−末端領域1〜3の結晶構造は、リガンド結合型および非結合型の両方が知られている(Olsonら,J.Biol.Chem.279:34000−34009(2004)、Olsonら,EMBO J.23:2019−2028(2004))。さらに、構造的に関連のあるCD−MPRも、リガンド結合型および非結合型が知られている(Olsonら,J.Biol.Chem.274:29889−29886(1999)、Olsonら,J.Biol.Chem.277:10156−10161(2002))。したがって、当業者は、その受容体構造情報を使用して、適当なオリゴ糖を選択することができる。
【0040】
オリゴ糖は、例えば、下記で示されるオリゴ糖1〜127を含む、上記で示された式I、II、III、IV、V、またはVIで表されるオリゴ糖のいずれか1つから選択することができる。式I〜IIIで表されるオリゴ糖は、分岐鎖の除去、単糖残基の除去および/または置換、および/または隣接する単糖残基間の結合(α1,2、α1,3、α1,4、もしくはα1,6など)の修飾により、式Bから従来の方法で誘導される。ある種の実施形態において、オリゴ糖は、式I〜VIのいずれか一つに関して、α1,2、α1,3、またはα1,6結合によって結合した、1本または両方の分岐鎖に、例えば1、2、または3つのさらなるマンノース残基を有することがある。
【0041】
オリゴ糖は、例えば1つ、2つまたは3つのM6P残基を有してよい。オリゴ糖は、合計で1、2、3、4、5、6、7、8、9、または10個の単糖残基を有してよい。他の実施形態において、オリゴ糖は、1、2、3、4、5、6、7、8、9、または10個のマンノース残基を有してよく、このうちいずれもリン酸化または非リン酸化されてよい。
【0042】
いくつかの実施形態において、オリゴ糖はオリゴ糖1〜96から選択され、これは式Iの種類である。
【0043】
【化8】
【0044】
【表1】
【0045】
【表2】
【0046】
【表3】
【0047】
いくつかの実施形態において、dはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖1および2、3および4、または95および96の混合物である)。
【0048】
いくつかの実施形態において、オリゴ糖はオリゴ糖97〜103から選択され、これは式IIの種類である。
【0049】
【化9】
【0050】
いくつかの実施形態において、fはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖97および98、99および100、または101および102の混合物である)。
【0051】
いくつかの実施形態において、オリゴ糖はオリゴ糖104〜127から選択され、これは式IIIの種類である。
【0052】
【化10】
【0053】
いくつかの実施形態において、iはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖104および105、106および107、または126および127の混合物である)。
【0054】
いくつかの実施形態において、オリゴ糖は、オリゴ糖128〜133から選択され、これは式IVの種類である。
【0055】
【化11】
【0056】
いくつかの実施形態において、kはαとβとの混合物である(すなわち、オリゴ糖は、例えば、オリゴ糖128および129、130および131、または132および133の混合物である)。
【0057】
いくつかの実施形態において、オリゴ糖はオリゴ糖134および135から選択され、これは式Vの種類である。
【0058】
【化12】
【0059】
いくつかの実施形態において、オリゴ糖はオリゴ糖134および135の混合物である。
【0060】
いくつかの実施形態において、オリゴ糖はオリゴ糖136であり、これは式VIの種類である。
【0061】
【化13】
【0062】
いくつかの実施形態において、オリゴ糖は、天然の供給源から単離することができる。天然の供給源から単離されたオリゴ糖は、関連するオリゴ糖の同種の混合物または異種の混合物であってよい。
【0063】
ある種の実施形態において、オリゴ糖は化学的および/または酵素的合成により製造される。いくつかの実施形態において、オリゴ糖は天然の供給源から単離されたオリゴ糖を、化学的または酵素的に修飾することで製造することができる(「半合成」)。
【0064】
オリゴ糖は、例えば図1〜7、Obsornら,Oligosaccharides:Their Synthesis and Biological Roles,Oxford University Press,2000、Wangら(eds),Synthesis of Carbohydrates through Biotechnology,American Chemical Society,2004、Seeberger,Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries,Wiley−Interscience,2001、Driguezら,Glycoscience:Synthesis of Oligosaccharides and Glycoconjugates,Springer,1999、Duffelsら,Chem.Eur.J.6:1416−1430(2000)、Hojoら,Current Prot.Peptide Sci.1:23−48(2000)、Seebergerら,Nature 446:1046−1051(2007)、Seebergerら,Nature Rev.Drug Discov.4:751−763(2005)、Srivastanaら,Carbohydrate Res.161:195−210(1987)、およびHagiharaら,Chem.Rec.6:290−302(2006)と、米国特許第5,324,663号、第6,156,547号、第6,573,337号、第6,723,843号、第7,019,131号、および第7,160,517号で説明されたように、化学的および/または酵素的に合成することができる。
【0065】
いくつかの実施形態において、オリゴ糖は連続して単糖を追加することで合成することができる。ある種の実施形態において、選択的保護および脱保護により、既存の糖の特定の位置(例えば2−O、3−O、4−O、または6−O)に、単糖を追加することができる。例えば、オリゴ糖82を、図1の逆合成解析および図2〜7に記載の合成スキームに記述されたように、合成することができる。いくつかの実施形態において、ビルディングブロック2aを、図3、5、および6の合成スキームのビルディングブロック2に置換することができる。ビルディングブロック2aが使われていれば、七糖段階でビルディングブロック2のベンジリデン基の除去を避けることができる。
【0066】
マンノース残基を、例えば米国特許第6,905,856号で説明されたように、酵素的にリン酸化することができる。ある種の実施形態において、1、2、または3つのM6P残基を、Berkowitzら,Org.Lett.6:4921−4924(2004)で説明されたように、例えば、マロニルエーテル、マロン酸、およびリン酸など、加水分解酵素耐性M6P模倣体で置き換えることができる。
【0067】
ある種の実施形態において、リンカーは、αまたはβ結合を介して糖に結合することができる。いくつかの実施形態において、β結合は、Crichら,Tetrahedron,54:8321−8348(1998)、Kimら,J.Am.Chem.Soc.,130:8537−8547(2008)、Srivasta et al,Tetrahedron Letters,35:3269−3272(1979)、Hodosiら,J.Am.Chem.Soc.,119:2335−2336(1997)、Nicolaouら,J.Am.Chem.Soc.,119:9057−9058(1997)に記載された方法により形成することができる。1つの実施形態において、不活性の脱離基を含むリンカーと反応させることができる中間体を形成するため、ジブチルスズオキシドを用いて、β結合を形成することができる。
【0068】
1つの実施形態は、式VII:
【0069】
【化14】
【0070】
[式中、
R1は、水素、ヒドロキシル、任意に置換された低級アルキル、ホスフェート、サルフェート、−OR7、保護基、および糖から選択され;
R2、R3、R4およびR5は、それぞれ独立して、水素、サルフェート、ヒドロキシル、−OR8、保護基、および糖から選択され;
R6は、水素、ヒドロキシル、カルボキシル、アルコキシカルボニル、アミノ、アミド、アルキアミノ(alkyamino)、アミノアルキル、アミノキシ、ヒドラジド、ヒドラジン、任意に置換されたアルケニル、および任意に置換されたC2−C6アルキルから選択され;
R7およびR8は、それぞれ独立して、アセチルおよび任意に置換された低級アルキルから選択され;ならびに
nは、1〜10の整数である]
で表される化合物を製造する方法であって、以下のステップ:
a)式VIII:
【0071】
【化15】
【0072】
[式中、
R1からR5は、上に定義された通りであり;ならびに、
R9およびR10は、R9およびR10のうちの1つが、ヒドロキシルのとき、もう一方が水素であるように、水素およびヒドロキシルから選択される]
で表される化合物を、式R11R12(Sn=O)で表される化合物で処理して、式IX:
【0073】
【化16】
【0074】
[式中、
R1からR5は、上で定義された通りであり;ならびに、
R11およびR12はそれぞれ独立に、非置換アルキルから選択され、またはR11およびR12は一緒になって、非置換アルキレンから選択される]
で表される化合物を形成し;
b)式IXの化合物を、任意に金属ハロゲン化物の存在下、式R6−(CH2)n−L:
[式中、
R6およびnは、上で定義された通りであり;ならびに、
Lはハロゲンである]
で表される化合物で処理して式VIIの化合物を形成すること
を含む、前記方法を提供する。
【0075】
「任意の」または「任意に」は、引き続き記述される事象または状況が起こる場合がある、または起こらない場合があること、明細書が事象または状況が起こる例、および起こらない例を含むことを意味する。例えば、「任意に置換されたアルキル」は、下記で定義されるような「アルキル」および「置換アルキル」の両方を含む。1または2以上の置換基を含むどんな基に関しても、かかる基が立体的に現実的でない、合成的に実行不可能である、および/または本質的に不安定な、いかなる置換または置換形式を取り込むことを意図しないことは、当業者により理解される。
【0076】
「アルキル」は、指定された数の炭素原子を有する直鎖および分岐鎖を含み、通常1〜20個の炭素原子を有し、例えば1〜8個の炭素原子を有し、例えば1〜6個の炭素原子を有する。例えば、C1−C6アルキルは、1〜6個の炭素原子の直鎖および分岐鎖の両方のアルキルを含む。アルキル基の例としては、メチル、エチル、プロピル、イソプロピル、n−ブチル、sec−ブチル、tert−ブチル、ペンチル、2−ペンチル、イソペンチル、ネオペンチル、ヘキシル、2−ヘキシル、3−ヘキシル、3−メチルペンチルなどが挙げられる。アルキレンは、アルキルの別の集合であり、アルキルと同じ残基を表すが、2箇所の結合部を有する。アルキレン基は、通常2〜20個の炭素原子、例えば2〜8個の炭素原子、例えば2〜6個の炭素原子を有する。例えば、C0アルキレンは、1つの共有結合を示し、C1アルキレンはメチレン基である。具体的な数の炭素を有するアルキル残基が指定されるとき、その数の炭素を有するすべての幾何異性体が含まれることを意図する。したがって、例えば、「ブチル」はn−ブチル、sec−ブチル、イソブチル、およびt−ブチルを含むことを意味し、「プロピル」は、n−プロピルおよびイソプロピルを含む。「低級アルキル」は1〜4個の炭素を有するアルキル基を示す。
【0077】
「アルケニル」は、親となるアルキルの隣り合う炭素原子から、1分子の水素分子が取り除かれることにより生成される、少なくとも1つの炭素−炭素二重結合を有する、不飽和の分岐または直鎖アルキルを示す。この基は、二重結合がシスまたはトランスのどちらの配置でもよい。例示的なアルキル基には、エテニル;プロプ−1−エン−1−イル、プロプ−1−エン−2−イル、プロプ−2−エン−1−イル(アリル)、プロプ−2−エン−2−イルなどのプロペニル;ブト−1−エン−1−イル、ブト−1−エン−2−イル、2−メチル−プロプ−1−エン−1−イル、ブト−2−エン−1−イル、ブト−2−エン−1−イル、ブト−2−エン−2−イル、ブタ−1,3−ジエン−1−イル、ブタ−1,3−ジエン−2−イルなどのブテニルなどを含むがこれに限定されない。いくつかの実施形態において、アルケニル基は2〜20個の炭素原子を有し、別の実施形態において、2〜6個の炭素原子を有する。
【0078】
本明細書で使用される「置換」という語は、指定された原子または基の1または2以上の水素のいずれかが、指示された基から選ばれたもので置き換わり、ただし指定された原子の通常の原子価を超えないことを意味する。置換基がオキソ(すなわち、=O)であるとき、原子の2つの水素が置き換わる。置換基および/または変数の組合せは、かかる組合せが安定した化合物または有用な合成中間物をもたらす場合に限り、許容される。安定した化合物または安定した構造は、反応混合物からの単離、および少なくとも実際の有用性を有する医薬としての、その後の処方でも存在するほど、十分強固である化合物を示唆するものである。他に断りが無い限り、置換基はそのコア構造に名付けられる。例えば、(シクロアルキル)アルキルが置換基の候補として列挙されるとき、この置換基のコア構造への結合位置は、アルキル部分にあることが、理解されるべきである。
【0079】
他に明らかに定義されない限り、「置換アルキル」という語は、1または2以上の水素原子が、以下から独立に選択される置換基によって置換されるアルキルのことである:
−Ra、−ORb、−O(C1−C2アルキル)O−(例えば、メチレンジオキシ−)、−SRb、−NRbRc、ハロ、シアノ、オキソ、ニトロ、サルフェート、ホスフェート、−CORb、−CO2Rb、−CONRbRc、−OCORb、−OCO2Ra、−OCONRbRc、−NRcCORb、−NRcCO2Ra、−NRcCONRbRc、−SORa、−SO2Ra、−SO2NRbRc、−NRcSO2Ra、エチレングリコール、およびポリエチレングリコール(PEG)。
ここでRaは、任意に置換されたC1−C6アルキル、任意に置換されたアリール、および任意に置換されたヘテロアリールから選択され;
Rbは、水素、−NH2、−NHRc、任意に置換されたC1−C6アルキル、任意に置換されたアリール、および任意に置換されたヘテロアリールから選択され;
Rcは、水素および任意に置換されたC1−C4アルキルから選択され;または
RbおよびRc、並びにそれらが結合する窒素原子は、任意に置換されたヘテロシクロアルキル基を形成し;ならびに
各任意に置換された基は、非置換でよく、あるいは、C1−C4アルキル、アリール、ヘテロアリール、アリール−C1−C4アルキル−、ヘテロアリール−C1−C4アルキル−、C1−C4ハロアルキル−、−OC1−C4アルキル、−OC1−C4アルキルフェニル、−C1−C4アルキル−OH、−OC1−C4ハロアルキル、ハロ、−OH、−NH2、−C1−C4アルキル−NH2、−N(C1−C4アルキル)(C1−C4アルキル)、−NH(C1−C4アルキル)、−N(C1−C4アルキル)(C1−C4アルキルフェニル)、−NH(C1−C4アルキルフェニル)、シアノ、ニトロ、オキソ(ヘテロアリールの置換基として)、−CO2H、−C(O)OC1−C4アルキル、−CON(C1−C4アルキル)(C1−C4アルキル)、−CONH(C1−C4アルキル)、−CONH2、−NHC(O)(C1−C4アルキル)、−NHC(O)(フェニル)、−N(C1−C4アルキル)C(O)(C1−C4アルキル)、−N(C1−C4アルキル)C(O)(フェニル)、−C(O)C1−C4アルキル、−C(O)C1−C4フェニル、−C(O)C1−C4ハロアルキル、−OC(O)C1−C4アルキル、−SO2(C1−C4アルキル)、−SO2(フェニル)、−SO2(C1−C4ハロアルキル)、−SO2NH2、−SO2NH(C1−C4アルキル)、−SO2NH(フェニル)、−NHSO2(C1−C4アルキル)、−NHSO2(フェニル)、および−NHSO2(C1−C4ハロアルキル)から独立して選択される、1または2以上の置換基で独立して置換されてよい。
【0080】
いくつかの実施形態において、R1は、水素、任意に置換された低級アルキル、ホスフェート、サルフェート、−OR7、保護基、および糖から選択される。追加の実施形態において、R2、R3、R4、およびR5は、それぞれ独立に、水素、サルフェート、ヒドロキシル、−OR8、保護基、および糖から選択される。ある種の実施形態において、R7およびR8はそれぞれ独立に、アセチルおよび任意に置換された低級アルキルから選択される。いくつかの実施形態において、R1、R2、R3、R4、およびR5のいずれかまたはすべては、選択的に除去することができる保護基から選択することができ、ベンジル、シリル、およびトリチルエーテル、並びに酢酸以外のエステルが含まれる。いくつかの実施形態において、R1からR5のうちの2つは、一緒になって、保護基を形成する。1つの実施形態において、R1は、−O−ベンジルおよび−OCPh3から選択される。別の実施形態において、R2からR5のうちの少なくとも1つは、−O−ベンジルである。いくつかの実施形態において、R1、R2、およびR4は、それぞれ−O−ベンジルである。さらなる実施形態において、R1は−OCPh3であり、R4は−O−ベンジルである。1つの実施形態において、R11およびR12は、一緒になって、ヘキサメチレンを形成する。別の実施形態において、R11およびR12は、それぞれイソプロピルである。さらに別の実施形態において、R11およびR12は、それぞれヘキシルである。
【0081】
追加の実施形態において、式VIIIの化合物は、任意に保護されたマンノース、ラムノース、イドース、およびアルトロースから選択される。1つの実施形態において、式VIIIの化合物は、任意に保護されたマンノースである。ある状況において、マンノースを、C−3、C−4、またはC−6位の1又は2以上を保護してもよい。いくつかの例において、単一の保護基は、2つの位置に結合することができる。例えば、ベンジリデン基は、C−4およびC−6位の両方を保護するために使用することができる。
【0082】
一般に、式IXの化合物と、強い求電子性または交差反応性でない糖のいかなる保護基も使用することができる。適当な保護基には、任意に置換されたベンジルエーテル、トリチルエーテル、アリルエーテル、またはシリルエーテルなどのエーテル;任意に置換された酢酸、安息香酸、クロロ酢酸、ピバル酸、またはレブリン酸などのエステル;ベンジリデン、イソプロピリデンおよびブタンジアセタールを含むアセタールなどが挙げられる。さらに、保護基を、カルバミン酸およびウレタンから選択することができる。いくつかの実施形態において、例えばベンジル、シリル、およびトリチルエーテル、ならびに酢酸以外のエステルなどの保護基を、選択的に除去することができる。保護基の位置および正体は、所望の最終生成物によって変えることができる。当業者に公知の追加の保護基を、本明細書で記述された実施形態に従って使用することができる。
【0083】
1つの実施形態において、式VIIIの化合物は、式R11R12(Sn=O)で表される化合物で処理され、式R11およびR12は、それぞれ独立に、非置換アルキルから選択され、またはR11およびR12は一緒になって、非置換アルキレンから選択される。いくつかの実施形態において、R11およびR12はブチルである。1つの実施形態において、式R11R12(Sn=O)で表される化合物を、トルエン、ベンゼン、ジメチルホルムアミド、イソプロパノール、メタノール、またはキシレンなどの溶媒中で、式VIIIの化合物と反応させる。いくつかの実施形態において、昇温、任意に還流下で、反応を実施して、式IXの化合物を形成する。いくつかの実施形態において、反応混合物を少なくとも40、50、60、70、または80℃に加熱する。追加の実施形態において、式R11R12(Sn=O)で表される化合物を、少なくとも1、2、5、10、15、または20時間、式VIIIの化合物と反応させる。
【0084】
1つの実施形態において、式IXの化合物を式R6−(CH2)−Lで表される化合物で、任意に金属ハロゲン化物の存在下で処理する。いくつかの実施形態において、R6は水素、ヒドロキシル、カルボキシル、アルコキシカルボニル、アミノ、アミド、アルキアミノ(alkyamino)、アミノアルキル、アミノキシ、ヒドラジド、ヒドラジン、任意に置換されたアルケニル、および任意に置換されたC2−C6アルキルから選択される。追加の実施形態において、nは1〜10の整数である。ある種の実施形態において、nは2、3、4、5、または6であり、R6はC1−C4アルコキシカルボニルである。1つの実施形態において、nは3であり、R6はメトキシカルボニルである。いくつかの実施形態において、Lは活性化されない脱離基である。活性化された脱離基の例としては、トリフレート、スルホネート、トシレート、および他の類似の基が挙げられる。いくつかの実施形態において、低い反応性の脱離基を、アリル基などの隣接基によって活性化することができる。ある種の実施形態において、R6は脱離基を活性化する置換基を含まない。いくつかの実施形態において、Lはブロミド、クロリド、またはヨージドである。1つの実施形態において、Lはブロミドである。追加の実施形態において、式R6−(CH2)n−Lの化合物は、4−ブロモ酪酸メチルである。
【0085】
1つの実施形態において、式VIIIの化合物は、任意に保護されたマンノースであり、式R6−(CH2)n−Lの化合物は、4−ブロモ酪酸メチルである。別の実施形態において、式VIIIの化合物は、3,4,6−トリ−O−ベンジル−D−マンノースおよび3−O−アリル−6−O−トリチル−D−マンノースから選択される。
【0086】
いくつかの実施形態において、式IXの化合物を式R6−(CH2)n−Lで表される化合物で、金属ハロゲン化物の存在下で処理する。金属ハロゲン化物のある種の実施形態は、金属フッ化物を含む。いくつかの実施形態において、金属フッ化物は、フッ化セシウム、フッ化ナトリウム、フッ化カルシウム、フッ化マグネシウム、フッ化リチウム、およびフッ化カリウムから選択される。1つの実施形態において、金属ハロゲン化物はフッ化セシウムである。いくつかの実施形態において、式IXの化合物を式R6−(CH2)n−Lの化合物で処理することは、テトラアルキルアンモニウムハライドの添加をさらに含む。いくつかの例において、テトラアルキルアンモニウムハライドは、テトラブチルアンモニウムヨージドである。追加の実施形態において、金属ハロゲン化物を反応に使用することができる。金属ハロゲン化物の例としては、ヨウ化ナトリウムなどのアルカリ金属ヨウ化物が挙げられる。
【0087】
1つの実施形態において、極性非プロトン性溶媒中で、式IXの化合物を、式R6−(CH2)n−Lの化合物と混合することができる。かかる溶媒には、当業者に公知の中でも、ジメチルホルムアミド、ジメチルアセトアミド、ジメチルスルホキシド、ニトロメタン、ヘキサメチルリン酸アミド、N−メチルピロリドン、アセトン、アセトニトリル、酢酸エチル、およびメチルエチルケトンが挙げられる。
【0088】
ある種の実施形態において、式IXの化合物は、式R6−(CH2)n−Lの化合物と、室温で混合することができる。他の実施形態において、反応物が組み合わされ、少なくとも50、60、70、または80℃に加熱されて、式VIIの化合物を形成する。さらなる実施形態において、混合物は少なくとも1、2、5、10、15、または20時間加熱される。
【0089】
いくつかの実施形態において、本明細書に記載された方法は、少なくとも50、60、70、80、90、95、または99%で立体特異的生成物をもたらす。追加の実施形態において、立体特異的生成物の収率は、最大可能収率の少なくとも50、60、70、75、80、85、90、95、または99%である。ある種の実施形態において、β−結合生成物とα−結合生成物との比率は、少なくとも10:1、20:1、30:1、40:1、50:1、または100:1である。
【0090】
1つの実施形態において、式VIIの化合物を大規模に製造することができる。いくつかの実施形態において、「大規模」は、出発物質、中間体または試薬を少なくとも50、100、500、または1000グラム使用することを示す。追加の実施形態において、「大規模」は、出発物質、中間体または試薬を、少なくとも10、25、50、100、250、または500kg使用することを含む。
【0091】
ジブチルスズオキシド試薬を使用して、式Aの化合物を製造する例示的な合成スキームを図8に示す。
【0092】
B.タンパク質
本明細書で記述されたオリゴ糖−タンパク複合体は、単離されたタンパク質、および組み換えられた、または合成的に生産されたタンパク質を含む、全ての純粋なタンパク質、部分的に精製されたタンパク質、あるいはその断片を含み得る。「純粋な」、「精製された」、および「単離された」という語は、天然の環境中の実質的に遊離の分子を示す。例として、純粋なタンパク質は、由来する細胞または組織の供給源の、実質的に遊離の細胞物質および/または他のタンパク質である。この語は例えば、少なくとも70〜80%、80〜90%、90〜95%、あるいは少なくとも95%、96%、97%、98%、99%、または100%(w/w)純粋である調製物に関する。
【0093】
他の実施形態において、タンパク質は、pH1〜7の範囲、例えば1〜3、2〜5、3〜6、4〜5、5〜6、または4〜6などで、活性アッセイによって測定された、最適な活性を有する酵素であってよい。例えば、酵素はpH4〜6の範囲で、最適pHを有し得る。
【0094】
いくつかの実施形態において、タンパク質は、1〜8の範囲、例えば1〜3、2〜5、3〜8、4〜5、5〜6、4〜6、5〜8、6〜8、または7〜8の範囲の等電点(pI)を有する酵素であり得る。タンパク質のpIを、例えばゲル等電点電気泳動を用いて測定することができる。
【0095】
ある種の実施形態において、タンパク質自体は少なくとも1つのオリゴ糖を有する(すなわち、糖タンパク質である)。個々の実施形態において、タンパク質は、治療用糖タンパク質である。例えば、治療用糖タンパク質は、例えば表1に列挙されたリソソーム加水分解酵素のうちの1つなど、ERT酵素を含むリソソーム酵素であってよい。ある種の実施形態において、リソソーム酵素は、例えばα−グルコシダーゼ(GAA)、α−ガラクトシダーゼA、酸性スフィンゴミエリナーゼ、およびα−L−イズロニダーゼから選択される。個々の実施形態において、リソソーム酵素はGAAである。
【0096】
【表4】
【0097】
いくつかの実施形態において、タンパク質は少なくとも1、2、3、4、5、またはそれ以上のN−結合型またはO−結合型オリゴ糖を有する糖タンパク質であってよい。他の実施形態において、タンパク質は、N−結合型またはO−結合型糖鎖形成のための、1、2、3、4、5、またはそれ以上のコンセンサス部位を有することができ、そしてその少なくとも1つがグリコシル化される。
【0098】
ある種の実施形態において、タンパク質は受容体のリガンドであってよい。例えばいくつかの実施形態において、タンパク質は、例えばマンノースまたはマンノース−6−リン酸などの糖を認識する受容体と結合する糖タンパク質であってよい。個々の実施形態において、糖タンパク質は、例えばアシアロ糖タンパク質受容体、CI−MPR、CD−MPR、またはマンノース受容体と結合することができる。
【0099】
好適なタンパク質配列は当業者に周知である。当業者は、異なる種の配列などを含む関連配列を比較することにより、保存領域および重要な機能的モチーフを容易に同定することができる。保存されたアミノ酸は、活性のためにより重要である可能性が高く、反対に、保存されていないアミノ酸は、変異(variation)を許容する可能性の高いポリペプチド領域を示す。この指針に従って、当業者はただの通常業務によって機能的変異体(functional variant)を容易に同定することができる。さらに、結晶構造が公知である場合に、当業者は結晶構造を分析して、構造および/または機能に重要であり、したがって変異(mutation)に対する許容性が低いと考えられるアミノ酸を同定することができる。当業者はまた、変異(variation)を許容し得るアミノ酸を同定することもできる。さらに当業者は,公知の構造−機能相関の観点で、可能性のある変異(mutation)を評価することができる。
【0100】
例えば、α−ガラクトシダーゼの配列および構造は周知である。例えば、Garmanら,J.Mol.Biol.,337:319−335(2004)、Garmanら,Mol.Genet.Metabol.,77:3−11(2002)、Matsuzawaら,Hum.Genet.117:317−328(2005)を参照のこと。また、GenBank Accession No.X05790も参照のこと。別の例において、GAAの配列は周知である(例えば、Martiniukら,Proc.Natl.Acad.Sci.USA 83:9641−9644(1986)、Hoefslootら,Biochem.J.272:493−497(1990)、Morelandら,J.Biol.Chem.280:6780−6791(2005)を参照のこと)。GenBank Accession No.NM_000152も参照のこと。さらに、E.coliの対応するα−グリコシダーゼの結晶構造は決定されており、構造的見識を他のα−グリコシダーゼに提供することができる。Loveringら,J.Biol.Chem.280:2105−2115(2005)を参照のこと。3つめの例において、酸性スフィンゴミエリナーゼの配列は、酸性スフィンゴミエリナーゼの配列および構造の重要な特徴であるとして周知である(例えば、Lansmannら,Eur.J.Biochem.270:1076−1088(2003)を参照のこと)。例えば、Setoら,Protein Sci.13:3172−3186(2004)、Qiuら,J.Biol.Chem.278:32744−32752(2003)、Takahashiら,Tohoku J.Exp.Med.206:333−340(2005)を参照のこと。GenBank Accession No.AI587087も参照のこと。さらに別の例において、α−L−イズロニダーゼの配列は、α−L−イズロニダーゼの重要な特徴のため、周知である(例えば、Scottら,Proc.Natl.Acad.Sci USA 88:9695−9699(1991)、Scottら,Genomics 13:1311−1313(1992)を参照のこと)。例えば、Scottら,Hum.Mutat.6:288−302(1995)、Rempelら,Mol.Genet.Metab.85:28−37(2005)、Durandら,Glycobiology 7:277−284(1997)、Beesleyら,Hum.Genet.109:503−511(2001)、Brooksら,Glycobiology 11:741−750(2001)、Niemanら,Biochemistry 42:8054−8065(2003)を参照のこと。さらなる例において、イズロン酸−2−スルファターゼの配列は、疾患を引き起こす変異体(mutation)であるとして周知である。例えば、Flomenら,Hum.Mol.Genet.2:5−10(1993)、Robertsら,J.Med.Genet.26:309−313(1989)、Wilsonら,Proc.Natl.Acad.Sci.USA 87:8531−8535(1990)、Wilsonら,Genomics 17:773−775(1993)、Sukegwa−Hayasakaら,J.Inherit.Metab.Dis 29:755−761(2006)およびその中の参考文献を参考のこと。イズロン酸−2−スルファターゼの構造はモデル化されている。例えば、Kimら,Hum.Mutat.21:193−201(2003)を参照のこと。別の例において、N−アセチルガラクトサミン−4−スルファターゼ(アリールスルファターゼB)の配列および構造は、疾患を引き起こす変異体(mutation)であるとして公知である。例えば、Litjensら,Hum.Mut.1:397−402(1992)、Petersら,J.Biol.Chem.265:3374−3381(1990)、Schuchmanら,Genomics 6:149−158(1990)、Bondら,Structure 15:277−289(1997)を参照のこと。
【0101】
C.リンカー
ある種の実施形態において、本発明のオリゴ糖−タンパク複合体は、複合体のオリゴ糖成分およびタンパク質成分との間にリンカーを含む。他の実施形態において、複合体は、リンカーを含まない。リンカーを含む実施形態において、CI−MPRへのオリゴ糖の結合を妨げず、および/またはタンパク質の活性(例えば、酵素的活性を含む)を阻まない限り、当業者に公知の任意の適当なリンカーを使用することができる。例えば、リンカーは、米国特許第4,671,958号、第4,867,973号、第5,691,154号、第5,846,728号、第6,472,506号、第6,541,669号、第7,141,676号、第7,176,185号、第7,232,805号、または米国特許出願公開第2006/0228348号で開示されたリンカーの1つであってよい。いくつかの実施形態において、リンカーは国際公開2008/089403号で開示されたリンカーから選択される。
【0102】
いくつかの実施形態において、リンカーは次式を有することができる。
【0103】
【化17】
【0104】
[式中、
Zは、任意に置換されたアルキル、アルケニル、アルキニル、アリール、エチレングリコール、ポリエチレングリコール(PEG)ヘテロアリール、およびヘテロシクリルから選択され、Pは、水素またはアミノ保護基から選択される]。
【0105】
本明細書で使用される、アミノオキシ化合物(例えば、アルキル、アルケニル、アルキニル、アリール、ヘテロアリール、ヘテロシクリル、アシルオキシ、アルコキシ、アリールオキシ、およびヘテロシクリルオキシなど)のいかなる化学基も、他に述べられない限り、置換または非置換でよく、1または2以上のヘテロ原子、または化学基により中断される場合がある。中断するヘテロ原子には、窒素、酸素、および硫黄が含まれる。置換基または中断する化学基を、例えば、アシル、アシルアミノ、アシルオキシ、アルケニル、アルコキシ、アルキル、アルキニル、アミド、アミノ、アリール、アリールオキシ、アジド、カルバモイル、カルボアルコキシ、カルボキシ、シアノ、シクロアルキル、ホルミル、グアニジノ、ハロ、ヘテロアリール、ヘテロシクリル、ヒドロキシ、イミノアミノ、ニトロ、オキソ、ホスホンアミノ、スルフィニル、スルホンアミノ、スルホナート、スルホニル、チオ、チオアシルアミノ、チオウレイド、およびウレイドから選択することができる。置換基それ自体は、置換または非置換でよく、例えば、窒素、硫黄、および酸素など、1または2以上のヘテロ原子で中断されているかまたは終結している場合がある。
【0106】
1つの実施形態において、リンカーは:
【0107】
【化18】
【0108】
との反応により形成され得る。
【0109】
追加の実施形態において、リンカーは次式を有することができる
【0110】
【化19】
【0111】
[式中、ZおよびPは、上で定義される通りである]。
【0112】
別の実施形態において、リンカーは、ジスルフィド結合を含むことができる。ジスルフィドリンカーを、例えばシステインを介して、タンパク質骨格にオリゴ糖を結合させるのに使用することができる。1つの実施形態において、リンカーは
【0113】
【化20】
【0114】
を含む、またはこれとの反応から形成することができる。
【0115】
一般に、複合体のオリゴ糖成分およびタンパク質成分間の立体障害を避けるよう、CI−MPRへのオリゴ糖の結合、および/またはタンパク質の活性(例えば、酵素的活性を含む)を妨げないようにリンカーを適当な長さにすることができ。例えば、リンカーは1〜100、1〜60、5〜60、5〜40、2〜50、2〜20、5〜10、または5〜20個の直鎖の原子を含むことができ、このリンカーは、エステル、アミド、ヒドラゾン、オキシム、セミカルバゾン、エーテル、チオエーテル、ホスホロチオ酸、ホスホン酸、チオエステル、および/またはジスルフィド結合により、タンパク質およびオリゴ糖に結合する。リンカーの残りの直鎖原子は、例えば、炭素、酸素、窒素、および硫黄から選択され、この原子のうちのいずれも、任意に炭素環、ヘテロ環、アリールまたはヘテロアリール環に含まれていてよい。リンカーの直鎖炭素原子は、任意にハロ、ヒドロキシ、ニトロ、ハロアルキル、アルキル、アルカリル、アリール、アラルキル、アルコキシ、アリールオキシ、アミノ、アシルアミノ、アルキルカルバモイル、アリールカルバモイル、アミノアルキル、アルコキシカルボニル、カルボキシ、ヒドロキシアルキル、アルカンスルホニル、アレンスルホニル、アルカンスルホンアミド、アレンスルホンアミド、アルキルスルホンアミド、アルキルカルボニル、アシルオキシ、シアノおよびウレイドから選択される置換基で置換されていてよい。リンカーの直鎖窒素原子は、任意にアシル、スルホニル、アルキル、アルカリル、アリール、アラルキル、アルコキシカルボニルで置換されていてよい。リンカーの直鎖硫黄原子は、任意に酸化されていてよい。
【0116】
ある種の実施形態において、例えば米国特許出願公開第2006/0228348号および米国特許第4,867,973号、第7,176,185号、第7,232,805号に開示されたように、リンカーを切断することができる。いくつかの実施形態において、リソソーム条件下で、リンカーを切断することができる。
【0117】
D.オリゴ糖−タンパク複合体の製造方法
例えば、式I、II、III、IV、V、またはVIのオリゴ糖を含む複合体など、本発明の複合体は、当業者に公知のいずれかの方法により製造することができる。これら方法のいずれかにおいて、適当なリンカーは、オリゴ糖またはタンパク質のどちらか、あるいはオリゴ糖およびタンパク質の両方に存在することができる。例えば、複合体を、例えばZhuら,Biochem.J.389:619−628(2005)、Zhuら,J.Biol.Chem.279:50336−50341(2004)、米国特許第5,153,312号、第5,212,298号、第5,280,113号、第5,306,492号、第5,521,290号、第7,001,994号、米国特許仮出願第60/885,457号、または第60/885,471号に記述されたように製造することができる。
【0118】
ある種の実施形態において、オリゴ糖はシステインまたはリシンなど、タンパク質のアミノ酸に結合することができる。例えば、糖はスクシンイミジル4−ホルミル安息香酸で、タンパク質のリシン残基を修飾することで、リシンを介して結合することができる。さらに、糖はトラウト試薬またはN−スクシンイミジル−3−(2−ピリジルジチオ)プロピオナート(SPDP)などのジスルフィド、もしくはN−スクシンイミジル−S−アセチルチオ酢酸(SATA)などの保護チオ−ルを含むリンカーで、リシンを修飾することにより、リシンを介して結合することができる。
【0119】
追加の実施形態において、オリゴ糖は糖タンパク質のグリカンに結合することができる。1つの実施形態において、オリゴ糖はグリカンのシアル酸残基に結合することができる。他の実施形態において、オリゴ糖は、グリカンのマンノース、フコース、ガラクトース、および/またはシアル酸残基に結合することができる。ガラクトースを介した結合について、まず、糖タンパク質をシアリダーゼで処理して、シアル酸残基を取り除き、その後オリゴ糖と反応させる前にガラクトースオキシダーゼで処理することができる。
【0120】
例えば、オリゴ糖−タンパク複合体は、(例えば、アミン、チオール、カルボン酸、ヒドロキシルを含む)タンパク質に存在し得るいずれかの官能基、および/または、タンパク質へ導入され得るいずれかの官能基を、オリゴ糖上の好適な第二の官能基と反応させることによって製造され得る。官能基の導入方法は、当技術分野で周知である。例えば、少なくとも1つのカルボニル基を有する糖タンパク質は、例えば、過ヨウ素酸塩(例えば、過ヨウ素酸ナトリウム)またはガラクトースオキシダーゼで、糖タンパク質を酸化することにより得られる。別の例において、カルボニル基は、例えばWangら,Proc.Natl.Acad.Sci.USA 100:56−61(2003)に記載されるように、拡張遺伝暗号を有する発現系を使用して導入され得る。また、例えば、米国特許出願公開第2006/0228348号には、糖タンパク質への反応基の導入について記述されており、参照のこと。
【0121】
いくつかの実施形態において、糖タンパク質は、カルボニル−反応基を含むリンカーで修飾されたオリゴ糖と結合する前に、過ヨウ素酸塩で酸化される。カルボニル−反応基の例としては、アミノオキシ、ヒドラジン、またはヒドラジドなどが挙げられる。ある種の実施形態において、糖タンパク質は、約1、2、3、4、5、7.5、10、または22.5mMの過ヨウ素酸塩で酸化される。ある種の実施形態において、糖タンパク質は、糖タンパク質グリカンのシアル酸残基を酸化し、フコースおよびマンノースの酸化を最小限にするのに十分な条件下で酸化される。例示的実施形態において、過ヨウ素酸塩の濃度は、約2、3、4、または5mM未満とした。1つの実施形態において、過ヨウ素酸塩は過ヨウ素酸ナトリウムである。
【0122】
ある種の実施形態において、結合の間に形成されるタンパク凝集体は、様々なクロマトグラフィー法を用いて、取り除くことができる。1つの実施形態において、疎水性相互作用クロマトグラフィー(HIC)を用いることができる。HICカラムの例としては、Butyl650Cおよび650M、Hexyl650C、Phenyl6FF、Capto OctylおよびCapto Phenylが挙げられる。他の実施形態としては、凝集体を銅、ニッケル、コバルト、または水銀などの金属キレートクロマトグラフィーにより取り除くことができる。1つの実施形態において、銅カラムを、結合・溶出モード(bind−and−elute)またはフロースルーモードで使用することができる。例示的な溶出緩衝液として、グリシンまたはイミダゾールが挙げられる。いくつかの実施形態において、凝集体は10、20、30、40、50、60、70、80、または90%減少する。追加の実施形態において、複合体は0.5、1、1.5、2、2.5、または3%未満の凝集体を含む。
【0123】
E.複合体
複合体のオリゴ糖およびタンパク質成分は、例えば、本明細書で記述されるオリゴ糖およびタンパク質のいずれかであってよい。ある種の実施形態において、オリゴ糖−タンパク複合体は、オリゴ糖−糖タンパク複合体である。いくつかの実施形態において、オリゴ糖−タンパク複合体は、オリゴ糖−リソソーム酵素複合体である。
【0124】
いくつかの実施形態において、複合体は、式I〜VIのオリゴ糖から選択されるオリゴ糖を含む。ある種の実施形態において、複合体は糖タンパク質1につき、オリゴ糖を平均で1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、または1〜2、1〜3、1〜4、1〜6、2〜4、2〜10、2〜12、4〜6、3〜8、5〜6、5〜10、5〜15、5〜20、10〜15、10〜20、12〜15、12〜18、または15〜20分子含む。いくつかの実施形態において、複合体はタンパク質1分子につき、オリゴ糖を少なくとも4、5、6、7、8、9、または10分子含む。追加の実施形態において、複合体はタンパク質1モルにつき、M6Pを少なくとも2、3、4、5、6、7、8、9、または10モル含む。
【0125】
ある種の実施形態において、複合体は複合体化されていないタンパク質と比較して、完全な活性(酵素活性など)を示す。他の実施形態において、複合体は複合体化されていないタンパク質と比較して、少なくとも例えば、50、60、70、75、80、85、90、92、94、95、96、97、98、または99%の活性を示す場合がある。酵素活性を含む活性を測定するアッセイは、当技術分野で周知である。例えば、Eisenthalら,Enzyme Assays:A Practical Approach,Oxford University Press:New York,2002を参照のこと。リソソーム酵素の活性を測定するアッセイは、例えば、Liら,Clin.Chem.50:1785−1796(2004)、Civalleroら,Clin Chim.Acta 372:98−102(2006)に記述されている。GAAの活性を測定する例示的なアッセイは、実施例6に記述されている。van Diggelenら,J.Inherit.Metab.Dis.28:733−741(2005)(酸性スフィンゴミエリナーゼの活性のアッセイを記述)、Downingら,Plant Biotechnol.4:169−181(2006)(α−L−イズロニダーゼの活性のアッセイを記述)、Voznyiら,J.Inherit.Metab.Dis.24:675−80(2001)(イズロン酸−2−スルファターゼ活性のアッセイを記述)、Murrayら,Mol.Genet.Metab.90:307−312(2007)(α−ガラクトシダーゼA活性のアッセイを記述)、Brooksら,J.Inher.Metab.Dis.14:5−12(1991)(N−アセチルガラクトサミン−4−スルファターゼ活性のアッセイを記述)も参照のこと。
【0126】
ある種の実施形態において、複合体は、対応する複合体化されていないタンパク質より、標的細胞(例えば、CI−MPRを介するエンドサイト−シス)によって、より効果的に取り込まれる。例えば、複合体は複合体化されていないタンパク質より、所与の期間に、例えば少なくとも10%、15%、20%、25%、30%、35%、40%、50%、60%、70%、80%、90%、93%、95%、96%、97%、98%、99%、100%、110%、120%、130%、140%、150%、または200%(mol/mol)、より効果的に取り込まれる場合がある。他の実施形態において、所与の期間に、非結合タンパク質と比較して、目的とする細胞型(例えば、L6筋芽細胞またはヒトポンペ病線維芽細胞(NIGMS Human Genetic Cell Repository、Cat.No.GM20005))により、少なくとも2、3、4、5、6、7、8、9、10、20、30、40、50、100、または1000倍(mol/mol)の複合体が取り込まれることがある。基準とされる期間は、例えば、10分、30分、45分、または1、2、3、5、6、12、24、48、または72時間、もしくはそれ以上でよい。L6筋芽細胞のin vitroでの取り込みは、例えば、実施例6およびZhuら,J.Biol.Chem.279:50336−50341(2004)に記述された通りに測定することができる。
【0127】
ある種の実施形態において、複合体化されていないタンパク質と比較して、複合体のCI−MPRへの結合の増加が示される。例えば、複合体化されたタンパク質及び複合体化されていないタンパク質の結合定数または解離定数を比較することにより測定されるとおり、例えば、複合体化されたタンパク質は、複合体化されていないタンパク質と比較して、少なくとも2、3、4、5、10、20、30、40、50、100、1,000、または10,000倍、CI−MPRへの親和性の改善を示し得る。CI−MPRへの結合は、例えば、実施例5およびZhuら,J.Biol.Chem.279:50336−50341(2004)に記述された通りに測定することができる。
【0128】
ある種の実施形態において、複合体は、複合体化されていないタンパク質と比較して、マンノース受容体による取り込みの増加を示さない。あるいは、複合体化されていないタンパク質と比較して、マンノース受容体による取り込みが、5、10、15、20、30、40、または50%未満である。ラット肺胞マクロファージ細胞のマンノース受容体による取り込みは、例えば、Zhuら,Biochem.J.389:619−628(2005)に記述されたように、in vitroで測定することができる。
【0129】
ある種の実施形態において、好適な動物モデルにおける、代謝的に欠損した酵素の蓄積した基質レベルに関して、複合体は、例えば少なくとも2、3、4、5、10、20、30、40、50、100、または1000倍の減少を示す場合がある。例えば、ポンペ病のマウスモデルにおけるグリコーゲンレベルの減少は、例えばZhuら,J.Biol.Chem.279:50336−50341(2004)に記述されたように測定することができる。あるいは、例えばKikuchiら,J.Clin.Invest.101:827−33(1998)に記述された、ポンペ病のウズラモデルを使用することができる。別の例として、肝臓および脾臓中に蓄積されたグリコサミノグリカンの減少を、Kakkisら,Mol.Genet.Metab.72:199−208(2001)に記述されたように、ムコ多糖症I型のネコモデルで測定することができる。さらに、グロボトリアオシルセラミドレベルの減少を、例えば、Ioannouら,Am.J.Hum.Genet.68:14−25(2001)に記述されたように、ファブリー病のマウスモデルで測定することができる。さらに別の例において、スフィンゴミエリンレベルの減少を、例えば、Horinouchiら,Nat.Genet.10:288−293(1995)に記述されたように、ニーマンピック病のA型およびB型のマウスモデルで測定することができる。
【0130】
II.医薬組成物
いくつかの実施形態において、本発明は、それを必要とする被験体のリソソーム蓄積症を治療する医薬の製造における、(1)タンパク質、および(2)式I〜VIのいずれか1つのオリゴ糖を含む、オリゴ糖−タンパク複合体の使用を提供する。
【0131】
本明細書で記述された医薬組成物は、上記で記述されたようなオリゴ糖−タンパク複合体、および充填剤、膨張剤、崩壊剤、緩衝剤、安定剤、または賦形剤など、少なくとも1つの添加剤を含む。いくつかの実施形態において、本発明の医薬組成物は、式I〜VIのいずれか1つののオリゴ糖、およびリソソーム酵素を含む、複合体を含む。
【0132】
標準の医薬処方技術は、当技術者に周知である(例えば、2005Physician’s Desk Reference(登録商標),Thomson Healthcare:Montvale,NJ,2004;Remington:The Science and Practice of Pharmacy,20th ed.,Gennadoら,Eds.Lippincott Williams&Wilkins:Philadelphia,PA,2000を参照のこと)。好適な医薬添加剤には、例えば、マンニトール、デンプン、グルコース、ラクトース、ショ糖、ゼラチン、麦芽、米、小麦粉、石灰、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。組成物はまた、pH緩衝試薬、および湿潤剤または乳化剤を含んでもよい。この組成物は、保存剤を含んでも含まなくてもよい。
【0133】
いくつかの実施形態において、α−ガラクトシダーゼA複合体を含む医薬組成物は、例えば、マンニトール、リン酸一ナトリウム一水和物、および/またはリン酸水素二ナトリウム七水和物など、1または2以上の賦形剤を含むことができる。いくつかの実施形態において、α−グルコシダーゼの複合体を含む医薬組成物は、マンニトール、ポリソルベート80、リン酸水素二ナトリウム七水和物、およびリン酸一ナトリウム一水和物の内、1または2以上のものを含むことができる。別の実施形態において、α−グルコシダーゼの複合体を含む医薬組成物は、グリシン2%まで、マンニトール2%まで、ポリソルベート80を0.01%までを備えた10mMヒスチジンpH6.5を含むことができる。
【0134】
医薬組成物は、単独の活性化合物として、または他の化合物、組成物、もしくは生物学的物質との組合せでも、本明細書に記述された複合体のいずれかを含むことができる。例えば、医薬組成物は、LSDおよび/またはLSDに関連する副作用の治療に有用な、1または2以上の小分子も含むことができる。いくつかの実施形態において、組成物はミグルスタット(miglustat)、および/または例えば米国特許出願公開第2003/0050299号、第2003/0153768号、第2005/0222244号、または第2005/0267094号に記述される1又は2以上の化合物を含むことができる。いくつかの実施形態において、医薬組成物は、1又は2以上の免疫抑制剤も含むことができる。
【0135】
医薬組成物の処方は、目的とする投与方法およびその他の要因に応じて変更してもよい(例えば、Roweら,Handbook of Pharmaceutical Excipients,4th ed.,APhA Publications,2003を参照のこと)。いくつかの実施形態において、組成物を、滅菌注射用蒸留水(米国薬局方)と再構成して、静脈注射による投与のため、滅菌した非発熱性のオフ−ホワイトの凍結乾燥した固形または粉末であってよい。他の実施形態において、組成物を滅菌した非発熱性の溶液であってよい。
【0136】
本明細書で記述された医薬組成物の投与は、いかなる個々の送達システムに限定されず、非経口(皮下、静脈内、頭蓋内、髄内、関節内、筋内、くも膜下、もしくは腹腔内注射を含む)、経皮、または経口(例えば、カプセル、懸濁剤、または錠剤)を挙げることができるがそれには限定されない。個体へは、単回投与または反復投与、様々な生理学的に許容される塩形態のいずれか、および/または医薬組成物の一部として、許容される医薬担体および/または添加剤と共に投与することができる。
【0137】
医薬的に許容される塩には、例えば、酢酸アニオン、ベンゼンスルホン酸アニオン、安息香酸アニオン、重炭酸アニオン、重酒石酸アニオン、ブロミドアニオン、カルシウムエデテート(calcium edetate)アニオン、カンシル酸アニオン、炭酸アニオン、クロリドアニオン、クエン酸アニオン、二塩酸、エデト酸アニオン、エジシル酸アニオン、エストル酸(estolate)アニオン、エシル酸アニオン、フマル酸アニオン、グルセプチン酸アニオン、グルコン酸アニオン、グルタミン酸アニオン、グリコリルアルサニレート(glycollylarsanilate)、ヘキシルレソルシネート(hexylresorcinate)アニオン、ヒドラバミン(hydrabamin)、臭化水素、塩酸、ヒドロキシナフトエート(hydroxynaphthoate)アニオン、ヨージドアニオン、イセチオン酸アニオン、乳酸アニオン、ラクトビオン酸酸アニオン、リンゴ酸アニオン、マレイン酸アニオン、マンデル酸アニオン、メシル酸アニオン、メチル臭化物、メチル硝酸アニオン、メチル硫酸アニオン、ムチン酸アニオン、ナプシル酸アニオン、硝酸アニオン、パモン酸アニオン(エンボネート(embonate))、パントテン酸アニオン、リン酸アニオン/二リン酸アニオン、ポリガラクツロ酸アニオン、サリチル酸アニオン、ステアリン酸アニオン、塩基性酢酸アニオン、コハク酸アニオン、硫酸アニオン、タンニン酸アニオン、酒石酸アニオン、およびテオクル酸アニオン/トリエチオダイドアニオン;ベンザチン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、メグルミン、およびプロカインの(有機)カチオン;並びにアルミニウム、カルシウム、リチウム、マグネシウム、カリウム、ナトリウム、及び亜鉛の(金属)カチオンが挙げられる。医薬的に許容される塩には、例えばBergeら,J.Pharm.Sci.66:1−19(1977)に記述された塩も含まれる。
【0138】
本明細書に記述される複合体は、治療有効量で投与される。一般に治療有効量は、被験体の年齢、全体的な状態、および性別、ならびに被験体の病状の重症度に応じて変更され得る。用量は医師により決定され、治療の実際の効果に合わせて、必要であれば調節することができる。この化合物の毒性および治療効果は、in vitroおよび/またはin vivoで標準的な医薬的手順により決定することができる。毒性および治療効果の用量比率は、治療指数(または治療可能比)であり、比率LD50/ED50として表すことができ、LD50は全個体の50%致死量であり、ED50は全個体の50%に治療効果がある用量である。本発明の複合体は、例えば、少なくとも1、1.5、2、3、4、5、6、7、8、9、10、および20の治療指数を示す場合がある。
【0139】
in vitroアッセイおよび動物試験により得られたデータを、例えば、ヒトにおける用量範囲を決めるのに使用することができる。この化合物の用量は、好ましくは、低い毒性、わずかな毒性、または無毒でED50を含む循環する濃度の範囲内である。使用する剤形および利用する投与経路によって、用量を変えることができる。どの複合体の治療有効量も、in vitroアッセイから、最初に推定することができる。in vitro実験で測定された、IC50値(すなわち、最大数の半数の症状を阻害する試験複合体の濃度)を含む、循環する血漿濃度範囲を実現するため、動物モデルにおいて、用量を決めることができる。血漿中の程度を、例えば、高速液体クロマトグラフィーまたは適当な酵素活性アッセイにより測定することができる。どの特定用量の効果も適当な評価項目のバイオアッセイにより、測定できる。
【0140】
他で指示されない限り、症状の重症度および疾患の進行により、ほぼ1μg/kg〜500mg/kgの用量で、複合体を投与することができる。例えば、外来診療で、例えば1日、2日、3日、4日、5日、6日、7日、8日、9日、10日またはそれ以上の日数おきに、あるいは例えば週1回、週2回、月1回、または1カ月2回の投与でゆっくりした静脈注射により、複合体を投与してよい。化合物の適当な治療有効用量は、治療する臨床医によって選択され、おおよそ1μg/kg〜500mg/kg、1μg/kg〜10mg/kg、1μg/kg〜1mg/kg、10μg/kg〜1mg/kg、10μg/kg〜100μg/kg、100μg/kg〜1mg/kg、および500μg/kg〜5mg/kgの範囲である。いくつかの実施形態において、適当な治療用量は、例えば0.1、0.25、0.5、0.75、1、5、10、15、20、30、40、50、60、70、および100mg/kgから選択される。
【0141】
α−ガラクトシダーゼAを含む複合体を、例えば、2週間に1回または4週間に1回、1.0mg/kg体重の用量で、10、13、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、または33mg/時未満あるいは等しい注入速度で、静脈注射により投与することができる。別の例において、α−グルコシダーゼを含む複合体を、例えば、2週間または4週間に1回、20mg/kgまたは40mg/kgの用量で、おおよそ例えば、1、2、3、4、5、6、7、8、9、または10時間にわたって、静脈注射により投与することができる。いくつかの実施形態において、α−グルコシダーゼの投与速度は、例えば1mg/kg/時で始め、それから例えば30分ごとに2mg/kg/時にし、注入速度に対する被験体の耐性を確立した後、例えば7mg/kg/時である最大まで増やすことができる。N−アセチルガラクトサミン−4−スルファターゼを含む複合体を、例えば、毎1.0mg/kg体重の用量で、毎週、おおよそ例えば1、2、3、4、5、6、7、8、9、または10時間にわたって、静脈注入により投与することができる。さらに具体的な用量の例としては、Physicians’ Desk Reference(登録商標)に記載されている。
【0142】
III.リソソーム蓄積症の治療方法
いくつかの実施形態において、本発明は例えば表1に開示されたリソソーム蓄積症の治療方法を提供する。いくつかの実施形態において、本発明はさらに、マンノース−6−リン酸を含むオリゴ糖と結合することにより、タンパク質をリソソームに向ける方法を提供する。
【0143】
ある種の実施形態において、当該方法は、治療有効量で本発明のオリゴ糖−タンパク複合体を、リソソーム蓄積症である被験体(例えば、ヒト、ネコ、イヌ、マウス、またはラットなどの哺乳動物、あるいは例えばウズラなどの鳥類を含む)に、投与することを含む。オリゴ糖−タンパク複合体は、式I〜VIのいずれかのオリゴ糖など、マンノース−6−リン酸を含むオリゴ糖と、リソソーム酵素(例えば、表1に列挙したリソソーム酵素)などの糖タンパク質との複合体であり得る。1つの実施形態において、当該方法は、本発明の複合体の少なくとも1つを含む医薬組成物を、それを必要とする被験体に投与することを含む。
【0144】
ある種の実施形態において、当該方法は、(1)タンパク質、および(2)式I〜VIのいずれかのオリゴ糖など、マンノース−6−リン酸を含むオリゴ糖を含む複合体を、1または2以上の他の治療剤(therapy)と投与することを含む。1または2以上の他の治療剤は、当該複合体の投与と同時に(混合された製剤としての同時投与を含む)、投与前に、または投与後に投与され得る。
【0145】
いくつかの実施形態において、当該方法は、解熱剤、抗ヒスタミン剤、および/または免疫抑制剤で、(本明細書で記述された複合体での治療前、治療後、または治療中に)被験体を治療することを含む。いくつかの実施形態において、注入に付随する反応を減少、または防止するために、被験体は、オリゴ糖−糖タンパク複合体で治療される前に、解熱剤、抗ヒスタミン剤、および/または免疫抑制剤で治療され得る。例えば、被験体は、アセトアミノフェン、アザチオプリン、シクロホスファミド、シクロスポリンA、ジフェンヒドラミン、メトトレキサート、ミコフェノール酸モフェチル、経口ステロイド、またはラパマイシンのうちの1または2以上で前治療され得る。
【0146】
いくつかの実施形態において、当該方法はアセトアミノフェン、アザチオプリン、シクロホスファミド、シクロスポリンA、ジフェンヒドラミン、メトトレキサート、ミコフェノール酸モフェチル、経口ステロイド、またはラパマイシンのうちの1または2以上で、例えば、t=0(複合体を投与する時間)および/またはt=12、24、36、48、60、72、96、120および144時間で、あるいはおおよそこの時間で、例えば最初の1、2、3、4、5、6、7、8、9、10回またはそれ以上での複合体治療回数で、被験体を治療することを含む。例えば、いくつかの実施例において、ファブリー病またはポンペ病である被験体は、メトトレキサート(例えば、1kgあたり0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2、3、4、5、6、8、10、12、15、25、30、35、40、50、60、70、80mgまたはそれ以上のメトトレキサート)で、例えばt=0、24、および48時間で、またはおおよそこの時間で、例えば最初の1、2、3、4、5、6、7、8週間での複合体の治療で、治療され得る。いくつかの実施形態において、複合体への免疫寛容が、シクロスポリンAおよびアザチオプリンでの治療によって、例えばムコ多糖症I型などのリソソーム蓄積症である被験体に誘導されることがある。例えば、Kakkisら,Proc.Natl.Acad.Sci.U.S.A.101:829−834(2004)に記述されたように、被験体はシクロスポリンAおよびアザチオプリンで治療されてもよい。
【0147】
いくつかの実施形態において、当該方法は、リソソーム蓄積症の治療のために行われる小分子治療および遺伝子治療を含む、小分子治療および/または遺伝子治療で、(複合体での治療前、治療後、または治療中に)被験体を治療することを含む。小分子治療は、ミグルスタットおよび/または例えば、米国特許出願公開第2003/0050299号、第2003/0153768号、第2005/0222244号、および第2005/0267094号に記述された1または2以上の化合物の投与を含んでよい。遺伝子治療は、例えば米国特許第5,952,516号、第6,066,626号、第6,071,890号、第6,287,857号、および米国特許出願公開第2003/0087868号に記述されたように実施することができる。
【0148】
「治療」、「治療方法」という語、およびその同族語は、治療処置および予防/防止対策の両方を示す。したがって、治療を必要とする人として、すでに特定のリソソーム蓄積症を有する個体、および疾患の危険性がある人(すなわち、最終的にその疾患にかかる、または疾患のある種の症状に陥りそうな人)を含んでよい。
【0149】
治療方法は、症状の予防もしくは緩和、または他の所望の生物学的成果をもたらし、臨床兆候の改善もしくは発症遅延、代謝的欠損酵素の活性増加、および/または代謝的欠損酵素の基質の蓄積が減少したレベルにより、評価することができる。
【0150】
いくつかの実施形態において、当該方法は、(1)リソソーム酵素、および(2)式I〜VIのいずれかのオリゴ糖を含む複合体を被験体に投与することを含み、これにより、内在性活性と比べて、例えば少なくとも10%、15%、20%、25%、30%、35%、40%、50%、60%、70%、80%、90%、または100%、被験体内の欠損したリソソーム酵素活性が増加する。いくつかの実施形態において、当該方法は、(1)リソソーム酵素、および(2)式I〜VIのいずれかのオリゴ糖を含む複合体を被験体に投与することを含み、これにより内在性活性と比べて、例えば少なくとも2、3、4、5、6、7、8、9、10、20、30、40、50、100、または1000倍、被験体内の欠損した酵素活性が増加する。増加した酵素活性を、例えば臨床症状の縮小、または適当な臨床的もしくは生物学的アッセイによって、測定することができる。
【0151】
いくつかの実施形態において、当該方法は、GAAを含む本発明の複合体を被験体に投与することを含み、これによって、ポンペ病(酸性α−グルコシダーゼ欠損症、酸性マルターゼ欠損症、グリコーゲン蓄積症II型、糖原病II型、およびリソソーマルα−グルコシダーゼ欠損症としても知られる)を治療する。ある種の実施形態において、組換えヒトGAA(rhGAA)はチャイニーズハムスター卵巣(CHO)細胞を用いて製造される。追加の実施形態において、rhGAAは、オリゴ糖103;103および式Aの混合物;オリゴ糖128、129、130、131、132、133、または136;並びにそれらの任意の組合せから選択されるオリゴ糖と複合体化され得る。1つの実施形態において、オリゴ糖は、オリゴ糖128および136から選択される。追加の実施形態において、GAA1モルにつき、少なくとも5モルの式Aを含む複合体が患者に投与される。
【0152】
ある種の実施形態において、患者は、ポンペ病と診断されている成人である。他の実施形態において、患者は幼児または子供である。
【0153】
GAA活性の増加は、例えば心筋細胞、骨格筋細胞、または皮膚線維芽細胞におけるリソソームグリコーゲンの蓄積の減少を、生化学的(例えば、Zhuら,J.Biol.Chem.279:50336−50341(2004)を参照のこと)または組織学的に観察することにより測定され得る。GAA活性はまた、例えば筋生検サンプル、培養皮膚線維芽細胞、リンパ球、乾燥血斑で測定され得る。乾燥血斑アッセイは、例えば、Umpathysivamら,Clin.Chem.47:1378−1383(2001)およびLiら,Clin.Chem.50:1785−1796(2004)に記述されている。ポンペ病の治療は、例えばクレアチニンキナーゼの血清レベル、運動機能の増幅率(例えば、アルバータ乳幼児運動発達検査法によって評価される)、エコー図で測定される左室心筋重量係数の変化、および心電図で測定される心臓電気活性によって、評価することもできる。GAA複合体の投与はまた、心肥大、心筋症、日中の眠気、労作性呼吸困難、成長障害、摂食困難、「筋緊張低下(floppiness)」、歩行障害、頭痛、低血圧、臓器肥大症(心臓、舌、肝臓の肥大など)、脊柱前弯症、平衡感覚障害、腰痛、朝の頭痛、筋力低下、呼吸不全、翼状肩甲、脊柱側弯症、深部腱反射の低下、睡眠時無呼吸、呼吸器感染症に対する感受性、および嘔吐など、ポンペ病の1または2以上の症状の縮小をもたらし得る。
【0154】
他の実施形態において、当該方法は、α−ガラクトシダーゼAを含む複合体を、被験体に投与することでファブリー病を治療することを含む。ファブリー病、またはアンダーソン−ファブリー病は、α−ガラクトシダーゼAの欠損を特徴とする、希少でX連鎖性のリソソーム蓄積症であり、内臓組織と、内皮、外皮および筋細胞のリソソームに、グロボトリアオシルセラミド(GL3)およびその他の中性スフィンゴ糖脂質の蓄積を引き起こす。血管系への中性スフィンゴ糖脂質の蓄積は、血管の狭窄および拡張、最終的には虚血および梗塞を引き起こす。
【0155】
α−ガラクトシダーゼAを含む本発明の複合体の投与は、例えば、肢端感覚異常、狭心症、角化血管腫、不整脈、歩行失調、四肢の灼熱痛ならびに/ヒリヒリする痛み、白内障、寒冷不耐症、伝導異常、角膜渦濁(corneal whorling)、冠動脈疾患、認知症、うつ病、下痢、心室拡張、めまい、心肥大、心筋症、複視、構音障害、疲労、赤血球沈降速度の上昇を伴う発熱、聴覚問題、心疾患、心臓弁疾患、高熱不耐性、片側運動失調、片側不全麻痺、発汗減少症、発汗不全、梗塞、虚血、関節痛、腎疾患、左室肥大、水晶体異常、レンズ混濁、脂肪尿、筋力低下、心筋梗塞、嘔気、眼振、痛み(例えば、全身に広がる激痛)、多渇症、タンパク尿、食後痛、腎不全、網膜異常、耳鳴り、胃痛、ST−T変化、脳卒中、尿毒症、弁膜症、めまい、嘔吐および虚弱を含む、1または2以上のファブリー病の臨床症状の縮小をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、例えば、血漿、涙液、白血球、生検組織、または培養皮膚線維芽細胞で、α−ガラクトシダーゼA活性の増加をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、複屈折性の脂肪球の増加の減少(例えば、少なくとも10%減少)または消失という、組織学的所見をもたらし得る。それはまた、尿沈渣の脂肪球減少、血清クレアチニンレベルまたはクレアチニンクリアランスによって測定した腎機能の改善、およびタンパク尿の減少をもたらし得る。α−ガラクトシダーゼA複合体の投与はまた、腎臓、心臓、および皮膚の毛細血管内皮のGL3含有物の減少をもたらし得る。ファブリー病治療の効果を測定する追加のアッセイは、例えば、MacDermottら,J.Med.Genet.38:750−760(2001)に記載されている。
【0156】
他の実施形態において、当該方法は、酸性スフィンゴミエリナーゼを含む本発明の複合体を、被験体に投与することを含み、これにより、ニーマンピックA病またはニーマンピックB病、あるいは酸性スフィンゴミエリナーゼ欠損症を治療する。酸性スフィンゴミエリナーゼ複合体の投与は、例えば、異常コレステロールレベル、異常脂質レベル、運動失調、血液異常、眼内のさくらんぼ赤色斑、頻繁な肺感染、成長遅延、肝脾腫大症、血小板数低下、リンパ節腫脹症、末梢神経障害、肺機能の問題、息切れ、皮膚色素変化、または黄色腫症を含む、1または2以上のニーマンピックA病またはニーマンピックB病に関する、臨床症状の減少をもたらし得る。いくつかの実施形態において、複合体を頭蓋内に投与することができる。
【0157】
他の実施形態において、当該方法は、α−L−イズロニダーゼを含む、本発明の複合体を、被験体に投与することを含み、これによりムコ多糖症I型(例えば、MPSI型のハーラーおよびハーラー−シャイエ型を含む)を治療する。α−L−イズロニダーゼ複合体の投与は、例えば、大動脈弁逆流、大動脈弁狭窄症、毛根管症候群、慢性鼻炎、伝音難聴、便秘、角膜混濁、発育遅延、下痢、腹部膨張、腰椎後弯症、突背、肝脾腫大症、水頭症、鼠径ヘルニア、脊柱後弯症、精神遅滞、僧帽弁逆流、僧帽弁狭窄症、夜盲症、開放隅角線内症、手の機能低下、進行性関節症、反復呼吸感染、呼吸不全、網膜変性、脊柱側弯症、感音難聴、激しい背中の痛み、鼻漏、睡眠時無呼吸、脊髄圧迫、母指球の萎縮、へそヘルニア、上気道の合併症など、1種または複数のMPSI型の臨床症状の減少をもたらし得る。
【0158】
さらに別の実施形態において、当該方法は、イズロン酸−2−スルファターゼを含む本発明の複合体を、被験体に投与することを含み、これにより、ムコ多糖症II型(ハンター病)を治療する。イズロン酸−2−スルファターゼ複合体の投与は、例えば、腎臓弁膜症、心肺機能不全、毛根管症候群、慢性下痢、慢性の乳頭浮腫、特徴的顔貌、角膜混濁、冠状動脈狭窄、難聴、異形症、異骨症、耳感染、聴覚障害、肝脾腫大症、水頭症、鼠径ヘルニア、関節硬直、後側弯症、精神遅滞、心筋症、心筋肥厚、肺高血圧、網膜機能不全、骨格異常、へそヘルニア、上気道感染、弁機能不全を含む、1または2以上のMPSII型の臨床症状の減少をもたらし得る。
【0159】
さらに他の実施形態において、当該方法はN−アセチルガラクトサミン−4−スルファターゼ(アリールスルファターゼB)を含む、本発明の複合体を被験体に投与することを含み、これによりムコ多糖症VI型(マロトー−ラミー症候群)を治療する。N−アセチルガラクトサミン−4−スルファターゼ複合体の投与は、例えば、失明、心臓異常、心肺疾患、特徴的顔貌、角膜混濁、耳感染、成長遅延、肝腫大、肝脾腫大症、関節変形、神経絞やく症候群、呼吸困難、骨格変形、脊髄圧迫、脾腫、関節の硬直、上気道閉塞を含む、1種または複数のMPSVI型の臨床症状の減少をもたらし得る。
【0160】
前述および以下の説明は、例示および説明のためのみであり、特許請求されている本発明を限定しない。
【実施例】
【0161】
実施例1.オリゴ糖合成の一般的手順
A.グリコシル化
グリコシル化反応を、ドナーである糖およびアクセプターである糖を結合させることによる、標準的な方法を用いて実施した。簡潔に言えば、他に指示されない限り、乾燥窒素下、加熱して活性化したモレキュラーシーブ4Aの存在下で、無水DCMにドナーおよびアクセプター化合物を溶解させた。この溶液を冷却し、0℃で〜30分間保存し、続いてTMSOTf(1eq)をゆっくり添加した。反応をヘキサン/EtOAcを用いて、TLC(シリカゲル)で確認し、TEAまたはヒューニッヒ塩基(1.05eq)を用いて停止した。混合物をろ過し、シロップになるまで濃縮し、指示されない限り、ヘキサン/EtOAcグラジエントを用いたフラッシュカラムクロマトグラフィーによって精製した。
【0162】
B.酸触媒脱アセチル化
ある種の実施形態において、アセチル化化合物は、グリコシル化または他の修飾、例えばリン酸化の前に、脱アセチル化される。以下の実施例において、塩化アセチルを冷(0℃)乾燥メタノールへ加えることによって、塩化水素が生成させた。濃縮した溶液を、DCM/メタノール中のアセチル化化合物の1:3溶液に加えた。溶液のメタノール成分に関して、最終濃度は3%w/vであった。脱アセチル化反応は、1級のアセテートについては、a)約18時間、2級のアセテートについては、b)約48から64時間で行った。反応を、TEAまたはヒューニッヒ塩基で停止し、指示されない限り、続いてDCMまたはEtOAcを水で抽出した。
【0163】
C.リン酸化
いくつかの例において、糖は部位特異的なリン酸化を受ける。室温で乾燥アセトニトリルの糖溶液に、5−メチルテトラゾール(3.4eq)を加え、この混合物を30分間、乾燥窒素下で撹拌した。ジベンジルジイソプロピルホスホルアミダイト(1個のOH基につき1.7eq)を加え、反応が完了するまで(約60分)撹拌した。反応をヘキサン/EtOAcを用いて、TLC(シリカゲル)により確認した。溶液を氷/水で15分冷却し、30%w/v過酸化水素(2eq)を加えた。約60分後、反応が完結し、過剰の飽和チオ硫酸ナトリウムを加えた。混合物をガム状になるまで濃縮し、EtOAcに溶解させ、半飽和食塩水で洗浄し、硫酸ナトリウムで乾燥させた。他で指示されない限り、残渣をヘキサン/EtOAcグラジエントを用いて、フラッシュクロマトグラフィーで精製した。
【0164】
実施例2.二糖アミノオキシアセトアミドプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシドの合成(17)
【0165】
【化21】
【0166】
アリルα−D−マンノシドをPekariら,J.Org.Chem.66:7432(2001)に記述されている方法にしたがって製造した。無水メタノール(100mL)中のアリル−α−D−マンノシド(8.68g、39.4mmol)に、2,3−ブタンジオン(3.63mL、86.1mmol)、トリメチルオルトホルメート(16mL、146mmol)、および10−(+)−カンファースルホン酸(1.37g、5.9mmol)を添加した。混合物を還流下で、9時間、乾燥窒素下で加熱した。反応混合物をTEA(1mL)で停止し、赤いシロップ状になるまで濃縮し、40〜80%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、アリル3−O,4−O−[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド1を白色固体として得た(3.89g、29.4%)。
【0167】
化合物1(2.65g、8mmol)を乾燥ピリジン(20mL)へ溶解し、その溶液を氷/水槽で冷却し、t−ブチルジフェニルシリルクロリド(2.28mL、8.7mmol)を加えた。18時間後、ピリジン(20mL)および無水酢酸(1.6mL、16mmol)を加え、この溶液を50℃まで16時間加熱し、その後、シロップ状になるまで濃縮し、トルエンと共に留去した。残渣をEtOAc(60mL)に溶解させ、1M HCl(2x50mL)、飽和炭酸水素ナトリウム(50mL)で洗浄し、硫酸ナトリウムで乾燥した。混合物をろ過し、真空下で濃縮し、溶液を濃縮し、アリル2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド2をシロップとして得た(5.0g)。
【0168】
塩化パラジウム(II)(0.425g、2.4mmol)を、2(5.0g、8mmol)の無水メタノール(25mL)溶液に撹拌しながら加えた。約3時間後、反応をTEA(0.75mL、4.8mmol)で停止した。メタノールを減圧下で取り除き、残渣を10〜50%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーで精製し、2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド3を、白色泡状物質として得た(2.72g、59.2%)。
【0169】
トリクロロアセトニトリル(1.75mL、17.4mmol)およびDBU(0.05mL,0.35mmol)を3(1.0g、1.74mmol)の乾燥DCM(1mL)溶液に添加した。約75分後、溶液をEtOAc/ヘキサン(0〜30%)を用いて、シリカゲルカラムフラッシュカラムクロマトグラフィーで直接精製し、2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノーストリクロロアセトイミデート4を、白色泡状物質として得た(0.98g、78.2%)。ドナーである4(0.98g、1.36mmol)およびアクセプターである3−N−ベンジルオキシカルボニルアミノプロパノール(0.284g、1.36mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2−O−アセチル−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド5を、白色泡状物質として得た(0.66g、64%)。
【0170】
無水メタノール(5mL)中の5(0.66g、0.87mmol)の溶液に、メタノール(0.05mL、0.22mmol)中の25%w/vナトリウムメトキシドの溶液を加えた。約1時間後、反応を氷酢酸(0.025mL)で停止し、シロップ状にまるまで濃縮した。生成物をDCM(10mL)に溶解させ、半飽和食塩水(5mL)で洗浄し、硫酸ナトリウムで乾燥して、白色泡状物質であるN−ベンジルオキシカルボニルアミノプロピル6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド6を得た(0.58g、92%)。
【0171】
【化22】
【0172】
アリルα−D−マンノシド(3.15g、14.3mmol)を乾燥ピリジン(20mL)に溶解させ、氷/水槽で冷却した。t−ブチルジフェニルシリルクロリド(4.03mL、15.7mmol)を加え、この溶液を室温になるまで放置した。18時間撹拌し、ベンゾイルクロリド(5.93mL、51.5mmol)を加え、24時間後、反応を水(3mL)で停止し、30分撹拌した。溶液を真空下で濃縮し、トルエンで分離した(3x50mL)。残渣をEtOAc(100mL)に溶解させ、冷却した1M HCl(50mL)、半飽和食塩水(50mL)、半飽和炭酸水素ナトリウム(50mL)、半飽和食塩水(50mL)で洗浄し、硫酸ナトリウムで乾燥し、ろ過し、シロップ状になるまで濃縮した。0〜50%EtOAc/ヘキサンを用いてシリカゲルフラッシュカラムクロマトグラフィーで精製し、アリル2,3,4−トリ−O−ベンゾイル−6−O−t−ブチルジフェニルシリル−α−D−マンノシド7を、白色泡状物質として得た(8.62g、78.2%)。
【0173】
氷酢酸(11.1mL、18.26mmol)および1M テトラブチルアンモニウムフルオリド(18.26mL、18.26mmol)を7(12.77g、16.6mmol)の乾燥THF(50mL)溶液に加えた。80分で、氷酢酸(0.15mL、25mmol)および1M テトラブチルアンモニウムフルオリド(2.5mL、25mmol)を加え、90分で追加の氷酢酸(0.25mL、4.15mmol)および1M テトラブチルアンモニウムフルオリド(4.15mL、4.15mmol)を加えた。2時間後、溶液を半分の容量まで濃縮し、EtOAc(150mL)で希釈した。溶液を半飽和食塩水(2x150mL)および半飽和重炭酸ナトリウム(200mL)で洗浄し、濃縮し、残渣を10〜50%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーで精製し、アリル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド8を、白色泡状物質として得た(6.75g、76.4%)。化合物8(6.75g、ymmol)を、一般的なリン酸化手順に従って変換し、アリル2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシド9を、白色泡状物質として得た(9.52g、95%)。
【0174】
塩化パラジウム(II)(0.638g、3.6mmol)を9(9.52g、12.1mmol)の乾燥メタノール(50mL)溶液に加えた。約5時間後、さらにパラジウム(II)(0.145g)を加えて、混合物を18時間保存した。この溶液をろ過し、濃縮し、残渣を20〜70%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノース10を、白色泡状物質として得た(3.8g、42,5%)。
【0175】
トリクロロアセトニトリル(5.06mL、5.1mmol)およびDBU(0.15mL、1mmol)を10(3.8g)の乾燥DCM溶液に、乾燥窒素下、0℃で加えた。約90分後、溶液を10〜60%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより直接精製し、2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシドトリクロロアセトイミデート11を、白色泡状物質として得た(3.3g、72.1%)。
【0176】
【化23】
【0177】
ドナーである6(2.62g、2.97mmol)およびアクセプターである11(1.92g、2.7mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2−O−[2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシル]−6−O−t−ブチルジフェニルシリル−3−O,4−O[ジメトキシブタン−2’,3’−ジイル]−α−D−マンノシド12を、白色泡状物質として得た(1.27g、32.2%)。
【0178】
【化24】
【0179】
TFA/水 19:1 v/v(8.4mL)を、DCM(8mL)中の12(2.1g、1.44mmol)の氷/水槽で冷却した溶液に加えた。約2時間後、出発物質はTLCによって示されたように消費された。エタノール(25mL)をこの溶液に加え、その後濃縮し、エタノール(3x25mL)と共に留去した。残渣を乾燥メタノール(10mL)に溶解させ、氷/水槽で冷却した。塩化アセチル(0.4mL)を加えて、3%w/vのHCl溶液を得た。この溶液を室温まで放置して温めた。約2時間後、出発物質はTLCに示されたように消費された。反応をトリエチルアミン(1mL)で停止し、濃縮し、残渣を30〜100%EtOAc/ヘキサンを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、白色泡状物質として13を得た(0.758g、47.9%)。無水メタノール(10mL)中の13(0.758g、0.69mmol)の溶液に、25% w/v ナトリウムメトキシドのメタノール(0.15mL)溶液を加えた。約1時間後、出発物質はtlcに示されたように消費された。反応を1M HClで停止し、14を得た。この溶液に氷酢酸(25μL)、湿10% Pd/C(0.1g)を加え、水素バルーンを取り付けた。6時間反応させたあと、生成物を5%硫酸/EtOHで炭化させたが、UV活性はなかった。混合物をろ過し、油状物になるまで濃縮し、その後水(10mL)に溶解させ、凍結乾燥して、3−アミノプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシド15を得た(0.300g、13から91.3%)。
【0180】
0.1MのNaOH溶液(2mL)を水(8.5mL)中の15(0.1g、0.21mmol)の溶液に加え、その後、THF(8.5mL)中のN−t−ブトキシカルボニル−アミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.14g、0.42mmol)を加えた。18時間後、この溶液を2MのHClでpH4に調節し、この溶液をDCM(3x10mL)で抽出した。水層を凍結乾燥して、N−t−ブトキシカルボニル−アミノオキシアセトアミドプロピル2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシド16を得た(0.12g)。化合物16(0.12g)をTFA/DCM1:1(10mL)に溶解し、その溶液を約30分間攪拌し、その後油状物になるまで濃縮した。これを水(5mL)に溶解させ、生成物を凍結乾燥して、固体(0.45g)を得た。この固体をBiogel(登録商標)P2を用いて精製し、水で溶出させて、17を得た(0.063g)。
【0181】
実施例3.三糖(35)の合成
【0182】
【化25】
【0183】
2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノーストリクロロアセトアミデート18を、Yamazakiら,Carb.Res.201:31(1990)に記述されたように製造した。
【0184】
【化26】
【0185】
6−O−アセチル−3,4,6−トリ−O−ベンゾイル−α−D−マンノーストリクロロアセトイミデート19を、Hengら,J.Carb.Chem.20:285(2001)に記述されたように製造した。
【0186】
【化27】
【0187】
ドナー19(5.0g、7.4mmol)およびアクセプターN−9−フルオレニルメチルカルボニルアミノプロパノール(2.41g、8.1mmol)を一般的なグリコシル化手順に従って変換し、白色泡状物質であるN−9−フルオレニルメチルカルボニルアミノプロピル6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド29を得た(4.0g、66.4%)。
【0188】
【化28】
【0189】
化合物29(4.0g、4.9mmol)を、2級アセテートの酸触媒脱アセチル化の一般的な手順(b)に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド30を、白色泡状物質として得た(3.3g、87.3%)。ドナー18(3.27g、5.16mmol)およびアクセプター30(3.3g、4.3mmol)を、一般的なグリコシル化手順に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル6−O−[2−O−アセチル−3,4,6−トリ−O−ベンジルマンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド31を白色泡状物質として得た(4.94g、92.7%)。
【0190】
【化29】
【0191】
化合物31(4.94g、3.96mmol)を、2級アセテートの酸触媒脱アセチル化の一般的な手順(b)に従って変換し、N−9−フルオレニルメチルカルボニルアミノプロピル6−O−[3,4,6−トリ−O−ベンジルマンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド32を白色泡状物質として得た(1.73g、36%)。化合物11を、実施例2の方法に従って製造した。ドナー11(3.37g、3.74mmol)およびアクセプター32(1.73g、1.44mmol)を、一般的なグリコシル化手順に従って変換し、白色泡状物質として33を得た(1.2g、43.1%)。
【0192】
【化30】
【0193】
無水THF(10mL)中の33(1.2g、0.62mmol)の溶液に、ドデシルチオ−ル(1.48mL、6.2mmol)およびジアザビシクロウンデカ−7−エン(DBU)(0.093mL、0.62mmol)を加えた。約18時間後、出発物質はtlcに示されたように消費された。この溶液をシロップ状になるまで濃縮し、0〜20%メタノール/DCMを用いて、シリカゲルフレッシュカラムクロマトグラフィーで精製した。生成物に、メタノール/水 1:1(20mL)および酢酸(25μL)を加え、Pd/C(0.1g)および水素のバルーンを取り付けた。18時間後、この溶液をセライトでろ過し、白色泡状物質になるまで濃縮した。この泡状物質を乾燥メタノール(10mL)、およびメタノール(0.15mL)の25%w/vナトリウムメトキシドの溶液に溶解させ、6時間後、この溶液を濃縮して、水(10mL)に入れ、DCM(10mL)で洗浄した。水層を凍結乾燥して、アミノプロピル6−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド34を、二ナトリウム塩として得た(0.27g、33から63.5%)。
【0194】
水/DMSO 1:1溶液(10mL)中の34(0.17g、0.3mmol)に、DMSO(2mL)中のN−t−ブトキシカルボニルアミノ−オキシアセチル2,3,5,6−テトラフルオロフェニレート(0.34g、1.14mmol)の溶液およびDMSO(1mL)中の3−ヒドロキシ−1,2,3−ベンゾトリアジン−4(3H)−オン(DHBT)(0.09g、0.6mmol)の溶液を加えた。24時間後、この溶液を,sephadexサイズ排除樹脂を用いて精製した。画分をシリカゲルプレート上で炭化させることで確認し、選択した画分をまとめ、凍結乾燥して固体を得た。TFA/DCM(8mL)に溶解させ、60分撹拌し、その後油状物になるまで濃縮した。水(5mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して35を得た(0.033g、34から16.4%)。
【0195】
実施例4:四糖の合成
A.アミノオキシアセトアミド1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸(28)
【0196】
【化31】
【0197】
化合物19を、実施例3に記述された方法に従って製造した。ドナー19(15.49g、24.3mmol)およびアクセプターである3−N−9−フルオレニルメトキシカルボニルアミノプロパノール(8.7g、29.19mmol)を、一般のグリコシル化手順に従って変換し、N−9−フルオレニルメトキシカルボニルアミノプロピル2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノシド20を、白色泡状物質として得た(10.55g、55%)。
【0198】
【化32】
【0199】
乾燥DCM(50mL)および無水メタノール(100mL)中の20(10.5g、ymmol)の溶液に、約30分の間、塩化アセチル(4.8mL、63mmol)を滴加し、2.3%w/vHClのメタノール溶液を得た。約18時間後、反応をヒューニッヒ塩基(9.81mL、63mmol)で停止し、さらに1.5mL加えた。この溶液をシロップ状になるまで濃縮し、クロロホルム(2x50mL)と共に留去し、DCM(100mL)に溶解させ、半飽和食塩水(100mL)で洗浄し、硫酸ナトリウムで乾燥した。この溶液を濃縮し、0〜100%EtOAc/ヘキサンで、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、白色泡状物質である、N−9−フルオレニルメトキシカルボニルアミノプロピル3,4,6−トリ−O−ベンジル−α−D−マンノシド21を得た(6.11g、61.2%)。ドナー21(6.11g、13.4mmol)およびアクセプター11(7.1g、7.9mmol)を一般的なグリコシル化手順に従って変換し、N−9−フルオレニルメトキシカルボニルアミノプロピル2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシド22を白色泡状物質として得た(5.8g、50.1%)。
【0200】
無水THF溶液(75mL)中の22(5.8g、3.96mmol)に、ドデシルチオ−ル(9.53mL、40mmol)およびDBU(0.6mL、4mmol)を加えた。約4時間後、反応をメタノール塩酸(5.6mL、8mmol)で停止し、シロップ状になるまで濃縮した。生成物をジエチルエーテル中で粉砕し、3−アミノプロピル2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシド塩酸塩23をゴム状物質(gum)として得た(3.77g、74.5%)。
【0201】
【化33】
【0202】
乾燥アセトニトリル(50mL)中の23(3.77g、2.95mmol)の溶液に、N−ベンジルオキシカルボニルグルタミン酸(0.3661g、1.3mmol)、N−ヒドロキシベンゾトリアゾール(HOBt)(0.4g、2.95mmol)、DBU(4.6mL、4mmol)、および1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド(EDC)(0.75g、8.85mmol)を加えた。16時間後、ヒューニッヒ塩基(0.25mL)を加え、続いてさらにEDC(0.75g、8.85mmol)を加えた。21時間後、この溶液をシロップ状になるまで濃縮し、0〜50%DCMに対して、10%2−プロパノール/DCMを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、N−ベンジルオキシカルボニルアミノ1,5−ジ−[3−アミドプロピル2−O−[2,3,4−トリ−O−ベンゾイル−6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシル]グルタミン酸24を白色泡状物質として得た(0.97g、11.2%)。
【0203】
無水DCM(20mL)およびメタノール(25mL)中の24(0.912g、0.33mmol)の溶液に、25%w/vナトリウムメトキシドのメタノール(0.09mL、0.41mmol)溶液を加えた。約6時間半後、反応を1M HCl(0.41mL、0.41mmol)で停止し、シロップ状になるまで濃縮し、0〜100%DCMに対して10%2−プロパノール/DCMを用いて、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、N−ベンジルオキシカルボニルアミノ1,5−ジ−3−アミノプロピル[2−O−[6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−α−D−マンノシル]グルタミン酸25を得た(0.306g、44.4%)。
【0204】
THF/水 2:1 v/v(75mL)中の25(0.3g、0.144mmol)の撹拌溶液に、10%湿Pd/C(0.052g)および水素バルーンを取り付けた。16時間後、氷酢酸(25μL)を加え、新鮮な水素バルーンを取り付けた。6時間後、さらに10%Pd/C(0.04g)を追加し、さらに水素を追加した。18時間後、新鮮な5%Pd/C(0.05g)を追加し、さらに水素を追加した。24時間後、生成物を5%硫酸/EtOHで炭化したが、UV活性はなかった。混合物を、セライトを通してろ過し、パッドを洗浄し、約30%まで濃縮して、THFを除去した。その後凍結乾燥して1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸26(0.121g、77.9%)を得た。
【0205】
DMSO(1mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.19g、0.57mmol)の溶液およびDMSO(1mL)中の3,4−ジヒドロ−3−ヒドロキシ−4−オキソ−1,2,3−ベンゾトリアジン(DHBT)(0.052g、0.3mmol)の溶液を、水/DMSO 1:1(7.5mL)中の26(0.16g、0.15mmol)の溶液に加えた。18時間後、この溶液をsephadexサイズ排除樹脂で精製した。シリカゲル上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥してN−t−ブトキシカルボニルアミノオキシアセトアミド1,5−ジ−3−アミドプロピル[2−O−[6−O−ホスホリル−α−D−マンノシル]−α−D−マンノシル]グルタミン酸27を得た(0.095g、42.1%)。化合物27に、TFA/DCM 1:1(8mL)を加え、混合物を溶解するまで(約60分)撹拌し、その後油状物になるまで濃縮した。水(10mL)を加えて、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して28を得た(0.048g、54.2%)。
【0206】
B.アミノオキシアセトアミドプロピル6−O−([α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド(47)
【0207】
【化34】
【0208】
化合物11を実施例2に記述された通りに製造した。ドナー11(5.0g、7.4mmol)およびアクセプターである3−N−ベンジルオキシカルボニルアミノプロパノール(1.93g、9.25mmol)を実施例1の一般的なグリコシル化手順に従って変換し、白色泡状物質を得た。生成物を、1級アセテートを酸性触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル2,3,4−トリ−O−ベンゾイル−α−D−マンノシド36を白色泡状物質として得た。
【0209】
【化35】
【0210】
ドナー19(4.47g、6.6mmol)およびアクセプター36(3.6g、5.3mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド37を白色泡状物質として得た(3.8g、63.3%)。化合物37(3.8g、3.17mmol)を、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換して、N−ベンジルオキシカルボニルアミノプロピル6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド38を白色泡状物質として得た(3.5g、95.3%)。
【0211】
化合物18を、Yamazakiら,Carb.Res.201:31(1990)に記述された方法に従って製造した。ドナー18(2.41g、3.75mmol)およびアクセプター38(3.5g、3mmol)を、実施例1の一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド39が油状物として得られた(5.63g)。化合物39(5.83g)を、2級アセテート(実施例1を参照のこと)を酸触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニル−アミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド40を白色泡状物質として得た(2.0g、38から41.9%)。ドナー19(1.07g、1.63mmol)およびアクセプター41(2.0g、1.3mmol)を、一般的なグリコシル化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド42を白色泡状物質として得た(1.4g、50.8%)。化合物42(1.4g、0.066mmol)を、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換し、N−ベンジルオキシカルボニル−アミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド43を白色泡状物質として得た(1.0g、80%)。化合物43(1.0g、0.048mmol)を一般的なリン酸化手順に従って変換し、N−ベンジルオキシカルボニルアミノプロピル6−O−([2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,3,4−トリ−O−ベンゾイル−α−D−マンノシド44を白色泡状物質として得た(0.9g、80.7%)。
【0212】
【化36】
【0213】
無水メタノール(20mL)中の44(0.9g、0.039mmol)の溶液に、メタノール(0.09mL、0.4mmol)中の25% w/v ナトリウムメトキシドの溶液を加えた。約7時間後、反応を1M HCl(0.5mL)、10%のPd/C(0.2g)、および水(10mL)で停止した。混合物を24時間、水素バルーン下で保持した。この混合物をセライトでろ過し、EtOAc(20mL)で洗浄した。溶液を凍結乾燥して、白色固体であるN−ベンジルオキシカルボニル−アミノプロピル6−O−([α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジル−ホスホリル−α−D−マンノシル])−α―D−マンノシド45を得た(0.23g、44から74.7%)。
【0214】
DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.375g,1.14mmol)の溶液およびDMSO(1mL)中のDHBT(0.1g、0.6mmol)の溶液を水/DMSO1:1(10mL)中の45(0.23g、0.3mmol)の溶液に加えた。24時間後、溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥した。生成物を、DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.375g、1.14mmol)の溶液およびDMSO(1mL)中のDHBT(0.1g、0.6mmol)の溶液を用いて、水/DMSO 1:1(10mL)中で再アシル化した。24時間後、この溶液をsephadexサイズ排除樹脂で精製し、画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノオキシアセトアミドプロピル6−O−([α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−α−D−マンノシド46を得た(0.11g、37.5%)。化合物46をTFA/DCM(8mL)に溶解させ、追加した。この溶液を60分間撹拌し、その後油状物になるまで濃縮した。水(5mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択された画分をまとめ、凍結乾燥して、47を得た(0.07g、70.7%)。
【0215】
実施例5:β−結合型六糖の合成
A.メチルクロトニル3,4,6−トリ−O−ベンジル−β−D−マンノシド(51)
【0216】
【化37】
【0217】
2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノースを、Mayerら,Eur J.Org.Chem.10:2563(1999)に記述の通りに製造した。メタノール(0.5mL)中の25%w/vナトリウムメトキシドを無水メタノール(50mL)中の2−O−アセチル−3,4,6−トリ−O−ベンジル−α−D−マンノース(8.0g、23.3mmol)の溶液に加えた。約2時間後、薄層クロマトグラフィー(TLC)で示されたように、出発物質が消費された。この反応を、Amberlite IR120(H+)樹脂で停止し、ろ過し、シロップ状になるまで濃縮して、3,4,6−トリ−O−ベンジル−α−D−マンノース50を得た(6.67g、99%)。化合物50にトルエン(150mL)、続いてジブチルスズオキシド(3.88g、16.28mmol)を加え、Dean−Starkコンデンサを用いながら混合物を還流下で3時間加熱した。得られた溶液を冷却し、シロップ状になるまで濃縮し、乾燥DMF(50mL)に溶解させた。フッ化セシウム(2.28g、12.1mmol)、テトラブチルアンモニウムヨージド(5.47g、14.8mmol)、および4−ブロモクロトン酸メチル(2.46mL、22.2mmol、工業用グレード)を、混合物に加え、約60℃で18時間加熱した。混合物を放冷し、ろ過して固体を取り除いた。イソプロピルエーテル/EtOAc 3.7:1(380mL)で希釈し、半飽和のチオ硫酸ナトリウム(240mL)で洗浄した。水相をイソプロピルエーテル/EtOAc 3.7:1(2x190mL)で抽出し、有機層を合わせて濃縮した。シロップをイソプロパノール(2x25mL)と共に留去し、0〜50%EtOAc/ヘキサンを用いて、フラッシュカラムクロマトグラフィーで精製し、51をシロップ状として得た(4.58g、56.4%)。13C−NMR(100MHz)1J1C,1H(100MHz)、157Hz(β<160Hz、α>170Hz)
【0218】
B.メチルブチリル3,4,6−トリ−O−ベンジル−β−D−マンノシド(52)
化合物50を実施例2に記載の通りに製造した。化合物50(9.4g、21mmol)にトルエン(200mL)、続いてジブチルスズオキシド(5.49g、22mmol)を添加し、Dean−Starkコンデンサを用いながら、混合物を還流下で23時間加熱した。得られた溶液を冷却し、シロップ状になるまで濃縮し、乾燥DMF(100mL)に溶解させた。4−ブロモ酪酸メチル(4.23mL、32mmol)、テトラブチルアンモニウムヨージド(1.94g、5.25mmol)、フッ化セシウム(3.91g、25.5mmol)を添加し、混合物を60℃で2時間加熱し、その後18時間常温とした。混合物を放冷し、セライトでろ過し、EtOAc(50mL)で洗浄した。ゴム状物質になるまで濃縮し、トルエン(3x40mL)と共に留去し、シリカに吸着させ、0〜70%EtOAc/ヘキサンを用いて、フラッシュクロマトグラフィーで精製して、52を油状物として得た(9.11g、78.6%)。13C−NMR(100MHz)1J1C,1H(100MHz)、157.2Hz(β<160Hz、α>170Hz)。α−結合の生成物は観察されなかった。
【0219】
【化38】
【0220】
C.メチルブチリル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド(57)
化合物52を実施例2に記述の通りに製造し、6−O−アセチル−3,4,6−トリ−O−ベンゾイル−α−D−マンノーストリクロロアセトイミデート19を、Hengら,J.Carb.Chem.20:285(2001)の方法に従って製造した。ドナー19(4.29g、6.36mmol)およびアクセプター52(2.9g、5.3mmol)を、実施例1の一般的なグリコシル化手順に従って変換して、メチルブチリル2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド53を白色泡状物質として得た(4.41g、78.5%)。化合物53(4.41g、4.1mmol)を実施例1に記述された、1級アセテートを酸触媒脱アセチル化する一般的な手順に従って変換して、メチルブチリル2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド54を白色泡状物質として得た(2.25g、53.7%)。化合物54(2.25g、2.2mmol)を実施例1の一般的なリン酸化手順に従って変換して、メチルブチリル2−O−[2,3,4−トリ−O−ベンゾイル−6−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド55を白色泡状物質として得た(2.2g、77.7%)。
【0221】
【化39】
【0222】
55を脱保護するため、メタノール(0.2mL)中の25% w/v ナトリウムメトキシドの溶液を、無水メタノール(20mL)中の55(2.2g、1.7mmol)の溶液に加えた。約24時間後、出発物質が消費されたことをTLCが示した。反応をAmberlite IR120(H+)で停止し、シロップ状になるまで濃縮した。溶液を濃縮し、シリカゲルフラッシュカラムクロマトグラフィーで精製して、メチルブチリル2−O−[6−O−ジベンジルホスホリル−α−D−マンノシル]−3,4,6−トリ−O−ベンジル−β−D−マンノシド56を白色泡状物質として得た(1.10g、67%)。
【0223】
THF/水(20mL)中の56(1.10g、1.15mmol)の撹拌溶液に、氷酢酸(25μL)を加え、10%湿Pd/C(0.1g)および水素バルーンを取り付けた。24時間反応後、生成物を5%硫酸/EtOHで炭化したが、UV下では見えなかった。混合物をセライトでろ過し、パッドを水(20mL)で洗浄した。溶液を濃縮し、真空下で乾燥して、メチルブチリル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド57を得た(0.6g、98%)。
【0224】
【化40】
【0225】
D.アミノオキシアセトアミドヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド(60)
ヒドラジン一水和物(0.44mL、5.75mmol)をメタノール(20mL)中の57(0.6g、1.15mmol)の溶液に撹拌しながら加えた。30分後、水(5mL)を加え、この溶液を18時間撹拌した。さらにヒドラジン(0.44mL、5.75mmol)を加えて、混合物を120時間撹拌した。この溶液を約25%の容積まで濃縮し、水(2x10mL)と共に留去し、生成物を凍結乾燥して、ヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド58を、オフ−ホワイトの固体として得た(0.6g、99%)。
【0226】
DMSO/水 1:1(10mL)中の58(0.2g、0.38mmol)に、DMSO(2mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(0.49g、1.52mmol)の溶液およびDMSO(2mL)中のDHBT(0.125g、0.76mmol)の溶液を加えた。18時間後、この溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノ−オキシアセトアミドヒドラジドブチル2−O−[6−O−ホスホリル−α−D−マンノシル]−β−D−マンノシド59をオフ−ホワイトの固体として得た(0.1g、37.8%)。化合物59をTFA/DCM1:1(8mL)に溶解させた。この溶液を約60分間撹拌し、その後油状物になるまで濃縮した。水(10mL)を加えて、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、60をオフ−ホワイトの固体として得た(0.045g、52.5%)。
【0227】
【化41】
【0228】
E.メチルブチリル3−O−アリル−6−O−トリチル−β−D−マンノシド(64)の大規模合成
100L CH2Cl2中の50.0kg(128.09mol)d−マンノースペンタアセテートの溶液を26.8kg(243.2mol、1.90eq.)チオフェノールおよび27.3kg(192.3mol、1.50eq.)三フッ化ホウ素ジエチルエーテルで処理し、得られた溶液を22℃で40時間撹拌し、HPLC分析によって、反応が完了したことを判断した。その後115Lの5N NaOH水を、撹拌した反応容器に注意深く入れ、分離した層および有機層を46Lの5N NaOHでもう一度洗浄した。CH2Cl2を蒸留により減圧下で除去し、残渣を60℃で100kgのイソプロパノールに再溶解させた。9℃まで冷却し、生成物F1が結晶化し、それをろ過して単離し、続いてイソプロパノールで洗浄して、35.8kgを得た(63%)。
【0229】
35.80kgのF1(88.28mol)を、143kgのMeOHに22℃で懸濁し、0.73kgの、30%ナトリウムメトキシドのメタノール溶液(4.05mol、0.046eq.)で処理し、透明な溶液を得た。TLC分析により、反応が完了したことを判断されたのち、0.49kgの酢酸(8.14mol、0.09eq.)を加え、溶媒を減圧下で除去した。残渣をトルエンに懸濁し、再び減圧下で濃縮し、最後にアセトンで処理し、生成物であるフェニル−α−D−チオマンノシドを結晶化した。ろ過後、洗浄および乾燥すると、19.65kgの収量(89%)で得られた。
【0230】
ピリジン(43.8kg)中のフェニル−α−D−チオマンノシド(19.25kg、70.69mol)の溶液をトルエン(89kg)中のトリフェニルメチルクロリド(19.7kg、70.66mol)の溶液に40℃で加え、22時間撹拌した。HPLC分析により反応が完了したことが判断された後、溶媒を減圧下で取り除き、残渣をトルエンに溶解させ、再濃縮した。さらにトルエンで希釈した後、溶液を水で1度洗浄した。ヘキサン(840L)およびジイソプロピルエーテル(250L)の混合物にこのトルエン溶液を加えて、生成物を沈殿させ、ろ過および乾燥すると、フェニル6−O−トリチル−1−チオ−α−D−マンノシド61が得られた(32.30kg、89%)。
【0231】
【化42】
【0232】
トルエン(500kg)中の61(30.0kg、58.29mol)およびジブチルスズオキシド(20.3kg、81.5mol)の混合物を還流下で、2時間、濃縮した溶媒の蒸気から水が分離しなくなるまで加熱した。この溶液を40℃に冷却し、DMF(34kg)を加えた。全溶媒のほぼ半分を減圧下で除去し、DMF(216kg)を加え、この溶液をほぼ半分の容量になるまで再び濃縮した。さらにDMF(250kg)を加え、続いて、フッ化セシウム(8.9kg、58.59mol)、DMF(65kg)中のテトラブチルアンモニウムヨージド(23.6kg、63.89mol)の溶液、およびアリルブロミド(21.1kg、174.4mol)を加えた。得られた混合物を50℃で15時間撹拌した。HPLC分析により、反応が完了したことが判断された後、ろ過によって反応混合物から固体を取り除き、ろ液をジイソプロピルエーテル(136kg)および酢酸エチル(30kg)の混合物、続いて、10% w/w チオ硫酸ナトリウム水溶液(300kg)で処理した。相を分離した後、下相を4回ジイソプロピルエーテル(136kg)および酢酸エチル(30kg)の混合物で再抽出し、まとめた上相を水(150kg)で3回洗浄した。上相を減圧下で濃縮し、残渣を75℃でエタノール(160kg)に溶解させた。0℃に冷却すると、生成物は結晶化し、ろ過で単離することができた。フィルターの固形物を洗浄し、乾燥させた後、フェニル3−O−アリル−6−O−トリチル−1−チオ−α−D−マンノシド62を得た(15kg、46%)。
【0233】
【化43】
【0234】
THF(63kg)およびピリジン(18kg、227.5mol)の混合物中の62(12.5kg、22.53mol)の溶液を、水(10.7kg)中のトルエンスルホン酸一水和物(16.7kg、87.79mol)で処理し、続いてN−クロロスクシンイミド(9.6kg、71.89mol)の水(17kg)およびTHF(83kg)の混合物溶液で、15℃で処理した。得られた混合物を22℃に温め、3時間撹拌した。HPLC分析により、反応が完了したことが判断された後、水(15kg)中のチオ硫酸ナトリウム(4.6kg、29.11mol)の溶液を、反応混合物に加えた。相を分離し、上相を減圧下で濃縮し、残渣をトルエンに溶解させ、再び濃縮した。残渣を酢酸エチルに再溶解させ、水、その後16% w/w 塩化ナトリウム水溶液で洗浄した。酢酸エチルを留去後、粗生成物を52kg超のシリカゲルを用いたカラムクロマトグラフィーにより精製し、3〜10% v/v 酢酸エチル/トルエンのグラジエントで溶出して、3−O−アリル−6−トリチル−α−D−マンノース63を得た(7.7kg、73%)。
【0235】
【化44】
【0236】
メタノール(61kg)中の63(7.7kg、16.65mol)およびジブチルスズオキシド(4.56kg、18.32mol)の混合物を、濁った溶液が得られるまで、還流下で加熱し、その後さらに1時間加熱した。この溶液を25℃に冷却し、全溶媒のほぼ半分を減圧下で除去し、DMF(33kg)を加え、この溶液をほぼ半分の容量まで再び濃縮した。さらにDMF(15kg)を加え、続いてフッ化セシウム(2.53kg、16.65mol)、DMF(17kg)中のテトラブチルアンモニウムヨージド(6.15kg、16.65mol)、および4−ブロモ酪酸メチル(4.52kg、24.97mol)を加えた。得られた混合物を80℃で5時間撹拌した。固体を反応混合物からろ過で除去し、ろ液をジイソプロピルエーテル(41kg)、酢酸エチル(14kg)、10% w/w チオ硫酸ナトリウム水溶液(77kg)の混合物で処理した。相の分離後、下相をジイソプロピルエーテル(41kg)および酢酸エチル(51kg)の混合物で再抽出し、まとめた上相を水(39kg)で洗浄した。上相を減圧下で濃縮して、残渣をメタノール(35kg)に溶解させた。
【0237】
反応収率を改善するため、結合反応のプロセスを繰り返した。溶解した残渣を再び減圧下で濃縮し、その後メタノール(122kg)で希釈した。メタノール(60L)を再び留去し、得られた溶液をジブチルスズオキシド(2.28kg、9.16mol)で処理し、混合物を2時間、還流下で加熱した。溶液を29℃に冷却し、全溶媒のほぼ半分を減圧下で留去し、DMF(37kg)を加えて、溶液を再び容量のおよそ半分まで濃縮した。さらにDMF(15kg)を加え、続いてフッ化セシウム(1.26kg、8.29mol)、DMF(17kg)中のテトラブチルアンモニウムヨージド(3.7kg、10.02mol)の溶液、および4−ブロモ酪酸メチル(3.06kg、16.90mol)を加えて、得られた混合物を80℃で2時間撹拌した。HPLC分析により反応が完了したことを判断した後、固体をろ過によって反応混合物から除去し、ろ液をジイソプロピルエーテル(25kg)、酢酸エチル(32kg)、および10% w/w チオ硫酸ナトリウム水溶液(77kg)の混合物で処理した。相の分離後、下相をジイソプロピルエーテル(25kg)および酢酸エチル(32kg)の混合物で再抽出し、まとめた上相を水(39kg)で洗浄した。溶液を減圧下で濃縮し、残渣をトルエン(43kg)に再溶解し、最終的にほぼ30Lの容量まで濃縮した。その後、64の粗メチルエステルを、50kgのシリカゲルを用いたカラムクロマトグラフィーにより精製し、5〜30% v/v 酢酸エチル/トルエンのグラジエントで溶出した。
【0238】
精製したメチルエステルのメタノール溶液(50kg)を、30% w/w 水酸化ナトリウム水溶液(3.29kg)およびメタノール(7.7kg)の混合物で処理し、得られた溶液を14時間撹拌した。HPLC分析により、反応が完了していることを判断し、反応混合物をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物、続いて水(77kg)で処理した。二相混合物を1.2μmのフィルターカートリッジに通し、相を分離した。下相をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物で処理し、下相のpHを、5% w/w クエン酸水溶液(38L)を添加して4.5〜5まで下げた。相を分離し、下相をジイソプロピルエーテル(41kg)および酢酸エチル(14kg)の混合物で抽出した。まとめた上相を水(39kg)で洗浄し、その後減圧下で濃縮した。残渣をジイソプロピルエーテル(39kg)と混合し、溶媒を部分的に濃縮して、約20Lの最終容量が得られ、生成物を結晶化し、ろ過により単離することができた。フィルターの固形物を洗浄し、乾燥した後、(3−O−アリル−6−O−トリチル−β−D−マンノシル)−4−ブタン酸64が得られた(4.7kg、51.5%)。
【0239】
【化45】
【0240】
F.四糖中間体(69)の大規模合成
化合物64をパートEの方法に従って製造した。64の保護基をさらなるグリコシル化反応の前に修飾した。THF(14kg)中の64(4.25kg、7.75mol)の溶液を、THF(45kg)中の60%水素化ナトリウム分散体(1.55kg、38.75mol)の撹拌スラリーに注意深く加え、得られた懸濁液を水素が発生しなくなるまで撹拌した。THF(2kg)中のテトラブチルアンモニウムヨージド(0.29g、0.78mol)の懸濁液、続いてベンジルブロミド(9.2kg、53.79mol)を、反応容器に入れた。混合物を22℃で46時間撹拌し、その後30℃、12時間、および35℃、48時間撹拌した。HPLC分析により反応が完了したことを確認した後、混合物を0℃に冷却し、無水メタノール(0.7kg、21.87mol)、続いて30% w/wの、ナトリウムメトキシド(2.1kg、11.66mol)のメタノール溶液を注意深く入れた。その後、酢酸(1.4kg)、続いてトリエチルアミン(9.4kg、92.89mol)を加え、混合物を18時間撹拌した。得られた懸濁液に、水(31kg)を加え、2相に分離した。上相を減圧下で濃縮し、残渣をトルエンに溶かし、再び最終容量が約20Lになるまで濃縮した。粗生成物を42kgのシリカゲルでカラムクロマトグラフィーにより精製し、5〜15%の酢酸エチル/ヘキサンのグラジエントで溶出して、メチルブチリル3−O−アリル−2,4−ジ−O−ベンジル−6−O−トリチル−β−D−マンノシド65を、酢酸エチル溶液として得た(4.4kg、77%)。
【0241】
【化46】
【0242】
37℃のメタノール(19kg)中の65(4.4kg、5.92mol)の溶液に、メタノール(6.3kg)中のトルエンスルホン酸一水和物(0.9kg、4.73mol)の溶液を加え、得られた混合物を1時間撹拌した。HPLC分析により、反応が完了したことを判断した後、トリエチルアミン(1.5kg、14.82mol)を入れ、この溶液を減圧下で濃縮した。その後、トルエン(28kg)を加え、溶液を水(32kg)で洗浄した。相を分離し、上相を減圧下で濃縮した。粗生成物をシリカゲル(32kg)を用いたシリカゲルクロマトグラフィーで、9%、その後17%、その後50% v/v 酢酸エチル/トルエンのグラジエントで溶出させて精製し、メチルブチリル3−O−アリル−2,4−ジ−O−ベンジル−β−D−マンノシド66を、トルエン溶液として得た(2.67kg、90%)。
【0243】
【化47】
【0244】
3.73kg(5.49mol、1.10eq.)の19および2.50kg(4.99mol)の66を34kgの乾燥トルエンに溶解させた。そのうち、約10Lを減圧下留去した。その後、溶液を0℃に冷却し、反応温度を<5℃に維持しつつ、22g(0.099mol、0.02eq.)のTMSOTfを滴加処理した。添加終了後、0℃で1時間撹拌した。HPLC分析により反応が完了したことを判断した後、混合物に30g(0.296mol、0.06eq.)のEt3Nを加えて中和した。ヘキサン(22L)を追加し、得られた懸濁液をろ過し、ろ液を33Lの水で洗浄し、減圧下で濃縮した。この残渣を10Lのトルエンに溶解させ、再度濃縮した。このプロセスを2回以上繰り返した。粗生成物のカラムクロマトグラフィー精製は、50kgシリカゲルで行い、9〜13% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、4.23kg、83%である化合物67を、トルエン溶液として得た。
【0245】
【化48】
【0246】
4.23kg(4.16mol)の67の、5.6LのCH2Cl2および40LのMeOH溶液に、0.72kgの10%Pd/Cを加え、続いて0.12kg(0.63mmol、0.15eq.)のトルエンスルホン酸一水和物溶液を加え、混合物を22℃で24時間撹拌した。HPLC分析により反応が完了したことを判断した後、パラジウム/炭素をろ過により除去し、さらに精製することなく、ろ液を次の工程で使用した。
【0247】
ろ液に4L MeOH中の2.97kg(15.6mol、3.75eq.)トルエンスルホン酸の溶液を加え、得られた混合物を16時間、22℃で撹拌した。HPLC分析により、反応が完了したことを判断した後、混合物を0℃に冷却し、1.64kg(16.2mol、3.89eq.)のトリエチルアミンを加えて中和した。この溶液を減圧下で濃縮し、残渣を82LのMTBEおよび32Lの水に分配した。有機相を減圧下で濃縮し、10Lのトルエンで希釈し、再濃縮した。この手順を2回以上繰り返した。粗生成物のカラムクロマトグラフィー精製を、38kgシリカゲルで行い、23〜26% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、2.59kgの化合物68をトルエン溶液として得た。(67から67%)。
【0248】
【化49】
【0249】
2.90kg(3.10mol)の68および5.24kg(8.23mol、2.65eq.)の18の、30kg乾燥トルエン溶液を、0℃で、0.049kg(0.185mol、0.06eq.)のTBDMSOTfで処理し、0℃で4時間撹拌した。HPLC分析により反応が完了していることを判断した後、0.104kg(1.03mol、0.128eq.)のトリエチルアミン、続いて33Lのヘキサンを加えた。得られた懸濁液をろ過し、ろ液を28Lの水、その後28Lの5%Na2CO3水溶液で洗浄した。トルエン相を減圧下で濃縮し、粗生成物を58kgシリカゲルのカラムクロマトグラフィーで精製し、9〜20% v/v EtOAc/ヘキサン:トルエン1:1のグラジエントで溶出して、4.40kgの四糖中間体69をトルエン溶液として得た(75%)。
【0250】
【化50】
【0251】
G.保護六糖(73)の合成
四糖中間体69を、実施例7に記述された方法に従って製造した。9.7g(5.15mmol)の69の、60ml CH2Cl2溶液に、100mlのMeOHを加え、続いて、混合物の温度を30℃未満に抑えながら、5.7N HClの1,4−ジオキサン溶液を27ml(0.154mol、30eq.)滴加した。その後、反応混合物を22℃で40時間撹拌した。HPLC分析により反応が完了したことを判断した後、混合物の温度が25℃未満に維持されるように、32ml(22.9mmol、44.7eq.)のトリエチルアミンを慎重に加えた。水(250ml)およびトルエン(200ml)を加え、混合物を振とうし、相を分離した。下相を50mlトルエンで再抽出し、まとめた上相を50mlの水で洗浄し、減圧下で濃縮した。粗生成物を100gのシリカゲルを用いてカラムクロマトグラフィー精製し、30〜50% v/v EtOAc/ヘキサンのグラジエントで溶出して、メチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル])−2,4−ジ−O−ベンジル−β−D−マンノシド70を得た(6.35g、69%)。
【0252】
【化51】
【0253】
1.0g(0.56mmol)の70および0.95g(1.40mmol、2.5eq.)の19の、9ml乾燥トルエン溶液を、0℃で、0.02g(0.075mmol、0.14eq.)のTBDMSOTfで処理し、0℃で1時間撹拌した。HPLC分析で反応が完了したことを判断した後、22ml(0.158mmol、0.28eq.)のトリエチルアミンを加えた。得られた混合物を、10mlの水で2度洗浄し、減圧下で濃縮した。15gシリカゲルのカラムクロマトグラフィーにより、粗生成物の精製を行い、15〜25% v/v EtOAc/ヘキサン:トルエン 1:1のグラジエントで溶出して、1.75gの化合物メチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−アセチル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド71を、残留トルエンを含む油状物として得た。
【0254】
【化52】
【0255】
10.0g(3.5mmol)の71の、40mL 1,4−ジオキサン溶液を、60mlのMeOHで処理し、その後3.25g(14.0mmol、4eq.)の(+)−カンファースルホン酸で処理し、得られた溶液を100時間、22℃で撹拌した。反応の終了をHPLC分析で判断した後、3ml(21.5mmol、6.2eq.)のトリエチルアミンを加え、溶媒を減圧下で除去した。残渣を200mlのMTBEに溶解させ、200mlのH2Oと共に振とうした。相を分離し、上相を減圧下で濃縮した。粗生成物のクロマトグラフィー精製は、114gのシリカゲルを用い、14〜25% v/v EtOAc/トルエンのグラジエントで溶出して、8.24gのメチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンゾイル−α−D−マンノシル]−2−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド72を、残留トルエンを含む油状物として得た。
【0256】
【化53】
【0257】
0.965g(0.35mmol)の72の、4g乾燥アセトニトリル溶液に、0.083g(0.70mmol、2eq.)の4,5−ジシアノイミダゾールを加え、続いて0.315g(0.91mmol、2.6eq.)のジベンジルジイソプロピルホスホルアミダイトを加え、混合物を23℃で1時間撹拌した。反応の終了をTLC分析で判断した後、0.5mlの水を加え、この溶液を15分撹拌した。水(9.5ml)および10mlのMTBEを加え、得られた混合物を振とうし、相を分離し、下相を10mlのMTBEと振とうした。上相をまとめて、減圧下で濃縮して、無色の油状物を得た。この残渣をCH2Cl2に溶解させ、−20℃に冷却し、0.247g(1.13mmol、3.2eq.)の70% 3−クロロ過安息香酸で処理した。TLC分析で反応の終了を判断した後、10mlの10%チオ硫酸ナトリウム水溶液を加え、混合物を23℃まで昇温した。下相を分離し、10mlの水と振とうし、減圧下で濃縮した。粗生成物を19gのシリカゲルでカラムクロマトグラフィーにより精製し、25〜50% v/v EtOAc/ヘキサンのグラジエントで溶出して、0.83g(72%)のメチルブチリル3−O−([3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−(6−O−[2,3,4−トリ−O−ベンゾイル−α−D−マンノシル]−6−O−[3,4,6−トリ−O−ベンジル−α−D−マンノシル]−2−O−[6−O−ジベンジルホスホリル−2,3,4−トリ−O−ベンゾイル−α−D−マンノシル])−2,4−ジ−O−ベンゾイル−β−D−マンノシド73を、無色の油状物として得た。
【0258】
【化54】
【0259】
H. アミノオキシアセトアミドヒドラジド 3−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−(6−O−[α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−β−D−マンノシド(77)の合成
化合物73を、実施例8に記述された方法に従って製造した。氷酢酸(100μL)をメタノール/THF 1:1(600mL)中の73(64g、19.6mmol)に加え、生成物をH−cube(登録商標)を用いて、20%Pd(OH)2/Cを使用し、50℃、H2圧50bar、および流速6mL/分で触媒上を再循環させて水素化した。20時間後に反応の完了をTLCで確認し、この溶液を濃縮して、74を泡状物質として得た(39.88g、93%)。
【0260】
【化55】
【0261】
メタノール(180mL)を74(33.8g、15.5mmol)に、溶解するまで撹拌しながら加え、この溶液を氷/水槽で15分間冷却した。この溶液に64%ヒドラジン一水和物(94ml、1.24mol)を、撹拌しながら加えた。30分後、水(120mL)を加え、この溶液を室温になるまで放置し、18時間保存した。この溶液を約100mLまで濃縮し、水と共に留去し(2x100mL)、最終溶液を水で約180mLに調節した。この溶液をDCM(2x100mL)で抽出し、その後3分割した60mLをsephadexサイズ排除カラムで分離した。もっとも純粋な物質を含む画分をまとめ、凍結乾燥して、75を得た(15.5g、80%)。
【0262】
【化56】
【0263】
DMSO(20mL)を水(30mL)中の75(2.5g、2.0mmol)にゆっくり加え、その後DMSO(6mL)中のN−t−ブトキシカルボニルアミノオキシアセチル2,3,5,6−テトラフルオロフェニレート(2.58g、7.6mmol)およびDMSO(4mL)中のDHBT(0.65g、4mmol)を加えた。18時間後、溶液をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、N−t−ブトキシカルボニルアミノオキシアセトアミドヒドラジドブチリル3−O−([α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−(6−O−[α−D−マンノシル]−6−O−[α−D−マンノシル]−2−O−[6−O−ホスホリル−α−D−マンノシル])−β−D−マンノシド76(2.57g、90%)を得た。DCM(30mL)、次にTFA(16mL)を化合物76(2.57g、1.8mmol)に加えた。混合物が溶解するまで(約60分)撹拌し、その後油状物になるまで濃縮した。水(20mL)を加え、生成物をsephadexサイズ排除樹脂で精製した。シリカゲルプレート上で炭化させることにより画分を確認し、選択した画分をまとめ、凍結乾燥して、77を得た(1.6g、67.1%)。
【0264】
【化57】
【0265】
実施例6:ジスルフィドリンカーを有する六糖の合成
A.遊離酸形態の六糖の製造
無水MeOHを化合物73に加え、続いてNaOMeを加え、4〜18時間インキュベートした。反応を氷酢酸で停止し、この溶液をシロップ状になるまで濃縮して、78を得た。
【0266】
【化58】
【0267】
化合物78をTHF/メタノール1:1に溶解し、Pd/C−H2で水素化した。溶液を固体になるまで濃縮し、水に溶解させ、NaOH水溶液でけん化した。pHを約4に調整し、Sephadex G−10で精製して、遊離酸81を得た。
【0268】
【化59】
【0269】
B.ジスルフィドリンカーの結合
異なる方法で製造された(Biomiraから入手された)化合物81の粗画分を、過剰なトリエチルアミン(TEA)と混合することで、TEA塩に変換し、続いて、移動相として30%アセトニトリル、0.1%ジカルボン酸TEAを用いて、Superdex Peptide(GEヘルスケア)でクロマトグラフィーを行った。まとめた画分を凍結乾燥し、グリカン:NEA:EDAC:NHS:HOBt:TEA(1:1:1.5:1:1:1 mol:mol)を含む反応中で、NEAと結合させ、軽く撹拌しながら一晩インキュベートした。一部の生成物(0.5mg)を、前回のようにSuperdex Peptideでクロマトグラフィーを行い、凍結乾燥して82を得た(0.28mg)。
【0270】
【化60】
【0271】
実施例7:α−グルコシダーゼ複合体の合成および酸化の最適化
A.複合体化
オリゴ糖を組換えヒト酸性α−グルコシダーゼ(rhGAA)と結合させて、NeoGAAを形成した。主にr^hGAAのシアル酸残基を介して結合したオリゴ糖との複合体を「SAM」とし、ガラクトース残基を介して結合したものを「GAM」とする。
【0272】
NeoGAAβSAM6を、Zhuら,Biochem J,389(Pt3):619−628(2005)に記述されたように、基本的に製造した。この実験で使用するrhGAAのサンプルを、タンパク質1モルにつき、シアル酸を約5.2モル有するように、単糖組成物分析により見出した。簡潔に述べると、5mg/mLのrhGAA(Genzyme Corp.)を100mMの酢酸ナトリウムpH5.6de緩衝液交換し、その後過ヨウ素酸ナトリウム(2、7.5、または22.5mM)と、氷上で、遮光で30分反応させた。反応を2%(vol/vol)になるように、グリセロールを添加することで停止した。酸化反応から小さい分子量の副生物を取り除くため、酸化rhGAAを緩衝液交換し、化合物77(図9に示されるように、タンパク質に対して0〜120倍のモル比)と37℃で6時間複合体化させた。すべての複合体を25mMリン酸ナトリウムpH6.25に緩衝液交換した。これは2%のマンニトールおよび0.005%のTween−80を含む。
【0273】
類似のNeoGAA複合体を、SAM2(化合物17、実施例2)、SAM3(化合物35、実施例3)、SAM4(化合物28、実施例4A)、直鎖SAM4(化合物47、実施例4B)、およびαSAM6(オリゴ糖103)を用いて、7.5mMの過ヨウ素酸塩を使用し、オリゴ糖とrhGAAのモル比を変えることで製造した。
【0274】
別の複合体化方法も実施した。具体的には、アミノキシ、ヒドラジドまたはチオ−ル反応性リンカーのいずれかを有する六糖を、Cys374、リシン、シアル酸、またはガラクトース残基を介して、rhGAAに結合させた。
【0275】
rhGAAのリシン残基を4−ホルミル安息香酸スクシンイミジル(SFB;Solulink Corp.)で修飾し、続いてオリゴ糖と複合体化させることにより、リシンの複合体化を実施した。簡潔に述べれば、rhGAAをまず、150mMの塩化ナトリウムを含む50mMリン酸ナトリウムpH7.2に緩衝液交換した。その後、SFBとGAAとのモル比を20:1として新たに製造された(SFB)で、緩衝化されたrhGAAを処理した。当該混合物を室温で30分インキュベートし、それを100mMの酢酸ナトリウムpH5.5に緩衝液交換し、室温、2時間でヒドラジド六糖へと複合体化させたか、またはSFBで修飾されたGAAを、100mMの酢酸ナトリウムpH5.6に緩衝液交換し、37℃、6時間で、アミノキシ六糖へ複合体化させた。
【0276】
システイン系の複合体化を、チオール反応性NEA−六糖82(実施例6I)と反応させることで実施した。NEA−修飾された六糖82を、水中で再構成し、50mMリン酸ナトリウムおよび50mMヒドロキシアミンpH7.2中で、2時間25℃で、rhGAAと(rhGAAに対するネオグリカンのモル比15:1)インキュベートした。50mMのリン酸ナトリウムpH4.1で、pHを6.2に調整し、インキュベーションを一晩続けた。生成物を25mMリン酸ナトリウムpH6.2に対する遠心ダイアフィルトレーション(centrifugal diafiltration)によって精製した。1molに対して1mol未満のM6Pが導入された。
【0277】
Cys374を介する直接的な複合体化は成功しなかったが、ホモ二官能基性のチオ−ル特異的試薬である、19.9Åのスペーサーアームを有する1,4−ジ−(3’−[2−ピリジルジチオ]−プロピオンアミド)ブタン(DPDPB)が試験され、オリゴ糖との複合体化の前に、溶媒接近がより可能な374位のチオ−ル基を提供した。60倍モル過剰のDPDPBを、共溶媒として10%DMSOまたは10%プロパノールのいずれかの存在下で、rhGAAと反応させた。これは、光散乱によって検出されるように、強い凝集を引き起こした。20%アセトニトリルの存在下での反応はまた、凝集を示したが、反応混合物の限外ろ過液の344nmにおける吸光度は、システインの定量的修飾と一致した。10%までのアセトニトリル濃度の減少は、凝集量を減少させたが、修飾の程度は低かった。
【0278】
別のチオ−ルに基づいた方法を、リシン残基のチオ−ル基の導入により実施した。4時間25℃、リン酸ナトリウムpH6.2中で100倍モル過剰のSATA−dPEG4−NHS(Quanta Biodesign)と酵素反応させることにより、リシン残基に保護チオ−ルが導入された。そしてそれを、同じ緩衝液に対する一晩の透析により精製した。次に、精製された生成物を、システインを基にした複合体に関して、上記で記述された条件下で、NEA−オリゴ糖82と反応させて、リシン−チオ−ル複合体を得た。これは、Man−6P含有量の約10倍の増加を示した(約5個のグリカンが複合体化された)。
【0279】
ヒドラジドとのリシン複合体の安定性は、無傷のタンパク分子量およびM6P含有量を測定することで、37℃、14日まで評価された。50%を超えるネオグリカンが14日間で消失したように、この複合体は安定ではない。リシンを介したアミノキシ複合体を、上記のように、rhGAAに対して0、16.6、25、33および40モル過剰の六糖を用いて製造した。全リシンの約31%だけが複合体化(または5個のネオグリカンが複合体化)されたが、当該複合体化は、16.6倍モル過剰で飽和した。高い凝集レベルが、数種の調製物においても観察された。PEG化されたSFBを試験したが、凝集の減少は見られなかった。
【0280】
2%マンニトールおよび0.005%Tween−80を含む、25mMリン酸ナトリウム、pH6.25中で、37℃で6時間、Clostridium perfringens由来のシアリダーゼ20mU/mgで、rhGAAをまず前処理することにより、ガラクトース複合体化(GAM)を実施した。ジシアリル化後、タンパク質を同一緩衝液中、37℃で一晩、1〜10μg/mgのガラクトースオキシダーゼ(GAO)および2U/mgのカタラーゼ(Sigma)で処理した。その後、Poros50D(陰イオン交換)クロマトグラフィーを用いて再精製して、ノイラミニダーゼおよびカタラーゼを除去した。両方の酵素で処理した生成物を、等量のdH2Oで希釈し、その後、10mMのリン酸ナトリウム緩衝液、pH6.9で事前に平衡状態にあるPoros50Dカラムにアプライした。カラムを10mMの酢酸ナトリウム緩衝液、pH5.0で洗浄し、rhGAAを150mM酢酸ナトリウム緩衝液、pH5.0で溶出させ、そして37℃で6時間、様々なモル比でアミノキシ六糖と複合体化させた。
【0281】
GAM複合体化は、GAAに対して16.6倍モル過剰の六糖で飽和し、約6〜7個のグリカンが複合体化された。凝集レベルは低かった。脱シアリル化の後、シアル酸は検出されなかった。一方、ガラクトースオキシダーゼ処理後にガラクトースもほとんど検出されなかった。いくつかの実例において、20〜30%のガラクトース残基が過剰酸化され、オリゴ糖に結合していないガラクツロン酸を生成した。GAOを滴定すると、1μg/mg超でGAOはグリカン複合体を減少させることを示した。GAOが2μg/mgGAOより多いと、ガラクツロン酸の過剰酸化産物量の明らかな増加がみられた。複合体の最大量は、rhGAA1mgにつき、1〜2μgGAOで実現した(図10E−GAOの滴定後における、GAM複合体の、Man−6P、Gal、GalAを含む単糖の含有量)。GAA 1mgにつき、GAOを0.5〜2μg使用すると、多くの複合体化が、タンパクのMan−6P含有量として観察された。より少ないまたはより多くのGAOを使用すると、より少ないガラクトース、またはより多いGalAのいずれかが生じた。
【0282】
NeoGAAに複合体化したビス−M6P六糖グリカンの量を、M6P含有量分析およびMALDI−TOFにより定量した。M6Pの定量に関して、サンプルをAmicon4、50,000MWCO遠心式フィルターユニットを用いて、5回ろ過を行って、緩衝液交換し、潜在的に過剰のグリカンを除去した。各80マイクログラムのrhGAAまたはNeoGAAサンプルを、6.75MのTFAで1時間半、100℃で加水分解した。サンプルを冷却し、Speed Vacで乾燥させ、200μLの蒸留水で再構成した。再構成したサンプルを再び、Speed Vacで乾燥させ、200μLの50mMクエン酸pH2.0で再構成した。サンプルをS Mini Hカートリッジ(Sartorius)でろ過し、これをクエン酸ナトリウムpH2.0で平衡化して、加水分解物から不純物を除去した。リボース−5−リン酸を、内部標準として、すべてのサンプルおよび標準に追加した。50μLの加水分解物をDionex HPLCに注入し、パルスアンペロメトリック検出機付き高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)によって分析した。加水分解した標準のM6Pで構築した、検量線を用いて定量を行った。その後、複合体化の程度を、グリカン1モルあたり、M6P 2モルの公知のモル比を基に計算した。
【0283】
MALDI−TOF MS分析を、直列モードでVoyagerDE−PRO質量分析計を用いて実施した。0.1%ギ酸水溶液に1:5で希釈し、すべてのサンプルおよび標準で実施し、続いて飽和シナピン酸の50%アセトニトリル/0.1%TFA溶液に1:1で希釈して行った。この混合物の1μLを標的に利用した。サンプル、レファレンス、BSA補正コントロールの3通りで分析した。二点校正を(M+H)+およびBSAの二量体イオンを用いて行った。各NeoGAAサンプルの複合体化の程度は、測定されたグリカン分子量が1323g/モルであることを考慮して、サンプルおよび酸化rhGAAコントロール(グリカンは追加されていない)間の分子量の違いを基に計算した。
【0284】
図9Aは、上記の二糖、三糖、四糖、および六糖複合体を用いた実験の結果を示す。
【0285】
図9Bは、異なる量の過ヨウ素酸塩で製造されたβSAM6複合体の結果を提供する。複合体化反応の飽和を実現するのに必要なオリゴ糖レベルは、酸化工程中に使用される過ヨウ素酸塩の量に比例した(図9B、上のパネル)。2mM過ヨウ素酸塩を用いて、シアル酸約5.2モルを有するrhGAAのサンプルは、タンパク質に対してほぼ25倍モル過剰の六糖で(シアル酸に対して4.8倍モル過剰で)飽和に達した。7.5mMの過ヨウ素酸塩を用いて、グリカンの33倍モル過剰で飽和に達した。グリカンの120倍モル過剰では、飽和に達したが、22.5mMの過ヨウ素酸塩で酸化したrhGAAでは実現しなかった。実現した複合体化の最大レベルは、異なるレベルの過ヨウ素酸塩で製造したサンプルでも異なった。7.5および22.5mMの過ヨウ素酸塩を用いて、タンパク質1モルにつき、それぞれ約8.5および10.5モルのグリカンが組み込まれた。2mM過ヨウ素酸塩での酸化の後、実現可能な複合体化レベルは、タンパク質1モルにつき約5モルのグリカンであり、これは出発物質中のシアル酸残基の数に近い。
【0286】
グリカンの滴定実験を、タンパク質1モルにつき、最初のシアル酸レベルが約7.2モルであるrhGAAを用いて、2mM過ヨウ素酸塩で繰り返した(図9B、下のパネル)。タンパク質1モルにつき、約7モルのグリカンの複合体化レベルは、タンパク質に対して33倍モル過剰以上のグリカンで達成された(シアル酸と比較して、約4.6倍モル過剰)。
【0287】
B.凝集の減少
特定の複合体化方法は、タンパク質の凝集を引き起こす。neoGAAにおいて、凝集を減少させるための2つの方法が開発された。1)様々なHICクロマトグラフィー担体を用いた、疎水性相互作用クロマトグラフィー(HIC)、および2)金属キレート化である。
【0288】
NeoGAAの3gのバッチを調製し、凝集除去に関して、HICおよび銅カラムを評価するために使用した。フロースルーモードで評価したHICカラムは:Butyl650Cおよび650M、Hexyl650C、Phenyl6FF、Capto Octyl、及びCapto Phenylであった。HexylおよびCapto Phenylで、87.5%および90.4%の回収率、並びにそれぞれ3.2%(初期レベル)から1.4%、および3.9%(初期レベル)から1.6%までの凝集減少という同等の結果を示した。表2を参照のこと。
【0289】
【表5】
【0290】
銅キレートカラム(GEまたはTosoh)の操作条件も、フロースルーまたは結合−溶出モードのどちらかで確立した。銅を充填した7mlの金属キレートFFカラム(I.D.、7ml)にまず、10mg/mlの複合体化したGAAをロードして、結合・溶出モードで評価した。室温で、溶出緩衝液として175mMグリシン、100mM酢酸、pH5.5でカラムから溶出されたとき、87%のNeoGAAが回収され、凝集体は1.2%であった。満足な回収率でカラムから溶出されるために、8℃にて175mMより高いグリシンが必要であった。フロースルーモードにおいては(表3)、溶出緩衝液として、150mMグリシン、100mM酢酸塩、pH5.5を用いて、92%の優れた回収率が実現され、凝集体は3.2から1.2%に減少した。
【0291】
【表6】
【0292】
イミダゾール(7.5、8および10mM)も、金属キレート6FFカラムの溶出緩衝液として試された。カラムを溶出するためには、イミダゾールが約8mM必要であった。イミダゾールはカラムから銅を溶出しないため、カラムをコンディショニングする、またはカラムの上方にEDTAで空きスペースを作る必要がなかった。NeoGAA 1mlにつき15mgのカラム容量を達成した。
【0293】
銅を充填したTosoAF−キレート650Mカラムも評価した。結合・溶出モードにおいて、15mg/mlのカラム容量が達成され、8mMのグリシンを用いて、94.1%の溶出および1.2%の凝集体であった。フロースルーモードにおいて、33.6mg/mlの容量を実現した。この溶出緩衝液において、50mMのグリシンで90.6%の回収率、1.2%の凝集体であった。
【0294】
C.オリゴ糖の分析
これらの実験によれば、>2mMの過ヨウ素酸塩の使用で、タンパク質中の出発時レベルのシアル酸レベルを超えたNeoGAAグリカンの取り込みが起こり、これは非シアル酸部分が酸化されたことを示す。過ヨウ素酸塩による、他の炭水化物部位での酸化レベルを測定するため、一連の過ヨウ素酸塩の滴定実験を実施し、他の単糖残基のレベルを観察した。
【0295】
シアル酸含有量を測定するため、サンプルを0.5Mのギ酸を用いて、80℃で1時間、酸加水分解させた。放出されたシアル酸を50〜180mM酢酸ナトリウムグラジエントを用いて、100mM水酸化ナトリウム中で、20分以上、Dionex CarboPac PA1カラムで、パルスアンペロメトリック検出付きの、高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)によって分析した。結果を、rhGAAまたはNeoGAA 1モルにつき、シアル酸(NANAまたはNGNA)のモルとして表し、真正の商用シアル酸標準物の検量線から測定した。
【0296】
フコース、ガラクトース、GlcNAc、およびマンノースを含む、中性単糖のレベルを、110℃で2時間、1MのTFA中で、100μgのrhGAAまたはNeoGAAを加水分解することで測定した。加水分解に続いて、チューブを氷上で冷却し、1分10,000rpmで遠心分離し、Speed Vacにより蒸発乾固した。放出された単糖を250μLの水に再懸濁させ、ボルテックスし、Millipore Ultrafree−MCフィルターチューブを用いてろ過した(10,000MWCO)。放出された単糖を、CarboPacPA1カラムで、パルスアンペロメトリック検出付きの高pH陰イオン交換クロマトグラフィー(HPAEC−PAD)により分析した。定量は、加水分解した単糖の検量線を用いて、同様の方法で行った。
【0297】
上記の実験結果を図10Aに示す。結果から、シアル酸が最も酸化を受けやすい単糖であると示唆された。低レベルのフコース破壊は2mMの過ヨウ素酸塩で検出することができ、その量は5mM以上で測定および再現することができた。マンノースのわずかな酸化は、7.5mM以上の過ヨウ素酸塩でだけ検出することができた。
【0298】
酸化シアル酸、フコース、およびマンノースの存在を確認するため、フラグメンテーション質量分析を、7.5mM過ヨウ素酸塩を用いて酸化したrhGAAから放出されたオリゴ糖に関して実施した。rhGAAおよびNeoGAA中のN−結合型オリゴ糖を、50mMリン酸ナトリウムpH7.0および10mMB−メルカプトエタノール中で、PNGaseFを用いて、一晩かけて分解させて、放出させた。放出されたオリゴ糖を、水を変更しながら、生物的透析(MWCO500Da)により不要なものを取り除いた。透析したサンプルをspeed−vac濃縮機で乾燥し、10mMギ酸アンモニウムpH4.0の50%アセトニトリルおよび50%水の溶液の110μLで再構成した。サンプルをアセトニトリル−水グラジエントおよび10mMギ酸アンモニウムpH4.0で、インラインMS検出器(QStar quadrupole飛行時間型、LCT Premier飛行時間型、およびLTQ直線イオントラップ機)を用いた、TSKGelAmide−80カラム(2x100mmへ100μL注入、5μm粒子サイズ)を使って分析した。
【0299】
オリゴ糖構造を分析するため、オリゴ糖を乾燥し、アントラニル酸(AA)で蛍光標識した。AA標識したオリゴ糖を、アセトニトリル/水グラジエントを用いて、蛍光検出機を使用し、TSKゲルアミド80カラムで、順相HPLCにより分離した。MSフラグメンテーション分析を、陽イオンモードで、LTQ XL直線イオントラップマススペクトロメータを用いて、インラインで行った。スペクトルを400〜2,000m/zでスキャンし、衝突エネルギーは35に(初期設定値、本文に示されない限り)、activationQは0.25に合わせた。
【0300】
シアル酸:シアル酸の完全酸化(C7,8およびC8,9結合の両方で)は、質量で62ダルトンの減少をもたらす。フコースおよびマンノースの酸化は、最初に2ダルトン減少(C2,3またはC3,4結合の酸化)させ、続いて残りのビシナル・ジオールの酸化で、30ダルトン減少する。AAを有する炭酸水素アルデヒドの還元的アミノ化は、酸素の消失を招き、1AAを追加するにたびに、正味121.1ダルトンの分子量の増加が引き起こされる。シアル酸、フコース、およびマンノースを酸化後の、rhGAAオリゴ糖における、理論的な分子量および観察された分子量の変化を表4に示す。
【0301】
【表7】
【0302】
酸化およびAA−誘導型酸化シアル酸、フコース、およびマンノースオリゴ糖種に対応する様々なイオンが観測された。放出されたすべてのオリゴ糖の還元末端のGlcNAcでの誘導化に加えて、AAは存在する反応性アルデヒド種の数と比例して、つまり、酸化シアル酸とでは1:1、酸化マンノースおよびフコースでは2:1で誘導された。
【0303】
図10Bの質量スペクトルは、シアル酸のC7位の酸化およびAA誘導を有するrhGAAオリゴ糖に対応する4つのイオンの検出を示す。シアル酸の酸化に関する2つのイオン(m/z1057および1130)を、さらなるMS/MSフラグメンテーション分析のために選択した。m/z 1057のフラグメーションパターンを図10Cに示し、各イオンは仮定された物質が注釈されている。フラグメンテーションイオンm/z 716、1032、1235、1397、1584、および1747は、MS3分析から選択された。MS3スペクトルは、二分岐鎖グリカンの末端の酸化およびAA標識シアル酸を含む、仮定されたオリゴ糖構造と一致した。特に、m/z 351のフラグメントの放出により、シアル酸のC7型へのAAの結合を確認した。全サンプルの分析において、C7型の酸化されたシアル酸だけが観察された。C8位で酸化されたシアル酸の存在に関する証拠は観察されなかった。
【0304】
フコース酸化:表5にAA−誘導化、酸化A1FおよびA2Fの理論質量および観測質量を列挙する。
【0305】
【表8】
【0306】
m/z1250.4の親イオンのフラグメンテーションパターンを図10Dに示し、酸化シアル酸および酸化フコースを有する、AA標識、一シアル酸、一本鎖、フコシル化コアオリゴ糖で予測されたものと一致する。主なフラグメントイオンはm/z 716、1397、1621、および1783で観測された。これらのイオンは仮定された物質を確認するため、MS3分析のために選択された。m/z 1621および1783のMS3スペクトルにおいて、イオンフラグメンテーションパターンから、親イオンから195ダルトンが消失していることを確認した(それぞれ、親イオン1621および1783から1426および1588まで)。この分子量の変化は、酸化フコースのC1および酸素原子間の切断と一致し、これはC4に結合した誘導体化されたアントラニル酸の消失をもたらす。加えて、さらなるフラグメンテーション、およびC3に結合した別の誘導体化アントラニル酸の消失が、親イオンから386ダルトンの消失として観測した(それぞれ、親イオン1621および1783由来で、1235および1397m/z)。
【0307】
酸化rhGAAオリゴ糖A1FのMSフラグメンテーションパターンは、アントラニル酸が結合したフコースのC3、4の誘導体化を示し、これにより過ヨウ素酸塩による酸化が起こることを確認した。C2−C3結合の酸化の証拠は観察されなかった。ビス−M6P六糖グリカンの酸化rhGAAへの結合は、縮合反応の間に、正味1305.3ダルトンの増加をもたらした。
【0308】
複合体化したオリゴ糖構造の一体性を確認するため、放出された天然オリゴ糖の高精度MS分析を実施した。2および7.5mMの過ヨウ素酸塩で製造したrhGAAおよびNeoGAA SAM6由来のオリゴ糖を、PNGase Fを使用して放出し、10mMのギ酸アンモニウムpH4.0を有するアセトニトリル−水グラジエントで、順相HPLC(TSKゲルアミド−80カラム)により放出した。グリカンの質量分析検出を、インライン、陰イオンモード、QStarまたはLCT飛行時間型質量分析装置を用いて実施した。
【0309】
次の表に、高性能MS/TOF分析を基に、2および7.5mMの過ヨウ素酸塩で酸化したrhGAA由来の天然N−結合型オリゴ糖ピークの同定、並びに2および7.5mMの過ヨウ素酸塩で製造されたSAM6における天然N−結合型オリゴ糖ピークの同定の概要を示す。「Ox」は、酸化の部位数を、「Conj」は複合体化されたビス−M6P六糖グリカンの数を示す。理論質量は理論のオリゴ糖構造のモノアイソトピック分子量から計算され、対応する電荷状態の理論m/zおよび観察m/zを示す。
【0310】
【表9】
【0311】
【表10】
【0312】
【表11】
【0313】
【表12】
【0314】
【表13】
【0315】
20ppm超の質量精度がすべてのオリゴ糖種に関して観測された。MSの結果は、シアル酸の酸化が1mM以上の過ヨウ素酸塩で完了することを示す単糖組成物分析と一致した。マンノースおよびフコースの一部の酸化も明らかであった。酸化マンノースおよび/またはフコースにつき、1および2モルのグリカンの結合に相当するイオンが観測され、これはAAで観測されたように、それぞれの両方のアルデヒド種がグリカンに対して反応性であることを示唆する。
【0316】
高マンノース構造(オリゴマンノース5および6)のいくつかの結合を、2および7.5mMの過ヨウ素酸塩で処理した物質で検出した。2mMの過ヨウ素酸塩で結合した物質において、0または1個複合体化されたグリカンがA1(モノシアル化)種で観測され、0、1、2、および3個複合体化されたグリカンに相当するイオンが、7.5mM SAM6のA1種で観測された。この結果から、7.5mM(2mMではなく)過ヨウ素酸塩を用いて、コア・マンノースおよび/またはガラクトース残基の幾らかの酸化および複合体化が、A1構造で起こることが示唆される。
【0317】
A2およびA2F種(二シアル酸、二分岐鎖、±フコース)について、モノ−およびビ−複合体化種が、2および7.5mMの過ヨウ素酸塩サンプルで観察された。酸化マンノースとのトリ−複合体化の証拠は、7.5mM過ヨウ素酸塩処理サンプルでのみ観察され、2mM処理では観察されず、高い過ヨウ素酸塩濃度のフコースを介した複合体化と一致した。
【0318】
図11Aは、rhGAAおよびNeoGAAから放出されたオリゴ糖のHPLC分析を示す。rhGAAコントロールに関して、N−結合型オリゴ糖種の大部分は、11〜13分に溶出し、これはリン酸化も複合体化もされないオリゴ糖に相当する領域である。NeoGAAオリゴ糖について、ビ−複合体化(bi−conjugated)オリゴ糖種は19〜20分、モノ−複合体化(mono−conjugated)オリゴ糖種は15〜18分、および酸化/無修飾オリゴ糖は10〜13分で溶出した。7.5mM過ヨウ素酸塩で生成されたSAM6サンプルについて、そのオリゴ糖のほぼ半分がビ−複合体化種に相当する領域で溶出し、2mM過ヨウ素酸塩処理したサンプルのオリゴ糖のおおよそ3分の1はビ−複合体であった。これらのサンプルの溶出分析結果は、MALDI−TOFおよびマンノース−6−リン酸含有量分析によって測定された複合体化レベル(SAM6を生成する2および7.5mM過ヨウ素酸塩に関して、NeoGAA1モルあたり、複合体化されたグリカンそれぞれ約7および9モル)に一致した。
【0319】
存在するオリゴ糖構造のより優れた、可視化および定性比較を提供するため、放出されたオリゴ糖を、パルスアンペロメトリック検出付きの高pH陰イオン交換クロマトグラフィーに(HPAEC−PAD)より分析した。サンプルを酢酸ナトリウム/100mM水酸化ナトリウムグラジエントを用いて、Dionex CarboPac PA100カラムで、HPAEC−PADにより分析した。オリゴ糖ピーク物質を、オフラインフラクションコレクション、透析vs.水、およびインラインMS分析を用いた順相HPLCによる分析によって確認した。
【0320】
図11Bは、(MSで測定された)ピーク物質を含む代表的なHPAEC−PADデータを示す。図11Cは、2および7.5mMの過ヨウ素酸塩処理したrhGAAおよびNeoGAASAM6サンプルのHPAEC−PADによるオリゴ糖のプロファイリングを示す。
【0321】
D.過ヨウ素酸塩処理後のrhGAAタンパク質骨格の分析
NeoGAAのタンパク質骨格のタンパク質修飾を調べるため、ペプチドマッピングLC/MSを実施した。0、2、7.5および22.5mMの過ヨウ素酸塩を用いて製造したNeoGAASAM6は、トリプシンを用いて調製され、LCT飛行時間型質量分析器を用いて逆相HPLCにより分析した。システイン、メチオニン、トリプトファン、チロシン、およびヒスチジン残基の酸化、ならびにアスパラギンの脱アミドなど、可能性のあるペプチド修飾を、BioPharmalynxソフトウェアを使用して評価した。検出された有意な修飾は、いくつかの異なる部位のメチオニン酸化のみであった。0、2、および22.5mM過ヨウ素酸塩処理後の、ペプチドT13(メチオニン172および173を含む)の酸化レベルを図12に示す。
【0322】
有意なレベルの酸化が、メチオニン残基122、172および173で見出された。これらのメチオニン残基の酸化を、LC/MS/MS分析により確認した。低レベルの酸化も、メチオニン363で、過ヨウ素酸塩に依存した方法により起こることが観察された。過ヨウ素酸塩の酸化を最小限にする試みにおいて、過ヨウ素酸塩の濃度は、1〜7.5mMのレベルで滴定され、LC/MSによりモニターされた最も感受性の高い部位(ペプチドT13)で酸化した。有意なレベルのメチオニン酸化は、1mMより高い過ヨウ素酸塩濃度で観測され、これによりrhGAAのメチオニンがシアル酸残基と同じくらい酸化に対する感受性が高いことが示された。
【0323】
GAM複合体化の間に、GAOによるガラクトース酸化は、Met172/173の約26%の酸化をもたらした。カタラーゼが2および50ユニット/mgGAAで酸化反応中に含まれた場合、メチオニン酸化は排除された。
【0324】
実施例8:GAA複合体のin vitro特性評価
NeoGAA SAM2を、実施例2の化合物17および7.5mM過ヨウ素酸塩を用いて、実施例7に記述されたように製造した。ガラクトース結合NeoGAA GAM2を二糖17と結合させる前に、ガラクトースオキシダーゼでrhGAAを処理することで製造した。同様に、三糖NeoGAA SAM3(化合物35、実施例3)、四糖NeoGAAs SAM4(化合物28、実施例4A)、および直鎖SAM4(化合物47、実施例4B)も製造した。さらに、六糖NeoGAAβ SAM6を、実施例7に記述されたように、2および7.5mM過ヨウ素酸塩を用いて、化合物77を用いて製造した。追加の六糖複合体αSAM6(α結合、シアル酸残基を介して複合体化されている)およびGAM6(ガラクトース残基を介して複合体化されている)が同様に製造された。
【0325】
A.特異的活性:
活性分析を、rhGAAおよびNeoGAAによって触媒される、合成基質p−ニトロフェニル−D−α−グルコピラノシド(p−NP)の加水分解の比率をモニターすることで実施した。放出される発色団を、アルカリ条件下で、400nmの吸光度で測定した。1単位の活性を、37℃、定義されたアッセイ条件下で、1分あたり、1μmolのp−ニトロフェニル−D−α−グルコピラノシドをp−ニトロフェノールに加水分解するのに必要な酵素量として定義する。
【0326】
NeoGAA SAM2、SAM3、SAM4、直鎖SAM4、αSAM6、およびβSAM6の特異的活性を、図13に示す。さらに、SAM法 vs.GAM法を用いて製造した複合体の特異的活性を評価した。GAAに対するM6Pの数と、小規模および大規模で製造されたNeoGAA複合体の特異的活性との間には逆相関(p<0.001)があり、GAAあたり16.6倍モル過剰のオリゴ糖であった。複合体化(2.5〜33倍モル過剰のオリゴ糖)の間にNeoGAA濃度が滴定される別の実験において、GAA活性の消失がまた、SAMまたはGAM複合体におけるM6P含量の増大と共に観察された。49〜81%のGAA活性を有するSAM(コントロールと比較)について、様々なNeoGAAが1タンパク質につきオリゴ糖を6〜8分子含有した。GAM複合体は67〜92%活性を有し、1タンパク質あたり4〜6個のオリゴ糖を含有した。
【0327】
B.M6P受容体結合:
複合体の機能的効果は、BiacoreおよびM6P受容体アフィニティ−カラムHPLCによって、NeoGAAの可溶性カチオン非依存性マンノース−6−リン酸受容体(sCIMPR)への結合をモニターすること、およびL6筋芽細胞における取り込みによって評価した。ウシ血清から精製した、sCIMPRは、A〜Xの細胞外ドメインを含み、膜透過部分を欠いている。
【0328】
Biacore分析に関して、sCIMPRをCM−5チップにアミン結合させ、NeoGAAサンプルの10μg/mLを表面に固定し、マンノース−6−リン酸の濃度を増加させて溶出した。結合したNeoGAAの50%を置き換えるのに必要なM6Pの濃度として、親和性を定量した(EC50)。当該方法を、受容体親和性の結合および酸化の効果をモニターするのに使用した。
【0329】
図14にBiacore分析の結果を示す。sCIMPRを固定化したBiacoreチップ上に10μg/mLのNeoGAAを注入すると、約250RUのレスポンスの増加を引き起こし、同じ量のrhGAAでは、約100RUの偏差を引き起こした。さらに、rhGAAよりNeoGAAを溶出するのに、ほぼ10倍高いM6P濃度が必要であった(rhGAAおよびNeoGAAについて、それぞれほぼ0.1vs.1.0mMのEC50値)。EC50値と、2および7.5mM過ヨウ素酸塩を用いて製造したNeoGAAの結合レベルには、直線的な関係が見られた(R−2乗>0.95)。調査した結合範囲において(1.6〜4.7モルのグリカン/モル、NeoGAA、2mMの過ヨウ素酸塩で製造、および1.0〜8.5モルのグリカン/モル、7.5mMのNaIO4で製造)、結合親和性に関する複合化レベルの効果は、2および7.5mM過ヨウ素酸塩で製造されたNeoGAAに類似していた。
【0330】
PorosEP樹脂にsCIMPRを固定化し、その後分析用HPLCカラムにパッキングすることで調製された、M6P受容体カラムを使用して、M6P受容体結合をHPLCにより評価した。rhGAAおよびNeoGAAを、0.25、0.85、5、および20mMのM6Pを用いて溶出した(図15AおよびB)。1つの実験において、SAM2およびGAM2を、βSAM6およびGAM6と比較した(図15C)。SAM6およびGAM6では、物質の大部分が溶出される前に(>95%の、カラムに結合した複合体)、20mMのM6Pが必要であった。SAM2およびGAM2は強固には結合せず、5mMのM6Pで大部分が溶出した(>95%がカラムに結合)。
【0331】
さらに、NeoGAA複合体SAM2、SAM3、SAM4、直鎖SAM4、およびαSAM6を、M6P受容体結合に関して評価した(図15D)。SAM2、SAM3、および両方のSAM4複合体の大部分が、5mMのM6Pで溶出したが、両方のSAM6複合体は20mMのM6Pが必要であった。
【0332】
種々の量が複合を有する、NeoGAA複合体を評価した(図15E)。結合画分のNeoGAAの割合は、複合体化されたNeoGAA1モルあたり、>2.0モルのグリカンを有する製造物に関して、一定して>95%だった。複合体化レベルの低いわずかな画分において(1.0〜1.7モルのグリカン)、結合画分は75〜90%であった。NeoGAAに関するM6Pカラム分析データも、rhGAAより高親和性種の量が高いことを示した。具体的には、結合rhGAAの大部分は、0.2mMのM6Pで溶出され、カラムからNeoGAAを溶出するには、20mMのM6Pが必要であった。高親和性種の割合に関して、複合化レベルの全体的な効果は、2.0〜7.5mM過ヨウ化酸塩処理に類似していた。
【0333】
図15Fは、さまざまな量のM6Pの効果を示す。SAM6複合体は、以下のM6P量、SAM6−1(rhGAA 1モルにつき、M6P 4.9モル)、SAM6−2(GAA 1モルにつき、M6P 7.4モル)、SAM6−3(rhGAA 1モルにつき、M6P 10.5モル)、SAM6−4(rhGAA 1モルにつき、M6P 11.2モル)およびSAM6−5(rhGAA 1モルにつき、M6P 16.6モル)を含有した。統計的有意性を^、*および***で示し、それぞれ100mg/kgのrhGAA、SAM6−1、SAM6−3と比較したp<0.05を表す。
【0334】
C.L6筋芽細胞による細胞内移行
L6筋芽細胞の取込みアッセイを、Zhuら,J.Biol.Chem.279:50336−50341(2004)に記述の通りに実施し、カチオン非依存性マンノース−6−リン酸受容体(CIMPR)経路を介して、rhGAAおよびNeoGAAが筋芽細胞を標的とすることが明らかにされた。L6筋芽細胞の取込みアッセイにおいて、rhGAAおよびNeoGAAを、L6筋芽細胞を含むウェル中の+/−5mM M6Pの培地に加え、あらかじめ決定された一晩にてインキュベートした。インキュベート後、細胞を溶解し、4−MUグルコシド基質を用いた活性、およびマイクロBCAアッセイによるタンパク質濃度を分析して、酵素の用量反応曲線を作成した。
【0335】
L6筋芽細胞の取込みアッセイの結果を、図16に示す。SAM2およびGAM2複合体は、無修飾のrhGAAより優位に高い取込みを示したが、二リン酸化されたSAM6またはGAM6複合体ほど高くはなかった(図16、上図)。NeoGAAのSAM2、SAM3、SAM4、直鎖SAM4、およびαSAM6の取込み試験も行った(図16、下図)。同様の実験において、実施例7のリシン−チオ−ル複合体は、約8倍の取込み増加を引き起こした。
【0336】
実施例9:新規GAA複合体のin vivoでの効果
ある種のNeoGAA複合体のin vivoでの効果を、Rabenら,J.Biol.Chem.273(30):19086−92(1998)に記述された、GAAノックアウトマウスモデルを用いて調査した。以下の通り、それぞれ6匹のマウス群を4週間にわたり1週間に1回処置した。
【0337】
【表14】
【0338】
サンプルを心臓、大腿四頭筋、および上腕三頭筋から集め、組織のグリコーゲン含有量を測定した。SAM2、SAM4およびSAM6動物の結果を、それぞれ図17、図18、および図19に示す。SAM6複合体を用いて12匹の動物群で、実験を繰り返し、SAM6複合体が、未修飾のrhGAAより5倍強力であることを確認した。
【0339】
SAMおよびGAM六糖複合体を、溶媒、20、60、または100mg/kgのrhGAA、あるいは4、12、または20mg/kgのSAM6またはGAM6を4週間、1週間に1回摂取した6匹のマウス群を用いて比較した。心臓、大腿四頭筋、上腕三頭筋、隔膜、および腰筋を摘出し、グリコーゲン含有量を分析した。図20に、この試験結果を示す。薬物動態および薬力学試験については、3〜6カ月齢の30匹のGAAノックアウトマウス(オス15匹、メス15匹)を、Charles River Laboratories、ウィルミントン、MAから入手した。各用量群にはオス5匹、メス5匹が含まれる。動物をグループにして飼育し、恒湿25℃、12時間の明暗周期を維持した。すべての動物は、飼料(PicoLab(登録商標)Rodent Diet20)および水を自由に摂取することができるようにした。各用量につき計10匹のマウスとするため、動物を1グループにつきオス5匹、メス5匹の3用量グループに無作為に分けた。各グループには、20mg/kgで、rhGAA、GAMおよびSAM複合体の単回静脈内投与を行った。薬物動態分析用の血液サンプルを、意識のあるマウスの後眼窩静脈叢を介して、投与後、5、15、30、60、120、240、および480分で回収した。血清中のrhGAA濃度を、GAA活性アッセイを用いて測定した。結果を図20Bに示す。
【0340】
実施例10:酸性スフィンゴミエリナーゼ複合体の合成
バキュロウイルス発現系またはチャイニーズハムスター卵巣細胞で発現した組換えヒト酸性スフィンゴミエリナーゼ(rhASM)は、遊離チオ−ル基を有するC−末端システインを有する。Lansmannら,Eur.J.Biochem.270:1076−1088(2003)およびQiuら,J.Biol.Chem.278:32744−32752(2003)を参照のこと。rhASMは、遊離チオ−ル基を介して、オリゴ糖1〜127のいずれかと結合することができ、このオリゴ糖は、米国特許仮出願第60/885,457号、または実施例6に記載された方法によると、リンカーおよびチオ−ル反応性基を含む。
【0341】
実施例11:α−L−イズロニダーゼ複合体の合成
α−L−イズロニダーゼは、オリゴ糖1〜127のいずれかと結合し、このオリゴ糖は、Leeら,Pharm.Res.20:818−825(2003)に記載された方法によると、プロピオンアルデヒド反応性基を含むリンカーを含む。α−L−イズロニダーゼおよびオリゴ糖は、室温、pH5.5、1日間で、還元剤としてシアノ水素化ホウ素ナトリウムの存在下で結合する。その後、小分子が透析または膜分離により、反応混合物から除去される。
【0342】
本明細書で引用されるすべての参考文献は、その全体が参照により本明細書に組み込まれる。参照により組み込まれる参考文献および特許または特許出願が、本明細書に含まれる発明に相反する場合、本明細書があらゆる相反する構成要素に取って代わる。
【0343】
本明細書および請求項で使用される成分、反応条件などの量を表す全数字は、「約」という語により、すべての場合で修飾されるものとして理解されるべきであり、約は例えば±5%を意味する。したがって、反対に指示されない限り、明細書および添付請求項に明記される数値パラメーターは、本明細書によって試みられた所望の特性によって変化する概数である。最低限でも、請求の範囲に相当する原理の応用を限定するいかなる意図は無く、各数値パラメーターは、有効数字の数および概数で表す方法の観点で解釈されるべきである。
【0344】
本発明の範囲および趣旨から逸脱することなく、当業者に明らかな、本発明の多くの修飾および変形を行うことができる。本明細書で記載された具体的な実施形態は、例示のためにのみ記載するものであり、いかなる方法でも限定されるものではない。本明細書および実施例は、例示としてのみの目的であり、本発明の本来の範囲および趣旨は以下の請求項により示される。
【特許請求の範囲】
【請求項1】
(1)タンパク質、および(2)式I〜VI:
【化1】
【化2】
[式中、
aは、α1,2;α1,3;α1,4;およびα1,6から選択され;
bは、α1,2;α1,3;およびα1,4から選択され;
cは、α1,2;α1,3;α1,4;およびα1,6から選択され;
dは、α、β、およびαとβとの混合物から選択され;
eは、α1,2;α1,3;α1,4;およびα1,6から選択され、
fは、α、β、およびαとβとの混合物から選択され;
gは、α1,2;α1,3;およびα1,4から選択され;
hは、α1,2;α1,3;α1,4;およびα1,6から選択され;
iは、α、β、およびαとβとの混合物から選択され;
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;
lは、α、β、およびαとβとの混合物から選択され;
mは、α、β、およびαとβとの混合物から選択され;
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、および、オキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで任意に置換され、且つN、O、またはSから選択される1または2以上のヘテロ原子で任意に中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4であり;並びに、
xが2もしくは3であり、またはyが2、3、もしくは4であるとき、式IVまたは式VIの各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択され;
ただし、eがα1,6であるとき、fはα、およびαとβとの混合物から選択される]
のいずれか1つのオリゴ糖を含む、オリゴ糖−タンパク複合体。
【請求項2】
前記オリゴ糖が2個のマンノース−6−リン酸残基を有する、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項3】
前記オリゴ糖が3個のマンノース−6−リン酸残基を有する、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項4】
前記オリゴ糖がオリゴ糖82である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項5】
前記オリゴ糖がオリゴ糖128、オリゴ糖129、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項6】
前記オリゴ糖がオリゴ糖130、オリゴ糖131、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項7】
前記オリゴ糖がオリゴ糖132、オリゴ糖133、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項8】
前記オリゴ糖がオリゴ糖136である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項9】
前記タンパク質が糖タンパク質である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項10】
前記糖タンパク質がリソソーム酵素である、請求項9に記載のオリゴ糖−タンパク複合体。
【請求項11】
前記リソソーム酵素が、酸性α−グルコシダーゼ、α−ガラクトシダーゼA、酸性スフィンゴミエリナーゼ、α−L−イズロニダーゼ、イズロン酸−2−スルファターゼ、またはN−アセチルガラクトサミン−4−スルファターゼである、請求項10に記載のオリゴ糖−タンパク複合体。
【請求項12】
前記リソソーム酵素が、酸性α−グルコシダーゼである、請求項10に記載のオリゴ糖−タンパク複合体。
【請求項13】
請求項1に記載のオリゴ糖−タンパク複合体および賦形剤を含む、医薬組成物。
【請求項14】
それを必要とする被験体のリソソーム蓄積症を治療するための医薬の製造における、請求項1に記載の複合体の使用。
【請求項15】
リソソーム蓄積症の治療方法であって、(1)リソソーム酵素、および(2)式I〜VI:
【化3】
【化4】
[式中、
aは、α1,2;α1,3;α1,4;およびα1,6から選択され;
bは、α1,2;α1,3;およびα1,4から選択され;
cは、α1,2;α1,3;α1,4;およびα1,6から選択され;
dは、α、β、およびαとβとの混合物から選択され;
eは、α1,2;α1,3;α1,4;およびα1,6から選択され;
fは、α、β、およびαとβとの混合物から選択され;
gは、α1,2;α1,3;およびα1,4から選択され;
hは、α1,2;α1,3;α1,4;およびα1,6から選択され;
iは、α、β、およびαとβとの混合物から選択され;
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;
lは、α、β、およびαとβとの混合物から選択され;
mは、α、β、およびαとβとの混合物から選択され;
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、および、オキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで任意に置換され、且つN、O、またはSから選択される1または2以上のヘテロ原子で任意に中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4であり;並びに、
xが2もしくは3であり、またはyが2、3、もしくは4であるとき、式IVまたは式VIの各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択され;
ただし、eがα1,6であるとき、fはα、およびαとβとの混合物から選択される]
のいずれか1つのオリゴ糖を含む、オリゴ糖−糖タンパク複合体を哺乳動物に投与することを含む、前記方法。
【請求項16】
前記リソソーム蓄積症が、ファブリー病、ポンペ病、ニーマン−ピックA病、ニーマン−ピックB病、ムコ多糖症I型、ムコ多糖症II型、およびムコ多糖症VI型から選択される、請求項15に記載の方法。
【請求項17】
前記リソソーム蓄積症がポンペ病である、請求項16に記載の方法。
【請求項18】
前記オリゴ糖−糖タンパク複合体で処理する前、処理後、または処理中に前記哺乳動物へ、メトトレキサートを投与することをさらに含む、請求項15に記載の方法。
【請求項1】
(1)タンパク質、および(2)式I〜VI:
【化1】
【化2】
[式中、
aは、α1,2;α1,3;α1,4;およびα1,6から選択され;
bは、α1,2;α1,3;およびα1,4から選択され;
cは、α1,2;α1,3;α1,4;およびα1,6から選択され;
dは、α、β、およびαとβとの混合物から選択され;
eは、α1,2;α1,3;α1,4;およびα1,6から選択され、
fは、α、β、およびαとβとの混合物から選択され;
gは、α1,2;α1,3;およびα1,4から選択され;
hは、α1,2;α1,3;α1,4;およびα1,6から選択され;
iは、α、β、およびαとβとの混合物から選択され;
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;
lは、α、β、およびαとβとの混合物から選択され;
mは、α、β、およびαとβとの混合物から選択され;
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、および、オキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで任意に置換され、且つN、O、またはSから選択される1または2以上のヘテロ原子で任意に中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4であり;並びに、
xが2もしくは3であり、またはyが2、3、もしくは4であるとき、式IVまたは式VIの各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択され;
ただし、eがα1,6であるとき、fはα、およびαとβとの混合物から選択される]
のいずれか1つのオリゴ糖を含む、オリゴ糖−タンパク複合体。
【請求項2】
前記オリゴ糖が2個のマンノース−6−リン酸残基を有する、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項3】
前記オリゴ糖が3個のマンノース−6−リン酸残基を有する、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項4】
前記オリゴ糖がオリゴ糖82である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項5】
前記オリゴ糖がオリゴ糖128、オリゴ糖129、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項6】
前記オリゴ糖がオリゴ糖130、オリゴ糖131、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項7】
前記オリゴ糖がオリゴ糖132、オリゴ糖133、またはそれらの混合物である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項8】
前記オリゴ糖がオリゴ糖136である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項9】
前記タンパク質が糖タンパク質である、請求項1に記載のオリゴ糖−タンパク複合体。
【請求項10】
前記糖タンパク質がリソソーム酵素である、請求項9に記載のオリゴ糖−タンパク複合体。
【請求項11】
前記リソソーム酵素が、酸性α−グルコシダーゼ、α−ガラクトシダーゼA、酸性スフィンゴミエリナーゼ、α−L−イズロニダーゼ、イズロン酸−2−スルファターゼ、またはN−アセチルガラクトサミン−4−スルファターゼである、請求項10に記載のオリゴ糖−タンパク複合体。
【請求項12】
前記リソソーム酵素が、酸性α−グルコシダーゼである、請求項10に記載のオリゴ糖−タンパク複合体。
【請求項13】
請求項1に記載のオリゴ糖−タンパク複合体および賦形剤を含む、医薬組成物。
【請求項14】
それを必要とする被験体のリソソーム蓄積症を治療するための医薬の製造における、請求項1に記載の複合体の使用。
【請求項15】
リソソーム蓄積症の治療方法であって、(1)リソソーム酵素、および(2)式I〜VI:
【化3】
【化4】
[式中、
aは、α1,2;α1,3;α1,4;およびα1,6から選択され;
bは、α1,2;α1,3;およびα1,4から選択され;
cは、α1,2;α1,3;α1,4;およびα1,6から選択され;
dは、α、β、およびαとβとの混合物から選択され;
eは、α1,2;α1,3;α1,4;およびα1,6から選択され;
fは、α、β、およびαとβとの混合物から選択され;
gは、α1,2;α1,3;およびα1,4から選択され;
hは、α1,2;α1,3;α1,4;およびα1,6から選択され;
iは、α、β、およびαとβとの混合物から選択され;
jは、α1,2であり;
kは、α、β、およびαとβとの混合物から選択され;
xは、1、2、または3であり;
lは、α、β、およびαとβとの混合物から選択され;
mは、α、β、およびαとβとの混合物から選択され;
RxおよびRyは、それぞれ独立に、ポリエチレングリコール、および、オキソ、ニトロ、ハロ、カルボキシル、シアノ、または低級アルキルで任意に置換され、且つN、O、またはSから選択される1または2以上のヘテロ原子で任意に中断されたC1−C10アルキルから選択され;
zは、0、1、2、3、または4であり;並びに、
xが2もしくは3であり、またはyが2、3、もしくは4であるとき、式IVまたは式VIの各マンノース間の結合は、α1,2;α1,3;α1,4;およびα1,6から選択され;
ただし、eがα1,6であるとき、fはα、およびαとβとの混合物から選択される]
のいずれか1つのオリゴ糖を含む、オリゴ糖−糖タンパク複合体を哺乳動物に投与することを含む、前記方法。
【請求項16】
前記リソソーム蓄積症が、ファブリー病、ポンペ病、ニーマン−ピックA病、ニーマン−ピックB病、ムコ多糖症I型、ムコ多糖症II型、およびムコ多糖症VI型から選択される、請求項15に記載の方法。
【請求項17】
前記リソソーム蓄積症がポンペ病である、請求項16に記載の方法。
【請求項18】
前記オリゴ糖−糖タンパク複合体で処理する前、処理後、または処理中に前記哺乳動物へ、メトトレキサートを投与することをさらに含む、請求項15に記載の方法。
【図1】

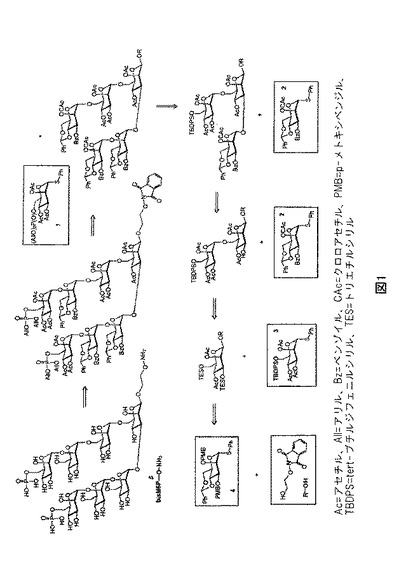
【図2】
【図3】
【図4】
【図5】
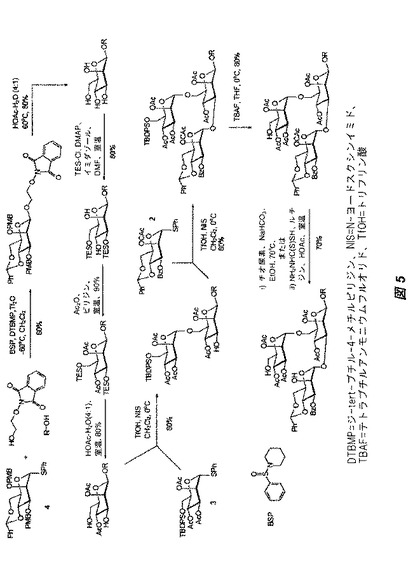
【図6】
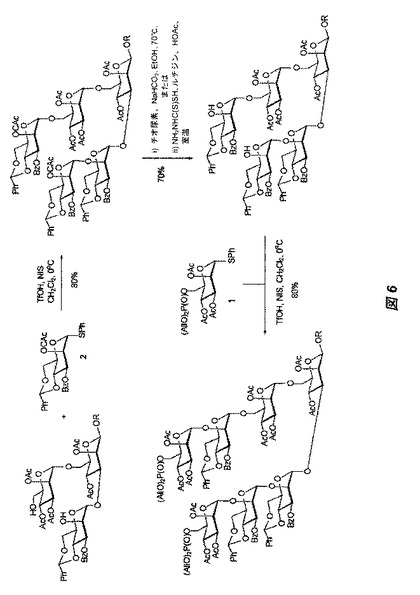
【図7】
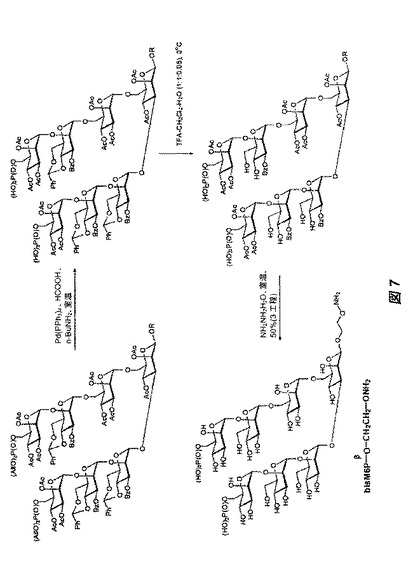
【図8A】
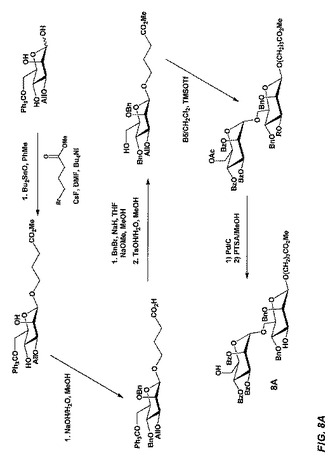
【図8B】
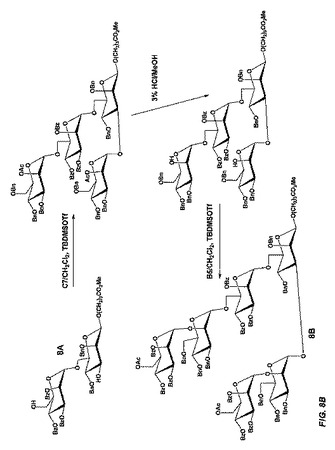
【図8C】
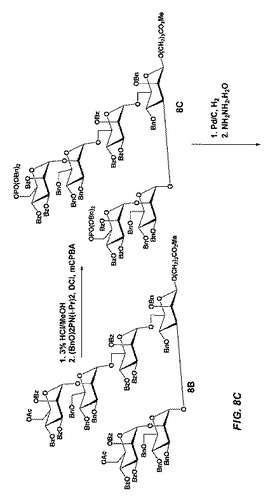
【図8D】
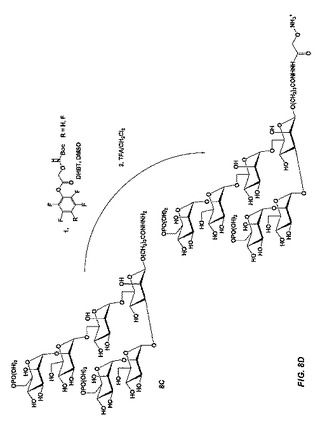
【図8E】
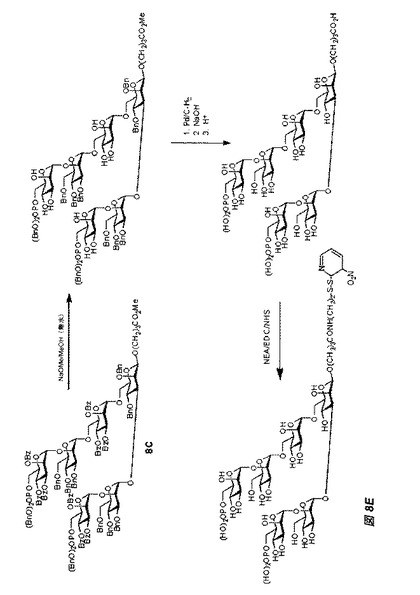
【図9A】
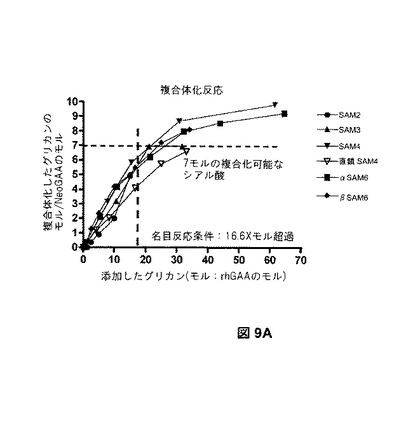
【図9B】
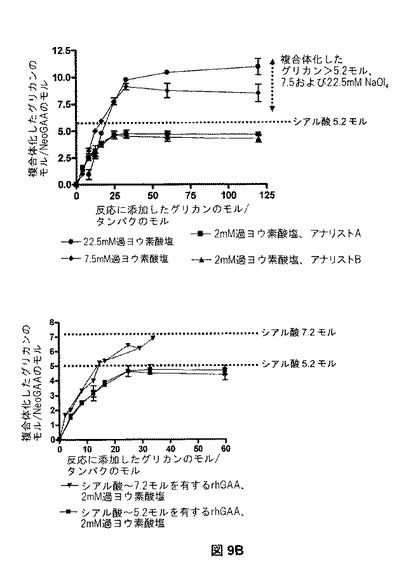
【図10A】
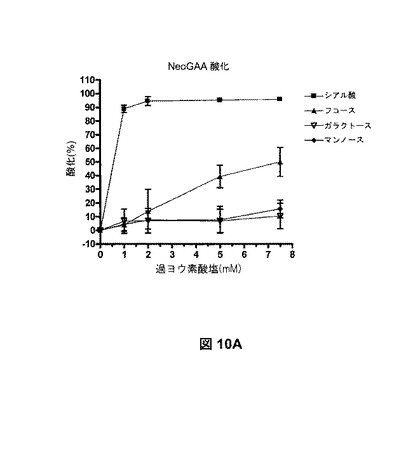
【図10B】
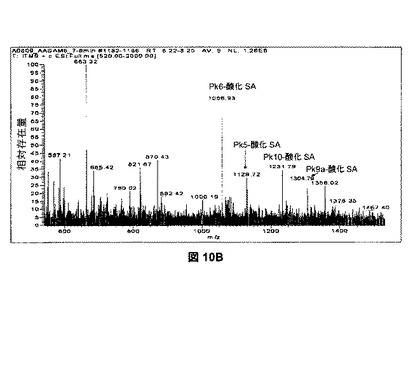
【図10C】
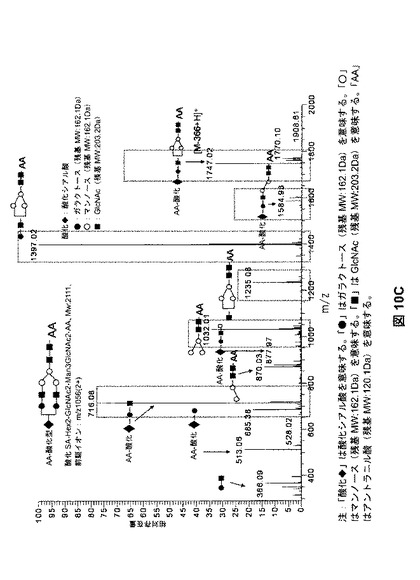
【図10D】
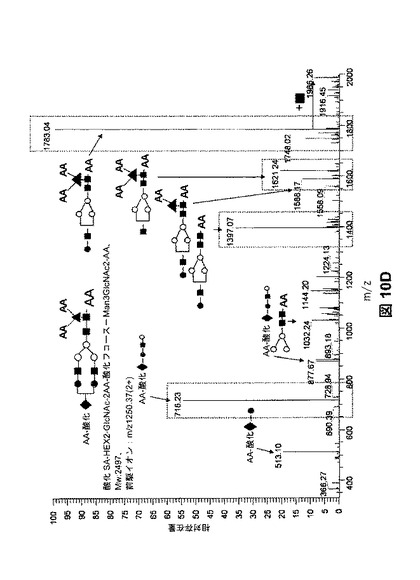
【図10E】
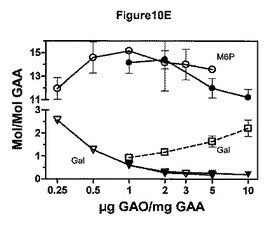
【図11A】
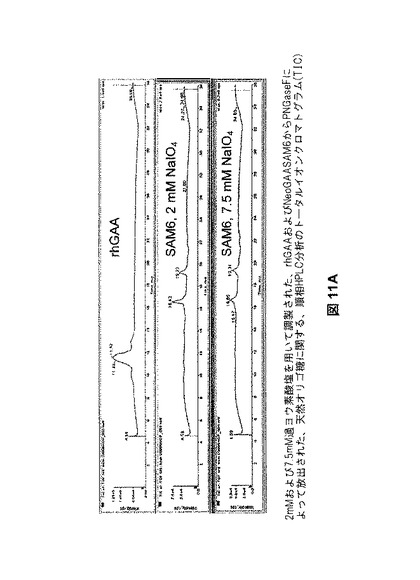
【図11B】
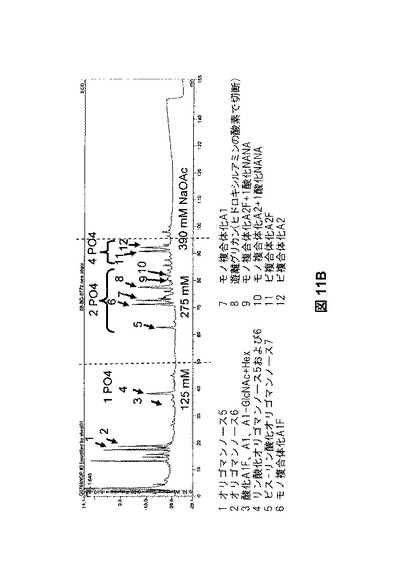
【図11C】
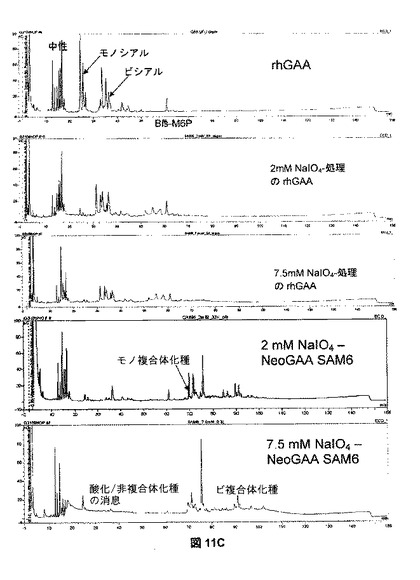
【図12】
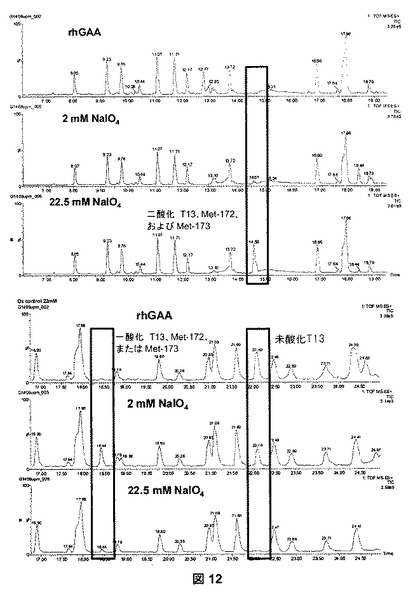
【図13A】
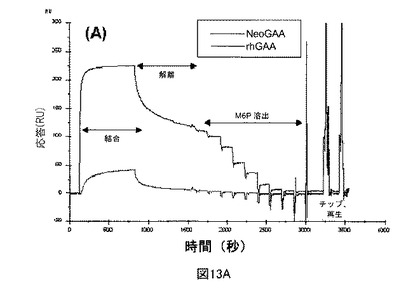
【図13B】
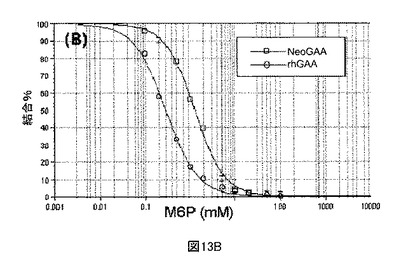
【図13C】
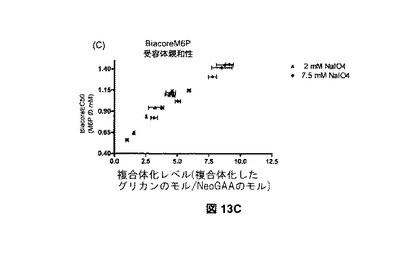
【図14】
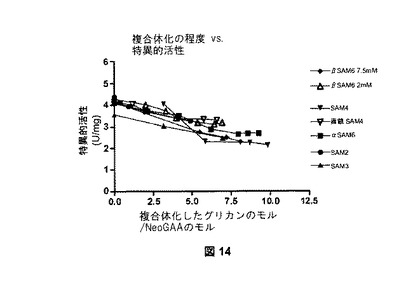
【図15A−C】
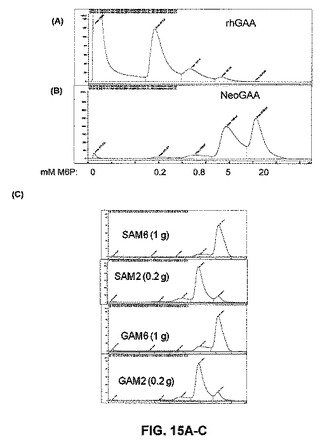
【図15D】
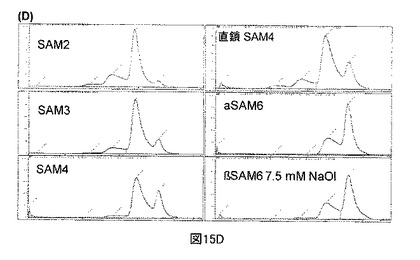
【図15E】
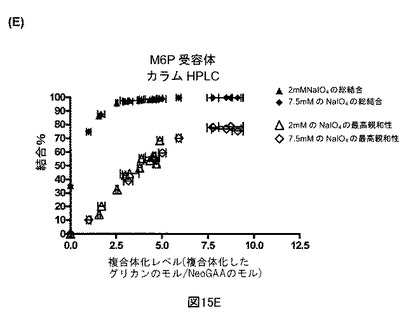
【図15F】
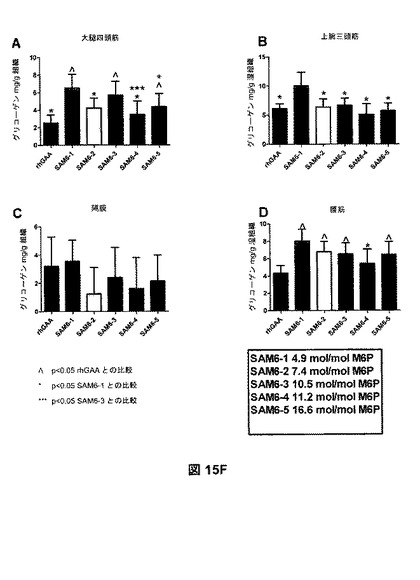
【図16】
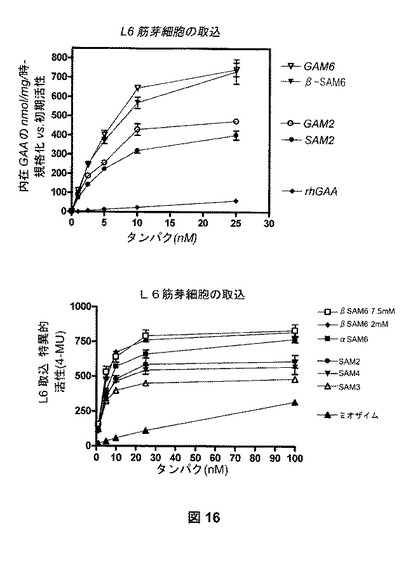
【図17】
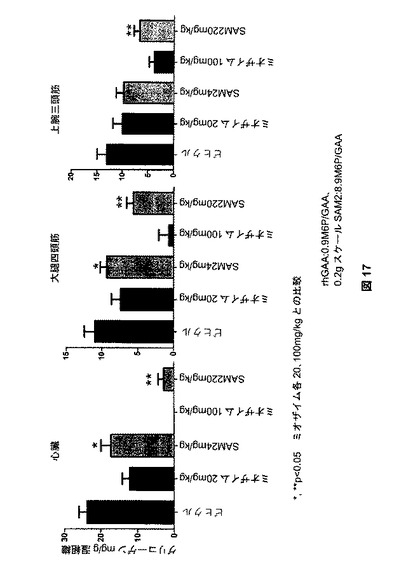
【図18】

【図19】
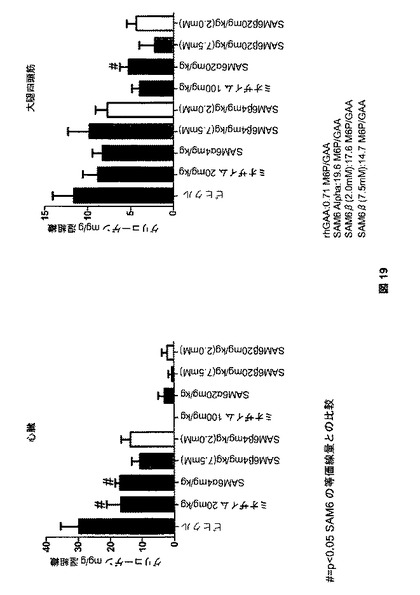
【図20】
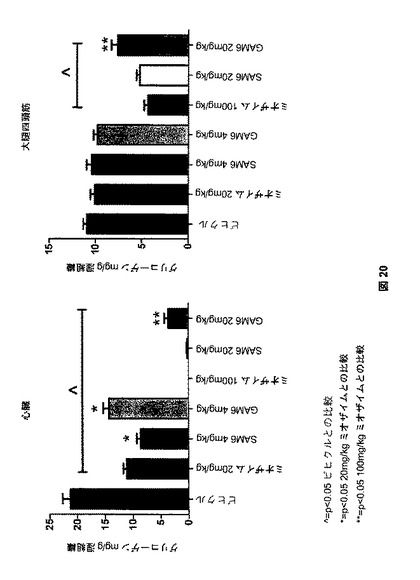
【図20B】
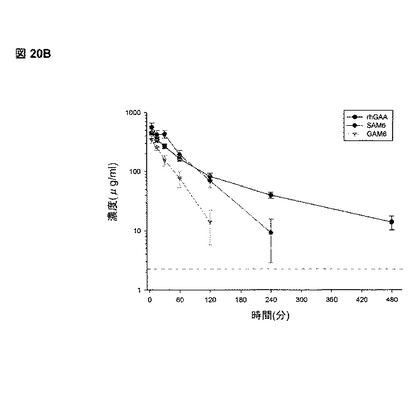
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図8C】
【図8D】
【図8E】
【図9A】
【図9B】
【図10A】
【図10B】
【図10C】
【図10D】
【図10E】
【図11A】
【図11B】
【図11C】
【図12】
【図13A】
【図13B】
【図13C】
【図14】
【図15A−C】
【図15D】
【図15E】
【図15F】
【図16】
【図17】
【図18】
【図19】
【図20】
【図20B】
【公表番号】特表2012−512313(P2012−512313A)
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願番号】特願2011−542288(P2011−542288)
【出願日】平成21年12月11日(2009.12.11)
【国際出願番号】PCT/US2009/067775
【国際公開番号】WO2010/075010
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(500034653)ジェンザイム・コーポレーション (37)
【Fターム(参考)】
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願日】平成21年12月11日(2009.12.11)
【国際出願番号】PCT/US2009/067775
【国際公開番号】WO2010/075010
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(500034653)ジェンザイム・コーポレーション (37)
【Fターム(参考)】
[ Back to top ]