
オリゴ糖、その調製方法並びに使用、及び同含有薬学的組成物
本発明は、オリゴ糖、該オリゴ糖の調製方法及び使用、並びに該オリゴ糖を含有する薬学的組成物に関する。とりわけ、本発明は、特に、転移形成を予防及び阻害するための、癌治療で使用できるオリゴ糖に関する。本発明のオリゴ糖は、例えば、初期の乳癌、肺癌、前立腺癌、結腸癌又は膵臓癌で使用できる。前記オリゴ糖は、皮下、経口又は静脈内の各投与が可能である。更に、前記オリゴ糖は、単独で、又は、例えば、ドセタキセル若しくはパクリタキセルなどの細胞障害剤などの他の抗癌剤とともに使用できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規化学化合物、特に、新規オリゴ糖に関し、これらを調製する方法、これらの使用、及びこれらを含有する薬学的組成物に関する。これらのオリゴ糖は、癌の治療、特に、転移形成の予防及び阻害に対し、有効である。
【0002】
より具体的には、第1の態様において、本発明は、本来抗血栓特性を有する多糖類を解重合する方法に関する。
【背景技術】
【0003】
抗血栓特性を有する多糖類の解重合するための方法は公知である。これらの方法の一般的な態様は:
−出発多糖類を医薬製品として使用する際に起こる、副作用を制限するために、更に低い平均分子量のオリゴ糖を得ることを目的とし;
−解重合後、満足のいく抗血栓活性を維持することである。
【0004】
ヘパリン又はエノキサパリン、チンザパリン若しくはフラグミンなどの低分子量のヘパリンなどの抗血栓特性を有する市販の多糖類は、全てヘパラナーゼ阻害剤である。
【0005】
正常な生理学的状態のもとでは、細胞が酵素のヘパラナーゼを発現する。この酵素により、間接的に、有糸分裂誘発、新血管形成及び組織修復を調節することが可能となる。ヘパラナーゼの作用機序の1つは、へパラン硫酸プロテオグリカン(HSPG)を開裂することである。このグリコサミノグリカンは、内皮細胞の表面に存在し、基底膜(細胞外マトリックス)の結合を確保している。へパラン硫酸プロテオグリカンの開裂は、FGF2などの成長因子の放出をもたらす。この成長因子の放出は、有糸分裂誘発及び血管新生にとって必要である。しかし、これは、これらの生物学的機序を引き起こすのに十分でなく、タンパク質のアロステリック修飾を起こし、その受容体との相互作用を促進するためには、FGF2が、グリコサミノグリカンに結合する必要がある。実際、HSPGの開裂を通して、ヘパラナーゼは、FGF2に結合するヘパラン硫酸フラグメントを生成し、その受容体との相互作用を促進し、こうして、上述の生物学的機序を誘導する。細胞外マトリックスにおけるHSPGの開裂及び毛細血管の脱構造化により、細胞遊出が可能になる。
【0006】
ヘパラナーゼは、腫瘍細胞によって過剰発現され、それ故、その転移及び血管新形成を促進する。これらの現象は、癌性腫瘍の増殖及び生存にとって不可欠である。
【0007】
ヘパラン硫酸に構造的に類似のグリコサミノグリカンであるヘパリンは、ヘパラナーゼの強力な阻害剤であることが知られている。この効力は、長い間、特異的なATIII−結合配列の存在に起因すると考えられている。実際に、この配列は、ヘパラン硫酸においてはそれほどでもないが、これらの2つのグリコサミノグリカンに共通している。ヘパラナーゼ開裂域を下記に表す:
【0008】
【化15】

【0009】
このα−エンドグリコシダーゼによる開裂点は、最小のATIII−結合配列の中央に位置している。従って、抗血栓及びヘパラナーゼ阻害特性が密接に関連すると考えることができる(実際に、ヘパリンは、ヘパリン硫酸に対する競合的基質である。)。その結果、抗転移物質として、このグリコサミノグリカンを使用するのは困難である。実際、その強力な抗凝血性質が、治療の限度を制限し、大量出血などの深刻な副作用を引き起こす。更に、特定の場合において、ヘパリンの反復注入は、致命的転帰をもたらす血小板減少症を引き起こす可能性がある(ヘパリン及び血小板第4因子(PF4)の結合に関する免疫反応)。
【0010】
抗移転活性との相関関係において、オリゴ糖の構造及びそれらの抗ヘパラナーゼ特性の関連を強調した文書はほとんどない。従って、Bitan,et al.(Isr.J.Med.Sci.1995;31:106−118)は、ヘパラナーゼ阻害ヘパリン種により肺メラノーマの定着化阻害に必要とされる構造条件を特定する:ヘパラナーゼは、16又はそれ以上の糖を含有するヘパリン断片により効果的に阻害される(要約;図2、p110;図3、p111;p116、右側コラム、第2文)。六糖は、ヘパラナーゼ阻害剤として劣っていると記述されている(図8、p115)。加えて、少なくとも4000ダルトン以上の分子量を有する分子を用いた場合にのみ、ヘパラナーゼの阻害が可能になると言われている(要約:p116、右側コラム、第4文)。しかしながら、ヘパラナーゼの阻害性を決定する方法は、様々な試験品の存在下で細胞外マトリックスを分解する細胞能力を評価するので、間接的な方法である(p108;右側コラム、第2段落「Degradation of Sulfated Proteoglycans(硫酸化プロテオグリカンの分解)」)。従って、この方法は、ヘパラナーゼに対し特異的ではない。更に、前述の全生成物は、ヘパリンを化学的に開裂する方法(亜硝酸)により取得される(p108;左側コラム、第2段落「Heparin−Derived Oligosaccharides(ヘパリン由来のオリゴ糖)」。
【0011】
次も参照されたい。
【0012】
Vlodavski I,et al.Modulation of neovascularization and metastasis by species of heparin,Heparin and related Polysaccharides,D.A.Lane,et al.,Editor,Plenum Press, New York, 1992;
Parish CR,et al.Evidence that sulphated polysaccharides inhibit tumor metastasis by blocking tumor−cell−derived heparanases, Int. J. Cancer 40:511−518,1987。
【0013】
現在のところ、商業的に許容される解決策がないため、抗転移化合物に対する必要性は非常に大きい。現在公知のヘパラナーゼ阻害性多糖及びオリゴ糖は、天然原料(ヘパリン)又は、事実上、実行するのが困難な方法(いくつかの低分子量のヘパリン)から直接的に生じたもので、特に、治療を受ける患者に出血の恐れがある場合において、抗癌治療に適さない際立った抗血栓成分を示す。
【発明の開示】
【発明が解決しようとする課題】
【0014】
従って、現在の問題点の1つは、容易かつ再現性のある方法を通して、基本的に抗血栓活性のない、顕著な抗ヘパラナーゼ活性を示す生成物を得ることである。
【課題を解決するための手段】
【0015】
この目的のために、驚くべきことに、特に、Xa及びIIa因子の阻害性のために、その抗血栓活性が実質的に消滅するまで(<35IU/mg)、多糖類がヘパラナーゼ1で解重合される、抗血栓特性を本来有する多糖類を解重合するための新規の方法により、顕著な抗ヘパラナーゼ活性を保存する生成物を取得することが可能であると明らかと成った。
【0016】
それ故に、この方法では、同時にその抗血栓要素を除去する一方で、多糖類の抗ヘパラナーゼ部位濃縮生成物を取得するための効果的な手段を構成する。本方法は、その抗血栓活性が20IU/mg未満になるまで、多糖類の解重合が遂行される場合に、より有利に使用される。
【0017】
とりわけ、5000Da未満、好ましくは、3000Da未満の平均分子量に達するまで、解重合が遂行される。あらゆる予想に反し、容易に実行可能な、この方法によって得られた生成物は、特に、優れたヘパラナーゼ阻害剤である六糖を含有する。加えて、これらの六糖は、一般的に1000ないし2000ダルトンであるので、平均分子量が4000ダルトンよりもかなり低い。多糖は、好ましくはヘパリンである。六糖画分の混合物が、硫酸化六糖ΔIs−Isid−Isid及びΔIs−Isid−IIsgluを実質的に含まなくなるまで、解重合は有利に続行される。
【0018】
酵素は、一般的に、「生理学的」条件下、すなわち、それらが抽出される生物中で、インビボで正常に機能する条件下で使用される(特に、pH、温度、イオン強度、場合により物理的補因子(可視光など)又は化学的補因子(補酵素など)。多くの酵素は、普通、生理学的温度を超える温度、例えば45〜50℃などで使用できる。本発明者らの状況では、あらゆる予想に反し、許容される選択性及び反応速度の状態下、好ましくは10〜20℃、特に16℃の温度で解重合が起こることが可能であり、解重合反応中に形成される、ヘパラナーゼ阻害化合物をできるだけ適切に維持することを観察した。更に、これにより、反応終了時の反応媒体中のヘパリナーゼ1の最終濃度を限定することが可能となる。実際に、ヘパラナーゼ1に対する最適反応温度を下回る温度(25ないし45℃とすることができる。)で、その反応を実行することにより、反応過程で、酵素の過剰な数の添加を防ぐことができる。酵素は、一般的に、基質欠乏以外の原因による反応速度の低下が観測される際に添加される。従って、特に、限られた酵素が存在するため、比較的低い反応温度を用いることにより、その後に実施される場合がある精製段階を間接的に促進することが可能である。それ故に、結果として、5〜40℃、好ましくは10〜20℃の温度で、解重合を実行できる。
【0019】
特に、二糖及び四糖などの、解重合中に形成された低分子量のオリゴ糖を取り除くことができるようにするために、本発明の方法では、pH8未満及びpH5超で、多糖類の解重合の生成物が、ゲル浸透クロマトグラフィーによって精製される工程を用いて、有利に遂行される。
【0020】
本発明の方法は、固定相(例えば、シリカ)が、(i)C18−グラフト化され、且つ(ii)セチルトリメチルアンモニウム(CTA−SAX)でグラフト化された逆相という、高性能液体クロマトグラフィ(HPLC)による、後の精製段階を有利に含む。
【0021】
前記方法は、また、好ましくは、200〜250nmの間で実質的に透明になる電解質を水溶液中に含有する移動相の使用を有利に含む、第1の脱塩段階も含む。許容される電解液は、NaClを含むが、200〜250nMでUV検出器とともに使用する場合には、過塩素酸、メタンスルホン酸塩又はNaなどのアルカリ金属のリン酸塩の使用が望ましい。第1の脱塩段階に対して許容される固定相は、陰イオン交換樹脂である。特に好ましい樹脂は、Sepharose Q(R)樹脂が望ましい。
【0022】
前記方法は、好ましくは、例えば、Sephadex G10(R)型の分子排除ゲルを好ましく使用する、第2の脱塩段階も含むことが可能である。
【0023】
HPLCカラムから排出されたときに収集される画分中に存在する本発明の生成物を検出する別の解決策は、必要に応じて、回折装置を使用することからなり得る。
【0024】
別の許容される脱塩技術には、例えば、重合体膜を使用する、浸透圧技術の使用を含む。
【0025】
第2の態様において、本発明は、第1の態様に従う方法によって得られた生成物に関する。
【0026】
Petitou,et al.J.Biol. Chem.(1988)、263(18)、8685−8690は、ヘパラナーゼ1を用いたヘパリンの部分解重合の方法により取得される生成物から単離された、ナトリウム塩形態の式ΔIs−IIaidu−IIsgluの六糖を開示している。この生成物は、抗血栓活性を示さないと説明されている。該生成物の他の性質については実証されていない。
【0027】
第3の態様において、本発明は以下の式(I)の生成物に関する。
【0028】
【化16】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択され、
n=0、R=SO3M及びM=Naの生成物は除かれる。)
【0029】
意外にも、MがLi、Na及びK、好ましくはNaから選択される場合には、本発明の第3態様に係る生成物が、より優れた物理化学的性質を示すことが観察されている。特に、溶解度及び安定度が向上している。
【0030】
式(I)の好ましい生成物は、n=0の生成物である。
【0031】
第4の態様において、本発明は、六糖に関する。
【0032】
下記の式(Ia)の生成物:
【0033】
【化17】
は、その第4態様に従った本発明に沿う。
【0034】
下記の式(1b)の生成物:
【0035】
【化18】
は、その第4態様に従った本発明に沿う。
【0036】
下記の式(Ic)の生成物:
【0037】
【化19】
は、その第4態様に従った本発明に沿う。
【0038】
下記の式(Id)の生成物:
【0039】
【化20】
は、その第4態様に従った本発明に沿う。
【0040】
下記の式(Ie)の生成物:
【0041】
【化21】
は、その第4態様に従った本発明に沿う。
【0042】
下記の式(If)の生成物:
【0043】
【化22】
は、その第4態様に従った本発明に沿う。
【0044】
下記の式(Ig)の生成物:
【0045】
【化23】
は、その第4態様に従った本発明に沿う。
【0046】
下記の式(Ih)の生成物:
【0047】
【化24】
は、その第4態様に従った本発明に沿う。
【0048】
下記の式(Ij)の生成物:
【0049】
【化25】
は、その第4態様に従った本発明に沿う。
【0050】
下記の式(Ik)の生成物:
【0051】
【化26】
は、その第4態様に従った本発明に沿う。
【0052】
下記の式(Im)の生成物:
【0053】
【化27】
は、その第4態様に従った本発明に沿う。
【0054】
第5の態様において、本発明は、特に癌、とりわけ乳癌、肺癌、前立腺癌、結腸癌又は膵臓癌に関連する、細胞増殖を調節するための、前記第2の態様から第4の態様に係るいずれか1つの態様の生成物の使用に関する。
【0055】
本発明の第5の態様に従った生成物の使用は、細胞増殖が、転移過程に関連しており、また、疾患の初期段階で使用される場合において、特に有利である。
【0056】
本発明の第5の態様に従った生成物の使用は、第2の抗癌生成物、特に、細胞毒性生成物と組み合わせると、特に有利である。
【0057】
第2の抗癌生成物は、シスプラシン又はオキサリプラチンなどのプラチナ誘導体、ドセタキセル又はパクリタキセルなどのタキソイド、5−FU、カペシタビン又はゲムシタビンなどのプリン塩基誘導体又はピリミジン塩基誘導体、ビンクリスチン又はビンブラスチンなどのビンカ、マスタード、スタウロスポリン、エリプチシン又はイリノテカン、トポテカンなどのカンプトテシンなどの縮合芳香族複素環、CA4Pなどのコンブレタスタチン、並びにリン酸コルチノールなどのコルヒチン誘導体から、有利に選択される。
【0058】
第2の抗癌生成物は、好ましくは、ドセタキセル、オキサリプラチン又はイリノテカンである。
【0059】
様々な市販の低分子量ヘパリンによるヘパラナーゼの阻害が研究されると、それらは全てヘパラナーゼを阻害することが明白になる(エノキサパリン、チンザパリン、フラグミン等)。しかし、本発明者らは、新しい超低分子量ヘプリン(ULMWH)(WO02/08295及び国際出願PCT/FR03/02960、未公開)は、エノキサパリンよりもATIIIに対する親和性を伴う更に多くの配列を含むが、ヘパラナーゼを阻害しないということを観察できた。その結果、上記の理論に対する矛盾がここに存在する。
【0060】
本発明に従って使用される方法は、特に、Is−IIa−IIs構造の、IsoATIIである六糖の形成をもたらし、下記は六糖Is−IIa−IIs(IsoATIII)である:
【0061】
【化28】
【0062】
下記の結果は、この六糖のIso ATIIIが非常に優れたヘパラナーゼの阻害剤であることを示す。また、ATIIIに対する親和性を持たず、従って、抗血栓活性を欠くという多大な有利さを有する。本発明の大きな利点は、ヘパリノイドの抗血栓特性を、それらのヘパラナーゼ阻害性質から分離することである。ヘパリン及びLMWHと比較すると、六糖Iso ATIIIの治療限界は大きく増大し、抗転移剤として使用できる可能を秘めている。本発明において、本発明者らは、それ自体の使用及び六糖のIso ATIIIのみの調製又はヘパラナーゼ1を用いたヘパリンの調節された解重合から生じる、他の六糖との混合物としての調製を権利として請求する。更に、aXa活性が消滅するまで、ヘパラナーゼ1を用いてヘパリンを解重合するという着想、及び抗転移剤としてのこの混合物の使用を権利として請求する。この場合、これは、非抗血栓性の、具体的には、ヘパラナーゼ阻害性のLMWHとなる。
【0063】
本発明は、単離された又は混合物としての、以下の生成物調製及びの抗転移剤としての使用を権利として請求する。
【0064】
【化29】
【0065】
実験の部
GPC
2つのTSK Super SW2000カラム(300×4.6mm)及び1つのTSK Super ガードカラム(35×4.6mm)(TOSOH BIOSEP)を用いて、ゲル排除クロマトグラフィーを実行する。UV域232nmにおいて、吸光光度分析法により、検出を行なう。移動相は、0.1M酢酸アンモニウムである。注入された量は5μlである。
【0066】
CTA−SAXクロマトグラフィー
CTA−SAX方法により、HPLCのモニタリングを実行する。使用したカラムは、45℃で、0.2ml/分にて、4時間、水/メタノール(68/32)v/vの混合物中の、1mM セチルトリメチルアンモニウムリン酸水素溶液の浸透により、セチルトリメチルアンモニウムがその上に吸着された、3μm粒子のHypersil BDS(150×2.1mm)である。
【0067】
この種のカラム上での分離条件は次のとおりである:グラフト化されたカラムの温度を40℃に維持する。メタンスルホン酸を添加することにより、溶媒AをpH3に調節した水で、溶出勾配を行なう。溶媒Bは、pHを2.6に調節されたメタンスルホン酸アンモニウムの2N溶液である。その勾配溶出は、次のとおりである。
【0068】
【表1】
【0069】
使用される検出は、232nmでのUV領域における吸光光度分析法である。また、アセチル化されたオリゴ糖に対し特異的な検出として、202〜247nmも使用される。
【0070】
CTA−SAX上でのセミ分取クロマトグラフィー
45℃で、2ml/分にて、4時間、水/メタノール(68/32)v/vの混合物中の、1mM セチルトリメチルアンモニウムリン酸水素溶液の浸透により、セチルトリメチルアンモニウム鎖がその上にグラフト化された、5μm粒子のHypersil BDSカラム(250×20mm)上のクロマトグラフィー。室温で分離を実行する。使用される溶出勾配:溶媒Aは、HClを添加することによってpHを2.5にした水である。溶媒Bは、pH2.5に調節された、2N NaCl溶液である。
【0071】
【表2】
【0072】
232nmでのUV域において、検出を行なう。100mgの六糖画分を、各分離において注入できる。
【0073】
六糖Iso ATIIIの調製
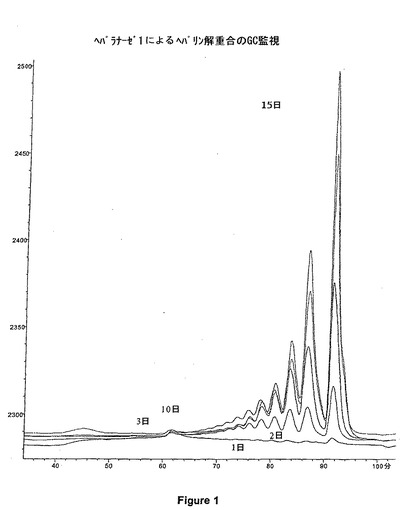
ヘパラナーゼ1を用いたヘパリンのATIIIの親和性部位の開裂によって、六糖ΔIs−IIaid−IIsglu(六糖iso ATIII)を得る。ヘパラナーゼ1を用いたヘパリンの解重合は、内因性溶解性であり:それらの非還元末端が不飽和のオリゴ糖の混合物をもたらす。反応終了後、二糖、四糖及び六糖の混合物を得る。ヘパリンの最も硫酸化された全領域を開裂し、二糖及び四糖へ転換する。アセチル化された部分のみが、六糖形態を維持し、特に、−GlcNS(6S又は6OH)−IdoA−GlcNAc(6S又は6OH)−GlcA−GlcNS(3S又は3OH,6S又は6OH)−型の鎖を維持する。
【0074】
【化30】
【0075】
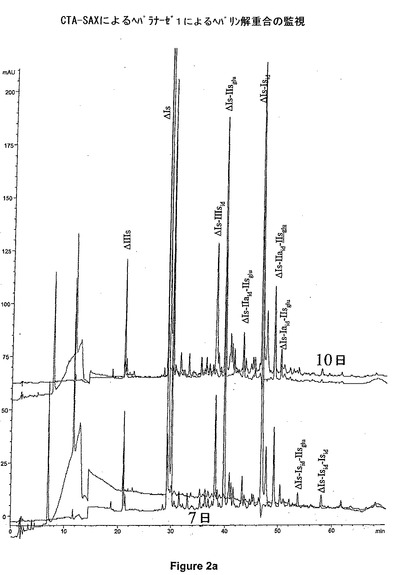
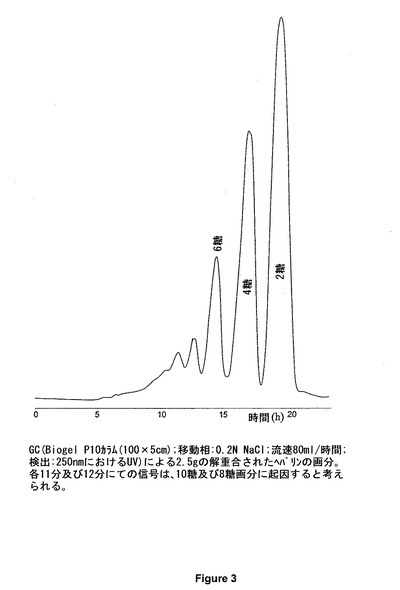
ヘパリンの解重合を次の条件下にて実行する:ブタの粘膜から得たヘパリン3gを、pH7に調節された、0.2M NaCl、0.02%BSA、5mM Na2HPO4の溶液30mL中に溶解する。解重合温度は16℃である。ヘパラナーゼ1の2IUを最初に導入する。7日後、ヘパリナーゼ1の追加単位を添加する。15日後、ヘパリンの解重合を終了したとみなす。その反応は、TSK Super SW 2000カラム(図1)上、又はCTA−SAXカラム(図2a)上のどちらかの分析用GCによってモニタリングする。サイズが八糖よりも大きなオリゴ糖の割合を制限し、前記混合物中の、2つの主な硫酸化された六糖であるΔIs−Isid−Isid及びΔIs−Isid−IIsgluを、四糖に解重合した際に、酵素反応が、十分促進されたと見なすことができる。酵素反応が終わると、0.2μmの膜を通して前記溶液をろ過し、次いで、2段階で、0.2N NaClの移動相が循環する、Biogel P10(Bio Rad)が充填されたGCカラム上に注入される(図3)。六糖のΔIs−1Iaid−IIsgluは、アルカリ溶媒中で非常に脆弱である:六糖のΔIs−1Iaid−IIsgluは、その3−O−硫酸化末端のグルコサミンを失い、pHが8を超えるとすぐに五糖ΔIs−IIaid−GIcAに転換される。従って、全六糖画分を、僅かに酸性化することが(pHが5〜6)非常に重要である。図4には、全六糖画分のクロマトグラムを示されている。
【0076】
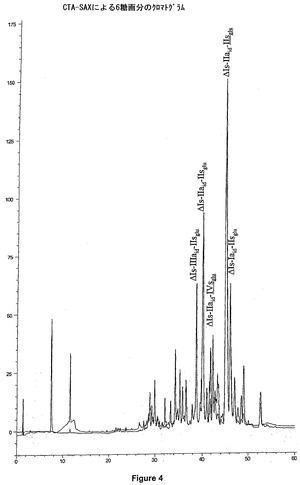
最終相は、CTA−SAXでグラフトされた、Hypersil BDS C18(5μm)が充填された、25×2.1cmのカラム上におけるセミ分取分離からなる(図5)。前記画分は、HPLCにより制御されている。セミ分取クロマトグラフィーで使用される移動相が、塩化ナトリウム溶液であるので、試料の最終の脱塩を調製する必要がある。これは、2段階で実行される。NaClの95%を取り除く第1段階は、水中で1/10に希釈された後、カラム上でそれらをろ過することによって、Q−Sepharose High Flow陰イオン交換相(Parmacia)(40×2.6cmカラム)上で、前記単離された六糖を含有する画分を再濃縮することからなる。前記六糖を、最少量(約50ml)で、1.5N NaClO4溶液を用いて溶出し、過塩素酸六糖溶液を得る。
【0077】
最終脱塩の第2段階は、前段階で、Sephadex G10カラム(100×7cm)上で得た過塩素酸六糖の溶液を注入して実行する。232nmにおけるUV検出及び塩の検出を可能にする伝導測定器により、モニタリングを行なう。
【0078】
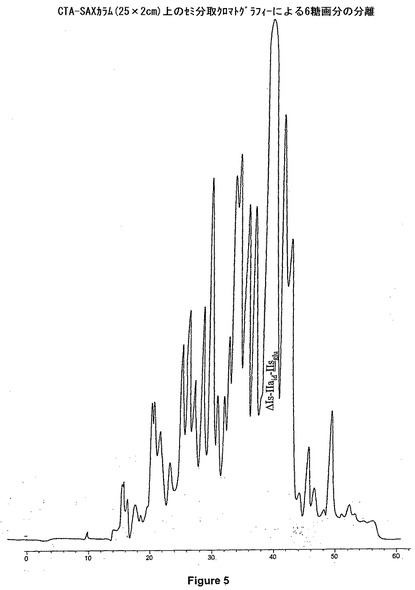
六糖と過塩素酸の分離の質が不十分である場合、この工程を繰返す必要があり得る。次いで、六糖の溶液を凍結乾燥させる。このようにして、ナトリウム塩形態の六糖ΔIs−IIaid−IIsglu108mgを得る。HPLC純度は92%となる(図6)。
【0079】
ヘパラナーゼ生物学的活性アッセイ:
ヘパラナーゼの阻害能力に関するHexa Iso ATIII関連の評価を、次のように実行した。
【0080】
放射線標識されたヘパリン/ヘパラン硫酸(HS)を、ヘパラナーゼで分解し、ゲル浸透クロマトグラフィー(FPLC)及び液体シンチレーションによって収集された画分を数えることにより測定可能な、低分子量のHS断片を生成する。
【0081】
ブタ腸粘膜からの未分画ヘパリン(ナトリウム塩)(等級Ia、183USP/mg)を、Sigma Biochemicals (Deisenhofen、ドイツ)から得た。
【0082】
ヘパリチナーゼ(HPリアーゼ(EC 4.2.2.8))を、Seigaku(東京、日本)から得た。
【0083】
TSK4000を、Toso Haasから得て、Pharmacia/LKB(Freiburg、ドイツ)から、プレカラムを備えたSepharose Qカラムを得た。
【0084】
代謝標識によって35−Sで標識されたヘパラン硫酸塩(プロテオグリカン)を調製するために、子宮線維芽細胞株を使用した。この細胞株は、シンデカン及びグリピカンなどの、様々なヘパラン硫酸プロテオグリカン(HS−PG)を比較的大量に生成することが示されている(Drzeniek,et al.Blood93:2884−2897,1999)。
【0085】
組織培養培地中の33μCi/mlの35−S−硫酸塩の存在下で24時間、約1×106細胞/mlの細胞密度で、細胞をインキュベートすることにより、この標識化を行なう。次いで、上清を収集し、プロテアーゼ阻害剤であるPMSF(フッ化フェニルメチルスルホニル)(1mmol/l)を添加する。試料は、ヘパラン硫酸プロテオグリカンを比較的大量に含有するので、またヘパラナーゼ酵素の特異性のために、コンドロイチン硫酸及びデルマタン硫酸(プロテオグリカン)の除去が必要でなく、HS−PGをSepharose Q上で、陰イオン交換クロマトグラフィーにより精製する。
【0086】
ヘパラナーゼを、フィコール勾配操作による、多形核細胞(PMN)で濃縮された、ヒト末梢血白血球(PBL、バッフィコート)から単離した。その単離したPMNの濃度を2.5×107細胞/mlに調節し、1時間、4℃で、インキュベートする。次いで、ヘパラナーゼを含有する上清を収集し、pHを6.2(クエン酸−リン酸バッファ20mM)に調節し、すぐに使用するか、又は−20℃にて、アリコートで、凍結保存する。
【0087】
約2200cpm/ml(cpm=計数毎分)に調節された、35−S−標識されたヘパラン硫酸(プロテオグリカン)200μLを、ヘパラナーゼを含有するPMN上清1μlで、37℃にて、18時間、インキュベートする。上記で得た、混合物200μlを、TSK4000ゲル浸透クロマトグラフィーカラム(FPLC)上で試料を採取し、その画分を収集して、液体シンチレーションカウントによって分析する。下記の式に従って、その分解量を測定した。
【0088】
%分解=[[Σ計数(cpm)画分20−33(HEP)−Σ計数(cpm)画分20−33(CONT)]/[総合計数(cpm)画分12−33(CONT)]]×100。
【0089】
例えば、%分解を下記のとおりに計算する:ヘパラナーゼでの処理後の試料の画分20−33における計数(cpm)から、対照試料のバックグランドノイズ計数(cpm)(画分20−33)を差し引いた合計を、カラムに用いられる総計数(画分12−33)で割る。補正因子を使用して、2200計数/cpmにおける、クロマトグラフィーの様々な回数の総計数を標準化した。その結果を%分解として得る。阻害アッセイにて、対照試料(ヘパラナーゼを伴う。)の分解を100%(分解)に固定し、その%阻害値を、これに基づき計算した。スルファターゼ活性の修正は、スルファターゼ活性が検出されない可能性があるため、必要でない。
【0090】
次のヘパラナーゼ阻害剤:未分画ヘパリン(UF−H)及びHexa Iso ATIIIを、3つの異なる濃度にて、上記のプロトコルでアッセイした。その比較は、重量基準で実行した。データを、ヘパラナーゼ活性の%阻害として表す。
【0091】
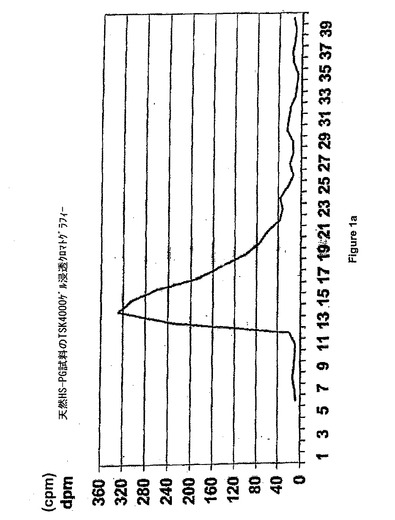
結果
まず、ヘパラナーゼアッセイを、この研究の必要性に応じて最適化した。実用的な理由から、分解アッセイにおけるインキュベーション時間を18時間の時点で確立した。標識化の効率性及びヘパラン硫酸(プロテオグリカン)の含有量に応じて、ヘパラン硫酸(プロテオグリカン)総計数を、約2200cpm/試料に固定し、ヘパラン硫酸(プロテオグリカン)の1バッチで全アッセイの実行を可能にした。図1aは、天然試料のTSK4000ゲル浸透クロマトグラフィーを示す。図1bは、その試料の分子分布における、ヘパラナーゼ誘起シフトを示す。次いで、ヘパラン硫酸プロテオグリカン約80%を分解できるヘパラナーゼ量を決定する(ヘパラン硫酸プロテオグリカン約35%及びコンドロイチン/デルマタン硫酸プロテオグリカン約65%を含有する試料)。従って、10−80%の範囲の分解は、比較的直線状であり、阻害剤の効果の決定において許容される。図1cは、97.3%の阻害で、ヘパラナーゼ活性の1μg/mlの未分画ヘパリン(UFH)の効果を示す。
【0092】
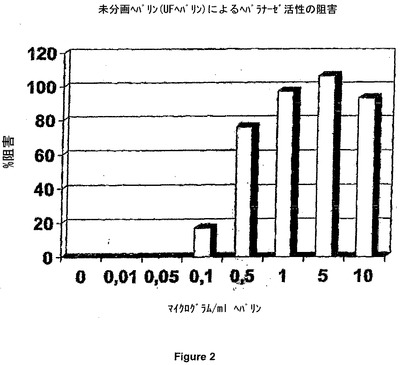
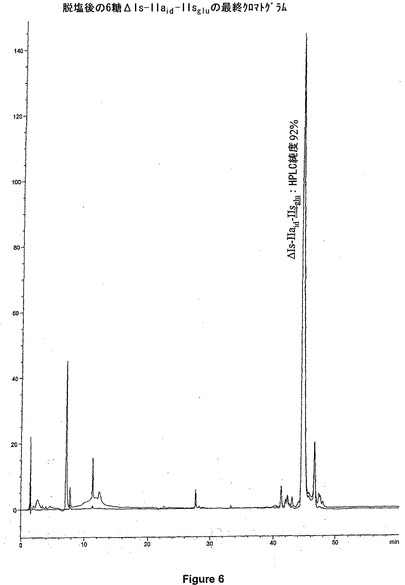
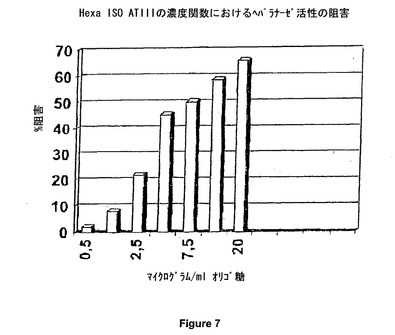
アッセイ状態を決定した後、ブタ腸粘膜から得た未分画ヘパリン(UFH)の効果を測定した。図2は、用量依存的な阻害を示す。ヘパラナーゼ活性の実質的に完全な阻害が、1μg/ml(最終濃度)の未分画ヘパリン(UFH)濃度にて、観察された。図7は、Hexa Iso ATIIIによる用量依存阻害を示す。これらのデータに基づいて、Hexa Iso ATIIIが、強度のヘパラナーゼ阻害活性を示すと結論付けることが可能である。
【0093】
次の刊行物の内容を、参照により、本明細書に組み込む。
【0094】
C.R.Parish,et al.Biochim.Biophys, Acta1471(2001)99−108。
【0095】
M.Bartlett,et al.Immunol.Cell Biol.73(1995)113−124。
【0096】
I.Vlodavsky,et al.IMAJ2(2000)37−45。
【0097】
Y.Matzner,et al.J.Clin.Invest.76(1985)、1306−1313。
【0098】
Z.Drzeniek,et al.Blood(1999)2884−2879。
【0099】
オリゴ糖を豊富に含有した他の分画は、ヘパラナーゼ1による、ヘパリンの分解生成物から単離できる。従って、六糖の場合、単一のCTA−SAXクロマトグラフの精製で十分である。この方法では、セチルトリメチルアンモニウム鎖が、45℃にて、2ml/分で、4時間、水−メタノールの混合物(68−32)v/v中の、セチルトリメチルアンモニウムリン酸水素の1mM溶液の浸透によりその上にグラフト化された、5μm粒子のHypersil BDS(250×20mm)カラムを使用する。
【0100】
室温で分離を実行する。次の勾配溶出を使用する:溶媒Aは、HClの添加によってpHを2.5に調節した水である。溶媒Bは、pHを2.5に調節したNaClの2N溶液である。
【0101】
【表3】
【0102】
検出をUV領域の232nmで行う。六糖画分100mgを、各分離において、注入できる。
【0103】
八糖及び十糖の画分の精製は、六糖画分の精製よりも更に複雑であり、一般的に、それはIonPac(R)AS11カラム(250×20mm)(Dionex)上で、更なる精製を必要とする。分離は室温で実行する。勾配溶出を使用する。溶媒Aは、過塩素酸の添加によりpH3に調節した水である。溶媒Bは、pH3に調節されたNaClO4の1M溶液である。
【0104】
【表4】
【0105】
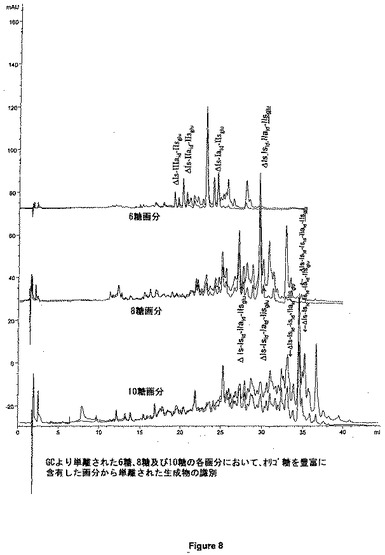
図8は、GCより単離された各六糖、八糖及び十糖において、オリゴ糖を豊富に含有した画分から単離された生成物の同定が可能である。
【0106】
酵素システムにおけるヘパラナーゼ阻害剤の活性評価
フォンダパリヌクスを分解する能力により、ヘパラナーゼの活性を決定する。フォンダパリヌクスの濃度を、その抗Xa因子の活性によって決定する。
【0107】
A.物質及び方法
ヘパラナーゼは、Sanofi−Synthelabo(Labege、フランス)によって、調製される。
【0108】
Xa因子をアッセイするための試薬は、Chromogenix(Montpellier、フランス)から販売されている。
【0109】
本発明の化合物の漸増濃度、ヘパラナーゼ阻害剤(可変希釈量:1nM〜10μM)を、ヘパラナーゼの一定濃度と混合する(各バッチにおいて、先行実験により、添加された0.45μg/mlのフォンダパリヌクスの分解に対し、十分な酵素活性を決定できる。)。37℃で5分後、その混合物をフォンダパリヌクスと接触させ、37℃で、1時間、放置する。95℃で、5分間加熱することにより、その反応を停止する。残存のフォンダパリヌクスの濃度を、Xa因子及びその特異的な発色基質(参照番号S2222)を添加することにより測定する。
【0110】
次の手順に従い、様々な混合物を調製する:
a)反応混合物
酢酸ナトリウムバッファ(0.2M、pH4.2)50μlを、フォンダパリヌクス(0.45μg/ml)50μl及びヘパラナーゼ溶液59μlと混合する。混合物を37℃で、1時間、インキュベートし、次いで95℃で、5分間、インキュベートする。pHは、2.4から7となる。次いで、反応混合物100μlを、175mM NaCl及び75mM EDTAを含有するpH14の50mMトリスバッファ50μlのと混合する。
【0111】
フォンダパリヌクスの抗Xa因子活性を、次の方法で測定する。
【0112】
b)フォンダパリヌクスの抗Xa因子活性のアッセイ
段階a)で得た溶液100μlを、AT(0.5μg/ml)100μlと混合する。この混合物を37℃で、2分間、維持し、次いで、Xa因子(7nkat/ml)100μlを添加する。その混合物を37℃で、2分間、維持し、発色基質(参照番号:S2222)(1mM)100μlを添加する。この混合物を37℃で、2分間、維持し、次いで酢酸(50%)100μlを添加する。
【0113】
光学濃度を405nmで読み取る。
【0114】
阻害剤なしで対照に対し、%阻害を決定する。%阻害曲線により、IC50を算出できる。
【0115】
【表5】
【図面の簡単な説明】
【0116】
【図1】ヘパラナーゼ1によるヘパリン解重合(GC監視)。
【図1a】天然HSPGαゲル浸透クロマトグラフ。
【図1b】ヘパラナーゼ処理後のゲル浸透クロマトグラフ。
【図1c】未分画ヘパリンのゲル浸透クロマトグラフ。
【図2】未分画ヘパリンのヘパラナーゼ阻害活性。
【図2a】ヘパラナーゼ1によるヘパリン解重合(CTA−SAX監視)。
【図3】解重合されたヘパリンの各画分。
【図4】解重合されたヘパリンの六糖画分。
【図5】六糖画分の半分取分離。
【図6】脱塩後の六糖画分分析。
【図7】Hexa Iso ATIIIのヘパラナーゼ阻害活性。
【図8】単離された各糖画分のGC分析。
【技術分野】
【0001】
本発明は、新規化学化合物、特に、新規オリゴ糖に関し、これらを調製する方法、これらの使用、及びこれらを含有する薬学的組成物に関する。これらのオリゴ糖は、癌の治療、特に、転移形成の予防及び阻害に対し、有効である。
【0002】
より具体的には、第1の態様において、本発明は、本来抗血栓特性を有する多糖類を解重合する方法に関する。
【背景技術】
【0003】
抗血栓特性を有する多糖類の解重合するための方法は公知である。これらの方法の一般的な態様は:
−出発多糖類を医薬製品として使用する際に起こる、副作用を制限するために、更に低い平均分子量のオリゴ糖を得ることを目的とし;
−解重合後、満足のいく抗血栓活性を維持することである。
【0004】
ヘパリン又はエノキサパリン、チンザパリン若しくはフラグミンなどの低分子量のヘパリンなどの抗血栓特性を有する市販の多糖類は、全てヘパラナーゼ阻害剤である。
【0005】
正常な生理学的状態のもとでは、細胞が酵素のヘパラナーゼを発現する。この酵素により、間接的に、有糸分裂誘発、新血管形成及び組織修復を調節することが可能となる。ヘパラナーゼの作用機序の1つは、へパラン硫酸プロテオグリカン(HSPG)を開裂することである。このグリコサミノグリカンは、内皮細胞の表面に存在し、基底膜(細胞外マトリックス)の結合を確保している。へパラン硫酸プロテオグリカンの開裂は、FGF2などの成長因子の放出をもたらす。この成長因子の放出は、有糸分裂誘発及び血管新生にとって必要である。しかし、これは、これらの生物学的機序を引き起こすのに十分でなく、タンパク質のアロステリック修飾を起こし、その受容体との相互作用を促進するためには、FGF2が、グリコサミノグリカンに結合する必要がある。実際、HSPGの開裂を通して、ヘパラナーゼは、FGF2に結合するヘパラン硫酸フラグメントを生成し、その受容体との相互作用を促進し、こうして、上述の生物学的機序を誘導する。細胞外マトリックスにおけるHSPGの開裂及び毛細血管の脱構造化により、細胞遊出が可能になる。
【0006】
ヘパラナーゼは、腫瘍細胞によって過剰発現され、それ故、その転移及び血管新形成を促進する。これらの現象は、癌性腫瘍の増殖及び生存にとって不可欠である。
【0007】
ヘパラン硫酸に構造的に類似のグリコサミノグリカンであるヘパリンは、ヘパラナーゼの強力な阻害剤であることが知られている。この効力は、長い間、特異的なATIII−結合配列の存在に起因すると考えられている。実際に、この配列は、ヘパラン硫酸においてはそれほどでもないが、これらの2つのグリコサミノグリカンに共通している。ヘパラナーゼ開裂域を下記に表す:
【0008】
【化15】
【0009】
このα−エンドグリコシダーゼによる開裂点は、最小のATIII−結合配列の中央に位置している。従って、抗血栓及びヘパラナーゼ阻害特性が密接に関連すると考えることができる(実際に、ヘパリンは、ヘパリン硫酸に対する競合的基質である。)。その結果、抗転移物質として、このグリコサミノグリカンを使用するのは困難である。実際、その強力な抗凝血性質が、治療の限度を制限し、大量出血などの深刻な副作用を引き起こす。更に、特定の場合において、ヘパリンの反復注入は、致命的転帰をもたらす血小板減少症を引き起こす可能性がある(ヘパリン及び血小板第4因子(PF4)の結合に関する免疫反応)。
【0010】
抗移転活性との相関関係において、オリゴ糖の構造及びそれらの抗ヘパラナーゼ特性の関連を強調した文書はほとんどない。従って、Bitan,et al.(Isr.J.Med.Sci.1995;31:106−118)は、ヘパラナーゼ阻害ヘパリン種により肺メラノーマの定着化阻害に必要とされる構造条件を特定する:ヘパラナーゼは、16又はそれ以上の糖を含有するヘパリン断片により効果的に阻害される(要約;図2、p110;図3、p111;p116、右側コラム、第2文)。六糖は、ヘパラナーゼ阻害剤として劣っていると記述されている(図8、p115)。加えて、少なくとも4000ダルトン以上の分子量を有する分子を用いた場合にのみ、ヘパラナーゼの阻害が可能になると言われている(要約:p116、右側コラム、第4文)。しかしながら、ヘパラナーゼの阻害性を決定する方法は、様々な試験品の存在下で細胞外マトリックスを分解する細胞能力を評価するので、間接的な方法である(p108;右側コラム、第2段落「Degradation of Sulfated Proteoglycans(硫酸化プロテオグリカンの分解)」)。従って、この方法は、ヘパラナーゼに対し特異的ではない。更に、前述の全生成物は、ヘパリンを化学的に開裂する方法(亜硝酸)により取得される(p108;左側コラム、第2段落「Heparin−Derived Oligosaccharides(ヘパリン由来のオリゴ糖)」。
【0011】
次も参照されたい。
【0012】
Vlodavski I,et al.Modulation of neovascularization and metastasis by species of heparin,Heparin and related Polysaccharides,D.A.Lane,et al.,Editor,Plenum Press, New York, 1992;
Parish CR,et al.Evidence that sulphated polysaccharides inhibit tumor metastasis by blocking tumor−cell−derived heparanases, Int. J. Cancer 40:511−518,1987。
【0013】
現在のところ、商業的に許容される解決策がないため、抗転移化合物に対する必要性は非常に大きい。現在公知のヘパラナーゼ阻害性多糖及びオリゴ糖は、天然原料(ヘパリン)又は、事実上、実行するのが困難な方法(いくつかの低分子量のヘパリン)から直接的に生じたもので、特に、治療を受ける患者に出血の恐れがある場合において、抗癌治療に適さない際立った抗血栓成分を示す。
【発明の開示】
【発明が解決しようとする課題】
【0014】
従って、現在の問題点の1つは、容易かつ再現性のある方法を通して、基本的に抗血栓活性のない、顕著な抗ヘパラナーゼ活性を示す生成物を得ることである。
【課題を解決するための手段】
【0015】
この目的のために、驚くべきことに、特に、Xa及びIIa因子の阻害性のために、その抗血栓活性が実質的に消滅するまで(<35IU/mg)、多糖類がヘパラナーゼ1で解重合される、抗血栓特性を本来有する多糖類を解重合するための新規の方法により、顕著な抗ヘパラナーゼ活性を保存する生成物を取得することが可能であると明らかと成った。
【0016】
それ故に、この方法では、同時にその抗血栓要素を除去する一方で、多糖類の抗ヘパラナーゼ部位濃縮生成物を取得するための効果的な手段を構成する。本方法は、その抗血栓活性が20IU/mg未満になるまで、多糖類の解重合が遂行される場合に、より有利に使用される。
【0017】
とりわけ、5000Da未満、好ましくは、3000Da未満の平均分子量に達するまで、解重合が遂行される。あらゆる予想に反し、容易に実行可能な、この方法によって得られた生成物は、特に、優れたヘパラナーゼ阻害剤である六糖を含有する。加えて、これらの六糖は、一般的に1000ないし2000ダルトンであるので、平均分子量が4000ダルトンよりもかなり低い。多糖は、好ましくはヘパリンである。六糖画分の混合物が、硫酸化六糖ΔIs−Isid−Isid及びΔIs−Isid−IIsgluを実質的に含まなくなるまで、解重合は有利に続行される。
【0018】
酵素は、一般的に、「生理学的」条件下、すなわち、それらが抽出される生物中で、インビボで正常に機能する条件下で使用される(特に、pH、温度、イオン強度、場合により物理的補因子(可視光など)又は化学的補因子(補酵素など)。多くの酵素は、普通、生理学的温度を超える温度、例えば45〜50℃などで使用できる。本発明者らの状況では、あらゆる予想に反し、許容される選択性及び反応速度の状態下、好ましくは10〜20℃、特に16℃の温度で解重合が起こることが可能であり、解重合反応中に形成される、ヘパラナーゼ阻害化合物をできるだけ適切に維持することを観察した。更に、これにより、反応終了時の反応媒体中のヘパリナーゼ1の最終濃度を限定することが可能となる。実際に、ヘパラナーゼ1に対する最適反応温度を下回る温度(25ないし45℃とすることができる。)で、その反応を実行することにより、反応過程で、酵素の過剰な数の添加を防ぐことができる。酵素は、一般的に、基質欠乏以外の原因による反応速度の低下が観測される際に添加される。従って、特に、限られた酵素が存在するため、比較的低い反応温度を用いることにより、その後に実施される場合がある精製段階を間接的に促進することが可能である。それ故に、結果として、5〜40℃、好ましくは10〜20℃の温度で、解重合を実行できる。
【0019】
特に、二糖及び四糖などの、解重合中に形成された低分子量のオリゴ糖を取り除くことができるようにするために、本発明の方法では、pH8未満及びpH5超で、多糖類の解重合の生成物が、ゲル浸透クロマトグラフィーによって精製される工程を用いて、有利に遂行される。
【0020】
本発明の方法は、固定相(例えば、シリカ)が、(i)C18−グラフト化され、且つ(ii)セチルトリメチルアンモニウム(CTA−SAX)でグラフト化された逆相という、高性能液体クロマトグラフィ(HPLC)による、後の精製段階を有利に含む。
【0021】
前記方法は、また、好ましくは、200〜250nmの間で実質的に透明になる電解質を水溶液中に含有する移動相の使用を有利に含む、第1の脱塩段階も含む。許容される電解液は、NaClを含むが、200〜250nMでUV検出器とともに使用する場合には、過塩素酸、メタンスルホン酸塩又はNaなどのアルカリ金属のリン酸塩の使用が望ましい。第1の脱塩段階に対して許容される固定相は、陰イオン交換樹脂である。特に好ましい樹脂は、Sepharose Q(R)樹脂が望ましい。
【0022】
前記方法は、好ましくは、例えば、Sephadex G10(R)型の分子排除ゲルを好ましく使用する、第2の脱塩段階も含むことが可能である。
【0023】
HPLCカラムから排出されたときに収集される画分中に存在する本発明の生成物を検出する別の解決策は、必要に応じて、回折装置を使用することからなり得る。
【0024】
別の許容される脱塩技術には、例えば、重合体膜を使用する、浸透圧技術の使用を含む。
【0025】
第2の態様において、本発明は、第1の態様に従う方法によって得られた生成物に関する。
【0026】
Petitou,et al.J.Biol. Chem.(1988)、263(18)、8685−8690は、ヘパラナーゼ1を用いたヘパリンの部分解重合の方法により取得される生成物から単離された、ナトリウム塩形態の式ΔIs−IIaidu−IIsgluの六糖を開示している。この生成物は、抗血栓活性を示さないと説明されている。該生成物の他の性質については実証されていない。
【0027】
第3の態様において、本発明は以下の式(I)の生成物に関する。
【0028】
【化16】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択され、
n=0、R=SO3M及びM=Naの生成物は除かれる。)
【0029】
意外にも、MがLi、Na及びK、好ましくはNaから選択される場合には、本発明の第3態様に係る生成物が、より優れた物理化学的性質を示すことが観察されている。特に、溶解度及び安定度が向上している。
【0030】
式(I)の好ましい生成物は、n=0の生成物である。
【0031】
第4の態様において、本発明は、六糖に関する。
【0032】
下記の式(Ia)の生成物:
【0033】
【化17】
は、その第4態様に従った本発明に沿う。
【0034】
下記の式(1b)の生成物:
【0035】
【化18】
は、その第4態様に従った本発明に沿う。
【0036】
下記の式(Ic)の生成物:
【0037】
【化19】
は、その第4態様に従った本発明に沿う。
【0038】
下記の式(Id)の生成物:
【0039】
【化20】
は、その第4態様に従った本発明に沿う。
【0040】
下記の式(Ie)の生成物:
【0041】
【化21】
は、その第4態様に従った本発明に沿う。
【0042】
下記の式(If)の生成物:
【0043】
【化22】
は、その第4態様に従った本発明に沿う。
【0044】
下記の式(Ig)の生成物:
【0045】
【化23】
は、その第4態様に従った本発明に沿う。
【0046】
下記の式(Ih)の生成物:
【0047】
【化24】
は、その第4態様に従った本発明に沿う。
【0048】
下記の式(Ij)の生成物:
【0049】
【化25】
は、その第4態様に従った本発明に沿う。
【0050】
下記の式(Ik)の生成物:
【0051】
【化26】
は、その第4態様に従った本発明に沿う。
【0052】
下記の式(Im)の生成物:
【0053】
【化27】
は、その第4態様に従った本発明に沿う。
【0054】
第5の態様において、本発明は、特に癌、とりわけ乳癌、肺癌、前立腺癌、結腸癌又は膵臓癌に関連する、細胞増殖を調節するための、前記第2の態様から第4の態様に係るいずれか1つの態様の生成物の使用に関する。
【0055】
本発明の第5の態様に従った生成物の使用は、細胞増殖が、転移過程に関連しており、また、疾患の初期段階で使用される場合において、特に有利である。
【0056】
本発明の第5の態様に従った生成物の使用は、第2の抗癌生成物、特に、細胞毒性生成物と組み合わせると、特に有利である。
【0057】
第2の抗癌生成物は、シスプラシン又はオキサリプラチンなどのプラチナ誘導体、ドセタキセル又はパクリタキセルなどのタキソイド、5−FU、カペシタビン又はゲムシタビンなどのプリン塩基誘導体又はピリミジン塩基誘導体、ビンクリスチン又はビンブラスチンなどのビンカ、マスタード、スタウロスポリン、エリプチシン又はイリノテカン、トポテカンなどのカンプトテシンなどの縮合芳香族複素環、CA4Pなどのコンブレタスタチン、並びにリン酸コルチノールなどのコルヒチン誘導体から、有利に選択される。
【0058】
第2の抗癌生成物は、好ましくは、ドセタキセル、オキサリプラチン又はイリノテカンである。
【0059】
様々な市販の低分子量ヘパリンによるヘパラナーゼの阻害が研究されると、それらは全てヘパラナーゼを阻害することが明白になる(エノキサパリン、チンザパリン、フラグミン等)。しかし、本発明者らは、新しい超低分子量ヘプリン(ULMWH)(WO02/08295及び国際出願PCT/FR03/02960、未公開)は、エノキサパリンよりもATIIIに対する親和性を伴う更に多くの配列を含むが、ヘパラナーゼを阻害しないということを観察できた。その結果、上記の理論に対する矛盾がここに存在する。
【0060】
本発明に従って使用される方法は、特に、Is−IIa−IIs構造の、IsoATIIである六糖の形成をもたらし、下記は六糖Is−IIa−IIs(IsoATIII)である:
【0061】
【化28】
【0062】
下記の結果は、この六糖のIso ATIIIが非常に優れたヘパラナーゼの阻害剤であることを示す。また、ATIIIに対する親和性を持たず、従って、抗血栓活性を欠くという多大な有利さを有する。本発明の大きな利点は、ヘパリノイドの抗血栓特性を、それらのヘパラナーゼ阻害性質から分離することである。ヘパリン及びLMWHと比較すると、六糖Iso ATIIIの治療限界は大きく増大し、抗転移剤として使用できる可能を秘めている。本発明において、本発明者らは、それ自体の使用及び六糖のIso ATIIIのみの調製又はヘパラナーゼ1を用いたヘパリンの調節された解重合から生じる、他の六糖との混合物としての調製を権利として請求する。更に、aXa活性が消滅するまで、ヘパラナーゼ1を用いてヘパリンを解重合するという着想、及び抗転移剤としてのこの混合物の使用を権利として請求する。この場合、これは、非抗血栓性の、具体的には、ヘパラナーゼ阻害性のLMWHとなる。
【0063】
本発明は、単離された又は混合物としての、以下の生成物調製及びの抗転移剤としての使用を権利として請求する。
【0064】
【化29】
【0065】
実験の部
GPC
2つのTSK Super SW2000カラム(300×4.6mm)及び1つのTSK Super ガードカラム(35×4.6mm)(TOSOH BIOSEP)を用いて、ゲル排除クロマトグラフィーを実行する。UV域232nmにおいて、吸光光度分析法により、検出を行なう。移動相は、0.1M酢酸アンモニウムである。注入された量は5μlである。
【0066】
CTA−SAXクロマトグラフィー
CTA−SAX方法により、HPLCのモニタリングを実行する。使用したカラムは、45℃で、0.2ml/分にて、4時間、水/メタノール(68/32)v/vの混合物中の、1mM セチルトリメチルアンモニウムリン酸水素溶液の浸透により、セチルトリメチルアンモニウムがその上に吸着された、3μm粒子のHypersil BDS(150×2.1mm)である。
【0067】
この種のカラム上での分離条件は次のとおりである:グラフト化されたカラムの温度を40℃に維持する。メタンスルホン酸を添加することにより、溶媒AをpH3に調節した水で、溶出勾配を行なう。溶媒Bは、pHを2.6に調節されたメタンスルホン酸アンモニウムの2N溶液である。その勾配溶出は、次のとおりである。
【0068】
【表1】
【0069】
使用される検出は、232nmでのUV領域における吸光光度分析法である。また、アセチル化されたオリゴ糖に対し特異的な検出として、202〜247nmも使用される。
【0070】
CTA−SAX上でのセミ分取クロマトグラフィー
45℃で、2ml/分にて、4時間、水/メタノール(68/32)v/vの混合物中の、1mM セチルトリメチルアンモニウムリン酸水素溶液の浸透により、セチルトリメチルアンモニウム鎖がその上にグラフト化された、5μm粒子のHypersil BDSカラム(250×20mm)上のクロマトグラフィー。室温で分離を実行する。使用される溶出勾配:溶媒Aは、HClを添加することによってpHを2.5にした水である。溶媒Bは、pH2.5に調節された、2N NaCl溶液である。
【0071】
【表2】
【0072】
232nmでのUV域において、検出を行なう。100mgの六糖画分を、各分離において注入できる。
【0073】
六糖Iso ATIIIの調製
ヘパラナーゼ1を用いたヘパリンのATIIIの親和性部位の開裂によって、六糖ΔIs−IIaid−IIsglu(六糖iso ATIII)を得る。ヘパラナーゼ1を用いたヘパリンの解重合は、内因性溶解性であり:それらの非還元末端が不飽和のオリゴ糖の混合物をもたらす。反応終了後、二糖、四糖及び六糖の混合物を得る。ヘパリンの最も硫酸化された全領域を開裂し、二糖及び四糖へ転換する。アセチル化された部分のみが、六糖形態を維持し、特に、−GlcNS(6S又は6OH)−IdoA−GlcNAc(6S又は6OH)−GlcA−GlcNS(3S又は3OH,6S又は6OH)−型の鎖を維持する。
【0074】
【化30】
【0075】
ヘパリンの解重合を次の条件下にて実行する:ブタの粘膜から得たヘパリン3gを、pH7に調節された、0.2M NaCl、0.02%BSA、5mM Na2HPO4の溶液30mL中に溶解する。解重合温度は16℃である。ヘパラナーゼ1の2IUを最初に導入する。7日後、ヘパリナーゼ1の追加単位を添加する。15日後、ヘパリンの解重合を終了したとみなす。その反応は、TSK Super SW 2000カラム(図1)上、又はCTA−SAXカラム(図2a)上のどちらかの分析用GCによってモニタリングする。サイズが八糖よりも大きなオリゴ糖の割合を制限し、前記混合物中の、2つの主な硫酸化された六糖であるΔIs−Isid−Isid及びΔIs−Isid−IIsgluを、四糖に解重合した際に、酵素反応が、十分促進されたと見なすことができる。酵素反応が終わると、0.2μmの膜を通して前記溶液をろ過し、次いで、2段階で、0.2N NaClの移動相が循環する、Biogel P10(Bio Rad)が充填されたGCカラム上に注入される(図3)。六糖のΔIs−1Iaid−IIsgluは、アルカリ溶媒中で非常に脆弱である:六糖のΔIs−1Iaid−IIsgluは、その3−O−硫酸化末端のグルコサミンを失い、pHが8を超えるとすぐに五糖ΔIs−IIaid−GIcAに転換される。従って、全六糖画分を、僅かに酸性化することが(pHが5〜6)非常に重要である。図4には、全六糖画分のクロマトグラムを示されている。
【0076】
最終相は、CTA−SAXでグラフトされた、Hypersil BDS C18(5μm)が充填された、25×2.1cmのカラム上におけるセミ分取分離からなる(図5)。前記画分は、HPLCにより制御されている。セミ分取クロマトグラフィーで使用される移動相が、塩化ナトリウム溶液であるので、試料の最終の脱塩を調製する必要がある。これは、2段階で実行される。NaClの95%を取り除く第1段階は、水中で1/10に希釈された後、カラム上でそれらをろ過することによって、Q−Sepharose High Flow陰イオン交換相(Parmacia)(40×2.6cmカラム)上で、前記単離された六糖を含有する画分を再濃縮することからなる。前記六糖を、最少量(約50ml)で、1.5N NaClO4溶液を用いて溶出し、過塩素酸六糖溶液を得る。
【0077】
最終脱塩の第2段階は、前段階で、Sephadex G10カラム(100×7cm)上で得た過塩素酸六糖の溶液を注入して実行する。232nmにおけるUV検出及び塩の検出を可能にする伝導測定器により、モニタリングを行なう。
【0078】
六糖と過塩素酸の分離の質が不十分である場合、この工程を繰返す必要があり得る。次いで、六糖の溶液を凍結乾燥させる。このようにして、ナトリウム塩形態の六糖ΔIs−IIaid−IIsglu108mgを得る。HPLC純度は92%となる(図6)。
【0079】
ヘパラナーゼ生物学的活性アッセイ:
ヘパラナーゼの阻害能力に関するHexa Iso ATIII関連の評価を、次のように実行した。
【0080】
放射線標識されたヘパリン/ヘパラン硫酸(HS)を、ヘパラナーゼで分解し、ゲル浸透クロマトグラフィー(FPLC)及び液体シンチレーションによって収集された画分を数えることにより測定可能な、低分子量のHS断片を生成する。
【0081】
ブタ腸粘膜からの未分画ヘパリン(ナトリウム塩)(等級Ia、183USP/mg)を、Sigma Biochemicals (Deisenhofen、ドイツ)から得た。
【0082】
ヘパリチナーゼ(HPリアーゼ(EC 4.2.2.8))を、Seigaku(東京、日本)から得た。
【0083】
TSK4000を、Toso Haasから得て、Pharmacia/LKB(Freiburg、ドイツ)から、プレカラムを備えたSepharose Qカラムを得た。
【0084】
代謝標識によって35−Sで標識されたヘパラン硫酸塩(プロテオグリカン)を調製するために、子宮線維芽細胞株を使用した。この細胞株は、シンデカン及びグリピカンなどの、様々なヘパラン硫酸プロテオグリカン(HS−PG)を比較的大量に生成することが示されている(Drzeniek,et al.Blood93:2884−2897,1999)。
【0085】
組織培養培地中の33μCi/mlの35−S−硫酸塩の存在下で24時間、約1×106細胞/mlの細胞密度で、細胞をインキュベートすることにより、この標識化を行なう。次いで、上清を収集し、プロテアーゼ阻害剤であるPMSF(フッ化フェニルメチルスルホニル)(1mmol/l)を添加する。試料は、ヘパラン硫酸プロテオグリカンを比較的大量に含有するので、またヘパラナーゼ酵素の特異性のために、コンドロイチン硫酸及びデルマタン硫酸(プロテオグリカン)の除去が必要でなく、HS−PGをSepharose Q上で、陰イオン交換クロマトグラフィーにより精製する。
【0086】
ヘパラナーゼを、フィコール勾配操作による、多形核細胞(PMN)で濃縮された、ヒト末梢血白血球(PBL、バッフィコート)から単離した。その単離したPMNの濃度を2.5×107細胞/mlに調節し、1時間、4℃で、インキュベートする。次いで、ヘパラナーゼを含有する上清を収集し、pHを6.2(クエン酸−リン酸バッファ20mM)に調節し、すぐに使用するか、又は−20℃にて、アリコートで、凍結保存する。
【0087】
約2200cpm/ml(cpm=計数毎分)に調節された、35−S−標識されたヘパラン硫酸(プロテオグリカン)200μLを、ヘパラナーゼを含有するPMN上清1μlで、37℃にて、18時間、インキュベートする。上記で得た、混合物200μlを、TSK4000ゲル浸透クロマトグラフィーカラム(FPLC)上で試料を採取し、その画分を収集して、液体シンチレーションカウントによって分析する。下記の式に従って、その分解量を測定した。
【0088】
%分解=[[Σ計数(cpm)画分20−33(HEP)−Σ計数(cpm)画分20−33(CONT)]/[総合計数(cpm)画分12−33(CONT)]]×100。
【0089】
例えば、%分解を下記のとおりに計算する:ヘパラナーゼでの処理後の試料の画分20−33における計数(cpm)から、対照試料のバックグランドノイズ計数(cpm)(画分20−33)を差し引いた合計を、カラムに用いられる総計数(画分12−33)で割る。補正因子を使用して、2200計数/cpmにおける、クロマトグラフィーの様々な回数の総計数を標準化した。その結果を%分解として得る。阻害アッセイにて、対照試料(ヘパラナーゼを伴う。)の分解を100%(分解)に固定し、その%阻害値を、これに基づき計算した。スルファターゼ活性の修正は、スルファターゼ活性が検出されない可能性があるため、必要でない。
【0090】
次のヘパラナーゼ阻害剤:未分画ヘパリン(UF−H)及びHexa Iso ATIIIを、3つの異なる濃度にて、上記のプロトコルでアッセイした。その比較は、重量基準で実行した。データを、ヘパラナーゼ活性の%阻害として表す。
【0091】
結果
まず、ヘパラナーゼアッセイを、この研究の必要性に応じて最適化した。実用的な理由から、分解アッセイにおけるインキュベーション時間を18時間の時点で確立した。標識化の効率性及びヘパラン硫酸(プロテオグリカン)の含有量に応じて、ヘパラン硫酸(プロテオグリカン)総計数を、約2200cpm/試料に固定し、ヘパラン硫酸(プロテオグリカン)の1バッチで全アッセイの実行を可能にした。図1aは、天然試料のTSK4000ゲル浸透クロマトグラフィーを示す。図1bは、その試料の分子分布における、ヘパラナーゼ誘起シフトを示す。次いで、ヘパラン硫酸プロテオグリカン約80%を分解できるヘパラナーゼ量を決定する(ヘパラン硫酸プロテオグリカン約35%及びコンドロイチン/デルマタン硫酸プロテオグリカン約65%を含有する試料)。従って、10−80%の範囲の分解は、比較的直線状であり、阻害剤の効果の決定において許容される。図1cは、97.3%の阻害で、ヘパラナーゼ活性の1μg/mlの未分画ヘパリン(UFH)の効果を示す。
【0092】
アッセイ状態を決定した後、ブタ腸粘膜から得た未分画ヘパリン(UFH)の効果を測定した。図2は、用量依存的な阻害を示す。ヘパラナーゼ活性の実質的に完全な阻害が、1μg/ml(最終濃度)の未分画ヘパリン(UFH)濃度にて、観察された。図7は、Hexa Iso ATIIIによる用量依存阻害を示す。これらのデータに基づいて、Hexa Iso ATIIIが、強度のヘパラナーゼ阻害活性を示すと結論付けることが可能である。
【0093】
次の刊行物の内容を、参照により、本明細書に組み込む。
【0094】
C.R.Parish,et al.Biochim.Biophys, Acta1471(2001)99−108。
【0095】
M.Bartlett,et al.Immunol.Cell Biol.73(1995)113−124。
【0096】
I.Vlodavsky,et al.IMAJ2(2000)37−45。
【0097】
Y.Matzner,et al.J.Clin.Invest.76(1985)、1306−1313。
【0098】
Z.Drzeniek,et al.Blood(1999)2884−2879。
【0099】
オリゴ糖を豊富に含有した他の分画は、ヘパラナーゼ1による、ヘパリンの分解生成物から単離できる。従って、六糖の場合、単一のCTA−SAXクロマトグラフの精製で十分である。この方法では、セチルトリメチルアンモニウム鎖が、45℃にて、2ml/分で、4時間、水−メタノールの混合物(68−32)v/v中の、セチルトリメチルアンモニウムリン酸水素の1mM溶液の浸透によりその上にグラフト化された、5μm粒子のHypersil BDS(250×20mm)カラムを使用する。
【0100】
室温で分離を実行する。次の勾配溶出を使用する:溶媒Aは、HClの添加によってpHを2.5に調節した水である。溶媒Bは、pHを2.5に調節したNaClの2N溶液である。
【0101】
【表3】
【0102】
検出をUV領域の232nmで行う。六糖画分100mgを、各分離において、注入できる。
【0103】
八糖及び十糖の画分の精製は、六糖画分の精製よりも更に複雑であり、一般的に、それはIonPac(R)AS11カラム(250×20mm)(Dionex)上で、更なる精製を必要とする。分離は室温で実行する。勾配溶出を使用する。溶媒Aは、過塩素酸の添加によりpH3に調節した水である。溶媒Bは、pH3に調節されたNaClO4の1M溶液である。
【0104】
【表4】
【0105】
図8は、GCより単離された各六糖、八糖及び十糖において、オリゴ糖を豊富に含有した画分から単離された生成物の同定が可能である。
【0106】
酵素システムにおけるヘパラナーゼ阻害剤の活性評価
フォンダパリヌクスを分解する能力により、ヘパラナーゼの活性を決定する。フォンダパリヌクスの濃度を、その抗Xa因子の活性によって決定する。
【0107】
A.物質及び方法
ヘパラナーゼは、Sanofi−Synthelabo(Labege、フランス)によって、調製される。
【0108】
Xa因子をアッセイするための試薬は、Chromogenix(Montpellier、フランス)から販売されている。
【0109】
本発明の化合物の漸増濃度、ヘパラナーゼ阻害剤(可変希釈量:1nM〜10μM)を、ヘパラナーゼの一定濃度と混合する(各バッチにおいて、先行実験により、添加された0.45μg/mlのフォンダパリヌクスの分解に対し、十分な酵素活性を決定できる。)。37℃で5分後、その混合物をフォンダパリヌクスと接触させ、37℃で、1時間、放置する。95℃で、5分間加熱することにより、その反応を停止する。残存のフォンダパリヌクスの濃度を、Xa因子及びその特異的な発色基質(参照番号S2222)を添加することにより測定する。
【0110】
次の手順に従い、様々な混合物を調製する:
a)反応混合物
酢酸ナトリウムバッファ(0.2M、pH4.2)50μlを、フォンダパリヌクス(0.45μg/ml)50μl及びヘパラナーゼ溶液59μlと混合する。混合物を37℃で、1時間、インキュベートし、次いで95℃で、5分間、インキュベートする。pHは、2.4から7となる。次いで、反応混合物100μlを、175mM NaCl及び75mM EDTAを含有するpH14の50mMトリスバッファ50μlのと混合する。
【0111】
フォンダパリヌクスの抗Xa因子活性を、次の方法で測定する。
【0112】
b)フォンダパリヌクスの抗Xa因子活性のアッセイ
段階a)で得た溶液100μlを、AT(0.5μg/ml)100μlと混合する。この混合物を37℃で、2分間、維持し、次いで、Xa因子(7nkat/ml)100μlを添加する。その混合物を37℃で、2分間、維持し、発色基質(参照番号:S2222)(1mM)100μlを添加する。この混合物を37℃で、2分間、維持し、次いで酢酸(50%)100μlを添加する。
【0113】
光学濃度を405nmで読み取る。
【0114】
阻害剤なしで対照に対し、%阻害を決定する。%阻害曲線により、IC50を算出できる。
【0115】
【表5】
【図面の簡単な説明】
【0116】
【図1】ヘパラナーゼ1によるヘパリン解重合(GC監視)。
【図1a】天然HSPGαゲル浸透クロマトグラフ。
【図1b】ヘパラナーゼ処理後のゲル浸透クロマトグラフ。
【図1c】未分画ヘパリンのゲル浸透クロマトグラフ。
【図2】未分画ヘパリンのヘパラナーゼ阻害活性。
【図2a】ヘパラナーゼ1によるヘパリン解重合(CTA−SAX監視)。
【図3】解重合されたヘパリンの各画分。
【図4】解重合されたヘパリンの六糖画分。
【図5】六糖画分の半分取分離。
【図6】脱塩後の六糖画分分析。
【図7】Hexa Iso ATIIIのヘパラナーゼ阻害活性。
【図8】単離された各糖画分のGC分析。
【特許請求の範囲】
【請求項1】
特に、Xa因子及びIIa因子の阻害による、抗血栓活性が、実質的に消滅されるまで(<35IU/mg)、多糖類がヘパリナーゼ1で解重合される段階を含むことを特徴とする、抗血栓特性を有する多糖を解重合する方法。
【請求項2】
多糖が、ヘパリンであることを特徴とする、請求項1に記載の方法。
【請求項3】
混合物が、硫酸化六糖ΔIs−Isid−Isid及びΔIs−Isid−IIsgluが実質的に存在しない六糖画分を含むまで、解重合が遂行されることを特徴とする、請求項1又は請求項2に記載の方法。
【請求項4】
解重合が、5ないし40℃の温度で実施されることを特徴とする、請求項1から3のいずれか一項に記載の方法。
【請求項5】
解重合が、10ないし20℃の温度で実施されることを特徴とする、請求項4に記載の方法。
【請求項6】
5000Da未満の平均分子量が達成されるまで解重合が遂行されることを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項7】
3000Da未満の平均分子量が達成されるまで解重合が遂行されることを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項8】
多糖の解重合の生成物が、pH8未満かつ5超で、ゲル浸透クロマトグラフィーにより精製される段階も含むことを特徴とする、請求項1又は請求項2に記載の方法。
【請求項9】
固定相が、(i)C18−グラフト化され、且つ(ii)セチルトリメチルアンモニウム(CTA−SAX)でグラフト化された逆相である、高速液体クロマトグラフィーによる精製段階も含むことを特徴とする、請求項8に記載の方法。
【請求項10】
脱塩段階も含むことを特徴とする、請求項9に記載の方法。
【請求項11】
脱塩段階が、200nmと250nmの間で実質的に透明な水溶液中に電解質を含有する移動相の使用を含む、請求項10に記載の方法。
【請求項12】
電解質が、Naなどのアルカリ金属の、過塩素酸塩、メタンスルホン酸塩又はリン酸塩から選択されることを特徴とする、請求項11に記載の方法。
【請求項13】
脱塩が、例えば、Sepharose Q(R)型の、陰イオン交換樹脂を使用して実施されることを特徴とする、請求項12に記載の方法。
【請求項14】
特に、Sephadex G10(R)型の、分子排除ゲルを使用する、第2の脱塩段階を含むことを特徴とする、請求項10から13のいずれか一項に記載の方法。
【請求項15】
請求項1から14のいずれか一項に記載の方法により得られる生成物。
【請求項16】
式(I)の生成物
【化1】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択される。)
ただし、n=0、R=SO3M及びM=Naの生成物は除かれる。
【請求項17】
Mが、Li、Na及びKから選択されることを特徴とする、請求項16に記載の生成物。
【請求項18】
n=0であることを特徴とする、請求項16又は請求項17に記載の生成物。
【請求項19】
M=Naであることを特徴とする、請求項15又は請求項16に記載の生成物。
【請求項20】
下記の式(Ia)の、請求項19に記載の生成物。
【化2】
【請求項21】
下記の式(Ib)の、請求項16に記載の生成物。
【化3】
【請求項22】
下記の式(Ic)の、請求項16に記載の生成物。
【化4】
【請求項23】
下記の式(Id)の、請求項16に記載の生成物。
【化5】
【請求項24】
下記の式(Ie)の、請求項16に記載の生成物。
【化6】
【請求項25】
下記の式(If)の、請求項16に記載の生成物。
【化7】
【請求項26】
下記の式(Ig)の、請求項16に記載の生成物。
【化8】
【請求項27】
下記の式(Ih)の、請求項16に記載の生成物。
【化9】
【請求項28】
下記の式(Ij)の、請求項16に記載の生成物。
【化10】
【請求項29】
下記の式(Ik)の、請求項16に記載の生成物。
【化11】
【請求項30】
下記の式(Im)の、請求項16に記載の生成物。
【化12】
【請求項31】
ヘパラナーゼ阻害剤としての、式(I)
【化13】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択される。)
の生成物の使用。
【請求項32】
【化14】
であることを特徴とする、請求項31に記載の式(I)の生成物の使用。
【請求項33】
式Ia、Ib、Ic、Id、Ie、If、Ig、Ih、Ij、Ik及びImの生成物から選択されることを特徴とする、請求項31に記載の式(I)の生成物の使用。
【請求項34】
細胞増殖を調節するための、請求項31から33のいずれか一項に記載の生成物の使用。
【請求項35】
細胞増殖が癌に関連することを特徴とする、請求項34に記載の生成物の使用。
【請求項36】
細胞増殖が転移過程に関連することを特徴とする、請求項35に記載の生成物の使用。
【請求項37】
細胞分裂停止剤としての、請求項35又は請求項36に記載の生成物の使用。
【請求項38】
疾患の初期段階で実施されることを特徴とする、請求項36又は請求項37に記載の生成物の使用。
【請求項39】
第2の抗癌生成物と組み合わされた、請求項37及び38のいずれか一項に記載の生成物の使用。
【請求項40】
第2の抗癌生成物が細胞毒性であることを特徴とする、請求項39に記載の生成物の使用。
【請求項41】
第2の抗癌生成物が、シスプラシン又はオキサリプラチンなどのプラチナ誘導体、ドセタキセル又はパクリタキセルなどのタキソイド、5−FU、カペシタビン又はゲムシタビンなどのプリン塩基又はピリミジン塩基誘導体、ビンクリスチン又はビンブラスチンなどのビンカ、マスタード、スタウロスポリン、エリプチシン又はイリノテカン、トポテカンなどのカンプトテシンなどの縮合芳香族複素環、CA4Pなどのコンブレタスタチン、並びにリン酸コルチノールなどのコルヒチン誘導体から選択されることを特徴とする、請求項40に記載の生成物の使用。
【請求項42】
第2の抗癌生成物が、ドセタキセル、オキサリプラチン又はイリノテカンであることを特徴とする、請求項41に記載の生成物の使用。
【請求項43】
癌が、乳癌、肺癌、前立腺癌、結腸癌又は膵臓癌であることを特徴とする、請求項34から42のいずれか一項に記載の生成物の使用。
【請求項1】
特に、Xa因子及びIIa因子の阻害による、抗血栓活性が、実質的に消滅されるまで(<35IU/mg)、多糖類がヘパリナーゼ1で解重合される段階を含むことを特徴とする、抗血栓特性を有する多糖を解重合する方法。
【請求項2】
多糖が、ヘパリンであることを特徴とする、請求項1に記載の方法。
【請求項3】
混合物が、硫酸化六糖ΔIs−Isid−Isid及びΔIs−Isid−IIsgluが実質的に存在しない六糖画分を含むまで、解重合が遂行されることを特徴とする、請求項1又は請求項2に記載の方法。
【請求項4】
解重合が、5ないし40℃の温度で実施されることを特徴とする、請求項1から3のいずれか一項に記載の方法。
【請求項5】
解重合が、10ないし20℃の温度で実施されることを特徴とする、請求項4に記載の方法。
【請求項6】
5000Da未満の平均分子量が達成されるまで解重合が遂行されることを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項7】
3000Da未満の平均分子量が達成されるまで解重合が遂行されることを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項8】
多糖の解重合の生成物が、pH8未満かつ5超で、ゲル浸透クロマトグラフィーにより精製される段階も含むことを特徴とする、請求項1又は請求項2に記載の方法。
【請求項9】
固定相が、(i)C18−グラフト化され、且つ(ii)セチルトリメチルアンモニウム(CTA−SAX)でグラフト化された逆相である、高速液体クロマトグラフィーによる精製段階も含むことを特徴とする、請求項8に記載の方法。
【請求項10】
脱塩段階も含むことを特徴とする、請求項9に記載の方法。
【請求項11】
脱塩段階が、200nmと250nmの間で実質的に透明な水溶液中に電解質を含有する移動相の使用を含む、請求項10に記載の方法。
【請求項12】
電解質が、Naなどのアルカリ金属の、過塩素酸塩、メタンスルホン酸塩又はリン酸塩から選択されることを特徴とする、請求項11に記載の方法。
【請求項13】
脱塩が、例えば、Sepharose Q(R)型の、陰イオン交換樹脂を使用して実施されることを特徴とする、請求項12に記載の方法。
【請求項14】
特に、Sephadex G10(R)型の、分子排除ゲルを使用する、第2の脱塩段階を含むことを特徴とする、請求項10から13のいずれか一項に記載の方法。
【請求項15】
請求項1から14のいずれか一項に記載の方法により得られる生成物。
【請求項16】
式(I)の生成物
【化1】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択される。)
ただし、n=0、R=SO3M及びM=Naの生成物は除かれる。
【請求項17】
Mが、Li、Na及びKから選択されることを特徴とする、請求項16に記載の生成物。
【請求項18】
n=0であることを特徴とする、請求項16又は請求項17に記載の生成物。
【請求項19】
M=Naであることを特徴とする、請求項15又は請求項16に記載の生成物。
【請求項20】
下記の式(Ia)の、請求項19に記載の生成物。
【化2】
【請求項21】
下記の式(Ib)の、請求項16に記載の生成物。
【化3】
【請求項22】
下記の式(Ic)の、請求項16に記載の生成物。
【化4】
【請求項23】
下記の式(Id)の、請求項16に記載の生成物。
【化5】
【請求項24】
下記の式(Ie)の、請求項16に記載の生成物。
【化6】
【請求項25】
下記の式(If)の、請求項16に記載の生成物。
【化7】
【請求項26】
下記の式(Ig)の、請求項16に記載の生成物。
【化8】
【請求項27】
下記の式(Ih)の、請求項16に記載の生成物。
【化9】
【請求項28】
下記の式(Ij)の、請求項16に記載の生成物。
【化10】
【請求項29】
下記の式(Ik)の、請求項16に記載の生成物。
【化11】
【請求項30】
下記の式(Im)の、請求項16に記載の生成物。
【化12】
【請求項31】
ヘパラナーゼ阻害剤としての、式(I)
【化13】
(式中、
Rは、H及びSO3Mから選択され、並びに
Mは、H、Li、Na及びKから選択される。)
の生成物の使用。
【請求項32】
【化14】
であることを特徴とする、請求項31に記載の式(I)の生成物の使用。
【請求項33】
式Ia、Ib、Ic、Id、Ie、If、Ig、Ih、Ij、Ik及びImの生成物から選択されることを特徴とする、請求項31に記載の式(I)の生成物の使用。
【請求項34】
細胞増殖を調節するための、請求項31から33のいずれか一項に記載の生成物の使用。
【請求項35】
細胞増殖が癌に関連することを特徴とする、請求項34に記載の生成物の使用。
【請求項36】
細胞増殖が転移過程に関連することを特徴とする、請求項35に記載の生成物の使用。
【請求項37】
細胞分裂停止剤としての、請求項35又は請求項36に記載の生成物の使用。
【請求項38】
疾患の初期段階で実施されることを特徴とする、請求項36又は請求項37に記載の生成物の使用。
【請求項39】
第2の抗癌生成物と組み合わされた、請求項37及び38のいずれか一項に記載の生成物の使用。
【請求項40】
第2の抗癌生成物が細胞毒性であることを特徴とする、請求項39に記載の生成物の使用。
【請求項41】
第2の抗癌生成物が、シスプラシン又はオキサリプラチンなどのプラチナ誘導体、ドセタキセル又はパクリタキセルなどのタキソイド、5−FU、カペシタビン又はゲムシタビンなどのプリン塩基又はピリミジン塩基誘導体、ビンクリスチン又はビンブラスチンなどのビンカ、マスタード、スタウロスポリン、エリプチシン又はイリノテカン、トポテカンなどのカンプトテシンなどの縮合芳香族複素環、CA4Pなどのコンブレタスタチン、並びにリン酸コルチノールなどのコルヒチン誘導体から選択されることを特徴とする、請求項40に記載の生成物の使用。
【請求項42】
第2の抗癌生成物が、ドセタキセル、オキサリプラチン又はイリノテカンであることを特徴とする、請求項41に記載の生成物の使用。
【請求項43】
癌が、乳癌、肺癌、前立腺癌、結腸癌又は膵臓癌であることを特徴とする、請求項34から42のいずれか一項に記載の生成物の使用。
【図1】

【図1a】
【図1b】

【図1c】

【図2】
【図2a】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図1a】
【図1b】
【図1c】
【図2】
【図2a】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2007−522823(P2007−522823A)
【公表日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願番号】特願2007−500254(P2007−500254)
【出願日】平成17年2月23日(2005.2.23)
【国際出願番号】PCT/FR2005/000431
【国際公開番号】WO2005/090591
【国際公開日】平成17年9月29日(2005.9.29)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
【公表日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願日】平成17年2月23日(2005.2.23)
【国際出願番号】PCT/FR2005/000431
【国際公開番号】WO2005/090591
【国際公開日】平成17年9月29日(2005.9.29)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
[ Back to top ]