
オリーブからの抽出物又はオリーブ・オイルの抽出後の残留物を含む固体成分からヒドロキティロソルを生成するプロセスと装置
【課題】 有機極性溶剤を使用せずに、オリーブからヒドロキティロソルを含む抽出物を溶離する方法を提供する。
【解決手段】 本発明のオリーブからヒドロキティロソルを含有する抽出物を生成する方法は、(a)水中で前記出発材料を酸性加水分解するステップと、前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、(b)前記(a)ステップの加水分解水溶液から懸濁した固体を除去するステップと、前記ステップにより、分離した水溶液を確保し、(c)前記(b)ステップで得た水溶液(A)を、第1のクロマトグラフィ・カラムに入れるステップと、前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、(d)前記クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップとを有する。
【解決手段】 本発明のオリーブからヒドロキティロソルを含有する抽出物を生成する方法は、(a)水中で前記出発材料を酸性加水分解するステップと、前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、(b)前記(a)ステップの加水分解水溶液から懸濁した固体を除去するステップと、前記ステップにより、分離した水溶液を確保し、(c)前記(b)ステップで得た水溶液(A)を、第1のクロマトグラフィ・カラムに入れるステップと、前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、(d)前記クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップとを有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、オリーブからの抽出物とオリーブ・オイルの抽出後の固体とから、ヒドロキティロソル(hydroxytyrosol)を製造する方法と装置に関する。具体的には、本発明は、ヒドロキティロソルを含有する抽出物を製造する方法に関する。このヒドロキティロソルを含有する抽出物は、食品、医薬品、化粧品の分野で、ヒドロキティロソルの原料として使用される。
【背景技術】
【0002】
オリーブは、いくつもの生物活性化合物(bioactive compounds)、特にポリフェノール(polyphenols)を含む。これらのポリフェノールの中でも、ヒドロキティロソルは、抗菌剤(antimicrobial)又はラジカル補足活性(radical scavenging activity)の分野で、きわだった生物学的な重要性を有する。
【0003】
ヒドロキティロソルは、以下の化学式を有する。

【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第6361803号明細書
【特許文献2】国際公開パンフレット第2004/005228号
【特許文献3】米国特許出願公開第2004/0102657号明細書
【特許文献4】ヨーロッパ特許1623960号明細書
【特許文献5】米国特許出願公開第2004/0176647号明細書
【0005】
オリーブオイル抽出後の残留物からヒドロキティロソルを製造する方法が、研究されてきた。ヒドロキティロソルの効率的な抽出方法は、非常に利益の多いものである。その理由は、特にオリーブオイル抽出後の残留物の中に存在するヒドロキティロソルとその前駆体が、オリーブ・オイルの製造過程でできる残留物の中に多くあるが、僅かな量しかオリーブオイル内には存在しない。エキストラ・バージン・オリーブ・オイルは、通常1−20ppmのヒドロキティロソルしか含有しない。
【0006】
本発明において、オリーブ・オイル抽出後の残留物には、オリーブの木の葉を含まない。木の葉は、オイル抽出前に除去されているからである。さらに、葉からのヒドロキティロソルの抽出は、オリーブ又はオリーブ残留物からの抽出よりも、様々な出発合成(とそれ故に様々な抽出問題)に直面する。
【0007】
本発明にとって重要なオリーブ・オイル抽出プロセスから得られたオリーブの残留物は、次の様に分類される。
ポマス(pomaces:搾りかす)、即ち、圧搾後の残留物、又は三相プロセスの圧搾後の残留物、あるいは二相プロセスの圧搾後の残留物を含む固体である。この固体状態においては、遠心分離のステップの間、オリーブの小片には水は添加されない。orujo(搾りかす)とalperujo(遠心分離後の残渣)とdefatted orujo(脱脂した搾りかす)は、相当量(45−70%)の水を含有する。抽出残留物は、絞りかすと、オリーブの乾燥固体とを含む。これらは、圧搾後のオイルの抽出後、15%未満の水しか含まず、残留オイルを含まない。
【0008】
本発明のグリーン・オリーブの抽出物は、オリーブ・オイル製造時の残留物の抽出物よりも、次の点で好ましい。ヒドロキティロソルを多く含み、ヒドロキシメチルフルフラール(hydroxymethylfurfural)の量が少ない点で好ましい。
【0009】
本明細書において、用語「ポマス(pomace:搾りかす)」あるいは「固体残留物(solid residue)」は、「搾りかす(orujo)」、「脱脂された搾りかす(defatted orujo)」、「搾りかす(orujillo)」あるいは「残渣(alperujo)」を意味する。本発明の好ましい出発材料は、オリーブで、より好ましくはグリーン・オリーブとポマスである。
【0010】
ポマス(又は食物からの水)の酸性加水分解(acid hydrolysis)を実行し、オレウロペイン分子(oleuropein molecule)内のエステル結合を切断し、ヒドロキティロソルを得ることは、公知である。
【0011】
特許文献1は、ヒドロキティロソルと他の化合物の抽出を、オリーブ・パルプ残留物を1時間(ex12)還流しながら中性状態あるいは酸性状態で加水分解して、行うことを開示している。この抽出された水溶液を、吸収XAD−7カラムに入れる。そこでメタノールで溶離し、抽出されたヒドロキティロソルを回収する。同文献の方法では、有機極性溶剤(organic polar solvents)を用いることが必要であり、しかし最低純度(a minimum purity grade)のヒドロキティロソルしか回収できない。それに加えて(ex.12)、メタノール溶液からのある種の不純物の凍結沈殿が必要である。極性水溶性溶剤(polar aqeous solvent)は、メタノール(methanol)、エタノール(ethanol)、アセトニトリル(acetonitrile)、アセトン(acetone)から選択される。有機極性溶剤は、エステル(esters)、アミド(amides)、ジメチル・スルホキシド(dimethyl sulfoxide)、ジオクサン(dioxane)、ジメチルホルムアミド(DMF:dimethylformamide)、あるいはその混合物から選択される。これらの溶剤の大部分は、毒性があり、所望のヒドロキティロソル生成物から完全に取り除くことは困難である。従って、使用された溶剤の痕跡が、何回かの純化ステップの後でも、最終生成物に見出されることがある。その結果、このプロセスで得られたヒドロキティロソルは、食品、化粧、薬品の分野で、安全に使用できない。更に、この最終生成物は、補強食品、特に食用オイルでの使用に適したものではなく、安全な食品を得るために、エタノールや酢酸(acetic acid)を、抽出物への添加剤として使用していない場合でも、食用に適したものにはならない。その結果得られた食料品(オイルとヒドロキティロソル(エタノールと酢酸と抽出物を含む))は、食品業界では受け入れ難いものである。
【0012】
特許文献2は、オリーブ・オイルの抽出により得られた植物性の水の室温での加水分解を開示する。このオリーブ・オイルの抽出物は、酸性化した植物性の水を、少なくとも2ヶ月(好ましくは6−12ヶ月)の間、植物性の水の中に本来存在するoleuropein(オレウロペイン)の少なくとも50%(好ましくは90%)が、ヒドロキティロソルに変わるまで、培養することにより、行った。この培養された植物性の水を、有機溶剤(例、酢酸エチル(ethyl acetate)で抽出するか、あるいは超臨界流体(supercritical fluid)(CO2)に接触させて、ヒドロキティロソル−リッチの化合物を生成する。このプロセスの主な問題点は、特に得られた最終生成物を食品、化粧品、医薬品として使用する場合、酸性化された植物性の水の培養に時間がかかることであり、回避すべき有機溶剤を使用することである。
【0013】
特許文献3は、高温(190−220℃)での水蒸気拡散プロセス(steam explosion process)で行う酸性加水分解を開示する。かくして得られた溶液は、活性化されていないイオン交換樹脂のカラム内で最初に部分的に純化し、その後XAD非イオン・カラムに入れる。そこからヒドロキティロソルを、メタノールあるいはエタノールで、溶離する。このプロセスは、このプロセス全体を考慮しても、ヒドロキティロソルの歩留まりが悪い。
【0014】
特許文献4は、ヒドロキティロソルとチロソール(tyrosol)を植物性の水(alpechin)から回収するプロセスを、濾過手段と後続の分離手段で実行することを開示する。この濾過手段は3つのユニットからなる複雑なプラントで行われる。チロソールを、その後プロトン性溶剤(protic solvent)(アルコール又は水)で酸化し、ヒドロキティロソルに変換して、準合成品(semi-synthetic)である最終生成物を得ている。更に同文献は、植物性の水(alpechin)のナノ濾過(nanofiltration)による濃縮(concentration)と、逆浸透圧化(reverse osmosis)を、中性又はアルカリ性のpHで行うことを開示(請求項4)している。しかし、このプロセスには、2つの重要な問題点がある。第1の問題点は、ヒドロキティロソルの劣化は、中性あるいはアルカリ性の状態で増加し、除去することが困難な不要な生成物が現れ、更に酸性加水分解が実行されないことに起因して、オレウロペイン・コンテンツが依然として高い点である。第2の問題点として、獲得可能な濃度係数(concentration factor)が低いことである。かくして、Ex‐1(表1を参照のこと)によると、ヒドロキティロソルの最大濃度は、1.2−1.6g/Lの範囲内である。
【0015】
特許文献5は、alperujoからフェノール(phenols)を抽出するプロセスを開示する。この抽出プロセスは、180‐240℃のオートクレーブ(加圧滅菌器)内で、水中で攪拌することにより行った。酸は添加せずに熱処理を行った結果、「アセチル基の遊離(liberation of acetyl groups)」と、その結果pHの減少が起こった(同文献の3項1段落目)。しかし、上記の条件で生成されたpHにおいては、オレウロペイン(oleuropein)の加水分解の完成には程遠い。このことは、2つの問題点がある。即ち、ヒドロキティロソルの歩留まりの低さと加水分解されたオレウロペインの残留である。これは、上記で議論したごとく、この様な抽出物を含む食料品は不味く、食料品への適用には不向きである。この生成物は、チロソールとヒドロキティロソルであり、これらはHPLC(high-performance liquid chromatography:高速液体クロマトグラフィ;混合物を分離する分析化学の手法)により、硫酸(sulphuric acid)/アセトニトリル溶離液(acetonitrile eluent)で、分離できる。上記の従来技術を要約すると、同文献のプロセスは、時間がかかり、複雑で、条件が厳しい。その結果、オレウロペインの量、即ち加水分解されない量、又は加水分解による副生成物の量が、多すぎる。例えば、ヒドロキシメチルフルフラール(hydroxymethylfurfural )が多く、後続の純化ステップが困難になる。
【0016】
副生成物の発生は、ヒドロキティロソル抽出物の生成プロセスにおいて、当初考えたよりも大きな問題であることが判った。このことは、上記の従来の文献からも明らかである。開始材料、特にオリーブとポマスは、中でもフェノール化合物(phenolic compounds)をグルコシド(glucoside)の形態で、あるいはエステルの形態で大量に含まれる。エステルは、オレウロペイン、ligustroside、verbascoside、幾つかのフラボノイド類(flavonoids)を含む。幾つかの他の天然化合物も存在する。それ故に、開始化合物が容易に劣化し、多量の副生成物になってしまう。
【0017】
副生成物を除去して、ヒドロキティロソルを純化しなければならないので、公知の方法では、有機溶剤を使用して、クロマトグラフィ・カラム(chromatographic columns)を用いる後続のプロセスの間、ヒドロキティロソルを純化する必要がある。有機溶剤の使用は、例えばメタノールの使用は、樹脂・カラム(resin column)から溶離(elute)する際には不都合なものである。得られた最終生成物を食品、化粧品、医薬品の分野で使用する場合には、特に不都合である。
【0018】
更に公知のヒドロキティロソル・リッチの抽出物は、食料品特に食用油の添加剤として使用するのに適したものではない。それ故に、以下に記載する特性を有するヒロキティロソルを含む抽出物を生成するニーズがある。即ち、この抽出物は、ヒドロキティロソル・リッチであり、開始生成物(例えばオレウロペインとverbascosides)の量が少なく、副生成物の量、特にヒドロキシメチルフルフラールの量が少なく、一般的に、砂糖や塩の量が極めて少ないものである。
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明の目的は、上記の問題点を解決することである。本発明は、オリーブあるいはオリーブの抽出物である副生成物から、ヒドロキティロソルを生成するプロセスを提供することである。しかも簡単で効率的で安価で、有機溶剤を含まない高純度の生成物を提供することである。食料や化粧品の添加物として使用するのに適したものを製造する方法を提供することである。
【課題を解決するための手段】
【0020】
この目的は、請求項1記載の本発明の手段により達成できる。即ち、出発材料から、ヒドロキティロソルを含有する抽出物を生成する方法において、前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択される。前記方法は、前記出発材料を酸性加水分解し、得られた溶液を純化しする。更に具体的には、
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する。
【0021】
本発明の方法は、開始材料の水中で酸性加水分解を以下の条件で行う。温度条件は、140℃未満で、好ましくは70℃−130℃の間で、更に好ましくは、110℃−130℃の間(還流温度以上)、である。圧力の掛かった状態で、例えば、連続的な殺菌システムで行う。圧力条件は、大気圧以上(ゲージ圧)で10‐20psiの範囲である。加水分解を受ける酸性混合物のpHは、1.6‐6.0の範囲内である。加水分解後の混合物は、従来公知の物理的方法で分離して、例えば、濾過と/又は遠心分離により分離して、加水分解後の水溶液から懸濁した固体を除去し、懸濁液中に固体の無い純化溶液を得る。
【0022】
開始材料は、オリーブ又はポマスである。即ち、上記に定義したオリーブ・オイルの抽出プロセスから得られた残留物である。好ましいオリーブは、グリーン・オリーブである。ポマスは、好ましくはオイルを含有しない。オイルは、加水分解のステップの前あるいは後の何れかで、公知の手段(例えば、脱脂orujoあるいはorujilloを準備するのに用いられるもの)により、あるいは珪藻土(diatomaceous earth)、あるいは他の濾過手段により除去する。
【0023】
本発明によれば、上記のステップ(酸性加水分解と純化(clarification))の後に、2つのステップが行われる。即ち、得られた生成物を、第1のクロマトグラフィ・カラム(酸で活性化した陰イオン交換樹脂を含む)に入れるステップと、前記第1のクロマトグラフィ・カラム内にある生成物を水で溶離するステップが行われる。
本発明の好まし実施例によれば、本発明は、更に純化ステップを実行する。この純化ステップは、前記の第1のクロマトグラフィ・カラム(陰イオン交換樹脂を含む)からの水溶離溶液を、第2のクロマトグラフィ・カラム(吸着性の非イオン性樹脂を含む)の内に入れるステップと、前記第2のクロマトグラフィ・カラム内に残った生成物を水で溶離するステップである。
【0024】
本発明の更なる態様によれば、流体生成物は、例えば、逆浸透圧濃縮(reverse osmosis concentration)方法で濃縮する。本発明の更なるステップによれば、クロマトグラフィの純化ステップと逆浸透圧濃縮ステップの後、得られた流体生成物を固体状にする。これは、例えば凍結乾燥(freeze-drying)、真空回転蒸発(vacuum rotoevaporation)、噴霧乾燥(spray drying)で行う。このステップは、マルトデキストリン(maltodextrin)のようなキャリア(担持流体)の有無を問わない。
【0025】
本発明の更なる目的は、上記のプロセスにより得られたヒドロキティロソルを含む抽出物である。この抽出物は、液体又は固体のいずれかの形態であり、ヒドロキティロソルを0.5%(w/w)以上含む。その純度は、40%以上で、好ましくは80%以上で、より好ましくは95%以上である。これは、280nmで測定したHPLCピーク面積(peak area)により決定した。これらの抽出物は、有機溶剤とヒドロキシメチルフルフラールを含まず、砂糖も塩もほぼ含まない。
【0026】
本発明の一態様によれば、本発明の方法により得られたヒドロキティロソルを含む流体生成物は、ヒドロキティロソルを35%(w/w)以上、好ましくは45%以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、35%以上である。
本発明のより好ましい態様によれば、本発明の方法により得られたヒドロキティロソルを含む固体生成物は、ヒドロキティロソルを20%(w/w)以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、20%以上である。
【0027】
本発明のより好ましい態様によれば、本発明の方法により得られたヒドロキティロソルを含む固体生成物は、ヒドロキティロソルを90%(w/w)以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、92%以上である。
【0028】
本発明の更なる目的は、上記したプロセスを実行する装置を提供することであり、この装置は、請求項13に記載されている。
本発明の装置又はプラントは、反応容器と、第1のクロマトグラフィ・カラムと、第2クロマトグラフィ・カラムとを含む。前記反応容器は、圧力のかかった状態で酸性の加水分解を実行する。前記クロマトグラフィ・カラムは、樹脂と、酸で活性化した陰イオン交換樹脂(弱塩基性(weakly basic))と、水溶離性の陰イオン交換樹脂を含む。第2クロマトグラフィ・カラムは、吸着性の非イオン樹脂を含む。この吸着性の非イオン樹脂は、マクロ網状の架橋された芳香族ポリマ・樹脂(macroreticular cross-link aromatic polymer resin)である
【0029】
本発明は、従来技術に対しいくつかの利点を提供する。加水分解ステップは、本発明により、水だけあるいはミネラル酸のみを用いて、特許請求の範囲に記載した還流温度(reflux temperture)以上で行い、加熱ステップと圧力ステップを組み合わせて行うことができる。圧力が、特許請求の範囲に記載された温度と共に加えられた場合に、その結果得られた加水分解は、約30分で終了するが、これには、副生成物の形成はなく、開始材料(オレウロペイン)を所望の最終生成物(ヒドロキティロソル)に完全に変換できる。本発明の加水分解ステップにより、短時間で極めて高い歩留まりで、ヒドロキティロソルを得ることができ、副生成物は全く生成されない。従来技術では、この様な副生成物は、取り除くことが困難であり、かなり量生成されていた。
【0030】
本発明の加水分解ステップは、更なる利点を有する。酸の添加と加熱を適宜組み合わせて実行することにより、高い変換率で加水分解を行うことができ、かつ有害な副生成物の形成を回避できる。その結果混合物が殺菌される。実際に、加水分解プロセスは、水溶液の殺菌を提供できる。即ち関連する生成物の殺菌が提供でき、これは極めて有益である。その理由は、ヒドロキティロソルを準備するプロセスで使用される開始材料(例えば、ポマス)は、通常既に処理された材料から得られ、一般的に純化−殺菌の前処理(depuration-sterilization pre-treatment)を行い、安全なヒドロキティロソル最終生成物を得ている。この純化−殺菌の前処理は、時には複雑な処理であり、プロセスの終了時に、更なる処理ステップを必要とする。その結果、所望の最終生成物の低い歩留まりとなる。本発明によれば、加水分解ステップにより、加水分解反応と、関連する必要な生成物の殺菌が同時に提供できる。かくして、開始材料の殺菌ステップを行う必要がなく、又更なる使用に適した加水分解された材料を提供できる。これは更なる殺菌処理を必要としない。
【0031】
これに加えて、加水分解ステップの後、純化ステップが行われる。この純化ステップは、加水分解ステップで得られた生成物を、少なくとも1つのクロマトグラフィ・カラム・システムに、あるいは2つのクロマトグラフィ・カラム・システムに入れ、このクロマトグラフィ・カラム内に残ったヒドロキティロソルを水で溶離する。この場合、加水分解ステップで生成された副生産物の量は、極めて少ないので、有機溶剤無しで、水単独で溶離、あるいは水と混合して溶離するようなクロマトグラフィ・カラムの使用が可能となる。この事は、有機溶剤は、ヒドロキティロソルとは接触せず、かくして純粋なヒドロキティロソルが得られる。これは、特に食品、化粧品、医薬品の分野での使用に適したものである。
【0032】
本発明の他の利点は、濃縮ステップに起因する。濃縮ステップにより、純化ステップの後のヒドロキティロソルの量を増やすことができる。この純化ステップは、液体抽出物、特に砂糖と塩を含まない液体抽出物を生成する。その結果、純化した流体抽出物の濃縮ステップの間、浸透圧の非常に重要な減少が得られ、かくして非常に高い濃縮係数(350倍まで)が可能となる。この濃縮ステップは、多くの量のヒドロキティロソルの含有により特徴付けられた「濃縮物(concentrate)」であり、これは更なる処理に直接使用できる。従来技術においては、濃縮ステップは通常存在せず、あった場合でも、生成物の濃縮係数は低い(2倍から5倍)。これは塩と砂糖を取り除く純化プロセスがないためである。ヒドロキティロソルの準備のプロセスにおては、処理された溶液/分散液内のヒドロキティロソルの濃度は常に低く、大量に処理する必要があり、かくして、最終生成物の歩留まりが下がる。
【0033】
本発明によれば、例えば、マルトデキストリン(maltodextrin:麦芽糖を含んだデキストリン;食品添加物)等のあらゆる担体(carrier)と混合していない最終固体生成物を得ることができることにより、最終使用意図に適し、法律上の要件に適した、純化したヒドロキティロソルを得ることができる。
【図面の簡単な説明】
【0034】
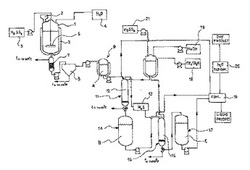
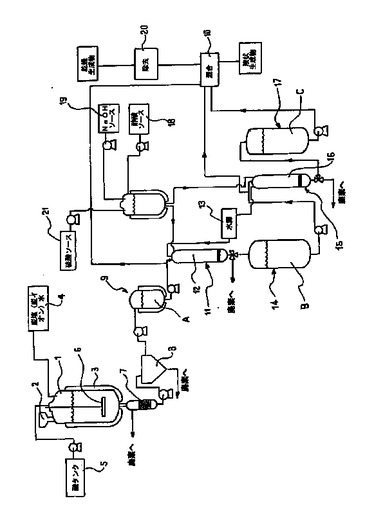
【図1】本発明の装置を表すブロック図。
【図2】本発明の第1実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【図3】本発明の第2実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【図4】本発明の第3実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【発明を実施するための形態】
【0035】
図1において、本発明の装置は、反応容器1を含む。好ましくは、連続する殺菌システムを含む。この殺菌システムで本発明の加水分解が行われる。
【0036】
上記したように、反応容器内で、加水分解を以下の条件で行う。温度は、140℃を超えることはなく、好ましくは70℃−130℃の間で、更に好ましくは(還流温度以上で)、110℃−130℃である。圧力条件は、ゲージ圧で(以下同様)、10‐20psi(68.94−137.8KPa)の範囲である。分解時間は15分−45分の間である。好ましくは、加水分解温度は、118℃から126℃の範囲内であり、最も好ましくは、120℃から121℃で、圧力は15psi(103.4KPa)で、30分間行われる。好ましい反応容器は、連続式の殺菌システムである。
【0037】
連続式の殺菌システムである反応容器1は、供給手段2を具備する。この供給手段2は、反応容器1に、開始材料であるオリーブ・オイル抽出後の残留物、即ち副生成物、言い換えるとポマス(即ち、圧搾したオリーブの固体残留物)を供給する。連続式の殺菌システムである反応容器1は、バッチ処理ではなく連続するプロセスで、出発材料を処理する。加熱手段3が、反応容器1を上記の温度に加熱する。加熱手段3は、反応容器1の周囲に巻かれた加熱ジャケットである。反応容器1は、脱塩(脱イオン)水4と攪拌手段6を具備する。この攪拌手段は、出発材料である混合水物を攪拌する。水と固体との比率は、1:1から4:1の間である。
【0038】
加水分解プロセスは、pHが1.0−6.0の間で行われ、好ましくは1.0−3.0の間で行われる。酸(好ましくは硫酸H2SO4)は、酸タンク5から得られる。
【0039】
特許請求の範囲に記載された酸性状態と温度の組み合わせにより、加水分解プロセスが行われる。この加水分解プロセスは、極めて速く、効率的で、30分間、120℃か−123℃で、15psiの圧力で行われる。更にこのプロセスにより、加水分解生成物が殺菌される。
【0040】
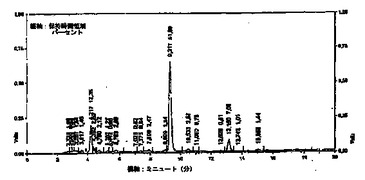
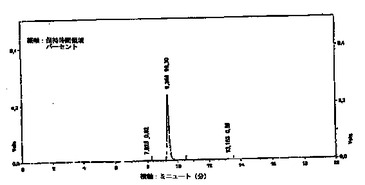
反応容器1の出口にフィルタ7が接続される。このフィルタ7は、ヒドロキティロソルや他のフェノール(phenols)を含む反応混合物から、固体を除去する。フィルタ処理された物は、その後更に分離手段好ましくは遠心分離機8に送られる。そこで更に固体が反応混合物から除去され、懸濁した固体のない流体、即ち後続の純化ステップに適した溶液を得る。遠心分離の後、分離した(不純物を除去した)流体は褐色をしており、貯蔵器9に貯蔵される。分離した流体Aは、ヒドロキティロソルと、オレウロペイン(oleuropein)の残留物と、少量のフェノールと、他の生成物とを含む。図2のクロマトグラフは、この流体中に含まれる検出した化合物の組成をパーセントを示す。この分析は、波長28nmのPHLCにより得られたものであり、9.317ミニュッツ(分)の大きなピークは、ヒドロキティロソルであり、13.150ミニュッツ(分)のピークは、チロソール(tyrosol)である。
【0041】
本発明の好ましい一実施例によれば、流体Aは、蒸発(evaporation)法、又は接線流濾過法(tangential flow filtration(TFF))(例、逆浸透圧システムを用いる)により濃縮される。この濃縮ステップは、流体Aに対し実行される。この濃縮ステップは、直接、あるいは好ましくは純化ステップの後で、クロマトグラフィ・カラムの手段で行う。この為に、貯蔵タンク即ち貯蔵器9が、濃縮手段10、TFFあるいは好ましくは逆浸透圧手段で、純化手段に接続される。
【0042】
純化手段は、一実施例では、単一の(クロマトグラフィ・)カラム・システムを含む。このカラム・システムは、酸で活性化された陰イオン交換樹脂(acid activated anion exchnage resin)から選択された樹脂のクロマトグラフィ・カラムを含む。本発明の他の実施例においては、純化手段は、前記の第1カラムに加えて、吸収性の非イオン性樹脂(absorbant non-ionic resin)から選択された樹脂の第2クロマトグラフィ・カラムを、更に含む。
【0043】
本発明の他の実施例においては、酸で活性化された陰イオン交換樹脂は、弱塩基性の陰イオン交換樹脂(weakly basic anion exchnage resin)である。吸収性の非イオン性樹脂は、マクロ網状の架橋された芳香族ポリマ(macroreticular cross-linked aromatic polymer)である。
【0044】
本発明のヒドロキティロソルの純化用クロマトグラフィ・樹脂として使用される陰イオン交換樹脂は、酸で活性化されている限り、それに限定されるない。弱塩基性の陰イオン交換樹脂の一例は、ポリアミン系樹脂(polyamine-type resin)(ポリアミン型キレーティング・樹脂を含む)を含む。その一例は、例えば、スチレン/ジビニルベンゼン・共重合体(styrene/divinylbenzene copolymer)とジエチレントリアミン(diethylenetriamine)の反応生成物、アリルアミン(allylamine)、ビニルアミン(vinylamine)等を主に含む化合物の重合化生成物(polymerization product)のような樹脂、アクリル・樹脂(acrylic resins)、例えば、ジビニルベンゼンの共重合体とアクリル酸あるいはメタクリル酸(methacrylic acid)あるいはジアチルアミンノプロピルアミン(dimethylaminopropylamine)を含むアミド化合物である。他の樹脂は、上記の弱塩基性の陰イオン交換樹脂が、強塩基性の交換基(strongly basic exchnage group)で、例えば、トリメチルアミン(tirmethylamine)とジメチルエタノールアミン(dimethylethanolamine)で部分的に置換したものである。より具体的には、従来公知の樹脂は、例えば、ダイヤイオン(Diaion:商標) WA10、WH20、WH21、WA30(三菱化学社製)、アンバーライト(Amberlite:商標)IRA−35、IRA−67(IRA−68)、IRA−93ZU、IRA−94S、IRA−478(ローム・アンド・ハース社製)、WGR−2(ダウ・ケミカル社製)等である。
【0045】
本発明の方法によれば、これらの樹脂は、使用される前に酸で、好ましくは、酢酸で活性化される。これらの樹脂は、特に本発明の方法に適したものである。低価格である点及びそれらがマイルドな状態で再生される点で適したものである。更に、樹脂の弱塩基性により、ヒドロキティロソル分子とチロソール分子の部分的な分離が容易となる。ヒドロキティロソル/チロソールのピーク率の比較と、図2、3のヒドロキティロソルのHPLC純度の観点からすると、本発明によるクロマトグラフィ手法による分離は、効率が良いことが判る。
【0046】
実際に、本発明の弱陰イオン交換樹脂の使用により、溶離(elution)プロセスの間、非常に類似した化合物の分離が可能となる。これは、強陰イオン交換樹脂を用いた従来技術では、不可能なことである。その理由は、この様な樹脂の溶離は、一般的にオール−オア−ナッシングのプロセスだからである。前述したように、本発明の更なる利点は、保持された生成物は、水で溶離でき、メタノールのような極性溶剤(polar solvent)を使用する必要がない点である。
【0047】
適宜の吸収性樹脂は、非イオン性で、疎水性で、マクロ網状の架橋された芳香族ポリマに基づく。この様な樹脂は、通常に芳香族ポリマ(aromatic polymer)、例えばスチレン(styrene)、ジビニルベンゼン共重合体(divinylbenzene copolymers)である。これらは、架橋(cross-linked)されている。この様な樹脂は公知であり、適宜の単量体(monomers)を重合させることにより生成される。本発明のヒドロキティロソルを純化するためのクロマトグラフィ用樹脂として使用される吸収性樹脂は、水で溶離(water eluted)される限り、特に限定されない。好ましい吸収性樹脂の一例は:アンバーライト(Amberlite:商標)XAD−4、XAD−7、XAD−1180、XAD−16、XAD−1600(ローム・アンド・ハース社から市販されている);XUS−40323.00、XUS−40285.00、XUS−40283.00(ダウ・ケミカル社から市販されている);SP−700、SP−825、SP−850、ダイヤイオン(Diaion:商標)HP10、HP20、HP30、HP40、DP50(三菱化学社から市販されている)である。
【0048】
この種の樹脂は、本発明にとって特に適したものである。その理由は、ヒドロキティロソルを吸収する非常に高い能力と、加水分解後に得られた溶液中の副生成物の量が少ないが故に、吸収されたヒドロキティロソルは、水で溶離することにより、回収できるからである。しかも、従来技術では必要とされた極性溶剤(例、メタノール、エタノール)を使用しないからである。吸収されたヒドロキティロソルは、大部分回収できる。
【0049】
図に示した実施例においては、本発明のプロセスは、クロマトグラフィ・カラムで2段階の純化を行う。
【0050】
流体Aは、ステップaとステップbから得られる。即ち流体Aは、加水分解ステップと固体分離ステップから得られる。流体Aは、クロマトグラフィ・カラム11内に投入される。このクロマトグラフィ・カラム11は、上記したように陰イオン交換樹脂12を含む。浸透圧物(permeate)が廃棄処理(図示せず)される。その後、水源13からの脱塩水が、クロマトグラフィ・カラム11に供給されて、保持された生成物を溶離し、この溶離された流体を、貯蔵器14に集める。かくして得られた流体生成物(流体B)は、ヒドロキティロソルの純度が、75%以上、通常80%以上である。この純度は、280nmでHPLCによるクロマトグラムにおけるピーク領域のパーセントで決定した。樹脂からのドロキティロソルの回収率は、85%以上、通常90%以上である。
【0051】
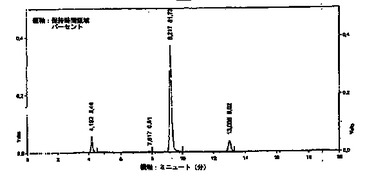
図3は、純化した流体生成物Bを得るためのHPLCクロマトグラム処理を示す。2回目の純化ステップにおいて、流体生成物Bは、クロマトグラフィ・カラム15内に注入される。このクロマトグラフィ・カラムは、非イオン性吸収樹脂16を含有する。浸透圧物質が廃棄処理装置に送られて、吸収されたヒドロキティロソルが、水源13からの脱塩(脱イオン)水の溶離により回収され、貯蔵器17に集められる。流体C(即ち貯蔵器17内に集められた流体)は、ヒドロキティロソルの純度は、90%以上、通常95%以上である。この純度は、280nmでHPLCによるクロマトグラムにおけるピーク領域のパーセントで決定した。樹脂から保持されたヒドロキティロソルの回収率は、90%以上、通常95%以上で、相当な量が回収される。図4は、得られた純化した流体CのHPLCクロマトグラムを示す。
【0052】
図1は、酢酸(CH3CO2H)のソース18とNaOHのソース19を示す。酢酸ソース18は、樹脂12の酸性活性化用のクロマトグラフィ・カラム11に接続される。NaOHソース19は、代わりにNaOHを再生するような他の適宜の装置でもよい。更に、樹脂16再生するNaOHソース19は、クロマトグラフィ・カラム15と硫酸ソース21に接続される。硫酸ソース21は、樹脂の表面活性化用である。
【0053】
流体Aと同様に、流体B、流体Cも、濃縮手段10で濃縮される。濃縮手段10は、例えば、蒸発手段、TFF手段、逆浸透圧手段である。この濃縮は、流体Bに、好ましくは流体Cに対し実行され、濃縮した流体生成物(即ち、抽出物)内で、ヒドロキティロソルの量が、10%になるまで、更には20%、35%、40%になるまで行われる。
【0054】
本発明のプロセスの更なるステップにおいて、前述したステップで得られた流体生成物を、乾燥手段20で乾燥して、固体状の最終生成物を生成する。乾燥手段20は、例えば、冷凍乾燥機(freeze-dryer)、真空回転蒸発器(vacuum rotoevaporator)、好ましくは噴霧乾燥機(spray-dryer)等である。この最終生成物の特徴は、乾燥された開始生成物(A、B、C)により様々であり、乾燥物質の純度は、45%−90%の範囲内にある(これは280nmでHPLC測定した)。
【0055】
この乾燥ステップは、流体Bと流体Cに対し実行されるが、その前にそれらは上記した方法で濃縮したものである。この乾燥ステップは、乾燥生成物の最終使用に適した担体(キャリア)を利用する。この担体は、マルトデキストリン(maltodextrines:麦芽糖を含んだデキストリン;食品添加物)、ラクトース(lactose:哺乳類の乳に存在する二糖)、レシチン(lecithins:動植物で量的に最も多いリン脂質;ホスファチジルコリン(phosphatidyl choline)ともいう)、カゼイン塩(caseinates:カゼインとカルシウムやナトリウムなどの金属との化合物)等である。
【0056】
乾燥技術は、従来公知であり、噴霧乾燥(通常、担体の使用を必要とする)と、凍結乾燥と、真空回転蒸発である。かくして得られた生成物は、ヒドロキティロソルの量が0.5%−10%の間であり、担体を使用した場合には、最大20%(w/w)である。このヒドロキティロソルの量は、担体を使用しない場合には、95%まで上がる。本発明により、純化された流体生成物(流体B特に流体C)が提供できる。この純化された流体生成物は、担体なしでも、蒸発で、乾燥粉末にできる程純度が高い。この事は、従来技術では不可能であった。
【0057】
本発明の実験例を具体的に説明する。
実験例1
オリーブ廃棄物(orujillo)からヒドロキティロソルの抽出、水相の純化
250gの乾燥したオリーブ廃棄物のサンプルを、838mlの脱塩(脱イオン)水と、16.7gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を、固体残留物からフィルタで濾過することにより分離した。この固相(個体物)は、フィルタ上に残ったものであるが、310mlの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約860mlあるが、その後、遠心分離で精製し、フィルタを通過した固体粒子を除去した。この固体除去の後、835mlの原水性抽出物(crude aqueous extract)を得た。この原水性抽出物は、HPLC純度が47.5%の1.41gのヒドロキティロソルを含む。
【0058】
実験例2
イオン交換によるヒドロキティロソルの純化
835mlの原水性抽出物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、ダイヤイオン(Diaion)WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。この溶離された相は、1.27gのヒドロキティロソルを含み、そのHPLC純度は80.85%であった。
【0059】
実験例3
イオン交換と吸収によるヒドロキティロソルの純化
835mlの原水性溶離物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、IRA−67を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%以上が回収されるまで、行われた。
【0060】
第1カラムから得られた溶離された相を、吸収性樹脂を含むクロマトグラフィ・カラムに入れた。これには、樹脂XAD−1180を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは少なくとも最初に入れたヒドロキティロソルの90%以上を回収するまで行った。この溶離された液相は、1.14gのヒドロキティロソルを含み、そのHPCLの純度は、約95.72%であった。
【0061】
実験例4
蒸発によるエンリッチにされたヒドロキティロソル原抽出物の濃縮
遠心分離するステップの前の実験例1で得られた水相(物)約860mlを蒸発法で濃縮して、最終的に193mlの容積を得た。その後、この水相を遠心分離で精製して、フィルタを通して、固体粒子を除去した。この固体除去の後、160mlの原水性抽出物は、1.41gのヒドロキティロソルを含み、そのHPLC純度は47.5%であった。
【0062】
実験例5
エンリッチにしたイオン交換により純化したヒドロキティロソル抽出物の、逆浸透圧方法による濃縮
80リットルの原水溶抽出液(実験例2によりパイロット・プラントで得られた150gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。この逆浸透圧装置は、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを10.8%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は80.53%であった。
【0063】
実験例6
イオン交換と吸収により純化したヒドロキティロソル溶離物の、逆浸透圧方法による濃縮
546リットルの原水溶抽出液(実験例3によりパイロット・プラントで得られた135gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。このパイロット・プラントは、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを12.20%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は95.27%であった。
【0064】
実験例7
純化ステップなしで、ヒドロキティロソル・リッチの原抽出物の噴霧乾燥
442mlの原水性抽出物(実験例1により得られた1.02gのヒドロキティロソルを含む)のサンプルを、100gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。100gのマルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリン(dextrose potato maltodextrin)を用いてもよい。蠕動ポンプ(peristaltic pump)を用いて噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。95gの褐色の粉末が得られた。その湿度は6.85%(karl Fischer:カール・フィッシャー滴定;各種物質の広範囲な水分測定に世界中で使用されている方法)であり、ヒドロキティロソルのリッチネスが0.98%であった。
【0065】
実験例8
ヒドロキティロソル・リッチの一部純化した水性抽出物の噴霧乾燥
290mlの水性抽出物(実験例2により得られた0.38gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、50gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。48.25gの灰色の粉末が得らた。その湿度は6.72%(karl Fischer)であり、ヒドロキティロソルのリッチネスが0.71%であった。
【0066】
実験例9
ヒドロキティロソル・リッチの純化した水性抽出物の噴霧乾燥
188mlの純化した水性抽出物(実験例3により得られた0.29gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、28.5gのマルトデキストリンとゆっくりと撹拌した。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が175℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。27.1gの白色の粉末が得られた。その湿度は5.45%(karl Fischer)であり、ヒドロキティロソルのリッチネスは0.97%であった。
【0067】
実験例10
ヒドロキティロソルが豊富に含まれる粉末の用意
1750lの水性抽出物(実験例3により得られた432gのヒドロキティロソルを含む)サンプルを、パイロットプラントで、実験例6により濃縮した。39.04%のヒドロキティロソルを含む濃縮物を得た。そのHPLC純度は95.60%であった。この濃縮溶液を、噴霧乾燥機に供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。375.84gの褐色の粉末が得られた。その湿度は4.35%(karl Fischer)であり、ヒドロキティロソルのリッチネスは94.74%であった。
【0068】
実験例11
オリーブの実からのオリーブの抽出物
25Kgのオリーブの実のサンプルを、50Lの脱塩(脱イオン)水と混合した。かくして得られた混合液を、数分間ブレンドした。その後636gの硫酸(98%)を添加した。この混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物からフィルタで濾過することにより分離した。この固相は、フィルタ上に残ったものであるが、12.5lの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約63Lあるが、その後、Kieselguhlar フィルタを通して濾過し、抽出されたオイルを除去した。このフィルタは、Celite 500珪藻土で予めコーティングしておいて。これにより、オイルを含有しない水相物を、56Lあるが、遠心分離で精製し、Kieselguhlar フィルタを通過した固体粒子を除去した。この固体除去の後、52Lの原水性抽出物(crude aqueous extract)を得た。これは、HPLC純度が50.5%の141gのヒドロキティロソルを含む。
【0069】
その後、原水性抽出物を、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で予め活性化しておいた。これには、IRA−67を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を、脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。
【0070】
第1クロマトグラフィ・カラムから得られた溶離された相を、吸収性樹脂を含むカラムに入れた。これには、樹脂XAD−1180を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは最初に入れたヒドロキティロソルの90%以上を回収するまで行った。
【0071】
この溶離された液相は、1.14gのヒドロキティロソルを含み、そのHPCL純度は、約96.7%であった。
【0072】
その後、パイロット・プラントで得られた461リットルの純化された溶離液(114gのヒドロキティロソルを含む)を、逆浸透圧のパイロット・プラントを用いて、濃縮し、10リットルの濃縮生成物にした。このパイロット・プラントは、2.5m2の高分子膜を具備する。この容量を、同一材料から形成した0.35m2の膜をその後用いて、ヒドロキティロソルを3.5%含むヒドロキティロソル濃縮物を得た。最終的に、RO濃縮物を、245ミリバールの真空条件で、78℃で回転蒸発(rotaevaporated)した。その結果、液体の状態で。オリーブの実の抽出物の10倍の濃度となり、その最終濃度は、37.2%でHPLC純度は、93.3%であった。
【0073】
実験例12
噴霧乾燥によるオリーブの実の抽出粉末の準備
260mlの純化した液状のオリーブ抽出物(実験例11により得られた19.5gのヒドロキティロソルを含む)のサンプルを、58gのマルトデキストリンとゆっくりと撹拌した。このマルトデキストリンは、260mlの脱塩水で予め溶解しておいた、ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。76gの白色の粉末が得られた。その湿度は5.4%(karl Fischer)であり、ヒドロキティロソルのリッチネスは、21.9%であった。
【0074】
実験例13
3相のポマス(脱脂搾りかす)からヒドロキティロソルの抽出、水相の純化
湿度が60.55%の475.5gの3相ポマスのサンプルを、800mlの脱塩(脱イオン)水と、26.36gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物から、600ミクロンのポリプロピレン製フィルタで濾過することにより分離した。濾過された水相約795mlを、蒸発させることにより濃縮して、最終的に343.8mlの容量を得た。その後、この水相を遠心分離で精製して、フィルタを通過した固体粒子を除去した。この固体除去の後、275mlの原水性抽出物(crude aqueous extract)を得た。これは、0.97gのヒドロキティロソルを含む。そのHPLC純度は47.5%であった。
【0075】
実験例14
3相のポマス(脱脂搾りかす)から得られたヒドロキティロソルのイオン交換と吸着による純化
275mlの原水性抽出物(実験例13により得られた0.97gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むカラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で予め活性化しておいた。これには、Diaion WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を、脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。
【0076】
第1カラムから得られた溶離された相を、吸収性樹脂を含むカラムに入れた。これには、Diaion HP20を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは、最初に入れたヒドロキティロソルの90%以上を回収するまで行った。
【0077】
この溶離された液相は、0.80gのヒドロキティロソルを含み、そのHPCL純度は、約95%以上であった。
【0078】
前述したように、本発明のプロセスは、オリーブ又はポマスから出発したヒドロキティロソルを含む抽出物を有機極性溶剤を使用せずに、準備する方法を提供する。本発明の方法により得られた生成物は、最終生成物とその中間生成物の両方とも、有機極性溶剤を含まない。この生成物は、砂糖も塩も含有しない。これは特許請求の範囲に記載した加水分解条件と純化手順の使用を組み合わせることにより達成できる。
【0079】
最終生成物は、その高純度と高いヒドロキティロソル含有特性により、特に担体溶剤(キャリア)と有機溶剤を含まない固体のヒドロキティロソル抽出物は、食品産業、化粧品産業、医薬産業の使用に適したものである。上記の濃度%は、全て重量%である。
【0080】
以上の説明は、本発明の一実施例に関するもので、この技術分野の当業者であれば、本発明の種々の変形例を考え得るが、それらはいずれも本発明の技術的範囲に包含される。特許請求の範囲の構成要素の後に記載した括弧内の番号は、図面の部品番号に態様し、発明の容易なる理解の為に付したものであり、発明を限定的に解釈するために用いてはならない。又、同一番号でも明細書と特許請求の範囲の部品名は必ずしも同一ではない。これは上記した理由による。用語「又は」に関して、例えば「A又はB」は、「Aのみ」、「Bのみ」ならず、「AとBの両方」を選択することも含む。特に記載のない限り、装置又は手段の数は、単数か複数かを問わない。
【符号の説明】
【0081】
1 反応容器
2 供給手段
3 加熱手段
4 脱塩(脱イオン)水
5 酸タンク
6 攪拌手段
7 フィルタ
8 遠心分離機
9 貯蔵器
10 濃縮手段
11 カラム
12 陰イオン交換樹脂
13 水源
14 貯蔵器
15 カラム
16 非イオン性吸収樹脂
17 貯蔵器
18 酢酸ソース
19 NaOHソース
20 乾燥手段
21 硫酸ソース
図2−4の翻訳
横軸:ミニュート(分)、
縦軸:保持時間領域パーセント
【技術分野】
【0001】
本発明は、オリーブからの抽出物とオリーブ・オイルの抽出後の固体とから、ヒドロキティロソル(hydroxytyrosol)を製造する方法と装置に関する。具体的には、本発明は、ヒドロキティロソルを含有する抽出物を製造する方法に関する。このヒドロキティロソルを含有する抽出物は、食品、医薬品、化粧品の分野で、ヒドロキティロソルの原料として使用される。
【背景技術】
【0002】
オリーブは、いくつもの生物活性化合物(bioactive compounds)、特にポリフェノール(polyphenols)を含む。これらのポリフェノールの中でも、ヒドロキティロソルは、抗菌剤(antimicrobial)又はラジカル補足活性(radical scavenging activity)の分野で、きわだった生物学的な重要性を有する。
【0003】
ヒドロキティロソルは、以下の化学式を有する。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第6361803号明細書
【特許文献2】国際公開パンフレット第2004/005228号
【特許文献3】米国特許出願公開第2004/0102657号明細書
【特許文献4】ヨーロッパ特許1623960号明細書
【特許文献5】米国特許出願公開第2004/0176647号明細書
【0005】
オリーブオイル抽出後の残留物からヒドロキティロソルを製造する方法が、研究されてきた。ヒドロキティロソルの効率的な抽出方法は、非常に利益の多いものである。その理由は、特にオリーブオイル抽出後の残留物の中に存在するヒドロキティロソルとその前駆体が、オリーブ・オイルの製造過程でできる残留物の中に多くあるが、僅かな量しかオリーブオイル内には存在しない。エキストラ・バージン・オリーブ・オイルは、通常1−20ppmのヒドロキティロソルしか含有しない。
【0006】
本発明において、オリーブ・オイル抽出後の残留物には、オリーブの木の葉を含まない。木の葉は、オイル抽出前に除去されているからである。さらに、葉からのヒドロキティロソルの抽出は、オリーブ又はオリーブ残留物からの抽出よりも、様々な出発合成(とそれ故に様々な抽出問題)に直面する。
【0007】
本発明にとって重要なオリーブ・オイル抽出プロセスから得られたオリーブの残留物は、次の様に分類される。
ポマス(pomaces:搾りかす)、即ち、圧搾後の残留物、又は三相プロセスの圧搾後の残留物、あるいは二相プロセスの圧搾後の残留物を含む固体である。この固体状態においては、遠心分離のステップの間、オリーブの小片には水は添加されない。orujo(搾りかす)とalperujo(遠心分離後の残渣)とdefatted orujo(脱脂した搾りかす)は、相当量(45−70%)の水を含有する。抽出残留物は、絞りかすと、オリーブの乾燥固体とを含む。これらは、圧搾後のオイルの抽出後、15%未満の水しか含まず、残留オイルを含まない。
【0008】
本発明のグリーン・オリーブの抽出物は、オリーブ・オイル製造時の残留物の抽出物よりも、次の点で好ましい。ヒドロキティロソルを多く含み、ヒドロキシメチルフルフラール(hydroxymethylfurfural)の量が少ない点で好ましい。
【0009】
本明細書において、用語「ポマス(pomace:搾りかす)」あるいは「固体残留物(solid residue)」は、「搾りかす(orujo)」、「脱脂された搾りかす(defatted orujo)」、「搾りかす(orujillo)」あるいは「残渣(alperujo)」を意味する。本発明の好ましい出発材料は、オリーブで、より好ましくはグリーン・オリーブとポマスである。
【0010】
ポマス(又は食物からの水)の酸性加水分解(acid hydrolysis)を実行し、オレウロペイン分子(oleuropein molecule)内のエステル結合を切断し、ヒドロキティロソルを得ることは、公知である。
【0011】
特許文献1は、ヒドロキティロソルと他の化合物の抽出を、オリーブ・パルプ残留物を1時間(ex12)還流しながら中性状態あるいは酸性状態で加水分解して、行うことを開示している。この抽出された水溶液を、吸収XAD−7カラムに入れる。そこでメタノールで溶離し、抽出されたヒドロキティロソルを回収する。同文献の方法では、有機極性溶剤(organic polar solvents)を用いることが必要であり、しかし最低純度(a minimum purity grade)のヒドロキティロソルしか回収できない。それに加えて(ex.12)、メタノール溶液からのある種の不純物の凍結沈殿が必要である。極性水溶性溶剤(polar aqeous solvent)は、メタノール(methanol)、エタノール(ethanol)、アセトニトリル(acetonitrile)、アセトン(acetone)から選択される。有機極性溶剤は、エステル(esters)、アミド(amides)、ジメチル・スルホキシド(dimethyl sulfoxide)、ジオクサン(dioxane)、ジメチルホルムアミド(DMF:dimethylformamide)、あるいはその混合物から選択される。これらの溶剤の大部分は、毒性があり、所望のヒドロキティロソル生成物から完全に取り除くことは困難である。従って、使用された溶剤の痕跡が、何回かの純化ステップの後でも、最終生成物に見出されることがある。その結果、このプロセスで得られたヒドロキティロソルは、食品、化粧、薬品の分野で、安全に使用できない。更に、この最終生成物は、補強食品、特に食用オイルでの使用に適したものではなく、安全な食品を得るために、エタノールや酢酸(acetic acid)を、抽出物への添加剤として使用していない場合でも、食用に適したものにはならない。その結果得られた食料品(オイルとヒドロキティロソル(エタノールと酢酸と抽出物を含む))は、食品業界では受け入れ難いものである。
【0012】
特許文献2は、オリーブ・オイルの抽出により得られた植物性の水の室温での加水分解を開示する。このオリーブ・オイルの抽出物は、酸性化した植物性の水を、少なくとも2ヶ月(好ましくは6−12ヶ月)の間、植物性の水の中に本来存在するoleuropein(オレウロペイン)の少なくとも50%(好ましくは90%)が、ヒドロキティロソルに変わるまで、培養することにより、行った。この培養された植物性の水を、有機溶剤(例、酢酸エチル(ethyl acetate)で抽出するか、あるいは超臨界流体(supercritical fluid)(CO2)に接触させて、ヒドロキティロソル−リッチの化合物を生成する。このプロセスの主な問題点は、特に得られた最終生成物を食品、化粧品、医薬品として使用する場合、酸性化された植物性の水の培養に時間がかかることであり、回避すべき有機溶剤を使用することである。
【0013】
特許文献3は、高温(190−220℃)での水蒸気拡散プロセス(steam explosion process)で行う酸性加水分解を開示する。かくして得られた溶液は、活性化されていないイオン交換樹脂のカラム内で最初に部分的に純化し、その後XAD非イオン・カラムに入れる。そこからヒドロキティロソルを、メタノールあるいはエタノールで、溶離する。このプロセスは、このプロセス全体を考慮しても、ヒドロキティロソルの歩留まりが悪い。
【0014】
特許文献4は、ヒドロキティロソルとチロソール(tyrosol)を植物性の水(alpechin)から回収するプロセスを、濾過手段と後続の分離手段で実行することを開示する。この濾過手段は3つのユニットからなる複雑なプラントで行われる。チロソールを、その後プロトン性溶剤(protic solvent)(アルコール又は水)で酸化し、ヒドロキティロソルに変換して、準合成品(semi-synthetic)である最終生成物を得ている。更に同文献は、植物性の水(alpechin)のナノ濾過(nanofiltration)による濃縮(concentration)と、逆浸透圧化(reverse osmosis)を、中性又はアルカリ性のpHで行うことを開示(請求項4)している。しかし、このプロセスには、2つの重要な問題点がある。第1の問題点は、ヒドロキティロソルの劣化は、中性あるいはアルカリ性の状態で増加し、除去することが困難な不要な生成物が現れ、更に酸性加水分解が実行されないことに起因して、オレウロペイン・コンテンツが依然として高い点である。第2の問題点として、獲得可能な濃度係数(concentration factor)が低いことである。かくして、Ex‐1(表1を参照のこと)によると、ヒドロキティロソルの最大濃度は、1.2−1.6g/Lの範囲内である。
【0015】
特許文献5は、alperujoからフェノール(phenols)を抽出するプロセスを開示する。この抽出プロセスは、180‐240℃のオートクレーブ(加圧滅菌器)内で、水中で攪拌することにより行った。酸は添加せずに熱処理を行った結果、「アセチル基の遊離(liberation of acetyl groups)」と、その結果pHの減少が起こった(同文献の3項1段落目)。しかし、上記の条件で生成されたpHにおいては、オレウロペイン(oleuropein)の加水分解の完成には程遠い。このことは、2つの問題点がある。即ち、ヒドロキティロソルの歩留まりの低さと加水分解されたオレウロペインの残留である。これは、上記で議論したごとく、この様な抽出物を含む食料品は不味く、食料品への適用には不向きである。この生成物は、チロソールとヒドロキティロソルであり、これらはHPLC(high-performance liquid chromatography:高速液体クロマトグラフィ;混合物を分離する分析化学の手法)により、硫酸(sulphuric acid)/アセトニトリル溶離液(acetonitrile eluent)で、分離できる。上記の従来技術を要約すると、同文献のプロセスは、時間がかかり、複雑で、条件が厳しい。その結果、オレウロペインの量、即ち加水分解されない量、又は加水分解による副生成物の量が、多すぎる。例えば、ヒドロキシメチルフルフラール(hydroxymethylfurfural )が多く、後続の純化ステップが困難になる。
【0016】
副生成物の発生は、ヒドロキティロソル抽出物の生成プロセスにおいて、当初考えたよりも大きな問題であることが判った。このことは、上記の従来の文献からも明らかである。開始材料、特にオリーブとポマスは、中でもフェノール化合物(phenolic compounds)をグルコシド(glucoside)の形態で、あるいはエステルの形態で大量に含まれる。エステルは、オレウロペイン、ligustroside、verbascoside、幾つかのフラボノイド類(flavonoids)を含む。幾つかの他の天然化合物も存在する。それ故に、開始化合物が容易に劣化し、多量の副生成物になってしまう。
【0017】
副生成物を除去して、ヒドロキティロソルを純化しなければならないので、公知の方法では、有機溶剤を使用して、クロマトグラフィ・カラム(chromatographic columns)を用いる後続のプロセスの間、ヒドロキティロソルを純化する必要がある。有機溶剤の使用は、例えばメタノールの使用は、樹脂・カラム(resin column)から溶離(elute)する際には不都合なものである。得られた最終生成物を食品、化粧品、医薬品の分野で使用する場合には、特に不都合である。
【0018】
更に公知のヒドロキティロソル・リッチの抽出物は、食料品特に食用油の添加剤として使用するのに適したものではない。それ故に、以下に記載する特性を有するヒロキティロソルを含む抽出物を生成するニーズがある。即ち、この抽出物は、ヒドロキティロソル・リッチであり、開始生成物(例えばオレウロペインとverbascosides)の量が少なく、副生成物の量、特にヒドロキシメチルフルフラールの量が少なく、一般的に、砂糖や塩の量が極めて少ないものである。
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明の目的は、上記の問題点を解決することである。本発明は、オリーブあるいはオリーブの抽出物である副生成物から、ヒドロキティロソルを生成するプロセスを提供することである。しかも簡単で効率的で安価で、有機溶剤を含まない高純度の生成物を提供することである。食料や化粧品の添加物として使用するのに適したものを製造する方法を提供することである。
【課題を解決するための手段】
【0020】
この目的は、請求項1記載の本発明の手段により達成できる。即ち、出発材料から、ヒドロキティロソルを含有する抽出物を生成する方法において、前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択される。前記方法は、前記出発材料を酸性加水分解し、得られた溶液を純化しする。更に具体的には、
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する。
【0021】
本発明の方法は、開始材料の水中で酸性加水分解を以下の条件で行う。温度条件は、140℃未満で、好ましくは70℃−130℃の間で、更に好ましくは、110℃−130℃の間(還流温度以上)、である。圧力の掛かった状態で、例えば、連続的な殺菌システムで行う。圧力条件は、大気圧以上(ゲージ圧)で10‐20psiの範囲である。加水分解を受ける酸性混合物のpHは、1.6‐6.0の範囲内である。加水分解後の混合物は、従来公知の物理的方法で分離して、例えば、濾過と/又は遠心分離により分離して、加水分解後の水溶液から懸濁した固体を除去し、懸濁液中に固体の無い純化溶液を得る。
【0022】
開始材料は、オリーブ又はポマスである。即ち、上記に定義したオリーブ・オイルの抽出プロセスから得られた残留物である。好ましいオリーブは、グリーン・オリーブである。ポマスは、好ましくはオイルを含有しない。オイルは、加水分解のステップの前あるいは後の何れかで、公知の手段(例えば、脱脂orujoあるいはorujilloを準備するのに用いられるもの)により、あるいは珪藻土(diatomaceous earth)、あるいは他の濾過手段により除去する。
【0023】
本発明によれば、上記のステップ(酸性加水分解と純化(clarification))の後に、2つのステップが行われる。即ち、得られた生成物を、第1のクロマトグラフィ・カラム(酸で活性化した陰イオン交換樹脂を含む)に入れるステップと、前記第1のクロマトグラフィ・カラム内にある生成物を水で溶離するステップが行われる。
本発明の好まし実施例によれば、本発明は、更に純化ステップを実行する。この純化ステップは、前記の第1のクロマトグラフィ・カラム(陰イオン交換樹脂を含む)からの水溶離溶液を、第2のクロマトグラフィ・カラム(吸着性の非イオン性樹脂を含む)の内に入れるステップと、前記第2のクロマトグラフィ・カラム内に残った生成物を水で溶離するステップである。
【0024】
本発明の更なる態様によれば、流体生成物は、例えば、逆浸透圧濃縮(reverse osmosis concentration)方法で濃縮する。本発明の更なるステップによれば、クロマトグラフィの純化ステップと逆浸透圧濃縮ステップの後、得られた流体生成物を固体状にする。これは、例えば凍結乾燥(freeze-drying)、真空回転蒸発(vacuum rotoevaporation)、噴霧乾燥(spray drying)で行う。このステップは、マルトデキストリン(maltodextrin)のようなキャリア(担持流体)の有無を問わない。
【0025】
本発明の更なる目的は、上記のプロセスにより得られたヒドロキティロソルを含む抽出物である。この抽出物は、液体又は固体のいずれかの形態であり、ヒドロキティロソルを0.5%(w/w)以上含む。その純度は、40%以上で、好ましくは80%以上で、より好ましくは95%以上である。これは、280nmで測定したHPLCピーク面積(peak area)により決定した。これらの抽出物は、有機溶剤とヒドロキシメチルフルフラールを含まず、砂糖も塩もほぼ含まない。
【0026】
本発明の一態様によれば、本発明の方法により得られたヒドロキティロソルを含む流体生成物は、ヒドロキティロソルを35%(w/w)以上、好ましくは45%以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、35%以上である。
本発明のより好ましい態様によれば、本発明の方法により得られたヒドロキティロソルを含む固体生成物は、ヒドロキティロソルを20%(w/w)以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、20%以上である。
【0027】
本発明のより好ましい態様によれば、本発明の方法により得られたヒドロキティロソルを含む固体生成物は、ヒドロキティロソルを90%(w/w)以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、92%以上である。
【0028】
本発明の更なる目的は、上記したプロセスを実行する装置を提供することであり、この装置は、請求項13に記載されている。
本発明の装置又はプラントは、反応容器と、第1のクロマトグラフィ・カラムと、第2クロマトグラフィ・カラムとを含む。前記反応容器は、圧力のかかった状態で酸性の加水分解を実行する。前記クロマトグラフィ・カラムは、樹脂と、酸で活性化した陰イオン交換樹脂(弱塩基性(weakly basic))と、水溶離性の陰イオン交換樹脂を含む。第2クロマトグラフィ・カラムは、吸着性の非イオン樹脂を含む。この吸着性の非イオン樹脂は、マクロ網状の架橋された芳香族ポリマ・樹脂(macroreticular cross-link aromatic polymer resin)である
【0029】
本発明は、従来技術に対しいくつかの利点を提供する。加水分解ステップは、本発明により、水だけあるいはミネラル酸のみを用いて、特許請求の範囲に記載した還流温度(reflux temperture)以上で行い、加熱ステップと圧力ステップを組み合わせて行うことができる。圧力が、特許請求の範囲に記載された温度と共に加えられた場合に、その結果得られた加水分解は、約30分で終了するが、これには、副生成物の形成はなく、開始材料(オレウロペイン)を所望の最終生成物(ヒドロキティロソル)に完全に変換できる。本発明の加水分解ステップにより、短時間で極めて高い歩留まりで、ヒドロキティロソルを得ることができ、副生成物は全く生成されない。従来技術では、この様な副生成物は、取り除くことが困難であり、かなり量生成されていた。
【0030】
本発明の加水分解ステップは、更なる利点を有する。酸の添加と加熱を適宜組み合わせて実行することにより、高い変換率で加水分解を行うことができ、かつ有害な副生成物の形成を回避できる。その結果混合物が殺菌される。実際に、加水分解プロセスは、水溶液の殺菌を提供できる。即ち関連する生成物の殺菌が提供でき、これは極めて有益である。その理由は、ヒドロキティロソルを準備するプロセスで使用される開始材料(例えば、ポマス)は、通常既に処理された材料から得られ、一般的に純化−殺菌の前処理(depuration-sterilization pre-treatment)を行い、安全なヒドロキティロソル最終生成物を得ている。この純化−殺菌の前処理は、時には複雑な処理であり、プロセスの終了時に、更なる処理ステップを必要とする。その結果、所望の最終生成物の低い歩留まりとなる。本発明によれば、加水分解ステップにより、加水分解反応と、関連する必要な生成物の殺菌が同時に提供できる。かくして、開始材料の殺菌ステップを行う必要がなく、又更なる使用に適した加水分解された材料を提供できる。これは更なる殺菌処理を必要としない。
【0031】
これに加えて、加水分解ステップの後、純化ステップが行われる。この純化ステップは、加水分解ステップで得られた生成物を、少なくとも1つのクロマトグラフィ・カラム・システムに、あるいは2つのクロマトグラフィ・カラム・システムに入れ、このクロマトグラフィ・カラム内に残ったヒドロキティロソルを水で溶離する。この場合、加水分解ステップで生成された副生産物の量は、極めて少ないので、有機溶剤無しで、水単独で溶離、あるいは水と混合して溶離するようなクロマトグラフィ・カラムの使用が可能となる。この事は、有機溶剤は、ヒドロキティロソルとは接触せず、かくして純粋なヒドロキティロソルが得られる。これは、特に食品、化粧品、医薬品の分野での使用に適したものである。
【0032】
本発明の他の利点は、濃縮ステップに起因する。濃縮ステップにより、純化ステップの後のヒドロキティロソルの量を増やすことができる。この純化ステップは、液体抽出物、特に砂糖と塩を含まない液体抽出物を生成する。その結果、純化した流体抽出物の濃縮ステップの間、浸透圧の非常に重要な減少が得られ、かくして非常に高い濃縮係数(350倍まで)が可能となる。この濃縮ステップは、多くの量のヒドロキティロソルの含有により特徴付けられた「濃縮物(concentrate)」であり、これは更なる処理に直接使用できる。従来技術においては、濃縮ステップは通常存在せず、あった場合でも、生成物の濃縮係数は低い(2倍から5倍)。これは塩と砂糖を取り除く純化プロセスがないためである。ヒドロキティロソルの準備のプロセスにおては、処理された溶液/分散液内のヒドロキティロソルの濃度は常に低く、大量に処理する必要があり、かくして、最終生成物の歩留まりが下がる。
【0033】
本発明によれば、例えば、マルトデキストリン(maltodextrin:麦芽糖を含んだデキストリン;食品添加物)等のあらゆる担体(carrier)と混合していない最終固体生成物を得ることができることにより、最終使用意図に適し、法律上の要件に適した、純化したヒドロキティロソルを得ることができる。
【図面の簡単な説明】
【0034】
【図1】本発明の装置を表すブロック図。
【図2】本発明の第1実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【図3】本発明の第2実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【図4】本発明の第3実施例によるヒドロキティロソルを含む生成物のクロマトグラムを表す図。
【発明を実施するための形態】
【0035】
図1において、本発明の装置は、反応容器1を含む。好ましくは、連続する殺菌システムを含む。この殺菌システムで本発明の加水分解が行われる。
【0036】
上記したように、反応容器内で、加水分解を以下の条件で行う。温度は、140℃を超えることはなく、好ましくは70℃−130℃の間で、更に好ましくは(還流温度以上で)、110℃−130℃である。圧力条件は、ゲージ圧で(以下同様)、10‐20psi(68.94−137.8KPa)の範囲である。分解時間は15分−45分の間である。好ましくは、加水分解温度は、118℃から126℃の範囲内であり、最も好ましくは、120℃から121℃で、圧力は15psi(103.4KPa)で、30分間行われる。好ましい反応容器は、連続式の殺菌システムである。
【0037】
連続式の殺菌システムである反応容器1は、供給手段2を具備する。この供給手段2は、反応容器1に、開始材料であるオリーブ・オイル抽出後の残留物、即ち副生成物、言い換えるとポマス(即ち、圧搾したオリーブの固体残留物)を供給する。連続式の殺菌システムである反応容器1は、バッチ処理ではなく連続するプロセスで、出発材料を処理する。加熱手段3が、反応容器1を上記の温度に加熱する。加熱手段3は、反応容器1の周囲に巻かれた加熱ジャケットである。反応容器1は、脱塩(脱イオン)水4と攪拌手段6を具備する。この攪拌手段は、出発材料である混合水物を攪拌する。水と固体との比率は、1:1から4:1の間である。
【0038】
加水分解プロセスは、pHが1.0−6.0の間で行われ、好ましくは1.0−3.0の間で行われる。酸(好ましくは硫酸H2SO4)は、酸タンク5から得られる。
【0039】
特許請求の範囲に記載された酸性状態と温度の組み合わせにより、加水分解プロセスが行われる。この加水分解プロセスは、極めて速く、効率的で、30分間、120℃か−123℃で、15psiの圧力で行われる。更にこのプロセスにより、加水分解生成物が殺菌される。
【0040】
反応容器1の出口にフィルタ7が接続される。このフィルタ7は、ヒドロキティロソルや他のフェノール(phenols)を含む反応混合物から、固体を除去する。フィルタ処理された物は、その後更に分離手段好ましくは遠心分離機8に送られる。そこで更に固体が反応混合物から除去され、懸濁した固体のない流体、即ち後続の純化ステップに適した溶液を得る。遠心分離の後、分離した(不純物を除去した)流体は褐色をしており、貯蔵器9に貯蔵される。分離した流体Aは、ヒドロキティロソルと、オレウロペイン(oleuropein)の残留物と、少量のフェノールと、他の生成物とを含む。図2のクロマトグラフは、この流体中に含まれる検出した化合物の組成をパーセントを示す。この分析は、波長28nmのPHLCにより得られたものであり、9.317ミニュッツ(分)の大きなピークは、ヒドロキティロソルであり、13.150ミニュッツ(分)のピークは、チロソール(tyrosol)である。
【0041】
本発明の好ましい一実施例によれば、流体Aは、蒸発(evaporation)法、又は接線流濾過法(tangential flow filtration(TFF))(例、逆浸透圧システムを用いる)により濃縮される。この濃縮ステップは、流体Aに対し実行される。この濃縮ステップは、直接、あるいは好ましくは純化ステップの後で、クロマトグラフィ・カラムの手段で行う。この為に、貯蔵タンク即ち貯蔵器9が、濃縮手段10、TFFあるいは好ましくは逆浸透圧手段で、純化手段に接続される。
【0042】
純化手段は、一実施例では、単一の(クロマトグラフィ・)カラム・システムを含む。このカラム・システムは、酸で活性化された陰イオン交換樹脂(acid activated anion exchnage resin)から選択された樹脂のクロマトグラフィ・カラムを含む。本発明の他の実施例においては、純化手段は、前記の第1カラムに加えて、吸収性の非イオン性樹脂(absorbant non-ionic resin)から選択された樹脂の第2クロマトグラフィ・カラムを、更に含む。
【0043】
本発明の他の実施例においては、酸で活性化された陰イオン交換樹脂は、弱塩基性の陰イオン交換樹脂(weakly basic anion exchnage resin)である。吸収性の非イオン性樹脂は、マクロ網状の架橋された芳香族ポリマ(macroreticular cross-linked aromatic polymer)である。
【0044】
本発明のヒドロキティロソルの純化用クロマトグラフィ・樹脂として使用される陰イオン交換樹脂は、酸で活性化されている限り、それに限定されるない。弱塩基性の陰イオン交換樹脂の一例は、ポリアミン系樹脂(polyamine-type resin)(ポリアミン型キレーティング・樹脂を含む)を含む。その一例は、例えば、スチレン/ジビニルベンゼン・共重合体(styrene/divinylbenzene copolymer)とジエチレントリアミン(diethylenetriamine)の反応生成物、アリルアミン(allylamine)、ビニルアミン(vinylamine)等を主に含む化合物の重合化生成物(polymerization product)のような樹脂、アクリル・樹脂(acrylic resins)、例えば、ジビニルベンゼンの共重合体とアクリル酸あるいはメタクリル酸(methacrylic acid)あるいはジアチルアミンノプロピルアミン(dimethylaminopropylamine)を含むアミド化合物である。他の樹脂は、上記の弱塩基性の陰イオン交換樹脂が、強塩基性の交換基(strongly basic exchnage group)で、例えば、トリメチルアミン(tirmethylamine)とジメチルエタノールアミン(dimethylethanolamine)で部分的に置換したものである。より具体的には、従来公知の樹脂は、例えば、ダイヤイオン(Diaion:商標) WA10、WH20、WH21、WA30(三菱化学社製)、アンバーライト(Amberlite:商標)IRA−35、IRA−67(IRA−68)、IRA−93ZU、IRA−94S、IRA−478(ローム・アンド・ハース社製)、WGR−2(ダウ・ケミカル社製)等である。
【0045】
本発明の方法によれば、これらの樹脂は、使用される前に酸で、好ましくは、酢酸で活性化される。これらの樹脂は、特に本発明の方法に適したものである。低価格である点及びそれらがマイルドな状態で再生される点で適したものである。更に、樹脂の弱塩基性により、ヒドロキティロソル分子とチロソール分子の部分的な分離が容易となる。ヒドロキティロソル/チロソールのピーク率の比較と、図2、3のヒドロキティロソルのHPLC純度の観点からすると、本発明によるクロマトグラフィ手法による分離は、効率が良いことが判る。
【0046】
実際に、本発明の弱陰イオン交換樹脂の使用により、溶離(elution)プロセスの間、非常に類似した化合物の分離が可能となる。これは、強陰イオン交換樹脂を用いた従来技術では、不可能なことである。その理由は、この様な樹脂の溶離は、一般的にオール−オア−ナッシングのプロセスだからである。前述したように、本発明の更なる利点は、保持された生成物は、水で溶離でき、メタノールのような極性溶剤(polar solvent)を使用する必要がない点である。
【0047】
適宜の吸収性樹脂は、非イオン性で、疎水性で、マクロ網状の架橋された芳香族ポリマに基づく。この様な樹脂は、通常に芳香族ポリマ(aromatic polymer)、例えばスチレン(styrene)、ジビニルベンゼン共重合体(divinylbenzene copolymers)である。これらは、架橋(cross-linked)されている。この様な樹脂は公知であり、適宜の単量体(monomers)を重合させることにより生成される。本発明のヒドロキティロソルを純化するためのクロマトグラフィ用樹脂として使用される吸収性樹脂は、水で溶離(water eluted)される限り、特に限定されない。好ましい吸収性樹脂の一例は:アンバーライト(Amberlite:商標)XAD−4、XAD−7、XAD−1180、XAD−16、XAD−1600(ローム・アンド・ハース社から市販されている);XUS−40323.00、XUS−40285.00、XUS−40283.00(ダウ・ケミカル社から市販されている);SP−700、SP−825、SP−850、ダイヤイオン(Diaion:商標)HP10、HP20、HP30、HP40、DP50(三菱化学社から市販されている)である。
【0048】
この種の樹脂は、本発明にとって特に適したものである。その理由は、ヒドロキティロソルを吸収する非常に高い能力と、加水分解後に得られた溶液中の副生成物の量が少ないが故に、吸収されたヒドロキティロソルは、水で溶離することにより、回収できるからである。しかも、従来技術では必要とされた極性溶剤(例、メタノール、エタノール)を使用しないからである。吸収されたヒドロキティロソルは、大部分回収できる。
【0049】
図に示した実施例においては、本発明のプロセスは、クロマトグラフィ・カラムで2段階の純化を行う。
【0050】
流体Aは、ステップaとステップbから得られる。即ち流体Aは、加水分解ステップと固体分離ステップから得られる。流体Aは、クロマトグラフィ・カラム11内に投入される。このクロマトグラフィ・カラム11は、上記したように陰イオン交換樹脂12を含む。浸透圧物(permeate)が廃棄処理(図示せず)される。その後、水源13からの脱塩水が、クロマトグラフィ・カラム11に供給されて、保持された生成物を溶離し、この溶離された流体を、貯蔵器14に集める。かくして得られた流体生成物(流体B)は、ヒドロキティロソルの純度が、75%以上、通常80%以上である。この純度は、280nmでHPLCによるクロマトグラムにおけるピーク領域のパーセントで決定した。樹脂からのドロキティロソルの回収率は、85%以上、通常90%以上である。
【0051】
図3は、純化した流体生成物Bを得るためのHPLCクロマトグラム処理を示す。2回目の純化ステップにおいて、流体生成物Bは、クロマトグラフィ・カラム15内に注入される。このクロマトグラフィ・カラムは、非イオン性吸収樹脂16を含有する。浸透圧物質が廃棄処理装置に送られて、吸収されたヒドロキティロソルが、水源13からの脱塩(脱イオン)水の溶離により回収され、貯蔵器17に集められる。流体C(即ち貯蔵器17内に集められた流体)は、ヒドロキティロソルの純度は、90%以上、通常95%以上である。この純度は、280nmでHPLCによるクロマトグラムにおけるピーク領域のパーセントで決定した。樹脂から保持されたヒドロキティロソルの回収率は、90%以上、通常95%以上で、相当な量が回収される。図4は、得られた純化した流体CのHPLCクロマトグラムを示す。
【0052】
図1は、酢酸(CH3CO2H)のソース18とNaOHのソース19を示す。酢酸ソース18は、樹脂12の酸性活性化用のクロマトグラフィ・カラム11に接続される。NaOHソース19は、代わりにNaOHを再生するような他の適宜の装置でもよい。更に、樹脂16再生するNaOHソース19は、クロマトグラフィ・カラム15と硫酸ソース21に接続される。硫酸ソース21は、樹脂の表面活性化用である。
【0053】
流体Aと同様に、流体B、流体Cも、濃縮手段10で濃縮される。濃縮手段10は、例えば、蒸発手段、TFF手段、逆浸透圧手段である。この濃縮は、流体Bに、好ましくは流体Cに対し実行され、濃縮した流体生成物(即ち、抽出物)内で、ヒドロキティロソルの量が、10%になるまで、更には20%、35%、40%になるまで行われる。
【0054】
本発明のプロセスの更なるステップにおいて、前述したステップで得られた流体生成物を、乾燥手段20で乾燥して、固体状の最終生成物を生成する。乾燥手段20は、例えば、冷凍乾燥機(freeze-dryer)、真空回転蒸発器(vacuum rotoevaporator)、好ましくは噴霧乾燥機(spray-dryer)等である。この最終生成物の特徴は、乾燥された開始生成物(A、B、C)により様々であり、乾燥物質の純度は、45%−90%の範囲内にある(これは280nmでHPLC測定した)。
【0055】
この乾燥ステップは、流体Bと流体Cに対し実行されるが、その前にそれらは上記した方法で濃縮したものである。この乾燥ステップは、乾燥生成物の最終使用に適した担体(キャリア)を利用する。この担体は、マルトデキストリン(maltodextrines:麦芽糖を含んだデキストリン;食品添加物)、ラクトース(lactose:哺乳類の乳に存在する二糖)、レシチン(lecithins:動植物で量的に最も多いリン脂質;ホスファチジルコリン(phosphatidyl choline)ともいう)、カゼイン塩(caseinates:カゼインとカルシウムやナトリウムなどの金属との化合物)等である。
【0056】
乾燥技術は、従来公知であり、噴霧乾燥(通常、担体の使用を必要とする)と、凍結乾燥と、真空回転蒸発である。かくして得られた生成物は、ヒドロキティロソルの量が0.5%−10%の間であり、担体を使用した場合には、最大20%(w/w)である。このヒドロキティロソルの量は、担体を使用しない場合には、95%まで上がる。本発明により、純化された流体生成物(流体B特に流体C)が提供できる。この純化された流体生成物は、担体なしでも、蒸発で、乾燥粉末にできる程純度が高い。この事は、従来技術では不可能であった。
【0057】
本発明の実験例を具体的に説明する。
実験例1
オリーブ廃棄物(orujillo)からヒドロキティロソルの抽出、水相の純化
250gの乾燥したオリーブ廃棄物のサンプルを、838mlの脱塩(脱イオン)水と、16.7gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を、固体残留物からフィルタで濾過することにより分離した。この固相(個体物)は、フィルタ上に残ったものであるが、310mlの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約860mlあるが、その後、遠心分離で精製し、フィルタを通過した固体粒子を除去した。この固体除去の後、835mlの原水性抽出物(crude aqueous extract)を得た。この原水性抽出物は、HPLC純度が47.5%の1.41gのヒドロキティロソルを含む。
【0058】
実験例2
イオン交換によるヒドロキティロソルの純化
835mlの原水性抽出物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、ダイヤイオン(Diaion)WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。この溶離された相は、1.27gのヒドロキティロソルを含み、そのHPLC純度は80.85%であった。
【0059】
実験例3
イオン交換と吸収によるヒドロキティロソルの純化
835mlの原水性溶離物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、IRA−67を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%以上が回収されるまで、行われた。
【0060】
第1カラムから得られた溶離された相を、吸収性樹脂を含むクロマトグラフィ・カラムに入れた。これには、樹脂XAD−1180を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは少なくとも最初に入れたヒドロキティロソルの90%以上を回収するまで行った。この溶離された液相は、1.14gのヒドロキティロソルを含み、そのHPCLの純度は、約95.72%であった。
【0061】
実験例4
蒸発によるエンリッチにされたヒドロキティロソル原抽出物の濃縮
遠心分離するステップの前の実験例1で得られた水相(物)約860mlを蒸発法で濃縮して、最終的に193mlの容積を得た。その後、この水相を遠心分離で精製して、フィルタを通して、固体粒子を除去した。この固体除去の後、160mlの原水性抽出物は、1.41gのヒドロキティロソルを含み、そのHPLC純度は47.5%であった。
【0062】
実験例5
エンリッチにしたイオン交換により純化したヒドロキティロソル抽出物の、逆浸透圧方法による濃縮
80リットルの原水溶抽出液(実験例2によりパイロット・プラントで得られた150gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。この逆浸透圧装置は、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを10.8%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は80.53%であった。
【0063】
実験例6
イオン交換と吸収により純化したヒドロキティロソル溶離物の、逆浸透圧方法による濃縮
546リットルの原水溶抽出液(実験例3によりパイロット・プラントで得られた135gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。このパイロット・プラントは、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを12.20%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は95.27%であった。
【0064】
実験例7
純化ステップなしで、ヒドロキティロソル・リッチの原抽出物の噴霧乾燥
442mlの原水性抽出物(実験例1により得られた1.02gのヒドロキティロソルを含む)のサンプルを、100gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。100gのマルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリン(dextrose potato maltodextrin)を用いてもよい。蠕動ポンプ(peristaltic pump)を用いて噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。95gの褐色の粉末が得られた。その湿度は6.85%(karl Fischer:カール・フィッシャー滴定;各種物質の広範囲な水分測定に世界中で使用されている方法)であり、ヒドロキティロソルのリッチネスが0.98%であった。
【0065】
実験例8
ヒドロキティロソル・リッチの一部純化した水性抽出物の噴霧乾燥
290mlの水性抽出物(実験例2により得られた0.38gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、50gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。48.25gの灰色の粉末が得らた。その湿度は6.72%(karl Fischer)であり、ヒドロキティロソルのリッチネスが0.71%であった。
【0066】
実験例9
ヒドロキティロソル・リッチの純化した水性抽出物の噴霧乾燥
188mlの純化した水性抽出物(実験例3により得られた0.29gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、28.5gのマルトデキストリンとゆっくりと撹拌した。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が175℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。27.1gの白色の粉末が得られた。その湿度は5.45%(karl Fischer)であり、ヒドロキティロソルのリッチネスは0.97%であった。
【0067】
実験例10
ヒドロキティロソルが豊富に含まれる粉末の用意
1750lの水性抽出物(実験例3により得られた432gのヒドロキティロソルを含む)サンプルを、パイロットプラントで、実験例6により濃縮した。39.04%のヒドロキティロソルを含む濃縮物を得た。そのHPLC純度は95.60%であった。この濃縮溶液を、噴霧乾燥機に供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。375.84gの褐色の粉末が得られた。その湿度は4.35%(karl Fischer)であり、ヒドロキティロソルのリッチネスは94.74%であった。
【0068】
実験例11
オリーブの実からのオリーブの抽出物
25Kgのオリーブの実のサンプルを、50Lの脱塩(脱イオン)水と混合した。かくして得られた混合液を、数分間ブレンドした。その後636gの硫酸(98%)を添加した。この混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物からフィルタで濾過することにより分離した。この固相は、フィルタ上に残ったものであるが、12.5lの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約63Lあるが、その後、Kieselguhlar フィルタを通して濾過し、抽出されたオイルを除去した。このフィルタは、Celite 500珪藻土で予めコーティングしておいて。これにより、オイルを含有しない水相物を、56Lあるが、遠心分離で精製し、Kieselguhlar フィルタを通過した固体粒子を除去した。この固体除去の後、52Lの原水性抽出物(crude aqueous extract)を得た。これは、HPLC純度が50.5%の141gのヒドロキティロソルを含む。
【0069】
その後、原水性抽出物を、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で予め活性化しておいた。これには、IRA−67を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を、脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。
【0070】
第1クロマトグラフィ・カラムから得られた溶離された相を、吸収性樹脂を含むカラムに入れた。これには、樹脂XAD−1180を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは最初に入れたヒドロキティロソルの90%以上を回収するまで行った。
【0071】
この溶離された液相は、1.14gのヒドロキティロソルを含み、そのHPCL純度は、約96.7%であった。
【0072】
その後、パイロット・プラントで得られた461リットルの純化された溶離液(114gのヒドロキティロソルを含む)を、逆浸透圧のパイロット・プラントを用いて、濃縮し、10リットルの濃縮生成物にした。このパイロット・プラントは、2.5m2の高分子膜を具備する。この容量を、同一材料から形成した0.35m2の膜をその後用いて、ヒドロキティロソルを3.5%含むヒドロキティロソル濃縮物を得た。最終的に、RO濃縮物を、245ミリバールの真空条件で、78℃で回転蒸発(rotaevaporated)した。その結果、液体の状態で。オリーブの実の抽出物の10倍の濃度となり、その最終濃度は、37.2%でHPLC純度は、93.3%であった。
【0073】
実験例12
噴霧乾燥によるオリーブの実の抽出粉末の準備
260mlの純化した液状のオリーブ抽出物(実験例11により得られた19.5gのヒドロキティロソルを含む)のサンプルを、58gのマルトデキストリンとゆっくりと撹拌した。このマルトデキストリンは、260mlの脱塩水で予め溶解しておいた、ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。76gの白色の粉末が得られた。その湿度は5.4%(karl Fischer)であり、ヒドロキティロソルのリッチネスは、21.9%であった。
【0074】
実験例13
3相のポマス(脱脂搾りかす)からヒドロキティロソルの抽出、水相の純化
湿度が60.55%の475.5gの3相ポマスのサンプルを、800mlの脱塩(脱イオン)水と、26.36gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物から、600ミクロンのポリプロピレン製フィルタで濾過することにより分離した。濾過された水相約795mlを、蒸発させることにより濃縮して、最終的に343.8mlの容量を得た。その後、この水相を遠心分離で精製して、フィルタを通過した固体粒子を除去した。この固体除去の後、275mlの原水性抽出物(crude aqueous extract)を得た。これは、0.97gのヒドロキティロソルを含む。そのHPLC純度は47.5%であった。
【0075】
実験例14
3相のポマス(脱脂搾りかす)から得られたヒドロキティロソルのイオン交換と吸着による純化
275mlの原水性抽出物(実験例13により得られた0.97gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むカラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で予め活性化しておいた。これには、Diaion WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を、脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。
【0076】
第1カラムから得られた溶離された相を、吸収性樹脂を含むカラムに入れた。これには、Diaion HP20を使用した。カラムの出口で回収された液相は、ヒドロキティロソルを含まない。その後、ヒドロキティロソルを樹脂から脱塩水で溶離した。これは、最初に入れたヒドロキティロソルの90%以上を回収するまで行った。
【0077】
この溶離された液相は、0.80gのヒドロキティロソルを含み、そのHPCL純度は、約95%以上であった。
【0078】
前述したように、本発明のプロセスは、オリーブ又はポマスから出発したヒドロキティロソルを含む抽出物を有機極性溶剤を使用せずに、準備する方法を提供する。本発明の方法により得られた生成物は、最終生成物とその中間生成物の両方とも、有機極性溶剤を含まない。この生成物は、砂糖も塩も含有しない。これは特許請求の範囲に記載した加水分解条件と純化手順の使用を組み合わせることにより達成できる。
【0079】
最終生成物は、その高純度と高いヒドロキティロソル含有特性により、特に担体溶剤(キャリア)と有機溶剤を含まない固体のヒドロキティロソル抽出物は、食品産業、化粧品産業、医薬産業の使用に適したものである。上記の濃度%は、全て重量%である。
【0080】
以上の説明は、本発明の一実施例に関するもので、この技術分野の当業者であれば、本発明の種々の変形例を考え得るが、それらはいずれも本発明の技術的範囲に包含される。特許請求の範囲の構成要素の後に記載した括弧内の番号は、図面の部品番号に態様し、発明の容易なる理解の為に付したものであり、発明を限定的に解釈するために用いてはならない。又、同一番号でも明細書と特許請求の範囲の部品名は必ずしも同一ではない。これは上記した理由による。用語「又は」に関して、例えば「A又はB」は、「Aのみ」、「Bのみ」ならず、「AとBの両方」を選択することも含む。特に記載のない限り、装置又は手段の数は、単数か複数かを問わない。
【符号の説明】
【0081】
1 反応容器
2 供給手段
3 加熱手段
4 脱塩(脱イオン)水
5 酸タンク
6 攪拌手段
7 フィルタ
8 遠心分離機
9 貯蔵器
10 濃縮手段
11 カラム
12 陰イオン交換樹脂
13 水源
14 貯蔵器
15 カラム
16 非イオン性吸収樹脂
17 貯蔵器
18 酢酸ソース
19 NaOHソース
20 乾燥手段
21 硫酸ソース
図2−4の翻訳
横軸:ミニュート(分)、
縦軸:保持時間領域パーセント
【特許請求の範囲】
【請求項1】
ある出発材料から、ヒドロキティロソルを含有する抽出物を生成する方法において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記方法は、前記出発材料を酸性加水分解し、得られた溶液を純化し、
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psi(137.8KPa)以下あり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する方法。
【請求項2】
(e)前記ステップ(d)で得られた溶液(B)を、第2クロマトグラフィ・カラムに入れるステップと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
(f)前記第2クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップと
をさらに有する
ことを特徴とする請求項1記載の方法。
【請求項3】
前記加水分解は、連続する殺菌システムで行われ、
前記加水分解の温度は、110℃−130℃であり、
前記加水分解のゲージ圧は、10−20psi(68.94−137.8KPa)である
ことを特徴とする請求項1又は2記載の方法。
【請求項4】
前記ステップ(a)は、15分−45分間行われる
ことを特徴とする請求項3記載の方法。
【請求項5】
前記ステップ(a)の温度は、118℃−126℃の範囲である
ことを特徴とする請求項3又は4記載の方法。
【請求項6】
(g)前記ステップ(d)で得られた溶液(B)、又は前記ステップ(f)で得られた溶液(C)を、逆浸透圧で濃縮ステップ、
を更に有する
ことを特徴とする請求項1−5の何れかに記載の方法。
【請求項7】
(h)前記生成物が固体状態となるまで、脱水するステップ、
を更に有する
ことを特徴とする請求項1−6の何れかに記載の方法。
【請求項8】
オイルを、前記出発材料から除去する、あるいは前記ステップ(b)で得られた溶液から除去する
ことを特徴とする請求項1−7の何れかに記載の方法。
【請求項9】
請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、80%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。
【請求項10】
請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを30%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。
【請求項11】
請求項1−7の何れかに記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、40%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。
【請求項12】
請求項10に記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを40%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。
【請求項13】
ある出発材料からヒドロキティロソルを含有する抽出物を生成する装置において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記装置は、前記出発材料を加水分解する手段を有し、
(a)水中で前記出発材料を酸性加水分解する手段と、
前記加水分解は、温度は70℃−140℃で、pHは1.0−6.0で、実行され、
(b)分離した水溶液を得る為、前記加水分解による生成物を処理する手段と、
(c)第1のクロマトグラフィ・カラムと、
前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する装置。
【請求項14】
(d)第2クロマトグラフィ・カラムと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
を有する
ことを特徴とする請求項13記載の装置。
【請求項15】
前記加水分解する手段は、連続する殺菌システムを有し、
前記連続する殺菌システムは、前記出発材料の連続する流れを圧力をかけながら処理する
ことを特徴とする請求項13又は14記載の装置。
【請求項16】
前記の酸で活性化した陰イオン交換樹脂は、弱塩基性の水溶離陰イオン交換樹脂であり、
前記吸収性の非イオン性樹脂は、マクロ網状の架橋された芳香族ポリマである
ことを特徴とする請求項14又は15記載の装置。
【請求項1】
ある出発材料から、ヒドロキティロソルを含有する抽出物を生成する方法において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記方法は、前記出発材料を酸性加水分解し、得られた溶液を純化し、
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psi(137.8KPa)以下あり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する方法。
【請求項2】
(e)前記ステップ(d)で得られた溶液(B)を、第2クロマトグラフィ・カラムに入れるステップと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
(f)前記第2クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップと
をさらに有する
ことを特徴とする請求項1記載の方法。
【請求項3】
前記加水分解は、連続する殺菌システムで行われ、
前記加水分解の温度は、110℃−130℃であり、
前記加水分解のゲージ圧は、10−20psi(68.94−137.8KPa)である
ことを特徴とする請求項1又は2記載の方法。
【請求項4】
前記ステップ(a)は、15分−45分間行われる
ことを特徴とする請求項3記載の方法。
【請求項5】
前記ステップ(a)の温度は、118℃−126℃の範囲である
ことを特徴とする請求項3又は4記載の方法。
【請求項6】
(g)前記ステップ(d)で得られた溶液(B)、又は前記ステップ(f)で得られた溶液(C)を、逆浸透圧で濃縮ステップ、
を更に有する
ことを特徴とする請求項1−5の何れかに記載の方法。
【請求項7】
(h)前記生成物が固体状態となるまで、脱水するステップ、
を更に有する
ことを特徴とする請求項1−6の何れかに記載の方法。
【請求項8】
オイルを、前記出発材料から除去する、あるいは前記ステップ(b)で得られた溶液から除去する
ことを特徴とする請求項1−7の何れかに記載の方法。
【請求項9】
請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、80%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。
【請求項10】
請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを30%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。
【請求項11】
請求項1−7の何れかに記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、40%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。
【請求項12】
請求項10に記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを40%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。
【請求項13】
ある出発材料からヒドロキティロソルを含有する抽出物を生成する装置において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記装置は、前記出発材料を加水分解する手段を有し、
(a)水中で前記出発材料を酸性加水分解する手段と、
前記加水分解は、温度は70℃−140℃で、pHは1.0−6.0で、実行され、
(b)分離した水溶液を得る為、前記加水分解による生成物を処理する手段と、
(c)第1のクロマトグラフィ・カラムと、
前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する装置。
【請求項14】
(d)第2クロマトグラフィ・カラムと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
を有する
ことを特徴とする請求項13記載の装置。
【請求項15】
前記加水分解する手段は、連続する殺菌システムを有し、
前記連続する殺菌システムは、前記出発材料の連続する流れを圧力をかけながら処理する
ことを特徴とする請求項13又は14記載の装置。
【請求項16】
前記の酸で活性化した陰イオン交換樹脂は、弱塩基性の水溶離陰イオン交換樹脂であり、
前記吸収性の非イオン性樹脂は、マクロ網状の架橋された芳香族ポリマである
ことを特徴とする請求項14又は15記載の装置。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2010−516750(P2010−516750A)
【公表日】平成22年5月20日(2010.5.20)
【国際特許分類】
【出願番号】特願2009−546832(P2009−546832)
【出願日】平成20年1月25日(2008.1.25)
【国際出願番号】PCT/IB2008/000173
【国際公開番号】WO2008/090460
【国際公開日】平成20年7月31日(2008.7.31)
【出願人】(509211022)プロベルテ ファーマ,エス.エー. (5)
【Fターム(参考)】
【公表日】平成22年5月20日(2010.5.20)
【国際特許分類】
【出願日】平成20年1月25日(2008.1.25)
【国際出願番号】PCT/IB2008/000173
【国際公開番号】WO2008/090460
【国際公開日】平成20年7月31日(2008.7.31)
【出願人】(509211022)プロベルテ ファーマ,エス.エー. (5)
【Fターム(参考)】
[ Back to top ]