
オレフィンの四量体化
本発明は、オレフィンの四量体化方法について記載しており、前記方法の生成物流は、30%を超える四量体オレフィンを含有する。本方法は、オレフィンの供給流を遷移金属化合物およびへテロ原子配位子を含有する触媒系と接触させる工程を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、オレフィン四量体化方法、オレフィンの四量体化用触媒系ならびにオレフィンの四量体化用触媒系のための配位子の同定および使用に関する。
【背景技術】
【0002】
本発明は、1−オクテンの生成を、有意な量のブテン、他のオクテン異性体、特定の高分子オリゴマーおよびポリエチレンの副生成を避けながら高い選択性で促進する方法および触媒系を規定する。この触媒系は、また、他のオレフィン、特にα(アルファ)−オレフィンの四量体化に使用することができる。
【0003】
1−オクテンの有用性はよく知られているにもかかわらず、1−オクテンを選択的に生成するエチレンの四量体化のための工業的に成功する方法は教示されていない。従来のエチレンのオリゴマー化技術が生成するのは、シュルツ−フローリーまたはポアソンの生成物分布のいずれかに従う一連のα−オレフィンである。定義上、これらの数学的分布は、形成することができる四量体の重量%を限定して生成物を分布させる。これに関連して、キレート配位子、好ましくは2−ジフェニルホスフィノ安息香酸(DPPBA)、ニッケル前駆物質、好ましくはNiCl2・6H2O、および触媒活性化剤、好ましくはテトラフェニルホウ酸ナトリウムを含むニッケル触媒が、1−オクテンを含有する直鎖状オレフィンの混合物を生じるエチレンのオリゴマー化の触媒作用をすることが従来技術(特許文献1)により知られている。直鎖状C8α−オレフィンに対する選択性は、19%であると言われている。同様に、類似の触媒系を使用するシェル高分子オレフィン法(the Shell Higher Olefins Process)(SHOP法、特許文献2および特許文献3)は、生成物の混合物中に11重量%の1−オクテンを一般的に生じることが報告されている(非特許文献1、非特許文献2および非特許文献3)。
【0004】
Gulf Oil Chemical Company(Chevron、例えば、特許文献4)およびEthyl Corporation(BP/Amoco、例えば、特許文献5)により独自に開発されたトリアルキルアンモニウム触媒に基づくチーグラータイプの技術もまた、エチレンをオリゴマー化するために工業的に使用されており、伝えられるところによれば13〜25重量%の1−オクテンを含有するオレフィンの混合物にしている(非特許文献1〜3)。
【0005】
従来技術は、また、リンおよび窒素の両方のヘテロ原子によるへテロ原子配位子を含有するクロム系触媒が、エチレンを三量体化して1−ヘキセンにする選択的触媒作用を及ぼすことを教示している。エチレンの三量体化のための上記へテロ原子配位子としては、ビス(2−ジエチルホスフィノ−エチル)アミン(特許文献6、これにより参照によりそのすべてを本明細書に組み込む)ならびに(o−メトキシフェニル)2PN(メチル)P(o−メトキシフェニル)2(特許文献7、これにより参照によりそのすべてを本明細書に組み込む)が挙げられる。これら触媒系および方法は、両方とも、1−ヘキセンの生成に非常に特異的であって、1−オクテンは不純物として生ずるのみである(一般的には、特許文献7により開示されているように、生成物の混合物の3重量%未満)。(o−メトキシフェニル)2PN(メチル)P(o−メトキシフェニル)2(特許文献7)に配位結合しているリンのヘテロ原子は、1つの窒素原子によって互いに隔たっている。その窒素原子は、少なくとも活性化剤が存在しなければクロムには配位せず、配位子にさらなる電子供与原子がなければ二座系であるものと考えられる。さらに、フェニル基のオルト位の極性または電子供与性置換基は、三座系の形成を助けることが論議されており、それは、一般に1−ヘキセン形成の選択性を高めるものと信じられている(非特許文献4:「このことにより、オルトメトキシ基がペンダントドナーとして作用し、クロム中心の配位の飽和を増すポテンシャルが重要な要素であると仮定した」を参照されたい)。特許文献7(実施例16)は、オレフィンの三量体化を用いるオクテンの製造方法および触媒系を教示している。この例においては、1−ブテンを2個のエチレン分子と共に三量体化して30%のオクテンを生じている。しかしながら、これらオクテンの性質は開示されておらず、本出願人は、それらは直鎖状および分枝状オクテンの混合物からなるものと考えている。
【0006】
従来技術は、一般に認められているエチレン三量体化のための7員メタラサイクル反応中間体(非特許文献5)の9員メタラサイクルへの拡大は起こりそうにない(非特許文献6および非特許文献7)ために、高い1−オクテンの選択性は達成され得ないことを教示している。9員環は、最も好適とは云えない中間サイズの環であり、したがって7員環と比較して疎まれるはずであることが論じられている(非特許文献6)。その上、「9員環が形成される場合は、それより11員環または13員環まで成長しそうである。言い換えると、多くのオクテンが期待されるのではなくて、かなりの量の(直鎖状の)デセンまたはドデセンの方がより理にかなっている。」ということも同じ著者により述べられている。
【0007】
正反対のことのその教示にもかかわらず、本出願人は、四量体化オレフィンを選択的に生成させる方法を見出した。本出願人は、さらに、リン原子上のヒドロカルビルまたはヘテロヒドロカルビル基に極性置換基のない窒素およびリンの両方のへテロ原子を有する混合へテロ原子配位子を含有するクロム系触媒は、しばしば70重量%を超える選択性でエチレンを選択的に四量体化して1−オクテンにするために使用することができることを見出した。この高度の1−オクテン選択性は、最高でも25重量%の1−オクテンを生じるだけの従来の1段法のエチレンオリゴマー化または三量体化技術によっては達成することができない。
【0008】
【特許文献1】米国特許第6184428号
【特許文献2】米国特許第3676523号
【特許文献3】米国特許第3635937号
【非特許文献1】Chem Systems PERP報告90−1
【非特許文献2】Chem Systems PERP報告93−6
【非特許文献3】Chem Systems PERP報告94/95S12
【特許文献4】ドイツ国特許第1443927号
【特許文献5】米国特許第3906053号
【特許文献6】国際公開03/053891
【特許文献7】国際公開02/04119
【非特許文献4】Chem.Commun.、2002、858〜859
【非特許文献5】Chem.Commun.、1989、674
【非特許文献6】Organometallics、2003、22、2564
【非特許文献7】Angew.Chem.Int.Ed.、2003、42(7)、808
【非特許文献8】D.E.Bergbreiter等、J.Am.Chem.Soc.、1987年、109、177〜179
【非特許文献9】C.Yuanyin等、Chinese J.React.Pol.、1992年、1(2)、152〜159
【非特許文献10】T.MonoiおよびY.Sasaki、J.Mol.Cat.A:Chem.、1987年、109、177〜179
【非特許文献11】Ewart等、J.Chem.Soc.1964、1543
【非特許文献12】Dossett、S.J.等、Chem.Commun.、2001、8、699
【非特許文献13】Balakrishna,M.S.等、J.Organomet.Chem.1990、390、2、203
【非特許文献14】Casalnuovo,A.L.等、J.Am.Chem.Soc.1994、116、22、9869
【非特許文献15】Rajanbabu,T.V.等、J.Org.Chem.1997、62、17、6012
【非特許文献16】Slawin,A.M.Z等、J.Chem.Soc.、Dalton Trans.2002、513
【非特許文献17】Schmidbaur,H.等、J.Organomet.Chem.1984、271、173
【非特許文献18】Balakrishna,M.S.等、lnorg.Chem.1993、32、5676
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明は、四量体生成物を選択的に生成する方法に関する。
本発明は、特に、エチレン等のオレフィンから1−オクテン等の四量体生成物を選択的に生成する方法に関する。
【0010】
本発明は、へテロ原子配位子を含有する遷移金属触媒系を使用して四量体生成物を選択的に生成する方法に関する。
【課題を解決するための手段】
【0011】
本発明の第1の態様によれば、オレフィンの四量体化方法であって、この四量体化方法の生成物はオレフィンであり、本方法の生成物流の30%を超えて生成する。
【0012】
本発明の第2の態様によれば、この四量体化方法は、オレフィンの供給流を遷移金属およびへテロ原子配位子を含む触媒系と接触させる工程を含み、この四量体化方法の生成物はオレフィンであり、本方法の生成物流の30%を超えて生成する。
本明細書において、%は重量%を表す。
【0013】
「四量体化」という用語は、一般に、4つの、好ましくは4つの同一のオレフィンモノマー単位が直鎖状および/または分枝状オレフィンを生ずる反応を指す。
【0014】
へテロ原子状のとは、同一であるかまたは異なっていてもよい、少なくとも2つのへテロ原子を含有する配位子を意味し、そのへテロ原子は、リン、ヒ素、アンチモン、硫黄、酸素、ビスマス、セレンまたは窒素から選択することができる。
【0015】
供給流は、四量体化するオレフィンを含んでおり、本発明による方法の中に連続方式またはバッチ方式で導入することができる。
生成物流は、四量体を含んでおり、その四量体は、本発明により連続方式またはバッチ方式で生成される。
【0016】
供給流は、α−オレフィンを含むことができ、生成物流は、少なくとも30%、好ましくは少なくとも35%の四量体化α−オレフィンモノマーを含むことができる。
【0017】
本方法は、α−オレフィンを四量体化する方法を含む。α−オレフィンという用語は、末端二重結合を有するすべての炭化水素化合物を意味する。この定義は、エチレン、プロピレン、1−ブテン、イソブチレン、1−ペンテン、1−ヘキセン、1−オクテン等を含む。
【0018】
本方法は、四量体α−オレフィン生成物を選択的に生じるα−オレフィンの四量体化の方法を含むことができる。
【0019】
オレフィンの供給流は、エチレンを含むことができ、生成物流は、少なくとも30%の1−オクテンを含むことができる。本方法は、エチレンの四量体化方法であり得る。
【0020】
本発明は、40%、50%、60%または70%のα−オレフィンを超える生成物流を生じさせるように、配位子、触媒系および/または処理条件を選択することを可能にする。生成物流の一層進んだ用途によっては、α−オレフィンのそのような高い選択性を有することが望ましい。
【0021】
オレフィン供給流は、エチレンを含むことができ、生成物流中の(C6+C8):(C4+C10)の比は、2.5:1を超えることができる。
オレフィン供給流は、エチレンを含むことができ、生成物流中のC8:C6の比は、1を超える。
【0022】
エチレンは、好ましくは10バールgを超え、より好ましくは30バールgを超える圧力で触媒系と接触させることができる。
【0023】
ヘテロ原子配位子は、次の一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、Bは、AとCの間の結合基であり、Rは、独立してホモまたはヘテロヒドロカルビル基のいずれかから選択され、nおよびmは、それぞれの価数ならびにAおよび/またはCの酸化状態により決まる)によって表すことができる。
【0024】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体であり得る。
電子供与体は、供与性の共有結合を含む化学結合の形成で使用される電子を供与する実体として定義される。
【0025】
ヘテロ原子配位子は、次の一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立してヒドロカルビルもしくはヘテロヒドロカルビルまたは置換ヒドロカルビルもしくは置換ヘテロヒドロカルビル基から選択される)によって表すことができる。
【0026】
ヘテロ原子配位子は、次の一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表すことができる。
【0027】
基R1、R2、R3およびR4のいずれもが、独立して、お互い同士の1つ以上、あるいは結合基Bと結合して、AおよびC、AおよびBまたはBおよびCと共に環構造を形成することができる。
【0028】
R1、R2、R3およびR4の1つ以上の置換基は、いずれも電子供与性でなくてもよい。
R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、芳香族の場合は、AまたはCに結合している原子と隣接する原子に置換基を有することができる。
【0029】
R1、R2、R3およびR4の1つ以上の各非電子供与性置換基は、非極性であり得る。IUPACは、非極性を不変の電気双極子モーメントを有さない実体として定義している。
【0030】
適当な非極性置換基は、メチル、エチル、プロピル、ブチル、イソプロピル、イソブチル、t−ブチル、ペンチル、ヘキシル、シクロペンチル、2−メチルシクロヘキシル、シクロヘキシル、シクロペンタジエニル、フェニル、ビフェニル、ナフチル、トリル、キシリル、メシチル、エテニル、プロペニルおよびベンジル基等であり得る。
【0031】
R1、R2、R3およびR4は、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択することができる。好ましくは、R1、R2、R3およびR4は、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択することができる。
【0032】
Bは、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合ならびに、メチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子またはハロゲンである)を含む基のいずれか1つから選択される。好ましくは、Bは、−N(R5)−であり、R5は、ヒドロカルビルまたは置換ヒドロカルビル基である。R5は、水素であるかまたは、アルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基またはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる群から選択することができる。好ましくは、R5は、イソプロピル、1−シクロヘキシルエチル、2−メチルシクロヘキシルまたは2−オクチル基であることができる。
【0033】
Bは、単一原子スペーサーであるように選択することができる。単一原子の結合スペーサーは、AおよびCに直接結合している置換または非置換原子として定義される。
【0034】
Aおよび/またはCは、独立して、S、Se、N、またはOで酸化され得る。
AおよびCは、独立して、リンまたはSもしくはSeもしくはNもしくはOで酸化されたリンであり得る。
【0035】
配位子は、また、複数の(R)nA−B−C(R)m単位を含有することができる。上記配位子の非限定的な例としては、デンドリマー配位子ならびに個々の単位が1つ以上のR基によるかまたは結合基Bのいずれかによって連結されている配位子が挙げられる。上記配位子のより具体的な非限定的な例としては、1,2−ジ−(N(P(フェニル)2)2)−ベンゼン、1,4−ジ−(N(P(フェニル)2)2)−ベンゼン、N(CH2CH2N(P(フェニル)2)2)3および1,4−ジ−(P(フェニル)N(メチル)P(フェニル)2)−ベンゼンが挙げられる。
【0036】
その配位子は、当業者に知られている手順および既刊文献に開示されている手順により調製することができる。配位子の例としては、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2がある。
【0037】
この触媒系は、活性化剤を含むことができ、本方法はへテロ原子配位子を遷移金属前駆物質および活性化剤と任意の順序で組合せる工程を含み得る。
【0038】
本方法は、遷移金属前駆物質およびヘテロ原子配位子からin situでヘテロ原子配位錯体を発生させる工程を含み得る。その方法は、ヘテロ原子配位子および遷移金属前駆物質を使用して調製し、予め形成しておいた配位錯体を反応混合物に加える工程、または遷移金属のヘテロ原子配位錯体がin situで発生するように、ヘテロ原子配位子および遷移金属前駆物質を反応器に別々に加える工程を含むことができる。ヘテロ原子配位錯体がin situで発生するとは、その錯体が、触媒反応が起こる媒体中で発生することを意味する。一般的には、ヘテロ原子配位錯体は、in situで発生する。一般的には、遷移金属前駆物質、およびヘテロ原子配位子は、約0.01:100〜10000:1、好ましくは約0.1:1〜10:1の金属/配位子の比を提供するように(in situおよびex situの両方で)合わせる。
【0039】
その遷移金属は、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択することができ、好ましくはクロムを選択する。
【0040】
ヘテロ原子配位子および活性化剤と混合することにより、本発明によりエチレンの四量体化の触媒作用を及ぼす遷移金属前駆物質は、単一の無機塩、もしくは有機塩、配位錯体もしくは有機金属錯体であって、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)−トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択することができる。好ましい遷移金属前駆物質としては、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)が挙げられる。
【0041】
ヘテロ原子配位子は、ポリマー鎖に付着させて、その結果、得られた遷移金属のヘテロ原子配位錯体を、高温においては可溶性であるが25℃では不溶性であるように変性することができる。このアプローチにより、その錯体を再使用のために反応混合物から回収することが可能であり、非特許文献8に記載されているように他の触媒に使用されてきた。同様に、これらの遷移金属錯体は、また、例えば、非特許文献9に、白金錯体の固定について示されているように、ヘテロ原子配位子を、シリカ、シリカゲル、ポリシロキサンまたはアルミナ等の骨格に結合することによって固定することもできる。
【0042】
本方法で使用するための活性化剤は、原則として、ヘテロ原子配位子および遷移金属前駆物質と組み合わせたときに活性触媒を生成する任意の化合物であり得る。活性化剤の混合物もまた使用することができる。適当な化合物としては、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)が挙げられる。
【0043】
適当な有機アルミニウム化合物としては、式AlR3で表され、Rが、独立して、C1〜C12のアルキル、酸素を含有する部分またはハロゲン化物である化合物、およびLiAlH4等の化合物等が挙げられる。例としては、トリメチルアルミニウム(TMA)、トリエチルアルミニウム(TEA)、トリイソブチルアルミニウム(TIBA)、トリ−n−オクチルアルミニウム、二塩化メチルアルミニウム、二塩化エチルアルミニウム、塩化ジメチルアルミニウム、塩化ジエチルアルミニウム、アルミニウムイソプロポキシド、エチルアルミニウムセスキクロリド、メチルアルミニウムセスキクロリド、およびアルミノキサンが挙げられる。アルミノキサンは、一般的には、アルキルアルミニウム化合物、例えばトリメチルアルミニウムに水を制御しながら添加することにより調製することができるオリゴマー化合物として当技術分野でよく知られている。この化合物は、直鎖状、環状、かご型またはそれらの混合物であり得る。異なるアルミノキサンの混合物もまた本方法で使用することができる。
【0044】
適当な有機ホウ素化合物の例としては、ボロキシン、NaBH4、トリエチルホウ素、トリス(ペンタフルオロフェニル)ホウ素、ホウ酸トリブチル等がある。
【0045】
活性化剤は、また、例えばナトリウムまたは亜鉛金属等、あるいは酸素等の還元剤または酸化剤として作用する化合物であるか、またはそれらを含有することもできる。
【0046】
活性化剤は、メチルアルミノキサン(MAO)およびエチルアルミノキサン(EAO)等のアルキルアルミノキサン、ならびに変性メチルアルミノキサン(MMAO)等の変性アルキルアルミノキサンから選択することができる。変性メチルアルミノキサン(Akzo Nobel社からの市販製品)は、メチル基に加えてイソブチル基またはn−オクチル基等の変性基を含有している。
【0047】
遷移金属およびアルミノキサンは、約1:1〜10000:1、好ましくは、約1:1〜1000:1、より好ましくは1:1〜300:1のAl/金属の比の割合で合わせることができる。
【0048】
本方法は、触媒系に、アルキルアルミノキサン1モル当たり0.01〜1000モルの間の量のトリアルキルアルミニウム化合物を添加する工程を含むことができる。
【0049】
アルミノキサンが、一般に、それらの調製に使用される対応するトリアルキルアルミニウム化合物の相当量を含有することは注目すべきことである。アルミノキサン中のこれらのトリアルキルアルミニウム化合物の存在は、それらの水との不完全な加水分解に起因するものと考えられる。この開示の中で示されるどのような量のトリアルキルアルミニウム化合物も、アルミノキサンに包含されるアルキルアルミニウム化合物に加えられるものである。
【0050】
本方法は、触媒系の成分を、オレフィンの存在下、−20℃〜250℃の間の任意の温度で混合する工程を含んでもよい。本出願人は、オレフィンの存在により触媒系が安定し得ることを見出した。
【0051】
本明細書に記載した触媒系の個々の成分は、同時にまたは任意の順序で連続的に、溶媒の存在下または不在下で、活性な触媒を生じさせるために合わせることができる。その触媒成分の混合は、−100℃〜250℃の間の任意の温度で行うことができる。触媒成分の混合中のオレフィンの存在は、改良された触媒性能をもたらす保護効果を一般に提供する。好ましい温度範囲は、20℃〜100℃の間である。
【0052】
本発明による触媒系またはその個々の成分を、担体材料、例えば、シリカ、アルミナ、MgCl2、ジルコニアまたはそれらの混合物に、あるいはポリマー、例えば、ポリエチレン、ポリプロピレン、ポリスチレン、またはポリ(アミノスチレン)に担持させることによって固定することもできる。触媒は、担体材料の存在下in situで形成させることができ、またはその担体に、同時もしくは連続して、1つ以上の触媒成分を予め含侵させるか、または混合することができる。場合によっては、その担体材料は、活性化剤の成分として作用させることもできる。このアプローチは、また、反応混合物からの再使用のための触媒の回収を容易にする。その概念は、例えば、非特許文献10に、クロム系のエチレン三量体化触媒での成功例が示されている。場合によっては、その担体は、また、例えば、上記担体がアルミノキサン官能基を含有する場合、またはその担体がアルミノキサンと同様の化学的機能を果たすことが可能な場合は触媒成分として作用することができ、それは例えばIOLA(商標)(Grace Davison社製市販製品)の場合である。
【0053】
本明細書に記載されている反応生成物は、開示されている触媒系を使用し、不活性溶媒の存在下または不在下の均一液相反応により、および/または触媒系がほとんどないか、またはない溶解性を示す形であるスラリー反応、および/または2相液/液反応、および/または試薬そのものおよび/または生成物のオレフィンが主要な媒体としての役割を果たすバルク相反応、および/または気相反応により、従来の装置および接触技術を使用して調製することができる。
【0054】
本方法は、また、不活性溶媒中で行うことができる。活性化剤と反応しない任意の不活性溶媒を使用することができる。これらの不活性溶媒としては、任意の飽和脂肪族炭化水素、不飽和脂肪族炭化水素および芳香族炭化水素ならびにハロゲン化炭化水素を挙げることができる。代表的な溶媒としては、非限定で、ベンゼン、トルエン、キシレン、クメン、ヘプタン、メチルシクロヘキサン、メチルシクロペンタン、シクロヘキサン、1−ヘキセン、1−オクテン、イオン性液体等を挙げることができる。
【0055】
本方法は、大気圧から500バールgの圧力で行うことができる。10〜70バールgの範囲のエチレン圧が好ましい。特に好ましい圧力は、30〜50バールgの範囲である。
【0056】
本方法は、−100℃〜250℃の範囲の温度で行うことができる。15℃〜130℃の範囲の温度が好ましい。特に好ましい温度は、35℃〜100℃の範囲である。
【0057】
本発明の好ましい実施形態においては、へテロ原子配位錯体および反応条件は、エチレンからの1−オクテンの収率が30重量%を超え、好ましくは35重量%を超えるように選択する。これに関して、収率とは、形成された全体の反応生成物100g当たりの形成された1−オクテンのグラム数を指す。
【0058】
1−オクテンに加えて、本方法は、また、へテロ原子配位子の性質および反応条件によって、様々な量の1−ブテン、1−ヘキセン、メチルシクロペンタン、メチレンシクロペンタン、プロピルシクロペンタン、プロピレンシクロペンタン、特定の高分子オリゴマーおよびポリエチレンも生成することができる。これら生成物のいくつかは、従来のエチレンのオリゴマー化および三量体化技術によっては、本発明で見られる収率で形成させることはできない。
【0059】
触媒、その個々の成分、試薬、溶媒および反応生成物は、一般に1回限りの基礎原料として採用されているが、これらの材料はいずれも、生産コストを最低限にするために、ある程度リサイクルすることが可能であるし、実際に好ましい。
【0060】
本方法は、任意のタイプの反応器を含む工場設備で行うことができる。上記反応器の例としては、非限定的に、バッチ式反応器、半バッチ式反応器および連続式反応器が挙げられる。その工場設備は、a)反応器、b)オレフィン反応物および触媒系のためのこの反応器への少なくとも1つの流入路、c)オリゴマー化反応生成物のためのこの反応器からの放流管路、およびd)触媒系が、本明細書に記載した遷移金属前駆物質および活性化剤のヘテロ原子配位錯体を含んでいる所望のオリゴマー化反応生成物を分離するための少なくとも1つの分離器を組み合わせて含むことができる。
【0061】
本方法の他の実施形態においては、反応器と分離器とを組み合わせて、同時に起こる反応生成物の形成、およびこれら化合物の反応器からの分離を容易にすることができる。この工程原理は、一般に反応蒸留として知られている。触媒系が溶媒または反応生成物中で溶解性を示さず、反応器の生成物、溶媒および未反応オレフィンと共に反応器から出ないように反応器中に固定される場合、その工程原理は一般に触媒蒸留として知られている。
【0062】
本発明のさらなる態様によれば、上で記したようにオレフィンの四量体化のための触媒系が提供される。その触媒系は、上で記したヘテロ原子配位子および遷移金属を含む。その触媒系は、また、上で記した活性化剤も含むことができる。
【0063】
ヘテロ原子配位子は、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、Bは、AとCの間の結合基であり、Rは、独立してホモまたはヘテロヒドロカルビル基のいずれかから選択され、nおよびmは、それぞれの価数ならびにAおよび/またはCの酸化状態により決まる)によって表される。
【0064】
Aおよび/またはCは、遷移金属と配位するための潜在的電子供与体であり得る。
ヘテロ原子配位子は、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立してヒドロカルビルもしくはヘテロヒドロカルビルまたは置換ヒドロカルビルもしくは置換ヘテロヒドロカルビル基から選択される)によって表すことができる。
【0065】
ヘテロ原子配位子は、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表すことができる。
【0066】
基R1、R2、R3およびR4のいずれもが、独立して、お互い同士の1つ以上、あるいは結合基Bと結合して、AおよびC、AおよびBまたはBおよびCと共に環構造を形成することができる。
【0067】
R1、R2、R3およびR4の1つ以上の置換基は、いずれも電子供与性でなくてもよい。
R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、芳香族の場合は、AまたはCに結合している原子と隣接する原子に置換基を有することができる。エチレンが四量体化される場合、特にAまたはCに結合している原子と隣接している芳香族基の原子に置換基が存在しない場合は、立体的かさを有する単一原子のスペーサーは、1−オクテンに向けた選択性を助長するように本出願人には思われる。各非電子供与性置換基は、非極性であり得る。このこともまた1−オクテンに向けた選択性を助長するようである。
【0068】
適当な非極性置換基は、メチル、エチル、プロピル、ブチル、イソプロピル、イソブチル、t−ブチル、ペンチル、ヘキシル、シクロペンチル、2−メチルシクロヘキシル、シクロヘキシル、シクロペンタジエニル、フェニル、ビフェニル、ナフチル、トリル、キシリル、メシチル、エテニル、プロペニルおよびベンジル基等であり得る。
【0069】
R1、R2、R3およびR4は、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択することができる。好ましくは、R1、R2、R3およびR4は、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択することができる。
【0070】
Bは、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;ならびに、メチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(式中、R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子またはハロゲンである)を含む基のいずれか1つから選択される。好ましくは、Bは、−N(R5)−であり、R5は、ヒドロカルビルまたは置換ヒドロカルビル基である。R5は、水素であるか、または、アルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基またはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる群から選択することができる。好ましくは、R5は、イソプロピル、1−シクロヘキシルエチル、2−メチルシクロヘキシルまたは2−オクチル基であり得る。
【0071】
Bは、単一原子スペーサーであるように選択することができる。本出願人は、AとCの間の上記単一原子スペーサーは、一般に四量体化触媒の選択性を増すことを見出した。
【0072】
Aおよび/またはCは、独立して、S、Se、N、またはOで酸化され得る。AおよびCは、独立して、リンまたはS、Se、NもしくはOで酸化されたリンであり得る。
【0073】
配位子は、また、複数の(R)nA−B−C(R)m単位を含有することができる。上記配位子の非限定的な例としては、個々の単位が1つ以上のR基によるかまたは結合基Bのいずれかによって連結されている配位子が挙げられる。上記配位子のより具体的な非限定的な例としては、1,2−ジ−(N(P(フェニル)2)2)−ベンゼン、1,4−ジ−(N(P(フェニル)2)2)−ベンゼン、N(CH2CH2N(P(フェニル)2)2)3および1,4−ジ−(P(フェニル)N(メチル)P(フェニル)2)−ベンゼンが挙げられる。
【0074】
配位子は、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si[(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれかの1つ以上から選択することができる。
【0075】
遷移金属は、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択することができ、好ましくはクロムである。
【0076】
遷移金属は、単一の無機塩もしくは有機塩、配位錯体もしくは有機金属錯体から選択される遷移金属前駆物質から誘導することができ、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)−トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択することができる。好ましい遷移金属前駆物質としては、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)が挙げられる。
【0077】
遷移金属前駆物質およびヘテロ原子配位子は、約0.01:100〜10000:1、好ましくは約0.1:1〜10:1の金属/配位子の比を有することができる。
【0078】
活性化剤は、原則として、ヘテロ原子配位子および遷移金属前駆物質と組み合わせたときに活性触媒を生成する任意の化合物であり得る。活性化剤の混合物もまた使用することができる。適当な化合物としては、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)が挙げられる。
【0079】
活性化剤は、メチルアルミノキサン(MAO)およびエチルアルミノキサン(EAO)等のアルキルアルミノキサン、ならびに変性メチルアルミノキサン(MMAO)等の変性アルキルアルミノキサンから選択することができる。変性メチルアルミノキサン(Akzo Nobel社からの市販製品)は、メチル基に加えて、イソブチル基またはn−オクチル基等の変性基を含有している。遷移金属およびアルミノキサンは、お互いに対して、約1:1〜10000:1、好ましくは、約1:1〜1000:1、より好ましくは1:1〜300:1のAl/金属の比の割合とすることができる。
【0080】
触媒系は、また、アルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を含むことができる。
【0081】
本発明のさらなる態様によれば、オレフィンの四量体化のための上記の触媒系のための上記の配位子が提供される。
【0082】
本発明は、また、オレフィンの四量体化の方法および触媒系での使用に適する配位子の同定および使用にも及ぶ。
【発明を実施するための最良の形態】
【0083】
本発明を、ここで以下の非限定的な実施例に関して説明する。実施例の個々の成分は、省略または置き換えることができ、必ずしも理想的ではないけれども本発明をなおも実施することができ、よって、これらの成分は、本発明の作用に必須であるとみなすべきではない。
【0084】
以下の実施例において、すべての手順は、予め乾燥した試薬を使用して不活性条件下で行った。化学物質は、他に記述がない限り、Sigma−AldrichまたはStrem Chemicalsから入手した。トリアルキルアルミニウムおよびアルミノキサン化合物ならびにそれらの溶液は、すべてCrompton Gmbh、Akzo NobelおよびAlbemarle Corporationから入手した。すべての実施例において、下記実施例に記載した触媒の調製において使用したMAOのモル量を計算するために、メチルアルミノキサン(MAO)のモル質量は、(CH3−Al−O)単位に対応して、58.016g/モルであるとみなした。同様に、エチルアルミノキサン(EAO)のモル質量は、(CH3CH2−Al−O)成分に対応して、72.042g/モルであり、トリメチルアルミニウムとトリイソブチルアルミニウムの70:30の混合物から調製した変性メチルアルミノキサンのそれは、(Me0.70isoBu0.30−Al−O)単位に対応して70.7g/モルとみなした。エチレンオリゴマー化生成物は、GC−MSおよびGC−FIDにより分析した。
【0085】
混合へテロ原子PNP配位子は、(a)非特許文献11、(b)非特許文献12、(c)非特許文献13に記載されているようにして、アミンとホスフィンクロリドR2PClとを反応させることによって作製した。それぞれのホスフィンクロリドR2PClは、非文献14、非特許文献15に記載されているようにして調製した。(フェニル)2PN(メチル)N(メチル)P(フェニル)2配位子は、非特許文献16に従い調製した。(フェニル)2PN(SiMe3)P(フェニル)2配位子に対しては、非特許文献17を用いた。配位子(フェニル)2P(=E)N(イソプロピル)P(フェニル)2(E=S、Se)は、非特許文献18に記載されているようにして調製した。
【実施例1】
【0086】
(フェニル)2PN(イソプロピル)P(フェニル)2配位子の調製
実施例1a):N,N−ジイソプロピルホスホルアミドジクロリドの調製
トルエン(80ml)中のジイソプロピルアミン(70ml、0.50モル)をPCl3(21.87ml、0.25モル)のトルエン(80ml)中の溶液に−10℃で加えた。その混合物を2時間攪拌し、続いて室温まで温まるままにした。その溶液をさらに1時間攪拌し、その後、セライトのパッドを通して濾過した。溶媒を除去して、生成物(35g、0.17モル、68%)を得た。31P{H}NMR:170ppm
【0087】
実施例1b):臭化フェニルマグネシウムの調製
マグネシウムの削りくず(9.11g、0.375モル)を、THF(100ml)中の4−ブロモベンゼン(7.90ml、75mmol)で処理した。激しい反応が起こりそれを氷浴で冷却した。反応が終息したところでその反応混合物を還流下で2時間加熱しグリニャール試薬を生じさせた。
【0088】
実施例1c):ビス(フェニル)塩化リンの調製
前記グリニャール試薬を、THF(100ml)中のN,N−ジイソプロピルホスホルアミドジクロリド(6.64ml、36mmol)に0℃で加えた。室温で一晩攪拌した後、その混合物をシクロヘキサン(200ml)で希釈し、ドライのHClガスをその溶液中に0.5時間吹き込んだ。沈殿物を濾過した後溶媒を除去し、ホスフィンの塩化物と臭素化物の混合物を80%の収率で得た。この粗生成物を単離することなくすべてを次の工程で使用した。
【0089】
実施例1d):(フェニル)2PN(イソプロピル)P(フェニル)2配位子の調製
DCM(80ml)およびトリエチルアミン(15ml)中の粗製ビス(フェニル)塩化リン(粗製反応混合物からの計算で28.8mmol)の0℃の溶液に、イソプロピルアミン(1.11ml、13mmol)を加えた。その反応物を30分間攪拌し、その後氷浴を取り外した。全部で14時間攪拌した後、その溶液を濾過して形成されたトリエチルアンモニウム塩を取り出した。その生成物を結晶化した後90%の収率で単離した。31P{H}NMR:49.0ppm(幅広の一重線)。
【実施例2】
【0090】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、29.0mgの(フェニル)2PN(メチル)P(フェニル)2(0.073mmol)の5mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の15mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の80℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を85℃に制御し、一方エチレン圧を30バールgに維持した。ガス同伴攪拌機を使用し、1100RPMの混合速度により初めから終わりまで完全な混合を確保した。反応は、60分後にエチレンの反応器への供給を打ち切ることによって終了し、反応器を10℃より下まで冷却した。過剰のエチレンをオートクレーブから解放したのち、オートクレーブに含有されていた液体をエタノールで、続いて水中の10%の塩酸で急冷した。液相のGC−FIDによる分析のための内部標準としてノナンを加えた。有機層の少量の試料を無水硫酸ナトリウムにより乾燥し、続いてGC−FIDにより分析した。有機層の残りを濾過して固体の生成物を単離した。これらの固体生成物を一晩100℃のオーブンで乾燥し、続いて重量を測定した。全体の生成物の質量は、31.86gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例3】
【0091】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、22.4mgの(フェニル)2PN(メチル)P(フェニル)2(0.056mmol)の5mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の15mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の80℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を85℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、28.76gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例4】
【0092】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.3mgの(フェニル)2PN(メチル)P(フェニル)2(0.066mmol)の3mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の17mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、47.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例5】
【0093】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(ペンチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.0mgの(フェニル)2PN(ペンチル)P(フェニル)2(0.074mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、74.84gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例6】
【0094】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(ベンジル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.7mgの(フェニル)2PN(ベンジル)P(フェニル)2(0.065mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、22.08gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例7】
【0095】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、34.9mgの(フェニル)2PN(フェニル)P(フェニル)2(0.076mmol)の10mlのトルエン中の溶液を、13.5mgのCrCl3(テトラヒドロフラン)3(0.036mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、48.21gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例8】
【0096】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(p−メトキシ−フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.6mgの(フェニル)2PN(p−メトキシ−フェニル)P(フェニル)2(0.062mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、7.01gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例9】
【0097】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(p−t−ブチル−フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、29.3mgの(フェニル)2PN(p−t−ブチル−フェニル)P(フェニル)2(0.062mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、62.15gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例10】
【0098】
Cr(2−エトキシへキサン酸)3、(フェニル)2PN(アリル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、27.6mgの(フェニル)2PN(アリル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、22.8mgのCr(2−エトキシへキサン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、12.68gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例11】
【0099】
Cr(アセチルアセトン酸)3、(フェニル)2PN[(CH2)3Si(OMe)3]P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、36.1mgの(フェニル)2PN[(CH2)3Si(OMe)3]P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、72.96gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例12】
【0100】
Cr(アセチルアセトン酸)3、(フェニル)2PN[(CH2)3−N−モルホリン]P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、33.8mgの(フェニル)2PN[(CH2)3−N−モルホリン]P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、22.2gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例13】
【0101】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.061mmol)の10mlのトルエン中の溶液を、11.6mgのCrCl3(テトラヒドロフラン)3(0.031mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、56.44gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例14】
【0102】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、17.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.04mmol)の10mlのトルエン中の溶液を、7.5mgのCrCl3(テトラヒドロフラン)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、4.0mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を43℃に維持し、一方エチレン圧を30バールgに保った。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、39.98gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例15】
【0103】
Cr(2−エチルヘキサン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、18.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.022mmol)の10mlのトルエン中の溶液を、7.6mgのCr(2−エチルヘキサン酸)3(0.011mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、3.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を50分後に終了し、上の実施例2の手順を使用した。生成物の量は、64.71gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例16】
【0104】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を14分後に終了し、上の実施例2の手順を使用した。生成物の量は、75.80gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例17】
【0105】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびEAO/TMAを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)、EAO(エチルアルミノキサン、33mmol)およびTMA(トリメチルアルミニウム、8.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を37分後に終了し、上の実施例2の手順を使用した。生成物の量は、29.03gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例18】
【0106】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMMAOを使用するエチレンの四量体化反応
シュレンク容器内で、17.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.04mmol)の10mlのトルエン中の溶液を、7.0mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMMAO(変性メチルアルミノキサン、Akzo Nobel MMAO−3A、6.0mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、74.11gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例19】
【0107】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2および担持されたMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。3.9gの担持されたMAO(SiO2上のMAO、Crompton、11.3mmolのMAOを含有)を30mlのトルエンに懸濁させ、続いてトルエン(50ml)とTMA(トリメチルアルミニウム、3.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。触媒溶液を続いて圧力反応器に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、43.61gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例20】
【0108】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、18.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.044mmol)の6.4mlのクメン中の溶液を、7.7mgのCr(アセチルアセトン酸)3(0.022mmol)の8mlのクメン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてクメン(180ml)とMAO(メチルアルミノキサン、4.4mmol、10%トルエン溶液)の40℃の混合物を含有する1000mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を25分後に終了し、上の実施例2の手順を使用した。生成物の量は、118.78gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例21】
【0109】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのエチルベンゼン中の溶液を、7.0mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのエチルベンゼン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてエチルベンゼン(76ml)とMAO(メチルアルミノキサン、4.0mmol、7%トルエン溶液)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を10分後に終了し、上の実施例2の手順を使用した。生成物の量は、70.6gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例22】
【0110】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、5.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.014mmol)の10mlのシクロヘキサン中の溶液を、3.5mgのCr(アセチルアセトン酸)3(0.01mmol)の10mlのシクロヘキサン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、2.0mmol、7%トルエン溶液)の溶液を、45℃のシクロヘキサン(170ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を39分後に終了し、上の実施例2の手順を使用した。生成物の量は、307.30gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例23】
【0111】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.6mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのクメン中の溶液を、7.4mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのクメン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、2.8mmol、7%トルエン溶液)の溶液を、45℃のクメン(180ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を75分後に終了し、上の実施例2の手順を使用した。生成物の量は、308.83gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例24】
【0112】
Cr(アセチルアセトン酸)3、(2−ナフチル)2PN(メチル)P(2−ナフチル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、39.6mgの(2−ナフチル)2PN(メチル)P(2−ナフチル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に維持し、一方エチレン圧を30バールgに保った。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、45.18gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例25】
【0113】
Cr(アセチルアセトン酸)3、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、47.0mgの(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、26.41gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例26】
【0114】
Cr(アセチルアセトン酸)3、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を65バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、52.34gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例27】
【0115】
Cr(アセチルアセトン酸)3、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、80.59gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例28】
【0116】
Cr(アセチルアセトン酸)3、(o−エチルフェニル)(Ph)PN(イソプロピル)PPh2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(o−エチルフェニル)(Ph)PN(イソプロピル)PPh2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を14分後に終了し、上の実施例2の手順を使用した。生成物の量は、63.78gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例29】
【0117】
Cr(アセチルアセトン酸)3、(フェニル)2P(=S)N(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.3mgの(フェニル)2P(=S)N(イソプロピル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、33.06gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例30】
【0118】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.6mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのクメン中の溶液を、7.4mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、4.0mmol、7%トルエン溶液)の溶液を、45℃のクメン(80ml)と1−オクテン(80ml)の混合物を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を45分後に終了し、上の実施例2の手順を使用した。生成物の量は、405.87gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例31】
【0119】
Cr(アセチルアセトン酸)3、(フェニル)2PN(メチル)N(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.3mgの(フェニル)2PN(メチル)N(メチル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その液体生成物の量は、22.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例32】
【0120】
Cr(アセチルアセトン酸)3、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、37.2mgの(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、14.7gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例33】
【0121】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、5.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.015mmol)の10mlのシクロヘキサン中の溶液を、3.8mgのCr(アセチルアセトン酸)3(0.011mmol)の10mlのシクロヘキサン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。1.8mmolのMAO(メチルアルミノキサン、7%トルエン溶液)を加え、その混合物を5分間攪拌した。この溶液を、45℃のシクロヘキサン(180ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、297.69gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例34】
【0122】
Cr(アセチルアセトン酸)3、(フェニル)2PN(SiMe3)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、39.8mgの(フェニル)2PN(SiMe3)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、26.9gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例35】
【0123】
Cr(アセチルアセトン酸)3、[(フェニル2P)2NCH2CH2]NおよびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、62.5mgの[(フェニル2P)2NCH2CH2]N(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、2.5gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例36】
【0124】
Cr(アセチルアセトン酸)3、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.7mgの(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)(0.026mmol)の10mlのトルエン中の溶液を、7.7mgのCr(アセチルアセトン酸)3(0.022mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、6.6mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、55.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例37】
【0125】
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2の調製
(フェニル)2PN(フェニル)P(フェニル)2(0.273g、0.591mmol)およびCrCl3(thf)3(0.206g、0.550mmol)を、トルエン(25ml)中にとり、80℃に一晩加熱して、青い粉末の沈殿を生じさせた。室温まで冷却した後、トルエンを沈殿から濾別し、生成物を石油エーテル(10ml)で2回洗浄した。真空下の乾燥により、0.303g(89%)の収量を得た。C60H50N2P4Cr2Cl6に対する計算値(測定値):C 58.13(57.98)%;H 4.07(3.97)%;N 2.26(2.12)%。磁気モーメント Cr1原子当たり4.06BM(二量体1個当たり5.74BM)。図1は、単結晶X線解析により得られた錯体の構造を示す。
【実施例38】
【0126】
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2およびMAOを使用するエチレンの四量体化反応
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2(0.0125g、Crが0.020mmol)のトルエン20ml中の懸濁液を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、4.61gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例39】
【0127】
(エチル)2PN(メチル)P(エチル)2配位子の調製
トルエン(25ml)中のメチルアミン(3.1mlの2M溶液、6.2mmol)を、トルエン(15ml)およびトリエチルアミン(5ml)中のクロロジエチルホスフィン(1.582g、12.7mmol)の溶液にゆっくりと加えた。その混合物を一晩攪拌した後、ガラス繊維のフィルターを通して濾過した。溶媒を真空下で除去し、10mlの水を加えた。その生成物を石油エーテルで抽出し(3×5ml)有機層を合わせた。溶媒を真空下で除去することにより、1.046g(81%)の生成物の収量を得た。31P{H}NMR:68ppm。
【実施例40】
【0128】
Cr(2−エチルヘキサン酸)3、(エチル)2PN(メチル)P(エチル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを10ml、0.020mmol)および(エチル)2PN(メチル)P(エチル)2の溶液(トルエン中0.005Mを4.1ml、0.0205mmol)を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、2.26gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例41】
【0129】
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2の調製
(エチル)2PN(メチル)P(エチル)2(0.362g、1.75mmol)およびCrCl3(thf)3(0.594g、1.58mmol)を使用して実施例38の手順に従った。0.520g(90%)の収量を得た。C18H46N2P4Cr2Cl6に対する計算値(測定値):C 29.57(29.62)%;H 6.34(6.45)%;N 3.83(3.87)%。磁気モーメント Cr1原子当たり3.86BM(二量体1個当たり5.46BM)。
【実施例42】
【0130】
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2およびMAOを使用するエチレンの四量体化反応
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2(0.0075g、Crが0.020mmol)のトルエン10ml中の懸濁液を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、3.06gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例43】
【0131】
(エチル)2PN(イソプロピル)P(フェニル)2配位子の調製
トルエン(15ml)中のN−(ジフェニルホスフィノ)メチルアミン(1.870g、7.69mmol)を、トルエン(20ml)およびトリエチルアミン(5ml)中のクロロジエチルホスフィン(0.986g、7.92mmol)の溶液にゆっくりと加えた。その混合物を一晩攪拌した後、ガラス繊維のフィルターを通して濾過した。溶媒を真空下で除去し、10mlの水を加えた。その生成物を石油エーテルで抽出し(3×5ml)有機層を合わせた。溶媒を真空下で除去することにより、2.200g(86%)の生成物の収量を得た。31P{H}NMR:49、43ppm。
【実施例44】
【0132】
Cr(2−エチルヘキサン酸)3、(フェニル)2PN(イソプロピル)P(エチル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを10ml、0.020mmol)および(フェニル)2PN(イソプロピル)P(エチル)2の溶液(トルエン中0.004Mを5ml、0.020mmol)を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、10.83gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例45】
【0133】
Cr(2−エチルヘキサン酸)3、(フェニル)(エチル)PN(メチル)P(エチル)(フェニル)およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを15ml、0.030mmol)および(フェニル)(エチル)PN(メチル)P(エチル)(フェニル)の溶液(トルエン中0.00365Mを9ml、0.033mmol)を、45℃でトルエン(100ml)およびMAO(9.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、0.897gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例46】
【0134】
Cr(2−エチルヘキサン酸)3、(フェニル)(エチル)PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを15ml、0.030mmol)および(フェニル)(エチル)PN(イソプロピル)P(フェニル)の溶液(9mlトルエン中0.034mmol)を、45℃でトルエン(100ml)およびMAO(9.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、13.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例47】
【0135】
Cr(アセチルアセトン酸)3、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、33.4mgの(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、8.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例48】
【0136】
Cr(アセチルアセトン酸)3、(フェニル)2PCH2CH2P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.3mgの(フェニル)2PCH2CH2P(フェニル)2(0.198mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、21.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【0137】
【表1】

【図面の簡単な説明】
【0138】
【図1】単結晶X線解析により得られた錯体の構造を表す模式図である。
【技術分野】
【0001】
本発明は、オレフィン四量体化方法、オレフィンの四量体化用触媒系ならびにオレフィンの四量体化用触媒系のための配位子の同定および使用に関する。
【背景技術】
【0002】
本発明は、1−オクテンの生成を、有意な量のブテン、他のオクテン異性体、特定の高分子オリゴマーおよびポリエチレンの副生成を避けながら高い選択性で促進する方法および触媒系を規定する。この触媒系は、また、他のオレフィン、特にα(アルファ)−オレフィンの四量体化に使用することができる。
【0003】
1−オクテンの有用性はよく知られているにもかかわらず、1−オクテンを選択的に生成するエチレンの四量体化のための工業的に成功する方法は教示されていない。従来のエチレンのオリゴマー化技術が生成するのは、シュルツ−フローリーまたはポアソンの生成物分布のいずれかに従う一連のα−オレフィンである。定義上、これらの数学的分布は、形成することができる四量体の重量%を限定して生成物を分布させる。これに関連して、キレート配位子、好ましくは2−ジフェニルホスフィノ安息香酸(DPPBA)、ニッケル前駆物質、好ましくはNiCl2・6H2O、および触媒活性化剤、好ましくはテトラフェニルホウ酸ナトリウムを含むニッケル触媒が、1−オクテンを含有する直鎖状オレフィンの混合物を生じるエチレンのオリゴマー化の触媒作用をすることが従来技術(特許文献1)により知られている。直鎖状C8α−オレフィンに対する選択性は、19%であると言われている。同様に、類似の触媒系を使用するシェル高分子オレフィン法(the Shell Higher Olefins Process)(SHOP法、特許文献2および特許文献3)は、生成物の混合物中に11重量%の1−オクテンを一般的に生じることが報告されている(非特許文献1、非特許文献2および非特許文献3)。
【0004】
Gulf Oil Chemical Company(Chevron、例えば、特許文献4)およびEthyl Corporation(BP/Amoco、例えば、特許文献5)により独自に開発されたトリアルキルアンモニウム触媒に基づくチーグラータイプの技術もまた、エチレンをオリゴマー化するために工業的に使用されており、伝えられるところによれば13〜25重量%の1−オクテンを含有するオレフィンの混合物にしている(非特許文献1〜3)。
【0005】
従来技術は、また、リンおよび窒素の両方のヘテロ原子によるへテロ原子配位子を含有するクロム系触媒が、エチレンを三量体化して1−ヘキセンにする選択的触媒作用を及ぼすことを教示している。エチレンの三量体化のための上記へテロ原子配位子としては、ビス(2−ジエチルホスフィノ−エチル)アミン(特許文献6、これにより参照によりそのすべてを本明細書に組み込む)ならびに(o−メトキシフェニル)2PN(メチル)P(o−メトキシフェニル)2(特許文献7、これにより参照によりそのすべてを本明細書に組み込む)が挙げられる。これら触媒系および方法は、両方とも、1−ヘキセンの生成に非常に特異的であって、1−オクテンは不純物として生ずるのみである(一般的には、特許文献7により開示されているように、生成物の混合物の3重量%未満)。(o−メトキシフェニル)2PN(メチル)P(o−メトキシフェニル)2(特許文献7)に配位結合しているリンのヘテロ原子は、1つの窒素原子によって互いに隔たっている。その窒素原子は、少なくとも活性化剤が存在しなければクロムには配位せず、配位子にさらなる電子供与原子がなければ二座系であるものと考えられる。さらに、フェニル基のオルト位の極性または電子供与性置換基は、三座系の形成を助けることが論議されており、それは、一般に1−ヘキセン形成の選択性を高めるものと信じられている(非特許文献4:「このことにより、オルトメトキシ基がペンダントドナーとして作用し、クロム中心の配位の飽和を増すポテンシャルが重要な要素であると仮定した」を参照されたい)。特許文献7(実施例16)は、オレフィンの三量体化を用いるオクテンの製造方法および触媒系を教示している。この例においては、1−ブテンを2個のエチレン分子と共に三量体化して30%のオクテンを生じている。しかしながら、これらオクテンの性質は開示されておらず、本出願人は、それらは直鎖状および分枝状オクテンの混合物からなるものと考えている。
【0006】
従来技術は、一般に認められているエチレン三量体化のための7員メタラサイクル反応中間体(非特許文献5)の9員メタラサイクルへの拡大は起こりそうにない(非特許文献6および非特許文献7)ために、高い1−オクテンの選択性は達成され得ないことを教示している。9員環は、最も好適とは云えない中間サイズの環であり、したがって7員環と比較して疎まれるはずであることが論じられている(非特許文献6)。その上、「9員環が形成される場合は、それより11員環または13員環まで成長しそうである。言い換えると、多くのオクテンが期待されるのではなくて、かなりの量の(直鎖状の)デセンまたはドデセンの方がより理にかなっている。」ということも同じ著者により述べられている。
【0007】
正反対のことのその教示にもかかわらず、本出願人は、四量体化オレフィンを選択的に生成させる方法を見出した。本出願人は、さらに、リン原子上のヒドロカルビルまたはヘテロヒドロカルビル基に極性置換基のない窒素およびリンの両方のへテロ原子を有する混合へテロ原子配位子を含有するクロム系触媒は、しばしば70重量%を超える選択性でエチレンを選択的に四量体化して1−オクテンにするために使用することができることを見出した。この高度の1−オクテン選択性は、最高でも25重量%の1−オクテンを生じるだけの従来の1段法のエチレンオリゴマー化または三量体化技術によっては達成することができない。
【0008】
【特許文献1】米国特許第6184428号
【特許文献2】米国特許第3676523号
【特許文献3】米国特許第3635937号
【非特許文献1】Chem Systems PERP報告90−1
【非特許文献2】Chem Systems PERP報告93−6
【非特許文献3】Chem Systems PERP報告94/95S12
【特許文献4】ドイツ国特許第1443927号
【特許文献5】米国特許第3906053号
【特許文献6】国際公開03/053891
【特許文献7】国際公開02/04119
【非特許文献4】Chem.Commun.、2002、858〜859
【非特許文献5】Chem.Commun.、1989、674
【非特許文献6】Organometallics、2003、22、2564
【非特許文献7】Angew.Chem.Int.Ed.、2003、42(7)、808
【非特許文献8】D.E.Bergbreiter等、J.Am.Chem.Soc.、1987年、109、177〜179
【非特許文献9】C.Yuanyin等、Chinese J.React.Pol.、1992年、1(2)、152〜159
【非特許文献10】T.MonoiおよびY.Sasaki、J.Mol.Cat.A:Chem.、1987年、109、177〜179
【非特許文献11】Ewart等、J.Chem.Soc.1964、1543
【非特許文献12】Dossett、S.J.等、Chem.Commun.、2001、8、699
【非特許文献13】Balakrishna,M.S.等、J.Organomet.Chem.1990、390、2、203
【非特許文献14】Casalnuovo,A.L.等、J.Am.Chem.Soc.1994、116、22、9869
【非特許文献15】Rajanbabu,T.V.等、J.Org.Chem.1997、62、17、6012
【非特許文献16】Slawin,A.M.Z等、J.Chem.Soc.、Dalton Trans.2002、513
【非特許文献17】Schmidbaur,H.等、J.Organomet.Chem.1984、271、173
【非特許文献18】Balakrishna,M.S.等、lnorg.Chem.1993、32、5676
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明は、四量体生成物を選択的に生成する方法に関する。
本発明は、特に、エチレン等のオレフィンから1−オクテン等の四量体生成物を選択的に生成する方法に関する。
【0010】
本発明は、へテロ原子配位子を含有する遷移金属触媒系を使用して四量体生成物を選択的に生成する方法に関する。
【課題を解決するための手段】
【0011】
本発明の第1の態様によれば、オレフィンの四量体化方法であって、この四量体化方法の生成物はオレフィンであり、本方法の生成物流の30%を超えて生成する。
【0012】
本発明の第2の態様によれば、この四量体化方法は、オレフィンの供給流を遷移金属およびへテロ原子配位子を含む触媒系と接触させる工程を含み、この四量体化方法の生成物はオレフィンであり、本方法の生成物流の30%を超えて生成する。
本明細書において、%は重量%を表す。
【0013】
「四量体化」という用語は、一般に、4つの、好ましくは4つの同一のオレフィンモノマー単位が直鎖状および/または分枝状オレフィンを生ずる反応を指す。
【0014】
へテロ原子状のとは、同一であるかまたは異なっていてもよい、少なくとも2つのへテロ原子を含有する配位子を意味し、そのへテロ原子は、リン、ヒ素、アンチモン、硫黄、酸素、ビスマス、セレンまたは窒素から選択することができる。
【0015】
供給流は、四量体化するオレフィンを含んでおり、本発明による方法の中に連続方式またはバッチ方式で導入することができる。
生成物流は、四量体を含んでおり、その四量体は、本発明により連続方式またはバッチ方式で生成される。
【0016】
供給流は、α−オレフィンを含むことができ、生成物流は、少なくとも30%、好ましくは少なくとも35%の四量体化α−オレフィンモノマーを含むことができる。
【0017】
本方法は、α−オレフィンを四量体化する方法を含む。α−オレフィンという用語は、末端二重結合を有するすべての炭化水素化合物を意味する。この定義は、エチレン、プロピレン、1−ブテン、イソブチレン、1−ペンテン、1−ヘキセン、1−オクテン等を含む。
【0018】
本方法は、四量体α−オレフィン生成物を選択的に生じるα−オレフィンの四量体化の方法を含むことができる。
【0019】
オレフィンの供給流は、エチレンを含むことができ、生成物流は、少なくとも30%の1−オクテンを含むことができる。本方法は、エチレンの四量体化方法であり得る。
【0020】
本発明は、40%、50%、60%または70%のα−オレフィンを超える生成物流を生じさせるように、配位子、触媒系および/または処理条件を選択することを可能にする。生成物流の一層進んだ用途によっては、α−オレフィンのそのような高い選択性を有することが望ましい。
【0021】
オレフィン供給流は、エチレンを含むことができ、生成物流中の(C6+C8):(C4+C10)の比は、2.5:1を超えることができる。
オレフィン供給流は、エチレンを含むことができ、生成物流中のC8:C6の比は、1を超える。
【0022】
エチレンは、好ましくは10バールgを超え、より好ましくは30バールgを超える圧力で触媒系と接触させることができる。
【0023】
ヘテロ原子配位子は、次の一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、Bは、AとCの間の結合基であり、Rは、独立してホモまたはヘテロヒドロカルビル基のいずれかから選択され、nおよびmは、それぞれの価数ならびにAおよび/またはCの酸化状態により決まる)によって表すことができる。
【0024】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体であり得る。
電子供与体は、供与性の共有結合を含む化学結合の形成で使用される電子を供与する実体として定義される。
【0025】
ヘテロ原子配位子は、次の一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立してヒドロカルビルもしくはヘテロヒドロカルビルまたは置換ヒドロカルビルもしくは置換ヘテロヒドロカルビル基から選択される)によって表すことができる。
【0026】
ヘテロ原子配位子は、次の一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表すことができる。
【0027】
基R1、R2、R3およびR4のいずれもが、独立して、お互い同士の1つ以上、あるいは結合基Bと結合して、AおよびC、AおよびBまたはBおよびCと共に環構造を形成することができる。
【0028】
R1、R2、R3およびR4の1つ以上の置換基は、いずれも電子供与性でなくてもよい。
R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、芳香族の場合は、AまたはCに結合している原子と隣接する原子に置換基を有することができる。
【0029】
R1、R2、R3およびR4の1つ以上の各非電子供与性置換基は、非極性であり得る。IUPACは、非極性を不変の電気双極子モーメントを有さない実体として定義している。
【0030】
適当な非極性置換基は、メチル、エチル、プロピル、ブチル、イソプロピル、イソブチル、t−ブチル、ペンチル、ヘキシル、シクロペンチル、2−メチルシクロヘキシル、シクロヘキシル、シクロペンタジエニル、フェニル、ビフェニル、ナフチル、トリル、キシリル、メシチル、エテニル、プロペニルおよびベンジル基等であり得る。
【0031】
R1、R2、R3およびR4は、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択することができる。好ましくは、R1、R2、R3およびR4は、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択することができる。
【0032】
Bは、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合ならびに、メチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子またはハロゲンである)を含む基のいずれか1つから選択される。好ましくは、Bは、−N(R5)−であり、R5は、ヒドロカルビルまたは置換ヒドロカルビル基である。R5は、水素であるかまたは、アルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基またはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる群から選択することができる。好ましくは、R5は、イソプロピル、1−シクロヘキシルエチル、2−メチルシクロヘキシルまたは2−オクチル基であることができる。
【0033】
Bは、単一原子スペーサーであるように選択することができる。単一原子の結合スペーサーは、AおよびCに直接結合している置換または非置換原子として定義される。
【0034】
Aおよび/またはCは、独立して、S、Se、N、またはOで酸化され得る。
AおよびCは、独立して、リンまたはSもしくはSeもしくはNもしくはOで酸化されたリンであり得る。
【0035】
配位子は、また、複数の(R)nA−B−C(R)m単位を含有することができる。上記配位子の非限定的な例としては、デンドリマー配位子ならびに個々の単位が1つ以上のR基によるかまたは結合基Bのいずれかによって連結されている配位子が挙げられる。上記配位子のより具体的な非限定的な例としては、1,2−ジ−(N(P(フェニル)2)2)−ベンゼン、1,4−ジ−(N(P(フェニル)2)2)−ベンゼン、N(CH2CH2N(P(フェニル)2)2)3および1,4−ジ−(P(フェニル)N(メチル)P(フェニル)2)−ベンゼンが挙げられる。
【0036】
その配位子は、当業者に知られている手順および既刊文献に開示されている手順により調製することができる。配位子の例としては、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2がある。
【0037】
この触媒系は、活性化剤を含むことができ、本方法はへテロ原子配位子を遷移金属前駆物質および活性化剤と任意の順序で組合せる工程を含み得る。
【0038】
本方法は、遷移金属前駆物質およびヘテロ原子配位子からin situでヘテロ原子配位錯体を発生させる工程を含み得る。その方法は、ヘテロ原子配位子および遷移金属前駆物質を使用して調製し、予め形成しておいた配位錯体を反応混合物に加える工程、または遷移金属のヘテロ原子配位錯体がin situで発生するように、ヘテロ原子配位子および遷移金属前駆物質を反応器に別々に加える工程を含むことができる。ヘテロ原子配位錯体がin situで発生するとは、その錯体が、触媒反応が起こる媒体中で発生することを意味する。一般的には、ヘテロ原子配位錯体は、in situで発生する。一般的には、遷移金属前駆物質、およびヘテロ原子配位子は、約0.01:100〜10000:1、好ましくは約0.1:1〜10:1の金属/配位子の比を提供するように(in situおよびex situの両方で)合わせる。
【0039】
その遷移金属は、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択することができ、好ましくはクロムを選択する。
【0040】
ヘテロ原子配位子および活性化剤と混合することにより、本発明によりエチレンの四量体化の触媒作用を及ぼす遷移金属前駆物質は、単一の無機塩、もしくは有機塩、配位錯体もしくは有機金属錯体であって、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)−トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択することができる。好ましい遷移金属前駆物質としては、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)が挙げられる。
【0041】
ヘテロ原子配位子は、ポリマー鎖に付着させて、その結果、得られた遷移金属のヘテロ原子配位錯体を、高温においては可溶性であるが25℃では不溶性であるように変性することができる。このアプローチにより、その錯体を再使用のために反応混合物から回収することが可能であり、非特許文献8に記載されているように他の触媒に使用されてきた。同様に、これらの遷移金属錯体は、また、例えば、非特許文献9に、白金錯体の固定について示されているように、ヘテロ原子配位子を、シリカ、シリカゲル、ポリシロキサンまたはアルミナ等の骨格に結合することによって固定することもできる。
【0042】
本方法で使用するための活性化剤は、原則として、ヘテロ原子配位子および遷移金属前駆物質と組み合わせたときに活性触媒を生成する任意の化合物であり得る。活性化剤の混合物もまた使用することができる。適当な化合物としては、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)が挙げられる。
【0043】
適当な有機アルミニウム化合物としては、式AlR3で表され、Rが、独立して、C1〜C12のアルキル、酸素を含有する部分またはハロゲン化物である化合物、およびLiAlH4等の化合物等が挙げられる。例としては、トリメチルアルミニウム(TMA)、トリエチルアルミニウム(TEA)、トリイソブチルアルミニウム(TIBA)、トリ−n−オクチルアルミニウム、二塩化メチルアルミニウム、二塩化エチルアルミニウム、塩化ジメチルアルミニウム、塩化ジエチルアルミニウム、アルミニウムイソプロポキシド、エチルアルミニウムセスキクロリド、メチルアルミニウムセスキクロリド、およびアルミノキサンが挙げられる。アルミノキサンは、一般的には、アルキルアルミニウム化合物、例えばトリメチルアルミニウムに水を制御しながら添加することにより調製することができるオリゴマー化合物として当技術分野でよく知られている。この化合物は、直鎖状、環状、かご型またはそれらの混合物であり得る。異なるアルミノキサンの混合物もまた本方法で使用することができる。
【0044】
適当な有機ホウ素化合物の例としては、ボロキシン、NaBH4、トリエチルホウ素、トリス(ペンタフルオロフェニル)ホウ素、ホウ酸トリブチル等がある。
【0045】
活性化剤は、また、例えばナトリウムまたは亜鉛金属等、あるいは酸素等の還元剤または酸化剤として作用する化合物であるか、またはそれらを含有することもできる。
【0046】
活性化剤は、メチルアルミノキサン(MAO)およびエチルアルミノキサン(EAO)等のアルキルアルミノキサン、ならびに変性メチルアルミノキサン(MMAO)等の変性アルキルアルミノキサンから選択することができる。変性メチルアルミノキサン(Akzo Nobel社からの市販製品)は、メチル基に加えてイソブチル基またはn−オクチル基等の変性基を含有している。
【0047】
遷移金属およびアルミノキサンは、約1:1〜10000:1、好ましくは、約1:1〜1000:1、より好ましくは1:1〜300:1のAl/金属の比の割合で合わせることができる。
【0048】
本方法は、触媒系に、アルキルアルミノキサン1モル当たり0.01〜1000モルの間の量のトリアルキルアルミニウム化合物を添加する工程を含むことができる。
【0049】
アルミノキサンが、一般に、それらの調製に使用される対応するトリアルキルアルミニウム化合物の相当量を含有することは注目すべきことである。アルミノキサン中のこれらのトリアルキルアルミニウム化合物の存在は、それらの水との不完全な加水分解に起因するものと考えられる。この開示の中で示されるどのような量のトリアルキルアルミニウム化合物も、アルミノキサンに包含されるアルキルアルミニウム化合物に加えられるものである。
【0050】
本方法は、触媒系の成分を、オレフィンの存在下、−20℃〜250℃の間の任意の温度で混合する工程を含んでもよい。本出願人は、オレフィンの存在により触媒系が安定し得ることを見出した。
【0051】
本明細書に記載した触媒系の個々の成分は、同時にまたは任意の順序で連続的に、溶媒の存在下または不在下で、活性な触媒を生じさせるために合わせることができる。その触媒成分の混合は、−100℃〜250℃の間の任意の温度で行うことができる。触媒成分の混合中のオレフィンの存在は、改良された触媒性能をもたらす保護効果を一般に提供する。好ましい温度範囲は、20℃〜100℃の間である。
【0052】
本発明による触媒系またはその個々の成分を、担体材料、例えば、シリカ、アルミナ、MgCl2、ジルコニアまたはそれらの混合物に、あるいはポリマー、例えば、ポリエチレン、ポリプロピレン、ポリスチレン、またはポリ(アミノスチレン)に担持させることによって固定することもできる。触媒は、担体材料の存在下in situで形成させることができ、またはその担体に、同時もしくは連続して、1つ以上の触媒成分を予め含侵させるか、または混合することができる。場合によっては、その担体材料は、活性化剤の成分として作用させることもできる。このアプローチは、また、反応混合物からの再使用のための触媒の回収を容易にする。その概念は、例えば、非特許文献10に、クロム系のエチレン三量体化触媒での成功例が示されている。場合によっては、その担体は、また、例えば、上記担体がアルミノキサン官能基を含有する場合、またはその担体がアルミノキサンと同様の化学的機能を果たすことが可能な場合は触媒成分として作用することができ、それは例えばIOLA(商標)(Grace Davison社製市販製品)の場合である。
【0053】
本明細書に記載されている反応生成物は、開示されている触媒系を使用し、不活性溶媒の存在下または不在下の均一液相反応により、および/または触媒系がほとんどないか、またはない溶解性を示す形であるスラリー反応、および/または2相液/液反応、および/または試薬そのものおよび/または生成物のオレフィンが主要な媒体としての役割を果たすバルク相反応、および/または気相反応により、従来の装置および接触技術を使用して調製することができる。
【0054】
本方法は、また、不活性溶媒中で行うことができる。活性化剤と反応しない任意の不活性溶媒を使用することができる。これらの不活性溶媒としては、任意の飽和脂肪族炭化水素、不飽和脂肪族炭化水素および芳香族炭化水素ならびにハロゲン化炭化水素を挙げることができる。代表的な溶媒としては、非限定で、ベンゼン、トルエン、キシレン、クメン、ヘプタン、メチルシクロヘキサン、メチルシクロペンタン、シクロヘキサン、1−ヘキセン、1−オクテン、イオン性液体等を挙げることができる。
【0055】
本方法は、大気圧から500バールgの圧力で行うことができる。10〜70バールgの範囲のエチレン圧が好ましい。特に好ましい圧力は、30〜50バールgの範囲である。
【0056】
本方法は、−100℃〜250℃の範囲の温度で行うことができる。15℃〜130℃の範囲の温度が好ましい。特に好ましい温度は、35℃〜100℃の範囲である。
【0057】
本発明の好ましい実施形態においては、へテロ原子配位錯体および反応条件は、エチレンからの1−オクテンの収率が30重量%を超え、好ましくは35重量%を超えるように選択する。これに関して、収率とは、形成された全体の反応生成物100g当たりの形成された1−オクテンのグラム数を指す。
【0058】
1−オクテンに加えて、本方法は、また、へテロ原子配位子の性質および反応条件によって、様々な量の1−ブテン、1−ヘキセン、メチルシクロペンタン、メチレンシクロペンタン、プロピルシクロペンタン、プロピレンシクロペンタン、特定の高分子オリゴマーおよびポリエチレンも生成することができる。これら生成物のいくつかは、従来のエチレンのオリゴマー化および三量体化技術によっては、本発明で見られる収率で形成させることはできない。
【0059】
触媒、その個々の成分、試薬、溶媒および反応生成物は、一般に1回限りの基礎原料として採用されているが、これらの材料はいずれも、生産コストを最低限にするために、ある程度リサイクルすることが可能であるし、実際に好ましい。
【0060】
本方法は、任意のタイプの反応器を含む工場設備で行うことができる。上記反応器の例としては、非限定的に、バッチ式反応器、半バッチ式反応器および連続式反応器が挙げられる。その工場設備は、a)反応器、b)オレフィン反応物および触媒系のためのこの反応器への少なくとも1つの流入路、c)オリゴマー化反応生成物のためのこの反応器からの放流管路、およびd)触媒系が、本明細書に記載した遷移金属前駆物質および活性化剤のヘテロ原子配位錯体を含んでいる所望のオリゴマー化反応生成物を分離するための少なくとも1つの分離器を組み合わせて含むことができる。
【0061】
本方法の他の実施形態においては、反応器と分離器とを組み合わせて、同時に起こる反応生成物の形成、およびこれら化合物の反応器からの分離を容易にすることができる。この工程原理は、一般に反応蒸留として知られている。触媒系が溶媒または反応生成物中で溶解性を示さず、反応器の生成物、溶媒および未反応オレフィンと共に反応器から出ないように反応器中に固定される場合、その工程原理は一般に触媒蒸留として知られている。
【0062】
本発明のさらなる態様によれば、上で記したようにオレフィンの四量体化のための触媒系が提供される。その触媒系は、上で記したヘテロ原子配位子および遷移金属を含む。その触媒系は、また、上で記した活性化剤も含むことができる。
【0063】
ヘテロ原子配位子は、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、Bは、AとCの間の結合基であり、Rは、独立してホモまたはヘテロヒドロカルビル基のいずれかから選択され、nおよびmは、それぞれの価数ならびにAおよび/またはCの酸化状態により決まる)によって表される。
【0064】
Aおよび/またはCは、遷移金属と配位するための潜在的電子供与体であり得る。
ヘテロ原子配位子は、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立してヒドロカルビルもしくはヘテロヒドロカルビルまたは置換ヒドロカルビルもしくは置換ヘテロヒドロカルビル基から選択される)によって表すことができる。
【0065】
ヘテロ原子配位子は、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、Bは、AとCの間の結合基であり、R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表すことができる。
【0066】
基R1、R2、R3およびR4のいずれもが、独立して、お互い同士の1つ以上、あるいは結合基Bと結合して、AおよびC、AおよびBまたはBおよびCと共に環構造を形成することができる。
【0067】
R1、R2、R3およびR4の1つ以上の置換基は、いずれも電子供与性でなくてもよい。
R1、R2、R3およびR4は、独立して、非芳香族基または複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、芳香族の場合は、AまたはCに結合している原子と隣接する原子に置換基を有することができる。エチレンが四量体化される場合、特にAまたはCに結合している原子と隣接している芳香族基の原子に置換基が存在しない場合は、立体的かさを有する単一原子のスペーサーは、1−オクテンに向けた選択性を助長するように本出願人には思われる。各非電子供与性置換基は、非極性であり得る。このこともまた1−オクテンに向けた選択性を助長するようである。
【0068】
適当な非極性置換基は、メチル、エチル、プロピル、ブチル、イソプロピル、イソブチル、t−ブチル、ペンチル、ヘキシル、シクロペンチル、2−メチルシクロヘキシル、シクロヘキシル、シクロペンタジエニル、フェニル、ビフェニル、ナフチル、トリル、キシリル、メシチル、エテニル、プロペニルおよびベンジル基等であり得る。
【0069】
R1、R2、R3およびR4は、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択することができる。好ましくは、R1、R2、R3およびR4は、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択することができる。
【0070】
Bは、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;ならびに、メチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(式中、R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子またはハロゲンである)を含む基のいずれか1つから選択される。好ましくは、Bは、−N(R5)−であり、R5は、ヒドロカルビルまたは置換ヒドロカルビル基である。R5は、水素であるか、または、アルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基またはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる群から選択することができる。好ましくは、R5は、イソプロピル、1−シクロヘキシルエチル、2−メチルシクロヘキシルまたは2−オクチル基であり得る。
【0071】
Bは、単一原子スペーサーであるように選択することができる。本出願人は、AとCの間の上記単一原子スペーサーは、一般に四量体化触媒の選択性を増すことを見出した。
【0072】
Aおよび/またはCは、独立して、S、Se、N、またはOで酸化され得る。AおよびCは、独立して、リンまたはS、Se、NもしくはOで酸化されたリンであり得る。
【0073】
配位子は、また、複数の(R)nA−B−C(R)m単位を含有することができる。上記配位子の非限定的な例としては、個々の単位が1つ以上のR基によるかまたは結合基Bのいずれかによって連結されている配位子が挙げられる。上記配位子のより具体的な非限定的な例としては、1,2−ジ−(N(P(フェニル)2)2)−ベンゼン、1,4−ジ−(N(P(フェニル)2)2)−ベンゼン、N(CH2CH2N(P(フェニル)2)2)3および1,4−ジ−(P(フェニル)N(メチル)P(フェニル)2)−ベンゼンが挙げられる。
【0074】
配位子は、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si[(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれかの1つ以上から選択することができる。
【0075】
遷移金属は、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択することができ、好ましくはクロムである。
【0076】
遷移金属は、単一の無機塩もしくは有機塩、配位錯体もしくは有機金属錯体から選択される遷移金属前駆物質から誘導することができ、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)−トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択することができる。好ましい遷移金属前駆物質としては、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)が挙げられる。
【0077】
遷移金属前駆物質およびヘテロ原子配位子は、約0.01:100〜10000:1、好ましくは約0.1:1〜10:1の金属/配位子の比を有することができる。
【0078】
活性化剤は、原則として、ヘテロ原子配位子および遷移金属前駆物質と組み合わせたときに活性触媒を生成する任意の化合物であり得る。活性化剤の混合物もまた使用することができる。適当な化合物としては、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)が挙げられる。
【0079】
活性化剤は、メチルアルミノキサン(MAO)およびエチルアルミノキサン(EAO)等のアルキルアルミノキサン、ならびに変性メチルアルミノキサン(MMAO)等の変性アルキルアルミノキサンから選択することができる。変性メチルアルミノキサン(Akzo Nobel社からの市販製品)は、メチル基に加えて、イソブチル基またはn−オクチル基等の変性基を含有している。遷移金属およびアルミノキサンは、お互いに対して、約1:1〜10000:1、好ましくは、約1:1〜1000:1、より好ましくは1:1〜300:1のAl/金属の比の割合とすることができる。
【0080】
触媒系は、また、アルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を含むことができる。
【0081】
本発明のさらなる態様によれば、オレフィンの四量体化のための上記の触媒系のための上記の配位子が提供される。
【0082】
本発明は、また、オレフィンの四量体化の方法および触媒系での使用に適する配位子の同定および使用にも及ぶ。
【発明を実施するための最良の形態】
【0083】
本発明を、ここで以下の非限定的な実施例に関して説明する。実施例の個々の成分は、省略または置き換えることができ、必ずしも理想的ではないけれども本発明をなおも実施することができ、よって、これらの成分は、本発明の作用に必須であるとみなすべきではない。
【0084】
以下の実施例において、すべての手順は、予め乾燥した試薬を使用して不活性条件下で行った。化学物質は、他に記述がない限り、Sigma−AldrichまたはStrem Chemicalsから入手した。トリアルキルアルミニウムおよびアルミノキサン化合物ならびにそれらの溶液は、すべてCrompton Gmbh、Akzo NobelおよびAlbemarle Corporationから入手した。すべての実施例において、下記実施例に記載した触媒の調製において使用したMAOのモル量を計算するために、メチルアルミノキサン(MAO)のモル質量は、(CH3−Al−O)単位に対応して、58.016g/モルであるとみなした。同様に、エチルアルミノキサン(EAO)のモル質量は、(CH3CH2−Al−O)成分に対応して、72.042g/モルであり、トリメチルアルミニウムとトリイソブチルアルミニウムの70:30の混合物から調製した変性メチルアルミノキサンのそれは、(Me0.70isoBu0.30−Al−O)単位に対応して70.7g/モルとみなした。エチレンオリゴマー化生成物は、GC−MSおよびGC−FIDにより分析した。
【0085】
混合へテロ原子PNP配位子は、(a)非特許文献11、(b)非特許文献12、(c)非特許文献13に記載されているようにして、アミンとホスフィンクロリドR2PClとを反応させることによって作製した。それぞれのホスフィンクロリドR2PClは、非文献14、非特許文献15に記載されているようにして調製した。(フェニル)2PN(メチル)N(メチル)P(フェニル)2配位子は、非特許文献16に従い調製した。(フェニル)2PN(SiMe3)P(フェニル)2配位子に対しては、非特許文献17を用いた。配位子(フェニル)2P(=E)N(イソプロピル)P(フェニル)2(E=S、Se)は、非特許文献18に記載されているようにして調製した。
【実施例1】
【0086】
(フェニル)2PN(イソプロピル)P(フェニル)2配位子の調製
実施例1a):N,N−ジイソプロピルホスホルアミドジクロリドの調製
トルエン(80ml)中のジイソプロピルアミン(70ml、0.50モル)をPCl3(21.87ml、0.25モル)のトルエン(80ml)中の溶液に−10℃で加えた。その混合物を2時間攪拌し、続いて室温まで温まるままにした。その溶液をさらに1時間攪拌し、その後、セライトのパッドを通して濾過した。溶媒を除去して、生成物(35g、0.17モル、68%)を得た。31P{H}NMR:170ppm
【0087】
実施例1b):臭化フェニルマグネシウムの調製
マグネシウムの削りくず(9.11g、0.375モル)を、THF(100ml)中の4−ブロモベンゼン(7.90ml、75mmol)で処理した。激しい反応が起こりそれを氷浴で冷却した。反応が終息したところでその反応混合物を還流下で2時間加熱しグリニャール試薬を生じさせた。
【0088】
実施例1c):ビス(フェニル)塩化リンの調製
前記グリニャール試薬を、THF(100ml)中のN,N−ジイソプロピルホスホルアミドジクロリド(6.64ml、36mmol)に0℃で加えた。室温で一晩攪拌した後、その混合物をシクロヘキサン(200ml)で希釈し、ドライのHClガスをその溶液中に0.5時間吹き込んだ。沈殿物を濾過した後溶媒を除去し、ホスフィンの塩化物と臭素化物の混合物を80%の収率で得た。この粗生成物を単離することなくすべてを次の工程で使用した。
【0089】
実施例1d):(フェニル)2PN(イソプロピル)P(フェニル)2配位子の調製
DCM(80ml)およびトリエチルアミン(15ml)中の粗製ビス(フェニル)塩化リン(粗製反応混合物からの計算で28.8mmol)の0℃の溶液に、イソプロピルアミン(1.11ml、13mmol)を加えた。その反応物を30分間攪拌し、その後氷浴を取り外した。全部で14時間攪拌した後、その溶液を濾過して形成されたトリエチルアンモニウム塩を取り出した。その生成物を結晶化した後90%の収率で単離した。31P{H}NMR:49.0ppm(幅広の一重線)。
【実施例2】
【0090】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、29.0mgの(フェニル)2PN(メチル)P(フェニル)2(0.073mmol)の5mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の15mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の80℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を85℃に制御し、一方エチレン圧を30バールgに維持した。ガス同伴攪拌機を使用し、1100RPMの混合速度により初めから終わりまで完全な混合を確保した。反応は、60分後にエチレンの反応器への供給を打ち切ることによって終了し、反応器を10℃より下まで冷却した。過剰のエチレンをオートクレーブから解放したのち、オートクレーブに含有されていた液体をエタノールで、続いて水中の10%の塩酸で急冷した。液相のGC−FIDによる分析のための内部標準としてノナンを加えた。有機層の少量の試料を無水硫酸ナトリウムにより乾燥し、続いてGC−FIDにより分析した。有機層の残りを濾過して固体の生成物を単離した。これらの固体生成物を一晩100℃のオーブンで乾燥し、続いて重量を測定した。全体の生成物の質量は、31.86gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例3】
【0091】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、22.4mgの(フェニル)2PN(メチル)P(フェニル)2(0.056mmol)の5mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の15mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の80℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を85℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、28.76gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例4】
【0092】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.3mgの(フェニル)2PN(メチル)P(フェニル)2(0.066mmol)の3mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の17mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、47.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例5】
【0093】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(ペンチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.0mgの(フェニル)2PN(ペンチル)P(フェニル)2(0.074mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、74.84gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例6】
【0094】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(ベンジル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.7mgの(フェニル)2PN(ベンジル)P(フェニル)2(0.065mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、22.08gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例7】
【0095】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、34.9mgの(フェニル)2PN(フェニル)P(フェニル)2(0.076mmol)の10mlのトルエン中の溶液を、13.5mgのCrCl3(テトラヒドロフラン)3(0.036mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、48.21gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例8】
【0096】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(p−メトキシ−フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.6mgの(フェニル)2PN(p−メトキシ−フェニル)P(フェニル)2(0.062mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、7.01gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例9】
【0097】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(p−t−ブチル−フェニル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、29.3mgの(フェニル)2PN(p−t−ブチル−フェニル)P(フェニル)2(0.062mmol)の10mlのトルエン中の溶液を、12.4mgのCrCl3(テトラヒドロフラン)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、62.15gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例10】
【0098】
Cr(2−エトキシへキサン酸)3、(フェニル)2PN(アリル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、27.6mgの(フェニル)2PN(アリル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、22.8mgのCr(2−エトキシへキサン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、12.68gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例11】
【0099】
Cr(アセチルアセトン酸)3、(フェニル)2PN[(CH2)3Si(OMe)3]P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、36.1mgの(フェニル)2PN[(CH2)3Si(OMe)3]P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、72.96gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例12】
【0100】
Cr(アセチルアセトン酸)3、(フェニル)2PN[(CH2)3−N−モルホリン]P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、33.8mgの(フェニル)2PN[(CH2)3−N−モルホリン]P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、22.2gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例13】
【0101】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.061mmol)の10mlのトルエン中の溶液を、11.6mgのCrCl3(テトラヒドロフラン)3(0.031mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、10.6mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を180分後に終了し、上の実施例2の手順を使用した。生成物の量は、56.44gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例14】
【0102】
CrCl3(テトラヒドロフラン)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、17.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.04mmol)の10mlのトルエン中の溶液を、7.5mgのCrCl3(テトラヒドロフラン)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、4.0mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を43℃に維持し、一方エチレン圧を30バールgに保った。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、39.98gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例15】
【0103】
Cr(2−エチルヘキサン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、18.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.022mmol)の10mlのトルエン中の溶液を、7.6mgのCr(2−エチルヘキサン酸)3(0.011mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、3.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を50分後に終了し、上の実施例2の手順を使用した。生成物の量は、64.71gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例16】
【0104】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を14分後に終了し、上の実施例2の手順を使用した。生成物の量は、75.80gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例17】
【0105】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびEAO/TMAを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)、EAO(エチルアルミノキサン、33mmol)およびTMA(トリメチルアルミニウム、8.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を37分後に終了し、上の実施例2の手順を使用した。生成物の量は、29.03gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例18】
【0106】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMMAOを使用するエチレンの四量体化反応
シュレンク容器内で、17.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.04mmol)の10mlのトルエン中の溶液を、7.0mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMMAO(変性メチルアルミノキサン、Akzo Nobel MMAO−3A、6.0mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、74.11gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例19】
【0107】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2および担持されたMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.2mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。3.9gの担持されたMAO(SiO2上のMAO、Crompton、11.3mmolのMAOを含有)を30mlのトルエンに懸濁させ、続いてトルエン(50ml)とTMA(トリメチルアルミニウム、3.3mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。触媒溶液を続いて圧力反応器に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、43.61gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例20】
【0108】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、18.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.044mmol)の6.4mlのクメン中の溶液を、7.7mgのCr(アセチルアセトン酸)3(0.022mmol)の8mlのクメン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてクメン(180ml)とMAO(メチルアルミノキサン、4.4mmol、10%トルエン溶液)の40℃の混合物を含有する1000mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を25分後に終了し、上の実施例2の手順を使用した。生成物の量は、118.78gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例21】
【0109】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.1mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのエチルベンゼン中の溶液を、7.0mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのエチルベンゼン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてエチルベンゼン(76ml)とMAO(メチルアルミノキサン、4.0mmol、7%トルエン溶液)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を10分後に終了し、上の実施例2の手順を使用した。生成物の量は、70.6gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例22】
【0110】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、5.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.014mmol)の10mlのシクロヘキサン中の溶液を、3.5mgのCr(アセチルアセトン酸)3(0.01mmol)の10mlのシクロヘキサン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、2.0mmol、7%トルエン溶液)の溶液を、45℃のシクロヘキサン(170ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を39分後に終了し、上の実施例2の手順を使用した。生成物の量は、307.30gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例23】
【0111】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.6mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのクメン中の溶液を、7.4mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのクメン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、2.8mmol、7%トルエン溶液)の溶液を、45℃のクメン(180ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を75分後に終了し、上の実施例2の手順を使用した。生成物の量は、308.83gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例24】
【0112】
Cr(アセチルアセトン酸)3、(2−ナフチル)2PN(メチル)P(2−ナフチル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、39.6mgの(2−ナフチル)2PN(メチル)P(2−ナフチル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に維持し、一方エチレン圧を30バールgに保った。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、45.18gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例25】
【0113】
Cr(アセチルアセトン酸)3、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、47.0mgの(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、26.41gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例26】
【0114】
Cr(アセチルアセトン酸)3、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を65バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、52.34gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例27】
【0115】
Cr(アセチルアセトン酸)3、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を45バールgに維持した。反応を15分後に終了し、上の実施例2の手順を使用した。生成物の量は、80.59gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例28】
【0116】
Cr(アセチルアセトン酸)3、(o−エチルフェニル)(Ph)PN(イソプロピル)PPh2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.1mgの(o−エチルフェニル)(Ph)PN(イソプロピル)PPh2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を14分後に終了し、上の実施例2の手順を使用した。生成物の量は、63.78gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例29】
【0117】
Cr(アセチルアセトン酸)3、(フェニル)2P(=S)N(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、30.3mgの(フェニル)2P(=S)N(イソプロピル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。生成物の量は、33.06gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例30】
【0118】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.6mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.026mmol)の10mlのクメン中の溶液を、7.4mgのCr(アセチルアセトン酸)3(0.02mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。この溶液およびMAO(メチルアルミノキサン、4.0mmol、7%トルエン溶液)の溶液を、45℃のクメン(80ml)と1−オクテン(80ml)の混合物を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を45分後に終了し、上の実施例2の手順を使用した。生成物の量は、405.87gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例31】
【0119】
Cr(アセチルアセトン酸)3、(フェニル)2PN(メチル)N(メチル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、28.3mgの(フェニル)2PN(メチル)N(メチル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その液体生成物の量は、22.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例32】
【0120】
Cr(アセチルアセトン酸)3、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、37.2mgの(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、14.7gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例33】
【0121】
Cr(アセチルアセトン酸)3、(フェニル)2PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、5.8mgの(フェニル)2PN(イソプロピル)P(フェニル)2(0.015mmol)の10mlのシクロヘキサン中の溶液を、3.8mgのCr(アセチルアセトン酸)3(0.011mmol)の10mlのシクロヘキサン中の溶液に加えた。その混合物を周囲温度で5分間攪拌した。1.8mmolのMAO(メチルアルミノキサン、7%トルエン溶液)を加え、その混合物を5分間攪拌した。この溶液を、45℃のシクロヘキサン(180ml)を含有し、40バールに加圧した1000mlの圧力反応器(オートクレーブ)にビュレットから加えた。添加後、エチレン圧を45バールgに維持し、温度を45℃に制御した。反応を60分後に終了し、上の実施例2の手順を使用した。生成物の量は、297.69gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例34】
【0122】
Cr(アセチルアセトン酸)3、(フェニル)2PN(SiMe3)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、39.8mgの(フェニル)2PN(SiMe3)P(フェニル)2(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、26.9gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例35】
【0123】
Cr(アセチルアセトン酸)3、[(フェニル2P)2NCH2CH2]NおよびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、62.5mgの[(フェニル2P)2NCH2CH2]N(0.066mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の60℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を65℃に制御し、一方エチレン圧を30バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、2.5gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例36】
【0124】
Cr(アセチルアセトン酸)3、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、11.7mgの(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)(0.026mmol)の10mlのトルエン中の溶液を、7.7mgのCr(アセチルアセトン酸)3(0.022mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、6.6mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、55.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例37】
【0125】
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2の調製
(フェニル)2PN(フェニル)P(フェニル)2(0.273g、0.591mmol)およびCrCl3(thf)3(0.206g、0.550mmol)を、トルエン(25ml)中にとり、80℃に一晩加熱して、青い粉末の沈殿を生じさせた。室温まで冷却した後、トルエンを沈殿から濾別し、生成物を石油エーテル(10ml)で2回洗浄した。真空下の乾燥により、0.303g(89%)の収量を得た。C60H50N2P4Cr2Cl6に対する計算値(測定値):C 58.13(57.98)%;H 4.07(3.97)%;N 2.26(2.12)%。磁気モーメント Cr1原子当たり4.06BM(二量体1個当たり5.74BM)。図1は、単結晶X線解析により得られた錯体の構造を示す。
【実施例38】
【0126】
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2およびMAOを使用するエチレンの四量体化反応
[Cr{(フェニル)2PN(フェニル)P(フェニル)2}Cl2(μ−Cl)]2(0.0125g、Crが0.020mmol)のトルエン20ml中の懸濁液を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、4.61gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例39】
【0127】
(エチル)2PN(メチル)P(エチル)2配位子の調製
トルエン(25ml)中のメチルアミン(3.1mlの2M溶液、6.2mmol)を、トルエン(15ml)およびトリエチルアミン(5ml)中のクロロジエチルホスフィン(1.582g、12.7mmol)の溶液にゆっくりと加えた。その混合物を一晩攪拌した後、ガラス繊維のフィルターを通して濾過した。溶媒を真空下で除去し、10mlの水を加えた。その生成物を石油エーテルで抽出し(3×5ml)有機層を合わせた。溶媒を真空下で除去することにより、1.046g(81%)の生成物の収量を得た。31P{H}NMR:68ppm。
【実施例40】
【0128】
Cr(2−エチルヘキサン酸)3、(エチル)2PN(メチル)P(エチル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを10ml、0.020mmol)および(エチル)2PN(メチル)P(エチル)2の溶液(トルエン中0.005Mを4.1ml、0.0205mmol)を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、2.26gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例41】
【0129】
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2の調製
(エチル)2PN(メチル)P(エチル)2(0.362g、1.75mmol)およびCrCl3(thf)3(0.594g、1.58mmol)を使用して実施例38の手順に従った。0.520g(90%)の収量を得た。C18H46N2P4Cr2Cl6に対する計算値(測定値):C 29.57(29.62)%;H 6.34(6.45)%;N 3.83(3.87)%。磁気モーメント Cr1原子当たり3.86BM(二量体1個当たり5.46BM)。
【実施例42】
【0130】
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2およびMAOを使用するエチレンの四量体化反応
[Cr{(エチル)2PN(メチル)P(エチル)2}Cl2(μ−Cl)]2(0.0075g、Crが0.020mmol)のトルエン10ml中の懸濁液を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、3.06gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例43】
【0131】
(エチル)2PN(イソプロピル)P(フェニル)2配位子の調製
トルエン(15ml)中のN−(ジフェニルホスフィノ)メチルアミン(1.870g、7.69mmol)を、トルエン(20ml)およびトリエチルアミン(5ml)中のクロロジエチルホスフィン(0.986g、7.92mmol)の溶液にゆっくりと加えた。その混合物を一晩攪拌した後、ガラス繊維のフィルターを通して濾過した。溶媒を真空下で除去し、10mlの水を加えた。その生成物を石油エーテルで抽出し(3×5ml)有機層を合わせた。溶媒を真空下で除去することにより、2.200g(86%)の生成物の収量を得た。31P{H}NMR:49、43ppm。
【実施例44】
【0132】
Cr(2−エチルヘキサン酸)3、(フェニル)2PN(イソプロピル)P(エチル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを10ml、0.020mmol)および(フェニル)2PN(イソプロピル)P(エチル)2の溶液(トルエン中0.004Mを5ml、0.020mmol)を、45℃でトルエン(100ml)およびMAO(6.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、10.83gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例45】
【0133】
Cr(2−エチルヘキサン酸)3、(フェニル)(エチル)PN(メチル)P(エチル)(フェニル)およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを15ml、0.030mmol)および(フェニル)(エチル)PN(メチル)P(エチル)(フェニル)の溶液(トルエン中0.00365Mを9ml、0.033mmol)を、45℃でトルエン(100ml)およびMAO(9.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、0.897gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例46】
【0134】
Cr(2−エチルヘキサン酸)3、(フェニル)(エチル)PN(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
Cr(2−エチルヘキサン酸)3の溶液(トルエン中0.002Mを15ml、0.030mmol)および(フェニル)(エチル)PN(イソプロピル)P(フェニル)の溶液(9mlトルエン中0.034mmol)を、45℃でトルエン(100ml)およびMAO(9.0mmol)を含有する300mlの圧力反応器(オートクレーブ)に加えた。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、13.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例47】
【0135】
Cr(アセチルアセトン酸)3、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、33.4mgの(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2(0.066mmol)の15mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(75ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を45バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、8.45gであった。この実施例の生成物の分布は、表1にまとめられている。
【実施例48】
【0136】
Cr(アセチルアセトン酸)3、(フェニル)2PCH2CH2P(フェニル)2およびMAOを使用するエチレンの四量体化反応
シュレンク容器内で、26.3mgの(フェニル)2PCH2CH2P(フェニル)2(0.198mmol)の10mlのトルエン中の溶液を、11.5mgのCr(アセチルアセトン酸)3(0.033mmol)の10mlのトルエン中の溶液に加えた。その混合物を周囲温度で5分間攪拌し、続いてトルエン(80ml)とMAO(メチルアルミノキサン、9.9mmol)の40℃の混合物を含有する300mlの圧力反応器(オートクレーブ)に移した。その圧力反応器にエチレンを充填し、その後反応器の温度を45℃に制御し、一方エチレン圧を40バールgに維持した。反応を30分後に終了し、上の実施例2の手順を使用した。その生成物の量は、21.23gであった。この実施例の生成物の分布は、表1にまとめられている。
【0137】
【表1】
【図面の簡単な説明】
【0138】
【図1】単結晶X線解析により得られた錯体の構造を表す模式図である。
【特許請求の範囲】
【請求項1】
生成物流が30%を超える四量体オレフィンを含有するオレフィンの四量体化方法。
【請求項2】
オレフィンの供給流を、遷移金属化合物およびへテロ原子配位子を含有する触媒系と接触させる工程を有する請求項1に記載の方法。
【請求項3】
前記供給流がα−オレフィンを含み、前記生成物流が少なくとも30%の四量体化α−オレフィンモノマーを含む請求項1または2に記載の方法。
【請求項4】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも30%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項5】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも40%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項6】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも50%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項7】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも60%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項8】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも70%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項9】
前記オレフィン供給流がエチレンを含み、前記生成物流中の(C6+C8):(C4+C10)の比が2.5:1を超える請求項1〜8のいずれかに記載の方法。
【請求項10】
前記オレフィン供給流がエチレンを含み、前記生成物流中のC8:C6の比が1を超える請求項1〜9のいずれかに記載の方法。
【請求項11】
エチレンを、10バールgを超える圧力で前記触媒系と接触させる請求項4〜10のいずれかに記載の方法。
【請求項12】
ヘテロ原子配位子が、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、BはAとCの間の結合基であり、2つのRは同じか異なるものであり、各Rは独立して、ホモまたはヘテロヒドロカルビル基のいずれかから選択され、各Rに対するnおよびmは独立して、AとCのそれぞれの価数および酸化状態により決まる)によって表される請求項1〜11のいずれかに記載の方法。
【請求項13】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体である請求項12に記載の方法。
【請求項14】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、ヒドロカルビル、ヘテロヒドロカルビル、置換ヒドロカルビルまたは置換ヘテロヒドロカルビル基から選択される)によって表される請求項12または13に記載の方法。
【請求項15】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表される請求項14に記載の方法。
【請求項16】
前記配位子が、複数の(R)nA−B−C(R)mを含有する請求項12に記載の方法。
【請求項17】
R1、R2、R3およびR4の1つ以上のいずれかの置換基が電子供与性ではない請求項15に記載の方法。
【請求項18】
R1、R2、R3およびR4が独立して、複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、AまたはCに結合している原子と隣接する原子に置換基を有する請求項15〜17のいずれかに記載の方法。
【請求項19】
いずれかの電子非供与性置換基が非極性である請求項17または18に記載の方法。
【請求項20】
Bが、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;およびメチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子もしくはハロゲンである)からなる群のいずれか1つから選択される請求項12〜19のいずれかに記載の方法。
【請求項21】
Bを単一原子スペーサーであるように選択する請求項12〜20のいずれかに記載の方法。
【請求項22】
Bを、−N(R5)−(R5は、水素であるかまたはアルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基もしくはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる基から選択される)であるように選択する請求項12〜21のいずれかに記載の方法。
【請求項23】
Aおよび/またはCを、独立して、S、Se、NまたはOで酸化する(Aおよび/またはCの価数は、上記酸化を可能とする)請求項12〜22のいずれかに記載の方法。
【請求項24】
AおよびCが、独立して、リンまたはS、Se、NもしくはOで酸化されたリンである請求項12〜23のいずれかに記載の方法。
【請求項25】
R1、R2、R3およびR4を、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択する請求項14〜22および24のいずれかに記載の方法。
【請求項26】
R1、R2、R3およびR4を、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択する請求項25に記載の方法。
【請求項27】
前記配位子を、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれか1つから選択する請求項1〜22、24〜26のいずれかに記載の方法。
【請求項28】
へテロ原子配位子を遷移金属前駆物質および活性化剤と任意の順序で合わせる工程を有する請求項1〜27のいずれかに記載の方法。
【請求項29】
へテロ原子配位子および遷移金属前駆物質を用いて調製した、予め形成された配位錯体を、活性化剤を含有する反応混合物に加える工程を有する請求項1〜28のいずれかに記載の方法。
【請求項30】
遷移金属前駆物質およびへテロ原子配位子からin situでヘテロ原子配位錯体を生成させる工程を有する請求項28に記載の方法。
【請求項31】
前記遷移金属を、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択する請求項2〜30のいずれかに記載の方法。
【請求項32】
前記遷移金属がクロムである請求項2〜30のいずれかに記載の方法。
【請求項33】
前記遷移金属前駆物質を、無機塩、有機塩、配位錯体および有機金属錯体からなる群から選択する請求項28〜30のいずれかに記載の方法。
【請求項34】
前記遷移金属前駆物質を、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択する請求項33に記載の方法。
【請求項35】
前記遷移金属を、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)から選択される錯体から選択する請求項28〜34のいずれかに記載の方法。
【請求項36】
遷移金属前駆物質に由来する遷移金属とへテロ原子配位子とを、約0.01:100〜10000:1の金属/配位子の比となるように合わせた請求項28〜35のいずれかに記載の方法。
【請求項37】
前記遷移金属前駆物質とへテロ原子配位子とを、約0.1:1から10:1の金属/配位子の比となるように合わせた請求項36に記載の方法。
【請求項38】
前記触媒系が、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)からなる群のいずれか1つから選択される活性化剤を含む請求項28〜37のいずれかに記載の方法。
【請求項39】
前記活性化剤をアルキルアルミノキサン類から選択する請求項28〜38のいずれかに記載の方法。
【請求項40】
アルキルアルミノキサンまたはその混合物を、メチルアルミノキサン(MAO)、エチルアルミノキサン(EAO)および変性アルキルアルミノキサン類(MMAO)からなる群から選択する請求項39に記載の方法。
【請求項41】
約1:1から10000:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項39または40に記載の方法。
【請求項42】
約1:1〜1000:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項41に記載の方法。
【請求項43】
約1:1〜300:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項42に記載の方法。
【請求項44】
前記触媒系に、アルキルアルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を加える工程を有する請求項39〜43のいずれかに記載の方法。
【請求項45】
オレフィンの存在下、−20℃〜250℃の間の任意の温度で触媒系の成分を混合する工程を有する請求項2〜44のいずれかに記載の方法。
【請求項46】
温度範囲が20℃〜100℃の間である請求項45に記載の方法。
【請求項47】
メチルシクロペンタンおよびメチレンシクロペンタンが生成物として形成され、独立して前記方法の生成物流の少なくとも1%を成す請求項1〜46のいずれかに記載の方法。
【請求項48】
遷移金属およびへテロ原子配位子を含む四量体化触媒系。
【請求項49】
ヘテロ原子配位子が、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、BはAとCの間の結合基であり、2つのRは同じか異なるものであり、各Rは独立して、ホモまたはヘテロヒドロカルビル基のいずれかから選択され、各Rに対するnおよびmは独立して、AとCのそれぞれの価数および酸化状態により決まる)によって表される請求項48に記載の触媒系。
【請求項50】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体である請求項49に記載の触媒系。
【請求項51】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、ヒドロカルビル、ヘテロヒドロカルビル、置換ヒドロカルビルまたは置換ヘテロヒドロカルビル基から選択される)によって表される請求項49または50に記載の触媒系。
【請求項52】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表される請求項51に記載の触媒系。
【請求項53】
前記配位子が複数の(R)nA−B−C(R)mを含有する請求項49に記載の触媒系。
【請求項54】
R1、R2、R3およびR4の1つ以上のいずれかの置換基が電子供与性ではない請求項52または53に記載の触媒系。
【請求項55】
R1、R2、R3およびR4が独立して、複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、AまたはCに結合している原子と隣接する原子に置換基を有する請求項51〜54のいずれかに記載の触媒系。
【請求項56】
いずれかの電子非供与性置換基が非極性である請求項54または55に記載の触媒系。
【請求項57】
Bが、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;およびメチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビル、置換ヒドロカルビル、置換へテロ原子またはハロゲンである)からなる群のいずれか1つから選択される請求項49〜56のいずれかに記載の触媒系。
【請求項58】
Bを単一原子スペーサーであるように選択する請求項49〜57のいずれかに記載の触媒系。
【請求項59】
Bを、−N(R5)−(R5は、水素であるかまたはアルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基もしくはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる基から選択される)であるように選択する請求項58に記載の触媒系。
【請求項60】
Aおよび/またはCが独立して、S、Se、NまたはOで酸化されている(Aおよび/またはCの価数は上記酸化を可能とする)請求項49〜59のいずれかに記載の触媒系。
【請求項61】
AおよびCが独立して、リンまたはS、Se、NもしくはOで酸化されたリンである請求項49〜60のいずれかに記載の触媒系。
【請求項62】
R1、R2、R3およびR4を、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択する請求項51〜61のいずれかに記載の触媒系。
【請求項63】
R1、R2、R3およびR4を独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択する請求項51〜62のいずれかに記載の触媒系。
【請求項64】
前記配位子を、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれか1つから選択する請求項49〜59、61〜63のいずれかに記載の触媒系。
【請求項65】
前記遷移金属を、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択する請求項49〜64のいずれかに記載の触媒系。
【請求項66】
前記遷移金属がクロムである請求項49〜65のいずれかに記載の触媒系。
【請求項67】
前記遷移金属が、無機塩、有機塩、配位錯体および有機金属錯体からなる群から選択される遷移金属前駆物質に由来するものである請求項66に記載の触媒系。
【請求項68】
前記遷移金属前駆物質を、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群から選択する請求項67に記載の触媒系。
【請求項69】
前記遷移金属を、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)から選択される錯体から選択する請求項48〜68のいずれかに記載の触媒系。
【請求項70】
遷移金属前駆物質に由来する遷移金属とへテロ原子配位子とが、約0.01:100〜10000:1の金属/配位子の比を有する請求項67〜68のいずれかに記載の触媒系。
【請求項71】
前記遷移金属前駆物質とへテロ原子配位子とを、約0.1:1〜10:1の金属/配位子の比となるように合わせた請求項70に記載の触媒系。
【請求項72】
活性化剤を含む請求項48〜71のいずれかに記載の触媒系。
【請求項73】
前記活性化剤が、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)からなる群のいずれか1つから選択される請求項72に記載の触媒系。
【請求項74】
前記活性化剤をアルキルアルミノキサン類から選択する請求項72に記載の触媒系。
【請求項75】
アルキルアルミノキサン、またはその混合物を、メチルアルミノキサン(MAO)、エチルアルミノキサン(EAO)および変性アルキルアルミノキサン類(MMAO)からなる群から選択する請求項74に記載の方法。
【請求項76】
前記遷移金属と前記アルミノキサンとを、約1:1〜10000:1のAl/金属の比となるように互いに対する割合で合わせる請求項74または75に記載の触媒系。
【請求項77】
前記遷移金属と前記アルミノキサンとを、約1:1〜1000:1のAl/金属の比となる割合で合わせる請求項76に記載の触媒系。
【請求項78】
前記遷移金属と前記アルミノキサンとを、約1:1〜300:1のAl/金属の比となる割合で合わせる請求項77に記載の方法。
【請求項79】
アルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を含む請求項74〜78のいずれかに記載の触媒系。
【請求項80】
請求項48〜79のいずれかに記載の四量体化触媒系のオレフィンの四量体化のための使用。
【請求項81】
請求項48〜78のいずれかに記載の四量体化触媒系のエチレンの四量体化のための使用。
【請求項82】
請求項1〜47のいずれかに記載の四量体化方法のための配位子の使用。
【請求項83】
請求項48〜79のいずれかに記載の四量体化触媒系のための配位子の使用。
【請求項84】
本明細書に実質的に記載されているオレフィンの四量体化方法。
【請求項85】
本明細書に実質的に記載されているオレフィンの四量体化触媒系。
【請求項1】
生成物流が30%を超える四量体オレフィンを含有するオレフィンの四量体化方法。
【請求項2】
オレフィンの供給流を、遷移金属化合物およびへテロ原子配位子を含有する触媒系と接触させる工程を有する請求項1に記載の方法。
【請求項3】
前記供給流がα−オレフィンを含み、前記生成物流が少なくとも30%の四量体化α−オレフィンモノマーを含む請求項1または2に記載の方法。
【請求項4】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも30%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項5】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも40%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項6】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも50%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項7】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも60%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項8】
前記オレフィン供給流がエチレンを含み、前記生成物流が少なくとも70%の1−オクテンを含む請求項1〜3のいずれかに記載の方法。
【請求項9】
前記オレフィン供給流がエチレンを含み、前記生成物流中の(C6+C8):(C4+C10)の比が2.5:1を超える請求項1〜8のいずれかに記載の方法。
【請求項10】
前記オレフィン供給流がエチレンを含み、前記生成物流中のC8:C6の比が1を超える請求項1〜9のいずれかに記載の方法。
【請求項11】
エチレンを、10バールgを超える圧力で前記触媒系と接触させる請求項4〜10のいずれかに記載の方法。
【請求項12】
ヘテロ原子配位子が、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、BはAとCの間の結合基であり、2つのRは同じか異なるものであり、各Rは独立して、ホモまたはヘテロヒドロカルビル基のいずれかから選択され、各Rに対するnおよびmは独立して、AとCのそれぞれの価数および酸化状態により決まる)によって表される請求項1〜11のいずれかに記載の方法。
【請求項13】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体である請求項12に記載の方法。
【請求項14】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、ヒドロカルビル、ヘテロヒドロカルビル、置換ヒドロカルビルまたは置換ヘテロヒドロカルビル基から選択される)によって表される請求項12または13に記載の方法。
【請求項15】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表される請求項14に記載の方法。
【請求項16】
前記配位子が、複数の(R)nA−B−C(R)mを含有する請求項12に記載の方法。
【請求項17】
R1、R2、R3およびR4の1つ以上のいずれかの置換基が電子供与性ではない請求項15に記載の方法。
【請求項18】
R1、R2、R3およびR4が独立して、複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、AまたはCに結合している原子と隣接する原子に置換基を有する請求項15〜17のいずれかに記載の方法。
【請求項19】
いずれかの電子非供与性置換基が非極性である請求項17または18に記載の方法。
【請求項20】
Bが、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;およびメチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビルもしくは置換ヒドロカルビル、置換へテロ原子もしくはハロゲンである)からなる群のいずれか1つから選択される請求項12〜19のいずれかに記載の方法。
【請求項21】
Bを単一原子スペーサーであるように選択する請求項12〜20のいずれかに記載の方法。
【請求項22】
Bを、−N(R5)−(R5は、水素であるかまたはアルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基もしくはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる基から選択される)であるように選択する請求項12〜21のいずれかに記載の方法。
【請求項23】
Aおよび/またはCを、独立して、S、Se、NまたはOで酸化する(Aおよび/またはCの価数は、上記酸化を可能とする)請求項12〜22のいずれかに記載の方法。
【請求項24】
AおよびCが、独立して、リンまたはS、Se、NもしくはOで酸化されたリンである請求項12〜23のいずれかに記載の方法。
【請求項25】
R1、R2、R3およびR4を、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択する請求項14〜22および24のいずれかに記載の方法。
【請求項26】
R1、R2、R3およびR4を、独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択する請求項25に記載の方法。
【請求項27】
前記配位子を、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれか1つから選択する請求項1〜22、24〜26のいずれかに記載の方法。
【請求項28】
へテロ原子配位子を遷移金属前駆物質および活性化剤と任意の順序で合わせる工程を有する請求項1〜27のいずれかに記載の方法。
【請求項29】
へテロ原子配位子および遷移金属前駆物質を用いて調製した、予め形成された配位錯体を、活性化剤を含有する反応混合物に加える工程を有する請求項1〜28のいずれかに記載の方法。
【請求項30】
遷移金属前駆物質およびへテロ原子配位子からin situでヘテロ原子配位錯体を生成させる工程を有する請求項28に記載の方法。
【請求項31】
前記遷移金属を、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択する請求項2〜30のいずれかに記載の方法。
【請求項32】
前記遷移金属がクロムである請求項2〜30のいずれかに記載の方法。
【請求項33】
前記遷移金属前駆物質を、無機塩、有機塩、配位錯体および有機金属錯体からなる群から選択する請求項28〜30のいずれかに記載の方法。
【請求項34】
前記遷移金属前駆物質を、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群のいずれか1つから選択する請求項33に記載の方法。
【請求項35】
前記遷移金属を、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)から選択される錯体から選択する請求項28〜34のいずれかに記載の方法。
【請求項36】
遷移金属前駆物質に由来する遷移金属とへテロ原子配位子とを、約0.01:100〜10000:1の金属/配位子の比となるように合わせた請求項28〜35のいずれかに記載の方法。
【請求項37】
前記遷移金属前駆物質とへテロ原子配位子とを、約0.1:1から10:1の金属/配位子の比となるように合わせた請求項36に記載の方法。
【請求項38】
前記触媒系が、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)からなる群のいずれか1つから選択される活性化剤を含む請求項28〜37のいずれかに記載の方法。
【請求項39】
前記活性化剤をアルキルアルミノキサン類から選択する請求項28〜38のいずれかに記載の方法。
【請求項40】
アルキルアルミノキサンまたはその混合物を、メチルアルミノキサン(MAO)、エチルアルミノキサン(EAO)および変性アルキルアルミノキサン類(MMAO)からなる群から選択する請求項39に記載の方法。
【請求項41】
約1:1から10000:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項39または40に記載の方法。
【請求項42】
約1:1〜1000:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項41に記載の方法。
【請求項43】
約1:1〜300:1のAl/金属の比となる割合で遷移金属とアルミノキサンとを合わせる請求項42に記載の方法。
【請求項44】
前記触媒系に、アルキルアルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を加える工程を有する請求項39〜43のいずれかに記載の方法。
【請求項45】
オレフィンの存在下、−20℃〜250℃の間の任意の温度で触媒系の成分を混合する工程を有する請求項2〜44のいずれかに記載の方法。
【請求項46】
温度範囲が20℃〜100℃の間である請求項45に記載の方法。
【請求項47】
メチルシクロペンタンおよびメチレンシクロペンタンが生成物として形成され、独立して前記方法の生成物流の少なくとも1%を成す請求項1〜46のいずれかに記載の方法。
【請求項48】
遷移金属およびへテロ原子配位子を含む四量体化触媒系。
【請求項49】
ヘテロ原子配位子が、一般式(R)nA−B−C(R)m(式中、AおよびCは、独立して、リン、ヒ素、アンチモン、酸素、ビスマス、硫黄、セレン、および窒素からなる群から選択され、BはAとCの間の結合基であり、2つのRは同じか異なるものであり、各Rは独立して、ホモまたはヘテロヒドロカルビル基のいずれかから選択され、各Rに対するnおよびmは独立して、AとCのそれぞれの価数および酸化状態により決まる)によって表される請求項48に記載の触媒系。
【請求項50】
Aおよび/またはCが遷移金属と配位するための潜在的電子供与体である請求項49に記載の触媒系。
【請求項51】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、ヒドロカルビル、ヘテロヒドロカルビル、置換ヒドロカルビルまたは置換ヘテロヒドロカルビル基から選択される)によって表される請求項49または50に記載の触媒系。
【請求項52】
ヘテロ原子配位子が、一般式(R1)(R2)A−B−C(R3)(R4)(式中、AおよびCは独立して、リン、ヒ素、アンチモン、ビスマスおよび窒素からなる群から選択され、BはAとCの間の結合基であり、R1、R2、R3およびR4は独立して、非芳香族基または複素環式芳香族を含む芳香族基である)によって表される請求項51に記載の触媒系。
【請求項53】
前記配位子が複数の(R)nA−B−C(R)mを含有する請求項49に記載の触媒系。
【請求項54】
R1、R2、R3およびR4の1つ以上のいずれかの置換基が電子供与性ではない請求項52または53に記載の触媒系。
【請求項55】
R1、R2、R3およびR4が独立して、複素環式芳香族を含む芳香族基であり、すべてではない基R1、R2、R3およびR4が、AまたはCに結合している原子と隣接する原子に置換基を有する請求項51〜54のいずれかに記載の触媒系。
【請求項56】
いずれかの電子非供与性置換基が非極性である請求項54または55に記載の触媒系。
【請求項57】
Bが、ヒドロカルビル、置換ヒドロカルビル、ヘテロヒドロカルビルおよび置換ヘテロヒドロカルビルを含む有機結合基;単一原子結合を含む無機結合基;イオン結合;およびメチレン、ジメチルメチレン、1,2−エタン、1,2−フェニレン、1,2−プロパン、1,2−カテコール、1,2−ジメチルヒドラジン、−B(R5)−、−Si(R5)2−、−P(R5)−および−N(R5)−(R5は、水素、ヒドロカルビル、置換ヒドロカルビル、置換へテロ原子またはハロゲンである)からなる群のいずれか1つから選択される請求項49〜56のいずれかに記載の触媒系。
【請求項58】
Bを単一原子スペーサーであるように選択する請求項49〜57のいずれかに記載の触媒系。
【請求項59】
Bを、−N(R5)−(R5は、水素であるかまたはアルキル、置換アルキル、アリール、置換アリール、アリールオキシ、置換アリールオキシ、ハロゲン、ニトロ、アルコキシカルボニル、カルボニルオキシ、アルコキシ、アミノカルボニル、カルボニルアミノ、ジアルキルアミノ、シリル基もしくはそれらの誘導体、およびこれら置換基のいずれかにより置換されているアリールからなる基から選択される)であるように選択する請求項58に記載の触媒系。
【請求項60】
Aおよび/またはCが独立して、S、Se、NまたはOで酸化されている(Aおよび/またはCの価数は上記酸化を可能とする)請求項49〜59のいずれかに記載の触媒系。
【請求項61】
AおよびCが独立して、リンまたはS、Se、NもしくはOで酸化されたリンである請求項49〜60のいずれかに記載の触媒系。
【請求項62】
R1、R2、R3およびR4を、独立して、ベンジル、フェニル、トリル、キシリル、メシチル、ビフェニル、ナフチル、アントラセニル、メトキシ、エトキシ、フェノキシ、トリルオキシ、ジメチルアミノ、ジエチルアミノ、メチルエチルアミノ、チオフェニル、ピリジル、チオエチル、チオフェノキシ、トリメチルシリル、ジメチルヒドラジル、メチル、エチル、エテニル、プロピル、ブチル、プロペニル、プロピニル、シクロペンチル、シクロヘキシル、フェロセニルおよびテトラヒドロフラニル基からなる群から選択する請求項51〜61のいずれかに記載の触媒系。
【請求項63】
R1、R2、R3およびR4を独立して、フェニル、トリル、ビフェニル、ナフチル、チオフェニルおよびエチル基からなる群から選択する請求項51〜62のいずれかに記載の触媒系。
【請求項64】
前記配位子を、(フェニル)2PN(メチル)P(フェニル)2、(フェニル)2PN(ペンチル)P(フェニル)2、(フェニル)2PN(フェニル)P(フェニル)2、(フェニル)2PN(p−メトキシフェニル)P(フェニル)2、(フェニル)2PN(p−tブチルフェニル)P(フェニル)2、(フェニル)2PN((CH2)3−N−モルホリン)P(フェニル)2、(フェニル)2PN(Si(CH3)3)P(フェニル)2、(((フェニル)2P)2NCH2CH2)N、(エチル)2PN(メチル)P(エチル)2、(エチル)2PN(イソプロピル)P(フェニル)2、(エチル)(フェニル)PN(メチル)P(エチル)(フェニル)、(エチル)(フェニル)PN(イソプロピル)P(フェニル)2、(フェニル)2P(=Se)N(イソプロピル)P(フェニル)2、(フェニル)2PCH2CH2P(フェニル)2、(o−エチルフェニル)(フェニル)PN(イソプロピル)P(フェニル)2、(o−メチルフェニル)2PN(イソプロピル)P(o−メチルフェニル)(フェニル)、(フェニル)2PN(ベンジル)P(フェニル)2、(フェニル)2PN(1−シクロヘキシル−エチル)P(フェニル)2、(フェニル)2PN[CH2CH2CH2Si(OMe3)]P(フェニル)2、(フェニル)2PN(シクロヘキシル)P(フェニル)2、(フェニル)2PN(2−メチルシクロヘキシル)P(フェニル)2、(フェニル)2PN(アリル)P(フェニル)2、(2−ナフチル)2PN(メチル)P(2−ナフチル)2、(p−ビフェニル)2PN(メチル)P(p−ビフェニル)2、(p−メチルフェニル)2PN(メチル)P(p−メチルフェニル)2、(2−チオフェニル)2PN(メチル)P(2−チオフェニル)2、(フェニル)2PN(メチル)N(メチル)P(フェニル)2、(m−メチルフェニル)2PN(メチル)P(m−メチルフェニル)2、(フェニル)2PN(イソプロピル)P(フェニル)2および(フェニル)2P(=S)N(イソプロピル)P(フェニル)2からなる群のいずれか1つから選択する請求項49〜59、61〜63のいずれかに記載の触媒系。
【請求項65】
前記遷移金属を、クロム、モリブデン、タングステン、チタン、タンタル、バナジウムおよびジルコニウムからなる群のいずれか1つから選択する請求項49〜64のいずれかに記載の触媒系。
【請求項66】
前記遷移金属がクロムである請求項49〜65のいずれかに記載の触媒系。
【請求項67】
前記遷移金属が、無機塩、有機塩、配位錯体および有機金属錯体からなる群から選択される遷移金属前駆物質に由来するものである請求項66に記載の触媒系。
【請求項68】
前記遷移金属前駆物質を、クロムトリクロリドトリス−テトラヒドロフラン錯体、(ベンゼン)トリカルボニルクロム、オクタン酸クロム(III)、アセチルアセトン酸クロム(III)、ヘキサカルボニルクロムおよび2−エチルヘキサン酸クロム(III)からなる群から選択する請求項67に記載の触媒系。
【請求項69】
前記遷移金属を、アセチルアセトン酸クロム(III)および2−エチルヘキサン酸クロム(III)から選択される錯体から選択する請求項48〜68のいずれかに記載の触媒系。
【請求項70】
遷移金属前駆物質に由来する遷移金属とへテロ原子配位子とが、約0.01:100〜10000:1の金属/配位子の比を有する請求項67〜68のいずれかに記載の触媒系。
【請求項71】
前記遷移金属前駆物質とへテロ原子配位子とを、約0.1:1〜10:1の金属/配位子の比となるように合わせた請求項70に記載の触媒系。
【請求項72】
活性化剤を含む請求項48〜71のいずれかに記載の触媒系。
【請求項73】
前記活性化剤が、有機アルミニウム化合物、有機ホウ素化合物、有機塩(臭化メチルリチウムおよび臭化メチルマグネシウム等)、無機酸および無機塩(テトラフルオロホウ酸エーテラート、テトラフルオロホウ酸銀およびヘキサフルオロアンチモン酸ナトリウム等)からなる群のいずれか1つから選択される請求項72に記載の触媒系。
【請求項74】
前記活性化剤をアルキルアルミノキサン類から選択する請求項72に記載の触媒系。
【請求項75】
アルキルアルミノキサン、またはその混合物を、メチルアルミノキサン(MAO)、エチルアルミノキサン(EAO)および変性アルキルアルミノキサン類(MMAO)からなる群から選択する請求項74に記載の方法。
【請求項76】
前記遷移金属と前記アルミノキサンとを、約1:1〜10000:1のAl/金属の比となるように互いに対する割合で合わせる請求項74または75に記載の触媒系。
【請求項77】
前記遷移金属と前記アルミノキサンとを、約1:1〜1000:1のAl/金属の比となる割合で合わせる請求項76に記載の触媒系。
【請求項78】
前記遷移金属と前記アルミノキサンとを、約1:1〜300:1のAl/金属の比となる割合で合わせる請求項77に記載の方法。
【請求項79】
アルミノキサン1モル当たり0.01〜100モルの間の量のトリアルキルアルミニウム化合物を含む請求項74〜78のいずれかに記載の触媒系。
【請求項80】
請求項48〜79のいずれかに記載の四量体化触媒系のオレフィンの四量体化のための使用。
【請求項81】
請求項48〜78のいずれかに記載の四量体化触媒系のエチレンの四量体化のための使用。
【請求項82】
請求項1〜47のいずれかに記載の四量体化方法のための配位子の使用。
【請求項83】
請求項48〜79のいずれかに記載の四量体化触媒系のための配位子の使用。
【請求項84】
本明細書に実質的に記載されているオレフィンの四量体化方法。
【請求項85】
本明細書に実質的に記載されているオレフィンの四量体化触媒系。
【図1】

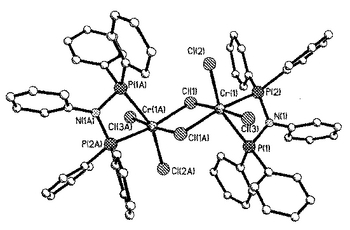
【公表番号】特表2006−517528(P2006−517528A)
【公表日】平成18年7月27日(2006.7.27)
【国際特許分類】
【出願番号】特願2005−511969(P2005−511969)
【出願日】平成15年12月19日(2003.12.19)
【国際出願番号】PCT/ZA2003/000187
【国際公開番号】WO2004/056479
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(505232829)サソル テクノロジー (ピーティーワイ) リミテッド (4)
【Fターム(参考)】
【公表日】平成18年7月27日(2006.7.27)
【国際特許分類】
【出願日】平成15年12月19日(2003.12.19)
【国際出願番号】PCT/ZA2003/000187
【国際公開番号】WO2004/056479
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(505232829)サソル テクノロジー (ピーティーワイ) リミテッド (4)
【Fターム(参考)】
[ Back to top ]