
カビの発現誘導型プロモーター
【課題】本発明は、真菌の生育に影響しない薬剤(例えばプロベナゾール)の添加により誘導されるプロモーター、及びその利用方法を提供することを課題とする。
【解決手段】本発明者らは、上記の課題を解決するために、まず複数の薬剤についてそれぞれ育成に影響しない濃度範囲を決定し、育成に影響しない低濃度の薬剤を用いて、これらの薬剤を与えたときの遺伝子発現プロファイルを、マイクロアレイを用いて取得した。次に、選択された遺伝子のプロモーター領域に核移行型GFPを結合したポリヌクレオチドを含むベクターをいもち病菌に導入し、低濃度のプロベナゾール存在下でGFPの発現が菌糸や付着器で誘導されることを確認した。その結果、本発明者らは真菌の生育に影響しない薬剤を低濃度添加した時に、いもち病菌の遺伝子発現を特異的に制御するプロモーターを新たに見出した。
【解決手段】本発明者らは、上記の課題を解決するために、まず複数の薬剤についてそれぞれ育成に影響しない濃度範囲を決定し、育成に影響しない低濃度の薬剤を用いて、これらの薬剤を与えたときの遺伝子発現プロファイルを、マイクロアレイを用いて取得した。次に、選択された遺伝子のプロモーター領域に核移行型GFPを結合したポリヌクレオチドを含むベクターをいもち病菌に導入し、低濃度のプロベナゾール存在下でGFPの発現が菌糸や付着器で誘導されることを確認した。その結果、本発明者らは真菌の生育に影響しない薬剤を低濃度添加した時に、いもち病菌の遺伝子発現を特異的に制御するプロモーターを新たに見出した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、真菌の生育に影響しない薬剤(例えばプロベナゾール)の添加により誘導されるプロモーター、及びその利用方法に関する。
【背景技術】
【0002】
糸状菌において目的遺伝子の機能解析は、遺伝子の欠損株もしくは破壊株や変異株を作製し、その形質を解析することにより行われる。遺伝子欠損株や破壊株は栄養要求マーカー遺伝子や薬剤耐性附与遺伝子と目的遺伝子の全部もしくは一部を置換することにより作製される。そのため、目的遺伝子が菌の生育に必至な致死遺伝子であった場合には遺伝子破壊株や欠損株は取得できない。また、ある条件下での遺伝子の機能の解析には遺伝子破壊株や欠損株は利用できない。そこで、解析対象の遺伝子の本来のプロモーター部分を条件特異的に誘導可能なプロモーターに置換し、特定の条件下で遺伝子の発現のオン・オフを制御することにより遺伝子の機能を解析するといった方法がとられている。
【0003】
これまでに、糸状菌の条件特異的誘導型プロモーターとしては、アルコール添加により遺伝子発現を誘導するアルコール脱水素酵素(alcA)のプロモーター(非特許文献1)、澱粉やマルトース等で発現が誘導されるα-アミラーゼ遺伝子(amyB)のプロモーター(非特許文献2)、チアミンで発現が誘導されるThiA (非特許文献3)などのアスペルギルス属菌由来のプロモーターが知られている。しかし、これらのプロモーターは培地に栄養成分を加えるものであるため、菌によっては通常の培養に用いる培地がこれらの誘導成分を含んでおり、これらのプロモーターが条件特異的に機能したかの確認が困難となるという問題があった。
【0004】
これらのことから、菌の生育に影響を与えない薬剤の添加により、条件特異的に遺伝子発現を誘導するプロモーターが求められていた。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Waring RB, et al., Gene, 79, 119-130 (1989)
【非特許文献2】Tada S, et al., Mol. Gen. Genet., 229, 301-306 (1991)
【非特許文献3】Shoji JY, et al., FEMS Microbiol. Lett., 244, 41-46 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、このような状況に鑑みてなされたものであり、真菌の生育に影響しない薬剤(例えばプロベナゾール)の添加により誘導されるプロモーター、及びその利用方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記の課題を解決するために鋭意検討した。
本発明者らは、まず複数の薬剤についてそれぞれ育成に影響しない濃度範囲を決定し、育成に影響しない低濃度の薬剤(1〜100ppm)を用いて、これらの薬剤を与えたときの遺伝子発現プロファイルを、マイクロアレイを用いて取得した。
【0008】
いもち病菌であるマグナポルテ属菌((マグナポルテ・グリセア(Magnaporthe grisea)、マグナポルテ・オリゼ(Magnaporthe oryzae));無性世代属名 ピリキュラリア(Pyricuralia)属)をはじめとしてカビでは、遺伝子発現解析に用いることができる誘導型プロモーターが少なく、特に薬剤によってドライブされるプロモーターはほとんど知られていない。そこで、菌の育成に影響を与えない低濃度の薬剤(プロベナゾール等)を菌糸に与えた時に特異的に誘導される遺伝子のうち(表6)、付着器形成によっても誘導される遺伝子を除いた。さらに複数の薬剤添加時のマイクロアレイデータを利用して他の薬剤でも誘導される遺伝子を除き、残った遺伝子(図1)をプロベナゾール特異的に発現する遺伝子の候補とした。
【0009】
候補遺伝子のプロベナゾールによる発現をRT-PCRで確認後、プロベナゾールで誘導される遺伝子として、MGG_04304(retinol dehydrogenase 8ホモログ:RDH8)及びMGG_10913(retinol dehydrogenase 12ホモログ:RDH12)を選択した(図2)。
【0010】
次に、核移行型GFPを結合したポリヌクレオチドを含むベクター(図6)にいもち病菌のRDH8及びRDH12遺伝子のプロモーター領域を結合し、本ベクターのいもち病菌導入株を作成した結果、低濃度(10 ppm)のプロベナゾール存在下でGFPの発現が菌糸や付着器で誘導されることを確認した(図7、9:RDH8プロモーターによるGFPのプロベナゾール特異的発現を示す、図8、10:RDH12プロモーターによるGFPのプロベナゾール特異的発現を示す)。
【0011】
即ち、本発明者らは、真菌の生育に影響しない薬剤(例えばプロベナゾール)を低濃度(例えば、1ppm以上)添加した時に、いもち病菌の遺伝子発現を特異的に制御するプロモーターを新たに見出し、本発明を完成するに至った。
【0012】
より具体的には、下記〔1〕〜〔10〕の発明を提供するものである。
〔1〕下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNA。
(a)配列番号:1又は2に記載の塩基配列からなるDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
〔2〕真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有することを特徴とする、〔1〕に記載のDNA。
〔3〕前記真菌の生育に影響しない薬剤がプロベナゾールであることを特徴とする、〔1〕または〔2〕に記載のDNA。
〔4〕前記真菌の生育に影響しない薬剤を、1ppm以上添加することによりプロモーター活性が誘導されることを特徴とする、〔1〕〜〔3〕のいずれかに記載のDNA。
〔5〕〔1〕〜〔4〕のいずれかに記載のDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNA。
〔6〕〔1〕〜〔5〕のいずれかに記載のDNAを含むベクター。
〔7〕〔1〕〜〔5〕のいずれかに記載のDNA、または〔6〕に記載のベクターを含む、形質転換細胞。
〔8〕真菌である、〔7〕に記載の形質転換細胞。
〔9〕真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法。
(1)〔5〕に記載のDNA、または〔6〕に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程
〔10〕下記の工程(a)〜(c)を含む、〔1〕に記載のDNAのプロモーター活性を調節する化合物のスクリーニング方法。
(a)〔1〕に記載のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【発明の効果】
【0013】
本発明の薬剤誘導型プロモーターの下流で目的遺伝子を結合させて糸状菌に導入すれば、生育を阻害することなく、真菌の遺伝子発現を低濃度の薬剤で制御することが可能となる。
また、真菌の生育および植物体の生育に影響しない薬剤、例えばプロベナゾールは植物に吸収されるため、抵抗性を誘導しない低濃度のプロベナゾールをあらかじめ与えた植物に、プロモーターの下流でドライブされた遺伝子が組み込まれた真菌を接種することにより、植物細胞内で特異的に菌の遺伝子発現を誘導させることが可能となる。
【図面の簡単な説明】
【0014】
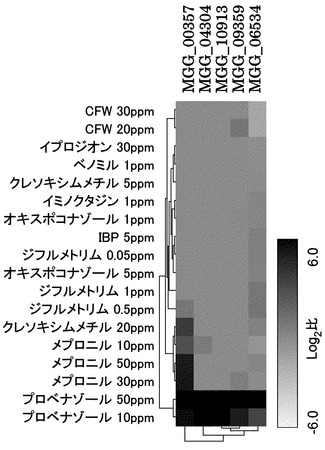
【図1】プロベナゾール50ppm処理で上昇する上位10遺伝子に含まれる5つのデヒドロゲナーゼ遺伝子における各種薬剤処理マイクロアレイ結果でのヒートマップ図を示す図面である。プロベナゾール処理でのみで、 5つのデヒドロゲナーゼ遺伝子の発現が増加している。
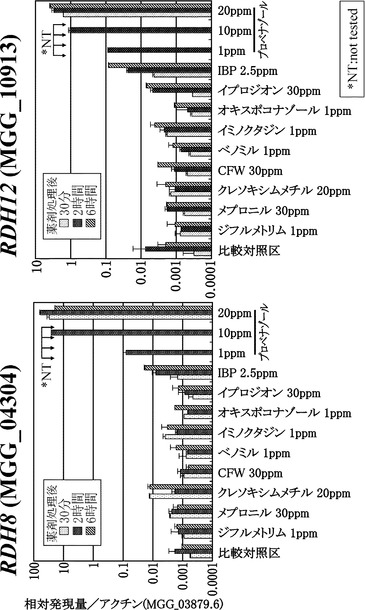
【図2】各種薬剤処理下でのRDH8及びRDH12遺伝子の発現検討結果を示す図面である。横軸は、添加処理を行う化合物の種類を示し、縦軸はアクチンに対する標的遺伝子の相対発現量を示す。
【図3】RDH8の5’末端領域の制限酵素地図を示す図である。
【図4】RDH12の5’末端領域の制限酵素地図を示す図である。
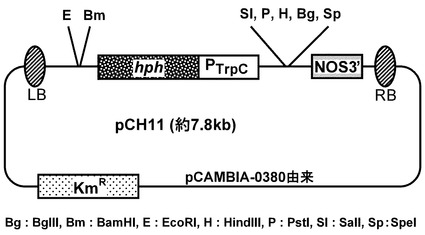
【図5】pCH11ベクターマップを示す図である。hph:ハイグロマイシンB耐性遺伝子、Km:カナマイシン耐性遺伝子、PtrpC:Aspergillus nidulans由来の糸状菌恒常的発現用トリプトファンCプロモーター、NOS3’:ターミネーター配列、RB、LB:染色体に組み込まれるT-DNAのボーダー配列を示す。ベクターマップの下に、制限酵素の種類を示す。
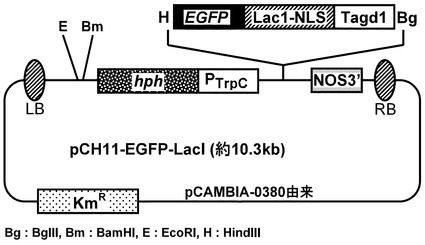
【図6】pCH11-EGFP-Laclベクターマップを示す図である。hph:ハイグロマイシンB耐性遺伝子、Km:カナマイシン耐性遺伝子、PtrpC:糸状菌恒常的発現用トリプトファンCプロモーター、EGFP :緑色蛍光タンパクenhanced green fluorescent protein、LacI:大腸菌由来核移行型ラクトースオペロン転写抑制遺伝子、TagdA:アスペルギルス オリゼΑ-グルコシダーゼ遺伝子agdA ターミネーター、NOS3’:ターミネーター配列、RB、LB:染色体に組み込まれるT-DNAのボーダー配列を示す。ベクターマップの下に、制限酵素の種類を示す。
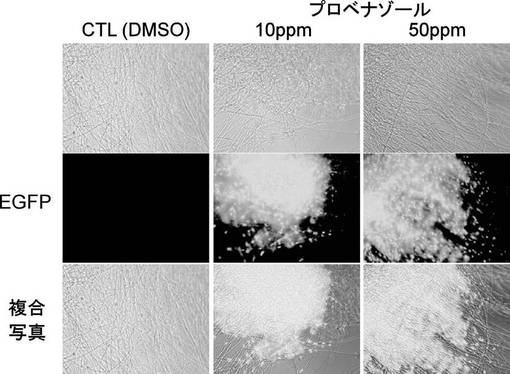
【図7】RDH8-EGFP株の栄養菌糸育成時(YG液体培地)におけるプロベナゾールによる発現誘導の蛍光レポーター観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【図8】RDH12-EGFP株の栄養菌糸育成時(YG液体培地)におけるプロベナゾールによる発現誘導の蛍光レポーター観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
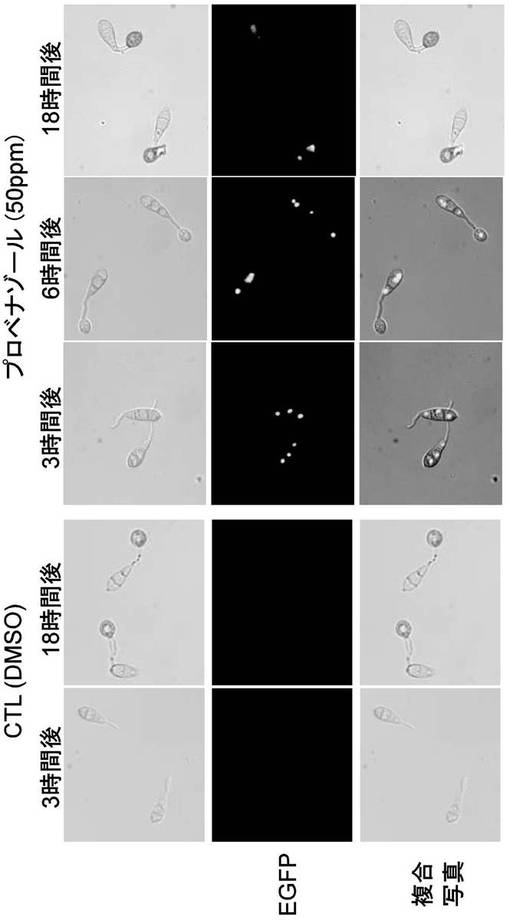
【図9】RDH8-EGFP株の感染器官形成時における蛍光観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【図10】RDH12-EGFP株の付着器形成時における蛍光観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【発明を実施するための形態】
【0015】
本発明は、真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有するDNAを提供する。本発明のDNAの好ましい態様としては、下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNAである。
(a)配列番号:1又は2に記載のプロモーター活性を有するDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
【0016】
本発明の「プロモーター活性を有するDNA(プロモーターDNA)」とは、DNAを鋳型としたmRNAの合成(転写)の開始に必要な特定塩基配列を含むDNAを意味し、自然界に存在するDNAの他、組換えなどの人工的な改変操作により作成されたDNAを含む。
【0017】
プロモーター活性は、当業者においては公知の方法(例えば、後述のレポーター遺伝子を用いて該遺伝子の発現を指標に測定する方法)によって適宜、評価することができる。
【0018】
本発明のプロモーターDNAは、配列番号:1又は2に記載の塩基配列からなるDNAだけでなく、配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなり、かつプロモーターとして作用する能力を有するDNA、または、配列番号:1又は2に記載の塩基配列において、その3'末端に翻訳効率を上げる塩基配列などを付加したものや、プロモーター活性を失うことなく、その5'末端を欠失したものを含む。当該、配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列は、配列番号:1又は2と、50、60、70、80%又は90%以上(具体的には91、92、93、94、95、96、97、98、99%以上)の配列相同性を有していることが好ましい。
【0019】
上記DNAを調製するために、当業者によりよく知られた方法としては、ハイブリダイゼーション技術(Southern, EM., J Mol Biol, 1975, 98, 503.)やポリメラーゼ連鎖反応(PCR)技術(Saiki, RK. et al., Science, 1985, 230, 1350.、Saiki, RK. et al., Science, 1988, 239, 487.)の他に、例えば、該DNAに対し、site-directed mutagenesis法(Kramer, W. & Fritz, HJ., Methods Enzymol, 1987, 154, 350.)により変異を導入する方法が挙げられる。
【0020】
本発明において欠失、置換等の変異が導入される塩基の数は、変異を導入されたDNAがプロモーター活性を有する限り、特に制限されないが、通常、20塩基対以内、好ましくは10塩基対以内、より好ましくは5塩基対以内、最も好ましくは3塩基対以内である。
【0021】
さらに、本発明のプロモーターDNAは、配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAを含む。ここで、ストリンジェントな条件とは、特に制限されるものではないが、例えば42℃、2×SSC(300mM NaCl、30mMクエン酸)、0.1%SDSの条件であり、好ましくは50℃、2×SSC 、0.1%SDSの条件であり、さらに好ましくは、65℃、0.1×SSCおよび0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。ハイブリダイゼーションのストリンジェンシーに影響する要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。当該、配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAは、配列番号:1又は2と、50、60、70、80%又は90%以上(具体的には91、92、93、94、95、96、97、98、99%以上)の配列相同性を有していることが好ましい。
【0022】
本発明において、「真菌の生育に影響しない薬剤」とは特に制限されるものではないが、好ましくは植物抵抗性誘導剤であるアシベンゾラルSメチルやチアジニルおよびイソチアニル、特に好ましくはプロベナゾールを例示することが出来る。
【0023】
本発明において、「真菌の生育に影響しない薬剤」の添加濃度は特に制限されるものではないが、1ppm以上の濃度で添加することが好ましい。ここで、「1ppm以上の濃度」とは、例えば、1ppm、5ppm、10ppm、20ppm、30ppm、50ppm、100ppmの濃度を例示することが出来る。
【0024】
さらに本発明は、本発明のプロモーター活性を有するDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNAを提供する。本発明において外来遺伝子とは、特に制限されず、所望の遺伝子を用いることができる。
【0025】
上記外来遺伝子としては、例えば、ルシフェラーゼ遺伝子、β-グルクロニダーゼ遺伝子(GUS)及び緑色蛍光遺伝子GFP、赤色蛍光遺伝子RFP、黄色蛍光遺伝子YFP等を例示することができる。
【0026】
また、本発明の上記DNAは、外来遺伝子に加えてさらにターミネーターが連結した構造であってもよい。該ターミネーターは、通常、真菌細胞内で機能するターミネーターを指し、本発明のプロモーターの近傍に配置されるDNA配列であり、例えば、アスペルギルス・ニジュランス(Aspergillus nidulans)由来のトリプトファンC(trpC)遺伝子ターミネーター、同菌由来のα-glucosidase (agdA)遺伝子ターミネーター、サッカロミセス・セルビシエ(Saccharomyces cerevisiae)由来のCYC1ターミネーター等を例示することができるが、ターミネーターとしての機能を有するものであれば、これらに特に制限されない。
【0027】
本発明において「機能的に連結」とは、本発明のプロモーターDNAの制御下にある外来遺伝子が、本発明のプロモーターDNAからの転写を受けるように、該プロモーターDNAと結合している状態を指す。プロモーター活性を有するDNAおよび外来遺伝子を「機能的に連結」させることは、当業者においては一般的な遺伝子工学技術を用いて、簡便に行い得ることである。
【0028】
また本発明は、本発明のプロモーターDNAを含むベクター、本発明のDNAの制御下に外来遺伝子が機能的に結合した構造を有するDNAを含むベクター、および、本発明のプロモーターの下流に遺伝子挿入部位を有する構造のDNAを含むベクター、並びに、上記ベクターにさらにターミネーターを担持するベクターを提供する。
【0029】
本発明のベクターは、通常、本発明のプロモーターDNAを各種細胞内で複製可能なベクターに挿入したものである。この複製可能なベクターとしては、公知の種々のベクターを用いることができる。例えば、pUC誘導体などの大腸菌で増幅可能なベクター、pPZP2H-lacなどの大腸菌とアグロバクテリウムの双方で増幅可能なシャトルベクターなどが挙げられる。なお、本発明のプロモーターDNAをベクターに挿入する方法としては、当業者に公知の方法を適用することができる。
【0030】
本発明のプロモーターDNAを導入可能な真菌としては、特に制限されないが、例えば、マグナポルテ(Magnaporthe)属、フザリウム(Fusarium)属、コレトトリカム(Colletotrichum)属、アルタナリア(Alternaria)属、コクリオボラス(Cochliobolus)属、アスペルギルス(Aspergillus)属、ボトリティス(Botrytis)属等を挙げることができる。
【0031】
また本発明は、本発明のプロモーターDNA、または該DNAを有するベクターを含む、形質転換細胞を提供する。
【0032】
本発明の真菌細胞は、本発明のDNAもしくはベクターを真菌細胞に導入したものである。真菌細胞としては、例えば、マグナポルテ属、フザリウム属、コレトトリカム属、アルタナリア属、コクリオボラス属、アスペルギルス属、及びボトリティス属等を例示することが出来る。
【0033】
本発明のDNAもしくはベクターを宿主真菌細胞中に導入するために、さまざまな手法を用いることができる。これらの手法には、形質転換因子としてアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)または、アグロバクテリウム・リゾゲネス(Agrobacterium rhizogenes)を用いたT-DNAによる真菌細胞の形質転換、プロトプラストへの直接導入(インジェクション法、エレクトロポレーション法など)、パーティクルガン法などや、その他の公知の方法が含まれる。
【0034】
プロトプラストへの直接導入では、通常、特別に必要とされるベクターはない。例えば、pUC誘導体のような単純なプラスミドを用いることができる。目的の遺伝子を真菌細胞に導入する方法によっては、他のDNA配列が必要になることもある。例えばTiまたはRiプラスミドを真菌細胞の形質転換に用いる場合には、TiおよびRiプラスミドのT-DNA領域の少なくとも右端の配列、大抵は両側の端の配列を、導入されるべき遺伝子の隣接領域となるように接続しなければならない。
【0035】
アグロバクテリウム属菌を形質転換に用いる場合には、導入すべき遺伝子を、特別のプラスミド、すなわち中間ベクターまたはバイナリーベクターの中にクローニングする必要がある。中間ベクターはアグロバクテリウム属菌の中では複製されない。中間ベクターは、ヘルパープラスミドあるいはエレクトロポレーションによってアグロバクテリウム属菌の中に移行される。中間ベクターは、T-DNAの配列と相同な領域をもつため、相同的組換えによって、アグロバクテリウム属菌のTiまたはRiプラスミド中に取り込まれる。宿主として使われるアグロバクテリウム属菌には、vir領域が含まれている必要がある。通常TiまたはRiプラスミドにvir領域が含まれており、その働きにより、T-DNAを真菌細胞に移行させることができる。
【0036】
一方、バイナリーベクターはアグロバクテリウム属菌の中で複製、維持され得るので、ヘルパープラスミドあるいはエレクトロポレーション法あるいは凍結溶解法によってアグロバクテリウム属菌中に取り込まれると、宿主のvir領域の働きによって、バイナリーベクター上のT-DNAを真菌細胞に移行させることができる。
【0037】
なお、このようにして得られた中間ベクターまたはバイナリーベクター、およびこれを含む大腸菌、アグロバクテリウム属菌、又はいもち病菌等の真菌も本発明の対象である。
【0038】
また、本発明のDNAもしくはベクターの導入によって形質転換された真菌細胞を効率的に選択するために、上記ベクターは、適当な選抜マーカー遺伝子を含む、もしくは選抜マーカー遺伝子を含むプラスミドベクターとともに真菌細胞へ導入することが好ましい。この目的に使用される選抜マーカー遺伝子は、例えば、抗生物質ハイグロマイシン耐性であるハイグロマイシンホスホトランスフェラーゼ遺伝子、ブラストサイジンS耐性遺伝子、カナマイシンまたはゲンタマイシン耐性であるネオマイシンホスホトランスフェラーゼ、および除草剤ホスフィノスリシン(ビアラホス)耐性であるアセチルトランスフェラーゼ遺伝子(bar)等を挙げることができる。
【0039】
上記ベクターを導入した真菌細胞は、導入した選抜マーカーに応じた選抜用薬剤を含む選抜用培地に置床し培養する。これにより、形質転換された真菌細胞を得ることができる。
【0040】
なお、形質転換真菌中の導入された外来DNAまたは核酸の存在は、公知のPCR法やサザンハイブリダイゼーション法によって、または真菌中の核酸の塩基配列を解析することによって確認することができる。
【0041】
本発明のDNAには、天然あるいは単離・精製されたゲノムDNA、及び化学合成DNAが含まれる。ゲノムDNAの調製は、当業者にとって常套手段を利用して行うことが可能である。
【0042】
本発明のDNAは、目的とする真菌、例えば、いもち病菌(マグナポルテ・グリセア:Magnaporthe grisea、マグナポルテ・オリゼ:Magnaporthe oryzae)の組織よりゲノムDNAを抽出し精製し、得られたDNAを鋳型としてPCRによって単離することができる。
【0043】
本発明における、配列番号:1又は2に記載の塩基配列からなるDNA、及びこれとストリンジェントな条件下でハイブリダイズするプロモーター活性を有するDNAを単離するためには、例えば、配列番号:1又は2に記載の塩基配列からなるDNA上の配列であって、本発明のプロモーターDNAを増幅するためのプライマーセットを用いることができる。このプライマーセットを用いて、真菌のゲノムDNAを鋳型としてPCRを行い、その後、得られた増幅DNA断片をプローブとして用いて、同じ真菌のゲノムライブラリーをスクリーニングすることができる。
【0044】
PCRは、市販のキットおよび装置の製造者の指針に基づいて行うか、当業者に周知の手法で行い得る。遺伝子ライブラリーの作製法、および遺伝子のクローニング法なども当業者に周知である。得られた遺伝子の塩基配列は、当該分野で公知のヌクレオチド配列解析法または市販されている自動シーケンサーを利用して決定し得る。PCR技術やハイブリダイゼーション技術によって単離し得る、配列番号:1又は2に記載の塩基配列からなるDNAとハイブリダイズするDNAもまた、本発明のDNAに含まれる。
【0045】
また本発明は、真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法を提供する。
(1)請求項5に記載のDNA、または請求項6に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程。
【0046】
さらに本発明は、下記の工程(a)〜(c)を含む、本発明のプロモーターDNAのプロモーター活性を調節する化合物のスクリーニング方法を提供する。
(a)本発明のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【0047】
本発明のスクリーニング方法に用いられる被験化合物としては、特に制限はなく、例えば、天然化合物、有機化合物、無機化合物、タンパク質、ペプチド等の単一化合物、並びに、化合物ライブラリー、遺伝子ライブラリーの発現産物、細胞抽出物、細胞培養上清、発酵真菌産生物、海洋生物抽出物、植物抽出物、原核細胞抽出物、真核単細胞抽出物もしくは動物細胞抽出物等を挙げることができる。
【0048】
本スクリーニング方法においては、まず、本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる。
【0049】
本発明において、「機能的に結合した」とは、本発明のプロモーターDNAに転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、本発明のプロモーターDNAとレポーター遺伝子とが結合していることをいう。本スクリーニング方法における「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞」として、例えば、上記DNAを含むベクターを導入した細胞を挙げることができる。該ベクターは、当業者に周知の方法により作製することができる。ベクターの細胞への導入は、一般的な方法、例えば、ポリエチレングリコール法、リン酸カルシウム沈殿法、電気パルス穿孔法、リポフェクタミン法、マイクロインジェクション法等によって実施することができる。
【0050】
また、「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞」には、染色体に該DNAが挿入された細胞も含まれる。染色体へのDNAの挿入は、当業者に一般的に用いられる方法、例えば、相同組み換えを利用した遺伝子導入法により行うことができる。
【0051】
本方法における「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞抽出液」とは、例えば、市販の試験管内転写翻訳キットに含まれる細胞抽出液に、本発明のプロモーターDNAとレポーター遺伝子とが機能的に結合した構造を有するDNAを添加したものを挙げることができる。
【0052】
本スクリーニング方法における「接触」は、本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞の培養液に被験化合物を添加する、または該DNAを含む上記の市販された細胞抽出液に被験化合物を添加することにより行うことができる。被験化合物がタンパク質の場合には、例えば、該タンパク質をコードするDNAを含むベクターを、該細胞へ導入する、または該ベクターを該細胞抽出液に添加することで行うことも可能である。
【0053】
本スクリーニング方法においては、次いで、該レポーター遺伝子の発現レベルを測定する。レポーター遺伝子の発現レベルは、当業者においては、該レポーター遺伝子の種類を考慮して、測定することができる。
【0054】
本スクリーニング方法においては、被験化合物の非存在下において測定した場合(対照)と比較して、被験化合物がレポーター遺伝子の発現レベルを変化させた場合に、被験化合物が本発明のDNAのプロモーター活性を調節する化合物であると判定される。
【0055】
さらに、本発明においては、上記スクリーニング方法を利用して、複数の被験化合物について、本発明のDNAのプロモーター活性を調節するか否かを評価し、プロモーター活性を調節する化合物を選択することにより、効率的にプロモーター活性を調節する化合物をスクリーニングすることができる。
【実施例】
【0056】
以下、実施例を用いて本発明をさらに具体的に説明する。ただし、本発明の技術的範囲はこれらの実施例に限定されるものではない。
〔実施例1〕誘導型プロモーター候補遺伝子の選抜方法
植物病原糸状菌であるいもち病菌に対して低濃度ではほとんど抗菌活性を示さない化合物プロベナゾールを用いて、当該化合物の暴露で顕著な応答性を示す候補遺伝子の選抜を以下の方法により行った。
【0057】
(1)トータルRNA抽出
まず、100mlのYG液体培地を入れた300ml容三角フラスコにいもち病菌マグナポルテ・グリセア(Magnaporthe grisea)野生株Guy11株の分生子を約2×106個分生子になるように接種し、26℃、48時間緩やかに振とう培養した。その後、表1で示した一般的な農業用薬剤で知られている各種化合物を、表記してある濃度添加し、さらに26℃で2時間培養した。本薬剤処理を行った菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0058】
【表1】

【0059】
(2)マイクロアレイ
マイクロアレイに用いるDNAチップとして、マグナポルテ・グリセアの10,000遺伝子から設計した60merのオリゴプローブをインクジェット方式でスポットした独自のマイクロアレイを使用した。抽出した薬剤処理区をtestサンプルに、薬剤無処理区を比較対象にCyanine3及びCyanine5を用いた二色法により行った。一回の実験区ごとにCyanine3とCyanine5のカラースワップを行う事により計2回の反復実験を行った。
【0060】
(3)RNAの標識(Cyanine5、Cyanine3)
RNAのCyanine3及びCyanine5標識はmRNAを用いて行い、トータルRNAからのmRNAの精製はOligotex-dT30<super> mRNA purification Kit(タカラバイオ社製)を用い、本キットに添付のマニュアルに従った。mRNAからのcDNAの作製及び標識はCyScribe cDNA Post Labelling Kit(GE helthcare)を用い、本キットに添付のマニュアルに従い行った。
【0061】
(4)ハイブリダイゼーション
Cyanine3及びCyanine5標識済みcDNA混合サンプルに、表2に示したハイブリダイゼーションバッファーを60μl加え溶解後、95℃で5分熱変性を行った。その後30分間室温で保持した後、DNAチップ上に滴下し、カバーガラスをのせた。カバーガラスをのせたDNAチップは温水上に浮かべたhumidity chamber上に置き42℃、一晩インキュベーションすることによりハイブリダイゼーションを行った。
【0062】
【表2】
【0063】
(5)ポストハイブリダイゼーション
42℃で一晩インキュベーション後、DNAチップは表3に示したウォッシュ バッファー1中でカバーガラスをはずした後、さらに新しいウォッシュバッファー1で15分、次いで表4に示したウォッシュバッファー2で5分、表5に示したウォッシュバッファー3で5分の順に振とうすることにより洗浄を行った。その後、さらにdH2O中で2分間洗浄後800×g、2分の遠心により水分を飛ばしGenePix 4000B(Axon instrument)を用いてスポットのスキャンを行った。
【0064】
【表3】
【0065】
【表4】
【0066】
【表5】
【0067】
マイクロアレイの結果、いもち病菌の生育にほとんど影響を及ぼさない化合物プロベナゾールを10ppmおよび50ppmの濃度処理したときにデヒドロゲナーゼと推定される遺伝子の増加が目立って確認された。プロベナゾールを50ppm処理したときに発現が上昇する遺伝子の上位10遺伝子の中には5つのデヒドロゲナーゼ遺伝子が含まれていた(表6)。表6中のBROAD IDナンバーとは、BROAD INSTITUTEのMagnaporthe grisea Database(http://www.broadinstitute.org/annotation/genome/magnaporthe_grisea/MultiHome.html)において、各遺伝子に付与されたナンバーである。本発明において、MGG_の頭文字から始まるナンバーは、このBROAD IDナンバーのことを示す。BROAD IDナンバーが付与された各遺伝子は、上記データベースにおいて、ゲノム上の位置、全長、転写産物、配列情報等を調べることが可能である。
【0068】
誘導プロモーターとしては、その誘導体処理によって生育阻害を示すことがなく、化合物の暴露に対して顕著な発現増加がみられること、かつ他の化合物の暴露に対しては発現の誘導されないことが望ましい。そこで、そのデヒドロゲナーゼ遺伝子の5つについてヒートマップを描きその発現誘導とその特異性を図1に示した。これを見ると、プロベナゾール処理によって発現が顕著に誘導された5つのデヒドロゲナーゼ遺伝子のほとんどがプロベナゾールでのみ発現が増加しており、プロベナゾールに対する特異性が高いことがわかる。IDナンバーMGG_00357の遺伝子はプロベナゾール以外のいくつかの化合物処理によっても遺伝子発現の増加が観察されるが、これらの化合物を処理した場合のいもち菌の菌糸生育は強く阻害を受けることから、プロベナゾールがこれらの遺伝子発現の誘導には効果的であることがわかる。そこで、これらの遺伝子をプロベナゾールによって誘導される誘導プロモーター候補とした。
【0069】
【表6】
【0070】
〔実施例2〕プロベナゾール誘導プロモーターの検証
図1に示した遺伝子が、プロベナゾールを処理したときに顕著な発現量の増加があり、かつ誘導プロモーターとして利用可能か否かの確認を行うため、MGG_04304(RDH8)及びMGG_10913(RDH12)遺伝子を例にその発現量の変化を定量RT-PCRを用いることによって検証した。
【0071】
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し26℃、48時間緩やかに振とう培養した。その後、以下の薬剤を各濃度添加し、さらに26℃で30分、2時間及び6時間培養した。薬剤処理に用いた化合物はプロベナゾールが1ppm、10ppmおよび20ppmとし、対照薬剤として呼吸鎖阻害剤としてジフルメトリム1ppm、メプロニル30ppm、クレソキシムメチル20ppm、細胞壁合成阻害剤としてCFW(カルコフロワーホワイト)30ppm、微小管生合成阻害剤としてベノミル1ppm、細胞膜機能阻害剤としてイミノクタジン1ppm、MAPキナーゼシグナリングに作用すると考えられるイプロジオン30ppm、エルゴステロール生合成阻害剤オキスポコナゾール・フマル酸塩1ppm、リン脂質合成阻害剤IBP(イプロベンフォス)2.5ppmを用いた。本薬剤処理により得た菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌、脱水後、乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0072】
(2)cDNAの合成
cDNAの合成には、トータルRNAを用いた。260 nmにおける吸光度からの計算値で5μg量のトータルRNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え100μlとした。これにRNase free DNase Iを5unit加え、37℃で1時間反応させ混在するゲノムDNAの分解を行った。その後、ExScript RT reagent Kit(タカラバイオ社製)を用いて添付マニュアルに従って逆転写反応を行い、cDNAを得た。
【0073】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit (Finnzymes社製)を用いた。スキャナー解析はDNA Engine OPTICON2(MJ Research社製)を、データ解析には解析ソフトOpticon Monitor2 ver.2.02(MJ Research社製)を用いた。反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40 サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でDissociation Curveの作成を行った。PCRは各cDNA、各標的遺伝子につき三連で行った。
【0074】
各化合物処理cDNAサンプルを用いてMGG_04304(RDH8)及びMGG_10913 (RDH12) 遺伝子の発現を経時的に調べた。またコントロールとしてActinタンパク質をコードしていると推定される遺伝子の転写量を調べた。それぞれ以下の特異的プライマーを用いた。
Actin-F:ATGCCATCGGAAAGACAGAC(配列番号:3)
Actin-R:CAGGAGTCGATCTCCAAAGC(配列番号:4)
RDH8_RTF:GGGCCATGATTGAGATAGTCAC(配列番号:5)
RDH8_RTR:GACAGCTCCTTGAACTCTTCCA(配列番号:6)
RDH12_RTF:AAGGAGAGCTGGCCCATATTAG(配列番号:7)
RDH12_RTR:GGTGGTAAAACTCGCCACTAAC(配列番号:8)
【0075】
PCR終了後、専用の解析ソフトOpticon Monitor2 ver.2.02(MJ Research社)を用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a) ΔCT値の算出(各cDNAの標的遺伝子とγ-actinとの差)
ΔCT = (y-x)
ここでxはCT (γ-actin)であり、yはCT(Target gene)である
b) ΔCT値の算出(あるcDNAのΔCTと基準とするcDNAのΔCTとの差)
ΔCT =ΔCT (C) -ΔCT (S)
ここで、CはComparative cDNAであり、SはStandard cDNAである
c) 相対発現量の算出
2-(ΔCT)
【0076】
RT-PCRの結果をMGG_04304(RDH8)については図2の左側に、MGG_10913 (RDH12)遺伝子については図2の右側にactin当たりの発現量をグラフ化したもので示した。いもち菌 野生株をプロベナゾールに曝すと、MGG_04304(RDH8)及びMGG_10913 (RDH12) 遺伝子の転写量は顕著に増加しており、MGG_04304及びMGG_10913は共に1ppm処理で、actin比で約0.1、無処理対照区(CTL)と比較して約100倍の発現増加を示した。また、処理濃度を濃くするにつれ発現量は増加し、20ppmでは処理時間に関わらずMGG_04304ではactin比で約80、無処理対照区(CTL)と比較して約8×104倍の増加を、MGG_10913の場合はactin比で約6、無処理対照区(CTL)と比較して約6×103倍の顕著な増加を示した。
【0077】
一方、プロベナゾール以外の化合物処理の場合はIBP(イプロベンフォス)、フルジオキソニル、プロシミドンなどで発現の誘導が見られたがその発現増加率はプロベナゾール1ppm処理の場合よりも低く、またこれらの化合物が菌糸生育阻害を示すことを考えるとMGG_04304(RDH8)及びMGG_10913 (RDH12)遺伝子がプロベナゾールでの遺伝子発現誘導に有効であることが確認された。
【0078】
〔実施例3〕誘導プロモーターによる遺伝子誘導のEGFPレポーターによる検出
プロベナゾールを暴露した時に顕著な遺伝子の発現の増加を示すMGG_04304(RDH8)及びMGG_10913 (RDH12)を例にして、本遺伝子の5’-UTR(5‘上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによってプロベナゾール処理による転写量の増加を可視化する菌株を作成した例を示す。
【0079】
(1)MGG_04304(RDH8)及びMGG_10913 (RDH12) 5'領域のクローニング
Broad Institute のマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からAOX 遺伝子の5'領域の塩基配列情報を取得し、その配列情報を参考にRDH8遺伝子のプライマーペアとして(5'- ATAAGGTGCGCGCAAAGGT -3'、配列番号:9、5'- CGCAAGCTTAGTTTGGATAGTCGTATGCA -3’、配列番号:10)をRDH12遺伝子のプライマーペアとして(5'- CTATCCCTTTAATTAACCGACC -3'、配列番号:11、5'- CGCAAGCTTTGGCAAGCTATCTTGTTT -3'、配列番号:12)のそれぞれ一組のオリゴヌクレオチドを設計した。なおプライマー内で、下線で示される塩基はHindIIIの配列を示す。これらのプライマーを用いてマグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は、PCR Thermal Cycler PERSONAL (タカラバイオ社製)を用いた。
【0080】
このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところRDH8は約1,200塩基対、RDH12は約1,500塩基対のDNA増幅が見られた。この増幅断片をアガロースゲル電気泳動に供したゲル中よりGENECLEAN IIIキット(Qbiogene社製)を用いてDNAを抽出し、これを挿入DNA断片とした。この挿入DNA断片をpGEM-T Easy Vector System(Promega社製)を用いてpGEM-T Easy Vectorに連結させ、連結DNA溶液を得た。連結DNA溶液10μlを氷中でよく冷却した後、氷上解凍したコンピテントセルJM109を100μl加え穏やかに撹拌し、氷中で20分間、続いて42℃で45秒ヒートショック処理を行った。これに、400μlのLB液体培地を加え、37℃で1時間培養後、100μg/mlのアンピシリンを添加したLB平板培地にまき、37℃で一晩培養した。目的のプラスミドDNAを用いて形質転換した大腸菌の単一のコロニーを3mlの100μg/mlのアンピシリンを添加したLB液体培地に植菌し、37℃で一晩振盪培養した。1.5mlの培養液を1.5mlのエッペンドルフチューブに移して15,000×gで1分間遠心分離し、沈殿を100μlの氷冷したTEG(25mM Tris-HCl、10mM EDTA、50mM Glucose、pH8.0)で懸濁し、これに200μlの0.2N NaOH-1%SDSを加えて穏やかに撹拌した後、150μlの3M NaOAc(pH5.2)を加えて混合した。これを15,000×g、4℃で5分間遠心分離し上清を回収し450μlのフェノール・クロロホルム・イソアミルアルコール(25:24:1)を加えて激しく撹拌した後、15,000×g、室温で5分間遠心分離し上層を回収した。この溶液に-20℃で氷冷した900μlのエタノールを加えて-20℃で10分間放置後、4℃にて15,000×gで5分間遠心分離した。沈殿を500μlの70%エタノールでリンスしたのち、乾燥させ、最後にRNase(100μg/ml)を含むTE(10mM Tris-HCl、1mM EDTA、pH8.0)50μlに溶解した。
【0081】
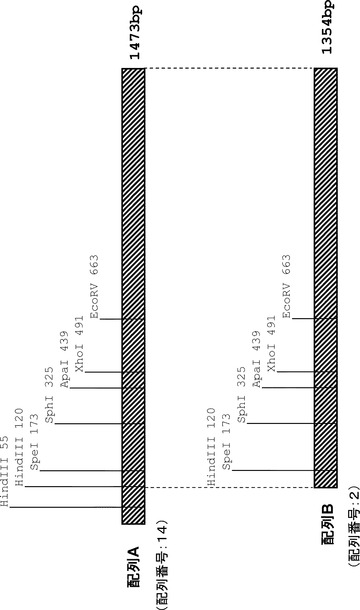
得られたプラスミド中の挿入DNA断片を添付のプロトコールに従い、ABI PRISMTM 377 DNA sequencing system (PE Biosystem社製)にて解析した。その結果、RDH8は1,218塩基対(配列番号:13、図3)、RDH12は1,473塩基対(配列番号:14、図4)からなることが明らかとなった。
また、本ベクターをpGEM-PrRDH8およびpGEM-PrRDH12とする。
これらのベクター中の挿入DNA断片中にはRDH8およびRDH12ともにRDH8のプライマー(配列番号:10)もしくはRDH12のプライマー(配列番号:12)に付加したHindIIIサイトとは別のHindIIIサイトを有していることから、このHindIIIサイトとRDH8のプライマー(配列番号:10)に付加してあるHindIIIサイトで切り出される1,152塩基対の配列(配列番号:1)をRDH8プロモーター領域とし、HindIIIサイトとRDH12のプライマー(配列番号:12)に付加してあるHindIIIサイトで切り出される1,354塩基対の配列(配列番号:2)をRDH12プロモーター領域とした。
【0082】
(2)EGFPベクターの作成
pCAMBIA-0380バイナリーベクター(CAMBIAより購入)を骨格にし、薬剤選択マーカーとしてハイグロマイシンB耐性遺伝子Hphを融合することにより作成した糸状菌形質転換ベクターpCH11(図5)をHindIII及びBglIIで消化し、そこにEGFP-LacI-TagdA (p3'SS d EGFPと大腸菌LacI:Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びAspergillus oryzae Α-グルコシダーゼ遺伝子agdA ターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)のHindIII-BglII断片を組み合すことによりEGFPベクター、pCH11-EGFP-LacIを作成した(図6)。
【0083】
(3)RDH8-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図6)プラスミドをHindIIIで消化し、そこにpGEM-PrRDH8をHindIII処理することにより得たMGG_04304(RDH8)遺伝子5'領域(配列番号:1)を定法により融合した。本レポーターアッセイプラスミドであるRDH8-EGFP-LacIレポーターベクターをpCH11-PrRDH8-EGFP-LacIと命名した。
【0084】
(4)RDH12-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図6)プラスミドをHindIIIで消化し、そこにpGEM-PrRDH12をHindIII処理することにより得たMGG_10913(RDH12)遺伝子5'領域(配列番号:2)を定法により融合した。本レポーターアッセイプラスミドであるRDH8-EGFP-LacIレポーターベクターをpCH11-PrRDH12-EGFP-LacIと命名した。
【0085】
(5)pCH11-PrRDH8-EGFP-LacIおよびpCH11-PrRDH12-EGFP-LacIプラスミドを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はアグロバクテリウム法(Gento Tsuji, Satoshi Fujii, Naoki Fujihara, Chika Hirose, Seiji Tsuge, Tomonori Shiraishi and Yasuyuki Kubo(2003):Agrobacterium tumefaciens-mediated transformation for random insertional mutagenesis in Colletorichum lagenarium. Journal of General Plant Pathology 69:230-239)を改良した方法を用いた。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。これらのベクターの挿入によって得られた形質転換株はpCH11-PrRDH8-EGFP-LacIの挿入によるものはRDH8-EGFP株、pCH11-PrRDH12-EGFP-LacIの挿入によるものはRDH12-EGFP株と命名した。
【0086】
(6)レポーター作動の確認
レポーター作動の確認は、栄養菌糸生育時および感染器官形成時の2つのステージで生育させたいもち病菌をプロベナゾールで処理した時のEGFPの蛍光を観察することによって行った。
a)栄養菌糸生育時でのレポーター動作確認の場合はマグナポルテ・グリセアRDH8-EGFP株およびRDH12-EGFP株の分生子を2×106個分生子/100mlになるように、100mlのYG液体培地を入れた300ml容坂口フラスコに接種し次の条件で培養した。
(条件1) 26℃、24時間培養
(条件2) 26℃、24時間培養した後、プロベナゾールを添加し、さらに26℃で6時間培養。
その後、各処理菌株の蛍光を、蛍光顕微鏡を用いて観察した。
b)感染器官形成時でのポーター動作確認の場合はマグナポルテ・グリセアRDH8-EGFP株およびRDH12-EGFP株の分生子1×105個/mlにプロベナゾールを添加した後、その分生子懸濁液40μlをカバーガラス上に滴下し、経時的に各処理菌株のEGFP蛍光を蛍光顕微鏡を用いて観察した。
【0087】
(7)EGFP蛍光観察
スライドガラスに培養後の栄養菌糸生育時および感染器官形成時の菌糸体をのせEGFPの蛍光観察を行った。いずれのステージにおいてもプロベナゾール処理においてのみ、核に局在した強いEGFPの蛍光が観察された(図7〜10)。以上の結果から、RDH8およびRDH12遺伝子のプロモーターはその下流の遺伝子を栄養菌糸生育時や感染器官形成時といった様々な生育ステージにおいても、プロベナゾール処理によって発現の誘導を制御することができることがわかった。また、そのRDH8およびRDH12遺伝子のプロモーターを用いたプロベナゾールによる発現制御は、今回のEGFP遺伝子のような他の遺伝子でRDH遺伝子を置き換えた場合もその誘導活性を維持しており、誘導プロモーターとして十分に利用可能であることが確認できた。
【技術分野】
【0001】
本発明は、真菌の生育に影響しない薬剤(例えばプロベナゾール)の添加により誘導されるプロモーター、及びその利用方法に関する。
【背景技術】
【0002】
糸状菌において目的遺伝子の機能解析は、遺伝子の欠損株もしくは破壊株や変異株を作製し、その形質を解析することにより行われる。遺伝子欠損株や破壊株は栄養要求マーカー遺伝子や薬剤耐性附与遺伝子と目的遺伝子の全部もしくは一部を置換することにより作製される。そのため、目的遺伝子が菌の生育に必至な致死遺伝子であった場合には遺伝子破壊株や欠損株は取得できない。また、ある条件下での遺伝子の機能の解析には遺伝子破壊株や欠損株は利用できない。そこで、解析対象の遺伝子の本来のプロモーター部分を条件特異的に誘導可能なプロモーターに置換し、特定の条件下で遺伝子の発現のオン・オフを制御することにより遺伝子の機能を解析するといった方法がとられている。
【0003】
これまでに、糸状菌の条件特異的誘導型プロモーターとしては、アルコール添加により遺伝子発現を誘導するアルコール脱水素酵素(alcA)のプロモーター(非特許文献1)、澱粉やマルトース等で発現が誘導されるα-アミラーゼ遺伝子(amyB)のプロモーター(非特許文献2)、チアミンで発現が誘導されるThiA (非特許文献3)などのアスペルギルス属菌由来のプロモーターが知られている。しかし、これらのプロモーターは培地に栄養成分を加えるものであるため、菌によっては通常の培養に用いる培地がこれらの誘導成分を含んでおり、これらのプロモーターが条件特異的に機能したかの確認が困難となるという問題があった。
【0004】
これらのことから、菌の生育に影響を与えない薬剤の添加により、条件特異的に遺伝子発現を誘導するプロモーターが求められていた。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Waring RB, et al., Gene, 79, 119-130 (1989)
【非特許文献2】Tada S, et al., Mol. Gen. Genet., 229, 301-306 (1991)
【非特許文献3】Shoji JY, et al., FEMS Microbiol. Lett., 244, 41-46 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、このような状況に鑑みてなされたものであり、真菌の生育に影響しない薬剤(例えばプロベナゾール)の添加により誘導されるプロモーター、及びその利用方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記の課題を解決するために鋭意検討した。
本発明者らは、まず複数の薬剤についてそれぞれ育成に影響しない濃度範囲を決定し、育成に影響しない低濃度の薬剤(1〜100ppm)を用いて、これらの薬剤を与えたときの遺伝子発現プロファイルを、マイクロアレイを用いて取得した。
【0008】
いもち病菌であるマグナポルテ属菌((マグナポルテ・グリセア(Magnaporthe grisea)、マグナポルテ・オリゼ(Magnaporthe oryzae));無性世代属名 ピリキュラリア(Pyricuralia)属)をはじめとしてカビでは、遺伝子発現解析に用いることができる誘導型プロモーターが少なく、特に薬剤によってドライブされるプロモーターはほとんど知られていない。そこで、菌の育成に影響を与えない低濃度の薬剤(プロベナゾール等)を菌糸に与えた時に特異的に誘導される遺伝子のうち(表6)、付着器形成によっても誘導される遺伝子を除いた。さらに複数の薬剤添加時のマイクロアレイデータを利用して他の薬剤でも誘導される遺伝子を除き、残った遺伝子(図1)をプロベナゾール特異的に発現する遺伝子の候補とした。
【0009】
候補遺伝子のプロベナゾールによる発現をRT-PCRで確認後、プロベナゾールで誘導される遺伝子として、MGG_04304(retinol dehydrogenase 8ホモログ:RDH8)及びMGG_10913(retinol dehydrogenase 12ホモログ:RDH12)を選択した(図2)。
【0010】
次に、核移行型GFPを結合したポリヌクレオチドを含むベクター(図6)にいもち病菌のRDH8及びRDH12遺伝子のプロモーター領域を結合し、本ベクターのいもち病菌導入株を作成した結果、低濃度(10 ppm)のプロベナゾール存在下でGFPの発現が菌糸や付着器で誘導されることを確認した(図7、9:RDH8プロモーターによるGFPのプロベナゾール特異的発現を示す、図8、10:RDH12プロモーターによるGFPのプロベナゾール特異的発現を示す)。
【0011】
即ち、本発明者らは、真菌の生育に影響しない薬剤(例えばプロベナゾール)を低濃度(例えば、1ppm以上)添加した時に、いもち病菌の遺伝子発現を特異的に制御するプロモーターを新たに見出し、本発明を完成するに至った。
【0012】
より具体的には、下記〔1〕〜〔10〕の発明を提供するものである。
〔1〕下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNA。
(a)配列番号:1又は2に記載の塩基配列からなるDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
〔2〕真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有することを特徴とする、〔1〕に記載のDNA。
〔3〕前記真菌の生育に影響しない薬剤がプロベナゾールであることを特徴とする、〔1〕または〔2〕に記載のDNA。
〔4〕前記真菌の生育に影響しない薬剤を、1ppm以上添加することによりプロモーター活性が誘導されることを特徴とする、〔1〕〜〔3〕のいずれかに記載のDNA。
〔5〕〔1〕〜〔4〕のいずれかに記載のDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNA。
〔6〕〔1〕〜〔5〕のいずれかに記載のDNAを含むベクター。
〔7〕〔1〕〜〔5〕のいずれかに記載のDNA、または〔6〕に記載のベクターを含む、形質転換細胞。
〔8〕真菌である、〔7〕に記載の形質転換細胞。
〔9〕真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法。
(1)〔5〕に記載のDNA、または〔6〕に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程
〔10〕下記の工程(a)〜(c)を含む、〔1〕に記載のDNAのプロモーター活性を調節する化合物のスクリーニング方法。
(a)〔1〕に記載のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【発明の効果】
【0013】
本発明の薬剤誘導型プロモーターの下流で目的遺伝子を結合させて糸状菌に導入すれば、生育を阻害することなく、真菌の遺伝子発現を低濃度の薬剤で制御することが可能となる。
また、真菌の生育および植物体の生育に影響しない薬剤、例えばプロベナゾールは植物に吸収されるため、抵抗性を誘導しない低濃度のプロベナゾールをあらかじめ与えた植物に、プロモーターの下流でドライブされた遺伝子が組み込まれた真菌を接種することにより、植物細胞内で特異的に菌の遺伝子発現を誘導させることが可能となる。
【図面の簡単な説明】
【0014】
【図1】プロベナゾール50ppm処理で上昇する上位10遺伝子に含まれる5つのデヒドロゲナーゼ遺伝子における各種薬剤処理マイクロアレイ結果でのヒートマップ図を示す図面である。プロベナゾール処理でのみで、 5つのデヒドロゲナーゼ遺伝子の発現が増加している。
【図2】各種薬剤処理下でのRDH8及びRDH12遺伝子の発現検討結果を示す図面である。横軸は、添加処理を行う化合物の種類を示し、縦軸はアクチンに対する標的遺伝子の相対発現量を示す。
【図3】RDH8の5’末端領域の制限酵素地図を示す図である。
【図4】RDH12の5’末端領域の制限酵素地図を示す図である。
【図5】pCH11ベクターマップを示す図である。hph:ハイグロマイシンB耐性遺伝子、Km:カナマイシン耐性遺伝子、PtrpC:Aspergillus nidulans由来の糸状菌恒常的発現用トリプトファンCプロモーター、NOS3’:ターミネーター配列、RB、LB:染色体に組み込まれるT-DNAのボーダー配列を示す。ベクターマップの下に、制限酵素の種類を示す。
【図6】pCH11-EGFP-Laclベクターマップを示す図である。hph:ハイグロマイシンB耐性遺伝子、Km:カナマイシン耐性遺伝子、PtrpC:糸状菌恒常的発現用トリプトファンCプロモーター、EGFP :緑色蛍光タンパクenhanced green fluorescent protein、LacI:大腸菌由来核移行型ラクトースオペロン転写抑制遺伝子、TagdA:アスペルギルス オリゼΑ-グルコシダーゼ遺伝子agdA ターミネーター、NOS3’:ターミネーター配列、RB、LB:染色体に組み込まれるT-DNAのボーダー配列を示す。ベクターマップの下に、制限酵素の種類を示す。
【図7】RDH8-EGFP株の栄養菌糸育成時(YG液体培地)におけるプロベナゾールによる発現誘導の蛍光レポーター観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【図8】RDH12-EGFP株の栄養菌糸育成時(YG液体培地)におけるプロベナゾールによる発現誘導の蛍光レポーター観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【図9】RDH8-EGFP株の感染器官形成時における蛍光観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【図10】RDH12-EGFP株の付着器形成時における蛍光観察の結果を示す写真である。上段の写真は、光学顕微鏡による菌糸の観察結果を示す。中段の写真は、蛍光顕微鏡によるEGFP発現確認の結果を示す。下段の写真は、上段と中段を複合させた写真である。
【発明を実施するための形態】
【0015】
本発明は、真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有するDNAを提供する。本発明のDNAの好ましい態様としては、下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNAである。
(a)配列番号:1又は2に記載のプロモーター活性を有するDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
【0016】
本発明の「プロモーター活性を有するDNA(プロモーターDNA)」とは、DNAを鋳型としたmRNAの合成(転写)の開始に必要な特定塩基配列を含むDNAを意味し、自然界に存在するDNAの他、組換えなどの人工的な改変操作により作成されたDNAを含む。
【0017】
プロモーター活性は、当業者においては公知の方法(例えば、後述のレポーター遺伝子を用いて該遺伝子の発現を指標に測定する方法)によって適宜、評価することができる。
【0018】
本発明のプロモーターDNAは、配列番号:1又は2に記載の塩基配列からなるDNAだけでなく、配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなり、かつプロモーターとして作用する能力を有するDNA、または、配列番号:1又は2に記載の塩基配列において、その3'末端に翻訳効率を上げる塩基配列などを付加したものや、プロモーター活性を失うことなく、その5'末端を欠失したものを含む。当該、配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列は、配列番号:1又は2と、50、60、70、80%又は90%以上(具体的には91、92、93、94、95、96、97、98、99%以上)の配列相同性を有していることが好ましい。
【0019】
上記DNAを調製するために、当業者によりよく知られた方法としては、ハイブリダイゼーション技術(Southern, EM., J Mol Biol, 1975, 98, 503.)やポリメラーゼ連鎖反応(PCR)技術(Saiki, RK. et al., Science, 1985, 230, 1350.、Saiki, RK. et al., Science, 1988, 239, 487.)の他に、例えば、該DNAに対し、site-directed mutagenesis法(Kramer, W. & Fritz, HJ., Methods Enzymol, 1987, 154, 350.)により変異を導入する方法が挙げられる。
【0020】
本発明において欠失、置換等の変異が導入される塩基の数は、変異を導入されたDNAがプロモーター活性を有する限り、特に制限されないが、通常、20塩基対以内、好ましくは10塩基対以内、より好ましくは5塩基対以内、最も好ましくは3塩基対以内である。
【0021】
さらに、本発明のプロモーターDNAは、配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAを含む。ここで、ストリンジェントな条件とは、特に制限されるものではないが、例えば42℃、2×SSC(300mM NaCl、30mMクエン酸)、0.1%SDSの条件であり、好ましくは50℃、2×SSC 、0.1%SDSの条件であり、さらに好ましくは、65℃、0.1×SSCおよび0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。ハイブリダイゼーションのストリンジェンシーに影響する要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。当該、配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAは、配列番号:1又は2と、50、60、70、80%又は90%以上(具体的には91、92、93、94、95、96、97、98、99%以上)の配列相同性を有していることが好ましい。
【0022】
本発明において、「真菌の生育に影響しない薬剤」とは特に制限されるものではないが、好ましくは植物抵抗性誘導剤であるアシベンゾラルSメチルやチアジニルおよびイソチアニル、特に好ましくはプロベナゾールを例示することが出来る。
【0023】
本発明において、「真菌の生育に影響しない薬剤」の添加濃度は特に制限されるものではないが、1ppm以上の濃度で添加することが好ましい。ここで、「1ppm以上の濃度」とは、例えば、1ppm、5ppm、10ppm、20ppm、30ppm、50ppm、100ppmの濃度を例示することが出来る。
【0024】
さらに本発明は、本発明のプロモーター活性を有するDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNAを提供する。本発明において外来遺伝子とは、特に制限されず、所望の遺伝子を用いることができる。
【0025】
上記外来遺伝子としては、例えば、ルシフェラーゼ遺伝子、β-グルクロニダーゼ遺伝子(GUS)及び緑色蛍光遺伝子GFP、赤色蛍光遺伝子RFP、黄色蛍光遺伝子YFP等を例示することができる。
【0026】
また、本発明の上記DNAは、外来遺伝子に加えてさらにターミネーターが連結した構造であってもよい。該ターミネーターは、通常、真菌細胞内で機能するターミネーターを指し、本発明のプロモーターの近傍に配置されるDNA配列であり、例えば、アスペルギルス・ニジュランス(Aspergillus nidulans)由来のトリプトファンC(trpC)遺伝子ターミネーター、同菌由来のα-glucosidase (agdA)遺伝子ターミネーター、サッカロミセス・セルビシエ(Saccharomyces cerevisiae)由来のCYC1ターミネーター等を例示することができるが、ターミネーターとしての機能を有するものであれば、これらに特に制限されない。
【0027】
本発明において「機能的に連結」とは、本発明のプロモーターDNAの制御下にある外来遺伝子が、本発明のプロモーターDNAからの転写を受けるように、該プロモーターDNAと結合している状態を指す。プロモーター活性を有するDNAおよび外来遺伝子を「機能的に連結」させることは、当業者においては一般的な遺伝子工学技術を用いて、簡便に行い得ることである。
【0028】
また本発明は、本発明のプロモーターDNAを含むベクター、本発明のDNAの制御下に外来遺伝子が機能的に結合した構造を有するDNAを含むベクター、および、本発明のプロモーターの下流に遺伝子挿入部位を有する構造のDNAを含むベクター、並びに、上記ベクターにさらにターミネーターを担持するベクターを提供する。
【0029】
本発明のベクターは、通常、本発明のプロモーターDNAを各種細胞内で複製可能なベクターに挿入したものである。この複製可能なベクターとしては、公知の種々のベクターを用いることができる。例えば、pUC誘導体などの大腸菌で増幅可能なベクター、pPZP2H-lacなどの大腸菌とアグロバクテリウムの双方で増幅可能なシャトルベクターなどが挙げられる。なお、本発明のプロモーターDNAをベクターに挿入する方法としては、当業者に公知の方法を適用することができる。
【0030】
本発明のプロモーターDNAを導入可能な真菌としては、特に制限されないが、例えば、マグナポルテ(Magnaporthe)属、フザリウム(Fusarium)属、コレトトリカム(Colletotrichum)属、アルタナリア(Alternaria)属、コクリオボラス(Cochliobolus)属、アスペルギルス(Aspergillus)属、ボトリティス(Botrytis)属等を挙げることができる。
【0031】
また本発明は、本発明のプロモーターDNA、または該DNAを有するベクターを含む、形質転換細胞を提供する。
【0032】
本発明の真菌細胞は、本発明のDNAもしくはベクターを真菌細胞に導入したものである。真菌細胞としては、例えば、マグナポルテ属、フザリウム属、コレトトリカム属、アルタナリア属、コクリオボラス属、アスペルギルス属、及びボトリティス属等を例示することが出来る。
【0033】
本発明のDNAもしくはベクターを宿主真菌細胞中に導入するために、さまざまな手法を用いることができる。これらの手法には、形質転換因子としてアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)または、アグロバクテリウム・リゾゲネス(Agrobacterium rhizogenes)を用いたT-DNAによる真菌細胞の形質転換、プロトプラストへの直接導入(インジェクション法、エレクトロポレーション法など)、パーティクルガン法などや、その他の公知の方法が含まれる。
【0034】
プロトプラストへの直接導入では、通常、特別に必要とされるベクターはない。例えば、pUC誘導体のような単純なプラスミドを用いることができる。目的の遺伝子を真菌細胞に導入する方法によっては、他のDNA配列が必要になることもある。例えばTiまたはRiプラスミドを真菌細胞の形質転換に用いる場合には、TiおよびRiプラスミドのT-DNA領域の少なくとも右端の配列、大抵は両側の端の配列を、導入されるべき遺伝子の隣接領域となるように接続しなければならない。
【0035】
アグロバクテリウム属菌を形質転換に用いる場合には、導入すべき遺伝子を、特別のプラスミド、すなわち中間ベクターまたはバイナリーベクターの中にクローニングする必要がある。中間ベクターはアグロバクテリウム属菌の中では複製されない。中間ベクターは、ヘルパープラスミドあるいはエレクトロポレーションによってアグロバクテリウム属菌の中に移行される。中間ベクターは、T-DNAの配列と相同な領域をもつため、相同的組換えによって、アグロバクテリウム属菌のTiまたはRiプラスミド中に取り込まれる。宿主として使われるアグロバクテリウム属菌には、vir領域が含まれている必要がある。通常TiまたはRiプラスミドにvir領域が含まれており、その働きにより、T-DNAを真菌細胞に移行させることができる。
【0036】
一方、バイナリーベクターはアグロバクテリウム属菌の中で複製、維持され得るので、ヘルパープラスミドあるいはエレクトロポレーション法あるいは凍結溶解法によってアグロバクテリウム属菌中に取り込まれると、宿主のvir領域の働きによって、バイナリーベクター上のT-DNAを真菌細胞に移行させることができる。
【0037】
なお、このようにして得られた中間ベクターまたはバイナリーベクター、およびこれを含む大腸菌、アグロバクテリウム属菌、又はいもち病菌等の真菌も本発明の対象である。
【0038】
また、本発明のDNAもしくはベクターの導入によって形質転換された真菌細胞を効率的に選択するために、上記ベクターは、適当な選抜マーカー遺伝子を含む、もしくは選抜マーカー遺伝子を含むプラスミドベクターとともに真菌細胞へ導入することが好ましい。この目的に使用される選抜マーカー遺伝子は、例えば、抗生物質ハイグロマイシン耐性であるハイグロマイシンホスホトランスフェラーゼ遺伝子、ブラストサイジンS耐性遺伝子、カナマイシンまたはゲンタマイシン耐性であるネオマイシンホスホトランスフェラーゼ、および除草剤ホスフィノスリシン(ビアラホス)耐性であるアセチルトランスフェラーゼ遺伝子(bar)等を挙げることができる。
【0039】
上記ベクターを導入した真菌細胞は、導入した選抜マーカーに応じた選抜用薬剤を含む選抜用培地に置床し培養する。これにより、形質転換された真菌細胞を得ることができる。
【0040】
なお、形質転換真菌中の導入された外来DNAまたは核酸の存在は、公知のPCR法やサザンハイブリダイゼーション法によって、または真菌中の核酸の塩基配列を解析することによって確認することができる。
【0041】
本発明のDNAには、天然あるいは単離・精製されたゲノムDNA、及び化学合成DNAが含まれる。ゲノムDNAの調製は、当業者にとって常套手段を利用して行うことが可能である。
【0042】
本発明のDNAは、目的とする真菌、例えば、いもち病菌(マグナポルテ・グリセア:Magnaporthe grisea、マグナポルテ・オリゼ:Magnaporthe oryzae)の組織よりゲノムDNAを抽出し精製し、得られたDNAを鋳型としてPCRによって単離することができる。
【0043】
本発明における、配列番号:1又は2に記載の塩基配列からなるDNA、及びこれとストリンジェントな条件下でハイブリダイズするプロモーター活性を有するDNAを単離するためには、例えば、配列番号:1又は2に記載の塩基配列からなるDNA上の配列であって、本発明のプロモーターDNAを増幅するためのプライマーセットを用いることができる。このプライマーセットを用いて、真菌のゲノムDNAを鋳型としてPCRを行い、その後、得られた増幅DNA断片をプローブとして用いて、同じ真菌のゲノムライブラリーをスクリーニングすることができる。
【0044】
PCRは、市販のキットおよび装置の製造者の指針に基づいて行うか、当業者に周知の手法で行い得る。遺伝子ライブラリーの作製法、および遺伝子のクローニング法なども当業者に周知である。得られた遺伝子の塩基配列は、当該分野で公知のヌクレオチド配列解析法または市販されている自動シーケンサーを利用して決定し得る。PCR技術やハイブリダイゼーション技術によって単離し得る、配列番号:1又は2に記載の塩基配列からなるDNAとハイブリダイズするDNAもまた、本発明のDNAに含まれる。
【0045】
また本発明は、真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法を提供する。
(1)請求項5に記載のDNA、または請求項6に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程。
【0046】
さらに本発明は、下記の工程(a)〜(c)を含む、本発明のプロモーターDNAのプロモーター活性を調節する化合物のスクリーニング方法を提供する。
(a)本発明のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【0047】
本発明のスクリーニング方法に用いられる被験化合物としては、特に制限はなく、例えば、天然化合物、有機化合物、無機化合物、タンパク質、ペプチド等の単一化合物、並びに、化合物ライブラリー、遺伝子ライブラリーの発現産物、細胞抽出物、細胞培養上清、発酵真菌産生物、海洋生物抽出物、植物抽出物、原核細胞抽出物、真核単細胞抽出物もしくは動物細胞抽出物等を挙げることができる。
【0048】
本スクリーニング方法においては、まず、本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる。
【0049】
本発明において、「機能的に結合した」とは、本発明のプロモーターDNAに転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、本発明のプロモーターDNAとレポーター遺伝子とが結合していることをいう。本スクリーニング方法における「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞」として、例えば、上記DNAを含むベクターを導入した細胞を挙げることができる。該ベクターは、当業者に周知の方法により作製することができる。ベクターの細胞への導入は、一般的な方法、例えば、ポリエチレングリコール法、リン酸カルシウム沈殿法、電気パルス穿孔法、リポフェクタミン法、マイクロインジェクション法等によって実施することができる。
【0050】
また、「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞」には、染色体に該DNAが挿入された細胞も含まれる。染色体へのDNAの挿入は、当業者に一般的に用いられる方法、例えば、相同組み換えを利用した遺伝子導入法により行うことができる。
【0051】
本方法における「本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞抽出液」とは、例えば、市販の試験管内転写翻訳キットに含まれる細胞抽出液に、本発明のプロモーターDNAとレポーター遺伝子とが機能的に結合した構造を有するDNAを添加したものを挙げることができる。
【0052】
本スクリーニング方法における「接触」は、本発明のプロモーターDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞の培養液に被験化合物を添加する、または該DNAを含む上記の市販された細胞抽出液に被験化合物を添加することにより行うことができる。被験化合物がタンパク質の場合には、例えば、該タンパク質をコードするDNAを含むベクターを、該細胞へ導入する、または該ベクターを該細胞抽出液に添加することで行うことも可能である。
【0053】
本スクリーニング方法においては、次いで、該レポーター遺伝子の発現レベルを測定する。レポーター遺伝子の発現レベルは、当業者においては、該レポーター遺伝子の種類を考慮して、測定することができる。
【0054】
本スクリーニング方法においては、被験化合物の非存在下において測定した場合(対照)と比較して、被験化合物がレポーター遺伝子の発現レベルを変化させた場合に、被験化合物が本発明のDNAのプロモーター活性を調節する化合物であると判定される。
【0055】
さらに、本発明においては、上記スクリーニング方法を利用して、複数の被験化合物について、本発明のDNAのプロモーター活性を調節するか否かを評価し、プロモーター活性を調節する化合物を選択することにより、効率的にプロモーター活性を調節する化合物をスクリーニングすることができる。
【実施例】
【0056】
以下、実施例を用いて本発明をさらに具体的に説明する。ただし、本発明の技術的範囲はこれらの実施例に限定されるものではない。
〔実施例1〕誘導型プロモーター候補遺伝子の選抜方法
植物病原糸状菌であるいもち病菌に対して低濃度ではほとんど抗菌活性を示さない化合物プロベナゾールを用いて、当該化合物の暴露で顕著な応答性を示す候補遺伝子の選抜を以下の方法により行った。
【0057】
(1)トータルRNA抽出
まず、100mlのYG液体培地を入れた300ml容三角フラスコにいもち病菌マグナポルテ・グリセア(Magnaporthe grisea)野生株Guy11株の分生子を約2×106個分生子になるように接種し、26℃、48時間緩やかに振とう培養した。その後、表1で示した一般的な農業用薬剤で知られている各種化合物を、表記してある濃度添加し、さらに26℃で2時間培養した。本薬剤処理を行った菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0058】
【表1】
【0059】
(2)マイクロアレイ
マイクロアレイに用いるDNAチップとして、マグナポルテ・グリセアの10,000遺伝子から設計した60merのオリゴプローブをインクジェット方式でスポットした独自のマイクロアレイを使用した。抽出した薬剤処理区をtestサンプルに、薬剤無処理区を比較対象にCyanine3及びCyanine5を用いた二色法により行った。一回の実験区ごとにCyanine3とCyanine5のカラースワップを行う事により計2回の反復実験を行った。
【0060】
(3)RNAの標識(Cyanine5、Cyanine3)
RNAのCyanine3及びCyanine5標識はmRNAを用いて行い、トータルRNAからのmRNAの精製はOligotex-dT30<super> mRNA purification Kit(タカラバイオ社製)を用い、本キットに添付のマニュアルに従った。mRNAからのcDNAの作製及び標識はCyScribe cDNA Post Labelling Kit(GE helthcare)を用い、本キットに添付のマニュアルに従い行った。
【0061】
(4)ハイブリダイゼーション
Cyanine3及びCyanine5標識済みcDNA混合サンプルに、表2に示したハイブリダイゼーションバッファーを60μl加え溶解後、95℃で5分熱変性を行った。その後30分間室温で保持した後、DNAチップ上に滴下し、カバーガラスをのせた。カバーガラスをのせたDNAチップは温水上に浮かべたhumidity chamber上に置き42℃、一晩インキュベーションすることによりハイブリダイゼーションを行った。
【0062】
【表2】
【0063】
(5)ポストハイブリダイゼーション
42℃で一晩インキュベーション後、DNAチップは表3に示したウォッシュ バッファー1中でカバーガラスをはずした後、さらに新しいウォッシュバッファー1で15分、次いで表4に示したウォッシュバッファー2で5分、表5に示したウォッシュバッファー3で5分の順に振とうすることにより洗浄を行った。その後、さらにdH2O中で2分間洗浄後800×g、2分の遠心により水分を飛ばしGenePix 4000B(Axon instrument)を用いてスポットのスキャンを行った。
【0064】
【表3】
【0065】
【表4】
【0066】
【表5】
【0067】
マイクロアレイの結果、いもち病菌の生育にほとんど影響を及ぼさない化合物プロベナゾールを10ppmおよび50ppmの濃度処理したときにデヒドロゲナーゼと推定される遺伝子の増加が目立って確認された。プロベナゾールを50ppm処理したときに発現が上昇する遺伝子の上位10遺伝子の中には5つのデヒドロゲナーゼ遺伝子が含まれていた(表6)。表6中のBROAD IDナンバーとは、BROAD INSTITUTEのMagnaporthe grisea Database(http://www.broadinstitute.org/annotation/genome/magnaporthe_grisea/MultiHome.html)において、各遺伝子に付与されたナンバーである。本発明において、MGG_の頭文字から始まるナンバーは、このBROAD IDナンバーのことを示す。BROAD IDナンバーが付与された各遺伝子は、上記データベースにおいて、ゲノム上の位置、全長、転写産物、配列情報等を調べることが可能である。
【0068】
誘導プロモーターとしては、その誘導体処理によって生育阻害を示すことがなく、化合物の暴露に対して顕著な発現増加がみられること、かつ他の化合物の暴露に対しては発現の誘導されないことが望ましい。そこで、そのデヒドロゲナーゼ遺伝子の5つについてヒートマップを描きその発現誘導とその特異性を図1に示した。これを見ると、プロベナゾール処理によって発現が顕著に誘導された5つのデヒドロゲナーゼ遺伝子のほとんどがプロベナゾールでのみ発現が増加しており、プロベナゾールに対する特異性が高いことがわかる。IDナンバーMGG_00357の遺伝子はプロベナゾール以外のいくつかの化合物処理によっても遺伝子発現の増加が観察されるが、これらの化合物を処理した場合のいもち菌の菌糸生育は強く阻害を受けることから、プロベナゾールがこれらの遺伝子発現の誘導には効果的であることがわかる。そこで、これらの遺伝子をプロベナゾールによって誘導される誘導プロモーター候補とした。
【0069】
【表6】
【0070】
〔実施例2〕プロベナゾール誘導プロモーターの検証
図1に示した遺伝子が、プロベナゾールを処理したときに顕著な発現量の増加があり、かつ誘導プロモーターとして利用可能か否かの確認を行うため、MGG_04304(RDH8)及びMGG_10913(RDH12)遺伝子を例にその発現量の変化を定量RT-PCRを用いることによって検証した。
【0071】
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し26℃、48時間緩やかに振とう培養した。その後、以下の薬剤を各濃度添加し、さらに26℃で30分、2時間及び6時間培養した。薬剤処理に用いた化合物はプロベナゾールが1ppm、10ppmおよび20ppmとし、対照薬剤として呼吸鎖阻害剤としてジフルメトリム1ppm、メプロニル30ppm、クレソキシムメチル20ppm、細胞壁合成阻害剤としてCFW(カルコフロワーホワイト)30ppm、微小管生合成阻害剤としてベノミル1ppm、細胞膜機能阻害剤としてイミノクタジン1ppm、MAPキナーゼシグナリングに作用すると考えられるイプロジオン30ppm、エルゴステロール生合成阻害剤オキスポコナゾール・フマル酸塩1ppm、リン脂質合成阻害剤IBP(イプロベンフォス)2.5ppmを用いた。本薬剤処理により得た菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌、脱水後、乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0072】
(2)cDNAの合成
cDNAの合成には、トータルRNAを用いた。260 nmにおける吸光度からの計算値で5μg量のトータルRNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え100μlとした。これにRNase free DNase Iを5unit加え、37℃で1時間反応させ混在するゲノムDNAの分解を行った。その後、ExScript RT reagent Kit(タカラバイオ社製)を用いて添付マニュアルに従って逆転写反応を行い、cDNAを得た。
【0073】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit (Finnzymes社製)を用いた。スキャナー解析はDNA Engine OPTICON2(MJ Research社製)を、データ解析には解析ソフトOpticon Monitor2 ver.2.02(MJ Research社製)を用いた。反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40 サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でDissociation Curveの作成を行った。PCRは各cDNA、各標的遺伝子につき三連で行った。
【0074】
各化合物処理cDNAサンプルを用いてMGG_04304(RDH8)及びMGG_10913 (RDH12) 遺伝子の発現を経時的に調べた。またコントロールとしてActinタンパク質をコードしていると推定される遺伝子の転写量を調べた。それぞれ以下の特異的プライマーを用いた。
Actin-F:ATGCCATCGGAAAGACAGAC(配列番号:3)
Actin-R:CAGGAGTCGATCTCCAAAGC(配列番号:4)
RDH8_RTF:GGGCCATGATTGAGATAGTCAC(配列番号:5)
RDH8_RTR:GACAGCTCCTTGAACTCTTCCA(配列番号:6)
RDH12_RTF:AAGGAGAGCTGGCCCATATTAG(配列番号:7)
RDH12_RTR:GGTGGTAAAACTCGCCACTAAC(配列番号:8)
【0075】
PCR終了後、専用の解析ソフトOpticon Monitor2 ver.2.02(MJ Research社)を用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a) ΔCT値の算出(各cDNAの標的遺伝子とγ-actinとの差)
ΔCT = (y-x)
ここでxはCT (γ-actin)であり、yはCT(Target gene)である
b) ΔCT値の算出(あるcDNAのΔCTと基準とするcDNAのΔCTとの差)
ΔCT =ΔCT (C) -ΔCT (S)
ここで、CはComparative cDNAであり、SはStandard cDNAである
c) 相対発現量の算出
2-(ΔCT)
【0076】
RT-PCRの結果をMGG_04304(RDH8)については図2の左側に、MGG_10913 (RDH12)遺伝子については図2の右側にactin当たりの発現量をグラフ化したもので示した。いもち菌 野生株をプロベナゾールに曝すと、MGG_04304(RDH8)及びMGG_10913 (RDH12) 遺伝子の転写量は顕著に増加しており、MGG_04304及びMGG_10913は共に1ppm処理で、actin比で約0.1、無処理対照区(CTL)と比較して約100倍の発現増加を示した。また、処理濃度を濃くするにつれ発現量は増加し、20ppmでは処理時間に関わらずMGG_04304ではactin比で約80、無処理対照区(CTL)と比較して約8×104倍の増加を、MGG_10913の場合はactin比で約6、無処理対照区(CTL)と比較して約6×103倍の顕著な増加を示した。
【0077】
一方、プロベナゾール以外の化合物処理の場合はIBP(イプロベンフォス)、フルジオキソニル、プロシミドンなどで発現の誘導が見られたがその発現増加率はプロベナゾール1ppm処理の場合よりも低く、またこれらの化合物が菌糸生育阻害を示すことを考えるとMGG_04304(RDH8)及びMGG_10913 (RDH12)遺伝子がプロベナゾールでの遺伝子発現誘導に有効であることが確認された。
【0078】
〔実施例3〕誘導プロモーターによる遺伝子誘導のEGFPレポーターによる検出
プロベナゾールを暴露した時に顕著な遺伝子の発現の増加を示すMGG_04304(RDH8)及びMGG_10913 (RDH12)を例にして、本遺伝子の5’-UTR(5‘上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによってプロベナゾール処理による転写量の増加を可視化する菌株を作成した例を示す。
【0079】
(1)MGG_04304(RDH8)及びMGG_10913 (RDH12) 5'領域のクローニング
Broad Institute のマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からAOX 遺伝子の5'領域の塩基配列情報を取得し、その配列情報を参考にRDH8遺伝子のプライマーペアとして(5'- ATAAGGTGCGCGCAAAGGT -3'、配列番号:9、5'- CGCAAGCTTAGTTTGGATAGTCGTATGCA -3’、配列番号:10)をRDH12遺伝子のプライマーペアとして(5'- CTATCCCTTTAATTAACCGACC -3'、配列番号:11、5'- CGCAAGCTTTGGCAAGCTATCTTGTTT -3'、配列番号:12)のそれぞれ一組のオリゴヌクレオチドを設計した。なおプライマー内で、下線で示される塩基はHindIIIの配列を示す。これらのプライマーを用いてマグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は、PCR Thermal Cycler PERSONAL (タカラバイオ社製)を用いた。
【0080】
このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところRDH8は約1,200塩基対、RDH12は約1,500塩基対のDNA増幅が見られた。この増幅断片をアガロースゲル電気泳動に供したゲル中よりGENECLEAN IIIキット(Qbiogene社製)を用いてDNAを抽出し、これを挿入DNA断片とした。この挿入DNA断片をpGEM-T Easy Vector System(Promega社製)を用いてpGEM-T Easy Vectorに連結させ、連結DNA溶液を得た。連結DNA溶液10μlを氷中でよく冷却した後、氷上解凍したコンピテントセルJM109を100μl加え穏やかに撹拌し、氷中で20分間、続いて42℃で45秒ヒートショック処理を行った。これに、400μlのLB液体培地を加え、37℃で1時間培養後、100μg/mlのアンピシリンを添加したLB平板培地にまき、37℃で一晩培養した。目的のプラスミドDNAを用いて形質転換した大腸菌の単一のコロニーを3mlの100μg/mlのアンピシリンを添加したLB液体培地に植菌し、37℃で一晩振盪培養した。1.5mlの培養液を1.5mlのエッペンドルフチューブに移して15,000×gで1分間遠心分離し、沈殿を100μlの氷冷したTEG(25mM Tris-HCl、10mM EDTA、50mM Glucose、pH8.0)で懸濁し、これに200μlの0.2N NaOH-1%SDSを加えて穏やかに撹拌した後、150μlの3M NaOAc(pH5.2)を加えて混合した。これを15,000×g、4℃で5分間遠心分離し上清を回収し450μlのフェノール・クロロホルム・イソアミルアルコール(25:24:1)を加えて激しく撹拌した後、15,000×g、室温で5分間遠心分離し上層を回収した。この溶液に-20℃で氷冷した900μlのエタノールを加えて-20℃で10分間放置後、4℃にて15,000×gで5分間遠心分離した。沈殿を500μlの70%エタノールでリンスしたのち、乾燥させ、最後にRNase(100μg/ml)を含むTE(10mM Tris-HCl、1mM EDTA、pH8.0)50μlに溶解した。
【0081】
得られたプラスミド中の挿入DNA断片を添付のプロトコールに従い、ABI PRISMTM 377 DNA sequencing system (PE Biosystem社製)にて解析した。その結果、RDH8は1,218塩基対(配列番号:13、図3)、RDH12は1,473塩基対(配列番号:14、図4)からなることが明らかとなった。
また、本ベクターをpGEM-PrRDH8およびpGEM-PrRDH12とする。
これらのベクター中の挿入DNA断片中にはRDH8およびRDH12ともにRDH8のプライマー(配列番号:10)もしくはRDH12のプライマー(配列番号:12)に付加したHindIIIサイトとは別のHindIIIサイトを有していることから、このHindIIIサイトとRDH8のプライマー(配列番号:10)に付加してあるHindIIIサイトで切り出される1,152塩基対の配列(配列番号:1)をRDH8プロモーター領域とし、HindIIIサイトとRDH12のプライマー(配列番号:12)に付加してあるHindIIIサイトで切り出される1,354塩基対の配列(配列番号:2)をRDH12プロモーター領域とした。
【0082】
(2)EGFPベクターの作成
pCAMBIA-0380バイナリーベクター(CAMBIAより購入)を骨格にし、薬剤選択マーカーとしてハイグロマイシンB耐性遺伝子Hphを融合することにより作成した糸状菌形質転換ベクターpCH11(図5)をHindIII及びBglIIで消化し、そこにEGFP-LacI-TagdA (p3'SS d EGFPと大腸菌LacI:Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びAspergillus oryzae Α-グルコシダーゼ遺伝子agdA ターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)のHindIII-BglII断片を組み合すことによりEGFPベクター、pCH11-EGFP-LacIを作成した(図6)。
【0083】
(3)RDH8-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図6)プラスミドをHindIIIで消化し、そこにpGEM-PrRDH8をHindIII処理することにより得たMGG_04304(RDH8)遺伝子5'領域(配列番号:1)を定法により融合した。本レポーターアッセイプラスミドであるRDH8-EGFP-LacIレポーターベクターをpCH11-PrRDH8-EGFP-LacIと命名した。
【0084】
(4)RDH12-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図6)プラスミドをHindIIIで消化し、そこにpGEM-PrRDH12をHindIII処理することにより得たMGG_10913(RDH12)遺伝子5'領域(配列番号:2)を定法により融合した。本レポーターアッセイプラスミドであるRDH8-EGFP-LacIレポーターベクターをpCH11-PrRDH12-EGFP-LacIと命名した。
【0085】
(5)pCH11-PrRDH8-EGFP-LacIおよびpCH11-PrRDH12-EGFP-LacIプラスミドを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はアグロバクテリウム法(Gento Tsuji, Satoshi Fujii, Naoki Fujihara, Chika Hirose, Seiji Tsuge, Tomonori Shiraishi and Yasuyuki Kubo(2003):Agrobacterium tumefaciens-mediated transformation for random insertional mutagenesis in Colletorichum lagenarium. Journal of General Plant Pathology 69:230-239)を改良した方法を用いた。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。これらのベクターの挿入によって得られた形質転換株はpCH11-PrRDH8-EGFP-LacIの挿入によるものはRDH8-EGFP株、pCH11-PrRDH12-EGFP-LacIの挿入によるものはRDH12-EGFP株と命名した。
【0086】
(6)レポーター作動の確認
レポーター作動の確認は、栄養菌糸生育時および感染器官形成時の2つのステージで生育させたいもち病菌をプロベナゾールで処理した時のEGFPの蛍光を観察することによって行った。
a)栄養菌糸生育時でのレポーター動作確認の場合はマグナポルテ・グリセアRDH8-EGFP株およびRDH12-EGFP株の分生子を2×106個分生子/100mlになるように、100mlのYG液体培地を入れた300ml容坂口フラスコに接種し次の条件で培養した。
(条件1) 26℃、24時間培養
(条件2) 26℃、24時間培養した後、プロベナゾールを添加し、さらに26℃で6時間培養。
その後、各処理菌株の蛍光を、蛍光顕微鏡を用いて観察した。
b)感染器官形成時でのポーター動作確認の場合はマグナポルテ・グリセアRDH8-EGFP株およびRDH12-EGFP株の分生子1×105個/mlにプロベナゾールを添加した後、その分生子懸濁液40μlをカバーガラス上に滴下し、経時的に各処理菌株のEGFP蛍光を蛍光顕微鏡を用いて観察した。
【0087】
(7)EGFP蛍光観察
スライドガラスに培養後の栄養菌糸生育時および感染器官形成時の菌糸体をのせEGFPの蛍光観察を行った。いずれのステージにおいてもプロベナゾール処理においてのみ、核に局在した強いEGFPの蛍光が観察された(図7〜10)。以上の結果から、RDH8およびRDH12遺伝子のプロモーターはその下流の遺伝子を栄養菌糸生育時や感染器官形成時といった様々な生育ステージにおいても、プロベナゾール処理によって発現の誘導を制御することができることがわかった。また、そのRDH8およびRDH12遺伝子のプロモーターを用いたプロベナゾールによる発現制御は、今回のEGFP遺伝子のような他の遺伝子でRDH遺伝子を置き換えた場合もその誘導活性を維持しており、誘導プロモーターとして十分に利用可能であることが確認できた。
【特許請求の範囲】
【請求項1】
下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNA。
(a)配列番号:1又は2に記載の塩基配列からなるDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
【請求項2】
真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有することを特徴とする、請求項1に記載のDNA。
【請求項3】
前記真菌の生育に影響しない薬剤がプロベナゾールであることを特徴とする、請求項1または2に記載のDNA。
【請求項4】
前記真菌の生育に影響しない薬剤を、1ppm以上添加することによりプロモーター活性が誘導されることを特徴とする、請求項1〜3のいずれかに記載のDNA。
【請求項5】
請求項1〜4のいずれかに記載のDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNA。
【請求項6】
請求項1〜5のいずれかに記載のDNAを含むベクター。
【請求項7】
請求項1〜5のいずれかに記載のDNA、または請求項6に記載のベクターを含む、形質転換細胞。
【請求項8】
真菌であることを特徴とする、請求項7に記載の形質転換細胞。
【請求項9】
真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法。
(1)請求項5に記載のDNA、または請求項6に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程
【請求項10】
下記の工程(a)〜(c)を含む、請求項1に記載のDNAのプロモーター活性を調節する化合物のスクリーニング方法。
(a)請求項1に記載のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【請求項1】
下記の(a)〜(c)のいずれかに記載のプロモーター活性を有するDNA。
(a)配列番号:1又は2に記載の塩基配列からなるDNA、
(b)配列番号:1又は2に記載の塩基配列において1もしくは複数の塩基が欠失、置換もしくは付加された塩基配列からなるDNA、及び
(c)配列番号:1又は2に記載の塩基配列からなるDNAとストリンジェントな条件下にてハイブリダイズするDNA
【請求項2】
真菌の生育に影響しない薬剤の添加により誘導されるプロモーター活性を有することを特徴とする、請求項1に記載のDNA。
【請求項3】
前記真菌の生育に影響しない薬剤がプロベナゾールであることを特徴とする、請求項1または2に記載のDNA。
【請求項4】
前記真菌の生育に影響しない薬剤を、1ppm以上添加することによりプロモーター活性が誘導されることを特徴とする、請求項1〜3のいずれかに記載のDNA。
【請求項5】
請求項1〜4のいずれかに記載のDNAの制御下に、外来遺伝子が機能的に結合した構造を有するDNA。
【請求項6】
請求項1〜5のいずれかに記載のDNAを含むベクター。
【請求項7】
請求項1〜5のいずれかに記載のDNA、または請求項6に記載のベクターを含む、形質転換細胞。
【請求項8】
真菌であることを特徴とする、請求項7に記載の形質転換細胞。
【請求項9】
真菌において外来遺伝子を発現させる方法であって、以下(1)及び(2)の工程を含む方法。
(1)請求項5に記載のDNA、または請求項6に記載のベクターを該真菌へ導入する工程、及び
(2)該真菌に、真菌の生育に影響しない薬剤を添加する工程
【請求項10】
下記の工程(a)〜(c)を含む、請求項1に記載のDNAのプロモーター活性を調節する化合物のスクリーニング方法。
(a)請求項1に記載のDNAの制御下に、レポーター遺伝子が機能的に結合した構造を有するDNAを含む細胞または細胞抽出液と、被験化合物を接触させる工程、
(b)該レポーター遺伝子の発現レベルを測定する工程、及び
(c)該レポーター遺伝子の発現レベルを変化させる化合物を選択する工程
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図8】

【図9】
【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2011−188758(P2011−188758A)
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【出願番号】特願2010−55441(P2010−55441)
【出願日】平成22年3月12日(2010.3.12)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度農業・食品産業技術総合研究機構生物系特定産業技術研究支援センター「生物系産業創出のための異分野融合研究支援事業(異分野融合研究開発型)」(モデル糸状菌情報を活用した抗いもち病菌剤探索システムの構築)産業技術力強化法第19条適用を受ける特許出願
【出願人】(501167644)独立行政法人農業生物資源研究所 (200)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【出願日】平成22年3月12日(2010.3.12)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度農業・食品産業技術総合研究機構生物系特定産業技術研究支援センター「生物系産業創出のための異分野融合研究支援事業(異分野融合研究開発型)」(モデル糸状菌情報を活用した抗いもち病菌剤探索システムの構築)産業技術力強化法第19条適用を受ける特許出願
【出願人】(501167644)独立行政法人農業生物資源研究所 (200)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
[ Back to top ]