
カラム内濃縮が可能な毛細管電気泳動を誘導結合プラズマに接続するためのインターフェース及び接続方法
【発明の詳細な説明】
【0001】
【産業上の利用分野】毛細管電気泳動(CE)と誘導結合プラズマ(ICP)を接続するための新しいインターフェースに関するものである。
【0002】
【従来の技術】金属元素の毒性は、化学種に依存することが多い。従って、金属元素の総量ならびにその化学種の分離および定量が生物および環境試料の分析で益々重要に成りつつある。クロマトグラフィ、原子分光測定法および質量分析法のような各種分析技術がこれらの分野で用いられるが、より良い結果を得るために、更に効率的で感度の良い方法が絶えず探し求められていた。
【0003】
【発明が解決しようとする課題】毛細管電気泳動(CE)は、高価で複雑な装置を必要としない、強力で簡単な方法である。この方法は環境、生物学、臨床を含む広い研究分野のサンプルの分析に用いられている。一般に、極く小量のサンプルしかCEに導入できないので、殆どの化学種に対して濃度の点で満足のいく検出限界を得るのは難しい。従って、高感度で高選択性の検出器の開発は、CEが実用になった後の、非常に重要かつ、やり甲斐のある仕事である。現在までのところ、オンカラムでUVおよび蛍光検出器が広く用いられている。しかしながら、これらの検出器の感度は、普通10−5から10−6Mの間である。質量分析、レーザー誘導蛍光分析、電流分析、伝導度分析、化学発光およびレーザーラマン法といった、他の幾つかの検出技術が、CEで分離された検体の高感度、高選択性の検出方法を提供するために研究されてきた。
【0004】誘導結合プラズマ発光測定法(Inductively Coupled Plasma Atomic Emission Spectrometry,ICP−AES)および質量分析法(Mass Spectrometry,MS)は、非常に感度が良く元素選択的検出技術である。これらの技術は、生物試料、環境試料および金属結合蛋白中の微量元素種を定量するために、高速液体クロマトグラフィおよび容積排除クロマトグラフィに接続されている。最近、これらの技術はCEと接続され、非常に有用な検出器であることが分かった。CE−ICP−AES/MSを使って仕事をする時の課題は、CEとICPを結合する効率的なインターフェースを設計することである。従来型の同軸型ニューマティックネブライザー(CPCN)および直接注入ネブライザー(DIN)に結合できるインターフェースが開発されたが、これらインターフェースは、どれも余りにも複雑で、他種のネブライザーに必ずしも接続して使用出来るわけではない。
【0005】
【課題を解決するための手段】本発明は、非常に簡単で殆どあらゆる種類のネブライザーに接続して使えるインターフェースを提供するものであり、3つの濾斗状チューブで構成されており、それぞれのチューブの尖った方の端が集まってY字状を形成し、他方の端が電気泳動毛細管、補助毛細管、およびネブライザーのサンプリングチューブにそれぞれ接続されていることを特徴とする。このインターフェースは、ネブライザーの外部に独立しており、ネブライザーのサンプリングチューブに直接結合させることができるものであり、サンプルはCEで分離され、次いでネブライザー中の高速アルゴンガス気流による自己吸引によりネブライザーに吸入される。このインターフェースは、CPCNおよびマイクロコンセントリックネブライザー(MCN)にうまく接続でき、満足できる感度および効率的分離が得られる。かくして、本発明はまた、電気泳動回路を形成する三又分岐を構成する電気泳動毛細管と補助毛細管とを有し、ネブライザーによる吸引またはペリスタチューブによる送液により電解液の流れを実現し、印可電圧により分離されたイオンを含む電解液の一部を直接導入することを特徴とする、電気泳動による分離されたイオンをネブライザーに直接導入する方法を提供するものである。
【0006】CEでは、検出感度を改善するために各種のオンカラム濃縮技術が用いられる。そうした技術の1つは、大量の低濃度のサンプルプラグを電気泳動毛細管に導入し、高電圧を印加してサンプルプラグの端に検体サンプルを濃縮することである。本発明では、この技術も利用され、感度の改善がもたらされた。大量のサンプルが毛細管に導入され、長いサンプルプラグが形成され、そこでは低濃度のために低伝導度となるので、良好な感度および効率的な分離度を得るために内径の大きな毛細管を用いることができる。また、大きなサンプルプラグでは0.05MのHNO3溶液を電気泳動電解質として用いることができる。
【0007】
【実施例】試薬および物質. 酢酸ナトリウム3水和物(純度99.0%、和光純薬工業)および硝酸(EL、比重1.38、関東化学)を電解質として用いた。K2Cr2O7(純度99.5%、和光純薬工業)、CrCl3・6H2O(純度99.5%、和光純薬工業)、FeCl2(純度97.0%、和光純薬工業)、FeCl3・6H2O(純度99.9%、和光純薬工業)、CuSO4・5H2O(純度99.5%、純正化学)およびエチレンジアミン四酢酸2水素2ナトリウム(純度99.5%、純正化学)を分析種として用いた。全ての溶液は、物質の適量を脱イオン水に溶解して製した。
【0008】毛細管電気泳動. 外径が375μmで、内径がそれぞれ100μm(長さ120cm)と内径150μm(長さ160cm)の2種類の溶融シリカ毛細管(GLサイエンスジャパン)を電気泳動毛細管として、また、長さが40cm、内径が150μmの毛細管を補助毛細管として用いた。高圧直流電源は、松定プレシジョン(株)製で、30kVまでの電圧及び200μA電流を供給できる。外径0.1mmの白金線を電極として用いた。
【0009】ネブライザー. 2種の市販ネブライザーをテストした。CPCN(グラスエクスパンジョンプロプリエータリリミテッド、オーストラリア)およびMCN(CETACテクノロジー、オマハ、ネブラスカ、USA)である。インターフェースは、ネブライザーのサンプリングチューブに直接接続した。ネブライザー毎にキャリヤーガスの流速を種々変えて最適の感度および分解能を得るようにした。
【0010】誘導結合プラズマ発光分光器. アナリティカルリサーチラボラトリーズ(ARL)のMAXIMIII光学発光分光測定器(FISSON製)を用いた。標準トーチMAXIM ICPを、製造業者の推奨通りに、中間のガス流速0.8l/分、外側ガス流速13l/分で使用した。電力は1.1kWであった。
【0011】スプレイチャンバー. 2種類のスプレイチャンバーをMCNおよびCPCNに接続してテストした。MCNの場合には、内部にインパクトビーズのない、円錐形で自作したスプレイチャンバーを用いた。CPCNの場合には、インパクトビーズのある正規のMAXIMタイプのスプレイチャンバー及び、MCNで使用したのと同じスプレイチャンバーを試験した。
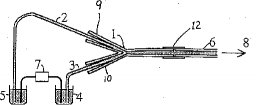
【0012】インターフェース. 図1は、CE−ICP−AES用インターフェースであり、このインターフェース1は、石英製であり、その第1の腕9は、電気泳動毛細管2の出口に接続し、第2の腕10は、補給電解質溶液を吸い上げる補助毛細管3に接続されている。この補助毛細管のもう一方の端は、補給電解質として用いられている0.05MのHNO3溶液中に浸されている。インターフェースの第3の腕11は、適当なチューブ例えばペリスタルチックポンプ用チューブによってネブライザー8のサンプリングチューブ6に直結している。図中12は、左右のチューブを結合するコネクターである。毛細管は直接インターフェースの各腕に挿入出来るので何ら前処理を必要としない。キャリヤーガスのバルブを開くと、ネブライザー内の高速アルゴンガス気流によって生じる圧力差によって、補給電解質溶液と電解質溶液またはサンプル溶液が、電気泳動毛細管および補助毛細管に吸い上げられる。2種類の溶液は、2つの腕の接合部で混合し、次いで、ネブライザーの中に吸引される。補給電解質溶液は、直流電源のアース側に接続しており、緩衝電解質溶液は、直流電源の陰極に接続している。負の高電圧によって電解質に自己吸引の流れに反対方向の電気浸透圧による流れを起こさせ、かくしてサンプルは、正の分極電圧を印加した時に比べて長時間毛細管の中に留まることになる。
【0013】サンプル注入. サンプルは、キャリヤーガスの流れによって生じる自己吸引によって直接毛細管に導入される。サンプリングの前および後には、空気を電気泳動毛細管の中に自己吸引して導入することのないように、補助毛細管は、インターフェースから外しておくか、またはキャリヤーガスのバルブを閉じて自己吸引作用がなくなるようにしておかなければならない。サンプルの容積は、2つの方法で測定できる。1つは、サンプリング時間の測定によるものであり、もう1つは、サンプリングの前後に蒸留水の重量を測定するものである。これら2つの方法を用いて測定したサンプリング容積は、それぞれ0.00314ml/分および0.00328ml/分と計算された。これらの測定は、内径150μm(長さ160cm)の毛細管を電気泳動毛細管として用い、MCNを0.25l/分のキャリヤーガス流量で操作して行なった。こうした条件下では、補給溶液の流速は、0.0136ml/分と計算され、この値は、サンプル流速の4倍以上である。このことは、サンプルが補給溶液で4倍以上に希釈されることを意味する。
【0014】CE−ICP−AESのインターフェースの特徴. ここに記載されているCE−ICP−AESのインターフェースの場合、支持電解質溶液は、2つの目的を果たしている。第1に、支持電解質溶液は、完全な電気泳動回路が出来上がるように電気泳動毛細管の出口(アース側)のところでの連続的かつ安定な電気的接触を保つために用いられる。第2に、支持電解質溶液は、ネブライザーに吸引される混合溶液のpH値の調整に使うことができる。一般に、高pH緩衝溶液が電解質溶液として用いられることは、しばしばある。しかし、アルカリ性のpH環境では、殆どの金属イオンは、水酸化物として存在し、それらは、ネブライザーの毛細管壁に容易に吸着されるか、濃度が高過ぎれば沈殿する。このことは、特にMCNを用いた場合には、ネブライザーを詰まらせることになりかねない。もし、0.04Mの酢酸ナトリウムを支持液および電解質溶液とし、そして、Cr2O72−およびCr3+(Cr3+の一部はCr(OH)3の形で存在しているであろう。)をこのシステムで分析すれば、サンプリング2回ないし3回毎にMCNを0.05MのHNO3で3分間ほど洗浄しなければならない。もし、0.04Mの酢酸ナトリウムの替わりに0.05MのHNO3を支持溶液として用いれば、詰まりが起きることは殆どない。補助毛細管の内径は、支持溶液の吸い上げ容積、電気泳動電流、および、電気泳動毛細管と補助毛細管との間の電圧分布に影響する。補助毛細管の内径が大きければ、支持液の吸い上げ容積が増し、目的化学種の希釈度が大きくなる。反対に、補助毛細管の内径が小さければ、希釈度を下げられる。しかし、このことは、インターフェースとネブライザーとの間の液体流速を下げ、それによってピークの幅を広げることになる。従って、この実験では、妥協が必要であり、内径150μmの毛細管が補助毛細管として用いられた。電気泳動電流は、支持液の濃度を変更することによって調整することもでき、従って、電気泳動毛細管と補助毛細管との間の電圧分布も同様に調整することができる。
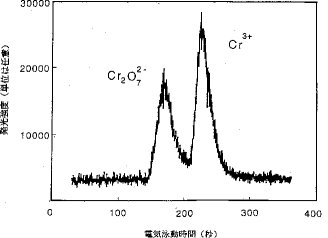
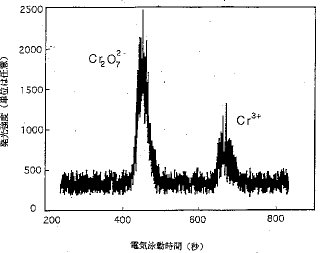
【0015】CE−ICP−AESのインターフェースの評価. このインターフェースおよびこのシステムを評価するために若干の予備テストが行なわれた。図2および図3は、それぞれCr2O72−およびCr3+を含むテスト溶液についてCE−CPCN−ICP−AES法およびCE−MCN−ICP−AES法によって得られた電気泳動図である。このうち図2は、全濃度:100μg/ml、化学種:Cr3+およびCr2O72−(1:1)、緩衝電解質溶液:0.04Mの酢酸ナトリウム(pH 7.2)、高電圧:15kV、毛細管の内径:100μm、サンプルプラグの長さ:電気泳動毛細管の25%の場合、図3は、全濃度:1μg/ml、化学種:Cr3+とCr2O72−(1:1)、緩衝電解質溶液:0.05MのHNO3、高電圧:8kV、毛細管の内径:150μm、サンプルプラグの長さ:電気泳動毛細管の30%の場合である。どちらの実験においても自作したスプレーチャンバーが用いられた。どちらの場合でも、Cr2O72−およびCr3+の完全分離が観測された。これらの結果は、このインターフェースがCPCNにもMCNにも適しており、CEとICP−AESを接続するのに間違いなく利用できることを明確に示すものである。
【0016】MCNはCPCNよりもずっと高いネブライザー効率を持っているので、CE−MCN−ICP−AES法の感度は、CE−CPCN−ICP−AESの感度よりも遥かに大きい。キャリヤーガス気流によって起きる電気泳動毛細管中の流速が、この2つのシステム間に分解能の差を生じるのである。この流速がCE−CPCN−ICP−AES法におけるよりも遥かに遅いCE−MCN−ICP−AES法では、8kVで約12分で完全な分離が得られる。しかし、CE−CPCN−ICP−AES法では、約4分間15kVでやっとベースライン分離が得られるに過ぎない。2つのピークの高さもこの流速に関連している。流速が低下すると、分離が良くなり、Cr3+のピーク高は減少するがCr2O72−のピーク高は殆ど変化しない。これは、高電荷カチオン(Cr3+)が毛細管壁に捕捉され易く、流速が遅いと、速い時に比べてサンプルが電気泳動毛細管中に長い時間留まることになるからである。このことが、高電荷カチオンの毛細管壁への吸着を増強することになる。電気泳動電圧として陽電圧を用いると、Cr3+がCr2O72−よりも早く毛細管から溶出する。陰電圧の時よりも陽電圧の時の方がCr3+が毛細管中に留まる時間が短いので、Cr3+のシグナル強度は、陰電圧で得られるシグナル強度よりも強い。CE−CPCN−ICP−AES法については、正規のMAXIMタイプのインパクトビーズ付のスプレーチャンバーもテストした。その感度は、現場合わせのインパクトビーズなしのスプレーチャンバーでの感度の少なくとも1/10以下であった。この装置を日常業務の目的で使う場合は、サンプルを2ml/分の割合でネブライザーに導入する。正規のMAXIMタイプのスプレーチャンバーは、大きな液滴がプラズマ中に入るのを防止し、ICPのエアロゾル溶媒による過負荷を防止できる。しかし、このシステムでは、極小量のサンプルしかネブライザーに導入されず、したがって、プラズマの溶媒過負荷を心配する必要はない。
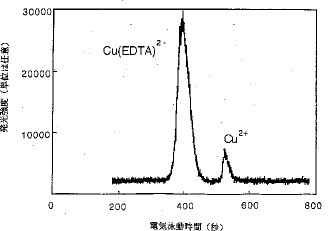
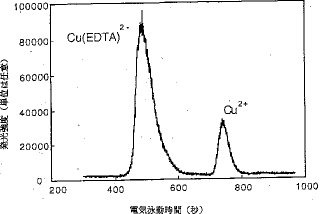
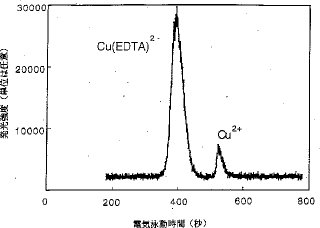
【0017】カラム上濃縮と分離. CE法では、サンプリング容積は、分離および感度に影響する非常に重要な因子である。一般に、サンプリング容積は、電気泳動毛細管の全長の10%以下である。本発明は、カラム上濃縮技術を用いており、普通よりも多量のサンプルが毛細管に導入された。図4および図5は、サンプリング容量を変化した時の銅の電気泳動図である。このうち、図4R>4は、全濃度:10μg/7ml、化学種:Cu2+とCu(EDTA)2−(1:1)、緩衝電解質溶液:0.05MのHNO3、高電圧:8kV、毛細管の内径:150μm、 サンプルプラグの長さ:電気泳動毛細管の5%の場合、図5は、サンプルプラグの長さが電気泳動毛細管の全長の30%で、その他の条件は図4での条件と同じの場合のものである。Cu2+およびCu(EDTA)2−についての図4と図5を比較すると、注入容積の増加が、ピーク幅に顕著な悪影響を及ぼすことなく、ピーク高および分解能をいずれも増加することが明らかである。
【0018】カラム上濃縮操作を以下に説明する。緩衝電解質溶液中よりもサンプルプラグ中にずっと高い電場勾配が生じる。その理由は、サンプルプラグ中の荷電粒子の濃度が、緩衝電解質溶液中よりもずっと低いからである。この(高い電場勾配)ため、サンプルプラグ中の荷電粒子は緩衝電解質溶液中の荷電粒子よりもずっと高速で動く。しかしながら、荷電粒子が緩衝電解質溶液に到達すれば、その領域にはより低い電場勾配しか存在しないので、移動速度は低下する。このことが、サンプル溶液と緩衝電解質溶液との境界に大量の荷電粒子の集積を齎らす。Cu2+とCu(EDTA)2−の運動方向は反対であるから、Cu2+とCu(EDTA)2−とは、サンプルプラグの別々の端に集積する。かくして、Cu2+とCu(EDTA)2−の分離が達成される。相対的に長いサンプルプラグが、ずっと良い分離度及びより多くの荷電粒子のサンプル溶液・緩衝電解質溶液境界での集積を引き起こすことになる。Cr3+およびCr2O72−の分離についも全く同じ事が言える。
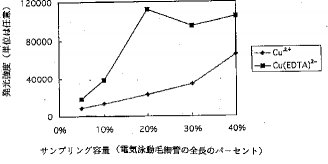
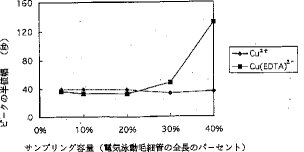
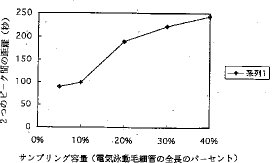
【0019】図6、図7および図8は、それぞれ、注入容積とピーク高、注入容積とピーク幅および注入容積と分解能の関係を示している。Cu2+は、Cu(EDTA)2−よりも長時間毛細管の中に留まるので、Cu2+は、より長い時間電気泳動作用を受けて毛細管中に濃縮される。従って、Cu2+のピーク幅は殆ど変化しないが、ピーク高は、注入容積が毛細管の全長の5%から40%まで増加する時、連続的に増加する。それとは対照的に、Cu(EDTA)2−のピーク幅は、注入容積が毛細管の全長の30%を超えると劇的に増加する。しかしながら、30%および40%注入容積でのピーク面積は、20%注入容積でのピーク面積よりも大きいけれども、30%および40%注入容積でのピーク高は、20%注入容積でのピーク高よりも低い。検体の毛細管内滞留時間の延長を齎らすキャリヤーガスの流速低下及び/または電気泳動電圧の増加は、Cu(EDTA)2−のピーク幅を幾分縮小すると期待される。しかしながら、キャリヤーガスの低流速はまた、ピーク幅を拡張させることもあり、また、高電圧は、陽イオンの毛細管壁への吸着を誘発し、シグナル強度を減少させる。
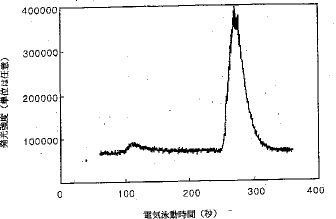
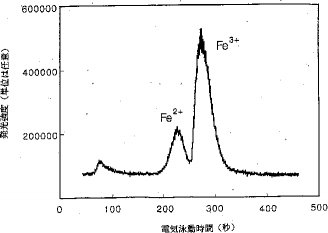
【0020】カラム上濃縮法では、比較的長いサンプルプラグが、低濃度用の反対極性を持った2種の荷電粒子を分離するのに適していると考えられた。何故ならば、これらの荷電粒子は、高電圧が印加されるとサンプルプラグの両端へと移動するからである。高電圧を印加した時に同じ方向に移動するFe3+およびFe2+の分離に対する注入容積の影響も研究した。若干の興味ある現象が観測された。比較的短いサンプルプラグ(電気泳動毛細管の全長の5%から25%)が毛細管に挿入された場合には、2つのピークが得られる。図9は、25%のサンプルプラグの場合の電気泳動図であり、図10は、38%のサンプルプラグの場合の電気泳動図である。後者の場合には、図10に示されているとおり、3つのピークが現われた。第2おび第3のピークはFe2+とFe3+であると推定されている。これらのピークを同定し、この現象を説明するために更に研究が続けられている。
【0021】電気泳動毛細管の内径と電解質. 電気泳動毛細管の内径もまた、分離効率および感度に影響する。一方では、内径の大きな毛細管は、大量のサンプルを導入出来る利点があり、したがって、より低い検出限界が期待できるが、他方、このことは、電気泳動電流の増加を招き、低分解能をきたす恐れがある。2種類の毛細管をテストした。1つは内径100μmの毛細管、もう1つは内径150μmの毛細管である。サンプル溶液の濃度が低く、比較的長いサンプルプラグが毛細管に挿入された時は、電気泳動電流は、低伝導度のサンプルプラグによって決まる。従って、効率的な分離は、このシステムでは内径150μmの毛細管で得られた。全濃度が10μg/mlの銅またはクロムを含む溶液の場合、Cr2O72−からのCr3+の分離またはCu(EDTA)2−からのCu2+の分離は、電気泳動電解質として0.05MCのHNO3または0.04Mの酢酸ナトリウムの何れを使用しても、サンプルプラグの長さが毛細管の全長の1%よりも短い時は、内径150μmの毛細管では不可能であった。サンプル溶液の全濃度が100μg/mlに増大し、サンプルプラグの長さが毛細管の全長の5%になった場合でもなお、内径150μmの毛細管で分離することは難しかった。内径100μmの毛細管では、同一条件で同じような結果を得るには、ずっと短いサンプルプラグ及びずっと高い濃度が必要であった。
【0022】2種類の電気泳動電解質、すなわち0.04Mの酢酸ナトリウムおよび0.05MのHNO3をテストした。0.04Mの酢酸ナトリウム溶液の伝導度は0.05MのHNO3の伝導度よりも遥かに低いので、0.04Mの酢酸ナトリウムに対する電気泳動電流は、0.05MのHNO3に対する電気泳動電流よりも低い。クロムまたは銅の全濃度が10μg/mlで、サンプルプラグの長さが毛細管の全長の5%というような、低濃度サンプルで比較的長いサンプルプラグの場合には、内径150μmまたは内径100μmのいずれの毛細管を用いても、Cr2O72−からCr3+を、またCu(EDTA)2−からCu2+を分離する電気泳動電解質として0.05MのHNO3を使用することができる。しかし、サンプル溶液の全濃度が100μg/mlの時は、同一条件のもとで効率的な分離を得ることは不可能であった。この場合には、内径100μmの毛細管と0.04Mの酢酸ナトリウムが効率的な分離を達成するのに有効であることが分かった。従って、電気泳動毛細管の内径および電気泳動電解質の選択は、サンプル溶液の濃度およびサンプルプラグの長さを考慮に入れなければならないということが結論できる。インターフェースをMCNと接続した場合には、高電解質濃度の緩衝溶液によって惹起されるMCNの機能停止(目詰まり)を回避することができるので、0.05MのHNO3を電気泳動電解質として用いることが望ましい。
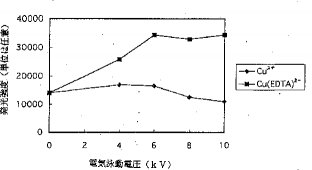
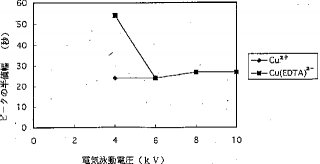
【0023】ピーク高およびピーク幅に及ぼす電気泳動電圧の影響. 電気泳動電圧が陽イオンのピーク高およびピーク幅に与える影響は、陰イオンに与える影響とは異なっており、図11および図12に示す通りである。20%サンプルプラグが毛細管に挿入された時には、陽イオンのピーク高はカラム上濃度(濃縮)および陽イオンの毛細管壁への吸着の両方の影響を受ける。Cu2+とCu(EDTA)2−のシグナル強度は、電圧を印加しない時は等しい。しかし、高電圧を印加した場合には、Cu2+のピーク高は4kVまでは電気泳動電圧に比例して増加し、ついで、電気泳動電圧が大きくなると共に減少する。Cu(EDTA)2−のピーク高は、6kVまで増加し続け、それ以後は、ピークの高さおよび幅は、電気泳動電圧が増大しても殆ど変化を示さない。図12に示されているように、Cu2+は,Cu(EDTA)2−よりも長時間毛細管の中に留まることができるので、濃縮しかつ良好なピーク形を得るためには、4kVで充分であるが、Cu(EDTA)2−に対しては6kVが必要である。高電圧は、陽イオンと陰イオンに異なった影響を持っているので、異なる化学種のピーク高またはピーク面積の比は、化学種の比(量比)と同じではない。従って、正確な結果を得るには、較正を行なわねばならない。図13は、0.5μg/mlのCu2+およびCu(EDTA)2−の電気泳動図を示している。Cu2+のピーク高はCu(EDTA)2−のピーク高よりもずっと低いことが分かる。
【0024】検出限界. 或る元素の検出限界は、ここでは、検体波長におけるバックグラウンド強度の標準偏差の3倍(3σ)に等しい実質シグナルを生じる濃度として定義された。18種の元素の検出限界が、電圧無印加、サンプルはプラズマに連続導入で、CE−MCN−ICP−AESシステムおよび従来型の自己吸引MCN−ICP−AESを用いて得られている。それらの値が表1に纏められている。
【0025】
【表1】

【0026】CE−MCN−ICPでは、内径150μmの毛細管が、電気泳動毛細管および補助毛細管のどちらにも使用された。CE−ICP−AESシステムでは、従来型のICP−AESにおけるよりも、ずっと小容積のサンプルがプラズマに導入されるので、CE−ICP−AESで得られる検出限界は、ICP−AESでの検出限界の1−4倍高い(大きい)。CE−ICP−AES分析では、シグナル強度は、カラム上濃縮技術を利用することによって数倍改善でき、従って高電圧無印加で得られる値よりも低い検出限界が、この技術によって期待できる。
【0027】上記の結果は、本発明のインターフェースが、毛細管電気泳動をICP−AESに接続するのに有効であることを示している。このインターフェースは、容易且つ直接に、従来型の同軸型ニューマティックネブライザーおよびマイクロコンセントリックネブライザーに接続することができる。このCE−ICP−AESシステムによって、Cu2+とCu(EDTA)2−、Cr3+とCr2O72−およびFe3+とFe2+をそれぞれ含むサンプルについて、効率的な分離ならびに十分な感度が得られた。カラム上濃縮技術によって、CE−ICP−AESの感度および分離効率を改善することができる。この実験では、内径150μmの毛細管しか使用しなかったけれども、もっとサイズの大きな毛細管を利用すれば効率的な分離とより高い感度を得ることが可能と思われる。さらに、カラム上濃縮技術を利用すれば、Cu2+とCu(EDTA)2−、Cr3+とCr2O72−、およびFe3+とFe2+を分離する電解質として0.05MのHNO3(pH約1.3)が利用できる。
【図面の簡単な説明】
【図1】毛細管電気泳動を誘導結合プラズマ分光測定法に接続するためのインターフェースの略図。
【図2】CE−CPCN−ICP−AES法で得られたクロムの電気泳動図。
【図3】CE−MCN−ICP−AES法で得られたクロムの電気泳動図。
【図4】CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【図5】サンプルプラグの長さが電気泳動毛細管の全長の30%の時の、CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【図6】注入容積がピーク高に及ぼす影響を示す。
【図7】注入容積がCu2+およびCu(EDTA)2−のピークの半値幅(fullwidth at half−maximum height(FWHM))に及ぼす影響を示す。
【図8】注入容積が分解能に及ぼす影響を示す。
【図9】CE−CPCN−ICP−AES法で得られた鉄の電気泳動図。
【図10】サンプルプラグの長さが電気泳動毛細管の全長の38%の時のCE−CPCN−ICP−AES法で得られた鉄の電気泳動図。
【図11】電圧がピーク高に及ぼす影響。
【図12】電圧が半値幅(FWHM)に及ぼす影響。
【図13】CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【符号の説明】
1 インターフェース
2 電気泳動毛細管
3 補助毛細管
4 補給電解質
5 緩衝電解質
6 ネブライザーのサンプリングチューブ
7 直流電源
8 ネブライザー
9 第1の腕
10 第2の腕
11 第3の腕
12 コネクター
【0001】
【産業上の利用分野】毛細管電気泳動(CE)と誘導結合プラズマ(ICP)を接続するための新しいインターフェースに関するものである。
【0002】
【従来の技術】金属元素の毒性は、化学種に依存することが多い。従って、金属元素の総量ならびにその化学種の分離および定量が生物および環境試料の分析で益々重要に成りつつある。クロマトグラフィ、原子分光測定法および質量分析法のような各種分析技術がこれらの分野で用いられるが、より良い結果を得るために、更に効率的で感度の良い方法が絶えず探し求められていた。
【0003】
【発明が解決しようとする課題】毛細管電気泳動(CE)は、高価で複雑な装置を必要としない、強力で簡単な方法である。この方法は環境、生物学、臨床を含む広い研究分野のサンプルの分析に用いられている。一般に、極く小量のサンプルしかCEに導入できないので、殆どの化学種に対して濃度の点で満足のいく検出限界を得るのは難しい。従って、高感度で高選択性の検出器の開発は、CEが実用になった後の、非常に重要かつ、やり甲斐のある仕事である。現在までのところ、オンカラムでUVおよび蛍光検出器が広く用いられている。しかしながら、これらの検出器の感度は、普通10−5から10−6Mの間である。質量分析、レーザー誘導蛍光分析、電流分析、伝導度分析、化学発光およびレーザーラマン法といった、他の幾つかの検出技術が、CEで分離された検体の高感度、高選択性の検出方法を提供するために研究されてきた。
【0004】誘導結合プラズマ発光測定法(Inductively Coupled Plasma Atomic Emission Spectrometry,ICP−AES)および質量分析法(Mass Spectrometry,MS)は、非常に感度が良く元素選択的検出技術である。これらの技術は、生物試料、環境試料および金属結合蛋白中の微量元素種を定量するために、高速液体クロマトグラフィおよび容積排除クロマトグラフィに接続されている。最近、これらの技術はCEと接続され、非常に有用な検出器であることが分かった。CE−ICP−AES/MSを使って仕事をする時の課題は、CEとICPを結合する効率的なインターフェースを設計することである。従来型の同軸型ニューマティックネブライザー(CPCN)および直接注入ネブライザー(DIN)に結合できるインターフェースが開発されたが、これらインターフェースは、どれも余りにも複雑で、他種のネブライザーに必ずしも接続して使用出来るわけではない。
【0005】
【課題を解決するための手段】本発明は、非常に簡単で殆どあらゆる種類のネブライザーに接続して使えるインターフェースを提供するものであり、3つの濾斗状チューブで構成されており、それぞれのチューブの尖った方の端が集まってY字状を形成し、他方の端が電気泳動毛細管、補助毛細管、およびネブライザーのサンプリングチューブにそれぞれ接続されていることを特徴とする。このインターフェースは、ネブライザーの外部に独立しており、ネブライザーのサンプリングチューブに直接結合させることができるものであり、サンプルはCEで分離され、次いでネブライザー中の高速アルゴンガス気流による自己吸引によりネブライザーに吸入される。このインターフェースは、CPCNおよびマイクロコンセントリックネブライザー(MCN)にうまく接続でき、満足できる感度および効率的分離が得られる。かくして、本発明はまた、電気泳動回路を形成する三又分岐を構成する電気泳動毛細管と補助毛細管とを有し、ネブライザーによる吸引またはペリスタチューブによる送液により電解液の流れを実現し、印可電圧により分離されたイオンを含む電解液の一部を直接導入することを特徴とする、電気泳動による分離されたイオンをネブライザーに直接導入する方法を提供するものである。
【0006】CEでは、検出感度を改善するために各種のオンカラム濃縮技術が用いられる。そうした技術の1つは、大量の低濃度のサンプルプラグを電気泳動毛細管に導入し、高電圧を印加してサンプルプラグの端に検体サンプルを濃縮することである。本発明では、この技術も利用され、感度の改善がもたらされた。大量のサンプルが毛細管に導入され、長いサンプルプラグが形成され、そこでは低濃度のために低伝導度となるので、良好な感度および効率的な分離度を得るために内径の大きな毛細管を用いることができる。また、大きなサンプルプラグでは0.05MのHNO3溶液を電気泳動電解質として用いることができる。
【0007】
【実施例】試薬および物質. 酢酸ナトリウム3水和物(純度99.0%、和光純薬工業)および硝酸(EL、比重1.38、関東化学)を電解質として用いた。K2Cr2O7(純度99.5%、和光純薬工業)、CrCl3・6H2O(純度99.5%、和光純薬工業)、FeCl2(純度97.0%、和光純薬工業)、FeCl3・6H2O(純度99.9%、和光純薬工業)、CuSO4・5H2O(純度99.5%、純正化学)およびエチレンジアミン四酢酸2水素2ナトリウム(純度99.5%、純正化学)を分析種として用いた。全ての溶液は、物質の適量を脱イオン水に溶解して製した。
【0008】毛細管電気泳動. 外径が375μmで、内径がそれぞれ100μm(長さ120cm)と内径150μm(長さ160cm)の2種類の溶融シリカ毛細管(GLサイエンスジャパン)を電気泳動毛細管として、また、長さが40cm、内径が150μmの毛細管を補助毛細管として用いた。高圧直流電源は、松定プレシジョン(株)製で、30kVまでの電圧及び200μA電流を供給できる。外径0.1mmの白金線を電極として用いた。
【0009】ネブライザー. 2種の市販ネブライザーをテストした。CPCN(グラスエクスパンジョンプロプリエータリリミテッド、オーストラリア)およびMCN(CETACテクノロジー、オマハ、ネブラスカ、USA)である。インターフェースは、ネブライザーのサンプリングチューブに直接接続した。ネブライザー毎にキャリヤーガスの流速を種々変えて最適の感度および分解能を得るようにした。
【0010】誘導結合プラズマ発光分光器. アナリティカルリサーチラボラトリーズ(ARL)のMAXIMIII光学発光分光測定器(FISSON製)を用いた。標準トーチMAXIM ICPを、製造業者の推奨通りに、中間のガス流速0.8l/分、外側ガス流速13l/分で使用した。電力は1.1kWであった。
【0011】スプレイチャンバー. 2種類のスプレイチャンバーをMCNおよびCPCNに接続してテストした。MCNの場合には、内部にインパクトビーズのない、円錐形で自作したスプレイチャンバーを用いた。CPCNの場合には、インパクトビーズのある正規のMAXIMタイプのスプレイチャンバー及び、MCNで使用したのと同じスプレイチャンバーを試験した。
【0012】インターフェース. 図1は、CE−ICP−AES用インターフェースであり、このインターフェース1は、石英製であり、その第1の腕9は、電気泳動毛細管2の出口に接続し、第2の腕10は、補給電解質溶液を吸い上げる補助毛細管3に接続されている。この補助毛細管のもう一方の端は、補給電解質として用いられている0.05MのHNO3溶液中に浸されている。インターフェースの第3の腕11は、適当なチューブ例えばペリスタルチックポンプ用チューブによってネブライザー8のサンプリングチューブ6に直結している。図中12は、左右のチューブを結合するコネクターである。毛細管は直接インターフェースの各腕に挿入出来るので何ら前処理を必要としない。キャリヤーガスのバルブを開くと、ネブライザー内の高速アルゴンガス気流によって生じる圧力差によって、補給電解質溶液と電解質溶液またはサンプル溶液が、電気泳動毛細管および補助毛細管に吸い上げられる。2種類の溶液は、2つの腕の接合部で混合し、次いで、ネブライザーの中に吸引される。補給電解質溶液は、直流電源のアース側に接続しており、緩衝電解質溶液は、直流電源の陰極に接続している。負の高電圧によって電解質に自己吸引の流れに反対方向の電気浸透圧による流れを起こさせ、かくしてサンプルは、正の分極電圧を印加した時に比べて長時間毛細管の中に留まることになる。
【0013】サンプル注入. サンプルは、キャリヤーガスの流れによって生じる自己吸引によって直接毛細管に導入される。サンプリングの前および後には、空気を電気泳動毛細管の中に自己吸引して導入することのないように、補助毛細管は、インターフェースから外しておくか、またはキャリヤーガスのバルブを閉じて自己吸引作用がなくなるようにしておかなければならない。サンプルの容積は、2つの方法で測定できる。1つは、サンプリング時間の測定によるものであり、もう1つは、サンプリングの前後に蒸留水の重量を測定するものである。これら2つの方法を用いて測定したサンプリング容積は、それぞれ0.00314ml/分および0.00328ml/分と計算された。これらの測定は、内径150μm(長さ160cm)の毛細管を電気泳動毛細管として用い、MCNを0.25l/分のキャリヤーガス流量で操作して行なった。こうした条件下では、補給溶液の流速は、0.0136ml/分と計算され、この値は、サンプル流速の4倍以上である。このことは、サンプルが補給溶液で4倍以上に希釈されることを意味する。
【0014】CE−ICP−AESのインターフェースの特徴. ここに記載されているCE−ICP−AESのインターフェースの場合、支持電解質溶液は、2つの目的を果たしている。第1に、支持電解質溶液は、完全な電気泳動回路が出来上がるように電気泳動毛細管の出口(アース側)のところでの連続的かつ安定な電気的接触を保つために用いられる。第2に、支持電解質溶液は、ネブライザーに吸引される混合溶液のpH値の調整に使うことができる。一般に、高pH緩衝溶液が電解質溶液として用いられることは、しばしばある。しかし、アルカリ性のpH環境では、殆どの金属イオンは、水酸化物として存在し、それらは、ネブライザーの毛細管壁に容易に吸着されるか、濃度が高過ぎれば沈殿する。このことは、特にMCNを用いた場合には、ネブライザーを詰まらせることになりかねない。もし、0.04Mの酢酸ナトリウムを支持液および電解質溶液とし、そして、Cr2O72−およびCr3+(Cr3+の一部はCr(OH)3の形で存在しているであろう。)をこのシステムで分析すれば、サンプリング2回ないし3回毎にMCNを0.05MのHNO3で3分間ほど洗浄しなければならない。もし、0.04Mの酢酸ナトリウムの替わりに0.05MのHNO3を支持溶液として用いれば、詰まりが起きることは殆どない。補助毛細管の内径は、支持溶液の吸い上げ容積、電気泳動電流、および、電気泳動毛細管と補助毛細管との間の電圧分布に影響する。補助毛細管の内径が大きければ、支持液の吸い上げ容積が増し、目的化学種の希釈度が大きくなる。反対に、補助毛細管の内径が小さければ、希釈度を下げられる。しかし、このことは、インターフェースとネブライザーとの間の液体流速を下げ、それによってピークの幅を広げることになる。従って、この実験では、妥協が必要であり、内径150μmの毛細管が補助毛細管として用いられた。電気泳動電流は、支持液の濃度を変更することによって調整することもでき、従って、電気泳動毛細管と補助毛細管との間の電圧分布も同様に調整することができる。
【0015】CE−ICP−AESのインターフェースの評価. このインターフェースおよびこのシステムを評価するために若干の予備テストが行なわれた。図2および図3は、それぞれCr2O72−およびCr3+を含むテスト溶液についてCE−CPCN−ICP−AES法およびCE−MCN−ICP−AES法によって得られた電気泳動図である。このうち図2は、全濃度:100μg/ml、化学種:Cr3+およびCr2O72−(1:1)、緩衝電解質溶液:0.04Mの酢酸ナトリウム(pH 7.2)、高電圧:15kV、毛細管の内径:100μm、サンプルプラグの長さ:電気泳動毛細管の25%の場合、図3は、全濃度:1μg/ml、化学種:Cr3+とCr2O72−(1:1)、緩衝電解質溶液:0.05MのHNO3、高電圧:8kV、毛細管の内径:150μm、サンプルプラグの長さ:電気泳動毛細管の30%の場合である。どちらの実験においても自作したスプレーチャンバーが用いられた。どちらの場合でも、Cr2O72−およびCr3+の完全分離が観測された。これらの結果は、このインターフェースがCPCNにもMCNにも適しており、CEとICP−AESを接続するのに間違いなく利用できることを明確に示すものである。
【0016】MCNはCPCNよりもずっと高いネブライザー効率を持っているので、CE−MCN−ICP−AES法の感度は、CE−CPCN−ICP−AESの感度よりも遥かに大きい。キャリヤーガス気流によって起きる電気泳動毛細管中の流速が、この2つのシステム間に分解能の差を生じるのである。この流速がCE−CPCN−ICP−AES法におけるよりも遥かに遅いCE−MCN−ICP−AES法では、8kVで約12分で完全な分離が得られる。しかし、CE−CPCN−ICP−AES法では、約4分間15kVでやっとベースライン分離が得られるに過ぎない。2つのピークの高さもこの流速に関連している。流速が低下すると、分離が良くなり、Cr3+のピーク高は減少するがCr2O72−のピーク高は殆ど変化しない。これは、高電荷カチオン(Cr3+)が毛細管壁に捕捉され易く、流速が遅いと、速い時に比べてサンプルが電気泳動毛細管中に長い時間留まることになるからである。このことが、高電荷カチオンの毛細管壁への吸着を増強することになる。電気泳動電圧として陽電圧を用いると、Cr3+がCr2O72−よりも早く毛細管から溶出する。陰電圧の時よりも陽電圧の時の方がCr3+が毛細管中に留まる時間が短いので、Cr3+のシグナル強度は、陰電圧で得られるシグナル強度よりも強い。CE−CPCN−ICP−AES法については、正規のMAXIMタイプのインパクトビーズ付のスプレーチャンバーもテストした。その感度は、現場合わせのインパクトビーズなしのスプレーチャンバーでの感度の少なくとも1/10以下であった。この装置を日常業務の目的で使う場合は、サンプルを2ml/分の割合でネブライザーに導入する。正規のMAXIMタイプのスプレーチャンバーは、大きな液滴がプラズマ中に入るのを防止し、ICPのエアロゾル溶媒による過負荷を防止できる。しかし、このシステムでは、極小量のサンプルしかネブライザーに導入されず、したがって、プラズマの溶媒過負荷を心配する必要はない。
【0017】カラム上濃縮と分離. CE法では、サンプリング容積は、分離および感度に影響する非常に重要な因子である。一般に、サンプリング容積は、電気泳動毛細管の全長の10%以下である。本発明は、カラム上濃縮技術を用いており、普通よりも多量のサンプルが毛細管に導入された。図4および図5は、サンプリング容量を変化した時の銅の電気泳動図である。このうち、図4R>4は、全濃度:10μg/7ml、化学種:Cu2+とCu(EDTA)2−(1:1)、緩衝電解質溶液:0.05MのHNO3、高電圧:8kV、毛細管の内径:150μm、 サンプルプラグの長さ:電気泳動毛細管の5%の場合、図5は、サンプルプラグの長さが電気泳動毛細管の全長の30%で、その他の条件は図4での条件と同じの場合のものである。Cu2+およびCu(EDTA)2−についての図4と図5を比較すると、注入容積の増加が、ピーク幅に顕著な悪影響を及ぼすことなく、ピーク高および分解能をいずれも増加することが明らかである。
【0018】カラム上濃縮操作を以下に説明する。緩衝電解質溶液中よりもサンプルプラグ中にずっと高い電場勾配が生じる。その理由は、サンプルプラグ中の荷電粒子の濃度が、緩衝電解質溶液中よりもずっと低いからである。この(高い電場勾配)ため、サンプルプラグ中の荷電粒子は緩衝電解質溶液中の荷電粒子よりもずっと高速で動く。しかしながら、荷電粒子が緩衝電解質溶液に到達すれば、その領域にはより低い電場勾配しか存在しないので、移動速度は低下する。このことが、サンプル溶液と緩衝電解質溶液との境界に大量の荷電粒子の集積を齎らす。Cu2+とCu(EDTA)2−の運動方向は反対であるから、Cu2+とCu(EDTA)2−とは、サンプルプラグの別々の端に集積する。かくして、Cu2+とCu(EDTA)2−の分離が達成される。相対的に長いサンプルプラグが、ずっと良い分離度及びより多くの荷電粒子のサンプル溶液・緩衝電解質溶液境界での集積を引き起こすことになる。Cr3+およびCr2O72−の分離についも全く同じ事が言える。
【0019】図6、図7および図8は、それぞれ、注入容積とピーク高、注入容積とピーク幅および注入容積と分解能の関係を示している。Cu2+は、Cu(EDTA)2−よりも長時間毛細管の中に留まるので、Cu2+は、より長い時間電気泳動作用を受けて毛細管中に濃縮される。従って、Cu2+のピーク幅は殆ど変化しないが、ピーク高は、注入容積が毛細管の全長の5%から40%まで増加する時、連続的に増加する。それとは対照的に、Cu(EDTA)2−のピーク幅は、注入容積が毛細管の全長の30%を超えると劇的に増加する。しかしながら、30%および40%注入容積でのピーク面積は、20%注入容積でのピーク面積よりも大きいけれども、30%および40%注入容積でのピーク高は、20%注入容積でのピーク高よりも低い。検体の毛細管内滞留時間の延長を齎らすキャリヤーガスの流速低下及び/または電気泳動電圧の増加は、Cu(EDTA)2−のピーク幅を幾分縮小すると期待される。しかしながら、キャリヤーガスの低流速はまた、ピーク幅を拡張させることもあり、また、高電圧は、陽イオンの毛細管壁への吸着を誘発し、シグナル強度を減少させる。
【0020】カラム上濃縮法では、比較的長いサンプルプラグが、低濃度用の反対極性を持った2種の荷電粒子を分離するのに適していると考えられた。何故ならば、これらの荷電粒子は、高電圧が印加されるとサンプルプラグの両端へと移動するからである。高電圧を印加した時に同じ方向に移動するFe3+およびFe2+の分離に対する注入容積の影響も研究した。若干の興味ある現象が観測された。比較的短いサンプルプラグ(電気泳動毛細管の全長の5%から25%)が毛細管に挿入された場合には、2つのピークが得られる。図9は、25%のサンプルプラグの場合の電気泳動図であり、図10は、38%のサンプルプラグの場合の電気泳動図である。後者の場合には、図10に示されているとおり、3つのピークが現われた。第2おび第3のピークはFe2+とFe3+であると推定されている。これらのピークを同定し、この現象を説明するために更に研究が続けられている。
【0021】電気泳動毛細管の内径と電解質. 電気泳動毛細管の内径もまた、分離効率および感度に影響する。一方では、内径の大きな毛細管は、大量のサンプルを導入出来る利点があり、したがって、より低い検出限界が期待できるが、他方、このことは、電気泳動電流の増加を招き、低分解能をきたす恐れがある。2種類の毛細管をテストした。1つは内径100μmの毛細管、もう1つは内径150μmの毛細管である。サンプル溶液の濃度が低く、比較的長いサンプルプラグが毛細管に挿入された時は、電気泳動電流は、低伝導度のサンプルプラグによって決まる。従って、効率的な分離は、このシステムでは内径150μmの毛細管で得られた。全濃度が10μg/mlの銅またはクロムを含む溶液の場合、Cr2O72−からのCr3+の分離またはCu(EDTA)2−からのCu2+の分離は、電気泳動電解質として0.05MCのHNO3または0.04Mの酢酸ナトリウムの何れを使用しても、サンプルプラグの長さが毛細管の全長の1%よりも短い時は、内径150μmの毛細管では不可能であった。サンプル溶液の全濃度が100μg/mlに増大し、サンプルプラグの長さが毛細管の全長の5%になった場合でもなお、内径150μmの毛細管で分離することは難しかった。内径100μmの毛細管では、同一条件で同じような結果を得るには、ずっと短いサンプルプラグ及びずっと高い濃度が必要であった。
【0022】2種類の電気泳動電解質、すなわち0.04Mの酢酸ナトリウムおよび0.05MのHNO3をテストした。0.04Mの酢酸ナトリウム溶液の伝導度は0.05MのHNO3の伝導度よりも遥かに低いので、0.04Mの酢酸ナトリウムに対する電気泳動電流は、0.05MのHNO3に対する電気泳動電流よりも低い。クロムまたは銅の全濃度が10μg/mlで、サンプルプラグの長さが毛細管の全長の5%というような、低濃度サンプルで比較的長いサンプルプラグの場合には、内径150μmまたは内径100μmのいずれの毛細管を用いても、Cr2O72−からCr3+を、またCu(EDTA)2−からCu2+を分離する電気泳動電解質として0.05MのHNO3を使用することができる。しかし、サンプル溶液の全濃度が100μg/mlの時は、同一条件のもとで効率的な分離を得ることは不可能であった。この場合には、内径100μmの毛細管と0.04Mの酢酸ナトリウムが効率的な分離を達成するのに有効であることが分かった。従って、電気泳動毛細管の内径および電気泳動電解質の選択は、サンプル溶液の濃度およびサンプルプラグの長さを考慮に入れなければならないということが結論できる。インターフェースをMCNと接続した場合には、高電解質濃度の緩衝溶液によって惹起されるMCNの機能停止(目詰まり)を回避することができるので、0.05MのHNO3を電気泳動電解質として用いることが望ましい。
【0023】ピーク高およびピーク幅に及ぼす電気泳動電圧の影響. 電気泳動電圧が陽イオンのピーク高およびピーク幅に与える影響は、陰イオンに与える影響とは異なっており、図11および図12に示す通りである。20%サンプルプラグが毛細管に挿入された時には、陽イオンのピーク高はカラム上濃度(濃縮)および陽イオンの毛細管壁への吸着の両方の影響を受ける。Cu2+とCu(EDTA)2−のシグナル強度は、電圧を印加しない時は等しい。しかし、高電圧を印加した場合には、Cu2+のピーク高は4kVまでは電気泳動電圧に比例して増加し、ついで、電気泳動電圧が大きくなると共に減少する。Cu(EDTA)2−のピーク高は、6kVまで増加し続け、それ以後は、ピークの高さおよび幅は、電気泳動電圧が増大しても殆ど変化を示さない。図12に示されているように、Cu2+は,Cu(EDTA)2−よりも長時間毛細管の中に留まることができるので、濃縮しかつ良好なピーク形を得るためには、4kVで充分であるが、Cu(EDTA)2−に対しては6kVが必要である。高電圧は、陽イオンと陰イオンに異なった影響を持っているので、異なる化学種のピーク高またはピーク面積の比は、化学種の比(量比)と同じではない。従って、正確な結果を得るには、較正を行なわねばならない。図13は、0.5μg/mlのCu2+およびCu(EDTA)2−の電気泳動図を示している。Cu2+のピーク高はCu(EDTA)2−のピーク高よりもずっと低いことが分かる。
【0024】検出限界. 或る元素の検出限界は、ここでは、検体波長におけるバックグラウンド強度の標準偏差の3倍(3σ)に等しい実質シグナルを生じる濃度として定義された。18種の元素の検出限界が、電圧無印加、サンプルはプラズマに連続導入で、CE−MCN−ICP−AESシステムおよび従来型の自己吸引MCN−ICP−AESを用いて得られている。それらの値が表1に纏められている。
【0025】
【表1】
【0026】CE−MCN−ICPでは、内径150μmの毛細管が、電気泳動毛細管および補助毛細管のどちらにも使用された。CE−ICP−AESシステムでは、従来型のICP−AESにおけるよりも、ずっと小容積のサンプルがプラズマに導入されるので、CE−ICP−AESで得られる検出限界は、ICP−AESでの検出限界の1−4倍高い(大きい)。CE−ICP−AES分析では、シグナル強度は、カラム上濃縮技術を利用することによって数倍改善でき、従って高電圧無印加で得られる値よりも低い検出限界が、この技術によって期待できる。
【0027】上記の結果は、本発明のインターフェースが、毛細管電気泳動をICP−AESに接続するのに有効であることを示している。このインターフェースは、容易且つ直接に、従来型の同軸型ニューマティックネブライザーおよびマイクロコンセントリックネブライザーに接続することができる。このCE−ICP−AESシステムによって、Cu2+とCu(EDTA)2−、Cr3+とCr2O72−およびFe3+とFe2+をそれぞれ含むサンプルについて、効率的な分離ならびに十分な感度が得られた。カラム上濃縮技術によって、CE−ICP−AESの感度および分離効率を改善することができる。この実験では、内径150μmの毛細管しか使用しなかったけれども、もっとサイズの大きな毛細管を利用すれば効率的な分離とより高い感度を得ることが可能と思われる。さらに、カラム上濃縮技術を利用すれば、Cu2+とCu(EDTA)2−、Cr3+とCr2O72−、およびFe3+とFe2+を分離する電解質として0.05MのHNO3(pH約1.3)が利用できる。
【図面の簡単な説明】
【図1】毛細管電気泳動を誘導結合プラズマ分光測定法に接続するためのインターフェースの略図。
【図2】CE−CPCN−ICP−AES法で得られたクロムの電気泳動図。
【図3】CE−MCN−ICP−AES法で得られたクロムの電気泳動図。
【図4】CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【図5】サンプルプラグの長さが電気泳動毛細管の全長の30%の時の、CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【図6】注入容積がピーク高に及ぼす影響を示す。
【図7】注入容積がCu2+およびCu(EDTA)2−のピークの半値幅(fullwidth at half−maximum height(FWHM))に及ぼす影響を示す。
【図8】注入容積が分解能に及ぼす影響を示す。
【図9】CE−CPCN−ICP−AES法で得られた鉄の電気泳動図。
【図10】サンプルプラグの長さが電気泳動毛細管の全長の38%の時のCE−CPCN−ICP−AES法で得られた鉄の電気泳動図。
【図11】電圧がピーク高に及ぼす影響。
【図12】電圧が半値幅(FWHM)に及ぼす影響。
【図13】CE−MCN−ICP−AES法で得られた銅の電気泳動図。
【符号の説明】
1 インターフェース
2 電気泳動毛細管
3 補助毛細管
4 補給電解質
5 緩衝電解質
6 ネブライザーのサンプリングチューブ
7 直流電源
8 ネブライザー
9 第1の腕
10 第2の腕
11 第3の腕
12 コネクター
【特許請求の範囲】
【請求項1】 3つの濾斗状チューブで構成されており、それぞれのチューブの尖った方の端が集まってY字状を形成し、他方の端が電気泳動毛細管、補助毛細管、およびネブライザーのサンプリングチューブにそれぞれ接続されていることを特徴とする、毛細管電気泳動を誘導結合プラズマ測定に接続するためのインターフェイス。
【請求項2】 電気泳動回路を形成する三又分岐を構成する電気泳動毛細管と補助毛細管とを有し、ネブライザーによる吸引またはペリスタチューブによる送液により電解液の流れを実現し、印可電圧により分離されたイオンを含む電解液の一部を直接導入することを特徴とする、電気泳動による分離されたイオンをネブライザーに直接導入する方法。
【請求項1】 3つの濾斗状チューブで構成されており、それぞれのチューブの尖った方の端が集まってY字状を形成し、他方の端が電気泳動毛細管、補助毛細管、およびネブライザーのサンプリングチューブにそれぞれ接続されていることを特徴とする、毛細管電気泳動を誘導結合プラズマ測定に接続するためのインターフェイス。
【請求項2】 電気泳動回路を形成する三又分岐を構成する電気泳動毛細管と補助毛細管とを有し、ネブライザーによる吸引またはペリスタチューブによる送液により電解液の流れを実現し、印可電圧により分離されたイオンを含む電解液の一部を直接導入することを特徴とする、電気泳動による分離されたイオンをネブライザーに直接導入する方法。
【図 1】

【図 2】
【図 3】
【図 4】
【図 5】
【図 6】
【図 7】
【図 8】
【図 9】
【図 10】
【図 11】
【図 12】
【図 13】
【図 2】
【図 3】
【図 4】
【図 5】
【図 6】
【図 7】
【図 8】
【図 9】
【図 10】
【図 11】
【図 12】
【図 13】
【特許番号】第2838200号
【登録日】平成10年(1998)10月16日
【発行日】平成10年(1998)12月16日
【国際特許分類】
【出願番号】特願平8−353825
【出願日】平成8年(1996)11月29日
【公開番号】特開平10−160706
【公開日】平成10年(1998)6月19日
【審査請求日】平成8年(1996)11月29日
【出願人】(591014709)農林水産省農業環境技術研究所長 (4)
【登録日】平成10年(1998)10月16日
【発行日】平成10年(1998)12月16日
【国際特許分類】
【出願日】平成8年(1996)11月29日
【公開番号】特開平10−160706
【公開日】平成10年(1998)6月19日
【審査請求日】平成8年(1996)11月29日
【出願人】(591014709)農林水産省農業環境技術研究所長 (4)
[ Back to top ]