
カリウムチャネル変異サッカロミセスセレビシエ酵母および真核細胞カリウムチャネルスクリーニングのためのその使用
【課題】 真核細胞カリウムチャネルのインヒビターおよび活性化物質を特定する新規の方法の提供。
【解決手段】 真核細胞カリウムチャネルの阻害物質を特定する方法であって、(a)3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い、(b)真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ、(c)前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして(d)前記真核細胞カリウムチャネルに対する被検物質の作用を決定する方法、ならびにそのような変異サッカロミセスセレビシエ細胞、そのような変異サッカロミセスセレビシエ細胞の製造および使用。
【解決手段】 真核細胞カリウムチャネルの阻害物質を特定する方法であって、(a)3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い、(b)真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ、(c)前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして(d)前記真核細胞カリウムチャネルに対する被検物質の作用を決定する方法、ならびにそのような変異サッカロミセスセレビシエ細胞、そのような変異サッカロミセスセレビシエ細胞の製造および使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、変異サッカロミセスセレビシエ(Saccharomyces cerevisiae)細胞を用いる真核細胞カリウムチャネルの阻害物質および活性化物質を特定する方法に関する。前記変異細胞の内在性カリウムチャネルTRK1、TRK2およびTOK1の機能は発現されないが、前記細胞は研究対象の真核細胞カリウムチャネルを異種発現させる。本発明はまた、TRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞、並びにこれら変異サッカロミセスセレビシエ細胞の作製および使用に関する。
【背景技術】
【0002】
細胞は各々、約6〜8nmの厚さを有する形質膜に包まれている。この膜は細胞の容積を決定し、細胞の内容物をその外界から分離させる。全ての生物膜は連結された脂質分子の二重層で構成され、前記二重層は多様な膜蛋白質を含んでいる。前記脂質二重層が生物膜の基本的構造を決定し、一方、前記蛋白質は生物膜の機能の大半に必要である。その疎水性内層のために、脂質二重層はほとんどの極性分子にとって非浸透性障壁として作用する。レセプター、イオンチャネルおよびおよびトランスポーターのような膜蛋白質のみがイオン流入および極性分子通過の制御を可能にする(Alberts et al.,1995)。したがって蛋白質は細胞内部および細胞周囲の多様なイオン濃度に寄与し、栄養物の進入および分解産物の排出を支配する。膜蛋白質のほとんどは、イオンチャネルのように形質膜の隙間を繰り返し埋め、したがってこれらは内在性膜蛋白質群に属する。これらの蛋白質は、疎水性領域(これは脂質二重層に広がる)および親水性部分(これらは膜のいずれかの面側の水性媒体に露出している)の両方を有する。イオンチャネルは全ての細胞に見出され、さらに神経細胞では活動電位の発生に必要である(Alberts et al.,1995)。イオンチャネルは、それらのイオン選択性の違いを基準に、さらにそれらの開閉メカニズムの相違を参考に区別することができる。
【0003】
カリウムチャネルは、興奮性および非興奮性細胞の両方で見出される不変的な膜蛋白質である(概略については以下を参照されたい:L.Y. Jan et al.,1997)。開放カリウムチャネルは膜電位をカリウム平衡電位に近づけ、その結果、閾電位から外れて活動電位を惹起させる。したがってカリウムチャネルは休止膜電位を強化し細胞を再分極させ、このようにして活動電位の頻度の長さを決定する(M.C. Sanguinetti et al.,1997; A.A. Wilde
et al.,1997; Q. Wang et al.,1998)。これらの機能のために、カリウムチャネルはまた多くの病態の発生の分子的原因を構成し、したがって治療薬剤の開発の重要な標的である。
【0004】
サッカロミセスセレビシエは3つのカリウムチャネル、すなわちTRK1、TRK2およびTOK1を有する。カリウムチャネルTRK1(YJL129c)は、“主要促進因子”カリウム透過酵素族に属し、高親和性カリウムトランスポーターであり媒体から細胞内へのカリウムイオン流入に必要である(R.F. Gaber et al.,1998; C.H. Ko et al.,1990; C.H. Ko et al.,1991)。欠失変異体Δtrk1は生存可能であり、少なくとも10mMK+で強く分極している(R.F. Gaber et al.,1988; R. Madrid et al.,1998)。Δtrk1株は1mMK+では生存できない(R.F. Gaber et.al.,1988)。
【0005】
カリウムチャネルTRK2(YKR050w)はまた“主要促進因子”カリウム透過酵素族に属し、低親和性カリウムトランスポーターで媒体から細胞内へのカリウムの流入に必要である(C.H. Ko et al.,1990; C.H. Ko et al.,1991; R. Madrid et al.,1998)。Δtrk2欠失変異体の表現型はΔtrk1変異体の場合ほど顕著ではない。Δtrk2株はまた1mMK+で
生存できる(C.H. Ko et al.,1990; R. Madrid et al.,1998)。
【0006】
カリウムチャネルTOK1(DVK1またはYORKとしても知られている)は、媒体から細胞内へのカリウムイオンの流入に必要である(K.A. Ketchum et al.,1995; C. Fairman et al.,1999)。しかしながら、イオン流入の方向は逆転可能で、したがって培養条件に応じて反対方向を取ることができる(C. Fairman et al.,1999)。
【0007】
その欠失変異体Δtrk1Δtrk2は既に繰り返して報告されている(C.H. Ko et al.,1990;
C.H. Ko et al.,1991; R. Madrid et al.,1998; C. Fairman et al.,1999)。
【0008】
これまで前記変異体は、表現型の相補性による高等真核細胞のK+チャネルの特定および記述のために用いられてきた。これまでに報告されれたものは、内方向整流チャネルKAT1cDNA(Arabidopsis thaliana)、HKT1cDNA(Triticum aestivum)、IRK1(Mus musculus)およびHKT1K+/Na+トランスポーター(Triticum aestivum)による相補性である(W. Tang et al.,1995; F.W. Smith et al.,1995; S.A. Goldstein et al.,1996; R.L. Nakamura et al.,1997)。さらに、TOK1およびキイロショウジョウバエ(Drosophila melanogaster)由来のその同族体ORK1の酵母細胞での過剰発現は、Δtrk1Δtrk2変異体の発育不全を相補できることが報告された(C. Fairman et al.,1999)。
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、例えばヒトのチャネルHERG1またはKv1.5は5mMのKClでΔtrk1Δtrk2の致死的表現型を相補できず、したがってスクリーニングできないので、多数の真核細胞のカリウムチャネルおよびカリウムチャネルの作用を改変できる物質の特定に関する研究が困難になっている。
【課題を解決するための手段】
【0010】
本発明は真核細胞のカリウムチャネルの阻害物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の影響を決定する。
【0011】
本方法で用いる変異サッカロミセスセレビシエ細胞では、遺伝子TRK1、TRK2およびTOK1(配列番号1、配列番号2および配列番号3)は、好ましくはノックアウトによってスイッチが切られており(Δtrk1、Δtrk2、 Δtok1)、好ましくは遺伝子の大部分が欠失している。
【0012】
本方法で用いる真核細胞カリウムチャネルは、調べようとしているカリウムチャネル、その阻害物質または活性化物質を特定しようとしているチャネルである。
例えば、真核細胞カリウムチャネルは、ヒトHERG1、ヒトKv1.5、ヒトROMK2またはgpIRK1(モルモット)チャネルである。真核細胞カリウムチャネルは、好ましくは問題のカリウムチャネルの天然の配列、例えば配列番号4、配列番号5、配列番号7(ROMK2)または配列番号6の配列の1つによってコードされるものを有する。しかしながら、カリウムチャネルの天然の配列はまた改変、例えば変異させたものでもよい。
【0013】
好ましくは、真核細胞カリウムチャネルをコードするヌクレオチド配列は、酵母の発現プラスミド、例えばp423GPD3、またはベクター(例えばpRS42xまたはpRS32xシリーズ)に組み込まれ、前記組換え発現プラスミドは変異サッカロミセスセレビシエ細胞に導入される。
【0014】
前記方法は、真核細胞カリウムチャネルに対して作用を及ぼす物質を特定することを目的とする。これらの物質は変異サッカロミセスセレビシエ細胞の増殖を抑制する。異種発現された真核細胞カリウムチャネルを抑制する被検物質は、前記変異サッカロミセスセレビシエ細胞は内在性カリウムチャネルを発現しないので、その分裂および成長を非常に困難にさせるか、または遅らせるか、または本発明の実施態様では致死させる。
【0015】
被検物質の作用は、例えば光学密度を600nmで測定するか、または変異サッカロミセスセレビシエ細胞で構成的に発現される増殖レポーターを用いて直接測定することができる。前記構成的に発現される増殖レポーターは、好ましくは、それ自体蛍光もしくは化学発光を示す蛋白質、または蛍光もしくは化学発光シグナルを生じる反応に加わる蛋白質をコードする。前記増殖レポーターをコードする配列は、好ましくはベクター由来である。適当な増殖レポーターは、例えばβ−ガラクトシダーゼのLacZ遺伝子または酸性ホスファターゼPH03(両方とも構成的酵母プロモーターの制御下で発現される)である。測定可能な蛍光または化学発光によって変異サッカロミセスセレビシエ細胞数に関する結果が得られる。蛍光もしくは化学発光が全くないかまたは少ない場合は、問題のサンプルに含まれる変異サッカロミセスセレビシエ細胞の数が少ないことになる。変異サッカロミセスセレビシエ細胞の数が少ない場合は、被検物質は真核細胞のカリウムチャネルに対して抑制作用を有することになる。
【0016】
前述の方法は特に容易な自動化を可能にし、平行的に多数の物質の検査を実施することができる。本発明の特定の実施態様では、2つまたは3つ以上の方法が比較的態様で実施され、その場合2つまたは3つ以上のサッカロミセスセレビシエ細胞が比較的態様で分析される。これらの変異サッカロミセスセレビシエ細胞は好ましくは同じ量の被検物質と一緒にインキュベートされるが、前記細胞は問題の真核細胞カリウムチャネルを種々の程度に発現する。本発明のまた別の実施態様では、問題の真核細胞カリウムチャネルの発現は同程度であるが、異なる量の被検物質と一緒にインキュベートされた変異サッカロミセスセレビシエ細胞が比較的態様で分析される。
【0017】
本発明の特徴はまた、内在性カリウムチャネルTRK1、TRK2およびTOK1が発現されない変異サッカロミセスセレビシエ細胞である。さらに別の特徴は、遺伝子TRK1、TRK2およびTOK1のスイッチが切られた変異サッカロミセスセレビシエ細胞に関する。前記遺伝子は、好ましくはそれら全体または一部分がノックアウトによって除去されている。さらにまた別の特徴は、特許手続き上の微生物の寄託の国際的承認に関するブダペスト条約に従いドイツ微生物および培養細胞コレクション(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Mascheroder Weg 1b, D-38124 Braunschweig)に寄託された変異サッカロミセスセレビシエに関する(寄託番号DSM13197)。
【0018】
本発明の特徴は真核細胞カリウムチャネルを異種発現する変異サッカロミセスセレビシエ細胞に関する。前記真核細胞カリウムチャネルは、好ましくはヒトカリウムチャネル(例えばHERG1、Kv1.5)、またはgpIRK1またはヒトKv4.3[Genbank Accession Number AF187963]、TASK[Genbank Accession Number AF006823]またはROMK2[Genbank Accession Number U12542]で、前記カリウムチャネルは天然の配列を有していてもよいし、また変異していてもよい。
【0019】
本発明はまた変異サッカロミセスセレビシエ細胞の製造方法に関する。前記細胞はカリウムチャネルTRK1、TRK2およびTOK1を発現せず、遺伝子TRK1、TRK2およびTOK1はノックアウトにより破壊されているかまたは欠失している。
本変異サッカロミセスセレビシエ細胞は、例えば、真核細胞カリウムチャネルの活性を抑制または活性化させる物質を特定する方法で用いることができる。また前記細胞は、例えば毒性物質の決定に用いることができる検査キットの部分でもよい。
【0020】
本発明はまた真核細胞のカリウムチャネルの活性化物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュヘ゛ートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の作用を決定する。
【0021】
本発明はさらにまた真核細胞のカリウムチャネルの活性化物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を前記真核細胞カリウムチャネルの阻害物質の存在下で被検物質と一緒にインキュベートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の作用を決定する。
【0022】
本発明はまた医薬の製造方法に関し、本方法では、
(a) 真核細胞カリウムチャネルの阻害物質を特定し;
(b) 前記阻害物質を公知の化学的方法によって製造または単離し;さらに
(c) 生理学的に許容できる添加物を前記阻害物質に添加する。
【0023】
本発明はまた医薬の製造方法に関し、本方法では、
(a) 真核細胞カリウムチャネルの活性化物質を特定し;
(b) 前記活性化物質を公知の化学的方法によって製造または単離し;さらに
(c) 生理学的に許容できる添加物を前記活性化物質に添加する。
【0024】
添付図面において:
図1は、三重ノックアウトを証明するための診断PCRを示す。ゲルの段/レーンの説明については実施例2の三重ノックアウトを参照されたい。
図2は、規定のKCl濃度を含むpH6.5のDPM培地でのYM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の増殖を示す。
図3は、5mMのKCl+2mMのRbClを含むDPM培地(pH6.5)でのYM189株およびYM190株(Δtrk1Δtrk2)並びにYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【0025】
図4は、5mMのKCl+2mMのCsClを含むDPM培地(pH6.5)でのYM189株およびYM191株(Δtrk1Δtrk2)並びにYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
図5は、5mMのKCl+1mMのRbClを含むDPM培地(pH6.5)でのYM19
4株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。(“KON”=コントロール)
図6は、5mMのKCl+1mMのCsClを含むDPM培地(pH6.5)でのYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。(“KON”=コントロール)
【0026】
図7は、活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mMKCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度の測定のために、市販のLacZレポーター系pYX232(Ingenius, Cat.No.MBV-032-10)で分析酵母株を形質転換した。LacZレポーター遺伝子の発現は、トリオースホスフェートイソメラーゼ遺伝子のための構成的サッカロミセスセレビシエプロモーターTPIの制御下にあった。LacZ酵素活性は、市販のアッセイ系(TROPIX)を用いて24時間後の化学発光を検出することによって測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。2つの別個のアッセイを別の日に互いに別々に実施した。
【0027】
図8は、活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度は、620nmの波長で光学密度を測定することによって38時間増殖後に測定した(出発培養密度:0.03OD620)。値は各事例で4つの測定値の平均±SDに一致する。
【0028】
図9は、0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mM KCl培地におけるサッカロミセスセレビシエの増殖を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度の測定のために、市販のLacZレポーター系pYX232(Ingenius, Cat.No.MBV-032-10)で分析酵母株を形質転換した。LacZレポーター遺伝子の発現は、トリオースホスフェートイソメラーゼ遺伝子のための構成的サッカロミセスセレビシエプロモーターTPIの制御下にあった。LacZ酵素活性は、市販のアッセイ系(TROPIX)を用いて24時間後の化学発光を検出することによって測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。2つの別個のアッセイを別の日に互いに別々に実施した。
【0029】
図10は、0.5mMのKClまたは80mMのKClが存在する96ウェルELISAプレートのDPM培地におけるサッカロミセスセレビシエ野生型株の増殖を示す。
それぞれの事例で阻害物質、ジプラシドンおよびピモジドを最終濃度30μMで用いた。細胞密度は、620nmの波長で光学密度を測定することによって24時間増殖後に測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。
【0030】
図11は、5mMのKClおよび活性化物質として0.5mMのCsClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルHERG1の発現を示す。
1:陰性コントロールとしてブランクベクターp423GPDが発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。2:陽性コントロールとしてp423GPD−TRK1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。3:p423GPD−HERG1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。4:p423GPD−HERG1が
発現している二重変異体Δtrk1Δtrk2の増殖。使用したベクターおよび構築物は本明細書で説明されている(段落0039以下および段落0043以下の配列を参照されたい)。
【0031】
図12は、5mMのKClおよび活性化物質として2mMのRbClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルKv1.5の発現を示す。
1:陰性コントロールとしてブランクベクターp423GPDが発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。2:陽性コントロールとしてp423GPD−TRK1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。3:p423GPD−Kv1.5が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。4:p423GPD−Kv1.5が発現している二重変異体Δtrk1Δtrk2の増殖。使用したベクターおよび構築物は本願明細書で説明されている(段落0039以下および段落0043以下の配列を参照されたい)。
【0032】
図13は、96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルROMK2の発現および陰性コントロールとしての酵母ベクターp423GPDの発現を示す。
細胞密度は、620nmの波長で光学密度を測定することによって24時間増殖後に測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。
図14は、p423GPD−ROMK2のプラスミドマップを示す。
【図面の簡単な説明】
【0033】
【図1】三重ノックアウトを証明するための診断PCRを示す。
【図2】規定のKCl濃度を含むpH6.5のDPM培地でのYM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図3】5mMのKCl+2mMのRbClを含むDPM培地(pH6.5)でのYM189株およびYM190株(Δtrk1Δtrk2)並びにYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図4】5mMのKCl+2mMのCsClを含むDPM培地(pH6.5)でのYM189株およびYM191株(Δtrk1Δtrk2)並びにYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図5】5mMのKCl+1mMのRbClを含むDPM培地(pH6.5)でのYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図6】5mMのKCl+1mMのCsClを含むDPM培地(pH6.5)でのYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図7】活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
【図8】活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
【図9】0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mMKCl培地におけるサッカロミセスセレビシエの増殖を示す。
【図10】0.5mMのKClまたは80mMのKClが存在する96ウェルELISAプレートのDPM培地におけるサッカロミセスセレビシエ野生型株の増殖を示す。
【図11】5mMのKClおよび活性化物質として0.5mMのCsClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルHERG1の発現を示す。
【図12】5mMのKClおよび活性化物質として2mMのRbClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルKv1.5の発現を示す。
【図13】96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルROMK2の発現および陰性コントロールとしての酵母ベクターp423GPDの発現を示す。
【図14】p423GPD−ROMK2のプラスミドマップを示す。
【実施例】
【0034】
材料および株
培地:
YPD(完全酵母培地):1%バクト(Bacto)酵母抽出物、2%バクトペプトン、2%バクト寒天、2%グルコース。
SC(合成完全)培地:0.67%バクト酵母窒素ベース、アミノ酸、2%グルコース。
胞子形成培地:1%酢酸カリウム、アミノ酸。
5−FOA培地:0.67%バクト酵母窒素ベース、アミノ酸、ウラシル(50μg/mL)、2%糖(ガラクトースまたはグルコース)、0.1%5−FOA。
全ての培地は次の文献に記載されている:(G.R. Fink et al.,1991)。
【0035】
アミノ酸ドロップアウトミックス:
L−アラニン2g;L−アルギニン2g;L−アスパラギン*H2O 2.27g;L−アスパラギン酸2g;L−システイン*HCl 2.6g;L−グルタミン2g;L−グルタミン酸2g;グリシン2g;ミオイノシトール2g;L−イソロイシン2g;L−メチオニン2g;PABA0.2g;L−フェニルアラニン2g;L−セリン2g;L−スレオニン2g;L−チロシン2g;L−バリン2g。
【0036】
マーカーアミノ酸用ストック溶液:
mM g/L
アデニン(100x) 30 5.53 加熱(60℃未満)
ロイシン(60x) 100 13.12 加熱
リジン(100x) 100 18.26 −
ヒスチジン(200x) 60 12.57 −
トリプトファン(100x) 40 8.17 −
ウラシル(100x) 20 2.24 0.5%NaHCO3溶液中で加熱
【0037】
ビタミン原液(50mL):
ビオチン20μg/L;パントテン酸カルシウム40μg/L;チアミン40μg/L
。
【0038】
限定カリウム培地(DPM):1.5L用(2×原液):
(NH4)2HPO4 8mM 3.2g
(NH4)2SO4 29mM 11.5g
MgSO4 2mM 0.8g(または1M原液6mL)
CaCl2 0.2mM 90μg(または0.5M原液1.2mL)
ビタミン原液 120μL
アミノ酸ドロップアウトミックス 6g
リジン 100倍原液330mL
アデニン 0.9mM 100倍原液30mL
→上記をHClでpH6.5(または他のpH)としオートクレーブする。
グルコース 2% 40%原液より
KCl 1M原液より
必須アミノ酸(Lys/Adeは除外)は原液から
寒天
【0039】
緩衝液および溶液:
TE緩衝液:トリス/HCl(pH7.5)10mM;EDTA(pH8.0)1mM;
TAE緩衝液:トリス40mM;EDTA1mM;酢酸0.2mM;
SSC緩衝液(20x):NaCl 3M;クエン酸ナトリウム*2H2O 0.3M;
ゲルローディング緩衝液:ブロムフェノールブルー0.05%(w/v);スクロース40%(w/v);EDTA(pH8.0)0.1M;SDS 0.5%(w/v)
ハイブリダイゼーション緩衝液:SSC 5x;SDS 0.1%(w/v);硫酸デキストラン5%(w/v);停止試薬1:20;
緩衝液A(無菌):トリス−HCl 100mM;NaCl(pH9.5) 300mM;
脱プリン溶液:HCl 0.25M;
変性溶液:NaCl 1.5M;NaOH 0.5M;
中和溶液:NaCl 1.5M;トリス(pH8.0)0.5M。
【0040】
オリゴヌクレオチド(PCRプライマー):

【0041】
【0042】
ベクター:
細菌ベクター
【0043】
酵母ベクター
【0044】
株:
細菌株:DH5α;ワンショット(One ShotTM) TOP10(Invitrogen)
酵母株:
この実験のために作製した全ての酵母株は以下の二倍体野生型株をベースにしている:W303MATa/α ade2.his3−11−15,leu2−3−112,trp1−1,ura3−1,can1−100;ATCC#208352
【0045】
【0046】
以下の酵母株を作製した:
【0047】
【0048】
カリウムチャネルクローン:
A)
系統名 KCNA5
別称 Kv1.5,(HK2,HPCN1)
種類 電圧開閉カリウムチャネル、shaker関連サブファミリー
(メンバー番号5)、遅延整流チャネル
染色体上の位置 12p13.32−p13.31
登録番号 NID g4504818
蛋白質 613アミノ酸、67kD
組織内分布 心臓、膵臓ランゲルハンス島およびインスリノーマ
同族体 mKcna5(Mus musculus)、70%がhHCN4と相同
参考文献 (S.L. Roberds et al.,1991; M.E. Curran et al.,1992;
D.J. Snyders et al.,1993)
【0049】
B)
系統名 HCN2
別称 BCNG2(脳環状ヌクレオチド開閉チャネル)、HAC1
種類 過分極−活性化および環状ヌクレオチド開閉カリウムチャネ
ル、電圧開閉カリウムチャネルのサブファミリーに属する
染色体上の位置 19p13.3
登録番号 NID g4996893
蛋白質 889アミノ酸
機能 ペースメーカー
組織内分布 脳、心臓
同族体 mHcn2(Mus musculus)
参考文献 (A. Ludwig et al.,1999)
【0050】
C)
系統名 KCNH2
別称 HERG1(より長いスプライス変種)
種類 電圧開閉カリウムチャネル、eag関連サブファミリー
(メンバー番号2)
染色体上の位置 7q35−q36
登録番号 NID g4557728
特性 K+チャネル調節因子1によってチャネル活性化が促進
参考文献 (M. Taglialatela et al.,1998; T. Itoh et al.,1998)
【0051】
D)
系統名 KCNJ2(モルモット)
別称 Kir2.1,IRK1
種類 内向き整流カリウムチャネル
組織内分布 脳、心臓、肺臓、腎臓、胎盤、骨格筋
参考文献 (W. Tang et al.,1995)
【0052】
方法:
ROMK2(後記の“ROMK2の配列”を参照されたい)
【0053】
PCR:
パワースクリプト(Powerscript)ポリメラーゼ(PAN Biotech)用プロトコル:
ローワー(lower)試薬用ミックス(ホットスタートプロトコル)(25μL):
3μLのH2O;2.5μLの10×オプチパフォーム(OptiPerform(登録商標))III緩衝液(pH9.2);10μLの1.25mM dNTP(=200μM);
1.5μLの前進プライマー(20pmol/μL);1.5μLの逆方向プライマー(20pmol/μL);1.5μLの50mg MgCl2(=1.25mM);5μLの5×オプチザイム(OptiZyme(登録商標))エンハンサー
【0054】
アッパー試薬用混合物(35μL):
23μLのH2O;3.5μLの10×オプチパフォーム(OptiPerform(登録商標))III緩衝液;1.5μLの50mM MgCl2;0.5μLのパワースクリプトDNAポリメラーゼ;7μLの5×オプチザイム(OptiZyme(登録商標))エンハンサー。
【0055】
PCRプログラム(ホットスタート):
1.94℃で1分
2.94℃で1分
3.50〜55℃(プライマーによる)で1.5分
4.69〜72℃(ポリメラーゼによる)で4分
5.2番から27回繰り返す
6.4℃∞
7.終了
【0056】
アンプリタック(AmpliTaq)ポリメラーゼ(Perkin Elmer)用プロトコル:
アッパー試薬用ミックス(ホットスタートプロトコル)(50μL):
18.1μLのH2O;4.2μLの10×緩衝液II;16.7μLのdNTP;2.5μLの前進プライマー;2.5μLの逆方向プライマー;6μLの25mM MgCl2(=1.5mM)。
ローワー試薬用ミックス(50μL):
42μLのH2O;5μLの10×緩衝液II;1μLのアンプリタック(AmpliTaq)ポリメラーゼ;2μLの鋳型。
【0057】
DNAの精製
PCR反応物の精製:PCR増幅生成物の精製はキット(High Pure PCR Product Purification Kit(Roche))を用いて実施した。
フェノール抽出:サンプル容積をTE緩衝液で200μLにする。200μLのフェノール/クロロホルム/イソアミルアルコール(25:24:1)を加え、混合し、最大速度で1分間遠心する。上部相を新しいエッペンドルフ試験管に移し、200μLのクロロホルム/イソアミルアルコールを加え、混合し1分間遠心する。上部相を取り出しエタノールで沈殿させる。
【0058】
エタノール沈殿:約200μLのサンプル容積に5μLの5M NaClおよび20μLの3M NaAc(pH5.7)をピペットで加える。2.5容の100%エタノールを添加して混合し、少なくとも30またはそれ以上−20℃で静置する。4℃で10分間遠心し、ペレットを170μLの70%冷エタノールで洗浄する。3分間遠心し、ペレットを37℃で乾燥させ、さらに30μLのH2Oに再懸濁させる。
【0059】
大腸菌(E. coli)のプラスミドDNAの単離:大腸菌オーバーナイト培養のプラスミドDNAの単離はキアゲン(Qiagen)のプロトコル(QIApre Spin Miniprep Kit Orotocl)を用いて実施した。
【0060】
サッカロミセスセレビシエのDNAの調製:
前記酵母細胞を10mLのYPDで一晩30℃でインキュベートし、次の朝、3000rpmで10分間遠心し、ペレットを1Mソルビトール、0.1MEDTA(pH7.5)の500μLに再懸濁し、エッペンドルフ試験管に移す。50μLのザイモラーゼ(zymolase)(ソルビトール/EDTA中に5mg/mL)を添加し、37℃で1時間インキュベートし、さらに1分間遠心する。50mMトリス、20mMのEDTA(pH7.4)500μLにペレットを再懸濁する。50μLの10%SDSを添加し、完全に混合し、さらに65℃で30分間インキュベートする。5MのKAc200μLを加え、氷上に1時間置き、1
0分間遠心する。上清(約650μL)を新しいエッペンドルフ試験管に移し、1容量のイソプロパノールを加え、穏やかに混合し、さらに5分間静置する。簡単に遠心するか、または沈殿したDNAをガラスのかぎ棒で抜き取り、空気中で前記ペレットを乾燥させる。ペレットまたはDNAを150μLのTE緩衝液に再懸濁し、65℃で10分間溶解させる。
DNAクローニング:全てのDNAクローニングは標準的なプロトコルにしたがって実施した。
【0061】
酵母の形質転換(酢酸リチウム法):
形質転換しようとする酵母株を5mLの適当な培養液中で一晩30℃でシェーカー上でインキュベートする。次の朝前記オーバーナイト培養を適当な培養液で希釈し(OD600=0.40.5)、さらに2時間シェーカー上で30℃でインキュベートする(OD600=0.40.8)。2500rpmで3分間遠心し、25mLの滅菌H2Oでペレットを洗浄する。2500rpmで3分間遠心し、1mLのLITE(100mMのLiAc,TE(pH7.5))に再懸濁し、懸濁液をエッペンドルフ試験管に移す。RTで5分間インキュベートし、15秒間遠心する(Quickspin)。100mMのLiAc 1mLでペレットを洗浄し(quick-spin)、細胞濃度に応じてペレットを200400μLの100mM LiAcに再懸濁して50μLの部分標本に分ける。
【0062】
表示の順番どおりに以下を加える:
240μLのPEG(50%)、ピペットで穏やかに懸濁物を混合する;
36μLの1M LiAc、ピペットで穏やかに懸濁物を混合する;
10μLの一本鎖精子DNA(−20℃で保存;使用前に80−90℃で10分間加熱してから氷に移す);
2〜3μgのプラスミドDNA(またはノックアウトトランスフォーメーションの場合には8〜10μLのミニプレップ(Miniprep))、ピペットで穏やかに懸濁物を混合し;
形質転換反応物をオーバーヘッド回転装置で低速で30℃30分間インキュベートする;
42℃15分間形質転換反応;
クィックスピンで遠心、ペレットを200μLのTE緩衝液に再懸濁(ノックアウトの場合は300μLのYPDにペレットを再懸濁)し、オーバーヘッド回転装置で30℃で4時間インキュベートする;
寒天平板当たり100μL(ノックアウトのばあいは全反応物)を平板培養し、30℃で3〜4日間インキュベート。
【0063】
配列決定:ABIPRISM(登録商標) red.プロトコルII/AmpliTaq(登録商標) FS1/4BigDyeターミネーター
【0064】
反応物:
プレミックス 2μL
DNA鋳型
ssDNA 50ng
dsDNA 250ng
PCR生成物(0.2−5kB) 10−50ng
プライマー 3〜10pmol
H2Oで最終容積 10μL
【0065】
サーモサイクラープロトコル(25サイクル):
1.96℃で15秒間
2.96℃で15秒間
3.55℃で10秒間
4.60℃で4分間
5.2.に戻って24サイクル
6.4℃∞
7.終了
【0066】
精製反応(セントリセップスピンカラム(Centri Sep Spin Colum)(Princeton Separations)):
カラムを750μLのH2Oで30分間予め膨潤させ、液体を排出させて2分間3000rpmで遠心する。反応物をH2Oで20μLにしてカラムに適用し、2分間3000rpmで遠心する。
サンプル適用:配列決定試験管に、4μLのセントリセップ(Centri Sep)溶出液+20μLのTSR(鋳型抑制試薬)、90℃で2分間。
【0067】
サザンブロット:
DNAプローブを適当な制限酵素で消化、ゲル電気泳動で分離し、ゲルから抽出する。ゲノムDNAを適当な制限酵素で一晩消化し、ゲル電気泳動で分離する(1%アガロースゲル)。
【0068】
ゲルの前処理:アガロースゲルからローディングウェルを取り出す。アガロースゲルを0.25MのHClで15分間脱プリン処理し、続いて蒸留水で2回洗浄する。アガロースゲルを0,5MのNaOHで30分間変性させ、ヴァキュームブロッターモデル785(Vaccum Blotter Model 785, BioRad)を用いてビニールシートの中央に移し、ウィンドー(ウィンドーシール)を切り取り、ナイロン膜と濾紙の端を切り揃え(各々の事例でゲルより0.5cm小さい)、ナイロン膜の端を蒸留水で湿らし(各々の事例でビニールシートの小窓より0.5cm広い)、続いてナイロン膜および濾紙をトランスファー溶液で湿らす。
【0069】
装置の構築(下から上へ):
ベースユニット、真空土台部、多孔性真空スラブ、濾紙、ナイロン膜、ビニールウィンドー、アガロースゲル、最終フレーム、蓋。
バイオラッド(BioRad)真空ポンプを10分間予熱し、真空を適用する(16932Pa(5インチHg))。
端に沿ってゲルを穏やかに圧迫する。
トランスファー溶液(約1L、10×SSC)を上部液だめに入れる;移動時間:90分;真空のスイッチを切り、ナイロン膜を取り出し、5分間2×SSCで洗浄し、続いて濾紙の間で風乾させる。DNA固定:ナイロン膜をUV透過性の密着フィルム上に置き、陽性コントロールとして端にプローブを適用する;UVストラタリンカーに入れ架橋を開始させる(1200000J→0);膜は粘着フィルム内またはワットマン濾紙の間で室温または4℃で保存することができる。
【0070】
ジーンイメージ・ランダムプライムラベリングモジュール(Amersham):
DNAプローブの標識:DNAプローブを96℃で5分間変性させ(ヒートショック)、その後氷上に置く。10μLの反応ミックス(ヌクレオチドミックス(5×)、トリス−HCl(pH7.8)中のフルオレセイン−11−dUTP,dATP,dCTP,dGTPおよびdTTP、2−メルカトエタノールおよびMgCl2);5μLのプライマー(Random Nonamers)、1μLの酵素溶液(クレノーフラグメント、5単位/mL);22μLの変性DNAプローブ;12μLのH2O。2時間37℃でインキュベートし、2μLのO.5M EDTA(=20mM)を添加し、部分標本を−20℃で保存する。標識効率の確認:5×ヌクレオチドミックスをTE緩衝液で1/5、1/10、1/25、1/50、1/100、1/250および1/500に希釈し;ナイロン膜の細片に5μLの
DNAプローブを5μLの1/5希釈と一緒に適用してさっと吸収させ、予め温めておいた2×SSCで60℃で15分間洗浄し;1/5希釈を除いた残りの溶液を参照用膜細片に適用し、両方の膜細片をUV光の下で観察し、サンプルの濃さを決定する。
【0071】
ハイブリダイゼーション:ナイロン膜(ブロット)を温かいハイブリダイゼーション緩衝液(0.3mL/cm2)で、回転オーブンで60℃2時間予備ハイブリダイズさせ;緩衝液を除きその10mLを残す。DNAプローブを変性させ(96℃で5分間、続いて氷上で冷却);プローブを前記10mLの緩衝液とともにブロット上に加え、回転オーブンで60℃で一晩ハイブリダイズさせる。
【0072】
洗浄工程:
温かい1×SSC,0.1%(w/v)SDS中でプラットフォームシェーカー上で15分;温かい0.5×SSC,0.1%(w/v)SDS中でプラットフォームシェーカー上で15分。
【0073】
ジーンイメージ・CDP−スターディテクションモジュール(Amersham):
停止および抗体反応:シェーカーの上で、緩衝液A中の停止試薬1/10希釈中で1時間室温でブロットをインキュベートする。抗体溶液(アルカリ性ホスファターゼ結合抗フルオレセイン抗体、5000×)を0.5%(w/v)BSA/緩衝液Aでブロットシールと一緒に金属薄片中で希釈し、さらにシェーカー上で1時間室温でインキュベートする。10分間緩衝液A中の0.3%トゥイーン(Tween)20で3回洗浄して未結合抗体溶液を除去する。
【0074】
シグナル発生および検出:洗浄緩衝液を排出し、ブロットを密着フィルム上に置き、5mLの検出試薬を適用し、2〜5分反応させ、再度排液し(アルカリ性ホスファターゼは発光を引き起こす);密着フィルムで包み、暗室の赤色光の中でフィルム(ハイパーフィルム(Hyperfilm(登録商標)MP, Amersham)を用い、フィルムカセット(BioMax, Kodak)中で0.5〜2時間露光させ、現像してスキャンする。ブロットは4℃で密着フィルム中で保存できる。
【0075】
実施例1:特異的欠失カセットの構築
欠失は全て標準的な方法によって実施した(G.R. Fink et al.,1991; A. Wach et al.,1994; U. Guldener et al.,1996; A.L. Goldstein et al.,1999)。
各々約500bpのフラグメント(それぞれは遺伝子の初めと終わりの領域をもつ)を以下のプライマーを用いてPCRにより増幅させた:TRK1の場合はTRK1-FL-BamHI-Fo、 TRK1-FL-PstI-Re、 TRK1-FL-PstI-FoおよびTRK1-FL-XhoI-Re、 TRK2の場合はTRK2-DEL-5-Fo-B、 TRK2-DEL-5-Re、 TRK2-DEL-3-FoおよびTRK2-DEL-3-Re、 並びにTOK1の場合はTOK1-DEL-5-Fo、 TOK1-DEL-5-Re、 TOK1-DEL-3-FoおよびTOK1-DEL-3-Re(2.3章参照)。増幅させた末端は後の酵母ゲノムへの正確な組み込みを可能にする。酵母株w303a/αまたはw303a/αΔtrk1はDNA鋳型として機能する。
【0076】
実施例2:単一、二重、三重変異体の構築
実施例2a:単一ノックアウト
TRK1、TRK2およびTOK1のための構築欠失カセットでそれぞれ二倍体酵母株YM96(MATa/MATα)を形質転換した。ゲノムへの欠失カセットの組み込みは、(−)URA/Glcでtrk1変異体(YM123/124)を、さらにYPD/ジェニチシンでtrk2変異体(YM158−161)およびtok1変異体(YM154−157)を増殖させることによって確認した。なぜならば、TRK1欠失カセットのURA3マーカーは(−)URA培地での増殖を可能にし、TRK2またはTOK1のKANマーカーはゲネチシン(geneticin)での増殖を可能にするからである(G.R. Fink et
al.,1991)。陽性コロニーを胞子形成平板にレプリカ平板培養により移し、ここで栄養成長を行わずに18−24時間後にMATa/MATα二倍体細胞は胞子を形成する。それらをザイモラーゼで処理し、再びYPD上で増殖させた後、解剖顕微鏡を用いていくつかのコロニーの四分子を個々の4つの胞子に分割する。
【0077】
胞子コロニーの交配型を交配テスター株との交配によって決定した(G.R. Fink et al.,1991)。欠失カセットの有無についての選別は、trk1については−URA培地、trk2およびtok1についてはゲネチシン含有培地でのレプリカ平板培養によって実施した。酵母のDNA調製によって形質転換体のゲノムDNAを得た後で、結果を診断用PCRおよびサザンブロットによって確認した。
【0078】
実施例2b:二重ノックアウト
TOK1欠失カセットで一倍体Δtrk1酵母株YM123およびYM124を形質転換し、YPD/ゲネチシンでの増殖によりTOK1欠失カセットの組み込みについて選別した。結果は診断用PCRおよびサザンブロットにより確認した。グリセロール培養は以下の株を用いて実施した:(+)URA3、(+)KAN)(Δtrk1Δtok1)株(YM140、YM141、YM143およびYM144)。単一コロニーをパッチとして画線培養し、5−FOA上にレプリカ培養し、TRK1欠失カセットからURA3マーカーおよびhisGリピートを排除したコロニーを選別した(G.R. Fink et al.,1991)。したがって、ウラシル合成のためのURA3遺伝子(TRK1で)を欠いたコロニーはいずれも(−)URA/Glcでは増殖せず、一方、TOK1欠失カセット中の耐性遺伝子により全てのコロニーがYPD/gen上で生存する。ゲノムからKanマーカーを除去するために、(−)URA3変異体をプラスミドpSH47で形質転換した。前記pSH47にはCreリコンビナーゼおよびウラシル合成(URA3)のための遺伝子が存在する。陽性形質転換体は(−)URA/Glcで増殖し、したがって(−)URA/Gal液体培地でのインキュベーションによってCreリコンビナーゼを誘発することが可能であった。この方法で、Kanマーカーは1つのloxPリピートとともに排除され、1つのloxPは保持される。
【0079】
オーバーナイト培養でOD600=5となった後で、1:10000および1:50000の希釈を(−)URA/Galで平板培養した。YPD/genでレプリカ培養した単一コロニーパッチは増殖を示さなかった(これはKanマーカーの排除は成功であったことを示している)。プラスミドpSH47を除去するために、その後、細胞を5−FOAで2回選別を繰り返した。グリセロール培養は(−)ura(−)kan(Δtrk1Δtok1)株(YM162、YM163およびYM164)を用いて実施した。
【0080】
実施例2c:三重ノックアウト
YPDでのオーバーナイト培養を単一Δtrk1Δtok1コロニー(YM162およびYM164)を用いて開始し、次の日、BsiWI/SpeI消化TRK2欠失カセットで形質転換し、YPD/KCl/ジェネチシンで平板培養した。酵母DNAを調製した後、この三重ノックアウトを診断用PCRおよびサザンブロットで確認した。
【0081】
【0082】
実施例3:ヒトカリウムチャネルのサブクローニング並びに二重および三重変異体の形質転換
ヒト遺伝子HERG、HCN2、Kv1.5および陽性コントロールとしてTRK1、IRK1(モルモット)をそれらを含むプラスミド(HERGはpcDNAのBamHI間に、 HCN2はpTLNのNcoI/XhoI間に、 Kv1.5はpcDNA3.1(-)のNheI/EcoRI間に、 IRK1はpSGEMのBamHI/EcoRI間に存在)から制限酵素を用いて切断して切り出し、ゲル電気泳動によって分離し、さらにゲルから抽出した。個々のヒトカリウムチャネルを酵母ベクターp423−GPD3(D. Mumberg et al.,1995; V. Ronicke et al.,1997)に連結し、大腸菌を形質転換した。プラスミド調製物のコントロール消化および配列決定によって前記のヒト遺伝子が組み込まれたクローンを特定することができた。続いてこれらのプラスミドでΔtrk1Δtrk2二重ノックアウト(YM168)およびΔtrk1Δtrk2Δtok1三重ノックアウト(YM182)を形質転換し、 (−)HIS/80mMKCl上で平板培養した。
【0083】
実施例4:ノックアウト株の性状決定
実施例4a:種々のK+濃度およびpH値の培養平板上での二重および三重変異体の増殖
種々のノックアウトのカリウム要求の相違を比較するために、酵母株YM182、YM168およびYM97(WT)を、種々のK+濃度およびpH値をもつDPM平板でインキュベートした。この目的のために、グリセロール培養のパッチを先ず最初に100mM KCl/pH6.5に画線培養した。2日間増殖させた後、50mM、30mMおよび5mM KClでレプリカ平板培養を実施した。
この実験によって、YM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の両株は50mMおよび30mMのKClで生存可能であることが示された。さらに、YM182株はYM168株よりも30mMのKClの存在下でより良好に増殖することが判明した。この2つの株のいずれも、野生型株YM97とは対照的に5mMのKClの存在下で生存できなかった。
【0084】
pH依存性を調べるために、前記3株をさらに100mMおよび5mM KCl/pH5.0並びに100mMおよび5mM KCl/pH4.0上でレプリカ平板培養した。この実験は、
YM168およびYM182はいずれもpH4.0では100mM KClでも5mM KClでも生存できないことを示した。pH5.0で100mM KClでは、YM168の増殖欠損はYM182の場合よりも激しい。ベクターpRS416GALのTRK1の発現は、YM168株(Δtrk1Δtrk2)およびYM182(Δtrk1Δtrk2Δtok1)の増殖欠損を完全に補完する。
【0085】
実施例4b:種々のK+濃度の液体培地での二重および三重変異体の増殖
YM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)(以後の全ての実験はこれらを基準にしている)の性状を調べるために、これら酵母株の液体培養中の増殖態様を調べた。最初に、オーバーナイト培養をDPM/80mM KCl中で開始させ、次の朝、DPM/5mM KClおよびDPM/15mM KClで前記培養をOD=0.05にした。それぞれ規定の間隔後に600nmでの光学密度を光度計により決定した。
これらの実験によって、YM182株の増殖欠損は、5mM KClおよび15mM KClではYM168よりも強くないことが判明した。
【0086】
実施例5:二重および三重ノックアウトのヒトカリウムチャネルの性状決定
実施例5a:培養平板のK+欠乏の補完能力
YM168(Δtrk1Δtrk2)およびYM182(Δtrk1Δtrk2Δtok1)の各株を、酵母発現ベクターとしてp423−GPD3中のヒトカリウムチャネルKv1.5((D. Fedida et al.,1998);YM190およびYM195)およびHERG1((D. Fedida et al.,1998);YM191およびYM196)でそれぞれ形質転換した。gpIRK1(W. Tang et al.,1995);YM193およびYM198)は酵母発現ベクターp423−GPD3中の陽性コントロールとして機能した(D. Mumberg et al.,1995; V. Ronicke et al.,1997)。ブランクベクターp423−GPD3(YM189およびYM194)は陰性コントロールとして機能した。形質転換酵母株を(−)HIS/80mM KCl上で平板培養した。この後、単一コロニーのパッチをDPM/5mM KCl(pH6.5)上でレプリカ平板培養し、カリウム欠乏を補完する能力を調べた。
【0087】
これらの実験によって、p423−GPD3の陽性コントロールgpIRK(YM193およびYM198)は完全に二重および三重ノックアウトの増殖欠損を補完することが示された。陰性コントロールとしてのブランクベクターp423−GDP3(YM189およびYM194)はこの増殖欠損を補完することができない。ヒトカリウムチャネルKv1.5は三重ノックアウトの増殖欠損を補完するが、陽性コントロールgpIRKよりはるかに効率が悪い。さらにまた、ヒトカリウムチャネルKv1.5は二重ノックアウトΔtrk1Δtrk2を補完しない。与えられた実験条件下では、HERGチャネルは二重および三重ノックアウトの増殖欠損を補完しない。
【0088】
実施例5b:活性化物質が存在する培養平板での増殖
種々のカリウムチャネルに対する活性化物質の作用を示すために、上記の株を以下の特定の活性化物質を含む培地中でインキュベートした。
Kv1.5:Rb+は過分極相を延長させる。これは、内向きのK+の流れはより長く続き、増殖欠損を補完する可能性を高めることを意味している。
HERG:Cs+は過分極相を延長させる。これは、内向きのK+の流れはより長く続き、増殖欠損を補完する可能性を高めることを意味している。このチャネルはCs+によって抑制される。
IRK1:Cs+はこのチャネルを阻害する。
p423−GPD3−Kv1.5を用いた実験によって、ヒトKv1.5チャネルは、2mM RbClの存在下でΔtrk1Δtrk2Δtok1変異体の増殖欠損を完全に補完できることが示された(図3)。Δtrk1Δtrk2変異体の増殖欠損の補完は極めて効率が低い(図3)。これは実施例6aで示される結果と一致する。
p423−GPD3−HERGを用いた実験によって、ヒトHERG1チャネルは、2mMCsClの存在下でΔtrk1Δtrk2Δtok1変異体の増殖欠損を完全に補完できることが示された(図4)。Δtrk1Δtrk2変異体の増殖欠損の補完は極めて効率が低い(図4)。これは実施例6aで示される結果と一致する。
【0089】
実施例5c:液体培養液中のRbClの存在下におけるΔtrk1Δtrk2Δtok1変異体のKv1.5による補完
酵母株YM194およびYM195を1mMのRbClを含むDPM/−HIS/5mMKCl中で液体培地における増殖態様の相違について調べた。この目的のために、10mLのオーバーナイト培養をDPM/−HIS/80mM KCl中で開始し、次の朝対応する培養液を用いてOD600を0.05にした(最終容積:20mL)。600nmでの光学密度は規定の間隔で光度計により測定した。
これらの実験は、TRK1、TRK2およびTOK1を欠く酵母株におけるベクターp423−GPD3のKv1.5の発現は前記によってもたらされた増殖欠損を補完することができることを明瞭に示した。
さらに別の実験で、Kv1.5およびgpIRKによる増殖欠損の補完は2mMのCsClの存在下で抑制されることが示された。
【0090】
実施例5d:液体培地中のCsCl存在下でのΔtrk1Δtrk2Δtok1変異体におけるHERG1チャネルによる補完
酵母株YM194およびYM196を1mMのCsClを含むDPM/−HIS/5mM KCl中で液体培地における増殖態様の相違について調べた。この目的のために、10mLのオーバーナイト培養をDPM/−HIS/80mM KCl中で開始し、次の朝対応する培養液を用いてOD600を0.05にした(最終容積:20mL)。600nmでの光学密度は規定の間隔で光度計により測定した。
これらの実験は、TRK1、TRK2およびTOK1を欠く酵母株におけるベクターp423−GPD3のHERG1の発現は前記によってもたらされた増殖欠損を補完できることを明瞭に示した。
【0091】
実施例6:
三重変異体Δtrk1Δtrk2Δtok1における全ての増殖アッセイは、各事例で表示したpHおよびカリウム濃度で増殖培地DPM(規定カリウム培地)で実施した。
ヒトHERG1K+チャネルの阻害物質として用いられた物質は、テルフェナジン(α−(4−tert−ブチルフェニル)−4−(α−ヒドロキシ−αフェニルベンジル)−1−ピペリジンブタノール;HMR)、 ピモジド(1−(4,4−bis(P−フルオロフェニル)ブチル)−4−(2−オキソ−1−ベンゾイミダゾリニル)−ピペリジン;Sigma, Cat.No.P100)、ジプラシドン(5−(2−[4−(1,2−ベンゾイソチアゾール−3−イル)ピペラジノ]−エチル)−6−クロロ−1,3−ジヒドロ−2H−インドール−2−オン;HMR)、ロラタジン(エチル4−(8−クロロ−5,6−ジヒドロ−11H−ベンゾ[5,6]シクロヘプタ[1,2−b−]ピペリジン−11−イリデン)−1−ピペリジンカルボキシレート;HMR)およびセルティンドール(1−(2−[4−[5−クロロ−1−(4−フルオロフェニル)−1H−インドール−3−イル]−1−ピペリジニル]エチル)−2−イミダゾリジノン;HMR)であった(E. Richelson, 1996; E. Richelson, 1999; E. Delpon et. al.,1999; T. Kobayashi et al.,2000; M.D. Drich et al.,2000)。カリウムチャネルに対して抑制作用をもたないはずの物質、ジフェニルヒドラミン(Sigma, Cat.No.D3630)およびフェキソフェナジン(4−[ヒドロキシ−4−[4−(ヒドロキシジフェニルメチル)−1−ピペリジニル]ブチル]−α,α−ジメチルベンゼン酢酸ヒドロクロリド;HMR)(M. Taglialatela et al.,1999; L.M. DuBuske, 1999)もまた用いた。全ての物質はDMSOに溶解し、最終濃度30μMで使用した。コントロールとして、前記物質を含まない0.5%の同じ最終濃度のDMSOを有する細胞、
DMSOが添加されていない細胞、物質が添加されていない細胞を測定した。
【0092】
図1および2に示したように、ヒトHERG1チャネルは、5mM KClのみを含む培地で三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができる。テルフェナジン、ピモジド、ジプラシドン、セルティンドールおよびロラタジンの存在下では、ヒトHERG1チャネルは、もはや5mM KClのみを含む培地では三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができない。
【0093】
実施例7:
酵母の3つの内在性カリウムチャネル蛋白質を全て発現する野生型株をテルフェナジン、ピモジド、ジフェニルヒドラミン、ジプラシドン、ロラタジン、フェキソフェナジンおよびセルティンドールとともにインキュベートしたとき、テルフェナジン、ロラタジンおよびセルティンドールはヒトHERG1チャネルの特異的阻害物質であることが示された(図9)。
【0094】
この結果によれば、ピモジドおよびジプラシドンはむしろ非特異的阻害物質であると考えるべきである。このことは、これらの物質はおそらくヒトHERG1チャネルだけでなくサッカロミセスセレビシエの内在性カリウムチャネルもまた抑制することを意味している。しかしながら、この結果は、これら物質について認められた抑制作用は、酵母細胞の増殖に必須の他の物質の抑制に起因する可能性を排除することができなかった。この可能性を調べるために、80mMのKClを含む増殖培地でこれらの物質の作用も調べた。
【0095】
これらの結果(図10)によって、ピモジドは、非特異的態様で内在性カリウムチャネルTRK1およびTRK2の活性を抑制することが明らかになった。より高いカリウム濃度で抑制作用が認められないことによって、ピモジドは酵母細胞で不変的な毒性作用をもたないと結論することができる。対照的に、ジプラシドンはより高いカリウム濃度でさえ酵母細胞の増殖を抑制し、したがってサッカロミセスセレビシエに対する毒性作用をもつことが明らかになった。この作用に必須である酵母の標的蛋白質はまだ特定されていない。
結論として、上記の実験は、前述の系によって、ヒトカリウムチャネルを特異的に抑制する物質を実際にサッカロミセスセレビシエで特定することができることを示した。
前記の結果は図10に示されている。
【0096】
実施例8:
ヒトカリウムチャネルHERG1およびKv1.5は二重変異体Δtrk1Δtrk2の増殖欠損を補完しない(図11および図12)。
結果:図11および図12
図11および12は、ヒトカリウムチャネルHERG1およびKv1.5は二重変異体Δtrk1Δtrk2の増殖欠損を補完しないことを示している(各事例について図11および12の第4の部分)。陰性コントロール、すなわち三重変異体Δtrk1Δtrk2Δtok1におけるブランクベクターp423GPD(各事例について図11および12の第1の部分)と比較したとき、増殖が改善されていないことが示されている。二重変異体Δtrk1Δtrk2の陰性コントロールp423GPDは表示されていないが、三重変異体Δtrk1Δtrk2Δtok1の陰性コントロールp423GPDと異ならない。対照的に、ヒトカリウムチャネルHERG1およびKv1.5は三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完する(各事例について、図11および12の第三の部分)。
【0097】
実施例9:
ヒトカリウムチャネルROMK2((M.E. Shuck et al.,1994; J.H. Bock et al.,1997);配列番号31、hROMK2)を酵母ベクターp423GPDでサブクローニングし、
三重変異体Δtrk1Δtrk2Δtok1を形質転換した。この実験によって、前記ヒトカリウムチャネルもまた三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができることが示された。
このヒトカリウムチャネルが二重変異体Δtrk1Δtrk2の増殖欠損を補完できるか否かはまだ調べられていない。ROMK2チャネルを特異的に抑制する物質はまだ判明していない。
前記の結果は図13に示されている。
【0098】
【表1】
【0099】
【表2】
【0100】
【表3】
【0101】
【表4】
【0102】
【表5】
【0103】
【表6】
【0104】
【表7】
【0105】
【表8】
【0106】
【表9】
【0107】
【表10】
【0108】
【表11】
【0109】
【表12】
【0110】
【表13】
【0111】
【表14】
【0112】
【表15】
【0113】
〔参考文献〕
Curran, M. E., Landes, G. M., and Keating, M. T. Molecular cloning, characterization, and genomic localization of a human potassium channel gene. Genomics 12:
729-737. (1992)
Dascal, N., Schreibmayer, W., Lim, N. F., Wang, W., Chavkin, C., DiMagno, L., Labarca, C., Kieffer, B. L., Gaveriaux-Ruff, C., and Trollinger, D. Atrial G pr
otein-activated K+ channel: expression cloning and molecular properties. Proc. Natl. Acad. Sci. U.S.A. 90: 10235-1O239. (1993)
Fairman, C., Zhou, X., and Kung, C. Potassium uptake through the TOK1 K+ channel in the budding yeast. J. Membr. Biol. 168: 149-157. (1999)
Fedida, D., Chen, F. S., and Zhang, X. The 1997 Stevenson Award Lecture. Cardiac K+ channel gating: cloned delayed rectifier mechanisms and drug modulation. Can. J. Physiol. Pharmacol. 76: 77-89.(1998)
Fink, G. R. and Guthrie, C. Guide to Yeast Genetics and Molecular Biology. Guthrie, C. and Fink, G. R. (194). 1991. Academic Presss, Inc. Methods in Enzymology. Ref Type: Book, Whole
Gaber, R. F., Styles, C. A., and Fink, G. R. TRK1 encodes a plasma membrane protein required for high-affinity potassium transport in Saccharomyces cerevisiae. Mol. Cell. Biol. 8: 2848-2859. (1988)
Goldstein, A. L. and McCusker, J. H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae [In Process Citation] Yeast.
15: 1541-1553.(1999)
Goldstein, S. A., Price, L. A., Rosenthal, D. N., and Pausch, M. H. ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae [published erratum appears in
Proc Natl Acad Sci USA 1999 Jan 5; 96(1) :318] Proc. Natl. Acad. Sci. U.S.A. 93: 13256-13261. (1996)
Guldener, U., Heck, S., Fielder, T., Beinhauer, J., and Hegemann, J. H. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic. Acids. Res. 24: 2519-2524. (1996)
Ikeda, K., Kobayashi, K., Kobayashi, T., Ichikawa, T., Kumanishi, T., Kishida,
H., Yano, R., and Manabe, T. Functional coupling of the nociceptin/ orphanin FQ
receptor with the G-protein-activated K+ (GIRK) channel. Brain Res. Mol. Brain Res. 45: 117-126. (1997)
Itoh, T., Tanaka, T., Nagai, R., Kamiya, T., Sawayama, T., Nakayama, T., Tomoike, H., Sakurada, H., Yazaki, Y., and Nakamura, Y. Genomic organization and mutational analysis of HERG, a gene responsible for familial long QT syndrome. Hum. Genet. 102: 435-439. (1998)
Jan, L. Y. and Jan, Y. N. Cloned potassium channels from eukaryotes and prokaryotes. Annu. Rev. Neurosci. 20 : 91-123 : 91-123. (1997)
Jelacic, T. M., Sims, S. M., and Clapham, D. E. Functional expression and characterization of G-protein-gated inwardly rectifying K+ channels containing GIRK3. J. Membr. Biol. 169: 123-129. (1999)
Ketchum, K. A., Joiner, W. J., Sellers, A. J., Kaczmarek, L. K., and Goldstein, S. A. A new family of outwardly rectifying potassium channel proteins with two
pore domains in tandem. Nature 376: 690-695. (1995)
Ko, C. H., Buckley, A. M., and Gaber, R. F. TRK2 is required for low affinity K+ transport in Saccharomyces cerevisiae. Genetics 125 : 305-312. (1990)
Ko, C. H. and Gaber, R. F. TRK1 and TRK2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 4266-4273. (1991)
Kubo, Y., Reuveny, E., Slesinger, P. A., Jan, Y. N., and Jan, L. Y. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel [see comments] Nature 364: 802-806. (1993)
Ludwig, A., Zong, X., Stieber, J., Hullin, R., Hofmann, F., and Biel, M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 18 : 2323-2329. (1999)
Madrid, R., Gomez, M. J., Ramos, J., and Rodriguez-Navarro, A. Ectopic potassium uptake in trk1 trk2 mutants of Saccharomyces cerevisiae correlates with a highly hyperpolarized membrane potential. J. Biol. Chem. 273: 14838-14844. (1998)
Main, M. J., Brown, J., Brown, S., Fraser, N. J., and Foord, S. M. The CGRP receptor can couple via pertussis toxin sensitive and insensitive G proteins. FEBS
Lett. 441 : 6-10. (1998)
Mumberg, D., Muller, R., and Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119-122. (1995)
Myers, A. M., Pape, L. K., and Tzagoloff, A. Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae. EMB0 J. 4: 2087-2092. (1985)
Nakamura, R. L., Anderson, J. A., and Gaber, R. F. Determination of key structural requirements of a K+ channel pore. J. Biol. Chem. 272: 1011-1O18. (1997)
Roberds, S. L. and Tamkun, M. M. Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proc. Natl. Acad. Sci. U.S.A. 88: 1798-1802. (1991)
Ronicke, V., Graulich, W., Mumberg, D., Muller, R., and Funk, M. Use of conditional promoters for expression of heterologous proteins in Saccharomyces cerevisiae. Methods Enzymol. 283: 313-22: 313-322. (1997)
Sanguinetti, M. C. and Zou, A. Molecular physiology of cardiac delayed rectifier K+ channels. Heart Vessels Suppl 12: 170-172. (1997)
Schreibmayer, W., Dessauer, C. W., Vorobiov, D., Gilman, A. G., Lester, H. A.,
Davidson, N., and Dascal, N. Inhibition of an inwardly rectifying K+ channel by
G-protein alpha-subunits. Nature 380: 624-627. (1996)
Smith, F. W., Ealing, P. M., Hawkesford, M. J., and Clarkson, D.T. Plant members of a family of sulfate transporters reveal functional subtypes. Proc. Natl. Acad. Sci. U.S.A. 92: 9373-9377. (1995)
Snyders, D. J., Tamkun, M. M., and Bennett, P.B.A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analisis after stable mammalian cell culture expression. J. Gen. Physiol. 101: 513-543. (1993)
Taglialatela, M., Castaldo, P., Pannaccione, A., Giorgio, G., and Annunziato, L. Human ether-a-gogo related gene (HERG) K+ channels as pharmacological targets: present and future implications. Biochem. Pharmacol. 55: 1741-1746. (1998)
Tang, W., Ruknudin, A., Yang, W. P., Shaw, S. Y., Knickerbocker, A., and Kurtz, S. Functional expression of a vertebrate inwardly rectifying K+ channel in yeast. Mol. Biol. Cell 6: 1231-124O. (1995)
Wach, A., Brachat, A., Pohlmann, R., and Philippsen, P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 10: 1793-18O8. (1994)
Wang, Q., Chen, Q., and Towbin, J. A. Genetics, molecular mechanisms and management of long QT syndrome. Ann. Med. 30: 58-65. (1998)
Wilde, A. A. and Veldkamp, M. W. lon channels, the QT interval, and arrhythmias. Pacing. Clin. Electrophysiol. 20: 2048-2051. (1997)
Wischmeyer, E., Doring, F., Spauschus, A., Thomzig, A., Veh, R., and Karschin,
A. Subunit interactions in the assembly of neuronal Kir3.0 inwardly rectifying K+ channels. Mol. Cell Neurosci. 9: 194-206. (1997)
Yamada, M., Inanobe, A., and Kurachi, Y. G protein regulation of potassium ion
channels. Pharmacol. Rev. 50: 723-760. (1998)
Bock, J. H., Shuck, M. E., Benjamin, C. W., Chee, M., Bienkowski, M. J., and Slightom, J. L. Nucleotide sequence analysis of the human KCNJ1 potassium channel
locus Gene 188: 9-16. (1997)
Delpon, E., Valenzuela, C., and Tamargo, J. Blockade of cardiac potassium and other channels by antihistamines Drug Saf 21 Suppl 1: 11-8; discussion 81-7: 11-18. (1999)
Drici, M. D. and Barhanin, J. Cardiac K+ channels and drug-acquired long QT syndrome Therapie 55: 185-193. (2000)
DuBuske, L. M. Second-generation antihistamines: the risk of ventricular arrhythmias Clin. Ther. 21: 281-295. (1999)
Itoh, T., Tanaka, T., Nagai, R., Kikuchi, K., Ogawa, S., Okada, S., Yamagata, S., Yano, K., Yazaki, Y., and Nakamura, Y. Genomic organization and mutational analysis of KVLQT1, a gene responsible for familial long QT syndrome Hum. Genet. 103: 290-294. (1998)
Kobayashi, T., Ikeda, K., and Kumanishi, T. Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K(+) (GIRK) channels expressed in Xenopus oocytes Br. J. Pharmacol. 129: 1716-1722.(2OOO)
Richelson, E. Preclinical pharmacology of neuroleptics: focus on new generation compounds J. Clin. Psychiatry 57 Suppl 11: 4-11: 4-11. (1996)
Richelson, E. Receptor pharmacology of neuroleptics: relation to clinical effects [see comments] J. Clin. Psychiatry 60 Suppl 10: 5-14: 5-14. (1999)
Shuck, M. E., Bock, J. H., Benjamin, C. W., Tsai, T. D., Lee, K. S., Slightom,
J. L., and Bienkowski, M. J. Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel J. Biol. Chem. 269: 24261-24270. (1994)
Taglialatela, M., Castaldo, P., Pannaccione, A., Giorgio, G., Genovese, A., Marone, G., and Annunziato, L. Cardiac ion channels and antihistamines: possible mechanisms of cardiotoxicity Clin. Exp. Allergy 29 Suppl 3: 182-9: 182-189. (1999)
【0114】
【0115】
【技術分野】
【0001】
本発明は、変異サッカロミセスセレビシエ(Saccharomyces cerevisiae)細胞を用いる真核細胞カリウムチャネルの阻害物質および活性化物質を特定する方法に関する。前記変異細胞の内在性カリウムチャネルTRK1、TRK2およびTOK1の機能は発現されないが、前記細胞は研究対象の真核細胞カリウムチャネルを異種発現させる。本発明はまた、TRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞、並びにこれら変異サッカロミセスセレビシエ細胞の作製および使用に関する。
【背景技術】
【0002】
細胞は各々、約6〜8nmの厚さを有する形質膜に包まれている。この膜は細胞の容積を決定し、細胞の内容物をその外界から分離させる。全ての生物膜は連結された脂質分子の二重層で構成され、前記二重層は多様な膜蛋白質を含んでいる。前記脂質二重層が生物膜の基本的構造を決定し、一方、前記蛋白質は生物膜の機能の大半に必要である。その疎水性内層のために、脂質二重層はほとんどの極性分子にとって非浸透性障壁として作用する。レセプター、イオンチャネルおよびおよびトランスポーターのような膜蛋白質のみがイオン流入および極性分子通過の制御を可能にする(Alberts et al.,1995)。したがって蛋白質は細胞内部および細胞周囲の多様なイオン濃度に寄与し、栄養物の進入および分解産物の排出を支配する。膜蛋白質のほとんどは、イオンチャネルのように形質膜の隙間を繰り返し埋め、したがってこれらは内在性膜蛋白質群に属する。これらの蛋白質は、疎水性領域(これは脂質二重層に広がる)および親水性部分(これらは膜のいずれかの面側の水性媒体に露出している)の両方を有する。イオンチャネルは全ての細胞に見出され、さらに神経細胞では活動電位の発生に必要である(Alberts et al.,1995)。イオンチャネルは、それらのイオン選択性の違いを基準に、さらにそれらの開閉メカニズムの相違を参考に区別することができる。
【0003】
カリウムチャネルは、興奮性および非興奮性細胞の両方で見出される不変的な膜蛋白質である(概略については以下を参照されたい:L.Y. Jan et al.,1997)。開放カリウムチャネルは膜電位をカリウム平衡電位に近づけ、その結果、閾電位から外れて活動電位を惹起させる。したがってカリウムチャネルは休止膜電位を強化し細胞を再分極させ、このようにして活動電位の頻度の長さを決定する(M.C. Sanguinetti et al.,1997; A.A. Wilde
et al.,1997; Q. Wang et al.,1998)。これらの機能のために、カリウムチャネルはまた多くの病態の発生の分子的原因を構成し、したがって治療薬剤の開発の重要な標的である。
【0004】
サッカロミセスセレビシエは3つのカリウムチャネル、すなわちTRK1、TRK2およびTOK1を有する。カリウムチャネルTRK1(YJL129c)は、“主要促進因子”カリウム透過酵素族に属し、高親和性カリウムトランスポーターであり媒体から細胞内へのカリウムイオン流入に必要である(R.F. Gaber et al.,1998; C.H. Ko et al.,1990; C.H. Ko et al.,1991)。欠失変異体Δtrk1は生存可能であり、少なくとも10mMK+で強く分極している(R.F. Gaber et al.,1988; R. Madrid et al.,1998)。Δtrk1株は1mMK+では生存できない(R.F. Gaber et.al.,1988)。
【0005】
カリウムチャネルTRK2(YKR050w)はまた“主要促進因子”カリウム透過酵素族に属し、低親和性カリウムトランスポーターで媒体から細胞内へのカリウムの流入に必要である(C.H. Ko et al.,1990; C.H. Ko et al.,1991; R. Madrid et al.,1998)。Δtrk2欠失変異体の表現型はΔtrk1変異体の場合ほど顕著ではない。Δtrk2株はまた1mMK+で
生存できる(C.H. Ko et al.,1990; R. Madrid et al.,1998)。
【0006】
カリウムチャネルTOK1(DVK1またはYORKとしても知られている)は、媒体から細胞内へのカリウムイオンの流入に必要である(K.A. Ketchum et al.,1995; C. Fairman et al.,1999)。しかしながら、イオン流入の方向は逆転可能で、したがって培養条件に応じて反対方向を取ることができる(C. Fairman et al.,1999)。
【0007】
その欠失変異体Δtrk1Δtrk2は既に繰り返して報告されている(C.H. Ko et al.,1990;
C.H. Ko et al.,1991; R. Madrid et al.,1998; C. Fairman et al.,1999)。
【0008】
これまで前記変異体は、表現型の相補性による高等真核細胞のK+チャネルの特定および記述のために用いられてきた。これまでに報告されれたものは、内方向整流チャネルKAT1cDNA(Arabidopsis thaliana)、HKT1cDNA(Triticum aestivum)、IRK1(Mus musculus)およびHKT1K+/Na+トランスポーター(Triticum aestivum)による相補性である(W. Tang et al.,1995; F.W. Smith et al.,1995; S.A. Goldstein et al.,1996; R.L. Nakamura et al.,1997)。さらに、TOK1およびキイロショウジョウバエ(Drosophila melanogaster)由来のその同族体ORK1の酵母細胞での過剰発現は、Δtrk1Δtrk2変異体の発育不全を相補できることが報告された(C. Fairman et al.,1999)。
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、例えばヒトのチャネルHERG1またはKv1.5は5mMのKClでΔtrk1Δtrk2の致死的表現型を相補できず、したがってスクリーニングできないので、多数の真核細胞のカリウムチャネルおよびカリウムチャネルの作用を改変できる物質の特定に関する研究が困難になっている。
【課題を解決するための手段】
【0010】
本発明は真核細胞のカリウムチャネルの阻害物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の影響を決定する。
【0011】
本方法で用いる変異サッカロミセスセレビシエ細胞では、遺伝子TRK1、TRK2およびTOK1(配列番号1、配列番号2および配列番号3)は、好ましくはノックアウトによってスイッチが切られており(Δtrk1、Δtrk2、 Δtok1)、好ましくは遺伝子の大部分が欠失している。
【0012】
本方法で用いる真核細胞カリウムチャネルは、調べようとしているカリウムチャネル、その阻害物質または活性化物質を特定しようとしているチャネルである。
例えば、真核細胞カリウムチャネルは、ヒトHERG1、ヒトKv1.5、ヒトROMK2またはgpIRK1(モルモット)チャネルである。真核細胞カリウムチャネルは、好ましくは問題のカリウムチャネルの天然の配列、例えば配列番号4、配列番号5、配列番号7(ROMK2)または配列番号6の配列の1つによってコードされるものを有する。しかしながら、カリウムチャネルの天然の配列はまた改変、例えば変異させたものでもよい。
【0013】
好ましくは、真核細胞カリウムチャネルをコードするヌクレオチド配列は、酵母の発現プラスミド、例えばp423GPD3、またはベクター(例えばpRS42xまたはpRS32xシリーズ)に組み込まれ、前記組換え発現プラスミドは変異サッカロミセスセレビシエ細胞に導入される。
【0014】
前記方法は、真核細胞カリウムチャネルに対して作用を及ぼす物質を特定することを目的とする。これらの物質は変異サッカロミセスセレビシエ細胞の増殖を抑制する。異種発現された真核細胞カリウムチャネルを抑制する被検物質は、前記変異サッカロミセスセレビシエ細胞は内在性カリウムチャネルを発現しないので、その分裂および成長を非常に困難にさせるか、または遅らせるか、または本発明の実施態様では致死させる。
【0015】
被検物質の作用は、例えば光学密度を600nmで測定するか、または変異サッカロミセスセレビシエ細胞で構成的に発現される増殖レポーターを用いて直接測定することができる。前記構成的に発現される増殖レポーターは、好ましくは、それ自体蛍光もしくは化学発光を示す蛋白質、または蛍光もしくは化学発光シグナルを生じる反応に加わる蛋白質をコードする。前記増殖レポーターをコードする配列は、好ましくはベクター由来である。適当な増殖レポーターは、例えばβ−ガラクトシダーゼのLacZ遺伝子または酸性ホスファターゼPH03(両方とも構成的酵母プロモーターの制御下で発現される)である。測定可能な蛍光または化学発光によって変異サッカロミセスセレビシエ細胞数に関する結果が得られる。蛍光もしくは化学発光が全くないかまたは少ない場合は、問題のサンプルに含まれる変異サッカロミセスセレビシエ細胞の数が少ないことになる。変異サッカロミセスセレビシエ細胞の数が少ない場合は、被検物質は真核細胞のカリウムチャネルに対して抑制作用を有することになる。
【0016】
前述の方法は特に容易な自動化を可能にし、平行的に多数の物質の検査を実施することができる。本発明の特定の実施態様では、2つまたは3つ以上の方法が比較的態様で実施され、その場合2つまたは3つ以上のサッカロミセスセレビシエ細胞が比較的態様で分析される。これらの変異サッカロミセスセレビシエ細胞は好ましくは同じ量の被検物質と一緒にインキュベートされるが、前記細胞は問題の真核細胞カリウムチャネルを種々の程度に発現する。本発明のまた別の実施態様では、問題の真核細胞カリウムチャネルの発現は同程度であるが、異なる量の被検物質と一緒にインキュベートされた変異サッカロミセスセレビシエ細胞が比較的態様で分析される。
【0017】
本発明の特徴はまた、内在性カリウムチャネルTRK1、TRK2およびTOK1が発現されない変異サッカロミセスセレビシエ細胞である。さらに別の特徴は、遺伝子TRK1、TRK2およびTOK1のスイッチが切られた変異サッカロミセスセレビシエ細胞に関する。前記遺伝子は、好ましくはそれら全体または一部分がノックアウトによって除去されている。さらにまた別の特徴は、特許手続き上の微生物の寄託の国際的承認に関するブダペスト条約に従いドイツ微生物および培養細胞コレクション(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Mascheroder Weg 1b, D-38124 Braunschweig)に寄託された変異サッカロミセスセレビシエに関する(寄託番号DSM13197)。
【0018】
本発明の特徴は真核細胞カリウムチャネルを異種発現する変異サッカロミセスセレビシエ細胞に関する。前記真核細胞カリウムチャネルは、好ましくはヒトカリウムチャネル(例えばHERG1、Kv1.5)、またはgpIRK1またはヒトKv4.3[Genbank Accession Number AF187963]、TASK[Genbank Accession Number AF006823]またはROMK2[Genbank Accession Number U12542]で、前記カリウムチャネルは天然の配列を有していてもよいし、また変異していてもよい。
【0019】
本発明はまた変異サッカロミセスセレビシエ細胞の製造方法に関する。前記細胞はカリウムチャネルTRK1、TRK2およびTOK1を発現せず、遺伝子TRK1、TRK2およびTOK1はノックアウトにより破壊されているかまたは欠失している。
本変異サッカロミセスセレビシエ細胞は、例えば、真核細胞カリウムチャネルの活性を抑制または活性化させる物質を特定する方法で用いることができる。また前記細胞は、例えば毒性物質の決定に用いることができる検査キットの部分でもよい。
【0020】
本発明はまた真核細胞のカリウムチャネルの活性化物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュヘ゛ートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の作用を決定する。
【0021】
本発明はさらにまた真核細胞のカリウムチャネルの活性化物質を特定する方法に関し、本方法では、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い;
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ;
(c) 前記変異サッカロミセスセレビシエ細胞を前記真核細胞カリウムチャネルの阻害物質の存在下で被検物質と一緒にインキュベートし;さらに
(d) 前記真核細胞カリウムチャネルに対する前記被検物質の作用を決定する。
【0022】
本発明はまた医薬の製造方法に関し、本方法では、
(a) 真核細胞カリウムチャネルの阻害物質を特定し;
(b) 前記阻害物質を公知の化学的方法によって製造または単離し;さらに
(c) 生理学的に許容できる添加物を前記阻害物質に添加する。
【0023】
本発明はまた医薬の製造方法に関し、本方法では、
(a) 真核細胞カリウムチャネルの活性化物質を特定し;
(b) 前記活性化物質を公知の化学的方法によって製造または単離し;さらに
(c) 生理学的に許容できる添加物を前記活性化物質に添加する。
【0024】
添付図面において:
図1は、三重ノックアウトを証明するための診断PCRを示す。ゲルの段/レーンの説明については実施例2の三重ノックアウトを参照されたい。
図2は、規定のKCl濃度を含むpH6.5のDPM培地でのYM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の増殖を示す。
図3は、5mMのKCl+2mMのRbClを含むDPM培地(pH6.5)でのYM189株およびYM190株(Δtrk1Δtrk2)並びにYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【0025】
図4は、5mMのKCl+2mMのCsClを含むDPM培地(pH6.5)でのYM189株およびYM191株(Δtrk1Δtrk2)並びにYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
図5は、5mMのKCl+1mMのRbClを含むDPM培地(pH6.5)でのYM19
4株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。(“KON”=コントロール)
図6は、5mMのKCl+1mMのCsClを含むDPM培地(pH6.5)でのYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。(“KON”=コントロール)
【0026】
図7は、活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mMKCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度の測定のために、市販のLacZレポーター系pYX232(Ingenius, Cat.No.MBV-032-10)で分析酵母株を形質転換した。LacZレポーター遺伝子の発現は、トリオースホスフェートイソメラーゼ遺伝子のための構成的サッカロミセスセレビシエプロモーターTPIの制御下にあった。LacZ酵素活性は、市販のアッセイ系(TROPIX)を用いて24時間後の化学発光を検出することによって測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。2つの別個のアッセイを別の日に互いに別々に実施した。
【0027】
図8は、活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度は、620nmの波長で光学密度を測定することによって38時間増殖後に測定した(出発培養密度:0.03OD620)。値は各事例で4つの測定値の平均±SDに一致する。
【0028】
図9は、0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mM KCl培地におけるサッカロミセスセレビシエの増殖を示す。
それぞれの事例で種々の阻害物質を最終濃度30μMで用いた。細胞密度の測定のために、市販のLacZレポーター系pYX232(Ingenius, Cat.No.MBV-032-10)で分析酵母株を形質転換した。LacZレポーター遺伝子の発現は、トリオースホスフェートイソメラーゼ遺伝子のための構成的サッカロミセスセレビシエプロモーターTPIの制御下にあった。LacZ酵素活性は、市販のアッセイ系(TROPIX)を用いて24時間後の化学発光を検出することによって測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。2つの別個のアッセイを別の日に互いに別々に実施した。
【0029】
図10は、0.5mMのKClまたは80mMのKClが存在する96ウェルELISAプレートのDPM培地におけるサッカロミセスセレビシエ野生型株の増殖を示す。
それぞれの事例で阻害物質、ジプラシドンおよびピモジドを最終濃度30μMで用いた。細胞密度は、620nmの波長で光学密度を測定することによって24時間増殖後に測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。
【0030】
図11は、5mMのKClおよび活性化物質として0.5mMのCsClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルHERG1の発現を示す。
1:陰性コントロールとしてブランクベクターp423GPDが発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。2:陽性コントロールとしてp423GPD−TRK1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。3:p423GPD−HERG1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。4:p423GPD−HERG1が
発現している二重変異体Δtrk1Δtrk2の増殖。使用したベクターおよび構築物は本明細書で説明されている(段落0039以下および段落0043以下の配列を参照されたい)。
【0031】
図12は、5mMのKClおよび活性化物質として2mMのRbClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルKv1.5の発現を示す。
1:陰性コントロールとしてブランクベクターp423GPDが発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。2:陽性コントロールとしてp423GPD−TRK1が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。3:p423GPD−Kv1.5が発現している三重変異体Δtrk1Δtrk2Δtok1の増殖。4:p423GPD−Kv1.5が発現している二重変異体Δtrk1Δtrk2の増殖。使用したベクターおよび構築物は本願明細書で説明されている(段落0039以下および段落0043以下の配列を参照されたい)。
【0032】
図13は、96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルROMK2の発現および陰性コントロールとしての酵母ベクターp423GPDの発現を示す。
細胞密度は、620nmの波長で光学密度を測定することによって24時間増殖後に測定した(出発培養密度:0.01OD620)。値は各事例で4つの測定値の平均±SDに一致する。
図14は、p423GPD−ROMK2のプラスミドマップを示す。
【図面の簡単な説明】
【0033】
【図1】三重ノックアウトを証明するための診断PCRを示す。
【図2】規定のKCl濃度を含むpH6.5のDPM培地でのYM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図3】5mMのKCl+2mMのRbClを含むDPM培地(pH6.5)でのYM189株およびYM190株(Δtrk1Δtrk2)並びにYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図4】5mMのKCl+2mMのCsClを含むDPM培地(pH6.5)でのYM189株およびYM191株(Δtrk1Δtrk2)並びにYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図5】5mMのKCl+1mMのRbClを含むDPM培地(pH6.5)でのYM194株およびYM195株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図6】5mMのKCl+1mMのCsClを含むDPM培地(pH6.5)でのYM194株およびYM196株(Δtrk1Δtrk2Δtok1)の増殖を示す。
【図7】活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
【図8】活性化物質として0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルHERG1の発現を示す。
【図9】0.5mMのCsClが存在する96ウェルELISAプレートのDPM−HIS/−TRP5mMKCl培地におけるサッカロミセスセレビシエの増殖を示す。
【図10】0.5mMのKClまたは80mMのKClが存在する96ウェルELISAプレートのDPM培地におけるサッカロミセスセレビシエ野生型株の増殖を示す。
【図11】5mMのKClおよび活性化物質として0.5mMのCsClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルHERG1の発現を示す。
【図12】5mMのKClおよび活性化物質として2mMのRbClが存在するDPM−HIS培地における三重変異体Δtrk1Δtrk2Δtok1および二重変異体Δtrk1Δtrk2におけるヒトカリウムチャネルKv1.5の発現を示す。
【図13】96ウェルELISAプレートのDPM−HIS5mM KCl培地の三重変異体Δtrk1Δtrk2Δtok1におけるヒトカリウムチャネルROMK2の発現および陰性コントロールとしての酵母ベクターp423GPDの発現を示す。
【図14】p423GPD−ROMK2のプラスミドマップを示す。
【実施例】
【0034】
材料および株
培地:
YPD(完全酵母培地):1%バクト(Bacto)酵母抽出物、2%バクトペプトン、2%バクト寒天、2%グルコース。
SC(合成完全)培地:0.67%バクト酵母窒素ベース、アミノ酸、2%グルコース。
胞子形成培地:1%酢酸カリウム、アミノ酸。
5−FOA培地:0.67%バクト酵母窒素ベース、アミノ酸、ウラシル(50μg/mL)、2%糖(ガラクトースまたはグルコース)、0.1%5−FOA。
全ての培地は次の文献に記載されている:(G.R. Fink et al.,1991)。
【0035】
アミノ酸ドロップアウトミックス:
L−アラニン2g;L−アルギニン2g;L−アスパラギン*H2O 2.27g;L−アスパラギン酸2g;L−システイン*HCl 2.6g;L−グルタミン2g;L−グルタミン酸2g;グリシン2g;ミオイノシトール2g;L−イソロイシン2g;L−メチオニン2g;PABA0.2g;L−フェニルアラニン2g;L−セリン2g;L−スレオニン2g;L−チロシン2g;L−バリン2g。
【0036】
マーカーアミノ酸用ストック溶液:
mM g/L
アデニン(100x) 30 5.53 加熱(60℃未満)
ロイシン(60x) 100 13.12 加熱
リジン(100x) 100 18.26 −
ヒスチジン(200x) 60 12.57 −
トリプトファン(100x) 40 8.17 −
ウラシル(100x) 20 2.24 0.5%NaHCO3溶液中で加熱
【0037】
ビタミン原液(50mL):
ビオチン20μg/L;パントテン酸カルシウム40μg/L;チアミン40μg/L
。
【0038】
限定カリウム培地(DPM):1.5L用(2×原液):
(NH4)2HPO4 8mM 3.2g
(NH4)2SO4 29mM 11.5g
MgSO4 2mM 0.8g(または1M原液6mL)
CaCl2 0.2mM 90μg(または0.5M原液1.2mL)
ビタミン原液 120μL
アミノ酸ドロップアウトミックス 6g
リジン 100倍原液330mL
アデニン 0.9mM 100倍原液30mL
→上記をHClでpH6.5(または他のpH)としオートクレーブする。
グルコース 2% 40%原液より
KCl 1M原液より
必須アミノ酸(Lys/Adeは除外)は原液から
寒天
【0039】
緩衝液および溶液:
TE緩衝液:トリス/HCl(pH7.5)10mM;EDTA(pH8.0)1mM;
TAE緩衝液:トリス40mM;EDTA1mM;酢酸0.2mM;
SSC緩衝液(20x):NaCl 3M;クエン酸ナトリウム*2H2O 0.3M;
ゲルローディング緩衝液:ブロムフェノールブルー0.05%(w/v);スクロース40%(w/v);EDTA(pH8.0)0.1M;SDS 0.5%(w/v)
ハイブリダイゼーション緩衝液:SSC 5x;SDS 0.1%(w/v);硫酸デキストラン5%(w/v);停止試薬1:20;
緩衝液A(無菌):トリス−HCl 100mM;NaCl(pH9.5) 300mM;
脱プリン溶液:HCl 0.25M;
変性溶液:NaCl 1.5M;NaOH 0.5M;
中和溶液:NaCl 1.5M;トリス(pH8.0)0.5M。
【0040】
オリゴヌクレオチド(PCRプライマー):
【0041】
【0042】
ベクター:
細菌ベクター
【0043】
酵母ベクター
【0044】
株:
細菌株:DH5α;ワンショット(One ShotTM) TOP10(Invitrogen)
酵母株:
この実験のために作製した全ての酵母株は以下の二倍体野生型株をベースにしている:W303MATa/α ade2.his3−11−15,leu2−3−112,trp1−1,ura3−1,can1−100;ATCC#208352
【0045】
【0046】
以下の酵母株を作製した:
【0047】
【0048】
カリウムチャネルクローン:
A)
系統名 KCNA5
別称 Kv1.5,(HK2,HPCN1)
種類 電圧開閉カリウムチャネル、shaker関連サブファミリー
(メンバー番号5)、遅延整流チャネル
染色体上の位置 12p13.32−p13.31
登録番号 NID g4504818
蛋白質 613アミノ酸、67kD
組織内分布 心臓、膵臓ランゲルハンス島およびインスリノーマ
同族体 mKcna5(Mus musculus)、70%がhHCN4と相同
参考文献 (S.L. Roberds et al.,1991; M.E. Curran et al.,1992;
D.J. Snyders et al.,1993)
【0049】
B)
系統名 HCN2
別称 BCNG2(脳環状ヌクレオチド開閉チャネル)、HAC1
種類 過分極−活性化および環状ヌクレオチド開閉カリウムチャネ
ル、電圧開閉カリウムチャネルのサブファミリーに属する
染色体上の位置 19p13.3
登録番号 NID g4996893
蛋白質 889アミノ酸
機能 ペースメーカー
組織内分布 脳、心臓
同族体 mHcn2(Mus musculus)
参考文献 (A. Ludwig et al.,1999)
【0050】
C)
系統名 KCNH2
別称 HERG1(より長いスプライス変種)
種類 電圧開閉カリウムチャネル、eag関連サブファミリー
(メンバー番号2)
染色体上の位置 7q35−q36
登録番号 NID g4557728
特性 K+チャネル調節因子1によってチャネル活性化が促進
参考文献 (M. Taglialatela et al.,1998; T. Itoh et al.,1998)
【0051】
D)
系統名 KCNJ2(モルモット)
別称 Kir2.1,IRK1
種類 内向き整流カリウムチャネル
組織内分布 脳、心臓、肺臓、腎臓、胎盤、骨格筋
参考文献 (W. Tang et al.,1995)
【0052】
方法:
ROMK2(後記の“ROMK2の配列”を参照されたい)
【0053】
PCR:
パワースクリプト(Powerscript)ポリメラーゼ(PAN Biotech)用プロトコル:
ローワー(lower)試薬用ミックス(ホットスタートプロトコル)(25μL):
3μLのH2O;2.5μLの10×オプチパフォーム(OptiPerform(登録商標))III緩衝液(pH9.2);10μLの1.25mM dNTP(=200μM);
1.5μLの前進プライマー(20pmol/μL);1.5μLの逆方向プライマー(20pmol/μL);1.5μLの50mg MgCl2(=1.25mM);5μLの5×オプチザイム(OptiZyme(登録商標))エンハンサー
【0054】
アッパー試薬用混合物(35μL):
23μLのH2O;3.5μLの10×オプチパフォーム(OptiPerform(登録商標))III緩衝液;1.5μLの50mM MgCl2;0.5μLのパワースクリプトDNAポリメラーゼ;7μLの5×オプチザイム(OptiZyme(登録商標))エンハンサー。
【0055】
PCRプログラム(ホットスタート):
1.94℃で1分
2.94℃で1分
3.50〜55℃(プライマーによる)で1.5分
4.69〜72℃(ポリメラーゼによる)で4分
5.2番から27回繰り返す
6.4℃∞
7.終了
【0056】
アンプリタック(AmpliTaq)ポリメラーゼ(Perkin Elmer)用プロトコル:
アッパー試薬用ミックス(ホットスタートプロトコル)(50μL):
18.1μLのH2O;4.2μLの10×緩衝液II;16.7μLのdNTP;2.5μLの前進プライマー;2.5μLの逆方向プライマー;6μLの25mM MgCl2(=1.5mM)。
ローワー試薬用ミックス(50μL):
42μLのH2O;5μLの10×緩衝液II;1μLのアンプリタック(AmpliTaq)ポリメラーゼ;2μLの鋳型。
【0057】
DNAの精製
PCR反応物の精製:PCR増幅生成物の精製はキット(High Pure PCR Product Purification Kit(Roche))を用いて実施した。
フェノール抽出:サンプル容積をTE緩衝液で200μLにする。200μLのフェノール/クロロホルム/イソアミルアルコール(25:24:1)を加え、混合し、最大速度で1分間遠心する。上部相を新しいエッペンドルフ試験管に移し、200μLのクロロホルム/イソアミルアルコールを加え、混合し1分間遠心する。上部相を取り出しエタノールで沈殿させる。
【0058】
エタノール沈殿:約200μLのサンプル容積に5μLの5M NaClおよび20μLの3M NaAc(pH5.7)をピペットで加える。2.5容の100%エタノールを添加して混合し、少なくとも30またはそれ以上−20℃で静置する。4℃で10分間遠心し、ペレットを170μLの70%冷エタノールで洗浄する。3分間遠心し、ペレットを37℃で乾燥させ、さらに30μLのH2Oに再懸濁させる。
【0059】
大腸菌(E. coli)のプラスミドDNAの単離:大腸菌オーバーナイト培養のプラスミドDNAの単離はキアゲン(Qiagen)のプロトコル(QIApre Spin Miniprep Kit Orotocl)を用いて実施した。
【0060】
サッカロミセスセレビシエのDNAの調製:
前記酵母細胞を10mLのYPDで一晩30℃でインキュベートし、次の朝、3000rpmで10分間遠心し、ペレットを1Mソルビトール、0.1MEDTA(pH7.5)の500μLに再懸濁し、エッペンドルフ試験管に移す。50μLのザイモラーゼ(zymolase)(ソルビトール/EDTA中に5mg/mL)を添加し、37℃で1時間インキュベートし、さらに1分間遠心する。50mMトリス、20mMのEDTA(pH7.4)500μLにペレットを再懸濁する。50μLの10%SDSを添加し、完全に混合し、さらに65℃で30分間インキュベートする。5MのKAc200μLを加え、氷上に1時間置き、1
0分間遠心する。上清(約650μL)を新しいエッペンドルフ試験管に移し、1容量のイソプロパノールを加え、穏やかに混合し、さらに5分間静置する。簡単に遠心するか、または沈殿したDNAをガラスのかぎ棒で抜き取り、空気中で前記ペレットを乾燥させる。ペレットまたはDNAを150μLのTE緩衝液に再懸濁し、65℃で10分間溶解させる。
DNAクローニング:全てのDNAクローニングは標準的なプロトコルにしたがって実施した。
【0061】
酵母の形質転換(酢酸リチウム法):
形質転換しようとする酵母株を5mLの適当な培養液中で一晩30℃でシェーカー上でインキュベートする。次の朝前記オーバーナイト培養を適当な培養液で希釈し(OD600=0.40.5)、さらに2時間シェーカー上で30℃でインキュベートする(OD600=0.40.8)。2500rpmで3分間遠心し、25mLの滅菌H2Oでペレットを洗浄する。2500rpmで3分間遠心し、1mLのLITE(100mMのLiAc,TE(pH7.5))に再懸濁し、懸濁液をエッペンドルフ試験管に移す。RTで5分間インキュベートし、15秒間遠心する(Quickspin)。100mMのLiAc 1mLでペレットを洗浄し(quick-spin)、細胞濃度に応じてペレットを200400μLの100mM LiAcに再懸濁して50μLの部分標本に分ける。
【0062】
表示の順番どおりに以下を加える:
240μLのPEG(50%)、ピペットで穏やかに懸濁物を混合する;
36μLの1M LiAc、ピペットで穏やかに懸濁物を混合する;
10μLの一本鎖精子DNA(−20℃で保存;使用前に80−90℃で10分間加熱してから氷に移す);
2〜3μgのプラスミドDNA(またはノックアウトトランスフォーメーションの場合には8〜10μLのミニプレップ(Miniprep))、ピペットで穏やかに懸濁物を混合し;
形質転換反応物をオーバーヘッド回転装置で低速で30℃30分間インキュベートする;
42℃15分間形質転換反応;
クィックスピンで遠心、ペレットを200μLのTE緩衝液に再懸濁(ノックアウトの場合は300μLのYPDにペレットを再懸濁)し、オーバーヘッド回転装置で30℃で4時間インキュベートする;
寒天平板当たり100μL(ノックアウトのばあいは全反応物)を平板培養し、30℃で3〜4日間インキュベート。
【0063】
配列決定:ABIPRISM(登録商標) red.プロトコルII/AmpliTaq(登録商標) FS1/4BigDyeターミネーター
【0064】
反応物:
プレミックス 2μL
DNA鋳型
ssDNA 50ng
dsDNA 250ng
PCR生成物(0.2−5kB) 10−50ng
プライマー 3〜10pmol
H2Oで最終容積 10μL
【0065】
サーモサイクラープロトコル(25サイクル):
1.96℃で15秒間
2.96℃で15秒間
3.55℃で10秒間
4.60℃で4分間
5.2.に戻って24サイクル
6.4℃∞
7.終了
【0066】
精製反応(セントリセップスピンカラム(Centri Sep Spin Colum)(Princeton Separations)):
カラムを750μLのH2Oで30分間予め膨潤させ、液体を排出させて2分間3000rpmで遠心する。反応物をH2Oで20μLにしてカラムに適用し、2分間3000rpmで遠心する。
サンプル適用:配列決定試験管に、4μLのセントリセップ(Centri Sep)溶出液+20μLのTSR(鋳型抑制試薬)、90℃で2分間。
【0067】
サザンブロット:
DNAプローブを適当な制限酵素で消化、ゲル電気泳動で分離し、ゲルから抽出する。ゲノムDNAを適当な制限酵素で一晩消化し、ゲル電気泳動で分離する(1%アガロースゲル)。
【0068】
ゲルの前処理:アガロースゲルからローディングウェルを取り出す。アガロースゲルを0.25MのHClで15分間脱プリン処理し、続いて蒸留水で2回洗浄する。アガロースゲルを0,5MのNaOHで30分間変性させ、ヴァキュームブロッターモデル785(Vaccum Blotter Model 785, BioRad)を用いてビニールシートの中央に移し、ウィンドー(ウィンドーシール)を切り取り、ナイロン膜と濾紙の端を切り揃え(各々の事例でゲルより0.5cm小さい)、ナイロン膜の端を蒸留水で湿らし(各々の事例でビニールシートの小窓より0.5cm広い)、続いてナイロン膜および濾紙をトランスファー溶液で湿らす。
【0069】
装置の構築(下から上へ):
ベースユニット、真空土台部、多孔性真空スラブ、濾紙、ナイロン膜、ビニールウィンドー、アガロースゲル、最終フレーム、蓋。
バイオラッド(BioRad)真空ポンプを10分間予熱し、真空を適用する(16932Pa(5インチHg))。
端に沿ってゲルを穏やかに圧迫する。
トランスファー溶液(約1L、10×SSC)を上部液だめに入れる;移動時間:90分;真空のスイッチを切り、ナイロン膜を取り出し、5分間2×SSCで洗浄し、続いて濾紙の間で風乾させる。DNA固定:ナイロン膜をUV透過性の密着フィルム上に置き、陽性コントロールとして端にプローブを適用する;UVストラタリンカーに入れ架橋を開始させる(1200000J→0);膜は粘着フィルム内またはワットマン濾紙の間で室温または4℃で保存することができる。
【0070】
ジーンイメージ・ランダムプライムラベリングモジュール(Amersham):
DNAプローブの標識:DNAプローブを96℃で5分間変性させ(ヒートショック)、その後氷上に置く。10μLの反応ミックス(ヌクレオチドミックス(5×)、トリス−HCl(pH7.8)中のフルオレセイン−11−dUTP,dATP,dCTP,dGTPおよびdTTP、2−メルカトエタノールおよびMgCl2);5μLのプライマー(Random Nonamers)、1μLの酵素溶液(クレノーフラグメント、5単位/mL);22μLの変性DNAプローブ;12μLのH2O。2時間37℃でインキュベートし、2μLのO.5M EDTA(=20mM)を添加し、部分標本を−20℃で保存する。標識効率の確認:5×ヌクレオチドミックスをTE緩衝液で1/5、1/10、1/25、1/50、1/100、1/250および1/500に希釈し;ナイロン膜の細片に5μLの
DNAプローブを5μLの1/5希釈と一緒に適用してさっと吸収させ、予め温めておいた2×SSCで60℃で15分間洗浄し;1/5希釈を除いた残りの溶液を参照用膜細片に適用し、両方の膜細片をUV光の下で観察し、サンプルの濃さを決定する。
【0071】
ハイブリダイゼーション:ナイロン膜(ブロット)を温かいハイブリダイゼーション緩衝液(0.3mL/cm2)で、回転オーブンで60℃2時間予備ハイブリダイズさせ;緩衝液を除きその10mLを残す。DNAプローブを変性させ(96℃で5分間、続いて氷上で冷却);プローブを前記10mLの緩衝液とともにブロット上に加え、回転オーブンで60℃で一晩ハイブリダイズさせる。
【0072】
洗浄工程:
温かい1×SSC,0.1%(w/v)SDS中でプラットフォームシェーカー上で15分;温かい0.5×SSC,0.1%(w/v)SDS中でプラットフォームシェーカー上で15分。
【0073】
ジーンイメージ・CDP−スターディテクションモジュール(Amersham):
停止および抗体反応:シェーカーの上で、緩衝液A中の停止試薬1/10希釈中で1時間室温でブロットをインキュベートする。抗体溶液(アルカリ性ホスファターゼ結合抗フルオレセイン抗体、5000×)を0.5%(w/v)BSA/緩衝液Aでブロットシールと一緒に金属薄片中で希釈し、さらにシェーカー上で1時間室温でインキュベートする。10分間緩衝液A中の0.3%トゥイーン(Tween)20で3回洗浄して未結合抗体溶液を除去する。
【0074】
シグナル発生および検出:洗浄緩衝液を排出し、ブロットを密着フィルム上に置き、5mLの検出試薬を適用し、2〜5分反応させ、再度排液し(アルカリ性ホスファターゼは発光を引き起こす);密着フィルムで包み、暗室の赤色光の中でフィルム(ハイパーフィルム(Hyperfilm(登録商標)MP, Amersham)を用い、フィルムカセット(BioMax, Kodak)中で0.5〜2時間露光させ、現像してスキャンする。ブロットは4℃で密着フィルム中で保存できる。
【0075】
実施例1:特異的欠失カセットの構築
欠失は全て標準的な方法によって実施した(G.R. Fink et al.,1991; A. Wach et al.,1994; U. Guldener et al.,1996; A.L. Goldstein et al.,1999)。
各々約500bpのフラグメント(それぞれは遺伝子の初めと終わりの領域をもつ)を以下のプライマーを用いてPCRにより増幅させた:TRK1の場合はTRK1-FL-BamHI-Fo、 TRK1-FL-PstI-Re、 TRK1-FL-PstI-FoおよびTRK1-FL-XhoI-Re、 TRK2の場合はTRK2-DEL-5-Fo-B、 TRK2-DEL-5-Re、 TRK2-DEL-3-FoおよびTRK2-DEL-3-Re、 並びにTOK1の場合はTOK1-DEL-5-Fo、 TOK1-DEL-5-Re、 TOK1-DEL-3-FoおよびTOK1-DEL-3-Re(2.3章参照)。増幅させた末端は後の酵母ゲノムへの正確な組み込みを可能にする。酵母株w303a/αまたはw303a/αΔtrk1はDNA鋳型として機能する。
【0076】
実施例2:単一、二重、三重変異体の構築
実施例2a:単一ノックアウト
TRK1、TRK2およびTOK1のための構築欠失カセットでそれぞれ二倍体酵母株YM96(MATa/MATα)を形質転換した。ゲノムへの欠失カセットの組み込みは、(−)URA/Glcでtrk1変異体(YM123/124)を、さらにYPD/ジェニチシンでtrk2変異体(YM158−161)およびtok1変異体(YM154−157)を増殖させることによって確認した。なぜならば、TRK1欠失カセットのURA3マーカーは(−)URA培地での増殖を可能にし、TRK2またはTOK1のKANマーカーはゲネチシン(geneticin)での増殖を可能にするからである(G.R. Fink et
al.,1991)。陽性コロニーを胞子形成平板にレプリカ平板培養により移し、ここで栄養成長を行わずに18−24時間後にMATa/MATα二倍体細胞は胞子を形成する。それらをザイモラーゼで処理し、再びYPD上で増殖させた後、解剖顕微鏡を用いていくつかのコロニーの四分子を個々の4つの胞子に分割する。
【0077】
胞子コロニーの交配型を交配テスター株との交配によって決定した(G.R. Fink et al.,1991)。欠失カセットの有無についての選別は、trk1については−URA培地、trk2およびtok1についてはゲネチシン含有培地でのレプリカ平板培養によって実施した。酵母のDNA調製によって形質転換体のゲノムDNAを得た後で、結果を診断用PCRおよびサザンブロットによって確認した。
【0078】
実施例2b:二重ノックアウト
TOK1欠失カセットで一倍体Δtrk1酵母株YM123およびYM124を形質転換し、YPD/ゲネチシンでの増殖によりTOK1欠失カセットの組み込みについて選別した。結果は診断用PCRおよびサザンブロットにより確認した。グリセロール培養は以下の株を用いて実施した:(+)URA3、(+)KAN)(Δtrk1Δtok1)株(YM140、YM141、YM143およびYM144)。単一コロニーをパッチとして画線培養し、5−FOA上にレプリカ培養し、TRK1欠失カセットからURA3マーカーおよびhisGリピートを排除したコロニーを選別した(G.R. Fink et al.,1991)。したがって、ウラシル合成のためのURA3遺伝子(TRK1で)を欠いたコロニーはいずれも(−)URA/Glcでは増殖せず、一方、TOK1欠失カセット中の耐性遺伝子により全てのコロニーがYPD/gen上で生存する。ゲノムからKanマーカーを除去するために、(−)URA3変異体をプラスミドpSH47で形質転換した。前記pSH47にはCreリコンビナーゼおよびウラシル合成(URA3)のための遺伝子が存在する。陽性形質転換体は(−)URA/Glcで増殖し、したがって(−)URA/Gal液体培地でのインキュベーションによってCreリコンビナーゼを誘発することが可能であった。この方法で、Kanマーカーは1つのloxPリピートとともに排除され、1つのloxPは保持される。
【0079】
オーバーナイト培養でOD600=5となった後で、1:10000および1:50000の希釈を(−)URA/Galで平板培養した。YPD/genでレプリカ培養した単一コロニーパッチは増殖を示さなかった(これはKanマーカーの排除は成功であったことを示している)。プラスミドpSH47を除去するために、その後、細胞を5−FOAで2回選別を繰り返した。グリセロール培養は(−)ura(−)kan(Δtrk1Δtok1)株(YM162、YM163およびYM164)を用いて実施した。
【0080】
実施例2c:三重ノックアウト
YPDでのオーバーナイト培養を単一Δtrk1Δtok1コロニー(YM162およびYM164)を用いて開始し、次の日、BsiWI/SpeI消化TRK2欠失カセットで形質転換し、YPD/KCl/ジェネチシンで平板培養した。酵母DNAを調製した後、この三重ノックアウトを診断用PCRおよびサザンブロットで確認した。
【0081】
【0082】
実施例3:ヒトカリウムチャネルのサブクローニング並びに二重および三重変異体の形質転換
ヒト遺伝子HERG、HCN2、Kv1.5および陽性コントロールとしてTRK1、IRK1(モルモット)をそれらを含むプラスミド(HERGはpcDNAのBamHI間に、 HCN2はpTLNのNcoI/XhoI間に、 Kv1.5はpcDNA3.1(-)のNheI/EcoRI間に、 IRK1はpSGEMのBamHI/EcoRI間に存在)から制限酵素を用いて切断して切り出し、ゲル電気泳動によって分離し、さらにゲルから抽出した。個々のヒトカリウムチャネルを酵母ベクターp423−GPD3(D. Mumberg et al.,1995; V. Ronicke et al.,1997)に連結し、大腸菌を形質転換した。プラスミド調製物のコントロール消化および配列決定によって前記のヒト遺伝子が組み込まれたクローンを特定することができた。続いてこれらのプラスミドでΔtrk1Δtrk2二重ノックアウト(YM168)およびΔtrk1Δtrk2Δtok1三重ノックアウト(YM182)を形質転換し、 (−)HIS/80mMKCl上で平板培養した。
【0083】
実施例4:ノックアウト株の性状決定
実施例4a:種々のK+濃度およびpH値の培養平板上での二重および三重変異体の増殖
種々のノックアウトのカリウム要求の相違を比較するために、酵母株YM182、YM168およびYM97(WT)を、種々のK+濃度およびpH値をもつDPM平板でインキュベートした。この目的のために、グリセロール培養のパッチを先ず最初に100mM KCl/pH6.5に画線培養した。2日間増殖させた後、50mM、30mMおよび5mM KClでレプリカ平板培養を実施した。
この実験によって、YM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)の両株は50mMおよび30mMのKClで生存可能であることが示された。さらに、YM182株はYM168株よりも30mMのKClの存在下でより良好に増殖することが判明した。この2つの株のいずれも、野生型株YM97とは対照的に5mMのKClの存在下で生存できなかった。
【0084】
pH依存性を調べるために、前記3株をさらに100mMおよび5mM KCl/pH5.0並びに100mMおよび5mM KCl/pH4.0上でレプリカ平板培養した。この実験は、
YM168およびYM182はいずれもpH4.0では100mM KClでも5mM KClでも生存できないことを示した。pH5.0で100mM KClでは、YM168の増殖欠損はYM182の場合よりも激しい。ベクターpRS416GALのTRK1の発現は、YM168株(Δtrk1Δtrk2)およびYM182(Δtrk1Δtrk2Δtok1)の増殖欠損を完全に補完する。
【0085】
実施例4b:種々のK+濃度の液体培地での二重および三重変異体の増殖
YM168株(Δtrk1Δtrk2)およびYM182株(Δtrk1Δtrk2Δtok1)(以後の全ての実験はこれらを基準にしている)の性状を調べるために、これら酵母株の液体培養中の増殖態様を調べた。最初に、オーバーナイト培養をDPM/80mM KCl中で開始させ、次の朝、DPM/5mM KClおよびDPM/15mM KClで前記培養をOD=0.05にした。それぞれ規定の間隔後に600nmでの光学密度を光度計により決定した。
これらの実験によって、YM182株の増殖欠損は、5mM KClおよび15mM KClではYM168よりも強くないことが判明した。
【0086】
実施例5:二重および三重ノックアウトのヒトカリウムチャネルの性状決定
実施例5a:培養平板のK+欠乏の補完能力
YM168(Δtrk1Δtrk2)およびYM182(Δtrk1Δtrk2Δtok1)の各株を、酵母発現ベクターとしてp423−GPD3中のヒトカリウムチャネルKv1.5((D. Fedida et al.,1998);YM190およびYM195)およびHERG1((D. Fedida et al.,1998);YM191およびYM196)でそれぞれ形質転換した。gpIRK1(W. Tang et al.,1995);YM193およびYM198)は酵母発現ベクターp423−GPD3中の陽性コントロールとして機能した(D. Mumberg et al.,1995; V. Ronicke et al.,1997)。ブランクベクターp423−GPD3(YM189およびYM194)は陰性コントロールとして機能した。形質転換酵母株を(−)HIS/80mM KCl上で平板培養した。この後、単一コロニーのパッチをDPM/5mM KCl(pH6.5)上でレプリカ平板培養し、カリウム欠乏を補完する能力を調べた。
【0087】
これらの実験によって、p423−GPD3の陽性コントロールgpIRK(YM193およびYM198)は完全に二重および三重ノックアウトの増殖欠損を補完することが示された。陰性コントロールとしてのブランクベクターp423−GDP3(YM189およびYM194)はこの増殖欠損を補完することができない。ヒトカリウムチャネルKv1.5は三重ノックアウトの増殖欠損を補完するが、陽性コントロールgpIRKよりはるかに効率が悪い。さらにまた、ヒトカリウムチャネルKv1.5は二重ノックアウトΔtrk1Δtrk2を補完しない。与えられた実験条件下では、HERGチャネルは二重および三重ノックアウトの増殖欠損を補完しない。
【0088】
実施例5b:活性化物質が存在する培養平板での増殖
種々のカリウムチャネルに対する活性化物質の作用を示すために、上記の株を以下の特定の活性化物質を含む培地中でインキュベートした。
Kv1.5:Rb+は過分極相を延長させる。これは、内向きのK+の流れはより長く続き、増殖欠損を補完する可能性を高めることを意味している。
HERG:Cs+は過分極相を延長させる。これは、内向きのK+の流れはより長く続き、増殖欠損を補完する可能性を高めることを意味している。このチャネルはCs+によって抑制される。
IRK1:Cs+はこのチャネルを阻害する。
p423−GPD3−Kv1.5を用いた実験によって、ヒトKv1.5チャネルは、2mM RbClの存在下でΔtrk1Δtrk2Δtok1変異体の増殖欠損を完全に補完できることが示された(図3)。Δtrk1Δtrk2変異体の増殖欠損の補完は極めて効率が低い(図3)。これは実施例6aで示される結果と一致する。
p423−GPD3−HERGを用いた実験によって、ヒトHERG1チャネルは、2mMCsClの存在下でΔtrk1Δtrk2Δtok1変異体の増殖欠損を完全に補完できることが示された(図4)。Δtrk1Δtrk2変異体の増殖欠損の補完は極めて効率が低い(図4)。これは実施例6aで示される結果と一致する。
【0089】
実施例5c:液体培養液中のRbClの存在下におけるΔtrk1Δtrk2Δtok1変異体のKv1.5による補完
酵母株YM194およびYM195を1mMのRbClを含むDPM/−HIS/5mMKCl中で液体培地における増殖態様の相違について調べた。この目的のために、10mLのオーバーナイト培養をDPM/−HIS/80mM KCl中で開始し、次の朝対応する培養液を用いてOD600を0.05にした(最終容積:20mL)。600nmでの光学密度は規定の間隔で光度計により測定した。
これらの実験は、TRK1、TRK2およびTOK1を欠く酵母株におけるベクターp423−GPD3のKv1.5の発現は前記によってもたらされた増殖欠損を補完することができることを明瞭に示した。
さらに別の実験で、Kv1.5およびgpIRKによる増殖欠損の補完は2mMのCsClの存在下で抑制されることが示された。
【0090】
実施例5d:液体培地中のCsCl存在下でのΔtrk1Δtrk2Δtok1変異体におけるHERG1チャネルによる補完
酵母株YM194およびYM196を1mMのCsClを含むDPM/−HIS/5mM KCl中で液体培地における増殖態様の相違について調べた。この目的のために、10mLのオーバーナイト培養をDPM/−HIS/80mM KCl中で開始し、次の朝対応する培養液を用いてOD600を0.05にした(最終容積:20mL)。600nmでの光学密度は規定の間隔で光度計により測定した。
これらの実験は、TRK1、TRK2およびTOK1を欠く酵母株におけるベクターp423−GPD3のHERG1の発現は前記によってもたらされた増殖欠損を補完できることを明瞭に示した。
【0091】
実施例6:
三重変異体Δtrk1Δtrk2Δtok1における全ての増殖アッセイは、各事例で表示したpHおよびカリウム濃度で増殖培地DPM(規定カリウム培地)で実施した。
ヒトHERG1K+チャネルの阻害物質として用いられた物質は、テルフェナジン(α−(4−tert−ブチルフェニル)−4−(α−ヒドロキシ−αフェニルベンジル)−1−ピペリジンブタノール;HMR)、 ピモジド(1−(4,4−bis(P−フルオロフェニル)ブチル)−4−(2−オキソ−1−ベンゾイミダゾリニル)−ピペリジン;Sigma, Cat.No.P100)、ジプラシドン(5−(2−[4−(1,2−ベンゾイソチアゾール−3−イル)ピペラジノ]−エチル)−6−クロロ−1,3−ジヒドロ−2H−インドール−2−オン;HMR)、ロラタジン(エチル4−(8−クロロ−5,6−ジヒドロ−11H−ベンゾ[5,6]シクロヘプタ[1,2−b−]ピペリジン−11−イリデン)−1−ピペリジンカルボキシレート;HMR)およびセルティンドール(1−(2−[4−[5−クロロ−1−(4−フルオロフェニル)−1H−インドール−3−イル]−1−ピペリジニル]エチル)−2−イミダゾリジノン;HMR)であった(E. Richelson, 1996; E. Richelson, 1999; E. Delpon et. al.,1999; T. Kobayashi et al.,2000; M.D. Drich et al.,2000)。カリウムチャネルに対して抑制作用をもたないはずの物質、ジフェニルヒドラミン(Sigma, Cat.No.D3630)およびフェキソフェナジン(4−[ヒドロキシ−4−[4−(ヒドロキシジフェニルメチル)−1−ピペリジニル]ブチル]−α,α−ジメチルベンゼン酢酸ヒドロクロリド;HMR)(M. Taglialatela et al.,1999; L.M. DuBuske, 1999)もまた用いた。全ての物質はDMSOに溶解し、最終濃度30μMで使用した。コントロールとして、前記物質を含まない0.5%の同じ最終濃度のDMSOを有する細胞、
DMSOが添加されていない細胞、物質が添加されていない細胞を測定した。
【0092】
図1および2に示したように、ヒトHERG1チャネルは、5mM KClのみを含む培地で三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができる。テルフェナジン、ピモジド、ジプラシドン、セルティンドールおよびロラタジンの存在下では、ヒトHERG1チャネルは、もはや5mM KClのみを含む培地では三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができない。
【0093】
実施例7:
酵母の3つの内在性カリウムチャネル蛋白質を全て発現する野生型株をテルフェナジン、ピモジド、ジフェニルヒドラミン、ジプラシドン、ロラタジン、フェキソフェナジンおよびセルティンドールとともにインキュベートしたとき、テルフェナジン、ロラタジンおよびセルティンドールはヒトHERG1チャネルの特異的阻害物質であることが示された(図9)。
【0094】
この結果によれば、ピモジドおよびジプラシドンはむしろ非特異的阻害物質であると考えるべきである。このことは、これらの物質はおそらくヒトHERG1チャネルだけでなくサッカロミセスセレビシエの内在性カリウムチャネルもまた抑制することを意味している。しかしながら、この結果は、これら物質について認められた抑制作用は、酵母細胞の増殖に必須の他の物質の抑制に起因する可能性を排除することができなかった。この可能性を調べるために、80mMのKClを含む増殖培地でこれらの物質の作用も調べた。
【0095】
これらの結果(図10)によって、ピモジドは、非特異的態様で内在性カリウムチャネルTRK1およびTRK2の活性を抑制することが明らかになった。より高いカリウム濃度で抑制作用が認められないことによって、ピモジドは酵母細胞で不変的な毒性作用をもたないと結論することができる。対照的に、ジプラシドンはより高いカリウム濃度でさえ酵母細胞の増殖を抑制し、したがってサッカロミセスセレビシエに対する毒性作用をもつことが明らかになった。この作用に必須である酵母の標的蛋白質はまだ特定されていない。
結論として、上記の実験は、前述の系によって、ヒトカリウムチャネルを特異的に抑制する物質を実際にサッカロミセスセレビシエで特定することができることを示した。
前記の結果は図10に示されている。
【0096】
実施例8:
ヒトカリウムチャネルHERG1およびKv1.5は二重変異体Δtrk1Δtrk2の増殖欠損を補完しない(図11および図12)。
結果:図11および図12
図11および12は、ヒトカリウムチャネルHERG1およびKv1.5は二重変異体Δtrk1Δtrk2の増殖欠損を補完しないことを示している(各事例について図11および12の第4の部分)。陰性コントロール、すなわち三重変異体Δtrk1Δtrk2Δtok1におけるブランクベクターp423GPD(各事例について図11および12の第1の部分)と比較したとき、増殖が改善されていないことが示されている。二重変異体Δtrk1Δtrk2の陰性コントロールp423GPDは表示されていないが、三重変異体Δtrk1Δtrk2Δtok1の陰性コントロールp423GPDと異ならない。対照的に、ヒトカリウムチャネルHERG1およびKv1.5は三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完する(各事例について、図11および12の第三の部分)。
【0097】
実施例9:
ヒトカリウムチャネルROMK2((M.E. Shuck et al.,1994; J.H. Bock et al.,1997);配列番号31、hROMK2)を酵母ベクターp423GPDでサブクローニングし、
三重変異体Δtrk1Δtrk2Δtok1を形質転換した。この実験によって、前記ヒトカリウムチャネルもまた三重変異体Δtrk1Δtrk2Δtok1の増殖欠損を補完することができることが示された。
このヒトカリウムチャネルが二重変異体Δtrk1Δtrk2の増殖欠損を補完できるか否かはまだ調べられていない。ROMK2チャネルを特異的に抑制する物質はまだ判明していない。
前記の結果は図13に示されている。
【0098】
【表1】
【0099】
【表2】
【0100】
【表3】
【0101】
【表4】
【0102】
【表5】
【0103】
【表6】
【0104】
【表7】
【0105】
【表8】
【0106】
【表9】
【0107】
【表10】
【0108】
【表11】
【0109】
【表12】
【0110】
【表13】
【0111】
【表14】
【0112】
【表15】
【0113】
〔参考文献〕
Curran, M. E., Landes, G. M., and Keating, M. T. Molecular cloning, characterization, and genomic localization of a human potassium channel gene. Genomics 12:
729-737. (1992)
Dascal, N., Schreibmayer, W., Lim, N. F., Wang, W., Chavkin, C., DiMagno, L., Labarca, C., Kieffer, B. L., Gaveriaux-Ruff, C., and Trollinger, D. Atrial G pr
otein-activated K+ channel: expression cloning and molecular properties. Proc. Natl. Acad. Sci. U.S.A. 90: 10235-1O239. (1993)
Fairman, C., Zhou, X., and Kung, C. Potassium uptake through the TOK1 K+ channel in the budding yeast. J. Membr. Biol. 168: 149-157. (1999)
Fedida, D., Chen, F. S., and Zhang, X. The 1997 Stevenson Award Lecture. Cardiac K+ channel gating: cloned delayed rectifier mechanisms and drug modulation. Can. J. Physiol. Pharmacol. 76: 77-89.(1998)
Fink, G. R. and Guthrie, C. Guide to Yeast Genetics and Molecular Biology. Guthrie, C. and Fink, G. R. (194). 1991. Academic Presss, Inc. Methods in Enzymology. Ref Type: Book, Whole
Gaber, R. F., Styles, C. A., and Fink, G. R. TRK1 encodes a plasma membrane protein required for high-affinity potassium transport in Saccharomyces cerevisiae. Mol. Cell. Biol. 8: 2848-2859. (1988)
Goldstein, A. L. and McCusker, J. H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae [In Process Citation] Yeast.
15: 1541-1553.(1999)
Goldstein, S. A., Price, L. A., Rosenthal, D. N., and Pausch, M. H. ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae [published erratum appears in
Proc Natl Acad Sci USA 1999 Jan 5; 96(1) :318] Proc. Natl. Acad. Sci. U.S.A. 93: 13256-13261. (1996)
Guldener, U., Heck, S., Fielder, T., Beinhauer, J., and Hegemann, J. H. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic. Acids. Res. 24: 2519-2524. (1996)
Ikeda, K., Kobayashi, K., Kobayashi, T., Ichikawa, T., Kumanishi, T., Kishida,
H., Yano, R., and Manabe, T. Functional coupling of the nociceptin/ orphanin FQ
receptor with the G-protein-activated K+ (GIRK) channel. Brain Res. Mol. Brain Res. 45: 117-126. (1997)
Itoh, T., Tanaka, T., Nagai, R., Kamiya, T., Sawayama, T., Nakayama, T., Tomoike, H., Sakurada, H., Yazaki, Y., and Nakamura, Y. Genomic organization and mutational analysis of HERG, a gene responsible for familial long QT syndrome. Hum. Genet. 102: 435-439. (1998)
Jan, L. Y. and Jan, Y. N. Cloned potassium channels from eukaryotes and prokaryotes. Annu. Rev. Neurosci. 20 : 91-123 : 91-123. (1997)
Jelacic, T. M., Sims, S. M., and Clapham, D. E. Functional expression and characterization of G-protein-gated inwardly rectifying K+ channels containing GIRK3. J. Membr. Biol. 169: 123-129. (1999)
Ketchum, K. A., Joiner, W. J., Sellers, A. J., Kaczmarek, L. K., and Goldstein, S. A. A new family of outwardly rectifying potassium channel proteins with two
pore domains in tandem. Nature 376: 690-695. (1995)
Ko, C. H., Buckley, A. M., and Gaber, R. F. TRK2 is required for low affinity K+ transport in Saccharomyces cerevisiae. Genetics 125 : 305-312. (1990)
Ko, C. H. and Gaber, R. F. TRK1 and TRK2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 4266-4273. (1991)
Kubo, Y., Reuveny, E., Slesinger, P. A., Jan, Y. N., and Jan, L. Y. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel [see comments] Nature 364: 802-806. (1993)
Ludwig, A., Zong, X., Stieber, J., Hullin, R., Hofmann, F., and Biel, M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 18 : 2323-2329. (1999)
Madrid, R., Gomez, M. J., Ramos, J., and Rodriguez-Navarro, A. Ectopic potassium uptake in trk1 trk2 mutants of Saccharomyces cerevisiae correlates with a highly hyperpolarized membrane potential. J. Biol. Chem. 273: 14838-14844. (1998)
Main, M. J., Brown, J., Brown, S., Fraser, N. J., and Foord, S. M. The CGRP receptor can couple via pertussis toxin sensitive and insensitive G proteins. FEBS
Lett. 441 : 6-10. (1998)
Mumberg, D., Muller, R., and Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119-122. (1995)
Myers, A. M., Pape, L. K., and Tzagoloff, A. Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae. EMB0 J. 4: 2087-2092. (1985)
Nakamura, R. L., Anderson, J. A., and Gaber, R. F. Determination of key structural requirements of a K+ channel pore. J. Biol. Chem. 272: 1011-1O18. (1997)
Roberds, S. L. and Tamkun, M. M. Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proc. Natl. Acad. Sci. U.S.A. 88: 1798-1802. (1991)
Ronicke, V., Graulich, W., Mumberg, D., Muller, R., and Funk, M. Use of conditional promoters for expression of heterologous proteins in Saccharomyces cerevisiae. Methods Enzymol. 283: 313-22: 313-322. (1997)
Sanguinetti, M. C. and Zou, A. Molecular physiology of cardiac delayed rectifier K+ channels. Heart Vessels Suppl 12: 170-172. (1997)
Schreibmayer, W., Dessauer, C. W., Vorobiov, D., Gilman, A. G., Lester, H. A.,
Davidson, N., and Dascal, N. Inhibition of an inwardly rectifying K+ channel by
G-protein alpha-subunits. Nature 380: 624-627. (1996)
Smith, F. W., Ealing, P. M., Hawkesford, M. J., and Clarkson, D.T. Plant members of a family of sulfate transporters reveal functional subtypes. Proc. Natl. Acad. Sci. U.S.A. 92: 9373-9377. (1995)
Snyders, D. J., Tamkun, M. M., and Bennett, P.B.A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analisis after stable mammalian cell culture expression. J. Gen. Physiol. 101: 513-543. (1993)
Taglialatela, M., Castaldo, P., Pannaccione, A., Giorgio, G., and Annunziato, L. Human ether-a-gogo related gene (HERG) K+ channels as pharmacological targets: present and future implications. Biochem. Pharmacol. 55: 1741-1746. (1998)
Tang, W., Ruknudin, A., Yang, W. P., Shaw, S. Y., Knickerbocker, A., and Kurtz, S. Functional expression of a vertebrate inwardly rectifying K+ channel in yeast. Mol. Biol. Cell 6: 1231-124O. (1995)
Wach, A., Brachat, A., Pohlmann, R., and Philippsen, P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 10: 1793-18O8. (1994)
Wang, Q., Chen, Q., and Towbin, J. A. Genetics, molecular mechanisms and management of long QT syndrome. Ann. Med. 30: 58-65. (1998)
Wilde, A. A. and Veldkamp, M. W. lon channels, the QT interval, and arrhythmias. Pacing. Clin. Electrophysiol. 20: 2048-2051. (1997)
Wischmeyer, E., Doring, F., Spauschus, A., Thomzig, A., Veh, R., and Karschin,
A. Subunit interactions in the assembly of neuronal Kir3.0 inwardly rectifying K+ channels. Mol. Cell Neurosci. 9: 194-206. (1997)
Yamada, M., Inanobe, A., and Kurachi, Y. G protein regulation of potassium ion
channels. Pharmacol. Rev. 50: 723-760. (1998)
Bock, J. H., Shuck, M. E., Benjamin, C. W., Chee, M., Bienkowski, M. J., and Slightom, J. L. Nucleotide sequence analysis of the human KCNJ1 potassium channel
locus Gene 188: 9-16. (1997)
Delpon, E., Valenzuela, C., and Tamargo, J. Blockade of cardiac potassium and other channels by antihistamines Drug Saf 21 Suppl 1: 11-8; discussion 81-7: 11-18. (1999)
Drici, M. D. and Barhanin, J. Cardiac K+ channels and drug-acquired long QT syndrome Therapie 55: 185-193. (2000)
DuBuske, L. M. Second-generation antihistamines: the risk of ventricular arrhythmias Clin. Ther. 21: 281-295. (1999)
Itoh, T., Tanaka, T., Nagai, R., Kikuchi, K., Ogawa, S., Okada, S., Yamagata, S., Yano, K., Yazaki, Y., and Nakamura, Y. Genomic organization and mutational analysis of KVLQT1, a gene responsible for familial long QT syndrome Hum. Genet. 103: 290-294. (1998)
Kobayashi, T., Ikeda, K., and Kumanishi, T. Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K(+) (GIRK) channels expressed in Xenopus oocytes Br. J. Pharmacol. 129: 1716-1722.(2OOO)
Richelson, E. Preclinical pharmacology of neuroleptics: focus on new generation compounds J. Clin. Psychiatry 57 Suppl 11: 4-11: 4-11. (1996)
Richelson, E. Receptor pharmacology of neuroleptics: relation to clinical effects [see comments] J. Clin. Psychiatry 60 Suppl 10: 5-14: 5-14. (1999)
Shuck, M. E., Bock, J. H., Benjamin, C. W., Tsai, T. D., Lee, K. S., Slightom,
J. L., and Bienkowski, M. J. Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel J. Biol. Chem. 269: 24261-24270. (1994)
Taglialatela, M., Castaldo, P., Pannaccione, A., Giorgio, G., Genovese, A., Marone, G., and Annunziato, L. Cardiac ion channels and antihistamines: possible mechanisms of cardiotoxicity Clin. Exp. Allergy 29 Suppl 3: 182-9: 182-189. (1999)
【0114】
【0115】
【特許請求の範囲】
【請求項1】
真核細胞カリウムチャネルの阻害物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項2】
変異サッカロミセスセレビシエ細胞(Δtrk1、Δtrk2、 Δtok1)で遺伝子TRK1、TRK2およびTOK1のスイッチが切られている請求項1に記載の方法。
【請求項3】
真核細胞カリウムチャネルがヒトカリウムチャネルである請求項1または2のいずれかに記載の方法。
【請求項4】
真核細胞カリウムチャネルがHERG1、Kv1.5またはgpIRK1である請求項1〜3のいずれかに記載の方法。
【請求項5】
真核細胞カリウムチャネルが変異している請求項1〜4のいずれかに記載の方法。
【請求項6】
真核細胞カリウムチャネルが酵母の発現プラスミドに存在する請求項1〜5のいずれかに記載の方法。
【請求項7】
変異サッカロミセスセレビシエ細胞が増殖レポーターを構成的に発現する請求項1〜6のいずれかに記載の方法。
【請求項8】
真核細胞カリウムチャネルに対して作用を及ぼす被検物質が変異サッカロミセスセレビシエ細胞の増殖を抑制する請求項1〜7のいずれかに記載の方法。
【請求項9】
真核細胞カリウムチャネルに対する被検物質の作用が、変異サッカロミセスセレビシエ細胞の細胞数を測定することによって決定される請求項1〜7のいずれかに記載の方法。
【請求項10】
細胞数が、構成的に発現された増殖レポーターの蛍光または化学発光により決定される請求項9に記載の方法。
【請求項11】
内在性カリウムチャネルTRK1、TRK2およびTOK1が発現されない変異サッカロミセスセレビシエ細胞。
【請求項12】
遺伝子TRK1、TRK2およびTOK1のスイッチが切られている変異サッカロミセスセレビシエ細胞。
【請求項13】
DSM13197として寄託された変異サッカロミセスセレビシエ細胞。
【請求項14】
サッカロミセスセレビシエ細胞が真核細胞カリウムチャネルを異種発現する請求項11〜13のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項15】
真核細胞カリウムチャネルがヒトカリウムチャネルである請求項11〜14のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項16】
真核細胞カリウムチャネルがHERG1、Kv1.5またはgpIRK1である請求項11〜15のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項17】
真核細胞カリウムチャネルが変異している請求項11〜16のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項18】
カリウムチャネルTRK1、TRK2およびTOK1を発現せず、前記遺伝子TRK1、TRK2およびTOK1がノックアウトによって破壊されている変異サッカロミセスセレビシエ細胞の作製方法。
【請求項19】
真核細胞カリウムチャネルの活性を抑制する物質を特定するための、請求項11〜17のいずれかに記載の変異サッカロミセスセレビシエ細胞の使用。
【請求項20】
真核細胞カリウムチャネルの活性化物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項21】
真核細胞カリウムチャネルの活性化物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を前記真核細胞カリウムチャネルの阻害物質の存在下で被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項22】
請求項11〜17のいずれかに記載の変異サッカロミセスセレビシエ細胞を含む検査キット。
【請求項23】
(a) 真核細胞カリウムチャネルの阻害物質を請求項1〜10のいずれかに記載の方法を利用して特定し
(b) 前記阻害物質を知られている化学的方法によって製造または単離し、そして
(c) 生理学的に許容できる添加物を前記阻害物質に添加することからなる医薬の製造方法。
【請求項24】
(a) 請求項20または21に記載の方法を利用して真核細胞カリウムチャネルの活性化物質を特定し
(b) 前記活性化物質を知られている化学的方法によって製造または単離し、そして
(c) 生理学的に許容できる添加物を前記活性化物質に添加することからなる医薬の製造方法。
【請求項1】
真核細胞カリウムチャネルの阻害物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項2】
変異サッカロミセスセレビシエ細胞(Δtrk1、Δtrk2、 Δtok1)で遺伝子TRK1、TRK2およびTOK1のスイッチが切られている請求項1に記載の方法。
【請求項3】
真核細胞カリウムチャネルがヒトカリウムチャネルである請求項1または2のいずれかに記載の方法。
【請求項4】
真核細胞カリウムチャネルがHERG1、Kv1.5またはgpIRK1である請求項1〜3のいずれかに記載の方法。
【請求項5】
真核細胞カリウムチャネルが変異している請求項1〜4のいずれかに記載の方法。
【請求項6】
真核細胞カリウムチャネルが酵母の発現プラスミドに存在する請求項1〜5のいずれかに記載の方法。
【請求項7】
変異サッカロミセスセレビシエ細胞が増殖レポーターを構成的に発現する請求項1〜6のいずれかに記載の方法。
【請求項8】
真核細胞カリウムチャネルに対して作用を及ぼす被検物質が変異サッカロミセスセレビシエ細胞の増殖を抑制する請求項1〜7のいずれかに記載の方法。
【請求項9】
真核細胞カリウムチャネルに対する被検物質の作用が、変異サッカロミセスセレビシエ細胞の細胞数を測定することによって決定される請求項1〜7のいずれかに記載の方法。
【請求項10】
細胞数が、構成的に発現された増殖レポーターの蛍光または化学発光により決定される請求項9に記載の方法。
【請求項11】
内在性カリウムチャネルTRK1、TRK2およびTOK1が発現されない変異サッカロミセスセレビシエ細胞。
【請求項12】
遺伝子TRK1、TRK2およびTOK1のスイッチが切られている変異サッカロミセスセレビシエ細胞。
【請求項13】
DSM13197として寄託された変異サッカロミセスセレビシエ細胞。
【請求項14】
サッカロミセスセレビシエ細胞が真核細胞カリウムチャネルを異種発現する請求項11〜13のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項15】
真核細胞カリウムチャネルがヒトカリウムチャネルである請求項11〜14のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項16】
真核細胞カリウムチャネルがHERG1、Kv1.5またはgpIRK1である請求項11〜15のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項17】
真核細胞カリウムチャネルが変異している請求項11〜16のいずれかに記載の変異サッカロミセスセレビシエ細胞。
【請求項18】
カリウムチャネルTRK1、TRK2およびTOK1を発現せず、前記遺伝子TRK1、TRK2およびTOK1がノックアウトによって破壊されている変異サッカロミセスセレビシエ細胞の作製方法。
【請求項19】
真核細胞カリウムチャネルの活性を抑制する物質を特定するための、請求項11〜17のいずれかに記載の変異サッカロミセスセレビシエ細胞の使用。
【請求項20】
真核細胞カリウムチャネルの活性化物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項21】
真核細胞カリウムチャネルの活性化物質を特定する方法であって、
(a) 3つの内在性カリウムチャネルTRK1、TRK2およびTOK1を発現しない変異サッカロミセスセレビシエ細胞を用い
(b) 真核細胞カリウムチャネルを前記変異サッカロミセスセレビシエ細胞で異種発現させ
(c) 前記変異サッカロミセスセレビシエ細胞を前記真核細胞カリウムチャネルの阻害物質の存在下で被検物質と一緒にインキュベートし、そして
(d) 前記真核細胞カリウムチャネルに対する被検物質の作用を決定する、上記方法。
【請求項22】
請求項11〜17のいずれかに記載の変異サッカロミセスセレビシエ細胞を含む検査キット。
【請求項23】
(a) 真核細胞カリウムチャネルの阻害物質を請求項1〜10のいずれかに記載の方法を利用して特定し
(b) 前記阻害物質を知られている化学的方法によって製造または単離し、そして
(c) 生理学的に許容できる添加物を前記阻害物質に添加することからなる医薬の製造方法。
【請求項24】
(a) 請求項20または21に記載の方法を利用して真核細胞カリウムチャネルの活性化物質を特定し
(b) 前記活性化物質を知られている化学的方法によって製造または単離し、そして
(c) 生理学的に許容できる添加物を前記活性化物質に添加することからなる医薬の製造方法。
【図1】

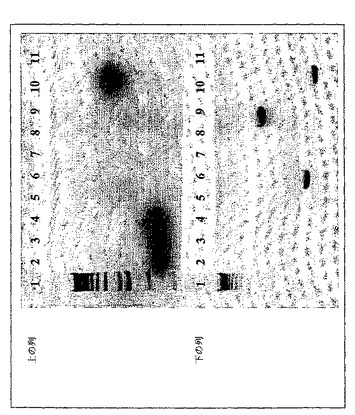
【図2】
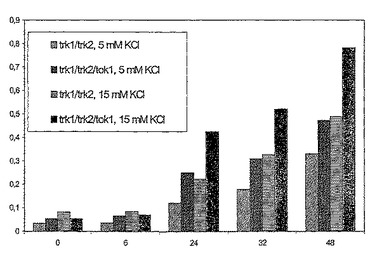
【図3】

【図4】
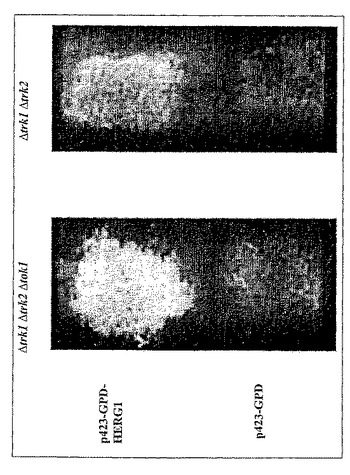
【図5】
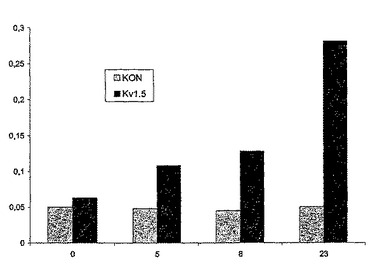
【図6】
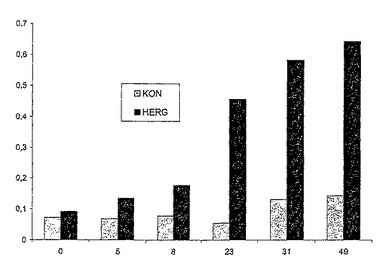
【図7】
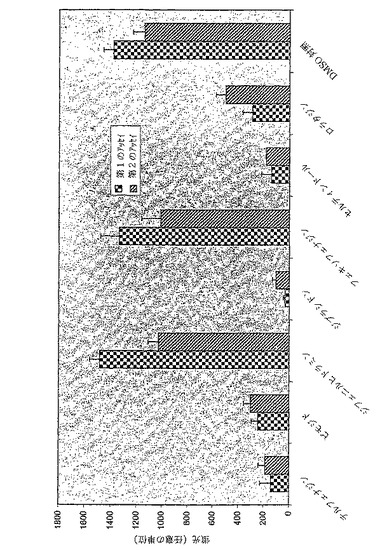
【図8】

【図9】
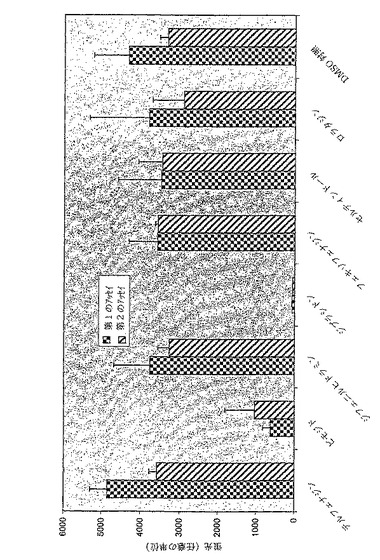
【図10】
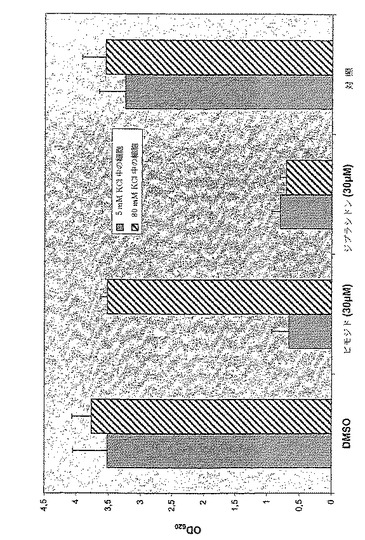
【図11】
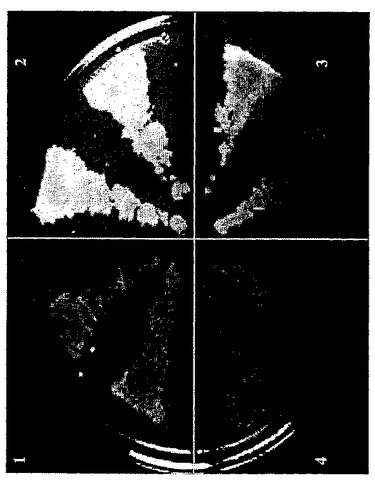
【図12】

【図13】
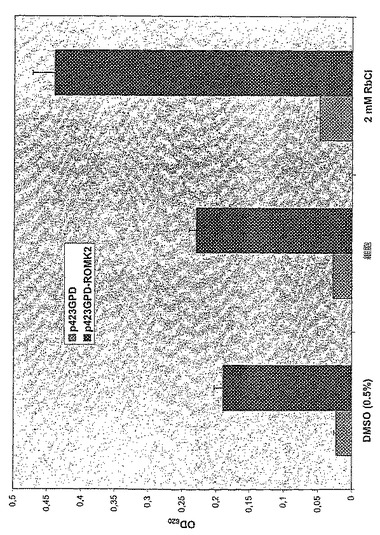
【図14】
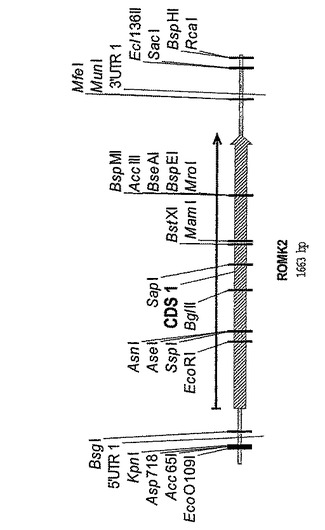
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2012−10709(P2012−10709A)
【公開日】平成24年1月19日(2012.1.19)
【国際特許分類】
【出願番号】特願2011−191229(P2011−191229)
【出願日】平成23年9月2日(2011.9.2)
【分割の表示】特願2001−551103(P2001−551103)の分割
【原出願日】平成13年1月5日(2001.1.5)
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
【公開日】平成24年1月19日(2012.1.19)
【国際特許分類】
【出願日】平成23年9月2日(2011.9.2)
【分割の表示】特願2001−551103(P2001−551103)の分割
【原出願日】平成13年1月5日(2001.1.5)
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
[ Back to top ]