
カーボンナノチューブの還元的官能基化
本発明は、カーボンナノチューブの官能基化(誘導体化)の新規なプロセス、並びに、延長として、フラーレン及び他の炭素表面に関する。一般に、このようなプロセスは還元的経路を含む。幾つかの具体例においては、カーボンナノチューブは、無水液体アンモニア中でアルカリ金属及び有機ハロゲン化物と反応する。他の具体例においては、無水液体アンモニア中でカーボンナノチューブをアルカリ金属及びモノマー種と反応させることによって、ポリマーをカーボンナノチューブ側壁から成長させる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、海軍研究局(the Office of Naval Research)の助成第N00014−01−1−0789号からの援助によりなされた。
関連出願に対するクロスリファレンス
【0002】
本出願は、2004年3月12日に出願された米国仮出願第60/552,550号;及び2004年9月17日に出願された第60/611,045号に対する優先権を請求する。
【0003】
本発明は一般にカーボンナノチューブに関し、特に還元的経路を経てカーボンナノチューブを誘導体化する方法に関する。
【背景技術】
【0004】
カーボンナノチューブ(CNTs、akaフラーレンパイプ)は、概念上それ自体の上に巻き上がり、フラーレンキャップによって末端が閉じたグラフェン(graphene)シートを含むナノスケール炭素構造である。単層カーボンナノチューブ(SWNTs)は、単一のこのようなグラフェン円筒のみを含むが、多層ナノチューブは、ロシアの入れ子人形のものに類似した仕方で1つが別のものの内部に入れ子になっている2つ以上の同軸グラフェン層で作られている。1993年におけるその最初の製造(Iijima et al., Nature, 1993, 363, 603; Bethune et al., Nature, 1993, 363, 605; Endo et al., Phys. Chem. Solids. 1993, 54, 1841)以来、SWNTsは、その類のない機械的、光学的、電子的、及び他の性質が理由となって広範に研究されてきた。例えば、SWNTsの注目すべき引張強さは、強化繊維及びポリマーナノコンポジットにおけるその使用をもたらした(Zhu et al., Nano Lett. 2003, 3, 1107及びこの中の参考文献)。CNTsの他の既存の及び潜在的な用途に関しては、Baughman et al, Science, 2002, 297, 787-792を参照されたい。
【0005】
SWNTsは通常自己組織化して、凝集体または束になり、ここで最高数百までのチューブがファンデルワールス力で結合する。医用生体用途を含む多くの用途の場合、こうした束からの個々のナノチューブの分離は不可欠である。このような分離は、加工及び操作にとって必要な一般的な有機溶媒及び/または水中のナノチューブの分散及び可溶化を改良する。SWNT表面の共有結合性修飾は一般に、ナノチューブの溶解度/懸濁性及び加工性を改良することによってこの問題を解決するのを助ける。ナノチューブ末端の化学的官能基化は一般に、こうした材料の電子的及びバルク性質を変化させないが、側壁官能基化は、ナノチューブの固有の性質を確かにかなり変更し(Chen et al, Science, 1998, 282, 95-98; Mickelson et al, Chem. Phys. Lett., 1998, 296, 188-194)、典型的にその溶解度/懸濁性により大きな影響を及ぼす(Boul et al, Chem. Phys. Lett., 1999, 310, 367-372)。しかしながら、化学のこの新たな分野における記録された結果の範囲は、主にナノチューブの現在の高コストが理由となって限定されている。
【0006】
SWNT側壁の修飾において直面するさらなる問題は、その比較的に不満足な反応性に関連している−主として、反応性のより高いフラーレンを基準としてナノチューブ壁のはるかに低い湾曲(M. S. Dresselhaus, G. Dresselhaus, P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996, Vol. 1)、並びにグラフェン壁に付着した官能基の数及びサイズが増大すると共に管状構造内部の歪みが増大することが理由となっている。ナノチューブの骨組みを構成する炭素原子の全てのsp2−結合状態は、付加タイプ反応の優勢な発生を促進する。こうした反応の最も良く特徴付けられた例は、ニトレン、アゾメチンイリド及びジアゾニウム塩から発生したアリールラジカルのSWNTsへの付加を含む(V. N. Khabashesku, J. L. Margrave, Chemistry of Carbon Nanotubes in Encyclopedia of Nanoscience and Nanotechnology, Ed. H. S. Nalwa, American Scientific Publishers, 2004; Bahr et al., J. Mater. Chem., 2002, 12, 1952; Holzinger et al., Angew. Chem. Int. Ed., 2001, 40, 4002)。SWNTsの他の報告されている側壁官能基化は、有機ラジカル(Peng et al, Chem. Commun., 2003, 362; Ying et al, Org. Lett, 2003, 5, 1471; Peng et al, J.Am, Chem. Soc, 2003, 125, 15174)及びビンジェル反応(Bingel reaction)(Coleman et al, J. Am. Chem. Soc, 2003, 125, 8722)を含む。加えて、水素をSWNTsの側壁に付加することは、バーチ還元の条件下で起きると報告されている(Pekker et al, J. Phys. Chem. B, 2001, 105, 7938)。
【0007】
個々のCNTsの直径及びキラリティーは、整数“n”及び“m”によって説明され、ここで、(n,m)は、概念上巻き上がってチューブを形成するグラフェンシートに沿ったベクトルである。|n−m|=3q(ここで、qはゼロでない整数である)である場合、CNTは半金属である(バンドギャップはミリeVのオーダー)。n−m=0である場合、CNTは真の金属であり、バンドギャップは0eVであり、“アームチェア”ナノチューブと呼ばれる。n−mの全ての他の組合せは、半導性CNTであり、バンドギャップは典型的に0.3〜1.0eVの範囲内である。O'Connell et al, Science, 2002, 297, 593を参照されたい。本明細書において使用するCNT“タイプ”は、(n,m)ベクトルによって説明されるこのような電子的タイプ(すなわち、金属性、半金属性、及び半導性)を指す。本明細書において使用するCNT“種”は、個別の(n,m)値を有するCNTsを指す。本明細書において使用するCNT“組成物”は、ナノチューブタイプ及び種の点でのCNT集団の構成を指す。
【0008】
全ての周知のCNT製造方法は、半導性、半金属性、及び金属性の電子的タイプのCNT集団を多分散させることをもたらす。M. S. Dresselhaus, G. Dresselhaus, P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996; Bronikowski et al., Journal of Vacuum Science & Technology A 2001, 19, 1800-1805; R. Saito, G. Dresselhaus, M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial College Press, London, 1998を参照されたい。従って、CNTs及びSWNTsの広範囲に及ぶ利用に対する主要な障害は特に、電子構造によるその操作である(Avouris, Ace. Chem. Res. 2002, 35, 1026-1034)。しかしながら、最近、CNTsをその電子構造(すなわち、電子的タイプ)に基づいて選択的に官能基化する方法が報告されている(Strano et al, Science, 2003, 301, 1519-1522;2004年7月29日に出願され、共通に譲渡された同時係属国際特許出願PCT/US04/24507)。このような報告において、金属性CNTsはジアゾニウム種と優先的に反応して、金属性及び半導性CNTsの混合物の部分的官能基化を経て金属性(半金属性を含む)及び半導性CNTsの分離または分別を可能にすることが認められている。CNTタイプ及び種、並びにその光学的確認の詳細な検討に関しては、Bachilo et al., Science, 2002, 298, 2361-2366; and Weisman et al, Nano. Lett, 2003, 3, 1235-1238を参照されたい。
【0009】
カーボンナノチューブの側壁を化学的に誘導体化することにおけるこのような上記に説明した進歩にもかかわらず、大部分のこのようなプロセスは、誘導体化プロセスの最中にカーボンナノチューブの超音波処理を必要とする。この音波処理は潜在的に、試料中の多くのナノチューブを損傷し得る。従って、より穏やかな条件下でカーボンナノチューブを誘導体化する方法は非常に有益であると思われる。
【発明の開示】
【0010】
本発明は、カーボンナノチューブの官能基化(すなわち、化学的誘導体化)の新規なプロセス(方法)、並びに、その延長上に、フラーレン及び他の炭素表面(すなわち、一般に無機炭素材料)に関する。一般に、このようなプロセスは還元的経路を含む。幾つかの具体例においては、カーボンナノチューブは、無水液体アンモニア中でアルカリ金属及び有機ハロゲン化物と反応する。他の具体例においては、無水液体アンモニア中でカーボンナノチューブをアルカリ金属及びモノマー種と反応させることによって、ポリマーをインシトゥでカーボンナノチューブ側壁から成長させる。
【0011】
幾つかの具体例においては、本発明は、炭素材料を官能基化する方法に関し、このような方法は:(a)炭素材料を液体アンモニアと合わせて、混合物を形成する工程と;(b)ある量のアルカリ金属を混合物中に溶解させて、還元的混合物を形成する工程と;(c)有機部分及びハロゲン化物部分を含む有機ハロゲン化物を還元的混合物に加え、その結果、有機ハロゲン化物の有機部分が炭素材料に付加して誘導体化炭素材料を形成するようにする工程と;を含む。様々なクエンチング(未反応のアルカリ金属を中和するための)、酸性化(未蒸発のアンモニアを中和するための)、ろ過(誘導体化炭素材料を集めるための)、及び洗浄(望まれない種を誘導体化炭素材料から除去するための)工程のうちの1つ以上を、所望により実行することができる。一般に、このような炭素材料としては、カーボンナノチューブ、ナノスクロール(nanoscroll)、フラーレン、ダイヤモンド、アセチレン型炭素(acetylenic carbon)、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せが挙げられるがこれらに限定されるものではない。
【0012】
幾つかの具体例においては、本発明は、ポリマーをインシトゥで炭素表面から成長させる方法に関し、このような方法は:(a)炭素材料を液体アンモニアと合わせて、混合物を形成する工程と;(b)ある量のアルカリ金属を混合物中に溶解させて、還元的混合物を形成する工程と;(c)ある量の少なくとも1つのモノマー種を還元的混合物に加え、その結果、モノマーが炭素材料の上に/において重合するようにする工程と;を含む。様々なクエンチング、酸性化、ろ過、及び洗浄工程のうちの1つ以上を、所望により実行することができる。このようなインシトゥ重合は、炭素材料及びポリマーを含む複合材料を与えることができる。幾つかの具体例においては、モノマーはアニオン重合を受けることが認められている。幾つかのまたは他の具体例においては、モノマーはラジカル重合を受けることが認められている。上記の官能基化反応におけるように、このような炭素材料としては、カーボンナノチューブ、ナノスクロール、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せが挙げられるがこれらに限定されるものではない。
【0013】
上記に説明した具体例の変形例は、アンモニアの代わりに、またはこれに加えて無水アミンのような他の溶媒の使用を含む。
【0014】
本発明の方法は、このタイプの側壁官能基化の同様の方法が存在しないという点で新規である。その上、このような方法は、カーボンナノチューブの側壁を誘導体化する穏やかな(すなわち、超音波処理を必要としない)拡大縮小できるプロセスに対する当分野において認識されている必要を満たす。
【0015】
上文は、下記の本発明の詳細な説明がより良く理解されるために、本発明の特徴をかなり広く略述した。本発明の請求の範囲の主題を形成する本発明のさらなる特徴及び利点を下文で説明する。
【0016】
本発明及びその利点のより完全な理解のために、ここから添付図面と共に以下の説明を参照する。
【発明を実施するための最良の形態】
【0017】
本発明は、カーボンナノチューブの官能基化(誘導体化)の新規なプロセス、並びに、延長として、フラーレン及び他の炭素表面に関する。一般に、このようなプロセスは還元的経路を含む。幾つかの具体例においては、カーボンナノチューブは、無水アンモニア中でアルカリ金属及び有機ハロゲン化物と反応する。他の具体例においては、無水アンモニア中でカーボンナノチューブをアルカリ金属及びモノマー種と反応させることによって、ポリマーをカーボンナノチューブ側壁から成長させる。
【0018】
本発明によれば、カーボンナノチューブ(CNTs)としては、単層カーボンナノチューブ(SWNTs)、二層カーボンナノチューブ(DWNTs)、多層カーボンナノチューブ(MWNTs)、小直径カーボンナノチューブ、及びこれらの組合せが挙げられるがこれらに限定されるものではない。小直径カーボンナノチューブは、本明細書において、層の数にかかわらず直径最大約3nmを有するカーボンナノチューブと定義される。CNTsの全ての製造方法は、炭素質不純物を有する製品を与える。加えて、SWNTsの大部分の製造方法及びMWNTsの多くの製造方法は、不純物として生成物中に残る金属触媒を使用する。本明細書において説明する実施例は一般に、単層カーボンナノチューブ(SWNTs)を用いて行われたが、本明細書において説明する方法及び組成物は一般に、任意の周知の方法によって製造された全てのカーボンナノチューブに適用できる−但しこれらは、その反応性が理由となって本明細書において説明する化学の影響を受けやすいことは理解されるはずである。その上、ナノチューブは、本明細書において説明する化学修飾にさらされる前に、切断、長さ分類、キラリティー分類、精製等を含む任意の数の合成後連続工程にさらされることができる。
【0019】
CNTsの場合特に、官能基化は、溶媒中の改良されたレベルのナノチューブ懸濁性/分散性及び/または溶解度をもたらすことができる。真の溶解度は、溶媒中のCNTsの再凝集は、熱力学的根拠で、連続した溶媒和状態よりも容易ではない状態であることに注意されたい。それはそれとして、安定な懸濁液は、広範囲のプロセスのためのCNTsの操作を適切に可能にすることができる。
【0020】
本発明によれば、アルカリ金属は、Li、Na、及びKである。本発明によれば、有機ハロゲン化物(R−X)は、有機部分(R−)及びハロゲン化物部分(−X)を含む。有機部分は、本発明の具体例に従って有機ハロゲン化物を適切に提供する任意の有機官能性とすることができる。適切な有機官能性としては、アルキル−、アリール−、アリル−、ベンジル−、及びこれらの組合せが挙げられるがこれらに限定されるものではない。ハロゲン化物部分は、−F、−Cl、−Br、−I、及びこれらの組合せからなる群から選択される。
官能基化反応
【0021】
幾つかの具体例においては、本発明は、以下の工程を含むプロセス(方法)に関する:(a)カーボンナノチューブまたは他の炭素材料を、ドライアイス凝縮器を備えた火炎乾燥三首丸底フラスコ(または他の適切な容器)中に置く工程と;(b)NH3ガスをフラスコ中に凝縮させ、次に非常に小さな部片のアルカリ金属を加え、続いてある量のハロゲン化アルキル/アリールを加える工程と;(c)反応を一晩進行させる工程と;(d)反応混合物をアルコール、水を用いてクエンチし、次に鉱酸を用いて酸性化する工程と;(e)内容物をメンブランフィルターを通してろ過し、エタノール、次にヘキサンを用いて十分に洗浄して、副産物を除去する工程。このようなプロセスは、超音波処理が必要な特徴ではないという点で類がない。
【0022】
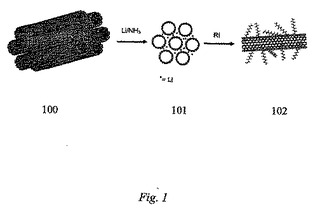
カーボンナノチューブ、または一般に任意の黒鉛質表面が官能基化されることを可能にする本発明の具体例である還元的経路のより詳細な説明が、便利に次に続く:カーボンナノチューブを火炎乾燥三首丸底フラスコに加えた。アンモニアを次にフラスコ中に凝縮させ、続いて小さな部片のリチウム金属を加えた。ハロゲン化アルキル/アリールを次に加え、アンモニアを蒸発させながら反応を一晩進行させた。反応混合物を次に氷浴中で冷却し、メタノールを徐々に加えることによってクエンチした。水を次に徐々に加え、続いて混合物が酸性になるまで10%HClを加えた。内容物を、0.2μm−ポアサイズPTFE(テフロン(登録商標)(Teflon(登録商標)))メンブランフィルターを使用してろ過した。ドデシル化ナノチューブのラマンスペクトルは、例えば、一般的な有機溶媒中の高い溶解度をもたらす並外れた官能基化を示す。ドデシル化単層カーボンナノチューブ(SWNTs)の原子間力顕微鏡のイメージングは、かなりの官能基化を示す単層カーボンナノチューブ束のかなりの剥脱を意味する極めて多量の個々のカーボンナノチューブを明らかにする。本発明の範囲は、以下のものを越えて拡大するが、このようにカーボンナノチューブに実験的に付着する官能基は、次のものを含む:n−ドデシル−、n−ブチル−、t−ブチル−、n−オクチル−、及びベンジル−。図1は、上記に説明した具体例の例を示し、ここで、SWNT束100は、無水液体アンモニア(NH3)中でLiによって還元されて、還元され挿入され/部分的に剥脱した束101を与える。この挿入され/部分的に剥脱した束101は次に有機ハロゲン化物(RI)と反応して、官能基化SWNTs102を与える。
【0023】
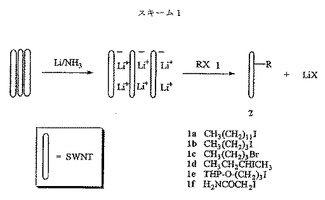
上記に説明した官能基化反応を図2(スキーム1)に概略で示し、ここで、SWNTsは、無水液体アンモニア中でリチウム(または他の適切なアルカリ金属)を用いてまず挿入/還元され、次に有機ハロゲン化物1と反応して、官能基化SWNTs2a〜fを与え、有機ハロゲン化物1a〜fは例として提供される。理論によって束縛されることを意図するものではないが、脱結束(debundling)は、リチウム(または他のアルカリ金属)による広範な挿入の点で説明することができ、図2に示すように、負に帯電したチューブの間に分散したリチウムイオンをもたらす。リチウムを液体アンモニア中のナノチューブの懸濁液に加えるにつれて、溶媒和電子に関連する強い青色は急速に消失し、SWNTsへの電子移動は容易なプロセスであることを示唆する。ラマン分光法を使用した予備実験は、炭素/リチウム比は2〜3であることを示唆する。ハロゲン化アルキルを加えることは、容易に解離してハロゲン化物及びアルキルラジカルを与えると思われるラジカルアニオンの形成をもたらすと思われる。理論によって束縛されることを意図するものではないが、ろ液のガスクロモタグラフィー(gas chromotagraphy)−質量分析法(GC−MS)の分析は、ラジカル経路のための強力な証拠を提供する(実施例7を参照されたい)。加えて、ケトンのカルボニル基への電子移動によって形成されたラジカルアニオンも、SWNTsの側壁に容易に付加することができる。
【0024】
カーボンナノチューブの官能基化は、これを一般的な有機溶媒中に可溶にする方法を提供し、従ってこれを複合体形成及び紡糸して繊維にするために修正可能にする。例えば、アルキル化SWNTsは、溶媒の例えばクロロホルム(CHCl3)、テトラハイドフラン(tetrahydofuran)(THF)、及びN,N−ジメチルホルムアミド(DMF)中の比較的に高いレベルの溶解度を示す。他の黒鉛質様表面の例えばカーボンブラックの官能基化は、様々なエラストマー及びポリマー中のその分散を促進する。加えて、本発明の具体例に従ってCNTsを官能基化する方法は、典型的に超音波処理を含む既存の従来技術の官能基化方法よりも一般により穏やかであり(すなわち、超音波処理を必要としない)、CNTsへのより少ない損傷を引き起こす。
【0025】
このような上記に説明した還元的官能基化方法は、様々な官能基(例えば、アルキル−、アリール−、アリル−、及びベンジル−)をカーボンナノチューブの側壁に付加することを可能にする。幾つかの具体例においては、こうした基のさらなる(その後の)官能基化が可能である。幾つかの具体例においては、様々な無水アミンを、無水アンモニアの代わりに、またはこれに加えて溶媒として使用してよい。試薬の量及びタイプ、反応時間等に、並びにクエンチング、酸性化、ろ過、及び洗浄の工程に、かなりの変化が存在する。加えて、本発明の方法は、はるかに大きな量に拡張可能である。
重合反応
【0026】
幾つかのまたは他の具体例においては、本発明は、以下の工程を含むプロセスに関する:(a)カーボンナノチューブを、ドライアイス凝縮器を備えた火炎乾燥三つ口フラスコ中に置く工程と;(b)NH3ガスをフラスコ中に凝縮させ、次に小さな部片のアルカリ金属を加え、続いてモノマー(例えば、アルキン/アルケン)を加える工程と;(c)反応を一晩進行させる工程と;(d)反応混合物をアルコールを用いてクエンチし、続いて鉱酸を用いて酸性化する工程と;(e)内容物をメンブランフィルターを通してろ過し、アルコール、次にヘキサンを用いて十分に洗浄して、副産物を除去する工程。一般に、適切なモノマーは、不飽和の領域(すなわち、二重若しくは三重結合、またはオリゴマーエチレンオキシド)を含む。
【0027】
カーボンナノチューブまたは一般に任意の黒鉛質表面の側壁に付着したポリマー鎖を形成するための、還元的経路を経るモノマー材料のインシトゥ重合に対処する本発明のさらに別の具体例であるこのような上記に説明した重合方法の詳細な説明が、次に続く:カーボンナノチューブを火炎乾燥三つ口フラスコに加えた。アンモニアを次にフラスコ中に凝縮させ、続いてリチウム金属を加えた。メタクリル酸メチル、アクリロニトリル、及びアセチレンを含む様々なモノマーを次に反応フラスコに加え、アンモニアが蒸発させながら一晩放置した。エタノールを次に加え、続いて反応混合物が酸性になるまで10%HClを加えた。内容物を分液漏斗に移し、官能基化カーボンナノチューブをクロロホルム中に抽出した。抽出物を、水/エタノール混合物を用いて洗浄し、0.2μmPTFE(ポリテトラフロウロエチレン(polytetraflouroethylene))メンブランフィルターを通してろ過した。
【0028】
インシトゥ重合を経て炭素材料/ポリマー複合体を製造するこのような上記に説明した方法にかなりの変化が存在する。様々なモノマー種または種の組合せを使用できる。加えて、試薬の量及びタイプ、反応時間等に、並びにクエンチング、酸性化、ろ過、及び洗浄の工程に変化が存在する。
【0029】
CNTs表面のインシトゥ重合の場合には、カーボンナノチューブはポリマーに化学的に結合し、これは、多機能性のさらなる見込みを有する軽量複合体系のための機械的強化として役立つことができる。本明細書において説明するもののような方法を使用して、様々なポリマーをナノチューブに化学的に付着させることができ、こうした付着したポリマー鎖のさらなる官能基化が可能である。
【0030】
理論によって束縛されることを意図するものではないが、少なくとも本発明の幾つかの具体例においては、バーチタイプ還元が起き、ここで、CNTsまたは他の炭素材料は、アルカリ金属によって表面において還元されると考えられている。この還元された表面は次に、有機(例えば、アルキル/アリール)ハロゲン化物と反応して、官能基化CNTsを形成することができるか、またはインシトゥ重合のためのモノマーと反応することができる。加えて、CNTsを含む具体例においては、超音波処理の無い場合のナノチューブ(特にSWNTs)の脱結束は、アルカリ金属による広範な挿入の点で説明することができ、負に帯電したナノチューブの間に分散したアルカリイオンをもたらす。官能基化反応に関して上記に言及したように、実験的に、リチウム金属を使用する場合、リチウムを液体アンモニア中のナノチューブの懸濁液に加えるにつれて、溶媒和電子に関連する強い青色は急速に消失し、SWNTsへの電子移動は容易なプロセスであることを示唆する。
【0031】
以下の実施例を、本発明の特定の具体例を証明するために提供する。下記の実施例において開示する方法は、単に本発明の模範的な具体例を表すことは、当業者であれば了解されるはずである。しかしながら、当業者であれば、本開示を考慮して、本発明の精神及び範囲から逸脱することなく、説明され依然として同様なまたは類似の結果を得る具体例に多くの変更を行い得ることは了解されよう。
【実施例】
【0032】
実施例l
この実施例は、式1(Eq.1)に表す、臭化n−ブチルの存在下でのLi/NH3還元によるカーボンナノチューブのアルキル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)3−Br→n−ブチル化SWNTs(Eq.1)
【0033】
撹拌子を備え、ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の(綿毛状、製造したまま)単層カーボンナノチューブ(SWNTs)を置いた。アルゴン(Ar)を用いてフラスコをフラッシングした後、これをドライアイス/アセトン浴中に浸漬し、NH3ガス(〜60mL)をその中で凝縮させた。次に、これに300mgのLi金属を少量ずつ加えた。Liを加えた直後に、混合物は色が深青色に変わった。5分後に、0.53mLの臭化n−ブチルを加えた。氷浴を除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。翌朝、フラスコが氷浴中にある間にまず10mLのメタノール、次に20mLの水を加えることによって、反応を完成させた。フラスコの内容物を、10%HClを用いて酸性化し、次に0.2μmポアサイズポリテトラフルオロエチレン(PTFE)メンブランフィルター上でろ過した。フィルター上に残った材料を20mLのエチルアルコール中に置き、20min音波処理し、次に再度0.2μmPTFEメンブランフィルター上でろ過した。次に、同じプロセスを、ベンゼン及びテトラヒドロフラン(THF)を用いて繰り返した。このように得られたバッキーペーパー(bucky paper)(SWNTs)を、それに続くラマン分析のために使用した。
【0034】
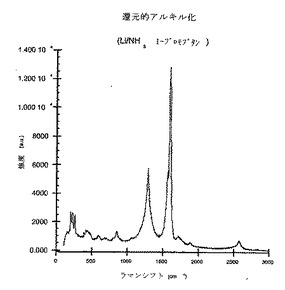
ラマン分析は、説明したように反応が進行してカーボンナノチューブを誘導体化することを示すかなりの無秩序なピーク(disorder peak)を明らかにする。官能基付加は、官能基が付加するナノチューブケージ内部の炭素を、支配的には性質がsp2から支配的には性質がsp3へと変化させる、という事実から無秩序なピークの増大が生じることに留意されたい。このラマン分析を図3に表す。
実施例2
【0035】
この実施例は、Eq.2によって示される、塩化ベンジルの存在下でのLi/NH3還元によるカーボンナノチューブのベンジル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+(C6H5)−CH2−Cl→ベンジル化SWNTs(Eq.2)
【0036】
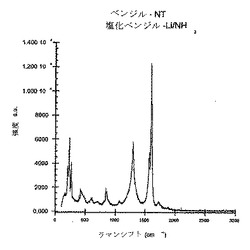
この反応を、実施例1において説明したものと同様の仕方で実行した。火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次に、このフラスコに〜60mLのNH3を凝縮させ、続いて非常に小さな部片のLi金属を加えた(合計〜462mg)。Liを完全に加えた後、〜2.108g(1.92mL)の塩化ベンジルを、シリンジでフラスコに加えた。フラスコの下の氷浴をそれに続いて除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加え、次に10%HClを用いて酸性化することによって、反応を完成させた。0.2μmPTFEメンブランフィルター上でエタノールを用いて材料を繰り返し洗浄してバッキーペーパーを与え、これをさらなる分析のために使用した。図4に示すように、ラマン分析は、かなりの無秩序なピークを明らかにした。
実施例3
【0037】
この実施例は、Eq.3によって示される、n−ドデシルイオダイドの存在下でのLi/NH3還元によるカーボンナノチューブのドデシル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)11−I→n−ドデシル化SWNTs(Eq.3)
【0038】
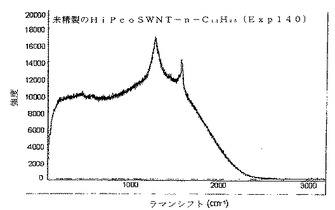
ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。Arを用いてフラスコをフラッシングした後、NH3ガス(〜60mL)をその中で凝縮させた。これにLi金属を少量ずつ加えた(合計〜462mg)。Liを加えた直後に、混合物は色が深青色に変わった。5分後に、4.933g(4.107mL)のヨードドデカンを加えた。氷浴を除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。翌朝、フラスコが氷浴中にある間にまず10mLのメタノール、次に20mLの水を加えることによって、反応を完成させた。次にこれを、酸性になるまで10%HClを用いて酸性化した。これを、それに続いて0.2μmポアサイズPTFEメンブランフィルター上でろ過した。フィルター上の材料を次に30mLのエタノール中で音波処理し、再度ろ過し、次にエタノール/ヘキサン混合物(2:1)を用いて洗浄した。メンブランフィルター上の得られた材料は、CHCl3中の並外れた溶解度を有することが決定された。このように得られたバッキーペーパー(SWNTsマット)を、それに続くラマン分析のために使用し、図5に示すようにかなりの無秩序なピークを明らかにした。
実施例4
【0039】
この実施例は、Eq.4によって示される、n−オクチルイオダイドの存在下でのLi/NH3還元によるカーボンナノチューブのオクチル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)7−Br→n−オクチル化SWNTs(Eq.4)
【0040】
ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次にフラスコに〜60mLのNH3ガスを凝縮させ、続いて非常に小さな部片のLi金属(合計462mg)を加えた。リチウムを加えた後、〜3.00mL(3.998g)のn−オクチルイオダイドをシリンジで加えた。NH3を徐々に蒸発させながら反応を一晩進行させた。次に、混合物中の反応を、10mLのアルコールを徐々に加えながらクエンチし、この間フラスコを氷浴中に維持した。次に、20mLの水を徐々に加え、続いて酸性になるまで10%HClを加えた。内容物を0.2μmPTFEメンブランフィルター上でろ過した。ろ紙上に残った材料を、アルコール、続いてヘキサンを用いて繰り返し洗浄した。
実施例5
【0041】
この実施例は、Eq.5によって示される、ヨウ化t−ブチルの存在下でのLi/NH3還元によるカーボンナノチューブのtert−ブチル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+(CH3)3C−I→t−ブチル化SWNTs(Eq.5)
【0042】
火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次に、このフラスコに〜70mLのNH3を凝縮させ、続いて非常に小さな部片のLi金属を加えた(合計〜462mg)。Liを完全に加えた後、〜3.065g(1.99mL)のヨウ化t−ブチルを、シリンジでフラスコに加えた。フラスコの下の氷浴をそれに続いて除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加え、次に10%HClを用いて酸性化することによって、反応を完成させた。0.2μmPTFEメンブランフィルター上でエタノールを用いて材料を繰り返し洗浄してバッキーペーパーを与え、これをさらなる分析のために使用した。
実施例6
【0043】
この実施例は、Eq.6によって示される、n−ドデシルイオダイドの存在下でのL1/NH3還元による、精製済みカーボンナノチューブのドデシル化を示すのに役立つ。
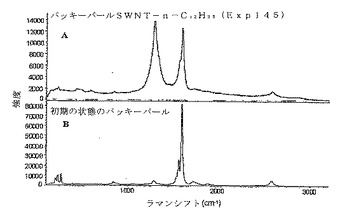
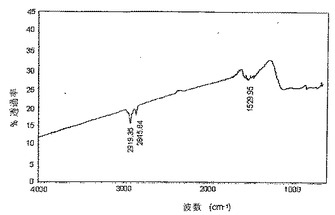
SWNTs(精製済み)+Li/NH3+CH3−(CH2)11−I→n−ドデシル化SWNTs(Eq.6)
【0044】
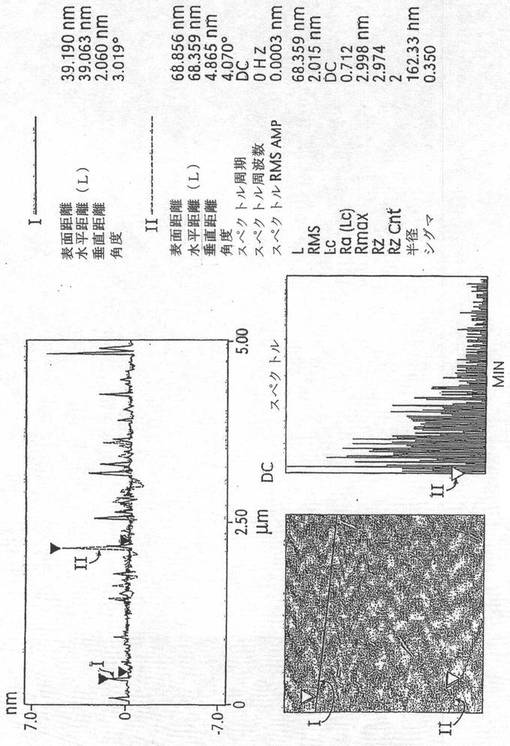
比較のために、実施例3において説明したプロセスを、精製済みSWNTsを使用して実行した。特に、使用した精製済みSWNTsは、“バッキーパール(bucky pearl)”として周知の高密度タイプだった。図6に示すように、ラマン分析は、本発明の誘導体化プロセスは、未精製のSWNT材料に作用するのと少なくとも同様に精製済みSWNT材料に作用することを示唆する。図7は、この実施例において製造したドデシル化SWNTsの原子間力顕微鏡法(AFM)像であり、図8は、対応するセクション分析(section analysis)である。図7及び8の両方は、SWNTsのかなりの脱結束(そのロープ化状態からの)及び極めて多量の個々のSWNTsを明らかにする−かなりの誘導体化を示す。図9は、ドデシル化SWNTsのFTIRスペクトルであり、図10(A)及び(B)は、ドデシル化SWNTsの高分解能透過型電子顕微鏡法(HRTEM)像であり、個々のナノチューブ及び広範な脱結束を示す。
実施例7
【0045】
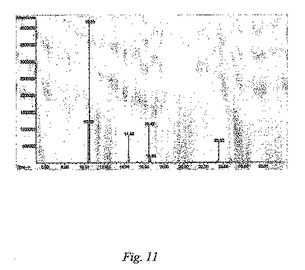
この実施例は、本発明の反応のための提案されるラジカル経路のためのGC−MSの証拠に関して詳述する。
【0046】
図11は、n−ドデシル化SWNTs(実施例6)の製造における副産物を表す質量ピークのGC−MSクロマトグラムを表し(温度プロフィル:50℃で2分保持、10℃/分で280℃にランプ、280℃で5分保持)、ここで:
T=10.38min:n−C12H24(M+168)
T=10.51min:n−C12H26(M+170)
T=14.43min:n−C12H24(M+168)の転位
T=16.49min:C21H44(M+296)
T=23.52min:n−C24H50(M+338)
【0047】
従って、n−ドデシルイオダイドをアルキル化試薬として使用する場合、n−C12H26、n−C12H24及びn−C24H5Oが、主要な副産物として形成される。n−C12H26及びn−C12H24は、ドデシルラジカルの不均化から生じると思われるが、n−C24H50は、ドデシルラジカルの二量化によって形成される。
実施例8
【0048】
この実施例は、生成物2a〜fの熱重量分析(TGA)を示す。
【0049】
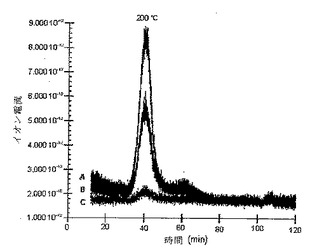
SWNTの共有結合官能基化のためのさらなる証拠は、電子衝撃イオン化モードで動作する質量分析計(MS)による揮発性生成物のオンライン監視と接続された100〜800℃の範囲のn−ブチル化SWNTsの熱重量分析(TGA)によって提供された。これはTGA−MS分析と呼ばれる。約200℃でのn−ブチル基の発生は、m/z57{CH3CH2CH2CH2}及び56{CH3CH2CH=CH2}での主要なピーク並びにm/z58{CH3CH2CH2CH3}でのより小さなピークによって示される。こうした結果を図12に提出する。TGA分析はまた、官能基化度の尺度を提供する。重量損失及び炭素/アルキル基比を下記の表1に提出する。
【表1】
実施例9
【0050】
この実施例は、本発明の方法に従って製造した官能基化SWNTsの熱安定性範囲を示すのに役立つ。
【0051】
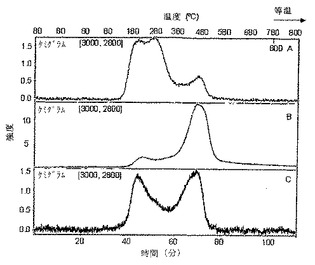
n−ドデシル化SWNTsを、アルゴン雰囲気中、温度800℃にして熱分解した。インシトゥでプロセスを調査するために、熱分解を、フーリエ変換赤外(FT−IR)分光光度計に結合した熱重量分析(TGA)装置の炉中で行った。試料を80℃で30分保持し、10℃min−1で800℃にランプし、次に等温的に10分間、800℃で保持した。熱分解の最中に試料から放出されたガス状種をFT−IR分光光度計中に供給し、熱分解プロセスの最中の濃度変化を時間及び/または温度に関して監視した。図13を参照すると、異なるアルカリ金属を使用して製造したn−ドデシル化SWNTsは、TGA−FTIRにおいてかなり異なるプロフィルを示す。リチウム(トレースA)を使用した反応は、熱分解プロセスの最中に2つの主要なピークを示す:約160〜300℃での主要なピーク、及び約480℃での小さなピーク。ナトリウム(トレースB)を使用すると、かなり異なるプロフィルを生じる:約180〜280℃での1つの小さなピーク及び380〜530℃で現れる主要なピーク。カリウムの場合(トレースC)、2つの異なる温度領域で同様の強度の2つのピークが示される。
実施例10
【0052】
この実施例は、Eq.7によって表す、リチオ化SWNTsによって開始されるメタクリル酸メチルの重合を示すのに役立つ。
SWNTs(精製済み)+Li/NΗ3+CH2=C(CH3)COOCH3→SWNTs−PMMA(Eq.7)
【0053】
メタクリル酸メチルを、5%NaOHを用いて2回、水を用いて2回洗浄し、次に無水MgSO4、CaH2上で乾燥し、次に減圧蒸留した。20mgの精製済み単層カーボンナノチューブを含む密閉された火炎乾燥100mL三首丸底フラスコを脱気し、アルゴンを3回再充填した。約70mLのNH3を次にフラスコ中に凝縮させ、続いてわずかに青色が残るまで小さな部片のリチウム金属(合計〜10mg)を加えた。約3.500g(3.7mL)のメタクリル酸メチルを次に、シリンジでフラスコに加えた。氷浴を次に除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加えることによって、反応を完成させた。10%HClを用いて酸性化した後に、ナノチューブをヘキサン中に抽出し、水を用いて数回洗浄した。ヘキサン層を次に0.2μmPTFEメンブランフィルターを通してろ過し、エタノール、クロロホルムを用いて洗浄し、次に真空オーブン(80℃)中で一晩乾燥した。
【0054】
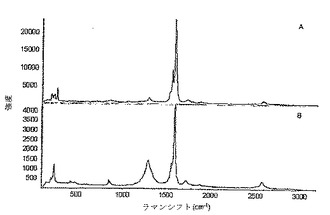
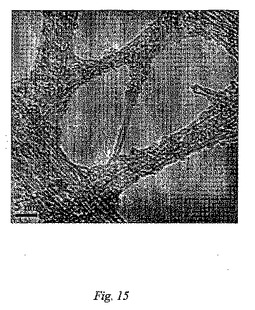
図14(A)及び(B)は、初期の状態のSWNTs(A)及びSWNTs−PMMA(B)のラマンスペクトル(780nm励起)である。このラマン分析は、メタクリル酸メチルの重合は、SWNTsの側壁において/に実現したという証拠を提供する。SWNTs−PMMAの透過型電子顕微鏡法(TEM)像(図15)は、ナノチューブの一部分はポリマーで被覆されたことを示す。図16(A)及び(B)は、SWNTs−PMMAの走査型電子顕微鏡(SEM)像を示し、ここで(B)は、(A)よりも高い倍率である。
【0055】
本明細書において参照する全ての特許及び刊行物を、本明細書によって参考文献によって取り入れる。上記に説明した具体例の上記に説明した構造、機能、及び作動の幾つかは本発明を実施する必要は無く、単に模範的な具体例または具体例の完成のために説明に含ませていることは理解されよう。加えて、上記に説明し、参照した特許及び刊行物において述べる特定の構造、機能、及び作動を本発明と共に実施することができるが、これらはその実施に不可欠では無いことは理解されよう。従って、添付の請求の範囲によって定義する本発明の精神及び範囲から実際に逸脱することなく、本発明を特に説明したものとは異なって実施してよいことは理解されるはずである。
【図面の簡単な説明】
【0056】
【図1】SWNT束100は、無水液体アンモニア(NH3)中でLiによって還元されて、還元され挿入され/剥脱した束101を与え、この挿入され/剥脱した束101は次に有機ハロゲン化物(RI)と反応して、官能基化SWNTs102を与える具体例を図で示す。
【図2】模範的な有機ハロゲン化物1a〜fに関して、図1に表した具体例を概略で示す。
【図3】本発明の方法に従ってブチル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図4】本発明の方法に従ってベンジル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図5】本発明の方法に従ってドデシル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図6】図6(A)は、ドデシル化バッキーパール(A)試料のラマンスペクトル(780nm励起)である。 図6(B)は、純粋な(B)試料のラマンスペクトル(780nm励起)である。
【図7】ドデシル化された精製済みのSWNTsのAFM像である。
【図8】図7に示したAFM像のセクション分析である。
【図9】本発明の具体例に従って製造した、ドデシル化された精製済みのSWNTsのFTIRスペクトルである。
【図10】図10(A)は、個々のナノチューブ及び広範な脱結束を示すドデシル化された精製済みのSWNTsの高分解能透過型電子顕微鏡法(HRTEM)像である。 図10(B)は、個々のナノチューブ及び広範な脱結束を示すドデシル化された精製済みのSWNTsの高分解能透過型電子顕微鏡法(HRTEM)像である。
【図11】ドデシル化SWNTs2aの製造における副産物を表す質量ピークのGC−MSクロマトグラムを表す(温度プロフィル:50℃で2分保持、10℃/分で280℃にランプ、280℃で5分保持)。
【図12】n−ブチル化SWNTsからの発生生成物のTGA−MS分析を表し、ここで、m/zイオン(A)57CH3CH2CH2CH2、(B)56CH3CH2CH=CH2、(C)58CH3CH2CH2CH3の場合のイオン電流対時間のプロットを図に示す。
【図13】3つの異なるアルカリ金属(A)リチウムから;(B)ナトリウムから;(C)カリウムから;によって官能基化されたn−ドデシル化SWNTsのケミグラム(chemigram)を表し、ここで、化学的検出はTGA−FTIRによって提供される。
【図14】図14(A)は、初期の状態のSWNTs(A)のラマンスペクトル(780nm励起)である。 図14(B)は、SWNTs−PMMA(B)のラマンスペクトル(780nm励起)である。
【図15】SWNTs−PMMAのTEM像である。
【図16】図16(A)は、異なる倍率でのSWNTs−PMMAのSEM像である。 図16(B)は、異なる倍率でのSWNTs−PMMAのSEM像である。
【技術分野】
【0001】
本発明は、海軍研究局(the Office of Naval Research)の助成第N00014−01−1−0789号からの援助によりなされた。
関連出願に対するクロスリファレンス
【0002】
本出願は、2004年3月12日に出願された米国仮出願第60/552,550号;及び2004年9月17日に出願された第60/611,045号に対する優先権を請求する。
【0003】
本発明は一般にカーボンナノチューブに関し、特に還元的経路を経てカーボンナノチューブを誘導体化する方法に関する。
【背景技術】
【0004】
カーボンナノチューブ(CNTs、akaフラーレンパイプ)は、概念上それ自体の上に巻き上がり、フラーレンキャップによって末端が閉じたグラフェン(graphene)シートを含むナノスケール炭素構造である。単層カーボンナノチューブ(SWNTs)は、単一のこのようなグラフェン円筒のみを含むが、多層ナノチューブは、ロシアの入れ子人形のものに類似した仕方で1つが別のものの内部に入れ子になっている2つ以上の同軸グラフェン層で作られている。1993年におけるその最初の製造(Iijima et al., Nature, 1993, 363, 603; Bethune et al., Nature, 1993, 363, 605; Endo et al., Phys. Chem. Solids. 1993, 54, 1841)以来、SWNTsは、その類のない機械的、光学的、電子的、及び他の性質が理由となって広範に研究されてきた。例えば、SWNTsの注目すべき引張強さは、強化繊維及びポリマーナノコンポジットにおけるその使用をもたらした(Zhu et al., Nano Lett. 2003, 3, 1107及びこの中の参考文献)。CNTsの他の既存の及び潜在的な用途に関しては、Baughman et al, Science, 2002, 297, 787-792を参照されたい。
【0005】
SWNTsは通常自己組織化して、凝集体または束になり、ここで最高数百までのチューブがファンデルワールス力で結合する。医用生体用途を含む多くの用途の場合、こうした束からの個々のナノチューブの分離は不可欠である。このような分離は、加工及び操作にとって必要な一般的な有機溶媒及び/または水中のナノチューブの分散及び可溶化を改良する。SWNT表面の共有結合性修飾は一般に、ナノチューブの溶解度/懸濁性及び加工性を改良することによってこの問題を解決するのを助ける。ナノチューブ末端の化学的官能基化は一般に、こうした材料の電子的及びバルク性質を変化させないが、側壁官能基化は、ナノチューブの固有の性質を確かにかなり変更し(Chen et al, Science, 1998, 282, 95-98; Mickelson et al, Chem. Phys. Lett., 1998, 296, 188-194)、典型的にその溶解度/懸濁性により大きな影響を及ぼす(Boul et al, Chem. Phys. Lett., 1999, 310, 367-372)。しかしながら、化学のこの新たな分野における記録された結果の範囲は、主にナノチューブの現在の高コストが理由となって限定されている。
【0006】
SWNT側壁の修飾において直面するさらなる問題は、その比較的に不満足な反応性に関連している−主として、反応性のより高いフラーレンを基準としてナノチューブ壁のはるかに低い湾曲(M. S. Dresselhaus, G. Dresselhaus, P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996, Vol. 1)、並びにグラフェン壁に付着した官能基の数及びサイズが増大すると共に管状構造内部の歪みが増大することが理由となっている。ナノチューブの骨組みを構成する炭素原子の全てのsp2−結合状態は、付加タイプ反応の優勢な発生を促進する。こうした反応の最も良く特徴付けられた例は、ニトレン、アゾメチンイリド及びジアゾニウム塩から発生したアリールラジカルのSWNTsへの付加を含む(V. N. Khabashesku, J. L. Margrave, Chemistry of Carbon Nanotubes in Encyclopedia of Nanoscience and Nanotechnology, Ed. H. S. Nalwa, American Scientific Publishers, 2004; Bahr et al., J. Mater. Chem., 2002, 12, 1952; Holzinger et al., Angew. Chem. Int. Ed., 2001, 40, 4002)。SWNTsの他の報告されている側壁官能基化は、有機ラジカル(Peng et al, Chem. Commun., 2003, 362; Ying et al, Org. Lett, 2003, 5, 1471; Peng et al, J.Am, Chem. Soc, 2003, 125, 15174)及びビンジェル反応(Bingel reaction)(Coleman et al, J. Am. Chem. Soc, 2003, 125, 8722)を含む。加えて、水素をSWNTsの側壁に付加することは、バーチ還元の条件下で起きると報告されている(Pekker et al, J. Phys. Chem. B, 2001, 105, 7938)。
【0007】
個々のCNTsの直径及びキラリティーは、整数“n”及び“m”によって説明され、ここで、(n,m)は、概念上巻き上がってチューブを形成するグラフェンシートに沿ったベクトルである。|n−m|=3q(ここで、qはゼロでない整数である)である場合、CNTは半金属である(バンドギャップはミリeVのオーダー)。n−m=0である場合、CNTは真の金属であり、バンドギャップは0eVであり、“アームチェア”ナノチューブと呼ばれる。n−mの全ての他の組合せは、半導性CNTであり、バンドギャップは典型的に0.3〜1.0eVの範囲内である。O'Connell et al, Science, 2002, 297, 593を参照されたい。本明細書において使用するCNT“タイプ”は、(n,m)ベクトルによって説明されるこのような電子的タイプ(すなわち、金属性、半金属性、及び半導性)を指す。本明細書において使用するCNT“種”は、個別の(n,m)値を有するCNTsを指す。本明細書において使用するCNT“組成物”は、ナノチューブタイプ及び種の点でのCNT集団の構成を指す。
【0008】
全ての周知のCNT製造方法は、半導性、半金属性、及び金属性の電子的タイプのCNT集団を多分散させることをもたらす。M. S. Dresselhaus, G. Dresselhaus, P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996; Bronikowski et al., Journal of Vacuum Science & Technology A 2001, 19, 1800-1805; R. Saito, G. Dresselhaus, M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial College Press, London, 1998を参照されたい。従って、CNTs及びSWNTsの広範囲に及ぶ利用に対する主要な障害は特に、電子構造によるその操作である(Avouris, Ace. Chem. Res. 2002, 35, 1026-1034)。しかしながら、最近、CNTsをその電子構造(すなわち、電子的タイプ)に基づいて選択的に官能基化する方法が報告されている(Strano et al, Science, 2003, 301, 1519-1522;2004年7月29日に出願され、共通に譲渡された同時係属国際特許出願PCT/US04/24507)。このような報告において、金属性CNTsはジアゾニウム種と優先的に反応して、金属性及び半導性CNTsの混合物の部分的官能基化を経て金属性(半金属性を含む)及び半導性CNTsの分離または分別を可能にすることが認められている。CNTタイプ及び種、並びにその光学的確認の詳細な検討に関しては、Bachilo et al., Science, 2002, 298, 2361-2366; and Weisman et al, Nano. Lett, 2003, 3, 1235-1238を参照されたい。
【0009】
カーボンナノチューブの側壁を化学的に誘導体化することにおけるこのような上記に説明した進歩にもかかわらず、大部分のこのようなプロセスは、誘導体化プロセスの最中にカーボンナノチューブの超音波処理を必要とする。この音波処理は潜在的に、試料中の多くのナノチューブを損傷し得る。従って、より穏やかな条件下でカーボンナノチューブを誘導体化する方法は非常に有益であると思われる。
【発明の開示】
【0010】
本発明は、カーボンナノチューブの官能基化(すなわち、化学的誘導体化)の新規なプロセス(方法)、並びに、その延長上に、フラーレン及び他の炭素表面(すなわち、一般に無機炭素材料)に関する。一般に、このようなプロセスは還元的経路を含む。幾つかの具体例においては、カーボンナノチューブは、無水液体アンモニア中でアルカリ金属及び有機ハロゲン化物と反応する。他の具体例においては、無水液体アンモニア中でカーボンナノチューブをアルカリ金属及びモノマー種と反応させることによって、ポリマーをインシトゥでカーボンナノチューブ側壁から成長させる。
【0011】
幾つかの具体例においては、本発明は、炭素材料を官能基化する方法に関し、このような方法は:(a)炭素材料を液体アンモニアと合わせて、混合物を形成する工程と;(b)ある量のアルカリ金属を混合物中に溶解させて、還元的混合物を形成する工程と;(c)有機部分及びハロゲン化物部分を含む有機ハロゲン化物を還元的混合物に加え、その結果、有機ハロゲン化物の有機部分が炭素材料に付加して誘導体化炭素材料を形成するようにする工程と;を含む。様々なクエンチング(未反応のアルカリ金属を中和するための)、酸性化(未蒸発のアンモニアを中和するための)、ろ過(誘導体化炭素材料を集めるための)、及び洗浄(望まれない種を誘導体化炭素材料から除去するための)工程のうちの1つ以上を、所望により実行することができる。一般に、このような炭素材料としては、カーボンナノチューブ、ナノスクロール(nanoscroll)、フラーレン、ダイヤモンド、アセチレン型炭素(acetylenic carbon)、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せが挙げられるがこれらに限定されるものではない。
【0012】
幾つかの具体例においては、本発明は、ポリマーをインシトゥで炭素表面から成長させる方法に関し、このような方法は:(a)炭素材料を液体アンモニアと合わせて、混合物を形成する工程と;(b)ある量のアルカリ金属を混合物中に溶解させて、還元的混合物を形成する工程と;(c)ある量の少なくとも1つのモノマー種を還元的混合物に加え、その結果、モノマーが炭素材料の上に/において重合するようにする工程と;を含む。様々なクエンチング、酸性化、ろ過、及び洗浄工程のうちの1つ以上を、所望により実行することができる。このようなインシトゥ重合は、炭素材料及びポリマーを含む複合材料を与えることができる。幾つかの具体例においては、モノマーはアニオン重合を受けることが認められている。幾つかのまたは他の具体例においては、モノマーはラジカル重合を受けることが認められている。上記の官能基化反応におけるように、このような炭素材料としては、カーボンナノチューブ、ナノスクロール、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せが挙げられるがこれらに限定されるものではない。
【0013】
上記に説明した具体例の変形例は、アンモニアの代わりに、またはこれに加えて無水アミンのような他の溶媒の使用を含む。
【0014】
本発明の方法は、このタイプの側壁官能基化の同様の方法が存在しないという点で新規である。その上、このような方法は、カーボンナノチューブの側壁を誘導体化する穏やかな(すなわち、超音波処理を必要としない)拡大縮小できるプロセスに対する当分野において認識されている必要を満たす。
【0015】
上文は、下記の本発明の詳細な説明がより良く理解されるために、本発明の特徴をかなり広く略述した。本発明の請求の範囲の主題を形成する本発明のさらなる特徴及び利点を下文で説明する。
【0016】
本発明及びその利点のより完全な理解のために、ここから添付図面と共に以下の説明を参照する。
【発明を実施するための最良の形態】
【0017】
本発明は、カーボンナノチューブの官能基化(誘導体化)の新規なプロセス、並びに、延長として、フラーレン及び他の炭素表面に関する。一般に、このようなプロセスは還元的経路を含む。幾つかの具体例においては、カーボンナノチューブは、無水アンモニア中でアルカリ金属及び有機ハロゲン化物と反応する。他の具体例においては、無水アンモニア中でカーボンナノチューブをアルカリ金属及びモノマー種と反応させることによって、ポリマーをカーボンナノチューブ側壁から成長させる。
【0018】
本発明によれば、カーボンナノチューブ(CNTs)としては、単層カーボンナノチューブ(SWNTs)、二層カーボンナノチューブ(DWNTs)、多層カーボンナノチューブ(MWNTs)、小直径カーボンナノチューブ、及びこれらの組合せが挙げられるがこれらに限定されるものではない。小直径カーボンナノチューブは、本明細書において、層の数にかかわらず直径最大約3nmを有するカーボンナノチューブと定義される。CNTsの全ての製造方法は、炭素質不純物を有する製品を与える。加えて、SWNTsの大部分の製造方法及びMWNTsの多くの製造方法は、不純物として生成物中に残る金属触媒を使用する。本明細書において説明する実施例は一般に、単層カーボンナノチューブ(SWNTs)を用いて行われたが、本明細書において説明する方法及び組成物は一般に、任意の周知の方法によって製造された全てのカーボンナノチューブに適用できる−但しこれらは、その反応性が理由となって本明細書において説明する化学の影響を受けやすいことは理解されるはずである。その上、ナノチューブは、本明細書において説明する化学修飾にさらされる前に、切断、長さ分類、キラリティー分類、精製等を含む任意の数の合成後連続工程にさらされることができる。
【0019】
CNTsの場合特に、官能基化は、溶媒中の改良されたレベルのナノチューブ懸濁性/分散性及び/または溶解度をもたらすことができる。真の溶解度は、溶媒中のCNTsの再凝集は、熱力学的根拠で、連続した溶媒和状態よりも容易ではない状態であることに注意されたい。それはそれとして、安定な懸濁液は、広範囲のプロセスのためのCNTsの操作を適切に可能にすることができる。
【0020】
本発明によれば、アルカリ金属は、Li、Na、及びKである。本発明によれば、有機ハロゲン化物(R−X)は、有機部分(R−)及びハロゲン化物部分(−X)を含む。有機部分は、本発明の具体例に従って有機ハロゲン化物を適切に提供する任意の有機官能性とすることができる。適切な有機官能性としては、アルキル−、アリール−、アリル−、ベンジル−、及びこれらの組合せが挙げられるがこれらに限定されるものではない。ハロゲン化物部分は、−F、−Cl、−Br、−I、及びこれらの組合せからなる群から選択される。
官能基化反応
【0021】
幾つかの具体例においては、本発明は、以下の工程を含むプロセス(方法)に関する:(a)カーボンナノチューブまたは他の炭素材料を、ドライアイス凝縮器を備えた火炎乾燥三首丸底フラスコ(または他の適切な容器)中に置く工程と;(b)NH3ガスをフラスコ中に凝縮させ、次に非常に小さな部片のアルカリ金属を加え、続いてある量のハロゲン化アルキル/アリールを加える工程と;(c)反応を一晩進行させる工程と;(d)反応混合物をアルコール、水を用いてクエンチし、次に鉱酸を用いて酸性化する工程と;(e)内容物をメンブランフィルターを通してろ過し、エタノール、次にヘキサンを用いて十分に洗浄して、副産物を除去する工程。このようなプロセスは、超音波処理が必要な特徴ではないという点で類がない。
【0022】
カーボンナノチューブ、または一般に任意の黒鉛質表面が官能基化されることを可能にする本発明の具体例である還元的経路のより詳細な説明が、便利に次に続く:カーボンナノチューブを火炎乾燥三首丸底フラスコに加えた。アンモニアを次にフラスコ中に凝縮させ、続いて小さな部片のリチウム金属を加えた。ハロゲン化アルキル/アリールを次に加え、アンモニアを蒸発させながら反応を一晩進行させた。反応混合物を次に氷浴中で冷却し、メタノールを徐々に加えることによってクエンチした。水を次に徐々に加え、続いて混合物が酸性になるまで10%HClを加えた。内容物を、0.2μm−ポアサイズPTFE(テフロン(登録商標)(Teflon(登録商標)))メンブランフィルターを使用してろ過した。ドデシル化ナノチューブのラマンスペクトルは、例えば、一般的な有機溶媒中の高い溶解度をもたらす並外れた官能基化を示す。ドデシル化単層カーボンナノチューブ(SWNTs)の原子間力顕微鏡のイメージングは、かなりの官能基化を示す単層カーボンナノチューブ束のかなりの剥脱を意味する極めて多量の個々のカーボンナノチューブを明らかにする。本発明の範囲は、以下のものを越えて拡大するが、このようにカーボンナノチューブに実験的に付着する官能基は、次のものを含む:n−ドデシル−、n−ブチル−、t−ブチル−、n−オクチル−、及びベンジル−。図1は、上記に説明した具体例の例を示し、ここで、SWNT束100は、無水液体アンモニア(NH3)中でLiによって還元されて、還元され挿入され/部分的に剥脱した束101を与える。この挿入され/部分的に剥脱した束101は次に有機ハロゲン化物(RI)と反応して、官能基化SWNTs102を与える。
【0023】
上記に説明した官能基化反応を図2(スキーム1)に概略で示し、ここで、SWNTsは、無水液体アンモニア中でリチウム(または他の適切なアルカリ金属)を用いてまず挿入/還元され、次に有機ハロゲン化物1と反応して、官能基化SWNTs2a〜fを与え、有機ハロゲン化物1a〜fは例として提供される。理論によって束縛されることを意図するものではないが、脱結束(debundling)は、リチウム(または他のアルカリ金属)による広範な挿入の点で説明することができ、図2に示すように、負に帯電したチューブの間に分散したリチウムイオンをもたらす。リチウムを液体アンモニア中のナノチューブの懸濁液に加えるにつれて、溶媒和電子に関連する強い青色は急速に消失し、SWNTsへの電子移動は容易なプロセスであることを示唆する。ラマン分光法を使用した予備実験は、炭素/リチウム比は2〜3であることを示唆する。ハロゲン化アルキルを加えることは、容易に解離してハロゲン化物及びアルキルラジカルを与えると思われるラジカルアニオンの形成をもたらすと思われる。理論によって束縛されることを意図するものではないが、ろ液のガスクロモタグラフィー(gas chromotagraphy)−質量分析法(GC−MS)の分析は、ラジカル経路のための強力な証拠を提供する(実施例7を参照されたい)。加えて、ケトンのカルボニル基への電子移動によって形成されたラジカルアニオンも、SWNTsの側壁に容易に付加することができる。
【0024】
カーボンナノチューブの官能基化は、これを一般的な有機溶媒中に可溶にする方法を提供し、従ってこれを複合体形成及び紡糸して繊維にするために修正可能にする。例えば、アルキル化SWNTsは、溶媒の例えばクロロホルム(CHCl3)、テトラハイドフラン(tetrahydofuran)(THF)、及びN,N−ジメチルホルムアミド(DMF)中の比較的に高いレベルの溶解度を示す。他の黒鉛質様表面の例えばカーボンブラックの官能基化は、様々なエラストマー及びポリマー中のその分散を促進する。加えて、本発明の具体例に従ってCNTsを官能基化する方法は、典型的に超音波処理を含む既存の従来技術の官能基化方法よりも一般により穏やかであり(すなわち、超音波処理を必要としない)、CNTsへのより少ない損傷を引き起こす。
【0025】
このような上記に説明した還元的官能基化方法は、様々な官能基(例えば、アルキル−、アリール−、アリル−、及びベンジル−)をカーボンナノチューブの側壁に付加することを可能にする。幾つかの具体例においては、こうした基のさらなる(その後の)官能基化が可能である。幾つかの具体例においては、様々な無水アミンを、無水アンモニアの代わりに、またはこれに加えて溶媒として使用してよい。試薬の量及びタイプ、反応時間等に、並びにクエンチング、酸性化、ろ過、及び洗浄の工程に、かなりの変化が存在する。加えて、本発明の方法は、はるかに大きな量に拡張可能である。
重合反応
【0026】
幾つかのまたは他の具体例においては、本発明は、以下の工程を含むプロセスに関する:(a)カーボンナノチューブを、ドライアイス凝縮器を備えた火炎乾燥三つ口フラスコ中に置く工程と;(b)NH3ガスをフラスコ中に凝縮させ、次に小さな部片のアルカリ金属を加え、続いてモノマー(例えば、アルキン/アルケン)を加える工程と;(c)反応を一晩進行させる工程と;(d)反応混合物をアルコールを用いてクエンチし、続いて鉱酸を用いて酸性化する工程と;(e)内容物をメンブランフィルターを通してろ過し、アルコール、次にヘキサンを用いて十分に洗浄して、副産物を除去する工程。一般に、適切なモノマーは、不飽和の領域(すなわち、二重若しくは三重結合、またはオリゴマーエチレンオキシド)を含む。
【0027】
カーボンナノチューブまたは一般に任意の黒鉛質表面の側壁に付着したポリマー鎖を形成するための、還元的経路を経るモノマー材料のインシトゥ重合に対処する本発明のさらに別の具体例であるこのような上記に説明した重合方法の詳細な説明が、次に続く:カーボンナノチューブを火炎乾燥三つ口フラスコに加えた。アンモニアを次にフラスコ中に凝縮させ、続いてリチウム金属を加えた。メタクリル酸メチル、アクリロニトリル、及びアセチレンを含む様々なモノマーを次に反応フラスコに加え、アンモニアが蒸発させながら一晩放置した。エタノールを次に加え、続いて反応混合物が酸性になるまで10%HClを加えた。内容物を分液漏斗に移し、官能基化カーボンナノチューブをクロロホルム中に抽出した。抽出物を、水/エタノール混合物を用いて洗浄し、0.2μmPTFE(ポリテトラフロウロエチレン(polytetraflouroethylene))メンブランフィルターを通してろ過した。
【0028】
インシトゥ重合を経て炭素材料/ポリマー複合体を製造するこのような上記に説明した方法にかなりの変化が存在する。様々なモノマー種または種の組合せを使用できる。加えて、試薬の量及びタイプ、反応時間等に、並びにクエンチング、酸性化、ろ過、及び洗浄の工程に変化が存在する。
【0029】
CNTs表面のインシトゥ重合の場合には、カーボンナノチューブはポリマーに化学的に結合し、これは、多機能性のさらなる見込みを有する軽量複合体系のための機械的強化として役立つことができる。本明細書において説明するもののような方法を使用して、様々なポリマーをナノチューブに化学的に付着させることができ、こうした付着したポリマー鎖のさらなる官能基化が可能である。
【0030】
理論によって束縛されることを意図するものではないが、少なくとも本発明の幾つかの具体例においては、バーチタイプ還元が起き、ここで、CNTsまたは他の炭素材料は、アルカリ金属によって表面において還元されると考えられている。この還元された表面は次に、有機(例えば、アルキル/アリール)ハロゲン化物と反応して、官能基化CNTsを形成することができるか、またはインシトゥ重合のためのモノマーと反応することができる。加えて、CNTsを含む具体例においては、超音波処理の無い場合のナノチューブ(特にSWNTs)の脱結束は、アルカリ金属による広範な挿入の点で説明することができ、負に帯電したナノチューブの間に分散したアルカリイオンをもたらす。官能基化反応に関して上記に言及したように、実験的に、リチウム金属を使用する場合、リチウムを液体アンモニア中のナノチューブの懸濁液に加えるにつれて、溶媒和電子に関連する強い青色は急速に消失し、SWNTsへの電子移動は容易なプロセスであることを示唆する。
【0031】
以下の実施例を、本発明の特定の具体例を証明するために提供する。下記の実施例において開示する方法は、単に本発明の模範的な具体例を表すことは、当業者であれば了解されるはずである。しかしながら、当業者であれば、本開示を考慮して、本発明の精神及び範囲から逸脱することなく、説明され依然として同様なまたは類似の結果を得る具体例に多くの変更を行い得ることは了解されよう。
【実施例】
【0032】
実施例l
この実施例は、式1(Eq.1)に表す、臭化n−ブチルの存在下でのLi/NH3還元によるカーボンナノチューブのアルキル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)3−Br→n−ブチル化SWNTs(Eq.1)
【0033】
撹拌子を備え、ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の(綿毛状、製造したまま)単層カーボンナノチューブ(SWNTs)を置いた。アルゴン(Ar)を用いてフラスコをフラッシングした後、これをドライアイス/アセトン浴中に浸漬し、NH3ガス(〜60mL)をその中で凝縮させた。次に、これに300mgのLi金属を少量ずつ加えた。Liを加えた直後に、混合物は色が深青色に変わった。5分後に、0.53mLの臭化n−ブチルを加えた。氷浴を除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。翌朝、フラスコが氷浴中にある間にまず10mLのメタノール、次に20mLの水を加えることによって、反応を完成させた。フラスコの内容物を、10%HClを用いて酸性化し、次に0.2μmポアサイズポリテトラフルオロエチレン(PTFE)メンブランフィルター上でろ過した。フィルター上に残った材料を20mLのエチルアルコール中に置き、20min音波処理し、次に再度0.2μmPTFEメンブランフィルター上でろ過した。次に、同じプロセスを、ベンゼン及びテトラヒドロフラン(THF)を用いて繰り返した。このように得られたバッキーペーパー(bucky paper)(SWNTs)を、それに続くラマン分析のために使用した。
【0034】
ラマン分析は、説明したように反応が進行してカーボンナノチューブを誘導体化することを示すかなりの無秩序なピーク(disorder peak)を明らかにする。官能基付加は、官能基が付加するナノチューブケージ内部の炭素を、支配的には性質がsp2から支配的には性質がsp3へと変化させる、という事実から無秩序なピークの増大が生じることに留意されたい。このラマン分析を図3に表す。
実施例2
【0035】
この実施例は、Eq.2によって示される、塩化ベンジルの存在下でのLi/NH3還元によるカーボンナノチューブのベンジル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+(C6H5)−CH2−Cl→ベンジル化SWNTs(Eq.2)
【0036】
この反応を、実施例1において説明したものと同様の仕方で実行した。火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次に、このフラスコに〜60mLのNH3を凝縮させ、続いて非常に小さな部片のLi金属を加えた(合計〜462mg)。Liを完全に加えた後、〜2.108g(1.92mL)の塩化ベンジルを、シリンジでフラスコに加えた。フラスコの下の氷浴をそれに続いて除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加え、次に10%HClを用いて酸性化することによって、反応を完成させた。0.2μmPTFEメンブランフィルター上でエタノールを用いて材料を繰り返し洗浄してバッキーペーパーを与え、これをさらなる分析のために使用した。図4に示すように、ラマン分析は、かなりの無秩序なピークを明らかにした。
実施例3
【0037】
この実施例は、Eq.3によって示される、n−ドデシルイオダイドの存在下でのLi/NH3還元によるカーボンナノチューブのドデシル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)11−I→n−ドデシル化SWNTs(Eq.3)
【0038】
ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。Arを用いてフラスコをフラッシングした後、NH3ガス(〜60mL)をその中で凝縮させた。これにLi金属を少量ずつ加えた(合計〜462mg)。Liを加えた直後に、混合物は色が深青色に変わった。5分後に、4.933g(4.107mL)のヨードドデカンを加えた。氷浴を除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。翌朝、フラスコが氷浴中にある間にまず10mLのメタノール、次に20mLの水を加えることによって、反応を完成させた。次にこれを、酸性になるまで10%HClを用いて酸性化した。これを、それに続いて0.2μmポアサイズPTFEメンブランフィルター上でろ過した。フィルター上の材料を次に30mLのエタノール中で音波処理し、再度ろ過し、次にエタノール/ヘキサン混合物(2:1)を用いて洗浄した。メンブランフィルター上の得られた材料は、CHCl3中の並外れた溶解度を有することが決定された。このように得られたバッキーペーパー(SWNTsマット)を、それに続くラマン分析のために使用し、図5に示すようにかなりの無秩序なピークを明らかにした。
実施例4
【0039】
この実施例は、Eq.4によって示される、n−オクチルイオダイドの存在下でのLi/NH3還元によるカーボンナノチューブのオクチル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+CH3−(CH2)7−Br→n−オクチル化SWNTs(Eq.4)
【0040】
ドライアイス凝縮器を取り付けた火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次にフラスコに〜60mLのNH3ガスを凝縮させ、続いて非常に小さな部片のLi金属(合計462mg)を加えた。リチウムを加えた後、〜3.00mL(3.998g)のn−オクチルイオダイドをシリンジで加えた。NH3を徐々に蒸発させながら反応を一晩進行させた。次に、混合物中の反応を、10mLのアルコールを徐々に加えながらクエンチし、この間フラスコを氷浴中に維持した。次に、20mLの水を徐々に加え、続いて酸性になるまで10%HClを加えた。内容物を0.2μmPTFEメンブランフィルター上でろ過した。ろ紙上に残った材料を、アルコール、続いてヘキサンを用いて繰り返し洗浄した。
実施例5
【0041】
この実施例は、Eq.5によって示される、ヨウ化t−ブチルの存在下でのLi/NH3還元によるカーボンナノチューブのtert−ブチル化を示すのに役立つ。
SWNTs(未精製)+Li/NH3+(CH3)3C−I→t−ブチル化SWNTs(Eq.5)
【0042】
火炎乾燥100mL三首丸底フラスコ中に、20mgの未精製の単層カーボンナノチューブを置いた。次に、このフラスコに〜70mLのNH3を凝縮させ、続いて非常に小さな部片のLi金属を加えた(合計〜462mg)。Liを完全に加えた後、〜3.065g(1.99mL)のヨウ化t−ブチルを、シリンジでフラスコに加えた。フラスコの下の氷浴をそれに続いて除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加え、次に10%HClを用いて酸性化することによって、反応を完成させた。0.2μmPTFEメンブランフィルター上でエタノールを用いて材料を繰り返し洗浄してバッキーペーパーを与え、これをさらなる分析のために使用した。
実施例6
【0043】
この実施例は、Eq.6によって示される、n−ドデシルイオダイドの存在下でのL1/NH3還元による、精製済みカーボンナノチューブのドデシル化を示すのに役立つ。
SWNTs(精製済み)+Li/NH3+CH3−(CH2)11−I→n−ドデシル化SWNTs(Eq.6)
【0044】
比較のために、実施例3において説明したプロセスを、精製済みSWNTsを使用して実行した。特に、使用した精製済みSWNTsは、“バッキーパール(bucky pearl)”として周知の高密度タイプだった。図6に示すように、ラマン分析は、本発明の誘導体化プロセスは、未精製のSWNT材料に作用するのと少なくとも同様に精製済みSWNT材料に作用することを示唆する。図7は、この実施例において製造したドデシル化SWNTsの原子間力顕微鏡法(AFM)像であり、図8は、対応するセクション分析(section analysis)である。図7及び8の両方は、SWNTsのかなりの脱結束(そのロープ化状態からの)及び極めて多量の個々のSWNTsを明らかにする−かなりの誘導体化を示す。図9は、ドデシル化SWNTsのFTIRスペクトルであり、図10(A)及び(B)は、ドデシル化SWNTsの高分解能透過型電子顕微鏡法(HRTEM)像であり、個々のナノチューブ及び広範な脱結束を示す。
実施例7
【0045】
この実施例は、本発明の反応のための提案されるラジカル経路のためのGC−MSの証拠に関して詳述する。
【0046】
図11は、n−ドデシル化SWNTs(実施例6)の製造における副産物を表す質量ピークのGC−MSクロマトグラムを表し(温度プロフィル:50℃で2分保持、10℃/分で280℃にランプ、280℃で5分保持)、ここで:
T=10.38min:n−C12H24(M+168)
T=10.51min:n−C12H26(M+170)
T=14.43min:n−C12H24(M+168)の転位
T=16.49min:C21H44(M+296)
T=23.52min:n−C24H50(M+338)
【0047】
従って、n−ドデシルイオダイドをアルキル化試薬として使用する場合、n−C12H26、n−C12H24及びn−C24H5Oが、主要な副産物として形成される。n−C12H26及びn−C12H24は、ドデシルラジカルの不均化から生じると思われるが、n−C24H50は、ドデシルラジカルの二量化によって形成される。
実施例8
【0048】
この実施例は、生成物2a〜fの熱重量分析(TGA)を示す。
【0049】
SWNTの共有結合官能基化のためのさらなる証拠は、電子衝撃イオン化モードで動作する質量分析計(MS)による揮発性生成物のオンライン監視と接続された100〜800℃の範囲のn−ブチル化SWNTsの熱重量分析(TGA)によって提供された。これはTGA−MS分析と呼ばれる。約200℃でのn−ブチル基の発生は、m/z57{CH3CH2CH2CH2}及び56{CH3CH2CH=CH2}での主要なピーク並びにm/z58{CH3CH2CH2CH3}でのより小さなピークによって示される。こうした結果を図12に提出する。TGA分析はまた、官能基化度の尺度を提供する。重量損失及び炭素/アルキル基比を下記の表1に提出する。
【表1】
実施例9
【0050】
この実施例は、本発明の方法に従って製造した官能基化SWNTsの熱安定性範囲を示すのに役立つ。
【0051】
n−ドデシル化SWNTsを、アルゴン雰囲気中、温度800℃にして熱分解した。インシトゥでプロセスを調査するために、熱分解を、フーリエ変換赤外(FT−IR)分光光度計に結合した熱重量分析(TGA)装置の炉中で行った。試料を80℃で30分保持し、10℃min−1で800℃にランプし、次に等温的に10分間、800℃で保持した。熱分解の最中に試料から放出されたガス状種をFT−IR分光光度計中に供給し、熱分解プロセスの最中の濃度変化を時間及び/または温度に関して監視した。図13を参照すると、異なるアルカリ金属を使用して製造したn−ドデシル化SWNTsは、TGA−FTIRにおいてかなり異なるプロフィルを示す。リチウム(トレースA)を使用した反応は、熱分解プロセスの最中に2つの主要なピークを示す:約160〜300℃での主要なピーク、及び約480℃での小さなピーク。ナトリウム(トレースB)を使用すると、かなり異なるプロフィルを生じる:約180〜280℃での1つの小さなピーク及び380〜530℃で現れる主要なピーク。カリウムの場合(トレースC)、2つの異なる温度領域で同様の強度の2つのピークが示される。
実施例10
【0052】
この実施例は、Eq.7によって表す、リチオ化SWNTsによって開始されるメタクリル酸メチルの重合を示すのに役立つ。
SWNTs(精製済み)+Li/NΗ3+CH2=C(CH3)COOCH3→SWNTs−PMMA(Eq.7)
【0053】
メタクリル酸メチルを、5%NaOHを用いて2回、水を用いて2回洗浄し、次に無水MgSO4、CaH2上で乾燥し、次に減圧蒸留した。20mgの精製済み単層カーボンナノチューブを含む密閉された火炎乾燥100mL三首丸底フラスコを脱気し、アルゴンを3回再充填した。約70mLのNH3を次にフラスコ中に凝縮させ、続いてわずかに青色が残るまで小さな部片のリチウム金属(合計〜10mg)を加えた。約3.500g(3.7mL)のメタクリル酸メチルを次に、シリンジでフラスコに加えた。氷浴を次に除去し、NH3を徐々に蒸発させながら反応を一晩進行させた。まず反応混合物を10mLのメタノールを用いてクエンチし、続いて20mLのH2Oを加えることによって、反応を完成させた。10%HClを用いて酸性化した後に、ナノチューブをヘキサン中に抽出し、水を用いて数回洗浄した。ヘキサン層を次に0.2μmPTFEメンブランフィルターを通してろ過し、エタノール、クロロホルムを用いて洗浄し、次に真空オーブン(80℃)中で一晩乾燥した。
【0054】
図14(A)及び(B)は、初期の状態のSWNTs(A)及びSWNTs−PMMA(B)のラマンスペクトル(780nm励起)である。このラマン分析は、メタクリル酸メチルの重合は、SWNTsの側壁において/に実現したという証拠を提供する。SWNTs−PMMAの透過型電子顕微鏡法(TEM)像(図15)は、ナノチューブの一部分はポリマーで被覆されたことを示す。図16(A)及び(B)は、SWNTs−PMMAの走査型電子顕微鏡(SEM)像を示し、ここで(B)は、(A)よりも高い倍率である。
【0055】
本明細書において参照する全ての特許及び刊行物を、本明細書によって参考文献によって取り入れる。上記に説明した具体例の上記に説明した構造、機能、及び作動の幾つかは本発明を実施する必要は無く、単に模範的な具体例または具体例の完成のために説明に含ませていることは理解されよう。加えて、上記に説明し、参照した特許及び刊行物において述べる特定の構造、機能、及び作動を本発明と共に実施することができるが、これらはその実施に不可欠では無いことは理解されよう。従って、添付の請求の範囲によって定義する本発明の精神及び範囲から実際に逸脱することなく、本発明を特に説明したものとは異なって実施してよいことは理解されるはずである。
【図面の簡単な説明】
【0056】
【図1】SWNT束100は、無水液体アンモニア(NH3)中でLiによって還元されて、還元され挿入され/剥脱した束101を与え、この挿入され/剥脱した束101は次に有機ハロゲン化物(RI)と反応して、官能基化SWNTs102を与える具体例を図で示す。
【図2】模範的な有機ハロゲン化物1a〜fに関して、図1に表した具体例を概略で示す。
【図3】本発明の方法に従ってブチル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図4】本発明の方法に従ってベンジル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図5】本発明の方法に従ってドデシル化された未精製のSWNTsのラマンスペクトル(780nm励起)である。
【図6】図6(A)は、ドデシル化バッキーパール(A)試料のラマンスペクトル(780nm励起)である。 図6(B)は、純粋な(B)試料のラマンスペクトル(780nm励起)である。
【図7】ドデシル化された精製済みのSWNTsのAFM像である。
【図8】図7に示したAFM像のセクション分析である。
【図9】本発明の具体例に従って製造した、ドデシル化された精製済みのSWNTsのFTIRスペクトルである。
【図10】図10(A)は、個々のナノチューブ及び広範な脱結束を示すドデシル化された精製済みのSWNTsの高分解能透過型電子顕微鏡法(HRTEM)像である。 図10(B)は、個々のナノチューブ及び広範な脱結束を示すドデシル化された精製済みのSWNTsの高分解能透過型電子顕微鏡法(HRTEM)像である。
【図11】ドデシル化SWNTs2aの製造における副産物を表す質量ピークのGC−MSクロマトグラムを表す(温度プロフィル:50℃で2分保持、10℃/分で280℃にランプ、280℃で5分保持)。
【図12】n−ブチル化SWNTsからの発生生成物のTGA−MS分析を表し、ここで、m/zイオン(A)57CH3CH2CH2CH2、(B)56CH3CH2CH=CH2、(C)58CH3CH2CH2CH3の場合のイオン電流対時間のプロットを図に示す。
【図13】3つの異なるアルカリ金属(A)リチウムから;(B)ナトリウムから;(C)カリウムから;によって官能基化されたn−ドデシル化SWNTsのケミグラム(chemigram)を表し、ここで、化学的検出はTGA−FTIRによって提供される。
【図14】図14(A)は、初期の状態のSWNTs(A)のラマンスペクトル(780nm励起)である。 図14(B)は、SWNTs−PMMA(B)のラマンスペクトル(780nm励起)である。
【図15】SWNTs−PMMAのTEM像である。
【図16】図16(A)は、異なる倍率でのSWNTs−PMMAのSEM像である。 図16(B)は、異なる倍率でのSWNTs−PMMAのSEM像である。
【特許請求の範囲】
【請求項1】
(a)炭素材料を無水液体アンモニアと合わせて、混合物を形成する工程と;
(b)ある量のアルカリ金属を前記混合物中に溶解させて、還元的混合物を形成する工程と;
(c)有機部分及びハロゲン化物部分を含む有機ハロゲン化物を前記還元的混合物に加え、その結果、前記有機ハロゲン化物の前記有機部分が前記炭素材料に付加して誘導体化炭素材料を形成するようにする工程と;を含む方法。
【請求項2】
過剰のアルカリ金属を中和するためのクエンチング工程をさらに含み、該クエンチング工程は、前記過剰のアルカリ金属を、アルコール、水、及びこれらの組合せからなる群から選択される種と反応させて、アルカリ酸化物、水酸化アルカリ、及びこれらの組合せからなる群から選択される中和済みの種を形成することを含む、請求項1に記載の方法。
【請求項3】
未蒸発のアンモニアを中和するための酸性化工程をさらに含む、請求項2に記載の方法。
【請求項4】
前記誘導体化炭素材料を集めるためのろ過工程をさらに含む、請求項1〜2、または3に記載の方法。
【請求項5】
集めた誘導体化炭素材料から他の種を除去するための洗浄工程をさらに含む、請求項4に記載の方法。
【請求項6】
前記炭素材料は、カーボンナノチューブ、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せからなる群から選択される、請求項1〜4、または5に記載の方法。
【請求項7】
前記炭素材料は単層カーボンナノチューブである、請求項1〜4、または5に記載の方法。
【請求項8】
前記炭素材料は小直径カーボンナノチューブである、請求項1〜4、または5に記載の方法。
【請求項9】
前記アルカリ金属は、リチウム、ナトリウム、カリウム、及びこれらの組合せからなる群から選択される、請求項1〜7、または8に記載の方法。
【請求項10】
前記有機ハロゲン化物の前記ハロゲン化物部分は、−F、−Cl、−Br、−I、及びこれらの組合せからなる群から選択される、請求項1〜8、または9に記載の方法。
【請求項11】
前記有機ハロゲン化物の前記有機部分は、アルキル−、アリール−、アリル−、ベンジル−、及びこれらの組合せからなる群から選択される、請求項1〜9、または10に記載の方法。
【請求項12】
(a)炭素材料を無水液体アンモニアと合わせて、混合物を形成する工程と;
(b)ある量のアルカリ金属を前記混合物中に溶解させて、還元的混合物を形成する工程と;
(c)ある量の少なくとも1つのモノマー種を前記還元的混合物に加え、その結果、前記モノマーが前記炭素材料に重合し、付着して、複合材料を形成するようにする工程と;を含む方法。
【請求項13】
過剰のアルカリ金属を中和するためのクエンチング工程をさらに含み、該クエンチング工程は、前記過剰のアルカリ金属を、アルコール、水、及びこれらの組合せからなる群から選択される種と反応させて、アルカリ酸化物、水酸化アルカリ、及びこれらの組合せからなる群から選択される中和済みの種を形成することを含む、請求項12に記載の方法。
【請求項14】
未蒸発のアンモニアを中和するための酸性化工程をさらに含む、請求項13に記載の方法。
【請求項15】
前記複合材料を集めるためのろ過工程をさらに含む、請求項12〜13、または14に記載の方法。
【請求項16】
集めた複合材料から他の種を除去するための洗浄工程をさらに含む、請求項15に記載の方法。
【請求項17】
前記炭素材料は、カーボンナノチューブ、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せからなる群から選択される、請求項12〜15、または16に記載の方法。
【請求項18】
前記炭素材料は単層カーボンナノチューブである、請求項17に記載の方法。
【請求項19】
前記炭素材料は小直径カーボンナノチューブである、請求項17に記載の方法。
【請求項20】
前記アルカリ金属は、リチウム、ナトリウム、カリウム、及びこれらの組合せからなる群から選択される、請求項12〜18、または19に記載の方法。
【請求項21】
前記モノマーは、ビニルモノマー、アセチレン、オリゴマーエチレンオキシド、及びこれらの組合せからなる群から選択される、請求項12〜19、または20に記載の方法。
【請求項22】
前記モノマーは、遊離基開始反応を受けることができる、請求項12〜20、または21に記載の方法。
【請求項23】
前記モノマーは、アニオン開始を受けることができる、請求項12〜20、または21に記載の方法。
【請求項24】
前記モノマーは、アルケン、アルキン、オリゴマーエチレンオキシド、及びこれらの組合せからなる群から選択される、請求項12〜22、または23に記載の方法。
【請求項1】
(a)炭素材料を無水液体アンモニアと合わせて、混合物を形成する工程と;
(b)ある量のアルカリ金属を前記混合物中に溶解させて、還元的混合物を形成する工程と;
(c)有機部分及びハロゲン化物部分を含む有機ハロゲン化物を前記還元的混合物に加え、その結果、前記有機ハロゲン化物の前記有機部分が前記炭素材料に付加して誘導体化炭素材料を形成するようにする工程と;を含む方法。
【請求項2】
過剰のアルカリ金属を中和するためのクエンチング工程をさらに含み、該クエンチング工程は、前記過剰のアルカリ金属を、アルコール、水、及びこれらの組合せからなる群から選択される種と反応させて、アルカリ酸化物、水酸化アルカリ、及びこれらの組合せからなる群から選択される中和済みの種を形成することを含む、請求項1に記載の方法。
【請求項3】
未蒸発のアンモニアを中和するための酸性化工程をさらに含む、請求項2に記載の方法。
【請求項4】
前記誘導体化炭素材料を集めるためのろ過工程をさらに含む、請求項1〜2、または3に記載の方法。
【請求項5】
集めた誘導体化炭素材料から他の種を除去するための洗浄工程をさらに含む、請求項4に記載の方法。
【請求項6】
前記炭素材料は、カーボンナノチューブ、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せからなる群から選択される、請求項1〜4、または5に記載の方法。
【請求項7】
前記炭素材料は単層カーボンナノチューブである、請求項1〜4、または5に記載の方法。
【請求項8】
前記炭素材料は小直径カーボンナノチューブである、請求項1〜4、または5に記載の方法。
【請求項9】
前記アルカリ金属は、リチウム、ナトリウム、カリウム、及びこれらの組合せからなる群から選択される、請求項1〜7、または8に記載の方法。
【請求項10】
前記有機ハロゲン化物の前記ハロゲン化物部分は、−F、−Cl、−Br、−I、及びこれらの組合せからなる群から選択される、請求項1〜8、または9に記載の方法。
【請求項11】
前記有機ハロゲン化物の前記有機部分は、アルキル−、アリール−、アリル−、ベンジル−、及びこれらの組合せからなる群から選択される、請求項1〜9、または10に記載の方法。
【請求項12】
(a)炭素材料を無水液体アンモニアと合わせて、混合物を形成する工程と;
(b)ある量のアルカリ金属を前記混合物中に溶解させて、還元的混合物を形成する工程と;
(c)ある量の少なくとも1つのモノマー種を前記還元的混合物に加え、その結果、前記モノマーが前記炭素材料に重合し、付着して、複合材料を形成するようにする工程と;を含む方法。
【請求項13】
過剰のアルカリ金属を中和するためのクエンチング工程をさらに含み、該クエンチング工程は、前記過剰のアルカリ金属を、アルコール、水、及びこれらの組合せからなる群から選択される種と反応させて、アルカリ酸化物、水酸化アルカリ、及びこれらの組合せからなる群から選択される中和済みの種を形成することを含む、請求項12に記載の方法。
【請求項14】
未蒸発のアンモニアを中和するための酸性化工程をさらに含む、請求項13に記載の方法。
【請求項15】
前記複合材料を集めるためのろ過工程をさらに含む、請求項12〜13、または14に記載の方法。
【請求項16】
集めた複合材料から他の種を除去するための洗浄工程をさらに含む、請求項15に記載の方法。
【請求項17】
前記炭素材料は、カーボンナノチューブ、フラーレン、ダイヤモンド、アセチレン型炭素、カーボンブラック、活性炭、黒鉛質炭素、及びこれらの組合せからなる群から選択される、請求項12〜15、または16に記載の方法。
【請求項18】
前記炭素材料は単層カーボンナノチューブである、請求項17に記載の方法。
【請求項19】
前記炭素材料は小直径カーボンナノチューブである、請求項17に記載の方法。
【請求項20】
前記アルカリ金属は、リチウム、ナトリウム、カリウム、及びこれらの組合せからなる群から選択される、請求項12〜18、または19に記載の方法。
【請求項21】
前記モノマーは、ビニルモノマー、アセチレン、オリゴマーエチレンオキシド、及びこれらの組合せからなる群から選択される、請求項12〜19、または20に記載の方法。
【請求項22】
前記モノマーは、遊離基開始反応を受けることができる、請求項12〜20、または21に記載の方法。
【請求項23】
前記モノマーは、アニオン開始を受けることができる、請求項12〜20、または21に記載の方法。
【請求項24】
前記モノマーは、アルケン、アルキン、オリゴマーエチレンオキシド、及びこれらの組合せからなる群から選択される、請求項12〜22、または23に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】

【図7】

【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図7】
【図8】
【公表番号】特表2007−530400(P2007−530400A)
【公表日】平成19年11月1日(2007.11.1)
【国際特許分類】
【出願番号】特願2007−503081(P2007−503081)
【出願日】平成17年3月11日(2005.3.11)
【国際出願番号】PCT/US2005/008303
【国際公開番号】WO2005/090233
【国際公開日】平成17年9月29日(2005.9.29)
【出願人】(501105635)ウィリアム・マーシュ・ライス・ユニバーシティ (26)
【Fターム(参考)】
【公表日】平成19年11月1日(2007.11.1)
【国際特許分類】
【出願日】平成17年3月11日(2005.3.11)
【国際出願番号】PCT/US2005/008303
【国際公開番号】WO2005/090233
【国際公開日】平成17年9月29日(2005.9.29)
【出願人】(501105635)ウィリアム・マーシュ・ライス・ユニバーシティ (26)
【Fターム(参考)】
[ Back to top ]