
ガボキサドールおよびPAT1阻害薬またはOAT阻害薬を含む医薬組成物
本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。本発明は、さらに、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを示す組成物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。本発明は、さらに、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物に関する。
【背景技術】
【0002】
ガボキサドール(4,5,6,7−テトラヒドロイソキサゾロ[5,4−c]ピリジン−3−オール)(THIP)は、欧州特許第0000338号(特許文献1)および欧州特許第0840601号(特許文献2)に記載されており、これまでに、睡眠障害の治療において、また、うつ病の前臨床モデルにおいて大きな可能性が示されている(国際公開第2004112786号(特許文献3))。ガボキサドールは、以下の一般式:
【0003】
【化1】

を有する。
【0004】
ガボキサドールは、当技術分野で周知の方法を用いて調製できる。例えば、欧州特許第0000338号(特許文献1)および国際公開第2005023820号(特許文献4)に開示されているように調製できる。
【0005】
国際公開第02094225号(特許文献5)では、ガボキサドールを含有し速放性のプロファイルを有する成形された固形の医薬単位剤形の調製に使用できる、顆粒状のガボキサドール含有調製物が開示されている。
【0006】
WO0122941(特許文献6)では、ガボキサドールを含有する溶融造粒組成物(melt granulated composition)、および前記組成物から調製される放出調節剤形が開示されている。
【0007】
ガボキサドール速放性製剤の治療的投与においては、迅速に溶解する結果、投与後間もなく血漿ガボキサドールレベルが急速に高まり、次いで、ガボキサドールが代謝されるかまたは排出されるにつれ数時間にわたり血漿レベルが低下した後、治療的レベルを下回る血漿レベルに達する。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】欧州特許第0000338号
【特許文献2】欧州特許第0840601号
【特許文献3】国際公開第2004112786号
【特許文献4】国際公開第2005023820号
【特許文献5】国際公開第02094225号
【特許文献6】国際公開第0122941号
【非特許文献】
【0009】
【非特許文献1】Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁
【非特許文献2】Lucas, M. L.ら、1975、Proc R.Soc Lond.B Biol Sci.、192巻、1106、39〜48頁
【非特許文献3】Boll, M.ら、2002、J Biol Chem.、277巻、25、22966〜22973頁
【非特許文献4】Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁
【非特許文献5】Koepsell, H.ら、2004、Pflugers.Arch.、447巻、5、666〜676頁
【非特許文献6】Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁
【非特許文献7】Burckhardt, B. C.ら、2003、Rev Physiol Biochem Pharmacol.、146巻、95〜158頁
【非特許文献8】Pritchard, J. B.ら、1999、J Biol Chem.、274巻、47、33382〜33387頁
【非特許文献9】Pavlova, A.ら、2000、Am.J Physiol Renal Physiol.、278巻、4、F635〜F643頁
【非特許文献10】Sekine, T.ら、2006、Am.J Physiol Renal Physiol.、290巻、2、F251〜F261頁
【非特許文献11】Wright, S. H.ら、2004、Am.J Physiol Renal Physiol.、287巻、3、F442〜F451頁
【非特許文献12】Gabrielsson and Weiner、2007、Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications、第4版、CRC Press、Baco Raton、FL ISBN 978-9-1976-5100-4
【非特許文献13】Remington、The Science and Practice of Pharmacy、第21版、Lippincott Williams & Wilkins (2005)
【発明の概要】
【発明が解決しようとする課題】
【0010】
薬理学的および生理学的なプロセスの中には、最適な治療効果に到達するために、治療上適切な血漿レベルでの長期にわたる暴露を要すると考えられるものがある。そのため、治療上適切な血漿レベルで長期にわたり暴露することが可能なガボキサドールの医薬剤形が必要である。さらに、Tmaxがより遅くおよび/またはCmaxが低下しており、加えて、場合によりAUCが増加している血漿プロファイルを提供するガボキサドールの医薬剤形が必要である。
【0011】
今回、驚くべきことに、ガボキサドールの吸収を変化させ、それにより、ピーク濃度を最低限とし、Tmaxを延長し、特別な状況においてはさらに薬物動態プロファイルの排出相(elimination phase)を延長する(すなわち、AUCを高める)ことが実証されたガボキサドール製剤を調製することが可能であることが見出された。
【課題を解決するための手段】
【0012】
一態様では、本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。
【0013】
別の態様では、本発明は、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物に関する。
【図面の簡単な説明】
【0014】
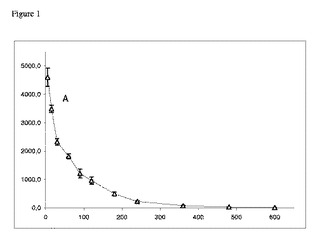
【図1】イヌにおけるIV投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。2.5mg/kgガボキサドール(A、(△))の静脈内注射後の血漿ガボキサドール濃度対時間プロファイル。血液試料は、薬物投与後5分、15分、30分、60分、90分、120分、180分、240分、360分、480分および600分の時点で収集した。示してあるのは、6匹のイヌ(n=6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
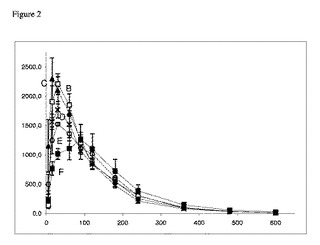
【図2】イヌにおけるPO投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。2.5mg/kgガボキサドール(B、(□))のPO投与後のビーグル犬の血漿ガボキサドール濃度対時間プロファイル。同じ用量を、2.5mg/kg Trp(C、(▲))、10.0mg/kg Trp(D、(×))、50.0mg/kg Trp(E、(○))および150.0mg/kg Trp(F、(■))との併用でも投与した。Trpは、ガボキサドールと同じ時点での併用投与として投与した。試料は、薬物投与後5分、15分、30分、60分、90分、120分、180分、240分、360分、480分および600分の時点で収集した。示してあるのは、6匹のイヌ(n=6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
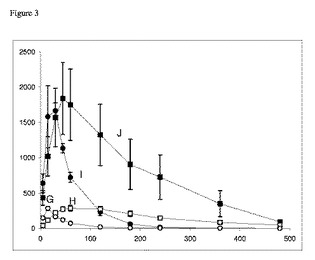
【図3】ラットにおけるPO投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。ガボキサドールのPO投与後のラットの血漿ガボキサドール濃度対時間プロファイル。投与した用量は、0.5mg/kg(G、(○))もしくは5.0mg/kg(H、(●))のガボキサドール単独、または、200.0mg/kg 5−HTPのプレ・インキュベーションとの併用、それぞれ(I、(□))もしくは(J、(■))であった。200.0mg/kg 5−HTPは、ガボキサドールの前30分のプレ・インキュベーションとして投与した。試料は、薬物投与後5分、15分、30分、45分、60分、120分、240分、360分および480分の時点で収集した。示してあるのは、5〜6匹のラット(n=5〜6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【図4】ラットにおけるIV投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。ガボキサドールのIV投与後のラットの血漿ガボキサドール濃度対時間プロファイル。ラットは、2.5mg/kgガボキサドール(K、(△))の静脈内注射を受けた。同じ用量を、200.0mg/kg 5−HTP(L、(▲))との併用でも投与した。200.0mg/kg 5−HTPは、ガボキサドールの前30分のプレ・インキュベーションとして投与した。試料は、薬物投与後5分、15分、30分、45分、60分、120分、240分、360分および480分の時点で収集した。示してあるのは、5〜6匹のラット(n=5〜6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【発明を実施するための形態】
【0015】
本発明者らは、原発性不眠症に罹患している一部の患者においてガボキサドールの速放性製剤を治療的投与すると、用量依存的な有害事象が生じることを見出した。観察されたこの有害事象は、平均Cmaxとほぼ同時に発生し、投与後数時間後に消失したことから、この有害事象はCmaxと相関がある。ガボキサドールの速放性製剤を用いた場合に観察された有害事象としては、眩暈、悪心、嘔吐、傾眠、振戦、倦怠、鎮静およびいくつかの精神医学的な有害事象が挙げられる。有害事象のさらなる分析により、本発明者らは、平均Cmaxを低下させることにより、および/または、平均Tmaxを長くすることにより、こうした有害事象は稀で穏やかになり、精神医学的な有害事象は生じないことを見出した。
【0016】
本発明者らは、ガボキサドールおよび1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物を調製して、ガボキサドールの吸収調節製剤を提供することが可能であることを見出した。本発明によれば、この医薬組成物中で使用されるガボキサドール、1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬の量を変化させることにより、ガボキサドールのCmax、Tmaxおよび場合によりAUCを調節することが可能である。本発明による組成物は、以下の利点の1つまたは複数をもたらす:血漿ガボキサドールレベルの急速な上昇を回避または減少できる、より遅いTmaxおよび/または低下したCmaxを有するガボキサドールの薬物動態プロファイルが達成でき、このプロファイルには、場合によりAUCの増加を加えることができる。したがって、以下の課題の1つまたは複数は、本発明により解決できる:治療上適切な血漿レベルに達しながら血漿ガボキサドールレベルの急速な増加に伴う影響を回避または減少できる、および/または治療上適切な血漿レベルはより長期間にわたり維持されるため、ガボキサドール投与間の時間間隔を速放性製剤と比較して長くすることができる。したがって、本発明によれば、ガボキサドールを含む医薬組成物であって、大部分の有害事象を伴う血漿レベルに達することなく、また、状況によっては長期間にわたり治療上適切な血漿レベルに達することが可能な医薬組成物が提供される。
【0017】
したがって、本発明は、ガボキサドールまたは薬学上許容可能なその塩を含む医薬組成物であって、ガボキサドールの速放性製剤と比較して平均Cmaxを低下させながらも治療上適切な血漿ガボキサドールレベルを実現する組成物に関する。
【0018】
いかなる特定の理論により拘束されるものでもないが、本発明による組成物中のPAT1阻害薬は、消化管からのガボキサドールの吸収速度を低下させることにより、ガボキサドールの吸収を調節するとの仮説が立てられる。さらには、いくらかの、おそらくは全てのPAT1阻害薬、およびOAT基質またはOAT阻害薬は、腎臓中の1つまたは複数の有機陰イオン輸送体(OAT)と相互作用し、および/または腎臓の血流を減少させ、それにより腎臓からのガボキサドールの排出速度も低下させることにより、血漿ガボキサドールレベルを、より長期間にわたり治療上適切なレベルにするとの仮説も立てられる。
【0019】
ヒトプロトン依存性アミノ酸輸送体1(hPAT1)は、2003年にCaco−2細胞からクローニングされた(Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。この輸送体は、溶質キャリアーファミリー(solute carrier family)SLC36に属し、4つのうちの第一のもの(SLC36A1)である。PAT3およびPAT4はオーファン輸送体であるが、PAT2は、肺、心臓、腎臓、筋肉、精巣、脾臓、副腎、胸腺および坐骨神経の組織中で主に発現する。分析から、ヒト組織においてhPAT1のmRNAが遍在性に発現することが発見されており、このmRNAはヒト消化管全体にわたって検出されており小腸で最も多く発現していることから、この輸送体は、腸管の孔の長さで基質を吸収するのに適切なものとなっている(Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。hPAT1経由でのアミノ酸輸送は、腸の中の酸性の微小気候により、頂端膜を隔てて構築されるプロトン(H+)の顕著な濃度勾配により活発化する(Lucas, M. L.ら、1975、Proc R.Soc Lond.B Biol Sci.、192巻、1106、39〜48頁(非特許文献2))。
【0020】
Caco−2細胞株は、ヒト小腸の上皮のモデルとして使用できる。プロトン依存性アミノ酸輸送体は、これまでに、この生体外モデルにおいて徹底的に、また、ある程度までは、トランスフェクトされた細胞系においても特徴付けられている(Boll, M.ら、2002、J Biol Chem.、277巻、25、22966〜22973頁(非特許文献3)、Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。競合アッセイならびに転位実験により、PAT1との相互作用について多様な化合物が試験されている。こうしたin vitroでの特徴付けによれば、PAT1基質は、(21〜28日目齢)Caco−2細胞の単層を超えて輸送され、膜を隔てたpH勾配に伴ってフラックスが増加する化合物を指す。さらに、高濃度の別のPAT1基質(L−プロリンであってもよいと考えられるが、これに限定されない)を加えることにより、この輸送は阻害されるはずである。
【0021】
PAT1阻害薬は、Caco−2細胞の単層を超えてのPAT1基質の輸送を減少させる化合物を指す。この阻害薬は、基質ポケット中の輸送体と結合するか否かにより、競合的または非競合的な様式で作用することができる。
【0022】
古典的なPAT1基質は、β−アラニンおよびAIB(α−(メチルアミノ)−イソ酪酸)のようないくつかのβ−アミノ酸に加えて、グリシン、アラニン、セリンおよびプロリンのような小型で非分枝状の双性イオン性α−アミノ酸、ならびに、GABA(γ−アミノ酪酸)のような少数のγ−アミノ酸である(Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁(非特許文献4))。生体異物の中には、hPAT1基質、例えば神経調節性かつ抗菌性の物質であるD−シクロセリンに囲まれていることが実証されているものがある。さらに、いくつかのGABA受容体遮断薬および再取込み阻害薬、ならびに、癌および線維性疾患の治療に用いられるプロリン類似体も、PAT1により輸送される(Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁(非特許文献4))。
【0023】
可能性のある競合的なPAT1阻害薬としては、グリシン、L−アラニン、D−アラニン、L−セリン、D−セリン、L−プロリン、D−プロリン、GABA(γ−アミノ酪酸)、サルコシン、ベタイン、N−メチル−L−アラニン(AIB(α−(メチルアミノ)−イソ酪酸))、D−シクロセリン、β−アラニン、ビガバトリン、グバシン、TACA(トランス−4−アミノクロトン酸)が挙げられるが、これらに限定されない。
可能性のあるPAT1阻害薬は、5−ヒドロキシトリプトファン(5−HTP)、セロトニン(5−HT)、L−トリプトファン(Trp)、トリプタミン、インドール−3−プロピオン酸であってもよいと考えられるが、これらに限定されない。
【0024】
有機陰イオン輸送体(OAT)は、1997年に同定された。この輸送体は、SLC22遺伝子ファミリーに属し(Koepsell, H.ら、2004、Pflugers.Arch.、447巻、5、666〜676頁(非特許文献5))、顕著かつ広範な基質特異性を特徴とする。現時点で公知の輸送体としては、腎臓内に主に局在するOAT1−4およびURAT1が挙げられる(Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁(非特許文献6))ことから、いくつかの刊行物では、生体異物および薬物の腎臓分泌に対するこの輸送体の寄与に注目している(総説については、Burckhardt, B. C.ら、2003、Rev Physiol Biochem Pharmacol.、146巻、95〜158頁(非特許文献7)を参照のこと)。脳内、特に脈絡膜叢および血液脳関門(Pritchard, J. B.ら、1999、J Biol Chem.、274巻、47、33382〜33387頁(非特許文献8))、胚の発生の異なる段階での目、骨格筋およびいくつかの臓器(Pavlova, A.ら、2000、Am.J Physiol Renal Physiol.、278巻、4、F635〜F643頁(非特許文献9))内においても発現が報告されている。OATは、基質転位の活発化にATP加水分解を直接には利用しない。OATファミリーのうち全てではなくても大部分のものは、陰イオン交換体として作用する。すなわち、そうした輸送体は、細胞中への有機陰イオンの取込みを、細胞からの別の有機陰イオンの放出と結び付ける。それにより、OATは、陰イオン(例えば、α−ケトグルタル酸イオン、乳酸イオンおよびニコチン酸イオン)の既存の細胞内>細胞外勾配を利用して、負の膜電位に逆らって、有機陰イオンの上り坂取込み(uphill uptake)を駆動する。腎臓の近位尿細管においては、OATは、乳酸イオン、ニコチン酸イオンおよびα−ケトグルタル酸イオンの細胞内>細胞外勾配を構築および維持するNa+駆動型のモノカルボン酸輸送体およびジカルボン酸輸送体と機能的に結び付く(Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁(非特許文献6))。
【0025】
典型的なOAT基質の分子量は最大400〜500であり(Sekine, T.ら、2006、Am.J Physiol Renal Physiol.、290巻、2、F251〜F261頁(非特許文献10)、Wright, S. H.ら、2004、Am.J Physiol Renal Physiol.、287巻、3、F442〜F451頁(非特許文献11))、その特異性は、以下を非限定的に含むOATにより輸送されている化学構造体という点で非常に広範である:キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン(carboxyflurescein)、ベンジルペニシリン、セファドロキシル、セファマドール(Cefamadole)、セファゾリン、セフォペラゾン、セフォタミム(Cefotamime)、セファレキシン、セファロチン、セフラジン、アシロビル(Acylovir)、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル(Valacylovir)、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク(Dilofenac)、ジフルシナル(Diflusinal)、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート(Mefanamate)、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダク(Sulidac)。本明細書では、OAT基質は、OATのmRNAをトランスフェクトされた卵母細胞中に、制御状況(control situation)と比較して顕著に増加した速度で輸送される化合物により定義される。
【0026】
定義
Cmaxは、実験中に予測される、血漿中薬物の最大濃度(ng*ml1)と定義される。Tmaxは、Cmaxに達すると予測される時間(min)と定義される。AUCは、薬物投与から薬物が排出するまでの、血漿薬物濃度−時間曲線下の総面積(ng*min*ml−1)である。曲線下面積は、クリアランスに支配される。クリアランスは、含有された薬物が完全に取り除かれている血液または血漿の単位時間当たりの体積(ml*h−1*kg−2)と定義される。単位時間当たりに排出される体内薬物量に関する排出速度定数は、薬物が排出される速度(h−1)と定義される(Gabrielsson and Weiner、2007、Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications、第4版、CRC Press、Baco Raton、FL ISBN 978-9-1976-5100-4(非特許文献12))。
【0027】
用語「PK」は、薬物動態プロファイルを指す。
【0028】
本明細書中で使用する場合、用語「対象」は、ヒトおよび動物など任意の温血種を指す。ガボキサドールで治療されることになるヒトなどの対象は、実際には、ヒト集団、男性または女性の任意の対象であってもよく、小児、成人または高齢者に分けてもよい。このような患者群の任意の1つは、本発明の一実施形態に関する。
【0029】
本明細書中で使用する場合、用語「治療する(こと)」または「治療」は、疾患もしくは状態を患っているまたは疾患もしくは状態に罹りやすいと考えられるが当該疾患もしくは状態の臨床症状もしくは潜在症状を未だ経験もしくは示していない対象において当該疾患もしくは状態の臨床症状の出現を予防または遅延させることを指す。「治療する(こと)」または「治療」は、疾患もしくは状態を阻害すること、すなわち、その発症、または、その少なくとも1つの臨床症状もしくは潜在症状を抑止または低減することも指す。「治療する(こと)」または「治療」は、さらに、疾患もしくは状態を緩和すること、すなわち、疾患もしくは状態またはその臨床症状もしくは潜在症状の少なくとも1つの軽減をもたらすことを指す。治療されることになる対象の利益は、統計的に有意であるか、あるいは、対象および/または医師に少なくとも知覚できるものであるか、そのいずれかである。但し、予防的(防止的)および治療的(治癒的)な治療は、本発明の2つの独立した実施形態である。
【0030】
本明細書中で使用する場合、用語「薬学上許容可能な」は、「一般に安全とみなされる」分子体(molecular entity)および組成物、例えば、生理学的に忍容でき、ヒトに投与した際、胃の不調など、アレルギー反応または同様の有害な反応を典型的に生じさせないものを指す。別の実施形態では、この用語は、FDAによる市販前審査(premarket review)および認可を受けている、連邦食品医薬品化粧品法の第204条(s)項および409条の下のGRASリストまたは同様のリスト、動物、より詳細にはヒトにおける使用についての米国薬局方または別の一般に認識された薬局方として、連邦政府または州政府の規制当局により認可された分子体および組成物を指す。
【0031】
第1の態様によれば、本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。本発明の第1の態様の一実施形態では、この組成物は、1つまたは複数のPAT1阻害薬を含むが、OAT阻害薬を含まない。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数のOAT阻害薬を含むが、PAT1阻害薬を含まない。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数のPAT1阻害薬および1つまたは複数のOAT阻害薬を両方とも含む。本発明の第1の態様の別の実施形態では、ガボキサドールは、酸付加塩、または双性イオン水和物(zwitter ion hydrate)もしくは双性イオン無水物(zwitter ion anhydrate)の形態である。本発明の第1の態様の別の実施形態では、ガボキサドールは、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である。本発明の第1の態様の別の実施形態では、ガボキサドールの量は、0.5mg〜50mgの範囲である。本発明の第1の態様の別の実施形態では、この組成物は、経口投与用の形態である。本発明の第1の態様の別の実施形態では、この組成物は、錠剤またはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である。本発明の第1の態様の別の実施形態では、前記ガボキサドールは結晶性である。本発明の第1の態様の別の実施形態では、PAT1はヒトPAT1である。本発明の第1の態様の別の実施形態では、PAT1阻害薬は、5−ヒドロキシトリプトファン(5−HTP)、L−プロリン、D−プロリン、サルコシン、L−アラニン、D−アラニン、N−メチル−L−アラニン、N−メチル−D−アラニン、α−(メチルアミノ)−イソ酪酸、ベタイン、D−シクロセリン、L−シクロセリン、β−アラニン、セロトニン、L−トリプトファン、D−トリプトファン、トリプタミン、インドール−3−プロピオン酸から選択される。本発明の第1の態様の別の実施形態では、PAT1阻害薬の量は、約0.5〜約3000mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約750mg、約1000mg、約1250mg、約1500mg、約1750mg、約2000mg、約2250mg、約2500mg、約2750mgまたは約3000mgである。本発明の第1の態様の別の実施形態では、OATはヒトOATである。本発明の第1の態様の別の実施形態では、OAT阻害薬は、キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン、ベンジルペニシリン、セファドロキシル、セファマドール、セファゾリン、セフォペラゾン、セフォタミム、セファレキシン、セファロチン、セフラジン、アシロビル、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク、ジフルシナル、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダクから選択される。本発明の第1の態様の別の実施形態では、OAT阻害薬の量は、約0.5〜約500mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mgまたは約500mgである。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数の賦形剤を含む。本発明の第1の態様の別の実施形態では、この組成物は、セロトニン再取込み阻害薬である化合物、または細胞外のセロトニンレベルの上昇をもたらす任意の他の化合物を含む。本発明の第1の態様の別の実施形態では、セロトニン取込み阻害薬は、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミンまたはこれらの化合物のいずれかの薬学上許容可能な塩から選択される。本発明の第1の態様の別の実施形態では、セロトニン取込み阻害薬は、塩基として、またはシュウ酸塩、臭化水素酸塩もしくは塩酸塩など薬学上許容可能なその塩としてのエスシタロプラムである。
【0032】
第2の態様によれば、本発明は、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを示す組成物に関する。本発明の第2の態様の一実施形態では、前記平均Tmaxは、約25分超、30分超、35分超、40分超、45分超、50分超、55分超、60分超、65分超、70分超または75分超である。本発明の第2の態様の別の実施形態では、この組成物は、約2250ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、前記平均Cmaxは、約2000ng/ml未満、約1750ng/ml未満、約1500ng/ml未満、約1250ng/ml未満、約1000ng/ml未満、約750ng/ml未満、約500ng/ml未満、約250ng/ml未満、約200ng/ml未満または約100ng/ml未満である。本発明の第2の態様の別の実施形態では、この組成物は、約8.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、前記平均AUC0〜∞は、約16.000ng・min・ml−1超、20.000ng・min・ml−1超、40.000ng・min・ml−1超、80.000ng・min・ml−1超、120.000ng・min・ml−1超または200.000ng・min・ml−1超である。本発明の第2の態様の別の実施形態では、クリアランスは40ml/min未満である。本発明の第2の態様の別の実施形態では、前記クリアランスは、30ml/min未満、20ml/min未満、10ml/min未満または5ml/min未満である。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約2mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約100ng/ml未満の平均Cmaxおよび約8.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約4mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約200ng/ml未満の平均Cmaxおよび約16.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約5mgまたは薬学上許容可能なその塩を含み、約20分時間より長い平均Tmax、約250ng/ml未満の平均Cmaxおよび約20.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0033】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約10mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約500ng/ml未満の平均Cmaxおよび約40.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0034】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約20mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約1000ng/ml未満の平均Cmaxおよび約80.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0035】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約30mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約1500ng/ml未満の平均Cmaxおよび約120.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0036】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約50mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約2500ng/ml未満の平均Cmaxおよび約200.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0037】
本発明の第2の態様の別の実施形態では、クリアランスは40ml/min未満であり、AUCは200.000ng・min・ml−1より高い。
【0038】
本発明の第2の態様の別の実施形態では、前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞は、この組成物をイヌに投与した際に得られ、前記クリアランスは、この組成物をイヌまたはラットに投与した際に得られる。
【0039】
本発明の第2の態様の別の実施形態では、前記平均Tmaxは、約30分より長い。本発明の第2の態様の別の実施形態では、この組成物は、約300ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、ガボキサドールの量は、約2.5mg、約5mgまたは約10mgから選択される。本発明の第2の態様の別の実施形態では、ガボキサドールの量は2.5mgであり、平均Cmaxは約40ng/ml未満、例えば、約35ng/ml、約30ng/ml、約25ng/mlまたは約20ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、ガボキサドールの量は5mgであり、平均Cmaxは約85ng/ml未満、例えば、約80ng/ml、約75ng/ml、約70ng/mlまたは約65ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、ガボキサドールの量は10mgであり、平均Cmaxは約150ng/ml未満、例えば、約145ng/ml、約140ng/ml、約135ng/mlまたは約130ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、前記平均Tmaxおよび平均Cmaxは、この組成物をヒトに投与した際に得られる。本発明の第2の態様の別の実施形態では、ガボキサドールは、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である。本発明の第2の態様の別の実施形態では、ガボキサドールは、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である。本発明の第2の態様の別の実施形態では、この組成物は、経口投与用の形態である。本発明の第2の態様の別の実施形態では、この組成物は、錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である。本発明の第2の態様の別の実施形態では、前記ガボキサドールは結晶性である。本発明の第2の態様の別の実施形態では、この組成物は、1つまたは複数の賦形剤を含む。
【0040】
本発明の一実施形態では、この医薬組成物は、ガボキサドールの速放性製剤の場合に観察されるCmaxの80%(75%、70%または65%など)に相当する平均Cmaxを示す。さらに本発明は、ガボキサドールまたは薬学上許容可能なその塩を含む医薬組成物であって、ガボキサドールの速放性製剤の場合に観察されるものより長い平均Tmaxを示しながらもなお治療上適切な血漿ガボキサドールレベルを提供する組成物に関する。
【0041】
さらなる一実施形態では、化合物は、PAT1およびOATを両方とも阻害する。
【0042】
本発明の組成物をイヌに投与した際に前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞が得られるさらなる一実施形態では、前記イヌはビーグルであり、前記ビーグルは、前記組成物の投与前20〜24時間(h)絶食させる。
【0043】
本発明の組成物をイヌに投与した際に前記クリアランスが得られるさらなる一実施形態では、前記イヌはビーグルであり、前記ビーグルは、前記組成物の投与前20〜24時間(h)絶食させる。
【0044】
本発明の組成物をラットに投与した際に前記クリアランスが得られるさらなる一実施形態では、前記ラットはオスのSprague−Dawleyラット(Charles River Laboratories、Wilmington、MA、USA)であり、前記ラットは、前記組成物の投与前16〜20時間までは、標準食および水で維持される。
【0045】
さらなる一実施形態では、本発明の医薬組成物は、原発性不眠症などの睡眠障害、または、大うつ病などのうつ病の治療用である。
【0046】
この明細書を通じ、「ガボキサドール」は、遊離塩基(双性イオン)、薬学上許容可能な塩(例えば、薬学上許容可能な酸付加塩)、こうした塩基または塩の水和物または溶媒和物、ならびに無水物、またさらには非晶質形態または結晶形態など、この化合物の一切の形態を包含することを意図したものである。
【0047】
さらなる一実施形態では、ガボキサドールは、双性イオン、典型的にはその水和物から選択されるが、無水物も適している。適当な一実施形態は、双性イオン一水和物である。
【0048】
さらなる一実施形態では、ガボキサドールは、酸付加塩、典型的には薬学上許容可能な酸付加塩から選択される。適当な一実施形態は、マレイン酸、フマル酸、安息香酸、アスコルビン酸、コハク酸、シュウ酸、ビスメチレンサリチル酸、メタンスルホン酸、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン酸、乳酸、リンゴ酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリン酸、パルミチン酸、イタコン酸、グリコール酸、p−アミノ安息香酸、グルタミン酸、ベンゼンスルホン酸またはテオフィリン酢酸の付加塩のいずれか1つなどの有機酸付加塩、ならびに、8−ハロテオフィリン、例えば8−ブロモテオフィリンである。別の適当な実施形態は、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸または硝酸の付加塩のいずれか1つなどの無機酸付加塩である。
【0049】
別の実施形態では、ガボキサドールは、塩酸塩、臭化水素酸塩または双性イオン一水和物の形態である。
【0050】
さらなる一実施形態では、ガボキサドールは、結晶性の塩酸塩、結晶性の臭化水素酸塩または結晶性の双性イオン一水和物など結晶性である。
【0051】
さらなる一実施形態では、本発明の医薬組成物は、ヒドロキシプロピルメチルセルロース(例えばMetolose 90SH−15.000およびMetolose 90SH−100.000)など親水性のセルロースエーテル・ポリマーを実際に含有する。
【0052】
本発明による酸付加塩は、不活性溶媒中の酸でガボキサドールを処理し、次いで、公知の方法による析出、単離、場合により再結晶化、所望により、湿式もしくは乾式の粉砕または別の好都合な方法による結晶性の生成物の微粒子化、または、溶媒−乳化法(solvent−emulsification process)からの粒子の調製により得ることができる。適当な方法は、例えば欧州特許第0000338号(特許文献1)に記載されている。
【0053】
ガボキサドールの塩の析出は、典型的には、不活性溶媒中、例えば、アルコール(例えば、エタノール、2−プロパノールおよびn−プロパノール)などの不活性な極性溶媒中で実施されるが、水、または水と不活性溶媒との混合物を使用してもよい。
【0054】
ガボキサドールは、経口投与用の固形形態(典型的には錠剤またはカプセル剤)などの経口投与用の形態として、または経口投与用の液体形態として投与してもよい。ガボキサドールは、速放性剤形、または制御放出剤形もしくは持続放出剤形の形態で投与してもよい。一実施形態によれば、この剤形は、睡眠導入量未満の量で、ガボキサドールを制御放出または持続放出する。ガボキサドールは、約0.1〜約150mg/日、約0.2〜約100mg/日、約0.5〜約50mg/日、約0.1〜約50mg/日、約1〜約15mg/日、または約2〜約5mg/日の量で活性成分を含有する錠剤またはカプセル剤などの単位投与形態で、便利に経口投与してもよい。典型的には、この医薬組成物は、約0.5mg〜約20mg、例えば、約0.5mg、約1mg、約1.5mg、約2mg、約2.5mg、約3mg、約3.5mg、約4mg、約4.5mg、約5mg、約5.5mg、約6mg、約6.5mg、約7mg、約7.5mg、約8mg、約8.5mg、約9mg、約9.5mg、約10mg、約10.5mg、約11mg、約11.5mg、約12mg、約12.5mg、約13mg、約13.5mg、約14mg、約14.5mg、約15mg、約15.5mg、約16mg、約16.5mg、約17mg、約17.5mg、約18mg、約18.5mg、約19mg、約19.5mgまたは約20mgなどのガボキサドールを含む。ガボキサドールの量は、遊離塩基(双性イオン)の形態に基づいて計算する。
【0055】
一実施形態では、ガボキサドールは、約2.5mg〜約20mgの用量を用いて1日1回(例えば午前または午後)投与する。別の実施形態では、ガボキサドールは1日2回投与する。
【0056】
本発明によれば、ガボキサドールまたは薬学上許容可能なその塩は、任意の適当な方式で、例えば、経口的または非経口的に投与してもよく、そのような投与に適した任意の形態、例えば、錠剤、カプセル剤、散剤、シロップ剤または液剤または注射用分散剤の形態で提供してもよい。別の実施形態では、また、本発明の目的によれば、ガボキサドールは、固形の医薬体(pharmaceutical entity)の形態で、適切には錠剤またはカプセル剤として、または注射用の懸濁剤、液剤または分散剤の形態で、投与する。加えて、ガボキサドールは、佐剤および/または希釈剤など薬学上許容可能な担体と共に投与してもよい。
【0057】
本発明はさらに、ガボキサドールと、セロトニン再取込み阻害薬(SRI)である化合物または細胞外5−HTの上昇をもたらす任意の他の化合物と、場合により、薬学上許容可能な担体または希釈剤とを含む医薬組成物またはキットにも関する。
【0058】
一実施形態では、SRIは、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、デュロキセチン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミンから選択される。念のため明確にしておくと、こうしたSRIのそれぞれは、個々の実施形態を構成し、個々の請求項の主題である場合がある。
【0059】
選択的なセロトニン再取込み阻害薬(SSRI)という用語は、ドパミン輸送体およびノルアドレナリン輸送体よりセロトニン輸送体で強い阻害効果を有するモノアミン輸送体阻害薬を意味する。
【0060】
選択的なセロトニン再取込み阻害薬(SSRI)は、本発明により使用される最も好ましいセロトニン再取込み阻害薬に含まれる。したがって、さらなる一実施形態では、SRIは、シタロプラム、エスシタロプラム、フルオキセチン、フルボキサミン、セルトラリンまたはパロキセチンなどのSSRIから選択される。
【0061】
シタロプラムは、好ましくは、臭化水素酸塩の形態で、または塩基として、エスシタロプラムはシュウ酸塩の形態で、フルオキセチン、セルトラリンおよびパロキセチンは塩酸塩の形態で、フルボキサミンはマレイン酸塩の形態で、使用される。
【0062】
本明細書中でこれまでに具体的に言及しているSSRIを含むセロトニン再取込み阻害薬は、分子量および活性の両方において異なる。結果として、併用療法において使用されるセロトニン再取込み阻害薬の量は、前記セロトニン再取込み阻害薬の性質による。本発明の一実施形態では、セロトニン再取込み阻害薬または細胞外5−HTレベルの増加をもたらす化合物は、化合物を単独で使用する際に必要な用量より低用量で投与される。別の実施形態では、セロトニン再取込み阻害薬または細胞外5−HTレベルの増加をもたらす化合物は、通常の用量で投与される。
【0063】
さらなる一実施形態では、ガボキサドールと、セロトニン再取込み阻害薬(SRI)である化合物または細胞外5−HTの上昇をもたらす任意の他の化合物と、場合により、薬学上許容可能な担体または希釈剤とを含む医薬組成物は、経口投与用の固形形態(典型的には錠剤またはカプセル剤)などの経口投与用の形態として、または経口投与用の液体形態として投与できる。この組成物は、速放性剤形の形態または制御放出剤形もしくは持続放出剤形の形態で投与できる。
【0064】
固形または液体の医薬調製物の調製の方法は、当技術分野で周知である。例えば、Remington、The Science and Practice of Pharmacy、第21版、Lippincott Williams & Wilkins (2005)(非特許文献13)を参照のこと。したがって、錠剤は、活性成分を、通常の担体(佐剤および/または希釈剤など)など当技術分野で公知の賦形剤と混合し、次いで、打錠機中でその混合物を圧縮することにより調製できる。佐剤および/または希釈剤の非限定例としては、トウモロコシ・デンプン、乳糖、マンニトール、リン酸カルシウム、微結晶性セルロース、タルク、ステアリン酸マグネシウム、ゼラチン、ゴムなどが挙げられる。活性成分と適合することを条件に、着色剤、アロマおよび保存剤など任意の他の佐剤または添加剤を使用してもよい。
【0065】
本明細書において引用および検討する全ての非特許参考文献、特許および特許出願は、参照によりその全体が、また、それぞれが参照により個別に組み込まれるのと同程度に、本明細書中に組み込まれる。
【0066】
参考文献リスト
1. Boll, M. et al. (2004) Pflugers.Arch. Vol. 447. 5. 776-779
2. Boll, M. et al. (2002) J Biol Chem. Vol. 277. 25. 22966-22973
3. Burckhardt, B. C. et al. (2003) Rev Physiol Biochem Pharmacol. Vol. 146. 95-158
4. Chen, Z. et al. (2003) J Physiol. Vol. 546. Pt 2. 349-361
5. Gabrielsson, J. and Weiner, D. (2007) Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications, 4th ed., CRC Press, Baco Raton, FL ISBN 978-9-1976-5100-4
6. Koepsell, H. et al. (2004) Pflugers.Arch. Vol. 447. 5. 666-676
7. Lucas, M. L. et al. (1975) Proc R.Soc Lond.B Biol Sci. Vol. 192. 1106. 39-48
8. Metzner, L. et al. (2006) Amino.Acids. Vol. 31. 2. 111-117
9. Pavlova, A. et al. (2000) Am.J Physiol Renal Physiol. Vol. 278. 4. F635-F643
10. Pritchard, J. B. et al. (1999) J Biol Chem. Vol. 274. 47. 33382-33387
11. Rizwan, A. N. et al. (2007) Pharm.Res. Vol. 24. 3. 450-470
12. Sekine, T. et al. (2006) Am.J Physiol Renal Physiol. Vol. 290. 2. F251-F261
13. Wright, S. H. et al. (2004) Am.J Physiol Renal Physiol. Vol. 287. 3. F442-F451
【実施例】
【0067】
例1
この例では、ビーグル犬において実施した試験で得られたデータについて記載する。
【0068】
材料
4,5,6,7−テトラヒドロイソキサゾロ[5,4−c]ピリジン−3−オール塩酸塩/C6H8N2O2,HCl、(ガボキサドール塩酸塩)および第二置換体(deuto substituted form)C6H4D4N2O2、HCL、(第二ガボキサドール塩酸塩(deuto−gaboxadol hydrochloride))は、H.Lundbeckから供給された。5−ヒドロキシ−L−トリプトファン(5−HTP)、L−トリプトファン(Trp)、L−プロリン(Pro)、アセトニトリル(ACN)およびメタノールは、Sigma−Aldrich(St.Louis、MO、USA)から入手した。酢酸はMERCK製であった。ヘパリン、5000IE/a.e./mlは、LEO(Ballerup、Dk)から購入した。
【0069】
方法
in vivo試験
試験開始に先立ち、デンマーク法務省により任命された動物愛護委員会(Animal Welfare Committee)によりプロトコールの承認を受け、全ての動物の手術は、EC指令86/609/EEC、動物実験を規制するデンマークの法律、ならびに、実験動物の管理および使用のためのNIHガイドライン(NIH Guidelines for the Care and Use of Laboratory Animals)に従って実施した。十分成長した6匹のオスのビーグル犬(体重15.9〜21.7kg)を選び、ローマ式の4分割デザイン(roman quadrant design)に配置し、ガボキサドール塩酸塩製剤全6種をランダムに、6週間にわたり投与するように割り付けた。イヌは、実験開始前20〜24時間絶食させ、投与10時間後に給餌を再開した。ガボキサドール1回分を、静脈内注射(1.0ml/kg)として、または軟らかいチューブを用いた胃の中への直接経口強制投与(5.0ml/kg)により与えられる経口溶液として、そのいずれかで投与した。全てのイヌに2.5mg/kgのガボキサドールを投与した。2つの化合物の同時併用投与を確実にするために、経口製剤は、ガボキサドールに加え、0mg/kg、2.5mg/kg、10.0mg/kg、50.0mg/kgまたは150.0mg/kgのトリプトファンを含有していた。全ての溶液をpH5.2に調節し、Vapro蒸気圧浸透圧計(モデル552O、Wescor Inc.、Logan、UT、USA)を用いて浸透圧モル濃度を調べ、グルコースを用いて静脈内用の溶液を等モル浸透圧濃度(isoosmolarity)に調節した。個別の静脈穿刺により、橈側皮静脈から血液試料2.0mlを採取し、抗凝血剤として200IEヘパリンを含有するエッペンドルフ管中に収集した。試料は、ガボキサドール投与前、ならびに、ガボキサドール投与の5分後、15分後、30分後、60分後、90分後および2時間後、3時間後、4時間後、6時間後、8時間後および10時間後に収集した。15分間、2200g、4〜8℃での遠心分離により速やかに血漿を採取し、−80℃で保管してからさらなる分析を実施した。ガボキサドールの各投与日の後、動物には6日のウォッシュアウト期間を与えた。
【0070】
WinNonlinで薬物動態(PK)を評価した。静脈内投与した動物の血漿濃度曲線を2コンパートメント・モデルに当てはめ、経口投与した動物のデータは、非コンパートメント・モデルにおいて分析した。統計分析はSigma Statで行った。
【0071】
HPLCおよびMS/MS検出による定量分析。
液体抽出により、血漿およびHBSS+試料からガボキサドールを抽出した。HBSS+100μl(試料20μlに精製水80μlを加えた)または血漿試料100μlを内部標準(intern standard)(d4−ガボキサドール)25μlおよび精製水25μlと混合した。冷アセトニトリル400μlの添加により、タンパク質沈降を実施した。10,000gで15分間の遠心分離の後、上澄み425μlをガラス管に移し、蒸発させて、窒素下、45℃で乾燥させた。試料をメタノール/アセトニトリル(30:70)80μl中で分離し、10分間回転混合して、3000rpmで3分間遠心分離してから、培地用のウェル・プレートに移し、10℃のオートサンプラー中に置いた。次いで、親水性の相互作用クロマトグラフィー(HILIC−クロマトグラフィー)により抽出試料中のガボキサドール濃度を定量化し、その後、MS/MS検出を行った。LCシステムは、Agilent1100シリーズのポンプ付き脱気装置で構成され、CTC Analyticsのインターフェースからコンピューターにデータが転送され、試料はPeltier ThermostatおよびHTC Palオートサンプラーで操作した。クロマトグラフィー分離用にはPhenomenex製のAsahipakアミノ・カラム、(NH2P−50、150×2mm)を使用し、移動相は20.0mM酢酸アンモニウムpH=4:アセトニトリル(30:70)、流速0.2ml/minとした。試料20μlをカラムに注入し、これを室温で維持した。合計通液時間は10分であり、最初の5分の溶出分は廃棄するようにした。カラムでのガボキサドールの溶出時間はおよそ8分であった。使用したMS/MSシステムは、Turbo Ion SprayおよびTurbo V源を装備したSciex API4000MS/MS検出装置(Applied Biosystems)から成るものであった。陰イオン化モードで検出を実施し、多重反応モニタリング(MRM)により、ガボキサドール(前駆物質139.1Da、生成物110.1Da)およびd4−ガボキサドール(前駆物質143.0、生成物112.2Da)を測定した。シグナルは、0.5〜2500.0ng/mlの間の線形であり、この手順による定量化の限界は0.5ng/mlであった。ソフトウェアは、Analyst(商標)のものであった(Applied Biosystem、バージョン4.0)。
【0072】
結果および考察
血漿濃度対時間プロファイルを図1および2に示す。
【0073】
表1.例1において動物から得られた薬物動態パラメーター。1kg当たりガボキサドール2.5mgをIV(A)投与およびPO(B〜F)投与。データは、平均値±SEM、n=6を表す。
【表1】
【0074】
イヌにおける経口投与後のガボキサドールのバイオアベイラビリティーFaは、85.3±5.7%であることがわかった(表1)。2.5〜150mg/kgトリプトファンの経口併用投与では、ガボキサドールのAUCは有意に変化せず、この製剤の平均の相対バイオアベイラビリティーは、75.0%(10mg/kgトリプトファン)〜86.1%(2.5mg/kgトリプトファン)の間で変動した。同様に、ガボキサドールの排出速度定数(ke)およびクリアランス(CL)は、トリプトファンの併用投与によって変化しなかった。しかし、トリプトファン併用投与により、ガボキサドールの最大血漿濃度Cmaxは、150mg/kgトリプトファンの不在下および存在下では、2502ng/mlから1419ng/mlへ57%低下した(p<0.001)。さらに、最大血漿濃度への到達に要する時間Tmaxは、0.46時間から1.5時間に増えた(p<0.01)。5つの投与群のCmax値の変化は、ガボキサドールとトリプトファンとの間に直接の相互作用があることを明確に示している。
【0075】
これらのデータに基づけば、Trpはガボキサドールの吸収プロファイルに影響を及ぼすことが明らかである。この効果には、PAT1輸送体と相互作用する2つの化合物が介在する、すなわち、Trp用量が高い状況下では、結合部位の多くがTrpにより占められるのと同様、ガボキサドールは、PAT1により輸送できないと考えられる。PAT1を阻害するか、またはPAT1の基質である化合物を併用投与すると、結果としてガボキサドールの吸収プロファイルを調節できる。
例2
【0076】
この例では、ラットにおいて実施した試験から得られたデータを記載する。
【0077】
材料
例1と同じ
【0078】
実験方法
以下を除き、例1と同じ:
経口製剤
ガボキサドール0.05mgまたは0.5mgならびに5−HTP 0.0mgまたは20.0mgを精製水(1ml当たり)に室温で溶解し、超音波下で10分間、氷の上に置いた。この製剤をNaOH/HClでpH4〜5に調節し、マンニトールを加えて等張にした。全ての溶液のpHを、pH4.0超5.0未満に調節し、マンニトールを加えて浸透圧モル濃度を280mmol/kgに調節した。
in vivoでの実験
220〜240グラムのオスのSprague−Dawleyラット(Charles River Laboratories、Wilmington、MA、USA)を収容し、実験に入る前に7日間馴化した。投与の16〜20時間前まではラットを標準食および水で維持し、実験を実施する前に完全な胃内容排出を確実にするために、この時点で食餌を回収した。水は、実験開始まで、また、2時間後に再度、動物が摂取可能であった。各動物は、静脈内製剤または経口製剤のいずれか一方を摂取するようにランダムに割り付けた。
【0079】
ラットの6つの並行群(n=6)に、0.5mg/kgまたは5.0mg/kgのガボキサドールの等張溶液を、生理食塩水または200.0mg/kg 5−HTPと共に、経口強制投与により投与した(10.0ml/kg)。ガボキサドール溶液の30分前に、5−HTPの懸濁液をプレ・インキュベーションとして30分投与した。
【0080】
個別の静脈穿刺により、尾静脈から血液試料0.2mlを採取し、20IEヘパリンを含有する血漿収集チューブ中に収集した。試料は、ガボキサドール投与後5分、15分、30分、45分、60分の時点、および、2時間後、3時間後、4時間後、6時間後、8時間後に収集した。3.600gで10分間の遠心分離により血漿を速やかに採取し、−80℃で保管してから、さらなる分析を実施した。実験の終了時に、動物を安楽死させた。
【0081】
結果および考察
血漿濃度対時間プロファイルを図3に示す。
【0082】
表2.例2において動物から得られた薬物動態パラメーター、1kg当たりガボキサドール0.5mgおよび5.0mgのPO投与。データは、平均値±SEM、n=6を表す。
【表2】
【0083】
図3および表2において見られるように、経口投与後のガボキサドールの吸収は、大部分は投与後15〜20分以内に生じたが、その理由は、血漿ガボキサドール濃度がこの期間中に増加したからである。15〜20分後、ガボキサドールの排出が増加していくと、血漿ガボキサドール濃度は低下した。1kg当たりガボキサドール0.5mgの前にプレ・インキュベーションとして1kg当たり5−HTP 200mgを投与すると、血漿ガボキサドール濃度ピーク時間(Tmax)は、16分から63分へと遅くなった。ラットに1kg当たりガボキサドール5.0mgを投与すると、5−HTPでのプレ・インキュベーション後、Tmaxは20分から43分へと遅くなった。最大血漿濃度Cmaxは、5−HTPのプレ・インキュベーションにより変化しないようであった。
【0084】
動物にガボキサドールおよびPAT1阻害薬5−HTPを投与すると、AUCは、対照動物(5−HTP非投与)と比較して増加した。5−HTP(200mg/kg)投与は、ガボキサドール(5.0mg/kgまたは0.5mg/kg)投与の40倍または400倍高く、AUCは対照群と比較して330%および540%増加した。AUCは、排出速度の低下により増加し得る。ガボキサドール排出速度定数は、5−HTPが存在する場合には約25%に低下した。
【0085】
まとめると、ガボキサドールの吸収は、PAT阻害薬5HTPの併用投与により変化するようである。さらに、ガボキサドールの排出は、PAT、OAT、または、5−HTPが相互作用する他の輸送体との相互作用に影響されるようである。
例3
【0086】
材料
例1と同じ
【0087】
実験方法
以下を除き、例2と同じ:
静脈内製剤
ガボキサドール0.25mgならびに5−HTP 0.0mgまたは10.0mgを精製水(1ml当たり)に室温で溶解し、超音波下で10分間、氷の上に置いた。静脈内注射に使用するガボキサドール溶液を、0.45μmのフィルターを通して濾過した。
【0088】
2.5mg/kgガボキサドールの尾静脈中への静脈内注射の30分前に、経口強制投与により、動物に100.0mg/kg 5−HTPまたは生理食塩水を投与した(5.0ml/kg)。
【0089】
結果および考察
血漿濃度対時間プロファイルを図4に示す。
【0090】
表3.例3において動物から得られた薬物動態パラメーター、1kg当たりガボキサドール2.5mgのIV投与。データは、平均値±SEM、n=6を表す。
【表3】
【0091】
5−HTPでプレ・インキュベートしたIV群(L群)の血漿プロファイルは、ガボキサドールのみを投与された群(K群)のラットのものと異なった。K群のAUCはL群のほぼ3倍の大きさであり、それはおそらく、排出速度定数Keがより小さいことが原因であった(表3)。こうした結果から、5−HTPは、OATまたは他の輸送体との相互作用によりガボキサドールのクリアランスを妨げることが示唆される。
【0092】
G群(1kg当たりガボキサドール0.5mg)のAUCがK群(1kg当たりガボキサドール2.5mg)の5倍の大きさであり、また、J群(5mg/kg)のAUCがG群のほぼ10倍の大きさであったように、ガボキサドールについてはAUC−用量の線形性が観察された。
【技術分野】
【0001】
本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。本発明は、さらに、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物に関する。
【背景技術】
【0002】
ガボキサドール(4,5,6,7−テトラヒドロイソキサゾロ[5,4−c]ピリジン−3−オール)(THIP)は、欧州特許第0000338号(特許文献1)および欧州特許第0840601号(特許文献2)に記載されており、これまでに、睡眠障害の治療において、また、うつ病の前臨床モデルにおいて大きな可能性が示されている(国際公開第2004112786号(特許文献3))。ガボキサドールは、以下の一般式:
【0003】
【化1】
を有する。
【0004】
ガボキサドールは、当技術分野で周知の方法を用いて調製できる。例えば、欧州特許第0000338号(特許文献1)および国際公開第2005023820号(特許文献4)に開示されているように調製できる。
【0005】
国際公開第02094225号(特許文献5)では、ガボキサドールを含有し速放性のプロファイルを有する成形された固形の医薬単位剤形の調製に使用できる、顆粒状のガボキサドール含有調製物が開示されている。
【0006】
WO0122941(特許文献6)では、ガボキサドールを含有する溶融造粒組成物(melt granulated composition)、および前記組成物から調製される放出調節剤形が開示されている。
【0007】
ガボキサドール速放性製剤の治療的投与においては、迅速に溶解する結果、投与後間もなく血漿ガボキサドールレベルが急速に高まり、次いで、ガボキサドールが代謝されるかまたは排出されるにつれ数時間にわたり血漿レベルが低下した後、治療的レベルを下回る血漿レベルに達する。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】欧州特許第0000338号
【特許文献2】欧州特許第0840601号
【特許文献3】国際公開第2004112786号
【特許文献4】国際公開第2005023820号
【特許文献5】国際公開第02094225号
【特許文献6】国際公開第0122941号
【非特許文献】
【0009】
【非特許文献1】Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁
【非特許文献2】Lucas, M. L.ら、1975、Proc R.Soc Lond.B Biol Sci.、192巻、1106、39〜48頁
【非特許文献3】Boll, M.ら、2002、J Biol Chem.、277巻、25、22966〜22973頁
【非特許文献4】Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁
【非特許文献5】Koepsell, H.ら、2004、Pflugers.Arch.、447巻、5、666〜676頁
【非特許文献6】Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁
【非特許文献7】Burckhardt, B. C.ら、2003、Rev Physiol Biochem Pharmacol.、146巻、95〜158頁
【非特許文献8】Pritchard, J. B.ら、1999、J Biol Chem.、274巻、47、33382〜33387頁
【非特許文献9】Pavlova, A.ら、2000、Am.J Physiol Renal Physiol.、278巻、4、F635〜F643頁
【非特許文献10】Sekine, T.ら、2006、Am.J Physiol Renal Physiol.、290巻、2、F251〜F261頁
【非特許文献11】Wright, S. H.ら、2004、Am.J Physiol Renal Physiol.、287巻、3、F442〜F451頁
【非特許文献12】Gabrielsson and Weiner、2007、Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications、第4版、CRC Press、Baco Raton、FL ISBN 978-9-1976-5100-4
【非特許文献13】Remington、The Science and Practice of Pharmacy、第21版、Lippincott Williams & Wilkins (2005)
【発明の概要】
【発明が解決しようとする課題】
【0010】
薬理学的および生理学的なプロセスの中には、最適な治療効果に到達するために、治療上適切な血漿レベルでの長期にわたる暴露を要すると考えられるものがある。そのため、治療上適切な血漿レベルで長期にわたり暴露することが可能なガボキサドールの医薬剤形が必要である。さらに、Tmaxがより遅くおよび/またはCmaxが低下しており、加えて、場合によりAUCが増加している血漿プロファイルを提供するガボキサドールの医薬剤形が必要である。
【0011】
今回、驚くべきことに、ガボキサドールの吸収を変化させ、それにより、ピーク濃度を最低限とし、Tmaxを延長し、特別な状況においてはさらに薬物動態プロファイルの排出相(elimination phase)を延長する(すなわち、AUCを高める)ことが実証されたガボキサドール製剤を調製することが可能であることが見出された。
【課題を解決するための手段】
【0012】
一態様では、本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。
【0013】
別の態様では、本発明は、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物に関する。
【図面の簡単な説明】
【0014】
【図1】イヌにおけるIV投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。2.5mg/kgガボキサドール(A、(△))の静脈内注射後の血漿ガボキサドール濃度対時間プロファイル。血液試料は、薬物投与後5分、15分、30分、60分、90分、120分、180分、240分、360分、480分および600分の時点で収集した。示してあるのは、6匹のイヌ(n=6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【図2】イヌにおけるPO投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。2.5mg/kgガボキサドール(B、(□))のPO投与後のビーグル犬の血漿ガボキサドール濃度対時間プロファイル。同じ用量を、2.5mg/kg Trp(C、(▲))、10.0mg/kg Trp(D、(×))、50.0mg/kg Trp(E、(○))および150.0mg/kg Trp(F、(■))との併用でも投与した。Trpは、ガボキサドールと同じ時点での併用投与として投与した。試料は、薬物投与後5分、15分、30分、60分、90分、120分、180分、240分、360分、480分および600分の時点で収集した。示してあるのは、6匹のイヌ(n=6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【図3】ラットにおけるPO投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。ガボキサドールのPO投与後のラットの血漿ガボキサドール濃度対時間プロファイル。投与した用量は、0.5mg/kg(G、(○))もしくは5.0mg/kg(H、(●))のガボキサドール単独、または、200.0mg/kg 5−HTPのプレ・インキュベーションとの併用、それぞれ(I、(□))もしくは(J、(■))であった。200.0mg/kg 5−HTPは、ガボキサドールの前30分のプレ・インキュベーションとして投与した。試料は、薬物投与後5分、15分、30分、45分、60分、120分、240分、360分および480分の時点で収集した。示してあるのは、5〜6匹のラット(n=5〜6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【図4】ラットにおけるIV投与後の血漿ガボキサドール濃度対時間プロファイルを示すグラフである。ガボキサドールのIV投与後のラットの血漿ガボキサドール濃度対時間プロファイル。ラットは、2.5mg/kgガボキサドール(K、(△))の静脈内注射を受けた。同じ用量を、200.0mg/kg 5−HTP(L、(▲))との併用でも投与した。200.0mg/kg 5−HTPは、ガボキサドールの前30分のプレ・インキュベーションとして投与した。試料は、薬物投与後5分、15分、30分、45分、60分、120分、240分、360分および480分の時点で収集した。示してあるのは、5〜6匹のラット(n=5〜6)の平均値±S.E.M.である。Y軸:血漿ガボキサドール濃度(ng/ml)。X軸:時間(分)。
【発明を実施するための形態】
【0015】
本発明者らは、原発性不眠症に罹患している一部の患者においてガボキサドールの速放性製剤を治療的投与すると、用量依存的な有害事象が生じることを見出した。観察されたこの有害事象は、平均Cmaxとほぼ同時に発生し、投与後数時間後に消失したことから、この有害事象はCmaxと相関がある。ガボキサドールの速放性製剤を用いた場合に観察された有害事象としては、眩暈、悪心、嘔吐、傾眠、振戦、倦怠、鎮静およびいくつかの精神医学的な有害事象が挙げられる。有害事象のさらなる分析により、本発明者らは、平均Cmaxを低下させることにより、および/または、平均Tmaxを長くすることにより、こうした有害事象は稀で穏やかになり、精神医学的な有害事象は生じないことを見出した。
【0016】
本発明者らは、ガボキサドールおよび1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物を調製して、ガボキサドールの吸収調節製剤を提供することが可能であることを見出した。本発明によれば、この医薬組成物中で使用されるガボキサドール、1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬の量を変化させることにより、ガボキサドールのCmax、Tmaxおよび場合によりAUCを調節することが可能である。本発明による組成物は、以下の利点の1つまたは複数をもたらす:血漿ガボキサドールレベルの急速な上昇を回避または減少できる、より遅いTmaxおよび/または低下したCmaxを有するガボキサドールの薬物動態プロファイルが達成でき、このプロファイルには、場合によりAUCの増加を加えることができる。したがって、以下の課題の1つまたは複数は、本発明により解決できる:治療上適切な血漿レベルに達しながら血漿ガボキサドールレベルの急速な増加に伴う影響を回避または減少できる、および/または治療上適切な血漿レベルはより長期間にわたり維持されるため、ガボキサドール投与間の時間間隔を速放性製剤と比較して長くすることができる。したがって、本発明によれば、ガボキサドールを含む医薬組成物であって、大部分の有害事象を伴う血漿レベルに達することなく、また、状況によっては長期間にわたり治療上適切な血漿レベルに達することが可能な医薬組成物が提供される。
【0017】
したがって、本発明は、ガボキサドールまたは薬学上許容可能なその塩を含む医薬組成物であって、ガボキサドールの速放性製剤と比較して平均Cmaxを低下させながらも治療上適切な血漿ガボキサドールレベルを実現する組成物に関する。
【0018】
いかなる特定の理論により拘束されるものでもないが、本発明による組成物中のPAT1阻害薬は、消化管からのガボキサドールの吸収速度を低下させることにより、ガボキサドールの吸収を調節するとの仮説が立てられる。さらには、いくらかの、おそらくは全てのPAT1阻害薬、およびOAT基質またはOAT阻害薬は、腎臓中の1つまたは複数の有機陰イオン輸送体(OAT)と相互作用し、および/または腎臓の血流を減少させ、それにより腎臓からのガボキサドールの排出速度も低下させることにより、血漿ガボキサドールレベルを、より長期間にわたり治療上適切なレベルにするとの仮説も立てられる。
【0019】
ヒトプロトン依存性アミノ酸輸送体1(hPAT1)は、2003年にCaco−2細胞からクローニングされた(Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。この輸送体は、溶質キャリアーファミリー(solute carrier family)SLC36に属し、4つのうちの第一のもの(SLC36A1)である。PAT3およびPAT4はオーファン輸送体であるが、PAT2は、肺、心臓、腎臓、筋肉、精巣、脾臓、副腎、胸腺および坐骨神経の組織中で主に発現する。分析から、ヒト組織においてhPAT1のmRNAが遍在性に発現することが発見されており、このmRNAはヒト消化管全体にわたって検出されており小腸で最も多く発現していることから、この輸送体は、腸管の孔の長さで基質を吸収するのに適切なものとなっている(Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。hPAT1経由でのアミノ酸輸送は、腸の中の酸性の微小気候により、頂端膜を隔てて構築されるプロトン(H+)の顕著な濃度勾配により活発化する(Lucas, M. L.ら、1975、Proc R.Soc Lond.B Biol Sci.、192巻、1106、39〜48頁(非特許文献2))。
【0020】
Caco−2細胞株は、ヒト小腸の上皮のモデルとして使用できる。プロトン依存性アミノ酸輸送体は、これまでに、この生体外モデルにおいて徹底的に、また、ある程度までは、トランスフェクトされた細胞系においても特徴付けられている(Boll, M.ら、2002、J Biol Chem.、277巻、25、22966〜22973頁(非特許文献3)、Chen, Z.ら、2003、J Physiol.、546巻、パート2、349〜361頁(非特許文献1))。競合アッセイならびに転位実験により、PAT1との相互作用について多様な化合物が試験されている。こうしたin vitroでの特徴付けによれば、PAT1基質は、(21〜28日目齢)Caco−2細胞の単層を超えて輸送され、膜を隔てたpH勾配に伴ってフラックスが増加する化合物を指す。さらに、高濃度の別のPAT1基質(L−プロリンであってもよいと考えられるが、これに限定されない)を加えることにより、この輸送は阻害されるはずである。
【0021】
PAT1阻害薬は、Caco−2細胞の単層を超えてのPAT1基質の輸送を減少させる化合物を指す。この阻害薬は、基質ポケット中の輸送体と結合するか否かにより、競合的または非競合的な様式で作用することができる。
【0022】
古典的なPAT1基質は、β−アラニンおよびAIB(α−(メチルアミノ)−イソ酪酸)のようないくつかのβ−アミノ酸に加えて、グリシン、アラニン、セリンおよびプロリンのような小型で非分枝状の双性イオン性α−アミノ酸、ならびに、GABA(γ−アミノ酪酸)のような少数のγ−アミノ酸である(Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁(非特許文献4))。生体異物の中には、hPAT1基質、例えば神経調節性かつ抗菌性の物質であるD−シクロセリンに囲まれていることが実証されているものがある。さらに、いくつかのGABA受容体遮断薬および再取込み阻害薬、ならびに、癌および線維性疾患の治療に用いられるプロリン類似体も、PAT1により輸送される(Metzner, L.ら、2006、Amino.Acids.、31巻、2、111〜117頁(非特許文献4))。
【0023】
可能性のある競合的なPAT1阻害薬としては、グリシン、L−アラニン、D−アラニン、L−セリン、D−セリン、L−プロリン、D−プロリン、GABA(γ−アミノ酪酸)、サルコシン、ベタイン、N−メチル−L−アラニン(AIB(α−(メチルアミノ)−イソ酪酸))、D−シクロセリン、β−アラニン、ビガバトリン、グバシン、TACA(トランス−4−アミノクロトン酸)が挙げられるが、これらに限定されない。
可能性のあるPAT1阻害薬は、5−ヒドロキシトリプトファン(5−HTP)、セロトニン(5−HT)、L−トリプトファン(Trp)、トリプタミン、インドール−3−プロピオン酸であってもよいと考えられるが、これらに限定されない。
【0024】
有機陰イオン輸送体(OAT)は、1997年に同定された。この輸送体は、SLC22遺伝子ファミリーに属し(Koepsell, H.ら、2004、Pflugers.Arch.、447巻、5、666〜676頁(非特許文献5))、顕著かつ広範な基質特異性を特徴とする。現時点で公知の輸送体としては、腎臓内に主に局在するOAT1−4およびURAT1が挙げられる(Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁(非特許文献6))ことから、いくつかの刊行物では、生体異物および薬物の腎臓分泌に対するこの輸送体の寄与に注目している(総説については、Burckhardt, B. C.ら、2003、Rev Physiol Biochem Pharmacol.、146巻、95〜158頁(非特許文献7)を参照のこと)。脳内、特に脈絡膜叢および血液脳関門(Pritchard, J. B.ら、1999、J Biol Chem.、274巻、47、33382〜33387頁(非特許文献8))、胚の発生の異なる段階での目、骨格筋およびいくつかの臓器(Pavlova, A.ら、2000、Am.J Physiol Renal Physiol.、278巻、4、F635〜F643頁(非特許文献9))内においても発現が報告されている。OATは、基質転位の活発化にATP加水分解を直接には利用しない。OATファミリーのうち全てではなくても大部分のものは、陰イオン交換体として作用する。すなわち、そうした輸送体は、細胞中への有機陰イオンの取込みを、細胞からの別の有機陰イオンの放出と結び付ける。それにより、OATは、陰イオン(例えば、α−ケトグルタル酸イオン、乳酸イオンおよびニコチン酸イオン)の既存の細胞内>細胞外勾配を利用して、負の膜電位に逆らって、有機陰イオンの上り坂取込み(uphill uptake)を駆動する。腎臓の近位尿細管においては、OATは、乳酸イオン、ニコチン酸イオンおよびα−ケトグルタル酸イオンの細胞内>細胞外勾配を構築および維持するNa+駆動型のモノカルボン酸輸送体およびジカルボン酸輸送体と機能的に結び付く(Rizwan, A. N.ら、2007、Pharm.Res.、24巻、3、450〜470頁(非特許文献6))。
【0025】
典型的なOAT基質の分子量は最大400〜500であり(Sekine, T.ら、2006、Am.J Physiol Renal Physiol.、290巻、2、F251〜F261頁(非特許文献10)、Wright, S. H.ら、2004、Am.J Physiol Renal Physiol.、287巻、3、F442〜F451頁(非特許文献11))、その特異性は、以下を非限定的に含むOATにより輸送されている化学構造体という点で非常に広範である:キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン(carboxyflurescein)、ベンジルペニシリン、セファドロキシル、セファマドール(Cefamadole)、セファゾリン、セフォペラゾン、セフォタミム(Cefotamime)、セファレキシン、セファロチン、セフラジン、アシロビル(Acylovir)、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル(Valacylovir)、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク(Dilofenac)、ジフルシナル(Diflusinal)、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート(Mefanamate)、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダク(Sulidac)。本明細書では、OAT基質は、OATのmRNAをトランスフェクトされた卵母細胞中に、制御状況(control situation)と比較して顕著に増加した速度で輸送される化合物により定義される。
【0026】
定義
Cmaxは、実験中に予測される、血漿中薬物の最大濃度(ng*ml1)と定義される。Tmaxは、Cmaxに達すると予測される時間(min)と定義される。AUCは、薬物投与から薬物が排出するまでの、血漿薬物濃度−時間曲線下の総面積(ng*min*ml−1)である。曲線下面積は、クリアランスに支配される。クリアランスは、含有された薬物が完全に取り除かれている血液または血漿の単位時間当たりの体積(ml*h−1*kg−2)と定義される。単位時間当たりに排出される体内薬物量に関する排出速度定数は、薬物が排出される速度(h−1)と定義される(Gabrielsson and Weiner、2007、Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications、第4版、CRC Press、Baco Raton、FL ISBN 978-9-1976-5100-4(非特許文献12))。
【0027】
用語「PK」は、薬物動態プロファイルを指す。
【0028】
本明細書中で使用する場合、用語「対象」は、ヒトおよび動物など任意の温血種を指す。ガボキサドールで治療されることになるヒトなどの対象は、実際には、ヒト集団、男性または女性の任意の対象であってもよく、小児、成人または高齢者に分けてもよい。このような患者群の任意の1つは、本発明の一実施形態に関する。
【0029】
本明細書中で使用する場合、用語「治療する(こと)」または「治療」は、疾患もしくは状態を患っているまたは疾患もしくは状態に罹りやすいと考えられるが当該疾患もしくは状態の臨床症状もしくは潜在症状を未だ経験もしくは示していない対象において当該疾患もしくは状態の臨床症状の出現を予防または遅延させることを指す。「治療する(こと)」または「治療」は、疾患もしくは状態を阻害すること、すなわち、その発症、または、その少なくとも1つの臨床症状もしくは潜在症状を抑止または低減することも指す。「治療する(こと)」または「治療」は、さらに、疾患もしくは状態を緩和すること、すなわち、疾患もしくは状態またはその臨床症状もしくは潜在症状の少なくとも1つの軽減をもたらすことを指す。治療されることになる対象の利益は、統計的に有意であるか、あるいは、対象および/または医師に少なくとも知覚できるものであるか、そのいずれかである。但し、予防的(防止的)および治療的(治癒的)な治療は、本発明の2つの独立した実施形態である。
【0030】
本明細書中で使用する場合、用語「薬学上許容可能な」は、「一般に安全とみなされる」分子体(molecular entity)および組成物、例えば、生理学的に忍容でき、ヒトに投与した際、胃の不調など、アレルギー反応または同様の有害な反応を典型的に生じさせないものを指す。別の実施形態では、この用語は、FDAによる市販前審査(premarket review)および認可を受けている、連邦食品医薬品化粧品法の第204条(s)項および409条の下のGRASリストまたは同様のリスト、動物、より詳細にはヒトにおける使用についての米国薬局方または別の一般に認識された薬局方として、連邦政府または州政府の規制当局により認可された分子体および組成物を指す。
【0031】
第1の態様によれば、本発明は、ガボキサドールまたは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物に関する。本発明の第1の態様の一実施形態では、この組成物は、1つまたは複数のPAT1阻害薬を含むが、OAT阻害薬を含まない。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数のOAT阻害薬を含むが、PAT1阻害薬を含まない。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数のPAT1阻害薬および1つまたは複数のOAT阻害薬を両方とも含む。本発明の第1の態様の別の実施形態では、ガボキサドールは、酸付加塩、または双性イオン水和物(zwitter ion hydrate)もしくは双性イオン無水物(zwitter ion anhydrate)の形態である。本発明の第1の態様の別の実施形態では、ガボキサドールは、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である。本発明の第1の態様の別の実施形態では、ガボキサドールの量は、0.5mg〜50mgの範囲である。本発明の第1の態様の別の実施形態では、この組成物は、経口投与用の形態である。本発明の第1の態様の別の実施形態では、この組成物は、錠剤またはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である。本発明の第1の態様の別の実施形態では、前記ガボキサドールは結晶性である。本発明の第1の態様の別の実施形態では、PAT1はヒトPAT1である。本発明の第1の態様の別の実施形態では、PAT1阻害薬は、5−ヒドロキシトリプトファン(5−HTP)、L−プロリン、D−プロリン、サルコシン、L−アラニン、D−アラニン、N−メチル−L−アラニン、N−メチル−D−アラニン、α−(メチルアミノ)−イソ酪酸、ベタイン、D−シクロセリン、L−シクロセリン、β−アラニン、セロトニン、L−トリプトファン、D−トリプトファン、トリプタミン、インドール−3−プロピオン酸から選択される。本発明の第1の態様の別の実施形態では、PAT1阻害薬の量は、約0.5〜約3000mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約750mg、約1000mg、約1250mg、約1500mg、約1750mg、約2000mg、約2250mg、約2500mg、約2750mgまたは約3000mgである。本発明の第1の態様の別の実施形態では、OATはヒトOATである。本発明の第1の態様の別の実施形態では、OAT阻害薬は、キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン、ベンジルペニシリン、セファドロキシル、セファマドール、セファゾリン、セフォペラゾン、セフォタミム、セファレキシン、セファロチン、セフラジン、アシロビル、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク、ジフルシナル、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダクから選択される。本発明の第1の態様の別の実施形態では、OAT阻害薬の量は、約0.5〜約500mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mgまたは約500mgである。本発明の第1の態様の別の実施形態では、この組成物は、1つまたは複数の賦形剤を含む。本発明の第1の態様の別の実施形態では、この組成物は、セロトニン再取込み阻害薬である化合物、または細胞外のセロトニンレベルの上昇をもたらす任意の他の化合物を含む。本発明の第1の態様の別の実施形態では、セロトニン取込み阻害薬は、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミンまたはこれらの化合物のいずれかの薬学上許容可能な塩から選択される。本発明の第1の態様の別の実施形態では、セロトニン取込み阻害薬は、塩基として、またはシュウ酸塩、臭化水素酸塩もしくは塩酸塩など薬学上許容可能なその塩としてのエスシタロプラムである。
【0032】
第2の態様によれば、本発明は、ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを示す組成物に関する。本発明の第2の態様の一実施形態では、前記平均Tmaxは、約25分超、30分超、35分超、40分超、45分超、50分超、55分超、60分超、65分超、70分超または75分超である。本発明の第2の態様の別の実施形態では、この組成物は、約2250ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、前記平均Cmaxは、約2000ng/ml未満、約1750ng/ml未満、約1500ng/ml未満、約1250ng/ml未満、約1000ng/ml未満、約750ng/ml未満、約500ng/ml未満、約250ng/ml未満、約200ng/ml未満または約100ng/ml未満である。本発明の第2の態様の別の実施形態では、この組成物は、約8.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、前記平均AUC0〜∞は、約16.000ng・min・ml−1超、20.000ng・min・ml−1超、40.000ng・min・ml−1超、80.000ng・min・ml−1超、120.000ng・min・ml−1超または200.000ng・min・ml−1超である。本発明の第2の態様の別の実施形態では、クリアランスは40ml/min未満である。本発明の第2の態様の別の実施形態では、前記クリアランスは、30ml/min未満、20ml/min未満、10ml/min未満または5ml/min未満である。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約2mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約100ng/ml未満の平均Cmaxおよび約8.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約4mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約200ng/ml未満の平均Cmaxおよび約16.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約5mgまたは薬学上許容可能なその塩を含み、約20分時間より長い平均Tmax、約250ng/ml未満の平均Cmaxおよび約20.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0033】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約10mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約500ng/ml未満の平均Cmaxおよび約40.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0034】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約20mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約1000ng/ml未満の平均Cmaxおよび約80.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0035】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約30mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約1500ng/ml未満の平均Cmaxおよび約120.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0036】
本発明の第2の態様の別の実施形態では、この組成物は、ガボキサドール約50mgまたは薬学上許容可能なその塩を含み、約20分超の平均Tmax、約2500ng/ml未満の平均Cmaxおよび約200.000ng・min・ml−1超の平均AUC0〜∞を含むin vivo血漿プロファイルを示す。
【0037】
本発明の第2の態様の別の実施形態では、クリアランスは40ml/min未満であり、AUCは200.000ng・min・ml−1より高い。
【0038】
本発明の第2の態様の別の実施形態では、前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞は、この組成物をイヌに投与した際に得られ、前記クリアランスは、この組成物をイヌまたはラットに投与した際に得られる。
【0039】
本発明の第2の態様の別の実施形態では、前記平均Tmaxは、約30分より長い。本発明の第2の態様の別の実施形態では、この組成物は、約300ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを示す。本発明の第2の態様の別の実施形態では、ガボキサドールの量は、約2.5mg、約5mgまたは約10mgから選択される。本発明の第2の態様の別の実施形態では、ガボキサドールの量は2.5mgであり、平均Cmaxは約40ng/ml未満、例えば、約35ng/ml、約30ng/ml、約25ng/mlまたは約20ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、ガボキサドールの量は5mgであり、平均Cmaxは約85ng/ml未満、例えば、約80ng/ml、約75ng/ml、約70ng/mlまたは約65ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、ガボキサドールの量は10mgであり、平均Cmaxは約150ng/ml未満、例えば、約145ng/ml、約140ng/ml、約135ng/mlまたは約130ng/mlであり、平均Tmaxは約1時間より長く、例えば、1.5時間、2時間または2.5時間である。本発明の第2の態様の別の実施形態では、前記平均Tmaxおよび平均Cmaxは、この組成物をヒトに投与した際に得られる。本発明の第2の態様の別の実施形態では、ガボキサドールは、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である。本発明の第2の態様の別の実施形態では、ガボキサドールは、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である。本発明の第2の態様の別の実施形態では、この組成物は、経口投与用の形態である。本発明の第2の態様の別の実施形態では、この組成物は、錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である。本発明の第2の態様の別の実施形態では、前記ガボキサドールは結晶性である。本発明の第2の態様の別の実施形態では、この組成物は、1つまたは複数の賦形剤を含む。
【0040】
本発明の一実施形態では、この医薬組成物は、ガボキサドールの速放性製剤の場合に観察されるCmaxの80%(75%、70%または65%など)に相当する平均Cmaxを示す。さらに本発明は、ガボキサドールまたは薬学上許容可能なその塩を含む医薬組成物であって、ガボキサドールの速放性製剤の場合に観察されるものより長い平均Tmaxを示しながらもなお治療上適切な血漿ガボキサドールレベルを提供する組成物に関する。
【0041】
さらなる一実施形態では、化合物は、PAT1およびOATを両方とも阻害する。
【0042】
本発明の組成物をイヌに投与した際に前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞が得られるさらなる一実施形態では、前記イヌはビーグルであり、前記ビーグルは、前記組成物の投与前20〜24時間(h)絶食させる。
【0043】
本発明の組成物をイヌに投与した際に前記クリアランスが得られるさらなる一実施形態では、前記イヌはビーグルであり、前記ビーグルは、前記組成物の投与前20〜24時間(h)絶食させる。
【0044】
本発明の組成物をラットに投与した際に前記クリアランスが得られるさらなる一実施形態では、前記ラットはオスのSprague−Dawleyラット(Charles River Laboratories、Wilmington、MA、USA)であり、前記ラットは、前記組成物の投与前16〜20時間までは、標準食および水で維持される。
【0045】
さらなる一実施形態では、本発明の医薬組成物は、原発性不眠症などの睡眠障害、または、大うつ病などのうつ病の治療用である。
【0046】
この明細書を通じ、「ガボキサドール」は、遊離塩基(双性イオン)、薬学上許容可能な塩(例えば、薬学上許容可能な酸付加塩)、こうした塩基または塩の水和物または溶媒和物、ならびに無水物、またさらには非晶質形態または結晶形態など、この化合物の一切の形態を包含することを意図したものである。
【0047】
さらなる一実施形態では、ガボキサドールは、双性イオン、典型的にはその水和物から選択されるが、無水物も適している。適当な一実施形態は、双性イオン一水和物である。
【0048】
さらなる一実施形態では、ガボキサドールは、酸付加塩、典型的には薬学上許容可能な酸付加塩から選択される。適当な一実施形態は、マレイン酸、フマル酸、安息香酸、アスコルビン酸、コハク酸、シュウ酸、ビスメチレンサリチル酸、メタンスルホン酸、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン酸、乳酸、リンゴ酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリン酸、パルミチン酸、イタコン酸、グリコール酸、p−アミノ安息香酸、グルタミン酸、ベンゼンスルホン酸またはテオフィリン酢酸の付加塩のいずれか1つなどの有機酸付加塩、ならびに、8−ハロテオフィリン、例えば8−ブロモテオフィリンである。別の適当な実施形態は、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸または硝酸の付加塩のいずれか1つなどの無機酸付加塩である。
【0049】
別の実施形態では、ガボキサドールは、塩酸塩、臭化水素酸塩または双性イオン一水和物の形態である。
【0050】
さらなる一実施形態では、ガボキサドールは、結晶性の塩酸塩、結晶性の臭化水素酸塩または結晶性の双性イオン一水和物など結晶性である。
【0051】
さらなる一実施形態では、本発明の医薬組成物は、ヒドロキシプロピルメチルセルロース(例えばMetolose 90SH−15.000およびMetolose 90SH−100.000)など親水性のセルロースエーテル・ポリマーを実際に含有する。
【0052】
本発明による酸付加塩は、不活性溶媒中の酸でガボキサドールを処理し、次いで、公知の方法による析出、単離、場合により再結晶化、所望により、湿式もしくは乾式の粉砕または別の好都合な方法による結晶性の生成物の微粒子化、または、溶媒−乳化法(solvent−emulsification process)からの粒子の調製により得ることができる。適当な方法は、例えば欧州特許第0000338号(特許文献1)に記載されている。
【0053】
ガボキサドールの塩の析出は、典型的には、不活性溶媒中、例えば、アルコール(例えば、エタノール、2−プロパノールおよびn−プロパノール)などの不活性な極性溶媒中で実施されるが、水、または水と不活性溶媒との混合物を使用してもよい。
【0054】
ガボキサドールは、経口投与用の固形形態(典型的には錠剤またはカプセル剤)などの経口投与用の形態として、または経口投与用の液体形態として投与してもよい。ガボキサドールは、速放性剤形、または制御放出剤形もしくは持続放出剤形の形態で投与してもよい。一実施形態によれば、この剤形は、睡眠導入量未満の量で、ガボキサドールを制御放出または持続放出する。ガボキサドールは、約0.1〜約150mg/日、約0.2〜約100mg/日、約0.5〜約50mg/日、約0.1〜約50mg/日、約1〜約15mg/日、または約2〜約5mg/日の量で活性成分を含有する錠剤またはカプセル剤などの単位投与形態で、便利に経口投与してもよい。典型的には、この医薬組成物は、約0.5mg〜約20mg、例えば、約0.5mg、約1mg、約1.5mg、約2mg、約2.5mg、約3mg、約3.5mg、約4mg、約4.5mg、約5mg、約5.5mg、約6mg、約6.5mg、約7mg、約7.5mg、約8mg、約8.5mg、約9mg、約9.5mg、約10mg、約10.5mg、約11mg、約11.5mg、約12mg、約12.5mg、約13mg、約13.5mg、約14mg、約14.5mg、約15mg、約15.5mg、約16mg、約16.5mg、約17mg、約17.5mg、約18mg、約18.5mg、約19mg、約19.5mgまたは約20mgなどのガボキサドールを含む。ガボキサドールの量は、遊離塩基(双性イオン)の形態に基づいて計算する。
【0055】
一実施形態では、ガボキサドールは、約2.5mg〜約20mgの用量を用いて1日1回(例えば午前または午後)投与する。別の実施形態では、ガボキサドールは1日2回投与する。
【0056】
本発明によれば、ガボキサドールまたは薬学上許容可能なその塩は、任意の適当な方式で、例えば、経口的または非経口的に投与してもよく、そのような投与に適した任意の形態、例えば、錠剤、カプセル剤、散剤、シロップ剤または液剤または注射用分散剤の形態で提供してもよい。別の実施形態では、また、本発明の目的によれば、ガボキサドールは、固形の医薬体(pharmaceutical entity)の形態で、適切には錠剤またはカプセル剤として、または注射用の懸濁剤、液剤または分散剤の形態で、投与する。加えて、ガボキサドールは、佐剤および/または希釈剤など薬学上許容可能な担体と共に投与してもよい。
【0057】
本発明はさらに、ガボキサドールと、セロトニン再取込み阻害薬(SRI)である化合物または細胞外5−HTの上昇をもたらす任意の他の化合物と、場合により、薬学上許容可能な担体または希釈剤とを含む医薬組成物またはキットにも関する。
【0058】
一実施形態では、SRIは、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、デュロキセチン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミンから選択される。念のため明確にしておくと、こうしたSRIのそれぞれは、個々の実施形態を構成し、個々の請求項の主題である場合がある。
【0059】
選択的なセロトニン再取込み阻害薬(SSRI)という用語は、ドパミン輸送体およびノルアドレナリン輸送体よりセロトニン輸送体で強い阻害効果を有するモノアミン輸送体阻害薬を意味する。
【0060】
選択的なセロトニン再取込み阻害薬(SSRI)は、本発明により使用される最も好ましいセロトニン再取込み阻害薬に含まれる。したがって、さらなる一実施形態では、SRIは、シタロプラム、エスシタロプラム、フルオキセチン、フルボキサミン、セルトラリンまたはパロキセチンなどのSSRIから選択される。
【0061】
シタロプラムは、好ましくは、臭化水素酸塩の形態で、または塩基として、エスシタロプラムはシュウ酸塩の形態で、フルオキセチン、セルトラリンおよびパロキセチンは塩酸塩の形態で、フルボキサミンはマレイン酸塩の形態で、使用される。
【0062】
本明細書中でこれまでに具体的に言及しているSSRIを含むセロトニン再取込み阻害薬は、分子量および活性の両方において異なる。結果として、併用療法において使用されるセロトニン再取込み阻害薬の量は、前記セロトニン再取込み阻害薬の性質による。本発明の一実施形態では、セロトニン再取込み阻害薬または細胞外5−HTレベルの増加をもたらす化合物は、化合物を単独で使用する際に必要な用量より低用量で投与される。別の実施形態では、セロトニン再取込み阻害薬または細胞外5−HTレベルの増加をもたらす化合物は、通常の用量で投与される。
【0063】
さらなる一実施形態では、ガボキサドールと、セロトニン再取込み阻害薬(SRI)である化合物または細胞外5−HTの上昇をもたらす任意の他の化合物と、場合により、薬学上許容可能な担体または希釈剤とを含む医薬組成物は、経口投与用の固形形態(典型的には錠剤またはカプセル剤)などの経口投与用の形態として、または経口投与用の液体形態として投与できる。この組成物は、速放性剤形の形態または制御放出剤形もしくは持続放出剤形の形態で投与できる。
【0064】
固形または液体の医薬調製物の調製の方法は、当技術分野で周知である。例えば、Remington、The Science and Practice of Pharmacy、第21版、Lippincott Williams & Wilkins (2005)(非特許文献13)を参照のこと。したがって、錠剤は、活性成分を、通常の担体(佐剤および/または希釈剤など)など当技術分野で公知の賦形剤と混合し、次いで、打錠機中でその混合物を圧縮することにより調製できる。佐剤および/または希釈剤の非限定例としては、トウモロコシ・デンプン、乳糖、マンニトール、リン酸カルシウム、微結晶性セルロース、タルク、ステアリン酸マグネシウム、ゼラチン、ゴムなどが挙げられる。活性成分と適合することを条件に、着色剤、アロマおよび保存剤など任意の他の佐剤または添加剤を使用してもよい。
【0065】
本明細書において引用および検討する全ての非特許参考文献、特許および特許出願は、参照によりその全体が、また、それぞれが参照により個別に組み込まれるのと同程度に、本明細書中に組み込まれる。
【0066】
参考文献リスト
1. Boll, M. et al. (2004) Pflugers.Arch. Vol. 447. 5. 776-779
2. Boll, M. et al. (2002) J Biol Chem. Vol. 277. 25. 22966-22973
3. Burckhardt, B. C. et al. (2003) Rev Physiol Biochem Pharmacol. Vol. 146. 95-158
4. Chen, Z. et al. (2003) J Physiol. Vol. 546. Pt 2. 349-361
5. Gabrielsson, J. and Weiner, D. (2007) Pharmacokinetic and Pharmacodynamic Data Analysis, Concepts and Applications, 4th ed., CRC Press, Baco Raton, FL ISBN 978-9-1976-5100-4
6. Koepsell, H. et al. (2004) Pflugers.Arch. Vol. 447. 5. 666-676
7. Lucas, M. L. et al. (1975) Proc R.Soc Lond.B Biol Sci. Vol. 192. 1106. 39-48
8. Metzner, L. et al. (2006) Amino.Acids. Vol. 31. 2. 111-117
9. Pavlova, A. et al. (2000) Am.J Physiol Renal Physiol. Vol. 278. 4. F635-F643
10. Pritchard, J. B. et al. (1999) J Biol Chem. Vol. 274. 47. 33382-33387
11. Rizwan, A. N. et al. (2007) Pharm.Res. Vol. 24. 3. 450-470
12. Sekine, T. et al. (2006) Am.J Physiol Renal Physiol. Vol. 290. 2. F251-F261
13. Wright, S. H. et al. (2004) Am.J Physiol Renal Physiol. Vol. 287. 3. F442-F451
【実施例】
【0067】
例1
この例では、ビーグル犬において実施した試験で得られたデータについて記載する。
【0068】
材料
4,5,6,7−テトラヒドロイソキサゾロ[5,4−c]ピリジン−3−オール塩酸塩/C6H8N2O2,HCl、(ガボキサドール塩酸塩)および第二置換体(deuto substituted form)C6H4D4N2O2、HCL、(第二ガボキサドール塩酸塩(deuto−gaboxadol hydrochloride))は、H.Lundbeckから供給された。5−ヒドロキシ−L−トリプトファン(5−HTP)、L−トリプトファン(Trp)、L−プロリン(Pro)、アセトニトリル(ACN)およびメタノールは、Sigma−Aldrich(St.Louis、MO、USA)から入手した。酢酸はMERCK製であった。ヘパリン、5000IE/a.e./mlは、LEO(Ballerup、Dk)から購入した。
【0069】
方法
in vivo試験
試験開始に先立ち、デンマーク法務省により任命された動物愛護委員会(Animal Welfare Committee)によりプロトコールの承認を受け、全ての動物の手術は、EC指令86/609/EEC、動物実験を規制するデンマークの法律、ならびに、実験動物の管理および使用のためのNIHガイドライン(NIH Guidelines for the Care and Use of Laboratory Animals)に従って実施した。十分成長した6匹のオスのビーグル犬(体重15.9〜21.7kg)を選び、ローマ式の4分割デザイン(roman quadrant design)に配置し、ガボキサドール塩酸塩製剤全6種をランダムに、6週間にわたり投与するように割り付けた。イヌは、実験開始前20〜24時間絶食させ、投与10時間後に給餌を再開した。ガボキサドール1回分を、静脈内注射(1.0ml/kg)として、または軟らかいチューブを用いた胃の中への直接経口強制投与(5.0ml/kg)により与えられる経口溶液として、そのいずれかで投与した。全てのイヌに2.5mg/kgのガボキサドールを投与した。2つの化合物の同時併用投与を確実にするために、経口製剤は、ガボキサドールに加え、0mg/kg、2.5mg/kg、10.0mg/kg、50.0mg/kgまたは150.0mg/kgのトリプトファンを含有していた。全ての溶液をpH5.2に調節し、Vapro蒸気圧浸透圧計(モデル552O、Wescor Inc.、Logan、UT、USA)を用いて浸透圧モル濃度を調べ、グルコースを用いて静脈内用の溶液を等モル浸透圧濃度(isoosmolarity)に調節した。個別の静脈穿刺により、橈側皮静脈から血液試料2.0mlを採取し、抗凝血剤として200IEヘパリンを含有するエッペンドルフ管中に収集した。試料は、ガボキサドール投与前、ならびに、ガボキサドール投与の5分後、15分後、30分後、60分後、90分後および2時間後、3時間後、4時間後、6時間後、8時間後および10時間後に収集した。15分間、2200g、4〜8℃での遠心分離により速やかに血漿を採取し、−80℃で保管してからさらなる分析を実施した。ガボキサドールの各投与日の後、動物には6日のウォッシュアウト期間を与えた。
【0070】
WinNonlinで薬物動態(PK)を評価した。静脈内投与した動物の血漿濃度曲線を2コンパートメント・モデルに当てはめ、経口投与した動物のデータは、非コンパートメント・モデルにおいて分析した。統計分析はSigma Statで行った。
【0071】
HPLCおよびMS/MS検出による定量分析。
液体抽出により、血漿およびHBSS+試料からガボキサドールを抽出した。HBSS+100μl(試料20μlに精製水80μlを加えた)または血漿試料100μlを内部標準(intern standard)(d4−ガボキサドール)25μlおよび精製水25μlと混合した。冷アセトニトリル400μlの添加により、タンパク質沈降を実施した。10,000gで15分間の遠心分離の後、上澄み425μlをガラス管に移し、蒸発させて、窒素下、45℃で乾燥させた。試料をメタノール/アセトニトリル(30:70)80μl中で分離し、10分間回転混合して、3000rpmで3分間遠心分離してから、培地用のウェル・プレートに移し、10℃のオートサンプラー中に置いた。次いで、親水性の相互作用クロマトグラフィー(HILIC−クロマトグラフィー)により抽出試料中のガボキサドール濃度を定量化し、その後、MS/MS検出を行った。LCシステムは、Agilent1100シリーズのポンプ付き脱気装置で構成され、CTC Analyticsのインターフェースからコンピューターにデータが転送され、試料はPeltier ThermostatおよびHTC Palオートサンプラーで操作した。クロマトグラフィー分離用にはPhenomenex製のAsahipakアミノ・カラム、(NH2P−50、150×2mm)を使用し、移動相は20.0mM酢酸アンモニウムpH=4:アセトニトリル(30:70)、流速0.2ml/minとした。試料20μlをカラムに注入し、これを室温で維持した。合計通液時間は10分であり、最初の5分の溶出分は廃棄するようにした。カラムでのガボキサドールの溶出時間はおよそ8分であった。使用したMS/MSシステムは、Turbo Ion SprayおよびTurbo V源を装備したSciex API4000MS/MS検出装置(Applied Biosystems)から成るものであった。陰イオン化モードで検出を実施し、多重反応モニタリング(MRM)により、ガボキサドール(前駆物質139.1Da、生成物110.1Da)およびd4−ガボキサドール(前駆物質143.0、生成物112.2Da)を測定した。シグナルは、0.5〜2500.0ng/mlの間の線形であり、この手順による定量化の限界は0.5ng/mlであった。ソフトウェアは、Analyst(商標)のものであった(Applied Biosystem、バージョン4.0)。
【0072】
結果および考察
血漿濃度対時間プロファイルを図1および2に示す。
【0073】
表1.例1において動物から得られた薬物動態パラメーター。1kg当たりガボキサドール2.5mgをIV(A)投与およびPO(B〜F)投与。データは、平均値±SEM、n=6を表す。
【表1】
【0074】
イヌにおける経口投与後のガボキサドールのバイオアベイラビリティーFaは、85.3±5.7%であることがわかった(表1)。2.5〜150mg/kgトリプトファンの経口併用投与では、ガボキサドールのAUCは有意に変化せず、この製剤の平均の相対バイオアベイラビリティーは、75.0%(10mg/kgトリプトファン)〜86.1%(2.5mg/kgトリプトファン)の間で変動した。同様に、ガボキサドールの排出速度定数(ke)およびクリアランス(CL)は、トリプトファンの併用投与によって変化しなかった。しかし、トリプトファン併用投与により、ガボキサドールの最大血漿濃度Cmaxは、150mg/kgトリプトファンの不在下および存在下では、2502ng/mlから1419ng/mlへ57%低下した(p<0.001)。さらに、最大血漿濃度への到達に要する時間Tmaxは、0.46時間から1.5時間に増えた(p<0.01)。5つの投与群のCmax値の変化は、ガボキサドールとトリプトファンとの間に直接の相互作用があることを明確に示している。
【0075】
これらのデータに基づけば、Trpはガボキサドールの吸収プロファイルに影響を及ぼすことが明らかである。この効果には、PAT1輸送体と相互作用する2つの化合物が介在する、すなわち、Trp用量が高い状況下では、結合部位の多くがTrpにより占められるのと同様、ガボキサドールは、PAT1により輸送できないと考えられる。PAT1を阻害するか、またはPAT1の基質である化合物を併用投与すると、結果としてガボキサドールの吸収プロファイルを調節できる。
例2
【0076】
この例では、ラットにおいて実施した試験から得られたデータを記載する。
【0077】
材料
例1と同じ
【0078】
実験方法
以下を除き、例1と同じ:
経口製剤
ガボキサドール0.05mgまたは0.5mgならびに5−HTP 0.0mgまたは20.0mgを精製水(1ml当たり)に室温で溶解し、超音波下で10分間、氷の上に置いた。この製剤をNaOH/HClでpH4〜5に調節し、マンニトールを加えて等張にした。全ての溶液のpHを、pH4.0超5.0未満に調節し、マンニトールを加えて浸透圧モル濃度を280mmol/kgに調節した。
in vivoでの実験
220〜240グラムのオスのSprague−Dawleyラット(Charles River Laboratories、Wilmington、MA、USA)を収容し、実験に入る前に7日間馴化した。投与の16〜20時間前まではラットを標準食および水で維持し、実験を実施する前に完全な胃内容排出を確実にするために、この時点で食餌を回収した。水は、実験開始まで、また、2時間後に再度、動物が摂取可能であった。各動物は、静脈内製剤または経口製剤のいずれか一方を摂取するようにランダムに割り付けた。
【0079】
ラットの6つの並行群(n=6)に、0.5mg/kgまたは5.0mg/kgのガボキサドールの等張溶液を、生理食塩水または200.0mg/kg 5−HTPと共に、経口強制投与により投与した(10.0ml/kg)。ガボキサドール溶液の30分前に、5−HTPの懸濁液をプレ・インキュベーションとして30分投与した。
【0080】
個別の静脈穿刺により、尾静脈から血液試料0.2mlを採取し、20IEヘパリンを含有する血漿収集チューブ中に収集した。試料は、ガボキサドール投与後5分、15分、30分、45分、60分の時点、および、2時間後、3時間後、4時間後、6時間後、8時間後に収集した。3.600gで10分間の遠心分離により血漿を速やかに採取し、−80℃で保管してから、さらなる分析を実施した。実験の終了時に、動物を安楽死させた。
【0081】
結果および考察
血漿濃度対時間プロファイルを図3に示す。
【0082】
表2.例2において動物から得られた薬物動態パラメーター、1kg当たりガボキサドール0.5mgおよび5.0mgのPO投与。データは、平均値±SEM、n=6を表す。
【表2】
【0083】
図3および表2において見られるように、経口投与後のガボキサドールの吸収は、大部分は投与後15〜20分以内に生じたが、その理由は、血漿ガボキサドール濃度がこの期間中に増加したからである。15〜20分後、ガボキサドールの排出が増加していくと、血漿ガボキサドール濃度は低下した。1kg当たりガボキサドール0.5mgの前にプレ・インキュベーションとして1kg当たり5−HTP 200mgを投与すると、血漿ガボキサドール濃度ピーク時間(Tmax)は、16分から63分へと遅くなった。ラットに1kg当たりガボキサドール5.0mgを投与すると、5−HTPでのプレ・インキュベーション後、Tmaxは20分から43分へと遅くなった。最大血漿濃度Cmaxは、5−HTPのプレ・インキュベーションにより変化しないようであった。
【0084】
動物にガボキサドールおよびPAT1阻害薬5−HTPを投与すると、AUCは、対照動物(5−HTP非投与)と比較して増加した。5−HTP(200mg/kg)投与は、ガボキサドール(5.0mg/kgまたは0.5mg/kg)投与の40倍または400倍高く、AUCは対照群と比較して330%および540%増加した。AUCは、排出速度の低下により増加し得る。ガボキサドール排出速度定数は、5−HTPが存在する場合には約25%に低下した。
【0085】
まとめると、ガボキサドールの吸収は、PAT阻害薬5HTPの併用投与により変化するようである。さらに、ガボキサドールの排出は、PAT、OAT、または、5−HTPが相互作用する他の輸送体との相互作用に影響されるようである。
例3
【0086】
材料
例1と同じ
【0087】
実験方法
以下を除き、例2と同じ:
静脈内製剤
ガボキサドール0.25mgならびに5−HTP 0.0mgまたは10.0mgを精製水(1ml当たり)に室温で溶解し、超音波下で10分間、氷の上に置いた。静脈内注射に使用するガボキサドール溶液を、0.45μmのフィルターを通して濾過した。
【0088】
2.5mg/kgガボキサドールの尾静脈中への静脈内注射の30分前に、経口強制投与により、動物に100.0mg/kg 5−HTPまたは生理食塩水を投与した(5.0ml/kg)。
【0089】
結果および考察
血漿濃度対時間プロファイルを図4に示す。
【0090】
表3.例3において動物から得られた薬物動態パラメーター、1kg当たりガボキサドール2.5mgのIV投与。データは、平均値±SEM、n=6を表す。
【表3】
【0091】
5−HTPでプレ・インキュベートしたIV群(L群)の血漿プロファイルは、ガボキサドールのみを投与された群(K群)のラットのものと異なった。K群のAUCはL群のほぼ3倍の大きさであり、それはおそらく、排出速度定数Keがより小さいことが原因であった(表3)。こうした結果から、5−HTPは、OATまたは他の輸送体との相互作用によりガボキサドールのクリアランスを妨げることが示唆される。
【0092】
G群(1kg当たりガボキサドール0.5mg)のAUCがK群(1kg当たりガボキサドール2.5mg)の5倍の大きさであり、また、J群(5mg/kg)のAUCがG群のほぼ10倍の大きさであったように、ガボキサドールについてはAUC−用量の線形性が観察された。
【特許請求の範囲】
【請求項1】
ガボキサドール(gaboxadol)もしくは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物。
【請求項2】
1つまたは複数のPAT1阻害薬を含むがOAT阻害薬を含まない、請求項1に記載の組成物。
【請求項3】
1つまたは複数のOAT阻害薬を含むがPAT1阻害薬を含まない、請求項1に記載の組成物。
【請求項4】
1つまたは複数のPAT1阻害薬および1つまたは複数のOAT阻害薬を両方とも含む、請求項1に記載の組成物。
【請求項5】
ガボキサドールが、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である、請求項1〜4のいずれか一つに記載の組成物。
【請求項6】
ガボキサドールが、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である、請求項1〜5のいずれか一つに記載の組成物。
【請求項7】
ガボキサドールの量が0.5mg〜50mgの範囲である、請求項1〜6のいずれか一つに記載の組成物。
【請求項8】
経口投与用の形態である、請求項1〜7のいずれか一つに記載の組成物。
【請求項9】
錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である、請求項1〜8のいずれか一つに記載の組成物。
【請求項10】
前記ガボキサドールが結晶性である、請求項1〜9のいずれか一つに記載の組成物。
【請求項11】
PAT1がヒトPAT1である、請求項1または2および4〜10のいずれか一つに記載の組成物。
【請求項12】
前記PAT1阻害薬が、5−ヒドロキシトリプトファン(5−HTP)、L−プロリン、D−プロリン、サルコシン、L−アラニン、D−アラニン、N−メチル−L−アラニン、N−メチル−D−アラニン、α−(メチルアミノ)−イソ酪酸、ベタイン、D−シクロセリン、L−シクロセリン、β−アラニン、セロトニン、L−トリプトファン、D−トリプトファン、トリプタミン、インドール−3−プロピオン酸から選択される、請求項1または2および4〜11のいずれか一つに記載の組成物。
【請求項13】
PAT1阻害薬の量が、約0.5〜約3000mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約750mg、約1000mg、約1250mg、約1500mg、約1750mg、約2000mg、約2250mg、約2500mg、約2750mgまたは約3000mgである、請求項1または2および4〜12のいずれか一つに記載の組成物。
【請求項14】
OATがヒトOATである、請求項1および3〜10のいずれか一つに記載の組成物。
【請求項15】
前記OAT阻害薬が、キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン(6‐carboxyflurescein)、ベンジルペニシリン、セファドロキシル、セファマドール、セファゾリン、セフォペラゾン、セフォタミム(Cefotamime)、セファレキシン、セファロチン、セフラジン、アシロビル、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク、ジフルシナル、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダク(Sulidac)から選択される、請求項1および3〜11のいずれか一つに記載の組成物。
【請求項16】
OAT阻害薬の量が、約0.5〜約500mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mgまたは約500mgである、請求項1および3〜12のいずれか一つに記載の組成物。
【請求項17】
1つまたは複数の賦形剤を含む、請求項1〜16のいずれか一つに記載の組成物。
【請求項18】
セロトニン再取込み阻害薬である化合物、または細胞外のセロトニンレベルの上昇をもたらす任意の他の化合物を含む、請求項1〜17のいずれか一つに記載の組成物。
【請求項19】
前記セロトニン取込み阻害薬が、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミン、またはこれらの化合物のいずれかの薬学上許容可能な塩から選択される、請求項18に記載の組成物。
【請求項20】
前記セロトニン取込み阻害薬が、塩基としての、またはシュウ酸塩、臭化水素酸塩もしくは塩酸塩など薬学上許容可能なその塩としてのエスシタロプラムである、請求項18または19のいずれか一つに記載の組成物。
【請求項21】
ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物。
【請求項22】
前記平均Tmaxが、約25分超、30分超、35分超、40分超、45分超、50分超、55分超、60分超、65分超、70分超または75分超である、請求項21に記載の組成物。
【請求項23】
約2250ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを提供する、請求項21または22のいずれか一つに記載の組成物。
【請求項24】
前記平均Cmaxが、約2000ng/ml未満、約1750ng/ml未満、約1500ng/ml未満、約1250ng/ml未満、約1000ng/ml未満、約750ng/ml未満、約500ng/ml未満、約250ng/ml未満、約200ng/ml未満または約100ng/ml未満である、請求項23に記載の組成物。
【請求項25】
約8.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21〜24のいずれか一つに記載の組成物。
【請求項26】
前記平均AUC0〜∞が、約16.000ng・min・ml−1超、20.000ng・min・ml−1超、40.000ng・min・ml−1超、80.000ng・min・ml−1超、120.000ng・min・ml−1超または200.000ng・min・ml−1超である、請求項25に記載の組成物。
【請求項27】
クリアランスが40ml/min未満である、請求項21〜26のいずれか一つに記載の組成物。
【請求項28】
前記クリアランスが、30ml/min未満、20ml/min未満、10ml/min未満または5ml/min未満である、請求項27に記載の組成物。
【請求項29】
ガボキサドール約2mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約100ng/ml未満の平均Cmaxおよび約8.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項30】
ガボキサドール約4mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約200ng/ml未満の平均Cmaxおよび約16.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項31】
ガボキサドール約5mgまたは薬学上許容可能なその塩を含み、約20分時間より大きい平均Tmax、約250ng/ml未満の平均Cmaxおよび約20.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項32】
ガボキサドール約10mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約500ng/ml未満の平均Cmaxおよび約40.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項33】
ガボキサドール約20mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約1000ng/ml未満の平均Cmaxおよび約80.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項34】
ガボキサドール約30mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約1500ng/ml未満の平均Cmaxおよび約120.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項35】
ガボキサドール約50mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約2500ng/ml未満の平均Cmaxおよび約200.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項36】
前記クリアランスが40ml/min未満であり、前記AUCが200.000ng・min・ml−1より高い、請求項21に記載の組成物。
【請求項37】
前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞が、前記組成物をイヌに投与した際に得られ、前記クリアランスが、前記組成物をイヌまたはラットに投与した際に得られる、請求項21〜35のいずれか一つに記載の組成物。
【請求項38】
前記平均Tmaxが約30分より長い、請求項21に記載の組成物。
【請求項39】
約300ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを提供する、請求項21または38のいずれか一つに記載の組成物。
【請求項40】
ガボキサドールの量が、約2.5mg、約5mgまたは約10mgから選択される、請求項21または38または39のいずれか一つに記載の組成物。
【請求項41】
ガボキサドールの量が2.5mgであり、平均Cmaxが約40ng/ml未満、例えば、約35ng/ml、約30ng/ml、約25ng/mlまたは約20ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項42】
ガボキサドールの量が5mgであり、平均Cmaxが約85ng/ml未満、例えば、約80ng/ml、約75ng/ml、約70ng/mlまたは約65ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項43】
ガボキサドールの量が10mgであり、平均Cmaxが約150ng/ml未満、例えば、約145ng/ml、約140ng/ml、約135ng/mlまたは約130ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項44】
前記平均Tmaxおよび平均Cmaxが、前記組成物をヒトに投与した際に得られる、請求項38〜43のいずれか一つに記載の組成物。
【請求項45】
ガボキサドールが、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である、請求項21〜44のいずれか一つに記載の組成物。
【請求項46】
ガボキサドールが、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である、請求項21〜45のいずれか一つに記載の組成物。
【請求項47】
経口投与用の形態である、請求項21〜46のいずれか一つに記載の組成物。
【請求項48】
錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である、請求項21〜47のいずれか一つに記載の組成物。
【請求項49】
前記ガボキサドールが結晶性である、請求項21〜48のいずれか一つに記載の組成物。
【請求項50】
1つまたは複数の賦形剤を含む、請求項21〜49のいずれか一つに記載の組成物。
【請求項1】
ガボキサドール(gaboxadol)もしくは薬学上許容可能なその塩および1つもしくは複数のPAT1阻害薬および/または1つもしくは複数のOAT阻害薬を含む医薬組成物。
【請求項2】
1つまたは複数のPAT1阻害薬を含むがOAT阻害薬を含まない、請求項1に記載の組成物。
【請求項3】
1つまたは複数のOAT阻害薬を含むがPAT1阻害薬を含まない、請求項1に記載の組成物。
【請求項4】
1つまたは複数のPAT1阻害薬および1つまたは複数のOAT阻害薬を両方とも含む、請求項1に記載の組成物。
【請求項5】
ガボキサドールが、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である、請求項1〜4のいずれか一つに記載の組成物。
【請求項6】
ガボキサドールが、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である、請求項1〜5のいずれか一つに記載の組成物。
【請求項7】
ガボキサドールの量が0.5mg〜50mgの範囲である、請求項1〜6のいずれか一つに記載の組成物。
【請求項8】
経口投与用の形態である、請求項1〜7のいずれか一つに記載の組成物。
【請求項9】
錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である、請求項1〜8のいずれか一つに記載の組成物。
【請求項10】
前記ガボキサドールが結晶性である、請求項1〜9のいずれか一つに記載の組成物。
【請求項11】
PAT1がヒトPAT1である、請求項1または2および4〜10のいずれか一つに記載の組成物。
【請求項12】
前記PAT1阻害薬が、5−ヒドロキシトリプトファン(5−HTP)、L−プロリン、D−プロリン、サルコシン、L−アラニン、D−アラニン、N−メチル−L−アラニン、N−メチル−D−アラニン、α−(メチルアミノ)−イソ酪酸、ベタイン、D−シクロセリン、L−シクロセリン、β−アラニン、セロトニン、L−トリプトファン、D−トリプトファン、トリプタミン、インドール−3−プロピオン酸から選択される、請求項1または2および4〜11のいずれか一つに記載の組成物。
【請求項13】
PAT1阻害薬の量が、約0.5〜約3000mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約750mg、約1000mg、約1250mg、約1500mg、約1750mg、約2000mg、約2250mg、約2500mg、約2750mgまたは約3000mgである、請求項1または2および4〜12のいずれか一つに記載の組成物。
【請求項14】
OATがヒトOATである、請求項1および3〜10のいずれか一つに記載の組成物。
【請求項15】
前記OAT阻害薬が、キヌレン酸、キサンツレン酸、5−ヒドロキシインドール酢酸、p−アミノ馬尿酸、6−カルボキシフルレセイン(6‐carboxyflurescein)、ベンジルペニシリン、セファドロキシル、セファマドール、セファゾリン、セフォペラゾン、セフォタミム(Cefotamime)、セファレキシン、セファロチン、セフラジン、アシロビル、アデフォビル、シドフォビル、ガンシクロビル、テノフォビル、バラシロビル、ジドブジン、アセタゾラミド、ブメタニド、クロロチアジド、エタクリン酸、フロセミド、ヒドロクロロチアジド、メタゾラミド、トリクロロメチアジド、アセトアミノフェン、アセチルサリチル酸ジロフェナク、ジフルシナル、エトドラク、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ロキソプロフェン、メファナメート、ナプロキセン、フェナセチン、ピロキシカム、サリチル酸、スリダク(Sulidac)から選択される、請求項1および3〜11のいずれか一つに記載の組成物。
【請求項16】
OAT阻害薬の量が、約0.5〜約500mgの範囲、例えば、約1mg、約5mg、約10mg、約25mg、約50mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mgまたは約500mgである、請求項1および3〜12のいずれか一つに記載の組成物。
【請求項17】
1つまたは複数の賦形剤を含む、請求項1〜16のいずれか一つに記載の組成物。
【請求項18】
セロトニン再取込み阻害薬である化合物、または細胞外のセロトニンレベルの上昇をもたらす任意の他の化合物を含む、請求項1〜17のいずれか一つに記載の組成物。
【請求項19】
前記セロトニン取込み阻害薬が、シタロプラム、エスシタロプラム、フルオキセチン、セルトラリン、パロキセチン、フルボキサミン、ベンラファキシン、デュロキセチン、ダポキセチン、ネファゾドン、イミプラミン、フェモキセチンおよびクロミプラミン、またはこれらの化合物のいずれかの薬学上許容可能な塩から選択される、請求項18に記載の組成物。
【請求項20】
前記セロトニン取込み阻害薬が、塩基としての、またはシュウ酸塩、臭化水素酸塩もしくは塩酸塩など薬学上許容可能なその塩としてのエスシタロプラムである、請求項18または19のいずれか一つに記載の組成物。
【請求項21】
ガボキサドール約0.5mg〜約50mgまたは薬学上許容可能なその塩を含む医薬組成物であって、約20分より長い平均Tmaxを含むin vivo血漿プロファイルを提供する組成物。
【請求項22】
前記平均Tmaxが、約25分超、30分超、35分超、40分超、45分超、50分超、55分超、60分超、65分超、70分超または75分超である、請求項21に記載の組成物。
【請求項23】
約2250ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを提供する、請求項21または22のいずれか一つに記載の組成物。
【請求項24】
前記平均Cmaxが、約2000ng/ml未満、約1750ng/ml未満、約1500ng/ml未満、約1250ng/ml未満、約1000ng/ml未満、約750ng/ml未満、約500ng/ml未満、約250ng/ml未満、約200ng/ml未満または約100ng/ml未満である、請求項23に記載の組成物。
【請求項25】
約8.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21〜24のいずれか一つに記載の組成物。
【請求項26】
前記平均AUC0〜∞が、約16.000ng・min・ml−1超、20.000ng・min・ml−1超、40.000ng・min・ml−1超、80.000ng・min・ml−1超、120.000ng・min・ml−1超または200.000ng・min・ml−1超である、請求項25に記載の組成物。
【請求項27】
クリアランスが40ml/min未満である、請求項21〜26のいずれか一つに記載の組成物。
【請求項28】
前記クリアランスが、30ml/min未満、20ml/min未満、10ml/min未満または5ml/min未満である、請求項27に記載の組成物。
【請求項29】
ガボキサドール約2mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約100ng/ml未満の平均Cmaxおよび約8.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項30】
ガボキサドール約4mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約200ng/ml未満の平均Cmaxおよび約16.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項31】
ガボキサドール約5mgまたは薬学上許容可能なその塩を含み、約20分時間より大きい平均Tmax、約250ng/ml未満の平均Cmaxおよび約20.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項32】
ガボキサドール約10mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約500ng/ml未満の平均Cmaxおよび約40.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項33】
ガボキサドール約20mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約1000ng/ml未満の平均Cmaxおよび約80.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項34】
ガボキサドール約30mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約1500ng/ml未満の平均Cmaxおよび約120.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項35】
ガボキサドール約50mgまたは薬学上許容可能なその塩を含み、約20分より大きい平均Tmax、約2500ng/ml未満の平均Cmaxおよび約200.000ng・min・ml−1より大きい平均AUC0〜∞を含むin vivo血漿プロファイルを提供する、請求項21に記載の組成物。
【請求項36】
前記クリアランスが40ml/min未満であり、前記AUCが200.000ng・min・ml−1より高い、請求項21に記載の組成物。
【請求項37】
前記平均Tmax、平均Cmaxおよび/または平均AUC0〜∞が、前記組成物をイヌに投与した際に得られ、前記クリアランスが、前記組成物をイヌまたはラットに投与した際に得られる、請求項21〜35のいずれか一つに記載の組成物。
【請求項38】
前記平均Tmaxが約30分より長い、請求項21に記載の組成物。
【請求項39】
約300ng/ml未満の平均Cmaxを含むin vivo血漿プロファイルを提供する、請求項21または38のいずれか一つに記載の組成物。
【請求項40】
ガボキサドールの量が、約2.5mg、約5mgまたは約10mgから選択される、請求項21または38または39のいずれか一つに記載の組成物。
【請求項41】
ガボキサドールの量が2.5mgであり、平均Cmaxが約40ng/ml未満、例えば、約35ng/ml、約30ng/ml、約25ng/mlまたは約20ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項42】
ガボキサドールの量が5mgであり、平均Cmaxが約85ng/ml未満、例えば、約80ng/ml、約75ng/ml、約70ng/mlまたは約65ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項43】
ガボキサドールの量が10mgであり、平均Cmaxが約150ng/ml未満、例えば、約145ng/ml、約140ng/ml、約135ng/mlまたは約130ng/mlであり、平均Tmaxが約1時間より長く、例えば、1.5時間、2時間または2.5時間である、請求項21または38〜40のいずれか一つに記載の組成物。
【請求項44】
前記平均Tmaxおよび平均Cmaxが、前記組成物をヒトに投与した際に得られる、請求項38〜43のいずれか一つに記載の組成物。
【請求項45】
ガボキサドールが、酸付加塩、または双性イオン水和物もしくは双性イオン無水物の形態である、請求項21〜44のいずれか一つに記載の組成物。
【請求項46】
ガボキサドールが、塩酸塩もしくは臭化水素酸塩から選択される薬学上許容可能な酸付加塩の形態、または双性イオン一水和物の形態である、請求項21〜45のいずれか一つに記載の組成物。
【請求項47】
経口投与用の形態である、請求項21〜46のいずれか一つに記載の組成物。
【請求項48】
錠剤もしくはカプセル剤など経口投与用の固形形態、または経口投与用の液体形態である、請求項21〜47のいずれか一つに記載の組成物。
【請求項49】
前記ガボキサドールが結晶性である、請求項21〜48のいずれか一つに記載の組成物。
【請求項50】
1つまたは複数の賦形剤を含む、請求項21〜49のいずれか一つに記載の組成物。
【図1】

【図2】
【図3】
【図4】

【図2】
【図3】
【図4】
【公表番号】特表2012−501301(P2012−501301A)
【公表日】平成24年1月19日(2012.1.19)
【国際特許分類】
【出願番号】特願2011−524183(P2011−524183)
【出願日】平成20年10月28日(2008.10.28)
【国際出願番号】PCT/DK2008/050264
【国際公開番号】WO2009/056146
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
【公表日】平成24年1月19日(2012.1.19)
【国際特許分類】
【出願日】平成20年10月28日(2008.10.28)
【国際出願番号】PCT/DK2008/050264
【国際公開番号】WO2009/056146
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
[ Back to top ]