
ガラクチュロン酸還元酵素をコードする遺伝子
【課題】新規なD−ガラクチュロン酸還元酵素をコードする遺伝子を提供する。
【解決手段】タイ地酒オウ(Ou)の酒もと(Cocha)から、低温適応酵母であるCryptococcus diffluens FC11株を分離し、当該酵母株から10〜30℃の低温においても比較的高い活性を示すD−ガラクチュロン酸還元酵素をコードする配列番号1で示される塩基配列を有する遺伝子を得た。
【解決手段】タイ地酒オウ(Ou)の酒もと(Cocha)から、低温適応酵母であるCryptococcus diffluens FC11株を分離し、当該酵母株から10〜30℃の低温においても比較的高い活性を示すD−ガラクチュロン酸還元酵素をコードする配列番号1で示される塩基配列を有する遺伝子を得た。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はガラクチュロン酸還元酵素をコードする遺伝子に関する。
【背景技術】
【0002】
ペクチンは野菜や果実に多く含まれているが、その再利用に関して、例えばりんご搾汁残渣に含まれるペクチンは、その約70%が農作物の肥料、家畜の飼料、増粘多糖類、ゲル化剤、乳タンパク安定剤などの食品添加物に再利用されるにすぎず、残る約30%のペクチンが廃棄されているとの報告がある。また、ペクチンはりんご以外の果物、特に柑橘類の搾汁残渣にも多く含まれるが、その再利用率はりんごの場合よりもさらに低いといわれている。このようにペクチンの再利用は肥料等の安価な利用かつ食品産業など限られた産業における利用にとどまっており、ペクチンを幅広い産業でバイオマス資源として再利用することは天然資源の確保の面からも、ごみ処理という環境浄化の面からも注目に値する。
【0003】
ペクチンは植物細胞壁・特に果実の細胞壁に多く含まれる非セルロース性多糖であり、ホモガラクツロナン、ラムノガラクツロナン−I、ラムノガラクツロナン−IIという3つの特徴的な構造ドメインから形成される複合多糖である。このうち、ホモガラクツロナンはペクチンの主要構成糖として知られており、ペクチンの約70%を占める。ホモガラクツロナンの中でもっとも一般的な構造はガラクチュロン酸のみがα−1,4−結合したポリガラクチュロン酸であり、一部のガラクチュロン酸にはそのカルボキシル基がメチルエステル化されたものやその水酸基がアセチル化されたものが存在している。
【0004】
ポリガラクチュロン酸は様々な微生物によって分解される。例えば、植物寄生性を示すバクテリア、例えば一部の糸状菌は、ガラクチュロン酸をピルビン酸とグリセルアルデヒド3リン酸にまで代謝する。この代謝は、真性細菌によく見られる代謝系であるエントナードウトロフ(ED)経路を利用することによって行われている。また、Kluyveromyces属酵母など、酵母においても、ポリガラクチュロン酸のグルコシド結合を加水分解するポリガラクチュロン酸分解酵素(ポリガラクチュロナーゼ)を生産する種はいくつか知られている(例えば非特許文献1や2参照)。
【0005】
しかしながら、ガラクチュロン酸をピルビン酸とグリセルアルデヒド3リン酸にまで代謝するED経路は真核生物では確認されておらず、酵母ではポリガラクチュロン酸から分解されたガラクチュロン酸を利用することはできないと考えられていた。事実、Kluyveromyces属酵母をはじめとしたポリガラクチュロナーゼ生産酵母において、ポリガラクチュロナーゼ分解産物であるガラクチュロン酸が資化されたという報告例はない。そして、ガラクチュロン酸を資化する酵母を利用することができれば、ペクチンの再資源化においても有効な手段を与えると考えられる。
【0006】
ガラクチュロン酸を還元する酵素として、これまでのところカビの一種であるHypocreajecorina株から得られたL−ガラクチュロン酸脱水素酵素が報告されているが(非特許文献3)、酵母を起源とするものは知られていない。しかも、低温においても活性を維持するものは知られていない。
【0007】
一方、近年では、食品の熟成等に用いられている低温機能性酵素への期待が高まっており、その低温適応性微生物が着目されている。
【0008】
このような状況下において、本願発明者らは、タイ地酒オウ(Ou)の酒もと(Cocha)から、低温適応酵母であるCryptococcus diffluens FC11株を分離し、当該酵母株から10〜30℃の低温においても比較的高い活性を示すD−ガラクチュロン酸還元酵素を見いだした(非特許文献4、5)
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Hirose, N., Kishida, K., Kawasaki, H. and Sakai, T. (1999) Purification and characterization of an endo-polygalacturonase from a mutant of Saccharomyces cerevisiae., Bioscience Biotechnology and Biochemistry vol.63, 1100-1103
【非特許文献2】Sienkstele, R., Bartkeviciute, D. and Sasnauskas K. (1999) Cloning,targeted disruption and heterologous expression of the Kluyveromyces marxianus endo-polygalacturonase gene (EPG1). Yeast vol.15, 311-322
【非特許文献3】Kuorelahti, S., Kalkkinen, N., Penttila, M., Londesborough, J., Richard, P., Identification in the mold Hypocrea jecorina of the first fungal D-galacturonic acid reductase, Biochemistry vol. 44: 11234-11240 (2005)
【非特許文献4】Kishida M., Seike Y., Kawasaki H. (2009) Identification and characterization of psychrophilic yeast newly isolated from fermentative source (Loog-pang) of traditional drink in Thailand. Biocontrol Science Vol.14, p119-122.
【非特許文献5】岸田正夫ら、「低温適応酵母が生産するD−ガラクチュロン酸還元酵素」 第60回日本生物工学回大会 講演要旨集、p133
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は上記D−ガラクチュロン酸還元酵素のアミノ酸配列を解析するとともにそれをコードする遺伝子を見いだし、工業的な生産を可能とすべく当該酵素をコードするcDNAを提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明に係るポリヌクレオチドは、D−ガラクチュロン酸還元酵素をコードする遺伝子であって、配列番号1に示される塩基配列を含むポリヌクレオチドである。
【発明の効果】
【0012】
本発明によると、低温でも高活性を示すD−ガラクチュロン酸還元酵素をコードする遺伝子が提供される。これにより当該酵素の工業的生産が可能となり、当該酵素を有効利用しうる新規な用途が期待される。
【図面の簡単な説明】
【0013】
【図1】図1はガラクチュロン酸還元酵素の活性測定方法を示す図である。
【図2】図2はDEAE-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムである。
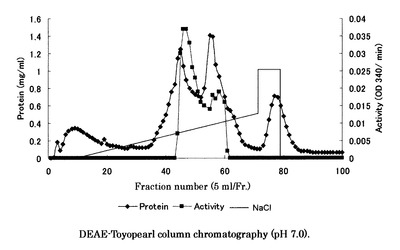
【図3】図3はRed-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムである。
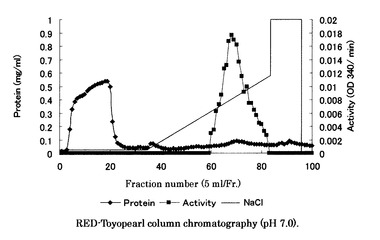
【図4】図4はMono Q カラムクロマトグラフィーにより得られたクロマトグラムである。
【図5】図5は各精製段階における酵素液の電気泳動画像である。
【図6】図6はゲルより抽出したペプチド断片のHPLCの結果を示す図である。
【図7】図7はTOPO TAクローニングベクターpCR2.1-TOPOの遺伝子地図である。
【図8】図8は3'-RACE法により得られた約550bpのcDNA部分遺伝子の泳動画像である。
【図9】図9は3'-RACE法により得られた約550bpのcDNA部分遺伝子の塩基配列を示す図である。
【図10】図10はカセットPCR法により得られた5´領域ゲノムDNA断片の電気泳動画像である。
【図11】図11はカセットPCR法により得られた5´領域ゲノムDNA断片の塩基配列を示す図である。
【図12】図12は決定されたD−ガラクチュロン産還元酵素の全遺伝子の塩基配列を示す図である。
【図13】図13は決定されたD−ガラクチュロン産還元酵素をコードするcDNAの塩基配列を示す図である。
【図14】図14は決定されたD−ガラクチュロン産還元酵素のアミノ酸配列を示す図である。
【発明を実施するための形態】
【0014】
本発明の遺伝子がコードするD−ガラクチュロン酸還元酵素は、上記のようにタイ地酒オウ(Ou)の酒もと(Cocha)から分離された低温適応酵母であるCryptococcus diffluens FC11株(寄託番号:NITE P-611、寄託日:平成20年7月18日)から取得される。当該酵素はD−ガラクチュロン酸を還元してL−ガラクトン酸を生成する。当該酵素は特願2008−285976号に開示されており、その記載が本願において援用される。その酵素の特徴として、特に10〜30℃の低温においても、至適温度における活性の60%程度の活性効率で反応を行わせることができ、室温条件でもガラクチュロン酸をL−ガラクトン酸に変換できることが挙げられる。当該酵素は、以下の理化学的特徴を有する。
【0015】
(1)温度安定性及びpH安定性
20〜45℃及びpH6.0〜8.4の環境下で安定である。
(2)至適温度及び至適pH
至適温度は40℃付近、至適pHは6.3付近である。
(3)基質特異性
ガラクチュロン酸、グルクロン酸に作用するが、ガラクトース、グルコース、マンノース、ソルビトール、グルコン酸には作用しない。
(4)金属による影響
各1mMのCu2+、Fe2+、Pb2+によって阻害を受けるが、各1mMのMg2+、Co2+、Mn2+、Na+、Ca2+、Ni2+、K+、Ba2+、Zn2+によって阻害を受けない。
(5)電子供与体
NADPH特異的である
【0016】
本発明に係る遺伝子は、上記ガラクチュロン酸還元酵素をコードするcDNAであって、配列番号1に示された塩基配列を有する。本発明においては、上記ガラクチュロン酸還元酵素を生産できる限りにおいて、配列番号1に示される塩基配列から、1〜数十個の塩基が欠失・置換・挿入された塩基配列からなるか、相同性が80%以上、好ましくは90%以上、望ましくは95%以上である塩基配列からなる遺伝子であってもよい。
【0017】
本発明に係る遺伝子は下記実施例において説明した方法により取得されるが、一般の遺伝子工学的手法により合成することもできる。また、本発明の遺伝子は、一般的な遺伝子工学的手法によって、その5´末端側にプロモータ領域、3´末端側にターミネータ領域などタンパク質発現のために必要な領域が結合された上で発現ベクターに組み込まれ、その後大腸菌など適当な宿主を形質転換して目的とする酵素の生産に利用される。
【実施例1】
【0018】
次に、本発明の遺伝子について下記の実施例に基づいて詳細に説明する。
〔ガラクチュロン酸還元酵素遺伝子のクローニング〕
1.菌の培養
単離された C. diffluens OPU-FC11株を使用した。種培養にはGYP培地(Yeast extract1%、Peptone1%、Glucose1%、pH5.0)、本培養にはガラクチュロン酸培地(Yeast extract1%、Peptone1%、D-Galacturonic acid (GalA)0.2%、pH5.0)を用いた。GYP培地を入れた短試験管に、菌を1白金耳植菌し、30℃1日間振盪培養した。その培養液を更に、種培養培地9本に50mlずつ植菌し、30℃で1日間振盪培養した。その培養液を500ml容坂口フラスコに200mlのガラクチュロン酸培地を入れた本培養培地20本に2mlずつ植菌し、計4Lを30℃で2日間振盪培養した。
【0019】
2.ガラクチュロン酸還元酵素の精製
2−1.粗酵素液の調整
培養菌体を遠心分離(10000rpm、10分)で集菌し、20mMリン酸カリウム緩衝液(以下「KPB」という)(pH7.0)で洗菌後、同緩衝液で懸濁した。懸濁菌体はOHTAKE WORKSのFRENCH PRESSを用いて1500kg/cm2で1回破砕を行った。破砕した菌体を遠心分離(10000rpm、10分)により、上清画分と沈殿(細胞残渣)画分に分離した。上清画分を10mMKPBで十分に透析したものを粗酵素として用いた。
【0020】
2−2.酵素の精製
得られた粗酵素をDEAE-Toyopearlによるカラムクロマトグラフィーにて精製した。4℃下にて、酵素液を20mMKPB(pH7.0)で平衡化したDEAE-Toyopearl (東ソー社;φ3×15cm)に供し、NaCl0〜0.5Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、5mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。酵素活性は、D−ガラクチュロン酸の還元に伴うNADPHの酸化減少量を、NADPHの特異吸収波長(OD340)の初速度変化を測定することによって求めた(図1)。DEAE-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムを図2に示す。活性の強い45〜60本目のフラクションを回収した。
【0021】
次に、DEAE-Toyopearl カラムクロマトグラフィーにより得られた活性画分を回収し、10mMKPB(pH7.0)で透析した後、同緩衝液で平衡化したRed-Toyopearl (東ソー社;φ2×4cm)に供し、NaCl0〜0.6Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、5mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。Red-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムを図3に示す。SDS−PAGEで活性のあった各フラクションの精製純度を確認後、夾雑タンパクが少ない61〜69本目のフラクションを回収した。SDS−PAGEは、Current Protocols in Molecular Biology(WILEY INTERSCIENCE社製)に記載されている方法に従った。このとき使用されたSDS−ポリアクリルアミドゲルの組成を表1に示す。分子量マーカーはXL-Lader Broad(APRO社製)が用いられた。酵素液とその1/3量のサンプルバッファー(表2)を混合し、100℃、5分間熱処理して調製した試料を20mAで電気泳動後、クマシーブルー染色を行った。
【0022】
【表1】

【0023】
【表2】
【0024】
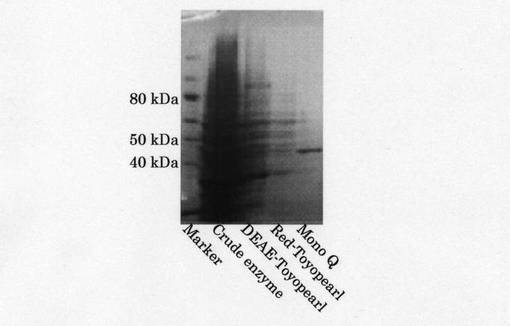
Red-Toyopearlカラムクロマトグラフィーにより得られた活性画分を回収し、10mMKPB(pH7.0)で透析した後、同緩衝液で平衡化したMono Q HR 5/5パックカラム (Amersham Pharmacia Biotech社製)に供し、NaCl〜0.4Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、1mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。Mono Q カラムクロマトグラフィーにより得られたクロマトグラムを図4に示す。上記SDS−PAGEによりMono Qカラムクロマトグラフィー後の活性画分の精製純度を調べた。79〜86本目のフラクションに約45kDaの電気泳動的に単一のバンドを確認することができた。各精製段階の酵素液の泳動写真による画像を図5に示す。
【0025】
3.内部アミノ酸配列の決定
3−1.プロテインシーケンサーによるN末端アミノ酸配列の解析
上記培養菌体を遠心分離 (10000rpm、10分)で集菌し、回収した菌体を適量1.5mlのエッペンチューブに入れ、ビーズ(直径0.1mm)1.0gと10mMKPB(pH7.0)を100μl加え、BIORUPTOR(コスモバイオ社製)を用いて、出力250W、インターバル30秒の条件で20分間超音波破砕を行った。その後、遠心分離(15000rpm、5分)し、上清を粗酵素液として回収した。
【0026】
この粗酵素液30μlをSDS−PAGEに供し、クマシーブルー染色後、酵素をHorizBLOT(ATTO社製)を用いてセミドライブロッティングを行い、PVDF膜に吸着させた。詳細な方法は説明書に従った。そして、プロテインシーケンサー PPSQ-33AでN末端アミノ酸配列解析を行った。SDS−PAGEは上記2.酵素の精製の項に記載の方法に従った。
【0027】
3−2.LC/IT/TOF−MSによるアミノ酸配列の解析
3−1.で得られた粗酵素液5μlをSDS−PAGEに供し、銀染色(Silver Stain, Wako Pure Chemicals Co)を行った。次にバンドを切り出し、下記の脱色、脱水、還元アルキル化、トリプシン消化を行い、分析用試料を得た。トリプシンペプチドのアミノ酸配列は、エレクトロスプレイイオン化ソース(NanoFrontier L; Hitachi High-Technologies, Ltd, Tokyo)を備え付けたLC/IT/TOFMSとNCBIデータベース(NCBInr 20080523) に接続したMascot serch software (http://www.matrixscience.com)によって分析した。
【0028】
(脱色)
1.100μlの脱色液を切り出したゲルに加え20分間室温で攪拌する。
2.100μlのMilliQ水を加え室温で10分間攪拌する。
3.溶媒を除去し、ゲルが完全に除去されるまで1.及び2.の操作を3回繰り返す。
【0029】
(脱水)
1.25mMの炭酸水素ナトリウムを含む50%アセトニトリルを上記脱色後の残留物に加え5分間室温で攪拌する。
2.アセトニトリルを除去し、Speed Vacuumにより10分間乾燥させる。
【0030】
(還元アルキル化)
1.上記乾燥物に25mMの炭酸水素ナトリウムを含む10mMジチオトレイトールを100μl加え、窒素雰囲気下において56℃、60分間攪拌する。
2.溶媒を除去し、100μlの25mMの炭酸水素ナトリウムを加え、室温にて10分間攪拌する。
3.25mMの炭酸水素ナトリウムを含む55mMヨードアセトアミド100μlを加え、暗所室温にて45分間攪拌する。
4.溶媒を除去し、2.の工程を繰り返し、溶媒を除去する。
【0031】
(トリプシン消化)
1.上記で得られた試料に、25mMの炭酸水素ナトリウムを含む50%アセトニトリルを加え、室温で10分間攪拌する。
2.溶媒を除去し、1.の工程を繰り返し、溶媒を除去する。
3.Speed Vacuumにより15分間乾燥させる。
4.50μlのトリプシンゴールド(Promega社製;10μg/ml25mMの炭酸水素ナトリウム)を加え、37℃で1晩インキュベートする。
5.他の試験管に4.のトリプシン溶液を移し、100μlの25mMの炭酸水素ナトリウムを加え、室温で15分間攪拌する。
6.溶液を他の試験管に移し、5%のホルマリンを含む50%アセトニトリルを加え、室温で30分間攪拌する。
7.上記5.及び6.の工程を再度繰り返す。
8.全ての溶液を集め(290μL)、Speed Vacuumにより約20μLまで濃縮する。
9.濃縮液をCosmospin filter G(Nakalai tesque社製)に移し、5,000rpm、3分間遠心分離する。
【0032】
3−3.プロテインシーケンサーによる内部配列の解析
上記2.酵素の精製の項で得られた精製酵素30μlをSDS−PAGEに供し、クマシーブルー染色後、目的の45kDaバンド部分を切り出し、島津テクノリサーチ社に委託解析した。そこでは切り出したバンドを還元アルキル化、Lysyl Endopeptidase処理後In-gel digestion法により抽出したペプチド断片をHPLCにおいて精製分取を行った。そして、分取したペプチド断片を、プロテインシーケンサー PPSQ-33Aでアミノ酸配列解析を行った。得られた配列情報はNCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)を用いて検索した。得られた配列情報はNCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)を用いて検索した。
【0033】
3−4.アミノ酸配列の決定
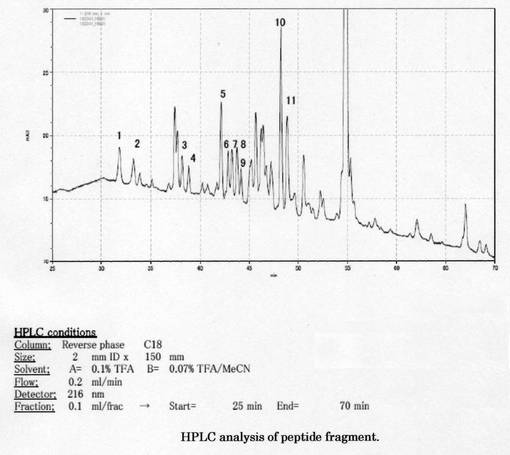
プロテインシーケンサーによるN末端アミノ酸配列の解析、LC/IT/TOFMSによるアミノ酸配列の解析では共にアミノ酸配列を決定することはできなかった。プロテインシーケンサーによる内部配列の解析で、決定することができた。ゲルより抽出したペプチド断片のHPLCの結果を図6に示す。これらの断片のうち、断片1、5、10(Fr−1、5、10) をプロテインシーケンサーPPSQ-33Aにてアミノ酸配列解析を行った。そのアミノ酸配列の結果を表3に示す。
【0034】
【表3】
上記のように3つの断片のアミノ酸配列が決定された。得られたアミノ酸配列を、NCBI BLASTでデータベース (Non-redundant protein sequences) との相同性検索したところ、3つの断片のアミノ酸配列がそれぞれ、糸状菌Pyrenophora tritici-repentisのD−ガラクチュロン酸還元酵素 (ACCESSION : XP_001930351)の部分アミノ酸配列と80%ほどの相同性を示した。よって得られたアミノ酸配列情報は正しいものであると考えられた。なお、断片Fr−1のN末端アミノ酸はGlu又はTyrの何れかであり、断片Fr−5のN末端アミノ酸はLys又はGlnの何れかであり、それに隣接するアミノ酸はGly又はHisの何れかであり、断片Fr−10のN末端アミノ酸はGly又はHisの何れかであるが、いずれも確定できなかった。また、断片Fr−5の10番目のアミノ酸も確定できなかった。
【0035】
4.3'-RACE法によるcDNA部分遺伝子のクローニングおよび塩基配列解析
4−1.酵素活性のタイムコース
ガラクチュロン酸還元酵素のmRNAを獲得するために、酵素活性の培養時間におけるタイムコースを測定した。上記2.1に示すガラクチュロン酸培地を100ml入れた500ml容坂口フラスコに、種培養したC. diffluens FC 11株を1ml植菌し、30℃で振騰培養した。そして回収できる菌体量まで培養した後、タンパク量あたりのガラクチュロン酸還元酵素活性が次第に強くなっている培養時間を確認するまで、2時間ごとに菌体を回収し比活性を測定した。菌体は遠心分離(10000rpm、10分間)で集菌し、上記3−1.プロテインシーケンサーによるN末端アミノ酸配列の解析の項に記載の方法で破砕し、酵素活性を測定した(結果は図示せず)。
【0036】
4−2.C. diffluens FC 11株からのTotal RNAの抽出とmRNAの精製
菌体は、タンパク量あたりのガラクチュロン酸還元酵素活性が次第に強くなっている11時間培養のものを使用した。培養菌体を遠心分離(10000 rpm、10分間)で集菌し、その菌体を、180℃で乾熱滅菌したメタルコーンMC−0212と共に破砕用チューブにいれ、液体窒素によって完全に凍結させた。その後、マルチビーズショッカー(安井器械社製)を用いて、1700rpm、20秒の操作を1回行うことで破砕した。Total RNA抽出はRNAqueous(AMBION社製)を用いて行った。方法は付属説明書に従った。混入したDNAを完全に除去するためOligotexTM-dt30 mRNA Purification kit(タカラバイオ社製)を用い、説明書に従ってmRNAを精製した。
【0037】
4−3.一本鎖cDNAの合成
抽出したRNAを用いて表4に示す試料を調製した。これを65℃で5分間インキュベートし、氷冷後、表5に示すcDNA合成反応液を調製した。30℃で30分間の前処理、続いて42℃で60分間反応し、99℃で5分間処理して反応を停止させた後、氷冷した。
【0038】
【表4】
【0039】
【表5】
【0040】
4−4.PCR
5´側のプライマーには、タンパク質の内部アミノ酸配列(表3)から作製した混合プライマーを使用した(表6)。また、3´側はT20APプライマーに相補するAP1およびAP2-2プライマーを用いた(表7)。PCRの特異性を高める為に、内部配列情報を基にしてnested PCRを行った。PCRには、Blend Ta(TOYOBO社製)を使用した。反応液組成とPCR条件は表に示す。
【0041】
【表6】
【0042】
【表7】
【表8】
【0043】
4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換
増幅したPCR産物をアガロース電気泳動に供した後、目的断片を切り出して、GFX PCR DNA and Gel Band Purification Kit(GE ヘルスケアバイオサイエンス)を用いて精製した。精製後、TOPO TA cloning Kit(invitrogen社製)を用いてpCR2.1-TOPO(図7)へ導入した。ライゲーション条件はキット説明書に従った。ライゲーション後、大腸菌を形質転換して、生じた白コロニーからコロニーダイレクトPCR法(使用プライマー:M13Forward、M13Reverse)により、DNA断片が挿入されているコロニーを選抜し、プラスミドを抽出した。獲得したプラスミドを鋳型としてDNAシーケンスを行った。シーケンス時のプライマーは、M13Forward、M13Reverse(表9)を用いた。
【0044】
【表9】
【0045】
形質転換は次の方法に従った。ライゲーション後の反応液を用いてE. coli HB101の形質転換を行った。コンピテントセルはHB101 Competent cells(タカラバイオ社製)を使用した。コンピテントセル10μlとプラスミドDNAをおだやかに混合し、20分間氷中で放置した後、42℃で45秒間の熱ショックを与えた。氷冷後、LB液体培地(Yeast extract0.5%、Peptone1.0%、NaCl0.5%、pH7.0)を100μl加え、37℃で60分間静置培養し、試料をLBAプレート(LB液体培地に寒天1.5%を加え、120℃、20分のオートクレーブを行った後、50℃まで冷却し、アンピシリンを50mg/mlとなるように添加したもの)に塗布した後、37℃で一晩静置培養した。
【0046】
また、コロニーダイレクトPCRは、生育したコロニーを鋳型としてPCRを行って、DNA断片が挿入されているコロニーを選抜した。反応液組成とPCR条件は表10に示す。プライマーは目的とする遺伝子に適応したものを用いた。プラスミド抽出は、大腸菌を50μg/mlのアンピシリンを含むLB液体培地に植菌し、37℃で一晩培養した。菌体を回収後、アルカリSDS法により行った。
【0047】
【表10】
【0048】
4−6.結果
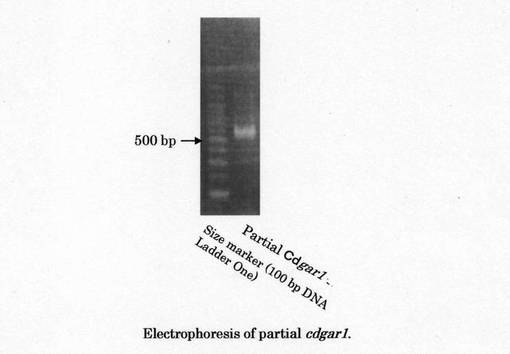
FC11株のmRNAから合成した一本鎖cDNAを鋳型とした3'-RACE法によって、約550bpのcDNA部分遺伝子を獲得した(図8)。サイズマーカーには100bp DNAラダーワン(ナカライテスク社製)を使用した。この断片を上記のとおりpCR2.1-TOPOへ導入し、HB101 Competent cellsを形質転換し、プラスミド取得後に当該部分遺伝子の塩基配列を解読した。塩基配列決定はタカラバイオ株式会社に依頼した。塩基配列の解析は遺伝子解析ソフトGENETYX(Software Development)を用いて行った。当該部分cDNAのシーケンス結果を図9に示す。その塩基配列は配列番号13に、それから得られる部分遺伝子のアミノ酸配列は配列番号14に示される。このアミノ酸配列は、ガラクチュロン酸還元酵素のアミノ酸配列276番目のグリシンからC末端までに該当する。
【0049】
5.カセットPCR法による5´領域ゲノムDNAのクローニング及び塩基配列解析
5−1.ゲノムDNAの調製
菌体は、上記4−1.酵素活性のタイムコースの項に準じて培養した。ただし、培養時間は菌体量の多い2日目のものを用いた。その培養菌体を遠心分離(10000rpm、10分間)で集菌し、乳鉢に液体窒素を注いで乳鉢を十分に冷やした後に集菌した菌体を入れ、乳棒ですりつぶした。ゲノム抽出はDNeasy Plant Maxi Kit(QIAGEN社製)を用いて行った。方法は付属説明書に従った。
【0050】
5−2.鋳型の調製
上記で調製したゲノムDNAを各種制限酵素(EcoR I、Hind III、Pst I、Sal I、Xba I (TOYOBO社製):20U)で、37℃、3時間の酵素処理を行った。制限酵素を熱失活させた後、PCR in vitro Cloning Kit(タカラバイオ社製)に添付の各種カセットとライゲーションを行い、カセットPCRの鋳型を調製した。詳細な方法は説明書に従った。
【0051】
5−3.PCR法
3´側には上記4.3'-RACE法で得られたcDNA塩基配列を基に作製したプライマーgar cas-1、gal cas-2を、5´側には各種カセット部分と相補するcassette c1、cassette c2を用いた(表11)。調製した各種カセットDNAを鋳型としnested PCRを行った。PCR反応液組成および条件は表12に示した。
【0052】
【表11】
【0053】
【表12】
【0054】
5−4.DNAシーケンス
上記4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換の項に記載の方法に準じて行った。
【0055】
5−5.結果
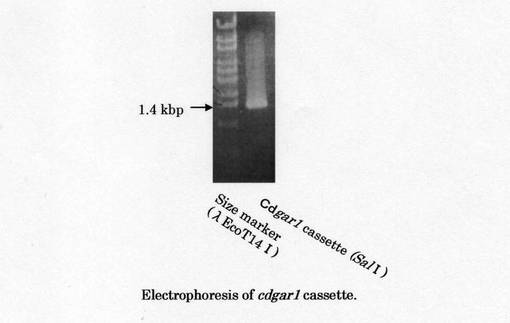
上記4−6.の結果を基に作製したプライマーと、鋳型としてカセット付ゲノムを用いて、カセットPCR法により、5´領域ゲノムDNA断片の獲得を試みた。鋳型にSalIcassetteを用いた場合に、約1.4kbpのDNA断片を獲得した(図10) 。この断片を精製し、pCR2.1-TOPOへ導入し、HB101 Competent cellsを形質転換した。プラスミド取得後塩基配列を解読した。シーケンス結果を図11及び配列番号19に示す。
【0056】
6.cDNA及び遺伝子全長の塩基配列解析
上記3'-RACE法とカセットPCR法から決定したDNA配列情報を基に、cDNA及び遺伝子全長の塩基配列解析を行った。獲得した塩基配列とそれをアミノ酸情報にした配列を、それぞれBLAST検索した。その結果、塩基配列は相同性のあるものを見つけることはできなかったが、アミノ酸配列はCryptococcus neoformansをはじめとする生物の様々な酸化還元酵素、A. nigerや P. tritici-repentis、H. jecorinaのガラクチュロン酸還元酵素と54〜64%の高い相同性を示した。このことから全アミノ酸配列も、カビのガラクチュロン酸還元酵素と高い相同性を示すことが予想された。また、カセットPCR法によって5´領域ゲノムDNAの塩基配列が取得された。ガラクチュロン酸還元酵素の分子量45kDaから考えると、Cdgar1の開始コドンまでの塩基配列情報を獲得できていると考えられた。また、5´領域ゲノムDNAの塩基配列情報からGT-AGルールに則ってイントロン予測、アミノ酸配列情報の相同性検索を行った結果、アミノ酸配列情報とイントロンを予測できた。また、N末端アミノ酸をコードする塩基配列情報の予測もできた。これらの情報と3'-RACE法で獲得した情報を元に作製したプライマーを用いて、cDNAを鋳型としてCdgar1全長を取得した。また、前記プライマーを用いてPCR法によりATGから終止コドン部まで増幅した上で、cDNAの5´上流領域(135塩基)を決定し全遺伝子全長の塩基配列を決定した。
【0057】
6−1.PCR法
カセットPCR法で決定した塩基配列をアミノ酸情報に変換後BLASTにて相同性検索し、N末端アミノ酸配列推定、そこからN末端プライマーを作製した。C末端プライマーは3'-RACE法で得られた塩基配列を元に作製した。4−3.の項で調製した一本鎖cDNAを鋳型としてPCR法を行った。また、5−1.の項で調製したゲノムDNAを鋳型としてPCR法を行った。各PCRにおいてプライマーには表13に示すプライマーを、PCRにはPhusion DNA ポリメラーゼを使用した。PCR反応液組成および条件は表14に示した。なお、ゲノムDNAの場合はcDNA代わりにゲノムDNA(0.1μg/μl)を使用した。
【表13】
【表14】
【0058】
6−2.DNAシーケンス
上記4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換の項に記載の方法と同様にして行った。
【0059】
6−3.配列決定
上記DNAシーケンスの結果から、Cryptococcus diffluens FC11株から得られるガラクチュロン酸還元酵素のゲノムDNAの塩基配列が決定され、それらは図12及び配列番号2で示された。また、D−ガラクチュロン酸還元酵素(CdGar1)をコードするcDNA塩基配列(CdGar1)は図13及び配列番号1で示されると共にD−ガラクチュロン酸還元酵素(CdGar1)のアミノ酸配列は図14及び配列番号22で示される。
【産業上の利用可能性】
【0060】
本発明によると新規なガラクチュロン酸還元酵素の量産が容易になる。この酵素を利用することにより、ペクチンの有効利用を促進することができる。
【技術分野】
【0001】
本発明はガラクチュロン酸還元酵素をコードする遺伝子に関する。
【背景技術】
【0002】
ペクチンは野菜や果実に多く含まれているが、その再利用に関して、例えばりんご搾汁残渣に含まれるペクチンは、その約70%が農作物の肥料、家畜の飼料、増粘多糖類、ゲル化剤、乳タンパク安定剤などの食品添加物に再利用されるにすぎず、残る約30%のペクチンが廃棄されているとの報告がある。また、ペクチンはりんご以外の果物、特に柑橘類の搾汁残渣にも多く含まれるが、その再利用率はりんごの場合よりもさらに低いといわれている。このようにペクチンの再利用は肥料等の安価な利用かつ食品産業など限られた産業における利用にとどまっており、ペクチンを幅広い産業でバイオマス資源として再利用することは天然資源の確保の面からも、ごみ処理という環境浄化の面からも注目に値する。
【0003】
ペクチンは植物細胞壁・特に果実の細胞壁に多く含まれる非セルロース性多糖であり、ホモガラクツロナン、ラムノガラクツロナン−I、ラムノガラクツロナン−IIという3つの特徴的な構造ドメインから形成される複合多糖である。このうち、ホモガラクツロナンはペクチンの主要構成糖として知られており、ペクチンの約70%を占める。ホモガラクツロナンの中でもっとも一般的な構造はガラクチュロン酸のみがα−1,4−結合したポリガラクチュロン酸であり、一部のガラクチュロン酸にはそのカルボキシル基がメチルエステル化されたものやその水酸基がアセチル化されたものが存在している。
【0004】
ポリガラクチュロン酸は様々な微生物によって分解される。例えば、植物寄生性を示すバクテリア、例えば一部の糸状菌は、ガラクチュロン酸をピルビン酸とグリセルアルデヒド3リン酸にまで代謝する。この代謝は、真性細菌によく見られる代謝系であるエントナードウトロフ(ED)経路を利用することによって行われている。また、Kluyveromyces属酵母など、酵母においても、ポリガラクチュロン酸のグルコシド結合を加水分解するポリガラクチュロン酸分解酵素(ポリガラクチュロナーゼ)を生産する種はいくつか知られている(例えば非特許文献1や2参照)。
【0005】
しかしながら、ガラクチュロン酸をピルビン酸とグリセルアルデヒド3リン酸にまで代謝するED経路は真核生物では確認されておらず、酵母ではポリガラクチュロン酸から分解されたガラクチュロン酸を利用することはできないと考えられていた。事実、Kluyveromyces属酵母をはじめとしたポリガラクチュロナーゼ生産酵母において、ポリガラクチュロナーゼ分解産物であるガラクチュロン酸が資化されたという報告例はない。そして、ガラクチュロン酸を資化する酵母を利用することができれば、ペクチンの再資源化においても有効な手段を与えると考えられる。
【0006】
ガラクチュロン酸を還元する酵素として、これまでのところカビの一種であるHypocreajecorina株から得られたL−ガラクチュロン酸脱水素酵素が報告されているが(非特許文献3)、酵母を起源とするものは知られていない。しかも、低温においても活性を維持するものは知られていない。
【0007】
一方、近年では、食品の熟成等に用いられている低温機能性酵素への期待が高まっており、その低温適応性微生物が着目されている。
【0008】
このような状況下において、本願発明者らは、タイ地酒オウ(Ou)の酒もと(Cocha)から、低温適応酵母であるCryptococcus diffluens FC11株を分離し、当該酵母株から10〜30℃の低温においても比較的高い活性を示すD−ガラクチュロン酸還元酵素を見いだした(非特許文献4、5)
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Hirose, N., Kishida, K., Kawasaki, H. and Sakai, T. (1999) Purification and characterization of an endo-polygalacturonase from a mutant of Saccharomyces cerevisiae., Bioscience Biotechnology and Biochemistry vol.63, 1100-1103
【非特許文献2】Sienkstele, R., Bartkeviciute, D. and Sasnauskas K. (1999) Cloning,targeted disruption and heterologous expression of the Kluyveromyces marxianus endo-polygalacturonase gene (EPG1). Yeast vol.15, 311-322
【非特許文献3】Kuorelahti, S., Kalkkinen, N., Penttila, M., Londesborough, J., Richard, P., Identification in the mold Hypocrea jecorina of the first fungal D-galacturonic acid reductase, Biochemistry vol. 44: 11234-11240 (2005)
【非特許文献4】Kishida M., Seike Y., Kawasaki H. (2009) Identification and characterization of psychrophilic yeast newly isolated from fermentative source (Loog-pang) of traditional drink in Thailand. Biocontrol Science Vol.14, p119-122.
【非特許文献5】岸田正夫ら、「低温適応酵母が生産するD−ガラクチュロン酸還元酵素」 第60回日本生物工学回大会 講演要旨集、p133
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は上記D−ガラクチュロン酸還元酵素のアミノ酸配列を解析するとともにそれをコードする遺伝子を見いだし、工業的な生産を可能とすべく当該酵素をコードするcDNAを提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明に係るポリヌクレオチドは、D−ガラクチュロン酸還元酵素をコードする遺伝子であって、配列番号1に示される塩基配列を含むポリヌクレオチドである。
【発明の効果】
【0012】
本発明によると、低温でも高活性を示すD−ガラクチュロン酸還元酵素をコードする遺伝子が提供される。これにより当該酵素の工業的生産が可能となり、当該酵素を有効利用しうる新規な用途が期待される。
【図面の簡単な説明】
【0013】
【図1】図1はガラクチュロン酸還元酵素の活性測定方法を示す図である。
【図2】図2はDEAE-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムである。
【図3】図3はRed-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムである。
【図4】図4はMono Q カラムクロマトグラフィーにより得られたクロマトグラムである。
【図5】図5は各精製段階における酵素液の電気泳動画像である。
【図6】図6はゲルより抽出したペプチド断片のHPLCの結果を示す図である。
【図7】図7はTOPO TAクローニングベクターpCR2.1-TOPOの遺伝子地図である。
【図8】図8は3'-RACE法により得られた約550bpのcDNA部分遺伝子の泳動画像である。
【図9】図9は3'-RACE法により得られた約550bpのcDNA部分遺伝子の塩基配列を示す図である。
【図10】図10はカセットPCR法により得られた5´領域ゲノムDNA断片の電気泳動画像である。
【図11】図11はカセットPCR法により得られた5´領域ゲノムDNA断片の塩基配列を示す図である。
【図12】図12は決定されたD−ガラクチュロン産還元酵素の全遺伝子の塩基配列を示す図である。
【図13】図13は決定されたD−ガラクチュロン産還元酵素をコードするcDNAの塩基配列を示す図である。
【図14】図14は決定されたD−ガラクチュロン産還元酵素のアミノ酸配列を示す図である。
【発明を実施するための形態】
【0014】
本発明の遺伝子がコードするD−ガラクチュロン酸還元酵素は、上記のようにタイ地酒オウ(Ou)の酒もと(Cocha)から分離された低温適応酵母であるCryptococcus diffluens FC11株(寄託番号:NITE P-611、寄託日:平成20年7月18日)から取得される。当該酵素はD−ガラクチュロン酸を還元してL−ガラクトン酸を生成する。当該酵素は特願2008−285976号に開示されており、その記載が本願において援用される。その酵素の特徴として、特に10〜30℃の低温においても、至適温度における活性の60%程度の活性効率で反応を行わせることができ、室温条件でもガラクチュロン酸をL−ガラクトン酸に変換できることが挙げられる。当該酵素は、以下の理化学的特徴を有する。
【0015】
(1)温度安定性及びpH安定性
20〜45℃及びpH6.0〜8.4の環境下で安定である。
(2)至適温度及び至適pH
至適温度は40℃付近、至適pHは6.3付近である。
(3)基質特異性
ガラクチュロン酸、グルクロン酸に作用するが、ガラクトース、グルコース、マンノース、ソルビトール、グルコン酸には作用しない。
(4)金属による影響
各1mMのCu2+、Fe2+、Pb2+によって阻害を受けるが、各1mMのMg2+、Co2+、Mn2+、Na+、Ca2+、Ni2+、K+、Ba2+、Zn2+によって阻害を受けない。
(5)電子供与体
NADPH特異的である
【0016】
本発明に係る遺伝子は、上記ガラクチュロン酸還元酵素をコードするcDNAであって、配列番号1に示された塩基配列を有する。本発明においては、上記ガラクチュロン酸還元酵素を生産できる限りにおいて、配列番号1に示される塩基配列から、1〜数十個の塩基が欠失・置換・挿入された塩基配列からなるか、相同性が80%以上、好ましくは90%以上、望ましくは95%以上である塩基配列からなる遺伝子であってもよい。
【0017】
本発明に係る遺伝子は下記実施例において説明した方法により取得されるが、一般の遺伝子工学的手法により合成することもできる。また、本発明の遺伝子は、一般的な遺伝子工学的手法によって、その5´末端側にプロモータ領域、3´末端側にターミネータ領域などタンパク質発現のために必要な領域が結合された上で発現ベクターに組み込まれ、その後大腸菌など適当な宿主を形質転換して目的とする酵素の生産に利用される。
【実施例1】
【0018】
次に、本発明の遺伝子について下記の実施例に基づいて詳細に説明する。
〔ガラクチュロン酸還元酵素遺伝子のクローニング〕
1.菌の培養
単離された C. diffluens OPU-FC11株を使用した。種培養にはGYP培地(Yeast extract1%、Peptone1%、Glucose1%、pH5.0)、本培養にはガラクチュロン酸培地(Yeast extract1%、Peptone1%、D-Galacturonic acid (GalA)0.2%、pH5.0)を用いた。GYP培地を入れた短試験管に、菌を1白金耳植菌し、30℃1日間振盪培養した。その培養液を更に、種培養培地9本に50mlずつ植菌し、30℃で1日間振盪培養した。その培養液を500ml容坂口フラスコに200mlのガラクチュロン酸培地を入れた本培養培地20本に2mlずつ植菌し、計4Lを30℃で2日間振盪培養した。
【0019】
2.ガラクチュロン酸還元酵素の精製
2−1.粗酵素液の調整
培養菌体を遠心分離(10000rpm、10分)で集菌し、20mMリン酸カリウム緩衝液(以下「KPB」という)(pH7.0)で洗菌後、同緩衝液で懸濁した。懸濁菌体はOHTAKE WORKSのFRENCH PRESSを用いて1500kg/cm2で1回破砕を行った。破砕した菌体を遠心分離(10000rpm、10分)により、上清画分と沈殿(細胞残渣)画分に分離した。上清画分を10mMKPBで十分に透析したものを粗酵素として用いた。
【0020】
2−2.酵素の精製
得られた粗酵素をDEAE-Toyopearlによるカラムクロマトグラフィーにて精製した。4℃下にて、酵素液を20mMKPB(pH7.0)で平衡化したDEAE-Toyopearl (東ソー社;φ3×15cm)に供し、NaCl0〜0.5Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、5mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。酵素活性は、D−ガラクチュロン酸の還元に伴うNADPHの酸化減少量を、NADPHの特異吸収波長(OD340)の初速度変化を測定することによって求めた(図1)。DEAE-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムを図2に示す。活性の強い45〜60本目のフラクションを回収した。
【0021】
次に、DEAE-Toyopearl カラムクロマトグラフィーにより得られた活性画分を回収し、10mMKPB(pH7.0)で透析した後、同緩衝液で平衡化したRed-Toyopearl (東ソー社;φ2×4cm)に供し、NaCl0〜0.6Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、5mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。Red-Toyopearlカラムクロマトグラフィーにより得られたクロマトグラムを図3に示す。SDS−PAGEで活性のあった各フラクションの精製純度を確認後、夾雑タンパクが少ない61〜69本目のフラクションを回収した。SDS−PAGEは、Current Protocols in Molecular Biology(WILEY INTERSCIENCE社製)に記載されている方法に従った。このとき使用されたSDS−ポリアクリルアミドゲルの組成を表1に示す。分子量マーカーはXL-Lader Broad(APRO社製)が用いられた。酵素液とその1/3量のサンプルバッファー(表2)を混合し、100℃、5分間熱処理して調製した試料を20mAで電気泳動後、クマシーブルー染色を行った。
【0022】
【表1】
【0023】
【表2】
【0024】
Red-Toyopearlカラムクロマトグラフィーにより得られた活性画分を回収し、10mMKPB(pH7.0)で透析した後、同緩衝液で平衡化したMono Q HR 5/5パックカラム (Amersham Pharmacia Biotech社製)に供し、NaCl〜0.4Mの直線濃度勾配法により溶出した。流速は1.0ml/minで行い、1mlずつ分画を行った。その後、それぞれのフラクションのタンパク質量と活性を測定した。Mono Q カラムクロマトグラフィーにより得られたクロマトグラムを図4に示す。上記SDS−PAGEによりMono Qカラムクロマトグラフィー後の活性画分の精製純度を調べた。79〜86本目のフラクションに約45kDaの電気泳動的に単一のバンドを確認することができた。各精製段階の酵素液の泳動写真による画像を図5に示す。
【0025】
3.内部アミノ酸配列の決定
3−1.プロテインシーケンサーによるN末端アミノ酸配列の解析
上記培養菌体を遠心分離 (10000rpm、10分)で集菌し、回収した菌体を適量1.5mlのエッペンチューブに入れ、ビーズ(直径0.1mm)1.0gと10mMKPB(pH7.0)を100μl加え、BIORUPTOR(コスモバイオ社製)を用いて、出力250W、インターバル30秒の条件で20分間超音波破砕を行った。その後、遠心分離(15000rpm、5分)し、上清を粗酵素液として回収した。
【0026】
この粗酵素液30μlをSDS−PAGEに供し、クマシーブルー染色後、酵素をHorizBLOT(ATTO社製)を用いてセミドライブロッティングを行い、PVDF膜に吸着させた。詳細な方法は説明書に従った。そして、プロテインシーケンサー PPSQ-33AでN末端アミノ酸配列解析を行った。SDS−PAGEは上記2.酵素の精製の項に記載の方法に従った。
【0027】
3−2.LC/IT/TOF−MSによるアミノ酸配列の解析
3−1.で得られた粗酵素液5μlをSDS−PAGEに供し、銀染色(Silver Stain, Wako Pure Chemicals Co)を行った。次にバンドを切り出し、下記の脱色、脱水、還元アルキル化、トリプシン消化を行い、分析用試料を得た。トリプシンペプチドのアミノ酸配列は、エレクトロスプレイイオン化ソース(NanoFrontier L; Hitachi High-Technologies, Ltd, Tokyo)を備え付けたLC/IT/TOFMSとNCBIデータベース(NCBInr 20080523) に接続したMascot serch software (http://www.matrixscience.com)によって分析した。
【0028】
(脱色)
1.100μlの脱色液を切り出したゲルに加え20分間室温で攪拌する。
2.100μlのMilliQ水を加え室温で10分間攪拌する。
3.溶媒を除去し、ゲルが完全に除去されるまで1.及び2.の操作を3回繰り返す。
【0029】
(脱水)
1.25mMの炭酸水素ナトリウムを含む50%アセトニトリルを上記脱色後の残留物に加え5分間室温で攪拌する。
2.アセトニトリルを除去し、Speed Vacuumにより10分間乾燥させる。
【0030】
(還元アルキル化)
1.上記乾燥物に25mMの炭酸水素ナトリウムを含む10mMジチオトレイトールを100μl加え、窒素雰囲気下において56℃、60分間攪拌する。
2.溶媒を除去し、100μlの25mMの炭酸水素ナトリウムを加え、室温にて10分間攪拌する。
3.25mMの炭酸水素ナトリウムを含む55mMヨードアセトアミド100μlを加え、暗所室温にて45分間攪拌する。
4.溶媒を除去し、2.の工程を繰り返し、溶媒を除去する。
【0031】
(トリプシン消化)
1.上記で得られた試料に、25mMの炭酸水素ナトリウムを含む50%アセトニトリルを加え、室温で10分間攪拌する。
2.溶媒を除去し、1.の工程を繰り返し、溶媒を除去する。
3.Speed Vacuumにより15分間乾燥させる。
4.50μlのトリプシンゴールド(Promega社製;10μg/ml25mMの炭酸水素ナトリウム)を加え、37℃で1晩インキュベートする。
5.他の試験管に4.のトリプシン溶液を移し、100μlの25mMの炭酸水素ナトリウムを加え、室温で15分間攪拌する。
6.溶液を他の試験管に移し、5%のホルマリンを含む50%アセトニトリルを加え、室温で30分間攪拌する。
7.上記5.及び6.の工程を再度繰り返す。
8.全ての溶液を集め(290μL)、Speed Vacuumにより約20μLまで濃縮する。
9.濃縮液をCosmospin filter G(Nakalai tesque社製)に移し、5,000rpm、3分間遠心分離する。
【0032】
3−3.プロテインシーケンサーによる内部配列の解析
上記2.酵素の精製の項で得られた精製酵素30μlをSDS−PAGEに供し、クマシーブルー染色後、目的の45kDaバンド部分を切り出し、島津テクノリサーチ社に委託解析した。そこでは切り出したバンドを還元アルキル化、Lysyl Endopeptidase処理後In-gel digestion法により抽出したペプチド断片をHPLCにおいて精製分取を行った。そして、分取したペプチド断片を、プロテインシーケンサー PPSQ-33Aでアミノ酸配列解析を行った。得られた配列情報はNCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)を用いて検索した。得られた配列情報はNCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)を用いて検索した。
【0033】
3−4.アミノ酸配列の決定
プロテインシーケンサーによるN末端アミノ酸配列の解析、LC/IT/TOFMSによるアミノ酸配列の解析では共にアミノ酸配列を決定することはできなかった。プロテインシーケンサーによる内部配列の解析で、決定することができた。ゲルより抽出したペプチド断片のHPLCの結果を図6に示す。これらの断片のうち、断片1、5、10(Fr−1、5、10) をプロテインシーケンサーPPSQ-33Aにてアミノ酸配列解析を行った。そのアミノ酸配列の結果を表3に示す。
【0034】
【表3】
上記のように3つの断片のアミノ酸配列が決定された。得られたアミノ酸配列を、NCBI BLASTでデータベース (Non-redundant protein sequences) との相同性検索したところ、3つの断片のアミノ酸配列がそれぞれ、糸状菌Pyrenophora tritici-repentisのD−ガラクチュロン酸還元酵素 (ACCESSION : XP_001930351)の部分アミノ酸配列と80%ほどの相同性を示した。よって得られたアミノ酸配列情報は正しいものであると考えられた。なお、断片Fr−1のN末端アミノ酸はGlu又はTyrの何れかであり、断片Fr−5のN末端アミノ酸はLys又はGlnの何れかであり、それに隣接するアミノ酸はGly又はHisの何れかであり、断片Fr−10のN末端アミノ酸はGly又はHisの何れかであるが、いずれも確定できなかった。また、断片Fr−5の10番目のアミノ酸も確定できなかった。
【0035】
4.3'-RACE法によるcDNA部分遺伝子のクローニングおよび塩基配列解析
4−1.酵素活性のタイムコース
ガラクチュロン酸還元酵素のmRNAを獲得するために、酵素活性の培養時間におけるタイムコースを測定した。上記2.1に示すガラクチュロン酸培地を100ml入れた500ml容坂口フラスコに、種培養したC. diffluens FC 11株を1ml植菌し、30℃で振騰培養した。そして回収できる菌体量まで培養した後、タンパク量あたりのガラクチュロン酸還元酵素活性が次第に強くなっている培養時間を確認するまで、2時間ごとに菌体を回収し比活性を測定した。菌体は遠心分離(10000rpm、10分間)で集菌し、上記3−1.プロテインシーケンサーによるN末端アミノ酸配列の解析の項に記載の方法で破砕し、酵素活性を測定した(結果は図示せず)。
【0036】
4−2.C. diffluens FC 11株からのTotal RNAの抽出とmRNAの精製
菌体は、タンパク量あたりのガラクチュロン酸還元酵素活性が次第に強くなっている11時間培養のものを使用した。培養菌体を遠心分離(10000 rpm、10分間)で集菌し、その菌体を、180℃で乾熱滅菌したメタルコーンMC−0212と共に破砕用チューブにいれ、液体窒素によって完全に凍結させた。その後、マルチビーズショッカー(安井器械社製)を用いて、1700rpm、20秒の操作を1回行うことで破砕した。Total RNA抽出はRNAqueous(AMBION社製)を用いて行った。方法は付属説明書に従った。混入したDNAを完全に除去するためOligotexTM-dt30 mRNA Purification kit(タカラバイオ社製)を用い、説明書に従ってmRNAを精製した。
【0037】
4−3.一本鎖cDNAの合成
抽出したRNAを用いて表4に示す試料を調製した。これを65℃で5分間インキュベートし、氷冷後、表5に示すcDNA合成反応液を調製した。30℃で30分間の前処理、続いて42℃で60分間反応し、99℃で5分間処理して反応を停止させた後、氷冷した。
【0038】
【表4】
【0039】
【表5】
【0040】
4−4.PCR
5´側のプライマーには、タンパク質の内部アミノ酸配列(表3)から作製した混合プライマーを使用した(表6)。また、3´側はT20APプライマーに相補するAP1およびAP2-2プライマーを用いた(表7)。PCRの特異性を高める為に、内部配列情報を基にしてnested PCRを行った。PCRには、Blend Ta(TOYOBO社製)を使用した。反応液組成とPCR条件は表に示す。
【0041】
【表6】
【0042】
【表7】
【表8】
【0043】
4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換
増幅したPCR産物をアガロース電気泳動に供した後、目的断片を切り出して、GFX PCR DNA and Gel Band Purification Kit(GE ヘルスケアバイオサイエンス)を用いて精製した。精製後、TOPO TA cloning Kit(invitrogen社製)を用いてpCR2.1-TOPO(図7)へ導入した。ライゲーション条件はキット説明書に従った。ライゲーション後、大腸菌を形質転換して、生じた白コロニーからコロニーダイレクトPCR法(使用プライマー:M13Forward、M13Reverse)により、DNA断片が挿入されているコロニーを選抜し、プラスミドを抽出した。獲得したプラスミドを鋳型としてDNAシーケンスを行った。シーケンス時のプライマーは、M13Forward、M13Reverse(表9)を用いた。
【0044】
【表9】
【0045】
形質転換は次の方法に従った。ライゲーション後の反応液を用いてE. coli HB101の形質転換を行った。コンピテントセルはHB101 Competent cells(タカラバイオ社製)を使用した。コンピテントセル10μlとプラスミドDNAをおだやかに混合し、20分間氷中で放置した後、42℃で45秒間の熱ショックを与えた。氷冷後、LB液体培地(Yeast extract0.5%、Peptone1.0%、NaCl0.5%、pH7.0)を100μl加え、37℃で60分間静置培養し、試料をLBAプレート(LB液体培地に寒天1.5%を加え、120℃、20分のオートクレーブを行った後、50℃まで冷却し、アンピシリンを50mg/mlとなるように添加したもの)に塗布した後、37℃で一晩静置培養した。
【0046】
また、コロニーダイレクトPCRは、生育したコロニーを鋳型としてPCRを行って、DNA断片が挿入されているコロニーを選抜した。反応液組成とPCR条件は表10に示す。プライマーは目的とする遺伝子に適応したものを用いた。プラスミド抽出は、大腸菌を50μg/mlのアンピシリンを含むLB液体培地に植菌し、37℃で一晩培養した。菌体を回収後、アルカリSDS法により行った。
【0047】
【表10】
【0048】
4−6.結果
FC11株のmRNAから合成した一本鎖cDNAを鋳型とした3'-RACE法によって、約550bpのcDNA部分遺伝子を獲得した(図8)。サイズマーカーには100bp DNAラダーワン(ナカライテスク社製)を使用した。この断片を上記のとおりpCR2.1-TOPOへ導入し、HB101 Competent cellsを形質転換し、プラスミド取得後に当該部分遺伝子の塩基配列を解読した。塩基配列決定はタカラバイオ株式会社に依頼した。塩基配列の解析は遺伝子解析ソフトGENETYX(Software Development)を用いて行った。当該部分cDNAのシーケンス結果を図9に示す。その塩基配列は配列番号13に、それから得られる部分遺伝子のアミノ酸配列は配列番号14に示される。このアミノ酸配列は、ガラクチュロン酸還元酵素のアミノ酸配列276番目のグリシンからC末端までに該当する。
【0049】
5.カセットPCR法による5´領域ゲノムDNAのクローニング及び塩基配列解析
5−1.ゲノムDNAの調製
菌体は、上記4−1.酵素活性のタイムコースの項に準じて培養した。ただし、培養時間は菌体量の多い2日目のものを用いた。その培養菌体を遠心分離(10000rpm、10分間)で集菌し、乳鉢に液体窒素を注いで乳鉢を十分に冷やした後に集菌した菌体を入れ、乳棒ですりつぶした。ゲノム抽出はDNeasy Plant Maxi Kit(QIAGEN社製)を用いて行った。方法は付属説明書に従った。
【0050】
5−2.鋳型の調製
上記で調製したゲノムDNAを各種制限酵素(EcoR I、Hind III、Pst I、Sal I、Xba I (TOYOBO社製):20U)で、37℃、3時間の酵素処理を行った。制限酵素を熱失活させた後、PCR in vitro Cloning Kit(タカラバイオ社製)に添付の各種カセットとライゲーションを行い、カセットPCRの鋳型を調製した。詳細な方法は説明書に従った。
【0051】
5−3.PCR法
3´側には上記4.3'-RACE法で得られたcDNA塩基配列を基に作製したプライマーgar cas-1、gal cas-2を、5´側には各種カセット部分と相補するcassette c1、cassette c2を用いた(表11)。調製した各種カセットDNAを鋳型としnested PCRを行った。PCR反応液組成および条件は表12に示した。
【0052】
【表11】
【0053】
【表12】
【0054】
5−4.DNAシーケンス
上記4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換の項に記載の方法に準じて行った。
【0055】
5−5.結果
上記4−6.の結果を基に作製したプライマーと、鋳型としてカセット付ゲノムを用いて、カセットPCR法により、5´領域ゲノムDNA断片の獲得を試みた。鋳型にSalIcassetteを用いた場合に、約1.4kbpのDNA断片を獲得した(図10) 。この断片を精製し、pCR2.1-TOPOへ導入し、HB101 Competent cellsを形質転換した。プラスミド取得後塩基配列を解読した。シーケンス結果を図11及び配列番号19に示す。
【0056】
6.cDNA及び遺伝子全長の塩基配列解析
上記3'-RACE法とカセットPCR法から決定したDNA配列情報を基に、cDNA及び遺伝子全長の塩基配列解析を行った。獲得した塩基配列とそれをアミノ酸情報にした配列を、それぞれBLAST検索した。その結果、塩基配列は相同性のあるものを見つけることはできなかったが、アミノ酸配列はCryptococcus neoformansをはじめとする生物の様々な酸化還元酵素、A. nigerや P. tritici-repentis、H. jecorinaのガラクチュロン酸還元酵素と54〜64%の高い相同性を示した。このことから全アミノ酸配列も、カビのガラクチュロン酸還元酵素と高い相同性を示すことが予想された。また、カセットPCR法によって5´領域ゲノムDNAの塩基配列が取得された。ガラクチュロン酸還元酵素の分子量45kDaから考えると、Cdgar1の開始コドンまでの塩基配列情報を獲得できていると考えられた。また、5´領域ゲノムDNAの塩基配列情報からGT-AGルールに則ってイントロン予測、アミノ酸配列情報の相同性検索を行った結果、アミノ酸配列情報とイントロンを予測できた。また、N末端アミノ酸をコードする塩基配列情報の予測もできた。これらの情報と3'-RACE法で獲得した情報を元に作製したプライマーを用いて、cDNAを鋳型としてCdgar1全長を取得した。また、前記プライマーを用いてPCR法によりATGから終止コドン部まで増幅した上で、cDNAの5´上流領域(135塩基)を決定し全遺伝子全長の塩基配列を決定した。
【0057】
6−1.PCR法
カセットPCR法で決定した塩基配列をアミノ酸情報に変換後BLASTにて相同性検索し、N末端アミノ酸配列推定、そこからN末端プライマーを作製した。C末端プライマーは3'-RACE法で得られた塩基配列を元に作製した。4−3.の項で調製した一本鎖cDNAを鋳型としてPCR法を行った。また、5−1.の項で調製したゲノムDNAを鋳型としてPCR法を行った。各PCRにおいてプライマーには表13に示すプライマーを、PCRにはPhusion DNA ポリメラーゼを使用した。PCR反応液組成および条件は表14に示した。なお、ゲノムDNAの場合はcDNA代わりにゲノムDNA(0.1μg/μl)を使用した。
【表13】
【表14】
【0058】
6−2.DNAシーケンス
上記4−5.TOPO TAクローニングベクターへのライゲーションおよび形質転換の項に記載の方法と同様にして行った。
【0059】
6−3.配列決定
上記DNAシーケンスの結果から、Cryptococcus diffluens FC11株から得られるガラクチュロン酸還元酵素のゲノムDNAの塩基配列が決定され、それらは図12及び配列番号2で示された。また、D−ガラクチュロン酸還元酵素(CdGar1)をコードするcDNA塩基配列(CdGar1)は図13及び配列番号1で示されると共にD−ガラクチュロン酸還元酵素(CdGar1)のアミノ酸配列は図14及び配列番号22で示される。
【産業上の利用可能性】
【0060】
本発明によると新規なガラクチュロン酸還元酵素の量産が容易になる。この酵素を利用することにより、ペクチンの有効利用を促進することができる。
【特許請求の範囲】
【請求項1】
配列番号22で示されるアミノ酸配列を有するポリペプチドをコードする単離されたポリヌクレオチド。
【請求項2】
配列番号1で示される塩基配列を含むポリヌクレオチド。
【請求項1】
配列番号22で示されるアミノ酸配列を有するポリペプチドをコードする単離されたポリヌクレオチド。
【請求項2】
配列番号1で示される塩基配列を含むポリヌクレオチド。
【図1】


【図2】
【図3】
【図4】
【図7】
【図9】

【図11】

【図12】

【図13】

【図14】

【図5】
【図6】
【図8】
【図10】
【図2】
【図3】
【図4】
【図7】
【図9】
【図11】
【図12】
【図13】
【図14】
【図5】
【図6】
【図8】
【図10】
【公開番号】特開2011−200212(P2011−200212A)
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願番号】特願2010−73693(P2010−73693)
【出願日】平成22年3月26日(2010.3.26)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願日】平成22年3月26日(2010.3.26)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
[ Back to top ]