
キシレン異性化触媒系とその使用方法
【課題】芳香環飽和に対するエチレン飽和比が3,500よりも大きい値を示す触媒系を提供する。
【解決手段】本触媒系は2成分を含み、この各成分が約1〜約12の拘束係数(Constraint Index)を有する結晶モレキュラーシーブおよび第VIII族金属に有効な量を含む。本触媒系は、特にエチルベンゼン変換/キシレン異性化反応に利用することができる。本触媒系は、競争的イオン交換により第VIII族金属をモレキュラーシーブに取り込むことにより調製することができる。
【解決手段】本触媒系は2成分を含み、この各成分が約1〜約12の拘束係数(Constraint Index)を有する結晶モレキュラーシーブおよび第VIII族金属に有効な量を含む。本触媒系は、特にエチルベンゼン変換/キシレン異性化反応に利用することができる。本触媒系は、競争的イオン交換により第VIII族金属をモレキュラーシーブに取り込むことにより調製することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、低い芳香環飽和と組み合わされた高いエチレン飽和活性を示す触媒系に関する。また、本発明は前記触媒系を用いたキシレンの異性化およびエチルベンゼンの変換のためのプロセスに関する。
【背景技術】
【0002】
パラ−キシレンは、通常は選択的溶媒抽出による石油ナフサ、特に改質ガソリンのような原料物質から分離された、C8芳香族の混合物から誘導され得る貴重な化学原料である。これら原料からの前記C8芳香族留分は、かなり広範な組成を有するが、通常は10〜32重量パーセントのエチルベンゼン、および約50重量パーセントのメタ異性体およびそれぞれ25重量パーセントのパラおよびオルト異性体とに分離される残りのキシレンからなる。
【0003】
個々の異性体生産物は、適当な物理的方法によって天然由来の混合物から分離することができる。エチルベンゼンは、費用のかかる操作であるが、分別蒸留により分離することができる。オルト−キシレンは分別蒸留により分離することができ、商業的にはその方法で生産されている。パラ−キシレンは分別析出、選択的吸着(例えば、パーレックス(商標)プロセス)、または膜分離により混合異性体から分離することができる。
【0004】
パラ−キシレンの商業的使用が増大するにつれて、所望のパラ−異性体の収率を上げるために物理的分離と他のキシレンアイソマーの化学的異性化とを組合せることが益々重要になっている。しかしエチレンベンゼンの沸点がパラ−キシレンおよびメタ−キシレンの沸点と非常に接近しているために、蒸留によるC8芳香族供給原料からのエチルベンゼンの完全な除去は実際的ではない。したがって、全ての商業的キシレン異性化プロセスの重要な特徴は、原料のエチルベンゼンを有用な副生物に変換し、同時にキシレンから他の化合物への全ての変換を最小限に抑える性能である。
【0005】
1つの商業的に成功したキシレン変換プロセスは米国特許4,899.011号に記載されている。ここでは、そのパラ−キシレン含量が涸渇したC8芳香族原料は2成分触媒系に接触させられる。第1触媒成分は脱エチル化により選択的にエチルベンゼンを変換してベンゼンを生成し、エチレンはエタンに変換される。一方、第2触媒成分は選択的にキシレンを異性化してパラ−キシレン含量を熱平衡値またはその近くまで上昇させる。第1触媒成分は、120℃において30%の平衡性能のオルト−キシレンを吸着する性能に基づいて50分を超えるオルト−キシレン吸着時間および0.64±0.11kPa(4.5±0.8mm水銀)オルト−キシレン分圧を有し、1〜12の束縛指数(Constraint Index)を有するゼオライトを含む。一方、第2成分は同条件下で10分未満のオルト−キシレン吸着時間を有する束縛指数1〜12のゼオライトを含む。好ましい態様において、第1触媒成分は1ミクロン以上の結晶サイズを有するZSM−5であり、第2触媒成分は0.02〜0.05ミクロンの結晶サイズを有するZSM−5である。また各触媒成分は水素化金属をも含む。
【0006】
米国特許4,899,011号のプロセスに関する改善は米国特許5,689,027号に記述されている。ここで、前記2成分系における第1触媒成分は、011特許で引用される同試験条件下において1200分を超える程度にまでそのオルト−キシレン吸着時間を増加させるために、コークス化により、又はより好ましくはシリカの表面コーティングの蓄積により予備選択活性化されている。そのような系を用いると、011特許のプロセスで得られるよりも著しく低いキシレン損失で、高いエチルベンゼン変換率が達成されることが見出された。ここでも、027特許で用いられる触媒成分は水素化金属を含む。
【0007】
011特許及び027特許のプロセスで用いられる希金属含有ゼオライト触媒の1つの製造方法は米国特許再公表31,919号に開示され、ゼオライトの結晶化後であって、最終触媒粒子の形成前でゼオライトの焼成または水蒸気処理のいずれもの前に、ゼオライトを含むカチオン形状の希金属を取り込むことを含む。希金属がプラチナの場合、919特許の実施例により相対的に低いキシレン損失で改善されたエチルベンゼン変換が得られることが証明される。
【発明の概要】
【発明が解決しようとする課題】
【0008】
上記のような最近の進歩にもかかわらず、平均して低いキシレン損失を達成するエチルベンゼン変換/キシレン異性化用の触媒を欲する継続した要求が依然としてある。したがって、例えば、プラチナ含有触媒はエチレン飽和には有効だが、それらは芳香環飽和も触媒する。さらに、芳香環飽和は低温で熱力学的に増強され、このため、触媒の予備スルフィド化、又はたとえ上昇した温度が生産予定および/またはサイクル時間悪影響を及ぼしても、上昇した温度における操作が必要とされる。
【課題を解決するための手段】
【0009】
<発明の要約>
本発明においては、芳香環飽和に対するエチレン飽和比が3,500を超える触媒系が提供される。本触媒系は2成分を含む。各成分は、約1〜約12の束縛指数(Constraint Index)を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属(米国式分類)を含む。
【0010】
好ましくは触媒系は10,000より大きな、より好ましくは20,000より大きな、さらにより好ましくは25,000より大きな、最も好ましくは30,000より大きな芳香環飽和に対するエチレン飽和比を示す。
【0011】
もう1つの態様において、競争的なイオン交換により第VIII族金属をモレキュラーシーブに取り込むことによって2成分触媒系を生産するプロセスが提供される。本プロセスは、モレキュラーシーブへの第VIII族金属のイオン交換に有効な条件下で、例えばアンモニウムカチオンのような非水素化金属カチオンおよびプラチナカチオンのような第VIII族金属(日本式では第10族)カチオンを含む水溶液を用いて、モレキュラーシーブを接触させることにより実施される。水溶液中の第VIII族金属に対する非水素化金属カチオンのモル比は、約500〜約6000の範囲内である。
【0012】
さらなる態様において、本発明は、エチルベンゼンおよびキシレンを含む原料の異性化のためのプロセスを提供する。本プロセスは、有効な条件下で原料を10,000より大きな芳香環飽和に対するエチレン飽和比を示す触媒系に接触させることにより実施されるプロセスであって、
(a)約1〜約12の束縛指数を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属を含む第1成分を用いて、水素存在下でエチルベンゼン変換条件下で、原料を接触させる工程、および
(b)約1〜約12の束縛指数を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属を用いて、キシレン異性化条件下で工程(a)のエチルベンゼンの涸渇した流出物を接触させる工程、
を含むプロセスである。
【図面の簡単な説明】
【0013】
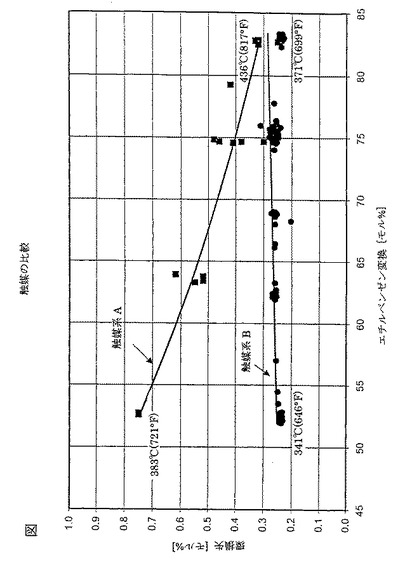
【図1】実施例2の触媒系について、芳香環損失[モル%]に対してプロットされたエチルベンゼン変換[モル%]のグラフを示す図である。
【発明を実施するための形態】
【0014】
<発明の詳細な説明>
本発明において、本触媒系用の芳香環飽和に対するエチレン飽和比は以下の式により決定される。
【0015】
【数1】

【0016】
上記式において示される値は343℃(650°F)の温度、10h-1の重量時間当たり空間速度、1の水素/炭化水素モル比、および1,653kPa−a(225psig)全体圧の圧力で決定される。用いられる原料は、13.0重量%エチルベンゼン、1.0重量%パラ−キシレン、67.0重量%メタ−キシレン、および19.0重量%オルト−キシレンからなるC8芳香族原料である。
【0017】
<原料>
一般に、任意のエチルベンゼンおよびキシレンを含む芳香族C8混合物が本発明のプロセスへの原料として用いられる。一般的に、そのような混合物は、典型的に、約5〜60重量%の範囲のエチルベンゼン含量、約0〜35重量%の範囲のオルト−キシレン含量、約20〜95重量%の範囲のメタ−キシレン含量、および約0〜15重量%の範囲のパラ−キシレンを有する。上記の芳香族C8混合物に加えて、前記原料は非芳香族炭化水素、すなわち約30重量%までの量のナフセンおよびパラフィンを含有することができる。好ましい態様において本発明は、例えば石油ナフサを触媒で改質して得られるC8芳香族混合物を加工して、減少したエチルベンゼン含量および増加したパラ−キシレン含量の混合物を得る手段を提供する。本発明は、特にパラ−キシレン含量の少ないC8芳香族混合物を処理してパラ−キシレン濃度をほぼ熱平衡レベルまで上昇させるのに有効である。
【0018】
本発明のプロセスは、2〜60重量%のエチルベンゼン、例えば約4〜20重量%エチルベンゼンを含むC8芳香族ストリームの異性化に特に好適である。この範囲は、改質ガソリンおよび熱分解ガソリン単位から誘導されるストリームのエチルベンゼン濃度をカバーする。本発明の触媒は、非抽出C8芳香族ストリームに存在するタイプのノルマルおよび分枝パラフィンの熱分解に適した高い活性をもつことができる。
【0019】
<触媒系>
前記触媒系は、例えば原料ストリーム中のエチルベンゼンの選択的脱エチル化および脱エチル化により生成されたエチレンのエタンへの変換のような、エチルベンゼンを変換する第1の機能を有する第1成分、および原料中のキシレンを選択的に異性化する第2の成分を含む。前記第1触媒成分は、約1〜約12の束縛指数および有効量の第VIII族金属を有する結晶性モレキュラーシーブを含む。第1成分は通常、原料中のキシレンのある種の異性化にも効果がある。第2成分は、約1〜約12の束縛指数および有効量の第VIII族金属を有する結晶性モレキュラーシーブを含む。
【0020】
前記触媒系の第1成分は、通常、C8芳香族原料のキシレン成分の異性化に有効な第2成分について上流に当たる。この態様において、第1成分はエチルベンゼン変換の所望のレベルを達成するのに十分な量で用いられ、一般に全触媒系の量の約5%より多い量であり、例えば10%より多く、例えば25%より多く、例えば50%より多く、例えば55%より多く、例えば60%より多く、例えば75%より多く、例えば80%より多い。
【0021】
第1および第2成分に用いることができるモレキュラーシーブの具体例としては、大孔モレキュラーシーブおよび中孔モレキュラーシーブが含まれる。これらのモレキュラーシーブは、バエルロッシャー(Ch.Baerlocher)、メイヤー(W.H.Meier)およびオルソン(D.H.Olson)編「ゼオライト編成型のアトラス」(エルゼビエ社、第5編、2001年)に記述されている。大孔モレキュラーシーブは、一般に約7Åより大きい孔サイズを有する。好適な大孔モレキュラーシーブの具体例としては、AEL、MOR、および*BEA構造型が含まれる。具体的な大孔モレキュラーシーブの具体例としては、ベータ(Beta)およびモルデナイト(mordenite)が含まれる。中孔モレキュラーシーブは、一般に約5Å〜約7Åの孔サイズを有する。好適な中孔モレキュラーシーブの具体例としては、AEL、MFI、MEL、MTW、MWW、TON、MTT、FERおよびMFS構造型(ゼオライト命名に関するIUPAC委員会)を有するものが含まれる。好ましいモレキュラーシーブは12以上のアルミナに対するシリカのモル比を有するアルミニウムケイ酸の形態である。具体的な中孔モレキュラーシーブの具体例としては、SAPO−11、例えばMCM−22、MCM−49およびMCM−56のようなMCM−22族のモレキュラーシーブ、ZSM−5、ZSM−11、ZSM−12、ZSM−22、ZSM−23、ZSM−34、ZSM−35、ZSM−48およびZSM−57が含まれる。
【0022】
第1および第2成分のそれぞれのモレキュラーシーブは第VIII族金属に結合している。この第VIII族金属としては、プラチナ、パラジウム、イリジウム、ルテニウム、ロジウム、オスミウム、ニッケル、および鉄が含まれる。前記モレキュラーシーブに結合した第VIII族金属は、通常、希金属になる。この貴金属はプラチナ、パラジウム、イリジウム、ルテニウム、ロジウム、オスミウムである。好ましくは、ブラチナはモレキュラーシーブに結合している。第VIII族金属としては、元素状態(すなわち、ゼロ価)またはオキサイド、スルフィド、ハライド、カルボキシレートなどのような幾つかの他の触媒的に活性な形態の金属も含むことが意図されている。前記第VIII族金属は遊離金属(すなわち、ゼロ価)状態の成分に存在することは必ずしも必要ではなく、金属のオキサイド、ハイドロキサイド、またはスルフィドのような化合物として存在することもできると考えられている。前記第VIII族金属は、例えばこの成分がオキサイドまたはハイドロキサイドの状態の場合には、好ましくは還元状態である。前記第VIII族金属の還元された結合価状態は、水素のような還元剤が反応原料に含有されている場合、反応進行中にin situで得ることができる。
【0023】
前記第VIII族金属は、通常、競争的なイオン交換により第1および第2成分中に取り込むことになる。競争的なイオン交換により、モレキュラーシーブにおける第VIII族金属の良好なアキシャル分布が達成される。
【0024】
本発明はいずれの操作理論にも限定されることが意図されていないが、ほとんどの第VIII族金属がモレキュラーシーブ内の孔にきめ細かく分散されるため、触媒系の低い芳香環飽和を伴った高いエチレン飽和活性の利点が得られる。前記第VIII族金属粒子がモレキュラーシーブの孔内にある場合、遷移状態の選択性のために、すなわち飽和した芳香族の反応遷移状態がモレキュラーシーブ中で形成されるには大きすぎるため、芳香族飽和は起こりえない。さらに、第VIII族金属粒子の高分散性のために、芳香族が1つ以上の第VIII族金属原子と接触することが保障される。提唱される理論の如何にかかわらず、前記プロセスはそれらに開示された改善された特性を有する。
【0025】
競争的なイオン交換には、非水素化金属カチオンがモレキュラーシーブにおいて交換可能なイオンとして第VIII族金属カチオンと競合することを利用することが含まれる。競争的なイオン交換は、第1および第2成分のモレキュラーシーブを、例えばプラチナカチオンのような第VIII族金属カチオンの予め決められた量および例えばアンモニウムカチオンのような非水素化金属カチオンの予め決められた量を含む供給水溶液と接触させることにより実施することができる。
【0026】
供給溶液における第VIII族カチオンに対する非水素化金属カチオンの比率は、供給溶液のpH、前記成分の固有の酸性度、およびモレキュラーシーブと結合した第VIII族金属の量を含む多くの因子により変動することになる。前記供給溶液は、第VIII族カチオンに対する非水素化金属カチオンのモル比が約500〜6000の範囲となるように、通常調製される。好ましくは、第VIII族カチオンに対する非水素化金属カチオンのモル比は約700〜2000の範囲である。より好ましくは、第VIII族カチオンに対する非水素化金属カチオンのモル比は約900〜1100の範囲である。供給溶液に存在する非水素化金属カチオンおよび第VIII族カチオンの量は、最終触媒に含有される第VIII族金属の所望量に依存する。
【0027】
通常、供給溶液のpHは4〜10に維持される。上段床成分(エチルベンゼン変換に有効な成分)のモレキュラーシーブがシリカで選択活性化(selectivated)されている場合、供給水溶液のpHは、通常7以下のpHに維持され、好ましくは約6.5〜7の範囲に維持される。供給プロセスにおける供給溶液pHの調整は通常、水酸化アンモニウムを含む水溶液を用いて行われる。
【0028】
供給溶液用の第VIII族金属カチオンの具体例には、塩化プラチナ酸、塩化プラチナ、および硝酸テトラアミンプラチナ(II)および塩化ペンタアミンクロロイリジウム(III)のようなテトラアミンプラチナおよびテトラアミンパラジウム錯体が含まれる。供給溶液用の非水素化金属カチオンの好適な具体例には、アンモニウムのハロゲン化物または硝酸塩が含まれる。金属の取り込み後、前記触媒は通常、水で洗浄され、乾燥および焼成される。
【0029】
供給溶液用の非水素化金属カチオンの好適な具体例にはアンモニウムカチオンが含まれる。
【0030】
第1および第2成分に存在する第VIII族金属の量は前記触媒成分の重量に基づいて例えば0.001〜約10重量%の範囲に変動させることができる。
【0031】
第1触媒成分に関して、第VIII族金属は、好ましくは約0.001〜約0.05重量%、より好ましくは約0.01〜0.04重量%の範囲で存在することになる。もちろん、これは金属の性質により変動することになる。第VIII族金属がプラチナの場合、第1触媒成分に存在する第VIII族金属の量は、好ましくは全触媒成分の約0.03重量%である。
【0032】
第2触媒成分に関して、第VIII族金属は、好ましくは約0.001〜約0.03重量%、例えば約0.0075〜0.02重量%の範囲で存在することになる。もちろん、これは成分の性質により変動することになる。第VIII族金属がプラチナの場合、第2触媒成分に存在する量は、好ましくは全触媒成分の約0.01重量%である。
【0033】
本発明のプロセスの実施において、第1および第2触媒成分のいずれか又は両方と、本プロセスの温度および他の条件に耐えられる他の物質を一緒に調製してもよい。そのような基材物質には、粘土、シリカ、および/または金属酸化物のような無機酸化物物質が含まれる。前記金属酸化物は、天然由来でも、ゼラチン状析出物でも、シリカおよび金属酸化物の混合物を含むゲルの形態でもよい。モレキュラーシーブと混成できる天然由来の粘土には、モンモリロナイト(montmorillonite)およびカオリン(kaolin)族のものが含まれる。これらの族には、主な無機構成がハロイサイト(halloysite)、カオリナイト(kaolinite)、ディッカイト(dickite)、ナクライト(nacrite)およびアナウクサイト(anauxite)であるディクシー(Dixie)、マクナミー(McNamee)、ジョージアおよびフロリダ粘土またはその他として広く知られているスベントナイト(subbentonite)およびカオリンが含まれる。そのような粘土は、元々採掘された状態で、または初めに焼成、酸処理または化学修飾された状態で用いることができる。
【0034】
前述の物質に加えて、本発明で用いられるモレキュラーシーブは、シリカ−アルミナ−トリア(thoria)、シリカ−アルミナ−ジルコニア、シリカ−アルミナ−マグネシア、およびシリカ−マグネシア−ジルコニアのような3成分化合物ばかりでなく、アルミナ、シリカ−アルミナ、シリカ−マグネシア、シリカ−ジルコニア、シリカ−トリア、シリカ−ベリリア、シリカ−チタニアのような多孔性の基材物質と混成することができる。これらの成分の混合物も用いることができる。さらに、前記モレキュラーシーブは米国特許6,198,013号に記述された方法を用いたゼオライト基材物質と混成することができる。本文献の全内容は、参照により本発明に取り込まれる。好ましくは、結合剤はシリカである。
【0035】
無水ベースのモレキュラーシーブ成分および無機酸化物基材の相対比率は、約1〜約99重量%の範囲、より普通には乾燥混成物の約10〜約80重量%の範囲のモレキュラーシーブ含量で広く変動し得る。
【0036】
本発明の触媒系の第1および第2成分は、第1成分が原料ストリームのエチルベンゼンを選択的にベンゼンへ脱エチル化し、一方、第2成分が原料のキシレンを選択的に異性化することを確保する多くの重要な側面において相互に異なることになる。これらの異なる性質を以下に記述する。
【0037】
例えば、本発明の触媒系の各成分は通常、互いに相容れないキシレン分散特性を示すことになる。これらの特性は、120℃および0.64±0.11kPa(4.5±0.8mm水銀)のオルト−キシレン分圧において30%平衡能のオルト−キシレンを吸着するのに必要な時間(分)を知ることにより決定することができる。本試験は米国特許4,117,026号、4,159,282号、およびRe.31,782号に記述されている。オルト−キシレンの平衡能は本発明では、モレキュラーシーブ100g当たりキシレン1gより大きいものと定義される。本発明の触媒系において、エチルベンゼン変換に有効な第1触媒成分は、好ましくは約50より大きなオルト−キシレン吸着時間(分)、好ましくは約1,200より大きく10,000分未満のオルト−キシレン吸着時間(分)を有する。一方、第2の異性化成分は、好ましくは約50分未満のオルト−キシレン吸着時間、好ましくは約10分未満のオルト−キシレン吸着時間を有する。
【0038】
<エチルベンゼン変換成分>
前記エチルベンゼン変換成分は、好ましくは約50より大きなオルト−キシレン吸着時間(分)、好ましくは約1,200より大きく10,000分未満のオルト−キシレン吸着時間(分)を有する。所望のキシレン分散特性は多くの方法により達成することができる。50分の最小値における又はこれに近いオルト−キシレン分散時間では、前記触媒中で用いられるモレキュラーシーブの大きな結晶形態、すなわち1ミクロンより大きな平均結晶サイズを有するものを選択すれば十分である。しかしながら、より高い分散性を達成するためには、用いられるプロセス条件下で不活性な、シリカのようなコークスおよび/または酸化物の層を触媒粒子の表面に析出させることにより選択活性化することが望ましいといえる。前記成分粒子が選択活性化される場合、大結晶サイズおよび中結晶サイズのいずれのモレキュラーシーブも第1成分に用いることができる。
【0039】
前記第1成分のモレキュラーシーブは、好ましくは第2触媒のモレキュラーシーブよりも高い酸活性を有する。好ましくは、前記第1触媒成分のモレキュラーシーブは第2成分のα値の少なくとも2倍のα値を有する。第2成分は通常30以上のα値を有する。α値の測定操作は米国特許3,354,078号、ジャーナル・オブ・キャタリシス、第4巻、527頁(1965年)、第6巻、278頁(1966年)、第61巻、395頁(1980年)に記述され、これらのその記述は参照により本発明に取り込まれる。本発明で用いられる試験の実験条件には、ジャーナル・オブ・キャタリシス、第61巻、395頁に詳細に記述されるように538℃の定常温度および可変流速が含まれる。より高いα値はより活性な熱分解触媒に対応する。
【0040】
好ましくは、第1成分は水蒸気処理されていない。水蒸気処理が第1成分のα値を上述の値に低下させるために用いられる場合、この水蒸気処理は典型的には、約100℃〜約600℃、例えば約175℃〜約325℃の温度で、約1%〜約100%の蒸気、例えば約50%〜約100%の蒸気を含む雰囲気において、約69Pa−a〜約345kPa−a(0.01psia〜約50psia)の圧力で、約0.1〜約24時間、例えば約3〜約6時間の間、第1成分を加熱することにより行われる。
【0041】
前記第1成分がシリカで選択活性化される場合、これは前記触媒を液体担体において有機ケイ素化合物を用いて1又は複数の処理を施すことにより簡便に行われる。さらに、各処理に続いて、処理された物質が酸素含有雰囲気、例えば空気中において焼成される。このような多工程の選択活性化の操作は米国特許5,476,823号に記述され、本文献の全ての内容は参照により本発明に取り込まれる。好ましくは、第1成分には、2〜4工程のシリカ選択活性化処理が施される。シリカ選択活性化される触媒が結合剤を含む場合、シリカのような非酸性の結合剤を用いることが好ましい。
【0042】
前記第1触媒成分の選択活性化に用いられる有機ケイ素化合物は、例えば、シリコーン、シロキサン、シラン、またはこれらの混合物であり得る。これらの有機ケイ素化合物は分子当たり2以上のケイ素原子をもつことができる。これらの有機ケイ素化合物は、液体担体媒体と組み合わせて溶解性または液体に変換できるなら、純粋形態で固体であり得る。予備選択活性化剤として用いられるシリコーン、シロキサン、またはシラン化合物の分子量は約80〜約20,000の範囲であり得、好ましくはおよそ150〜10,000の範囲内である。予備選択活性化シリコーン化合物には、ジメチルシリコーン、ジエチルシリコーン、フェニルメチルシリコーン、メチル水素シリコーン、エチル水素シリコーン、フェニル水素シリコーン、メチルエチルシリコーン、フェニルエチルシリコーン、ジフェニルシリコーン、メチルトリフルオロプロピルシリコーン、エチルトリフルオロプロピルシリコーン、ポリジメチルシリコーン、テトラクロロフェニルメチルシリコーン、テトラクロロフェニルエチルシリコーン、テトラクロロフェニル水素シリコーン、テトラクロロフェニルフェニルシリコーン、メチルビニルシリコーン、およびエチルビニルシリコーンが含まれる。前記予備選択活性化シリコーン、シロキサン、またはシラン化合物は直線状である必要はなく、例えばヘキサメチルシクロトリシロキサン、オクタメチルシクロテトラシロキサン、ヘキサフェニルシクロトリシロキサン、およびオクタフェニルシクロテトラシロキサンのような環状でもよい。これらの化合物の混合物も、他の官能基を持つシリコーンと同様に、予備選択活性化剤として用いることができる。
【0043】
好ましくは、モレキュラーシーブの予備選択活性化に用いられる有機ケイ素化合物の動力学的直径は、有機ケイ素化合物のモレキュラーシーブ孔への進入およびそれに伴うモレキュラーシーブの内部活性の低下を回避するために、モレキュラーシーブの直径よりも長い。
【0044】
好ましい有機ケイ素予備選択活性化剤には、特に予備選択活性化剤が有機担体に溶解し、または水溶性担体中で乳化する場合、ジメチルフェニルメチルシロキサン(例えば、Dow−550)およびフェニルメチルポリシロキサン(例えば、Dow−710)が含まれる。Dow−550およびDow−710はミシガン州、ミッドランドのダウ・ケミカル社から入手できる。
【0045】
好ましくは、前記有機ケイ素化合物用の液体担体は、分子当たり5以上、特に7以上の炭素原子を有する直鎖状、分枝状、または環状の炭化水素、例えばヘプタン、オクタン、ノナン、またはウンデカンのようなアルカンのような有機化合物である。これら有機化合物、例えばアルカンの沸点は約70℃より高くてもよい。水素熱分解循環油のような低揮発性有機化合物の混合物は担体として用いることができる。特に好ましい有機担体はデカンおよびドデカンである。
【0046】
有機ケイ素化合物の各含浸に続いて、前記触媒は200℃を超えるが、モレキュラーシーブの結晶が悪影響を受ける温度未満で、約0.2℃/分〜5℃/分の速度で焼成される。この焼成温度は一般に600℃未満であり、好ましくはおよそ350〜550℃の範囲内である。焼成温度における焼成の時間は1〜24時間、例えば2〜6時間であり得る。好ましくは、前記触媒は3回の選択活性化操作に付される。
【0047】
シリカ選択活性化に追加して、またはこれに替えて、前記第1触媒成分はコークス選択活性化に付すこともできる。この任意のコークス選択活性化は、典型的に前記触媒を熱分解性有機化合物の分解温度を超えて上昇した温度で、モレキュラーシーブの結晶が悪影響を受ける温度未満で、当該熱分解性有機化合物と接触させることを含む。この接触温度は、例えば約650℃未満でもよい。このコークス選択活性化プロセスに用いることができる有機物質には、具体例を挙げれば、パラフィン、シクロパラフィン、オレフィン、シクロオレフィン、および芳香族のような炭化水素、アルコール、アルデヒド、エーテル、ケトン、およびフェノールのような酸素含有有機化合物、およびフラン、チオフェン、ピロール、およびピリジンのようなヘテロ環化合物が含まれる。水素助原料を、過剰なコークスの生成を止めるために用いることができる。コークス選択活性化に関する更に詳細な事項は米国特許4,117,026号に記述されている。シリカ選択活性化に続いてコークス選択活性化を組み合わせることにより、特定のキシレン分散性を獲得するために必要とされる有機ケイ素含浸処理工程の数を減らすことができる。
【0048】
<異性化成分>
前記触媒系の第2成分はC8芳香族を含む原料中のキシレンの異性化に有効である。前記第2成分は、好ましくは約50分未満のオルト−キシレン吸着時間を有し、この時間はさらに好ましくは約10分未満である。これは、この成分中に0.02〜0.05ミクロンの平均結晶サイズを有する小結晶サイズのモレキュラーシーブを用いることにより達成される。前記触媒系の第二成分のモレキュラーシーブは、典型的には約30以上のα値を有することになる。
【0049】
好ましくは、前記第2モレキュラーシーブは、このモレキュラーシーブを用いて第VIII族金属の取り込みの前に、所望のα値を達成するために水蒸気処理が施される。
【0050】
<プロセス条件>
本発明のプロセスにおいて用いられる条件は狭い範囲になるように定義されるものではないが、一般に、約204〜約540℃(400〜1,000°F)の温度、約100〜約7,000kPa−a(0〜1,000psig)の圧力、0.1〜200hr-1の重量時間当たり空間速度(WHSV)、約0.2〜約10の炭化水素(HC)に対する水素(H2)モル比が含まれる。好ましくは、前記条件には、約343〜約413℃(650〜775°F)の温度、約445〜約2,860kPa−a(50〜400psig)の圧力、約3〜50hr-1のWHSV、約0.7〜約3のHCに対するH2モル比が含まれる。
【0051】
一般に、本発明のプロセスは上述の触媒系を含む固定床において実施される。好ましい態様において、前記触媒系の第1および第2成分は単一反応槽における連続床に存在する。すなわち、エチルベンゼン変換に有効な本発明のプロセスで用いられる触媒系の成分は第1床を形成し、一方、キシレン異性化に有効な前記触媒系の他の成分は前記第1床下流の第2床を形成する。前記原料は、好ましくは軽量気体を途中で分離することなく第1から第2床へ直列に流れる。この配置に替えて、第1および第2床は、分離した反応槽、所望ならば異なるプロセス条件で操作が可能な反応槽に配置することができる。本発明の第1および第2触媒成分の前後に、追加の触媒床を設けることともできる。
【0052】
前記変換プロセスの後に、前記異性化生成物はパラ−キシレンおよび/または他の望ましい1または複数のキシレンを単離するために処理することができる。したがって、例えば、前記異性化生成物は結晶化剤、膜分離単位、または選択的吸着単位のような様々なパラ−キシレン回収単位に供給することができ、最終的にパラ−キシレンを単離および回収することができる。残留異性化物からはC8よりも軽い生成物を取り除くことができる。残留異性化物中のC8よりも重い生成物は、さらに加工し、分画して取り出すことができる。パラ−キシレンが取り除かれたC8画分は異性化装置に再循環することができる。
【0053】
本発明のプロセスの1つの結果は、熱平衡未満の量のパラ−キシレンを含有する原料の混合キシレン成分を、異性化装置からの生成物が熱平衡に少なくとも近い量のパラ−キシレンを含む程度まで変換することである。
【0054】
本発明のプロセスのもう1つの結果は、最小限のキシレン損失で混合キシレン原料に含まれるエチルベンゼンの高い割合での変換である。例えば、50重量%より高いエチルベンゼン変換レベルが、2重量%未満のキシレン損失レベルで達成することができる。
【実施例】
【0055】
以下、実施例に基づき、本発明についてさらに詳細に説明する。なお、本発明は下記実施例に限定されるものではない。
【0056】
<比較例1>
芳香環族飽和に対するエチレン飽和の評価を2成分触媒系を用いて実施した。この2成分触媒系は40重量%の上段床成分および60重量%の下段床成分を含んだ。プラチナを初期湿性含浸(incipient wetness impregnation)により上段床成分に取り込み、プラチナを混合(mulling)しながら下段床成分に取り込んだ。
【0057】
前記2成分触媒系の上段床成分は中結晶サイズを有するZSM−5より形成した。前記ZSM−5は、65重量%のZSM−5および35重量%のシリカ結合剤の重量比でシリカ結合剤と混成した。このシリカ結合ZSM−5を従来法により1/16”直径円筒形粒子に押出し、次にデカン中7.8重量%ジメチルフェニルメチルポリシロキサンを用いた4連続含浸処理を含む多段階シリカ選択活性化操作を施した。各含浸の後に溶媒は取替え、前記触媒は窒素中で焼成し、次に空気中で538℃に加熱した。次に、硝酸テトラアミンプラチナ(II)を用いた初期湿性含浸、続いて乾燥および空気焼成により前記選択活性化された触媒にプラチナを取り込んだ。次に前記触媒をα値158まで水蒸気処理した。得られた触媒は0.1重量%のプナチナを含有した。
【0058】
前記下段床成分は小結晶サイズを有するZSM−5より形成した。前記ZSM−5は、50重量%のZSM−5および50重量%のアルミナ結合剤の重量比でアルミナ結合剤と混成した。このアルミナ結合ZSM−5を従来法により0.16cm(1/16”)直径円筒形粒子に押出した。すなわち、塩化テトラアミンプラチナの形態で0.1重量%のプラチナを用いZSM−5およびアルミナ結合剤物質を混合しながら添加し、押出した。前記混合は米国特許Re.31,919号に記述された技術を用いて実施した。次に押出物は乾燥および空気中で焼成を行った。次に前記触媒はα値18まで水蒸気処理した。前記下段床成分は0.1重量%のプラチナを含有した。
【0059】
前記2成分触媒系は、429℃(805°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0060】
前記評価で用いた原料は、0.7重量%非芳香族、0.6重量%トルエン、18.7重量%エチルベンゼン、0.6重量%パラ−キシレン、61.7重量%メタ−キシレン、16.7重量%オルト−キシレン、1重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの2倍モルの硫黄を用いて399℃(750°F)および1,825kPa−a(250psig)において予備スルフィド化し、次に11日間429℃(805°F)、10h-1WHSV、0.9:1のH2:HC、および1,384kPa−a(186psig)の全体圧力で脱エッジング(de-edged)を行った。その結果を下記表Iに示す。
【0061】
【表1】
【0062】
<比較例2>
芳香族環飽和に対するエチレン飽和の評価を2成分触媒系を用いて実施した。この2成分触媒系は30重量%の上段床成分および70重量%の下段床成分を含んだ。プラチナをイオン交換により上段床成分に取り込み、プラチナをイオン交換により下段床成分に取り込んだ。
【0063】
前記上段床成分は、前記触媒成分が3工程の選択活性化操作に付され、500のα値を有し、前記プラチナがイオン交換により前記選択活性化された触媒に取り込まれることを除き、比較例1の上段床と同様に調製された。前記イオン交換は500mLの水に硝酸テトラアミンプラチナ(II)(0.030gのプラチナ)を溶解し、次にこの溶液を80℃(176°F)まで加熱して行った。次に、100gの前記シリコン選択活性化触媒をこの溶液に浸した。前記溶液は8時間循環した。前記触媒を溶液から取り出し、蒸留水で洗浄、121℃(250°F)で乾燥、354℃(660°F)で1時間空気中で焼成した。前記上段床成分は0.03重量%のプラチナを含有し、500のα値を有した。
【0064】
前記下段床成分は、既にα値108に水蒸気処理された80重量%ZSM−5および20重量%シリカ結合剤を含む押出物を形成することにより調製された。次に、硝酸テトラアミンプラチナ(II)(0.02gのプラチナ)を600mLの蒸留水に溶解し、この溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH8〜9に調整した。前記プラチナ含有溶液を3時間前記触媒上に循環させた。前記溶液のpHは、10重量%水酸化アンモニウムを含む溶液を用いて取り込みの間pH8〜9に維持した。前記触媒を溶液から取り出し、蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記下段床成分は0.01重量%のプラチナを含有し、108のα値を有した。
【0065】
前記2成分触媒系は、376℃(709°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0066】
前記評価で用いた原料は、0.6重量%非芳香族、1.4重量%トルエン、14.7重量%エチルベンゼン、1.3重量%パラ−キシレン、62.8重量%メタ−キシレン、18.8重量%オルト−キシレン、0.4重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの20倍モルの硫黄を用いて357℃(675°F)および1,653kPa−a(225psig)において予備スルフィド化し、次に16日間399℃(750°F)、10h-1WHSV、0.6:1のH2:HC、および1,466kPa−a(198psig)の全体圧力で脱エッジング(de-edged)を行った。その試験結果を下記表IIに示す。
【0067】
【表2】
【0068】
<実施例1>
芳香族環飽和に対するエチレン飽和の評価を2成分触媒系について実施した。この2成分触媒系は30重量%の上段床(第1)成分および70重量%の下段床(第2)成分を含んだ。プラチナを競争的イオン交換により上段床成分および下段床成分に取り込んだ。
【0069】
前記上段床成分は、前記プラチナが競争的イオン交換により前記選択活性化された触媒に取り込まれることを除き、比較例2の上段床と同様に調製された。前記触媒に取り込まれたプラチナ量は0.03重量%であった。前記競争的イオン交換は、初めに250gの上段床成分を300mLカラムに充填することにより実施した。前記触媒はカラムを通して湿った空気を通過させることにより湿気をもたせた。次に、0.05N硝酸アンモニウムを含む溶液をカラムを通して循環し、前記溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH6.5〜7.0に調整した。硝酸テトラアミンプラチナ(II)(250mLの蒸留水に0.075gのプラチナが溶解された)溶液を4時間をかけて硝酸アンモニウムタンクに加えた。プラチナ交換溶液におけるプラチナに対する硝酸アンモニウムのモル比は976であった。前記プラチナ交換溶液は約48時間にわたり触媒床を通して循環した。この間、pHは10重量%水酸化アンモニウムを含む溶液を用いて継続的にpH6.5〜7.0に維持された。前記触媒は蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記上段床成分は0.03重量%のプラチナを含有した。
【0070】
前記下段床成分は、前記プラチナが競争的イオン交換により前記触媒に取り込まれたことを除き、比較例2の下段床と同様に調製された。取り込まれたプラチナ量は0.01重量%であった。前記競争的イオン交換は、初めに1,298gの下段床成分を1Lカラムに充填することにより実施した。その後、前記成分はカラムを通して湿った空気を通過させることにより湿気をもたせた。次に、0.05N硝酸アンモニウム溶液3,894gをカラムを通して循環し、前記溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH8〜9に調整した。硝酸テトラアミンプラチナ(II)(250mLの蒸留水に0.13gのプラチナが溶解された)溶液を4時間をかけて硝酸アンモニウムタンクに加えた。プラチナに対する硝酸アンモニウムのモル比は2,921であった。前記プラチナ交換溶液は約12時間にわたり前記成分上を循環した。この間、前記pHは10重量%水酸化アンモニウムを含む溶液を用いて継続的に維持された。前記成分は蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記下段床成分は0.01重量%のプラチナを含有した。
【0071】
前記2成分触媒系について、357℃(675°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0072】
前記評価で用いた原料は、0.7重量%非芳香族、1.4重量%トルエン、14.6重量%エチルベンゼン、1.3重量%パラ−キシレン、63.1重量%メタ−キシレン、18.8重量%オルト−キシレン、0.1重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの20倍モルの硫黄を用いて357℃(675°F)および1,653kPa−a(225psig)において予備スルフィド化し、次に3日間413℃(775°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力で脱エッジング(de-edged)を行った。その結果を下記表IIIに示す。
【0073】
【表3】
【0074】
表IIIの結果は、前記触媒が低い芳香族環損失の下に高いエチレン飽和で優れたエチルベンゼン変換を行うことを示している。
【0075】
<実施例2>
触媒系Aおよび触媒系Bをエチルベンゼン変換および芳香族環損失について評価した。
【0076】
触媒系Aは、それが50重量%の上段床および50重量%の下段床を含むことを除き、比較例1の触媒系と同様である。触媒系Aは10h-1WHSV、1:1のH2:HC、1,653kPa−a(225psig)の全体圧力、および381℃(721°F)〜436℃(817°F)の温度範囲の条件下で運転された。本試験で用いた原料は、10.3重量%エチルベンゼン、1.2重量%パラ−キシレン、61.8重量%メタ−キシレン、26.7重量%オルト−キシレンからなるC8芳香族原料である。
【0077】
触媒系Bの上段および下段床は、それが25重量%の上段床および75重量%の下段床を含むことを除き、実施例1と同様である。触媒系Bは10h-1WHSV、1:1のH2:HC、1,653kPa−a(225psig)の全体圧力、および341℃(646°F)〜371℃(699°F)の温度範囲の条件下で運転された。本試験の実施前に、触媒系Bは、2日間404℃(760°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力で脱エッジング(de-edged)を行った。本試験で用いた原料は、0.3重量%非芳香族、0.4重量%トルエン、12.9重量%エチルベンゼン、1.2重量%パラ−キシレン、66.5重量%メタ−キシレン、18.4重量%オルト−キシレン、0.3重量%の炭素数9以上の芳香族からなるC8芳香族原料である。
【0078】
本試験の結果は図に示した。
【0079】
図における結果は、触媒系Bが低い変換温度で低い芳香族環損失が低いことを示している。
【技術分野】
【0001】
本発明は、低い芳香環飽和と組み合わされた高いエチレン飽和活性を示す触媒系に関する。また、本発明は前記触媒系を用いたキシレンの異性化およびエチルベンゼンの変換のためのプロセスに関する。
【背景技術】
【0002】
パラ−キシレンは、通常は選択的溶媒抽出による石油ナフサ、特に改質ガソリンのような原料物質から分離された、C8芳香族の混合物から誘導され得る貴重な化学原料である。これら原料からの前記C8芳香族留分は、かなり広範な組成を有するが、通常は10〜32重量パーセントのエチルベンゼン、および約50重量パーセントのメタ異性体およびそれぞれ25重量パーセントのパラおよびオルト異性体とに分離される残りのキシレンからなる。
【0003】
個々の異性体生産物は、適当な物理的方法によって天然由来の混合物から分離することができる。エチルベンゼンは、費用のかかる操作であるが、分別蒸留により分離することができる。オルト−キシレンは分別蒸留により分離することができ、商業的にはその方法で生産されている。パラ−キシレンは分別析出、選択的吸着(例えば、パーレックス(商標)プロセス)、または膜分離により混合異性体から分離することができる。
【0004】
パラ−キシレンの商業的使用が増大するにつれて、所望のパラ−異性体の収率を上げるために物理的分離と他のキシレンアイソマーの化学的異性化とを組合せることが益々重要になっている。しかしエチレンベンゼンの沸点がパラ−キシレンおよびメタ−キシレンの沸点と非常に接近しているために、蒸留によるC8芳香族供給原料からのエチルベンゼンの完全な除去は実際的ではない。したがって、全ての商業的キシレン異性化プロセスの重要な特徴は、原料のエチルベンゼンを有用な副生物に変換し、同時にキシレンから他の化合物への全ての変換を最小限に抑える性能である。
【0005】
1つの商業的に成功したキシレン変換プロセスは米国特許4,899.011号に記載されている。ここでは、そのパラ−キシレン含量が涸渇したC8芳香族原料は2成分触媒系に接触させられる。第1触媒成分は脱エチル化により選択的にエチルベンゼンを変換してベンゼンを生成し、エチレンはエタンに変換される。一方、第2触媒成分は選択的にキシレンを異性化してパラ−キシレン含量を熱平衡値またはその近くまで上昇させる。第1触媒成分は、120℃において30%の平衡性能のオルト−キシレンを吸着する性能に基づいて50分を超えるオルト−キシレン吸着時間および0.64±0.11kPa(4.5±0.8mm水銀)オルト−キシレン分圧を有し、1〜12の束縛指数(Constraint Index)を有するゼオライトを含む。一方、第2成分は同条件下で10分未満のオルト−キシレン吸着時間を有する束縛指数1〜12のゼオライトを含む。好ましい態様において、第1触媒成分は1ミクロン以上の結晶サイズを有するZSM−5であり、第2触媒成分は0.02〜0.05ミクロンの結晶サイズを有するZSM−5である。また各触媒成分は水素化金属をも含む。
【0006】
米国特許4,899,011号のプロセスに関する改善は米国特許5,689,027号に記述されている。ここで、前記2成分系における第1触媒成分は、011特許で引用される同試験条件下において1200分を超える程度にまでそのオルト−キシレン吸着時間を増加させるために、コークス化により、又はより好ましくはシリカの表面コーティングの蓄積により予備選択活性化されている。そのような系を用いると、011特許のプロセスで得られるよりも著しく低いキシレン損失で、高いエチルベンゼン変換率が達成されることが見出された。ここでも、027特許で用いられる触媒成分は水素化金属を含む。
【0007】
011特許及び027特許のプロセスで用いられる希金属含有ゼオライト触媒の1つの製造方法は米国特許再公表31,919号に開示され、ゼオライトの結晶化後であって、最終触媒粒子の形成前でゼオライトの焼成または水蒸気処理のいずれもの前に、ゼオライトを含むカチオン形状の希金属を取り込むことを含む。希金属がプラチナの場合、919特許の実施例により相対的に低いキシレン損失で改善されたエチルベンゼン変換が得られることが証明される。
【発明の概要】
【発明が解決しようとする課題】
【0008】
上記のような最近の進歩にもかかわらず、平均して低いキシレン損失を達成するエチルベンゼン変換/キシレン異性化用の触媒を欲する継続した要求が依然としてある。したがって、例えば、プラチナ含有触媒はエチレン飽和には有効だが、それらは芳香環飽和も触媒する。さらに、芳香環飽和は低温で熱力学的に増強され、このため、触媒の予備スルフィド化、又はたとえ上昇した温度が生産予定および/またはサイクル時間悪影響を及ぼしても、上昇した温度における操作が必要とされる。
【課題を解決するための手段】
【0009】
<発明の要約>
本発明においては、芳香環飽和に対するエチレン飽和比が3,500を超える触媒系が提供される。本触媒系は2成分を含む。各成分は、約1〜約12の束縛指数(Constraint Index)を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属(米国式分類)を含む。
【0010】
好ましくは触媒系は10,000より大きな、より好ましくは20,000より大きな、さらにより好ましくは25,000より大きな、最も好ましくは30,000より大きな芳香環飽和に対するエチレン飽和比を示す。
【0011】
もう1つの態様において、競争的なイオン交換により第VIII族金属をモレキュラーシーブに取り込むことによって2成分触媒系を生産するプロセスが提供される。本プロセスは、モレキュラーシーブへの第VIII族金属のイオン交換に有効な条件下で、例えばアンモニウムカチオンのような非水素化金属カチオンおよびプラチナカチオンのような第VIII族金属(日本式では第10族)カチオンを含む水溶液を用いて、モレキュラーシーブを接触させることにより実施される。水溶液中の第VIII族金属に対する非水素化金属カチオンのモル比は、約500〜約6000の範囲内である。
【0012】
さらなる態様において、本発明は、エチルベンゼンおよびキシレンを含む原料の異性化のためのプロセスを提供する。本プロセスは、有効な条件下で原料を10,000より大きな芳香環飽和に対するエチレン飽和比を示す触媒系に接触させることにより実施されるプロセスであって、
(a)約1〜約12の束縛指数を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属を含む第1成分を用いて、水素存在下でエチルベンゼン変換条件下で、原料を接触させる工程、および
(b)約1〜約12の束縛指数を有する結晶性モレキュラーシーブおよび有効量の第VIII族金属を用いて、キシレン異性化条件下で工程(a)のエチルベンゼンの涸渇した流出物を接触させる工程、
を含むプロセスである。
【図面の簡単な説明】
【0013】
【図1】実施例2の触媒系について、芳香環損失[モル%]に対してプロットされたエチルベンゼン変換[モル%]のグラフを示す図である。
【発明を実施するための形態】
【0014】
<発明の詳細な説明>
本発明において、本触媒系用の芳香環飽和に対するエチレン飽和比は以下の式により決定される。
【0015】
【数1】
【0016】
上記式において示される値は343℃(650°F)の温度、10h-1の重量時間当たり空間速度、1の水素/炭化水素モル比、および1,653kPa−a(225psig)全体圧の圧力で決定される。用いられる原料は、13.0重量%エチルベンゼン、1.0重量%パラ−キシレン、67.0重量%メタ−キシレン、および19.0重量%オルト−キシレンからなるC8芳香族原料である。
【0017】
<原料>
一般に、任意のエチルベンゼンおよびキシレンを含む芳香族C8混合物が本発明のプロセスへの原料として用いられる。一般的に、そのような混合物は、典型的に、約5〜60重量%の範囲のエチルベンゼン含量、約0〜35重量%の範囲のオルト−キシレン含量、約20〜95重量%の範囲のメタ−キシレン含量、および約0〜15重量%の範囲のパラ−キシレンを有する。上記の芳香族C8混合物に加えて、前記原料は非芳香族炭化水素、すなわち約30重量%までの量のナフセンおよびパラフィンを含有することができる。好ましい態様において本発明は、例えば石油ナフサを触媒で改質して得られるC8芳香族混合物を加工して、減少したエチルベンゼン含量および増加したパラ−キシレン含量の混合物を得る手段を提供する。本発明は、特にパラ−キシレン含量の少ないC8芳香族混合物を処理してパラ−キシレン濃度をほぼ熱平衡レベルまで上昇させるのに有効である。
【0018】
本発明のプロセスは、2〜60重量%のエチルベンゼン、例えば約4〜20重量%エチルベンゼンを含むC8芳香族ストリームの異性化に特に好適である。この範囲は、改質ガソリンおよび熱分解ガソリン単位から誘導されるストリームのエチルベンゼン濃度をカバーする。本発明の触媒は、非抽出C8芳香族ストリームに存在するタイプのノルマルおよび分枝パラフィンの熱分解に適した高い活性をもつことができる。
【0019】
<触媒系>
前記触媒系は、例えば原料ストリーム中のエチルベンゼンの選択的脱エチル化および脱エチル化により生成されたエチレンのエタンへの変換のような、エチルベンゼンを変換する第1の機能を有する第1成分、および原料中のキシレンを選択的に異性化する第2の成分を含む。前記第1触媒成分は、約1〜約12の束縛指数および有効量の第VIII族金属を有する結晶性モレキュラーシーブを含む。第1成分は通常、原料中のキシレンのある種の異性化にも効果がある。第2成分は、約1〜約12の束縛指数および有効量の第VIII族金属を有する結晶性モレキュラーシーブを含む。
【0020】
前記触媒系の第1成分は、通常、C8芳香族原料のキシレン成分の異性化に有効な第2成分について上流に当たる。この態様において、第1成分はエチルベンゼン変換の所望のレベルを達成するのに十分な量で用いられ、一般に全触媒系の量の約5%より多い量であり、例えば10%より多く、例えば25%より多く、例えば50%より多く、例えば55%より多く、例えば60%より多く、例えば75%より多く、例えば80%より多い。
【0021】
第1および第2成分に用いることができるモレキュラーシーブの具体例としては、大孔モレキュラーシーブおよび中孔モレキュラーシーブが含まれる。これらのモレキュラーシーブは、バエルロッシャー(Ch.Baerlocher)、メイヤー(W.H.Meier)およびオルソン(D.H.Olson)編「ゼオライト編成型のアトラス」(エルゼビエ社、第5編、2001年)に記述されている。大孔モレキュラーシーブは、一般に約7Åより大きい孔サイズを有する。好適な大孔モレキュラーシーブの具体例としては、AEL、MOR、および*BEA構造型が含まれる。具体的な大孔モレキュラーシーブの具体例としては、ベータ(Beta)およびモルデナイト(mordenite)が含まれる。中孔モレキュラーシーブは、一般に約5Å〜約7Åの孔サイズを有する。好適な中孔モレキュラーシーブの具体例としては、AEL、MFI、MEL、MTW、MWW、TON、MTT、FERおよびMFS構造型(ゼオライト命名に関するIUPAC委員会)を有するものが含まれる。好ましいモレキュラーシーブは12以上のアルミナに対するシリカのモル比を有するアルミニウムケイ酸の形態である。具体的な中孔モレキュラーシーブの具体例としては、SAPO−11、例えばMCM−22、MCM−49およびMCM−56のようなMCM−22族のモレキュラーシーブ、ZSM−5、ZSM−11、ZSM−12、ZSM−22、ZSM−23、ZSM−34、ZSM−35、ZSM−48およびZSM−57が含まれる。
【0022】
第1および第2成分のそれぞれのモレキュラーシーブは第VIII族金属に結合している。この第VIII族金属としては、プラチナ、パラジウム、イリジウム、ルテニウム、ロジウム、オスミウム、ニッケル、および鉄が含まれる。前記モレキュラーシーブに結合した第VIII族金属は、通常、希金属になる。この貴金属はプラチナ、パラジウム、イリジウム、ルテニウム、ロジウム、オスミウムである。好ましくは、ブラチナはモレキュラーシーブに結合している。第VIII族金属としては、元素状態(すなわち、ゼロ価)またはオキサイド、スルフィド、ハライド、カルボキシレートなどのような幾つかの他の触媒的に活性な形態の金属も含むことが意図されている。前記第VIII族金属は遊離金属(すなわち、ゼロ価)状態の成分に存在することは必ずしも必要ではなく、金属のオキサイド、ハイドロキサイド、またはスルフィドのような化合物として存在することもできると考えられている。前記第VIII族金属は、例えばこの成分がオキサイドまたはハイドロキサイドの状態の場合には、好ましくは還元状態である。前記第VIII族金属の還元された結合価状態は、水素のような還元剤が反応原料に含有されている場合、反応進行中にin situで得ることができる。
【0023】
前記第VIII族金属は、通常、競争的なイオン交換により第1および第2成分中に取り込むことになる。競争的なイオン交換により、モレキュラーシーブにおける第VIII族金属の良好なアキシャル分布が達成される。
【0024】
本発明はいずれの操作理論にも限定されることが意図されていないが、ほとんどの第VIII族金属がモレキュラーシーブ内の孔にきめ細かく分散されるため、触媒系の低い芳香環飽和を伴った高いエチレン飽和活性の利点が得られる。前記第VIII族金属粒子がモレキュラーシーブの孔内にある場合、遷移状態の選択性のために、すなわち飽和した芳香族の反応遷移状態がモレキュラーシーブ中で形成されるには大きすぎるため、芳香族飽和は起こりえない。さらに、第VIII族金属粒子の高分散性のために、芳香族が1つ以上の第VIII族金属原子と接触することが保障される。提唱される理論の如何にかかわらず、前記プロセスはそれらに開示された改善された特性を有する。
【0025】
競争的なイオン交換には、非水素化金属カチオンがモレキュラーシーブにおいて交換可能なイオンとして第VIII族金属カチオンと競合することを利用することが含まれる。競争的なイオン交換は、第1および第2成分のモレキュラーシーブを、例えばプラチナカチオンのような第VIII族金属カチオンの予め決められた量および例えばアンモニウムカチオンのような非水素化金属カチオンの予め決められた量を含む供給水溶液と接触させることにより実施することができる。
【0026】
供給溶液における第VIII族カチオンに対する非水素化金属カチオンの比率は、供給溶液のpH、前記成分の固有の酸性度、およびモレキュラーシーブと結合した第VIII族金属の量を含む多くの因子により変動することになる。前記供給溶液は、第VIII族カチオンに対する非水素化金属カチオンのモル比が約500〜6000の範囲となるように、通常調製される。好ましくは、第VIII族カチオンに対する非水素化金属カチオンのモル比は約700〜2000の範囲である。より好ましくは、第VIII族カチオンに対する非水素化金属カチオンのモル比は約900〜1100の範囲である。供給溶液に存在する非水素化金属カチオンおよび第VIII族カチオンの量は、最終触媒に含有される第VIII族金属の所望量に依存する。
【0027】
通常、供給溶液のpHは4〜10に維持される。上段床成分(エチルベンゼン変換に有効な成分)のモレキュラーシーブがシリカで選択活性化(selectivated)されている場合、供給水溶液のpHは、通常7以下のpHに維持され、好ましくは約6.5〜7の範囲に維持される。供給プロセスにおける供給溶液pHの調整は通常、水酸化アンモニウムを含む水溶液を用いて行われる。
【0028】
供給溶液用の第VIII族金属カチオンの具体例には、塩化プラチナ酸、塩化プラチナ、および硝酸テトラアミンプラチナ(II)および塩化ペンタアミンクロロイリジウム(III)のようなテトラアミンプラチナおよびテトラアミンパラジウム錯体が含まれる。供給溶液用の非水素化金属カチオンの好適な具体例には、アンモニウムのハロゲン化物または硝酸塩が含まれる。金属の取り込み後、前記触媒は通常、水で洗浄され、乾燥および焼成される。
【0029】
供給溶液用の非水素化金属カチオンの好適な具体例にはアンモニウムカチオンが含まれる。
【0030】
第1および第2成分に存在する第VIII族金属の量は前記触媒成分の重量に基づいて例えば0.001〜約10重量%の範囲に変動させることができる。
【0031】
第1触媒成分に関して、第VIII族金属は、好ましくは約0.001〜約0.05重量%、より好ましくは約0.01〜0.04重量%の範囲で存在することになる。もちろん、これは金属の性質により変動することになる。第VIII族金属がプラチナの場合、第1触媒成分に存在する第VIII族金属の量は、好ましくは全触媒成分の約0.03重量%である。
【0032】
第2触媒成分に関して、第VIII族金属は、好ましくは約0.001〜約0.03重量%、例えば約0.0075〜0.02重量%の範囲で存在することになる。もちろん、これは成分の性質により変動することになる。第VIII族金属がプラチナの場合、第2触媒成分に存在する量は、好ましくは全触媒成分の約0.01重量%である。
【0033】
本発明のプロセスの実施において、第1および第2触媒成分のいずれか又は両方と、本プロセスの温度および他の条件に耐えられる他の物質を一緒に調製してもよい。そのような基材物質には、粘土、シリカ、および/または金属酸化物のような無機酸化物物質が含まれる。前記金属酸化物は、天然由来でも、ゼラチン状析出物でも、シリカおよび金属酸化物の混合物を含むゲルの形態でもよい。モレキュラーシーブと混成できる天然由来の粘土には、モンモリロナイト(montmorillonite)およびカオリン(kaolin)族のものが含まれる。これらの族には、主な無機構成がハロイサイト(halloysite)、カオリナイト(kaolinite)、ディッカイト(dickite)、ナクライト(nacrite)およびアナウクサイト(anauxite)であるディクシー(Dixie)、マクナミー(McNamee)、ジョージアおよびフロリダ粘土またはその他として広く知られているスベントナイト(subbentonite)およびカオリンが含まれる。そのような粘土は、元々採掘された状態で、または初めに焼成、酸処理または化学修飾された状態で用いることができる。
【0034】
前述の物質に加えて、本発明で用いられるモレキュラーシーブは、シリカ−アルミナ−トリア(thoria)、シリカ−アルミナ−ジルコニア、シリカ−アルミナ−マグネシア、およびシリカ−マグネシア−ジルコニアのような3成分化合物ばかりでなく、アルミナ、シリカ−アルミナ、シリカ−マグネシア、シリカ−ジルコニア、シリカ−トリア、シリカ−ベリリア、シリカ−チタニアのような多孔性の基材物質と混成することができる。これらの成分の混合物も用いることができる。さらに、前記モレキュラーシーブは米国特許6,198,013号に記述された方法を用いたゼオライト基材物質と混成することができる。本文献の全内容は、参照により本発明に取り込まれる。好ましくは、結合剤はシリカである。
【0035】
無水ベースのモレキュラーシーブ成分および無機酸化物基材の相対比率は、約1〜約99重量%の範囲、より普通には乾燥混成物の約10〜約80重量%の範囲のモレキュラーシーブ含量で広く変動し得る。
【0036】
本発明の触媒系の第1および第2成分は、第1成分が原料ストリームのエチルベンゼンを選択的にベンゼンへ脱エチル化し、一方、第2成分が原料のキシレンを選択的に異性化することを確保する多くの重要な側面において相互に異なることになる。これらの異なる性質を以下に記述する。
【0037】
例えば、本発明の触媒系の各成分は通常、互いに相容れないキシレン分散特性を示すことになる。これらの特性は、120℃および0.64±0.11kPa(4.5±0.8mm水銀)のオルト−キシレン分圧において30%平衡能のオルト−キシレンを吸着するのに必要な時間(分)を知ることにより決定することができる。本試験は米国特許4,117,026号、4,159,282号、およびRe.31,782号に記述されている。オルト−キシレンの平衡能は本発明では、モレキュラーシーブ100g当たりキシレン1gより大きいものと定義される。本発明の触媒系において、エチルベンゼン変換に有効な第1触媒成分は、好ましくは約50より大きなオルト−キシレン吸着時間(分)、好ましくは約1,200より大きく10,000分未満のオルト−キシレン吸着時間(分)を有する。一方、第2の異性化成分は、好ましくは約50分未満のオルト−キシレン吸着時間、好ましくは約10分未満のオルト−キシレン吸着時間を有する。
【0038】
<エチルベンゼン変換成分>
前記エチルベンゼン変換成分は、好ましくは約50より大きなオルト−キシレン吸着時間(分)、好ましくは約1,200より大きく10,000分未満のオルト−キシレン吸着時間(分)を有する。所望のキシレン分散特性は多くの方法により達成することができる。50分の最小値における又はこれに近いオルト−キシレン分散時間では、前記触媒中で用いられるモレキュラーシーブの大きな結晶形態、すなわち1ミクロンより大きな平均結晶サイズを有するものを選択すれば十分である。しかしながら、より高い分散性を達成するためには、用いられるプロセス条件下で不活性な、シリカのようなコークスおよび/または酸化物の層を触媒粒子の表面に析出させることにより選択活性化することが望ましいといえる。前記成分粒子が選択活性化される場合、大結晶サイズおよび中結晶サイズのいずれのモレキュラーシーブも第1成分に用いることができる。
【0039】
前記第1成分のモレキュラーシーブは、好ましくは第2触媒のモレキュラーシーブよりも高い酸活性を有する。好ましくは、前記第1触媒成分のモレキュラーシーブは第2成分のα値の少なくとも2倍のα値を有する。第2成分は通常30以上のα値を有する。α値の測定操作は米国特許3,354,078号、ジャーナル・オブ・キャタリシス、第4巻、527頁(1965年)、第6巻、278頁(1966年)、第61巻、395頁(1980年)に記述され、これらのその記述は参照により本発明に取り込まれる。本発明で用いられる試験の実験条件には、ジャーナル・オブ・キャタリシス、第61巻、395頁に詳細に記述されるように538℃の定常温度および可変流速が含まれる。より高いα値はより活性な熱分解触媒に対応する。
【0040】
好ましくは、第1成分は水蒸気処理されていない。水蒸気処理が第1成分のα値を上述の値に低下させるために用いられる場合、この水蒸気処理は典型的には、約100℃〜約600℃、例えば約175℃〜約325℃の温度で、約1%〜約100%の蒸気、例えば約50%〜約100%の蒸気を含む雰囲気において、約69Pa−a〜約345kPa−a(0.01psia〜約50psia)の圧力で、約0.1〜約24時間、例えば約3〜約6時間の間、第1成分を加熱することにより行われる。
【0041】
前記第1成分がシリカで選択活性化される場合、これは前記触媒を液体担体において有機ケイ素化合物を用いて1又は複数の処理を施すことにより簡便に行われる。さらに、各処理に続いて、処理された物質が酸素含有雰囲気、例えば空気中において焼成される。このような多工程の選択活性化の操作は米国特許5,476,823号に記述され、本文献の全ての内容は参照により本発明に取り込まれる。好ましくは、第1成分には、2〜4工程のシリカ選択活性化処理が施される。シリカ選択活性化される触媒が結合剤を含む場合、シリカのような非酸性の結合剤を用いることが好ましい。
【0042】
前記第1触媒成分の選択活性化に用いられる有機ケイ素化合物は、例えば、シリコーン、シロキサン、シラン、またはこれらの混合物であり得る。これらの有機ケイ素化合物は分子当たり2以上のケイ素原子をもつことができる。これらの有機ケイ素化合物は、液体担体媒体と組み合わせて溶解性または液体に変換できるなら、純粋形態で固体であり得る。予備選択活性化剤として用いられるシリコーン、シロキサン、またはシラン化合物の分子量は約80〜約20,000の範囲であり得、好ましくはおよそ150〜10,000の範囲内である。予備選択活性化シリコーン化合物には、ジメチルシリコーン、ジエチルシリコーン、フェニルメチルシリコーン、メチル水素シリコーン、エチル水素シリコーン、フェニル水素シリコーン、メチルエチルシリコーン、フェニルエチルシリコーン、ジフェニルシリコーン、メチルトリフルオロプロピルシリコーン、エチルトリフルオロプロピルシリコーン、ポリジメチルシリコーン、テトラクロロフェニルメチルシリコーン、テトラクロロフェニルエチルシリコーン、テトラクロロフェニル水素シリコーン、テトラクロロフェニルフェニルシリコーン、メチルビニルシリコーン、およびエチルビニルシリコーンが含まれる。前記予備選択活性化シリコーン、シロキサン、またはシラン化合物は直線状である必要はなく、例えばヘキサメチルシクロトリシロキサン、オクタメチルシクロテトラシロキサン、ヘキサフェニルシクロトリシロキサン、およびオクタフェニルシクロテトラシロキサンのような環状でもよい。これらの化合物の混合物も、他の官能基を持つシリコーンと同様に、予備選択活性化剤として用いることができる。
【0043】
好ましくは、モレキュラーシーブの予備選択活性化に用いられる有機ケイ素化合物の動力学的直径は、有機ケイ素化合物のモレキュラーシーブ孔への進入およびそれに伴うモレキュラーシーブの内部活性の低下を回避するために、モレキュラーシーブの直径よりも長い。
【0044】
好ましい有機ケイ素予備選択活性化剤には、特に予備選択活性化剤が有機担体に溶解し、または水溶性担体中で乳化する場合、ジメチルフェニルメチルシロキサン(例えば、Dow−550)およびフェニルメチルポリシロキサン(例えば、Dow−710)が含まれる。Dow−550およびDow−710はミシガン州、ミッドランドのダウ・ケミカル社から入手できる。
【0045】
好ましくは、前記有機ケイ素化合物用の液体担体は、分子当たり5以上、特に7以上の炭素原子を有する直鎖状、分枝状、または環状の炭化水素、例えばヘプタン、オクタン、ノナン、またはウンデカンのようなアルカンのような有機化合物である。これら有機化合物、例えばアルカンの沸点は約70℃より高くてもよい。水素熱分解循環油のような低揮発性有機化合物の混合物は担体として用いることができる。特に好ましい有機担体はデカンおよびドデカンである。
【0046】
有機ケイ素化合物の各含浸に続いて、前記触媒は200℃を超えるが、モレキュラーシーブの結晶が悪影響を受ける温度未満で、約0.2℃/分〜5℃/分の速度で焼成される。この焼成温度は一般に600℃未満であり、好ましくはおよそ350〜550℃の範囲内である。焼成温度における焼成の時間は1〜24時間、例えば2〜6時間であり得る。好ましくは、前記触媒は3回の選択活性化操作に付される。
【0047】
シリカ選択活性化に追加して、またはこれに替えて、前記第1触媒成分はコークス選択活性化に付すこともできる。この任意のコークス選択活性化は、典型的に前記触媒を熱分解性有機化合物の分解温度を超えて上昇した温度で、モレキュラーシーブの結晶が悪影響を受ける温度未満で、当該熱分解性有機化合物と接触させることを含む。この接触温度は、例えば約650℃未満でもよい。このコークス選択活性化プロセスに用いることができる有機物質には、具体例を挙げれば、パラフィン、シクロパラフィン、オレフィン、シクロオレフィン、および芳香族のような炭化水素、アルコール、アルデヒド、エーテル、ケトン、およびフェノールのような酸素含有有機化合物、およびフラン、チオフェン、ピロール、およびピリジンのようなヘテロ環化合物が含まれる。水素助原料を、過剰なコークスの生成を止めるために用いることができる。コークス選択活性化に関する更に詳細な事項は米国特許4,117,026号に記述されている。シリカ選択活性化に続いてコークス選択活性化を組み合わせることにより、特定のキシレン分散性を獲得するために必要とされる有機ケイ素含浸処理工程の数を減らすことができる。
【0048】
<異性化成分>
前記触媒系の第2成分はC8芳香族を含む原料中のキシレンの異性化に有効である。前記第2成分は、好ましくは約50分未満のオルト−キシレン吸着時間を有し、この時間はさらに好ましくは約10分未満である。これは、この成分中に0.02〜0.05ミクロンの平均結晶サイズを有する小結晶サイズのモレキュラーシーブを用いることにより達成される。前記触媒系の第二成分のモレキュラーシーブは、典型的には約30以上のα値を有することになる。
【0049】
好ましくは、前記第2モレキュラーシーブは、このモレキュラーシーブを用いて第VIII族金属の取り込みの前に、所望のα値を達成するために水蒸気処理が施される。
【0050】
<プロセス条件>
本発明のプロセスにおいて用いられる条件は狭い範囲になるように定義されるものではないが、一般に、約204〜約540℃(400〜1,000°F)の温度、約100〜約7,000kPa−a(0〜1,000psig)の圧力、0.1〜200hr-1の重量時間当たり空間速度(WHSV)、約0.2〜約10の炭化水素(HC)に対する水素(H2)モル比が含まれる。好ましくは、前記条件には、約343〜約413℃(650〜775°F)の温度、約445〜約2,860kPa−a(50〜400psig)の圧力、約3〜50hr-1のWHSV、約0.7〜約3のHCに対するH2モル比が含まれる。
【0051】
一般に、本発明のプロセスは上述の触媒系を含む固定床において実施される。好ましい態様において、前記触媒系の第1および第2成分は単一反応槽における連続床に存在する。すなわち、エチルベンゼン変換に有効な本発明のプロセスで用いられる触媒系の成分は第1床を形成し、一方、キシレン異性化に有効な前記触媒系の他の成分は前記第1床下流の第2床を形成する。前記原料は、好ましくは軽量気体を途中で分離することなく第1から第2床へ直列に流れる。この配置に替えて、第1および第2床は、分離した反応槽、所望ならば異なるプロセス条件で操作が可能な反応槽に配置することができる。本発明の第1および第2触媒成分の前後に、追加の触媒床を設けることともできる。
【0052】
前記変換プロセスの後に、前記異性化生成物はパラ−キシレンおよび/または他の望ましい1または複数のキシレンを単離するために処理することができる。したがって、例えば、前記異性化生成物は結晶化剤、膜分離単位、または選択的吸着単位のような様々なパラ−キシレン回収単位に供給することができ、最終的にパラ−キシレンを単離および回収することができる。残留異性化物からはC8よりも軽い生成物を取り除くことができる。残留異性化物中のC8よりも重い生成物は、さらに加工し、分画して取り出すことができる。パラ−キシレンが取り除かれたC8画分は異性化装置に再循環することができる。
【0053】
本発明のプロセスの1つの結果は、熱平衡未満の量のパラ−キシレンを含有する原料の混合キシレン成分を、異性化装置からの生成物が熱平衡に少なくとも近い量のパラ−キシレンを含む程度まで変換することである。
【0054】
本発明のプロセスのもう1つの結果は、最小限のキシレン損失で混合キシレン原料に含まれるエチルベンゼンの高い割合での変換である。例えば、50重量%より高いエチルベンゼン変換レベルが、2重量%未満のキシレン損失レベルで達成することができる。
【実施例】
【0055】
以下、実施例に基づき、本発明についてさらに詳細に説明する。なお、本発明は下記実施例に限定されるものではない。
【0056】
<比較例1>
芳香環族飽和に対するエチレン飽和の評価を2成分触媒系を用いて実施した。この2成分触媒系は40重量%の上段床成分および60重量%の下段床成分を含んだ。プラチナを初期湿性含浸(incipient wetness impregnation)により上段床成分に取り込み、プラチナを混合(mulling)しながら下段床成分に取り込んだ。
【0057】
前記2成分触媒系の上段床成分は中結晶サイズを有するZSM−5より形成した。前記ZSM−5は、65重量%のZSM−5および35重量%のシリカ結合剤の重量比でシリカ結合剤と混成した。このシリカ結合ZSM−5を従来法により1/16”直径円筒形粒子に押出し、次にデカン中7.8重量%ジメチルフェニルメチルポリシロキサンを用いた4連続含浸処理を含む多段階シリカ選択活性化操作を施した。各含浸の後に溶媒は取替え、前記触媒は窒素中で焼成し、次に空気中で538℃に加熱した。次に、硝酸テトラアミンプラチナ(II)を用いた初期湿性含浸、続いて乾燥および空気焼成により前記選択活性化された触媒にプラチナを取り込んだ。次に前記触媒をα値158まで水蒸気処理した。得られた触媒は0.1重量%のプナチナを含有した。
【0058】
前記下段床成分は小結晶サイズを有するZSM−5より形成した。前記ZSM−5は、50重量%のZSM−5および50重量%のアルミナ結合剤の重量比でアルミナ結合剤と混成した。このアルミナ結合ZSM−5を従来法により0.16cm(1/16”)直径円筒形粒子に押出した。すなわち、塩化テトラアミンプラチナの形態で0.1重量%のプラチナを用いZSM−5およびアルミナ結合剤物質を混合しながら添加し、押出した。前記混合は米国特許Re.31,919号に記述された技術を用いて実施した。次に押出物は乾燥および空気中で焼成を行った。次に前記触媒はα値18まで水蒸気処理した。前記下段床成分は0.1重量%のプラチナを含有した。
【0059】
前記2成分触媒系は、429℃(805°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0060】
前記評価で用いた原料は、0.7重量%非芳香族、0.6重量%トルエン、18.7重量%エチルベンゼン、0.6重量%パラ−キシレン、61.7重量%メタ−キシレン、16.7重量%オルト−キシレン、1重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの2倍モルの硫黄を用いて399℃(750°F)および1,825kPa−a(250psig)において予備スルフィド化し、次に11日間429℃(805°F)、10h-1WHSV、0.9:1のH2:HC、および1,384kPa−a(186psig)の全体圧力で脱エッジング(de-edged)を行った。その結果を下記表Iに示す。
【0061】
【表1】
【0062】
<比較例2>
芳香族環飽和に対するエチレン飽和の評価を2成分触媒系を用いて実施した。この2成分触媒系は30重量%の上段床成分および70重量%の下段床成分を含んだ。プラチナをイオン交換により上段床成分に取り込み、プラチナをイオン交換により下段床成分に取り込んだ。
【0063】
前記上段床成分は、前記触媒成分が3工程の選択活性化操作に付され、500のα値を有し、前記プラチナがイオン交換により前記選択活性化された触媒に取り込まれることを除き、比較例1の上段床と同様に調製された。前記イオン交換は500mLの水に硝酸テトラアミンプラチナ(II)(0.030gのプラチナ)を溶解し、次にこの溶液を80℃(176°F)まで加熱して行った。次に、100gの前記シリコン選択活性化触媒をこの溶液に浸した。前記溶液は8時間循環した。前記触媒を溶液から取り出し、蒸留水で洗浄、121℃(250°F)で乾燥、354℃(660°F)で1時間空気中で焼成した。前記上段床成分は0.03重量%のプラチナを含有し、500のα値を有した。
【0064】
前記下段床成分は、既にα値108に水蒸気処理された80重量%ZSM−5および20重量%シリカ結合剤を含む押出物を形成することにより調製された。次に、硝酸テトラアミンプラチナ(II)(0.02gのプラチナ)を600mLの蒸留水に溶解し、この溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH8〜9に調整した。前記プラチナ含有溶液を3時間前記触媒上に循環させた。前記溶液のpHは、10重量%水酸化アンモニウムを含む溶液を用いて取り込みの間pH8〜9に維持した。前記触媒を溶液から取り出し、蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記下段床成分は0.01重量%のプラチナを含有し、108のα値を有した。
【0065】
前記2成分触媒系は、376℃(709°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0066】
前記評価で用いた原料は、0.6重量%非芳香族、1.4重量%トルエン、14.7重量%エチルベンゼン、1.3重量%パラ−キシレン、62.8重量%メタ−キシレン、18.8重量%オルト−キシレン、0.4重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの20倍モルの硫黄を用いて357℃(675°F)および1,653kPa−a(225psig)において予備スルフィド化し、次に16日間399℃(750°F)、10h-1WHSV、0.6:1のH2:HC、および1,466kPa−a(198psig)の全体圧力で脱エッジング(de-edged)を行った。その試験結果を下記表IIに示す。
【0067】
【表2】
【0068】
<実施例1>
芳香族環飽和に対するエチレン飽和の評価を2成分触媒系について実施した。この2成分触媒系は30重量%の上段床(第1)成分および70重量%の下段床(第2)成分を含んだ。プラチナを競争的イオン交換により上段床成分および下段床成分に取り込んだ。
【0069】
前記上段床成分は、前記プラチナが競争的イオン交換により前記選択活性化された触媒に取り込まれることを除き、比較例2の上段床と同様に調製された。前記触媒に取り込まれたプラチナ量は0.03重量%であった。前記競争的イオン交換は、初めに250gの上段床成分を300mLカラムに充填することにより実施した。前記触媒はカラムを通して湿った空気を通過させることにより湿気をもたせた。次に、0.05N硝酸アンモニウムを含む溶液をカラムを通して循環し、前記溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH6.5〜7.0に調整した。硝酸テトラアミンプラチナ(II)(250mLの蒸留水に0.075gのプラチナが溶解された)溶液を4時間をかけて硝酸アンモニウムタンクに加えた。プラチナ交換溶液におけるプラチナに対する硝酸アンモニウムのモル比は976であった。前記プラチナ交換溶液は約48時間にわたり触媒床を通して循環した。この間、pHは10重量%水酸化アンモニウムを含む溶液を用いて継続的にpH6.5〜7.0に維持された。前記触媒は蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記上段床成分は0.03重量%のプラチナを含有した。
【0070】
前記下段床成分は、前記プラチナが競争的イオン交換により前記触媒に取り込まれたことを除き、比較例2の下段床と同様に調製された。取り込まれたプラチナ量は0.01重量%であった。前記競争的イオン交換は、初めに1,298gの下段床成分を1Lカラムに充填することにより実施した。その後、前記成分はカラムを通して湿った空気を通過させることにより湿気をもたせた。次に、0.05N硝酸アンモニウム溶液3,894gをカラムを通して循環し、前記溶液のpHを10重量%水酸化アンモニウムを含む溶液を用いてpH8〜9に調整した。硝酸テトラアミンプラチナ(II)(250mLの蒸留水に0.13gのプラチナが溶解された)溶液を4時間をかけて硝酸アンモニウムタンクに加えた。プラチナに対する硝酸アンモニウムのモル比は2,921であった。前記プラチナ交換溶液は約12時間にわたり前記成分上を循環した。この間、前記pHは10重量%水酸化アンモニウムを含む溶液を用いて継続的に維持された。前記成分は蒸留水で洗浄、121℃(250°F)で乾燥、349℃(660°F)で3時間空気中で焼成した。前記下段床成分は0.01重量%のプラチナを含有した。
【0071】
前記2成分触媒系について、357℃(675°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力のマイクロユニットでエチルベンゼン変換を評価した。
【0072】
前記評価で用いた原料は、0.7重量%非芳香族、1.4重量%トルエン、14.6重量%エチルベンゼン、1.3重量%パラ−キシレン、63.1重量%メタ−キシレン、18.8重量%オルト−キシレン、0.1重量%の炭素数9以上の芳香族からなるC8芳香族原料である。前記触媒は、プラチナの20倍モルの硫黄を用いて357℃(675°F)および1,653kPa−a(225psig)において予備スルフィド化し、次に3日間413℃(775°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力で脱エッジング(de-edged)を行った。その結果を下記表IIIに示す。
【0073】
【表3】
【0074】
表IIIの結果は、前記触媒が低い芳香族環損失の下に高いエチレン飽和で優れたエチルベンゼン変換を行うことを示している。
【0075】
<実施例2>
触媒系Aおよび触媒系Bをエチルベンゼン変換および芳香族環損失について評価した。
【0076】
触媒系Aは、それが50重量%の上段床および50重量%の下段床を含むことを除き、比較例1の触媒系と同様である。触媒系Aは10h-1WHSV、1:1のH2:HC、1,653kPa−a(225psig)の全体圧力、および381℃(721°F)〜436℃(817°F)の温度範囲の条件下で運転された。本試験で用いた原料は、10.3重量%エチルベンゼン、1.2重量%パラ−キシレン、61.8重量%メタ−キシレン、26.7重量%オルト−キシレンからなるC8芳香族原料である。
【0077】
触媒系Bの上段および下段床は、それが25重量%の上段床および75重量%の下段床を含むことを除き、実施例1と同様である。触媒系Bは10h-1WHSV、1:1のH2:HC、1,653kPa−a(225psig)の全体圧力、および341℃(646°F)〜371℃(699°F)の温度範囲の条件下で運転された。本試験の実施前に、触媒系Bは、2日間404℃(760°F)、10h-1WHSV、1:1のH2:HC、および1,653kPa−a(225psig)の全体圧力で脱エッジング(de-edged)を行った。本試験で用いた原料は、0.3重量%非芳香族、0.4重量%トルエン、12.9重量%エチルベンゼン、1.2重量%パラ−キシレン、66.5重量%メタ−キシレン、18.4重量%オルト−キシレン、0.3重量%の炭素数9以上の芳香族からなるC8芳香族原料である。
【0078】
本試験の結果は図に示した。
【0079】
図における結果は、触媒系Bが低い変換温度で低い芳香族環損失が低いことを示している。
【特許請求の範囲】
【請求項1】
10,000より大きな芳香族環飽和に対するエチレン飽和比を示す2成分触媒系であって、
前記芳香族環飽和に対するエチレン飽和比が以下の式により決定され、
【数1】
(a)前記触媒系の容積に対し5%以上存在し、エチルベンゼン変換に有効であり、1〜12の束縛指数(Constraint Index)を有する第1モレキュラーシーブおよび有効量の第VIII族金属を含む第1成分、及び
(b)キシレン異性化に有効であり、1〜12の束縛指数(Constraint Index)を有する第2モレキュラーシーブ及び有効量の第VIII族金属を含む第2成分を含み、
前記第1成分が、前記第2成分のα値の2倍以上のα値を有し、
前記第1成分及び前記第2成分が、芳香族環飽和に対するエチレン飽和比が10,000より大きなると共にキシレン損失レベルが2重量%未満となるように選択される、2成分触媒系。
【請求項2】
前記第1成分、または前記第1成分および前記第2成分の両者が非アルミナ結合剤を含む、請求項1に記載の2成分触媒系。
【請求項3】
前記結合剤がシリカである、請求項1または2に記載の2成分触媒系。
【請求項4】
前記第1成分が、前記触媒系の容積で10%以上存在する、請求項1〜3のいずれか1項に記載の2成分触媒系。
【請求項5】
前記第1成分および前記第2成分に存在する前記第VIII族金属がプラチナである、請求項1〜4のいずれか1項に記載の2成分触媒系。
【請求項6】
前記第1モレキュラーシーブおよび前記第2モレキュラーシーブが、モルデナイト(mordenite)、ベータ(Beta)、MCM−22族モレキュラーシーブ、ZSM−5、ZSM−11、ZSM−12、ZSM−22、ZSM−23、ZSM−34、ZSM−35、ZSM−48、およびZSM−57からなる群から選択される、請求項1〜5のいずれか1項に記載の2成分触媒系。
【請求項7】
前記第2成分が30以上のα値を有する、請求項1〜6のいずれか1項に記載の2成分触媒系。
【請求項8】
前記第1成分に存在する前記第VIII族金属の量が、前記第1成分の重量に基づいて0.01〜0.04重量%の範囲内の量である、請求項1〜7のいずれか1項に記載の2成分触媒系。
【請求項9】
前記第2成分に存在する前記第VIII族金属の量が、前記第2成分の重量に基づいて0.0075〜0.02重量%の範囲内の量である、請求項1〜7のいずれか1項に記載の2成分触媒系。
【請求項10】
請求項1に記載の2成分触媒系を調製するプロセスであって、
(i)前記第1モレキュラーシーブを非水素化金属カチオンおよび第VIII族金属カチオンを含有する第1供給水溶液と接触することにより、第VIII族金属を前記第1成分の前記第1モレキュラーシーブ内へ0.01〜0.04重量%の範囲の量でイオン交換する工程であって、
前記第1モレキュラーシーブ内に前記第VIII族カチオンをイオン交換するのに有効な条件下における前記第1供給水溶液中の第VIII族カチオンに対する非水素化金属カチオンのモル比が500〜6,000の範囲である工程、および
(ii)前記第2モレキュラーシーブを非水素化金属カチオンおよび第VIII族金属カチオンを含有する第2供給水溶液と接触することにより、第VIII族金属を前記第2成分の前記第2モレキュラーシーブ内へ0.0075〜0.02重量%の範囲の量でイオン交換する工程であって、
前記第2モレキュラーシーブ内に前記第VIII族カチオンをイオン交換するのに有効な条件下における前記第1供給水溶液中の第VIII族カチオンに対する非水素化金属カチオンのモル比が500〜6,000の範囲である工程を含むプロセス。
【請求項11】
前記第1成分を、前記第1モレキュラーシーブをケイ素化合物と接触させて選択活性化されたモレキュラーシーブを得て、引き続き当該選択活性化されたモレキュラーシーブを焼成することを含む少なくとも1つの選択活性化操作に付すことにより修飾する、請求項10に記載のプロセス。
【請求項12】
前記第1成分が3回の選択活性化操作に付される、請求項11に記載のプロセス。
【請求項13】
前記第VIII族金属が、前記選択活性化操作の完了後に前記触媒系に取り込まれる、請求項11または12に記載のプロセス。
【請求項14】
前記第1および第2供給溶液のpHが10未満に維持される、請求項10〜13のいずれか1項に記載のプロセス。
【請求項15】
前記第1モレキュラーシーブがシリカを用いて選択活性化され、前記第1供給溶液のpHが7未満に維持されることを特徴とする請求項10〜13のいずれか1項に記載のプロセス。
【請求項1】
10,000より大きな芳香族環飽和に対するエチレン飽和比を示す2成分触媒系であって、
前記芳香族環飽和に対するエチレン飽和比が以下の式により決定され、
【数1】
(a)前記触媒系の容積に対し5%以上存在し、エチルベンゼン変換に有効であり、1〜12の束縛指数(Constraint Index)を有する第1モレキュラーシーブおよび有効量の第VIII族金属を含む第1成分、及び
(b)キシレン異性化に有効であり、1〜12の束縛指数(Constraint Index)を有する第2モレキュラーシーブ及び有効量の第VIII族金属を含む第2成分を含み、
前記第1成分が、前記第2成分のα値の2倍以上のα値を有し、
前記第1成分及び前記第2成分が、芳香族環飽和に対するエチレン飽和比が10,000より大きなると共にキシレン損失レベルが2重量%未満となるように選択される、2成分触媒系。
【請求項2】
前記第1成分、または前記第1成分および前記第2成分の両者が非アルミナ結合剤を含む、請求項1に記載の2成分触媒系。
【請求項3】
前記結合剤がシリカである、請求項1または2に記載の2成分触媒系。
【請求項4】
前記第1成分が、前記触媒系の容積で10%以上存在する、請求項1〜3のいずれか1項に記載の2成分触媒系。
【請求項5】
前記第1成分および前記第2成分に存在する前記第VIII族金属がプラチナである、請求項1〜4のいずれか1項に記載の2成分触媒系。
【請求項6】
前記第1モレキュラーシーブおよび前記第2モレキュラーシーブが、モルデナイト(mordenite)、ベータ(Beta)、MCM−22族モレキュラーシーブ、ZSM−5、ZSM−11、ZSM−12、ZSM−22、ZSM−23、ZSM−34、ZSM−35、ZSM−48、およびZSM−57からなる群から選択される、請求項1〜5のいずれか1項に記載の2成分触媒系。
【請求項7】
前記第2成分が30以上のα値を有する、請求項1〜6のいずれか1項に記載の2成分触媒系。
【請求項8】
前記第1成分に存在する前記第VIII族金属の量が、前記第1成分の重量に基づいて0.01〜0.04重量%の範囲内の量である、請求項1〜7のいずれか1項に記載の2成分触媒系。
【請求項9】
前記第2成分に存在する前記第VIII族金属の量が、前記第2成分の重量に基づいて0.0075〜0.02重量%の範囲内の量である、請求項1〜7のいずれか1項に記載の2成分触媒系。
【請求項10】
請求項1に記載の2成分触媒系を調製するプロセスであって、
(i)前記第1モレキュラーシーブを非水素化金属カチオンおよび第VIII族金属カチオンを含有する第1供給水溶液と接触することにより、第VIII族金属を前記第1成分の前記第1モレキュラーシーブ内へ0.01〜0.04重量%の範囲の量でイオン交換する工程であって、
前記第1モレキュラーシーブ内に前記第VIII族カチオンをイオン交換するのに有効な条件下における前記第1供給水溶液中の第VIII族カチオンに対する非水素化金属カチオンのモル比が500〜6,000の範囲である工程、および
(ii)前記第2モレキュラーシーブを非水素化金属カチオンおよび第VIII族金属カチオンを含有する第2供給水溶液と接触することにより、第VIII族金属を前記第2成分の前記第2モレキュラーシーブ内へ0.0075〜0.02重量%の範囲の量でイオン交換する工程であって、
前記第2モレキュラーシーブ内に前記第VIII族カチオンをイオン交換するのに有効な条件下における前記第1供給水溶液中の第VIII族カチオンに対する非水素化金属カチオンのモル比が500〜6,000の範囲である工程を含むプロセス。
【請求項11】
前記第1成分を、前記第1モレキュラーシーブをケイ素化合物と接触させて選択活性化されたモレキュラーシーブを得て、引き続き当該選択活性化されたモレキュラーシーブを焼成することを含む少なくとも1つの選択活性化操作に付すことにより修飾する、請求項10に記載のプロセス。
【請求項12】
前記第1成分が3回の選択活性化操作に付される、請求項11に記載のプロセス。
【請求項13】
前記第VIII族金属が、前記選択活性化操作の完了後に前記触媒系に取り込まれる、請求項11または12に記載のプロセス。
【請求項14】
前記第1および第2供給溶液のpHが10未満に維持される、請求項10〜13のいずれか1項に記載のプロセス。
【請求項15】
前記第1モレキュラーシーブがシリカを用いて選択活性化され、前記第1供給溶液のpHが7未満に維持されることを特徴とする請求項10〜13のいずれか1項に記載のプロセス。
【図1】

【公開番号】特開2012−110892(P2012−110892A)
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願番号】特願2011−282857(P2011−282857)
【出願日】平成23年12月26日(2011.12.26)
【分割の表示】特願2007−523553(P2007−523553)の分割
【原出願日】平成17年6月8日(2005.6.8)
【出願人】(599134676)エクソンモービル・ケミカル・パテンツ・インク (301)
【Fターム(参考)】
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願日】平成23年12月26日(2011.12.26)
【分割の表示】特願2007−523553(P2007−523553)の分割
【原出願日】平成17年6月8日(2005.6.8)
【出願人】(599134676)エクソンモービル・ケミカル・パテンツ・インク (301)
【Fターム(参考)】
[ Back to top ]