
キシロース代謝を改善した形質転換大腸菌
【課題】グルコース存在下のカタボライト抑制の影響を受けにくく,キシロース代謝が改善された大腸菌株の提供。
【解決手段】構成発現型のプロモーターおよびキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターを大腸菌株に導入することにより,培養初期からキシロース異性化酵素を発現させ,カタボライト抑制の影響を受けにくい形質転換大腸菌。加えて,キシロース異性化酵素に加え,一連するキシロースに関連する酵素やタンパクを発現させることにより,キシロース代謝をさらに改善した形質転換大腸菌。
【解決手段】構成発現型のプロモーターおよびキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターを大腸菌株に導入することにより,培養初期からキシロース異性化酵素を発現させ,カタボライト抑制の影響を受けにくい形質転換大腸菌。加えて,キシロース異性化酵素に加え,一連するキシロースに関連する酵素やタンパクを発現させることにより,キシロース代謝をさらに改善した形質転換大腸菌。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は,キシロース代謝を改善した形質転換大腸菌に関する。
【0002】
キシロースは,分子式がC5H10O5で表される分子量150.13の糖であり,単糖,五炭糖及びアルドースに分類される。自然界において,キシロースは単体ではほとんどみられないが,ヘミセルロースの一つであるキシランを構成するものとして植物に広く存在する。
【0003】
キシロースの代謝は,バイオマスからのエタノール生産を行う際に,エタノール生産効率を左右する重要なポイントとなってくる。
例えば,農産廃棄物や廃材由来の木質系バイオマスは,食糧とは競合しないことから利用が期待されるバイオマスである。この木質系バイオマスは,セルロースが約50%,ヘミセルロースが約30%,その他リグニンなどで構成されている。この木質系バイオマスをバイオマス・エタノール原料として利用する際,醸造酵母Saccharomyces cerevisiae(S. cerevisiae)が従来用いられてきた。このS. cerevisiaeは,セルロース由来のグルコースをエタノールへ変換することはできるものの,ヘミセルロース由来のキシロースをエタノールへ変換することが出来ない。そこで,木質系バイオマスから,より効率良くバイオマス・エタノールを生産するためには,ヘミセルロース由来のキシロースをもエタノール原料として利用することが求められるため,キシロースからのエタノール発酵が可能な菌株を用いる必要がある。
【0004】
このキシロースからのエタノール発酵が可能な菌株として,酵母の1種であるPichia stipitisやPachysolen tannophilusが知られている(非特許文献1)。加えて,形質転換に広く汎用されている大腸菌においてもこのような試みがなされており,例えば,太田らにより開発されたKO11株が知られている(特許文献1,特許文献2,非特許文献2)。
【0005】
また,菌株のキシロース代謝能を向上させる技術として,下記のような技術が開示されている。
特許文献3には,ペントース糖の利用および発酵に必要な酵素をコードする外来構造遺伝子を含む,安定なZymomonas組込み体の製造方法が開示されている。
特許文献4には,xylABFGHR遺伝子座などのキシロース資化酵素をコードする遺伝子の発現量が増加されたEscherichia属細菌を用いて,L-ヒスチジン,L-スレオニン,L-リジン,L-グルタミン酸およびL-トリプトファンなどのL-アミノ酸を生産する方法が開示されている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Delgenes J. P., MolettaR. and Navarro J.M. (1986) the effect of aeration on D-xylosefermentation by Pachysolen tannophilus,Pichia stipitis, Kluyveromyces marxianus and Cadida shenatae. Biotechnol. Lett. 1986 Nov; 8(12): 897-900
【非特許文献2】Ohta et al. (1991) Appl.Environ.Microbiol.57:893-900
【特許文献】
【0007】
【特許文献1】米国特許第5000000号明細書
【特許文献2】米国特許第5424202号明細書
【特許文献3】国際公開第2004/037973号
【特許文献4】特開2005−261433号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
非特許文献1には,エタノール発酵が可能な菌株として,酵母の1種であるPichia stipitisやPachysolen tannophilusに関する技術が開示されている。しかしながら,これらの菌株は約1% (v/v) 程度という僅かなエタノールしか生産できないことからエタノールの生産効率という点において,必ずしも十分な技術とはいえない。
また,特許文献1等に開示されているKO11株は,キシロースなどの五炭糖からも優れたエタノール生産能を有している。しかしながら,KO11株はグルコース存在下ではカタボライト抑制を受けるため,キシロースよりもグルコースを優先的に消費し,キシロースをあまり消費しないという技術的課題が存在する。
【0009】
特許文献3には,ペントース糖の利用および発酵に必要な酵素をコードする外来構造遺伝子を含む,安定なZymomonas組込み体の製造方法が開示されている。しかしながら,KO11株やキシローストランスポーターに関する遺伝子の導入についても何ら開示はない。さらには,カタボライト抑制についての開示もない。
【0010】
特許文献4には,xylABFGHR遺伝子座などのキシロース資化酵素をコードする遺伝子の発現量が増加されたEscherichia属細菌を用いて,L-ヒスチジン,L-スレオニン,L-リジン,L-グルタミン酸およびL-トリプトファンなどのL-アミノ酸を生産する方法が開示されている。
しかしながら,特許文献4に記載の技術に関しては,キシロース代謝を促進するため,xylオペロンの調節遺伝子であるxylRも含めてキシロース代謝に関連する一連のタンパクをコードする遺伝子をほとんどすべて導入し,その発現量を増加させようとする技術に過ぎないものである。よって,特許文献4に記載の技術は,結局は,グルコースによるキシロース代謝の抑制を受けてしまう技術にとどまり,培養液中のグルコースを大腸菌が消費した後にカタボライト抑制が解除されxylABFGH遺伝子が発現されるため,グルコースと同時にキシロースが消費されないという技術的課題を含むものである。
【0011】
かかるカタボライト抑制の問題は,KO11株のみにとどまらず形質転換に広く汎用されている大腸菌にも存在する問題である。すなわち,大腸菌にキシロース代謝関連遺伝子を単に導入したとしても,グルコース代謝によりキシロースオペロン調節遺伝子が作動し,キシロース代謝が抑制されるものと考えられる。
【0012】
上記事情を背景として,本発明では,グルコース存在下のカタボライト抑制の影響を受けにくく,キシロース代謝が改善された大腸菌株の開発を課題とする。
【課題を解決するための手段】
【0013】
発明者らは,大腸菌におけるキシロース代謝経路の分析を行った。大腸菌では,キシロースの輸送や代謝に関する種々のタンパクが存在するが,発明者は,それらの中でもキシロースの異性化を行う酵素に着目した。すなわち,大腸菌のキシロース異性化酵素は,キシロースとの基質親和性が極端に低く,触媒効率も低い。このため,大腸菌において,キシロース異性化の段階がキシロース代謝の律速段階になっていることに発明者は着目した。
【0014】
この知見を基に発明者は,構成発現型のプロモーターおよびキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターを大腸菌株に導入することにより,培養初期からキシロース異性化酵素を発現させ,カタボライト抑制の影響を受けにくい形質転換大腸菌の開発に成功した。加えて,発明者は更なる改良を行い,キシロース異性化酵素に加え,一連のキシロースに関連する酵素やタンパクを発現させることにより,キシロース代謝をさらに改善した形質転換大腸菌の開発に成功した。
【0015】
本発明は,以下から構成される。
【0016】
本発明の第1の構成は,構成発現型プロモーターとキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターが導入されたことを特徴とする形質転換大腸菌である。
本発明の第2の構成は,前記構成発現型プロモーターが,GAPDHプロモーターであることを特徴とする請求項1に記載の形質転換大腸菌株である。
本発明の第3の構成は,前記キシロース異性化酵素遺伝子が,xylAであることを特徴とする第1又は2の構成に記載の形質転換大腸菌である。
本発明の第4の構成は,前記プラスミド・ベクターに,さらにキシルロースリン酸化酵素遺伝子を挿入することを特徴とする第1ないし3の構成に記載の形質転換大腸菌株である。
本発明の第5の構成は,前記キシルロースリン酸化酵素遺伝子が,xylBであることを特徴とする第4の構成に記載の形質転換大腸菌である。
本発明の第6の構成は,前記プラスミド・ベクターに,さらにキシロース輸送タンパク遺伝子を挿入することを特徴とする第1ないし5の構成に記載の形質転換大腸菌である。
本発明の第7の構成は,前記キシロース輸送タンパク遺伝子が,xylF,xylG,xylHであることを特徴とする第6の構成に記載の形質転換大腸菌である。
本発明の第8の構成は,前記大腸菌が,KO11株であることを特徴とする請求項1ないし7に記載の形質転換大腸菌である。
【発明の効果】
【0017】
本発明により,グルコース存在下においてもカタボライト抑制の影響を受けにくく,キシロース代謝が改善された形質転換大腸菌の提供が可能となった。かかる形質転換大腸菌により,グルコース消費中のカタボライト抑制が緩和され,キシロースの代謝が改善される。ひいては,かかる形質転換大腸菌により,木質系および草本系バイオマスからのエタノール生産向上ないしエタノール生産方法の改善に期待できる。さらには,本発明にかかる形質転換大腸菌をさらに改良することにより,エタノール以外での利用,例えばアミノ酸発酵や乳酸発酵などについても期待できる。
【図面の簡単な説明】
【0018】
【図1】実施例1,実施例2のプラスミド・ベクターのMCS領域の遺伝子配置を示した図
【図2】プラスミド・ベクターpUC18の遺伝子配置を示した図
【発明を実施するための形態】
【0019】
以下,本発明にかかる形質転換大腸菌について,説明を行う。まず,本発明を構成する主な要素について説明を行う。
【0020】
本発明において,大腸菌とは,Escherichia属に属するグラム陰性桿菌として定義される。本発明では,この大腸菌に,構成発現型プロモーターおよびキシロース代謝関連酵素遺伝子を挿入したプラスミド・ベクターを導入することにより,本発明にかかる形質転換大腸菌を得ることができる。本発明で用いられる大腸菌は,特に限定する必要はないが,安全性等の観点から,O157等病原性を有する大腸菌を用いることは避けたほうがよい。好ましい大腸菌株としては,JM109,JM110,BL21,DH5αなどが挙げられる。
【0021】
本発明において構成発現型プロモーターとは,常時目的遺伝子を発現させるプロモーターとして定義される。かかる構成発現型プロモーターは,種々の観点から選択することができる。例えば,入手のしやすさ,Multiple cloning site(MCS領域)への挿入のしやすさなどである。好ましい構成発現型プロモーターとしては,グリセルアルデヒド3-リン酸脱水素酵素遺伝子(GAPDHプロモーター),ホスホグリセリン酸キナーゼプロモーター(PGKプロモーター)などを用いることができる。
【0022】
本発明においてキシロース異性化酵素遺伝子とは,キシロースをキシルロースに変換する酵素をコードする遺伝子として定義される。かかる場合,大腸菌やKO11株内においてキシロースを異性化する酵素を発現しうる限り,特に限定する必要はないが,大腸菌やKO11株において発現し,かつ,キシロースとの基質親和性が高い酵素を発現する遺伝子を選択することが好ましい。好ましくは,大腸菌由来や放線菌由来,乳酸菌由来のキシロース異性化酵素遺伝子を用いることができる。大腸菌由来としては,xylAを用いることが好ましい。また,放線菌由来としては,xylA,SCO1169,SGR_1070などを,乳酸菌由来としては,LAF_0402,LVIS_0183などを用いることができる。
【0023】
また,キシルロースリン酸化酵素遺伝子とは,キシルロースの5位をリン酸化し,キシルロース5リン酸に変換する酵素をコードする遺伝子として定義される。かかる場合,大腸菌やKO11株内においてキシルロースの5位をリン酸化する酵素を発現しうる限り,特に限定する必要はないが,大腸菌やKO11株において発現し,かつ,キシルロースのリン酸化能が高い酵素を発現する遺伝子を選択することが好ましい。好ましくは,xylBを用いることができる。
【0024】
さらに,キシロース輸送タンパク遺伝子とは,キシロースを細胞内に輸送する機能を果たすタンパクをコードする遺伝子として定義される。かかる場合,大腸菌やKO11株において,キシロースを細胞内に輸送する機能を果たしうる限り,特に限定する必要はなく,種々のキシローストランスポーターやキシロースシンポーターをコードする遺伝子を用いることができる。好ましくは,キシロースABCトランスポーターをコードするxylF,xylG,xylHの遺伝子群を用いることができる。また,キシロースシンポーターとして,xylEを用いることができる。
【0025】
本発明においてKO11株とは,エタノール発酵細菌Zymomonas mobilis由来のピルビン酸デカルボキシラーゼ遺伝子pdcとアルコールデヒドロゲナーゼ遺伝子adhBを大腸菌の染色体上のピルビン酸ギ酸リアーゼ遺伝子pfl内に組み込んだ菌株として定義される。かかるKO11株は,標準株ATCC 55124株として既に市販されており,これを用いればよい。
【0026】
続いて,本発明にかかる形質転換大腸菌の作製方法について説明する。
【0027】
本発明にかかる形質転換大腸菌は,プラスミド・ベクターに構成発現型プロモーターとキシロース異性化酵素遺伝子ないしはその他のキシロース関連遺伝子を挿入し,かかる遺伝子挿入プラスミド・ベクターを大腸菌にトランスフェクションすることにより得ることができる。
【0028】
用いるプラスミド・ベクターとしては,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入しうる限り特に限定する必要はなく,種々の観点から選択することができる。例えば,選択マーカーとして抗生物質耐性遺伝子を持つことや,MCS領域を有すること,大腸菌内で多コピーになること,入手のしやすさなどの観点から適宜選択することができる。かかる観点から,pUC18,pUC19,pBluescript IIなどを用いることが好ましい。
【0029】
プラスミド・ベクターに,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入する方法としては,通常用いられる方法を用いればよい。また,遺伝子を挿入したプラスミド・ベクターについても,通常用いられる方法により,大腸菌にトランスフェクションすることができる。以下,例を挙げて説明を行う。
【0030】
まず,構成発現型プロモーターおよびキシロース異性化酵素遺伝子など,挿入を行う遺伝子のインサートDNAを準備する。このインサートDNAは,大腸菌のDNA抽出物から所定のプライマーを用いてPCRにより増殖したものから準備してもよいし,人工的に合成したものから準備してもよい。これら準備された前段階のインサートDNAについては,必要に応じて,増殖・精製を行う。また,必要に応じて制限酵素処理ないし修飾処理を,プラスミド・ベクターとのライゲーションに先立って行うこともできる。
【0031】
続いて,プラスミド・ベクターを準備する。プラスミド・ベクターは市販されているものを用いてもよいし,ボイルプレップ法,アルカリプレップ法,アルカリCsCl法など種々のプラスミド分離法を用いて細胞から抽出・精製したものを用いてもよい。
プラスミド・ベクターは,Hind IIIやSal Iなど選択したプラスミド・ベクターに対応した種々の制限酵素を用いてMCSの切断を行い,必要に応じて脱リン酸化などの処理をさらに行ったうえで,準備されたインサートDNAとのライゲーションを行う。
ライゲーションの後,コンピテント・セルへのトランスフォーメーションを行う。このコンピテント・セルを,プラスミド・ベクターの抗生物質耐性遺伝子に従って,適切な抗生物質を含むLB培地プレートに塗り広げ,単一プラスミドを有するコロニーを形成させる。この形成されたコロニーについて,遺伝子挿入プラスミドのインサートDNAの配列や挿入方向を,通常用いられる分析方法,例えばコロニー・ダイレクトPCR法などで確認することにより,目的に合致した形質転換コロニーを得ることができる。この形質転換コロニーを培養・増殖し,プラスミド分離法を用いることにより,本発明に用いられる構成発現型プロモーターとキシロース異性化酵素遺伝子等を導入したプラスミド・ベクター(遺伝子挿入プラスミド・ベクター)を得ることができる。
【0032】
遺伝子挿入プラスミド・ベクターは,大腸菌に塩化カルシウム法やエレクトロポレーション法などにより大腸菌にトランスフェクションすることにより,本発明にかかる形質転換大腸菌を得ることができる。なお,KO11株についても,大腸菌と同様,トランスフェクションを行うことにより,形質転換KO11株を得ることができる。
【0033】
以上,本発明にかかる形質転換大腸菌を説明してきた。プラスミド・ベクターに導入する遺伝子については,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入することにより,本発明にかかる形質転換大腸菌を得ることができるが,キシルロースリン酸化酵素遺伝子,キシローストランスポーター遺伝子を発現させることにより,本発明にかかる形質転換大腸菌のキシロース代謝改善効果をより効果的にすることができる。さらには,構成発現型プロモーターとしてGAPDH promoterを用いることにより,さらに効果的なキシロース代謝改善効果を期待することができる。
【0034】
本発明により,グルコース存在下においてもカタボライト抑制の影響を受けにくく,キシロース代謝が改善された形質転換大腸菌の提供が可能となった。かかる形質転換大腸菌により,グルコース消費中のカタボライト抑制が緩和され,キシロースの代謝が改善される。ひいては,かかる形質転換大腸菌により,木質系および草本系バイオマスからのエタノール生産向上ないしエタノール生産方法の改善に期待できる。さらには,本発明にかかる形質転換大腸菌をさらに改良することにより,エタノール以外での利用,例えばアミノ酸発酵や乳酸発酵などについても期待できる。
【実施例】
【0035】
以下,本発明にかかる形質転換大腸菌について,KO11株を用いた場合を例に挙げて詳述するが,当然のことながら,本発明の内容は実施例に限定されるものではない。
【0036】
<<実施例1および実施例2,形質転換KO11株の作製>>
実施例1では,キシロース異性化酵素をコードするxylA遺伝子およびキシルロースリン酸化酵素をコードするxylB遺伝子を挿入したプラスミド・ベクターを作製し,KO11株へトランスフェクションを行った。
実施例2では,xylA遺伝子,xylB遺伝子に加え,キシローストランスポーターを構成する一連のタンパクをコードするxylF,xylG,xylHの遺伝子を挿入したプラスミド・ベクターを作製し,KO11株へトランスフェクションを行った。
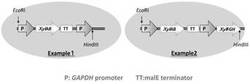
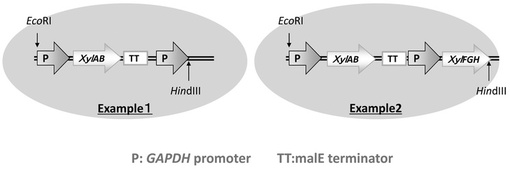
いずれの実施例についても,プラスミド・ベクターのMCS領域に,GAPDH promoterおよびmalE terminatorを合わせて導入した。図1に,実施例1および実施例2のプラスミド・ベクターのMCS領域における遺伝子配置を示す。
【0037】
<I.インサートDNAの準備>
1.大腸菌JM109(タカラバイオ株式会社製)を用いて,ゲノムDNAを抽出した。すなわち,LB液体培地にて培養した大腸菌JM109について,遠心分離を行った後,上清を除去し,細胞沈殿物を得た。得られた沈殿物をDNA抽出キット(株式会社ニッポンジーン製)のソリューションバッファーを用いて溶菌し,溶菌溶液を得た。この溶菌溶液について遠心分離後,上清を除去し,粗DNA沈殿物を得た。得られた粗DNA沈殿物を70%エタノールにてリンスし,RNaseAを加え反応させた後,CIA処理を行い,精製DNA沈殿物を行った。さらに得られた精製DNA沈殿物について,エタノール沈殿,PEG沈殿を行った後,70%エタノールにてリンスを行い,減圧乾燥後,超純水を加え溶解し,JM109ゲノムDNAを含むJM109ゲノムDNA抽出液とした。
【0038】
2.I−1で得られたJM109ゲノムDNA抽出液を用いて,下記プライマーによりPCRを行い,xylABのPCR産物を得た。すなわち,200μL PCRチューブに10×Reaction buffer 5μL,25 mM dNTPs溶液5μL,50mM MgCl2溶液2μL,下記プライマーを含む溶液各2.5μL,超純水30.5μL,JM109ゲノムDNA抽出液2μLを加え,最後にDNA polymerase 0.5μLを加えて全量20μLとした。この溶液をサーマル・サイクラーにセットし,94℃で5分間加温しDNA polymeraseを活性化した後,94℃で30秒間,51℃で30秒間,72℃で3.5分間の反応を25サイクル行った。反応後,さらに72℃で10分間反応させ,増幅末端にAを付加したPCR産物を得た。得られたPCR産物は,xylAB/Sac-Kpnとして以降用いた。
Forward primer
Sac-xylAB (se):5'-CTTGAGCTCATTACGACATCATCCATCAC-3'
Reverse primer
Kpn-xylAB(an):5'-CGGGTACCTTACGCCATTAATGGCAG-3'
【0039】
3.I−2と同様に,xylFGHのPCR産物を得た。すなわち,下記プライマーを含む反応溶液をサーマル・サイクラーにセットし,94℃で5分間加温しDNA polymeraseを活性化した後,94℃で30秒間,55℃で30秒間,72℃で3.5分間の反応を25サイクル行い,PCR産物を得た。得られたPCR産物は,xylFGH/Hind IIIとして以降用いた。
Forward primer
Pxo14:5'-CCCAAGCTTACTACAGAAGGCC-3'
Reverse primer
Pxo10:5'-CCCAAGCTTTCAAGAACGGCGTTTGG-3'
【0040】
4.I−2で得られたPCR産物,xylAB/Sac-Kpnを用い,TAクローニングにより,形質転換体を得た。すなわち,xylAB/Sac-Kpnとプラスミド・ベクターpCR2.1(Invitrogen),Ligation mix(ニッポンジーン)を混合,反応させ,PCR産物のプラスミド・ベクターpCR2.1へのライゲーションを行った。これを,E.coli JM109 competent cell(タカラバイオ株式会社製)に緩やかに加え撹拌することにより,トランスフォーメーションを行った。反応後のコンピテント・セルを培養した後,X-galならびにIPTGを塗布しておいたアンピシリン含有LBプレート培地にコンラージ棒を用いて塗布,培養を行った。
【0041】
5.I−4でプレート上に生じた白色コロニーの増幅断片有無をコロニー・ダイレクトPCR法により確認することにより,形質転換体の確認を行った。すなわち,1.5mLチューブに超純水103.2μL,10×Reaction buffer 16μL,25 mM dNTPs溶液16μL,50 mM MgCl2溶液8μL,M13 universal primer,M13 reversal primerをそれぞれ8μL,最後にDNA polymerase 0.8μLを加えプレミックスを作り,200μL PCRチューブに20μLずつ分注し,プレート上に生じた形質転換体のコロニーを楊枝で少量とり,チューブ内で懸濁させた後,サーマル・サイクラーにセットした。94℃で5分間加温し,DNA polymeraseを活性化した後,94℃で30秒間,50℃で30秒間,72℃で1分間の反応を25サイクル行った。この方法によりは,xylAB/Sac-Kpnは,約3000bp付近に遺伝子の増幅断片が確認されたものを形質転換体とした。
M13 universal primer:5'-GTTTTCCCAGTCACGACGTT
M13 reversal primer:5'-GGAAACAGCTATGACCATGA
【0042】
6.I−3で得られたPCR産物,xylFGH/Hind IIIについて,制限酵素反応とゲル抽出を行うことにより,インサートDNA・xylFGH/Hind IIIならびにベクターDNA・pUC18を得た。すなわち,2つの1.5mLチューブにそれぞれ10×H Buffer 5μL,プラスミド・ベクターpUC18とPCR産物xylFGH/Hind IIIを43μLずつ入れ,最後にSalI 2μLを,混合させ,37℃で16時間処理した。反応後さらに,それぞれフェノクロ処理,エタノール沈殿,リンス,減圧乾燥後超純水44μLに溶解後それぞれのチューブに10×B Bufferを5μL,Hind III 1μL 加え37℃で16時間処理した。そして,フェノクロ処理,エタノール沈殿,リンス減圧乾燥後xylFGH/Hind IIIを制限酵素消化したものはTE buffer 30μLに溶解し,プラスミド・ベクターpUC18を制限酵素消化したものは超純水30μLに溶解した。このうちxylFGH/Hind IIIを制限酵素消化したものを,2%アガロース・ゲルを用いて電気泳動した後,約3900bp付近のDNA断片をゲルよりカッターで切り出した。その後,QIAquick Gel Extraction Kitを用いてゲルよりDNAを抽出し,インサートDNA・xylFGH/Hind IIIとした。また,約2.7bpのバンドについても同様の操作を行い,ベクターDNA・pUC18とした。
7.なお,上記により得られたインサートDNAについては,I−4のTAクローニングおよびI−5のコロニー・ダイレクトPCR法と同様の作業により,約3900bp付近に遺伝子増幅断片が確認できるものを形質転換体とした。
【0043】
8.I−5およびI−7により得られた2つの形質転換体について,遺伝子の挿入方向と挿入遺伝子配列の確認を行った。すなわち,それぞれの形質転換体の遺伝子挿入プラスミド・ベクターDNAをミニプレップ法により抽出し,制限酵素処理およびアガロース・ゲル電気泳動を行い,インサートDNAの方向確認を行った。さらに,それぞれの形質転換体についてコロニー・ダイレクトPCR法により,シークエンス解析を行い,増幅されている遺伝子がそれぞれxylA,xylB,xylF,xylG,xylHであることを確認した。
【0044】
9.遺伝子の挿入方向および挿入遺伝子配列の確認を行ったI−5およびI−7により得られた2つの形質転換体について,ミニプレップ法により遺伝子挿入プラスミドの抽出を行った。さらに,I−5で得られた形質転換体の抽出プラスミドについて,Sac IとKpn Iによる制限酵素処理を行い約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,インサートDNA xylAB/Sac-Kpnを得た。同様に,I−7で得られた形質転換体の抽出プラスミドについて,Hind IIIによる制限酵素処理を行い,約3800bp付近のDNA断片をゲルから切り出し,抽出を行うことにより,インサートDNA xylFGH/Hind IIIを得た。
【0045】
<II.プラスミド・ベクターの準備>
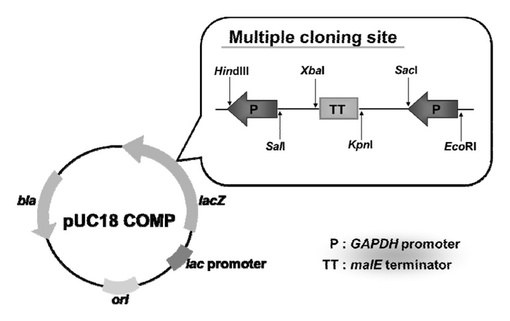
1.プラスミド・ベクターとして,上記の記載と同様の方法により,GAPDH遺伝子プロモーターおよびmalE遺伝子ターミネーターを導入したプラスミド・ベクターpUC18compの作製を行った。なお,それぞれのPCR産物を得るために下記のプライマーを用いた。図2に,プラスミド・ベクターpUC18compの遺伝子配置を示す。
(1) GAPDHプロモーター
Forward primer
Sal-Pgap (se):5'-ATTAGTCGACATTACGTGACTGATTCTAAC-3'
Reverse primer
Hind-Pgap (an):5'-CCCAAGCTTATATTCCACCAGCTATTTG-3',又は
Sac-Pgap (an):5'-CTTGAGCTCATATTCCACCAGCTATTTG-3'
(2) malEターミネーター
Forward primer
Kpn-malTT (se):5'-GAGGTACCAGATGTTGTTCTGCCAATG-3'
Reverse primer
Kpn-malTT (an):5'-GCTCTAGAGAGCACGAAAGAGAATTATC-3'
【0046】
<III.遺伝子挿入プラスミド・ベクターの作製>
1.プラスミド・ベクターpUC18compを制限酵素消化,ゲル抽出を行い,ベクターpUC18comp/Sac-Kpn,ベクターpUC18comp/Hind IIIを作製した。すなわち,プラスミド・ベクターpUC18compをSac I,Kpn Iにて処理を行い,2%アガロース・ゲルを用いて電気泳動した後,約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,ベクターpUC18comp/Sac-Kpnとした。同様に,Hind IIIで処理を行い脱リン酸化した後に,2%アガロース・ゲルを用いて電気泳動を行い約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,ベクターpUC18comp/Hind IIIとした。
【0047】
2.III−1で得られたベクターpUC18comp/Sac-KpnとI−9で得られたインサートDNAxylAB/Sac-Kpnについて,I−4と同様の方法により,ライゲーションを行い,コンピテント・セルにトランスフェクションをした後,コロニーを得た。得られたコロニーのうち,白色コロニーについて,I−5と同様に,遺伝子増幅の有無をコロニー・ダイレクトPCR法により確認することにより,形質転換体を得た。得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0048】
3.III−2と同様に,III−1で得られたベクターpUC18comp/Hind IIIとI−9で得られたインサートDNA xylFGH/Hind IIIのライゲーションを行い,コロニー・ダイレクトPCR法により遺伝子増幅の有無を確認することにより,形質転換体を得た。得られた形質転換体については,得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0049】
4.III−2で得られた形質転換体について,ミニプレップ法にて遺伝子挿入プラスミド・ベクターを抽出し,制限酵素処理を行い脱リン酸化した後,ゲル抽出により約3800bp付近のDNA断片を切り出し,ベクターDNA xylAB/Hind IIIとした。加えて,III−3で得られた形質転換体についても,ミニプレップ法にて遺伝子挿入プラスミド・ベクターを抽出し,制限酵素処理を行った後,ゲル抽出により約3800bp付近のDNA断片を切り出し,インサートDNA xylFGH/Hind IIIとした。これらにより得られた,ベクターDNA xylAB/Hind IIIとインサートDNA xylFGH/Hind IIIを,I−4と同様の方法により,ライゲーションを行い,コンピテント・セルにトランスフェクションをした後,コロニーを得た。得られたコロニーのうち,白色コロニーについて,I−5と同様に,遺伝子増幅の有無をコロニー・ダイレクトPCR法により確認することにより,約4000bp付近に遺伝子の増幅断片が確認されたものを形質転換体として得た。得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0050】
<IV.KO11株へのトランスフェクション>
1.KO11株について,コンピテント・セルとしての調製を行った。すなわち,試験管にLB液体培地5mLを作製しオートクレーブし,600μg/mLとなるようにクロラムフェニコールを加え,E. coli KO11株を植菌し,濁度がOD660≒0.6に達するまで37℃,60 rpmで回転振とう培養した。この培養液1.0mLをチューブに移し,6,000rpmで5 分間遠心分離した後,上清を捨てた。残ったペレットを滅菌水1mLに再懸濁した。この遠心分離,再懸濁の操作を計3回行い,菌体を洗浄し,さらに10% グリセロール溶液1mLに再懸濁した。この遠心分離,再懸濁の操作を計2回行い,菌体を洗浄し,最終的に10% グリセロール溶液に100μLに再懸濁したものをKO11株のコンピテント・セルとした。
【0051】
2.IV−1で調製したKO11株のコンピテント・セルに,所定の遺伝子挿入プラスミド・ベクターをエレクトロポレーション法によりトランスフェクションした。すなわち,III−2で得られた形質転換体からプラスミド・ベクターDNA・xylABを,IV−4で得られた形質転換体からプラスミド・ベクターxylAB・xylFGHを抽出し,それぞれの遺伝子挿入プラスミド・ベクター溶液をKO11株コンピテント・セルに加えて混合し,エレクトロポレーション・キュベット (0.2 cm) に移した。ジーン・パルサーIIおよびパルス・コントローラー PLUSを用いて,電圧 1.5 kV,抵抗 200,静電容量25μFでエレクトロポレーションを行った後,直ちにLB液体培地900μLを加え,1.5mL容チューブに移し,37℃で1時間回転振とう培養 (60 rpm) した。培養液100μLを, LB/Ampプレート培地にコンラージ棒を用いて塗布し,37℃で16時間培養した。プレート上に生じたコロニーを形質転換体とし,プレートは4℃で保存した。その後,コロニーを5mL LB/Amp液体培地に植菌し,37℃で16時間培養した。プラスミド・ベクターDNA・xylAB導入株を実施例1,プラスミド・ベクターxylAB・xylFGH導入株を実施例2として20%(w/v)グリセロール中で−84℃にて保存し,以降の評価に用いた。
【0052】
<<実施例1および実施例2,KO11株における糖消費およびエタノール生産能評価>>
実施例1および実施例2で作製した形質転換KO11株の糖消費およびエタノール生産能を評価するため,遺伝子導入を行っていないKO11株を比較例として実験を行った。
【0053】
<方法>
1.評価する際の培地としてM9混合糖培地を用いた。すなわち,74.8 gのグルコースと3.2 gのキシロースを含む水溶液32mLを加えた100mL三角フラスコをオートクレーブ滅菌した。オートクレーブ滅菌後順次下記材料を加え培地調製を行い,グルコースおよびキシロースの最終濃度をそれぞれ6% (w/v) と4% (w/v) とした培地をM9混合糖培地として用いた。
・グルコース
・キシロース
・滅菌水
・20×M9 salts…NH4Cl 20 g/L,KH2PO4 60 g/L,NaHPO4 120 g/L,NaCl 10g/Lとなるように蒸留水に溶解し,オートクレーブ滅菌(120℃,15分間)したものを用いた。
・20% (v/v) CSL…CSL原液と滅菌水を用いて終濃度が20% (v/v)となるように調製した。
・MgSO4・7H2O
・1 Mリン酸緩衝液…1 MのNa2HPO4・12H2O溶液に,30℃でpH 6.7となるまで1 MのKH2PO4溶液を添加し,オートクレーブ滅菌したものを用いた。
【0054】
2.実施例1および実施例2の形質転換KO11株について,クリーンベンチ内において,クロラムフェニコール入りLB液体培地ないしアンピシリン入りLB液体培地5mLに植菌を行い,37℃,60 rpmの条件下で16時間回転振とう培養した。この培養液100μLを,クリーンベンチ内において,LB液体培地150mLと各種抗生物質を含む500mL容三角フラスコに加え,37℃,150 rpmの条件下で24時間振とう培養した。この培養液を前培養液とした。前培養液を6,000 ×gで10分間遠心し菌体を回収後,本培養液としてM9混合糖培地に乾燥菌体重量が1 g/Lとなるように植菌し,濃硫酸を入れた発酵栓にて栓をした。発酵栓をした100mL容三角フラスコは,インキュベーターを用いて37℃,150 rpmの条件下で4日間発酵試験を行った。
【0055】
3.HPLCを用いて,各時間点におけるグルコースやキシロースの残存率等の分析を行った。すなわち,発酵試験を行った前記100mL容三角フラスコ中の懸濁液から1mLを各時間点において回収し,それらを15000 rpmの条件下で5分間遠心分離を行うことにより固形成分を沈殿させ上清を得た。得られた上清は,300μLずつ2本に分けエタノール600μLを加え混合した後,フィルターろ過したものを試料溶液とし,1本をエタノール濃度測定用,もう1本を培地糖濃度測定用としてHPLC分析を行った。なお,HPLC分析の分析条件は,下記であり,検出器としては示差屈折率検出器を用いた。
また,前記上清の分析と同様に,糖濃度の標準溶液の分析を合わせて行った。すなわち,0,2.5,5,7.5,10% (w/v) グルコース溶液,または0,2,4,6,8% (w/v) キシロース溶液300μLを用い同様の分析を行い,ピーク面積の比から培養液上清中の糖濃度を算出した。
・HPLC分析条件
カラム Asahipak NH2P-50 4E;直径4.0 mm ,長さ250 mm (Shodex,東京)
溶媒 アセトニトリル : 水=8 : 2 (v/v)
カラム温度 40℃
流速 0.75mL/min
【0056】
4.GC分析により,各時間点におけるエタノール濃度の分析を行った。すなわち,前述のように得られた上清300μLと等容量の5% (v/v) イソプロパノール (内部標準物質) を混合し,下記条件によるGC分析を行った。また,エタノール濃度の標準溶液として,5% (v/v) イソプロパノールと0,2,4,6,8,10% (v/v) エタノールをそれぞれ等容量混合したもの作製したものについても合わせて分析を行い,検出したピーク面積の比から培養液上清中のエタノール濃度を算出した。
・ 分析条件
カラム温度 150℃,検出器温度 250℃,気化室温度 250℃
カラム 液相 5% Thermon 1000
担体Sunpak-A直径3.2 mm 長さ2.1 m
流速 40mL/min
キャリアガス N2
【0057】
<結果>
1.HPLCによる糖濃度分析
(1) 各培養時間におけるM9混合糖培地中のキシロース残存率およびキシロース消費率の比較を表1に,グルコース残存率およびグルコース消費率の比較を表2に示す。なお,各糖における残存率および糖消費率は,下記式に従い算出を行った。
・糖残存率(%)=各時間点における培地中の糖濃度/0時間における培地中の糖濃度
×100
・糖消費率(%)=100−残糖率
(2) 実施例1において,キシロース残存率は発酵初期の12時間後や24時間後では95%以上とキシロース消費はあまり進んでいなかった。しかしながら,発酵36時間後では比較例とほぼ同じ水準までキシロース消費が進行し,発酵48時間後では66.2%と,比較例よりも約20%も多くキシロースを消費していた。一方,実施例2では,発酵初期は比較例と同様のキシロース消費が進行し,発酵36時間後では約11%,発酵48時間後では約19%,比較例よりも多くキシロースを消費していた。これらより,実施例1および実施例2は,実用化において現実的な培養時間と考えられる48時間で,比較例と比べて優れたキシロース代謝を示すことが分かった。
(3) また,発酵48時間後におけるグルコース残存率は,実施例1が18.7%,実施例2が16.9%であった。これらは,比較例のグルコース残存率と比べると,実施例1は約12%,実施例2では約10%の差であり,グルコース消費の差は比較的小さいことが分かった。
【0058】
【表1】

【0059】
【表2】
【0060】
2.GCによるエタノール濃度分析
(1) 各培養時間におけるM9混合糖培地中のエタノール濃度の比較を表3に示す。
(2) 発酵48時間後におけるエタノール濃度は,実施例1が4.15%,実施例2が4.18%であった。一方,比較例は4.07%であり,わずかではあるものの実施例においてエタノール生産の増加が見られた。
【0061】
【表3】
【技術分野】
【0001】
本発明は,キシロース代謝を改善した形質転換大腸菌に関する。
【0002】
キシロースは,分子式がC5H10O5で表される分子量150.13の糖であり,単糖,五炭糖及びアルドースに分類される。自然界において,キシロースは単体ではほとんどみられないが,ヘミセルロースの一つであるキシランを構成するものとして植物に広く存在する。
【0003】
キシロースの代謝は,バイオマスからのエタノール生産を行う際に,エタノール生産効率を左右する重要なポイントとなってくる。
例えば,農産廃棄物や廃材由来の木質系バイオマスは,食糧とは競合しないことから利用が期待されるバイオマスである。この木質系バイオマスは,セルロースが約50%,ヘミセルロースが約30%,その他リグニンなどで構成されている。この木質系バイオマスをバイオマス・エタノール原料として利用する際,醸造酵母Saccharomyces cerevisiae(S. cerevisiae)が従来用いられてきた。このS. cerevisiaeは,セルロース由来のグルコースをエタノールへ変換することはできるものの,ヘミセルロース由来のキシロースをエタノールへ変換することが出来ない。そこで,木質系バイオマスから,より効率良くバイオマス・エタノールを生産するためには,ヘミセルロース由来のキシロースをもエタノール原料として利用することが求められるため,キシロースからのエタノール発酵が可能な菌株を用いる必要がある。
【0004】
このキシロースからのエタノール発酵が可能な菌株として,酵母の1種であるPichia stipitisやPachysolen tannophilusが知られている(非特許文献1)。加えて,形質転換に広く汎用されている大腸菌においてもこのような試みがなされており,例えば,太田らにより開発されたKO11株が知られている(特許文献1,特許文献2,非特許文献2)。
【0005】
また,菌株のキシロース代謝能を向上させる技術として,下記のような技術が開示されている。
特許文献3には,ペントース糖の利用および発酵に必要な酵素をコードする外来構造遺伝子を含む,安定なZymomonas組込み体の製造方法が開示されている。
特許文献4には,xylABFGHR遺伝子座などのキシロース資化酵素をコードする遺伝子の発現量が増加されたEscherichia属細菌を用いて,L-ヒスチジン,L-スレオニン,L-リジン,L-グルタミン酸およびL-トリプトファンなどのL-アミノ酸を生産する方法が開示されている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Delgenes J. P., MolettaR. and Navarro J.M. (1986) the effect of aeration on D-xylosefermentation by Pachysolen tannophilus,Pichia stipitis, Kluyveromyces marxianus and Cadida shenatae. Biotechnol. Lett. 1986 Nov; 8(12): 897-900
【非特許文献2】Ohta et al. (1991) Appl.Environ.Microbiol.57:893-900
【特許文献】
【0007】
【特許文献1】米国特許第5000000号明細書
【特許文献2】米国特許第5424202号明細書
【特許文献3】国際公開第2004/037973号
【特許文献4】特開2005−261433号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
非特許文献1には,エタノール発酵が可能な菌株として,酵母の1種であるPichia stipitisやPachysolen tannophilusに関する技術が開示されている。しかしながら,これらの菌株は約1% (v/v) 程度という僅かなエタノールしか生産できないことからエタノールの生産効率という点において,必ずしも十分な技術とはいえない。
また,特許文献1等に開示されているKO11株は,キシロースなどの五炭糖からも優れたエタノール生産能を有している。しかしながら,KO11株はグルコース存在下ではカタボライト抑制を受けるため,キシロースよりもグルコースを優先的に消費し,キシロースをあまり消費しないという技術的課題が存在する。
【0009】
特許文献3には,ペントース糖の利用および発酵に必要な酵素をコードする外来構造遺伝子を含む,安定なZymomonas組込み体の製造方法が開示されている。しかしながら,KO11株やキシローストランスポーターに関する遺伝子の導入についても何ら開示はない。さらには,カタボライト抑制についての開示もない。
【0010】
特許文献4には,xylABFGHR遺伝子座などのキシロース資化酵素をコードする遺伝子の発現量が増加されたEscherichia属細菌を用いて,L-ヒスチジン,L-スレオニン,L-リジン,L-グルタミン酸およびL-トリプトファンなどのL-アミノ酸を生産する方法が開示されている。
しかしながら,特許文献4に記載の技術に関しては,キシロース代謝を促進するため,xylオペロンの調節遺伝子であるxylRも含めてキシロース代謝に関連する一連のタンパクをコードする遺伝子をほとんどすべて導入し,その発現量を増加させようとする技術に過ぎないものである。よって,特許文献4に記載の技術は,結局は,グルコースによるキシロース代謝の抑制を受けてしまう技術にとどまり,培養液中のグルコースを大腸菌が消費した後にカタボライト抑制が解除されxylABFGH遺伝子が発現されるため,グルコースと同時にキシロースが消費されないという技術的課題を含むものである。
【0011】
かかるカタボライト抑制の問題は,KO11株のみにとどまらず形質転換に広く汎用されている大腸菌にも存在する問題である。すなわち,大腸菌にキシロース代謝関連遺伝子を単に導入したとしても,グルコース代謝によりキシロースオペロン調節遺伝子が作動し,キシロース代謝が抑制されるものと考えられる。
【0012】
上記事情を背景として,本発明では,グルコース存在下のカタボライト抑制の影響を受けにくく,キシロース代謝が改善された大腸菌株の開発を課題とする。
【課題を解決するための手段】
【0013】
発明者らは,大腸菌におけるキシロース代謝経路の分析を行った。大腸菌では,キシロースの輸送や代謝に関する種々のタンパクが存在するが,発明者は,それらの中でもキシロースの異性化を行う酵素に着目した。すなわち,大腸菌のキシロース異性化酵素は,キシロースとの基質親和性が極端に低く,触媒効率も低い。このため,大腸菌において,キシロース異性化の段階がキシロース代謝の律速段階になっていることに発明者は着目した。
【0014】
この知見を基に発明者は,構成発現型のプロモーターおよびキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターを大腸菌株に導入することにより,培養初期からキシロース異性化酵素を発現させ,カタボライト抑制の影響を受けにくい形質転換大腸菌の開発に成功した。加えて,発明者は更なる改良を行い,キシロース異性化酵素に加え,一連のキシロースに関連する酵素やタンパクを発現させることにより,キシロース代謝をさらに改善した形質転換大腸菌の開発に成功した。
【0015】
本発明は,以下から構成される。
【0016】
本発明の第1の構成は,構成発現型プロモーターとキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターが導入されたことを特徴とする形質転換大腸菌である。
本発明の第2の構成は,前記構成発現型プロモーターが,GAPDHプロモーターであることを特徴とする請求項1に記載の形質転換大腸菌株である。
本発明の第3の構成は,前記キシロース異性化酵素遺伝子が,xylAであることを特徴とする第1又は2の構成に記載の形質転換大腸菌である。
本発明の第4の構成は,前記プラスミド・ベクターに,さらにキシルロースリン酸化酵素遺伝子を挿入することを特徴とする第1ないし3の構成に記載の形質転換大腸菌株である。
本発明の第5の構成は,前記キシルロースリン酸化酵素遺伝子が,xylBであることを特徴とする第4の構成に記載の形質転換大腸菌である。
本発明の第6の構成は,前記プラスミド・ベクターに,さらにキシロース輸送タンパク遺伝子を挿入することを特徴とする第1ないし5の構成に記載の形質転換大腸菌である。
本発明の第7の構成は,前記キシロース輸送タンパク遺伝子が,xylF,xylG,xylHであることを特徴とする第6の構成に記載の形質転換大腸菌である。
本発明の第8の構成は,前記大腸菌が,KO11株であることを特徴とする請求項1ないし7に記載の形質転換大腸菌である。
【発明の効果】
【0017】
本発明により,グルコース存在下においてもカタボライト抑制の影響を受けにくく,キシロース代謝が改善された形質転換大腸菌の提供が可能となった。かかる形質転換大腸菌により,グルコース消費中のカタボライト抑制が緩和され,キシロースの代謝が改善される。ひいては,かかる形質転換大腸菌により,木質系および草本系バイオマスからのエタノール生産向上ないしエタノール生産方法の改善に期待できる。さらには,本発明にかかる形質転換大腸菌をさらに改良することにより,エタノール以外での利用,例えばアミノ酸発酵や乳酸発酵などについても期待できる。
【図面の簡単な説明】
【0018】
【図1】実施例1,実施例2のプラスミド・ベクターのMCS領域の遺伝子配置を示した図
【図2】プラスミド・ベクターpUC18の遺伝子配置を示した図
【発明を実施するための形態】
【0019】
以下,本発明にかかる形質転換大腸菌について,説明を行う。まず,本発明を構成する主な要素について説明を行う。
【0020】
本発明において,大腸菌とは,Escherichia属に属するグラム陰性桿菌として定義される。本発明では,この大腸菌に,構成発現型プロモーターおよびキシロース代謝関連酵素遺伝子を挿入したプラスミド・ベクターを導入することにより,本発明にかかる形質転換大腸菌を得ることができる。本発明で用いられる大腸菌は,特に限定する必要はないが,安全性等の観点から,O157等病原性を有する大腸菌を用いることは避けたほうがよい。好ましい大腸菌株としては,JM109,JM110,BL21,DH5αなどが挙げられる。
【0021】
本発明において構成発現型プロモーターとは,常時目的遺伝子を発現させるプロモーターとして定義される。かかる構成発現型プロモーターは,種々の観点から選択することができる。例えば,入手のしやすさ,Multiple cloning site(MCS領域)への挿入のしやすさなどである。好ましい構成発現型プロモーターとしては,グリセルアルデヒド3-リン酸脱水素酵素遺伝子(GAPDHプロモーター),ホスホグリセリン酸キナーゼプロモーター(PGKプロモーター)などを用いることができる。
【0022】
本発明においてキシロース異性化酵素遺伝子とは,キシロースをキシルロースに変換する酵素をコードする遺伝子として定義される。かかる場合,大腸菌やKO11株内においてキシロースを異性化する酵素を発現しうる限り,特に限定する必要はないが,大腸菌やKO11株において発現し,かつ,キシロースとの基質親和性が高い酵素を発現する遺伝子を選択することが好ましい。好ましくは,大腸菌由来や放線菌由来,乳酸菌由来のキシロース異性化酵素遺伝子を用いることができる。大腸菌由来としては,xylAを用いることが好ましい。また,放線菌由来としては,xylA,SCO1169,SGR_1070などを,乳酸菌由来としては,LAF_0402,LVIS_0183などを用いることができる。
【0023】
また,キシルロースリン酸化酵素遺伝子とは,キシルロースの5位をリン酸化し,キシルロース5リン酸に変換する酵素をコードする遺伝子として定義される。かかる場合,大腸菌やKO11株内においてキシルロースの5位をリン酸化する酵素を発現しうる限り,特に限定する必要はないが,大腸菌やKO11株において発現し,かつ,キシルロースのリン酸化能が高い酵素を発現する遺伝子を選択することが好ましい。好ましくは,xylBを用いることができる。
【0024】
さらに,キシロース輸送タンパク遺伝子とは,キシロースを細胞内に輸送する機能を果たすタンパクをコードする遺伝子として定義される。かかる場合,大腸菌やKO11株において,キシロースを細胞内に輸送する機能を果たしうる限り,特に限定する必要はなく,種々のキシローストランスポーターやキシロースシンポーターをコードする遺伝子を用いることができる。好ましくは,キシロースABCトランスポーターをコードするxylF,xylG,xylHの遺伝子群を用いることができる。また,キシロースシンポーターとして,xylEを用いることができる。
【0025】
本発明においてKO11株とは,エタノール発酵細菌Zymomonas mobilis由来のピルビン酸デカルボキシラーゼ遺伝子pdcとアルコールデヒドロゲナーゼ遺伝子adhBを大腸菌の染色体上のピルビン酸ギ酸リアーゼ遺伝子pfl内に組み込んだ菌株として定義される。かかるKO11株は,標準株ATCC 55124株として既に市販されており,これを用いればよい。
【0026】
続いて,本発明にかかる形質転換大腸菌の作製方法について説明する。
【0027】
本発明にかかる形質転換大腸菌は,プラスミド・ベクターに構成発現型プロモーターとキシロース異性化酵素遺伝子ないしはその他のキシロース関連遺伝子を挿入し,かかる遺伝子挿入プラスミド・ベクターを大腸菌にトランスフェクションすることにより得ることができる。
【0028】
用いるプラスミド・ベクターとしては,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入しうる限り特に限定する必要はなく,種々の観点から選択することができる。例えば,選択マーカーとして抗生物質耐性遺伝子を持つことや,MCS領域を有すること,大腸菌内で多コピーになること,入手のしやすさなどの観点から適宜選択することができる。かかる観点から,pUC18,pUC19,pBluescript IIなどを用いることが好ましい。
【0029】
プラスミド・ベクターに,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入する方法としては,通常用いられる方法を用いればよい。また,遺伝子を挿入したプラスミド・ベクターについても,通常用いられる方法により,大腸菌にトランスフェクションすることができる。以下,例を挙げて説明を行う。
【0030】
まず,構成発現型プロモーターおよびキシロース異性化酵素遺伝子など,挿入を行う遺伝子のインサートDNAを準備する。このインサートDNAは,大腸菌のDNA抽出物から所定のプライマーを用いてPCRにより増殖したものから準備してもよいし,人工的に合成したものから準備してもよい。これら準備された前段階のインサートDNAについては,必要に応じて,増殖・精製を行う。また,必要に応じて制限酵素処理ないし修飾処理を,プラスミド・ベクターとのライゲーションに先立って行うこともできる。
【0031】
続いて,プラスミド・ベクターを準備する。プラスミド・ベクターは市販されているものを用いてもよいし,ボイルプレップ法,アルカリプレップ法,アルカリCsCl法など種々のプラスミド分離法を用いて細胞から抽出・精製したものを用いてもよい。
プラスミド・ベクターは,Hind IIIやSal Iなど選択したプラスミド・ベクターに対応した種々の制限酵素を用いてMCSの切断を行い,必要に応じて脱リン酸化などの処理をさらに行ったうえで,準備されたインサートDNAとのライゲーションを行う。
ライゲーションの後,コンピテント・セルへのトランスフォーメーションを行う。このコンピテント・セルを,プラスミド・ベクターの抗生物質耐性遺伝子に従って,適切な抗生物質を含むLB培地プレートに塗り広げ,単一プラスミドを有するコロニーを形成させる。この形成されたコロニーについて,遺伝子挿入プラスミドのインサートDNAの配列や挿入方向を,通常用いられる分析方法,例えばコロニー・ダイレクトPCR法などで確認することにより,目的に合致した形質転換コロニーを得ることができる。この形質転換コロニーを培養・増殖し,プラスミド分離法を用いることにより,本発明に用いられる構成発現型プロモーターとキシロース異性化酵素遺伝子等を導入したプラスミド・ベクター(遺伝子挿入プラスミド・ベクター)を得ることができる。
【0032】
遺伝子挿入プラスミド・ベクターは,大腸菌に塩化カルシウム法やエレクトロポレーション法などにより大腸菌にトランスフェクションすることにより,本発明にかかる形質転換大腸菌を得ることができる。なお,KO11株についても,大腸菌と同様,トランスフェクションを行うことにより,形質転換KO11株を得ることができる。
【0033】
以上,本発明にかかる形質転換大腸菌を説明してきた。プラスミド・ベクターに導入する遺伝子については,構成発現型プロモーターおよびキシロース異性化酵素遺伝子を挿入することにより,本発明にかかる形質転換大腸菌を得ることができるが,キシルロースリン酸化酵素遺伝子,キシローストランスポーター遺伝子を発現させることにより,本発明にかかる形質転換大腸菌のキシロース代謝改善効果をより効果的にすることができる。さらには,構成発現型プロモーターとしてGAPDH promoterを用いることにより,さらに効果的なキシロース代謝改善効果を期待することができる。
【0034】
本発明により,グルコース存在下においてもカタボライト抑制の影響を受けにくく,キシロース代謝が改善された形質転換大腸菌の提供が可能となった。かかる形質転換大腸菌により,グルコース消費中のカタボライト抑制が緩和され,キシロースの代謝が改善される。ひいては,かかる形質転換大腸菌により,木質系および草本系バイオマスからのエタノール生産向上ないしエタノール生産方法の改善に期待できる。さらには,本発明にかかる形質転換大腸菌をさらに改良することにより,エタノール以外での利用,例えばアミノ酸発酵や乳酸発酵などについても期待できる。
【実施例】
【0035】
以下,本発明にかかる形質転換大腸菌について,KO11株を用いた場合を例に挙げて詳述するが,当然のことながら,本発明の内容は実施例に限定されるものではない。
【0036】
<<実施例1および実施例2,形質転換KO11株の作製>>
実施例1では,キシロース異性化酵素をコードするxylA遺伝子およびキシルロースリン酸化酵素をコードするxylB遺伝子を挿入したプラスミド・ベクターを作製し,KO11株へトランスフェクションを行った。
実施例2では,xylA遺伝子,xylB遺伝子に加え,キシローストランスポーターを構成する一連のタンパクをコードするxylF,xylG,xylHの遺伝子を挿入したプラスミド・ベクターを作製し,KO11株へトランスフェクションを行った。
いずれの実施例についても,プラスミド・ベクターのMCS領域に,GAPDH promoterおよびmalE terminatorを合わせて導入した。図1に,実施例1および実施例2のプラスミド・ベクターのMCS領域における遺伝子配置を示す。
【0037】
<I.インサートDNAの準備>
1.大腸菌JM109(タカラバイオ株式会社製)を用いて,ゲノムDNAを抽出した。すなわち,LB液体培地にて培養した大腸菌JM109について,遠心分離を行った後,上清を除去し,細胞沈殿物を得た。得られた沈殿物をDNA抽出キット(株式会社ニッポンジーン製)のソリューションバッファーを用いて溶菌し,溶菌溶液を得た。この溶菌溶液について遠心分離後,上清を除去し,粗DNA沈殿物を得た。得られた粗DNA沈殿物を70%エタノールにてリンスし,RNaseAを加え反応させた後,CIA処理を行い,精製DNA沈殿物を行った。さらに得られた精製DNA沈殿物について,エタノール沈殿,PEG沈殿を行った後,70%エタノールにてリンスを行い,減圧乾燥後,超純水を加え溶解し,JM109ゲノムDNAを含むJM109ゲノムDNA抽出液とした。
【0038】
2.I−1で得られたJM109ゲノムDNA抽出液を用いて,下記プライマーによりPCRを行い,xylABのPCR産物を得た。すなわち,200μL PCRチューブに10×Reaction buffer 5μL,25 mM dNTPs溶液5μL,50mM MgCl2溶液2μL,下記プライマーを含む溶液各2.5μL,超純水30.5μL,JM109ゲノムDNA抽出液2μLを加え,最後にDNA polymerase 0.5μLを加えて全量20μLとした。この溶液をサーマル・サイクラーにセットし,94℃で5分間加温しDNA polymeraseを活性化した後,94℃で30秒間,51℃で30秒間,72℃で3.5分間の反応を25サイクル行った。反応後,さらに72℃で10分間反応させ,増幅末端にAを付加したPCR産物を得た。得られたPCR産物は,xylAB/Sac-Kpnとして以降用いた。
Forward primer
Sac-xylAB (se):5'-CTTGAGCTCATTACGACATCATCCATCAC-3'
Reverse primer
Kpn-xylAB(an):5'-CGGGTACCTTACGCCATTAATGGCAG-3'
【0039】
3.I−2と同様に,xylFGHのPCR産物を得た。すなわち,下記プライマーを含む反応溶液をサーマル・サイクラーにセットし,94℃で5分間加温しDNA polymeraseを活性化した後,94℃で30秒間,55℃で30秒間,72℃で3.5分間の反応を25サイクル行い,PCR産物を得た。得られたPCR産物は,xylFGH/Hind IIIとして以降用いた。
Forward primer
Pxo14:5'-CCCAAGCTTACTACAGAAGGCC-3'
Reverse primer
Pxo10:5'-CCCAAGCTTTCAAGAACGGCGTTTGG-3'
【0040】
4.I−2で得られたPCR産物,xylAB/Sac-Kpnを用い,TAクローニングにより,形質転換体を得た。すなわち,xylAB/Sac-Kpnとプラスミド・ベクターpCR2.1(Invitrogen),Ligation mix(ニッポンジーン)を混合,反応させ,PCR産物のプラスミド・ベクターpCR2.1へのライゲーションを行った。これを,E.coli JM109 competent cell(タカラバイオ株式会社製)に緩やかに加え撹拌することにより,トランスフォーメーションを行った。反応後のコンピテント・セルを培養した後,X-galならびにIPTGを塗布しておいたアンピシリン含有LBプレート培地にコンラージ棒を用いて塗布,培養を行った。
【0041】
5.I−4でプレート上に生じた白色コロニーの増幅断片有無をコロニー・ダイレクトPCR法により確認することにより,形質転換体の確認を行った。すなわち,1.5mLチューブに超純水103.2μL,10×Reaction buffer 16μL,25 mM dNTPs溶液16μL,50 mM MgCl2溶液8μL,M13 universal primer,M13 reversal primerをそれぞれ8μL,最後にDNA polymerase 0.8μLを加えプレミックスを作り,200μL PCRチューブに20μLずつ分注し,プレート上に生じた形質転換体のコロニーを楊枝で少量とり,チューブ内で懸濁させた後,サーマル・サイクラーにセットした。94℃で5分間加温し,DNA polymeraseを活性化した後,94℃で30秒間,50℃で30秒間,72℃で1分間の反応を25サイクル行った。この方法によりは,xylAB/Sac-Kpnは,約3000bp付近に遺伝子の増幅断片が確認されたものを形質転換体とした。
M13 universal primer:5'-GTTTTCCCAGTCACGACGTT
M13 reversal primer:5'-GGAAACAGCTATGACCATGA
【0042】
6.I−3で得られたPCR産物,xylFGH/Hind IIIについて,制限酵素反応とゲル抽出を行うことにより,インサートDNA・xylFGH/Hind IIIならびにベクターDNA・pUC18を得た。すなわち,2つの1.5mLチューブにそれぞれ10×H Buffer 5μL,プラスミド・ベクターpUC18とPCR産物xylFGH/Hind IIIを43μLずつ入れ,最後にSalI 2μLを,混合させ,37℃で16時間処理した。反応後さらに,それぞれフェノクロ処理,エタノール沈殿,リンス,減圧乾燥後超純水44μLに溶解後それぞれのチューブに10×B Bufferを5μL,Hind III 1μL 加え37℃で16時間処理した。そして,フェノクロ処理,エタノール沈殿,リンス減圧乾燥後xylFGH/Hind IIIを制限酵素消化したものはTE buffer 30μLに溶解し,プラスミド・ベクターpUC18を制限酵素消化したものは超純水30μLに溶解した。このうちxylFGH/Hind IIIを制限酵素消化したものを,2%アガロース・ゲルを用いて電気泳動した後,約3900bp付近のDNA断片をゲルよりカッターで切り出した。その後,QIAquick Gel Extraction Kitを用いてゲルよりDNAを抽出し,インサートDNA・xylFGH/Hind IIIとした。また,約2.7bpのバンドについても同様の操作を行い,ベクターDNA・pUC18とした。
7.なお,上記により得られたインサートDNAについては,I−4のTAクローニングおよびI−5のコロニー・ダイレクトPCR法と同様の作業により,約3900bp付近に遺伝子増幅断片が確認できるものを形質転換体とした。
【0043】
8.I−5およびI−7により得られた2つの形質転換体について,遺伝子の挿入方向と挿入遺伝子配列の確認を行った。すなわち,それぞれの形質転換体の遺伝子挿入プラスミド・ベクターDNAをミニプレップ法により抽出し,制限酵素処理およびアガロース・ゲル電気泳動を行い,インサートDNAの方向確認を行った。さらに,それぞれの形質転換体についてコロニー・ダイレクトPCR法により,シークエンス解析を行い,増幅されている遺伝子がそれぞれxylA,xylB,xylF,xylG,xylHであることを確認した。
【0044】
9.遺伝子の挿入方向および挿入遺伝子配列の確認を行ったI−5およびI−7により得られた2つの形質転換体について,ミニプレップ法により遺伝子挿入プラスミドの抽出を行った。さらに,I−5で得られた形質転換体の抽出プラスミドについて,Sac IとKpn Iによる制限酵素処理を行い約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,インサートDNA xylAB/Sac-Kpnを得た。同様に,I−7で得られた形質転換体の抽出プラスミドについて,Hind IIIによる制限酵素処理を行い,約3800bp付近のDNA断片をゲルから切り出し,抽出を行うことにより,インサートDNA xylFGH/Hind IIIを得た。
【0045】
<II.プラスミド・ベクターの準備>
1.プラスミド・ベクターとして,上記の記載と同様の方法により,GAPDH遺伝子プロモーターおよびmalE遺伝子ターミネーターを導入したプラスミド・ベクターpUC18compの作製を行った。なお,それぞれのPCR産物を得るために下記のプライマーを用いた。図2に,プラスミド・ベクターpUC18compの遺伝子配置を示す。
(1) GAPDHプロモーター
Forward primer
Sal-Pgap (se):5'-ATTAGTCGACATTACGTGACTGATTCTAAC-3'
Reverse primer
Hind-Pgap (an):5'-CCCAAGCTTATATTCCACCAGCTATTTG-3',又は
Sac-Pgap (an):5'-CTTGAGCTCATATTCCACCAGCTATTTG-3'
(2) malEターミネーター
Forward primer
Kpn-malTT (se):5'-GAGGTACCAGATGTTGTTCTGCCAATG-3'
Reverse primer
Kpn-malTT (an):5'-GCTCTAGAGAGCACGAAAGAGAATTATC-3'
【0046】
<III.遺伝子挿入プラスミド・ベクターの作製>
1.プラスミド・ベクターpUC18compを制限酵素消化,ゲル抽出を行い,ベクターpUC18comp/Sac-Kpn,ベクターpUC18comp/Hind IIIを作製した。すなわち,プラスミド・ベクターpUC18compをSac I,Kpn Iにて処理を行い,2%アガロース・ゲルを用いて電気泳動した後,約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,ベクターpUC18comp/Sac-Kpnとした。同様に,Hind IIIで処理を行い脱リン酸化した後に,2%アガロース・ゲルを用いて電気泳動を行い約3500bp付近のDNA断片をゲルよりカッターで切り出し,抽出を行うことにより,ベクターpUC18comp/Hind IIIとした。
【0047】
2.III−1で得られたベクターpUC18comp/Sac-KpnとI−9で得られたインサートDNAxylAB/Sac-Kpnについて,I−4と同様の方法により,ライゲーションを行い,コンピテント・セルにトランスフェクションをした後,コロニーを得た。得られたコロニーのうち,白色コロニーについて,I−5と同様に,遺伝子増幅の有無をコロニー・ダイレクトPCR法により確認することにより,形質転換体を得た。得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0048】
3.III−2と同様に,III−1で得られたベクターpUC18comp/Hind IIIとI−9で得られたインサートDNA xylFGH/Hind IIIのライゲーションを行い,コロニー・ダイレクトPCR法により遺伝子増幅の有無を確認することにより,形質転換体を得た。得られた形質転換体については,得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0049】
4.III−2で得られた形質転換体について,ミニプレップ法にて遺伝子挿入プラスミド・ベクターを抽出し,制限酵素処理を行い脱リン酸化した後,ゲル抽出により約3800bp付近のDNA断片を切り出し,ベクターDNA xylAB/Hind IIIとした。加えて,III−3で得られた形質転換体についても,ミニプレップ法にて遺伝子挿入プラスミド・ベクターを抽出し,制限酵素処理を行った後,ゲル抽出により約3800bp付近のDNA断片を切り出し,インサートDNA xylFGH/Hind IIIとした。これらにより得られた,ベクターDNA xylAB/Hind IIIとインサートDNA xylFGH/Hind IIIを,I−4と同様の方法により,ライゲーションを行い,コンピテント・セルにトランスフェクションをした後,コロニーを得た。得られたコロニーのうち,白色コロニーについて,I−5と同様に,遺伝子増幅の有無をコロニー・ダイレクトPCR法により確認することにより,約4000bp付近に遺伝子の増幅断片が確認されたものを形質転換体として得た。得られた形質転換体については,I−8と同様の方法により,インサート方向の確認を行った。
【0050】
<IV.KO11株へのトランスフェクション>
1.KO11株について,コンピテント・セルとしての調製を行った。すなわち,試験管にLB液体培地5mLを作製しオートクレーブし,600μg/mLとなるようにクロラムフェニコールを加え,E. coli KO11株を植菌し,濁度がOD660≒0.6に達するまで37℃,60 rpmで回転振とう培養した。この培養液1.0mLをチューブに移し,6,000rpmで5 分間遠心分離した後,上清を捨てた。残ったペレットを滅菌水1mLに再懸濁した。この遠心分離,再懸濁の操作を計3回行い,菌体を洗浄し,さらに10% グリセロール溶液1mLに再懸濁した。この遠心分離,再懸濁の操作を計2回行い,菌体を洗浄し,最終的に10% グリセロール溶液に100μLに再懸濁したものをKO11株のコンピテント・セルとした。
【0051】
2.IV−1で調製したKO11株のコンピテント・セルに,所定の遺伝子挿入プラスミド・ベクターをエレクトロポレーション法によりトランスフェクションした。すなわち,III−2で得られた形質転換体からプラスミド・ベクターDNA・xylABを,IV−4で得られた形質転換体からプラスミド・ベクターxylAB・xylFGHを抽出し,それぞれの遺伝子挿入プラスミド・ベクター溶液をKO11株コンピテント・セルに加えて混合し,エレクトロポレーション・キュベット (0.2 cm) に移した。ジーン・パルサーIIおよびパルス・コントローラー PLUSを用いて,電圧 1.5 kV,抵抗 200,静電容量25μFでエレクトロポレーションを行った後,直ちにLB液体培地900μLを加え,1.5mL容チューブに移し,37℃で1時間回転振とう培養 (60 rpm) した。培養液100μLを, LB/Ampプレート培地にコンラージ棒を用いて塗布し,37℃で16時間培養した。プレート上に生じたコロニーを形質転換体とし,プレートは4℃で保存した。その後,コロニーを5mL LB/Amp液体培地に植菌し,37℃で16時間培養した。プラスミド・ベクターDNA・xylAB導入株を実施例1,プラスミド・ベクターxylAB・xylFGH導入株を実施例2として20%(w/v)グリセロール中で−84℃にて保存し,以降の評価に用いた。
【0052】
<<実施例1および実施例2,KO11株における糖消費およびエタノール生産能評価>>
実施例1および実施例2で作製した形質転換KO11株の糖消費およびエタノール生産能を評価するため,遺伝子導入を行っていないKO11株を比較例として実験を行った。
【0053】
<方法>
1.評価する際の培地としてM9混合糖培地を用いた。すなわち,74.8 gのグルコースと3.2 gのキシロースを含む水溶液32mLを加えた100mL三角フラスコをオートクレーブ滅菌した。オートクレーブ滅菌後順次下記材料を加え培地調製を行い,グルコースおよびキシロースの最終濃度をそれぞれ6% (w/v) と4% (w/v) とした培地をM9混合糖培地として用いた。
・グルコース
・キシロース
・滅菌水
・20×M9 salts…NH4Cl 20 g/L,KH2PO4 60 g/L,NaHPO4 120 g/L,NaCl 10g/Lとなるように蒸留水に溶解し,オートクレーブ滅菌(120℃,15分間)したものを用いた。
・20% (v/v) CSL…CSL原液と滅菌水を用いて終濃度が20% (v/v)となるように調製した。
・MgSO4・7H2O
・1 Mリン酸緩衝液…1 MのNa2HPO4・12H2O溶液に,30℃でpH 6.7となるまで1 MのKH2PO4溶液を添加し,オートクレーブ滅菌したものを用いた。
【0054】
2.実施例1および実施例2の形質転換KO11株について,クリーンベンチ内において,クロラムフェニコール入りLB液体培地ないしアンピシリン入りLB液体培地5mLに植菌を行い,37℃,60 rpmの条件下で16時間回転振とう培養した。この培養液100μLを,クリーンベンチ内において,LB液体培地150mLと各種抗生物質を含む500mL容三角フラスコに加え,37℃,150 rpmの条件下で24時間振とう培養した。この培養液を前培養液とした。前培養液を6,000 ×gで10分間遠心し菌体を回収後,本培養液としてM9混合糖培地に乾燥菌体重量が1 g/Lとなるように植菌し,濃硫酸を入れた発酵栓にて栓をした。発酵栓をした100mL容三角フラスコは,インキュベーターを用いて37℃,150 rpmの条件下で4日間発酵試験を行った。
【0055】
3.HPLCを用いて,各時間点におけるグルコースやキシロースの残存率等の分析を行った。すなわち,発酵試験を行った前記100mL容三角フラスコ中の懸濁液から1mLを各時間点において回収し,それらを15000 rpmの条件下で5分間遠心分離を行うことにより固形成分を沈殿させ上清を得た。得られた上清は,300μLずつ2本に分けエタノール600μLを加え混合した後,フィルターろ過したものを試料溶液とし,1本をエタノール濃度測定用,もう1本を培地糖濃度測定用としてHPLC分析を行った。なお,HPLC分析の分析条件は,下記であり,検出器としては示差屈折率検出器を用いた。
また,前記上清の分析と同様に,糖濃度の標準溶液の分析を合わせて行った。すなわち,0,2.5,5,7.5,10% (w/v) グルコース溶液,または0,2,4,6,8% (w/v) キシロース溶液300μLを用い同様の分析を行い,ピーク面積の比から培養液上清中の糖濃度を算出した。
・HPLC分析条件
カラム Asahipak NH2P-50 4E;直径4.0 mm ,長さ250 mm (Shodex,東京)
溶媒 アセトニトリル : 水=8 : 2 (v/v)
カラム温度 40℃
流速 0.75mL/min
【0056】
4.GC分析により,各時間点におけるエタノール濃度の分析を行った。すなわち,前述のように得られた上清300μLと等容量の5% (v/v) イソプロパノール (内部標準物質) を混合し,下記条件によるGC分析を行った。また,エタノール濃度の標準溶液として,5% (v/v) イソプロパノールと0,2,4,6,8,10% (v/v) エタノールをそれぞれ等容量混合したもの作製したものについても合わせて分析を行い,検出したピーク面積の比から培養液上清中のエタノール濃度を算出した。
・ 分析条件
カラム温度 150℃,検出器温度 250℃,気化室温度 250℃
カラム 液相 5% Thermon 1000
担体Sunpak-A直径3.2 mm 長さ2.1 m
流速 40mL/min
キャリアガス N2
【0057】
<結果>
1.HPLCによる糖濃度分析
(1) 各培養時間におけるM9混合糖培地中のキシロース残存率およびキシロース消費率の比較を表1に,グルコース残存率およびグルコース消費率の比較を表2に示す。なお,各糖における残存率および糖消費率は,下記式に従い算出を行った。
・糖残存率(%)=各時間点における培地中の糖濃度/0時間における培地中の糖濃度
×100
・糖消費率(%)=100−残糖率
(2) 実施例1において,キシロース残存率は発酵初期の12時間後や24時間後では95%以上とキシロース消費はあまり進んでいなかった。しかしながら,発酵36時間後では比較例とほぼ同じ水準までキシロース消費が進行し,発酵48時間後では66.2%と,比較例よりも約20%も多くキシロースを消費していた。一方,実施例2では,発酵初期は比較例と同様のキシロース消費が進行し,発酵36時間後では約11%,発酵48時間後では約19%,比較例よりも多くキシロースを消費していた。これらより,実施例1および実施例2は,実用化において現実的な培養時間と考えられる48時間で,比較例と比べて優れたキシロース代謝を示すことが分かった。
(3) また,発酵48時間後におけるグルコース残存率は,実施例1が18.7%,実施例2が16.9%であった。これらは,比較例のグルコース残存率と比べると,実施例1は約12%,実施例2では約10%の差であり,グルコース消費の差は比較的小さいことが分かった。
【0058】
【表1】
【0059】
【表2】
【0060】
2.GCによるエタノール濃度分析
(1) 各培養時間におけるM9混合糖培地中のエタノール濃度の比較を表3に示す。
(2) 発酵48時間後におけるエタノール濃度は,実施例1が4.15%,実施例2が4.18%であった。一方,比較例は4.07%であり,わずかではあるものの実施例においてエタノール生産の増加が見られた。
【0061】
【表3】
【特許請求の範囲】
【請求項1】
構成発現型プロモーターとキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターが導入されたことを特徴とする形質転換大腸菌
【請求項2】
前記構成発現型プロモーターが,GAPDHプロモーターであることを特徴とする請求項1に記載の形質転換大腸菌
【請求項3】
前記キシロース異性化酵素遺伝子が,xylAであることを特徴とする請求項1又は2に記載の形質転換大腸菌
【請求項4】
前記プラスミド・ベクターに,さらにキシルロースリン酸化酵素遺伝子を挿入することを特徴とする請求項1ないし3に記載の形質転換大腸菌
【請求項5】
前記キシルロースリン酸化酵素遺伝子が,xylBであることを特徴とする請求項4に記載の形質転換大腸菌
【請求項6】
前記プラスミド・ベクターに,さらにキシロース輸送タンパク遺伝子を挿入することを特徴とする請求項1ないし5に記載の形質転換大腸菌
【請求項7】
前記キシロース輸送タンパク遺伝子が,xylF,xylG,xylHであることを特徴とする請求項6に記載の形質転換大腸菌
【請求項8】
前記大腸菌が,KO11株であることを特徴とする請求項1ないし7に記載の形質転換大腸菌
【請求項1】
構成発現型プロモーターとキシロース異性化酵素遺伝子を挿入したプラスミド・ベクターが導入されたことを特徴とする形質転換大腸菌
【請求項2】
前記構成発現型プロモーターが,GAPDHプロモーターであることを特徴とする請求項1に記載の形質転換大腸菌
【請求項3】
前記キシロース異性化酵素遺伝子が,xylAであることを特徴とする請求項1又は2に記載の形質転換大腸菌
【請求項4】
前記プラスミド・ベクターに,さらにキシルロースリン酸化酵素遺伝子を挿入することを特徴とする請求項1ないし3に記載の形質転換大腸菌
【請求項5】
前記キシルロースリン酸化酵素遺伝子が,xylBであることを特徴とする請求項4に記載の形質転換大腸菌
【請求項6】
前記プラスミド・ベクターに,さらにキシロース輸送タンパク遺伝子を挿入することを特徴とする請求項1ないし5に記載の形質転換大腸菌
【請求項7】
前記キシロース輸送タンパク遺伝子が,xylF,xylG,xylHであることを特徴とする請求項6に記載の形質転換大腸菌
【請求項8】
前記大腸菌が,KO11株であることを特徴とする請求項1ないし7に記載の形質転換大腸菌
【図1】

【図2】
【図2】
【公開番号】特開2012−170440(P2012−170440A)
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願番号】特願2011−38297(P2011−38297)
【出願日】平成23年2月24日(2011.2.24)
【出願人】(504224153)国立大学法人 宮崎大学 (239)
【Fターム(参考)】
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願日】平成23年2月24日(2011.2.24)
【出願人】(504224153)国立大学法人 宮崎大学 (239)
【Fターム(参考)】
[ Back to top ]