
キノリン−カルボキサミド化合物の結晶形態物
【課題】5−HT4受容体アゴニストとして有用なキノリノン−カルボキサミド化合物の結晶塩形態物を提供すること。
【解決手段】本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。本発明はまた、このような結晶塩形態物を含む製薬組成物、5HT4受容体活性に関連した疾患を治療するためにこのような結晶塩形態物を使用する方法、およびこのような結晶塩形態物の調製に有用な方法を提供する。
【解決手段】本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。本発明はまた、このような結晶塩形態物を含む製薬組成物、5HT4受容体活性に関連した疾患を治療するためにこのような結晶塩形態物を使用する方法、およびこのような結晶塩形態物の調製に有用な方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、5−HT4受容体アゴニストとして有用なキノリノン−カルボキサミド化合物の結晶塩形態物に関する。本発明はまた、このような結晶化合物を含む製薬組成物、5HT4受容体活性に媒介された病状を治療するためにこのような化合物を使用する方法、およびこのような化合物の調製に有用な方法に関する。
【背景技術】
【0002】
(当業界の状況)
本願と同一の譲受人に譲渡された2004年4月7日出願の米国特許仮出願第60/560,076号、および2005年4月6日出願の米国特許出願第11/100,113号は、胃腸管運動性低下の疾患の治療に有用であると予想される、新規なキノリノン−カルボキサミド化合物を開示している。特に、化合物、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−
ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、5−HT4受容体アゴニスト活性を示すも
のとして、上記出願に具体的に開示されている。1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒド
ロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの化学構造は式Iにより表される:
【0003】
【化1】

治療薬としてこの化合物を効果的に使用するために、容易に製造でき、許容できる化学的および物理的安定性を有する固体状態の塩形態物を有することが望ましいと考えられる。例えば、熱的に安定、例えば、約200℃を越える温度で安定であり、吸湿性でも潮解性でもなく、そのため処理および貯蔵が容易な塩形態物を有することがきわめて望ましいと考えられる。結晶固体は、製造品の純度および安定性が増加するので、非結晶形態物よりも一般的に好ましい。
【発明の概要】
【発明が解決しようとする課題】
【0004】
式Iの化合物の非結晶塩形態物についてはまだ報告されていない。したがって、吸湿性でも潮解性でもなく、好適な熱的安定性を示す式Iの化合物の安定な結晶塩形態物が必要とされている。
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。一態様において、本発明の結晶塩形態物は、式Iの化合物の結晶塩酸塩である。他の態様において、本発明の結晶塩形態物は、式Iの化合物の塩酸塩の結晶水和物である。
【0006】
驚くべきことに、本発明の結晶塩酸塩は、約200℃を越える温度で熱的に安定であり、室温で約2%と約90%との間の範囲の相対湿度に晒された場合に示す重量変化が約0.2%未満であることがわかった。さらに、本発明の結晶塩酸塩も、その水和物も、室温で90%までの相対湿度に晒された場合、潮解性ではない。
【0007】
他の使用法として、本発明の結晶塩形態物は、胃腸管の運動低下疾患を治療するための製薬組成物の調製に有用であることが予想される。したがって、その組成物の他の態様において、本発明は、製薬的に許容できる担体および1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を含む製薬組成物を提供する。
【0008】
本発明はまた、5−HT4受容体活性に関連した疾患または病態、例えば、胃腸管の運動低下疾患を治療する方法を提供し、該方法は、哺乳動物に、治療的有効量の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶質塩酸塩またはその溶媒和物を投与することを含む。
【0009】
他の方法態様において、本発明は、本発明の結晶塩酸塩を調製するための方法を提供し、該方法は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドを塩酸と接触させて反応混合物を形成し、該反応混合物から該結晶塩酸塩を単離することを含む。
本発明はまた、療法において、または薬剤として使用するため、本明細書に記載された本発明の結晶塩酸塩、ならびに、薬剤の製造において、特に哺乳動物における胃腸管の運動低下疾患を治療するための薬剤製造に関して、本発明の結晶塩酸塩の使用法を提供する。
例えば、本発明は以下の項目を提供する。
(項目1)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩またはその溶媒和物である、結晶塩形態物。
(項目2)
前記結晶塩形態物が結晶塩酸塩である、項目1に記載の結晶塩形態物。
(項目3)
前記結晶塩形態物が、4.41±0.2、8.82±0.2、9.08±0.2、11.21±0.2、14.40±0.2、16.42±0.2、17.35±0.2、17.61±0.2、18.14±0.2、19.04±0.2、19.95±0.2、20.20±0.2、21.23±0.2、22.13±0.2、22.48±0.2、22.83±0.2、24.16±0.2、25.37±0.2、25.56±0.2、26.22±0.2、27.33±0.2、29.08±0.2、および29.61±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする、項目2に記載の結晶塩形態物。
(項目4)
前記粉末x線回折パターンが、14.40±0.2、17.35±0.2、17.61±0.2、19.04±0.2、21.23±0.2、および22.13±0.2から選択される2θ値における2つ以上の回折ピークを含む、項目3に記載の結晶塩形態物。
(項目5)
前記結晶塩形態物は、ピーク位置が図1に示されたパターンのピーク位置に実質的に従う粉末x線回折パターンを特徴とする、項目2に記載の結晶塩形態物。
(項目6)
前記結晶塩形態物が、約230℃超の温度において、吸熱性熱流の最大値を示す示差走査熱量測定トレースを特徴とする、項目2に記載の結晶塩形態物。
(項目7)
前記結晶塩形態物が、図2に示された示差走査熱量測定トレースに実質的に従う示差走査熱量測定トレースを特徴とする、項目2に記載の結晶塩形態物。
(項目8)
前記結晶塩形態物が水和物である、項目1に記載の結晶塩形態物。
(項目9)
前記結晶塩形態物が、5.30±0.2、7.43±0.2、8.72±0.2、10.52±0.2、13.85±0.2、14.11±0.2、15.80±0.2、15.99±0.2、17.26±0.2、19.53±0.2、20.08±0.2、21.06±0.2、21.48±0.2、21.92±0.2、22.85±0.2、23.91±0.2、25.28±0.2、26.06±0.2、27.34±0.2、27.51±0.2、および29.67±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする、項目8に記載の結晶塩形態物。
(項目10)
前記粉末x線回折パターンが、10.52±0.2、13.85±0.2、15.80±0.2、17.26±0.2、および21.06±0.2から選択される2θ値における2つ以上の回折ピークを含む、項目9に記載の結晶塩形態物。
(項目11)
前記結晶塩形態物は、ピーク位置が図4に示されたパターンのピーク位置に実質的に従う粉末x線回折パターンを特徴とする、項目8に記載の結晶塩形態物。
(項目12)
前記結晶塩形態物が、図5に示された示差走査熱量測定トレースに実質的に従う示差走査熱量測定トレースを特徴とする、項目8に記載の結晶塩形態物。
(項目13)
製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む製薬組成物。
(項目14)
5−HT4受容体活性に関連した病状を有する哺乳動物を治療する方法であって、該方法は、治療的有効量の製薬組成物を前記哺乳動物に投与することを含み、該製薬組成物は、製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む、方法。
(項目15)
哺乳動物における胃腸管の運動低下障害を治療する方法であって、該方法は、治療的有効量の製薬組成物を前記哺乳動物に投与することを含み、該製薬組成物は、製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む、方法。
(項目16)
前記運動低下障害が、慢性便秘症、便秘優勢過敏性腸症候群、糖尿病性胃疾患および特発性胃疾患、ならびに機能性消化不良症から選択される、項目15に記載の方法。
(項目17)
治療において使用するための、項目1から12のいずれか一項に記載の結晶塩形態物。
(項目18)
薬剤を製造するための、項目1から12のいずれか一項に記載の結晶塩形態物の使用。
(項目19)
前記薬剤が、5−HT4受容体活性に関連した、哺乳動物における病状を治療するための薬剤である、項目18に記載の使用。
(項目20)
前記病状が、胃腸管の運動低下障害である、項目19に記載の使用。
(項目21)
前記病状が、慢性便秘症、便秘優勢過敏性腸症候群、糖尿病性胃疾患および特発性胃疾患、ならびに機能性消化不良症から選択される、項目19に記載の使用。
(項目22)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩を調製するためのプロセスであって、該プロセスは、以下:
(a)1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドと塩酸とを接触させて、反応混合物を形成すること;および
(b)前記反応混合物から前記結晶塩酸塩を単離すること、
を含む、プロセス。
(項目23)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩の結晶水和物を調製するためのプロセスであって、該プロセスは、以下:
(a)1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩を、1ミリリットル当たり約50ミリグラム超の濃度で水に溶解させ、懸濁液を形成すること;および
(b)前記懸濁液から前記結晶水和物を単離すること、
を含む、プロセス。
【図面の簡単な説明】
【0010】
本発明の種々の態様は、添付の図面を参照して例示される。
【図1】図1は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶質塩酸塩の粉末x線回折(PXRD)パターンを示している。
【図2】図2は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩に関する示差走査熱量測定(DSC)トレース(ボトムトレース、右側縦軸)および熱重量分析(TGA)トレース(トップ追跡、左側縦軸)を示している。
【図3】図3は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩に関する動的水分吸収(DMS)を示している。
【図4】図4は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩の結晶水和物の粉末x線回折(PXRD)パターンを示している。
【図5】図5は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物に関する示差走査熱量測定(DSC)トレース(トップトレース、左側縦軸)および熱重量分析(TGA)トレース(ボトムトレース、右側縦軸)を示している。
【図6】図6は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物に関する動的水分吸収(DMS)を示している。
【発明を実施するための形態】
【0011】
(発明の詳細な説明)
本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。
【0012】
定義
本発明の化合物、組成物および方法を記述する際、以下の用語は、別に指示されない限り、以下の意味を有する。
【0013】
用語の「治療的有効量」は、治療を必要としている患者に投与した際に治療をもたらす上で十分な量を意味する。
【0014】
本明細書に用いられる用語の「治療」は、哺乳動物(特にヒト)などの患者における疾患、障害、または病状の治療を意味し、以下を含む:
(a)疾患、障害、または病状の発現の予防、すなわち、患者の予防的処置;
(b)疾患、障害、または病状の寛解、すなわち、患者における疾患、障害、または病状を除去することまたは退行を生じさせること;
(c)疾患、障害、または病状の抑制、すなわち、患者における疾患、障害、または病状の発現の緩徐化または阻止;または
(d)患者における疾患、障害、または病状の症状の軽減。
【0015】
用語の「溶媒和物」は、一種または複数種の溶質分子、すなわち、本発明の化合物または製薬的に許容できるその塩および一種または複数種の溶媒分子により形成された複合体または凝集体を意味する。このような溶媒和物は、溶質と溶媒の実質的に一定のモル比を有する、典型的に結晶質の固体である。代表的な溶媒としては、例えば、水、メタノール、エタノール、イソプロパノール、酢酸などが挙げられる。溶媒が水の場合、形成された溶媒和物は特に水和物と称される。
【0016】
本明細書に用いられる用語の「結晶塩酸塩」は、結晶格子中に溶媒分子の実質的に一定のモル分率を含まない結晶質固体、すなわち、溶媒和物ではないものを意味する。本発明の溶媒和物、または特に水和物は明白に同定される。
【0017】
本明細書および添付の項目に用いられる単数形の「1つの」、「1個の」、「1つ」、および「前記」は、その内容が明らかに別を指示しない限り、複数の記述を含み得る。
【0018】
用語の「アミノ保護基」は、アミノ窒素における望ましくない反応を防ぐために好適な保護基を意味する。代表的なアミノ保護基としては、限定はしないが、ホルミル;アシル基、例えば、アセチルなどのアルカノイル基;tert−ブトキシカルボニル(Boc)などのアルコキシカルボニル基;ベンジルオキシカルボニル(Cbz)および9−フルオレニルメトキシカルボニル(Fmoc)などのアリールメトキシカルボニル基;ベンジル(Bn)、トリチル(Tr)、および1,1−ジ−(4’−メトキシフェニル)メチルなどのアリールメチル基;トリメチルシリル(TMS)およびtert−ブチルジメチルシリル(TBDMS)などのシリル基;などが挙げられる。
【0019】
活性剤
本発明の塩形態物、すなわち、式Iの化合物は、市販のAutoNomソフトウェア(MDL Information Systems、GmbH、フランクフルト、ドイツ国)を用いて、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドと命名される。(1S、3R、5R)の呼称は、二環式環系に結合した結合の相対的な方向を言う。あるいは、該化合物は、N−〔(3−エンド)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル〕−1−(1−メチルエチル)−2−オキソー1,2−ジヒドロ−3−キノリンカルボキサミドと表される。
【0020】
本発明の塩形態物
一態様において、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩を提供する。
【0021】
本発明の結晶塩酸塩は、式Iの化合物の1モル当量当たり、約0.9モル当量と約1.1モル当量との間など、式Iの化合物の1モル当量当たり、約0.8モル当量と約1.2モル当量との間の塩酸を典型的に含有する。
【0022】
活性剤に対する塩酸のモル比は、当業者に利用できる方法によって容易に決定できる。例えば、このようなモル比は、硝酸銀の標準溶液による滴定によって容易に決定できる。あるいは、該モル比を決定するために、元素分析、1H NMR、およびイオンクロマトグラフィ法が使用できる。
【0023】
一態様において、本発明の結晶塩酸塩は、4.41±0.2、8.82±0.2、9.08±0.2、11.21±0.2、14.40±0.2、16.42±0.2、17.35±0.2、17.61±0.2、18.14±0.2、19.04±0.2、19.95±0.2、20.20±0.2、21.23±0.2、22.13±0.2、22.48±0.2、22.83±0.2、24.16±0.2、25.37±0.2、25.56±0.2、26.22±0.2、27.33±0.2、29.08±0.2、および29.61±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折(PXRD)パターンを特徴とする。この態様において、該結晶形態物は特に、14.40±0.2、17.35±0.2、17.61±0.2、19.04±0.2、21.23±0.2、および22.13±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする。
【0024】
粉末x線回折の分野で十分知られているように、PXRDスペクトルのピーク位置は、サンプル調製および機器の幾何学的配置などの詳細などの実験的詳細に対して、相対的ピーク高さよりも相対的に感受性が低い。したがって、一態様において、式Iの化合物の結晶塩酸塩は、ピーク位置が実質的に図1に示されたものによる粉末x線回折パターンを特徴とする。
【0025】
また、本発明の結晶塩酸塩は、図2にしたがって、示されたように、約230℃から約260℃の範囲の吸熱性の熱流におけるピークを示す示差走査熱量測定(DSC)トレースにより実証される高温熱的安定性を特徴とする。さらに、熱重量分析(TGA)トレースで、約225℃以下での有意な熱的事象は示されない。
【0026】
さらに他の態様において、結晶塩酸塩は、約758、783、795、802、949、981、1149、1158、1217、1332、1377、1453、1467、1487、1525、1566、1575、1615、1672、および3197cm−1における有意な吸収バンドを示す赤外線吸収スペクトルを特徴とする。
【0027】
式Iの化合物の結晶塩酸塩は、図3に示されるように、例外的に低レベルの吸湿性(すなわち、室温で、2%相対湿度から90%相対湿度の湿度範囲で、約0.2%未満の重量増加)の可逆的吸着/脱離プロフィルを有することが実証されている。
【0028】
また、式Iの化合物の結晶塩酸塩は、長期間、高温および高湿に曝された際に安定であることが判明している。例えば、40℃および相対湿度75%で24週間貯蔵後のHPLCで、化学的分解は示されず、DSC、TGA、またはPXRDの結果にも検出できる変化はなかった。
【0029】
他の態様において、本発明は、式Iの化合物の塩酸塩の結晶水和物を提供する。
【0030】
一態様において、本発明の塩酸塩の結晶水和物は、5.30±0.2、7.43±0.2、8.72±0.2、10.52±0.2、13.85±0.2、14.11±0.2、15.80±0.2、15.99±0.2、17.26±0.2、19.53±0.2、20.08±0.2、21.06±0.2、21.48±0.2、21.92±0.2、22.85±0.2、23.91±0.2、25.28±0.2、26.06±0.2、27.34±0.2、27.51±0.2、および29.67±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折(PXRD)パターンを特徴とする。この態様において、該結晶形態物は特に、10.52±0.2、13.85±0.2、15.80±0.2、17.26±0.2、および21.06±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする。
【0031】
他の態様において、式Iの化合物の塩酸塩の結晶水和物は、ピーク位置が実質的に図4に示されたものによる粉末x線回折パターンを特徴とする。
【0032】
本発明の塩酸塩の結晶水和物はまた、図5に示されるように、約225℃から約250℃の範囲での結晶の融解で確認される吸熱性の熱流における実質的ピーク、および低温での幅広いまたは弱い吸熱を示す示差走査熱量測定(DSC)トレースを特徴とする。さらに、熱重量分析(TGA)トレースで、分解温度は約250℃以上であることが示されている。
【0033】
式Iの化合物の塩酸塩の結晶水和物は、図6に示されるように、約2%から約90%の相対湿度の全範囲にわたって、室温で可逆的吸着/脱離プロフィルを有することが実証されている。該結晶水和物は、約40%と約75%との間の相対湿度で、約0.25%未満の重量増加を示す。
【0034】
本発明のこれらの性質は、以下の実施例においてさらに例示されている。
【0035】
合成法
活性剤、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、下記の実施例に記載された方法を用いて、または本出願の背景の節に挙げられた、本願と同一の譲受人に譲渡された米国特許出願に記載された方法を用いて、容易に入手できる出発材料から調製できる。
【0036】
本発明の結晶質塩酸塩を調製するために、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドを、典型的に、約1モル当量から約1.2モル当量などの約1モル当量から約1.5モル当量の濃塩酸に接触させる。一般的にこの反応は、約20℃から約80℃の範囲の温度で、不活性希釈剤中で行われる。この反応に好適な不活性希釈剤としては、限定はしないが、エタノール、メタノール、イソプロパノール、酢酸エチル、アセトニトリル、トルエン、テトラヒドロフラン、およびそれらの組み合わせが挙げられる。
【0037】
反応が完了したら、本発明の結晶塩を、沈殿、濃縮、遠心分離などの任意の慣例的手段により、該反応混合物から単離する。
【0038】
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S
,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩を、約50mg/mL以上の濃度で水に溶解することにより、結晶水和物を調製でき、これにより提供される懸濁液から、得られた結晶水和物を慣例的手段により単離できる。
【0039】
製薬組成物
本発明の結晶塩酸塩形態物は典型的に、製薬組成物の形態で患者に投与される。このような製薬組成物は、限定はしないが、経口、経直腸、経膣、経鼻、吸入、局所(経皮を含む)および非経口の投与様式など、任意の許容できる投与経路によって患者に投与できる。
【0040】
したがって、本発明は、その組成物態様の1つにおいて、製薬的に許容できる担体または賦形剤ならびに式Iの化合物の結晶塩酸塩の治療的有効量を含む製薬組成物に関する。任意に、所望の場合、このような製薬組成物は、他の治療的および/または製剤化剤を含有してもよい。
【0041】
本発明の製薬組成物は典型的に、本発明の結晶塩の治療的有効量を含有する。典型的に、このような製薬組成物は、約5重量%から約60重量%の活性剤など、約1重量%から約70重量%の活性剤を含め、約0.1重量%から約95重量%の活性剤を含有する。
【0042】
本発明の製薬組成物中には、任意の慣例的な担体または賦形剤を使用できる。具体的な担体または賦形剤、もしくは担体または賦形剤の組み合わせの選択は、具体的な患者または病状または疾患状態のタイプの治療に用いられる投与様式に依存する。これに関して、具体的な投与様式に対して好適な製薬組成物の調製は、十分に製薬業界者の範囲内にある。また、そのような組成物のための成分は、例えば、Sigma、P.O.Box 14508、セントルイス、ミズーリ州 63178から市販されている。さらに例えば、慣例的な製剤化法は、Remington:The Science and Practice of Phrmacy、第20版、Lippincott Williams & White、バルチモア、メリーランド州(2000);およびH.C.Anselら、Pharmaceutical Dosage Forms and Drug Delivery Systems、第7版、Lippincott Williams & White、バルチモア、メリーランド州(1999)に記載されている。
【0043】
製薬的に許容できる担体として役立ち得る材料の代表例としては、限定はしないが、以下のものが挙げられる:(1)ラクトース、グルコース、およびスクロースなどの糖類;(2)トウモロコシ澱粉およびジャガイモ澱粉などの澱粉類;(3)微結晶セルロース、およびその誘導体など、カルボキシメチルセルロースナトリウム、エチルセルロースおよびセルロースアセテートなどのセルロース;(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)カカオバターおよび座剤ワックスなどの賦形剤;(9)落花生油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油および大豆油などの油類;(10)プロピレングリコールなどのグリコール類;(11)グリセリン、ソルビトール、マンニトールおよびポリエチレングリコールなどのポリオール類;(12)オレイン酸エチルおよびラウリン酸エチルなどのエステル類;(13)寒天;(14)水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;(15)アルギン酸;(16)パイロジェンのない水;(17)等張生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸緩衝液;および(21)製薬組成物に用いられる他の非毒性適合性物質。
【0044】
本発明の製薬組成物は、本発明の化合物を、製薬的に許容できる担体および一種または複数種の任意の成分と、十分に完全に混合またはブレンドすることによって典型的に調製される。必要または所望の場合、得られた均一にブレンドされた混合物を次に、慣例的な方法および装置を用いて、錠剤、カプセル剤、丸剤などに形状化または装填することができる。
【0045】
本発明の製薬組成物は、単位投与量形態にパッケージ化することが好ましい。用語の「単位投与量形態」とは、患者への投与に好適な物理的に個別の単位、すなわち、単独で、または1つまたは複数の追加の単位と組み合わせて、所望の治療的効果を生じさせるように計算された活性剤の予め決められた量を含有する各単位を言う。例えば、このような単位投与量形態は、カプセル剤、錠剤、丸剤などであり得る。
【0046】
好ましい一実施形態において、本発明の製薬組成物は、経口投与に好適である。経口投与に好適な製薬組成物は、カプセル剤、錠剤、丸剤、舐剤、カシェ剤、糖衣剤、散剤、顆粒剤の形態であり得るか;または水性もしくは非水性液体中の液剤もしくは懸濁剤;または水中油もしくは油中水液乳剤;またはエリキシル剤またはシロップ剤;などであり得;各々が活性剤として本発明の化合物の予め決められた量を含有する。
【0047】
固体投与形態(すなわち、カプセル剤、錠剤、丸剤など)における経口投与が意図される場合、本発明の製薬組成物は典型的に、活性剤としての本発明の化合物、およびクエン酸ナトリウムまたはリン酸二カルシウムなどの、一種または複数種の製薬的に許容できる担体を含む。任意に、または代替として、このような固体投与形態は、以下のものもまた含む;(1)澱粉、微結晶セルロース、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;(2)カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアラビアゴム;(3)グリセロールなどの湿潤剤;(4)寒天、炭酸カルシウム、ジャガイモ澱粉またはタピオカ澱粉、アルギン酸、一定のケイ酸塩、および/または炭酸ナトリウムなどの崩壊剤;(5)パラフィンなどの溶解遅延剤;(6)第四級アンモニウム化合物などの吸収促進剤;(7)セチルアルコールおよび/またはモノステアリン酸グリセロールなどの浸潤剤;(8)カオリンおよび/またはベントナイトなどの吸収剤;(9)タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはそれらの混合物;(10)着色剤;および(11)緩衝剤。
【0048】
本発明の製薬組成物中には、放出剤、浸潤剤、コーティング剤、甘味剤、風味剤ならびに香料、保存剤および抗酸化剤もまた存在し得る。製薬的に許容できる抗酸化剤の例としては以下のものが挙げられる:(1)アスコルビン酸、塩酸システイン、硫酸二ナトリウム、異性重硫酸ナトリウム重亜硫酸ナトリウムなどの水溶性抗酸化剤;(2)パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、プロピルガレート、アルファ−トコフェロールなどの油溶性抗酸化剤;(3)クエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート化剤。錠剤、カプセル剤、丸剤などのためのコーティング剤としては、セルロースアセテートフタレート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピルメチルセルロースフタレート、メタクリル酸−メタクリル酸エステルコポリマー、セルロースアセテートトリメリテート(CAT)、カルボキシメチルエチルセルロース(CMEC)、ヒドロキシプロピルメチルセルロースアセテートスクシネート(HPMCAS)など、腸溶コーティングに用いられるものが挙げられる。
【0049】
所望の場合、本発明の製薬組成物は、例えば、種々の比率でのヒドロキシメチルセルロース;または他のポリマーマトリックス、リポソームおよび/またはミクロスフェアを用いて、活性剤の緩徐な、または制御された放出を提供するように製剤化することもできる。
【0050】
また、本発明の製薬組成物は、任意に、隠蔽剤を含有でき、活性成分を、胃腸管のある一定の部分でのみ、または優先的に、任意に遅延様式で、放出するように製剤化できる。使用できる包埋組成物の例としては、ポリマー物質およびワックスが挙げられる。活性成分はまた、適切な場合、上記の賦形剤の一種または複数種と共に、マイクロカプセル化形態においてあり得る。
【0051】
経口投与用の好適な液体投与形態としては、例えば、製薬的に許容できる乳剤、マイクロ乳剤、液剤、懸濁剤、シロップ剤およびエリキシル剤が挙げられる。このような液体投与形態は典型的に、活性成分、ならびに、例えば、水または他の溶媒などの不活性希釈剤、可溶化剤、およびエチルアルコール、イソプロピルアルコール、エチルカルボネート、エチルアセテート、ベンジルアルコール、ベンジルベンゾエート、プロピレングリコール、1,3−ブチレングリコール、油類(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコール類およびソルビタンの脂肪酸エステル類およびそれらの混合物などの乳化剤を含む。懸濁剤は、活性成分に加えて、例えば、エトキシル化イソステアリルアルコール類、ポリオキシエチレンソルビトールならびにソルビタンエステル類、微結晶セルロース、アルミニウムメタヒドロキシド、ベントナイト、寒天およびトラガカント、ならびにそれらの混合物などの懸濁化剤を含有できる。
【0052】
あるいは、本発明の製薬組成物は、吸入による投与のために製剤化される。吸入による投与のための好適な製薬組成物は典型的に、エアロゾルまたは粉末の形態にある。このような組成物は一般的に、用量計測吸入器、ドライパウダー吸入器、ネブライザーまたは類似の送達デバイスなど、周知の送達デバイスを用いて投与される。
【0053】
減圧容器を用いて吸入により投与される場合、本発明の製薬組成物は典型的に、活性成分、ならびにジクロロジフルオロメタン、トリクロロフルオロメタン、ジ
クロロテトラフルオロエタン、二酸化炭素または他の好適な気体などの好適な噴射剤を含む。
【0054】
また、該製薬組成物は、本発明の化合物、ならびに粉末吸入器における使用にとって好適な粉末を含むカプセル剤またはカートリッジの形態にあってもよい。好適な粉末基材としては、例えば、ラクトースまたは澱粉が挙げられる。
【0055】
本発明の化合物はまた、公知の経皮送達系および賦形剤を用いて、経皮投与もできる。例えば、本発明の化合物を、プロピレングリコール、ポリエチレングリコールモノラウレート、アザシクロアルカン−2−オン類などの浸透増強剤と混合することができる。所望の場合、このような経皮組成物中に、ゲル化剤、乳化剤および緩衝剤などの追加の賦形剤を使用できる。
【0056】
以下の製剤は、本発明の典型的な製薬組成物を例示している。
【0057】
製剤例A
経口投与用のハードゼラチンカプセル剤は以下のとおり調製される:
【0058】
【表1】
典型的操作:成分を完全に混合してからハードゼラチンカプセルに充填する(1カプセル当たり260mgの組成物)。
【0059】
製剤例B
経口投与用のハードゼラチンカプセル剤は以下のとおり調製される:
【0060】
【表2】
典型的操作:成分を完全に混合してから、45号メッシュU.S.ふるいを通して、ハードゼラチンカプセルに充填する(1カプセル当たり200mgの組成物)。
【0061】
製剤例C
経口投与用のカプセル剤は以下のとおり調製される:
【0062】
【表3】
典型的操作:成分を完全に混合してからゼラチンカプセルに充填する(1カプセル当たり310mgの組成物)。
【0063】
製剤例D
経口投与用の錠剤は以下のとおり調製される:
【0064】
【表4】
典型的操作:該活性成分、澱粉およびセルロースを45番メッシュU.S.ふるいに通し、完全に混合する。得られた粉末とポリビニルピロリドンの溶液とを混合し、次いで、この混合物を14番メッシュU.S.ふるいに通す。このようにして作製された顆粒を50〜60℃で乾燥し、18番メッシュU.S.ふるいに通す。次いで、この顆粒に、カルボキシメチルセルロース澱粉、ステアリン酸マグネシウムおよびタルク(先に60番メッシュU.S.ふるいに通した)を加える。混合後、該混合物を錠剤機で圧縮し100mgの重量の錠剤を得る。
【0065】
製剤例E
経口投与用錠剤は以下のとおり調製される:
【0066】
【表5】
典型的操作:該成分を完全に混合してから圧縮して錠剤を形成する(1錠当たり440mgの組成物)。
【0067】
製剤例F
経口投与用単一刻み目入り錠剤は以下のとおり調製される:
【0068】
【表6】
典型的操作:該成分を完全に混合し、圧縮して単一刻み目入り錠剤を形成する(1錠当たり215mgの組成物)。
【0069】
製剤例G
経口投与用懸濁剤は以下のとおり調製される:
【0070】
【表7】
典型的操作:該成分を混合して、懸濁液10mL当たり活性成分10mgを含有する懸濁剤を形成する。
【0071】
製剤例H
吸入による投与のためのドライパウダーは以下のとおり調製される:
【0072】
【表8】
典型的操作:該活性成分を超微粉砕してからラクトースと混合する。次いで、このブレンドした混合物をゼラチン吸入カートリッジ内へ充填する。粉末吸入器を用いて、該カートリッジの内容物を投与する。
【0073】
製剤例I
計測用量吸入器内の吸入による投与のためのドライパウダーは以下のとおり調製される:
典型的操作:200mLの脱塩水中に溶解させた0.2gのレシチンから形成した溶液中に、10μm未満の平均サイズを有する超微粉砕粒子としての活性化合物10gを分散させることによって、本発明の塩を5重量%およびレシチンを0.1重量%含有する懸濁液を調製する。該懸濁液をスプレー乾燥し、得られた物質を、1.5μm未満の平均直径を有する粒子へと超微粉砕する。該粒子を、加圧した1,1,1,2−テトラフルオロエタンと共にカートリッジ内へ充填する。
【0074】
製剤例J
注射用製剤は以下のとおり調製される:
【0075】
【表9】
典型的操作:上記成分を混合し、0.5N HClまたは0.5N NaOHを用いて、pHを4±0.5に調整する。
【0076】
製剤例K
経口投与用のカプセル剤は以下のとおり調製される:
【0077】
【表10】
典型的操作:該成分を完全に混合してから、ゼラチンカプセル(サイズ#1、白、不透明)内へ充填する(1カプセル当たり264mgの組成物)。
【0078】
製剤例L
経口投与用のカプセル剤は以下のとおり調製される:
【0079】
【表11】
典型的操作:該成分を完全に混合してから、ゼラチンカプセル(サイズ#1、白、不透明)内へ充填する(1カプセル当たり148mgの組成物)。
【0080】
有用性
式Iの化合物、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、5−HT4受容体アゴニストであり、したがって、式Iの化合物の当該結晶塩形態物は、5−HT4受容体に媒介された、または5−HT4受容体活性に関連した病状、すなわち、5−HT4受容体アゴニストを用いる治療によって寛解する病状の治療に有用であることが予想される。このような病状としては、限定はしないが、過敏性腸症候群(IBS)、慢性便秘症、機能性消化不良症、胃内容排出遅延、逆流性食道炎(GERD)、胃不全麻痺、糖尿病性特発性胃疾患、術後腸閉塞症、腸偽性閉塞、および薬剤誘導移行遅延が挙げられる。また、いくつかの5−HT4受容体アゴニスト化合物は、認知障害、行動障害、気分障害、および自律神経機能の制御障害などの中枢神経系障害の治療に使用できる。
【0081】
特に、本発明の塩形態物は、胃腸(GI)管の運動性を高め、したがって、ヒトを含めた哺乳動物における、運動低下に起因するGI管の障害を治療する上で有用であることが予想される。このようなGI運動障害としては、例えば、慢性便秘症、便秘優勢過敏性腸症候群(C−IBS)、糖尿病性および特発性胃疾患、および機能性消化不良症が挙げられる。
【0082】
したがって、一態様において、本発明は、哺乳動物における胃腸管の運動を高める方法を提供し、該方法は、製薬的に許容できる担体および本発明の結晶塩を含む治療的有効量の製薬組成物を、哺乳動物に投与することを含む。
【0083】
5−HT4受容体に媒介されたGI管の運動低下障害または他の病態の治療に用いる場合、本発明の塩形態物は典型的に、一日単回投与で、または一日複数回投与で経口投与されるが、他の投与形態も使用できる。一投与当たりの活性剤の量または一日当たり投与される総量は、典型的には、治療される病態、選択された投与経路、投与される実際の化合物およびその相対的活性、年齢、体重、および個々の患者の応答、患者の症状の重症度などの関連状況に鑑みて、医師によって決定される。
【0084】
5−HT4受容体に媒介されたGI管の運動低下障害または他の障害を治療するための好適な用量は、活性剤の約0.0007mg/kg/日から約1mg/kg/日など、活性剤の約0.0007mg/kg/日から約20mg/kg/日の範囲であると考えられる。平均70kgのヒトでは、これは、1日当たり約0.05mg/kg/日から約70mg/kg/日の活性剤の量になる。
【0085】
本発明の一態様において、本発明の塩形態物は、慢性便秘症の治療に用いられる。慢性便秘症の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。慢性便秘症を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0086】
本発明の他の態様において、本発明の塩形態物は、過敏性腸症候群の治療に用いられる。便秘優勢過敏性腸症候群の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。便秘優勢過敏性腸症候群を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0087】
本発明の他の態様において、本発明の塩形態物は、糖尿病性および特発性胃疾患の治療に用いられる。糖尿病性および特発性胃疾患の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。糖尿病性胃疾患を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0088】
本発明のさらに他の態様において、本発明の塩形態物は、機能性消化不良症の治療に用いられる。機能性消化不良症の治療に用いられる場合、本発明の化合物は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。機能性消化不良症を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0089】
本発明はまた、5−HT4受容体活性に関連した疾患または病態を有する哺乳動物を治療する方法を提供し、該方法は、本発明の塩形態物、または本発明の塩形態を含む製薬組成物の治療的有効量を、哺乳動物に投与することを含む。
【0090】
上記のとおり、本発明の塩形態物は、5−HT4受容体アゴニストである。したがって、本発明はさらに、哺乳動物において、5−HT4受容体に作動する方法を提供し、該方法は、本発明の塩形態物を哺乳動物に投与することがを含む。
【0091】
本発明の塩酸塩形態物の性質、ならびに有用性は、当業者によく知られた種々のインビトロおよびインビボアッセイを用いて実証することができる。典型的なアッセイは、以下の実施例において、さらに詳細に記載されている。
【実施例】
【0092】
以下の合成的および生物学的実施例は、本発明を例示するために提供されており、決して本発明の範囲を限定するものとして考えてはならない。下記の実施例において、以下の略号は、別に指示されない限り、以下の意味をもつ。下記に定義されていない略号は、一般に認められている意味をもつものである。
【0093】
Boc = t−ブトキシカルボニル
(Boc)2O = ジ−t−ブチルジカルボネート
DCM = ジクロロメタン
DMF = N,N−ジメチルホルムアミド
DMSO = ジメチルスルホキシド
EtOAc = エチルアセテート
mCPBA = m−クロロ安息香酸
MeCN = アセトニトリル
MTBE = t−ブチルメチルエーテル
PyBop = ベンゾトリアゾール−1−イルオキシトリピロリジノ−ホスホニウムヘキサフルオロホスフェート
Rf = 保持因子
RT = 室温
TFA = トリフルオロ酢酸
THF = テトラヒドロフラン
試剤(第二級アミンを含む)および溶媒は、民間供給者(Aldrich、Fluka、Sigmaなど)から購入し、そのまま精製せずに使用した。反応は、別に注記しない限り、窒素雰囲気下で行った。反応混合物の経過は、薄層クロマトグラフィ(TLC)、分析用高性能液体クロマトグラフィ(anal.HPLC)、および質量分析によってモニターし、その詳細は、下記および反応の具体的な実施例において個別に提示されている。反応混合物は各反応において具体的に記載されているとおりに作製し;通常、抽出のほか温度および溶媒依存的な結晶化、沈殿などの精製法により精製した。また、反応混合物は分取HPLCによって規定通りに精製した。反応生成物の特性解析は、質量分析および1H−NMR分光法により、規定通りに実施した。NMR測定では、サンプルを重水素化溶媒(CD3OD、CDCl3、またはDMSO−d6)中に溶解し、1H−NMRスペクトルを、標準的な観察条件下、Varian Gemini2000機器(300MHz)によって得た。化合物の質量分析による同定は、Applied Biosystems(フォスターシティー、カリフォルニア州)モデルのAPI150EX機器またはAgilent(パロアルト、カリフォルニア州)モデルの1100LC/MSD機器による電気スプレー電離法(ESMS)によって実施した。水分含量は、Brinkmann(ウェストベリー、ニューヨーク)のMetrohm Karl Fischer Model 813クーロメーターを用いて、カールフィッシャー滴定法により判定した。
【0094】
調製1:(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステル
a.8−ベンジル−8−アザビシクロ〔3.2.1〕オクタン−3−オンの調製
水(170mL)に溶かした2,5−ジメトキシテトラヒドロフラン(82.2g、0.622モル)の不均一溶液に、濃塩酸(30mL)を攪拌しながら加えた。0℃(氷浴)に冷却した別のフラスコ内で、水(350mL)に溶かしたベンジルアミン(100g、0.933モル)の溶液に、濃塩酸(92mL)を徐々に加えた。2,5−ジメトキシテトラヒドロフラン溶液をおよそ20分間攪拌し、水(250mL)で希釈し、次に、ベンジルアミン溶液を加え、引き続き、水(400mL)に溶かした1,3−アセトンジカルボン酸(100g、0.684モル)の溶液を加え、次いで水(200mL)に溶かしたリン酸水素ナトリウム(44g、0.31モル)を加えた。40%NaOHを用いて、pHをpH1からpH約4.5に調整した。得られた混濁淡黄色溶液を一晩攪拌した。次いで、50%塩酸を用いて、該溶液をpH7.5からpH3に酸性化し、85℃に加熱し、2時間攪拌した。該溶液を室温まで冷却し、40%NaOHを用いてpH12に塩基性化し、ジクロロメタン(3×500mL)で抽出した。有機層を合せて、生理食塩水で洗浄し、乾燥し(MgSO4)、ろ過し、減圧下濃縮し、粘稠な褐色オイルとして、標題の粗中間体を得た。
【0095】
0℃で、メタノール(1000mL)に溶かした該粗中間体の溶液に、ジ−tert−ブチルジカーボネート(74.6g、0.342モル)を加えた。該溶液を室温まで温め、一晩攪拌した。減圧下、メタノールを除去し、得られたオイルをジクロロメタン(1000mL)に溶解させた。該中間体を1MのH3PO4(1000mL)中に抽出し、ジクロロメタン(3×250mL)で洗浄した。水層を、水性NaOHを用いて、pH12へ塩基性化し、ジクロロメタン(3×500mL)で抽出した。有機層を合せて乾燥し(MgSO4)、ろ過し、減圧下濃縮して、粘稠な淡褐色オイルとして、標題中間体を得た。
【0096】
【数1】
b.3−オキソ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステルの調製
EtOAc(300mL)に溶かした8−ベンジル−8−アザビシクロ〔3.2.1〕オクタン−3−オン(75g、0.348モル)の溶液に、EtOAc(300mL)に溶かしたジ−t−ブチルジカーボネート(83.6g、0.383モル、1.1当量)を加えた。得られた溶液およびすすぎ液(100mLのEtOAc)を、窒素流下、23gの水酸化パラジウム(20重量%のPd、乾燥ベース、炭素上、水で約50%湿性;例えば、Pearlmanの触媒)を含有する1LのParr水素化容器に加えた。該反応容器を脱気し(真空とN2を交互に5回)、60psiのH2ガスへと加圧した。該反応溶液を2日間攪拌し、適宜H2を再供給して、シリカ薄層クロマトグラフィでモニターして反応が完了するまで、H2圧を60psiに保たせた。次いで、該黒色溶液をCelite(登録商標)のパッドを通してろ過し、減圧下濃縮して、粘稠な黄色から橙色オイルとして、標題中間体を定量的に得た。これをそのまま処理せずに、次のステップに用いた。
【0097】
【数2】
c.(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステルの調製
メタノール(1L)に溶かした先のステップの生成物(75.4g、0.335モル)の溶液に、N2流下、機械的攪拌器により攪拌しながら、フマル酸アンモニウム(422.5g、6.7モル)、水(115mL)および活性炭素上パラジウム65g(乾燥ベース上10%、水で約50%湿性;DegussaタイプE101NE/W)を加えた。24時間後および48時間後、各々の時点で、追加部分のフマル酸アンモニウム(132g、2.1モル)を加えた。分析用HPLCによるモニターで反応の進行が停止したら、Celite(登録商標)(>500g)を加え、得られた濃厚懸濁液をろ過してから、採取した固体をメタノール(約500mL)ですすいだ。ろ液を合せて、全てのメタノールが除去されるまで、減圧下濃縮した。次いで、得られた混濁二相溶液をpH2で約1.5Lから2.0Lの最終容量まで1Mのリン酸で希釈し、ジクロロメタン(3×700mL)で洗浄した。水層を、40%の水性NaOHを用いて、pH12に塩基性化し、ジクロロメタン(3×700mL)で抽出した。有機層を合せてMgSO4で乾燥し、ろ過し、回転蒸発、次いで高真空により濃縮し、52g(70%)の標題中間体、一般に、N−Boc−エンド−3−アミノトロパンを、白色から淡黄色固体として得た。この生成物のエンドアミン対エキソアミンの異性体比は、1H−NMR分析に基づき、>99であった(分析用HPLCにより>96%の純度)。
【0098】
【数3】
分析用HPLC(無勾配方法;5分間にわたる90:10(A:B)に対する2:98(A:B):保持時間=2.14分)。
【0099】
調製2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸
先ず、水(2L)に溶かした2−アミノフェニルメタノール(255.2g、2.07モル)および酢酸(3.56mL、62ミリモル)の攪拌懸濁液に、室温で、アセトン(228.2mL、3.11モル)を加えた。4時間後、該懸濁液を0℃に冷却し、さらに2.5時間攪拌してからろ過した。該固体を採取し、水で洗浄し、湿潤固体を凍結乾燥により冷却、乾燥し、2,2−ジメチル−1,4−ジヒドロ−2H−ベンゾ〔1,3〕オキサジン(332.2g、98%)を、オフホワイトの固体として得た。
【0100】
【数4】
THF(1L)に溶かした2,2−ジメチル−1,4−ジヒドロ−2H−ベンゾ〔1,3〕オキサジン(125g、0.77モル)の溶液を、シンチレーション漏斗を介してろ過してから、0℃で、THF(800mL)中、1.0MのLiAlH4の攪拌溶液に、2.5時間かけて添加漏斗を介し、滴下により加えた。0℃で、Na2SO4・10H2O(110g)を、1.5時間かけて部分に分けて徐々に加えることによって、該反応をクエンチした。該反応混合物を一晩攪拌し、ろ過し、固体塩をTHFで完全に洗浄した。ろ液を減圧下濃縮し、2−イソプロピルアミノフェニルメタノール(120g、95%)を黄色オイルとして得た。
【0101】
【数5】
トルエン(800mL)に溶かした2−イソプロピルアミノフェニルメタノール(118g、0.71モル)の攪拌溶液に、二酸化マンガン(85%、182.6g、1.79モル)を加え、該反応混合物を117℃に4時間加熱した。該反応混合物を一晩、室温まで冷却させてから、Celiteのパッドを介してろ過し、これをトルエンで溶出した。ろ液を減圧下濃縮し、2−イソプロピルアミノベンズアルデヒド(105g、90%)を橙色オイルとして得た。
【0102】
【数6】
0℃で、メタノール(1L)に溶かした2−イソプロピルアミノベンズアルデヒド(105g、0.64モル)、酢酸(73.6mL、1.29モル)およびエチレンジアミン(43.0mL、0.64モル)の攪拌溶液に、2,2−ジメチル−〔1,3〕ジオキサン−4,6−ジオン、一般にMeldrumの酸(166.9g、1.16モル)を加えた。該反応混合物を0℃で1時間、次いで室温で一晩攪拌した。得られた懸濁液をろ過し、固体をメタノールで洗浄し採取しt、標題の中間体、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸を、オフホワイトの固体として得た。
【0103】
【数7】
(実施例1:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成)
a.{(1S,3R,5R)−3−[1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボニル〕アミノ]−8−アザ−ビシクロ〔3.2.1〕オクト−3−
イル}アミドの調製
3Lフラスコ内に、トルエン(1L)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸(112.4g、0.486モル、1.1当量)を懸濁させた。該混合物を85℃に加熱し、塩化チオニル(86.74g、0.729モル)を70分かけて滴下して加えた。該混合物を攪拌しながら95℃に1.5時間加熱してから、室温まで冷却させた。
【0104】
別の12Lフラスコに、トルエン(1L)に溶かした(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステル(100.0g、0.442モル、1当量)を懸濁させ、3MのNaOH(4当量)を加えた。該混合物を室温で10分間攪拌してから、約5℃に冷却させた。内部温度を10℃以下に保ちながら、40分かけて、酸クロリド溶液を攪拌しながら徐々に加えた。該混合物を3℃〜5℃で30分間攪拌し、一晩、層分離させた。トルエン層(約2.5L)を採取し、回転蒸発により、約半分(約1.2L)に濃縮し、次のステップに直接使用した。
【0105】
b.1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの調製
先のステップで調製したトルエン溶液(約1.2L)に、20℃で20分かけて、攪拌しながら、トリフルオロ酢酸(200mL)を加えた。該混合物を、20℃で2時間攪拌した。水(1.55L)を加え、該混合物を20℃で30分間攪拌した。30分後、該混合物は3層に分かれた。下層(約350mL)の粘稠褐色オイルが粗中間体を含有した。
【0106】
MTBE(2.8L)を入れた12Lフラスコに、該粗褐色オイルを、1℃〜2℃で1時間かけて攪拌しながら加えた。該懸濁液を同じ温度で1時間攪拌してからろ過した。ろ液をMTBE(2×300mL)で洗浄し、室温で4日間、真空下乾燥し、標題中間体のトリフルオロ酢酸塩(163.3g)を淡黄色粉末として得た。
【0107】
c.N−メチル−N−〔(S)−2−オキシラン−2−イルメチル〕メタンスルホンアミドの調製
12Lのフラスコに、水(1L)を入れ、次いでNaOH(水中50%、146.81g、1.835モル)を加えた。NaOHを含むビーカーを水(2×500mL)で洗浄し、洗浄液を該フラスコに加えた。該混合物を室温で10分間攪拌し、約8℃に冷却した。水(500mL)に溶かした(N−メチル)メタンスルホンアミド(200.2g、1.835モル)を5分間かけて加えた。該混合物を、3℃〜4℃で20時間攪拌した。ジクロロメタン(2L)を加え、該混合物を5℃〜10℃で30分間攪拌した。2層を10分間かけて分離させ、採取した。有機層(約2.5L)を12Lフラスコに戻し加えて、1MのH3PO4(800mL)および生理食塩水(800mL)で洗浄した。ジクロロメタンを回転蒸発により除去した。粗生成物にトルエン(400mL)を加え、回転蒸発により除去した。さらに3サイクルのトルエン処理の後、標題中間体を得(228.2g)、これをそのまま精製せずに次のステップに使用した。
【0108】
d.1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成
3Lのフラスコ内に、無水エタノール(400mL)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドトリフルオロアセテート(105.0g、0.232モル)を懸濁させた。この懸濁液に、無水エタノール(100mL)中に溶解させたNaOH(水中50%、0.243モル、1.05当量)を室温で加えた。NaOHを含むビーカーをエタノール(2×50mL)で洗浄し、洗浄液を該反応混合物に加えた。30分の攪拌後、無水エタノール(100mL)中のN−メチル−N−〔(S)−2−オキシラン−2−イルメチル〕メタンスルホンアミド(62.0g、1.5当量)を加えた。該混合物を2時間還流し、室温まで冷却し、標題化合物の種結晶を加えた。約5分の攪拌後、白色固体が形成した。該混合物を3℃〜5℃に冷却し、2時間攪拌した。白色固体をろ過し、湿潤ケークを冷無水エタノール(3×50mL)で洗浄した。該固体を、30℃で60時間、真空下乾燥し、標題化合物(93.8g、カールフィッシャー法による含水量は2.03%)を得た。
【0109】
【数8】
種結晶は、結晶化が自然に生じる、本実施例の方法による標題化合物の先の調製から得られた。
【0110】
(実施例2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成)
1Lフラスコ内に、無水エタノール(210mL)中、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−
2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド(34.7g、0.069モル)を懸濁
させた。室温で濃HCl(1.1当量)を攪拌しながら加えた。該混合物を30分間還流下攪拌し、室温まで冷却させて2時間攪拌した。固体をろ過し、湿潤ケークを冷無水エタノール(3×50mL)で洗浄した。該固体を、30℃で48時間、真空下乾燥し、標題化合物(34.5g、収率93.7%、カールフィッシャー法による含水量は0.13%)を得た。
【0111】
(実施例3:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物合成)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩(139mg、0.28ミリモル)を、注射用滅菌水(2mL)中に溶解させた。数時間経つと、該溶液は混濁した懸濁液になった。該懸濁液を攪拌し、周囲温度で一晩放置すると、白色沈殿が生じた。固体をろ過により採取し、周囲条件(およそ40〜50%の相対湿度)で2分間乾燥して、標題化合物(130mg、収率91%)を得た。
【0112】
(実施例4〜9:本発明の塩形態物の性質)
実施例2のとおり調製した、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(式Iの化合物)の結晶塩酸塩のサンプル、および実施例3のとおり調製した、式Iの化合物の塩酸塩の結晶水和物のサンプルを、粉末x線回折(PXRD)、示差走査熱量測定(DSC)、熱重量分析(TGA)赤外線分光法(IR)および元素分析により分析した。
【0113】
(実施例4:粉末x線回折)
Si(Li)ソリッドステート検出器と共に、1.542Å(45kV、40mA)でのCu Kα放射線を用い、Thermo ARL X−Ray Diffractometer Model XTRA(Thermo ARL SA、スイス国)により、粉末x線回折パターンを得た。この分析は、典型的に、2シータ角における2°から35°の範囲にわたって、1箇所当たり、0.03°の段階サイズで、2°/分の走査速度で実施した。分析のため、受領されたサンプル、または微粉末へと粉砕したサンプルのいずれかを、機器のカップ充填サンプルカップにはめ込むようにデザインされた注文の少容量挿入体内へ静かに入れた。±0.02°の2シータ角以内への機器補正は、シリコン金属標準と比較することにより、毎週検証した。本発明の結晶塩酸塩のサンプルおよび本発明の塩酸塩の水和物のサンプルに関する典型的なPXRDパターンは、それぞれ図1および図4に示されている。
【0114】
(実施例5:熱分析)
TA Instruments Model Q−100モジュールを用いて、示差走査熱量測定(DSC)を実施した。データを採取し、Q Series(商標)ソフトウェア用のTA Instruments Thermal Advantageを用いて分析した。約1mg〜10mgのサンプルを、ふた付きのアルミニウムパン内へ正確に量り入れた。5℃から、典型的には、265℃の、10℃/分の線形加熱ランプを用いて、サンプルを評価した。使用中、DSCセルを乾燥窒素でパージした。化合物Iの結晶塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なDSCトレースは、それぞれ、図2および図5に示されている。
【0115】
TA Instruments Model Q−500モジュールを用いて、熱重量分析(TGA)を実施した。データを採取し、Q Series(商標)ソフトウェア用のTA Instruments Thermal Advantageを用いて分析した。約1mg〜5mgの重量のサンプルを、白金クレードル上のアルミニウムパン内に入れ、10℃/分の線形加熱ランプを用いて、周囲温度から300℃を走査した。使用中、天秤およびファーネスを窒素でパージした。化合物Iの結晶質塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なTGAトレースもまた、それぞれ、図2および図5に示されている。
【0116】
図2のDSCトレースは、本発明の塩酸塩が、約230℃から約260℃の範囲において吸熱性の熱流が最大となる優れた熱安定性を有し、約225℃以下で有意な熱事象を有さないことを実証している。DSCトレースとTGAトレースとの比較により、本発明の塩酸塩が、約230℃以上の温度で、融解と分解を同時に受けることが示されている。
【0117】
当該水和物形態に関する図5におけるDSCトレースにより、約225℃から約250℃の範囲における該結晶が融解する吸熱性の熱流の実質的なピーク、ならびにより低温での幅広い、または弱い吸熱が示されている。DSCトレースとTGAトレースとの比較により、水和物結晶形態の分解は、融解転移の温度において有意ではないことが示されている。
【0118】
(実施例6:動的水分吸収の評価)
VTI大気微量天秤であるSGA−100システム(VTI社、ヒアレー、フロリダ州)を用いて、25℃で、動的水分吸収(DMS)評価を実施した。およそ5mg〜10mgのサンプルサイズを用い、湿度は、分析開始時、周囲の数値に設定した。典型的なDMS分析は、3つの走査:5%RH/段階の走査速度で、周囲湿度から2%相対湿度(RH)、2%RHから90%RH、90%RHから5%RHからなる。2分おきに質量を測定し、サンプルの質量が5つの連続箇所で0.02%以内に安定していた場合はRHを次の値(±5%RH)に変化させる。化合物Iの結晶塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なDMSトレースは、それぞれ、図3および図6に示されている。
【0119】
該塩酸塩は、2%RHから90%RHの範囲全体にわたる0.2%未満の重量変化で、可逆的な吸着/脱離を示す。該水和物形態は、該サンプルを周囲RHから2%RHへと乾燥させた際、乾燥検体が2%RHから40%RHに曝された際の水分率である、約2.3%の重量損失を有する吸着/脱離プロフィルを示す。該水和物形態は、40%RHから75%RHの範囲で、約0.25%未満の重量増加であった。
【0120】
(実施例7:赤外線分析)
Nicolet減衰全反射(ATR)サンプルホールダーを装備したAvatar 360 FT−IR分光器を用いて、4000cm−1から675cm−1の周波数範囲にわたって、赤外線(IR)吸収スペクトルを判定した。本発明の結晶塩酸塩サンプルに関する典型的なIR吸収スペクトルは、758±1、783±1、795±1、802±1、949±1、981±1、1149±1、1158±1、1217±1、1332±1、1377±1、1453±1、1467±1、1487±1、1525±1、1566±1、1575±1、1615±1、1672±1、および3197±1cm−1に有意な吸収バンドを有した。
【0121】
(実施例8:固体状態の安定性評価)
本発明の塩酸塩サンプルを、40℃、75%RHで、複数の開放ガラスバイアルに貯蔵した。特定の間隔で、代表的なバイアルの内容物を取り出し、化学的純度に関して、DSC、TGA、PXRD、およびHPLCにより分析した。貯蔵24週間後、DSCまたはTGAの温熱図にもPXRDパターンにも、検出可能な変化は存在しなかった。該貯蔵サンプルの化学的純度は99.6%であった。
【0122】
(実施例9:対イオンモル比の判定)
塩酸塩(HA)対1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(式Iの化合物)の対イオンモル比を、以下の式に従って算出した:
対イオン比=(WHA/MWHA)/(WI/MWI)
式中、WHAは、サンプル中のHClの重量パーセンテージであり、MWHAは、HClの分子量であり、MWIは、式Iの化合物の分子量(504.6amu)であり、WIは、以下の式に従って算出した、サンプル中の式Iの化合物の重量パーセンテージであり:
WI=100−WHX−WH2O−WRS
化合物Iが不純物を有さないという仮定で、式中、WH2Oは、水分含量の重量パーセンテージであり、WRSは残留溶媒の重量パーセンテージである。
【0123】
本発明の結晶塩酸塩のサンプルに関して、塩酸塩対化合物Iのモル比は、HCl(WHA)の重量パーセンテージの6.3%、および値、WH2O=0.26%およびWRS=0.47%を用いて、0.94:1と算出された。HCl含量は、硝酸銀の標準溶液による滴定により判定し、水分含量WH2Oは、電量的カールフィッシャー滴定により判定し、残留溶媒含量WRSは、ガスクロマトグラフィにより判定した。
【0124】
(比較実施例1:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドのクエン酸塩の合成)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(0.1g、0.2ミリモル)をエタノール(1mL)中に懸濁させた。この懸濁液に、エタノール(0.072mL、0.072ミリモル、0.33当量)に溶かしたクエン酸の1M溶液を加えた。該混合物を、清澄になるまで簡単に超音波処理し、ふたをしてから一晩放置した。ふたを取り、該混合物を、周囲条件下で、固体が見られるまで蒸発させた。次いで、該混合物に再度ふたをして、72時間放置した。得られた固体をろ過し、冷エタノールで洗浄し、標題化合物を固体として得た(74.3mg)。
【0125】
(比較実施例2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの酸性塩の合成)
比較実施例1の手順に従って、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの以下の酸性塩を、以下の酸(二番目の括弧内に生成物の重量)の指示された当量を用いて、固体形態で調製した:アジピン酸(0.5当量)(48.5mg);リン酸(0.5当量)(86.6mg);硫酸(0.5当量)(27.0mg);酒石酸(0.5当量)(66.3mg);リンゴ酸(0.5当量)(25.3mg);および臭化水素酸(1当量)(62.9mg)。
【0126】
(比較実施例3:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドのメタンスルホン酸塩の合成)
50%アセトニトリル/水(1mL)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの溶液に、エタノール(0.2mL、0.2ミリモル、1当量)に溶かしたメタンスルホン酸の1M溶液を加えた。次いで、該混合物を凍結させ、一晩凍結乾燥した。得られた固体を緩やかに温めながらイソプロパノール(1mL)中に溶解させ、冷却させた。得られた固体をろ過により採取し、冷イソプロパノールで洗浄し、標題化合物を固体として得た(90mg)。
【0127】
(比較実施例4:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの酸性塩の合成)
比較実施例3の手順に従って、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの以下の酸性塩を、以下の酸(二番目の括弧内に生成物の重量)の指示された当量を用いて、固体形態で調製した:フマル酸(1当量)(107.2mg);安息香酸(1当量)(105.2mg);および(R)−マンデル酸(1当量)(96.1mg)。
【0128】
(比較実施例5:比較実施例1〜4の化合物の性質)
比較実施例1〜4の式Iの化合物の検出塩酸塩を、PXRD、DSC、およびTGAにより分析した。全ての場合で、約100℃以下の温度で、TGAによる重量損失が見られたが、これは、溶媒の損失に起因する可能性が高いと考えられる。溶媒損失に起因する低温特性を除いて、DSCによって吸熱性熱流が見られた温度、ならびにPXRDによる結晶性の確認を、表Iに要約してあり、ここで、化学量論組成は、該酸性塩の調製に用いられた酸の当量数によって示されている。
【0129】
【表12】
アッセイ1:5−HT4(c)ヒト受容体に対する放射性リガンド結合アッセイ
a.膜調製5−HT4(c)
ヒト5−HT4(c)受容体cDNA(〔3H〕−GR113808膜放射性リガンド結合アッセイを用いて判定したBmax=約6.0pmol/mgタンパク質)を安定トランスフェクトしたHEK−293(ヒト胎児腎臓)細胞を、37℃において、5%CO2、加湿インキュベーター中、T−225フラスコ内で、10%ウシ胎仔血清(FBS)(GIBCO−Invitrogen社:Cat#10437)、2mMのL−グルタミンおよび(100ユニット)ペニシリン−(100μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:Cat#15140)を添加した、4,500mg/LのD−グルコースおよび塩酸ピリドキシン(GIBCO−Invitrogen社、カールスバッド、カリフォルニア州、カタログ番号11965)を含有するダルベッコー修飾イーグル培地(DMEM)中で増殖させた。培地へ800μg/mLのジェネテシン(GIBCO−Invitrogen社:カタログ番号10131)を添加することによって、細胞を継続的な選択圧下で増殖させた。
【0130】
細胞は、およそ60%〜80%の集密度(<35継代培養)まで増殖させた。採取20〜22時間前に、細胞を2回洗浄し、無血清DMEMを供給した。膜調製の全ての処置は氷上で実施した。25mLピペットを用いた緩やかな機械的攪拌と摩砕により、細胞単層を浮揚させた。細胞を1000rpm(5分)での遠心分離により採取した。
【0131】
膜調製のため、細胞ペレットを氷冷した50mMの4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)、pH7.4(膜調製緩衝液)に再懸濁し(30〜40 T225フラスコから40mL/全細胞産出)、氷上、ポリトロンディスラプター(19,2×10秒に設定)を用いて均一化した。得られた均一物を、4℃で5分間、1200gで遠心分離した。ペレットを捨て、上澄み液を、40,000g(20分)遠心分離した。該ペレットは、膜調製緩衝液を用いた再懸濁により1回洗浄し、40,000g(20分)で遠心分離した。最終ペレットを、50mMのHEPES、pH7.4(アッセイ緩衝液)に再懸濁させた(当量1T225フラスコ/1mL)。膜懸濁液のタンパク質濃度は、ブラッドフォードの方法(Bradford、1976年)により判定した。膜は−80℃で凍結保存した。
【0132】
b.放射性リガンド結合アッセイ
0.025%ウシ血清アルブミン(BSA)を含有する50mMのHEPES、pH7.4中、2μgの膜タンパク質を含有する400μLの全アッセイ容量において、1.1mLの96ディープウェルポリプロピレンアッセイプレート(Axygen)中で、放射性リガンド結合アッセイを実施した。0.001nM〜5.0nMの範囲で異なる8〜12の濃度で、〔3H〕−GR11308(Amersham社、バックス、英国:カタログ番号TRK944;比放射能、約82Ci/mmol)を用い、放射性リガンドのKd値決定のための飽和結合試験を実施した。0.15nMでの〔3H〕−GR113808および10pM〜100μMの範囲で異なる11の濃度の化合物により、化合物のpKi値決定のための置換アッセイを実施した。
【0133】
試験化合物は、DMSO中、10mMの原液として受領し、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、同じ緩衝液中に連続(1:5)希釈した。比特異的結合は、1μMの非標識GR113808の存在下で判定した。アッセイは室温で60分間インキュベートし、次いで、0.3%のポリエチレンイミンに予め浸漬した96ウェルGF/Bグラスファイバーフィルタープレート(Packard BioScience社、メリデン、コネチカット州)上での迅速ろ過により、結合反応を終了させた。フィルタープレートをろ過緩衝液(氷冷した50mMのHEPES、pH7.4)で3回洗浄し、非結合の放射能を除去した。プレートを乾燥し、各ウェルに、35μLのMicroscint−20液体シンチレーション液(Packard BioScience社、メリデン、コネチカット州)を加え、Packard Topcount液体シンチレーションカウンター(Packard BioScience社、メリデン、コネチカット州)において、プレートをカウントした。
【0134】
結合データは、1部位競合に関する3−パラメーターモデルを用いて、GraphPad Prism Software package(GraphPad Software社、サンジエゴ、カリフォルニア州)による非線形回帰解析によって解析した。BOTTOM(最小曲線)を、1μMのGR113808の存在下で決定した非特異的結合に関する値に調整した。Cheng−Prusoff式(ChengおよびPrusoff、Biochemical Pharmacology、1973年、22、3099−108頁):Ki=IC50/(1+〔L〕/Kd)、式中、〔L〕=〔3H〕−GR113808濃度、を用い、最良適合したIC50値、および放射性リガンドのKd値から、Prismにおいて、試験化合物に関するKi値を産出した。結果は、Ki値の負の十進法対数、pKiとして表される。
【0135】
このアッセイにおいて、より高いpKi値を有する試験化合物は、5−HT4受容体に対して、より高い結合親和性を有する。式Iの化合物は、このアッセイにおいて、約7.5超のpKi値を有した。
【0136】
アッセイ2:5−HT3Aヒト受容体に対する放射性リガンド結合アッセイ:受容体サブタイプ選択性の判定
a.膜調製5−HT3A
ヒト5−HT3A受容体cDNAを安定トランスフェクトしたHEK−293(ヒト胎児腎臓)細胞を、Michael Bruess博士(ボン大学、東ドイツ)から入手した(〔3H〕−GR65630膜放射性リガンド結合アッセイを用いて判定したBmax=約9.0pmol/mgタンパク質)。37℃において、5%CO2、加湿インキュベーター中、T−225フラスコまたは細胞材料内で、10%熱不活化ウシ胎仔血清(FBS)(Hyclone、ローガン、ユタ州:カタログ番号SH30070.03)、および(50ユニット)ペニシリン−(50μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:カタログ番号15140)を添加した、50%ダルベッコー修飾イーグル培地(DMEM)(GIBCO−Invitrogen社、カールスバッド、カリフォルニア州、カタログ番号11965)および50%ハムF12(GIBCO−Invitrogen社:カタログ番号11765)中で、細胞を増殖させた。
【0137】
細胞は、およそ70%〜80%の集密度(<35継代培養)まで増殖させた。膜調製の全ての処置は氷上で実施した。細胞を採取するために、培地を吸引し、Ca2+、Mg2+のないダルベッコー燐酸緩衝生理食塩水(dPBS)ですすいだ。緩やかな機械的攪拌により、細胞単層を浮揚させた。細胞を1000rpm(5分)での遠心分離により採取した。5−HT4(c)受容体を発現する膜に関して、上記のプロトコルに引き続いて膜調製の工程を行った。
【0138】
b.放射線リガンド結合アッセイ
0.025%BSAアッセイ緩衝液を含有する50mMのHEPES、pH7.4中、1.5〜2μgの膜タンパク質を含有する200μLの全アッセイ容量において、96ウェルポリプロピレンアッセイプレート中で、放射性リガンド結合アッセイを実施した。0.005nM〜20nMの範囲で12の異なる濃度で、〔3H〕−GR65630(PerkinElmer Life Sciences社、ボストン、マサチューセッツ州:カタログ番号NET1011;比放射能、約85Ci/mmol)を用い、放射性リガンドのKd値決定のための飽和結合試験を実施した。0.50nMでの〔3H〕−GR65630および10pMから100μMの範囲で11の異なる濃度の化合物により、化合物のpKi値決定のための置換アッセイを実施した。化合物は、DMSO中、10mMの原液として受領し(節3.1を参照)、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、連続(1:5)希釈してから同じ緩衝液中に作製した。非特異的結合は、10μMの非標識MDL72222の存在下で判定した。アッセイは室温で60分間インキュベートし、次いで、0.3%のポリエチレンイミンに予め浸漬した96ウェルGF/Bグラスファイバーフィルタープレート(Packard BioScience社、メリデン、コネチカット州)上での迅速ろ過により、結合反応を終了させた。フィルタープレートをろ過緩衝液(氷冷した50mMのHEPES、pH7.4)で3回洗浄し、非結合の放射能を除去した。プレートを乾燥し、各ウェルに、35μLのMicroscint−20液体シンチレーション液(Packard BioScience社、メリデン、コネチカット州)を加え、Packard Topcount液体シンチレーションカウンター(Packard BioScience社、メリデン、コネチカット州)において、プレートをカウントした。
【0139】
結合データは、上記の非線形回帰操作を用いて解析し、Ki値を決定した。BOTTOM(最小曲線)を、10μMのMDL72222の存在下で決定した非特異的結合に関する値に調整した。Cheng−Prusoff式中の量〔L〕を、〔3H〕−GR65630濃度として定義した。
【0140】
5−HT4受容体サブタイプに関し、5−HT4受容体サブタイプに対する選択性を、Ki(5−HT3A)/Ki(5−HT4(c))比として産出した。このアッセイにおいて、式Iの化合物は、約1000超の5−HT4/5−HT3受容体サブタイプ選択性を有した。
【0141】
アッセイ3:ヒト5−HT4(c)受容体を発現するHEK−293細胞による細胞全体cAMP蓄積フラッシュアッセイ
このアッセイでは、5−HT4受容体を発現するHEK−293細胞を種々の濃度の試験化合物に接触させた際に産生されるサイクリックAMPの量を測定することにより、試験化合物の機能的効力を判定した。
【0142】
a.細胞培養
クローン化ヒト5−HT4(c)受容体cDNAを安定トランスフェクトした、該受容体を発現するHEK−293(ヒト胎児腎臓)細胞を、2種の密度:(1)〔3H〕−GR113808膜放射性リガンド結合アッセイを用いて判定した約0.5〜0.6pmol/mgタンパク質の密度で;および(2)約6.0pmol/mgタンパク質の密度で調製した。37℃において、5%CO2、加湿インキュベーター中、T−225フラスコ内で、10%ウシ胎仔血清(FBS)(GIBCO−Iんヴぃtろげn社:カタログ番号10437)、および(100ユニット)ペニシリン−(100μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:カタログ番号15140)を添加した、4,500mg/LのD−グルコース(GIBCO−Invitrogen社、カタログ番号11965)を含有するダルベッコー修飾イーグル培地(DMEM)中で、該細胞を増殖させた。培地へジェネテシン(800μg/mL:GIBCO−Invitrogen社:カタログ番号10131)を添加することによって、細胞を継続的な選択圧下で増殖させた。
【0143】
b.細胞調製
細胞は、およそ60%〜80%の集密度まで増殖させた。アッセイの20〜22時間前に、細胞を2回洗浄し、4,500mg/LのD−グルコース(GIBCO−Invitrogen社、カタログ番号11965)を含有する無血清DMEMを供給した。該細胞を採取するために、培地を吸引し、各T−225フラスコに10mLのVersene(GIBCO−Invitrogen社、カタログ番号15040)を加えた。細胞を、室温で5分間インキュベートし、次いで、機械的攪拌によってフラスコから移動させた。該細胞懸濁液を等容量の予備加温した(37℃)dPBSを含有する遠心分離管に移し、1000rpmで5分間遠心分離した。上澄み液を捨て、ペレットを予備加温した(37℃)刺激緩衝液(2〜3のT−225フラスコ当たり、10mM当量)に再懸濁した。この時間を書きとめ、時間ゼロとして印づけた。Coulterカウンターにより細胞をカウントした(8μm以上をカウント、フラスコ収量は、1〜2×107細胞/フラスコ)。予備加温した(37℃)刺激緩衝液(フラッシュプレートキット内で提供されるような)中、5×105細胞/mlの濃度で、細胞を再懸濁させ、37℃で10分間予備インキュベートした。
【0144】
cAMPアッセイは、製造元の指示に従って、125I−cAMP(SMP004B、PerkinElmer Life Sciences社、ボストン、マサチューセッツ
州)により、Flashplate Adenylyl Cyclase Activation Assay Systemを用い、放射免疫アッセイ様式において実施した。
【0145】
細胞は上記のとおり増殖させ、調製した。該アッセイにおける最終細胞濃度は、25×103細胞/ウェルであり、最終アッセイ容量は、100μLであった。試験化合物は、DMSO中、10mMの原液として受領し、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、同じ緩衝液中に連続(1:5)希釈した。10pMから100μM(最終アッセイ濃度)の範囲の異なる11の濃度の化合物により、サイクリックAMP蓄積アッセイを実施した。各プレートについて、5−HT濃度−応答曲線(10pMから100μM)を含めた。該細胞を振とうしながら37℃で15分間インキュベートし、各ウェルに、100μlの氷冷検出用緩衝液(フラッシュプレートキット内で提供されるような)の添加によって該反応を終了させた。該プレートを密封し、4℃で一晩インキュベートした。Topcount(Packard Bioscience社、メリデン、コネチカット州)を用い、シンチレーション近接分光法により、結合した放射能を定量化した。
【0146】
製造元の使用説明書に提供された指示に従って、反応液の1mL当たり産生されたcAMPの量を、cAMP標準曲線から外挿した。3−パラメーターS字形用量応答モデル(単一条件つき傾斜)を用い、GraphPad Prism Softwareパッケージによる非線形回帰解析によって、データを解析した。効力データは、EC50値の負の十進法対数pEC50値として報告され、ここで、EC50は、最大応答の50%に関する有効濃度である。
【0147】
このアッセイでより高いpEC50値を示す試験化合物は、5−HT4受容体に作用するより高い効力を有する。約0.5〜0.6pmol/mgタンパク質の密度を有する細胞系(1)において、このアッセイにおいて試験された式Iの化合物は、約7.5超のpEC50値を有した。
【0148】
アッセイ4:hERG心臓カリウムチャネルを発現する細胞全体におけるカリウムイオン流阻害のインビトロ電圧固定アッセイ
hERGのcDNAを安定トランスフェクトしたCHO−K1細胞は、ウィスコンシン大学のGail Robertsonから入手した。細胞は必要時まで低温保存庫に保持した。10%ウシ胎仔活性および200μg/mLのジェネテシンを添加したダルベッコー修飾イーグル培地/F12中で、細胞を増殖させ、継代した。細胞全体電圧固定試験用に単離細胞が選択され得る濃度で、35mm2のディッシュ(2mLの培地を含有)中、ポリ−D−リシン(100μg/mL)をコーティングしたガラスのカバースリップ上に、細胞を接種した。該ディッシュは、37℃で、加湿、5%CO2の環境中に維持した。
【0149】
少なくとも7日ごとに、細胞外溶液を調製し、使用しない時は4℃に保存した。該細胞外溶液は、以下を含有した(mM):NaCl(137)、KCl(4)、CaCl2(1.8)、MgCl2(1)、グルコース(10)、4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)(10)、NaOHによりpH7.4。試験化合物の不在下または存在下での細胞外溶液をリザーバ内に入れ、そこからおよそ0.5mL/分で、該細胞外溶液を記録チャンバーに流入させた。細胞内溶液を調製し、分画し、使用日まで−20℃で保存した。細胞内溶液は、以下を含有した(mM):KCl(130)、MgCl2(1)、エチレングリコール−ビス(ベータ−アミノエチルエーテル)N,N,N’,N’−テトラ酢酸塩(EGTA)(5)、MgATP(5)、4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)(10)、KOHによりpH7.2。全ての実験は、室温(20〜22℃)で実施した。
【0150】
細胞を接種したカバースリップを記録チャンバーに移し、継続的に潅流した。該細胞とパッチ電極との間にギガオーム密封を形成した。安定なパッチが達成したら、開始保持電位を−80mVで、電圧固定様式で記録を始めた。安定な細胞全体電流の達成後、細胞を試験化合物に曝露させた。標準的な電圧プロトコルは:−80mVの保持電位から+20mVへ4.8秒間、5秒間−50mVに再分極させ、次いで、元の保持電位(−80mV)に戻す段階であった。この電圧プロトコルは15秒ごとに1回行った(0.067Hz)。再分極段階時のピーク電流振幅は、pClampソフトウェアを用いて判定した。細胞上に、3μMの濃度の試験化合物を5分間潅流させ、次いで化合物の不在下で5分間の洗浄時間をとった。最後に、該細胞の機能を試験するために、陽性対照(シサプリド、20nM)を潅流液に加えた。−80mVから+20mVへの段階は、hERGチャネルを活性化し、外向きの電流をもたらす。−50mVへ戻す段階は、該チャネルが不活化から回復し、非活性化するため、外向きのテール電流をもたらす。
【0151】
再分極段階時のピーク電流振幅は、pClampソフトウェアを用いて判定した。対照および試験物のデータは、Origin(登録商標)(OriginLab社、ノーサンプトン、マサチューセッツ州)に送出し、そこで、個々の電流振幅は化合物不在下の最初の電流振幅へと正規化された。各条件に関する正規化電流の平均および標準誤差を算出し、該実験の時間経過に対してプロットした。
【0152】
試験物または媒体対照(通常0.3%DMSO)のいずれかに5分間曝露後、観察されたK+流阻害間で比較を行った。2集団、独立t検定(Microcal Origin
v.6.0)を用いて、実験群間の統計的比較を実施した。p<0.05で差は有意と考えられた。
【0153】
このアッセイにおいて、カリウムイオン流の阻害パーセンテージが小さいほど、治療薬として用いた場合の試験化合物の心臓再分極化パターンを変化させる潜在能力はより小さくなる。このアッセイにおいて、3μMの濃度での式Iの化合物が試験され、約15%未満のカリウムイオン流の阻害を示した。
【0154】
アッセイ5:経口生物学的利用能のインビトロモデル:Caco−2浸透アッセイ
経口投与後、腸を通過して血流に入る試験化合物の能力をモデル化するために、Caco−2浸透アッセイを実施した。ヒト小腸単層の密着した接合を模倣してデザインされた細胞単層を溶液中の試験化合物が浸透する速度を判定した。
【0155】
Caco−2(大腸、腺癌;ヒト)細胞はATCC(American Type Culture Collection;ロックビル、メリーランド州)から入手した。浸透試験では、予め湿らせたトランスウェルポリカーボネートフィルター(Costar;ケンブリッジ、マサチューセッツ州)上に、63,000細胞/cm2の密度で細胞を接種した。培養液中、細胞単層は21日後に形成された。トランスウェルプレート内での細胞培養後、細胞単層を含有する膜をトランスウェルプレートから引き離し、拡散チャンバー(Costar;ケンブリッジ、マサチューセッツ州)内に挿入した。該拡散チャンバーを、温度制御用に循環外部サーモスタット制御の37℃水を備えた加熱ブロック内に挿入した。空気多枝管により拡散チャンバーの各半分に95%O2/5%CO2が送達され、攪拌されない境界層の減少に効果的な、細胞単層を横切る層流を作り出した。
【0156】
100μMの試験化合物濃度、および単層の完全性をモニターするための14C−マンニトールによって、浸透試験を実施した。全ての実験が、37℃で60分間行われた。チャンバーの供給側と受取側の双方から、0分、30分および60分でのサンプルを採取した。試験化合物およびマンニトールの濃度に関して、サンプルをHPLCまたは液体シンチレーションカウンティングにより分析した。cm/秒での浸透係数(Kp)を算出した。
【0157】
このアッセイにおいて、約10×10−6cm/秒超のKp値が好適な生物学的利用能を示すと考えられる。式Iの化合物をこのアッセイにおいて試験すると、約20×10−6cm/秒超のKp値を示した。
【0158】
アッセイ6:ラットにおける薬物動態試験
約5と約6の間のpHで、0.1%乳酸中、試験化合物の水溶液製剤を調製した。オスのSDラット(CD株、Charles River Laboratories、ウィルミントン、マサチューセッツ州)に、2.5mg/kgの用量で静脈内投与(IV)により、または5mg/kgの用量で経口胃管(PO)により、試験化合物を投与した。投与容量は、IVでは1mL/kg、PO投与では、2mL/kgであった。動物から、投与前に、ならびに投与後2分(IVのみ)、5分、15分、および30分、1時間、2時間、4時間、8時間、および24時間目に、連続血液サンプルを採取した。血漿中の試験化合物濃度は、1ng/mLの定量化下限で、液体クロマトグラフィ−質量分光分析(LC−MS/MS)(MDS SCIEX、API4000、Applied Biosystems、Foster City、カリフォルニア州)により判定した。
【0159】
標準的な薬物動態パラメーターは、WinNnlin(4.0.1版、Phrsight、マウンテンビュー、カリフォルニア州)を用いて非区画分析(IVにはモデル201、POにはモデル200)により評価した。血漿中試験化合物濃度の曲線における最大値対時間は、Cmaxと表される。曲線下面積対投与時間から最後の測定可能濃度までの時間曲線(AUC(0−t))を、線形台形則により算出した。経口生物学的利用能、すなわち、IV投与のAUC(0−t)に対するPO投与のAUC(0−t)の用量正規化比は:
F(%)=AUCPO/AUCIV×用量IV/用量PO×100%
として算出した。
【0160】
このアッセイにおいて、パラメーターCmax、AUC(0−t)、およびF(%)のより大きな値を示す試験化合物は、経口投与された際により大きな生物学的利用能を有することが予想される。式Iの化合物は、0.16μg/mLのCmax値、0.46μg・時間/mLおよびラットモデルにおける約19%の生物学的利用能(F(%))を有した。
【0161】
本発明をその特定の実施形態を参照して説明したが、本発明の真の精神および範囲から逸脱することなく、種々の変更ができ、等価物に置換できることは、当業者により理解されるはずである。また、本発明の目的、精神および範囲に対して、特定の状況、材料、物質組成、方法、処理、処理工程または工程に適合させるために、多くの修飾をなすことができる。このような修飾は全て、本明細書に添付した請求項の範囲内にあることが意図されている。また、本明細書において上記で引用した全ての刊行物、特許、および特許文書は、個々に参照として組み込まれているように、全体が本明細書に参照として組み込まれている。
【技術分野】
【0001】
(発明の分野)
本発明は、5−HT4受容体アゴニストとして有用なキノリノン−カルボキサミド化合物の結晶塩形態物に関する。本発明はまた、このような結晶化合物を含む製薬組成物、5HT4受容体活性に媒介された病状を治療するためにこのような化合物を使用する方法、およびこのような化合物の調製に有用な方法に関する。
【背景技術】
【0002】
(当業界の状況)
本願と同一の譲受人に譲渡された2004年4月7日出願の米国特許仮出願第60/560,076号、および2005年4月6日出願の米国特許出願第11/100,113号は、胃腸管運動性低下の疾患の治療に有用であると予想される、新規なキノリノン−カルボキサミド化合物を開示している。特に、化合物、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−
ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、5−HT4受容体アゴニスト活性を示すも
のとして、上記出願に具体的に開示されている。1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒド
ロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの化学構造は式Iにより表される:
【0003】
【化1】
治療薬としてこの化合物を効果的に使用するために、容易に製造でき、許容できる化学的および物理的安定性を有する固体状態の塩形態物を有することが望ましいと考えられる。例えば、熱的に安定、例えば、約200℃を越える温度で安定であり、吸湿性でも潮解性でもなく、そのため処理および貯蔵が容易な塩形態物を有することがきわめて望ましいと考えられる。結晶固体は、製造品の純度および安定性が増加するので、非結晶形態物よりも一般的に好ましい。
【発明の概要】
【発明が解決しようとする課題】
【0004】
式Iの化合物の非結晶塩形態物についてはまだ報告されていない。したがって、吸湿性でも潮解性でもなく、好適な熱的安定性を示す式Iの化合物の安定な結晶塩形態物が必要とされている。
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。一態様において、本発明の結晶塩形態物は、式Iの化合物の結晶塩酸塩である。他の態様において、本発明の結晶塩形態物は、式Iの化合物の塩酸塩の結晶水和物である。
【0006】
驚くべきことに、本発明の結晶塩酸塩は、約200℃を越える温度で熱的に安定であり、室温で約2%と約90%との間の範囲の相対湿度に晒された場合に示す重量変化が約0.2%未満であることがわかった。さらに、本発明の結晶塩酸塩も、その水和物も、室温で90%までの相対湿度に晒された場合、潮解性ではない。
【0007】
他の使用法として、本発明の結晶塩形態物は、胃腸管の運動低下疾患を治療するための製薬組成物の調製に有用であることが予想される。したがって、その組成物の他の態様において、本発明は、製薬的に許容できる担体および1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を含む製薬組成物を提供する。
【0008】
本発明はまた、5−HT4受容体活性に関連した疾患または病態、例えば、胃腸管の運動低下疾患を治療する方法を提供し、該方法は、哺乳動物に、治療的有効量の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶質塩酸塩またはその溶媒和物を投与することを含む。
【0009】
他の方法態様において、本発明は、本発明の結晶塩酸塩を調製するための方法を提供し、該方法は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドを塩酸と接触させて反応混合物を形成し、該反応混合物から該結晶塩酸塩を単離することを含む。
本発明はまた、療法において、または薬剤として使用するため、本明細書に記載された本発明の結晶塩酸塩、ならびに、薬剤の製造において、特に哺乳動物における胃腸管の運動低下疾患を治療するための薬剤製造に関して、本発明の結晶塩酸塩の使用法を提供する。
例えば、本発明は以下の項目を提供する。
(項目1)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩またはその溶媒和物である、結晶塩形態物。
(項目2)
前記結晶塩形態物が結晶塩酸塩である、項目1に記載の結晶塩形態物。
(項目3)
前記結晶塩形態物が、4.41±0.2、8.82±0.2、9.08±0.2、11.21±0.2、14.40±0.2、16.42±0.2、17.35±0.2、17.61±0.2、18.14±0.2、19.04±0.2、19.95±0.2、20.20±0.2、21.23±0.2、22.13±0.2、22.48±0.2、22.83±0.2、24.16±0.2、25.37±0.2、25.56±0.2、26.22±0.2、27.33±0.2、29.08±0.2、および29.61±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする、項目2に記載の結晶塩形態物。
(項目4)
前記粉末x線回折パターンが、14.40±0.2、17.35±0.2、17.61±0.2、19.04±0.2、21.23±0.2、および22.13±0.2から選択される2θ値における2つ以上の回折ピークを含む、項目3に記載の結晶塩形態物。
(項目5)
前記結晶塩形態物は、ピーク位置が図1に示されたパターンのピーク位置に実質的に従う粉末x線回折パターンを特徴とする、項目2に記載の結晶塩形態物。
(項目6)
前記結晶塩形態物が、約230℃超の温度において、吸熱性熱流の最大値を示す示差走査熱量測定トレースを特徴とする、項目2に記載の結晶塩形態物。
(項目7)
前記結晶塩形態物が、図2に示された示差走査熱量測定トレースに実質的に従う示差走査熱量測定トレースを特徴とする、項目2に記載の結晶塩形態物。
(項目8)
前記結晶塩形態物が水和物である、項目1に記載の結晶塩形態物。
(項目9)
前記結晶塩形態物が、5.30±0.2、7.43±0.2、8.72±0.2、10.52±0.2、13.85±0.2、14.11±0.2、15.80±0.2、15.99±0.2、17.26±0.2、19.53±0.2、20.08±0.2、21.06±0.2、21.48±0.2、21.92±0.2、22.85±0.2、23.91±0.2、25.28±0.2、26.06±0.2、27.34±0.2、27.51±0.2、および29.67±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする、項目8に記載の結晶塩形態物。
(項目10)
前記粉末x線回折パターンが、10.52±0.2、13.85±0.2、15.80±0.2、17.26±0.2、および21.06±0.2から選択される2θ値における2つ以上の回折ピークを含む、項目9に記載の結晶塩形態物。
(項目11)
前記結晶塩形態物は、ピーク位置が図4に示されたパターンのピーク位置に実質的に従う粉末x線回折パターンを特徴とする、項目8に記載の結晶塩形態物。
(項目12)
前記結晶塩形態物が、図5に示された示差走査熱量測定トレースに実質的に従う示差走査熱量測定トレースを特徴とする、項目8に記載の結晶塩形態物。
(項目13)
製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む製薬組成物。
(項目14)
5−HT4受容体活性に関連した病状を有する哺乳動物を治療する方法であって、該方法は、治療的有効量の製薬組成物を前記哺乳動物に投与することを含み、該製薬組成物は、製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む、方法。
(項目15)
哺乳動物における胃腸管の運動低下障害を治療する方法であって、該方法は、治療的有効量の製薬組成物を前記哺乳動物に投与することを含み、該製薬組成物は、製薬的に許容できる担体および項目1から12のいずれか一項に記載の結晶塩形態物を含む、方法。
(項目16)
前記運動低下障害が、慢性便秘症、便秘優勢過敏性腸症候群、糖尿病性胃疾患および特発性胃疾患、ならびに機能性消化不良症から選択される、項目15に記載の方法。
(項目17)
治療において使用するための、項目1から12のいずれか一項に記載の結晶塩形態物。
(項目18)
薬剤を製造するための、項目1から12のいずれか一項に記載の結晶塩形態物の使用。
(項目19)
前記薬剤が、5−HT4受容体活性に関連した、哺乳動物における病状を治療するための薬剤である、項目18に記載の使用。
(項目20)
前記病状が、胃腸管の運動低下障害である、項目19に記載の使用。
(項目21)
前記病状が、慢性便秘症、便秘優勢過敏性腸症候群、糖尿病性胃疾患および特発性胃疾患、ならびに機能性消化不良症から選択される、項目19に記載の使用。
(項目22)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩を調製するためのプロセスであって、該プロセスは、以下:
(a)1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドと塩酸とを接触させて、反応混合物を形成すること;および
(b)前記反応混合物から前記結晶塩酸塩を単離すること、
を含む、プロセス。
(項目23)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩の結晶水和物を調製するためのプロセスであって、該プロセスは、以下:
(a)1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩を、1ミリリットル当たり約50ミリグラム超の濃度で水に溶解させ、懸濁液を形成すること;および
(b)前記懸濁液から前記結晶水和物を単離すること、
を含む、プロセス。
【図面の簡単な説明】
【0010】
本発明の種々の態様は、添付の図面を参照して例示される。
【図1】図1は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶質塩酸塩の粉末x線回折(PXRD)パターンを示している。
【図2】図2は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩に関する示差走査熱量測定(DSC)トレース(ボトムトレース、右側縦軸)および熱重量分析(TGA)トレース(トップ追跡、左側縦軸)を示している。
【図3】図3は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩に関する動的水分吸収(DMS)を示している。
【図4】図4は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの塩酸塩の結晶水和物の粉末x線回折(PXRD)パターンを示している。
【図5】図5は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物に関する示差走査熱量測定(DSC)トレース(トップトレース、左側縦軸)および熱重量分析(TGA)トレース(ボトムトレース、右側縦軸)を示している。
【図6】図6は、本発明の1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物に関する動的水分吸収(DMS)を示している。
【発明を実施するための形態】
【0011】
(発明の詳細な説明)
本発明は、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩またはその溶媒和物を提供する。
【0012】
定義
本発明の化合物、組成物および方法を記述する際、以下の用語は、別に指示されない限り、以下の意味を有する。
【0013】
用語の「治療的有効量」は、治療を必要としている患者に投与した際に治療をもたらす上で十分な量を意味する。
【0014】
本明細書に用いられる用語の「治療」は、哺乳動物(特にヒト)などの患者における疾患、障害、または病状の治療を意味し、以下を含む:
(a)疾患、障害、または病状の発現の予防、すなわち、患者の予防的処置;
(b)疾患、障害、または病状の寛解、すなわち、患者における疾患、障害、または病状を除去することまたは退行を生じさせること;
(c)疾患、障害、または病状の抑制、すなわち、患者における疾患、障害、または病状の発現の緩徐化または阻止;または
(d)患者における疾患、障害、または病状の症状の軽減。
【0015】
用語の「溶媒和物」は、一種または複数種の溶質分子、すなわち、本発明の化合物または製薬的に許容できるその塩および一種または複数種の溶媒分子により形成された複合体または凝集体を意味する。このような溶媒和物は、溶質と溶媒の実質的に一定のモル比を有する、典型的に結晶質の固体である。代表的な溶媒としては、例えば、水、メタノール、エタノール、イソプロパノール、酢酸などが挙げられる。溶媒が水の場合、形成された溶媒和物は特に水和物と称される。
【0016】
本明細書に用いられる用語の「結晶塩酸塩」は、結晶格子中に溶媒分子の実質的に一定のモル分率を含まない結晶質固体、すなわち、溶媒和物ではないものを意味する。本発明の溶媒和物、または特に水和物は明白に同定される。
【0017】
本明細書および添付の項目に用いられる単数形の「1つの」、「1個の」、「1つ」、および「前記」は、その内容が明らかに別を指示しない限り、複数の記述を含み得る。
【0018】
用語の「アミノ保護基」は、アミノ窒素における望ましくない反応を防ぐために好適な保護基を意味する。代表的なアミノ保護基としては、限定はしないが、ホルミル;アシル基、例えば、アセチルなどのアルカノイル基;tert−ブトキシカルボニル(Boc)などのアルコキシカルボニル基;ベンジルオキシカルボニル(Cbz)および9−フルオレニルメトキシカルボニル(Fmoc)などのアリールメトキシカルボニル基;ベンジル(Bn)、トリチル(Tr)、および1,1−ジ−(4’−メトキシフェニル)メチルなどのアリールメチル基;トリメチルシリル(TMS)およびtert−ブチルジメチルシリル(TBDMS)などのシリル基;などが挙げられる。
【0019】
活性剤
本発明の塩形態物、すなわち、式Iの化合物は、市販のAutoNomソフトウェア(MDL Information Systems、GmbH、フランクフルト、ドイツ国)を用いて、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドと命名される。(1S、3R、5R)の呼称は、二環式環系に結合した結合の相対的な方向を言う。あるいは、該化合物は、N−〔(3−エンド)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル〕−1−(1−メチルエチル)−2−オキソー1,2−ジヒドロ−3−キノリンカルボキサミドと表される。
【0020】
本発明の塩形態物
一態様において、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの結晶塩酸塩を提供する。
【0021】
本発明の結晶塩酸塩は、式Iの化合物の1モル当量当たり、約0.9モル当量と約1.1モル当量との間など、式Iの化合物の1モル当量当たり、約0.8モル当量と約1.2モル当量との間の塩酸を典型的に含有する。
【0022】
活性剤に対する塩酸のモル比は、当業者に利用できる方法によって容易に決定できる。例えば、このようなモル比は、硝酸銀の標準溶液による滴定によって容易に決定できる。あるいは、該モル比を決定するために、元素分析、1H NMR、およびイオンクロマトグラフィ法が使用できる。
【0023】
一態様において、本発明の結晶塩酸塩は、4.41±0.2、8.82±0.2、9.08±0.2、11.21±0.2、14.40±0.2、16.42±0.2、17.35±0.2、17.61±0.2、18.14±0.2、19.04±0.2、19.95±0.2、20.20±0.2、21.23±0.2、22.13±0.2、22.48±0.2、22.83±0.2、24.16±0.2、25.37±0.2、25.56±0.2、26.22±0.2、27.33±0.2、29.08±0.2、および29.61±0.2から選択される2θ値における2つ以上の回折ピークを有する粉末x線回折(PXRD)パターンを特徴とする。この態様において、該結晶形態物は特に、14.40±0.2、17.35±0.2、17.61±0.2、19.04±0.2、21.23±0.2、および22.13±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする。
【0024】
粉末x線回折の分野で十分知られているように、PXRDスペクトルのピーク位置は、サンプル調製および機器の幾何学的配置などの詳細などの実験的詳細に対して、相対的ピーク高さよりも相対的に感受性が低い。したがって、一態様において、式Iの化合物の結晶塩酸塩は、ピーク位置が実質的に図1に示されたものによる粉末x線回折パターンを特徴とする。
【0025】
また、本発明の結晶塩酸塩は、図2にしたがって、示されたように、約230℃から約260℃の範囲の吸熱性の熱流におけるピークを示す示差走査熱量測定(DSC)トレースにより実証される高温熱的安定性を特徴とする。さらに、熱重量分析(TGA)トレースで、約225℃以下での有意な熱的事象は示されない。
【0026】
さらに他の態様において、結晶塩酸塩は、約758、783、795、802、949、981、1149、1158、1217、1332、1377、1453、1467、1487、1525、1566、1575、1615、1672、および3197cm−1における有意な吸収バンドを示す赤外線吸収スペクトルを特徴とする。
【0027】
式Iの化合物の結晶塩酸塩は、図3に示されるように、例外的に低レベルの吸湿性(すなわち、室温で、2%相対湿度から90%相対湿度の湿度範囲で、約0.2%未満の重量増加)の可逆的吸着/脱離プロフィルを有することが実証されている。
【0028】
また、式Iの化合物の結晶塩酸塩は、長期間、高温および高湿に曝された際に安定であることが判明している。例えば、40℃および相対湿度75%で24週間貯蔵後のHPLCで、化学的分解は示されず、DSC、TGA、またはPXRDの結果にも検出できる変化はなかった。
【0029】
他の態様において、本発明は、式Iの化合物の塩酸塩の結晶水和物を提供する。
【0030】
一態様において、本発明の塩酸塩の結晶水和物は、5.30±0.2、7.43±0.2、8.72±0.2、10.52±0.2、13.85±0.2、14.11±0.2、15.80±0.2、15.99±0.2、17.26±0.2、19.53±0.2、20.08±0.2、21.06±0.2、21.48±0.2、21.92±0.2、22.85±0.2、23.91±0.2、25.28±0.2、26.06±0.2、27.34±0.2、27.51±0.2、および29.67±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折(PXRD)パターンを特徴とする。この態様において、該結晶形態物は特に、10.52±0.2、13.85±0.2、15.80±0.2、17.26±0.2、および21.06±0.2から選択される、2θ値における2つ以上の回折ピークを有する粉末x線回折パターンを特徴とする。
【0031】
他の態様において、式Iの化合物の塩酸塩の結晶水和物は、ピーク位置が実質的に図4に示されたものによる粉末x線回折パターンを特徴とする。
【0032】
本発明の塩酸塩の結晶水和物はまた、図5に示されるように、約225℃から約250℃の範囲での結晶の融解で確認される吸熱性の熱流における実質的ピーク、および低温での幅広いまたは弱い吸熱を示す示差走査熱量測定(DSC)トレースを特徴とする。さらに、熱重量分析(TGA)トレースで、分解温度は約250℃以上であることが示されている。
【0033】
式Iの化合物の塩酸塩の結晶水和物は、図6に示されるように、約2%から約90%の相対湿度の全範囲にわたって、室温で可逆的吸着/脱離プロフィルを有することが実証されている。該結晶水和物は、約40%と約75%との間の相対湿度で、約0.25%未満の重量増加を示す。
【0034】
本発明のこれらの性質は、以下の実施例においてさらに例示されている。
【0035】
合成法
活性剤、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、下記の実施例に記載された方法を用いて、または本出願の背景の節に挙げられた、本願と同一の譲受人に譲渡された米国特許出願に記載された方法を用いて、容易に入手できる出発材料から調製できる。
【0036】
本発明の結晶質塩酸塩を調製するために、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドを、典型的に、約1モル当量から約1.2モル当量などの約1モル当量から約1.5モル当量の濃塩酸に接触させる。一般的にこの反応は、約20℃から約80℃の範囲の温度で、不活性希釈剤中で行われる。この反応に好適な不活性希釈剤としては、限定はしないが、エタノール、メタノール、イソプロパノール、酢酸エチル、アセトニトリル、トルエン、テトラヒドロフラン、およびそれらの組み合わせが挙げられる。
【0037】
反応が完了したら、本発明の結晶塩を、沈殿、濃縮、遠心分離などの任意の慣例的手段により、該反応混合物から単離する。
【0038】
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S
,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩を、約50mg/mL以上の濃度で水に溶解することにより、結晶水和物を調製でき、これにより提供される懸濁液から、得られた結晶水和物を慣例的手段により単離できる。
【0039】
製薬組成物
本発明の結晶塩酸塩形態物は典型的に、製薬組成物の形態で患者に投与される。このような製薬組成物は、限定はしないが、経口、経直腸、経膣、経鼻、吸入、局所(経皮を含む)および非経口の投与様式など、任意の許容できる投与経路によって患者に投与できる。
【0040】
したがって、本発明は、その組成物態様の1つにおいて、製薬的に許容できる担体または賦形剤ならびに式Iの化合物の結晶塩酸塩の治療的有効量を含む製薬組成物に関する。任意に、所望の場合、このような製薬組成物は、他の治療的および/または製剤化剤を含有してもよい。
【0041】
本発明の製薬組成物は典型的に、本発明の結晶塩の治療的有効量を含有する。典型的に、このような製薬組成物は、約5重量%から約60重量%の活性剤など、約1重量%から約70重量%の活性剤を含め、約0.1重量%から約95重量%の活性剤を含有する。
【0042】
本発明の製薬組成物中には、任意の慣例的な担体または賦形剤を使用できる。具体的な担体または賦形剤、もしくは担体または賦形剤の組み合わせの選択は、具体的な患者または病状または疾患状態のタイプの治療に用いられる投与様式に依存する。これに関して、具体的な投与様式に対して好適な製薬組成物の調製は、十分に製薬業界者の範囲内にある。また、そのような組成物のための成分は、例えば、Sigma、P.O.Box 14508、セントルイス、ミズーリ州 63178から市販されている。さらに例えば、慣例的な製剤化法は、Remington:The Science and Practice of Phrmacy、第20版、Lippincott Williams & White、バルチモア、メリーランド州(2000);およびH.C.Anselら、Pharmaceutical Dosage Forms and Drug Delivery Systems、第7版、Lippincott Williams & White、バルチモア、メリーランド州(1999)に記載されている。
【0043】
製薬的に許容できる担体として役立ち得る材料の代表例としては、限定はしないが、以下のものが挙げられる:(1)ラクトース、グルコース、およびスクロースなどの糖類;(2)トウモロコシ澱粉およびジャガイモ澱粉などの澱粉類;(3)微結晶セルロース、およびその誘導体など、カルボキシメチルセルロースナトリウム、エチルセルロースおよびセルロースアセテートなどのセルロース;(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)カカオバターおよび座剤ワックスなどの賦形剤;(9)落花生油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油および大豆油などの油類;(10)プロピレングリコールなどのグリコール類;(11)グリセリン、ソルビトール、マンニトールおよびポリエチレングリコールなどのポリオール類;(12)オレイン酸エチルおよびラウリン酸エチルなどのエステル類;(13)寒天;(14)水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;(15)アルギン酸;(16)パイロジェンのない水;(17)等張生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸緩衝液;および(21)製薬組成物に用いられる他の非毒性適合性物質。
【0044】
本発明の製薬組成物は、本発明の化合物を、製薬的に許容できる担体および一種または複数種の任意の成分と、十分に完全に混合またはブレンドすることによって典型的に調製される。必要または所望の場合、得られた均一にブレンドされた混合物を次に、慣例的な方法および装置を用いて、錠剤、カプセル剤、丸剤などに形状化または装填することができる。
【0045】
本発明の製薬組成物は、単位投与量形態にパッケージ化することが好ましい。用語の「単位投与量形態」とは、患者への投与に好適な物理的に個別の単位、すなわち、単独で、または1つまたは複数の追加の単位と組み合わせて、所望の治療的効果を生じさせるように計算された活性剤の予め決められた量を含有する各単位を言う。例えば、このような単位投与量形態は、カプセル剤、錠剤、丸剤などであり得る。
【0046】
好ましい一実施形態において、本発明の製薬組成物は、経口投与に好適である。経口投与に好適な製薬組成物は、カプセル剤、錠剤、丸剤、舐剤、カシェ剤、糖衣剤、散剤、顆粒剤の形態であり得るか;または水性もしくは非水性液体中の液剤もしくは懸濁剤;または水中油もしくは油中水液乳剤;またはエリキシル剤またはシロップ剤;などであり得;各々が活性剤として本発明の化合物の予め決められた量を含有する。
【0047】
固体投与形態(すなわち、カプセル剤、錠剤、丸剤など)における経口投与が意図される場合、本発明の製薬組成物は典型的に、活性剤としての本発明の化合物、およびクエン酸ナトリウムまたはリン酸二カルシウムなどの、一種または複数種の製薬的に許容できる担体を含む。任意に、または代替として、このような固体投与形態は、以下のものもまた含む;(1)澱粉、微結晶セルロース、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;(2)カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアラビアゴム;(3)グリセロールなどの湿潤剤;(4)寒天、炭酸カルシウム、ジャガイモ澱粉またはタピオカ澱粉、アルギン酸、一定のケイ酸塩、および/または炭酸ナトリウムなどの崩壊剤;(5)パラフィンなどの溶解遅延剤;(6)第四級アンモニウム化合物などの吸収促進剤;(7)セチルアルコールおよび/またはモノステアリン酸グリセロールなどの浸潤剤;(8)カオリンおよび/またはベントナイトなどの吸収剤;(9)タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはそれらの混合物;(10)着色剤;および(11)緩衝剤。
【0048】
本発明の製薬組成物中には、放出剤、浸潤剤、コーティング剤、甘味剤、風味剤ならびに香料、保存剤および抗酸化剤もまた存在し得る。製薬的に許容できる抗酸化剤の例としては以下のものが挙げられる:(1)アスコルビン酸、塩酸システイン、硫酸二ナトリウム、異性重硫酸ナトリウム重亜硫酸ナトリウムなどの水溶性抗酸化剤;(2)パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、プロピルガレート、アルファ−トコフェロールなどの油溶性抗酸化剤;(3)クエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート化剤。錠剤、カプセル剤、丸剤などのためのコーティング剤としては、セルロースアセテートフタレート(CAP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピルメチルセルロースフタレート、メタクリル酸−メタクリル酸エステルコポリマー、セルロースアセテートトリメリテート(CAT)、カルボキシメチルエチルセルロース(CMEC)、ヒドロキシプロピルメチルセルロースアセテートスクシネート(HPMCAS)など、腸溶コーティングに用いられるものが挙げられる。
【0049】
所望の場合、本発明の製薬組成物は、例えば、種々の比率でのヒドロキシメチルセルロース;または他のポリマーマトリックス、リポソームおよび/またはミクロスフェアを用いて、活性剤の緩徐な、または制御された放出を提供するように製剤化することもできる。
【0050】
また、本発明の製薬組成物は、任意に、隠蔽剤を含有でき、活性成分を、胃腸管のある一定の部分でのみ、または優先的に、任意に遅延様式で、放出するように製剤化できる。使用できる包埋組成物の例としては、ポリマー物質およびワックスが挙げられる。活性成分はまた、適切な場合、上記の賦形剤の一種または複数種と共に、マイクロカプセル化形態においてあり得る。
【0051】
経口投与用の好適な液体投与形態としては、例えば、製薬的に許容できる乳剤、マイクロ乳剤、液剤、懸濁剤、シロップ剤およびエリキシル剤が挙げられる。このような液体投与形態は典型的に、活性成分、ならびに、例えば、水または他の溶媒などの不活性希釈剤、可溶化剤、およびエチルアルコール、イソプロピルアルコール、エチルカルボネート、エチルアセテート、ベンジルアルコール、ベンジルベンゾエート、プロピレングリコール、1,3−ブチレングリコール、油類(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコール類およびソルビタンの脂肪酸エステル類およびそれらの混合物などの乳化剤を含む。懸濁剤は、活性成分に加えて、例えば、エトキシル化イソステアリルアルコール類、ポリオキシエチレンソルビトールならびにソルビタンエステル類、微結晶セルロース、アルミニウムメタヒドロキシド、ベントナイト、寒天およびトラガカント、ならびにそれらの混合物などの懸濁化剤を含有できる。
【0052】
あるいは、本発明の製薬組成物は、吸入による投与のために製剤化される。吸入による投与のための好適な製薬組成物は典型的に、エアロゾルまたは粉末の形態にある。このような組成物は一般的に、用量計測吸入器、ドライパウダー吸入器、ネブライザーまたは類似の送達デバイスなど、周知の送達デバイスを用いて投与される。
【0053】
減圧容器を用いて吸入により投与される場合、本発明の製薬組成物は典型的に、活性成分、ならびにジクロロジフルオロメタン、トリクロロフルオロメタン、ジ
クロロテトラフルオロエタン、二酸化炭素または他の好適な気体などの好適な噴射剤を含む。
【0054】
また、該製薬組成物は、本発明の化合物、ならびに粉末吸入器における使用にとって好適な粉末を含むカプセル剤またはカートリッジの形態にあってもよい。好適な粉末基材としては、例えば、ラクトースまたは澱粉が挙げられる。
【0055】
本発明の化合物はまた、公知の経皮送達系および賦形剤を用いて、経皮投与もできる。例えば、本発明の化合物を、プロピレングリコール、ポリエチレングリコールモノラウレート、アザシクロアルカン−2−オン類などの浸透増強剤と混合することができる。所望の場合、このような経皮組成物中に、ゲル化剤、乳化剤および緩衝剤などの追加の賦形剤を使用できる。
【0056】
以下の製剤は、本発明の典型的な製薬組成物を例示している。
【0057】
製剤例A
経口投与用のハードゼラチンカプセル剤は以下のとおり調製される:
【0058】
【表1】
典型的操作:成分を完全に混合してからハードゼラチンカプセルに充填する(1カプセル当たり260mgの組成物)。
【0059】
製剤例B
経口投与用のハードゼラチンカプセル剤は以下のとおり調製される:
【0060】
【表2】
典型的操作:成分を完全に混合してから、45号メッシュU.S.ふるいを通して、ハードゼラチンカプセルに充填する(1カプセル当たり200mgの組成物)。
【0061】
製剤例C
経口投与用のカプセル剤は以下のとおり調製される:
【0062】
【表3】
典型的操作:成分を完全に混合してからゼラチンカプセルに充填する(1カプセル当たり310mgの組成物)。
【0063】
製剤例D
経口投与用の錠剤は以下のとおり調製される:
【0064】
【表4】
典型的操作:該活性成分、澱粉およびセルロースを45番メッシュU.S.ふるいに通し、完全に混合する。得られた粉末とポリビニルピロリドンの溶液とを混合し、次いで、この混合物を14番メッシュU.S.ふるいに通す。このようにして作製された顆粒を50〜60℃で乾燥し、18番メッシュU.S.ふるいに通す。次いで、この顆粒に、カルボキシメチルセルロース澱粉、ステアリン酸マグネシウムおよびタルク(先に60番メッシュU.S.ふるいに通した)を加える。混合後、該混合物を錠剤機で圧縮し100mgの重量の錠剤を得る。
【0065】
製剤例E
経口投与用錠剤は以下のとおり調製される:
【0066】
【表5】
典型的操作:該成分を完全に混合してから圧縮して錠剤を形成する(1錠当たり440mgの組成物)。
【0067】
製剤例F
経口投与用単一刻み目入り錠剤は以下のとおり調製される:
【0068】
【表6】
典型的操作:該成分を完全に混合し、圧縮して単一刻み目入り錠剤を形成する(1錠当たり215mgの組成物)。
【0069】
製剤例G
経口投与用懸濁剤は以下のとおり調製される:
【0070】
【表7】
典型的操作:該成分を混合して、懸濁液10mL当たり活性成分10mgを含有する懸濁剤を形成する。
【0071】
製剤例H
吸入による投与のためのドライパウダーは以下のとおり調製される:
【0072】
【表8】
典型的操作:該活性成分を超微粉砕してからラクトースと混合する。次いで、このブレンドした混合物をゼラチン吸入カートリッジ内へ充填する。粉末吸入器を用いて、該カートリッジの内容物を投与する。
【0073】
製剤例I
計測用量吸入器内の吸入による投与のためのドライパウダーは以下のとおり調製される:
典型的操作:200mLの脱塩水中に溶解させた0.2gのレシチンから形成した溶液中に、10μm未満の平均サイズを有する超微粉砕粒子としての活性化合物10gを分散させることによって、本発明の塩を5重量%およびレシチンを0.1重量%含有する懸濁液を調製する。該懸濁液をスプレー乾燥し、得られた物質を、1.5μm未満の平均直径を有する粒子へと超微粉砕する。該粒子を、加圧した1,1,1,2−テトラフルオロエタンと共にカートリッジ内へ充填する。
【0074】
製剤例J
注射用製剤は以下のとおり調製される:
【0075】
【表9】
典型的操作:上記成分を混合し、0.5N HClまたは0.5N NaOHを用いて、pHを4±0.5に調整する。
【0076】
製剤例K
経口投与用のカプセル剤は以下のとおり調製される:
【0077】
【表10】
典型的操作:該成分を完全に混合してから、ゼラチンカプセル(サイズ#1、白、不透明)内へ充填する(1カプセル当たり264mgの組成物)。
【0078】
製剤例L
経口投与用のカプセル剤は以下のとおり調製される:
【0079】
【表11】
典型的操作:該成分を完全に混合してから、ゼラチンカプセル(サイズ#1、白、不透明)内へ充填する(1カプセル当たり148mgの組成物)。
【0080】
有用性
式Iの化合物、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドは、5−HT4受容体アゴニストであり、したがって、式Iの化合物の当該結晶塩形態物は、5−HT4受容体に媒介された、または5−HT4受容体活性に関連した病状、すなわち、5−HT4受容体アゴニストを用いる治療によって寛解する病状の治療に有用であることが予想される。このような病状としては、限定はしないが、過敏性腸症候群(IBS)、慢性便秘症、機能性消化不良症、胃内容排出遅延、逆流性食道炎(GERD)、胃不全麻痺、糖尿病性特発性胃疾患、術後腸閉塞症、腸偽性閉塞、および薬剤誘導移行遅延が挙げられる。また、いくつかの5−HT4受容体アゴニスト化合物は、認知障害、行動障害、気分障害、および自律神経機能の制御障害などの中枢神経系障害の治療に使用できる。
【0081】
特に、本発明の塩形態物は、胃腸(GI)管の運動性を高め、したがって、ヒトを含めた哺乳動物における、運動低下に起因するGI管の障害を治療する上で有用であることが予想される。このようなGI運動障害としては、例えば、慢性便秘症、便秘優勢過敏性腸症候群(C−IBS)、糖尿病性および特発性胃疾患、および機能性消化不良症が挙げられる。
【0082】
したがって、一態様において、本発明は、哺乳動物における胃腸管の運動を高める方法を提供し、該方法は、製薬的に許容できる担体および本発明の結晶塩を含む治療的有効量の製薬組成物を、哺乳動物に投与することを含む。
【0083】
5−HT4受容体に媒介されたGI管の運動低下障害または他の病態の治療に用いる場合、本発明の塩形態物は典型的に、一日単回投与で、または一日複数回投与で経口投与されるが、他の投与形態も使用できる。一投与当たりの活性剤の量または一日当たり投与される総量は、典型的には、治療される病態、選択された投与経路、投与される実際の化合物およびその相対的活性、年齢、体重、および個々の患者の応答、患者の症状の重症度などの関連状況に鑑みて、医師によって決定される。
【0084】
5−HT4受容体に媒介されたGI管の運動低下障害または他の障害を治療するための好適な用量は、活性剤の約0.0007mg/kg/日から約1mg/kg/日など、活性剤の約0.0007mg/kg/日から約20mg/kg/日の範囲であると考えられる。平均70kgのヒトでは、これは、1日当たり約0.05mg/kg/日から約70mg/kg/日の活性剤の量になる。
【0085】
本発明の一態様において、本発明の塩形態物は、慢性便秘症の治療に用いられる。慢性便秘症の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。慢性便秘症を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0086】
本発明の他の態様において、本発明の塩形態物は、過敏性腸症候群の治療に用いられる。便秘優勢過敏性腸症候群の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。便秘優勢過敏性腸症候群を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0087】
本発明の他の態様において、本発明の塩形態物は、糖尿病性および特発性胃疾患の治療に用いられる。糖尿病性および特発性胃疾患の治療に用いられる場合、本発明の塩は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。糖尿病性胃疾患を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0088】
本発明のさらに他の態様において、本発明の塩形態物は、機能性消化不良症の治療に用いられる。機能性消化不良症の治療に用いられる場合、本発明の化合物は、典型的に、一日単回投与で、または一日複数回投与で、経口投与される。機能性消化不良症を治療するための用量は、1日当たり、約0.05mgから約70mgの範囲であると考えられる。
【0089】
本発明はまた、5−HT4受容体活性に関連した疾患または病態を有する哺乳動物を治療する方法を提供し、該方法は、本発明の塩形態物、または本発明の塩形態を含む製薬組成物の治療的有効量を、哺乳動物に投与することを含む。
【0090】
上記のとおり、本発明の塩形態物は、5−HT4受容体アゴニストである。したがって、本発明はさらに、哺乳動物において、5−HT4受容体に作動する方法を提供し、該方法は、本発明の塩形態物を哺乳動物に投与することがを含む。
【0091】
本発明の塩酸塩形態物の性質、ならびに有用性は、当業者によく知られた種々のインビトロおよびインビボアッセイを用いて実証することができる。典型的なアッセイは、以下の実施例において、さらに詳細に記載されている。
【実施例】
【0092】
以下の合成的および生物学的実施例は、本発明を例示するために提供されており、決して本発明の範囲を限定するものとして考えてはならない。下記の実施例において、以下の略号は、別に指示されない限り、以下の意味をもつ。下記に定義されていない略号は、一般に認められている意味をもつものである。
【0093】
Boc = t−ブトキシカルボニル
(Boc)2O = ジ−t−ブチルジカルボネート
DCM = ジクロロメタン
DMF = N,N−ジメチルホルムアミド
DMSO = ジメチルスルホキシド
EtOAc = エチルアセテート
mCPBA = m−クロロ安息香酸
MeCN = アセトニトリル
MTBE = t−ブチルメチルエーテル
PyBop = ベンゾトリアゾール−1−イルオキシトリピロリジノ−ホスホニウムヘキサフルオロホスフェート
Rf = 保持因子
RT = 室温
TFA = トリフルオロ酢酸
THF = テトラヒドロフラン
試剤(第二級アミンを含む)および溶媒は、民間供給者(Aldrich、Fluka、Sigmaなど)から購入し、そのまま精製せずに使用した。反応は、別に注記しない限り、窒素雰囲気下で行った。反応混合物の経過は、薄層クロマトグラフィ(TLC)、分析用高性能液体クロマトグラフィ(anal.HPLC)、および質量分析によってモニターし、その詳細は、下記および反応の具体的な実施例において個別に提示されている。反応混合物は各反応において具体的に記載されているとおりに作製し;通常、抽出のほか温度および溶媒依存的な結晶化、沈殿などの精製法により精製した。また、反応混合物は分取HPLCによって規定通りに精製した。反応生成物の特性解析は、質量分析および1H−NMR分光法により、規定通りに実施した。NMR測定では、サンプルを重水素化溶媒(CD3OD、CDCl3、またはDMSO−d6)中に溶解し、1H−NMRスペクトルを、標準的な観察条件下、Varian Gemini2000機器(300MHz)によって得た。化合物の質量分析による同定は、Applied Biosystems(フォスターシティー、カリフォルニア州)モデルのAPI150EX機器またはAgilent(パロアルト、カリフォルニア州)モデルの1100LC/MSD機器による電気スプレー電離法(ESMS)によって実施した。水分含量は、Brinkmann(ウェストベリー、ニューヨーク)のMetrohm Karl Fischer Model 813クーロメーターを用いて、カールフィッシャー滴定法により判定した。
【0094】
調製1:(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステル
a.8−ベンジル−8−アザビシクロ〔3.2.1〕オクタン−3−オンの調製
水(170mL)に溶かした2,5−ジメトキシテトラヒドロフラン(82.2g、0.622モル)の不均一溶液に、濃塩酸(30mL)を攪拌しながら加えた。0℃(氷浴)に冷却した別のフラスコ内で、水(350mL)に溶かしたベンジルアミン(100g、0.933モル)の溶液に、濃塩酸(92mL)を徐々に加えた。2,5−ジメトキシテトラヒドロフラン溶液をおよそ20分間攪拌し、水(250mL)で希釈し、次に、ベンジルアミン溶液を加え、引き続き、水(400mL)に溶かした1,3−アセトンジカルボン酸(100g、0.684モル)の溶液を加え、次いで水(200mL)に溶かしたリン酸水素ナトリウム(44g、0.31モル)を加えた。40%NaOHを用いて、pHをpH1からpH約4.5に調整した。得られた混濁淡黄色溶液を一晩攪拌した。次いで、50%塩酸を用いて、該溶液をpH7.5からpH3に酸性化し、85℃に加熱し、2時間攪拌した。該溶液を室温まで冷却し、40%NaOHを用いてpH12に塩基性化し、ジクロロメタン(3×500mL)で抽出した。有機層を合せて、生理食塩水で洗浄し、乾燥し(MgSO4)、ろ過し、減圧下濃縮し、粘稠な褐色オイルとして、標題の粗中間体を得た。
【0095】
0℃で、メタノール(1000mL)に溶かした該粗中間体の溶液に、ジ−tert−ブチルジカーボネート(74.6g、0.342モル)を加えた。該溶液を室温まで温め、一晩攪拌した。減圧下、メタノールを除去し、得られたオイルをジクロロメタン(1000mL)に溶解させた。該中間体を1MのH3PO4(1000mL)中に抽出し、ジクロロメタン(3×250mL)で洗浄した。水層を、水性NaOHを用いて、pH12へ塩基性化し、ジクロロメタン(3×500mL)で抽出した。有機層を合せて乾燥し(MgSO4)、ろ過し、減圧下濃縮して、粘稠な淡褐色オイルとして、標題中間体を得た。
【0096】
【数1】
b.3−オキソ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステルの調製
EtOAc(300mL)に溶かした8−ベンジル−8−アザビシクロ〔3.2.1〕オクタン−3−オン(75g、0.348モル)の溶液に、EtOAc(300mL)に溶かしたジ−t−ブチルジカーボネート(83.6g、0.383モル、1.1当量)を加えた。得られた溶液およびすすぎ液(100mLのEtOAc)を、窒素流下、23gの水酸化パラジウム(20重量%のPd、乾燥ベース、炭素上、水で約50%湿性;例えば、Pearlmanの触媒)を含有する1LのParr水素化容器に加えた。該反応容器を脱気し(真空とN2を交互に5回)、60psiのH2ガスへと加圧した。該反応溶液を2日間攪拌し、適宜H2を再供給して、シリカ薄層クロマトグラフィでモニターして反応が完了するまで、H2圧を60psiに保たせた。次いで、該黒色溶液をCelite(登録商標)のパッドを通してろ過し、減圧下濃縮して、粘稠な黄色から橙色オイルとして、標題中間体を定量的に得た。これをそのまま処理せずに、次のステップに用いた。
【0097】
【数2】
c.(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステルの調製
メタノール(1L)に溶かした先のステップの生成物(75.4g、0.335モル)の溶液に、N2流下、機械的攪拌器により攪拌しながら、フマル酸アンモニウム(422.5g、6.7モル)、水(115mL)および活性炭素上パラジウム65g(乾燥ベース上10%、水で約50%湿性;DegussaタイプE101NE/W)を加えた。24時間後および48時間後、各々の時点で、追加部分のフマル酸アンモニウム(132g、2.1モル)を加えた。分析用HPLCによるモニターで反応の進行が停止したら、Celite(登録商標)(>500g)を加え、得られた濃厚懸濁液をろ過してから、採取した固体をメタノール(約500mL)ですすいだ。ろ液を合せて、全てのメタノールが除去されるまで、減圧下濃縮した。次いで、得られた混濁二相溶液をpH2で約1.5Lから2.0Lの最終容量まで1Mのリン酸で希釈し、ジクロロメタン(3×700mL)で洗浄した。水層を、40%の水性NaOHを用いて、pH12に塩基性化し、ジクロロメタン(3×700mL)で抽出した。有機層を合せてMgSO4で乾燥し、ろ過し、回転蒸発、次いで高真空により濃縮し、52g(70%)の標題中間体、一般に、N−Boc−エンド−3−アミノトロパンを、白色から淡黄色固体として得た。この生成物のエンドアミン対エキソアミンの異性体比は、1H−NMR分析に基づき、>99であった(分析用HPLCにより>96%の純度)。
【0098】
【数3】
分析用HPLC(無勾配方法;5分間にわたる90:10(A:B)に対する2:98(A:B):保持時間=2.14分)。
【0099】
調製2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸
先ず、水(2L)に溶かした2−アミノフェニルメタノール(255.2g、2.07モル)および酢酸(3.56mL、62ミリモル)の攪拌懸濁液に、室温で、アセトン(228.2mL、3.11モル)を加えた。4時間後、該懸濁液を0℃に冷却し、さらに2.5時間攪拌してからろ過した。該固体を採取し、水で洗浄し、湿潤固体を凍結乾燥により冷却、乾燥し、2,2−ジメチル−1,4−ジヒドロ−2H−ベンゾ〔1,3〕オキサジン(332.2g、98%)を、オフホワイトの固体として得た。
【0100】
【数4】
THF(1L)に溶かした2,2−ジメチル−1,4−ジヒドロ−2H−ベンゾ〔1,3〕オキサジン(125g、0.77モル)の溶液を、シンチレーション漏斗を介してろ過してから、0℃で、THF(800mL)中、1.0MのLiAlH4の攪拌溶液に、2.5時間かけて添加漏斗を介し、滴下により加えた。0℃で、Na2SO4・10H2O(110g)を、1.5時間かけて部分に分けて徐々に加えることによって、該反応をクエンチした。該反応混合物を一晩攪拌し、ろ過し、固体塩をTHFで完全に洗浄した。ろ液を減圧下濃縮し、2−イソプロピルアミノフェニルメタノール(120g、95%)を黄色オイルとして得た。
【0101】
【数5】
トルエン(800mL)に溶かした2−イソプロピルアミノフェニルメタノール(118g、0.71モル)の攪拌溶液に、二酸化マンガン(85%、182.6g、1.79モル)を加え、該反応混合物を117℃に4時間加熱した。該反応混合物を一晩、室温まで冷却させてから、Celiteのパッドを介してろ過し、これをトルエンで溶出した。ろ液を減圧下濃縮し、2−イソプロピルアミノベンズアルデヒド(105g、90%)を橙色オイルとして得た。
【0102】
【数6】
0℃で、メタノール(1L)に溶かした2−イソプロピルアミノベンズアルデヒド(105g、0.64モル)、酢酸(73.6mL、1.29モル)およびエチレンジアミン(43.0mL、0.64モル)の攪拌溶液に、2,2−ジメチル−〔1,3〕ジオキサン−4,6−ジオン、一般にMeldrumの酸(166.9g、1.16モル)を加えた。該反応混合物を0℃で1時間、次いで室温で一晩攪拌した。得られた懸濁液をろ過し、固体をメタノールで洗浄し採取しt、標題の中間体、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸を、オフホワイトの固体として得た。
【0103】
【数7】
(実施例1:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成)
a.{(1S,3R,5R)−3−[1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボニル〕アミノ]−8−アザ−ビシクロ〔3.2.1〕オクト−3−
イル}アミドの調製
3Lフラスコ内に、トルエン(1L)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸(112.4g、0.486モル、1.1当量)を懸濁させた。該混合物を85℃に加熱し、塩化チオニル(86.74g、0.729モル)を70分かけて滴下して加えた。該混合物を攪拌しながら95℃に1.5時間加熱してから、室温まで冷却させた。
【0104】
別の12Lフラスコに、トルエン(1L)に溶かした(1S,3R,5R)−3−アミノ−8−アザビシクロ〔3.2.1〕オクタン−8−カルボン酸t−ブチルエステル(100.0g、0.442モル、1当量)を懸濁させ、3MのNaOH(4当量)を加えた。該混合物を室温で10分間攪拌してから、約5℃に冷却させた。内部温度を10℃以下に保ちながら、40分かけて、酸クロリド溶液を攪拌しながら徐々に加えた。該混合物を3℃〜5℃で30分間攪拌し、一晩、層分離させた。トルエン層(約2.5L)を採取し、回転蒸発により、約半分(約1.2L)に濃縮し、次のステップに直接使用した。
【0105】
b.1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの調製
先のステップで調製したトルエン溶液(約1.2L)に、20℃で20分かけて、攪拌しながら、トリフルオロ酢酸(200mL)を加えた。該混合物を、20℃で2時間攪拌した。水(1.55L)を加え、該混合物を20℃で30分間攪拌した。30分後、該混合物は3層に分かれた。下層(約350mL)の粘稠褐色オイルが粗中間体を含有した。
【0106】
MTBE(2.8L)を入れた12Lフラスコに、該粗褐色オイルを、1℃〜2℃で1時間かけて攪拌しながら加えた。該懸濁液を同じ温度で1時間攪拌してからろ過した。ろ液をMTBE(2×300mL)で洗浄し、室温で4日間、真空下乾燥し、標題中間体のトリフルオロ酢酸塩(163.3g)を淡黄色粉末として得た。
【0107】
c.N−メチル−N−〔(S)−2−オキシラン−2−イルメチル〕メタンスルホンアミドの調製
12Lのフラスコに、水(1L)を入れ、次いでNaOH(水中50%、146.81g、1.835モル)を加えた。NaOHを含むビーカーを水(2×500mL)で洗浄し、洗浄液を該フラスコに加えた。該混合物を室温で10分間攪拌し、約8℃に冷却した。水(500mL)に溶かした(N−メチル)メタンスルホンアミド(200.2g、1.835モル)を5分間かけて加えた。該混合物を、3℃〜4℃で20時間攪拌した。ジクロロメタン(2L)を加え、該混合物を5℃〜10℃で30分間攪拌した。2層を10分間かけて分離させ、採取した。有機層(約2.5L)を12Lフラスコに戻し加えて、1MのH3PO4(800mL)および生理食塩水(800mL)で洗浄した。ジクロロメタンを回転蒸発により除去した。粗生成物にトルエン(400mL)を加え、回転蒸発により除去した。さらに3サイクルのトルエン処理の後、標題中間体を得(228.2g)、これをそのまま精製せずに次のステップに使用した。
【0108】
d.1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成
3Lのフラスコ内に、無水エタノール(400mL)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドトリフルオロアセテート(105.0g、0.232モル)を懸濁させた。この懸濁液に、無水エタノール(100mL)中に溶解させたNaOH(水中50%、0.243モル、1.05当量)を室温で加えた。NaOHを含むビーカーをエタノール(2×50mL)で洗浄し、洗浄液を該反応混合物に加えた。30分の攪拌後、無水エタノール(100mL)中のN−メチル−N−〔(S)−2−オキシラン−2−イルメチル〕メタンスルホンアミド(62.0g、1.5当量)を加えた。該混合物を2時間還流し、室温まで冷却し、標題化合物の種結晶を加えた。約5分の攪拌後、白色固体が形成した。該混合物を3℃〜5℃に冷却し、2時間攪拌した。白色固体をろ過し、湿潤ケークを冷無水エタノール(3×50mL)で洗浄した。該固体を、30℃で60時間、真空下乾燥し、標題化合物(93.8g、カールフィッシャー法による含水量は2.03%)を得た。
【0109】
【数8】
種結晶は、結晶化が自然に生じる、本実施例の方法による標題化合物の先の調製から得られた。
【0110】
(実施例2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミドの合成)
1Lフラスコ内に、無水エタノール(210mL)中、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−
2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド(34.7g、0.069モル)を懸濁
させた。室温で濃HCl(1.1当量)を攪拌しながら加えた。該混合物を30分間還流下攪拌し、室温まで冷却させて2時間攪拌した。固体をろ過し、湿潤ケークを冷無水エタノール(3×50mL)で洗浄した。該固体を、30℃で48時間、真空下乾燥し、標題化合物(34.5g、収率93.7%、カールフィッシャー法による含水量は0.13%)を得た。
【0111】
(実施例3:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩の結晶水和物合成)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザ−ビシクロ〔3.2.1〕オクト−3−イル}アミド塩酸塩(139mg、0.28ミリモル)を、注射用滅菌水(2mL)中に溶解させた。数時間経つと、該溶液は混濁した懸濁液になった。該懸濁液を攪拌し、周囲温度で一晩放置すると、白色沈殿が生じた。固体をろ過により採取し、周囲条件(およそ40〜50%の相対湿度)で2分間乾燥して、標題化合物(130mg、収率91%)を得た。
【0112】
(実施例4〜9:本発明の塩形態物の性質)
実施例2のとおり調製した、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(式Iの化合物)の結晶塩酸塩のサンプル、および実施例3のとおり調製した、式Iの化合物の塩酸塩の結晶水和物のサンプルを、粉末x線回折(PXRD)、示差走査熱量測定(DSC)、熱重量分析(TGA)赤外線分光法(IR)および元素分析により分析した。
【0113】
(実施例4:粉末x線回折)
Si(Li)ソリッドステート検出器と共に、1.542Å(45kV、40mA)でのCu Kα放射線を用い、Thermo ARL X−Ray Diffractometer Model XTRA(Thermo ARL SA、スイス国)により、粉末x線回折パターンを得た。この分析は、典型的に、2シータ角における2°から35°の範囲にわたって、1箇所当たり、0.03°の段階サイズで、2°/分の走査速度で実施した。分析のため、受領されたサンプル、または微粉末へと粉砕したサンプルのいずれかを、機器のカップ充填サンプルカップにはめ込むようにデザインされた注文の少容量挿入体内へ静かに入れた。±0.02°の2シータ角以内への機器補正は、シリコン金属標準と比較することにより、毎週検証した。本発明の結晶塩酸塩のサンプルおよび本発明の塩酸塩の水和物のサンプルに関する典型的なPXRDパターンは、それぞれ図1および図4に示されている。
【0114】
(実施例5:熱分析)
TA Instruments Model Q−100モジュールを用いて、示差走査熱量測定(DSC)を実施した。データを採取し、Q Series(商標)ソフトウェア用のTA Instruments Thermal Advantageを用いて分析した。約1mg〜10mgのサンプルを、ふた付きのアルミニウムパン内へ正確に量り入れた。5℃から、典型的には、265℃の、10℃/分の線形加熱ランプを用いて、サンプルを評価した。使用中、DSCセルを乾燥窒素でパージした。化合物Iの結晶塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なDSCトレースは、それぞれ、図2および図5に示されている。
【0115】
TA Instruments Model Q−500モジュールを用いて、熱重量分析(TGA)を実施した。データを採取し、Q Series(商標)ソフトウェア用のTA Instruments Thermal Advantageを用いて分析した。約1mg〜5mgの重量のサンプルを、白金クレードル上のアルミニウムパン内に入れ、10℃/分の線形加熱ランプを用いて、周囲温度から300℃を走査した。使用中、天秤およびファーネスを窒素でパージした。化合物Iの結晶質塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なTGAトレースもまた、それぞれ、図2および図5に示されている。
【0116】
図2のDSCトレースは、本発明の塩酸塩が、約230℃から約260℃の範囲において吸熱性の熱流が最大となる優れた熱安定性を有し、約225℃以下で有意な熱事象を有さないことを実証している。DSCトレースとTGAトレースとの比較により、本発明の塩酸塩が、約230℃以上の温度で、融解と分解を同時に受けることが示されている。
【0117】
当該水和物形態に関する図5におけるDSCトレースにより、約225℃から約250℃の範囲における該結晶が融解する吸熱性の熱流の実質的なピーク、ならびにより低温での幅広い、または弱い吸熱が示されている。DSCトレースとTGAトレースとの比較により、水和物結晶形態の分解は、融解転移の温度において有意ではないことが示されている。
【0118】
(実施例6:動的水分吸収の評価)
VTI大気微量天秤であるSGA−100システム(VTI社、ヒアレー、フロリダ州)を用いて、25℃で、動的水分吸収(DMS)評価を実施した。およそ5mg〜10mgのサンプルサイズを用い、湿度は、分析開始時、周囲の数値に設定した。典型的なDMS分析は、3つの走査:5%RH/段階の走査速度で、周囲湿度から2%相対湿度(RH)、2%RHから90%RH、90%RHから5%RHからなる。2分おきに質量を測定し、サンプルの質量が5つの連続箇所で0.02%以内に安定していた場合はRHを次の値(±5%RH)に変化させる。化合物Iの結晶塩酸塩のサンプル、および化合物Iの塩酸塩の結晶水和物のサンプルに関する典型的なDMSトレースは、それぞれ、図3および図6に示されている。
【0119】
該塩酸塩は、2%RHから90%RHの範囲全体にわたる0.2%未満の重量変化で、可逆的な吸着/脱離を示す。該水和物形態は、該サンプルを周囲RHから2%RHへと乾燥させた際、乾燥検体が2%RHから40%RHに曝された際の水分率である、約2.3%の重量損失を有する吸着/脱離プロフィルを示す。該水和物形態は、40%RHから75%RHの範囲で、約0.25%未満の重量増加であった。
【0120】
(実施例7:赤外線分析)
Nicolet減衰全反射(ATR)サンプルホールダーを装備したAvatar 360 FT−IR分光器を用いて、4000cm−1から675cm−1の周波数範囲にわたって、赤外線(IR)吸収スペクトルを判定した。本発明の結晶塩酸塩サンプルに関する典型的なIR吸収スペクトルは、758±1、783±1、795±1、802±1、949±1、981±1、1149±1、1158±1、1217±1、1332±1、1377±1、1453±1、1467±1、1487±1、1525±1、1566±1、1575±1、1615±1、1672±1、および3197±1cm−1に有意な吸収バンドを有した。
【0121】
(実施例8:固体状態の安定性評価)
本発明の塩酸塩サンプルを、40℃、75%RHで、複数の開放ガラスバイアルに貯蔵した。特定の間隔で、代表的なバイアルの内容物を取り出し、化学的純度に関して、DSC、TGA、PXRD、およびHPLCにより分析した。貯蔵24週間後、DSCまたはTGAの温熱図にもPXRDパターンにも、検出可能な変化は存在しなかった。該貯蔵サンプルの化学的純度は99.6%であった。
【0122】
(実施例9:対イオンモル比の判定)
塩酸塩(HA)対1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(式Iの化合物)の対イオンモル比を、以下の式に従って算出した:
対イオン比=(WHA/MWHA)/(WI/MWI)
式中、WHAは、サンプル中のHClの重量パーセンテージであり、MWHAは、HClの分子量であり、MWIは、式Iの化合物の分子量(504.6amu)であり、WIは、以下の式に従って算出した、サンプル中の式Iの化合物の重量パーセンテージであり:
WI=100−WHX−WH2O−WRS
化合物Iが不純物を有さないという仮定で、式中、WH2Oは、水分含量の重量パーセンテージであり、WRSは残留溶媒の重量パーセンテージである。
【0123】
本発明の結晶塩酸塩のサンプルに関して、塩酸塩対化合物Iのモル比は、HCl(WHA)の重量パーセンテージの6.3%、および値、WH2O=0.26%およびWRS=0.47%を用いて、0.94:1と算出された。HCl含量は、硝酸銀の標準溶液による滴定により判定し、水分含量WH2Oは、電量的カールフィッシャー滴定により判定し、残留溶媒含量WRSは、ガスクロマトグラフィにより判定した。
【0124】
(比較実施例1:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドのクエン酸塩の合成)
1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミド(0.1g、0.2ミリモル)をエタノール(1mL)中に懸濁させた。この懸濁液に、エタノール(0.072mL、0.072ミリモル、0.33当量)に溶かしたクエン酸の1M溶液を加えた。該混合物を、清澄になるまで簡単に超音波処理し、ふたをしてから一晩放置した。ふたを取り、該混合物を、周囲条件下で、固体が見られるまで蒸発させた。次いで、該混合物に再度ふたをして、72時間放置した。得られた固体をろ過し、冷エタノールで洗浄し、標題化合物を固体として得た(74.3mg)。
【0125】
(比較実施例2:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの酸性塩の合成)
比較実施例1の手順に従って、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの以下の酸性塩を、以下の酸(二番目の括弧内に生成物の重量)の指示された当量を用いて、固体形態で調製した:アジピン酸(0.5当量)(48.5mg);リン酸(0.5当量)(86.6mg);硫酸(0.5当量)(27.0mg);酒石酸(0.5当量)(66.3mg);リンゴ酸(0.5当量)(25.3mg);および臭化水素酸(1当量)(62.9mg)。
【0126】
(比較実施例3:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドのメタンスルホン酸塩の合成)
50%アセトニトリル/水(1mL)に溶かした1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの溶液に、エタノール(0.2mL、0.2ミリモル、1当量)に溶かしたメタンスルホン酸の1M溶液を加えた。次いで、該混合物を凍結させ、一晩凍結乾燥した。得られた固体を緩やかに温めながらイソプロパノール(1mL)中に溶解させ、冷却させた。得られた固体をろ過により採取し、冷イソプロパノールで洗浄し、標題化合物を固体として得た(90mg)。
【0127】
(比較実施例4:1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの酸性塩の合成)
比較実施例3の手順に従って、1−イソプロピル−2−オキソ−1,2−ジヒドロキノリン−3−カルボン酸{(1S,3R,5R)−8−〔(R)−2−ヒドロキシ−3−(メタンスルホニル−メチル−アミノ)プロピル〕−8−アザビシクロ〔3.2.1〕オクト−3−イル}アミドの以下の酸性塩を、以下の酸(二番目の括弧内に生成物の重量)の指示された当量を用いて、固体形態で調製した:フマル酸(1当量)(107.2mg);安息香酸(1当量)(105.2mg);および(R)−マンデル酸(1当量)(96.1mg)。
【0128】
(比較実施例5:比較実施例1〜4の化合物の性質)
比較実施例1〜4の式Iの化合物の検出塩酸塩を、PXRD、DSC、およびTGAにより分析した。全ての場合で、約100℃以下の温度で、TGAによる重量損失が見られたが、これは、溶媒の損失に起因する可能性が高いと考えられる。溶媒損失に起因する低温特性を除いて、DSCによって吸熱性熱流が見られた温度、ならびにPXRDによる結晶性の確認を、表Iに要約してあり、ここで、化学量論組成は、該酸性塩の調製に用いられた酸の当量数によって示されている。
【0129】
【表12】
アッセイ1:5−HT4(c)ヒト受容体に対する放射性リガンド結合アッセイ
a.膜調製5−HT4(c)
ヒト5−HT4(c)受容体cDNA(〔3H〕−GR113808膜放射性リガンド結合アッセイを用いて判定したBmax=約6.0pmol/mgタンパク質)を安定トランスフェクトしたHEK−293(ヒト胎児腎臓)細胞を、37℃において、5%CO2、加湿インキュベーター中、T−225フラスコ内で、10%ウシ胎仔血清(FBS)(GIBCO−Invitrogen社:Cat#10437)、2mMのL−グルタミンおよび(100ユニット)ペニシリン−(100μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:Cat#15140)を添加した、4,500mg/LのD−グルコースおよび塩酸ピリドキシン(GIBCO−Invitrogen社、カールスバッド、カリフォルニア州、カタログ番号11965)を含有するダルベッコー修飾イーグル培地(DMEM)中で増殖させた。培地へ800μg/mLのジェネテシン(GIBCO−Invitrogen社:カタログ番号10131)を添加することによって、細胞を継続的な選択圧下で増殖させた。
【0130】
細胞は、およそ60%〜80%の集密度(<35継代培養)まで増殖させた。採取20〜22時間前に、細胞を2回洗浄し、無血清DMEMを供給した。膜調製の全ての処置は氷上で実施した。25mLピペットを用いた緩やかな機械的攪拌と摩砕により、細胞単層を浮揚させた。細胞を1000rpm(5分)での遠心分離により採取した。
【0131】
膜調製のため、細胞ペレットを氷冷した50mMの4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)、pH7.4(膜調製緩衝液)に再懸濁し(30〜40 T225フラスコから40mL/全細胞産出)、氷上、ポリトロンディスラプター(19,2×10秒に設定)を用いて均一化した。得られた均一物を、4℃で5分間、1200gで遠心分離した。ペレットを捨て、上澄み液を、40,000g(20分)遠心分離した。該ペレットは、膜調製緩衝液を用いた再懸濁により1回洗浄し、40,000g(20分)で遠心分離した。最終ペレットを、50mMのHEPES、pH7.4(アッセイ緩衝液)に再懸濁させた(当量1T225フラスコ/1mL)。膜懸濁液のタンパク質濃度は、ブラッドフォードの方法(Bradford、1976年)により判定した。膜は−80℃で凍結保存した。
【0132】
b.放射性リガンド結合アッセイ
0.025%ウシ血清アルブミン(BSA)を含有する50mMのHEPES、pH7.4中、2μgの膜タンパク質を含有する400μLの全アッセイ容量において、1.1mLの96ディープウェルポリプロピレンアッセイプレート(Axygen)中で、放射性リガンド結合アッセイを実施した。0.001nM〜5.0nMの範囲で異なる8〜12の濃度で、〔3H〕−GR11308(Amersham社、バックス、英国:カタログ番号TRK944;比放射能、約82Ci/mmol)を用い、放射性リガンドのKd値決定のための飽和結合試験を実施した。0.15nMでの〔3H〕−GR113808および10pM〜100μMの範囲で異なる11の濃度の化合物により、化合物のpKi値決定のための置換アッセイを実施した。
【0133】
試験化合物は、DMSO中、10mMの原液として受領し、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、同じ緩衝液中に連続(1:5)希釈した。比特異的結合は、1μMの非標識GR113808の存在下で判定した。アッセイは室温で60分間インキュベートし、次いで、0.3%のポリエチレンイミンに予め浸漬した96ウェルGF/Bグラスファイバーフィルタープレート(Packard BioScience社、メリデン、コネチカット州)上での迅速ろ過により、結合反応を終了させた。フィルタープレートをろ過緩衝液(氷冷した50mMのHEPES、pH7.4)で3回洗浄し、非結合の放射能を除去した。プレートを乾燥し、各ウェルに、35μLのMicroscint−20液体シンチレーション液(Packard BioScience社、メリデン、コネチカット州)を加え、Packard Topcount液体シンチレーションカウンター(Packard BioScience社、メリデン、コネチカット州)において、プレートをカウントした。
【0134】
結合データは、1部位競合に関する3−パラメーターモデルを用いて、GraphPad Prism Software package(GraphPad Software社、サンジエゴ、カリフォルニア州)による非線形回帰解析によって解析した。BOTTOM(最小曲線)を、1μMのGR113808の存在下で決定した非特異的結合に関する値に調整した。Cheng−Prusoff式(ChengおよびPrusoff、Biochemical Pharmacology、1973年、22、3099−108頁):Ki=IC50/(1+〔L〕/Kd)、式中、〔L〕=〔3H〕−GR113808濃度、を用い、最良適合したIC50値、および放射性リガンドのKd値から、Prismにおいて、試験化合物に関するKi値を産出した。結果は、Ki値の負の十進法対数、pKiとして表される。
【0135】
このアッセイにおいて、より高いpKi値を有する試験化合物は、5−HT4受容体に対して、より高い結合親和性を有する。式Iの化合物は、このアッセイにおいて、約7.5超のpKi値を有した。
【0136】
アッセイ2:5−HT3Aヒト受容体に対する放射性リガンド結合アッセイ:受容体サブタイプ選択性の判定
a.膜調製5−HT3A
ヒト5−HT3A受容体cDNAを安定トランスフェクトしたHEK−293(ヒト胎児腎臓)細胞を、Michael Bruess博士(ボン大学、東ドイツ)から入手した(〔3H〕−GR65630膜放射性リガンド結合アッセイを用いて判定したBmax=約9.0pmol/mgタンパク質)。37℃において、5%CO2、加湿インキュベーター中、T−225フラスコまたは細胞材料内で、10%熱不活化ウシ胎仔血清(FBS)(Hyclone、ローガン、ユタ州:カタログ番号SH30070.03)、および(50ユニット)ペニシリン−(50μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:カタログ番号15140)を添加した、50%ダルベッコー修飾イーグル培地(DMEM)(GIBCO−Invitrogen社、カールスバッド、カリフォルニア州、カタログ番号11965)および50%ハムF12(GIBCO−Invitrogen社:カタログ番号11765)中で、細胞を増殖させた。
【0137】
細胞は、およそ70%〜80%の集密度(<35継代培養)まで増殖させた。膜調製の全ての処置は氷上で実施した。細胞を採取するために、培地を吸引し、Ca2+、Mg2+のないダルベッコー燐酸緩衝生理食塩水(dPBS)ですすいだ。緩やかな機械的攪拌により、細胞単層を浮揚させた。細胞を1000rpm(5分)での遠心分離により採取した。5−HT4(c)受容体を発現する膜に関して、上記のプロトコルに引き続いて膜調製の工程を行った。
【0138】
b.放射線リガンド結合アッセイ
0.025%BSAアッセイ緩衝液を含有する50mMのHEPES、pH7.4中、1.5〜2μgの膜タンパク質を含有する200μLの全アッセイ容量において、96ウェルポリプロピレンアッセイプレート中で、放射性リガンド結合アッセイを実施した。0.005nM〜20nMの範囲で12の異なる濃度で、〔3H〕−GR65630(PerkinElmer Life Sciences社、ボストン、マサチューセッツ州:カタログ番号NET1011;比放射能、約85Ci/mmol)を用い、放射性リガンドのKd値決定のための飽和結合試験を実施した。0.50nMでの〔3H〕−GR65630および10pMから100μMの範囲で11の異なる濃度の化合物により、化合物のpKi値決定のための置換アッセイを実施した。化合物は、DMSO中、10mMの原液として受領し(節3.1を参照)、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、連続(1:5)希釈してから同じ緩衝液中に作製した。非特異的結合は、10μMの非標識MDL72222の存在下で判定した。アッセイは室温で60分間インキュベートし、次いで、0.3%のポリエチレンイミンに予め浸漬した96ウェルGF/Bグラスファイバーフィルタープレート(Packard BioScience社、メリデン、コネチカット州)上での迅速ろ過により、結合反応を終了させた。フィルタープレートをろ過緩衝液(氷冷した50mMのHEPES、pH7.4)で3回洗浄し、非結合の放射能を除去した。プレートを乾燥し、各ウェルに、35μLのMicroscint−20液体シンチレーション液(Packard BioScience社、メリデン、コネチカット州)を加え、Packard Topcount液体シンチレーションカウンター(Packard BioScience社、メリデン、コネチカット州)において、プレートをカウントした。
【0139】
結合データは、上記の非線形回帰操作を用いて解析し、Ki値を決定した。BOTTOM(最小曲線)を、10μMのMDL72222の存在下で決定した非特異的結合に関する値に調整した。Cheng−Prusoff式中の量〔L〕を、〔3H〕−GR65630濃度として定義した。
【0140】
5−HT4受容体サブタイプに関し、5−HT4受容体サブタイプに対する選択性を、Ki(5−HT3A)/Ki(5−HT4(c))比として産出した。このアッセイにおいて、式Iの化合物は、約1000超の5−HT4/5−HT3受容体サブタイプ選択性を有した。
【0141】
アッセイ3:ヒト5−HT4(c)受容体を発現するHEK−293細胞による細胞全体cAMP蓄積フラッシュアッセイ
このアッセイでは、5−HT4受容体を発現するHEK−293細胞を種々の濃度の試験化合物に接触させた際に産生されるサイクリックAMPの量を測定することにより、試験化合物の機能的効力を判定した。
【0142】
a.細胞培養
クローン化ヒト5−HT4(c)受容体cDNAを安定トランスフェクトした、該受容体を発現するHEK−293(ヒト胎児腎臓)細胞を、2種の密度:(1)〔3H〕−GR113808膜放射性リガンド結合アッセイを用いて判定した約0.5〜0.6pmol/mgタンパク質の密度で;および(2)約6.0pmol/mgタンパク質の密度で調製した。37℃において、5%CO2、加湿インキュベーター中、T−225フラスコ内で、10%ウシ胎仔血清(FBS)(GIBCO−Iんヴぃtろげn社:カタログ番号10437)、および(100ユニット)ペニシリン−(100μg)ストレプトマイシン/ml(GIBCO−Invitrogen社:カタログ番号15140)を添加した、4,500mg/LのD−グルコース(GIBCO−Invitrogen社、カタログ番号11965)を含有するダルベッコー修飾イーグル培地(DMEM)中で、該細胞を増殖させた。培地へジェネテシン(800μg/mL:GIBCO−Invitrogen社:カタログ番号10131)を添加することによって、細胞を継続的な選択圧下で増殖させた。
【0143】
b.細胞調製
細胞は、およそ60%〜80%の集密度まで増殖させた。アッセイの20〜22時間前に、細胞を2回洗浄し、4,500mg/LのD−グルコース(GIBCO−Invitrogen社、カタログ番号11965)を含有する無血清DMEMを供給した。該細胞を採取するために、培地を吸引し、各T−225フラスコに10mLのVersene(GIBCO−Invitrogen社、カタログ番号15040)を加えた。細胞を、室温で5分間インキュベートし、次いで、機械的攪拌によってフラスコから移動させた。該細胞懸濁液を等容量の予備加温した(37℃)dPBSを含有する遠心分離管に移し、1000rpmで5分間遠心分離した。上澄み液を捨て、ペレットを予備加温した(37℃)刺激緩衝液(2〜3のT−225フラスコ当たり、10mM当量)に再懸濁した。この時間を書きとめ、時間ゼロとして印づけた。Coulterカウンターにより細胞をカウントした(8μm以上をカウント、フラスコ収量は、1〜2×107細胞/フラスコ)。予備加温した(37℃)刺激緩衝液(フラッシュプレートキット内で提供されるような)中、5×105細胞/mlの濃度で、細胞を再懸濁させ、37℃で10分間予備インキュベートした。
【0144】
cAMPアッセイは、製造元の指示に従って、125I−cAMP(SMP004B、PerkinElmer Life Sciences社、ボストン、マサチューセッツ
州)により、Flashplate Adenylyl Cyclase Activation Assay Systemを用い、放射免疫アッセイ様式において実施した。
【0145】
細胞は上記のとおり増殖させ、調製した。該アッセイにおける最終細胞濃度は、25×103細胞/ウェルであり、最終アッセイ容量は、100μLであった。試験化合物は、DMSO中、10mMの原液として受領し、25℃で、0.1%BSAを含有する50mMのHEPES、pH7.4中に400μMに希釈し、同じ緩衝液中に連続(1:5)希釈した。10pMから100μM(最終アッセイ濃度)の範囲の異なる11の濃度の化合物により、サイクリックAMP蓄積アッセイを実施した。各プレートについて、5−HT濃度−応答曲線(10pMから100μM)を含めた。該細胞を振とうしながら37℃で15分間インキュベートし、各ウェルに、100μlの氷冷検出用緩衝液(フラッシュプレートキット内で提供されるような)の添加によって該反応を終了させた。該プレートを密封し、4℃で一晩インキュベートした。Topcount(Packard Bioscience社、メリデン、コネチカット州)を用い、シンチレーション近接分光法により、結合した放射能を定量化した。
【0146】
製造元の使用説明書に提供された指示に従って、反応液の1mL当たり産生されたcAMPの量を、cAMP標準曲線から外挿した。3−パラメーターS字形用量応答モデル(単一条件つき傾斜)を用い、GraphPad Prism Softwareパッケージによる非線形回帰解析によって、データを解析した。効力データは、EC50値の負の十進法対数pEC50値として報告され、ここで、EC50は、最大応答の50%に関する有効濃度である。
【0147】
このアッセイでより高いpEC50値を示す試験化合物は、5−HT4受容体に作用するより高い効力を有する。約0.5〜0.6pmol/mgタンパク質の密度を有する細胞系(1)において、このアッセイにおいて試験された式Iの化合物は、約7.5超のpEC50値を有した。
【0148】
アッセイ4:hERG心臓カリウムチャネルを発現する細胞全体におけるカリウムイオン流阻害のインビトロ電圧固定アッセイ
hERGのcDNAを安定トランスフェクトしたCHO−K1細胞は、ウィスコンシン大学のGail Robertsonから入手した。細胞は必要時まで低温保存庫に保持した。10%ウシ胎仔活性および200μg/mLのジェネテシンを添加したダルベッコー修飾イーグル培地/F12中で、細胞を増殖させ、継代した。細胞全体電圧固定試験用に単離細胞が選択され得る濃度で、35mm2のディッシュ(2mLの培地を含有)中、ポリ−D−リシン(100μg/mL)をコーティングしたガラスのカバースリップ上に、細胞を接種した。該ディッシュは、37℃で、加湿、5%CO2の環境中に維持した。
【0149】
少なくとも7日ごとに、細胞外溶液を調製し、使用しない時は4℃に保存した。該細胞外溶液は、以下を含有した(mM):NaCl(137)、KCl(4)、CaCl2(1.8)、MgCl2(1)、グルコース(10)、4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)(10)、NaOHによりpH7.4。試験化合物の不在下または存在下での細胞外溶液をリザーバ内に入れ、そこからおよそ0.5mL/分で、該細胞外溶液を記録チャンバーに流入させた。細胞内溶液を調製し、分画し、使用日まで−20℃で保存した。細胞内溶液は、以下を含有した(mM):KCl(130)、MgCl2(1)、エチレングリコール−ビス(ベータ−アミノエチルエーテル)N,N,N’,N’−テトラ酢酸塩(EGTA)(5)、MgATP(5)、4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)(10)、KOHによりpH7.2。全ての実験は、室温(20〜22℃)で実施した。
【0150】
細胞を接種したカバースリップを記録チャンバーに移し、継続的に潅流した。該細胞とパッチ電極との間にギガオーム密封を形成した。安定なパッチが達成したら、開始保持電位を−80mVで、電圧固定様式で記録を始めた。安定な細胞全体電流の達成後、細胞を試験化合物に曝露させた。標準的な電圧プロトコルは:−80mVの保持電位から+20mVへ4.8秒間、5秒間−50mVに再分極させ、次いで、元の保持電位(−80mV)に戻す段階であった。この電圧プロトコルは15秒ごとに1回行った(0.067Hz)。再分極段階時のピーク電流振幅は、pClampソフトウェアを用いて判定した。細胞上に、3μMの濃度の試験化合物を5分間潅流させ、次いで化合物の不在下で5分間の洗浄時間をとった。最後に、該細胞の機能を試験するために、陽性対照(シサプリド、20nM)を潅流液に加えた。−80mVから+20mVへの段階は、hERGチャネルを活性化し、外向きの電流をもたらす。−50mVへ戻す段階は、該チャネルが不活化から回復し、非活性化するため、外向きのテール電流をもたらす。
【0151】
再分極段階時のピーク電流振幅は、pClampソフトウェアを用いて判定した。対照および試験物のデータは、Origin(登録商標)(OriginLab社、ノーサンプトン、マサチューセッツ州)に送出し、そこで、個々の電流振幅は化合物不在下の最初の電流振幅へと正規化された。各条件に関する正規化電流の平均および標準誤差を算出し、該実験の時間経過に対してプロットした。
【0152】
試験物または媒体対照(通常0.3%DMSO)のいずれかに5分間曝露後、観察されたK+流阻害間で比較を行った。2集団、独立t検定(Microcal Origin
v.6.0)を用いて、実験群間の統計的比較を実施した。p<0.05で差は有意と考えられた。
【0153】
このアッセイにおいて、カリウムイオン流の阻害パーセンテージが小さいほど、治療薬として用いた場合の試験化合物の心臓再分極化パターンを変化させる潜在能力はより小さくなる。このアッセイにおいて、3μMの濃度での式Iの化合物が試験され、約15%未満のカリウムイオン流の阻害を示した。
【0154】
アッセイ5:経口生物学的利用能のインビトロモデル:Caco−2浸透アッセイ
経口投与後、腸を通過して血流に入る試験化合物の能力をモデル化するために、Caco−2浸透アッセイを実施した。ヒト小腸単層の密着した接合を模倣してデザインされた細胞単層を溶液中の試験化合物が浸透する速度を判定した。
【0155】
Caco−2(大腸、腺癌;ヒト)細胞はATCC(American Type Culture Collection;ロックビル、メリーランド州)から入手した。浸透試験では、予め湿らせたトランスウェルポリカーボネートフィルター(Costar;ケンブリッジ、マサチューセッツ州)上に、63,000細胞/cm2の密度で細胞を接種した。培養液中、細胞単層は21日後に形成された。トランスウェルプレート内での細胞培養後、細胞単層を含有する膜をトランスウェルプレートから引き離し、拡散チャンバー(Costar;ケンブリッジ、マサチューセッツ州)内に挿入した。該拡散チャンバーを、温度制御用に循環外部サーモスタット制御の37℃水を備えた加熱ブロック内に挿入した。空気多枝管により拡散チャンバーの各半分に95%O2/5%CO2が送達され、攪拌されない境界層の減少に効果的な、細胞単層を横切る層流を作り出した。
【0156】
100μMの試験化合物濃度、および単層の完全性をモニターするための14C−マンニトールによって、浸透試験を実施した。全ての実験が、37℃で60分間行われた。チャンバーの供給側と受取側の双方から、0分、30分および60分でのサンプルを採取した。試験化合物およびマンニトールの濃度に関して、サンプルをHPLCまたは液体シンチレーションカウンティングにより分析した。cm/秒での浸透係数(Kp)を算出した。
【0157】
このアッセイにおいて、約10×10−6cm/秒超のKp値が好適な生物学的利用能を示すと考えられる。式Iの化合物をこのアッセイにおいて試験すると、約20×10−6cm/秒超のKp値を示した。
【0158】
アッセイ6:ラットにおける薬物動態試験
約5と約6の間のpHで、0.1%乳酸中、試験化合物の水溶液製剤を調製した。オスのSDラット(CD株、Charles River Laboratories、ウィルミントン、マサチューセッツ州)に、2.5mg/kgの用量で静脈内投与(IV)により、または5mg/kgの用量で経口胃管(PO)により、試験化合物を投与した。投与容量は、IVでは1mL/kg、PO投与では、2mL/kgであった。動物から、投与前に、ならびに投与後2分(IVのみ)、5分、15分、および30分、1時間、2時間、4時間、8時間、および24時間目に、連続血液サンプルを採取した。血漿中の試験化合物濃度は、1ng/mLの定量化下限で、液体クロマトグラフィ−質量分光分析(LC−MS/MS)(MDS SCIEX、API4000、Applied Biosystems、Foster City、カリフォルニア州)により判定した。
【0159】
標準的な薬物動態パラメーターは、WinNnlin(4.0.1版、Phrsight、マウンテンビュー、カリフォルニア州)を用いて非区画分析(IVにはモデル201、POにはモデル200)により評価した。血漿中試験化合物濃度の曲線における最大値対時間は、Cmaxと表される。曲線下面積対投与時間から最後の測定可能濃度までの時間曲線(AUC(0−t))を、線形台形則により算出した。経口生物学的利用能、すなわち、IV投与のAUC(0−t)に対するPO投与のAUC(0−t)の用量正規化比は:
F(%)=AUCPO/AUCIV×用量IV/用量PO×100%
として算出した。
【0160】
このアッセイにおいて、パラメーターCmax、AUC(0−t)、およびF(%)のより大きな値を示す試験化合物は、経口投与された際により大きな生物学的利用能を有することが予想される。式Iの化合物は、0.16μg/mLのCmax値、0.46μg・時間/mLおよびラットモデルにおける約19%の生物学的利用能(F(%))を有した。
【0161】
本発明をその特定の実施形態を参照して説明したが、本発明の真の精神および範囲から逸脱することなく、種々の変更ができ、等価物に置換できることは、当業者により理解されるはずである。また、本発明の目的、精神および範囲に対して、特定の状況、材料、物質組成、方法、処理、処理工程または工程に適合させるために、多くの修飾をなすことができる。このような修飾は全て、本明細書に添付した請求項の範囲内にあることが意図されている。また、本明細書において上記で引用した全ての刊行物、特許、および特許文書は、個々に参照として組み込まれているように、全体が本明細書に参照として組み込まれている。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

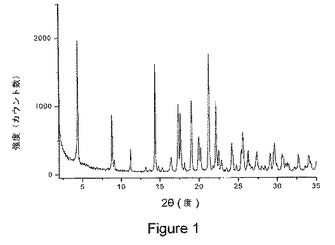
【図2】
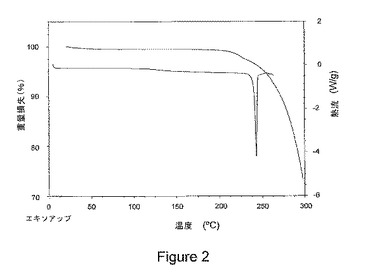
【図3】
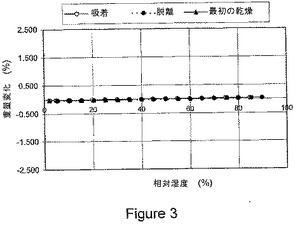
【図4】
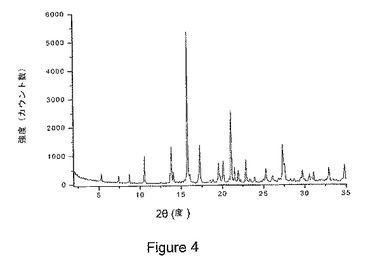
【図5】
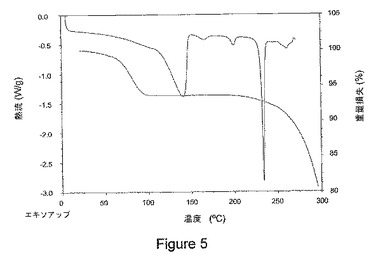
【図6】
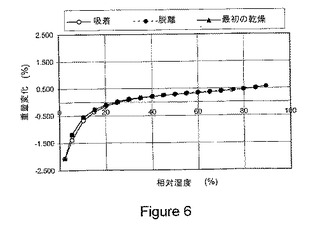
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−153725(P2012−153725A)
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−113464(P2012−113464)
【出願日】平成24年5月17日(2012.5.17)
【分割の表示】特願2008−505559(P2008−505559)の分割
【原出願日】平成18年4月5日(2006.4.5)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願番号】特願2012−113464(P2012−113464)
【出願日】平成24年5月17日(2012.5.17)
【分割の表示】特願2008−505559(P2008−505559)の分割
【原出願日】平成18年4月5日(2006.4.5)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
[ Back to top ]