
キノリン化合物の結晶形及びその製造法
【課題】 安定性に優れた結晶性ピタバスタチンカルシウム原薬製造方法を提供する。
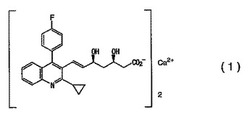
【解決手段】 式(1)
【化1】

で表される化合物(ピタバスタチンカルシウム)を製造するに際し、水分を5〜15%に調節しすること及び結晶形を結晶形態Aに制御することにより、安定性に優れた原薬を得た。
【解決手段】 式(1)
【化1】
で表される化合物(ピタバスタチンカルシウム)を製造するに際し、水分を5〜15%に調節しすること及び結晶形を結晶形態Aに制御することにより、安定性に優れた原薬を得た。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、HMG-CoA還元酵素阻害剤として高脂血症の治療に有用な、化学名 Monocalcium
bis[(3R,5S,6E)-7-(2-cyclopropyl-4-(4-fluorophenyl)-3-quinolyl)-3,5-dihydroxy-6-heptenoate]によって知られている結晶性形態のピタバスタチンカルシウム、その製造法、及びこの該化合物と医薬的に許容し得る担体を含有する医薬組成物に関するものである。
【0002】
詳細には、5〜15%(W/W)の水分を含有することを特徴とし、安定性などの面から医薬品原薬として有用な結晶性形態のピタバスタチンカルシウム、その製造法、及びそれを含む医薬組成物に関する。
【背景技術】
【0003】
ピタバスタチンカルシウム(特許文献1、2及び3参照。)は抗高脂血症治療薬として上市されており、その製造法としては、光学活性α−メチルベンルアミンを用いて光学分割する製造法(特許文献4及び非特許文献1参照。)が既に報告されている。
【0004】
【化1】
【0005】
原料である式(3)で表される化合物の製造法としては、光学異性体分離カラムを用いたクロマト分離(特許文献5参照。)、不斉合成(特許文献6及び7参照。)あるいはキラルシントン(特許文献8参照。)を用いて製造できる式(4)で表される化合物を化学的にシン還元する方法、式(4)で表される化合物を生物学的にシン還元する方法(特許文献9参照。)、及び酵素を利用した光学分割法(特許文献10参照。)が知られている。
【0006】
【化2】
(式中、RはC1−4アルキル基を表わす。)
【0007】
【化3】
(式中、RはC1−4アルキル基を表わす。)
【特許文献1】特開平1−279866号公報
【特許文献2】欧州公開特許304063号公報
【特許文献3】米国特許5011930号公報
【特許文献4】特開平5−148237号公報
【特許文献5】国際公開第95/23125号パンフレット
【特許文献6】国際公開第03/042180号パンフレット
【特許文献7】特開平8−092217号公報
【特許文献8】特開平8−127585号公報
【特許文献9】特開2002-300897号公報
【特許文献10】特開平13−352996号公報
【非特許文献1】Bioorganic & Medicinal Chemistry Letters, 9 (1999) p.2977
【発明の開示】
【発明が解決しようとする課題】
【0008】
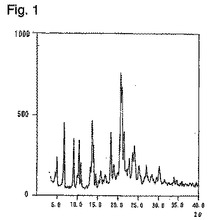
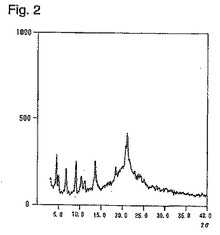
医薬品の原薬としては、高品質及び保存上から安定な結晶性形態を有することが望ましく、さらに大規模な製造にも耐えられることが要求される。ところが、従来のピタバスタチンカルシウムの製造法においては、水分値や結晶形に関する記載が全くない。ピタバスタチンカルシウム(結晶性形態A)に、一般的に行なわれるような乾燥を実施すると、乾燥前は、図1で示すような粉末X線回折図示したものが、水分が4%以下になったところで図2に示すようにアモルファスに近い状態まで結晶性が低下することが判明した。さらに、アモルファス化したピタバスタチンカルシウムは表1に示す如く、保存中の安定性が極めて悪くなることも明らかとなった。
【0009】
【表1】
本発明が解決しようとする課題は、特別な貯蔵条件でなくとも安定なピタバスタチンカルシウムの結晶性原薬を提供することであり、さらに工業的大量製造を可能にすることである。
【課題を解決するための手段】
【0010】
本発明者らは、水分と原薬安定性の相関について鋭意検討を行なった結果、原薬に含まれる水分量を特定の範囲にコントロールすることで、ピタバスタチンカルシウムの安定性が格段に向上することを見出した。さらに、水分が同等で結晶形が異なる形態を3種類見出し、その中で、CuKα放射線を使用して測定した粉末X線回折図によって特徴づけられる結晶(結晶性形態A)が、最も医薬品の原薬として好ましいことを見出し、本発明を完成させた。
【0011】
即ち、本発明は、下記の要旨を有するものである。
1. 式(1)
【0012】
【化4】
で表される化合物であり、5〜15%の水分を含み、CuKα放射線を使用して測定するX線粉末解析において、30.16°の回折角(2θ)に、相対強度が25%より大きなピークを有することを特徴とする結晶(結晶性形態A)。
2. 水又は60%以上の水を含んだC1−4アルコールに溶解された式(2)
【0013】
【化5】
(式中、M+はアルカリ金属イオンを表わす。)で表される化合物に、カルシウム化合物を添加することを特徴とする、上記1に記載の結晶(結晶性形態A)の製造法。
3. 水分値を5〜15%に調整することを特徴とする、上記1に記載の結晶(結晶性形態A)の原薬の製造法。
4. 上記1に記載の結晶(結晶性形態A)を含有することを特徴とする医薬組成物。
【0014】
結晶形態A以外の2種類を結晶形態B及び結晶形態Cと略記するが、これらはいずれも結晶形態Aに特徴的な回折角10.40°、13.20°及び30.16°のピークが存在しないことから、結晶多形であることが明らかにされる。これらは、ろ過性が悪く、厳密な乾燥条件が必要であり(乾燥中の結晶形転移)、NaClなどの無機物が混入する危険性を有し、更に結晶形制御の再現性が必ずしも得られないことが明らかであった。したがって、工業的製造法の観点からは欠点が多く、医薬品の原薬としては結晶形態Aが最も優れている。
【発明を実施するための最良の形態】
【0015】
以下、更に詳細に本発明を説明する。
【0016】
結晶性形態Aのピタバスタチンカルシウムは、その粉末X線回折パターンによって特徴付けることができる。
────────────────────────────────
回折角(2θ) d-面間隔 相対強度
(°) (>25%)
────────────────────────────────
4.96 17.7999 35.9
6.72 13.1423 55.1
9.08 9.7314 33.3
10.40 8.4991 34.8
10.88 8.1248 27.3
13.20 6.7020 27.8
13.60 6.5053 48.8
13.96 6.3387 60.0
18.32 4.8386 56.7
20.68 4.2915 100.0
21.52 4.1259 57.4
23.64 3.7604 41.3
24.12 3.6866 45.0
27.00 3.2996 28.5
30.16 2.9607 30.6
────────────────────────────────
装置
粉末X線回折測定装置:MXLabo(マックサイエンス製)
線源:Cu、波長:1.54056A、ゴニオメータ:縦型ゴニオメータ
モノクロメータ:使用、補助装置:なし、管電圧:50.0Kv、管電流:30.0mA
測定方法:
測定前に、シリコン(標準物質)を用いてX−線管アラインメントを検査する。
試料約100mgをガラス試料板にのせ平坦にした後、以下の条件にて測定する。
データ範囲:3.0400〜40.0000deg、データ点数:925
スキャン軸:2θ/θ、θ軸角度:設定なし
サンプリング間隔:0.0400deg、スキャン速度:4.800deg/min
本発明は、また、ピタバスタチンカルシウムを結晶性形態Aに制御するための製造法を提供する。
【0017】
【化6】
【0018】
原料は式(2)に示すピタバスタチンのアルカリ金属塩であり、アルカリ金属としてはリチウム、ナトリウム、カリウム等を挙げることができ、ナトリウムが好ましい。
カルシウム化合物としては塩化カルシウム、酢酸カルシウムなどが好ましく、使用量は式(2)の化合物に対して0.3倍モル〜3倍モル、好ましくは0.5〜2倍モルの範囲である。
【0019】
式(2)のピタバスタチンのアルカリ金属塩は必ずしも単離される必要はなく、例えば式(3)の化合物などを加水分解する反応に連続してCa塩を製造することもできる。
【0020】
【化7】
【0021】
使用する溶媒としては、水又は60%以上の水を含んだC1−4アルコールが好ましい。C1−4アルコールとしては、メチルアルコール、エチルアルコール、n-プロピルアルコール、イソプロピルアルコール、n-ブチルアルコール、イソブチルアルコール、sec-ブチルアルコール及びtert-ブチルアルコール等を挙げることができる。
【0022】
溶媒の使用量は、式(2)で表される化合物の使用量に対して、3〜100質量倍の範囲であり、5〜30質量倍の範囲が好ましい。
晶析温度は特に限定されないが、−10〜70℃の範囲であり、好ましくは−5〜40℃の範囲であり、更に好ましくは0〜20℃の範囲である。
晶析時間は特に限定されないが、30分〜15時間程度行えば十分である。
結晶を析出させる際の方法としては、静置で行う方法、攪拌下で行う方法等が挙げられるが、攪拌下で行うのが好ましい。
また、必要に応じて結晶形態Aの種晶を使用してもよい。
【0023】
析出した結晶を濾過し、乾燥するが、水分の調整が本発明において極めて重要である。乾燥温度は特に限定されないが、好ましくは15〜40℃の範囲である。
水分値は、最終的に5〜15%(W/W)の範囲になるよう調整されるが、好ましくは7〜15%(W/W)、より好ましくは7〜13%(W/W)、最も好ましくは9〜13%(W/W)の範囲である。
得られたピタバスタチンカルシウムは粉砕された後、医薬品用の原薬として使用される。
【0024】
本発明に係る化合物の投与形態としては、注射剤(皮下、静脈内、筋肉内、腹腔内注射)、軟膏剤、坐剤、エアゾール剤等による非経口投与又は錠剤、カプセル剤、顆粒剤、丸剤、シロップ剤、液剤、乳剤、懸濁液剤等による経口投与をあげることができる。
本発明に係る化合物を含有する上記の医薬的又は獣医薬的組成物は、全組成物の重量に対して、本発明に係る化合物を約0.001〜30%、好ましくは、約0.01〜10%を含有する。
本発明に係る化合物に又は該化合物を含有する組成物に加えて、他の医薬的に又は獣医薬的に活性な化合物を含ませることができる。
【0025】
本発明に係る化合物の臨床的投与量は、年令、体重、患者の感受性、症状の程度等により異なるが、通常効果的な投与量は、成人一日当たり0.003〜100mg、好ましくは、0.01〜10mg程度である。しかし必要により上記の範囲外の量を用いることもできる。
【0026】
本発明に係る化合物は、製薬の慣用手段によって投与用に製剤化される。
即ち、経口投与用の錠剤、カプセル剤、顆粒剤、丸剤は、賦形剤、例えば白糖、乳糖、ブドウ糖、でんぷん、マンニット;結合剤、例えばヒドロキシプロピルセルロース、シロップ、アラビアゴム、ゼラチン、ソルビット、トラガント、メチルセルロース、ポリビニルピロリドン;崩壊剤、例えばでんぷん、カルボキシメチルセルロース又はそのカルシウム塩、微結晶セルロース、ポリエチレングリコール;滑沢剤、例えばタルク、ステアリン酸マグネシウム又はカルシウム、シリカ;潤滑剤、例えばラウリル酸ナトリウム、グリセロール等を使用して調製される。
【0027】
注射剤、液剤、乳剤、懸濁剤、シロップ剤及びエアゾール剤は、活性成分の溶剤、例えば水、エチルアルコール、イソプロピルアルコール、プロピレングリコール、1,3−ブチレングリコール、ポリエチレングリコール;界面活性剤、例えばソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン脂肪酸エステル、水素添加ヒマシ油のポリオキシエチレンエーテル、レシチン;懸濁剤、例えばカルボキシメチルナトリウム塩、メチルセルロース等のセルロース誘導体、トラガント、アラビアゴム等の天然ゴム類;保存剤、例えばパラオキシ安息香酸のエステル、塩化ベンザルコニウム、ソルビン酸塩等を使用して調製される。
【0028】
経皮吸収型製剤である軟膏には、例えば白色ワセリン、流動パラフィン、高級アルコール、マクロゴール軟膏、親水軟膏、水性ゲル基剤等が用いられる。
坐剤は、例えばカカオ脂、ポリエチレングリコール、ラノリン、脂肪酸トリグリセライド、ココナット油、ポリソルベート等を使用して調製される。
【実施例】
【0029】
次に実施例を挙げて本発明を具体的に説明するが、本発明の範囲はこれらに限定されるものではない。
尚、実施例に使用した化合物(5)は、WO95/23125号公報に記載の方法に従って製造した。
【0030】
実施例1
【0031】
【化8】
【0032】
2.71kg(6.03mol)の化合物(5)を、50kgのエタノールに撹拌しながら溶解し、均一溶液であることを確認した上で、58.5kgの水を加えた。-3〜3℃に冷却した後、2mol/リットル(L)水酸化ナトリウム水溶液の3.37Lを滴下した後、続けて同温度で3時間撹拌し、加水分解反応を完結させた。全量の水酸化ナトリウム水溶液を反応系に送り込むため、4.70kgの水を使用した。
【0033】
反応混合物を減圧下に蒸留して溶媒を留去し、52.2kgのエタノール/水を除去後、内温を10〜20℃に調整した。得られた濃縮液中に、別途調製しておいた塩化カルシウム水溶液(95%CaCl2 775g/水39.3kg、6.63mol)を2時間かけて滴下した。全量の塩化カルシウム水溶液を反応系に送り込むため、4.70kgの水を使用した。滴下終了後、同温度で12時間撹拌を継続し、析出した結晶を濾取した。結晶を72.3kgの水で洗浄後、乾燥器内で減圧下40℃にて、品温に注意しながら、水分値が10%になるまで乾燥することにより、2.80kg(収率95%)のピタバスタチンカルシウムを白色の結晶として得た。
粉末X線回折を測定して、この結晶が結晶形態Aであることを確認した。
【産業上の利用可能性】
【0034】
本発明により、安定性に優れたピタバスタチンカルシウム結晶性原薬の工業的な製造法が確立された。
【図面の簡単な説明】
【0035】
【図1】水分値が8.78%である結晶性形態Aの粉末X線回折図である。
【図2】図1で使用した結晶を乾燥し、水分値を3.76%とした際の粉末X線回折図である。
【技術分野】
【0001】
本発明は、HMG-CoA還元酵素阻害剤として高脂血症の治療に有用な、化学名 Monocalcium
bis[(3R,5S,6E)-7-(2-cyclopropyl-4-(4-fluorophenyl)-3-quinolyl)-3,5-dihydroxy-6-heptenoate]によって知られている結晶性形態のピタバスタチンカルシウム、その製造法、及びこの該化合物と医薬的に許容し得る担体を含有する医薬組成物に関するものである。
【0002】
詳細には、5〜15%(W/W)の水分を含有することを特徴とし、安定性などの面から医薬品原薬として有用な結晶性形態のピタバスタチンカルシウム、その製造法、及びそれを含む医薬組成物に関する。
【背景技術】
【0003】
ピタバスタチンカルシウム(特許文献1、2及び3参照。)は抗高脂血症治療薬として上市されており、その製造法としては、光学活性α−メチルベンルアミンを用いて光学分割する製造法(特許文献4及び非特許文献1参照。)が既に報告されている。
【0004】
【化1】
【0005】
原料である式(3)で表される化合物の製造法としては、光学異性体分離カラムを用いたクロマト分離(特許文献5参照。)、不斉合成(特許文献6及び7参照。)あるいはキラルシントン(特許文献8参照。)を用いて製造できる式(4)で表される化合物を化学的にシン還元する方法、式(4)で表される化合物を生物学的にシン還元する方法(特許文献9参照。)、及び酵素を利用した光学分割法(特許文献10参照。)が知られている。
【0006】
【化2】
(式中、RはC1−4アルキル基を表わす。)
【0007】
【化3】
(式中、RはC1−4アルキル基を表わす。)
【特許文献1】特開平1−279866号公報
【特許文献2】欧州公開特許304063号公報
【特許文献3】米国特許5011930号公報
【特許文献4】特開平5−148237号公報
【特許文献5】国際公開第95/23125号パンフレット
【特許文献6】国際公開第03/042180号パンフレット
【特許文献7】特開平8−092217号公報
【特許文献8】特開平8−127585号公報
【特許文献9】特開2002-300897号公報
【特許文献10】特開平13−352996号公報
【非特許文献1】Bioorganic & Medicinal Chemistry Letters, 9 (1999) p.2977
【発明の開示】
【発明が解決しようとする課題】
【0008】
医薬品の原薬としては、高品質及び保存上から安定な結晶性形態を有することが望ましく、さらに大規模な製造にも耐えられることが要求される。ところが、従来のピタバスタチンカルシウムの製造法においては、水分値や結晶形に関する記載が全くない。ピタバスタチンカルシウム(結晶性形態A)に、一般的に行なわれるような乾燥を実施すると、乾燥前は、図1で示すような粉末X線回折図示したものが、水分が4%以下になったところで図2に示すようにアモルファスに近い状態まで結晶性が低下することが判明した。さらに、アモルファス化したピタバスタチンカルシウムは表1に示す如く、保存中の安定性が極めて悪くなることも明らかとなった。
【0009】
【表1】
本発明が解決しようとする課題は、特別な貯蔵条件でなくとも安定なピタバスタチンカルシウムの結晶性原薬を提供することであり、さらに工業的大量製造を可能にすることである。
【課題を解決するための手段】
【0010】
本発明者らは、水分と原薬安定性の相関について鋭意検討を行なった結果、原薬に含まれる水分量を特定の範囲にコントロールすることで、ピタバスタチンカルシウムの安定性が格段に向上することを見出した。さらに、水分が同等で結晶形が異なる形態を3種類見出し、その中で、CuKα放射線を使用して測定した粉末X線回折図によって特徴づけられる結晶(結晶性形態A)が、最も医薬品の原薬として好ましいことを見出し、本発明を完成させた。
【0011】
即ち、本発明は、下記の要旨を有するものである。
1. 式(1)
【0012】
【化4】
で表される化合物であり、5〜15%の水分を含み、CuKα放射線を使用して測定するX線粉末解析において、30.16°の回折角(2θ)に、相対強度が25%より大きなピークを有することを特徴とする結晶(結晶性形態A)。
2. 水又は60%以上の水を含んだC1−4アルコールに溶解された式(2)
【0013】
【化5】
(式中、M+はアルカリ金属イオンを表わす。)で表される化合物に、カルシウム化合物を添加することを特徴とする、上記1に記載の結晶(結晶性形態A)の製造法。
3. 水分値を5〜15%に調整することを特徴とする、上記1に記載の結晶(結晶性形態A)の原薬の製造法。
4. 上記1に記載の結晶(結晶性形態A)を含有することを特徴とする医薬組成物。
【0014】
結晶形態A以外の2種類を結晶形態B及び結晶形態Cと略記するが、これらはいずれも結晶形態Aに特徴的な回折角10.40°、13.20°及び30.16°のピークが存在しないことから、結晶多形であることが明らかにされる。これらは、ろ過性が悪く、厳密な乾燥条件が必要であり(乾燥中の結晶形転移)、NaClなどの無機物が混入する危険性を有し、更に結晶形制御の再現性が必ずしも得られないことが明らかであった。したがって、工業的製造法の観点からは欠点が多く、医薬品の原薬としては結晶形態Aが最も優れている。
【発明を実施するための最良の形態】
【0015】
以下、更に詳細に本発明を説明する。
【0016】
結晶性形態Aのピタバスタチンカルシウムは、その粉末X線回折パターンによって特徴付けることができる。
────────────────────────────────
回折角(2θ) d-面間隔 相対強度
(°) (>25%)
────────────────────────────────
4.96 17.7999 35.9
6.72 13.1423 55.1
9.08 9.7314 33.3
10.40 8.4991 34.8
10.88 8.1248 27.3
13.20 6.7020 27.8
13.60 6.5053 48.8
13.96 6.3387 60.0
18.32 4.8386 56.7
20.68 4.2915 100.0
21.52 4.1259 57.4
23.64 3.7604 41.3
24.12 3.6866 45.0
27.00 3.2996 28.5
30.16 2.9607 30.6
────────────────────────────────
装置
粉末X線回折測定装置:MXLabo(マックサイエンス製)
線源:Cu、波長:1.54056A、ゴニオメータ:縦型ゴニオメータ
モノクロメータ:使用、補助装置:なし、管電圧:50.0Kv、管電流:30.0mA
測定方法:
測定前に、シリコン(標準物質)を用いてX−線管アラインメントを検査する。
試料約100mgをガラス試料板にのせ平坦にした後、以下の条件にて測定する。
データ範囲:3.0400〜40.0000deg、データ点数:925
スキャン軸:2θ/θ、θ軸角度:設定なし
サンプリング間隔:0.0400deg、スキャン速度:4.800deg/min
本発明は、また、ピタバスタチンカルシウムを結晶性形態Aに制御するための製造法を提供する。
【0017】
【化6】
【0018】
原料は式(2)に示すピタバスタチンのアルカリ金属塩であり、アルカリ金属としてはリチウム、ナトリウム、カリウム等を挙げることができ、ナトリウムが好ましい。
カルシウム化合物としては塩化カルシウム、酢酸カルシウムなどが好ましく、使用量は式(2)の化合物に対して0.3倍モル〜3倍モル、好ましくは0.5〜2倍モルの範囲である。
【0019】
式(2)のピタバスタチンのアルカリ金属塩は必ずしも単離される必要はなく、例えば式(3)の化合物などを加水分解する反応に連続してCa塩を製造することもできる。
【0020】
【化7】
【0021】
使用する溶媒としては、水又は60%以上の水を含んだC1−4アルコールが好ましい。C1−4アルコールとしては、メチルアルコール、エチルアルコール、n-プロピルアルコール、イソプロピルアルコール、n-ブチルアルコール、イソブチルアルコール、sec-ブチルアルコール及びtert-ブチルアルコール等を挙げることができる。
【0022】
溶媒の使用量は、式(2)で表される化合物の使用量に対して、3〜100質量倍の範囲であり、5〜30質量倍の範囲が好ましい。
晶析温度は特に限定されないが、−10〜70℃の範囲であり、好ましくは−5〜40℃の範囲であり、更に好ましくは0〜20℃の範囲である。
晶析時間は特に限定されないが、30分〜15時間程度行えば十分である。
結晶を析出させる際の方法としては、静置で行う方法、攪拌下で行う方法等が挙げられるが、攪拌下で行うのが好ましい。
また、必要に応じて結晶形態Aの種晶を使用してもよい。
【0023】
析出した結晶を濾過し、乾燥するが、水分の調整が本発明において極めて重要である。乾燥温度は特に限定されないが、好ましくは15〜40℃の範囲である。
水分値は、最終的に5〜15%(W/W)の範囲になるよう調整されるが、好ましくは7〜15%(W/W)、より好ましくは7〜13%(W/W)、最も好ましくは9〜13%(W/W)の範囲である。
得られたピタバスタチンカルシウムは粉砕された後、医薬品用の原薬として使用される。
【0024】
本発明に係る化合物の投与形態としては、注射剤(皮下、静脈内、筋肉内、腹腔内注射)、軟膏剤、坐剤、エアゾール剤等による非経口投与又は錠剤、カプセル剤、顆粒剤、丸剤、シロップ剤、液剤、乳剤、懸濁液剤等による経口投与をあげることができる。
本発明に係る化合物を含有する上記の医薬的又は獣医薬的組成物は、全組成物の重量に対して、本発明に係る化合物を約0.001〜30%、好ましくは、約0.01〜10%を含有する。
本発明に係る化合物に又は該化合物を含有する組成物に加えて、他の医薬的に又は獣医薬的に活性な化合物を含ませることができる。
【0025】
本発明に係る化合物の臨床的投与量は、年令、体重、患者の感受性、症状の程度等により異なるが、通常効果的な投与量は、成人一日当たり0.003〜100mg、好ましくは、0.01〜10mg程度である。しかし必要により上記の範囲外の量を用いることもできる。
【0026】
本発明に係る化合物は、製薬の慣用手段によって投与用に製剤化される。
即ち、経口投与用の錠剤、カプセル剤、顆粒剤、丸剤は、賦形剤、例えば白糖、乳糖、ブドウ糖、でんぷん、マンニット;結合剤、例えばヒドロキシプロピルセルロース、シロップ、アラビアゴム、ゼラチン、ソルビット、トラガント、メチルセルロース、ポリビニルピロリドン;崩壊剤、例えばでんぷん、カルボキシメチルセルロース又はそのカルシウム塩、微結晶セルロース、ポリエチレングリコール;滑沢剤、例えばタルク、ステアリン酸マグネシウム又はカルシウム、シリカ;潤滑剤、例えばラウリル酸ナトリウム、グリセロール等を使用して調製される。
【0027】
注射剤、液剤、乳剤、懸濁剤、シロップ剤及びエアゾール剤は、活性成分の溶剤、例えば水、エチルアルコール、イソプロピルアルコール、プロピレングリコール、1,3−ブチレングリコール、ポリエチレングリコール;界面活性剤、例えばソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン脂肪酸エステル、水素添加ヒマシ油のポリオキシエチレンエーテル、レシチン;懸濁剤、例えばカルボキシメチルナトリウム塩、メチルセルロース等のセルロース誘導体、トラガント、アラビアゴム等の天然ゴム類;保存剤、例えばパラオキシ安息香酸のエステル、塩化ベンザルコニウム、ソルビン酸塩等を使用して調製される。
【0028】
経皮吸収型製剤である軟膏には、例えば白色ワセリン、流動パラフィン、高級アルコール、マクロゴール軟膏、親水軟膏、水性ゲル基剤等が用いられる。
坐剤は、例えばカカオ脂、ポリエチレングリコール、ラノリン、脂肪酸トリグリセライド、ココナット油、ポリソルベート等を使用して調製される。
【実施例】
【0029】
次に実施例を挙げて本発明を具体的に説明するが、本発明の範囲はこれらに限定されるものではない。
尚、実施例に使用した化合物(5)は、WO95/23125号公報に記載の方法に従って製造した。
【0030】
実施例1
【0031】
【化8】
【0032】
2.71kg(6.03mol)の化合物(5)を、50kgのエタノールに撹拌しながら溶解し、均一溶液であることを確認した上で、58.5kgの水を加えた。-3〜3℃に冷却した後、2mol/リットル(L)水酸化ナトリウム水溶液の3.37Lを滴下した後、続けて同温度で3時間撹拌し、加水分解反応を完結させた。全量の水酸化ナトリウム水溶液を反応系に送り込むため、4.70kgの水を使用した。
【0033】
反応混合物を減圧下に蒸留して溶媒を留去し、52.2kgのエタノール/水を除去後、内温を10〜20℃に調整した。得られた濃縮液中に、別途調製しておいた塩化カルシウム水溶液(95%CaCl2 775g/水39.3kg、6.63mol)を2時間かけて滴下した。全量の塩化カルシウム水溶液を反応系に送り込むため、4.70kgの水を使用した。滴下終了後、同温度で12時間撹拌を継続し、析出した結晶を濾取した。結晶を72.3kgの水で洗浄後、乾燥器内で減圧下40℃にて、品温に注意しながら、水分値が10%になるまで乾燥することにより、2.80kg(収率95%)のピタバスタチンカルシウムを白色の結晶として得た。
粉末X線回折を測定して、この結晶が結晶形態Aであることを確認した。
【産業上の利用可能性】
【0034】
本発明により、安定性に優れたピタバスタチンカルシウム結晶性原薬の工業的な製造法が確立された。
【図面の簡単な説明】
【0035】
【図1】水分値が8.78%である結晶性形態Aの粉末X線回折図である。
【図2】図1で使用した結晶を乾燥し、水分値を3.76%とした際の粉末X線回折図である。
【特許請求の範囲】
【請求項1】
式(1)
【化1】
で表される化合物であり、5〜15%の水分を含み、CuKα放射線を使用して測定するX線粉末解析において、30.16°の回折角(2θ)に、相対強度が25%より大きなピークを有することを特徴とする結晶(結晶性形態A)。
【請求項2】
水又は60%以上の水を含んだC1−4アルコールに溶解された式(2)
【化2】
(式中、M+はアルカリ金属イオンを表わす。)で表される化合物に、カルシウム化合物を添加することを特徴とする、請求項1に記載の結晶(結晶性形態A)の製造法。
【請求項3】
水分値を5〜15%に調整することを特徴とする、請求項1に記載の結晶(結晶性形態A)の原薬の製造法。
【請求項4】
請求項1に記載の結晶(結晶性形態A)を含有することを特徴とする医薬組成物。
【請求項1】
式(1)
【化1】
で表される化合物であり、5〜15%の水分を含み、CuKα放射線を使用して測定するX線粉末解析において、30.16°の回折角(2θ)に、相対強度が25%より大きなピークを有することを特徴とする結晶(結晶性形態A)。
【請求項2】
水又は60%以上の水を含んだC1−4アルコールに溶解された式(2)
【化2】
(式中、M+はアルカリ金属イオンを表わす。)で表される化合物に、カルシウム化合物を添加することを特徴とする、請求項1に記載の結晶(結晶性形態A)の製造法。
【請求項3】
水分値を5〜15%に調整することを特徴とする、請求項1に記載の結晶(結晶性形態A)の原薬の製造法。
【請求項4】
請求項1に記載の結晶(結晶性形態A)を含有することを特徴とする医薬組成物。
【図1】

【図2】
【図2】
【公表番号】特表2007−516952(P2007−516952A)
【公表日】平成19年6月28日(2007.6.28)
【国際特許分類】
【出願番号】特願2006−520594(P2006−520594)
【出願日】平成16年12月17日(2004.12.17)
【国際出願番号】PCT/JP2004/019451
【国際公開番号】WO2005/063711
【国際公開日】平成17年7月14日(2005.7.14)
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
【公表日】平成19年6月28日(2007.6.28)
【国際特許分類】
【出願日】平成16年12月17日(2004.12.17)
【国際出願番号】PCT/JP2004/019451
【国際公開番号】WO2005/063711
【国際公開日】平成17年7月14日(2005.7.14)
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
[ Back to top ]