
キラルシアノヒドリンの酵素的加水分解によりキラルα−ヒドロキシカルボン酸を製造する方法
本発明は、(R)−又は(S)−シアノヒドリンを、ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 の存在下の酵素的加水分解により、それぞれ(R)−又は(S)−α−ヒドロキシカルボン酸に変換することを特徴とする、結晶性のキラルα−ヒドロキシカルボン酸の製造方法に関する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
光学活性α−ヒドロキシカルボン酸は、例えば、食品添加物として、又は、医薬用活性化合物、ビタミン及び液晶の製造において用いられる。
【0002】
これらの光学活性α−ヒドロキシカルボン酸はまた、有利なことに、例えば Effenberger 他,Angew. Chem.,95(1983),No.1,50頁に従って、他の方法では調製が極めて困難なN−置換光学活性α−アミノ酸に変換することができる。
【0003】
キラルα−ヒドロキシカルボン酸は、今日では、化学的に、又は発酵によって、又は酵素的に得ることができる。
【0004】
従って、文献には、キラルα−ヒドロキシカルボン酸を合成する多くの種々の方法が開示されている。
【0005】
例えば、適当な微生物の添加により、シアノヒドリンのラセミ体を加水分解して、所望のキラルα−ヒドロキシカルボン酸を得ることができる。
【0006】
アルカリゲネス(Alcaligenes)属、シュードモナス(Pseudomonas)属、アシネトバクター(Acinetobacter)属、ロードコッカス(Rhodococcus)属、カンジダ(Candida)属等の種々の微生物を用いる、シアノヒドリンのラセミ体からのキラルα−ヒドロキシカルボン酸の製造、特に光学活性な乳酸又はマンデル酸の製造が、例えば、欧州特許第0449684号明細書、欧州特許第0527553号明細書、欧州特許第0610048号明細書等に記載されている。
【0007】
このような従来技術からは、ラセミ体のシアノヒドリンをニトリラーゼの存在下で共役α−ヒドロキシカルボン酸に酵素的に加水分解するとき、酵素が短時間のうちに失活し、その結果、所望のα−ヒドロキシカルボン酸が、通常、低い収率及び濃度でしか得られないという問題が生じていることも分かる。このことは、シアノヒドリンを共役のα−ヒドロキシアミドに変換するニトリルヒドラターゼの使用にも当てはまる。このヒドロキシアミドは、共役のα−ヒドロキシカルボン酸に更に変換することができる。
【0008】
例えば Angew. Chem.,1994,106,1615頁以降からは、光学活性シアノヒドリンを、ラセミ化を行わずに濃塩酸で加水分解して、共役のキラルα−ヒドロキシカルボン酸を得ることができることも分かる。そのようにして製造されたキラルα−ヒドロキシカルボン酸の光学純度は、用いたキラルシアノヒドリンの光学純度に一致する。このことは、用いるキラルシアノヒドリンを、シアニド基を共役のアルデヒド又はケトンに酵素触媒で付加することにより in situ で得、単離又は精製を行わずに更に処理する場合にも当てはまる。
【0009】
この反応の欠点は、反応性の高い基質が分解し、腐食を引き起こすことである。
【0010】
本発明は、キラルシアノヒドリンと同程度の極性を有するニトリルを、穏和かつ効率的な方法を用いて、シアノヒドリンとほぼ同じエナンチオマー純度を有する共役のキラルヒドロキシカルボン酸に変換することができる方法を見出すことを目的としていた。
【0011】
意外なことに、この目的は、ロードコッカス属に属する特殊な細菌を用いることによって達成された。
【0012】
すなわち、本発明は、キラルα−ヒドロキシカルボン酸を製造する方法であって、(R)−又は(S)−シアノヒドリンを、ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 の存在下で酵素的加水分解を行うことにより、共役の(R)−又は(S)−α−ヒドロキシカルボン酸に変換することを含む方法に関する。
【0013】
本発明の方法において、(R)−及び(S)−シアノヒドリンは、光学純度が>99%eeまでの(R)−及び(S)−α−ヒドロキシカルボン酸に変換される。
【0014】
対応するアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加によって得られる(R)−及び(S)−シアノヒドリンは、出発化合物となる。
【0015】
対応するアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加は、従来技術に準じて、例えば、欧州特許第0951561号明細書、欧州特許第0927766号明細書、欧州特許第0632130号明細書、欧州特許第0547655号明細書、欧州特許第0326063号明細書等の方法に準じて行うことができる。
【0016】
好適な出発化合物は、従来技術において挙げられているアルデヒド又はケトンである。
【0017】
好適なアルデヒドの例としては、脂肪族、芳香族又は複素芳香族アルデヒドがある。脂肪族アルデヒドとは、飽和又は不飽和の脂肪族の直鎖、分枝鎖又は環状アルデヒドを意味するものとする。好ましい脂肪族アルデヒドは、特に2個〜18個、特に好ましくは2個〜12個の炭素原子を有する、飽和又はモノ不飽和若しくはポリ不飽和の直鎖アルデヒドである。アルデヒドは、C−C二重結合を有しても、C−C三重結合を有してもよい。アルデヒドは、置換されていなくても、また、反応条件下で不活性である基によって一置換又は多置換されていてもよい。例えば、置換されていてもよいアリール基又はヘテロアリール基(例えば、フェニル基又はインドリル基)、或いは、C1−C6アルキル基、置換されていてもよいシクロアルキル基(これは、O、S、P又はNの群より選ばれる1つ又は複数のヘテロ原子を有してもよい)、ハロゲン基、エーテル基、アルコール基、アシル基、カルボン酸基、カルボン酸エステル基、ニトロ基又はアジド基によって一置換又は多置換されていてもよい。
【0018】
芳香族又は複素芳香族アルデヒドの例としては、ベンズアルデヒド、又は種々の置換基で置換されたベンズアルデヒド(例えば、2−クロロベンズアルデヒド、3,4−ジフルオロベンズアルデヒド、4−メチルベンズアルデヒド、3−フェノキシベンズアルデヒド、4−フルオロ−3−フェノキシベンズアルデヒド)があり、更に、フルフラール、アントラセン−9−カルボアルデヒド、フラン−3−カルボアルデヒド、インドール−3−カルボアルデヒド、ナフタレン−1−カルボアルデヒド、フタルジアルデヒド、ピラゾール−3−カルボアルデヒド、ピロール−2−カルボアルデヒド、チオフェン−2−カルボアルデヒド、イソフタルアルデヒド、ピリジンアルデヒド等がある。
【0019】
ケトンの例としては、カルボニル炭素原子が不均等に置換されている脂肪族、芳香族又は複素芳香族ケトンがある。脂肪族ケトンとは、直鎖、分枝鎖又は環状ケトンを意味するものとする。ケトンは、飽和であっても、モノ不飽和又はポリ不飽和であってもよい。ケトンは、置換されていなくても、また、反応条件下で不活性である基によって一置換又は多置換されていてもよい。例えば、置換されていてもよいアリール基又はヘテロアリール基(例えば、フェニル基又はインドリル基)、或いは、ハロゲン基、エーテル基、アルコール基、アシル基、カルボン酸基、カルボン酸エステル基、ニトロ基又はアジド基によって一置換又は多置換されていてもよい。
【0020】
芳香族ケトン又は複素芳香族ケトンの例としては、アセトフェノン、インドリルアセトン等がある。
【0021】
好ましいのは、式:
【化1】

[式中、R1及びR2は相互に独立して、H、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいC1−C6アルキルラジカル若しくはC1−C6アルケニルラジカル、又は、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいフェニルラジカルである。但し、R1及びR2が共にHであることはない。]
の(R)−又は(S)−シアノヒドリンである。
【0022】
反応条件下で不活性である置換基として好ましいのは、例えば、ハロゲン(例えば、フッ素、臭素及び塩素)、C1−C6アルキル若しくはC1−C6アルコキシ、エーテル、エステル、アセタール、又は置換されていてもよいフェニル若しくはフェニルオキシである。
【0023】
本発明の方法における適当な化合物は、特に好ましくは、(R)−又は(S)−シアノヒドリンであり、例えば、(R)−又は(S)−2−ヒドロキシ−4−フェニルブチロニトリル、(R)−又は(S)−2−クロロマンデロニトリル、(R)−又は(S)−マンデロニトリル、(R)−又は(S)−4−メチルマンデロニトリル、(R)−又は(S)−3−フェノキシマンデロニトリル、(R)−又は(S)−2−ヒドロキシ−2−メチルヘプタンニトリル、(R)−又は(S)−2−ヒドロキシ−2−フェニルプロピオニトリル、(R)−又は(S)−2−ヒドロキシ−3−ペンテンニトリル、(R)−又は(S)−1−ヒドロキシシクロヘキサンニトリル、及び(R)−又は(S)−アセトフェノンシアノヒドリンである。
【0024】
次いで、対応する(R)−又は(S)−シアノヒドリンを、本発明に従って酵素的に加水分解する。
【0025】
酵素的加水分解は、ロードコッカス・エリスロポレス NCIMB 11540 の存在下、本発明に従って行う。
【0026】
ロードコッカス・エリスロポレス NCIMB 11540 は、意外なことに、上記のシアノヒドリンと同様の極性を有するニトリルのニトリル官能基を加水分解することができる、利用可能なニトリルヒドラターゼ/アミダーゼ酵素系を有することを特徴とする微生物であった。
【0027】
ロードコッカス・エリスロポレス NCIMB 11540 のニトリルヒドラターゼ/アミダーゼ酵素系を用いると、最初のステップにおいて、キラルシアノヒドリンが、ニトリルヒドラターゼにより共役のキラルヒドロキシアミドに加水分解され、次の加水分解ステップにおいて、キラルヒドロキシアミドが、アミダーゼにより、対応するキラルα−ヒドロキシカルボン酸に変換される。
【0028】
微生物は、本発明の方法では、任意の所望の形態で、例えば、粉砕細胞、粗酵素若しくは精製酵素、組換え酵素、固定化細胞若しくは固定化酵素、凍結乾燥細胞、又は「休止細胞」の形態で用いることができる。
【0029】
組換え酵素、休止細胞又は凍結乾燥細胞を用いるのが好ましく、組換え酵素又は休止細胞を用いるのが特に好ましい。
【0030】
ニトリルヒドラターゼ/アミダーゼ活性を有する、ロードコッカス・エリスロポレス NCIMB 11540 細胞の調製物を直接用いる代わりに、適当な微生物(例えば、大腸菌(E. coli)、ピキア・パストリス(Pichia pastoris)、サッカロマイセス(Saccharomyces)、アスペルギルス(Aspergillus)、K. Lactis 等)において発現させた組換え調製物を用いるのもよい。対応する遺伝子は、プラスミド構築物を用いて、適当な宿主細胞に、例えば、大腸菌、ピキア・パストリス、サッカロマイセス、アスペルギルス又は K. Lactis の宿主細胞に導入する。誘導可能なプロモーターを選ぶことによって、ニトリルヒドラターゼだけでなく、アミダーゼも、活性な形態で過剰発現させることができる。アミダーゼの場合、ロードコッカス細胞の対応する発酵の場合よりもはるかに高い活性レベルを達成することができる。
【0031】
次いで、微生物を、所望の形態で水性溶媒(例えば、水又は緩衝溶液)に懸濁する。好適な緩衝溶液は、例えば、リン酸緩衝液(例えば、K/Naリン酸緩衝液)、PBS緩衝液、酪酸緩衝液、クエン酸塩溶液等である。
【0032】
用いる緩衝溶液のpHは、4.5〜11、好ましくは5.5〜8.5の範囲内にあるのがよい。
【0033】
次いで、得られた懸濁物を、対応するキラルシアノヒドリンと混合する。キラルシアノヒドリンは、限定された水溶性を有する脂溶性化合物であるので、シアノヒドリンを水性溶媒に溶解させるためには、共溶媒として可溶化剤を用いる必要がある。
【0034】
適当な可溶化剤は、例えば、有機溶媒、界面活性剤、相間移動触媒等である。
【0035】
本発明の方法における共溶媒として適当な有機溶媒は、第1に、基質を十分に溶解することができ、第2に、酵素活性に対する悪影響が可能な限り小さいものである。
【0036】
このような有機溶媒の例としては、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド(DMF)、C1−C6アルコール(例えば、メタノール、エタノール、イソプロパノール、1−ブタノール、2−ブタノール、tert−ブタノール又は1−ペンタノール)、トルエン又はtert−ブチルメチルエーテル(TBME)、或いはそれらの混合物がある。
【0037】
用いる共溶媒としては、DMSO、DMF、エタノール、イソプロパノール又はそれらの混合物が好ましく、DMSO又はDMFが特に好ましい。
【0038】
共溶媒の割合は、反応溶液の全体積を基準として、0.5〜20体積%の範囲内にあるのがよい。
【0039】
共溶媒の割合は、好ましくは1〜15体積%、特に好ましくは2〜10体積%である。
【0040】
本発明の方法における反応液中の基質濃度は、(反応溶液の全体積を基準として、)1〜100g/lの範囲内にあるのがよい。十分に高い基質濃度が許容されることは、本発明の酵素的加水分解を実験規模で用いるための基本的前提条件である。
【0041】
基質濃度としては、50g/l以下が好ましく、25g/l以下が特に好ましい。
【0042】
反応可能な基質濃度は、用いる酵素量に依存する。効率的な定量的反応のためには、最初の加水分解ステップを極めて迅速に行って、シアノヒドリンの分解及びこれに伴うラセミ化を避けなければならず、そのため、比較的大きい細胞密度が必要となる。
【0043】
この場合、反応系の十分な混合を確実に行うことに留意する必要がある。
【0044】
細胞量又は酵素量は、用いる形態における微生物の活性に依存し、更に、基質濃度及び共溶媒にも依存する。
【0045】
反応混合物のpHは、4.5〜11、好ましくは5.5〜8の範囲内にあるのがよい。
【0046】
また、必要に応じて、適当な酸又は酸性塩(例えば、リン酸、ホウ酸、クエン酸等)を反応混合物に加えて、pHを調節することができる。
【0047】
本発明の酵素的加水分解は、10〜60℃、好ましくは15〜50℃、特に好ましくは20〜45℃の範囲内の温度で行う。
【0048】
加水分解を行って、所望のキラルα−ヒドロキシカルボン酸を得た後、キラルα−ヒドロキシカルボン酸は、既知の技術によって、例えば、細胞の遠心分離、又はHCl(例えば、pH2)による酸性化後の生成物の抽出によって、また、必要に応じて更に、活性炭濾過及び再結晶による精製を行うことによって、反応混合物から単離される。
【0049】
従って、ロードコッカス・エリスロポレス NCIMB 11540 を本発明に従って用いれば、極性のニトリル(例えば、キラルシアノヒドリン)が、穏和な条件下で簡便かつ効率的に、ラセミ化が生じることなく、共役のキラルα−ヒドロキシカルボン酸に変換される。所望のα−ヒドロキシカルボン酸が、用いたシアノヒドリンのee値に応じて、99%超までの高い光学純度で、かつ98%超までの高い収率で得られる。
【0050】
(実施例1:生体触媒の製造)
ロードコッカス・エリスロポレス NCIMB 11540 のバイオマスを製造するために、複合的な標準培地(培地A、表1参照)を用いた。菌株を、培地Aを用いた寒天プレート上に保持した(15g/lの寒天を用いた固化)。プレートを、パラフィルムで側面から包むことによって密封し、冷蔵庫に4℃で保存した。
【0051】
液体培養物の増殖を、250mlの培地Aを用いて、シケイン(chicane)付きの1000ml三角フラスコ内で、30℃及び130rpmで行った。
【0052】
変形例I(前培養なし): 寒天プレートのバイオマスの約半分を、5mlの滅菌生理食塩水に懸濁した。細胞懸濁物を250mlの培地に加えた。
【0053】
変形例II(前培養あり): 前培養のために、寒天プレートのバイオマスの一部を5mlの滅菌生理食塩水に懸濁した。細胞懸濁物を100mlの培地に加えた(=前培養)。20〜24時間の増殖後、この前培養物の5mlを250mlの培地に加えた。
【0054】
細胞を、0〜4℃で、30分間、約3000rpmの遠心分離によって集めた。細胞をK/Naリン酸緩衝液(50mM、pH6.5)で1回洗浄した。次いで、細胞を新しい緩衝液に再懸濁し、急速冷凍後に凍結乾燥し(凍結乾燥細胞を用いる反応、実施例2)、或いは、この細胞懸濁物(培養体積の約6〜8%)を生体触媒反応にそのまま用いた(休止細胞を用いる反応、実施例3)。
【0055】
【表1】
【0056】
(実施例2:分析スケールで凍結乾燥細胞を用いる反応)
31.6mg、52.6mg及び105.2mgの凍結乾燥細胞を、130rpm、20〜25℃で約1時間、10mlのリン酸緩衝液(50mM、pH6.5)中で再水和した。この細胞懸濁物の475μl分を1.5mlエッペンドルフ反応容器に移し、2−ヒドロキシ−4−フェニルブチロニトリルの約200mM基質DMSO溶液25μlと混合した(細胞3mg、5mg及び10mg/ml;基質濃度約10mM;5%DMSO)。反応を、Thermomixer 内で30℃及び1000rpmで行った。0分、2分、4分、6分、8分、10分、15分、20分、30分、60分及び120分後の各時点で、1個のエッペンドルフ反応容器を0.5mlの1N HClと混合した。遠心分離(5分、13000rpm)及び対応する希釈の後、シアノヒドリン、ヒドロキシアミド及びヒドロキシ酸の濃度をHPLCによって測定した。
【0057】
【表2】
【0058】
(実施例3:休止細胞及び凍結乾燥細胞を用いる酵素的加水分解)
生体触媒を、実施例1、変形例Iと同様にして作製した(培養フラスコ2つ)。20時間後(OD546=3.5及び1.8)、細胞を、発酵液の10ml分×4から遠心分離し、K/Na−PO4緩衝液(pH6.5、50mM)で1回洗浄した。細胞サンプルのうちの2つを活性測定前に凍結乾燥し、残りの2つを休止細胞として用いた。
【0059】
細胞を1.8mlのK/Na−PO4緩衝液(pH6.5、50mM)に再懸濁した(凍結乾燥細胞を再水和のために1時間振盪した)。200mM基質DMSO溶液200μlを加える(基質濃度約20mM)ことによって反応を開始させ、振盪キャビネット内で30℃及び130rpmで反応を行った。30分、60分及び17時間後、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。ここでは、基質及び2つの生成物のみを考慮した。基質としては、(R)−2−クロロマンデロニトリル(ee>99%)を用いた。結果を表3に示す。
【0060】
【表3】
【0061】
(実施例4:異なる基質濃度を用いる酵素的加水分解)
実験4.1:
実験4.1では、反応を、3種の基質濃度(2.2g/l、6.6g/l、13.2g/l)を用いて、分析スケール(反応体積 1ml)で行った。
【0062】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後(OD546 6.1)に集めた。発酵溶液10ml分×8から得られる細胞を培養チューブ中で遠心分離した。得られた細胞集団を各々、2mlのK/Na−リン酸緩衝液(pH6.8、50mM)で1回洗浄した。
【0063】
2個のチューブの内容物を、乾燥重量を測定するために凍結乾燥した。
重量: 1. 凍結乾燥細胞37mg/発酵溶液10ml 2. 30mg
(この量は、下記の反応における1ml当たりの細胞の使用量にほぼ相当する。)
【0064】
残りの6個のチューブの内容物を950μlの緩衝液に再懸濁し(OD 約40)、エッペンドルフ容器に移した。これらの細胞懸濁物の各々に、種々の濃度の基質溶液50μlを加えた(3種の濃度、並列バッチ、共溶媒として5%DMSO)。エッペンドルフ容器を、Thermomixer 上で30℃及び1000rpmで振盪した。変換率をモニターするために、各々の場合に、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。
【0065】
下記の濃度の(R)−2−クロロマンデロニトリルを用いた。
a. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル11mg(約260mM) バッチ内の基質濃度:2.2g/l(13.1mM)
b. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル33mg(約290mM) バッチ内の基質濃度:6.6g/l(39.4mM)
c. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル66mg(約1580mM) バッチ内の基質濃度:13.2g/l(78.8mM)
【0066】
表4.1では、バッチa〜cが、2−クロロマンデル酸の形成(%)に関して比較されている。すべてのバッチにおいて、ヒドロキシ酸が定量的に形成された。比較的高い基質濃度も、問題なく許容されることが分かった。
【0067】
【表4】
【0068】
実験4.2:
実験4.2では、反応を、2種の異なる基質濃度(10g/l、20g/l)を用いて、5mlスケールで行った。
【0069】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2l)、19時間後(OD546 8.4)に集めた。細胞を約140mlの緩衝液に再懸濁した(休止細胞、OD546 52)。各々の場合に、この細胞懸濁物の4.75mlを酵素反応に用いた。
【0070】
2種の異なる濃度の(R)−2−クロロマンデロニトリルを並列バッチ内で調べた。250μlの基質溶液を加えることによって反応を開始させ、振盪キャビネット内の培養チューブ中で30℃及び130rpmで行った。変換率をモニターするために、各々の場合に、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。
【0071】
a. (R)−2−クロロマンデロニトリル50mg、DMSO250μlに溶解([S]=60mM、10g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してヒドロキシアミドを形成し、2時間後には、約40%のヒドロキシ酸が形成された。20時間後、反応は定量的であった。
【0072】
b. (R)−2−クロロマンデロニトリル100mg、DMSO250μlに溶解([S]=120mM、20g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、18時間後には、36%のヒドロキシ酸が形成された。43時間後には、41%のヒドロキシ酸が形成され、その後は、更なる反応が起こらなかった。
【0073】
実験4.3:
実験4.3では、反応を、10g/l及び15g/lの(R)−2−クロロマンデロニトリルを用いて行った。更に、変換完了後、形成されたヒドロキシ酸のeeを測定した。
【0074】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2.75l)、20時間後に集めた。細胞を約200mlの緩衝液に再懸濁した(休止細胞、OD546 44)。各々の場合に、この細胞懸濁物の4.85mlを酵素反応に用いた。
【0075】
2種の異なる濃度の(R)−2−クロロマンデロニトリルを並列バッチ内で調べた。150μlの基質溶液を加えることによって反応を開始させ、振盪キャビネット内の培養チューブ中で40℃及び150rpmで行った。変換率をHPLCによってモニターした。変換完了時に、ヒドロキシ酸を酸性化して抽出し、eeを測定した。
【0076】
a. (R)−2−クロロマンデロニトリル50mg、DMSO150μlに溶解([S]=60mM、10g/l、共溶媒:3%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、80%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0077】
b. (R)−2−クロロマンデロニトリル75mg、DMSO150μlに溶解([S]=90mM、15g/l、共溶媒:3%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、約60%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee=99%)。
【0078】
(実施例5:異なる共溶媒を用いる酵素的加水分解)
実験5.1:
50mlスケールの反応において、DMSOを、共溶媒としてEtOHと比較した。5%の共溶媒を用いたが、基質濃度は(R)−2−クロロマンデロニトリル4g/lにすぎなかった。
【0079】
発酵培地250ml分×8(−80ml、実験4.1参照)(OD 約6.1)からのバイオマスを20時間後に集めた。細胞を100mlのK/Na−リン酸緩衝液(pH6.5、50mM)に懸濁した。この細胞懸濁物(OD 60)を酵素反応に用いた。DMSO又はEtOHに溶解した(R)−2−クロロマンデロニトリルの反応を、100ml磨りガラス連結三角フラスコ内で150rpm及び30℃で行った。変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。変換完了後、生成物のeeを測定した。
【0080】
a. 細胞懸濁物50mgを、DMSO2300μl及び0.1%H3PO4200μlに溶解した(R)−2−クロロマンデロニトリル(>99%)200mgと混合した。
基質濃度:4g/l(24mM)、共溶媒として5%DMSO
(R)−(2)−クロロマンデル酸の生成物ee:97%
【0081】
b. 細胞懸濁物50mgを、EtOH2300μl及び0.1%H3PO4200μlに溶解した(R)−2−クロロマンデロニトリル(>99%)200mgと混合した。
基質濃度:4g/l(24mM)、共溶媒として5%EtOH
(R)−(2)−クロロマンデル酸の生成物ee:>99%
【0082】
実験5.2:
ここでは、溶媒DMSO、EtOH及びiPrOHを5%の割合で再び用いた。基質濃度は(R)−2−クロロマンデロニトリル10g/lであった。反応を5mlスケールで行った。
【0083】
生体触媒を、実施例4、実験4.2に従って作製した(OD546 8.4)。各々の場合に、4.75mlの細胞懸濁物(休止細胞、OD546 52)を酵素反応に用いた。DMSO、EtOH及びi−PrOHに溶解した(R)−2−クロロマンデロニトリル(>99%)の反応を、培養チューブ中で150rpm及び30℃で行った(並列バッチ)。
【0084】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。変換完了後、生成物のeeを測定した。
【0085】
a. (R)−2−クロロマンデロニトリル50mg、DMSO250μlに溶解([S]=60mM、10g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してヒドロキシアミドを形成し、2時間後には、約40%のヒドロキシ酸が形成された。20時間後、反応は定量的であった(生成物ee=95%)。
【0086】
b. (R)−2−クロロマンデロニトリル50mg、EtOH250μlに溶解([S]=60mM、10g/l、共溶媒:5%EtOH)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、3時間後には、35%のヒドロキシ酸が形成された。19時間後には、ヒドロキシ酸を形成する加水分解は94%完了し、28時間後には、反応は事実上完了した(生成物ee=97%)。
【0087】
c. (R)−2−クロロマンデロニトリル50mg、i−PrOH250μlに溶解([S]=60mM、10g/l、共溶媒:5%i−PrOH)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、3時間後には、8%のヒドロキシ酸が形成された。44時間後には、64%のヒドロキシ酸が形成された(生成物ee=92.3)。
【0088】
(実施例6:異なる温度における酵素反応)
実験6.1:
実験6.1では、反応の経過を、30℃、35℃及び40℃の反応温度で比較した。バッチ反応を、(R)−2−クロロマンデロニトリル10g/lの基質濃度を用いて、5mlスケールで行った。反応完了後、生成物のeeを測定した。
【0089】
生体触媒を、実施例4、実験4.2に従って作製した(OD546 8.4)。各々の場合に、4.75mlの細胞懸濁物(休止細胞、OD546 52)を酵素反応に用いた。DMSO250μlに溶解した(R)−2−クロロマンデロニトリル(>99%)50mg([S]=60mM、10g/l)の反応を、培養チューブ中、150rpmで3種の異なる温度(30℃、35℃、40℃)で行った(並列バッチ)。
【0090】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。反応完了後、生成物のeeを測定した。
【0091】
a. T=30℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、42%のヒドロキシ酸が形成され、約20時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=95%)。
【0092】
b. T=35℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、65%のヒドロキシ酸が形成され、19時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=96.5%)。
【0093】
c. T=40℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、86%のヒドロキシ酸が形成され、19時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=97.9%)。
【0094】
実験6.2:
実験6.2では、温度を50℃に上げた。
【0095】
生体触媒を、実施例4、実験4.3に従って作製した(OD546 44)。各々の場合に、4.85mlの細胞懸濁物(休止細胞、OD546 44)を酵素反応に用いた。DMSO(3%)150μlに溶解した(R)−2−クロロマンデロニトリル(>99%)50mg([S]=60mM、10g/l)の反応を、培養チューブ中、150rpmで3種の異なる温度(30℃、40℃、50℃)で行った(並列バッチ)。
【0096】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。反応完了後、生成物のeeを測定した。
【0097】
a. T=30℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、42%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0098】
b. T=40℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、80%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0099】
c. T=50℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、92%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0100】
(実施例7:セミ分取スケールで行う、ロードコッカス・エリスロポレス NCIMB 11540 の存在下のアルデヒドのシアノヒドリンの反応)
すべての反応について、K/Na−リン酸緩衝液(50mM、pH6.5)を用いた。反応をHPLCで追った。サンプル採取後、生体触媒反応を停止させるために、サンプル容量分を1N HClと混合した(並列サンプル)。遠心分離(5分、13000rpm)後、サンプルをHPLC溶離液で希釈した。
【0101】
精密な分析のために、バイオマスを、4℃及び3000rpmで30分間遠心分離し、蒸留H2Oで1回洗浄した。上清を1N HClでpH2に酸性化した後、TBMEで3〜4回抽出した。
【0102】
実験7.1:
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 5.9)に集めた。細胞を緩衝液に再懸濁して、約180mlにした(休止細胞、OD546 80)。
【0103】
(R)−2−クロロマンデロニトリル(ee>99%)0.6gをDMSO1.5mlに溶解し、この細胞懸濁物の60mlと混合した。加水分解を、振盪キャビネット内で30℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、17時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.73g(109%)
生成物ee:>99%
【0104】
実験7.2:
実験7.2では、いくつかの反応パラメーターを変えた。標準的な条件は、10g/lの基質、及び共溶媒としてのDMSO(ここでは2.5%)であった。別のバッチ反応を、15g/lの基質を用い、更には10g/lの基質、及び共溶媒としてのDMFを用いて行った。
【0105】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 6.8)に集めた。細胞を緩衝液に再懸濁して、約190mlの緩衝液を得た(休止細胞、OD546 69)。3種の反応を行った。
【0106】
バッチA: (R)−2−クロロマンデロニトリル(ee>99%)0.3gをDMSO750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.31g(93%)
生成物ee:>99%
【0107】
バッチB: (R)−2−クロロマンデロニトリル(ee>99%)0.3gをDMF750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.30g(90%)
生成物ee:98.5%
【0108】
バッチC: (R)−2−クロロマンデロニトリル(ee>99%)0.45gをDMSO750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が事実上完了した。
粗収量:0.45g(90%)
生成物ee:>99%
【0109】
実験7.3:
ここでは、異なる基質濃度を有する2つのバッチ(バッチA 10g/l;バッチB 15g/l)で反応を行った。両方の反応は実質的に同一の速度で進行し、2時間後には完了した。
【0110】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 1.2)に集めた。細胞を約180mlの緩衝液に再懸濁した(休止細胞、OD546 70)。2種の反応を行った。
【0111】
バッチA: (R)−2−クロロマンデロニトリル(ee>99%)0.8gをDMSO1.6mlに溶解し、細胞懸濁物(80ml)に加えた。加水分解を、振盪キャビネット内で50℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、2時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.85g(95%)
生成物ee:>99%
【0112】
バッチB: (R)−2−クロロマンデロニトリル(ee>99%)1.2gをDMSO1.6mlに溶解し、細胞懸濁物(80ml)に加えた。加水分解を、振盪キャビネット内で50℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、2時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:1.26g(94%)
生成物ee:98.9%
【0113】
実験7.4:
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2.5l)、20時間後(OD546 6.9)に集めた。細胞を約160mlの緩衝液に再懸濁した(休止細胞、OD546 63)。
【0114】
(R)−2−クロロマンデロニトリル(ee>99%)1.3gをDMSO2.5mlに溶解し、この細胞懸濁物の140mlに加えた。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、3時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:1.43g(98%)
生成物ee:>99%
【0115】
実験7.5:
この反応では、1gのマンデロニトリルを8g/lの基質濃度で加水分解して、対応するヒドロキシ酸を得た。
【0116】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後(OD546 8.4)に集めた。細胞を約120mlの緩衝液に再懸濁した(休止細胞、OD546 74)。生体触媒を超低温冷凍した後、反応を一晩行った。
【0117】
(R)−(+)−マンデロニトリル1.0gをDMSO2.4mlに溶解し、細胞懸濁物(120ml)に加えた。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−マンデル酸を生成する反応が完了した。
粗収量:1.16g(100%)
生成物ee:93%
【0118】
(実施例8:セミ分取スケールで行う、ロードコッカス・エリスロポレス NCIMB 11540 の存在下のケトンのシアノヒドリンの反応)
K/Na−リン酸緩衝液(50mM、pH6.5)を用いた。反応をTLCで追った。
【0119】
精密な分析のために、バイオマスを、4℃及び6000rpmで20分間遠心分離し、蒸留H2Oで1回洗浄した。上清を1N HClでpH2に酸性化した後、TBMEで3〜4回抽出した。
【0120】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後に集めた。細胞を約60mlの緩衝液に再懸濁した(休止細胞、OD546 60)。
【0121】
(S)−アセトフェノンシアノヒドリン(アセトフェノン 25%、ee 94%)300mgをDMSO1mlに溶解し、細胞懸濁物に加えた。20時間後には、反応が完了し(TLCによる)、生成物(1−フェニルエタノール及び微量の他の不純物を含有)を抽出した。反応は、エナンチオマー純度を喪失することなく進行した。
粗収量:357mg
【0122】
(実施例9:大腸菌における組換え発現による、置換シアノヒドリンの加水分解のためのニトリルヒドラターゼ及びアミダーゼの酵素調製物の作製)
ニトリルヒドラターゼを発現させるために、また、アミダーゼを発現させるために、pMS470 プラスミド系を用いた。このプラスミドは、複製されたエレメントに加えて、選択可能なアンピシリン抵抗性を有し、また、誘導性 tac プロモーターによって、クローン化されたオープンリーディングフレームの制御された過剰発現を可能にする Lac リプレッサー遺伝子 lac Iを有する。
【0123】
ロードコッカス・エリスロポレス NCIMB 11540 のニトリルヒドラターゼのための発現プラスミド:
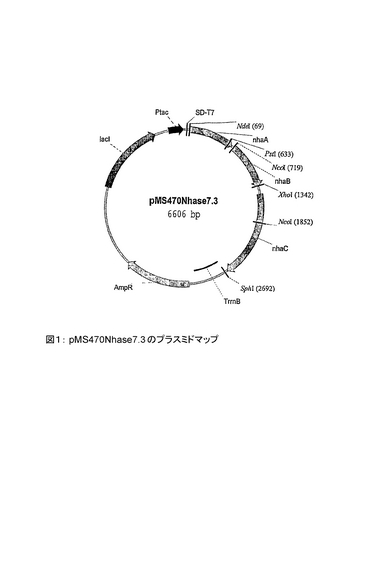
プラスミドマップが図1に示されている。このプラスミドは、pMS470Nhase7.3 という名称を有する。このプラスミドは、ニトリルヒドラターゼの2つの遺伝子領域(α−及びβ−サブユニット)に加えて、アクチベータータンパク質をコードする第3のオープンリーディングフレームも有する。
【0124】
ロードコッカス・エリスロポレス NCIMB 11540 のアミダーゼのための発現プラスミド:
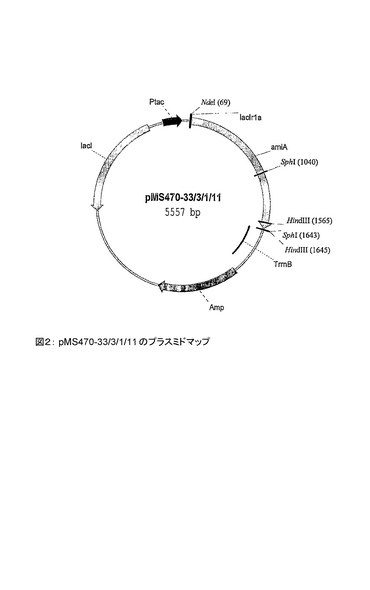
ニトリルヒドラターゼの場合とは異なり、ロードコッカス・エリスロポレス NCIMB 11540 のアミダーゼの発現に必要なのは1つのリーディングフレームのみである。これを、プラスミド pMS470-33/3/1/11 の、tac プロモーターの下流側にクローン化した。図2は、この構築物のプラスミドマップを示す。
【0125】
組換えニトリルヒドラターゼ及び組換えアミダーゼの発酵:
2つの酵素の発酵はすべて、pMS470 系における酵素の過剰発現のために開発された一般的なプロトコールによって行った。
【0126】
ここで、サンプルは、
・一晩培養した培養物(ONC)から、LB培地及び抗生物質を含有する振盪フラスコ内の主培養液に接種し、
・対数期まで増殖させ、
・0.8〜1.5のOD600(600nmにおける光学濃度)で、IPTG(イソプロピルチオガラクトピラノシド)で誘導し、
・更に18時間誘導し(タンパク質発現)、
・集め(遠心分離)、破砕した(超音波)。
【0127】
ニトリルヒドラターゼの発現:
pMS470Nhase7.3(又は pMSNhasetactac7.3)によって形質転換された大腸菌B BL21細胞をLB−アンピシリンプレート上で単離し、100mlのLB−アンピシリン培地のONCに個々のコロニーを接種した。翌朝、0.01〜0.03のOD600(Beckmann光度計)に達するように、1000mlシケイン付フラスコ内の、250mlのLB−アンピシリン培地からなる主培養液に接種した。25℃よりも高い温度では専ら不溶性の封入体が形成されるので、増殖温度を25℃に制御した。誘導密度(OD600=1、Beckmann光度計)に到達した後、IPTGを0.1mMの濃度まで加えることによって培養物を誘導した。更に、培地に0.1mMクエン酸鉄(III)アンモニウムを補充した。>4のOD600に達した後、培養物を集め(約3000×gで15分間遠心分離)、約100mlのPBS緩衝液で1回洗浄した。その後、細胞ペレットをPBS緩衝液に再懸濁し(総容量約5ml)、超音波プローブ(BRANSON Sonifier 250、出力60%に設定、一定周波数で処理、1回30秒を5回、1回ごとに冷却のために1分間休止)を用いて破砕した(破砕を顕微鏡で目視して確認)。得られた粗ライセートを、下記に列挙する条件の下でメタクリロニトリルを基質として用いて分析したところ、約100〜250U/ml(pMSNhasetactac7.3 については約350〜500U/ml)の典型的な活性を有していた。
【0128】
保存のために、ライセートを−20℃で貯蔵した。室温で貯蔵すると、活性が急速に失われる。
【0129】
活性測定: 粗ライセートを、活性測定の直前にPBS緩衝液で1:10に希釈した。PBS緩衝液を溶媒とする40mMメタクリロニトリル溶液1.4mlを希釈ライセート20μlと混合し、28℃でインキュベートした(Eppendorf Thermomixer 5436)。0分、1分、2分、5分、10分及び15分の時点で、200μlのサンプルを抜き取り、これらのサンプル中の酵素反応を800μlの0.17%リン酸によって直ちに停止させた。遠心分離(16000×g、10分)の後、サンプルを224nmで分光光度法により分析した(Perkin Elmer UV/VIS分光計 Lambda Bio)。吸光の増大がメタクリルアミドの濃度の増大と相関することが明らかとなった。なお、0.57l・mmol−1・cm−1のε値を基準として用いた。
【0130】
アミダーゼの発現:
pMS470-33/3/1/11 によって形質転換された大腸菌B BL21細胞をLB−アンピシリンプレート上で単離し、100mlのLB−アンピシリン培地のONCに個々のコロニーを接種した。翌朝、0.01〜0.03のOD600(Beckmann光度計)に達するように、1000mlシケイン付フラスコ内の、250mlのSOC−アンピシリン培地からなる主培養液に接種した。37℃で発酵させると、専ら不溶性の、かつ不活性なタンパク質が形成されるので、増殖温度を30℃に制御した。誘導密度(OD600=1、Beckmann光度計)に到達した後、IPTGを0.3mMの濃度まで加えることによって培養物を誘導した。16時間の誘導の後、細胞を集め(約3000×gで10分間遠心分離)、リン酸ナトリウム緩衝液(0.1M、pH=7)で洗浄した。得られたペレットを洗浄緩衝液に再懸濁し(総容量約5ml)、超音波プローブ(BRANSON Sonifier 250、出力60%に設定、一定周波数で処理、1回30秒を5回、1回ごとに冷却のために1分間休止)を用いて破砕した(冷却負荷は終始一定)(破砕を顕微鏡で目視して確認)。このようにして得られた粗ライセートを、−20℃で凍結保存した。ライセートは、PBS緩衝液中のアセトアミド(40mM)を基質として用いて37℃で測定した(インドフェノールブルー法による遊離アンモニウムの測定)ところ、約75U/mlの活性を有していた。
【0131】
アミダーゼ活性の測定: 下記の溶液を用いた。
基質溶液:40mMアセトアミドのPBS溶液
溶液A:10%(w/v)フェノールのエタノール(95%)溶液
溶液B:0.5%(w/v)ニトロプルシドナトリウムのddH2O溶液
溶液C:クエン酸三ナトリウム100g及び水酸化ナトリウム5gが水550mlに溶解した溶液
溶液D:市販の通常の次亜塩素酸ナトリウム溶液600mlが、1000mlになるように希釈された溶液
アンモニウム標準液:0μg/l、80μg/l、120μg/l、200μg/l、280μg/l及び400μg/lの硫酸アンモニウム水溶液
【0132】
1.4mlの基質溶液を30℃で10μlの酵素希釈物(PBS中1:10)と共にインキュベートした(Eppendorf Thermomixer 5436)。0分、1分、2分、5分、10分及び15分後に、サンプル100μl中の酵素反応を、20μlの溶液Aを用いて停止させた。最後のサンプルを抜き取った後、得られた溶液を400μlの水で希釈した。更に、較正のために、アンモニウム標準溶液(各500ml)を20μlの溶液Aと混合した。その後直ちに、サンプルに、また、標準液に、溶液B20μl、及び、4部の溶液Cと1部の溶液Dとの混合液50μlをピペットで加えた。ボルテックスによって十分に混合されるようにした。得られたサンプル及び標準液を37℃で15分間放置した。すべてのサンプル及び標準液を水で希釈(1:10)した後、得られた青色の呈色を、分光光度計(Perkin Elmer UV/VIS分光計 Lambda Bio)を用いて640nmで定量した。青色の呈色の経時的な増大と標準液の吸光度との相関関係を求めることによって、活性(1分当たりの遊離アンモニウムのμmol数)を逆算することができる。
【0133】
(実施例10:組換え酵素を用いる加水分解)
これらの反応では、ロードコッカス・エリスロポレス NCIMB 11540 のクローン化ニトリルヒドラターゼを大腸菌クローン7.3(実施例9に従って作製)の粗ライセートとして用いた。
【0134】
手順:
50μlの粗ライセートを425μlの緩衝液(K/Na−PO4緩衝液、pH7、50mM)で希釈し、DMSOを溶媒とする約220mMの基質溶液25μlと混合した(タンパク質5mg/ml、基質濃度約10mM、5%DMSO)。反応を、Thermomixer 内で30℃及び1000rpmで行った。0分、2分、4分、6分、8分、10分、15分、20分、30分、60分及び120分後、各々の場合に、1個のエッペンドルフ容器を0.5mlの1N HClと混合した。遠心分離(5分、13000rpm)及び対応する希釈の後、シアノヒドリン及びヒドロキシアミドの濃度をHPLCによって測定した。ニトリルヒドラターゼの活性をヒドロキシアミドの形成速度(初期範囲における傾き)から求めた。標準基質(活性100%)として、2−ヒドロキシ−4−フェニルブチロニトリルを用いた。その他の基質の加水分解における活性を、2−ヒドロキシ−4−フェニルブチロニトリルに対する活性と比較した(表5)。
【0135】
結果:
2−ヒドロキシ−4−フェニルブチロニトリルの加水分解における、大腸菌クローン7.3の粗ライセート中のニトリルヒドラターゼの活性は、約0.3μmol・mg−1・min−1であった。表5では、異なる基質に対する活性が比較されている。
【0136】
【表5】
【0137】
(セミ分取スケールで行う(R)−2−クロロマンデロニトリルの反応)
ロードコッカス・エリスロポレス NCIMB 11540 由来のクローン化ニトリルヒドラターゼ(大腸菌クローン7.3、実施例9に従って作製)の粗ライセート50mlを100mlの緩衝液(K/Na−PO4緩衝液、50mM、pH6.5)で希釈した。(R)−2−クロロマンデロニトリル(ee>99%)1.0gがDMSO1.5mlに溶解した溶液を加えた後、懸濁物を150rpm及び30℃で振盪した。反応完了後、細胞破片を遠心分離し、生成物を、4日間にわたってCH2Cl2で連続抽出した。
粗生成物のee:>99%
精製後の収量:0.91g(82%)
【図面の簡単な説明】
【0138】
【図1】pMS470Nhase7.3 のプラスミドマップある。
【図2】pMS470-33/3/1/11 のプラスミドマップある。
【発明の詳細な説明】
【0001】
光学活性α−ヒドロキシカルボン酸は、例えば、食品添加物として、又は、医薬用活性化合物、ビタミン及び液晶の製造において用いられる。
【0002】
これらの光学活性α−ヒドロキシカルボン酸はまた、有利なことに、例えば Effenberger 他,Angew. Chem.,95(1983),No.1,50頁に従って、他の方法では調製が極めて困難なN−置換光学活性α−アミノ酸に変換することができる。
【0003】
キラルα−ヒドロキシカルボン酸は、今日では、化学的に、又は発酵によって、又は酵素的に得ることができる。
【0004】
従って、文献には、キラルα−ヒドロキシカルボン酸を合成する多くの種々の方法が開示されている。
【0005】
例えば、適当な微生物の添加により、シアノヒドリンのラセミ体を加水分解して、所望のキラルα−ヒドロキシカルボン酸を得ることができる。
【0006】
アルカリゲネス(Alcaligenes)属、シュードモナス(Pseudomonas)属、アシネトバクター(Acinetobacter)属、ロードコッカス(Rhodococcus)属、カンジダ(Candida)属等の種々の微生物を用いる、シアノヒドリンのラセミ体からのキラルα−ヒドロキシカルボン酸の製造、特に光学活性な乳酸又はマンデル酸の製造が、例えば、欧州特許第0449684号明細書、欧州特許第0527553号明細書、欧州特許第0610048号明細書等に記載されている。
【0007】
このような従来技術からは、ラセミ体のシアノヒドリンをニトリラーゼの存在下で共役α−ヒドロキシカルボン酸に酵素的に加水分解するとき、酵素が短時間のうちに失活し、その結果、所望のα−ヒドロキシカルボン酸が、通常、低い収率及び濃度でしか得られないという問題が生じていることも分かる。このことは、シアノヒドリンを共役のα−ヒドロキシアミドに変換するニトリルヒドラターゼの使用にも当てはまる。このヒドロキシアミドは、共役のα−ヒドロキシカルボン酸に更に変換することができる。
【0008】
例えば Angew. Chem.,1994,106,1615頁以降からは、光学活性シアノヒドリンを、ラセミ化を行わずに濃塩酸で加水分解して、共役のキラルα−ヒドロキシカルボン酸を得ることができることも分かる。そのようにして製造されたキラルα−ヒドロキシカルボン酸の光学純度は、用いたキラルシアノヒドリンの光学純度に一致する。このことは、用いるキラルシアノヒドリンを、シアニド基を共役のアルデヒド又はケトンに酵素触媒で付加することにより in situ で得、単離又は精製を行わずに更に処理する場合にも当てはまる。
【0009】
この反応の欠点は、反応性の高い基質が分解し、腐食を引き起こすことである。
【0010】
本発明は、キラルシアノヒドリンと同程度の極性を有するニトリルを、穏和かつ効率的な方法を用いて、シアノヒドリンとほぼ同じエナンチオマー純度を有する共役のキラルヒドロキシカルボン酸に変換することができる方法を見出すことを目的としていた。
【0011】
意外なことに、この目的は、ロードコッカス属に属する特殊な細菌を用いることによって達成された。
【0012】
すなわち、本発明は、キラルα−ヒドロキシカルボン酸を製造する方法であって、(R)−又は(S)−シアノヒドリンを、ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 の存在下で酵素的加水分解を行うことにより、共役の(R)−又は(S)−α−ヒドロキシカルボン酸に変換することを含む方法に関する。
【0013】
本発明の方法において、(R)−及び(S)−シアノヒドリンは、光学純度が>99%eeまでの(R)−及び(S)−α−ヒドロキシカルボン酸に変換される。
【0014】
対応するアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加によって得られる(R)−及び(S)−シアノヒドリンは、出発化合物となる。
【0015】
対応するアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加は、従来技術に準じて、例えば、欧州特許第0951561号明細書、欧州特許第0927766号明細書、欧州特許第0632130号明細書、欧州特許第0547655号明細書、欧州特許第0326063号明細書等の方法に準じて行うことができる。
【0016】
好適な出発化合物は、従来技術において挙げられているアルデヒド又はケトンである。
【0017】
好適なアルデヒドの例としては、脂肪族、芳香族又は複素芳香族アルデヒドがある。脂肪族アルデヒドとは、飽和又は不飽和の脂肪族の直鎖、分枝鎖又は環状アルデヒドを意味するものとする。好ましい脂肪族アルデヒドは、特に2個〜18個、特に好ましくは2個〜12個の炭素原子を有する、飽和又はモノ不飽和若しくはポリ不飽和の直鎖アルデヒドである。アルデヒドは、C−C二重結合を有しても、C−C三重結合を有してもよい。アルデヒドは、置換されていなくても、また、反応条件下で不活性である基によって一置換又は多置換されていてもよい。例えば、置換されていてもよいアリール基又はヘテロアリール基(例えば、フェニル基又はインドリル基)、或いは、C1−C6アルキル基、置換されていてもよいシクロアルキル基(これは、O、S、P又はNの群より選ばれる1つ又は複数のヘテロ原子を有してもよい)、ハロゲン基、エーテル基、アルコール基、アシル基、カルボン酸基、カルボン酸エステル基、ニトロ基又はアジド基によって一置換又は多置換されていてもよい。
【0018】
芳香族又は複素芳香族アルデヒドの例としては、ベンズアルデヒド、又は種々の置換基で置換されたベンズアルデヒド(例えば、2−クロロベンズアルデヒド、3,4−ジフルオロベンズアルデヒド、4−メチルベンズアルデヒド、3−フェノキシベンズアルデヒド、4−フルオロ−3−フェノキシベンズアルデヒド)があり、更に、フルフラール、アントラセン−9−カルボアルデヒド、フラン−3−カルボアルデヒド、インドール−3−カルボアルデヒド、ナフタレン−1−カルボアルデヒド、フタルジアルデヒド、ピラゾール−3−カルボアルデヒド、ピロール−2−カルボアルデヒド、チオフェン−2−カルボアルデヒド、イソフタルアルデヒド、ピリジンアルデヒド等がある。
【0019】
ケトンの例としては、カルボニル炭素原子が不均等に置換されている脂肪族、芳香族又は複素芳香族ケトンがある。脂肪族ケトンとは、直鎖、分枝鎖又は環状ケトンを意味するものとする。ケトンは、飽和であっても、モノ不飽和又はポリ不飽和であってもよい。ケトンは、置換されていなくても、また、反応条件下で不活性である基によって一置換又は多置換されていてもよい。例えば、置換されていてもよいアリール基又はヘテロアリール基(例えば、フェニル基又はインドリル基)、或いは、ハロゲン基、エーテル基、アルコール基、アシル基、カルボン酸基、カルボン酸エステル基、ニトロ基又はアジド基によって一置換又は多置換されていてもよい。
【0020】
芳香族ケトン又は複素芳香族ケトンの例としては、アセトフェノン、インドリルアセトン等がある。
【0021】
好ましいのは、式:
【化1】
[式中、R1及びR2は相互に独立して、H、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいC1−C6アルキルラジカル若しくはC1−C6アルケニルラジカル、又は、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいフェニルラジカルである。但し、R1及びR2が共にHであることはない。]
の(R)−又は(S)−シアノヒドリンである。
【0022】
反応条件下で不活性である置換基として好ましいのは、例えば、ハロゲン(例えば、フッ素、臭素及び塩素)、C1−C6アルキル若しくはC1−C6アルコキシ、エーテル、エステル、アセタール、又は置換されていてもよいフェニル若しくはフェニルオキシである。
【0023】
本発明の方法における適当な化合物は、特に好ましくは、(R)−又は(S)−シアノヒドリンであり、例えば、(R)−又は(S)−2−ヒドロキシ−4−フェニルブチロニトリル、(R)−又は(S)−2−クロロマンデロニトリル、(R)−又は(S)−マンデロニトリル、(R)−又は(S)−4−メチルマンデロニトリル、(R)−又は(S)−3−フェノキシマンデロニトリル、(R)−又は(S)−2−ヒドロキシ−2−メチルヘプタンニトリル、(R)−又は(S)−2−ヒドロキシ−2−フェニルプロピオニトリル、(R)−又は(S)−2−ヒドロキシ−3−ペンテンニトリル、(R)−又は(S)−1−ヒドロキシシクロヘキサンニトリル、及び(R)−又は(S)−アセトフェノンシアノヒドリンである。
【0024】
次いで、対応する(R)−又は(S)−シアノヒドリンを、本発明に従って酵素的に加水分解する。
【0025】
酵素的加水分解は、ロードコッカス・エリスロポレス NCIMB 11540 の存在下、本発明に従って行う。
【0026】
ロードコッカス・エリスロポレス NCIMB 11540 は、意外なことに、上記のシアノヒドリンと同様の極性を有するニトリルのニトリル官能基を加水分解することができる、利用可能なニトリルヒドラターゼ/アミダーゼ酵素系を有することを特徴とする微生物であった。
【0027】
ロードコッカス・エリスロポレス NCIMB 11540 のニトリルヒドラターゼ/アミダーゼ酵素系を用いると、最初のステップにおいて、キラルシアノヒドリンが、ニトリルヒドラターゼにより共役のキラルヒドロキシアミドに加水分解され、次の加水分解ステップにおいて、キラルヒドロキシアミドが、アミダーゼにより、対応するキラルα−ヒドロキシカルボン酸に変換される。
【0028】
微生物は、本発明の方法では、任意の所望の形態で、例えば、粉砕細胞、粗酵素若しくは精製酵素、組換え酵素、固定化細胞若しくは固定化酵素、凍結乾燥細胞、又は「休止細胞」の形態で用いることができる。
【0029】
組換え酵素、休止細胞又は凍結乾燥細胞を用いるのが好ましく、組換え酵素又は休止細胞を用いるのが特に好ましい。
【0030】
ニトリルヒドラターゼ/アミダーゼ活性を有する、ロードコッカス・エリスロポレス NCIMB 11540 細胞の調製物を直接用いる代わりに、適当な微生物(例えば、大腸菌(E. coli)、ピキア・パストリス(Pichia pastoris)、サッカロマイセス(Saccharomyces)、アスペルギルス(Aspergillus)、K. Lactis 等)において発現させた組換え調製物を用いるのもよい。対応する遺伝子は、プラスミド構築物を用いて、適当な宿主細胞に、例えば、大腸菌、ピキア・パストリス、サッカロマイセス、アスペルギルス又は K. Lactis の宿主細胞に導入する。誘導可能なプロモーターを選ぶことによって、ニトリルヒドラターゼだけでなく、アミダーゼも、活性な形態で過剰発現させることができる。アミダーゼの場合、ロードコッカス細胞の対応する発酵の場合よりもはるかに高い活性レベルを達成することができる。
【0031】
次いで、微生物を、所望の形態で水性溶媒(例えば、水又は緩衝溶液)に懸濁する。好適な緩衝溶液は、例えば、リン酸緩衝液(例えば、K/Naリン酸緩衝液)、PBS緩衝液、酪酸緩衝液、クエン酸塩溶液等である。
【0032】
用いる緩衝溶液のpHは、4.5〜11、好ましくは5.5〜8.5の範囲内にあるのがよい。
【0033】
次いで、得られた懸濁物を、対応するキラルシアノヒドリンと混合する。キラルシアノヒドリンは、限定された水溶性を有する脂溶性化合物であるので、シアノヒドリンを水性溶媒に溶解させるためには、共溶媒として可溶化剤を用いる必要がある。
【0034】
適当な可溶化剤は、例えば、有機溶媒、界面活性剤、相間移動触媒等である。
【0035】
本発明の方法における共溶媒として適当な有機溶媒は、第1に、基質を十分に溶解することができ、第2に、酵素活性に対する悪影響が可能な限り小さいものである。
【0036】
このような有機溶媒の例としては、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド(DMF)、C1−C6アルコール(例えば、メタノール、エタノール、イソプロパノール、1−ブタノール、2−ブタノール、tert−ブタノール又は1−ペンタノール)、トルエン又はtert−ブチルメチルエーテル(TBME)、或いはそれらの混合物がある。
【0037】
用いる共溶媒としては、DMSO、DMF、エタノール、イソプロパノール又はそれらの混合物が好ましく、DMSO又はDMFが特に好ましい。
【0038】
共溶媒の割合は、反応溶液の全体積を基準として、0.5〜20体積%の範囲内にあるのがよい。
【0039】
共溶媒の割合は、好ましくは1〜15体積%、特に好ましくは2〜10体積%である。
【0040】
本発明の方法における反応液中の基質濃度は、(反応溶液の全体積を基準として、)1〜100g/lの範囲内にあるのがよい。十分に高い基質濃度が許容されることは、本発明の酵素的加水分解を実験規模で用いるための基本的前提条件である。
【0041】
基質濃度としては、50g/l以下が好ましく、25g/l以下が特に好ましい。
【0042】
反応可能な基質濃度は、用いる酵素量に依存する。効率的な定量的反応のためには、最初の加水分解ステップを極めて迅速に行って、シアノヒドリンの分解及びこれに伴うラセミ化を避けなければならず、そのため、比較的大きい細胞密度が必要となる。
【0043】
この場合、反応系の十分な混合を確実に行うことに留意する必要がある。
【0044】
細胞量又は酵素量は、用いる形態における微生物の活性に依存し、更に、基質濃度及び共溶媒にも依存する。
【0045】
反応混合物のpHは、4.5〜11、好ましくは5.5〜8の範囲内にあるのがよい。
【0046】
また、必要に応じて、適当な酸又は酸性塩(例えば、リン酸、ホウ酸、クエン酸等)を反応混合物に加えて、pHを調節することができる。
【0047】
本発明の酵素的加水分解は、10〜60℃、好ましくは15〜50℃、特に好ましくは20〜45℃の範囲内の温度で行う。
【0048】
加水分解を行って、所望のキラルα−ヒドロキシカルボン酸を得た後、キラルα−ヒドロキシカルボン酸は、既知の技術によって、例えば、細胞の遠心分離、又はHCl(例えば、pH2)による酸性化後の生成物の抽出によって、また、必要に応じて更に、活性炭濾過及び再結晶による精製を行うことによって、反応混合物から単離される。
【0049】
従って、ロードコッカス・エリスロポレス NCIMB 11540 を本発明に従って用いれば、極性のニトリル(例えば、キラルシアノヒドリン)が、穏和な条件下で簡便かつ効率的に、ラセミ化が生じることなく、共役のキラルα−ヒドロキシカルボン酸に変換される。所望のα−ヒドロキシカルボン酸が、用いたシアノヒドリンのee値に応じて、99%超までの高い光学純度で、かつ98%超までの高い収率で得られる。
【0050】
(実施例1:生体触媒の製造)
ロードコッカス・エリスロポレス NCIMB 11540 のバイオマスを製造するために、複合的な標準培地(培地A、表1参照)を用いた。菌株を、培地Aを用いた寒天プレート上に保持した(15g/lの寒天を用いた固化)。プレートを、パラフィルムで側面から包むことによって密封し、冷蔵庫に4℃で保存した。
【0051】
液体培養物の増殖を、250mlの培地Aを用いて、シケイン(chicane)付きの1000ml三角フラスコ内で、30℃及び130rpmで行った。
【0052】
変形例I(前培養なし): 寒天プレートのバイオマスの約半分を、5mlの滅菌生理食塩水に懸濁した。細胞懸濁物を250mlの培地に加えた。
【0053】
変形例II(前培養あり): 前培養のために、寒天プレートのバイオマスの一部を5mlの滅菌生理食塩水に懸濁した。細胞懸濁物を100mlの培地に加えた(=前培養)。20〜24時間の増殖後、この前培養物の5mlを250mlの培地に加えた。
【0054】
細胞を、0〜4℃で、30分間、約3000rpmの遠心分離によって集めた。細胞をK/Naリン酸緩衝液(50mM、pH6.5)で1回洗浄した。次いで、細胞を新しい緩衝液に再懸濁し、急速冷凍後に凍結乾燥し(凍結乾燥細胞を用いる反応、実施例2)、或いは、この細胞懸濁物(培養体積の約6〜8%)を生体触媒反応にそのまま用いた(休止細胞を用いる反応、実施例3)。
【0055】
【表1】
【0056】
(実施例2:分析スケールで凍結乾燥細胞を用いる反応)
31.6mg、52.6mg及び105.2mgの凍結乾燥細胞を、130rpm、20〜25℃で約1時間、10mlのリン酸緩衝液(50mM、pH6.5)中で再水和した。この細胞懸濁物の475μl分を1.5mlエッペンドルフ反応容器に移し、2−ヒドロキシ−4−フェニルブチロニトリルの約200mM基質DMSO溶液25μlと混合した(細胞3mg、5mg及び10mg/ml;基質濃度約10mM;5%DMSO)。反応を、Thermomixer 内で30℃及び1000rpmで行った。0分、2分、4分、6分、8分、10分、15分、20分、30分、60分及び120分後の各時点で、1個のエッペンドルフ反応容器を0.5mlの1N HClと混合した。遠心分離(5分、13000rpm)及び対応する希釈の後、シアノヒドリン、ヒドロキシアミド及びヒドロキシ酸の濃度をHPLCによって測定した。
【0057】
【表2】
【0058】
(実施例3:休止細胞及び凍結乾燥細胞を用いる酵素的加水分解)
生体触媒を、実施例1、変形例Iと同様にして作製した(培養フラスコ2つ)。20時間後(OD546=3.5及び1.8)、細胞を、発酵液の10ml分×4から遠心分離し、K/Na−PO4緩衝液(pH6.5、50mM)で1回洗浄した。細胞サンプルのうちの2つを活性測定前に凍結乾燥し、残りの2つを休止細胞として用いた。
【0059】
細胞を1.8mlのK/Na−PO4緩衝液(pH6.5、50mM)に再懸濁した(凍結乾燥細胞を再水和のために1時間振盪した)。200mM基質DMSO溶液200μlを加える(基質濃度約20mM)ことによって反応を開始させ、振盪キャビネット内で30℃及び130rpmで反応を行った。30分、60分及び17時間後、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。ここでは、基質及び2つの生成物のみを考慮した。基質としては、(R)−2−クロロマンデロニトリル(ee>99%)を用いた。結果を表3に示す。
【0060】
【表3】
【0061】
(実施例4:異なる基質濃度を用いる酵素的加水分解)
実験4.1:
実験4.1では、反応を、3種の基質濃度(2.2g/l、6.6g/l、13.2g/l)を用いて、分析スケール(反応体積 1ml)で行った。
【0062】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後(OD546 6.1)に集めた。発酵溶液10ml分×8から得られる細胞を培養チューブ中で遠心分離した。得られた細胞集団を各々、2mlのK/Na−リン酸緩衝液(pH6.8、50mM)で1回洗浄した。
【0063】
2個のチューブの内容物を、乾燥重量を測定するために凍結乾燥した。
重量: 1. 凍結乾燥細胞37mg/発酵溶液10ml 2. 30mg
(この量は、下記の反応における1ml当たりの細胞の使用量にほぼ相当する。)
【0064】
残りの6個のチューブの内容物を950μlの緩衝液に再懸濁し(OD 約40)、エッペンドルフ容器に移した。これらの細胞懸濁物の各々に、種々の濃度の基質溶液50μlを加えた(3種の濃度、並列バッチ、共溶媒として5%DMSO)。エッペンドルフ容器を、Thermomixer 上で30℃及び1000rpmで振盪した。変換率をモニターするために、各々の場合に、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。
【0065】
下記の濃度の(R)−2−クロロマンデロニトリルを用いた。
a. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル11mg(約260mM) バッチ内の基質濃度:2.2g/l(13.1mM)
b. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル33mg(約290mM) バッチ内の基質濃度:6.6g/l(39.4mM)
c. 基質溶液:DMSO250μl中に(R)−2−クロロマンデロニトリル66mg(約1580mM) バッチ内の基質濃度:13.2g/l(78.8mM)
【0066】
表4.1では、バッチa〜cが、2−クロロマンデル酸の形成(%)に関して比較されている。すべてのバッチにおいて、ヒドロキシ酸が定量的に形成された。比較的高い基質濃度も、問題なく許容されることが分かった。
【0067】
【表4】
【0068】
実験4.2:
実験4.2では、反応を、2種の異なる基質濃度(10g/l、20g/l)を用いて、5mlスケールで行った。
【0069】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2l)、19時間後(OD546 8.4)に集めた。細胞を約140mlの緩衝液に再懸濁した(休止細胞、OD546 52)。各々の場合に、この細胞懸濁物の4.75mlを酵素反応に用いた。
【0070】
2種の異なる濃度の(R)−2−クロロマンデロニトリルを並列バッチ内で調べた。250μlの基質溶液を加えることによって反応を開始させ、振盪キャビネット内の培養チューブ中で30℃及び130rpmで行った。変換率をモニターするために、各々の場合に、200μlを抜き取り、200μlの1N HClと混合した。遠心分離(5分、13000rpm)及び希釈の後、変換率をHPLCによって測定した。
【0071】
a. (R)−2−クロロマンデロニトリル50mg、DMSO250μlに溶解([S]=60mM、10g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してヒドロキシアミドを形成し、2時間後には、約40%のヒドロキシ酸が形成された。20時間後、反応は定量的であった。
【0072】
b. (R)−2−クロロマンデロニトリル100mg、DMSO250μlに溶解([S]=120mM、20g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、18時間後には、36%のヒドロキシ酸が形成された。43時間後には、41%のヒドロキシ酸が形成され、その後は、更なる反応が起こらなかった。
【0073】
実験4.3:
実験4.3では、反応を、10g/l及び15g/lの(R)−2−クロロマンデロニトリルを用いて行った。更に、変換完了後、形成されたヒドロキシ酸のeeを測定した。
【0074】
生体触媒を実施例1、変形例IIに従って作製し(発酵培地2.75l)、20時間後に集めた。細胞を約200mlの緩衝液に再懸濁した(休止細胞、OD546 44)。各々の場合に、この細胞懸濁物の4.85mlを酵素反応に用いた。
【0075】
2種の異なる濃度の(R)−2−クロロマンデロニトリルを並列バッチ内で調べた。150μlの基質溶液を加えることによって反応を開始させ、振盪キャビネット内の培養チューブ中で40℃及び150rpmで行った。変換率をHPLCによってモニターした。変換完了時に、ヒドロキシ酸を酸性化して抽出し、eeを測定した。
【0076】
a. (R)−2−クロロマンデロニトリル50mg、DMSO150μlに溶解([S]=60mM、10g/l、共溶媒:3%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、80%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0077】
b. (R)−2−クロロマンデロニトリル75mg、DMSO150μlに溶解([S]=90mM、15g/l、共溶媒:3%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、約60%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee=99%)。
【0078】
(実施例5:異なる共溶媒を用いる酵素的加水分解)
実験5.1:
50mlスケールの反応において、DMSOを、共溶媒としてEtOHと比較した。5%の共溶媒を用いたが、基質濃度は(R)−2−クロロマンデロニトリル4g/lにすぎなかった。
【0079】
発酵培地250ml分×8(−80ml、実験4.1参照)(OD 約6.1)からのバイオマスを20時間後に集めた。細胞を100mlのK/Na−リン酸緩衝液(pH6.5、50mM)に懸濁した。この細胞懸濁物(OD 60)を酵素反応に用いた。DMSO又はEtOHに溶解した(R)−2−クロロマンデロニトリルの反応を、100ml磨りガラス連結三角フラスコ内で150rpm及び30℃で行った。変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。変換完了後、生成物のeeを測定した。
【0080】
a. 細胞懸濁物50mgを、DMSO2300μl及び0.1%H3PO4200μlに溶解した(R)−2−クロロマンデロニトリル(>99%)200mgと混合した。
基質濃度:4g/l(24mM)、共溶媒として5%DMSO
(R)−(2)−クロロマンデル酸の生成物ee:97%
【0081】
b. 細胞懸濁物50mgを、EtOH2300μl及び0.1%H3PO4200μlに溶解した(R)−2−クロロマンデロニトリル(>99%)200mgと混合した。
基質濃度:4g/l(24mM)、共溶媒として5%EtOH
(R)−(2)−クロロマンデル酸の生成物ee:>99%
【0082】
実験5.2:
ここでは、溶媒DMSO、EtOH及びiPrOHを5%の割合で再び用いた。基質濃度は(R)−2−クロロマンデロニトリル10g/lであった。反応を5mlスケールで行った。
【0083】
生体触媒を、実施例4、実験4.2に従って作製した(OD546 8.4)。各々の場合に、4.75mlの細胞懸濁物(休止細胞、OD546 52)を酵素反応に用いた。DMSO、EtOH及びi−PrOHに溶解した(R)−2−クロロマンデロニトリル(>99%)の反応を、培養チューブ中で150rpm及び30℃で行った(並列バッチ)。
【0084】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。変換完了後、生成物のeeを測定した。
【0085】
a. (R)−2−クロロマンデロニトリル50mg、DMSO250μlに溶解([S]=60mM、10g/l、共溶媒:5%DMSO)
早くも30分後には、シアノヒドリンのすべてが反応してヒドロキシアミドを形成し、2時間後には、約40%のヒドロキシ酸が形成された。20時間後、反応は定量的であった(生成物ee=95%)。
【0086】
b. (R)−2−クロロマンデロニトリル50mg、EtOH250μlに溶解([S]=60mM、10g/l、共溶媒:5%EtOH)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、3時間後には、35%のヒドロキシ酸が形成された。19時間後には、ヒドロキシ酸を形成する加水分解は94%完了し、28時間後には、反応は事実上完了した(生成物ee=97%)。
【0087】
c. (R)−2−クロロマンデロニトリル50mg、i−PrOH250μlに溶解([S]=60mM、10g/l、共溶媒:5%i−PrOH)
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、3時間後には、8%のヒドロキシ酸が形成された。44時間後には、64%のヒドロキシ酸が形成された(生成物ee=92.3)。
【0088】
(実施例6:異なる温度における酵素反応)
実験6.1:
実験6.1では、反応の経過を、30℃、35℃及び40℃の反応温度で比較した。バッチ反応を、(R)−2−クロロマンデロニトリル10g/lの基質濃度を用いて、5mlスケールで行った。反応完了後、生成物のeeを測定した。
【0089】
生体触媒を、実施例4、実験4.2に従って作製した(OD546 8.4)。各々の場合に、4.75mlの細胞懸濁物(休止細胞、OD546 52)を酵素反応に用いた。DMSO250μlに溶解した(R)−2−クロロマンデロニトリル(>99%)50mg([S]=60mM、10g/l)の反応を、培養チューブ中、150rpmで3種の異なる温度(30℃、35℃、40℃)で行った(並列バッチ)。
【0090】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。反応完了後、生成物のeeを測定した。
【0091】
a. T=30℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、42%のヒドロキシ酸が形成され、約20時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=95%)。
【0092】
b. T=35℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、65%のヒドロキシ酸が形成され、19時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=96.5%)。
【0093】
c. T=40℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、86%のヒドロキシ酸が形成され、19時間後には、(R)−2−クロロマンデル酸を形成する加水分解は完了した(生成物ee=97.9%)。
【0094】
実験6.2:
実験6.2では、温度を50℃に上げた。
【0095】
生体触媒を、実施例4、実験4.3に従って作製した(OD546 44)。各々の場合に、4.85mlの細胞懸濁物(休止細胞、OD546 44)を酵素反応に用いた。DMSO(3%)150μlに溶解した(R)−2−クロロマンデロニトリル(>99%)50mg([S]=60mM、10g/l)の反応を、培養チューブ中、150rpmで3種の異なる温度(30℃、40℃、50℃)で行った(並列バッチ)。
【0096】
変換率をHPLCによってモニターするために、各々の場合に、測定前に、200μlのサンプルを200μlの1N HClと混合し、遠心分離し(5分、13000rpm)、希釈した。反応完了後、生成物のeeを測定した。
【0097】
a. T=30℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、42%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0098】
b. T=40℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、80%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0099】
c. T=50℃
早くも30分後には、シアノヒドリンのすべてが反応してアミドを形成し、2時間後には、92%のヒドロキシ酸が形成され、19時間後には、ヒドロキシ酸を形成する加水分解は完了した(生成物ee>99%)。
【0100】
(実施例7:セミ分取スケールで行う、ロードコッカス・エリスロポレス NCIMB 11540 の存在下のアルデヒドのシアノヒドリンの反応)
すべての反応について、K/Na−リン酸緩衝液(50mM、pH6.5)を用いた。反応をHPLCで追った。サンプル採取後、生体触媒反応を停止させるために、サンプル容量分を1N HClと混合した(並列サンプル)。遠心分離(5分、13000rpm)後、サンプルをHPLC溶離液で希釈した。
【0101】
精密な分析のために、バイオマスを、4℃及び3000rpmで30分間遠心分離し、蒸留H2Oで1回洗浄した。上清を1N HClでpH2に酸性化した後、TBMEで3〜4回抽出した。
【0102】
実験7.1:
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 5.9)に集めた。細胞を緩衝液に再懸濁して、約180mlにした(休止細胞、OD546 80)。
【0103】
(R)−2−クロロマンデロニトリル(ee>99%)0.6gをDMSO1.5mlに溶解し、この細胞懸濁物の60mlと混合した。加水分解を、振盪キャビネット内で30℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、17時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.73g(109%)
生成物ee:>99%
【0104】
実験7.2:
実験7.2では、いくつかの反応パラメーターを変えた。標準的な条件は、10g/lの基質、及び共溶媒としてのDMSO(ここでは2.5%)であった。別のバッチ反応を、15g/lの基質を用い、更には10g/lの基質、及び共溶媒としてのDMFを用いて行った。
【0105】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 6.8)に集めた。細胞を緩衝液に再懸濁して、約190mlの緩衝液を得た(休止細胞、OD546 69)。3種の反応を行った。
【0106】
バッチA: (R)−2−クロロマンデロニトリル(ee>99%)0.3gをDMSO750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.31g(93%)
生成物ee:>99%
【0107】
バッチB: (R)−2−クロロマンデロニトリル(ee>99%)0.3gをDMF750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.30g(90%)
生成物ee:98.5%
【0108】
バッチC: (R)−2−クロロマンデロニトリル(ee>99%)0.45gをDMSO750μlに溶解し、この細胞懸濁物の30mlと混合した。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−2−クロロマンデル酸を生成する反応が事実上完了した。
粗収量:0.45g(90%)
生成物ee:>99%
【0109】
実験7.3:
ここでは、異なる基質濃度を有する2つのバッチ(バッチA 10g/l;バッチB 15g/l)で反応を行った。両方の反応は実質的に同一の速度で進行し、2時間後には完了した。
【0110】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地3l)、20時間後(OD546 1.2)に集めた。細胞を約180mlの緩衝液に再懸濁した(休止細胞、OD546 70)。2種の反応を行った。
【0111】
バッチA: (R)−2−クロロマンデロニトリル(ee>99%)0.8gをDMSO1.6mlに溶解し、細胞懸濁物(80ml)に加えた。加水分解を、振盪キャビネット内で50℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、2時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:0.85g(95%)
生成物ee:>99%
【0112】
バッチB: (R)−2−クロロマンデロニトリル(ee>99%)1.2gをDMSO1.6mlに溶解し、細胞懸濁物(80ml)に加えた。加水分解を、振盪キャビネット内で50℃及び150rpmで行った。30分後には、シアノヒドリンが完全に加水分解され、2時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:1.26g(94%)
生成物ee:98.9%
【0113】
実験7.4:
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2.5l)、20時間後(OD546 6.9)に集めた。細胞を約160mlの緩衝液に再懸濁した(休止細胞、OD546 63)。
【0114】
(R)−2−クロロマンデロニトリル(ee>99%)1.3gをDMSO2.5mlに溶解し、この細胞懸濁物の140mlに加えた。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、3時間後には、(R)−2−クロロマンデル酸を生成する反応が完了した。
粗収量:1.43g(98%)
生成物ee:>99%
【0115】
実験7.5:
この反応では、1gのマンデロニトリルを8g/lの基質濃度で加水分解して、対応するヒドロキシ酸を得た。
【0116】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後(OD546 8.4)に集めた。細胞を約120mlの緩衝液に再懸濁した(休止細胞、OD546 74)。生体触媒を超低温冷凍した後、反応を一晩行った。
【0117】
(R)−(+)−マンデロニトリル1.0gをDMSO2.4mlに溶解し、細胞懸濁物(120ml)に加えた。加水分解を、振盪キャビネット内で40℃及び150rpmで行った。15分後には、シアノヒドリンが完全に加水分解され、5時間後には、(R)−マンデル酸を生成する反応が完了した。
粗収量:1.16g(100%)
生成物ee:93%
【0118】
(実施例8:セミ分取スケールで行う、ロードコッカス・エリスロポレス NCIMB 11540 の存在下のケトンのシアノヒドリンの反応)
K/Na−リン酸緩衝液(50mM、pH6.5)を用いた。反応をTLCで追った。
【0119】
精密な分析のために、バイオマスを、4℃及び6000rpmで20分間遠心分離し、蒸留H2Oで1回洗浄した。上清を1N HClでpH2に酸性化した後、TBMEで3〜4回抽出した。
【0120】
生体触媒を、実施例1、変形例IIに従って作製し(発酵培地2l)、20時間後に集めた。細胞を約60mlの緩衝液に再懸濁した(休止細胞、OD546 60)。
【0121】
(S)−アセトフェノンシアノヒドリン(アセトフェノン 25%、ee 94%)300mgをDMSO1mlに溶解し、細胞懸濁物に加えた。20時間後には、反応が完了し(TLCによる)、生成物(1−フェニルエタノール及び微量の他の不純物を含有)を抽出した。反応は、エナンチオマー純度を喪失することなく進行した。
粗収量:357mg
【0122】
(実施例9:大腸菌における組換え発現による、置換シアノヒドリンの加水分解のためのニトリルヒドラターゼ及びアミダーゼの酵素調製物の作製)
ニトリルヒドラターゼを発現させるために、また、アミダーゼを発現させるために、pMS470 プラスミド系を用いた。このプラスミドは、複製されたエレメントに加えて、選択可能なアンピシリン抵抗性を有し、また、誘導性 tac プロモーターによって、クローン化されたオープンリーディングフレームの制御された過剰発現を可能にする Lac リプレッサー遺伝子 lac Iを有する。
【0123】
ロードコッカス・エリスロポレス NCIMB 11540 のニトリルヒドラターゼのための発現プラスミド:
プラスミドマップが図1に示されている。このプラスミドは、pMS470Nhase7.3 という名称を有する。このプラスミドは、ニトリルヒドラターゼの2つの遺伝子領域(α−及びβ−サブユニット)に加えて、アクチベータータンパク質をコードする第3のオープンリーディングフレームも有する。
【0124】
ロードコッカス・エリスロポレス NCIMB 11540 のアミダーゼのための発現プラスミド:
ニトリルヒドラターゼの場合とは異なり、ロードコッカス・エリスロポレス NCIMB 11540 のアミダーゼの発現に必要なのは1つのリーディングフレームのみである。これを、プラスミド pMS470-33/3/1/11 の、tac プロモーターの下流側にクローン化した。図2は、この構築物のプラスミドマップを示す。
【0125】
組換えニトリルヒドラターゼ及び組換えアミダーゼの発酵:
2つの酵素の発酵はすべて、pMS470 系における酵素の過剰発現のために開発された一般的なプロトコールによって行った。
【0126】
ここで、サンプルは、
・一晩培養した培養物(ONC)から、LB培地及び抗生物質を含有する振盪フラスコ内の主培養液に接種し、
・対数期まで増殖させ、
・0.8〜1.5のOD600(600nmにおける光学濃度)で、IPTG(イソプロピルチオガラクトピラノシド)で誘導し、
・更に18時間誘導し(タンパク質発現)、
・集め(遠心分離)、破砕した(超音波)。
【0127】
ニトリルヒドラターゼの発現:
pMS470Nhase7.3(又は pMSNhasetactac7.3)によって形質転換された大腸菌B BL21細胞をLB−アンピシリンプレート上で単離し、100mlのLB−アンピシリン培地のONCに個々のコロニーを接種した。翌朝、0.01〜0.03のOD600(Beckmann光度計)に達するように、1000mlシケイン付フラスコ内の、250mlのLB−アンピシリン培地からなる主培養液に接種した。25℃よりも高い温度では専ら不溶性の封入体が形成されるので、増殖温度を25℃に制御した。誘導密度(OD600=1、Beckmann光度計)に到達した後、IPTGを0.1mMの濃度まで加えることによって培養物を誘導した。更に、培地に0.1mMクエン酸鉄(III)アンモニウムを補充した。>4のOD600に達した後、培養物を集め(約3000×gで15分間遠心分離)、約100mlのPBS緩衝液で1回洗浄した。その後、細胞ペレットをPBS緩衝液に再懸濁し(総容量約5ml)、超音波プローブ(BRANSON Sonifier 250、出力60%に設定、一定周波数で処理、1回30秒を5回、1回ごとに冷却のために1分間休止)を用いて破砕した(破砕を顕微鏡で目視して確認)。得られた粗ライセートを、下記に列挙する条件の下でメタクリロニトリルを基質として用いて分析したところ、約100〜250U/ml(pMSNhasetactac7.3 については約350〜500U/ml)の典型的な活性を有していた。
【0128】
保存のために、ライセートを−20℃で貯蔵した。室温で貯蔵すると、活性が急速に失われる。
【0129】
活性測定: 粗ライセートを、活性測定の直前にPBS緩衝液で1:10に希釈した。PBS緩衝液を溶媒とする40mMメタクリロニトリル溶液1.4mlを希釈ライセート20μlと混合し、28℃でインキュベートした(Eppendorf Thermomixer 5436)。0分、1分、2分、5分、10分及び15分の時点で、200μlのサンプルを抜き取り、これらのサンプル中の酵素反応を800μlの0.17%リン酸によって直ちに停止させた。遠心分離(16000×g、10分)の後、サンプルを224nmで分光光度法により分析した(Perkin Elmer UV/VIS分光計 Lambda Bio)。吸光の増大がメタクリルアミドの濃度の増大と相関することが明らかとなった。なお、0.57l・mmol−1・cm−1のε値を基準として用いた。
【0130】
アミダーゼの発現:
pMS470-33/3/1/11 によって形質転換された大腸菌B BL21細胞をLB−アンピシリンプレート上で単離し、100mlのLB−アンピシリン培地のONCに個々のコロニーを接種した。翌朝、0.01〜0.03のOD600(Beckmann光度計)に達するように、1000mlシケイン付フラスコ内の、250mlのSOC−アンピシリン培地からなる主培養液に接種した。37℃で発酵させると、専ら不溶性の、かつ不活性なタンパク質が形成されるので、増殖温度を30℃に制御した。誘導密度(OD600=1、Beckmann光度計)に到達した後、IPTGを0.3mMの濃度まで加えることによって培養物を誘導した。16時間の誘導の後、細胞を集め(約3000×gで10分間遠心分離)、リン酸ナトリウム緩衝液(0.1M、pH=7)で洗浄した。得られたペレットを洗浄緩衝液に再懸濁し(総容量約5ml)、超音波プローブ(BRANSON Sonifier 250、出力60%に設定、一定周波数で処理、1回30秒を5回、1回ごとに冷却のために1分間休止)を用いて破砕した(冷却負荷は終始一定)(破砕を顕微鏡で目視して確認)。このようにして得られた粗ライセートを、−20℃で凍結保存した。ライセートは、PBS緩衝液中のアセトアミド(40mM)を基質として用いて37℃で測定した(インドフェノールブルー法による遊離アンモニウムの測定)ところ、約75U/mlの活性を有していた。
【0131】
アミダーゼ活性の測定: 下記の溶液を用いた。
基質溶液:40mMアセトアミドのPBS溶液
溶液A:10%(w/v)フェノールのエタノール(95%)溶液
溶液B:0.5%(w/v)ニトロプルシドナトリウムのddH2O溶液
溶液C:クエン酸三ナトリウム100g及び水酸化ナトリウム5gが水550mlに溶解した溶液
溶液D:市販の通常の次亜塩素酸ナトリウム溶液600mlが、1000mlになるように希釈された溶液
アンモニウム標準液:0μg/l、80μg/l、120μg/l、200μg/l、280μg/l及び400μg/lの硫酸アンモニウム水溶液
【0132】
1.4mlの基質溶液を30℃で10μlの酵素希釈物(PBS中1:10)と共にインキュベートした(Eppendorf Thermomixer 5436)。0分、1分、2分、5分、10分及び15分後に、サンプル100μl中の酵素反応を、20μlの溶液Aを用いて停止させた。最後のサンプルを抜き取った後、得られた溶液を400μlの水で希釈した。更に、較正のために、アンモニウム標準溶液(各500ml)を20μlの溶液Aと混合した。その後直ちに、サンプルに、また、標準液に、溶液B20μl、及び、4部の溶液Cと1部の溶液Dとの混合液50μlをピペットで加えた。ボルテックスによって十分に混合されるようにした。得られたサンプル及び標準液を37℃で15分間放置した。すべてのサンプル及び標準液を水で希釈(1:10)した後、得られた青色の呈色を、分光光度計(Perkin Elmer UV/VIS分光計 Lambda Bio)を用いて640nmで定量した。青色の呈色の経時的な増大と標準液の吸光度との相関関係を求めることによって、活性(1分当たりの遊離アンモニウムのμmol数)を逆算することができる。
【0133】
(実施例10:組換え酵素を用いる加水分解)
これらの反応では、ロードコッカス・エリスロポレス NCIMB 11540 のクローン化ニトリルヒドラターゼを大腸菌クローン7.3(実施例9に従って作製)の粗ライセートとして用いた。
【0134】
手順:
50μlの粗ライセートを425μlの緩衝液(K/Na−PO4緩衝液、pH7、50mM)で希釈し、DMSOを溶媒とする約220mMの基質溶液25μlと混合した(タンパク質5mg/ml、基質濃度約10mM、5%DMSO)。反応を、Thermomixer 内で30℃及び1000rpmで行った。0分、2分、4分、6分、8分、10分、15分、20分、30分、60分及び120分後、各々の場合に、1個のエッペンドルフ容器を0.5mlの1N HClと混合した。遠心分離(5分、13000rpm)及び対応する希釈の後、シアノヒドリン及びヒドロキシアミドの濃度をHPLCによって測定した。ニトリルヒドラターゼの活性をヒドロキシアミドの形成速度(初期範囲における傾き)から求めた。標準基質(活性100%)として、2−ヒドロキシ−4−フェニルブチロニトリルを用いた。その他の基質の加水分解における活性を、2−ヒドロキシ−4−フェニルブチロニトリルに対する活性と比較した(表5)。
【0135】
結果:
2−ヒドロキシ−4−フェニルブチロニトリルの加水分解における、大腸菌クローン7.3の粗ライセート中のニトリルヒドラターゼの活性は、約0.3μmol・mg−1・min−1であった。表5では、異なる基質に対する活性が比較されている。
【0136】
【表5】
【0137】
(セミ分取スケールで行う(R)−2−クロロマンデロニトリルの反応)
ロードコッカス・エリスロポレス NCIMB 11540 由来のクローン化ニトリルヒドラターゼ(大腸菌クローン7.3、実施例9に従って作製)の粗ライセート50mlを100mlの緩衝液(K/Na−PO4緩衝液、50mM、pH6.5)で希釈した。(R)−2−クロロマンデロニトリル(ee>99%)1.0gがDMSO1.5mlに溶解した溶液を加えた後、懸濁物を150rpm及び30℃で振盪した。反応完了後、細胞破片を遠心分離し、生成物を、4日間にわたってCH2Cl2で連続抽出した。
粗生成物のee:>99%
精製後の収量:0.91g(82%)
【図面の簡単な説明】
【0138】
【図1】pMS470Nhase7.3 のプラスミドマップある。
【図2】pMS470-33/3/1/11 のプラスミドマップある。
【特許請求の範囲】
【請求項1】
キラルα−ヒドロキシカルボン酸を製造する方法であって、(R)−又は(S)−シアノヒドリンを、ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 の存在下で酵素的加水分解を行うことにより、共役の(R)−又は(S)−α−ヒドロキシカルボン酸に変換することを含む方法。
【請求項2】
対応する脂肪族、芳香族又は複素芳香族のアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加によって得られる(R)−又は(S)−シアノヒドリンを出発物質として用いることを特徴とする、請求項1に記載の方法。
【請求項3】
式:
【化1】
[式中、R1及びR2は相互に独立して、H、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいC1−C6アルキルラジカル若しくはC1−C6アルケニルラジカル、又は、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいフェニルラジカルである。但し、R1及びR2が共にHであることはない。]
の(R)−又は(S)−シアノヒドリンを用いることを特徴とする、請求項1に記載の方法。
【請求項4】
微生物ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 を、粉砕細胞、粗酵素若しくは精製酵素、組換え酵素、固定化細胞若しくは固定化酵素、凍結乾燥細胞、又は「休止細胞」の形態で用いることを特徴とする、請求項1に記載の方法。
【請求項5】
微生物ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 を水性溶媒に懸濁し、得られた懸濁物を、共溶媒としての可溶化剤の存在下、対応するキラルシアノヒドリンと混合することを特徴とする、請求項1に記載の方法。
【請求項6】
可溶化剤として、有機溶媒、界面活性剤又は相間移動触媒を用いることを特徴とする、請求項5に記載の方法。
【請求項7】
有機溶媒として、DMSO、DMF、C1−C6アルコール、TMBE又はそれらの混合物を用いることを特徴とする、請求項6に記載の方法。
【請求項8】
共溶媒の割合が、反応溶液の全体積を基準として、0.5〜20体積%の範囲内にあることを特徴とする、請求項5に記載の方法。
【請求項9】
反応混合物のpHが4.5〜11の範囲内にあることを特徴とする、請求項1に記載の方法。
【請求項10】
加水分解を10〜60℃の範囲内の温度で行うことを特徴とする、請求項1に記載の方法。
【請求項11】
組換え酵素として、適当な宿主細胞における pMS470 プラスミド系の発現によって得られる酵素を用いることを特徴とする、請求項4に記載の方法。
【請求項1】
キラルα−ヒドロキシカルボン酸を製造する方法であって、(R)−又は(S)−シアノヒドリンを、ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 の存在下で酵素的加水分解を行うことにより、共役の(R)−又は(S)−α−ヒドロキシカルボン酸に変換することを含む方法。
【請求項2】
対応する脂肪族、芳香族又は複素芳香族のアルデヒド又はケトンへの、シアニド基の酵素的な、又は化学的に触媒された付加によって得られる(R)−又は(S)−シアノヒドリンを出発物質として用いることを特徴とする、請求項1に記載の方法。
【請求項3】
式:
【化1】
[式中、R1及びR2は相互に独立して、H、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいC1−C6アルキルラジカル若しくはC1−C6アルケニルラジカル、又は、反応条件下で不活性である置換基によって一置換又は多置換されていてもよいフェニルラジカルである。但し、R1及びR2が共にHであることはない。]
の(R)−又は(S)−シアノヒドリンを用いることを特徴とする、請求項1に記載の方法。
【請求項4】
微生物ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 を、粉砕細胞、粗酵素若しくは精製酵素、組換え酵素、固定化細胞若しくは固定化酵素、凍結乾燥細胞、又は「休止細胞」の形態で用いることを特徴とする、請求項1に記載の方法。
【請求項5】
微生物ロードコッカス・エリスロポレス(Rhodococcus erythropolis)NCIMB 11540 を水性溶媒に懸濁し、得られた懸濁物を、共溶媒としての可溶化剤の存在下、対応するキラルシアノヒドリンと混合することを特徴とする、請求項1に記載の方法。
【請求項6】
可溶化剤として、有機溶媒、界面活性剤又は相間移動触媒を用いることを特徴とする、請求項5に記載の方法。
【請求項7】
有機溶媒として、DMSO、DMF、C1−C6アルコール、TMBE又はそれらの混合物を用いることを特徴とする、請求項6に記載の方法。
【請求項8】
共溶媒の割合が、反応溶液の全体積を基準として、0.5〜20体積%の範囲内にあることを特徴とする、請求項5に記載の方法。
【請求項9】
反応混合物のpHが4.5〜11の範囲内にあることを特徴とする、請求項1に記載の方法。
【請求項10】
加水分解を10〜60℃の範囲内の温度で行うことを特徴とする、請求項1に記載の方法。
【請求項11】
組換え酵素として、適当な宿主細胞における pMS470 プラスミド系の発現によって得られる酵素を用いることを特徴とする、請求項4に記載の方法。
【図1】

【図2】
【図2】
【公表番号】特表2006−525795(P2006−525795A)
【公表日】平成18年11月16日(2006.11.16)
【国際特許分類】
【出願番号】特願2006−501668(P2006−501668)
【出願日】平成16年1月30日(2004.1.30)
【国際出願番号】PCT/EP2004/000859
【国際公開番号】WO2004/076385
【国際公開日】平成16年9月10日(2004.9.10)
【出願人】(505326209)ディーエスエム ファイン ケミカルズ オーストリア エヌエフジー ゲーエムベーハー アンド コーポレイション カーゲー (13)
【Fターム(参考)】
【公表日】平成18年11月16日(2006.11.16)
【国際特許分類】
【出願日】平成16年1月30日(2004.1.30)
【国際出願番号】PCT/EP2004/000859
【国際公開番号】WO2004/076385
【国際公開日】平成16年9月10日(2004.9.10)
【出願人】(505326209)ディーエスエム ファイン ケミカルズ オーストリア エヌエフジー ゲーエムベーハー アンド コーポレイション カーゲー (13)
【Fターム(参考)】
[ Back to top ]