
クリアランス速度を高めるための二重特異性抗体点変異
(a)CH2ドメインに1以上のアミノ酸変異を有するIgG由来のヒトヒンジ定常領域、(b)2つのscFv、および(c)2つのFvを含む変異二重特異性抗体が構築されている。この種の抗体はクリアランスの増強を示し、それはプレターゲッティング法に関して特に有用であることが分かっっている。
【発明の詳細な説明】
【発明の背景】
【0001】
発明の分野
本発明は、対応する親二重特異性抗体(bsAb)よりも迅速に患者の身体から除去される変異二重特異性抗体(bsAb)に関する。特に、本発明は、IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbに関し、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。
【0002】
関連技術
標的部位の検出は、検出剤のシグナル対バックグラウンド比が高いと有利である。治療は、標的部位で治療剤ができる限り多く確実に付着すること、および取り込みおよび結合の持続時間が適度に長いことが有利となる。ターゲッティング率および標的部位への薬剤送達量を向上させるためには、選択的局在性のためにターゲッティング部分と結合した診断剤または治療剤を含んでなるターゲッティングベクターの使用が長く知られている。
【0003】
ターゲッティングベクターの例としては、抗体または抗体フラグメント、細胞または組織特異的ペプチド、ならびにホルモンおよびその他の受容体結合分子などのターゲッティング部分の診断剤または治療剤複合体が挙げられる。例えば、病理細胞および正常細胞に関連する、ならびに病原体微生物に関連する種々の決定基に対する抗体が、多種多様な病状または病変の検出および治療に用いられている。これらの方法では、例えば、Hansen et al., 米国特許第3,927,193号明細書およびGoldenberg, 米国特許第4,331,647号明細書、同第4,348,376号明細書、同第4,361,544号明細書、同第4,468,457号明細書、同第4,444,744号明細書、同第4,460,459号明細書、同第4,460,561号明細書、同第4,624,846号明細書および同第4,818,709号明細書(引用することによりその全開示内容を本明細書の一部とする)に記載のように、ターゲッティング抗体を直接適当な検出剤または治療剤と結合させる。
【0004】
ダイレクトターゲッティング法、すなわち、診断剤または治療剤(「有効な薬剤」)がターゲッティング部分と直接結合される方法で遭遇する問題は、複合体の比較的小さな部分は標的部位と結合するが、複合体の大部分は循環系中に残ったままであり、何らかの方法で標的複合体の機能を損なうことである。診断複合体の場合には、例えば、ラジオイムノシンチグラフィーまたは磁気共鳴イメージング複合体、循環系中に残る非標的化複合体が、バックグラウンドを増加させて解像度を低下させることがある。抗体などの長期間循環するターゲッティング部分に付着した毒性の高い治療剤、例えば放射性同位元素、薬物または毒素を有する治療用複合体の場合、循環する複合体は骨髄毒性または全身性副作用などの宿主に許容されない毒性となり得る。
【0005】
プレターゲッティング法は、検出剤または治療剤の標的:バックグラウンド比を増加させるために開発されてきた。プレターゲッティングおよびビオチン/アビジンアプローチの例は、例えば、Goodwin et al., 米国特許第4,863,713号明細書;Goodwin et al., J. Nucl. Med. 29:226, 1988; Hnatowich et al., J. Nucl. Med. 28:1294, 1987; Oehr et al., J. Nucl. Med. 29:728, 1988; Klibanov et al., J. Nucl. Med. 29:1951, 1988; Sinitsyn et al., J. Nucl. Med. 30:66, 1989; Kalofonos et al., J. Nucl. Med. 31:1791, 1990; Schechter et al., Int. J. Cancer 48:167, 1991; Paganelli et al., Cancer Res. 51:5960, 1991; Paganelli et al., Nucl. Med. Commun. 12:211, 1991;米国特許第5,256,395号明細書; Stickney et al., Cancer Res. 51:6650, 1991; Yuan et al., Cancer Res. 51:3119, 1991;米国特許第6,077,499号明細書;米国出願番号09/597,580;同10/361,026;同09/337,756;同09/823,746;同10/116,116;同09/382,186;同10/150,654;米国特許第6,090,381号明細書;同第6,472,511号明細書;米国出願番号10/114,315;米国仮出願番号60/386,411;同60/345,641;同60/3328,835;同60/426,379;米国出願番号09/823,746;同09/337,756;および米国仮出願番号60/342,103に記載されている。なお、これらは総て引用することによりその全開示内容を本明細書の一部とする。
【0006】
プレターゲッティング法では、一次ターゲッティング種(診断剤または治療剤と結合していないもの)を投与する。一次ターゲッティング種は、標的部位と結合するターゲッティング部分、および標的化可能な構築物の結合部位と結合可能な結合部分を含む。一次ターゲッティング種が十分に付着すれば、標的化可能な構築物を投与する。標的化可能な構築物は、一次ターゲッティング種の利用可能な結合部位を認識する結合部位および診断剤または治療剤を含む。
【0007】
プレターゲッティングは、ダイレクトターゲッティング法の使用にある一定の利点を与える手法である。例えば、治療、例えば放射線免疫治療のための標的部位への放射性核種のin vivo送達にプレターゲッティング法を用いると、放射免疫複合体の長期循環により生じる骨髄毒性が減少する。これは、放射性同位元素が、多くの場合に長期間循環する種である第一のターゲッティング分子と直接結合するよりも迅速にクリアランスされる、分子量の低いキレートとして送達されるためである。
【0008】
プレターゲッティング法で遭遇する問題は、循環している一次ターゲッティング種(標的部位と結合していない一次ターゲッティング種)が、標的化可能な複合体の、(一次ターゲッティング種の結合部分を介して)標的部位と結合しているターゲッティング種との結合を妨害することである。従って、循環している一次ターゲッティング種の量を最小限に抑える方法が必要とされる。
【0009】
循環している一次ターゲッティング種の量を最小限に抑える試みはいくつかなされてきた。この目標を達成する一つの方法は、身体からのクリアランス速度の速い一次ターゲッティング種を調製することである。例えば、Ward et al.(米国特許第6,165,745号明細書)はマウス由来の変異IgG1を合成し、Hornick et al.「The Journal of Nuclear Medicine 11 355-362 (2000)」は変異キメラTNT−3抗体を合成した。これらの変異抗体は、本発明による変異bsAbとは異なる。違いの一つは、本発明による変異bsAbは二重特異性抗体であるのに対し、Hornick et al.およびWard et al.の抗体は単一特異性抗体である点である。二重特異性抗体は単一特異性抗体に比べて異なる特性を有することから、この違いは有意である。本変異bsAbとWard et al.のマウス抗体とのもう一つの違いは、Ward et al.のマウス抗体にはエフェクター機能がないことである。従って、Ward et al.の抗体は、本変異bsAbのように補体を固定することまたは効果的なADCC(抗体依存性細胞傷害)を果たすことができない。
【発明の概要】
【0010】
IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbを提供することが本発明の目的であり、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。いくつかの実施態様においては、FvおよびscFvはCDRグラフト化マウスまたはヒト化成分である。別の実施態様では、FvおよびscFvはヒトまたはヒト化成分である。いくつかの実施態様において、ヒンジ定常領域はイソロイシン253のアラニンへの変異を含む。本発明はまた、FvがhMN14−IgG、ヒト化クラスIII、抗−CEA mAbに由来し(米国特許第5,874,540号明細書参照)、scFvは734scFvであり、ヒンジ定常領域で253番目のイソロイシンがアラニンへ変異している変異bsAbを提供する。
【発明の具体的説明】
【0011】
特に断りのない限り、可算名詞("a"または"an")は1つ以上のものを意味する。
【0012】
I.概要
本発明は、IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbに関し、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。本発明による変異bsAbは、対応する親bsAbよりも迅速に患者の身体から除去される。二重特異性抗体は、1999年6月22日出願の米国出願番号09/337,756に開示されている。プレターゲッティング法に用いると、循環している一次ターゲッティング種(標的部位と結合していない変異bsAb)の量が最小限度に抑えられる。さらに、血中で捕捉される標的化可能な構築物の量も最小限度に抑えられる。
【0013】
ヒトヒンジ定常領域はエフェクター機能を含み得る。抗体分子のFc部分は、細胞溶解を引き起こす作用をするようメカニズムを設定する補体結合反応およびADCC(抗体依存性細胞傷害)などのエフェクター機能を提供する。しかし、治療機能のためにはこのFc部分は必要でなく、アポトーシスなどの他のメカニズムが機能を果たす可能性がある。従って、生得的なADCC、アポトーシス誘発および補体活性化/溶解が達成され得る。
【0014】
scFvは標的化可能な構築物の結合部位に対して特異的である。標的化可能な構築物は担体部分および少なくとも1ユニットの認識可能なハプテンから成る。認識可能なハプテンの例としては、限定されるものではないが、ヒスタミンスクシニルグリシン(HSG)、DTPAおよびフルオレセインイソチオシアネートが挙げられる。標的化可能な構築物は、罹患組織の治療または同定に有用な種々の薬剤と結合してもよい。結合される薬剤の例としては、限定されるものではないが、キレート剤、金属キレート錯体、薬物、毒素(例えば、リシン、アブリン、リボヌクレアーゼ、DNアーゼI、ブドウ球菌内毒素−A、アメリカヤマゴボウ抗ウイルスタンパク質、ゲロニン、ジフテリア毒、シュードモナス外毒素、およびシュードモナス内毒素)およびその他のエフェクター分子が挙げられる。結合に好適な薬物としては、ドキソルビシン類似体、SN−38、エトポシド、メトトレキサート、6−メルカプトプリンまたはリン酸エトポシド、カリチェアマイシン(calicheamicin)、パクリタキセル、2−ピロリノドキソルビシン、CC−1067、およびアドゼレシンまたはその組み合わせが挙げられる。薬物の例としては、ナイトロジェンマスタード、エチレンイミン誘導体、スルホン酸アルキル、ニトロソウレア、トリアゼン、葉酸類似体、アントラサイクリン、タキサン、COX−2阻害剤、ピリミジン類似体、プリン類似体、抗生物質、酵素、エピポドフィロトキシン、プラチナ錯体、ビンカアルカロイド、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、アンタゴニスト、エンドスタチン、タキソール、カンプトセシン、ドキソルビシン、およびそれらの類似体、ならびにそれらの組み合わせが挙げられる。さらに、プロドラッグの活性化または薬物の標的特異的傷害性の増加に有用な酵素を標的化可能な構築物と結合させることもできる。よって、標的化可能な構築物に対して反応性のあるscFvを含む変異bsAbの使用によって、各適用ごとに新規なbsAbを産生させることなく、種々の治療および診断適用を実施することが可能となった。
【0015】
さらに、本発明は、哺乳類における標的細胞、組織または病原体の検出または治療のための方法を包含し、IgG由来のヒトヒンジ定常領域、2つのFvおよび2つのscFvを含み、ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む、有効量の変異bsAbを投与することを含んでなる。本明細書において「病原体」とは、限定されるものではないが、菌類(例えば、ヒストプラズマ・カプスラーツム、ブラストミセス・デルマチチジス、コクシジオイデス・イミティスおよびカンジダ種)、ウイルス(例えば、ヒト免疫不全ウイルス(HIV)、ヘルペスウイルス、サイトメガロウイルス、狂犬病ウイルス、インフルエンザウイルス、B型肝炎ウイルス、センダイウイルス、ネコ白血病ウイルス、レオウイルス、ポリオウイルス、ヒト血清パルボ様ウイルス、シミアンウイルス40、呼吸器多核体(RS)ウイルス、マウス乳癌ウイルス、水疱−帯状疱疹ウイルス、デングウイルス、風疹ウイルス、麻疹ウイルス、アデノウイルス、ヒトT細胞白血病ウイルス、エプスタイン−バーウイルス、マウス白血病ウイルス、流行性耳下腺炎ウイルス、水疱性口内炎ウイルス、シンドビスウイルス、リンパ球性脈絡髄膜炎ウイルス、疣贅ウイルスおよびブルータングウイルス)、寄生虫、微生物(例えば、リケッチア)および細菌(例えば、ストレプトコッカス・アガラクチエ、レジュネラ・ニューモフィラ、化膿連鎖球菌、大腸菌、淋菌、髄膜炎菌、肺炎双球菌、B型インフルエンザ菌、梅毒トレポネーマ、ライム病スピロヘータ、緑膿菌、緑膿菌、ウシ流産菌、結核菌、炭疽菌の胞子および破傷風毒素)をさす。米国特許第5,332,567号明細書を参照されたい。
【0016】
本明細書において「抗体」とは、全長(すなわち、天然に存在する、または正常免疫グロブリン遺伝子フラグメント組換え法により形成される)免疫グロブリン分子(例えばIgG抗体)、または抗体フラグメントのような、免疫グロブリン分子の免疫学的に活性な(すなわち、特異的に結合する)部分をさす。抗体という語は、キメラ抗体、CDRグラフト化(ヒト化)抗体、および完全なヒト抗体を包含する。「IgG」という語は、抗原に対して産生され、抗原と特異的に結合することができる抗体、すなわち免疫グロブリンGを意味する。抗体はAbと省略される。モノクローナル抗体はmAbと省略される。
【0017】
「ヒト抗体」は、抗原刺激に応答して特定のヒト抗体を産生するように「操作された」トランスジェニックマウスから得られる抗体である。この技術では、ヒトH鎖およびL鎖の遺伝子座のエレメントが、内在性H鎖およびL鎖遺伝子座が標的化破壊されている胚幹細胞株由来のマウス系統に導入される。トランスジェニックマウスは、ヒト抗原に特異的なヒト抗体を合成することができ、このマウスを用いてヒト抗体分泌ハイブリドーマを作製することができる。トランスジェニックマウスからヒト抗体を得る方法は、Green et al., Nature Genet. 7:13 (1994)、Lonberg et al., Nature 368:856 (1994)、およびTaylor et al., Int. Immun. 6:579 (1994)に記載されている。完全ヒト抗体はまた、遺伝子または染色体トランスフェクション法、ならびにファージディスプレー技術によって構築でき、これらは全て当技術分野で公知である。例えば、未免疫のドナー由来の免疫グロブリン可変ドメイン遺伝子レパートリーからのヒト抗体およびそのフラグメントのin vitro作製に関してはMcCafferty et al., Nature 348:552-553 (1990)を参照。この技術では、抗体可変ドメイン遺伝子がフレーム内で糸状バクテリオファージの主要または微量コートタンパク質遺伝子にクローニングされ、ファージ粒子表面上に機能的抗体フラグメントとして提示される。この糸状粒子はファージゲノムの単鎖DNAコピーを含むため、抗体の機能特性に基づいて選択すれば、それらの特性を示す抗体をコードする遺伝子が選択されることになる。このように、ファージはB細胞の特性のいくつかを模倣する。ファージディスプレー法は多様な形式で行うことが可能であり、総説としては、例えば、Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993)を参照されたい。
【0018】
ヒト抗体はまた、in vitro活性化B細胞でも産生し得る。引用することによりその全開示内容が本明細書の一部とされる米国特許第5,567,610号明細書および同第5,229,275明細書を参照されたい。
【0019】
抗体フラグメントは、F(ab’)2、F(ab)2、Fab’、Fab、Fv、scFvなどの抗体の一部分である。構造にかかわらず、抗体フラグメントは無傷の抗体によって認識される同じ抗原と結合する。例えば、抗CEAモノクローナル抗体フラグメントはCEAのエピトープと結合する。
【0020】
「抗体フラグメント」という語は、特定の抗原に結合して複合体を形成することにより抗体のようにふるまう合成または遺伝子組み換えタンパク質も含む。例えば、抗体フラグメントには、L鎖可変領域からなる単離フラグメント、H鎖およびL鎖の可変領域からなる「Fv」フラグメント、L鎖およびH鎖可変領域がペプチドリンカーによって接続されている組み換え単鎖ポリペプチド分子(「scFvタンパク質」)、ならびに超可変領域に類似したアミノ酸残基からなる最小認識ユニットが含まれる。
【0021】
キメラ抗体は、齧歯類抗体などの第1の種由来の可変ドメインおよび相補性決定領域を含む組み換えタンパク質であり、抗体分子のH鎖およびL鎖定常領域はヒト抗体などの第2の種に由来する。
【0022】
ヒト化抗体は、モノクローナル抗体の相補性決定領域が、マウス免疫グロブリンなどの第1の種の免疫グロブリンのH鎖およびL鎖可変領域からヒトH鎖およびL鎖可変ドメイン中に移されている組み換えタンパク質であり、抗体分子のH鎖およびL鎖定常領域はヒト抗体に由来する。ヒト化抗体はまたCDRグラフト化抗体と呼ばれる。
【0023】
本明細書において、「二重特異性抗体」とは、2つの異なる部分、すなわち標的組織および標的化可能な構築物と結合可能な抗体をさす。
【0024】
本明細書において、治療剤は、特定の投与計画に従って本発明による抗体と組み合わせて被験体へ投与される、または抗体部分と結合して治療に有用な複合体を形成する分子または原子である。治療剤の例としては、薬物、毒素、ホルモン、酵素、免疫調節剤、キレート剤、ホウ素化合物、光活性薬または色素、および放射性同位元素が挙げられる。免疫調節剤の例は、サイトカイン、幹細胞増殖因子、リンホトキシン、造血因子、コロニー刺激因子(CSF)、インターフェロン(IFN)、エリスロポエチン、トロンボポエチン、およびそれらの組み合わせからなる群から選択される。特に有用なものは腫瘍壊死因子(TNF)などのリンホトキシン、インターロイキン(IL)などの造血因子、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)などのコロニー刺激因子、インターフェロン−α、−βまたは−γなどのインターフェロン、ならびに「S1因子」と呼ばれる幹細胞増殖因子である。より具体的には、IL−1、IL−2、IL−3、IL−6、IL−10、IL−12、IL−18、インターフェロン−γ、TNF−αまたはそれらの組み合わせなどの免疫調節剤が本発明において有用である。「scFv」とは、抗体のL鎖およびH鎖の可変領域がペプチドリンカーによって接続されている組み換え単鎖ポリペプチド分子を意味する。
【0025】
「Fv」とは、H鎖およびL鎖の可変領域からなるフラグメントを意味する。
【0026】
「組み換え宿主」は、クローニングベクターまたは発現ベクターのいずれかを含む任意の原核細胞または真核細胞であってよい。この語はまた、それらの原核細胞または真核細胞、ならびに宿主細胞または宿主細胞の細胞の染色体またはゲノム中にクローン化された遺伝子を含むように遺伝子組み換えされたトランスジェニック動物も含む。好適な哺乳類宿主細胞には、SP2/0細胞およびNS0細胞のような骨髄腫細胞、ならびにチャイニーズハムスター卵巣(CHO)細胞、ハイブリドーマ細胞株および抗体発現に有用な他の哺乳類宿主細胞を含む。また、mAbおよび他の融合タンパク質の発現に特に有用なものはWO 0063403 A2に開示されたヒト細胞株PER.C6であり、従来の哺乳類細胞株CHO、COS、 Vero、 HeLa、 BHKおよびSP2細胞系統に比べて2〜200倍の組換えタンパク質を産生する。修飾された免疫機構を有する特別なトランスジェニック動物は完全なヒト抗体を作製するために特に有用である。
【0027】
抗原はいずれの抗原であってもよい。抗原の例は、細胞表面または腫瘍関連抗原、あるいは微生物または寄生虫に関連する抗原、あるいは罹患組織または自己免疫疾患に関与するBまたはT細胞などの疾病を導く細胞種、あるいは心疾患または神経疾患の標的抗原 (例えば、前の場合にはアテローム斑または塞栓、および後の場合にはアルツハイマー病に関連するものなどのアミロイド)がある。本明細書において「組織」とは、当業者がそれを意味すると理解するであろう組織を意味する。本発明において提案されるように、組織は、体組織または体液(例えば血液細胞)の個々の細胞もしくは細胞群、または細胞培養物も意味する。さらに、組織は被験体内にあっても、生検組織であっても、または被験者から摘出した組織であってもよい。この組織はまた、身体の器官の全てまたは一部であってもよい。さらに、この組織は、切除と本発明による方法との間に保存工程を含まずに被験体から取り出されたばかりの組織であるという点で「新鮮」であると言える。組織はまた、本発明による方法の適用前に、限定されるものではないが、凍結、急速凍結、パラフィン包埋および組織固定をはじめとする標準の組織調製技術によって保存してもよい。
【0028】
「標的組織」とは、標的化可能な複合体が送達されうる系、器官、組織、細胞、受容体または細胞小器官である。本発明の治療的態様では、標的組織は、感染しているか、機能障害性であるか、置換されているか、または異所的である(例えば、感染細胞、癌細胞、子宮内膜症など)。骨髄など正常な組織もまた、治療的介入が必要であれば、これらの方法で切除してもよい。本発明の診断的態様では、標的組織を検出することが望ましい。
【0029】
本明細書において「被験体」とは、限定されるものではないが、ヒトおよびその他の霊長類、齧歯類(例えばマウス、ラット、およびモルモット)、ウサギ類(lagamorphs)(例えばウサギ)、ウシ類(例えばウシ)、ヒツジ類(例えばヒツジ)、ヤギ類(例えばヤギ)、ブタ類(例えばブタ)、ウマ類(例えばウマ)、イヌ類(例えばイヌ)、ネコ類(例えばネコ)、家禽類(例えばニワトリ、シチメンチョウ、アヒル、ガチョウ、その他のキジ類の鳥など)をはじめとするいずれの動物(すなわち、脊椎動物および無脊椎動物)、ならびに、限定されるものではないが、有蹄類(例えばシカ)、クマ類、魚類、ウサギ類、齧歯類、鳥類などの野生動物もさす。この語は特定の齢または性別に限定するものではない。よって、成体および新生児(仔)被験体、ならびに胎児(仔)は、雌雄にかかわらず、この語に包含される。
【0030】
本明細書において「親bsAb」という語は、親bsAbのヒンジ定常領域がCH2−CH3ドメイン境界領域で1以上のアミノ酸変異を含まないことを除いて、あらゆる点で変異bsAbに類似したbsAbを意味するのに使用する。
【0031】
本明細書において「ヒンジ定常領域」は、IgGのCl、CH1、ヒンジ、CH2およびCH3領域を含む。H鎖定常領域はCH1、ヒンジ、CH2およびCH3領域を含むが、L鎖定常領域はCl領域を含む。
【0032】
II.変異二重特異性抗体
変異bsAbのFvは抗体に由来し、標的組織と特異的に結合する。Fvの例は、「Anti-CD20 Antibodies And Fusion Proteins Thereof And Methods Of Use」と題された、代理人整理番号18733/1073、米国仮出願番号60/356,132、同60/416,232および代理人整理番号18733/1155(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどの抗CD20抗体;クラスIII抗癌胎児性抗原抗体(抗CEA抗体)である、米国出願番号5,874,540(引用することによりその全開示内容を本明細書の一部とする)に開示のものなどのhMN−14抗体;米国出願番号10/116,116(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのMu−9抗体;米国仮出願番号60/360,259(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのLL1抗体;米国仮出願番号60/399,707(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのAFP抗体;「Monoclonal Antibody cPAM4」と題された、代理人整理番号18733/1102の米国仮出願(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのPAM4抗体;米国仮出願番号60/360,229(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのRS7抗体;ならびに米国特許第5,789,554号明細書および同第6,187,287号明細書ならびに米国出願番号09/741,843および同09/988,013(引用することによりその全開示内容を本明細書の一部とする)に開示のものなどのCD22抗体由来のものである。造血器腫瘍および固形腫瘍のその他の多くの腫瘍関連抗原が当業者に公知であり、引用出願にも含められているように、(限定されるものではないが)CD15、CD19、CD20、CD21、CD22、CD23、CD25、CD40、CD45、CD66、CD74、CD80、Ii、Ia、HLA−DR、PSMA、PSA、前立腺酸性ホスファターゼ、テネイシン、Le(y)、AFP、HCG、CEA、CSAp、PAM4、MUC1、MUC2、MUC3、MUC4、EGP−1、EGP−2、EGFR、HER2/neu、インスリン成長因子受容体、S100、VEGF、胎盤成長因子(PlGF)、胎盤アルカリ性ホスファターゼ、壊死産物、癌遺伝子産物などが挙げられる。hMN−14のH鎖cDNAおよびアミノ酸配列を図1に示し、hMN−14のL鎖cDNAおよびアミノ酸配列を図2に示す。
【0033】
FvをコードするcDNAは、ヒンジ定常領域をコードするベクターへ挿入してもよい。発現ベクターの例として、IgG1のヒンジ定常領域のアミノ酸をコードするpdHL2がGillies S.D., Lo KM, and Wesolowski, J. J. Immunol Methods 125 191-202 (1989)およびLosman, M.J. et al. Cancer Supplement 80 2660-2666 (1997)によって報告されており、本発明による変異二重特異性抗体の構築に用いてよい。
【0034】
Fvはマウス抗体、CDRグラフト化(ヒト化)抗体、またはヒト抗体由来であってよい。Fvはヒトモノクローナル抗体、ヒトFvライブラリーを含むトランスジェニックマウス、またはファージ/リボソームヒトIgGライブラリーから得ることができる。
【0035】
FvがCDRグラフト化抗体に由来する場合、適当な可変領域枠組み構造配列を、抗原結合領域がそれに由来するドナー抗体のクラスまたは種類を考慮して用いてよい。用いるヒト枠組み構造の種類がドナー抗体と同じまたは類似のクラスまたは種類であることが好ましい。枠組み構造は、ドナー抗体配列との相同性を、特にCDRに空間的に近接または隣接する位置で最大化または最適化するように選択すると有利である。CDRグラフト化抗体を構築するために用いてよいヒト枠組み構造の例として、LAY、POM、TUR、TEI、KOL、NEWM、REIおよびEU(Kabat et al, 1987)がある。KOLおよびNEWMはH鎖構築に好適である。REIはL鎖構築に好適であり、EUはH鎖およびL鎖双方の構築に好適である。
【0036】
CDRグラフト化抗体のL鎖またはH鎖可変領域は、必要に応じてヒトL鎖またはH鎖定常ドメインと融合させてもよい(本明細書において「H鎖定常ドメイン」とは、特に指定のない限り、ヒンジ領域を含むものと理解される)。CDRグラフト化抗体のヒト定常ドメインは、それが存在する場合は、抗体の予定される機能、特に、必要とされうるエフェクター機能を考慮して選択してもよい。例えば、CDRグラフト化抗体が治療目的を意図し、抗体のエフェクター機能を必要とする場合には、IgG1およびIgG3イソ型ドメインを用いてよい。あるいは、CDRグラフト化抗体が抗体エフェクター機能を必要としない目的を意図する場合、例えばイメージング、診断または細胞毒性の標的化目的である場合は、IgG2およびIgG4イソ型ドメインを用いてよい。L鎖可変領域と融合してよいL鎖ヒト定常ドメインとしては、ヒトλまたは、特に、ヒトκ鎖が挙げられる。
【0037】
二重特異性変異抗体のヒンジ定常領域は、CH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。言い換えれば、二重特異性変異抗体のヒトヒンジ定常領域を二重特異性親抗体のヒトヒンジ定常領域と比較すると、その領域は1以上のアミノ酸が異なっている。
【0038】
変異は、例えば置換アミノ酸が電荷または大きさなど類似の構造または化学特性を有する「保存的」変化を包含する(例えばロイシンのイソロイシンによる置換)。変異はまた、例えば「非保存的」変化(例えばグリシンのトリプトファンによる置換)も包含する。
【0039】
好ましい実施態様では、253番目の位置のアミノ酸(Edelmanの付番方式に従う)が変異している。この位置での変異の例としては、イソロイシンのアラニンによる置換がある。いくつかの実施態様では、253番目のアミノ酸が、変異bsAbのクリアランスの薬物動態が253番目の位置のアミノ酸がアラニンへ変化している場合に認められるものと同等となるアミノ酸へ変異している。
【0040】
一つの実施態様では、二重特異性変異抗体のヒンジ定常領域はヒトIgG1のアミノ酸配列を含んでなる。H鎖のCl、CH1、ヒンジ、CH2およびCH3領域をコードするアミノ酸は、図1のアミノ酸番号139〜468として示され、Cl鎖をコードするアミノ酸は図2のアミノ酸番号128〜232として示される。イソロイシン253を特定するために用いた番号体系は、Edelman et al.がEu H鎖およびL鎖の開示中で用いた番号体系の開示に一致することに留意されたい(Edelman et al. Biochemistry 63, 78-85 (1969))。
【0041】
二重特異性変異抗体のscFv成分は、標的化可能な構築物と特異的に結合する。いずれのscFv成分の使用も本発明で意図される。好ましいscFv成分は679scFv(マウス抗HSG由来)および734scFv(マウス抗−diDTPA由来)である。scFvはマウス、CDRグラフト化(ヒト化)またはヒトであってよい。
【0042】
scFvがCDRグラフト化抗体に由来する場合、適当な可変領域枠組み構造配列を、抗原結合領域がそれに由来するドナー抗体のクラスまたは種を考慮して用いてよい。用いるヒト枠組み構造の種類がドナー抗体と同じまたは類似のクラスまたは種類であることが好ましい。枠組み構造は、ドナー抗体配列との相同性を、特にCDRに空間的に近接または隣接する位置で最大化または最適化するように選択すると有利である。CDRグラフト化抗体を構築するために用いてよいヒト枠組み構造の例としては、LAY、POM、TUR、TEI、KOL、NEWM、REIおよびEU(Kabat et al, 1987)がある。KOLおよびNEWMはH鎖構築に好適である。REIはL鎖構築に好適であり、EUはH鎖およびL鎖双方の構築に好適である。
【0043】
CDRグラフト化抗体のL鎖またはH鎖可変領域は、必要に応じてヒトL鎖またはH鎖定常ドメインと融合させてもよい(本明細書において「H鎖定常ドメイン」とは、特に断りのない限り、ヒンジ領域を含むものと理解される)。CDRグラフト化抗体のヒト定常ドメインは、それが存在する場合は、抗体の予定される機能、特に、必要とされうるエフェクター機能を考慮して選択してもよい。例えば、CDRグラフト化抗体が治療目的を意図し、抗体のエフェクター機能が必要とされる場合には、IgG1およびIgG3イソ型ドメインを用いてよい。あるいは、CDRグラフト化抗体が抗体エフェクター機能が必要とされない目的を意図する場合、例えばイメージング、診断または細胞毒性の標的化目的には、IgG2およびIgG4イソ型ドメインを用いてよい。L鎖可変領域と融合してよいL鎖ヒト定常ドメインには、ヒトλまたは、特に、ヒトκ鎖が挙げられる。
【0044】
好ましい変異bsAbはhMN−14IgGI253A−(734scFv)2である。この変異bsAbでは、FvはhMN−14IgGに由来し、scFvは734scFV(マウス抗diDTPA由来)であり、ヒンジ定常領域はヒトIgG1のアミノ酸配列を含む。
【0045】
本発明の一つの実施態様では、変異bsAbと標的化可能な構築物との間に一対一結合の相互作用が得られる。例えば、本発明による変異bsAbが、2つのDTPA部位を含む二価の標的化可能な構築物IMP192と相互作用する場合、1つのbsAbが1つのIMP192と結合する。この相互作用は実施例3で説明する。
【0046】
III.変異bsAbを標的化可能な構築物
いくつかの実施態様では、本発明による変異bsAbは標的化可能な構築物と結合する。変異bsAbのscFvが標的化可能な構築物と結合するのが好ましい。標的化可能な構築物は多様な構造であり得るが、十分な免疫応答を誘起することだけでなく、急速なin vivoクリアランスに関しても選択される。本発明における使用のための標的化可能な構築物の例としては、引用することによりその全開示内容が本明細書の一部とされる1999年6月22日出願の米国出願番号09/337,756、および2001年4月3日出願の米国出願番号09/823,746に記載されている。
【0047】
疎水性薬剤は強い免疫応答を誘起するのに最良であるのに対して、親水性の薬剤は急速なin vivoクリアランスに好ましく、従って、必要な疎水性と親水性とのバランスを確立するべきである。これは、多くの有機体部分の固有の疎水性を相殺するため親水性のキレート剤の使用に頼ることによって一部達成されている。また、反対の溶液特性、例えばそのうちの一部は疎水性で一部は親水性であるアミノ酸を含むペプチドを有する標的化可能な構築物のサブユニットを選択してもよい。ペプチドの他に、炭水化物を用いてもよい。
【0048】
わずか2アミノ酸残基しかないペプチドを用いてもよく、キレート剤などのその他の部分と共役するならば、2〜10残基が好ましい。リンカーは分子量の低い複合体であるべきであり、分子量は50,000ダルトン未満であることが好ましく、約20,000ダルトン、10,000ダルトンまたは5,000ダルトン未満が有利であり、キレート中に金属イオンを含む。例えば、公知のペプチドDTPA−Tyr−Lys(DTPA)−OH(式中、DTPAはジエチレントリアミン五酢酸である)を用いて、この分子のインジウム−DTPA部分に対する抗体が産生されている。しかし、インジウム非含有分子を用いることによって、また適当なスクリーニング工程によって、チロシル−リシンジペプチドに対する新規なAbを作出することができる。通常、ペプチドDOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2[式中、DOTAは1,4,7,10−テトラアザシクロドデカン四酢酸であり、HSGは式:
【化1】

のヒスタミンスクシニルグリシル群である]、などの抗原性ペプチドは4以上の残基を有する。
【0049】
金属非含有ペプチドを免疫原として用い、結果として生じるAbをPhe−Lys−Tyr−Lys主鎖に対する反応性をスクリーニングしてもよい。
【0050】
本発明はまた、最終のbsAb/リンカー系とともに用いた場合、リンカー部分を認識するscFv成分が完全に特異的であることを確実にするため、非天然アミノ酸、例えばD−アミノ酸の主鎖構造への組み込みも意図する。本発明はさらに、非天然アミノ酸およびペプチドから構築されたものなどのその他の主鎖構造も意図する。
【0051】
免疫原として用いられるペプチドは、固相支持体ならびに反復直交性の脱保護およびカップリングの標準技術を用いて自動ペプチド合成装置で便宜に合成される。ペプチド中の遊離アミノ基は、後にキレート錯体に用いられるが、アセチル基などの標準の保護基を用いてブロッキングするのが有利である。このような保護基は当業者に公知であろう。Greene and Wuts Protective Groups in Organic Synthesis, 1999 (John Wiley and Sons, N.Y.)を参照されたい。ペプチドを後の変異bsAbの使用のために準備する場合、in vivo カルボキシペプチダーゼ活性を阻害するために、樹脂から切り取って対応するC末端アミドを生成すると有利である。
【0052】
免疫原のハプテンは、免疫原性の認識部分、例えば化学ハプテンを含んでなる。化学ハプテン、好ましくはHSGまたはDTPAハプテンの使用により、抗体に対するリンカーの高い特異性が発揮される。これは、HSGまたはDTPAハプテンに対して作製された抗体が公知であり、この抗体のscFv部分が変異bsAbへ容易に組み込まれるために生じる。よって、リンカーと、付着したハプテンとの結合はscFv成分に対して特異性が高いと思われる。
【0053】
標的化可能な構築物は一価または二価であってよく、二価のペプチドが好ましいペプチドである。標的化可能な構築物の一例はIMP192(Ac−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(TscG−Cys−)−NH2)である。IMP192は診断用にはTc−99mとIn−111の双方と結合し、治療用にはRe−188とRe−186の双方と結合する。IMP192もまた、チロシンを含む二価のDTPA−ペプチドである。
【0054】
本発明による方法において、標的化可能な構築物は、罹患組織の検出に有用な1以上の放射性同位元素を含んでよい。特に有用な診断用放射性核種としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gd、177Lu、32P、188Re、90Y、またはその他のγ-、β-、または陽電子放射体が挙げられ、エネルギー範囲は20〜4,000keVであることが好ましく、25〜4,000keVがより好ましく、20〜1,000keVがさらに好ましく、70〜700keVがなお一層好ましい。
【0055】
本発明による方法において、標的化可能な構築物は、罹患組織の検出に有用な1以上の放射性同位元素を含んでよい。特に有用な治療用放射性核種としては、限定されるものではないが、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acが挙げられる。治療用の放射性核種はエネルギー範囲が60〜700keVであることが好ましい。
【0056】
本発明による方法において、標的化可能な構築物は、磁気共鳴イメージング(MRI)に用いる1以上の造影剤を含んでよい。限定されるものではないが、標的化可能な化合物は、例えば、Mn、Fe、およびGdなどの1以上の常磁性イオンを含む。
【0057】
本発明による方法において、標的化可能な構築物は、超音波イメージングに用いる1以上の造影剤を含んでよい。限定されるものではないが、標的化可能な構築物は、例えば、1以上の超音波イメージング剤を含む。このような一つの実施態様では、標的化可能な構築物は、リポソーム脂質膜の外面に二価DTPA−ペプチドが共有結合したリポソームである。所望により、該リポソームはガス充填されてよい。
【0058】
IV.キレート部分
リンカー部分に親水性のキレート部分が存在することにより急速なin vivoクリアランスが確実となる。親水性に加え、キレート剤はその金属結合特性で選択され、自由に変えられる。というのも、少なくともそのbsAbエピトープがペプチドの一部分であるか、または非キレート化学ハプテンであるリンカーには、金属−キレート錯体の認識はもはや問題ではないからである。
【0059】
特に有用な金属−キレートの組み合わせとしては、2−ベンジル−DTPAおよびそのモノメチルおよびシクロヘキシル類似体が挙げられ、放射性イメージングおよびRAITに47Sc、52Fe、55Co、67Ga、68Ga、111In、89Zr、90Y、161Tb、177Lu、212Bi、213Bi、および225Acとともに用いられる。同じキレート剤は、Mn、Fe、およびGdなどの非放射性金属とキレート化した場合、本発明による変異bsAbとともに用いるとMRIに使用できる。NOTA(1,4,7−トリアザ−シクロノナン−N,N’,N’’−三酢酸)、DOTA、およびTETA(p−ブロモアセトアミド−ベンジル−テトラエチルアミン四酢酸)などの大環状キレート剤は種々の金属および放射性金属とともに使用され、最も特にそれぞれGa、YおよびCuの放射性核種とともに使用される。
【0060】
DTPAおよびDOTA系キレート剤は、リガンドにカルボン酸塩またはアミン基など硬い塩基のキレート性官能基が含まれる場合に、硬い酸のカチオン、特にIIa族およびIIIa族の金属カチオンをキレート化するのに最も効果的である。このような金属−キレート錯体は、環の大きさを目的の金属に調製することによって非常に安定させることができる。大環状ポリエーテルなどその他の環状キレート剤は、RAITで223Raなどの核種の安定した結合のために注目される。ポルフィリンキレート剤を多数の放射性金属とともに用いてもよく、bsAbに導かれる免疫光線療法のためのある種の非放射性金属錯体としても有用である。1種類以上のキレート剤を担体と結合させて複数の金属イオン、例えば、非放射性イオン、診断用放射性核種および/または治療用放射性核種と結合させてよい。特に有用な治療用放射性核種としては、限定されるものではないが、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acが挙げられる。特に有用な診断用放射性核種としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gdおよび175Luが挙げられる。
【0061】
米国特許第5,753,206号明細書に開示のものなどのキレート剤、特にチオセミ−カルバゾニルグリオキシルシステイン(Tscg−Cys)およびチオセミカルバジニル−アセチルシステイン(Tsca−Cys)キレート剤は、Tc、Re、Biおよびその他の遷移金属の軟らかい酸のカチオン類、ランタニド類、ならびに軟らかい塩基のリガンド、特に硫黄またはリンを含むリガンドと緊密に結合するアクチニド類との結合に用いると有利である。1種類以上のキレート剤を、ペプチド、例えばDTPAまたは類似のキレート剤、例えばIn(III)カチオンのキレート剤、およびチオールを含むキレート剤、例えばTcカチオンのTscg−Cysと結合させると有用である。di−DTPAハプテンの抗体は公知であり(Barbet‘395、前掲)、ターゲッティング抗体と容易に結合してbsAbを形成するため、放射性同位元素をターゲッティングするためのプレターゲッティングプロトコールで、ペプチドハプテンを非放射性diDTPAキレート剤および別のキレート剤を放射性同位元素との結合に使用することができる。このようなペプチドの一例はAc−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(Tscg−Cys−)−NH2である。このペプチドはIn(III)を予め付加し、その後99m−Tcカチオンで標識することができ、In(III)イオンはDTPAに選択的にキレート化され、Tcカチオンはチオールを含むTscg−Cysと選択的に結合する。NOTA、DOTA、TETAなどのその他の硬い酸のキレート剤は、DTPA基と置換することができ、抗di−DTPA Mabの産生に用いるものと類似の技術を用いてそれらに特異的なMabを作製することができる。
【0062】
当然のことながら、2種類の異なる硬い酸または軟らかい酸のキレート剤を、例えば異なるキレート環サイズのリンカーへ組み込んで、異なるカチオンサイズ、キレート環の幾何学、およびカチオンの好ましい複合イオン構造のため、2種類の異なる硬い酸または軟らかい酸のカチオンと選択的に結合させることができる。これによって、その一方または双方が放射性であるかまたはMRI増強に有用である2種類の異なる金属を、プレターゲッティングされたbsAbよる最終捕捉のためのリンカーへ組み込むことが可能となる。
【0063】
好ましいキレート剤としては、NOTA、DOTAおよびTscgならびにその組み合わせが挙げられる。このようなキレート剤は、次の構造:
(a)DOTA−Phe−Lys(HSG)−D−Tyr−Lys(HSG)−NH2;
(b)DOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2;
(c)Ac−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys)−NH2;
【化2】
に例示されるようなキレート剤−ペプチド複合体モチーフへ組み込まれている。
【0064】
上記キレート剤−ペプチド複合体(d)および(e)は、68Gaと結合することが示されており、従って陽電子放射断層撮影法(PET)への適用に有用である。
【0065】
キレート剤は、下記の実施例でより詳細に考察される標準の化学反応を用いてリンカー部分と結合される。要するに、ペプチドAc−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys−)−NH2の合成は、ペプチドシンセサイザーのRinkアミド樹脂へAloc−Lys(Fmoc)−OHが最初に付着することによって達成される。本明細書で用いられる保護基の省略形「Aloc」および「Fmoc」はアリールオキシカルボニルおよびフルオレニルメチルオキシカルボニルの基をさす。次にFmoc−Cys(Trt)−OHおよびTscGを標準のFmoc自動合成プロトコールを用いてリシンの側鎖へ加えて次のペプチド:Aloc−Lys(Tscg−Cys(Trt)−rink樹脂を形成した。次にAloc基を取り除いた。次にペプチド合成をシンセサイザーで継続して次のペプチド:(Lys(Aloc)−D−Tyr−Lys(Aloc)−Lys(Tscg−Cys(Trt)−)−rink樹脂を形成した。その後N末端をアシル化し、Aloc保護基の側鎖を取り除いた。生じたペプチドを次に、カイザー試験を用いて樹脂がアミンに検査陰性となるまで活性化N−トリチル−HSG−OHで処理した。Karacay et al. Bioconjugate Chem. 11:842-854 (2000)参照。Ac−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys−)−NH2の合成、ならびにDOTA−Phe−Lys(HSG)−D−Tyr−Lys(HSG)−NH2;およびDOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2の合成はより詳細に下に記載されている。
【0066】
V.金属キレート調製のための一般法
キレート剤−ペプチド複合体は固形物と同じく長期間保存し得る。それらは計量して金属結合反応のための単位用量とし、固体、水溶液または半水溶液、冷凍溶液あるいは凍結乾燥調製物の単位用量として保存してよい。それらを周知の手順によって標識してもよい。一般的に、硬い酸のカチオンを便宜な塩の溶液として導入し、それを硬い酸のキレート剤、およびあるいは軟らかい酸のキレート剤にとる。しかし、軟らかい酸のカチオンを後で添加すると軟らかい酸のキレート剤がそれと結合し、そこでキレート化する可能性のある硬い酸のカチオンをどれも置換してしまう。例えば、たとえ過剰な量の非放射性111InCl3の存在下でも、99m−Tc(V)グルコヘプトネートまたはin situで塩化第一錫およびNa99m−TcO4を用いて生成したTcカチオンでの標識は、軟らかい酸のキレート剤上で量的に進行する。その他の186Re、188Re、213Biなどの軟らかい酸のカチオン、ならびにMn、Co、Ni、Pb、Cu、Cd、Au、Fe、Ag(一価)、ZnおよびHg、特に64Cuおよび67Cuなどの二価または三価のカチオンは、放射免疫診断または放射免疫治療に有用なものもいくつかあるが、類似の方法によってリンカーペプチド上に付加することができる。Reカチオンもin situでペルレナートおよび錫イオンから生成でき、または予め還元されたレニウムグルコヘプトネートまたはその他のトランスキレート剤を用いることもできる。ペルレナートの還元にはTcの還元に必要とされるよりも多い錫イオン(一般的に終濃度200mg/mL超)が必要であるので、ジスルフィド−環化ペプチド中に存在するものなどの感受性の高いジスルフィド結合を高レベルの錫イオンが還元しないことを確実にするため特別な注意が必要である。レニウムでの放射性標識の際は、Tc−99mで用いるのと類似の手順が用いられる。Tscg−Cys−リガンドのReO金属錯体の好ましい調製方法は、ペプチドをReOCl3(P(Ph3)2と反応させることによるが、ReO(エチレンジアミン)2などのその他の還元種を用いることも可能である。
【0067】
VI.抗体の作製方法
ペプチド主鎖に対する抗体は周知のAb産生法によって作製する。例えば、フロイント完全アジュバントでの(ペプチド)n−KLH(式中、KLHはキーホルリンペットヘモシアニンであり、nは1〜30である)などの免疫原の注射の後、フロイントの不完全アジュバント中に懸濁した同じ免疫原を免疫反応性のある動物へ2回注射し、静脈からの抗原の追加免疫3日後に脾臓細胞を回収する。回収した脾臓細胞を次にSp2/0−Ag14骨髄腫細胞と融合させ、生じたクローンの培養上清を直接結合ELISA法を用いて抗ペプチド反応性について分析した。生成したAbの優れた選択性は当初の免疫原のペプチドフラグメントを用いて分析できる。これらのフラグメントは自動ペプチドシンセサイザーを用いて容易に調製できる。Abの産生のため、酵素の欠乏したハイブリドーマを単離して融合した細胞株の選択を可能にする。この技術はまた、1以上のリンカーを含むキレート、例えばIn(III)−DTPAキレートに対する抗体を作製するのに用いることができる。In(III)−di−DTPAのモノクローナルマウス抗体が公知である(Barbet‘395, 前掲)。
【0068】
本発明で用いられる変異二重特異性抗体は、マーカー物質のように種々の細胞表面または細胞内の腫瘍関連抗原に特異的である。このようなマーカーは、腫瘍により産生された物質、もしくは細胞質中、核中であろうと種々の細胞小器官あるいは細胞の下部構造中であろうと、腫瘍部位、腫瘍細胞表面上または腫瘍細胞内に集積する物質であってよい。このような腫瘍関連マーカーのの中には、Herberman, "Immunodiagnosis of Cancer", in Fleisher ed., "The Clinical Biochemistry of Cancer", page 347 (American Association of Clinical Chemists, 1979) および米国特許第4,150,149号明細書;同第4,361,544号明細書;および4,444,744号明細書に開示されているものがある。
【0069】
腫瘍関連マーカーは、Herberman(前掲)によって、腫瘍胎児抗原、胎盤の抗原、発癌または腫瘍ウイルス関連抗原、組織関連抗原、器官関連抗原、異所性ホルモンおよび正常な抗原またはその変異形態をはじめとするいくつかのカテゴリーに分類されている。時折、腫瘍関連マーカーのサブユニットを用いて腫瘍特異性の高い抗体、例えば、米国特許第4,361,644号明細書および同第4,444,744号明細書に開示のように非腫瘍物質に対する交差反応性が非常に低減された抗体の産生を刺激するヒト絨毛性ゴナドトロピン(HCG)のβサブユニットまたは癌胎児抗原(CEA)のγ領域を作製することが有利である。
【0070】
注目されるもう一つのマーカーは、トランスメンブランアクチベーターおよびCAML相互作用子(TACI)である。Yu et al. Nat. Immunol. 1:252-256 (2000)参照。要するに、TACIはB細胞の悪性腫瘍(例えばリンパ腫)のマーカーである。さらに、TACIおよびB細胞成熟抗原(BCMA)は、腫瘍壊死因子ホモログである増殖誘発リガンド(APRIL)によって結合することが知られている。APRILは、一次B細胞およびT細胞のin vitroでの増殖を刺激し、B細胞のin vivoでの集積により脾臓重量を増加させる。APRILはまた、受容体結合に関してTALL−I(BLySまたはBAFFとも呼ばれる)と競合する。可溶性BCMAおよびTACIは、APRILの結合を特異的に阻止し、APRILに刺激される一次B細胞の増殖を阻害する。BCMA−Fcはまた、マウスにおいてキーホルリンペットヘモシアニンおよびニューモバックスに対する抗体の産生を阻害し、BCMAおよび/またはTACIを介するAPRILおよび/またはTALL−Iのシグナル伝達が、液性免疫の生成に必要なことを示している。よって、2リガンド−2受容体経路からのAPRIL−TALL−IおよびBCMA−TACIは、B細胞およびT細胞機能の刺激に関与する。
【0071】
免疫原に対する最初の抗体を作製した後、抗体を配列決定し、その後組換え技術によって生産すればよい。マウス抗体および抗体フラグメントのヒト化およびキメラ化は、当業者に周知である。例えば、ヒト化モノクローナル抗体はマウス相補性決定領域をマウス免疫グロブリンのH鎖およびL鎖可変領域からヒト可変ドメインへ移すことによって作出し、その後、マウス対応物の枠組み構造領域中の ヒト残基を置換する。ヒト化モノクローナル抗体由来の抗体成分の使用は、マウス定常領域の免疫原性に関連する潜在的な問題を未然に防ぐ。マウス免疫グロブリン可変ドメインのクローニングの一般的技術は、例えば、引用することによりその全開示内容が本明細書の一部とされるOrlandi et al., Proc. Nat'l Acad. Sci. USA 86: 3833 (1989)の刊行物に記載されている。ヒト化Mabを作出する技術は、例えば、引用することによりその全開示内容が本明細書の一部とされるJones et al., Nature 321: 522 (1986), Riechmann et al., Nature 332: 323 (1988), Verhoeyen et al., Science 239: 1534 (1988), Carter et al., Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech. 12: 437 (1992), and Singer et al., J. Immun. 150: 2844 (1993)に記載されている。
【0072】
あるいは、完全なヒト抗体は、トランスジェニック非ヒト動物から得ることができる。例えば、Mendez et al., Nature Genetics, 15: 146-156 (1997); 米国特許第5,633,425号明細書を参照されたい。例えば、ヒト抗体は、ヒト免疫グロブリン遺伝子座を有するトランスジェニックマウスから回収できる。このマウス体液性免疫機構は、内在性免疫グロブリン遺伝子を不活性化して、ヒト免疫グロブリン遺伝子座を導入することによりヒト化されている。このヒト免疫グロブリン遺伝子座は非常に複雑で、あわせてヒトゲノムのほぼ0.2%を占める多数の不連続セグメントを含んでなる。トランスジェニックマウスの適当な抗体レパートリー産生を確実するために、ヒトH鎖およびL鎖遺伝子座の大部分をマウスゲノムへ導入しなければならない。これは、生殖細胞系構成における、ヒトH鎖またはL鎖免疫グロブリン遺伝子座を含む酵母人工染色体(YAC)の形成から始まる段階的なプロセスで達成される。各々の挿入体はほぼ1Mbのサイズなので、YAC構築物は免疫グロブリン遺伝子座の重複フラグメントの相同的組換えを必要とする。2つのYACは、1つがH鎖遺伝子座を含み、1つがL鎖遺伝子座を含み、YAC含有酵母スフェロプラスト(spheroblast)マウス胚幹細胞を融合させて個別にマウスへ導入される。次いで、胚幹細胞クローンをマウス胚盤胞に微量注入する。その結果得られたキメラ雄動物の生殖細胞系統を介するYAC伝達能力に関してスクリーニングし、マウス抗体産生を欠損しているマウスを繁殖させた。一種はヒトH鎖の遺伝子座を含み、もう一種はヒトL鎖遺伝子座を含む二種のトランスジェニック系統のマウスを繁殖させることによって、免疫化に反応してヒト抗体を産生する子孫をつくる。
【0073】
再配列されていないヒト免疫グロブリン遺伝子もまた、微小核体媒介性染色体移入法(MMCT)によってマウス胚幹細胞へ導入できる。例えば、Tomizuka et al., Nature Genetics, 16: 133 (1997)を参照されたい。この方法論ではヒト染色体を含む微小核体をマウス胚幹細胞と融合させる。移入された染色体は安定して保持され、成体キメラは適当な組織特異的発現を示す。
【0074】
あるいは、本発明による抗体または抗体フラグメントは、コンビナトリアル免疫グロブリンライブラリーから単離したヒト抗体フラグメントに由来するものであってもよい。例えば、引用することによりその全開示内容が本明細書の一部とされるBarbas et al., METHODS: A Companion to Methods in Enzymology 2: 119 (1991),およびWinter et al., Ann. Rev. Immunol. 12: 433 (1994)を参照されたい。B細胞を不死化させてモノクローナル抗体を産生することに関連する困難の多くは、ファージディスプレーを用いて抗体フラグメントを操作し大腸菌に発現させることによって克服できる。高親和性の回復を確実にするために、モノクローナル抗体コンビナトリアル免疫グロブリンライブラリーは大きなレパートリーを含まなければならない。一般的戦略では、免疫マウスのリンパ球または脾臓細胞から得たmRNAを利用して、逆転写酵素を用いてcDNAを合成する。H鎖およびL鎖遺伝子をPCRによって個別に増幅し、ファージクローニングベクターに連結する。一方はH鎖遺伝子を含み、他方はL鎖遺伝子を含む二種類の異なるライブラリーを作製する。ファージDNAを各ライブラリーから単離し、H鎖およびL鎖配列を連結し、パッケージングしてコンビナトリアルライブラリーを形成する。各々のファージはH鎖およびL鎖cDNAの無作為の組み合わせを含み、大腸菌に感染させた際に感染細胞中で抗体鎖の発現を指示する。注目される抗原を認識する抗体を同定するため、ファージライブラリーをプレーティングし、プラーク中に存在する抗体分子を膜へ移す。この膜を放射性標識した抗原でインキュベートし、次に洗浄して過剰な非結合リガンドを取り除く。オートラジオグラムの放射性スポットが抗原と結合している抗体を含むプラークを識別する。ヒト免疫グロブリンファージライブラリーの作出に有用なクローニングおよび発現ベクターを、例えば、STRATAGENE Cloning Systems (La Jolla, CA)から得ることができる。
【0075】
類似の手法を用いて高親和性scFvが得られる。例えば、Vaughn et al., Nat. Biotechnol., 14: 309-314 (1996)を参照されたい。広いレパートリーを有するscFvライブラリーは、全て公知のVH、VkおよびVλ遺伝子ファミリーに対応するPCRプライマーを用いて非免疫ヒトドナー由来のV遺伝子を単離することにより構築できる。増幅の後、VkおよびVλプールを合わせて一つのプールとする。これらのフラグメントをファージミドベクターへ連結する。scFvリンカー、(Gly4、Ser)3をその後、VLフラグメントのファージミド上流へ連結する。VHおよびリンカー−VLフラグメントを増幅させ、JH領域に構築する。結果として生じるVH−リンカー−VLフラグメントをファージミドベクターへ連結する。ファージミドライブラリーを、上記のようにフィルターか、またはイムノチューブ(Nunc; Maxisorp)を用いて選別する。類似の結果は、免疫ウサギのリンパ球または脾臓細胞由来のコンビナトリアル免疫グロブリンライブラリーを構築し、ピキア・パストリス(P. pastoris)でscFv構築物を発現させることによって達成することができる。例えば、Ridder et al., Biotechnology, 13: 255-260 (1995)を参照されたい。さらに、適当なscFvを単離した後、結合親和性が高くおよび解離率の低い抗体フラグメントが、CDR3変異誘発および鎖のシャッフリングなどの親和性成熟プロセスによって得られる。例えば、Jackson et al., Br. J. Cancer, 78: 181-188 (1998); Osbourn et al., Immunotechnology, 2: 181-196 (1996)を参照されたい。
【0076】
種々の組換え法を用いて二重特異性抗体および抗体フラグメントを作出できる。例えば、二重特異性抗体および抗体フラグメントをトランスジェニック家畜の乳汁中で産生できる。例えば、Colman, A., Biochem. Soc. Symp., 63: 141-147, 1998;米国特許第5,827,690号明細書を参照されたい。対をなす免疫グロブリンH鎖およびL鎖をコードするDNAセグメントをそれぞれ含む二つのDNA構築物を調製する。このフラグメントを、哺乳類上皮細胞中で優先的に発現されるプロモーター配列を含む発現ベクターへクローニングする。例としては、限定されるものではないが、ウサギ、ウシおよびヒツジのカゼイン遺伝子、ウシα−ラクトグロブリン遺伝子、ヒツジβ−ラクトグロブリン遺伝子、およびマウスホエー酸タンパク質遺伝子由来のプロモーターが挙げられる。好ましくは、挿入されたフラグメントの3’部位に哺乳類特異的遺伝子由来の同族のゲノム配列が隣接する。これによりポリアデニル化部位および転写安定化配列が提供される。この発現カセットを受精した哺乳類の卵子の前核に同時注入し、次いでレシピエント雌の子宮に着床させて妊娠させる。出産後、双方のトランス遺伝子の存在についてサザン分析でその後代をスクリーニングする。抗体が存在するためには、H鎖およびL鎖遺伝子双方が同時に同じ細胞中で発現されなければならない。トランスジェニック雌動物から得た乳汁を当技術分野で公知の標準的免疫学的方法により、該抗体または抗体フラグメントの存在と機能性を分析する。この抗体は当技術分野で公知の標準的方法により、乳汁から精製することができる。
【0077】
キメラAbは、マウスL鎖可変およびH鎖可変ドメインをコードするcDNAフラグメントをヒト抗体由来Cドメインをコードするフラグメントに連結することによって構築される。Cドメインは抗原結合を導かないため、キメラ抗体は元のマウスAbと同じ抗原特異性を保持するが、配列でヒト抗体により近くなる。キメラAbはなおマウス配列を一部含み、また一方、依然として免疫原性である可能性がある。ヒト化Abは、抗体を認識するのに必要なマウスアミノ酸のみを含む。この産物はヒト抗体枠組み構造中へマウス相補性決定領域由来のアミノ酸を形成することによって構築される。
【0078】
VII.変異二重特異性抗体の設計および発現のための一般法
種々の変異誘発技術を用いて本発明による変異bsAbを構築してよい。当業者であればこのような技術に精通しているであろう。例えば、変異bsAbの発現ベクターは、変異したHCフラグメントを構築し、このフラグメントを、親bsAbの発現ベクターへサブクローニングして対応する野生型フラグメントを置換し、宿主細胞にベクターをトランスフェクトして得てもよい。
【0079】
親bsAbの発現ベクターを得るために、当業者は容易に利用できる技術を用いることができる。このような技術のうちのいくつかが、引用することによりその全開示内容が本明細書の一部とされる1999年6月22日出願の米国出願番号09/337,756に開示されている。要するに、hMN14IgG−(734 scFv)2などの親bsAbの発現ベクターを構築するため、単鎖734Fv(734scFv)をコードする遺伝子セグメントを構築すればよい。この734scFvセグメントは、短く柔軟なリンカー(sL)(Coloma & Morrison 1997 p.787/id)をコードするDNAフラグメントを介してヒトγ鎖遺伝子の3’末端と結合してよく、その結果CH1−ヒンジ−CH2−CH3−sL−734scFv(CH−scFv)の融合遺伝子配列となる。このCH−scFv融合遺伝子セグメントは、次に同じくhMN−14 L鎖遺伝子セグメントを含む発現ベクターhMN14pdHL2中で、hMN−14 VHの配列、ならびにトランスフェクタントの選択のため、またその結果生じたトランスフェクトされた配列の増幅のためのdhfr遺伝子と結合させることができる(Dorai & Moore 1987 p. 815/id and Gillies, Lo et al. 1989 p. 131/id)。hMN14IgG−(734scFv)2(bsAb2pdHL2)をコードするベクターは、融合bsAbの発現のためのSp2/0骨髄腫細胞へトランスフェクトすればよい。bsAb、hMN14IgG−(734scFv)2は、アフィニティークロマトグラフィーによって培養上清から精製し、SDS−PAGEによって分析することができる。親または変異bsAb内の異なる結合部分の免疫反応性を評価するため、競合ELISA結合アッセイを行ってもよい。
【0080】
その他の特異性および個別の変異bsAbを有するIgG−scFvのbsAbは、IgGおよび/またはscFvの可変領域配列だけを、その他のAbのもので置換することによって作製することができる。CDRグラフト化変異bsAbは、IgGまたはscFvの可変領域配列だけをCDRグラフト化Abで置換することによって作出することができる。一般に、この「CDRグラフト化」技術は、マウスCDR、ヒト可変領域枠組み構造およびヒト定常領域からなる組換え、医薬抗体の作製に適用されてきた(例えばRiechmann, L. et al, (1988) Nature, 332, 323-327)。このような「再構成」または「ヒト化」抗体はキメラ抗体よりもマウスの含有部分が少なく、ヒトFc依存性エフェクター機能の刺激に必要なヒト定常領域を保持する。その結果、CDRグラフト化抗体は、ヒトへ投与した時にキメラ抗体よりもHAMA反応を惹起する可能性は低く、循環系中のそれらの半減期はヒト自然抗体のものに近づくはずであり、それらの診断および治療価値は向上する。
【0081】
実際には、元のマウス抗体の特異性を保持している有効なヒト化抗体を作製するためには、単にCDRを置換するだけでは十分でないことが普通である。さらに、少数の重要なマウス抗体残基をヒト可変領域中へ組み込む必要がある。このような残基の同定は、元のマウス抗体およびアクセプターであるヒト抗体の双方の構造に依存する。英国特許出願番号9019812.8(引用することによりその全開示内容を本明細書の一部とする)は、有効な抗原結合を促進するのに十分な外来残基の置換の最低数を同定する方法を開示している。本発明の一つの実施態様では、変異融合タンパク質のFvおよびscFvはCDRグラフト化マウスFvおよびscFvである。本発明の別の実施態様では、変異融合タンパク質のFvおよびscFvはヒト化されている。一つの実施態様では、Fvは734scFvに由来するものであり、scFvは734scFvである。本発明の好ましい実施態様では、変異融合タンパク質はhMN−14IgGI253A−(734scFv)2である。
【0082】
VIII.変異bsAbの投与方法
本発明は、米国特許第6,126,916号明細書;同第6,077,499号明細書;同第6,010,680号明細書;同第5,776,095号明細書;同第5,776,094号明細書;同第5,776,093号明細書;同第5,772,981号明細書;同第5,753,206号明細書;同第5,746,996号明細書;同第5,697,902号明細書;同第5,328,679号明細書;同第5,128,119号明細書;同第5,101,827号明細書;および同第4,735,210号明細書に記載の方法を用いて、正常組織および器官の処置および/またはイメージングにおける、本発明による二重特異性抗体および標的化可能な構築物の使用を意図する。さらなる方法は1999年6月22日出願の米国出願番号09/337,756、および2001年4月3日出願の米国出願番号09/823,746に記載されている。本明細書において「組織」とは、限定されるものではないが、卵巣、胸腺、副甲状腺または脾臓由来の組織をはじめとする組織をさす。本発明による変異bsAbで治療可能な疾患および状態の例としては、免疫調節障害、自己免疫疾患、臓器移植拒絶症または移植片対宿主病が挙げられる。B細胞を標的化する抗体を用いる自己免疫疾患の免疫療法はWO00/74718mに記載されており、このクレームは米国仮出願番号60/138,284号に優先するものである(引用することによりその全内容を本明細書の一部とする)。自己免疫疾患の例としては、急性特発性血小板減少性紫斑病、慢性特発性血小板減少性紫斑病、皮膚筋炎、シドナム舞踏病、重症筋無力症、全身性紅斑性狼瘡、狼瘡腎炎、リウマチ熱、多腺性症候群、水疱性類天瘡、真性糖尿病、ヘノッホ−シェーンライン紫斑病、溶血性連鎖球菌感染後腎炎、結節性紅斑らい、高安動脈炎、アジソン病、慢性関節リウマチ、多発性硬化症、類肉腫症、潰瘍性大腸炎、多形性紅斑、IgA腎症、結節性多発性動脈炎、強直性脊椎炎、グッドパスチャー症候群、閉塞性血栓血管炎、シェーグレン症候群、原発性胆汁性肝硬変、橋本甲状腺炎、甲状腺中毒症、強皮症、慢性活動性肝炎、多発性筋炎/皮膚筋炎、多発性軟骨炎、尋常性天疱瘡、ウェジナー肉芽腫、膜性腎症、筋萎縮性側索硬化症、脊髄ろう、巨細胞動脈炎/多筋痛、悪性貧血、急速進行性糸球体腎炎、および繊維性肺胞炎がある。
【0083】
本発明による変異bsAbは、一次ターゲッティング種としてプレターゲッティング法に使用してもよい。プレターゲッティング法では、変異bsAbを投与する。一次ターゲッティング種が十分に付着したら、標的化可能な構築物を投与する。標的化可能な構築物は、一次ターゲッティング種および診断剤または治療剤の利用可能な結合部位を認識する結合部位を含んでなる。標的化可能な構築物の例は上述したとおりである。試薬の用量およびタイミングは、当業者には容易に決定できるものであり、用いる試薬の特有の性質に依存する。プレターゲッティング法はクリアランス剤を用いて行っても、用いずに行ってもよい。
【0084】
bsAbが罹患組織に対して標的化するのに十分な時間が経過したら、診断剤を投与する。診断剤の投与の後、イメージングを行うことができる。腫瘍は、適当な波長光が送達される種々の構造を直接または間接に調べることによって体腔中の腫瘍を検出することができ、その後回収できる。体のどの部位の病巣であっても、非電離放射線が送達され、それらの構造から再捕捉される限り、調べることができる。例えば、高解像度、非侵襲性のイメージング技術であるPETをヒト疾病の視覚化のために本発明による抗体とともに使用できる。PETでは、F−18を陽電子放射体として用いる場合、陽電子崩壊減衰中に生じる511keVのγ光子が検出される。
【0085】
本発明は、一般に25〜600keVのγ粒子および/または陽電子を放射する診断剤の使用を意図する。このような薬剤の例としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154〜158Gdおよび175Luが挙げられる。
【0086】
術中/内視鏡プローブを用いる検出も、本発明による変異bsAbおよびI−125標識されたペプチドである標的化可能な構築物を利用する方法において意図される。このような方法は、引用することによりその全開示内容が本明細書の一部とされる米国特許第5,716,595号明細書および同第6,096,289号明細書に開示されている。
【0087】
本変異bsAbは、米国特許第6,096,289号明細書;同第4,331,647号明細書;同第4,818,709号明細書;同第4,348,376号明細書;同第4,361,544号明細書;同第4,444,744号明細書;同第5,851,527号明細書に論じられている光線力学療法(PDT)に用いることができる。
【0088】
PDTでは、感光剤、例えばジヘマトポルフィリンエーテルなどのヘマトポルフィリン誘導体を被験体へ投与する。抗腫瘍活性は、例えば630nmの光を用いて惹起する。膜が日光によって増感しない、長い波長に有用なものをはじめとする代わりの感光剤が利用できる。このような感光剤の例としては、限定されるものではないが、ベンゾポルフィリン一酸環A(BPD−MA)、錫エチオプルプリン(SnET2)、スルホン化アルミニウムフタロシアニン(AlSPc)およびルテチウムタキサフィリン(Lutex)が挙げられる。
【0089】
さらに、PDTでは、診断剤を例えば全身に注射し、レーザー誘起蛍光を用いて内視鏡により光活性化剤の付着している癌の部位を検出する。例えば、これは初期肺腫瘍の蛍光気管支鏡検査に適用されている。Doiron et al. Chest 76:32 (1979)。別の例では、抗体および抗体フラグメントを単光子放出に用いることができる。例えば、Tc−99m標識した診断剤を、本発明による抗体または抗体フラグメントの投与の後に被験体へ投与してよい。その後被験体を、単光子放出コンピュータ断層撮影画像を生じるγ線カメラでスキャンし、病巣または腫瘍部位を決定する。
【0090】
治療上有用な免疫複合体は、光活性薬または色素を抗体複合体と結合させることにより得ることができる。蛍光またはその他の色素体、あるいはポルフィリンなどの色素は可視光に感受性であり、好適な光線を病巣に向けることにより病巣の検出および治療に用いられてきた。治療では、これは光照射、光療法、または光線力学療法と呼ばれている(Jori et al. (eds.), Photodynamic Therapy of Tumors and Other Diseases (Libreria Progetto 1985); van den Bergh, Chem. Britain 22:430 (1986))。さらに、光療法を達するためには、モノクローナル抗体を光活性色素と結合させる(Mew et al., J. Immunol. 130:1473 (1983); idem., Cancer Res. 45:4380 (1985); Oseroff et al., Proc. Natl. Acad. Sci. USA 83:8744 (1986); idem., Photochem. Photobiol. 46:83 (1987); Hasan et al., Prog. Clin. Biol. Res. 288:471 (1989); Tatsuta et al., Lasers Surg. Med. 9:422 (1989); Pelegrin et al., Cancer 67:2529 (1991))。しかし、これらの初期の研究には、内視鏡での治療、特に抗体フラグメントまたはサブフラグメントを用いるものの使用は含まれていない。従って、本発明は、光活性薬または色素を含んでなる免疫複合体の使用を意図する。
【0091】
リンカー部分はまた、標的部位のプロドラッグを活性化できる、または身体の解毒経路を制御することにより通常の治療の効果を改善することのできる酵素と結合させてもよい。bsAbの投与に続いて、リンカー部分と結合した酵素、bsAbの2番目のアーム(scFv成分)に認識される分子量の低いハプテンを投与する。酵素を標的部位へプレターゲッティングした後、標的部位で作用することが知られている細胞傷害性の薬物を注入する。この薬物は哺乳類の通常の解毒プロセスにより解毒されるものであればよい。例えば、この薬物は、肝臓毒性の低いグルクロニドに変換され得る。解毒された中間体は、次に、標的部位でプレターゲッティングされた酵素によってより有毒な形態へ再変換することができる。あるいは、投与されたプロドラッグを、プレターゲッティングされた酵素により有効な薬物へと変換することもできる。プレターゲッティングされた酵素は、解毒された薬物を再利用することにより治療の効果を向上させる。この手法は、いずれの酵素−薬物対を用いる場合にも適用することができる。
【0092】
抗癌治療に有用なある種の細胞傷害性薬物は、比較的血清に不溶性である。結合されない形態では非常に有毒なものも中にはあるが、プロドラッグへ変換することによりその毒性は相当に低減される。難溶性の薬物をより可溶性の複合体、例えばグルクロニド、親水性酸とのエステルまたは親水性アミンとのアミドへ変換すると、その薬物の血清の水性相での可溶性、ならびに静脈、動脈、または細胞壁毛細官を通過し、腫瘍を浸している間質液まで到達する能力が改善される。プロドラッグの開裂により、可溶性の低い薬物が標的部位に置かれる。このようなプロドラッグから薬物への変換の多くの例が、Hansen米国特許第5,851,527号明細書に開示されている。
【0093】
肝臓で芳香族または脂環式アルコール、チオール、フェノールおよびアミンなどのある種の有毒物質がグルクロニドへ変換されることは、それらを解毒し、より尿中に排泄しやすくする身体の手段である。このような物質へ変換できるある種の抗腫瘍薬は、エピルビシン、ドキソルビシンの4−エピマー(アドリアマイシン)であり、これはアントラサイクリン配糖体でヒトβ−D−グルクロニダーゼの基質であることが示されている。例えば、Arcamone Cancer Res. 45:5995 (1985)を参照されたい。極性基のより少ないその他の類似体は、より脂肪親和性であると考えられ、このような手法に大いに有望である。芳香族または脂環式アルコール、チオールまたはアミン基を含むその他の薬物または毒素は、このような複合体形成のための候補である。このような薬物、またはその他のそのプロドラッグ形態は、本発明による部位特異的増強法に好適な候補である。
【0094】
プロドラッグCPT−11(イリノテカン)はin vivoでカルボキシルエステラーゼによって活性代謝物SN−38に変換される。従って、本発明の一つの適用例は、腫瘍およびハプテン(例えばdi−DTPA)に対して標的化したbsAbを用い、その後にdi−DTPA−カルボキシルエステラーゼ複合体を注射することである。好適な腫瘍対バックグラウンド局在比に達したならば、CPT−11を投与する。腫瘍局在性カルボキシルエステラーゼは、腫瘍でCPT−11をSN−38へ変換するのに役立つ。可溶性が低いために、活性のあるSN−38は腫瘍近傍に残ったままとなり、その結果、標的とされる抗原について陰性の隣接する腫瘍細胞へ効果を発揮する。これは本方法のさらなる利点である。カルボキシルエステラーゼの変更形態は既に文献に記載されており、本発明の範囲内にある。例えば、Potter et al., Cancer Res. 58:2646-2651 (1998)およびPotter et al., Cancer Res. 58:3627-3632 (1998)を参照されたい。
【0095】
エトポシドは、そのグルクロニド形成によって大部分が解毒される、広く用いられる制癌剤であり、本発明の範囲内にある。例えば、Hande et al. Cancer Res. 48:1829-1834 (1988)を参照されたい。グルクロニド複合体は、細胞傷害性薬物から製造でき、mAb−グルクロニダーゼ複合体を用いてプレターゲッティングした腫瘍の治療時に注入できる。例えば、Wang et al. Cancer Res. 52:4484-4491 (1992)を参照されたい。従って、このような複合体も本明細書に記載のプレターゲッティング手法とともに使用できる。同様に、ダウノマイシンおよびドキソルビシンの誘導体に基づいて設計されたプロドラッグは、カルボキシルエステラーゼおよびグルクロニダーゼとの併用について記載されている。例えば、Bakina et al. J. Med Chem. 40:4013-4018 (1997)を参照されたい。本発明の範囲内で使用できるプロドラッグ/酵素対のその他の例としては、限定されるものではないが、フェノールマスタードおよびβ−グルクロニダーゼのヒドロキシ誘導体のグルクロニドプロドラッグ;フェノールマスタードまたはCPT−11およびカルボキシペプチダーゼ;メトトレキサート置換α−アミノ酸およびカルボキシペプチダーゼA;6−メルカプトプリンおよびドキソルビシンなどの薬物のペニシリンまたはセファロスポリン複合体およびβ−ラクタマーゼ;リン酸エトポシドおよびアルカリホスファターゼが挙げられる。
【0096】
あるいは、標的部位でプロドラッグを活性化させるか、または身体の解毒経路を制御することにより通常の治療の効力を向上させる能力のある酵素は、ハプテンと結合させてもよい。プレターゲッティングbsAbの投与の後に酵素−ハプテン複合体を被験体へ投与し、標的部位に向ける。酵素が標的部位に局在した後、標的部位で作用することが知られている細胞傷害性薬物、またはin situでプレターゲッティングされた酵素によって薬物へ変換されるそのプロドラッグ形態を注射する。上記で論じたように、この薬物は、哺乳類の通常の解毒プロセスを用いて、解毒されて毒性の低い中間体、最も一般的にはグルクロニドを形成する。解毒された中間体、例えばグルクロニドは、プレターゲッティングされた酵素によってそのより有毒な形態へ再変換され、従って標的部位での細胞傷害性が高くなる。この結果、薬物が再利用される。同様に、投与したプロドラッグを通常の生物学的プロセスによって有効な薬物へ変換できる。プレターゲッティングされた酵素は、解毒された薬物を再利用することにより治療の効力を高める。このアプローチは、どの酵素−薬物対を用いる場合にも適用できる。
【0097】
本発明はさらに、ホウ素中性子捕捉療法(Boron Neutron Capture Therapy, BNCT)プロトコールに照らした、本発明によるbsAbおよび診断剤の使用を意図する。BNCTは、腫瘍に局在する10B原子の中性子照射によって電離放射線を腫瘍細胞へ送達するように設計された二元システムである。BNCTは、安定したイソ型である同位体標識10B(自然界には19.8%の割合で存在)が熱中性子照射されてα粒子および7Li原子核を生じる際に起こる核反応に基づく。これらの粒子の飛程は約1細胞径であり、結果として線形の高エネルギー転移を生じる。この核反応で生じる飛程の短い1.7MeVのα粒子がほんのわずかあれば、細胞核を標的とし、それを破壊するのに十分である。BNCTで癌治療を成功させるには高濃度の10Bを腫瘍部位に局在させるが、標的でない器官は基本的にホウ素を含まないままにしておく方法が必要である。BNCTのためのプレターゲッティングbsAbを用いて被験体内で腫瘍を治療する組成物および方法は、同時係属の特許出願第09/205,243号に記載され、本発明の目的のために容易に修正できる。
【0098】
本発明による変異bsAbのscFv成分もまた、酵素に特異的なものとすることができる点に留意すべきである。
【0099】
変異bsAbの投与と標的化可能な構築物の投与との間で加えるクリアランス剤を用いてもよい。本発明者らは新規な機構作用を有するクリアランス剤、つまりbsAbの疾病ターゲッティングアームに標的化されたグリコシル化抗イディオタイプのFab’フラグメントを本発明に用い得ることを見出した。抗−CEA(MN14Ab)x抗−ペプチドbsAbを得、疾病標的に最大限まで付着させる。残余bsAbを除去するため、WI2と称する、MN−14に対する抗イディオタイプAbを、好ましくはグリコシル化Fab’フラグメントとして得る。クリアランス剤は一価の様式でbsAbと結合するが、それに付属するグリコシル残基は全複合体を急速な代謝が起こる肝臓へ向ける。その後、リンカー部分に関連する治療が患者に対して行われる。bsAbのMN−14アームに対するWI2 Abは高親和性で、クリアランス機構はその他の開示される機構とは異なる(Goodwin et al.同上参照)。それは、WI2−Fab’が一価の部分であるため、架橋を伴わないからである。
【0100】
本変異bsAbはまた、超音波イメージング法でも使用できる。造影剤などの超音波増強剤は、二価のDTPAペプチドなどの標的化可能な構築物と結合してもよい。限定されるものではないが、リポソーム、好ましくはガス充填したリポソームなどの増強剤を用いてよい。この方法では、変異bsAbが最初に投与され、その後リポソーム−標的化可能な構築物複合体が投与される。Maresca, G. et al., Eur J. Radiol. Suppl. 2 S171-178 (1998); Demos, Sm. Et al. J. Drug Target 5 507-518 (1998);およびUnger, E. et al., Am J. Cardiol. 81 58G-61G (1998)を参照されたい。
【0101】
変異二重特異性抗体は、多成分治療法の一成分として投与してよい。変異二重特異性抗体は、疾病または症状を治療するために用いられる少なくとも1種類の治療剤の投与前、投与中、または投与後に投与してよい。
【0102】
プレターゲッティング法における変異bsAbの使用例は、プレターゲッティング法における親bsAbの使用との比較において、実施例2に説明されている。このデータにより、親bsAbと比較した、本発明による変異bsAbのクリアランス速度の上昇が説明される。さらに、該データにより、変異bsAbが用いられる場合と比較して、親bsAbが用いられる場合に、より大量の標的化可能な構築物が血中で捕捉されることが説明される。
【0103】
図5および6は、親bsAb、125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング法のデータを示す。図7は、変異bsAb、125I−hMN−14IgGI253A−(734scFv)2に関するプレターゲッティング法のデータを示す。125I標識によって、身体の異なる領域に存在するbsAbの量を決定することができる。図5および7のデータ比較は、変異bsAbが親bsAbよりも速く身体から消失することを示す。例えば、親bsAbで4日間のプレターゲッティング後(図5)、およびIMP−192の注射後3時間の、腫瘍および血液の%ID/gは、それぞれ19.21±7.318および3.73±0.75であった。これに対して、変異bsAbで4日間のプレターゲッティング後(図5)、およびIMP−192の注射後3時間の、腫瘍および血液の%ID/gは、それぞれ2.42±0.78および0.07±0.01であった。
【0104】
図5および7の125Iの腫瘍対血液比の比較(図5および7の「血液」の欄参照)によれば、変異bsAbに関して高いシグナル対バックグラウンド比が達成されたことが実証される。親bsAbでのプレターゲッティングの6日後でさえ(図4参照)、腫瘍対血液比は、変異bsAbでのプレターゲッティングの4日後よりもはるかに低い。
【0105】
99mTc標識によって、身体の異なる領域に存在する標的化可能な構築物の量を決定することができる。IMP−192(99mTc標識した標的化可能な構築物)の%ID/gの比較は、腫瘍対血液比が変異bsAbを用いるプレターゲッティング法に関してはるかに高いことを示す。この結果は、変異bsAbに関するプレターゲッティング法において血中で捕捉された標的化可能な構築物のほうが少ないことを説明する。親bsAbを用いた場合(図5および6参照)、99mTc標識した標的化可能な構築物は、腫瘍部位に現れるよりもむしろ血中で捕捉される。したがって、低い腫瘍対血液比が認められる。例えば、99mTc標識した標的化可能な構築物の腫瘍対血液比は、図5(親bsAb)の左側、「血液」の欄に示される。注射後3時間の腫瘍対血液比は0.24±0.05である。それに対して、図5(変異bsAb)では注射後3時間の腫瘍対血液比は3.52±1.45である。
【0106】
IX.その他の適用例
本発明は、米国特許第5,716,595号明細書および同第6,096,289号明細書に記載されているように、上記に論じた変異bsAbおよびリンカー部分と結合した治療剤の手術中、血管内での使用、ならびに内視鏡による腫瘍および病巣の検出、生検および治療を包含する。
【0107】
本発明による変異bsAbは、治療またはイメージング目的だけでなく、in vitroで研究を行う際の補助としても用いることができる。例えば、本発明によるbsAbをin vitroで用いることにより、標的化可能な構築物が1以上のbsAbを含む安定した複合体を形成できるかどうかを確かめることができる。このようなアッセイは、bsAbを含む安定した複合体を形成する標的化可能な構築物の同定の際に当業者の助けとなる。これによって、次に当業者は治療剤および/またはイメージング剤として優れていると思われる標的化可能な構築物を同定することができる。
【0108】
アッセイは、当該の標的化可能な構築物と少なくとも2モル当量の変異bsAbとを組み合わせて行うのが有利である。インキュベーションの後、混合物をサイズ排除HPLCで分析して該構築物がbsAbと結合しているかどうかを判断する。あるいは、種々のbsAb溶液を標準的な96ウェルプレートへ置くという、標準的なコンビナトリアル(組み合わせ)法を用いてアッセイを行う。各ウェルには、標的化可能な構築物の溶液を加える。インキュベーションおよび分析を受けて、どの構築物がどのbsAbと最も良く結合するかが容易に判断できる。
【0109】
当然のことながら、変異bsAbの標的化可能な構築物への添加順序は重要ではない。つまり、変異bsAbを構築物へ加えてよく、逆もまた同じである。同様に、変異bsAbも構築物も溶液中にある必要はない。つまり、それらは溶液中で加えてもそのまま加えてもよく、そのうち最も便宜なものであってよい。最後に、結合が確立さえすれば、結合の分析方法は重大ではない。よって、限定されるものではないが、FABMS、高磁界NMR、またはサイズ排除HPLCと併せた、またはその代わりとなるその他の適当な方法をはじめとする標準的分析法を用いて結合を分析してよい。
【実施例】
【0110】
材料および方法
734scFvの設計および構築
734scFvを、sL−Vλ−L−VHの配置を有するよう設計した。式中、sLは短く柔軟なリンカー、Gly−Gly−Gly−Ser(Coloma & Morrison, Nat. Biotechnol. 15:159-163 (1997))であり、hMN−14IgG H鎖と734scFvとの間の結合の役割を果たし、Lは3反復のGly−Gly−Gly−Gly−Ser(Huston, Levinson, et al. PNAS 85:5879-5883 (1988))からなる734のVλおよびVHとの間の長いリンカーである。プライマー対734VLscFv5’(Cys)/734VLscFv3’および734VHscFv5’/734VHscFv3’(SacI)を用いて734のそれぞれのVλおよびVH配列を増幅した。得られたDNA産物を、制限酵素消化および連結によって734scFv遺伝子へと組み立て、DNAシーケンシングによって配列を確認した。
734VLscFv5’(Cys):5’-TT CTC TCT GCA GAG CCC AAA TCT TGT GGT GGC GGT TCA CAG CTG GTT GTG ACT CAG-3’;
734VLscFv3’:5’-A GCC TCC GCC TCC TGA TCC GCC ACC TCC TAA GAT CTT CAG TTT GGT TCC-3’;
734VHscFv5’:5’-CC GGA GGC GGT GGG AGT GAG GTG AAA CTG CAG GAG-3’;
734VHscFv3’(SacI):5’-AA CCT TGA GCT CGG CCG TCG CAC TCA TGA GGA GAC GGT GAC CG-3’。
【0111】
hMN−14IgG−(734scFv)2の発現ベクターの構築
734scFvをヒトH鎖定常領域(HC)のC末端へ結合させるため、新規なプライマー対、734scFv2−5’および734scFv−3’を合成し、734scFvをコードするDNAの増幅に用いた。プライマー734scFv2−5’は、734scFvとヒトHCのC末端とのフレーム内結合のための正確な配列をもたらした。得られたDNAフラグメントをヒトHC配列と連結し、HC−734scFvをコードする構築物を形成した。次にhMN−14の発現ベクター、hMN−14pdHL2中の正常ヒトHCをコードするDNAフラグメントを、HC−734scFvフラグメントで置換し、その結果、融合構築体の発現ベクターhMN−14IgG−(734scFv)2pdHL2を得た。
734scFv2-5’:5’-TCC CCG GGT AAA GGT GGC GGT TCA CAG CTG-3’;
734scFv-3’:5’-GAG CTC GGC CGT CGC AC-3’。
【0112】
変異融合bsAb、hMN−14IgG(I253A)−(734scFv)2の構築
イソロイシン253をヒトHC鎖のCH2ドメインに置いた。I253A変異をhMN−14IgG−(734scFv)2に導入するため、CH1および部分的にCH2ドメインをコードする挿入DNAフラグメントを含むプラスミドベクターCH1kbpKSを、オリゴヌクレオチドに誘導される部位特異的変異誘発に用いた。オリゴヌクレオチドI253ACH2は、CH2中の野生型配列KDTLM253ISRTPEをKDTLM253ASRTPEへ変換するが、変異誘発プライマーとして設計および合成されている。変異誘発は、Sculptor IVM system(Amersham, Arlington Heights, IL)を製造業者の仕様書に従って用いることにより達成された。ジデオキシDNAシーケンシングによって配列が検証された後、変異したHCフラグメントをhMN−14IgG−(734scFv)2pdHL2へサブクローニングして対応する野生型フラグメントを置換し、その結果、変異融合bsAbの発現ベクター、hMN−14IgG(I253A)−(734scFv)2pdHL2を得た。
I253ACH2:5’-AAG GAC ACC CTC ATG GCT AGC CGG ACC CCT GAG-3’。
【0113】
bsAbの発現および産生
発現ベクターを、〜30μgのSalIで線状化したDNAを用いて2〜5×106個の細胞がトランスフェクトされるエレクトロポレーションによってSp2/0細胞へトランスフェクトし、96ウェル細胞培養プレートにプレーティングした。2日後、最終濃度0.025〜0.075μMのメトトレキサート(MTX)をトランスフェクタントの選択のため細胞培養培地に加えた。MTX耐性コロニーが2〜3週で出現し、これをELISAでヒトIgGの分泌についてスクリーニングした。要するに、残存コロニー由来の細胞培養上清を、ヤギ抗ヒトIgG F(ab’)2特異的抗体をコーティングしたELISAプレートのマイクロウェル中で1時間インキュベートした。次にペルオキシダーゼ結合ヤギ抗ヒトIgG Fcフラグメント特異的抗体を加え、ウェル中で1時間インキュベートした。上清中のヒトIgGの存在は、o−フェニレンジアミン二塩酸塩0.4mg/mlおよびH2O2 0.0125%を含む基質溶液を添加することにより明らかにした。陽性クローンから最もAbを産生するものを決定し、選択し、さらに拡張した。hMN−14IgG−(734scFv)2およびhMN−14IgG(I253A)−(734scFv)2を、タンパク質AまたはDTPAカラムのいずれかでのアフィニティークロマトグラフィーにより細胞培養上清から精製した。
【0114】
Ac−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(TscG−Cys−)−NH2 (IMP192)の合成:
第一のアミノ酸、Aloc−Lys(Fmoc)−OHをペプチドシンセサイザー上でRinkアミド樹脂0.21mmolへ付着させ、その後、標準のFmoc自動合成プロトコールを用いてTc−99mリガンド結合残基Fmoc−Cys(Trt)−OHおよびTscGをリシンの側鎖へ添加して次のペプチド:Aloc−Lys(TscG−Cys(Trt)−rink樹脂を形成した。次に、CH2Cl2 10mL、氷酢酸0.75mLおよびジイソプロピルエチルアミン2.5ml中に溶解したPd[P(Ph)3]4 100mgを含む溶液8mLで樹脂を処理してAloc基を取り除いた。次に樹脂混合物をトリブチルスズヒドリド0.8mlで処理し、60分間ボルテックスで混合した。次にペプチド合成をシンセサイザーで継続して次のペプチド:Lys(Aloc)−Tyr−Lys(Aloc)−Lys(TscG−Cys−)−rink樹脂を製造した。樹脂をDMF 10mL、無水酢酸3mL、およびジイソプロピルエチルアミン6mLを含む溶液8mLで60分間ボルテックスで混合することによりN末端をアセチル化した。次にAloc保護基の側鎖を上記の通り除去し、標準のFmoc脱保護プロトコールを用いて樹脂をピペリジンで処理して樹脂に残っている可能性のある酢酸を除去した。活性化DTPAおよびDTPAの添加:DTPA5gをメタノール中1.0Mテトラブチル水酸化アンモニウム40mLに溶解した。このメタノールを高圧下で除去して粘性の油状物質を得た。この油状物質をDMF50mLに溶解し、揮発性の溶媒をロータリーエバポレーターで高圧下で除去した。DMFによる処理をさらに2回繰り返した。次に粘性の油状物質をDMF50mlに溶解し、HBTU5gと混合した。次に活性化DTPA溶液のアリコート8mlを14時間ボルテックスで混合した樹脂に加えた。このDTPAによる処理は、カイザー試験を用いて樹脂がアミンに検査陰性となるまで繰り返した。
【0115】
切断および精製:次にペプチドを、TFA30ml、トリイソプロピルシラン1ml、およびエタンジチオール1mlからなる溶液8mlで60分間(mm→min?)処理することによって樹脂から切断した。粗切断ペプチドをエーテル30mlを注入して沈殿させ、遠心分離により回収した。次にこのペプチドを4×30cm Waters分取C−18 Delta−Pakカラム(15μm、100Å)を用いる逆相HPLCによって精製した。HPLC画分を回収し、凍結乾燥させてESMS(MH±1590)により所望の産物を含む画分を得た。
【0116】
キットの作成:このペプチドをペプチド78μg、非放射性InCl3 0.92mg、塩化第一錫100μg、ゲンチシン酸3mg、およびHPCD(再構成時の10%)を含む凍結乾燥キットを構築した。
【0117】
放射性ラベリング
抗体タンパク質60μgをクロラミン−T法(Greenwood, Hunter, et al., Biochem. J. 89 11-123 (1963))を用いてI−125で標識し、NAP−5脱塩カラム(Pharmacia, Piscataway, NJ)を用いて精製した。
【0118】
Tc−99m標識したIMP−192を準備するため、IMP−192 50μgを含むキットを20mCi過酸化テクネチウムを含む生理食塩水1.5mlで再構成した。再構成したキットを室温で10分間インキュベートし、次に沸騰水浴中で15分間加熱した。
【0119】
実施例1:ヒト結腸腫瘍を有するマウスでの125I−hMN−14IgGI253A−(734scFv)2および125I−hMN−14IgG−(734scFv)2の体内分布
実験の手順
125I−hMN−14IgG−(734scFv)2および125I−hMN−14IgGI253A−(734scFv)2の簡単な体内分布パターンを評価した。GW39ヒト結腸癌異種移植片を有する雌ヌードマウス群へ、20μg(5μCi)/マウスの125I標識した親または変異bsAbを静脈注射により投与した。マウスを注射後の計画した時点で安楽死させた、その器官を取り出し、重さを測り、I−125放射能を計数した。
【0120】
GW−39ヒト結腸腫瘍細胞株を、他で記載のように(Tu, et al. Tumour Biology 9:212-220 (1988))ヌードマウス中で連続的な皮下異種移植片として増殖させた。
【0121】
結果
125I標識したhMN−14IgG−(734scFv)2およびhMN−14IgGI253A(734scFv)2変異体の腫瘍組織および正常組織での体内分布を、注射後1、2、3および4日のヒト結腸腫瘍を有するマウスで検査した。結果を図3および4に示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)として表される。
【0122】
hMN−14IgGI253A(734scFv)2の腫瘍への取り込みは、hMN−14IgG−(734scFv)2よりも有意に低かった。hMN−14IgGI253A(734scFv)2のクリアランス速度の増加は、肝臓、脾臓、腎臓、肺、胃、小腸、大腸および血液などの正常な組織でも見られた。図3および4参照。hMN−14IgGI253A(734scFv)2のクリアランスの加速は、肝臓、脾臓、腎臓、肺、胃、小腸、大腸および血液などの多くの正常組織の腫瘍対器官比を高くした。さらに、hMN−14IgGI253A(734scFv)2 変異体の注射後1〜4日の腫瘍対血液比は、hMN−14IgG−(734scFv)2の腫瘍対血液比と比較して速い速度で増加した。
【0123】
実施例2:ヒト結腸腫瘍を有するマウスでの125I−hMN−14IgGI253A−(734scFv)2および125I−hMN−14IgG−(734scFv)2のプレターゲッティング
実験の手順
変異および親bsAbのプレターゲッティングの体内分布パターンを評価した。GW39ヒト結腸癌異種移植片を有する雌ヌードマウス群へ、20μg(5μCi)/マウスの125I標識した変異または親bsAbを静脈注射により投与した。変異または親bsAbの注射の後、予め定めたクリアランス時間の間、bsAbが腫瘍部位に局在し、循環から取り除かれるのを待った。99mTc標識した二価のDTPAペプチド、IMP−192を次に静脈注射した。このマウスを、ペプチド注射後の種々の時点で屠殺し、器官を取り出し、重さを測り、I−125およびTc−99m双方の放射能を計数した。
【0124】
GW−39ヒト結腸腫瘍細胞株を、他で記載のように(Tu, et al. Tumour Biology 9:212-220 (1988))ヌードマウス中で連続的な皮下異種移植片として増殖させた。
【0125】
結果
125I標識したhMN−14IgGI253A−(734scFv)2および125I標識したhMN−14IgG−(734scFv)2の腫瘍組織および正常組織での体内分布を、9mTc標識した二価のDTPAペプチド、IMP−192の注射後3、6、および24時間のヒト結腸腫瘍を有するマウスで検査した。IMP−192の注射の前に、変異または親bsAbでのプレターゲッティングを4日間行なった。125I標識した変異および親bsAbの腫瘍組織および組織での体内分布を図5〜7に示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)として表される。さらに、IMP−192(99mTc標識した二価のDTPAペプチド)の腫瘍組織および組織での体内分布を図5〜7に示す。変異bsAbのクリアランスの加速が認められる。さらに、親bsAbでのプレターゲッティングと比較して、変異bsAbでプレターゲッティングの後により高い腫瘍対血液比が認められる。変異融合タンパク質でのプレターゲッティング後よりも親融合タンパク質でのプレターゲッティング後により多くのDTPA-ペプチドが血中に捕捉されたことが注目される。
【0126】
当業者には、本発明による組成物および方法に対して種々の改変および変更をなしうることが明らかである。よって、本発明は、添付のクレームおよびそれらの均等物の範囲内に入る限り、このような改変および変更を含むものとする。
【0127】
上記に引用した全ての文献の開示は、各々が個別に引用することにより本明細書の一部とされるのと同程度に、引用することにより明白にその全開示内容を本明細書の一部とする。
【0128】
実施例3:In−DTPA含有ペプチドとhMN−14IgGI253A−(734scFv)2との結合
In−DTPAペプチドと抗In−DTPA抗体hMN−14IgG(1253A)−(734scFv)2との結合を、サイズ排除HPLCおよびBiacore Xを用いるアフィニティー遮断研究によって調査した。
【0129】
HPLCを用いる結合分析
IMP192キットをTc−99m 20.9mCiで標識した。このキットからのアリコートを希釈し、次のモル比(ペプチド/ab)1:5、1:1、および20:1でhMN−14IgG(1253A)−(734scFv)2と混合した。このペプチド/抗体混合物、ペプチド単独および抗体単独を、0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムで試験した。HPLCの痕跡(図8〜12)は本質的にただ1種類のペプチド/抗体複合体が形成されていることを示す。hMN−14IgGI253A−(734scFv)2 の既知の基準は約9.41分でカラムから溶出する(図8)。Tc−99m IMP192の既知の基準は約14.85分でカラムから溶出する(図9)。hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の1:1混合物をカラムに適用した場合、約9.56分に1回だけピークが認められた(図10)。それに対して、hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の1:5混合物をカラムに適用した場合、2回の大きなピークが認められ、1回は約9.56分(hMN−14IgGI253A−(734scFv)2)そしてもう1回は約14.80分(Tc−99m IMP192)であった(図11)。hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の20:1混合物をカラムに適用した場合、9.56分で1回だけピークが認められた(図12)。
【0130】
実施例4:臨床試験
実施例4A. 結腸ポリープ患者のポリープを除去すると、それが悪性であると分かった。CATスキャンは腫瘍を明示できなかったが、3ヵ月後、患者の血中のCEAレベルが上昇。この患者に10mgのhMN14−IgG[734−scFv]2を静脈点滴により投与する。3日後、患者に40mCiのTc−99mで標識した二価のペプチドIMP192を投与する。翌日、この患者に放射性シンチグラフィーを行い、ポリープの切除部位に近接する結節にただ一箇所つの活性部位が認められる。この結節を切除し、この患者はその後10年間罹患しなかった。
【0131】
実施例4B. 結腸癌患者の一次腫瘍を切除した。2年後、この患者は血中のCEAレベルの上昇を示し、CATスキャンによって肝臓に切除不可能な複数の小さな転移が示される。患者に100mgのhMN14−IgG[734−scFv]2を静脈点滴により投与する。3日後、患者に160mCiのI−131で標識した二価のDTPAペプチド、IMP156を静脈点滴により投与する。CEAの血中レベルは徐々に正常範囲まで降下した。CATスキャンによって転移のいくつかが消散したことが示され、残りの病巣は9ヶ月間増殖しなかった。
【0132】
当業者には、本発明による組成物および方法に対して種々の改変および変更をなしうることが明らかである。よって、本発明は、添付のクレームおよびそれらの均等物の範囲内に入る限り、このような改変および変更を含むものとする。
【0133】
上記に引用した全ての文献、特許、および特許出願の開示は、各々が個別に引用することにより本明細書の一部とされるのと同程度に、引用することにより明白にその全開示内容を本明細書の一部とする。
【図面の簡単な説明】
【0134】
【図1】hMN−14のH鎖cDNAおよびアミノ酸配列を示す。VH、CH1、ヒンジ、CH2およびCH3領域が示されている。274番のアミノ酸のイソロイシンは、Edelman, et al.のナンバリング方法によればイソロイシン253に相当する。Edelman et al. Biochemistry 63, 78-85 (1969)を参照されたい。
【図2】hMN−14のL鎖cDNAおよびアミノ酸配列を示す。VkおよびCK領域が示されている。
【図3】ヒト結腸腫瘍を有するマウスでの、注射後1、2、3および4日のhMN−14IgGI253A−(734scFv)2の体内分布を示す。「I253A」とは253番目のイソロイシンがアラニンへ変わっていることをさす。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示した。
【図4】ヒト結腸腫瘍を有するマウスでの、注射後1、2、3および4日のhMN−14IgG−(734scFv)2の体内分布を示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示した。
【図5】125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識された二価のDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後4日間、125I−hMN−14IgG−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc−標識された標的化可能な構築物のデータを示す。
【図6】125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識されたdiDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後6日間、125I−hMN−14IgG−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc−標識された標的化可能な構築物のデータを示す。
【図7】125I−hMN−14IgGI253A−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識されたdiDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後4日間、125I−hMN−14IgGI253A−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc標識された標的化可能な構築物のデータを示す。
【図8】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2の既知の標準溶出プロフィールを示す。
【図9】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのTc−99m IMP192の既知の標準溶出プロフィールを示す。
【図10】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の1:1混合物の既知の標準溶出プロフィールを示す。
【図11】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の1:5混合物の既知の標準溶出プロフィールを示す。
【図12】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の20:1混合物の既知の標準溶出プロフィールを示す。
【発明の背景】
【0001】
発明の分野
本発明は、対応する親二重特異性抗体(bsAb)よりも迅速に患者の身体から除去される変異二重特異性抗体(bsAb)に関する。特に、本発明は、IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbに関し、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。
【0002】
関連技術
標的部位の検出は、検出剤のシグナル対バックグラウンド比が高いと有利である。治療は、標的部位で治療剤ができる限り多く確実に付着すること、および取り込みおよび結合の持続時間が適度に長いことが有利となる。ターゲッティング率および標的部位への薬剤送達量を向上させるためには、選択的局在性のためにターゲッティング部分と結合した診断剤または治療剤を含んでなるターゲッティングベクターの使用が長く知られている。
【0003】
ターゲッティングベクターの例としては、抗体または抗体フラグメント、細胞または組織特異的ペプチド、ならびにホルモンおよびその他の受容体結合分子などのターゲッティング部分の診断剤または治療剤複合体が挙げられる。例えば、病理細胞および正常細胞に関連する、ならびに病原体微生物に関連する種々の決定基に対する抗体が、多種多様な病状または病変の検出および治療に用いられている。これらの方法では、例えば、Hansen et al., 米国特許第3,927,193号明細書およびGoldenberg, 米国特許第4,331,647号明細書、同第4,348,376号明細書、同第4,361,544号明細書、同第4,468,457号明細書、同第4,444,744号明細書、同第4,460,459号明細書、同第4,460,561号明細書、同第4,624,846号明細書および同第4,818,709号明細書(引用することによりその全開示内容を本明細書の一部とする)に記載のように、ターゲッティング抗体を直接適当な検出剤または治療剤と結合させる。
【0004】
ダイレクトターゲッティング法、すなわち、診断剤または治療剤(「有効な薬剤」)がターゲッティング部分と直接結合される方法で遭遇する問題は、複合体の比較的小さな部分は標的部位と結合するが、複合体の大部分は循環系中に残ったままであり、何らかの方法で標的複合体の機能を損なうことである。診断複合体の場合には、例えば、ラジオイムノシンチグラフィーまたは磁気共鳴イメージング複合体、循環系中に残る非標的化複合体が、バックグラウンドを増加させて解像度を低下させることがある。抗体などの長期間循環するターゲッティング部分に付着した毒性の高い治療剤、例えば放射性同位元素、薬物または毒素を有する治療用複合体の場合、循環する複合体は骨髄毒性または全身性副作用などの宿主に許容されない毒性となり得る。
【0005】
プレターゲッティング法は、検出剤または治療剤の標的:バックグラウンド比を増加させるために開発されてきた。プレターゲッティングおよびビオチン/アビジンアプローチの例は、例えば、Goodwin et al., 米国特許第4,863,713号明細書;Goodwin et al., J. Nucl. Med. 29:226, 1988; Hnatowich et al., J. Nucl. Med. 28:1294, 1987; Oehr et al., J. Nucl. Med. 29:728, 1988; Klibanov et al., J. Nucl. Med. 29:1951, 1988; Sinitsyn et al., J. Nucl. Med. 30:66, 1989; Kalofonos et al., J. Nucl. Med. 31:1791, 1990; Schechter et al., Int. J. Cancer 48:167, 1991; Paganelli et al., Cancer Res. 51:5960, 1991; Paganelli et al., Nucl. Med. Commun. 12:211, 1991;米国特許第5,256,395号明細書; Stickney et al., Cancer Res. 51:6650, 1991; Yuan et al., Cancer Res. 51:3119, 1991;米国特許第6,077,499号明細書;米国出願番号09/597,580;同10/361,026;同09/337,756;同09/823,746;同10/116,116;同09/382,186;同10/150,654;米国特許第6,090,381号明細書;同第6,472,511号明細書;米国出願番号10/114,315;米国仮出願番号60/386,411;同60/345,641;同60/3328,835;同60/426,379;米国出願番号09/823,746;同09/337,756;および米国仮出願番号60/342,103に記載されている。なお、これらは総て引用することによりその全開示内容を本明細書の一部とする。
【0006】
プレターゲッティング法では、一次ターゲッティング種(診断剤または治療剤と結合していないもの)を投与する。一次ターゲッティング種は、標的部位と結合するターゲッティング部分、および標的化可能な構築物の結合部位と結合可能な結合部分を含む。一次ターゲッティング種が十分に付着すれば、標的化可能な構築物を投与する。標的化可能な構築物は、一次ターゲッティング種の利用可能な結合部位を認識する結合部位および診断剤または治療剤を含む。
【0007】
プレターゲッティングは、ダイレクトターゲッティング法の使用にある一定の利点を与える手法である。例えば、治療、例えば放射線免疫治療のための標的部位への放射性核種のin vivo送達にプレターゲッティング法を用いると、放射免疫複合体の長期循環により生じる骨髄毒性が減少する。これは、放射性同位元素が、多くの場合に長期間循環する種である第一のターゲッティング分子と直接結合するよりも迅速にクリアランスされる、分子量の低いキレートとして送達されるためである。
【0008】
プレターゲッティング法で遭遇する問題は、循環している一次ターゲッティング種(標的部位と結合していない一次ターゲッティング種)が、標的化可能な複合体の、(一次ターゲッティング種の結合部分を介して)標的部位と結合しているターゲッティング種との結合を妨害することである。従って、循環している一次ターゲッティング種の量を最小限に抑える方法が必要とされる。
【0009】
循環している一次ターゲッティング種の量を最小限に抑える試みはいくつかなされてきた。この目標を達成する一つの方法は、身体からのクリアランス速度の速い一次ターゲッティング種を調製することである。例えば、Ward et al.(米国特許第6,165,745号明細書)はマウス由来の変異IgG1を合成し、Hornick et al.「The Journal of Nuclear Medicine 11 355-362 (2000)」は変異キメラTNT−3抗体を合成した。これらの変異抗体は、本発明による変異bsAbとは異なる。違いの一つは、本発明による変異bsAbは二重特異性抗体であるのに対し、Hornick et al.およびWard et al.の抗体は単一特異性抗体である点である。二重特異性抗体は単一特異性抗体に比べて異なる特性を有することから、この違いは有意である。本変異bsAbとWard et al.のマウス抗体とのもう一つの違いは、Ward et al.のマウス抗体にはエフェクター機能がないことである。従って、Ward et al.の抗体は、本変異bsAbのように補体を固定することまたは効果的なADCC(抗体依存性細胞傷害)を果たすことができない。
【発明の概要】
【0010】
IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbを提供することが本発明の目的であり、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。いくつかの実施態様においては、FvおよびscFvはCDRグラフト化マウスまたはヒト化成分である。別の実施態様では、FvおよびscFvはヒトまたはヒト化成分である。いくつかの実施態様において、ヒンジ定常領域はイソロイシン253のアラニンへの変異を含む。本発明はまた、FvがhMN14−IgG、ヒト化クラスIII、抗−CEA mAbに由来し(米国特許第5,874,540号明細書参照)、scFvは734scFvであり、ヒンジ定常領域で253番目のイソロイシンがアラニンへ変異している変異bsAbを提供する。
【発明の具体的説明】
【0011】
特に断りのない限り、可算名詞("a"または"an")は1つ以上のものを意味する。
【0012】
I.概要
本発明は、IgG由来の1つのヒトヒンジ定常領域、2つのscFvおよび2つのFvを含む変異bsAbに関し、該ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。本発明による変異bsAbは、対応する親bsAbよりも迅速に患者の身体から除去される。二重特異性抗体は、1999年6月22日出願の米国出願番号09/337,756に開示されている。プレターゲッティング法に用いると、循環している一次ターゲッティング種(標的部位と結合していない変異bsAb)の量が最小限度に抑えられる。さらに、血中で捕捉される標的化可能な構築物の量も最小限度に抑えられる。
【0013】
ヒトヒンジ定常領域はエフェクター機能を含み得る。抗体分子のFc部分は、細胞溶解を引き起こす作用をするようメカニズムを設定する補体結合反応およびADCC(抗体依存性細胞傷害)などのエフェクター機能を提供する。しかし、治療機能のためにはこのFc部分は必要でなく、アポトーシスなどの他のメカニズムが機能を果たす可能性がある。従って、生得的なADCC、アポトーシス誘発および補体活性化/溶解が達成され得る。
【0014】
scFvは標的化可能な構築物の結合部位に対して特異的である。標的化可能な構築物は担体部分および少なくとも1ユニットの認識可能なハプテンから成る。認識可能なハプテンの例としては、限定されるものではないが、ヒスタミンスクシニルグリシン(HSG)、DTPAおよびフルオレセインイソチオシアネートが挙げられる。標的化可能な構築物は、罹患組織の治療または同定に有用な種々の薬剤と結合してもよい。結合される薬剤の例としては、限定されるものではないが、キレート剤、金属キレート錯体、薬物、毒素(例えば、リシン、アブリン、リボヌクレアーゼ、DNアーゼI、ブドウ球菌内毒素−A、アメリカヤマゴボウ抗ウイルスタンパク質、ゲロニン、ジフテリア毒、シュードモナス外毒素、およびシュードモナス内毒素)およびその他のエフェクター分子が挙げられる。結合に好適な薬物としては、ドキソルビシン類似体、SN−38、エトポシド、メトトレキサート、6−メルカプトプリンまたはリン酸エトポシド、カリチェアマイシン(calicheamicin)、パクリタキセル、2−ピロリノドキソルビシン、CC−1067、およびアドゼレシンまたはその組み合わせが挙げられる。薬物の例としては、ナイトロジェンマスタード、エチレンイミン誘導体、スルホン酸アルキル、ニトロソウレア、トリアゼン、葉酸類似体、アントラサイクリン、タキサン、COX−2阻害剤、ピリミジン類似体、プリン類似体、抗生物質、酵素、エピポドフィロトキシン、プラチナ錯体、ビンカアルカロイド、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、アンタゴニスト、エンドスタチン、タキソール、カンプトセシン、ドキソルビシン、およびそれらの類似体、ならびにそれらの組み合わせが挙げられる。さらに、プロドラッグの活性化または薬物の標的特異的傷害性の増加に有用な酵素を標的化可能な構築物と結合させることもできる。よって、標的化可能な構築物に対して反応性のあるscFvを含む変異bsAbの使用によって、各適用ごとに新規なbsAbを産生させることなく、種々の治療および診断適用を実施することが可能となった。
【0015】
さらに、本発明は、哺乳類における標的細胞、組織または病原体の検出または治療のための方法を包含し、IgG由来のヒトヒンジ定常領域、2つのFvおよび2つのscFvを含み、ヒンジ定常領域はCH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む、有効量の変異bsAbを投与することを含んでなる。本明細書において「病原体」とは、限定されるものではないが、菌類(例えば、ヒストプラズマ・カプスラーツム、ブラストミセス・デルマチチジス、コクシジオイデス・イミティスおよびカンジダ種)、ウイルス(例えば、ヒト免疫不全ウイルス(HIV)、ヘルペスウイルス、サイトメガロウイルス、狂犬病ウイルス、インフルエンザウイルス、B型肝炎ウイルス、センダイウイルス、ネコ白血病ウイルス、レオウイルス、ポリオウイルス、ヒト血清パルボ様ウイルス、シミアンウイルス40、呼吸器多核体(RS)ウイルス、マウス乳癌ウイルス、水疱−帯状疱疹ウイルス、デングウイルス、風疹ウイルス、麻疹ウイルス、アデノウイルス、ヒトT細胞白血病ウイルス、エプスタイン−バーウイルス、マウス白血病ウイルス、流行性耳下腺炎ウイルス、水疱性口内炎ウイルス、シンドビスウイルス、リンパ球性脈絡髄膜炎ウイルス、疣贅ウイルスおよびブルータングウイルス)、寄生虫、微生物(例えば、リケッチア)および細菌(例えば、ストレプトコッカス・アガラクチエ、レジュネラ・ニューモフィラ、化膿連鎖球菌、大腸菌、淋菌、髄膜炎菌、肺炎双球菌、B型インフルエンザ菌、梅毒トレポネーマ、ライム病スピロヘータ、緑膿菌、緑膿菌、ウシ流産菌、結核菌、炭疽菌の胞子および破傷風毒素)をさす。米国特許第5,332,567号明細書を参照されたい。
【0016】
本明細書において「抗体」とは、全長(すなわち、天然に存在する、または正常免疫グロブリン遺伝子フラグメント組換え法により形成される)免疫グロブリン分子(例えばIgG抗体)、または抗体フラグメントのような、免疫グロブリン分子の免疫学的に活性な(すなわち、特異的に結合する)部分をさす。抗体という語は、キメラ抗体、CDRグラフト化(ヒト化)抗体、および完全なヒト抗体を包含する。「IgG」という語は、抗原に対して産生され、抗原と特異的に結合することができる抗体、すなわち免疫グロブリンGを意味する。抗体はAbと省略される。モノクローナル抗体はmAbと省略される。
【0017】
「ヒト抗体」は、抗原刺激に応答して特定のヒト抗体を産生するように「操作された」トランスジェニックマウスから得られる抗体である。この技術では、ヒトH鎖およびL鎖の遺伝子座のエレメントが、内在性H鎖およびL鎖遺伝子座が標的化破壊されている胚幹細胞株由来のマウス系統に導入される。トランスジェニックマウスは、ヒト抗原に特異的なヒト抗体を合成することができ、このマウスを用いてヒト抗体分泌ハイブリドーマを作製することができる。トランスジェニックマウスからヒト抗体を得る方法は、Green et al., Nature Genet. 7:13 (1994)、Lonberg et al., Nature 368:856 (1994)、およびTaylor et al., Int. Immun. 6:579 (1994)に記載されている。完全ヒト抗体はまた、遺伝子または染色体トランスフェクション法、ならびにファージディスプレー技術によって構築でき、これらは全て当技術分野で公知である。例えば、未免疫のドナー由来の免疫グロブリン可変ドメイン遺伝子レパートリーからのヒト抗体およびそのフラグメントのin vitro作製に関してはMcCafferty et al., Nature 348:552-553 (1990)を参照。この技術では、抗体可変ドメイン遺伝子がフレーム内で糸状バクテリオファージの主要または微量コートタンパク質遺伝子にクローニングされ、ファージ粒子表面上に機能的抗体フラグメントとして提示される。この糸状粒子はファージゲノムの単鎖DNAコピーを含むため、抗体の機能特性に基づいて選択すれば、それらの特性を示す抗体をコードする遺伝子が選択されることになる。このように、ファージはB細胞の特性のいくつかを模倣する。ファージディスプレー法は多様な形式で行うことが可能であり、総説としては、例えば、Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993)を参照されたい。
【0018】
ヒト抗体はまた、in vitro活性化B細胞でも産生し得る。引用することによりその全開示内容が本明細書の一部とされる米国特許第5,567,610号明細書および同第5,229,275明細書を参照されたい。
【0019】
抗体フラグメントは、F(ab’)2、F(ab)2、Fab’、Fab、Fv、scFvなどの抗体の一部分である。構造にかかわらず、抗体フラグメントは無傷の抗体によって認識される同じ抗原と結合する。例えば、抗CEAモノクローナル抗体フラグメントはCEAのエピトープと結合する。
【0020】
「抗体フラグメント」という語は、特定の抗原に結合して複合体を形成することにより抗体のようにふるまう合成または遺伝子組み換えタンパク質も含む。例えば、抗体フラグメントには、L鎖可変領域からなる単離フラグメント、H鎖およびL鎖の可変領域からなる「Fv」フラグメント、L鎖およびH鎖可変領域がペプチドリンカーによって接続されている組み換え単鎖ポリペプチド分子(「scFvタンパク質」)、ならびに超可変領域に類似したアミノ酸残基からなる最小認識ユニットが含まれる。
【0021】
キメラ抗体は、齧歯類抗体などの第1の種由来の可変ドメインおよび相補性決定領域を含む組み換えタンパク質であり、抗体分子のH鎖およびL鎖定常領域はヒト抗体などの第2の種に由来する。
【0022】
ヒト化抗体は、モノクローナル抗体の相補性決定領域が、マウス免疫グロブリンなどの第1の種の免疫グロブリンのH鎖およびL鎖可変領域からヒトH鎖およびL鎖可変ドメイン中に移されている組み換えタンパク質であり、抗体分子のH鎖およびL鎖定常領域はヒト抗体に由来する。ヒト化抗体はまたCDRグラフト化抗体と呼ばれる。
【0023】
本明細書において、「二重特異性抗体」とは、2つの異なる部分、すなわち標的組織および標的化可能な構築物と結合可能な抗体をさす。
【0024】
本明細書において、治療剤は、特定の投与計画に従って本発明による抗体と組み合わせて被験体へ投与される、または抗体部分と結合して治療に有用な複合体を形成する分子または原子である。治療剤の例としては、薬物、毒素、ホルモン、酵素、免疫調節剤、キレート剤、ホウ素化合物、光活性薬または色素、および放射性同位元素が挙げられる。免疫調節剤の例は、サイトカイン、幹細胞増殖因子、リンホトキシン、造血因子、コロニー刺激因子(CSF)、インターフェロン(IFN)、エリスロポエチン、トロンボポエチン、およびそれらの組み合わせからなる群から選択される。特に有用なものは腫瘍壊死因子(TNF)などのリンホトキシン、インターロイキン(IL)などの造血因子、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)などのコロニー刺激因子、インターフェロン−α、−βまたは−γなどのインターフェロン、ならびに「S1因子」と呼ばれる幹細胞増殖因子である。より具体的には、IL−1、IL−2、IL−3、IL−6、IL−10、IL−12、IL−18、インターフェロン−γ、TNF−αまたはそれらの組み合わせなどの免疫調節剤が本発明において有用である。「scFv」とは、抗体のL鎖およびH鎖の可変領域がペプチドリンカーによって接続されている組み換え単鎖ポリペプチド分子を意味する。
【0025】
「Fv」とは、H鎖およびL鎖の可変領域からなるフラグメントを意味する。
【0026】
「組み換え宿主」は、クローニングベクターまたは発現ベクターのいずれかを含む任意の原核細胞または真核細胞であってよい。この語はまた、それらの原核細胞または真核細胞、ならびに宿主細胞または宿主細胞の細胞の染色体またはゲノム中にクローン化された遺伝子を含むように遺伝子組み換えされたトランスジェニック動物も含む。好適な哺乳類宿主細胞には、SP2/0細胞およびNS0細胞のような骨髄腫細胞、ならびにチャイニーズハムスター卵巣(CHO)細胞、ハイブリドーマ細胞株および抗体発現に有用な他の哺乳類宿主細胞を含む。また、mAbおよび他の融合タンパク質の発現に特に有用なものはWO 0063403 A2に開示されたヒト細胞株PER.C6であり、従来の哺乳類細胞株CHO、COS、 Vero、 HeLa、 BHKおよびSP2細胞系統に比べて2〜200倍の組換えタンパク質を産生する。修飾された免疫機構を有する特別なトランスジェニック動物は完全なヒト抗体を作製するために特に有用である。
【0027】
抗原はいずれの抗原であってもよい。抗原の例は、細胞表面または腫瘍関連抗原、あるいは微生物または寄生虫に関連する抗原、あるいは罹患組織または自己免疫疾患に関与するBまたはT細胞などの疾病を導く細胞種、あるいは心疾患または神経疾患の標的抗原 (例えば、前の場合にはアテローム斑または塞栓、および後の場合にはアルツハイマー病に関連するものなどのアミロイド)がある。本明細書において「組織」とは、当業者がそれを意味すると理解するであろう組織を意味する。本発明において提案されるように、組織は、体組織または体液(例えば血液細胞)の個々の細胞もしくは細胞群、または細胞培養物も意味する。さらに、組織は被験体内にあっても、生検組織であっても、または被験者から摘出した組織であってもよい。この組織はまた、身体の器官の全てまたは一部であってもよい。さらに、この組織は、切除と本発明による方法との間に保存工程を含まずに被験体から取り出されたばかりの組織であるという点で「新鮮」であると言える。組織はまた、本発明による方法の適用前に、限定されるものではないが、凍結、急速凍結、パラフィン包埋および組織固定をはじめとする標準の組織調製技術によって保存してもよい。
【0028】
「標的組織」とは、標的化可能な複合体が送達されうる系、器官、組織、細胞、受容体または細胞小器官である。本発明の治療的態様では、標的組織は、感染しているか、機能障害性であるか、置換されているか、または異所的である(例えば、感染細胞、癌細胞、子宮内膜症など)。骨髄など正常な組織もまた、治療的介入が必要であれば、これらの方法で切除してもよい。本発明の診断的態様では、標的組織を検出することが望ましい。
【0029】
本明細書において「被験体」とは、限定されるものではないが、ヒトおよびその他の霊長類、齧歯類(例えばマウス、ラット、およびモルモット)、ウサギ類(lagamorphs)(例えばウサギ)、ウシ類(例えばウシ)、ヒツジ類(例えばヒツジ)、ヤギ類(例えばヤギ)、ブタ類(例えばブタ)、ウマ類(例えばウマ)、イヌ類(例えばイヌ)、ネコ類(例えばネコ)、家禽類(例えばニワトリ、シチメンチョウ、アヒル、ガチョウ、その他のキジ類の鳥など)をはじめとするいずれの動物(すなわち、脊椎動物および無脊椎動物)、ならびに、限定されるものではないが、有蹄類(例えばシカ)、クマ類、魚類、ウサギ類、齧歯類、鳥類などの野生動物もさす。この語は特定の齢または性別に限定するものではない。よって、成体および新生児(仔)被験体、ならびに胎児(仔)は、雌雄にかかわらず、この語に包含される。
【0030】
本明細書において「親bsAb」という語は、親bsAbのヒンジ定常領域がCH2−CH3ドメイン境界領域で1以上のアミノ酸変異を含まないことを除いて、あらゆる点で変異bsAbに類似したbsAbを意味するのに使用する。
【0031】
本明細書において「ヒンジ定常領域」は、IgGのCl、CH1、ヒンジ、CH2およびCH3領域を含む。H鎖定常領域はCH1、ヒンジ、CH2およびCH3領域を含むが、L鎖定常領域はCl領域を含む。
【0032】
II.変異二重特異性抗体
変異bsAbのFvは抗体に由来し、標的組織と特異的に結合する。Fvの例は、「Anti-CD20 Antibodies And Fusion Proteins Thereof And Methods Of Use」と題された、代理人整理番号18733/1073、米国仮出願番号60/356,132、同60/416,232および代理人整理番号18733/1155(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどの抗CD20抗体;クラスIII抗癌胎児性抗原抗体(抗CEA抗体)である、米国出願番号5,874,540(引用することによりその全開示内容を本明細書の一部とする)に開示のものなどのhMN−14抗体;米国出願番号10/116,116(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのMu−9抗体;米国仮出願番号60/360,259(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのLL1抗体;米国仮出願番号60/399,707(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのAFP抗体;「Monoclonal Antibody cPAM4」と題された、代理人整理番号18733/1102の米国仮出願(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのPAM4抗体;米国仮出願番号60/360,229(引用することによりその全開示内容を本明細書の一部とする)に記載のものなどのRS7抗体;ならびに米国特許第5,789,554号明細書および同第6,187,287号明細書ならびに米国出願番号09/741,843および同09/988,013(引用することによりその全開示内容を本明細書の一部とする)に開示のものなどのCD22抗体由来のものである。造血器腫瘍および固形腫瘍のその他の多くの腫瘍関連抗原が当業者に公知であり、引用出願にも含められているように、(限定されるものではないが)CD15、CD19、CD20、CD21、CD22、CD23、CD25、CD40、CD45、CD66、CD74、CD80、Ii、Ia、HLA−DR、PSMA、PSA、前立腺酸性ホスファターゼ、テネイシン、Le(y)、AFP、HCG、CEA、CSAp、PAM4、MUC1、MUC2、MUC3、MUC4、EGP−1、EGP−2、EGFR、HER2/neu、インスリン成長因子受容体、S100、VEGF、胎盤成長因子(PlGF)、胎盤アルカリ性ホスファターゼ、壊死産物、癌遺伝子産物などが挙げられる。hMN−14のH鎖cDNAおよびアミノ酸配列を図1に示し、hMN−14のL鎖cDNAおよびアミノ酸配列を図2に示す。
【0033】
FvをコードするcDNAは、ヒンジ定常領域をコードするベクターへ挿入してもよい。発現ベクターの例として、IgG1のヒンジ定常領域のアミノ酸をコードするpdHL2がGillies S.D., Lo KM, and Wesolowski, J. J. Immunol Methods 125 191-202 (1989)およびLosman, M.J. et al. Cancer Supplement 80 2660-2666 (1997)によって報告されており、本発明による変異二重特異性抗体の構築に用いてよい。
【0034】
Fvはマウス抗体、CDRグラフト化(ヒト化)抗体、またはヒト抗体由来であってよい。Fvはヒトモノクローナル抗体、ヒトFvライブラリーを含むトランスジェニックマウス、またはファージ/リボソームヒトIgGライブラリーから得ることができる。
【0035】
FvがCDRグラフト化抗体に由来する場合、適当な可変領域枠組み構造配列を、抗原結合領域がそれに由来するドナー抗体のクラスまたは種類を考慮して用いてよい。用いるヒト枠組み構造の種類がドナー抗体と同じまたは類似のクラスまたは種類であることが好ましい。枠組み構造は、ドナー抗体配列との相同性を、特にCDRに空間的に近接または隣接する位置で最大化または最適化するように選択すると有利である。CDRグラフト化抗体を構築するために用いてよいヒト枠組み構造の例として、LAY、POM、TUR、TEI、KOL、NEWM、REIおよびEU(Kabat et al, 1987)がある。KOLおよびNEWMはH鎖構築に好適である。REIはL鎖構築に好適であり、EUはH鎖およびL鎖双方の構築に好適である。
【0036】
CDRグラフト化抗体のL鎖またはH鎖可変領域は、必要に応じてヒトL鎖またはH鎖定常ドメインと融合させてもよい(本明細書において「H鎖定常ドメイン」とは、特に指定のない限り、ヒンジ領域を含むものと理解される)。CDRグラフト化抗体のヒト定常ドメインは、それが存在する場合は、抗体の予定される機能、特に、必要とされうるエフェクター機能を考慮して選択してもよい。例えば、CDRグラフト化抗体が治療目的を意図し、抗体のエフェクター機能を必要とする場合には、IgG1およびIgG3イソ型ドメインを用いてよい。あるいは、CDRグラフト化抗体が抗体エフェクター機能を必要としない目的を意図する場合、例えばイメージング、診断または細胞毒性の標的化目的である場合は、IgG2およびIgG4イソ型ドメインを用いてよい。L鎖可変領域と融合してよいL鎖ヒト定常ドメインとしては、ヒトλまたは、特に、ヒトκ鎖が挙げられる。
【0037】
二重特異性変異抗体のヒンジ定常領域は、CH2−CH3ドメイン境界領域に1以上のアミノ酸変異を含む。言い換えれば、二重特異性変異抗体のヒトヒンジ定常領域を二重特異性親抗体のヒトヒンジ定常領域と比較すると、その領域は1以上のアミノ酸が異なっている。
【0038】
変異は、例えば置換アミノ酸が電荷または大きさなど類似の構造または化学特性を有する「保存的」変化を包含する(例えばロイシンのイソロイシンによる置換)。変異はまた、例えば「非保存的」変化(例えばグリシンのトリプトファンによる置換)も包含する。
【0039】
好ましい実施態様では、253番目の位置のアミノ酸(Edelmanの付番方式に従う)が変異している。この位置での変異の例としては、イソロイシンのアラニンによる置換がある。いくつかの実施態様では、253番目のアミノ酸が、変異bsAbのクリアランスの薬物動態が253番目の位置のアミノ酸がアラニンへ変化している場合に認められるものと同等となるアミノ酸へ変異している。
【0040】
一つの実施態様では、二重特異性変異抗体のヒンジ定常領域はヒトIgG1のアミノ酸配列を含んでなる。H鎖のCl、CH1、ヒンジ、CH2およびCH3領域をコードするアミノ酸は、図1のアミノ酸番号139〜468として示され、Cl鎖をコードするアミノ酸は図2のアミノ酸番号128〜232として示される。イソロイシン253を特定するために用いた番号体系は、Edelman et al.がEu H鎖およびL鎖の開示中で用いた番号体系の開示に一致することに留意されたい(Edelman et al. Biochemistry 63, 78-85 (1969))。
【0041】
二重特異性変異抗体のscFv成分は、標的化可能な構築物と特異的に結合する。いずれのscFv成分の使用も本発明で意図される。好ましいscFv成分は679scFv(マウス抗HSG由来)および734scFv(マウス抗−diDTPA由来)である。scFvはマウス、CDRグラフト化(ヒト化)またはヒトであってよい。
【0042】
scFvがCDRグラフト化抗体に由来する場合、適当な可変領域枠組み構造配列を、抗原結合領域がそれに由来するドナー抗体のクラスまたは種を考慮して用いてよい。用いるヒト枠組み構造の種類がドナー抗体と同じまたは類似のクラスまたは種類であることが好ましい。枠組み構造は、ドナー抗体配列との相同性を、特にCDRに空間的に近接または隣接する位置で最大化または最適化するように選択すると有利である。CDRグラフト化抗体を構築するために用いてよいヒト枠組み構造の例としては、LAY、POM、TUR、TEI、KOL、NEWM、REIおよびEU(Kabat et al, 1987)がある。KOLおよびNEWMはH鎖構築に好適である。REIはL鎖構築に好適であり、EUはH鎖およびL鎖双方の構築に好適である。
【0043】
CDRグラフト化抗体のL鎖またはH鎖可変領域は、必要に応じてヒトL鎖またはH鎖定常ドメインと融合させてもよい(本明細書において「H鎖定常ドメイン」とは、特に断りのない限り、ヒンジ領域を含むものと理解される)。CDRグラフト化抗体のヒト定常ドメインは、それが存在する場合は、抗体の予定される機能、特に、必要とされうるエフェクター機能を考慮して選択してもよい。例えば、CDRグラフト化抗体が治療目的を意図し、抗体のエフェクター機能が必要とされる場合には、IgG1およびIgG3イソ型ドメインを用いてよい。あるいは、CDRグラフト化抗体が抗体エフェクター機能が必要とされない目的を意図する場合、例えばイメージング、診断または細胞毒性の標的化目的には、IgG2およびIgG4イソ型ドメインを用いてよい。L鎖可変領域と融合してよいL鎖ヒト定常ドメインには、ヒトλまたは、特に、ヒトκ鎖が挙げられる。
【0044】
好ましい変異bsAbはhMN−14IgGI253A−(734scFv)2である。この変異bsAbでは、FvはhMN−14IgGに由来し、scFvは734scFV(マウス抗diDTPA由来)であり、ヒンジ定常領域はヒトIgG1のアミノ酸配列を含む。
【0045】
本発明の一つの実施態様では、変異bsAbと標的化可能な構築物との間に一対一結合の相互作用が得られる。例えば、本発明による変異bsAbが、2つのDTPA部位を含む二価の標的化可能な構築物IMP192と相互作用する場合、1つのbsAbが1つのIMP192と結合する。この相互作用は実施例3で説明する。
【0046】
III.変異bsAbを標的化可能な構築物
いくつかの実施態様では、本発明による変異bsAbは標的化可能な構築物と結合する。変異bsAbのscFvが標的化可能な構築物と結合するのが好ましい。標的化可能な構築物は多様な構造であり得るが、十分な免疫応答を誘起することだけでなく、急速なin vivoクリアランスに関しても選択される。本発明における使用のための標的化可能な構築物の例としては、引用することによりその全開示内容が本明細書の一部とされる1999年6月22日出願の米国出願番号09/337,756、および2001年4月3日出願の米国出願番号09/823,746に記載されている。
【0047】
疎水性薬剤は強い免疫応答を誘起するのに最良であるのに対して、親水性の薬剤は急速なin vivoクリアランスに好ましく、従って、必要な疎水性と親水性とのバランスを確立するべきである。これは、多くの有機体部分の固有の疎水性を相殺するため親水性のキレート剤の使用に頼ることによって一部達成されている。また、反対の溶液特性、例えばそのうちの一部は疎水性で一部は親水性であるアミノ酸を含むペプチドを有する標的化可能な構築物のサブユニットを選択してもよい。ペプチドの他に、炭水化物を用いてもよい。
【0048】
わずか2アミノ酸残基しかないペプチドを用いてもよく、キレート剤などのその他の部分と共役するならば、2〜10残基が好ましい。リンカーは分子量の低い複合体であるべきであり、分子量は50,000ダルトン未満であることが好ましく、約20,000ダルトン、10,000ダルトンまたは5,000ダルトン未満が有利であり、キレート中に金属イオンを含む。例えば、公知のペプチドDTPA−Tyr−Lys(DTPA)−OH(式中、DTPAはジエチレントリアミン五酢酸である)を用いて、この分子のインジウム−DTPA部分に対する抗体が産生されている。しかし、インジウム非含有分子を用いることによって、また適当なスクリーニング工程によって、チロシル−リシンジペプチドに対する新規なAbを作出することができる。通常、ペプチドDOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2[式中、DOTAは1,4,7,10−テトラアザシクロドデカン四酢酸であり、HSGは式:
【化1】
のヒスタミンスクシニルグリシル群である]、などの抗原性ペプチドは4以上の残基を有する。
【0049】
金属非含有ペプチドを免疫原として用い、結果として生じるAbをPhe−Lys−Tyr−Lys主鎖に対する反応性をスクリーニングしてもよい。
【0050】
本発明はまた、最終のbsAb/リンカー系とともに用いた場合、リンカー部分を認識するscFv成分が完全に特異的であることを確実にするため、非天然アミノ酸、例えばD−アミノ酸の主鎖構造への組み込みも意図する。本発明はさらに、非天然アミノ酸およびペプチドから構築されたものなどのその他の主鎖構造も意図する。
【0051】
免疫原として用いられるペプチドは、固相支持体ならびに反復直交性の脱保護およびカップリングの標準技術を用いて自動ペプチド合成装置で便宜に合成される。ペプチド中の遊離アミノ基は、後にキレート錯体に用いられるが、アセチル基などの標準の保護基を用いてブロッキングするのが有利である。このような保護基は当業者に公知であろう。Greene and Wuts Protective Groups in Organic Synthesis, 1999 (John Wiley and Sons, N.Y.)を参照されたい。ペプチドを後の変異bsAbの使用のために準備する場合、in vivo カルボキシペプチダーゼ活性を阻害するために、樹脂から切り取って対応するC末端アミドを生成すると有利である。
【0052】
免疫原のハプテンは、免疫原性の認識部分、例えば化学ハプテンを含んでなる。化学ハプテン、好ましくはHSGまたはDTPAハプテンの使用により、抗体に対するリンカーの高い特異性が発揮される。これは、HSGまたはDTPAハプテンに対して作製された抗体が公知であり、この抗体のscFv部分が変異bsAbへ容易に組み込まれるために生じる。よって、リンカーと、付着したハプテンとの結合はscFv成分に対して特異性が高いと思われる。
【0053】
標的化可能な構築物は一価または二価であってよく、二価のペプチドが好ましいペプチドである。標的化可能な構築物の一例はIMP192(Ac−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(TscG−Cys−)−NH2)である。IMP192は診断用にはTc−99mとIn−111の双方と結合し、治療用にはRe−188とRe−186の双方と結合する。IMP192もまた、チロシンを含む二価のDTPA−ペプチドである。
【0054】
本発明による方法において、標的化可能な構築物は、罹患組織の検出に有用な1以上の放射性同位元素を含んでよい。特に有用な診断用放射性核種としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gd、177Lu、32P、188Re、90Y、またはその他のγ-、β-、または陽電子放射体が挙げられ、エネルギー範囲は20〜4,000keVであることが好ましく、25〜4,000keVがより好ましく、20〜1,000keVがさらに好ましく、70〜700keVがなお一層好ましい。
【0055】
本発明による方法において、標的化可能な構築物は、罹患組織の検出に有用な1以上の放射性同位元素を含んでよい。特に有用な治療用放射性核種としては、限定されるものではないが、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acが挙げられる。治療用の放射性核種はエネルギー範囲が60〜700keVであることが好ましい。
【0056】
本発明による方法において、標的化可能な構築物は、磁気共鳴イメージング(MRI)に用いる1以上の造影剤を含んでよい。限定されるものではないが、標的化可能な化合物は、例えば、Mn、Fe、およびGdなどの1以上の常磁性イオンを含む。
【0057】
本発明による方法において、標的化可能な構築物は、超音波イメージングに用いる1以上の造影剤を含んでよい。限定されるものではないが、標的化可能な構築物は、例えば、1以上の超音波イメージング剤を含む。このような一つの実施態様では、標的化可能な構築物は、リポソーム脂質膜の外面に二価DTPA−ペプチドが共有結合したリポソームである。所望により、該リポソームはガス充填されてよい。
【0058】
IV.キレート部分
リンカー部分に親水性のキレート部分が存在することにより急速なin vivoクリアランスが確実となる。親水性に加え、キレート剤はその金属結合特性で選択され、自由に変えられる。というのも、少なくともそのbsAbエピトープがペプチドの一部分であるか、または非キレート化学ハプテンであるリンカーには、金属−キレート錯体の認識はもはや問題ではないからである。
【0059】
特に有用な金属−キレートの組み合わせとしては、2−ベンジル−DTPAおよびそのモノメチルおよびシクロヘキシル類似体が挙げられ、放射性イメージングおよびRAITに47Sc、52Fe、55Co、67Ga、68Ga、111In、89Zr、90Y、161Tb、177Lu、212Bi、213Bi、および225Acとともに用いられる。同じキレート剤は、Mn、Fe、およびGdなどの非放射性金属とキレート化した場合、本発明による変異bsAbとともに用いるとMRIに使用できる。NOTA(1,4,7−トリアザ−シクロノナン−N,N’,N’’−三酢酸)、DOTA、およびTETA(p−ブロモアセトアミド−ベンジル−テトラエチルアミン四酢酸)などの大環状キレート剤は種々の金属および放射性金属とともに使用され、最も特にそれぞれGa、YおよびCuの放射性核種とともに使用される。
【0060】
DTPAおよびDOTA系キレート剤は、リガンドにカルボン酸塩またはアミン基など硬い塩基のキレート性官能基が含まれる場合に、硬い酸のカチオン、特にIIa族およびIIIa族の金属カチオンをキレート化するのに最も効果的である。このような金属−キレート錯体は、環の大きさを目的の金属に調製することによって非常に安定させることができる。大環状ポリエーテルなどその他の環状キレート剤は、RAITで223Raなどの核種の安定した結合のために注目される。ポルフィリンキレート剤を多数の放射性金属とともに用いてもよく、bsAbに導かれる免疫光線療法のためのある種の非放射性金属錯体としても有用である。1種類以上のキレート剤を担体と結合させて複数の金属イオン、例えば、非放射性イオン、診断用放射性核種および/または治療用放射性核種と結合させてよい。特に有用な治療用放射性核種としては、限定されるものではないが、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acが挙げられる。特に有用な診断用放射性核種としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gdおよび175Luが挙げられる。
【0061】
米国特許第5,753,206号明細書に開示のものなどのキレート剤、特にチオセミ−カルバゾニルグリオキシルシステイン(Tscg−Cys)およびチオセミカルバジニル−アセチルシステイン(Tsca−Cys)キレート剤は、Tc、Re、Biおよびその他の遷移金属の軟らかい酸のカチオン類、ランタニド類、ならびに軟らかい塩基のリガンド、特に硫黄またはリンを含むリガンドと緊密に結合するアクチニド類との結合に用いると有利である。1種類以上のキレート剤を、ペプチド、例えばDTPAまたは類似のキレート剤、例えばIn(III)カチオンのキレート剤、およびチオールを含むキレート剤、例えばTcカチオンのTscg−Cysと結合させると有用である。di−DTPAハプテンの抗体は公知であり(Barbet‘395、前掲)、ターゲッティング抗体と容易に結合してbsAbを形成するため、放射性同位元素をターゲッティングするためのプレターゲッティングプロトコールで、ペプチドハプテンを非放射性diDTPAキレート剤および別のキレート剤を放射性同位元素との結合に使用することができる。このようなペプチドの一例はAc−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(Tscg−Cys−)−NH2である。このペプチドはIn(III)を予め付加し、その後99m−Tcカチオンで標識することができ、In(III)イオンはDTPAに選択的にキレート化され、Tcカチオンはチオールを含むTscg−Cysと選択的に結合する。NOTA、DOTA、TETAなどのその他の硬い酸のキレート剤は、DTPA基と置換することができ、抗di−DTPA Mabの産生に用いるものと類似の技術を用いてそれらに特異的なMabを作製することができる。
【0062】
当然のことながら、2種類の異なる硬い酸または軟らかい酸のキレート剤を、例えば異なるキレート環サイズのリンカーへ組み込んで、異なるカチオンサイズ、キレート環の幾何学、およびカチオンの好ましい複合イオン構造のため、2種類の異なる硬い酸または軟らかい酸のカチオンと選択的に結合させることができる。これによって、その一方または双方が放射性であるかまたはMRI増強に有用である2種類の異なる金属を、プレターゲッティングされたbsAbよる最終捕捉のためのリンカーへ組み込むことが可能となる。
【0063】
好ましいキレート剤としては、NOTA、DOTAおよびTscgならびにその組み合わせが挙げられる。このようなキレート剤は、次の構造:
(a)DOTA−Phe−Lys(HSG)−D−Tyr−Lys(HSG)−NH2;
(b)DOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2;
(c)Ac−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys)−NH2;
【化2】
に例示されるようなキレート剤−ペプチド複合体モチーフへ組み込まれている。
【0064】
上記キレート剤−ペプチド複合体(d)および(e)は、68Gaと結合することが示されており、従って陽電子放射断層撮影法(PET)への適用に有用である。
【0065】
キレート剤は、下記の実施例でより詳細に考察される標準の化学反応を用いてリンカー部分と結合される。要するに、ペプチドAc−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys−)−NH2の合成は、ペプチドシンセサイザーのRinkアミド樹脂へAloc−Lys(Fmoc)−OHが最初に付着することによって達成される。本明細書で用いられる保護基の省略形「Aloc」および「Fmoc」はアリールオキシカルボニルおよびフルオレニルメチルオキシカルボニルの基をさす。次にFmoc−Cys(Trt)−OHおよびTscGを標準のFmoc自動合成プロトコールを用いてリシンの側鎖へ加えて次のペプチド:Aloc−Lys(Tscg−Cys(Trt)−rink樹脂を形成した。次にAloc基を取り除いた。次にペプチド合成をシンセサイザーで継続して次のペプチド:(Lys(Aloc)−D−Tyr−Lys(Aloc)−Lys(Tscg−Cys(Trt)−)−rink樹脂を形成した。その後N末端をアシル化し、Aloc保護基の側鎖を取り除いた。生じたペプチドを次に、カイザー試験を用いて樹脂がアミンに検査陰性となるまで活性化N−トリチル−HSG−OHで処理した。Karacay et al. Bioconjugate Chem. 11:842-854 (2000)参照。Ac−Lys(HSG)D−Tyr−Lys(HSG)−Lys(Tscg−Cys−)−NH2の合成、ならびにDOTA−Phe−Lys(HSG)−D−Tyr−Lys(HSG)−NH2;およびDOTA−Phe−Lys(HSG)−Tyr−Lys(HSG)−NH2の合成はより詳細に下に記載されている。
【0066】
V.金属キレート調製のための一般法
キレート剤−ペプチド複合体は固形物と同じく長期間保存し得る。それらは計量して金属結合反応のための単位用量とし、固体、水溶液または半水溶液、冷凍溶液あるいは凍結乾燥調製物の単位用量として保存してよい。それらを周知の手順によって標識してもよい。一般的に、硬い酸のカチオンを便宜な塩の溶液として導入し、それを硬い酸のキレート剤、およびあるいは軟らかい酸のキレート剤にとる。しかし、軟らかい酸のカチオンを後で添加すると軟らかい酸のキレート剤がそれと結合し、そこでキレート化する可能性のある硬い酸のカチオンをどれも置換してしまう。例えば、たとえ過剰な量の非放射性111InCl3の存在下でも、99m−Tc(V)グルコヘプトネートまたはin situで塩化第一錫およびNa99m−TcO4を用いて生成したTcカチオンでの標識は、軟らかい酸のキレート剤上で量的に進行する。その他の186Re、188Re、213Biなどの軟らかい酸のカチオン、ならびにMn、Co、Ni、Pb、Cu、Cd、Au、Fe、Ag(一価)、ZnおよびHg、特に64Cuおよび67Cuなどの二価または三価のカチオンは、放射免疫診断または放射免疫治療に有用なものもいくつかあるが、類似の方法によってリンカーペプチド上に付加することができる。Reカチオンもin situでペルレナートおよび錫イオンから生成でき、または予め還元されたレニウムグルコヘプトネートまたはその他のトランスキレート剤を用いることもできる。ペルレナートの還元にはTcの還元に必要とされるよりも多い錫イオン(一般的に終濃度200mg/mL超)が必要であるので、ジスルフィド−環化ペプチド中に存在するものなどの感受性の高いジスルフィド結合を高レベルの錫イオンが還元しないことを確実にするため特別な注意が必要である。レニウムでの放射性標識の際は、Tc−99mで用いるのと類似の手順が用いられる。Tscg−Cys−リガンドのReO金属錯体の好ましい調製方法は、ペプチドをReOCl3(P(Ph3)2と反応させることによるが、ReO(エチレンジアミン)2などのその他の還元種を用いることも可能である。
【0067】
VI.抗体の作製方法
ペプチド主鎖に対する抗体は周知のAb産生法によって作製する。例えば、フロイント完全アジュバントでの(ペプチド)n−KLH(式中、KLHはキーホルリンペットヘモシアニンであり、nは1〜30である)などの免疫原の注射の後、フロイントの不完全アジュバント中に懸濁した同じ免疫原を免疫反応性のある動物へ2回注射し、静脈からの抗原の追加免疫3日後に脾臓細胞を回収する。回収した脾臓細胞を次にSp2/0−Ag14骨髄腫細胞と融合させ、生じたクローンの培養上清を直接結合ELISA法を用いて抗ペプチド反応性について分析した。生成したAbの優れた選択性は当初の免疫原のペプチドフラグメントを用いて分析できる。これらのフラグメントは自動ペプチドシンセサイザーを用いて容易に調製できる。Abの産生のため、酵素の欠乏したハイブリドーマを単離して融合した細胞株の選択を可能にする。この技術はまた、1以上のリンカーを含むキレート、例えばIn(III)−DTPAキレートに対する抗体を作製するのに用いることができる。In(III)−di−DTPAのモノクローナルマウス抗体が公知である(Barbet‘395, 前掲)。
【0068】
本発明で用いられる変異二重特異性抗体は、マーカー物質のように種々の細胞表面または細胞内の腫瘍関連抗原に特異的である。このようなマーカーは、腫瘍により産生された物質、もしくは細胞質中、核中であろうと種々の細胞小器官あるいは細胞の下部構造中であろうと、腫瘍部位、腫瘍細胞表面上または腫瘍細胞内に集積する物質であってよい。このような腫瘍関連マーカーのの中には、Herberman, "Immunodiagnosis of Cancer", in Fleisher ed., "The Clinical Biochemistry of Cancer", page 347 (American Association of Clinical Chemists, 1979) および米国特許第4,150,149号明細書;同第4,361,544号明細書;および4,444,744号明細書に開示されているものがある。
【0069】
腫瘍関連マーカーは、Herberman(前掲)によって、腫瘍胎児抗原、胎盤の抗原、発癌または腫瘍ウイルス関連抗原、組織関連抗原、器官関連抗原、異所性ホルモンおよび正常な抗原またはその変異形態をはじめとするいくつかのカテゴリーに分類されている。時折、腫瘍関連マーカーのサブユニットを用いて腫瘍特異性の高い抗体、例えば、米国特許第4,361,644号明細書および同第4,444,744号明細書に開示のように非腫瘍物質に対する交差反応性が非常に低減された抗体の産生を刺激するヒト絨毛性ゴナドトロピン(HCG)のβサブユニットまたは癌胎児抗原(CEA)のγ領域を作製することが有利である。
【0070】
注目されるもう一つのマーカーは、トランスメンブランアクチベーターおよびCAML相互作用子(TACI)である。Yu et al. Nat. Immunol. 1:252-256 (2000)参照。要するに、TACIはB細胞の悪性腫瘍(例えばリンパ腫)のマーカーである。さらに、TACIおよびB細胞成熟抗原(BCMA)は、腫瘍壊死因子ホモログである増殖誘発リガンド(APRIL)によって結合することが知られている。APRILは、一次B細胞およびT細胞のin vitroでの増殖を刺激し、B細胞のin vivoでの集積により脾臓重量を増加させる。APRILはまた、受容体結合に関してTALL−I(BLySまたはBAFFとも呼ばれる)と競合する。可溶性BCMAおよびTACIは、APRILの結合を特異的に阻止し、APRILに刺激される一次B細胞の増殖を阻害する。BCMA−Fcはまた、マウスにおいてキーホルリンペットヘモシアニンおよびニューモバックスに対する抗体の産生を阻害し、BCMAおよび/またはTACIを介するAPRILおよび/またはTALL−Iのシグナル伝達が、液性免疫の生成に必要なことを示している。よって、2リガンド−2受容体経路からのAPRIL−TALL−IおよびBCMA−TACIは、B細胞およびT細胞機能の刺激に関与する。
【0071】
免疫原に対する最初の抗体を作製した後、抗体を配列決定し、その後組換え技術によって生産すればよい。マウス抗体および抗体フラグメントのヒト化およびキメラ化は、当業者に周知である。例えば、ヒト化モノクローナル抗体はマウス相補性決定領域をマウス免疫グロブリンのH鎖およびL鎖可変領域からヒト可変ドメインへ移すことによって作出し、その後、マウス対応物の枠組み構造領域中の ヒト残基を置換する。ヒト化モノクローナル抗体由来の抗体成分の使用は、マウス定常領域の免疫原性に関連する潜在的な問題を未然に防ぐ。マウス免疫グロブリン可変ドメインのクローニングの一般的技術は、例えば、引用することによりその全開示内容が本明細書の一部とされるOrlandi et al., Proc. Nat'l Acad. Sci. USA 86: 3833 (1989)の刊行物に記載されている。ヒト化Mabを作出する技術は、例えば、引用することによりその全開示内容が本明細書の一部とされるJones et al., Nature 321: 522 (1986), Riechmann et al., Nature 332: 323 (1988), Verhoeyen et al., Science 239: 1534 (1988), Carter et al., Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech. 12: 437 (1992), and Singer et al., J. Immun. 150: 2844 (1993)に記載されている。
【0072】
あるいは、完全なヒト抗体は、トランスジェニック非ヒト動物から得ることができる。例えば、Mendez et al., Nature Genetics, 15: 146-156 (1997); 米国特許第5,633,425号明細書を参照されたい。例えば、ヒト抗体は、ヒト免疫グロブリン遺伝子座を有するトランスジェニックマウスから回収できる。このマウス体液性免疫機構は、内在性免疫グロブリン遺伝子を不活性化して、ヒト免疫グロブリン遺伝子座を導入することによりヒト化されている。このヒト免疫グロブリン遺伝子座は非常に複雑で、あわせてヒトゲノムのほぼ0.2%を占める多数の不連続セグメントを含んでなる。トランスジェニックマウスの適当な抗体レパートリー産生を確実するために、ヒトH鎖およびL鎖遺伝子座の大部分をマウスゲノムへ導入しなければならない。これは、生殖細胞系構成における、ヒトH鎖またはL鎖免疫グロブリン遺伝子座を含む酵母人工染色体(YAC)の形成から始まる段階的なプロセスで達成される。各々の挿入体はほぼ1Mbのサイズなので、YAC構築物は免疫グロブリン遺伝子座の重複フラグメントの相同的組換えを必要とする。2つのYACは、1つがH鎖遺伝子座を含み、1つがL鎖遺伝子座を含み、YAC含有酵母スフェロプラスト(spheroblast)マウス胚幹細胞を融合させて個別にマウスへ導入される。次いで、胚幹細胞クローンをマウス胚盤胞に微量注入する。その結果得られたキメラ雄動物の生殖細胞系統を介するYAC伝達能力に関してスクリーニングし、マウス抗体産生を欠損しているマウスを繁殖させた。一種はヒトH鎖の遺伝子座を含み、もう一種はヒトL鎖遺伝子座を含む二種のトランスジェニック系統のマウスを繁殖させることによって、免疫化に反応してヒト抗体を産生する子孫をつくる。
【0073】
再配列されていないヒト免疫グロブリン遺伝子もまた、微小核体媒介性染色体移入法(MMCT)によってマウス胚幹細胞へ導入できる。例えば、Tomizuka et al., Nature Genetics, 16: 133 (1997)を参照されたい。この方法論ではヒト染色体を含む微小核体をマウス胚幹細胞と融合させる。移入された染色体は安定して保持され、成体キメラは適当な組織特異的発現を示す。
【0074】
あるいは、本発明による抗体または抗体フラグメントは、コンビナトリアル免疫グロブリンライブラリーから単離したヒト抗体フラグメントに由来するものであってもよい。例えば、引用することによりその全開示内容が本明細書の一部とされるBarbas et al., METHODS: A Companion to Methods in Enzymology 2: 119 (1991),およびWinter et al., Ann. Rev. Immunol. 12: 433 (1994)を参照されたい。B細胞を不死化させてモノクローナル抗体を産生することに関連する困難の多くは、ファージディスプレーを用いて抗体フラグメントを操作し大腸菌に発現させることによって克服できる。高親和性の回復を確実にするために、モノクローナル抗体コンビナトリアル免疫グロブリンライブラリーは大きなレパートリーを含まなければならない。一般的戦略では、免疫マウスのリンパ球または脾臓細胞から得たmRNAを利用して、逆転写酵素を用いてcDNAを合成する。H鎖およびL鎖遺伝子をPCRによって個別に増幅し、ファージクローニングベクターに連結する。一方はH鎖遺伝子を含み、他方はL鎖遺伝子を含む二種類の異なるライブラリーを作製する。ファージDNAを各ライブラリーから単離し、H鎖およびL鎖配列を連結し、パッケージングしてコンビナトリアルライブラリーを形成する。各々のファージはH鎖およびL鎖cDNAの無作為の組み合わせを含み、大腸菌に感染させた際に感染細胞中で抗体鎖の発現を指示する。注目される抗原を認識する抗体を同定するため、ファージライブラリーをプレーティングし、プラーク中に存在する抗体分子を膜へ移す。この膜を放射性標識した抗原でインキュベートし、次に洗浄して過剰な非結合リガンドを取り除く。オートラジオグラムの放射性スポットが抗原と結合している抗体を含むプラークを識別する。ヒト免疫グロブリンファージライブラリーの作出に有用なクローニングおよび発現ベクターを、例えば、STRATAGENE Cloning Systems (La Jolla, CA)から得ることができる。
【0075】
類似の手法を用いて高親和性scFvが得られる。例えば、Vaughn et al., Nat. Biotechnol., 14: 309-314 (1996)を参照されたい。広いレパートリーを有するscFvライブラリーは、全て公知のVH、VkおよびVλ遺伝子ファミリーに対応するPCRプライマーを用いて非免疫ヒトドナー由来のV遺伝子を単離することにより構築できる。増幅の後、VkおよびVλプールを合わせて一つのプールとする。これらのフラグメントをファージミドベクターへ連結する。scFvリンカー、(Gly4、Ser)3をその後、VLフラグメントのファージミド上流へ連結する。VHおよびリンカー−VLフラグメントを増幅させ、JH領域に構築する。結果として生じるVH−リンカー−VLフラグメントをファージミドベクターへ連結する。ファージミドライブラリーを、上記のようにフィルターか、またはイムノチューブ(Nunc; Maxisorp)を用いて選別する。類似の結果は、免疫ウサギのリンパ球または脾臓細胞由来のコンビナトリアル免疫グロブリンライブラリーを構築し、ピキア・パストリス(P. pastoris)でscFv構築物を発現させることによって達成することができる。例えば、Ridder et al., Biotechnology, 13: 255-260 (1995)を参照されたい。さらに、適当なscFvを単離した後、結合親和性が高くおよび解離率の低い抗体フラグメントが、CDR3変異誘発および鎖のシャッフリングなどの親和性成熟プロセスによって得られる。例えば、Jackson et al., Br. J. Cancer, 78: 181-188 (1998); Osbourn et al., Immunotechnology, 2: 181-196 (1996)を参照されたい。
【0076】
種々の組換え法を用いて二重特異性抗体および抗体フラグメントを作出できる。例えば、二重特異性抗体および抗体フラグメントをトランスジェニック家畜の乳汁中で産生できる。例えば、Colman, A., Biochem. Soc. Symp., 63: 141-147, 1998;米国特許第5,827,690号明細書を参照されたい。対をなす免疫グロブリンH鎖およびL鎖をコードするDNAセグメントをそれぞれ含む二つのDNA構築物を調製する。このフラグメントを、哺乳類上皮細胞中で優先的に発現されるプロモーター配列を含む発現ベクターへクローニングする。例としては、限定されるものではないが、ウサギ、ウシおよびヒツジのカゼイン遺伝子、ウシα−ラクトグロブリン遺伝子、ヒツジβ−ラクトグロブリン遺伝子、およびマウスホエー酸タンパク質遺伝子由来のプロモーターが挙げられる。好ましくは、挿入されたフラグメントの3’部位に哺乳類特異的遺伝子由来の同族のゲノム配列が隣接する。これによりポリアデニル化部位および転写安定化配列が提供される。この発現カセットを受精した哺乳類の卵子の前核に同時注入し、次いでレシピエント雌の子宮に着床させて妊娠させる。出産後、双方のトランス遺伝子の存在についてサザン分析でその後代をスクリーニングする。抗体が存在するためには、H鎖およびL鎖遺伝子双方が同時に同じ細胞中で発現されなければならない。トランスジェニック雌動物から得た乳汁を当技術分野で公知の標準的免疫学的方法により、該抗体または抗体フラグメントの存在と機能性を分析する。この抗体は当技術分野で公知の標準的方法により、乳汁から精製することができる。
【0077】
キメラAbは、マウスL鎖可変およびH鎖可変ドメインをコードするcDNAフラグメントをヒト抗体由来Cドメインをコードするフラグメントに連結することによって構築される。Cドメインは抗原結合を導かないため、キメラ抗体は元のマウスAbと同じ抗原特異性を保持するが、配列でヒト抗体により近くなる。キメラAbはなおマウス配列を一部含み、また一方、依然として免疫原性である可能性がある。ヒト化Abは、抗体を認識するのに必要なマウスアミノ酸のみを含む。この産物はヒト抗体枠組み構造中へマウス相補性決定領域由来のアミノ酸を形成することによって構築される。
【0078】
VII.変異二重特異性抗体の設計および発現のための一般法
種々の変異誘発技術を用いて本発明による変異bsAbを構築してよい。当業者であればこのような技術に精通しているであろう。例えば、変異bsAbの発現ベクターは、変異したHCフラグメントを構築し、このフラグメントを、親bsAbの発現ベクターへサブクローニングして対応する野生型フラグメントを置換し、宿主細胞にベクターをトランスフェクトして得てもよい。
【0079】
親bsAbの発現ベクターを得るために、当業者は容易に利用できる技術を用いることができる。このような技術のうちのいくつかが、引用することによりその全開示内容が本明細書の一部とされる1999年6月22日出願の米国出願番号09/337,756に開示されている。要するに、hMN14IgG−(734 scFv)2などの親bsAbの発現ベクターを構築するため、単鎖734Fv(734scFv)をコードする遺伝子セグメントを構築すればよい。この734scFvセグメントは、短く柔軟なリンカー(sL)(Coloma & Morrison 1997 p.787/id)をコードするDNAフラグメントを介してヒトγ鎖遺伝子の3’末端と結合してよく、その結果CH1−ヒンジ−CH2−CH3−sL−734scFv(CH−scFv)の融合遺伝子配列となる。このCH−scFv融合遺伝子セグメントは、次に同じくhMN−14 L鎖遺伝子セグメントを含む発現ベクターhMN14pdHL2中で、hMN−14 VHの配列、ならびにトランスフェクタントの選択のため、またその結果生じたトランスフェクトされた配列の増幅のためのdhfr遺伝子と結合させることができる(Dorai & Moore 1987 p. 815/id and Gillies, Lo et al. 1989 p. 131/id)。hMN14IgG−(734scFv)2(bsAb2pdHL2)をコードするベクターは、融合bsAbの発現のためのSp2/0骨髄腫細胞へトランスフェクトすればよい。bsAb、hMN14IgG−(734scFv)2は、アフィニティークロマトグラフィーによって培養上清から精製し、SDS−PAGEによって分析することができる。親または変異bsAb内の異なる結合部分の免疫反応性を評価するため、競合ELISA結合アッセイを行ってもよい。
【0080】
その他の特異性および個別の変異bsAbを有するIgG−scFvのbsAbは、IgGおよび/またはscFvの可変領域配列だけを、その他のAbのもので置換することによって作製することができる。CDRグラフト化変異bsAbは、IgGまたはscFvの可変領域配列だけをCDRグラフト化Abで置換することによって作出することができる。一般に、この「CDRグラフト化」技術は、マウスCDR、ヒト可変領域枠組み構造およびヒト定常領域からなる組換え、医薬抗体の作製に適用されてきた(例えばRiechmann, L. et al, (1988) Nature, 332, 323-327)。このような「再構成」または「ヒト化」抗体はキメラ抗体よりもマウスの含有部分が少なく、ヒトFc依存性エフェクター機能の刺激に必要なヒト定常領域を保持する。その結果、CDRグラフト化抗体は、ヒトへ投与した時にキメラ抗体よりもHAMA反応を惹起する可能性は低く、循環系中のそれらの半減期はヒト自然抗体のものに近づくはずであり、それらの診断および治療価値は向上する。
【0081】
実際には、元のマウス抗体の特異性を保持している有効なヒト化抗体を作製するためには、単にCDRを置換するだけでは十分でないことが普通である。さらに、少数の重要なマウス抗体残基をヒト可変領域中へ組み込む必要がある。このような残基の同定は、元のマウス抗体およびアクセプターであるヒト抗体の双方の構造に依存する。英国特許出願番号9019812.8(引用することによりその全開示内容を本明細書の一部とする)は、有効な抗原結合を促進するのに十分な外来残基の置換の最低数を同定する方法を開示している。本発明の一つの実施態様では、変異融合タンパク質のFvおよびscFvはCDRグラフト化マウスFvおよびscFvである。本発明の別の実施態様では、変異融合タンパク質のFvおよびscFvはヒト化されている。一つの実施態様では、Fvは734scFvに由来するものであり、scFvは734scFvである。本発明の好ましい実施態様では、変異融合タンパク質はhMN−14IgGI253A−(734scFv)2である。
【0082】
VIII.変異bsAbの投与方法
本発明は、米国特許第6,126,916号明細書;同第6,077,499号明細書;同第6,010,680号明細書;同第5,776,095号明細書;同第5,776,094号明細書;同第5,776,093号明細書;同第5,772,981号明細書;同第5,753,206号明細書;同第5,746,996号明細書;同第5,697,902号明細書;同第5,328,679号明細書;同第5,128,119号明細書;同第5,101,827号明細書;および同第4,735,210号明細書に記載の方法を用いて、正常組織および器官の処置および/またはイメージングにおける、本発明による二重特異性抗体および標的化可能な構築物の使用を意図する。さらなる方法は1999年6月22日出願の米国出願番号09/337,756、および2001年4月3日出願の米国出願番号09/823,746に記載されている。本明細書において「組織」とは、限定されるものではないが、卵巣、胸腺、副甲状腺または脾臓由来の組織をはじめとする組織をさす。本発明による変異bsAbで治療可能な疾患および状態の例としては、免疫調節障害、自己免疫疾患、臓器移植拒絶症または移植片対宿主病が挙げられる。B細胞を標的化する抗体を用いる自己免疫疾患の免疫療法はWO00/74718mに記載されており、このクレームは米国仮出願番号60/138,284号に優先するものである(引用することによりその全内容を本明細書の一部とする)。自己免疫疾患の例としては、急性特発性血小板減少性紫斑病、慢性特発性血小板減少性紫斑病、皮膚筋炎、シドナム舞踏病、重症筋無力症、全身性紅斑性狼瘡、狼瘡腎炎、リウマチ熱、多腺性症候群、水疱性類天瘡、真性糖尿病、ヘノッホ−シェーンライン紫斑病、溶血性連鎖球菌感染後腎炎、結節性紅斑らい、高安動脈炎、アジソン病、慢性関節リウマチ、多発性硬化症、類肉腫症、潰瘍性大腸炎、多形性紅斑、IgA腎症、結節性多発性動脈炎、強直性脊椎炎、グッドパスチャー症候群、閉塞性血栓血管炎、シェーグレン症候群、原発性胆汁性肝硬変、橋本甲状腺炎、甲状腺中毒症、強皮症、慢性活動性肝炎、多発性筋炎/皮膚筋炎、多発性軟骨炎、尋常性天疱瘡、ウェジナー肉芽腫、膜性腎症、筋萎縮性側索硬化症、脊髄ろう、巨細胞動脈炎/多筋痛、悪性貧血、急速進行性糸球体腎炎、および繊維性肺胞炎がある。
【0083】
本発明による変異bsAbは、一次ターゲッティング種としてプレターゲッティング法に使用してもよい。プレターゲッティング法では、変異bsAbを投与する。一次ターゲッティング種が十分に付着したら、標的化可能な構築物を投与する。標的化可能な構築物は、一次ターゲッティング種および診断剤または治療剤の利用可能な結合部位を認識する結合部位を含んでなる。標的化可能な構築物の例は上述したとおりである。試薬の用量およびタイミングは、当業者には容易に決定できるものであり、用いる試薬の特有の性質に依存する。プレターゲッティング法はクリアランス剤を用いて行っても、用いずに行ってもよい。
【0084】
bsAbが罹患組織に対して標的化するのに十分な時間が経過したら、診断剤を投与する。診断剤の投与の後、イメージングを行うことができる。腫瘍は、適当な波長光が送達される種々の構造を直接または間接に調べることによって体腔中の腫瘍を検出することができ、その後回収できる。体のどの部位の病巣であっても、非電離放射線が送達され、それらの構造から再捕捉される限り、調べることができる。例えば、高解像度、非侵襲性のイメージング技術であるPETをヒト疾病の視覚化のために本発明による抗体とともに使用できる。PETでは、F−18を陽電子放射体として用いる場合、陽電子崩壊減衰中に生じる511keVのγ光子が検出される。
【0085】
本発明は、一般に25〜600keVのγ粒子および/または陽電子を放射する診断剤の使用を意図する。このような薬剤の例としては、限定されるものではないが、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154〜158Gdおよび175Luが挙げられる。
【0086】
術中/内視鏡プローブを用いる検出も、本発明による変異bsAbおよびI−125標識されたペプチドである標的化可能な構築物を利用する方法において意図される。このような方法は、引用することによりその全開示内容が本明細書の一部とされる米国特許第5,716,595号明細書および同第6,096,289号明細書に開示されている。
【0087】
本変異bsAbは、米国特許第6,096,289号明細書;同第4,331,647号明細書;同第4,818,709号明細書;同第4,348,376号明細書;同第4,361,544号明細書;同第4,444,744号明細書;同第5,851,527号明細書に論じられている光線力学療法(PDT)に用いることができる。
【0088】
PDTでは、感光剤、例えばジヘマトポルフィリンエーテルなどのヘマトポルフィリン誘導体を被験体へ投与する。抗腫瘍活性は、例えば630nmの光を用いて惹起する。膜が日光によって増感しない、長い波長に有用なものをはじめとする代わりの感光剤が利用できる。このような感光剤の例としては、限定されるものではないが、ベンゾポルフィリン一酸環A(BPD−MA)、錫エチオプルプリン(SnET2)、スルホン化アルミニウムフタロシアニン(AlSPc)およびルテチウムタキサフィリン(Lutex)が挙げられる。
【0089】
さらに、PDTでは、診断剤を例えば全身に注射し、レーザー誘起蛍光を用いて内視鏡により光活性化剤の付着している癌の部位を検出する。例えば、これは初期肺腫瘍の蛍光気管支鏡検査に適用されている。Doiron et al. Chest 76:32 (1979)。別の例では、抗体および抗体フラグメントを単光子放出に用いることができる。例えば、Tc−99m標識した診断剤を、本発明による抗体または抗体フラグメントの投与の後に被験体へ投与してよい。その後被験体を、単光子放出コンピュータ断層撮影画像を生じるγ線カメラでスキャンし、病巣または腫瘍部位を決定する。
【0090】
治療上有用な免疫複合体は、光活性薬または色素を抗体複合体と結合させることにより得ることができる。蛍光またはその他の色素体、あるいはポルフィリンなどの色素は可視光に感受性であり、好適な光線を病巣に向けることにより病巣の検出および治療に用いられてきた。治療では、これは光照射、光療法、または光線力学療法と呼ばれている(Jori et al. (eds.), Photodynamic Therapy of Tumors and Other Diseases (Libreria Progetto 1985); van den Bergh, Chem. Britain 22:430 (1986))。さらに、光療法を達するためには、モノクローナル抗体を光活性色素と結合させる(Mew et al., J. Immunol. 130:1473 (1983); idem., Cancer Res. 45:4380 (1985); Oseroff et al., Proc. Natl. Acad. Sci. USA 83:8744 (1986); idem., Photochem. Photobiol. 46:83 (1987); Hasan et al., Prog. Clin. Biol. Res. 288:471 (1989); Tatsuta et al., Lasers Surg. Med. 9:422 (1989); Pelegrin et al., Cancer 67:2529 (1991))。しかし、これらの初期の研究には、内視鏡での治療、特に抗体フラグメントまたはサブフラグメントを用いるものの使用は含まれていない。従って、本発明は、光活性薬または色素を含んでなる免疫複合体の使用を意図する。
【0091】
リンカー部分はまた、標的部位のプロドラッグを活性化できる、または身体の解毒経路を制御することにより通常の治療の効果を改善することのできる酵素と結合させてもよい。bsAbの投与に続いて、リンカー部分と結合した酵素、bsAbの2番目のアーム(scFv成分)に認識される分子量の低いハプテンを投与する。酵素を標的部位へプレターゲッティングした後、標的部位で作用することが知られている細胞傷害性の薬物を注入する。この薬物は哺乳類の通常の解毒プロセスにより解毒されるものであればよい。例えば、この薬物は、肝臓毒性の低いグルクロニドに変換され得る。解毒された中間体は、次に、標的部位でプレターゲッティングされた酵素によってより有毒な形態へ再変換することができる。あるいは、投与されたプロドラッグを、プレターゲッティングされた酵素により有効な薬物へと変換することもできる。プレターゲッティングされた酵素は、解毒された薬物を再利用することにより治療の効果を向上させる。この手法は、いずれの酵素−薬物対を用いる場合にも適用することができる。
【0092】
抗癌治療に有用なある種の細胞傷害性薬物は、比較的血清に不溶性である。結合されない形態では非常に有毒なものも中にはあるが、プロドラッグへ変換することによりその毒性は相当に低減される。難溶性の薬物をより可溶性の複合体、例えばグルクロニド、親水性酸とのエステルまたは親水性アミンとのアミドへ変換すると、その薬物の血清の水性相での可溶性、ならびに静脈、動脈、または細胞壁毛細官を通過し、腫瘍を浸している間質液まで到達する能力が改善される。プロドラッグの開裂により、可溶性の低い薬物が標的部位に置かれる。このようなプロドラッグから薬物への変換の多くの例が、Hansen米国特許第5,851,527号明細書に開示されている。
【0093】
肝臓で芳香族または脂環式アルコール、チオール、フェノールおよびアミンなどのある種の有毒物質がグルクロニドへ変換されることは、それらを解毒し、より尿中に排泄しやすくする身体の手段である。このような物質へ変換できるある種の抗腫瘍薬は、エピルビシン、ドキソルビシンの4−エピマー(アドリアマイシン)であり、これはアントラサイクリン配糖体でヒトβ−D−グルクロニダーゼの基質であることが示されている。例えば、Arcamone Cancer Res. 45:5995 (1985)を参照されたい。極性基のより少ないその他の類似体は、より脂肪親和性であると考えられ、このような手法に大いに有望である。芳香族または脂環式アルコール、チオールまたはアミン基を含むその他の薬物または毒素は、このような複合体形成のための候補である。このような薬物、またはその他のそのプロドラッグ形態は、本発明による部位特異的増強法に好適な候補である。
【0094】
プロドラッグCPT−11(イリノテカン)はin vivoでカルボキシルエステラーゼによって活性代謝物SN−38に変換される。従って、本発明の一つの適用例は、腫瘍およびハプテン(例えばdi−DTPA)に対して標的化したbsAbを用い、その後にdi−DTPA−カルボキシルエステラーゼ複合体を注射することである。好適な腫瘍対バックグラウンド局在比に達したならば、CPT−11を投与する。腫瘍局在性カルボキシルエステラーゼは、腫瘍でCPT−11をSN−38へ変換するのに役立つ。可溶性が低いために、活性のあるSN−38は腫瘍近傍に残ったままとなり、その結果、標的とされる抗原について陰性の隣接する腫瘍細胞へ効果を発揮する。これは本方法のさらなる利点である。カルボキシルエステラーゼの変更形態は既に文献に記載されており、本発明の範囲内にある。例えば、Potter et al., Cancer Res. 58:2646-2651 (1998)およびPotter et al., Cancer Res. 58:3627-3632 (1998)を参照されたい。
【0095】
エトポシドは、そのグルクロニド形成によって大部分が解毒される、広く用いられる制癌剤であり、本発明の範囲内にある。例えば、Hande et al. Cancer Res. 48:1829-1834 (1988)を参照されたい。グルクロニド複合体は、細胞傷害性薬物から製造でき、mAb−グルクロニダーゼ複合体を用いてプレターゲッティングした腫瘍の治療時に注入できる。例えば、Wang et al. Cancer Res. 52:4484-4491 (1992)を参照されたい。従って、このような複合体も本明細書に記載のプレターゲッティング手法とともに使用できる。同様に、ダウノマイシンおよびドキソルビシンの誘導体に基づいて設計されたプロドラッグは、カルボキシルエステラーゼおよびグルクロニダーゼとの併用について記載されている。例えば、Bakina et al. J. Med Chem. 40:4013-4018 (1997)を参照されたい。本発明の範囲内で使用できるプロドラッグ/酵素対のその他の例としては、限定されるものではないが、フェノールマスタードおよびβ−グルクロニダーゼのヒドロキシ誘導体のグルクロニドプロドラッグ;フェノールマスタードまたはCPT−11およびカルボキシペプチダーゼ;メトトレキサート置換α−アミノ酸およびカルボキシペプチダーゼA;6−メルカプトプリンおよびドキソルビシンなどの薬物のペニシリンまたはセファロスポリン複合体およびβ−ラクタマーゼ;リン酸エトポシドおよびアルカリホスファターゼが挙げられる。
【0096】
あるいは、標的部位でプロドラッグを活性化させるか、または身体の解毒経路を制御することにより通常の治療の効力を向上させる能力のある酵素は、ハプテンと結合させてもよい。プレターゲッティングbsAbの投与の後に酵素−ハプテン複合体を被験体へ投与し、標的部位に向ける。酵素が標的部位に局在した後、標的部位で作用することが知られている細胞傷害性薬物、またはin situでプレターゲッティングされた酵素によって薬物へ変換されるそのプロドラッグ形態を注射する。上記で論じたように、この薬物は、哺乳類の通常の解毒プロセスを用いて、解毒されて毒性の低い中間体、最も一般的にはグルクロニドを形成する。解毒された中間体、例えばグルクロニドは、プレターゲッティングされた酵素によってそのより有毒な形態へ再変換され、従って標的部位での細胞傷害性が高くなる。この結果、薬物が再利用される。同様に、投与したプロドラッグを通常の生物学的プロセスによって有効な薬物へ変換できる。プレターゲッティングされた酵素は、解毒された薬物を再利用することにより治療の効力を高める。このアプローチは、どの酵素−薬物対を用いる場合にも適用できる。
【0097】
本発明はさらに、ホウ素中性子捕捉療法(Boron Neutron Capture Therapy, BNCT)プロトコールに照らした、本発明によるbsAbおよび診断剤の使用を意図する。BNCTは、腫瘍に局在する10B原子の中性子照射によって電離放射線を腫瘍細胞へ送達するように設計された二元システムである。BNCTは、安定したイソ型である同位体標識10B(自然界には19.8%の割合で存在)が熱中性子照射されてα粒子および7Li原子核を生じる際に起こる核反応に基づく。これらの粒子の飛程は約1細胞径であり、結果として線形の高エネルギー転移を生じる。この核反応で生じる飛程の短い1.7MeVのα粒子がほんのわずかあれば、細胞核を標的とし、それを破壊するのに十分である。BNCTで癌治療を成功させるには高濃度の10Bを腫瘍部位に局在させるが、標的でない器官は基本的にホウ素を含まないままにしておく方法が必要である。BNCTのためのプレターゲッティングbsAbを用いて被験体内で腫瘍を治療する組成物および方法は、同時係属の特許出願第09/205,243号に記載され、本発明の目的のために容易に修正できる。
【0098】
本発明による変異bsAbのscFv成分もまた、酵素に特異的なものとすることができる点に留意すべきである。
【0099】
変異bsAbの投与と標的化可能な構築物の投与との間で加えるクリアランス剤を用いてもよい。本発明者らは新規な機構作用を有するクリアランス剤、つまりbsAbの疾病ターゲッティングアームに標的化されたグリコシル化抗イディオタイプのFab’フラグメントを本発明に用い得ることを見出した。抗−CEA(MN14Ab)x抗−ペプチドbsAbを得、疾病標的に最大限まで付着させる。残余bsAbを除去するため、WI2と称する、MN−14に対する抗イディオタイプAbを、好ましくはグリコシル化Fab’フラグメントとして得る。クリアランス剤は一価の様式でbsAbと結合するが、それに付属するグリコシル残基は全複合体を急速な代謝が起こる肝臓へ向ける。その後、リンカー部分に関連する治療が患者に対して行われる。bsAbのMN−14アームに対するWI2 Abは高親和性で、クリアランス機構はその他の開示される機構とは異なる(Goodwin et al.同上参照)。それは、WI2−Fab’が一価の部分であるため、架橋を伴わないからである。
【0100】
本変異bsAbはまた、超音波イメージング法でも使用できる。造影剤などの超音波増強剤は、二価のDTPAペプチドなどの標的化可能な構築物と結合してもよい。限定されるものではないが、リポソーム、好ましくはガス充填したリポソームなどの増強剤を用いてよい。この方法では、変異bsAbが最初に投与され、その後リポソーム−標的化可能な構築物複合体が投与される。Maresca, G. et al., Eur J. Radiol. Suppl. 2 S171-178 (1998); Demos, Sm. Et al. J. Drug Target 5 507-518 (1998);およびUnger, E. et al., Am J. Cardiol. 81 58G-61G (1998)を参照されたい。
【0101】
変異二重特異性抗体は、多成分治療法の一成分として投与してよい。変異二重特異性抗体は、疾病または症状を治療するために用いられる少なくとも1種類の治療剤の投与前、投与中、または投与後に投与してよい。
【0102】
プレターゲッティング法における変異bsAbの使用例は、プレターゲッティング法における親bsAbの使用との比較において、実施例2に説明されている。このデータにより、親bsAbと比較した、本発明による変異bsAbのクリアランス速度の上昇が説明される。さらに、該データにより、変異bsAbが用いられる場合と比較して、親bsAbが用いられる場合に、より大量の標的化可能な構築物が血中で捕捉されることが説明される。
【0103】
図5および6は、親bsAb、125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング法のデータを示す。図7は、変異bsAb、125I−hMN−14IgGI253A−(734scFv)2に関するプレターゲッティング法のデータを示す。125I標識によって、身体の異なる領域に存在するbsAbの量を決定することができる。図5および7のデータ比較は、変異bsAbが親bsAbよりも速く身体から消失することを示す。例えば、親bsAbで4日間のプレターゲッティング後(図5)、およびIMP−192の注射後3時間の、腫瘍および血液の%ID/gは、それぞれ19.21±7.318および3.73±0.75であった。これに対して、変異bsAbで4日間のプレターゲッティング後(図5)、およびIMP−192の注射後3時間の、腫瘍および血液の%ID/gは、それぞれ2.42±0.78および0.07±0.01であった。
【0104】
図5および7の125Iの腫瘍対血液比の比較(図5および7の「血液」の欄参照)によれば、変異bsAbに関して高いシグナル対バックグラウンド比が達成されたことが実証される。親bsAbでのプレターゲッティングの6日後でさえ(図4参照)、腫瘍対血液比は、変異bsAbでのプレターゲッティングの4日後よりもはるかに低い。
【0105】
99mTc標識によって、身体の異なる領域に存在する標的化可能な構築物の量を決定することができる。IMP−192(99mTc標識した標的化可能な構築物)の%ID/gの比較は、腫瘍対血液比が変異bsAbを用いるプレターゲッティング法に関してはるかに高いことを示す。この結果は、変異bsAbに関するプレターゲッティング法において血中で捕捉された標的化可能な構築物のほうが少ないことを説明する。親bsAbを用いた場合(図5および6参照)、99mTc標識した標的化可能な構築物は、腫瘍部位に現れるよりもむしろ血中で捕捉される。したがって、低い腫瘍対血液比が認められる。例えば、99mTc標識した標的化可能な構築物の腫瘍対血液比は、図5(親bsAb)の左側、「血液」の欄に示される。注射後3時間の腫瘍対血液比は0.24±0.05である。それに対して、図5(変異bsAb)では注射後3時間の腫瘍対血液比は3.52±1.45である。
【0106】
IX.その他の適用例
本発明は、米国特許第5,716,595号明細書および同第6,096,289号明細書に記載されているように、上記に論じた変異bsAbおよびリンカー部分と結合した治療剤の手術中、血管内での使用、ならびに内視鏡による腫瘍および病巣の検出、生検および治療を包含する。
【0107】
本発明による変異bsAbは、治療またはイメージング目的だけでなく、in vitroで研究を行う際の補助としても用いることができる。例えば、本発明によるbsAbをin vitroで用いることにより、標的化可能な構築物が1以上のbsAbを含む安定した複合体を形成できるかどうかを確かめることができる。このようなアッセイは、bsAbを含む安定した複合体を形成する標的化可能な構築物の同定の際に当業者の助けとなる。これによって、次に当業者は治療剤および/またはイメージング剤として優れていると思われる標的化可能な構築物を同定することができる。
【0108】
アッセイは、当該の標的化可能な構築物と少なくとも2モル当量の変異bsAbとを組み合わせて行うのが有利である。インキュベーションの後、混合物をサイズ排除HPLCで分析して該構築物がbsAbと結合しているかどうかを判断する。あるいは、種々のbsAb溶液を標準的な96ウェルプレートへ置くという、標準的なコンビナトリアル(組み合わせ)法を用いてアッセイを行う。各ウェルには、標的化可能な構築物の溶液を加える。インキュベーションおよび分析を受けて、どの構築物がどのbsAbと最も良く結合するかが容易に判断できる。
【0109】
当然のことながら、変異bsAbの標的化可能な構築物への添加順序は重要ではない。つまり、変異bsAbを構築物へ加えてよく、逆もまた同じである。同様に、変異bsAbも構築物も溶液中にある必要はない。つまり、それらは溶液中で加えてもそのまま加えてもよく、そのうち最も便宜なものであってよい。最後に、結合が確立さえすれば、結合の分析方法は重大ではない。よって、限定されるものではないが、FABMS、高磁界NMR、またはサイズ排除HPLCと併せた、またはその代わりとなるその他の適当な方法をはじめとする標準的分析法を用いて結合を分析してよい。
【実施例】
【0110】
材料および方法
734scFvの設計および構築
734scFvを、sL−Vλ−L−VHの配置を有するよう設計した。式中、sLは短く柔軟なリンカー、Gly−Gly−Gly−Ser(Coloma & Morrison, Nat. Biotechnol. 15:159-163 (1997))であり、hMN−14IgG H鎖と734scFvとの間の結合の役割を果たし、Lは3反復のGly−Gly−Gly−Gly−Ser(Huston, Levinson, et al. PNAS 85:5879-5883 (1988))からなる734のVλおよびVHとの間の長いリンカーである。プライマー対734VLscFv5’(Cys)/734VLscFv3’および734VHscFv5’/734VHscFv3’(SacI)を用いて734のそれぞれのVλおよびVH配列を増幅した。得られたDNA産物を、制限酵素消化および連結によって734scFv遺伝子へと組み立て、DNAシーケンシングによって配列を確認した。
734VLscFv5’(Cys):5’-TT CTC TCT GCA GAG CCC AAA TCT TGT GGT GGC GGT TCA CAG CTG GTT GTG ACT CAG-3’;
734VLscFv3’:5’-A GCC TCC GCC TCC TGA TCC GCC ACC TCC TAA GAT CTT CAG TTT GGT TCC-3’;
734VHscFv5’:5’-CC GGA GGC GGT GGG AGT GAG GTG AAA CTG CAG GAG-3’;
734VHscFv3’(SacI):5’-AA CCT TGA GCT CGG CCG TCG CAC TCA TGA GGA GAC GGT GAC CG-3’。
【0111】
hMN−14IgG−(734scFv)2の発現ベクターの構築
734scFvをヒトH鎖定常領域(HC)のC末端へ結合させるため、新規なプライマー対、734scFv2−5’および734scFv−3’を合成し、734scFvをコードするDNAの増幅に用いた。プライマー734scFv2−5’は、734scFvとヒトHCのC末端とのフレーム内結合のための正確な配列をもたらした。得られたDNAフラグメントをヒトHC配列と連結し、HC−734scFvをコードする構築物を形成した。次にhMN−14の発現ベクター、hMN−14pdHL2中の正常ヒトHCをコードするDNAフラグメントを、HC−734scFvフラグメントで置換し、その結果、融合構築体の発現ベクターhMN−14IgG−(734scFv)2pdHL2を得た。
734scFv2-5’:5’-TCC CCG GGT AAA GGT GGC GGT TCA CAG CTG-3’;
734scFv-3’:5’-GAG CTC GGC CGT CGC AC-3’。
【0112】
変異融合bsAb、hMN−14IgG(I253A)−(734scFv)2の構築
イソロイシン253をヒトHC鎖のCH2ドメインに置いた。I253A変異をhMN−14IgG−(734scFv)2に導入するため、CH1および部分的にCH2ドメインをコードする挿入DNAフラグメントを含むプラスミドベクターCH1kbpKSを、オリゴヌクレオチドに誘導される部位特異的変異誘発に用いた。オリゴヌクレオチドI253ACH2は、CH2中の野生型配列KDTLM253ISRTPEをKDTLM253ASRTPEへ変換するが、変異誘発プライマーとして設計および合成されている。変異誘発は、Sculptor IVM system(Amersham, Arlington Heights, IL)を製造業者の仕様書に従って用いることにより達成された。ジデオキシDNAシーケンシングによって配列が検証された後、変異したHCフラグメントをhMN−14IgG−(734scFv)2pdHL2へサブクローニングして対応する野生型フラグメントを置換し、その結果、変異融合bsAbの発現ベクター、hMN−14IgG(I253A)−(734scFv)2pdHL2を得た。
I253ACH2:5’-AAG GAC ACC CTC ATG GCT AGC CGG ACC CCT GAG-3’。
【0113】
bsAbの発現および産生
発現ベクターを、〜30μgのSalIで線状化したDNAを用いて2〜5×106個の細胞がトランスフェクトされるエレクトロポレーションによってSp2/0細胞へトランスフェクトし、96ウェル細胞培養プレートにプレーティングした。2日後、最終濃度0.025〜0.075μMのメトトレキサート(MTX)をトランスフェクタントの選択のため細胞培養培地に加えた。MTX耐性コロニーが2〜3週で出現し、これをELISAでヒトIgGの分泌についてスクリーニングした。要するに、残存コロニー由来の細胞培養上清を、ヤギ抗ヒトIgG F(ab’)2特異的抗体をコーティングしたELISAプレートのマイクロウェル中で1時間インキュベートした。次にペルオキシダーゼ結合ヤギ抗ヒトIgG Fcフラグメント特異的抗体を加え、ウェル中で1時間インキュベートした。上清中のヒトIgGの存在は、o−フェニレンジアミン二塩酸塩0.4mg/mlおよびH2O2 0.0125%を含む基質溶液を添加することにより明らかにした。陽性クローンから最もAbを産生するものを決定し、選択し、さらに拡張した。hMN−14IgG−(734scFv)2およびhMN−14IgG(I253A)−(734scFv)2を、タンパク質AまたはDTPAカラムのいずれかでのアフィニティークロマトグラフィーにより細胞培養上清から精製した。
【0114】
Ac−Lys(DTPA)−Tyr−Lys(DTPA)−Lys(TscG−Cys−)−NH2 (IMP192)の合成:
第一のアミノ酸、Aloc−Lys(Fmoc)−OHをペプチドシンセサイザー上でRinkアミド樹脂0.21mmolへ付着させ、その後、標準のFmoc自動合成プロトコールを用いてTc−99mリガンド結合残基Fmoc−Cys(Trt)−OHおよびTscGをリシンの側鎖へ添加して次のペプチド:Aloc−Lys(TscG−Cys(Trt)−rink樹脂を形成した。次に、CH2Cl2 10mL、氷酢酸0.75mLおよびジイソプロピルエチルアミン2.5ml中に溶解したPd[P(Ph)3]4 100mgを含む溶液8mLで樹脂を処理してAloc基を取り除いた。次に樹脂混合物をトリブチルスズヒドリド0.8mlで処理し、60分間ボルテックスで混合した。次にペプチド合成をシンセサイザーで継続して次のペプチド:Lys(Aloc)−Tyr−Lys(Aloc)−Lys(TscG−Cys−)−rink樹脂を製造した。樹脂をDMF 10mL、無水酢酸3mL、およびジイソプロピルエチルアミン6mLを含む溶液8mLで60分間ボルテックスで混合することによりN末端をアセチル化した。次にAloc保護基の側鎖を上記の通り除去し、標準のFmoc脱保護プロトコールを用いて樹脂をピペリジンで処理して樹脂に残っている可能性のある酢酸を除去した。活性化DTPAおよびDTPAの添加:DTPA5gをメタノール中1.0Mテトラブチル水酸化アンモニウム40mLに溶解した。このメタノールを高圧下で除去して粘性の油状物質を得た。この油状物質をDMF50mLに溶解し、揮発性の溶媒をロータリーエバポレーターで高圧下で除去した。DMFによる処理をさらに2回繰り返した。次に粘性の油状物質をDMF50mlに溶解し、HBTU5gと混合した。次に活性化DTPA溶液のアリコート8mlを14時間ボルテックスで混合した樹脂に加えた。このDTPAによる処理は、カイザー試験を用いて樹脂がアミンに検査陰性となるまで繰り返した。
【0115】
切断および精製:次にペプチドを、TFA30ml、トリイソプロピルシラン1ml、およびエタンジチオール1mlからなる溶液8mlで60分間(mm→min?)処理することによって樹脂から切断した。粗切断ペプチドをエーテル30mlを注入して沈殿させ、遠心分離により回収した。次にこのペプチドを4×30cm Waters分取C−18 Delta−Pakカラム(15μm、100Å)を用いる逆相HPLCによって精製した。HPLC画分を回収し、凍結乾燥させてESMS(MH±1590)により所望の産物を含む画分を得た。
【0116】
キットの作成:このペプチドをペプチド78μg、非放射性InCl3 0.92mg、塩化第一錫100μg、ゲンチシン酸3mg、およびHPCD(再構成時の10%)を含む凍結乾燥キットを構築した。
【0117】
放射性ラベリング
抗体タンパク質60μgをクロラミン−T法(Greenwood, Hunter, et al., Biochem. J. 89 11-123 (1963))を用いてI−125で標識し、NAP−5脱塩カラム(Pharmacia, Piscataway, NJ)を用いて精製した。
【0118】
Tc−99m標識したIMP−192を準備するため、IMP−192 50μgを含むキットを20mCi過酸化テクネチウムを含む生理食塩水1.5mlで再構成した。再構成したキットを室温で10分間インキュベートし、次に沸騰水浴中で15分間加熱した。
【0119】
実施例1:ヒト結腸腫瘍を有するマウスでの125I−hMN−14IgGI253A−(734scFv)2および125I−hMN−14IgG−(734scFv)2の体内分布
実験の手順
125I−hMN−14IgG−(734scFv)2および125I−hMN−14IgGI253A−(734scFv)2の簡単な体内分布パターンを評価した。GW39ヒト結腸癌異種移植片を有する雌ヌードマウス群へ、20μg(5μCi)/マウスの125I標識した親または変異bsAbを静脈注射により投与した。マウスを注射後の計画した時点で安楽死させた、その器官を取り出し、重さを測り、I−125放射能を計数した。
【0120】
GW−39ヒト結腸腫瘍細胞株を、他で記載のように(Tu, et al. Tumour Biology 9:212-220 (1988))ヌードマウス中で連続的な皮下異種移植片として増殖させた。
【0121】
結果
125I標識したhMN−14IgG−(734scFv)2およびhMN−14IgGI253A(734scFv)2変異体の腫瘍組織および正常組織での体内分布を、注射後1、2、3および4日のヒト結腸腫瘍を有するマウスで検査した。結果を図3および4に示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)として表される。
【0122】
hMN−14IgGI253A(734scFv)2の腫瘍への取り込みは、hMN−14IgG−(734scFv)2よりも有意に低かった。hMN−14IgGI253A(734scFv)2のクリアランス速度の増加は、肝臓、脾臓、腎臓、肺、胃、小腸、大腸および血液などの正常な組織でも見られた。図3および4参照。hMN−14IgGI253A(734scFv)2のクリアランスの加速は、肝臓、脾臓、腎臓、肺、胃、小腸、大腸および血液などの多くの正常組織の腫瘍対器官比を高くした。さらに、hMN−14IgGI253A(734scFv)2 変異体の注射後1〜4日の腫瘍対血液比は、hMN−14IgG−(734scFv)2の腫瘍対血液比と比較して速い速度で増加した。
【0123】
実施例2:ヒト結腸腫瘍を有するマウスでの125I−hMN−14IgGI253A−(734scFv)2および125I−hMN−14IgG−(734scFv)2のプレターゲッティング
実験の手順
変異および親bsAbのプレターゲッティングの体内分布パターンを評価した。GW39ヒト結腸癌異種移植片を有する雌ヌードマウス群へ、20μg(5μCi)/マウスの125I標識した変異または親bsAbを静脈注射により投与した。変異または親bsAbの注射の後、予め定めたクリアランス時間の間、bsAbが腫瘍部位に局在し、循環から取り除かれるのを待った。99mTc標識した二価のDTPAペプチド、IMP−192を次に静脈注射した。このマウスを、ペプチド注射後の種々の時点で屠殺し、器官を取り出し、重さを測り、I−125およびTc−99m双方の放射能を計数した。
【0124】
GW−39ヒト結腸腫瘍細胞株を、他で記載のように(Tu, et al. Tumour Biology 9:212-220 (1988))ヌードマウス中で連続的な皮下異種移植片として増殖させた。
【0125】
結果
125I標識したhMN−14IgGI253A−(734scFv)2および125I標識したhMN−14IgG−(734scFv)2の腫瘍組織および正常組織での体内分布を、9mTc標識した二価のDTPAペプチド、IMP−192の注射後3、6、および24時間のヒト結腸腫瘍を有するマウスで検査した。IMP−192の注射の前に、変異または親bsAbでのプレターゲッティングを4日間行なった。125I標識した変異および親bsAbの腫瘍組織および組織での体内分布を図5〜7に示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)として表される。さらに、IMP−192(99mTc標識した二価のDTPAペプチド)の腫瘍組織および組織での体内分布を図5〜7に示す。変異bsAbのクリアランスの加速が認められる。さらに、親bsAbでのプレターゲッティングと比較して、変異bsAbでプレターゲッティングの後により高い腫瘍対血液比が認められる。変異融合タンパク質でのプレターゲッティング後よりも親融合タンパク質でのプレターゲッティング後により多くのDTPA-ペプチドが血中に捕捉されたことが注目される。
【0126】
当業者には、本発明による組成物および方法に対して種々の改変および変更をなしうることが明らかである。よって、本発明は、添付のクレームおよびそれらの均等物の範囲内に入る限り、このような改変および変更を含むものとする。
【0127】
上記に引用した全ての文献の開示は、各々が個別に引用することにより本明細書の一部とされるのと同程度に、引用することにより明白にその全開示内容を本明細書の一部とする。
【0128】
実施例3:In−DTPA含有ペプチドとhMN−14IgGI253A−(734scFv)2との結合
In−DTPAペプチドと抗In−DTPA抗体hMN−14IgG(1253A)−(734scFv)2との結合を、サイズ排除HPLCおよびBiacore Xを用いるアフィニティー遮断研究によって調査した。
【0129】
HPLCを用いる結合分析
IMP192キットをTc−99m 20.9mCiで標識した。このキットからのアリコートを希釈し、次のモル比(ペプチド/ab)1:5、1:1、および20:1でhMN−14IgG(1253A)−(734scFv)2と混合した。このペプチド/抗体混合物、ペプチド単独および抗体単独を、0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムで試験した。HPLCの痕跡(図8〜12)は本質的にただ1種類のペプチド/抗体複合体が形成されていることを示す。hMN−14IgGI253A−(734scFv)2 の既知の基準は約9.41分でカラムから溶出する(図8)。Tc−99m IMP192の既知の基準は約14.85分でカラムから溶出する(図9)。hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の1:1混合物をカラムに適用した場合、約9.56分に1回だけピークが認められた(図10)。それに対して、hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の1:5混合物をカラムに適用した場合、2回の大きなピークが認められ、1回は約9.56分(hMN−14IgGI253A−(734scFv)2)そしてもう1回は約14.80分(Tc−99m IMP192)であった(図11)。hMN−14IgGI253A−(734scFv)2対Tc−99m IMP192の20:1混合物をカラムに適用した場合、9.56分で1回だけピークが認められた(図12)。
【0130】
実施例4:臨床試験
実施例4A. 結腸ポリープ患者のポリープを除去すると、それが悪性であると分かった。CATスキャンは腫瘍を明示できなかったが、3ヵ月後、患者の血中のCEAレベルが上昇。この患者に10mgのhMN14−IgG[734−scFv]2を静脈点滴により投与する。3日後、患者に40mCiのTc−99mで標識した二価のペプチドIMP192を投与する。翌日、この患者に放射性シンチグラフィーを行い、ポリープの切除部位に近接する結節にただ一箇所つの活性部位が認められる。この結節を切除し、この患者はその後10年間罹患しなかった。
【0131】
実施例4B. 結腸癌患者の一次腫瘍を切除した。2年後、この患者は血中のCEAレベルの上昇を示し、CATスキャンによって肝臓に切除不可能な複数の小さな転移が示される。患者に100mgのhMN14−IgG[734−scFv]2を静脈点滴により投与する。3日後、患者に160mCiのI−131で標識した二価のDTPAペプチド、IMP156を静脈点滴により投与する。CEAの血中レベルは徐々に正常範囲まで降下した。CATスキャンによって転移のいくつかが消散したことが示され、残りの病巣は9ヶ月間増殖しなかった。
【0132】
当業者には、本発明による組成物および方法に対して種々の改変および変更をなしうることが明らかである。よって、本発明は、添付のクレームおよびそれらの均等物の範囲内に入る限り、このような改変および変更を含むものとする。
【0133】
上記に引用した全ての文献、特許、および特許出願の開示は、各々が個別に引用することにより本明細書の一部とされるのと同程度に、引用することにより明白にその全開示内容を本明細書の一部とする。
【図面の簡単な説明】
【0134】
【図1】hMN−14のH鎖cDNAおよびアミノ酸配列を示す。VH、CH1、ヒンジ、CH2およびCH3領域が示されている。274番のアミノ酸のイソロイシンは、Edelman, et al.のナンバリング方法によればイソロイシン253に相当する。Edelman et al. Biochemistry 63, 78-85 (1969)を参照されたい。
【図2】hMN−14のL鎖cDNAおよびアミノ酸配列を示す。VkおよびCK領域が示されている。
【図3】ヒト結腸腫瘍を有するマウスでの、注射後1、2、3および4日のhMN−14IgGI253A−(734scFv)2の体内分布を示す。「I253A」とは253番目のイソロイシンがアラニンへ変わっていることをさす。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示した。
【図4】ヒト結腸腫瘍を有するマウスでの、注射後1、2、3および4日のhMN−14IgG−(734scFv)2の体内分布を示す。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示した。
【図5】125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識された二価のDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後4日間、125I−hMN−14IgG−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc−標識された標的化可能な構築物のデータを示す。
【図6】125I−hMN−14IgG−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識されたdiDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後6日間、125I−hMN−14IgG−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc−標識された標的化可能な構築物のデータを示す。
【図7】125I−hMN−14IgGI253A−(734scFv)2に関するプレターゲッティング実験から得た体内分布データを示す。標的化可能な構築物は、Tc−99m標識されたdiDTPA、IMP−192であった。ヒト結腸腫瘍を有するマウスを、標的化可能な複合体の注入後4日間、125I−hMN−14IgGI253A−(734scFv)2でプレターゲッティングした。標的化可能な複合体の注入後3、6および24時間のデータを得た。データはグラムあたりの注射量の中央値のパーセンテージ(%ID/g)で示す。腫瘍対血液の比は「血液」の欄に示されている。表の左側は125I標識されたbsAbのデータを示し、表の右側は99mTc標識された標的化可能な構築物のデータを示す。
【図8】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2の既知の標準溶出プロフィールを示す。
【図9】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのTc−99m IMP192の既知の標準溶出プロフィールを示す。
【図10】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の1:1混合物の既知の標準溶出プロフィールを示す。
【図11】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の1:5混合物の既知の標準溶出プロフィールを示す。
【図12】0.2Mリン酸バッファーpH6.8を用いて1mL/分で溶出するBio−Sil SEC 250 300mm×7.8mm HPLCカラムでのhMN−14IgGI253A−(734scFv)2とTc−99m IMP192の20:1混合物の既知の標準溶出プロフィールを示す。
【特許請求の範囲】
【請求項1】
(a)IgG由来のヒトヒンジ定常領域、
(b)2つのscFv、および
(c)2つのFv
を少なくとも含んでなり、該定常領域がCH2ドメインに1以上のアミノ酸変異を含む、変異二重特異性抗体。
【請求項2】
前記scFvおよびFvが、CDRグラフト化マウスscFvおよびFvである、請求項1に記載の変異二重特異性抗体。
【請求項3】
前記scFvおよびFvが、ヒト化されたものまたはヒト由来のものである、請求項1に記載の変異二重特異性抗体。
【請求項4】
前記ヒンジ定常領域が、253番目のイソロイシンがアラニンで置換されているか、またはそのアミノ酸置換がアラニン置換と同等もしくはそれを超えて血液クリアランスを促進する、ロイシン以外のアミノ酸で置換されている変異を含む、請求項1に記載の変異二重特異性抗体。
【請求項5】
前記FvがhMN14−IgGに由来するものであり、前記scFvが734scFvである、請求項4に記載の変異二重特異性抗体。
【請求項6】
前記scFvが一価の標的化可能な構築物と結合する、請求項1に記載の変異二重特異性抗体。
【請求項7】
前記scFvが二価の標的化可能な構築物と結合する、請求項1に記載の変異二重特異性抗体。
【請求項8】
前記Fvが標的細胞上のエピトープと結合する、請求項1に記載の変異二重特異性抗体。
【請求項9】
疾病または疾病に至る可能性のある症状を、治療もしくは診断、または治療および診断する方法であって、
(A)Fvが前記疾病または前記症状に関連するマーカー物質に向けられている請求項1に記載の変異二重特異性抗体を被験体に投与する工程、
(B)所望により、被験体にクリアランス組成物を投与し、該組成物に非局在抗体または抗体フラグメントを循環系から除去させる工程、および
(C)二価ハプテンを含む標的化可能な構築物であって、両ハプテン部分が請求項1に記載の変異二重特異性の単一分子上の2つのscFvと結合するものであり、診断もしくは治療用カチオンおよび/または1以上のキレート化もしくは化学結合した治療剤もしくは診断剤をさらに含んでなる前記構築物を被験体に投与する工程
を含んでなる、方法。
【請求項10】
前記変異二重特異性抗体が、前記疾病または症状の治療に用いられる少なくとも1つの治療剤を投与する前、投与中、または投与した後に投与される、請求項9に記載の方法。
【請求項11】
前記標的化可能な構築物が酵素を含んでなるものであり、
a)プロドラッグ(前記酵素が標的部位で該プロドラッグを薬物へと変換することができる場合)、
b)被験体内で解毒されて毒性の低い中間体を形成することができる薬物(前記酵素が解毒された前記中間体を毒性型へと再変換し、これにより、標的部位で薬物の毒性を増強することができる場合)、または
c)被験体内で自然のプロセスを経て活性化され、毒性の低い中間体へと変換されることにより解毒されるプロドラッグ(前記酵素が解毒された前記中間体を毒性型へと再変換し、これにより、標的部位で薬物の毒性を増強することができる場合)
を被験体に投与する工程をさらに含んでなる、請求項9に記載の方法。
【請求項12】
前記プロドラッグが、エピルビシングルクロニド、CPT−11、エトポシドグルクロニド、ダウノマイシングルクロニドおよびドキソルビシングルクロニドからなる群から選択される、請求項11に記載の方法。
【請求項13】
前記診断剤が25〜4,000keVのγ粒子および/または陽電子を放射するものである、請求項9に記載の方法。
【請求項14】
前記診断剤が、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gd、177Lu、32P、188Re、および90Yまたはそれらの組み合わせからなる群から選択される、請求項9に記載の方法。
【請求項15】
前記放射性同位元素を用いて陽電子放射断層撮影法(PET)を行う、請求項9に記載の方法。
【請求項16】
前記標的化可能な構築物が、磁気共鳴イメージング(MRI)に用いる1以上の造影剤を含んでなる、請求項9に記載の方法。
【請求項17】
前記造影剤が、Mn、FeおよびGdからなる群から選択される、請求項16に記載の方法。
【請求項18】
前記標的化可能な構築物が、超音波イメージングで用いる1以上の造影剤を含んでなる、請求項9に記載の方法。
【請求項19】
前記標的化可能な構築物が、リポソーム脂質膜の外面に二価DTPA−ペプチドが共有結合したリポソームである、請求項18に記載の方法。
【請求項20】
前記リポソームがガス充填されている、請求項19に記載の方法。
【請求項21】
前記標的化可能な構築物が、罹患組織を死滅させるのに有用な1種以上の放射性同位元素を含んでなる、請求項9に記載の方法。
【請求項22】
前記放射性同位元素のエネルギー範囲が60〜700keVである、請求項21に記載の方法。
【請求項23】
前記放射性同位元素が、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acまたはそれらの組み合わせからなる群から選択される、請求項21に記載の方法。
【請求項24】
前記標的化可能な構築物が10B原子を含んでなるものであり、罹患組織に局在した前記ホウ素原子を照射し、それにより罹患組織のBNCTを果たす工程をさらに含んでなる、請求項21に記載の方法。
【請求項25】
前記治療剤が、薬物、毒素、ホルモン、酵素、免疫調節剤、キレート剤、ホウ素化合物、光活性薬、色素、または放射性同位元素である、請求項9に記載の方法。
【請求項26】
前記標的化可能な構築物が1種以上の毒素を含んでなる、請求項21に記載の方法。
【請求項27】
前記毒素が、リシン、アブリン、リボヌクレアーゼ、DNアーゼI、ブドウ球菌内毒素−A、アメリカヤマゴボウ抗ウイルスタンパク質、ゲロニン、ジフテリア毒、シュードモナス外毒素、およびシュードモナス内毒素またはそれらの組み合わせからなる群から選択される、請求項26に記載の方法。
【請求項28】
前記標的化可能な構築物が1種以上の薬物を含んでなる、請求項9に記載の方法。
【請求項29】
前記薬物が、ナイトロジェンマスタード、エチレンイミン誘導体、スルホン酸アルキル、ニトロソウレア、トリアゼン、葉酸類似体、アントラサイクリン、タキサン、COX−2阻害剤、ピリミジン類似体、プリン類似体、抗生物質、酵素、エピポドフィロトキシン、プラチナ錯体、ビンカアルカロイド、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、アンタゴニスト、エンドスタチン、タキソール、カンプトセシン、ドキソルビシンおよびそれらの類似体、ならびにそれらの組み合わせからなる群から選択される、請求項28に記載の方法。
【請求項30】
前記標的化可能な構築物が光線力学的療法のための1種以上の薬剤を含んでなる、請求項9に記載の方法。
【請求項31】
光線力学的療法のための前記薬剤が感光剤である、請求項30に記載の方法。
【請求項32】
前記感光剤が、ベンゾポルフィリン一酸環A(BPD−MA)、錫エチオプルプリン(SnET2)、スルホン化アルミニウムフタロシアニン(AlSPc)およびルテチウムタキサフィリン(Lutex)からなる群から選択される、請求項31に記載の方法。
【請求項33】
前記標的組織が腫瘍である、請求項9に記載の方法。
【請求項34】
前記標的組織が腫瘍である、請求項11に記載の方法。
【請求項35】
前記腫瘍が、結腸特異的抗原−p(CSAp)、癌胎児性抗原(CEA)、CD4、CD5、CD8、CD14、CD15、CD19、CD20、CD21、CD22、CD23、CD25、CD33、CD37、CD38、CD40、CD40L、CD46、CD52、CD54、CD66a−e、CD74、CD75、CD80、CD126、B7、HLA−DR、Ia、Ii、HM1.24、MUC1、MUC2、MUC3、MUC4、Tag−72、PSMA、EGP−1、EGP−2、PSA、AFP、HCG、HCG−β、PLAP、PAP、ヒストン、テネイシン、VEGF、PlGF、S100、EGFR、インスリン様成長因子、HER2/neu、臓器向性ホルモン、癌遺伝子産物、およびサイトケラチンからなる群から選択される抗原を産生するか、またはこれに関連するものである、請求項33または34に記載の方法。
【請求項36】
変異二重特異性抗体が、クラスIII抗CEA抗体のFvを組み込んでいる、請求項9または11に記載の方法。
【請求項37】
変異二重特異性抗体が、Mab 679のscFvを組み込んでいる、請求項9または11に記載の方法。
【請求項38】
前記疾病が、免疫調節障害、自己免疫疾患、臓器移植拒絶症、循環器病、神経疾患または移植片対宿主病である、請求項9または11に記載の方法。
【請求項39】
前記自己免疫疾患が、急性特発性血小板減少性紫斑病、慢性特発性血小板減少性紫斑病、皮膚筋炎、シドナム舞踏病、重症筋無力症、全身性紅斑性狼瘡、狼瘡腎炎、リウマチ熱、多腺性症候群、水疱性類天瘡、真性糖尿病、ヘノッホ−シェーンライン紫斑病、溶血性連鎖球菌感染後腎炎、結節性紅斑らい、高安動脈炎、アジソン病、慢性関節リウマチ、多発性硬化症、類肉腫症、潰瘍性大腸炎、多形性紅斑、IgA腎症、結節性多発性動脈炎、強直性脊椎炎、グッドパスチャー症候群、閉塞性血栓血管炎、シェーグレン症候群、原発性胆汁性肝硬変、橋本甲状腺炎、甲状腺中毒症、強皮症、慢性活動性肝炎、多発性筋炎/皮膚筋炎、多発性軟骨炎、尋常性天疱瘡、ウェジナー肉芽腫、膜性腎症、筋萎縮性側索硬化症、脊髄ろう、巨細胞動脈炎/多筋痛、悪性貧血、急速進行性糸球体腎炎、および繊維性肺胞炎からなる群から選択される、請求項38に記載の方法。
【請求項40】
前記疾患が、真菌、ウイルス、寄生虫または細菌により引き起こされるものであり、かつ、変異二重特異性のFvが、真菌、ウイルス、寄生虫または細菌を標的とするものである、請求項9または11に記載の方法。
【請求項41】
前記ウイルスが、ヒト免疫不全ウイルス(HIV)、ヘルペスウイルス、サイトメガロウイルス、狂犬病ウイルス、インフルエンザウイルス、B型肝炎ウイルス、センダイウイルス、ネコ白血病ウイルス、レオウイルス、ポリオウイルス、ヒト血清パルボ様ウイルス、シミアンウイルス40、呼吸器多核体(RS)ウイルス、マウス乳癌ウイルス、水疱−帯状疱疹ウイルス、デングウイルス、風疹ウイルス、麻疹ウイルス、アデノウイルス、ヒトT細胞白血病ウイルス、エプスタイン−バーウイルス、マウス白血病ウイルス、流行性耳下腺炎ウイルス、水疱性口内炎ウイルス、シンドビスウイルス、リンパ球性脈絡髄膜炎ウイルス、疣贅ウイルスおよびブルータングウイルスからなる群から選択される、請求項40に記載の方法。
【請求項42】
前記細菌が、炭疽菌(Anthrax bacillus)、ストレプトコッカス・アガラクチエ(Streptococcus agalactiae)、レジュネラ・ニューモフィラ(Legionella pneumophilia)、化膿連鎖球菌(Streptococcus pyogenes)、大腸菌(Escherichia coli)、淋菌(Neisseria gonorrhoeae)、髄膜炎菌(Neisseria meningitidis)、肺炎双球菌(Pneumococcus)、B型インフルエンザ菌(Hemophilis influenzae B)、梅毒トレポネーマ(Treponema pallidum)、ライム病スピロヘータ、緑膿菌(Pseudomonas aeruginosa)、らい菌(Mycobacterium leprae)、ウシ流産菌(Brucella abortus)、結核菌(Mycobacterium tuberculosis)および破傷風毒素からなる群から選択される、請求項40に記載の方法。
【請求項43】
前記病原体が原生動物である、請求項40に記載の方法。
【請求項44】
前記原生動物が、熱帯熱マラリア原虫(Plasmodium falciparum)、三日熱マラリア原虫(Plasmodium vivax)、トキソプラズマ(Toxoplasma gondii)、ランゲリ・トリパノソーマ(Trypanosoma rangeli)、クルーズ・トリパノソーマ(Trypanosoma cruzi)、ローデシア・トリパノソーマ(Trypanosoma rhodesiensei)、ブルセイ・トリパノソーマ(Trypanosoma brucei)、マンソン住血吸虫(Schistosoma mansoni)、日本住血吸虫(Schistosoma japanicum)、ウシバベシア(Babesia bovis)、エルメリア・テネラ(Elmeria tenella)、回旋糸状虫(Onchocerca volvulus)、熱帯リーシュマニア(Leishmania tropica)、旋毛虫(Trichinella spiralis)、回旋糸状虫(Onchocerca volvulus)、タイレリア・パルバ(Theileria parva)、胞状条虫(Taenia hydatigena)、テニア・オビス(Taenia ovis)、カギナシサナダ(Taenia saginata)、単胞条虫(Echinococcus granulosus)およびメソセストイデス・コルチ(Mesocestoides corti)からなる群から選択される、請求項43に記載の方法。
【請求項45】
前記寄生虫が蠕虫またはマラリア原虫である、請求項40に記載の方法。
【請求項46】
前記細菌がマイコプラズマである、請求項40に記載の方法。
【請求項47】
前記マイコプラズマが、マイコプラズマ・アルスリティディス(Mycoplasma arthritidis)、M.ヒオリニス(M. hyorhinis)、M.オーラル(M. orale)、M.アルギニニ(M. arginini)、アコレプラスマ・ライドラウィー(Acholeplasma laidlawii)、M.サリバルム(M. salivarum)、および肺炎マイコプラズマ(M. pneumoniae)からなる群から選択される、請求項46に記載の方法。
【請求項48】
前記真菌が、ヒストプラズマ・カプスラーツム(Histoplasma capsulatum)、ブラストミセス・デルマティティディス(Blastomyces dermatitidis)、コクシジオイデス・イミティス(Coccidioides immitis)、およびカンジダ種(Candida)からなる群から選択される、請求項40に記載の方法。
【請求項49】
組織が正常な異所組織である、請求項9または11に記載の方法。
【請求項50】
前記正常組織が、卵巣、胸腺、副甲状腺、骨髄、または脾臓に由来する組織である、請求項49に記載の方法。
【請求項51】
前記被験体が哺乳類である、請求項9または11に記載の方法。
【請求項52】
前記哺乳類被験体がヒトまたは霊長類である、請求項9または11に記載の方法。
【請求項53】
前記哺乳類被験体が、齧歯類、ウサギ類、ウシ類、ヒツジ類、ヤギ類、ブタ類、ウマ類、イヌ類、ネコ類、家禽類、有蹄類、およびクマ類からなる群から選択される、請求項51に記載の方法。
【請求項54】
罹患組織を同定するための術中診断のための、請求項9に記載の方法。
【請求項55】
罹患組織を同定するための内視鏡診断のための、請求項9に記載の方法。
【請求項56】
所定の診断または治療の前、同時または後に第二の治療剤が投与される、請求項9〜55のいずれか一項に記載の方法。
【請求項57】
第二の治療剤が、薬物、裸の抗体、免疫調節剤、または薬物、放射性同位元素、免疫調節剤もしくは毒素を担持する抗体複合体である、請求項56に記載の方法。
【請求項58】
被験体において罹患組織を治療または同定するのに有用なキットであって、
(A)請求項9に記載の変異二重特異性抗体、
(B)所望により、請求項9に記載のクリアランス剤、および
(C)請求項11に記載の標的化可能な構築物
を含んでなる、キット。
【請求項59】
被験体において罹患組織を治療または同定するのに有用なキットであって、
(A)請求項11に記載の変異二重特異性抗体、
(B)所望により、請求項11に記載のクリアランス剤、
(C)請求項11に記載の標的化可能な構築物、および
(D)請求項11に記載のプロドラッグ
を含んでなる、キット。
【請求項60】
前記免疫調節剤が、サイトカイン、幹細胞増殖因子、リンホトキシン、造血因子、コロニー刺激因子(CSF)、インターフェロン(IFN)、エリスロポエチン、トロンボポエチン、およびそれらの組み合わせからなる群から選択される、請求項25に記載の方法。
【請求項61】
前記リンホトキシンが腫瘍壊死因子(TNF)である、請求項60に記載の方法。
【請求項62】
前記造血系因子がインターロイキン(IL)である、請求項60に記載の方法。
【請求項63】
前記コロニー刺激因子が、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)である、請求項60に記載の方法。
【請求項64】
前記インターフェロンが、インターフェロン−α、−βまたは−γである、請求項60に記載の方法。
【請求項65】
前記幹細胞増殖因子がS1因子である、請求項60に記載の方法。
【請求項66】
前記免疫調節剤が、IL−1、IL−2、IL−3、IL−6、IL−10、IL−12、IL−18、インターフェロン−γ、TNF−αまたはそれらの組み合わせである、請求項25に記載の方法。
【請求項67】
前記神経疾患がアルツハイマー病である、請求項38に記載の方法。
【請求項1】
(a)IgG由来のヒトヒンジ定常領域、
(b)2つのscFv、および
(c)2つのFv
を少なくとも含んでなり、該定常領域がCH2ドメインに1以上のアミノ酸変異を含む、変異二重特異性抗体。
【請求項2】
前記scFvおよびFvが、CDRグラフト化マウスscFvおよびFvである、請求項1に記載の変異二重特異性抗体。
【請求項3】
前記scFvおよびFvが、ヒト化されたものまたはヒト由来のものである、請求項1に記載の変異二重特異性抗体。
【請求項4】
前記ヒンジ定常領域が、253番目のイソロイシンがアラニンで置換されているか、またはそのアミノ酸置換がアラニン置換と同等もしくはそれを超えて血液クリアランスを促進する、ロイシン以外のアミノ酸で置換されている変異を含む、請求項1に記載の変異二重特異性抗体。
【請求項5】
前記FvがhMN14−IgGに由来するものであり、前記scFvが734scFvである、請求項4に記載の変異二重特異性抗体。
【請求項6】
前記scFvが一価の標的化可能な構築物と結合する、請求項1に記載の変異二重特異性抗体。
【請求項7】
前記scFvが二価の標的化可能な構築物と結合する、請求項1に記載の変異二重特異性抗体。
【請求項8】
前記Fvが標的細胞上のエピトープと結合する、請求項1に記載の変異二重特異性抗体。
【請求項9】
疾病または疾病に至る可能性のある症状を、治療もしくは診断、または治療および診断する方法であって、
(A)Fvが前記疾病または前記症状に関連するマーカー物質に向けられている請求項1に記載の変異二重特異性抗体を被験体に投与する工程、
(B)所望により、被験体にクリアランス組成物を投与し、該組成物に非局在抗体または抗体フラグメントを循環系から除去させる工程、および
(C)二価ハプテンを含む標的化可能な構築物であって、両ハプテン部分が請求項1に記載の変異二重特異性の単一分子上の2つのscFvと結合するものであり、診断もしくは治療用カチオンおよび/または1以上のキレート化もしくは化学結合した治療剤もしくは診断剤をさらに含んでなる前記構築物を被験体に投与する工程
を含んでなる、方法。
【請求項10】
前記変異二重特異性抗体が、前記疾病または症状の治療に用いられる少なくとも1つの治療剤を投与する前、投与中、または投与した後に投与される、請求項9に記載の方法。
【請求項11】
前記標的化可能な構築物が酵素を含んでなるものであり、
a)プロドラッグ(前記酵素が標的部位で該プロドラッグを薬物へと変換することができる場合)、
b)被験体内で解毒されて毒性の低い中間体を形成することができる薬物(前記酵素が解毒された前記中間体を毒性型へと再変換し、これにより、標的部位で薬物の毒性を増強することができる場合)、または
c)被験体内で自然のプロセスを経て活性化され、毒性の低い中間体へと変換されることにより解毒されるプロドラッグ(前記酵素が解毒された前記中間体を毒性型へと再変換し、これにより、標的部位で薬物の毒性を増強することができる場合)
を被験体に投与する工程をさらに含んでなる、請求項9に記載の方法。
【請求項12】
前記プロドラッグが、エピルビシングルクロニド、CPT−11、エトポシドグルクロニド、ダウノマイシングルクロニドおよびドキソルビシングルクロニドからなる群から選択される、請求項11に記載の方法。
【請求項13】
前記診断剤が25〜4,000keVのγ粒子および/または陽電子を放射するものである、請求項9に記載の方法。
【請求項14】
前記診断剤が、18F、52Fe、62Cu、64Cu、67Cu、67Ga、68Ga、86Y、89Zr、94mTc、94Tc、99mTc、111In、123I、124I、125I、131I、154−158Gd、177Lu、32P、188Re、および90Yまたはそれらの組み合わせからなる群から選択される、請求項9に記載の方法。
【請求項15】
前記放射性同位元素を用いて陽電子放射断層撮影法(PET)を行う、請求項9に記載の方法。
【請求項16】
前記標的化可能な構築物が、磁気共鳴イメージング(MRI)に用いる1以上の造影剤を含んでなる、請求項9に記載の方法。
【請求項17】
前記造影剤が、Mn、FeおよびGdからなる群から選択される、請求項16に記載の方法。
【請求項18】
前記標的化可能な構築物が、超音波イメージングで用いる1以上の造影剤を含んでなる、請求項9に記載の方法。
【請求項19】
前記標的化可能な構築物が、リポソーム脂質膜の外面に二価DTPA−ペプチドが共有結合したリポソームである、請求項18に記載の方法。
【請求項20】
前記リポソームがガス充填されている、請求項19に記載の方法。
【請求項21】
前記標的化可能な構築物が、罹患組織を死滅させるのに有用な1種以上の放射性同位元素を含んでなる、請求項9に記載の方法。
【請求項22】
前記放射性同位元素のエネルギー範囲が60〜700keVである、請求項21に記載の方法。
【請求項23】
前記放射性同位元素が、32P、33P、47Sc、64Cu、67Cu、67Ga、90Y、111Ag、111In、125I、131I、142Pr、153Sm、161Tb、166Dy、166Ho、177Lu、186Re、188Re、189Re、212Pb、212Bi、213Bi、211At、223Raおよび225Acまたはそれらの組み合わせからなる群から選択される、請求項21に記載の方法。
【請求項24】
前記標的化可能な構築物が10B原子を含んでなるものであり、罹患組織に局在した前記ホウ素原子を照射し、それにより罹患組織のBNCTを果たす工程をさらに含んでなる、請求項21に記載の方法。
【請求項25】
前記治療剤が、薬物、毒素、ホルモン、酵素、免疫調節剤、キレート剤、ホウ素化合物、光活性薬、色素、または放射性同位元素である、請求項9に記載の方法。
【請求項26】
前記標的化可能な構築物が1種以上の毒素を含んでなる、請求項21に記載の方法。
【請求項27】
前記毒素が、リシン、アブリン、リボヌクレアーゼ、DNアーゼI、ブドウ球菌内毒素−A、アメリカヤマゴボウ抗ウイルスタンパク質、ゲロニン、ジフテリア毒、シュードモナス外毒素、およびシュードモナス内毒素またはそれらの組み合わせからなる群から選択される、請求項26に記載の方法。
【請求項28】
前記標的化可能な構築物が1種以上の薬物を含んでなる、請求項9に記載の方法。
【請求項29】
前記薬物が、ナイトロジェンマスタード、エチレンイミン誘導体、スルホン酸アルキル、ニトロソウレア、トリアゼン、葉酸類似体、アントラサイクリン、タキサン、COX−2阻害剤、ピリミジン類似体、プリン類似体、抗生物質、酵素、エピポドフィロトキシン、プラチナ錯体、ビンカアルカロイド、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、アンタゴニスト、エンドスタチン、タキソール、カンプトセシン、ドキソルビシンおよびそれらの類似体、ならびにそれらの組み合わせからなる群から選択される、請求項28に記載の方法。
【請求項30】
前記標的化可能な構築物が光線力学的療法のための1種以上の薬剤を含んでなる、請求項9に記載の方法。
【請求項31】
光線力学的療法のための前記薬剤が感光剤である、請求項30に記載の方法。
【請求項32】
前記感光剤が、ベンゾポルフィリン一酸環A(BPD−MA)、錫エチオプルプリン(SnET2)、スルホン化アルミニウムフタロシアニン(AlSPc)およびルテチウムタキサフィリン(Lutex)からなる群から選択される、請求項31に記載の方法。
【請求項33】
前記標的組織が腫瘍である、請求項9に記載の方法。
【請求項34】
前記標的組織が腫瘍である、請求項11に記載の方法。
【請求項35】
前記腫瘍が、結腸特異的抗原−p(CSAp)、癌胎児性抗原(CEA)、CD4、CD5、CD8、CD14、CD15、CD19、CD20、CD21、CD22、CD23、CD25、CD33、CD37、CD38、CD40、CD40L、CD46、CD52、CD54、CD66a−e、CD74、CD75、CD80、CD126、B7、HLA−DR、Ia、Ii、HM1.24、MUC1、MUC2、MUC3、MUC4、Tag−72、PSMA、EGP−1、EGP−2、PSA、AFP、HCG、HCG−β、PLAP、PAP、ヒストン、テネイシン、VEGF、PlGF、S100、EGFR、インスリン様成長因子、HER2/neu、臓器向性ホルモン、癌遺伝子産物、およびサイトケラチンからなる群から選択される抗原を産生するか、またはこれに関連するものである、請求項33または34に記載の方法。
【請求項36】
変異二重特異性抗体が、クラスIII抗CEA抗体のFvを組み込んでいる、請求項9または11に記載の方法。
【請求項37】
変異二重特異性抗体が、Mab 679のscFvを組み込んでいる、請求項9または11に記載の方法。
【請求項38】
前記疾病が、免疫調節障害、自己免疫疾患、臓器移植拒絶症、循環器病、神経疾患または移植片対宿主病である、請求項9または11に記載の方法。
【請求項39】
前記自己免疫疾患が、急性特発性血小板減少性紫斑病、慢性特発性血小板減少性紫斑病、皮膚筋炎、シドナム舞踏病、重症筋無力症、全身性紅斑性狼瘡、狼瘡腎炎、リウマチ熱、多腺性症候群、水疱性類天瘡、真性糖尿病、ヘノッホ−シェーンライン紫斑病、溶血性連鎖球菌感染後腎炎、結節性紅斑らい、高安動脈炎、アジソン病、慢性関節リウマチ、多発性硬化症、類肉腫症、潰瘍性大腸炎、多形性紅斑、IgA腎症、結節性多発性動脈炎、強直性脊椎炎、グッドパスチャー症候群、閉塞性血栓血管炎、シェーグレン症候群、原発性胆汁性肝硬変、橋本甲状腺炎、甲状腺中毒症、強皮症、慢性活動性肝炎、多発性筋炎/皮膚筋炎、多発性軟骨炎、尋常性天疱瘡、ウェジナー肉芽腫、膜性腎症、筋萎縮性側索硬化症、脊髄ろう、巨細胞動脈炎/多筋痛、悪性貧血、急速進行性糸球体腎炎、および繊維性肺胞炎からなる群から選択される、請求項38に記載の方法。
【請求項40】
前記疾患が、真菌、ウイルス、寄生虫または細菌により引き起こされるものであり、かつ、変異二重特異性のFvが、真菌、ウイルス、寄生虫または細菌を標的とするものである、請求項9または11に記載の方法。
【請求項41】
前記ウイルスが、ヒト免疫不全ウイルス(HIV)、ヘルペスウイルス、サイトメガロウイルス、狂犬病ウイルス、インフルエンザウイルス、B型肝炎ウイルス、センダイウイルス、ネコ白血病ウイルス、レオウイルス、ポリオウイルス、ヒト血清パルボ様ウイルス、シミアンウイルス40、呼吸器多核体(RS)ウイルス、マウス乳癌ウイルス、水疱−帯状疱疹ウイルス、デングウイルス、風疹ウイルス、麻疹ウイルス、アデノウイルス、ヒトT細胞白血病ウイルス、エプスタイン−バーウイルス、マウス白血病ウイルス、流行性耳下腺炎ウイルス、水疱性口内炎ウイルス、シンドビスウイルス、リンパ球性脈絡髄膜炎ウイルス、疣贅ウイルスおよびブルータングウイルスからなる群から選択される、請求項40に記載の方法。
【請求項42】
前記細菌が、炭疽菌(Anthrax bacillus)、ストレプトコッカス・アガラクチエ(Streptococcus agalactiae)、レジュネラ・ニューモフィラ(Legionella pneumophilia)、化膿連鎖球菌(Streptococcus pyogenes)、大腸菌(Escherichia coli)、淋菌(Neisseria gonorrhoeae)、髄膜炎菌(Neisseria meningitidis)、肺炎双球菌(Pneumococcus)、B型インフルエンザ菌(Hemophilis influenzae B)、梅毒トレポネーマ(Treponema pallidum)、ライム病スピロヘータ、緑膿菌(Pseudomonas aeruginosa)、らい菌(Mycobacterium leprae)、ウシ流産菌(Brucella abortus)、結核菌(Mycobacterium tuberculosis)および破傷風毒素からなる群から選択される、請求項40に記載の方法。
【請求項43】
前記病原体が原生動物である、請求項40に記載の方法。
【請求項44】
前記原生動物が、熱帯熱マラリア原虫(Plasmodium falciparum)、三日熱マラリア原虫(Plasmodium vivax)、トキソプラズマ(Toxoplasma gondii)、ランゲリ・トリパノソーマ(Trypanosoma rangeli)、クルーズ・トリパノソーマ(Trypanosoma cruzi)、ローデシア・トリパノソーマ(Trypanosoma rhodesiensei)、ブルセイ・トリパノソーマ(Trypanosoma brucei)、マンソン住血吸虫(Schistosoma mansoni)、日本住血吸虫(Schistosoma japanicum)、ウシバベシア(Babesia bovis)、エルメリア・テネラ(Elmeria tenella)、回旋糸状虫(Onchocerca volvulus)、熱帯リーシュマニア(Leishmania tropica)、旋毛虫(Trichinella spiralis)、回旋糸状虫(Onchocerca volvulus)、タイレリア・パルバ(Theileria parva)、胞状条虫(Taenia hydatigena)、テニア・オビス(Taenia ovis)、カギナシサナダ(Taenia saginata)、単胞条虫(Echinococcus granulosus)およびメソセストイデス・コルチ(Mesocestoides corti)からなる群から選択される、請求項43に記載の方法。
【請求項45】
前記寄生虫が蠕虫またはマラリア原虫である、請求項40に記載の方法。
【請求項46】
前記細菌がマイコプラズマである、請求項40に記載の方法。
【請求項47】
前記マイコプラズマが、マイコプラズマ・アルスリティディス(Mycoplasma arthritidis)、M.ヒオリニス(M. hyorhinis)、M.オーラル(M. orale)、M.アルギニニ(M. arginini)、アコレプラスマ・ライドラウィー(Acholeplasma laidlawii)、M.サリバルム(M. salivarum)、および肺炎マイコプラズマ(M. pneumoniae)からなる群から選択される、請求項46に記載の方法。
【請求項48】
前記真菌が、ヒストプラズマ・カプスラーツム(Histoplasma capsulatum)、ブラストミセス・デルマティティディス(Blastomyces dermatitidis)、コクシジオイデス・イミティス(Coccidioides immitis)、およびカンジダ種(Candida)からなる群から選択される、請求項40に記載の方法。
【請求項49】
組織が正常な異所組織である、請求項9または11に記載の方法。
【請求項50】
前記正常組織が、卵巣、胸腺、副甲状腺、骨髄、または脾臓に由来する組織である、請求項49に記載の方法。
【請求項51】
前記被験体が哺乳類である、請求項9または11に記載の方法。
【請求項52】
前記哺乳類被験体がヒトまたは霊長類である、請求項9または11に記載の方法。
【請求項53】
前記哺乳類被験体が、齧歯類、ウサギ類、ウシ類、ヒツジ類、ヤギ類、ブタ類、ウマ類、イヌ類、ネコ類、家禽類、有蹄類、およびクマ類からなる群から選択される、請求項51に記載の方法。
【請求項54】
罹患組織を同定するための術中診断のための、請求項9に記載の方法。
【請求項55】
罹患組織を同定するための内視鏡診断のための、請求項9に記載の方法。
【請求項56】
所定の診断または治療の前、同時または後に第二の治療剤が投与される、請求項9〜55のいずれか一項に記載の方法。
【請求項57】
第二の治療剤が、薬物、裸の抗体、免疫調節剤、または薬物、放射性同位元素、免疫調節剤もしくは毒素を担持する抗体複合体である、請求項56に記載の方法。
【請求項58】
被験体において罹患組織を治療または同定するのに有用なキットであって、
(A)請求項9に記載の変異二重特異性抗体、
(B)所望により、請求項9に記載のクリアランス剤、および
(C)請求項11に記載の標的化可能な構築物
を含んでなる、キット。
【請求項59】
被験体において罹患組織を治療または同定するのに有用なキットであって、
(A)請求項11に記載の変異二重特異性抗体、
(B)所望により、請求項11に記載のクリアランス剤、
(C)請求項11に記載の標的化可能な構築物、および
(D)請求項11に記載のプロドラッグ
を含んでなる、キット。
【請求項60】
前記免疫調節剤が、サイトカイン、幹細胞増殖因子、リンホトキシン、造血因子、コロニー刺激因子(CSF)、インターフェロン(IFN)、エリスロポエチン、トロンボポエチン、およびそれらの組み合わせからなる群から選択される、請求項25に記載の方法。
【請求項61】
前記リンホトキシンが腫瘍壊死因子(TNF)である、請求項60に記載の方法。
【請求項62】
前記造血系因子がインターロイキン(IL)である、請求項60に記載の方法。
【請求項63】
前記コロニー刺激因子が、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)である、請求項60に記載の方法。
【請求項64】
前記インターフェロンが、インターフェロン−α、−βまたは−γである、請求項60に記載の方法。
【請求項65】
前記幹細胞増殖因子がS1因子である、請求項60に記載の方法。
【請求項66】
前記免疫調節剤が、IL−1、IL−2、IL−3、IL−6、IL−10、IL−12、IL−18、インターフェロン−γ、TNF−αまたはそれらの組み合わせである、請求項25に記載の方法。
【請求項67】
前記神経疾患がアルツハイマー病である、請求項38に記載の方法。
【図1】


【図2】

【図3】

【図4】
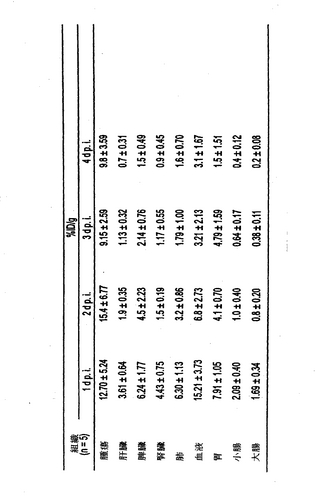
【図5】
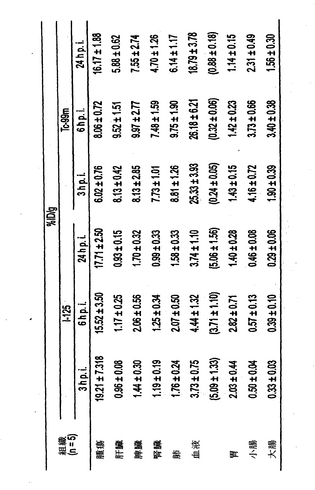
【図6】
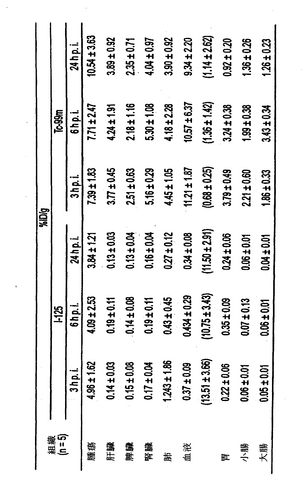
【図7】

【図8】

【図9】
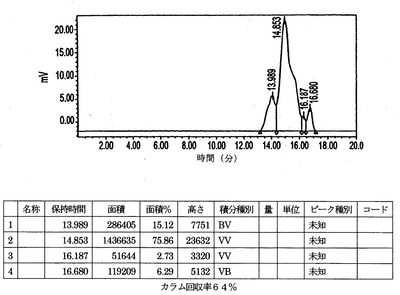
【図10】
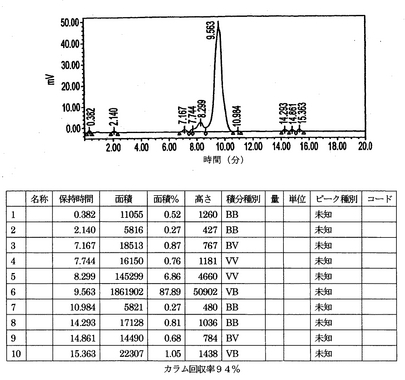
【図11】
【図12】
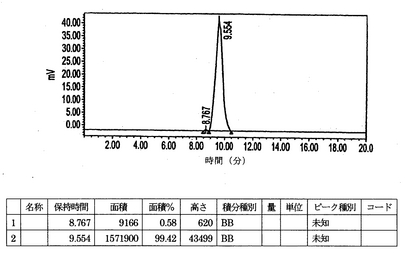
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2006−502091(P2006−502091A)
【公表日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願番号】特願2003−573034(P2003−573034)
【出願日】平成15年3月3日(2003.3.3)
【国際出願番号】PCT/GB2003/000871
【国際公開番号】WO2003/074569
【国際公開日】平成15年9月12日(2003.9.12)
【出願人】(504149971)イミューノメディクス、インコーポレイテッド (48)
【氏名又は名称原語表記】IMMUNOMEDICS, INC.
【Fターム(参考)】
【公表日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願日】平成15年3月3日(2003.3.3)
【国際出願番号】PCT/GB2003/000871
【国際公開番号】WO2003/074569
【国際公開日】平成15年9月12日(2003.9.12)
【出願人】(504149971)イミューノメディクス、インコーポレイテッド (48)
【氏名又は名称原語表記】IMMUNOMEDICS, INC.
【Fターム(参考)】
[ Back to top ]