
クロマン誘導体の製造方法
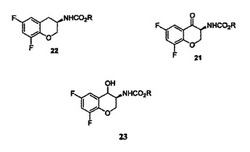
式(22)の化合物の製造方法であって、式(21)の化合物を還元して、式(23)の化合物を生成すること、続いて、C1〜C6のアルキルスルホン酸及び任意に塩素化溶媒を含む溶媒中で、式23の化合物を水素化分解することを含む。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、カルバミン酸エチルエステルを製造する方法に関する。このようなカルバミン酸エステルは、WO2004/033447に記載されているように、ドーパミン-β-ヒドロキシラーゼ阻害剤の製造に有用である。
【背景技術】
【0002】
下記のアミン8は、WO2004/033447に記載されるドーパミン-β-ヒドロキシラーゼ阻害剤の製造における有用な中間体である。これは、L-セリンメチルエステル塩酸塩(2)から出発して、そのN-トリチル誘導体と2,4-ジフルオロフェノールとを光延条件下で縮合し(Cherney, R. L.; Wang, L.の論文「N-フェニルフルオレニル又はN-トリチルセリンエステルを用いる効率的光延反応(Efficient Mitsunobu reactions with N-phenylfluorenyl or N-trityl serine esters)」J. Org. Chem. 1996, v. 61, N 7, p. 2544-2546)、続いて、脱保護し、得られるアミノ酸(4)をエトキシカルボニル化し、N保護された誘導体(20)をフリーデル-クラフツ環化し(McClure, D. E.; Arison, B. H.; Jones, J. H.; Baldwin, J. J.の論文「保護アミノ酸のフリーデル-クラフツ反応からのキラルα-アミノケトン(Chiral α-amino ketones from the Friedel-Crafts reaction of protected amino acids)」J. Org. Chem. 1981, v.46, N 11, p. 2431-2433)、かつエトキシカルボニルアミノケトン(21)を還元することによって合成できる。カルバミン酸エチル(22)のアルカリ加水分解によって8が得られる。
【化1】
【0003】
他の鏡像異性体を用いて、同様の反応を下記のように行うことができる。
【化2】
上のスキーム中の第2、3、4、5、及び6の化合物は、それぞれ化合物4'、20'、21'、22'及び8'である。
【0004】
アリールケトン(化合物21など)のメチレン化合物(化合物22など)への還元は、合成的に有用な反応であり、この変換のために様々な方法が開発されている。すなわち、クレメンゼン及びウォルフ-キッシュナー還元、接触水素化分解(Sarda, N., Grouiller, A., Pacheco, H.の論文(Nouvelle synthese de ramino-3-chromanne. Synthese et configuration absolue de ses enantiomeres)Tetrahedron Letters 1976, N 4, p. 271-272;Norlander, E. J., Njoroge, F. G., Payne, M. J., Warman, D.の論文「フリーデル-クラフツ合成のためのキラル試薬としてのN-(トリフルオロアセチル)-α-アミノ酸塩酸塩(N-(Trifluoroacetyl)-α-amino acid chlorides as chiral reagents for Friedel-Crafts synthesis)」J. Org. Chem. 1985, v. 50, N 19, p. 3481-3484;Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 49, N 32, p. 4107-4111)、Et3SiH/CF3COOH(Norlander, E. J., Njoroge, F. G., Payne, M. J., Warman, D.の論文「フリーデル-クラフツ合成のためのキラル試薬としてのN-(トリフルオロアセチル)-α-アミノ酸塩酸塩(N-(Trifluoroacetyl)-α-amino acid chlorides as chiral reagents for Friedel-Crafts synthesis)」J. Org. Chem. 1985, v. 50, N 19, p. 3481-3484;Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 49, N 32, p. 4107-4111)、Et3SiH/BF3Et2O(Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 1049, N 32, p. 4107-4111)、Et3SiH/TiCl4(Yato, M.; Homma, K.; Ishida, A.の論文「四塩化チタンによって媒介される芳香族ケトンの新規シラン還元:γ-及びδ-アリール置換アミノ酸の合成(New silane reduction of aromatic ketones mediated by titanium tetrachloride: a synthesis of γ- and δ-aryl substituted amino acids)」Heterocycles 1998, v. 49, p. 233-254)、NaBH4/BF3Et2O(Perry, P. J.; Pavlidis, V. H.; Coutts, I. G. C.の論文「ヨードトリメチルシランを用いたα,α-ジアリールアルコールの対応するアルカンへの高速還元(The rapid reduction of α,α-diaryl alcohols to the corresponding alkanes using iodotrimethylsilane)」Synth. Commun. 1996, v. 26, N 1, p. 101-111)である。上述の方法のいずれも、21から22へ変換して中間体のアルコール又はその出発物質との混合物を得ることに対して、有効性を提供するものではない。この種の変換に有効であることが周知の他の試薬、ヨードトリメチルシラン(Pettit, G. R.; Piatak, D. M.の論文、J. Org. Chem. 1962, v. 27, p. 2127)又はその等価物(Cain, G. A.; Holler, E. R.の論文「拡張した範囲でのその場のヨードトリメチルシラン媒介によるベンジルアルコールの選択的還元(Extended scope of in situ iodotrimethysilane mediated selective reduction of benzylic alcohols)」Chem. Commun. 2001, p. 1168-1169)などを用いて、中間体のアルコールを22に還元する試みは、どちらも作用しなかった。接触還元法を予め用いてネピカスタット中間体を製造する(米国特許第5,438,150号)ことに相当する、ある種のスキームを用いて、結果を改善することができる。
【化3】
【0005】
しかし、化合物22の収率は、50%を上回ることはなく、主な副生成物は、トリフルオロアセチル化されたアルコールに相当するものである。
【化4】
更に、トリフルオロ酢酸と硫酸との高腐食性の混合物を大量に用いることは、問題を引起こし得る。トリフルオロ酢酸を酢酸に置換える試みは、かなりの量のアセトキシ副生成物を伴い、低い収率となった。
【発明の概要】
【0006】
驚くべきことに、本発明者らは、メタンスルホン酸などのC1〜C6のアルキルスルホン酸、又は好ましくは、3容量のジクロロメタンなどの塩素化溶媒と1容量のメタンスルホン酸などのC1〜C6のアルキルスルホン酸との混合物中で還元を行い、21から22への完全な変換が得られることを見出した。
【0007】
しかし、化合物22の光学純度の分析によって、顕著なラセミ化が明らかとなった。そのラセミ化の原因は、3容量のジクロロメタンなどの塩素化溶媒と1容量のメタンスルホン酸との混合物中における、出発物質の鏡像異性の不安定性にあることが見出された。こうした結果に基づいて、光学純度における状態を改善するために、水素化ホウ素ナトリウムを用いて21から中間体のアルコールへ変換し、続いて3容量のジクロロメタンと1容量のメタンスルホン酸との混合物中で接触還元を行うことを含む、2工程の方法が案出された。該2工程の方法によって、再現性よく、94-99% eeという高収率の化合物22が得られた。
【図面の簡単な説明】
【0008】
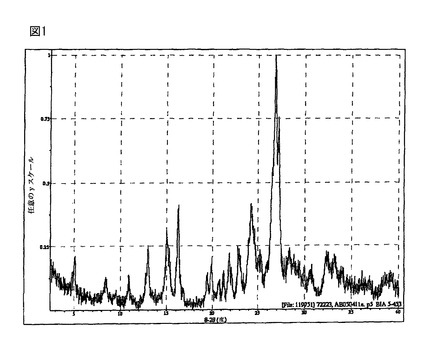
【図1】(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Aは、4.9、8.3、12.9、15.0、16.2、19.8、21.8、22.9及び24.2、26.8±0.2 °2θにピークを有するXRPDパターンを有する。図1に該XRPDパターンを示す。
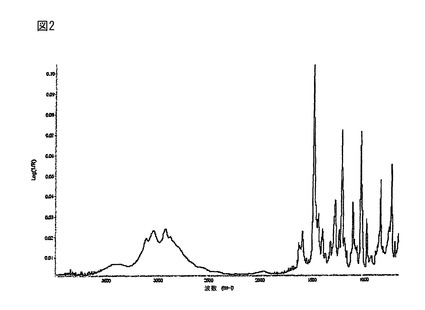
【図2】更に、(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Aは、3053.30、2939.70、1599.80、1491.90、1448.30、1406.10、1330.70、1287.60、1244.50、1220.70、1194.00、1117.50、1039.50、985.50、851.80、747.00及び713.70 cm-1に特有のFT-IRのピークを有する。図2に該FT-IRスペクトルを示す。
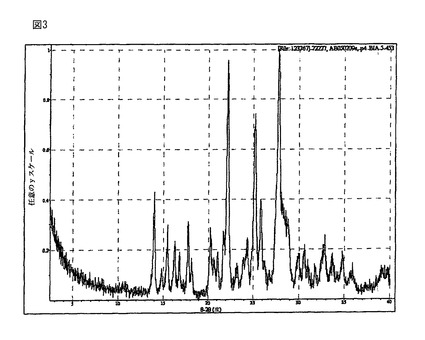
【図3】(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Cは、13.9、15.3、16.2、16.7、17.7、18.1、20.2、21.0、22.1、24.2、25.1及び25.7にピークを有するXRPDパターンを有する。図3に該XRPDパターンを示す。
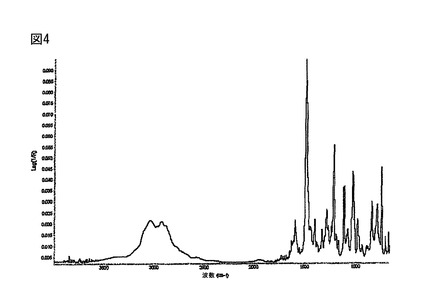
【図4】更に、(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Cは、3041.70、1596.50、1492、1403.40、1333.80、1290.90、1220.2、1173.20、1117.4、1078.10、1033.4、984.90、845.2、792.6、750.1及び713.20cm-1に特有のFT-IRのピークを有する。図4に該FT-IRスペクトルを示す。
【発明を実施するための形態】
【0009】
したがって、本発明の一態様によれば、式22の化合物の製造方法が提供され:
【化5】
該方法は、式21の化合物:
【化6】
を還元して、式23の化合物:
【化7】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23の化合物の水素化分解を行うことを含む。これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。
【0010】
好ましくは、Rは、エチル、メチル、tBu、又はベンジル、最も好ましくは、エチルである。
好ましくは、アルキルスルホン酸は、メタンスルホン酸である。
反応は、実質的に純粋なアルキルスルホン酸の溶媒中で行うことが可能であるが、塩素化溶媒及びC1〜C6のアルキルスルホン酸を含む溶媒混合物中で、該反応を行うことが好ましい。更に、該アルキルスルホン酸は、メタンスルホン酸であることが好ましい。塩素化溶媒とアルキルスルホン酸溶媒との比率は、10:1以下であることが好ましく、より好ましくは、5:1以下であり、更により好ましくは、4:1〜2:1の範囲内である。最も好ましくは、塩素化溶媒とアルキルスルホン酸溶媒との比率は、約3:1である。
【0011】
一般に、本方法の第1工程は、(R)-(6,8-ジフルオロ-4-オキソクロマン-3-イル)カルバミン酸エチルエステル(21)を還元することを含む。これは、適当な溶媒中で、水素化ホウ素、特に、水素化ホウ素ナトリウムなどのアルカリ金属の水素化ホウ素を用いて行うことができる。該溶媒は、低級アルコールとTHFとの任意の組み合わせとし得る。該低級アルコールは、炭素原子1〜6個を有する、直鎖又は分枝のアルコールであってよく;該低級アルコールは、エタノール、又は最も好ましくは、メタノールとし得る。好適には、この還元は、5〜25℃にて0.5〜4時間で行うことができる。
【0012】
該還元の工程の後に、中間体のアルコールの水素化分解が続き、好ましくは、パラジウム触媒上で行う(7% w/wの活性炭上の無水の10%のPdを使用できる。)。好適には、該反応は、溶媒混合物中、15〜100psiの水素圧及び20〜25℃の温度にて7〜26時間で行うことができる。1容量未満のメタンスルホン酸では、変換が低くなることが見出されている。
【0013】
塩素化溶媒とアルキルスルホン酸溶媒との比率が、5:1未満の場合、該反応には、100psiの水素圧及び15時間の反応時間とともに10% w/wのPdが要求されるだろう。このように、塩素化溶媒の量が多くなるほど、反応時間、水素圧、及びPdの必要量が増加する傾向がある。
有利には、該塩素化溶媒は、ハロゲン化アルキル、好ましくは、C1〜C6のハロゲン化アルキル、より好ましくは、ハロゲン化メチル又はエチルである。好ましくは、ハロゲン化物は、塩化物である。最も好ましくは、該塩素化溶媒は、ジクロロメタン又はジクロロメタン-クロロホルムの混合物である。あるいは、該塩素化溶媒は、1,2-ジクロロエタンであってよい。
【0014】
本発明の別の態様によれば、式8の化合物の製造方法が提供され、該方法は、上記の方法を用いて式22の化合物を形成し、続いて、式22の化合物をアルカリ加水分解することを含む。
該アルカリ加水分解の工程は、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びL-酒石酸の存在下で行うことができる。好ましくは、該アルカリ金属の水酸化物は、水酸化カリウムである。好ましくは、該アルコールは、メタノールである。
【0015】
本発明の別の態様によれば、式14の化合物の製造方法が提供され:
【化8】
例えば、上記の方法によって、式8の化合物を製造すること、次いで、式8の化合物と式13の化合物とを反応させることを含む。
【化9】
好ましくは、該反応は、アルカリ金属のイソチオシアン酸塩、好ましくは、チオシアン酸カリウム、及び有機溶媒、好ましくは、酢酸の存在下で行われる。
【0016】
本発明の別の態様によれば、式1の化合物の製造方法が提供され:
【化10】
上記の方法によって、式14の化合物を製造すること、次いで溶媒の存在下で、アルカリ金属水素化ホウ素、好ましくは、水素化ホウ素ナトリウム(NaBH4)を用いて、式14の化合物を式1の化合物に変換すること、続いて、HClを添加し、次いで、式1の化合物を回収することを含む。
好ましくは、化合物14の回収に使用される溶媒は、イソプロピルアルコール、水及びジクロロメタンの混合物である。
【0017】
本発明の更なる態様によれば、式4、4'、8、8'、20、20'、21、21'、23、及び23'の化合物が提供される。一実施態様において、実質的に単離された形態で、前記化合物が提供される。
下記の実施例を参照して、更に本発明を説明する
【実施例】
【0018】
(実施例1)
((R)-2-アミノ-3-(2,4-ジフルオロフェノキシ)プロピオン酸(4))
容器を窒素でパージし、続いて、L-セリンメチルエステル塩酸塩(25kg)及びジクロロメタン(400kg、300L)を加えた。グリコール冷却を用いて、該容器の内容物の温度を保持した(温度範囲15〜25℃)。トリエチルアミン(33.4kg)を45分かけて該容器に加えた。ジクロロメタン(265kg)中の塩化トリチル(45.7kg)の溶液を調製し、温度を15〜25℃に保持しながら、該溶液に3時間かけて加えた。得られた反応混合物を25〜30℃で6時間撹拌した。HPLC分析によって、反応の完了を確認した。
【0019】
水(263kg)を該容器に加え、該混合物を30分間撹拌し、30分静置した。下部の有機相を分離し、次いで上部の水相をDCM(90kg)で抽出した。あわせた有機相を容器に再度加え(水相を除去した後)、トルエン(450kg)を添加した。減圧蒸留を用いてDCMを蒸留した(35℃未満のベース温度及び大気圧を初期に用い、続いて、200mbarまで真空度を低下した。)。DCM/トルエン含有量について、留出物をガスクロマトグラフィーで測定した。
次いで、冷却水を用いて、該容器の内容物を30℃未満まで冷却し、該反応器を窒素でガス抜きした。該容器に2,4-ジフルオロフェノール(21.3kg)を加え、続いて、トリフェニルホシン(triphenylphoshine)(42.4kg)を加えた。該混合物を30分間撹拌した。反応温度を25〜30℃の範囲に保持しながら、アゾジカルボン酸ジイソプロピル(DIAD、40.9kg)を3時間30分かけて加えた。該反応混合物を、更に4時間撹拌した後、試料を採取した。反応はHPLCで分析し、出発物質は見られなかった。
【0020】
6Nの塩酸(400kg)を容器中の反応混合物に加え、該混合物を還流までゆっくりと温めた(実測温度=79.3℃)。該混合物を更に4時間還流で保持した後、60〜65℃まで冷却した。該混合物を60〜65℃で1時間静置した後に、下部の水相を分離した。有機相を60〜65℃で2Nの塩酸(20kg)で抽出し、水相を最初の下部の水相とあわせた。
該あわせた水相を20〜30℃まで冷却し、次いで、32% w/wの水酸化ナトリウム溶液(294.5kg使用)を用いて、pH6.8〜7.2にpHを調節した。得られた懸濁液を1時間撹拌し、pHを調べ、適宜6.8〜7.2に調節した。固形物を濾過し、濾過ケーキを水(175L)で洗浄した。次いで、濾紙上の固形物をアセトン(140kg)で再度スラリー化し、濾過した。固形物を可能な限り乾燥させて取り出し(21.3kg湿潤重量)、次いで、40〜45℃/100〜60mbarで乾燥した。
収量=15.25kg(乾燥生成物)。
【0021】
(実施例2)
((R)-(6,8-ジフルオロ-4-オキソクロマン-3-イル)カルバミン酸エチルエステル(21))
容器を窒素でパージし、1Mの水酸化ナトリウム(120kg)を加え、続いて、化合物4(15.25kg)を加えた。クロロギ酸エチル(9.5kg)を該容器に加え、該混合物を0〜10℃まで冷却した。1Nの水酸化ナトリウム(35kg)を用いて、該混合物のpHを8.9〜9.1に調節した。得られた混合物を0〜10℃で2時間撹拌した。該混合物から試料を採取し、薄層クロマトグラフィー(TLC)で分析した。反応は未完了であった。更に14.5kgの1Nの水酸化ナトリウムを添加した。反応が完了したことをTLCで確認した。
ジクロロメタン(265kg)を該容器に加えた。冷却しながら6Mの塩酸(20kg)を用いて、pHを0.9〜1.2に調節した。得られた混合物を更なる時間撹拌した。pHを調べたところ、更なる調節は必要なかった。1時間静置した後、下部の有機相を取除いた。該容器を空にした後、有機相を2Mの塩酸(60kg)及び飽和鹹水(60kg)で洗浄した。
【0022】
次いで、該有機相を容器に加え、減圧蒸留を用いて、およそ100Lまで容量を減少させた。温度が35℃を上回って上昇しないようにした(初期に大気圧を用い、続いて、380mbarまで減圧した。)。このストリップ工程の目的は、有機相を乾燥させることにある。溶媒のストリップに5時間要した。該混合物のカールフィッシャー分析から、水は0.1445%であった。
次いで、該混合物を冷却し、ドラムへ移した。次いで、該容器に五塩化リン(16kg)及びジクロロメタン(265kg)を加えた。懸濁液を0〜2℃まで冷却した。次いで、反応温度を0〜5℃の範囲に保持しながら、アミンを保護した溶液を3時間かけて加えた。得られた反応混合物を、0〜5℃の温度範囲で、更に4時間撹拌した。粗製のDCM溶液の中間体を清潔な乾燥ドラムに加えた。
【0023】
容器にジクロロメタン(265kg)及び塩化アルミニウム(25.1kg)を加え、該懸濁液を3〜5℃まで冷却した。3〜8℃の温度範囲を保持しながら、ドラムにかけた溶液を該容器に加えた。添加には1.5時間を要し、次いで、該混合物を更に6時間撹拌した。一方、およそ10% w/wの塩酸溶液(水285kg及び濃HCl31kg)を第2の容器で調製し、0℃まで冷した。10℃未満の温度を保持しながら、第1の容器の溶液を冷した酸溶液に注いで、反応を停止させた。実質的に固形の塩化アルミニウムは残留しなかった;少量の固形物が存在するだけであった。反応停止に3.25時間要し、続いて、1時間撹拌し、1時間静置した。有機相を分離して、残った水相をジクロロメタン(60kg)で抽出した。該有機相をあわせて、2Mの塩酸(230kg)、水(230kg)、10%の炭酸水素塩溶液(230kg)で洗浄し、続いて、飽和鹹水(95kg)で洗浄した。該有機相を、溶媒濃縮のために、容器に再び加え、次いで、20リッターのロータリーエバポレーターでの蒸発のために、25リッターの容器に移した。生成物を、湿潤した固形物になるまで蒸発させてトレーに移すことができるようにし、該生成物を真空オーブン中30℃で乾燥させ、残留している微量の溶媒を除去した。
収量=13.2kg、キラルHPLCによる鏡像体過剰率99.6%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量4μl、流れ:0.3ml/分、溶出液:30:70 MeOH:IPA。
【0024】
(実施例3)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
メタノール(265ml)中の化合物21(36g、132.7mmol)の懸濁液に、水素化ホウ素ナトリウム(5.5g、144.7mmol)を、5〜10℃の範囲の反応温度を保持しながら、20分間に、分割して加えた。該混合物を30分間で20℃まで温めて、水(20ml)を分割して加え、該混合物を減圧下で蒸発乾固した。残渣を酢酸エチル(250ml)と10%の鹹水(250ml)との間で分配した。有機相を乾燥させ(MgSO4)、減圧下で蒸発乾固した。得られるオイル(37.5g)を、ジクロロメタン(260ml)及びメタンスルホン酸(87ml)の混合物中、10%のPd/C(2.6g)上、100psiの水素圧及び20〜25℃にて7時間、水素化した。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(65ml)、水(65ml)及びジクロロメタン(65ml)で洗浄した。撹拌及び氷冷して30℃未満の温度を保持しながら、4NのNaOH(348ml)を濾液に添加した。有機相を鹹水(100ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固した。収量28g(82%)、キラルHPLCによる鏡像体過剰率98%。AB:次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0025】
(実施例4)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
容器を窒素でパージした。化合物21(13.2kg)を容器に加え、続いて、メタノール(155kg)を加えた。該懸濁液を5〜10℃に冷却し、次いで、反応温度を5〜10℃の範囲に保持しながら、水素化ホウ素ナトリウム(2.1kg)を該容器に分割して(200g)加えた。ガスの発生を観察し、発泡が停止した後に次の水素化ホウ素ナトリウムを添加するようにした。水素化ホウ素ナトリウムの添加に4時間要した。該反応混合物を、5〜10℃の範囲に温度を保持しながら、4時間撹拌した。採取した試料からは出発物質の残留は見られなかった。
【0026】
アセトン(20kg)を加えて反応停止させ、該混合物を更に2時間撹拌した。減圧蒸留を用いて、メタノール/アセトンの容量を減少させた(45℃以下のベース温度及び大気圧を初期に用い、続いて、170mbarまで真空度を低下した)。ストリップに6.5時間要し、次いで、該濃縮溶液を25リッターの容器に移して、実験室にて更に蒸発させた。重量21.9 kgの濃厚な、粘着性の残渣が得られた。該残渣をジクロロメタン(300kg)に溶解し、有機混合物を10% の鹹水溶液(100kg)で抽出した。次いで、該鹹水溶液をジクロロメタン(100kg)で逆抽出した。あわせた有機相を容器に加えた。該容器を窒素で0.5 barまで2回パージした。触媒である木炭(1.25kg乾燥基準)上の10%のパラジウムを、該容器に加え、続いて、メタンスルホン酸(72 kg)を加えた。最終圧力を調べながら、該容器を窒素で0.5 barまで加圧した。次いで、該容器を水素で3回0.5 barまで加圧した。撹拌機を始動し、圧力を1.2barまで増加させた。該混合物を26時間水素化させた。窒素でパージした後、該混合物をCelite(商標)を用いて濾過した。有機相を分離したままにした。次いで、Celite(商標)及び触媒を水(100kg)で洗浄した。清潔な容器に水を再び入れ、冷却しながら、極めてゆっくりとジクロロメタン相を添加した。有機相と水相との混合において、大きな発熱が観察された。2相を混合すると、該混合物を5〜10℃に冷却した後、4Nの水酸化ナトリウム(およそ172 kg添加)でpHを10に調節した。冷却を必要とし、pHの調節には2.5時間要した。混合物をCelite(商標)を用いて濾過し、水(30kg)で洗浄した。該混合物を容器に入れて静置し、次いで、有機相を除去した。減圧蒸留を使用して、1.5時間かけて有機相の容量を60Lまで減少させた(40℃以下のベース温度及び大気圧を初期に用い、続いて、200mbarまで真空度を低下した。)。次いで、生成物を単離するために、20リッターのロータリーエバポレーターに濃縮物を移した。微量の溶媒をも乾燥させるために、生成物を、真空下、30℃でオーブンに設置した。収量7.87kg、キラルHPLCによる鏡像体過剰率94.1%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0027】
(実施例5)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
化合物21(30g)をメタノール(250ml)に取り、反応温度を5〜10℃の範囲に保持しながら、水素化ホウ素ナトリウム(4.8g)を分割して加えた。全ての水素化ホウ素ナトリウムを添加した後、溶液を観察した。該混合物を室温で2時間撹拌した。該反応溶液にアセトン(20ml)を加えて反応を停止させ、該混合物を一晩撹拌した。留出物中のメタノール含有量が0.7%(GC)に達するまで、クロロホルムを周期的に加えて、メタノールを大気圧で蒸留し、全体の容量が250mlになるようにクロロホルムで調節した。該混合物を10%の鹹水(200ml)で洗浄し、有機相を分析し(カールフィッシャー=0.20%の水)、ジクロロメタン(100ml)及びメタンスルホン酸(117g)と混合し、10%のPd/C(2.93g)上、5bar及び25℃で16時間、水素化した。HPLCによると、反応は完了していなかったので、更に53gのメタンスルホン酸を添加し、更に8時間水素化を続けた。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(65ml)、水(65ml)及びジクロロメタン(60ml)で洗浄した。撹拌及び氷冷して30℃未満の温度を保持しながら、4NのNaOH(340ml)を濾液に添加した。有機相を鹹水(100ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固した。収量23.5g(83%)、キラルHPLCによる鏡像体過剰率97%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0028】
(実施例6)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
メタノール(0.5ml)とTHF(0.5ml)との混合物中の化合物21(0.14g、0.5mmol)の懸濁液に、反応温度を5〜10℃の範囲に維持しながら、水素化ホウ素ナトリウム(0.04g、1mmol)を加えた。該混合物を30分間で20℃まで温めた。水(1ml)を分割して加え、該混合物を減圧下で乾燥するまで蒸発させた。残渣を酢酸エチル(2ml)と10%の鹹水(2ml)との間で分配し、有機相を乾燥させ(MgSO4)、減圧下で蒸発乾固した。ジクロロメタン(1ml)及びメタンスルホン酸(0.33ml)の混合物中、10%のPd/C(0.01g)上、100psiの水素圧、20〜25℃にて16時間、得られたオイル(0.13g)を水素化した。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(2ml)、水(2ml)及びジクロロメタン(2ml)で洗浄した。撹拌及び30℃未満の温度を維持するための氷冷をしながら、4NのNaOH(1.5ml)を濾液に添加した。有機相を鹹水(2ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固させた。収量0.087g(69%)、キラルHPLCによる鏡像体過剰率96%。AB:次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0029】
(実施例7)
((R)-6,8-ジフルオロクロマン-3-イルアミンL-酒石酸塩(8))
容器を窒素でパージし、次いで、化合物22(13.7kg)を加え、続いて、メタノール(86kg)を加えた。該混合物を63〜65℃まで温めた。反応温度を63〜65℃の範囲に保持しながら、水酸化カリウム(40%、51kg)溶液を一度に加え、15分後、第2の40%の水酸化カリウム(44kg)を30分間かけて添加した。反応温度を65〜70℃の範囲に保持しながら、得られた混合物を更に24時間撹拌した。次いで、該混合物を冷却し、水を加えた(44kg)。該混合物を再度加熱し、減圧蒸留を用いてメタノールを除去した(70℃以下のベース温度及び大気圧を初期に用い、続いて、125mbarまで真空度を低下した。)。蒸留には5.5時間要した。該反応混合物を30℃まで冷却し、残留する水相をトルエン(152kg)で2回抽出した。あわせた有機相を減圧蒸留によって、およそ6OLの容量まで減少させた(3.5時間)。変性エタノールを加え(395kg)、トルエンのレベルが2%未満になるまで(標準的なGC分析を用いて測定した。)蒸留を続けた。容器の内容物を20℃まで冷却し、元の変性エタノールの容量になるように、新鮮な変性エタノールで容量を調節した。水(72kg)中のL-酒石酸(8.86kg)の溶液を調製し、反応温度を15〜25℃の範囲に保持しながら、この溶液を1時間かけて加えた。得られた沈殿物を1時間撹拌した。次いで、容器の内容物を濾過し、濾過ケーキを変性エタノール(200L)で洗浄した。生成物を60〜100mbarの真空にて45〜50℃で乾燥した。収量=15.1kg、キラルHPLCによる鏡像体過剰率99.0%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:0.2%のtert-ブチルアミンを含む70:30 MeOH:IPA。
【0030】
(実施例8)
((R)-2-{2-[3-(6,8-ジフルオロクロマン-3-イル)-2-チオキソ-2,3-ジヒドロ-1H-イミダゾール-4-イル]エチルイソインドール-1,3-ジオン(14)の製造)
【化11】
【表1】
【0031】
反応器に固体試薬を入れて、酢酸を一度に加え、該混合物を窒素下で撹拌しながら105〜110℃に加熱し、これら条件下で2時間保持した。約90℃で加熱しながら、水(24mL)をゆっくりと加えた(結晶化が起こった。)。該懸濁液を撹拌しながら氷浴で冷却し、氷中で0.5時間撹拌した。水(24mL)をゆっくりと加え、1時間撹拌を続けた。沈殿物を収集し、AcOH-水(1:1 v/v)及び水で洗浄し、次いで、真空中50〜60℃で乾燥した。得られる固形物(5.25g、99%)を、還流下でIPA(48mL)とDCM(72mL)との混合物に溶解させた。不溶性の物質(K酒石酸塩)を濾過し、ロータバップで約50℃及び500mbarにて結晶化が起こるまで、濾液を蒸発させた。次いで、加熱を止め、残圧を測定するための負圧計を用いて、蒸発を続けて残留のDCMを除去した。懸濁液を冷蔵庫に一晩放置し、結晶を収集し、IPAで洗浄した後に、真空中50℃にて乾燥した。収量3.40g(64%)。
【0032】
(実施例9)
((R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩(BIA 5-453)(1)の製造、形態A製造のための改良)
【化12】
【表2】
【0033】
2-プロパノール、水、及びDCMの混合物中の14の懸濁液に、18℃にて1分間に、撹拌しながら、NaBH4を一度に加えた(温度は1時間後に27.5℃まで上昇した。)。該混合物を18〜20℃で16時間撹拌した(1時間でほとんど透明な溶液)。該混合物を氷浴にて冷却し、温度を10℃未満に保持しながら、6NのHCl(39.6mL、237.6mmol)を滴加した。該混合物を15分間撹拌し、固形物を濾過し、濾過ケーキをDCM(300mL)で洗浄した(4.9gの固形物を得た。)。5NのNaOH(60mL)を母液に添加し、該混合物を15分間撹拌した。有機の上の層を分離し、鹹水で洗浄し、最小量の固形物を濾過して除去した。得られる透明溶液に6NのHCl(40ml)を添加し、蒸気温度が76〜78℃に達するまでDCMを蒸留して除去し、該混合物を還流下で1.5時間撹拌し、室温まで冷却した。水(300mL)を加え、IPAをロータバップで除去した(420mL収集された)。次いで、残渣をEtOAc-石油エーテル(2:1 v/v)混合物(200及び100mL)で洗浄した。2回の洗浄後、水相で結晶化を開始した。6NのHCl(40mL)を添加し、該懸濁液を撹拌しながら1時間、氷中で冷却した。沈殿物を収集し、冷した3NのHCl(75mL)及び冷したIPA(50mL)で洗浄し、次いで、真空中50℃で乾燥した。収量11.58g(73%)。
【0034】
(実施例10)
((R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩(BIA 5-453)(1)の製造、形態C製造のための改良)
【化13】
【表3】
【0035】
2-プロパノール、水、及びDCMの混合物中の14の懸濁液に、20℃にて1分間に撹拌しながら、NaBH4を一度に加えた(温度は22℃まで上昇した。)。該混合物を20℃で16時間撹拌し(0.5時間後、透明な溶液が生成された。)、6NのHClを滴加した。加熱温度が76〜78℃に達するまでDCMを蒸留し、該混合物を還流下で1.5時間撹拌し、室温まで冷却した。水(30mL)を加えた。2-プロパノールをロータバップで除去し、残渣をEtOAc-石油エーテル(2:1 v/v)混合物(2x20mL)で2回洗浄した。DCM中の10%の2-プロパノールの溶液(40mL)を、撹拌しながら水層に加え、続いて、5NのNaOHを添加してpH9〜10にした。有機層を分離し、乾燥し(MgSO4)、蒸発乾固した。無水EtOH(15mL)と無水EtOH(15mL)中3MのHCl(1.5mL、混合物のpHは約2)との混合物に、残渣を加熱しながら溶解させた。得られる溶液を65〜70℃で2時間撹拌し、結晶を収集し、EtOHで洗浄し、真空中40℃で乾燥した。収量1.12g(71%)。
【0036】
(X線粉末回折(XRPD))
次のShimadzu社のパラメータを使用して、形態A及びCのXRPDのピークリストを生成した。
【表4】
【0037】
(FT-IR分光)
赤外スペクトルは、Ever-Glo 中/遠IR源、広範囲の臭化カリウム(KBr)ビームスプリッタ、及び重水素化硫酸グリシン(DTGS)検出器を備えた、Magna-IR 860(登録商標)フーリエ変換赤外(FT-IR)分光光度計(Thermo Nicolet社)で取得した。サンダードームアクセサリーを使用して試料採取を行った。バックグラウンドのデータセットをクリーンなGe結晶から得た。Log 1/R(反射率)スペクトルは、互いに対してこれら2つのデータセットの比率をとることによって得られた。波長の校正は、ポリスチレンを用いて行った。更なるパラメータは下記である。
【表5】
【技術分野】
【0001】
本発明は、カルバミン酸エチルエステルを製造する方法に関する。このようなカルバミン酸エステルは、WO2004/033447に記載されているように、ドーパミン-β-ヒドロキシラーゼ阻害剤の製造に有用である。
【背景技術】
【0002】
下記のアミン8は、WO2004/033447に記載されるドーパミン-β-ヒドロキシラーゼ阻害剤の製造における有用な中間体である。これは、L-セリンメチルエステル塩酸塩(2)から出発して、そのN-トリチル誘導体と2,4-ジフルオロフェノールとを光延条件下で縮合し(Cherney, R. L.; Wang, L.の論文「N-フェニルフルオレニル又はN-トリチルセリンエステルを用いる効率的光延反応(Efficient Mitsunobu reactions with N-phenylfluorenyl or N-trityl serine esters)」J. Org. Chem. 1996, v. 61, N 7, p. 2544-2546)、続いて、脱保護し、得られるアミノ酸(4)をエトキシカルボニル化し、N保護された誘導体(20)をフリーデル-クラフツ環化し(McClure, D. E.; Arison, B. H.; Jones, J. H.; Baldwin, J. J.の論文「保護アミノ酸のフリーデル-クラフツ反応からのキラルα-アミノケトン(Chiral α-amino ketones from the Friedel-Crafts reaction of protected amino acids)」J. Org. Chem. 1981, v.46, N 11, p. 2431-2433)、かつエトキシカルボニルアミノケトン(21)を還元することによって合成できる。カルバミン酸エチル(22)のアルカリ加水分解によって8が得られる。
【化1】
【0003】
他の鏡像異性体を用いて、同様の反応を下記のように行うことができる。
【化2】
上のスキーム中の第2、3、4、5、及び6の化合物は、それぞれ化合物4'、20'、21'、22'及び8'である。
【0004】
アリールケトン(化合物21など)のメチレン化合物(化合物22など)への還元は、合成的に有用な反応であり、この変換のために様々な方法が開発されている。すなわち、クレメンゼン及びウォルフ-キッシュナー還元、接触水素化分解(Sarda, N., Grouiller, A., Pacheco, H.の論文(Nouvelle synthese de ramino-3-chromanne. Synthese et configuration absolue de ses enantiomeres)Tetrahedron Letters 1976, N 4, p. 271-272;Norlander, E. J., Njoroge, F. G., Payne, M. J., Warman, D.の論文「フリーデル-クラフツ合成のためのキラル試薬としてのN-(トリフルオロアセチル)-α-アミノ酸塩酸塩(N-(Trifluoroacetyl)-α-amino acid chlorides as chiral reagents for Friedel-Crafts synthesis)」J. Org. Chem. 1985, v. 50, N 19, p. 3481-3484;Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 49, N 32, p. 4107-4111)、Et3SiH/CF3COOH(Norlander, E. J., Njoroge, F. G., Payne, M. J., Warman, D.の論文「フリーデル-クラフツ合成のためのキラル試薬としてのN-(トリフルオロアセチル)-α-アミノ酸塩酸塩(N-(Trifluoroacetyl)-α-amino acid chlorides as chiral reagents for Friedel-Crafts synthesis)」J. Org. Chem. 1985, v. 50, N 19, p. 3481-3484;Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 49, N 32, p. 4107-4111)、Et3SiH/BF3Et2O(Norlander, E. J., Payne, M. J., Njoroge, F. G., Balk, M. A., Laikos, G. D., Vishvanath, V. M.の論文「N-(トリフルオロアセチル)-α-アミノ酸塩酸塩を用いるフリーデル-クラフツアシル化 β-アリールアルキルアミン及び3-置換1,2,3,4-テトラヒドロイソキノリンの製造への応用(Friedel-Crafts acylation with N-(trifluoroacetyl)-α-amino acid chlorides. Application to the preparation of β-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines)」J. Org. Chem. 1984, v. 1049, N 32, p. 4107-4111)、Et3SiH/TiCl4(Yato, M.; Homma, K.; Ishida, A.の論文「四塩化チタンによって媒介される芳香族ケトンの新規シラン還元:γ-及びδ-アリール置換アミノ酸の合成(New silane reduction of aromatic ketones mediated by titanium tetrachloride: a synthesis of γ- and δ-aryl substituted amino acids)」Heterocycles 1998, v. 49, p. 233-254)、NaBH4/BF3Et2O(Perry, P. J.; Pavlidis, V. H.; Coutts, I. G. C.の論文「ヨードトリメチルシランを用いたα,α-ジアリールアルコールの対応するアルカンへの高速還元(The rapid reduction of α,α-diaryl alcohols to the corresponding alkanes using iodotrimethylsilane)」Synth. Commun. 1996, v. 26, N 1, p. 101-111)である。上述の方法のいずれも、21から22へ変換して中間体のアルコール又はその出発物質との混合物を得ることに対して、有効性を提供するものではない。この種の変換に有効であることが周知の他の試薬、ヨードトリメチルシラン(Pettit, G. R.; Piatak, D. M.の論文、J. Org. Chem. 1962, v. 27, p. 2127)又はその等価物(Cain, G. A.; Holler, E. R.の論文「拡張した範囲でのその場のヨードトリメチルシラン媒介によるベンジルアルコールの選択的還元(Extended scope of in situ iodotrimethysilane mediated selective reduction of benzylic alcohols)」Chem. Commun. 2001, p. 1168-1169)などを用いて、中間体のアルコールを22に還元する試みは、どちらも作用しなかった。接触還元法を予め用いてネピカスタット中間体を製造する(米国特許第5,438,150号)ことに相当する、ある種のスキームを用いて、結果を改善することができる。
【化3】
【0005】
しかし、化合物22の収率は、50%を上回ることはなく、主な副生成物は、トリフルオロアセチル化されたアルコールに相当するものである。
【化4】
更に、トリフルオロ酢酸と硫酸との高腐食性の混合物を大量に用いることは、問題を引起こし得る。トリフルオロ酢酸を酢酸に置換える試みは、かなりの量のアセトキシ副生成物を伴い、低い収率となった。
【発明の概要】
【0006】
驚くべきことに、本発明者らは、メタンスルホン酸などのC1〜C6のアルキルスルホン酸、又は好ましくは、3容量のジクロロメタンなどの塩素化溶媒と1容量のメタンスルホン酸などのC1〜C6のアルキルスルホン酸との混合物中で還元を行い、21から22への完全な変換が得られることを見出した。
【0007】
しかし、化合物22の光学純度の分析によって、顕著なラセミ化が明らかとなった。そのラセミ化の原因は、3容量のジクロロメタンなどの塩素化溶媒と1容量のメタンスルホン酸との混合物中における、出発物質の鏡像異性の不安定性にあることが見出された。こうした結果に基づいて、光学純度における状態を改善するために、水素化ホウ素ナトリウムを用いて21から中間体のアルコールへ変換し、続いて3容量のジクロロメタンと1容量のメタンスルホン酸との混合物中で接触還元を行うことを含む、2工程の方法が案出された。該2工程の方法によって、再現性よく、94-99% eeという高収率の化合物22が得られた。
【図面の簡単な説明】
【0008】
【図1】(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Aは、4.9、8.3、12.9、15.0、16.2、19.8、21.8、22.9及び24.2、26.8±0.2 °2θにピークを有するXRPDパターンを有する。図1に該XRPDパターンを示す。
【図2】更に、(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Aは、3053.30、2939.70、1599.80、1491.90、1448.30、1406.10、1330.70、1287.60、1244.50、1220.70、1194.00、1117.50、1039.50、985.50、851.80、747.00及び713.70 cm-1に特有のFT-IRのピークを有する。図2に該FT-IRスペクトルを示す。
【図3】(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Cは、13.9、15.3、16.2、16.7、17.7、18.1、20.2、21.0、22.1、24.2、25.1及び25.7にピークを有するXRPDパターンを有する。図3に該XRPDパターンを示す。
【図4】更に、(R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩の結晶形態Cは、3041.70、1596.50、1492、1403.40、1333.80、1290.90、1220.2、1173.20、1117.4、1078.10、1033.4、984.90、845.2、792.6、750.1及び713.20cm-1に特有のFT-IRのピークを有する。図4に該FT-IRスペクトルを示す。
【発明を実施するための形態】
【0009】
したがって、本発明の一態様によれば、式22の化合物の製造方法が提供され:
【化5】
該方法は、式21の化合物:
【化6】
を還元して、式23の化合物:
【化7】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23の化合物の水素化分解を行うことを含む。これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。
【0010】
好ましくは、Rは、エチル、メチル、tBu、又はベンジル、最も好ましくは、エチルである。
好ましくは、アルキルスルホン酸は、メタンスルホン酸である。
反応は、実質的に純粋なアルキルスルホン酸の溶媒中で行うことが可能であるが、塩素化溶媒及びC1〜C6のアルキルスルホン酸を含む溶媒混合物中で、該反応を行うことが好ましい。更に、該アルキルスルホン酸は、メタンスルホン酸であることが好ましい。塩素化溶媒とアルキルスルホン酸溶媒との比率は、10:1以下であることが好ましく、より好ましくは、5:1以下であり、更により好ましくは、4:1〜2:1の範囲内である。最も好ましくは、塩素化溶媒とアルキルスルホン酸溶媒との比率は、約3:1である。
【0011】
一般に、本方法の第1工程は、(R)-(6,8-ジフルオロ-4-オキソクロマン-3-イル)カルバミン酸エチルエステル(21)を還元することを含む。これは、適当な溶媒中で、水素化ホウ素、特に、水素化ホウ素ナトリウムなどのアルカリ金属の水素化ホウ素を用いて行うことができる。該溶媒は、低級アルコールとTHFとの任意の組み合わせとし得る。該低級アルコールは、炭素原子1〜6個を有する、直鎖又は分枝のアルコールであってよく;該低級アルコールは、エタノール、又は最も好ましくは、メタノールとし得る。好適には、この還元は、5〜25℃にて0.5〜4時間で行うことができる。
【0012】
該還元の工程の後に、中間体のアルコールの水素化分解が続き、好ましくは、パラジウム触媒上で行う(7% w/wの活性炭上の無水の10%のPdを使用できる。)。好適には、該反応は、溶媒混合物中、15〜100psiの水素圧及び20〜25℃の温度にて7〜26時間で行うことができる。1容量未満のメタンスルホン酸では、変換が低くなることが見出されている。
【0013】
塩素化溶媒とアルキルスルホン酸溶媒との比率が、5:1未満の場合、該反応には、100psiの水素圧及び15時間の反応時間とともに10% w/wのPdが要求されるだろう。このように、塩素化溶媒の量が多くなるほど、反応時間、水素圧、及びPdの必要量が増加する傾向がある。
有利には、該塩素化溶媒は、ハロゲン化アルキル、好ましくは、C1〜C6のハロゲン化アルキル、より好ましくは、ハロゲン化メチル又はエチルである。好ましくは、ハロゲン化物は、塩化物である。最も好ましくは、該塩素化溶媒は、ジクロロメタン又はジクロロメタン-クロロホルムの混合物である。あるいは、該塩素化溶媒は、1,2-ジクロロエタンであってよい。
【0014】
本発明の別の態様によれば、式8の化合物の製造方法が提供され、該方法は、上記の方法を用いて式22の化合物を形成し、続いて、式22の化合物をアルカリ加水分解することを含む。
該アルカリ加水分解の工程は、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びL-酒石酸の存在下で行うことができる。好ましくは、該アルカリ金属の水酸化物は、水酸化カリウムである。好ましくは、該アルコールは、メタノールである。
【0015】
本発明の別の態様によれば、式14の化合物の製造方法が提供され:
【化8】
例えば、上記の方法によって、式8の化合物を製造すること、次いで、式8の化合物と式13の化合物とを反応させることを含む。
【化9】
好ましくは、該反応は、アルカリ金属のイソチオシアン酸塩、好ましくは、チオシアン酸カリウム、及び有機溶媒、好ましくは、酢酸の存在下で行われる。
【0016】
本発明の別の態様によれば、式1の化合物の製造方法が提供され:
【化10】
上記の方法によって、式14の化合物を製造すること、次いで溶媒の存在下で、アルカリ金属水素化ホウ素、好ましくは、水素化ホウ素ナトリウム(NaBH4)を用いて、式14の化合物を式1の化合物に変換すること、続いて、HClを添加し、次いで、式1の化合物を回収することを含む。
好ましくは、化合物14の回収に使用される溶媒は、イソプロピルアルコール、水及びジクロロメタンの混合物である。
【0017】
本発明の更なる態様によれば、式4、4'、8、8'、20、20'、21、21'、23、及び23'の化合物が提供される。一実施態様において、実質的に単離された形態で、前記化合物が提供される。
下記の実施例を参照して、更に本発明を説明する
【実施例】
【0018】
(実施例1)
((R)-2-アミノ-3-(2,4-ジフルオロフェノキシ)プロピオン酸(4))
容器を窒素でパージし、続いて、L-セリンメチルエステル塩酸塩(25kg)及びジクロロメタン(400kg、300L)を加えた。グリコール冷却を用いて、該容器の内容物の温度を保持した(温度範囲15〜25℃)。トリエチルアミン(33.4kg)を45分かけて該容器に加えた。ジクロロメタン(265kg)中の塩化トリチル(45.7kg)の溶液を調製し、温度を15〜25℃に保持しながら、該溶液に3時間かけて加えた。得られた反応混合物を25〜30℃で6時間撹拌した。HPLC分析によって、反応の完了を確認した。
【0019】
水(263kg)を該容器に加え、該混合物を30分間撹拌し、30分静置した。下部の有機相を分離し、次いで上部の水相をDCM(90kg)で抽出した。あわせた有機相を容器に再度加え(水相を除去した後)、トルエン(450kg)を添加した。減圧蒸留を用いてDCMを蒸留した(35℃未満のベース温度及び大気圧を初期に用い、続いて、200mbarまで真空度を低下した。)。DCM/トルエン含有量について、留出物をガスクロマトグラフィーで測定した。
次いで、冷却水を用いて、該容器の内容物を30℃未満まで冷却し、該反応器を窒素でガス抜きした。該容器に2,4-ジフルオロフェノール(21.3kg)を加え、続いて、トリフェニルホシン(triphenylphoshine)(42.4kg)を加えた。該混合物を30分間撹拌した。反応温度を25〜30℃の範囲に保持しながら、アゾジカルボン酸ジイソプロピル(DIAD、40.9kg)を3時間30分かけて加えた。該反応混合物を、更に4時間撹拌した後、試料を採取した。反応はHPLCで分析し、出発物質は見られなかった。
【0020】
6Nの塩酸(400kg)を容器中の反応混合物に加え、該混合物を還流までゆっくりと温めた(実測温度=79.3℃)。該混合物を更に4時間還流で保持した後、60〜65℃まで冷却した。該混合物を60〜65℃で1時間静置した後に、下部の水相を分離した。有機相を60〜65℃で2Nの塩酸(20kg)で抽出し、水相を最初の下部の水相とあわせた。
該あわせた水相を20〜30℃まで冷却し、次いで、32% w/wの水酸化ナトリウム溶液(294.5kg使用)を用いて、pH6.8〜7.2にpHを調節した。得られた懸濁液を1時間撹拌し、pHを調べ、適宜6.8〜7.2に調節した。固形物を濾過し、濾過ケーキを水(175L)で洗浄した。次いで、濾紙上の固形物をアセトン(140kg)で再度スラリー化し、濾過した。固形物を可能な限り乾燥させて取り出し(21.3kg湿潤重量)、次いで、40〜45℃/100〜60mbarで乾燥した。
収量=15.25kg(乾燥生成物)。
【0021】
(実施例2)
((R)-(6,8-ジフルオロ-4-オキソクロマン-3-イル)カルバミン酸エチルエステル(21))
容器を窒素でパージし、1Mの水酸化ナトリウム(120kg)を加え、続いて、化合物4(15.25kg)を加えた。クロロギ酸エチル(9.5kg)を該容器に加え、該混合物を0〜10℃まで冷却した。1Nの水酸化ナトリウム(35kg)を用いて、該混合物のpHを8.9〜9.1に調節した。得られた混合物を0〜10℃で2時間撹拌した。該混合物から試料を採取し、薄層クロマトグラフィー(TLC)で分析した。反応は未完了であった。更に14.5kgの1Nの水酸化ナトリウムを添加した。反応が完了したことをTLCで確認した。
ジクロロメタン(265kg)を該容器に加えた。冷却しながら6Mの塩酸(20kg)を用いて、pHを0.9〜1.2に調節した。得られた混合物を更なる時間撹拌した。pHを調べたところ、更なる調節は必要なかった。1時間静置した後、下部の有機相を取除いた。該容器を空にした後、有機相を2Mの塩酸(60kg)及び飽和鹹水(60kg)で洗浄した。
【0022】
次いで、該有機相を容器に加え、減圧蒸留を用いて、およそ100Lまで容量を減少させた。温度が35℃を上回って上昇しないようにした(初期に大気圧を用い、続いて、380mbarまで減圧した。)。このストリップ工程の目的は、有機相を乾燥させることにある。溶媒のストリップに5時間要した。該混合物のカールフィッシャー分析から、水は0.1445%であった。
次いで、該混合物を冷却し、ドラムへ移した。次いで、該容器に五塩化リン(16kg)及びジクロロメタン(265kg)を加えた。懸濁液を0〜2℃まで冷却した。次いで、反応温度を0〜5℃の範囲に保持しながら、アミンを保護した溶液を3時間かけて加えた。得られた反応混合物を、0〜5℃の温度範囲で、更に4時間撹拌した。粗製のDCM溶液の中間体を清潔な乾燥ドラムに加えた。
【0023】
容器にジクロロメタン(265kg)及び塩化アルミニウム(25.1kg)を加え、該懸濁液を3〜5℃まで冷却した。3〜8℃の温度範囲を保持しながら、ドラムにかけた溶液を該容器に加えた。添加には1.5時間を要し、次いで、該混合物を更に6時間撹拌した。一方、およそ10% w/wの塩酸溶液(水285kg及び濃HCl31kg)を第2の容器で調製し、0℃まで冷した。10℃未満の温度を保持しながら、第1の容器の溶液を冷した酸溶液に注いで、反応を停止させた。実質的に固形の塩化アルミニウムは残留しなかった;少量の固形物が存在するだけであった。反応停止に3.25時間要し、続いて、1時間撹拌し、1時間静置した。有機相を分離して、残った水相をジクロロメタン(60kg)で抽出した。該有機相をあわせて、2Mの塩酸(230kg)、水(230kg)、10%の炭酸水素塩溶液(230kg)で洗浄し、続いて、飽和鹹水(95kg)で洗浄した。該有機相を、溶媒濃縮のために、容器に再び加え、次いで、20リッターのロータリーエバポレーターでの蒸発のために、25リッターの容器に移した。生成物を、湿潤した固形物になるまで蒸発させてトレーに移すことができるようにし、該生成物を真空オーブン中30℃で乾燥させ、残留している微量の溶媒を除去した。
収量=13.2kg、キラルHPLCによる鏡像体過剰率99.6%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量4μl、流れ:0.3ml/分、溶出液:30:70 MeOH:IPA。
【0024】
(実施例3)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
メタノール(265ml)中の化合物21(36g、132.7mmol)の懸濁液に、水素化ホウ素ナトリウム(5.5g、144.7mmol)を、5〜10℃の範囲の反応温度を保持しながら、20分間に、分割して加えた。該混合物を30分間で20℃まで温めて、水(20ml)を分割して加え、該混合物を減圧下で蒸発乾固した。残渣を酢酸エチル(250ml)と10%の鹹水(250ml)との間で分配した。有機相を乾燥させ(MgSO4)、減圧下で蒸発乾固した。得られるオイル(37.5g)を、ジクロロメタン(260ml)及びメタンスルホン酸(87ml)の混合物中、10%のPd/C(2.6g)上、100psiの水素圧及び20〜25℃にて7時間、水素化した。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(65ml)、水(65ml)及びジクロロメタン(65ml)で洗浄した。撹拌及び氷冷して30℃未満の温度を保持しながら、4NのNaOH(348ml)を濾液に添加した。有機相を鹹水(100ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固した。収量28g(82%)、キラルHPLCによる鏡像体過剰率98%。AB:次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0025】
(実施例4)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
容器を窒素でパージした。化合物21(13.2kg)を容器に加え、続いて、メタノール(155kg)を加えた。該懸濁液を5〜10℃に冷却し、次いで、反応温度を5〜10℃の範囲に保持しながら、水素化ホウ素ナトリウム(2.1kg)を該容器に分割して(200g)加えた。ガスの発生を観察し、発泡が停止した後に次の水素化ホウ素ナトリウムを添加するようにした。水素化ホウ素ナトリウムの添加に4時間要した。該反応混合物を、5〜10℃の範囲に温度を保持しながら、4時間撹拌した。採取した試料からは出発物質の残留は見られなかった。
【0026】
アセトン(20kg)を加えて反応停止させ、該混合物を更に2時間撹拌した。減圧蒸留を用いて、メタノール/アセトンの容量を減少させた(45℃以下のベース温度及び大気圧を初期に用い、続いて、170mbarまで真空度を低下した)。ストリップに6.5時間要し、次いで、該濃縮溶液を25リッターの容器に移して、実験室にて更に蒸発させた。重量21.9 kgの濃厚な、粘着性の残渣が得られた。該残渣をジクロロメタン(300kg)に溶解し、有機混合物を10% の鹹水溶液(100kg)で抽出した。次いで、該鹹水溶液をジクロロメタン(100kg)で逆抽出した。あわせた有機相を容器に加えた。該容器を窒素で0.5 barまで2回パージした。触媒である木炭(1.25kg乾燥基準)上の10%のパラジウムを、該容器に加え、続いて、メタンスルホン酸(72 kg)を加えた。最終圧力を調べながら、該容器を窒素で0.5 barまで加圧した。次いで、該容器を水素で3回0.5 barまで加圧した。撹拌機を始動し、圧力を1.2barまで増加させた。該混合物を26時間水素化させた。窒素でパージした後、該混合物をCelite(商標)を用いて濾過した。有機相を分離したままにした。次いで、Celite(商標)及び触媒を水(100kg)で洗浄した。清潔な容器に水を再び入れ、冷却しながら、極めてゆっくりとジクロロメタン相を添加した。有機相と水相との混合において、大きな発熱が観察された。2相を混合すると、該混合物を5〜10℃に冷却した後、4Nの水酸化ナトリウム(およそ172 kg添加)でpHを10に調節した。冷却を必要とし、pHの調節には2.5時間要した。混合物をCelite(商標)を用いて濾過し、水(30kg)で洗浄した。該混合物を容器に入れて静置し、次いで、有機相を除去した。減圧蒸留を使用して、1.5時間かけて有機相の容量を60Lまで減少させた(40℃以下のベース温度及び大気圧を初期に用い、続いて、200mbarまで真空度を低下した。)。次いで、生成物を単離するために、20リッターのロータリーエバポレーターに濃縮物を移した。微量の溶媒をも乾燥させるために、生成物を、真空下、30℃でオーブンに設置した。収量7.87kg、キラルHPLCによる鏡像体過剰率94.1%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0027】
(実施例5)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
化合物21(30g)をメタノール(250ml)に取り、反応温度を5〜10℃の範囲に保持しながら、水素化ホウ素ナトリウム(4.8g)を分割して加えた。全ての水素化ホウ素ナトリウムを添加した後、溶液を観察した。該混合物を室温で2時間撹拌した。該反応溶液にアセトン(20ml)を加えて反応を停止させ、該混合物を一晩撹拌した。留出物中のメタノール含有量が0.7%(GC)に達するまで、クロロホルムを周期的に加えて、メタノールを大気圧で蒸留し、全体の容量が250mlになるようにクロロホルムで調節した。該混合物を10%の鹹水(200ml)で洗浄し、有機相を分析し(カールフィッシャー=0.20%の水)、ジクロロメタン(100ml)及びメタンスルホン酸(117g)と混合し、10%のPd/C(2.93g)上、5bar及び25℃で16時間、水素化した。HPLCによると、反応は完了していなかったので、更に53gのメタンスルホン酸を添加し、更に8時間水素化を続けた。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(65ml)、水(65ml)及びジクロロメタン(60ml)で洗浄した。撹拌及び氷冷して30℃未満の温度を保持しながら、4NのNaOH(340ml)を濾液に添加した。有機相を鹹水(100ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固した。収量23.5g(83%)、キラルHPLCによる鏡像体過剰率97%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0028】
(実施例6)
((R)-(6,8-ジフルオロクロマン-3-イル)カルバミン酸エチルエステル(22))
メタノール(0.5ml)とTHF(0.5ml)との混合物中の化合物21(0.14g、0.5mmol)の懸濁液に、反応温度を5〜10℃の範囲に維持しながら、水素化ホウ素ナトリウム(0.04g、1mmol)を加えた。該混合物を30分間で20℃まで温めた。水(1ml)を分割して加え、該混合物を減圧下で乾燥するまで蒸発させた。残渣を酢酸エチル(2ml)と10%の鹹水(2ml)との間で分配し、有機相を乾燥させ(MgSO4)、減圧下で蒸発乾固した。ジクロロメタン(1ml)及びメタンスルホン酸(0.33ml)の混合物中、10%のPd/C(0.01g)上、100psiの水素圧、20〜25℃にて16時間、得られたオイル(0.13g)を水素化した。触媒をCelite(商標)で濾過し、Celite(商標)パッドをジクロロメタン(2ml)、水(2ml)及びジクロロメタン(2ml)で洗浄した。撹拌及び30℃未満の温度を維持するための氷冷をしながら、4NのNaOH(1.5ml)を濾液に添加した。有機相を鹹水(2ml)で洗浄し、乾燥させ(MgSO4)、蒸発乾固させた。収量0.087g(69%)、キラルHPLCによる鏡像体過剰率96%。AB:次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:70:30 MeOH:IPA。
【0029】
(実施例7)
((R)-6,8-ジフルオロクロマン-3-イルアミンL-酒石酸塩(8))
容器を窒素でパージし、次いで、化合物22(13.7kg)を加え、続いて、メタノール(86kg)を加えた。該混合物を63〜65℃まで温めた。反応温度を63〜65℃の範囲に保持しながら、水酸化カリウム(40%、51kg)溶液を一度に加え、15分後、第2の40%の水酸化カリウム(44kg)を30分間かけて添加した。反応温度を65〜70℃の範囲に保持しながら、得られた混合物を更に24時間撹拌した。次いで、該混合物を冷却し、水を加えた(44kg)。該混合物を再度加熱し、減圧蒸留を用いてメタノールを除去した(70℃以下のベース温度及び大気圧を初期に用い、続いて、125mbarまで真空度を低下した。)。蒸留には5.5時間要した。該反応混合物を30℃まで冷却し、残留する水相をトルエン(152kg)で2回抽出した。あわせた有機相を減圧蒸留によって、およそ6OLの容量まで減少させた(3.5時間)。変性エタノールを加え(395kg)、トルエンのレベルが2%未満になるまで(標準的なGC分析を用いて測定した。)蒸留を続けた。容器の内容物を20℃まで冷却し、元の変性エタノールの容量になるように、新鮮な変性エタノールで容量を調節した。水(72kg)中のL-酒石酸(8.86kg)の溶液を調製し、反応温度を15〜25℃の範囲に保持しながら、この溶液を1時間かけて加えた。得られた沈殿物を1時間撹拌した。次いで、容器の内容物を濾過し、濾過ケーキを変性エタノール(200L)で洗浄した。生成物を60〜100mbarの真空にて45〜50℃で乾燥した。収量=15.1kg、キラルHPLCによる鏡像体過剰率99.0%。次の条件をキラルHPLCに用いた:カラムChiralPak AD-H、波長210nm、注入量20μl、流れ:0.5ml/分、溶出液:0.2%のtert-ブチルアミンを含む70:30 MeOH:IPA。
【0030】
(実施例8)
((R)-2-{2-[3-(6,8-ジフルオロクロマン-3-イル)-2-チオキソ-2,3-ジヒドロ-1H-イミダゾール-4-イル]エチルイソインドール-1,3-ジオン(14)の製造)
【化11】
【表1】
【0031】
反応器に固体試薬を入れて、酢酸を一度に加え、該混合物を窒素下で撹拌しながら105〜110℃に加熱し、これら条件下で2時間保持した。約90℃で加熱しながら、水(24mL)をゆっくりと加えた(結晶化が起こった。)。該懸濁液を撹拌しながら氷浴で冷却し、氷中で0.5時間撹拌した。水(24mL)をゆっくりと加え、1時間撹拌を続けた。沈殿物を収集し、AcOH-水(1:1 v/v)及び水で洗浄し、次いで、真空中50〜60℃で乾燥した。得られる固形物(5.25g、99%)を、還流下でIPA(48mL)とDCM(72mL)との混合物に溶解させた。不溶性の物質(K酒石酸塩)を濾過し、ロータバップで約50℃及び500mbarにて結晶化が起こるまで、濾液を蒸発させた。次いで、加熱を止め、残圧を測定するための負圧計を用いて、蒸発を続けて残留のDCMを除去した。懸濁液を冷蔵庫に一晩放置し、結晶を収集し、IPAで洗浄した後に、真空中50℃にて乾燥した。収量3.40g(64%)。
【0032】
(実施例9)
((R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩(BIA 5-453)(1)の製造、形態A製造のための改良)
【化12】
【表2】
【0033】
2-プロパノール、水、及びDCMの混合物中の14の懸濁液に、18℃にて1分間に、撹拌しながら、NaBH4を一度に加えた(温度は1時間後に27.5℃まで上昇した。)。該混合物を18〜20℃で16時間撹拌した(1時間でほとんど透明な溶液)。該混合物を氷浴にて冷却し、温度を10℃未満に保持しながら、6NのHCl(39.6mL、237.6mmol)を滴加した。該混合物を15分間撹拌し、固形物を濾過し、濾過ケーキをDCM(300mL)で洗浄した(4.9gの固形物を得た。)。5NのNaOH(60mL)を母液に添加し、該混合物を15分間撹拌した。有機の上の層を分離し、鹹水で洗浄し、最小量の固形物を濾過して除去した。得られる透明溶液に6NのHCl(40ml)を添加し、蒸気温度が76〜78℃に達するまでDCMを蒸留して除去し、該混合物を還流下で1.5時間撹拌し、室温まで冷却した。水(300mL)を加え、IPAをロータバップで除去した(420mL収集された)。次いで、残渣をEtOAc-石油エーテル(2:1 v/v)混合物(200及び100mL)で洗浄した。2回の洗浄後、水相で結晶化を開始した。6NのHCl(40mL)を添加し、該懸濁液を撹拌しながら1時間、氷中で冷却した。沈殿物を収集し、冷した3NのHCl(75mL)及び冷したIPA(50mL)で洗浄し、次いで、真空中50℃で乾燥した。収量11.58g(73%)。
【0034】
(実施例10)
((R)-5-(2-アミノエチル)-l-(6,8-ジフルオロクロマン-3-イル)-1,3-ジヒドロイミダゾール-2-チオン塩酸塩(BIA 5-453)(1)の製造、形態C製造のための改良)
【化13】
【表3】
【0035】
2-プロパノール、水、及びDCMの混合物中の14の懸濁液に、20℃にて1分間に撹拌しながら、NaBH4を一度に加えた(温度は22℃まで上昇した。)。該混合物を20℃で16時間撹拌し(0.5時間後、透明な溶液が生成された。)、6NのHClを滴加した。加熱温度が76〜78℃に達するまでDCMを蒸留し、該混合物を還流下で1.5時間撹拌し、室温まで冷却した。水(30mL)を加えた。2-プロパノールをロータバップで除去し、残渣をEtOAc-石油エーテル(2:1 v/v)混合物(2x20mL)で2回洗浄した。DCM中の10%の2-プロパノールの溶液(40mL)を、撹拌しながら水層に加え、続いて、5NのNaOHを添加してpH9〜10にした。有機層を分離し、乾燥し(MgSO4)、蒸発乾固した。無水EtOH(15mL)と無水EtOH(15mL)中3MのHCl(1.5mL、混合物のpHは約2)との混合物に、残渣を加熱しながら溶解させた。得られる溶液を65〜70℃で2時間撹拌し、結晶を収集し、EtOHで洗浄し、真空中40℃で乾燥した。収量1.12g(71%)。
【0036】
(X線粉末回折(XRPD))
次のShimadzu社のパラメータを使用して、形態A及びCのXRPDのピークリストを生成した。
【表4】
【0037】
(FT-IR分光)
赤外スペクトルは、Ever-Glo 中/遠IR源、広範囲の臭化カリウム(KBr)ビームスプリッタ、及び重水素化硫酸グリシン(DTGS)検出器を備えた、Magna-IR 860(登録商標)フーリエ変換赤外(FT-IR)分光光度計(Thermo Nicolet社)で取得した。サンダードームアクセサリーを使用して試料採取を行った。バックグラウンドのデータセットをクリーンなGe結晶から得た。Log 1/R(反射率)スペクトルは、互いに対してこれら2つのデータセットの比率をとることによって得られた。波長の校正は、ポリスチレンを用いて行った。更なるパラメータは下記である。
【表5】
【特許請求の範囲】
【請求項1】
式22の化合物の製造方法であって:
【化1】
式21の化合物:
【化2】
を還元して、式23の化合物:
【化3】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23の化合物の水素化分解を行うことを含み、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項2】
式22'の化合物の製造方法であって:
【化4】
式21'の化合物:
【化5】
を還元して、式23'の化合物:
【化6】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23'の化合物の水素化分解を行うことを含み、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項3】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項1又は2記載の方法。
【請求項4】
前記C1〜C6のアルキルスルホン酸が、メタンスルホン酸である、請求項1、2又は3記載の方法。
【請求項5】
前記水素化分解に使用される溶媒が、塩素化溶媒を更に含む、請求項1〜4のいずれか一項記載の方法。
【請求項6】
前記塩素化溶媒とC1〜C6のアルキルスルホン酸との比率が、10:1以下である、請求項5記載の方法。
【請求項7】
前記塩素化溶媒とC1〜C6のアルキルスルホン酸との比率が、約3:1である、請求項5記載の方法。
【請求項8】
前記式21又は21'の化合物の還元が、適当な溶媒中で水素化ホウ素化合物を使用して行われる、請求項1〜7のいずれか一項記載の方法。
【請求項9】
前記水素化ホウ素化合物が、水素化ホウ素ナトリウムである、請求項8記載の方法。
【請求項10】
前記溶媒が、低級アルコールとTHFとの任意の組み合わせである、請求項8又は9記載の方法。
【請求項11】
前記溶媒が、メタノールとTHFとの任意の組み合わせである、請求項8又は9記載の方法。
【請求項12】
前記水素化分解の工程が、触媒の存在下で行われる、請求項1〜11のいずれか一項記載の方法。
【請求項13】
前記触媒が、パラジウム触媒である、請求項12記載の方法。
【請求項14】
前記水素化分解の工程が、水素雰囲気下で行われる、請求項1〜13のいずれか一項記載の方法。
【請求項15】
前記水素化分解に使用される溶媒が、ハロゲン化アルキルとメタンスルホン酸との組み合わせを含む、請求項1〜14のいずれか一項記載の方法。
【請求項16】
前記水素化分解に使用される溶媒が、ジクロロメタンとメタンスルホン酸との組み合わせを含む、請求項1〜15のいずれか一項記載の方法。
【請求項17】
式8の化合物の製造方法であって:
【化7】
式22の化合物:
【化8】
をアルカリ加水分解させることを含む、前記方法。
【請求項18】
前記式22の化合物が、請求項1又は3〜16のいずれか一項記載の方法によって形成される、請求項17記載の方法。
【請求項19】
前記アルカリ加水分解の工程が、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びL-酒石酸の存在下で行われる、請求項17又は18記載の方法。
【請求項20】
式8'の化合物の製造方法であって:
【化9】
式22'の化合物:
【化10】
をアルカリ加水分解させることを含む、前記方法。
【請求項21】
前記式22'の化合物が、請求項2〜16のいずれか一項記載の方法によって形成される、請求項20記載の方法。
【請求項22】
前記アルカリ加水分解の工程が、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びD-酒石酸の存在下で行われる、請求項20又は21記載の方法。
【請求項23】
前記アルカリ金属の水酸化物が、水酸化カリウムであり、前記アルコールが、メタノールである、請求項19又は22記載の方法。
【請求項24】
式14の化合物の製造方法であって:
【化11】
請求項17、18、19又は23のいずれか一項記載の方法によって、式8の化合物を形成すること、次いで、式8の化合物と式13の化合物:
【化12】
とを反応させることを含む、前記方法。
【請求項25】
式14'の化合物の製造方法であって:
【化13】
請求項20、21、22又は23のいずれか一項記載の方法によって、式8'の化合物を形成すること、次いで、式8'の化合物と式13の化合物:
【化14】
とを反応させることを含む、前記方法。
【請求項26】
前記反応が、アルカリ金属のイソチオシアン酸塩及び有機酸の存在下で行われる、請求項24又は25記載の方法。
【請求項27】
前記アルカリ金属のイソチオシアン酸塩が、イソチオシアン酸カリウムである、請求項26記載の方法。
【請求項28】
前記有機酸が、酢酸である、請求項26又は27記載の方法。
【請求項29】
式1の化合物を製造する方法であって:
【化15】
請求項24、26、27又は28のいずれか一項記載の方法によって、式14の化合物を製造すること、次いで、溶媒の存在下でアルカリ金属水素化ホウ素を用いて、式14の化合物を式1の化合物に変換すること、続いて、HClを添加し、次いで、式1の化合物を回収することを含む、前記方法。
【請求項30】
式1'の化合物を製造する方法であって:
【化16】
請求項25、26、27又は28のいずれか一項記載の方法によって、式14'の化合物を製造すること、次いで、溶媒の存在下でアルカリ金属水素化ホウ素を用いて、式14'の化合物を式1'の化合物に変換すること、続いて、HClを添加し、次いで、式1'の化合物を回収することを含む、前記方法。
【請求項31】
前記アルカリ金属水素化ホウ素が、水素化ホウ素ナトリウム(NaBH4)である、請求項29又は30記載の方法。
【請求項32】
前記化合物14又は14'の変換に使用される溶媒が、イソプロピルアルコール、水及びジクロロメタンの混合物である、請求項29、30又は31記載の方法。
【請求項33】
式22の化合物の製造方法であって:
【化17】
式21の化合物:
【化18】
を水素化分解して、式22の化合物を生成することを含み、該水素化分解が、3容量の塩素化溶媒と1容量のC1〜C6のアルキルスルホン酸、好ましくはメタンスルホン酸との組み合わせを含む溶媒混合物中で行われ、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項34】
式22'の化合物の製造方法であって:
【化19】
式21'の化合物:
【化20】
を水素化分解して、式22'の化合物を生成することを含み、該水素化分解が、3容量の塩素化溶媒と1容量のC1〜C6のアルキルスルホン酸、好ましくはメタンスルホン酸との組み合わせを含む溶媒混合物中で行われ、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であって、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項35】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項33又は34記載の方法。
【請求項36】
式4の化合物又はその医薬として許容し得る塩:
【化21】
。
【請求項37】
式4'の化合物又はその医薬として許容し得る塩:
【化22】
。
【請求項38】
式20の化合物又はその医薬として許容し得る塩:
【化23】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項39】
式20'の化合物又はその医薬として許容し得る塩:
【化24】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項40】
式21の化合物又はその医薬として許容し得る塩:
【化25】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項41】
式21'の化合物又はその医薬として許容し得る塩:
【化26】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項42】
式23の化合物又はその医薬として許容し得る塩:
【化27】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項43】
式23'の化合物又はその医薬として許容し得る塩:
【化28】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項44】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項36〜41のいずれか一項記載の方法。
【請求項45】
式8の化合物:
【化29】
。
【請求項46】
式8'の化合物:
【化30】
。
【請求項1】
式22の化合物の製造方法であって:
【化1】
式21の化合物:
【化2】
を還元して、式23の化合物:
【化3】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23の化合物の水素化分解を行うことを含み、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項2】
式22'の化合物の製造方法であって:
【化4】
式21'の化合物:
【化5】
を還元して、式23'の化合物:
【化6】
を生成し、続いて、C1〜C6のアルキルスルホン酸を含む溶媒中で、式23'の化合物の水素化分解を行うことを含み、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項3】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項1又は2記載の方法。
【請求項4】
前記C1〜C6のアルキルスルホン酸が、メタンスルホン酸である、請求項1、2又は3記載の方法。
【請求項5】
前記水素化分解に使用される溶媒が、塩素化溶媒を更に含む、請求項1〜4のいずれか一項記載の方法。
【請求項6】
前記塩素化溶媒とC1〜C6のアルキルスルホン酸との比率が、10:1以下である、請求項5記載の方法。
【請求項7】
前記塩素化溶媒とC1〜C6のアルキルスルホン酸との比率が、約3:1である、請求項5記載の方法。
【請求項8】
前記式21又は21'の化合物の還元が、適当な溶媒中で水素化ホウ素化合物を使用して行われる、請求項1〜7のいずれか一項記載の方法。
【請求項9】
前記水素化ホウ素化合物が、水素化ホウ素ナトリウムである、請求項8記載の方法。
【請求項10】
前記溶媒が、低級アルコールとTHFとの任意の組み合わせである、請求項8又は9記載の方法。
【請求項11】
前記溶媒が、メタノールとTHFとの任意の組み合わせである、請求項8又は9記載の方法。
【請求項12】
前記水素化分解の工程が、触媒の存在下で行われる、請求項1〜11のいずれか一項記載の方法。
【請求項13】
前記触媒が、パラジウム触媒である、請求項12記載の方法。
【請求項14】
前記水素化分解の工程が、水素雰囲気下で行われる、請求項1〜13のいずれか一項記載の方法。
【請求項15】
前記水素化分解に使用される溶媒が、ハロゲン化アルキルとメタンスルホン酸との組み合わせを含む、請求項1〜14のいずれか一項記載の方法。
【請求項16】
前記水素化分解に使用される溶媒が、ジクロロメタンとメタンスルホン酸との組み合わせを含む、請求項1〜15のいずれか一項記載の方法。
【請求項17】
式8の化合物の製造方法であって:
【化7】
式22の化合物:
【化8】
をアルカリ加水分解させることを含む、前記方法。
【請求項18】
前記式22の化合物が、請求項1又は3〜16のいずれか一項記載の方法によって形成される、請求項17記載の方法。
【請求項19】
前記アルカリ加水分解の工程が、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びL-酒石酸の存在下で行われる、請求項17又は18記載の方法。
【請求項20】
式8'の化合物の製造方法であって:
【化9】
式22'の化合物:
【化10】
をアルカリ加水分解させることを含む、前記方法。
【請求項21】
前記式22'の化合物が、請求項2〜16のいずれか一項記載の方法によって形成される、請求項20記載の方法。
【請求項22】
前記アルカリ加水分解の工程が、アルカリ土類金属の水酸化物又はアルカリ金属の水酸化物;炭素原子1〜6個を有するアルコール;及びD-酒石酸の存在下で行われる、請求項20又は21記載の方法。
【請求項23】
前記アルカリ金属の水酸化物が、水酸化カリウムであり、前記アルコールが、メタノールである、請求項19又は22記載の方法。
【請求項24】
式14の化合物の製造方法であって:
【化11】
請求項17、18、19又は23のいずれか一項記載の方法によって、式8の化合物を形成すること、次いで、式8の化合物と式13の化合物:
【化12】
とを反応させることを含む、前記方法。
【請求項25】
式14'の化合物の製造方法であって:
【化13】
請求項20、21、22又は23のいずれか一項記載の方法によって、式8'の化合物を形成すること、次いで、式8'の化合物と式13の化合物:
【化14】
とを反応させることを含む、前記方法。
【請求項26】
前記反応が、アルカリ金属のイソチオシアン酸塩及び有機酸の存在下で行われる、請求項24又は25記載の方法。
【請求項27】
前記アルカリ金属のイソチオシアン酸塩が、イソチオシアン酸カリウムである、請求項26記載の方法。
【請求項28】
前記有機酸が、酢酸である、請求項26又は27記載の方法。
【請求項29】
式1の化合物を製造する方法であって:
【化15】
請求項24、26、27又は28のいずれか一項記載の方法によって、式14の化合物を製造すること、次いで、溶媒の存在下でアルカリ金属水素化ホウ素を用いて、式14の化合物を式1の化合物に変換すること、続いて、HClを添加し、次いで、式1の化合物を回収することを含む、前記方法。
【請求項30】
式1'の化合物を製造する方法であって:
【化16】
請求項25、26、27又は28のいずれか一項記載の方法によって、式14'の化合物を製造すること、次いで、溶媒の存在下でアルカリ金属水素化ホウ素を用いて、式14'の化合物を式1'の化合物に変換すること、続いて、HClを添加し、次いで、式1'の化合物を回収することを含む、前記方法。
【請求項31】
前記アルカリ金属水素化ホウ素が、水素化ホウ素ナトリウム(NaBH4)である、請求項29又は30記載の方法。
【請求項32】
前記化合物14又は14'の変換に使用される溶媒が、イソプロピルアルコール、水及びジクロロメタンの混合物である、請求項29、30又は31記載の方法。
【請求項33】
式22の化合物の製造方法であって:
【化17】
式21の化合物:
【化18】
を水素化分解して、式22の化合物を生成することを含み、該水素化分解が、3容量の塩素化溶媒と1容量のC1〜C6のアルキルスルホン酸、好ましくはメタンスルホン酸との組み合わせを含む溶媒混合物中で行われ、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項34】
式22'の化合物の製造方法であって:
【化19】
式21'の化合物:
【化20】
を水素化分解して、式22'の化合物を生成することを含み、該水素化分解が、3容量の塩素化溶媒と1容量のC1〜C6のアルキルスルホン酸、好ましくはメタンスルホン酸との組み合わせを含む溶媒混合物中で行われ、これらの式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であって、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する、前記方法。
【請求項35】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項33又は34記載の方法。
【請求項36】
式4の化合物又はその医薬として許容し得る塩:
【化21】
。
【請求項37】
式4'の化合物又はその医薬として許容し得る塩:
【化22】
。
【請求項38】
式20の化合物又はその医薬として許容し得る塩:
【化23】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項39】
式20'の化合物又はその医薬として許容し得る塩:
【化24】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項40】
式21の化合物又はその医薬として許容し得る塩:
【化25】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項41】
式21'の化合物又はその医薬として許容し得る塩:
【化26】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項42】
式23の化合物又はその医薬として許容し得る塩:
【化27】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項43】
式23'の化合物又はその医薬として許容し得る塩:
【化28】
(式中、Rはアルキル又はアリールであり:用語アルキルは、炭素原子1〜6個を含む、直鎖又は分枝であり、アリール、アルコキシ、ハロゲン、アルコキシカルボニル、又はヒドロキシカルボニル基によって任意に置換されている炭化水素鎖を意味し;用語アリールは、アルキルオキシ、ハロゲン、又はニトロ基によって任意に置換されているフェニル又はナフチル基を意味し;かつ用語ハロゲンは、フッ素、塩素、臭素、又はヨウ素を意味する。)。
【請求項44】
前記Rが、エチル、メチル、tBu、又はベンジルである、請求項36〜41のいずれか一項記載の方法。
【請求項45】
式8の化合物:
【化29】
。
【請求項46】
式8'の化合物:
【化30】
。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2010−527997(P2010−527997A)
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願番号】特願2010−509294(P2010−509294)
【出願日】平成20年5月21日(2008.5.21)
【国際出願番号】PCT/PT2008/000023
【国際公開番号】WO2008/143540
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(596095518)バイアル−ポルテラ アンド シーエー,エス.エー. (25)
【Fターム(参考)】
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願日】平成20年5月21日(2008.5.21)
【国際出願番号】PCT/PT2008/000023
【国際公開番号】WO2008/143540
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(596095518)バイアル−ポルテラ アンド シーエー,エス.エー. (25)
【Fターム(参考)】
[ Back to top ]