
グリメピリド誘導及びインスリン誘導グリコシルホスファチジルイノシトール特異的ホスホリパーゼC制御
【課題】哺乳類GPI−PLC(グリコシルホスファチジルイノシトール特異的ホスホリパーゼC)の活性を調節する化合物を提供することを目的とする。
【解決手段】本発明は、式I:
【化1】

(式中、R1、R2、R3、R4及びR5は明細書中に定義されるとおりである)の化合物、全てのその立体異性体の形態、及び全ての比率のそれらの混合物並びに生理的に許容されるその塩を提供する。
【解決手段】本発明は、式I:
【化1】
(式中、R1、R2、R3、R4及びR5は明細書中に定義されるとおりである)の化合物、全てのその立体異性体の形態、及び全ての比率のそれらの混合物並びに生理的に許容されるその塩を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、哺乳類GPI−PLC(グリコシルホスファチジルイノシトール特異的ホスホリパーゼC)の活性を調節する、異なる方法に関する。
【背景技術】
【0002】
哺乳類原形質膜GPI−PLCに特異的な阻害剤、GPI2350の合成は、細菌やトリパノソーマの(G)PI−PLCの(結晶)構造、基質の必要条件、GPI認識及び切断機構についての背景的知識や、更にこれまでそれらについて記載された阻害剤に基づいていた。トリパノソーマのGPI―PLCに対しては、GPI及びPIは、それぞれ、効率的な及び貧弱な基質である一方で、細菌のPI−PLCに対してはその反対である。後者は、80残基のトリパノソーマのGPI−PLCにタンパク質配列が類似した領域を有している(“Kuppe et al., (1989), J. Bacteriol. 171:6077-6083")。GPI認識の解析のために、B.cereusからのPI−PLCの三次元構造が、グルコサミニル(α1→6)−ミオ−イノシトール(GMI)との複合体で2.2オングストロームの分解能で最近決定され、GMIのミオ−イノシトール部分は遊離のミオ−イノシトールと同じ位置を占有しているのに対して、グルコサミン部分は触媒部位の入り口で溶媒に露出した状態にあることが明らかにされた(“Heinz et al.,(1995), EMBO J. 14:3855-3863" 及び“Heinz et al., (1996), Biochemistry 35:9496-9504")。コア四糖類の残りの部分は、殆んどPI−PLCと接触がなく、このことはGPIアンカー内に受入れられたコアグリカンの顕著な構造多様性を説明しているのであろう。これらをもとにして、現時点の実験データは、細菌のPI−PLCによるPI及びGPIの、更に、トリパノソーマのGPI−PLCによるGPIの切断の触媒機構は全て類似していることを示唆している。しかしながら、哺乳類PI−PLCがセカンドメッセンジャー、イノシトール三リン酸、を生成して、GPIアンカーを受入れることができず、細菌のPI−PLCがリン酸化PIを切断することができないことを考慮すると、このことが脂肪細胞原形質膜GPI−PLCに対して正しいかどうかは知られていなかった。
【0003】
細菌及びトリパノソーマの(G)PI−PLCの基質必要条件は、以前、PI類縁体及びGMI誘導体で試験されていた。触媒作用には、イノシトールの2位に遊離のOH基が必要とされる。T.brucei由来のミリスチン酸エステル含有VSGを用いた研究で、(G)PI−PLCは、GMIの2−デオキシ−イノシトール類縁体によって競合的に阻止され、イノシトール−2−OHは、触媒作用には必要であるが、基質認識に重要ではないことが示された。更に、基質認識には、イノシトールの1位に荷電したホスホリル基(即ち、ホスホネート又はホスホジエステル)が必要である(“Morris, J. C., et al.,
(1996), J. Biol. Chem. 271:15468-15477”)。それ故、イノシトール−1位及びイノシトール−2位の両方のOH基は触媒作用に関与しているが、なお、ホスホリル基のみが基質認識に必要であるようにみえる。興味深いことに、グルコサミン(α1→6)−イノシトール−1,2−環状リン酸は、トリパノソーマGPI−PLCに対してGMI−1−リン酸よりも、よりよい阻害剤であるが、細菌のPI−PLCに対してはそうでないことが判明したが、このことは環状型が前者に対して生成物類縁体として作用するのであろうことを示唆している。更に、GMI−1−リン酸のホスホネート誘導体は、十中八九切断できない基質類縁体であるので、より強力な阻害剤であることが判った。これらのデータは、細菌のPI−PLC作用について提案された2ステップ・メカニズムに合致している。つまり、PIは、先ず切断されて環状イノシトール−1,2−リン酸(cIP)構造を産生し、次いで、これがイノシトール−1−リン酸に加水分解される。興味深いことに、cIP構造は、トリパノソーマのGPI−PLC触媒作用で、第2ステップ(脱環化)で
はなく第1ステップ(環化)の作用を示す、T.brucei由来のGPI−PLCへの暴露を受けて、トリパノソーマのVSGのいわゆる交差反応決定基として免疫学的に同定することができる。一過性又は安定したcIP形成に必要な条件は、ヒト赤血球AChEでのように、イノシトール残基の1位又は2位がパルミトイル化されたGPIアンカーはホスホリパーゼ切断に抵抗性である、という発見と一致している。
【0004】
GMI−1−ドデシルホスホネートは、やはり膜と会合しているトリパノソーマのGPI−PLCに対して相当するヘキシル誘導体よりもかなり阻害的であることが判った(“Morris, J. C. et al. (1995), J. Biol. Chem. 270: 2517-2524")。興味深いことに、イノシトールに炭水化物の置換基(即ち、グルコサミン)がないと、つまりイノシトール−1−リン酸の切断されない類縁体では、ミオ−イノシトール−1−O−ドデシルリン酸によって例示されるように、トリパノソーマのGPI−PLCを阻害することが見出された。この型の阻害剤の効率は、トリパノソーマのGPI−PLCを10〜90μMのIC50で競合的に阻害する、2−フルオロ置換体を有する2−デオキシ−イノシトール−1−O−ドデシルホスホネートの2位の置換体でかなり増加した(“Morris, J. C. et al. (1996), J. Biol. Chem. 271: 15468-15477")。これまでに報告されたGPI−PLCの最も強力な阻害剤は、2−デオキシ−イノシトールの2位にフルオロ基を、1位にドデシルホスホネートの両者を有しており、ミオ−イノシトール−1−O−ドデシル−ホスホン酸よりも少なくとも5倍より阻害的である(“Morris, J. C. et al. (1998), Biochem. Biophys. Res. Commun. 244: 873-867")。興味深いことは、B.cereus及びT.brucei由来の(G)PI−PLCのこれら化合物のいくつかによる差別的な阻害は、二つの酵素が(G)PI−PLCの機械的なサブクラスを表すと論じていることである。
【0005】
残念ながら、類似のデータが哺乳類のGPI−PLCについて未だに得られていない。驚くべきことに、新しく合成されたミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(GPI−2350)は、細菌及び脂肪細胞のGPI−PLCの強力な阻害剤であることが判った。
【0006】
アルカリ性ホスファターゼ(aP)が細菌のホスファチジルイノシトール特異的ホスホリパーゼC(PI−PLC)によって膜二重層から放出されるという最初の観察は、グリコシルホスファチジルイノシトール(GPI)脂質への共有結合を含むタンパク質の別の型の膜付着の同定へと導いた。
【0007】
GPIアンカーの最初の完全な構造は、トリパノソーマ・ブルセイ由来の変異体表面グリコプロテイン(VSG)について明らかにされた。
【0008】
コア四糖類は、3つのマンノース残基と非アセチル化グルコサミンからなり、後者の一末端はホスホエタノールアミン架橋を経てタンパク質部分にアミド結合しており、他の末端はホスファチジルイノシトール(PI)の6−ヒドロキシル基にグリコシド結合的に結合している。PIは、それぞれジアシルグリセロール及びホスファチジン酸を放出する特異性C及びDの(G)PI−特異的ホスホリパーゼ[G]PI−PLC/Dによって切断され、タンパク質部分の末端(ホスホノ)イノシトールグリカン(PIG)構造を残す。
【0009】
それ以来、種々の起源の細菌PI−PLCが、GPIアンカー型タンパク質(GPI−タンパク質)を検出するのに一般的に使用されている。
【0010】
GPIアンカー型VSGからなる防護的表面被膜のGPI−PLCによる脂肪分解放出は、T.bruceiにとっては、宿主の免疫系を逃れるために抗原変異を達成するために必要であろうと推測される。
【0011】
殆んどの哺乳類細胞及び組織は、大多数が原形質膜の外小葉に埋め込まれているGPIアンカーを有しているGPI−タンパク質を発現するので、内因性の(G)PI/PI−PLC/Dが、それら細胞の表面発現の特異的ダウン・レギュレーションを調節し、同時に循環における可溶性タンパク質部分の増加を調節することができる。
【0012】
GPI−タンパク質の可溶性型は、5−ヌクレオチダーゼ(5’−Nuc)、Thy−1、アルカリ性ホスファターゼ(aP)及びCD16受容体のように血流内を循環していることが検出された。GPI−PLCは、又、ヒト好中球、ウシ脳、ラット腸及びヒト上皮性悪性腫瘍細胞株において同定することができた。ラット肝からのGPI−PLCは、均質性を示すまで精製された。内因性GPI−PLCは、ブタの最大の小管から腎ジペプチダーゼを放出する能力を有していると記載されていた。しかしながら、GPI−PLCの構造又は遺伝子の解明は未だされていない。哺乳類GPI−PLCが膜依存性であるのに対して、哺乳類GPI−PLDは、異なった種(ヒト、ラット、ウシ)及び器官(胎盤、脳、肝、血清)の組織材料の全ての型から回収することができる。
【0013】
GPI−PLDの主な役割は、多分、リソソームへのエンドサイトーシス及びトラフィッキングの後のGPIタンパク質のGPIアンカーの分解である。哺乳類の組織は、血清中及び細胞表面において、異なった機能性を持って活性である二つのはっきりと識別できるGPI−PLDを持っている。
【0014】
酵母及び齧歯類脂肪細胞の5’−Nuc及びaPのようないくつかのGPIタンパク質は、インビトロ及びインビボでGPI−PLCによるGPIアンカーの脂肪分解により細胞表面から可溶性のものとして放出されているようにはみえない。
【0015】
付加的な温和な塩及び/又はトリプシン切断が、顆粒画分/細胞から分離の後に、可溶性画分/培地中のいくつかの脂肪分解的に切断されたGPIタンパク質のタンパク質部分の回収に必要とされるのは、受容体タンパク質の存在を示唆している。
【0016】
これらの場合において、GPI−PLCの活性はGPI−タンパク質の局在又はトポロジーに影響しないが、代わりに、両親媒性から親水性の形態への変換の途中で機能的(触媒/結合)特性を変化する。無処置のGPIアンカーの存在は、それに付いているタンパク質部分の立体構造及び挙動に影響している。
【0017】
5’−Nuc及びGce1の場合には、触媒及び結合効率は、膜に包埋された又は界面活性剤ミセル又はリポソームに再構築された完全な形のものと比較すると、脂肪分解的に切断されたGPIタンパク質において増加した。
【0018】
GPIタンパク質の脂肪分解プロセシングが細胞壁の生合成の間ある役割を演じているような、酵母におけるグルコースのような栄養シグナルによって、及び齧歯類の脂肪細胞、ミオサイト及びヒト内皮細胞におけるグルコースやある種のホルモン、増殖因子及び薬物(例えば、インスリン、グリメピリド)によって、真核生物において、多くのGPI−PLが、アップレギュレートされている。
【発明の概要】
【発明が解決しようとする課題】
【0019】
グリメピリド、抗糖尿病薬は、膵細胞からインスリン放出を刺激することによって、更に、少しの程度であるが、筋肉細胞におけるグルコース輸送の活性化及び脂肪細胞における脂肪分解の阻害のような、末梢組織での代謝的インスリン作用を模倣することによって、主に血糖を低下させる。
【0020】
スルホニルウレア、グリメピリド、の血糖低下効果は、部分的には、IRS−PI3K経路のインスリン受容体非依存性活性化を経た脂肪細胞及び筋肉細胞における非酸化的グルコース代謝の刺激によって引き起こされる。単離したラット脂肪細胞において、グリメピリドの作用の分子機構は、アシル化された非受容体チロシンキナーゼ、pp59Lyn、
及びいくつかのGPIタンパク質のような脂質ラフト関連シグナリング成分の再分配及びそれに伴っておこる活性化、並びに、インスリンによって中程度に活性化もされる原形質膜グリコシルホスファチジルイノシトール特異的GPI−PLCの促進に関与していることが明らかにされている。
【0021】
グリメピリドは、膵臓β細胞からのインスリン放出を促進するスルホニルウレア剤であり、膵臓外の機構を経て作用するであろう。グリメピリドは、血糖症が食事及び運動だけではコントロールされない2型(非インスリン依存性)糖尿病患者に1日1回投与され、続発性スルホニルウレア障害の患者にインスリンと併用されることもある。
【0022】
グリメピリドの最大の血糖低下効果は、投与後最初の4時間に起こる。グリメピリドは、グリベンクラミド(グリブリド)よりも心臓血管変動に対する重篤な影響はより少ない。薬物動態は高齢の患者又は腎疾患や肝臓疾患のそれら患者において主として不変である。グリメピリドの薬物相互作用は殆んど報告されていない。
【0023】
2型糖尿病患者において、グリメピリドの有効投与範囲は、0.5から8mg/日であり、4mg/日と8mg/日の投与間に有効性の差が殆んどない。グリメピリドは、1年間の試験において、グリベンクラミド及びグリピジドの有効性と類似していた。しかしながら、グリメピリドは最初の数週間にわたる治療でグリピジドよりもより速く血糖を低下するようである。グリメピリド及びグリクラジドが、14週間の試験で基準線に良好に血糖コントロールができている患者で比較され、効果に差がないことが指摘された。グリメピリドとインスリンの併用は、絶食時血糖標的レベルが≦7.8mmol/Lに達する続発性スルホニルウレア障害の患者の治療において、インスリンとプラセボの併用と同程度の効果があったが、グリメピリドを用いることにより血糖症に対してより低いインスリン投与量とより速い効果が見られた。
【0024】
グリメピリド単独療法は、一般的に良好な耐容性を有するが、治療期間1年以下(≦1年)の患者の10から20%、及び6ヶ月間同時にインスリンを投与された患者の50%以下(≦50%)において、低血糖症を発症した。集計した治験データは、グリメピリドは、グリベンクラミドよりも低血糖症の発生率が低く、特に治療第1ヵ月目において低いであろうことが示唆された。投与量は、通常1mg/日で開始され、1から2週間の間隔で血糖コントロールのために通常の投与範囲1から4mg/日に漸増する(英国では最大6mg/日又は米国では8mg/日)。
【0025】
グリメピリドは、主に膵臓β細胞からのインスリンの放出の促進によって、及び僅かな程度で、筋肉細胞におけるグルコース輸送の活性化及び脂肪細胞における脂肪分解の阻害のような、末梢組織における代謝性インスリン作用を模倣することによって、グルコースを低下させる。
【0026】
グリメピリドは、初代培養又は培養した齧歯類脂肪細胞を薬理学的濃度で処理すると、GPI−PLCの活性化によって、5’−Nuc、aP及びGce1のようなGPIタンパク質サブセットの両親媒性から親水性への変換を強力に誘導することが明らかにされた。
【0027】
本発明において初めて開示されたイノシトール誘導体、GPI−2350は、高力価(
IC50=0.2〜10μM)及び選択性で細菌、トリパノソーマ及び血清GPI−PLC及びGPI−PLDを阻害する。GPI−2350は、無処置のラット脂肪細胞のGPI−PLCを殆んど完全にダウンレギュレートする。GPI−PLCは、代謝性インスリンシグナリングでは役割を演じない一方で、GPI−PLCの活性化は、界面活性剤不溶性の糖脂質に富む脂質ラフト領域(DIG)からインスリン受容体基質1(IRS−1)へのインスリン受容体非依存性クロストークを経由するグリメピリドのインスリン模倣効果には必須である。
【0028】
脂肪細胞のような多くの最終分化細胞の原形質膜に多数発現しているDIGは、シグナル伝達並びにタンパク質及び脂質のトラフィッキング及び選別を包含する膜媒介生物学的過程のためのプラットフォームとして役目を果たしている、特殊な膜ミクロドメインである。
【0029】
それらは、外部原形質小葉中のコレステロール及びスフィンゴ(糖)脂質、及び内小葉の飽和アシル鎖を有するリン脂質及びコレステロールに濃縮されていて、二重層内に液状の秩序相を形成している。DIGは、冷状態で1%トライトンX−100に不溶性であり、ショ糖勾配遠心分離法で低浮遊密度であることを特徴とする。これらの判断基準に基づいて、ある種のGPIアンカーされ、アシル化した、膜貫通シグナリングタンパク質が、原形質膜のDIG対非DIG領域に豊富にあることが見出された。更に、高コレステロール含有のDIG(hcDIG)及び低コレステロール含有のDIG(lcDIG)は、それぞれ、それらの低浮遊密度及び高浮遊密度に基いて互いに区別することができる。
【0030】
hcDIGからlcDIGへの、GPIアンカーされ、そしてアシル化されたある種のシグナリングタンパク質の刺激依存性再分配は、GPI−2350によってブロックされた。
【0031】
GPI−2350は、脂質ラフト関連GPI−PLC(IC50=5〜10μM)による無処置のラット脂肪細胞由来の、Gce1及び5’−Nucのような、GPIタンパク質の基本的及びグリメピリド/インスリン誘発脂肪分解放出を減少させた。GPI−2350(50μM)によるGPI−PLCの阻害は、(i)pp59Lyn及びGce1のカベオリンからの解離、(ii)hcDIGからlcDIGへの再分配、(iii)pp59Lyn及びIRS−1のチロシンリン酸化、(iv)グルコース輸送の促進及び(v)グリメピリドに応答する脂肪分解の阻害、の殆んど完全な遮断を導く。
【0032】
GPI−PLCのインスリン活性化は、脂質ラフト分布に対する適度な効果を有し、及び、それ(例えば、IRS−1のチロシンリン酸化及び脂肪分解の阻害)が僅かに減少するだけであったので、あったとしても代謝性インスリンシグナリングにおける軽微な役割がGPI−2350のみの存在下で明らかにされた。
【0033】
グリメピリドに誘導されたGPI−PLCによって生成する脂肪分解的に切断されたGPIタンパク質は、それらの非切断両親媒性のもの及びpp59Lynのように、lcDIGに再分配するよりもむしろhcDIGと会合して残っている。
【0034】
ラット脂肪細胞において再分配され、活性化されたpp59LynによるIRSのチロシンリン酸化を経るインスリンシグナリングカスケードへのグリメピリドのクロストークは、hcDIGが関与するGPI−PLCの活性化を必要とする。
【課題を解決するための手段】
【0035】
本発明は、哺乳類のGPI−PLCの活性を調節する化合物の同定方法であって、
a)哺乳類細胞をグリメピリドと共にインキュベートし;
b)a)の細胞のhcDIGを調製し;
c)b)からのhcDIGを化合物と共にインキュベートし;そして
d)c)のhcDIGからのGPI−PLCの活性を定量する;
ことを含む方法に関する。
【0036】
方法のステップa)に記載の哺乳類細胞は、齧歯類又はイヌの細胞に関する。齧歯類は、例えば、マウス、ラット又はモルモットである。本方法は、ヒト、又は例えばチンパンジー、ゴリラ又はボノボのような類人猿の細胞にも関する。そのような細胞は、例えば、膵臓細胞、筋肉細胞、肝細胞、腎細胞、脳細胞、脂肪細胞である。哺乳類細胞は、細胞培養によっても提供することができる。細胞を取得し、処理し、収穫し、加工するために、通常の技術が使われる(例えば、Current Protocols in Cell Biology, John Wiley & Sons; 0-471-24108-3-Looseleaf; 0-471-24105-9-CD-ROM)。
【0037】
本発明の方法のステップc)に記載のhcDIGからのGPI−PLCの活性を定量するには、hcDIGからpp59Lynの解離を測定することによって、又はhcDIGからlcDIGへのpp59Lyn及び/又はGce1の再分配を測定することによって、又はpp59Lyn及び/又はIRS−1のリン酸化の変化を測定することによって、又はグルコース輸送の促進及び/又は脂質分解の阻害を測定することによって実行される。
【0038】
本発明は、更に、前に開示したように本発明の方法によって同定することができる化合物に関する。そのような化合物は、例えば、本発明に開示されているような式Iを有する化合物又はその誘導体である。
【0039】
本発明は、又、全ての立体異性体の形態における、および全ての比率のその混合物である式Iの化合物:
【化1】
(式中、R1、R2、R3及びR4は、互いに独立して、OH又はFのいずれかであり、そしてR5は(C1−C20)−アルキル又は(C1−C20)−アルケニルである)及び生理的に耐容されるその塩に関する。
【0040】
本発明は、又、R1、R2、R3及び/又はR4のいずれか2つが互いに独立してFである、前記に記載の式Iの化合物に関する。
【0041】
本発明は、又、R1、R2、R3及びR4が各々の場合においてOHである、前記に記載の式Iの化合物に関する。
【0042】
本発明は、又、R5がC12−アルキルである、前記に記載の式Iの化合物に関する。
【0043】
本発明のそのような化合物は、例えば、全てのその立体異性体の形態を含む、以下の式を有する。
【化2】
【0044】
又は、本発明のそのような化合物は、例えば、以下の式を有する。
【化3】
【0045】
そのような本発明の化合物は、例えば、その全ての立体異性体の形態を含む、ミオ−イノシトール−1,2−シクロドデシルホスホン酸であり得る。
【0046】
本発明の記載において、用語(C1−C20)−アルキルは、メチル、エチル、プロピル、ブチル、ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、デュオデシル、トレデシル、クアトルデシル、クインデシル、セスデシル、セプタデシル、オクタデシル、ノナデシル、エイコシルの全ての立体異性体の立体構造を有する、全ての直鎖状又は分枝鎖状の化合物に関するものとする。
【0047】
本発明の記載において、用語(C2−C20)−アルケニルは、エチレン、プロピレン、
ブチレン、ペンチレン、ヘキシレン、ヘプチレン、オクチレン、ノニレン、デシレン、ウンデシレン、デュオデシレン、トレデシレン、クアトルデシレン、クインデシレン、セスデシレン、セプタデシレン、オクタデシレン、ノナデシレン、エイコシレンの全ての立体異性体の立体構造を有する、全ての直鎖状又は分枝鎖状の化合物を包含するものとする。
【0048】
本発明は、更に、ミオ−イノシトール−1,2−シクロドデシルホスホン酸の製造方法であって、
a)ラセミ体の1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトールをリン酸化してテトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を得ること;及び
b)テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を接触水素化すること;
を含む方法に関する。
【0049】
該製造方法は、前に特定したようなR1、R2、R3、R4及び/又はR5を含む適切な出発物質を使用することによって、本発明の別のいずれかの化合物の製造に適用し得るものである。
【0050】
本発明は、又、式Iを有する少なくとも1つの化合物及び/又は生理的に耐容される塩及び/又はそのプロドラッグ及び医薬として許容される担体を含む医薬組成物に関する。
【0051】
本発明は、更に、糖尿病の治療のために使用される薬剤の副作用を変えるための、式Iの化合物及び/又は生理的に耐容されるその塩及び/又はそのプロドラッグの使用に関する。そのような薬剤は、例えば、グリメピリドである。副作用は、例えば、グリメピリドの誤った投与量に起因する低血糖である。
【0052】
本発明は、又、糖尿病の治療のために使用される薬剤(例えば、グリメピリド)の副作用の治療用の医薬組成物を製造するための、式Iを有する化合物の使用に関する。
本発明は、又、脂肪細胞の原形質膜を横切るシグナル伝達の間にDIG中のシグナル伝達タンパク質を同定するための、又は、GPI−PLC及び/又は依存性のシグナル伝達因子の哺乳類の変異体を同定及び特性付けるための、式Iの化合物の使用に関する。
【0053】
本発明は、又、グリメピリドの活性を調節する化合物の同定方法であって、
a)哺乳類細胞をグリメピリドと化合物との混合物と共にインキュベートし;
b)a)の細胞のhcDIGを調製し;そして
c)b)のhcDIGからのGPI−PLCの活性を定量する;
ことを含む方法に関する。
【0054】
本発明の記載における化合物は、化学合成によって製造されるものか、天然源から単離されるもののいずれかであって、50と50,000ダルトンの間の分子量を有する、いずれかの有機及び/又は炭水化物化合物を意味するものとする。
【0055】
グリメピリドの活性の調節とは、活性が、促進されるか、阻害されるか、活性をある一定のレベルに安定させるという意味で保持されるか、を意味するものとする。
【0056】
グリメピリドの活性を調節する化合物の同定方法のステップa)における哺乳類細胞は、例えばラット又はマウスのような齧歯類の細胞、イヌの細胞又はヒトの細胞を包含するものとする。そのような細胞は、例えば、膵臓細胞、筋肉細胞、肝細胞、腎細胞、脳細胞又は脂肪細胞であり得る。又、使用することができるのは、細胞培養(例えば、初代細胞培養)の細胞である。グリメピリドの活性を調節する化合物の同定方法のステップc)に記載のGPI−PLCの活性の定量は、hcDIGからのpp59Lynの解離を測定することによって、又はhcDIGからlcDIGのpp59Lyn及び/又はGce1再分配を測定することによって、又はpp59Lyn及び/又はIRS−1のリン酸化の変化を測定することによって、又はグルコース輸送の促進及び/又は脂質分解の阻害を測定することによって、達成することができる。
【0057】
グリメピリドの活性を調節する化合物の同定方法のステップc)に記載のhcDIGか
らのGPI−PLCの活性が弱められている場合は、同定された化合物がグリメピリドの活性を不活化しているか又は減少させている。GPI−PLCの活性が増強されている場合は、同定された化合物がグリメピリドの活性を促進しているか、又は支持している。
【0058】
医薬として許容される陰イオンを有する式Iの化合物の塩は、医薬として許容される塩の製造又は精製のための、及び/又は非治療、例えばインビトロでの応用において使用するための有用な中間体として本発明の範囲内に、同様に包含される。
【0059】
式Iの化合物の塩は、当業者によく知られている通常の方法を用いて製造することができる。塩は、例えば、式Iの化合物を、溶媒又は希釈剤中で、無機若しくは有機酸又は塩基と組み合わせることによって製造することができる。
【0060】
本明細書で使用される用語「生理的に活性な誘導体」は、例えば、ヒトのような哺乳類に投与されると、式Iの化合物又はその活性代謝物を(直接的に、又は間接的に)形成することができる、本発明の式Iの化合物の生理的に許容される誘導体のいずれか、例えばエステル、に関する。
【0061】
生理的に活性な誘導体は、又、本発明の化合物のプロドラッグを包含する。そのようなプロドラッグは、本発明の化合物にインビボで代謝され得る。これらのプロドラッグはそれ自体活性又は不活性であることができる。
【0062】
本発明の化合物は、種々の多形相、例えば、非晶性及び結晶性多形相として存在することができる。本発明の化合物の全ての多形相は、本発明の範囲内に包含され、本発明の更なる態様である。
【0063】
以下、「式Iの化合物(類)」についての全ての言及は、上に記載した式Iの化合物/化合物類、並びに、本明細書に記載したそれらの塩、溶媒和物及び生理的に活性な誘導体を意味する。
【0064】
所望の生物学的効果を達成するために必要な式Iの化合物の量は、多くの因子、例えば、選択された特異的化合物、目的とする使用、投与様式及び患者の臨床状態に依存している。一般的に、1日投与量は、体重1キログラム当たり1日につき0.3mgから100mg(典型的には3mgから50mg)の範囲内、例えば、3〜10mg/kg/日である。静脈内投与量は、例えば、0.3mgから1.0mg/kgの範囲であり、これは1分当たり体重1キログラムにつき10ngから100ngの注入として適切に投与することができる。これらの目的のための適切な注入溶液は、例えば、1ミリリットル当たり0.1ngから10mg、典型的には1ngから10mgを含有することができる。個々の投与量は、例えば、1mgから10gの活性化合物を含有することができる。それ故、注射用アンプルは、例えば、1mgから100mg、を含有することができ、そして、例えば、錠剤又はカプセルのような経口投与可能な個々の剤形は、例えば、1.0から1,000mg、典型的には10から600mgを含有することができる。医薬として許容される塩の場合、上記した重量の詳細は、塩から由来するジヒドロチアゾリウムイオンの重量と関連している。上に挙げた状態の予防又は治療のために、式Iの化合物は、化合物としてそれ自体使用することができるが、それらは耐容される賦形剤との医薬組成物の形態で存在するのが好ましい。賦形剤は、勿論、それが組成物の別の構成物と相性がいいこと、及び患者の健康に有害でないことという意味において、耐容性でなければならない。賦形剤は、固体又は液体又は両者であり得、好ましくは、例えば、活性化合物の0.05質量%から95質量%までを含有することができる錠剤として、個々の投与量として化合物と処方される。更に医薬として活性な物質も、式(I)の更なる化合物を包含して存在し得る。本発明による医薬組成物は、本質的に成分を薬理学的に許容される賦形剤及び/又は助剤
と混合することから成る、公知の薬学的方法の一つによって製造することができる。
【0065】
本発明による医薬組成物は、個々の場合における最も適切な投与方法が、治療される状態の性質及び重症度、並びに各々の場合に使用される式(I)による化合物の性質に依存するものの、口腔、直腸、局所、経口(例えば、舌下)及び非経口(例えば、皮下、筋肉内、皮内又は静脈内)投与に適したものである。糖衣をかけた剤形及び糖衣をかけた遅延放出剤形も、又、本発明の範囲内に包含される。耐酸性及び腸溶性剤形が好ましい。好適な腸溶性コーティングとしては、酢酸フタル酸セルロース、ポリ酢酸ビニルフタル酸塩、フタル酸ヒドロキシプロピルメチルセルロース及びメタクリル酸及びメタクリル酸メチルのアニオン性ポリマーが挙げられる。
【0066】
経口投与用に好適な医薬化合物は、例えば、各々の場合において式(I)の化合物の一定量を含有する、カプセル、カシェ剤、トローチ剤又は錠剤のような分離された単位で存在することができ、粉剤又は顆粒剤として、水性又は非水性の溶液又は懸濁液として、又は水中油又は油中水エマルジョンとして、分離された単位で存在することができる。既に挙げたように、これらの組成物は、活性化合物と(1つ又はそれ以上の付加的成分からなり得る)賦形剤が接触されるステップを包含するいかなる好適な薬学的方法によっても製造することができる。一般的に、組成物は、活性化合物を液体及び/又は微細に分割した固体の賦形剤と均一及び均質に混合し、その後、必要により生成物を成形することにより、製造される。かくして、例えば、錠剤は、化合物の粉末又は顆粒を加圧又は成型によって、もし適切ならば、1つ又はそれ以上の追加の成分と共に、製造することができる。圧縮した錠剤は、化合物を、例えば粉末又は顆粒のような自由に流れる形態で、適切な場合は結合剤、滑沢剤、不活性な希釈剤及び/又は1つの(多くの)界面活性/分散剤と混合して、好適な装置で錠剤化することによって製造することができる。成形された錠剤は、粉体化合物を、不活性液体希釈剤でぬらし、好適な機械で成型することによって製造することができる。
【0067】
経口(舌下)投与に好適な医薬組成物は、式Iの化合物と香味料、通常ショ糖及びアラビアゴム又はトラガカントゴムを含有するトローチ錠、及びゼラチン及びグリセリン又はショ糖及びアラビアゴムのような不活性基剤中に化合物を含んだ芳香錠を包含する。
【0068】
好適な非経口投与用医薬組成物は、好ましくは、意図した被投与者の血液と好ましくは等張である、式Iの化合物の滅菌水性製剤を包含する。これらの製剤は、投与は注射として皮下、筋肉内又は経皮で行うことができるが、好ましくは、静注で投与される。これらの製剤は、好ましくは、化合物を水と混合し、得られた溶液を滅菌し、血液と等張にすることによって製造することができる。本発明の注射できる組成物は、一般的に、活性化合物を0.1から5質量%含有している。
【0069】
直腸投与に好適な医薬組成物は、好ましくは、個々の投与量の座薬として存在する。それらは、式(I)の化合物を1又はそれ以上の通常の固体賦形剤、例えば、カカオバターと混合し、得られた混合物を成形することによって製造することができる。
【0070】
皮膚に局所適用する好適な医薬組成物は、好ましくは、軟膏、クリーム、ローション、ペースト、スプレー、エーロゾル又はオイルとして存在する。使用することのできる賦形剤は、ワセリン、ラノリン、ポリエチレングリコール、アルコール及びこれらの物質の2つ又はそれ以上の組合せである。活性化合物は、一般的に、組成物の0.1から15質量
%の濃度、例えば、0.5から2質量%の濃度で存在する。
【0071】
経皮投与も、又、可能である。経皮投与用に好適な医薬組成物は、患者の表皮と長期間密接に接触するのに適している個々のパッチとして存在することができる。そのようなパ
ッチは、場合によっては緩衝性水性溶液中の活性化合物を、接着剤中に溶解及び/又は分散させて、又はポリマー中に分散させて、好適に含有している。適切な活性化合物濃度は、約1%から35%、好ましくは約3%から15%である。特別な可能性として、活性化合物は、例えば、Pharmaceutical Research, 2(6): 318 (1986) に記載されているように、エレクトロトランスポート又はイオントフォレーゼによって放出することができる。
【図面の簡単な説明】
【0072】
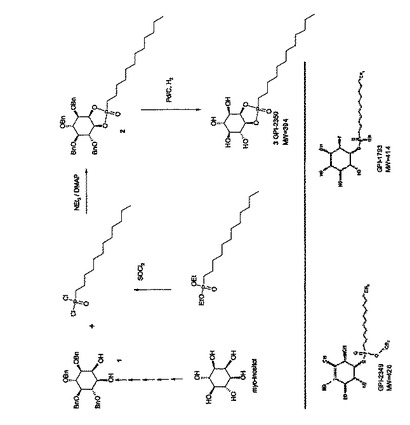
【図1】(G)PI−PLC阻害剤の構造及び合成を示す。材料と方法を参照。
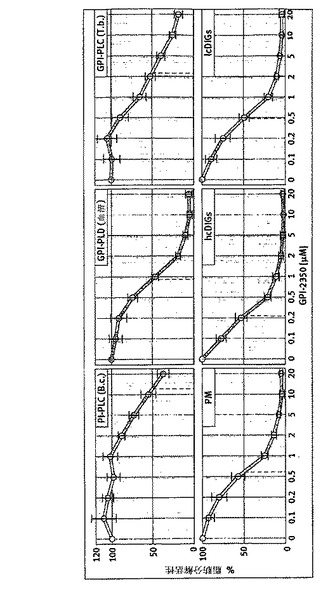
【図2】GPI−2350の力価を示す。B.cereus、ラット血清及びT.brucei由来の精製(G)PI−PLC/D、並びに単離したラット脂肪細胞由来の総原形質膜(PM)及びhc/lcDIGをGPI−2350の濃度を増加しながらインキュベートし(10分間、30℃)、次いでTX−114分配によってAChE及びaPの両親媒性から親水性への変換につきアッセイした(材料と方法を参照)。脂肪分解活性は、GPI−2350の非存在下における対照反応(100%と設定)の%を計算した。少なくとも4回の独立したインキュベーションを行い、AChE及びap活性を各3回測定して、平均+SDを得た。
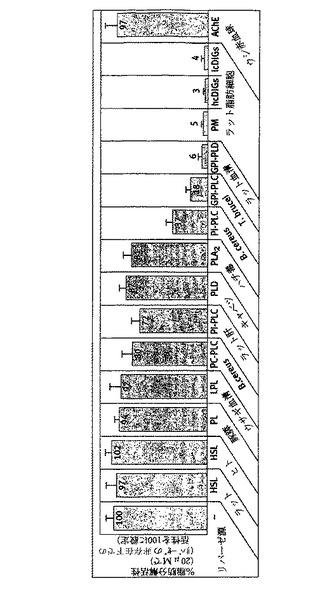
【図3】GPI−2350の選択性を示す。記載したように異なる由来の種々の(部分的に)精製した又は組換えリパーゼ(HSL、ホルモン感受性リパーゼ;PL、膵リパーゼ;LPL、リポプロテインリパーゼ;PC−PLC、ホスファチジルコリン特異的ホスホリパーゼC;PI−PLC、ホスファチジルイノシトール特異的ホスホリパーゼC;PLD、ホスホリパーゼD;PLA2、ホスホリパーゼA2;GPI−PLC、グリコシルホスファチジルイノシトール特異的PLC/D及びAChE(アセチルコリンエステラーゼ))、及び単離したラット脂肪細胞由来の総原形質膜(PM)及びhc/lcDIGを標準的なアッセイプロトコールに従ってGPI−2350(50μM)の非存在下又は存在下でアッセイした(材料と方法を参照)。少なくとも3回独立してインキュベーションを行い、各2回測定して、平均+SDを得た。
【図4】インスリン/グリメピリド誘導GPI−PLCによるGPIタンパク質の切断に対するGPI−2350の効果を示す。パネルA及びB.hcDIGを、未処理ラット脂肪細胞から調製し、次いで、記載したようにして、GPI−2350の非存在下又は存在下でインキュベートし(5分間、37℃)(パネルA、50μM;パネルB、濃度を増加させる)、続いてインスリン(10μM)又はグリメピリド(20μM)の無し又は有りで処理した(120分間、30℃)。パネルC及びD。単離したラット脂肪細胞を、記載したようにしてインスリン(10μM)又はグリメピリド(20μM)の非存在下又は存在下でインキュベートした(15分間、37℃)。hcDIGを調製し、次いでGPI−2350とインキュベート(120分間、30℃)した(パネルC、50μM;パネルD、濃度を増加させる)。パネルE、F及びG。単離したラット脂肪細胞を、記載したようにして、GPI−2350の非存在下又は存在下(パネルE及びG、50μM;パネルF、濃度を増加させる)でインキュベートし(5分間、37℃)、次いでインスリン(10nM)又はグリメピリド(20μM)の無し又は有りで処理した(90分間、37℃)。hcDIGを調製した。パネルA〜D。hcDIGのGPI−PLC活性を、外因基質として5’−Nucを用い、その親水性型への変換として測定した。パネルE〜G。hcDIGのTX−114枯渇相で回収したタンパク質を、親水性5’−NucおよびaPにつき免疫ブロット処理し、又はフォトアフィニティ標識によって親水性Gce1を分析した。3回の独立した細胞インキュベーションを行い、それぞれ活性測定及びゲル・ランを2回(代表例1つを示す)行って、平均+SEを得た。
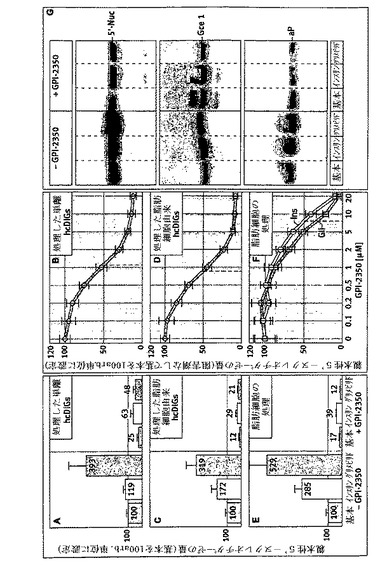
【図5】DIG内のシグナル伝達タンパク質のグリメピリド誘発再分配に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、記載したようにして、GPI−2350の非存在下又は存在下(最終濃度、50μM)で処理し(5分間、37℃)、次いでグリメピリド(50μM)の非存在下又は存在下でインキュベートした(120分間、37℃)。hcDIG及びlcDIGを調製し、それぞれ免疫ブロット法及びフォトアフィニティ標識によって、pp59Lyn、5’−Nuc、カベオリン1、インスリン受容体βサブユニット(IRβ)、グルコーストランスポーター4(Glut4)、及びGce1の存在についてアッセイした(ゲルの添付書類を参照)。hc/lcDIGと共に回収したpp59Lyn、Gce1及び5’−Nucの量は、GPI−2350の非存在下(100と設定)における対照反応(基本)と比較して示した。少なくとも3回の独立した細胞インキュベーションを行い、それぞれゲル・ラン2回(代表例1つを示す)を行って、平均+SDを得た。
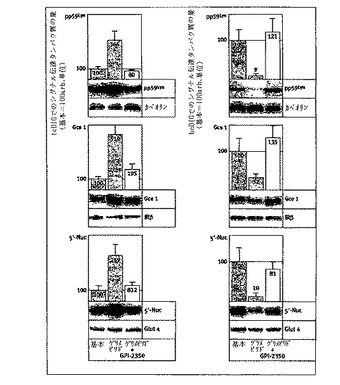
【図6】カベオリン由来のシグナル伝達タンパク質のグリメピリド/インスリン誘発解離に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、GPI−2350(左パネル、50μM;右パネル、濃度増加)の無し又は有りで処理し(5分間、37℃)、次いでインスリン(10nM)又はグリメピリド(20μM)の非存在又は存在下でインキュベート(120分間、37℃)した。hcDIGを脂肪細胞から調製し、可溶化した。可溶化hcDIGから調製したカベオリン1免疫沈降物を、それぞれ免疫ブロット法及びフォトアフィニティ標識によってpp59Lyn及びGce1の存在につきアッセイした。pp59Lyn及びGce1の量は、相同免疫ブロット法(ゲルの添付書類を参照)によって免疫沈降カベオリン1の回収につき補正した後に、対照反応において(パネルA、基本を100に設定)、又はインスリン又はグリメピリド促進状態において(パネルB、100に設定)GPI−2350の非存在と比較して示した。少なくとも4回の独立した細胞インキュベーションを行い、それぞれにつきゲル・ランを2回(代表例1つを示す)行って、平均+SDを得た。
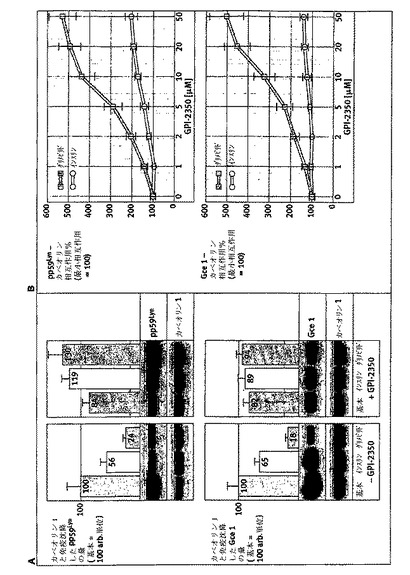
【図7】シグナル伝達タンパク質のグリメピリド/インスリン誘導活性化に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、GPI−2350(パネルA、50μM;パネルB、濃度を増加)の無し又は有りで処理(5分間、37℃)し、次いでインスリン(10nM)又はグリメピリド(20μM)の非存在下又は存在下でインキュベート(120分間、37℃)した。IRS−1及びpp59Lynは、脱脂した核の次にある下層(材料と方法を参照)から免疫沈降し、可溶化しhc/lcDIGとそれぞれ結合し、次いでホスホチロシンにつき免疫ブロット法にかけた。チロシンリン酸化IRS−1及びpp59Lynの量は、相同免疫ブロット法(ゲルの添付書類を参照)によって免疫沈降IRS−1及びpp59Lynの回収につき補正した後に、対照反応において(パネルA、基本を100に設定)、又はインスリン又はグリメピリド促進状態において(パネルB、それぞれの場合において100に設定)、GPI−2350の非存在のものと比較して示した。少なくとも4回の独立した細胞インキュベーションを行い、それぞれにつきゲル・ラン2回(代表例1つを示す)を行って、平均+SDを得た。
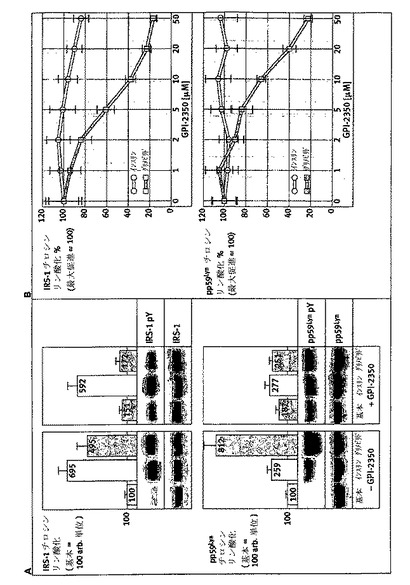
【図8】グリメピリド/インスリン誘導代謝活性に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、記載されたように、GPI−2350(パネルA、50μM;パネルB、濃度を増加;パネルC、5μM、50μM)の無し又は有りで処理(5分間、37℃)し、次いでインスリン(Ins、10nM)又はグリメピリド(Gli、20μM又は濃度を増加)の非存在下又は存在下でインキュベート(15分間、37℃)した。次いで脂肪細胞を、グルコース輸送及びイソプロテレノール誘導脂肪分解についてアッセイした。活性は、対照反応において(パネルA、基本)、又はインスリン又はグリメピリド促進状態において(パネルB及びC、100に設定)、GPI−2350の非存在のものと比較して示した。少なくとも5回の独立した細胞インキュベーションを行い、それぞれについて活性測定を3回行って、平均+SDを得た。
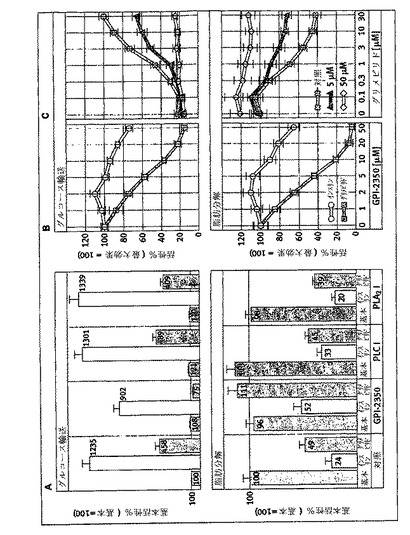
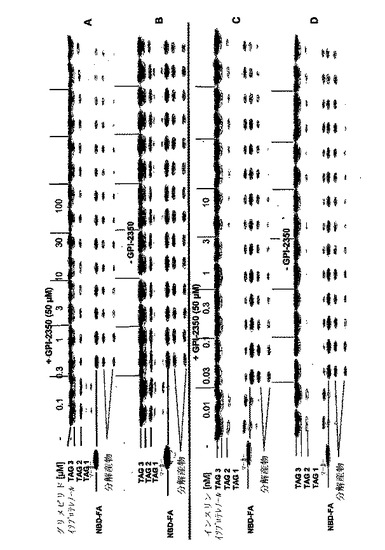
【図9】単離したラット脂肪細胞を0.5mM・NBD−FAで標識し(60分間、37℃)、洗滌し、次いでグリメピリド(パネルA、B)又はインスリン(パネルC、D)の濃度を増加して作用させる前に、GPI−2350の非存在下(パネルB、D)又は存在下(50μM;パネルA、C)でインキュベート(5分間、37℃)した。記載したようにして、イソプロテレノール(1μM)の無し又は有りで、更にインキュベーション(120分間、37℃)した後、総細胞懸濁液を、クロロホルム/ヘプタン/メタノール/0.1N・HCl(3/3/2/1、容積比)で抽出した。有機層を、マーカーとしてNBD−FAを同時に使用し、薄層クロマトグラフィー(ジエチルエーテル/石油エーテル/酢酸、78/22/1、容積比)及び蛍光イメージングにより分析した。2回の独立した標識化とインキュベーションを行った典型的な実験を示すが、同じような結果が繰り返された。TAG:トリアシルグリセロール。
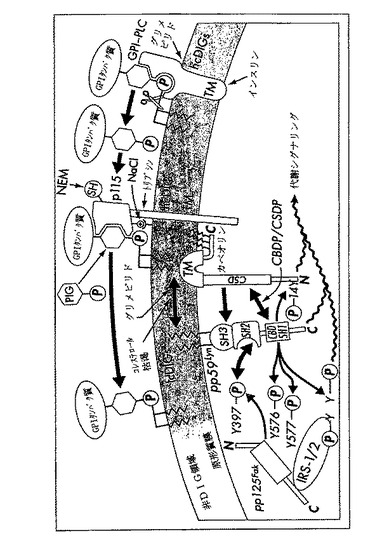
【図10】ラット脂肪細胞原形質膜の非DIG領域、hcDIG及びlcDIGの間のGPIタンパク質の再分配の機構についてのワーキングモデル、インスリン、グリメピリド及びGPI−PLC及び推定GPIタンパク質受容体、p115、を包含するコレステロール枯渇による制御、並びにカベオリン、pp125Fak及びpp59Lynを経由するIRS−1への代謝シグナリングの下流へのカップリングを示す。GPI−PLC及びp115の膜貫通領域(TM)を経由するhcDIGにおけるトポロジー、膜配向及び固定(anchorage)の型は、仮説に基づいたものである。しかし、DIGの細胞外小葉に面した活性部位及び結合部位のそれぞれは、大多数のGPIタンパク質の明らかにされた細胞表面位置を基にして強く示唆されているものである。カベオリンは、カルボキシル末端におけるフック様TMと3重のパルミトイル化の両者によってhcDIGの、アミノ末端おける2重アシル化によってpp59Lynの、原形質膜小葉中に埋め込まれている。
【実施例】
【0073】
材料及び方法:
ヒト組換えインスリン、PIG41(“Frick, W. et al., (1998), Biochemistry 37,
13421-13436" に開示されている)及びグリメピリド(商品名アマリール、Amaryl)は、Aventis Pharma Germany (Frankfurt, Germany)の薬化学・合成部から入手した。12−((7−ニトロベンズ−2−オキサ−1,3−ジアゾール−4−イル)アミノ)ドデカン酸(NBD−FA)は、Mueller, et al., (1997), Biochem. Biophys. Acta 1347: 23-39の記載に従って合成された。コラゲナーゼ(Worthington, CLS, type I, 250単位/mg)は、Biochrom (Berlin, Germany)により提供された。牛乳由来リポタンパク質リパーゼ(LPL、アフィニティ精製)、ホスホリパーゼA2(ミツバチ毒)、粗ブタ膵リパーゼ(PL)、組換えPC及びPI−PLC(Bacillus cereus)、粗PLD(キャベツ)及び脱脂BSA(画分V)は、Sigma/Aldrich (Deisenhofen, Germany)から購入し、粗PI−PLC(ラット肝)及びプロテイナーゼ阻害剤は、Roche Molecular Biochemicals (Mannheim, Germany)から購入した。組換えGPI−PLC(Trypanosma brucei)は、Oxford Glycosystems (Oxford, UK)から得た。PLA2(AACOCF3)及びPLC(U73122)の阻害剤は、Tocris (Avonmouth, UK)から入手した。Bisbodipy-C11-PCは、Molecular Probes (Eugene, OR)から購入した。脂質は、Avanti Polar Lipids (Birmingham, AL)から購入した。カベオリン1(クローンC060)の免疫沈降用抗体及び、カベオリン1(ウサギ)及びpp59Lyn(クローン32)の免疫ブロッティング用抗体はTransduction Laboratories (Lexington, KY)から入手した。ホスホチロシン(クローン4G10)の免疫ブロッティング用抗体は、Upstate Biotechnology (Lake Placid, NY)によって供された。(昆虫細胞で発現した総ヒト組換えタンパク質に対する)IRS−1の免疫ブロッティング用抗体(ウサギ、アフィニティ精製)、5’−Nuc(ラット)及びaP(ウシ)は、Biotrend (Cologne, Germany)によって製造された。ECL Renaissanceケミルミネッセンス検出キットは、NEN/DuPont (Bad Homburg, Germany)から得た。スプラーグドーリーラットは、Charles-River Laboratories (UK)から供給された。
【0074】
GPI−2350の合成(図1の図式も参照):
出発物質として、ラセミ体1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトール(以下、化合物1)は、Zhai, H.-X. et al., (1995), Tetrahedron Lett. 36: 7403-7406に記載の方法によって製造された。化合物1は、次いで、トリエチルアミン及びジメチルアミノピリジンの存在下、ドデシルホスホン酸ジクロライドでリン酸化し、テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(以下、化合物2)を得た。10%パラジウム炭で接触水素化することによって、化合物2を脱ベンジ
ル化し、ミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(以下、化合物3、又はGPI−2350)をジアステレオマーの混合物として得た。化合物2(図1)の合成のために、1g(1.8mmol)の化合物1を、メチレンクロリド60ml及びトリエチルアミン1mlに溶解し、ジメチルアミノピリジン200mg及びドデシルホスホン酸ジクロライド1g(10mmol)を加えた。この反応溶液を45分間室温で放置した。次いで、酢酸エチル50mlを加え、混合物をシリカゲルで濾過した。溶媒を濃縮後、残留物をフラッシュクロマトグラフィー(n−ヘプタン:酢酸エチル=1:1、v/v)で精製した。化合物2の収量(収率):580mg(43%)、白色非晶質固体。TLC:n−ヘプタン/酢酸エチル(1/1、v/v)、Rf=0.7。MS:(M+Li)+=761.4、計算値C60H59O7P、M=754.9。化合物3(図1)の合成のために、505mg(0.67mmol)の化合物2を酢酸エチル5mlとメタノール15mlの混合物に溶解した。パラジウム(10%)炭700mgを加えた後、反応混合物を水素添加(6時間、1気圧H2)した。パラジウムをシリカゲルで濾過し、メタノール100mlで洗浄した。溶媒を濃縮後、残留物をフラッシュクロマトグラフィー(メチレンクロリド/メタノール、5/1、v/v)によって精製した。化合物3の収量(収率):179mg(68%)、白色非晶質固体。TLC:メチレンクロリド/メタノール(5/1、v/v)、Rf=0.15。MS:(M+Li)+=401.2、計算値C18H35O7P、M=394.4。化合物3は、塩基性溶媒よりも酸性溶媒下の方が不安定である。メタノール中では何日も安定であり、乾燥非晶質固体は4℃で何年も安定である。
【0075】
ラット脂肪細胞の調製及びインキュベーション:
雄性ラット(120〜140g、自由給餌、文献53を参照)の精巣上体脂肪体から消化によって単離した脂肪細胞を、1%(w/v)BSA含有KRH(20mMHepes/KOH、pH7.4、1.2mM・KH2PO4、140mM・NaCl、4.7mM・KCl、2.5mM・CaCl2、1.2mM・MgSO4)で2回洗浄し、次いで、100μg/mlゲンタマイシン、100nM・1−メチル−2−フェニルエチルアデノシン、0.5U/mlアデノシンデアミナーゼ、1mMピルビン酸ナトリウム及び5mM・D−グルコースを補充した同じ培地中で、GPI−2350(DMSO中に10mM保存溶液として調製し、インビトロ及び脂肪細胞培養における最終DMSO濃度は、いずれの阻害剤濃度でも0.5%で一定に保たれた)、インスリン又はグリメピリド(“Mueller et al.,
(1994), J. Cell. Biol. 126:1267-1276" に記載されたようにして調製した)の存在下又は非存在下で、37℃にて振とう水浴中で、5%CO2/95%O2で一定でバブリングしながら、インキュベーション容積1ml当たりパックした細胞容積100μlの最終力価(少量の一定量(アリコート)を毛細管ヘマトクリットチューブに吸引し、脂肪細胞による懸濁液の分画占有を定量するためにマイクロヘマトクリット遠心機で60秒遠心して決定する;10%サイトクリットは約1.5×106細胞/mlに相当する)で、指定された期間インキュベートした。
【0076】
原形質膜及びlc/hcDIGの調製:
原形質膜及びDIGを、溶解バッファー(25mMモルポリノエタンスルフォン酸(MES)、pH6.0、140mM・NaCl、2mM・EDTA、0.5mM・EGTA、0.25Mショ糖、50mM・NaF、5mMピロリン酸ナトリウム、10mMグリセロール−3−リン酸、1mMオルトバナジウム酸ナトリウム及びプロテアーゼ阻害剤)中、モーター駆動テフロン・イン・ガラス・ホモジナイザー(10ストローク)を用いてホモジナイズし、前処理し洗浄した脂肪細胞のポストニュークレアー・インフラネイタント(postnuclear infranatant)から調製した。原形質膜を脱脂したポストニュークレアー・インフラネイタントの分画遠心法によって得、前に記載した(48、53)ようにしてショ糖及びパーコールクッションを通して2回逐次遠心によって精製し、最後に25mMトリス/HCl(pH7.4)、0.25Mショ糖、1mM・EDTAに2mgタンパク質/mlで懸濁した。hc/lcDIGを、“Mueller, G. et al., (2002), Mol.
Med. 8: 120-136" に報告されたようにして界面活性剤法及び不連続ショ糖勾配遠心分離によって得た。15〜22%(画分4〜5)及び28〜35%(画分8〜9)ショ糖インターフェースの光散乱乳白色バンドを、屈折率を用いた濃度測定で、それぞれ、hcDIG及びlcDIGとして集めた。集めた勾配画分を25mM・MES(pH6.5)、1%トライトンX−100、150mM・NaClで3倍希釈した後、遠心分離(50,000×g、30分、4℃)によりhcDIG及びlcDIGを収集し、次いで、“Mueller, G. et al., (2002), Mol. Med. 8: 120-136" 及び/又は“Mueller, G. et al., (2001), Mol. Cell. Biol. 21: 4553-4587" に記載されているように適切なマーカーの豊富な部分/乏しい部分により特徴づけ(免疫ブロット法、酵素アッセイ法)、或いはトライトンX−114分画法(下記を参照)にかけた。カベオリンの免疫沈降のために、DIGを10mMトリス/HCl(pH7.4)、150mM・NaCl、1%トライトンX−100、60mMβ−オクチルグルコシド、0.3%デオキシコール酸、5mM・EDTA、0.5mM・EGTA、1mMオルトバナジウム酸ナトリウム、50mM・NaF、1μMミクロシスチン及びプロテアーゼ阻害剤の溶液に可溶化し(1時間、4℃)、遠心分離した(50,000×g、30分間)。直接免疫ブロット法のために、DIGを2倍のLaemmli試料バッファーに可溶化し遠心分離した(10,000×5、5分間)。上清を使用した。
【0077】
GPI−PLC/Dアッセイ:
GPI−PLD(ラット血清)を、“Hari et al., (1997), Biochim. Biophys. Acta 1355: 293-302" に従って、200mMトリス/マレイン酸(pH7.0)及び1%NonidetP−40溶液100μl中のヒト胎盤aP溶液(10mM・Hepes/NaOH、pH7.0、150mM・NaCl中100単位/ml)10μlと10分間、37℃にてインキュベーションし、次いで0.4mlの氷冷停止バッファー(10mM・Hepes/NaOH、pH7.0、150mM・NaCl、0.1mM・MgCl2及び0.01mM酢酸亜鉛)を添加して反応を停止することによってアッセイした。GPI−PLC(T.brucei)を、“Mensa-Wilmot et al., (1995), Methods Enzymol. 250: 641-655" の記載のようにして、50mMトリス/HCl(pH8.0)、0.25%Nonidet/P−40及び5mM・EDTA溶液50μl中のひと胎盤aP溶液(上記を参照)5μlと、30分間、37℃にてインキュベーションし、次いで停止バッファー0.45mlを添加して反応を停止することによってアッセイした。PI−PLC(B.cereus)及び脂肪細胞GPI−PLC(5〜25μg原形質膜又はhc/lcDIG)を、20mM・Hepes/KOH(pH7.8)、144mM・NaCl、0.1%TX−100、0.2mM・MgCl2溶液100μl中のウシ赤血球アセチルコリンエステラーゼ(AChE)溶液10μl(10mMトリス/HCl、pH7.4、144mM・NaCl、0.1%TX−100中12単位/ml)と1時間、25℃にてインキュベーションし、次いで氷酢酸5μl及び次いで10mMトリス/HCl(pH7.4)、144mM・NaCl、0.4mlを加えて反応を停止することによってアッセイした。各反応混合物を、TX−114分配にかけた(以下を参照)。GPI−PLC/D活性は、TX−114消耗相中で測定された親水性aP又はAChEの活性と分配前に測定された総活性の比率から計算され、そして(G)PI−PLC/Dを欠いたブランクのインキュベーションで現れたTX−114消耗相(総活性の10〜20%の割合を占める)中の非酵素バックグランドに対して補正された。
【0078】
その他のリパーゼアッセイ:
ホルモン感受性リパーゼ(HSL)及びリポプロテインリパーゼ(LPL)は、“Vertesy et al., (2002), Journal Antibiotics 55:480-494" に記載されたようにして、放射能標識トリオレイン液滴エマルジョンを使用して測定された。膵リパーゼ(PL)は、5mMトリス/HCl(pH6.5)、6mMタウロデオキシコール酸ナトリウム、150mM・NaCl、1mM・CaCl2溶液2ml中の0.25mmolのトリブチリンを、
記録pHスタット(1,000rpm、25℃で撹拌)を用いてpHを6.5に調節しつつ、同じバッファー中のブタPL及びコリパーゼとインキュベーションすることによって定量した。PC−PLCは、10mMジパルミトイルレシチン0.2ml、10mM・SDC0.1ml及び0.03M・CaCl20.1mlをバクテリアPC−PLC(50mMトリス/HCl、pH7.5、0.1%BSA)と、10分間、37℃にてインキュベーションすることでアッセイした。反応は、50%TCAを0.1ml、次いでクロロホルム/メタノール(66/33/1、容量比)を2.5ml加えることによって停止した。遠心分離(1,500×g、15分)後、メタノール/水相(約1.33ml)の上部0.2ml部を除き、60%HCIO40.5mlを補充し、次に170℃で1時間加熱して、最終的に無機リン酸塩を分析した。哺乳類PI−PLCは、10mM・PI0.1ml、0.8%デオキシコール酸ナトリウム0.1ml、0.1%BSA、100mMホウ酸ナトリウム(pH7.5)0.2ml、及びラット肝PI−PLCを20分間、37℃でインキュベーションすることによって測定した。反応を停止し、PC−PLCで記載したようにして更に処理した。PLDを40mM・Hepes/KOH(pH6.0)、4mMCaCl2及びキャベツPLD溶液250μl中1.6mM(U−14C)ホスファチジルコリン(約2,000−4,000dpm/nmol)でアッセイした。30分間、37℃にてインキュベーションの後、反応を、キャリアーホスファチジン酸含有クロロホルム/メタノール(2/1、v/v)5mlを加えて停止した。水可溶性物質を除去した後、最後の洗浄低クロロホルム相中に含有される脂質を、加熱活性化したシリカゲルプレートに転送し、第1次元方向にクロロホルム/メタノール/アンモニア(65/35/4、体積比)を使用し、第2次元方向にクロロホルム/アセトン/メタノール/酢酸/水(50/20/10/10/5、体積比)で2次元的に分離した。放射性ホスファチジン酸を蛍光イメージングにより検出した。PLA2を、1−パルミトイル−2−パルミトイル−sn−グリセロール−3−ホスホコリン/ビスボジピ−C11−PC/ホスファチジル−グリセロール/コレステロール(10/0.05/2/3、体積比)からなる単層リポソーム基質で、“Kim, T.-S. et al., (1997), J. Biol. Chem. 272:2542-2550" に従って定量した。濃度−反応曲線を、Marquardt-Levenberg非線形最小二乗演算手法を使用してフィットさせた。対数・直線軸にプロットするとき、この式はシグモイド曲線となる(Sigma Plot Software,
Jandel Scientific)。
【0079】
カベオリンの免疫沈降:
可溶性DIG(上記を参照、5〜20μgタンパク質)は、プロテインA/G−セファロースでインキュベーションし、次いで遠心分離(10,000×g、5分間)することにより事前に確かめた。上清を、10mMトリス/HCl(pH7.4)、150mM・NaCl、1%TX−100の1ml中で、プロテインA/G−セファロースに前もって吸着させた抗カベオリン1抗体と4℃にて1時間インキュベートした。免疫複合体を、同じバッファーで2回、次いでTX−100不含のバッファーで2回洗浄し、最後にβ−メルカプトエタノールなしで行うSDS−PAGEにかけた。免疫沈降カベオリンの回収は、膜の細片化に次いで、同じブロットの抗カベオリン抗体による均一免疫ブロッティングを行うことによって正常化された。
【0080】
免疫ブロット法:
SDS−PAGEによって分離されたポリペプチドを、“Mueller, G. et al., (2001), Mol. Cell. Biol. 21: 4553-4567”に記載されたように半乾燥法を用いてポリビニリデンジフルオライド膜に移した。洗浄した膜を抗カベオリン1抗体(1:2,000)、抗pp59Lyn抗体(1:1,250)、抗aP抗体(1:500)、抗5’−Nuc抗体(1:750)および抗IRS−1抗体(1:2,000)を15℃にて4時間インキュベートした。洗浄した膜を、西洋ワサビペルオキシダーゼを結合した、ヤギ抗マウスIgG2次抗体(1:2,000)又はヤギ抗ウサギIgG2次抗体(1:4,000)とインキュベートした。標識したタンパク質を増強したケミルミネセンスによって可視化した。
【0081】
TX−114分配:
ペレット化したhc/lcDIG(10〜50μgタンパク質)又は(G)PI−PLC反応混合物(0.5ml)を、それぞれ氷冷TX−114(1%)、25mMトリス/HCl(pH7.4)、144mM・NaCl溶液1ml中に懸濁することにより、又は氷冷TX−114(2%)0.5mlと混合することにより、“Bordier, C., (1981), J.
Biol, Chem. 272: 2542-2550" に記載された、TX−114が豊富な相とTX−114が枯渇した相との間の分配を使用して、両親媒性タンパク質及び親水性タンパク質に分離した。氷上で1時間インキュベーションした後、混合物を氷上で0.25Mショ糖及び25mMトリス/HCl(pH7.4)溶液0.4mlのクッション上に重層した。相分離を37℃に暖め、次いで遠心分離(10,000×g、1分間)することによって誘導した。下のTX−114が豊富な相の再抽出後、集めた上のTX−114が枯渇した相の一定量を5’−Nuc、aP及びAChE活性について測定するか、又は、SDS−PAGE分析用に沈殿(15%ポリエチレングリコール4,000)させた。
【0082】
グルコース輸送アッセイ:
グルコース輸送を、50μl部分中、最終濃度50μM(0.33μCi/ml)の2
−デオキシ−D−(2,6−3H)グルコースとともに、洗浄した10,000〜20,0
00個の脂肪細胞を、20μMサイトカラシンBの存在下又は非存在下において20分間、37℃でインキュベーションすることにより、“Mueller, G. et al., (1994), J. Cell Biol. 126: 1267-1276" に記載されたようにして定量した。
【0083】
種々の手順:
電気せん孔法は、“Mueller, G. et al., (2000), Mol. Cell. Biol. 20: 4708-4723"
に記載されたようにして実施した。脂肪分解は、“Mueller, G. et al., (2003) ,Biochimie 85: 1245-1256" に記載されたようにして、前もって標識され、イソプロテレノール刺激をされた脂肪細胞からの、グリセロール又は蛍光NBD−FAの放出として測定した。Gce1は、“Mueller, G. et al., (1994), Biochemistry 33: 12149-12159" に記載されたようにして、可溶化原形質膜又はhc/lcDIG(10〜50μgタンパク質)を8−N3−[32P]cAMPで光標識化し、次いで蛍光イメージングすることによって検出した。5’−Nuc、aP及びAChE活性は、“Eliakim, R. et al., (1990), Biochim. Biophys. Acta 1355: 293-302"に従って測定した。タンパク質は、Pierce (Rockford, IL)のBCAタンパク質測定キット、及び較正基準としてBSAを使用して測定した。SDS−PAGEは、前もって成型されたゲル(Novex, San Diego, CA;10%Bis−Tris分離ゲル、モルホリノプロパンスルホン酸−SDSランニングバッファー)を使用して実施された。ルミイメージ(lumiimage)は、ルミイメージャー(LumiImager)ソフトウエア(Roche Diagnostics)を使用したルミイメージャーで評価した。リン光イメージ及び蛍光イメージは、Storm 860 PhosphorImager system (Molecular Dynamics, Gelsenkirchen, Germany)を使用して処理し定量化した。図は、Adobe Photoshopソフトウエア(Adobe Systems, Mountain View, CA)を使用して構築した。
【0084】
GPI−2350は、高力価及び選択性を有する種々の起源の(G)PI−PLC/Dを阻害する:
(精製した、又は、未精製の)B.cereusPI−PLC、T.bruceiGPI−PLC及びラット血清GPI−PLDに対するGPI−2350の効果を、適切な条件下(低又は高界面活性剤濃度)で、GPI−2350の濃度を増加しつつ、部分的に精製した可溶化GPIタンパク質と共にそれらをインキュベーションすることによって観察した。次いで、総GPIタンパク質に対する、消化混合物中に脂肪分解的に切断されたGPIアンカーを内蔵する親水性GPIタンパク質の比率を、TX−114分配で界面活性剤が枯渇している相、及び分配前の総消化混合物、それぞれにおける酵素活性の測定によ
って定量した(図2)。GPI−2350は、細菌、トリパノソーマ及び血清(G)PI−PLC/Dを、それぞれ10、2及び1μMのIC50で、明らかに阻害した。GPI−2350は、又、酵素源として可溶化原形質膜、lcDIG又はhcDIGを、及び基質としてAChEを使用すると、ラット脂肪細胞GPI−PLCを0.2〜0.5μMのIC50で遮断した。脂肪細胞GPI−PLCの切断特異性を明らかにするため、本発明者らは、ミオ−[14C]−イノシトールで代謝的に標識されたサッカロマイセスセレヴィシエスフェロブラストから調製したGPIタンパク質、Gce1を使用した。hcDIGとのインキュベーションで、親水性及び14C標識されたGce1が生成され、それはcIPに対して生じる抗CRD抗体と濃度及び時間依存的に免疫沈降した(データは示さない)。脂肪酸アシル鎖を欠きcIPを内蔵するGPIタンパク質の、hcDIG依存的な生成により、ラット脂肪細胞の原形質膜にGPI−PLCの発現が確認された。
【0085】
次に、GPI−2350の選択性を試験した。種々の起源からのいくつかの天然リパーゼ(HSL、PL、LPL)及び異なった特異性(A2、C、D)のPC/PI特異的ホ
スホリパーゼそしてウシAChE(これらは全ていわゆる触媒三点セットを経て作動することが知られている)は、細菌、トリパノソーマ、血清及びラット脂肪細胞GPI−PLC/Dを60から95%遮断する濃度(50μM)下では有意な影響は受けなかった(図3)。cIPの代わりにオープン(open)を内蔵するGPI−2350の二つの誘導体2−デオキシ−2−フルオロ−scyllo−イノシトール−1−O−ドデシル−ホスホン酸(GPI−1793)及びミオ−イノシトール−1−O−ドデシル−ホスホン酸メチルエステル(GPI−2349)は、試験した最高濃度で脂肪細胞GPI−PLCに対して、それぞれ温和から非常に温和な効果を有するだけであったが、しかし、細菌及びトリパノソーマ(G)PI−PLCを同程度の力価で阻害した(表1)。これらの知見は、安定した又は一過性の1,2−環状リン酸結合の生成を含む触媒機構における類似性を示しているが、しかしながら、細菌、トリパノソーマ及び脂肪細胞(G)PI−PLCの間の基質認識における幾分かの相異も示している。後者がcIP中間体を経由して作動、予備的な動態試験に基づいていると仮定すると、GPI−2350は拮抗阻害剤(Ki=3〜20μM)として作用することを仮定しなければならない。このことは、中性リパーゼ及び哺乳類PC/PI特異的ホスホリパーゼはGPI構造を認識しないので、その顕著な選択性を説明できるであろう。拮抗的作用機構に一致して、GPI−2350による細菌及びトリパノソーマ(G)PI−PLCの半値阻害は、過剰の新鮮なGPIタンパク質基質との反応混合物の10倍希釈において完全に逆転した。対照的に、再単離及び充分な洗浄に先立って50μMGPI−2350で処理した脂肪細胞hcDIGは、GPI−2350非存在下でインキュベーションすると、精製した可溶化AChEに対しては、低い脂肪分解的切断のみを呈した(データは示さない)。このことは、両親媒性GPI−2350のhcDIGとの相互作用により説明することができるであろう。あるいは、GPI−2350は、遷移状態類似体として作用し、脂肪細胞GPI−PLCの活性部位への(一過性の)共有結合を受けるのであろう。GPI−2350の環状リン酸結合が(G)PI−PLC/Dによって開環されるかどうかを知ることは、興味あることであろう。
【0086】
GPI−2350は、単離されたDIG及び無処置の脂肪細胞においてGPIタンパク質のインスリン誘導及びグリメピリド誘導切断をブロックする:
GPI−2350及びGPI−PLCの両者が別々の複合体(それぞれ、界面活性剤ミセル及びDIG)で存在するとき、インビトロにおける脂肪細胞原形質膜GPI−PLCによるGPIタンパク質の切断に対するGPI−2350の介入の観察に基づいて、次に、基質としての内因性又は外因性GPIタンパク質の無処置の脂肪細胞及び単離されたDIGにおける基本的及び促進されたGPI−PLC活性に対するGPI−2350の効果について試験した(図4)。単離されたラット脂肪細胞において、GPI−PLC活性(内因性5’−Nuc、Gce1及びaPの切断として、それらの両親媒性から親水性への変換の観察が伴う)は、グリメピリド及びインスリンの両者によって(それぞれ、5倍及
び3倍まで)促進された(パネルE、G)。促進は、これらの前処置した脂肪細胞から調製されたhcDIGにおいて部分的に保存されていた(パネルC)(それぞれ、3倍及び1.7倍)。GPI−PLCは、グリメピリドで処理されなかった脂肪細胞から調製されたhcDIGの直接インキュベーションにより際だって活性化されたが、インスリンではそうでなかった(パネルA)。GPI−2350は、基本よりも低く、そして単離されたhcDIG及び未処理の脂肪細胞の両者において同じようなレベルで、濃度依存的に基本のGPI−PLC活性を減少させ、同様にグリメピリド及びインスリン誘導GPI−PLC活性を減少させた(図4、全てのパネル)。阻害剤がインスリン又はグリメピリドとのインキュベーションより前に、及びインキュベーションの間に存在していたので、明らかにGPI−2350は、GPI−PLCの活性を妨害しない(パネルC、E)。未処理の脂肪細胞でアッセイする場合、GPI−PLCの阻害のIC50は(5〜10μM、パネルF)は、単離された未処理のhcDIGに比較してより高く(1μM、パネルB、D)、同様に可溶化hcDIG及び原形質膜(0.2〜0.5μM、図2)に比較して、より高かった。このことは、単離されたDIGと比較して未処理の脂肪細胞の原形質膜においてDIGの中に埋め込まれた場合、GPI−2350に対してGPI−PLCの触媒部位の接近し易さが損傷されたことを反映しているのであろう。しかし、GPI−PLCの主要な基質、GPI脂質及びタンパク質アンカーは原形質膜の外小葉に局在化しており、そしてGPI−2350はGPI−PLCの触媒部位の(拮抗)阻害剤として作用しているようであるので(上記を参照)、その両親媒性を基本として、むしろ高いと予測されている、GPI−2350の細胞透過性の必要性は、多分除外することができる。むしろ、DIGの液体定序ドメインよりもむしろ非DIG領域の無秩序ドメインへ選択的に分配される界面活性剤に類似している脂肪細胞原形質膜(総細胞表面の80〜90%を含む)の非DIG領域に、GPI−2350はそのドデシル鎖を経て自然に分配されることが考えられる。
【0087】
DIG及び原形質膜の単離に使用された実験的条件下で、単離された総原形質膜中のGPI−PLC活性の65〜85%(外因性AChEの切断として測定)が、脂肪細胞の処理に関係なくDIG(即ち、hcDIGプラスlcDIG)により回復した。同様の回復が、典型的なDIGマーカータンパク質、カベオリン1(70〜90%)(免疫ブロット法により定量した)、及びDIG常在性タンパク質、p115(55〜75%)(合成PIGの結合により定量した)について観察された。更に、区別を付けて処理した脂肪細胞からDIG(hcDIGプラスlcDIG)で回復したaP、5’−Nuc及びGce1の総量(両親媒性プラス親水性のもの)は、おおよそ一定(基準を100%と設定して90〜140%)であって、このことは種々の条件下でDIGの単離手順の再現性を明示している。単離されたhcDIGは、lcDIG及び非DIG領域よりも比較的低いタンパク質含量により特徴づけられているので、GPI−PLCは、残りの原形質膜に対してhcDIGが非常に豊富であり、GPIタンパク質基質の局在化と一致している。
【0088】
DIG内にGPIアンカーされた及びアシル化されたシグナル伝達タンパク質の再分配にはラット脂肪細胞におけるGPI−PLC作用を必要とする:
次に、GPIタンパク質の脂肪分解的切断の阻害が、脂肪細胞原形質膜のDIG内でGPIアンカーされた及びアシル化されたシグナル伝達タンパク質の局在及び刺激依存性再分配に影響するかどうかを研究した。このために、グリメピリドで処理する前にGPI−2350の非存在下又は存在下でインキュベートした脂肪細胞からhc/lcDIGを調製し、次いでいくつかのGPIアンカーされた又はアシル化されたシグナル伝達タンパク質の存在につき分析した(図5)。基本の脂肪細胞に比べてhcDIGの量の70から90%の損失に相当する、lcDIGの量の3から5倍の増加に見られるように、グリメピリドに反応したhcDIGからlcDIGへのpp59Lyn、Gce1及び5’−Nucのしっかりした再分配は、GPI−2350の最大有効濃度の存在下で殆んど完全に止められた。対照的に、lcDIGに対してhcDIGにおいて、hcDIG常駐タンパク質
であるカベオリン1及びIRβが比較的豊富であること、並びにhcDIG及びlcDIGにおいてグルコース輸送体イソ型4(Glut4)が同様に多量であることの両者は、グリメピリドに影響されず、GPI−2350の存在下で顕著には変化しなかった(図5)。このことはGPI−PLCの強力な阻害は、DIGの構造における一般的且つ非特異的変化をもたらすよりはむしろhcDIGからlcDIGへの、Gce1及びpp59Lynのような、ある種の脂質修飾シグナル伝達タンパク質の刺激依存性転移を、特異的に妨げることを明らかにしている。
【0089】
次に、GPI−PLCの作用とGPIタンパク質の再分配の間の推定因果関係、即ち脂肪分解的切断及び/又は非切断GPIタンパク質は、刺激された単離ラット脂肪細胞において、実際にlcDIGに転移するのか否か、の根底にあるメカニズムを研究した。このために、ラット脂肪細胞をGPI−2350と共に、又は、無しにインキュベートし、インスリン又はグリメピリドを作用させた。総原形質膜、hcDIG及びlcDIGから回収されたGPIタンパク質、即ち5’−Nuc、aP及びGce1の親水性型及び両親媒性型の量は、TX−114分配によって定量された(表2)。インスリン及びより強力なグリメピリドでの処理は、GPI−2350によって完全にブロックされた原形質膜の親水性GPIタンパク質の増加をもたらし、効率的なGPI−PLC阻害を示している。インスリン及びグリメピリドの両者に応じて、親水性GPIタンパク質の量の増加が、もっぱらhcDIGにおいてのみ検出され、同じ位置での両親媒性型の減少を伴っていた。これらの変化は、GPI−2350の存在下で更に低い基本値に逆転し、この両親媒性から親水性への変換という脂肪分解性を示していた。それ故、hcDIGでのインスリン/グリメピリド促進GPI−PLCによって生成する親水性GPIタンパク質は、GPIアンカーによらない分子機構を経てhcDIGに関連して残る(表2)。興味深いことに、hcDIGからlcDIGへの5’−Nuc、Gce1及びaPのグリメピリド誘導再分配(図5参照)は非切断型に限定され、両親媒性においては3から4倍上昇したレベルであるが、lcDIGで回復した親水性GPIタンパク質では変らない低レベルであることを反映していたが、それにもかかわらず、GPI−2350によって完全に除去されていた(表2)。対照的に、親水性5’−Nuc、Gce1及びaPのインスリン誘導生成は、lcDIGで両親媒性の対応物の非常にゆるやかな増加(有意差の限界)を伴ったのみであったが、それは再びGPI−2350の存在で排除された。試験した3つのGPIタンパク質全てが、ほんの少しの量的差異だけで、もっぱら、刺激依存性脂肪分解的切断と両親媒性型の再分配との間に相関性を示した(表2)。総合すると、hcDIGで脂肪分解的に切断されたGPIタンパク質の生成及び蓄積のためのGPI−PLC活性化が必要である(グリメピリド)。
【0090】
GPI−PLCの阻害は、GPIアンカー型及びアシル化シグナル伝達タンパク質のカベオリンからの解離を妨げる:
ラット脂肪細胞におけるhcDIGからlcDIGへのシグナル伝達タンパク質の再分配は、最近、カベオリン1からの解離と活性化を伴うことが示されている。次に、GPI−PLCを阻害することによるラット脂肪細胞における、カベオリンからの解離/活性化と、GPIタンパク質の刺激依存性脂質分解的切断との間の推定因果関係、及び、それに続いて、Gce1とpp59Lynのカベオリン1との相互関係、及び後者のキナーゼ活性の解析が研究された(図6)。単離及び可溶化されたhcDIGから調製されたカベオリン1免疫沈降物からのpp59Lyn及びGce1の両者のインスリン誘発最大損失(35〜45%)及びグリメピリド誘発最大損失(75〜85%)最大損失は、5〜10μMのIC50(図6、パネルB)を有するGPI−2350(50μM:図6、パネルA)によって完全に妨げられた。この力価は、未処理の脂肪細胞におけるGPI−2350によるGPI−PLC阻害に類似している(図3)。非受容体チロシンキナーゼのようなシグナル伝達タンパク質のカベオリン結合ドメイン(CBD)へのカベオリン1の足場(scaffolding)ドメイン(CSD)の結合、及び、逆に、CBDへの結合からのCSDの除去は
、全ての場合ではないが多くの場合、インビトロアッセイにおけるシグナル伝達タンパク質の不活化及び活性化のそれぞれの引き金を引くことが示された。このことは、原形質膜DIGで作動しているシグナル伝達における、CSD−CBD相互関係の調節機能を暗示している。
【0091】
ラット脂肪細胞におけるGPI−PLCの阻害は、グリメピリドの代謝活性をダウンレギュレートするがインスリンはされない:
次いで、インスリン及びグリメピリドに反応したIRS−1チロシンリン酸化におけるGPI−PLC阻害の差動効果が、インスリン標的細胞における代謝活性に反映するか否かを研究した。このために、単離したラット脂肪細胞を、どちらかの刺激で作用させる前にGPI−2350で前処理し、次いでグルコース輸送及びイソプロテレノール誘導脂肪分解についてアッセイした(図8)。グリメピリドによるグルコース輸送の促進及びイソプロテレノール誘導脂肪分解の阻害は、GPI−2350の存在下、濃度依存的様式(IC50=2〜5μM)で対照値(50μMで)よりも低く損なわれた。従って、グリメピリド濃度−反応曲線は右に移動し、阻害剤がない場合と比較してGPI−2350(5μM)によるGPI−PLCの最大半量阻害の途中で平坦になった。対照的に、グルコース輸送のインスリンによる促進及びイソプロテレノール誘導脂肪分解の阻害は、グリメピリドに比較して2から3倍、より顕著であるが、50μMで、GPI−2350のみによってそれぞれ最大25及び33%減少した(図8)。これらのGPI−PLC阻害の差動効果は、蛍光脂肪酸誘導体で前もって標識したラット脂肪細胞からのグリセロールに代わってNBD−FAの放出として測定した場合、グリメピリド及びインスリンによるイソプロテレノール誘導脂肪分解の阻害に対するGPI−2350の効果の分析によって確認された(図9)。グリメピリド(IC50=3μM)の濃度増加に反応してイソプロテレノール誘導脂肪細胞から放出された酸化分解産物を包含するNBD−FAの量の減少は、50μMグリメピリドの存在で完全に消失された(パネルA、B)。対照的に、NBD−FA(及び分解産物)放出のインスリン阻害は、インスリンに対する見かけのIC50の少しの増加(0.1対0.3μM)を伴うGPI−2350(50μM)のみによって、最大25〜30%損なわれた。GPI−PLC阻害に対するこれらGPI−2350媒介効果の特異性は、ラット肝PI−PLC及びハチ毒PLA2の阻害剤の損傷によって、基本的な及びグリメピリド調節グルコース輸送及び脂肪分解に顕著に影響を与えることで確認された(図8)。
【0092】
最後に、DIG内でGPIタンパク質の再分配に依存性であるか、無関係であるかのいずれかである、グリメピリド以外のインスリン模倣刺激のシグナル伝達におけるGPI−PLCの関与について調べた。ラット脂肪細胞において、m−β−CDを用いたコレステロール枯渇によるhcDIGの崩壊、トリプシン/塩/NEM処理によるPIG受容体p115の不活化又は合成リガンドPIG41でのp115の占拠は、lcDIGでの5’−Nucの顕著な増加及びグルコース輸送活性化を導いた(表3)。対照的に、オルトバナジウム酸ナトリウム及び合成CBDPはlcDIGへの5’−Nuc転位の同時誘導なしにグルコース輸送を促進した。このことは、よく知られている、チロシンホスファターゼ阻害剤、バナジウム酸塩によるインスリン受容体及びIRS−1脱リン酸化、及びCBDPによるpp59Lyn及びIRS−1チロシンリン酸化の直接促進といった作用機序に一致している。これらの刺激によるIRS−1チロシンリン酸化でもなくグルコース輸送でもない促進(表3)は、GPI−2350の存在下で顕著に影響され、それらは全てGPI−PLCの下流の点でグルコース輸送システムにクロストークしていることを暗示している。
【0093】
表の説明
表1:(G)PI−PLC阻害剤の特徴。
表2:hcDIG、lcDIG及び総原形質膜における脂肪分解的切断及び非切断GPI
タンパク質の局在化に対する、インスリン、グリメピリド及びGPI−2350の効果。表3:ラット脂肪細胞における種々のインスリン模倣刺激によって誘導される5’−Nuc転位及びグルコース輸送に対するGPI−2350の効果。
【0094】
表についての説明
表1:ラット脂肪細胞由来のhcDIGのGPI−PLC、T.brucei由来GPI−PLC及びB.cereus由来PI−PLCを、適切な条件下で指示した阻害剤の増加する濃度(0.05μM〜1mM)のない場合とある場合における、可溶化した及び
部分精製ウシ赤血球AChE又はヒト胎盤aPとインキュベートした(材料と方法を参照)。切断率は、親水性AChEの量及びTX−114分配の際TX−114枯渇相で回復したaP活性から計算した。少なくとも3回の独立したインキュベーションと分配/4回の酵素アッセイ、から得られた平均+SDを記した。n.a.:該当なし。
【0095】
表2:単離したラット脂肪細胞を、GPI−2350(50μM)なし又はありで処理し(5分間、37℃)、次いで指示したように、グリメピリド(20μM)又はインスリン(10nM)の非存在下又は存在下にインキュベートした(120分間、37℃)。総原形質膜(PM)、hcDIG及びlcDIGを調製し、次いでTX−114分配にかけた。TX−114豊富相及び枯渇相を5’−Nuc及びaP(活性測定)及びGce1(光親和性標識)についてアッセイした。両親媒性及び親水性GPIタンパク質の量は、GPI−2350の非存在下に基本の脂肪細胞から調製した、総原形質膜及びhcDIGの両親媒性型のそれぞれに対して計算した(各100arb.単位に設定した)。hcDIGプラスlcDIGからの親水性及び両親媒性GPIタンパク質の回収は、種々のインキュベーション条件下で同程度であった。少なくとも3回の独立した細胞インキュベーションを行い、それぞれ活性測定及びゲル・ランを2回行って、平均±SDを得た。
【0096】
表3:単離したラット脂肪細胞を、メチル−β−シクロデキストリン(m−β−CD、10mM、50分間)、CBDP(300μM、電気穿孔法、次いで、洗浄及びそれに続く30分間のインキュベーション)、PIG41(10μM、15分間)、オルトバナジウム酸ナトリウム(1mM、15分間)、トリプシン(10μg/ml、15分間、次いで0.5M・NaCl処理及びそれに続く洗浄)及びN−エチルマレイミド(1mM、5
分間、次いでDTTの添加)の存在下でインキュベートした。脂肪細胞懸濁液の半分にGPI−2350(最終濃度50μM)を加え、更にインキュベーション(60分間、37℃)の後、細胞の一部分を、単離したlcDIG中の5’−Nuc活性についてアッセイした。基本的なインキュベーション中の5’−Nucの増加を100arb.単位に設定した。細胞の別の部分は(15分間のインキュベーション後)、基本のインキュベーションについて100arb.単位に設定したグルコース輸送につきアッセイした。少なくとも3回独立した細胞インキュベーションを行い、それぞれ3回ずつ活性を測定して、平均+SDを得た。
【0097】
【表1】
【0098】
【表2】
【0099】
【表3】
【技術分野】
【0001】
本発明は、哺乳類GPI−PLC(グリコシルホスファチジルイノシトール特異的ホスホリパーゼC)の活性を調節する、異なる方法に関する。
【背景技術】
【0002】
哺乳類原形質膜GPI−PLCに特異的な阻害剤、GPI2350の合成は、細菌やトリパノソーマの(G)PI−PLCの(結晶)構造、基質の必要条件、GPI認識及び切断機構についての背景的知識や、更にこれまでそれらについて記載された阻害剤に基づいていた。トリパノソーマのGPI―PLCに対しては、GPI及びPIは、それぞれ、効率的な及び貧弱な基質である一方で、細菌のPI−PLCに対してはその反対である。後者は、80残基のトリパノソーマのGPI−PLCにタンパク質配列が類似した領域を有している(“Kuppe et al., (1989), J. Bacteriol. 171:6077-6083")。GPI認識の解析のために、B.cereusからのPI−PLCの三次元構造が、グルコサミニル(α1→6)−ミオ−イノシトール(GMI)との複合体で2.2オングストロームの分解能で最近決定され、GMIのミオ−イノシトール部分は遊離のミオ−イノシトールと同じ位置を占有しているのに対して、グルコサミン部分は触媒部位の入り口で溶媒に露出した状態にあることが明らかにされた(“Heinz et al.,(1995), EMBO J. 14:3855-3863" 及び“Heinz et al., (1996), Biochemistry 35:9496-9504")。コア四糖類の残りの部分は、殆んどPI−PLCと接触がなく、このことはGPIアンカー内に受入れられたコアグリカンの顕著な構造多様性を説明しているのであろう。これらをもとにして、現時点の実験データは、細菌のPI−PLCによるPI及びGPIの、更に、トリパノソーマのGPI−PLCによるGPIの切断の触媒機構は全て類似していることを示唆している。しかしながら、哺乳類PI−PLCがセカンドメッセンジャー、イノシトール三リン酸、を生成して、GPIアンカーを受入れることができず、細菌のPI−PLCがリン酸化PIを切断することができないことを考慮すると、このことが脂肪細胞原形質膜GPI−PLCに対して正しいかどうかは知られていなかった。
【0003】
細菌及びトリパノソーマの(G)PI−PLCの基質必要条件は、以前、PI類縁体及びGMI誘導体で試験されていた。触媒作用には、イノシトールの2位に遊離のOH基が必要とされる。T.brucei由来のミリスチン酸エステル含有VSGを用いた研究で、(G)PI−PLCは、GMIの2−デオキシ−イノシトール類縁体によって競合的に阻止され、イノシトール−2−OHは、触媒作用には必要であるが、基質認識に重要ではないことが示された。更に、基質認識には、イノシトールの1位に荷電したホスホリル基(即ち、ホスホネート又はホスホジエステル)が必要である(“Morris, J. C., et al.,
(1996), J. Biol. Chem. 271:15468-15477”)。それ故、イノシトール−1位及びイノシトール−2位の両方のOH基は触媒作用に関与しているが、なお、ホスホリル基のみが基質認識に必要であるようにみえる。興味深いことに、グルコサミン(α1→6)−イノシトール−1,2−環状リン酸は、トリパノソーマGPI−PLCに対してGMI−1−リン酸よりも、よりよい阻害剤であるが、細菌のPI−PLCに対してはそうでないことが判明したが、このことは環状型が前者に対して生成物類縁体として作用するのであろうことを示唆している。更に、GMI−1−リン酸のホスホネート誘導体は、十中八九切断できない基質類縁体であるので、より強力な阻害剤であることが判った。これらのデータは、細菌のPI−PLC作用について提案された2ステップ・メカニズムに合致している。つまり、PIは、先ず切断されて環状イノシトール−1,2−リン酸(cIP)構造を産生し、次いで、これがイノシトール−1−リン酸に加水分解される。興味深いことに、cIP構造は、トリパノソーマのGPI−PLC触媒作用で、第2ステップ(脱環化)で
はなく第1ステップ(環化)の作用を示す、T.brucei由来のGPI−PLCへの暴露を受けて、トリパノソーマのVSGのいわゆる交差反応決定基として免疫学的に同定することができる。一過性又は安定したcIP形成に必要な条件は、ヒト赤血球AChEでのように、イノシトール残基の1位又は2位がパルミトイル化されたGPIアンカーはホスホリパーゼ切断に抵抗性である、という発見と一致している。
【0004】
GMI−1−ドデシルホスホネートは、やはり膜と会合しているトリパノソーマのGPI−PLCに対して相当するヘキシル誘導体よりもかなり阻害的であることが判った(“Morris, J. C. et al. (1995), J. Biol. Chem. 270: 2517-2524")。興味深いことに、イノシトールに炭水化物の置換基(即ち、グルコサミン)がないと、つまりイノシトール−1−リン酸の切断されない類縁体では、ミオ−イノシトール−1−O−ドデシルリン酸によって例示されるように、トリパノソーマのGPI−PLCを阻害することが見出された。この型の阻害剤の効率は、トリパノソーマのGPI−PLCを10〜90μMのIC50で競合的に阻害する、2−フルオロ置換体を有する2−デオキシ−イノシトール−1−O−ドデシルホスホネートの2位の置換体でかなり増加した(“Morris, J. C. et al. (1996), J. Biol. Chem. 271: 15468-15477")。これまでに報告されたGPI−PLCの最も強力な阻害剤は、2−デオキシ−イノシトールの2位にフルオロ基を、1位にドデシルホスホネートの両者を有しており、ミオ−イノシトール−1−O−ドデシル−ホスホン酸よりも少なくとも5倍より阻害的である(“Morris, J. C. et al. (1998), Biochem. Biophys. Res. Commun. 244: 873-867")。興味深いことは、B.cereus及びT.brucei由来の(G)PI−PLCのこれら化合物のいくつかによる差別的な阻害は、二つの酵素が(G)PI−PLCの機械的なサブクラスを表すと論じていることである。
【0005】
残念ながら、類似のデータが哺乳類のGPI−PLCについて未だに得られていない。驚くべきことに、新しく合成されたミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(GPI−2350)は、細菌及び脂肪細胞のGPI−PLCの強力な阻害剤であることが判った。
【0006】
アルカリ性ホスファターゼ(aP)が細菌のホスファチジルイノシトール特異的ホスホリパーゼC(PI−PLC)によって膜二重層から放出されるという最初の観察は、グリコシルホスファチジルイノシトール(GPI)脂質への共有結合を含むタンパク質の別の型の膜付着の同定へと導いた。
【0007】
GPIアンカーの最初の完全な構造は、トリパノソーマ・ブルセイ由来の変異体表面グリコプロテイン(VSG)について明らかにされた。
【0008】
コア四糖類は、3つのマンノース残基と非アセチル化グルコサミンからなり、後者の一末端はホスホエタノールアミン架橋を経てタンパク質部分にアミド結合しており、他の末端はホスファチジルイノシトール(PI)の6−ヒドロキシル基にグリコシド結合的に結合している。PIは、それぞれジアシルグリセロール及びホスファチジン酸を放出する特異性C及びDの(G)PI−特異的ホスホリパーゼ[G]PI−PLC/Dによって切断され、タンパク質部分の末端(ホスホノ)イノシトールグリカン(PIG)構造を残す。
【0009】
それ以来、種々の起源の細菌PI−PLCが、GPIアンカー型タンパク質(GPI−タンパク質)を検出するのに一般的に使用されている。
【0010】
GPIアンカー型VSGからなる防護的表面被膜のGPI−PLCによる脂肪分解放出は、T.bruceiにとっては、宿主の免疫系を逃れるために抗原変異を達成するために必要であろうと推測される。
【0011】
殆んどの哺乳類細胞及び組織は、大多数が原形質膜の外小葉に埋め込まれているGPIアンカーを有しているGPI−タンパク質を発現するので、内因性の(G)PI/PI−PLC/Dが、それら細胞の表面発現の特異的ダウン・レギュレーションを調節し、同時に循環における可溶性タンパク質部分の増加を調節することができる。
【0012】
GPI−タンパク質の可溶性型は、5−ヌクレオチダーゼ(5’−Nuc)、Thy−1、アルカリ性ホスファターゼ(aP)及びCD16受容体のように血流内を循環していることが検出された。GPI−PLCは、又、ヒト好中球、ウシ脳、ラット腸及びヒト上皮性悪性腫瘍細胞株において同定することができた。ラット肝からのGPI−PLCは、均質性を示すまで精製された。内因性GPI−PLCは、ブタの最大の小管から腎ジペプチダーゼを放出する能力を有していると記載されていた。しかしながら、GPI−PLCの構造又は遺伝子の解明は未だされていない。哺乳類GPI−PLCが膜依存性であるのに対して、哺乳類GPI−PLDは、異なった種(ヒト、ラット、ウシ)及び器官(胎盤、脳、肝、血清)の組織材料の全ての型から回収することができる。
【0013】
GPI−PLDの主な役割は、多分、リソソームへのエンドサイトーシス及びトラフィッキングの後のGPIタンパク質のGPIアンカーの分解である。哺乳類の組織は、血清中及び細胞表面において、異なった機能性を持って活性である二つのはっきりと識別できるGPI−PLDを持っている。
【0014】
酵母及び齧歯類脂肪細胞の5’−Nuc及びaPのようないくつかのGPIタンパク質は、インビトロ及びインビボでGPI−PLCによるGPIアンカーの脂肪分解により細胞表面から可溶性のものとして放出されているようにはみえない。
【0015】
付加的な温和な塩及び/又はトリプシン切断が、顆粒画分/細胞から分離の後に、可溶性画分/培地中のいくつかの脂肪分解的に切断されたGPIタンパク質のタンパク質部分の回収に必要とされるのは、受容体タンパク質の存在を示唆している。
【0016】
これらの場合において、GPI−PLCの活性はGPI−タンパク質の局在又はトポロジーに影響しないが、代わりに、両親媒性から親水性の形態への変換の途中で機能的(触媒/結合)特性を変化する。無処置のGPIアンカーの存在は、それに付いているタンパク質部分の立体構造及び挙動に影響している。
【0017】
5’−Nuc及びGce1の場合には、触媒及び結合効率は、膜に包埋された又は界面活性剤ミセル又はリポソームに再構築された完全な形のものと比較すると、脂肪分解的に切断されたGPIタンパク質において増加した。
【0018】
GPIタンパク質の脂肪分解プロセシングが細胞壁の生合成の間ある役割を演じているような、酵母におけるグルコースのような栄養シグナルによって、及び齧歯類の脂肪細胞、ミオサイト及びヒト内皮細胞におけるグルコースやある種のホルモン、増殖因子及び薬物(例えば、インスリン、グリメピリド)によって、真核生物において、多くのGPI−PLが、アップレギュレートされている。
【発明の概要】
【発明が解決しようとする課題】
【0019】
グリメピリド、抗糖尿病薬は、膵細胞からインスリン放出を刺激することによって、更に、少しの程度であるが、筋肉細胞におけるグルコース輸送の活性化及び脂肪細胞における脂肪分解の阻害のような、末梢組織での代謝的インスリン作用を模倣することによって、主に血糖を低下させる。
【0020】
スルホニルウレア、グリメピリド、の血糖低下効果は、部分的には、IRS−PI3K経路のインスリン受容体非依存性活性化を経た脂肪細胞及び筋肉細胞における非酸化的グルコース代謝の刺激によって引き起こされる。単離したラット脂肪細胞において、グリメピリドの作用の分子機構は、アシル化された非受容体チロシンキナーゼ、pp59Lyn、
及びいくつかのGPIタンパク質のような脂質ラフト関連シグナリング成分の再分配及びそれに伴っておこる活性化、並びに、インスリンによって中程度に活性化もされる原形質膜グリコシルホスファチジルイノシトール特異的GPI−PLCの促進に関与していることが明らかにされている。
【0021】
グリメピリドは、膵臓β細胞からのインスリン放出を促進するスルホニルウレア剤であり、膵臓外の機構を経て作用するであろう。グリメピリドは、血糖症が食事及び運動だけではコントロールされない2型(非インスリン依存性)糖尿病患者に1日1回投与され、続発性スルホニルウレア障害の患者にインスリンと併用されることもある。
【0022】
グリメピリドの最大の血糖低下効果は、投与後最初の4時間に起こる。グリメピリドは、グリベンクラミド(グリブリド)よりも心臓血管変動に対する重篤な影響はより少ない。薬物動態は高齢の患者又は腎疾患や肝臓疾患のそれら患者において主として不変である。グリメピリドの薬物相互作用は殆んど報告されていない。
【0023】
2型糖尿病患者において、グリメピリドの有効投与範囲は、0.5から8mg/日であり、4mg/日と8mg/日の投与間に有効性の差が殆んどない。グリメピリドは、1年間の試験において、グリベンクラミド及びグリピジドの有効性と類似していた。しかしながら、グリメピリドは最初の数週間にわたる治療でグリピジドよりもより速く血糖を低下するようである。グリメピリド及びグリクラジドが、14週間の試験で基準線に良好に血糖コントロールができている患者で比較され、効果に差がないことが指摘された。グリメピリドとインスリンの併用は、絶食時血糖標的レベルが≦7.8mmol/Lに達する続発性スルホニルウレア障害の患者の治療において、インスリンとプラセボの併用と同程度の効果があったが、グリメピリドを用いることにより血糖症に対してより低いインスリン投与量とより速い効果が見られた。
【0024】
グリメピリド単独療法は、一般的に良好な耐容性を有するが、治療期間1年以下(≦1年)の患者の10から20%、及び6ヶ月間同時にインスリンを投与された患者の50%以下(≦50%)において、低血糖症を発症した。集計した治験データは、グリメピリドは、グリベンクラミドよりも低血糖症の発生率が低く、特に治療第1ヵ月目において低いであろうことが示唆された。投与量は、通常1mg/日で開始され、1から2週間の間隔で血糖コントロールのために通常の投与範囲1から4mg/日に漸増する(英国では最大6mg/日又は米国では8mg/日)。
【0025】
グリメピリドは、主に膵臓β細胞からのインスリンの放出の促進によって、及び僅かな程度で、筋肉細胞におけるグルコース輸送の活性化及び脂肪細胞における脂肪分解の阻害のような、末梢組織における代謝性インスリン作用を模倣することによって、グルコースを低下させる。
【0026】
グリメピリドは、初代培養又は培養した齧歯類脂肪細胞を薬理学的濃度で処理すると、GPI−PLCの活性化によって、5’−Nuc、aP及びGce1のようなGPIタンパク質サブセットの両親媒性から親水性への変換を強力に誘導することが明らかにされた。
【0027】
本発明において初めて開示されたイノシトール誘導体、GPI−2350は、高力価(
IC50=0.2〜10μM)及び選択性で細菌、トリパノソーマ及び血清GPI−PLC及びGPI−PLDを阻害する。GPI−2350は、無処置のラット脂肪細胞のGPI−PLCを殆んど完全にダウンレギュレートする。GPI−PLCは、代謝性インスリンシグナリングでは役割を演じない一方で、GPI−PLCの活性化は、界面活性剤不溶性の糖脂質に富む脂質ラフト領域(DIG)からインスリン受容体基質1(IRS−1)へのインスリン受容体非依存性クロストークを経由するグリメピリドのインスリン模倣効果には必須である。
【0028】
脂肪細胞のような多くの最終分化細胞の原形質膜に多数発現しているDIGは、シグナル伝達並びにタンパク質及び脂質のトラフィッキング及び選別を包含する膜媒介生物学的過程のためのプラットフォームとして役目を果たしている、特殊な膜ミクロドメインである。
【0029】
それらは、外部原形質小葉中のコレステロール及びスフィンゴ(糖)脂質、及び内小葉の飽和アシル鎖を有するリン脂質及びコレステロールに濃縮されていて、二重層内に液状の秩序相を形成している。DIGは、冷状態で1%トライトンX−100に不溶性であり、ショ糖勾配遠心分離法で低浮遊密度であることを特徴とする。これらの判断基準に基づいて、ある種のGPIアンカーされ、アシル化した、膜貫通シグナリングタンパク質が、原形質膜のDIG対非DIG領域に豊富にあることが見出された。更に、高コレステロール含有のDIG(hcDIG)及び低コレステロール含有のDIG(lcDIG)は、それぞれ、それらの低浮遊密度及び高浮遊密度に基いて互いに区別することができる。
【0030】
hcDIGからlcDIGへの、GPIアンカーされ、そしてアシル化されたある種のシグナリングタンパク質の刺激依存性再分配は、GPI−2350によってブロックされた。
【0031】
GPI−2350は、脂質ラフト関連GPI−PLC(IC50=5〜10μM)による無処置のラット脂肪細胞由来の、Gce1及び5’−Nucのような、GPIタンパク質の基本的及びグリメピリド/インスリン誘発脂肪分解放出を減少させた。GPI−2350(50μM)によるGPI−PLCの阻害は、(i)pp59Lyn及びGce1のカベオリンからの解離、(ii)hcDIGからlcDIGへの再分配、(iii)pp59Lyn及びIRS−1のチロシンリン酸化、(iv)グルコース輸送の促進及び(v)グリメピリドに応答する脂肪分解の阻害、の殆んど完全な遮断を導く。
【0032】
GPI−PLCのインスリン活性化は、脂質ラフト分布に対する適度な効果を有し、及び、それ(例えば、IRS−1のチロシンリン酸化及び脂肪分解の阻害)が僅かに減少するだけであったので、あったとしても代謝性インスリンシグナリングにおける軽微な役割がGPI−2350のみの存在下で明らかにされた。
【0033】
グリメピリドに誘導されたGPI−PLCによって生成する脂肪分解的に切断されたGPIタンパク質は、それらの非切断両親媒性のもの及びpp59Lynのように、lcDIGに再分配するよりもむしろhcDIGと会合して残っている。
【0034】
ラット脂肪細胞において再分配され、活性化されたpp59LynによるIRSのチロシンリン酸化を経るインスリンシグナリングカスケードへのグリメピリドのクロストークは、hcDIGが関与するGPI−PLCの活性化を必要とする。
【課題を解決するための手段】
【0035】
本発明は、哺乳類のGPI−PLCの活性を調節する化合物の同定方法であって、
a)哺乳類細胞をグリメピリドと共にインキュベートし;
b)a)の細胞のhcDIGを調製し;
c)b)からのhcDIGを化合物と共にインキュベートし;そして
d)c)のhcDIGからのGPI−PLCの活性を定量する;
ことを含む方法に関する。
【0036】
方法のステップa)に記載の哺乳類細胞は、齧歯類又はイヌの細胞に関する。齧歯類は、例えば、マウス、ラット又はモルモットである。本方法は、ヒト、又は例えばチンパンジー、ゴリラ又はボノボのような類人猿の細胞にも関する。そのような細胞は、例えば、膵臓細胞、筋肉細胞、肝細胞、腎細胞、脳細胞、脂肪細胞である。哺乳類細胞は、細胞培養によっても提供することができる。細胞を取得し、処理し、収穫し、加工するために、通常の技術が使われる(例えば、Current Protocols in Cell Biology, John Wiley & Sons; 0-471-24108-3-Looseleaf; 0-471-24105-9-CD-ROM)。
【0037】
本発明の方法のステップc)に記載のhcDIGからのGPI−PLCの活性を定量するには、hcDIGからpp59Lynの解離を測定することによって、又はhcDIGからlcDIGへのpp59Lyn及び/又はGce1の再分配を測定することによって、又はpp59Lyn及び/又はIRS−1のリン酸化の変化を測定することによって、又はグルコース輸送の促進及び/又は脂質分解の阻害を測定することによって実行される。
【0038】
本発明は、更に、前に開示したように本発明の方法によって同定することができる化合物に関する。そのような化合物は、例えば、本発明に開示されているような式Iを有する化合物又はその誘導体である。
【0039】
本発明は、又、全ての立体異性体の形態における、および全ての比率のその混合物である式Iの化合物:
【化1】
(式中、R1、R2、R3及びR4は、互いに独立して、OH又はFのいずれかであり、そしてR5は(C1−C20)−アルキル又は(C1−C20)−アルケニルである)及び生理的に耐容されるその塩に関する。
【0040】
本発明は、又、R1、R2、R3及び/又はR4のいずれか2つが互いに独立してFである、前記に記載の式Iの化合物に関する。
【0041】
本発明は、又、R1、R2、R3及びR4が各々の場合においてOHである、前記に記載の式Iの化合物に関する。
【0042】
本発明は、又、R5がC12−アルキルである、前記に記載の式Iの化合物に関する。
【0043】
本発明のそのような化合物は、例えば、全てのその立体異性体の形態を含む、以下の式を有する。
【化2】
【0044】
又は、本発明のそのような化合物は、例えば、以下の式を有する。
【化3】
【0045】
そのような本発明の化合物は、例えば、その全ての立体異性体の形態を含む、ミオ−イノシトール−1,2−シクロドデシルホスホン酸であり得る。
【0046】
本発明の記載において、用語(C1−C20)−アルキルは、メチル、エチル、プロピル、ブチル、ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、デュオデシル、トレデシル、クアトルデシル、クインデシル、セスデシル、セプタデシル、オクタデシル、ノナデシル、エイコシルの全ての立体異性体の立体構造を有する、全ての直鎖状又は分枝鎖状の化合物に関するものとする。
【0047】
本発明の記載において、用語(C2−C20)−アルケニルは、エチレン、プロピレン、
ブチレン、ペンチレン、ヘキシレン、ヘプチレン、オクチレン、ノニレン、デシレン、ウンデシレン、デュオデシレン、トレデシレン、クアトルデシレン、クインデシレン、セスデシレン、セプタデシレン、オクタデシレン、ノナデシレン、エイコシレンの全ての立体異性体の立体構造を有する、全ての直鎖状又は分枝鎖状の化合物を包含するものとする。
【0048】
本発明は、更に、ミオ−イノシトール−1,2−シクロドデシルホスホン酸の製造方法であって、
a)ラセミ体の1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトールをリン酸化してテトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を得ること;及び
b)テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を接触水素化すること;
を含む方法に関する。
【0049】
該製造方法は、前に特定したようなR1、R2、R3、R4及び/又はR5を含む適切な出発物質を使用することによって、本発明の別のいずれかの化合物の製造に適用し得るものである。
【0050】
本発明は、又、式Iを有する少なくとも1つの化合物及び/又は生理的に耐容される塩及び/又はそのプロドラッグ及び医薬として許容される担体を含む医薬組成物に関する。
【0051】
本発明は、更に、糖尿病の治療のために使用される薬剤の副作用を変えるための、式Iの化合物及び/又は生理的に耐容されるその塩及び/又はそのプロドラッグの使用に関する。そのような薬剤は、例えば、グリメピリドである。副作用は、例えば、グリメピリドの誤った投与量に起因する低血糖である。
【0052】
本発明は、又、糖尿病の治療のために使用される薬剤(例えば、グリメピリド)の副作用の治療用の医薬組成物を製造するための、式Iを有する化合物の使用に関する。
本発明は、又、脂肪細胞の原形質膜を横切るシグナル伝達の間にDIG中のシグナル伝達タンパク質を同定するための、又は、GPI−PLC及び/又は依存性のシグナル伝達因子の哺乳類の変異体を同定及び特性付けるための、式Iの化合物の使用に関する。
【0053】
本発明は、又、グリメピリドの活性を調節する化合物の同定方法であって、
a)哺乳類細胞をグリメピリドと化合物との混合物と共にインキュベートし;
b)a)の細胞のhcDIGを調製し;そして
c)b)のhcDIGからのGPI−PLCの活性を定量する;
ことを含む方法に関する。
【0054】
本発明の記載における化合物は、化学合成によって製造されるものか、天然源から単離されるもののいずれかであって、50と50,000ダルトンの間の分子量を有する、いずれかの有機及び/又は炭水化物化合物を意味するものとする。
【0055】
グリメピリドの活性の調節とは、活性が、促進されるか、阻害されるか、活性をある一定のレベルに安定させるという意味で保持されるか、を意味するものとする。
【0056】
グリメピリドの活性を調節する化合物の同定方法のステップa)における哺乳類細胞は、例えばラット又はマウスのような齧歯類の細胞、イヌの細胞又はヒトの細胞を包含するものとする。そのような細胞は、例えば、膵臓細胞、筋肉細胞、肝細胞、腎細胞、脳細胞又は脂肪細胞であり得る。又、使用することができるのは、細胞培養(例えば、初代細胞培養)の細胞である。グリメピリドの活性を調節する化合物の同定方法のステップc)に記載のGPI−PLCの活性の定量は、hcDIGからのpp59Lynの解離を測定することによって、又はhcDIGからlcDIGのpp59Lyn及び/又はGce1再分配を測定することによって、又はpp59Lyn及び/又はIRS−1のリン酸化の変化を測定することによって、又はグルコース輸送の促進及び/又は脂質分解の阻害を測定することによって、達成することができる。
【0057】
グリメピリドの活性を調節する化合物の同定方法のステップc)に記載のhcDIGか
らのGPI−PLCの活性が弱められている場合は、同定された化合物がグリメピリドの活性を不活化しているか又は減少させている。GPI−PLCの活性が増強されている場合は、同定された化合物がグリメピリドの活性を促進しているか、又は支持している。
【0058】
医薬として許容される陰イオンを有する式Iの化合物の塩は、医薬として許容される塩の製造又は精製のための、及び/又は非治療、例えばインビトロでの応用において使用するための有用な中間体として本発明の範囲内に、同様に包含される。
【0059】
式Iの化合物の塩は、当業者によく知られている通常の方法を用いて製造することができる。塩は、例えば、式Iの化合物を、溶媒又は希釈剤中で、無機若しくは有機酸又は塩基と組み合わせることによって製造することができる。
【0060】
本明細書で使用される用語「生理的に活性な誘導体」は、例えば、ヒトのような哺乳類に投与されると、式Iの化合物又はその活性代謝物を(直接的に、又は間接的に)形成することができる、本発明の式Iの化合物の生理的に許容される誘導体のいずれか、例えばエステル、に関する。
【0061】
生理的に活性な誘導体は、又、本発明の化合物のプロドラッグを包含する。そのようなプロドラッグは、本発明の化合物にインビボで代謝され得る。これらのプロドラッグはそれ自体活性又は不活性であることができる。
【0062】
本発明の化合物は、種々の多形相、例えば、非晶性及び結晶性多形相として存在することができる。本発明の化合物の全ての多形相は、本発明の範囲内に包含され、本発明の更なる態様である。
【0063】
以下、「式Iの化合物(類)」についての全ての言及は、上に記載した式Iの化合物/化合物類、並びに、本明細書に記載したそれらの塩、溶媒和物及び生理的に活性な誘導体を意味する。
【0064】
所望の生物学的効果を達成するために必要な式Iの化合物の量は、多くの因子、例えば、選択された特異的化合物、目的とする使用、投与様式及び患者の臨床状態に依存している。一般的に、1日投与量は、体重1キログラム当たり1日につき0.3mgから100mg(典型的には3mgから50mg)の範囲内、例えば、3〜10mg/kg/日である。静脈内投与量は、例えば、0.3mgから1.0mg/kgの範囲であり、これは1分当たり体重1キログラムにつき10ngから100ngの注入として適切に投与することができる。これらの目的のための適切な注入溶液は、例えば、1ミリリットル当たり0.1ngから10mg、典型的には1ngから10mgを含有することができる。個々の投与量は、例えば、1mgから10gの活性化合物を含有することができる。それ故、注射用アンプルは、例えば、1mgから100mg、を含有することができ、そして、例えば、錠剤又はカプセルのような経口投与可能な個々の剤形は、例えば、1.0から1,000mg、典型的には10から600mgを含有することができる。医薬として許容される塩の場合、上記した重量の詳細は、塩から由来するジヒドロチアゾリウムイオンの重量と関連している。上に挙げた状態の予防又は治療のために、式Iの化合物は、化合物としてそれ自体使用することができるが、それらは耐容される賦形剤との医薬組成物の形態で存在するのが好ましい。賦形剤は、勿論、それが組成物の別の構成物と相性がいいこと、及び患者の健康に有害でないことという意味において、耐容性でなければならない。賦形剤は、固体又は液体又は両者であり得、好ましくは、例えば、活性化合物の0.05質量%から95質量%までを含有することができる錠剤として、個々の投与量として化合物と処方される。更に医薬として活性な物質も、式(I)の更なる化合物を包含して存在し得る。本発明による医薬組成物は、本質的に成分を薬理学的に許容される賦形剤及び/又は助剤
と混合することから成る、公知の薬学的方法の一つによって製造することができる。
【0065】
本発明による医薬組成物は、個々の場合における最も適切な投与方法が、治療される状態の性質及び重症度、並びに各々の場合に使用される式(I)による化合物の性質に依存するものの、口腔、直腸、局所、経口(例えば、舌下)及び非経口(例えば、皮下、筋肉内、皮内又は静脈内)投与に適したものである。糖衣をかけた剤形及び糖衣をかけた遅延放出剤形も、又、本発明の範囲内に包含される。耐酸性及び腸溶性剤形が好ましい。好適な腸溶性コーティングとしては、酢酸フタル酸セルロース、ポリ酢酸ビニルフタル酸塩、フタル酸ヒドロキシプロピルメチルセルロース及びメタクリル酸及びメタクリル酸メチルのアニオン性ポリマーが挙げられる。
【0066】
経口投与用に好適な医薬化合物は、例えば、各々の場合において式(I)の化合物の一定量を含有する、カプセル、カシェ剤、トローチ剤又は錠剤のような分離された単位で存在することができ、粉剤又は顆粒剤として、水性又は非水性の溶液又は懸濁液として、又は水中油又は油中水エマルジョンとして、分離された単位で存在することができる。既に挙げたように、これらの組成物は、活性化合物と(1つ又はそれ以上の付加的成分からなり得る)賦形剤が接触されるステップを包含するいかなる好適な薬学的方法によっても製造することができる。一般的に、組成物は、活性化合物を液体及び/又は微細に分割した固体の賦形剤と均一及び均質に混合し、その後、必要により生成物を成形することにより、製造される。かくして、例えば、錠剤は、化合物の粉末又は顆粒を加圧又は成型によって、もし適切ならば、1つ又はそれ以上の追加の成分と共に、製造することができる。圧縮した錠剤は、化合物を、例えば粉末又は顆粒のような自由に流れる形態で、適切な場合は結合剤、滑沢剤、不活性な希釈剤及び/又は1つの(多くの)界面活性/分散剤と混合して、好適な装置で錠剤化することによって製造することができる。成形された錠剤は、粉体化合物を、不活性液体希釈剤でぬらし、好適な機械で成型することによって製造することができる。
【0067】
経口(舌下)投与に好適な医薬組成物は、式Iの化合物と香味料、通常ショ糖及びアラビアゴム又はトラガカントゴムを含有するトローチ錠、及びゼラチン及びグリセリン又はショ糖及びアラビアゴムのような不活性基剤中に化合物を含んだ芳香錠を包含する。
【0068】
好適な非経口投与用医薬組成物は、好ましくは、意図した被投与者の血液と好ましくは等張である、式Iの化合物の滅菌水性製剤を包含する。これらの製剤は、投与は注射として皮下、筋肉内又は経皮で行うことができるが、好ましくは、静注で投与される。これらの製剤は、好ましくは、化合物を水と混合し、得られた溶液を滅菌し、血液と等張にすることによって製造することができる。本発明の注射できる組成物は、一般的に、活性化合物を0.1から5質量%含有している。
【0069】
直腸投与に好適な医薬組成物は、好ましくは、個々の投与量の座薬として存在する。それらは、式(I)の化合物を1又はそれ以上の通常の固体賦形剤、例えば、カカオバターと混合し、得られた混合物を成形することによって製造することができる。
【0070】
皮膚に局所適用する好適な医薬組成物は、好ましくは、軟膏、クリーム、ローション、ペースト、スプレー、エーロゾル又はオイルとして存在する。使用することのできる賦形剤は、ワセリン、ラノリン、ポリエチレングリコール、アルコール及びこれらの物質の2つ又はそれ以上の組合せである。活性化合物は、一般的に、組成物の0.1から15質量
%の濃度、例えば、0.5から2質量%の濃度で存在する。
【0071】
経皮投与も、又、可能である。経皮投与用に好適な医薬組成物は、患者の表皮と長期間密接に接触するのに適している個々のパッチとして存在することができる。そのようなパ
ッチは、場合によっては緩衝性水性溶液中の活性化合物を、接着剤中に溶解及び/又は分散させて、又はポリマー中に分散させて、好適に含有している。適切な活性化合物濃度は、約1%から35%、好ましくは約3%から15%である。特別な可能性として、活性化合物は、例えば、Pharmaceutical Research, 2(6): 318 (1986) に記載されているように、エレクトロトランスポート又はイオントフォレーゼによって放出することができる。
【図面の簡単な説明】
【0072】
【図1】(G)PI−PLC阻害剤の構造及び合成を示す。材料と方法を参照。
【図2】GPI−2350の力価を示す。B.cereus、ラット血清及びT.brucei由来の精製(G)PI−PLC/D、並びに単離したラット脂肪細胞由来の総原形質膜(PM)及びhc/lcDIGをGPI−2350の濃度を増加しながらインキュベートし(10分間、30℃)、次いでTX−114分配によってAChE及びaPの両親媒性から親水性への変換につきアッセイした(材料と方法を参照)。脂肪分解活性は、GPI−2350の非存在下における対照反応(100%と設定)の%を計算した。少なくとも4回の独立したインキュベーションを行い、AChE及びap活性を各3回測定して、平均+SDを得た。
【図3】GPI−2350の選択性を示す。記載したように異なる由来の種々の(部分的に)精製した又は組換えリパーゼ(HSL、ホルモン感受性リパーゼ;PL、膵リパーゼ;LPL、リポプロテインリパーゼ;PC−PLC、ホスファチジルコリン特異的ホスホリパーゼC;PI−PLC、ホスファチジルイノシトール特異的ホスホリパーゼC;PLD、ホスホリパーゼD;PLA2、ホスホリパーゼA2;GPI−PLC、グリコシルホスファチジルイノシトール特異的PLC/D及びAChE(アセチルコリンエステラーゼ))、及び単離したラット脂肪細胞由来の総原形質膜(PM)及びhc/lcDIGを標準的なアッセイプロトコールに従ってGPI−2350(50μM)の非存在下又は存在下でアッセイした(材料と方法を参照)。少なくとも3回独立してインキュベーションを行い、各2回測定して、平均+SDを得た。
【図4】インスリン/グリメピリド誘導GPI−PLCによるGPIタンパク質の切断に対するGPI−2350の効果を示す。パネルA及びB.hcDIGを、未処理ラット脂肪細胞から調製し、次いで、記載したようにして、GPI−2350の非存在下又は存在下でインキュベートし(5分間、37℃)(パネルA、50μM;パネルB、濃度を増加させる)、続いてインスリン(10μM)又はグリメピリド(20μM)の無し又は有りで処理した(120分間、30℃)。パネルC及びD。単離したラット脂肪細胞を、記載したようにしてインスリン(10μM)又はグリメピリド(20μM)の非存在下又は存在下でインキュベートした(15分間、37℃)。hcDIGを調製し、次いでGPI−2350とインキュベート(120分間、30℃)した(パネルC、50μM;パネルD、濃度を増加させる)。パネルE、F及びG。単離したラット脂肪細胞を、記載したようにして、GPI−2350の非存在下又は存在下(パネルE及びG、50μM;パネルF、濃度を増加させる)でインキュベートし(5分間、37℃)、次いでインスリン(10nM)又はグリメピリド(20μM)の無し又は有りで処理した(90分間、37℃)。hcDIGを調製した。パネルA〜D。hcDIGのGPI−PLC活性を、外因基質として5’−Nucを用い、その親水性型への変換として測定した。パネルE〜G。hcDIGのTX−114枯渇相で回収したタンパク質を、親水性5’−NucおよびaPにつき免疫ブロット処理し、又はフォトアフィニティ標識によって親水性Gce1を分析した。3回の独立した細胞インキュベーションを行い、それぞれ活性測定及びゲル・ランを2回(代表例1つを示す)行って、平均+SEを得た。
【図5】DIG内のシグナル伝達タンパク質のグリメピリド誘発再分配に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、記載したようにして、GPI−2350の非存在下又は存在下(最終濃度、50μM)で処理し(5分間、37℃)、次いでグリメピリド(50μM)の非存在下又は存在下でインキュベートした(120分間、37℃)。hcDIG及びlcDIGを調製し、それぞれ免疫ブロット法及びフォトアフィニティ標識によって、pp59Lyn、5’−Nuc、カベオリン1、インスリン受容体βサブユニット(IRβ)、グルコーストランスポーター4(Glut4)、及びGce1の存在についてアッセイした(ゲルの添付書類を参照)。hc/lcDIGと共に回収したpp59Lyn、Gce1及び5’−Nucの量は、GPI−2350の非存在下(100と設定)における対照反応(基本)と比較して示した。少なくとも3回の独立した細胞インキュベーションを行い、それぞれゲル・ラン2回(代表例1つを示す)を行って、平均+SDを得た。
【図6】カベオリン由来のシグナル伝達タンパク質のグリメピリド/インスリン誘発解離に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、GPI−2350(左パネル、50μM;右パネル、濃度増加)の無し又は有りで処理し(5分間、37℃)、次いでインスリン(10nM)又はグリメピリド(20μM)の非存在又は存在下でインキュベート(120分間、37℃)した。hcDIGを脂肪細胞から調製し、可溶化した。可溶化hcDIGから調製したカベオリン1免疫沈降物を、それぞれ免疫ブロット法及びフォトアフィニティ標識によってpp59Lyn及びGce1の存在につきアッセイした。pp59Lyn及びGce1の量は、相同免疫ブロット法(ゲルの添付書類を参照)によって免疫沈降カベオリン1の回収につき補正した後に、対照反応において(パネルA、基本を100に設定)、又はインスリン又はグリメピリド促進状態において(パネルB、100に設定)GPI−2350の非存在と比較して示した。少なくとも4回の独立した細胞インキュベーションを行い、それぞれにつきゲル・ランを2回(代表例1つを示す)行って、平均+SDを得た。
【図7】シグナル伝達タンパク質のグリメピリド/インスリン誘導活性化に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、GPI−2350(パネルA、50μM;パネルB、濃度を増加)の無し又は有りで処理(5分間、37℃)し、次いでインスリン(10nM)又はグリメピリド(20μM)の非存在下又は存在下でインキュベート(120分間、37℃)した。IRS−1及びpp59Lynは、脱脂した核の次にある下層(材料と方法を参照)から免疫沈降し、可溶化しhc/lcDIGとそれぞれ結合し、次いでホスホチロシンにつき免疫ブロット法にかけた。チロシンリン酸化IRS−1及びpp59Lynの量は、相同免疫ブロット法(ゲルの添付書類を参照)によって免疫沈降IRS−1及びpp59Lynの回収につき補正した後に、対照反応において(パネルA、基本を100に設定)、又はインスリン又はグリメピリド促進状態において(パネルB、それぞれの場合において100に設定)、GPI−2350の非存在のものと比較して示した。少なくとも4回の独立した細胞インキュベーションを行い、それぞれにつきゲル・ラン2回(代表例1つを示す)を行って、平均+SDを得た。
【図8】グリメピリド/インスリン誘導代謝活性に対するGPI−2350の効果を示す。単離したラット脂肪細胞を、記載されたように、GPI−2350(パネルA、50μM;パネルB、濃度を増加;パネルC、5μM、50μM)の無し又は有りで処理(5分間、37℃)し、次いでインスリン(Ins、10nM)又はグリメピリド(Gli、20μM又は濃度を増加)の非存在下又は存在下でインキュベート(15分間、37℃)した。次いで脂肪細胞を、グルコース輸送及びイソプロテレノール誘導脂肪分解についてアッセイした。活性は、対照反応において(パネルA、基本)、又はインスリン又はグリメピリド促進状態において(パネルB及びC、100に設定)、GPI−2350の非存在のものと比較して示した。少なくとも5回の独立した細胞インキュベーションを行い、それぞれについて活性測定を3回行って、平均+SDを得た。
【図9】単離したラット脂肪細胞を0.5mM・NBD−FAで標識し(60分間、37℃)、洗滌し、次いでグリメピリド(パネルA、B)又はインスリン(パネルC、D)の濃度を増加して作用させる前に、GPI−2350の非存在下(パネルB、D)又は存在下(50μM;パネルA、C)でインキュベート(5分間、37℃)した。記載したようにして、イソプロテレノール(1μM)の無し又は有りで、更にインキュベーション(120分間、37℃)した後、総細胞懸濁液を、クロロホルム/ヘプタン/メタノール/0.1N・HCl(3/3/2/1、容積比)で抽出した。有機層を、マーカーとしてNBD−FAを同時に使用し、薄層クロマトグラフィー(ジエチルエーテル/石油エーテル/酢酸、78/22/1、容積比)及び蛍光イメージングにより分析した。2回の独立した標識化とインキュベーションを行った典型的な実験を示すが、同じような結果が繰り返された。TAG:トリアシルグリセロール。
【図10】ラット脂肪細胞原形質膜の非DIG領域、hcDIG及びlcDIGの間のGPIタンパク質の再分配の機構についてのワーキングモデル、インスリン、グリメピリド及びGPI−PLC及び推定GPIタンパク質受容体、p115、を包含するコレステロール枯渇による制御、並びにカベオリン、pp125Fak及びpp59Lynを経由するIRS−1への代謝シグナリングの下流へのカップリングを示す。GPI−PLC及びp115の膜貫通領域(TM)を経由するhcDIGにおけるトポロジー、膜配向及び固定(anchorage)の型は、仮説に基づいたものである。しかし、DIGの細胞外小葉に面した活性部位及び結合部位のそれぞれは、大多数のGPIタンパク質の明らかにされた細胞表面位置を基にして強く示唆されているものである。カベオリンは、カルボキシル末端におけるフック様TMと3重のパルミトイル化の両者によってhcDIGの、アミノ末端おける2重アシル化によってpp59Lynの、原形質膜小葉中に埋め込まれている。
【実施例】
【0073】
材料及び方法:
ヒト組換えインスリン、PIG41(“Frick, W. et al., (1998), Biochemistry 37,
13421-13436" に開示されている)及びグリメピリド(商品名アマリール、Amaryl)は、Aventis Pharma Germany (Frankfurt, Germany)の薬化学・合成部から入手した。12−((7−ニトロベンズ−2−オキサ−1,3−ジアゾール−4−イル)アミノ)ドデカン酸(NBD−FA)は、Mueller, et al., (1997), Biochem. Biophys. Acta 1347: 23-39の記載に従って合成された。コラゲナーゼ(Worthington, CLS, type I, 250単位/mg)は、Biochrom (Berlin, Germany)により提供された。牛乳由来リポタンパク質リパーゼ(LPL、アフィニティ精製)、ホスホリパーゼA2(ミツバチ毒)、粗ブタ膵リパーゼ(PL)、組換えPC及びPI−PLC(Bacillus cereus)、粗PLD(キャベツ)及び脱脂BSA(画分V)は、Sigma/Aldrich (Deisenhofen, Germany)から購入し、粗PI−PLC(ラット肝)及びプロテイナーゼ阻害剤は、Roche Molecular Biochemicals (Mannheim, Germany)から購入した。組換えGPI−PLC(Trypanosma brucei)は、Oxford Glycosystems (Oxford, UK)から得た。PLA2(AACOCF3)及びPLC(U73122)の阻害剤は、Tocris (Avonmouth, UK)から入手した。Bisbodipy-C11-PCは、Molecular Probes (Eugene, OR)から購入した。脂質は、Avanti Polar Lipids (Birmingham, AL)から購入した。カベオリン1(クローンC060)の免疫沈降用抗体及び、カベオリン1(ウサギ)及びpp59Lyn(クローン32)の免疫ブロッティング用抗体はTransduction Laboratories (Lexington, KY)から入手した。ホスホチロシン(クローン4G10)の免疫ブロッティング用抗体は、Upstate Biotechnology (Lake Placid, NY)によって供された。(昆虫細胞で発現した総ヒト組換えタンパク質に対する)IRS−1の免疫ブロッティング用抗体(ウサギ、アフィニティ精製)、5’−Nuc(ラット)及びaP(ウシ)は、Biotrend (Cologne, Germany)によって製造された。ECL Renaissanceケミルミネッセンス検出キットは、NEN/DuPont (Bad Homburg, Germany)から得た。スプラーグドーリーラットは、Charles-River Laboratories (UK)から供給された。
【0074】
GPI−2350の合成(図1の図式も参照):
出発物質として、ラセミ体1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトール(以下、化合物1)は、Zhai, H.-X. et al., (1995), Tetrahedron Lett. 36: 7403-7406に記載の方法によって製造された。化合物1は、次いで、トリエチルアミン及びジメチルアミノピリジンの存在下、ドデシルホスホン酸ジクロライドでリン酸化し、テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(以下、化合物2)を得た。10%パラジウム炭で接触水素化することによって、化合物2を脱ベンジ
ル化し、ミオ−イノシトール−1,2−シクロ−ドデシルホスホン酸(以下、化合物3、又はGPI−2350)をジアステレオマーの混合物として得た。化合物2(図1)の合成のために、1g(1.8mmol)の化合物1を、メチレンクロリド60ml及びトリエチルアミン1mlに溶解し、ジメチルアミノピリジン200mg及びドデシルホスホン酸ジクロライド1g(10mmol)を加えた。この反応溶液を45分間室温で放置した。次いで、酢酸エチル50mlを加え、混合物をシリカゲルで濾過した。溶媒を濃縮後、残留物をフラッシュクロマトグラフィー(n−ヘプタン:酢酸エチル=1:1、v/v)で精製した。化合物2の収量(収率):580mg(43%)、白色非晶質固体。TLC:n−ヘプタン/酢酸エチル(1/1、v/v)、Rf=0.7。MS:(M+Li)+=761.4、計算値C60H59O7P、M=754.9。化合物3(図1)の合成のために、505mg(0.67mmol)の化合物2を酢酸エチル5mlとメタノール15mlの混合物に溶解した。パラジウム(10%)炭700mgを加えた後、反応混合物を水素添加(6時間、1気圧H2)した。パラジウムをシリカゲルで濾過し、メタノール100mlで洗浄した。溶媒を濃縮後、残留物をフラッシュクロマトグラフィー(メチレンクロリド/メタノール、5/1、v/v)によって精製した。化合物3の収量(収率):179mg(68%)、白色非晶質固体。TLC:メチレンクロリド/メタノール(5/1、v/v)、Rf=0.15。MS:(M+Li)+=401.2、計算値C18H35O7P、M=394.4。化合物3は、塩基性溶媒よりも酸性溶媒下の方が不安定である。メタノール中では何日も安定であり、乾燥非晶質固体は4℃で何年も安定である。
【0075】
ラット脂肪細胞の調製及びインキュベーション:
雄性ラット(120〜140g、自由給餌、文献53を参照)の精巣上体脂肪体から消化によって単離した脂肪細胞を、1%(w/v)BSA含有KRH(20mMHepes/KOH、pH7.4、1.2mM・KH2PO4、140mM・NaCl、4.7mM・KCl、2.5mM・CaCl2、1.2mM・MgSO4)で2回洗浄し、次いで、100μg/mlゲンタマイシン、100nM・1−メチル−2−フェニルエチルアデノシン、0.5U/mlアデノシンデアミナーゼ、1mMピルビン酸ナトリウム及び5mM・D−グルコースを補充した同じ培地中で、GPI−2350(DMSO中に10mM保存溶液として調製し、インビトロ及び脂肪細胞培養における最終DMSO濃度は、いずれの阻害剤濃度でも0.5%で一定に保たれた)、インスリン又はグリメピリド(“Mueller et al.,
(1994), J. Cell. Biol. 126:1267-1276" に記載されたようにして調製した)の存在下又は非存在下で、37℃にて振とう水浴中で、5%CO2/95%O2で一定でバブリングしながら、インキュベーション容積1ml当たりパックした細胞容積100μlの最終力価(少量の一定量(アリコート)を毛細管ヘマトクリットチューブに吸引し、脂肪細胞による懸濁液の分画占有を定量するためにマイクロヘマトクリット遠心機で60秒遠心して決定する;10%サイトクリットは約1.5×106細胞/mlに相当する)で、指定された期間インキュベートした。
【0076】
原形質膜及びlc/hcDIGの調製:
原形質膜及びDIGを、溶解バッファー(25mMモルポリノエタンスルフォン酸(MES)、pH6.0、140mM・NaCl、2mM・EDTA、0.5mM・EGTA、0.25Mショ糖、50mM・NaF、5mMピロリン酸ナトリウム、10mMグリセロール−3−リン酸、1mMオルトバナジウム酸ナトリウム及びプロテアーゼ阻害剤)中、モーター駆動テフロン・イン・ガラス・ホモジナイザー(10ストローク)を用いてホモジナイズし、前処理し洗浄した脂肪細胞のポストニュークレアー・インフラネイタント(postnuclear infranatant)から調製した。原形質膜を脱脂したポストニュークレアー・インフラネイタントの分画遠心法によって得、前に記載した(48、53)ようにしてショ糖及びパーコールクッションを通して2回逐次遠心によって精製し、最後に25mMトリス/HCl(pH7.4)、0.25Mショ糖、1mM・EDTAに2mgタンパク質/mlで懸濁した。hc/lcDIGを、“Mueller, G. et al., (2002), Mol.
Med. 8: 120-136" に報告されたようにして界面活性剤法及び不連続ショ糖勾配遠心分離によって得た。15〜22%(画分4〜5)及び28〜35%(画分8〜9)ショ糖インターフェースの光散乱乳白色バンドを、屈折率を用いた濃度測定で、それぞれ、hcDIG及びlcDIGとして集めた。集めた勾配画分を25mM・MES(pH6.5)、1%トライトンX−100、150mM・NaClで3倍希釈した後、遠心分離(50,000×g、30分、4℃)によりhcDIG及びlcDIGを収集し、次いで、“Mueller, G. et al., (2002), Mol. Med. 8: 120-136" 及び/又は“Mueller, G. et al., (2001), Mol. Cell. Biol. 21: 4553-4587" に記載されているように適切なマーカーの豊富な部分/乏しい部分により特徴づけ(免疫ブロット法、酵素アッセイ法)、或いはトライトンX−114分画法(下記を参照)にかけた。カベオリンの免疫沈降のために、DIGを10mMトリス/HCl(pH7.4)、150mM・NaCl、1%トライトンX−100、60mMβ−オクチルグルコシド、0.3%デオキシコール酸、5mM・EDTA、0.5mM・EGTA、1mMオルトバナジウム酸ナトリウム、50mM・NaF、1μMミクロシスチン及びプロテアーゼ阻害剤の溶液に可溶化し(1時間、4℃)、遠心分離した(50,000×g、30分間)。直接免疫ブロット法のために、DIGを2倍のLaemmli試料バッファーに可溶化し遠心分離した(10,000×5、5分間)。上清を使用した。
【0077】
GPI−PLC/Dアッセイ:
GPI−PLD(ラット血清)を、“Hari et al., (1997), Biochim. Biophys. Acta 1355: 293-302" に従って、200mMトリス/マレイン酸(pH7.0)及び1%NonidetP−40溶液100μl中のヒト胎盤aP溶液(10mM・Hepes/NaOH、pH7.0、150mM・NaCl中100単位/ml)10μlと10分間、37℃にてインキュベーションし、次いで0.4mlの氷冷停止バッファー(10mM・Hepes/NaOH、pH7.0、150mM・NaCl、0.1mM・MgCl2及び0.01mM酢酸亜鉛)を添加して反応を停止することによってアッセイした。GPI−PLC(T.brucei)を、“Mensa-Wilmot et al., (1995), Methods Enzymol. 250: 641-655" の記載のようにして、50mMトリス/HCl(pH8.0)、0.25%Nonidet/P−40及び5mM・EDTA溶液50μl中のひと胎盤aP溶液(上記を参照)5μlと、30分間、37℃にてインキュベーションし、次いで停止バッファー0.45mlを添加して反応を停止することによってアッセイした。PI−PLC(B.cereus)及び脂肪細胞GPI−PLC(5〜25μg原形質膜又はhc/lcDIG)を、20mM・Hepes/KOH(pH7.8)、144mM・NaCl、0.1%TX−100、0.2mM・MgCl2溶液100μl中のウシ赤血球アセチルコリンエステラーゼ(AChE)溶液10μl(10mMトリス/HCl、pH7.4、144mM・NaCl、0.1%TX−100中12単位/ml)と1時間、25℃にてインキュベーションし、次いで氷酢酸5μl及び次いで10mMトリス/HCl(pH7.4)、144mM・NaCl、0.4mlを加えて反応を停止することによってアッセイした。各反応混合物を、TX−114分配にかけた(以下を参照)。GPI−PLC/D活性は、TX−114消耗相中で測定された親水性aP又はAChEの活性と分配前に測定された総活性の比率から計算され、そして(G)PI−PLC/Dを欠いたブランクのインキュベーションで現れたTX−114消耗相(総活性の10〜20%の割合を占める)中の非酵素バックグランドに対して補正された。
【0078】
その他のリパーゼアッセイ:
ホルモン感受性リパーゼ(HSL)及びリポプロテインリパーゼ(LPL)は、“Vertesy et al., (2002), Journal Antibiotics 55:480-494" に記載されたようにして、放射能標識トリオレイン液滴エマルジョンを使用して測定された。膵リパーゼ(PL)は、5mMトリス/HCl(pH6.5)、6mMタウロデオキシコール酸ナトリウム、150mM・NaCl、1mM・CaCl2溶液2ml中の0.25mmolのトリブチリンを、
記録pHスタット(1,000rpm、25℃で撹拌)を用いてpHを6.5に調節しつつ、同じバッファー中のブタPL及びコリパーゼとインキュベーションすることによって定量した。PC−PLCは、10mMジパルミトイルレシチン0.2ml、10mM・SDC0.1ml及び0.03M・CaCl20.1mlをバクテリアPC−PLC(50mMトリス/HCl、pH7.5、0.1%BSA)と、10分間、37℃にてインキュベーションすることでアッセイした。反応は、50%TCAを0.1ml、次いでクロロホルム/メタノール(66/33/1、容量比)を2.5ml加えることによって停止した。遠心分離(1,500×g、15分)後、メタノール/水相(約1.33ml)の上部0.2ml部を除き、60%HCIO40.5mlを補充し、次に170℃で1時間加熱して、最終的に無機リン酸塩を分析した。哺乳類PI−PLCは、10mM・PI0.1ml、0.8%デオキシコール酸ナトリウム0.1ml、0.1%BSA、100mMホウ酸ナトリウム(pH7.5)0.2ml、及びラット肝PI−PLCを20分間、37℃でインキュベーションすることによって測定した。反応を停止し、PC−PLCで記載したようにして更に処理した。PLDを40mM・Hepes/KOH(pH6.0)、4mMCaCl2及びキャベツPLD溶液250μl中1.6mM(U−14C)ホスファチジルコリン(約2,000−4,000dpm/nmol)でアッセイした。30分間、37℃にてインキュベーションの後、反応を、キャリアーホスファチジン酸含有クロロホルム/メタノール(2/1、v/v)5mlを加えて停止した。水可溶性物質を除去した後、最後の洗浄低クロロホルム相中に含有される脂質を、加熱活性化したシリカゲルプレートに転送し、第1次元方向にクロロホルム/メタノール/アンモニア(65/35/4、体積比)を使用し、第2次元方向にクロロホルム/アセトン/メタノール/酢酸/水(50/20/10/10/5、体積比)で2次元的に分離した。放射性ホスファチジン酸を蛍光イメージングにより検出した。PLA2を、1−パルミトイル−2−パルミトイル−sn−グリセロール−3−ホスホコリン/ビスボジピ−C11−PC/ホスファチジル−グリセロール/コレステロール(10/0.05/2/3、体積比)からなる単層リポソーム基質で、“Kim, T.-S. et al., (1997), J. Biol. Chem. 272:2542-2550" に従って定量した。濃度−反応曲線を、Marquardt-Levenberg非線形最小二乗演算手法を使用してフィットさせた。対数・直線軸にプロットするとき、この式はシグモイド曲線となる(Sigma Plot Software,
Jandel Scientific)。
【0079】
カベオリンの免疫沈降:
可溶性DIG(上記を参照、5〜20μgタンパク質)は、プロテインA/G−セファロースでインキュベーションし、次いで遠心分離(10,000×g、5分間)することにより事前に確かめた。上清を、10mMトリス/HCl(pH7.4)、150mM・NaCl、1%TX−100の1ml中で、プロテインA/G−セファロースに前もって吸着させた抗カベオリン1抗体と4℃にて1時間インキュベートした。免疫複合体を、同じバッファーで2回、次いでTX−100不含のバッファーで2回洗浄し、最後にβ−メルカプトエタノールなしで行うSDS−PAGEにかけた。免疫沈降カベオリンの回収は、膜の細片化に次いで、同じブロットの抗カベオリン抗体による均一免疫ブロッティングを行うことによって正常化された。
【0080】
免疫ブロット法:
SDS−PAGEによって分離されたポリペプチドを、“Mueller, G. et al., (2001), Mol. Cell. Biol. 21: 4553-4567”に記載されたように半乾燥法を用いてポリビニリデンジフルオライド膜に移した。洗浄した膜を抗カベオリン1抗体(1:2,000)、抗pp59Lyn抗体(1:1,250)、抗aP抗体(1:500)、抗5’−Nuc抗体(1:750)および抗IRS−1抗体(1:2,000)を15℃にて4時間インキュベートした。洗浄した膜を、西洋ワサビペルオキシダーゼを結合した、ヤギ抗マウスIgG2次抗体(1:2,000)又はヤギ抗ウサギIgG2次抗体(1:4,000)とインキュベートした。標識したタンパク質を増強したケミルミネセンスによって可視化した。
【0081】
TX−114分配:
ペレット化したhc/lcDIG(10〜50μgタンパク質)又は(G)PI−PLC反応混合物(0.5ml)を、それぞれ氷冷TX−114(1%)、25mMトリス/HCl(pH7.4)、144mM・NaCl溶液1ml中に懸濁することにより、又は氷冷TX−114(2%)0.5mlと混合することにより、“Bordier, C., (1981), J.
Biol, Chem. 272: 2542-2550" に記載された、TX−114が豊富な相とTX−114が枯渇した相との間の分配を使用して、両親媒性タンパク質及び親水性タンパク質に分離した。氷上で1時間インキュベーションした後、混合物を氷上で0.25Mショ糖及び25mMトリス/HCl(pH7.4)溶液0.4mlのクッション上に重層した。相分離を37℃に暖め、次いで遠心分離(10,000×g、1分間)することによって誘導した。下のTX−114が豊富な相の再抽出後、集めた上のTX−114が枯渇した相の一定量を5’−Nuc、aP及びAChE活性について測定するか、又は、SDS−PAGE分析用に沈殿(15%ポリエチレングリコール4,000)させた。
【0082】
グルコース輸送アッセイ:
グルコース輸送を、50μl部分中、最終濃度50μM(0.33μCi/ml)の2
−デオキシ−D−(2,6−3H)グルコースとともに、洗浄した10,000〜20,0
00個の脂肪細胞を、20μMサイトカラシンBの存在下又は非存在下において20分間、37℃でインキュベーションすることにより、“Mueller, G. et al., (1994), J. Cell Biol. 126: 1267-1276" に記載されたようにして定量した。
【0083】
種々の手順:
電気せん孔法は、“Mueller, G. et al., (2000), Mol. Cell. Biol. 20: 4708-4723"
に記載されたようにして実施した。脂肪分解は、“Mueller, G. et al., (2003) ,Biochimie 85: 1245-1256" に記載されたようにして、前もって標識され、イソプロテレノール刺激をされた脂肪細胞からの、グリセロール又は蛍光NBD−FAの放出として測定した。Gce1は、“Mueller, G. et al., (1994), Biochemistry 33: 12149-12159" に記載されたようにして、可溶化原形質膜又はhc/lcDIG(10〜50μgタンパク質)を8−N3−[32P]cAMPで光標識化し、次いで蛍光イメージングすることによって検出した。5’−Nuc、aP及びAChE活性は、“Eliakim, R. et al., (1990), Biochim. Biophys. Acta 1355: 293-302"に従って測定した。タンパク質は、Pierce (Rockford, IL)のBCAタンパク質測定キット、及び較正基準としてBSAを使用して測定した。SDS−PAGEは、前もって成型されたゲル(Novex, San Diego, CA;10%Bis−Tris分離ゲル、モルホリノプロパンスルホン酸−SDSランニングバッファー)を使用して実施された。ルミイメージ(lumiimage)は、ルミイメージャー(LumiImager)ソフトウエア(Roche Diagnostics)を使用したルミイメージャーで評価した。リン光イメージ及び蛍光イメージは、Storm 860 PhosphorImager system (Molecular Dynamics, Gelsenkirchen, Germany)を使用して処理し定量化した。図は、Adobe Photoshopソフトウエア(Adobe Systems, Mountain View, CA)を使用して構築した。
【0084】
GPI−2350は、高力価及び選択性を有する種々の起源の(G)PI−PLC/Dを阻害する:
(精製した、又は、未精製の)B.cereusPI−PLC、T.bruceiGPI−PLC及びラット血清GPI−PLDに対するGPI−2350の効果を、適切な条件下(低又は高界面活性剤濃度)で、GPI−2350の濃度を増加しつつ、部分的に精製した可溶化GPIタンパク質と共にそれらをインキュベーションすることによって観察した。次いで、総GPIタンパク質に対する、消化混合物中に脂肪分解的に切断されたGPIアンカーを内蔵する親水性GPIタンパク質の比率を、TX−114分配で界面活性剤が枯渇している相、及び分配前の総消化混合物、それぞれにおける酵素活性の測定によ
って定量した(図2)。GPI−2350は、細菌、トリパノソーマ及び血清(G)PI−PLC/Dを、それぞれ10、2及び1μMのIC50で、明らかに阻害した。GPI−2350は、又、酵素源として可溶化原形質膜、lcDIG又はhcDIGを、及び基質としてAChEを使用すると、ラット脂肪細胞GPI−PLCを0.2〜0.5μMのIC50で遮断した。脂肪細胞GPI−PLCの切断特異性を明らかにするため、本発明者らは、ミオ−[14C]−イノシトールで代謝的に標識されたサッカロマイセスセレヴィシエスフェロブラストから調製したGPIタンパク質、Gce1を使用した。hcDIGとのインキュベーションで、親水性及び14C標識されたGce1が生成され、それはcIPに対して生じる抗CRD抗体と濃度及び時間依存的に免疫沈降した(データは示さない)。脂肪酸アシル鎖を欠きcIPを内蔵するGPIタンパク質の、hcDIG依存的な生成により、ラット脂肪細胞の原形質膜にGPI−PLCの発現が確認された。
【0085】
次に、GPI−2350の選択性を試験した。種々の起源からのいくつかの天然リパーゼ(HSL、PL、LPL)及び異なった特異性(A2、C、D)のPC/PI特異的ホ
スホリパーゼそしてウシAChE(これらは全ていわゆる触媒三点セットを経て作動することが知られている)は、細菌、トリパノソーマ、血清及びラット脂肪細胞GPI−PLC/Dを60から95%遮断する濃度(50μM)下では有意な影響は受けなかった(図3)。cIPの代わりにオープン(open)を内蔵するGPI−2350の二つの誘導体2−デオキシ−2−フルオロ−scyllo−イノシトール−1−O−ドデシル−ホスホン酸(GPI−1793)及びミオ−イノシトール−1−O−ドデシル−ホスホン酸メチルエステル(GPI−2349)は、試験した最高濃度で脂肪細胞GPI−PLCに対して、それぞれ温和から非常に温和な効果を有するだけであったが、しかし、細菌及びトリパノソーマ(G)PI−PLCを同程度の力価で阻害した(表1)。これらの知見は、安定した又は一過性の1,2−環状リン酸結合の生成を含む触媒機構における類似性を示しているが、しかしながら、細菌、トリパノソーマ及び脂肪細胞(G)PI−PLCの間の基質認識における幾分かの相異も示している。後者がcIP中間体を経由して作動、予備的な動態試験に基づいていると仮定すると、GPI−2350は拮抗阻害剤(Ki=3〜20μM)として作用することを仮定しなければならない。このことは、中性リパーゼ及び哺乳類PC/PI特異的ホスホリパーゼはGPI構造を認識しないので、その顕著な選択性を説明できるであろう。拮抗的作用機構に一致して、GPI−2350による細菌及びトリパノソーマ(G)PI−PLCの半値阻害は、過剰の新鮮なGPIタンパク質基質との反応混合物の10倍希釈において完全に逆転した。対照的に、再単離及び充分な洗浄に先立って50μMGPI−2350で処理した脂肪細胞hcDIGは、GPI−2350非存在下でインキュベーションすると、精製した可溶化AChEに対しては、低い脂肪分解的切断のみを呈した(データは示さない)。このことは、両親媒性GPI−2350のhcDIGとの相互作用により説明することができるであろう。あるいは、GPI−2350は、遷移状態類似体として作用し、脂肪細胞GPI−PLCの活性部位への(一過性の)共有結合を受けるのであろう。GPI−2350の環状リン酸結合が(G)PI−PLC/Dによって開環されるかどうかを知ることは、興味あることであろう。
【0086】
GPI−2350は、単離されたDIG及び無処置の脂肪細胞においてGPIタンパク質のインスリン誘導及びグリメピリド誘導切断をブロックする:
GPI−2350及びGPI−PLCの両者が別々の複合体(それぞれ、界面活性剤ミセル及びDIG)で存在するとき、インビトロにおける脂肪細胞原形質膜GPI−PLCによるGPIタンパク質の切断に対するGPI−2350の介入の観察に基づいて、次に、基質としての内因性又は外因性GPIタンパク質の無処置の脂肪細胞及び単離されたDIGにおける基本的及び促進されたGPI−PLC活性に対するGPI−2350の効果について試験した(図4)。単離されたラット脂肪細胞において、GPI−PLC活性(内因性5’−Nuc、Gce1及びaPの切断として、それらの両親媒性から親水性への変換の観察が伴う)は、グリメピリド及びインスリンの両者によって(それぞれ、5倍及
び3倍まで)促進された(パネルE、G)。促進は、これらの前処置した脂肪細胞から調製されたhcDIGにおいて部分的に保存されていた(パネルC)(それぞれ、3倍及び1.7倍)。GPI−PLCは、グリメピリドで処理されなかった脂肪細胞から調製されたhcDIGの直接インキュベーションにより際だって活性化されたが、インスリンではそうでなかった(パネルA)。GPI−2350は、基本よりも低く、そして単離されたhcDIG及び未処理の脂肪細胞の両者において同じようなレベルで、濃度依存的に基本のGPI−PLC活性を減少させ、同様にグリメピリド及びインスリン誘導GPI−PLC活性を減少させた(図4、全てのパネル)。阻害剤がインスリン又はグリメピリドとのインキュベーションより前に、及びインキュベーションの間に存在していたので、明らかにGPI−2350は、GPI−PLCの活性を妨害しない(パネルC、E)。未処理の脂肪細胞でアッセイする場合、GPI−PLCの阻害のIC50は(5〜10μM、パネルF)は、単離された未処理のhcDIGに比較してより高く(1μM、パネルB、D)、同様に可溶化hcDIG及び原形質膜(0.2〜0.5μM、図2)に比較して、より高かった。このことは、単離されたDIGと比較して未処理の脂肪細胞の原形質膜においてDIGの中に埋め込まれた場合、GPI−2350に対してGPI−PLCの触媒部位の接近し易さが損傷されたことを反映しているのであろう。しかし、GPI−PLCの主要な基質、GPI脂質及びタンパク質アンカーは原形質膜の外小葉に局在化しており、そしてGPI−2350はGPI−PLCの触媒部位の(拮抗)阻害剤として作用しているようであるので(上記を参照)、その両親媒性を基本として、むしろ高いと予測されている、GPI−2350の細胞透過性の必要性は、多分除外することができる。むしろ、DIGの液体定序ドメインよりもむしろ非DIG領域の無秩序ドメインへ選択的に分配される界面活性剤に類似している脂肪細胞原形質膜(総細胞表面の80〜90%を含む)の非DIG領域に、GPI−2350はそのドデシル鎖を経て自然に分配されることが考えられる。
【0087】
DIG及び原形質膜の単離に使用された実験的条件下で、単離された総原形質膜中のGPI−PLC活性の65〜85%(外因性AChEの切断として測定)が、脂肪細胞の処理に関係なくDIG(即ち、hcDIGプラスlcDIG)により回復した。同様の回復が、典型的なDIGマーカータンパク質、カベオリン1(70〜90%)(免疫ブロット法により定量した)、及びDIG常在性タンパク質、p115(55〜75%)(合成PIGの結合により定量した)について観察された。更に、区別を付けて処理した脂肪細胞からDIG(hcDIGプラスlcDIG)で回復したaP、5’−Nuc及びGce1の総量(両親媒性プラス親水性のもの)は、おおよそ一定(基準を100%と設定して90〜140%)であって、このことは種々の条件下でDIGの単離手順の再現性を明示している。単離されたhcDIGは、lcDIG及び非DIG領域よりも比較的低いタンパク質含量により特徴づけられているので、GPI−PLCは、残りの原形質膜に対してhcDIGが非常に豊富であり、GPIタンパク質基質の局在化と一致している。
【0088】
DIG内にGPIアンカーされた及びアシル化されたシグナル伝達タンパク質の再分配にはラット脂肪細胞におけるGPI−PLC作用を必要とする:
次に、GPIタンパク質の脂肪分解的切断の阻害が、脂肪細胞原形質膜のDIG内でGPIアンカーされた及びアシル化されたシグナル伝達タンパク質の局在及び刺激依存性再分配に影響するかどうかを研究した。このために、グリメピリドで処理する前にGPI−2350の非存在下又は存在下でインキュベートした脂肪細胞からhc/lcDIGを調製し、次いでいくつかのGPIアンカーされた又はアシル化されたシグナル伝達タンパク質の存在につき分析した(図5)。基本の脂肪細胞に比べてhcDIGの量の70から90%の損失に相当する、lcDIGの量の3から5倍の増加に見られるように、グリメピリドに反応したhcDIGからlcDIGへのpp59Lyn、Gce1及び5’−Nucのしっかりした再分配は、GPI−2350の最大有効濃度の存在下で殆んど完全に止められた。対照的に、lcDIGに対してhcDIGにおいて、hcDIG常駐タンパク質
であるカベオリン1及びIRβが比較的豊富であること、並びにhcDIG及びlcDIGにおいてグルコース輸送体イソ型4(Glut4)が同様に多量であることの両者は、グリメピリドに影響されず、GPI−2350の存在下で顕著には変化しなかった(図5)。このことはGPI−PLCの強力な阻害は、DIGの構造における一般的且つ非特異的変化をもたらすよりはむしろhcDIGからlcDIGへの、Gce1及びpp59Lynのような、ある種の脂質修飾シグナル伝達タンパク質の刺激依存性転移を、特異的に妨げることを明らかにしている。
【0089】
次に、GPI−PLCの作用とGPIタンパク質の再分配の間の推定因果関係、即ち脂肪分解的切断及び/又は非切断GPIタンパク質は、刺激された単離ラット脂肪細胞において、実際にlcDIGに転移するのか否か、の根底にあるメカニズムを研究した。このために、ラット脂肪細胞をGPI−2350と共に、又は、無しにインキュベートし、インスリン又はグリメピリドを作用させた。総原形質膜、hcDIG及びlcDIGから回収されたGPIタンパク質、即ち5’−Nuc、aP及びGce1の親水性型及び両親媒性型の量は、TX−114分配によって定量された(表2)。インスリン及びより強力なグリメピリドでの処理は、GPI−2350によって完全にブロックされた原形質膜の親水性GPIタンパク質の増加をもたらし、効率的なGPI−PLC阻害を示している。インスリン及びグリメピリドの両者に応じて、親水性GPIタンパク質の量の増加が、もっぱらhcDIGにおいてのみ検出され、同じ位置での両親媒性型の減少を伴っていた。これらの変化は、GPI−2350の存在下で更に低い基本値に逆転し、この両親媒性から親水性への変換という脂肪分解性を示していた。それ故、hcDIGでのインスリン/グリメピリド促進GPI−PLCによって生成する親水性GPIタンパク質は、GPIアンカーによらない分子機構を経てhcDIGに関連して残る(表2)。興味深いことに、hcDIGからlcDIGへの5’−Nuc、Gce1及びaPのグリメピリド誘導再分配(図5参照)は非切断型に限定され、両親媒性においては3から4倍上昇したレベルであるが、lcDIGで回復した親水性GPIタンパク質では変らない低レベルであることを反映していたが、それにもかかわらず、GPI−2350によって完全に除去されていた(表2)。対照的に、親水性5’−Nuc、Gce1及びaPのインスリン誘導生成は、lcDIGで両親媒性の対応物の非常にゆるやかな増加(有意差の限界)を伴ったのみであったが、それは再びGPI−2350の存在で排除された。試験した3つのGPIタンパク質全てが、ほんの少しの量的差異だけで、もっぱら、刺激依存性脂肪分解的切断と両親媒性型の再分配との間に相関性を示した(表2)。総合すると、hcDIGで脂肪分解的に切断されたGPIタンパク質の生成及び蓄積のためのGPI−PLC活性化が必要である(グリメピリド)。
【0090】
GPI−PLCの阻害は、GPIアンカー型及びアシル化シグナル伝達タンパク質のカベオリンからの解離を妨げる:
ラット脂肪細胞におけるhcDIGからlcDIGへのシグナル伝達タンパク質の再分配は、最近、カベオリン1からの解離と活性化を伴うことが示されている。次に、GPI−PLCを阻害することによるラット脂肪細胞における、カベオリンからの解離/活性化と、GPIタンパク質の刺激依存性脂質分解的切断との間の推定因果関係、及び、それに続いて、Gce1とpp59Lynのカベオリン1との相互関係、及び後者のキナーゼ活性の解析が研究された(図6)。単離及び可溶化されたhcDIGから調製されたカベオリン1免疫沈降物からのpp59Lyn及びGce1の両者のインスリン誘発最大損失(35〜45%)及びグリメピリド誘発最大損失(75〜85%)最大損失は、5〜10μMのIC50(図6、パネルB)を有するGPI−2350(50μM:図6、パネルA)によって完全に妨げられた。この力価は、未処理の脂肪細胞におけるGPI−2350によるGPI−PLC阻害に類似している(図3)。非受容体チロシンキナーゼのようなシグナル伝達タンパク質のカベオリン結合ドメイン(CBD)へのカベオリン1の足場(scaffolding)ドメイン(CSD)の結合、及び、逆に、CBDへの結合からのCSDの除去は
、全ての場合ではないが多くの場合、インビトロアッセイにおけるシグナル伝達タンパク質の不活化及び活性化のそれぞれの引き金を引くことが示された。このことは、原形質膜DIGで作動しているシグナル伝達における、CSD−CBD相互関係の調節機能を暗示している。
【0091】
ラット脂肪細胞におけるGPI−PLCの阻害は、グリメピリドの代謝活性をダウンレギュレートするがインスリンはされない:
次いで、インスリン及びグリメピリドに反応したIRS−1チロシンリン酸化におけるGPI−PLC阻害の差動効果が、インスリン標的細胞における代謝活性に反映するか否かを研究した。このために、単離したラット脂肪細胞を、どちらかの刺激で作用させる前にGPI−2350で前処理し、次いでグルコース輸送及びイソプロテレノール誘導脂肪分解についてアッセイした(図8)。グリメピリドによるグルコース輸送の促進及びイソプロテレノール誘導脂肪分解の阻害は、GPI−2350の存在下、濃度依存的様式(IC50=2〜5μM)で対照値(50μMで)よりも低く損なわれた。従って、グリメピリド濃度−反応曲線は右に移動し、阻害剤がない場合と比較してGPI−2350(5μM)によるGPI−PLCの最大半量阻害の途中で平坦になった。対照的に、グルコース輸送のインスリンによる促進及びイソプロテレノール誘導脂肪分解の阻害は、グリメピリドに比較して2から3倍、より顕著であるが、50μMで、GPI−2350のみによってそれぞれ最大25及び33%減少した(図8)。これらのGPI−PLC阻害の差動効果は、蛍光脂肪酸誘導体で前もって標識したラット脂肪細胞からのグリセロールに代わってNBD−FAの放出として測定した場合、グリメピリド及びインスリンによるイソプロテレノール誘導脂肪分解の阻害に対するGPI−2350の効果の分析によって確認された(図9)。グリメピリド(IC50=3μM)の濃度増加に反応してイソプロテレノール誘導脂肪細胞から放出された酸化分解産物を包含するNBD−FAの量の減少は、50μMグリメピリドの存在で完全に消失された(パネルA、B)。対照的に、NBD−FA(及び分解産物)放出のインスリン阻害は、インスリンに対する見かけのIC50の少しの増加(0.1対0.3μM)を伴うGPI−2350(50μM)のみによって、最大25〜30%損なわれた。GPI−PLC阻害に対するこれらGPI−2350媒介効果の特異性は、ラット肝PI−PLC及びハチ毒PLA2の阻害剤の損傷によって、基本的な及びグリメピリド調節グルコース輸送及び脂肪分解に顕著に影響を与えることで確認された(図8)。
【0092】
最後に、DIG内でGPIタンパク質の再分配に依存性であるか、無関係であるかのいずれかである、グリメピリド以外のインスリン模倣刺激のシグナル伝達におけるGPI−PLCの関与について調べた。ラット脂肪細胞において、m−β−CDを用いたコレステロール枯渇によるhcDIGの崩壊、トリプシン/塩/NEM処理によるPIG受容体p115の不活化又は合成リガンドPIG41でのp115の占拠は、lcDIGでの5’−Nucの顕著な増加及びグルコース輸送活性化を導いた(表3)。対照的に、オルトバナジウム酸ナトリウム及び合成CBDPはlcDIGへの5’−Nuc転位の同時誘導なしにグルコース輸送を促進した。このことは、よく知られている、チロシンホスファターゼ阻害剤、バナジウム酸塩によるインスリン受容体及びIRS−1脱リン酸化、及びCBDPによるpp59Lyn及びIRS−1チロシンリン酸化の直接促進といった作用機序に一致している。これらの刺激によるIRS−1チロシンリン酸化でもなくグルコース輸送でもない促進(表3)は、GPI−2350の存在下で顕著に影響され、それらは全てGPI−PLCの下流の点でグルコース輸送システムにクロストークしていることを暗示している。
【0093】
表の説明
表1:(G)PI−PLC阻害剤の特徴。
表2:hcDIG、lcDIG及び総原形質膜における脂肪分解的切断及び非切断GPI
タンパク質の局在化に対する、インスリン、グリメピリド及びGPI−2350の効果。表3:ラット脂肪細胞における種々のインスリン模倣刺激によって誘導される5’−Nuc転位及びグルコース輸送に対するGPI−2350の効果。
【0094】
表についての説明
表1:ラット脂肪細胞由来のhcDIGのGPI−PLC、T.brucei由来GPI−PLC及びB.cereus由来PI−PLCを、適切な条件下で指示した阻害剤の増加する濃度(0.05μM〜1mM)のない場合とある場合における、可溶化した及び
部分精製ウシ赤血球AChE又はヒト胎盤aPとインキュベートした(材料と方法を参照)。切断率は、親水性AChEの量及びTX−114分配の際TX−114枯渇相で回復したaP活性から計算した。少なくとも3回の独立したインキュベーションと分配/4回の酵素アッセイ、から得られた平均+SDを記した。n.a.:該当なし。
【0095】
表2:単離したラット脂肪細胞を、GPI−2350(50μM)なし又はありで処理し(5分間、37℃)、次いで指示したように、グリメピリド(20μM)又はインスリン(10nM)の非存在下又は存在下にインキュベートした(120分間、37℃)。総原形質膜(PM)、hcDIG及びlcDIGを調製し、次いでTX−114分配にかけた。TX−114豊富相及び枯渇相を5’−Nuc及びaP(活性測定)及びGce1(光親和性標識)についてアッセイした。両親媒性及び親水性GPIタンパク質の量は、GPI−2350の非存在下に基本の脂肪細胞から調製した、総原形質膜及びhcDIGの両親媒性型のそれぞれに対して計算した(各100arb.単位に設定した)。hcDIGプラスlcDIGからの親水性及び両親媒性GPIタンパク質の回収は、種々のインキュベーション条件下で同程度であった。少なくとも3回の独立した細胞インキュベーションを行い、それぞれ活性測定及びゲル・ランを2回行って、平均±SDを得た。
【0096】
表3:単離したラット脂肪細胞を、メチル−β−シクロデキストリン(m−β−CD、10mM、50分間)、CBDP(300μM、電気穿孔法、次いで、洗浄及びそれに続く30分間のインキュベーション)、PIG41(10μM、15分間)、オルトバナジウム酸ナトリウム(1mM、15分間)、トリプシン(10μg/ml、15分間、次いで0.5M・NaCl処理及びそれに続く洗浄)及びN−エチルマレイミド(1mM、5
分間、次いでDTTの添加)の存在下でインキュベートした。脂肪細胞懸濁液の半分にGPI−2350(最終濃度50μM)を加え、更にインキュベーション(60分間、37℃)の後、細胞の一部分を、単離したlcDIG中の5’−Nuc活性についてアッセイした。基本的なインキュベーション中の5’−Nucの増加を100arb.単位に設定した。細胞の別の部分は(15分間のインキュベーション後)、基本のインキュベーションについて100arb.単位に設定したグルコース輸送につきアッセイした。少なくとも3回独立した細胞インキュベーションを行い、それぞれ3回ずつ活性を測定して、平均+SDを得た。
【0097】
【表1】
【0098】
【表2】
【0099】
【表3】
【特許請求の範囲】
【請求項1】
式I:
【化1】
(式中、R1、R2、R3及びR4は、互いに独立して、H又はOH又はFのいずれかであり、そしてR5は、(C1−C20)−アルキル又は(C1−C20)−アルケニルである)の化
合物、全てのその立体異性体の形態、及び全ての比率のそれらの混合物並びに生理的に許容されるその塩。
【請求項2】
R1、R2、R3及びR4のいずれか2つが互いに独立してFである、請求項1に記載の式Iの化合物。
【請求項3】
R1、R2、R3及びR4がOHである、請求項1に記載の式Iの化合物。
【請求項4】
R5がC12−アルキルである、請求項1〜3のいずれか1項に記載の式Iの化合物。
【請求項5】
a)ラセミ体の1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトールをリン酸化してテトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を得;
b)テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を接触水素化してそれぞれの化合物を得る;
ことを含む、請求項1〜4のいずれか1項に記載の化合物の製造方法。
【請求項6】
請求項1〜4のいずれか1項に記載の少なくとも1つの式Iの化合物及び/又は生理的に許容される塩及び/又はそのプロドラッグ、及び医薬として許容される担体を含む医薬組成物。
【請求項7】
糖尿病の治療のために使用される薬剤の副作用を変えるか又はそれを治療するための医薬の製造における、請求項1〜4のいずれか1項に記載の式Iの化合物及び/又は生理的に許容されるその塩及び/又はそのプロドラッグの使用。
【請求項8】
薬剤がグリメピリドである、請求項7に記載の式Iの化合物の使用。
【請求項1】
式I:
【化1】
(式中、R1、R2、R3及びR4は、互いに独立して、H又はOH又はFのいずれかであり、そしてR5は、(C1−C20)−アルキル又は(C1−C20)−アルケニルである)の化
合物、全てのその立体異性体の形態、及び全ての比率のそれらの混合物並びに生理的に許容されるその塩。
【請求項2】
R1、R2、R3及びR4のいずれか2つが互いに独立してFである、請求項1に記載の式Iの化合物。
【請求項3】
R1、R2、R3及びR4がOHである、請求項1に記載の式Iの化合物。
【請求項4】
R5がC12−アルキルである、請求項1〜3のいずれか1項に記載の式Iの化合物。
【請求項5】
a)ラセミ体の1,4,5,6−テトラ−O−ベンジル−ミオ−イノシトールをリン酸化してテトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を得;
b)テトラ−O−ベンジル−ミオ−イノシトール−1,2−シクロドデシルホスホン酸を接触水素化してそれぞれの化合物を得る;
ことを含む、請求項1〜4のいずれか1項に記載の化合物の製造方法。
【請求項6】
請求項1〜4のいずれか1項に記載の少なくとも1つの式Iの化合物及び/又は生理的に許容される塩及び/又はそのプロドラッグ、及び医薬として許容される担体を含む医薬組成物。
【請求項7】
糖尿病の治療のために使用される薬剤の副作用を変えるか又はそれを治療するための医薬の製造における、請求項1〜4のいずれか1項に記載の式Iの化合物及び/又は生理的に許容されるその塩及び/又はそのプロドラッグの使用。
【請求項8】
薬剤がグリメピリドである、請求項7に記載の式Iの化合物の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2011−140492(P2011−140492A)
【公開日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願番号】特願2011−8777(P2011−8777)
【出願日】平成23年1月19日(2011.1.19)
【分割の表示】特願2006−553524(P2006−553524)の分割
【原出願日】平成17年2月16日(2005.2.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
【公開日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願日】平成23年1月19日(2011.1.19)
【分割の表示】特願2006−553524(P2006−553524)の分割
【原出願日】平成17年2月16日(2005.2.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
[ Back to top ]