
グルコースをα−1,4−グルカンに変換する方法
【課題】グルコースからα−グルカンを効率よく製造する方法を提供すること。
【解決手段】グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、反応開始時の該溶液のpHは、pH5.7〜pH12であり、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【解決手段】グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、反応開始時の該溶液のpHは、pH5.7〜pH12であり、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、グルコースからα−グルカンを製造する方法に関する。特定の実施形態では、本発明は、セロビオースからα−グルカンを製造する方法に関する。
【背景技術】
【0002】
人間は、デンプンなどのα−グルカンを消化してエネルギー源として利用している。そのため、α−グルカンは、人類にとってエネルギー源として非常に重要である。さらに、α−グルカンは、食品産業をはじめ、医薬、化粧品、化学工業、製紙、繊維などにおける原料としても幅広く利用されている。特に、アミロースは、種々の機能を有するので、幅広い分野での利用が期待されている。
【0003】
近年、人口増加により食糧危機が問題視されており、植物の生産するデンプンだけでは将来エネルギー源が不足すると予想されている。
【0004】
一方、人間は、セルロースなどのβ−グルカンを消化できないので、エネルギー源として利用することができず、β−グルカンは、食物繊維成分としてのみ利用されている。それゆえ、β−グルカンを食糧危機問題の解決には利用できない。しかし、β−グルカンの年間生産量は、デンプンの約2万倍と推定されており、枯渇の心配はない。そのため、β−グルカンを、人間がエネルギー源とし得る物質に変換する種々の試みが行われている。
【0005】
例えば、セルロースをグルコースまで分解し、エタノール醗酵に利用することが検討されている。グルコースは、セルロース以外にも、種々の糖類を変換することにより入手することができる。グルコースは安価で大量に入手可能な原料である。グルコースは人間によって代謝され得るが、甘すぎるため、エネルギー源として大量に摂取することができない。また、グルコースは極めて水溶性が高いため、水分を含む食品中では固形物としてのテクスチャー(食感)を持つことができない。
【0006】
他方、α−グルコースの直鎖状ポリマーであるα−グルカン(例えば、デンプン)は、通常人間によって摂食されており、大量摂取することが可能である。それゆえ、グルコースを原料として、α−グルカンのようなテクスチャーを持ちかつ甘くない素材を合成することができれば、非常に有用である。
【0007】
さらに、上記のように、セルロースはバイオマス資源として豊富に存在し、枯渇の心配がない。そのため、セルロース資源を原料としたα−グルコースポリマーの合成技術が開発されれば食糧危機問題の解決に大きな貢献ができる。しかし、これまでにそのような技術は開示されていない。
【0008】
本願発明で利用する種々の酵素反応の従来技術について以下に説明する。
【0009】
図1の(1)に示すように、グルコースおよびポリリン酸にポリホスフェートグルコキナーゼ(PGK)を作用させると、グルコース−6−リン酸(G6−P)が生成する。この反応は公知であり、例えば、非特許文献1および2に記載されている。
【0010】
図1の(2)に示すように、グルコースまたはグルコース−1,6−ビスホスフェートをコファクターとし、G6−Pにホスホグルコムターゼ(PGM)が作用することによりグルコース−1−リン酸(G1−P)が生成する。この反応は公知であるが、これは、植物体内におけるデンプン合成反応や動物の肝臓や筋肉細胞あるいは微生物におけるグリコーゲン合成反応の一部であり、進行するためにエネルギーを必要とし、in vitroにおけるG1−P合成方法としての先行技術はない。逆にG1−PにPGMを作用させることによりG6−Pを生成する技術は多く存在する。PGMによる酵素反応は平衡反応であるが、G6−Pが非常に安定であるために、99%以上がG6−Pとなる(非特許文献3を参照のこと)。すなわち、(2)の反応は、G6−P生成側に非常に大きく傾いている。このことを利用して、G1−P濃度をG6−P経由で測定する方法が確立されている(非特許文献4を参照のこと)。しかし、G6−PからG1−Pを生成することはin vitroでは行なわれていなかった。G6−Pから得られたG1−Pを基質としてアミロースを合成することはまったく考えられてこなかった。
【0011】
図1の(3)に示すように、G1−PとプライマーにGPを作用させることによりアミロースが生産される。この技術は公知であり、例えば、非特許文献5および特許文献1に記載されている。特許文献1に記載される反応の概略を以下に示す:
【0012】
【化1】

この反応において、他の酵素が存在しなければ、α−グルカンはアミロースとなり、ブランチングエンザイム(BE)、ディスプロポーショネーティングエンザイム(MalQ)などの他の酵素が存在すると、α−グルカンは、デンプン、グリコーゲンなどとなる。
【0013】
グルコースを出発原料とし、PGKとPGMとGPという3種類の酵素を同時に作用させる1つの反応系で直接アミロースを生産する方法は知られていない。
【0014】
セロビオースの利用に関しては、セロビオースを原料に、1つの反応系でα−グルカンを製造する技術が知られている(特許文献2)。特許文献2の方法では、セロビオースの一部をG1−Pとグルコースに変換し、得られたG1−Pにα−1,4−グルカンホスホリラーゼを作用させてα−グルカンに変換している。特許文献2の方法では、2つの平衡反応をカップリングさせるため、リン酸はリサイクルされて蓄積しないが、グルコースは反応に利用されないため、反応中に蓄積し続ける。それゆえ、特許文献2の方法では、平衡反応をα−グルカン合成側に向かわせる目的で、グルコースをフラクトースに変換するかもしくはグルコースを反応系から消去する方法をとっている。そのため、特許文献2の方法では、α−グルカンに変換されるグルコース源はG1−Pに由来するグルコース部分のみであり、セロビオースに対するα−グルカン収率は最大でも50%以下であった。
【特許文献1】国際特許公開第02/097107号パンフレット
【特許文献2】国際特許公開第2005/05681号パンフレット
【非特許文献1】Simon J. Pilkis, Irene T. Weber, Robert W. Harrison, Graeme I. Bell (1994) Glucokinase : Structural analysis of a protein involved in susceptibility to diabetes. J. boil. chem. Vol. 269. No. 35. pp. 21925−21928
【非特許文献2】Shotaro Tanaka, Sun−Og Lee, Kazuhiro Hamaoka, Junichi Kato, Noboru Takiguchi, Kazunori Nakamura, Hisao Ohtake, Akio Kuroda (2003) Strictly polyphosphate−dependent glucokinase in a polyphosphate−accumulating bacterium, Microlunatus phosphovorus. J. Bacteriol. Vol. 185. No. 18. pp. 5654−5656
【非特許文献3】William J. Ray, Jr., and Gertrude A. Roscelli (1964)The phosphoglucomutase pathway. J. boil. chem.Vol. 239. No. 11. pp. 3935−3941
【非特許文献4】G. Michael (1974) D−Glucose 1−phosphate in methods of enzymatic analysis 2nd English ed. (H. U. Bergmeyer, ed) Vol. 4, pp.185−191, Academic Press, New York
【非特許文献5】Micheal J. Gidley and Paul V. Bulpin (1989) Aggregation of amylase in Aqueous system: The effect of chain length on phase behavior and aggregation kinetics. Macromolecules, 22, pp. 341−346
【発明の開示】
【発明が解決しようとする課題】
【0015】
本発明は、上記問題点の解決を意図するものであり、グルコースからα−グルカンを製造する方法を提供することを目的とする。
【課題を解決するための手段】
【0016】
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、ポリホスフェートグルコキナーゼおよびポリリン酸を用いてグルコースをほぼ100%、G6−Pに変換し、生成したG6−PをG1−Pに変換することにより、これまでアミロース合成の阻害剤となっていた副産物グルコースを除去するのではなく、積極的に利用できることを見出した。本発明によれば、グルコースを基質としてアミロース合成に利用することができ、セロビオースを理論的に100%アミロースに変換できる。本発明者らは、この知見に基づいて本発明を完成させた。
【0017】
本発明により、グルコースおよびポリリン酸を基質とし、3種類の酵素(ポリホスフェートグルコキナーゼ、ホスホグルコムターゼおよびα−1,4−グルカンホスホリラーゼ)を1つの反応溶液中で作用させることを含む、アミロースを合成する方法が提供される。
【0018】
グルコースを原料としてアミロースを合成する技術はこれまでにない。従来の方法でグルコースからアミロースを合成するためには、(1)グルコースとポリリン酸にPGKを作用させてG6−Pを得て;(2)得られたG6−PにPGMを作用させてG1−Pを生成し、そして(3)得られたG1−PにGPを作用させてアミロースを合成する。この3段階の方法の理論上のアミロース合成収率は、主に(2)および(3)の平衡反応の収率に基づく。(2)の反応(PGMによるG6−P⇔G1−P平衡反応)は、G6−P側に大きく偏っており、その比率はG6−P : G1−P =94.2 : 5.8である。(3)の反応(GPによるアミロース合成反応)では、アミロース : G1−P=73:27である。また、(1)の反応は不可逆反応であり、ポリリン酸濃度を調節することにより、ほぼ100%のグルコースがG6−Pに変換され得る。以上のことから、計算すると(1)の反応と、(2)の反応と、(3)の反応とをそれぞれ別個の工程で行なった場合の理論上のアミロース収率は、グルコースに対して約4.2%と算出される。
【0019】
グルコースからアミロースができないかまたは収率が非常に低い原因は2点考えられる。1点目は、反応溶液中の遊離リン酸濃度とG1−P濃度との比率の影響である。2点目は、このグルコースからのアミロース合成が平衡収支に逆らった反応であることである。本発明ではグルコースからアミロースを合成するために、溶液中のグルコースと遊離リン酸との濃度比を調節することと、物質のポテンシャルエネルギーに逆らいアミロースを獲得するためにアミロースの老化(沈澱)を利用することで問題解決を図った。
【0020】
まず1点目について、アミロースが得られない原因は、(3)の反応(GPによるアミロース合成反応)がG1−Pと遊離のリン酸との濃度比に大きく影響されることによる。アミロース合成反応に対するG1−P/リン酸濃度比の影響を検討したところ、遊離リン酸の濃度がG1−P濃度の4倍以上のときアミロースが合成されないことがわかった。本発明の方法においては、一般に、最初の反応液にリン酸を意図的に添加しない。しかし、一般に、通常の方法で工業的に製造されるポリリン酸、あるいは一般に工業的に市販されているポリリン酸には、重合していないモノマーのリン酸、すなわち遊離のリン酸が不純物として少量含まれている。そのため、リン酸を意図的に添加しなくても、(1)の反応で用いるポリリン酸として、工業的に入手可能なポリリン酸を用いると、その中に少量の不純物として遊離のリン酸が含まれていることが多い。この、ポリリン酸溶液由来の遊離リン酸の濃度がグルコースからアミロースを合成するときの中間生成物として生じるG1−P濃度の4倍を超えるとアミロースが合成されない。そのため、ポリリン酸溶液由来の遊離リン酸が生成されるG1−P濃度の4倍濃度より小さくなるようにグルコース濃度およびポリリン酸濃度を調節することが好ましい。
【0021】
2点目の原因はG6−Pが非常に安定であることに起因する。(2)の平衡反応は上記したようにG6−P側に大きく偏り、平衡反応は「酵素ハンドブック」丸尾文治、田宮信雄監修 朝倉書店によるとG6−P生成側に大きくよることが記載されている。それに対して(3)の平衡反応ではアミロース合成側に偏っている。従って、(2)の反応と(3)の反応を1つの溶液中で同時に行うと、G1−PからG6−Pへの転移反応と、G1−Pからアミロースへの合成反応が競合すると考えられる。(2)の反応および(3)の反応は平衡反応であり、同時に反応させた場合、ひとつの平衡反応と見ることができる。このときの平衡収支は、G6−Pとアミロースのポテンシャルエネルギーに依存する。実験例2に示すように、G6−Pとアミロースではどちらが安定しているのかを検討するために、低濃度アミロースの溶解液にPGM、GPおよびリン酸を加えて反応させた。その結果、図6に示すように、アミロースの95%以上がG6−PとG1−Pに分解された。この結果より、溶液に溶けている状態のアミロースでは、PGMとGPとの平衡反応により、G6−Pへの変換方向に強くシフトされることが確認され、アミロースと比べてもG6−Pが安定であると考えられる。
【0022】
しかし、本願発明の方法においては、400mMのグルコースを原料とし、ポリリン酸および酵素の濃度、pH等を適切に調整することにより、7%程度のアミロース合成収率を得ることができた。この値は、PGM−GP平衡状態におけるアミロースの割合を超える。この原因はアミロースの老化現象に起因すると考える。老化しやすい条件として基質濃度を500mMに上げアミロース合成反応をおこなうとアミロース収率は19.6%にまで上昇した。さらにアミロース合成反応の経時的な測定によって酵素反応が平衡に達した後、老化によるものと考えられるアミロースの増加を測定できた。よって、グルコースからアミロースを合成するためには、合成したアミロースを老化させることが非常に有用であると考えられる。
【0023】
さらに、本願発明の方法を使用することにより、セロビオースから効率よくα−グルカンに変換することが可能となった。上記のように、従来の方法では、セロビオースの一部をG1−Pとグルコースに変換し、得られたG1−Pにα−1,4−グルカンホスホリラーゼを作用させることにより、α−グルカンを得ていた。従来の方法では、グルコースは蓄積し続けるので、平衡反応をα−グルカン合成側に向かわせる目的で、グルコースをフラクトースに変換するかもしくは反応系から消去する方法をとっている。そのため、α−グルカンに変換されるグルコース源はG1−Pに由来するグルコース部分のみであり、セロビオースに対するα−グルカン収率は最大でも50%以下であった。
【0024】
我々は、ポリホスフェートグルコキナーゼとポリリン酸を用いてグルコースを100%G6−Pに変換して、生成したG6−PをG1−Pに変換することで、これまでアミロース合成の阻害となっていた副産物グルコースを消去するのではなく、積極的に基質としてアミロース合成に利用する技術を検討し、理論的にセロビオースを100%アミロースに変換できる技術を完成させた。
【0025】
上記目的を達成するために、本発明は、例えば、以下の手段を提供する:
(項目1)
グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、
反応開始時の該溶液のpHは、pH5.7〜pH12であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【0026】
(項目2)
前記反応開始時の前記溶液中のグルコース濃度が200mM〜2000mMである、請求項1に記載の方法。
【0027】
(項目3)
前記反応開始時の前記溶液中のグルコース濃度が500mM〜2000mMである、請求項1に記載の方法。
【0028】
(項目4)
前記反応開始時の前記溶液のpHが、pH7.4〜pH11である、請求項1〜3のいずれか1項に記載の方法。
【0029】
(項目5)
前記α−グルカンがアミロースである、項目1〜4のいずれか1項に記載の方法。
【0030】
(項目6)
前記溶液が、ゲスト化合物を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目1〜5のいずれか1項に記載の方法。
【0031】
(項目7)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目6に記載の方法。
【0032】
(項目8)
グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース−6−リン酸濃度は、20mM〜2000mMであり、
反応開始時の該溶液のpHは、pH6.7〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)が0.4以下である、方法。
【0033】
(項目9)
前記反応開始時の前記溶液中のグルコース−6−リン酸濃度が100mM〜2000mMである、請求項8に記載の方法。
【0034】
(項目10)
前記反応開始時の前記溶液のpHが、pH7.3〜pH11である、請求項8または9に記載の方法。
【0035】
(項目11)
前記α−グルカンがアミロースである、項目8〜10のいずれか1項に記載の方法。
【0036】
(項目12)
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目8〜11のいずれか1項に記載の方法。
【0037】
(項目13)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目12に記載の方法。
【0038】
(項目14)
セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法であって、
反応開始時の該溶液中のセロビオース濃度は、100mM〜350mMであり、
反応開始時の該溶液のpHは、pH5〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)が2.0以下である、方法。
【0039】
(項目15)
前記反応開始時の前記溶液中のセロビオース濃度が120mM〜350mMである、請求項14に記載の方法。
【0040】
(項目16)
前記反応開始時の前記溶液のpHが、pH5.6〜pH11である、請求項14または15に記載の方法。
【0041】
(項目17)
前記α−グルカンが、アミロースである、項目14〜16のいずれか1項に記載の方法。
【0042】
(項目18)
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目14〜17のいずれか1項に記載の方法。
【0043】
(項目19)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目18に記載の方法。
【発明の効果】
【0044】
本願発明の方法により、大量摂取が不可能なグルコースを、大量摂取可能な食品へと変換できる。また、本発明は、食糧危機問題の解決にも大きく貢献する。
【0045】
特定の実施形態では、本願発明の方法により、非消化性のセルロースを、可食性の食品へと変換できる。また、本発明は、食糧危機問題およびゴミ問題の解決にも大きく貢献する。
【0046】
グルコースとアミロースとでエネルギー状態を比較すると、アミロースの方がエネルギー準位が高い。そのため、グルコースからアミロースを合成するのは、エネルギー論的に困難である。
【0047】
図1の(1)、(2)、(3)の反応を個別に反応させた場合、(1)の収率はほぼ100%であり、(2)の収率は約7%であり、(3)の収率は約70%である。そのため、グルコースからのアミロースの理論収率は、約5%である(100×0.058×(0.73)=4.2(%))である。それに対して、(1)〜(3)の反応を1つの溶液中で同時に反応させた場合、(a)溶液のpHを調整することおよび(b)溶液中の基質の濃度を高くすること、によって、高いアミロース収率を上げることができる。
【0048】
詳細には、反応溶液の初期pH(反応開始時の溶液のpH)を好ましくはpH7.4〜pH11に、さらに好ましくはpH7.4〜8.5にする。反応で使用する各酵素の代表的な至適pHは、PGKが約pH7.0であり、PGMが約pH7.5であり、そしてGPが約pH5.6である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHをpH7.5からpH11に調整することにより、約15%以上のアミロース収率を得られる。
【0049】
さらに、基質濃度を高くすることにより、アミロースが老化して沈澱し、反応系から外れやすくなる。アミロースが沈澱すると反応系内のアミロース濃度が低下し、その結果、GPによるアミロース合成が促進される。それに伴い、PGM平衡反応によってG1−Pが生成される。合成アミロースを効率よく老化させることにより、G1−Pが連続的に供給され、アミロース収率を約20%程度まで向上させることができる。これは、3つの反応を個別に反応させた場合の理論上のアミロース収率の約5倍であり、極めて顕著な効果である。
【0050】
さらに、ゲスト物質の存在下で合成反応を行なうことにより、アミロースの沈澱形成が促進される。特定のゲスト物質の存在下では、アミロース収率を約35%まで向上させることができた。
【0051】
特許文献2に記載される方法では、セロビオースにセロビオースホスホリラーゼ(CBP)を作用させると、目的の生成物G1−Pの他に同量の副生成物グルコース(Glc)が生成される。そのため、特許文献2に記載の方法では、アミロースの収率の理論的上限は、50%であった。実際にはCBPの平衡反応がセロビオース合成側に有利であるため、アミロースの収率は16%程度であり、ムタロターゼ、グルコースオキシダーゼおよびペルオキシダーゼをグルコースに作用させて副生成物のGlcを除去することでアミロースの最大収率は32.4%まであがった。
【0052】
それに対して、本願発明において、セロビオースからのアミロースの合成反応と(1)および(2)の反応を組み合わせた場合、グルコースをアミロース合成に利用することが可能となり、その結果、アミロース収率の理論的上限を100%にすることができる。本明細書中に記載した実施例においては、反応溶液の初期pHを調整することにより、実際に最大66.8%のアミロース収率を得ることができた。
【発明を実施するための最良の形態】
【0053】
以下、本発明を詳細に説明する。
【0054】
本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当該分野で通常用いられる意味で用いられることが理解されるべきである。
【0055】
本明細書中では「α−グルカン」とは、D−グルコースを構成単位とする糖であって、α−1,4−グルコシド結合によって連結された糖単位を少なくとも2糖単位以上有する糖をいう。α−グルカンは、直鎖状、分岐状または環状の分子であり得る。直鎖状α−グルカンとα−1,4−グルカンとは同義語である。直鎖状α−グルカンでは、α−1,4−グルコシド結合によってのみ糖単位の間が連結されている。α−1,6−グルコシド結合を1つ以上含むα−グルカンは、分岐状α−グルカンである。α−グルカンは、好ましくは、直鎖状の部分をある程度含む。分岐のない直鎖状α−グルカンがより好ましい。本発明で製造されるα−グルカンは、好ましくは、アミロース、環状構造を有するグルカンまたは分岐構造を有するグルカンであり、より好ましくはアミロースである。1分子のα−グルカンに含まれる糖単位の数を、このα−グルカンの重合度という。
【0056】
α−グルカンは、場合によっては、分岐の数(すなわち、α−1,6−グルコシド結合の数)が少ないことが好ましい。このような場合、分岐の数は、代表的には0〜10000個、好ましくは0〜1000個、より好ましくは0〜500個、さらに好ましくは0〜100個、さらに好ましくは0〜50個、さらに好ましくは0〜25個、さらに好ましくは0個である。
【0057】
本発明の方法によって製造される分岐状α−グルカンでは、α−1,6−グルコシド結合を1としたときのα−1,6−グルコシド結合の数に対するα−1,4−グルコシド結合の数の比は、好ましくは1〜10000であり、より好ましくは10〜5000であり、さらに好ましくは50〜1000であり、さらに好ましくは100〜500である。
【0058】
α−1,6−グルコシド結合は、α−グルカン中に無秩序に分布していてもよいし、均質に分布していてもよい。α−グルカン中に糖単位で5個以上の直鎖状部分ができる程度の分布であることが好ましい。
【0059】
α−グルカンは、D−グルコースのみから構成されていてもよいし、α−グルカンの性質を損なわない程度に修飾された誘導体であってもよい。修飾されていないことが好ましい。α−グルカンの性質を損なわない程度の修飾としては、エステル化、エーテル化、架橋などが挙げられるが、これらに限定されない。これらの修飾は、当該分野で公知の方法に従って行われ得る。
【0060】
α−グルカンは、代表的には約1×103以上、好ましくは約5×103以上、より好ましくは約1×104以上、さらに好ましくは約5×104以上、さらに好ましくは約1×105以上の分子量を有する。α−グルカンは、代表的には約1×106以下、好ましくは約5×105以下、さらに好ましくは約1×105以下の分子量を有する。
【0061】
当業者は、本発明の製造方法で用いられる基質(例えば、プライマー、グルコース、グルコース−6−リン酸、セロビオースなど)の量、酵素の量、反応時間などを適宜設定することによって所望の分子量のα−グルカンが得られることを容易に理解する。
【0062】
<α−グルカンの製造に用いる材料>
一つの実施形態では、本発明の製造方法では、例えば、グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を用いる。
【0063】
別の実施形態では、本発明の製造方法では、例えば、グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を用いる。
【0064】
さらに別の実施形態では、本発明の製造方法では、例えば、セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を用いる。
【0065】
これらの方法で用いる溶液の調製においては、例えば、これらの各物質と、緩衝剤および溶媒を主な材料として用いる。これらの材料は通常、反応開始時に全て添加されるが、反応の途中でこれらのうちの任意の材料を追加して添加してもよい。
【0066】
本発明の製造方法では、溶液中にゲスト化合物をさらに含み得る。
【0067】
本発明の製造方法では、必要に応じて、枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素を用いることができる。枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素は、目的とするα−グルカンの構造に応じて、本発明の製造方法の最初から溶液中に添加してもよく、途中から溶液中に添加してもよい。
【0068】
(1.グルコース)
グルコースは、C6H12O6で示される、分子量約180の単糖である。グルコースは、代表的なアルドヘキソースであり、天然に広く分布する単糖である。グルコースは、甘い果実および他の植物組織中に遊離型で多量に存在する。グルコースは、スクロース、マルトース、ラクトースなどのオリゴ糖;澱粉、セルロース、デキストラン、ラミナリン、グリコーゲンなどの多糖;種々の配糖体などの構成成分として多量に存在する。これらの物質を酸で加水分解するとD−グルコースを生じる。本発明で使用するグルコースは、純粋なものであってもよく、ある程度不純なものであってもよい。グルコースは、グルコースを構成成分とする種々の物質を分解することによって入手されたものであってもよい。例えば、セルロースは、グルコースがβ結合で連結したものであり、セルラーゼによって加水分解することにより、グルコースが生成される。例えば、デンプンをアミラーゼなどの酵素によって加水分解することにより、グルコースが生成される。さらに、フラクトースをグルコースイソメラーゼによって変換することにより、グルコースが生成される。本発明で使用されるグルコースは、特定の実施形態では、グルコースとして溶液に添加されることが好ましい。別の実施形態では、分解または変換されることによってグルコースを生じる物質を反応溶液中に添加することによって、グルコースが提供されてもよい。
【0069】
(2.ポリリン酸)
本明細書において使用される場合、用語「ポリリン酸(polyphosphate)」とは、ポリリン酸(polyphosphoric acid)およびその塩をいう。酸である狭義のポリリン酸は、オルトリン酸の脱水縮合によって生じる直鎖状高分子リン酸である。狭義のポリリン酸は、一般式Hn+2PnO3n+1(n=2以上の整数)によって示される。塩であるポリリン酸(すなわち、ポリリン酸塩)は、一般式MIn+2PnO3n+1(n=2以上の整数)によって示される。本明細書において、特に断りの無い限り、「ポリリン酸」は、広義のポリリン酸、すなわち狭義のポリリン酸およびその塩を示す。狭義のポリリン酸の例としては、二リン酸、三リン酸、四リン酸、五リン酸などが挙げられる。狭義のポリリン酸は、例えば、(1)リン酸を加熱する方法、または(2)リン酸に五酸化二リンを添加する方法によって製造され得る。一般に、それぞれのポリリン酸を純品として製造することは困難であるので、種々の重合度のポリリン酸の混合物が市販されている。ポリリン酸の水溶液は容易に加水分解して低分子量のポリリン酸を経て、最終的にはオルトリン酸になる。本発明で使用されるポリリン酸の重合度は、好ましくは2以上であり、より好ましくは5以上である。本発明で使用されるポリリン酸の重合度は、好ましくは20以下であり、より好ましくは12以下である。
【0070】
ポリリン酸もまた、陽イオンを含む水溶液中で使用される。この場合、酸であるポリリン酸と塩であるポリリン酸とポリリン酸のイオンとが水溶液中で共存するので、酸であるポリリン酸と塩であるポリリン酸とは区別をしにくい。そのため、塩であるポリリン酸は、ポリリン酸と同様に使用され得る。本発明において、ポリリン酸塩は、好ましくはポリリン酸の任意の塩であり、より好ましくはポリリン酸の金属塩である。ポリリン酸塩の好ましい具体例としては、ポリリン酸ナトリウム、ポリリン酸カリウム、ポリリン酸アンモニウム、ポリリン酸カルシウム、ポリリン酸第2鉄などが挙げられる。市販のポリリン酸ナトリウムは、一般に、トリポリリン酸ナトリウムを主成分とし、少量のピロリン酸塩、トリメタリン酸塩および非結晶性の高重合リン酸塩が混在する。市販のポリリン酸カリウムは、一般に、トリポリリン酸カリウム(K5P3O10)を主成分とする。
【0071】
ポリリン酸塩は、ポリリン酸ナトリウム単独で、ポリリン酸カリウム単独で、またはポリリン酸ナトリウムとポリリン酸カリウムとの混合物として用いられてもよい。
【0072】
(3.プライマー)
本明細書中で使用される場合、用語「プライマー」とは、α−1,4−グルコシド結合で糖単位が結合できる遊離部分を1個以上有する分子をいう。プライマーは、α−グルカンの合成においてグリコシド残基を付加するための出発物質として作用する。なお、本明細書中では、グリコシド残基とグルコース残基とは交換可能に使用され得る。プライマーは、G1−Pのグリコシド残基のアクセプターとして作用する分子ともいうことができる。プライマーは、α−1,4−グルコシド結合で糖単位が結合できる遊離部分を1個以上有すれば、他の部分は糖以外の部分によって形成されていてもよい。本発明の方法では、反応開始時に含まれるプライマーに対して1つのグリコシド残基がα−1,4結合で転移することによって、このプライマーよりも重合度が1大きいα−グルカンが形成される。形成されたこのα−グルカンは、同じ溶液中で再度アクセプターとして作用することができる。このようにして、本発明の方法では、プライマーに対してグリコシド残基がα−1,4−グルコシド結合で順次結合されて、任意の重合度のα−グルカンが合成される。プライマーとしては、α−1,4−グルカンホスホリラーゼによって糖単位が付加され得る任意の糖が挙げられる。
【0073】
プライマーは、本発明の反応の出発物質として作用し得ればよく、例えば、本発明の方法によって合成されたα−グルカンをプライマーとして用いて、本発明の方法によってα−1,4−グルコシド鎖を再度伸長することも可能である。
【0074】
ただし、後述するゲスト化合物を用いる場合には、そのゲスト化合物を包接しない化合物をプライマーとして用いることが好ましい。
【0075】
プライマーは、α−1,4−グルコシド結合のみを含むα−1,4−グルカンであっても、α−1,6−グルコシド結合を部分的に有してもよい。当業者は、所望のグルカンに応じて、適切なプライマーを容易に選択し得る。直鎖状のアミロースを合成する場合には、α−1,4−グルコシド結合のみを含むα−1,4−グルカンをプライマーとして用いれば、枝切り酵素などを用いずに直鎖状アミロースを合成できるので好ましい。
【0076】
プライマーの例としては、マルトオリゴ糖、アミロース、アミロペクチン、グリコーゲン、デキストリン、プルラン、カップリングシュガー、澱粉およびこれらの誘導体が挙げられる。
【0077】
マルトオリゴ糖は、本明細書中では、約2個〜約10個のグルコースが脱水縮合して生じた物質であって、α−1,4結合によって連結された物質をいう。マルトオリゴ糖は、好ましくは約3個〜約10個の糖単位、より好ましくは約4個〜約10個の糖単位、さらに好ましくは約5個〜約10個の糖単位を有する。マルトオリゴ糖の例としては、マルトース、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオース、マルトオクタオース、マルトノナオース、マルトデカオースなどのマルトオリゴ糖が挙げられる。1つの実施形態では、マルトオリゴ糖は、好ましくはマルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオースまたはマルトヘプタオースであり、より好ましくはマルトテトラオース、マルトペンタオース、マルトヘキサオースまたはマルトヘプタオースであり、さらに好ましくはマルトテトラオースである。マルトオリゴ糖は、単品であってもよいし、複数のマルトオリゴ糖の混合物であってもよい。コストが低いため、マルトオリゴ糖の混合物が好ましい。1つの実施態様では、マルトオリゴ糖の混合物は、重合度4以上のマルトオリゴ糖に加えて、マルトトリオース、マルトースおよびグルコースのうちの少なくとも1つを含有する。オリゴ糖は、直鎖状のオリゴ糖であってもよいし、分枝状のオリゴ糖であってもよい。オリゴ糖は、その分子内に、環状部分を有し得る。本発明では、直鎖状のオリゴ糖が好ましい。
【0078】
アミロースとは、α−1,4結合によって連結されたグルコース単位から構成される直鎖分子である。アミロースは、天然の澱粉中に含まれる。
【0079】
アミロペクチンとは、α−1,4結合によって連結されたグルコース単位に、α1,6結合でグルコース単位が連結された、分枝状分子である。アミロペクチンは天然の澱粉中に含まれる。アミロペクチンとしては、例えば、アミロペクチン100%からなるワキシーコーンスターチが用いられ得る。例えば、重合度が約1×105程度以上のアミロペクチンが原料として用いられ得る。
【0080】
グリコーゲンは、グルコースから構成されるグルカンの一種であり、高頻度の枝分かれを有するグルカンである。グリコーゲンは、動植物の貯蔵多糖としてほとんどあらゆる細胞に顆粒状態で広く分布している。グリコーゲンは、植物中では、例えば、トウモロコシの種子などに存在する。グリコーゲンは、代表的には、グルコースのα−1,4−結合の糖鎖に対して、グルコースおよそ3単位おきに1本程度の割合で、平均重合度12〜18のグルコースのα−1,4−結合の糖鎖がα−1,6−結合で結合している。また、α−1,6−結合で結合している分枝にも同様にグルコースのα−1,4−結合の糖鎖がα−1,6−結合で結合している。そのため、グリコーゲンは網状構造を形成する。
【0081】
グリコーゲンの分子量は代表的には約1×105〜約1×108であり、好ましくは約1×106〜約1×107である。
【0082】
プルランは、マルトトリオースが規則正しく、階段状にα−1,6−結合した、分子量約10万〜約30万(例えば、約20万)のグルカンである。プルランは、例えば、澱粉を原料として黒酵母Aureobasidium pullulansを培養することにより製造される。プルランは、例えば、林原商事から入手され得る。
【0083】
カップリングシュガーは、ショ糖、グルコシルスクロース、マルトシルスクロースを主成分とする混合物である。カップリングシュガーは、例えば、ショ糖と澱粉との混合溶液にBacillus megateriumなどが産生するサイクロデキストリングルカノトランスフェラーゼを作用させることにより製造される。カップリングシュガーは、例えば、林原商事から入手され得る。
【0084】
澱粉は、アミロースとアミロペクチンとの混合物である。澱粉としては、通常市販されている澱粉であればどのような澱粉でも用いられ得る。澱粉に含まれるアミロースとアミロペクチンとの比率は、澱粉を産生する植物の種類によって異なる。モチゴメ、モチトウモロコシなどの有する澱粉のほとんどはアミロペクチンである。他方、アミロースのみからなり、かつアミロペクチンを含まない澱粉は、通常の植物からは得られない。
【0085】
澱粉は、天然の澱粉、澱粉分解物および化工澱粉に区分される。
【0086】
天然の澱粉は、原料により、いも類澱粉および穀類澱粉に分けられる。いも類澱粉の例としては、馬鈴薯澱粉、タピオカ澱粉、甘藷澱粉、くず澱粉、およびわらび澱粉などが挙げられる。穀類澱粉の例としては、コーンスターチ、小麦澱粉、および米澱粉などが挙げられる。天然の澱粉の例は、澱粉を生産する植物の品種改良の結果、アミロースの含量を50%〜70%まで高めたハイアミロース澱粉(例えば、ハイアミロースコーンスターチ)である。天然の澱粉の別の例は、澱粉を生産する植物の品種改良の結果、アミロースを含まないワキシー澱粉である。
【0087】
可溶性澱粉は、天然の澱粉に種々の処理を施すことにより得られる、水溶性の澱粉をいう。
【0088】
化工澱粉は、天然の澱粉に加水分解、エステル化、またはα化などの処理を施して、より利用しやすい性質を持たせた澱粉である。糊化開始温度、糊の粘度、糊の透明度、老化安定性などを様々な組み合わせで有する幅広い種類の化工澱粉が入手可能である。化工澱粉の種類には種々ある。このような澱粉の例は、澱粉の糊化温度以下において澱粉粒子を酸に浸漬することにより、澱粉分子は切断するが、澱粉粒子は破壊していない澱粉である。
【0089】
澱粉分解物は、澱粉に酵素処理または加水分解などの処理を施して得られる、処理前よりも分子量が小さいオリゴ糖もしくは多糖である。澱粉分解物の例としては、澱粉枝切り酵素分解物、澱粉ホスホリラーゼ分解物および澱粉部分加水分解物が挙げられる。
【0090】
澱粉枝切り酵素分解物は、澱粉に枝切り酵素を作用させることによって得られる。枝切り酵素の作用時間を種々に変更することによって、任意の程度に分岐部分(すなわち、α−1,6−グルコシド結合)が切断された澱粉枝切り酵素分解物が得られる。枝切り酵素分解物の例としては、糖単位数4〜10000のうちα−1,6−グルコシド結合を1個〜20個有する分解物、糖単位数3〜500のα−1,6−グルコシド結合を全く有さない分解物、マルトオリゴ糖およびアミロースが挙げられる。澱粉枝切り酵素分解物の場合、分解された澱粉の種類によって得られる分解物の分子量の分布が異なり得る。澱粉枝切り酵素分解物は、種々の長さの糖鎖の混合物であり得る。
【0091】
澱粉ホスホリラーゼ分解物は、澱粉にα−1,4−グルカンホスホリラーゼ(ホスホリラーゼともいう)を作用させることによって得られる。α−1,4−グルカンホスホリラーゼは、澱粉の非還元性末端からグルコース残基を1糖単位ずつ他の基質へと転移させる。α−1,4−グルカンホスホリラーゼは、α−1,6−グルコシド結合を切断することができないので、α−1,4−グルカンホスホリラーゼを澱粉に充分に長時間作用させると、α−1,6−グルコシド結合の部分で切断が終わった分解物が得られる。本発明では、澱粉ホスホリラーゼ分解物の有する糖単位数は、好ましくは約10〜約100,000、より好ましくは約50〜約50,000、さらにより好ましくは約100〜約10,000である。澱粉ホスホリラーゼ分解物は、分解された澱粉の種類によって得られる分解産物の分子量の分布が異なり得る。澱粉ホスホリラーゼ分解物は、種々の長さの糖鎖の混合物であり得る。
【0092】
デキストリンおよび澱粉部分加水分解物は、澱粉を、酸、アルカリ、酵素などの作用によって部分的に分解して得られる分解物をいう。本発明では、デキストリンおよび澱粉部分加水分解物の有する糖単位数は、好ましくは約10〜約100,000、より好ましくは約50〜約50,000、さらにより好ましくは約100〜約10,000である。デキストリンおよび澱粉部分加水分解物の場合、分解された澱粉の種類によって得られる分解産物の分子量の分布が異なり得る。デキストリンおよび澱粉部分加水分解物は、種々の長さを持つ糖鎖の混合物であり得る。
【0093】
澱粉は、可溶性澱粉、ワキシー澱粉、ハイアミロース澱粉、澱粉枝切り酵素分解物、澱粉ホスホリラーゼ分解物、澱粉部分加水分解物、化工澱粉、およびこれらの誘導体からなる群から選択されることが好ましい。
【0094】
本発明の方法では、上記各種糖の誘導体は、プライマーとして用いられ得る。例えば、上記糖のアルコール性水酸基の少なくとも1つが、ヒドロキシアルキル化、アルキル化、アセチル化、カルボキシメチル化、硫酸化、あるいはリン酸化された誘導体などが用いられ得る。さらに、これらの2種以上の誘導体の混合物が原料として用いられ得る。
【0095】
(4.ポリホスフェートグルコキナーゼ)
ポリホスフェートグルコキナーゼ(EC:2.7.1.63)とは、重合度nのポリリン酸(polyphosphate)のリン酸基をD−グルコースに転移して重合度n−1のポリリン酸とグルコース−6−リン酸を生成する反応を触媒する酵素をいう。この反応は以下のように記載される:
(リン酸)n+D−グルコース→(リン酸)n−1+D−グルコース−6−リン酸
ポリホスフェートグルコキナーゼは、ポリホスフェート−グルコースホスホトランスフェラーゼ、ポリホスフェート:D−グルコース6−ホスホトランスフェラーゼまたはPGKとも呼ばれる。
【0096】
ポリホスフェートグルコキナーゼは、多くの微生物に存在する。ポリホスフェートグルコキナーゼを産生する微生物の例としては、Microlunatus phosphovolas、Mycobacterium属細菌(例えば、M.phlei、M.tuberculosis、M.bovis)、Corynebacterium属細菌(例えばC.diphtheriae、C.xerosis、C.efficiens)、Propionibacterium shermanii、Streptomyces coelicolorなどが挙げられる。本発明においては、任意の生物由来のポリホスフェートグルコキナーゼを使用し得る。本発明においては、Microlunatus phosphovolas由来のポリホスフェートグルコキナーゼを用いることが好ましい。
【0097】
また、至適pHの観点からは、約5.5以上の至適pHを有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適pHは、より好ましくは約6.0以上であり、さらに好ましくは約6.5以上である。また、約10以下の至適pHを有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適pHは、より好ましくは約8.0以下であり、さらに好ましくは約7.5以下である。
【0098】
また、ポリホスフェートグルコキナーゼの反応至適温度は、可能な限り高いことが好ましい。至適温度の観点からは、約35℃以上の至適温度を有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適温度は、より好ましくは約40℃以上であり、さらに好ましくは約45℃以上である。当該分野においては、至適温度の高い種々のポリホスフェートグルコキナーゼが公知である。高温条件で生育する細菌の有する酵素は、一般的に反応至適温度が高温である。例えば、Thermobifida fuscaの生育温度は、約50℃であるので、Thermobifida fuscaの有する酵素は一般的に、反応至適温度が約50℃であるか、または少なくとも約50℃での反応性が非常に高い。Thermobifida fuscaの好適な生育条件は、約50℃にてpH7.4であり、このポリホスフェートグルコキナーゼを産生するThermobifida fuscaは、ATCCから購入することができる。Thermobifida fusca由来のポリホスフェートグルコキナーゼのヌクレオチド配列を配列番号9に示し、Thermobifida fusca由来のポリホスフェートグルコキナーゼのアミノ酸配列を配列番号10に示す。ポリホスフェートグルコキナーゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0099】
反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。ポリホスフェートグルコキナーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0100】
(5.ホスホグルコムターゼ)
ホスホグルコムターゼ(EC:2.7.5.1)とは、α−D−グルコース1,6−二リン酸とα−D−グルコース1−リン酸から、α−D−グルコース6−リン酸とα−D−グルコースを生成する反応を触媒する酵素をいう。この反応は以下のように記載される:
【0101】
【化2】
ホスホグルコムターゼは、α−ホスフェートグルコムターゼ、α−D−グルコース−1,6−ビスホスフェート:α−D−グルコース−1−ホスフェートホスホトランスフェラーゼまたはPGMとも呼ばれる。
【0102】
ホスホグルコムターゼは、糖代謝の主経路に位置する酵素のためほとんどすべての生物に存在する。ホスホグルコムターゼは、微生物、動物および植物に存在する。ホスホグルコムターゼを産生する生物の例としては、例えば、ウサギ、ヒラメ、サメ、ウシ、E.coli、酵母、馬鈴薯(Potato)が挙げられる。本発明においては、任意の生物由来のホスホグルコムターゼを使用し得る。本発明においては、E.coli由来のホスホグルコムターゼを使用することが好ましい。
【0103】
また、至適pHの観点からは、約6.0以上の至適pHを有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適pHは、より好ましくは約6.5以上であり、さらに好ましくは約7.0以上である。また、約10以下の至適pHを有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適pHは、より好ましくは約8.5以下であり、さらに好ましくは約8.0以下である。
【0104】
また、至適温度の観点からは、約35℃以上の至適温度を有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適温度は、より好ましくは約40℃以上であり、さらに好ましくは約45℃以上である。当該分野においては、至適温度の高い種々のホスホグルコムターゼが公知である。高温条件で生育する細菌の有する酵素は、一般的に、反応至適温度が高温である。例えば、Thermus thermophilusの生育温度は、約75℃であるので、Thermus thermophilusの有する酵素は、一般的に、反応至適温度が約75℃であるか、または少なくとも約75℃での反応性が非常に高い。Thermus thermophilusの好適な生育条件は、約75℃にてpH7.5であり、このホスホグルコムターゼを産生するThermus thermophilusは、ATCCから購入することができる。Thermus thermophilus由来のホスホグルコムターゼのヌクレオチド配列を配列番号11に示し、Thermus thermophilus由来のホスホグルコムターゼのアミノ酸配列を配列番号12に示す。Thermotoga lettingae生育温度は、約65℃であるので、Thermotoga lettingaeの有する酵素は、一般的に、反応至適温度が約65℃であるか、または少なくとも約65℃での反応性が非常に高い。Thermotoga lettingaeの好適な生育条件は、約65℃にてpH7.5であり、このホスホグルコムターゼを産生するThermotoga lettingaeは、ATCCから購入することができる。Thermotoga lettingae由来のホスホグルコムターゼのヌクレオチド配列を配列番号13に示し、Thermotoga lettingae由来のホスホグルコムターゼのアミノ酸配列を配列番号14に示す。ホスホグルコムターゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0105】
反応開始時の溶液中に含まれるホスホグルコムターゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるホスホグルコムターゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるホスホグルコムターゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。ホスホグルコムターゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0106】
(6.α−1,4−グルカンホスホリラーゼ)
α−1,4−グルカンホスホリラーゼ(EC:2.4.1.1)とは、α−1,4−グルカン(重合度n)の加リン酸分解による、α−1,4−グルカン(重合度n−1)とα−D−グルコース−1−リン酸との産生を触媒する酵素の総称であり、ホスホリラーゼ、スターチホスホリラーゼ、グリコーゲンホスホリラーゼ、マルトデキストリンホスホリラーゼなどと呼ばれる場合もある。α−1,4−グルカンホスホリラーゼは、加リン酸分解の逆反応である、α−1,4−グルカン(重合度n−1)およびα−D−グルコース−1−リン酸からα−1,4−グルカン(重合度n)を合成する反応をも触媒し得る。反応がどちらの方向に進むかは、基質の量に依存する。生体内では、無機リン酸の量が多いので、α−1,4−グルカンホスホリラーゼは加リン酸分解の方向に反応が進む。本発明の方法では、無機リン酸は、β−1,4−グルカンの加リン酸分解に使われ、反応溶液中に含まれる無機リン酸の量が少ないので、α−グルカンの合成の方向に反応が進む。
【0107】
α−1,4−グルカンホスホリラーゼは、デンプンまたはグリコーゲンを貯蔵し得る種々の植物、動物および微生物中に普遍的に存在すると考えられる。
【0108】
α−1,4−グルカンホスホリラーゼを産生する植物の例としては、藻類、ジャガイモ(馬鈴薯ともいう)、サツマイモ(甘藷ともいう)、ヤマイモ、サトイモ、キャッサバなどの芋類、キャベツ、ホウレンソウなどの野菜類、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、アワなどの穀類、えんどう豆、大豆、小豆、うずら豆などの豆類などが挙げられる。
【0109】
α−1,4−グルカンホスホリラーゼを産生する動物の例としては、ヒト、ウサギ、ラット、ブタなどの哺乳類などが挙げられる。
【0110】
α−1,4−グルカンホスホリラーゼを産生する微生物の例としては、Thermus
aquaticus、Bacillus stearothermophilus、Deinococcus radiodurans、Thermococcus litoralis、Streptomyces coelicolor、Pyrococcus
horikoshi、Mycobacterium tuberculosis、Thermotoga maritima、Aquifex aeolicus、Methanococcus Jannaschii、Pseudomonas aeruginosa、Chlamydia pneumoniae、Chlorella vulgaris、Agrobacterium tumefaciens、Clostridium
pasteurianum、Klebsiella pneumoniae、Synecococcus sp.、Synechocystis sp.、E.coli、Neurospora crassa、Saccharomyces cerevisiae、Chlamydomonas sp.などが挙げられる。α−1,4−グルカンホスホリラーゼを産生する生物はこれらに限定されない。
【0111】
本発明で用いられるα−1,4−グルカンホスホリラーゼは、ジャガイモ、Thermus aquaticus、Bacillus stearothermophilusに由来することが好ましく、ジャガイモに由来することがより好ましい。特定の実施形態では、本発明で用いられるα−1,4−グルカンホスホリラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高いα−1,4−グルカンホスホリラーゼは、例えば、高度好熱細菌に由来し得る。
【0112】
また、至適pHの観点からは、約4.0以上の至適pHを有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適pHは、より好ましくは約4.5以上であり、さらに好ましくは約5.0以上である。また、約7.0以下の至適pHを有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適pHは、より好ましくは約6.5以下であり、さらに好ましくは約6.0以下である。
【0113】
本発明においては、後述するとおり、反応開始時点における反応溶液のpHを弱アルカリ性側にすることが好ましいが、その場合、α−1,4−グルカンホスホリラーゼの至適pHと比較して、多少の差が生じることになる。反応開始時点における反応溶液のpHと、α−1,4−グルカンホスホリラーゼの至適pHとの差は、約0.5以上とすることが可能であり、約1.0以上とすることも可能であり、さらには約1.5以上とすることも可能である。反応開始時点における反応溶液のpHと、α−1,4−グルカンホスホリラーゼの至適pHとの差は、好ましくは約5.5以下であり、より好ましくは約4.0以下であり、さらに好ましくは約2.5以下である。
【0114】
また、至適温度の観点からは、約30℃以上の至適温度を有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適温度は、より好ましくは約35℃以上であり、さらに好ましくは約40℃以上である。当該分野においては、至適温度の高い種々のα−1,4−グルカンホスホリラーゼが公知である。例えば、Aquifex aeolicus由来のα−1,4−グルカンホスホリラーゼの至適温度は約90℃であり、至適pHは5.6である。このα−1,4−グルカンホスホリラーゼを産生するAquifex aeolicusは、例えば、ATCCから入手可能である。α−1,4−グルカンホスホリラーゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0115】
反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。α−1,4−グルカンホスホリラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0116】
(7.ゲスト化合物)
アミロースなどの直鎖状α−グルカン、および分岐状α−グルカンのうちの直鎖状の部分がゲスト化合物を包接することが公知である。すなわち、これらのα−グルカンは、ホスト化合物として作用することが公知である。これらのα−グルカンは、ゲスト化合物を包接すると沈澱しやすくなる。そのため、本発明の方法においては、反応溶液中にゲスト化合物を含むことが好ましい。ゲスト化合物は、反応開始時から溶液中に含有されてもよく、あるいは、ある程度反応が進んでから溶液中に添加されてもよい。
【0117】
本明細書中で使用される場合、用語「ゲスト化合物」とは、アミロースによって包接される化合物をいう。ゲスト化合物がアミロースによって包接された場合に得られる包接化合物が難溶性または不溶性であるものが好ましい。
【0118】
ゲスト化合物は、好ましくは、プライマーによって包接されない化合物である。
【0119】
本発明で好ましく使用され得るゲスト化合物の例としては、デカン酸(カプリン酸)、ラウリン酸、ドデシル硫酸ナトリウム、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートおよび3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートなどが挙げられる。ゲスト化合物は、好ましくは、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートであり、より好ましくは3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである。
【0120】
(8.グルコース−6−リン酸、グルコース−1−リン酸、または無機リン酸)
本発明において、出発物質としてグルコース−6−リン酸を使用することが可能である。本明細書において使用される場合、用語「グルコース−6−リン酸(glucose−6−phosphate)とは、グルコース−6−リン酸(C6H13O9P;glucose−6−phosphoric acid)およびその塩をいう。グルコース−6−リン酸は好ましくは、狭義のグルコース−6−リン酸(C6H13O9P)の任意の金属塩であり、より好ましくはグルコース−1−リン酸(C6H13O9P)の任意のアルカリ金属塩である。狭義のグルコース−6−リン酸は、グルコースの6位の炭素にリン酸基が結合している物質である。グルコース−6−リン酸の好ましい具体例としては、グルコース−6−リン酸二ナトリウム、グルコース−6−リン酸二カリウム、グルコース−6−リン酸(C6H13O9P)、などが挙げられる。本明細書において、特に断りの無い限り、グルコース−6−リン酸は、広義のグルコース−6−リン酸、すなわち狭義のグルコース−6−リン酸(C6H13O9P)およびその塩を示す。
【0121】
反応開始時の反応溶液は、1種類のグルコース−6−リン酸を含有してもよく、複数種類のグルコース−6−リン酸を含有してもよい。
【0122】
本明細書において使用される場合、用語「グルコース−1−リン酸(glucose−1−phosphate)」とは、グルコース−1−リン酸(C6H13O9P;glucose−6−phosphoric acid)およびその塩をいう。グルコース−1−リン酸は好ましくは、狭義のグルコース−1−リン酸(C6H13O9P)の任意の金属塩であり、より好ましくはグルコース−1−リン酸(C6H13O9P)の任意のアルカリ金属塩である。グルコース−1−リン酸の好ましい具体例としては、グルコース−1−リン酸二ナトリウム、グルコース−1−リン酸二カリウム、グルコース−1−リン酸(C6H13O9P)、などが挙げられる。本明細書において、括弧書きで化学式を書いていないグルコース−1−リン酸は、広義のグルコース−1−リン酸、すなわち狭義のグルコース−1−リン酸(C6H13O9P)およびその塩を示す。
【0123】
反応開始時の反応溶液は、1種類のグルコース−1−リン酸を含有してもよく、複数種類のグルコース−1−リン酸を含有してもよい。
【0124】
本明細書において使用される場合、用語「無機リン酸」とは、リン酸のあらゆる種類のイオンおよび塩の総称である。例えば、無機リン酸は、セロビオースからグルコース−1−リン酸を合成する反応においてリン酸基質を供与する。すなわち、無機リン酸は、この反応においてリン酸源として作用する。ここでリン酸基質とは、グルコース−1−リン酸のリン酸部分(moiety)の原料となる物質をいう。β−1,4−グルカンホスホリラーゼによって触媒されるβ−1,4−グルカン加リン酸分解において、無機リン酸はリン酸イオンの形態で基質として作用していると考えられる。当該分野ではこの基質を慣習的に無機リン酸というので、本明細書中でも、この基質を無機リン酸という。無機リン酸には、リン酸およびリン酸の無機塩が含まれる。通常、無機リン酸は、アルカリ金属イオンなどの陽イオンを含む水中で使用される。この場合、リン酸とリン酸塩とリン酸イオンとは平衡状態になるので、リン酸とリン酸塩とは区別をしにくい。従って、便宜上、リン酸とリン酸塩とを合わせて無機リン酸という。本発明において、無機リン酸は好ましくは、リン酸の任意の金属塩であり、より好ましくはリン酸のアルカリ金属塩である。無機リン酸の好ましい具体例としては、リン酸二水素ナトリウム、リン酸水素二ナトリウム、リン酸三ナトリウム、リン酸二水素カリウム、リン酸水素二カリウム、リン酸三カリウム、リン酸(H3PO4)、リン酸二水素アンモニウム、リン酸水素二アンモニウムなどが挙げられる。
【0125】
反応開始時の反応溶液は、1種類の無機リン酸を含有してもよく、複数種類の無機リン酸を含有してもよい。
【0126】
無機リン酸は、例えば、ポリリン酸(例えば、ピロリン酸、三リン酸および四リン酸)のようなリン酸縮合体またはその塩を、物理的、化学的または酵素反応などによって分解したものを反応溶液に添加することによって提供され得る。ポリリン酸は、通常、少量の無機リン酸を不純物として含有する。そのため、α−グルカンの合成には無機リン酸が必要であるが、市販のポリリン酸を添加すれば、無機リン酸を添加する必要がない場合がある。
【0127】
(9.グルコースイソメラーゼ(EC:5.3.1.5))
本発明の方法において、出発物質としてフルクトースを使用することも可能である。他の酵素反応の副産物としてフルクトースが得られる場合、本発明ではこのフルクトースを有効に利用することができる。
【0128】
出発物質としてフルクトースを使用する実施形態では、反応溶液中にグルコースイソメラーゼをさらに含むことが好ましい。溶液中にグルコースイソメラーゼを含むことにより、フルクトースをグルコースへと変換できる。
【0129】
本発明の製造方法で用いられ得るグルコースイソメラーゼは、D−グルコースとD−フルクトースとの相互変換を触媒し得る酵素である。グルコースイソメラーゼは、D−キシロースとD−キシルロースとの相互変換をも触媒し得るので、キシロースイソメラーゼとも呼ばれる。
【0130】
グルコースイソメラーゼは、微生物、動物および植物に存在する。グルコースイソメラーゼを産生する微生物の例としては、Streptomyces rubiginosus、Streptomyces olivochromogenes、Streptomyces murinus、Streptomyces violaceoniger、Streptomyces diastaticus、Streptomyces albus、Streptomyces sp.、Escherichia coli、Bacteroides xylanolyticus、Arthrobacter sp.、Candida boidinii、Clostridium thermosulfurogenes、Clostridium thermohydrosulfuricum、Thermoanaerobacterium saccharolyticum、Thermoanaerobacter sp.、Thermotoga neapolitana、Thermus aquaticus、Lactobacillus brevis、Lactobacillus xylosus、Agrobacterium tumefaciens、Bacillus sp.、Actinoplanes missouriensisおよびParacolobacterium aerogenoidesが挙げられる。グルコースイソメラーゼを産生する動物の例としては、Trypanosoma bruceiが挙げられる。グルコースイソメラーゼは、植物由来であってもよい。グルコースイソメラーゼを産生する生物はこれらに限定されない。
【0131】
本発明で用いられ得るグルコースイソメラーゼは、Streptomyces rubiginosusまたはBacillus sp.に由来することが好ましく、Streptomyces rubiginosusに由来することがより好ましい。本発明で用いられるグルコースイソメラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高いグルコースイソメラーゼは、例えば、高度好熱細菌に由来し得る。
【0132】
反応開始時の溶液中に含まれるグルコースイソメラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中のフルクトース1gに対して、好ましくは約0.01U以上であり、より好ましくは約0.05U以上であり、さらに好ましくは約0.1U以上であり、特に好ましくは約0.5U以上であり、最も好ましくは約1U以上である。好ましい実施形態では、反応開始時の溶液中のフルクトース1gに対して、好ましくは約500U以下であり、より好ましくは約100U以下であり、さらに好ましくは約50U以下であり、特に好ましくは約10U以下であり、最も好ましくは約5U以下である。グルコースイソメラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0133】
(10.β−1,4−グルカンホスホリラーゼ)
本発明において、出発物質としてβ−1,4−グルカンを使用することも可能である。出発物質としてβ−1,4−グルカンを使用する実施形態では、反応溶液中にβ−1,4−グルカンホスホリラーゼを含むことが好ましい。
【0134】
本明細書中では、「β−1,4−グルカンホスホリラーゼ」とは、β−1,4−グルカンの非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う任意の酵素をいう。β−1,4−グルカンホスホリラーゼは、加リン酸分解の逆反応であるβ−1,4−グルカン合成反応をも触媒し得る。反応がどちらの方向に進むかは、基質の量に依存するが、この反応は、β−1,4−グルカン合成反応の方向に進みやすい傾向がある。β−1,4−グルカンホスホリラーゼによって触媒される反応は、次式により示される:
【0135】
【化3】
なお、この式において、出発時のβ−1,4−グルカンの重合度が2の場合、β−1,4−グルカンの代わりにグルコースが得られる。
【0136】
β−1,4−グルカンホスホリラーゼは好ましくは、セロビオースホスホリラーゼ(EC:2.4.1.20)またはセロデキストリンホスホリラーゼ(EC:2.4.1.49)である。
【0137】
セロビオースホスホリラーゼは、セロビオースの非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う酵素をいう。セロビオースホスホリラーゼによって触媒される反応は、次式により示される:
【0138】
【化4】
セロデキストリンホスホリラーゼは、重合度3以上のセロオリゴ糖の非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う酵素をいう。セロオリゴ糖は、セロデキストリンとも呼ばれる。セロデキストリンホスホリラーゼによって触媒される反応は、次式により示される:
【0139】
【化5】
本発明の方法においては、β−1,4−グルカンがセロビオースである場合、β−1,4−グルカンホスホリラーゼとしてセロビオースホスホリラーゼを用いることが好ましい。本発明の方法においては、β−1,4−グルカンがセロオリゴ糖である場合、β−1,4−グルカンホスホリラーゼとしてセロデキストリンホスホリラーゼを用いることが好ましい。本発明の方法においてはまた、β−1,4−グルカンがセロオリゴ糖である場合、β−1,4−グルカンホスホリラーゼとしてセロビオースホスホリラーゼおよびセロデキストリンホスホリラーゼを用いることが好ましい。この場合、セロデキストリンホスホリラーゼの作用によってセロオリゴ糖が分解されることによって生じたグルコース−1−リン酸がα−グルカン合成に使用され、かつ最終的に生じたセロビオースをセロビオースホスホリラーゼによって分解し得るので、セロオリゴ糖からα−グルカンの合成速度がより速くなる。
【0140】
出発物質としてβ−1,4−グルカンホスホリラーゼを使用する実施形態では、反応用液中に少なくともセロビオースホスホリラーゼを含有することが好ましい。セロビオースによって記載される上記反応によって生成されるグルコースは、ポリホスフェートグルコキナーゼおよびホスホグルコムターゼによって触媒される反応によりG1−Pへと変換されるため、β−1,4−グルカン(特に、セロビオース)を効率よく利用できる。
【0141】
β−1,4−グルカンホスホリラーゼは、自然界では種々の生物に含まれる。β−1,4−グルカンホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。β−1,4−グルカンホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される。β−1,4−グルカンホスホリラーゼは、植物由来であってもよい。
【0142】
セロビオースホスホリラーゼは、自然界では種々の生物に含まれる。セロビオースホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga
neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。セロビオースホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択され、より好ましくはClostridium thermocellumまたはCellvibrio gilvusであり、最も好ましくはClostridium thermocellumである。セロビオースホスホリラーゼは、植物由来であってもよい。
【0143】
セロデキストリンホスホリラーゼは、自然界では種々の生物に含まれる。セロデキストリンホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。セロデキストリンホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択され、より好ましくはClostridium thermocellumまたはCellulomonas sp.であり、最も好ましくはClostridium thermocellumである。セロデキストリンホスホリラーゼホスホリラーゼは、植物由来であってもよい。
【0144】
β−1,4−グルカンホスホリラーゼ(好ましくはセロビオースホスホリラーゼまたはセロデキストリンホスホリラーゼ、最も好ましくはセロビオースホスホリラーゼ)は、β−1,4−グルカンホスホリラーゼ(好ましくはセロビオースホスホリラーゼまたはセロデキストリンホスホリラーゼ、最も好ましくはセロビオースホスホリラーゼ)を産生する任意の生物由来であり得る。β−1,4−グルカンホスホリラーゼは、ある程度の耐熱性を有することが好ましい。β−1,4−グルカンホスホリラーゼは、耐熱性が高ければ高いほど好ましい。例えば、β−1,4−グルカンホスホリラーゼを1.4mMの2−メルカプトエタノールを含む50mMリン酸緩衝液(pH7.5)中で55℃にて20分間加熱した場合に加熱前のβ−1,4−グルカンホスホリラーゼの活性の50%以上の活性を保持するものであることが好ましく、60%以上の活性を保持するものであることがより好ましく、70%以上の活性を保持するものであることがさらに好ましく、80%以上の活性を保持するものであることが特に好ましく、85%以上の活性を保持するものであることが最も好ましい。β−1,4−グルカンホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来である。
【0145】
β−1,4−グルカンホスホリラーゼがセロビオースホスホリラーゼである場合、セロビオースホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga
maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来であり、より好ましくはClostridium thermocellumまたはCellvibrio gilvus由来であり、最も好ましくはClostridium thermocellum由来である。
【0146】
β−1,4−グルカンホスホリラーゼがセロビオースホスホリラーゼである場合、セロビオースホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga
maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来であり、より好ましくはClostridium thermocellumまたはCellulomonas sp.由来であり、最も好ましくはClostridium thermocellum由来である。
【0147】
反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のβ−1,4−グルカンの1gに対して、好ましくは約0.01U以上であり、より好ましくは約0.05U以上であり、さらに好ましくは約0.1U以上であり、特に好ましくは約0.5U以上であり、最も好ましくは約1U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のβ−1,4−グルカンの1gに対して、好ましくは約1,000U以下であり、より好ましくは約500U以下であり、さらに好ましくは約100U以下であり、特に好ましくは約50U以下であり、最も好ましくは約7U以下である。β−1,4−グルカンホスホリラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0148】
(11.アミラーゼ)
本発明の方法において、出発物質としてデンプンを使用することも可能である。デンプンを出発物質として使用する実施形態では、アミラーゼなどのデンプン分解酵素は、アミロース合成を行う反応溶液とは別の溶液に含まれることが好ましい。生成したアミロースが分解されてしまうからである。アミラーゼを使用する場合は、デンプンをアミラーゼで分解してグルコースを得た後に、その反応液を加熱してアミラーゼを失活させることが好ましい。
【0149】
アミラーゼはデンプンを加水分解する酵素の総称である。アミラーゼには、非還元性末端から特定数のグルコース単位を切り離していくエキソ型と、ほぼランダムに近い分解様式をもつエンド型がある。本発明においてアミラーゼを使用する場合には、エキソ型およびエンド型のいずれのアミラーゼを使用してもよい。アミラーゼの例としては、エキソ−1,4−α−D−グルコシダーゼ(EC3.2.1.3;グルコシダーゼとも呼ばれる)、β−アミラーゼ(EC3.2.1.3)、エキソ−イソマルトトリヒドロラーゼ(EC3.2.1.95)、エキソ−マルトテトラオヒドロラーゼ(EC3.2.1.60)、エキソ−マルトヘキサオヒドロラーゼ(EC3.2.1.98)が挙げられる。
【0150】
(12.β−1,4−グルカン)
本発明において、出発物質としてβ−1,4−グルカンを使用することが可能である。本明細書中では「β−1,4−グルカン」とは、D−グルコースを構成単位とする糖であって、β−1,4−グルコシド結合によって連結された糖単位を少なくとも2糖単位以上有する糖をいう。β−1,4−グルカンは、直鎖状の分子であり得る。直鎖状β−グルカンとβ−1,4−グルカンとセルロースとは同義語である。直鎖状β−グルカンでは、β−1,4−グルコシド結合によってのみ糖単位の間が連結されている。1分子のβ−1,4−グルカンに含まれる糖単位の数を、このβ−1,4−グルカンの重合度という。β−1,4−グルカンの重合度は、好ましくは、約2〜約10であり、より好ましくは約2〜約8であり、より好ましくは約2〜約5である。重合度が約2〜約10のβ−1,4−グルカンを、セロオリゴ糖ともいう。重合度が2のβ−1,4−グルカンを特に、セロビオースという。重合度が3のβ−1,4−グルカンをセロトリオースという。重合度が4のβ−1,4−グルカンをセロテトラオースという。β−1,4−グルカンの重合度が低いほど溶解度が高く、取り扱いが容易であるので、重合度の低いβ−1,4−グルカンがより好ましい。β−1,4−グルカンは、あらゆる植物中に存在する。β−1,4−グルカンは、植物から単離されたまま未改変のものであってもよく、植物から単離したものを化学的または酵素的に処理することによって得られたものであってもよい。β−1,4−グルカンはまた、古紙、建材、古布などの廃棄物から再生されるセルロースまたはそれから調製されたものであってもよい。例えば、植物から単離した高分子量のセルロースに対してセルラーゼを作用させることによって、より低分子量のセロオリゴ糖が得られる。植物からセロオリゴ糖を大量に生産する方法は当該分野で公知である。このような文献の例としては、特開2001−95594号公報が挙げられる。β−1,4−グルカンは、β−1,4−グルカンを含む植物破砕液から精製β−1,4−グルカンに至るいずれの生成段階のものとして提供されてもよい。本発明の方法で使用されるβ−1,4−グルカンは、純粋なものであることが好ましい。しかし、本発明で用いる酵素の作用を阻害しない限り、任意の他の夾雑物を含んでいてもよい。
【0151】
出発物質としてβ−1,4−グルカンを使用する実施形態において、反応開始時の溶液中に含まれるβ−1,4−グルカンの濃度は、好ましくは約0.1重量%以上であり、より好ましくは約0.5重量%以上であり、さらに好ましくは約1重量%以上であり、特に好ましくは約2重量%以上であり、最も好ましくは約3重量%以上である。出発物質としてβ−1,4−グルカンを使用する実施形態において、反応開始時の溶液中に含まれるβ−1,4−グルカンの濃度は、好ましくは約40重量%以下であり、より好ましくは約30重量%以下であり、さらに好ましくは約20重量%以下であり、特に好ましくは約15重量%以下であり、最も好ましくは約12重量%以下である。なお、本明細書中でβ−1,4−グルカンの濃度は、Weight/Volumeで、すなわち、
(β−1,4−グルカンの重量)×100/(溶液の容量)
で計算する。β−1,4−グルカンの重量が多すぎると、溶液中に未反応のβ−1,4−グルカンが析出する場合がある。β−1,4−グルカンの使用量が少なすぎると、高温での反応において、反応自体は起こるものの、収率が低下する場合がある。
【0152】
(13.枝切り酵素)
本発明の方法において、プライマーとしてα−1,6−グルコシド結合を含有するものを用いる場合などの、生成物に分岐が生じる場合には、必要に応じて、枝切り酵素を用いることができる。
【0153】
本発明で用いられ得る枝切り酵素は、α−1,6−グルコシド結合を切断し得る酵素である。枝切り酵素は、アミロペクチンおよびグリコーゲンにともによく作用するイソアミラーゼ(EC 3.2.1.68)と、アミロペクチン、グリコーゲンおよびプルランに作用するα−デキストリンエンド−1,6−α−グルコシダーゼ(プルラナーゼともいう)(EC 3.2.1.41)との2つに分類される。
【0154】
枝切り酵素は、微生物および植物に存在する。枝切り酵素を産生する微生物の例としては、Saccharomyces cerevisiae、Chlamydomonas
sp.、Bacillus brevis、Bacillus acidopullulyticus、Bacillus macerans、Bacillus stearothermophilus、Bacillus circulans、Thermus
aquaticus、Klebsiella pneumoniae、Thermoactinomyces thalpophilus、Thermoanaerobacter ethanolicus、Pseudomonas amyloderamosaなどが挙げられる。枝切り酵素を産生する植物の例としては、ジャガイモ、サツマイモ、トウモロコシ、イネ、コムギ、オオムギ、オートムギ、サトウダイコンなどが挙げられる。枝切り酵素を産生する生物はこれらに限定されない。
【0155】
本発明で用いられ得る枝切り酵素は、Klebsiella pneumoniae、Bacillus brevis、Bacillus acidopullulyticus、Pseudomonas amyloderamosaに由来することが好ましく、Klebsiella pneumoniae、Pseudomonas amyloderamosaに由来することがより好ましい。本発明で用いられる枝切り酵素は、反応至適温度が高いことが好ましい。反応至適温度が高い枝切り酵素は、例えば、高度好熱細菌に由来し得る。
【0156】
(14.ブランチングエンザイム(EC.2.4.1.18))
本発明の方法において、生成物に分岐を生じさせることが所望される場合には、必要に応じて、ブランチングエンザイムを用いることができる。
【0157】
本発明で用いられ得るブランチングエンザイムは、α−1,4−グルカン鎖の一部をこのα−1,4−グルカン鎖のうちのあるグルコース残基の6位に転移して分枝を作り得る酵素である。ブランチングエンザイムは、1,4−α−グルカン分枝酵素、枝つくり酵素またはQ酵素とも呼ばれる。
【0158】
ブランチングエンザイムは、微生物、動物、および植物に存在する。ブランチングエンザイムを産生する微生物の例としては、Bacillus stearothermophilus、Bacillus subtilis、Bacillus caldolyticus、Bacillus licheniformis、Bacillus amyloliquefaciens、Bacillus coagulans、Bacillus caldovelox、Bacillus thermocatenulatus、Bacillus smithii、Bacillus megaterium、Bacillus brevis、Alkalophillic Bacillus sp.、Streptomyces coelicolor、Aquifex aeolicus、Synechosystis sp.、E.coli、Agrobacteirum tumefaciens、Thermus aquaticus、Rhodothermus obamensis、Neurospora crassa、酵母などが挙げられる。ブランチングエンザイムを産生する動物の例としてはヒト、ウサギ、ラット、ブタなどの哺乳類が挙げられる。ブランチングエンザイムを産生する植物の例としては、藻類、ジャガイモ、サツマイモ、ヤマイモ、キャッサバなどの芋類、ホウレンソウなどの野菜類、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、アワなどの穀類、えんどう豆、大豆、小豆、うずら豆などの豆類などが挙げられる。ブランチングエンザイムを産生する生物はこれらに限定されない。
【0159】
本発明で用いられ得るブランチングエンザイムは、ジャガイモ、Bacillus stearothermophilus、Aquifex aeolicusに由来することが好ましく、Bacillus stearothermophilus、Aquifex aeolicusに由来することがより好ましい。本発明で用いられるブランチングエンザイムは、反応至適温度が高いことが好ましい。反応至適温度が高いブランチングエンザイムは、例えば、高度好熱細菌に由来し得る。
【0160】
(15.4−α−グルカノトランスフェラーゼ(EC.2.4.1.25))
本発明の方法において、生成物に環状構造を生じさせる場合には、必要に応じて、4−α−グルカノトランスフェラーゼを用いることができる。
【0161】
本発明で用いられ得る4−α−グルカノトランスフェラーゼは、ディスプロポーショネーティングエンザイム、D−酵素、アミロマルターゼ、不均化酵素などとも呼ばれ、マルトオリゴ糖の糖転移反応(不均一化反応)を触媒し得る酵素である。4−α−グルカノトランスフェラーゼは、供与体分子の非還元末端からグルコシル基あるいは、マルトシルもしくはマルトオリゴシルユニットを受容体分子の非還元末端に転移する酵素である。従って、酵素反応は、最初に与えられたマルトオリゴ糖の重合度の不均一化をもたらす。供与体分子と受容体分子とが同一の場合は、分子内転移が生じ、その結果、環状構造をもつ生成物が得られる。
【0162】
4−α−グルカノトランスフェラーゼは、微生物および植物に存在する。4−α−グルカノトランスフェラーゼを産生する微生物の例としては、Aquifex aeolicus、Streptococcus pneumoniae、Clostridium butylicum、Deinococcus radiodurans、Haemophilus influenzae、Mycobacterium tuberculosis、Thermococcus litralis、Thermotoga maritima、Thermotoga neapolitana、Chlamydia psittaci、Pyrococcus sp.、Dictyoglomus thermophilum、Borrelia burgdorferi、Synechosystis sp.、E.coli、Thermus aquaticusなどが挙げられる。4−α−グルカノトランスフェラーゼを産生する植物の例としては、ジャガイモ、サツマイモ、ヤマイモ、キャッサバなどの芋類、トウモロコシ、イネ、コムギ、などの穀類、えんどう豆、大豆、などの豆類などが挙げられる。4−α−グルカノトランスフェラーゼを産生する生物はこれらに限定されない。
【0163】
本発明で用いられ得る4−α−グルカノトランスフェラーゼは、ジャガイモ、Thermus aquaticus、Thermococcus litralisに由来することが好ましく、ジャガイモ、Thermus aquaticusに由来することがより好ましい。本発明で用いられる4−α−グルカノトランスフェラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高い4−α−グルカノトランスフェラーゼは、例えば、高度好熱細菌に由来し得る。
【0164】
(16.グリコーゲンデブランチングエンザイム(EC.2.4.1.25/EC.3.2.1.33))
本発明の方法において、生成物に環状構造を生じさせる場合には、必要に応じて、グリコーゲンデブランチングエンザイムを用いることができる。
【0165】
本発明で用いられ得るグリコーゲンデブランチングエンザイムは、α−1,6−グルコシダーゼ活性と、4−α−グルカノトランスフェラーゼ活性との2種類の活性をもつ酵素である。グリコーゲンデブランチングエンザイムが持つ、4−α−グルカノトランスフェラーゼ活性により、環状構造を持つ生成物が得られる。
【0166】
グリコーゲンデブランチングエンザイムは、微生物および動物に存在する。グリコーゲンデブランチングエンザイムを産生する微生物の例としては、酵母などが挙げられる。グリコーゲンデブランチングエンザイムを産生する動物の例としては、ヒト、ウサギ、ラット、ブタなどの哺乳類が挙げられる。グリコーゲンデブランチングエンザイムを産生する生物はこれらに限定されない。
【0167】
本発明で用いられ得るグリコーゲンデブランチングエンザイムは、酵母に由来することが好ましい。本発明で用いられるグリコーゲンデブランチングエンザイムは、反応至適温度が高いことが好ましい。反応至適温度が高いグリコーゲンデブランチングエンザイムは、例えば、タンパク質工学的手法により、中温で作用し得る酵素に改変を加えることで得られる。
【0168】
(17.本発明で使用される酵素についての一般的説明)
本明細書中では、酵素がある生物に「由来する」とは、その生物が天然で有するその酵素のアミノ酸配列と同じアミノ酸配列を有することをいう。ある生物の有するある酵素をその生物から直接単離した場合も、遺伝子組換え技術を用いてその生物の有するアミノ酸配列と同じアミノ酸配列を有する酵素を別の生物から得た場合も、その生物に由来するという。人工合成された遺伝子であっても同じ配列を有する場合はその生物に由来するという。場合によっては、それらの場合を含めて、その生物から得られた酵素という。例えば、あの生物から入手したある酵素をコードする核酸分子を大腸菌に導入して、その大腸菌からその酵素を単離する場合も、その酵素は「その生物に由来する」または「その生物から得られた」という。
【0169】
本発明で用いられる各種酵素は、各酵素について記載したような、自然界に存在する、その酵素を産生する生物から直接単離された酵素であってよい。あるいは、本発明で用いられる各種酵素は、その酵素を産生する生物から単離した、その酵素をコードする核酸分子を用いて組換えされた微生物(例えば、細菌、真菌など)から単離した酵素であってもよい。
【0170】
本発明においては、天然の酵素と同等以上の酵素活性を有するのであれば、そのアミノ酸配列の一部が改変された酵素を使用してもよい。改変された酵素が使用される場合、この改変された酵素は、天然の酵素に対して、好ましくは少なくとも約80%以上のアミノ酸配列同一性を有し、より好ましくは少なくとも約90%以上のアミノ酸配列同一性を有し、最も好ましくは約95%以上のアミノ酸配列同一性を有する。好ましくは、この改変された酵素は、天然の酵素に対して1または数個のアミノ酸の置換、欠失または付加を有する。このような置換、欠失または付加は、保存的なものであることが好ましい。このような置換、欠失または付加は、酵素の非活性部位に存在することが好ましい。天然の酵素活性に悪影響を及ぼさずにアミノ酸の置換、欠失または付加をする方法は、当業者に公知である。好ましくは、この改変された酵素は、天然の酵素と比較して約90%以上の活性を有し、より好ましくは約95%以上の活性を有し、さらに好ましくは約100%以上の活性を有する。このような改変された酵素は、当業者に公知の方法によって作製され得る。
【0171】
本発明の方法で用いられる各種酵素は、例えば、以下のようにして調製され得る。まず、その酵素を産生する微生物(例えば、細菌、真菌など)を培養する。この微生物は、その酵素を直接生産する微生物であってもよい。また、その酵素をコードする核酸分子をクローン化し、得られた核酸分子で、その酵素の発現に有利な微生物(例えば、細菌、真菌など)を組換えして、組換えされた微生物を得、得られた微生物からその酵素を得てもよい。
【0172】
各種酵素をコードする核酸分子での組換えに用いられる微生物は、各種酵素の発現の容易さ、培養の容易さ、増殖の速さ、安全性などの種々の条件を考慮して容易に選択され得る。アミラーゼは、生成されたグルカンを分解してしまうので、本発明のアミロース合成方法に使用される各種酵素は、夾雑物としてアミラーゼを含まないことが好ましい。従って、アミラーゼを産生しないかまたは低レベルでしか発現しない微生物(例えば、細菌、真菌など)を遺伝子組換えに用いることが好ましい。各種酵素の遺伝子組換えのためには、大腸菌または枯草菌のような中温菌を用いることが好ましい。アミラーゼを産生しないかまたは低レベルでしか発現しない微生物(例えば、細菌、真菌など)を用いて産生される各種酵素は、アミラーゼを実質的に含まないため、本発明の方法での使用に好ましい。
【0173】
クローン化した遺伝子での微生物(例えば、細菌、真菌など)の遺伝子組換えは、当業者に周知の方法に従って行われ得る。クローン化した遺伝子を用いる場合、この遺伝子を、構成性プロモーターまたは誘導性プロモーターに作動可能に連結することが好ましい。「作動可能に連結する」とは、プロモーターと遺伝子とが、そのプロモーターによって遺伝子の発現が調節されるように連結されることをいう。誘導性プロモーターを用いる場合、培養を、誘導条件下で行うことが好ましい。種々の誘導性プロモーターは当業者に公知である。
【0174】
クローン化した遺伝子について、生産される各種酵素が菌体外に分泌されるように、シグナルペプチドをコードするヌクレオチド配列をこの遺伝子に連結し得る。シグナルペプチドをコードするヌクレオチド配列は当業者に公知である。
【0175】
当業者は、各種酵素を生産するために、微生物(例えば、細菌、真菌など)の培養の条件を適切に設定し得る。微生物の培養に適切な培地、各誘導性プロモーターに適切な誘導条件などは当業者に公知である。
【0176】
例えば、発現された酵素が形質転換細胞内に蓄積する場合、形質転換細胞を適切な条件下で培養した後、培養物を遠心分離または濾過することによって細胞を回収し、次いで適切な緩衝液に懸濁する。次いで超音波処理などにより細胞を破砕した後、遠心分離もしくは濾過することによって上清を得る。あるいは、発現された酵素が形質転換細胞外に分泌される場合、このようにして形質転換細胞を培養した後、培養物を遠心分離または濾過することによって細胞を分離して上清を得る。目的の酵素が形質転換細胞内に蓄積する場合も、形質転換細胞外に分泌される場合も、このようにして得られた酵素含有上清を通常の手段(例えば、塩析法、溶媒沈澱、限外濾過)を用いて濃縮し、酵素を含む画分を得る。この画分を濾過、あるいは遠心分離、脱塩処理などの処理を行い粗酵素液を得る。さらにこの粗酵素液を、凍結乾燥、等電点電気泳動、イオン交換クロマトグラフィー、晶出などの通常の酵素の精製手段を適宜組み合わせることによって、比活性が向上した粗酵素あるいは精製酵素が得られる。α−アミラーゼなどのグルカンを加水分解する酵素が含まれていなければ、粗酵素をそのまま、例えば、α−グルカンの製造に用い得る。
【0177】
各種酵素は、精製されていても未精製であってもよい。各種酵素は、固定化されていても固定化されていなくともよい。各種酵素は、固定化されることが好ましい。固定化の方法としては、担体結合法(たとえば、共有結合法、イオン結合法、または物理的吸着法)、架橋法または包括法(格子型またはマイクロカプセル型)など、当業者に周知の方法が使用され得る。各種酵素は、担体上に固定化されていることが好ましい。
【0178】
(18.溶媒)
本発明の方法に用いる溶媒は、本発明で用いられる各種酵素の酵素活性を損なわない溶媒であれば任意の溶媒であり得る。
【0179】
なお、グルカンを生成する反応が進行し得る限り、溶媒が本発明の方法に用いる材料を完全に溶解する必要はない。例えば、酵素が固体の担体上に担持されている場合には、酵素が溶媒中に溶解する必要はない。さらに、グルコースなどの反応材料も全てが溶解している必要はなく、反応が進行し得る程度の材料の一部が溶解していればよい。
【0180】
代表的な溶媒は、水である。溶媒は、各種酵素を調製する際に得られる細胞破砕液のうちの水分であってもよい。
【0181】
水は、軟水、中間水および硬水のいずれであってもよい。硬水とは、硬度20°以上の水をいい、中間水とは、硬度10°以上20°未満の水をいい、軟水とは、硬度10°未満の水をいう。水は、好ましくは軟水または中間水であり、より好ましくは軟水である。
【0182】
(19.他の成分)
本発明で使用される溶液中には、目的の反応を妨害しない限り、任意の他の物質を含み得る。このような物質の例としては、緩衝剤、各種酵素を産生する微生物(例えば、細菌、真菌など)の成分、塩類、培地成分などが挙げられる。
【0183】
<α−グルカンの製造>
1つの実施形態では、本発明のα−グルカンは、グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法によって製造される。
【0184】
別の実施形態では、本発明のα−グルカンは、グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法によって製造される。
【0185】
別の実施形態では、本発明のα−グルカンは、セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0186】
別の実施形態では、本発明のα−グルカンは、フルクトースと、ポリリン酸と、プライマーと、グルコースイソメラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0187】
別の実施形態では、本発明のα−グルカンは、デンプンをアミラーゼによってグルコースに分解する工程;アミラーゼを失活させる工程;ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0188】
図15に、これらの方法をまとめて記載する。本発明の最も基本的な方法は、G6−PからG1−Pを経てアミロース、デンプン、グリコーゲンなどのα−グルカンを製造する方法である。G6−Pは、グルコースおよびポリリン酸にPGKを作用させることにより生成され得る。グルコースは、フルクトースにグルコースイソメラーゼを作用させることによって生成され得る。グルコースはまた、セルロースにセルラーゼ、セロビオースホスホリラーゼなどを作用させることによって生成され得る。グルコースはまた、デンプンにアミラーゼを作用させることによって生成され得る。生成物であるα−グルカンを分解してしまうアミラーゼを使用する方法を除けば、これらのグルコースを生成する方法は、グルコースからG6−Pを経て、次いでG1−Pを経てα−グルカンを製造する方法と同一の溶液中で同時に行われ得る。
【0189】
本発明の製造方法においては例えば、まず、溶液を調製する。溶液は、例えば、適切な溶媒に、反応に使用する各種物質を添加することにより調製され得る。あるいは、溶液は、反応に使用する各種物質をそれぞれ含む溶液を混合することによって調製してもよい。あるいは、溶液は、反応に使用する各種物質のうちのいくつかの成分を含む溶液に固体状の他の成分を混合することによって調製してもよい。本発明の製造方法で用いられる溶液には、酵素反応を阻害しない限り、必要に応じて、pHを調整する目的で任意の緩衝剤を加えてもよい。この溶液のpHは、酵素反応を過度に阻害しない限り、任意のpHであり得る。pH値は、好ましくは約5.7〜約11であり、より好ましくは約7.4〜約8.5である。pHは、反応に用いる酵素の至適pHに合わせて適切に設定され得る。溶液の塩濃度もまた、酵素反応を過度に阻害しない限り、任意の塩濃度であり得る。PGMはMgイオンを必要とするため、MgCl2などのMgイオンを放出する物質を溶液に添加することが好ましい。Mgイオンを放出する物質の、反応開始時の溶液中での濃度は、好ましくは約20mM以上であり、より好ましくは約30mM以上であり、さらに好ましくは約40mM以上である。Mgイオンを放出する物質の、反応開始時の溶液中での濃度は、好ましくは約100mM以下であり、より好ましくは約80mM以下であり、さらに好ましくは約60mM以下である。
【0190】
また、この溶液には、必要に応じて枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素を添加してもよい。これらの酵素は、α−グルカン合成反応の開始時に添加されてもよく、反応の途中に添加されてもよく、また、反応が終了した後に添加されてもよい。
【0191】
さらに、この溶液には、ゲスト化合物を添加してもよい。ゲスト化合物は、反応開始時にこの溶液中に含有されてもよく、あるいは、反応がある程度進んだ時点で反応溶液に添加されてもよい。ゲスト化合物としてN−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物としてN−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネートを使用する場合、この化合物の濃度が10mMであると、コントロール(ゲスト化合物を添加しない場合)の約1.5倍の収率を得ることができるという顕著な効果が得られる。
【0192】
ゲスト化合物として3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物として3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度が1mM〜10mMであると、コントロール(ゲスト化合物を添加しない場合)の約1.8倍〜約2倍の収率を得ることができるという顕著な効果が得られる。
【0193】
ゲスト化合物として3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物として3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度が10mMであると、コントロール(ゲスト化合物を添加しない場合)の約2.7倍の収率を得ることができるという顕著な効果が得られる。
【0194】
次いで、溶液を、当該分野で公知の方法によって必要に応じて加熱することにより、反応させる。溶液の温度は、本発明の効果が得られる限り、任意の温度であり、添加した酵素がその活性を示す温度である。例えば、耐熱性酵素を用い、反応温度をその耐熱酵素に最適な温度にすることによって、添加した耐熱性酵素以外の混入した酵素の活性を抑え得る。この反応工程における溶液の温度は、所定の反応時間後に反応前のこの溶液に含まれる各種酵素の活性の約50%以上、より好ましくは約80%以上の活性が残る温度であることが好ましい。反応に使用する各種酵素の反応至適温度がいずれも約30℃〜40℃の範囲内である場合、反応温度は、好ましくは約25℃以上であり、より好ましくは約30℃以上であり;反応温度は、好ましくは約50℃以下であり、より好ましくは約40℃以下である。反応に使用する各種酵素がいずれも耐熱性酵素である場合、反応温度は、その耐熱性酵素の反応至適温度の±10℃以内にあることが好ましく、±5℃以内にあることが好ましい。反応に使用する各種酵素の反応至適温度が幅広い範囲にわたる場合、反応温度は、その範囲内で適切に設定され得る。反応に使用する各種酵素の反応至適温度が±5℃以内にあることが好ましく、ほぼ同じであることが好ましい。
【0195】
グルコースを出発物質とする実施形態では、反応溶液の初期pH(反応開始時の溶液のpH)は、好ましくは約5.7以上であり、より好ましくは約7.4以上である。グルコースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12.0以下であり、より好ましくは約11.0以下であり、さらに好ましくは約8.5以下である。反応で使用する各酵素の代表的な至適pHは、Microlunatus phosphovolas由来のPGKが約pH7.0であり、Escherichia coli由来のPGMが約pH7.5であり、そしてGPが約pH5.5である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約20%程度のアミロース収率を得られる。
【0196】
G6−Pを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約7.3以上であり、より好ましくは約7.5以上である。G6−Pを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12.0以下であり、より好ましくは約11.0以下であり、さらに好ましくは約8.5以下である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約25%以上のアミロース収率を得られる。
【0197】
セロビオースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約5.6以上であり、より好ましくは約7.5以上である。セロビオースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12以下であり、より好ましくは約8.5以下である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約62%以上のアミロース収率を得られる。
【0198】
グルコースを出発物質とする実施形態では、反応溶液の初期グルコース濃度(反応開始時の溶液のグルコース濃度)は、好ましくは約100mM以上であり、より好ましくは約200mM以上であり、特に好ましくは約400mM以上であり、最も好ましくは約500mM以上である。グルコースを出発材料として用いる場合、溶液中に含まれるグルコースの濃度に特に上限はないが、例えば、約2.5M以下、約2M以下、約1.5M以下、約1M以下、約750mM以下などであり得る。グルコースの量が多すぎると、反応に適切な量のポリリン酸を溶解することができず、収率が低下する場合がある。グルコースの使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0199】
グルコース−6−リン酸を出発物質とする実施形態では、反応溶液の初期グルコース−6−リン酸濃度(反応開始時の溶液のグルコース−6−リン酸濃度)は、好ましくは約20mM以上であり、より好ましくは約100mM以上であり、特に好ましくは約200mM以上である。グルコース−6−リン酸を出発材料として用いる場合、溶液中に含まれるグルコース−6−リン酸の濃度に特に上限はないが、例えば、約2M以下、約1.5M以下、約1M以下、約750mM以下などであり得る。グルコース−6−リン酸の量が多すぎると溶解することができず、収率が低下する場合がある。グルコース−6−リン酸の使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0200】
セロビオースを出発物質とする実施形態では、反応溶液の初期セロビオース濃度(反応開始時の溶液のセロビオース濃度)は、好ましくは約100mM以上であり、より好ましくは約150mM以上であり、特に好ましくは約175mM以上である。セロビオースを出発材料として用いる場合、溶液中に含まれるセロビオースの濃度に特に上限はないが、例えば、約1.0M以下、約750mM以下、約500mM以下、約250mM以下、約125mM以下などであり得る。セロビオースの量が多すぎると、セロビオースそのものや反応に適切な量のポリリン酸を溶解することができず、収率が低下する場合がある。セロビオースの使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0201】
グルコースを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が、好ましくは約0.15以下であり、より好ましくは約0.03以下である。
【0202】
G6−Pを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/G6−P)が、好ましくは約0.4以下であり、より好ましくは約0である。
【0203】
セロビオースを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/CB)が、好ましくは約0〜4であり、より好ましくは約0.008〜2であり、更に好ましくは0.028〜0.810であり、最も好ましくは0.056〜0.162である。
【0204】
反応時間は、反応温度、反応により生産されるグルカンの分子量および酵素の残存活性を考慮して、任意の時間で設定され得る。反応時間は、好ましくは約1時間以上であり、より好ましくは約2時間以上であり、さらに好ましくは約3時間以上である。反応時間は、好ましくは約100時間以下であり、より好ましくは約72時間以下であり、さらに好ましくは約54時間以下であり、特に好ましくは約36時間以下であり、最も好ましくは約24時間以下である。
【0205】
加熱は、どのような手段を用いて行ってもよいが、溶液全体に均質に熱が伝わるように、攪拌を行いながら加熱することが好ましい。溶液は、例えば、温水ジャケットと攪拌装置を備えたステンレス製反応タンクの中に入れられて攪拌される。
【0206】
本発明の方法ではまた、反応がある程度進んだ段階で、各種原料のうちの少なくとも1つを反応溶液に追加してもよい。
【0207】
このようにして、α−グルカンを含有する溶液が生産される。
【0208】
反応終了後、溶液は、必要に応じて例えば、100℃にて10分間加熱することによって溶液中の酵素を失活させ得る。あるいは、酵素を失活させる処理を行うことなく後の工程を行ってもよい。溶液は、そのまま保存されてもよいし、生産されたグルカンを単離するために処理されてもよい。
【0209】
<精製方法>
生産されたα−グルカンは、必要に応じて精製され得る。精製することにより除去される不純物の例は、グルコースである。α−グルカンの精製法の例としては、有機溶媒を用いる方法(T.J.Schochら、J.American Chemical Society,64,2957(1942))および有機溶媒を用いない方法がある。
【0210】
有機溶媒を用いる精製に使用され得る有機溶媒の例としては、アセトン、n−アミルアルコール、ペンタゾール、n−プロピルアルコール、n−ヘキシルアルコール、2−エチル−1−ブタノール、2−エチル−1−ヘキサノール、ラウリルアルコール、シクロヘキサノール、n−ブチルアルコール、3−ペンタノール、4−メチル−2−ペンタノール、d,l−ボルネオール、α−テルピネオール、イソブチルアルコール、sec−ブチルアルコール、2−メチル−1−ブタノール、イソアミルアルコール、tert−アミルアルコール、メントール、メタノール、エタノールおよびエーテルが挙げられる。
【0211】
有機溶媒を用いない精製方法の例を、以下に示す。
【0212】
(1)α−グルカン生産反応後、反応溶液を冷却することによりα−グルカンを沈澱させ、そして沈澱したα−グルカンを、膜分画、濾過、遠心分離などの一般的な固液分離方法により精製する方法;
(2)α−グルカン生産反応の間もしくはα−グルカン生産反応後に反応溶液を冷却してα−グルカンをゲル化し、ゲル化したα−グルカンを回収し、そしてゲル化したα−グルカンから、グルコースを、水による洗浄、凍結融解、ろ過などの操作によって除去する方法;ならびに
(3)α−グルカン生産反応後、水に溶解しているα−グルカンを沈澱させずに、限外ろ過膜を用いた膜分画もしくはクロマトグラフィーに供してグルコースを除去する方法。
【0213】
精製に使用され得る限外濾過膜の例としては、分画分子量約1,000〜約100,000、好ましくは約5,000〜約50,000、より好ましくは約10,000〜約30,000の限外濾過膜(ダイセル製UF膜ユニット)が挙げられる。
【0214】
クロマトグラフィーに使用され得る担体の例としては、ゲル濾過クロマトグラフィー用担体、配位子交換クロマトグラフィー用担体、イオン交換クロマトグラフィー用担体および疎水クロマトグラフィー用担体が挙げられる。
【実施例】
【0215】
以下の実施例により本発明をさらに詳細に説明する。本発明は以下の実施例のみに限定されない。
【0216】
(1.測定方法および計算方法)
本発明における各種酵素の活性および得られるα−グルカンの収率を、以下の測定方法によって測定した。
【0217】
(1.1 ポリホスフェートグルコキナーゼの活性測定法)
200mM グルコース、100mM MgCl2、8%ポリリン酸の混合液25μlと適切に希釈した酵素液25μlを加えて50μlの混合溶液として反応を開始させる。この混合溶液を45℃で15分間インキュベートして反応させた後、100℃で10分間保持することにより酵素を失活させる。続いて20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを酵素反応溶液に添加して混合する。この溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。この方法により、1分間に1μmolのグルコース−6−リン酸を生成する活性をポリホスフェートグルコキナーゼ1単位とする。
【0218】
(1.2 ホスホグルコムターゼの活性測定法)
200mM グルコース−1−リン酸、100mM MgCl2、20mM Cofactor (グルコース またはグルコース−1,6−ビスホスフェート)の混合液25μlと適切に希釈した酵素液25μlを加えて50μlの混合溶液として反応を開始させる。この混合溶液を45℃で15分間インキュベートして反応させた後、100℃で10分間保持することにより酵素を失活させる。続いて20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを酵素反応溶液に添加して混合する。この溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。この方法により、1分間に1μmolのグルコース−6−リン酸を生成する活性をホスホグルコムターゼ1単位とする。
【0219】
(1.3 α−1,4−グルカンホスホリラーゼの活性測定法)
50μlの4%クラスターデキストリン水溶液と50μlの50mMグルコース−1−リン酸ナトリウム水溶液とを混合し、さらに適切に希釈した酵素液100μlを加えて200μlの混合物として反応を開始させる。この混合物を37℃で15分間インキュベートして反応を進行させた後、800μlのモリブデン試薬(15mM モリブデン酸アンモニウム、100mM 酢酸亜鉛)を混合し、反応を停止させる。続いて200μlの568mMアスコルビン酸(pH5.0)を加えて攪拌し、反応系を得る。この反応系を、30℃で20分間保持した後、分光光度計を用いて850nmでの吸光度を測定する。濃度既知の無機リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中の無機リン酸を求める。この方法により、1分間に1μmolの無機リン酸を生成する活性を、α−1,4−グルカンホスホリラーゼ1単位とする。
【0220】
(1.4 セロビオースホスホリラーゼの活性測定法)
25μlの40mMセロビオース水溶液と25μlの40mMリン酸ナトリウム水溶液(pH7.5)とを混合し、さらに適切に希釈した酵素液(試料)50μlを加えて100μlの混合物として反応を開始させる。この混合物を37℃で10分間インキュベートすることにより反応を進行させた後、100℃で10分間保持することによって酵素を失活させる。続いて600μlの1M Tris−塩酸緩衝液(pH7.0)および300μlの発色試薬(CII発色試薬(和光純薬社製))をこの混合液に添加して混合し、30℃で20分間保持した後、505nmでの吸光度を測定する。濃度既知のグルコース水溶液を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース量を求める。セロビオースホスホリラーゼ1単位とは、上記方法により20mMセロビオースから1分間に1μmolのグルコースを生成する酵素量と定義する。
【0221】
(1.5 得られるα−グルカンの収率の計算方法)
本発明の製造方法によるα−グルカンの収率を、得られたα−グルカン中に取り込まれたグルコース残基のモル数が、最初に添加された初発物質のグルコース当量でのモル数(S;グルコース当量のモル数)(例えば、グルコースを出発物質とする場合はグルコースのモル数、G6−Pを出発物質とする場合はG6−Pのモル数、二糖のセロビオースを出発物質とする場合はセロビオースのモル数の2倍量)の何%にあたるかによって計算した。反応終了後、反応溶液を100℃で10分間加熱して酵素を失活させた。次いで、反応溶液を適切に希釈し、充分量のα−アミラーゼと充分量のグルコアミラーゼを添加して40℃で6時間インキュベートすることによって溶液中のα−グルカンをグルコースにまで分解した。α−アミラーゼおよびグルコアミラーゼによる処理を行った溶液のグルコース量(G1;モル数)、およびこの処理を行ってない溶液のグルコース量(G2;モル数)を測定した。グルコース当量でG3モルのプライマーを使用したとすると、Gx=(G1−G2−G3)によって、α−グルカンに取り込まれたグルコース量(収量;モル数)が計算できる。このグルコース量を初発物質のモル数で除算して100倍することにより、収率を計算した。この計算式を次式に示す。
【0222】
【数1】
(1.6 グルコース濃度測定方法)
適切に希釈した試料100μlに600μlの1M Tris−塩酸緩衝液(pH7.0)および300μlの発色試薬(CII発色試薬(和光純薬社製))を添加して混合し、37℃、20分間保持した後、505nmでの吸光度を測定する。濃度既知のグルコース水溶液を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース量を求める。
【0223】
(1.7 リン酸濃度測定方法)
適切に希釈した試料200μlに800μlのモリブデン試薬(15mM モリブデン酸アンモニウム、100mM 酢酸亜鉛)および200μlの568mMアスコルビン酸(pH5.0)を加えて攪拌し、30℃で20分間保持した後、分光光度計を用いて850nmでの吸光度を測定する。濃度既知の無機リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中の無機リン酸を求める。
【0224】
(1.8 G1−P濃度測定方法)
適切に希釈した試料50μlに20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、15μM グルコース−1,6−ビスホスフェート、3μg/ml ウサギ筋肉由来ホスホグルコムターゼ、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを添加して混合する。この混合溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−1−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−1−リン酸濃度を求める。
【0225】
(1.9 G6−P濃度測定方法)
適切に希釈した試料50μlに20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを添加して混合する。この混合溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。
【0226】
(2.酵素の調製)
本発明の実験例および実施例で用いた各種酵素を、以下の方法によって調製した。
【0227】
(2.1 組換えポリホスフェートグルコキナーゼ(PGK)の調製方法)
Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのヌクレオチド配列(配列番号1)を合成し、発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−PGKを得た。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。
【0228】
その後、得られた大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で大腸菌の増殖したコロニーを選択することにより、Microlunatus phosphovolas由来ポリホスフェートグルコキナーゼ遺伝子が導入された大腸菌を得た。それらの大腸菌についてポリホスフェートグルコキナーゼを発現していることを酵素活性測定によって確認した。
【0229】
この大腸菌を、抗生物質カナマイシンを含むTB培地(1.2%トリプトン、2.4%酵母エキス(ともにDifco社製)、0.231%リン酸二水素カリウム、1.254%リン酸水素二カリウム、0.4%グリセリン)600mLに接種し、100rpmで振盪させながら37℃、24時間培養した。次いで、この培養液を5,000rpmにて10分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、180mLの50mM MOPS buffer (pH7)中に懸濁し、次いで1%リゾチームを20mL加え溶菌させた。溶菌後、10,000rpmにて10分間遠心分離し、ポリホスフェートグルコキナーゼを含む沈澱を得た。この沈澱を再度180mLの50mM MOPS buffer(pH7)中に懸濁し、酵素保持のため20mLグリセリン(final10%)を加えた。この懸濁液中には357U/mLのポリホスフェートグルコキナーゼが含まれていた。
【0230】
なお、得られた組換えポリホスフェートグルコキナーゼの反応至適温度は65℃であり、反応至適pHは約7.0である。
【0231】
(2.2 組換えホスホグルコムターゼ(PGM)の調製方法)
Escherichia coliに由来するホスホグルコムターゼのヌクレオチド配列(配列番号3)を合成し、発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−PGKを得た。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。
【0232】
その後、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で大腸菌の増殖したコロニーを選択することにより、Escherichia coli由来ホスホグルコムターゼ遺伝子が導入された大腸菌を得た。それらの大腸菌についてホスホグルコムターゼを発現していることを酵素活性測定によって確認した。
【0233】
この大腸菌を、抗生物質カナマイシンを含むTB培地(1.2%トリプトン、2.4%酵母エキス(ともにDifco社製)、0.231%リン酸二水素カリウム、1.254%リン酸水素二カリウム、0.4%グリセリン)600mLに接種し、100rpmで振盪させながら37℃、24時間培養した。次いで、この培養液を5,000rpmにて10分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、180mLの50mM MOPS buffer (pH7)中に懸濁し、次いで1%リゾチームを20mL加え溶菌させた。溶菌後、10,000rpmにて10分間遠心分離し、ホスホグルコムターゼを含む酵素溶液を得た。この酵素溶液には500U/mLのホスホグルコムターゼが含まれていた。
【0234】
なお、得られた組換えホスホグルコムターゼの反応至適温度は60℃であり、反応至適pHは約7.5である。
【0235】
(2.3 組換え馬鈴薯α−1,4−グルカンホスホリラーゼ(GP)の調製方法)
馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子(Nakanoら、Journal of Biochemistry(Tokyo)106(1989)691;配列番号5)を選択マーカー遺伝子Amprとともに発現ベクターpET3d(STRATAGENE社製)に組み込み、プラスミドpET−PGP113を得た。このプラスミドでは、α−1,4−グルカンホスホリラーゼ遺伝子を、イソプロピル−β−D−チオガラクトピラノシド(IPTG)誘導性プロモーターの制御下に作動可能に連結した。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。この大腸菌を、抗生物質アンピシリンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で増殖した大腸菌を選択することにより、馬鈴薯由来α−1,4−グルカンホスホリラーゼ遺伝子が導入された大腸菌を得た。得られた大腸菌がα−1,4−グルカンホスホリラーゼ遺伝子を含むことを、導入された遺伝子の配列を解析することによって確認した。また、得られた大腸菌がα−1,4−グルカンホスホリラーゼを発現していることを、活性測定によって確認した。
【0236】
この大腸菌を、抗生物質アンピシリンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム)1リットルに接種し、120rpmで振盪させながら37℃で3時間振盪培養した。その後、IPTGを0.1mM、ピリドキシンを1mMになるようにそれぞれこの培地に添加し、22℃でさらに20時間振盪培養した。次いで、この培養液を5,000rpmにて5分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、50mlの0.05%のTriton X−100を含む20mM トリス−塩酸緩衝液(pH7.0)中に懸濁し、次いで超音波処理により破砕し、菌体破砕液50mlを得た。この破砕液中には、4.7U/mgのα−1,4−グルカンホスホリラーゼが含まれていた。
【0237】
この菌体破砕液を、55℃で30分間加熱した。加熱後、8,500rpmにて20分間遠心分離し、不溶性のタンパク質などを除去して上清を得た。得られた上清を、あらかじめ平衡化しておいた陰イオン交換樹脂Q−Sepharoseに流してα−1,4−グルカンホスホリラーゼを樹脂に吸着させた。樹脂を、200mM塩化ナトリウムを含む緩衝液で洗浄して不純物を除去した。続いて、タンパク質を300mM塩化ナトリウムを含む緩衝液で溶出させ、組換えα−1,4−グルカンホスホリラーゼ酵素溶液とした。
【0238】
なお、得られた組換えα−1,4−グルカンホスホリラーゼの反応至適温度は40℃であり、反応至適pHは約5.6である。
【0239】
(2.4 組換えセロビオースホスホリラーゼの調製方法)
Clostridium thermocellumの染色体遺伝子を抽出し、これをテンプレートとした。以下の2種の合成DNAプライマー:
合成DNAプライマー1:5’ aaactctagaaataattttgtttaactttaagaaggagatataccatggagttcggtttttttgatgat 3’(配列番号7)および
合成DNAプライマー2:5’ aaactcgagaattacttcaactttgtgagtcttt 3’(配列番号8)
を用い、
98℃で1分間、55℃で1分間、68℃で3分間の順で30サイクル加熱
の条件下でPCRを行うことにより、CBP遺伝子を含む領域を増幅させた。増幅した遺伝子を選択マーカー遺伝子Kmrとともに発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−CBP1を得た。このプラスミドでは、セロビオースホスホリラーゼ遺伝子を、イソプロピル−β−D−チオガラクトピラノシド(IPTG)誘導性プロモーターの制御下に作動可能に連結した。
【0240】
このプラスミドを、大腸菌BL21(DE3)pLysS(STRATAGENE社製)に、コンピテントセル法により導入した。この大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン(Difco社製)、0.5%酵母エキス(Difco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で増殖した大腸菌を選択することにより、Clostridium thermocellum由来セロビオースホスホリラーゼ遺伝子が導入された大腸菌を得た。
【0241】
得られた大腸菌がセロビオースホスホリラーゼ遺伝子を含むことを、導入された遺伝子の配列を解析することによって確認した。また、得られた大腸菌がセロビオースホスホリラーゼを発現していることを、活性測定によって確認した。
【0242】
この大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム)1リットルに接種し、120rpmで振盪させながら37℃で3時間振盪培養した。その後、IPTGを1.0mMになるようにこの培地に添加し、37℃でさらに8時間振盪培養した。次いで、この培養液を5,000rpmにて5分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、50mlの1.4mMの2−メルカプトエタノールを含む50mMリン酸緩衝液(pH7.5)中に懸濁し、次いで超音波処理により破砕し、菌体破砕液50mlを得た。この破砕液中には、132U/mlのセロビオースホスホリラーゼが含まれていた。
【0243】
この菌体破砕液を、55℃で20分間加熱した。加熱後、8,500rpmにて20分間遠心分離し、不溶性のタンパク質などを除去して上清を得た。得られた上清を、あらかじめ平衡化しておいたHis−Tag吸着樹脂Ni−NTA agarose(QIAGEN社製)に流してセロビオースホスホリラーゼをこの樹脂に吸着させた。この樹脂を、300mM塩化ナトリウムと20mMイミダゾールおよび1.4mM2−メルカプトエタノール含む緩衝液で洗浄して不純物を除去した。続いて、タンパク質を300mM塩化ナトリウムと150mMイミダゾールおよび1.4mM 2−メルカプトエタノールを含む緩衝液で溶出させ、組換えセロビオースホスホリラーゼ酵素溶液とした。
【0244】
なお、得られた組換えセロビオースホスホリラーゼの反応至適温度は60℃であり、反応至適pHは6.0である。
【0245】
(実施例1:グルコースからのアミロースの合成)
以下の表1に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを7.0に調整し、そして45℃で24時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約20mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、約0.04であった。
【0246】
【表1】
グルコース(Glc)濃度、グルコース−6−リン酸(G6−P)濃度、グルコース−1−リン酸(G1−P)濃度、およびアミロース収率を経時的に測定した。結果を以下の表2および図2に示す。
【0247】
【表2】
反応開始から12時間以降G6−P濃度とG1−P濃度がほぼ一定になったことから、溶液中の酵素反応は、反応開始から12時間で平衡に達したと考えられる。それに対して、アミロース収率は増加し、グルコース濃度が減少した。反応開始から12時間以降のアミロースの増加はグルコース減少量にほぼ一致している。この結果は、酵素反応が平衡に達した後アミロースが老化することで反応系外に出てそれに伴い平衡状態を保つためにGlcが消費され、反応がアミロース合成側にシフトすることによってアミロースが合成されたことを示すと考えられる。
【0248】
表2および図2においては、反応開始時のグルコース濃度を100%(mM/mM)とし、グルコース濃度、G6−P濃度、G1−P濃度およびアミロース収率をそれぞれ、反応開始時のグルコース濃度(mM)に対する比率として示した。
【0249】
(実施例2:反応開始時の反応溶液のpH)
反応開始時の溶液のpHを5.2〜12に変化させたこと以外は、上記表1の組成と同じ組成の溶液を調製し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。合成されたアミロースの濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、いずれの条件でも約12mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、いずれの条件でも約0.03であった。
【0250】
反応開始時点で測定したpHの値と、アミロースの収率の結果を以下の表3および図3に示す。
【0251】
【表3】
反応溶液の初期pHを弱アルカリ側以上にすることでアミロース合成収率が上がり、約20%の値が得られた。初期pHが5.6までの酸性側ではアミロースが酵素合成されず、弱酸性から中性付近では低収率でアミロースが得られた。初期pH7.1のとき収率9.1%であった。pH7.4からpH11ではアミロース収率15%以上を示し、pH7.4からpH8.5では約19%のアミロース収率を示した。初期pH7.7のとき収率19.6%であった。
【0252】
(実施例3:反応溶液中の遊離リン酸とグルコースとのモル比(Pi/Glc)の検討)
溶液中の遊離のリン酸濃度はアミロース合成反応に大きく影響を与える。直接的にはGP酵素の平衡反応に影響する。この平衡反応ではアミロースとともに副生成物としてリン酸が生成されるためリン酸濃度は高くなるとアミロース合成側への反応が進まなくなる。反応初期には溶液中にリン酸の存在しないことが望ましいが、GlcからG6−Pへの変換反応に必要なポリリン酸には遊離のリン酸が含まれてしまう。そのため、アミロース合成可能な初期遊離リン酸/Glc濃度比(Pi/Glc)を検討した。
【0253】
以下の表4に示す濃度の無機リン酸を添加したこと以外は表1の組成の溶液を作製して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約8に調整した。合成されたアミロースの量を測定し、収率を計算した。添加した無機リン酸の量、無機リン酸とグルコースとのモル比(Pi/Glc)、アミロースの合成濃度(mM)および収率(%)を以下の表4に示す。アミロースの収率(%)とPi/Glcとの関係を図4に示す。
【0254】
【表4】
この結果、グルコースの約0.2倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。
【0255】
(実験例1:グルコースからのG6−Pの生成反応)
先行技術(1)の反応の方向を確認するために、以下の実験を行った。
【0256】
以下の表5に記載の組成の溶液を作成し、反応開始時点でのこの溶液のpHを7に調整し、45℃で24時間インキュベートすることにより、G6−Pを生成させた。
【0257】
【表5】
生成されたG6−Pの濃度を測定し、G6−Pの収率(変換率)(%)を計算した。結果を図5に示す。この結果、先行技術の(1)の反応は不可逆反応であり、グルコースに対して90%以上がG6−Pに変換されることが確認された。
【0258】
(実験例2:G6−PとG1−Pとアミロースとの平衡反応)
アミロースをMOPS(pH7)バッファーに加えて100℃で熱処理して完全に溶解し、その後、45℃に保ったまま、以下の表6に記載の組成になるようにPGM、GP、リン酸などを加え、反応開始時点でのこの溶液のpHを7に調整し、そして4時間インキュベートして、平衡反応を行なった。
【0259】
【表6】
反応途中でサンプリングした。アミロース、G1−PおよびG6−Pの濃度を測定し、反応開始時のアミロースのモル数を100%としたときのアミロース、G1−P、G6−PおよびG1−P+G6−Pの濃度を計算した。結果を図6に示す。その結果、アミロースの95%以上がG6−PとG1−Pに分解された。2種類の酵素PGM−GPの平衡反応はG6−P側に非常に有利で、完全に溶解したアミロースに対してPGMとGPを作用させたとき反応開始から4時間後ではG6−P:88.5%、G1−P:6.8%、アミロース:4.7%の割合で存在した。
【0260】
(実施例4:アミロース合成)
以下の表7に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約12mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、0.03であった。
【0261】
【表7】
アミロースの合成濃度を測定し、収率を計算した。その結果、収率は19.6%であった。上記の実験例2によれば、平衡状態でのアミロース収率が約4.7%であった。これに対し、本実施例では、500mMのグルコースを基質としたアミロース合成の収率が19.6%であった。この値は、本実施例によって得られるアミロースの収率が、PGM−GP平衡反応によって得られるアミロース収率を大きく上回ることを示す。本実施例によってPGM−GP平衡反応によるアミロース収率よりも高い値が得られた理由は、アミロースの老化によって反応系外に出たアミロースが蓄積されたためであると考えられる。
【0262】
(実施例5:出発グルコース濃度を変えた場合のアミロース収率の変化)
グルコース濃度を変化させ、それに合わせて酵素量、プライマー濃度およびポリリン酸濃度を比例して増加させたこと以外は、表1と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約0.5〜約50.2であり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、約0.03であった。
【0263】
結果を以下の表8および図7Aおよび図7Bに示す。
【0264】
【表8】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図7A)。また、グルコース濃度が増加するに従ってアミロースの収量も増加した(図7B)。
【0265】
(実施例6:G6−Pを出発原料として使用した、ゲスト化合物の存在下でのアミロース合成)
以下の表9に記載の組成をベースとして以下の表10に記載の量で各種ゲスト化合物を添加した組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、0mMであり、遊離リン酸のモル濃度とG6−Pのモル濃度との比率(Pi/G6−P)は、0であった。コントロールは、ゲスト化合物を添加しない比較例6である。合成されたアミロース量を測定し、収率を計算した。
【0266】
【表9】
結果を以下の表10および図8に示す。
【0267】
【表10】
アミロースは脂肪酸などを包接する作用を持ち、包接される側の物質(ゲスト化合物)の種類によっては沈澱しやすくなる。アミロース合成反応溶液中にゲスト化合物を添加し、この沈澱促進作用を利用することで、アミロース収率を上げることができる。G6−Pからアミロースを合成するとき、アミロース収率は20%程度であるが、ゲスト化合物を加えることで有意に収率が上昇した。ゲスト化合物SB3−16を10mM添加したときアミロース収率は53.3%まで上がった。
【0268】
(実施例7−1〜7−3:セロビオースからのアミロース合成)
以下の表11に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを約7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。比較例7として、PGK、PGMおよびポリリン酸を含まず、リン酸を含む組成でアミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例7−1では約15mMであり、実施例7−2および7−3では約10mMであり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例7−1では約0.15であり、実施例7−2では約0.10、7−3では約0.057であった。合成されたアミロースの濃度を測定し、収率を計算した。
【0269】
【表11】
結果を以下の表12に示す。
【0270】
【表12】
セロビオースを出発原料としたときのアミロース合成収率は従来の方法(比較例7)では16.3%であったが、PGK、PGMを共存させることで約40%まで上昇し(実施例7−1)、さらに基質濃度を上げ、初期pHを弱アルカリ側にすることでアミロース収率が66.8%にあがった(実施例7−3)。
【0271】
(実施例8−1から8−3:種々のpH条件下での、セロビオースを出発物質としたアミロース合成)
反応開始時のpHの影響を調べるために、以下の表13に記載の組成の溶液を調製し、反応開始時の溶液のpHをそれぞれ5.6、7.0または7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例8−1から8−3では約10mMであり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例8−1および8−2では約0.10であり、実施例3では約0.057であった。合成されたアミロースの量を測定し、収率を計算した。
【0272】
反応開始時点で測定したpHの値と、収率の結果を以下の表14および図9に示す。
【0273】
【表13】
【0274】
【表14】
この結果、反応溶液の初期pH値が高くなるほど、アミロース収率が上がることがわかった。
【0275】
(実施例9:反応溶液中の遊離リン酸とセロビオースとのモル比(Pi/CB)の検討)
以下の表15に示す組成の溶液を作製して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約7.8に調整した。合成されたアミロースの濃度を測定し、収率を計算した。添加した無機リン酸の量、無機リン酸とセロビオースとのモル比(Pi/CB)、アミロースの収率(%)を以下の表16に示す。結果を図10に示す。
【0276】
【表15】
【0277】
【表16】
この結果、セロビオースの約4倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。
【0278】
(実施例10:出発セロビオース濃度を変えた場合のアミロース収率の変化)
セロビオース濃度を変化させ、それに合わせて酵素量およびプライマー濃度を比例して増加させたことと実施例10−1のポリリン酸濃度が1%であること以外は、実施例7−2と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース量を測定し、収率を計算した。実施例10−2は、実施例7−2と同じ条件での反応であり、実施例10−3は、実施例7−3と同じ条件での反応であった。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例10−1では約2.8であり、実施例10−2および10−3では約11.2あり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例10−1では0.140であり、実施例10−2では約0.112であり、実施例10−3では0.064であった。
【0279】
結果を以下の表17および図11に示す。
【0280】
【表17】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図11)。
【0281】
(実施例11:種々のpH条件下での、G6−Pを出発物質としたアミロース合成)
反応開始時の溶液のpHを6.7〜8.5に変化させたこと以外は上記表9と同じ組成の溶液を調製し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、いずれの条件でも0mMであり、遊離リン酸のモル濃度とG6−Pのモル濃度との比率(Pi/G6−P)は、いずれの条件でも0であった。合成されたアミロースの濃度を測定し、収率を計算した。
【0282】
反応開始時点で測定したpHの値と、収率の結果を以下の表18および図12に示す。
【0283】
【表18】
反応溶液の初期pHを弱アルカリ側以上にすることでアミロース合成収率があがり約30%の値を得た。初期pHが6.7までの酸性側ではアミロースがほとんど合成されず、弱酸性から中性付近では低収率でアミロースを得た。初期pH7.1のとき収率6.37%であった。pH7.3からpH8.5ではアミロース収率10%以上を示し、pH7.6からpH8.5では約24%以上のアミロース収率を示した。初期pH8.4のとき収率30.51%であった。
【0284】
(実施例12:反応溶液中の遊離リン酸とグルコース−6−リン酸とのモル比(Pi/G6−P)の検討)
以下の表19に示す濃度の無機リン酸を添加したこと以外は表9の組成の溶液を作成して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約8に調整した。合成されたアミロースの濃度を測定し、収率を計算した。添加した無機リン酸の濃度、無機リン酸とグルコース−6−リン酸とのモル比(Pi/G6−P)、アミロースの濃度(mM)および収率(%)を以下の表19に示す。結果を図13に示す。
【0285】
【表19】
この結果、グルコース−6−リン酸の約0.5倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。また、遊離リン酸の濃度が高いほど、収率が下がった。従って、この反応では、遊離リン酸をできる限り添加しないことが好ましい。
【0286】
(実施例13:出発グルコース−6−リン酸濃度を変えた場合のアミロース収率の変化)
グルコース−6−リン酸濃度を変化させ、それに合わせて酵素量およびプライマー濃度を比例して増加させたこと以外は、表9と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約0であり、遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)は、約0であった。
【0287】
結果を以下の表20、図14Aおよび図14Bに示す。
【0288】
【表20】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図14A)。
【産業上の利用可能性】
【0289】
本発明により、安価で容易に入手可能なグルコースからα−グルカンを効率よく製造することができる。
【0290】
以上のように、本発明の好ましい実施形態を用いて本発明を例示してきたが、本発明は、この実施形態に限定して解釈されるべきものではない。本発明は、特許請求の範囲によってのみその範囲が解釈されるべきであることが理解される。当業者は、本発明の具体的な好ましい実施形態の記載から、本発明の記載および技術常識に基づいて等価な範囲を実施することができることが理解される。本明細書において引用した特許、特許出願および文献は、その内容自体が具体的に本明細書に記載されているのと同様にその内容が本明細書に対する参考として援用されるべきであることが理解される。
【図面の簡単な説明】
【0291】
【図1】図1は、本願発明で利用する種々の酵素反応の概略を示す図である。
【図2】図2は、グルコースを出発物質とした場合の、グルコース(Glc)量、グルコース−6−リン酸(G6−P)量、グルコース−1−リン酸(G−1−P)量、およびアミロースの合成量の経時変化を示すグラフである。
【図3】図3は、グルコースを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図4】図4は、グルコースを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/グルコース濃度比によるアミロース収率の変化を示すグラフである。
【図5】図5は、グルコースおよびポリホスフェートグルコキナーゼを含む溶液をインキュベートした場合にグルコース−6−リン酸に変換された割合を示すグラフである。
【図6】図6は、アミロース、ホスホグルコムターゼおよびα−1,4−グルカンホスホリラーゼを含む溶液をインキュベートした場合のアミロース量、グルコース−1−リン酸量、グルコース−6−リン酸量および(グルコース−1−リン酸量とグルコース−6−リン酸量との合計)を示すグラフである。
【図7A】図7Aは、グルコースを出発物質とした場合の、グルコース濃度とアミロースの収率(%)との関係を示すグラフである。
【図7B】図7Bは、グルコースを出発物質とした場合の、グルコース濃度とアミロースの収量(mM)との関係を示すグラフである。
【図8】図8は、種々の包接化合物を使用し、グルコースを出発物質とした場合の、アミロースの収率を示すグラフである。
【図9】図9は、セロビオースを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図10】図10は、セロビオースを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/セロビオース濃度比によるアミロース収率の変化を示すグラフである。
【図11】図11は、G6−Pを出発物質とした場合の、セロビオース濃度とアミロースの収率(%)との関係を示すグラフである。
【図12】図12は、G6−Pを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図13】図13は、G6−Pを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/G6−P濃度比によるアミロース収率の変化を示すグラフである。
【図14A】図14Aは、G6−Pを出発物質とした場合の、G6−P濃度とアミロースの収率(%)との関係を示すグラフである。
【図14B】図14Bは、グルコースを出発物質とした場合の、G6−P濃度とアミロースの収量(mM)との関係を示すグラフである。
【図15】図15は、本発明で使用され得る種々の方法をまとめて示す模式図である。
【配列表フリーテキスト】
【0292】
配列番号1:Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのヌクレオチド配列;
配列番号2:Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのアミノ酸配列;
配列番号3:Escherichia coliに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号4:Escherichia coliに由来するホスホグルコムターゼのアミノ酸配列;
配列番号5:馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子のヌクレオチド配列;
配列番号6:馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子のアミノ酸配列;
配列番号7:合成DNAプライマー1のヌクレオチド配列;
配列番号8:合成DNAプライマー2のヌクレオチド配列;
配列番号9:Thermobifida fuscaに由来するポリホスフェートグルコキナーゼのヌクレオチド配列;
配列番号10:Thermobifida fuscaに由来するポリホスフェートグルコキナーゼのアミノ酸配列;
配列番号11:Thermus thermophilusに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号12:Thermus thermophilusに由来するホスホグルコムターゼのアミノ酸配列;
配列番号13:Thermotoga lettingaeに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号14:Thermotoga lettingae由来のホスホグルコムターゼのアミノ酸配列。
【技術分野】
【0001】
本発明は、グルコースからα−グルカンを製造する方法に関する。特定の実施形態では、本発明は、セロビオースからα−グルカンを製造する方法に関する。
【背景技術】
【0002】
人間は、デンプンなどのα−グルカンを消化してエネルギー源として利用している。そのため、α−グルカンは、人類にとってエネルギー源として非常に重要である。さらに、α−グルカンは、食品産業をはじめ、医薬、化粧品、化学工業、製紙、繊維などにおける原料としても幅広く利用されている。特に、アミロースは、種々の機能を有するので、幅広い分野での利用が期待されている。
【0003】
近年、人口増加により食糧危機が問題視されており、植物の生産するデンプンだけでは将来エネルギー源が不足すると予想されている。
【0004】
一方、人間は、セルロースなどのβ−グルカンを消化できないので、エネルギー源として利用することができず、β−グルカンは、食物繊維成分としてのみ利用されている。それゆえ、β−グルカンを食糧危機問題の解決には利用できない。しかし、β−グルカンの年間生産量は、デンプンの約2万倍と推定されており、枯渇の心配はない。そのため、β−グルカンを、人間がエネルギー源とし得る物質に変換する種々の試みが行われている。
【0005】
例えば、セルロースをグルコースまで分解し、エタノール醗酵に利用することが検討されている。グルコースは、セルロース以外にも、種々の糖類を変換することにより入手することができる。グルコースは安価で大量に入手可能な原料である。グルコースは人間によって代謝され得るが、甘すぎるため、エネルギー源として大量に摂取することができない。また、グルコースは極めて水溶性が高いため、水分を含む食品中では固形物としてのテクスチャー(食感)を持つことができない。
【0006】
他方、α−グルコースの直鎖状ポリマーであるα−グルカン(例えば、デンプン)は、通常人間によって摂食されており、大量摂取することが可能である。それゆえ、グルコースを原料として、α−グルカンのようなテクスチャーを持ちかつ甘くない素材を合成することができれば、非常に有用である。
【0007】
さらに、上記のように、セルロースはバイオマス資源として豊富に存在し、枯渇の心配がない。そのため、セルロース資源を原料としたα−グルコースポリマーの合成技術が開発されれば食糧危機問題の解決に大きな貢献ができる。しかし、これまでにそのような技術は開示されていない。
【0008】
本願発明で利用する種々の酵素反応の従来技術について以下に説明する。
【0009】
図1の(1)に示すように、グルコースおよびポリリン酸にポリホスフェートグルコキナーゼ(PGK)を作用させると、グルコース−6−リン酸(G6−P)が生成する。この反応は公知であり、例えば、非特許文献1および2に記載されている。
【0010】
図1の(2)に示すように、グルコースまたはグルコース−1,6−ビスホスフェートをコファクターとし、G6−Pにホスホグルコムターゼ(PGM)が作用することによりグルコース−1−リン酸(G1−P)が生成する。この反応は公知であるが、これは、植物体内におけるデンプン合成反応や動物の肝臓や筋肉細胞あるいは微生物におけるグリコーゲン合成反応の一部であり、進行するためにエネルギーを必要とし、in vitroにおけるG1−P合成方法としての先行技術はない。逆にG1−PにPGMを作用させることによりG6−Pを生成する技術は多く存在する。PGMによる酵素反応は平衡反応であるが、G6−Pが非常に安定であるために、99%以上がG6−Pとなる(非特許文献3を参照のこと)。すなわち、(2)の反応は、G6−P生成側に非常に大きく傾いている。このことを利用して、G1−P濃度をG6−P経由で測定する方法が確立されている(非特許文献4を参照のこと)。しかし、G6−PからG1−Pを生成することはin vitroでは行なわれていなかった。G6−Pから得られたG1−Pを基質としてアミロースを合成することはまったく考えられてこなかった。
【0011】
図1の(3)に示すように、G1−PとプライマーにGPを作用させることによりアミロースが生産される。この技術は公知であり、例えば、非特許文献5および特許文献1に記載されている。特許文献1に記載される反応の概略を以下に示す:
【0012】
【化1】
この反応において、他の酵素が存在しなければ、α−グルカンはアミロースとなり、ブランチングエンザイム(BE)、ディスプロポーショネーティングエンザイム(MalQ)などの他の酵素が存在すると、α−グルカンは、デンプン、グリコーゲンなどとなる。
【0013】
グルコースを出発原料とし、PGKとPGMとGPという3種類の酵素を同時に作用させる1つの反応系で直接アミロースを生産する方法は知られていない。
【0014】
セロビオースの利用に関しては、セロビオースを原料に、1つの反応系でα−グルカンを製造する技術が知られている(特許文献2)。特許文献2の方法では、セロビオースの一部をG1−Pとグルコースに変換し、得られたG1−Pにα−1,4−グルカンホスホリラーゼを作用させてα−グルカンに変換している。特許文献2の方法では、2つの平衡反応をカップリングさせるため、リン酸はリサイクルされて蓄積しないが、グルコースは反応に利用されないため、反応中に蓄積し続ける。それゆえ、特許文献2の方法では、平衡反応をα−グルカン合成側に向かわせる目的で、グルコースをフラクトースに変換するかもしくはグルコースを反応系から消去する方法をとっている。そのため、特許文献2の方法では、α−グルカンに変換されるグルコース源はG1−Pに由来するグルコース部分のみであり、セロビオースに対するα−グルカン収率は最大でも50%以下であった。
【特許文献1】国際特許公開第02/097107号パンフレット
【特許文献2】国際特許公開第2005/05681号パンフレット
【非特許文献1】Simon J. Pilkis, Irene T. Weber, Robert W. Harrison, Graeme I. Bell (1994) Glucokinase : Structural analysis of a protein involved in susceptibility to diabetes. J. boil. chem. Vol. 269. No. 35. pp. 21925−21928
【非特許文献2】Shotaro Tanaka, Sun−Og Lee, Kazuhiro Hamaoka, Junichi Kato, Noboru Takiguchi, Kazunori Nakamura, Hisao Ohtake, Akio Kuroda (2003) Strictly polyphosphate−dependent glucokinase in a polyphosphate−accumulating bacterium, Microlunatus phosphovorus. J. Bacteriol. Vol. 185. No. 18. pp. 5654−5656
【非特許文献3】William J. Ray, Jr., and Gertrude A. Roscelli (1964)The phosphoglucomutase pathway. J. boil. chem.Vol. 239. No. 11. pp. 3935−3941
【非特許文献4】G. Michael (1974) D−Glucose 1−phosphate in methods of enzymatic analysis 2nd English ed. (H. U. Bergmeyer, ed) Vol. 4, pp.185−191, Academic Press, New York
【非特許文献5】Micheal J. Gidley and Paul V. Bulpin (1989) Aggregation of amylase in Aqueous system: The effect of chain length on phase behavior and aggregation kinetics. Macromolecules, 22, pp. 341−346
【発明の開示】
【発明が解決しようとする課題】
【0015】
本発明は、上記問題点の解決を意図するものであり、グルコースからα−グルカンを製造する方法を提供することを目的とする。
【課題を解決するための手段】
【0016】
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、ポリホスフェートグルコキナーゼおよびポリリン酸を用いてグルコースをほぼ100%、G6−Pに変換し、生成したG6−PをG1−Pに変換することにより、これまでアミロース合成の阻害剤となっていた副産物グルコースを除去するのではなく、積極的に利用できることを見出した。本発明によれば、グルコースを基質としてアミロース合成に利用することができ、セロビオースを理論的に100%アミロースに変換できる。本発明者らは、この知見に基づいて本発明を完成させた。
【0017】
本発明により、グルコースおよびポリリン酸を基質とし、3種類の酵素(ポリホスフェートグルコキナーゼ、ホスホグルコムターゼおよびα−1,4−グルカンホスホリラーゼ)を1つの反応溶液中で作用させることを含む、アミロースを合成する方法が提供される。
【0018】
グルコースを原料としてアミロースを合成する技術はこれまでにない。従来の方法でグルコースからアミロースを合成するためには、(1)グルコースとポリリン酸にPGKを作用させてG6−Pを得て;(2)得られたG6−PにPGMを作用させてG1−Pを生成し、そして(3)得られたG1−PにGPを作用させてアミロースを合成する。この3段階の方法の理論上のアミロース合成収率は、主に(2)および(3)の平衡反応の収率に基づく。(2)の反応(PGMによるG6−P⇔G1−P平衡反応)は、G6−P側に大きく偏っており、その比率はG6−P : G1−P =94.2 : 5.8である。(3)の反応(GPによるアミロース合成反応)では、アミロース : G1−P=73:27である。また、(1)の反応は不可逆反応であり、ポリリン酸濃度を調節することにより、ほぼ100%のグルコースがG6−Pに変換され得る。以上のことから、計算すると(1)の反応と、(2)の反応と、(3)の反応とをそれぞれ別個の工程で行なった場合の理論上のアミロース収率は、グルコースに対して約4.2%と算出される。
【0019】
グルコースからアミロースができないかまたは収率が非常に低い原因は2点考えられる。1点目は、反応溶液中の遊離リン酸濃度とG1−P濃度との比率の影響である。2点目は、このグルコースからのアミロース合成が平衡収支に逆らった反応であることである。本発明ではグルコースからアミロースを合成するために、溶液中のグルコースと遊離リン酸との濃度比を調節することと、物質のポテンシャルエネルギーに逆らいアミロースを獲得するためにアミロースの老化(沈澱)を利用することで問題解決を図った。
【0020】
まず1点目について、アミロースが得られない原因は、(3)の反応(GPによるアミロース合成反応)がG1−Pと遊離のリン酸との濃度比に大きく影響されることによる。アミロース合成反応に対するG1−P/リン酸濃度比の影響を検討したところ、遊離リン酸の濃度がG1−P濃度の4倍以上のときアミロースが合成されないことがわかった。本発明の方法においては、一般に、最初の反応液にリン酸を意図的に添加しない。しかし、一般に、通常の方法で工業的に製造されるポリリン酸、あるいは一般に工業的に市販されているポリリン酸には、重合していないモノマーのリン酸、すなわち遊離のリン酸が不純物として少量含まれている。そのため、リン酸を意図的に添加しなくても、(1)の反応で用いるポリリン酸として、工業的に入手可能なポリリン酸を用いると、その中に少量の不純物として遊離のリン酸が含まれていることが多い。この、ポリリン酸溶液由来の遊離リン酸の濃度がグルコースからアミロースを合成するときの中間生成物として生じるG1−P濃度の4倍を超えるとアミロースが合成されない。そのため、ポリリン酸溶液由来の遊離リン酸が生成されるG1−P濃度の4倍濃度より小さくなるようにグルコース濃度およびポリリン酸濃度を調節することが好ましい。
【0021】
2点目の原因はG6−Pが非常に安定であることに起因する。(2)の平衡反応は上記したようにG6−P側に大きく偏り、平衡反応は「酵素ハンドブック」丸尾文治、田宮信雄監修 朝倉書店によるとG6−P生成側に大きくよることが記載されている。それに対して(3)の平衡反応ではアミロース合成側に偏っている。従って、(2)の反応と(3)の反応を1つの溶液中で同時に行うと、G1−PからG6−Pへの転移反応と、G1−Pからアミロースへの合成反応が競合すると考えられる。(2)の反応および(3)の反応は平衡反応であり、同時に反応させた場合、ひとつの平衡反応と見ることができる。このときの平衡収支は、G6−Pとアミロースのポテンシャルエネルギーに依存する。実験例2に示すように、G6−Pとアミロースではどちらが安定しているのかを検討するために、低濃度アミロースの溶解液にPGM、GPおよびリン酸を加えて反応させた。その結果、図6に示すように、アミロースの95%以上がG6−PとG1−Pに分解された。この結果より、溶液に溶けている状態のアミロースでは、PGMとGPとの平衡反応により、G6−Pへの変換方向に強くシフトされることが確認され、アミロースと比べてもG6−Pが安定であると考えられる。
【0022】
しかし、本願発明の方法においては、400mMのグルコースを原料とし、ポリリン酸および酵素の濃度、pH等を適切に調整することにより、7%程度のアミロース合成収率を得ることができた。この値は、PGM−GP平衡状態におけるアミロースの割合を超える。この原因はアミロースの老化現象に起因すると考える。老化しやすい条件として基質濃度を500mMに上げアミロース合成反応をおこなうとアミロース収率は19.6%にまで上昇した。さらにアミロース合成反応の経時的な測定によって酵素反応が平衡に達した後、老化によるものと考えられるアミロースの増加を測定できた。よって、グルコースからアミロースを合成するためには、合成したアミロースを老化させることが非常に有用であると考えられる。
【0023】
さらに、本願発明の方法を使用することにより、セロビオースから効率よくα−グルカンに変換することが可能となった。上記のように、従来の方法では、セロビオースの一部をG1−Pとグルコースに変換し、得られたG1−Pにα−1,4−グルカンホスホリラーゼを作用させることにより、α−グルカンを得ていた。従来の方法では、グルコースは蓄積し続けるので、平衡反応をα−グルカン合成側に向かわせる目的で、グルコースをフラクトースに変換するかもしくは反応系から消去する方法をとっている。そのため、α−グルカンに変換されるグルコース源はG1−Pに由来するグルコース部分のみであり、セロビオースに対するα−グルカン収率は最大でも50%以下であった。
【0024】
我々は、ポリホスフェートグルコキナーゼとポリリン酸を用いてグルコースを100%G6−Pに変換して、生成したG6−PをG1−Pに変換することで、これまでアミロース合成の阻害となっていた副産物グルコースを消去するのではなく、積極的に基質としてアミロース合成に利用する技術を検討し、理論的にセロビオースを100%アミロースに変換できる技術を完成させた。
【0025】
上記目的を達成するために、本発明は、例えば、以下の手段を提供する:
(項目1)
グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、
反応開始時の該溶液のpHは、pH5.7〜pH12であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【0026】
(項目2)
前記反応開始時の前記溶液中のグルコース濃度が200mM〜2000mMである、請求項1に記載の方法。
【0027】
(項目3)
前記反応開始時の前記溶液中のグルコース濃度が500mM〜2000mMである、請求項1に記載の方法。
【0028】
(項目4)
前記反応開始時の前記溶液のpHが、pH7.4〜pH11である、請求項1〜3のいずれか1項に記載の方法。
【0029】
(項目5)
前記α−グルカンがアミロースである、項目1〜4のいずれか1項に記載の方法。
【0030】
(項目6)
前記溶液が、ゲスト化合物を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目1〜5のいずれか1項に記載の方法。
【0031】
(項目7)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目6に記載の方法。
【0032】
(項目8)
グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース−6−リン酸濃度は、20mM〜2000mMであり、
反応開始時の該溶液のpHは、pH6.7〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)が0.4以下である、方法。
【0033】
(項目9)
前記反応開始時の前記溶液中のグルコース−6−リン酸濃度が100mM〜2000mMである、請求項8に記載の方法。
【0034】
(項目10)
前記反応開始時の前記溶液のpHが、pH7.3〜pH11である、請求項8または9に記載の方法。
【0035】
(項目11)
前記α−グルカンがアミロースである、項目8〜10のいずれか1項に記載の方法。
【0036】
(項目12)
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目8〜11のいずれか1項に記載の方法。
【0037】
(項目13)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目12に記載の方法。
【0038】
(項目14)
セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法であって、
反応開始時の該溶液中のセロビオース濃度は、100mM〜350mMであり、
反応開始時の該溶液のpHは、pH5〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)が2.0以下である、方法。
【0039】
(項目15)
前記反応開始時の前記溶液中のセロビオース濃度が120mM〜350mMである、請求項14に記載の方法。
【0040】
(項目16)
前記反応開始時の前記溶液のpHが、pH5.6〜pH11である、請求項14または15に記載の方法。
【0041】
(項目17)
前記α−グルカンが、アミロースである、項目14〜16のいずれか1項に記載の方法。
【0042】
(項目18)
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、項目14〜17のいずれか1項に記載の方法。
【0043】
(項目19)
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、項目18に記載の方法。
【発明の効果】
【0044】
本願発明の方法により、大量摂取が不可能なグルコースを、大量摂取可能な食品へと変換できる。また、本発明は、食糧危機問題の解決にも大きく貢献する。
【0045】
特定の実施形態では、本願発明の方法により、非消化性のセルロースを、可食性の食品へと変換できる。また、本発明は、食糧危機問題およびゴミ問題の解決にも大きく貢献する。
【0046】
グルコースとアミロースとでエネルギー状態を比較すると、アミロースの方がエネルギー準位が高い。そのため、グルコースからアミロースを合成するのは、エネルギー論的に困難である。
【0047】
図1の(1)、(2)、(3)の反応を個別に反応させた場合、(1)の収率はほぼ100%であり、(2)の収率は約7%であり、(3)の収率は約70%である。そのため、グルコースからのアミロースの理論収率は、約5%である(100×0.058×(0.73)=4.2(%))である。それに対して、(1)〜(3)の反応を1つの溶液中で同時に反応させた場合、(a)溶液のpHを調整することおよび(b)溶液中の基質の濃度を高くすること、によって、高いアミロース収率を上げることができる。
【0048】
詳細には、反応溶液の初期pH(反応開始時の溶液のpH)を好ましくはpH7.4〜pH11に、さらに好ましくはpH7.4〜8.5にする。反応で使用する各酵素の代表的な至適pHは、PGKが約pH7.0であり、PGMが約pH7.5であり、そしてGPが約pH5.6である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHをpH7.5からpH11に調整することにより、約15%以上のアミロース収率を得られる。
【0049】
さらに、基質濃度を高くすることにより、アミロースが老化して沈澱し、反応系から外れやすくなる。アミロースが沈澱すると反応系内のアミロース濃度が低下し、その結果、GPによるアミロース合成が促進される。それに伴い、PGM平衡反応によってG1−Pが生成される。合成アミロースを効率よく老化させることにより、G1−Pが連続的に供給され、アミロース収率を約20%程度まで向上させることができる。これは、3つの反応を個別に反応させた場合の理論上のアミロース収率の約5倍であり、極めて顕著な効果である。
【0050】
さらに、ゲスト物質の存在下で合成反応を行なうことにより、アミロースの沈澱形成が促進される。特定のゲスト物質の存在下では、アミロース収率を約35%まで向上させることができた。
【0051】
特許文献2に記載される方法では、セロビオースにセロビオースホスホリラーゼ(CBP)を作用させると、目的の生成物G1−Pの他に同量の副生成物グルコース(Glc)が生成される。そのため、特許文献2に記載の方法では、アミロースの収率の理論的上限は、50%であった。実際にはCBPの平衡反応がセロビオース合成側に有利であるため、アミロースの収率は16%程度であり、ムタロターゼ、グルコースオキシダーゼおよびペルオキシダーゼをグルコースに作用させて副生成物のGlcを除去することでアミロースの最大収率は32.4%まであがった。
【0052】
それに対して、本願発明において、セロビオースからのアミロースの合成反応と(1)および(2)の反応を組み合わせた場合、グルコースをアミロース合成に利用することが可能となり、その結果、アミロース収率の理論的上限を100%にすることができる。本明細書中に記載した実施例においては、反応溶液の初期pHを調整することにより、実際に最大66.8%のアミロース収率を得ることができた。
【発明を実施するための最良の形態】
【0053】
以下、本発明を詳細に説明する。
【0054】
本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当該分野で通常用いられる意味で用いられることが理解されるべきである。
【0055】
本明細書中では「α−グルカン」とは、D−グルコースを構成単位とする糖であって、α−1,4−グルコシド結合によって連結された糖単位を少なくとも2糖単位以上有する糖をいう。α−グルカンは、直鎖状、分岐状または環状の分子であり得る。直鎖状α−グルカンとα−1,4−グルカンとは同義語である。直鎖状α−グルカンでは、α−1,4−グルコシド結合によってのみ糖単位の間が連結されている。α−1,6−グルコシド結合を1つ以上含むα−グルカンは、分岐状α−グルカンである。α−グルカンは、好ましくは、直鎖状の部分をある程度含む。分岐のない直鎖状α−グルカンがより好ましい。本発明で製造されるα−グルカンは、好ましくは、アミロース、環状構造を有するグルカンまたは分岐構造を有するグルカンであり、より好ましくはアミロースである。1分子のα−グルカンに含まれる糖単位の数を、このα−グルカンの重合度という。
【0056】
α−グルカンは、場合によっては、分岐の数(すなわち、α−1,6−グルコシド結合の数)が少ないことが好ましい。このような場合、分岐の数は、代表的には0〜10000個、好ましくは0〜1000個、より好ましくは0〜500個、さらに好ましくは0〜100個、さらに好ましくは0〜50個、さらに好ましくは0〜25個、さらに好ましくは0個である。
【0057】
本発明の方法によって製造される分岐状α−グルカンでは、α−1,6−グルコシド結合を1としたときのα−1,6−グルコシド結合の数に対するα−1,4−グルコシド結合の数の比は、好ましくは1〜10000であり、より好ましくは10〜5000であり、さらに好ましくは50〜1000であり、さらに好ましくは100〜500である。
【0058】
α−1,6−グルコシド結合は、α−グルカン中に無秩序に分布していてもよいし、均質に分布していてもよい。α−グルカン中に糖単位で5個以上の直鎖状部分ができる程度の分布であることが好ましい。
【0059】
α−グルカンは、D−グルコースのみから構成されていてもよいし、α−グルカンの性質を損なわない程度に修飾された誘導体であってもよい。修飾されていないことが好ましい。α−グルカンの性質を損なわない程度の修飾としては、エステル化、エーテル化、架橋などが挙げられるが、これらに限定されない。これらの修飾は、当該分野で公知の方法に従って行われ得る。
【0060】
α−グルカンは、代表的には約1×103以上、好ましくは約5×103以上、より好ましくは約1×104以上、さらに好ましくは約5×104以上、さらに好ましくは約1×105以上の分子量を有する。α−グルカンは、代表的には約1×106以下、好ましくは約5×105以下、さらに好ましくは約1×105以下の分子量を有する。
【0061】
当業者は、本発明の製造方法で用いられる基質(例えば、プライマー、グルコース、グルコース−6−リン酸、セロビオースなど)の量、酵素の量、反応時間などを適宜設定することによって所望の分子量のα−グルカンが得られることを容易に理解する。
【0062】
<α−グルカンの製造に用いる材料>
一つの実施形態では、本発明の製造方法では、例えば、グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を用いる。
【0063】
別の実施形態では、本発明の製造方法では、例えば、グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を用いる。
【0064】
さらに別の実施形態では、本発明の製造方法では、例えば、セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を用いる。
【0065】
これらの方法で用いる溶液の調製においては、例えば、これらの各物質と、緩衝剤および溶媒を主な材料として用いる。これらの材料は通常、反応開始時に全て添加されるが、反応の途中でこれらのうちの任意の材料を追加して添加してもよい。
【0066】
本発明の製造方法では、溶液中にゲスト化合物をさらに含み得る。
【0067】
本発明の製造方法では、必要に応じて、枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素を用いることができる。枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素は、目的とするα−グルカンの構造に応じて、本発明の製造方法の最初から溶液中に添加してもよく、途中から溶液中に添加してもよい。
【0068】
(1.グルコース)
グルコースは、C6H12O6で示される、分子量約180の単糖である。グルコースは、代表的なアルドヘキソースであり、天然に広く分布する単糖である。グルコースは、甘い果実および他の植物組織中に遊離型で多量に存在する。グルコースは、スクロース、マルトース、ラクトースなどのオリゴ糖;澱粉、セルロース、デキストラン、ラミナリン、グリコーゲンなどの多糖;種々の配糖体などの構成成分として多量に存在する。これらの物質を酸で加水分解するとD−グルコースを生じる。本発明で使用するグルコースは、純粋なものであってもよく、ある程度不純なものであってもよい。グルコースは、グルコースを構成成分とする種々の物質を分解することによって入手されたものであってもよい。例えば、セルロースは、グルコースがβ結合で連結したものであり、セルラーゼによって加水分解することにより、グルコースが生成される。例えば、デンプンをアミラーゼなどの酵素によって加水分解することにより、グルコースが生成される。さらに、フラクトースをグルコースイソメラーゼによって変換することにより、グルコースが生成される。本発明で使用されるグルコースは、特定の実施形態では、グルコースとして溶液に添加されることが好ましい。別の実施形態では、分解または変換されることによってグルコースを生じる物質を反応溶液中に添加することによって、グルコースが提供されてもよい。
【0069】
(2.ポリリン酸)
本明細書において使用される場合、用語「ポリリン酸(polyphosphate)」とは、ポリリン酸(polyphosphoric acid)およびその塩をいう。酸である狭義のポリリン酸は、オルトリン酸の脱水縮合によって生じる直鎖状高分子リン酸である。狭義のポリリン酸は、一般式Hn+2PnO3n+1(n=2以上の整数)によって示される。塩であるポリリン酸(すなわち、ポリリン酸塩)は、一般式MIn+2PnO3n+1(n=2以上の整数)によって示される。本明細書において、特に断りの無い限り、「ポリリン酸」は、広義のポリリン酸、すなわち狭義のポリリン酸およびその塩を示す。狭義のポリリン酸の例としては、二リン酸、三リン酸、四リン酸、五リン酸などが挙げられる。狭義のポリリン酸は、例えば、(1)リン酸を加熱する方法、または(2)リン酸に五酸化二リンを添加する方法によって製造され得る。一般に、それぞれのポリリン酸を純品として製造することは困難であるので、種々の重合度のポリリン酸の混合物が市販されている。ポリリン酸の水溶液は容易に加水分解して低分子量のポリリン酸を経て、最終的にはオルトリン酸になる。本発明で使用されるポリリン酸の重合度は、好ましくは2以上であり、より好ましくは5以上である。本発明で使用されるポリリン酸の重合度は、好ましくは20以下であり、より好ましくは12以下である。
【0070】
ポリリン酸もまた、陽イオンを含む水溶液中で使用される。この場合、酸であるポリリン酸と塩であるポリリン酸とポリリン酸のイオンとが水溶液中で共存するので、酸であるポリリン酸と塩であるポリリン酸とは区別をしにくい。そのため、塩であるポリリン酸は、ポリリン酸と同様に使用され得る。本発明において、ポリリン酸塩は、好ましくはポリリン酸の任意の塩であり、より好ましくはポリリン酸の金属塩である。ポリリン酸塩の好ましい具体例としては、ポリリン酸ナトリウム、ポリリン酸カリウム、ポリリン酸アンモニウム、ポリリン酸カルシウム、ポリリン酸第2鉄などが挙げられる。市販のポリリン酸ナトリウムは、一般に、トリポリリン酸ナトリウムを主成分とし、少量のピロリン酸塩、トリメタリン酸塩および非結晶性の高重合リン酸塩が混在する。市販のポリリン酸カリウムは、一般に、トリポリリン酸カリウム(K5P3O10)を主成分とする。
【0071】
ポリリン酸塩は、ポリリン酸ナトリウム単独で、ポリリン酸カリウム単独で、またはポリリン酸ナトリウムとポリリン酸カリウムとの混合物として用いられてもよい。
【0072】
(3.プライマー)
本明細書中で使用される場合、用語「プライマー」とは、α−1,4−グルコシド結合で糖単位が結合できる遊離部分を1個以上有する分子をいう。プライマーは、α−グルカンの合成においてグリコシド残基を付加するための出発物質として作用する。なお、本明細書中では、グリコシド残基とグルコース残基とは交換可能に使用され得る。プライマーは、G1−Pのグリコシド残基のアクセプターとして作用する分子ともいうことができる。プライマーは、α−1,4−グルコシド結合で糖単位が結合できる遊離部分を1個以上有すれば、他の部分は糖以外の部分によって形成されていてもよい。本発明の方法では、反応開始時に含まれるプライマーに対して1つのグリコシド残基がα−1,4結合で転移することによって、このプライマーよりも重合度が1大きいα−グルカンが形成される。形成されたこのα−グルカンは、同じ溶液中で再度アクセプターとして作用することができる。このようにして、本発明の方法では、プライマーに対してグリコシド残基がα−1,4−グルコシド結合で順次結合されて、任意の重合度のα−グルカンが合成される。プライマーとしては、α−1,4−グルカンホスホリラーゼによって糖単位が付加され得る任意の糖が挙げられる。
【0073】
プライマーは、本発明の反応の出発物質として作用し得ればよく、例えば、本発明の方法によって合成されたα−グルカンをプライマーとして用いて、本発明の方法によってα−1,4−グルコシド鎖を再度伸長することも可能である。
【0074】
ただし、後述するゲスト化合物を用いる場合には、そのゲスト化合物を包接しない化合物をプライマーとして用いることが好ましい。
【0075】
プライマーは、α−1,4−グルコシド結合のみを含むα−1,4−グルカンであっても、α−1,6−グルコシド結合を部分的に有してもよい。当業者は、所望のグルカンに応じて、適切なプライマーを容易に選択し得る。直鎖状のアミロースを合成する場合には、α−1,4−グルコシド結合のみを含むα−1,4−グルカンをプライマーとして用いれば、枝切り酵素などを用いずに直鎖状アミロースを合成できるので好ましい。
【0076】
プライマーの例としては、マルトオリゴ糖、アミロース、アミロペクチン、グリコーゲン、デキストリン、プルラン、カップリングシュガー、澱粉およびこれらの誘導体が挙げられる。
【0077】
マルトオリゴ糖は、本明細書中では、約2個〜約10個のグルコースが脱水縮合して生じた物質であって、α−1,4結合によって連結された物質をいう。マルトオリゴ糖は、好ましくは約3個〜約10個の糖単位、より好ましくは約4個〜約10個の糖単位、さらに好ましくは約5個〜約10個の糖単位を有する。マルトオリゴ糖の例としては、マルトース、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオース、マルトオクタオース、マルトノナオース、マルトデカオースなどのマルトオリゴ糖が挙げられる。1つの実施形態では、マルトオリゴ糖は、好ましくはマルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオースまたはマルトヘプタオースであり、より好ましくはマルトテトラオース、マルトペンタオース、マルトヘキサオースまたはマルトヘプタオースであり、さらに好ましくはマルトテトラオースである。マルトオリゴ糖は、単品であってもよいし、複数のマルトオリゴ糖の混合物であってもよい。コストが低いため、マルトオリゴ糖の混合物が好ましい。1つの実施態様では、マルトオリゴ糖の混合物は、重合度4以上のマルトオリゴ糖に加えて、マルトトリオース、マルトースおよびグルコースのうちの少なくとも1つを含有する。オリゴ糖は、直鎖状のオリゴ糖であってもよいし、分枝状のオリゴ糖であってもよい。オリゴ糖は、その分子内に、環状部分を有し得る。本発明では、直鎖状のオリゴ糖が好ましい。
【0078】
アミロースとは、α−1,4結合によって連結されたグルコース単位から構成される直鎖分子である。アミロースは、天然の澱粉中に含まれる。
【0079】
アミロペクチンとは、α−1,4結合によって連結されたグルコース単位に、α1,6結合でグルコース単位が連結された、分枝状分子である。アミロペクチンは天然の澱粉中に含まれる。アミロペクチンとしては、例えば、アミロペクチン100%からなるワキシーコーンスターチが用いられ得る。例えば、重合度が約1×105程度以上のアミロペクチンが原料として用いられ得る。
【0080】
グリコーゲンは、グルコースから構成されるグルカンの一種であり、高頻度の枝分かれを有するグルカンである。グリコーゲンは、動植物の貯蔵多糖としてほとんどあらゆる細胞に顆粒状態で広く分布している。グリコーゲンは、植物中では、例えば、トウモロコシの種子などに存在する。グリコーゲンは、代表的には、グルコースのα−1,4−結合の糖鎖に対して、グルコースおよそ3単位おきに1本程度の割合で、平均重合度12〜18のグルコースのα−1,4−結合の糖鎖がα−1,6−結合で結合している。また、α−1,6−結合で結合している分枝にも同様にグルコースのα−1,4−結合の糖鎖がα−1,6−結合で結合している。そのため、グリコーゲンは網状構造を形成する。
【0081】
グリコーゲンの分子量は代表的には約1×105〜約1×108であり、好ましくは約1×106〜約1×107である。
【0082】
プルランは、マルトトリオースが規則正しく、階段状にα−1,6−結合した、分子量約10万〜約30万(例えば、約20万)のグルカンである。プルランは、例えば、澱粉を原料として黒酵母Aureobasidium pullulansを培養することにより製造される。プルランは、例えば、林原商事から入手され得る。
【0083】
カップリングシュガーは、ショ糖、グルコシルスクロース、マルトシルスクロースを主成分とする混合物である。カップリングシュガーは、例えば、ショ糖と澱粉との混合溶液にBacillus megateriumなどが産生するサイクロデキストリングルカノトランスフェラーゼを作用させることにより製造される。カップリングシュガーは、例えば、林原商事から入手され得る。
【0084】
澱粉は、アミロースとアミロペクチンとの混合物である。澱粉としては、通常市販されている澱粉であればどのような澱粉でも用いられ得る。澱粉に含まれるアミロースとアミロペクチンとの比率は、澱粉を産生する植物の種類によって異なる。モチゴメ、モチトウモロコシなどの有する澱粉のほとんどはアミロペクチンである。他方、アミロースのみからなり、かつアミロペクチンを含まない澱粉は、通常の植物からは得られない。
【0085】
澱粉は、天然の澱粉、澱粉分解物および化工澱粉に区分される。
【0086】
天然の澱粉は、原料により、いも類澱粉および穀類澱粉に分けられる。いも類澱粉の例としては、馬鈴薯澱粉、タピオカ澱粉、甘藷澱粉、くず澱粉、およびわらび澱粉などが挙げられる。穀類澱粉の例としては、コーンスターチ、小麦澱粉、および米澱粉などが挙げられる。天然の澱粉の例は、澱粉を生産する植物の品種改良の結果、アミロースの含量を50%〜70%まで高めたハイアミロース澱粉(例えば、ハイアミロースコーンスターチ)である。天然の澱粉の別の例は、澱粉を生産する植物の品種改良の結果、アミロースを含まないワキシー澱粉である。
【0087】
可溶性澱粉は、天然の澱粉に種々の処理を施すことにより得られる、水溶性の澱粉をいう。
【0088】
化工澱粉は、天然の澱粉に加水分解、エステル化、またはα化などの処理を施して、より利用しやすい性質を持たせた澱粉である。糊化開始温度、糊の粘度、糊の透明度、老化安定性などを様々な組み合わせで有する幅広い種類の化工澱粉が入手可能である。化工澱粉の種類には種々ある。このような澱粉の例は、澱粉の糊化温度以下において澱粉粒子を酸に浸漬することにより、澱粉分子は切断するが、澱粉粒子は破壊していない澱粉である。
【0089】
澱粉分解物は、澱粉に酵素処理または加水分解などの処理を施して得られる、処理前よりも分子量が小さいオリゴ糖もしくは多糖である。澱粉分解物の例としては、澱粉枝切り酵素分解物、澱粉ホスホリラーゼ分解物および澱粉部分加水分解物が挙げられる。
【0090】
澱粉枝切り酵素分解物は、澱粉に枝切り酵素を作用させることによって得られる。枝切り酵素の作用時間を種々に変更することによって、任意の程度に分岐部分(すなわち、α−1,6−グルコシド結合)が切断された澱粉枝切り酵素分解物が得られる。枝切り酵素分解物の例としては、糖単位数4〜10000のうちα−1,6−グルコシド結合を1個〜20個有する分解物、糖単位数3〜500のα−1,6−グルコシド結合を全く有さない分解物、マルトオリゴ糖およびアミロースが挙げられる。澱粉枝切り酵素分解物の場合、分解された澱粉の種類によって得られる分解物の分子量の分布が異なり得る。澱粉枝切り酵素分解物は、種々の長さの糖鎖の混合物であり得る。
【0091】
澱粉ホスホリラーゼ分解物は、澱粉にα−1,4−グルカンホスホリラーゼ(ホスホリラーゼともいう)を作用させることによって得られる。α−1,4−グルカンホスホリラーゼは、澱粉の非還元性末端からグルコース残基を1糖単位ずつ他の基質へと転移させる。α−1,4−グルカンホスホリラーゼは、α−1,6−グルコシド結合を切断することができないので、α−1,4−グルカンホスホリラーゼを澱粉に充分に長時間作用させると、α−1,6−グルコシド結合の部分で切断が終わった分解物が得られる。本発明では、澱粉ホスホリラーゼ分解物の有する糖単位数は、好ましくは約10〜約100,000、より好ましくは約50〜約50,000、さらにより好ましくは約100〜約10,000である。澱粉ホスホリラーゼ分解物は、分解された澱粉の種類によって得られる分解産物の分子量の分布が異なり得る。澱粉ホスホリラーゼ分解物は、種々の長さの糖鎖の混合物であり得る。
【0092】
デキストリンおよび澱粉部分加水分解物は、澱粉を、酸、アルカリ、酵素などの作用によって部分的に分解して得られる分解物をいう。本発明では、デキストリンおよび澱粉部分加水分解物の有する糖単位数は、好ましくは約10〜約100,000、より好ましくは約50〜約50,000、さらにより好ましくは約100〜約10,000である。デキストリンおよび澱粉部分加水分解物の場合、分解された澱粉の種類によって得られる分解産物の分子量の分布が異なり得る。デキストリンおよび澱粉部分加水分解物は、種々の長さを持つ糖鎖の混合物であり得る。
【0093】
澱粉は、可溶性澱粉、ワキシー澱粉、ハイアミロース澱粉、澱粉枝切り酵素分解物、澱粉ホスホリラーゼ分解物、澱粉部分加水分解物、化工澱粉、およびこれらの誘導体からなる群から選択されることが好ましい。
【0094】
本発明の方法では、上記各種糖の誘導体は、プライマーとして用いられ得る。例えば、上記糖のアルコール性水酸基の少なくとも1つが、ヒドロキシアルキル化、アルキル化、アセチル化、カルボキシメチル化、硫酸化、あるいはリン酸化された誘導体などが用いられ得る。さらに、これらの2種以上の誘導体の混合物が原料として用いられ得る。
【0095】
(4.ポリホスフェートグルコキナーゼ)
ポリホスフェートグルコキナーゼ(EC:2.7.1.63)とは、重合度nのポリリン酸(polyphosphate)のリン酸基をD−グルコースに転移して重合度n−1のポリリン酸とグルコース−6−リン酸を生成する反応を触媒する酵素をいう。この反応は以下のように記載される:
(リン酸)n+D−グルコース→(リン酸)n−1+D−グルコース−6−リン酸
ポリホスフェートグルコキナーゼは、ポリホスフェート−グルコースホスホトランスフェラーゼ、ポリホスフェート:D−グルコース6−ホスホトランスフェラーゼまたはPGKとも呼ばれる。
【0096】
ポリホスフェートグルコキナーゼは、多くの微生物に存在する。ポリホスフェートグルコキナーゼを産生する微生物の例としては、Microlunatus phosphovolas、Mycobacterium属細菌(例えば、M.phlei、M.tuberculosis、M.bovis)、Corynebacterium属細菌(例えばC.diphtheriae、C.xerosis、C.efficiens)、Propionibacterium shermanii、Streptomyces coelicolorなどが挙げられる。本発明においては、任意の生物由来のポリホスフェートグルコキナーゼを使用し得る。本発明においては、Microlunatus phosphovolas由来のポリホスフェートグルコキナーゼを用いることが好ましい。
【0097】
また、至適pHの観点からは、約5.5以上の至適pHを有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適pHは、より好ましくは約6.0以上であり、さらに好ましくは約6.5以上である。また、約10以下の至適pHを有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適pHは、より好ましくは約8.0以下であり、さらに好ましくは約7.5以下である。
【0098】
また、ポリホスフェートグルコキナーゼの反応至適温度は、可能な限り高いことが好ましい。至適温度の観点からは、約35℃以上の至適温度を有するポリホスフェートグルコキナーゼが好ましい。ポリホスフェートグルコキナーゼの至適温度は、より好ましくは約40℃以上であり、さらに好ましくは約45℃以上である。当該分野においては、至適温度の高い種々のポリホスフェートグルコキナーゼが公知である。高温条件で生育する細菌の有する酵素は、一般的に反応至適温度が高温である。例えば、Thermobifida fuscaの生育温度は、約50℃であるので、Thermobifida fuscaの有する酵素は一般的に、反応至適温度が約50℃であるか、または少なくとも約50℃での反応性が非常に高い。Thermobifida fuscaの好適な生育条件は、約50℃にてpH7.4であり、このポリホスフェートグルコキナーゼを産生するThermobifida fuscaは、ATCCから購入することができる。Thermobifida fusca由来のポリホスフェートグルコキナーゼのヌクレオチド配列を配列番号9に示し、Thermobifida fusca由来のポリホスフェートグルコキナーゼのアミノ酸配列を配列番号10に示す。ポリホスフェートグルコキナーゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0099】
反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるポリホスフェートグルコキナーゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。ポリホスフェートグルコキナーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0100】
(5.ホスホグルコムターゼ)
ホスホグルコムターゼ(EC:2.7.5.1)とは、α−D−グルコース1,6−二リン酸とα−D−グルコース1−リン酸から、α−D−グルコース6−リン酸とα−D−グルコースを生成する反応を触媒する酵素をいう。この反応は以下のように記載される:
【0101】
【化2】
ホスホグルコムターゼは、α−ホスフェートグルコムターゼ、α−D−グルコース−1,6−ビスホスフェート:α−D−グルコース−1−ホスフェートホスホトランスフェラーゼまたはPGMとも呼ばれる。
【0102】
ホスホグルコムターゼは、糖代謝の主経路に位置する酵素のためほとんどすべての生物に存在する。ホスホグルコムターゼは、微生物、動物および植物に存在する。ホスホグルコムターゼを産生する生物の例としては、例えば、ウサギ、ヒラメ、サメ、ウシ、E.coli、酵母、馬鈴薯(Potato)が挙げられる。本発明においては、任意の生物由来のホスホグルコムターゼを使用し得る。本発明においては、E.coli由来のホスホグルコムターゼを使用することが好ましい。
【0103】
また、至適pHの観点からは、約6.0以上の至適pHを有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適pHは、より好ましくは約6.5以上であり、さらに好ましくは約7.0以上である。また、約10以下の至適pHを有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適pHは、より好ましくは約8.5以下であり、さらに好ましくは約8.0以下である。
【0104】
また、至適温度の観点からは、約35℃以上の至適温度を有するホスホグルコムターゼが好ましい。ホスホグルコムターゼの至適温度は、より好ましくは約40℃以上であり、さらに好ましくは約45℃以上である。当該分野においては、至適温度の高い種々のホスホグルコムターゼが公知である。高温条件で生育する細菌の有する酵素は、一般的に、反応至適温度が高温である。例えば、Thermus thermophilusの生育温度は、約75℃であるので、Thermus thermophilusの有する酵素は、一般的に、反応至適温度が約75℃であるか、または少なくとも約75℃での反応性が非常に高い。Thermus thermophilusの好適な生育条件は、約75℃にてpH7.5であり、このホスホグルコムターゼを産生するThermus thermophilusは、ATCCから購入することができる。Thermus thermophilus由来のホスホグルコムターゼのヌクレオチド配列を配列番号11に示し、Thermus thermophilus由来のホスホグルコムターゼのアミノ酸配列を配列番号12に示す。Thermotoga lettingae生育温度は、約65℃であるので、Thermotoga lettingaeの有する酵素は、一般的に、反応至適温度が約65℃であるか、または少なくとも約65℃での反応性が非常に高い。Thermotoga lettingaeの好適な生育条件は、約65℃にてpH7.5であり、このホスホグルコムターゼを産生するThermotoga lettingaeは、ATCCから購入することができる。Thermotoga lettingae由来のホスホグルコムターゼのヌクレオチド配列を配列番号13に示し、Thermotoga lettingae由来のホスホグルコムターゼのアミノ酸配列を配列番号14に示す。ホスホグルコムターゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0105】
反応開始時の溶液中に含まれるホスホグルコムターゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるホスホグルコムターゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるホスホグルコムターゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。ホスホグルコムターゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0106】
(6.α−1,4−グルカンホスホリラーゼ)
α−1,4−グルカンホスホリラーゼ(EC:2.4.1.1)とは、α−1,4−グルカン(重合度n)の加リン酸分解による、α−1,4−グルカン(重合度n−1)とα−D−グルコース−1−リン酸との産生を触媒する酵素の総称であり、ホスホリラーゼ、スターチホスホリラーゼ、グリコーゲンホスホリラーゼ、マルトデキストリンホスホリラーゼなどと呼ばれる場合もある。α−1,4−グルカンホスホリラーゼは、加リン酸分解の逆反応である、α−1,4−グルカン(重合度n−1)およびα−D−グルコース−1−リン酸からα−1,4−グルカン(重合度n)を合成する反応をも触媒し得る。反応がどちらの方向に進むかは、基質の量に依存する。生体内では、無機リン酸の量が多いので、α−1,4−グルカンホスホリラーゼは加リン酸分解の方向に反応が進む。本発明の方法では、無機リン酸は、β−1,4−グルカンの加リン酸分解に使われ、反応溶液中に含まれる無機リン酸の量が少ないので、α−グルカンの合成の方向に反応が進む。
【0107】
α−1,4−グルカンホスホリラーゼは、デンプンまたはグリコーゲンを貯蔵し得る種々の植物、動物および微生物中に普遍的に存在すると考えられる。
【0108】
α−1,4−グルカンホスホリラーゼを産生する植物の例としては、藻類、ジャガイモ(馬鈴薯ともいう)、サツマイモ(甘藷ともいう)、ヤマイモ、サトイモ、キャッサバなどの芋類、キャベツ、ホウレンソウなどの野菜類、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、アワなどの穀類、えんどう豆、大豆、小豆、うずら豆などの豆類などが挙げられる。
【0109】
α−1,4−グルカンホスホリラーゼを産生する動物の例としては、ヒト、ウサギ、ラット、ブタなどの哺乳類などが挙げられる。
【0110】
α−1,4−グルカンホスホリラーゼを産生する微生物の例としては、Thermus
aquaticus、Bacillus stearothermophilus、Deinococcus radiodurans、Thermococcus litoralis、Streptomyces coelicolor、Pyrococcus
horikoshi、Mycobacterium tuberculosis、Thermotoga maritima、Aquifex aeolicus、Methanococcus Jannaschii、Pseudomonas aeruginosa、Chlamydia pneumoniae、Chlorella vulgaris、Agrobacterium tumefaciens、Clostridium
pasteurianum、Klebsiella pneumoniae、Synecococcus sp.、Synechocystis sp.、E.coli、Neurospora crassa、Saccharomyces cerevisiae、Chlamydomonas sp.などが挙げられる。α−1,4−グルカンホスホリラーゼを産生する生物はこれらに限定されない。
【0111】
本発明で用いられるα−1,4−グルカンホスホリラーゼは、ジャガイモ、Thermus aquaticus、Bacillus stearothermophilusに由来することが好ましく、ジャガイモに由来することがより好ましい。特定の実施形態では、本発明で用いられるα−1,4−グルカンホスホリラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高いα−1,4−グルカンホスホリラーゼは、例えば、高度好熱細菌に由来し得る。
【0112】
また、至適pHの観点からは、約4.0以上の至適pHを有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適pHは、より好ましくは約4.5以上であり、さらに好ましくは約5.0以上である。また、約7.0以下の至適pHを有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適pHは、より好ましくは約6.5以下であり、さらに好ましくは約6.0以下である。
【0113】
本発明においては、後述するとおり、反応開始時点における反応溶液のpHを弱アルカリ性側にすることが好ましいが、その場合、α−1,4−グルカンホスホリラーゼの至適pHと比較して、多少の差が生じることになる。反応開始時点における反応溶液のpHと、α−1,4−グルカンホスホリラーゼの至適pHとの差は、約0.5以上とすることが可能であり、約1.0以上とすることも可能であり、さらには約1.5以上とすることも可能である。反応開始時点における反応溶液のpHと、α−1,4−グルカンホスホリラーゼの至適pHとの差は、好ましくは約5.5以下であり、より好ましくは約4.0以下であり、さらに好ましくは約2.5以下である。
【0114】
また、至適温度の観点からは、約30℃以上の至適温度を有するα−1,4−グルカンホスホリラーゼが好ましい。α−1,4−グルカンホスホリラーゼの至適温度は、より好ましくは約35℃以上であり、さらに好ましくは約40℃以上である。当該分野においては、至適温度の高い種々のα−1,4−グルカンホスホリラーゼが公知である。例えば、Aquifex aeolicus由来のα−1,4−グルカンホスホリラーゼの至適温度は約90℃であり、至適pHは5.6である。このα−1,4−グルカンホスホリラーゼを産生するAquifex aeolicusは、例えば、ATCCから入手可能である。α−1,4−グルカンホスホリラーゼの至適温度に特に上限はなく、至適温度は例えば、約80℃以下、約75℃以下、約70℃以下、約65℃以下、約60℃以下、約55℃以下、約50℃以下、約45℃以下、約40℃以下などであり得る。至適温度が非常に高い酵素については、その酵素を入手することが困難である場合がある。
【0115】
反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のグルコース1gに対して、好ましくは約1U以上であり、より好ましくは約10U以上であり、さらに好ましくは約50U以上であり、特に好ましくは約100U以上であり、最も好ましくは約150U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるα−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のグルコースに対して、好ましくは約1500U以下であり、より好ましくは約1000U以下であり、さらに好ましくは約500U以下であり、特に好ましくは約300U以下であり、最も好ましくは約200U以下である。α−1,4−グルカンホスホリラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0116】
(7.ゲスト化合物)
アミロースなどの直鎖状α−グルカン、および分岐状α−グルカンのうちの直鎖状の部分がゲスト化合物を包接することが公知である。すなわち、これらのα−グルカンは、ホスト化合物として作用することが公知である。これらのα−グルカンは、ゲスト化合物を包接すると沈澱しやすくなる。そのため、本発明の方法においては、反応溶液中にゲスト化合物を含むことが好ましい。ゲスト化合物は、反応開始時から溶液中に含有されてもよく、あるいは、ある程度反応が進んでから溶液中に添加されてもよい。
【0117】
本明細書中で使用される場合、用語「ゲスト化合物」とは、アミロースによって包接される化合物をいう。ゲスト化合物がアミロースによって包接された場合に得られる包接化合物が難溶性または不溶性であるものが好ましい。
【0118】
ゲスト化合物は、好ましくは、プライマーによって包接されない化合物である。
【0119】
本発明で好ましく使用され得るゲスト化合物の例としては、デカン酸(カプリン酸)、ラウリン酸、ドデシル硫酸ナトリウム、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートおよび3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートなどが挙げられる。ゲスト化合物は、好ましくは、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートであり、より好ましくは3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである。
【0120】
(8.グルコース−6−リン酸、グルコース−1−リン酸、または無機リン酸)
本発明において、出発物質としてグルコース−6−リン酸を使用することが可能である。本明細書において使用される場合、用語「グルコース−6−リン酸(glucose−6−phosphate)とは、グルコース−6−リン酸(C6H13O9P;glucose−6−phosphoric acid)およびその塩をいう。グルコース−6−リン酸は好ましくは、狭義のグルコース−6−リン酸(C6H13O9P)の任意の金属塩であり、より好ましくはグルコース−1−リン酸(C6H13O9P)の任意のアルカリ金属塩である。狭義のグルコース−6−リン酸は、グルコースの6位の炭素にリン酸基が結合している物質である。グルコース−6−リン酸の好ましい具体例としては、グルコース−6−リン酸二ナトリウム、グルコース−6−リン酸二カリウム、グルコース−6−リン酸(C6H13O9P)、などが挙げられる。本明細書において、特に断りの無い限り、グルコース−6−リン酸は、広義のグルコース−6−リン酸、すなわち狭義のグルコース−6−リン酸(C6H13O9P)およびその塩を示す。
【0121】
反応開始時の反応溶液は、1種類のグルコース−6−リン酸を含有してもよく、複数種類のグルコース−6−リン酸を含有してもよい。
【0122】
本明細書において使用される場合、用語「グルコース−1−リン酸(glucose−1−phosphate)」とは、グルコース−1−リン酸(C6H13O9P;glucose−6−phosphoric acid)およびその塩をいう。グルコース−1−リン酸は好ましくは、狭義のグルコース−1−リン酸(C6H13O9P)の任意の金属塩であり、より好ましくはグルコース−1−リン酸(C6H13O9P)の任意のアルカリ金属塩である。グルコース−1−リン酸の好ましい具体例としては、グルコース−1−リン酸二ナトリウム、グルコース−1−リン酸二カリウム、グルコース−1−リン酸(C6H13O9P)、などが挙げられる。本明細書において、括弧書きで化学式を書いていないグルコース−1−リン酸は、広義のグルコース−1−リン酸、すなわち狭義のグルコース−1−リン酸(C6H13O9P)およびその塩を示す。
【0123】
反応開始時の反応溶液は、1種類のグルコース−1−リン酸を含有してもよく、複数種類のグルコース−1−リン酸を含有してもよい。
【0124】
本明細書において使用される場合、用語「無機リン酸」とは、リン酸のあらゆる種類のイオンおよび塩の総称である。例えば、無機リン酸は、セロビオースからグルコース−1−リン酸を合成する反応においてリン酸基質を供与する。すなわち、無機リン酸は、この反応においてリン酸源として作用する。ここでリン酸基質とは、グルコース−1−リン酸のリン酸部分(moiety)の原料となる物質をいう。β−1,4−グルカンホスホリラーゼによって触媒されるβ−1,4−グルカン加リン酸分解において、無機リン酸はリン酸イオンの形態で基質として作用していると考えられる。当該分野ではこの基質を慣習的に無機リン酸というので、本明細書中でも、この基質を無機リン酸という。無機リン酸には、リン酸およびリン酸の無機塩が含まれる。通常、無機リン酸は、アルカリ金属イオンなどの陽イオンを含む水中で使用される。この場合、リン酸とリン酸塩とリン酸イオンとは平衡状態になるので、リン酸とリン酸塩とは区別をしにくい。従って、便宜上、リン酸とリン酸塩とを合わせて無機リン酸という。本発明において、無機リン酸は好ましくは、リン酸の任意の金属塩であり、より好ましくはリン酸のアルカリ金属塩である。無機リン酸の好ましい具体例としては、リン酸二水素ナトリウム、リン酸水素二ナトリウム、リン酸三ナトリウム、リン酸二水素カリウム、リン酸水素二カリウム、リン酸三カリウム、リン酸(H3PO4)、リン酸二水素アンモニウム、リン酸水素二アンモニウムなどが挙げられる。
【0125】
反応開始時の反応溶液は、1種類の無機リン酸を含有してもよく、複数種類の無機リン酸を含有してもよい。
【0126】
無機リン酸は、例えば、ポリリン酸(例えば、ピロリン酸、三リン酸および四リン酸)のようなリン酸縮合体またはその塩を、物理的、化学的または酵素反応などによって分解したものを反応溶液に添加することによって提供され得る。ポリリン酸は、通常、少量の無機リン酸を不純物として含有する。そのため、α−グルカンの合成には無機リン酸が必要であるが、市販のポリリン酸を添加すれば、無機リン酸を添加する必要がない場合がある。
【0127】
(9.グルコースイソメラーゼ(EC:5.3.1.5))
本発明の方法において、出発物質としてフルクトースを使用することも可能である。他の酵素反応の副産物としてフルクトースが得られる場合、本発明ではこのフルクトースを有効に利用することができる。
【0128】
出発物質としてフルクトースを使用する実施形態では、反応溶液中にグルコースイソメラーゼをさらに含むことが好ましい。溶液中にグルコースイソメラーゼを含むことにより、フルクトースをグルコースへと変換できる。
【0129】
本発明の製造方法で用いられ得るグルコースイソメラーゼは、D−グルコースとD−フルクトースとの相互変換を触媒し得る酵素である。グルコースイソメラーゼは、D−キシロースとD−キシルロースとの相互変換をも触媒し得るので、キシロースイソメラーゼとも呼ばれる。
【0130】
グルコースイソメラーゼは、微生物、動物および植物に存在する。グルコースイソメラーゼを産生する微生物の例としては、Streptomyces rubiginosus、Streptomyces olivochromogenes、Streptomyces murinus、Streptomyces violaceoniger、Streptomyces diastaticus、Streptomyces albus、Streptomyces sp.、Escherichia coli、Bacteroides xylanolyticus、Arthrobacter sp.、Candida boidinii、Clostridium thermosulfurogenes、Clostridium thermohydrosulfuricum、Thermoanaerobacterium saccharolyticum、Thermoanaerobacter sp.、Thermotoga neapolitana、Thermus aquaticus、Lactobacillus brevis、Lactobacillus xylosus、Agrobacterium tumefaciens、Bacillus sp.、Actinoplanes missouriensisおよびParacolobacterium aerogenoidesが挙げられる。グルコースイソメラーゼを産生する動物の例としては、Trypanosoma bruceiが挙げられる。グルコースイソメラーゼは、植物由来であってもよい。グルコースイソメラーゼを産生する生物はこれらに限定されない。
【0131】
本発明で用いられ得るグルコースイソメラーゼは、Streptomyces rubiginosusまたはBacillus sp.に由来することが好ましく、Streptomyces rubiginosusに由来することがより好ましい。本発明で用いられるグルコースイソメラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高いグルコースイソメラーゼは、例えば、高度好熱細菌に由来し得る。
【0132】
反応開始時の溶液中に含まれるグルコースイソメラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中のフルクトース1gに対して、好ましくは約0.01U以上であり、より好ましくは約0.05U以上であり、さらに好ましくは約0.1U以上であり、特に好ましくは約0.5U以上であり、最も好ましくは約1U以上である。好ましい実施形態では、反応開始時の溶液中のフルクトース1gに対して、好ましくは約500U以下であり、より好ましくは約100U以下であり、さらに好ましくは約50U以下であり、特に好ましくは約10U以下であり、最も好ましくは約5U以下である。グルコースイソメラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0133】
(10.β−1,4−グルカンホスホリラーゼ)
本発明において、出発物質としてβ−1,4−グルカンを使用することも可能である。出発物質としてβ−1,4−グルカンを使用する実施形態では、反応溶液中にβ−1,4−グルカンホスホリラーゼを含むことが好ましい。
【0134】
本明細書中では、「β−1,4−グルカンホスホリラーゼ」とは、β−1,4−グルカンの非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う任意の酵素をいう。β−1,4−グルカンホスホリラーゼは、加リン酸分解の逆反応であるβ−1,4−グルカン合成反応をも触媒し得る。反応がどちらの方向に進むかは、基質の量に依存するが、この反応は、β−1,4−グルカン合成反応の方向に進みやすい傾向がある。β−1,4−グルカンホスホリラーゼによって触媒される反応は、次式により示される:
【0135】
【化3】
なお、この式において、出発時のβ−1,4−グルカンの重合度が2の場合、β−1,4−グルカンの代わりにグルコースが得られる。
【0136】
β−1,4−グルカンホスホリラーゼは好ましくは、セロビオースホスホリラーゼ(EC:2.4.1.20)またはセロデキストリンホスホリラーゼ(EC:2.4.1.49)である。
【0137】
セロビオースホスホリラーゼは、セロビオースの非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う酵素をいう。セロビオースホスホリラーゼによって触媒される反応は、次式により示される:
【0138】
【化4】
セロデキストリンホスホリラーゼは、重合度3以上のセロオリゴ糖の非還元末端側グルコース残基をリン酸基に転移して加リン酸分解を行う酵素をいう。セロオリゴ糖は、セロデキストリンとも呼ばれる。セロデキストリンホスホリラーゼによって触媒される反応は、次式により示される:
【0139】
【化5】
本発明の方法においては、β−1,4−グルカンがセロビオースである場合、β−1,4−グルカンホスホリラーゼとしてセロビオースホスホリラーゼを用いることが好ましい。本発明の方法においては、β−1,4−グルカンがセロオリゴ糖である場合、β−1,4−グルカンホスホリラーゼとしてセロデキストリンホスホリラーゼを用いることが好ましい。本発明の方法においてはまた、β−1,4−グルカンがセロオリゴ糖である場合、β−1,4−グルカンホスホリラーゼとしてセロビオースホスホリラーゼおよびセロデキストリンホスホリラーゼを用いることが好ましい。この場合、セロデキストリンホスホリラーゼの作用によってセロオリゴ糖が分解されることによって生じたグルコース−1−リン酸がα−グルカン合成に使用され、かつ最終的に生じたセロビオースをセロビオースホスホリラーゼによって分解し得るので、セロオリゴ糖からα−グルカンの合成速度がより速くなる。
【0140】
出発物質としてβ−1,4−グルカンホスホリラーゼを使用する実施形態では、反応用液中に少なくともセロビオースホスホリラーゼを含有することが好ましい。セロビオースによって記載される上記反応によって生成されるグルコースは、ポリホスフェートグルコキナーゼおよびホスホグルコムターゼによって触媒される反応によりG1−Pへと変換されるため、β−1,4−グルカン(特に、セロビオース)を効率よく利用できる。
【0141】
β−1,4−グルカンホスホリラーゼは、自然界では種々の生物に含まれる。β−1,4−グルカンホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。β−1,4−グルカンホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される。β−1,4−グルカンホスホリラーゼは、植物由来であってもよい。
【0142】
セロビオースホスホリラーゼは、自然界では種々の生物に含まれる。セロビオースホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga
neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。セロビオースホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択され、より好ましくはClostridium thermocellumまたはCellvibrio gilvusであり、最も好ましくはClostridium thermocellumである。セロビオースホスホリラーゼは、植物由来であってもよい。
【0143】
セロデキストリンホスホリラーゼは、自然界では種々の生物に含まれる。セロデキストリンホスホリラーゼを産生する生物の例としては、Clostridium属の生物(例えば、Clostridium thermocellumおよびClostridium sterocorarium)、Cellvibrio属の生物(例えば、Cellvibrio gilvus)、Thermotoga属の生物(例えば、Thermotoga neapolitanaおよびThermotoga maritima)、Ruminococcas属の生物(例えば、Ruminococcas flavofaciens)、Fomes属の生物(例えば、Fomes annos)、Cellulomonas属の生物およびErwinia属の生物が挙げられる。セロデキストリンホスホリラーゼを産生する生物は好ましくは、Clostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択され、より好ましくはClostridium thermocellumまたはCellulomonas sp.であり、最も好ましくはClostridium thermocellumである。セロデキストリンホスホリラーゼホスホリラーゼは、植物由来であってもよい。
【0144】
β−1,4−グルカンホスホリラーゼ(好ましくはセロビオースホスホリラーゼまたはセロデキストリンホスホリラーゼ、最も好ましくはセロビオースホスホリラーゼ)は、β−1,4−グルカンホスホリラーゼ(好ましくはセロビオースホスホリラーゼまたはセロデキストリンホスホリラーゼ、最も好ましくはセロビオースホスホリラーゼ)を産生する任意の生物由来であり得る。β−1,4−グルカンホスホリラーゼは、ある程度の耐熱性を有することが好ましい。β−1,4−グルカンホスホリラーゼは、耐熱性が高ければ高いほど好ましい。例えば、β−1,4−グルカンホスホリラーゼを1.4mMの2−メルカプトエタノールを含む50mMリン酸緩衝液(pH7.5)中で55℃にて20分間加熱した場合に加熱前のβ−1,4−グルカンホスホリラーゼの活性の50%以上の活性を保持するものであることが好ましく、60%以上の活性を保持するものであることがより好ましく、70%以上の活性を保持するものであることがさらに好ましく、80%以上の活性を保持するものであることが特に好ましく、85%以上の活性を保持するものであることが最も好ましい。β−1,4−グルカンホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来である。
【0145】
β−1,4−グルカンホスホリラーゼがセロビオースホスホリラーゼである場合、セロビオースホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga
maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来であり、より好ましくはClostridium thermocellumまたはCellvibrio gilvus由来であり、最も好ましくはClostridium thermocellum由来である。
【0146】
β−1,4−グルカンホスホリラーゼがセロビオースホスホリラーゼである場合、セロビオースホスホリラーゼは、好ましくはClostridium thermocellum、Clostridium sterocorarium、Cellvibrio gilvus、Thermotoga neapolitana、Thermotoga
maritima、Ruminococcas flavofaciens、Fomes annos、Cellulomonas sp.、Erwinia sp.からなる群より選択される細菌由来であり、より好ましくはClostridium thermocellumまたはCellulomonas sp.由来であり、最も好ましくはClostridium thermocellum由来である。
【0147】
反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、目的の反応を進行させるに充分な量であればよい。好ましい実施形態では、反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のβ−1,4−グルカンの1gに対して、好ましくは約0.01U以上であり、より好ましくは約0.05U以上であり、さらに好ましくは約0.1U以上であり、特に好ましくは約0.5U以上であり、最も好ましくは約1U以上である。好ましい実施形態では、反応開始時の溶液中に含まれるβ−1,4−グルカンホスホリラーゼの量は、反応開始時の溶液中のβ−1,4−グルカンの1gに対して、好ましくは約1,000U以下であり、より好ましくは約500U以下であり、さらに好ましくは約100U以下であり、特に好ましくは約50U以下であり、最も好ましくは約7U以下である。β−1,4−グルカンホスホリラーゼの重量が多すぎると、反応中に変性した酵素が凝集しやすくなる場合がある。使用量が少なすぎると、反応自体は起こるものの、グルカンの収率が低下する場合がある。
【0148】
(11.アミラーゼ)
本発明の方法において、出発物質としてデンプンを使用することも可能である。デンプンを出発物質として使用する実施形態では、アミラーゼなどのデンプン分解酵素は、アミロース合成を行う反応溶液とは別の溶液に含まれることが好ましい。生成したアミロースが分解されてしまうからである。アミラーゼを使用する場合は、デンプンをアミラーゼで分解してグルコースを得た後に、その反応液を加熱してアミラーゼを失活させることが好ましい。
【0149】
アミラーゼはデンプンを加水分解する酵素の総称である。アミラーゼには、非還元性末端から特定数のグルコース単位を切り離していくエキソ型と、ほぼランダムに近い分解様式をもつエンド型がある。本発明においてアミラーゼを使用する場合には、エキソ型およびエンド型のいずれのアミラーゼを使用してもよい。アミラーゼの例としては、エキソ−1,4−α−D−グルコシダーゼ(EC3.2.1.3;グルコシダーゼとも呼ばれる)、β−アミラーゼ(EC3.2.1.3)、エキソ−イソマルトトリヒドロラーゼ(EC3.2.1.95)、エキソ−マルトテトラオヒドロラーゼ(EC3.2.1.60)、エキソ−マルトヘキサオヒドロラーゼ(EC3.2.1.98)が挙げられる。
【0150】
(12.β−1,4−グルカン)
本発明において、出発物質としてβ−1,4−グルカンを使用することが可能である。本明細書中では「β−1,4−グルカン」とは、D−グルコースを構成単位とする糖であって、β−1,4−グルコシド結合によって連結された糖単位を少なくとも2糖単位以上有する糖をいう。β−1,4−グルカンは、直鎖状の分子であり得る。直鎖状β−グルカンとβ−1,4−グルカンとセルロースとは同義語である。直鎖状β−グルカンでは、β−1,4−グルコシド結合によってのみ糖単位の間が連結されている。1分子のβ−1,4−グルカンに含まれる糖単位の数を、このβ−1,4−グルカンの重合度という。β−1,4−グルカンの重合度は、好ましくは、約2〜約10であり、より好ましくは約2〜約8であり、より好ましくは約2〜約5である。重合度が約2〜約10のβ−1,4−グルカンを、セロオリゴ糖ともいう。重合度が2のβ−1,4−グルカンを特に、セロビオースという。重合度が3のβ−1,4−グルカンをセロトリオースという。重合度が4のβ−1,4−グルカンをセロテトラオースという。β−1,4−グルカンの重合度が低いほど溶解度が高く、取り扱いが容易であるので、重合度の低いβ−1,4−グルカンがより好ましい。β−1,4−グルカンは、あらゆる植物中に存在する。β−1,4−グルカンは、植物から単離されたまま未改変のものであってもよく、植物から単離したものを化学的または酵素的に処理することによって得られたものであってもよい。β−1,4−グルカンはまた、古紙、建材、古布などの廃棄物から再生されるセルロースまたはそれから調製されたものであってもよい。例えば、植物から単離した高分子量のセルロースに対してセルラーゼを作用させることによって、より低分子量のセロオリゴ糖が得られる。植物からセロオリゴ糖を大量に生産する方法は当該分野で公知である。このような文献の例としては、特開2001−95594号公報が挙げられる。β−1,4−グルカンは、β−1,4−グルカンを含む植物破砕液から精製β−1,4−グルカンに至るいずれの生成段階のものとして提供されてもよい。本発明の方法で使用されるβ−1,4−グルカンは、純粋なものであることが好ましい。しかし、本発明で用いる酵素の作用を阻害しない限り、任意の他の夾雑物を含んでいてもよい。
【0151】
出発物質としてβ−1,4−グルカンを使用する実施形態において、反応開始時の溶液中に含まれるβ−1,4−グルカンの濃度は、好ましくは約0.1重量%以上であり、より好ましくは約0.5重量%以上であり、さらに好ましくは約1重量%以上であり、特に好ましくは約2重量%以上であり、最も好ましくは約3重量%以上である。出発物質としてβ−1,4−グルカンを使用する実施形態において、反応開始時の溶液中に含まれるβ−1,4−グルカンの濃度は、好ましくは約40重量%以下であり、より好ましくは約30重量%以下であり、さらに好ましくは約20重量%以下であり、特に好ましくは約15重量%以下であり、最も好ましくは約12重量%以下である。なお、本明細書中でβ−1,4−グルカンの濃度は、Weight/Volumeで、すなわち、
(β−1,4−グルカンの重量)×100/(溶液の容量)
で計算する。β−1,4−グルカンの重量が多すぎると、溶液中に未反応のβ−1,4−グルカンが析出する場合がある。β−1,4−グルカンの使用量が少なすぎると、高温での反応において、反応自体は起こるものの、収率が低下する場合がある。
【0152】
(13.枝切り酵素)
本発明の方法において、プライマーとしてα−1,6−グルコシド結合を含有するものを用いる場合などの、生成物に分岐が生じる場合には、必要に応じて、枝切り酵素を用いることができる。
【0153】
本発明で用いられ得る枝切り酵素は、α−1,6−グルコシド結合を切断し得る酵素である。枝切り酵素は、アミロペクチンおよびグリコーゲンにともによく作用するイソアミラーゼ(EC 3.2.1.68)と、アミロペクチン、グリコーゲンおよびプルランに作用するα−デキストリンエンド−1,6−α−グルコシダーゼ(プルラナーゼともいう)(EC 3.2.1.41)との2つに分類される。
【0154】
枝切り酵素は、微生物および植物に存在する。枝切り酵素を産生する微生物の例としては、Saccharomyces cerevisiae、Chlamydomonas
sp.、Bacillus brevis、Bacillus acidopullulyticus、Bacillus macerans、Bacillus stearothermophilus、Bacillus circulans、Thermus
aquaticus、Klebsiella pneumoniae、Thermoactinomyces thalpophilus、Thermoanaerobacter ethanolicus、Pseudomonas amyloderamosaなどが挙げられる。枝切り酵素を産生する植物の例としては、ジャガイモ、サツマイモ、トウモロコシ、イネ、コムギ、オオムギ、オートムギ、サトウダイコンなどが挙げられる。枝切り酵素を産生する生物はこれらに限定されない。
【0155】
本発明で用いられ得る枝切り酵素は、Klebsiella pneumoniae、Bacillus brevis、Bacillus acidopullulyticus、Pseudomonas amyloderamosaに由来することが好ましく、Klebsiella pneumoniae、Pseudomonas amyloderamosaに由来することがより好ましい。本発明で用いられる枝切り酵素は、反応至適温度が高いことが好ましい。反応至適温度が高い枝切り酵素は、例えば、高度好熱細菌に由来し得る。
【0156】
(14.ブランチングエンザイム(EC.2.4.1.18))
本発明の方法において、生成物に分岐を生じさせることが所望される場合には、必要に応じて、ブランチングエンザイムを用いることができる。
【0157】
本発明で用いられ得るブランチングエンザイムは、α−1,4−グルカン鎖の一部をこのα−1,4−グルカン鎖のうちのあるグルコース残基の6位に転移して分枝を作り得る酵素である。ブランチングエンザイムは、1,4−α−グルカン分枝酵素、枝つくり酵素またはQ酵素とも呼ばれる。
【0158】
ブランチングエンザイムは、微生物、動物、および植物に存在する。ブランチングエンザイムを産生する微生物の例としては、Bacillus stearothermophilus、Bacillus subtilis、Bacillus caldolyticus、Bacillus licheniformis、Bacillus amyloliquefaciens、Bacillus coagulans、Bacillus caldovelox、Bacillus thermocatenulatus、Bacillus smithii、Bacillus megaterium、Bacillus brevis、Alkalophillic Bacillus sp.、Streptomyces coelicolor、Aquifex aeolicus、Synechosystis sp.、E.coli、Agrobacteirum tumefaciens、Thermus aquaticus、Rhodothermus obamensis、Neurospora crassa、酵母などが挙げられる。ブランチングエンザイムを産生する動物の例としてはヒト、ウサギ、ラット、ブタなどの哺乳類が挙げられる。ブランチングエンザイムを産生する植物の例としては、藻類、ジャガイモ、サツマイモ、ヤマイモ、キャッサバなどの芋類、ホウレンソウなどの野菜類、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、アワなどの穀類、えんどう豆、大豆、小豆、うずら豆などの豆類などが挙げられる。ブランチングエンザイムを産生する生物はこれらに限定されない。
【0159】
本発明で用いられ得るブランチングエンザイムは、ジャガイモ、Bacillus stearothermophilus、Aquifex aeolicusに由来することが好ましく、Bacillus stearothermophilus、Aquifex aeolicusに由来することがより好ましい。本発明で用いられるブランチングエンザイムは、反応至適温度が高いことが好ましい。反応至適温度が高いブランチングエンザイムは、例えば、高度好熱細菌に由来し得る。
【0160】
(15.4−α−グルカノトランスフェラーゼ(EC.2.4.1.25))
本発明の方法において、生成物に環状構造を生じさせる場合には、必要に応じて、4−α−グルカノトランスフェラーゼを用いることができる。
【0161】
本発明で用いられ得る4−α−グルカノトランスフェラーゼは、ディスプロポーショネーティングエンザイム、D−酵素、アミロマルターゼ、不均化酵素などとも呼ばれ、マルトオリゴ糖の糖転移反応(不均一化反応)を触媒し得る酵素である。4−α−グルカノトランスフェラーゼは、供与体分子の非還元末端からグルコシル基あるいは、マルトシルもしくはマルトオリゴシルユニットを受容体分子の非還元末端に転移する酵素である。従って、酵素反応は、最初に与えられたマルトオリゴ糖の重合度の不均一化をもたらす。供与体分子と受容体分子とが同一の場合は、分子内転移が生じ、その結果、環状構造をもつ生成物が得られる。
【0162】
4−α−グルカノトランスフェラーゼは、微生物および植物に存在する。4−α−グルカノトランスフェラーゼを産生する微生物の例としては、Aquifex aeolicus、Streptococcus pneumoniae、Clostridium butylicum、Deinococcus radiodurans、Haemophilus influenzae、Mycobacterium tuberculosis、Thermococcus litralis、Thermotoga maritima、Thermotoga neapolitana、Chlamydia psittaci、Pyrococcus sp.、Dictyoglomus thermophilum、Borrelia burgdorferi、Synechosystis sp.、E.coli、Thermus aquaticusなどが挙げられる。4−α−グルカノトランスフェラーゼを産生する植物の例としては、ジャガイモ、サツマイモ、ヤマイモ、キャッサバなどの芋類、トウモロコシ、イネ、コムギ、などの穀類、えんどう豆、大豆、などの豆類などが挙げられる。4−α−グルカノトランスフェラーゼを産生する生物はこれらに限定されない。
【0163】
本発明で用いられ得る4−α−グルカノトランスフェラーゼは、ジャガイモ、Thermus aquaticus、Thermococcus litralisに由来することが好ましく、ジャガイモ、Thermus aquaticusに由来することがより好ましい。本発明で用いられる4−α−グルカノトランスフェラーゼは、反応至適温度が高いことが好ましい。反応至適温度が高い4−α−グルカノトランスフェラーゼは、例えば、高度好熱細菌に由来し得る。
【0164】
(16.グリコーゲンデブランチングエンザイム(EC.2.4.1.25/EC.3.2.1.33))
本発明の方法において、生成物に環状構造を生じさせる場合には、必要に応じて、グリコーゲンデブランチングエンザイムを用いることができる。
【0165】
本発明で用いられ得るグリコーゲンデブランチングエンザイムは、α−1,6−グルコシダーゼ活性と、4−α−グルカノトランスフェラーゼ活性との2種類の活性をもつ酵素である。グリコーゲンデブランチングエンザイムが持つ、4−α−グルカノトランスフェラーゼ活性により、環状構造を持つ生成物が得られる。
【0166】
グリコーゲンデブランチングエンザイムは、微生物および動物に存在する。グリコーゲンデブランチングエンザイムを産生する微生物の例としては、酵母などが挙げられる。グリコーゲンデブランチングエンザイムを産生する動物の例としては、ヒト、ウサギ、ラット、ブタなどの哺乳類が挙げられる。グリコーゲンデブランチングエンザイムを産生する生物はこれらに限定されない。
【0167】
本発明で用いられ得るグリコーゲンデブランチングエンザイムは、酵母に由来することが好ましい。本発明で用いられるグリコーゲンデブランチングエンザイムは、反応至適温度が高いことが好ましい。反応至適温度が高いグリコーゲンデブランチングエンザイムは、例えば、タンパク質工学的手法により、中温で作用し得る酵素に改変を加えることで得られる。
【0168】
(17.本発明で使用される酵素についての一般的説明)
本明細書中では、酵素がある生物に「由来する」とは、その生物が天然で有するその酵素のアミノ酸配列と同じアミノ酸配列を有することをいう。ある生物の有するある酵素をその生物から直接単離した場合も、遺伝子組換え技術を用いてその生物の有するアミノ酸配列と同じアミノ酸配列を有する酵素を別の生物から得た場合も、その生物に由来するという。人工合成された遺伝子であっても同じ配列を有する場合はその生物に由来するという。場合によっては、それらの場合を含めて、その生物から得られた酵素という。例えば、あの生物から入手したある酵素をコードする核酸分子を大腸菌に導入して、その大腸菌からその酵素を単離する場合も、その酵素は「その生物に由来する」または「その生物から得られた」という。
【0169】
本発明で用いられる各種酵素は、各酵素について記載したような、自然界に存在する、その酵素を産生する生物から直接単離された酵素であってよい。あるいは、本発明で用いられる各種酵素は、その酵素を産生する生物から単離した、その酵素をコードする核酸分子を用いて組換えされた微生物(例えば、細菌、真菌など)から単離した酵素であってもよい。
【0170】
本発明においては、天然の酵素と同等以上の酵素活性を有するのであれば、そのアミノ酸配列の一部が改変された酵素を使用してもよい。改変された酵素が使用される場合、この改変された酵素は、天然の酵素に対して、好ましくは少なくとも約80%以上のアミノ酸配列同一性を有し、より好ましくは少なくとも約90%以上のアミノ酸配列同一性を有し、最も好ましくは約95%以上のアミノ酸配列同一性を有する。好ましくは、この改変された酵素は、天然の酵素に対して1または数個のアミノ酸の置換、欠失または付加を有する。このような置換、欠失または付加は、保存的なものであることが好ましい。このような置換、欠失または付加は、酵素の非活性部位に存在することが好ましい。天然の酵素活性に悪影響を及ぼさずにアミノ酸の置換、欠失または付加をする方法は、当業者に公知である。好ましくは、この改変された酵素は、天然の酵素と比較して約90%以上の活性を有し、より好ましくは約95%以上の活性を有し、さらに好ましくは約100%以上の活性を有する。このような改変された酵素は、当業者に公知の方法によって作製され得る。
【0171】
本発明の方法で用いられる各種酵素は、例えば、以下のようにして調製され得る。まず、その酵素を産生する微生物(例えば、細菌、真菌など)を培養する。この微生物は、その酵素を直接生産する微生物であってもよい。また、その酵素をコードする核酸分子をクローン化し、得られた核酸分子で、その酵素の発現に有利な微生物(例えば、細菌、真菌など)を組換えして、組換えされた微生物を得、得られた微生物からその酵素を得てもよい。
【0172】
各種酵素をコードする核酸分子での組換えに用いられる微生物は、各種酵素の発現の容易さ、培養の容易さ、増殖の速さ、安全性などの種々の条件を考慮して容易に選択され得る。アミラーゼは、生成されたグルカンを分解してしまうので、本発明のアミロース合成方法に使用される各種酵素は、夾雑物としてアミラーゼを含まないことが好ましい。従って、アミラーゼを産生しないかまたは低レベルでしか発現しない微生物(例えば、細菌、真菌など)を遺伝子組換えに用いることが好ましい。各種酵素の遺伝子組換えのためには、大腸菌または枯草菌のような中温菌を用いることが好ましい。アミラーゼを産生しないかまたは低レベルでしか発現しない微生物(例えば、細菌、真菌など)を用いて産生される各種酵素は、アミラーゼを実質的に含まないため、本発明の方法での使用に好ましい。
【0173】
クローン化した遺伝子での微生物(例えば、細菌、真菌など)の遺伝子組換えは、当業者に周知の方法に従って行われ得る。クローン化した遺伝子を用いる場合、この遺伝子を、構成性プロモーターまたは誘導性プロモーターに作動可能に連結することが好ましい。「作動可能に連結する」とは、プロモーターと遺伝子とが、そのプロモーターによって遺伝子の発現が調節されるように連結されることをいう。誘導性プロモーターを用いる場合、培養を、誘導条件下で行うことが好ましい。種々の誘導性プロモーターは当業者に公知である。
【0174】
クローン化した遺伝子について、生産される各種酵素が菌体外に分泌されるように、シグナルペプチドをコードするヌクレオチド配列をこの遺伝子に連結し得る。シグナルペプチドをコードするヌクレオチド配列は当業者に公知である。
【0175】
当業者は、各種酵素を生産するために、微生物(例えば、細菌、真菌など)の培養の条件を適切に設定し得る。微生物の培養に適切な培地、各誘導性プロモーターに適切な誘導条件などは当業者に公知である。
【0176】
例えば、発現された酵素が形質転換細胞内に蓄積する場合、形質転換細胞を適切な条件下で培養した後、培養物を遠心分離または濾過することによって細胞を回収し、次いで適切な緩衝液に懸濁する。次いで超音波処理などにより細胞を破砕した後、遠心分離もしくは濾過することによって上清を得る。あるいは、発現された酵素が形質転換細胞外に分泌される場合、このようにして形質転換細胞を培養した後、培養物を遠心分離または濾過することによって細胞を分離して上清を得る。目的の酵素が形質転換細胞内に蓄積する場合も、形質転換細胞外に分泌される場合も、このようにして得られた酵素含有上清を通常の手段(例えば、塩析法、溶媒沈澱、限外濾過)を用いて濃縮し、酵素を含む画分を得る。この画分を濾過、あるいは遠心分離、脱塩処理などの処理を行い粗酵素液を得る。さらにこの粗酵素液を、凍結乾燥、等電点電気泳動、イオン交換クロマトグラフィー、晶出などの通常の酵素の精製手段を適宜組み合わせることによって、比活性が向上した粗酵素あるいは精製酵素が得られる。α−アミラーゼなどのグルカンを加水分解する酵素が含まれていなければ、粗酵素をそのまま、例えば、α−グルカンの製造に用い得る。
【0177】
各種酵素は、精製されていても未精製であってもよい。各種酵素は、固定化されていても固定化されていなくともよい。各種酵素は、固定化されることが好ましい。固定化の方法としては、担体結合法(たとえば、共有結合法、イオン結合法、または物理的吸着法)、架橋法または包括法(格子型またはマイクロカプセル型)など、当業者に周知の方法が使用され得る。各種酵素は、担体上に固定化されていることが好ましい。
【0178】
(18.溶媒)
本発明の方法に用いる溶媒は、本発明で用いられる各種酵素の酵素活性を損なわない溶媒であれば任意の溶媒であり得る。
【0179】
なお、グルカンを生成する反応が進行し得る限り、溶媒が本発明の方法に用いる材料を完全に溶解する必要はない。例えば、酵素が固体の担体上に担持されている場合には、酵素が溶媒中に溶解する必要はない。さらに、グルコースなどの反応材料も全てが溶解している必要はなく、反応が進行し得る程度の材料の一部が溶解していればよい。
【0180】
代表的な溶媒は、水である。溶媒は、各種酵素を調製する際に得られる細胞破砕液のうちの水分であってもよい。
【0181】
水は、軟水、中間水および硬水のいずれであってもよい。硬水とは、硬度20°以上の水をいい、中間水とは、硬度10°以上20°未満の水をいい、軟水とは、硬度10°未満の水をいう。水は、好ましくは軟水または中間水であり、より好ましくは軟水である。
【0182】
(19.他の成分)
本発明で使用される溶液中には、目的の反応を妨害しない限り、任意の他の物質を含み得る。このような物質の例としては、緩衝剤、各種酵素を産生する微生物(例えば、細菌、真菌など)の成分、塩類、培地成分などが挙げられる。
【0183】
<α−グルカンの製造>
1つの実施形態では、本発明のα−グルカンは、グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法によって製造される。
【0184】
別の実施形態では、本発明のα−グルカンは、グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法によって製造される。
【0185】
別の実施形態では、本発明のα−グルカンは、セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0186】
別の実施形態では、本発明のα−グルカンは、フルクトースと、ポリリン酸と、プライマーと、グルコースイソメラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0187】
別の実施形態では、本発明のα−グルカンは、デンプンをアミラーゼによってグルコースに分解する工程;アミラーゼを失活させる工程;ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法によって製造される。
【0188】
図15に、これらの方法をまとめて記載する。本発明の最も基本的な方法は、G6−PからG1−Pを経てアミロース、デンプン、グリコーゲンなどのα−グルカンを製造する方法である。G6−Pは、グルコースおよびポリリン酸にPGKを作用させることにより生成され得る。グルコースは、フルクトースにグルコースイソメラーゼを作用させることによって生成され得る。グルコースはまた、セルロースにセルラーゼ、セロビオースホスホリラーゼなどを作用させることによって生成され得る。グルコースはまた、デンプンにアミラーゼを作用させることによって生成され得る。生成物であるα−グルカンを分解してしまうアミラーゼを使用する方法を除けば、これらのグルコースを生成する方法は、グルコースからG6−Pを経て、次いでG1−Pを経てα−グルカンを製造する方法と同一の溶液中で同時に行われ得る。
【0189】
本発明の製造方法においては例えば、まず、溶液を調製する。溶液は、例えば、適切な溶媒に、反応に使用する各種物質を添加することにより調製され得る。あるいは、溶液は、反応に使用する各種物質をそれぞれ含む溶液を混合することによって調製してもよい。あるいは、溶液は、反応に使用する各種物質のうちのいくつかの成分を含む溶液に固体状の他の成分を混合することによって調製してもよい。本発明の製造方法で用いられる溶液には、酵素反応を阻害しない限り、必要に応じて、pHを調整する目的で任意の緩衝剤を加えてもよい。この溶液のpHは、酵素反応を過度に阻害しない限り、任意のpHであり得る。pH値は、好ましくは約5.7〜約11であり、より好ましくは約7.4〜約8.5である。pHは、反応に用いる酵素の至適pHに合わせて適切に設定され得る。溶液の塩濃度もまた、酵素反応を過度に阻害しない限り、任意の塩濃度であり得る。PGMはMgイオンを必要とするため、MgCl2などのMgイオンを放出する物質を溶液に添加することが好ましい。Mgイオンを放出する物質の、反応開始時の溶液中での濃度は、好ましくは約20mM以上であり、より好ましくは約30mM以上であり、さらに好ましくは約40mM以上である。Mgイオンを放出する物質の、反応開始時の溶液中での濃度は、好ましくは約100mM以下であり、より好ましくは約80mM以下であり、さらに好ましくは約60mM以下である。
【0190】
また、この溶液には、必要に応じて枝切り酵素、ブランチングエンザイム、4−α−グルカノトランスフェラーゼおよびグリコーゲンデブランチングエンザイムからなる群より選択される酵素を添加してもよい。これらの酵素は、α−グルカン合成反応の開始時に添加されてもよく、反応の途中に添加されてもよく、また、反応が終了した後に添加されてもよい。
【0191】
さらに、この溶液には、ゲスト化合物を添加してもよい。ゲスト化合物は、反応開始時にこの溶液中に含有されてもよく、あるいは、反応がある程度進んだ時点で反応溶液に添加されてもよい。ゲスト化合物としてN−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物としてN−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネートを使用する場合、この化合物の濃度が10mMであると、コントロール(ゲスト化合物を添加しない場合)の約1.5倍の収率を得ることができるという顕著な効果が得られる。
【0192】
ゲスト化合物として3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物として3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度が1mM〜10mMであると、コントロール(ゲスト化合物を添加しない場合)の約1.8倍〜約2倍の収率を得ることができるという顕著な効果が得られる。
【0193】
ゲスト化合物として3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度は、好ましくは約0.1mM以上であり、より好ましくは約1mM以上であり、さらに好ましくは約10mM以上であり;この化合物の濃度は、好ましくは約50mM以下であり、より好ましくは約20mM以下であり、さらに好ましくは約15mM以下である。ゲスト化合物として3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートを使用する場合、この化合物の濃度が10mMであると、コントロール(ゲスト化合物を添加しない場合)の約2.7倍の収率を得ることができるという顕著な効果が得られる。
【0194】
次いで、溶液を、当該分野で公知の方法によって必要に応じて加熱することにより、反応させる。溶液の温度は、本発明の効果が得られる限り、任意の温度であり、添加した酵素がその活性を示す温度である。例えば、耐熱性酵素を用い、反応温度をその耐熱酵素に最適な温度にすることによって、添加した耐熱性酵素以外の混入した酵素の活性を抑え得る。この反応工程における溶液の温度は、所定の反応時間後に反応前のこの溶液に含まれる各種酵素の活性の約50%以上、より好ましくは約80%以上の活性が残る温度であることが好ましい。反応に使用する各種酵素の反応至適温度がいずれも約30℃〜40℃の範囲内である場合、反応温度は、好ましくは約25℃以上であり、より好ましくは約30℃以上であり;反応温度は、好ましくは約50℃以下であり、より好ましくは約40℃以下である。反応に使用する各種酵素がいずれも耐熱性酵素である場合、反応温度は、その耐熱性酵素の反応至適温度の±10℃以内にあることが好ましく、±5℃以内にあることが好ましい。反応に使用する各種酵素の反応至適温度が幅広い範囲にわたる場合、反応温度は、その範囲内で適切に設定され得る。反応に使用する各種酵素の反応至適温度が±5℃以内にあることが好ましく、ほぼ同じであることが好ましい。
【0195】
グルコースを出発物質とする実施形態では、反応溶液の初期pH(反応開始時の溶液のpH)は、好ましくは約5.7以上であり、より好ましくは約7.4以上である。グルコースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12.0以下であり、より好ましくは約11.0以下であり、さらに好ましくは約8.5以下である。反応で使用する各酵素の代表的な至適pHは、Microlunatus phosphovolas由来のPGKが約pH7.0であり、Escherichia coli由来のPGMが約pH7.5であり、そしてGPが約pH5.5である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約20%程度のアミロース収率を得られる。
【0196】
G6−Pを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約7.3以上であり、より好ましくは約7.5以上である。G6−Pを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12.0以下であり、より好ましくは約11.0以下であり、さらに好ましくは約8.5以下である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約25%以上のアミロース収率を得られる。
【0197】
セロビオースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約5.6以上であり、より好ましくは約7.5以上である。セロビオースを出発物質とする実施形態では、反応溶液の初期pHは、好ましくは約12以下であり、より好ましくは約8.5以下である。初期pHが低すぎるとアミロース合成収率が低くなるかまたはアミロースが合成されない。反応溶液の初期pHを適切に調整することにより、約62%以上のアミロース収率を得られる。
【0198】
グルコースを出発物質とする実施形態では、反応溶液の初期グルコース濃度(反応開始時の溶液のグルコース濃度)は、好ましくは約100mM以上であり、より好ましくは約200mM以上であり、特に好ましくは約400mM以上であり、最も好ましくは約500mM以上である。グルコースを出発材料として用いる場合、溶液中に含まれるグルコースの濃度に特に上限はないが、例えば、約2.5M以下、約2M以下、約1.5M以下、約1M以下、約750mM以下などであり得る。グルコースの量が多すぎると、反応に適切な量のポリリン酸を溶解することができず、収率が低下する場合がある。グルコースの使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0199】
グルコース−6−リン酸を出発物質とする実施形態では、反応溶液の初期グルコース−6−リン酸濃度(反応開始時の溶液のグルコース−6−リン酸濃度)は、好ましくは約20mM以上であり、より好ましくは約100mM以上であり、特に好ましくは約200mM以上である。グルコース−6−リン酸を出発材料として用いる場合、溶液中に含まれるグルコース−6−リン酸の濃度に特に上限はないが、例えば、約2M以下、約1.5M以下、約1M以下、約750mM以下などであり得る。グルコース−6−リン酸の量が多すぎると溶解することができず、収率が低下する場合がある。グルコース−6−リン酸の使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0200】
セロビオースを出発物質とする実施形態では、反応溶液の初期セロビオース濃度(反応開始時の溶液のセロビオース濃度)は、好ましくは約100mM以上であり、より好ましくは約150mM以上であり、特に好ましくは約175mM以上である。セロビオースを出発材料として用いる場合、溶液中に含まれるセロビオースの濃度に特に上限はないが、例えば、約1.0M以下、約750mM以下、約500mM以下、約250mM以下、約125mM以下などであり得る。セロビオースの量が多すぎると、セロビオースそのものや反応に適切な量のポリリン酸を溶解することができず、収率が低下する場合がある。セロビオースの使用量が少なすぎると、アミロース合成の方向に反応が進まない場合がある。
【0201】
グルコースを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が、好ましくは約0.15以下であり、より好ましくは約0.03以下である。
【0202】
G6−Pを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/G6−P)が、好ましくは約0.4以下であり、より好ましくは約0である。
【0203】
セロビオースを出発物質とする実施形態では、反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/CB)が、好ましくは約0〜4であり、より好ましくは約0.008〜2であり、更に好ましくは0.028〜0.810であり、最も好ましくは0.056〜0.162である。
【0204】
反応時間は、反応温度、反応により生産されるグルカンの分子量および酵素の残存活性を考慮して、任意の時間で設定され得る。反応時間は、好ましくは約1時間以上であり、より好ましくは約2時間以上であり、さらに好ましくは約3時間以上である。反応時間は、好ましくは約100時間以下であり、より好ましくは約72時間以下であり、さらに好ましくは約54時間以下であり、特に好ましくは約36時間以下であり、最も好ましくは約24時間以下である。
【0205】
加熱は、どのような手段を用いて行ってもよいが、溶液全体に均質に熱が伝わるように、攪拌を行いながら加熱することが好ましい。溶液は、例えば、温水ジャケットと攪拌装置を備えたステンレス製反応タンクの中に入れられて攪拌される。
【0206】
本発明の方法ではまた、反応がある程度進んだ段階で、各種原料のうちの少なくとも1つを反応溶液に追加してもよい。
【0207】
このようにして、α−グルカンを含有する溶液が生産される。
【0208】
反応終了後、溶液は、必要に応じて例えば、100℃にて10分間加熱することによって溶液中の酵素を失活させ得る。あるいは、酵素を失活させる処理を行うことなく後の工程を行ってもよい。溶液は、そのまま保存されてもよいし、生産されたグルカンを単離するために処理されてもよい。
【0209】
<精製方法>
生産されたα−グルカンは、必要に応じて精製され得る。精製することにより除去される不純物の例は、グルコースである。α−グルカンの精製法の例としては、有機溶媒を用いる方法(T.J.Schochら、J.American Chemical Society,64,2957(1942))および有機溶媒を用いない方法がある。
【0210】
有機溶媒を用いる精製に使用され得る有機溶媒の例としては、アセトン、n−アミルアルコール、ペンタゾール、n−プロピルアルコール、n−ヘキシルアルコール、2−エチル−1−ブタノール、2−エチル−1−ヘキサノール、ラウリルアルコール、シクロヘキサノール、n−ブチルアルコール、3−ペンタノール、4−メチル−2−ペンタノール、d,l−ボルネオール、α−テルピネオール、イソブチルアルコール、sec−ブチルアルコール、2−メチル−1−ブタノール、イソアミルアルコール、tert−アミルアルコール、メントール、メタノール、エタノールおよびエーテルが挙げられる。
【0211】
有機溶媒を用いない精製方法の例を、以下に示す。
【0212】
(1)α−グルカン生産反応後、反応溶液を冷却することによりα−グルカンを沈澱させ、そして沈澱したα−グルカンを、膜分画、濾過、遠心分離などの一般的な固液分離方法により精製する方法;
(2)α−グルカン生産反応の間もしくはα−グルカン生産反応後に反応溶液を冷却してα−グルカンをゲル化し、ゲル化したα−グルカンを回収し、そしてゲル化したα−グルカンから、グルコースを、水による洗浄、凍結融解、ろ過などの操作によって除去する方法;ならびに
(3)α−グルカン生産反応後、水に溶解しているα−グルカンを沈澱させずに、限外ろ過膜を用いた膜分画もしくはクロマトグラフィーに供してグルコースを除去する方法。
【0213】
精製に使用され得る限外濾過膜の例としては、分画分子量約1,000〜約100,000、好ましくは約5,000〜約50,000、より好ましくは約10,000〜約30,000の限外濾過膜(ダイセル製UF膜ユニット)が挙げられる。
【0214】
クロマトグラフィーに使用され得る担体の例としては、ゲル濾過クロマトグラフィー用担体、配位子交換クロマトグラフィー用担体、イオン交換クロマトグラフィー用担体および疎水クロマトグラフィー用担体が挙げられる。
【実施例】
【0215】
以下の実施例により本発明をさらに詳細に説明する。本発明は以下の実施例のみに限定されない。
【0216】
(1.測定方法および計算方法)
本発明における各種酵素の活性および得られるα−グルカンの収率を、以下の測定方法によって測定した。
【0217】
(1.1 ポリホスフェートグルコキナーゼの活性測定法)
200mM グルコース、100mM MgCl2、8%ポリリン酸の混合液25μlと適切に希釈した酵素液25μlを加えて50μlの混合溶液として反応を開始させる。この混合溶液を45℃で15分間インキュベートして反応させた後、100℃で10分間保持することにより酵素を失活させる。続いて20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを酵素反応溶液に添加して混合する。この溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。この方法により、1分間に1μmolのグルコース−6−リン酸を生成する活性をポリホスフェートグルコキナーゼ1単位とする。
【0218】
(1.2 ホスホグルコムターゼの活性測定法)
200mM グルコース−1−リン酸、100mM MgCl2、20mM Cofactor (グルコース またはグルコース−1,6−ビスホスフェート)の混合液25μlと適切に希釈した酵素液25μlを加えて50μlの混合溶液として反応を開始させる。この混合溶液を45℃で15分間インキュベートして反応させた後、100℃で10分間保持することにより酵素を失活させる。続いて20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを酵素反応溶液に添加して混合する。この溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。この方法により、1分間に1μmolのグルコース−6−リン酸を生成する活性をホスホグルコムターゼ1単位とする。
【0219】
(1.3 α−1,4−グルカンホスホリラーゼの活性測定法)
50μlの4%クラスターデキストリン水溶液と50μlの50mMグルコース−1−リン酸ナトリウム水溶液とを混合し、さらに適切に希釈した酵素液100μlを加えて200μlの混合物として反応を開始させる。この混合物を37℃で15分間インキュベートして反応を進行させた後、800μlのモリブデン試薬(15mM モリブデン酸アンモニウム、100mM 酢酸亜鉛)を混合し、反応を停止させる。続いて200μlの568mMアスコルビン酸(pH5.0)を加えて攪拌し、反応系を得る。この反応系を、30℃で20分間保持した後、分光光度計を用いて850nmでの吸光度を測定する。濃度既知の無機リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中の無機リン酸を求める。この方法により、1分間に1μmolの無機リン酸を生成する活性を、α−1,4−グルカンホスホリラーゼ1単位とする。
【0220】
(1.4 セロビオースホスホリラーゼの活性測定法)
25μlの40mMセロビオース水溶液と25μlの40mMリン酸ナトリウム水溶液(pH7.5)とを混合し、さらに適切に希釈した酵素液(試料)50μlを加えて100μlの混合物として反応を開始させる。この混合物を37℃で10分間インキュベートすることにより反応を進行させた後、100℃で10分間保持することによって酵素を失活させる。続いて600μlの1M Tris−塩酸緩衝液(pH7.0)および300μlの発色試薬(CII発色試薬(和光純薬社製))をこの混合液に添加して混合し、30℃で20分間保持した後、505nmでの吸光度を測定する。濃度既知のグルコース水溶液を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース量を求める。セロビオースホスホリラーゼ1単位とは、上記方法により20mMセロビオースから1分間に1μmolのグルコースを生成する酵素量と定義する。
【0221】
(1.5 得られるα−グルカンの収率の計算方法)
本発明の製造方法によるα−グルカンの収率を、得られたα−グルカン中に取り込まれたグルコース残基のモル数が、最初に添加された初発物質のグルコース当量でのモル数(S;グルコース当量のモル数)(例えば、グルコースを出発物質とする場合はグルコースのモル数、G6−Pを出発物質とする場合はG6−Pのモル数、二糖のセロビオースを出発物質とする場合はセロビオースのモル数の2倍量)の何%にあたるかによって計算した。反応終了後、反応溶液を100℃で10分間加熱して酵素を失活させた。次いで、反応溶液を適切に希釈し、充分量のα−アミラーゼと充分量のグルコアミラーゼを添加して40℃で6時間インキュベートすることによって溶液中のα−グルカンをグルコースにまで分解した。α−アミラーゼおよびグルコアミラーゼによる処理を行った溶液のグルコース量(G1;モル数)、およびこの処理を行ってない溶液のグルコース量(G2;モル数)を測定した。グルコース当量でG3モルのプライマーを使用したとすると、Gx=(G1−G2−G3)によって、α−グルカンに取り込まれたグルコース量(収量;モル数)が計算できる。このグルコース量を初発物質のモル数で除算して100倍することにより、収率を計算した。この計算式を次式に示す。
【0222】
【数1】
(1.6 グルコース濃度測定方法)
適切に希釈した試料100μlに600μlの1M Tris−塩酸緩衝液(pH7.0)および300μlの発色試薬(CII発色試薬(和光純薬社製))を添加して混合し、37℃、20分間保持した後、505nmでの吸光度を測定する。濃度既知のグルコース水溶液を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース量を求める。
【0223】
(1.7 リン酸濃度測定方法)
適切に希釈した試料200μlに800μlのモリブデン試薬(15mM モリブデン酸アンモニウム、100mM 酢酸亜鉛)および200μlの568mMアスコルビン酸(pH5.0)を加えて攪拌し、30℃で20分間保持した後、分光光度計を用いて850nmでの吸光度を測定する。濃度既知の無機リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中の無機リン酸を求める。
【0224】
(1.8 G1−P濃度測定方法)
適切に希釈した試料50μlに20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、15μM グルコース−1,6−ビスホスフェート、3μg/ml ウサギ筋肉由来ホスホグルコムターゼ、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを添加して混合する。この混合溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−1−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−1−リン酸濃度を求める。
【0225】
(1.9 G6−P濃度測定方法)
適切に希釈した試料50μlに20mM Tris−HCl(pH7) 550μlおよび3mM NADP+、15mM MgCl2、3mM EDTA、6.16μg/ml グルコース−6−ホスホデヒドロゲナーゼ、200mM Tris−HCl (pH7)を含む混合溶液300μlを添加して混合する。この混合溶液を30℃、30分間保持した後、分光光度計を用いて340nmでの吸光度を測定する。濃度既知のグルコース−6−リン酸を用いて同様に吸光度を測定し、標準曲線を作成する。この標準曲線に試料で得られた吸光度を当てはめ、試料中のグルコース−6−リン酸濃度を求める。
【0226】
(2.酵素の調製)
本発明の実験例および実施例で用いた各種酵素を、以下の方法によって調製した。
【0227】
(2.1 組換えポリホスフェートグルコキナーゼ(PGK)の調製方法)
Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのヌクレオチド配列(配列番号1)を合成し、発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−PGKを得た。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。
【0228】
その後、得られた大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で大腸菌の増殖したコロニーを選択することにより、Microlunatus phosphovolas由来ポリホスフェートグルコキナーゼ遺伝子が導入された大腸菌を得た。それらの大腸菌についてポリホスフェートグルコキナーゼを発現していることを酵素活性測定によって確認した。
【0229】
この大腸菌を、抗生物質カナマイシンを含むTB培地(1.2%トリプトン、2.4%酵母エキス(ともにDifco社製)、0.231%リン酸二水素カリウム、1.254%リン酸水素二カリウム、0.4%グリセリン)600mLに接種し、100rpmで振盪させながら37℃、24時間培養した。次いで、この培養液を5,000rpmにて10分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、180mLの50mM MOPS buffer (pH7)中に懸濁し、次いで1%リゾチームを20mL加え溶菌させた。溶菌後、10,000rpmにて10分間遠心分離し、ポリホスフェートグルコキナーゼを含む沈澱を得た。この沈澱を再度180mLの50mM MOPS buffer(pH7)中に懸濁し、酵素保持のため20mLグリセリン(final10%)を加えた。この懸濁液中には357U/mLのポリホスフェートグルコキナーゼが含まれていた。
【0230】
なお、得られた組換えポリホスフェートグルコキナーゼの反応至適温度は65℃であり、反応至適pHは約7.0である。
【0231】
(2.2 組換えホスホグルコムターゼ(PGM)の調製方法)
Escherichia coliに由来するホスホグルコムターゼのヌクレオチド配列(配列番号3)を合成し、発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−PGKを得た。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。
【0232】
その後、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で大腸菌の増殖したコロニーを選択することにより、Escherichia coli由来ホスホグルコムターゼ遺伝子が導入された大腸菌を得た。それらの大腸菌についてホスホグルコムターゼを発現していることを酵素活性測定によって確認した。
【0233】
この大腸菌を、抗生物質カナマイシンを含むTB培地(1.2%トリプトン、2.4%酵母エキス(ともにDifco社製)、0.231%リン酸二水素カリウム、1.254%リン酸水素二カリウム、0.4%グリセリン)600mLに接種し、100rpmで振盪させながら37℃、24時間培養した。次いで、この培養液を5,000rpmにて10分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、180mLの50mM MOPS buffer (pH7)中に懸濁し、次いで1%リゾチームを20mL加え溶菌させた。溶菌後、10,000rpmにて10分間遠心分離し、ホスホグルコムターゼを含む酵素溶液を得た。この酵素溶液には500U/mLのホスホグルコムターゼが含まれていた。
【0234】
なお、得られた組換えホスホグルコムターゼの反応至適温度は60℃であり、反応至適pHは約7.5である。
【0235】
(2.3 組換え馬鈴薯α−1,4−グルカンホスホリラーゼ(GP)の調製方法)
馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子(Nakanoら、Journal of Biochemistry(Tokyo)106(1989)691;配列番号5)を選択マーカー遺伝子Amprとともに発現ベクターpET3d(STRATAGENE社製)に組み込み、プラスミドpET−PGP113を得た。このプラスミドでは、α−1,4−グルカンホスホリラーゼ遺伝子を、イソプロピル−β−D−チオガラクトピラノシド(IPTG)誘導性プロモーターの制御下に作動可能に連結した。このプラスミドを、大腸菌BL21(DE3)(STRATAGENE社製)に、コンピテントセル法により導入した。この大腸菌を、抗生物質アンピシリンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で増殖した大腸菌を選択することにより、馬鈴薯由来α−1,4−グルカンホスホリラーゼ遺伝子が導入された大腸菌を得た。得られた大腸菌がα−1,4−グルカンホスホリラーゼ遺伝子を含むことを、導入された遺伝子の配列を解析することによって確認した。また、得られた大腸菌がα−1,4−グルカンホスホリラーゼを発現していることを、活性測定によって確認した。
【0236】
この大腸菌を、抗生物質アンピシリンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム)1リットルに接種し、120rpmで振盪させながら37℃で3時間振盪培養した。その後、IPTGを0.1mM、ピリドキシンを1mMになるようにそれぞれこの培地に添加し、22℃でさらに20時間振盪培養した。次いで、この培養液を5,000rpmにて5分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、50mlの0.05%のTriton X−100を含む20mM トリス−塩酸緩衝液(pH7.0)中に懸濁し、次いで超音波処理により破砕し、菌体破砕液50mlを得た。この破砕液中には、4.7U/mgのα−1,4−グルカンホスホリラーゼが含まれていた。
【0237】
この菌体破砕液を、55℃で30分間加熱した。加熱後、8,500rpmにて20分間遠心分離し、不溶性のタンパク質などを除去して上清を得た。得られた上清を、あらかじめ平衡化しておいた陰イオン交換樹脂Q−Sepharoseに流してα−1,4−グルカンホスホリラーゼを樹脂に吸着させた。樹脂を、200mM塩化ナトリウムを含む緩衝液で洗浄して不純物を除去した。続いて、タンパク質を300mM塩化ナトリウムを含む緩衝液で溶出させ、組換えα−1,4−グルカンホスホリラーゼ酵素溶液とした。
【0238】
なお、得られた組換えα−1,4−グルカンホスホリラーゼの反応至適温度は40℃であり、反応至適pHは約5.6である。
【0239】
(2.4 組換えセロビオースホスホリラーゼの調製方法)
Clostridium thermocellumの染色体遺伝子を抽出し、これをテンプレートとした。以下の2種の合成DNAプライマー:
合成DNAプライマー1:5’ aaactctagaaataattttgtttaactttaagaaggagatataccatggagttcggtttttttgatgat 3’(配列番号7)および
合成DNAプライマー2:5’ aaactcgagaattacttcaactttgtgagtcttt 3’(配列番号8)
を用い、
98℃で1分間、55℃で1分間、68℃で3分間の順で30サイクル加熱
の条件下でPCRを行うことにより、CBP遺伝子を含む領域を増幅させた。増幅した遺伝子を選択マーカー遺伝子Kmrとともに発現ベクターpET28a(STRATAGENE社製)に組み込み、プラスミドpET28a−CBP1を得た。このプラスミドでは、セロビオースホスホリラーゼ遺伝子を、イソプロピル−β−D−チオガラクトピラノシド(IPTG)誘導性プロモーターの制御下に作動可能に連結した。
【0240】
このプラスミドを、大腸菌BL21(DE3)pLysS(STRATAGENE社製)に、コンピテントセル法により導入した。この大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン(Difco社製)、0.5%酵母エキス(Difco社製)、1%塩化ナトリウム、1.5%寒天))を含むプレートにプレーティングして、37℃で一晩培養した。このプレート上で増殖した大腸菌を選択することにより、Clostridium thermocellum由来セロビオースホスホリラーゼ遺伝子が導入された大腸菌を得た。
【0241】
得られた大腸菌がセロビオースホスホリラーゼ遺伝子を含むことを、導入された遺伝子の配列を解析することによって確認した。また、得られた大腸菌がセロビオースホスホリラーゼを発現していることを、活性測定によって確認した。
【0242】
この大腸菌を、抗生物質カナマイシンを含むLB培地(1%トリプトン、0.5%酵母エキス(ともにDifco社製)、1%塩化ナトリウム)1リットルに接種し、120rpmで振盪させながら37℃で3時間振盪培養した。その後、IPTGを1.0mMになるようにこの培地に添加し、37℃でさらに8時間振盪培養した。次いで、この培養液を5,000rpmにて5分間遠心分離して、大腸菌の菌体を収集した。得られた菌体を、50mlの1.4mMの2−メルカプトエタノールを含む50mMリン酸緩衝液(pH7.5)中に懸濁し、次いで超音波処理により破砕し、菌体破砕液50mlを得た。この破砕液中には、132U/mlのセロビオースホスホリラーゼが含まれていた。
【0243】
この菌体破砕液を、55℃で20分間加熱した。加熱後、8,500rpmにて20分間遠心分離し、不溶性のタンパク質などを除去して上清を得た。得られた上清を、あらかじめ平衡化しておいたHis−Tag吸着樹脂Ni−NTA agarose(QIAGEN社製)に流してセロビオースホスホリラーゼをこの樹脂に吸着させた。この樹脂を、300mM塩化ナトリウムと20mMイミダゾールおよび1.4mM2−メルカプトエタノール含む緩衝液で洗浄して不純物を除去した。続いて、タンパク質を300mM塩化ナトリウムと150mMイミダゾールおよび1.4mM 2−メルカプトエタノールを含む緩衝液で溶出させ、組換えセロビオースホスホリラーゼ酵素溶液とした。
【0244】
なお、得られた組換えセロビオースホスホリラーゼの反応至適温度は60℃であり、反応至適pHは6.0である。
【0245】
(実施例1:グルコースからのアミロースの合成)
以下の表1に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを7.0に調整し、そして45℃で24時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約20mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、約0.04であった。
【0246】
【表1】
グルコース(Glc)濃度、グルコース−6−リン酸(G6−P)濃度、グルコース−1−リン酸(G1−P)濃度、およびアミロース収率を経時的に測定した。結果を以下の表2および図2に示す。
【0247】
【表2】
反応開始から12時間以降G6−P濃度とG1−P濃度がほぼ一定になったことから、溶液中の酵素反応は、反応開始から12時間で平衡に達したと考えられる。それに対して、アミロース収率は増加し、グルコース濃度が減少した。反応開始から12時間以降のアミロースの増加はグルコース減少量にほぼ一致している。この結果は、酵素反応が平衡に達した後アミロースが老化することで反応系外に出てそれに伴い平衡状態を保つためにGlcが消費され、反応がアミロース合成側にシフトすることによってアミロースが合成されたことを示すと考えられる。
【0248】
表2および図2においては、反応開始時のグルコース濃度を100%(mM/mM)とし、グルコース濃度、G6−P濃度、G1−P濃度およびアミロース収率をそれぞれ、反応開始時のグルコース濃度(mM)に対する比率として示した。
【0249】
(実施例2:反応開始時の反応溶液のpH)
反応開始時の溶液のpHを5.2〜12に変化させたこと以外は、上記表1の組成と同じ組成の溶液を調製し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。合成されたアミロースの濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、いずれの条件でも約12mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、いずれの条件でも約0.03であった。
【0250】
反応開始時点で測定したpHの値と、アミロースの収率の結果を以下の表3および図3に示す。
【0251】
【表3】
反応溶液の初期pHを弱アルカリ側以上にすることでアミロース合成収率が上がり、約20%の値が得られた。初期pHが5.6までの酸性側ではアミロースが酵素合成されず、弱酸性から中性付近では低収率でアミロースが得られた。初期pH7.1のとき収率9.1%であった。pH7.4からpH11ではアミロース収率15%以上を示し、pH7.4からpH8.5では約19%のアミロース収率を示した。初期pH7.7のとき収率19.6%であった。
【0252】
(実施例3:反応溶液中の遊離リン酸とグルコースとのモル比(Pi/Glc)の検討)
溶液中の遊離のリン酸濃度はアミロース合成反応に大きく影響を与える。直接的にはGP酵素の平衡反応に影響する。この平衡反応ではアミロースとともに副生成物としてリン酸が生成されるためリン酸濃度は高くなるとアミロース合成側への反応が進まなくなる。反応初期には溶液中にリン酸の存在しないことが望ましいが、GlcからG6−Pへの変換反応に必要なポリリン酸には遊離のリン酸が含まれてしまう。そのため、アミロース合成可能な初期遊離リン酸/Glc濃度比(Pi/Glc)を検討した。
【0253】
以下の表4に示す濃度の無機リン酸を添加したこと以外は表1の組成の溶液を作製して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約8に調整した。合成されたアミロースの量を測定し、収率を計算した。添加した無機リン酸の量、無機リン酸とグルコースとのモル比(Pi/Glc)、アミロースの合成濃度(mM)および収率(%)を以下の表4に示す。アミロースの収率(%)とPi/Glcとの関係を図4に示す。
【0254】
【表4】
この結果、グルコースの約0.2倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。
【0255】
(実験例1:グルコースからのG6−Pの生成反応)
先行技術(1)の反応の方向を確認するために、以下の実験を行った。
【0256】
以下の表5に記載の組成の溶液を作成し、反応開始時点でのこの溶液のpHを7に調整し、45℃で24時間インキュベートすることにより、G6−Pを生成させた。
【0257】
【表5】
生成されたG6−Pの濃度を測定し、G6−Pの収率(変換率)(%)を計算した。結果を図5に示す。この結果、先行技術の(1)の反応は不可逆反応であり、グルコースに対して90%以上がG6−Pに変換されることが確認された。
【0258】
(実験例2:G6−PとG1−Pとアミロースとの平衡反応)
アミロースをMOPS(pH7)バッファーに加えて100℃で熱処理して完全に溶解し、その後、45℃に保ったまま、以下の表6に記載の組成になるようにPGM、GP、リン酸などを加え、反応開始時点でのこの溶液のpHを7に調整し、そして4時間インキュベートして、平衡反応を行なった。
【0259】
【表6】
反応途中でサンプリングした。アミロース、G1−PおよびG6−Pの濃度を測定し、反応開始時のアミロースのモル数を100%としたときのアミロース、G1−P、G6−PおよびG1−P+G6−Pの濃度を計算した。結果を図6に示す。その結果、アミロースの95%以上がG6−PとG1−Pに分解された。2種類の酵素PGM−GPの平衡反応はG6−P側に非常に有利で、完全に溶解したアミロースに対してPGMとGPを作用させたとき反応開始から4時間後ではG6−P:88.5%、G1−P:6.8%、アミロース:4.7%の割合で存在した。
【0260】
(実施例4:アミロース合成)
以下の表7に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約12mMであり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、0.03であった。
【0261】
【表7】
アミロースの合成濃度を測定し、収率を計算した。その結果、収率は19.6%であった。上記の実験例2によれば、平衡状態でのアミロース収率が約4.7%であった。これに対し、本実施例では、500mMのグルコースを基質としたアミロース合成の収率が19.6%であった。この値は、本実施例によって得られるアミロースの収率が、PGM−GP平衡反応によって得られるアミロース収率を大きく上回ることを示す。本実施例によってPGM−GP平衡反応によるアミロース収率よりも高い値が得られた理由は、アミロースの老化によって反応系外に出たアミロースが蓄積されたためであると考えられる。
【0262】
(実施例5:出発グルコース濃度を変えた場合のアミロース収率の変化)
グルコース濃度を変化させ、それに合わせて酵素量、プライマー濃度およびポリリン酸濃度を比例して増加させたこと以外は、表1と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約0.5〜約50.2であり、遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)は、約0.03であった。
【0263】
結果を以下の表8および図7Aおよび図7Bに示す。
【0264】
【表8】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図7A)。また、グルコース濃度が増加するに従ってアミロースの収量も増加した(図7B)。
【0265】
(実施例6:G6−Pを出発原料として使用した、ゲスト化合物の存在下でのアミロース合成)
以下の表9に記載の組成をベースとして以下の表10に記載の量で各種ゲスト化合物を添加した組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、0mMであり、遊離リン酸のモル濃度とG6−Pのモル濃度との比率(Pi/G6−P)は、0であった。コントロールは、ゲスト化合物を添加しない比較例6である。合成されたアミロース量を測定し、収率を計算した。
【0266】
【表9】
結果を以下の表10および図8に示す。
【0267】
【表10】
アミロースは脂肪酸などを包接する作用を持ち、包接される側の物質(ゲスト化合物)の種類によっては沈澱しやすくなる。アミロース合成反応溶液中にゲスト化合物を添加し、この沈澱促進作用を利用することで、アミロース収率を上げることができる。G6−Pからアミロースを合成するとき、アミロース収率は20%程度であるが、ゲスト化合物を加えることで有意に収率が上昇した。ゲスト化合物SB3−16を10mM添加したときアミロース収率は53.3%まで上がった。
【0268】
(実施例7−1〜7−3:セロビオースからのアミロース合成)
以下の表11に記載の組成の溶液を調製し、反応開始時点でのこの溶液のpHを約7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。比較例7として、PGK、PGMおよびポリリン酸を含まず、リン酸を含む組成でアミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例7−1では約15mMであり、実施例7−2および7−3では約10mMであり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例7−1では約0.15であり、実施例7−2では約0.10、7−3では約0.057であった。合成されたアミロースの濃度を測定し、収率を計算した。
【0269】
【表11】
結果を以下の表12に示す。
【0270】
【表12】
セロビオースを出発原料としたときのアミロース合成収率は従来の方法(比較例7)では16.3%であったが、PGK、PGMを共存させることで約40%まで上昇し(実施例7−1)、さらに基質濃度を上げ、初期pHを弱アルカリ側にすることでアミロース収率が66.8%にあがった(実施例7−3)。
【0271】
(実施例8−1から8−3:種々のpH条件下での、セロビオースを出発物質としたアミロース合成)
反応開始時のpHの影響を調べるために、以下の表13に記載の組成の溶液を調製し、反応開始時の溶液のpHをそれぞれ5.6、7.0または7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例8−1から8−3では約10mMであり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例8−1および8−2では約0.10であり、実施例3では約0.057であった。合成されたアミロースの量を測定し、収率を計算した。
【0272】
反応開始時点で測定したpHの値と、収率の結果を以下の表14および図9に示す。
【0273】
【表13】
【0274】
【表14】
この結果、反応溶液の初期pH値が高くなるほど、アミロース収率が上がることがわかった。
【0275】
(実施例9:反応溶液中の遊離リン酸とセロビオースとのモル比(Pi/CB)の検討)
以下の表15に示す組成の溶液を作製して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約7.8に調整した。合成されたアミロースの濃度を測定し、収率を計算した。添加した無機リン酸の量、無機リン酸とセロビオースとのモル比(Pi/CB)、アミロースの収率(%)を以下の表16に示す。結果を図10に示す。
【0276】
【表15】
【0277】
【表16】
この結果、セロビオースの約4倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。
【0278】
(実施例10:出発セロビオース濃度を変えた場合のアミロース収率の変化)
セロビオース濃度を変化させ、それに合わせて酵素量およびプライマー濃度を比例して増加させたことと実施例10−1のポリリン酸濃度が1%であること以外は、実施例7−2と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを7.8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース量を測定し、収率を計算した。実施例10−2は、実施例7−2と同じ条件での反応であり、実施例10−3は、実施例7−3と同じ条件での反応であった。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、実施例10−1では約2.8であり、実施例10−2および10−3では約11.2あり、遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)は、実施例10−1では0.140であり、実施例10−2では約0.112であり、実施例10−3では0.064であった。
【0279】
結果を以下の表17および図11に示す。
【0280】
【表17】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図11)。
【0281】
(実施例11:種々のpH条件下での、G6−Pを出発物質としたアミロース合成)
反応開始時の溶液のpHを6.7〜8.5に変化させたこと以外は上記表9と同じ組成の溶液を調製し、そして45℃で16時間インキュベートすることにより、アミロースを合成させた。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、いずれの条件でも0mMであり、遊離リン酸のモル濃度とG6−Pのモル濃度との比率(Pi/G6−P)は、いずれの条件でも0であった。合成されたアミロースの濃度を測定し、収率を計算した。
【0282】
反応開始時点で測定したpHの値と、収率の結果を以下の表18および図12に示す。
【0283】
【表18】
反応溶液の初期pHを弱アルカリ側以上にすることでアミロース合成収率があがり約30%の値を得た。初期pHが6.7までの酸性側ではアミロースがほとんど合成されず、弱酸性から中性付近では低収率でアミロースを得た。初期pH7.1のとき収率6.37%であった。pH7.3からpH8.5ではアミロース収率10%以上を示し、pH7.6からpH8.5では約24%以上のアミロース収率を示した。初期pH8.4のとき収率30.51%であった。
【0284】
(実施例12:反応溶液中の遊離リン酸とグルコース−6−リン酸とのモル比(Pi/G6−P)の検討)
以下の表19に示す濃度の無機リン酸を添加したこと以外は表9の組成の溶液を作成して45℃で16時間反応させてアミロースを合成した。反応開始時点でのこの溶液のpHを約8に調整した。合成されたアミロースの濃度を測定し、収率を計算した。添加した無機リン酸の濃度、無機リン酸とグルコース−6−リン酸とのモル比(Pi/G6−P)、アミロースの濃度(mM)および収率(%)を以下の表19に示す。結果を図13に示す。
【0285】
【表19】
この結果、グルコース−6−リン酸の約0.5倍濃度の遊離のリン酸が含まれるとアミロース合成が阻害された。また、遊離リン酸の濃度が高いほど、収率が下がった。従って、この反応では、遊離リン酸をできる限り添加しないことが好ましい。
【0286】
(実施例13:出発グルコース−6−リン酸濃度を変えた場合のアミロース収率の変化)
グルコース−6−リン酸濃度を変化させ、それに合わせて酵素量およびプライマー濃度を比例して増加させたこと以外は、表9と同じ組成の溶液を調製し、反応開始時点でのこの溶液のpHを8に調整し、そして45℃で16時間インキュベートすることにより、アミロースを合成した。合成されたアミロース濃度を測定し、収率を計算した。反応開始時の溶液中の遊離リン酸の濃度を測定したところ、約0であり、遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)は、約0であった。
【0287】
結果を以下の表20、図14Aおよび図14Bに示す。
【0288】
【表20】
基質濃度を上昇させるとそれに伴い溶液中のアミロース濃度も上昇するので、アミロースが老化しやすい状態になる。そのため、高濃度基質からのアミロース合成ではアミロース収率が高くなる傾向にある(図14A)。
【産業上の利用可能性】
【0289】
本発明により、安価で容易に入手可能なグルコースからα−グルカンを効率よく製造することができる。
【0290】
以上のように、本発明の好ましい実施形態を用いて本発明を例示してきたが、本発明は、この実施形態に限定して解釈されるべきものではない。本発明は、特許請求の範囲によってのみその範囲が解釈されるべきであることが理解される。当業者は、本発明の具体的な好ましい実施形態の記載から、本発明の記載および技術常識に基づいて等価な範囲を実施することができることが理解される。本明細書において引用した特許、特許出願および文献は、その内容自体が具体的に本明細書に記載されているのと同様にその内容が本明細書に対する参考として援用されるべきであることが理解される。
【図面の簡単な説明】
【0291】
【図1】図1は、本願発明で利用する種々の酵素反応の概略を示す図である。
【図2】図2は、グルコースを出発物質とした場合の、グルコース(Glc)量、グルコース−6−リン酸(G6−P)量、グルコース−1−リン酸(G−1−P)量、およびアミロースの合成量の経時変化を示すグラフである。
【図3】図3は、グルコースを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図4】図4は、グルコースを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/グルコース濃度比によるアミロース収率の変化を示すグラフである。
【図5】図5は、グルコースおよびポリホスフェートグルコキナーゼを含む溶液をインキュベートした場合にグルコース−6−リン酸に変換された割合を示すグラフである。
【図6】図6は、アミロース、ホスホグルコムターゼおよびα−1,4−グルカンホスホリラーゼを含む溶液をインキュベートした場合のアミロース量、グルコース−1−リン酸量、グルコース−6−リン酸量および(グルコース−1−リン酸量とグルコース−6−リン酸量との合計)を示すグラフである。
【図7A】図7Aは、グルコースを出発物質とした場合の、グルコース濃度とアミロースの収率(%)との関係を示すグラフである。
【図7B】図7Bは、グルコースを出発物質とした場合の、グルコース濃度とアミロースの収量(mM)との関係を示すグラフである。
【図8】図8は、種々の包接化合物を使用し、グルコースを出発物質とした場合の、アミロースの収率を示すグラフである。
【図9】図9は、セロビオースを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図10】図10は、セロビオースを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/セロビオース濃度比によるアミロース収率の変化を示すグラフである。
【図11】図11は、G6−Pを出発物質とした場合の、セロビオース濃度とアミロースの収率(%)との関係を示すグラフである。
【図12】図12は、G6−Pを出発物質とした場合の、種々のpHでのアミロースの合成量を示すグラフである。
【図13】図13は、G6−Pを出発物質とした場合の、反応開始時の溶液中の無機リン酸濃度/G6−P濃度比によるアミロース収率の変化を示すグラフである。
【図14A】図14Aは、G6−Pを出発物質とした場合の、G6−P濃度とアミロースの収率(%)との関係を示すグラフである。
【図14B】図14Bは、グルコースを出発物質とした場合の、G6−P濃度とアミロースの収量(mM)との関係を示すグラフである。
【図15】図15は、本発明で使用され得る種々の方法をまとめて示す模式図である。
【配列表フリーテキスト】
【0292】
配列番号1:Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのヌクレオチド配列;
配列番号2:Microlunatus phosphovolasに由来するポリホスフェートグルコキナーゼのアミノ酸配列;
配列番号3:Escherichia coliに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号4:Escherichia coliに由来するホスホグルコムターゼのアミノ酸配列;
配列番号5:馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子のヌクレオチド配列;
配列番号6:馬鈴薯α−1,4−グルカンホスホリラーゼ遺伝子のアミノ酸配列;
配列番号7:合成DNAプライマー1のヌクレオチド配列;
配列番号8:合成DNAプライマー2のヌクレオチド配列;
配列番号9:Thermobifida fuscaに由来するポリホスフェートグルコキナーゼのヌクレオチド配列;
配列番号10:Thermobifida fuscaに由来するポリホスフェートグルコキナーゼのアミノ酸配列;
配列番号11:Thermus thermophilusに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号12:Thermus thermophilusに由来するホスホグルコムターゼのアミノ酸配列;
配列番号13:Thermotoga lettingaeに由来するホスホグルコムターゼのヌクレオチド配列;
配列番号14:Thermotoga lettingae由来のホスホグルコムターゼのアミノ酸配列。
【特許請求の範囲】
【請求項1】
グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、
反応開始時の該溶液のpHは、pH5.7〜pH12であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【請求項2】
前記反応開始時の前記溶液中のグルコース濃度が200mM〜2000mMである、請求項1に記載の方法。
【請求項3】
前記反応開始時の前記溶液中のグルコース濃度が500mM〜2000mMである、請求項1に記載の方法。
【請求項4】
前記反応開始時の前記溶液のpHが、pH7.4〜pH11である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記α−グルカンがアミロースである、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記溶液が、ゲスト化合物を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項6に記載の方法。
【請求項8】
グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース−6−リン酸濃度は、20mM〜2000mMであり、
反応開始時の該溶液のpHは、pH6.7〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)が0.4以下である、方法。
【請求項9】
前記反応開始時の前記溶液中のグルコース−6−リン酸濃度が100mM〜2000mMである、請求項8に記載の方法。
【請求項10】
前記反応開始時の前記溶液のpHが、pH7.3〜pH11である、請求項8または9に記載の方法。
【請求項11】
前記α−グルカンがアミロースである、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項8〜11のいずれか1項に記載の方法。
【請求項13】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項12に記載の方法。
【請求項14】
セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法であって、
反応開始時の該溶液中のセロビオース濃度は、100mM〜350mMであり、
反応開始時の該溶液のpHは、pH5〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)が2.0以下である、方法。
【請求項15】
前記反応開始時の前記溶液中のセロビオース濃度が120mM〜350mMである、請求項14に記載の方法。
【請求項16】
前記反応開始時の前記溶液のpHが、pH5.6〜pH11である、請求項14または15に記載の方法。
【請求項17】
前記α−グルカンが、アミロースである、請求項14〜16のいずれか1項に記載の方法。
【請求項18】
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項14〜17のいずれか1項に記載の方法。
【請求項19】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項18に記載の方法。
【請求項1】
グルコースと、ポリリン酸と、プライマーと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース濃度は、100mM〜2000mMであり、
反応開始時の該溶液のpHは、pH5.7〜pH12であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコースのモル濃度との比率(Pi/Glc)が0.15以下である、方法。
【請求項2】
前記反応開始時の前記溶液中のグルコース濃度が200mM〜2000mMである、請求項1に記載の方法。
【請求項3】
前記反応開始時の前記溶液中のグルコース濃度が500mM〜2000mMである、請求項1に記載の方法。
【請求項4】
前記反応開始時の前記溶液のpHが、pH7.4〜pH11である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記α−グルカンがアミロースである、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記溶液が、ゲスト化合物を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項6に記載の方法。
【請求項8】
グルコース−6−リン酸と、プライマーと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼとを含む溶液を反応させてα−グルカンを製造する工程を包含する方法であって、
反応開始時の該溶液中のグルコース−6−リン酸濃度は、20mM〜2000mMであり、
反応開始時の該溶液のpHは、pH6.7〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とグルコース−6−リン酸のモル濃度との比率(Pi/G6−P)が0.4以下である、方法。
【請求項9】
前記反応開始時の前記溶液中のグルコース−6−リン酸濃度が100mM〜2000mMである、請求項8に記載の方法。
【請求項10】
前記反応開始時の前記溶液のpHが、pH7.3〜pH11である、請求項8または9に記載の方法。
【請求項11】
前記α−グルカンがアミロースである、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項8〜11のいずれか1項に記載の方法。
【請求項13】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項12に記載の方法。
【請求項14】
セロビオースと、ポリリン酸と、プライマーと、セロビオースホスホリラーゼと、ポリホスフェートグルコキナーゼと、ホスホグルコムターゼと、α−1,4−グルカンホスホリラーゼを含む溶液を反応させて、α−グルカンを生産する工程を包含する方法であって、
反応開始時の該溶液中のセロビオース濃度は、100mM〜350mMであり、
反応開始時の該溶液のpHは、pH5〜pH11であり、
反応開始時の該溶液中の遊離リン酸のモル濃度とセロビオースのモル濃度との比率(Pi/CB)が2.0以下である、方法。
【請求項15】
前記反応開始時の前記溶液中のセロビオース濃度が120mM〜350mMである、請求項14に記載の方法。
【請求項16】
前記反応開始時の前記溶液のpHが、pH5.6〜pH11である、請求項14または15に記載の方法。
【請求項17】
前記α−グルカンが、アミロースである、請求項14〜16のいずれか1項に記載の方法。
【請求項18】
前記溶液が、ゲスト物質を含み、該ゲスト化合物は前記製造されるα−グルカンに包接され得る化合物であり、
前記製造工程において得られるα−グルカンと該ゲスト化合物との包接化合物を形成させる、請求項14〜17のいずれか1項に記載の方法。
【請求項19】
前記ゲスト化合物が、N−ドデシル−N,N−ジメチル−3−アンモニオ−1−プロパンスルホネート、3−(N,N−ジメチルミリスチルアンモニオ)プロパンスルホネートまたは3−(N,N−ジメチルパルミチル−アンモニオ)プロパンスルホネートである、請求項18に記載の方法。
【図1】

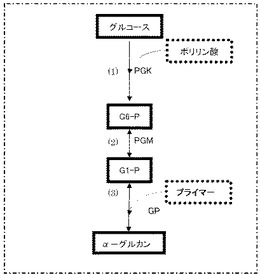
【図2】
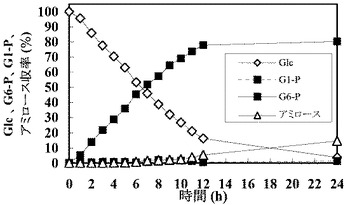
【図3】
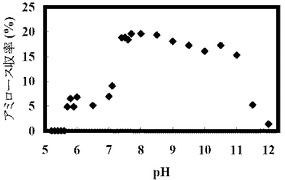
【図4】

【図5】
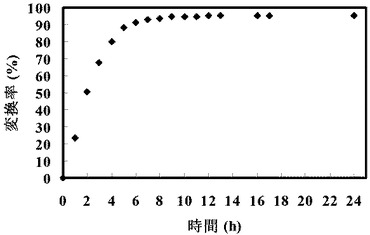
【図6】
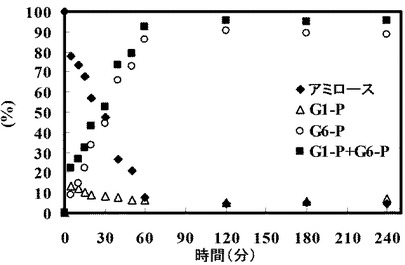
【図7A】
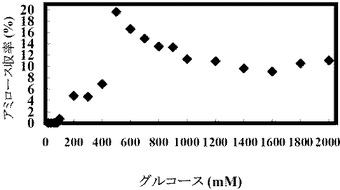
【図7B】
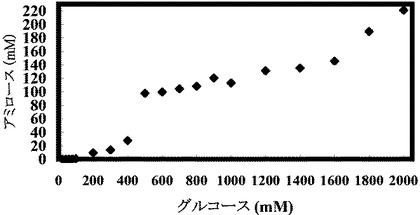
【図8】
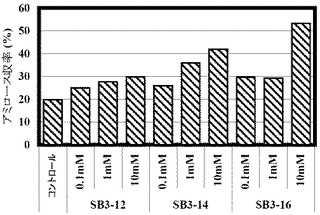
【図9】
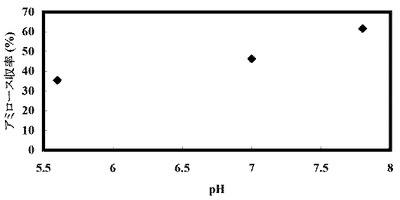
【図10】
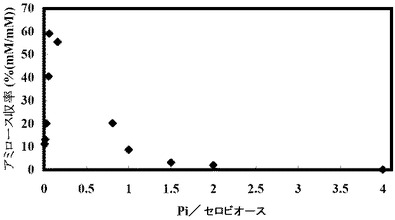
【図11】
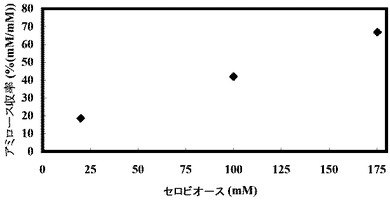
【図12】
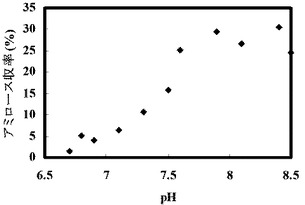
【図13】
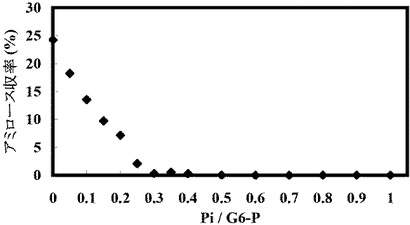
【図14A】
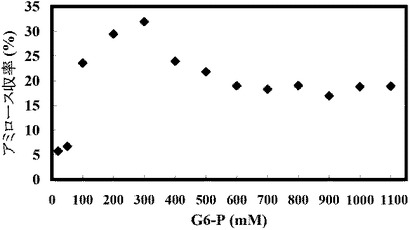
【図14B】
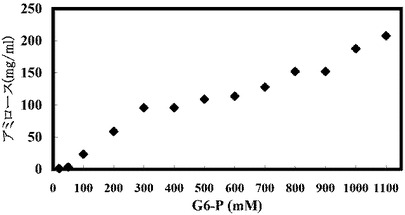
【図15】
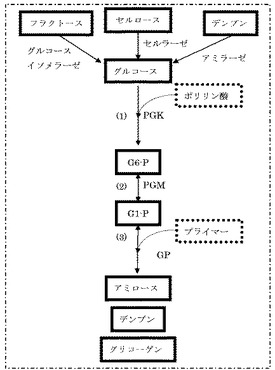
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15】
【公開番号】特開2010−148407(P2010−148407A)
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2008−328854(P2008−328854)
【出願日】平成20年12月24日(2008.12.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)「国等の委託研究の成果に係る特許出願(平成20年度生物系特定産業技術研究支援センター「生物系産業創出のための異分野融合研究支援事業」産業技術力強化法第19条の適用を受けるもの)」
【出願人】(000000228)江崎グリコ株式会社 (187)
【Fターム(参考)】
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願日】平成20年12月24日(2008.12.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)「国等の委託研究の成果に係る特許出願(平成20年度生物系特定産業技術研究支援センター「生物系産業創出のための異分野融合研究支援事業」産業技術力強化法第19条の適用を受けるもの)」
【出願人】(000000228)江崎グリコ株式会社 (187)
【Fターム(参考)】
[ Back to top ]