
グルコース脱水素酵素
【課題】ボトリティス・バシアナ由来のグルコース脱水素酵素遺伝子を特定、単離し、効率的に組み換え酵素を製造する、グルコース脱水素酵素の効率的な製造方法を提供するものである。
【解決手段】以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【解決手段】以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、グルコース脱水素酵素及びその製造方法、並びにグルコース脱水素酵素を用いたグルコースの測定方法、グルコース測定試薬組成物及びグルコース測定用バイオセンサに関する。
【背景技術】
【0002】
血液中のグルコース濃度の迅速かつ正確な測定は、糖尿病を診断するうえで重要である。グルコースの測定法としては、化学法と酵素法があるが、酵素法が特異性、安全性の点で優れている。当該酵素法の中でも、検体の微量化、測定時間の短縮、装置の小型化の点から、電気化学的バイオセンサが有利である。
【0003】
そのようなバイオセンサに使用可能な酵素として、グルコース酸化酵素が知られている。しかし、グルコース酸化酵素は、血中の溶存酸素により測定誤差が生じるという問題があるため、いくつかのグルコース脱水素酵素が開発されてきた。グルコース脱水素酵素のうち、フラビン結合型グルコース脱水素酵素は、補酵素の添加を必要としないこと、溶存酸素の影響を受けないこと及び基質特異性に優れることから、グルコースバイオセンサ用の酵素として注目されている(特許文献1〜4)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開2004/058958号パンフレット
【特許文献2】国際公開2006/101239号パンフレット
【特許文献3】国際公開2008/001903号パンフレット
【特許文献4】国際公開2010/140431号パンフレット
【非特許文献】
【0005】
【非特許文献1】Mol. Plant Pathol. 5(1),p17-27,2004
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、グルコースバイオセンサ用の酵素として有用なグルコース脱水素酵素、これを用いたグルコースバイオセンサ及び当該グルコース脱水素酵素遺伝子を特定、単離し、効率的に組み換え酵素を製造する方法を提供することである。
【課題を解決するための手段】
【0007】
そこで本発明者は、糸状菌由来のグルコース脱水素酵素に着目し、ボトリオティニア・フケリアナ由来のグルコース脱水素酵素遺伝子を単離し、さらにその組み換えベクターを大腸菌、糸状菌、酵母等の微生物に導入して組み換えグルコース脱水素酵素を製造し、その酵素活性、アミノ酸配列、グルコースセンサへの利用性等について検討した。その結果、ボトリオティニア・フケリアナ由来の配列番号51で示されるアミノ酸配列を有する酵素は、グルコース酸化酵素(GOD)であるとされていたが(非特許文献1)、本発明により得られた組み換え酵素はグルコース酸化酵素ではなくフラビン結合型グルコース脱水素酵素であり、かつそのアミノ酸配列は従来推定されていたものとは異なり、配列番号5で示されるものであることを見出した。従って、本発明により得られたフラビン結合型グルコース脱水素酵素は、溶存酸素の影響を受けないことからグルコースバイオセンサ用の酵素として有用であることを見出した。さらには、種々の組み換え酵素のうち、形質転換真核細胞により得られた糖ポリペプチドは、形質転換大腸菌により得られた糖鎖を有さないポリペプチドに比べて、バイオセンサ用酵素として有用であることを見出し、本発明を完成した。
【0008】
すなわち、本発明は、以下の〔1〕〜〔12〕を提供するものである。
〔1〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔2〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を含有するグルコース測定試薬組成物:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔3〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコースの測定方法:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔4〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔5〕ボトリオティニア・フケリアナ由来の組み換えグルコース脱水素酵素遺伝子を用いた形質転換真核細胞で発現させたものである〔4〕記載のフラビン結合型グルコース脱水素酵素。
〔6〕以下の(a)又は(b)のポリヌクレオチド:
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
〔7〕以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチド:
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
〔8〕〔6〕又は〔7〕に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチド。
〔9〕シグナル配列が真核細胞由来タンパクのシグナル配列である、〔8〕記載のポリヌクレオチド。
〔10〕〔6〕〜〔9〕の何れか1項に記載のポリヌクレオチドを含む組み換えベクター。
〔11〕〔10〕記載の組み換えベクターを用いることによって作成された形質転換真核細胞。
〔12〕〔11〕記載の形質転換真核細胞を培養し、得られた培養物からグルコース脱水素酵素を採取することを特徴とするグルコース脱水素酵素の製造方法。
【発明の効果】
【0009】
本発明の糖ポリペプチドは、フラビン結合型グルコース脱水素酵素であり、溶存酸素の影響を受けずにグルコースを正確に測定できるグルコースバイオセンサに有用である。また、本発明のポリヌクレオチドを利用することにより、グルコースに対する基質認識性に優れ、しかもマルトースに対する作用性が低いという優れた特性を有するグルコース脱水素酵素を均質かつ大量に生産することが可能となる。
【図面の簡単な説明】
【0010】
【図1】本発明グルコース脱水素酵素によるグルコース量測定結果を示す図である。
【発明を実施するための形態】
【0011】
本発明において、「グルコース脱水素酵素」とは、電子受容体存在下で、グルコースの1位の水酸基を脱水素(酸化)する反応を触媒し、グルコースへの作用性に対してマルトースへの作用性が5%以下である可溶性の蛋白質を意味し、該酵素は以下の性質を特徴とする。
1)フラビンアデニンジヌクレオチド(FAD)を補酵素とする、
2)酸素を電子受容体としない、
3)基質濃度333mMにおいて、グルコースへの作用性を100%とした場合にマルトースへの作用性が5%以下、及び
4)酵素タンパクのポリペプチドの分子量が60〜70kDaである。
酵素タンパクのポリペプチドの分子量とは、真核細胞で発現させた糖鎖修飾された組み換え酵素を糖鎖切断処理し、SDS−ポリアクリルアミド電気泳動に供した際の分子量である。尚、真核細胞で発現した組み換え酵素は、宿主、培養条件等の影響で糖鎖付加量は変化するため、80〜200kDa程度の分子量になり得る。
また、本発明において、「糖ポリペプチド」とは、糖鎖が付加したポリペプチドをいう。
【0012】
本発明のポリヌクレオチドは、以下の(a)又は(b)のポリヌクレオチドである。
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【0013】
更に、本発明のポリヌクレオチドは、以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチドである。
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
【0014】
前記のポリヌクレオチド(b)又はポリペプチド(d)としては、塩基配列(a)又はアミノ酸配列(c)と少なくとも80%以上の相同性を有する塩基配列又はアミノ酸配列を有するものが挙げられる。ここで、少なくとも80%の相同性を有する塩基配列又はアミノ酸配列とは、夫々比較対象となる基準配列の全長にわたり、少なくとも80%の同一性を示し、好ましくは少なくとも85%、より好ましくは少なくとも90%、更に好ましくは少なくとも95%の同一性を有する各配列をいう。このような配列の同一性パーセンテージは、基準配列を照会配列として比較するアルゴリズムをもった公開又は市販されているソフトウエアを用いて計算することができる。例として、BLAST、FASTA、又はGENETYX(ソフトウエア開発社製)などを用いることができ、これらはデフォルトパラメーターで使用することができる。
【0015】
本発明において、ポリヌクレオチド間のハイブリダイズに際しての「ストリンジェントな条件下でハイブリダイズ」の具体的な条件とは、例えば、50%ホルムアミド、5×SSC(150mM 塩化ナトリウム、15mM クエン酸三ナトリウム、10mM リン酸ナトリウム、1mM エチレンジアミン四酢酸、pH7.2)、5×デンハート(Denhardt’s)溶液、0.1% SDS、10% デキストラン硫酸及び100μg/mLの変性サケ精子DNAで42℃インキュベーションした後、フィルターを0.2×SSC中42℃で洗浄することを例示することができる。
【0016】
尚、本発明において、「ポリヌクレオチド」とは、プリン又はピリミジンが糖にβ-N-グリコシド結合したヌクレオシドのリン酸エステル(ATP(アデノシン三リン酸)、GTP(グアノシン三リン酸)、CTP(シチジン三リン酸)、UTP(ウリジン三リン酸);又はdATP(デオキシアデノシン三リン酸)、dGTP(デオキシグアノシン三リン酸)、dCTP(デオキシシチジン三リン酸)、dTTP(デオキシチミジン三リン酸))が100個以上結合した分子を言い、具体的にはグルコース脱水素酵素をコードする染色体DNA、染色体DNAから転写されたmRNA、mRNAから合成されたcDNA及び、それらを鋳型としてPCR増幅したポリヌクレオチドを含む。「オリゴヌクレオチド」とはヌクレオチドが2-99個連結した分子を言う。また「ポリペプチド」とは、アミド結合(ペプチド結合)又は非天然の残基連結によって互いに結合した30個以上のアミノ酸残基から構成された分子を意味する。「糖ポリペプチド」とは、前記ポリペプチドに糖鎖が付加した分子を意味する。
【0017】
本発明のポリヌクレオチド(遺伝子)の最も具体的な態様は、配列番号4又は配列番号6の塩基配列を含むポリヌクレオチドである。配列番号6に代表される染色体DNAであるポリヌクレオチドは、例えばボトリオティニア・フケリアナ株から染色体DNAライブラリーを調製し、特許文献2に記載のアスペルギルス・テレウス(Aspergillus terreus)由来グルコース脱水素酵素のアミノ酸配列及びアスペルギルス・オリゼ(Aspergillus oryzae)由来グルコース脱水素酵素のアミノ酸配列又はその他のグルコース脱水素酵素のアミノ酸配列に基づいて作成した複数のオリゴヌクレオチドプローブを用いて当業者に公知の方法によって上記染色体DNAライブラリーをスクリーニングすることによって取得することができる。
【0018】
プローブの標識は、当業者に公知の任意の方法、例えば、ラジオアイソトープ(RI)法又は非RI法によって行うことができるが、非RI法を用いることが好ましい。非RI法としては、蛍光標識法、ビオチン標識法、化学発光法等が挙げられるが、蛍光標識法を用いることが好ましい。蛍光物質としては、オリゴヌクレオチドの塩基部分と結合できるものを適宜に選択して用いることができるが、シアニン色素(例えば、Cy DyeTMシリーズのCy3、Cy5等)、ローダミン6G試薬、N-アセトキシ-N2-アセチルアミノフルオレン(AAF)、AAIF(AAFのヨウ素誘導体)などを使用することができる。
【0019】
或いは、配列番号4に代表されるcDNAであるポリヌクレオチドは、例えば、本明細書の実施例に具体的に記載されているように、染色体DNAからイントロンを含む目的のポリヌクレオチドを取得した後、PCRによってイントロンを削除して得ることができる他、例えば、cDNAライブラリーを鋳型とし、上記で作成したオリゴヌクレオチドプライマー(プローブ)のセットを用いた当業者に公知の各種PCR法によって得ることができ、又は、ボトリオティニア・フケリアナ株から抽出した全RNAもしくはmRNAを鋳型とするRT−PCR法によっても得ることができる。尚、PCRにおけるプライマー使用の最終濃度が約0.1から約1μMになるよう留意することも必要である。プライマー設計においては、市販のソフトウエア、例えばOligoTM(National Bioscience Inc.(米国)製)、GENETYX(ソフトウエア開発社製)等を用いることもできる。
【0020】
尚、このようなオリゴヌクレオチドプローブやオリゴヌクレオチドプライマーセットは、例えば本発明のポリヌクレオチドであるcDNAを適当な制限酵素で切断して作成することもできる。
【0021】
更に、本発明のポリヌクレオチドは、〔6〕又は〔7〕に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチドである。シグナル配列をコードする塩基配列は、グルコース脱水素酵素活性を有する糖ポリペプチドを効率良く分泌できるシグナル配列をコードする塩基配列であれば良く、例えば真核細胞由来タンパクのシグナル配列をコードする塩基配列が好ましく、ボトリオティニア属又はアスペルギルス属由来タンパクのシグナル配列をコードする塩基配列がより好ましい。更に、該ポリヌクレオチドにより形質転換する宿主と同属由来タンパクのシグナル配列をコードする塩基配列や、グルコース脱水素酵素のシグナル配列をコードする塩基配列が例示できる。塩基配列によりコードされるシグナル配列は、例えば特許文献2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素配列と分泌タンパクのアミノ酸配列とを比較することや、シグナル配列予測サイト(Signal p:http://www.cbs.dtu.dk/services/SignalP/)を用いて分泌タンパクのアミノ酸配列から推定でき、更に1〜数個のアミノ酸が置換、欠失又は付加されたシグナル配列でも良い。具体例としては配列番号8、配列番号10、配列番号12及び配列番号14に記載の配列が例示でき、これらの配列又はこれらの配列を数アミノ酸欠失させた配列を適宜組み合わせた配列でも良い。
【0022】
シグナル配列をコードする塩基配列を有するポリヌクレオチドの具体例としては、配列番号1、配列番号3、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46及び配列番号48に記載のポリヌクレオチドが例示できるが、グルコース脱水素酵素活性を有するタンパク質をコードする部位は、イントロンを含んでいても良い。
【0023】
又、(a)〜(d)のポリヌクレオチドは、例えば、前記のボトリオティニア・フケリアナ由来のグルコース脱水素酵素cDNAを、公知のミューテーション導入法や変異導入PCR法等によって改変して作成することができる。更に、NBRC7185株以外のボトリオティニア・フケリアナ株の染色体DNAやそのcDNAライブラリーから、配列番号1のヌクレオチド配列情報に基づいて作成したオリゴヌクレオチドを用いるプローブハイブリダイゼーション法によって取得することができる。ハイブリダイゼーションに際して、ストリンジェント条件を様々に変化させることによって、上記ポリヌクレオチドを取得することができる。ストリンジェント条件は、ハイブリダイゼーション及び洗浄工程における塩濃度、有機溶媒(ホルムアルデヒド等)の濃度、温度条件等によって規定され、例えば、米国特許No.6,100,037号明細書等に開示されているような、当業者らに周知の様々な条件を採用することができる。
【0024】
更に、文献(例えばCarruthers(1982)Cold Spring Harbor Symp. Quant. Biol. 47:411-418; Adams(1983)J. Am. Chem. Soc. 105:661; Belousov(1997)Nucleic Acid Res. 25:3440-3444; Frenkel(1995)Free Radic. Biol. Med. 19:373-380; Blommers(1994)Biochemistry 33:7886-7896; Narang(1979)Meth. Enzymol. 68:90; Brown(1979)Meth. Enzymol. 68:109; Beaucage(1981)Tetra. Lett. 22:1859; 米国特許第4,458,066号)に記載されているような周知の化学合成技術により、in vitroにおいて本発明のポリヌクレオチドを合成することができる。
【0025】
本発明の組み換えベクターは、クローニングベクター又は発現ベクターであり、インサートとしてのポリヌクレオチドの種類や、その使用目的等に応じて適宜のものを使用する。例えば、cDNA又はそのORF領域をインサートとしてグルコース脱水素酵素を生産する場合には、in vitro転写用の発現ベクターや、酵母、カビなどの糸状菌、昆虫細胞、哺乳動物細胞等の真核細胞のそれぞれに適した発現ベクターを使用することもできる。
【0026】
本発明の形質転換細胞としては、例えば、酵母、カビ、昆虫細胞、哺乳動物細胞等の真核細胞を使用することができる。これらの形質転換細胞は、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法など公知の方法によって組み換えベクターを細胞に導入することによって調製することができる。組み換えベクター及び形質転換細胞の具体例として、下記実施例に示した組み換えベクターと、このベクターによる形質転換カビ、形質転換酵母が挙げられる。
【0027】
グルコース脱水素酵素を真核細胞で発現させて生産させる場合には、前記ポリヌクレオチドを、プロモーター、スプライシング領域、ポリ(A)付加部位等を有する真核細胞用発現ベクターに挿入して組み換えベクターを作成し、真核細胞内に導入すれば、グルコース脱水素酵素を真核細胞で生産することができる。プラスミドのような状態で細胞内に維持することもできるし、染色体中に組みこませて維持することもできる。発現ベクターとしては、pKA1、pCDM8、pSVK3、pSVL、pBK−CMV、pBK−RSV、EBVベクター、pRS、pYE82などが例示できる。また、pIND/V5−His、pFLAG−CMV−2、pEGFP−N1、pEGFP−C1などを発現ベクターとして用いれば、Hisタグ、FLAGタグ、GFPなど各種タグを付加した融合蛋白質としてグルコース脱水素酵素ポリペプチドを発現させることもできる。真核細胞としては、サル腎臓細胞COS−7、チャイニーズハムスター卵巣細胞CHOなどの哺乳動物培養細胞、出芽酵母、分裂酵母、カビ、カイコ細胞、アフリカツメガエル卵細胞などが一般に用いられるが、グルコース脱水素酵素を発現できるものであれば、いかなる真核細胞でもよい。発現ベクターを真核細胞に導入するには、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法など公知の方法を用いることができる。真核細胞で発現させて生産させる場合の挿入遺伝子はイントロンを含んでいても含んでいなくても良く、例えば配列番号1、配列番号3、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46又は配列番号48に記載のシグナル配列を含む遺伝子配列が好ましいが、ベクター側に分泌シグナル配列がある場合は、配列番号4又は配列番号6に記載の遺伝子配列を挿入すれば良く、菌体内に組み換えタンパクを生産させたい場合は、配列番号4又は配列番号6に記載の遺伝子配列に開始コドンATGを付加したポリヌクレオチドを挿入すれば良いが、菌体外に分泌生産することが好ましい。真核細胞では、本発明の組み換えグルコース脱水素酵素の発現効率が高く、好ましくは培養液1mLあたり少なくとも20U、より好ましくは培養液1mLあたり少なくとも50U、更に好ましくは培養液1mLあたり少なくとも80U、特に好ましくは培養液1mLあたり少なくとも100U、最も好ましくは培養液1mLあたり少なくとも120Uである。更に真核細胞では、原核細胞での組み換え製造時に問題となる封入体を作ることが無く、組み換え酵素を安定して高発現させることができる。加えて、培養物から得られる粗酵素の比活性が高く、夾雑蛋白質が少ないため、精製工程が簡便で精製コストが抑えられ、高収率で精製酵素が得られる。
【0028】
グルコース脱水素酵素を真核細胞で発現させた後、培養物(菌体、もしくは菌体外に分泌された酵素を含む培養液、培地組成物等)から目的蛋白質を単離精製するためには、公知の分離操作を組み合わせて行うことができる。例えば、尿素などの変性剤や界面活性剤による処理、熱処理、pH処理、超音波処理、酵素消化、塩析や溶媒沈殿法、透析、遠心分離、限外濾過、ゲル濾過、SDS−PAGE、等電点電気泳動、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトグラフィー(タグ配列を利用した方法及びグルコース脱水素酵素に特異的なポリクローナル抗体、モノクローナル抗体を用いる方法も含む)、などが挙げられる。
【0029】
また、グルコース脱水素酵素は、本発明のポリヌクレオチド(cDNA又はその翻訳領域)を用いた組み換えDNA技術によって取得できる。例えば前記ポリヌクレオチドを有するベクターからin vitro転写によってRNAを調製し、これを鋳型としてin vitro翻訳を行うことによりin vitroでグルコース脱水素酵素を作成することができる。またポリヌクレオチドを公知の方法により適当な発現ベクターに組み換えれば、酵母、カビ、昆虫細胞、哺乳動物細胞等の真核細胞で、ポリヌクレオチドがコードしているグルコース脱水素酵素を大量に発現させる事ができる。また宿主に対応して、同一アミノ酸配列であるが、コドンユーセージを最適化したポリヌクレオチドを導入しても良い。
【0030】
グルコース脱水素酵素をin vitro発現させて生産させる場合には、前記のポリヌクレオチドを、RNAポリメラーゼが結合できるプロモーターを有するベクターに挿入して組み換えベクターを作成し、このベクターを、プロモーターに対応するRNAポリメラーゼを含むウサギ網状赤血球溶解物や小麦胚芽抽出物などのin vitro翻訳系に添加すれば、グルコース脱水素酵素をin vitroで生産することができる。RNAポリメラーゼが結合できるプロモーターとしては、T3、T7、SP6などが例示できる。これらのプロモーターを含むベクターとしては、pKA1、pCDM8、pT3/T718、pT7/319、pBluescriptIIなどが例示できる。
【0031】
本発明のグルコース脱水素酵素は、以下の(e)又は(f)の糖ポリペプチドからなるグルコース脱水素酵素である。
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)アミノ酸配列(e)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【0032】
本発明のグルコース脱水素酵素の最も具体的な態様は、配列番号5で示されるアミノ酸配列からなる糖ポリペプチドである。グルコース脱水素酵素活性があれば、配列番号5に記載のアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなる糖ポリペプチドでも良く、1〜数個とは、好ましくは多くとも100個、より好ましくは多くとも80個、更に好ましくは多くとも60個、特に好ましくは多くとも40個である。
例として、配列番号2、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47又は配列番号49に記載のアミノ酸配列からなる糖ポリペプチドを有するグルコース脱水素酵素が例示できる。
また、本発明のグルコース脱水素酵素には、配列番号5のアミノ酸配列と少なくとも80%の相同性、より好ましくは85%以上の相同性、さらに好ましくは90%以上の相同性、さらに好ましくは95%以上の相同性を有するアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチドが含まれる。
【0033】
本発明のグルコース脱水素酵素は、電子受容体存在下でグルコースを脱水素する反応を触媒する酵素であるから、この反応による変化が利用できる用途であれば、特に制限されない。例えば、生体物質を含む試料中のグルコースの測定及び測定用試薬、消去用試薬へ使用するなどの医療分野、臨床分野への使用が可能であり、フラビン結合型グルコース脱水素酵素を使用した物質生産においても使用可能である。
【0034】
本発明のグルコース脱水素酵素は、グルコース測定試薬組成物に用いることができる。本発明の測定試薬組成物は、本発明の糖ポリペプチドからなるグルコース脱水素酵素に、牛血清アルブミン(BSA)若しくは卵白アルブミン、該酵素と作用性のない糖類若しくは糖アルコール類、カルボキシル基含有化合物、アルカリ土類金属化合物、アンモニウム塩、硫酸塩又はタンパク質等から成る群より選ばれる熱安定化剤、又は緩衝剤等の当業者に公知の他の任意成分を適宜含有させ、該酵素や試薬成分の熱安定性や保存安定性を高めることができる。更に、被験試料中に存在する、測定に影響を与える夾雑物質の影響を抑える公知の物質を該測定試薬組成物に含ませることができる。加えて、該組成物が溶解した際に該酵素が作用し易いpHとなるように、該組成物に緩衝剤を含有させることができる。
【0035】
本発明のグルコース脱水素酵素は、試料液中のグルコース濃度を測定するグルコースセンサとしてのバイオセンサに用いることができる。本発明のバイオセンサは、酵素として本発明の糖ポリペプチドからなるグルコース脱水素酵素を反応層に使用したセンサであればよい。例えば、該バイオセンサは、絶縁性基板上にスクリーン印刷や蒸着などの方法を利用して電極系を形成し、更に酸化還元酵素と電子受容体とを含む測定試薬を備えることによって作製される。このバイオセンサの測定試薬に基質を含む試料液を接触させると、測定試薬が溶解して酵素と基質が反応し、これにともなって電子受容体が還元される。酵素反応終了後、還元された電子受容体を電気化学的に酸化させ、このとき、このバイオセンサは得られる酸化電流値から試料液中の基質濃度を測定することが可能である。この他に、発色強度又はpH変化などを検知する方式のバイオセンサも構築可能である。これらのバイオセンサにより、測定対象物質を基質とする酵素を選択することによって、様々な物質の測定が可能である。
【0036】
バイオセンサの電子受容体としては、電子の授受能に優れた物質を用いることができる。電子の授受能に優れた物質とは、一般的に「電子伝達体」、「メディエータ」あるいは「酸化還元媒介剤」と呼ばれる化学物質やタンパク質性の電子メディエータであり、これらに該当する化学物質として、例えば、特表2002−526759に挙げられた電子伝達体や酸化還元媒介剤などを利用してもよい。
【0037】
更に本発明のグルコース脱水素酵素は、バイオ電池に用いることができる。本発明のバイオ電池は、酸化反応を行うアノード極及び還元反応を行うカソード極から構成され、必要に応じてアノードとカソードを隔離する電解質層を含んで構成される。上記の電子メディエータ及び本発明の酵素を含む酵素電極をアノード電極に使用し、基質を酸化することによって生じた電子を電極に取り出すと共に、プロトンを発生させる。一方、カソード側には、一般的にカソード電極に使用される酵素を使用すれば良く、例えばラッカーゼ、アスコルビン酸オキシダーゼ又はビリルビンオキシダーゼを使用し、アノード側で発生させたプロトンを酸素と反応させることによって水を生成させる。電極としては、カーボン、金、白金等、一般的にバイオ電池に使用される電極を用いることができる。
【0038】
[酵素活性測定法]
以下の手順に従って各溶液を混合し、吸光度を測定し、GLD活性を調べた。
100mMリン酸カリウム緩衝液(pH6.0)1.00mL、1M D−グルコース溶液1.00mL、超純水0.61mL、3mM 2,6−ジクロロフェノールインドフェノール(以下DCIPという)0.14mL及び3mM 1−メトキシ−5−メチルフェナジウムメチルサルフェイト(以下1−m−PMSという)0.20mLを混合し、37℃で10分間保温後、酵素サンプル0.05mLを添加し、反応を開始した。反応開始時から5分間、酵素反応の進行に伴う600nmにおける吸光度の1分間あたりの減少量(ΔA600)を測定し、直線部分から式1に従いGLD活性を算出した。この際、GLD活性は、37℃、pH6.0で1分間に1μmolのDCIPを還元する酵素量を1Uと定義した。尚、式中の3.0は反応試薬+酵素溶液の液量(mL)、10.8はpH6.0におけるDCIPのモル吸光係数(mM-1cm-1)、1.0はセルの光路長(cm)、0.05は酵素溶液の液量(mL)、ΔA600blankは酵素の希釈に用いた溶液を酵素溶液の代わりに添加して反応開始した場合の600nmにおける吸光度の1分間あたりの減少量、dfは希釈倍率を表す。
【0039】
【数1】

【0040】
本酵素のタンパク濃度の測定においては、該酵素を、好ましくは終濃度0.2〜0.9 mg/mL になるように適宜希釈して用いる。本発明におけるタンパク濃度は、日本バイオ・ラッド(株)から購入できるタンパク濃度測定キットであるBio−Rad Protein Assayを用い、取扱説明書に従って、牛血清アルブミン(BSA,和光純薬工業(株)製,生化学用)を標準物質として作成した検量線から換算して求めることができる。
【0041】
尚、本発明を実施するために使用する様々な技術は、特にその出典を明示した技術を除いては、公知の文献等に基づいて当業者であれば容易かつ確実に実施可能である。例えば、遺伝子工学及び分子生物学的技術はSambrook and Maniatis, in Molecular Cloning-A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 1989; Ausubel, F. M. et al., Current Protocols in Molecular Biology, John Wiley & Sons, New York, N.Y, 1995などに記載の方法あるいはそこで引用された文献記載の方法又はそれらと実質的に同様な方法や改変法に基づき実施可能である。さらに、この発明における用語は基本的にはIUPAC-IUB Commission on Biochemical Nomenclatureによるものであり、あるいは当該分野において慣用的に使用される用語の意味に基づくものである。
【実施例】
【0042】
以下、実施例に則して本発明を更に詳しく説明する。尚、本発明の技術的範囲はこれらの記載によって何等制限されるものではない。又、本明細書中に引用される文献に記載された内容は、本明細書の一部として本明細書の開示内容を構成するものである。
[実施例1]
(ボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)活性の確認)
ポテトデキストロースブロス(Difco社製)2.4%の培地100mLを500mL容の坂口フラスコに入れ、シリコ栓をし、121℃、20分間オートクレーブした。冷却したこの液体培地に、ボトリオティニア・フケリアナ(Botryotinia fuckeliana)NBRC7185株を接種し、25℃で216時間、通気撹拌の条件で菌体を培養した結果、培養上清に、培養液1mL当たり0.21U/mLのグルコース脱水素酵素活性が確認された。尚、グルコース酸化酵素(GOD)活性は検出されなかった。
GOD活性は、以下の方法で測定した。100mMリン酸カリウム緩衝液(pH7.0)1.00mL、25mM 4−アミノアンチピリン0.10mL、420mMフェノール0.10mL、ペルオキシダーゼ(100units/mL)0.10mL、超純水0.65mL、D−グルコース1.00mLを混合し、37℃で5分間保温後、酵素サンプル0.05mLを添加し、反応を開始した。反応開始時から酵素反応の進行に伴う500nmにおける吸光度の1分間あたりの増加量(ΔA500)を測定し、式2に従いGOD活性を算出した。この際GOD活性は、37℃、pH7.0で、1分間に1μmolの過酸化水素を生成する酵素量を1Uと定義した。尚、式中の3.0は反応試薬+酵素溶液の液量(mL)、10.66は本測定条件におけるモル吸光係数(mM-1cm-1)、0.5は1モルの過酸化水素の生成量に対するキノン型色素の生成量、1.0はセルの光路長(cm)、0.05は酵素溶液の液量(mL)、ΔA500blankは酵素の希釈に用いた溶液を酵素溶液の代わりに添加して反応開始した場合の500nmにおける吸光度の1分間あたりの増加量、dfは希釈倍率を表す。
【0043】
【数2】
【0044】
[実施例2]
(真核細胞による組み換えボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)の発現)
(1)菌体培養
グルコース(ナカライ社製)1%(W/V)、脱脂大豆(昭和産業社製)2%(W/V)、コーンスティープリカー(サンエイ糖化社製)0.5%(W/V)、硫酸マグネシウム七水和物(ナカライ社製)0.1%(W/V)及び水からなる液体培地をpH6.0に調整し、100mLを500mL容の坂口フラスコに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、ボトリオティニア・フケリアナNBRC7185株を接種し、25℃で90時間振とう培養した後、吸引ろ過により湿菌体を回収した。
【0045】
(2)染色体DNAの抽出
(1)で得られた菌体のうち湿菌体0.25gを液体窒素により凍結した後、粉砕し、常法により染色体DNAを抽出した。
【0046】
(3)BfGLD遺伝子の取得
(2)で取得したDNAを鋳型とし、BfGLD遺伝子をPCR増幅した。プライマーは、本発明者らによって既に解明されていた複数のGLD遺伝子配列から共通配列を解析し、その共通配列を基に縮重プライマーを設計して遺伝子断片を取得し、最終的に下記のプライマー:primer1F(配列番号15)及びprimer2R(配列番号16)を用いて、目的のGLD遺伝子全長(イントロンを含む)を含む約1.8kbpのDNA断片を取得した。
primer1F:(TGACCAATTCCGCAGCTCGTCAAA)ATGTATCGTTTACTCTCTACATTTG(配列番号15)
(括弧内:転写増強因子)
primer2R:((GCTATCCTGTTACGCTTCTAGA))GCATGCCTAAATGTCCTCCTTGATCAAATCT(配列番号16)
(二重括弧内:pSENSベクター配列、下線部:制限酵素部位(SphI))
primer3F:((CCGTCCTCCAAGTTA))GTCGAC(TGACCAATTCCGCAGCTCGTCAAA)(配列番号17)
(二重括弧内:pSENSベクター配列、下線部:制限酵素部位(SalI)、括弧内:転写増強因子)
【0047】
(4)BfGLD遺伝子を含むベクターの調製
公知文献1(Aspergillus属の異種遺伝子発現系、峰時俊貴、化学と生物、38、12、831−838、2000)に記載してあるアスペルギルス・オリゼ由来のアミラーゼ系の改良プロモーターを使用し、その下流に(3)で得られたDNA断片を鋳型として上記のプライマー:primer3F(配列番号17)及び上記のプライマー:primer2Rを用いて増幅したGLD遺伝子を結合させることで、該遺伝子が発現可能なプラスミドベクターを調製した。この発現用プラスミドベクターを大腸菌JM109株に導入して形質転換し、得られた形質転換体を培養して、集菌した菌体から、Illustra plasmid−prep MINI Flow Kit(GEヘルスケア社製)を用いてプラスミドを抽出した。該プラスミド中のインサートの配列解析を行ったところ、イントロンを含むBfGLD遺伝子(配列番号3)が確認できた。
【0048】
(5)形質転換体の取得
(4)で抽出したプラスミドを用いて、公知文献2(Biosci. Biotech. Biochem.,61(8),1367−1369,1997)及び3(清酒用麹菌の遺伝子操作技術、五味勝也、醸協、494−502、2000)に記載の方法に準じて、BfGLDを生産する組み換えカビ(アスペルギルス・オリゼ)を作製し、得られた組み換え株をCzapek−Dox固体培地で純化した。使用する宿主としては、アスペルギルス・オリゼNS4株を使用した。本菌株は、公知文献2にあるように、1997年(平成9年)に醸造試験所で育種され、転写因子の解析、各種酵素の高生産株の育種などに利用され、分譲されているものが入手可能である。
【0049】
(6)組み換えカビ由来BfGLDの確認
パインデックス2%(松谷化学工業社製)(w/v)、トリプトン1%(BD社製)(w/v)、リン酸二水素カリウム0.5%(ナカライテスク社製)(w/v)、硫酸マグネシウム七水和物0.05%(w/v)(ナカライテスク社製)及び水からなる液体培地10mLを太試験管(22mm×200mm)に入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、(5)で取得した形質転換体を植菌し、30℃で3〜4日間振とう培養した。培養終了後、遠心して上清を回収し、組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)とした。
【0050】
前述の酵素活性測定法に従い組み換えSc(+)BfGLDのグルコース脱水素酵素活性(U/mL)を測定したところ、培養液1mL当たり、3日目が133U/mL、4日目が98U/mLのグルコース脱水素酵素活性を確認できた。尚、グルコース酸化酵素活性は確認できなかった。また、本発明の組み換えBfGLDは、基質濃度333mMにおいて、D−グルコースに対する活性を100%とした場合に、マルトース対する反応性が1.3%であった。
【0051】
(6)酵素の精製
D−グルコース1%(w/v)、大豆粉2%(w/v)、コーンスティープリカー0.5%(w/v)、硫酸マグネシウム七水和物0.1%(w/v)及び水からなる液体培地(pH7.0)50mLを200mL容の三角フラスコに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、(5)で取得した形質転換体を植菌し、25℃で3日間振とう培養して種培養液とした。前記と同様の培地組成で消泡剤を添加した培地3.5Lを5L容ジャーファーメンターに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、種培養液を50mL植菌し、30℃、300rpm、1v/v/mで7日間培養した。培養終了後、培養液をろ布でろ過し、回収したろ液を遠心して上清を回収し、更にメンブレンフィルター(10μm、アドバンテック社製)でろ過して培養上清を回収し、分画分子量8,000の限外ろ過膜(ミリポア社製)で濃縮して粗酵素液とした。粗酵素液の比活性は1,170U/mgだった。
【0052】
前記粗酵素液を、50%飽和硫酸アンモニウム溶液(pH5.0)になるように調整し、4℃で一晩放置後、遠心分離して上清を回収した。
該上清を、50%飽和硫酸アンモニウムを含む50mM酢酸ナトリウム緩衝液(pH5.0)で予め平衡化したTOYOPEARL Butyl−650C(東ソー社製)カラム(φ7.5cm×17.7cm)に通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から50mM酢酸ナトリウム緩衝液(pH5.0)へのグラジエント溶出法で酵素を溶出させて、活性画分を回収した。回収した活性画分を、限外濾過膜で濃縮後、脱塩し、1mM酢酸ナトリウム緩衝液(pH5.0)と平衡化させ、同緩衝液で予め平衡化したDEAEセルファインA−500m(チッソ社製)カラム(φ6.0cm×21.9cm)に通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から200mM酢酸ナトリウム緩衝液(pH5.0)へのグラジエント溶出法で酵素を溶出させて、活性画分を回収した。回収した活性画分を、分画分子量8,000の限外ろ過膜で濃縮後、水置換して組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)の精製酵素とした。該精製酵素の比活性は2,100U/mgだった。
更に、糖鎖切断前後の組み換えBfGLDをSDS−ポリアクリルアミド電気泳動に供して確認を行った。詳細には、組み換えSc(+)BfGLD5μLと1%SDS及び2%β―メルカプトエタノールを含む0.4Mリン酸カリウム緩衝液(pH6.0)5μLを混合し、100℃で3分間熱処理を行った。糖鎖切断処理として、熱処理後のサンプルにエンドグリコシダーゼH(ロシュ製)を10μL(50mU)添加し、37℃で18時間反応させた。糖鎖切断処理前後のサンプルをSDS−ポリアクリルアミド電気泳動に供し、分子量マーカーより分子量を求めたところ、糖鎖切断前の酵素が90〜100kDa、糖鎖切断後の酵素が60〜70kDaの組み換えBfGLDを確認できた。
【0053】
[参考例1]
(原核細胞による組み換えボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)の発現)
(1)BfGLD遺伝子のクローニング
実施例2の(4)で抽出したプラスミドを鋳型とし、BfGLD遺伝子をPCR増幅した。予想シグナル配列をコードする遺伝子配列を含まないBfGLD(16AA(−)BfGLD)遺伝子を得るために、野生型BfGLDの予想N末端からC末端をコードする遺伝子を増幅できる下記のprimer4F_BfGLD_Sig(-)/Sac(配列番号18)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いてPCRを行った。予想シグナル配列をコードする遺伝子配列を含まないポリヌクレオチドの取得は、分泌シグナル配列をコードする遺伝子配列を含む外来遺伝子を用いて大腸菌で組み換えタンパクを発現させる場合は、組み換えタンパクがペリプラズムに移行されるため生産性が悪いことから、組み換えタンパクを効率よく回収したい場合は、一般的にシグナル配列をコードする遺伝子配列を削除した配列を用いることが知られているためである。尚、PCRは、DNAポリメラーゼ、ExTaq(タカラバイオ社製)を使用し、反応条件は[94℃/30秒→50℃/1分→72℃/2分]×30サイクルとした。野生型BfGLDの予想N末端は、BfGLDの全長アミノ酸配列(配列番号2)と国際公開2006/101239号パンフレットの配列番号2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素(AtGLD)との配列同一性を調べ、野生型AtGLDのN末端である国際公開2006/101239号パンフレットの配列番号2の20番目以降のアミノ酸配列から、野生型BfGLDのN末端は、配列番号1記載の17番目以降のアミノ酸配列であると予測した。
primer4F_BfGLD_Sig(-)/Sac:AAAGAGCTCGAGCACCGACTCTACCTTAAA(配列番号18)
(下線部:制限酵素部位(SacI))
primer5R_BfGLD/Hind:CCCAAGCTTCTAAATGTCCTCCTTGATC(配列番号19)
(下線部:制限酵素部位(HindIII))
PCRにより、46bpのイントロンを含み、予想シグナル配列をコードする48bpの塩基配列を含まない配列にプライマー付加配列が付加したDNA断片を取得した。
【0054】
次いで、イントロンを削除するため、前記で得られたDNA断片を鋳型として、primer4F_BfGLD_Sig(-)/Sac(配列番号18)及び下記のprimer6R_Bf_up(int)(配列番号20)のプライマーセット又は下記のprimer7F_Bf_(int)down(配列番号21)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いて各々前記PCR条件でPCR増幅した。
primer6R_Bf_up(int):GGGTGTATGCCATTCCATTGATAGTACTAGTCCCT(配列番号20)
primer7F_Bf_(int)down:GAATGGCATACACCCGAGCCGAAGAT(配列番号21)
得られた2つの増幅配列を1つのPCR増幅チューブに入れ、primer4F_BfGLD_Sig(-)/Sac(配列番号18)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いて前記PCR条件でPCR増幅し、イントロンを含まず、予想シグナル配列をコードする48bpの塩基配列を含まない16AA(−)BfGLD遺伝子にプライマー付加配列が付加したDNA断片を取得した。
【0055】
(2)BfGLD遺伝子を含むベクターの調製及び形質転換体の取得
pUC18ベクター(タカラバイオ社製)を制限酵素SacIとHindIIIで開裂し、同制限酵素処理したPCR増幅DNA断片をベクターにライゲーションし、大腸菌JM109株に導入して形質転換した。得られた形質転換体のうち6クローンよりプラスミドDNAを調製し、SacIとHindIIIで処理したところ全てのクローンで目的のサイズの断片が確認できた。そのうち1クローンについてプラスミドを調製してそのインサートの配列決定を行ったところ、イントロンを含まず、予想シグナル配列をコードする48bpの塩基配列を含まない配列番号50に記載の16AA(−)BfGLD遺伝子が確認された。
【0056】
(3)組み換え大腸菌由来BfGLDの確認
LB液体培地10mLを太試験管に入れ、シリコ栓をし、121℃、20分間オートクレーブした。冷却したこの液体培地に、50μg/mLアンピシリンを加え、(4)で作成した形質転換体を接種し、37℃で振とう培養した。培養液のOD600が約0.4〜0.5となった時点で、IPTGを1mM添加し、更に18時間振とう培養を行った。培養終了後、菌体を遠心により集め、20mMリン酸カリウム緩衝液(pH7.0)に懸濁した。超音波破砕装置を用いて菌体を破砕後、遠心して無細胞抽出液を調製した。前述の酵素活性測定法に従い組み換えBfGLD糖鎖無しサンプル(Sc(−)BfGLD)のグルコース脱水素酵素活性(U/mL)を測定したところ、培養液1mL当たり、0.703U/mLのグルコース脱水素酵素活性を確認できた。尚、グルコース酸化酵素活性は確認できなかった。
【0057】
(4)組み換えSc(−)BfGLDの精製
(2)で作成した形質転換体を、50μg/mLアンピシリンを含むLB液体培地10mLに植菌し、37℃で7時間振とう培養した後、50μg/mLアンピシリンを含むLB液体培地50mL入りの250mL容のマイヤーフラスコに植菌し、37℃で17.5時間振とう培養し、前培養液とした。50μg/mLアンピシリンを含むLB液体培地1.5L入りの2L容のジャーファーメンターに前培養液50mLを植菌し、37℃で培養し、培養液のOD600が約0.4〜0.5となった時点で、IPTGを1mM添加し、25.5時間通気撹拌条件で培養を行った。培養終了後、遠心により集めた菌体を、20mMリン酸カリウム緩衝液(pH6.0)に懸濁し、超音波破砕装置を用いて菌体を破砕後、遠心して無細胞抽出液を調製した。該無細胞抽出液の比活性は0.0182U/mgだった。前記無細胞抽出液を20mM リン酸カリウム緩衝液(pH7.5)中で透析し、同緩衝液で平衡化したDEAE−セルロファインA−500カラムに通液し、溶出液を集めた。前記溶出液を5mM リン酸カリウム緩衝液(pH7.5)中で透析し、同緩衝液で平衡化したDEAE−セルロファインA−500カラムに通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から0.3M 塩化カリウムを含む同緩衝液へのグラジエント溶出法で酵素を溶出させて活性画分を集めた。活性画分を分画分子量10000の限外濾過膜で濃縮し、脱塩後20mMリン酸カリウム緩衝液(pH6.0)に置換し、Sc(−)BfGLDの精製酵素とした。
更に、組み換えSc(−)BfGLDをSDS−ポリアクリルアミド電気泳動に供し、分子量マーカーより分子量を求めたところ約60kDaの組み換えSc(−)BfGLDを確認できた。
【0058】
[実施例3]
(組み換えBfGLDの公知配列との比較)
シグナル配列部分を含むBfGLDのアミノ酸配列は配列番号2に記載の通りだったが、該配列を用いてBLAST検索したところ、驚いたことにボトリオティニア・フケリアナ由来グルコース酸化酵素のアミノ酸配列と一致していた。しかし、本発明で得られたBfGLDは、実施例2の(6)に記載の通りグルコース脱水素酵素活性を有しており、グルコース酸化酵素活性は全くみられなかった。ボトリオティニア・フケリアナ由来グルコース酸化酵素のアミノ酸配列は公知文献4(Molecular Plant Pathology(2004)5(1),17−27)に記載されており、Botrytis cinerea(=Botryotinia fuckeliana)のセルフクローニングを行っていることが記載されている。更に該文献中に記載の公知文献5(Physiological and Molecular Plant Pathology(1998)53,123―132)に、野生型のBotrytis cinerea(=Botryotinia fuckeliana)由来グルコース酸化酵素の特性が記載されているが、該酵素はサブユニット分子量が35kDaで、ゲルろ過分子量が約160kDaの4量体酵素であることが記載されている。これに対して本発明の組み換えBfGLDは、SDS−ポリアクリルアミド電気泳動における分子量が、真核細胞で組み換えた場合、糖鎖切断前の酵素が90〜100kDa、糖鎖切断後の酵素が60〜70kDaのグルコース脱水素酵素である。例えば、公知文献6(蛋白質核酸酵素(2004)49(5),625-633,Review)には、キサンチン脱水素酵素が、プロテアーゼによる部分分解でキサンチン酸化酵素へ変換されることが記載されている。つまり、立体構造が取られた後に、プロテアーゼにより分解されており、キサンチン脱水素酵素とキサンチン酸化酵素は同じ遺伝子に由来している。これらのことから、本発明のボトリオティニア・フケリアナ由来グルコース脱水素酵素とボトリオティニア・フケリアナ由来グルコース酸化酵素は、遺伝子配列から推測されるアミノ酸配列は同じ配列であっても、ボトリオティニア・フケリアナ由来グルコース脱水素酵素はサブユニット分子量が糖鎖切断前の酵素で90〜100kDa、糖鎖切断後の酵素で60〜70kDaであり、かつグルコース脱水素酵素活性を有しており、一方、論文記載のボトリオティニア・フケリアナ由来グルコース酸化酵素はサブユニット分子量が35kDaで、かつグルコース酸化酵素活性を有しており、構造、機能とも全く異なる酵素であり、両者の違いは、立体構造を取った後にプロテアーゼによる分解等何らかの修飾を受けるか否かによると推察される。特に、アスペルギルス属で組み換えた酵素においては、プロテアーゼ修飾による活性の変換は見られなかったため、アスペルギルス属で組み換え製造することは、組み換えBfGLDを効率的に製造できる有意義な方法と思われる。
【0059】
[実施例4]
(電極によるグルコースの測定)
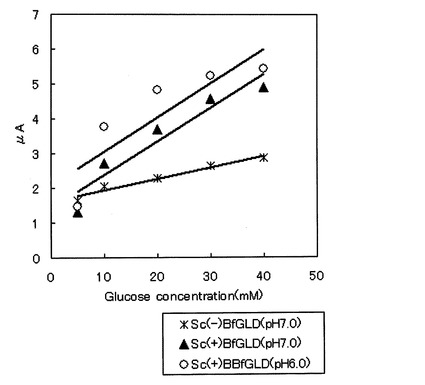
組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)を使用し、電極によるD−グルコースの測定を行った。酵素0.16Uを搭載したグラッシーカーボン(GC)電極を用いて、グルコース濃度に対する応答電流値を測定した。電解セル中に、50mM リン酸ナトリウムバッファー(pH6.0又はpH7.0)1.8mL及び1M ヘキサシアノ鉄(III)酸カリウム(フェリシアン化カリウム)水溶液0.2mLを添加した。GC電極をポテンショスタットBAS100B/W(BAS製)に接続し、37℃で溶液を撹拌し、銀塩化銀参照電極に対して+500mVを印加した。これらの系に終濃度が10mMになるように1M D−グルコース溶液を5μl添加し、定常状態の電流値を測定した。更に各終濃度になるように1M D−グルコース溶液を添加し電流値を測定するという作業を、繰り返し、最終的に各グルコース濃度(5、10、20、30、40mM)の電流値を測定した。これをプロットしたところ、検量線が作成できた(図1)。尚、50mM リン酸ナトリウムバッファー(pH7.0)を用いて組み換えBfGLD糖鎖無しサンプル(Sc(−)BfGLD)についても同様に電流値を測定した結果、Sc(+)BfGLDの方が、Sc(−)BfGLDより約2倍のセンサ感度を示した。
【0060】
[実施例5]
(N末端解析)
組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)のN末端を解析したところ、STLNYであることが明らかになった。つまり、MYRLLSTFAVASLAAASTDの19アミノ酸がシグナル配列であり、該酵素は、翻訳後のペプチドからシグナルペプチダーゼによる修飾で、該19アミノ酸が削除された形で存在していることが分かった。
【0061】
[実施例6]
(シグナル配列変異)
組み換えカビによる組み換えBfGLD製造において、分泌シグナルの変異を行った。
下記に記載したプライマーを合成し、実施例2の(4)で取得したプラスミドベクターを鋳型として、シグナル配列を置換したBfGLD遺伝子をPCR法により増幅した。
primer8_Bf-Atsignal1-7AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))TTTGCTGTAGCCTCTTTGGCTGCAG(配列番号22)
primer9_Bf-Atsignal2-F:((ATGTTGGGAAAGCTCTCCTTCCTCAGTGCCCTGTCCCTGGCAGTGGCGGCACCTTTG))(配列番号23)
primer10_Bf-eno-Atsignal-F:(TGACCAATTCCGCAGCTCGTCAAA)((ATGTTGGGAAAGCTCTCCTTCCTCA))(配列番号24)
primer11_Bf-infusion-F:CTCCAAGTTAGTCGAC(TGACCAATTCCGCAGCTCGTCAAA)(配列番号25)
primer12_Bf-infusion-R:CGCTTCTAGAGCATGCCTAAATGTCCTCCTTGATCAAATC(配列番号26)
primer13_Bf-Aosig-short1-7AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))TTTGCTGTAGCCTCTTTGGCTGCA(配列番号27)
primer14_Bf-Aosig-short2-F:((ATGCTCTTCTCACTGGCATTCCTGAGTGCCCTGTCGCTGGCCACGGCA))(配列番号28)
primer15_Bf-Aosignal1-7AA-F:((TCGCTGGCCACGGCATCACCGGCTGGACGGGCC))TTTGCTGTAGCCTCTTTGGCTGCAGCTAGCACC(配列番号29)
primer16_Bf-Aosignal2-F:((ATGCTCTTCTCACTGGCATTCCTGAGTGCCCTGTCGCTGGCCACGGCATCACCGGCTGGACGGGCC))(配列番号30)
primer17_Bf-eno-Aosignal-F:(TGACCAATTCCGCAGCTCGTCAAA)((ATGCTCTTCTCACTGGCATTCCTGA))(配列番号31)
primer18_Bf-Atsignal1-16AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))AGCACCGACTCTACCTTAAACTATG(配列番号32)
primer19_Bf-Aosig1-short1-16AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))AGCACCGACTCTACCTTAAACTATG(配列番号33)
primer20_Bf-Atsignal1-19AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))TCTACCTTAAACTATGATTATATCATCG(配列番号34)
primer21_Bf-Aosig1-short1-19AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))TCTACCTTAAACTATGATTATATCATCG(配列番号35)
(FはForward、RはReverse、下線部:制限酵素切断部位、二重括弧内:各シグナル配列、括弧内:enoA 5'-UTR、その他:ORF)
尚、上記プライマーは、国際公開2006/101239号パンフレットの配列番号2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素(AtGLD)のシグナル配列情報(該公報記載の配列番号2の1から19番目のアミノ酸配列:配列番号10)及びシグナル配列予測サイト(Signal p:http://www.cbs.dtu.dk/services/SignalP/)を用いて推定したアスペルギルス・オリゼ由来グルコース脱水素酵素(AoGLD)のシグナル配列を元にデザインした。尚、AoGLDのシグナル配列は、2種類(短め:配列番号12及び長め:配列番号14)の配列を予想した。
【0062】
1.BfGLDのシグナル配列のうち7アミノ酸を削除して別シグナルを付加するためのクローニング
1−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer8_Bf-Atsignal1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-7AA(−)BfGLD遺伝子として配列番号36に示し、該配列にコードされるアミノ酸配列を配列番号37に示した。
【0063】
1−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer13_Bf-Aosig-short1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-7AA(−)BfGLD遺伝子として配列番号38に示し、該配列にコードされるアミノ酸配列を配列番号39に示した。
【0064】
1−(2)L.AoGLDの予想長めシグナル配列22アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAoGLDの予想長めシグナル配列22アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号13に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想長めシグナル配列をコードする塩基配列66bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer15_Bf-Aosignal1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer16_Bf-Aosignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAoGLDの予想長めシグナル配列(配列番号14)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigL-7AA(−)BfGLD遺伝子として配列番号40に示し、該配列にコードされるアミノ酸配列を配列番号41に示した。
【0065】
2.BfGLDのシグナル配列のうち16アミノ酸を削除して別シグナルを付加するためのクローニング
2−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから16番目までのアミノ酸(MYRLLSTFAVASLAAA)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち48番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer18_Bf-Atsignal1-16AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の16アミノ酸(MYRLLSTFAVASLAAA)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-16AA(−)BfGLD遺伝子として配列番号42に示し、該配列にコードされるアミノ酸配列を配列番号43に示した。
【0066】
2−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから16番目までのアミノ酸(MYRLLSTFAVASLAAA)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち48番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer19_Bf-Aosig-short1-16AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の16アミノ酸(MYRLLSTFAVASLAAA)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-16AA(−)BfGLD遺伝子として配列番号44に示し、該配列にコードされるアミノ酸配列を配列番号45に示した。
【0067】
3.BfGLDの全シグナル配列19アミノ酸を削除して別シグナルを付加するためのクローニング
3−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDの全シグナル配列、つまりメチオニンから19番目までのアミノ酸(MYRLLSTFAVASLAAASTD)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち57番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer20_Bf-Atsignal1-19AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)の全シグナル配列19アミノ酸(MYRLLSTFAVASLAAASTD)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-19AA(−)BfGLD遺伝子として配列番号46に示し、該配列にコードされるアミノ酸配列を配列番号47に示した。
【0068】
3−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDの全シグナル配列、つまりメチオニンから19番目までのアミノ酸(MYRLLSTFAVASLAAASTD)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち57番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer21_Bf-Aosig-short1-19AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)の全シグナル配列19アミノ酸(MYRLLSTFAVASLAAASTD)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-19AA(−)BfGLD遺伝子として配列番号48に示し、該配列にコードされるアミノ酸配列を配列番号49に示した。
【0069】
2.ベクターの調整
得られた各PCR産物を、In-Fusion Advantage PCRクローニングキット(タカラバイオ社製)を用いて、公知文献1に記載してあるアスペルギルス・オリゼ由来のアミラーゼ系の改良プロモーターの下流に結合させる事で、各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子が発現可能なベクターを各々調製した。
【0070】
これらの発現用ベクターを各々大腸菌JM109株に導入して形質転換し、得られた形質転換体を各々培養して、集菌した菌体から、Illustra plasmid-prep MINI Flow Kit(GEヘルスケア社製)を用いて、各プラスミドを抽出し、インサートの配列解析を各々行ったところ、各プラスミドで各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子が確認できた。
【0071】
3.形質転換体の取得
実施例2の(5)と同様の方法で、各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子を導入した組み換えカビを各々作製し、Czapek-Dox固体培地で各形質転換体を純化した。パインデックス2%(松谷化学工業社製)(W/V)、トリプトン1%(BD社製)(W/V)、リン酸二水素カリウム0.5%(ナカライテスク社製)(W/V)、硫酸マグネシウム七水和物0.05%(W/V)(ナカライテスク社製)及び水からなる液体培地10mLを太試験管(22mm×200mm)に入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、取得した各形質転換体を1遺伝子につき数本ずつ植菌し、30℃で4日間振とう培養した。3日目及び4日目にサンプリングした培養上清を遠心(3,000×g、20分)し、沈殿を取り除いたものを各酵素サンプルとした。
【0072】
前述の酵素活性測定法に従い各酵素サンプルのグルコース脱水素酵素活性(U/mL)を測定した結果、同一遺伝子につき活性の高かった2サンプルの平均値が下記の表1のとおりとなった。
【0073】
【表1】
【0074】
本酵素サンプルのうち、Atsig-7AA(−)BfGLD遺伝子に由来する酵素サンプルについて、N末端を解析したところ、STLNYであることが明らかになった。つまり、野生型のオリジナルシグナル配列の一部を削除して、他シグナル配列を付加しても、酵素のN末端は野生型と同じSTLNYとなっていることが分かった。
【0075】
以上の結果より、BfGLDを組み換え製造する場合、BfGLDのシグナル配列をコードする遺伝子の一部又は全部を、他のシグナル配列をコードする遺伝子と置換しても野生型と同じBfGLDを製造できることが明らかになった。
【技術分野】
【0001】
本発明は、グルコース脱水素酵素及びその製造方法、並びにグルコース脱水素酵素を用いたグルコースの測定方法、グルコース測定試薬組成物及びグルコース測定用バイオセンサに関する。
【背景技術】
【0002】
血液中のグルコース濃度の迅速かつ正確な測定は、糖尿病を診断するうえで重要である。グルコースの測定法としては、化学法と酵素法があるが、酵素法が特異性、安全性の点で優れている。当該酵素法の中でも、検体の微量化、測定時間の短縮、装置の小型化の点から、電気化学的バイオセンサが有利である。
【0003】
そのようなバイオセンサに使用可能な酵素として、グルコース酸化酵素が知られている。しかし、グルコース酸化酵素は、血中の溶存酸素により測定誤差が生じるという問題があるため、いくつかのグルコース脱水素酵素が開発されてきた。グルコース脱水素酵素のうち、フラビン結合型グルコース脱水素酵素は、補酵素の添加を必要としないこと、溶存酸素の影響を受けないこと及び基質特異性に優れることから、グルコースバイオセンサ用の酵素として注目されている(特許文献1〜4)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】国際公開2004/058958号パンフレット
【特許文献2】国際公開2006/101239号パンフレット
【特許文献3】国際公開2008/001903号パンフレット
【特許文献4】国際公開2010/140431号パンフレット
【非特許文献】
【0005】
【非特許文献1】Mol. Plant Pathol. 5(1),p17-27,2004
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、グルコースバイオセンサ用の酵素として有用なグルコース脱水素酵素、これを用いたグルコースバイオセンサ及び当該グルコース脱水素酵素遺伝子を特定、単離し、効率的に組み換え酵素を製造する方法を提供することである。
【課題を解決するための手段】
【0007】
そこで本発明者は、糸状菌由来のグルコース脱水素酵素に着目し、ボトリオティニア・フケリアナ由来のグルコース脱水素酵素遺伝子を単離し、さらにその組み換えベクターを大腸菌、糸状菌、酵母等の微生物に導入して組み換えグルコース脱水素酵素を製造し、その酵素活性、アミノ酸配列、グルコースセンサへの利用性等について検討した。その結果、ボトリオティニア・フケリアナ由来の配列番号51で示されるアミノ酸配列を有する酵素は、グルコース酸化酵素(GOD)であるとされていたが(非特許文献1)、本発明により得られた組み換え酵素はグルコース酸化酵素ではなくフラビン結合型グルコース脱水素酵素であり、かつそのアミノ酸配列は従来推定されていたものとは異なり、配列番号5で示されるものであることを見出した。従って、本発明により得られたフラビン結合型グルコース脱水素酵素は、溶存酸素の影響を受けないことからグルコースバイオセンサ用の酵素として有用であることを見出した。さらには、種々の組み換え酵素のうち、形質転換真核細胞により得られた糖ポリペプチドは、形質転換大腸菌により得られた糖鎖を有さないポリペプチドに比べて、バイオセンサ用酵素として有用であることを見出し、本発明を完成した。
【0008】
すなわち、本発明は、以下の〔1〕〜〔12〕を提供するものである。
〔1〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔2〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を含有するグルコース測定試薬組成物:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔3〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコースの測定方法:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔4〕以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
〔5〕ボトリオティニア・フケリアナ由来の組み換えグルコース脱水素酵素遺伝子を用いた形質転換真核細胞で発現させたものである〔4〕記載のフラビン結合型グルコース脱水素酵素。
〔6〕以下の(a)又は(b)のポリヌクレオチド:
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
〔7〕以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチド:
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
〔8〕〔6〕又は〔7〕に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチド。
〔9〕シグナル配列が真核細胞由来タンパクのシグナル配列である、〔8〕記載のポリヌクレオチド。
〔10〕〔6〕〜〔9〕の何れか1項に記載のポリヌクレオチドを含む組み換えベクター。
〔11〕〔10〕記載の組み換えベクターを用いることによって作成された形質転換真核細胞。
〔12〕〔11〕記載の形質転換真核細胞を培養し、得られた培養物からグルコース脱水素酵素を採取することを特徴とするグルコース脱水素酵素の製造方法。
【発明の効果】
【0009】
本発明の糖ポリペプチドは、フラビン結合型グルコース脱水素酵素であり、溶存酸素の影響を受けずにグルコースを正確に測定できるグルコースバイオセンサに有用である。また、本発明のポリヌクレオチドを利用することにより、グルコースに対する基質認識性に優れ、しかもマルトースに対する作用性が低いという優れた特性を有するグルコース脱水素酵素を均質かつ大量に生産することが可能となる。
【図面の簡単な説明】
【0010】
【図1】本発明グルコース脱水素酵素によるグルコース量測定結果を示す図である。
【発明を実施するための形態】
【0011】
本発明において、「グルコース脱水素酵素」とは、電子受容体存在下で、グルコースの1位の水酸基を脱水素(酸化)する反応を触媒し、グルコースへの作用性に対してマルトースへの作用性が5%以下である可溶性の蛋白質を意味し、該酵素は以下の性質を特徴とする。
1)フラビンアデニンジヌクレオチド(FAD)を補酵素とする、
2)酸素を電子受容体としない、
3)基質濃度333mMにおいて、グルコースへの作用性を100%とした場合にマルトースへの作用性が5%以下、及び
4)酵素タンパクのポリペプチドの分子量が60〜70kDaである。
酵素タンパクのポリペプチドの分子量とは、真核細胞で発現させた糖鎖修飾された組み換え酵素を糖鎖切断処理し、SDS−ポリアクリルアミド電気泳動に供した際の分子量である。尚、真核細胞で発現した組み換え酵素は、宿主、培養条件等の影響で糖鎖付加量は変化するため、80〜200kDa程度の分子量になり得る。
また、本発明において、「糖ポリペプチド」とは、糖鎖が付加したポリペプチドをいう。
【0012】
本発明のポリヌクレオチドは、以下の(a)又は(b)のポリヌクレオチドである。
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【0013】
更に、本発明のポリヌクレオチドは、以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチドである。
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
【0014】
前記のポリヌクレオチド(b)又はポリペプチド(d)としては、塩基配列(a)又はアミノ酸配列(c)と少なくとも80%以上の相同性を有する塩基配列又はアミノ酸配列を有するものが挙げられる。ここで、少なくとも80%の相同性を有する塩基配列又はアミノ酸配列とは、夫々比較対象となる基準配列の全長にわたり、少なくとも80%の同一性を示し、好ましくは少なくとも85%、より好ましくは少なくとも90%、更に好ましくは少なくとも95%の同一性を有する各配列をいう。このような配列の同一性パーセンテージは、基準配列を照会配列として比較するアルゴリズムをもった公開又は市販されているソフトウエアを用いて計算することができる。例として、BLAST、FASTA、又はGENETYX(ソフトウエア開発社製)などを用いることができ、これらはデフォルトパラメーターで使用することができる。
【0015】
本発明において、ポリヌクレオチド間のハイブリダイズに際しての「ストリンジェントな条件下でハイブリダイズ」の具体的な条件とは、例えば、50%ホルムアミド、5×SSC(150mM 塩化ナトリウム、15mM クエン酸三ナトリウム、10mM リン酸ナトリウム、1mM エチレンジアミン四酢酸、pH7.2)、5×デンハート(Denhardt’s)溶液、0.1% SDS、10% デキストラン硫酸及び100μg/mLの変性サケ精子DNAで42℃インキュベーションした後、フィルターを0.2×SSC中42℃で洗浄することを例示することができる。
【0016】
尚、本発明において、「ポリヌクレオチド」とは、プリン又はピリミジンが糖にβ-N-グリコシド結合したヌクレオシドのリン酸エステル(ATP(アデノシン三リン酸)、GTP(グアノシン三リン酸)、CTP(シチジン三リン酸)、UTP(ウリジン三リン酸);又はdATP(デオキシアデノシン三リン酸)、dGTP(デオキシグアノシン三リン酸)、dCTP(デオキシシチジン三リン酸)、dTTP(デオキシチミジン三リン酸))が100個以上結合した分子を言い、具体的にはグルコース脱水素酵素をコードする染色体DNA、染色体DNAから転写されたmRNA、mRNAから合成されたcDNA及び、それらを鋳型としてPCR増幅したポリヌクレオチドを含む。「オリゴヌクレオチド」とはヌクレオチドが2-99個連結した分子を言う。また「ポリペプチド」とは、アミド結合(ペプチド結合)又は非天然の残基連結によって互いに結合した30個以上のアミノ酸残基から構成された分子を意味する。「糖ポリペプチド」とは、前記ポリペプチドに糖鎖が付加した分子を意味する。
【0017】
本発明のポリヌクレオチド(遺伝子)の最も具体的な態様は、配列番号4又は配列番号6の塩基配列を含むポリヌクレオチドである。配列番号6に代表される染色体DNAであるポリヌクレオチドは、例えばボトリオティニア・フケリアナ株から染色体DNAライブラリーを調製し、特許文献2に記載のアスペルギルス・テレウス(Aspergillus terreus)由来グルコース脱水素酵素のアミノ酸配列及びアスペルギルス・オリゼ(Aspergillus oryzae)由来グルコース脱水素酵素のアミノ酸配列又はその他のグルコース脱水素酵素のアミノ酸配列に基づいて作成した複数のオリゴヌクレオチドプローブを用いて当業者に公知の方法によって上記染色体DNAライブラリーをスクリーニングすることによって取得することができる。
【0018】
プローブの標識は、当業者に公知の任意の方法、例えば、ラジオアイソトープ(RI)法又は非RI法によって行うことができるが、非RI法を用いることが好ましい。非RI法としては、蛍光標識法、ビオチン標識法、化学発光法等が挙げられるが、蛍光標識法を用いることが好ましい。蛍光物質としては、オリゴヌクレオチドの塩基部分と結合できるものを適宜に選択して用いることができるが、シアニン色素(例えば、Cy DyeTMシリーズのCy3、Cy5等)、ローダミン6G試薬、N-アセトキシ-N2-アセチルアミノフルオレン(AAF)、AAIF(AAFのヨウ素誘導体)などを使用することができる。
【0019】
或いは、配列番号4に代表されるcDNAであるポリヌクレオチドは、例えば、本明細書の実施例に具体的に記載されているように、染色体DNAからイントロンを含む目的のポリヌクレオチドを取得した後、PCRによってイントロンを削除して得ることができる他、例えば、cDNAライブラリーを鋳型とし、上記で作成したオリゴヌクレオチドプライマー(プローブ)のセットを用いた当業者に公知の各種PCR法によって得ることができ、又は、ボトリオティニア・フケリアナ株から抽出した全RNAもしくはmRNAを鋳型とするRT−PCR法によっても得ることができる。尚、PCRにおけるプライマー使用の最終濃度が約0.1から約1μMになるよう留意することも必要である。プライマー設計においては、市販のソフトウエア、例えばOligoTM(National Bioscience Inc.(米国)製)、GENETYX(ソフトウエア開発社製)等を用いることもできる。
【0020】
尚、このようなオリゴヌクレオチドプローブやオリゴヌクレオチドプライマーセットは、例えば本発明のポリヌクレオチドであるcDNAを適当な制限酵素で切断して作成することもできる。
【0021】
更に、本発明のポリヌクレオチドは、〔6〕又は〔7〕に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチドである。シグナル配列をコードする塩基配列は、グルコース脱水素酵素活性を有する糖ポリペプチドを効率良く分泌できるシグナル配列をコードする塩基配列であれば良く、例えば真核細胞由来タンパクのシグナル配列をコードする塩基配列が好ましく、ボトリオティニア属又はアスペルギルス属由来タンパクのシグナル配列をコードする塩基配列がより好ましい。更に、該ポリヌクレオチドにより形質転換する宿主と同属由来タンパクのシグナル配列をコードする塩基配列や、グルコース脱水素酵素のシグナル配列をコードする塩基配列が例示できる。塩基配列によりコードされるシグナル配列は、例えば特許文献2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素配列と分泌タンパクのアミノ酸配列とを比較することや、シグナル配列予測サイト(Signal p:http://www.cbs.dtu.dk/services/SignalP/)を用いて分泌タンパクのアミノ酸配列から推定でき、更に1〜数個のアミノ酸が置換、欠失又は付加されたシグナル配列でも良い。具体例としては配列番号8、配列番号10、配列番号12及び配列番号14に記載の配列が例示でき、これらの配列又はこれらの配列を数アミノ酸欠失させた配列を適宜組み合わせた配列でも良い。
【0022】
シグナル配列をコードする塩基配列を有するポリヌクレオチドの具体例としては、配列番号1、配列番号3、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46及び配列番号48に記載のポリヌクレオチドが例示できるが、グルコース脱水素酵素活性を有するタンパク質をコードする部位は、イントロンを含んでいても良い。
【0023】
又、(a)〜(d)のポリヌクレオチドは、例えば、前記のボトリオティニア・フケリアナ由来のグルコース脱水素酵素cDNAを、公知のミューテーション導入法や変異導入PCR法等によって改変して作成することができる。更に、NBRC7185株以外のボトリオティニア・フケリアナ株の染色体DNAやそのcDNAライブラリーから、配列番号1のヌクレオチド配列情報に基づいて作成したオリゴヌクレオチドを用いるプローブハイブリダイゼーション法によって取得することができる。ハイブリダイゼーションに際して、ストリンジェント条件を様々に変化させることによって、上記ポリヌクレオチドを取得することができる。ストリンジェント条件は、ハイブリダイゼーション及び洗浄工程における塩濃度、有機溶媒(ホルムアルデヒド等)の濃度、温度条件等によって規定され、例えば、米国特許No.6,100,037号明細書等に開示されているような、当業者らに周知の様々な条件を採用することができる。
【0024】
更に、文献(例えばCarruthers(1982)Cold Spring Harbor Symp. Quant. Biol. 47:411-418; Adams(1983)J. Am. Chem. Soc. 105:661; Belousov(1997)Nucleic Acid Res. 25:3440-3444; Frenkel(1995)Free Radic. Biol. Med. 19:373-380; Blommers(1994)Biochemistry 33:7886-7896; Narang(1979)Meth. Enzymol. 68:90; Brown(1979)Meth. Enzymol. 68:109; Beaucage(1981)Tetra. Lett. 22:1859; 米国特許第4,458,066号)に記載されているような周知の化学合成技術により、in vitroにおいて本発明のポリヌクレオチドを合成することができる。
【0025】
本発明の組み換えベクターは、クローニングベクター又は発現ベクターであり、インサートとしてのポリヌクレオチドの種類や、その使用目的等に応じて適宜のものを使用する。例えば、cDNA又はそのORF領域をインサートとしてグルコース脱水素酵素を生産する場合には、in vitro転写用の発現ベクターや、酵母、カビなどの糸状菌、昆虫細胞、哺乳動物細胞等の真核細胞のそれぞれに適した発現ベクターを使用することもできる。
【0026】
本発明の形質転換細胞としては、例えば、酵母、カビ、昆虫細胞、哺乳動物細胞等の真核細胞を使用することができる。これらの形質転換細胞は、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法など公知の方法によって組み換えベクターを細胞に導入することによって調製することができる。組み換えベクター及び形質転換細胞の具体例として、下記実施例に示した組み換えベクターと、このベクターによる形質転換カビ、形質転換酵母が挙げられる。
【0027】
グルコース脱水素酵素を真核細胞で発現させて生産させる場合には、前記ポリヌクレオチドを、プロモーター、スプライシング領域、ポリ(A)付加部位等を有する真核細胞用発現ベクターに挿入して組み換えベクターを作成し、真核細胞内に導入すれば、グルコース脱水素酵素を真核細胞で生産することができる。プラスミドのような状態で細胞内に維持することもできるし、染色体中に組みこませて維持することもできる。発現ベクターとしては、pKA1、pCDM8、pSVK3、pSVL、pBK−CMV、pBK−RSV、EBVベクター、pRS、pYE82などが例示できる。また、pIND/V5−His、pFLAG−CMV−2、pEGFP−N1、pEGFP−C1などを発現ベクターとして用いれば、Hisタグ、FLAGタグ、GFPなど各種タグを付加した融合蛋白質としてグルコース脱水素酵素ポリペプチドを発現させることもできる。真核細胞としては、サル腎臓細胞COS−7、チャイニーズハムスター卵巣細胞CHOなどの哺乳動物培養細胞、出芽酵母、分裂酵母、カビ、カイコ細胞、アフリカツメガエル卵細胞などが一般に用いられるが、グルコース脱水素酵素を発現できるものであれば、いかなる真核細胞でもよい。発現ベクターを真核細胞に導入するには、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法など公知の方法を用いることができる。真核細胞で発現させて生産させる場合の挿入遺伝子はイントロンを含んでいても含んでいなくても良く、例えば配列番号1、配列番号3、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46又は配列番号48に記載のシグナル配列を含む遺伝子配列が好ましいが、ベクター側に分泌シグナル配列がある場合は、配列番号4又は配列番号6に記載の遺伝子配列を挿入すれば良く、菌体内に組み換えタンパクを生産させたい場合は、配列番号4又は配列番号6に記載の遺伝子配列に開始コドンATGを付加したポリヌクレオチドを挿入すれば良いが、菌体外に分泌生産することが好ましい。真核細胞では、本発明の組み換えグルコース脱水素酵素の発現効率が高く、好ましくは培養液1mLあたり少なくとも20U、より好ましくは培養液1mLあたり少なくとも50U、更に好ましくは培養液1mLあたり少なくとも80U、特に好ましくは培養液1mLあたり少なくとも100U、最も好ましくは培養液1mLあたり少なくとも120Uである。更に真核細胞では、原核細胞での組み換え製造時に問題となる封入体を作ることが無く、組み換え酵素を安定して高発現させることができる。加えて、培養物から得られる粗酵素の比活性が高く、夾雑蛋白質が少ないため、精製工程が簡便で精製コストが抑えられ、高収率で精製酵素が得られる。
【0028】
グルコース脱水素酵素を真核細胞で発現させた後、培養物(菌体、もしくは菌体外に分泌された酵素を含む培養液、培地組成物等)から目的蛋白質を単離精製するためには、公知の分離操作を組み合わせて行うことができる。例えば、尿素などの変性剤や界面活性剤による処理、熱処理、pH処理、超音波処理、酵素消化、塩析や溶媒沈殿法、透析、遠心分離、限外濾過、ゲル濾過、SDS−PAGE、等電点電気泳動、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトグラフィー(タグ配列を利用した方法及びグルコース脱水素酵素に特異的なポリクローナル抗体、モノクローナル抗体を用いる方法も含む)、などが挙げられる。
【0029】
また、グルコース脱水素酵素は、本発明のポリヌクレオチド(cDNA又はその翻訳領域)を用いた組み換えDNA技術によって取得できる。例えば前記ポリヌクレオチドを有するベクターからin vitro転写によってRNAを調製し、これを鋳型としてin vitro翻訳を行うことによりin vitroでグルコース脱水素酵素を作成することができる。またポリヌクレオチドを公知の方法により適当な発現ベクターに組み換えれば、酵母、カビ、昆虫細胞、哺乳動物細胞等の真核細胞で、ポリヌクレオチドがコードしているグルコース脱水素酵素を大量に発現させる事ができる。また宿主に対応して、同一アミノ酸配列であるが、コドンユーセージを最適化したポリヌクレオチドを導入しても良い。
【0030】
グルコース脱水素酵素をin vitro発現させて生産させる場合には、前記のポリヌクレオチドを、RNAポリメラーゼが結合できるプロモーターを有するベクターに挿入して組み換えベクターを作成し、このベクターを、プロモーターに対応するRNAポリメラーゼを含むウサギ網状赤血球溶解物や小麦胚芽抽出物などのin vitro翻訳系に添加すれば、グルコース脱水素酵素をin vitroで生産することができる。RNAポリメラーゼが結合できるプロモーターとしては、T3、T7、SP6などが例示できる。これらのプロモーターを含むベクターとしては、pKA1、pCDM8、pT3/T718、pT7/319、pBluescriptIIなどが例示できる。
【0031】
本発明のグルコース脱水素酵素は、以下の(e)又は(f)の糖ポリペプチドからなるグルコース脱水素酵素である。
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)アミノ酸配列(e)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【0032】
本発明のグルコース脱水素酵素の最も具体的な態様は、配列番号5で示されるアミノ酸配列からなる糖ポリペプチドである。グルコース脱水素酵素活性があれば、配列番号5に記載のアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなる糖ポリペプチドでも良く、1〜数個とは、好ましくは多くとも100個、より好ましくは多くとも80個、更に好ましくは多くとも60個、特に好ましくは多くとも40個である。
例として、配列番号2、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47又は配列番号49に記載のアミノ酸配列からなる糖ポリペプチドを有するグルコース脱水素酵素が例示できる。
また、本発明のグルコース脱水素酵素には、配列番号5のアミノ酸配列と少なくとも80%の相同性、より好ましくは85%以上の相同性、さらに好ましくは90%以上の相同性、さらに好ましくは95%以上の相同性を有するアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチドが含まれる。
【0033】
本発明のグルコース脱水素酵素は、電子受容体存在下でグルコースを脱水素する反応を触媒する酵素であるから、この反応による変化が利用できる用途であれば、特に制限されない。例えば、生体物質を含む試料中のグルコースの測定及び測定用試薬、消去用試薬へ使用するなどの医療分野、臨床分野への使用が可能であり、フラビン結合型グルコース脱水素酵素を使用した物質生産においても使用可能である。
【0034】
本発明のグルコース脱水素酵素は、グルコース測定試薬組成物に用いることができる。本発明の測定試薬組成物は、本発明の糖ポリペプチドからなるグルコース脱水素酵素に、牛血清アルブミン(BSA)若しくは卵白アルブミン、該酵素と作用性のない糖類若しくは糖アルコール類、カルボキシル基含有化合物、アルカリ土類金属化合物、アンモニウム塩、硫酸塩又はタンパク質等から成る群より選ばれる熱安定化剤、又は緩衝剤等の当業者に公知の他の任意成分を適宜含有させ、該酵素や試薬成分の熱安定性や保存安定性を高めることができる。更に、被験試料中に存在する、測定に影響を与える夾雑物質の影響を抑える公知の物質を該測定試薬組成物に含ませることができる。加えて、該組成物が溶解した際に該酵素が作用し易いpHとなるように、該組成物に緩衝剤を含有させることができる。
【0035】
本発明のグルコース脱水素酵素は、試料液中のグルコース濃度を測定するグルコースセンサとしてのバイオセンサに用いることができる。本発明のバイオセンサは、酵素として本発明の糖ポリペプチドからなるグルコース脱水素酵素を反応層に使用したセンサであればよい。例えば、該バイオセンサは、絶縁性基板上にスクリーン印刷や蒸着などの方法を利用して電極系を形成し、更に酸化還元酵素と電子受容体とを含む測定試薬を備えることによって作製される。このバイオセンサの測定試薬に基質を含む試料液を接触させると、測定試薬が溶解して酵素と基質が反応し、これにともなって電子受容体が還元される。酵素反応終了後、還元された電子受容体を電気化学的に酸化させ、このとき、このバイオセンサは得られる酸化電流値から試料液中の基質濃度を測定することが可能である。この他に、発色強度又はpH変化などを検知する方式のバイオセンサも構築可能である。これらのバイオセンサにより、測定対象物質を基質とする酵素を選択することによって、様々な物質の測定が可能である。
【0036】
バイオセンサの電子受容体としては、電子の授受能に優れた物質を用いることができる。電子の授受能に優れた物質とは、一般的に「電子伝達体」、「メディエータ」あるいは「酸化還元媒介剤」と呼ばれる化学物質やタンパク質性の電子メディエータであり、これらに該当する化学物質として、例えば、特表2002−526759に挙げられた電子伝達体や酸化還元媒介剤などを利用してもよい。
【0037】
更に本発明のグルコース脱水素酵素は、バイオ電池に用いることができる。本発明のバイオ電池は、酸化反応を行うアノード極及び還元反応を行うカソード極から構成され、必要に応じてアノードとカソードを隔離する電解質層を含んで構成される。上記の電子メディエータ及び本発明の酵素を含む酵素電極をアノード電極に使用し、基質を酸化することによって生じた電子を電極に取り出すと共に、プロトンを発生させる。一方、カソード側には、一般的にカソード電極に使用される酵素を使用すれば良く、例えばラッカーゼ、アスコルビン酸オキシダーゼ又はビリルビンオキシダーゼを使用し、アノード側で発生させたプロトンを酸素と反応させることによって水を生成させる。電極としては、カーボン、金、白金等、一般的にバイオ電池に使用される電極を用いることができる。
【0038】
[酵素活性測定法]
以下の手順に従って各溶液を混合し、吸光度を測定し、GLD活性を調べた。
100mMリン酸カリウム緩衝液(pH6.0)1.00mL、1M D−グルコース溶液1.00mL、超純水0.61mL、3mM 2,6−ジクロロフェノールインドフェノール(以下DCIPという)0.14mL及び3mM 1−メトキシ−5−メチルフェナジウムメチルサルフェイト(以下1−m−PMSという)0.20mLを混合し、37℃で10分間保温後、酵素サンプル0.05mLを添加し、反応を開始した。反応開始時から5分間、酵素反応の進行に伴う600nmにおける吸光度の1分間あたりの減少量(ΔA600)を測定し、直線部分から式1に従いGLD活性を算出した。この際、GLD活性は、37℃、pH6.0で1分間に1μmolのDCIPを還元する酵素量を1Uと定義した。尚、式中の3.0は反応試薬+酵素溶液の液量(mL)、10.8はpH6.0におけるDCIPのモル吸光係数(mM-1cm-1)、1.0はセルの光路長(cm)、0.05は酵素溶液の液量(mL)、ΔA600blankは酵素の希釈に用いた溶液を酵素溶液の代わりに添加して反応開始した場合の600nmにおける吸光度の1分間あたりの減少量、dfは希釈倍率を表す。
【0039】
【数1】
【0040】
本酵素のタンパク濃度の測定においては、該酵素を、好ましくは終濃度0.2〜0.9 mg/mL になるように適宜希釈して用いる。本発明におけるタンパク濃度は、日本バイオ・ラッド(株)から購入できるタンパク濃度測定キットであるBio−Rad Protein Assayを用い、取扱説明書に従って、牛血清アルブミン(BSA,和光純薬工業(株)製,生化学用)を標準物質として作成した検量線から換算して求めることができる。
【0041】
尚、本発明を実施するために使用する様々な技術は、特にその出典を明示した技術を除いては、公知の文献等に基づいて当業者であれば容易かつ確実に実施可能である。例えば、遺伝子工学及び分子生物学的技術はSambrook and Maniatis, in Molecular Cloning-A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 1989; Ausubel, F. M. et al., Current Protocols in Molecular Biology, John Wiley & Sons, New York, N.Y, 1995などに記載の方法あるいはそこで引用された文献記載の方法又はそれらと実質的に同様な方法や改変法に基づき実施可能である。さらに、この発明における用語は基本的にはIUPAC-IUB Commission on Biochemical Nomenclatureによるものであり、あるいは当該分野において慣用的に使用される用語の意味に基づくものである。
【実施例】
【0042】
以下、実施例に則して本発明を更に詳しく説明する。尚、本発明の技術的範囲はこれらの記載によって何等制限されるものではない。又、本明細書中に引用される文献に記載された内容は、本明細書の一部として本明細書の開示内容を構成するものである。
[実施例1]
(ボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)活性の確認)
ポテトデキストロースブロス(Difco社製)2.4%の培地100mLを500mL容の坂口フラスコに入れ、シリコ栓をし、121℃、20分間オートクレーブした。冷却したこの液体培地に、ボトリオティニア・フケリアナ(Botryotinia fuckeliana)NBRC7185株を接種し、25℃で216時間、通気撹拌の条件で菌体を培養した結果、培養上清に、培養液1mL当たり0.21U/mLのグルコース脱水素酵素活性が確認された。尚、グルコース酸化酵素(GOD)活性は検出されなかった。
GOD活性は、以下の方法で測定した。100mMリン酸カリウム緩衝液(pH7.0)1.00mL、25mM 4−アミノアンチピリン0.10mL、420mMフェノール0.10mL、ペルオキシダーゼ(100units/mL)0.10mL、超純水0.65mL、D−グルコース1.00mLを混合し、37℃で5分間保温後、酵素サンプル0.05mLを添加し、反応を開始した。反応開始時から酵素反応の進行に伴う500nmにおける吸光度の1分間あたりの増加量(ΔA500)を測定し、式2に従いGOD活性を算出した。この際GOD活性は、37℃、pH7.0で、1分間に1μmolの過酸化水素を生成する酵素量を1Uと定義した。尚、式中の3.0は反応試薬+酵素溶液の液量(mL)、10.66は本測定条件におけるモル吸光係数(mM-1cm-1)、0.5は1モルの過酸化水素の生成量に対するキノン型色素の生成量、1.0はセルの光路長(cm)、0.05は酵素溶液の液量(mL)、ΔA500blankは酵素の希釈に用いた溶液を酵素溶液の代わりに添加して反応開始した場合の500nmにおける吸光度の1分間あたりの増加量、dfは希釈倍率を表す。
【0043】
【数2】
【0044】
[実施例2]
(真核細胞による組み換えボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)の発現)
(1)菌体培養
グルコース(ナカライ社製)1%(W/V)、脱脂大豆(昭和産業社製)2%(W/V)、コーンスティープリカー(サンエイ糖化社製)0.5%(W/V)、硫酸マグネシウム七水和物(ナカライ社製)0.1%(W/V)及び水からなる液体培地をpH6.0に調整し、100mLを500mL容の坂口フラスコに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、ボトリオティニア・フケリアナNBRC7185株を接種し、25℃で90時間振とう培養した後、吸引ろ過により湿菌体を回収した。
【0045】
(2)染色体DNAの抽出
(1)で得られた菌体のうち湿菌体0.25gを液体窒素により凍結した後、粉砕し、常法により染色体DNAを抽出した。
【0046】
(3)BfGLD遺伝子の取得
(2)で取得したDNAを鋳型とし、BfGLD遺伝子をPCR増幅した。プライマーは、本発明者らによって既に解明されていた複数のGLD遺伝子配列から共通配列を解析し、その共通配列を基に縮重プライマーを設計して遺伝子断片を取得し、最終的に下記のプライマー:primer1F(配列番号15)及びprimer2R(配列番号16)を用いて、目的のGLD遺伝子全長(イントロンを含む)を含む約1.8kbpのDNA断片を取得した。
primer1F:(TGACCAATTCCGCAGCTCGTCAAA)ATGTATCGTTTACTCTCTACATTTG(配列番号15)
(括弧内:転写増強因子)
primer2R:((GCTATCCTGTTACGCTTCTAGA))GCATGCCTAAATGTCCTCCTTGATCAAATCT(配列番号16)
(二重括弧内:pSENSベクター配列、下線部:制限酵素部位(SphI))
primer3F:((CCGTCCTCCAAGTTA))GTCGAC(TGACCAATTCCGCAGCTCGTCAAA)(配列番号17)
(二重括弧内:pSENSベクター配列、下線部:制限酵素部位(SalI)、括弧内:転写増強因子)
【0047】
(4)BfGLD遺伝子を含むベクターの調製
公知文献1(Aspergillus属の異種遺伝子発現系、峰時俊貴、化学と生物、38、12、831−838、2000)に記載してあるアスペルギルス・オリゼ由来のアミラーゼ系の改良プロモーターを使用し、その下流に(3)で得られたDNA断片を鋳型として上記のプライマー:primer3F(配列番号17)及び上記のプライマー:primer2Rを用いて増幅したGLD遺伝子を結合させることで、該遺伝子が発現可能なプラスミドベクターを調製した。この発現用プラスミドベクターを大腸菌JM109株に導入して形質転換し、得られた形質転換体を培養して、集菌した菌体から、Illustra plasmid−prep MINI Flow Kit(GEヘルスケア社製)を用いてプラスミドを抽出した。該プラスミド中のインサートの配列解析を行ったところ、イントロンを含むBfGLD遺伝子(配列番号3)が確認できた。
【0048】
(5)形質転換体の取得
(4)で抽出したプラスミドを用いて、公知文献2(Biosci. Biotech. Biochem.,61(8),1367−1369,1997)及び3(清酒用麹菌の遺伝子操作技術、五味勝也、醸協、494−502、2000)に記載の方法に準じて、BfGLDを生産する組み換えカビ(アスペルギルス・オリゼ)を作製し、得られた組み換え株をCzapek−Dox固体培地で純化した。使用する宿主としては、アスペルギルス・オリゼNS4株を使用した。本菌株は、公知文献2にあるように、1997年(平成9年)に醸造試験所で育種され、転写因子の解析、各種酵素の高生産株の育種などに利用され、分譲されているものが入手可能である。
【0049】
(6)組み換えカビ由来BfGLDの確認
パインデックス2%(松谷化学工業社製)(w/v)、トリプトン1%(BD社製)(w/v)、リン酸二水素カリウム0.5%(ナカライテスク社製)(w/v)、硫酸マグネシウム七水和物0.05%(w/v)(ナカライテスク社製)及び水からなる液体培地10mLを太試験管(22mm×200mm)に入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、(5)で取得した形質転換体を植菌し、30℃で3〜4日間振とう培養した。培養終了後、遠心して上清を回収し、組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)とした。
【0050】
前述の酵素活性測定法に従い組み換えSc(+)BfGLDのグルコース脱水素酵素活性(U/mL)を測定したところ、培養液1mL当たり、3日目が133U/mL、4日目が98U/mLのグルコース脱水素酵素活性を確認できた。尚、グルコース酸化酵素活性は確認できなかった。また、本発明の組み換えBfGLDは、基質濃度333mMにおいて、D−グルコースに対する活性を100%とした場合に、マルトース対する反応性が1.3%であった。
【0051】
(6)酵素の精製
D−グルコース1%(w/v)、大豆粉2%(w/v)、コーンスティープリカー0.5%(w/v)、硫酸マグネシウム七水和物0.1%(w/v)及び水からなる液体培地(pH7.0)50mLを200mL容の三角フラスコに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、(5)で取得した形質転換体を植菌し、25℃で3日間振とう培養して種培養液とした。前記と同様の培地組成で消泡剤を添加した培地3.5Lを5L容ジャーファーメンターに入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、種培養液を50mL植菌し、30℃、300rpm、1v/v/mで7日間培養した。培養終了後、培養液をろ布でろ過し、回収したろ液を遠心して上清を回収し、更にメンブレンフィルター(10μm、アドバンテック社製)でろ過して培養上清を回収し、分画分子量8,000の限外ろ過膜(ミリポア社製)で濃縮して粗酵素液とした。粗酵素液の比活性は1,170U/mgだった。
【0052】
前記粗酵素液を、50%飽和硫酸アンモニウム溶液(pH5.0)になるように調整し、4℃で一晩放置後、遠心分離して上清を回収した。
該上清を、50%飽和硫酸アンモニウムを含む50mM酢酸ナトリウム緩衝液(pH5.0)で予め平衡化したTOYOPEARL Butyl−650C(東ソー社製)カラム(φ7.5cm×17.7cm)に通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から50mM酢酸ナトリウム緩衝液(pH5.0)へのグラジエント溶出法で酵素を溶出させて、活性画分を回収した。回収した活性画分を、限外濾過膜で濃縮後、脱塩し、1mM酢酸ナトリウム緩衝液(pH5.0)と平衡化させ、同緩衝液で予め平衡化したDEAEセルファインA−500m(チッソ社製)カラム(φ6.0cm×21.9cm)に通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から200mM酢酸ナトリウム緩衝液(pH5.0)へのグラジエント溶出法で酵素を溶出させて、活性画分を回収した。回収した活性画分を、分画分子量8,000の限外ろ過膜で濃縮後、水置換して組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)の精製酵素とした。該精製酵素の比活性は2,100U/mgだった。
更に、糖鎖切断前後の組み換えBfGLDをSDS−ポリアクリルアミド電気泳動に供して確認を行った。詳細には、組み換えSc(+)BfGLD5μLと1%SDS及び2%β―メルカプトエタノールを含む0.4Mリン酸カリウム緩衝液(pH6.0)5μLを混合し、100℃で3分間熱処理を行った。糖鎖切断処理として、熱処理後のサンプルにエンドグリコシダーゼH(ロシュ製)を10μL(50mU)添加し、37℃で18時間反応させた。糖鎖切断処理前後のサンプルをSDS−ポリアクリルアミド電気泳動に供し、分子量マーカーより分子量を求めたところ、糖鎖切断前の酵素が90〜100kDa、糖鎖切断後の酵素が60〜70kDaの組み換えBfGLDを確認できた。
【0053】
[参考例1]
(原核細胞による組み換えボトリオティニア・フケリアナ由来グルコース脱水素酵素(BfGLD)の発現)
(1)BfGLD遺伝子のクローニング
実施例2の(4)で抽出したプラスミドを鋳型とし、BfGLD遺伝子をPCR増幅した。予想シグナル配列をコードする遺伝子配列を含まないBfGLD(16AA(−)BfGLD)遺伝子を得るために、野生型BfGLDの予想N末端からC末端をコードする遺伝子を増幅できる下記のprimer4F_BfGLD_Sig(-)/Sac(配列番号18)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いてPCRを行った。予想シグナル配列をコードする遺伝子配列を含まないポリヌクレオチドの取得は、分泌シグナル配列をコードする遺伝子配列を含む外来遺伝子を用いて大腸菌で組み換えタンパクを発現させる場合は、組み換えタンパクがペリプラズムに移行されるため生産性が悪いことから、組み換えタンパクを効率よく回収したい場合は、一般的にシグナル配列をコードする遺伝子配列を削除した配列を用いることが知られているためである。尚、PCRは、DNAポリメラーゼ、ExTaq(タカラバイオ社製)を使用し、反応条件は[94℃/30秒→50℃/1分→72℃/2分]×30サイクルとした。野生型BfGLDの予想N末端は、BfGLDの全長アミノ酸配列(配列番号2)と国際公開2006/101239号パンフレットの配列番号2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素(AtGLD)との配列同一性を調べ、野生型AtGLDのN末端である国際公開2006/101239号パンフレットの配列番号2の20番目以降のアミノ酸配列から、野生型BfGLDのN末端は、配列番号1記載の17番目以降のアミノ酸配列であると予測した。
primer4F_BfGLD_Sig(-)/Sac:AAAGAGCTCGAGCACCGACTCTACCTTAAA(配列番号18)
(下線部:制限酵素部位(SacI))
primer5R_BfGLD/Hind:CCCAAGCTTCTAAATGTCCTCCTTGATC(配列番号19)
(下線部:制限酵素部位(HindIII))
PCRにより、46bpのイントロンを含み、予想シグナル配列をコードする48bpの塩基配列を含まない配列にプライマー付加配列が付加したDNA断片を取得した。
【0054】
次いで、イントロンを削除するため、前記で得られたDNA断片を鋳型として、primer4F_BfGLD_Sig(-)/Sac(配列番号18)及び下記のprimer6R_Bf_up(int)(配列番号20)のプライマーセット又は下記のprimer7F_Bf_(int)down(配列番号21)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いて各々前記PCR条件でPCR増幅した。
primer6R_Bf_up(int):GGGTGTATGCCATTCCATTGATAGTACTAGTCCCT(配列番号20)
primer7F_Bf_(int)down:GAATGGCATACACCCGAGCCGAAGAT(配列番号21)
得られた2つの増幅配列を1つのPCR増幅チューブに入れ、primer4F_BfGLD_Sig(-)/Sac(配列番号18)及びprimer5R_BfGLD/Hind(配列番号19)のプライマーセットを用いて前記PCR条件でPCR増幅し、イントロンを含まず、予想シグナル配列をコードする48bpの塩基配列を含まない16AA(−)BfGLD遺伝子にプライマー付加配列が付加したDNA断片を取得した。
【0055】
(2)BfGLD遺伝子を含むベクターの調製及び形質転換体の取得
pUC18ベクター(タカラバイオ社製)を制限酵素SacIとHindIIIで開裂し、同制限酵素処理したPCR増幅DNA断片をベクターにライゲーションし、大腸菌JM109株に導入して形質転換した。得られた形質転換体のうち6クローンよりプラスミドDNAを調製し、SacIとHindIIIで処理したところ全てのクローンで目的のサイズの断片が確認できた。そのうち1クローンについてプラスミドを調製してそのインサートの配列決定を行ったところ、イントロンを含まず、予想シグナル配列をコードする48bpの塩基配列を含まない配列番号50に記載の16AA(−)BfGLD遺伝子が確認された。
【0056】
(3)組み換え大腸菌由来BfGLDの確認
LB液体培地10mLを太試験管に入れ、シリコ栓をし、121℃、20分間オートクレーブした。冷却したこの液体培地に、50μg/mLアンピシリンを加え、(4)で作成した形質転換体を接種し、37℃で振とう培養した。培養液のOD600が約0.4〜0.5となった時点で、IPTGを1mM添加し、更に18時間振とう培養を行った。培養終了後、菌体を遠心により集め、20mMリン酸カリウム緩衝液(pH7.0)に懸濁した。超音波破砕装置を用いて菌体を破砕後、遠心して無細胞抽出液を調製した。前述の酵素活性測定法に従い組み換えBfGLD糖鎖無しサンプル(Sc(−)BfGLD)のグルコース脱水素酵素活性(U/mL)を測定したところ、培養液1mL当たり、0.703U/mLのグルコース脱水素酵素活性を確認できた。尚、グルコース酸化酵素活性は確認できなかった。
【0057】
(4)組み換えSc(−)BfGLDの精製
(2)で作成した形質転換体を、50μg/mLアンピシリンを含むLB液体培地10mLに植菌し、37℃で7時間振とう培養した後、50μg/mLアンピシリンを含むLB液体培地50mL入りの250mL容のマイヤーフラスコに植菌し、37℃で17.5時間振とう培養し、前培養液とした。50μg/mLアンピシリンを含むLB液体培地1.5L入りの2L容のジャーファーメンターに前培養液50mLを植菌し、37℃で培養し、培養液のOD600が約0.4〜0.5となった時点で、IPTGを1mM添加し、25.5時間通気撹拌条件で培養を行った。培養終了後、遠心により集めた菌体を、20mMリン酸カリウム緩衝液(pH6.0)に懸濁し、超音波破砕装置を用いて菌体を破砕後、遠心して無細胞抽出液を調製した。該無細胞抽出液の比活性は0.0182U/mgだった。前記無細胞抽出液を20mM リン酸カリウム緩衝液(pH7.5)中で透析し、同緩衝液で平衡化したDEAE−セルロファインA−500カラムに通液し、溶出液を集めた。前記溶出液を5mM リン酸カリウム緩衝液(pH7.5)中で透析し、同緩衝液で平衡化したDEAE−セルロファインA−500カラムに通液して酵素を吸着させた。該カラムを同緩衝液で洗浄した後、同緩衝液から0.3M 塩化カリウムを含む同緩衝液へのグラジエント溶出法で酵素を溶出させて活性画分を集めた。活性画分を分画分子量10000の限外濾過膜で濃縮し、脱塩後20mMリン酸カリウム緩衝液(pH6.0)に置換し、Sc(−)BfGLDの精製酵素とした。
更に、組み換えSc(−)BfGLDをSDS−ポリアクリルアミド電気泳動に供し、分子量マーカーより分子量を求めたところ約60kDaの組み換えSc(−)BfGLDを確認できた。
【0058】
[実施例3]
(組み換えBfGLDの公知配列との比較)
シグナル配列部分を含むBfGLDのアミノ酸配列は配列番号2に記載の通りだったが、該配列を用いてBLAST検索したところ、驚いたことにボトリオティニア・フケリアナ由来グルコース酸化酵素のアミノ酸配列と一致していた。しかし、本発明で得られたBfGLDは、実施例2の(6)に記載の通りグルコース脱水素酵素活性を有しており、グルコース酸化酵素活性は全くみられなかった。ボトリオティニア・フケリアナ由来グルコース酸化酵素のアミノ酸配列は公知文献4(Molecular Plant Pathology(2004)5(1),17−27)に記載されており、Botrytis cinerea(=Botryotinia fuckeliana)のセルフクローニングを行っていることが記載されている。更に該文献中に記載の公知文献5(Physiological and Molecular Plant Pathology(1998)53,123―132)に、野生型のBotrytis cinerea(=Botryotinia fuckeliana)由来グルコース酸化酵素の特性が記載されているが、該酵素はサブユニット分子量が35kDaで、ゲルろ過分子量が約160kDaの4量体酵素であることが記載されている。これに対して本発明の組み換えBfGLDは、SDS−ポリアクリルアミド電気泳動における分子量が、真核細胞で組み換えた場合、糖鎖切断前の酵素が90〜100kDa、糖鎖切断後の酵素が60〜70kDaのグルコース脱水素酵素である。例えば、公知文献6(蛋白質核酸酵素(2004)49(5),625-633,Review)には、キサンチン脱水素酵素が、プロテアーゼによる部分分解でキサンチン酸化酵素へ変換されることが記載されている。つまり、立体構造が取られた後に、プロテアーゼにより分解されており、キサンチン脱水素酵素とキサンチン酸化酵素は同じ遺伝子に由来している。これらのことから、本発明のボトリオティニア・フケリアナ由来グルコース脱水素酵素とボトリオティニア・フケリアナ由来グルコース酸化酵素は、遺伝子配列から推測されるアミノ酸配列は同じ配列であっても、ボトリオティニア・フケリアナ由来グルコース脱水素酵素はサブユニット分子量が糖鎖切断前の酵素で90〜100kDa、糖鎖切断後の酵素で60〜70kDaであり、かつグルコース脱水素酵素活性を有しており、一方、論文記載のボトリオティニア・フケリアナ由来グルコース酸化酵素はサブユニット分子量が35kDaで、かつグルコース酸化酵素活性を有しており、構造、機能とも全く異なる酵素であり、両者の違いは、立体構造を取った後にプロテアーゼによる分解等何らかの修飾を受けるか否かによると推察される。特に、アスペルギルス属で組み換えた酵素においては、プロテアーゼ修飾による活性の変換は見られなかったため、アスペルギルス属で組み換え製造することは、組み換えBfGLDを効率的に製造できる有意義な方法と思われる。
【0059】
[実施例4]
(電極によるグルコースの測定)
組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)を使用し、電極によるD−グルコースの測定を行った。酵素0.16Uを搭載したグラッシーカーボン(GC)電極を用いて、グルコース濃度に対する応答電流値を測定した。電解セル中に、50mM リン酸ナトリウムバッファー(pH6.0又はpH7.0)1.8mL及び1M ヘキサシアノ鉄(III)酸カリウム(フェリシアン化カリウム)水溶液0.2mLを添加した。GC電極をポテンショスタットBAS100B/W(BAS製)に接続し、37℃で溶液を撹拌し、銀塩化銀参照電極に対して+500mVを印加した。これらの系に終濃度が10mMになるように1M D−グルコース溶液を5μl添加し、定常状態の電流値を測定した。更に各終濃度になるように1M D−グルコース溶液を添加し電流値を測定するという作業を、繰り返し、最終的に各グルコース濃度(5、10、20、30、40mM)の電流値を測定した。これをプロットしたところ、検量線が作成できた(図1)。尚、50mM リン酸ナトリウムバッファー(pH7.0)を用いて組み換えBfGLD糖鎖無しサンプル(Sc(−)BfGLD)についても同様に電流値を測定した結果、Sc(+)BfGLDの方が、Sc(−)BfGLDより約2倍のセンサ感度を示した。
【0060】
[実施例5]
(N末端解析)
組み換えBfGLD糖鎖有りサンプル(Sc(+)BfGLD)のN末端を解析したところ、STLNYであることが明らかになった。つまり、MYRLLSTFAVASLAAASTDの19アミノ酸がシグナル配列であり、該酵素は、翻訳後のペプチドからシグナルペプチダーゼによる修飾で、該19アミノ酸が削除された形で存在していることが分かった。
【0061】
[実施例6]
(シグナル配列変異)
組み換えカビによる組み換えBfGLD製造において、分泌シグナルの変異を行った。
下記に記載したプライマーを合成し、実施例2の(4)で取得したプラスミドベクターを鋳型として、シグナル配列を置換したBfGLD遺伝子をPCR法により増幅した。
primer8_Bf-Atsignal1-7AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))TTTGCTGTAGCCTCTTTGGCTGCAG(配列番号22)
primer9_Bf-Atsignal2-F:((ATGTTGGGAAAGCTCTCCTTCCTCAGTGCCCTGTCCCTGGCAGTGGCGGCACCTTTG))(配列番号23)
primer10_Bf-eno-Atsignal-F:(TGACCAATTCCGCAGCTCGTCAAA)((ATGTTGGGAAAGCTCTCCTTCCTCA))(配列番号24)
primer11_Bf-infusion-F:CTCCAAGTTAGTCGAC(TGACCAATTCCGCAGCTCGTCAAA)(配列番号25)
primer12_Bf-infusion-R:CGCTTCTAGAGCATGCCTAAATGTCCTCCTTGATCAAATC(配列番号26)
primer13_Bf-Aosig-short1-7AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))TTTGCTGTAGCCTCTTTGGCTGCA(配列番号27)
primer14_Bf-Aosig-short2-F:((ATGCTCTTCTCACTGGCATTCCTGAGTGCCCTGTCGCTGGCCACGGCA))(配列番号28)
primer15_Bf-Aosignal1-7AA-F:((TCGCTGGCCACGGCATCACCGGCTGGACGGGCC))TTTGCTGTAGCCTCTTTGGCTGCAGCTAGCACC(配列番号29)
primer16_Bf-Aosignal2-F:((ATGCTCTTCTCACTGGCATTCCTGAGTGCCCTGTCGCTGGCCACGGCATCACCGGCTGGACGGGCC))(配列番号30)
primer17_Bf-eno-Aosignal-F:(TGACCAATTCCGCAGCTCGTCAAA)((ATGCTCTTCTCACTGGCATTCCTGA))(配列番号31)
primer18_Bf-Atsignal1-16AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))AGCACCGACTCTACCTTAAACTATG(配列番号32)
primer19_Bf-Aosig1-short1-16AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))AGCACCGACTCTACCTTAAACTATG(配列番号33)
primer20_Bf-Atsignal1-19AA-F:((CCCTGTCCCTGGCAGTGGCGGCACCTTTG))TCTACCTTAAACTATGATTATATCATCG(配列番号34)
primer21_Bf-Aosig1-short1-19AA-F:((AGTGCCCTGTCGCTGGCCACGGCA))TCTACCTTAAACTATGATTATATCATCG(配列番号35)
(FはForward、RはReverse、下線部:制限酵素切断部位、二重括弧内:各シグナル配列、括弧内:enoA 5'-UTR、その他:ORF)
尚、上記プライマーは、国際公開2006/101239号パンフレットの配列番号2に記載のアスペルギルス・テレウス由来グルコース脱水素酵素(AtGLD)のシグナル配列情報(該公報記載の配列番号2の1から19番目のアミノ酸配列:配列番号10)及びシグナル配列予測サイト(Signal p:http://www.cbs.dtu.dk/services/SignalP/)を用いて推定したアスペルギルス・オリゼ由来グルコース脱水素酵素(AoGLD)のシグナル配列を元にデザインした。尚、AoGLDのシグナル配列は、2種類(短め:配列番号12及び長め:配列番号14)の配列を予想した。
【0062】
1.BfGLDのシグナル配列のうち7アミノ酸を削除して別シグナルを付加するためのクローニング
1−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer8_Bf-Atsignal1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-7AA(−)BfGLD遺伝子として配列番号36に示し、該配列にコードされるアミノ酸配列を配列番号37に示した。
【0063】
1−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer13_Bf-Aosig-short1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-7AA(−)BfGLD遺伝子として配列番号38に示し、該配列にコードされるアミノ酸配列を配列番号39に示した。
【0064】
1−(2)L.AoGLDの予想長めシグナル配列22アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから7番目までのアミノ酸(MYRLLST)を削除してAoGLDの予想長めシグナル配列22アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち21番目までの塩基を、配列番号13に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想長めシグナル配列をコードする塩基配列66bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer15_Bf-Aosignal1-7AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer16_Bf-Aosignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の7アミノ酸(MYRLLST)をAoGLDの予想長めシグナル配列(配列番号14)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigL-7AA(−)BfGLD遺伝子として配列番号40に示し、該配列にコードされるアミノ酸配列を配列番号41に示した。
【0065】
2.BfGLDのシグナル配列のうち16アミノ酸を削除して別シグナルを付加するためのクローニング
2−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから16番目までのアミノ酸(MYRLLSTFAVASLAAA)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち48番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer18_Bf-Atsignal1-16AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の16アミノ酸(MYRLLSTFAVASLAAA)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-16AA(−)BfGLD遺伝子として配列番号42に示し、該配列にコードされるアミノ酸配列を配列番号43に示した。
【0066】
2−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDのシグナル配列のうちメチオニンから16番目までのアミノ酸(MYRLLSTFAVASLAAA)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち48番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer19_Bf-Aosig-short1-16AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)のシグナル配列部分の16アミノ酸(MYRLLSTFAVASLAAA)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-16AA(−)BfGLD遺伝子として配列番号44に示し、該配列にコードされるアミノ酸配列を配列番号45に示した。
【0067】
3.BfGLDの全シグナル配列19アミノ酸を削除して別シグナルを付加するためのクローニング
3−(1)AtGLDのシグナル配列19アミノ酸を付加するためのクローニング
BfGLDの全シグナル配列、つまりメチオニンから19番目までのアミノ酸(MYRLLSTFAVASLAAASTD)を削除してAtGLDのシグナル配列19アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち57番目までの塩基を、配列番号9に記載の、アスペルギルス・テレウス由来グルコース脱水素酵素のシグナル配列をコードする塩基配列57bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer20_Bf-Atsignal1-19AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer9_Bf-Atsignal2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer10_Bf-eno-Atsignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)の全シグナル配列19アミノ酸(MYRLLSTFAVASLAAASTD)をAtGLDのシグナル配列(配列番号10)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAtsig-19AA(−)BfGLD遺伝子として配列番号46に示し、該配列にコードされるアミノ酸配列を配列番号47に示した。
【0068】
3−(2)S.AoGLDの予想短めシグナル配列16アミノ酸を付加するためのクローニング
BfGLDの全シグナル配列、つまりメチオニンから19番目までのアミノ酸(MYRLLSTFAVASLAAASTD)を削除してAoGLDの予想短めシグナル配列16アミノ酸を付加するために、イントロンを含む全長BfGLD遺伝子のシグナル配列部分をコードする塩基配列のうち57番目までの塩基を、配列番号11に記載の、アスペルギルス・オリゼ由来グルコース脱水素酵素の予想短めシグナル配列をコードする塩基配列48bpに置換するために、PCRを行った。PCRは4段階で行った。まず、1段階目として、実施例2の(4)で抽出したプラスミドを鋳型として、primer21_Bf-Aosig-short1-19AA-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。次に、2段階目として、1段階目のPCR産物を鋳型として、primer14_Bf-Aosig-short2-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。続いて、3段階目として、2段階目のPCR産物を鋳型として、primer17_Bf-eno-Aosignal-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。最後に、4段階目として、3段階目のPCR産物を鋳型として、primer11_Bf-infusion-Fとprimer12_Bf-infusion-Rとを用いたPCRを行った。得られたPCR産物は、BfGLDの全長アミノ酸配列(配列番号2)の全シグナル配列19アミノ酸(MYRLLSTFAVASLAAASTD)をAoGLDの予想短めシグナル配列(配列番号12)に置換した配列をコードしている、イントロンを含むポリヌクレオチドである。該配列のイントロンを含まない配列をAosigS-19AA(−)BfGLD遺伝子として配列番号48に示し、該配列にコードされるアミノ酸配列を配列番号49に示した。
【0069】
2.ベクターの調整
得られた各PCR産物を、In-Fusion Advantage PCRクローニングキット(タカラバイオ社製)を用いて、公知文献1に記載してあるアスペルギルス・オリゼ由来のアミラーゼ系の改良プロモーターの下流に結合させる事で、各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子が発現可能なベクターを各々調製した。
【0070】
これらの発現用ベクターを各々大腸菌JM109株に導入して形質転換し、得られた形質転換体を各々培養して、集菌した菌体から、Illustra plasmid-prep MINI Flow Kit(GEヘルスケア社製)を用いて、各プラスミドを抽出し、インサートの配列解析を各々行ったところ、各プラスミドで各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子が確認できた。
【0071】
3.形質転換体の取得
実施例2の(5)と同様の方法で、各々イントロンを含むAtsig-7AA(−)BfGLD遺伝子、AosigS-7AA(−)BfGLD遺伝子、AosigL-7AA(−)BfGLD遺伝子、Atsig-16AA(−)BfGLD遺伝子、AosigS-16AA(−)BfGLD遺伝子、Atsig-19AA(−)BfGLD遺伝子及びAosigS-19AA(−)BfGLD遺伝子を導入した組み換えカビを各々作製し、Czapek-Dox固体培地で各形質転換体を純化した。パインデックス2%(松谷化学工業社製)(W/V)、トリプトン1%(BD社製)(W/V)、リン酸二水素カリウム0.5%(ナカライテスク社製)(W/V)、硫酸マグネシウム七水和物0.05%(W/V)(ナカライテスク社製)及び水からなる液体培地10mLを太試験管(22mm×200mm)に入れ、121℃、20分間オートクレーブした。冷却したこの液体培地に、取得した各形質転換体を1遺伝子につき数本ずつ植菌し、30℃で4日間振とう培養した。3日目及び4日目にサンプリングした培養上清を遠心(3,000×g、20分)し、沈殿を取り除いたものを各酵素サンプルとした。
【0072】
前述の酵素活性測定法に従い各酵素サンプルのグルコース脱水素酵素活性(U/mL)を測定した結果、同一遺伝子につき活性の高かった2サンプルの平均値が下記の表1のとおりとなった。
【0073】
【表1】
【0074】
本酵素サンプルのうち、Atsig-7AA(−)BfGLD遺伝子に由来する酵素サンプルについて、N末端を解析したところ、STLNYであることが明らかになった。つまり、野生型のオリジナルシグナル配列の一部を削除して、他シグナル配列を付加しても、酵素のN末端は野生型と同じSTLNYとなっていることが分かった。
【0075】
以上の結果より、BfGLDを組み換え製造する場合、BfGLDのシグナル配列をコードする遺伝子の一部又は全部を、他のシグナル配列をコードする遺伝子と置換しても野生型と同じBfGLDを製造できることが明らかになった。
【特許請求の範囲】
【請求項1】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項2】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を含有するグルコース測定試薬組成物:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項3】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコースの測定方法:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項4】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項5】
ボトリオティニア・フケリアナ由来の組み換えグルコース脱水素酵素遺伝子を用いた形質転換真核細胞で発現させたものである請求項4記載のフラビン結合型グルコース脱水素酵素。
【請求項6】
以下の(a)又は(b)のポリヌクレオチド:
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【請求項7】
以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチド:
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
【請求項8】
請求項6又は7に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチド。
【請求項9】
シグナル配列が真核細胞由来タンパクのシグナル配列である、請求項8記載のポリヌクレオチド。
【請求項10】
請求項6〜9の何れか1項に記載のポリヌクレオチドを含む組み換えベクター。
【請求項11】
請求項10記載の組み換えベクターを用いることによって作成された形質転換真核細胞。
【請求項12】
請求項11記載の形質転換真核細胞を培養し、得られた培養物からグルコース脱水素酵素を採取することを特徴とするグルコース脱水素酵素の製造方法。
【請求項1】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコース測定用バイオセンサ:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項2】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を含有するグルコース測定試薬組成物:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項3】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素を使用することを特徴とするグルコースの測定方法:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項4】
以下の(e)又は(f)の糖ポリペプチドからなるフラビン結合型グルコース脱水素酵素:
(e)配列番号5で示されるアミノ酸配列からなる糖ポリペプチド、
(f)配列番号5で示されるアミノ酸配列において1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有する糖ポリペプチド。
【請求項5】
ボトリオティニア・フケリアナ由来の組み換えグルコース脱水素酵素遺伝子を用いた形質転換真核細胞で発現させたものである請求項4記載のフラビン結合型グルコース脱水素酵素。
【請求項6】
以下の(a)又は(b)のポリヌクレオチド:
(a)配列番号4又は配列番号6で示される塩基配列からなるポリヌクレオチド、
(b)塩基配列(a)からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつグルコース脱水素酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【請求項7】
以下の(c)又は(d)のポリペプチドをコードするポリヌクレオチド:
(c)配列番号5で示されるアミノ酸配列からなるポリペプチド、
(d)アミノ酸配列(c)のアミノ酸配列において、1〜数個のアミノ酸が置換、欠失又は付加されたアミノ酸配列からなり、かつグルコース脱水素酵素活性を有するポリペプチド。
【請求項8】
請求項6又は7に記載のポリヌクレオチドの上流に、シグナル配列をコードする塩基配列を有するポリヌクレオチド。
【請求項9】
シグナル配列が真核細胞由来タンパクのシグナル配列である、請求項8記載のポリヌクレオチド。
【請求項10】
請求項6〜9の何れか1項に記載のポリヌクレオチドを含む組み換えベクター。
【請求項11】
請求項10記載の組み換えベクターを用いることによって作成された形質転換真核細胞。
【請求項12】
請求項11記載の形質転換真核細胞を培養し、得られた培養物からグルコース脱水素酵素を採取することを特徴とするグルコース脱水素酵素の製造方法。
【図1】

【公開番号】特開2013−90620(P2013−90620A)
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2012−136112(P2012−136112)
【出願日】平成24年6月15日(2012.6.15)
【出願人】(000210067)池田食研株式会社 (35)
【Fターム(参考)】
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成24年6月15日(2012.6.15)
【出願人】(000210067)池田食研株式会社 (35)
【Fターム(参考)】
[ Back to top ]