
ゲムシタビンの高立体選択的な新規合成プロセス及び中間体
本発明は、以下の反応を含む、工業化生産に適したゲムシタビンの高立体選択的な新規合成プロセスを提供する。また、肝心な中間体化合物も開示する。基G1、G2、G3、G4及びG5の定義の詳しくは明細書に参照する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗腫瘍性ヌクレオチド類代謝拮抗薬の化学合成プロセスに関するものであり、具体的には、ゲムシタビンの高立体選択的な合成プロセス及び中間体に関するものである。
【背景技術】
【0002】
ゲムシタビンは、細胞複製を阻害するジフルオロヌクレオチド類代謝拮抗薬抗腫瘍薬で、デオキシシチジンの水溶性類似物であり、リボヌクレオチド還元酵素に対して、当該酵素の基質と一致の代替物であり、この酵素はDNAの合成と修復において、必要なデオキシリボヌクレオチドの合成に対して非常に重要である。
【0003】
ゲムシタビンは、化学名が2’-デオキシ-2’,2’-ジフルオロシチジンであり、化学構造が下記式2で表される。
【0004】
【化1】
【0005】
Hertelらは米国特許(US)第4808614号に初めてゲムシタビン化合物を開示したと同時に、以下のような当該化合物の合成ルートも開示した。
【0006】
【化2】
【0007】
この合成ルートにおいて、メチル-2,2-ジフルオロ-3-水酸基-3-(2,2-ジメチル-ジオキソラン-4-イル)プロピオネートを合成する時のみに、シリカゲルカラムにより3-R-ヒドロキシ産物を分離することにかかわり、その後の反応がいずれも反応の立体化学にかかわっていない。
【0008】
Chou Ta-Senは、米国特許(US)第5401861号及び欧州特許(EP)第0577303号に、予めα-アノマーを主とする2-デオキシ-2,2-ジフルオロ-D-リボフラノシル-3,5-ジ-O-ベンゾイルメタンスルホネートを製造することによって、β-アノマーを主とするゲムシタビンを製造するもう一つの合成ルートを開示した。反応は、低温(-78℃)でα-アノマーを主とするメタンスルホネート中間体を製造した後、3〜20倍以上(モル)のシリル基で保護されたシトシンとの反応により、立体選択的にβ-アノマーゲムシタビンを主とするものを合成することを特徴としている。なお、以上の特許文献には、3、5位の水酸基保護基はベンゾイル基で、また、α-アノマーを主とするメタンスルホネートを製造する反応は低温反応であり、反応条件は過酷で、大規模の工業的普及及び応用に適しない。
【0009】
また、Lee Jaeheonらは国際特許出願WO2006/009353に以下のような中間体の合成ルートを開示した。
【0010】
【化3】
【0011】
そして、Lee Jaeheonらは国際特許出願WO2006/011713に以下の合成ルートを開示した。
【0012】
【化4】
【0013】
以上の合成ルートにおいて、P1はBz又はBiPhC(O)-で、P2は-P(O)(OPh)2である。以上の反応によりα-アノマーに富んだハロゲン化中間体が得られ、これにより、立体選択的にゲムシタビンを合成することに有益な中間体を提供できる。しかし、実験により、WO2006/009353に記載のプロセスにおいてカリウム塩化合物を製造する後処理の時、低温で迅速に、乾燥まで反応液における有機溶媒を濃縮する必要があり、そうでないと、産物(カリウム塩)はひどく分解し、純度と収率に影響を与えることを見出し、これは工業的操作において実現されにくい。また、ハロゲン化プロセスには、過剰なハロゲン化水素を使用しなければならないから、毒性が大きくて、労働者保護に不利であり、排ガス、排水、固形廃棄物などの処理が困難で、環境汚染を引き起こしやすくなる。
【0014】
つまり、従来の技術では、ゲムシタビンの合成ルートに関しては、依然として前記した欠点がある。現在、反応条件が温和で、環境汚染が少なく、立体選択性が高い、ゲムシタビンの合成プロセスの開発が望まれている。
【発明の概要】
【0015】
本発明者は、ゲムシタビンの合成プロセスを検討する過程に、驚いたことに、新規なゲムシタビンの合成ルートを見出し、前記した従来の技術の欠点を克服し、高収率で高立体選択的にゲムシタビン及びその塩酸塩を得ることができる。
【0016】
本発明は、高立体選択的にゲムシタビンを合成するプロセスを提供することを目的とする。
【0017】
また、本発明は、ゲムシタビンを合成する中間体を提供することを目的とする。
【0018】
具体的には、本発明は、以下の反応を含む、ゲムシタビンの合成プロセスを提供する。
【0019】
【化5】
【0020】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である
【0021】
【化6】
【0022】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在(すなわち、ベンゼン環は直接カルボニル基と連結すること)であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基である、
G4とG5は、それぞれ独立に、C1〜C7トリアルキルシリル基、好ましくはトリメチルシリル基、イソプロピルジメチルシリル基、メチルジイソプロピルシリル基、トリイソプロピルシリル基又はt-ブチルジメチルシリル基、より好もしくはトリメチルシリル;t-ブトキシカルボニル基、カルボベンゾキシ基又は9-フルオレニルメトキシカルボニル基(Fmoc);或いは、ホルミル基、アセチル基、プロピオニル基、イソプロピオニル基、ブチリル基、イソブチリル基、ピバロイル等から選ばれる基である。]
好ましくは、前記反応が、α-アノマーに富んだ式11の化合物と式12の化合物を反応し、β-アノマーに富んだ式13の化合物を得るものである。
【0023】
前記反応は、具体的には、式12の化合物をα-アノマーに富んだ式11の化合物に対して所定のモル比で有機溶媒に溶解させ、40〜300°Cまで加熱し、これに、20時間以内に、予め有機溶媒に溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、保温しながら、10分〜20時間反応させて、β-アノマーに富んだ式13の化合物を得るものである。
【0024】
前記所定のモル比とは、前記α-アノマーに富んだ式11の化合物に対する前記式12の化合物のモル比であり、当該モル比が1〜20程度であり、好ましくは1.5〜15程度である。
【0025】
前記α-アノマーに富んだ式11の化合物とは、そのα:βが1:1以上である。
【0026】
前記有機溶媒は、沸点が70℃以上のハロゲン化炭化水素類、ベンゼン類、エーテル類等の溶媒から選ばれるものでもよい。好ましくは、1,2-ジクロロエタン、トルエン、キシレン、置換ベンゼン、アニソール、ジフェニルエーテル、又は置換ジフェニルエーテル等の一種又は2種以上の混合物である。
【0027】
前記、式12の化合物を有機溶媒に溶解させて40〜250℃に加熱するのは、該当温度が、100〜150℃が好ましく、110〜150℃が特に好ましい。
【0028】
前記、20時間以内に、予め有機溶媒に溶解した式11の化合物を滴下するのは、滴下時間が4〜7時間であることが好ましい。
【0029】
前記、滴下が終わった後保温しながら10分〜20時間反応させるのは、反応時間が3〜6時間であることが好ましい。
【0030】
具体的には、本発明の1つの実施形態において、式12の化合物を式11の化合物に対してモル比で2.5キシレンに溶解させ、100℃まで昇温し、5時間以内に、予めキシレンに溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、昇温し、3時間反応し、β-アノマーに富んだ式13の化合物を得る。
【0031】
本発明の他の実施形態において、式12の化合物を式11の化合物に対してモル比で10トルエンに溶解させ、還流するまで攪拌昇温し、3時間以内に、予めトルエンに溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、5時間反応し、β-アノマーに富んだ式13の化合物を得る。
【0032】
ここで、前記プロセスはさらに式13の化合物を脱保護して、式2の化合物を得る工程を含むことができる。
【0033】
【化7】
【0034】
反応の条件は、窒素雰囲気中で、乾燥したメタノール/アンモニア溶液に式13の化合物を加え、0℃〜65℃で撹拌しながら、1〜30時間反応させ、乾燥まで減圧濃縮し、水を添加して溶解させ、一種類の有機溶媒で有機不純物を抽出して除去し、乾燥まで水相を濃縮して、式2の化合物を得るように選択される。
【0035】
前記乾燥したメタノール/アンモニア溶液は、濃度が5〜16%であることが好ましい。
【0036】
前記、保温撹拌し2〜30時間反応させるのは、好ましい反応温度が20〜50℃であり、好ましい反応時間が15〜20時間である。
【0037】
前記、一種類の有機溶媒で有機不純物を抽出して除去するのは、当該有機溶媒が、メチルベンゾエートを溶解できる、かつ水と混ざらない低沸点溶媒のいずれかから選ばれ、ジクロロメタン又はエチルアセテートが好ましい。
【0038】
ここに述べたプロセスは、さらに式2の化合物と塩酸とを反応させ、塩酸塩を得る工程を含むことができる。
【0039】
【化8】
【0040】
反応条件は、式2の化合物を一種類の有機溶媒に加え、-10〜50℃でpHが1.5〜2.5になるように濃塩酸を滴下し、低温で10分〜8時間、結晶を育成し、吸引濾過し、有機溶媒で濾過ケーキを洗浄して、ゲムシタビン塩酸塩を得ることである。
【0041】
ここで、前記、式2の化合物を一種類の有機溶媒に加えるのは、当該有機溶媒がゲムシタビン塩酸塩を溶解しにくい有機溶媒のいずれかから選ばれ、メタノール、エタノール、イソプロピルアルコール、アセトンの一種又は2種以上の混合物が好ましい。
【0042】
前記、低温で10分〜8時間、結晶を育成するのは、低温とは、-10〜25℃であり、好ましくは0〜5℃であり、結晶を育成する時間が2〜5時間であることが好ましい。
【0043】
一方、本発明は下記式で表される化合物を提供している。
【0044】
【化9】
【0045】
[ただし、式中、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【0046】
【化10】
【0047】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
好ましくは、G1とG2が、それぞれ独立に、ベンゾイル基、フェニルアセチル基、ビフェニルカルボニル基、ビフェニルアセチル基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基である、ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基である。]
好ましい中間体化合物は以下の化合物を含む。
【0048】
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-ベンジルスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート。
【0049】
また、本発明が提供した中間体は、さらにα-アノマーに富んだ式11の化合物の混合物を含み、好ましくは、当該混合物に、α:βが1:1以上である。
【0050】
また、本発明は、式11の化合物の合成方法を提供し、その合成ルートがルート1に示す。
【0051】
【化11】
【0052】
合成ルート1において、まず、酸スカベンジャの存在下で、有機溶媒に式3の化合物とアシル化剤を反応させ、水酸基が保護された式4の化合物を製造した。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
【0053】
次は、トリフルオロ酢酸と水の存在下で、アセトニトリルに式4の化合物を加水分解させて、式5の化合物を生成し、その後、還流により式5の化合物を脱水してラクトン化し、式6の化合物を得た。
【0054】
次いで、酸スカベンジャの存在下で、有機溶媒に式6の化合物とアシル化剤を反応させ、5’位の水酸基が保護された式7の化合物を製造し、アセトアセテートとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
【0055】
最後に、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。そのスルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれ、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-ニトロベンゼンスルホニル基である。
【0056】
ただし、G1、G2及びG3の定義は前記した通りである。
【0057】
式11の化合物の水酸基保護基は同じである場合、以下の合成ルート2を経て合成することができる。
【0058】
【化12】
【0059】
ただし、G1とG2は同じ基であり、以下の構造で定義された基である。
【0060】
【化13】
【0061】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、またはp-ニトロベンゼンスルホニル基である。
【0062】
合成ルート2において、まず、酸スカベンジャの存在下で、有機溶媒に式9の化合物とアシル化剤を反応させ、水酸基が保護された式7の化合物を製造し、トルエンとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-アミノピリジンが好ましい。
【0063】
次いで、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。
【0064】
従来の技術に比べて、本発明の優れた技術効果は以下の面に表されている。
【0065】
1.本発明はゲムシタビンを合成するプロセスのステップを減少した
本発明は、適宜な保護基を選択することにより、高収率で立体選択的にゲムシタビンを合成でき、従来の技術において反応ステップが多く、立体選択性が低いという欠点を克服した。
【0066】
本発明は、新規な中間体化合物11と保護されたシチジンとの縮合を利用して、β異性体に富んだ化合物13が得られ、そして、本発明は縮合反応中、式11の化合物を保護されたシチジン系に滴下し、保護されたシチジンが常に高濃度状態で反応させることが保証され、2.5モル当量比のみを使用する場合、生成したゲムシタビン(β:α)比率が3.5:1に達し、この比率は(US)第5371210号において3.0モル当量を使用して生成した異性体1.3:1の比率よりはるかに高く、かつ20.0モル比を使用して生成した異性体4:1の比率に相当する。また、本発明は、シチジンの使用量を大きく減少させ、メリットが明らかに見える。また、WO2006/071090に比べて、臭化物と保護されたシチジンが縮合した後、さらに多量溶媒でブロモトリメチルシランを抽出するという不具合を避けることができ、しかも環境負荷を減らすことができる。脱保護して塩を生成する合計五つのステップの反応で塩酸ゲムシタビンを得る(合計収率:35.9%)。産物の品質はUSP28及びEP5.6版の要求に合っている。
【0067】
WO2006/011713には、t−ブチルリチウムアルミニウムハイドライドで化合物7を還元した後、ジフェニルオキシホスホリルクロリドと反応して、化合物XIVを製造し、さらに臭化させ、化合物XVを生成し、次いで、異性体の比(α:β)が1:8.8(89.9%)に達すように、さらに15〜20モル当量の保護されたシチジンを使用する必要があり、次いで、脱保護し、塩を生成する合計六つのステップの反応で塩酸ゲムシタビンを得る(合計収率:46.4%)(製品の純度:99.97%)。本発明の合成ルートは、反応ステップがより少なく、より産業化しやすく、より操作性がよい。
【0068】
2.本発明は、後処理が簡略化され、より工業化生産に有利である。
【0069】
ゲムシタビンを合成する従来の技術において、多数の後処理がカラムクロマトグラフィーを使用し、これは工業化生産に不利で、しかも製品のコストが増加してしまう。本発明のゲムシタビンを合成するプロセスは、ただ洗浄、再結晶のような簡単な後処理技術によって、高純度またはee値がより高い産物が得られる。
【0070】
3.反応の条件はより温和である。
【0071】
本発明のゲムシタビンを合成するプロセスの反応条件は、従来の技術に比べて、より温和である。-20°C以下の低温条件でなくても、β-アノマーに富んだ重要な中間体(即ち、式11の化合物)が得られ、最終の高立体選択的にゲムシタビン又はその塩酸塩を合成することに有利な条件を与える。
【図面の簡単な説明】
【0072】
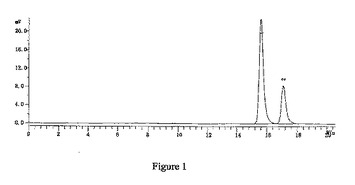
【図1】2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートのHPLCによる検出の結果(α:βは2.4:1)を示す図である。
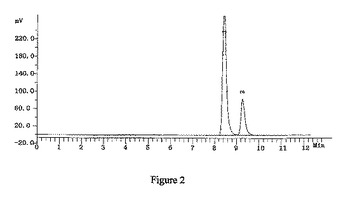
【図2】2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートのHPLCによる検出の結果(α:βは2.5:1)を示す図である。
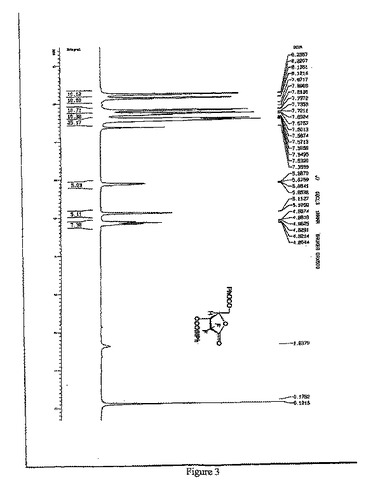
【図3】D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)ベンゾエートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
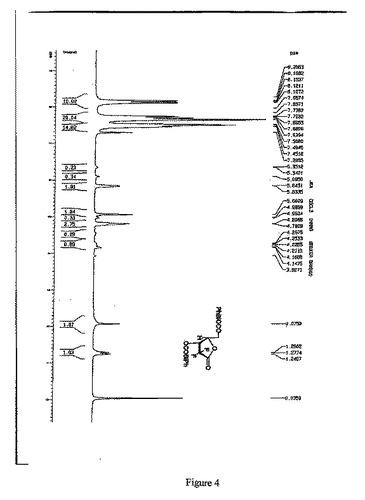
【図4】D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)ベンゾエートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
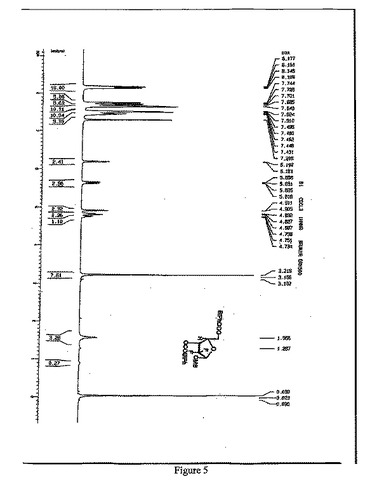
【図5】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
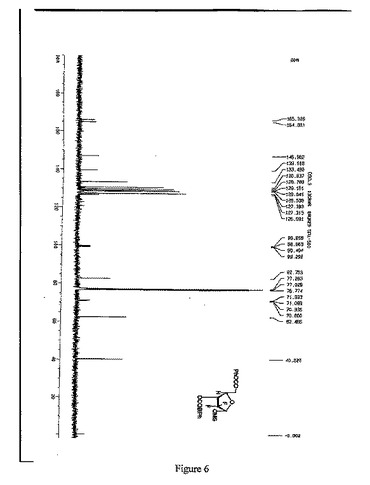
【図6】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz,CDCl3)スペクトルを示す図である。
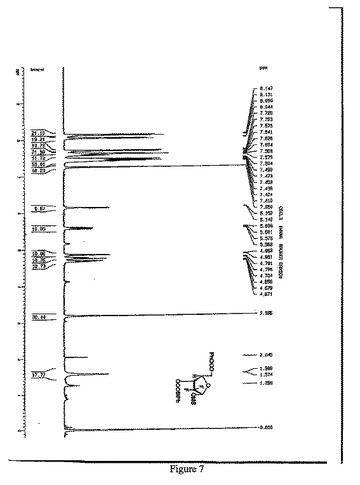
【図7】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
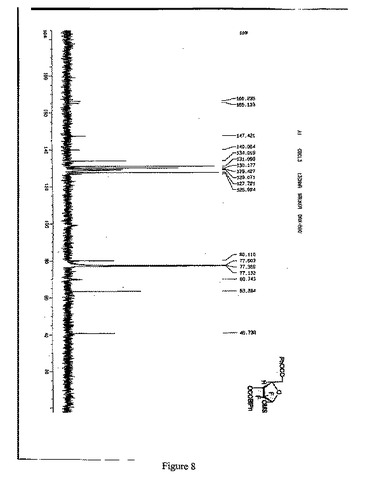
【図8】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz, CDCl3)スペクトルを示す図である。
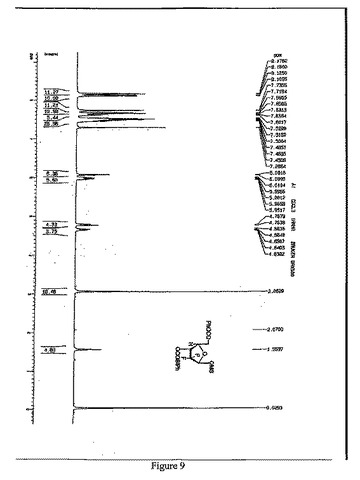
【図9】1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
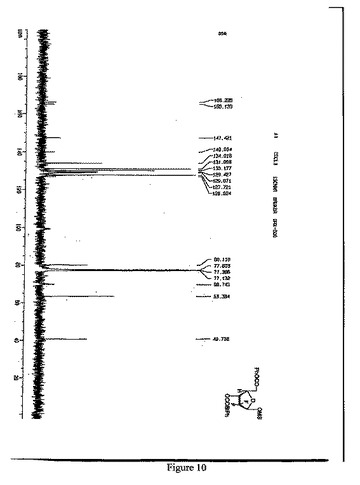
【図10】1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz, CDCl3)スペクトルを示す図である。
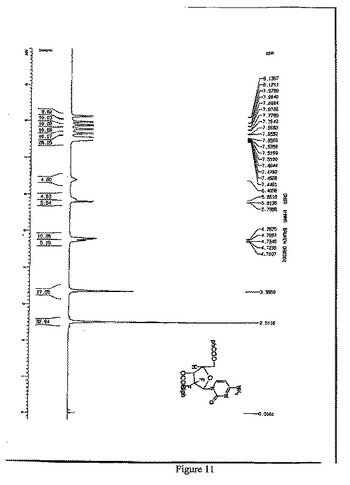
【図11】1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アミノピリミジン-2-オンの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【発明を実施するための形態】
【0073】
以下、実施例によって本発明をより詳細に説明する。以下の実施例が、本発明が実施できることを説明するために過ぎず、従来の技術によって本発明へのいかなる変更や改変も本発明の請求の範囲に包含されることは、当業者には明らかである。
【0074】
HPLCにより式11の化合物、式13の化合物を分析するのに、いずれもPhenomenex Luna C18(4.6×250mm, 5μm)カラムを用い、アセトニトリル/1%トリエチルアミン(リン酸でpHを7.0に調整する)水溶液(80:20,v/v)を移動相とし、流速を1.0ml/minとする。
【0075】
〔実施例1〕
2,2-ジフルオロ-3-(4-ビフェニルカルボニル)オキソ-3-(2,2-ジメチル-[1,3]ジオキソラン-4イル)プロピオネート(化合物4)の製造
窒素雰囲気中、四口フラスコに化合物3を290.0g加え、ジクロロメタン2900mlで溶解させた後、ピリジン117.2mlを添加した。10分攪拌した。約0.5時間かけてビフェニルカルボニルクロリド296.8gを徐々に添加し、その過程中、温度が20〜25℃になるように制御されている。その後、室温で、6時間攪拌・反応した。1N塩酸950mlで洗浄し、5%NaHCO3溶液950mlで洗浄し、そして、飽和NaCl溶液950mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、490.0g産物を得た。(3R/3S=3:1)収率:98.9%
【0076】
〔実施例2〕
2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3(4-フェニル)ベンゾエート(化合物6)の製造
アセトニトリル2500mlに2,2-ジフルオロ-3-(4-ビフェニルカルボニル)オキソ-3-(2,2-ジメチル-[1,3]ジオキソラン-4イル)プロピオネート(化合物4)495.0gを加え、攪拌溶解した後、トリフルオロ酢酸14.5mlと蒸留水81mlを添加し、還流するように攪拌昇温し、3時間反応させ、還流に代えて蒸留し(常圧蒸留)、反応液500mlを蒸かすごとに、無水トルエン500mlを追加した。蒸留速度を500ml/15分程度にコントロールした。反応液の温度が100℃に達し、乾燥まで反応液を減圧濃縮させ、次いで、アセトアセテート及びn−ヘキサンで再結晶させ、化合物6を317.5g得た。(3R/3S=3:1)収率:80.0%
【0077】
〔実施例3〕
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)ベンゾエート(化合物7)の製造
窒素雰囲気中、室温で、5L四口フラスコにD-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3(4-フェニル)ベンゾエート(化合物6)200gを加え、ジクロロメタン2000mlで溶解させた後、ピリジン59.5mlを添加し、10分攪拌した。室温で、ベンゾイルクロリド96.0gのジクロロメタン溶液(480ml)を滴下し、滴下終わってから、室温で、6時間攪拌・反応した。1N塩酸950mlで洗浄し、5%NaHCO3溶液950mlで洗浄し、そして、飽和NaCl溶液950mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮し、トルエンとn−ヘキサンで再結晶させ、異性体を除き、白色の固体化合物7を170g得た。(3R:3S=50:1)収率:65.0%
1H-NMR(500MHz,CDCl3)(図3参照):δ8.23(d,J=8.0 Hz,2H),8.13(d,J=7.3 Hz,2H), 7.79-7.35(m,10H), 5.88-5.85(m,1H), 5.12-5.10(m,1H),4.88-4.80(m,2H).
前記と同じ方法で以下の化合物を製造した。
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-フェニルアセチル-3-(4-フェニル)ベンゾエート
(合計収率:48.7%;3R:3S=38:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-フェニルアセテート
(合計収率:46.5%;3R:3S=42:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)フェニルアセテート
(合計収率:43.5%;3R:3S=35:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-フェニルアセチル-3-ベンゾエート
(合計収率:45.0%;3R:3S=43:1)
【0078】
〔実施例4〕
1-オキソ-2-デオキシ-2,2-ジフルオロ-フラノース(化合物9)の製造
四口フラスコに化合物3を290g加え、MeCN2700mlで溶解させ、攪拌した後、蒸留水81ml、CF3COOH 14.5mlを添加し、昇温しながら、激しく還流させ、3時間反応させた。還流に代えて蒸留し(常圧蒸留)、反応液500mlを蒸かすごとに、無水トルエン500mlを追加した。反応液の温度が100℃に達するまで蒸留速度を500ml/15分程度にコントロールした。反応が終了した後、乾燥まで反応液を減圧濃縮して、赤褐色油状化合物9を200g得た。収率:100.0%
【0079】
〔実施例5〕
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)ベンゾエート(化合物7)の製造
窒素雰囲気中、化合物9(200g)をジクロロメタン2000mlに溶解させた後、DMAP34.8g、ピリジン257.5mlを添加した。10分攪拌した。約0.5時間かけてビフェニルカルボニルクロリド617.0gを徐々に添加し、その過程中、温度が20〜25℃になるように制御されている。その後、室温で、6時間攪拌・反応した。1N塩酸1800mlで洗浄し、5%NaHCO3溶液1800mlで洗浄し、そして、飽和NaCl溶液1800mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、トルエンとn−ヘキサンで再結晶させ、白色の固体化合物7 を358g得た。(3R:3S=45:1)収率:57.0%
1H-NMR(500MHz,CDCl3)(図4参照):
δ8.26-7.28(m,18H),5.84-5.83(m,1H),5.09-5.07(m,1H),4.82-4.79(m,2H).
前記と同じ方法で以下の化合物を製造した。
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)フェニルアセテート
(合計収率:52.0%;3R:3S=20:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-フェニルアセテート
(合計収率:55.5%;3R:3S=46:1)
【0080】
〔実施例6〕
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)の製造
窒素雰囲気中、THF630mlにトリt−ブチルリチウムアルミニウムハイドライド59.8gを加え、-18℃まで冷却し、徐々に化合物7を添加し、保温しながら、2時間攪拌・反応させた。5℃以下で、緩やかに1N HClを2500ml添加し、二塩化メタン(600ml×3)で抽出して、有機相を分取し、10%炭酸ナトリウム、水で洗浄し、有機相を分取して、無水硫酸ナトリウムで乾燥させた。吸引濾過して、乾燥までろ液を減圧濃縮させた。ジクロロメタン950mlを添加し、溶解させ、トリエチルアミン41.7mlを添加し、0〜5℃まで降温し、メチルスルホニルクロリド23.2mlのジクロロメタン溶液(50ml)を滴下し、保温しながら、2時間攪拌・反応させ、1N HCl、10%炭酸ナトリウム及び水で洗浄し、有機相を分取して無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、アルコールで精製して、白色の固体100.6g (α:β=2.4:1)を得た(HPLCによる検出の結果は図1を参照)。収率:85.0%
本製品は、カラムクロマトグラフィー(アセトアセテート及びn−ヘキサンを溶離液とする)により精製され、単純なα-アノマーメタンスルホネートとβ-アノマーメタンスルホネートを得た。
α-アノマーメタンスルホネートmp:154.5〜156.5℃
1H-NMR(500MHz,CDCl3)(図5参照):
δ8.17-8.13(m,4H), 7.74-7.64(m,8H), 7.52-7.43 (m,7H), 6.18(d,J=5.5Hz,1H), 5.63(dd,J=3.5,16.5Hz,1H), 4.91-4.73(m,2H),3.22(s,3H),
13C-NMR(125MHz, CDCl3) (図6参照):
δ166.5,165.3, 147.4,146.6, 140.2, 140.0, 131.0, 130.7, 129.4, 129.3, 128.8, 128.6, 128.2, 127.7, 127.6, 127.5, 127.0, 122(q,CF2), 99.9,83.0,71.6,62.9,40.6.
質量スペクトルFAB 609(M+1);元素分析C32H26F2O8S 理論値:C:85.69% H:5.84% F:8.47% 実測値:C:85.71% H:5.80% F:8.49%
前記と同じ方法で以下の化合物を製造する。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート
(収率:79.8%;α:β=2.2:1)
N,N-ジメチル-アミノピリジンを酸スカベンジャとする。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート
(収率:66.5%;α:β=1.5:1)
(2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p−ニトロベンゼンスルホネート
(収率:69.0%;α:β=1.7:1)
【0081】
〔実施例7〕
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)の製造
窒素雰囲気中、THF 600mlにトリt−ブチルリチウムアルミニウムハイドライド59.8gを加え、-18℃に冷却し、化合物7のTHF溶液(450ml)を滴下し、保温しながら、2時間攪拌・反応させた。1N HClを2400ml添加し、反応を終了させ、二塩化メタン(500ml×3)で抽出して、有機相を分取し、10%炭酸ナトリウム、水で洗浄し、有機相を分取して無水硫酸ナトリウムで乾燥させた。吸引濾過して、乾燥までろ液を減圧濃縮させた。ジクロロメタン950mlを添加して溶解させ、トリエチルアミン41.7mlを添加し、0-5℃まで降温し、メチルスルホニルクロリド23.2mlのジクロロメタン(50ml)溶液を滴下し、保温しながら、2時間攪拌・反応させ、1N HCl、10%炭酸ナトリウム及び水で洗浄し、有機相を分取して無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮し、アルコールで精製して、白色の固体88.2g (α:β=2.5:1)を得た(HPLCによる検出の結果は図2を参照)。収率:83.0%
本製品は、カラムクロマトグラフィー(エチルアセテート及びn−ヘキサンを溶離液とする)により精製され、単純なα-アノマーメタンスルホネートとβ-アノマーメタンスルホネートを得た。
α-アノマーメタンスルホネート mp:136.0〜139.0℃
1H-NMR(500MHz,CDCl3)(図7参照):
δ8.11(d,J=8.0Hz,2H),8.04(d,J=7.5Hz,2H), 7.72-7.41 (m,10H), 7.60-7.57(m,1H), 6.15 (d,J=5.0Hz,1H) , 5.60-5.56(m,1H), 4.79-4.67 (m,2H), 3.18(s,3H)
13C-NMR(125MHz,CDCl3)(図8参照):
δ165.9, 164.9,146.9, 139.6,133.5,130.6, 129.8, 129.1, 129.0, 128.5, 127.4, 127.3, 126.1,122(q,CF2),99.7,82.7, 71.0,62.5,40.2
質量スペクトルFAB 533(M+1);元素分析C26H22F2O8S 理論値:C:83.85% H:5.95% F:10.20% 実測値:C:83.88% H:5.91% F:10.21%
β-アノマーメタンスルホネート mp:162.0〜163.5℃
1H-NMR(500MHz,CDCl3)(図9参照):
δ8.16(d,J=8.0Hz,2H),8.11(d,J=7.5Hz,2H), 7.73-7.45(m,10H), 7.60-7.57(m,1H), 6.07(d,J=7.7Hz,1H),6.01-5.95(m,1H),4.78-4.62(m,2H),3.06(s,3H)
13C-NMR(125MHz, CDCl3) (図10参照):
δ166.2,165.1,147.4,140.0,134.0,131.1,130.0,129.4,129.0,127.7,126.9,122(q,CF2),99.7,80.1,69.7,63.4,40.7
前記と同じ方法で以下の化合物を製造する。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート
(収率:80.2%;α:β=2.1:1)
N,N-ジメチル-アミノピリジンを酸スカベンジャとする。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-ベンジルスルホネート
(収率:69.3%;α:β=1.6:1)
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-p−ニトロベンゼンスルホネート
(収率:70.5%;α:β=1.7:1)
【0082】
〔実施例8〕
1-(2’-デオキシ-2’,2’-ジフルオロ-3,5-ジ(4-フェニル)ベンゾイル-D-アラビノフラノース-4-アミノピリミジン-2-オン(化合物13)の製造
窒素雰囲気中、シチジン100gをキシレン550mlに加え、そして、硫酸アンモニウム0.5g、ヘキサメチルジシラザン283mlを加え、還流するまで昇温し、3時間反応させ、乾燥まで減圧濃縮し、キシレン600mlを添加し、100℃まで昇温し、5時間以内に2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)(α:β=2.4:1)219gのキシレン溶液(1100ml)を滴下し、滴下終わってから、還流しながら、3時間反応させた(α:β=1:1.8)。40℃まで降温し、水(500ml×3)、5%炭酸水素ナトリウム500ml及び塩水で洗浄し、乾燥まで有機相を濃縮し、アルコールで再結晶して、α-アノマー異性体を除去し、目標化合物119.0g (α:β=1:40)を得た。収率:53.0%
前記と同じ方法で以下の化合物を製造する。
1-(2’-デオキシ-2’,2’-ジフルオロ-3,5-ジ(4-フェニル)ベンゾイル-D-アラビノフラノース-4-アセチルアミノピリミジン-2-オン
(収率:49.5%;α:β=1:35)
【0083】
〔実施例9〕
1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アミノピリミジン-2-オン(化合物13)の製造
窒素雰囲気中、シチジン400gをトルエン2200mlに加え、そして、硫酸アンモニウム2.0g、ヘキサメチルジシラザン1130mlを加え、還流するまで昇温し、3時間反応させ、乾燥まで減圧濃縮し、キシレン2400mlを添加して溶解させ、100℃まで昇温し、3時間以内に2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)(α:β=2.5:1)122gのトルエン溶液(600ml)を滴下し、滴下終わってから、還流しながら、5時間反応させた(α:β=1:2.1)。60℃まで降温し、メタノール780mlを滴下し、10分攪拌した後、2N HClを780ml滴下し、その結果、大量の固体が析出し、保温しながら、20分攪拌し、室温まで降温し、吸収濾過し、2N HCl(1000ml×3)で濾過ケーキを洗浄して、シチジンを除き、さらにアルコールで精製して、α-アノマー異性体を除去し、目標化合物78.7g (α:β=1:35)を得た。収率:55.0%
β-アノマー化合物13 mp:247.5-248.5°C。
1H-NMR(500MHz,CDCl3)(図11参照):
δ8.13(d,J=7.5Hz,2H), 7.97(d,J=7.5Hz,2H) , 7.87(d,J=7.5Hz,2H) , 7.77 (d,J=7.3Hz,2H) , 7.66(d,J=6.5Hz,2H) , 7.55-7.44(m,6H) , 6.40(s,br,1H), 5.86(s,br.1H) , 4.78-4.71(m,3H).
前記と同じ方法で以下の化合物を製造する。
1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アセチルアミノピリミジン-2-オン
(収率:51.5%;α:β=1:40)
【0084】
〔実施例10〕
2’-デオキシ-2’,2’-ジフルオロシチジン(化合物2)の製造
窒素雰囲気中、化合物13をメタノール/アンモニア溶液に加え、室温で、攪拌して、一晩反応させた。乾燥まで減圧濃縮し、水で溶解させ、エチルアセテートで抽出し、水相を濃縮して、ゲムシタビン18.7gを得た。(純度: 97.0% , ee 99.5%) 収率:88.5%
【0085】
〔実施例11〕
塩酸ゲムシタビンの製造
化合物2をイソプロピルアルコールに溶解させ、0〜5℃まで降温し、pHが2になるように濃塩酸を滴下して、保温しながら、2時間結晶を育成し、吸引濾過し、イソプロピルアルコール100mlで濾過ケーキを洗浄して、ゲムシタビン塩酸塩51.2gを得た。(純度 : 99.8%) 収率:90.0%
【技術分野】
【0001】
本発明は、抗腫瘍性ヌクレオチド類代謝拮抗薬の化学合成プロセスに関するものであり、具体的には、ゲムシタビンの高立体選択的な合成プロセス及び中間体に関するものである。
【背景技術】
【0002】
ゲムシタビンは、細胞複製を阻害するジフルオロヌクレオチド類代謝拮抗薬抗腫瘍薬で、デオキシシチジンの水溶性類似物であり、リボヌクレオチド還元酵素に対して、当該酵素の基質と一致の代替物であり、この酵素はDNAの合成と修復において、必要なデオキシリボヌクレオチドの合成に対して非常に重要である。
【0003】
ゲムシタビンは、化学名が2’-デオキシ-2’,2’-ジフルオロシチジンであり、化学構造が下記式2で表される。
【0004】
【化1】
【0005】
Hertelらは米国特許(US)第4808614号に初めてゲムシタビン化合物を開示したと同時に、以下のような当該化合物の合成ルートも開示した。
【0006】
【化2】
【0007】
この合成ルートにおいて、メチル-2,2-ジフルオロ-3-水酸基-3-(2,2-ジメチル-ジオキソラン-4-イル)プロピオネートを合成する時のみに、シリカゲルカラムにより3-R-ヒドロキシ産物を分離することにかかわり、その後の反応がいずれも反応の立体化学にかかわっていない。
【0008】
Chou Ta-Senは、米国特許(US)第5401861号及び欧州特許(EP)第0577303号に、予めα-アノマーを主とする2-デオキシ-2,2-ジフルオロ-D-リボフラノシル-3,5-ジ-O-ベンゾイルメタンスルホネートを製造することによって、β-アノマーを主とするゲムシタビンを製造するもう一つの合成ルートを開示した。反応は、低温(-78℃)でα-アノマーを主とするメタンスルホネート中間体を製造した後、3〜20倍以上(モル)のシリル基で保護されたシトシンとの反応により、立体選択的にβ-アノマーゲムシタビンを主とするものを合成することを特徴としている。なお、以上の特許文献には、3、5位の水酸基保護基はベンゾイル基で、また、α-アノマーを主とするメタンスルホネートを製造する反応は低温反応であり、反応条件は過酷で、大規模の工業的普及及び応用に適しない。
【0009】
また、Lee Jaeheonらは国際特許出願WO2006/009353に以下のような中間体の合成ルートを開示した。
【0010】
【化3】
【0011】
そして、Lee Jaeheonらは国際特許出願WO2006/011713に以下の合成ルートを開示した。
【0012】
【化4】
【0013】
以上の合成ルートにおいて、P1はBz又はBiPhC(O)-で、P2は-P(O)(OPh)2である。以上の反応によりα-アノマーに富んだハロゲン化中間体が得られ、これにより、立体選択的にゲムシタビンを合成することに有益な中間体を提供できる。しかし、実験により、WO2006/009353に記載のプロセスにおいてカリウム塩化合物を製造する後処理の時、低温で迅速に、乾燥まで反応液における有機溶媒を濃縮する必要があり、そうでないと、産物(カリウム塩)はひどく分解し、純度と収率に影響を与えることを見出し、これは工業的操作において実現されにくい。また、ハロゲン化プロセスには、過剰なハロゲン化水素を使用しなければならないから、毒性が大きくて、労働者保護に不利であり、排ガス、排水、固形廃棄物などの処理が困難で、環境汚染を引き起こしやすくなる。
【0014】
つまり、従来の技術では、ゲムシタビンの合成ルートに関しては、依然として前記した欠点がある。現在、反応条件が温和で、環境汚染が少なく、立体選択性が高い、ゲムシタビンの合成プロセスの開発が望まれている。
【発明の概要】
【0015】
本発明者は、ゲムシタビンの合成プロセスを検討する過程に、驚いたことに、新規なゲムシタビンの合成ルートを見出し、前記した従来の技術の欠点を克服し、高収率で高立体選択的にゲムシタビン及びその塩酸塩を得ることができる。
【0016】
本発明は、高立体選択的にゲムシタビンを合成するプロセスを提供することを目的とする。
【0017】
また、本発明は、ゲムシタビンを合成する中間体を提供することを目的とする。
【0018】
具体的には、本発明は、以下の反応を含む、ゲムシタビンの合成プロセスを提供する。
【0019】
【化5】
【0020】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である
【0021】
【化6】
【0022】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在(すなわち、ベンゼン環は直接カルボニル基と連結すること)であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基である、
G4とG5は、それぞれ独立に、C1〜C7トリアルキルシリル基、好ましくはトリメチルシリル基、イソプロピルジメチルシリル基、メチルジイソプロピルシリル基、トリイソプロピルシリル基又はt-ブチルジメチルシリル基、より好もしくはトリメチルシリル;t-ブトキシカルボニル基、カルボベンゾキシ基又は9-フルオレニルメトキシカルボニル基(Fmoc);或いは、ホルミル基、アセチル基、プロピオニル基、イソプロピオニル基、ブチリル基、イソブチリル基、ピバロイル等から選ばれる基である。]
好ましくは、前記反応が、α-アノマーに富んだ式11の化合物と式12の化合物を反応し、β-アノマーに富んだ式13の化合物を得るものである。
【0023】
前記反応は、具体的には、式12の化合物をα-アノマーに富んだ式11の化合物に対して所定のモル比で有機溶媒に溶解させ、40〜300°Cまで加熱し、これに、20時間以内に、予め有機溶媒に溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、保温しながら、10分〜20時間反応させて、β-アノマーに富んだ式13の化合物を得るものである。
【0024】
前記所定のモル比とは、前記α-アノマーに富んだ式11の化合物に対する前記式12の化合物のモル比であり、当該モル比が1〜20程度であり、好ましくは1.5〜15程度である。
【0025】
前記α-アノマーに富んだ式11の化合物とは、そのα:βが1:1以上である。
【0026】
前記有機溶媒は、沸点が70℃以上のハロゲン化炭化水素類、ベンゼン類、エーテル類等の溶媒から選ばれるものでもよい。好ましくは、1,2-ジクロロエタン、トルエン、キシレン、置換ベンゼン、アニソール、ジフェニルエーテル、又は置換ジフェニルエーテル等の一種又は2種以上の混合物である。
【0027】
前記、式12の化合物を有機溶媒に溶解させて40〜250℃に加熱するのは、該当温度が、100〜150℃が好ましく、110〜150℃が特に好ましい。
【0028】
前記、20時間以内に、予め有機溶媒に溶解した式11の化合物を滴下するのは、滴下時間が4〜7時間であることが好ましい。
【0029】
前記、滴下が終わった後保温しながら10分〜20時間反応させるのは、反応時間が3〜6時間であることが好ましい。
【0030】
具体的には、本発明の1つの実施形態において、式12の化合物を式11の化合物に対してモル比で2.5キシレンに溶解させ、100℃まで昇温し、5時間以内に、予めキシレンに溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、昇温し、3時間反応し、β-アノマーに富んだ式13の化合物を得る。
【0031】
本発明の他の実施形態において、式12の化合物を式11の化合物に対してモル比で10トルエンに溶解させ、還流するまで攪拌昇温し、3時間以内に、予めトルエンに溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、5時間反応し、β-アノマーに富んだ式13の化合物を得る。
【0032】
ここで、前記プロセスはさらに式13の化合物を脱保護して、式2の化合物を得る工程を含むことができる。
【0033】
【化7】
【0034】
反応の条件は、窒素雰囲気中で、乾燥したメタノール/アンモニア溶液に式13の化合物を加え、0℃〜65℃で撹拌しながら、1〜30時間反応させ、乾燥まで減圧濃縮し、水を添加して溶解させ、一種類の有機溶媒で有機不純物を抽出して除去し、乾燥まで水相を濃縮して、式2の化合物を得るように選択される。
【0035】
前記乾燥したメタノール/アンモニア溶液は、濃度が5〜16%であることが好ましい。
【0036】
前記、保温撹拌し2〜30時間反応させるのは、好ましい反応温度が20〜50℃であり、好ましい反応時間が15〜20時間である。
【0037】
前記、一種類の有機溶媒で有機不純物を抽出して除去するのは、当該有機溶媒が、メチルベンゾエートを溶解できる、かつ水と混ざらない低沸点溶媒のいずれかから選ばれ、ジクロロメタン又はエチルアセテートが好ましい。
【0038】
ここに述べたプロセスは、さらに式2の化合物と塩酸とを反応させ、塩酸塩を得る工程を含むことができる。
【0039】
【化8】
【0040】
反応条件は、式2の化合物を一種類の有機溶媒に加え、-10〜50℃でpHが1.5〜2.5になるように濃塩酸を滴下し、低温で10分〜8時間、結晶を育成し、吸引濾過し、有機溶媒で濾過ケーキを洗浄して、ゲムシタビン塩酸塩を得ることである。
【0041】
ここで、前記、式2の化合物を一種類の有機溶媒に加えるのは、当該有機溶媒がゲムシタビン塩酸塩を溶解しにくい有機溶媒のいずれかから選ばれ、メタノール、エタノール、イソプロピルアルコール、アセトンの一種又は2種以上の混合物が好ましい。
【0042】
前記、低温で10分〜8時間、結晶を育成するのは、低温とは、-10〜25℃であり、好ましくは0〜5℃であり、結晶を育成する時間が2〜5時間であることが好ましい。
【0043】
一方、本発明は下記式で表される化合物を提供している。
【0044】
【化9】
【0045】
[ただし、式中、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【0046】
【化10】
【0047】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
好ましくは、G1とG2が、それぞれ独立に、ベンゾイル基、フェニルアセチル基、ビフェニルカルボニル基、ビフェニルアセチル基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基である、ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基である。]
好ましい中間体化合物は以下の化合物を含む。
【0048】
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-ベンジルスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート。
【0049】
また、本発明が提供した中間体は、さらにα-アノマーに富んだ式11の化合物の混合物を含み、好ましくは、当該混合物に、α:βが1:1以上である。
【0050】
また、本発明は、式11の化合物の合成方法を提供し、その合成ルートがルート1に示す。
【0051】
【化11】
【0052】
合成ルート1において、まず、酸スカベンジャの存在下で、有機溶媒に式3の化合物とアシル化剤を反応させ、水酸基が保護された式4の化合物を製造した。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
【0053】
次は、トリフルオロ酢酸と水の存在下で、アセトニトリルに式4の化合物を加水分解させて、式5の化合物を生成し、その後、還流により式5の化合物を脱水してラクトン化し、式6の化合物を得た。
【0054】
次いで、酸スカベンジャの存在下で、有機溶媒に式6の化合物とアシル化剤を反応させ、5’位の水酸基が保護された式7の化合物を製造し、アセトアセテートとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
【0055】
最後に、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。そのスルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれ、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-ニトロベンゼンスルホニル基である。
【0056】
ただし、G1、G2及びG3の定義は前記した通りである。
【0057】
式11の化合物の水酸基保護基は同じである場合、以下の合成ルート2を経て合成することができる。
【0058】
【化12】
【0059】
ただし、G1とG2は同じ基であり、以下の構造で定義された基である。
【0060】
【化13】
【0061】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、好ましくは不存在、或いは、-CH2-又は-CH2CH2-である、
R2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲン(フッ素、塩素、臭素、ヨード)で置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、またはp-ニトロベンゼンスルホニル基である。
【0062】
合成ルート2において、まず、酸スカベンジャの存在下で、有機溶媒に式9の化合物とアシル化剤を反応させ、水酸基が保護された式7の化合物を製造し、トルエンとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-アミノピリジンが好ましい。
【0063】
次いで、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。
【0064】
従来の技術に比べて、本発明の優れた技術効果は以下の面に表されている。
【0065】
1.本発明はゲムシタビンを合成するプロセスのステップを減少した
本発明は、適宜な保護基を選択することにより、高収率で立体選択的にゲムシタビンを合成でき、従来の技術において反応ステップが多く、立体選択性が低いという欠点を克服した。
【0066】
本発明は、新規な中間体化合物11と保護されたシチジンとの縮合を利用して、β異性体に富んだ化合物13が得られ、そして、本発明は縮合反応中、式11の化合物を保護されたシチジン系に滴下し、保護されたシチジンが常に高濃度状態で反応させることが保証され、2.5モル当量比のみを使用する場合、生成したゲムシタビン(β:α)比率が3.5:1に達し、この比率は(US)第5371210号において3.0モル当量を使用して生成した異性体1.3:1の比率よりはるかに高く、かつ20.0モル比を使用して生成した異性体4:1の比率に相当する。また、本発明は、シチジンの使用量を大きく減少させ、メリットが明らかに見える。また、WO2006/071090に比べて、臭化物と保護されたシチジンが縮合した後、さらに多量溶媒でブロモトリメチルシランを抽出するという不具合を避けることができ、しかも環境負荷を減らすことができる。脱保護して塩を生成する合計五つのステップの反応で塩酸ゲムシタビンを得る(合計収率:35.9%)。産物の品質はUSP28及びEP5.6版の要求に合っている。
【0067】
WO2006/011713には、t−ブチルリチウムアルミニウムハイドライドで化合物7を還元した後、ジフェニルオキシホスホリルクロリドと反応して、化合物XIVを製造し、さらに臭化させ、化合物XVを生成し、次いで、異性体の比(α:β)が1:8.8(89.9%)に達すように、さらに15〜20モル当量の保護されたシチジンを使用する必要があり、次いで、脱保護し、塩を生成する合計六つのステップの反応で塩酸ゲムシタビンを得る(合計収率:46.4%)(製品の純度:99.97%)。本発明の合成ルートは、反応ステップがより少なく、より産業化しやすく、より操作性がよい。
【0068】
2.本発明は、後処理が簡略化され、より工業化生産に有利である。
【0069】
ゲムシタビンを合成する従来の技術において、多数の後処理がカラムクロマトグラフィーを使用し、これは工業化生産に不利で、しかも製品のコストが増加してしまう。本発明のゲムシタビンを合成するプロセスは、ただ洗浄、再結晶のような簡単な後処理技術によって、高純度またはee値がより高い産物が得られる。
【0070】
3.反応の条件はより温和である。
【0071】
本発明のゲムシタビンを合成するプロセスの反応条件は、従来の技術に比べて、より温和である。-20°C以下の低温条件でなくても、β-アノマーに富んだ重要な中間体(即ち、式11の化合物)が得られ、最終の高立体選択的にゲムシタビン又はその塩酸塩を合成することに有利な条件を与える。
【図面の簡単な説明】
【0072】
【図1】2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートのHPLCによる検出の結果(α:βは2.4:1)を示す図である。
【図2】2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートのHPLCによる検出の結果(α:βは2.5:1)を示す図である。
【図3】D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)ベンゾエートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【図4】D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)ベンゾエートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【図5】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【図6】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz,CDCl3)スペクトルを示す図である。
【図7】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【図8】1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz, CDCl3)スペクトルを示す図である。
【図9】1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【図10】1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネートの13C-NMR(125MHz, CDCl3)スペクトルを示す図である。
【図11】1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アミノピリミジン-2-オンの1H-NMR(500MHz,CDCl3)スペクトルを示す図である。
【発明を実施するための形態】
【0073】
以下、実施例によって本発明をより詳細に説明する。以下の実施例が、本発明が実施できることを説明するために過ぎず、従来の技術によって本発明へのいかなる変更や改変も本発明の請求の範囲に包含されることは、当業者には明らかである。
【0074】
HPLCにより式11の化合物、式13の化合物を分析するのに、いずれもPhenomenex Luna C18(4.6×250mm, 5μm)カラムを用い、アセトニトリル/1%トリエチルアミン(リン酸でpHを7.0に調整する)水溶液(80:20,v/v)を移動相とし、流速を1.0ml/minとする。
【0075】
〔実施例1〕
2,2-ジフルオロ-3-(4-ビフェニルカルボニル)オキソ-3-(2,2-ジメチル-[1,3]ジオキソラン-4イル)プロピオネート(化合物4)の製造
窒素雰囲気中、四口フラスコに化合物3を290.0g加え、ジクロロメタン2900mlで溶解させた後、ピリジン117.2mlを添加した。10分攪拌した。約0.5時間かけてビフェニルカルボニルクロリド296.8gを徐々に添加し、その過程中、温度が20〜25℃になるように制御されている。その後、室温で、6時間攪拌・反応した。1N塩酸950mlで洗浄し、5%NaHCO3溶液950mlで洗浄し、そして、飽和NaCl溶液950mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、490.0g産物を得た。(3R/3S=3:1)収率:98.9%
【0076】
〔実施例2〕
2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3(4-フェニル)ベンゾエート(化合物6)の製造
アセトニトリル2500mlに2,2-ジフルオロ-3-(4-ビフェニルカルボニル)オキソ-3-(2,2-ジメチル-[1,3]ジオキソラン-4イル)プロピオネート(化合物4)495.0gを加え、攪拌溶解した後、トリフルオロ酢酸14.5mlと蒸留水81mlを添加し、還流するように攪拌昇温し、3時間反応させ、還流に代えて蒸留し(常圧蒸留)、反応液500mlを蒸かすごとに、無水トルエン500mlを追加した。蒸留速度を500ml/15分程度にコントロールした。反応液の温度が100℃に達し、乾燥まで反応液を減圧濃縮させ、次いで、アセトアセテート及びn−ヘキサンで再結晶させ、化合物6を317.5g得た。(3R/3S=3:1)収率:80.0%
【0077】
〔実施例3〕
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)ベンゾエート(化合物7)の製造
窒素雰囲気中、室温で、5L四口フラスコにD-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3(4-フェニル)ベンゾエート(化合物6)200gを加え、ジクロロメタン2000mlで溶解させた後、ピリジン59.5mlを添加し、10分攪拌した。室温で、ベンゾイルクロリド96.0gのジクロロメタン溶液(480ml)を滴下し、滴下終わってから、室温で、6時間攪拌・反応した。1N塩酸950mlで洗浄し、5%NaHCO3溶液950mlで洗浄し、そして、飽和NaCl溶液950mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮し、トルエンとn−ヘキサンで再結晶させ、異性体を除き、白色の固体化合物7を170g得た。(3R:3S=50:1)収率:65.0%
1H-NMR(500MHz,CDCl3)(図3参照):δ8.23(d,J=8.0 Hz,2H),8.13(d,J=7.3 Hz,2H), 7.79-7.35(m,10H), 5.88-5.85(m,1H), 5.12-5.10(m,1H),4.88-4.80(m,2H).
前記と同じ方法で以下の化合物を製造した。
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-フェニルアセチル-3-(4-フェニル)ベンゾエート
(合計収率:48.7%;3R:3S=38:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-フェニルアセテート
(合計収率:46.5%;3R:3S=42:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-ベンゾイル-3-(4-フェニル)フェニルアセテート
(合計収率:43.5%;3R:3S=35:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-5-フェニルアセチル-3-ベンゾエート
(合計収率:45.0%;3R:3S=43:1)
【0078】
〔実施例4〕
1-オキソ-2-デオキシ-2,2-ジフルオロ-フラノース(化合物9)の製造
四口フラスコに化合物3を290g加え、MeCN2700mlで溶解させ、攪拌した後、蒸留水81ml、CF3COOH 14.5mlを添加し、昇温しながら、激しく還流させ、3時間反応させた。還流に代えて蒸留し(常圧蒸留)、反応液500mlを蒸かすごとに、無水トルエン500mlを追加した。反応液の温度が100℃に達するまで蒸留速度を500ml/15分程度にコントロールした。反応が終了した後、乾燥まで反応液を減圧濃縮して、赤褐色油状化合物9を200g得た。収率:100.0%
【0079】
〔実施例5〕
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)ベンゾエート(化合物7)の製造
窒素雰囲気中、化合物9(200g)をジクロロメタン2000mlに溶解させた後、DMAP34.8g、ピリジン257.5mlを添加した。10分攪拌した。約0.5時間かけてビフェニルカルボニルクロリド617.0gを徐々に添加し、その過程中、温度が20〜25℃になるように制御されている。その後、室温で、6時間攪拌・反応した。1N塩酸1800mlで洗浄し、5%NaHCO3溶液1800mlで洗浄し、そして、飽和NaCl溶液1800mlで洗浄した。有機相を分取し、無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、トルエンとn−ヘキサンで再結晶させ、白色の固体化合物7 を358g得た。(3R:3S=45:1)収率:57.0%
1H-NMR(500MHz,CDCl3)(図4参照):
δ8.26-7.28(m,18H),5.84-5.83(m,1H),5.09-5.07(m,1H),4.82-4.79(m,2H).
前記と同じ方法で以下の化合物を製造した。
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-(4-フェニル)フェニルアセテート
(合計収率:52.0%;3R:3S=20:1)
D-エリスロ-2-デオキシ-2,2-ジフルオロ-フラノース-1-オキソ-3,5-ジ-フェニルアセテート
(合計収率:55.5%;3R:3S=46:1)
【0080】
〔実施例6〕
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)の製造
窒素雰囲気中、THF630mlにトリt−ブチルリチウムアルミニウムハイドライド59.8gを加え、-18℃まで冷却し、徐々に化合物7を添加し、保温しながら、2時間攪拌・反応させた。5℃以下で、緩やかに1N HClを2500ml添加し、二塩化メタン(600ml×3)で抽出して、有機相を分取し、10%炭酸ナトリウム、水で洗浄し、有機相を分取して、無水硫酸ナトリウムで乾燥させた。吸引濾過して、乾燥までろ液を減圧濃縮させた。ジクロロメタン950mlを添加し、溶解させ、トリエチルアミン41.7mlを添加し、0〜5℃まで降温し、メチルスルホニルクロリド23.2mlのジクロロメタン溶液(50ml)を滴下し、保温しながら、2時間攪拌・反応させ、1N HCl、10%炭酸ナトリウム及び水で洗浄し、有機相を分取して無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮して、アルコールで精製して、白色の固体100.6g (α:β=2.4:1)を得た(HPLCによる検出の結果は図1を参照)。収率:85.0%
本製品は、カラムクロマトグラフィー(アセトアセテート及びn−ヘキサンを溶離液とする)により精製され、単純なα-アノマーメタンスルホネートとβ-アノマーメタンスルホネートを得た。
α-アノマーメタンスルホネートmp:154.5〜156.5℃
1H-NMR(500MHz,CDCl3)(図5参照):
δ8.17-8.13(m,4H), 7.74-7.64(m,8H), 7.52-7.43 (m,7H), 6.18(d,J=5.5Hz,1H), 5.63(dd,J=3.5,16.5Hz,1H), 4.91-4.73(m,2H),3.22(s,3H),
13C-NMR(125MHz, CDCl3) (図6参照):
δ166.5,165.3, 147.4,146.6, 140.2, 140.0, 131.0, 130.7, 129.4, 129.3, 128.8, 128.6, 128.2, 127.7, 127.6, 127.5, 127.0, 122(q,CF2), 99.9,83.0,71.6,62.9,40.6.
質量スペクトルFAB 609(M+1);元素分析C32H26F2O8S 理論値:C:85.69% H:5.84% F:8.47% 実測値:C:85.71% H:5.80% F:8.49%
前記と同じ方法で以下の化合物を製造する。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート
(収率:79.8%;α:β=2.2:1)
N,N-ジメチル-アミノピリジンを酸スカベンジャとする。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート
(収率:66.5%;α:β=1.5:1)
(2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p−ニトロベンゼンスルホネート
(収率:69.0%;α:β=1.7:1)
【0081】
〔実施例7〕
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)の製造
窒素雰囲気中、THF 600mlにトリt−ブチルリチウムアルミニウムハイドライド59.8gを加え、-18℃に冷却し、化合物7のTHF溶液(450ml)を滴下し、保温しながら、2時間攪拌・反応させた。1N HClを2400ml添加し、反応を終了させ、二塩化メタン(500ml×3)で抽出して、有機相を分取し、10%炭酸ナトリウム、水で洗浄し、有機相を分取して無水硫酸ナトリウムで乾燥させた。吸引濾過して、乾燥までろ液を減圧濃縮させた。ジクロロメタン950mlを添加して溶解させ、トリエチルアミン41.7mlを添加し、0-5℃まで降温し、メチルスルホニルクロリド23.2mlのジクロロメタン(50ml)溶液を滴下し、保温しながら、2時間攪拌・反応させ、1N HCl、10%炭酸ナトリウム及び水で洗浄し、有機相を分取して無水Na2SO4で乾燥し、吸引濾過し、乾燥まで濾液を減圧濃縮し、アルコールで精製して、白色の固体88.2g (α:β=2.5:1)を得た(HPLCによる検出の結果は図2を参照)。収率:83.0%
本製品は、カラムクロマトグラフィー(エチルアセテート及びn−ヘキサンを溶離液とする)により精製され、単純なα-アノマーメタンスルホネートとβ-アノマーメタンスルホネートを得た。
α-アノマーメタンスルホネート mp:136.0〜139.0℃
1H-NMR(500MHz,CDCl3)(図7参照):
δ8.11(d,J=8.0Hz,2H),8.04(d,J=7.5Hz,2H), 7.72-7.41 (m,10H), 7.60-7.57(m,1H), 6.15 (d,J=5.0Hz,1H) , 5.60-5.56(m,1H), 4.79-4.67 (m,2H), 3.18(s,3H)
13C-NMR(125MHz,CDCl3)(図8参照):
δ165.9, 164.9,146.9, 139.6,133.5,130.6, 129.8, 129.1, 129.0, 128.5, 127.4, 127.3, 126.1,122(q,CF2),99.7,82.7, 71.0,62.5,40.2
質量スペクトルFAB 533(M+1);元素分析C26H22F2O8S 理論値:C:83.85% H:5.95% F:10.20% 実測値:C:83.88% H:5.91% F:10.21%
β-アノマーメタンスルホネート mp:162.0〜163.5℃
1H-NMR(500MHz,CDCl3)(図9参照):
δ8.16(d,J=8.0Hz,2H),8.11(d,J=7.5Hz,2H), 7.73-7.45(m,10H), 7.60-7.57(m,1H), 6.07(d,J=7.7Hz,1H),6.01-5.95(m,1H),4.78-4.62(m,2H),3.06(s,3H)
13C-NMR(125MHz, CDCl3) (図10参照):
δ166.2,165.1,147.4,140.0,134.0,131.1,130.0,129.4,129.0,127.7,126.9,122(q,CF2),99.7,80.1,69.7,63.4,40.7
前記と同じ方法で以下の化合物を製造する。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート
(収率:80.2%;α:β=2.1:1)
N,N-ジメチル-アミノピリジンを酸スカベンジャとする。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-ベンジルスルホネート
(収率:69.3%;α:β=1.6:1)
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-ジ-(4-フェニル)ベンゾエート-1-p−ニトロベンゼンスルホネート
(収率:70.5%;α:β=1.7:1)
【0082】
〔実施例8〕
1-(2’-デオキシ-2’,2’-ジフルオロ-3,5-ジ(4-フェニル)ベンゾイル-D-アラビノフラノース-4-アミノピリミジン-2-オン(化合物13)の製造
窒素雰囲気中、シチジン100gをキシレン550mlに加え、そして、硫酸アンモニウム0.5g、ヘキサメチルジシラザン283mlを加え、還流するまで昇温し、3時間反応させ、乾燥まで減圧濃縮し、キシレン600mlを添加し、100℃まで昇温し、5時間以内に2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)(α:β=2.4:1)219gのキシレン溶液(1100ml)を滴下し、滴下終わってから、還流しながら、3時間反応させた(α:β=1:1.8)。40℃まで降温し、水(500ml×3)、5%炭酸水素ナトリウム500ml及び塩水で洗浄し、乾燥まで有機相を濃縮し、アルコールで再結晶して、α-アノマー異性体を除去し、目標化合物119.0g (α:β=1:40)を得た。収率:53.0%
前記と同じ方法で以下の化合物を製造する。
1-(2’-デオキシ-2’,2’-ジフルオロ-3,5-ジ(4-フェニル)ベンゾイル-D-アラビノフラノース-4-アセチルアミノピリミジン-2-オン
(収率:49.5%;α:β=1:35)
【0083】
〔実施例9〕
1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アミノピリミジン-2-オン(化合物13)の製造
窒素雰囲気中、シチジン400gをトルエン2200mlに加え、そして、硫酸アンモニウム2.0g、ヘキサメチルジシラザン1130mlを加え、還流するまで昇温し、3時間反応させ、乾燥まで減圧濃縮し、キシレン2400mlを添加して溶解させ、100℃まで昇温し、3時間以内に2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート(化合物11)(α:β=2.5:1)122gのトルエン溶液(600ml)を滴下し、滴下終わってから、還流しながら、5時間反応させた(α:β=1:2.1)。60℃まで降温し、メタノール780mlを滴下し、10分攪拌した後、2N HClを780ml滴下し、その結果、大量の固体が析出し、保温しながら、20分攪拌し、室温まで降温し、吸収濾過し、2N HCl(1000ml×3)で濾過ケーキを洗浄して、シチジンを除き、さらにアルコールで精製して、α-アノマー異性体を除去し、目標化合物78.7g (α:β=1:35)を得た。収率:55.0%
β-アノマー化合物13 mp:247.5-248.5°C。
1H-NMR(500MHz,CDCl3)(図11参照):
δ8.13(d,J=7.5Hz,2H), 7.97(d,J=7.5Hz,2H) , 7.87(d,J=7.5Hz,2H) , 7.77 (d,J=7.3Hz,2H) , 7.66(d,J=6.5Hz,2H) , 7.55-7.44(m,6H) , 6.40(s,br,1H), 5.86(s,br.1H) , 4.78-4.71(m,3H).
前記と同じ方法で以下の化合物を製造する。
1-(2’-デオキシ-2’,2’-ジフルオロ-5-ベンゾイル-3-(4-フェニル)ベンゾエート-D-アラビノフラノース-4-アセチルアミノピリミジン-2-オン
(収率:51.5%;α:β=1:40)
【0084】
〔実施例10〕
2’-デオキシ-2’,2’-ジフルオロシチジン(化合物2)の製造
窒素雰囲気中、化合物13をメタノール/アンモニア溶液に加え、室温で、攪拌して、一晩反応させた。乾燥まで減圧濃縮し、水で溶解させ、エチルアセテートで抽出し、水相を濃縮して、ゲムシタビン18.7gを得た。(純度: 97.0% , ee 99.5%) 収率:88.5%
【0085】
〔実施例11〕
塩酸ゲムシタビンの製造
化合物2をイソプロピルアルコールに溶解させ、0〜5℃まで降温し、pHが2になるように濃塩酸を滴下して、保温しながら、2時間結晶を育成し、吸引濾過し、イソプロピルアルコール100mlで濾過ケーキを洗浄して、ゲムシタビン塩酸塩51.2gを得た。(純度 : 99.8%) 収率:90.0%
【特許請求の範囲】
【請求項1】
以下の反応を含む、ゲムシタビン又はその塩酸塩の合成プロセス。
【化1】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【化2】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
条件としては、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲンで置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基である、
G4とG5は、それぞれ独立に、C1〜C7トリアルキルシリル基;t-ブトキシカルボニル基、カルボベンゾキシ基又は9-フルオレニルメトキシカルボニル基;或いは、ホルミル基、アセチル基、プロピオニル基、イソプロピオニル基、ブチリル基、イソブチリル基、ピバロイルから選ばれる基である。]
【請求項2】
前記反応が、α-アノマーに富んだ式11の化合物と式12の化合物を反応して、β-アノマーに富んだ式13の化合物を得るものである請求項1に記載のプロセス。
【請求項3】
G2がビフェニルカルボニル基である請求項1に記載のプロセス。
【請求項4】
G1とG2がいずれもビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項3に記載のプロセス。
【請求項5】
前記反応が、式12の化合物をα-アノマーに富んだ式11の化合物に対して所定のモル比で有機溶媒に溶解させ、40〜300℃まで加熱し、20時間以内に、予め有機溶媒に溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、保温しながら10分〜20時間反応させて、β-アノマーに富んだ式13の化合物を得るものである請求項1〜4いずれかに記載のプロセス。
【請求項6】
前記有機溶媒が、1,2-ジクロロエタン、トルエン、キシレン、置換ベンゼン、アニソール、ジフェニルエーテル、又は置換ジフェニルエーテルからなる1種又は2種以上の混合物であり、反応温度が110〜130℃である請求項5に記載のプロセス。
【請求項7】
さらに以下の脱保護反応を含む、請求項1〜4いずれかに記載のプロセス。
【化3】
[ただし、反応条件は、メタノール/アンモニア溶液に行うものである。]
【請求項8】
さらにゲムシタビンと塩酸を反応して塩酸ゲムシタビンを生成する請求項6に記載のプロセス。
【請求項9】
そのα-アノマー又はβ-アノマー、或いはこれらの混合物を含む下記式で表される化合物。
【化4】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【化5】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基からなる群から選ばれる基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲンで置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基である。]
【請求項10】
G1とG2が、それぞれ独立に、ベンソイル基、フェニルアセチル基、ビフェニルカルボニル基、ビフェニルアセチル基である、
なお、G1とG2のうち、少なくとも一つのR2がフェニル基又は置換フェニル基から選ばれる基である、
G3はメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基から選ばれる基である請求項9に記載の化合物。
【請求項11】
G1とG2がいずれもビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項9に記載の化合物。
【請求項12】
G1がベンゾイル基であり、G2がビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項9に記載の化合物。
【請求項13】
以下の化合物から選ばれた請求項9〜12のいずれかに記載の化合物。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-ベンジルスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート。
【請求項14】
以下の合成ルート1を経て合成できる請求項9に記載の化合物の製造方法。
【化6】
[まず、酸スカベンジャの存在下で、有機溶媒に式3の化合物とアシル化剤を反応させ、水酸基が保護された式4の化合物を製造し、アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-アミノピリジンが好ましい。
次は、トリフルオロ酢酸と水の存在下で、アセトニトリルに式4の化合物を加水分解させて、式5の化合物を生成し、その後、還流により式5の化合物を脱水してラクトン化し、式6の化合物を得た。
次いで、酸スカベンジャの存在下で、有機溶媒に式6の化合物とアシル化剤を反応させ、5’位の水酸基が保護された式7の化合物を製造し、エチルアセテートとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
最後に、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。そのスルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-ニトロベンゼンスルホニル基である。
ただし、G1、G2及びG3の定義は請求項9に説明した通りである。]
【請求項15】
式11の化合物の水酸基保護基が同じである場合、以下の合成ルート2を経て合成できる請求項9に記載の化合物の製造方法。
【化7】
[まず、酸スカベンジャの存在下で、有機溶媒に式9の化合物とアシル化剤を反応させ、水酸基が保護された式7の化合物を製造し、トルエンとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
次いで、一種類の還元剤の存在下で式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ式11の化合物を得た。前記スルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれる基である。
ここで、G1とG3は同じ基であり、且つG1、G2とG3の定義は請求項9に説明した通りである。]
【請求項1】
以下の反応を含む、ゲムシタビン又はその塩酸塩の合成プロセス。
【化1】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【化2】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基から選ばれる基である、
条件としては、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲンで置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基である、
G4とG5は、それぞれ独立に、C1〜C7トリアルキルシリル基;t-ブトキシカルボニル基、カルボベンゾキシ基又は9-フルオレニルメトキシカルボニル基;或いは、ホルミル基、アセチル基、プロピオニル基、イソプロピオニル基、ブチリル基、イソブチリル基、ピバロイルから選ばれる基である。]
【請求項2】
前記反応が、α-アノマーに富んだ式11の化合物と式12の化合物を反応して、β-アノマーに富んだ式13の化合物を得るものである請求項1に記載のプロセス。
【請求項3】
G2がビフェニルカルボニル基である請求項1に記載のプロセス。
【請求項4】
G1とG2がいずれもビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項3に記載のプロセス。
【請求項5】
前記反応が、式12の化合物をα-アノマーに富んだ式11の化合物に対して所定のモル比で有機溶媒に溶解させ、40〜300℃まで加熱し、20時間以内に、予め有機溶媒に溶解しておいたα-アノマーに富んだ式11の化合物溶液を滴下し、滴下終わってから、保温しながら10分〜20時間反応させて、β-アノマーに富んだ式13の化合物を得るものである請求項1〜4いずれかに記載のプロセス。
【請求項6】
前記有機溶媒が、1,2-ジクロロエタン、トルエン、キシレン、置換ベンゼン、アニソール、ジフェニルエーテル、又は置換ジフェニルエーテルからなる1種又は2種以上の混合物であり、反応温度が110〜130℃である請求項5に記載のプロセス。
【請求項7】
さらに以下の脱保護反応を含む、請求項1〜4いずれかに記載のプロセス。
【化3】
[ただし、反応条件は、メタノール/アンモニア溶液に行うものである。]
【請求項8】
さらにゲムシタビンと塩酸を反応して塩酸ゲムシタビンを生成する請求項6に記載のプロセス。
【請求項9】
そのα-アノマー又はβ-アノマー、或いはこれらの混合物を含む下記式で表される化合物。
【化4】
[ただし、置換基G1とG2は、それぞれ独立に、以下の一般式で定義される基である、
【化5】
式中、R1はC1〜C3アルキル基から選ばれる基、又は不存在であり、
R2は水素、C1〜C4アルキル基、フェニル基又は置換フェニル基からなる群から選ばれる基である、
なお、G1とG2のうち、少なくとも一つのR2はフェニル基又は置換フェニル基から選ばれる基である、
ここに述べた置換フェニル基はC1〜C4アルキル基、又はハロゲンで置換されたフェニル基である、
G3はC1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基又は置換アリールスルホニル基から選ばれる基である。]
【請求項10】
G1とG2が、それぞれ独立に、ベンソイル基、フェニルアセチル基、ビフェニルカルボニル基、ビフェニルアセチル基である、
なお、G1とG2のうち、少なくとも一つのR2がフェニル基又は置換フェニル基から選ばれる基である、
G3はメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-トルエンスルホニル基、p-ニトロベンゼンスルホニル基から選ばれる基である請求項9に記載の化合物。
【請求項11】
G1とG2がいずれもビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項9に記載の化合物。
【請求項12】
G1がベンゾイル基であり、G2がビフェニルカルボニル基であり、G3がメチルスルホニル基である請求項9に記載の化合物。
【請求項13】
以下の化合物から選ばれた請求項9〜12のいずれかに記載の化合物。
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-ベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-3,5-ジ-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1α-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
1β-2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-メタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-エタンスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-ベンジルスルホネート;
2-デオキシ-2,2-ジフルオロ-D-アラビノフラノース-5-ベンゾエート-3-(4-フェニル)ベンゾエート-1-p-ニトロベンゼンスルホネート。
【請求項14】
以下の合成ルート1を経て合成できる請求項9に記載の化合物の製造方法。
【化6】
[まず、酸スカベンジャの存在下で、有機溶媒に式3の化合物とアシル化剤を反応させ、水酸基が保護された式4の化合物を製造し、アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-アミノピリジンが好ましい。
次は、トリフルオロ酢酸と水の存在下で、アセトニトリルに式4の化合物を加水分解させて、式5の化合物を生成し、その後、還流により式5の化合物を脱水してラクトン化し、式6の化合物を得た。
次いで、酸スカベンジャの存在下で、有機溶媒に式6の化合物とアシル化剤を反応させ、5’位の水酸基が保護された式7の化合物を製造し、エチルアセテートとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
最後に、一種類の還元剤の存在下で、式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ(α:βは2 - 2.5:1に達する)式11の化合物を得た。そのスルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれる基であり、好ましくはメチルスルホニル基、エチルスルホニル基、ベンジルスルホニル基、p-ニトロベンゼンスルホニル基である。
ただし、G1、G2及びG3の定義は請求項9に説明した通りである。]
【請求項15】
式11の化合物の水酸基保護基が同じである場合、以下の合成ルート2を経て合成できる請求項9に記載の化合物の製造方法。
【化7】
[まず、酸スカベンジャの存在下で、有機溶媒に式9の化合物とアシル化剤を反応させ、水酸基が保護された式7の化合物を製造し、トルエンとn-ヘキサンにより精製して、単純なエリスロ構造を得た。アシル化剤は、ベンゾイルクロリド、フェニルアセチルクロリド、ビフェニルカルボニルクロリド及びビフェニルアセチルクロリドが好ましく、酸スカベンジャは、ピリジン、トリエチルアミン及びN,N-ジメチル-4-アミノピリジンが好ましい。
次いで、一種類の還元剤の存在下で式7の化合物を還元して、式10の化合物を得た後、有機溶媒に式10の化合物とスルホン化剤及び酸スカベンジャを反応して、α-アノマーに富んだ式11の化合物を得た。前記スルホン化剤は、C1〜C4アルキルスルホニル基、アリールスルホニル基、置換アルキルスルホニル基と置換アリールスルホニル基から選ばれる基である。
ここで、G1とG3は同じ基であり、且つG1、G2とG3の定義は請求項9に説明した通りである。]
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2010−528060(P2010−528060A)
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願番号】特願2010−509653(P2010−509653)
【出願日】平成19年9月7日(2007.9.7)
【国際出願番号】PCT/CN2007/002672
【国際公開番号】WO2008/144970
【国際公開日】平成20年12月4日(2008.12.4)
【出願人】(509330138)ナンジン キャベンディッシュ バイオ−エンジニアリング テクノロジー カンパニー,リミテッド (4)
【氏名又は名称原語表記】Nanjing Cavendish Bio−Engineering Technology Co.,Ltd.
【住所又は居所原語表記】No.6 Maiyue Road,Maigaoqiao Pioneering Park,Qixia District,Nanjing,Jiangsu 210028,China
【出願人】(509330149)
【氏名又は名称原語表記】XU,Yongxiang
【住所又は居所原語表記】Room 105,No.9,Yijing Garden,Gulou Digest,Nanjing,Jiangsu 210001,China
【Fターム(参考)】
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願日】平成19年9月7日(2007.9.7)
【国際出願番号】PCT/CN2007/002672
【国際公開番号】WO2008/144970
【国際公開日】平成20年12月4日(2008.12.4)
【出願人】(509330138)ナンジン キャベンディッシュ バイオ−エンジニアリング テクノロジー カンパニー,リミテッド (4)
【氏名又は名称原語表記】Nanjing Cavendish Bio−Engineering Technology Co.,Ltd.
【住所又は居所原語表記】No.6 Maiyue Road,Maigaoqiao Pioneering Park,Qixia District,Nanjing,Jiangsu 210028,China
【出願人】(509330149)
【氏名又は名称原語表記】XU,Yongxiang
【住所又は居所原語表記】Room 105,No.9,Yijing Garden,Gulou Digest,Nanjing,Jiangsu 210001,China
【Fターム(参考)】
[ Back to top ]