
コアギュロゲン原料及びその製造方法、それを用いた生物由来の生理活性物質の測定方法及び測定装置
【課題】LAL試薬、あるいは、生物由来の生理活性物質に汚染されたLAL試薬等におけるコアギュロゲンの機能を維持したまま凝固酵素活性を不可逆的に不活性化し、試薬に利用可能なコアギュロゲン原料を取得する技術を提供する。
【解決手段】LAL試薬をある所定温度で所定時間に亘って加熱処理することにより、LAL試薬中の酵素活性のみを不可逆的に失活させる。その際、活性化した凝固酵素により加水分解されコアギュリンとなってゲル化や凝集反応を惹起するという、コアギュロゲン本来の活性は維持させる。
【解決手段】LAL試薬をある所定温度で所定時間に亘って加熱処理することにより、LAL試薬中の酵素活性のみを不可逆的に失活させる。その際、活性化した凝固酵素により加水分解されコアギュリンとなってゲル化や凝集反応を惹起するという、コアギュロゲン本来の活性は維持させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エンドトキシンやβ−D−グルカンなどの生物由来の生理活性物質の検出または濃度測定を、迅速または高感度に計測するための試薬、その製造法、ならびにそれを用いた測定方法及び測定装置に関する。
【背景技術】
【0002】
エンドトキシンはグラム陰性菌の細胞壁に存在するリポ多糖であり、最も代表的な発熱性物質である。このエンドトキシンに汚染された輸液、注射薬剤、血液などが人体に入ると、発熱やショックなどの重篤な副作用を惹起するおそれがある。このため、上記の薬剤などは、エンドトキシンにより汚染されることが無いように管理することが義務付けられている。
【0003】
ところで、カブトガニの血球抽出物(以下、「LAL : Limulus amoebocyte lysate」ともいう。)の中には、エンドトキシンによって活性化される酵素であるセリンプロテアーゼが存在する。そして、LALとエンドトキシンとが反応する際には、エンドトキシンの量に応じて活性化されたセリンプロテアーゼによる酵素カスケードによって、LAL中に存在するコアギュロゲンがコアギュリンへと加水分解されて会合し、不溶性のゲルが生成される。このLALの特性を用いて、エンドトキシンを高感度に検出することが可能である。
【0004】
また、β−D−グルカンは真菌に特徴的な細胞膜を構成しているポリサッカライド(多糖体)である。β−D−グルカンを測定することによりカンジダやスペルギルス、クリプトコッカスのような一般の臨床でよく見られる真菌のみならず、稀な真菌も含む広範囲で真菌感染症のスクリーニングなどに有効である。
【0005】
β−D−グルカンの測定においても、カブトガニの血球抽出成分がβ−D−グルカンによって凝固(ゲル凝固)する特性を利用して、β−D−グルカンを高感度に検出することが可能である。
【0006】
このエンドトキシンやβ−D−グルカンなどの、カブトガニの血球抽出成分によって検出可能な生物由来の生理活性物質(以下、所定生理活性物質ともいう)の検出または濃度測定を行う方法としては、所定生理活性物質の検出または濃度測定(以下、単純に「所定生理活性物質の測定」ともいう。)をすべき試料とLALを元に製造された試薬(LAL試薬)とを混和した混和液を静置し、一定時間後に容器を転倒させて、試料の垂れ落ちの有無によりゲル化したかどうかを判定し、試料に一定濃度以上のエンドトキシンが含まれるか否かを調べる半定量的なゲル化法がある。また、LAL試薬と所定生理活性物質との反応によるゲルの生成に伴う試料の濁りを経時的に計測して解析する比濁法や、酵素カスケードにより加水分解されて発色する合成基質を用いる比色法などがある。
【0007】
上記の比濁法によって所定生理活性物質の測定を行う場合には、乾熱滅菌処理されたガラス製測定セルに測定試料とLAL試薬との混和液を生成させる。そして、混和液のゲル化を外部から光学的に測定する。しかしながら、比濁法においては特に所定生理活性物質の濃度が低い試料において混和液がゲル化するまでに非常に多くの時間を要する場合がある。これに対し、所定生理活性物質の短時間測定が可能な方法が求められている。測定試料とLAL試薬との混和液を例えば磁性攪拌子を用いて攪拌することにより、ゲル微粒子を生成せしめ、ゲル粒子により散乱されるレーザー光の強度、あるいは、混和液を透過す
る光の強度から、試料中の所定生理活性物質の存在を短時間で測定できるレーザー散乱粒子計測法等が提案されている。
【0008】
LAL試薬はカブトガニの血球抽出物を主原料として製造される。このため、製造上ある確率でエンドトキシンやβ−D−グルカンが混入してしまい、試薬として利用できない廃棄物となる可能性がある。また、エンドトキシン、あるいは、β−D−グルカンのいずれの測定法も、測定試薬の原料が有限資源であるカブトガニの血球抽出物であるため、試薬の使用量を減少させ、あるいは、試薬製造上のロスを低減させるといった取り組みが必要になる。
【0009】
試薬の使用量を減少させる試みとしては、単純に試料容量を減少させることはもちろん、測定感度を高めて結果的に測定回数を低減することも効果的である。一方、製造上のロスを減らすにはエンドトキシンやβ−D−グルカンによる汚染をいかに抑制するかということが重要である。さらに、製造中に試薬として使用できなくなった原料からエンドトキシンやβ−D−グルカンを除去して再利用したり、補助的な試薬の添加物として利用したりすることなども考えねばならない。補助的な試薬の添加物として利用する場合、試薬の粘性の調整剤としての役割や、ゲル化・凝集での中心的役割を担うコアギュロゲン原料としての利用などが考えられる。
【0010】
しかしながら、例えばエンドトキシンに汚染されて使用できなくなった原料中のエンドトキシンを除去するだけでは既に活性化してしまった酵素群を除去することは不可能である。原料中の酵素活性のみを不活性化してコアギュロゲンの機能、すなわち、活性化された凝固酵素により加水分解されてコアギュリンとなり、ゲル化・凝集反応を生じさせる機能を維持した原料の製造方法は報告されていなかった。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特許第2667695号公報
【特許文献2】特開2004−061314号公報
【特許文献3】特開平10−293129号公報
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は上述の問題点に鑑みて案出されたものであり、その目的とするところは、LAL試薬、あるいは、生物由来の生理活性物質に汚染されたLAL試薬等におけるコアギュロゲンの機能を維持したまま凝固酵素活性を不可逆的に不活性化し、試薬に利用可能なコアギュロゲン原料を取得する技術を提供することである。
【課題を解決するための手段】
【0013】
上記の課題を解決するための本発明は、若干の所定生理活性物質の混入を許容するLAL試薬をある温度条件下において加熱処理することにより、LAL試薬中の酵素活性のみを不可逆的に失活させたことを最大の特徴とする。本発明においては、その際、活性化した凝固酵素により加水分解されコアギュリンとなってゲル化・凝集反応を惹起するという、コアギュロゲン本来の活性は維持させる。
【0014】
より詳しくは、本発明は、所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料の製造方法であって、
LALを所定温度で所定時間に亘り加熱処理することで、該LAL中の前記酵素群の少なくとも一部を失活させるとともにコアギュロゲンの活性は維持させることを特徴とする。
【0015】
これによれば、LAL中の酵素カスケードを構成する酵素群の少なくとも一部を失活させることで酵素カスケードの発生を抑止し、エンドトキシンやβ−D−グルカンなどの汚染によっても加水分解してコアギュリンになりづらいコアギュロゲン原料を取得することが可能となる。
【0016】
また、これによれば、エンドトキシンやβ−D−グルカンなどの汚染により使用不可となったLAL試薬のコアギュロゲンを、機能を維持したまま取得することができるので、カブトガニの血球から得られる有限の資源をより有効に活用することが可能となる。
【0017】
ここで、所定の酵素群とは例えばLAL中のC因子、B因子、G因子、凝固酵素前駆体など、エンドトキシンやβ−D−グルカンなどにより酵素カスケードを生じ最終的にコアギュロゲンを加水分解する凝固酵素を生じる原因となる酵素類を意味している。すなわち、LAL中の全ての酵素を失活させずとも、少なくとも一部の酵素が失活し、エンドトキシンやβ−D−グルカンなどによっても、最終的にコアギュロゲンを加水分解する凝固酵素が生じないようになれば本発明の目的は達成できる。
【0018】
また、上記において生物由来の所定の生理活性物質とは、LAL中の所定の酵素群を活性化させ酵素カスケードを生じせしめ、最終的にコアギュロゲンを加水分解する凝固酵素を生じせしめる特性を有する生理活性物質を意味しており、例としては上述のとおり、エンドトキシンやβ−D−グルカンが挙げられる。但し、同等の特性を有する他の生理活性物質をも含む趣旨であり、上述の2物質に限定するものではない。
【0019】
なお、上記においてLALが溶液として供給される場合には、所定温度は60℃以上としてもよい。ここで、発明者の鋭意研究により、溶液としてのLAL試薬を60℃以上に加熱処理することで、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、LALが溶液として供給される場合に所定温度を60℃以上とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならないコアギュロゲン原料を取得することが可能となる。
【0020】
また、上記においてLALが溶液として供給される場合には、所定温度は80℃以下としてもよい。ここで、発明者の鋭意研究により、LAL試薬を80℃より高い温度まで加熱処理して酵素活性を失活させた場合には、LAL中の蛋白質の変性により試薬が白濁してしまうことが分かってきた。そうすると、所定生理活性物質の検出や濃度測定を、試薬の透過光や散乱光によって光学的な手法によって行う目的には適さなくなってしまうおそれがある。従って本発明においては、LALが溶液として供給される場合には所定温度を80℃以下とすることで、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となる。
【0021】
さらに、上記においてLALが溶液として供給される場合には、所定時間は10分以上8時間以下としてもよい。ここで、溶液としてのLAL試薬を60℃以上に加熱処理した場合、加熱時間は10分程度で、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、所定時間を10分以上とすることで、広い範囲の加熱温度でセリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができる。また、加熱時間があまりに長い場合には、低温でもLAL中の蛋白質が変性して白濁が生じるおそれがある。従って本発明では、所定時間を10分以上8時間以下
とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならず、光学的な測定に適したコアギュロゲン原料を取得することが可能となる。
【0022】
さらに、上記においてLALが凍結乾燥体として供給される場合には、所定温度は100℃以上250℃以下としてもよい。ここで、LALが凍結乾燥体として供給される場合には、溶液として供給される場合と比較してより高い温度で加熱処理する必要があることが分かってきた。この場合には、LAL試薬を100℃以上250℃以下とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならず、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となる。
【0023】
また、この場合には、所定時間は300分以上としてもよい。ここで、LALが凍結乾燥体として供給される場合には、LAL試薬を120℃で300分に亘って加熱することで、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、所定時間を300分以上とすることで、より広い範囲の加熱温度でより確実に、エンドトキシンやβ−D−グルカンなどの汚染によっても加水分解してコアギュリンにならないコアギュロゲン原料を取得することが可能となる。
【0024】
また、本発明は、所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料であって、
LALを所定温度で所定時間に亘り加熱処理することで、前記LAL中の前記酵素群の少なくとも一部を失活させたことを特徴とするコアギュロゲン原料であってもよい。
【0025】
このコアギュロゲン原料においては、所定生理活性物質との反応で酵素カスケードを生じる酵素群が失活されているので、少量の所定生理活性物質による汚染によりコアギュロゲンが加水分解されてコアギュリンとなることを抑止でき、よりハンドリングの良いコアギュロゲン原料を得ることができる。なお、この場合においても、LALが溶液として供給される場合には、所定温度は60℃以上としてもよい。また、80℃以下としてもよい。さらに、所定時間は10分以上8時間以下としてもよい。一方、LALが凍結乾燥体として供給される場合には、所定温度は100℃以上250℃以下としてもよい。また、所定時間は300分以上としてもよい。
【0026】
また、本発明においては、上記で得られたコアギュロゲン原料と、加熱処理されていないLALとを混合することで、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたことを特徴とする、前記生理活性物質の検出または前記生理活性物質の濃度測定に使用されるLAL試薬としてもよい。
【0027】
すなわち、所定の酵素群を失活させたコアギュリン原料を、熱処理をしていないLALに加えることで、通常よりコアギュロゲンの濃度の高いLAL試薬を調製することができる。これにより、所定生理活性物質との反応により活性化された酵素群によって加水分解されコアギュリンとなるコアギュロゲンの量を相対的に増加させることができる。従って、所定生理活性物質によるLAL試薬のゲル化をより顕著にすることができ、所定生理活性物質の測定をより高感度にすることができる。
【0028】
また、本発明においては、上記で得られたコアギュロゲン原料におけるコアギュロゲンを、該コアギュロゲンより大径の多数の微粒子の表面に結合または吸着させて調製された
ことを特徴とするエンドトキシンの測定用のコアギュロゲン結合マイクロビーズとしてもよい。
【0029】
ここで、コアギュロゲンを例えば樹脂製の微粒子の上に結合または吸着させた状態のLALにエンドトキシンなどの所定生理活性物質を作用させると、LAL単体に所定生理活性物質を作用させた場合と比較して、より大きな凝集塊を早期に生成することが分かってきた。これは、微粒子上のコアギュロゲンがLAL中の酵素カスケードにより加水分解されコアギュリンとなり、これらが微粒子同士を会合させることによる。また、この凝集反応は試料の濁りや色の影響を受けづらいこと、さらには、この凝集反応はLAL単体で惹起される凝集反応よりも、非常に強大であることも明確になってきている。
【0030】
本発明においては、この現象を利用し、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させた試薬(以下、「コアギュロゲン結合マイクロビーズ」ともいう。)を調製した。このコアギュロゲン結合マイクロビーズとLALとを混合させた試薬にエンドトキシンを含む試料を作用させることで、LAL中にもともと存在したコアギュロゲンがコアギュリンとなり凝集するとともに、微粒子の上に結合または吸着されたコアギュロゲンがコアギュリンとなり微粒子同士を会合させてより大きな凝集塊を早期に生成させる。これによれば、LAL試薬と試料との混和液におけるゲル粒子の生成を大幅に促進することができる。その結果、所定生理活性物質の検出または濃度の測定を、より迅速且つ、高感度にすることができる。
【0031】
なお、LAL中に含まれるコアギュロゲンを微粒子の上に結合または吸着させる際には、まず、蛋白質を結合、吸着することが可能な官能基を微粒子の表面に存在せしめる。そして、従来の方法によれば、その状態でLALを作用させて、コアギュロゲンを微粒子表面に化学的に結合させるか、静電的、親水的、疎水的に吸着させる。その際のLAL中のは長時間に及ぶため、従来の方法では、微粒子や使用する試薬類、水などに微量に混在する所定生理活性物質が蛋白質における酵素群と反応し、コアギュロゲンをコアギュリンに加水分解して、コアギュロゲンと微粒子の結合・吸着反応中に微粒子の凝集が開始してしまうことが考えられる。そしてこの場合には、酵素の活性化によって試薬中のコアギュロゲンが加水分解されて消費されてしまうおそれがある。
【0032】
このような不都合に対し、従来は、コアギュロゲンを微粒子の表面に結合または吸着させる際には、LAL試薬と微粒子の懸濁液とを混合するとともに、LAL中の酵素群と所定生理活性物質との反応を抑制する抑制剤を添加する必要があり、コアギュロゲンを微粒子の表面に結合または吸着させる作業が複雑でコスト的にも不利な状況であった。これに対し本発明では、コアギュロゲン原料の調製過程でLAL中の酵素群を失活させるため、抑制剤を添加する工程を省略することが可能である。これにより、より簡単にまたはより低廉なコストで、コアギュロゲンを微粒子の上に結合または吸着させた試薬を調製することが可能である。
【0033】
また、本発明は、上記のコアギュロゲン原料と、加熱処理されていないLALとを混合することによって、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたLAL試薬と、前記所定の生理活性物質を含む試料とを混和させ、
前記所定の生理活性物質によって前記LAL試薬中の酵素群が活性化されて生じる酵素カスケードによって濃度が高められた前記コアギュロゲンがコアギュリンに加水分解されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法であってもよい。
【0034】
この生物由来の生理活性物質の測定方法によれば、より高い濃度のコアギュロゲンを用いて、所定生理活性物質の測定を行うことができる。従って、所定生理活性物質とLAL
との反応によるゲル化をより顕著にすることができ、所定生理活性物質の測定をより高感度にすることができる。
【0035】
また、本発明は、上記のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記加熱処理されていないLALにおける酵素群が活性化されて生じる酵素カスケードによって前記微粒子の表面に結合または吸着したコアギュロゲンがコアギュリンに加水分解され、前記微粒子同士が架橋されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法であってもよい。
【0036】
この所定生理活性物質の測定方法によれば、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させたコアギュロゲン結合マイクロビーズと加熱処理されていないLALとを混合させた試薬にエンドトキシンを含む試料を作用させる。このことで、LAL中にもともと存在したコアギュロゲンがコアギュリンとなり凝集するとともに、微粒子の上に結合または吸着されたコアギュロゲンがコアギュリンとなり微粒子同士を会合させてより大きな凝集塊を早期に生成させる。これによれば、LAL試薬と試料との混和液におけるゲル粒子の生成を大幅に促進することができる。その結果、所定生理活性物質の検出または濃度の測定を、より迅速且つ、高感度にすることができる。
【0037】
また、本発明は、上記のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料との混和液を光の入射可能に保持し、前記混和液における反応を進行させる混和液保持手段と、
前記混和液保持手段中の前記混和液を攪拌する攪拌手段と、
前記混和液保持手段中の混和液に光を入射する光入射手段と、
前記入射光の前記混和液における透過光を受光し電気信号に変換する受光手段と、
前記受光手段において変換された電気信号から取得される前記混和液の透過率より前記試料中の前記生理活性物質の濃度を導出する導出手段と、
を備えることを特徴とする生物由来の生理活性物質の攪拌比濁測定装置であってもよい。
【0038】
本発明においては、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させたコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、所定生理活性物質を含む試料とを混和させ混和液保持手段に入れる。そして、この混和液を攪拌手段により攪拌することで、所定生理活性物質で活性化された凝固酵素によるコアギュロゲンの加水分解及び、加水分解で得られたコアギュリンによる微粒子の会合を促進する。
【0039】
そして、光入射手段から入射された光のうち、上記混和液を透過して受光素子に届いた光の強度を測定する。さらに、導出手段においては上記混和液の透過率より所定生理活性物質の濃度が導出される。
【0040】
ここにおいて、微粒子の上に結合または吸着されたコアギュロゲンの加水分解が進みコアギュリンが生成されると、コアギュリンが微粒子同士を会合させてより大きな凝集塊を早期に生成させる。ここで、コアギュロゲン結合マイクロビーズと加熱処理されていないLALと所定生理活性物質を含む試料との混和液は、混和時点では、多量のマイクロビーズの微粒子が分散しているために強く濁っている。そして、コアギュリンの生成による微粒子の会合が進行すると微粒子の濃度が急激に減少するために混和液の透過率が急激に上昇する。
【0041】
本発明における攪拌比濁測定装置では、通常の比濁法に係る測定装置と異なり、コアギュリンの生成に伴う微粒子の会合に起因する透過率の急激な上昇を測定するため、混和液を攪拌することによる反応の促進効果と相まって、測定時間を大幅に短縮できるとともに、非常に高感度に所定生理活性物質の測定を行うことができる。
【0042】
なお、上記した本発明の課題を解決する手段については、可能なかぎり組み合わせて用いることができる。
【発明の効果】
【0043】
本発明にあっては、LAL試薬、あるいは、生物由来の生理活性物質に汚染されたLAL試薬等におけるコアギュロゲンの機能を維持したまま凝固酵素活性を不可逆的に不活性化し、試薬に利用可能なコアギュロゲン原料を取得することが可能となる。
【図面の簡単な説明】
【0044】
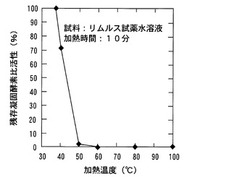
【図1】本発明の実施例におけるLAL試薬水溶液の加熱温度と残存凝固酵素比活性との関係を示すグラフである。
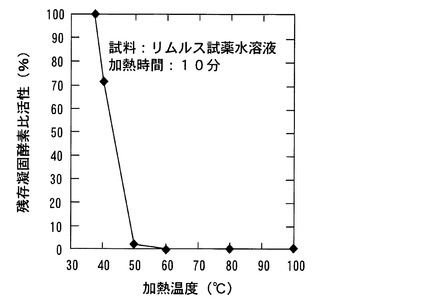
【図2】本発明の実施例におけるLAL試薬凍結乾燥体の加熱時間と残存凝固酵素比活性との関係を示すグラフである。
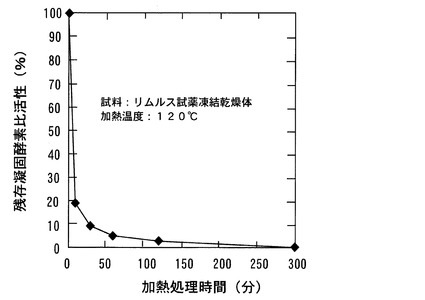
【図3】本発明の実施例における比濁計測装置の概略構成を示す図である。
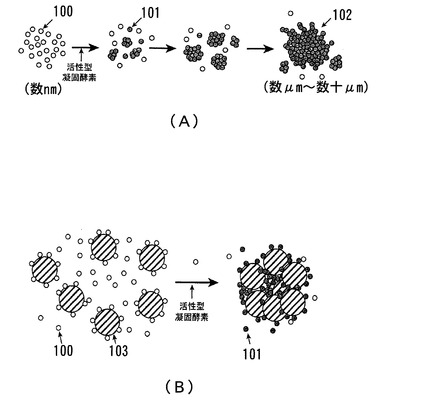
【図4】活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程とコアギュロゲン結合マイクロビーズの凝集過程とを説明するための図である。
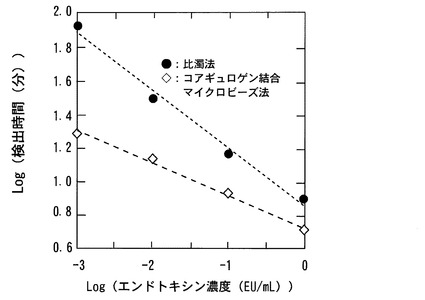
【図5】コアギュロゲン結合マイクロビーズ法と比濁法におけるエンドトキシン用量反応を比較したグラフである。
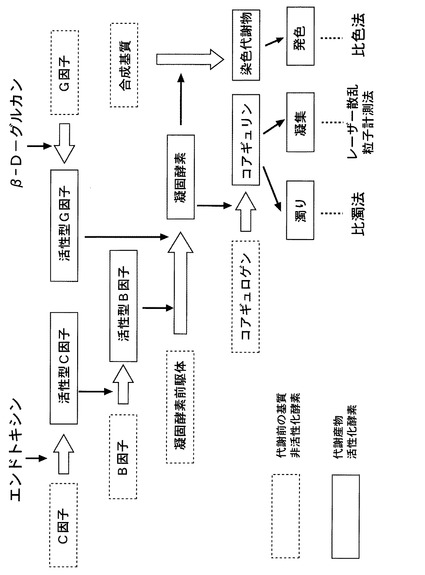
【図6】エンドトキシンまたはβ―D−グルカンにより、LALがゲル化する過程及び、その検出方法について説明するための概略図である。
【発明を実施するための形態】
【0045】
LALとエンドトキシンとが反応してゲルが生成される過程はよく調べられている。すなわち、図6に示すように、エンドトキシンがLAL中のセリンプロテアーゼであるC因子に結合すると、C因子は活性化して活性型C因子となる、活性型C因子はLAL中の別のセリンプロテアーゼであるB因子を加水分解して活性化させ活性化B因子とする。この活性化B因子は直ちにLAL中の凝固酵素の前駆体を加水分解して凝固酵素とし、さらに、この凝固酵素がLAL中のコアギュロゲンを加水分解してコアギュリンを生成する。そして、生成したコアギュリンが互いに会合して不溶性のゲルをさらに生成し、LAL全体がこれに巻き込まれてゲル化すると考えられている。
【0046】
また、同様にβ−D−グルカンがLAL中のG因子に結合すると、G因子は活性化して活性型G因子となる、活性型G因子はLAL中の凝固酵素の前駆体を加水分解して凝固酵素とする。その結果、エンドトキシンとLALとの反応と同様、コアギュリンが生成され、生成したコアギュリンが互いに会合して不溶性のゲルをさらに生成する。
【0047】
この一連の反応は哺乳動物に見られるクリスマス因子やトロンビンなどのセリンプロテアーゼを介したフィブリンゲルの生成過程に類似している。このような酵素カスケード反応はごく少量の活性化因子であっても、その後のカスケードを連鎖して活性化していくために非常に強い増幅作用を有する。従って、LALを用いた所定生理活性物質の測定法によれば、サブピコグラム/mLオーダーのきわめて微量の所定生理活性物質を検出することが可能になっている。
【0048】
所定生理活性物質を定量することが可能な測定法としては前述のように比濁法、ならびに、レーザー光散乱粒子計測法が挙げられる。これらの測定法はこのLALの酵素カスケ
ード反応によって生成されるコアギュリンの会合物を前者は試料の濁りとして、後者は系内に生成されるゲルの微粒子として検出することで、高感度な測定を可能にしている。
【0049】
特にレーザー光散乱粒子計測法では、系内に生成されたゲルの微粒子を直接測定するため、比濁法よりも高感度であり、且つ、一般的にLALと検体からなる試料を強制的に攪拌するので、比濁法と比較して短時間でゲルの生成を検出できる。
【0050】
また、エンドトキシンの別の測定法として比色法がある。これは図6に示すように、LALの酵素カスケード反応を利用しつつも、コアギュリンゲルによる試料の濁りを測定するのではなく、凝固酵素により加水分解を受け発色する合成基質を利用して、合成基質を含んだLALと検体とを反応させ、その吸光度変化を測定する方法である。この比色法においては、系内に生成されていく発色物質の濃度を測定するので、試料におけるゲルの生成を測定する比濁法やレーザー光散乱粒子計測法と比較すると、短時間で低濃度の所定生理活性物質を測定することができる。
【0051】
ところで、上記した各々の測定法でエンドトキシンやβ−D−グルカンを測定するためのLAL試薬を製造する過程でエンドトキシン、あるいは、β−D−グルカンが混入すると、混入した物質の量にもよるが、試薬の製造途中で原料がゲル化してしまって試薬として使用不可能となる場合がある。その際の混入量がたとえ僅かであっても、既に述べたようにLAL試薬は酵素のカスケードによる増幅作用を持つため、試薬の調製に長時間を要する場合には、試薬全体がゲル化してしまうこともあり得る。
【0052】
エンドトキシンに関してはポリミキシンBやポリリジンなどの吸着物質が知られており、LAL試薬に適用することにより混入したエンドトキシンを除去することも可能である。しかしながら、既に活性化してしまったC因子、B因子、ならびに、凝固酵素は酵素カスケードの最下流のコアギュロゲンを加水分解する方向に働き続けるため、エンドトキシンを除去するだけではLAL試薬のゲル化を防止できない。このことはβ−D−グルカンについても同様である。
【0053】
以下に、エンドトキシン、あるいは、β−D−グルカンがごく微量混入しても、製造途中でゲル化することを抑制できるLAL試薬、あるいは、LALを素材としたコアギュロゲン原料の製造法の例を示す。しかしながら本発明のLAL試薬及び、コアギュロゲン原料の製造法は、以下の例に限定されるものではない。
【0054】
本発明に係るコアギュロゲン原料の素材としてはカブトガニの血球抽出物、それを用いて製造されたLAL試薬、ならびに、それらにごく微量のエンドトキシン、あるいは、β−D−グルカンが混入したために試薬として利用できなくなった廃棄物などを用いることができる。しかし、本発明の目的が機能を持ったコアギュロゲン原料の製造にあることから、既にコアギュロゲンの大多数がコアギュリンとなりゲル化してしまったものは除外する。これらの素材に対して必要に応じてエンドトキシンを吸着する物質、β−D−グルカンを吸着する物質、エンドトキシンの作用を失活させる物質、β−D−グルカンの作用を失活させる物質の少なくとも一つ以上を添加してもよい。
【0055】
エンドトキシンを吸着する物質としてはポリミキシンB、ポリリジン、ポリオルチニン、ポリエチレンイミンなど、あるいは、C因子自体やC因子がエンドトキシンを結合する部位を含む蛋白質やポリペプチド、エンドトキシンを結合する抗体などを例示できる。エンドトキシンを失活させる物質としては、鉄イオン、アルミニウムイオン、クロムイオン、ニッケルイオン、コバルトイオン、マンガンイオンを含む水溶液、ならびに、これらを供給する金属片などを例示できる。
【0056】
同様に、β−D−グルカンと結合する物質としてはレクチン、あるいは、G因子自体あるいはG因子がβ−D−グルカンを結合する部位を含む蛋白質やポリペプチド、β−D−グルカンを結合する抗体などを例示できる。また、β−D−グルカンを不活性化するためには、素材の水素イオン濃度を下げることにより、β−D−グルカンの溶解性を下げて析出させてしまうことも考えられる。
【0057】
このように必要に応じて最適な添加物を混合させた素材に対して、次に加熱処理を行う。この加熱処理を適当な加熱温度、加熱時間で行うことにより、素材中の所定の酵素群であるC因子、活性型C因子、B因子、活性型B因子、G因子、活性型G因子、凝固酵素前駆体、凝固酵素の少なくとも一部を不可逆的に不活性化させる。そして、エンドトキシンやβ−D−グルカンとの反応によって凝固酵素が生じないようにする。そうすれば、たとえエンドトキシンやβ−D−グルカンに汚染されても、加水分解してコアギュリンとなってゲル化することのないコアギュロゲン原料を得ることができる。
【0058】
この場合の加熱処理は素材の状態により、加熱温度と加熱時間を適宜調整すればよい。例えば、素材が溶液の場合、反応させる温度が40℃以上140℃以下であることが好ましく、60℃以上100℃以下であることがさらに好ましい。好ましい加熱時間は加熱温度によって大きく異なるが、30秒以上72時間以下であることが好ましく、10分以上8時間以下であることがさらに好ましい。
【0059】
加熱温度を高くしすぎたり、加熱時間を長くしすぎたりすると、LAL試薬あるいは、LAL中の蛋白質が変性して白濁してしまい、試料の濁りを測定する方法には使用が困難になったり、コアギュロゲンが変性して機能を失ってしまったりする可能性があるので注意を要する。
【0060】
一方、素材が、既に凍結乾燥体となっているLAL試薬である場合には、素材が溶液の場合と異なり、より高い加熱温度とより長い加熱時間が必要となる。加熱温度は80℃以上300℃以下が好ましく、100℃以上250℃以下がさらに好ましい。好ましい加熱時間は水溶液の場合と同様に加熱温度により異なるが、30秒以上72時間以下が好ましく、10分以上8時間以下がさらに好ましい。
【0061】
このように加熱処理した素材は必要に応じて種々の金属塩あるいはアンモニウム塩、酸、アルカリ、さらには、緩衝液などによりpHを調整したり、必要に応じて界面活性剤や糖類などを添加しても良い。
【0062】
このように製造したコアギュロゲン原料はそのままではエンドトキシンやβ−D−グルカンを作用させてもゲル化、あるいは、凝集反応を起こすことはない。しかしながら、活性化した凝固酵素を作用させるか、あるいは、加熱処理していないLAL試薬と混和後にエンドトキシンやβ−D−グルカンを作用させることにより速やかにコアギュリンへと加水分解されゲル化、あるいは、凝集反応が惹起される。
【0063】
また、本発明により製造したコアギュロゲン原料はエンドトキシンやβ−D−グルカンの測定感度の向上、測定時間の短縮、測定利便性の向上などに役立てることが可能である。すなわち、本発明のコアギュロゲン原料を、加熱処理していないLAL試薬と混和することにより、通常よりもコアギュロゲン濃度の高いLAL試薬を調製することが可能である。このLAL試薬にエンドトキシンやβ−D−グルカンを作用させることによって、より迅速で高感度な測定が可能となる。
【0064】
また、例えば、本発明によるコアギュロゲン原料をポリスチレンラテックスの微小な粒子の表面に結合または吸着させた状態のLAL試薬を調製することにより、従来の比濁法
によるエンドトキシン測定よりも大幅な時間短縮が可能な測定法が実現できる。
【0065】
すなわち、コアギュロゲン原料をポリスチレンラテックスの微粒子(以下、ビーズともいう)の表面に結合または吸着させたLAL試薬に、エンドトキシンやβ−D−グルカンによって活性化された凝固酵素を作用させると、コアギュロゲンが酵素カスケードにより加水分解されコアギュリンとなり、これらが微粒子同士を会合させる。従って、本発明により取得されたコアギュロゲン原料におけるコアギュロゲンを微粒子に結合または吸着しておくことで、より効率的に微粒子同士を会合させることが可能となり、より早期に凝集塊を生成させることが可能となる。
【0066】
本発明のコアギュロゲン原料におけるコアギュロゲンをビーズに結合させるには、ビーズが持つ電荷で吸着させる方法、ビーズのイオン的性質を利用する方法、ビーズ表面にコアギュロゲン原料中の蛋白質と反応可能な官能基を形成し、その官能基と蛋白質中のアミノ基、あるいは、カルボキシル基を利用して化学的に結合させる方法などが考えられるが、どのような方法を用いても良い。
【0067】
ところで、従来、LALの素材を改変してこのようにビーズに結合または吸着させるような操作をすると、製造にかかる時間が数時間以上という長期に及ぶため、他の素材中に微量に混入したエンドトキシンやβ−D−グルカンが影響したり、あるいは、作業中にそれらの生理活性物質が微量に混入したりして、原料がゲル化してしまい、そのままでは目的の物質を得ることができない場合があった。
【0068】
これに対して、従来は、原料に対して各種の酵素阻害剤を添加する対策が考えられた。この酵素阻害剤としては、Diisopropylfluorophosphate、Benzamidine、Phenylmethanesulfonyl fluoride、4-(2-Aminoethyl)-benzenesulfonyl fluoride、6-Amidino-2-naphthyl-4-guanidinobenzoatedimethanesulfonate、p-Amidinophenylmethylsulfonyl fluoride、Aprotinin、Antipain、Leupeptin、Ecotin、PPACK(Phe-Pro-Arg-chloromethylketone)、α2-Macroglobulin、Trypsin inhibitorなどを例示することができる。
【0069】
しかしながら、本発明のコアギュロゲン原料を用いた場合は、LAL中の酵素機能のみが不可逆的に不活性化されているとともにコアギュロゲン活性は維持されている。従って、上記のような酵素阻害剤を添加するまでもなく、ビーズへの結合または吸着操作中に素材中に混入したエンドトキシンやβ−D−グルカンによる、原料のゲル化を防止することができる。
【0070】
以下に、本発明のコアギュロゲン原料の製造方法の詳細について示す。製造例は製造方法の一例を示したものであって、本発明に係る製造方法は以下の製造例の条件に限定されるものではない。
【0071】
〔製造例1〕
LAL試薬の凍結乾燥体(リムルスES−IIシングルテストワコー、和光純薬製、以下ES−IIと略す。)に所定量(0.15mL)の注射用蒸留水(大塚製薬製)を加えてボルテックスミキサーで混和した。その後、予め所定温度(後述)に加熱しておいたアルミブロックヒーター(HDB−1N、アズワン製、以下特に断らない限りアルミブロックヒーターとしてこの機種を使用する。)にセットして所定時間(後述)の加熱を行った。加熱後すぐに氷中で冷却しコアギュロゲン原料の水溶液を得た。
【0072】
〔製造例2〕
LAL試薬(ES−II)の凍結乾燥体をそのまま、予め120℃に加熱しておいたアルミブロックヒーターにセットして所定時間(後述)の加熱を行った。加熱後すぐに氷中
で冷却しコアギュロゲン原料の凍結乾燥体を得た。
【0073】
〔製造例3〕
製造例1、ならびに製造例2で得られたコアギュロゲン原料の酵素活性の不可逆的な失活とコアギュロゲン機能の維持とを、エンドトキシンの定量が可能なレーザー散乱粒子計測法を用いて評価した。レーザー散乱粒子計測法においては、φ7mm、長さ50mmのガラス製で内部に試料を攪拌するためのステンレス製の攪拌子(φ1mm、長さ5mm)を内在した専用のガラス容器を使用した。この容器をエンドトキシンフリーにするために、容器の開口部をアルミ箔で覆い、さらに、20本ずつアルミ箔で小分けに梱包したものを鉄製の乾熱処理缶に詰め、250℃で3時間加熱処理して、エンドトキシンを熱分解した。このように乾熱処理によりエンドトキシンフリーにした測定容器を製造し試験に使用した。
【0074】
〔製造例4〕
本発明によるコアギュロゲン原料のコアギュロゲン機能、すなわち、活性化した凝固酵素によりコアギュロゲンが加水分解されてゲル化するか、あるいは、凝集反応を示すかを調べるために、凝固酵素の活性化物を以下のようにして得た。すなわち、加熱処理していないLAL試薬(ES−II)に1.0EU/mLの濃度のエンドトキシン水溶液200μLを作用させて、製造例3により製造した測定容器にLAL試薬とエンドトキシン水溶液の混合物を移注し、37℃に保温しながら容器内部のステンレス攪拌子を容器底面方向に配置した攪拌器により回転して反応を進行させた。
【0075】
レーザー光散乱粒子計測法では、エンドトキシンによりゲル化したLAL試薬が攪拌により微小な凝集塊として出現し、その凝集塊の出現時間と試料中のエンドトキシンの濃度は両対数プロットで直線に乗ることが知られている。この条件の場合、約6分で凝集が検出されたが、凝集検出後も攪拌を続け、測定開始から20分の時点で容器を装置から取り出し、容器内部のゲル凝集物を全量回収し、エンドトキシンフリーのディスポーザブル遠心管に移注して、15000rpm(ローター半径7cm)で2分間遠心して、コアギュリンポリマーが主成分であるゲル成分を沈殿させた。別の方法により、遠心上清にはコアギュロゲンが含まれていないことを確認した。この製造法により、エンドトキシンによって活性化した凝固酵素を調製した。
【0076】
〔製造例5〕
LAL試薬(ES−II)に所定濃度(後述)のエンドトキシン水溶液0.2mLを加えてボルテックスミキサーで混和した後、予め、100℃に加熱しておいたアルミブロックヒーターにセットして20分間の加熱を行った。加熱後すぐに氷中で冷却しエンドトキシンが混入したコアギュロゲン原料の水溶液を得た。
【0077】
〔製造例6〕
表面がカルボキシル化されたポリスチレンラテックス微粒子(Polybeads Carboxylate Microspheres、0.45μm、固形分2.63%、Polysciences Inc.製、以下、カルボキシルビーズと略す。)の懸濁液0.5mLをエンドトキシンフリーのスクリューキャップ付き遠心管(容量2.0mL)にて15000rpmで5分間遠心し、上澄みを除去した。次いで、注射用蒸留水を加えて2.0mLにし、懸濁させた後に再び遠心して上澄みを除去した。この遠心操作による上澄み除去をもう一度繰り返した後、注射用蒸留水1mLに再懸濁した後、エンドトキシンフリーのスクリューキャップ付き遠心管(容量15mL)に移注した。さらに、4mLの注射用蒸留水を加えた後にオートクレーブ(121℃、20分)を掛け、ビーズに混入するエンドトキシンを不活性化ならびに除去した。
【0078】
オートクレーブ後のカルボキシルビーズを容量2mLのスクリューキャップ付き遠心管
に移注し、上述した要領で遠心と上澄み除去、さらに、注射用蒸留水への再懸濁をセットとする洗浄操作を合計2回行った。次に、注射用蒸留水で溶解し、濃度が20mg/mLになるように調製した水溶性カルボジイミド(WSC、同仁化学製)水溶液10mLに0.1M酢酸バッファ(pH4.98)2mLを混合し、これを孔径φ0.2μmの滅菌用フィルタ(ミリポア製)に通し、このうちの0.5mLに上記のカルボキシルビーズの沈殿物を再懸濁させた。
【0079】
また、LAL試薬(ES−II)の凍結乾燥体に注射用蒸留水(大塚製薬製)0.5mLを加えて溶解し、60℃で10分間加熱をおこなった。この加熱処理により得られた本発明のコアギュロゲン原料0.5mLと上述の方法で調製したカルボキシルビーズ懸濁液0.5mL、さらに、1.0%になるように調製した非イオン性界面活性剤であるTriton
X−100水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)を10μL混合し、室温で2時間反応させた。
【0080】
この反応により、カルボキシルビーズのカルボキシル基がカルボジイミドで活性化された後、コアギュロゲンのアミノ基とアミド結合を起こして、ビーズ表面にコアギュロゲンが化学的に結合される。反応後、上記のように遠心、上澄み除去、注射用蒸留水による再懸濁を2回おこなって反応物を洗浄した。次いで、0.25Mのアミノエタノール水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)を100μL加えて、室温で20分間攪拌して、カルボキシルビーズ上の活性化したカルボキシル基のうち未反応のものを不活性化させた。その後、同様に注射用蒸留水で合計3回洗浄し、5mLの注射用蒸留水に再懸濁させ、上述の1.0%のTriton X−100水溶液と2%のアジ化ナトリウム水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)をそれぞれ50μLずつ加えてコアギュロゲン結合マイクロビーズを得た。
【0081】
以下に、本発明の実施例について説明する。
【0082】
〔実施例1〕
製造例1において、アルミブロックヒーターの指示温度を40℃、50℃、60℃、70℃、80℃、100℃、ならびに、120℃のいずれかの条件において、加熱時間を10分にして製造したコアギュロゲン原料から100μLをとり、製造例3で製造した測定容器に入れた。そこに、2.0EU/mLの濃度のエンドトキシン水溶液100μLを加えて作用させ、レーザー散乱粒子計測装置(PA−200、興和製、以下PA−200と略す。)で測定を行った。加熱処理なしの場合の測定濃度(C=1.0EU/mL)と、加熱したLAL試薬での測定濃度(S)から、加熱された後に残存する酵素活性である残存凝固酵素比活性(A)を以下の式で定義して導出した。
A=S/C×100(%)・・・・・(1)
【0083】
図1には、得られた残存凝固酵素比活性(A)とLAL試薬水溶液の加熱温度との関係を示す。図1に示したように、加熱時間を10分とした場合には、加熱温度が60℃以上になると酵素活性が完全に消失することが分かった。また、図示しないが、加熱温度が高いほど製造後のコアギュロゲン原料水溶液は白濁する現象が観察された。例えば、120℃で加熱してコアギュロゲン原料水溶液を製造した場合、かなりの部分が熱変性によるものと見られる白濁と不溶物に変化してしまっており、実使用には適さないことが判った。
【0084】
〔実施例2〕
製造例1において、アルミブロックヒーターの指示温度を40℃、50℃、60℃、70℃、80℃、100℃、ならびに、120℃のいずれかの条件において、加熱時間を10分にして製造したコアギュロゲン原料(150μL)に、製造例4で得られた活性化した凝固酵素水溶液50μLを加えて作用させ、ボルテックスミキサーで5秒間混和させた
。その試料を製造例3で製造した測定容器に移注し、レーザー散乱粒子計測装置(PA−200)で凝集開始時間の測定を行った。
【0085】
表1にそれぞれの加熱温度に対する凝集開始時間を示したが、いずれの加熱温度においても活性化した凝固酵素水溶液を添加してから3分程度で凝集が開始しており、コアギュロゲンの機能は加熱による影響を殆ど受けていないことが示された。また、120℃という高温で処理した場合には、LAL試薬中の大部分の蛋白質は熱変性して凝固してしまうものの、その中に残存するコアギュロゲン自体は機能を維持しており、活性化した凝固酵素水溶液の添加から4分程度で凝集が開始した。このことから、コアギュロゲンは非常に加熱に強いことが示された。また、表1から分かるように、LAL試薬を80℃以下で加熱処理して酵素活性を失活させた場合には、LAL試薬に白濁は殆ど見られなかった。従って、LAL試薬を80℃以下で加熱処理して酵素活性を失活させることで、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となることが分かった。
【0086】
【表1】

【0087】
〔実施例3〕
製造例2において、加熱時間を10分、30分、60分、120分、ならびに、300分のいずれかの時間にして、LAL試薬(リムルスES−IIシングルテストワコー、和光純薬製)を開封せずにそのまま加熱処理したコアギュロゲン原料の凍結乾燥体に1.0EU/mLのエンドトキシン水溶液(200μL)を添加した。そして、ボルテックスミキサーで5秒間混和した後、製造例3により製造した測定容器に移注して、レーザー光散乱粒子計測装置(PA−200)にて、加熱処理の後に残存する酵素の活性を調べた。そして、上述の式(1)で定義される残存凝固酵素比活性(%)を算出した。
【0088】
図2には本実施例で得られた残存凝固酵素比活性と、LAL試薬凍結乾燥体の加熱処理時間との関係を示す。図2に示したように、実施例1の場合と比較して、高温下で長時間の加熱処理を行っても、なかなか、酵素活性が完全に消失しないことが分かる。しかしながら、120℃で300分の加熱処理により、残存凝固酵素比活性は0.2%程度に減少した。このように製造したコアギュロゲン原料では、原料を水に溶解させたときの水溶液の色がやや黄変する傾向が見られたが、白濁や凝固不溶物の発生はなく、水に速やかに溶解した。
【0089】
〔実施例4〕
実施例4においては、予めエンドトキシンが混入した状態でLAL試薬を加熱処理した場合の、混入したエンドトキシンの濃度と加熱処理による酵素の不活性化の程度との関係を調べた。製造例5において、予め混入させておくエンドトキシン水溶液のエンドトキシン濃度10、1、0.1EU/mLとして、それぞれの濃度のエンドトキシンが混入したコアギュロゲン原料を得た。
【0090】
次に、本実施例においては、上記のエンドトキシンが混入したコアギュロゲン原料と比較するためのコントロールコアギュロゲン原料を、エンドトキシンを含まない注射用蒸留水(大塚製薬製)0.2mLで溶解したLAL試薬を製造例5に示した手順で加熱することで調製した。さらに、エンドトキシン水溶液の希釈系列を作り、それぞれ200μLを加熱処理していないLAL試薬(ES−II)に入れてボルテックスミキサーで5秒間攪拌した後、それぞれの試料から100μLを取り、上記のコントロールコアギュロゲン原料100μLと混和し、レーザー光散乱粒子計測装置(PA−200)にてエンドトキシン濃度の検量線を得た。
【0091】
次に、加熱処理していないLAL試薬(ES−II)を注射用蒸留水(大塚製薬製)200μLで溶解し、そのうちの100μLを上記の予めエンドトキシンが混入したそれぞれのコアギュロゲン原料100μLとボルテックスミキサーで5秒間混和させた。そして、レーザー光散乱粒子計測装置(PA−200)でエンドトキシン濃度を上記で得られた検量線に照らし合わせて算出した。得られた結果を表2に示した。表2に示すように、いずれの試料においても測定されたエンドトキシン濃度は、実際に混入したエンドトキシン濃度より低かった。そして、表中の加熱後の活性残存率が示すように、得られたエンドトキシン濃度の実際の値からの低下の度合は、混入させたエンドトキシンの濃度が低いほど大きかった。このように、予め混入させたエンドトキシンの濃度が低いほど加熱処理による酵素の不活性化の効果が大きいことが示された。
【0092】
【表2】
【0093】
〔実施例5〕
製造例6により製造されたコアギュロゲン結合マイクロビーズを用いてエンドトキシン水溶液の希釈系列中のエンドトキシン濃度を攪拌比濁法を用いて測定した(以下、この測定法をコアギュロゲン結合マクロビーズ法とする。)。攪拌比濁法は従来法の比濁法と異なり、試料を攪拌しつつ、試料の透過率の変化の度合からエンドトキシンの濃度を測定する。
【0094】
図3には、本実施例の攪拌比濁測定装置としての比濁計測装置1の概略構成を示す。本実施例の攪拌比濁法においては、試料は混和液保持手段としての専用のガラス製容器2に移注する。ガラス製容器2の周囲を囲うように保温器5が設けられている。この保温器5の内部には図示しない電熱線が備えられており、この電熱線に通電されることにより、ガラス製容器2を約37℃に保温するようになっている。このガラス製容器2の中にはステンレス製の攪拌子3が備えられている。この攪拌子3は、ガラス製容器2の下部に設置された攪拌器4の作用によってガラス製容器2の中で回転する。すなわち、攪拌器4はモータ4aとモータ4aの出力軸に設けられた永久磁石4bとからなっている。そして、モータ4aに通電されることで永久磁石4bが回転する。この永久磁石4bからの磁界が回転するために、ステンレス製の攪拌子3が回転磁界の作用で回転する。この攪拌子3と攪拌器4とは攪拌手段に相当する。
【0095】
なお、比濁計測装置1には光入射手段としての光源6と受光手段としての受光素子9が
設置されている。光源6から出射した光はアパーチャ7を通過した後、保温器5に設けられた入射孔5aを通過してガラス製容器2中の試料に入射される。ガラス製容器2中の試料を透過した光は保温器5に設けられた出射孔5bから出射され、アパーチャ8を通過して受光素子9に照射される。受光素子9では、受光した光の強度に応じた光電信号を出力する。この光電信号の出力より、導出手段としての演算装置10において試料の透過率が算出される。
【0096】
熱処理していないLAL試薬(ES−II)2本を200μLの注射用蒸留水に溶解させ、そこに、製造例6で製造したコアギュロゲン結合マイクロビーズ50μLを入れてボルテックスミキサーで5秒間攪拌しコアギュロゲン結合マイクロビーズ−LAL試薬混合物を得た。エンドトキシン水溶液の希釈系列、すなわち、2、0.2、0.02、0.002EU/mLのいずれか50μLと上記のコアギュロゲン結合マイクロビーズ−LAL試薬混合物50μLを上記の製造例3に準じて製造したガラス製容器2に入れて比濁計測装置1にて測定を行った。
【0097】
コアギュロゲン結合マイクロビーズ法では、作用させたエンドトキシンによりLAL試薬中の凝固酵素が活性化されると、同じくLAL試薬中に含まれるコアギュロゲンがコアギュリンとなり、同時に、コアギュロゲン結合マイクロビーズ上のコアギュロゲンも加水分解されてコアギュリンとなるが、ビーズがこれらのコアギュリンにより架橋されて大きな凝集塊を形成する。試料には、当初、多量の単体ビーズが含まれており強く濁っているが、凝集により単体のビーズ濃度が急激に減少するため、試料の光透過率が上昇する。コアギュロゲン結合マイクロビーズ法ではこの光の透過率の急激な上昇により凝集塊の発生開始時間を決定する。
【0098】
図4には、従来の活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程と、コアギュロゲン結合マイクロビーズ法におけるコアギュロゲンのゲル化・凝集過程とを示す。図4(A)に示したのは従来の活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程である。図に示すように、粒径にして数nm程度のコアギュロゲン100が活性型凝固酵素によって加水分解されてコアギュリン101となる。これらのコアギュリン101は凝集して多量体となり攪拌比濁法においてはゲル粒子を形成する。その後、時間の経過とともにコアギュリン101の凝集はさらに進行しゲル粒子の粒径は徐々に大きくなる。そして、比較的長いラグタイムが経過した時点で測定可能な数μm〜数十μmの粒径のゲル粒子102となる。
【0099】
これに対し、コアギュロゲン結合マイクロビーズ法では、図4(B)に示すように、ビーズ103上に結合したコアギュロゲン100に活性型凝固酵素が作用すると、ビーズ103上のコアギュロゲン100が酵素カスケードによって加水分解されてコアギュリン101となり、コアギュリン101が凝集する過程ではコアギュリン101がビーズ103同士を会合させる。このことにより、コアギュリン101とビーズ103を主成分とする粒子の径が早急に大きくなり、短時間のラグタイムにおいて測定可能な粒径を得ることとなる。なお、この凝集反応はLAL単体で惹起される凝集反応よりも、非常に強大であることも明確になってきている。
【0100】
また、本実施例においては、コアギュロゲン結合マイクロビーズ法を用いた場合と比較するために比濁法によってもエンドトキシン濃度測定を行った。比濁法では、熱処理していないLAL試薬に注射用水100μL、エンドトキシン水溶液希釈系列のいずれかの濃度の試料を100μL入れて比濁法装置(トキシノメータET−2000、和光純薬製)で測定を行った。コアギュロゲン結合マイクロビーズ法と比濁法の比較結果を図5に示したが、両測定法とも横軸にエンドトキシン濃度、縦軸に検出時間を取ると、その両軸対数プロットは直線に近似された。近似曲線はコアギュロゲン結合マイクロビーズ法がY=−
0.190X+0.738、比濁法ではY=−0.349X+0.869であることから、コアギュロゲン結合マイクロビーズ法が比濁法に比べて大幅に測定時間を短縮可能であること、作用させるエンドトキシンの濃度が低いほど測定に要する時間の差が大きく開いてくることが示された。測定に要する実時間などを表3にまとめた。
【0101】
【表3】
【0102】
本実施例において、ビーズ103の素材は特に限定されるものではないが、ポリスチレンラテックス樹脂の他、シリカ、シリコン樹脂、セルロース樹脂、ポリビニルアルコール樹脂、ハイドロキシアパタイトなどが例示でき、ポリスチレンラテックス樹脂、シリカ、セルロース樹脂が望ましい。
【0103】
また、ビーズ103の大きさは、凝集により早期に光学的に検出可能となる条件と、調製時のハンドリングの容易さ、系内への分散のし易さなどから、0.05μmから50μmの範囲であるものが用いられる。ビーズ103の表面にコアギュロゲンを結合させるには、静電的、親水的、あるいは、疎水的にコアギュロゲンを吸着させる方法と、化学的に結合させる方法とが考えられる。
【符号の説明】
【0104】
1・・・比濁計測装置
2・・・ガラス製容器
3・・・攪拌子
4・・・攪拌器
4a・・・モータ
4b・・・磁石
5・・・保温器
5a・・・入射孔
5b・・・出射孔
6・・・光源
7・・・アパーチャ
8・・・アパーチャ
9・・・受光素子
10・・・演算装置
100・・・コアギュロゲン
101・・・コアギュリン
102・・・凝集粒子
103・・・ビーズ
【技術分野】
【0001】
本発明は、エンドトキシンやβ−D−グルカンなどの生物由来の生理活性物質の検出または濃度測定を、迅速または高感度に計測するための試薬、その製造法、ならびにそれを用いた測定方法及び測定装置に関する。
【背景技術】
【0002】
エンドトキシンはグラム陰性菌の細胞壁に存在するリポ多糖であり、最も代表的な発熱性物質である。このエンドトキシンに汚染された輸液、注射薬剤、血液などが人体に入ると、発熱やショックなどの重篤な副作用を惹起するおそれがある。このため、上記の薬剤などは、エンドトキシンにより汚染されることが無いように管理することが義務付けられている。
【0003】
ところで、カブトガニの血球抽出物(以下、「LAL : Limulus amoebocyte lysate」ともいう。)の中には、エンドトキシンによって活性化される酵素であるセリンプロテアーゼが存在する。そして、LALとエンドトキシンとが反応する際には、エンドトキシンの量に応じて活性化されたセリンプロテアーゼによる酵素カスケードによって、LAL中に存在するコアギュロゲンがコアギュリンへと加水分解されて会合し、不溶性のゲルが生成される。このLALの特性を用いて、エンドトキシンを高感度に検出することが可能である。
【0004】
また、β−D−グルカンは真菌に特徴的な細胞膜を構成しているポリサッカライド(多糖体)である。β−D−グルカンを測定することによりカンジダやスペルギルス、クリプトコッカスのような一般の臨床でよく見られる真菌のみならず、稀な真菌も含む広範囲で真菌感染症のスクリーニングなどに有効である。
【0005】
β−D−グルカンの測定においても、カブトガニの血球抽出成分がβ−D−グルカンによって凝固(ゲル凝固)する特性を利用して、β−D−グルカンを高感度に検出することが可能である。
【0006】
このエンドトキシンやβ−D−グルカンなどの、カブトガニの血球抽出成分によって検出可能な生物由来の生理活性物質(以下、所定生理活性物質ともいう)の検出または濃度測定を行う方法としては、所定生理活性物質の検出または濃度測定(以下、単純に「所定生理活性物質の測定」ともいう。)をすべき試料とLALを元に製造された試薬(LAL試薬)とを混和した混和液を静置し、一定時間後に容器を転倒させて、試料の垂れ落ちの有無によりゲル化したかどうかを判定し、試料に一定濃度以上のエンドトキシンが含まれるか否かを調べる半定量的なゲル化法がある。また、LAL試薬と所定生理活性物質との反応によるゲルの生成に伴う試料の濁りを経時的に計測して解析する比濁法や、酵素カスケードにより加水分解されて発色する合成基質を用いる比色法などがある。
【0007】
上記の比濁法によって所定生理活性物質の測定を行う場合には、乾熱滅菌処理されたガラス製測定セルに測定試料とLAL試薬との混和液を生成させる。そして、混和液のゲル化を外部から光学的に測定する。しかしながら、比濁法においては特に所定生理活性物質の濃度が低い試料において混和液がゲル化するまでに非常に多くの時間を要する場合がある。これに対し、所定生理活性物質の短時間測定が可能な方法が求められている。測定試料とLAL試薬との混和液を例えば磁性攪拌子を用いて攪拌することにより、ゲル微粒子を生成せしめ、ゲル粒子により散乱されるレーザー光の強度、あるいは、混和液を透過す
る光の強度から、試料中の所定生理活性物質の存在を短時間で測定できるレーザー散乱粒子計測法等が提案されている。
【0008】
LAL試薬はカブトガニの血球抽出物を主原料として製造される。このため、製造上ある確率でエンドトキシンやβ−D−グルカンが混入してしまい、試薬として利用できない廃棄物となる可能性がある。また、エンドトキシン、あるいは、β−D−グルカンのいずれの測定法も、測定試薬の原料が有限資源であるカブトガニの血球抽出物であるため、試薬の使用量を減少させ、あるいは、試薬製造上のロスを低減させるといった取り組みが必要になる。
【0009】
試薬の使用量を減少させる試みとしては、単純に試料容量を減少させることはもちろん、測定感度を高めて結果的に測定回数を低減することも効果的である。一方、製造上のロスを減らすにはエンドトキシンやβ−D−グルカンによる汚染をいかに抑制するかということが重要である。さらに、製造中に試薬として使用できなくなった原料からエンドトキシンやβ−D−グルカンを除去して再利用したり、補助的な試薬の添加物として利用したりすることなども考えねばならない。補助的な試薬の添加物として利用する場合、試薬の粘性の調整剤としての役割や、ゲル化・凝集での中心的役割を担うコアギュロゲン原料としての利用などが考えられる。
【0010】
しかしながら、例えばエンドトキシンに汚染されて使用できなくなった原料中のエンドトキシンを除去するだけでは既に活性化してしまった酵素群を除去することは不可能である。原料中の酵素活性のみを不活性化してコアギュロゲンの機能、すなわち、活性化された凝固酵素により加水分解されてコアギュリンとなり、ゲル化・凝集反応を生じさせる機能を維持した原料の製造方法は報告されていなかった。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特許第2667695号公報
【特許文献2】特開2004−061314号公報
【特許文献3】特開平10−293129号公報
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は上述の問題点に鑑みて案出されたものであり、その目的とするところは、LAL試薬、あるいは、生物由来の生理活性物質に汚染されたLAL試薬等におけるコアギュロゲンの機能を維持したまま凝固酵素活性を不可逆的に不活性化し、試薬に利用可能なコアギュロゲン原料を取得する技術を提供することである。
【課題を解決するための手段】
【0013】
上記の課題を解決するための本発明は、若干の所定生理活性物質の混入を許容するLAL試薬をある温度条件下において加熱処理することにより、LAL試薬中の酵素活性のみを不可逆的に失活させたことを最大の特徴とする。本発明においては、その際、活性化した凝固酵素により加水分解されコアギュリンとなってゲル化・凝集反応を惹起するという、コアギュロゲン本来の活性は維持させる。
【0014】
より詳しくは、本発明は、所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料の製造方法であって、
LALを所定温度で所定時間に亘り加熱処理することで、該LAL中の前記酵素群の少なくとも一部を失活させるとともにコアギュロゲンの活性は維持させることを特徴とする。
【0015】
これによれば、LAL中の酵素カスケードを構成する酵素群の少なくとも一部を失活させることで酵素カスケードの発生を抑止し、エンドトキシンやβ−D−グルカンなどの汚染によっても加水分解してコアギュリンになりづらいコアギュロゲン原料を取得することが可能となる。
【0016】
また、これによれば、エンドトキシンやβ−D−グルカンなどの汚染により使用不可となったLAL試薬のコアギュロゲンを、機能を維持したまま取得することができるので、カブトガニの血球から得られる有限の資源をより有効に活用することが可能となる。
【0017】
ここで、所定の酵素群とは例えばLAL中のC因子、B因子、G因子、凝固酵素前駆体など、エンドトキシンやβ−D−グルカンなどにより酵素カスケードを生じ最終的にコアギュロゲンを加水分解する凝固酵素を生じる原因となる酵素類を意味している。すなわち、LAL中の全ての酵素を失活させずとも、少なくとも一部の酵素が失活し、エンドトキシンやβ−D−グルカンなどによっても、最終的にコアギュロゲンを加水分解する凝固酵素が生じないようになれば本発明の目的は達成できる。
【0018】
また、上記において生物由来の所定の生理活性物質とは、LAL中の所定の酵素群を活性化させ酵素カスケードを生じせしめ、最終的にコアギュロゲンを加水分解する凝固酵素を生じせしめる特性を有する生理活性物質を意味しており、例としては上述のとおり、エンドトキシンやβ−D−グルカンが挙げられる。但し、同等の特性を有する他の生理活性物質をも含む趣旨であり、上述の2物質に限定するものではない。
【0019】
なお、上記においてLALが溶液として供給される場合には、所定温度は60℃以上としてもよい。ここで、発明者の鋭意研究により、溶液としてのLAL試薬を60℃以上に加熱処理することで、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、LALが溶液として供給される場合に所定温度を60℃以上とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならないコアギュロゲン原料を取得することが可能となる。
【0020】
また、上記においてLALが溶液として供給される場合には、所定温度は80℃以下としてもよい。ここで、発明者の鋭意研究により、LAL試薬を80℃より高い温度まで加熱処理して酵素活性を失活させた場合には、LAL中の蛋白質の変性により試薬が白濁してしまうことが分かってきた。そうすると、所定生理活性物質の検出や濃度測定を、試薬の透過光や散乱光によって光学的な手法によって行う目的には適さなくなってしまうおそれがある。従って本発明においては、LALが溶液として供給される場合には所定温度を80℃以下とすることで、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となる。
【0021】
さらに、上記においてLALが溶液として供給される場合には、所定時間は10分以上8時間以下としてもよい。ここで、溶液としてのLAL試薬を60℃以上に加熱処理した場合、加熱時間は10分程度で、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、所定時間を10分以上とすることで、広い範囲の加熱温度でセリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができる。また、加熱時間があまりに長い場合には、低温でもLAL中の蛋白質が変性して白濁が生じるおそれがある。従って本発明では、所定時間を10分以上8時間以下
とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならず、光学的な測定に適したコアギュロゲン原料を取得することが可能となる。
【0022】
さらに、上記においてLALが凍結乾燥体として供給される場合には、所定温度は100℃以上250℃以下としてもよい。ここで、LALが凍結乾燥体として供給される場合には、溶液として供給される場合と比較してより高い温度で加熱処理する必要があることが分かってきた。この場合には、LAL試薬を100℃以上250℃以下とすることで、より確実に、エンドトキシンやβ−D−グルカンなどに汚染されても加水分解してコアギュリンにならず、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となる。
【0023】
また、この場合には、所定時間は300分以上としてもよい。ここで、LALが凍結乾燥体として供給される場合には、LAL試薬を120℃で300分に亘って加熱することで、セリンプロテアーゼ等の酵素群の酵素活性を略完全に失活させることができることが分かってきた。従って、所定時間を300分以上とすることで、より広い範囲の加熱温度でより確実に、エンドトキシンやβ−D−グルカンなどの汚染によっても加水分解してコアギュリンにならないコアギュロゲン原料を取得することが可能となる。
【0024】
また、本発明は、所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料であって、
LALを所定温度で所定時間に亘り加熱処理することで、前記LAL中の前記酵素群の少なくとも一部を失活させたことを特徴とするコアギュロゲン原料であってもよい。
【0025】
このコアギュロゲン原料においては、所定生理活性物質との反応で酵素カスケードを生じる酵素群が失活されているので、少量の所定生理活性物質による汚染によりコアギュロゲンが加水分解されてコアギュリンとなることを抑止でき、よりハンドリングの良いコアギュロゲン原料を得ることができる。なお、この場合においても、LALが溶液として供給される場合には、所定温度は60℃以上としてもよい。また、80℃以下としてもよい。さらに、所定時間は10分以上8時間以下としてもよい。一方、LALが凍結乾燥体として供給される場合には、所定温度は100℃以上250℃以下としてもよい。また、所定時間は300分以上としてもよい。
【0026】
また、本発明においては、上記で得られたコアギュロゲン原料と、加熱処理されていないLALとを混合することで、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたことを特徴とする、前記生理活性物質の検出または前記生理活性物質の濃度測定に使用されるLAL試薬としてもよい。
【0027】
すなわち、所定の酵素群を失活させたコアギュリン原料を、熱処理をしていないLALに加えることで、通常よりコアギュロゲンの濃度の高いLAL試薬を調製することができる。これにより、所定生理活性物質との反応により活性化された酵素群によって加水分解されコアギュリンとなるコアギュロゲンの量を相対的に増加させることができる。従って、所定生理活性物質によるLAL試薬のゲル化をより顕著にすることができ、所定生理活性物質の測定をより高感度にすることができる。
【0028】
また、本発明においては、上記で得られたコアギュロゲン原料におけるコアギュロゲンを、該コアギュロゲンより大径の多数の微粒子の表面に結合または吸着させて調製された
ことを特徴とするエンドトキシンの測定用のコアギュロゲン結合マイクロビーズとしてもよい。
【0029】
ここで、コアギュロゲンを例えば樹脂製の微粒子の上に結合または吸着させた状態のLALにエンドトキシンなどの所定生理活性物質を作用させると、LAL単体に所定生理活性物質を作用させた場合と比較して、より大きな凝集塊を早期に生成することが分かってきた。これは、微粒子上のコアギュロゲンがLAL中の酵素カスケードにより加水分解されコアギュリンとなり、これらが微粒子同士を会合させることによる。また、この凝集反応は試料の濁りや色の影響を受けづらいこと、さらには、この凝集反応はLAL単体で惹起される凝集反応よりも、非常に強大であることも明確になってきている。
【0030】
本発明においては、この現象を利用し、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させた試薬(以下、「コアギュロゲン結合マイクロビーズ」ともいう。)を調製した。このコアギュロゲン結合マイクロビーズとLALとを混合させた試薬にエンドトキシンを含む試料を作用させることで、LAL中にもともと存在したコアギュロゲンがコアギュリンとなり凝集するとともに、微粒子の上に結合または吸着されたコアギュロゲンがコアギュリンとなり微粒子同士を会合させてより大きな凝集塊を早期に生成させる。これによれば、LAL試薬と試料との混和液におけるゲル粒子の生成を大幅に促進することができる。その結果、所定生理活性物質の検出または濃度の測定を、より迅速且つ、高感度にすることができる。
【0031】
なお、LAL中に含まれるコアギュロゲンを微粒子の上に結合または吸着させる際には、まず、蛋白質を結合、吸着することが可能な官能基を微粒子の表面に存在せしめる。そして、従来の方法によれば、その状態でLALを作用させて、コアギュロゲンを微粒子表面に化学的に結合させるか、静電的、親水的、疎水的に吸着させる。その際のLAL中のは長時間に及ぶため、従来の方法では、微粒子や使用する試薬類、水などに微量に混在する所定生理活性物質が蛋白質における酵素群と反応し、コアギュロゲンをコアギュリンに加水分解して、コアギュロゲンと微粒子の結合・吸着反応中に微粒子の凝集が開始してしまうことが考えられる。そしてこの場合には、酵素の活性化によって試薬中のコアギュロゲンが加水分解されて消費されてしまうおそれがある。
【0032】
このような不都合に対し、従来は、コアギュロゲンを微粒子の表面に結合または吸着させる際には、LAL試薬と微粒子の懸濁液とを混合するとともに、LAL中の酵素群と所定生理活性物質との反応を抑制する抑制剤を添加する必要があり、コアギュロゲンを微粒子の表面に結合または吸着させる作業が複雑でコスト的にも不利な状況であった。これに対し本発明では、コアギュロゲン原料の調製過程でLAL中の酵素群を失活させるため、抑制剤を添加する工程を省略することが可能である。これにより、より簡単にまたはより低廉なコストで、コアギュロゲンを微粒子の上に結合または吸着させた試薬を調製することが可能である。
【0033】
また、本発明は、上記のコアギュロゲン原料と、加熱処理されていないLALとを混合することによって、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたLAL試薬と、前記所定の生理活性物質を含む試料とを混和させ、
前記所定の生理活性物質によって前記LAL試薬中の酵素群が活性化されて生じる酵素カスケードによって濃度が高められた前記コアギュロゲンがコアギュリンに加水分解されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法であってもよい。
【0034】
この生物由来の生理活性物質の測定方法によれば、より高い濃度のコアギュロゲンを用いて、所定生理活性物質の測定を行うことができる。従って、所定生理活性物質とLAL
との反応によるゲル化をより顕著にすることができ、所定生理活性物質の測定をより高感度にすることができる。
【0035】
また、本発明は、上記のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記加熱処理されていないLALにおける酵素群が活性化されて生じる酵素カスケードによって前記微粒子の表面に結合または吸着したコアギュロゲンがコアギュリンに加水分解され、前記微粒子同士が架橋されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法であってもよい。
【0036】
この所定生理活性物質の測定方法によれば、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させたコアギュロゲン結合マイクロビーズと加熱処理されていないLALとを混合させた試薬にエンドトキシンを含む試料を作用させる。このことで、LAL中にもともと存在したコアギュロゲンがコアギュリンとなり凝集するとともに、微粒子の上に結合または吸着されたコアギュロゲンがコアギュリンとなり微粒子同士を会合させてより大きな凝集塊を早期に生成させる。これによれば、LAL試薬と試料との混和液におけるゲル粒子の生成を大幅に促進することができる。その結果、所定生理活性物質の検出または濃度の測定を、より迅速且つ、高感度にすることができる。
【0037】
また、本発明は、上記のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料との混和液を光の入射可能に保持し、前記混和液における反応を進行させる混和液保持手段と、
前記混和液保持手段中の前記混和液を攪拌する攪拌手段と、
前記混和液保持手段中の混和液に光を入射する光入射手段と、
前記入射光の前記混和液における透過光を受光し電気信号に変換する受光手段と、
前記受光手段において変換された電気信号から取得される前記混和液の透過率より前記試料中の前記生理活性物質の濃度を導出する導出手段と、
を備えることを特徴とする生物由来の生理活性物質の攪拌比濁測定装置であってもよい。
【0038】
本発明においては、LAL中の酵素群を失活させて得られたコアギュロゲン原料を微粒子の上に結合または吸着させたコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、所定生理活性物質を含む試料とを混和させ混和液保持手段に入れる。そして、この混和液を攪拌手段により攪拌することで、所定生理活性物質で活性化された凝固酵素によるコアギュロゲンの加水分解及び、加水分解で得られたコアギュリンによる微粒子の会合を促進する。
【0039】
そして、光入射手段から入射された光のうち、上記混和液を透過して受光素子に届いた光の強度を測定する。さらに、導出手段においては上記混和液の透過率より所定生理活性物質の濃度が導出される。
【0040】
ここにおいて、微粒子の上に結合または吸着されたコアギュロゲンの加水分解が進みコアギュリンが生成されると、コアギュリンが微粒子同士を会合させてより大きな凝集塊を早期に生成させる。ここで、コアギュロゲン結合マイクロビーズと加熱処理されていないLALと所定生理活性物質を含む試料との混和液は、混和時点では、多量のマイクロビーズの微粒子が分散しているために強く濁っている。そして、コアギュリンの生成による微粒子の会合が進行すると微粒子の濃度が急激に減少するために混和液の透過率が急激に上昇する。
【0041】
本発明における攪拌比濁測定装置では、通常の比濁法に係る測定装置と異なり、コアギュリンの生成に伴う微粒子の会合に起因する透過率の急激な上昇を測定するため、混和液を攪拌することによる反応の促進効果と相まって、測定時間を大幅に短縮できるとともに、非常に高感度に所定生理活性物質の測定を行うことができる。
【0042】
なお、上記した本発明の課題を解決する手段については、可能なかぎり組み合わせて用いることができる。
【発明の効果】
【0043】
本発明にあっては、LAL試薬、あるいは、生物由来の生理活性物質に汚染されたLAL試薬等におけるコアギュロゲンの機能を維持したまま凝固酵素活性を不可逆的に不活性化し、試薬に利用可能なコアギュロゲン原料を取得することが可能となる。
【図面の簡単な説明】
【0044】
【図1】本発明の実施例におけるLAL試薬水溶液の加熱温度と残存凝固酵素比活性との関係を示すグラフである。
【図2】本発明の実施例におけるLAL試薬凍結乾燥体の加熱時間と残存凝固酵素比活性との関係を示すグラフである。
【図3】本発明の実施例における比濁計測装置の概略構成を示す図である。
【図4】活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程とコアギュロゲン結合マイクロビーズの凝集過程とを説明するための図である。
【図5】コアギュロゲン結合マイクロビーズ法と比濁法におけるエンドトキシン用量反応を比較したグラフである。
【図6】エンドトキシンまたはβ―D−グルカンにより、LALがゲル化する過程及び、その検出方法について説明するための概略図である。
【発明を実施するための形態】
【0045】
LALとエンドトキシンとが反応してゲルが生成される過程はよく調べられている。すなわち、図6に示すように、エンドトキシンがLAL中のセリンプロテアーゼであるC因子に結合すると、C因子は活性化して活性型C因子となる、活性型C因子はLAL中の別のセリンプロテアーゼであるB因子を加水分解して活性化させ活性化B因子とする。この活性化B因子は直ちにLAL中の凝固酵素の前駆体を加水分解して凝固酵素とし、さらに、この凝固酵素がLAL中のコアギュロゲンを加水分解してコアギュリンを生成する。そして、生成したコアギュリンが互いに会合して不溶性のゲルをさらに生成し、LAL全体がこれに巻き込まれてゲル化すると考えられている。
【0046】
また、同様にβ−D−グルカンがLAL中のG因子に結合すると、G因子は活性化して活性型G因子となる、活性型G因子はLAL中の凝固酵素の前駆体を加水分解して凝固酵素とする。その結果、エンドトキシンとLALとの反応と同様、コアギュリンが生成され、生成したコアギュリンが互いに会合して不溶性のゲルをさらに生成する。
【0047】
この一連の反応は哺乳動物に見られるクリスマス因子やトロンビンなどのセリンプロテアーゼを介したフィブリンゲルの生成過程に類似している。このような酵素カスケード反応はごく少量の活性化因子であっても、その後のカスケードを連鎖して活性化していくために非常に強い増幅作用を有する。従って、LALを用いた所定生理活性物質の測定法によれば、サブピコグラム/mLオーダーのきわめて微量の所定生理活性物質を検出することが可能になっている。
【0048】
所定生理活性物質を定量することが可能な測定法としては前述のように比濁法、ならびに、レーザー光散乱粒子計測法が挙げられる。これらの測定法はこのLALの酵素カスケ
ード反応によって生成されるコアギュリンの会合物を前者は試料の濁りとして、後者は系内に生成されるゲルの微粒子として検出することで、高感度な測定を可能にしている。
【0049】
特にレーザー光散乱粒子計測法では、系内に生成されたゲルの微粒子を直接測定するため、比濁法よりも高感度であり、且つ、一般的にLALと検体からなる試料を強制的に攪拌するので、比濁法と比較して短時間でゲルの生成を検出できる。
【0050】
また、エンドトキシンの別の測定法として比色法がある。これは図6に示すように、LALの酵素カスケード反応を利用しつつも、コアギュリンゲルによる試料の濁りを測定するのではなく、凝固酵素により加水分解を受け発色する合成基質を利用して、合成基質を含んだLALと検体とを反応させ、その吸光度変化を測定する方法である。この比色法においては、系内に生成されていく発色物質の濃度を測定するので、試料におけるゲルの生成を測定する比濁法やレーザー光散乱粒子計測法と比較すると、短時間で低濃度の所定生理活性物質を測定することができる。
【0051】
ところで、上記した各々の測定法でエンドトキシンやβ−D−グルカンを測定するためのLAL試薬を製造する過程でエンドトキシン、あるいは、β−D−グルカンが混入すると、混入した物質の量にもよるが、試薬の製造途中で原料がゲル化してしまって試薬として使用不可能となる場合がある。その際の混入量がたとえ僅かであっても、既に述べたようにLAL試薬は酵素のカスケードによる増幅作用を持つため、試薬の調製に長時間を要する場合には、試薬全体がゲル化してしまうこともあり得る。
【0052】
エンドトキシンに関してはポリミキシンBやポリリジンなどの吸着物質が知られており、LAL試薬に適用することにより混入したエンドトキシンを除去することも可能である。しかしながら、既に活性化してしまったC因子、B因子、ならびに、凝固酵素は酵素カスケードの最下流のコアギュロゲンを加水分解する方向に働き続けるため、エンドトキシンを除去するだけではLAL試薬のゲル化を防止できない。このことはβ−D−グルカンについても同様である。
【0053】
以下に、エンドトキシン、あるいは、β−D−グルカンがごく微量混入しても、製造途中でゲル化することを抑制できるLAL試薬、あるいは、LALを素材としたコアギュロゲン原料の製造法の例を示す。しかしながら本発明のLAL試薬及び、コアギュロゲン原料の製造法は、以下の例に限定されるものではない。
【0054】
本発明に係るコアギュロゲン原料の素材としてはカブトガニの血球抽出物、それを用いて製造されたLAL試薬、ならびに、それらにごく微量のエンドトキシン、あるいは、β−D−グルカンが混入したために試薬として利用できなくなった廃棄物などを用いることができる。しかし、本発明の目的が機能を持ったコアギュロゲン原料の製造にあることから、既にコアギュロゲンの大多数がコアギュリンとなりゲル化してしまったものは除外する。これらの素材に対して必要に応じてエンドトキシンを吸着する物質、β−D−グルカンを吸着する物質、エンドトキシンの作用を失活させる物質、β−D−グルカンの作用を失活させる物質の少なくとも一つ以上を添加してもよい。
【0055】
エンドトキシンを吸着する物質としてはポリミキシンB、ポリリジン、ポリオルチニン、ポリエチレンイミンなど、あるいは、C因子自体やC因子がエンドトキシンを結合する部位を含む蛋白質やポリペプチド、エンドトキシンを結合する抗体などを例示できる。エンドトキシンを失活させる物質としては、鉄イオン、アルミニウムイオン、クロムイオン、ニッケルイオン、コバルトイオン、マンガンイオンを含む水溶液、ならびに、これらを供給する金属片などを例示できる。
【0056】
同様に、β−D−グルカンと結合する物質としてはレクチン、あるいは、G因子自体あるいはG因子がβ−D−グルカンを結合する部位を含む蛋白質やポリペプチド、β−D−グルカンを結合する抗体などを例示できる。また、β−D−グルカンを不活性化するためには、素材の水素イオン濃度を下げることにより、β−D−グルカンの溶解性を下げて析出させてしまうことも考えられる。
【0057】
このように必要に応じて最適な添加物を混合させた素材に対して、次に加熱処理を行う。この加熱処理を適当な加熱温度、加熱時間で行うことにより、素材中の所定の酵素群であるC因子、活性型C因子、B因子、活性型B因子、G因子、活性型G因子、凝固酵素前駆体、凝固酵素の少なくとも一部を不可逆的に不活性化させる。そして、エンドトキシンやβ−D−グルカンとの反応によって凝固酵素が生じないようにする。そうすれば、たとえエンドトキシンやβ−D−グルカンに汚染されても、加水分解してコアギュリンとなってゲル化することのないコアギュロゲン原料を得ることができる。
【0058】
この場合の加熱処理は素材の状態により、加熱温度と加熱時間を適宜調整すればよい。例えば、素材が溶液の場合、反応させる温度が40℃以上140℃以下であることが好ましく、60℃以上100℃以下であることがさらに好ましい。好ましい加熱時間は加熱温度によって大きく異なるが、30秒以上72時間以下であることが好ましく、10分以上8時間以下であることがさらに好ましい。
【0059】
加熱温度を高くしすぎたり、加熱時間を長くしすぎたりすると、LAL試薬あるいは、LAL中の蛋白質が変性して白濁してしまい、試料の濁りを測定する方法には使用が困難になったり、コアギュロゲンが変性して機能を失ってしまったりする可能性があるので注意を要する。
【0060】
一方、素材が、既に凍結乾燥体となっているLAL試薬である場合には、素材が溶液の場合と異なり、より高い加熱温度とより長い加熱時間が必要となる。加熱温度は80℃以上300℃以下が好ましく、100℃以上250℃以下がさらに好ましい。好ましい加熱時間は水溶液の場合と同様に加熱温度により異なるが、30秒以上72時間以下が好ましく、10分以上8時間以下がさらに好ましい。
【0061】
このように加熱処理した素材は必要に応じて種々の金属塩あるいはアンモニウム塩、酸、アルカリ、さらには、緩衝液などによりpHを調整したり、必要に応じて界面活性剤や糖類などを添加しても良い。
【0062】
このように製造したコアギュロゲン原料はそのままではエンドトキシンやβ−D−グルカンを作用させてもゲル化、あるいは、凝集反応を起こすことはない。しかしながら、活性化した凝固酵素を作用させるか、あるいは、加熱処理していないLAL試薬と混和後にエンドトキシンやβ−D−グルカンを作用させることにより速やかにコアギュリンへと加水分解されゲル化、あるいは、凝集反応が惹起される。
【0063】
また、本発明により製造したコアギュロゲン原料はエンドトキシンやβ−D−グルカンの測定感度の向上、測定時間の短縮、測定利便性の向上などに役立てることが可能である。すなわち、本発明のコアギュロゲン原料を、加熱処理していないLAL試薬と混和することにより、通常よりもコアギュロゲン濃度の高いLAL試薬を調製することが可能である。このLAL試薬にエンドトキシンやβ−D−グルカンを作用させることによって、より迅速で高感度な測定が可能となる。
【0064】
また、例えば、本発明によるコアギュロゲン原料をポリスチレンラテックスの微小な粒子の表面に結合または吸着させた状態のLAL試薬を調製することにより、従来の比濁法
によるエンドトキシン測定よりも大幅な時間短縮が可能な測定法が実現できる。
【0065】
すなわち、コアギュロゲン原料をポリスチレンラテックスの微粒子(以下、ビーズともいう)の表面に結合または吸着させたLAL試薬に、エンドトキシンやβ−D−グルカンによって活性化された凝固酵素を作用させると、コアギュロゲンが酵素カスケードにより加水分解されコアギュリンとなり、これらが微粒子同士を会合させる。従って、本発明により取得されたコアギュロゲン原料におけるコアギュロゲンを微粒子に結合または吸着しておくことで、より効率的に微粒子同士を会合させることが可能となり、より早期に凝集塊を生成させることが可能となる。
【0066】
本発明のコアギュロゲン原料におけるコアギュロゲンをビーズに結合させるには、ビーズが持つ電荷で吸着させる方法、ビーズのイオン的性質を利用する方法、ビーズ表面にコアギュロゲン原料中の蛋白質と反応可能な官能基を形成し、その官能基と蛋白質中のアミノ基、あるいは、カルボキシル基を利用して化学的に結合させる方法などが考えられるが、どのような方法を用いても良い。
【0067】
ところで、従来、LALの素材を改変してこのようにビーズに結合または吸着させるような操作をすると、製造にかかる時間が数時間以上という長期に及ぶため、他の素材中に微量に混入したエンドトキシンやβ−D−グルカンが影響したり、あるいは、作業中にそれらの生理活性物質が微量に混入したりして、原料がゲル化してしまい、そのままでは目的の物質を得ることができない場合があった。
【0068】
これに対して、従来は、原料に対して各種の酵素阻害剤を添加する対策が考えられた。この酵素阻害剤としては、Diisopropylfluorophosphate、Benzamidine、Phenylmethanesulfonyl fluoride、4-(2-Aminoethyl)-benzenesulfonyl fluoride、6-Amidino-2-naphthyl-4-guanidinobenzoatedimethanesulfonate、p-Amidinophenylmethylsulfonyl fluoride、Aprotinin、Antipain、Leupeptin、Ecotin、PPACK(Phe-Pro-Arg-chloromethylketone)、α2-Macroglobulin、Trypsin inhibitorなどを例示することができる。
【0069】
しかしながら、本発明のコアギュロゲン原料を用いた場合は、LAL中の酵素機能のみが不可逆的に不活性化されているとともにコアギュロゲン活性は維持されている。従って、上記のような酵素阻害剤を添加するまでもなく、ビーズへの結合または吸着操作中に素材中に混入したエンドトキシンやβ−D−グルカンによる、原料のゲル化を防止することができる。
【0070】
以下に、本発明のコアギュロゲン原料の製造方法の詳細について示す。製造例は製造方法の一例を示したものであって、本発明に係る製造方法は以下の製造例の条件に限定されるものではない。
【0071】
〔製造例1〕
LAL試薬の凍結乾燥体(リムルスES−IIシングルテストワコー、和光純薬製、以下ES−IIと略す。)に所定量(0.15mL)の注射用蒸留水(大塚製薬製)を加えてボルテックスミキサーで混和した。その後、予め所定温度(後述)に加熱しておいたアルミブロックヒーター(HDB−1N、アズワン製、以下特に断らない限りアルミブロックヒーターとしてこの機種を使用する。)にセットして所定時間(後述)の加熱を行った。加熱後すぐに氷中で冷却しコアギュロゲン原料の水溶液を得た。
【0072】
〔製造例2〕
LAL試薬(ES−II)の凍結乾燥体をそのまま、予め120℃に加熱しておいたアルミブロックヒーターにセットして所定時間(後述)の加熱を行った。加熱後すぐに氷中
で冷却しコアギュロゲン原料の凍結乾燥体を得た。
【0073】
〔製造例3〕
製造例1、ならびに製造例2で得られたコアギュロゲン原料の酵素活性の不可逆的な失活とコアギュロゲン機能の維持とを、エンドトキシンの定量が可能なレーザー散乱粒子計測法を用いて評価した。レーザー散乱粒子計測法においては、φ7mm、長さ50mmのガラス製で内部に試料を攪拌するためのステンレス製の攪拌子(φ1mm、長さ5mm)を内在した専用のガラス容器を使用した。この容器をエンドトキシンフリーにするために、容器の開口部をアルミ箔で覆い、さらに、20本ずつアルミ箔で小分けに梱包したものを鉄製の乾熱処理缶に詰め、250℃で3時間加熱処理して、エンドトキシンを熱分解した。このように乾熱処理によりエンドトキシンフリーにした測定容器を製造し試験に使用した。
【0074】
〔製造例4〕
本発明によるコアギュロゲン原料のコアギュロゲン機能、すなわち、活性化した凝固酵素によりコアギュロゲンが加水分解されてゲル化するか、あるいは、凝集反応を示すかを調べるために、凝固酵素の活性化物を以下のようにして得た。すなわち、加熱処理していないLAL試薬(ES−II)に1.0EU/mLの濃度のエンドトキシン水溶液200μLを作用させて、製造例3により製造した測定容器にLAL試薬とエンドトキシン水溶液の混合物を移注し、37℃に保温しながら容器内部のステンレス攪拌子を容器底面方向に配置した攪拌器により回転して反応を進行させた。
【0075】
レーザー光散乱粒子計測法では、エンドトキシンによりゲル化したLAL試薬が攪拌により微小な凝集塊として出現し、その凝集塊の出現時間と試料中のエンドトキシンの濃度は両対数プロットで直線に乗ることが知られている。この条件の場合、約6分で凝集が検出されたが、凝集検出後も攪拌を続け、測定開始から20分の時点で容器を装置から取り出し、容器内部のゲル凝集物を全量回収し、エンドトキシンフリーのディスポーザブル遠心管に移注して、15000rpm(ローター半径7cm)で2分間遠心して、コアギュリンポリマーが主成分であるゲル成分を沈殿させた。別の方法により、遠心上清にはコアギュロゲンが含まれていないことを確認した。この製造法により、エンドトキシンによって活性化した凝固酵素を調製した。
【0076】
〔製造例5〕
LAL試薬(ES−II)に所定濃度(後述)のエンドトキシン水溶液0.2mLを加えてボルテックスミキサーで混和した後、予め、100℃に加熱しておいたアルミブロックヒーターにセットして20分間の加熱を行った。加熱後すぐに氷中で冷却しエンドトキシンが混入したコアギュロゲン原料の水溶液を得た。
【0077】
〔製造例6〕
表面がカルボキシル化されたポリスチレンラテックス微粒子(Polybeads Carboxylate Microspheres、0.45μm、固形分2.63%、Polysciences Inc.製、以下、カルボキシルビーズと略す。)の懸濁液0.5mLをエンドトキシンフリーのスクリューキャップ付き遠心管(容量2.0mL)にて15000rpmで5分間遠心し、上澄みを除去した。次いで、注射用蒸留水を加えて2.0mLにし、懸濁させた後に再び遠心して上澄みを除去した。この遠心操作による上澄み除去をもう一度繰り返した後、注射用蒸留水1mLに再懸濁した後、エンドトキシンフリーのスクリューキャップ付き遠心管(容量15mL)に移注した。さらに、4mLの注射用蒸留水を加えた後にオートクレーブ(121℃、20分)を掛け、ビーズに混入するエンドトキシンを不活性化ならびに除去した。
【0078】
オートクレーブ後のカルボキシルビーズを容量2mLのスクリューキャップ付き遠心管
に移注し、上述した要領で遠心と上澄み除去、さらに、注射用蒸留水への再懸濁をセットとする洗浄操作を合計2回行った。次に、注射用蒸留水で溶解し、濃度が20mg/mLになるように調製した水溶性カルボジイミド(WSC、同仁化学製)水溶液10mLに0.1M酢酸バッファ(pH4.98)2mLを混合し、これを孔径φ0.2μmの滅菌用フィルタ(ミリポア製)に通し、このうちの0.5mLに上記のカルボキシルビーズの沈殿物を再懸濁させた。
【0079】
また、LAL試薬(ES−II)の凍結乾燥体に注射用蒸留水(大塚製薬製)0.5mLを加えて溶解し、60℃で10分間加熱をおこなった。この加熱処理により得られた本発明のコアギュロゲン原料0.5mLと上述の方法で調製したカルボキシルビーズ懸濁液0.5mL、さらに、1.0%になるように調製した非イオン性界面活性剤であるTriton
X−100水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)を10μL混合し、室温で2時間反応させた。
【0080】
この反応により、カルボキシルビーズのカルボキシル基がカルボジイミドで活性化された後、コアギュロゲンのアミノ基とアミド結合を起こして、ビーズ表面にコアギュロゲンが化学的に結合される。反応後、上記のように遠心、上澄み除去、注射用蒸留水による再懸濁を2回おこなって反応物を洗浄した。次いで、0.25Mのアミノエタノール水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)を100μL加えて、室温で20分間攪拌して、カルボキシルビーズ上の活性化したカルボキシル基のうち未反応のものを不活性化させた。その後、同様に注射用蒸留水で合計3回洗浄し、5mLの注射用蒸留水に再懸濁させ、上述の1.0%のTriton X−100水溶液と2%のアジ化ナトリウム水溶液(注射用蒸留水で調製後、孔径φ0.2μmの滅菌用フィルタで濾過)をそれぞれ50μLずつ加えてコアギュロゲン結合マイクロビーズを得た。
【0081】
以下に、本発明の実施例について説明する。
【0082】
〔実施例1〕
製造例1において、アルミブロックヒーターの指示温度を40℃、50℃、60℃、70℃、80℃、100℃、ならびに、120℃のいずれかの条件において、加熱時間を10分にして製造したコアギュロゲン原料から100μLをとり、製造例3で製造した測定容器に入れた。そこに、2.0EU/mLの濃度のエンドトキシン水溶液100μLを加えて作用させ、レーザー散乱粒子計測装置(PA−200、興和製、以下PA−200と略す。)で測定を行った。加熱処理なしの場合の測定濃度(C=1.0EU/mL)と、加熱したLAL試薬での測定濃度(S)から、加熱された後に残存する酵素活性である残存凝固酵素比活性(A)を以下の式で定義して導出した。
A=S/C×100(%)・・・・・(1)
【0083】
図1には、得られた残存凝固酵素比活性(A)とLAL試薬水溶液の加熱温度との関係を示す。図1に示したように、加熱時間を10分とした場合には、加熱温度が60℃以上になると酵素活性が完全に消失することが分かった。また、図示しないが、加熱温度が高いほど製造後のコアギュロゲン原料水溶液は白濁する現象が観察された。例えば、120℃で加熱してコアギュロゲン原料水溶液を製造した場合、かなりの部分が熱変性によるものと見られる白濁と不溶物に変化してしまっており、実使用には適さないことが判った。
【0084】
〔実施例2〕
製造例1において、アルミブロックヒーターの指示温度を40℃、50℃、60℃、70℃、80℃、100℃、ならびに、120℃のいずれかの条件において、加熱時間を10分にして製造したコアギュロゲン原料(150μL)に、製造例4で得られた活性化した凝固酵素水溶液50μLを加えて作用させ、ボルテックスミキサーで5秒間混和させた
。その試料を製造例3で製造した測定容器に移注し、レーザー散乱粒子計測装置(PA−200)で凝集開始時間の測定を行った。
【0085】
表1にそれぞれの加熱温度に対する凝集開始時間を示したが、いずれの加熱温度においても活性化した凝固酵素水溶液を添加してから3分程度で凝集が開始しており、コアギュロゲンの機能は加熱による影響を殆ど受けていないことが示された。また、120℃という高温で処理した場合には、LAL試薬中の大部分の蛋白質は熱変性して凝固してしまうものの、その中に残存するコアギュロゲン自体は機能を維持しており、活性化した凝固酵素水溶液の添加から4分程度で凝集が開始した。このことから、コアギュロゲンは非常に加熱に強いことが示された。また、表1から分かるように、LAL試薬を80℃以下で加熱処理して酵素活性を失活させた場合には、LAL試薬に白濁は殆ど見られなかった。従って、LAL試薬を80℃以下で加熱処理して酵素活性を失活させることで、光学的な測定にも適した、より利用価値の高いコアギュロゲン原料を取得することが可能となることが分かった。
【0086】
【表1】
【0087】
〔実施例3〕
製造例2において、加熱時間を10分、30分、60分、120分、ならびに、300分のいずれかの時間にして、LAL試薬(リムルスES−IIシングルテストワコー、和光純薬製)を開封せずにそのまま加熱処理したコアギュロゲン原料の凍結乾燥体に1.0EU/mLのエンドトキシン水溶液(200μL)を添加した。そして、ボルテックスミキサーで5秒間混和した後、製造例3により製造した測定容器に移注して、レーザー光散乱粒子計測装置(PA−200)にて、加熱処理の後に残存する酵素の活性を調べた。そして、上述の式(1)で定義される残存凝固酵素比活性(%)を算出した。
【0088】
図2には本実施例で得られた残存凝固酵素比活性と、LAL試薬凍結乾燥体の加熱処理時間との関係を示す。図2に示したように、実施例1の場合と比較して、高温下で長時間の加熱処理を行っても、なかなか、酵素活性が完全に消失しないことが分かる。しかしながら、120℃で300分の加熱処理により、残存凝固酵素比活性は0.2%程度に減少した。このように製造したコアギュロゲン原料では、原料を水に溶解させたときの水溶液の色がやや黄変する傾向が見られたが、白濁や凝固不溶物の発生はなく、水に速やかに溶解した。
【0089】
〔実施例4〕
実施例4においては、予めエンドトキシンが混入した状態でLAL試薬を加熱処理した場合の、混入したエンドトキシンの濃度と加熱処理による酵素の不活性化の程度との関係を調べた。製造例5において、予め混入させておくエンドトキシン水溶液のエンドトキシン濃度10、1、0.1EU/mLとして、それぞれの濃度のエンドトキシンが混入したコアギュロゲン原料を得た。
【0090】
次に、本実施例においては、上記のエンドトキシンが混入したコアギュロゲン原料と比較するためのコントロールコアギュロゲン原料を、エンドトキシンを含まない注射用蒸留水(大塚製薬製)0.2mLで溶解したLAL試薬を製造例5に示した手順で加熱することで調製した。さらに、エンドトキシン水溶液の希釈系列を作り、それぞれ200μLを加熱処理していないLAL試薬(ES−II)に入れてボルテックスミキサーで5秒間攪拌した後、それぞれの試料から100μLを取り、上記のコントロールコアギュロゲン原料100μLと混和し、レーザー光散乱粒子計測装置(PA−200)にてエンドトキシン濃度の検量線を得た。
【0091】
次に、加熱処理していないLAL試薬(ES−II)を注射用蒸留水(大塚製薬製)200μLで溶解し、そのうちの100μLを上記の予めエンドトキシンが混入したそれぞれのコアギュロゲン原料100μLとボルテックスミキサーで5秒間混和させた。そして、レーザー光散乱粒子計測装置(PA−200)でエンドトキシン濃度を上記で得られた検量線に照らし合わせて算出した。得られた結果を表2に示した。表2に示すように、いずれの試料においても測定されたエンドトキシン濃度は、実際に混入したエンドトキシン濃度より低かった。そして、表中の加熱後の活性残存率が示すように、得られたエンドトキシン濃度の実際の値からの低下の度合は、混入させたエンドトキシンの濃度が低いほど大きかった。このように、予め混入させたエンドトキシンの濃度が低いほど加熱処理による酵素の不活性化の効果が大きいことが示された。
【0092】
【表2】
【0093】
〔実施例5〕
製造例6により製造されたコアギュロゲン結合マイクロビーズを用いてエンドトキシン水溶液の希釈系列中のエンドトキシン濃度を攪拌比濁法を用いて測定した(以下、この測定法をコアギュロゲン結合マクロビーズ法とする。)。攪拌比濁法は従来法の比濁法と異なり、試料を攪拌しつつ、試料の透過率の変化の度合からエンドトキシンの濃度を測定する。
【0094】
図3には、本実施例の攪拌比濁測定装置としての比濁計測装置1の概略構成を示す。本実施例の攪拌比濁法においては、試料は混和液保持手段としての専用のガラス製容器2に移注する。ガラス製容器2の周囲を囲うように保温器5が設けられている。この保温器5の内部には図示しない電熱線が備えられており、この電熱線に通電されることにより、ガラス製容器2を約37℃に保温するようになっている。このガラス製容器2の中にはステンレス製の攪拌子3が備えられている。この攪拌子3は、ガラス製容器2の下部に設置された攪拌器4の作用によってガラス製容器2の中で回転する。すなわち、攪拌器4はモータ4aとモータ4aの出力軸に設けられた永久磁石4bとからなっている。そして、モータ4aに通電されることで永久磁石4bが回転する。この永久磁石4bからの磁界が回転するために、ステンレス製の攪拌子3が回転磁界の作用で回転する。この攪拌子3と攪拌器4とは攪拌手段に相当する。
【0095】
なお、比濁計測装置1には光入射手段としての光源6と受光手段としての受光素子9が
設置されている。光源6から出射した光はアパーチャ7を通過した後、保温器5に設けられた入射孔5aを通過してガラス製容器2中の試料に入射される。ガラス製容器2中の試料を透過した光は保温器5に設けられた出射孔5bから出射され、アパーチャ8を通過して受光素子9に照射される。受光素子9では、受光した光の強度に応じた光電信号を出力する。この光電信号の出力より、導出手段としての演算装置10において試料の透過率が算出される。
【0096】
熱処理していないLAL試薬(ES−II)2本を200μLの注射用蒸留水に溶解させ、そこに、製造例6で製造したコアギュロゲン結合マイクロビーズ50μLを入れてボルテックスミキサーで5秒間攪拌しコアギュロゲン結合マイクロビーズ−LAL試薬混合物を得た。エンドトキシン水溶液の希釈系列、すなわち、2、0.2、0.02、0.002EU/mLのいずれか50μLと上記のコアギュロゲン結合マイクロビーズ−LAL試薬混合物50μLを上記の製造例3に準じて製造したガラス製容器2に入れて比濁計測装置1にて測定を行った。
【0097】
コアギュロゲン結合マイクロビーズ法では、作用させたエンドトキシンによりLAL試薬中の凝固酵素が活性化されると、同じくLAL試薬中に含まれるコアギュロゲンがコアギュリンとなり、同時に、コアギュロゲン結合マイクロビーズ上のコアギュロゲンも加水分解されてコアギュリンとなるが、ビーズがこれらのコアギュリンにより架橋されて大きな凝集塊を形成する。試料には、当初、多量の単体ビーズが含まれており強く濁っているが、凝集により単体のビーズ濃度が急激に減少するため、試料の光透過率が上昇する。コアギュロゲン結合マイクロビーズ法ではこの光の透過率の急激な上昇により凝集塊の発生開始時間を決定する。
【0098】
図4には、従来の活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程と、コアギュロゲン結合マイクロビーズ法におけるコアギュロゲンのゲル化・凝集過程とを示す。図4(A)に示したのは従来の活性型凝固酵素によるコアギュロゲンのゲル化・凝集過程である。図に示すように、粒径にして数nm程度のコアギュロゲン100が活性型凝固酵素によって加水分解されてコアギュリン101となる。これらのコアギュリン101は凝集して多量体となり攪拌比濁法においてはゲル粒子を形成する。その後、時間の経過とともにコアギュリン101の凝集はさらに進行しゲル粒子の粒径は徐々に大きくなる。そして、比較的長いラグタイムが経過した時点で測定可能な数μm〜数十μmの粒径のゲル粒子102となる。
【0099】
これに対し、コアギュロゲン結合マイクロビーズ法では、図4(B)に示すように、ビーズ103上に結合したコアギュロゲン100に活性型凝固酵素が作用すると、ビーズ103上のコアギュロゲン100が酵素カスケードによって加水分解されてコアギュリン101となり、コアギュリン101が凝集する過程ではコアギュリン101がビーズ103同士を会合させる。このことにより、コアギュリン101とビーズ103を主成分とする粒子の径が早急に大きくなり、短時間のラグタイムにおいて測定可能な粒径を得ることとなる。なお、この凝集反応はLAL単体で惹起される凝集反応よりも、非常に強大であることも明確になってきている。
【0100】
また、本実施例においては、コアギュロゲン結合マイクロビーズ法を用いた場合と比較するために比濁法によってもエンドトキシン濃度測定を行った。比濁法では、熱処理していないLAL試薬に注射用水100μL、エンドトキシン水溶液希釈系列のいずれかの濃度の試料を100μL入れて比濁法装置(トキシノメータET−2000、和光純薬製)で測定を行った。コアギュロゲン結合マイクロビーズ法と比濁法の比較結果を図5に示したが、両測定法とも横軸にエンドトキシン濃度、縦軸に検出時間を取ると、その両軸対数プロットは直線に近似された。近似曲線はコアギュロゲン結合マイクロビーズ法がY=−
0.190X+0.738、比濁法ではY=−0.349X+0.869であることから、コアギュロゲン結合マイクロビーズ法が比濁法に比べて大幅に測定時間を短縮可能であること、作用させるエンドトキシンの濃度が低いほど測定に要する時間の差が大きく開いてくることが示された。測定に要する実時間などを表3にまとめた。
【0101】
【表3】
【0102】
本実施例において、ビーズ103の素材は特に限定されるものではないが、ポリスチレンラテックス樹脂の他、シリカ、シリコン樹脂、セルロース樹脂、ポリビニルアルコール樹脂、ハイドロキシアパタイトなどが例示でき、ポリスチレンラテックス樹脂、シリカ、セルロース樹脂が望ましい。
【0103】
また、ビーズ103の大きさは、凝集により早期に光学的に検出可能となる条件と、調製時のハンドリングの容易さ、系内への分散のし易さなどから、0.05μmから50μmの範囲であるものが用いられる。ビーズ103の表面にコアギュロゲンを結合させるには、静電的、親水的、あるいは、疎水的にコアギュロゲンを吸着させる方法と、化学的に結合させる方法とが考えられる。
【符号の説明】
【0104】
1・・・比濁計測装置
2・・・ガラス製容器
3・・・攪拌子
4・・・攪拌器
4a・・・モータ
4b・・・磁石
5・・・保温器
5a・・・入射孔
5b・・・出射孔
6・・・光源
7・・・アパーチャ
8・・・アパーチャ
9・・・受光素子
10・・・演算装置
100・・・コアギュロゲン
101・・・コアギュリン
102・・・凝集粒子
103・・・ビーズ
【特許請求の範囲】
【請求項1】
所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料の製造方法であって、
LALを所定温度で所定時間に亘り加熱処理することで、該LAL中の前記酵素群の少なくとも一部を失活させるとともにコアギュロゲンの活性は維持させることを特徴とするコアギュロゲン原料の製造方法。
【請求項2】
前記LALは、溶液として供給され、
前記所定温度は60℃以上であることと特徴とする請求項1に記載のコアギュロゲン原料の製造方法。
【請求項3】
前記LALは、溶液として供給され、
前記所定温度は80℃以下であることと特徴とする請求項1または2に記載のコアギュロゲン原料の製造方法。
【請求項4】
前記LALは、溶液として供給され、
前記所定時間は10分以上8時間以下であることを特徴とする請求項1から3のいずれか一項に記載のコアギュロゲン原料の製造方法。
【請求項5】
前記LALは、凍結乾燥体として供給され、
前記所定温度は100℃以上250℃以下であることを特徴とする請求項1に記載のコアギュロゲン原料の製造方法。
【請求項6】
前記LALは、凍結乾燥体として供給され、
前記所定時間は300分以上であることを特徴とする請求項1または5に記載のコアギュロゲン原料の製造方法。
【請求項7】
所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料であって、
LALを所定温度で所定時間に亘り加熱処理することで、前記LAL中の前記酵素群の少なくとも一部を失活させたことを特徴とするコアギュロゲン原料。
【請求項8】
前記LALは、溶液として供給され、
前記所定温度は60℃以上であることと特徴とする請求項7に記載のコアギュロゲン原料。
【請求項9】
前記LALは、溶液として供給され、
前記所定温度は80℃以下であることと特徴とする請求項7または8に記載のコアギュロゲン原料。
【請求項10】
前記LALは、溶液として供給され、
前記所定時間は10分以上8時間以下であることを特徴とする請求項7から9のいずれか一項に記載のコアギュロゲン原料。
【請求項11】
前記LALは、凍結乾燥体として供給され、
前記所定温度は100℃以上250℃以下であることを特徴とする請求項7に記載のコアギュロゲン原料。
【請求項12】
前記LALは、凍結乾燥体として供給され、
前記所定時間は300分以上であることを特徴とする請求項7または11に記載のコアギュロゲン原料。
【請求項13】
請求項7から12のいずれか一項に記載のコアギュロゲン原料と、加熱処理されていないLALとを混合することで、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたことを特徴とする、前記生理活性物質の検出または前記生理活性物質の濃度測定に使用されるLAL試薬。
【請求項14】
請求項7から12のいずれか一項に記載のコアギュロゲン原料におけるコアギュロゲンを、該コアギュロゲンより大径の多数の微粒子の表面に結合または吸着させて調製されたことを特徴とするエンドトキシンの測定用のコアギュロゲン結合マイクロビーズ。
【請求項15】
請求項13に記載のLAL試薬と、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記LAL試薬中の酵素群が活性化されて生じる酵素カスケードによって、濃度が高められた前記コアギュロゲンがコアギュリンに加水分解されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法。
【請求項16】
請求項14に記載のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記加熱処理されていないLALにおける酵素群が活性化されて生じる酵素カスケードによって前記微粒子の表面に結合または吸着したコアギュロゲンがコアギュリンに加水分解され、前記微粒子同士が架橋されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法。
【請求項17】
請求項14に記載のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料との混和液を光の入射可能に保持し、前記混和液における反応を進行させる混和液保持手段と、
前記混和液保持手段中の前記混和液を攪拌する攪拌手段と、
前記混和液保持手段中の混和液に光を入射する光入射手段と、
前記入射光の前記混和液における透過光を受光し電気信号に変換する受光手段と、
前記受光手段において変換された電気信号から取得される前記混和液の透過率より前記試料中の前記生理活性物質の濃度を導出する導出手段と、
を備えることを特徴とする攪拌比濁測定装置。
【請求項1】
所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料の製造方法であって、
LALを所定温度で所定時間に亘り加熱処理することで、該LAL中の前記酵素群の少なくとも一部を失活させるとともにコアギュロゲンの活性は維持させることを特徴とするコアギュロゲン原料の製造方法。
【請求項2】
前記LALは、溶液として供給され、
前記所定温度は60℃以上であることと特徴とする請求項1に記載のコアギュロゲン原料の製造方法。
【請求項3】
前記LALは、溶液として供給され、
前記所定温度は80℃以下であることと特徴とする請求項1または2に記載のコアギュロゲン原料の製造方法。
【請求項4】
前記LALは、溶液として供給され、
前記所定時間は10分以上8時間以下であることを特徴とする請求項1から3のいずれか一項に記載のコアギュロゲン原料の製造方法。
【請求項5】
前記LALは、凍結乾燥体として供給され、
前記所定温度は100℃以上250℃以下であることを特徴とする請求項1に記載のコアギュロゲン原料の製造方法。
【請求項6】
前記LALは、凍結乾燥体として供給され、
前記所定時間は300分以上であることを特徴とする請求項1または5に記載のコアギュロゲン原料の製造方法。
【請求項7】
所定の酵素群とコアギュロゲンとを含んだカブトガニの血球抽出物であるLALと、生物由来の所定の生理活性物質とを反応させ、前記生理活性物質によって前記酵素群が活性化されて生じる酵素カスケードによってコアギュロゲンがコアギュリンに加水分解されることを利用して、前記生理活性物質を検出または前記生理活性物質の濃度を測定する際に使用される、コアギュロゲン原料であって、
LALを所定温度で所定時間に亘り加熱処理することで、前記LAL中の前記酵素群の少なくとも一部を失活させたことを特徴とするコアギュロゲン原料。
【請求項8】
前記LALは、溶液として供給され、
前記所定温度は60℃以上であることと特徴とする請求項7に記載のコアギュロゲン原料。
【請求項9】
前記LALは、溶液として供給され、
前記所定温度は80℃以下であることと特徴とする請求項7または8に記載のコアギュロゲン原料。
【請求項10】
前記LALは、溶液として供給され、
前記所定時間は10分以上8時間以下であることを特徴とする請求項7から9のいずれか一項に記載のコアギュロゲン原料。
【請求項11】
前記LALは、凍結乾燥体として供給され、
前記所定温度は100℃以上250℃以下であることを特徴とする請求項7に記載のコアギュロゲン原料。
【請求項12】
前記LALは、凍結乾燥体として供給され、
前記所定時間は300分以上であることを特徴とする請求項7または11に記載のコアギュロゲン原料。
【請求項13】
請求項7から12のいずれか一項に記載のコアギュロゲン原料と、加熱処理されていないLALとを混合することで、前記加熱処理されていないLALにおけるコアギュロゲン濃度を高めたことを特徴とする、前記生理活性物質の検出または前記生理活性物質の濃度測定に使用されるLAL試薬。
【請求項14】
請求項7から12のいずれか一項に記載のコアギュロゲン原料におけるコアギュロゲンを、該コアギュロゲンより大径の多数の微粒子の表面に結合または吸着させて調製されたことを特徴とするエンドトキシンの測定用のコアギュロゲン結合マイクロビーズ。
【請求項15】
請求項13に記載のLAL試薬と、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記LAL試薬中の酵素群が活性化されて生じる酵素カスケードによって、濃度が高められた前記コアギュロゲンがコアギュリンに加水分解されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法。
【請求項16】
請求項14に記載のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料とを混和させることで、
前記所定の生理活性物質によって前記加熱処理されていないLALにおける酵素群が活性化されて生じる酵素カスケードによって前記微粒子の表面に結合または吸着したコアギュロゲンがコアギュリンに加水分解され、前記微粒子同士が架橋されることを利用して、前記所定の生理活性物質を検出または前記所定の生理活性物質の濃度を測定することを特徴とする生物由来の生理活性物質の測定方法。
【請求項17】
請求項14に記載のコアギュロゲン結合マイクロビーズと、加熱処理されていないLALと、前記所定の生理活性物質を含む試料との混和液を光の入射可能に保持し、前記混和液における反応を進行させる混和液保持手段と、
前記混和液保持手段中の前記混和液を攪拌する攪拌手段と、
前記混和液保持手段中の混和液に光を入射する光入射手段と、
前記入射光の前記混和液における透過光を受光し電気信号に変換する受光手段と、
前記受光手段において変換された電気信号から取得される前記混和液の透過率より前記試料中の前記生理活性物質の濃度を導出する導出手段と、
を備えることを特徴とする攪拌比濁測定装置。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2010−190801(P2010−190801A)
【公開日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願番号】特願2009−37150(P2009−37150)
【出願日】平成21年2月19日(2009.2.19)
【出願人】(000163006)興和株式会社 (618)
【Fターム(参考)】
【公開日】平成22年9月2日(2010.9.2)
【国際特許分類】
【出願日】平成21年2月19日(2009.2.19)
【出願人】(000163006)興和株式会社 (618)
【Fターム(参考)】
[ Back to top ]