
コアシェルポリマ
【課題】耐熱性等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与える熱硬化性樹脂組成物に用いられるコアシェルポリマを提供する。
【解決手段】コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマ。
【解決手段】コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマ。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、熱硬化性樹脂組成物に用いられるコアシェルポリマに関する。
【背景技術】
【0002】
従来より、エポキシ樹脂の靭性を強化する方法としては、エポキシ樹脂組成物中にゴム成分あるいは熱可塑性樹脂などの改質剤を添加する方法が知られている。ゴム成分を添加する方法としては、反応性液状ゴム(CTBNなど)やニトリルゴムを添加する方法(例えば、特許文献1参照)が知られている。しかし、反応性液状ゴムは、一旦エポキシ樹脂へ溶解した後、硬化時に相分離するという過程を経るため、配合するエポキシ樹脂の種類や硬化条件の違いによって得られる硬化物のモルホロジーが変化し、所望の改質効果が得られなかったり、品質の再現性に問題があることに加え、硬化後のエポキシ樹脂相にゴム成分が一部溶解し残存するために、硬化物の弾性率やガラス転移温度(以下、Tgとも言う。)が低下してエポキシ樹脂製品の品質が低下する等の問題があった。さらに、エポキシ樹脂製品自体が大型で、製品作成のための硬化温度が部位によって異なると、部位により品質が異なってしまう虞もあった。
【0003】
ゴム成分の添加によって問題となるモルホロジー変化とその制御の問題を解決する方法として、エポキシ樹脂中でアクリル酸エステル等のモノマーを重合することで、エポキシ樹脂中にゴム状粒子が分散した組成物を得る方法が知られている(例えば、特許文献2参照)。しかしながら、上記方法でもゴム成分の一部が硬化後のエポキシ樹脂相に溶解することを避けられず、ガラス転移温度が低下する場合があり、品質的に十分なものとは言えなかった。
【0004】
一方、熱可塑性樹脂を添加する方法としては、ガラス転移温度の高い熱可塑性樹脂(いわゆるスーパーエンプラなど)が使用されることが周知である。この方法では、硬化物のガラス転移温度、耐熱性を保持したままある程度の靭性を付与することが可能であるが、一般に添加量を多く要するため、系の粘度が増加し、取り扱い性に問題があり、溶解などの煩雑な工程が必要となったり、モルホロジーのコントロールが必要であるという問題があった。
【0005】
また、エポキシ樹脂に不溶なゴム状重合体粒子を含むエポキシ樹脂組成物は、ゴム成分が硬化後のエポキシ樹脂相に溶解することがないため、耐熱性(ガラス転移温度)の低下を抑制することができる。この場合のゴム状重合体粒子はエポキシ樹脂中で重合したものではなく、あらかじめ重合しておいたものをエポキシ樹脂に混合したものである。このようなゴム状重合体粒子としては、いわゆるコアシェルポリマが代表的なものである(例えば、特許文献3若しくは特許文献4参照)。また、これらのコアシェルポリマは、一次粒子の集合体(凝集体)として、例えば、数十〜数百ミクロンのパウダー状で市販されており、エポキシ樹脂に混合するに際しては、これらを10μm未満に微粉末化したり、更に50〜200℃の温度で加熱攪拌、高速せん断攪拌、熱ロール、インターミキサー、ニーダーや三本ロール等の混錬機で入念に混合しなければ、混合したコアシェルポリマが容易に沈殿あるいは浮上して分離する問題がある。更に数時間に亘るような入念な混合混錬を経た後でも、混合したコアシェルポリマは一次粒子で分散せずに凝集しており、さらにエポキシ樹脂の種類によっては混合したコアシェルポリマが分離しやすいなどの問題や、分散安定剤などを添加する必要があるなど、満足できるものではない。更に、エポキシ樹脂中で実際に分散しているコアシェルポリマの大きさは一次粒子ではないため、コアシェルポリマ粒子の設計を最適化することが難しいなどの問題点があった。これらの背景から、市販されているパウダー状のコアシェルポリマは、エポキシ樹脂の強化剤として充分な性能を有していないのが現状であった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特公昭62−34251号公報
【特許文献2】特開昭59−138254号公報
【特許文献3】米国特許第3322852号明細書
【特許文献4】米国特許第3496250号明細書
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、上記のような従来技術が有するエポキシ樹脂強化に際しての種々の問題点を克服し、耐熱等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与える熱硬化性樹脂組成物に用いられるコアシェルポリマを提供することにある。
【課題を解決するための手段】
【0008】
すなわち、本発明は、(1)コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマに関する。
【0009】
また、本発明は、(2)シェル部が、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマからなる前記(1)記載のコアシェルポリマに関する。
【0010】
また、本発明は、(3)コア部が、ジエン系ゴム重合体、アクリル系ゴム重合体、シリコン系ゴム重合体及びオレフィン系ゴム重合体の中から選択される少なくとも1種であり、コア部のガラス転移温度が0℃以下である前記(1)又は(2)に記載のコアシェルポリマに関する。
【0011】
また、本発明は、(4)架橋微粒子である前記(1)〜(3)のいずれかに記載のコアシェルポリマに関する。
【0012】
また、本発明は、(5)粒子径が30〜500nmである前記(1)〜(4)のいずれかに記載のコアシェルポリマに関する。
【発明の効果】
【0013】
本発明によれば、耐熱性等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与える熱硬化性樹脂組成物に用いられるコアシェルポリマを提供することができる。
【図面の簡単な説明】
【0014】
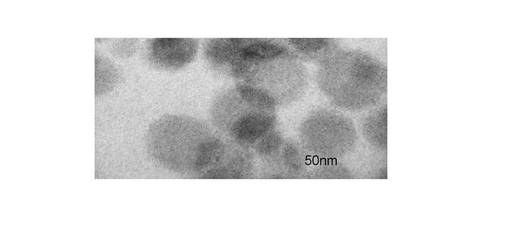
【図1】本発明の硬化物の透過電子顕微鏡像(TEM像)である。
【発明を実施するための形態】
【0015】
本発明の熱硬化性樹脂組成物は、エポキシ樹脂(A)と、シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有するコアシェルポリマ(B)と、硬化剤及び/または硬化触媒(C)を含むことを特徴とする。
【0016】
本発明に用いられるエポキシ樹脂(A)は、多層回路基板の層間絶縁膜あるいは平坦化膜、電子部品等の保護膜あるいは電気絶縁膜などに用いられるエポキシ樹脂であれば特に限定されないが、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、水添ビスフェノールA型エポキシ樹脂、水添ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビフェニル型エポキシ樹脂、ナフタレン型エポキシ樹脂、フルオレン型エポキシ樹脂、スピロ環型エポキシ樹脂、ビスフェノールアルカン類型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、オルソクレゾールノボラック型エポキシ樹脂、臭素化クレゾールノボラック型エポキシ樹脂、トリスヒドロキシメタン型エポキシ樹脂、テトラフェニロールエタン型エポキシ樹脂、脂環型エポキシ樹脂、アルコール型エポキシ樹脂、ブチルグリシジルエーテル、フェニルグリシジルエーテル、クレジルグリシジルエーテル、ノニルグリシジルエーテル、ジエチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリセリンポリグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、1,6−へキサンジオールジグリシジルエーテル、トリメチロールプロパントリグリシジルエーテル、ヘキサヒドロフタル酸ジグリシジルエーテル、脂肪酸変性エポキシ樹脂、トルイジン型エポキシ樹脂、アニリン型エポキシ樹脂、アミノフェノール型エポキシ樹脂、1,3−ビス(N,N−ジグリシジルアミノメチル)シクロヘキサン、ヒンダトイン型エポキシ樹脂、トリグリシジルイソシアヌレート、テトラグリシジルジアミノジフェニルメタン、ジフェニルエーテル型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ダイマー酸ジグリシジルエステル、ヘキサヒドロフタル酸ジグリシジルエステル、ダイマー酸ジグリシジルエーテル、シリコーン変性エポキシ樹脂、ケイ素含有エポキシ樹脂、ウレタン変性エポキシ樹脂、NBR変性エポキシ樹脂、CTBN変性エポキシ樹脂、エポキシ化ポリブタジエンなどが挙げられる。これらのなかでも、フェノールノボラック型エポキシ樹脂が好ましい。また、これらのエポキシ樹脂は1種単独でまたは2種以上を混合して使用してもよい。
【0017】
本発明で用いることのできるコアシェルポリマ(B)は、コア部(B−1)と、コア部の外層を形成するシェル部(B−2)より構成される。コア部(B−1)はエラストマーまたはゴム状のポリマを主成分とするポリマからなることが好ましく、シェル部(B−2)はコア部(B−1)にグラフト重合されたポリマ成分からことが好ましい。
【0018】
前記コア部(B−1)を構成するポリマはシェル部(B−2)とのグラフトを阻害するものでなければ特に制限は無く、例えば、ブタジエンゴム、スチレン−ブタジエンゴム、アクリロニトリル−ブタジエンゴム等のジエン系ゴム重合体;アクリル酸ブチルゴム、ブタジエン−アクリル酸ブチルゴム、アクリル酸2-エチルヘキシル−アクリル酸ブチルゴム、メタクリル酸2-エチルヘキシル−アクリル酸ブチルゴム、アクリル酸ステアリル−アクリル酸ブチルゴム、ジメチルシロキサン−アクリル酸ブチルゴム、シリコン系/アクリル酸ブチル複合ゴム等のアクリル系ゴム重合体;エチレン−プロピレンゴム、エチレン−プロピレン−ジエンゴム等のオレフィン系ゴム重合体;ポリジメチルシロキサン等のシリコン系ゴム重合体などが挙げられ、これらは、単独でまたは2種以上を混合して使用してもよい。これらのなかでも、スチレン−ブタジエンゴム、アクリロニトリル−ブタジエンゴム、アクリル酸2-エチルヘキシル−アクリル酸ブチルゴムなどが賞用される。
【0019】
前記コア部(B−1)は、ガラス転移温度が0℃以下であることが好ましく、
−10℃以下であることがより好ましい。前記コア部(B−1)のガラス転移温度が0℃を超える場合は、得られる熱硬化性樹脂組成物の熱膨張係数が小さくなり難い傾向にある。コア部(B−1)のガラス転移温度は、示差走査熱量法(DSC法)により測定することが出来る。
前記コアシェルポリマのシェル部は、ポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有するものである。側鎖の原子数が3未満である場合は、エポキシ樹脂と混合後のライフが短く、ある程度放置してから硬化すると、凝集物が多くなり外観・性能悪化等の問題が出る傾向にある。
【0020】
である。側鎖の原子数は、好ましくは3〜18、より好ましくは6〜12である。かかるシェル部(B−2)を構成するポリマは、コア部(B−1)を構成するポリマにグラフト重合されており、実質的にコア部(B−1)を構成するポリマと結合していることが好ましく、後述する有機溶媒及びエポキシ樹脂(A)に対して膨潤性、相溶性もしくは親和性を有するものが好ましい。シェル部(B−2)の理論ガラス転移温度は、好ましくは20℃以下、より好ましくは10℃以下、特に好ましくは0℃以下である。前記シェル部(B−2)の理論ガラス転移温度が20℃を超える場合は、得られる熱硬化性樹脂組成物の熱膨張係数が小さくなり難い傾向にある。シェル部(B−2)の理論ガラス転移温度は、下式から算出することができる。1/Tg=W1/T1+W2/T2+・・・Wn/Tn
式中のW1、W2・・・Wnは各モノマーの重量%(=(各モノマーの配合量/モノマー全重量)×100)であり、T1、T2・・・Tnは、各モノマーのホモポリマーのガラス転移温度(絶対温度)である。
【0021】
シェル部(B−2)を構成するポリマは、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマであることが好ましい。炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物としては、例えば、2−(メタ)アクリロイルオキシエチルコハク酸、2−(メタ)アクリロイルオキシエチルフタル酸、2−(メタ)アクリロイルオキシエチルマレイン酸、2−(メタ)アクリロイルオキシエチルヘキサヒドロフタル酸などが挙げられ、これらは、単独でまたは2種以上を混合して使用してもよい。これらのなかでも、2−アクリロイルオキシエチルコハク酸、2−アクリロイルオキシエチルフタル酸などが賞用される。
【0022】
上記化合物は、無水コハク酸、無水フタル酸、無水マレイン酸、無水ヘキサヒドロフタル酸などのジカルボン酸無水物と(メタ)アクリル酸2−ヒドロキシエチルと脱水反応させることにより得られる。シェル部としてエステル部分に(メタ)アクリロイルオキシアルキレン基を有する炭素数が1以下のジカルボン酸ハーフエステルを重合して得られるポリマを用いたコアシェルポリマは、エポキシ樹脂と混合後のライフが短く、ある程度放置してから硬化すると、凝集物が多くなり外観・性能悪化等の問題が出る傾向にある。
【0023】
また、良好なグラフト重合性と、エポキシ樹脂に対する親和性の双方を可能にできるという点から、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物と共重合可能な単量体を共重合してもよい。かかる共重合可能な単量体としては、例えば、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸プロピル、(メタ)アクリル酸n−ブチル、(メタ)アクリル酸i−ブチル、(メタ)アクリル酸t−ブチル、(メタ)アクリル酸ペンチル、(メタ)アクリル酸n−ヘキシル、(メタ)アクリル酸2−エチルヘキシル、(メタ)アクリル酸n−オクチル、(メタ)アクリル酸ドデシル、(メタ)アクリル酸オクタデシル、(メタ)アクリル酸ブトキシエチル、(メタ)アクリル酸フェニル、(メタ)アクリル酸ベンジル、(メタ)アクリル酸ナフチル、(メタ)アクリル酸グリシジル等の(メタ)アクリル酸エステル類;α−メチルスチレン、α−エチルスチレン、α−フルオロスチレン、α−クロルスチレン、α−ブロモスチレン、フルオロスチレン、クロロスチレン、ブロモスチレン、メチルスチレン、メトキシスチレン等の芳香族ビニル化合物;(メタ)アクリルアミド、N−ジメチル(メタ)アクリルアミド、N−ジエチル(メタ)アクリルアミド等の(メタ)アクリルアミド類;(メタ)アクリル酸カルシウム、(メタ)アクリル酸バリウム、(メタ)アクリル酸鉛、(メタ)アクリル酸アクリル酸すず、(メタ)アクリル酸亜鉛等の(メタ)アクリル酸金属塩等の不飽和脂肪酸;(メタ)アクリロニトリル等のシアン化ビニル化合物、(メタ)アクリル酸シクロペンチル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸メチルシクロヘキシル、(メタ)アクリル酸トリメチルシクロヘキシル、(メタ)アクリル酸ノルボルニル、(メタ)アクリル酸ノルボルニルメチル、(メタ)アクリル酸シアノノルボルニル、(メタ)アクリル酸イソボルニル、(メタ)アクリル酸ボルニル、(メタ)アクリル酸メンチル、(メタ)アクリル酸フェンチル、(メタ)アクリル酸アダマンチル、(メタ)アクリル酸ジメチルアダマンチル、(メタ)アクリル酸トリシクロ〔5.2.1.02,6〕デカ−8−イル、(メタ)アクリル酸トリシクロ〔5.2.1.02,6〕デカ−4−メチル、(メタ)アクリル酸シクロデシル等の脂環式炭化水素基を有する(メタ)アクリル酸エステル;N−メチルマレイミド、N−エチルマレイミド、N−プロピルマレイミド、N−i−プロピルマレイミド、N−ブチルマレイミド、N−i−ブチルマレイミド、N−t−ブチルマレイミド、N−ラウリルマレイミド、N−シクロヘキシルマレイミド、N−ベンジルマレイミド、N−フェニルマレイミド、N−(2−クロロフェニル)マレイミド、N−(4−クロロフェニル)マレイミド、N−(4−ブロモフェニル)フェニルマレイミド、N−(2−メチルフェニル)マレイミド、N−(2−エチルフェニルマレイミド、N−(2−メトキシフェニル)マレイミド、N−(2,4,6−トリメチルフェニル)マレイミド、N−(4−ベンジルフェニル)マレイミド、N−(2,4,6−トリブロモフェニル)マレイミド等のN−置換マレイミド;などが挙げられる。また、これらは1種又は2種以上で使用してもよい。これらのなかでもアクリル酸n−ブチル、アクリル酸2−エチルヘキシルなどが賞用される。
【0024】
本発明ではコアシェルポリマ(B)は架橋微粒子であることが好ましい。コアシェルポリマ(B)を架橋粒子とするために、シェル部(B−2)を形成する前記炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物は、分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体と共重合することが好ましい。かかる分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体としては、例えば、エチレングリコールジ(メタ)アクリレート、1,3−ブタンジオールジ(メタ)アクリレート、1,4−ブタンジオールジ(メタ)アクリレート、1,6−ヘキサンジオールジ(メタ)アクリレート、ジエチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、(メタ)アクリル変性ポリジメチルシロキサンなどの分子内に2個以上の(メタ)アクリロイル基を有する単量体;ジビニルベンゼンなどの分子内に2個以上のビニル基を有する単量体;ビスフェノールAジグリシジルエーテル(メタ)アクリル酸付加物、ビスフェノールAポリオキシエチレン付加ジグリシジルエーテル(メタ)アクリル酸付加物、ビスフェノールAポリオキプロピレン付加ジグリシジルエーテル(メタ)アクリル酸付加物、トリメチロールプロパン(メタ)アクリル酸安息香酸エステルなどの側鎖にベンゼン環を有する分子内に2個以上の(メタ)アクリロイル基を有する単量体;などが挙げられる。これらのなかでも、側鎖にベンゼン環を有する分子内に2個以上の(メタ)アクリロイル基を有する単量体は、有機溶媒及びエポキシ樹脂(A)に対して、相溶性もしくは親和性を高めることができる。これら単量体は一種類単独で又は二種以上を組み合わせて使用することができる。これら単量体のなかでも、これらのなかでも1,6−ヘキサンジオールジアクリレート、ジエチレングリコールジアクリレートなどが賞用される。
【0025】
分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体の使用量は、シェル部(B−2)を構成する単量体100重量部に対して1重量部以上であることが好ましく、2重量部以上であることがより好ましい。前記分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体の使用量が1重量部未満である場合は、架橋が不十分となる傾向がある。
【0026】
本発明で用いられるコアシェルポリマ(B)におけるコア部(B−1)/シェル部(B−2)の重量比は、30/70〜90/10の範囲であることが好ましく、40/60〜80/20の範囲にあることがより好ましい。前記コア部(B−1)/シェル部(B−2)の重量比が30/70を越える(コア部(B−1)の割合が30未満である)場合は、エポキシ樹脂(A)に対する靱性の改良効果が低下する傾向がある。前記コア部(B−1)/シェル部(B−2)の重量比が90/10を越える(シェル部(B−2)の割合が10未満である)場合は、取り扱い時に凝集し易く操作性に問題が生じるとともに期待する物性が得られない可能性がある。
【0027】
コアシェルポリマ(B)が架橋粒子である場合の粒子径は、30〜500nmであることが好ましく、40〜200nmであることがより好ましく、45〜150nmであることが特に好ましい。前記粒子径が30nm未満である場合は、30nmよりも小さい粒子径のコアを入手することが難しく量産性の観点から好ましくない傾向にあり、500nmを超える場合は、得られる熱硬化性樹脂組成物のガラス転移温度が低下する傾向にある。なお、前記架橋粒子の粒子径は、超微粒子粒度分布計(Leeds & Northrup製 MICROTRAC UPA150)を用いて測定できる。架橋粒子の粒子径を制御する方法としては特に限定されず、たとえば、乳化重合によりコアシェルポリマ(B)を合成する場合、使用する乳化剤の量により乳化重合中のミセルの数を制御して粒径をコントロールすることができる。
【0028】
本発明においてコアシェルポリマ(B)の配合量は、前記エポキシ樹脂(A)100重量部に対して、1〜100重量部であることが好ましく、5〜100重量部であることがより好ましい。前記コアシェルポリマ(B)の配合量を前記1〜100重量部の範囲にすることにより、得られる硬化物の強靱性が向上し、長期使用中に硬化部にクラックが発生しにくくなり、また、コアシェルポリマ(B)と他成分との相溶性が向上するとともに得られる硬化物の耐熱性が向上し易くなる。
【0029】
このようなコアシェルポリマ(B)の製造方法は特に制限無く、周知の方法、例えば、乳化重合法、懸濁重合法、マイクロサスペンジョン重合法などが挙げられる。これらのなかで乳化重合法による製造方法が好適である。
【0030】
コアシェルポリマ(B)を乳化重合法により製造する方法は、例えば、コア部(B−1)となるポリマ(b−1)をまず重合し、このポリマ(b−1)をシード粒子として所定量を別の重合容器に添加した後、シェル部(B−2)となるポリマ(b−2)を与える単量体を重合する方法、又は、ポリマ(b−1)を重合し、同一重合容器内でポリマ(b−2)を与える単量体を重合する方法などが挙げられる。ポリマ(b−2)を与える単量体混合物を仕込む方法としては、単量体混合物を全量一括で仕込んで重合する方法、単量体混合物の一部を重合した後、残りを連続的または断続的に添加する方法、単量体混合物を重合の始めから終わりまで連続的に添加する方法、またはこれらの仕込み方法を組み合わせる方法などが挙げられる。重合温度は通常、コア部で50〜90℃、シェル部で30〜90℃である。
コアシェルポリマ(B)を乳化重合法により製造するに際しては、界面活性剤を用いて水中に単量体類を乳化し、重合開始剤として過酸化物触媒やレドックス系触媒などのラジカル重合開始剤を用い、さらに必要に応じてメルカプタン系化合物やハロゲン化炭化水素などの分子量調節剤を添加して、重合を行い、ポリマエマルジョンを合成することができる。このポリマエマルジョンからポリマ析出させ乾燥することにより、コアシェルポリマ(B)を単離することができる。
【0031】
コアシェルポリマ(B)を乳化重合で製造する場合に用いる界面活性剤は、特に限定されないが、たとえば、アルキルベンゼンスルホン酸塩等のアニオン系界面活性剤;アルキルナフタレンスルホン酸塩、アルキルトリメチルアンモニウム塩、ジアルキルジメチルアンモニウム塩等のカチオン系界面活性剤;ポリオキシエチレンアルキルエーテル、ポリオキシエチレンアルキルアリルエーテル、ポリオキシエチレン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、脂肪酸モノグリセリド等のノニオン系界面活性剤および両性の界面活性剤;ならびに反応性乳化剤を用いることができる。これらの界面活性剤は1種単独で、または2種以上を混合して用いることができる。本発明の製造方法における上記界面活性剤の使用量は、単量体の使用量の合計100重量%に対して0.01〜25重量%が好ましい。前記界面活性剤の使用量が0.01重量%未満である場合は、重合反応中に凝集物が生成しやすい傾向にある。前記界面活性剤の使用量が25重量%を超える場合は、ポリマ微粒子を凝集させることが困難になり、また、残存界面活性剤が不純物として影響を及ぼす傾向にある。
【0032】
ポリマエマルジョンからポリマを析出させる方法としては、例えば、塩化カルシウム、硫酸マグネシウム、硫酸アルミニウムなどの多価金属塩、塩化ナトリウム、硫酸などの凝固剤を添加する方法;メタノール、エタノールなどのアルコール類、アセトン、メチルエチルケトンなどの有機溶剤を添加する方法;ノニオン系界面活性剤を用い、当該ノニオン系界面活性剤の曇点以下の温度で乳化重合を行った後、前記曇点以上に加熱する方法;ノニオン系界面活性剤を用いて乳化重合を行なった後、当該ノニオン系界面活性剤よりも低曇点のノニオン系界面活性剤、電解質アルコール、脂肪酸などを添加し加熱する方法;アニオン系及び/又はカチオン系界面活性剤と、ノニオン系界面活性剤を用いて乳化重合を行なった後、当該ノニオン系界面活性剤よりも低曇点のノニオン系界面活性剤あるいは電解質を添加し加熱する方法などが挙げられる。
【0033】
ポリマエマルジョンから析出したポリマ凝固物は、必要に応じて水洗、メタノール洗浄等を行なうことができる。次いでポリマ凝固物を、遠心脱水機等による脱水処理、所望により乾燥処理を行うことによりコアシェルポリマ(B)が単離される。ここで、脱水(乾燥)処理後におけるコアシェルポリマ(B)の水分含有率は、5重量%未満であることが好ましく、1重量%未満であることがより好ましい。前記水分含有率が1重量%未満であるコアシェルポリマ(B)は、接着剤などの用途に好適に使用することができる。
【0034】
本発明では、コアシェルポリマ(B)をエポキシ樹脂(A)と混合する方法としては、例えば、乳化重合により得られたポリマエマルジョンをエポキシ樹脂(A)に混合する方法、ポリマエマルジョンから単離したコアシェルポリマ(B)を所望の有機溶剤に予め分散した後、エポキシ樹脂(A)と混合する方法などが挙げられる。
【0035】
コアシェルポリマ(B)分散させるために用いる有機溶媒としては、特に制限されないが、例えば、エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート等のエチレングリコールモノアルキルエーテルアセテート類;プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコールモノブチルエーテル等のプロピレングリコールモノアルキルエーテル類;プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル、プロピレングリコールジプロピルエーテル、プロピレングリコールジブチルエーテル等のプロピレングリコールジアルキルエーテル類;プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート、プロピレングリコールモノブチルエーテルアセテート等のプロピレングリコールモノアルキルエーテルアセテート類;エチルセロソルブ、ブチルセロソルブ等のセロソルブ類;ブチルカルビトール等のカルビトール類;乳酸メチル、乳酸エチル、乳酸n−プロピル、乳酸イソプロピル等の乳酸エステル類;酢酸エチル、酢酸n−プロピル、酢酸イソプロピル、酢酸n−ブチル、酢酸イソブチル、酢酸n−アミル、酢酸イソアミル、プロピオン酸イソプロピル、プロピオン酸n−ブチル、プロピオン酸イソブチル等の脂肪族カルボン酸エステル類;3−メトキシプロピオン酸メチル、3−メトキシプロピオン酸エチル、3−エトキシプロピオン酸メチル、3−エトキシプロピオン酸エチル、ピルビン酸メチル、ピルビン酸エチル等の他のエステル類;トルエン、キシレン等の芳香族炭化水素類;2−ヘプタノン、3−ヘプタノン、4−ヘプタノン、シクロヘキサノン等のケトン類;N−ジメチルホルムアミド、N−メチルアセトアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン等のアミド類;γ−ブチロラクン等のラクトン類などを挙げることができる。これらの有機溶媒は、1種単独で、あるいは2種以上を混合して使用することができる。これら有機溶媒のなかでも、シェル部(B−2)を構成するポリマ(b−2)の溶解度パラメータと近い有機溶媒が好ましく、一般的にHansenやHoyの計算方法で用いられるLondon分散力項、双極子間力項、水素結合力項それぞれの差の2乗の和が16以下であることが好ましい。
【0036】
このコアシェルポリマ(B)に有機溶媒を分散させる方法としては、コアシェルポリマ(B)を有機溶媒中に投入し、「ホモミキサー」〔特殊機化工業株式会社製〕などを用いて、コアシェルポリマ(B)の粒径が0.5mm以下となるまで粉砕処理を行い、その後、「超音波ホモジナイザ」〔日本エマソン株式会社製〕、「ナノマイザー」〔吉田機械興業株式会社製〕などを用い、一次粒子まで分散を行うことが好ましい。
【0037】
本発明で用いられる硬化剤および硬化触媒(C)は特に制限されず、例えば、脂肪族または芳香族のアミン類、ポリアミド樹脂、カルボン酸類、酸無水物類、フェノール樹脂類、ポリスルフィフィド樹脂、ポリビニルフェノール類、ジシアンジアミド、二塩基酸ジヒドラジド、イミダゾール類、有機ボロン、有機ホスフィン、グアニジン類およびこれらの塩などが挙げられる。これらは1種単独で、または2種以上を組み合わせて用いることができる。また、硬化反応を促進する目的で、硬化触媒とともに硬化促進剤を併用することもできる。ここで、「硬化剤」とは、自ら架橋構造を形成するものであり、「硬化触媒」とは、自らは架橋構造を形成しないが、架橋反応を促進するものであり、「硬化促進剤」とは、硬化触媒の触媒作用を増大させるものである。
【0038】
前記(C)硬化剤および/または硬化触媒の配合量は、エポキシ樹脂(A)100重量部に対して、0.1〜20重量部であることが好ましく、0.5〜10重量部であることがより好ましい。
【0039】
本発明の熱硬化性樹脂組成物は、必要に応じて、有機溶剤、密着助剤、レベリング剤、無機充填剤、有機或いは高分子充填剤、導電性付与剤、滑剤、摺動性付与剤、着色剤、高分子添加剤、反応性希釈剤、濡れ性改良剤、界面活性剤、可塑剤、酸化防止剤、帯電防止剤、防カビ剤、調湿剤、難燃剤およびその他添加剤などを含有することもでき、これらの添加剤は本発明の効果を損なわない範囲の量を使用することができる。
【0040】
また、本発明の熱硬化性樹脂組成物は、本発明の効果を損なわない範囲で、上記エポキシ樹脂(A)以外の樹脂(以下、「その他の樹脂」ともいう)を含有することができる。その他の樹脂としては、例えば、フェノール性水酸基を有する樹脂、ポリイミド、アクリルポリマー、ポリスチレン系樹脂、ポリオレフィン系エラストマー、スチレンブタジエンエラストマー、シリコンエラストマー、トリレンジイソシアネート等のジイソシアネート化合物やそのブロック化物、高密度ポリエチレン、中密度ポリエチレン、ポリプロピレン、ポリカーボネート、ポリアリレート、ポリアミド、ポリアミドイミド、ポリスルホン、ポリエーテルスルホン、ポリエーテルケトン、ポリフェニレンスルフィド、(変性)ポリカルボジイミド、ポリエーテルイミド、ポリエステルイミド、変性ポリフェニレンオキシド、フェノール性水酸基を有する樹脂、オキセタン基を有する樹脂等の熱可塑性あるいは熱硬化性の樹脂等を挙げることができる。
【0041】
本発明に係る熱硬化性樹脂組成物は、各成分が良好な相溶性を示し、耐熱性等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与えることができる。本発明の硬化物において、前記コアシェルポリマ(B)は30〜500nmの粒子径でエポキシ樹脂相に分散しており、例えば、図1に示す透過電子顕微鏡像(TEM像)により確認することができる。
【0042】
従って、本発明の熱硬化性樹脂組成物は、特に、多層回路基板の層間絶縁膜あるいは平坦化膜、各種の電気機器や電子部品等の保護膜あるいは電気絶縁膜、各種電子材料用の接着剤、コンデンサーフィルムなどに極めて好適に用いることができる。また、半導体封止材料、アンダーフィル用材料あるいは液晶封止用材料などとしても好適に使用することができる。また、液状の前記熱硬化性樹脂組成物をガラスクロスなどに含浸させたのち乾燥したプリプレグ、あるいは無溶媒の前記熱硬化性樹脂組成物をガラスクロスなどに含浸させたプリプレグを、銅張り積層板などの積層材などとして用いることもできる。さらに、本発明の熱硬化性樹脂組成物は、たとえば、粉末、ペレット等の形態で、熱硬化性成形材料として用いることもできる。
【0043】
本発明に係る熱硬化性樹脂組成物を予め表面処理した適当な支持体に塗布して熱硬化性薄膜を成形し、この薄膜を支持体とともにラミネーターを用いて基材に転写した後、硬化することにより硬化物層と支持体層を有する基板を得ることができる。
【0044】
また、支持体として表面離型処理したフィルムを用いると、基材に転写後、支持体のみを剥離することにより、熱硬化性樹脂層を形成することができる。
【0045】
得られた熱硬化性フィルムは、電気機器や電子部品等の低応力接着フィルムなどとして用いることができる。
【実施例】
【0046】
以下、実施例により本発明を具体的に説明するが、これらは本発明の範囲を制限するものではない。
【0047】
実施例1
(1)ポリマエマルジョンの製造
4Lのガラス容器に純水1360g、界面活性剤として予め30%水溶液に希釈したエマルゲン109P(ノニオン性界面活性剤、花王株式会社製商品名)328g(正味98g)、及びNipol1562(アクリロニトリル−ブタジエンゴム、日本ゼオン社製品名、ガラス転移温度−20℃)800をg、反応器に入れて撹拌混合、窒素置換を行った。その後ブチルアクリレート280g、1,6−ヘキサンジオールジアクリレート15g、2−アクリロイルオキシエチルフタル酸31g、重合開始剤として、クメンハイドロパーオキサイド4gの混合物を入れ、攪拌しながら含浸を1時間行った。含浸終了後、40℃にて昇温した。昇温後、過硫酸カリウム0.15重量部を純水2重量部に溶解したものを添加し、60℃に昇温した。昇温後、ピロリン酸ナトリウム6g、硫酸鉄(II)0.008gを純水50gに溶解した物を加え6時間撹拌を続け、コアシェルポリマ粒子が分散したポリマエマルジョンを得た。このときの反応率は95%であった。得られたコアシェルポリマ(I)のコア部/シェル部の重量比は、50/50であった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−51℃であった。
【0048】
得られたポリマエマルジョン中のポリマ粒子の平均粒子径を、超微粒子粒度分布計(Leeds & Northrup製 MICROTRAC UPA150)を用いて、下記の処理を行ったエマルションの状態で測定した。
【0049】
得られたポリマエマルジョンを、純水で100倍に希釈し、超音波洗浄器(Leo Ultrasonic製 LEO−80:周波数46Hz、出力80W)を使用して、超音波を3分間照射することにより、ポリマ粒子を分散させた。このポリマ粒子が分散したエマルジョンを前記粒度分布計に入れ、粒子径を測定した。測定時間は3分間とした。
【0050】
測定条件
光源 :ダイオードレーザー(780nm、3mW)
測定レンジ:フルレンジ(0.0032〜6.5406μm)
粒子の性状:透明、球形粒子
分散媒 :水
付属のプログラム(マウンテック製 MICROTRAC Data Handling System SD−UPA150−100)によって、得られた測定データの解析を行ったところ、ポリマ粒子の50%平均粒子径は70nmであった。
【0051】
(2)ガラス織布含浸用樹脂組成物及び測定基板の作製と評価
ノボラックフェノール型エポキシ樹脂 N−770(商品名、大日本インキ化学株式会社製)30g、ノボラックフェノール樹脂 HP−850(商品名、日立化成工業株式会社製)16g、ジシアンジアミド(商品名、日本カーバイド株式会社製)0.04g、上記で合成したコアシェルポリマ(I)2.5g、硬化促進剤2PZ-CNS-PW(商品名、四国化成工業株式会社製)0.1g及びメチルエチルケトン(試薬)80gを混合して樹脂組成物を作製した。
【0052】
上記により得られた樹脂組成物を、厚みが0.2mmのガラス織布(秤量210g/m2)に含浸し、160℃で3分間加熱して半硬化(Bステージ状態)のプリプレグ(樹脂含有率55重量%)を得た。このプリプレグから半硬化樹脂を採取して粉末にし、テフロン(登録商標)シートをスペーサおよび離型シートとして175℃、90分、2.5MPaのプレス条件で硬化して厚さ0.2mmの樹脂板を作製し、測定基板を得た。
【0053】
得られた測定基板について弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
なお、弾性率はレオロジー社製の動的粘弾性スペクトロメータDVE−V4型を用い、チャック間距離20mm、周波数10Hz、昇温速度3℃/分の条件で、50℃における弾性率を測定した。熱膨張率は、Seiko Instruments社製のSSC/5200を用い、昇温速度2℃/分、測定モードを引張りモードとし、40℃〜120℃の値から測定した。ガラス転移温度は、弾性率と同条件で測定を行なった。
【0054】
実施例2
ポリマエマルジョンの製造において2−アクリロイルオキシエチルフタル酸31gを、2−アクリロイルオキシエチルコハク酸31gに変えたこと以外は、実施例1(1)と同様にポリマエマルジョンを製造した(反応率95%)。得られたコアシェルポリマ(II)のコア部/シェル部の重量比は、50/50であり、コアシェルポリマ(II)の粒子径は74nmであった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−50℃であった。
【0055】
次いで、コアシェルポリマ(I)に変えてコアシェルポリマ(II)を用いること以外は実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
実施例3
ポリマエマルジョンの製造において2−アクリロイルオキシエチルフタル酸の使用量を62gに変えたこと以外は、実施例1(1)と同様にポリマエマルジョンを製造した(反応率97%)。得られたコアシェルポリマ(III)のコア部/シェル部の重量比は、50/50であり、コアシェルポリマ(III)の粒子径は115nmであった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−50℃であった。
【0056】
次いで、コアシェルポリマ(I)に変えてコアシェルポリマ(III)を用いること以外は実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例1
コアシェルポリマ(I)に変えて、シェル部がアクリルガラス体でカルボン酸官能基を有するパラロイドEXL2655(商品名、ロームアンドハース株式会社製、平均粒子径0.2μm)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例2
コアシェルポリマ(I)に変えて、カルボン酸変性NBR粒子である非コアシェルのXER−91(商品名、JSR株式会社製、平均粒子径70nm)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例3
コアシェルポリマ(I)に変えて、液状ゴムであるCTBN(宇部興産株式会社製、カルボキシル基末端液状ゴム)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例4
コアシェルポリマ(I)を用いないこと以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
【表1】

【0057】
実施例1〜3は、コアシェルポリマ(I)を用いない比較例4と比べ弾性率及び熱膨張率を低減しつつ、ガラス転移温度を向上することができた。これに対し、比較例1〜3は弾性率及び熱膨張率に関しては低減できるが、ガラス転移温度が低下している。
【技術分野】
【0001】
本発明は、熱硬化性樹脂組成物に用いられるコアシェルポリマに関する。
【背景技術】
【0002】
従来より、エポキシ樹脂の靭性を強化する方法としては、エポキシ樹脂組成物中にゴム成分あるいは熱可塑性樹脂などの改質剤を添加する方法が知られている。ゴム成分を添加する方法としては、反応性液状ゴム(CTBNなど)やニトリルゴムを添加する方法(例えば、特許文献1参照)が知られている。しかし、反応性液状ゴムは、一旦エポキシ樹脂へ溶解した後、硬化時に相分離するという過程を経るため、配合するエポキシ樹脂の種類や硬化条件の違いによって得られる硬化物のモルホロジーが変化し、所望の改質効果が得られなかったり、品質の再現性に問題があることに加え、硬化後のエポキシ樹脂相にゴム成分が一部溶解し残存するために、硬化物の弾性率やガラス転移温度(以下、Tgとも言う。)が低下してエポキシ樹脂製品の品質が低下する等の問題があった。さらに、エポキシ樹脂製品自体が大型で、製品作成のための硬化温度が部位によって異なると、部位により品質が異なってしまう虞もあった。
【0003】
ゴム成分の添加によって問題となるモルホロジー変化とその制御の問題を解決する方法として、エポキシ樹脂中でアクリル酸エステル等のモノマーを重合することで、エポキシ樹脂中にゴム状粒子が分散した組成物を得る方法が知られている(例えば、特許文献2参照)。しかしながら、上記方法でもゴム成分の一部が硬化後のエポキシ樹脂相に溶解することを避けられず、ガラス転移温度が低下する場合があり、品質的に十分なものとは言えなかった。
【0004】
一方、熱可塑性樹脂を添加する方法としては、ガラス転移温度の高い熱可塑性樹脂(いわゆるスーパーエンプラなど)が使用されることが周知である。この方法では、硬化物のガラス転移温度、耐熱性を保持したままある程度の靭性を付与することが可能であるが、一般に添加量を多く要するため、系の粘度が増加し、取り扱い性に問題があり、溶解などの煩雑な工程が必要となったり、モルホロジーのコントロールが必要であるという問題があった。
【0005】
また、エポキシ樹脂に不溶なゴム状重合体粒子を含むエポキシ樹脂組成物は、ゴム成分が硬化後のエポキシ樹脂相に溶解することがないため、耐熱性(ガラス転移温度)の低下を抑制することができる。この場合のゴム状重合体粒子はエポキシ樹脂中で重合したものではなく、あらかじめ重合しておいたものをエポキシ樹脂に混合したものである。このようなゴム状重合体粒子としては、いわゆるコアシェルポリマが代表的なものである(例えば、特許文献3若しくは特許文献4参照)。また、これらのコアシェルポリマは、一次粒子の集合体(凝集体)として、例えば、数十〜数百ミクロンのパウダー状で市販されており、エポキシ樹脂に混合するに際しては、これらを10μm未満に微粉末化したり、更に50〜200℃の温度で加熱攪拌、高速せん断攪拌、熱ロール、インターミキサー、ニーダーや三本ロール等の混錬機で入念に混合しなければ、混合したコアシェルポリマが容易に沈殿あるいは浮上して分離する問題がある。更に数時間に亘るような入念な混合混錬を経た後でも、混合したコアシェルポリマは一次粒子で分散せずに凝集しており、さらにエポキシ樹脂の種類によっては混合したコアシェルポリマが分離しやすいなどの問題や、分散安定剤などを添加する必要があるなど、満足できるものではない。更に、エポキシ樹脂中で実際に分散しているコアシェルポリマの大きさは一次粒子ではないため、コアシェルポリマ粒子の設計を最適化することが難しいなどの問題点があった。これらの背景から、市販されているパウダー状のコアシェルポリマは、エポキシ樹脂の強化剤として充分な性能を有していないのが現状であった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特公昭62−34251号公報
【特許文献2】特開昭59−138254号公報
【特許文献3】米国特許第3322852号明細書
【特許文献4】米国特許第3496250号明細書
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、上記のような従来技術が有するエポキシ樹脂強化に際しての種々の問題点を克服し、耐熱等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与える熱硬化性樹脂組成物に用いられるコアシェルポリマを提供することにある。
【課題を解決するための手段】
【0008】
すなわち、本発明は、(1)コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマに関する。
【0009】
また、本発明は、(2)シェル部が、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマからなる前記(1)記載のコアシェルポリマに関する。
【0010】
また、本発明は、(3)コア部が、ジエン系ゴム重合体、アクリル系ゴム重合体、シリコン系ゴム重合体及びオレフィン系ゴム重合体の中から選択される少なくとも1種であり、コア部のガラス転移温度が0℃以下である前記(1)又は(2)に記載のコアシェルポリマに関する。
【0011】
また、本発明は、(4)架橋微粒子である前記(1)〜(3)のいずれかに記載のコアシェルポリマに関する。
【0012】
また、本発明は、(5)粒子径が30〜500nmである前記(1)〜(4)のいずれかに記載のコアシェルポリマに関する。
【発明の効果】
【0013】
本発明によれば、耐熱性等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与える熱硬化性樹脂組成物に用いられるコアシェルポリマを提供することができる。
【図面の簡単な説明】
【0014】
【図1】本発明の硬化物の透過電子顕微鏡像(TEM像)である。
【発明を実施するための形態】
【0015】
本発明の熱硬化性樹脂組成物は、エポキシ樹脂(A)と、シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有するコアシェルポリマ(B)と、硬化剤及び/または硬化触媒(C)を含むことを特徴とする。
【0016】
本発明に用いられるエポキシ樹脂(A)は、多層回路基板の層間絶縁膜あるいは平坦化膜、電子部品等の保護膜あるいは電気絶縁膜などに用いられるエポキシ樹脂であれば特に限定されないが、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、水添ビスフェノールA型エポキシ樹脂、水添ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビフェニル型エポキシ樹脂、ナフタレン型エポキシ樹脂、フルオレン型エポキシ樹脂、スピロ環型エポキシ樹脂、ビスフェノールアルカン類型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、オルソクレゾールノボラック型エポキシ樹脂、臭素化クレゾールノボラック型エポキシ樹脂、トリスヒドロキシメタン型エポキシ樹脂、テトラフェニロールエタン型エポキシ樹脂、脂環型エポキシ樹脂、アルコール型エポキシ樹脂、ブチルグリシジルエーテル、フェニルグリシジルエーテル、クレジルグリシジルエーテル、ノニルグリシジルエーテル、ジエチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリセリンポリグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、1,6−へキサンジオールジグリシジルエーテル、トリメチロールプロパントリグリシジルエーテル、ヘキサヒドロフタル酸ジグリシジルエーテル、脂肪酸変性エポキシ樹脂、トルイジン型エポキシ樹脂、アニリン型エポキシ樹脂、アミノフェノール型エポキシ樹脂、1,3−ビス(N,N−ジグリシジルアミノメチル)シクロヘキサン、ヒンダトイン型エポキシ樹脂、トリグリシジルイソシアヌレート、テトラグリシジルジアミノジフェニルメタン、ジフェニルエーテル型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ダイマー酸ジグリシジルエステル、ヘキサヒドロフタル酸ジグリシジルエステル、ダイマー酸ジグリシジルエーテル、シリコーン変性エポキシ樹脂、ケイ素含有エポキシ樹脂、ウレタン変性エポキシ樹脂、NBR変性エポキシ樹脂、CTBN変性エポキシ樹脂、エポキシ化ポリブタジエンなどが挙げられる。これらのなかでも、フェノールノボラック型エポキシ樹脂が好ましい。また、これらのエポキシ樹脂は1種単独でまたは2種以上を混合して使用してもよい。
【0017】
本発明で用いることのできるコアシェルポリマ(B)は、コア部(B−1)と、コア部の外層を形成するシェル部(B−2)より構成される。コア部(B−1)はエラストマーまたはゴム状のポリマを主成分とするポリマからなることが好ましく、シェル部(B−2)はコア部(B−1)にグラフト重合されたポリマ成分からことが好ましい。
【0018】
前記コア部(B−1)を構成するポリマはシェル部(B−2)とのグラフトを阻害するものでなければ特に制限は無く、例えば、ブタジエンゴム、スチレン−ブタジエンゴム、アクリロニトリル−ブタジエンゴム等のジエン系ゴム重合体;アクリル酸ブチルゴム、ブタジエン−アクリル酸ブチルゴム、アクリル酸2-エチルヘキシル−アクリル酸ブチルゴム、メタクリル酸2-エチルヘキシル−アクリル酸ブチルゴム、アクリル酸ステアリル−アクリル酸ブチルゴム、ジメチルシロキサン−アクリル酸ブチルゴム、シリコン系/アクリル酸ブチル複合ゴム等のアクリル系ゴム重合体;エチレン−プロピレンゴム、エチレン−プロピレン−ジエンゴム等のオレフィン系ゴム重合体;ポリジメチルシロキサン等のシリコン系ゴム重合体などが挙げられ、これらは、単独でまたは2種以上を混合して使用してもよい。これらのなかでも、スチレン−ブタジエンゴム、アクリロニトリル−ブタジエンゴム、アクリル酸2-エチルヘキシル−アクリル酸ブチルゴムなどが賞用される。
【0019】
前記コア部(B−1)は、ガラス転移温度が0℃以下であることが好ましく、
−10℃以下であることがより好ましい。前記コア部(B−1)のガラス転移温度が0℃を超える場合は、得られる熱硬化性樹脂組成物の熱膨張係数が小さくなり難い傾向にある。コア部(B−1)のガラス転移温度は、示差走査熱量法(DSC法)により測定することが出来る。
前記コアシェルポリマのシェル部は、ポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有するものである。側鎖の原子数が3未満である場合は、エポキシ樹脂と混合後のライフが短く、ある程度放置してから硬化すると、凝集物が多くなり外観・性能悪化等の問題が出る傾向にある。
【0020】
である。側鎖の原子数は、好ましくは3〜18、より好ましくは6〜12である。かかるシェル部(B−2)を構成するポリマは、コア部(B−1)を構成するポリマにグラフト重合されており、実質的にコア部(B−1)を構成するポリマと結合していることが好ましく、後述する有機溶媒及びエポキシ樹脂(A)に対して膨潤性、相溶性もしくは親和性を有するものが好ましい。シェル部(B−2)の理論ガラス転移温度は、好ましくは20℃以下、より好ましくは10℃以下、特に好ましくは0℃以下である。前記シェル部(B−2)の理論ガラス転移温度が20℃を超える場合は、得られる熱硬化性樹脂組成物の熱膨張係数が小さくなり難い傾向にある。シェル部(B−2)の理論ガラス転移温度は、下式から算出することができる。1/Tg=W1/T1+W2/T2+・・・Wn/Tn
式中のW1、W2・・・Wnは各モノマーの重量%(=(各モノマーの配合量/モノマー全重量)×100)であり、T1、T2・・・Tnは、各モノマーのホモポリマーのガラス転移温度(絶対温度)である。
【0021】
シェル部(B−2)を構成するポリマは、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマであることが好ましい。炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物としては、例えば、2−(メタ)アクリロイルオキシエチルコハク酸、2−(メタ)アクリロイルオキシエチルフタル酸、2−(メタ)アクリロイルオキシエチルマレイン酸、2−(メタ)アクリロイルオキシエチルヘキサヒドロフタル酸などが挙げられ、これらは、単独でまたは2種以上を混合して使用してもよい。これらのなかでも、2−アクリロイルオキシエチルコハク酸、2−アクリロイルオキシエチルフタル酸などが賞用される。
【0022】
上記化合物は、無水コハク酸、無水フタル酸、無水マレイン酸、無水ヘキサヒドロフタル酸などのジカルボン酸無水物と(メタ)アクリル酸2−ヒドロキシエチルと脱水反応させることにより得られる。シェル部としてエステル部分に(メタ)アクリロイルオキシアルキレン基を有する炭素数が1以下のジカルボン酸ハーフエステルを重合して得られるポリマを用いたコアシェルポリマは、エポキシ樹脂と混合後のライフが短く、ある程度放置してから硬化すると、凝集物が多くなり外観・性能悪化等の問題が出る傾向にある。
【0023】
また、良好なグラフト重合性と、エポキシ樹脂に対する親和性の双方を可能にできるという点から、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物と共重合可能な単量体を共重合してもよい。かかる共重合可能な単量体としては、例えば、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸プロピル、(メタ)アクリル酸n−ブチル、(メタ)アクリル酸i−ブチル、(メタ)アクリル酸t−ブチル、(メタ)アクリル酸ペンチル、(メタ)アクリル酸n−ヘキシル、(メタ)アクリル酸2−エチルヘキシル、(メタ)アクリル酸n−オクチル、(メタ)アクリル酸ドデシル、(メタ)アクリル酸オクタデシル、(メタ)アクリル酸ブトキシエチル、(メタ)アクリル酸フェニル、(メタ)アクリル酸ベンジル、(メタ)アクリル酸ナフチル、(メタ)アクリル酸グリシジル等の(メタ)アクリル酸エステル類;α−メチルスチレン、α−エチルスチレン、α−フルオロスチレン、α−クロルスチレン、α−ブロモスチレン、フルオロスチレン、クロロスチレン、ブロモスチレン、メチルスチレン、メトキシスチレン等の芳香族ビニル化合物;(メタ)アクリルアミド、N−ジメチル(メタ)アクリルアミド、N−ジエチル(メタ)アクリルアミド等の(メタ)アクリルアミド類;(メタ)アクリル酸カルシウム、(メタ)アクリル酸バリウム、(メタ)アクリル酸鉛、(メタ)アクリル酸アクリル酸すず、(メタ)アクリル酸亜鉛等の(メタ)アクリル酸金属塩等の不飽和脂肪酸;(メタ)アクリロニトリル等のシアン化ビニル化合物、(メタ)アクリル酸シクロペンチル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸メチルシクロヘキシル、(メタ)アクリル酸トリメチルシクロヘキシル、(メタ)アクリル酸ノルボルニル、(メタ)アクリル酸ノルボルニルメチル、(メタ)アクリル酸シアノノルボルニル、(メタ)アクリル酸イソボルニル、(メタ)アクリル酸ボルニル、(メタ)アクリル酸メンチル、(メタ)アクリル酸フェンチル、(メタ)アクリル酸アダマンチル、(メタ)アクリル酸ジメチルアダマンチル、(メタ)アクリル酸トリシクロ〔5.2.1.02,6〕デカ−8−イル、(メタ)アクリル酸トリシクロ〔5.2.1.02,6〕デカ−4−メチル、(メタ)アクリル酸シクロデシル等の脂環式炭化水素基を有する(メタ)アクリル酸エステル;N−メチルマレイミド、N−エチルマレイミド、N−プロピルマレイミド、N−i−プロピルマレイミド、N−ブチルマレイミド、N−i−ブチルマレイミド、N−t−ブチルマレイミド、N−ラウリルマレイミド、N−シクロヘキシルマレイミド、N−ベンジルマレイミド、N−フェニルマレイミド、N−(2−クロロフェニル)マレイミド、N−(4−クロロフェニル)マレイミド、N−(4−ブロモフェニル)フェニルマレイミド、N−(2−メチルフェニル)マレイミド、N−(2−エチルフェニルマレイミド、N−(2−メトキシフェニル)マレイミド、N−(2,4,6−トリメチルフェニル)マレイミド、N−(4−ベンジルフェニル)マレイミド、N−(2,4,6−トリブロモフェニル)マレイミド等のN−置換マレイミド;などが挙げられる。また、これらは1種又は2種以上で使用してもよい。これらのなかでもアクリル酸n−ブチル、アクリル酸2−エチルヘキシルなどが賞用される。
【0024】
本発明ではコアシェルポリマ(B)は架橋微粒子であることが好ましい。コアシェルポリマ(B)を架橋粒子とするために、シェル部(B−2)を形成する前記炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物は、分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体と共重合することが好ましい。かかる分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体としては、例えば、エチレングリコールジ(メタ)アクリレート、1,3−ブタンジオールジ(メタ)アクリレート、1,4−ブタンジオールジ(メタ)アクリレート、1,6−ヘキサンジオールジ(メタ)アクリレート、ジエチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、(メタ)アクリル変性ポリジメチルシロキサンなどの分子内に2個以上の(メタ)アクリロイル基を有する単量体;ジビニルベンゼンなどの分子内に2個以上のビニル基を有する単量体;ビスフェノールAジグリシジルエーテル(メタ)アクリル酸付加物、ビスフェノールAポリオキシエチレン付加ジグリシジルエーテル(メタ)アクリル酸付加物、ビスフェノールAポリオキプロピレン付加ジグリシジルエーテル(メタ)アクリル酸付加物、トリメチロールプロパン(メタ)アクリル酸安息香酸エステルなどの側鎖にベンゼン環を有する分子内に2個以上の(メタ)アクリロイル基を有する単量体;などが挙げられる。これらのなかでも、側鎖にベンゼン環を有する分子内に2個以上の(メタ)アクリロイル基を有する単量体は、有機溶媒及びエポキシ樹脂(A)に対して、相溶性もしくは親和性を高めることができる。これら単量体は一種類単独で又は二種以上を組み合わせて使用することができる。これら単量体のなかでも、これらのなかでも1,6−ヘキサンジオールジアクリレート、ジエチレングリコールジアクリレートなどが賞用される。
【0025】
分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体の使用量は、シェル部(B−2)を構成する単量体100重量部に対して1重量部以上であることが好ましく、2重量部以上であることがより好ましい。前記分子内に2個以上の(メタ)アクリロイル基又はビニル基を有する単量体の使用量が1重量部未満である場合は、架橋が不十分となる傾向がある。
【0026】
本発明で用いられるコアシェルポリマ(B)におけるコア部(B−1)/シェル部(B−2)の重量比は、30/70〜90/10の範囲であることが好ましく、40/60〜80/20の範囲にあることがより好ましい。前記コア部(B−1)/シェル部(B−2)の重量比が30/70を越える(コア部(B−1)の割合が30未満である)場合は、エポキシ樹脂(A)に対する靱性の改良効果が低下する傾向がある。前記コア部(B−1)/シェル部(B−2)の重量比が90/10を越える(シェル部(B−2)の割合が10未満である)場合は、取り扱い時に凝集し易く操作性に問題が生じるとともに期待する物性が得られない可能性がある。
【0027】
コアシェルポリマ(B)が架橋粒子である場合の粒子径は、30〜500nmであることが好ましく、40〜200nmであることがより好ましく、45〜150nmであることが特に好ましい。前記粒子径が30nm未満である場合は、30nmよりも小さい粒子径のコアを入手することが難しく量産性の観点から好ましくない傾向にあり、500nmを超える場合は、得られる熱硬化性樹脂組成物のガラス転移温度が低下する傾向にある。なお、前記架橋粒子の粒子径は、超微粒子粒度分布計(Leeds & Northrup製 MICROTRAC UPA150)を用いて測定できる。架橋粒子の粒子径を制御する方法としては特に限定されず、たとえば、乳化重合によりコアシェルポリマ(B)を合成する場合、使用する乳化剤の量により乳化重合中のミセルの数を制御して粒径をコントロールすることができる。
【0028】
本発明においてコアシェルポリマ(B)の配合量は、前記エポキシ樹脂(A)100重量部に対して、1〜100重量部であることが好ましく、5〜100重量部であることがより好ましい。前記コアシェルポリマ(B)の配合量を前記1〜100重量部の範囲にすることにより、得られる硬化物の強靱性が向上し、長期使用中に硬化部にクラックが発生しにくくなり、また、コアシェルポリマ(B)と他成分との相溶性が向上するとともに得られる硬化物の耐熱性が向上し易くなる。
【0029】
このようなコアシェルポリマ(B)の製造方法は特に制限無く、周知の方法、例えば、乳化重合法、懸濁重合法、マイクロサスペンジョン重合法などが挙げられる。これらのなかで乳化重合法による製造方法が好適である。
【0030】
コアシェルポリマ(B)を乳化重合法により製造する方法は、例えば、コア部(B−1)となるポリマ(b−1)をまず重合し、このポリマ(b−1)をシード粒子として所定量を別の重合容器に添加した後、シェル部(B−2)となるポリマ(b−2)を与える単量体を重合する方法、又は、ポリマ(b−1)を重合し、同一重合容器内でポリマ(b−2)を与える単量体を重合する方法などが挙げられる。ポリマ(b−2)を与える単量体混合物を仕込む方法としては、単量体混合物を全量一括で仕込んで重合する方法、単量体混合物の一部を重合した後、残りを連続的または断続的に添加する方法、単量体混合物を重合の始めから終わりまで連続的に添加する方法、またはこれらの仕込み方法を組み合わせる方法などが挙げられる。重合温度は通常、コア部で50〜90℃、シェル部で30〜90℃である。
コアシェルポリマ(B)を乳化重合法により製造するに際しては、界面活性剤を用いて水中に単量体類を乳化し、重合開始剤として過酸化物触媒やレドックス系触媒などのラジカル重合開始剤を用い、さらに必要に応じてメルカプタン系化合物やハロゲン化炭化水素などの分子量調節剤を添加して、重合を行い、ポリマエマルジョンを合成することができる。このポリマエマルジョンからポリマ析出させ乾燥することにより、コアシェルポリマ(B)を単離することができる。
【0031】
コアシェルポリマ(B)を乳化重合で製造する場合に用いる界面活性剤は、特に限定されないが、たとえば、アルキルベンゼンスルホン酸塩等のアニオン系界面活性剤;アルキルナフタレンスルホン酸塩、アルキルトリメチルアンモニウム塩、ジアルキルジメチルアンモニウム塩等のカチオン系界面活性剤;ポリオキシエチレンアルキルエーテル、ポリオキシエチレンアルキルアリルエーテル、ポリオキシエチレン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、脂肪酸モノグリセリド等のノニオン系界面活性剤および両性の界面活性剤;ならびに反応性乳化剤を用いることができる。これらの界面活性剤は1種単独で、または2種以上を混合して用いることができる。本発明の製造方法における上記界面活性剤の使用量は、単量体の使用量の合計100重量%に対して0.01〜25重量%が好ましい。前記界面活性剤の使用量が0.01重量%未満である場合は、重合反応中に凝集物が生成しやすい傾向にある。前記界面活性剤の使用量が25重量%を超える場合は、ポリマ微粒子を凝集させることが困難になり、また、残存界面活性剤が不純物として影響を及ぼす傾向にある。
【0032】
ポリマエマルジョンからポリマを析出させる方法としては、例えば、塩化カルシウム、硫酸マグネシウム、硫酸アルミニウムなどの多価金属塩、塩化ナトリウム、硫酸などの凝固剤を添加する方法;メタノール、エタノールなどのアルコール類、アセトン、メチルエチルケトンなどの有機溶剤を添加する方法;ノニオン系界面活性剤を用い、当該ノニオン系界面活性剤の曇点以下の温度で乳化重合を行った後、前記曇点以上に加熱する方法;ノニオン系界面活性剤を用いて乳化重合を行なった後、当該ノニオン系界面活性剤よりも低曇点のノニオン系界面活性剤、電解質アルコール、脂肪酸などを添加し加熱する方法;アニオン系及び/又はカチオン系界面活性剤と、ノニオン系界面活性剤を用いて乳化重合を行なった後、当該ノニオン系界面活性剤よりも低曇点のノニオン系界面活性剤あるいは電解質を添加し加熱する方法などが挙げられる。
【0033】
ポリマエマルジョンから析出したポリマ凝固物は、必要に応じて水洗、メタノール洗浄等を行なうことができる。次いでポリマ凝固物を、遠心脱水機等による脱水処理、所望により乾燥処理を行うことによりコアシェルポリマ(B)が単離される。ここで、脱水(乾燥)処理後におけるコアシェルポリマ(B)の水分含有率は、5重量%未満であることが好ましく、1重量%未満であることがより好ましい。前記水分含有率が1重量%未満であるコアシェルポリマ(B)は、接着剤などの用途に好適に使用することができる。
【0034】
本発明では、コアシェルポリマ(B)をエポキシ樹脂(A)と混合する方法としては、例えば、乳化重合により得られたポリマエマルジョンをエポキシ樹脂(A)に混合する方法、ポリマエマルジョンから単離したコアシェルポリマ(B)を所望の有機溶剤に予め分散した後、エポキシ樹脂(A)と混合する方法などが挙げられる。
【0035】
コアシェルポリマ(B)分散させるために用いる有機溶媒としては、特に制限されないが、例えば、エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート等のエチレングリコールモノアルキルエーテルアセテート類;プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコールモノブチルエーテル等のプロピレングリコールモノアルキルエーテル類;プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル、プロピレングリコールジプロピルエーテル、プロピレングリコールジブチルエーテル等のプロピレングリコールジアルキルエーテル類;プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート、プロピレングリコールモノブチルエーテルアセテート等のプロピレングリコールモノアルキルエーテルアセテート類;エチルセロソルブ、ブチルセロソルブ等のセロソルブ類;ブチルカルビトール等のカルビトール類;乳酸メチル、乳酸エチル、乳酸n−プロピル、乳酸イソプロピル等の乳酸エステル類;酢酸エチル、酢酸n−プロピル、酢酸イソプロピル、酢酸n−ブチル、酢酸イソブチル、酢酸n−アミル、酢酸イソアミル、プロピオン酸イソプロピル、プロピオン酸n−ブチル、プロピオン酸イソブチル等の脂肪族カルボン酸エステル類;3−メトキシプロピオン酸メチル、3−メトキシプロピオン酸エチル、3−エトキシプロピオン酸メチル、3−エトキシプロピオン酸エチル、ピルビン酸メチル、ピルビン酸エチル等の他のエステル類;トルエン、キシレン等の芳香族炭化水素類;2−ヘプタノン、3−ヘプタノン、4−ヘプタノン、シクロヘキサノン等のケトン類;N−ジメチルホルムアミド、N−メチルアセトアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン等のアミド類;γ−ブチロラクン等のラクトン類などを挙げることができる。これらの有機溶媒は、1種単独で、あるいは2種以上を混合して使用することができる。これら有機溶媒のなかでも、シェル部(B−2)を構成するポリマ(b−2)の溶解度パラメータと近い有機溶媒が好ましく、一般的にHansenやHoyの計算方法で用いられるLondon分散力項、双極子間力項、水素結合力項それぞれの差の2乗の和が16以下であることが好ましい。
【0036】
このコアシェルポリマ(B)に有機溶媒を分散させる方法としては、コアシェルポリマ(B)を有機溶媒中に投入し、「ホモミキサー」〔特殊機化工業株式会社製〕などを用いて、コアシェルポリマ(B)の粒径が0.5mm以下となるまで粉砕処理を行い、その後、「超音波ホモジナイザ」〔日本エマソン株式会社製〕、「ナノマイザー」〔吉田機械興業株式会社製〕などを用い、一次粒子まで分散を行うことが好ましい。
【0037】
本発明で用いられる硬化剤および硬化触媒(C)は特に制限されず、例えば、脂肪族または芳香族のアミン類、ポリアミド樹脂、カルボン酸類、酸無水物類、フェノール樹脂類、ポリスルフィフィド樹脂、ポリビニルフェノール類、ジシアンジアミド、二塩基酸ジヒドラジド、イミダゾール類、有機ボロン、有機ホスフィン、グアニジン類およびこれらの塩などが挙げられる。これらは1種単独で、または2種以上を組み合わせて用いることができる。また、硬化反応を促進する目的で、硬化触媒とともに硬化促進剤を併用することもできる。ここで、「硬化剤」とは、自ら架橋構造を形成するものであり、「硬化触媒」とは、自らは架橋構造を形成しないが、架橋反応を促進するものであり、「硬化促進剤」とは、硬化触媒の触媒作用を増大させるものである。
【0038】
前記(C)硬化剤および/または硬化触媒の配合量は、エポキシ樹脂(A)100重量部に対して、0.1〜20重量部であることが好ましく、0.5〜10重量部であることがより好ましい。
【0039】
本発明の熱硬化性樹脂組成物は、必要に応じて、有機溶剤、密着助剤、レベリング剤、無機充填剤、有機或いは高分子充填剤、導電性付与剤、滑剤、摺動性付与剤、着色剤、高分子添加剤、反応性希釈剤、濡れ性改良剤、界面活性剤、可塑剤、酸化防止剤、帯電防止剤、防カビ剤、調湿剤、難燃剤およびその他添加剤などを含有することもでき、これらの添加剤は本発明の効果を損なわない範囲の量を使用することができる。
【0040】
また、本発明の熱硬化性樹脂組成物は、本発明の効果を損なわない範囲で、上記エポキシ樹脂(A)以外の樹脂(以下、「その他の樹脂」ともいう)を含有することができる。その他の樹脂としては、例えば、フェノール性水酸基を有する樹脂、ポリイミド、アクリルポリマー、ポリスチレン系樹脂、ポリオレフィン系エラストマー、スチレンブタジエンエラストマー、シリコンエラストマー、トリレンジイソシアネート等のジイソシアネート化合物やそのブロック化物、高密度ポリエチレン、中密度ポリエチレン、ポリプロピレン、ポリカーボネート、ポリアリレート、ポリアミド、ポリアミドイミド、ポリスルホン、ポリエーテルスルホン、ポリエーテルケトン、ポリフェニレンスルフィド、(変性)ポリカルボジイミド、ポリエーテルイミド、ポリエステルイミド、変性ポリフェニレンオキシド、フェノール性水酸基を有する樹脂、オキセタン基を有する樹脂等の熱可塑性あるいは熱硬化性の樹脂等を挙げることができる。
【0041】
本発明に係る熱硬化性樹脂組成物は、各成分が良好な相溶性を示し、耐熱性等を損なうことなく、弾性率に優れた高い靭性を有する硬化物を与えることができる。本発明の硬化物において、前記コアシェルポリマ(B)は30〜500nmの粒子径でエポキシ樹脂相に分散しており、例えば、図1に示す透過電子顕微鏡像(TEM像)により確認することができる。
【0042】
従って、本発明の熱硬化性樹脂組成物は、特に、多層回路基板の層間絶縁膜あるいは平坦化膜、各種の電気機器や電子部品等の保護膜あるいは電気絶縁膜、各種電子材料用の接着剤、コンデンサーフィルムなどに極めて好適に用いることができる。また、半導体封止材料、アンダーフィル用材料あるいは液晶封止用材料などとしても好適に使用することができる。また、液状の前記熱硬化性樹脂組成物をガラスクロスなどに含浸させたのち乾燥したプリプレグ、あるいは無溶媒の前記熱硬化性樹脂組成物をガラスクロスなどに含浸させたプリプレグを、銅張り積層板などの積層材などとして用いることもできる。さらに、本発明の熱硬化性樹脂組成物は、たとえば、粉末、ペレット等の形態で、熱硬化性成形材料として用いることもできる。
【0043】
本発明に係る熱硬化性樹脂組成物を予め表面処理した適当な支持体に塗布して熱硬化性薄膜を成形し、この薄膜を支持体とともにラミネーターを用いて基材に転写した後、硬化することにより硬化物層と支持体層を有する基板を得ることができる。
【0044】
また、支持体として表面離型処理したフィルムを用いると、基材に転写後、支持体のみを剥離することにより、熱硬化性樹脂層を形成することができる。
【0045】
得られた熱硬化性フィルムは、電気機器や電子部品等の低応力接着フィルムなどとして用いることができる。
【実施例】
【0046】
以下、実施例により本発明を具体的に説明するが、これらは本発明の範囲を制限するものではない。
【0047】
実施例1
(1)ポリマエマルジョンの製造
4Lのガラス容器に純水1360g、界面活性剤として予め30%水溶液に希釈したエマルゲン109P(ノニオン性界面活性剤、花王株式会社製商品名)328g(正味98g)、及びNipol1562(アクリロニトリル−ブタジエンゴム、日本ゼオン社製品名、ガラス転移温度−20℃)800をg、反応器に入れて撹拌混合、窒素置換を行った。その後ブチルアクリレート280g、1,6−ヘキサンジオールジアクリレート15g、2−アクリロイルオキシエチルフタル酸31g、重合開始剤として、クメンハイドロパーオキサイド4gの混合物を入れ、攪拌しながら含浸を1時間行った。含浸終了後、40℃にて昇温した。昇温後、過硫酸カリウム0.15重量部を純水2重量部に溶解したものを添加し、60℃に昇温した。昇温後、ピロリン酸ナトリウム6g、硫酸鉄(II)0.008gを純水50gに溶解した物を加え6時間撹拌を続け、コアシェルポリマ粒子が分散したポリマエマルジョンを得た。このときの反応率は95%であった。得られたコアシェルポリマ(I)のコア部/シェル部の重量比は、50/50であった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−51℃であった。
【0048】
得られたポリマエマルジョン中のポリマ粒子の平均粒子径を、超微粒子粒度分布計(Leeds & Northrup製 MICROTRAC UPA150)を用いて、下記の処理を行ったエマルションの状態で測定した。
【0049】
得られたポリマエマルジョンを、純水で100倍に希釈し、超音波洗浄器(Leo Ultrasonic製 LEO−80:周波数46Hz、出力80W)を使用して、超音波を3分間照射することにより、ポリマ粒子を分散させた。このポリマ粒子が分散したエマルジョンを前記粒度分布計に入れ、粒子径を測定した。測定時間は3分間とした。
【0050】
測定条件
光源 :ダイオードレーザー(780nm、3mW)
測定レンジ:フルレンジ(0.0032〜6.5406μm)
粒子の性状:透明、球形粒子
分散媒 :水
付属のプログラム(マウンテック製 MICROTRAC Data Handling System SD−UPA150−100)によって、得られた測定データの解析を行ったところ、ポリマ粒子の50%平均粒子径は70nmであった。
【0051】
(2)ガラス織布含浸用樹脂組成物及び測定基板の作製と評価
ノボラックフェノール型エポキシ樹脂 N−770(商品名、大日本インキ化学株式会社製)30g、ノボラックフェノール樹脂 HP−850(商品名、日立化成工業株式会社製)16g、ジシアンジアミド(商品名、日本カーバイド株式会社製)0.04g、上記で合成したコアシェルポリマ(I)2.5g、硬化促進剤2PZ-CNS-PW(商品名、四国化成工業株式会社製)0.1g及びメチルエチルケトン(試薬)80gを混合して樹脂組成物を作製した。
【0052】
上記により得られた樹脂組成物を、厚みが0.2mmのガラス織布(秤量210g/m2)に含浸し、160℃で3分間加熱して半硬化(Bステージ状態)のプリプレグ(樹脂含有率55重量%)を得た。このプリプレグから半硬化樹脂を採取して粉末にし、テフロン(登録商標)シートをスペーサおよび離型シートとして175℃、90分、2.5MPaのプレス条件で硬化して厚さ0.2mmの樹脂板を作製し、測定基板を得た。
【0053】
得られた測定基板について弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
なお、弾性率はレオロジー社製の動的粘弾性スペクトロメータDVE−V4型を用い、チャック間距離20mm、周波数10Hz、昇温速度3℃/分の条件で、50℃における弾性率を測定した。熱膨張率は、Seiko Instruments社製のSSC/5200を用い、昇温速度2℃/分、測定モードを引張りモードとし、40℃〜120℃の値から測定した。ガラス転移温度は、弾性率と同条件で測定を行なった。
【0054】
実施例2
ポリマエマルジョンの製造において2−アクリロイルオキシエチルフタル酸31gを、2−アクリロイルオキシエチルコハク酸31gに変えたこと以外は、実施例1(1)と同様にポリマエマルジョンを製造した(反応率95%)。得られたコアシェルポリマ(II)のコア部/シェル部の重量比は、50/50であり、コアシェルポリマ(II)の粒子径は74nmであった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−50℃であった。
【0055】
次いで、コアシェルポリマ(I)に変えてコアシェルポリマ(II)を用いること以外は実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
実施例3
ポリマエマルジョンの製造において2−アクリロイルオキシエチルフタル酸の使用量を62gに変えたこと以外は、実施例1(1)と同様にポリマエマルジョンを製造した(反応率97%)。得られたコアシェルポリマ(III)のコア部/シェル部の重量比は、50/50であり、コアシェルポリマ(III)の粒子径は115nmであった。また、シェル部のガラス転移温度(理論ガラス転移温度)は−50℃であった。
【0056】
次いで、コアシェルポリマ(I)に変えてコアシェルポリマ(III)を用いること以外は実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例1
コアシェルポリマ(I)に変えて、シェル部がアクリルガラス体でカルボン酸官能基を有するパラロイドEXL2655(商品名、ロームアンドハース株式会社製、平均粒子径0.2μm)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例2
コアシェルポリマ(I)に変えて、カルボン酸変性NBR粒子である非コアシェルのXER−91(商品名、JSR株式会社製、平均粒子径70nm)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例3
コアシェルポリマ(I)に変えて、液状ゴムであるCTBN(宇部興産株式会社製、カルボキシル基末端液状ゴム)を用いること以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
比較例4
コアシェルポリマ(I)を用いないこと以外は、実施例1(2)と同様に操作して樹脂組成物及び測定基板を作製し、弾性率、熱膨張率、ガラス転移温度を測定した。結果を表1に示す。
【表1】
【0057】
実施例1〜3は、コアシェルポリマ(I)を用いない比較例4と比べ弾性率及び熱膨張率を低減しつつ、ガラス転移温度を向上することができた。これに対し、比較例1〜3は弾性率及び熱膨張率に関しては低減できるが、ガラス転移温度が低下している。
【特許請求の範囲】
【請求項1】
コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマ。
【請求項2】
シェル部が、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマからなる請求項1記載のコアシェルポリマ。
【請求項3】
コア部が、ジエン系ゴム重合体、アクリル系ゴム重合体、シリコン系ゴム重合体及びオレフィン系ゴム重合体の中から選択される少なくとも1種であり、コア部のガラス転移温度が0℃以下である請求項1又は2に記載のコアシェルポリマ。
【請求項4】
架橋微粒子である請求項1〜3のいずれか一項に記載のコアシェルポリマ。
【請求項5】
粒子径が30〜500nmである請求項1〜4のいずれか一項に記載のコアシェルポリマ。
【請求項1】
コア部と、該コア部の外層を形成するシェル部を有し、前記シェル部にポリマ主鎖から伸びる原子数3以上の側鎖を介して存在するカルボキシル基を有し、前記シェル部の理論ガラス転移温度が20℃以下である、エポキシ樹脂に混合して用いられるコアシェルポリマ。
【請求項2】
シェル部が、炭素数2以上のジカルボン酸のハーフエステルであってエステル部分に(メタ)アクリロイルオキシアルキレン基を有する化合物を重合して得られるポリマからなる請求項1記載のコアシェルポリマ。
【請求項3】
コア部が、ジエン系ゴム重合体、アクリル系ゴム重合体、シリコン系ゴム重合体及びオレフィン系ゴム重合体の中から選択される少なくとも1種であり、コア部のガラス転移温度が0℃以下である請求項1又は2に記載のコアシェルポリマ。
【請求項4】
架橋微粒子である請求項1〜3のいずれか一項に記載のコアシェルポリマ。
【請求項5】
粒子径が30〜500nmである請求項1〜4のいずれか一項に記載のコアシェルポリマ。
【図1】

【公開番号】特開2012−136713(P2012−136713A)
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2012−93010(P2012−93010)
【出願日】平成24年4月16日(2012.4.16)
【分割の表示】特願2007−139508(P2007−139508)の分割
【原出願日】平成19年5月25日(2007.5.25)
【出願人】(000004455)日立化成工業株式会社 (4,649)
【Fターム(参考)】
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願日】平成24年4月16日(2012.4.16)
【分割の表示】特願2007−139508(P2007−139508)の分割
【原出願日】平成19年5月25日(2007.5.25)
【出願人】(000004455)日立化成工業株式会社 (4,649)
【Fターム(参考)】
[ Back to top ]