
コンツラキン−G、そのアナログおよびそのための使用
【課題】コンツラキン−G、そのアナログおよびその使用を開示する。
【解決手段】コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体、この成熟ペプチドの前駆体をコードするcDNAクローン、および前駆ペプチド。さらに、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用。
【解決手段】コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体、この成熟ペプチドの前駆体をコードするcDNAクローン、および前駆ペプチド。さらに、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願に関する相互参照
本発明は、1998年10月20日付けで出願された米国仮特許出願シリアル番号60/105,015、1999年4月9日付けで出願されたシリアル番号60/128,561および1999年4月23日付けで出願されたシリアル番号60/130,661(各々をここに出典明示して本明細書の一部とみなす)に関する。
【0002】
本発明は、メリーランド州ベセスダの国立保健研究所(National Institute of Health)によって与えられたグラント番号GM−48677下で政府援助によりなされた。米国政府は本発明にある種の権利を有する。
【0003】
発明の背景
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G(Contulakin-G)、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【0004】
本発明の背景を例示し、特別な場合、当該実施に関するさらなる詳細を提供するために本明細書で用いられる刊行物および他の資料は、出典明示して本明細書の一部とみなし、利便のため、以下の本文中で数字で参照され、各々、添付の出典リストにおいて分類される。
【背景技術】
【0005】
コーヌス(Conus)属の軟体動物は、それらがユニークな捕食性のライフスタイルをなすことができるようにする毒液を生産する。被食体は、銛および皮下針の両方に機能し、使い捨ての中空歯である、高度に特殊化した毒液器官によって注入される毒液により不動にされる。
【0006】
生物体間の相互作用で、毒液を注入する動物およびその毒物を注入される犠牲者間のそれらより著しいものはほとんどない。毒液は、被食体を捕獲するための一義的武器または防御機構として用いることができる。これらの毒液の多くは、神経筋系の受容体およびイオンチャンネンを指向する分子を含有する。
【0007】
コーヌス毒液から単離されたいくつかのペプチドは、特徴付けられてきている。これらはニコチン性アセチルコリン受容体、筋ナトリウムチャンネルおよびニューロンカルシウムチャンネルをそれぞれ標的にするα−、μ−およびω−コノトキシンを含む(Oliveraら、1985)。また、バソプレッシンアナログであるコノプレシンも同定された(Cruzら、1987)。さらに、コナントキンと命名されたペプチドは、Conus geographusおよびConus tulipa(Menaら、1990;Haackら、1990)から単離された。これらのペプチドは、異常な年齢依存的生理学的効果を有し;2週齢より若いマウスにおいて睡眠様状態、3週齢より加齢したマウスにおいて活動亢進挙動を誘導する(Haackら、1990)。κ−コノトキシンの単離、構造および活性は、米国特許第5,633,347号に記載されている。最近、D−トリプトファンを含有するコントリファンと命名されたペプチドがConus radiatusから単離され(米国シリアル番号09/061,026)、ブロモ−トリプトファン・コノペプチドは、Conus imperialisおよびConus radiatusから単離された(米国シリアル番号08/785,534)。
前記のコノペプチドの活性を有するさらなるコノペプチドならびにさらなる活性を有するコノトキシンペプチドを同定することが所望される。
【発明の概要】
【発明が解決しようとする課題】
【0008】
発明の概要
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【課題を解決するための手段】
【0009】
1つの具体例において、本発明は、コンツラキン−G、コンツラキン−Gプロペプチドおよびこのペプチドをコードする核酸に指向される。コンツラキン−Gは、以下の式:Xaa1-Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Thr-Lys-Lys-Xaa2-Tyr-Ile-Leu(配列番号:1)[式中、Xaa1はピロ-Glu、Xaa2はプロリンまたはヒドロキシプロリンであって、Thr10はO−グリカンを含有するように修飾されている]を有する。好ましくは、X2はプロリンである。本発明により、グリカンとは、N-、S-またはO-結合した単糖、二糖、三糖、多糖またはオリゴ糖を意味し、それは、当該技術分野において知られた合成的または酵素的な方法によって天然または修飾したアミノ酸のいずれのヒドロキシ、アミノまたはチオール基にも結合できる。グリカンを構成する単糖には、D-アロース、D-アルトロース、D-グルコース、D-マンノース、D-グロース、D-イドース、D-ガラクトース、D-タロース、D-ガラクトサミン、D-グルコサミン、D-N-アセチル-グルコサミン(GlcNAc)、D-N-アセチル-グルコサミン(GalNAc)、D-フコースまたはD-アラビノースが含まれ得る。これらの糖は、構造的には、本明細書に記載のごとく、例えば、その組合せを含めた、1以上のO−スルフェート、O−ホスフェート、O−アセチルまたはシアル酸のごとき酸性基で修飾され得る。また、グリカンには、D−ペニシラミン 2,5およびそのハロゲン化誘導体またはポリプロピレングリコール誘導体のごとき類似するポリヒドロキシグループが含まれる。グリコシド結合は、ベータおよび1−4または1−3、好ましくは1−3である。グリカンとアミノ酸との間の結合は、アルファまたはベータ、好ましくは、アルファであってもよく、それは1−である。好ましいグリカンは、本明細書にさらに記載され、最も好ましいグリカンは、Gal(β1→3)GalNAc(α1→)である。
【0010】
第2の具体例において、本発明は、以下の一般式:
Xaa1-Xaa2-Xaa3-Xaa3-Gly-Gly-Xaa2-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa10-Ile-Leu(配列番号:2)
[式中、Xaa1はピロ-Glu、Glu、Gln、またはγ-カルボキシ-Glu;Xaa2はSer、ThrまたはS-グリカン修飾Cys;Xaa3はGluまたはγ-カルボキシ-Glu;Xaa4はAsn、N-グリカン修飾AsnまたはS-グリカン修飾Cys;Xaa5はAlaまたはGly;Xaa6はThr、Ser、S-グリカン修飾Cys、Tyrまたは(4-ヒドロキシメチル-Phe、4-ヒドロキシフェニル-Gly、2,6-ジメチル-Tyr、3-ニトロ-Tyrおよび5-アミノ-Tyrのごとき)いずれかのヒドロキシ含有非天然アミノ酸;Xaa7はLys、N-メチル-Lys、N,N-ジメチル-Lys、N,N,N-トリメチル-Lys、Arg、オルニチン、ホモアルギニンまたは(N-1-(2-ピラゾリニル)-Argのごとき)いずれかの非天然の塩基性アミノ酸;Xaa8はAla、Gly、Lys、N-メチル-Lys、N,N-ジメチル-Lys、N,N,N-トリメチル-Lys、Arg、オルニチン、ホモアルギニン、(N-1-(2-ピラゾリニル)-Argのごとき)いずれかの非天然の塩基性アミノ酸またはX-Lys、ここに、Xは(CH2)n、フェニル、−(CH2)m−(CH=CH)−(CH2)mHまたは−(CH2)m−(C=C)−(CH2)mH(式中、nは1〜4であって、mは0〜2である);Xaa9はProまたはヒドロキシ-Pro;およびXaa10はTyr、モノ-ヨード-Tyr、ジ-ヨード-Tyr、O-スルホ-Tyr、O-ホスホ-Tyr、ニトロ-Tyr、Trp、D-Trp、ブロモ-Trp、ブロモ-D-Trp、クロロ-Trp、クロロ-D-Trp、Phe、L-ネオ-Trpまたは(ニトロ-Phe、4-置換Phe(ここに、該置換基は、C1−C3アルキル、カルボニル、ヒドロキシメチル、スルホメチル、ハロ、フェニル、-CHO、−CN、-SO3Hおよび-NHAc、2,6-ジメチル-Tyrおよび5-アミノ-Tyrである)のごとき)いずれかの非天然の芳香族アミノ酸である]を有するジェネリック(generic)コンツラキン−Gに指向される。C末端には、遊離カルボキシル基が含まれ、アミド化され、アシル化され、グリカンが含まれ、またはアルデヒドが含まれる。C末端は、遊離カルボキシルを含むことが好ましい。このペプチドは、さらに、前記のごとき1以上のグリカンを含み得る。該グリカンは、残基2、7、8、10および16にて生じ得る。前記および他の非天然の塩基性アミノ酸、非天然のヒドロキシ含有アミノ酸または非天然の芳香族アミノ酸は、マサチューセッツ州ウスターのRSP Amino Acid Analogues、Inc.によっておよびそこから入手可能なBuilding Block Index、バージョン2.2(ここに出典明示して本明細書の一部とみなす)に記載されている。
【0011】
第3の具体例において、本発明は、コンツラキン−Gのアナログまたはジェネリックコンツラキン−Gに指向される。これらのアナログには、Thr10までおよびそれを含むコンツラキン−Gまたはジェネリックコンツラキン−GのN末端切形が含まれる。N末端切形がThr10を通る場合、Lys11はカルボキシル化修飾したリンカーを用いてN-グリコシル化される。このN-グリコシル化Lys11は、図1(Tothら、1999)に示されるごとく表示でき、ここに、R2、R3およびR4は本明細書の記載に同じである。これらの切形において、切形に近い残基は、グリコシル化セリンで置換されることが好ましい。さらなるアナログには、1〜9位の残基に代えて、Ser−O−グリカン、Thr-O−グリカンまたはCys-S-グリカンに置換されたペプチドが含まれる。
【0012】
第4の具体例において、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤として本明細書に記載されたペプチドの使用に指向される。この具体例の1つの態様において、無痛覚症は、本明細書に記載されたペプチドのうちの1種を用いて哺乳動物に誘導される。この具体例の第2の態様において、癲癇または痙攣は哺乳動物において治療される。この具体例の第3の態様において、精神分裂病は哺乳動物において治療される。この具体例の第4の態様において、遅発性異常運動症および急性ジストニー反応(acute dystonic reaction)は哺乳動物において治療される。この具体例の第5の態様において、炎症は哺乳動物において治療される。
【図面の簡単な説明】
【0013】
【図1】図1は、カルボキシル化修飾したリンカーを用いるLysのN-グリコシル化の構造を示す。
【図2】図2は、コンツラキン−GのThr10に結合した天然のO−グリカンを示す。
【図3】図3は、コンツラキン−Gの1以上の残基に結合できるグリカンのアナログを示す。
【図4】図4は、好ましいコアのO−グリカンを示す(Van de Steenら、1998)。ムチン型O−結合オリゴ糖は、GalNAc残基によってSerまたはThr(またはこのペプチドのヒドロキシル化残基)に結合する。この最初のGalNAc残基に結合した単糖形成用ブロックおよび該結合は、「コア・グリカン」を定義し、8種が同定されている。グリコシド結合の型(配向および結合性)は、各コア・グリカンにつき定義される。
【図5】図5は、コンツラキン−Gの精製を示す。Conus geographusからの粗凍結乾燥毒液の1グラムを抽出し、従前に記載されるごとく、Sephadex G-25に適用した(Oliveraら、1984)。麻痺性および睡眠性の活性を含む3つの連続する画分(Ve/Vo=1.37ないし1.41)をプールし、分取用逆相Vydac C18カラムに適用し、0.1%トリフルオロ酢酸中のアセトニトリルの勾配で溶出した。マウスにicv投与した場合、パネルA中の矢印によって示された成分は動揺および死亡を引き起こした。
【図6】図6は、天然のコンツラキン−G(286〜1886ダルトン)のナノ−ESI MS/MSスペクトルを示す(該MS/MS実験は、示された速記法(Mcluckeyら、1991)を用いて表示され、ここに、黒丸はm/z 1035[M+2H]2+前駆体を表し、矢印は、白丸の方向に向けられ、前駆体から生成したフラグメントを表す)。スペクトル上、糖アミノ酸の構造は、矢印が2つの部位を示し、それはMS/MSスペクトルにおいて観察された主要フラグメントイオンに導く(Craigら、1993)。
【図7A−7C】図7A−7Cは、有害な加熱によって得られた脊髄媒介(肢の引込め)および脊柱上媒介(後肢のなめ)の痛覚性行動に対するCGX−1063の用量応答を示す。データは、応答(図7Aおよび7B)または最初の落下(図7C)に対して秒単位で表示される。図7Aにおいて、50℃のホットプレート上に置いた後、最初の観察可能な応答までの潜在時間が示される。図7Bは、最初の後肢のなめに対する潜在時間を示す。図7Cは、加速回転棒(accelerating rotorod)上に置いた後の最初の落下までの潜在時間を示す(図7A〜7C、n=3〜10)。
【図8A−8B】図8A〜8Bは、持続性の痛みに対する痛覚性応答に対するCGX−1063の効果を示す。図8Aにおいて、データは動物がホルマリン注入した後肢をなめるのに費やした時間量として示される(n=7〜10動物/処置群)。髄腔内CGX−1063は、生理食塩水を髄腔内注射した対照に比較してホルマリン試験において第2相の痛覚性応答を用量依存的に低下させた。図8Bは、ホルマリン試験直後の加速回転棒からの最初の落下までの潜在時間を示す。
【図9】図9は、部分的な座骨神経結紮1週間後の機械的刺激に対する肢引込め閾値を示す。データは、較正されたファン・フレイ(von Frey)フィラメントで測定されたグラム単位の50%引込め閾値として表示される(群当たりn=3〜9の動物)。
【図10A−10B】図10A〜10Bは、テイル・フリック試験におけるCGX−1160(コンツラキン−G)、CGX−1063およびNTの比較を示す。3種の化合物の用量応答を図10Aに示す。図10Bは、各化合物につき試験された最高用量での効果時間を示す(CGX−1160=100ピコモル;CGX−1063=100ピコモル;NT=10ナノモル)。
【図11A−11B】図11A〜11Bは、ホルマリン試験の第1相(図11A)および第2相(図11B)に対するCGX−1160、CGX−1063およびNTの効果を示す。3種の化合物の全てが、ホルマリンのi.pl.後の痛覚性行動を用量依存的に低下させた。第2相(図11B)において、CGX−1160は、CGX−1063より10倍、NTより600〜700倍効力があった。
【図12A−12C】図12A〜12Cは、慢性炎症で誘導された機械的異痛症に対するCGX−1160、CGX−1063およびNTの効果を示す。括弧内の数字は、各々対応する対照値の百分率を示す。図12Aにおいて、CGX−1160は、CFA誘導された異痛を強力にかつ用量依存的に逆転させた。図12Bにおいて、CGX−1063は、CFA誘導された異痛を逆転させたが、CGX−1160よりこのモデルにおいて約100倍小さな効力であった。図12Cにおいて、NTは1000ピコモルにてCFA誘導された異痛を逆転させたが、100ピコモルでは逆転させず、CGX−1160より約10000倍小さな効力であった。
【図13A−13B】図13A〜13Bは、CGX−1160、CGX−1063およびNTの運動障害効果を示す。図13Aは、試験された最高用量(ホルマリン試験の第2相におけるED50の約100倍)での3種の化合物のピーク効果までの時間および効果時間を示す。図13Bは、運動障害に対する各化合物の用量応答を示す。
【図14A−14C】図14A〜14Cは、CGX−1160、CGX−1063およびNTの運動障害の用量効果、ピーク効果までの時間および期間を示す。図14Aは、CGX−1160がそのED50の100倍以上の用量だけに長時間作用型の運動障害を引き起こすことを示す。図14Bは、CGX−1063がそのED50の10倍以上の用量にて長時間作用型の運動障害を引き起こすことを示す。図14Cは、NTがそのED50の100倍以上の用量にて長時間作用型の運動障害を引き起こすことを示す。
【図15A−15B】図15A〜15Bは、体温変化に対するCGX−1160、CGX−1063およびNTの比較を示す。図15Aは、各化合物のピークまでの時間および期間を示し、図15Bは各化合物の用量応答を示す。
【図16A−16C】図16A〜16Cは、CGX−1160、CGX−1063およびNTの低体温の用量−効果および期間を示す。図16Aにおいて、CGX−1160は、ED50より100〜500倍大きい用量だけで低体温を引き起こした。図16Bは、CGX−1160は、ED50より10倍高い用量(100ピコモル)にてCGX−1063の長時間作用型の低体温効果を示す。図16Cにおいて、NTは、そのED50より10〜100倍高い用量にて低体温効果を有した。
【図17】図17は、通過距離によって測定されたD−アンフェタミン刺激運動活性に対するThr10−g コンツラキン−G(CGX−1160;100ピコモル i.c.v.)の効果を示す。略語:sal-sal:i.p.処置は生理食塩水であって、i.c.v.処置は生理食塩水である; amphet(3mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置は生理食塩水である;amphet(10mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(10mg/kg)であって、i.c.v.処置は生理食塩水である;sal-ctl:i.p.処置は生理食塩水であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル);amphet(3mg/kg)-ctl:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル)である。各バーは、群当たり3〜7匹のマウスの平均±SEMを示す。a:生理食塩水−生理食塩水処置された群(sal-sal)に対するP<0.05;b:アンフェタミン−生理食塩水処置された群(amphet(3mg/kg)-sal)に対するP<0.05。
【図18】図18は、移動に費やされた時間によって測定されたD−アンフェタミン刺激運動活性に対するThr10−g コンツラキン−G(CGX−1160;100ピコモル i.c.v.)の効果を示す。略語:sal-sal:i.p.処置は生理食塩水であって、i.c.v.処置は生理食塩水である; amphet(3mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置は生理食塩水である;amphet(10mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(10mg/kg)であって、i.c.v.処置は生理食塩水である;sal-ctl:i.p.処置は生理食塩水であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル);amphet(3mg/kg)-ctl:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル)である。各バーは、群当たり3〜7匹のマウスの平均±SEMを示す。a:生理食塩水−生理食塩水処置された群(sal-sal)に対するP<0.05;b:アンフェタミン−生理食塩水処置された群(amphet(3mg/kg)-sal)に対するP<0.05。
【図19】図19は、最小運動障害用量より完全に下の用量でのフリングス・マウスにおけるi.c.v.投与後の聴覚性発作に対するCGX−1160およびCGX−1063の用量依存的保護を示す。各点は、(少なくとも4匹のマウスの群における毒性の)パーセント保護を表す。
【図20】図20は、フリングス・マウスにおけるi.c.v.投与後の聴覚性発作のブロックにおけるCGX−1160の長時間持続型の効力を示す。ニューロテンシンは、5ナノモルまでのi.c.v.投与後に50%だけ有効であった。各点は、4匹のマウスの群におけるパーセント保護を表す。
【発明を実施するための形態】
【0014】
好ましい具体例の詳細な記載
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【0015】
本発明は、前記のコンツラキン−Gおよびコンツラキン−Gアナログに指向される。これらのペプチドは、該ペプチドの1以上で全部までのヒドロキシル部位にて単一または複数のグリカンの翻訳後修飾を含み得る。該グリカンは、本明細書の記載に同じである。コンツラキン−Gに結合した天然のO−グリカンは、図2に示される。図3は、コンツラキン−Gの1以上の残基に結合できるグリカンのアナログを示す。この図において、R1は化学的または酵素的のいずれかでグリカンで誘導体化できるアミノ酸であり;R2はOH、NH2、NHSO3Na、NHAc、O−スルフェート、O−ホスフェートまたはO−グリカンであり;R3はH、SO3、PO3、アセチル、シアル酸または単糖であり;R4はH、SO3、PO3、アセチルまたは単糖であり;R5はOH、NH2、NHSO3Na、NHAc、O−スルフェート、O−ホスフェート、O−単糖またはO−アセチルであり;R6はH、SO3、PO3、アセチルまたは単糖であり;R7はH、SO3、PO3、アセチルまたは単糖であり;R8はH、SO3、PO3、アセチルまたは単糖であり;nは0〜4であって、mは1〜4である。
【0016】
本明細書に開示されたコンツラキン−Gまたはアナログを修飾するのに用いることができる好ましいコア・グリカンは、図4に示される。さらに、本明細書に記載された単糖を用いてこれらのコアから分岐がなされてもよい。好ましいグリコシド結合は、本明細書に記載の単糖を用いて、図4のコア5および7によって特定され、さらに、GalNAc鋳型の3、4および6位のグリカンと相同する。いずれの遊離ヒドロキシ官能基もO−硫酸化、O−燐酸化またはO−アセチル化できる。
【0017】
グリコシル化コノペプチド(コンツラキン−GまたはCGX−1160)は非グリコシル化コノペプチド(Thr10−コンツラキン−GまたはCGX−1063)よりin vivoにて高い効力を有するが、これらのin vitroでの効力はほぼ同一である。グリコシル化は、受容体と良好に結合するのに、その作用部位へのコノペプチドの送達の増強および/またはコノペプチド分解の阻害に重要であり得る。
【0018】
さらに、本発明は、さらに本明細書に記載されたコンツラキン−GをコードするDNA配列に指向される。本発明は、本明細書のさらなる詳細において記載されたコンツラキン−Gのプロペプチドにさらに指向される。
本発明は、医薬として有用である新規な線状グリコシル化コンツラキン−Gおよびその誘導体に、その製法に、これらの化合物および医薬上許容される担体を含む医薬組成物に、および医薬上の治療方法に関する。本発明の新規な化合物は、中枢神経系用剤であり、それらの生物学的作用は、新規な「ニューロテンシン受容体上のコンツラキン−G結合部位」にて影響する。より詳細には、本発明の新規な化合物は、鎮痛剤、抗炎症剤、精神分裂病のごとき精神病を治療するための抗精神病剤であり、癲癇の確立された動物モデルにおいて強力な抗発作特性を示す。
【0019】
痛み:
骨変性疾患および癌のごとき疾患において生じ得るような慢性または難治性の痛みは、様々な鎮痛剤およびしばしばモルヒネのごときオピオイド化合物で治療される弱質疾患である。
【0020】
一般的には、痛みの知覚を管理する脳経路は、依然として不完全に理解されており、「痛覚の経路」といわれる脊髄への知覚の導入性シナプス接続は、いくらか詳細に報告されている。かかる経路の第1の脚において、末梢性部位から脊髄へ突出するC−およびA−線維は、痛覚シグナルを運ぶ。脊髄の背角における多シナプスの結合は、中脳水道周囲灰色領域を含めた脳の様々な領域までの痛みの感覚の中継および調節に係わる。無痛覚または痛みの知覚の縮小は、かかる痛覚の経路に沿った伝達を減少させることにより直接的に達成できる。鎮痛性アヘン剤は、エンドルフィンまたはエンケファリンの効果の模倣により作用し、それはC−またはA−線維末端にて前シナプス的にシナプス形成し、それらが発火した場合にはサブスタンスPを含めた神経伝達物質の放出を阻害すると思われている。また、脳からの下向経路は、C−およびA-線維発火に抑制的である。
【0021】
ある種の型の痛みは複雑な病因を持っている。例えば、神経障害的な痛みは、一般には、末梢神経の損傷または部分的な離断(transection)に起因する慢性の疾患である。この型の痛みは、知覚過敏症、または外部の有害な刺激に対する感受性の増強によって特徴づけられる。神経障害的な痛みの知覚過敏性成分は、全身に広がり、急性形態の痛みに応答するような同一の医薬介在に応答しない。
【0022】
モルヒネのごときオピオイド化合物は、多くの型の痛みにつき無痛覚を得るのに有効であるが、必ずしも有効とは限らなく、患者における耐性を引き起こしかねない。対象がオピオイド麻薬に耐性である場合、増加した服用量が満足な鎮痛性効果達成するのに必要である。これらの化合物は、生命を脅かし得る呼吸抑制のごとき副作用を引き起こしかねない。さらに、オピオイドは、頻繁に患者の身体的依存を生じる。依存性は、オピオイドの用量およびそれが対象によって摂取された期間と関係するようである。この理由のために、慢性的痛みの管理についての別法の治療は後に広く求められる。さらに、必要とされる鎮痛性化合物の用量を低下させるために、オピオイド治療についての置換物または添加物のいずれかとして機能する化合物は、痛み、特に慢性的な難治性の型の痛みの治療に利用性を有する。
【0023】
コンツラキン−Gは、ある種のニューロテンシン受容体上の部位で作用するすることが示され、また、ニューロテンシンは鎮痛作用を有することが示されている(Clineschmidtら、1979)ので、コンツラキン−G様コノペプチドは、痛みおよび関連疾患の治療に有用である。
【0024】
精神分裂病:精神分裂病は、第1に、ドーパミン受容体をブロックするフェノチアジンおよびブチロフェノンのごとき神経弛緩性化合物で現在治療される神経性疾患である。コンツラキン−Gがある種のニューロテンシン受容体上の部位にて作用することが示され、また、ニューロテンシン作用が精神分裂病の病因に関係付けられる(Nemerotfら、1992)ので、コンツラキン−G様コノペプチドが精神分裂病および関連疾患の治療に有用である。
【0025】
精神分裂病の治療に有用なコノペプチドのin vitroにての選択基準には以下が含まれる:a)コンツラキン−G部位の活性化;b)脳辺縁領域に局在化したコンツラキン−G結合部位に対する高親和性の可逆的な結合、およびc)脳領域、特に脳辺縁領域からのドーパミン放出の阻害。
【0026】
次いで、前記のin vitroでのスクリーニング・アッセイにおいて十分に高活性を示す化合物は、抗精神性化合物をスクリーニングするのに用いられる動物モデルにおいて試験される。
【0027】
遅発性異常運動症および他の急性ジストニー反応:遅発性異常運動症および急性ジストニー反応は、抗精神病治療の副作用として共通して生じる運動障害であり、ハロペリドールのごときドーパミン・アンタゴニストを使用する。これらの障害は、運動の制御に関連した脳のある領域、特に基底核のドーパミン受容体の過敏性によって特徴づけられる。現在、断続的な抗精神的治療は、障害の開始を回避する試みに用いられ、かかる障害は治療の中止によって処置される。
【0028】
遅発性異常運動症の治療のためのオメガ−コノペプチドの選択基準は、以下が含まれる:a)コンツラキン−G部位の活性化;b)コンツラキン−G部位に対する高親和性の可逆的な結合;c)線条体の脳領域および基底核の他の領域からのドーパミン放出の阻害、ならびにd)辺縁領域のドーパミン放出の阻害に対する基底核におけるドーパミン放出の阻害比率。
【0029】
次いで、in vitroでのスクリーニング・アッセイにおいて十分に高活性を示す化合物は、前記のラット線条体変化モデルにおいて試験される。かかる運動疾患を治療する方法に有用な化合物は、その病変の反対側の脳の側の線条に注射した場合、行動の変化を矯正する。
【0030】
炎症:炎症の神経性成分は、交感神経系のブロック、特にベータ−アドレナリン作動性受容体のブロックが炎症性関節損傷を低減するのに役立つという点で記載されている。炎症の治療に有用な化合物は、以下のin vitro特性を有することが期待されるであろう:a)新規なコンツラキン−G部位の活性化;b)コンツラキン−G結合部位に対する高親和性結合、およびc)神経組織からのノルエピネフリン放出の阻害。かかるin vitroスクリーニング・アッセイにおいて十分に高活性を示す化合物は、慢性関節リウマチの動物モデルにおいて試験される。
【0031】
癲癇:癲癇は、発作の症状発現の繰り返しによって特徴づけられる中枢神経系の障害を記述する一般用語である。かかる発作は、知覚、自律または運動の神経系を含むことができ、脳内の異常放電の存在によって電気生理学的に認識される。かかる異常な放電活性の病態生理は良好には理解されていない;しかしながら、GABA入力のごとき抑制性の神経入力の喪失が少なくともいくらかの癲癇発作に関係するという証拠がある。
【0032】
これらの薬物が、GABA受容体と関連する塩素イオン・チャンネルに対する効果を介してGABA作動性神経阻害を強めることが知られているので、癲癇の症状発現を抑制するか、あるいは阻害するベンゾジアゼピン(例えば、ジアゼパム)のある種の能力は、癲癇活性におけるGABA作動性病態生理の証拠であると幾分考えられる。他の抗癲癇化合物の生化学的効果には、電圧感受性のナトリウムまたはカリウムのチャンネルの阻害(フェニトイン)によって興奮性の膜の安定化、ならびにGABA作動性伝達の促進、興奮性の(グルタミン作動性)神経伝達の効果の阻害、および神経伝達物質放出の低下(フェノバルビタール)によって特徴付けられる神経機能の全般的低下が含まれる。
【0033】
癲癇の治療に有用な化合物は、以下のin vitro特性を有することが期待されるであろう:a)新規なコンツラキン−G部位の活性化;b)コンツラキン−Gコノペプチド結合部位に対する高親和性結合、およびc)神経組織からの興奮性神経伝達物質放出の阻害。かかるin vitroスクリーニング・アッセイにおいて十分に高活性を示す化合物は、癲癇の確立された動物モデルにおいて試験した。
【0034】
前記の特定の疾患に加えて、本発明のペプチド、誘導体およびアナログは、ニューロテンシン受容体に結合することが判明したので、これらの化合物もニューロテンシン受容体と関連する疾患、およびニューロテンシン様化合物または他の化合物が活性であることが示された疾患と関連して有用である。これらの活性には以下のものが含まれる:メタンフェタミン・アンタゴニスト、抗精神剤、大脳用薬剤、鎮痛剤、抗菌体内毒素ショック効果、プロテアーゼ阻害作用(抗トロンビン作用、抗プラスミン作用)、低血圧作用、抗DIC作用、抗アレルギー作用、創傷治癒作用、脳浮腫、肺浮腫、気管浮腫、血栓、動脈硬化症、やけどおよび高血圧症、(気管支喘息および花粉症のごとき)アレルギー疾患、外科手術時の傷害された組織部分のごとき鋭い外傷からの出血低減、交通事故等によって引き起こされた脳または他の組織の裂傷、ならびに腫脹の緩和および治癒では、外傷によって引き起こされた痛み炎症、鈍い外傷によって引き起こされた内出血の抑制、内出血を伴う浮腫および炎症、脳梗塞(例えば、脳血栓および脳塞栓)を含む虚血性脳疾患において判明した脳マトリックスへの血液成分の漏出を抑制することによる脳浮腫の抑制または改善、頭蓋内出血(例えば、脳出血および蜘蛛膜下出血)、一過性脳虚血発作、高血圧性脳疾患における急性の脳血管障害、やけどの抑制および改善、凍瘡、他の皮膚炎症および浮腫、上部気管炎症、喘息、鼻づまり、肺浮腫、ならびに環境化学物質、癌の化学療法剤、菌体内毒素および炎症性メディエーターのごとき血管内皮および粘膜を直接的に損傷する内因性および外来性因子によって引き起こされる炎症疾患。
【0035】
本発明のコノペプチドは、コーヌス毒液からの単離によって同定される。別法として、本発明のコノペプチドは、変性したプローブを含む通常の技術を用いて種々のコーヌス種のcDNAライブラリーをスクリーニングすることによって組換えDNA技術を用いて同定される。これらのプローブにハイブリダイズするクローンを分析して、評価されるべき特定のcDNAライブラリーにつきcDNAクローニング部位を近接するPCRプライマーを用いて決定されるように、最小サイズの要求、すなわち、(あるプロペプチドにつき)約300個のヌクレオチドを有するクローンに合うものを同定する。次いで、これらの最小サイズのクローンを配列決定する。次いで、配列をコノペプチドにつき前記の特性を有するペプチドの存在につき調べる。この方法によって同定されたペプチドの生物学的活性は、本明細書、米国特許第5,635,347号に記載のごとくまたは当該技術分野において、通常、試験される。
【0036】
これらのペプチドは、化学的に合成するのに十分小さい。コノペプチドの化学合成およびこれらの合成生成物の生物学的活性の表示に加えて、前述のコノペプチドを調製するための一般的な化学合成は後記される。また、これらのコノペプチドの様々なものは、米国特許第4,447,356号(Oliveraら、1984)、第5,514,774号(Oliveraら、1996)および第5,591,821号(Oliveraら、1997)(その開示をここに出典明示して本明細書の一部とみなす)に記載される技術を用いて特定のコーヌス種からの単離および精製によって得ることができる。
【0037】
本発明のコノペプチドは、イモガイ(cone snail)からの精製によって得ることができるが、個々のイモガイから得ることができるコノペプチド量は非常に少ないために、所望の実質的に純粋なコノペプチドは固相戦略を用いて化学合成によって商業的に価値のある量において最良に実践的に得られる。例えば、単一のイモガイからの収量は、約10マイクログラム以下のコノペプチドであるかもしれない。「実質的に純粋な」とは、該ペプチドが同一型の他の生物学的分子の実質的な不存在下にて存在することを意味し;好ましくは、少なくとも約85%純度および好ましくは少なくとも約95%純度の量にて好ましくは存在する。生物学上活性なコノペプチドの化学合成は、アミノ酸配列の正確な決定にもちろん依存する。かくして、本発明のコノペプチドは、単離され、合成され、および/または実質的に純粋であり得る。
【0038】
また、コノペプチドは、当該技術においてよく知られた組換えDNA技術によって産生できる。かかる技術はSambrookら(1989)によって記載されている。この方法において産生されたペプチドは、単離され、必要な場合には還元され、酸化されて、最終分子において存在するならば正確なジスルフィド結合を形成される。
【0039】
本発明のコノペプチドの中のジスルフィド結合を形成する1つの方法は冷室温または室温下にて長期間の線状ペプチドの空気酸化である。この手順の結果、生物学的に活性なジスルフィド結合したペプチドの実質的な量を創製できる。酸化されたペプチドを逆相高速液体クロマトグラフィー(HPLC)等を用いて分別して、異なる結合立体配置を有するペプチドを分離する。その後、これらの画分と天然の物質の溶出との比較、または単純なアッセイを用いてのいずれかによって、最大の生物学的効力につき正確な結合を有する特定の画分を容易に決定する。また、in vivoにて生じる架橋および/または再配置が生物学的に強力なコノペプチド分子を創製することが判明したために、1以上の画分を有する線状ペプチドまたは酸化生成物が時々、in vivo投与につき用いることができることが判明した。しかしながら、生物学的効力のほとんどない他の画分の存在から生じる希釈のために、多少高用量を必要とするかもしれない。
【0040】
排除固相技術により、部分的固相技術により、断片縮合または古典的溶液カップリングによるごとき適当な方法によって該ペプチドを合成する。
【0041】
通常の溶液相ペプチド合成法において、ペプチド鎖は、構成アミノ酸を所望の向きで成長しつつあるペプチド鎖に付加する一連のカップリング反応によって調製できる。中間体の続いての単離および精製と共に、溶液中で反応を行うための種々のカップリング試薬、例えば、ジシクロへキシルカルボジイミドまたはジイソプロピル−カルボニルジイミダゾール、種々の活性エステル、例えば、N−ヒドロキシフタルイミドまたはN−ヒドロキシスクシンイミドおよび種々の開裂試薬の使用は、よく知られた古典的ペプチド法である。古典的溶液合成法は、論文[Methoden der Organischen Chemie(Houben-Weyl): Synthese von Peptiden ](1974)に詳細に記載されている。排除固相合成の技術は、テキスト「Solid-Phase Peptide Synthesis」(StewartおよびYoung、1969)に記載され、米国特許第4,105,603号(Valeら、1978)の開示によって例示されている。断片縮合合成方法は、米国特許第3,972,859号(1976)の開示によって例示されている。他の利用できる合成は、米国特許第3,842,067号(1974)および米国特許第3,862,925号(1975)によって例示されている。γ−カルボキシグルタミン酸残基を含むペプチドの合成は、Rivierら(1987)、Nishiuchiら(1993)およびZhouら(1996)によって例示される。コノペプチドの合成は、米国特許第4,447,356号(Oliveraら、1984)、第5,514,774号(Oliveraら、1996)および第5,591,821号(Oliveraら、1997)に記載されている。
【0042】
かかる化学合成に共通しているのは、当該基が最後に取去されるまで、その部位で化学反応が起こるのを妨ぐ適当な保護基で種々のアミノ酸部位の不安定側鎖基を保護することである。また、通常、共通するのは、その原子団がカルボニル基で反応し、続いてα−アミノ保護基を選択的に除去して、その位置で次の反応を起こす間に、アミノ酸またはフラグメント上のα−アミノ基を保護することである。従って、かかる合成の工程として、不安定な側鎖を有する残基の種々のものに結合した適当な側鎖保護基を持つ、ペプチド鎖中の所望の配列に位置する各アミノ酸残基を含む中間化合物が生成されることが共通する。
【0043】
側鎖アミノ保護基の選択に関する限り、一般的には、合成の間のα−アミノ基の脱保護の間に除去されないものが選ばれる。しかしながら、いくつかのアミノ酸、例えばHisについては、保護は一般的に必要でない。ペプチドの合成に用いられるべき特定の側鎖保護基の選択において、一般的規則は次の通りである:(a)保護基は、好ましくはその保護特性を保持し、カップリング条件下で切断除去されず、(b)保護基は、合成の各工程にてα−アミノ保護基を取去するために選択された反応条件下で安定であるべきであり、(c)ペプチド鎖を望まないように改変しないであろう反応条件下で、所望のアミノ酸配列を含有する合成を完了するに際して、側鎖保護基は、除去できなければならない。
【0044】
組換えDNA技術を用いて、これらのペプチドの多く、または全てでさえ調製できる。しかしながら、ペプチドがそのように調製されない場合、当該分野で公知の他の同等の化学合成を前記のごとく用いることもできるが、それらは、好ましくはメリフィールドの固相合成法を用いて調製される。固相合成法は、保護されたα−アミノ酸を適当な樹脂にカップリングすることによって、ペプチドのC末端から開始される。かかる出発物質は、クロロメチル化樹脂もしくはヒドロキシメチル樹脂に対するエステル結合によって、またはベンズヒドリルアミン(BHA)もしくはパラメチルベンズヒドリルアミン(MBHA)樹脂に対するアミド結合によって、α−アミノ保護アミノ酸を結合させることによって調製できる。ヒドロキシメチル樹脂の調製法は、Bodanskyら(1966)によって記載されている。クロロメチル化樹脂は、Bio Rad Laboratories(Richmond、CA)およびLab. Systems, Inc.から商業的に入手可能である。かかる樹脂の調製法は、StewartおよびYoung(1969)によって記載されている。BHAおよびMBHA樹脂支持体は商業的に入手可能で、合成されるべき所望のポリペプチドがC末端にて非置換アミドを有する場合に一般的に使用される。かくして、固体樹脂支持体は、式−O−CH2−樹脂支持体、−NH−BHA−樹脂支持体または−NH−MBHA−樹脂支持体を有するもののごとき、当該分野で公知であるもののいずれかであってよい。非置換アミドが所望される場合、開裂が直接的にアミドを与えるのでBHAまたはMBHA樹脂の使用が好ましい。N−メチルアミドが所望される場合、それはN−メチルBHA樹脂から生成できる。他の置換アミドが所望なら、米国特許第4,569,967号(Kornreichら、1986)の教示が使用できるか、あるいはC末端にて遊離酸よりもなお他の基が所望ならば、Houben-Weylのテキスト(1974)に記載されたような古典的方法を用いてペプチドを合成するのが望ましいであろう。
【0045】
BocまたはFmocによって、および側鎖保護基によって保護されたC末端アミノ酸は、もし適当ならば、Horikiら(1978)に記載された手法に従って、C末端にて遊離酸を有するペプチドが合成されるべき場合、約60℃にて24時間撹拌しつつジメチルホルムアミド(DMF)中のKFを用いて、クロロメチル化樹脂にまずカップリングできる。樹脂支持体へのBoc保護アミノ酸のカップリングに続いて、塩化メチレン中のトリフルオロ酢酸(TFA)またはTFA単独を用いることによってα−アミノ保護基を取去する。脱保護は約0℃および室温の間の温度で行う。ジオキサン中のHClのごとき他の標準的開裂試薬および特異的α−アミノ保護基の除去の条件は、SchroderおよびLubke(1965)に記載されたごとくに用いることができる。
【0046】
α−アミノ保護基の除去後、残存するα−アミノおよび側鎖保護アミノ酸を所望の順序で段階にカップリングして、前記定義の中間体化合物を得るか、あるいは合成法中に別々に各アミノ酸を付加することに対する代替法として、それらのいくつかは、固相反応器に添加する前にもう1つにカップリングしてもよい。適当なカップリング試薬の選択は当業者の技量内のものである。特に、カップリング試薬として適当なのは、N,N’−ジシクロヘキシルカルボジイミド(HoBrまたはHoAtの存在下でのDCC、DIC、HBTU、HATU、TBTU)である。
【0047】
ペプチドの固相合成法において用いられる活性化試薬は、ペプチド分野でよく知られている。適当な活性化試薬の例は、N,N’−ジイソプロピルカルボジイミドおよびN−エチル−N’−(3−ジメチルアミノプロピル)カルボジイミドのごときカルボジイミドである。ペプチドカップリングにおける他の活性化試薬およびその使用は、SchroderおよびLubke(1965)およびKapoor(1970)によって記載されている。
【0048】
各保護アミノ酸またはアミノ酸配列は、約2倍以上過剰に固相反応器に導入され、カップリングは、ジメチルホルムアミド(DMF):CH2Cl2(1:1)またはDMFもしくはCH2Cl2単独の媒体中で行うことができる。中間体カップリングが生じる場合には、カップリング手法は、次のアミノ酸のカップリングに先立ってα−アミノ保護基の除去前に繰り返される。合成法の各工程におけるカップリング反応の成功は、もし手動で行うならば、Kaiserら(1970)によって記載されたごとくニンヒドリン反応によって好ましくはモニターされる。カップリング反応は、Rivierら(1978)によって報告されたごときプログラムを用いて、Beckman990自動合成機のように自動的に行うことができる。
【0049】
所望のアミノ酸配列が完成した後、中間体ペプチドは、液体フッ化水素またはTFA(Fmoc化学を用いるなら)のごとき試薬での処理によって樹脂支持体から取り去ることができ、これは樹脂からペプチドを開裂させるにのみならず、全ての残存する側鎖保護基を開裂させ、またもし先に除去されていなければN末端にてα−アミノ保護基を切断して、遊離酸の形態のペプチドを得る。もしMetが配列中に存在するならば、Boc保護基は、好ましくはHFで樹脂からペプチドを開裂させるに先立って、トリフルオロ酢酸(TFA)/エタンジチオールを用いまず取去して、可能なS−アルキル化を消失させる。開裂にフッ化水素またはTFAを用いる場合、アニソール、クレゾール、ジメチルスルフィドおよびメチルエチルスルフィドのごとき1以上の掃去剤を反応器に含める。
【0050】
ペプチド樹脂の一部の間のペプチドを環化するのとは対照的に、好ましくは線状ペプチドの環化を行ってCys残基間に結合を生じさせる。かかるジスルフィド環化結合を行うには、十分に保護されたペプチドを、当該分野でよく知られたように、アンモノリシスによってヒドロキシメチル化樹脂またはクロロメチル化樹脂支持体から開裂させて、十分に保護されたアミド中間体を得、しかる後、これを適当に環化し脱保護する。別法として、前記樹脂またはベンズヒドリルアミン(BHA)樹脂またはパラメチルベンズヒドリルアミン(MBHA)からのペプチドの開裂と同様に脱保護は、フッ化水素酸(HF)またはTFAで0℃にて起こすことができ、続いて前記したごとくに酸化する。環化のための適当な方法は、Cartierら(1996)によって記載された方法である。
【0051】
また、前記のコンツラキン−GまたはThr10−gコンツラキン−Gのムテイン、アナログまたは活性フラグメントは、ここに企図される。Hammerlandら(1992)参照。コノトキシンペプチドの誘導ムテイン、アナログまたは活性フラグメントは、米国特許第5,545,723号(特に、第2欄第50行ないし第3欄8行参照);米国特許第5,534,615号(特に、第19欄第45行ないし第22欄33行参照)および米国特許第5,364,769号(特に、第4欄第55行ないし第7欄26行参照)(各々を出典明示して本明細書の一部とみなす)に概説されたごとき保存的アミノ酸置換を含めた公知技術により合成できる。
【0052】
有効成分として本発明の化合物を含有する医薬組成物は伝統的医薬混合技術に従って調製できる。Remington`s Pharmaceutical Sciences、第18版(1990、Mack Publishing Co.、Easton、PA)参照。典型的には、有効成分の拮抗量を、医薬上許容される担体と混合されるであろう。担体は、例えば、静脈内、経口または非経口投与に望まれる製剤形態に応じて非常に種々の形態を取ることができる。
【0053】
経口投与については、該化合物は、カプセル剤、丸剤、錠剤、ロゼンジ、メルト剤、散剤、懸濁剤または乳液のごとき固体または液体製剤に処方できる。経口投薬形態の組成物を調製するにおいて、経口液体製剤(例えば、懸濁剤、エリキシル剤および水剤)の場合には、例えば、水、グリコール、油、アルコール、矯味剤、保存剤、着色剤、懸濁化剤等;経口固体製剤(例えば、散剤、カプセル剤および錠剤)の場合には、澱粉、糖、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等のごとき通常の医薬媒体のうちいずれを用いてもよい。投与の容易さのために、錠剤およびカプセル剤は最も有利な経口用量単位形態を表し、その場合に固形医薬担体が明らかに使用される。もし所望ならば、錠剤は標準的技術によって糖衣、腸溶性コーティングとすることができる。
【0054】
非経口投与については、該化合物は、医薬担体に溶解し、懸濁剤液のいずれかとして投与することができる。適当な担体の例は、水、生理的食塩水、デキストロース溶液、フラクトース溶液、エタノールまたは動物、植物また合成起源の油である。また、担体は、例えば保存剤、懸濁化剤、可溶化剤、緩衝剤等の他の成分を含んでもよい。該化合物が髄腔内投与されるべき場合、脳脊髄液に溶解してもよい。
【0055】
本発明による活性薬剤の投与は、いずれの適当な送達手段を用いても達成でき、
(a)ポンプ(例えば、Annals of Pharmacotherapy, 27:912(1993); Cancer, 41:1270(1993); Cancer Research, 44:1698(1984)参照)
(b)マイクロカプセル化(米国特許第4,352,883号;第4,353,888号および米国特許第5,084,350号参照)
(c)持続性放出ポリマー埋込み剤(米国特許第4,883,666号)
(d)マイクロカプセル化(米国特許第5,284,761号、第5,158,881号、第4,976,859号および第4,968,733号ならびに公開されたPCT特許出願WO92/19195、WO95/05452参照)
(e)CNSに対する裸のまたは非カプセル化の細胞移植(米国特許第5,082,670号および第5,618,531号参照)
(f)皮下、静脈内、動脈内、筋肉内もしくは他の適当な部位への注射、または
(g)カプセル剤、液剤、錠剤、丸剤または持続性放出製剤における経口投与
が含まれる。
【0056】
本発明の1つの具体例において、活性薬剤は、CNS、好ましくは、脳室、脳実質、髄腔または他の適当なCNS位置、最も好ましくは髄腔内に直接的に送達される。
【0057】
あるいは、ターゲティング治療を用いて、抗体または細胞特異的リガンドのごときターゲティング・システムの使用によって、さらにとりわけあるタイプの細胞に活性薬剤を送達できる。ターゲティングは、種々の理由、例えば、薬剤が許容できない毒性があるならば、余りにも高用量を必要とするならば、または標的細胞に入ることができないならば望ましい。
【0058】
また、ペプチドである活性薬剤は、活性薬剤をコードするDNA配列を患者の身体、とりわけ脊髄領域に移植するために設計された細胞に導入する細胞ベースの送達システムにて投与することもできる。適当な送達システムは、米国特許第5,550,050号およびPCT出願公開WO 92/19195、 WO 94/25503、 WO 95/01203、 WO 95/05452、 WO 96/02286、 WO 96/02646、 WO 96/40871、 WO 96/40959およびWO 97/12635に記載されている。適当なDNA配列は、開示された配列および公知の遺伝暗号に基づいて、各活性薬剤について合成により調製できる。
【0059】
活性薬剤は、好ましくは医療上の有効量にて投与される。投与される正確な量、および投与の速度および時間経過は、治療されるべき疾患の性質および重篤度に依存するであろう。治療の処方、例えば用量、時期等の決定は、一般医師または専門家の責任内にあり、典型的には、治療すべき疾患、個々の患者の状態、送達部位、投与方法および実務者に知られた他の因子が考慮される。技術およびプロトコールの例は、Remington`s Pharmaceutical Sciencesに見出される。典型的には、本発明の活性薬剤は、有効成分の約0.001μg/kgないし約500μg/kg、好ましくは約0.01μg/kgないし約100μg/kg、より好ましくは約0.1μg/kgないし約50μg/kgおよび最も好ましくは約1μg/kgないし約10μg/kgの用量範囲にてそれらの効果を示す。適当な用量は、1日当たり多回サブ用量(multiple sub−doses per day)で投与できる。典型的には、用量またはサブ用量は、単位投与形態当たり有効成分の約0.1μgないし約500μgを含有させることができる。より好ましい用量は、単位投与形態当たり有効成分の約0.5μgないし約100μgを含有させることとなろう。投薬は、一般的に低レベルにて開始され、所望の効果が達成されるまで増加させる。
【0060】
実施例
本発明は以下の実施例にさらに詳述され、例示的な方法によって提供されるが、それらは本発明を限定するものではない。当該分野でよく知られた標準的技術、または以下に特に記載された技術を利用する。用いられた略語は以下の通りである:Bop、ベンゾトリアゾイルオキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート;Boc、tertブチロキシカルボニル;Fmoc、9−フルオレニルメトキシカルボニル;Gal、ガラクトース;GalNAc、N-アセチルガラクトサミン;hNTR1、ヒト・ニューロテンシン1型受容体;Hex、ヘキソース;HexNAc、N-アセチルヘキソサミン;icv、脳室内;LSI、液体二次イオン化;MALD、マトリックス支援レーザー脱離;MS、質量分析;mNTR3、マウス・ニューロテンシン3型受容体;ナノ−ESI、ナノ−電子スプレー;NMP、N−メチルピロリドン;NMR、核磁気共鳴;ppm、100万分の1;rNTR1、ラット・ニューロテンシン1型受容体;rNTR2、ラット・ニューロテンシン2型受容体;RP−HPLC、逆相高速液体クロマトグラフィー。アミノ酸は、標準的な3または1文字略語によって示される。
【0061】
実施例1
コンツラキン−Gの初期分析についての実験手順
1.粗毒液。 Conus geographusの被検体は、フィリピンのMarinduque Isから採取した。粗毒液は、毒液管の切開によって得、次いで凍結乾燥し、−70℃にて貯蔵した。
【0062】
2.ペプチド精製。 凍結乾燥したC. geographus毒液(1g)を1.1%酢酸で抽出し、従前に記載された(Oliveraら、1984)ごとくSephadex G-25カラムのクロマトグラフィーに付した。マウスを不活発および不応答にするペプチドを分取用および半分取用ならびに分析用の逆相C18カラムでの一連のRP−HPLC精製によって精製した。0.1%トリフルオロ酢酸中のアセトニトリルの勾配を用いて、カラムからペプチドを溶出した。主要な種は、再度精製の後、さらに特徴付けした。略言すると、Conus geographusからの1グラムの粗製の凍結乾燥した毒液を抽出し、従前に記載された(Oliveraら、1984)ごとくSephadex G-25カラムに適用した。麻痺性および睡眠性の活性を含む3つの連続する画分(Ve/Vo=1.37ないし1.41)をプールし、分取用逆相Vydac C18カラムに適用し、0.1%トリフルオロ酢酸中のアセトニトリルの勾配で溶出した(図1)。図1中に矢印によって示された成分は、マウスにicv投与した場合、動揺および死を引き起こした。これを半分取用C18カラムに適用して、0.1%トリフルオロ酢酸中の12〜42%アセトニトリル勾配で溶出した。icv投与した場合、マウスを不応答にした成分をさらに0.1%トリフルオロ酢酸中の20.4%アセトニトリルにてアイソクラティクな溶出で精製した。該成分のアリコートをicv注射したマウスは、それ自身、5分間立直りが困難であり、12分間非常に不活発となった。約25〜30分において、マウスは、手足を伸ばし、その腹部を下にして横わった。
【0063】
3.生物学的活性. 典型的には、部分精製された天然ペプチドをicv投与したマウスは、まず5分後に立直りが困難であり、12分後に非常に不活発となって、次いで、30分後に腹部を下にして休息した。これらの徴候をアッセイとして用いて、精製中の生物学的活性ペプチドを同定した。
【0064】
4.酵素的加水分解. 約180ピコモルのペプチド(6μl)を50μlの50mMクエン酸塩/燐酸塩緩衝液(pH4.5)中の7mU β-ガルクトシダーゼ(ウシ精巣)(2μl)で32℃にて53時間インキュベートした。約60ピコモルの該ペプチド(2μl)を50μlの20mMカコジル酸(pH6.0)中の2mU O-グリコシダーゼ(Diplococcus pneumoniae)(2μl)で32℃にて19時間インキュベートした。
【0065】
5.化学的配列およびアミノ酸分析. 自動化学配列分析は、477A蛋白質シークエンサー(Applied Biosystems, Foster City. CA)で行った。アミノ酸分析は、プレカラム誘導体化を用いて行った。約500ピコモルのコンツラキン−Gを真空下にて濃HClと共に密封し、110℃にて24時間加水分解し、凍結乾燥し、次いでo−フタルアルデヒドで誘導体化した。次いで、誘導体化したアミノ酸をRP−HPLCで分析した。
【0066】
6.質量分析.マトリックス支援レーザー脱離(MALD)(Hillenkampら、1993)質量分析は、グリッドレス反射、N2レーザーおよび100MHzのディジタイザを取り付けた「Bruker REFLEX」(Bruker Daltonics, Billerica, MA)飛行時間(Cotter、1989)質量分析機を用いて測定した。+31kVの加速電圧、および1.16および30kVの間の反射器電圧は、ポスト・ソース・ディケイ(post source decay)(Spenglerら, 1992)測定につき使用された。(0.1%トリフルオロ酢酸中の)試料は、α−シアノ−4−ヒドロキシ桂皮酸と共に適用した。液体二次イオン化(LSI)(Barberら, 1982)質量分析は、10kV加速電圧、1000または3000分解能で操作されたJeol HX110(Jeol. Tokyo, Japan)二重焦点質量分析器を用いて測定した。(0.1%トリフルオロ酢酸および25%アセトニトリル中の)試料は、チオグリセロールおよびジチオトレイトールマトリックス中で混合した。ナノ−電子スプレー(ナノ−ESI)質量分析は、Esquire イオン捕捉質量分析計(Bruker Daltonics, Billerica, MA)を用いて測定した。0.1%トリフルオロ酢酸およびアセトニトリル水溶液中で集めたRP−HPLC精製試料をメタノール1%酢酸で希釈し、ナノスプレー毛細管に移し、分析した。質量の精度は、典型的には、使用した磁気セクター装置の分解電力設定に依存して、飛行時間装置では1000ppm、イオン捕捉装置では200ppmおよび二重焦点質量分析計では20〜100ppmより良好であった。
【0067】
7.コンツラキン−Gの合成. 固相の糖ペプチド合成は、Fmoc化学を用いて、チロシンおよびセリンではt−ブチルエーテル側鎖保護、リジンではN−t−Boc側鎖保護、グルタミン酸ではt−ブチルエステル側鎖保護を手動にて行った(保護アミノ酸はBachem, Torrance, CAから入手した)。Wang樹脂で出発して、アミノ酸をBop/ジイソプロピルエチルアミン/N-メチルピロリドン/ジクロロメタンでカップリングし(Stewartら, 1984; LeNguyenら, 1986)、次いでN−脱保護をN-メチルピロリドン/ピペリジンで成した(Stewartら, 1984; LeNguyenら, 1986)。Wang樹脂は、0.2ナノモル/gの置換でThe Salk Instituteにて調製した。最初の6個のアミノ酸のカップリング後、樹脂を過アセチル化したFmoc-Oβ-D-Galp-(1→3)-α-D-GalpNAc-(1→O )トレオニンでカップリングし、他に記載したごとく(Luningら, 1989)合成し、続いて、配列中の残りの9個のアミノ酸を単一カップリングをした。注意は、グリコシル化アミノ酸からの酢酸および酢酸塩不純物を取出すのに払われ;これには、溶出剤としてジクロロメタン−酢酸エチル4:1を用いるシリカゲルのクロマトグラフィー精製、濃縮およびベンゼンからの生成物の最終的な凍結乾燥が含まれた。非グリコシル化ペプチドは、Fmoc−トレオニン(Bachem, Torrance, CA)を用いて同様に合成した。樹脂は、切断条件(95%トリフルオロ酢酸/5%アニソール(Stewartら, 1984))に付し、糖ペプチドの場合には、得られた過アセチル化糖ペプチドをRP−HPLCで単離し、主成分m/z 2322.3(MALD分析)は所望の生成物(2322.0ダルトン)に一致した。凍結乾燥後、過アセチル化糖ペプチドを乾燥メタノール中の20μlのナトリウムメトキシド(Sigma, St Louis, MO)(50mM)で1分間処理し(糖上のO−アセチル基を除去し(Norbergら, 1994))、次いで−20℃にて凍結乾燥した。脱アセチル化試料を勾配制御器を装備したWaters Prep LC/System 500A、Waters Model 450 Variable Wavelength Detector および Vydac C18 15〜20μm粒子を充填した Waters 1000 PrepPack カートリッジチャンバーカラム(65.5 ×320 mm)に負荷した。流速条件:波長230nm、AUFS 2.0、フロー 100ml/分、勾配20〜60%B/60分間;(ここに、A緩衝液は水中0.1%トリフルオロ酢酸であって、B緩衝液は60%アセトニトリル水溶液中0.1%トリフルオロ酢酸である)。画分(200ml)を手動で集めた。m/z 2069.9(LSI分析)の主成分は、所望の生成物(2069.98ダルトン)に一致した。分取用RP−HPLC精製後、十分に精製したコンツラキン−Gは、分析特性および生物学的試験用に得られた。1H−NMRデータを含めた合成コンツラキン−Gのさらに多数の特徴付けが別に示されるであろう。
【0068】
8.共−溶出. 天然および合成のコンツラキン−Gを別々に分析し、2.1×150mmのVydac C18カラムおよび0%Bから40%Bへの0.5%/分間の勾配を用いてRP−HPLCで共溶出した(ここに、A緩衝液は、水中0.55%トリフルオロ酢酸であって、B緩衝液は90%アセトニトリル水溶液中0.55%トリフルオロ酢酸である)。
【0069】
9.結合試験. 非グリコシル化Thr10−コンツラキン−Gおよび合成コンツラキン−Gは、従前に記載された(Cusackら, 1993)ごとく、放射性リガンド結合アッセイにおける全てのピッペッティング工程につきBiomek 1000ロボット工学的ワークステーションを用いて、ヒト・ニューロテンシン1型受容体(hNTR1)でアッセイした。[3H]ニューロテンシン1−13を用い、非標識ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成的コンツラキン−Gの濃度を変更する競合結合アッセイをHEK293細胞系からの膜調製物で行った。非特異的結合は、1mlの合計容量のアッセイ試験管内の1μM非標識ニューロテンシン1−13を用いて測定した。インキュベーションは20℃にて30分間であった。アッセイは、冷0.9%NaCl(5×1.5ml)の添加、続いての0.2%ポリエチレンイミンで前処理したGF/Bフィルター細片を介する急速濾過によってルーチン的に終了した。結合アッセイの詳細は、以前に記載されている(Cusackら、1991)。データは、LIGANDプログラムを用いて分析した(Munsonら, 1980)。
非グリコシル化Thr10−コンツラキン−Gおよび合成的コンツラキン−Gは、ラット・ニューロテンシン1型および2型の受容体(rNTR1およびrNTR2)ならびにマウス・ニューロテンシン3型(mNTR3)で別々にアッセイされた。[125I−Tyr3]ニューロテンシン1−13を調製して、従前に記載(Saadoulら, 1984)のごとく精製した。rNTR1(Tanakaら, 1990)および(ラット脳cDNAライブラリー(Stratagene)をスクリーニングすることによってJ Mazellaの研究室においてクローン化された)rNTR2のいずれかを発現している安定な、トランスフェクトされたCHO細胞を10%ウシ胎仔血清および0.25mg/mlG418(Sigma, France)を含有するDMEM中で増殖させた。細胞膜のホモジネートは、最初に記載された(Chabryら, 1994)ごとく調製した。蛋白質濃度は、標準品として卵白アルブミンを用いるBio−Radの手法によって測定した。
【0070】
10.細胞膜に対する結合実験. 膜(NTR2では25μg、NTR1では10μg)を0.4nM[125I−Tyr3]ニューロテンシン1−13(2000Ci/ミリモル)を用い、ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成的コンツラキン−Gの濃度を増加させて、0.1%ウシ血清アルブミンおよび0.8mM 1−10フェナントロリンを含有する250μlの50mMトリス−HCl(pH7.5)中で25℃にて20分間インキュベートした。結合実験は、2mlの氷冷緩衝液の添加によって終了し、続いて酢酸セルロースフィルター(Sartorius)を通して濾過し、2回洗浄した。フィルターに保持した放射性活性は、γ−カウンターで計測した。
【0071】
11.可溶化抽出物に関する結合実験. CHAPS−可溶化抽出物(100μg)は、0.2nM[125I−Tyr3]ニューロテンシン1−13と共に、0.1% CHAPSを含有するトリス−グリセロール緩衝液の250μl中で0℃にて1時間インキュベートした。結合リガンドは、0.3%ポリエチレンイミンで前処理したGF/Bフィルターでの濾過によって遊離リガンドと分離した。フィルターは、3mlの氷冷緩衝液で急速洗浄し、次いで、放射活性を計測した。
【0072】
mNTR3に関する結合実験では、マウス脳からの膜ホモジネートを10%(w/v)グリセロール、0.1mMフェニルメチルスルホニルフルオリド、1μMペプスタチン、1mMインドアセトアミド、および5mM EDTA(トリス−グリセロール緩衝液)を含有する25mMトリス−HCl緩衝液(pH7.5)に懸濁した。可溶化は、0.125%CHSを含有する0.625%CHAPSを含むトリス−グリセロール緩衝液中にて10mg/mlの濃度にてホモジネートをインキュベートすることによって行った(Mazellaら、1988)。可溶化抽出物を100,000×gにて4℃で30分間遠心することによって回収し、直ちに用いるか、あるいは−20℃にて貯蔵した。
【0073】
12.ホスホイノシチド測定。 rNTR1またはNTR2を発現する細胞は、無血清HAMのF−10培地中の1μCiのmyo−[3H]イノシトール(ICN)の存在下にて15〜18時間12ウェルプレート中で増殖した。細胞は0.1%ウシ血清アルブミンを含有するpH7.5のEale緩衝液(25mM Hepes、25mM トリス、140mM NaCl、5mM KCl、1.8mM CaCl2、0.8mM MgCl2、5mMグルコース)で洗浄し、Eale緩衝液中の30mM LiClの900ml中で37℃にて15分間インキュベートした。次いで、ニューロテンシン1−13を15分間指示濃度にて添加した。反応は、pH5.5の750μlの氷冷10mM HCOOHによって停止させた。4℃にて30分後に、上清を集め、2.5mlの5mM NH4OHによって中和した。総[3H]ホスホイノシチド(PI)は、5mlの水ならびに4mlの40mMおよび1Mのギ酸アンモニウムで順次溶出することによるDowex AG-X8(Bio-Rad)(Van Rentcrghemら、1988)クロマトグラフィーで遊離[3H]イノシトールから分離した。1M画分中に含有する放射活性は、5mlのEcolume(ICN)の添加後に計測した。
【0074】
13.コンツラキン−GをコードするcDNAの同定。コンツラキン−Gをコードするクローンは、従前に記載(Colledgeら、1992)のConus geographus毒液管から得られたmRNAを用いて構築されたサイズ−分別されたcDNAから選択した。ライブラリーは、ペプチド(5'- ATR ATN GGY TTY TTN GT-3';配列番号:3)のアミノ酸番号10〜15に対応する特異的なプローブを用いてスクリーニングした。そのオリゴヌクレオチドを末端標識し、ハイブリダイズし、ポリメラーゼ鎖反応による第二のスクリーニングを従前に記載(Jimenezら、1996)のこのプローブにハイブリダイズした10個のクローンに対して行った。第二のスクリーニングにおいて同定されたクローンは、従前に記載(Monjeら、1993)のDNA配列決定のために調製した。核酸配列は、従前に記載(Jimenezら、1996)のSequenase バージョン2.0DNA配列決定キットについての標準的プロトコールに従って決定した。
【0075】
実施例2
コンツラキン−Gの精製
Conus geographus毒液の画分は、マウスを過剰に不活発とさせることで検出した。通常、伏せているマウスが棒で突つかれる場合、彼らは直ちに置き上がり、相当な距離を走る。図5に示されたConus geographusからの画分のi.c.v.注射に際して、彼らが起きる前にマウスを非常に多くの力で突かなければなく、起きた後に、彼らは1または2歩歩き、直ちに、再度伏せるだろう。この「不活発な行動」に続いて数工程で精製し、見掛け上均質のペプチドをさらに分析した。このペプチドは、コンツラキン−G(フィリピン語のtulakinは、基語tulak、押すことからの「押されるか突かねばならない」を意味する)と命名された。「G」は、ペプチドがConus geographusからのものであることを示す。
【0076】
実施例3
精製されたコンツラキン−Gの生化学的特徴付け
精製ペプチドの試みられたアミノ酸配列分析は、該ペプチドがN末端にてブロックされることを明らかにした。大部分のN末端的がブロックされたコーヌスペプチドは、第1位にてピログルタミン酸残基を持っているので、ペプチドをピログルタミン酸アミノペプチダーゼで処理した。この結果、保持時間をシフトさせ、ピログルタミン酸残基の除去を示唆した。酵素処理後、ピログルタミン酸残基の除去を確認する標準的Edman方法によって、配列Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Xaa-Lys-Lys-Pro-Tyr-Ile-Leu(配列番号:4)を得、ここに、Xaaは、アミノ酸が第9サイクルにおいて(第10位にて)指定されないが、トレオニン残基についての非常に低いシグナルが観察されたことを示す。アミノ酸分析は、該ペプチドの1つのトレオニン残基の存在と一致した。
【0077】
第10位のアミノ酸残基の性質を確認するために、該ペプチドをコードするcDNAクローンを単離した。該クローンによって明らかにされたヌクレオチド配列および推定アミノ酸配列を、表1ならびに、各々、配列番号:5および配列番号:6中に示した。直接的なEdman配列決定によって得られたコンツラキン−Gのアミノ酸配列は、(51〜66残基にて)クローンにおける唯一重要なオープンリーディングフレームのC末端の方向にコードされることが判明し;予測アミノ酸配列は、成熟ペプチドの10位(前駆体の残基60)がトレオニンについてのコドンによってコードされることを明らかとした。かくして、Edman配列決定は、クローニング結果と共に、修飾されたトレオニン残基が第10位に存在することを示唆した。
【0078】
【表1】

【0079】
精製コンツラキン−G画分の質量分析(MALD、LSIおよびナノ−ESI)は、表2に要約されるごとく様々な無傷の種を明らかとした。異なる種の強度のいくらかの変動は、異なるイオン化技術で観察され、それは各イオン化技術が持つバイアスにおける差(Craigら、1994)に生起された。次の分析において、発明者らは、調査されたイオン化技術のすべてで観察された無傷の質量M1=2069で主要な糖形態に集中した。第10残基でThrと推測された配列についての実測質量(2069ダルトン)および計算された質量との差は、365ダルトンであった。トレオニンの1つの可能な修飾がO−グリコシル化であるために、発明者らは、この質量差に基づき、未同定残基がヘキソース−N−アセチル−ヘキソサミン−トレオニン(Hex-HexNAc-Thr)であり、その結果、365.13ダルトンが付加されることを提案した。観察された質量(表2)は、提案された二糖結合したペプチドの[M1+H]+または[M1+2H]2+の計算されたモノアイソトピック質量(各々、2069.98または1035.5ダルトン)と一致する。強度のフラグメントイオンは、(p(χ3)10(Craigら、1993)と表示された)完全なHex-HexNAcグリカンの喪失または末端ヘキソース残基p(χ8)10の喪失に対応してコンツラキン−Gの二重荷電[M1+2H]2+の無傷な分子のナノ−ESI MS/MS質量分析(図6)において観察された。
【0080】
【表2】
【0081】
実施例4
Thr10がO−グリコシル化される証拠
天然のコンツラキン−Gはウシの精巣から単離されたβ−ガラクトシダーゼで処理した。この酵素は、複合糖質の非還元端からの末端のβ 1→3ガラクトピラノシル残基を優先的に加水分解した。天然試料のβ−ガラクトシダーゼ処理後、新しい成分をRP−HPLCで観察した。この成分を集めて、MALD−MSで分析し、その中の種はm/z1907にて観察された。質量差およびその酵素の特異性は遊離されるべき末端のガラクトース残基と一致した。β−ガラクトシダーゼ加水分解の結果に基づいて、発明者らは、グリカン部位が糖ペプチドのコアユニットとしてセリンまたはトレオニンに結合した二糖Gal(β1→3)GalNAc(α1→)を遊離させるO−グリコシダーゼ処理に感受性であろうと推理した。天然のコンツラキン−GのO−グリコシダーゼ処理は、酵素的加水分解混合物をRP−HPLCで分析した後に、事実、新しい種を生じさせた。新しい成分を集めて、MALD−MSで分析し、m/z1704の種は、Hex-HexNAcの喪失と一致して観察された(すなわち、該質量は、第10位での非修飾トレオニン残基を持つペプチドについて予測されたものと一致した)。酵素的加水分解の結果は、Gal(β1→3)GalNAc(α1→)グリカンの存在と一致する。O−グリコシダーゼおよびβ−ガラクトシダーゼの加水分解の結果に基づき、大部分の豊富な糖ペプチドの構造は:
【0082】
【化1】
【0083】
である。
【0084】
実施例5
非グリコシル化およびグリコシル化のコンツラキン−Gの合成
16個のアミノ酸の非グリコシル化ペプチドを化学的に合成した。合成物質は、RP−HPLCに対して酵素的にデス−グリコシル化したコンツラキン−Gと同一の保持時間を有することが判明した。また、Thr10に結合したGal(β1→3)GalNAc(α1→)を含有する16個のアミノ酸のグリコシル化コンツラキン−Gを合成した。この合成グリコシル化コンツラキン−Gは、RP−HPLCで天然のコンツラキン−Gと共溶出した。天然および合成のコンツラキン−Gの双方で観察されたポスト・ソース・ディケイ断片化スペクトルは、非常に小さな断片化パターンを示した。
【0085】
実施例6
合成のグリコシル化および非グリコシル化のコンツラキン−Gの生物学的効力
ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成コンツラキン−Gをicv投与した場合に、腸収縮、毛を口でそろえる/繕いの不存在、および尾部抑制の感受性の低下と共に、天然のコンツラキン−Gが当初に単離された運動制御の喪失が観察された徴候であった。これらの観察をより詳細に調べるために、表3に詳述したごとく、用量応答比較を行った。非グリコシル化Thr10−コンツラキン−Gアナログは、1ナノモル以上の用量にて活性であるが、300ピコモルの用量では不活性であった。対照的に、コンツラキン−Gは、30ピコモルの用量または約5ピコモル/gにて運動制御の喪失を得ることが判明した。
【0086】
【表3】
【0087】
コンツラキン−Gの6個のC末端アミノ酸は、ニューロテンシン1−13、ニューロメジン、キセニン(xenin)およびキセノプシン(xenopsin)のC末端のアミノ酸配列にかなり類似性を示す(図4参照)。コンツラキン−Gまたはニューロテンシン1−13のいずれかをicv投与した場合に観察された同様の徴候およびコンツラキン−Gおよびニューロテンシン1−13の間のかなりの相同性のために、発明者らは、多数のクーロン化されたニューロテンシン受容体に対するコンツラキン−Gの親和性を試験した。図5に示すごとく、非グリコシル化Thr10−コンツラキン−Gアナログは、ニューロテンシン1−13より10倍低くヒトニューロテンシン1型受容体(hNTR1)と結合し、他のNTRでも同等に低い親和性で結合することが判明した。コンツラキン−Gは、試験されたNTRの全てに対して、非グリコシル化Thr10−コンツラキン−Gアナログよりかなり低い親和性を示した。
【0088】
コンツラキン−Gおよび非グリコシル化Thr10−コンツラキン−Gアナログは共に、rNTRを発現するCHOに対して試験した場合にアゴニストとして作用した。rNTR2を発現するCHOでは、応答は観察されなかった。非グリコシル化Thr10−コンツラキン−Gアナログは、わずかに低い効力(0.6nM)を生じたが、ニューロテンシン1−13と比較して同様の効力であった。合成グリコシル化コンツラキン−Gの効力は、かなり低く(20〜30nM)、作動性の効力は、ニューロテンシン1−13で観察されたものの約半分であった。
【0089】
【表4】
【0090】
【表5】
【0091】
実施例7
コンツラキン−Gアナログの生物学的活性
コンツラキン−Gのいくつかペプチドアナログの生物学的活性をマウスのicv注射による前記と同様の方法において試験した。これらのペプチドは本明細書に記載のごとく合成し、以下のアナログが含まれた:
Ser10に関して天然のグリコシル化を含むSer10−コンツラキン−G(アナログA);および
Ser10に関して天然のグリコシル化を含むΔ1−9−Ser10−コンツラキン−G(アナログB)
アナログAは天然のコンツラキン−Gよりわずかに活性であることが判明した。また、アナログBは、100ピコモルの用量にて2週齡のマウスにおいて試験した場合に、アナログAと同一の活性、すなわち、開始および回復時間を有した。この試験において、マウスは、75分間後に彼ら自身では起きることが依然としてできなかった。1ナノモルおよび300ピコモルの用量での3週齡のマウスにおける試験の場合、同一活性は、そのアナログ間に見られ、これらのマウスは、100分間眠そうであった。これらの実験は、N末端アミノ酸残基を取り除いたグリコシル化コンツラキン−Gアナログが活性を保持することを示した。同様の結果は、Thr10に天然のグリコシル化を含むかまたは含まないSer6に関して天然のグリコシル化を含むΔ1−5−Ser6−コンツラキン−Gのごとき他のアナログについて達成された。これらの結果は、切形部位の付近のグリコシル化セリン残基の置換は、活性アナログを与えることを示す。
【0092】
前記に特徴付けられたコーヌスペプチドのコンツラキン−Gは、新規な生物学的特徴:コーヌスペプチドにおいて従前には判明していない翻訳後のO−グリコシル化トレオニンを有する。質量分析および特異的な酵素的加水分解を用いて、Thr10が二糖Gal(β1→3)GalNAc(α1→)で修飾されることが判明した。コンツラキン−Gの対応するグリコシル化および非グリコシル化形態を合成し、RP−HPLC共溶出およびMS断片化判定基準に基づいて天然分子のこの主要なグリコシル化形態の分子構造を確認した。質量分析で観察された他の微量の分子種の質量は、特徴付けられたオリゴヌクレオチド・コア・ユニット(Baenziger、1994)上の周辺部位にてグリカン構造変異体と一致した。
【0093】
コンツラキン−GをコードするcDNAクローンの分析は、コンツラキン−G前駆体のプレプロペプチドの組成が他のコーヌスペプチド前駆体(Olivcraら、1997)のものと同様であることを明らかとした。典型的なシグナル配列は見出され、直ちにコンツラキン−G配列に対するN末端は、2つの塩基性アミノ酸であり、それは蛋白質分解切断をおそらく知らせて、成熟ペプチドのN末端を生成する(グルタミン残基は自然にまたはグルタミニルシクラーゼ作用(Fisherら、1987)のためのいずれかでピログルタミン酸に環化するであろう)。大部分の点において、コンツラキン−G前駆体が他の全てのコーヌス毒液ペプチド前駆体と同一組成を有し、同一方法にてプロセシングされると予測されるので、クローンによって予測された10個のC末端アミノ酸は、毒液から精製されたコンツラキン−G中に存在しない。1つの可能性は、該クローンが、異なる変異体、例えば、代替的にスプライスされたものを表すことである。あるいは、C末端でのさらなる蛋白質のプロセシングは、成熟したコンツラキン−Gを精製するのに必要であろう。
【0094】
最近20年間にわたって、増加している数の生物学的に重要な糖ペプチドおよび糖蛋白質が同定されてきている。Pisanoら(Yoshidaら、1976)によって最初に同定されたベスプラキニン1(Vespulakinin 1)は、発明者らの知識によれば、コーヌス以外の毒液から単離された唯一の他のO−グリコシル化ペプチド毒素である。ベスプラキニン1(Vespulakinin 1)は、黄色ジャケットスズメバチ(yellow jacket wasp)のVespula maculifronsの毒液嚢から抽出された。該ペプチド(TAT*T*RRRGRPPGFSPFR-OH(配列番号:12)、ここに、星印はO−結合のグリコシル化トレオニン残基を示す)は、2つの連続部位のO−結合のグリコシル化を含む。ベスプラキニンのC末端は、ブラジキニン(RPPGFSPFR-OH(配列番号:13)の配列に同一であり、該ペプチドは、ブラジキニンによっても得られた多数の徴候を獲得することが判明した。従って、ベスプラキニンは、O−結合のグリコシル化ペプチド毒のもう1つの例であり、ここに、C末端は、哺乳動物の神経伝達物質受容体を標的とするようである。かくして、コンツラキン−Gおよびベスプラキニン1は、哺乳動物の神経ペプチドに高度に相同性を有する配列にグリコシル化したN末端延長を含む。K+チャンネル阻害剤のκA−コノトキシンSIVAは、ジスルフィド豊富なコーヌスペプチドに通常属し、その中に長いN末端尾部を有し、O−グリコシル化残基を有する(Craigら、1998)。
【0095】
大部分のコーヌスペプチドでは、特定の立体配置は、複数のジスルフィド結合またはγ−カルボキシグルタメート残基の適当な間隔のいずれかによって、安定化して、α−ヘリックスの形成を促す(Oliveraら、1990)。複数のジスルフィドのないコーヌスペプチドは、コノプレシン(conopressin)、コナントキン、コントリファン(contryphan)および現在のコンツラキン−Gを含めた大部分の折衷的な組の族を含む。コノプレシンは、恐らく、バソプレシン/オキシトシン族のペプチドに明らかに相同性である内因性の軟体動物ペプチドであり;コーヌス毒液管中より軟体動物組織中により広範に分布する。しかしながら、他のジスルフィド豊富でないペプチド(コナントキン、コントリファンおよびコンツラキン−G)は、通常ではない翻訳後修飾を示す特殊化された毒液ペプチドであろう。本明細書に記載のコンツラキン−GのO−グリコシル化トレオニン部位に加えて、グルタミン酸残基のγ−カルボキシル化およびトリプトファンの翻訳後のエピマー化および臭素化は、コナントキンおよびコントリファンに発見された。
【0096】
いくつかの系の証拠は、コンツラキン−Gが脊椎動物源から単離されるべきペプチドのニューロテンシン族の最初のメンバーであることと一致する。最初に、コンツラキン−GのC末端領域は、表4に示すごとく(脊椎動物からの全ての)ニューロテンシン族の他のメンバーに著しい程度の類似性を示す。さらに、コンツラキン−Gは、3つの既知のニューロテンシン受容体サブタイプに結合するのに競合し;また、コンツラキン−Gがクローン化されたニューロテンシン受容体に対するアゴニストとして作用するという証拠は、前記に示されている。しかしながら、最も説得力のあるのは、コンツラキン−Gがマウスに注射される場合に、ニューロテンシンの投与と同一の行動的徴候が得られることである。かくして、構造データ、結合データおよびin vivo行動的徴候は、全て、ニューロテンシン族のペプチドに対し、てのコンツラキン−Gの徴候と一致する。
【0097】
明らかに、コンツラキン−Gおよび非グリコシル化Thr10−コンツラキン−Gは共に生理学的な相対濃度(各々、20〜30および0.6nM)にてrNTR1アゴニストである。コンツラキン−Gおよび非グリコシル化アナログの双方の観察されたアゴニスト効果、ならびにIP蓄積アッセイを用いるrNTR2発現のCHO細胞に対するこれらのリガンドのいずれかのアゴニスト効果の不存在は、in vitro結合データと関連せず;両ペプチドは、それらのIC50結合親和性のかなり低い濃度(各々、524および79nM)にてアゴニストである。従って、最も推測されないのは、その見掛上低い結合親和性が与えられると、icv投与後の非グリコシル化アナログと比較したグリコシル化コンツラキン−Gの効力の増加である。
【0098】
かくして、グリカンの役割は、奇異なものである。in vitroにて、該グルカンは、結合親和性、作動性の能力または作動性の効力のいずれも増大しない。対照的に、in vitroにて、グリカンは、該ペプチドの効力をかなり増大する。1つの単純な説明は、非グリコシル化アナログと比較してコンツラキン−Gの効力の増大が安定性の増大のためであるということである。効力の増大の代替的な機構は、グリカンによって促進された作用部位への輸送である。加えて、グリコシル化ペプチドは、依然として規定されていないニューロテンシン受容体サブタイプに高親和性で作用できるか(Tylerら、1998)、あるいはニューロテンシン受容体サブタイプの特定の状態に選択的な高親和性を有し得る。さらに、もう1つの可能性は、相対的な標的となるニューロテンシン受容体が炭水化物結合部位と共に密接に局在化できるか、またはグリカンがある種のオピエートペプチドにつき提唱された機構の「アドレス標識」として機能できるということである。安定性の増大の仮説を支持する予備的データは得られ−コンツラキン−Gの蛋白質分解は、グリカン部位の存在によって阻害される。安定性の増大の結果、受容体での糖ペプチドの供給を増強する。しかしながら、O−グリコシル化によってもたらされたコンツラキン−Gのin vivo効力の増大は、前記に概説した可能性のより均衡化した評価を明らかに必要とする。
【0099】
実施例8
Thr10−コンツラキン−Gの鎮痛活性を評価するための材料および方法
1.急性の痛み(ホットプレート)。 Thr10−コンツラキン−G(CGX−1063)またはビヒクルを5μlの用量にて脳室内(icv)を介して投与した。投与15分間後に、動物を55℃にホットプレート上に置いた。脊髄的に媒介された行動応答の最初の応答(しりごみ)に対する潜在時間、および急性の痛みに対する中枢的に総合された運動応答の最初の後肢のなめを記録した。マウスは、応答が観察されないならば、60秒後にホットプレートから取出した。ホットプレート上に置く直前に、運動機能を加速回転棒からの最初の落下までの潜在時間を測定することによって試験した。
【0100】
2.持続性の痛み(ホルマリン試験)。 脊髄内(it)薬物注射をHyldonおよびWilcox(1980)に記載されたごとく行った。CGX−1063(10または100ピコモル)またはビヒクルを5μlの容量にて投与した。it注射の15分間後に、右の後肢(hindpaw)に20μlのホルマリンを注射した。動物は、鏡が背後に置かれた透明のプレキシガラスの筒に入れ、観察を促した。動物は、5分間当たり2分間よく観察し、動物が注射された肢をなめるのに費やす時間量を合計45〜50分間この方法にて記録した。結果は、5分間当たりの秒で表されるなめ時間として示した。実験の終わりにて、全動物を加速回転棒上に置き、最初の落下までの潜在時間を記録した。
【0101】
2.神経障害の痛み。部分的坐骨神経結紮モデルを用いて、神経障害の痛みにおけるCGX−1063の効力を評価した。神経損傷は、MalmbergおよびBasbaum(1998)の方法により生成した。動物をケタミン/キシラジン(xylazine)溶液で麻酔し、CGX−1063(100ピコモル)は結紮側に対する引込め閾値を劇的に増大させた(約6倍増加)。注目すべきことには、反対側の機械的閾値は、かなりには変更されなかった。偽手術の動物において、手術したまたは手術していない側の間の引込め閾値において差はなかった。髄腔内CGX−1063の後に、引込め閾値は、これらの動物の両方の後肢において均等に増加した。
【0102】
このデータは、CGX−1063が痛みの3つの通常用いられるモデル:急性、持続性/炎症性および神経障害性の痛みモデルにおいて潜在的な鎮痛特性を有することを示す。中枢的(icv)投与されたCGX−1063は、用量依存的に急性の痛みのホットプレートモデルにおける応答潜在時間を低下させ、低ピコモルないし高フェムトモルの範囲にて有効であった。予備的データは、このモデルにおけるCGX−1063の鎮痛効果がオピオイド機構を介して媒介されないことを示す。また、CGX−1063は、持続性/炎症性の痛みのホルマリンモデルにおいて痛覚活性を低下させるのに有効であった。CGX−1063は、ホルマリン試験の第2(炎症性)相を用量依存的に低下させたが、より低用量では第1相の活性を低下した。最後に、CGX−1063は、神経障害の痛みのモデルに十分な鎮痛活性を示した。このモデルにおける機械的な引込め閾値は、予め処置した値に比較して6倍近くに増加したが、非傷害の肢において感受性を改変しなく、これはCGX−1063が神経障害異痛を低下させたが、正常な感覚伝達に影響しないことをおそらく示す。
【0103】
実施例10
コンツラキン−Gの鎮痛活性を評価するための材料および方法
1.急性の痛み(テイル・フリック(tail-flick))。薬物(コンツラキン−G(CGX−1160)またはThr10−コンツラキン−G(CGX−1063))または生理食塩水(saline)をHyldenおよびWilcoxの方法(HyldenおよびWilcox、1980)に従い5μlの一定容量中にて脊髄内(i.t.)投与した。マウスは、タオルに穏やかに巻き、尾部を露出した。i.t.注射後の種々の時点にて、尾部を54℃に維持した水浴中に少し浸し、次いで活発な尾部引込めまでの時間を記録した。8秒間までに尾部引込めがなかったならば、尾部を取出し、組織損傷を防止した。
【0104】
2.持続性の痛み(ホルマリン試験)。 CGX−1063(1、10または100ピコモル)、ニューロテンシン(NT)(1、10、100または1000ピコモル)またはビヒクルを5μlの容量中でi.t.投与した。i.t.注射の15分間後に、右後肢に20μlの5%ホルマリンを注射した。動物を鏡が背後に置かれた透明のプレキシガラスの筒に入れ、観察を促した。動物は、5分間当たり2分間よく観察し、動物が注射された肢をなめるのに費やす時間量を合計45〜50分間この方法にて記録した。結果は、5分間当たりの秒で表されるなめ時間として示した。実験の終わりにて、全動物を加速回転棒上に置き、最初の落下までの潜在時間を記録した。
【0105】
3.慢性炎症性異痛症(CFAモデル)。マウスは、右後肢に20μlのCFAの足底注射(i.pl.)にて与え、それらのホームケージに戻した。3日後、マウスをワイヤメッシュ枠のプレキシガラスの筒に入れ、少なくとも60分間慣らせた。機械的な異痛症は、記載された(Chaplanら(1994))ごとき上下方法を用いて補正されたvonFreyフィラメントで評価し、50%の引込め閾値を計算した。そのシリーズにおいていずれのフィラメントに対しても応答しない動物は、後肢を曲げることなくして典型的に持ち上げるフィラメントであり、体重の約1/10に対応する3.6グラムの最大値を指定した。
【0106】
4.毒性試験。CGX−1160、CGX−1063およびNTの運動障害効果を正確に評価するために、50匹のマウスをi.t.にてCGX−1160またはCGX−1063(1、10、100、500および1000ピコモル)、NT(0.1、1、10および100ナノモル)または生理食塩水(n=3の各化合物の最高用量を除いて群当たりn=5)を受ける群に分けた。注射15分後
にて出発して、動物を加速回転棒上に置き、最初に落下までの潜在時間を記録した。動物は、30、60、120、240および300分(または、落下までの潜在時間が対照値にもどるまで)にて再試験した。また、直腸温を同一時点にてこれらの動物において記録した。
【0107】
実施例11
コンツラキン−Gの鎮痛活性
CGX−1160は、テイル・フリック潜在時間(図10A)を用量依存的に増大させ、ピーク効果の時間は≦30分間であった(試験された最も早い時間、図10B)。さらに、潜在時間の増加は持続的であり、注射5時間後にて引込め時間が上昇し、注射後24時間にてベースラインに戻った(図10B)。また、CGX−1063は、CGX−1160に対して、このモデルにおいて、引込めの潜在時間に変動しやすいが、用量依存的な増加を示し、次いで、最も適度な抗痛覚効力のみを示した。対照的に、NTは、テイル・フリックアッセイにおいて引込め潜在時間を有意には上昇させなかった(図10A〜10B)。
【0108】
試験された全化合物は、ホルマリン試験の両相に抗痛覚性特性を示したが、異なる効力であった。CGX−1160は、3つの化合物のうち最も効力があった。ホルマリン試験の第1相(図11A)において、CGX−1160は、約30〜40ピコモルのED50であったが、NTは約1ナノモルのED50を有した。CGX−1063は、第1相において、50%の抗痛覚閾値に達しなかった、しかしながら、この試験における不規則な用量応答は、このアッセイにおいて100ピコモル用量を繰り返しを保証した。ホルマリン試験の第2相において、3つの化合物の全ては肢なめ時間を用量依存的に低下させた(パーセント抗痛覚における増加のごとく図中に示す;図11B)。再度、CGX−1160は他の化合物より強力であり、1ピコモルのED50が見積られた。この化合物の低用量は、将来的に評価されて、より正確なED50を計算するのに必要な容量応答曲線を完成されるであろう。また、CGX−1063は、第2相における抗痛覚性行動を低下させるのに有効であり、ED50は10〜20ピコモルであると見積られた。NTは、いずれのコンツラキンより劇的に小さな効力であり、ED50は600〜700ピコモルと見積られた(図11B)。
【0109】
CGX−1160は、非常に強力でかつ用量依存的なCFA誘導の機械的異痛症の逆転を示した(図12A)。i.t.で与えられた100フェムトモルのCGX−1160は、CFA誘導の機械的異痛症を完全に逆転した。注目すべきことには、この用量にて、機械的異痛症に対する反対側の感受性は変化せず、慢性炎症のNT受容体における潜在的な片側変化を示した。高用量のCGX−1160において、CFA注射した肢および反対側の注射していない肢の双方における機械的な引込め閾値を劇的に上昇させた。図12Aおよび12Bにおいて、バー上の数字は、薬物前レベルに対する機械的閾値におけるパーセント増加を示す。示されたごとく、試験された全用量にて、CGX−1160は、注射されていない側に対し、CFA注射した側に対して非常に大きな抗異痛効果を有する。CGX−1063は、CGX−1160より小さな効力であったが、また、CFA誘導異痛症を完全に逆転させた(図12B)。最小の有効量は、10ピコモルであったが、しかしながら、CGX−1160とは異なり、この用量にて反対側は、プレ薬物ベースライン測定に対して上昇した。この試験において調べた他のモデルと一致して、NTは、1ナノモルにてCFAモデルにおいて有効性を示したが、100ピコモルでは有効性を示さなかった(図12C)。CGX−1160およびNTの他の用量は、将来的に調べて、これらの化合物についての正確なED50を決定するであろう。
【0110】
CGX−1160、−1063およびNTの全ては、運動障害および体温に対する用量依存的な効果を示した。3つの化合物の全てにつき、最大障害は、i.t.注射15分後(運動障害、図13A)、または30分後(低体温効果、図14A)であった。CGX−1063は、試験された低用量(1ピコモル、図13B)では運動毒性を持たないが、高用量にて動物はかなりの運動毒性を示した(推測されたTD50は10ピコモル、図13Bおよび15A)。10ピコモルにて、この毒性は30分間で終わり、60分までに消滅した。100ピコモルまたは1ナノモルが投与された場合に、動物は2〜3時間運動障害された(図14A)。CGX−1160は、運動障害を引き起こすににCGX−1063と同効力であった(推定されたTD50は10〜20ピコモルであった、図13B)。CGX−1063と同様に、高用量(100〜500×そのED50)にて、CGX−1063は、運動障害を示し、それは5時間後に消失した(図13Aおよび14B)。NT誘導運動障害についての推定されたTD50は、3ナノモルであった(図13B)。コンツラキンと同様に、高用量にて、NT誘導運動障害は、2〜4時間に終わった(図13Aおよび14C)。
【0111】
これらの化合物の低体温効果は、運動毒性に類似した。3つ全てが、体温の用量依存的低下を引き起こした。CGX−1160および−1063は同効力であり、TD50は100ピコモルと推定された(図15B)。しかしながら、この用量にて、CGX−1063は、体温低下を誘導し、2〜3時間で終わったが(図15Aおよび16A)、CGX−1160によって引き起こされた低体温効果は、60分間までに解消した(図15Aおよび16B)。最高用量のCGX−1160(500ピコモル、500×そのED50)にて、低体温効果は、注射後6時間まで解消しなかった(図16B)。NTは、コンツラキンに非常に類似する用量応答および経時変化を示した。低用量にて、NTは効果を有さず、短時間作用型の低体温効果を示した(図16C)。しかしながら、高用量(100ナノモル)にて、NTは劇的でかつ長時間作用型の低体温症を引き起こし、それは3時間までに解消した(15Aおよび16C)。
【0112】
本データは、CGX−1160およびCGX−1063は急性および慢性の痛みの種々の動物モデルにおいて有効な、効力があり、広スペクトルの鎮痛剤である。典型的には、CGX−1160は、CGX−1063より10倍の効力があり、NTより1000倍の効力がある(表6)。CGX−1160は、慢性炎症の痛みのモデルに特に効力があり、ここに、CGX−1160は、CFA注射を受けた肢にだけ機械的な引込め閾値を選択的に増加させるが、非注射の肢の閾値を変更しない。この発見は、慢性炎症が、炎症した肢に対応する抗痛覚性経路におけるNT受容体の再構成に導くことができることを示す。CGX−1160は、効力の増大を示すこれらの実験における唯一の化合物であったので、これはCGX−1160が選択性および特異性を有し得る受容体サブタイプの上方調節を示し得る。この化合物は、運動障害または低体温のいずれかより10〜100倍小さな用量にて抗痛覚を示し、一方、CGX−1063およびNTは、i.t.投与した場合のほぼ等用量にて、抗痛覚、運動障害および低体温症を引き起こすという発見は、CGX−1160サブタイプ選択性のこの仮説の支持内にある。100ピコモルにてi.t.で与えた場合(ホルマリン試験の第2相におけるそのED50の約10倍、図16A参照)には、CGX−1063は、CGX−1160(図16Bの10ピコモル用量を比較されたし)およびNT(図16Cの10ナノモル用量を比較されたし)の比較抗痛覚用量に対して長時間作用型の低体温を引き起こした。これは、CGX−1063がNTアナログの低体温効果に関連するNT受容体サブタイプに対して選択的であることを潜在的に示す。かくして、CGX−1160中のThr10のO−グリコシル化は、最近NTR2であると考えられている抗痛覚性NTRサブタイプに対する選択性ならびにペプチダーゼに対して代謝耐性を分け与えることができる。
【0113】
【表6】
【0114】
実施例12
コンツラキン−Gの抗精神病活性を評価するための材料および方法
1.材料。D−アンフェタミンは、Sigma(St. Louis、MO)から入手した。コンツラキン−G(CGX−1160;合成の16個のアミノ酸O−結合糖ペプチド)を上記のごとく合成した。
【0115】
2.動物。雄性CF−1マウス(30〜35グラム;Charles River Laboratories)を用いた。全動物は、12時間の明暗サイクルを持つ温度制御(23°±3℃)室にて飼育し、食餌および水を自由摂取させた。全動物は、実験用動物のヒューマンケアに対するPublic Health Service方針に従って安楽死させた。
【0116】
3.運動活性。動物は、透明のプラスチックケージ(40cm×22cm、深さ20cm)中に入れ、30分間馴化させた。次いで、動物は、10μl Hamiltonシリンジを通してフリーハンドで脳室内(i.c.v.)注射(5μl容積)によってコンツラキン−G(100ピコモル)または生理食塩水(ビヒクル)のいずれかを受けた。5分後に、動物は、腹腔内(i.p.)投与を介して生理食塩水または硫酸D−アンフェタミンを受けた。通過距離(cm)および移動に費やされた時間(秒)をVideomex-V 追跡システム(Columbus Instruments、Columbus、OH)を用いて30分間にわたりモニターした。全試験は、隔離された、薄暗い光の行動室において行った。
【0117】
4.統計量。データは、薬物を唯一の因子とする一元分散分析(ANOVA)を用いて分析し、続いて、個々の群の比較についてのNewman−Keuls多重比較検定を行い、統計学的有意差としてP<0.05を受入れた。統計学的解析は、GraphPad PRISMソフトウエア(バージョン2.01、GraphPad、San Diego、CA)で行った。
【0118】
実施例13
コンツラキン−Gの抗精神病活性
通過距離[F(4,21)=7.87、P<0.05]および移動に費やした時間[F(4,21)=6.17、P<0.05]の双方により測定された運動活性に対する薬物処置の有意な効果が、本試験において判明した。D−アンフェタミン投与の結果、通過距離および移動に費やした時間の双方が用量依存的に増加した(図17〜18)。コンツラキン−G(100ピコモルi.c.v.)でのマウスの前処置は、通過距離および移動に費やした時間におけるアンフェタミン刺激された(3mg/kg i.p.)増加をかなり低下させた。基礎的運動活性(通過距離および移動に費やした時間の双方)における低下は、コンツラキン−G(100ピコモル i.c.v.)での前処置後に見られたが、しかしながら、この低下は統計学的有意差には達しなかった。
【0119】
集中する系の証拠は、(Nemeroff et al.、1992に総説された)標準的な神経弛緩薬物が関連した副作用プロフィールなくして抗精神病活性を有し得ることを意味する。引き続いて、多くのグループが新規な抗精神病薬としてニューロテンシンアナログに集中した。コンツラキン−Gがニューロテンシンと相同性のC末端を共通し、in vivoおよびin vitroアッセイの双方にてニューロテンシンに似ているので、抗精神病効力の前臨床的スクリーニング推定D−アンフェタミンの刺激運動活性を阻害するためのコンツラキン−Gの能力を評価した。この例は、コンツラキン−Gでのマウスの前処置が、運動活性におけるアンフェタミン刺激の増加をかなり低下させることを示す。これらのデータは、コンツラキン−Gがニューロテンシンと同様の抗精神病活性を有することを示す。しかしながら、前記のごとく、ニューロテンシンは、ラットニューロテンシン受容体rNTR1(IC50:ニューロテンシンにつき3.2nM;コンツラキン−Gにつき524nM)およびrNTR2(IC50:ニューロテンシンにつき6.0nM)およびマウス・ニューロテンシン受容体mNTR3(IC50:ニューロテンシンにつき1.4nM;コンツラキン−Gにつき250nM)にてコンツラキン−Gよりはるかに大きな効力であるが、コンツラキン−Gはi.c.v.投与後のin vivoアッセイ(運動活性の視覚的に見積られた評価)にて1ないし2オーダー程度より効力があった。これらの結果は、コンツラキン−Gおよびニューロテンシンが、重なるが、区別される集団のニューロテンシン受容体サブタイプまたは活性化状態と相互作用できることを示す。かくして、コンツラキン−Gは、ニューロテンシンの制限する副作用を共有しなかったであろう。
【0120】
実施例14
コンツラキン−Gの抗痙攣活性を評価するための材料および方法
1.動物。雄性フリングス(Frings)(20〜25グラム)を12時間の明暗サイクルを持つ温度制御(23°±1℃)室にて飼育し、食餌および水を自由摂取させる。マウスは、HEW 刊行物(NIH)第8623号、「実験用動物の配慮および使用についてのガイド(Guide for the Care and Use of Laboratory Animals)」の推奨に一致する方法で飼育し、食餌させ、扱った。全マウスは、実験用動物のヒューマンケアに対するPublic Health Serviceの方針に従って安楽死させた。
【0121】
2.抗痙攣性の評価。フリングス・マウスを丸いプレキシガラスジャー(直径15cm、高さ18cm)に入れ、110デシベル(11KHz)の音響刺激に曝露した。次いで、マウスは、後肢強直性伸展の存在または不存在にて25秒間観察した。後肢強直性伸展を示さない動物は、保護されたと考えた。
【0122】
3.回転棒試験。運動障害は、6rpmにて回転する回転棒上にマウスを置くことによるピーク効果時間にて評価した。1分間に3度落下した動物を障害されたと考えた。
【0123】
実施例15
コンツラキン−Gの抗痙攣活性
コンツラキン−G(CGX−1160)およびThr10−コンツラキン−G(CGX−1063)は、i.c.v.投与後にフリングス・マウスにおける聴覚性発作を強力でかつ用量依存的にブロックした(図19)。痛みモデルにおける効力と同様に、CGX−1160はCGX−1063より効力があり、ED50は、各々、7.1ピコモルおよび27.0ピコモルであった(表7)。また、従前の試験に一致して、NTは、CGX−1160または−1063より劇的に小さな効力であった。NTの用量応答曲線が依然として完成していないので、NTは1ナノモルのi.c.v.投与後の50%保護を示した。運動毒性につき試験した場合、CGX−1160は、200ピコモルまでの用量にて50%毒性レベルに達しなかったが(図19)、一方、CGX−1063のTD50は、約375ピコモルであり、その結果、試験された用量につきPIが14と見積られた。
【0124】
別の実験において、CGX−1160のピーク効果までの時間および作用期間を調べた。100ピコモル(約14×ED50)のCGX−1160のi.c.v.投与は、i.c.v.注射30分後にて活性を示さず、60分後にて100%保護し、4時間にて依然として、試験された動物において50%保護を示した(図20)。
【0125】
本発明の方法および組成物が、種々の具体例の形式で組込むことができ、それらの少しだけが本明細書に開示されることは、認められるであろう。当業者ならば、他の具体例が本発明の範囲から逸脱しないことは明らかであろう。かくして、記載された具体例は例示的なものであり、限定として解釈すべきではない。
【0126】
参考文献リスト
Annals of Pharmacotherapy 27:912 (1993).
Araki, K.ら (1973). Chem. Pharm. Bull. (Tokyo) 21:2801-2804.
Barber, M.ら (1982). Anal. Chem. 54:645A-657A.
Benziger, J.U. (1994). Faseb. J. 8:1019-1025.
Cancer 41:1270 (1993).
Cancer Res. 44:1698 (1984).
Carraway, R.ら (1973). J. Biol. Chem. 248:6854-6861.
Cartier, G.E.ら (1996). J. Biol. Chem. 271:7522-7528.
Chabry, J.ら (1994). J. Neurochem. 63:19-27.
Chaplan, S.R.ら (1994). J. Neurosci. Methods 53:55-63.
Clineschmidt, B. V.ら (1979). Eur. J. Pharmacol. 54:129-139.
Colledge, C. J.ら (1992). Toxicon. 30:1111-1116.
Cotter, R. J. (1989). Biomed Mass Spectrom. 18:513-532.
Craig, A.G.ら (1993). Biol. Mass Spectrom. 22:31-44.
Craig, A.G.ら (1994). Biol Mass Spectrom. 23:519-528.
Craig, A.G.ら (1998). Biochemistry 37:16019-16025.
Cruz, L.J.ら (1987). Conus geographus toxins that discriminate between
neuronal and muscle sodium channels. J. Biol. Chem. 260:9280-9288.
Cusack, B.ら. (1993). J. Recept. Res. 13:123-134.
Cusack, B.ら (1991). Eur J. Pharmacol. 206:339-342.
Feurle, G.E.ら (1992). J. Biol. Chem. 267:22305-22309.
Fischer, W.ら (1987). Proc. Nat. Acad. Sci. USA 84:3628-3632.
Haack, J.A.ら (1990). Contryphan-T: a gamma-carboxyglutamate containing peptide with N-methyl-d-aspartate antagonist activity.
J. Biol. Chem. 265:6025-6029.
【0127】
Hammerland, I,.G.ら (1992). Eur. J. Pharmacol. 226:239-244.
Hillenkamp, F.ら (1993). Anal. Chem. 63:1193A-1203A.
Horiki, K.ら (1978). Chemistly Letters 165-68.
Hylden, J.L.K.ら(1980). Eur. J. Pharmacol.67:313-316.
Jimenez, E. C.ら (1996). J. Biol. Chem. 271:28002-28005.
Kaiserら (1970). Anal. Biochem. 34:595.
Kapoor(1970). J. Pharm. Sci. 59:1-27.
LeNguyen, D.およびRivier, J. (1986). Intl. J. Pep. Prot. 27:285-292.
Luning, B.ら(1989) . Glycoconjugate j. 6:5-19.
Malmberg, A.Bら (1998). Pain 76:215-222.
Mazella, J.ら (1988). J. Biol. Chem. 263:144-149.
Mcluckey, S.A.ら (1991). Anal. Chem. 63:375-383.
Mena, E.E.ら (1990). Contryphan-G: a novel peptide antagonist to the N-
methyl-l)-aspartic acid (NMDA) receptor. Neurosci. Lett. 118:241-244.
Methoden der 0rganischen Chemie (Houben-Weyl). Synthese von peptiden, E. Wunsch (編), Georg Thieme Verlag, Stuttgart, Ger. (1974).
Minamino, N.ら(1984). Biochem. Biophys. Res. Commun. 122: 542-549.
Monje, V. D.ら(1993). Neuropharmacology 32: 1141-1149.
Munson, P. J.ら (1980). Anal. Biochem. 107:220-239.
Nishiuchi, Y.ら (1993). Synthesis of gamma-carboxyglutamic acid-
containing peptides by the Boc strategy. Int. J. Pept. Protein
Res. 42:533-538.
Nemeroff, C. B.ら (1992). Ann. N. Y. Acad. Sci. 668:146-156.
Norberg, T.ら (1994). In: Methods in Enzymology, Y. C. Lee 編, Academic
Press, New York, NY, 87-107頁.
Olivera, B.M.ら (1984). Biochemistry 23:5087-5090.
【0128】
Olivera, B.M.ら (1985). Science 230:1338-1343.
Olivera, B.M.ら (1990). Science 249:257-263.
Olivera, B.M.ら (1997). Mol. Biol. Cell 8:2101-2109.
Remington's Pharmaceutical Sciences, 第18版, Mack Publishing Co.,
Easton, PA (1990).
Rivier, J.R.ら (1978). Biopolymers 17:1927-38.
Rivier, J.R.ら (1987). Biochem. 26:8508-8512.
Sadoul, J. L.ら (1984). Biochem Biophys. Res. Commun. 120:812-819.
Sambrook, J.ら (1989). Molecular Cloning: A Laboratory Manual, 第2版,
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
Schroderら (1965). The peptides 1:72-75, Academic Press, NY.
Spengler, B.ら (1992). Rapid Commun. Mass Spectrom. 6:105-108.
Stewart, J.M.ら (1984). In: Pierce Chemical Company, Rockford, IL.,
176頁.
Stewart, J.M.ら, Solid-Phase Peptide Synthesis, Freeman & Co.,
San Francisco, CA (1969).
Tanaka, K.ら (1990). Neuron 4, 847-854.
Toth, I.ら(1999). J. Med. Chem. (JMC ASAP web edition 22 Sept. 1999).
Van Renterghem, C.ら(1988). Biochem. Biophys. Res. Comm. 157:977-985.
Yoshida, H.ら (1976). Biochemistry 15:61-64.
Zhou L.M.ら (1996). Synthetic Analogues of Contryphan-G: NMDA
Antagonists Acting through a Novel Polyamine-Coupled Site.
J. Neuroehen1. 66:620-628.
【0129】
米国特許第3,972,859号.
米国特許第3,842,067号.
米国特許第3,862,925号.
米国特許第4,105,603号.
米国特許第4,352,883号.
米国特許第4,353,888号.
米国特許第4,447,356号.
米国特許第4,569,967号.
米国特許第4,883,666号.
米国特許第4,968,733号.
米国特許第4,976,859号.
米国特許第5,082,670号.
米国特許第5,084n350号.
米国特許第5,158,881号.
米国特許第5,284,761号.
米国特許第5,364,769号.
米国特許第5,514,774号.
米国特許第5,534,615号.
米国特許第5,545,723号.
米国特許第5,550,050号.
米国特許第5,591,821号.
米国特許第5,618,531号.
米国特許第5,633,347号.
【0130】
米国出願シリアル番号08/785,534.
米国出願シリアル番号09/061,026.
PCT公開出願WO 92/19195.
PCT公開出願WO 94/25503.
PCT公開出願WO 95/01203.
PCT公開出願WO 95/05452.
PCT公開出願WO 96/02286.
PCT公開出願WO 96/02646.
PCT公開出願WO 96/11698.
PCT公開出願WO 96/40871.
PCT公開出願WO 96/40959.
PCT公開出願WO 97/12635.
【技術分野】
【0001】
関連出願に関する相互参照
本発明は、1998年10月20日付けで出願された米国仮特許出願シリアル番号60/105,015、1999年4月9日付けで出願されたシリアル番号60/128,561および1999年4月23日付けで出願されたシリアル番号60/130,661(各々をここに出典明示して本明細書の一部とみなす)に関する。
【0002】
本発明は、メリーランド州ベセスダの国立保健研究所(National Institute of Health)によって与えられたグラント番号GM−48677下で政府援助によりなされた。米国政府は本発明にある種の権利を有する。
【0003】
発明の背景
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G(Contulakin-G)、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【0004】
本発明の背景を例示し、特別な場合、当該実施に関するさらなる詳細を提供するために本明細書で用いられる刊行物および他の資料は、出典明示して本明細書の一部とみなし、利便のため、以下の本文中で数字で参照され、各々、添付の出典リストにおいて分類される。
【背景技術】
【0005】
コーヌス(Conus)属の軟体動物は、それらがユニークな捕食性のライフスタイルをなすことができるようにする毒液を生産する。被食体は、銛および皮下針の両方に機能し、使い捨ての中空歯である、高度に特殊化した毒液器官によって注入される毒液により不動にされる。
【0006】
生物体間の相互作用で、毒液を注入する動物およびその毒物を注入される犠牲者間のそれらより著しいものはほとんどない。毒液は、被食体を捕獲するための一義的武器または防御機構として用いることができる。これらの毒液の多くは、神経筋系の受容体およびイオンチャンネンを指向する分子を含有する。
【0007】
コーヌス毒液から単離されたいくつかのペプチドは、特徴付けられてきている。これらはニコチン性アセチルコリン受容体、筋ナトリウムチャンネルおよびニューロンカルシウムチャンネルをそれぞれ標的にするα−、μ−およびω−コノトキシンを含む(Oliveraら、1985)。また、バソプレッシンアナログであるコノプレシンも同定された(Cruzら、1987)。さらに、コナントキンと命名されたペプチドは、Conus geographusおよびConus tulipa(Menaら、1990;Haackら、1990)から単離された。これらのペプチドは、異常な年齢依存的生理学的効果を有し;2週齢より若いマウスにおいて睡眠様状態、3週齢より加齢したマウスにおいて活動亢進挙動を誘導する(Haackら、1990)。κ−コノトキシンの単離、構造および活性は、米国特許第5,633,347号に記載されている。最近、D−トリプトファンを含有するコントリファンと命名されたペプチドがConus radiatusから単離され(米国シリアル番号09/061,026)、ブロモ−トリプトファン・コノペプチドは、Conus imperialisおよびConus radiatusから単離された(米国シリアル番号08/785,534)。
前記のコノペプチドの活性を有するさらなるコノペプチドならびにさらなる活性を有するコノトキシンペプチドを同定することが所望される。
【発明の概要】
【発明が解決しようとする課題】
【0008】
発明の概要
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【課題を解決するための手段】
【0009】
1つの具体例において、本発明は、コンツラキン−G、コンツラキン−Gプロペプチドおよびこのペプチドをコードする核酸に指向される。コンツラキン−Gは、以下の式:Xaa1-Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Thr-Lys-Lys-Xaa2-Tyr-Ile-Leu(配列番号:1)[式中、Xaa1はピロ-Glu、Xaa2はプロリンまたはヒドロキシプロリンであって、Thr10はO−グリカンを含有するように修飾されている]を有する。好ましくは、X2はプロリンである。本発明により、グリカンとは、N-、S-またはO-結合した単糖、二糖、三糖、多糖またはオリゴ糖を意味し、それは、当該技術分野において知られた合成的または酵素的な方法によって天然または修飾したアミノ酸のいずれのヒドロキシ、アミノまたはチオール基にも結合できる。グリカンを構成する単糖には、D-アロース、D-アルトロース、D-グルコース、D-マンノース、D-グロース、D-イドース、D-ガラクトース、D-タロース、D-ガラクトサミン、D-グルコサミン、D-N-アセチル-グルコサミン(GlcNAc)、D-N-アセチル-グルコサミン(GalNAc)、D-フコースまたはD-アラビノースが含まれ得る。これらの糖は、構造的には、本明細書に記載のごとく、例えば、その組合せを含めた、1以上のO−スルフェート、O−ホスフェート、O−アセチルまたはシアル酸のごとき酸性基で修飾され得る。また、グリカンには、D−ペニシラミン 2,5およびそのハロゲン化誘導体またはポリプロピレングリコール誘導体のごとき類似するポリヒドロキシグループが含まれる。グリコシド結合は、ベータおよび1−4または1−3、好ましくは1−3である。グリカンとアミノ酸との間の結合は、アルファまたはベータ、好ましくは、アルファであってもよく、それは1−である。好ましいグリカンは、本明細書にさらに記載され、最も好ましいグリカンは、Gal(β1→3)GalNAc(α1→)である。
【0010】
第2の具体例において、本発明は、以下の一般式:
Xaa1-Xaa2-Xaa3-Xaa3-Gly-Gly-Xaa2-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa10-Ile-Leu(配列番号:2)
[式中、Xaa1はピロ-Glu、Glu、Gln、またはγ-カルボキシ-Glu;Xaa2はSer、ThrまたはS-グリカン修飾Cys;Xaa3はGluまたはγ-カルボキシ-Glu;Xaa4はAsn、N-グリカン修飾AsnまたはS-グリカン修飾Cys;Xaa5はAlaまたはGly;Xaa6はThr、Ser、S-グリカン修飾Cys、Tyrまたは(4-ヒドロキシメチル-Phe、4-ヒドロキシフェニル-Gly、2,6-ジメチル-Tyr、3-ニトロ-Tyrおよび5-アミノ-Tyrのごとき)いずれかのヒドロキシ含有非天然アミノ酸;Xaa7はLys、N-メチル-Lys、N,N-ジメチル-Lys、N,N,N-トリメチル-Lys、Arg、オルニチン、ホモアルギニンまたは(N-1-(2-ピラゾリニル)-Argのごとき)いずれかの非天然の塩基性アミノ酸;Xaa8はAla、Gly、Lys、N-メチル-Lys、N,N-ジメチル-Lys、N,N,N-トリメチル-Lys、Arg、オルニチン、ホモアルギニン、(N-1-(2-ピラゾリニル)-Argのごとき)いずれかの非天然の塩基性アミノ酸またはX-Lys、ここに、Xは(CH2)n、フェニル、−(CH2)m−(CH=CH)−(CH2)mHまたは−(CH2)m−(C=C)−(CH2)mH(式中、nは1〜4であって、mは0〜2である);Xaa9はProまたはヒドロキシ-Pro;およびXaa10はTyr、モノ-ヨード-Tyr、ジ-ヨード-Tyr、O-スルホ-Tyr、O-ホスホ-Tyr、ニトロ-Tyr、Trp、D-Trp、ブロモ-Trp、ブロモ-D-Trp、クロロ-Trp、クロロ-D-Trp、Phe、L-ネオ-Trpまたは(ニトロ-Phe、4-置換Phe(ここに、該置換基は、C1−C3アルキル、カルボニル、ヒドロキシメチル、スルホメチル、ハロ、フェニル、-CHO、−CN、-SO3Hおよび-NHAc、2,6-ジメチル-Tyrおよび5-アミノ-Tyrである)のごとき)いずれかの非天然の芳香族アミノ酸である]を有するジェネリック(generic)コンツラキン−Gに指向される。C末端には、遊離カルボキシル基が含まれ、アミド化され、アシル化され、グリカンが含まれ、またはアルデヒドが含まれる。C末端は、遊離カルボキシルを含むことが好ましい。このペプチドは、さらに、前記のごとき1以上のグリカンを含み得る。該グリカンは、残基2、7、8、10および16にて生じ得る。前記および他の非天然の塩基性アミノ酸、非天然のヒドロキシ含有アミノ酸または非天然の芳香族アミノ酸は、マサチューセッツ州ウスターのRSP Amino Acid Analogues、Inc.によっておよびそこから入手可能なBuilding Block Index、バージョン2.2(ここに出典明示して本明細書の一部とみなす)に記載されている。
【0011】
第3の具体例において、本発明は、コンツラキン−Gのアナログまたはジェネリックコンツラキン−Gに指向される。これらのアナログには、Thr10までおよびそれを含むコンツラキン−Gまたはジェネリックコンツラキン−GのN末端切形が含まれる。N末端切形がThr10を通る場合、Lys11はカルボキシル化修飾したリンカーを用いてN-グリコシル化される。このN-グリコシル化Lys11は、図1(Tothら、1999)に示されるごとく表示でき、ここに、R2、R3およびR4は本明細書の記載に同じである。これらの切形において、切形に近い残基は、グリコシル化セリンで置換されることが好ましい。さらなるアナログには、1〜9位の残基に代えて、Ser−O−グリカン、Thr-O−グリカンまたはCys-S-グリカンに置換されたペプチドが含まれる。
【0012】
第4の具体例において、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤として本明細書に記載されたペプチドの使用に指向される。この具体例の1つの態様において、無痛覚症は、本明細書に記載されたペプチドのうちの1種を用いて哺乳動物に誘導される。この具体例の第2の態様において、癲癇または痙攣は哺乳動物において治療される。この具体例の第3の態様において、精神分裂病は哺乳動物において治療される。この具体例の第4の態様において、遅発性異常運動症および急性ジストニー反応(acute dystonic reaction)は哺乳動物において治療される。この具体例の第5の態様において、炎症は哺乳動物において治療される。
【図面の簡単な説明】
【0013】
【図1】図1は、カルボキシル化修飾したリンカーを用いるLysのN-グリコシル化の構造を示す。
【図2】図2は、コンツラキン−GのThr10に結合した天然のO−グリカンを示す。
【図3】図3は、コンツラキン−Gの1以上の残基に結合できるグリカンのアナログを示す。
【図4】図4は、好ましいコアのO−グリカンを示す(Van de Steenら、1998)。ムチン型O−結合オリゴ糖は、GalNAc残基によってSerまたはThr(またはこのペプチドのヒドロキシル化残基)に結合する。この最初のGalNAc残基に結合した単糖形成用ブロックおよび該結合は、「コア・グリカン」を定義し、8種が同定されている。グリコシド結合の型(配向および結合性)は、各コア・グリカンにつき定義される。
【図5】図5は、コンツラキン−Gの精製を示す。Conus geographusからの粗凍結乾燥毒液の1グラムを抽出し、従前に記載されるごとく、Sephadex G-25に適用した(Oliveraら、1984)。麻痺性および睡眠性の活性を含む3つの連続する画分(Ve/Vo=1.37ないし1.41)をプールし、分取用逆相Vydac C18カラムに適用し、0.1%トリフルオロ酢酸中のアセトニトリルの勾配で溶出した。マウスにicv投与した場合、パネルA中の矢印によって示された成分は動揺および死亡を引き起こした。
【図6】図6は、天然のコンツラキン−G(286〜1886ダルトン)のナノ−ESI MS/MSスペクトルを示す(該MS/MS実験は、示された速記法(Mcluckeyら、1991)を用いて表示され、ここに、黒丸はm/z 1035[M+2H]2+前駆体を表し、矢印は、白丸の方向に向けられ、前駆体から生成したフラグメントを表す)。スペクトル上、糖アミノ酸の構造は、矢印が2つの部位を示し、それはMS/MSスペクトルにおいて観察された主要フラグメントイオンに導く(Craigら、1993)。
【図7A−7C】図7A−7Cは、有害な加熱によって得られた脊髄媒介(肢の引込め)および脊柱上媒介(後肢のなめ)の痛覚性行動に対するCGX−1063の用量応答を示す。データは、応答(図7Aおよび7B)または最初の落下(図7C)に対して秒単位で表示される。図7Aにおいて、50℃のホットプレート上に置いた後、最初の観察可能な応答までの潜在時間が示される。図7Bは、最初の後肢のなめに対する潜在時間を示す。図7Cは、加速回転棒(accelerating rotorod)上に置いた後の最初の落下までの潜在時間を示す(図7A〜7C、n=3〜10)。
【図8A−8B】図8A〜8Bは、持続性の痛みに対する痛覚性応答に対するCGX−1063の効果を示す。図8Aにおいて、データは動物がホルマリン注入した後肢をなめるのに費やした時間量として示される(n=7〜10動物/処置群)。髄腔内CGX−1063は、生理食塩水を髄腔内注射した対照に比較してホルマリン試験において第2相の痛覚性応答を用量依存的に低下させた。図8Bは、ホルマリン試験直後の加速回転棒からの最初の落下までの潜在時間を示す。
【図9】図9は、部分的な座骨神経結紮1週間後の機械的刺激に対する肢引込め閾値を示す。データは、較正されたファン・フレイ(von Frey)フィラメントで測定されたグラム単位の50%引込め閾値として表示される(群当たりn=3〜9の動物)。
【図10A−10B】図10A〜10Bは、テイル・フリック試験におけるCGX−1160(コンツラキン−G)、CGX−1063およびNTの比較を示す。3種の化合物の用量応答を図10Aに示す。図10Bは、各化合物につき試験された最高用量での効果時間を示す(CGX−1160=100ピコモル;CGX−1063=100ピコモル;NT=10ナノモル)。
【図11A−11B】図11A〜11Bは、ホルマリン試験の第1相(図11A)および第2相(図11B)に対するCGX−1160、CGX−1063およびNTの効果を示す。3種の化合物の全てが、ホルマリンのi.pl.後の痛覚性行動を用量依存的に低下させた。第2相(図11B)において、CGX−1160は、CGX−1063より10倍、NTより600〜700倍効力があった。
【図12A−12C】図12A〜12Cは、慢性炎症で誘導された機械的異痛症に対するCGX−1160、CGX−1063およびNTの効果を示す。括弧内の数字は、各々対応する対照値の百分率を示す。図12Aにおいて、CGX−1160は、CFA誘導された異痛を強力にかつ用量依存的に逆転させた。図12Bにおいて、CGX−1063は、CFA誘導された異痛を逆転させたが、CGX−1160よりこのモデルにおいて約100倍小さな効力であった。図12Cにおいて、NTは1000ピコモルにてCFA誘導された異痛を逆転させたが、100ピコモルでは逆転させず、CGX−1160より約10000倍小さな効力であった。
【図13A−13B】図13A〜13Bは、CGX−1160、CGX−1063およびNTの運動障害効果を示す。図13Aは、試験された最高用量(ホルマリン試験の第2相におけるED50の約100倍)での3種の化合物のピーク効果までの時間および効果時間を示す。図13Bは、運動障害に対する各化合物の用量応答を示す。
【図14A−14C】図14A〜14Cは、CGX−1160、CGX−1063およびNTの運動障害の用量効果、ピーク効果までの時間および期間を示す。図14Aは、CGX−1160がそのED50の100倍以上の用量だけに長時間作用型の運動障害を引き起こすことを示す。図14Bは、CGX−1063がそのED50の10倍以上の用量にて長時間作用型の運動障害を引き起こすことを示す。図14Cは、NTがそのED50の100倍以上の用量にて長時間作用型の運動障害を引き起こすことを示す。
【図15A−15B】図15A〜15Bは、体温変化に対するCGX−1160、CGX−1063およびNTの比較を示す。図15Aは、各化合物のピークまでの時間および期間を示し、図15Bは各化合物の用量応答を示す。
【図16A−16C】図16A〜16Cは、CGX−1160、CGX−1063およびNTの低体温の用量−効果および期間を示す。図16Aにおいて、CGX−1160は、ED50より100〜500倍大きい用量だけで低体温を引き起こした。図16Bは、CGX−1160は、ED50より10倍高い用量(100ピコモル)にてCGX−1063の長時間作用型の低体温効果を示す。図16Cにおいて、NTは、そのED50より10〜100倍高い用量にて低体温効果を有した。
【図17】図17は、通過距離によって測定されたD−アンフェタミン刺激運動活性に対するThr10−g コンツラキン−G(CGX−1160;100ピコモル i.c.v.)の効果を示す。略語:sal-sal:i.p.処置は生理食塩水であって、i.c.v.処置は生理食塩水である; amphet(3mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置は生理食塩水である;amphet(10mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(10mg/kg)であって、i.c.v.処置は生理食塩水である;sal-ctl:i.p.処置は生理食塩水であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル);amphet(3mg/kg)-ctl:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル)である。各バーは、群当たり3〜7匹のマウスの平均±SEMを示す。a:生理食塩水−生理食塩水処置された群(sal-sal)に対するP<0.05;b:アンフェタミン−生理食塩水処置された群(amphet(3mg/kg)-sal)に対するP<0.05。
【図18】図18は、移動に費やされた時間によって測定されたD−アンフェタミン刺激運動活性に対するThr10−g コンツラキン−G(CGX−1160;100ピコモル i.c.v.)の効果を示す。略語:sal-sal:i.p.処置は生理食塩水であって、i.c.v.処置は生理食塩水である; amphet(3mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置は生理食塩水である;amphet(10mg/kg)-sal:i.p.処置は硫酸D-アンフェタミン(10mg/kg)であって、i.c.v.処置は生理食塩水である;sal-ctl:i.p.処置は生理食塩水であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル);amphet(3mg/kg)-ctl:i.p.処置は硫酸D-アンフェタミン(3mg/kg)であって、i.c.v.処置はThr10-g コンツラキン−G(100ピコモル)である。各バーは、群当たり3〜7匹のマウスの平均±SEMを示す。a:生理食塩水−生理食塩水処置された群(sal-sal)に対するP<0.05;b:アンフェタミン−生理食塩水処置された群(amphet(3mg/kg)-sal)に対するP<0.05。
【図19】図19は、最小運動障害用量より完全に下の用量でのフリングス・マウスにおけるi.c.v.投与後の聴覚性発作に対するCGX−1160およびCGX−1063の用量依存的保護を示す。各点は、(少なくとも4匹のマウスの群における毒性の)パーセント保護を表す。
【図20】図20は、フリングス・マウスにおけるi.c.v.投与後の聴覚性発作のブロックにおけるCGX−1160の長時間持続型の効力を示す。ニューロテンシンは、5ナノモルまでのi.c.v.投与後に50%だけ有効であった。各点は、4匹のマウスの群におけるパーセント保護を表す。
【発明を実施するための形態】
【0014】
好ましい具体例の詳細な記載
本発明は、(天然のグリコシル化ペプチドである)コンツラキン−G、デス−グリコシル化コンツラキン−G(Thr10−コンツラキン−Gという)およびその誘導体に、この成熟ペプチドの前駆体をコードするcDNAクローンに、および前駆ペプチドに指向される。さらに、本発明は、抗発作、抗炎症、抗ショック、抗血栓症、低血圧症、無痛覚症、抗精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、抗動脈硬化症、脈管および血管の障害、ならびに神経学的、神経薬理学的および神経精神薬理学的な障害のための治療剤としてのこのペプチドの使用に指向される。
【0015】
本発明は、前記のコンツラキン−Gおよびコンツラキン−Gアナログに指向される。これらのペプチドは、該ペプチドの1以上で全部までのヒドロキシル部位にて単一または複数のグリカンの翻訳後修飾を含み得る。該グリカンは、本明細書の記載に同じである。コンツラキン−Gに結合した天然のO−グリカンは、図2に示される。図3は、コンツラキン−Gの1以上の残基に結合できるグリカンのアナログを示す。この図において、R1は化学的または酵素的のいずれかでグリカンで誘導体化できるアミノ酸であり;R2はOH、NH2、NHSO3Na、NHAc、O−スルフェート、O−ホスフェートまたはO−グリカンであり;R3はH、SO3、PO3、アセチル、シアル酸または単糖であり;R4はH、SO3、PO3、アセチルまたは単糖であり;R5はOH、NH2、NHSO3Na、NHAc、O−スルフェート、O−ホスフェート、O−単糖またはO−アセチルであり;R6はH、SO3、PO3、アセチルまたは単糖であり;R7はH、SO3、PO3、アセチルまたは単糖であり;R8はH、SO3、PO3、アセチルまたは単糖であり;nは0〜4であって、mは1〜4である。
【0016】
本明細書に開示されたコンツラキン−Gまたはアナログを修飾するのに用いることができる好ましいコア・グリカンは、図4に示される。さらに、本明細書に記載された単糖を用いてこれらのコアから分岐がなされてもよい。好ましいグリコシド結合は、本明細書に記載の単糖を用いて、図4のコア5および7によって特定され、さらに、GalNAc鋳型の3、4および6位のグリカンと相同する。いずれの遊離ヒドロキシ官能基もO−硫酸化、O−燐酸化またはO−アセチル化できる。
【0017】
グリコシル化コノペプチド(コンツラキン−GまたはCGX−1160)は非グリコシル化コノペプチド(Thr10−コンツラキン−GまたはCGX−1063)よりin vivoにて高い効力を有するが、これらのin vitroでの効力はほぼ同一である。グリコシル化は、受容体と良好に結合するのに、その作用部位へのコノペプチドの送達の増強および/またはコノペプチド分解の阻害に重要であり得る。
【0018】
さらに、本発明は、さらに本明細書に記載されたコンツラキン−GをコードするDNA配列に指向される。本発明は、本明細書のさらなる詳細において記載されたコンツラキン−Gのプロペプチドにさらに指向される。
本発明は、医薬として有用である新規な線状グリコシル化コンツラキン−Gおよびその誘導体に、その製法に、これらの化合物および医薬上許容される担体を含む医薬組成物に、および医薬上の治療方法に関する。本発明の新規な化合物は、中枢神経系用剤であり、それらの生物学的作用は、新規な「ニューロテンシン受容体上のコンツラキン−G結合部位」にて影響する。より詳細には、本発明の新規な化合物は、鎮痛剤、抗炎症剤、精神分裂病のごとき精神病を治療するための抗精神病剤であり、癲癇の確立された動物モデルにおいて強力な抗発作特性を示す。
【0019】
痛み:
骨変性疾患および癌のごとき疾患において生じ得るような慢性または難治性の痛みは、様々な鎮痛剤およびしばしばモルヒネのごときオピオイド化合物で治療される弱質疾患である。
【0020】
一般的には、痛みの知覚を管理する脳経路は、依然として不完全に理解されており、「痛覚の経路」といわれる脊髄への知覚の導入性シナプス接続は、いくらか詳細に報告されている。かかる経路の第1の脚において、末梢性部位から脊髄へ突出するC−およびA−線維は、痛覚シグナルを運ぶ。脊髄の背角における多シナプスの結合は、中脳水道周囲灰色領域を含めた脳の様々な領域までの痛みの感覚の中継および調節に係わる。無痛覚または痛みの知覚の縮小は、かかる痛覚の経路に沿った伝達を減少させることにより直接的に達成できる。鎮痛性アヘン剤は、エンドルフィンまたはエンケファリンの効果の模倣により作用し、それはC−またはA−線維末端にて前シナプス的にシナプス形成し、それらが発火した場合にはサブスタンスPを含めた神経伝達物質の放出を阻害すると思われている。また、脳からの下向経路は、C−およびA-線維発火に抑制的である。
【0021】
ある種の型の痛みは複雑な病因を持っている。例えば、神経障害的な痛みは、一般には、末梢神経の損傷または部分的な離断(transection)に起因する慢性の疾患である。この型の痛みは、知覚過敏症、または外部の有害な刺激に対する感受性の増強によって特徴づけられる。神経障害的な痛みの知覚過敏性成分は、全身に広がり、急性形態の痛みに応答するような同一の医薬介在に応答しない。
【0022】
モルヒネのごときオピオイド化合物は、多くの型の痛みにつき無痛覚を得るのに有効であるが、必ずしも有効とは限らなく、患者における耐性を引き起こしかねない。対象がオピオイド麻薬に耐性である場合、増加した服用量が満足な鎮痛性効果達成するのに必要である。これらの化合物は、生命を脅かし得る呼吸抑制のごとき副作用を引き起こしかねない。さらに、オピオイドは、頻繁に患者の身体的依存を生じる。依存性は、オピオイドの用量およびそれが対象によって摂取された期間と関係するようである。この理由のために、慢性的痛みの管理についての別法の治療は後に広く求められる。さらに、必要とされる鎮痛性化合物の用量を低下させるために、オピオイド治療についての置換物または添加物のいずれかとして機能する化合物は、痛み、特に慢性的な難治性の型の痛みの治療に利用性を有する。
【0023】
コンツラキン−Gは、ある種のニューロテンシン受容体上の部位で作用するすることが示され、また、ニューロテンシンは鎮痛作用を有することが示されている(Clineschmidtら、1979)ので、コンツラキン−G様コノペプチドは、痛みおよび関連疾患の治療に有用である。
【0024】
精神分裂病:精神分裂病は、第1に、ドーパミン受容体をブロックするフェノチアジンおよびブチロフェノンのごとき神経弛緩性化合物で現在治療される神経性疾患である。コンツラキン−Gがある種のニューロテンシン受容体上の部位にて作用することが示され、また、ニューロテンシン作用が精神分裂病の病因に関係付けられる(Nemerotfら、1992)ので、コンツラキン−G様コノペプチドが精神分裂病および関連疾患の治療に有用である。
【0025】
精神分裂病の治療に有用なコノペプチドのin vitroにての選択基準には以下が含まれる:a)コンツラキン−G部位の活性化;b)脳辺縁領域に局在化したコンツラキン−G結合部位に対する高親和性の可逆的な結合、およびc)脳領域、特に脳辺縁領域からのドーパミン放出の阻害。
【0026】
次いで、前記のin vitroでのスクリーニング・アッセイにおいて十分に高活性を示す化合物は、抗精神性化合物をスクリーニングするのに用いられる動物モデルにおいて試験される。
【0027】
遅発性異常運動症および他の急性ジストニー反応:遅発性異常運動症および急性ジストニー反応は、抗精神病治療の副作用として共通して生じる運動障害であり、ハロペリドールのごときドーパミン・アンタゴニストを使用する。これらの障害は、運動の制御に関連した脳のある領域、特に基底核のドーパミン受容体の過敏性によって特徴づけられる。現在、断続的な抗精神的治療は、障害の開始を回避する試みに用いられ、かかる障害は治療の中止によって処置される。
【0028】
遅発性異常運動症の治療のためのオメガ−コノペプチドの選択基準は、以下が含まれる:a)コンツラキン−G部位の活性化;b)コンツラキン−G部位に対する高親和性の可逆的な結合;c)線条体の脳領域および基底核の他の領域からのドーパミン放出の阻害、ならびにd)辺縁領域のドーパミン放出の阻害に対する基底核におけるドーパミン放出の阻害比率。
【0029】
次いで、in vitroでのスクリーニング・アッセイにおいて十分に高活性を示す化合物は、前記のラット線条体変化モデルにおいて試験される。かかる運動疾患を治療する方法に有用な化合物は、その病変の反対側の脳の側の線条に注射した場合、行動の変化を矯正する。
【0030】
炎症:炎症の神経性成分は、交感神経系のブロック、特にベータ−アドレナリン作動性受容体のブロックが炎症性関節損傷を低減するのに役立つという点で記載されている。炎症の治療に有用な化合物は、以下のin vitro特性を有することが期待されるであろう:a)新規なコンツラキン−G部位の活性化;b)コンツラキン−G結合部位に対する高親和性結合、およびc)神経組織からのノルエピネフリン放出の阻害。かかるin vitroスクリーニング・アッセイにおいて十分に高活性を示す化合物は、慢性関節リウマチの動物モデルにおいて試験される。
【0031】
癲癇:癲癇は、発作の症状発現の繰り返しによって特徴づけられる中枢神経系の障害を記述する一般用語である。かかる発作は、知覚、自律または運動の神経系を含むことができ、脳内の異常放電の存在によって電気生理学的に認識される。かかる異常な放電活性の病態生理は良好には理解されていない;しかしながら、GABA入力のごとき抑制性の神経入力の喪失が少なくともいくらかの癲癇発作に関係するという証拠がある。
【0032】
これらの薬物が、GABA受容体と関連する塩素イオン・チャンネルに対する効果を介してGABA作動性神経阻害を強めることが知られているので、癲癇の症状発現を抑制するか、あるいは阻害するベンゾジアゼピン(例えば、ジアゼパム)のある種の能力は、癲癇活性におけるGABA作動性病態生理の証拠であると幾分考えられる。他の抗癲癇化合物の生化学的効果には、電圧感受性のナトリウムまたはカリウムのチャンネルの阻害(フェニトイン)によって興奮性の膜の安定化、ならびにGABA作動性伝達の促進、興奮性の(グルタミン作動性)神経伝達の効果の阻害、および神経伝達物質放出の低下(フェノバルビタール)によって特徴付けられる神経機能の全般的低下が含まれる。
【0033】
癲癇の治療に有用な化合物は、以下のin vitro特性を有することが期待されるであろう:a)新規なコンツラキン−G部位の活性化;b)コンツラキン−Gコノペプチド結合部位に対する高親和性結合、およびc)神経組織からの興奮性神経伝達物質放出の阻害。かかるin vitroスクリーニング・アッセイにおいて十分に高活性を示す化合物は、癲癇の確立された動物モデルにおいて試験した。
【0034】
前記の特定の疾患に加えて、本発明のペプチド、誘導体およびアナログは、ニューロテンシン受容体に結合することが判明したので、これらの化合物もニューロテンシン受容体と関連する疾患、およびニューロテンシン様化合物または他の化合物が活性であることが示された疾患と関連して有用である。これらの活性には以下のものが含まれる:メタンフェタミン・アンタゴニスト、抗精神剤、大脳用薬剤、鎮痛剤、抗菌体内毒素ショック効果、プロテアーゼ阻害作用(抗トロンビン作用、抗プラスミン作用)、低血圧作用、抗DIC作用、抗アレルギー作用、創傷治癒作用、脳浮腫、肺浮腫、気管浮腫、血栓、動脈硬化症、やけどおよび高血圧症、(気管支喘息および花粉症のごとき)アレルギー疾患、外科手術時の傷害された組織部分のごとき鋭い外傷からの出血低減、交通事故等によって引き起こされた脳または他の組織の裂傷、ならびに腫脹の緩和および治癒では、外傷によって引き起こされた痛み炎症、鈍い外傷によって引き起こされた内出血の抑制、内出血を伴う浮腫および炎症、脳梗塞(例えば、脳血栓および脳塞栓)を含む虚血性脳疾患において判明した脳マトリックスへの血液成分の漏出を抑制することによる脳浮腫の抑制または改善、頭蓋内出血(例えば、脳出血および蜘蛛膜下出血)、一過性脳虚血発作、高血圧性脳疾患における急性の脳血管障害、やけどの抑制および改善、凍瘡、他の皮膚炎症および浮腫、上部気管炎症、喘息、鼻づまり、肺浮腫、ならびに環境化学物質、癌の化学療法剤、菌体内毒素および炎症性メディエーターのごとき血管内皮および粘膜を直接的に損傷する内因性および外来性因子によって引き起こされる炎症疾患。
【0035】
本発明のコノペプチドは、コーヌス毒液からの単離によって同定される。別法として、本発明のコノペプチドは、変性したプローブを含む通常の技術を用いて種々のコーヌス種のcDNAライブラリーをスクリーニングすることによって組換えDNA技術を用いて同定される。これらのプローブにハイブリダイズするクローンを分析して、評価されるべき特定のcDNAライブラリーにつきcDNAクローニング部位を近接するPCRプライマーを用いて決定されるように、最小サイズの要求、すなわち、(あるプロペプチドにつき)約300個のヌクレオチドを有するクローンに合うものを同定する。次いで、これらの最小サイズのクローンを配列決定する。次いで、配列をコノペプチドにつき前記の特性を有するペプチドの存在につき調べる。この方法によって同定されたペプチドの生物学的活性は、本明細書、米国特許第5,635,347号に記載のごとくまたは当該技術分野において、通常、試験される。
【0036】
これらのペプチドは、化学的に合成するのに十分小さい。コノペプチドの化学合成およびこれらの合成生成物の生物学的活性の表示に加えて、前述のコノペプチドを調製するための一般的な化学合成は後記される。また、これらのコノペプチドの様々なものは、米国特許第4,447,356号(Oliveraら、1984)、第5,514,774号(Oliveraら、1996)および第5,591,821号(Oliveraら、1997)(その開示をここに出典明示して本明細書の一部とみなす)に記載される技術を用いて特定のコーヌス種からの単離および精製によって得ることができる。
【0037】
本発明のコノペプチドは、イモガイ(cone snail)からの精製によって得ることができるが、個々のイモガイから得ることができるコノペプチド量は非常に少ないために、所望の実質的に純粋なコノペプチドは固相戦略を用いて化学合成によって商業的に価値のある量において最良に実践的に得られる。例えば、単一のイモガイからの収量は、約10マイクログラム以下のコノペプチドであるかもしれない。「実質的に純粋な」とは、該ペプチドが同一型の他の生物学的分子の実質的な不存在下にて存在することを意味し;好ましくは、少なくとも約85%純度および好ましくは少なくとも約95%純度の量にて好ましくは存在する。生物学上活性なコノペプチドの化学合成は、アミノ酸配列の正確な決定にもちろん依存する。かくして、本発明のコノペプチドは、単離され、合成され、および/または実質的に純粋であり得る。
【0038】
また、コノペプチドは、当該技術においてよく知られた組換えDNA技術によって産生できる。かかる技術はSambrookら(1989)によって記載されている。この方法において産生されたペプチドは、単離され、必要な場合には還元され、酸化されて、最終分子において存在するならば正確なジスルフィド結合を形成される。
【0039】
本発明のコノペプチドの中のジスルフィド結合を形成する1つの方法は冷室温または室温下にて長期間の線状ペプチドの空気酸化である。この手順の結果、生物学的に活性なジスルフィド結合したペプチドの実質的な量を創製できる。酸化されたペプチドを逆相高速液体クロマトグラフィー(HPLC)等を用いて分別して、異なる結合立体配置を有するペプチドを分離する。その後、これらの画分と天然の物質の溶出との比較、または単純なアッセイを用いてのいずれかによって、最大の生物学的効力につき正確な結合を有する特定の画分を容易に決定する。また、in vivoにて生じる架橋および/または再配置が生物学的に強力なコノペプチド分子を創製することが判明したために、1以上の画分を有する線状ペプチドまたは酸化生成物が時々、in vivo投与につき用いることができることが判明した。しかしながら、生物学的効力のほとんどない他の画分の存在から生じる希釈のために、多少高用量を必要とするかもしれない。
【0040】
排除固相技術により、部分的固相技術により、断片縮合または古典的溶液カップリングによるごとき適当な方法によって該ペプチドを合成する。
【0041】
通常の溶液相ペプチド合成法において、ペプチド鎖は、構成アミノ酸を所望の向きで成長しつつあるペプチド鎖に付加する一連のカップリング反応によって調製できる。中間体の続いての単離および精製と共に、溶液中で反応を行うための種々のカップリング試薬、例えば、ジシクロへキシルカルボジイミドまたはジイソプロピル−カルボニルジイミダゾール、種々の活性エステル、例えば、N−ヒドロキシフタルイミドまたはN−ヒドロキシスクシンイミドおよび種々の開裂試薬の使用は、よく知られた古典的ペプチド法である。古典的溶液合成法は、論文[Methoden der Organischen Chemie(Houben-Weyl): Synthese von Peptiden ](1974)に詳細に記載されている。排除固相合成の技術は、テキスト「Solid-Phase Peptide Synthesis」(StewartおよびYoung、1969)に記載され、米国特許第4,105,603号(Valeら、1978)の開示によって例示されている。断片縮合合成方法は、米国特許第3,972,859号(1976)の開示によって例示されている。他の利用できる合成は、米国特許第3,842,067号(1974)および米国特許第3,862,925号(1975)によって例示されている。γ−カルボキシグルタミン酸残基を含むペプチドの合成は、Rivierら(1987)、Nishiuchiら(1993)およびZhouら(1996)によって例示される。コノペプチドの合成は、米国特許第4,447,356号(Oliveraら、1984)、第5,514,774号(Oliveraら、1996)および第5,591,821号(Oliveraら、1997)に記載されている。
【0042】
かかる化学合成に共通しているのは、当該基が最後に取去されるまで、その部位で化学反応が起こるのを妨ぐ適当な保護基で種々のアミノ酸部位の不安定側鎖基を保護することである。また、通常、共通するのは、その原子団がカルボニル基で反応し、続いてα−アミノ保護基を選択的に除去して、その位置で次の反応を起こす間に、アミノ酸またはフラグメント上のα−アミノ基を保護することである。従って、かかる合成の工程として、不安定な側鎖を有する残基の種々のものに結合した適当な側鎖保護基を持つ、ペプチド鎖中の所望の配列に位置する各アミノ酸残基を含む中間化合物が生成されることが共通する。
【0043】
側鎖アミノ保護基の選択に関する限り、一般的には、合成の間のα−アミノ基の脱保護の間に除去されないものが選ばれる。しかしながら、いくつかのアミノ酸、例えばHisについては、保護は一般的に必要でない。ペプチドの合成に用いられるべき特定の側鎖保護基の選択において、一般的規則は次の通りである:(a)保護基は、好ましくはその保護特性を保持し、カップリング条件下で切断除去されず、(b)保護基は、合成の各工程にてα−アミノ保護基を取去するために選択された反応条件下で安定であるべきであり、(c)ペプチド鎖を望まないように改変しないであろう反応条件下で、所望のアミノ酸配列を含有する合成を完了するに際して、側鎖保護基は、除去できなければならない。
【0044】
組換えDNA技術を用いて、これらのペプチドの多く、または全てでさえ調製できる。しかしながら、ペプチドがそのように調製されない場合、当該分野で公知の他の同等の化学合成を前記のごとく用いることもできるが、それらは、好ましくはメリフィールドの固相合成法を用いて調製される。固相合成法は、保護されたα−アミノ酸を適当な樹脂にカップリングすることによって、ペプチドのC末端から開始される。かかる出発物質は、クロロメチル化樹脂もしくはヒドロキシメチル樹脂に対するエステル結合によって、またはベンズヒドリルアミン(BHA)もしくはパラメチルベンズヒドリルアミン(MBHA)樹脂に対するアミド結合によって、α−アミノ保護アミノ酸を結合させることによって調製できる。ヒドロキシメチル樹脂の調製法は、Bodanskyら(1966)によって記載されている。クロロメチル化樹脂は、Bio Rad Laboratories(Richmond、CA)およびLab. Systems, Inc.から商業的に入手可能である。かかる樹脂の調製法は、StewartおよびYoung(1969)によって記載されている。BHAおよびMBHA樹脂支持体は商業的に入手可能で、合成されるべき所望のポリペプチドがC末端にて非置換アミドを有する場合に一般的に使用される。かくして、固体樹脂支持体は、式−O−CH2−樹脂支持体、−NH−BHA−樹脂支持体または−NH−MBHA−樹脂支持体を有するもののごとき、当該分野で公知であるもののいずれかであってよい。非置換アミドが所望される場合、開裂が直接的にアミドを与えるのでBHAまたはMBHA樹脂の使用が好ましい。N−メチルアミドが所望される場合、それはN−メチルBHA樹脂から生成できる。他の置換アミドが所望なら、米国特許第4,569,967号(Kornreichら、1986)の教示が使用できるか、あるいはC末端にて遊離酸よりもなお他の基が所望ならば、Houben-Weylのテキスト(1974)に記載されたような古典的方法を用いてペプチドを合成するのが望ましいであろう。
【0045】
BocまたはFmocによって、および側鎖保護基によって保護されたC末端アミノ酸は、もし適当ならば、Horikiら(1978)に記載された手法に従って、C末端にて遊離酸を有するペプチドが合成されるべき場合、約60℃にて24時間撹拌しつつジメチルホルムアミド(DMF)中のKFを用いて、クロロメチル化樹脂にまずカップリングできる。樹脂支持体へのBoc保護アミノ酸のカップリングに続いて、塩化メチレン中のトリフルオロ酢酸(TFA)またはTFA単独を用いることによってα−アミノ保護基を取去する。脱保護は約0℃および室温の間の温度で行う。ジオキサン中のHClのごとき他の標準的開裂試薬および特異的α−アミノ保護基の除去の条件は、SchroderおよびLubke(1965)に記載されたごとくに用いることができる。
【0046】
α−アミノ保護基の除去後、残存するα−アミノおよび側鎖保護アミノ酸を所望の順序で段階にカップリングして、前記定義の中間体化合物を得るか、あるいは合成法中に別々に各アミノ酸を付加することに対する代替法として、それらのいくつかは、固相反応器に添加する前にもう1つにカップリングしてもよい。適当なカップリング試薬の選択は当業者の技量内のものである。特に、カップリング試薬として適当なのは、N,N’−ジシクロヘキシルカルボジイミド(HoBrまたはHoAtの存在下でのDCC、DIC、HBTU、HATU、TBTU)である。
【0047】
ペプチドの固相合成法において用いられる活性化試薬は、ペプチド分野でよく知られている。適当な活性化試薬の例は、N,N’−ジイソプロピルカルボジイミドおよびN−エチル−N’−(3−ジメチルアミノプロピル)カルボジイミドのごときカルボジイミドである。ペプチドカップリングにおける他の活性化試薬およびその使用は、SchroderおよびLubke(1965)およびKapoor(1970)によって記載されている。
【0048】
各保護アミノ酸またはアミノ酸配列は、約2倍以上過剰に固相反応器に導入され、カップリングは、ジメチルホルムアミド(DMF):CH2Cl2(1:1)またはDMFもしくはCH2Cl2単独の媒体中で行うことができる。中間体カップリングが生じる場合には、カップリング手法は、次のアミノ酸のカップリングに先立ってα−アミノ保護基の除去前に繰り返される。合成法の各工程におけるカップリング反応の成功は、もし手動で行うならば、Kaiserら(1970)によって記載されたごとくニンヒドリン反応によって好ましくはモニターされる。カップリング反応は、Rivierら(1978)によって報告されたごときプログラムを用いて、Beckman990自動合成機のように自動的に行うことができる。
【0049】
所望のアミノ酸配列が完成した後、中間体ペプチドは、液体フッ化水素またはTFA(Fmoc化学を用いるなら)のごとき試薬での処理によって樹脂支持体から取り去ることができ、これは樹脂からペプチドを開裂させるにのみならず、全ての残存する側鎖保護基を開裂させ、またもし先に除去されていなければN末端にてα−アミノ保護基を切断して、遊離酸の形態のペプチドを得る。もしMetが配列中に存在するならば、Boc保護基は、好ましくはHFで樹脂からペプチドを開裂させるに先立って、トリフルオロ酢酸(TFA)/エタンジチオールを用いまず取去して、可能なS−アルキル化を消失させる。開裂にフッ化水素またはTFAを用いる場合、アニソール、クレゾール、ジメチルスルフィドおよびメチルエチルスルフィドのごとき1以上の掃去剤を反応器に含める。
【0050】
ペプチド樹脂の一部の間のペプチドを環化するのとは対照的に、好ましくは線状ペプチドの環化を行ってCys残基間に結合を生じさせる。かかるジスルフィド環化結合を行うには、十分に保護されたペプチドを、当該分野でよく知られたように、アンモノリシスによってヒドロキシメチル化樹脂またはクロロメチル化樹脂支持体から開裂させて、十分に保護されたアミド中間体を得、しかる後、これを適当に環化し脱保護する。別法として、前記樹脂またはベンズヒドリルアミン(BHA)樹脂またはパラメチルベンズヒドリルアミン(MBHA)からのペプチドの開裂と同様に脱保護は、フッ化水素酸(HF)またはTFAで0℃にて起こすことができ、続いて前記したごとくに酸化する。環化のための適当な方法は、Cartierら(1996)によって記載された方法である。
【0051】
また、前記のコンツラキン−GまたはThr10−gコンツラキン−Gのムテイン、アナログまたは活性フラグメントは、ここに企図される。Hammerlandら(1992)参照。コノトキシンペプチドの誘導ムテイン、アナログまたは活性フラグメントは、米国特許第5,545,723号(特に、第2欄第50行ないし第3欄8行参照);米国特許第5,534,615号(特に、第19欄第45行ないし第22欄33行参照)および米国特許第5,364,769号(特に、第4欄第55行ないし第7欄26行参照)(各々を出典明示して本明細書の一部とみなす)に概説されたごとき保存的アミノ酸置換を含めた公知技術により合成できる。
【0052】
有効成分として本発明の化合物を含有する医薬組成物は伝統的医薬混合技術に従って調製できる。Remington`s Pharmaceutical Sciences、第18版(1990、Mack Publishing Co.、Easton、PA)参照。典型的には、有効成分の拮抗量を、医薬上許容される担体と混合されるであろう。担体は、例えば、静脈内、経口または非経口投与に望まれる製剤形態に応じて非常に種々の形態を取ることができる。
【0053】
経口投与については、該化合物は、カプセル剤、丸剤、錠剤、ロゼンジ、メルト剤、散剤、懸濁剤または乳液のごとき固体または液体製剤に処方できる。経口投薬形態の組成物を調製するにおいて、経口液体製剤(例えば、懸濁剤、エリキシル剤および水剤)の場合には、例えば、水、グリコール、油、アルコール、矯味剤、保存剤、着色剤、懸濁化剤等;経口固体製剤(例えば、散剤、カプセル剤および錠剤)の場合には、澱粉、糖、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等のごとき通常の医薬媒体のうちいずれを用いてもよい。投与の容易さのために、錠剤およびカプセル剤は最も有利な経口用量単位形態を表し、その場合に固形医薬担体が明らかに使用される。もし所望ならば、錠剤は標準的技術によって糖衣、腸溶性コーティングとすることができる。
【0054】
非経口投与については、該化合物は、医薬担体に溶解し、懸濁剤液のいずれかとして投与することができる。適当な担体の例は、水、生理的食塩水、デキストロース溶液、フラクトース溶液、エタノールまたは動物、植物また合成起源の油である。また、担体は、例えば保存剤、懸濁化剤、可溶化剤、緩衝剤等の他の成分を含んでもよい。該化合物が髄腔内投与されるべき場合、脳脊髄液に溶解してもよい。
【0055】
本発明による活性薬剤の投与は、いずれの適当な送達手段を用いても達成でき、
(a)ポンプ(例えば、Annals of Pharmacotherapy, 27:912(1993); Cancer, 41:1270(1993); Cancer Research, 44:1698(1984)参照)
(b)マイクロカプセル化(米国特許第4,352,883号;第4,353,888号および米国特許第5,084,350号参照)
(c)持続性放出ポリマー埋込み剤(米国特許第4,883,666号)
(d)マイクロカプセル化(米国特許第5,284,761号、第5,158,881号、第4,976,859号および第4,968,733号ならびに公開されたPCT特許出願WO92/19195、WO95/05452参照)
(e)CNSに対する裸のまたは非カプセル化の細胞移植(米国特許第5,082,670号および第5,618,531号参照)
(f)皮下、静脈内、動脈内、筋肉内もしくは他の適当な部位への注射、または
(g)カプセル剤、液剤、錠剤、丸剤または持続性放出製剤における経口投与
が含まれる。
【0056】
本発明の1つの具体例において、活性薬剤は、CNS、好ましくは、脳室、脳実質、髄腔または他の適当なCNS位置、最も好ましくは髄腔内に直接的に送達される。
【0057】
あるいは、ターゲティング治療を用いて、抗体または細胞特異的リガンドのごときターゲティング・システムの使用によって、さらにとりわけあるタイプの細胞に活性薬剤を送達できる。ターゲティングは、種々の理由、例えば、薬剤が許容できない毒性があるならば、余りにも高用量を必要とするならば、または標的細胞に入ることができないならば望ましい。
【0058】
また、ペプチドである活性薬剤は、活性薬剤をコードするDNA配列を患者の身体、とりわけ脊髄領域に移植するために設計された細胞に導入する細胞ベースの送達システムにて投与することもできる。適当な送達システムは、米国特許第5,550,050号およびPCT出願公開WO 92/19195、 WO 94/25503、 WO 95/01203、 WO 95/05452、 WO 96/02286、 WO 96/02646、 WO 96/40871、 WO 96/40959およびWO 97/12635に記載されている。適当なDNA配列は、開示された配列および公知の遺伝暗号に基づいて、各活性薬剤について合成により調製できる。
【0059】
活性薬剤は、好ましくは医療上の有効量にて投与される。投与される正確な量、および投与の速度および時間経過は、治療されるべき疾患の性質および重篤度に依存するであろう。治療の処方、例えば用量、時期等の決定は、一般医師または専門家の責任内にあり、典型的には、治療すべき疾患、個々の患者の状態、送達部位、投与方法および実務者に知られた他の因子が考慮される。技術およびプロトコールの例は、Remington`s Pharmaceutical Sciencesに見出される。典型的には、本発明の活性薬剤は、有効成分の約0.001μg/kgないし約500μg/kg、好ましくは約0.01μg/kgないし約100μg/kg、より好ましくは約0.1μg/kgないし約50μg/kgおよび最も好ましくは約1μg/kgないし約10μg/kgの用量範囲にてそれらの効果を示す。適当な用量は、1日当たり多回サブ用量(multiple sub−doses per day)で投与できる。典型的には、用量またはサブ用量は、単位投与形態当たり有効成分の約0.1μgないし約500μgを含有させることができる。より好ましい用量は、単位投与形態当たり有効成分の約0.5μgないし約100μgを含有させることとなろう。投薬は、一般的に低レベルにて開始され、所望の効果が達成されるまで増加させる。
【0060】
実施例
本発明は以下の実施例にさらに詳述され、例示的な方法によって提供されるが、それらは本発明を限定するものではない。当該分野でよく知られた標準的技術、または以下に特に記載された技術を利用する。用いられた略語は以下の通りである:Bop、ベンゾトリアゾイルオキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート;Boc、tertブチロキシカルボニル;Fmoc、9−フルオレニルメトキシカルボニル;Gal、ガラクトース;GalNAc、N-アセチルガラクトサミン;hNTR1、ヒト・ニューロテンシン1型受容体;Hex、ヘキソース;HexNAc、N-アセチルヘキソサミン;icv、脳室内;LSI、液体二次イオン化;MALD、マトリックス支援レーザー脱離;MS、質量分析;mNTR3、マウス・ニューロテンシン3型受容体;ナノ−ESI、ナノ−電子スプレー;NMP、N−メチルピロリドン;NMR、核磁気共鳴;ppm、100万分の1;rNTR1、ラット・ニューロテンシン1型受容体;rNTR2、ラット・ニューロテンシン2型受容体;RP−HPLC、逆相高速液体クロマトグラフィー。アミノ酸は、標準的な3または1文字略語によって示される。
【0061】
実施例1
コンツラキン−Gの初期分析についての実験手順
1.粗毒液。 Conus geographusの被検体は、フィリピンのMarinduque Isから採取した。粗毒液は、毒液管の切開によって得、次いで凍結乾燥し、−70℃にて貯蔵した。
【0062】
2.ペプチド精製。 凍結乾燥したC. geographus毒液(1g)を1.1%酢酸で抽出し、従前に記載された(Oliveraら、1984)ごとくSephadex G-25カラムのクロマトグラフィーに付した。マウスを不活発および不応答にするペプチドを分取用および半分取用ならびに分析用の逆相C18カラムでの一連のRP−HPLC精製によって精製した。0.1%トリフルオロ酢酸中のアセトニトリルの勾配を用いて、カラムからペプチドを溶出した。主要な種は、再度精製の後、さらに特徴付けした。略言すると、Conus geographusからの1グラムの粗製の凍結乾燥した毒液を抽出し、従前に記載された(Oliveraら、1984)ごとくSephadex G-25カラムに適用した。麻痺性および睡眠性の活性を含む3つの連続する画分(Ve/Vo=1.37ないし1.41)をプールし、分取用逆相Vydac C18カラムに適用し、0.1%トリフルオロ酢酸中のアセトニトリルの勾配で溶出した(図1)。図1中に矢印によって示された成分は、マウスにicv投与した場合、動揺および死を引き起こした。これを半分取用C18カラムに適用して、0.1%トリフルオロ酢酸中の12〜42%アセトニトリル勾配で溶出した。icv投与した場合、マウスを不応答にした成分をさらに0.1%トリフルオロ酢酸中の20.4%アセトニトリルにてアイソクラティクな溶出で精製した。該成分のアリコートをicv注射したマウスは、それ自身、5分間立直りが困難であり、12分間非常に不活発となった。約25〜30分において、マウスは、手足を伸ばし、その腹部を下にして横わった。
【0063】
3.生物学的活性. 典型的には、部分精製された天然ペプチドをicv投与したマウスは、まず5分後に立直りが困難であり、12分後に非常に不活発となって、次いで、30分後に腹部を下にして休息した。これらの徴候をアッセイとして用いて、精製中の生物学的活性ペプチドを同定した。
【0064】
4.酵素的加水分解. 約180ピコモルのペプチド(6μl)を50μlの50mMクエン酸塩/燐酸塩緩衝液(pH4.5)中の7mU β-ガルクトシダーゼ(ウシ精巣)(2μl)で32℃にて53時間インキュベートした。約60ピコモルの該ペプチド(2μl)を50μlの20mMカコジル酸(pH6.0)中の2mU O-グリコシダーゼ(Diplococcus pneumoniae)(2μl)で32℃にて19時間インキュベートした。
【0065】
5.化学的配列およびアミノ酸分析. 自動化学配列分析は、477A蛋白質シークエンサー(Applied Biosystems, Foster City. CA)で行った。アミノ酸分析は、プレカラム誘導体化を用いて行った。約500ピコモルのコンツラキン−Gを真空下にて濃HClと共に密封し、110℃にて24時間加水分解し、凍結乾燥し、次いでo−フタルアルデヒドで誘導体化した。次いで、誘導体化したアミノ酸をRP−HPLCで分析した。
【0066】
6.質量分析.マトリックス支援レーザー脱離(MALD)(Hillenkampら、1993)質量分析は、グリッドレス反射、N2レーザーおよび100MHzのディジタイザを取り付けた「Bruker REFLEX」(Bruker Daltonics, Billerica, MA)飛行時間(Cotter、1989)質量分析機を用いて測定した。+31kVの加速電圧、および1.16および30kVの間の反射器電圧は、ポスト・ソース・ディケイ(post source decay)(Spenglerら, 1992)測定につき使用された。(0.1%トリフルオロ酢酸中の)試料は、α−シアノ−4−ヒドロキシ桂皮酸と共に適用した。液体二次イオン化(LSI)(Barberら, 1982)質量分析は、10kV加速電圧、1000または3000分解能で操作されたJeol HX110(Jeol. Tokyo, Japan)二重焦点質量分析器を用いて測定した。(0.1%トリフルオロ酢酸および25%アセトニトリル中の)試料は、チオグリセロールおよびジチオトレイトールマトリックス中で混合した。ナノ−電子スプレー(ナノ−ESI)質量分析は、Esquire イオン捕捉質量分析計(Bruker Daltonics, Billerica, MA)を用いて測定した。0.1%トリフルオロ酢酸およびアセトニトリル水溶液中で集めたRP−HPLC精製試料をメタノール1%酢酸で希釈し、ナノスプレー毛細管に移し、分析した。質量の精度は、典型的には、使用した磁気セクター装置の分解電力設定に依存して、飛行時間装置では1000ppm、イオン捕捉装置では200ppmおよび二重焦点質量分析計では20〜100ppmより良好であった。
【0067】
7.コンツラキン−Gの合成. 固相の糖ペプチド合成は、Fmoc化学を用いて、チロシンおよびセリンではt−ブチルエーテル側鎖保護、リジンではN−t−Boc側鎖保護、グルタミン酸ではt−ブチルエステル側鎖保護を手動にて行った(保護アミノ酸はBachem, Torrance, CAから入手した)。Wang樹脂で出発して、アミノ酸をBop/ジイソプロピルエチルアミン/N-メチルピロリドン/ジクロロメタンでカップリングし(Stewartら, 1984; LeNguyenら, 1986)、次いでN−脱保護をN-メチルピロリドン/ピペリジンで成した(Stewartら, 1984; LeNguyenら, 1986)。Wang樹脂は、0.2ナノモル/gの置換でThe Salk Instituteにて調製した。最初の6個のアミノ酸のカップリング後、樹脂を過アセチル化したFmoc-Oβ-D-Galp-(1→3)-α-D-GalpNAc-(1→O )トレオニンでカップリングし、他に記載したごとく(Luningら, 1989)合成し、続いて、配列中の残りの9個のアミノ酸を単一カップリングをした。注意は、グリコシル化アミノ酸からの酢酸および酢酸塩不純物を取出すのに払われ;これには、溶出剤としてジクロロメタン−酢酸エチル4:1を用いるシリカゲルのクロマトグラフィー精製、濃縮およびベンゼンからの生成物の最終的な凍結乾燥が含まれた。非グリコシル化ペプチドは、Fmoc−トレオニン(Bachem, Torrance, CA)を用いて同様に合成した。樹脂は、切断条件(95%トリフルオロ酢酸/5%アニソール(Stewartら, 1984))に付し、糖ペプチドの場合には、得られた過アセチル化糖ペプチドをRP−HPLCで単離し、主成分m/z 2322.3(MALD分析)は所望の生成物(2322.0ダルトン)に一致した。凍結乾燥後、過アセチル化糖ペプチドを乾燥メタノール中の20μlのナトリウムメトキシド(Sigma, St Louis, MO)(50mM)で1分間処理し(糖上のO−アセチル基を除去し(Norbergら, 1994))、次いで−20℃にて凍結乾燥した。脱アセチル化試料を勾配制御器を装備したWaters Prep LC/System 500A、Waters Model 450 Variable Wavelength Detector および Vydac C18 15〜20μm粒子を充填した Waters 1000 PrepPack カートリッジチャンバーカラム(65.5 ×320 mm)に負荷した。流速条件:波長230nm、AUFS 2.0、フロー 100ml/分、勾配20〜60%B/60分間;(ここに、A緩衝液は水中0.1%トリフルオロ酢酸であって、B緩衝液は60%アセトニトリル水溶液中0.1%トリフルオロ酢酸である)。画分(200ml)を手動で集めた。m/z 2069.9(LSI分析)の主成分は、所望の生成物(2069.98ダルトン)に一致した。分取用RP−HPLC精製後、十分に精製したコンツラキン−Gは、分析特性および生物学的試験用に得られた。1H−NMRデータを含めた合成コンツラキン−Gのさらに多数の特徴付けが別に示されるであろう。
【0068】
8.共−溶出. 天然および合成のコンツラキン−Gを別々に分析し、2.1×150mmのVydac C18カラムおよび0%Bから40%Bへの0.5%/分間の勾配を用いてRP−HPLCで共溶出した(ここに、A緩衝液は、水中0.55%トリフルオロ酢酸であって、B緩衝液は90%アセトニトリル水溶液中0.55%トリフルオロ酢酸である)。
【0069】
9.結合試験. 非グリコシル化Thr10−コンツラキン−Gおよび合成コンツラキン−Gは、従前に記載された(Cusackら, 1993)ごとく、放射性リガンド結合アッセイにおける全てのピッペッティング工程につきBiomek 1000ロボット工学的ワークステーションを用いて、ヒト・ニューロテンシン1型受容体(hNTR1)でアッセイした。[3H]ニューロテンシン1−13を用い、非標識ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成的コンツラキン−Gの濃度を変更する競合結合アッセイをHEK293細胞系からの膜調製物で行った。非特異的結合は、1mlの合計容量のアッセイ試験管内の1μM非標識ニューロテンシン1−13を用いて測定した。インキュベーションは20℃にて30分間であった。アッセイは、冷0.9%NaCl(5×1.5ml)の添加、続いての0.2%ポリエチレンイミンで前処理したGF/Bフィルター細片を介する急速濾過によってルーチン的に終了した。結合アッセイの詳細は、以前に記載されている(Cusackら、1991)。データは、LIGANDプログラムを用いて分析した(Munsonら, 1980)。
非グリコシル化Thr10−コンツラキン−Gおよび合成的コンツラキン−Gは、ラット・ニューロテンシン1型および2型の受容体(rNTR1およびrNTR2)ならびにマウス・ニューロテンシン3型(mNTR3)で別々にアッセイされた。[125I−Tyr3]ニューロテンシン1−13を調製して、従前に記載(Saadoulら, 1984)のごとく精製した。rNTR1(Tanakaら, 1990)および(ラット脳cDNAライブラリー(Stratagene)をスクリーニングすることによってJ Mazellaの研究室においてクローン化された)rNTR2のいずれかを発現している安定な、トランスフェクトされたCHO細胞を10%ウシ胎仔血清および0.25mg/mlG418(Sigma, France)を含有するDMEM中で増殖させた。細胞膜のホモジネートは、最初に記載された(Chabryら, 1994)ごとく調製した。蛋白質濃度は、標準品として卵白アルブミンを用いるBio−Radの手法によって測定した。
【0070】
10.細胞膜に対する結合実験. 膜(NTR2では25μg、NTR1では10μg)を0.4nM[125I−Tyr3]ニューロテンシン1−13(2000Ci/ミリモル)を用い、ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成的コンツラキン−Gの濃度を増加させて、0.1%ウシ血清アルブミンおよび0.8mM 1−10フェナントロリンを含有する250μlの50mMトリス−HCl(pH7.5)中で25℃にて20分間インキュベートした。結合実験は、2mlの氷冷緩衝液の添加によって終了し、続いて酢酸セルロースフィルター(Sartorius)を通して濾過し、2回洗浄した。フィルターに保持した放射性活性は、γ−カウンターで計測した。
【0071】
11.可溶化抽出物に関する結合実験. CHAPS−可溶化抽出物(100μg)は、0.2nM[125I−Tyr3]ニューロテンシン1−13と共に、0.1% CHAPSを含有するトリス−グリセロール緩衝液の250μl中で0℃にて1時間インキュベートした。結合リガンドは、0.3%ポリエチレンイミンで前処理したGF/Bフィルターでの濾過によって遊離リガンドと分離した。フィルターは、3mlの氷冷緩衝液で急速洗浄し、次いで、放射活性を計測した。
【0072】
mNTR3に関する結合実験では、マウス脳からの膜ホモジネートを10%(w/v)グリセロール、0.1mMフェニルメチルスルホニルフルオリド、1μMペプスタチン、1mMインドアセトアミド、および5mM EDTA(トリス−グリセロール緩衝液)を含有する25mMトリス−HCl緩衝液(pH7.5)に懸濁した。可溶化は、0.125%CHSを含有する0.625%CHAPSを含むトリス−グリセロール緩衝液中にて10mg/mlの濃度にてホモジネートをインキュベートすることによって行った(Mazellaら、1988)。可溶化抽出物を100,000×gにて4℃で30分間遠心することによって回収し、直ちに用いるか、あるいは−20℃にて貯蔵した。
【0073】
12.ホスホイノシチド測定。 rNTR1またはNTR2を発現する細胞は、無血清HAMのF−10培地中の1μCiのmyo−[3H]イノシトール(ICN)の存在下にて15〜18時間12ウェルプレート中で増殖した。細胞は0.1%ウシ血清アルブミンを含有するpH7.5のEale緩衝液(25mM Hepes、25mM トリス、140mM NaCl、5mM KCl、1.8mM CaCl2、0.8mM MgCl2、5mMグルコース)で洗浄し、Eale緩衝液中の30mM LiClの900ml中で37℃にて15分間インキュベートした。次いで、ニューロテンシン1−13を15分間指示濃度にて添加した。反応は、pH5.5の750μlの氷冷10mM HCOOHによって停止させた。4℃にて30分後に、上清を集め、2.5mlの5mM NH4OHによって中和した。総[3H]ホスホイノシチド(PI)は、5mlの水ならびに4mlの40mMおよび1Mのギ酸アンモニウムで順次溶出することによるDowex AG-X8(Bio-Rad)(Van Rentcrghemら、1988)クロマトグラフィーで遊離[3H]イノシトールから分離した。1M画分中に含有する放射活性は、5mlのEcolume(ICN)の添加後に計測した。
【0074】
13.コンツラキン−GをコードするcDNAの同定。コンツラキン−Gをコードするクローンは、従前に記載(Colledgeら、1992)のConus geographus毒液管から得られたmRNAを用いて構築されたサイズ−分別されたcDNAから選択した。ライブラリーは、ペプチド(5'- ATR ATN GGY TTY TTN GT-3';配列番号:3)のアミノ酸番号10〜15に対応する特異的なプローブを用いてスクリーニングした。そのオリゴヌクレオチドを末端標識し、ハイブリダイズし、ポリメラーゼ鎖反応による第二のスクリーニングを従前に記載(Jimenezら、1996)のこのプローブにハイブリダイズした10個のクローンに対して行った。第二のスクリーニングにおいて同定されたクローンは、従前に記載(Monjeら、1993)のDNA配列決定のために調製した。核酸配列は、従前に記載(Jimenezら、1996)のSequenase バージョン2.0DNA配列決定キットについての標準的プロトコールに従って決定した。
【0075】
実施例2
コンツラキン−Gの精製
Conus geographus毒液の画分は、マウスを過剰に不活発とさせることで検出した。通常、伏せているマウスが棒で突つかれる場合、彼らは直ちに置き上がり、相当な距離を走る。図5に示されたConus geographusからの画分のi.c.v.注射に際して、彼らが起きる前にマウスを非常に多くの力で突かなければなく、起きた後に、彼らは1または2歩歩き、直ちに、再度伏せるだろう。この「不活発な行動」に続いて数工程で精製し、見掛け上均質のペプチドをさらに分析した。このペプチドは、コンツラキン−G(フィリピン語のtulakinは、基語tulak、押すことからの「押されるか突かねばならない」を意味する)と命名された。「G」は、ペプチドがConus geographusからのものであることを示す。
【0076】
実施例3
精製されたコンツラキン−Gの生化学的特徴付け
精製ペプチドの試みられたアミノ酸配列分析は、該ペプチドがN末端にてブロックされることを明らかにした。大部分のN末端的がブロックされたコーヌスペプチドは、第1位にてピログルタミン酸残基を持っているので、ペプチドをピログルタミン酸アミノペプチダーゼで処理した。この結果、保持時間をシフトさせ、ピログルタミン酸残基の除去を示唆した。酵素処理後、ピログルタミン酸残基の除去を確認する標準的Edman方法によって、配列Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Xaa-Lys-Lys-Pro-Tyr-Ile-Leu(配列番号:4)を得、ここに、Xaaは、アミノ酸が第9サイクルにおいて(第10位にて)指定されないが、トレオニン残基についての非常に低いシグナルが観察されたことを示す。アミノ酸分析は、該ペプチドの1つのトレオニン残基の存在と一致した。
【0077】
第10位のアミノ酸残基の性質を確認するために、該ペプチドをコードするcDNAクローンを単離した。該クローンによって明らかにされたヌクレオチド配列および推定アミノ酸配列を、表1ならびに、各々、配列番号:5および配列番号:6中に示した。直接的なEdman配列決定によって得られたコンツラキン−Gのアミノ酸配列は、(51〜66残基にて)クローンにおける唯一重要なオープンリーディングフレームのC末端の方向にコードされることが判明し;予測アミノ酸配列は、成熟ペプチドの10位(前駆体の残基60)がトレオニンについてのコドンによってコードされることを明らかとした。かくして、Edman配列決定は、クローニング結果と共に、修飾されたトレオニン残基が第10位に存在することを示唆した。
【0078】
【表1】
【0079】
精製コンツラキン−G画分の質量分析(MALD、LSIおよびナノ−ESI)は、表2に要約されるごとく様々な無傷の種を明らかとした。異なる種の強度のいくらかの変動は、異なるイオン化技術で観察され、それは各イオン化技術が持つバイアスにおける差(Craigら、1994)に生起された。次の分析において、発明者らは、調査されたイオン化技術のすべてで観察された無傷の質量M1=2069で主要な糖形態に集中した。第10残基でThrと推測された配列についての実測質量(2069ダルトン)および計算された質量との差は、365ダルトンであった。トレオニンの1つの可能な修飾がO−グリコシル化であるために、発明者らは、この質量差に基づき、未同定残基がヘキソース−N−アセチル−ヘキソサミン−トレオニン(Hex-HexNAc-Thr)であり、その結果、365.13ダルトンが付加されることを提案した。観察された質量(表2)は、提案された二糖結合したペプチドの[M1+H]+または[M1+2H]2+の計算されたモノアイソトピック質量(各々、2069.98または1035.5ダルトン)と一致する。強度のフラグメントイオンは、(p(χ3)10(Craigら、1993)と表示された)完全なHex-HexNAcグリカンの喪失または末端ヘキソース残基p(χ8)10の喪失に対応してコンツラキン−Gの二重荷電[M1+2H]2+の無傷な分子のナノ−ESI MS/MS質量分析(図6)において観察された。
【0080】
【表2】
【0081】
実施例4
Thr10がO−グリコシル化される証拠
天然のコンツラキン−Gはウシの精巣から単離されたβ−ガラクトシダーゼで処理した。この酵素は、複合糖質の非還元端からの末端のβ 1→3ガラクトピラノシル残基を優先的に加水分解した。天然試料のβ−ガラクトシダーゼ処理後、新しい成分をRP−HPLCで観察した。この成分を集めて、MALD−MSで分析し、その中の種はm/z1907にて観察された。質量差およびその酵素の特異性は遊離されるべき末端のガラクトース残基と一致した。β−ガラクトシダーゼ加水分解の結果に基づいて、発明者らは、グリカン部位が糖ペプチドのコアユニットとしてセリンまたはトレオニンに結合した二糖Gal(β1→3)GalNAc(α1→)を遊離させるO−グリコシダーゼ処理に感受性であろうと推理した。天然のコンツラキン−GのO−グリコシダーゼ処理は、酵素的加水分解混合物をRP−HPLCで分析した後に、事実、新しい種を生じさせた。新しい成分を集めて、MALD−MSで分析し、m/z1704の種は、Hex-HexNAcの喪失と一致して観察された(すなわち、該質量は、第10位での非修飾トレオニン残基を持つペプチドについて予測されたものと一致した)。酵素的加水分解の結果は、Gal(β1→3)GalNAc(α1→)グリカンの存在と一致する。O−グリコシダーゼおよびβ−ガラクトシダーゼの加水分解の結果に基づき、大部分の豊富な糖ペプチドの構造は:
【0082】
【化1】
【0083】
である。
【0084】
実施例5
非グリコシル化およびグリコシル化のコンツラキン−Gの合成
16個のアミノ酸の非グリコシル化ペプチドを化学的に合成した。合成物質は、RP−HPLCに対して酵素的にデス−グリコシル化したコンツラキン−Gと同一の保持時間を有することが判明した。また、Thr10に結合したGal(β1→3)GalNAc(α1→)を含有する16個のアミノ酸のグリコシル化コンツラキン−Gを合成した。この合成グリコシル化コンツラキン−Gは、RP−HPLCで天然のコンツラキン−Gと共溶出した。天然および合成のコンツラキン−Gの双方で観察されたポスト・ソース・ディケイ断片化スペクトルは、非常に小さな断片化パターンを示した。
【0085】
実施例6
合成のグリコシル化および非グリコシル化のコンツラキン−Gの生物学的効力
ニューロテンシン1−13、非グリコシル化Thr10−コンツラキン−Gまたは合成コンツラキン−Gをicv投与した場合に、腸収縮、毛を口でそろえる/繕いの不存在、および尾部抑制の感受性の低下と共に、天然のコンツラキン−Gが当初に単離された運動制御の喪失が観察された徴候であった。これらの観察をより詳細に調べるために、表3に詳述したごとく、用量応答比較を行った。非グリコシル化Thr10−コンツラキン−Gアナログは、1ナノモル以上の用量にて活性であるが、300ピコモルの用量では不活性であった。対照的に、コンツラキン−Gは、30ピコモルの用量または約5ピコモル/gにて運動制御の喪失を得ることが判明した。
【0086】
【表3】
【0087】
コンツラキン−Gの6個のC末端アミノ酸は、ニューロテンシン1−13、ニューロメジン、キセニン(xenin)およびキセノプシン(xenopsin)のC末端のアミノ酸配列にかなり類似性を示す(図4参照)。コンツラキン−Gまたはニューロテンシン1−13のいずれかをicv投与した場合に観察された同様の徴候およびコンツラキン−Gおよびニューロテンシン1−13の間のかなりの相同性のために、発明者らは、多数のクーロン化されたニューロテンシン受容体に対するコンツラキン−Gの親和性を試験した。図5に示すごとく、非グリコシル化Thr10−コンツラキン−Gアナログは、ニューロテンシン1−13より10倍低くヒトニューロテンシン1型受容体(hNTR1)と結合し、他のNTRでも同等に低い親和性で結合することが判明した。コンツラキン−Gは、試験されたNTRの全てに対して、非グリコシル化Thr10−コンツラキン−Gアナログよりかなり低い親和性を示した。
【0088】
コンツラキン−Gおよび非グリコシル化Thr10−コンツラキン−Gアナログは共に、rNTRを発現するCHOに対して試験した場合にアゴニストとして作用した。rNTR2を発現するCHOでは、応答は観察されなかった。非グリコシル化Thr10−コンツラキン−Gアナログは、わずかに低い効力(0.6nM)を生じたが、ニューロテンシン1−13と比較して同様の効力であった。合成グリコシル化コンツラキン−Gの効力は、かなり低く(20〜30nM)、作動性の効力は、ニューロテンシン1−13で観察されたものの約半分であった。
【0089】
【表4】
【0090】
【表5】
【0091】
実施例7
コンツラキン−Gアナログの生物学的活性
コンツラキン−Gのいくつかペプチドアナログの生物学的活性をマウスのicv注射による前記と同様の方法において試験した。これらのペプチドは本明細書に記載のごとく合成し、以下のアナログが含まれた:
Ser10に関して天然のグリコシル化を含むSer10−コンツラキン−G(アナログA);および
Ser10に関して天然のグリコシル化を含むΔ1−9−Ser10−コンツラキン−G(アナログB)
アナログAは天然のコンツラキン−Gよりわずかに活性であることが判明した。また、アナログBは、100ピコモルの用量にて2週齡のマウスにおいて試験した場合に、アナログAと同一の活性、すなわち、開始および回復時間を有した。この試験において、マウスは、75分間後に彼ら自身では起きることが依然としてできなかった。1ナノモルおよび300ピコモルの用量での3週齡のマウスにおける試験の場合、同一活性は、そのアナログ間に見られ、これらのマウスは、100分間眠そうであった。これらの実験は、N末端アミノ酸残基を取り除いたグリコシル化コンツラキン−Gアナログが活性を保持することを示した。同様の結果は、Thr10に天然のグリコシル化を含むかまたは含まないSer6に関して天然のグリコシル化を含むΔ1−5−Ser6−コンツラキン−Gのごとき他のアナログについて達成された。これらの結果は、切形部位の付近のグリコシル化セリン残基の置換は、活性アナログを与えることを示す。
【0092】
前記に特徴付けられたコーヌスペプチドのコンツラキン−Gは、新規な生物学的特徴:コーヌスペプチドにおいて従前には判明していない翻訳後のO−グリコシル化トレオニンを有する。質量分析および特異的な酵素的加水分解を用いて、Thr10が二糖Gal(β1→3)GalNAc(α1→)で修飾されることが判明した。コンツラキン−Gの対応するグリコシル化および非グリコシル化形態を合成し、RP−HPLC共溶出およびMS断片化判定基準に基づいて天然分子のこの主要なグリコシル化形態の分子構造を確認した。質量分析で観察された他の微量の分子種の質量は、特徴付けられたオリゴヌクレオチド・コア・ユニット(Baenziger、1994)上の周辺部位にてグリカン構造変異体と一致した。
【0093】
コンツラキン−GをコードするcDNAクローンの分析は、コンツラキン−G前駆体のプレプロペプチドの組成が他のコーヌスペプチド前駆体(Olivcraら、1997)のものと同様であることを明らかとした。典型的なシグナル配列は見出され、直ちにコンツラキン−G配列に対するN末端は、2つの塩基性アミノ酸であり、それは蛋白質分解切断をおそらく知らせて、成熟ペプチドのN末端を生成する(グルタミン残基は自然にまたはグルタミニルシクラーゼ作用(Fisherら、1987)のためのいずれかでピログルタミン酸に環化するであろう)。大部分の点において、コンツラキン−G前駆体が他の全てのコーヌス毒液ペプチド前駆体と同一組成を有し、同一方法にてプロセシングされると予測されるので、クローンによって予測された10個のC末端アミノ酸は、毒液から精製されたコンツラキン−G中に存在しない。1つの可能性は、該クローンが、異なる変異体、例えば、代替的にスプライスされたものを表すことである。あるいは、C末端でのさらなる蛋白質のプロセシングは、成熟したコンツラキン−Gを精製するのに必要であろう。
【0094】
最近20年間にわたって、増加している数の生物学的に重要な糖ペプチドおよび糖蛋白質が同定されてきている。Pisanoら(Yoshidaら、1976)によって最初に同定されたベスプラキニン1(Vespulakinin 1)は、発明者らの知識によれば、コーヌス以外の毒液から単離された唯一の他のO−グリコシル化ペプチド毒素である。ベスプラキニン1(Vespulakinin 1)は、黄色ジャケットスズメバチ(yellow jacket wasp)のVespula maculifronsの毒液嚢から抽出された。該ペプチド(TAT*T*RRRGRPPGFSPFR-OH(配列番号:12)、ここに、星印はO−結合のグリコシル化トレオニン残基を示す)は、2つの連続部位のO−結合のグリコシル化を含む。ベスプラキニンのC末端は、ブラジキニン(RPPGFSPFR-OH(配列番号:13)の配列に同一であり、該ペプチドは、ブラジキニンによっても得られた多数の徴候を獲得することが判明した。従って、ベスプラキニンは、O−結合のグリコシル化ペプチド毒のもう1つの例であり、ここに、C末端は、哺乳動物の神経伝達物質受容体を標的とするようである。かくして、コンツラキン−Gおよびベスプラキニン1は、哺乳動物の神経ペプチドに高度に相同性を有する配列にグリコシル化したN末端延長を含む。K+チャンネル阻害剤のκA−コノトキシンSIVAは、ジスルフィド豊富なコーヌスペプチドに通常属し、その中に長いN末端尾部を有し、O−グリコシル化残基を有する(Craigら、1998)。
【0095】
大部分のコーヌスペプチドでは、特定の立体配置は、複数のジスルフィド結合またはγ−カルボキシグルタメート残基の適当な間隔のいずれかによって、安定化して、α−ヘリックスの形成を促す(Oliveraら、1990)。複数のジスルフィドのないコーヌスペプチドは、コノプレシン(conopressin)、コナントキン、コントリファン(contryphan)および現在のコンツラキン−Gを含めた大部分の折衷的な組の族を含む。コノプレシンは、恐らく、バソプレシン/オキシトシン族のペプチドに明らかに相同性である内因性の軟体動物ペプチドであり;コーヌス毒液管中より軟体動物組織中により広範に分布する。しかしながら、他のジスルフィド豊富でないペプチド(コナントキン、コントリファンおよびコンツラキン−G)は、通常ではない翻訳後修飾を示す特殊化された毒液ペプチドであろう。本明細書に記載のコンツラキン−GのO−グリコシル化トレオニン部位に加えて、グルタミン酸残基のγ−カルボキシル化およびトリプトファンの翻訳後のエピマー化および臭素化は、コナントキンおよびコントリファンに発見された。
【0096】
いくつかの系の証拠は、コンツラキン−Gが脊椎動物源から単離されるべきペプチドのニューロテンシン族の最初のメンバーであることと一致する。最初に、コンツラキン−GのC末端領域は、表4に示すごとく(脊椎動物からの全ての)ニューロテンシン族の他のメンバーに著しい程度の類似性を示す。さらに、コンツラキン−Gは、3つの既知のニューロテンシン受容体サブタイプに結合するのに競合し;また、コンツラキン−Gがクローン化されたニューロテンシン受容体に対するアゴニストとして作用するという証拠は、前記に示されている。しかしながら、最も説得力のあるのは、コンツラキン−Gがマウスに注射される場合に、ニューロテンシンの投与と同一の行動的徴候が得られることである。かくして、構造データ、結合データおよびin vivo行動的徴候は、全て、ニューロテンシン族のペプチドに対し、てのコンツラキン−Gの徴候と一致する。
【0097】
明らかに、コンツラキン−Gおよび非グリコシル化Thr10−コンツラキン−Gは共に生理学的な相対濃度(各々、20〜30および0.6nM)にてrNTR1アゴニストである。コンツラキン−Gおよび非グリコシル化アナログの双方の観察されたアゴニスト効果、ならびにIP蓄積アッセイを用いるrNTR2発現のCHO細胞に対するこれらのリガンドのいずれかのアゴニスト効果の不存在は、in vitro結合データと関連せず;両ペプチドは、それらのIC50結合親和性のかなり低い濃度(各々、524および79nM)にてアゴニストである。従って、最も推測されないのは、その見掛上低い結合親和性が与えられると、icv投与後の非グリコシル化アナログと比較したグリコシル化コンツラキン−Gの効力の増加である。
【0098】
かくして、グリカンの役割は、奇異なものである。in vitroにて、該グルカンは、結合親和性、作動性の能力または作動性の効力のいずれも増大しない。対照的に、in vitroにて、グリカンは、該ペプチドの効力をかなり増大する。1つの単純な説明は、非グリコシル化アナログと比較してコンツラキン−Gの効力の増大が安定性の増大のためであるということである。効力の増大の代替的な機構は、グリカンによって促進された作用部位への輸送である。加えて、グリコシル化ペプチドは、依然として規定されていないニューロテンシン受容体サブタイプに高親和性で作用できるか(Tylerら、1998)、あるいはニューロテンシン受容体サブタイプの特定の状態に選択的な高親和性を有し得る。さらに、もう1つの可能性は、相対的な標的となるニューロテンシン受容体が炭水化物結合部位と共に密接に局在化できるか、またはグリカンがある種のオピエートペプチドにつき提唱された機構の「アドレス標識」として機能できるということである。安定性の増大の仮説を支持する予備的データは得られ−コンツラキン−Gの蛋白質分解は、グリカン部位の存在によって阻害される。安定性の増大の結果、受容体での糖ペプチドの供給を増強する。しかしながら、O−グリコシル化によってもたらされたコンツラキン−Gのin vivo効力の増大は、前記に概説した可能性のより均衡化した評価を明らかに必要とする。
【0099】
実施例8
Thr10−コンツラキン−Gの鎮痛活性を評価するための材料および方法
1.急性の痛み(ホットプレート)。 Thr10−コンツラキン−G(CGX−1063)またはビヒクルを5μlの用量にて脳室内(icv)を介して投与した。投与15分間後に、動物を55℃にホットプレート上に置いた。脊髄的に媒介された行動応答の最初の応答(しりごみ)に対する潜在時間、および急性の痛みに対する中枢的に総合された運動応答の最初の後肢のなめを記録した。マウスは、応答が観察されないならば、60秒後にホットプレートから取出した。ホットプレート上に置く直前に、運動機能を加速回転棒からの最初の落下までの潜在時間を測定することによって試験した。
【0100】
2.持続性の痛み(ホルマリン試験)。 脊髄内(it)薬物注射をHyldonおよびWilcox(1980)に記載されたごとく行った。CGX−1063(10または100ピコモル)またはビヒクルを5μlの容量にて投与した。it注射の15分間後に、右の後肢(hindpaw)に20μlのホルマリンを注射した。動物は、鏡が背後に置かれた透明のプレキシガラスの筒に入れ、観察を促した。動物は、5分間当たり2分間よく観察し、動物が注射された肢をなめるのに費やす時間量を合計45〜50分間この方法にて記録した。結果は、5分間当たりの秒で表されるなめ時間として示した。実験の終わりにて、全動物を加速回転棒上に置き、最初の落下までの潜在時間を記録した。
【0101】
2.神経障害の痛み。部分的坐骨神経結紮モデルを用いて、神経障害の痛みにおけるCGX−1063の効力を評価した。神経損傷は、MalmbergおよびBasbaum(1998)の方法により生成した。動物をケタミン/キシラジン(xylazine)溶液で麻酔し、CGX−1063(100ピコモル)は結紮側に対する引込め閾値を劇的に増大させた(約6倍増加)。注目すべきことには、反対側の機械的閾値は、かなりには変更されなかった。偽手術の動物において、手術したまたは手術していない側の間の引込め閾値において差はなかった。髄腔内CGX−1063の後に、引込め閾値は、これらの動物の両方の後肢において均等に増加した。
【0102】
このデータは、CGX−1063が痛みの3つの通常用いられるモデル:急性、持続性/炎症性および神経障害性の痛みモデルにおいて潜在的な鎮痛特性を有することを示す。中枢的(icv)投与されたCGX−1063は、用量依存的に急性の痛みのホットプレートモデルにおける応答潜在時間を低下させ、低ピコモルないし高フェムトモルの範囲にて有効であった。予備的データは、このモデルにおけるCGX−1063の鎮痛効果がオピオイド機構を介して媒介されないことを示す。また、CGX−1063は、持続性/炎症性の痛みのホルマリンモデルにおいて痛覚活性を低下させるのに有効であった。CGX−1063は、ホルマリン試験の第2(炎症性)相を用量依存的に低下させたが、より低用量では第1相の活性を低下した。最後に、CGX−1063は、神経障害の痛みのモデルに十分な鎮痛活性を示した。このモデルにおける機械的な引込め閾値は、予め処置した値に比較して6倍近くに増加したが、非傷害の肢において感受性を改変しなく、これはCGX−1063が神経障害異痛を低下させたが、正常な感覚伝達に影響しないことをおそらく示す。
【0103】
実施例10
コンツラキン−Gの鎮痛活性を評価するための材料および方法
1.急性の痛み(テイル・フリック(tail-flick))。薬物(コンツラキン−G(CGX−1160)またはThr10−コンツラキン−G(CGX−1063))または生理食塩水(saline)をHyldenおよびWilcoxの方法(HyldenおよびWilcox、1980)に従い5μlの一定容量中にて脊髄内(i.t.)投与した。マウスは、タオルに穏やかに巻き、尾部を露出した。i.t.注射後の種々の時点にて、尾部を54℃に維持した水浴中に少し浸し、次いで活発な尾部引込めまでの時間を記録した。8秒間までに尾部引込めがなかったならば、尾部を取出し、組織損傷を防止した。
【0104】
2.持続性の痛み(ホルマリン試験)。 CGX−1063(1、10または100ピコモル)、ニューロテンシン(NT)(1、10、100または1000ピコモル)またはビヒクルを5μlの容量中でi.t.投与した。i.t.注射の15分間後に、右後肢に20μlの5%ホルマリンを注射した。動物を鏡が背後に置かれた透明のプレキシガラスの筒に入れ、観察を促した。動物は、5分間当たり2分間よく観察し、動物が注射された肢をなめるのに費やす時間量を合計45〜50分間この方法にて記録した。結果は、5分間当たりの秒で表されるなめ時間として示した。実験の終わりにて、全動物を加速回転棒上に置き、最初の落下までの潜在時間を記録した。
【0105】
3.慢性炎症性異痛症(CFAモデル)。マウスは、右後肢に20μlのCFAの足底注射(i.pl.)にて与え、それらのホームケージに戻した。3日後、マウスをワイヤメッシュ枠のプレキシガラスの筒に入れ、少なくとも60分間慣らせた。機械的な異痛症は、記載された(Chaplanら(1994))ごとき上下方法を用いて補正されたvonFreyフィラメントで評価し、50%の引込め閾値を計算した。そのシリーズにおいていずれのフィラメントに対しても応答しない動物は、後肢を曲げることなくして典型的に持ち上げるフィラメントであり、体重の約1/10に対応する3.6グラムの最大値を指定した。
【0106】
4.毒性試験。CGX−1160、CGX−1063およびNTの運動障害効果を正確に評価するために、50匹のマウスをi.t.にてCGX−1160またはCGX−1063(1、10、100、500および1000ピコモル)、NT(0.1、1、10および100ナノモル)または生理食塩水(n=3の各化合物の最高用量を除いて群当たりn=5)を受ける群に分けた。注射15分後
にて出発して、動物を加速回転棒上に置き、最初に落下までの潜在時間を記録した。動物は、30、60、120、240および300分(または、落下までの潜在時間が対照値にもどるまで)にて再試験した。また、直腸温を同一時点にてこれらの動物において記録した。
【0107】
実施例11
コンツラキン−Gの鎮痛活性
CGX−1160は、テイル・フリック潜在時間(図10A)を用量依存的に増大させ、ピーク効果の時間は≦30分間であった(試験された最も早い時間、図10B)。さらに、潜在時間の増加は持続的であり、注射5時間後にて引込め時間が上昇し、注射後24時間にてベースラインに戻った(図10B)。また、CGX−1063は、CGX−1160に対して、このモデルにおいて、引込めの潜在時間に変動しやすいが、用量依存的な増加を示し、次いで、最も適度な抗痛覚効力のみを示した。対照的に、NTは、テイル・フリックアッセイにおいて引込め潜在時間を有意には上昇させなかった(図10A〜10B)。
【0108】
試験された全化合物は、ホルマリン試験の両相に抗痛覚性特性を示したが、異なる効力であった。CGX−1160は、3つの化合物のうち最も効力があった。ホルマリン試験の第1相(図11A)において、CGX−1160は、約30〜40ピコモルのED50であったが、NTは約1ナノモルのED50を有した。CGX−1063は、第1相において、50%の抗痛覚閾値に達しなかった、しかしながら、この試験における不規則な用量応答は、このアッセイにおいて100ピコモル用量を繰り返しを保証した。ホルマリン試験の第2相において、3つの化合物の全ては肢なめ時間を用量依存的に低下させた(パーセント抗痛覚における増加のごとく図中に示す;図11B)。再度、CGX−1160は他の化合物より強力であり、1ピコモルのED50が見積られた。この化合物の低用量は、将来的に評価されて、より正確なED50を計算するのに必要な容量応答曲線を完成されるであろう。また、CGX−1063は、第2相における抗痛覚性行動を低下させるのに有効であり、ED50は10〜20ピコモルであると見積られた。NTは、いずれのコンツラキンより劇的に小さな効力であり、ED50は600〜700ピコモルと見積られた(図11B)。
【0109】
CGX−1160は、非常に強力でかつ用量依存的なCFA誘導の機械的異痛症の逆転を示した(図12A)。i.t.で与えられた100フェムトモルのCGX−1160は、CFA誘導の機械的異痛症を完全に逆転した。注目すべきことには、この用量にて、機械的異痛症に対する反対側の感受性は変化せず、慢性炎症のNT受容体における潜在的な片側変化を示した。高用量のCGX−1160において、CFA注射した肢および反対側の注射していない肢の双方における機械的な引込め閾値を劇的に上昇させた。図12Aおよび12Bにおいて、バー上の数字は、薬物前レベルに対する機械的閾値におけるパーセント増加を示す。示されたごとく、試験された全用量にて、CGX−1160は、注射されていない側に対し、CFA注射した側に対して非常に大きな抗異痛効果を有する。CGX−1063は、CGX−1160より小さな効力であったが、また、CFA誘導異痛症を完全に逆転させた(図12B)。最小の有効量は、10ピコモルであったが、しかしながら、CGX−1160とは異なり、この用量にて反対側は、プレ薬物ベースライン測定に対して上昇した。この試験において調べた他のモデルと一致して、NTは、1ナノモルにてCFAモデルにおいて有効性を示したが、100ピコモルでは有効性を示さなかった(図12C)。CGX−1160およびNTの他の用量は、将来的に調べて、これらの化合物についての正確なED50を決定するであろう。
【0110】
CGX−1160、−1063およびNTの全ては、運動障害および体温に対する用量依存的な効果を示した。3つの化合物の全てにつき、最大障害は、i.t.注射15分後(運動障害、図13A)、または30分後(低体温効果、図14A)であった。CGX−1063は、試験された低用量(1ピコモル、図13B)では運動毒性を持たないが、高用量にて動物はかなりの運動毒性を示した(推測されたTD50は10ピコモル、図13Bおよび15A)。10ピコモルにて、この毒性は30分間で終わり、60分までに消滅した。100ピコモルまたは1ナノモルが投与された場合に、動物は2〜3時間運動障害された(図14A)。CGX−1160は、運動障害を引き起こすににCGX−1063と同効力であった(推定されたTD50は10〜20ピコモルであった、図13B)。CGX−1063と同様に、高用量(100〜500×そのED50)にて、CGX−1063は、運動障害を示し、それは5時間後に消失した(図13Aおよび14B)。NT誘導運動障害についての推定されたTD50は、3ナノモルであった(図13B)。コンツラキンと同様に、高用量にて、NT誘導運動障害は、2〜4時間に終わった(図13Aおよび14C)。
【0111】
これらの化合物の低体温効果は、運動毒性に類似した。3つ全てが、体温の用量依存的低下を引き起こした。CGX−1160および−1063は同効力であり、TD50は100ピコモルと推定された(図15B)。しかしながら、この用量にて、CGX−1063は、体温低下を誘導し、2〜3時間で終わったが(図15Aおよび16A)、CGX−1160によって引き起こされた低体温効果は、60分間までに解消した(図15Aおよび16B)。最高用量のCGX−1160(500ピコモル、500×そのED50)にて、低体温効果は、注射後6時間まで解消しなかった(図16B)。NTは、コンツラキンに非常に類似する用量応答および経時変化を示した。低用量にて、NTは効果を有さず、短時間作用型の低体温効果を示した(図16C)。しかしながら、高用量(100ナノモル)にて、NTは劇的でかつ長時間作用型の低体温症を引き起こし、それは3時間までに解消した(15Aおよび16C)。
【0112】
本データは、CGX−1160およびCGX−1063は急性および慢性の痛みの種々の動物モデルにおいて有効な、効力があり、広スペクトルの鎮痛剤である。典型的には、CGX−1160は、CGX−1063より10倍の効力があり、NTより1000倍の効力がある(表6)。CGX−1160は、慢性炎症の痛みのモデルに特に効力があり、ここに、CGX−1160は、CFA注射を受けた肢にだけ機械的な引込め閾値を選択的に増加させるが、非注射の肢の閾値を変更しない。この発見は、慢性炎症が、炎症した肢に対応する抗痛覚性経路におけるNT受容体の再構成に導くことができることを示す。CGX−1160は、効力の増大を示すこれらの実験における唯一の化合物であったので、これはCGX−1160が選択性および特異性を有し得る受容体サブタイプの上方調節を示し得る。この化合物は、運動障害または低体温のいずれかより10〜100倍小さな用量にて抗痛覚を示し、一方、CGX−1063およびNTは、i.t.投与した場合のほぼ等用量にて、抗痛覚、運動障害および低体温症を引き起こすという発見は、CGX−1160サブタイプ選択性のこの仮説の支持内にある。100ピコモルにてi.t.で与えた場合(ホルマリン試験の第2相におけるそのED50の約10倍、図16A参照)には、CGX−1063は、CGX−1160(図16Bの10ピコモル用量を比較されたし)およびNT(図16Cの10ナノモル用量を比較されたし)の比較抗痛覚用量に対して長時間作用型の低体温を引き起こした。これは、CGX−1063がNTアナログの低体温効果に関連するNT受容体サブタイプに対して選択的であることを潜在的に示す。かくして、CGX−1160中のThr10のO−グリコシル化は、最近NTR2であると考えられている抗痛覚性NTRサブタイプに対する選択性ならびにペプチダーゼに対して代謝耐性を分け与えることができる。
【0113】
【表6】
【0114】
実施例12
コンツラキン−Gの抗精神病活性を評価するための材料および方法
1.材料。D−アンフェタミンは、Sigma(St. Louis、MO)から入手した。コンツラキン−G(CGX−1160;合成の16個のアミノ酸O−結合糖ペプチド)を上記のごとく合成した。
【0115】
2.動物。雄性CF−1マウス(30〜35グラム;Charles River Laboratories)を用いた。全動物は、12時間の明暗サイクルを持つ温度制御(23°±3℃)室にて飼育し、食餌および水を自由摂取させた。全動物は、実験用動物のヒューマンケアに対するPublic Health Service方針に従って安楽死させた。
【0116】
3.運動活性。動物は、透明のプラスチックケージ(40cm×22cm、深さ20cm)中に入れ、30分間馴化させた。次いで、動物は、10μl Hamiltonシリンジを通してフリーハンドで脳室内(i.c.v.)注射(5μl容積)によってコンツラキン−G(100ピコモル)または生理食塩水(ビヒクル)のいずれかを受けた。5分後に、動物は、腹腔内(i.p.)投与を介して生理食塩水または硫酸D−アンフェタミンを受けた。通過距離(cm)および移動に費やされた時間(秒)をVideomex-V 追跡システム(Columbus Instruments、Columbus、OH)を用いて30分間にわたりモニターした。全試験は、隔離された、薄暗い光の行動室において行った。
【0117】
4.統計量。データは、薬物を唯一の因子とする一元分散分析(ANOVA)を用いて分析し、続いて、個々の群の比較についてのNewman−Keuls多重比較検定を行い、統計学的有意差としてP<0.05を受入れた。統計学的解析は、GraphPad PRISMソフトウエア(バージョン2.01、GraphPad、San Diego、CA)で行った。
【0118】
実施例13
コンツラキン−Gの抗精神病活性
通過距離[F(4,21)=7.87、P<0.05]および移動に費やした時間[F(4,21)=6.17、P<0.05]の双方により測定された運動活性に対する薬物処置の有意な効果が、本試験において判明した。D−アンフェタミン投与の結果、通過距離および移動に費やした時間の双方が用量依存的に増加した(図17〜18)。コンツラキン−G(100ピコモルi.c.v.)でのマウスの前処置は、通過距離および移動に費やした時間におけるアンフェタミン刺激された(3mg/kg i.p.)増加をかなり低下させた。基礎的運動活性(通過距離および移動に費やした時間の双方)における低下は、コンツラキン−G(100ピコモル i.c.v.)での前処置後に見られたが、しかしながら、この低下は統計学的有意差には達しなかった。
【0119】
集中する系の証拠は、(Nemeroff et al.、1992に総説された)標準的な神経弛緩薬物が関連した副作用プロフィールなくして抗精神病活性を有し得ることを意味する。引き続いて、多くのグループが新規な抗精神病薬としてニューロテンシンアナログに集中した。コンツラキン−Gがニューロテンシンと相同性のC末端を共通し、in vivoおよびin vitroアッセイの双方にてニューロテンシンに似ているので、抗精神病効力の前臨床的スクリーニング推定D−アンフェタミンの刺激運動活性を阻害するためのコンツラキン−Gの能力を評価した。この例は、コンツラキン−Gでのマウスの前処置が、運動活性におけるアンフェタミン刺激の増加をかなり低下させることを示す。これらのデータは、コンツラキン−Gがニューロテンシンと同様の抗精神病活性を有することを示す。しかしながら、前記のごとく、ニューロテンシンは、ラットニューロテンシン受容体rNTR1(IC50:ニューロテンシンにつき3.2nM;コンツラキン−Gにつき524nM)およびrNTR2(IC50:ニューロテンシンにつき6.0nM)およびマウス・ニューロテンシン受容体mNTR3(IC50:ニューロテンシンにつき1.4nM;コンツラキン−Gにつき250nM)にてコンツラキン−Gよりはるかに大きな効力であるが、コンツラキン−Gはi.c.v.投与後のin vivoアッセイ(運動活性の視覚的に見積られた評価)にて1ないし2オーダー程度より効力があった。これらの結果は、コンツラキン−Gおよびニューロテンシンが、重なるが、区別される集団のニューロテンシン受容体サブタイプまたは活性化状態と相互作用できることを示す。かくして、コンツラキン−Gは、ニューロテンシンの制限する副作用を共有しなかったであろう。
【0120】
実施例14
コンツラキン−Gの抗痙攣活性を評価するための材料および方法
1.動物。雄性フリングス(Frings)(20〜25グラム)を12時間の明暗サイクルを持つ温度制御(23°±1℃)室にて飼育し、食餌および水を自由摂取させる。マウスは、HEW 刊行物(NIH)第8623号、「実験用動物の配慮および使用についてのガイド(Guide for the Care and Use of Laboratory Animals)」の推奨に一致する方法で飼育し、食餌させ、扱った。全マウスは、実験用動物のヒューマンケアに対するPublic Health Serviceの方針に従って安楽死させた。
【0121】
2.抗痙攣性の評価。フリングス・マウスを丸いプレキシガラスジャー(直径15cm、高さ18cm)に入れ、110デシベル(11KHz)の音響刺激に曝露した。次いで、マウスは、後肢強直性伸展の存在または不存在にて25秒間観察した。後肢強直性伸展を示さない動物は、保護されたと考えた。
【0122】
3.回転棒試験。運動障害は、6rpmにて回転する回転棒上にマウスを置くことによるピーク効果時間にて評価した。1分間に3度落下した動物を障害されたと考えた。
【0123】
実施例15
コンツラキン−Gの抗痙攣活性
コンツラキン−G(CGX−1160)およびThr10−コンツラキン−G(CGX−1063)は、i.c.v.投与後にフリングス・マウスにおける聴覚性発作を強力でかつ用量依存的にブロックした(図19)。痛みモデルにおける効力と同様に、CGX−1160はCGX−1063より効力があり、ED50は、各々、7.1ピコモルおよび27.0ピコモルであった(表7)。また、従前の試験に一致して、NTは、CGX−1160または−1063より劇的に小さな効力であった。NTの用量応答曲線が依然として完成していないので、NTは1ナノモルのi.c.v.投与後の50%保護を示した。運動毒性につき試験した場合、CGX−1160は、200ピコモルまでの用量にて50%毒性レベルに達しなかったが(図19)、一方、CGX−1063のTD50は、約375ピコモルであり、その結果、試験された用量につきPIが14と見積られた。
【0124】
別の実験において、CGX−1160のピーク効果までの時間および作用期間を調べた。100ピコモル(約14×ED50)のCGX−1160のi.c.v.投与は、i.c.v.注射30分後にて活性を示さず、60分後にて100%保護し、4時間にて依然として、試験された動物において50%保護を示した(図20)。
【0125】
本発明の方法および組成物が、種々の具体例の形式で組込むことができ、それらの少しだけが本明細書に開示されることは、認められるであろう。当業者ならば、他の具体例が本発明の範囲から逸脱しないことは明らかであろう。かくして、記載された具体例は例示的なものであり、限定として解釈すべきではない。
【0126】
参考文献リスト
Annals of Pharmacotherapy 27:912 (1993).
Araki, K.ら (1973). Chem. Pharm. Bull. (Tokyo) 21:2801-2804.
Barber, M.ら (1982). Anal. Chem. 54:645A-657A.
Benziger, J.U. (1994). Faseb. J. 8:1019-1025.
Cancer 41:1270 (1993).
Cancer Res. 44:1698 (1984).
Carraway, R.ら (1973). J. Biol. Chem. 248:6854-6861.
Cartier, G.E.ら (1996). J. Biol. Chem. 271:7522-7528.
Chabry, J.ら (1994). J. Neurochem. 63:19-27.
Chaplan, S.R.ら (1994). J. Neurosci. Methods 53:55-63.
Clineschmidt, B. V.ら (1979). Eur. J. Pharmacol. 54:129-139.
Colledge, C. J.ら (1992). Toxicon. 30:1111-1116.
Cotter, R. J. (1989). Biomed Mass Spectrom. 18:513-532.
Craig, A.G.ら (1993). Biol. Mass Spectrom. 22:31-44.
Craig, A.G.ら (1994). Biol Mass Spectrom. 23:519-528.
Craig, A.G.ら (1998). Biochemistry 37:16019-16025.
Cruz, L.J.ら (1987). Conus geographus toxins that discriminate between
neuronal and muscle sodium channels. J. Biol. Chem. 260:9280-9288.
Cusack, B.ら. (1993). J. Recept. Res. 13:123-134.
Cusack, B.ら (1991). Eur J. Pharmacol. 206:339-342.
Feurle, G.E.ら (1992). J. Biol. Chem. 267:22305-22309.
Fischer, W.ら (1987). Proc. Nat. Acad. Sci. USA 84:3628-3632.
Haack, J.A.ら (1990). Contryphan-T: a gamma-carboxyglutamate containing peptide with N-methyl-d-aspartate antagonist activity.
J. Biol. Chem. 265:6025-6029.
【0127】
Hammerland, I,.G.ら (1992). Eur. J. Pharmacol. 226:239-244.
Hillenkamp, F.ら (1993). Anal. Chem. 63:1193A-1203A.
Horiki, K.ら (1978). Chemistly Letters 165-68.
Hylden, J.L.K.ら(1980). Eur. J. Pharmacol.67:313-316.
Jimenez, E. C.ら (1996). J. Biol. Chem. 271:28002-28005.
Kaiserら (1970). Anal. Biochem. 34:595.
Kapoor(1970). J. Pharm. Sci. 59:1-27.
LeNguyen, D.およびRivier, J. (1986). Intl. J. Pep. Prot. 27:285-292.
Luning, B.ら(1989) . Glycoconjugate j. 6:5-19.
Malmberg, A.Bら (1998). Pain 76:215-222.
Mazella, J.ら (1988). J. Biol. Chem. 263:144-149.
Mcluckey, S.A.ら (1991). Anal. Chem. 63:375-383.
Mena, E.E.ら (1990). Contryphan-G: a novel peptide antagonist to the N-
methyl-l)-aspartic acid (NMDA) receptor. Neurosci. Lett. 118:241-244.
Methoden der 0rganischen Chemie (Houben-Weyl). Synthese von peptiden, E. Wunsch (編), Georg Thieme Verlag, Stuttgart, Ger. (1974).
Minamino, N.ら(1984). Biochem. Biophys. Res. Commun. 122: 542-549.
Monje, V. D.ら(1993). Neuropharmacology 32: 1141-1149.
Munson, P. J.ら (1980). Anal. Biochem. 107:220-239.
Nishiuchi, Y.ら (1993). Synthesis of gamma-carboxyglutamic acid-
containing peptides by the Boc strategy. Int. J. Pept. Protein
Res. 42:533-538.
Nemeroff, C. B.ら (1992). Ann. N. Y. Acad. Sci. 668:146-156.
Norberg, T.ら (1994). In: Methods in Enzymology, Y. C. Lee 編, Academic
Press, New York, NY, 87-107頁.
Olivera, B.M.ら (1984). Biochemistry 23:5087-5090.
【0128】
Olivera, B.M.ら (1985). Science 230:1338-1343.
Olivera, B.M.ら (1990). Science 249:257-263.
Olivera, B.M.ら (1997). Mol. Biol. Cell 8:2101-2109.
Remington's Pharmaceutical Sciences, 第18版, Mack Publishing Co.,
Easton, PA (1990).
Rivier, J.R.ら (1978). Biopolymers 17:1927-38.
Rivier, J.R.ら (1987). Biochem. 26:8508-8512.
Sadoul, J. L.ら (1984). Biochem Biophys. Res. Commun. 120:812-819.
Sambrook, J.ら (1989). Molecular Cloning: A Laboratory Manual, 第2版,
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
Schroderら (1965). The peptides 1:72-75, Academic Press, NY.
Spengler, B.ら (1992). Rapid Commun. Mass Spectrom. 6:105-108.
Stewart, J.M.ら (1984). In: Pierce Chemical Company, Rockford, IL.,
176頁.
Stewart, J.M.ら, Solid-Phase Peptide Synthesis, Freeman & Co.,
San Francisco, CA (1969).
Tanaka, K.ら (1990). Neuron 4, 847-854.
Toth, I.ら(1999). J. Med. Chem. (JMC ASAP web edition 22 Sept. 1999).
Van Renterghem, C.ら(1988). Biochem. Biophys. Res. Comm. 157:977-985.
Yoshida, H.ら (1976). Biochemistry 15:61-64.
Zhou L.M.ら (1996). Synthetic Analogues of Contryphan-G: NMDA
Antagonists Acting through a Novel Polyamine-Coupled Site.
J. Neuroehen1. 66:620-628.
【0129】
米国特許第3,972,859号.
米国特許第3,842,067号.
米国特許第3,862,925号.
米国特許第4,105,603号.
米国特許第4,352,883号.
米国特許第4,353,888号.
米国特許第4,447,356号.
米国特許第4,569,967号.
米国特許第4,883,666号.
米国特許第4,968,733号.
米国特許第4,976,859号.
米国特許第5,082,670号.
米国特許第5,084n350号.
米国特許第5,158,881号.
米国特許第5,284,761号.
米国特許第5,364,769号.
米国特許第5,514,774号.
米国特許第5,534,615号.
米国特許第5,545,723号.
米国特許第5,550,050号.
米国特許第5,591,821号.
米国特許第5,618,531号.
米国特許第5,633,347号.
【0130】
米国出願シリアル番号08/785,534.
米国出願シリアル番号09/061,026.
PCT公開出願WO 92/19195.
PCT公開出願WO 94/25503.
PCT公開出願WO 95/01203.
PCT公開出願WO 95/05452.
PCT公開出願WO 96/02286.
PCT公開出願WO 96/02646.
PCT公開出願WO 96/11698.
PCT公開出願WO 96/40871.
PCT公開出願WO 96/40959.
PCT公開出願WO 97/12635.
【特許請求の範囲】
【請求項1】
アミノ酸配列:
Xaa1-Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Thr-Lys-Lys-Xaa2-Tyr-Ile-Leu(配列番号:1)
[式中、Xaa1はピロ-Glu、Xaa2はプロリンまたはヒドロキシプロリンであって、Thr10はO−グリカンを含有するように修飾され、該O−グリカンは、Gal(β1→3)GalNAc(α1→)である]
を含む実質的に純粋なコンツラキン−G。
【請求項2】
Xaa2がプロリンである請求項1記載の実質的に純粋なコンツラキン−G。
【請求項3】
配列番号:6記載のアミノ酸配列を含むコンツラキン−G前駆体をコードする核酸を含むことを特徴とする単離核酸。
【請求項4】
配列番号:5記載のヌクレオチド配列を含むことを特徴とする請求項3記載の単離核酸。
【請求項5】
配列番号:6記載のアミノ酸配列を含むことを特徴とする単離コンツラキン−G前駆ペプチド。
【請求項6】
請求項1または2のコンツラキン−G、および医薬上許容される担体を含む、それを必要とする個人のニューロテンシン受容体に対するコンツラキン−G部位を調節するための医薬組成物。
【請求項7】
ニューロテンシン受容体の該調節が、発作、炎症、ショック、血栓症、高血圧症、痛み、精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、動脈硬化症、脈管拡張および血管拡張を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項8】
ニューロテンシン受容体の該調節が、癲癇、発作または痙攣を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項9】
ニューロテンシン受容体の該調節が、痛みを治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項10】
ニューロテンシン受容体の該調節が、精神分裂病を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項11】
ニューロテンシン受容体の該調節が、炎症を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項12】
ニューロテンシン受容体の該調節が、遅発性異常運動症または急性ジストニー反応(acute dystonic reaction)を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項1】
アミノ酸配列:
Xaa1-Ser-Glu-Glu-Gly-Gly-Ser-Asn-Ala-Thr-Lys-Lys-Xaa2-Tyr-Ile-Leu(配列番号:1)
[式中、Xaa1はピロ-Glu、Xaa2はプロリンまたはヒドロキシプロリンであって、Thr10はO−グリカンを含有するように修飾され、該O−グリカンは、Gal(β1→3)GalNAc(α1→)である]
を含む実質的に純粋なコンツラキン−G。
【請求項2】
Xaa2がプロリンである請求項1記載の実質的に純粋なコンツラキン−G。
【請求項3】
配列番号:6記載のアミノ酸配列を含むコンツラキン−G前駆体をコードする核酸を含むことを特徴とする単離核酸。
【請求項4】
配列番号:5記載のヌクレオチド配列を含むことを特徴とする請求項3記載の単離核酸。
【請求項5】
配列番号:6記載のアミノ酸配列を含むことを特徴とする単離コンツラキン−G前駆ペプチド。
【請求項6】
請求項1または2のコンツラキン−G、および医薬上許容される担体を含む、それを必要とする個人のニューロテンシン受容体に対するコンツラキン−G部位を調節するための医薬組成物。
【請求項7】
ニューロテンシン受容体の該調節が、発作、炎症、ショック、血栓症、高血圧症、痛み、精神病、パーキンソン病、胃腸障害、内因性鬱病、認識機能障害、不安、遅発性異常運動症、薬物依存症、パニック発作、躁病、過敏性腸症候群、下痢、潰瘍、胃腸腫瘍、トゥーレット症候群、ハンチントン舞踏病、血管性漏出、動脈硬化症、脈管拡張および血管拡張を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項8】
ニューロテンシン受容体の該調節が、癲癇、発作または痙攣を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項9】
ニューロテンシン受容体の該調節が、痛みを治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項10】
ニューロテンシン受容体の該調節が、精神分裂病を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項11】
ニューロテンシン受容体の該調節が、炎症を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【請求項12】
ニューロテンシン受容体の該調節が、遅発性異常運動症または急性ジストニー反応(acute dystonic reaction)を治療するのに用いられることを特徴とする請求項6記載の医薬組成物。
【図1】

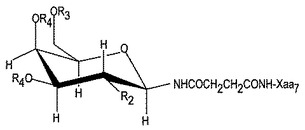
【図2】
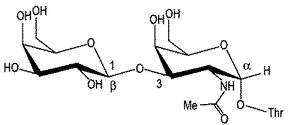
【図3】
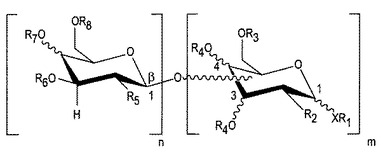
【図4】
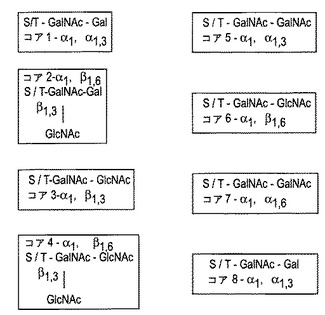
【図5】
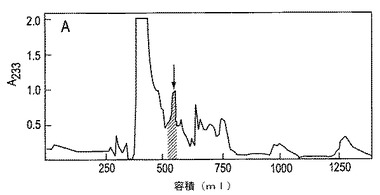
【図6】
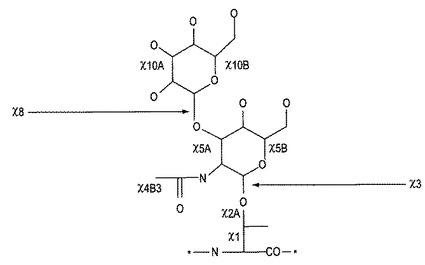
【図7A−7C】
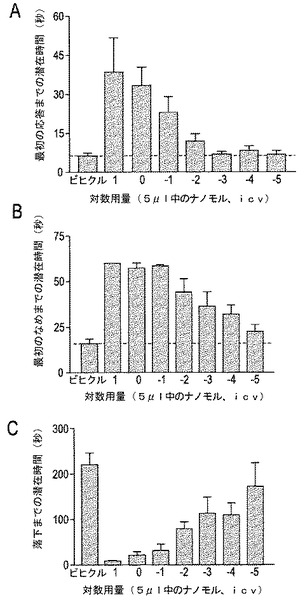
【図8A−8B】
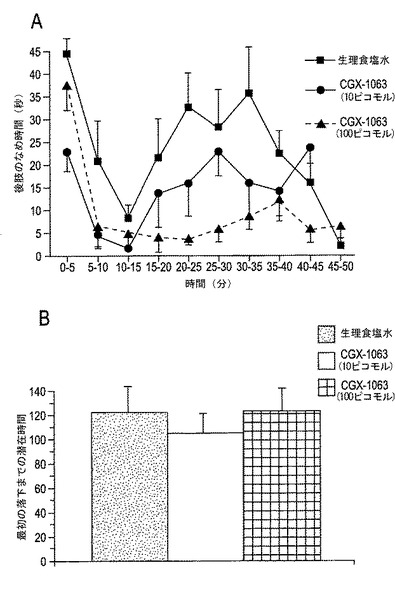
【図9】
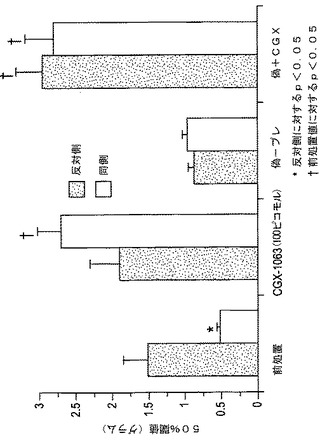
【図10A−10B】
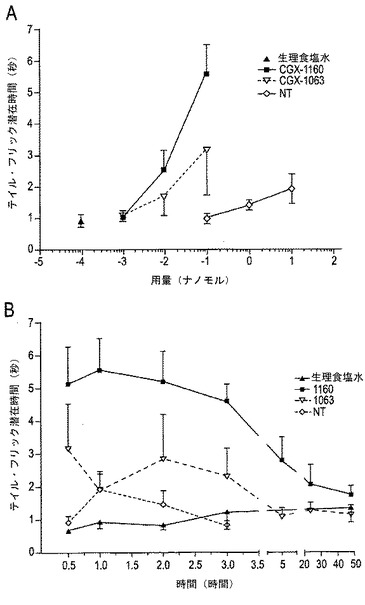
【図11A−11B】
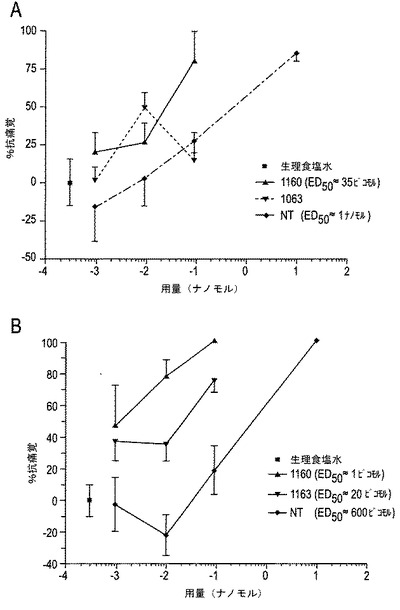
【図12A−12C】
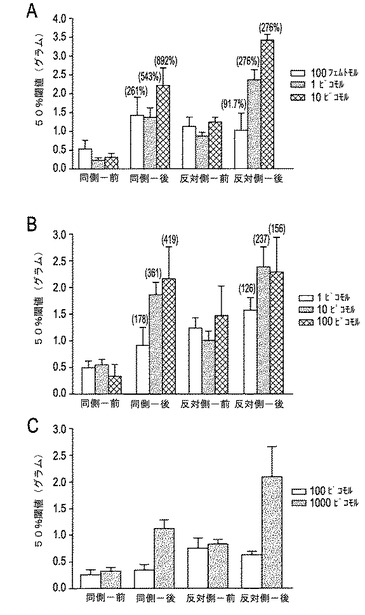
【図13A−13B】
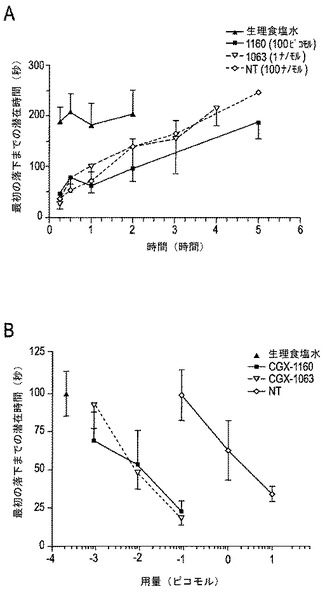
【図14A−14C】
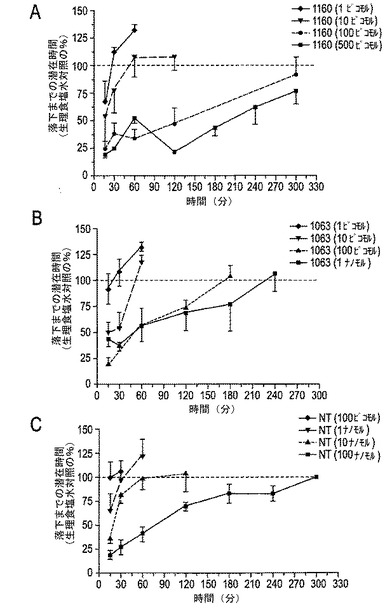
【図15A−15B】
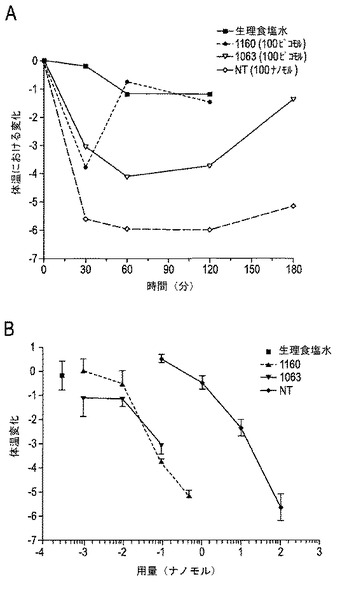
【図16A−16C】
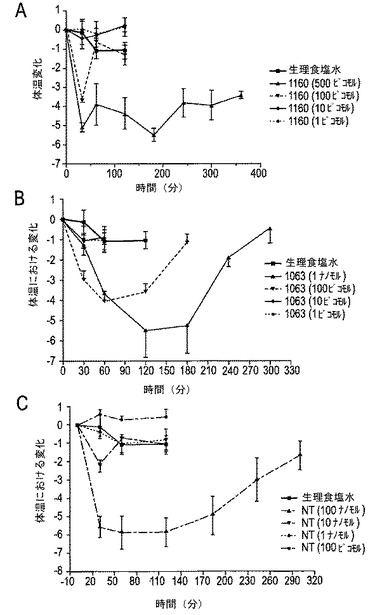
【図17】
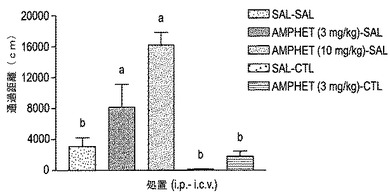
【図18】
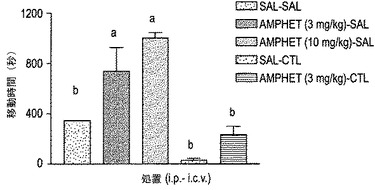
【図19】
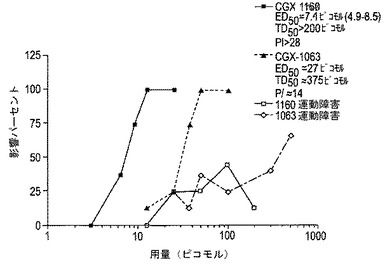
【図20】
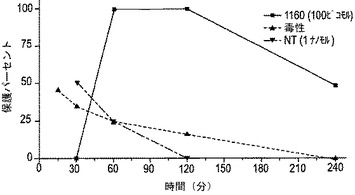
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A−7C】
【図8A−8B】
【図9】
【図10A−10B】
【図11A−11B】
【図12A−12C】
【図13A−13B】
【図14A−14C】
【図15A−15B】
【図16A−16C】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2011−24579(P2011−24579A)
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−172540(P2010−172540)
【出願日】平成22年7月30日(2010.7.30)
【分割の表示】特願2000−576865(P2000−576865)の分割
【原出願日】平成11年10月20日(1999.10.20)
【出願人】(500189481)コグネティックス・インコーポレイテッド (1)
【出願人】(500189230)ユニバーシティ・オブ・ユタ・リサーチ・ファウンデーション (11)
【出願人】(500189470)ソーク・インスティテュート (1)
【Fターム(参考)】
【公開日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願番号】特願2010−172540(P2010−172540)
【出願日】平成22年7月30日(2010.7.30)
【分割の表示】特願2000−576865(P2000−576865)の分割
【原出願日】平成11年10月20日(1999.10.20)
【出願人】(500189481)コグネティックス・インコーポレイテッド (1)
【出願人】(500189230)ユニバーシティ・オブ・ユタ・リサーチ・ファウンデーション (11)
【出願人】(500189470)ソーク・インスティテュート (1)
【Fターム(参考)】
[ Back to top ]