
ゴム変性硬質シリコーン樹脂及びその製造方法
【課題】破壊靱性が優れたシリコーン樹脂を提供すること。
【解決手段】(A)有機シリコーン樹脂、(II)式(I)の加水分解性前駆体及び(III)(II)から形成される水解物、からなる群から選ばれる有機ケイ素組成物と(B)シリコーンゴムとの共重合反応生成物を含むゴム変性硬質シリコーン樹脂であって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴムが存在するゴム変性硬質シリコーン樹脂。
【解決手段】(A)有機シリコーン樹脂、(II)式(I)の加水分解性前駆体及び(III)(II)から形成される水解物、からなる群から選ばれる有機ケイ素組成物と(B)シリコーンゴムとの共重合反応生成物を含むゴム変性硬質シリコーン樹脂であって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴムが存在するゴム変性硬質シリコーン樹脂。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、その構造中に導入されたシリコーンゴムを含むシリコーン樹脂を提供する。このゴム変性シリコーン樹脂及びそれから製造される強化複合材料は破壊靱性に著しい改良を示す。
【背景技術】
【0002】
有機ポリマー樹脂にゴムを添加することによりそれらの靱性を向上させることができる。そのような系は、Effects of Rubber Additions On The Fracture Toughness Of A Polyester Resin (Tetlow, P. D. et al. Proceedings of the Annual Technical Conference, 1979, Reinforced Plastics/Composites Institute The Society of the Plastics Industry, Inc. Vol. 34, 23F)及び1968年4月にサンフランシスコで発表されたCrack Toughened Polyester Resin Formulations (McGarry, F. J. et al., American Chemical Society Division of Organic Coating and Plastics Chemistry Vol. 28, No. 1, pp 526-36)に記載されている。
【0003】
種々のシリコーン組成物の靱性を向上させることに関する技術の現状は米国特許第5,034,061号明細書に開示されており、この特許明細書には透明な耐破砕性コーティングを形成するように改良されたシリコーン樹脂/流体ポリマーが教示されている。この組成物は不飽和オレフィン系官能基Rを有するR3 SiO1/2 単位及びSiO4/2 単位から本質的になるシリコーン樹脂コポリマー、ビニル官能基を有するポリジオルガノシロキサン流体、水素官能基を有するオルガノポリシロキサン架橋剤及び触媒を含む。前記組成物は白熱ガラスランプをコーティングするのに特に適するものとして開示されている。
【0004】
カナダ特許第691,206号明細書には、防振用のシリカ充填剤入りシリコーン樹脂/流体混合物の使用が開示されている。開示されているシリコーン樹脂/流体組成物の防振能は損失剪断弾性率G''に対する弾性剪断弾性率G’の比の測定値を通じて説明されている。この比の大きさは、その材料の振動吸収能に反比例するものであると示されている。その材料のG’/G''比は樹脂成分を用いずに調製された組成物と比較されている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記強化シリコーン組成物は弾性率が低い。これまで、硬質シリコーン樹脂の破壊靱性をうまく向上させることは達成されていない。シリコーン樹脂を記述する際に本明細書において使用する「硬質」なる用語は、樹脂材料が、その無充填状態でヤング率が少なくとも6.9×108 Paである特定の「剛性」を示すことを意味する。本明細書において「無充填」なる用語は、カーボン、ガラス繊維又はシリカのような強化用充填剤が樹脂に添加されていないことを意味する。
【0006】
硬質シリコーン樹脂は、それらの耐熱性及び防火性の利点を利用する用途に使用される。これらの特性は、シリコーン樹脂を、電気用積層板用の繊維強化複合材料、自動車部品、航空機及び船の構造用途に有用なものにしている。従来の未変性硬質シリコーン樹脂は非常に脆く、それらの用途はかなり限定されている。従って、脆性破壊が回避されねばならない用途での独特の防火性、電気的特性及び耐熱性の利用を可能にする本質的に向上した破壊靱性を有する硬質シリコーン樹脂が要求されている。
【課題を解決するための手段】
【0007】
我々は破壊靱性が改良されたゴム変性硬質シリコーン樹脂を発見した。我々のゴム変性硬質シリコーン樹脂は、
(A)(I)実験式:
【0008】
【化1】

【0009】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)実験式:
【0010】
【化2】
【0011】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は水素原子、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとから形成されるコポリマーを含むものであって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴム(B)が存在するものである。
【図面の簡単な説明】
【0012】
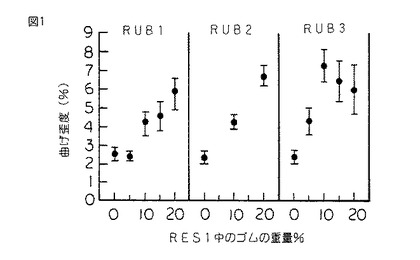
【図1】図1は、3つの異なる重合度を有する3種の異なるシリコーンゴム(RUB1、RUB2及びRUB3)により変性されたRES1、すなわちメチルフェニルシルセスキオキサン樹脂に関するシリコーンゴムの重量百分率の関数として表された破壊点曲げ歪度(%)(ASTM D 790に従う三点曲げから得られた)のグラフである。前記変性は縮合反応を用いて達成された。
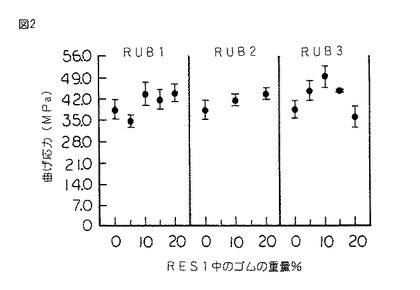
【図2】図2は、図1のシリコーンゴム変性樹脂のシリコーンゴムの重量百分率の関数として表された破壊点曲げ応力のグラフである。
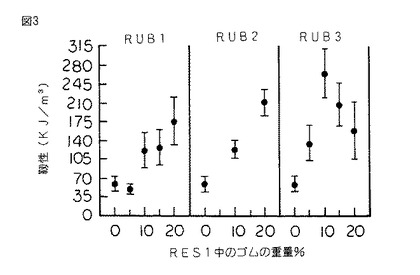
【図3】図3は、図1のシリコーンゴム変性樹脂のゴムの重量百分率の関数として表された靱性のグラフであって、応力−歪み曲線下の面積により測定されたものである。
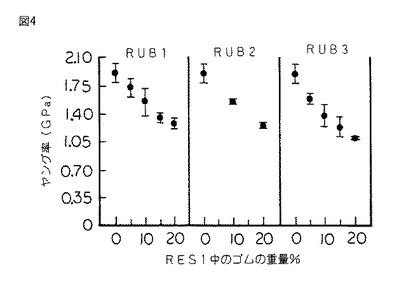
【図4】図4は、図1のシリコーンゴム変性樹脂のシリコーンゴムの百分率の関数として表されたヤング率のグラフである。
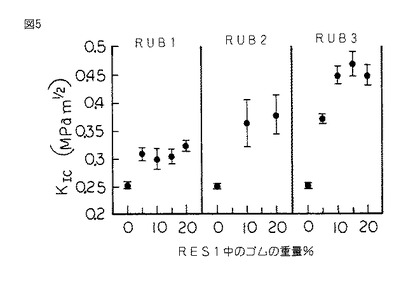
【図5】図5は、図1のシリコーンゴム変性樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された破壊靱性KIcのグラフである。
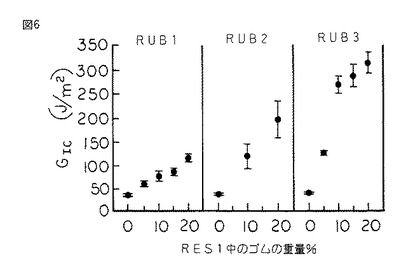
【図6】図6は、図1のシリコーンゴム変性樹脂の、ASTM D 5045 に従って決定され、シリコーンゴムの重量百分率の関数として表された臨界歪エネルギー放出率GIcのグラフである。
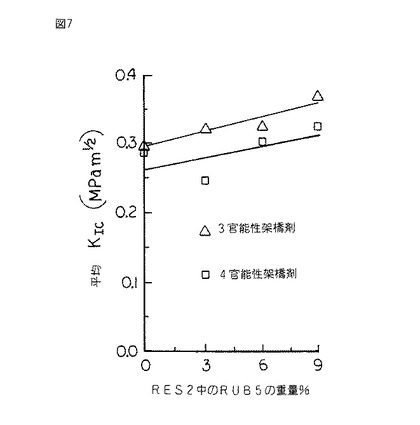
【図7】図7は、RUB5、すなわちジメチルビニルシロキシ末端ポリジメチルシロキサンにより変性されたRES2、すなわちビニル官能シルセスキオキサン樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された破壊靱性KIcのグラフである。前記変性は架橋剤及び付加硬化を用いて達成された。
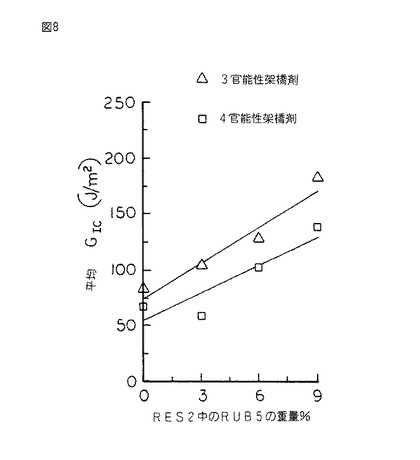
【図8】図8は、図7のシリコーンゴム変性樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された臨界歪エネルギー放出率GIcのグラフである。
【発明を実施するための形態】
【0013】
本発明の新規ゴム変性硬質シリコーン樹脂は共重合により製造される。この共重合は、縮合反応、付加反応又は遊離基重合を通じて実施される。
【0014】
共重合が縮合反応を通じて実施される場合、樹脂の最終硬化は加熱による共重合後に達成される。これにより共重合分子間でのさらなる縮合が起こる。
【0015】
従って、本発明は、
(1)次の成分:
(A)(I)実験式:
【0016】
【化3】
【0017】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂であって、その加水分解性前駆体(II)の加水分解及び縮合により形成される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)実験式:
【0018】
【化4】
【0019】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は、ヒドロキシル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルカルボキシル基及びアリールカルボキシル基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとを有機溶剤中に溶解させる工程;
(2)縮合触媒を加える工程;
(3)溶液からの共重合生成物の沈殿又はそれらのゲル化を引き起こすことなく成分(B)の全てを成分(A)と共重合させる工程;
(4)共重合した溶液から揮発分を除去する工程;及び
(5)硬化を引き起こすのに十分な温度に揮発分が除去された前記共重合した溶液を加熱する工程;
を含むゴム変性硬質シリコーン樹脂の製造方法を含む。
【0020】
従って、本発明の目的は、無充填状態で少なくとも6.9×108 Paのヤング率を有するゴム変性シリコーン樹脂を提供することである。
【0021】
本発明の別の目的は、未変性及び無充填の状態にある前記シリコーン樹脂と比較して、無充填状態で、Kicにより評価される増加した破壊靱性、Gicにより評価される増加した臨界歪エネルギー放出率、及び積分応力−歪靱性を有する樹脂を提供することである。
【0022】
本発明の他の目的は、上記基準を満たすそのような樹脂の製造方法を提供することである。
【0023】
本発明の特徴は、ゴム変性シリコーン樹脂のKIc及びGIcの値が、未変性及び無充填状態にあるシリコーン樹脂と比べて25%以上も増加することである。
【0024】
本発明のゴム変性硬質シリコーン樹脂は、(A)(I)有機シリコーン樹脂、(II)(I)の加水分解性前駆体及び(III)(II)から形成される加水分解生成物からなる群から選ばれる有機ケイ素組成物と(B)シリコーンゴムとから形成されるコポリマーを含む。
【0025】
有機シリコーン樹脂(I)は実験式:
【0026】
【化5】
【0027】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される。
【0028】
組成物(A)の有機シリコーン樹脂(I)はシルセスキオキサンであり、(I)の上記R1 、R2 及びR3 基をその構造の一部として含む加水分解性前駆体(II)の加水分解及び縮合により周知の方法で調製される。そのような加水分解性前駆体には、望ましい3次元樹脂構造を生じるオルガノトリアルコキシシラン及びオルガノトリハロシランのような三官能性シラン;又は末端キャップ剤として作用するトリオルガノモノアルコキシシラン、トリオルガノモノハロシラン、ジシロキサン及びジシラザンのような一官能性シランが含まれる。当業者は、ジオルガノジハロシラン及びジオルガノジアルコキシシランのような二官能性シラン、並びに少量のテトラハロシラン及びテトラアルコキシシランのような四官能性シランも樹脂前駆体として有用であることも認識するであろう。
【0029】
本発明の好ましい態様において、R1 、R2 及びR3 基の大部分は無官能性である。従って、これらの基は特許請求の範囲に記載のゴム変性硬質シリコーン樹脂を生成する共重合反応に関与せず、アルキル基、アリール基又はこれらの組み合わせである。最も好ましくは、これらの基はメチル、フェニル又はこれらの組み合わせである。
【0030】
本発明の第2成分は実験式:
【0031】
【化6】
【0032】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は、水素、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴム(B)である。
【0033】
R4 基がアルキル基及びアリール基のみを表すことは重要である。従って、R4 基は共重合反応に関与しない。しかしながら、各R5 は本発明のコポリマーを形成する共重合反応に関与する官能基である。
【0034】
(B)に関する式において、xはシリコーンゴムの平均非官能線状鎖長、すなわちR5 基間の平均鎖長を表す。従って、種々の重合度を有するシリコーンゴム(B)の混合物も上記実験式により表すことができる。本発明に使用されるシリコーンゴムの殆どは鎖の末端基にのみR5 基を有する。そのような場合、用語「重合度」(「DP」)はxの値である。しかしながら、DPなる用語は末端官能シロキシ基を包含しない。
【0035】
好ましい態様において、R4 基はメチル基、フェニル基又はこれらの組み合わせである。
【0036】
我々は、有機ケイ素組成物(A)のR1 、R2 及びR3 基の百分率が高く、シリコーンゴム(B)のR4 基が主としてメチルであるか又は主としてフェニルである場合に、有機ケイ素組成物(A)及びシリコーンゴム(B)は相溶性であり、前記ゴムは樹脂構造中に均質に分散することができることを見出した。
【0037】
我々は、シリコーンゴム(B)のDPが比較的大きく、例えば55〜200の間になる場合には、ゴムは樹脂から分離する傾向になり、明確に識別される二相系の形成が起こることも見出した。本発明の組成物を配合する際に、有機ケイ素組成物(A)及びゴム(B)は比較的低い固形分濃度(すなわち、極めて薄い濃度から30重量%)で有機溶剤中に溶解される。高濃度であるほど、樹脂(I)の形態にある有機ケイ素組成物(A)とゴム(B)との均質溶液を形成することは非常に困難である。
【0038】
2相系への分離は、硬化したゴム変性樹脂マトリックスにおいて時々観察されたが、そのような分離はそれが形成される有機溶液中では示されない。ある場合において、そのような硬化後分離は本発明のゴム変性樹脂の機械的特性を向上させる。さらに、1,000程度の高いDPを有するシリコーンゴム(B)を使用することもできる。
【0039】
本発明において、有機ケイ素組成物(A)及びシリコーンゴム(B)は、硬化したゴム変性硬質シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で存在する。我々は意外にも本発明のゴム変性シリコーン樹脂のヤング率はゴム含有率の増加に伴って減少することを見出した。従って、有機ケイ素組成物(A)及びゴム(B)の相対量は、硬化した樹脂の剛性が構造用用途に満足に使用できない程度に低下しないように選択される。シリコーンゴムの種類及び架橋剤の使用も本発明の変性樹脂のヤング率に影響を及ぼす。しかしながら、殆どの場合に、変性樹脂の30重量%を超える量のシリコーンゴムの添加は、上記のヤング率を下回るヤング率を有する組成物をもたらす。同様に、向上した靱性の有利な効果は、少なくとも2重量%のシリコーンゴム(B)が使用されない限り実現されない。
【0040】
一態様において、有機ケイ素組成物(A)は、シリコーンゴム(B)と共重合される前に、まず望ましい分子量を有するものに形成される有機シリコーン樹脂(I)である。まず有機溶剤中で樹脂(I)を、望ましい量の架橋剤及び触媒量の触媒と共に、シリコーンゴム(B)と配合することが望ましい。この組成物は、揮発分を除去され、金型に流し込まれ、その後に熱硬化される。ある場合に、揮発分の除去は金型中で実施される。揮発分の除去が高温で実施される場合には、硬化触媒の添加は冷却後に通常実施される。
【0041】
他の態様において、組成物は揮発分の除去工程後に射出成形されてよい。
【0042】
有機ケイ素組成物(A)とゴム(B)の間の縮合反応を利用する本発明の組成物において、最終硬化を実施する前に、まず有機ケイ素組成物(A)分子及びゴム(B)分子をもう一方と選択的に共重合させることができる。
【0043】
例えば、樹脂(I)の形態にある有機ケイ素組成物(A)及びシリコーンゴム(B)は、必要であれば望ましい量の架橋剤及び縮合触媒と共にトルエンのような有機溶剤中に溶解される。チタンテトラブトキシドのような比較的弱い縮合触媒を使用することが好ましい。90℃の温度でこの比較的弱い触媒は、有機ケイ素組成物(A)又はゴム(B)間のホモ重合を引き起こすことなく有機ケイ素組成物(A)、ゴム(B)及び必要であれば架橋剤の間の共重合を引き起こす。この反応は、ゴム(B)分子の全てが有機ケイ素組成物(A)分子と共重合する程度まで実施されることが好ましい。この結果は29Si核磁気共鳴分光分析(NMR)を使用して確認した。
【0044】
ジルコニウム及びハフニウムのアルコキシドも比較的弱い縮合触媒として同様に作用する。
【0045】
上記条件下ではホモ重合が起こりうる。しかしながら、形成されるコポリマーが有機溶剤に可溶のままであり、かつ、有機溶剤中で安定であるために実質的なホモ重合は起こらない。この「軽度」の共重合は軽度の増粘に類似する。軽度の共重合は触媒の濃度により調節され、また、その触媒強度及び軽度の共重合が実施される温度に関係する。
【0046】
「軽度」に共重合した状態にあるコポリマーは溶液中に留まる。その後、Dow Corning (商標)触媒15又はY-177 、コリンオクトエート及びオクタン酸亜鉛触媒(これらは両方ともミシガン州ミッドランド所在のDow Corning Corporation から市販入手可能)のような強い縮合触媒が「軽度に共重合した」溶液に添加される。この溶液は次にキャストされ、揮発分を除去され、熱が加えられることにより最終硬化して硬質状態になる。往々にして、最終硬化は圧力を加えることにより促進される。縮合硬化系において、最終硬化は樹脂(I)に結合している残留シラノール基の縮合を通じて達成される。
【0047】
シリコーンゴム(B)が加水分解性又は縮合性のR5 基を含む場合、ゴム(B)は有機シリコーン樹脂(I)の加水分解性前駆体(II)又は(II)の部分水解物(III)と配合され、次に軽度の共重合工程を達成するため及び樹脂の網状分子の成長を引き起こすために加水分解/縮合反応が実施される。軽度の共重合工程は、組成物がキャスティングに望ましい粘度に達するまで上記のような比較的弱い触媒により触媒される。その後、組成物は、揮発分が除去され、適切な強い縮合触媒が添加される。材料は次にキャストされ、最終的に熱硬化される。
【0048】
本発明のゴム変性シリコーン樹脂を得るために使用される方法に関わらず、硬化した組成物は望ましい剛性及び向上した破壊靱性を示す。
【0049】
本発明のコポリマーは架橋剤の作用を用いて又は用いずに形成され、縮合硬化組成物の場合には触媒の作用を用いて又は用いずに形成される。
【0050】
本発明のコポリマーを得るために使用される硬化機構の種類には、一般的に縮合硬化系、付加硬化系、及び遊離基重合系と呼ばれているものが含まれる。
【0051】
ビニル基のようなアルケニル基を含む系において、シリコーン樹脂(I)及びシリコーンゴム(B)に反応性官能基を付与するために、遊離基重合は実施可能な硬化系である。過酸化物のような遊離基開始剤が通常使用される。汎用されている開始剤としては過酸化アリールがある。よく知られているビニルに特異的な過酸化物開始剤は(CH3 )3 COOC(CH3 )3 である。
【0052】
縮合硬化系において、錫、チタン及び亜鉛の金属エステル又はアルコキシドが好ましい触媒である。チタンテトラブトキシド、ジブチル錫ジラウレート及びオクタン酸亜鉛がよく知られている例である。しかしながら、錫触媒は硬化した本発明のゴム変性硬質樹脂の熱安定性を低下させるものであって、高温用途に対しては概してその使用は避けられる。好ましさはさほどでもないが使用可能なものは、硫酸、リン酸、水酸化カリウム、水酸化セシウム、金属シラノラート及び焼石灰のような強酸及び強塩基である。しかしながら、これらの後者の触媒は、硬化した硬質シリコーン注型品の熱安定性を損なう場合もある。アミン又は金属カルボキシレート及び第4級塩基も有用である。水酸化テトラメチルアンモニウムのような特定の第4級塩基は、硬化温度に加熱された場合に揮発性副生成物を分解するために都合良い。従って、それらは本発明のシリコーン樹脂/ゴムコポリマーマトリックスから容易に除去される。
【0053】
揮発性副生成物が硬化機構により形成される場合には注意が払われねばならない。そのような揮発性副生成物は、注型品の欠陥として作用する気泡の形成により靱性を著しく低下させる原因となりうる。
【0054】
付加硬化系に対し、クロロ白金酸のような白金族触媒が往々にして使用される。実際に、適切な白金触媒には白金化合物又は白金錯体が含まれる。クロロ白金酸に加え、クロロ白金酸六水和物、カルステット触媒(すなわち、クロロ白金酸とsym −ジビニルテトラメチルジシロキサンの錯体)、ジクロロビス(トリフェニルホスフィン)白金(II)、塩化白金及び酸化白金が使用される。
【0055】
縮合硬化系において、シリコーン樹脂(I)とシリコーンゴム(B)の間の共重合及び共重合した系の最終硬化は、触媒の不在下で引き起こされる。しかしながら、そのような反応は高温に加熱することを必要とし、この理由から好ましくない。また、上記の軽度の共重合は触媒の助けをかりずに達成されない。
【0056】
本発明を実施するのに適する架橋剤にはシリコーンゴムの製造及び硬化において周知のものが含まれる。これらの架橋剤は、それらの化学式が少なくとも1個のケイ素原子を含むという意味においてシリコーンをベースとするものである。テトラエトキシシラン、メチルトリメトキシシラン、並びにメチルトリアセトキシ−、メチルトリオキシモ−及びテトラオキシモシランが周知の例である。架橋剤は反応性官能基間に比較的短いシロギン鎖を含む。本明細書で使用する場合に「架橋剤」なる用語は、シラン、及びシロキシ鎖のDPが6未満であるようなシロキサンに限定される。
【0057】
有機シリコーン樹脂を形成するために酸加水分解及び縮合を使用することによって、有意の濃度の立体障害を受けていない残留ヒドロキシル基を有する樹脂組成物が得られる。従って、この樹脂の残留ヒドロキシ官能基を使用する縮合反応は、縮合硬化系を通じて本発明のコポリマーを形成する都合のよい方法である。
【0058】
本発明の耐破壊性シリコーン樹脂組成物は、構造用部材又は耐力部材の製造に非常に有用であると期待される。さらに、本発明の組成物は繊維強化ゴム変性硬質樹脂のような複合材料における連続相として非常に有利に使用される。
【0059】
本発明の組成物は他の多くの用途に適用されてもよい。例えば、有機溶剤中に軽度に共重合した有機ケイ素組成物(A)及びゴム(B)を含む溶液は、基材上で硬化性コーティング組成物を形成し、この硬化性コーティング組成物は接着剤として作用する。同様に、本発明の組成物を硬化させてフィルム又はシート製品の形態にすることもできる。
【実施例】
【0060】
実施例−強化用第2相を含まない樹脂
縮合硬化系
最高20重量%までの種々の重量%で4つの異なる種類のシリコーンゴム(本明細書においてRUB1、RUB2、RUB3及びRUB4と示す)により第1樹脂(本明細書においてRES1と示す)を変性させた。以下で述べるようにトルエン中で種々の組成物を調製し、Teflon(商標)によりライニングされた四角形のアルミニウム金型に流し込み、110℃の減圧下で揮発分を除去し、次のスケジュールで硬化させた:95℃で48時間;110℃で24時間;120℃で24時間;130℃で24時間;140℃で24時間;150℃で12時間;175℃で6時間;200℃で12時間;230℃で3時間;及び260℃で5時間。この硬化スケジュールによって、亀裂及び気泡のような欠陥が存在しない硬化したスラブが確実に得られた。
【0061】
硬化したスラブから採取した、試験片をASTM D 790に従って破断するまで三点曲げ試験にかけ、そして曲げ強さ、歪度、ヤング率及び積分応力−歪靱性をシリコーンゴムの重量%の関数としてプロットした。(図1〜4を参照されたい。)
【0062】
硬化したスラブから採取される試験片を同様に作製し、片へりノッチ三点曲げモードでASTM D 5045 に従って試験し、破壊靱性Kic及び臨界歪エネルギー放出率GIcをシリコーンゴムの重量%の関数として決定した。(図5及び6を参照されたい。)
【0063】
RES1は実験式:
【0064】
【化7】
【0065】
(式中、Me及びPhはそれぞれメチル基及びフェニル基を表す。以下同様とする。)
により表される数平均分子量が1,300以下のシルセスキオキサン樹脂(ミシガン州ミッドランド所在のDow Corning Corporation から市販入手可能なDow Corning (商標)4-3136)であった。この樹脂は固形フレークの形態で入手可能なものである。
【0066】
前述のように、酸加水分解/縮合により形成されるシルセスキオキサン樹脂は概して有意の量の立体障害を受けていないヒドロキシル基を含む。上記実験式はそのような残留ヒドロキシル基(往々にして残留シラノール基と呼ばれる)を考慮していない。これらの基は本発明における共重合に関与する反応性基であるため、残留ヒドロキシル基を考慮して上記式を次式:
【0067】
【化8】
【0068】
のような適切な実験式に書き直すことができる。
【0069】
種々のゴムRUB1、RUB2、RUB3及びRUB4の各々を同様に作製した。酢酸カリウム触媒の存在下、130〜150℃の間の温度で揮発分を絶えず除去しながら、所定の望ましい平均重合度(DP)を有するジメチルシラノール末端ポリジオルガノシロキサンを化学量論的に過剰のテトラエチルオルトシリケートと反応させた。29Si核磁気共鳴分光分析(NMR)が末端ヒドロキシル基のトリエトキシ転化が完了することを示すまで加熱を続けた。未反応テトラエチルオルトシリケートはストリッピング及び加熱により除去した。
RUB1、RUB2、RUB3及びRUB4は、DPがそれぞれ6、14、55及び376であるものであった。
【0070】
十分な量のRES1及び所定量のシリコーンゴム(B)を望ましい比でトルエンに溶解させることによりRES1の変性を実施した。RUB1及びRUB2の場合に固形分50重量%の溶液を使用した。高DPゴムと樹脂の間の限られた相溶性のために、RES1とRUB3の場合に38重量%の最大固形分を使用した。また、同じ理由から、RES1とRUB4の場合に20重量%の最大固形分を使用した。その後、チタンテトラブトキシド触媒の形態にある比較的弱い縮合触媒を0.1〜1.0重量%の間の濃度で加えた。その後、反応混合物の温度は3時間で90℃に上昇し、続いて加熱することにより25時間還流させ、RES1及び種々のゴム(B)を軽度に共重合させた。次に、溶液をストリップして固形分50〜65重量%とし、室温に冷却し、そして次に0.20重量%の比較的強い縮合触媒(Dow Corning (商標)触媒15又はY-177 )を加えた。次にこの溶液から揮発分を除去し、上記のように注型してスラブとし、硬化させた。
【0071】
最後に、RES1をRUB3及びRUB4の組み合わせと共重合させた。RUB3及びRUB4の両方を各々5重量%の濃度で使用し、残りはRES1であった。この組成物は、RES1と10重量%のRUB3及びRES1と10重量%のRUB4の独立に調製された軽度に共重合した等しい部(固形分)の組み合わせをトルエン中で配合することにより調製した。その後、この溶液から揮発分を除去し、上記のように注型してスラブとし、硬化させた。
【0072】
付加硬化系
平均DPが9であるジメチルビニルシロキシ末端ポリジメチルシロキサン(RUB5)により最大15重量%までの種々の重量%で第2樹脂RES2、すなわちビニル官能シルセスキオキサン樹脂を変性させた。
【0073】
RUB5はペンシルヴェニア州チュリータウン所在のGelest, Inc.製の市販入手可能な製品であり、商標DMS-V05 のもとに市販されている。
【0074】
RES2は、フェニルトリクロロシランの酸触媒(HCl)される加水分解/縮合から周知の方法で調製した。次にこの反応生成物から酸を洗い落とし、水酸化カリウムの存在下で更に増粘させた。重合は、ジメチルビニルクロロシランのトルエン溶液の添加を通じて末端キャッピングすることにより停止させた。水酸化カリウムを中和し、溶液から濾過して取り除き、樹脂を固形分75重量%に濃縮した。RES2分子は1350〜1415のMnを有していた。
【0075】
RES2の実験式は次式:
【0076】
【化9】
【0077】
又は次式:
【0078】
【化10】
【0079】
のように表される。ここで、Viはビニル基を表し、以下同様とする。
【0080】
2つの異なる方法を使用してRES2のゴム変性を実施した。両方の方法において、化学量論的に過剰のSiH官能架橋剤を使用した。第1の方法において、架橋剤は三官能性のフェニルトリス(ジメチル水素シロキシ)シラン(United Chemical Technologies, Inc.から市販入手可能)であった。第2の方法において、架橋剤は四官能性のテトラキス(水素ジメチルシロキシ)シランであった。
【0081】
望ましい割合でRUB4をRES2のトルエン溶液に溶解させることによりRES2のトルエン溶液中で種々の組成物を調製した。その後、化学量論的に過剰の架橋剤をそれぞれ加えた。得られた溶液を50℃で1.5時間減圧脱気した。溶液を室温に冷却し、これに触媒量(1〜100重量ppm)のヘキサクロロ白金酸を加えた。Teflon(商標)によりライニングされた四角形のアルミニウム金型に得られた組成物を流し込み、140〜160℃で16時間硬化させ、金型から取り出し、200〜260℃で4〜8時間を要して後硬化させた。三官能性架橋剤を使用して組成物を硬化させる間に亀裂は観察されなかった。四官能性架橋剤を使用して組成物を硬化させる間に幾つかの亀裂が観察された。しかしながら、より低い硬化温度を用いることはそのような亀裂を減少させることに役立つことが見出された。
【0082】
硬化したスラブから採取される試験片を同様に作製し、片へりノッチ三点曲げモードでASTM D 5045 に従って試験し、破壊靱性KIc及び臨界歪エネルギー放出率GIcをシリコーンゴムの重量%の関数として決定した。(図7及び8を参照されたい。)
【0083】
縮合硬化系
上記試験から得られた結果は図1〜8にグラフにより示されている。RUB5によるRES1の変性は、結果が示されていない唯一の例である。図1〜6において、中心点は試験パラメーターの算術平均値を表し、縦棒はデーターの拡がりを表す。およそ5〜6つの試験片を各ゴム荷重度で使用した。
【0084】
図1を参照すると、RUB1及びRUB2(それぞれDP=6及び14)におけるようにシリコーンゴムが比較的低いDPを有する場合には、シリコーンゴムの添加量が多いほど破壊点歪度が増加することがわかる。しかしながら、RUB3(DP=55)におけるようにシリコーンゴムのDPが幾分高いと、破壊点曲げ歪度はシリコーンゴム10重量%でピークに達する。
【0085】
図2を参照すると、シリコーンゴム含有率の関数として表されている破壊点曲げ応力は、前記歪度の場合に類似する挙動を示す。同様に図3も、積分応力−歪により測定され、ゴム含有率の関数として表された靱性も同じ傾向にあることを示している。
【0086】
図4は、本発明のコポリマーに使用されるゴムのDPに関わらず全ての場合に、コポリマーのゴム含有率が増加すると組成物のヤング率又は「剛性」が低下することを示している。本発明は、構造用部材を製造するために使用される硬質シリコーンの破壊靱性を改良する。従って、少なくとも6.9×108 Paのヤング率を有するゴム変性シリコーン樹脂のみがそのような用途に必須の剛性を有すると考えられる。
【0087】
図5は、RES1の破壊靱性KIcが、RUB1(DP=6)のような比較的DPが低いシリコーンゴムとの共重合により僅かに増加したことを示している。RUB2(DP=14)のようなよりDPが高いシリコーンゴムとの共重合は、RES1の破壊靱性に非常に多大な影響を及ぼす。最後に、RUB3(DP=55)のようなよりDPが高いシリコーンゴムを使用して共重合が実施される場合には、シリコーンゴムの重量百分率の関数として表されるKIcの値は破壊歪、応力及び靱性の特性と同様な挙動を示す。KIcのピーク値は10〜20重量%の間のRUB3で生じる。
【0088】
図6は、本発明のコポリマー中のシリコーンゴムのDPに関わらず、RES1の臨界歪エネルギー放出率GIcはシリコーンゴムの量の増加に従って増加することを示している。それにもかかわらず、最も高い55のDPを有するRUB3を用いて実施した場合に増加率は最も大きい。
【0089】
RES1のKIc及びGIcの値は、本発明に従って樹脂がシリコーンゴムにより変性された場合により大きい。図5は、未変性RES1のKIcが0.25MPam1/2 であることを示している。RES1を15重量%のRUB3と共重合させた場合には、KIcの値は0.47MPam1/2 である。このことは、その未変性状態に比して破壊靱性が80%以上増加したことを表している。同様に、GIcは38J/m2 から260J/m2 に増加した。このことは臨界歪エネルギー放出率が585%以上増加したことを表している。
【0090】
本発明のよりいっそう注目すべき結果は、KIc及びGIcの増加が破壊点曲げ歪度の160%の増加、破壊点曲げ応力の32%の増加及び積分応力−歪靱性の350%の増加を伴うことである。これらの利点は全てヤング率のほんの僅かな減少(21%)を伴って得られた。従って、硬質シリコーン樹脂は意外にも本発明において改質され、剛性の実質的な低下を伴わずにかなり延性のある挙動を示す。
【0091】
上記のように、本発明に従ってRES1をRUB4と共重合させた。RUB4は共重合生成物中に2重量%存在していた。機械的試験にかけた場合に、シリコーンゴム変性樹脂は、破壊点曲げ歪度が3.65%、破壊点曲げ応力が44.4MPa、ヤング率が1.79GPa、靱性が108.4KJ/m3 、KIcが0.29MPam1/2 及びGIcが62.5J/m2 であることが分かった。このことは、未変性RES1に比して破壊靱性が16%以上増加し、臨界歪エネルギー放出率が63%以上増加したことを表している。また、破壊点曲げ歪度の48%の増加、破壊点曲げ応力の16%の増加及び積分応力−歪靱性の81%の増加が観測された。これらの利点は、ヤング率の値のほんの僅かな減少(すなわち、5%未満)を伴って得られた。
【0092】
また、上記のように、RES1をRUB3及びRUB4の組み合わせと共重合させた。RUB3及びRUB4の両方を各5重量%の濃度で使用し、残りはRES1であった。機械的試験にかけた場合に、シリコーンゴム変性樹脂は、破壊点曲げ歪度が5.06%、破壊点曲げ応力が40.6MPa、ヤング率が1.34GPa、積分応力−歪靱性が146.4KJ/m3 、KIcが0.52MPam1/2 及びGIcが282J/m2 であった。またこのことも、未変性RES1に比してKIcが100%増加し、そして臨界歪エネルギー放出率GIcが600%以上増加したことを表している。また、破壊点曲げ歪度の106%の増加、破壊点曲げ応力の6%の増加及び積分応力−歪靱性の145%の増加が観測された。これらの利点は、ヤング率の値のほんの僅かな減少(すなわち、29%未満)を伴って得られた。
【0093】
付加硬化系
図7及び8は、シリコーンゴムRUB5によるRES2の変性を示すものであって、ゴムの量の増加に伴ってKIc及びGIcの値が大きくなる結果を表している。しかしながら、これらから得られる利点は縮合硬化系において得られるものほど顕著ではない。それでもKIcの値の18%増加及びGIcの値の100以上の増加が観測された。
【0094】
水素官能価が異なる2種の異なる架橋剤を使用してRES2及びRUB5を組み合わせた。各場合において、化学量論的に過剰の架橋剤を使用した。図7及び8のグラフ上のデータは、三官能性架橋剤が所定のRUB5含有率でKIc及びGIcの値に多大な影響を及ぼすことを表している。従って、樹脂のシリコーンゴム変性の効果は架橋密度に左右され得る。
【0095】
架橋密度を考慮して上記データを解釈することは更に議論する価値がある。用いた系において、樹脂RES2及びシリコーンゴムRUB5の両方がビニル官能性であり、架橋剤は水素官能性であった。従って、触媒の存在下で架橋剤は、それらの間にRES2分子が存在しない2個以上のRUB5分子間;それらの間にRUB5分子が存在しない2個以上のRES2分子間;及びそれらの間にRUB5分子が存在するRES2分子間、に自由に結合を形成する。シリコーンゴム添加の有利な効果は、架橋剤により形成される樹脂間結合がシリコーンゴムの平均DPを減少させるのと同じ効果を有するために判明していない。
【0096】
実施例−連続相として本発明のゴム変性硬質シリコーン樹脂を使用するガラス布帛複合材料
複合材料は、1)連続マトリックス相、及び2)強化相、の少なくとも2つの相を含むものとして概して規定される。強化相は好ましくはモノフィラメント、チョップトファイバー及び織布からなる。そのような複合材料に連続相として本発明のゴム変性硬質シリコーン樹脂を使用する利点を調査した。
【0097】
未加工の(ヒートクリーニングされた)E−ガラス織物(タイプ7781)及び未変性RES1並びに10重量%のRUB3により変性されたRES1から2つの12プライ積層複合構造物を作製した。これら2つの複合構造物をそれぞれCOMP1及びCOMP2と呼ぶことにする。COMP1のガラス含有率は63重量%であり、COMP2のガラス含有率は59重量%であった。硬化したスラブは厚さが3.39〜3.56mmであった。
【0098】
硬化した複合材料のスラブから切り出された試験片に対して機械的試験を上記のように実施した。その結果を表1に記載する。
【0099】
【表1】
【0100】
表1の結果は、組成物を複合材料の製造に使用した場合にも本発明のゴム変性硬質シリコーン樹脂の有利な効果が実現されることを示している。
【技術分野】
【0001】
本発明は、その構造中に導入されたシリコーンゴムを含むシリコーン樹脂を提供する。このゴム変性シリコーン樹脂及びそれから製造される強化複合材料は破壊靱性に著しい改良を示す。
【背景技術】
【0002】
有機ポリマー樹脂にゴムを添加することによりそれらの靱性を向上させることができる。そのような系は、Effects of Rubber Additions On The Fracture Toughness Of A Polyester Resin (Tetlow, P. D. et al. Proceedings of the Annual Technical Conference, 1979, Reinforced Plastics/Composites Institute The Society of the Plastics Industry, Inc. Vol. 34, 23F)及び1968年4月にサンフランシスコで発表されたCrack Toughened Polyester Resin Formulations (McGarry, F. J. et al., American Chemical Society Division of Organic Coating and Plastics Chemistry Vol. 28, No. 1, pp 526-36)に記載されている。
【0003】
種々のシリコーン組成物の靱性を向上させることに関する技術の現状は米国特許第5,034,061号明細書に開示されており、この特許明細書には透明な耐破砕性コーティングを形成するように改良されたシリコーン樹脂/流体ポリマーが教示されている。この組成物は不飽和オレフィン系官能基Rを有するR3 SiO1/2 単位及びSiO4/2 単位から本質的になるシリコーン樹脂コポリマー、ビニル官能基を有するポリジオルガノシロキサン流体、水素官能基を有するオルガノポリシロキサン架橋剤及び触媒を含む。前記組成物は白熱ガラスランプをコーティングするのに特に適するものとして開示されている。
【0004】
カナダ特許第691,206号明細書には、防振用のシリカ充填剤入りシリコーン樹脂/流体混合物の使用が開示されている。開示されているシリコーン樹脂/流体組成物の防振能は損失剪断弾性率G''に対する弾性剪断弾性率G’の比の測定値を通じて説明されている。この比の大きさは、その材料の振動吸収能に反比例するものであると示されている。その材料のG’/G''比は樹脂成分を用いずに調製された組成物と比較されている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記強化シリコーン組成物は弾性率が低い。これまで、硬質シリコーン樹脂の破壊靱性をうまく向上させることは達成されていない。シリコーン樹脂を記述する際に本明細書において使用する「硬質」なる用語は、樹脂材料が、その無充填状態でヤング率が少なくとも6.9×108 Paである特定の「剛性」を示すことを意味する。本明細書において「無充填」なる用語は、カーボン、ガラス繊維又はシリカのような強化用充填剤が樹脂に添加されていないことを意味する。
【0006】
硬質シリコーン樹脂は、それらの耐熱性及び防火性の利点を利用する用途に使用される。これらの特性は、シリコーン樹脂を、電気用積層板用の繊維強化複合材料、自動車部品、航空機及び船の構造用途に有用なものにしている。従来の未変性硬質シリコーン樹脂は非常に脆く、それらの用途はかなり限定されている。従って、脆性破壊が回避されねばならない用途での独特の防火性、電気的特性及び耐熱性の利用を可能にする本質的に向上した破壊靱性を有する硬質シリコーン樹脂が要求されている。
【課題を解決するための手段】
【0007】
我々は破壊靱性が改良されたゴム変性硬質シリコーン樹脂を発見した。我々のゴム変性硬質シリコーン樹脂は、
(A)(I)実験式:
【0008】
【化1】
【0009】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)実験式:
【0010】
【化2】
【0011】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は水素原子、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとから形成されるコポリマーを含むものであって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴム(B)が存在するものである。
【図面の簡単な説明】
【0012】
【図1】図1は、3つの異なる重合度を有する3種の異なるシリコーンゴム(RUB1、RUB2及びRUB3)により変性されたRES1、すなわちメチルフェニルシルセスキオキサン樹脂に関するシリコーンゴムの重量百分率の関数として表された破壊点曲げ歪度(%)(ASTM D 790に従う三点曲げから得られた)のグラフである。前記変性は縮合反応を用いて達成された。
【図2】図2は、図1のシリコーンゴム変性樹脂のシリコーンゴムの重量百分率の関数として表された破壊点曲げ応力のグラフである。
【図3】図3は、図1のシリコーンゴム変性樹脂のゴムの重量百分率の関数として表された靱性のグラフであって、応力−歪み曲線下の面積により測定されたものである。
【図4】図4は、図1のシリコーンゴム変性樹脂のシリコーンゴムの百分率の関数として表されたヤング率のグラフである。
【図5】図5は、図1のシリコーンゴム変性樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された破壊靱性KIcのグラフである。
【図6】図6は、図1のシリコーンゴム変性樹脂の、ASTM D 5045 に従って決定され、シリコーンゴムの重量百分率の関数として表された臨界歪エネルギー放出率GIcのグラフである。
【図7】図7は、RUB5、すなわちジメチルビニルシロキシ末端ポリジメチルシロキサンにより変性されたRES2、すなわちビニル官能シルセスキオキサン樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された破壊靱性KIcのグラフである。前記変性は架橋剤及び付加硬化を用いて達成された。
【図8】図8は、図7のシリコーンゴム変性樹脂の、ASTM D 5045 により決定され、シリコーンゴムの重量百分率の関数として表された臨界歪エネルギー放出率GIcのグラフである。
【発明を実施するための形態】
【0013】
本発明の新規ゴム変性硬質シリコーン樹脂は共重合により製造される。この共重合は、縮合反応、付加反応又は遊離基重合を通じて実施される。
【0014】
共重合が縮合反応を通じて実施される場合、樹脂の最終硬化は加熱による共重合後に達成される。これにより共重合分子間でのさらなる縮合が起こる。
【0015】
従って、本発明は、
(1)次の成分:
(A)(I)実験式:
【0016】
【化3】
【0017】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂であって、その加水分解性前駆体(II)の加水分解及び縮合により形成される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)実験式:
【0018】
【化4】
【0019】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は、ヒドロキシル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルカルボキシル基及びアリールカルボキシル基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとを有機溶剤中に溶解させる工程;
(2)縮合触媒を加える工程;
(3)溶液からの共重合生成物の沈殿又はそれらのゲル化を引き起こすことなく成分(B)の全てを成分(A)と共重合させる工程;
(4)共重合した溶液から揮発分を除去する工程;及び
(5)硬化を引き起こすのに十分な温度に揮発分が除去された前記共重合した溶液を加熱する工程;
を含むゴム変性硬質シリコーン樹脂の製造方法を含む。
【0020】
従って、本発明の目的は、無充填状態で少なくとも6.9×108 Paのヤング率を有するゴム変性シリコーン樹脂を提供することである。
【0021】
本発明の別の目的は、未変性及び無充填の状態にある前記シリコーン樹脂と比較して、無充填状態で、Kicにより評価される増加した破壊靱性、Gicにより評価される増加した臨界歪エネルギー放出率、及び積分応力−歪靱性を有する樹脂を提供することである。
【0022】
本発明の他の目的は、上記基準を満たすそのような樹脂の製造方法を提供することである。
【0023】
本発明の特徴は、ゴム変性シリコーン樹脂のKIc及びGIcの値が、未変性及び無充填状態にあるシリコーン樹脂と比べて25%以上も増加することである。
【0024】
本発明のゴム変性硬質シリコーン樹脂は、(A)(I)有機シリコーン樹脂、(II)(I)の加水分解性前駆体及び(III)(II)から形成される加水分解生成物からなる群から選ばれる有機ケイ素組成物と(B)シリコーンゴムとから形成されるコポリマーを含む。
【0025】
有機シリコーン樹脂(I)は実験式:
【0026】
【化5】
【0027】
(式中、0.8≦(a+b+c)≦1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される。
【0028】
組成物(A)の有機シリコーン樹脂(I)はシルセスキオキサンであり、(I)の上記R1 、R2 及びR3 基をその構造の一部として含む加水分解性前駆体(II)の加水分解及び縮合により周知の方法で調製される。そのような加水分解性前駆体には、望ましい3次元樹脂構造を生じるオルガノトリアルコキシシラン及びオルガノトリハロシランのような三官能性シラン;又は末端キャップ剤として作用するトリオルガノモノアルコキシシラン、トリオルガノモノハロシラン、ジシロキサン及びジシラザンのような一官能性シランが含まれる。当業者は、ジオルガノジハロシラン及びジオルガノジアルコキシシランのような二官能性シラン、並びに少量のテトラハロシラン及びテトラアルコキシシランのような四官能性シランも樹脂前駆体として有用であることも認識するであろう。
【0029】
本発明の好ましい態様において、R1 、R2 及びR3 基の大部分は無官能性である。従って、これらの基は特許請求の範囲に記載のゴム変性硬質シリコーン樹脂を生成する共重合反応に関与せず、アルキル基、アリール基又はこれらの組み合わせである。最も好ましくは、これらの基はメチル、フェニル又はこれらの組み合わせである。
【0030】
本発明の第2成分は実験式:
【0031】
【化6】
【0032】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は、水素、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴム(B)である。
【0033】
R4 基がアルキル基及びアリール基のみを表すことは重要である。従って、R4 基は共重合反応に関与しない。しかしながら、各R5 は本発明のコポリマーを形成する共重合反応に関与する官能基である。
【0034】
(B)に関する式において、xはシリコーンゴムの平均非官能線状鎖長、すなわちR5 基間の平均鎖長を表す。従って、種々の重合度を有するシリコーンゴム(B)の混合物も上記実験式により表すことができる。本発明に使用されるシリコーンゴムの殆どは鎖の末端基にのみR5 基を有する。そのような場合、用語「重合度」(「DP」)はxの値である。しかしながら、DPなる用語は末端官能シロキシ基を包含しない。
【0035】
好ましい態様において、R4 基はメチル基、フェニル基又はこれらの組み合わせである。
【0036】
我々は、有機ケイ素組成物(A)のR1 、R2 及びR3 基の百分率が高く、シリコーンゴム(B)のR4 基が主としてメチルであるか又は主としてフェニルである場合に、有機ケイ素組成物(A)及びシリコーンゴム(B)は相溶性であり、前記ゴムは樹脂構造中に均質に分散することができることを見出した。
【0037】
我々は、シリコーンゴム(B)のDPが比較的大きく、例えば55〜200の間になる場合には、ゴムは樹脂から分離する傾向になり、明確に識別される二相系の形成が起こることも見出した。本発明の組成物を配合する際に、有機ケイ素組成物(A)及びゴム(B)は比較的低い固形分濃度(すなわち、極めて薄い濃度から30重量%)で有機溶剤中に溶解される。高濃度であるほど、樹脂(I)の形態にある有機ケイ素組成物(A)とゴム(B)との均質溶液を形成することは非常に困難である。
【0038】
2相系への分離は、硬化したゴム変性樹脂マトリックスにおいて時々観察されたが、そのような分離はそれが形成される有機溶液中では示されない。ある場合において、そのような硬化後分離は本発明のゴム変性樹脂の機械的特性を向上させる。さらに、1,000程度の高いDPを有するシリコーンゴム(B)を使用することもできる。
【0039】
本発明において、有機ケイ素組成物(A)及びシリコーンゴム(B)は、硬化したゴム変性硬質シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で存在する。我々は意外にも本発明のゴム変性シリコーン樹脂のヤング率はゴム含有率の増加に伴って減少することを見出した。従って、有機ケイ素組成物(A)及びゴム(B)の相対量は、硬化した樹脂の剛性が構造用用途に満足に使用できない程度に低下しないように選択される。シリコーンゴムの種類及び架橋剤の使用も本発明の変性樹脂のヤング率に影響を及ぼす。しかしながら、殆どの場合に、変性樹脂の30重量%を超える量のシリコーンゴムの添加は、上記のヤング率を下回るヤング率を有する組成物をもたらす。同様に、向上した靱性の有利な効果は、少なくとも2重量%のシリコーンゴム(B)が使用されない限り実現されない。
【0040】
一態様において、有機ケイ素組成物(A)は、シリコーンゴム(B)と共重合される前に、まず望ましい分子量を有するものに形成される有機シリコーン樹脂(I)である。まず有機溶剤中で樹脂(I)を、望ましい量の架橋剤及び触媒量の触媒と共に、シリコーンゴム(B)と配合することが望ましい。この組成物は、揮発分を除去され、金型に流し込まれ、その後に熱硬化される。ある場合に、揮発分の除去は金型中で実施される。揮発分の除去が高温で実施される場合には、硬化触媒の添加は冷却後に通常実施される。
【0041】
他の態様において、組成物は揮発分の除去工程後に射出成形されてよい。
【0042】
有機ケイ素組成物(A)とゴム(B)の間の縮合反応を利用する本発明の組成物において、最終硬化を実施する前に、まず有機ケイ素組成物(A)分子及びゴム(B)分子をもう一方と選択的に共重合させることができる。
【0043】
例えば、樹脂(I)の形態にある有機ケイ素組成物(A)及びシリコーンゴム(B)は、必要であれば望ましい量の架橋剤及び縮合触媒と共にトルエンのような有機溶剤中に溶解される。チタンテトラブトキシドのような比較的弱い縮合触媒を使用することが好ましい。90℃の温度でこの比較的弱い触媒は、有機ケイ素組成物(A)又はゴム(B)間のホモ重合を引き起こすことなく有機ケイ素組成物(A)、ゴム(B)及び必要であれば架橋剤の間の共重合を引き起こす。この反応は、ゴム(B)分子の全てが有機ケイ素組成物(A)分子と共重合する程度まで実施されることが好ましい。この結果は29Si核磁気共鳴分光分析(NMR)を使用して確認した。
【0044】
ジルコニウム及びハフニウムのアルコキシドも比較的弱い縮合触媒として同様に作用する。
【0045】
上記条件下ではホモ重合が起こりうる。しかしながら、形成されるコポリマーが有機溶剤に可溶のままであり、かつ、有機溶剤中で安定であるために実質的なホモ重合は起こらない。この「軽度」の共重合は軽度の増粘に類似する。軽度の共重合は触媒の濃度により調節され、また、その触媒強度及び軽度の共重合が実施される温度に関係する。
【0046】
「軽度」に共重合した状態にあるコポリマーは溶液中に留まる。その後、Dow Corning (商標)触媒15又はY-177 、コリンオクトエート及びオクタン酸亜鉛触媒(これらは両方ともミシガン州ミッドランド所在のDow Corning Corporation から市販入手可能)のような強い縮合触媒が「軽度に共重合した」溶液に添加される。この溶液は次にキャストされ、揮発分を除去され、熱が加えられることにより最終硬化して硬質状態になる。往々にして、最終硬化は圧力を加えることにより促進される。縮合硬化系において、最終硬化は樹脂(I)に結合している残留シラノール基の縮合を通じて達成される。
【0047】
シリコーンゴム(B)が加水分解性又は縮合性のR5 基を含む場合、ゴム(B)は有機シリコーン樹脂(I)の加水分解性前駆体(II)又は(II)の部分水解物(III)と配合され、次に軽度の共重合工程を達成するため及び樹脂の網状分子の成長を引き起こすために加水分解/縮合反応が実施される。軽度の共重合工程は、組成物がキャスティングに望ましい粘度に達するまで上記のような比較的弱い触媒により触媒される。その後、組成物は、揮発分が除去され、適切な強い縮合触媒が添加される。材料は次にキャストされ、最終的に熱硬化される。
【0048】
本発明のゴム変性シリコーン樹脂を得るために使用される方法に関わらず、硬化した組成物は望ましい剛性及び向上した破壊靱性を示す。
【0049】
本発明のコポリマーは架橋剤の作用を用いて又は用いずに形成され、縮合硬化組成物の場合には触媒の作用を用いて又は用いずに形成される。
【0050】
本発明のコポリマーを得るために使用される硬化機構の種類には、一般的に縮合硬化系、付加硬化系、及び遊離基重合系と呼ばれているものが含まれる。
【0051】
ビニル基のようなアルケニル基を含む系において、シリコーン樹脂(I)及びシリコーンゴム(B)に反応性官能基を付与するために、遊離基重合は実施可能な硬化系である。過酸化物のような遊離基開始剤が通常使用される。汎用されている開始剤としては過酸化アリールがある。よく知られているビニルに特異的な過酸化物開始剤は(CH3 )3 COOC(CH3 )3 である。
【0052】
縮合硬化系において、錫、チタン及び亜鉛の金属エステル又はアルコキシドが好ましい触媒である。チタンテトラブトキシド、ジブチル錫ジラウレート及びオクタン酸亜鉛がよく知られている例である。しかしながら、錫触媒は硬化した本発明のゴム変性硬質樹脂の熱安定性を低下させるものであって、高温用途に対しては概してその使用は避けられる。好ましさはさほどでもないが使用可能なものは、硫酸、リン酸、水酸化カリウム、水酸化セシウム、金属シラノラート及び焼石灰のような強酸及び強塩基である。しかしながら、これらの後者の触媒は、硬化した硬質シリコーン注型品の熱安定性を損なう場合もある。アミン又は金属カルボキシレート及び第4級塩基も有用である。水酸化テトラメチルアンモニウムのような特定の第4級塩基は、硬化温度に加熱された場合に揮発性副生成物を分解するために都合良い。従って、それらは本発明のシリコーン樹脂/ゴムコポリマーマトリックスから容易に除去される。
【0053】
揮発性副生成物が硬化機構により形成される場合には注意が払われねばならない。そのような揮発性副生成物は、注型品の欠陥として作用する気泡の形成により靱性を著しく低下させる原因となりうる。
【0054】
付加硬化系に対し、クロロ白金酸のような白金族触媒が往々にして使用される。実際に、適切な白金触媒には白金化合物又は白金錯体が含まれる。クロロ白金酸に加え、クロロ白金酸六水和物、カルステット触媒(すなわち、クロロ白金酸とsym −ジビニルテトラメチルジシロキサンの錯体)、ジクロロビス(トリフェニルホスフィン)白金(II)、塩化白金及び酸化白金が使用される。
【0055】
縮合硬化系において、シリコーン樹脂(I)とシリコーンゴム(B)の間の共重合及び共重合した系の最終硬化は、触媒の不在下で引き起こされる。しかしながら、そのような反応は高温に加熱することを必要とし、この理由から好ましくない。また、上記の軽度の共重合は触媒の助けをかりずに達成されない。
【0056】
本発明を実施するのに適する架橋剤にはシリコーンゴムの製造及び硬化において周知のものが含まれる。これらの架橋剤は、それらの化学式が少なくとも1個のケイ素原子を含むという意味においてシリコーンをベースとするものである。テトラエトキシシラン、メチルトリメトキシシラン、並びにメチルトリアセトキシ−、メチルトリオキシモ−及びテトラオキシモシランが周知の例である。架橋剤は反応性官能基間に比較的短いシロギン鎖を含む。本明細書で使用する場合に「架橋剤」なる用語は、シラン、及びシロキシ鎖のDPが6未満であるようなシロキサンに限定される。
【0057】
有機シリコーン樹脂を形成するために酸加水分解及び縮合を使用することによって、有意の濃度の立体障害を受けていない残留ヒドロキシル基を有する樹脂組成物が得られる。従って、この樹脂の残留ヒドロキシ官能基を使用する縮合反応は、縮合硬化系を通じて本発明のコポリマーを形成する都合のよい方法である。
【0058】
本発明の耐破壊性シリコーン樹脂組成物は、構造用部材又は耐力部材の製造に非常に有用であると期待される。さらに、本発明の組成物は繊維強化ゴム変性硬質樹脂のような複合材料における連続相として非常に有利に使用される。
【0059】
本発明の組成物は他の多くの用途に適用されてもよい。例えば、有機溶剤中に軽度に共重合した有機ケイ素組成物(A)及びゴム(B)を含む溶液は、基材上で硬化性コーティング組成物を形成し、この硬化性コーティング組成物は接着剤として作用する。同様に、本発明の組成物を硬化させてフィルム又はシート製品の形態にすることもできる。
【実施例】
【0060】
実施例−強化用第2相を含まない樹脂
縮合硬化系
最高20重量%までの種々の重量%で4つの異なる種類のシリコーンゴム(本明細書においてRUB1、RUB2、RUB3及びRUB4と示す)により第1樹脂(本明細書においてRES1と示す)を変性させた。以下で述べるようにトルエン中で種々の組成物を調製し、Teflon(商標)によりライニングされた四角形のアルミニウム金型に流し込み、110℃の減圧下で揮発分を除去し、次のスケジュールで硬化させた:95℃で48時間;110℃で24時間;120℃で24時間;130℃で24時間;140℃で24時間;150℃で12時間;175℃で6時間;200℃で12時間;230℃で3時間;及び260℃で5時間。この硬化スケジュールによって、亀裂及び気泡のような欠陥が存在しない硬化したスラブが確実に得られた。
【0061】
硬化したスラブから採取した、試験片をASTM D 790に従って破断するまで三点曲げ試験にかけ、そして曲げ強さ、歪度、ヤング率及び積分応力−歪靱性をシリコーンゴムの重量%の関数としてプロットした。(図1〜4を参照されたい。)
【0062】
硬化したスラブから採取される試験片を同様に作製し、片へりノッチ三点曲げモードでASTM D 5045 に従って試験し、破壊靱性Kic及び臨界歪エネルギー放出率GIcをシリコーンゴムの重量%の関数として決定した。(図5及び6を参照されたい。)
【0063】
RES1は実験式:
【0064】
【化7】
【0065】
(式中、Me及びPhはそれぞれメチル基及びフェニル基を表す。以下同様とする。)
により表される数平均分子量が1,300以下のシルセスキオキサン樹脂(ミシガン州ミッドランド所在のDow Corning Corporation から市販入手可能なDow Corning (商標)4-3136)であった。この樹脂は固形フレークの形態で入手可能なものである。
【0066】
前述のように、酸加水分解/縮合により形成されるシルセスキオキサン樹脂は概して有意の量の立体障害を受けていないヒドロキシル基を含む。上記実験式はそのような残留ヒドロキシル基(往々にして残留シラノール基と呼ばれる)を考慮していない。これらの基は本発明における共重合に関与する反応性基であるため、残留ヒドロキシル基を考慮して上記式を次式:
【0067】
【化8】
【0068】
のような適切な実験式に書き直すことができる。
【0069】
種々のゴムRUB1、RUB2、RUB3及びRUB4の各々を同様に作製した。酢酸カリウム触媒の存在下、130〜150℃の間の温度で揮発分を絶えず除去しながら、所定の望ましい平均重合度(DP)を有するジメチルシラノール末端ポリジオルガノシロキサンを化学量論的に過剰のテトラエチルオルトシリケートと反応させた。29Si核磁気共鳴分光分析(NMR)が末端ヒドロキシル基のトリエトキシ転化が完了することを示すまで加熱を続けた。未反応テトラエチルオルトシリケートはストリッピング及び加熱により除去した。
RUB1、RUB2、RUB3及びRUB4は、DPがそれぞれ6、14、55及び376であるものであった。
【0070】
十分な量のRES1及び所定量のシリコーンゴム(B)を望ましい比でトルエンに溶解させることによりRES1の変性を実施した。RUB1及びRUB2の場合に固形分50重量%の溶液を使用した。高DPゴムと樹脂の間の限られた相溶性のために、RES1とRUB3の場合に38重量%の最大固形分を使用した。また、同じ理由から、RES1とRUB4の場合に20重量%の最大固形分を使用した。その後、チタンテトラブトキシド触媒の形態にある比較的弱い縮合触媒を0.1〜1.0重量%の間の濃度で加えた。その後、反応混合物の温度は3時間で90℃に上昇し、続いて加熱することにより25時間還流させ、RES1及び種々のゴム(B)を軽度に共重合させた。次に、溶液をストリップして固形分50〜65重量%とし、室温に冷却し、そして次に0.20重量%の比較的強い縮合触媒(Dow Corning (商標)触媒15又はY-177 )を加えた。次にこの溶液から揮発分を除去し、上記のように注型してスラブとし、硬化させた。
【0071】
最後に、RES1をRUB3及びRUB4の組み合わせと共重合させた。RUB3及びRUB4の両方を各々5重量%の濃度で使用し、残りはRES1であった。この組成物は、RES1と10重量%のRUB3及びRES1と10重量%のRUB4の独立に調製された軽度に共重合した等しい部(固形分)の組み合わせをトルエン中で配合することにより調製した。その後、この溶液から揮発分を除去し、上記のように注型してスラブとし、硬化させた。
【0072】
付加硬化系
平均DPが9であるジメチルビニルシロキシ末端ポリジメチルシロキサン(RUB5)により最大15重量%までの種々の重量%で第2樹脂RES2、すなわちビニル官能シルセスキオキサン樹脂を変性させた。
【0073】
RUB5はペンシルヴェニア州チュリータウン所在のGelest, Inc.製の市販入手可能な製品であり、商標DMS-V05 のもとに市販されている。
【0074】
RES2は、フェニルトリクロロシランの酸触媒(HCl)される加水分解/縮合から周知の方法で調製した。次にこの反応生成物から酸を洗い落とし、水酸化カリウムの存在下で更に増粘させた。重合は、ジメチルビニルクロロシランのトルエン溶液の添加を通じて末端キャッピングすることにより停止させた。水酸化カリウムを中和し、溶液から濾過して取り除き、樹脂を固形分75重量%に濃縮した。RES2分子は1350〜1415のMnを有していた。
【0075】
RES2の実験式は次式:
【0076】
【化9】
【0077】
又は次式:
【0078】
【化10】
【0079】
のように表される。ここで、Viはビニル基を表し、以下同様とする。
【0080】
2つの異なる方法を使用してRES2のゴム変性を実施した。両方の方法において、化学量論的に過剰のSiH官能架橋剤を使用した。第1の方法において、架橋剤は三官能性のフェニルトリス(ジメチル水素シロキシ)シラン(United Chemical Technologies, Inc.から市販入手可能)であった。第2の方法において、架橋剤は四官能性のテトラキス(水素ジメチルシロキシ)シランであった。
【0081】
望ましい割合でRUB4をRES2のトルエン溶液に溶解させることによりRES2のトルエン溶液中で種々の組成物を調製した。その後、化学量論的に過剰の架橋剤をそれぞれ加えた。得られた溶液を50℃で1.5時間減圧脱気した。溶液を室温に冷却し、これに触媒量(1〜100重量ppm)のヘキサクロロ白金酸を加えた。Teflon(商標)によりライニングされた四角形のアルミニウム金型に得られた組成物を流し込み、140〜160℃で16時間硬化させ、金型から取り出し、200〜260℃で4〜8時間を要して後硬化させた。三官能性架橋剤を使用して組成物を硬化させる間に亀裂は観察されなかった。四官能性架橋剤を使用して組成物を硬化させる間に幾つかの亀裂が観察された。しかしながら、より低い硬化温度を用いることはそのような亀裂を減少させることに役立つことが見出された。
【0082】
硬化したスラブから採取される試験片を同様に作製し、片へりノッチ三点曲げモードでASTM D 5045 に従って試験し、破壊靱性KIc及び臨界歪エネルギー放出率GIcをシリコーンゴムの重量%の関数として決定した。(図7及び8を参照されたい。)
【0083】
縮合硬化系
上記試験から得られた結果は図1〜8にグラフにより示されている。RUB5によるRES1の変性は、結果が示されていない唯一の例である。図1〜6において、中心点は試験パラメーターの算術平均値を表し、縦棒はデーターの拡がりを表す。およそ5〜6つの試験片を各ゴム荷重度で使用した。
【0084】
図1を参照すると、RUB1及びRUB2(それぞれDP=6及び14)におけるようにシリコーンゴムが比較的低いDPを有する場合には、シリコーンゴムの添加量が多いほど破壊点歪度が増加することがわかる。しかしながら、RUB3(DP=55)におけるようにシリコーンゴムのDPが幾分高いと、破壊点曲げ歪度はシリコーンゴム10重量%でピークに達する。
【0085】
図2を参照すると、シリコーンゴム含有率の関数として表されている破壊点曲げ応力は、前記歪度の場合に類似する挙動を示す。同様に図3も、積分応力−歪により測定され、ゴム含有率の関数として表された靱性も同じ傾向にあることを示している。
【0086】
図4は、本発明のコポリマーに使用されるゴムのDPに関わらず全ての場合に、コポリマーのゴム含有率が増加すると組成物のヤング率又は「剛性」が低下することを示している。本発明は、構造用部材を製造するために使用される硬質シリコーンの破壊靱性を改良する。従って、少なくとも6.9×108 Paのヤング率を有するゴム変性シリコーン樹脂のみがそのような用途に必須の剛性を有すると考えられる。
【0087】
図5は、RES1の破壊靱性KIcが、RUB1(DP=6)のような比較的DPが低いシリコーンゴムとの共重合により僅かに増加したことを示している。RUB2(DP=14)のようなよりDPが高いシリコーンゴムとの共重合は、RES1の破壊靱性に非常に多大な影響を及ぼす。最後に、RUB3(DP=55)のようなよりDPが高いシリコーンゴムを使用して共重合が実施される場合には、シリコーンゴムの重量百分率の関数として表されるKIcの値は破壊歪、応力及び靱性の特性と同様な挙動を示す。KIcのピーク値は10〜20重量%の間のRUB3で生じる。
【0088】
図6は、本発明のコポリマー中のシリコーンゴムのDPに関わらず、RES1の臨界歪エネルギー放出率GIcはシリコーンゴムの量の増加に従って増加することを示している。それにもかかわらず、最も高い55のDPを有するRUB3を用いて実施した場合に増加率は最も大きい。
【0089】
RES1のKIc及びGIcの値は、本発明に従って樹脂がシリコーンゴムにより変性された場合により大きい。図5は、未変性RES1のKIcが0.25MPam1/2 であることを示している。RES1を15重量%のRUB3と共重合させた場合には、KIcの値は0.47MPam1/2 である。このことは、その未変性状態に比して破壊靱性が80%以上増加したことを表している。同様に、GIcは38J/m2 から260J/m2 に増加した。このことは臨界歪エネルギー放出率が585%以上増加したことを表している。
【0090】
本発明のよりいっそう注目すべき結果は、KIc及びGIcの増加が破壊点曲げ歪度の160%の増加、破壊点曲げ応力の32%の増加及び積分応力−歪靱性の350%の増加を伴うことである。これらの利点は全てヤング率のほんの僅かな減少(21%)を伴って得られた。従って、硬質シリコーン樹脂は意外にも本発明において改質され、剛性の実質的な低下を伴わずにかなり延性のある挙動を示す。
【0091】
上記のように、本発明に従ってRES1をRUB4と共重合させた。RUB4は共重合生成物中に2重量%存在していた。機械的試験にかけた場合に、シリコーンゴム変性樹脂は、破壊点曲げ歪度が3.65%、破壊点曲げ応力が44.4MPa、ヤング率が1.79GPa、靱性が108.4KJ/m3 、KIcが0.29MPam1/2 及びGIcが62.5J/m2 であることが分かった。このことは、未変性RES1に比して破壊靱性が16%以上増加し、臨界歪エネルギー放出率が63%以上増加したことを表している。また、破壊点曲げ歪度の48%の増加、破壊点曲げ応力の16%の増加及び積分応力−歪靱性の81%の増加が観測された。これらの利点は、ヤング率の値のほんの僅かな減少(すなわち、5%未満)を伴って得られた。
【0092】
また、上記のように、RES1をRUB3及びRUB4の組み合わせと共重合させた。RUB3及びRUB4の両方を各5重量%の濃度で使用し、残りはRES1であった。機械的試験にかけた場合に、シリコーンゴム変性樹脂は、破壊点曲げ歪度が5.06%、破壊点曲げ応力が40.6MPa、ヤング率が1.34GPa、積分応力−歪靱性が146.4KJ/m3 、KIcが0.52MPam1/2 及びGIcが282J/m2 であった。またこのことも、未変性RES1に比してKIcが100%増加し、そして臨界歪エネルギー放出率GIcが600%以上増加したことを表している。また、破壊点曲げ歪度の106%の増加、破壊点曲げ応力の6%の増加及び積分応力−歪靱性の145%の増加が観測された。これらの利点は、ヤング率の値のほんの僅かな減少(すなわち、29%未満)を伴って得られた。
【0093】
付加硬化系
図7及び8は、シリコーンゴムRUB5によるRES2の変性を示すものであって、ゴムの量の増加に伴ってKIc及びGIcの値が大きくなる結果を表している。しかしながら、これらから得られる利点は縮合硬化系において得られるものほど顕著ではない。それでもKIcの値の18%増加及びGIcの値の100以上の増加が観測された。
【0094】
水素官能価が異なる2種の異なる架橋剤を使用してRES2及びRUB5を組み合わせた。各場合において、化学量論的に過剰の架橋剤を使用した。図7及び8のグラフ上のデータは、三官能性架橋剤が所定のRUB5含有率でKIc及びGIcの値に多大な影響を及ぼすことを表している。従って、樹脂のシリコーンゴム変性の効果は架橋密度に左右され得る。
【0095】
架橋密度を考慮して上記データを解釈することは更に議論する価値がある。用いた系において、樹脂RES2及びシリコーンゴムRUB5の両方がビニル官能性であり、架橋剤は水素官能性であった。従って、触媒の存在下で架橋剤は、それらの間にRES2分子が存在しない2個以上のRUB5分子間;それらの間にRUB5分子が存在しない2個以上のRES2分子間;及びそれらの間にRUB5分子が存在するRES2分子間、に自由に結合を形成する。シリコーンゴム添加の有利な効果は、架橋剤により形成される樹脂間結合がシリコーンゴムの平均DPを減少させるのと同じ効果を有するために判明していない。
【0096】
実施例−連続相として本発明のゴム変性硬質シリコーン樹脂を使用するガラス布帛複合材料
複合材料は、1)連続マトリックス相、及び2)強化相、の少なくとも2つの相を含むものとして概して規定される。強化相は好ましくはモノフィラメント、チョップトファイバー及び織布からなる。そのような複合材料に連続相として本発明のゴム変性硬質シリコーン樹脂を使用する利点を調査した。
【0097】
未加工の(ヒートクリーニングされた)E−ガラス織物(タイプ7781)及び未変性RES1並びに10重量%のRUB3により変性されたRES1から2つの12プライ積層複合構造物を作製した。これら2つの複合構造物をそれぞれCOMP1及びCOMP2と呼ぶことにする。COMP1のガラス含有率は63重量%であり、COMP2のガラス含有率は59重量%であった。硬化したスラブは厚さが3.39〜3.56mmであった。
【0098】
硬化した複合材料のスラブから切り出された試験片に対して機械的試験を上記のように実施した。その結果を表1に記載する。
【0099】
【表1】
【0100】
表1の結果は、組成物を複合材料の製造に使用した場合にも本発明のゴム変性硬質シリコーン樹脂の有利な効果が実現されることを示している。
【特許請求の範囲】
【請求項1】
(A)(I)式:
【化1】
(式中、0.8≦(a+b+c)≦=1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)式:
【化2】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は水素原子、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとの共重合反応生成物を含むゴム変性硬質シリコーン樹脂であって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴム(B)が存在するゴム変性硬質シリコーン樹脂。
【請求項2】
少なくとも6.9×108 Paのヤング率を有するゴム変性硬質シリコーン樹脂の製造方法であって、
(1)請求項1に記載の有機シリコーン組成物を有機溶剤中に溶解させる工程;
(2)触媒を加える工程;
(3)溶液からの共重合生成物の沈殿又はそれらのゲル化を引き起こすことなく成分(B)の全てを請求項1に記載の成分(A)と共重合させる工程;
(4)共重合した溶液から揮発分を除去する工程;及び
(5)硬化を引き起こすのに十分な温度に揮発分が除去された前記共重合した溶液を加熱する工程;
を含む方法。
【請求項1】
(A)(I)式:
【化1】
(式中、0.8≦(a+b+c)≦=1.6であることを条件としてaは正の数値、b及びcは0又は正の数値であり、R1 、R2 及びR3 は、水素原子、ヒドロキシル基、アルキル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アリール基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルエーテル基、アリールエーテル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基である)により表される有機シリコーン樹脂;
(II)(I)の加水分解性前駆体;並びに
(III)(II)から形成される加水分解生成物;
からなる群から選ばれる有機ケイ素組成物と、
(B)式:
【化2】
(式中、各R4 はアルキル基又はアリール基から独立に選ばれる一価基であり、各R5 は水素原子、ヒドロキシル基、アルケニル基、アルコキシ基、オキシモ基、アルキルオキシモ基、アリールオキシモ基、アルキルエポキシド基、アリールエポキシド基、アルキルカルボキシル基、アリールカルボキシル基、アルキルアミド基、アリールアミド基、アルキルアミノ基及びアリールアミノ基からなる群から独立に選ばれる一価基であり、pは1、2又は3、qは1又は2、xは6以上の値、及びyは0〜10の値である)
により表されるシリコーンゴムとの共重合反応生成物を含むゴム変性硬質シリコーン樹脂であって、前記ゴム変性シリコーン樹脂が少なくとも6.9×108 Paのヤング率を有するような相対量で前記有機ケイ素組成物(A)及び前記シリコーンゴム(B)が存在するゴム変性硬質シリコーン樹脂。
【請求項2】
少なくとも6.9×108 Paのヤング率を有するゴム変性硬質シリコーン樹脂の製造方法であって、
(1)請求項1に記載の有機シリコーン組成物を有機溶剤中に溶解させる工程;
(2)触媒を加える工程;
(3)溶液からの共重合生成物の沈殿又はそれらのゲル化を引き起こすことなく成分(B)の全てを請求項1に記載の成分(A)と共重合させる工程;
(4)共重合した溶液から揮発分を除去する工程;及び
(5)硬化を引き起こすのに十分な温度に揮発分が除去された前記共重合した溶液を加熱する工程;
を含む方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2011−219768(P2011−219768A)
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−153138(P2011−153138)
【出願日】平成23年7月11日(2011.7.11)
【分割の表示】特願2008−272203(P2008−272203)の分割
【原出願日】平成10年1月5日(1998.1.5)
【出願人】(590001418)ダウ コーニング コーポレーション (166)
【氏名又は名称原語表記】DOW CORNING CORPORATION
【Fターム(参考)】
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【出願番号】特願2011−153138(P2011−153138)
【出願日】平成23年7月11日(2011.7.11)
【分割の表示】特願2008−272203(P2008−272203)の分割
【原出願日】平成10年1月5日(1998.1.5)
【出願人】(590001418)ダウ コーニング コーポレーション (166)
【氏名又は名称原語表記】DOW CORNING CORPORATION
【Fターム(参考)】
[ Back to top ]