
サイトカイン、ストレス、およびオンコプロテインに活性化されるヒト蛋白質キナーゼキナーゼ
【課題】ヒトマイトジェン活性化プロテイン(MAP)キナーゼキナーゼアイソフォーム(MKK)、およびその利用方法を提供する。
【解決手段】活性化転写因子2(ATF2)およびc-Junを含む他の因子の活性化を誘導する、ヒトMAPキナーゼp38およびJNKを活性化する独特のシグナル伝達経路を仲介するMKK。および、MKK機能または活性を調節する試薬の同定方法、ならびにMKKを介する疾病の治療におけるそのような試薬の利用法。
【解決手段】活性化転写因子2(ATF2)およびc-Junを含む他の因子の活性化を誘導する、ヒトMAPキナーゼp38およびJNKを活性化する独特のシグナル伝達経路を仲介するMKK。および、MKK機能または活性を調節する試薬の同定方法、ならびにMKKを介する疾病の治療におけるそのような試薬の利用法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は蛋白質キナーゼに関する。
【背景技術】
【0002】
マイトジェン活性化プロテイン(MAP)キナーゼは、細胞表面から核へのシグナル伝達の重要な仲介因子となっている。酵母では、SMK1、HOG1、MPK1、FUS3、およびKSS1を含む多数のMAPキナーゼが記述されている。哺乳類で同定されたMAPキナーゼは、細胞外シグナル制御MAPキナーゼ(ERK)、c-Junアミノ末端キナーゼ(JNK)、およびp38キナーゼ(Davis (1994) Trends Biochem. Sci. 19:470(非特許文献1))である。これらのMAPキナーゼアイソフォームは、スレオニンとチロシンの二重のリン酸化によって、活性化される。
【0003】
活性化転写因子2 (ATF2)、ATFa、およびcAMP応答配列結合蛋白質(CRE-BPa)は、多くの遺伝子のプロモーターにある類似配列に結合する関連した転写因子である(Ziff (1990) Trends in Genet. 6:69(非特許文献2))。これらの転写因子が結合すると、転写活性が上昇する。ATF2はオンコプロテインEla(LiuおよびGreen (1994) Nature 368:520(非特許文献3))、B型肝炎ウイルスX蛋白質(Maguireら(1991) Science 252:842(非特許文献4))、およびヒトT細胞白血病ウイルス1 tax蛋白質(WagnerおよびGreen (1993) Science 262:395(非特許文献5))を含むいくつかのウイルス蛋白質に結合する。またATF2は、癌抑制遺伝子産物Rb(Kimら(1992) Nature 358:331(非特許文献6))、高移動度群蛋白質HMG(I)Y(Duら(1993) Cell 74:887(非特許文献7))、ならびに転写因子である核NF- κB(Duら(1993) Cell 74:887(非特許文献7))およびc-Jun(BenbrookおよびJones (1990) Oncogene 5:295(非特許文献8))とも、相互作用する。
【非特許文献1】Davis (1994) Trends Biochem. Sci. 19:470
【非特許文献2】Ziff (1990) Trends in Genet. 6:69
【非特許文献3】LiuおよびGreen (1994) Nature 368:520
【非特許文献4】Maguireら(1991) Science 252:842
【非特許文献5】WagnerおよびGreen (1993) Science 262:395
【非特許文献6】Kimら(1992) Nature 358:331
【非特許文献7】Duら(1993) Cell 74:887
【非特許文献8】BenbrookおよびJones (1990) Oncogene 5:295
【発明の開示】
【0004】
本発明は、新しいグループのヒトマイトジェン活性化プロテインキナーゼキナーゼ(MKK)の同定と単離に基づく。本明細書に記述されるMKKアイソフォームMKK3、MKK6、MKK4(MKK4-α、-β、および-γを含む)、MKK7(マウスMKK7、ヒトMKK7、MKK7b、MKK7c、MKK7d、およびMKK7eを含む)は、セリン、スレオニン、およびチロシンキナーゼ活性を持つ。MKK3、MKK4、およびMKK6は、ヒトMAPキナーゼp38のThr180およびTyr182を特異的にリン酸化する。MKK4アイソフォームは、ヒトMAPキナーゼJNK(JNK1、JNK2、およびJNK5を含む)のThr183およびTyr185もリン酸化する。MKK7アイソフォームは、JNKのThr183およびTyr185をリン酸化する。
【0005】
したがって、本発明はヒトp38MAPキナーゼを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なヒトMKKポリペプチドを特徴とする。MKK3は、配列番号:2のアミノ酸配列を持つ。さらに本発明は、配列番号:4のアミノ酸配列を持ち、ヒトp38 MAPキナーゼを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つMKK6も含む。
【0006】
さらに本発明は、ヒトp38 MAPキナーゼおよびJNKを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なヒトMKKポリペプチドを特徴とする。MKK4アイソフォームのMKK4-αは、配列番号:6のアミノ酸配列を持つ。MKK4アイソフォームのMKK4-βは、配列番号:8のアミノ酸配列を持つ。MKK4アイソフォームのMKK4-γは、配列番号:10のアミノ酸配列を持つ。
【0007】
また、本発明はマイトジェン活性化プロテインキナーゼJNKを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なMKKポリペプチド(MKK7)をも特徴とする。MKKアイソフォームMKK7(マウス)およびMKK7(ヒト)は、それぞれ、配列番号:18および26のアミノ酸配列を持つ。MKK7アイソフォームのMKK7b、MKK7c、MKK7d、およびMKK7eは、それぞれ、配列番号:20、配列番号:28、配列番号:30および配列番号:32のアミノ酸配列を持つ。
【0008】
本明細書で使用される「マイトジェン活性化プロテインキナーゼキナーゼ」または「MKK」という用語は、ヒトマイトジェン活性化プロテインキナーゼのリン酸化および活性化という、特徴的な活性を持つ蛋白質キナーゼを意味する。MKKの例には、p38 MAPキナーゼのThr180およびTyr182を特異的にリン酸化し活性化するMKK3およびMKK6、p38 MAPキナーゼのThr180およびTyr182ならびにJNKのThr183およびTyr185を特異的にリン酸化し活性化するMKK4アイソフォーム、JNKのThr183およびTyr185を特異的にリン酸化するMKK7アイソフォームが含まれる。
【0009】
「MKK7」は、セリン、スレオニン、およびチロシンキナーゼ活性を持ち、マイトジェン活性化プロテイン(MAP)キナーゼJNKをリン酸化するがp38をリン酸化しない、マイトジェン活性化プロテインキナーゼキナーゼ(MKK)ポリペプチドの哺乳類アイソフォームである。
【0010】
本発明は、開示される特定のp38およびJNK MKK、ならびに本発明のMKKのポリヌクレオチドおよびアミノ酸配列から調製されるプローブまたは抗体を使用して同定および単離される近縁のMKKも含む。これは、例えば、ゲノム、cDNA、またはコンビナトリアルケミストリーのライブラリーを、開示されるMKKの核酸配列の全体または一部を持つプローブによってスクリーニングするなどの、標準的な技術を用いて実行できる。さらに本発明は、本明細書に記述されるMKKのアミノ酸配列の全体または一部を持つ合成ポリヌクレオチドも含む。
【0011】
「ポリペプチド」という用語は、長さまたは翻訳後修飾(例、グリコシル化またはリン酸化)に関わらず、任意のアミノ酸鎖を意味し、天然蛋白質ならびに合成または組換えのポリペプチドおよびペプチドを含む。
【0012】
ポリペプチドに関して用いる場合の「実質的に純粋」という用語は、天然に会合している蛋白質や天然に存在する有機分子を含まない、重量で少なくとも60%のポリペプチドを意味する。実質的に純粋なMKKポリペプチド(例、ヒト)は、重量で少なくとも75%、より好ましくは少なくとも90%、および最も好ましくは少なくとも99%、MKKポリペプチドである。実質的に純粋なMKKは、例えば、天然源からの抽出、MKKポリペプチドをコードする組換え核酸の発現、または蛋白質の化学合成によって得られる。純度は、例えば、カラムクロマトグラフィー、ポリアクリルアミドゲル電気泳動、またはHPLC分析のような、任意の適切な方法で測定できる。
【0013】
1つの局面において、本発明は、本発明のMKKをコードする単離ポリヌクレオチドを特徴とする。1つの態様では、ポリヌクレオチドは配列番号:1のヌクレオチド配列である。別の態様では、ポリヌクレオチドは、それぞれ配列番号:3、配列番号:5、配列番号:7、配列番号:9、配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、または配列番号:31のヌクレオチド配列である。
【0014】
本明細書で使用される「ポリヌクレオチド」とは、分離した断片、または、より大きな構築物の成分の形の、デオキシリボヌクレオチドまたはリボヌクレオチドの核酸配列を指す。本発明のポリペプチドの一部または全体をコードするDNAは、組換え転写ユニット中で発現できる合成遺伝子を提供するcDNA断片またはオリゴヌクレオチドから、組み立てることができる。本発明のポリヌクレオチド配列には、DNA、RNA、およびcDNA配列が含まれ、天然由来または当技術分野で公知の方法によって合成された合成配列であり得る。
【0015】
「単離された」ポリヌクレオチドとは、生物体の天然に存在するゲノムにおける配列から、何らかのやり方で分離された核酸分子である。したがって、「単離ポリヌクレオチド」という用語には、天然に存在しない任意の核酸分子が含まれる。したがって、この用語には、例えば、ベクター、自律的に複製するプラスミドもしくはウイルス、もしくは原核生物もしくは真核生物のゲノムDNAに組み込まれた組換えポリヌクレオチド、または他の配列とは独立した別個の分子として存在する組換えポリヌクレオチドが含まれる。また、別のポリペプチド配列をコードするハイブリッド遺伝子の一部である組換えDNAも含まれる。
【0016】
本発明の単離ポリヌクレオチド配列には、本明細書で説明するポリヌクレオチド配列に、ストリンジェントな条件下でハイブリダイズするポリヌクレオチド配列も含まれる。「ストリンジェントな条件」という用語は、本明細書に記述するような、ハイブリダイズするポリヌクレオチド配列間の特異性を保証するようなハイブリダイゼーション条件または、それよりも厳密な条件を意味する。当業者は、非特異的なハイブリダイゼーションの数を低下させ、相補性の高い配列のみが同定されるような、温度や塩濃度を含めたハイブリダイゼーション後の洗浄条件を選択することができる(Sambrookら(1989)「分子クローニング(Molecular Cloning)」第2版;Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY)。
【0017】
本発明の単離ポリヌクレオチド配列には、MKKをコードするポリヌクレオチドに相補的な配列(アンチセンス配列)も含まれる。アンチセンス核酸は、特定のmRNA分子の少なくとも一部に相補的なDNAまたはRNA分子である(Weintraub (1990) Scientific American 262:40)。本発明には、MKKポリペプチドの生産を阻害するすべてのアンチセンスポリヌクレオチドが含まれる。細胞中では、アンチセンス核酸は、対応するmRNAにハイブリダイズし、二重鎖分子を形成する。約15ヌクレオチドのアンチセンスオリゴマーは、簡単に合成され、標的MKK産生細胞に導入できるので、好ましい。アンチセンス法を用いて遺伝子の翻訳を阻害する方法は、当技術分野で公知であり、例えばMarcus-Sakura Anal. Biochem., 172:289 (1988)に記述されている。
【0018】
また、MKKのリボザイムヌクレオチド配列も、本発明に含まれる。リボザイムは、DNA制限エンドヌクレアーゼと同様な方法で、他の一本鎖RNAを特異的に切断する能力を持つRNA分子である。これらのRNAをコードするヌクレオチド配列を修飾することによって、RNA分子中の特異的なヌクレオチド配列を認識し、切断するように分子を操作できる(Cech (1988) J. Amer. Med. Assn. 260:3030)。この方法の大きな利点は、配列特異的であるため、特定の配列のmRNAのみが不活化されるということである。
【0019】
リボザイムには、テトラヒメナ型(Hasselhoff (1988) Nature 334:585)および「ハンマーヘッド」型の、2つの基本的なタイプがある。テトラヒメナ型リボザイムは、長さが4塩基の配列を認識するが、「ハンマーヘッド」型リボザイムは、長さが11〜18塩基の塩基配列を認識する。配列が長ければ長いほど、その配列が標的mRNA種のみに見られる可能性が高くなる。したがって、特定のmRNA種を不活化するためには、テトラヒメナ型リボザイムよりもハンマーヘッド型リボザイムの方が好ましく、短い認識配列よりも、18塩基の認識配列のほうが好ましい。
【0020】
MKKポリペプチドは、MKKポリペプチドのエピトープと免疫反応するか、結合する抗体を生産するためにも、使用できる。したがって、本発明の1つの局面は、本発明のMKKポリペプチドに対する抗体を特徴とする。本発明の抗体には、異なるエピトープ特異性を持つモノクローナル抗体のプールを含むポリクローナル抗体、および別個のモノクローナル抗体調製物が含まれる。モノクローナル抗体は、当技術分野で公知の方法によって、MKKポリペプチドの抗原含有断片から生産される(例えば、Kohlerら(1975) Nature 256:495参照)。
【0021】
本明細書で使用される「抗体」という用語は、エピトープ決定基に結合する能力のある、完全な分子、ならびにFa、F(ab')2、およびFvなどの、その断片を含む。MKKポリペプチドに特異的に結合する抗体は、完全なポリペプチドまたは目的の小さなペプチドを含む断片を、免疫抗原として使用して調製できる。動物の免疫に使用するポリペプチドまたはペプチドは、翻訳されたcDNA由来でも化学合成されたものでもよく、必要に応じてキャリア蛋白質に結合できる。ペプチドに化学的に結合されて一般的に使用されるキャリアには、ウシ血清アルブミンおよびチログロブリンが含まれる。その後、結合されたペプチドを用いて動物(例、マウス、ラットまたはウサギ)を免疫する。
【0022】
「特異的に結合する」分子(例、抗体)とは、例えばMKK7を含む生体試料などの試料中で、例えばMKK7のような特定のポリペプチドに結合するが、他の分子は実質的に認識しないか、または結合しない分子である。検出可能マーカーを含む化合物に結合した抗体(またはその断片)から成る構築物という表現には、化学的方法および組換え技術を含む、任意の技術により作製された構築物が含まれる。
【0023】
本発明は、MKKシグナル伝達経路の活性化を測定することによって、MKKを介する疾病のリスクを持つ患者を同定する方法も特徴とする。MKKシグナル伝達経路の活性化は、MKK合成;MKKアイソフォームの活性化;MKK基質p38またはJNKのアイソフォームの活性化;またはATF2、ATFa、CRE-BPaおよびc-Junのようなp38およびJNKの基質の活性化を、測定することにより決定できる。「JNK」または「JNKアイソフォーム」という用語には、JNK1、JNK2、およびJNK3が含まれる。本明細書で使用される「MKK基質」という用語には、MKKの基質およびMKKの基質の基質、例、p38、JNK、ATF2、およびc-Junも含まれる。
【0024】
1つの態様において、MKKシグナル伝達経路の活性化は、適当なMKKシグナル伝達経路基質(例、p38、JNKアイソフォーム、ATF2、ATFa、CRE-BPa、またはc-Junから選択される)の活性化を測定することによって決定される。MKK活性は、標識されたリン(例えば[32]Pまたは[33]P)の取り込みの割合を定量することによって決定される基質のリン酸化の割合によって測定される。これは、抗体のような、リン酸化に特異的な試薬を用いても測定できる。MKKの基質リン酸化の特異性は、p38の活性化、JNKの活性化、もしくはその両方を測定することによって、またはMKKリン酸化部位を持たない変異p38もしくはJNK分子を利用して調べられる。対照値と比較して基質のリン酸化が変化していることは、MKKシグナル伝達経路が変化したことを示し、患者のMKKを介する疾病のリスクが高まっていることを示す。MKKによるp38およびJNKの活性化は、MKKシグナル伝達の基質ATF2、またはATFaやCRE-BPaのような関連化合物と組み合わせたアッセイ法で検出できる。基質c-Junを用いても、活性化は検出できる。ATF2を解析に使用する際には、これは完全な蛋白質または完全な蛋白質の断片、例えば活性化ドメイン(残基1〜109、またはその一部)として存在する。ATF2は、MKK活性を測定する試験試料および[γ-32P]ATPと共に、ATF2がリン酸化されるのに十分な条件下でインキュベーションする。その後、ATF2を単離して、リン酸化の量を定量する。1つの特定の態様において、ATF2は免疫沈降によって単離され、SDS-PAGEによって解析され、オートラジオグラフィーによって検出される。
【0025】
別の態様において、MKKシグナル伝達経路の活性化は、試験試料中のMKK発現のレベルを測定することにより決定される。1つの特定の態様では、MKK発現のレベルは、ウエスタンブロット分析で測定される。試料中に存在する蛋白質は、ゲル電気泳動によって分別し、メンブレンに移し、MKKに対する標識抗体でプローブする。別の特定の態様では、MKK発現のレベルは、ノーザンブロット分析で測定する。細胞の総mRNAまたはリン酸化[ポリ(A)+]mRNAを試験試料から単離する。RNAは電気泳動によって分別し、メンブレンに移す。メンブレンは標識MKK cDNAでプローブする。別の態様いおいて、MKK発現は、発現したmRNAに対する定量的PCRによって測定される。
【0026】
本発明のMKKは、MKK活性を調節する試薬のスクリーニングに役立つ。MKKはリン酸化によって活性化される。したがって、1つの局面において、本発明は、試験試薬とMKKをインキュベートして、MKKの合成、リン酸化、機能、または活性に対する試験試薬の効果を測定することによって、MKK活性を調節する試薬を同定する方法を特徴とする。1つの態様において、試験試薬はMKKおよび[32]P-ATPと共にインキュベートされ、上述のようにMKKリン酸化の速度が決定される。別の態様では、MKKポリヌクレオチド発現ベクターによりトランスフェクトされた細胞と共に試験試薬をインキュベートし、上述のように、ノーザンブロット分析によってMKK転写に対する試験試薬の効果を測定する。別の態様では、MKK合成に対する試験試薬の効果は、MKKに対する抗体を用いてウエスタンブロット分析によって測定される。さらに別の態様では、MKK活性に対する試薬の効果は、試験試薬、[32]P-ATP、ならびにp38、JNK、およびATF2のうちの1つまたは複数を含むMKKシグナル伝達経路の基質と共に、MKKをインキュベートすることにより測定される。基質のリン酸化の割合は、上述のように決定される。
【0027】
「MKK活性の調節」という用語には、阻害または刺激効果が含まれる。
【0028】
本発明は、MKK活性を阻害する試薬のスクリーニングに特に有用である。そのような試薬は、例えば、炎症および酸化による損傷のような、MKKを介する疾病の治療または予防に役立つ。
【0029】
本発明はさらに、MKK活性を阻害する治療試薬の有効量を、必要としている患者に投与することにより、MKKを介する疾病を治療する方法を特徴とする。
【0030】
「MKKを介する疾病」とは、少なくとも一部は、MKKシグナル伝達経路の過剰な活性化の結果として起こる病的状態である。MKKシグナル伝達経路は、炎症およびストレスを含む、いくつかの要因により活性化される。MKKを介する疾病には、例えば、虚血性心疾患、熱または放射線(UV、X-線、γ、β等)による熱傷、腎不全、酸化ストレスまたはアルコールによる肝臓障害、呼吸障害症候群、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、およびその他の種類の炎症性疾病が含まれる。
【0031】
「治療試薬」とは、必要とする患者に投与された場合に、MKKを介する疾病に望ましい効果を与える任意の化合物または分子である。
【0032】
MKKを介する疾病には、増殖性疾患、特にストレスに関連した疾患がさらに含まれる。ストレスに関連したMKKを介する増殖性疾患の例には、乾癬、後天性免疫不全症候群、皮膚、骨髄、肺、肝臓、乳房、胃腸系、および尿生殖路の悪性腫瘍を含む、体の種々の組織の悪性腫瘍がある。好ましくは、治療試薬はMKKの活性または発現を阻害して、細胞の増殖を阻害するかアポトーシスを誘導する。
【0033】
「MKK活性を阻害する」治療試薬は、MKKを介するシグナル伝達経路に干渉する。例えば、治療試薬はMKKのプロテインキナーゼ活性を変化させたり、例えばMKK mRNAに結合できるアンチセンスポリヌクレオチドのように、MKKの転写または翻訳レベルを低下させたり、またはp38、JNK、もしくはATF2のMKKによるリン酸化を抑制することができ、その結果MKKを介するシグナル伝達経路を混乱させる。そのような試薬の例には、MKKポリペプチドに特異的に結合する抗体、およびMKKポリペプチド活性を競合阻害するMKKポリペプチド断片が含まれる。
【0034】
「MKK活性を増大させる」治療試薬は、MKKを介するシグナル伝達経路を補う。そのような試薬の例には、MKKポリペプチド自身が含まれ、これはMKKポリペプチドの発現不足、または変異MKKポリペプチドの発現に起因するMKKを介する疾病の場合に投与できる。さらに、MKKポリペプチドをコードするDNAの一部を、MKKポリペプチドの発現が不足している細胞に導入することもできる。
【0035】
「治療的有効量」とは、MKKを介する疾病に伴う症状を低下または予防するために十分な試薬の量である。
【0036】
本発明の方法によって同定されるMKKを介する疾病の治療のための治療試薬は、注射、注入、徐放性の注射または移植による非経口的方法を含む、当技術分野で公知のいくつもの方法で、患者の静脈内、腹腔内、筋肉内、皮下、または経皮的に投与される。表皮の疾患および上皮組織の疾患は、試薬の局所適用により治療される。試薬は、安定性と送達の効率を改善するために他の化合物と混合される(例、リポソーム、保存剤、またはジメチルスルホキシド(DMSO))。アンチセンス配列を含むポリヌクレオチド配列を、当技術分野で公知の技術を用いて治療用に投与し、MKKを介する疾病を患う患者の細胞中に導入することができる。これらの方法には、ウイルスベクター(例、レトロウイルス、アデノウイルス、ワクシニアウイルス、またはヘルペスウイルス)、コロイド分散、およびリポソームの使用が含まれる。
【0037】
本発明の材料は、MKKのレベルまたは活性の検出のためのキットの調製に、理想的に適している。したがって、本発明は、MKKに結合する抗体またはMKKポリヌクレオチドにハイブリダイズする核酸プローブ、および適当な緩衝液を含むキットを特徴とする。プローブまたはモノクローナル抗体は、MKKポリヌクレオチドまたは蛋白質への結合を検出するために標識することができる。1つの好ましい態様において、キットはMKKに対する標識抗体を特徴とする。
【0038】
特に定義しないかぎり、本明細書で使用される全ての技術および科学的用語は、本発明が属する技術分野の当業者が一般に理解するものと同一の意味を持つ。本発明の実施または試験には、本明細書に説明する方法および材料と類似または同等なものを使用することもできるが、適当な方法および材料を以下に記述する。本明細書に記載される出版物、特許出願、特許、および他の参考文献は全て、その全体が参照として組み入れられる。さらに、材料、方法、および実施例は説明のみのためであり、制限する意図はない。
【0039】
本発明の他の特徴および利点は、以下の詳細な説明および請求の範囲から明らかになると思われる。
【0040】
ヒトマイトジェン活性化プロテインキナーゼキナーゼ
本明細書に記述されるヒトMAPキナーゼキナーゼMKK3およびMKK4 (MKK3/4)、およびMKK7は、細胞表面から核までの特定の経路に沿った特異的なシグナルの伝達を仲介する。これらのシグナル伝達経路は、サイトカイン、UV照射、浸透圧ショック、および酸化ストレスのような要素によって開始する。MKK3/4、MKK6、およびMKK7が活性化されると、MAPキナーゼの活性化が起きる。p38はMKK3およびMKK4により活性化される。JNKはMKK4およびMKK7により活性化される。次に、p38およびJNKは、ATF2、ATFa、およびCRE-BPaのような関連する転写因子のグループを活性化する。次にこれらの転写因子は、特定の遺伝子の発現を活性化する。例えば、ATF2はヒトT細胞白血病ウイルス1(WagnerおよびGreen (1993) Science 262:395)、トランスフォーミング増殖因子b2(Kimら(1992)前記)、インターフェロンβ(Duら(1993) Cell 74:887)、およびE-セレクチン(DeLucaら(1994) J. Biol. Chem. 269:19193)の発現を活性化することが知られている。また、ATF2は、T細胞特異的エンハンサーの機能にも関係する(Georgopoulosら(1992) Mol. Cell. Biol. 12:747)。
【0041】
MAPキナーゼのJNKグループは、細胞が環境ストレスにさらされたり、炎症促進型サイトカインで細胞を処理すると活性化される(Guptaら(1994) EMBO J. 15:2760-2770; Derijardら(1991) Cell 76:1025-1037; Kyriakisら(1994) Nature 369:156-160; Slussら(1994) Mol. Cell. Biol. 14:8376-8384; Kallunkiら(1994) Genes & Dev. 8:2996-3007)。JNKシグナル伝達経路の標的には、転写因子ATF2およびc-jun(Whitmarsh & Davis (1996) J. Mol. Med. 74:589-607)が含まれる。これらの転写因子は、多くの遺伝子のプロモーター中のAP-1およびAP-1様部位に、ホモまたはヘテロダイマー複合体として結合するbZIPグループのメンバーである(Curran & Franza (1988) Cell 55:395-397)。JNKはATF2およびc-JunのNH2末端領域に結合し、各々の転写因子の活性化ドメイン内の2つの部位をリン酸化する(Derijardら(1994) Cell 76:1025-1037; van Damら(1995) EMBO J. 14:1798-1811; Livingstoneら(1995) EMBO J. 14:1785-1797)。このリン酸化によって、転写活性が上昇する(Whitmarsh、前記)。これらの生化学研究を合わせると、JNKシグナル伝達経路が、サイトカインおよび環境ストレスに応答したAP-1転写活性の調節に寄与していることが示される(Whitmarsh、前記)。JNKシグナル伝達経路がAP1転写活性の正常な調節に必要であることを示す遺伝的証拠が、この仮説を強く支持している(Yangら(1997) Proc. Natl. Acad. Sci. USA, 94:3004-3009)。
【0042】
JNKは、Thr-183およびTyr-185の二重のリン酸化によって活性化される(Derijard、前記)。MKK4(SEKIとしても知られる)は、JNKシグナル伝達経路の成分として同定された最初のMAPキナーゼキナーゼであった(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290; Sanchezら(1994) Nature 372:794-798)。MKK4がJNKをリン酸化し活性化することが生化学研究によって示されている(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290; Sanchezら(1994) Nature 372:794-798)。しかし、MKK4はp38 MAPキナーゼもリン酸化し活性化するので、MKK4の機能は、JNKシグナル伝達経路に限定されていない可能性がある(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290)。MKK4がJNKおよびp38 MAPキナーゼの両方を活性化するというこの特異性により、サイトカインや環境ストレスによって処理された細胞における、これらのMAPキナーゼの同格の活性化を担うと考えられる機構が提供される(Davis (1994) Trends Biochem. Sci. 19:470-473)。しかし、この同格の活性化は、常に観察されるとは限らない。例えば、肝臓におけるJNKの活性化は、p38 MAPキナーゼ活性の低下と相関している(Mendelsonら(1996) Proc. Natl. Acad. Sci. USA 93:12908-12913)。これらのデータは、MKK4の性質は、インビボにおけるJNKの調節を説明するには不十分であることを示唆している。
【0043】
ヒトMKKの単離は、実施例1、実施例22、Derijardら((1995) Science 267:682-685、参照として本明細書に組み入れられる)、およびRaingeaudら((1995) Mol. Cell. Biol. 16:1247-1255)に記述されている。酵母PBS2配列の特徴的な領域を用いて、ポリメラーゼ連鎖反応(PCR)プライマーがデザインされた。これらのプライマーを用いてヒト脳mRNAを増幅すると、特異的な産物が形成されたので、これらをプラスミドベクターにクローニングし、配列の決定を行った。ヒトプロテインキナーゼをコードする2つの異なる相補的DNA(cDNA)が同定された:1つは36 kD蛋白質(MKK3)をコードし、1つは44 kD蛋白質(MKK4)をコードしていた。MKK4には、α、β、およびγと同定された、NH2末端が僅かに異なる3つのアイソフォームが含まれている。MKK3(配列番号:2)、MKK4-α(配列番号:6)、MKK4-β(配列番号:8)、およびMKK4-γ(配列番号:10)のアミノ酸配列は、図1に示されている。MKK3(図4)、MKK6(図5)、MKK4-α(図6)、MKK4-β(図7)、およびMKK4−γ(図8)の核酸およびアミノ酸配列も示されている。MKK6は、MKK3とのクロスハイブリダイゼーションによって、ヒトの骨格筋ライブラリーから単離された。N末端の違いを除いて、MKK6はMKK3と相同性が高い。存在するヒトMKK3およびMKK4の他のアイソフォームは、実施例1に説明する方法によって同定できる。
【0044】
これらのヒトMKKアイソフォームの発現を、8人の成人組織から単離されたmRNAのノーザン(RNA)ブロット分析によって調べた(実施例2)。両方のプロテインキナーゼとも、ヒトの組織で広範囲に発現されており、骨格筋組織で最も発現が多いことが分かった。
【0045】
MKK3の基質特異性は、エピトーグタグを付けた組換えMAPキナーゼ(JNK1、p38およびERK2)を基質として用いたインビトロのリン酸化解析で調べた(実施例3)。MKK3はp38をリン酸化したが、JNK1またはERK2はリン酸化しなかった。p38のリン酸化アミノ酸分析によって、ホスホスレオニンおよびホスホチロシンの存在が示された。p38の変異分析によって、Thr180およびTyr182のリン酸化部位を、それぞれAlaおよびPheによって置換すると、p38のリン酸化が阻止されることが示された。これらの結果は、MKK3がインビトロでp38 MAPキナーゼキナーゼとして働くことを立証するものである。
【0046】
MKK4の基質特異性のインビトロ実験は、実施例4に記載されている。[γ-32P]ATP、およびJNK1、p38、またはERK2と共にインキュベーションされたMKK4は、p38およびJNK1の両方をリン酸化することが分かった。MKK4によるJNKおよびp38の活性化もまた、MKK4を野生型または変異型JNK1またはp38とインキュベーションすることにより調べられた。p38の基質ATF2は、各解析に含まれていた。MKK4は、MKK3よりも自己リン酸化が少ないことが分かった。MKK4は、活性化されたMAPキナーゼの基質でもあることが分かった。MKK3とは異なり、MKK4はJNK1も活性化することが分かった。野生型JNK1とインキュベートされたMKK4は、ATF2のリン酸化を上昇させたが、変異型JNK1とインキュベーションした場合には上昇させなかった。これらの結果は、MKK4が、MAPキナーゼのJNKサブグループもリン酸化するp38 MAPキナーゼキナーゼであることを立証する。
【0047】
UV刺激を受けたMKK3によるインビボのp38の活性化は、実施例5に記載されている。MKK3を発現する細胞を、UV照射の存在下または非存在下で露出した。免疫沈降によってMKK3を単離し、基質のp38またはJNKを用いてプロテインキナーゼアッセイ法を行なった。一部のアッセイ法では、p38およびJNKの基質として、ATF2が含まれていた。非活性化培養COS細胞由来のMKK3は、MKK3の基礎活性による、p38 MAPキナーゼのリン酸化を低レベルで誘導した。UV照射した細胞のMKK3は、p38 MAPキナーゼのリン酸化を増加させたがJNK1のリン酸化は増加させなかった。p38活性の上昇は、基質としてATF2を含めたアッセイ法でも検出された。これらの結果は、MKK3がUV照射によって活性化されることを立証する。
【0048】
MKK3およびMKK4の発現のp38活性に対する効果は、COS-1細胞で調べられた(実施例6)。p38およびMEK1、MKK3、またはMKK4をコードするベクターで細胞をトランスフェクトした。細胞の一部は、EGFまたはUV照射の処理をした。免疫沈降によってp38を単離し、[γ-32P]ATPおよびATF2によって活性を解析した。ERK活性化因子MEK1の発現は、p38によるATF2のリン酸化を変化させなかった。これとは対照的に、MKK3またはMKK4の発現は、p38 MAPキナーゼの活性を上昇させた。MKK3およびMKK4によるp38の活性化は、UV照射細胞で観察されたものと類似しており、EGF処理細胞で検出されるものよりもはるかに強かった。このようなインビトロの結果は、MKK3とMKK4がインビボでp38を活性化させるという証拠となる。
【0049】
JNK1によるATF2の調節の可能性を調べるために、一連の実験が行なわれた。これらの実験は、参照として本明細書に組み入れられるGuptaら(1995) Science 267:389-393に記述されている。ATF2のリン酸化に対するUV照射の効果が、エピトープタグを付けたJNK1の存在下および非存在下でトランスフェクトされたCOS-1細胞において調べられた(実施例7)。細胞にUV照射をし、JNK1およびJNK2は、基質ATF2を用いたゲル内プロテインキナーゼアッセイ法によって可視化した。JNK1およびJNK2は、UV照射をしたトランスフェクト細胞および非トランスフェクト細胞で検出された;しかし、JNK1レベルは、トランスフェクト細胞で高かった。これらの結果は、ATF2がJNK1およびJNK2プロテインキナーゼの基質であり、これらのプロテインキナーゼはUV線に露出された細胞で活性化されることを示す。
【0050】
JNK1によるATF2のリン酸化部位は、欠失分析によって調べられた(実施例8)。連続的なNH2末端ドメイン欠失GST-ATF2融合蛋白質を作製し、UV照射細胞から単離されたJNK1によるリン酸化を調べた。その結果、ATF2のNH2末端ドメインのリン酸化には、JNK1はATF2の残基1〜60の存在を必要とすることが示された。
【0051】
JNK1の結合に必要なATF2の残基も同様に調べた。JNK1を、固定化したATF2と共にインキュベートし、結合しなかったJNK1を念入りに洗浄して除去し、結合したJNK1を[γ-32P]ATPとのインキュベーションによって検出した。結果は、JNK1による結合とリン酸化のためには、ATF2の残基20〜60が必要なことを示している。ATF2と55 kD JNK2プロテインキナーゼとの間の同様な結合相互作用も観察された。
【0052】
JNK1によるリン酸化は、ATF2の電気泳動の移動度を低下させることが示された(実施例9)。全長ATF2分子(残基1〜505)のリン酸化アミノ酸分析によって、JNKはThrおよびSer残基の両方をリン酸化することが示された。ThrおよびSerリン酸化の主要な部位は、それぞれNH2およびCOOH末端ドメインに存在していた。NH2末端のリン酸化部位は、リン酸化ペプチドマッピングと変異解析によって、Thr69およびThr71と同定された。これらのThrリン酸化部位は、ATF2内でJNK結合に必要なサブドメイン(残基20〜60)とは別の領域に存在している。
【0053】
ATF2のリン酸化で見られる電気泳動の移動度の低下を、さらに詳しく調べられた(実施例10)。JNK1を発現するCHO細胞に、UV照射、炎症促進型サイトカインであるインターロイキン-1(IL-1)処理、または血清処理をして、JNK1を活性化した。JNK1に活性化されたATF2の電気泳動の移動度の低下は、UV照射とIL-1処理をした細胞で観察された。血清で細胞を処理すると、これよりも小さな効果が見られた。これらの結果は、ATF2がJNK1のインビボでの基質であることを示す。
【0054】
野生型(Thr69、71)ATF2およびリン酸化欠損(Ala69、71)ATF2分子の性質に対する、UV照射の効果を調べた(実施例11)。UVに露出すると、内因性および過剰発現した野生型のATF2の両方の電気泳動の移動度が低下した。電気泳動の移動度のこの変化は、ATF2のリン酸化の増加を伴っていた。電気泳動の移動度のシフトおよびリン酸化の増加のいずれも、ATF2内でThr69およびThr71をAlaで置換すると阻止された。この変異により、インビボにおけるATF2のThr残基のリン酸化も阻止された。
【0055】
GAL4 DNA結合ドメイン、および野生型または変異型ATF2からなる融合蛋白質の転写活性を調べた(実施例12)。ATF2のThr69および/またはThr71における点変異は、野生型分子と比較してATF2の転写活性を有意に低下させ、これらの部位におけるリン酸化が、活性に生理的に関連していることを示した。
【0056】
JNK1がATF2のNH2末端活性化ドメインに結合することから(実施例8に記述)、触媒作用の不活性なJNK1分子が、野生型JNK1分子のドミナントな阻害剤として機能することが示唆された。この仮説は、触媒作用の不活性なJNK1分子のATF2機能に対する効果を調べて、検証された(実施例13)。触媒作用の不活性なJNK1変異体は、Thr183およびTyr185活性化リン酸化部位を、それぞれAlaおよびPheで置換して作製された(Ala183、Phe185、「ドミナントネガティブ」と呼ばれる)。野生型JNK1の発現は、血清刺激されたATF2の転写活性を僅かに上昇させた。これとは対照的に、ドミナントネガティブJNK1は、対照および血清刺激されたATF2活性の両方を阻害した。この阻害効果は、JNK1変異体がATF2活性化ドメインに非生産的に結合し、ATF2のリン酸化を効果的に阻害することによる。
【0057】
癌抑制遺伝子産物Rbは、ATF2に結合し、ATF2に刺激される遺伝子発現を上昇させる(Kimら(1992) Nature 358:331)。同様に、アデノウイルスのオンコプロテインE1AはATF2のDNA結合ドメインと会合し、ATF2のNH2末端活性化ドメインが必要な機構によって、ATF2に刺激される遺伝子発現を上昇させる(LiuおよびGreen (1994) Nature 368:520)。ATF2の転写活性は、ルシフェラーゼレポーター遺伝子システムを用いて、野生型または変異型ATF2分子を発現する、対照、Rb処理、およびE1A処理細胞において調べられた(実施例14)。RbとE1Aは、野生型および変異型ATF2の両方の、ATF2に刺激される遺伝子発現を上昇させることが分かった。しかし、変異型ATF2では、野生型ATF2よりも、レポーター遺伝子発現のレベルは低かった。これらの結果を合わせると、最大の転写活性を得るためには、ATF2リン酸化(Thr69およびThr71での)に加えて、RbまたはE1Aのいずれかが必要であることが示される。したがって、RbおよびE1AはATF2のリン酸化と協調して働き、転写活性を制御している。
【0058】
P38活性化の作用を調べ、p38 MAPキナーゼ経路とERKおよびJNKシグナル伝達経路との関係を確立するために、一連の実験が行われた(Raingeaudら(1995) J. Biol. Chem. 270:7420、参照として本明細書に組み入れられる)。まず、ERKおよび/またはJNKグループのMAPキナーゼの基質であることが示されている蛋白質と共にp38をインキュベートすることにより、p38の基質特異性を調べた(実施例15)。本発明者らは、MBP(Ericksonら(1990) J. Biol. Chem. 265:19728)、EGF-R(Northwoodら(1991) J. Biol. Chem. 266:15266)、細胞質ホスホリパーゼA2 (cPLA2) (Linら(1993) Cell 72:269)、c-Myc(Alvarezら(1991) J. Biol. Chem. 266:15277)、IκB、c-Jun、および野生型(Thr69、71)または変異(Ala69、71)ATF2のリン酸化を調べた。p38はMBPおよびEGF-Rをリン酸化し、それよりも低いレベルでIκBをリン酸化したが、他のERK基質はリン酸化せず、このことからp38の基質特異性が、ERKおよびJNKグループのMAPキナーゼとは異なることが示される。野生型ATF2はp38の優れた基質であるが、変異型ATF2(Ala69、71)はそうでないことが判明した。
【0059】
p38によるATF2のリン酸化は、ポリアクリルアミドゲル電気泳動において、ATF2の電気泳動の移動度のシフトを伴った。本発明者らは、p38によるATF2のリン酸化部位は、JNK1と同じであるという仮説を、Thr69およびThr71をAla (Ala69、71)で置換することにより、検証した。p38は変異ATF2をリン酸化しないことが分かったが、これはp38がATF2のNH2末端活性化ドメイン内のThr69およびThr71をリン酸化することを示す。
【0060】
JNK1またはp38を発現する細胞の抽出物を、エピトープのみ(GST)またはGST- ATF2(活性化ドメインを含む残基1〜109)とインキュベートして、ATF2に対するJNKとp38の結合の比較を行なった(実施例16)。結合したプロテインキナーゼは、ウエスタンブロット分析によって検出した。結果は、p38とJNKの両方がATF2の活性化ドメインに結合することを示す。
【0061】
EGFおよびホルボールエステルは、ERKシグナル伝達経路の強力な活性化因子であり(EganおよびWeinberg (1993) Nature 365:781)、MAPキナーゼのERKサブグループを最大限に活性化する。しかし、これらの処理は、JNKプロテインキナーゼの活性を僅かに上昇させるのみである(Derijardら(1994) 上記;Hibiら(1993)上記)。EGFまたはホルボールエステル、およびUV照射、浸透圧ショック、インターロイキン-1、腫瘍壊死因子、およびLPSのp38活性に対する効果を全て試験した(実施例17)。重要なことに、EGFおよびホルボールエステルは、p38のプロテインキナーゼ活性を中程度にしか上昇させなかったが、環境ストレス(UV照射および浸透圧ショック)は、p38とJNKの両方の活性を著しく上昇させた。p38とJNKのいずれも、炎症促進型サイトカイン(TNFおよびIL-1)または内毒素LPSで処理された細胞において活性化された。これらの結果を合わせると、p38は、JNKと同様に、ストレスに誘導されるシグナル伝達経路によって活性化されることが示される。
【0062】
ERKおよびJNKは、それぞれ、モチーフThr-Glu-TyrおよびThr-Pro-Tyr内の2重のリン酸化によって活性化される。これとは対照的に、p38は関連配列Thr-Glu-Tyrを持っている。このモチーフがp38の活性化に関連しているかどうかを調べるために、Thr-Gly-TyrをAla-Gly-Pheで置換した効果を調べた(実施例18)。野生型(Thr180、Tyr182)または変異型p38(Ala180、Phe182)を発現する細胞に対するUV照射の効果を調べた。抗ホスホチロシン抗体を用いたウエスタンブロット分析では、UV照射を行なうとp38のTyrリン酸化が上昇することが示された。Tyrリン酸化の上昇は、[γ-32P]リン酸塩標識細胞から単離されたp38のホスホ化アミノ酸分析によって、確認された。この分析では、UV照射がp38のThrリン酸化を上昇させることも示された。重要なことに、Thr180およびTyr182のリン酸化の上昇は、Ala180/Phe182変異によって、阻止された。この結果は、UV照射が二重のリン酸化によるp38の活性化を増加させることを示す。
【0063】
最近、マイトジェンに誘導される二重特異性を持つフォスファターゼMKP1およびPAC1によって、ERK活性が調節されていることが示された(Wardら(1994) Nature 367:651)。二重リン酸化によりp38が活性化される(実施例18)ため、p38も二重特異性ホスファターゼによって調節されているという可能性が生じる。本発明者らは、p38 MAPキナーゼ活性に対するMKP1およびPAC1の効果を調べた(実施例19)。ヒトMKP1およびPAC1を発現する細胞を、UV照射処理をしたものとしないものとで、p38活性を測定した。PAC1またはMKP1の発現は、p38活性を阻害することが分かった。MKP1の阻害効果は、PAC1よりも大きかった。これとは対照に、触媒作用の不活性な変異ホスファターゼ(変異PAC1 Cys257/Ser)でトランスフェクトされた細胞は、p38 MAPキナーゼを阻害しなかった。これらの結果は、p38が二重特異性ホスファターゼPAC1およびMKP1によって調節されることを示す。
【0064】
p38 MAPキナーゼの細胞内分布は、間接免疫蛍光顕微鏡法によって調べた(実施例20)。エピトープタグを付けたp38 MAPキナーゼは、M2モノクローナル抗体を用いて検出した。エピトープタグを付けたp38 MAPキナーゼでトランスフェクトされた細胞の特異的な染色は、細胞表面、細胞質、および核で観察された。UV照射後には、細胞表面および核のp38 MAPキナーゼには著しい変化は観察されなかったが、細胞質p38 MAPキナーゼの核周囲領域への局在の増加が検出された。
【0065】
高浸透圧培地によるJNKの活性化を研究するために、一連の実験を行った(実施例21)。これらの実験は、参照として本明細書に組み入れられるGalcheva-Gargovaら(1994) Science 265:806に報告された。エピトープタグを付けたJNK1を発現するCHO細胞は、0〜1000 mMソルビトールとインキュベートされ、基質c-Junを用いて免疫複合体キナーゼ解析によって、JNK1活性が測定された。100 mMソルビトールで1時間インキュベートされた細胞で、JNK1活性の上昇が観察された。300 mMソルビトールへの露出では、5分以内にJNK1活性の上昇が観察された。最大の活性は、浸透圧ショックの15〜30分後に観察され、この後は、JNK1活性が徐々に低下した。浸透圧ショックによるJNKの活性化は、野生型(Thr183、Tyr185)または変異(Ala183、Phe185)JNK1を発現する細胞で調べた。JNK1活性は、300 mMソルビトールの存在下または非存在下での15分のインキュベーション後に測定された。野生型JNK1を発現する細胞では、JNK1活性の上昇が見られたが、変異JNK1を発現する細胞では見られなかった。これらの結果は、高浸透圧培地に露出された哺乳類培養細胞において、JNKシグナル伝達経路が活性化されることを示す。
【0066】
上述の実験の結果は、図3に示されており、これはERK、p38、およびJNK MAPキナーゼシグナル伝達経路を模式的に表わしている。細胞をEGFまたはホルボールエステルで処理すると、ERKが強力に活性化される。これとは対照的に、p38はこのような条件では僅かに活性化されるのみである(実施例15)。しかし、UV照射、浸透圧ショック、および炎症性サイトカインは、p38活性を著しく上昇させる。ERKとp38の活性化パターンのこのような違いは、これらのMAPキナーゼが異なるシグナル伝達経路によって調節されていることを示唆する。これらのシグナル伝達経路が別のものであることの分子的基礎は、ERKを活性化するMAPキナーゼキナーゼ(MEK1およびMEK2)とp38(MKK3、MKK4、およびMKK6)が異なるという証明によって立証される。
【0067】
マウスおよびヒトMKK7の単離は、実施例22に記述されている。ショウジョウバエMAPキナーゼキナーゼhep配列の特徴的な領域を用いて、ポリメラーゼ連鎖反応(PCR)のプライマーがデザインされた。これらのプライマーを用いてマウス精巣mRNAを増幅すると、特異的な産物が形成され、これはプラスミドベクターにクローニングされ、配列決定が行なわれた。hepに関連する1つの配列が同定され、マウス精巣ライブラリーのスクリーニングに使用された。プロテインキナーゼをコードする5つのDNA(cDNA)が同定された:1つはMAPプロテインキナーゼキナーゼ(MKK7)をコードしていた。他は種々のスプライシング変異体をコードしていた:MKK7b(部分配列が図11に記載されている)、MKK7c(図13)、MKK7d(図14)、MKK7e(図15)。MKK7(配列番号:18)およびhep(配列番号:21)の推定アミノ酸配列は図9に示されており、MAPキナーゼキナーゼMEK1(配列番号:11)、MEK2(配列番号:12)、MKK3(配列番号:2)、MKK4(配列番号:10)、MKK5(配列番号:22)、およびMKK6(配列番号:4)と比較されている。ヒトMKK7は、全長(マウス)MKK7 cDNAプローブを用いてヒトcDNAライブラリーをスクリーニングして、同定された。同定された部分配列(3'末端を欠く)は、マウスMKK7cと相同である。
【0068】
MKK7およびMKK4アイソフォームの発現は、8つのマウス組織から単離されたポリA+ mRNAのノーザン(RNA)ブロット分析によって調べた(実施例23)。両方のプロテインキナーゼが、広範囲で発現されていることが判明した。
【0069】
MKK7の基質特異性は、エピトープタグを付けた組換えMAPキナーゼ(JNK1、p38、およびERK2)を基質としたインビトロリン酸化アッセイ法で調べた(実施例24)。MKK7はJNKをリン酸化したが、p38もERK2もリン酸化しなかった。MKK7は、p38およびJNK1によってリン酸化された。
【0070】
MKK7は、インビボでJNKプロテインキナーゼを特異的に活性化することが分かった(実施例25)。CHO細胞に、エピトープタグを付けたMAPキナーゼ(JNK1、p38、またはERK2)と、空の発現ベクターまたはMKK1、MKK4、MKK6、もしくはMKK7をコードする発現ベクターとを、コトランスフェクションし、リン酸化反応の産物を分析した。MKK7はJNK1しか活性化しなかったが、その程度はMKK4よりも高かった。
【0071】
MKK7がAP-1転写活性を上昇させるかどうかを検証するために、コトランスフェクション解析を実施した(実施例26)。MKK7とJNKとを共に発現させると、AP-1レポーター遺伝子発現が上昇し、それはMKK4およびJNKで見られる上昇よりも大きかった。レポーター遺伝子にATF2を使った場合も、同様な結果が観察された。また、MKK7単独で、ATF2の発現を増加させることができた(図16)。
【0072】
MKKアイソフォームは、MKK活性を調節する試薬のスクリーニングに有用である。実施例の後の用途の項には、MKK活性を阻害または活性化する能力のある試薬の同定方法が記述されている。
【0073】
ヒトMKKアイソフォームおよびMKKが仲介するシグナル伝達経路を見出すことは、MKKを介する疾患の治療にとって、臨床的意義がある。MKKアイソフォームの1つの用途は、MKK-MAPキナーゼ-ATF2経路の活性化を阻害または予防できる試薬をスクリーニングするための方法における使用である。
【0074】
実施例
以下の実施例は、本発明を例証するためのもので、制限するものではない。
【0075】
実施例1. MKKプロテインキナーゼ
MKK3およびMKK4の一次配列は、ヒト胎児脳ライブラリーから単離されたcDNAクローンの配列から、推定された。
【0076】
PBS2(Brewsterら(1993) Science 259:1760; Maedaら(1994) Nature 369:242)の配列に基づいて、プライマーTTYTAYGGNGCNTTYTTYATHGA(配列番号:14)およびATBCTYTCNGGNGCCATKTA(配列番号:15)がデザインされた。プライマーは、ヒト脳mRNAを鋳型としたPCR反応で使用された。PBS2関連プロテインキナーゼの断片をコードする2つの配列が同定された。全長ヒトcDNAクローンは、ヒト胎児脳ライブラリーのスクリーニングで単離された(Derijardら(1995) Science 267:682-685)。cDNAクローンは、アプライド バイオシステムズ(Applied Biosystems)モデル373Aを用いて配列を決定した。MKK3(2030塩基対(bp))およびMKK4(3576 bp)について得られた最長のクローンは、これらのプロテインキナーゼのコード領域全体を含んでいた。
【0077】
MKK3(配列番号:2)およびMKK4-α(配列番号:6)の一次構造は、図1に示されている。インフレームの停止コドンがMKK3 cDNAの5'非翻訳領域に存在するが、MKK4 cDNAの5'領域にはない。表示されたMKK4蛋白質配列は、2番目のインフレーム開始コドンから開始する。
【0078】
これらの配列を、ヒトMAPキナーゼMEK1(配列番号:11)およびMEK2(配列番号:12)(ZhengおよびGuan (1993) J. Biol. Chem. 268:11435)ならびに酵母MAPキナーゼキナーゼPBS2(配列番号:13)(BoguslawaskiおよびPolazzi (1987) Proc. Natl. Acad. Sci. USA 84:5848)と比較した(図1)。キナーゼとヒトMKK3(サブドメインIとXIの間)の同一性および類似性は、BESTFITプログラム(バージョン7.2、Wisconsin genetics Computer Group)によって計算した(同一性の割合/類似性の割合):MEK1, 41%/63%, MEK2, 41%/62%; MKK4α, 52%/73%; およびPBS2, 40%/59%。キナーゼとヒトMKK4αの同一性および類似性は以下のとおりであると計算された(同一性の割合/類似性の割合):MEK1, 44%/63%, MEK2, 45%/61%; MKK3, 52%/73%; およびPBS2, 44%/58%。
【0079】
MKK3およびMKK4γのcDNA配列は、それぞれ寄託番号L36719およびL36870でジーンバンク(GenBank)に寄託された。MKK4γcDNA配列には、異なるスプライシング部位からインビボで生成するMKK4αおよびMKK4βのcDNA配列が含まれている。当業者は、寄託されたcDNA配列から、MKK3およびMKK4アイソフォームのアミノ酸配列を簡単に決定できる。
【0080】
実施例2. 成人組織におけるMKK3およびMKK4 mRNAの発現
ヒトの心臓、脳、胎盤、肺、肝臓、筋肉、腎臓、および膵臓組織から単離されたポリアデニル化[ポリ(A)+]mRNA (2μg)を用いて、ノーザンブロット分析が行われた。変性アガロースゲル電気泳動によってmRNAは分別され、ナイロンメンブレンに移された。ブロットは、[α-32P]ATP(デオキシアデノシン3リン酸)(Amersham International PLC)を用いたランダムプライミングによって標識されたMKK3およびMKK4 cDNAでプローブした。MKK3およびMKK4は、調べたすべての組織で発現されていた;MKK3およびMKK4の発現が最高だったのは、骨格筋組織だった。
【0081】
ヒトと酵母のMAPキナーゼキナーゼグループのメンバー間の関係は、樹状図で示されている(図2)。MKK3/4は、ヒトMAPキナーゼキナーゼの独特なサブグループを形成する。
【0082】
実施例3. MKK3によるp38 MAPキナーゼのインビトロリン酸化
GST-JNK1およびGST-ERK2は記述されている(Derijardら(1994)上記;Guptaら(1995) Science 267: 389; WartmannおよびDavis (1994) J. Biol. Chem. 269:6695、各々は参照として本明細書に組み入れられる)。GST-p38 MAPキナーゼは、発現ベクターpGSTag(Dressierら(1992) Biotechniques 13:866)およびp38 MAPキナーゼcDNAのコード領域を含むPCR断片から、調製された。GST-MKK3およびMKK4は、pGEX3X(Pharmacia-LKB Biotechnology)およびMKK3およびMKK4 cDNAのコード領域を含むPCR断片から、調製された。GST融合蛋白質は、GSHアガロース(SmithおよびJohnson (1988) Gene 67:31)を用いたアフィニティークロマトグラフィーによって、精製された。発現ベクターpCMV-Flag-JNK1およびpCMV-MEK1は、記述されている(Derijardら(1994)上記;WartmannおよびDavis (1994)上記)。プラスミドpCMV-Flag-p38 MAPキナーゼは、発現ベクターpCMV5(Anderssonら(1989) J. Biol. Chem. 264:8222)およびp38 MAPキナーゼcDNAから調製された。MKK3およびMKK4の発現ベクターは、cDNAをpCDNA3(Invitrogen)のポリリンカーにサブクローニングして、調製された。Flagエピトープ(Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys(配列番号:16);Immunex、ワシントン州シアトル)は、挿入重複PCR(Hoら(1989) Gene 77:51)によって、キナーゼのコドン1と2の間に挿入された。
【0083】
プロテインキナーゼアッセイ法は、キナーゼ緩衝液(25 mM 4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸、pH 7.4、25 mM β-グリセロリン酸塩、25 mM MgCl2、2 mMジチオスレイトール、および0.1 mM オルトバナジン酸塩)中で実行された。組換えGST-MKK3は、[γ-32P]ATPおよび緩衝液、GST-JNK1、GST-p38 MAPキナーゼ、またはGST-ERK2とインキュベートされた。アッセイ法は、最終容量25μlの中に、1 μgの基質蛋白質および50 μM [γ-32P]ATP(10 Ci/mmol)を添加して開始された。25℃で30分後に、レムリ(Laemmli)サンプル緩衝液を添加して反応を終了させた。SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)後に、オートラジオグラフィーによって基質蛋白質のリン酸化を調べた。部分酸加水分解および薄層クロマトグラフィーによって、ホスホアミノ酸分析を行った(Derijardら(1994)上記;Alvarezら(1991) J. Biol. Chem. 266:15277)。すべてのグループでMKK3の自己リン酸化が観察された。MKK3は、p38 MAPキナーゼをリン酸化したが、JNK1またはERK2はしなかった。
【0084】
同様な挿入重複PCR手法を用いて、p38のThr180およびTyr182を、それぞれAlaおよびPheで置換した。すべてのプラスミドの配列は、アプライド バイオシステムズ(Applied Biosystems)モデル373Aを用いた自動シーケンシングによって、確認された。GST-MKK3は[γ-32P]ATPおよび緩衝液、野生型GST-p38 MAPキナーゼ(TGY)または変異GST-p38 MAPキナーゼ(AGF)とインキュベートされた。リン酸化された蛋白質は、SDS-PAGEで解析され、オートラジオグラフィーで検出された。野生型p38のリン酸化のみが観察された。
【0085】
実施例4. MKK4によるJNKおよびp38 MAPキナーゼのインビトロでのリン酸化および活性化
プロテインキナーゼアッセイ法は、実施例3のように行なわれた。組換えGST-MKK4は、[γ-32P]ATPおよび緩衝液、GST-JNK1、GST-p38 MAPキナーゼ、またはGST-ERK2とインキュベートされた。JNK1およびp38は、リン酸化され、またJNK1およびp38と共にインキュベートされたMKK4もリン酸化された。
【0086】
GST-MKK4は[γ-32P]ATPおよび緩衝液、野生型JNK1(Thr183、Tyr185)、または変異GST-JNK1(Ala183、Phe185)とインキュベートした。各インキュベーションには、JNK1の基質ATF2(Guptaら(1995)上記)も含まれていた。ATF2はMKK4および野生型JNK1の存在下で、リン酸化された。これらの結果は、MKK4がp38とJNK1の両方をリン酸化し、活性化することを立証する。
【0087】
実施例5. UV刺激を受けたMKK3によるp38 MAPキナーゼのリン酸化および活性化
エピトープタグを付けたMKK3を、ウシ胎仔血清(5%)(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で維持したCOS-1細胞で発現させた。細胞は、製造元(Gibco-BRL)の推奨する方法にしたがって、リポフェクタミン試薬を用いてトランスフェクトし、記述されたようにUV照射またはEGFで処理した(Derijardら(1994)上記)。
【0088】
細胞は、UV-Cの存在下または非存在下に露出した(40 J/m2)。細胞は溶解緩衝液(20 mM Tris, pH 7.4, 1% 登録商標TRITON X-100, 10%グリセロール、137 mM NaCl, 2 mM EDTA, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸ナトリウム、1 mMフェニルメチルスルホニルフルオリド、およびロイペプチン(10μg/ml))で溶解し、4℃で100,000 x gで15分間遠心した。MKK3は免疫沈降によって単離された。エピトープタグを付けたプロテインキナーゼは、プロテインGセファロース(Pharmacia-LKB Biotechnology)に結合したFlagエピトープ(IBI-Kodak)に対するM2抗体と、4℃で1時間インキュベートした。免疫沈降物は、溶解緩衝液で2回、キナーゼ緩衝液で2回洗った。
【0089】
プロテインキナーゼアッセイ法は、基質GST-p38 MAPキナーゼまたはJNK1を用いて行なった。一部のアッセイ法にはATF2が含まれていた。MKK3による基礎レベルのp38 MAPキナーゼリン酸化が観察された。UV照射を行なうと、p38 MAPキナーゼのリン酸化が増加したが、JNK1は増加しなかった。p38 MAPキナーゼ活性が増加すると、ATF2のリン酸化が増加した。
【0090】
実施例6. MKK3またはMKK4を発現する細胞におけるp38 MAPキナーゼの活性化
COS-1細胞はエピトープタグを付けたp38 MAPキナーゼと、空の発現ベクターまたはMEK1、MKK3、またはMKK4αをコードする発現ベクターでトランスフェクトした。培養物の一部は、UV照射(40 J/m2)または10 nM EGFで処理した。p38 MAPキナーゼは、モノクローナル抗体M2による免疫沈降で単離し、プロテインキナーゼ活性は、[γ-32P]ATPおよびATF2を基質として用いて、免疫複合体で測定された。リン酸化反応の産物は、SDS-PAGE後にオートラジオグラフィーによって可視化した。対照のMEK1またはEGF処理群ではATF2はリン酸化されなかったが、MKK3、MKK4、およびUV照射群ではリン酸化された。ATF2のMKK3およびMKK4によるリン酸化は、UV照射細胞から単離されたp38 MAPキナーゼで見られるものと類似していた。
【0091】
実施例7. JNK1およびJNK2によるATF2のリン酸化
COS-1細胞は、ウシ血清アルブミン(5%)(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で維持された。[32] Pによる代謝標識は、[32]オルトリン酸塩(2 mCi/ml)(Dupont-NEN)を添加したリン酸塩フリーのダルベッコ変法イーグル培地(Flow Laboratories Inc.)中で、3時間細胞をインキュベートして行なった。COS-1細胞は、エピトープタグを付けたJNK1の存在下(JNK1)または非存在下(モック)でトランスフェクトした。JNK1 cDNAをコードするプラスミド発現ベクターは、以前に記述されている(Derijardら(1994) Cell 76:1025、参照として本明細書に組み入れられる)。プラスミドDNAは、リポフェクタミン法(Gibco-BRL)によって、COS-1細胞にトランスフェクトした。48時間インキュベーションした後、培養物の一部に40 J/m2のUV照射を行ない、37℃で1時間インキュベートした。
【0092】
細胞は、20 mM Tris, pH 7.5, 25 mMβ-グリセロリン酸塩、10%グリセロール、1% 登録商標Triton X-100, 0.5% (w/v) デオキシコール酸塩、0.1% (w/v) SDS, 0.137 M NaCl, 2 mM ピロリン酸塩、1 mMオルトバナジン酸塩、2 mM EDTA, 10μg/mlロイペプチンおよび1 mM PMSF中で溶解された。可溶性抽出物は、4℃で20分間微量遠心して調製された。組換えJNK1を抗原として調製されたウサギ抗血清を用いた反応によって、JNK1免疫沈降物も調製された。
【0093】
ゲル内プロテインキナーゼアッセイ法は、細胞溶解物とJNK1免疫沈降物を用いて、SDS-PAGE後に、プロテインキナーゼを再生、ゲル中での基質(GST-ATF2、残基1〜505)の重合、および[γ-32P]ATPとのインキュベーションによって実行した(Derijardら(1994)上記)。[32P]リン酸塩の取り込みは、オートラジオグラフィーによって可視化し、ホスホールイメージャー(Phosphorimager)およびイメージクアント(ImageQuant)ソフトウェア(Molecular Dynamics Inc.、カリフォルニア州サニーベール)によって定量した。細胞溶解物には、UV照射細胞から調製された抽出物中でATF2をリン酸化する46 kDおよび55 kDのプロテインキナーゼが存在することが示された。46 kDおよび55 kDプロテインキナーゼは、それぞれJNK1およびJNK2であると同定された。
【0094】
実施例8. JNK1のATF2への結合およびNH2末端活性化ドメインのリン酸化
ATF2のJNK1によるリン酸化部位は、ATF2の連続的なNH2末端ドメイン欠失を作製して調べた。ATF2をコードするプラスミド発現ベクター(pECE-ATF2)(LiuおよびGreen (1994) および(1990))は、記述されている。GST-ATF2融合蛋白質の細菌の発現ベクターは、ポリメラーゼ連鎖反応(PCR)によるATF2 cDNA断片をpGEX-3X (Pharmacia-LKB Biotechnology Inc.)へサブクローニングして、作製された。作製されたすべてのプラスミドの配列は、アプライドバイオシステムズ(Applied Biosystems)モデル373Aを用いた自動シーケンシングによって確認された。GST-ATF2蛋白質は、記述されたようにして精製され(SmithおよびJohnson (1988) Gene 67:31)、SDS-PAGEで解析され、クーマシーブルーで染色された。GST-ATF2融合蛋白質には、残基1〜505、1〜349、350〜505、1〜109、20〜109、40〜109、および60〜109が含まれていた。
【0095】
UV照射細胞から単離されたJNK1によるGST-ATF2融合蛋白質のリン酸化は、免疫複合体キナーゼアッセイ法によって、調べられた。免疫複合体キナーゼアッセイ法は、Flagエピトープタグを付けたJNK1およびモノクローナル抗体M2 (IBI-Kodak)を用いて、Derijardら(1994)上記、に記述されたようにして行なわれた。免疫複合体蛋白質キナーゼアッセイ法は、組換えJNK1を抗原として調製されたウサギ抗血清を用いても、行なわれた。細胞は、20 mM Tris, pH 7.5, 10%グリセロール、1% 登録商標Triton X-100, 0.137 M NaCl, 25 mMβ-グリセロリン酸塩、2 mM EDTA, 1 mMオルトバナジン酸塩、2 mM ピロリン酸塩、10μg/mlロイペプチン、および1 mM PMSF中で溶解された。JNK1は、JNKに対するウサギポリクローナル抗体、またはFlagエピトープに対するM2モノクローナル抗体に結合した、プロテインG-セファロースを用いて免疫沈降させた。ビーズは溶解緩衝液で3回、キナーゼ緩衝液(20 mM Hepes, pH 7.6, 20 mM MgCl2, 25 mMβ-グリセロリン酸塩、100μMオルソバナジン酸ナトリウム、2 mMジチオスレイトール)で1回洗った。キナーゼアッセイ法は、30μlのキナーゼ緩衝液中で1 μgの基質、20 μMアデノシン3リン酸、および10μCiの[γ-32P]ATPを用いて、25℃で10分間行なった。反応は、レムリ(Laemmli)サンプル緩衝液を添加して停止し、産物はSDS-PAGE(10%ゲル)で解析した。JNK1は、残基1〜505、1〜349、1〜109、20〜109、および40〜109を持つGST-ATF2融合蛋白質をリン酸化するが、60〜109はしなかった。これらの結果は、JNKによるリン酸化には、ATF2の残基1〜60が必要であることを示す。
【0096】
固定したGST-ATF2融合蛋白質の結合は、Hibiら((1993) Genes Dev. 7:2135、参照として本明細書に組み入れられる)に記述されるようにして、固相キナーゼアッセイ法によって調べられた。UV照射細胞のJNK1は、GSH-アガロースに結合したGST-ATF2融合蛋白質とインキュベートした。アガロースビーズは、未結合のJNK1を取り除くために、念入りに洗った。結合したJNK1プロテインキナーゼによるGST-ATF2融合蛋白質のリン酸化は、[γ-32P]ATPを添加して調べた。JNK1は、残基1〜505、1〜349、1〜109、20〜109、および40〜109を持つGST-ATF2融合蛋白質に結合し、JNK1のATF2への結合には残基20〜60が必要であることが示された。
【0097】
実施例9. JNK1によるATF2のNH2末端活性化ドメインのThr69およびThr71のリン酸化
野生型(Thr69、71)およびリン酸化欠損(Ala69、71)ATF2分子の性質に対するUV照射の影響が調べられた。モックトランスフェクトおよびJNK1でトランスフェクトしたCOS細胞を、40 J/m2 UV照射の有無の処理をした。エピトープタグを付けたJNK1は、モノクローナル抗体M2による免疫沈降で単離された。GST- ATF2(残基1〜109)は、上述のように免疫複合体キナーゼアッセイ法で調べた。GST- ATF2は、SDS-PAGEによって他の蛋白質から分離し、クーマシーブルー染色した。GST- ATF2のリン酸化は、オートラジオグラフィーで検出した。JNK1トランスフェクト細胞は、ATF2をリン酸化したが、モックトランスフェクト細胞はしなかった。JNK1によるATF2のリン酸化は、UV照射をした細胞の方が多かった。JNK1によるATF2のリン酸化は、電気泳動の移動度の低下を伴っていた。
【0098】
別の実験では、全長ATF2(残基1〜505)、NH2末端断片(残基1〜109)、およびCOOH末端断片(残基95〜505)を含むGST融合蛋白質が、JNK1によってリン酸化され、ホスホアミノ酸分析によって、リン酸化部位が分析された。ホスホペプチドマッピングおよびホスホアミノ酸分析に使用した方法は、記述されている(Alvarezら(1991) J. Biol. Chem. 266:15277)。ペプチドマップの水平方向は電気泳動で、垂直方向はクロマトグラフィーである。ホスホペプチドマッピングおよび変異分析によって、リン酸化のNH2末端部位は、Thr69およびThr71であると同定された。上述のように、位置指定突然変異導入法を実行してThr69およびThr71をAlaに置換した。変異ATF2のリン酸化は観察されなかった。
【0099】
実施例10. JNK活性化ATF2の電気泳動移動度の低下
CHO細胞は、5%ウシ血清アルブミン(Gibco-BRL)を添加したハム(Ham)のF12培地で維持された。上述のようにして、細胞を標識し、JNK1によってトランスフェクトした。CHO細胞はUV-C(40 J/m2)、IL-1α(10 ng/ml)(Genzyme)、またはウシ胎仔血清(20%)(Gibco-BRL)で処理した。細胞を回収する前に、37℃で30分インキュベートした。ATF2の電気泳動の移動度は、SDS-PAGE後に蛋白質免疫ブロット分析によって調べた。UV、IL-1、および血清で処理された細胞で、ATF2の電気泳動の移動度の変化が観察された。これらの結果は、JNK1による活性化は、ATF2の電気泳動の移動度の変化を伴うことを示し、さらに、ATF2がJNK1のインビボの基質であることを示唆する。
【0100】
実施例11. JNKの活性化後のATF2リン酸化の増加
COS-1細胞は、上述のようにATF2発現ベクターの存在下(ATF2)または非存在下(対照)で、トランスフェクトされた(Haiら(1989)上記)。細胞を40 J /m2 UV-Cに露出した効果を調べた。照射後、細胞を37℃で0または30分(対照)および0、15、30、45分(ATF2)インキュベートし、回収した。SDS-PAGEでのATF2の電気泳動の移動度は、上述のように蛋白質免疫ブロット分析によって調べた。ATF2トランスフェクト細胞では、ATF2は2つの電気泳動移動度を示したが、対照細胞ではそうではなかった。
【0101】
40 J/m2 UV-Cの処理または非処理後、37℃で30分インキュベートした[32]P標識細胞において、野生型(Thr69、71)ATF2および変異型(Ala69、71)ATF2のリン酸化状態を調べた(Haiら(1989)上記)。ATF2蛋白質は免疫沈降で単離され、SDS-PAGEおよびオートラジオグラフィーで分析した。リン酸化ATF2蛋白質は、上述のようにホスホアミノ酸分析によって調べた。両方の型のATF2がホスホセリンを含んでいたが、野生型ATF2のみがホスホスレオニンを含んでいた。
【0102】
トリプシンホスホペプチドマッピングを用いて、インビトロでJNK1によってリン酸化されたATF2と、COS-1細胞中でリン酸化されたATF2とを比較した。インビボとインビトロでリン酸化されたATF2を等量含む試料(ミックス)でも、マップを作製した。ATF2のThr69およびThr71に変異が導入されると、UV照射細胞から単離されたATF2のマップで、2つのリン酸化トリプシンペプチドが失われる。これらのホスホペプチドは、Thr69およびThr71を含むモノリン酸化およびビスリン酸化ペプチドに対応する。このホスホペプチドのいずれも、インビトロでJNK1によってリン酸化されたATF2のマップで観察された。
【0103】
実施例12. リン酸化部位Thr69およびThr71の変異による、ATF2刺激遺伝子発現の阻害
上述のように、ATF2およびGAL4 DNA結合ドメインを含む融合蛋白質を、CHO細胞で発現させた。GAL4- ATF2融合蛋白質の活性は、レポータープラスミドpG5E1bLuc (Sethら(1992) J. Biol. Chem. 267:24796、参照として本明細書に組み入れられる) とのコトランスフェクション解析で測定した。このレポータープラスミドには、最小プロモーター要素およびホタルルシフェラーゼ遺伝子の上流に5つのGAL4部位がクローニングされている。トランスフェクションの効率は、βガラクトシダーゼを発現する対照プラスミドでモニターした(pCH110; Pharmacia-LKB Biotechnology)。細胞抽出物中で検出されるルシフェラーゼおよびβガラクトシダーゼの活性は、3回の実験の平均の活性比として測定された(Guptaら(1993) Proc. Natl. Acad. Sci. USA 90:3216、参照として本明細書に組み入れられる)。表1に示す結果は、転写活性にはThr69およびThr71のリン酸化が重要であることを示す。
【0104】
【表1】

【0105】
実施例13. ATF2機能に対するドミナントネガティブJNK1変異体の影響
野生型(Thr183、Tyr185)または変異(Ala183、Phe185)JNK1でトランスフェクトされた血清処理CHO細胞中の、ATF2リン酸化部位Thr69およびThr72における点変異の影響を、ルシフェラーゼレポータープラスミド系を用いて決定した。対照実験は、モックトランスフェクト細胞を用いて行なった。CHO細胞は、18時間血清除去した後、血清の存在下または非存在下で4時間インキュベートした。野生型ATF2が発現されると、血清に刺激されるATF2転写活性がわずかに上昇した。反対に、変異JNK1は対照および血清刺激ATF2活性の両方を阻害した。
【0106】
実施例14. ATF2に刺激される遺伝子発現に対する、癌抑制遺伝子産物RbおよびアデノウイルスオンコプロテインE1aの影響
癌抑制遺伝子産物RbおよびアデノウイルスオンコプロテインE1Aの発現のATF2転写活性に対する影響は、上述のように、ルシフェラーゼレポータープラスミドおよびGAL4- ATF2 (残基1〜505)を用いて調べた。細胞は、野生型(Thr69、71)または変異(Ala69、71)ATF2でトランスフェクトされた。GAL4- ATF2の非存在下では、ルシフェラーゼ活性に対するRbまたはE1Aの効果は、検出されなかった。野生型および変異ATF2のいずれでも、RbおよびE1AはATF2に刺激される遺伝子発現を増加させることが分かった。しかし、変異ATF2によるレポーター遺伝子発現のレベルは、野生型ATF2よりも低かった。これらの結果は、最大の転写活性を得るためには、ATF2のリン酸化(Thr69およびThr71)に加え、RbまたはE1Aが必要なことを示す。
【0107】
実施例15. p38 MAPキナーゼの基質特異性
p38 MAPキナーゼの基質リン酸化は、細菌で発現されたp38 MAPキナーゼを、IκB、cMyc、EGF-R、細胞質ホスフォリパーゼA2 (cPLA2)、c-Jun、および変異ATF2(Thr69、71)ならびにATP [γ-32P](Raingeaudら(1995) J. Biol. Chem 270:7420、参照として本明細書に組み入れられる)とインキュベーションして調べた。GST-IκBは、バルチモア(D. Baltiomore)博士(マサチューセッツ工科大学)から提供された。GST-cMyc (Alvarezら(1991) J. Biol. Chem. 266:15277)、GST-EGF-R(残基647〜688)(Kolandら(1990) Biochem. Biophys. Res. Commun. 166:90)、およびGST-c-Jun (Derijardら(1994)上記)は、記述されている。リン酸化反応は、30分後にLaemmliサンプル緩衝液を添加して、停止した。リン酸化蛋白質は、SDS-PAGEで分離し、オートラジオグラフィーで検出した。基質蛋白質のリン酸化率は、ホスホールイメージャー(PhosphorImager)(Molecular Dynamics Inc.) 分析によって定量した。ATF2、MBP、EGF-R、およびIκBの相対的リン酸化は、それぞれ、1.0、0.23、0.04、および0.001だった。
【0108】
実施例16. ATF2に対するp38 MAPキナーゼの結合
エピトープタグを付けたJNK1およびp38 MAPキナーゼを発現する細胞抽出物を、GSHアガロースに固定した、ATF2の活性化ドメイン(残基1〜109)を含むGST融合蛋白質とインキュベートした。上清を取り除き、アガロースを念入りに洗った。上清およびアガロース結合画分のウエスタンブロット分析は、以下のように行なった。蛋白質はSDS-PAGEによって分画し、電気泳動によってイモビロン(Immobilon)-Pメンブレンに移し、ホスホチロシン(PY20)およびフラッグエピトープ(M2)に対するモノクローナル抗体で、プローブした。免疫複合体は増幅化学ルミネセンス(Amersham International PLC)を用いて検出した。対照実験は、固定したGSTを用いて行なった。
【0109】
実施例17. 炎症促進型サイトカインおよび環境ストレスによるp38 MAPキナーゼおよびJNK1の活性化
ホルボールエステル、EGF、UV照射、浸透圧ストレス、IL-1、腫瘍壊死因子(TNF)、およびLPSの、p38 MAPキナーゼおよびJNK1の活性に対する影響は、ATP [γ-32P]およびATF2を基質として用いた免疫複合体タンパク質キナーゼアッセイ法によって、測定した。TBFαおよびIL-1αは、ゲンザイム社(Genzyme Corp.)のものである。リポ多糖(LPS)は、記述されたようにして(Mathisonら(1988) J. Clin. Invest. 81:1925)、凍結乾燥した細菌サルモネラ ミネソタ(Salmonella minesota)Re595から単離された。ホルボールミリステートアセテートは、シグマ(Sigma)のものだった。EGFはマウスの唾液腺から精製された(Davis (1988) J. Biol. Chem. 263:9462)。キナーゼアッセイ法は、p38およびJNKの免疫沈降物を用いて行なった。免疫複合体は、キナーゼ緩衝液(上述)で2回洗い、アッセイ法は、最終容量25μl中で、1μgのATF2および50μMの[γ-32P]ATP(10 Ci/mmol)を添加して開始した。反応は、30℃、30分後に、レムリ(Laemmli)サンプル緩衝液を添加して停止した。ATF2のリン酸化は、SDS-PAGE後にオートラジオグラフィーによって調べ、ATF2のリン酸化率は、ホスホールイメージャー(PhosphorImager)分析によって定量した。
【0110】
結果は表2に示されている。HeLa細胞を10 nMのホルボールミリステートアセテートに露出すると、p38とJNK1が非常に弱く活性化された。同様に、10 nMのEGFで処理しても、p38とJNK1を非常に弱く活性化したのみだった。これとは対照的に、40 J/m2 UV-C、300 mMソルビトール、10 ng/mlインターロイキン-1、および10 ng/ml TNFαで処理すると、p38とJNK1活性が強く活性化された。p38の活性に対するLPSの効果は、ヒトCD14を発現するCHO細胞を用いて調べた。CHO細胞を10 ng/ml LPSに露出しても、p38とJNK1活性は僅かに活性化されただけだった。
【0111】
【表2】
【0112】
実施例18. TyrおよびThrの二重リン酸化によるp38 MAPキナーゼの活性化
野生型(Thr180、Tyr182)または変異(Ala180、Phe182)p38 MAPキナーゼを発現するCOS-1細胞を、UV-C (40 J/m2)の照射の有無で処理した。細胞はUV照射の有無で処理後、30分で回収した。対照実験は、モックトランスフェクト細胞を用いて行なった。エピトープタグをつけたp38 MAPキナーゼの発現レベルおよび、p38 MAPキナーゼのTyrリン酸化状態は、モノクローナル抗体M2およびホスホチロシンモノクローナル抗体PY20を用いたウエスタンブロット分析によって調べた。免疫複合体は、増幅化学ルミネセンスによって検出した。
【0113】
野生型および変異p38は、同様なレベルで発現されていた。ウエスタンブロット分析では、UV照射によってp38のTyrリン酸化が増加したことが示された。Tyrリン酸化の増加は、[32P]リン酸塩標識細胞から単離されたp38のホスホアミノ酸分析でも確認された。この結果では、UV照射がp38のThrリン酸化を増加させたことも示された。TyrおよびThrのリン酸化の増加は、変異p38によって阻害された。野生型および変異型のp38は、免疫沈降によってCOS-1細胞から単離された。プロテインキナーゼ活性は、[γ-32P]ATPおよびGST-ATF2を基質として使用して、免疫複合体中で測定された。リン酸化GST- ATF2は、SDS-PAGE後にオートラジオグラフィーによって検出された。UV照射によって、野生型p38の活性が著しく上昇したが、変異p38は触媒作用が不活性であることが分かった。これらの結果は、p38がThr-Gly-Tyrモチーフ内の二重のリン酸化によって活性化されることを示す。
【0114】
実施例19. MAPキナーゼホスファターゼはp38 MAPキナーゼ活性化を阻害する
細胞を40 J/m2のUV-Cの照射の有無で処理した。対照実験は、モックトランスフェクトした細胞(対照)、および触媒作用が不活性の変異ホスファターゼmPAC1 (Cys257/Ser)およびヒトMKP1でトランスフェクトした細胞を用いて行なった。p38 MAPキナーゼの活性は、[γ-32P]ATPおよびGST- ATF2を基質として用いた免疫複合体プロテインキナーゼアッセイ法で測定した。PAC1またはMKP1の発現は、p38によるリン酸化を阻害することが分かったが、これはp38が両特異性を持つホスファターゼPAC1およびMKP1によって調節され得ることを示す。
【0115】
実施例20. p38 MAPキナーゼの細胞内分布
エピトープタグを付けたp38 MAPキナーゼは、COS細胞で発現された。細胞は、40 J/m2 のUV照射の有無で処理した後、37℃で60分間インキュベートした。p38 MAPキナーゼは、モノクローナル抗体M2を用いた間接免疫蛍光によって検出した。デジタルイメージング顕微鏡法によって像を得て、画像復元処理を行った。
【0116】
免疫組織化学
カバースリップ(22 mm x 22 mm No.1; ゴールドシールカバーグラス; Becton-Dickinson)は、0.1 N HCl中で10分間煮沸による前処理をして、蒸留水ですすぎ、オートクレーブして、0.01% ポリ-L-リジン(Sigma; ミズーリ州セントルイス)でコートした。カバースリップは、35 mmマルチウェル組織培養プレート(Becton Dickinson、英国)の底に置いた。トランスフェクトされたCOS-1細胞をカバースリップ上に直接播き、5%ウシ胎仔血清(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で一晩接着させた。トランスフェクションの24時間後に、細胞を一度洗い、25 mM Hepes, pH 7.4、137 mM NaCl、6 mM KCl、1 mM MgCl2、1 mM CaCl2、10 mMグルコース中で37℃で30分間インキュベートした。リン酸緩衝生理食塩水で細胞を1度洗い、組織培養ウェルからカバースリップを取り出した。新しく調製したリン酸緩衝生理食塩水中の4%パラホルムアルデヒドで、22℃で15分間細胞を固定した。リン酸緩衝生理食塩水中の0.25% 登録商標Triton X-100で5分間処理して細胞を透過性にして、DWB溶液(150 mM NaCl、15 mMクエン酸ナトリウム、pH 7.0、2%ウマ血清、1%(w/v)ウシ血清アルブミン、0.05% 登録商標Triton X-100)で3回洗った。一次抗体(抗フラッグモノクローナル抗体M2、Eastman-Kodak Co.,コネチカット州ニューヘブン)は、DWBで250倍に希釈し、加湿環境下で22℃で1時間細胞に適用した。再び細胞を上述のように3回洗い、2次抗体であるフルオレセインイソチオシアネート結合ヤギ抗マウスIg(Kirkegaard & Perry Laboratories Inc.、メリーランド州ゲーサーズバーグ)は、250倍希釈して、加湿環境下で22℃で1時間適用した。その後、DWBで細胞を3回洗い、その後、免疫蛍光分析用に、ゲル−マウント(Gel-Mount)(Biomeda Corp.、カリフォルニア州フォスターシティ)によってスライド上にマウントした。対照実験は、観察された免疫蛍光の特異性を評価するために実行した。トランスフェクトされた細胞が、一次モノクローナル抗体M2の非存在下で染色されたトランスフェクト細胞や、モックトランスフェクトされた細胞では、蛍光は検出されなかった。
【0117】
デジタルイメージング顕微鏡法および画像復元
個々の細胞中の蛍光分布のデジタル画像は、既述のように、エピ蛍光用のゼイズ(Zeiss)IM-35顕微鏡上でニコン60x プラナポ(Planapo)対物レンズ(開口数=1.4)を用いて得た(Carringtonら(1990) 細胞生物学の非侵襲性技術(Non-invasive Techniques in Cell Biology)、Fosbett & Grinstein編、Wiley-Liss, NY; pp 53-72; Fayら(1989) J. Microsci. 153:133-149)。種々の焦平面の画像は、コンピュータ制御焦点機構および熱電流冷却電荷結合素子カメラ(モデル220;Photometrics Ltd.、アリゾナ州ツーソン)を用いて得た。試料の励起源への露出は、コンピュータ制御シャッターおよび波長選択システム(MVI、マサチューセッツ州エイボン)により決定した。電荷結合素子カメラおよび顕微鏡の機能は、マイクロコンピュータで制御され、カメラから得たデータはシリコン グラフィクス(Silicon Graphics)モデル4D/GTXワークステーション(カリフォルニア州マウンテンビュー)へ転送して画像処理をした。画像は、感度の不均一性および電荷結合素子検知器の暗電流のための補正をした。顕微鏡のぼやけの較正は、この装置の点広がり関数を、直径0.3μmの蛍光標識ラテックスビーズ(Molecular Probes Inc.)の0.125μm間隔の光学切片のシリーズとして測定することにより、決定した。画像復元アルゴリズムは、誤った問題の理論に基づいたもので、未処理画像よりも実質的に正確な細胞内の定量的色素密度値を得るものである(Carringtonら(1990)上記;Fayら(1989)上記)。画像処理後に、シリコン グラフィクス(Silicon Graphics)ワークステーション上のコンピュータグラフィックスソフトウェアを用いて、細胞の個々の光学切片を分析した。p38 MAPキナーゼは、細胞表面、細胞質、および核に観察された。照射後、細胞質p38の核周囲領域への局在が増加した。
【0118】
実施例21. 浸透圧ショックによるMKKシグナル伝達経路の活性化
CHO細胞を、プラスミドpCMV-Flag-Jnk1およびpRSV-Neoでコトランスフェクションした(Derijardら(1994)上記)。ジェネティシン(Gibco-BRL)を用いた選択によって、エピトープタグを付けたJnk1(Flag; Immunex Corp.)を発現する安定した細胞株を単離した。細胞を、0、100、150、300、600、または1000 mMソルビトールと37℃で1時間インキュベートした。細胞は溶解緩衝液(20 mM Tris, pH 7.4, 1% 登録商標TRITON X-100, 2 mM EDTA, 137 mM NaCl, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸塩、2 mMピロリン酸塩、10%グリセロール、1 mMフェニルメチルスルホニルフルオリド、および10μg/mlロイペプチン)中で回収し、4℃で100,000 x gで30分間遠心して可溶性抽出物を得た。エピトープタグを付けたJNK1は、モノクローナル抗体M2(Immunex Corp)を用いた免疫沈降によって単離した。免疫沈降物は、溶解緩衝液で念入りに洗った。免疫複合体キナーゼアッセイ法は、25μlの25 mM Hepes, pH 7.4, 25 mM MgCl2、25 mMβ-グリセロリン酸塩、2 mMジチオスレイトール、100μMオルトバナジン酸塩、および50μM ATP [γ-32P] (10μCi/mmol)中で、細菌で発現されたグルタチオン-S-トランスフェラーゼGST)融合c-Jun(残基1〜79)2.5μgを基質として使用して、行なった。c-Junのリン酸化はSDS-PAGE後に、オートラジオグラフィーおよびホスホールイメージャー(PhosphorImager)(Molecular Dynamics Inc.)分析によって調べた。すべての濃度のソルビトール処理で、JNK1の活性化が観察された。
【0119】
JNK1プロテインキナーゼ活性化の時間変化は、上述のように300 mMソルビトールを添加した培地中でインキュベートした細胞中で、測定された。ソルビトールへの露出後、5分以内にJNK1の活性の上昇が観察され、最大活性は15〜30分後に見られた。
【0120】
JNK1のリン酸化部位Thr183およびTyr185に変異を導入すると、浸透圧ショックによるJNK1プロテインキナーゼ活性の活性化が阻止された。CHO細胞をベクター、野生型JNK1(Thr183、Tyr185)、および変異JNK1(Ala183、Phe185)でトランスフェクトした。300 mMソルビトールの添加のある培地とない培地で15分間インキュベートしてから、上述のようにJNK1プロテインキナーゼ活性を測定した。JNK1の活性化は、野生型JNK1では見られたが、変異JNK1では見られなかった。
【0121】
実施例22. MKK7の分子クローニング
RT-PCRを用いて新規の哺乳類MAPキナーゼキナーゼの断片を同定した。このプロトコール用にデザインしたプライマーATNGCNGTNAARCARATG(配列番号:23)およびATNCKYTCNGGNGCCATRTA(配列番号:24)は、ショウジョウバエMAPキナーゼキナーゼhep(Gliseら(1995) Cell 83:451-461)の配列に基づいている。マウスの精巣mRNAは鋳型として使用された。マウス精巣mRNAのRT-PCR増幅後に、単一の生成物(461 bp)が検出された。配列分析により、このPCR産物が新規の哺乳類MAPキナーゼキナーゼの断片であることが同定された。全長マウスcDNAクローンは、マウス精巣ライブラリー(Stratagene Inc.)をスクリーニングして、単離された。このcDNAクローンの配列は、アプライドシステムズ(Applied Biosystems)モデル373Aを用いて決定した。配列分析により、7つのクローン群が、プロテインキナーゼと推定されるものをコードする単一の長いオープンリーディングフレームを含むと同定された(図9および図10;配列番号:17および配列番号:18)。インフレームの停止コドンがこれらのクローンの5'および3'領域に検出された。この配列は、プロテインキナーゼサブドメインI〜XIを含んでおり、MAPキナーゼキナーゼグループに関連している。この新規のプロテインキナーゼは、MKK7と命名された。MAPキナーゼキナーゼのサブドメインVIIIに存在するリン酸化を活性化する部位は、MKK7に保存されている。MKK7を他の哺乳類MAPキナーゼキナーゼグループメンバーと比較すると、MKK7がJNK活性化因子MKK4と近縁であることが示された。λファージライブラリーから単離された別の1つのcDNAクローンは、他の7つのクローンとは異なっていた。このクローンは、3'非翻訳領域およびMKK7のコード領域を含んでいたが、インフレームの停止コドンを欠く、異なる5'領域を持っていた。このクローンは、MKK7の異なるスプライシングを表わす(MKK7b;図11;配列番号:19)。MKK7b cDNAクローンは、その5'領域中に開始コドンを持たない;したがって、このcDNAは、単離された他のクローンと同じMKK7プロテインキナーゼをコードしている。しかし、MKK7b cDNAクローンが全長クローンでなければ、追加の5'配列がインフレームの開始コドンを含む可能性もある。もしそうならば、MKK7bはM-[?]-SPAPAPSQRAALQLPLANDGGSRSPSSESSPQHPTPPTRPRH- (配列番号:33)という配列をMKK7の開始メチオニン(図9)に融合させると予測される。ショウジョウバエのMAPキナーゼキナーゼhepはMKK7と実質的な配列類似性を持っているが、MKK7bのNH2末端部分は、hepプロテインキナーゼでは保存されていない。3つの別のクローンが、5'および3'領域が異なるMKK7のスプライシング変異体をコードしていた。これらのクローン(MKK7c(図13)、MKK7d(図14)、およびMKK7e(図15))は、5'および3'領域にインフレームの停止コドンが存在するため、全長である。
【0122】
全長のマウスMKK7 cDNAプローブで、ヒトcDNAライブラリーをスクリーニングした。単一のクローンが同定され、配列決定された。3'末端を持たない部分的MKK7配列が同定された(図12;配列番号:25、および配列番号:26)。配列は、マウスMKK7cと最も相同性が高かった。
【0123】
MKK7、MKK7b、hep、およびヒトMKK7のcDNAの配列は、それぞれ寄託番号U93030、U93031、U93032、AFOO319としてジーンバンク(Genbank)に寄託された。
【0124】
実施例23. MKK7の発現
MKK7の発現は、異なる組織から単離されたmRNAのノーザンブロット分析によって調べられた。分析は、異なる組織から単離され、変性アガロースゲル電気泳動により分画し、ナイロンメンブレン(Clontech)に移されたポリA+ mRNA(2μg)を用いて行なった。ブロットは、[α-32P]dATP(Amersham International PLC)を用いたランダムプライマーで標識したMKK4およびMKK7 cDNAでプローブした。
【0125】
MKK7はマウス組織で広範囲に発現されていることが分かった。精巣で2つのMKK7トランスクリプト(4.0 kbおよび1.6 kb)が検出されたのを除き、調べたすべての組織で、単一のMKK7トランスクリプト(約4.0 kb)が検出された。MKK7発現のレベルが最高だったのは、精巣だった。かなりのMKK7発現が、心臓、脳、肺、肝臓、および腎臓で観察された。これは、脳、肝臓、筋肉、心臓、および腎臓でかなりの量の発現が観察されるが、脳でレベルが最高であるMKK4の発現とは対照的だった。MKK4およびMKK7は、共に発現されるが、調べた組織によって、各MAPキナーゼキナーゼの相対的な量は、異なっていた。
【0126】
実施例24. インビトロにおける、MKK7によるJNKの特異的活性化
MKK7の特異性を調べるために、インビトロプロテインキナーゼアッセイ法を行なった。MKK7 cDNA(Eco RIおよびPvu II断片)をpGEX-5XL (Pharmacia-LKB) のEco RIおよびSma I部位にサブクローニングして、細菌のMKK7発現ベクターを調製した。アフィニティークロマトグラフィーによって、グルタチオン-S-トランスフェラーゼ(GST)融合蛋白質を精製した(SmithおよびJohnson (1988) Gene 67:31-40)。組換え蛋白質GST-ATF2 (Guptaら(1995) Science 267:389-393)、GST-cJun (Derijard (1994)上記)、GST-cMyc (Alvarezら(1991) J. Biol. Chem. 266:15277-15285)、GST-ERK2 (Sethら(1992) J. Biol. Chem. 267:24796-24804)、GST-p38 (Raingeaudら(1995) J. Biol. Chem. 270:7420-7426)、およびGST-JNK1 (Derijard (1994)上記)は記述されている。
【0127】
プロテインキナーゼアッセイ法は、キナーゼ緩衝液(25 mM 4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸塩(pH 7.4)、25 mM β-グリセロリン酸塩、25 mM MgCl2、2 mMジチオスレイトール、0.1 mM オルトバナジン酸塩)中で実行された。アッセイ法は、最終容量25μlの中に、1 μgの基質蛋白質および50 μM [γ-32P]ATP(10 Ci/mmol)を添加して開始された。25℃で30分後に、レムリ(Laemmli)サンプル緩衝液を添加して反応は終了した。SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)後に、オートラジオグラフィーによって基質蛋白質のリン酸化を調べた。
【0128】
組換えMAPキナーゼは、基質ATP [γ-32P] を用いて、GST(対照)またはGST-MKK7とインキュベートされた。細菌から精製された組換えMKK7の自己リン酸化は観察されなかった。組換えMKK7をMAPキナーゼとインキュベートすると、MKK7はJNK1をリン酸化するが、p38とERK2はリン酸化しないことが示された。MKK7はp38とJNK1によってリン酸化された。MAPキナーゼによるMAPキナーゼキナーゼの逆方向のリン酸化の意義は不明であるが、同様な逆方向リン酸化はMKK4(Derijard (1995)上記)およびショウジョウバエJNK活性化因子hep (Sluss (1996)上記)を用いたキナーゼアッセイ法でも、検出されている。
【0129】
MKK7によるJNK1のリン酸化がプロテインキナーゼ活性を上昇させたかどうかを調べるために、JNK基質としてATF2を使った実験を行った。GST-MKK7は、プロテインキナーゼアッセイ法で組換えJNK1とインキュベートされた。JNK活性は、各アッセイ法にJNKの基質ATF2を含めて、測定された。ATF2はMKK7によってリン酸化されないが、JNK1によって弱くリン酸化される。JNK1とMKK7をインキュベートすると、JNK1がリン酸化され、ATF2のリン酸化が大きく上昇した。このデータは、MKK7がJNK1をリン酸化し、活性化することを示す。この結論を確認するために、JNKの二重のリン酸化のモチーフThr-Pro-TyrをAla-Pro-Pheで置換した効果を調べた。MKK7は変異JNK1 (APF)蛋白質をリン酸化しなかった。さらに、MKK7は、変異JNK1プロテインキナーゼによるATF2リン酸化を上昇させなかった。したがって、MKK7はインビトロにおけるJNKの活性化因子である。
【0130】
実施例25. インビボにおけるMKK7によるJNKの特異的活性化
インビボにおけるMKK7の特異性を調べるために、コトランスフェクション解析を行なった。CHO細胞は、ウシ胎仔血清(5%;Gibco-BRL)を添加したダルベッコ変法イーグル培地で維持された。細胞は、製造元(Gibco-BRL)の推奨する方法にしたがって、リポフェクタミン試薬を用いてトランスフェクトした(Derijardら(1994)上記)。細胞に、エピトープタグを付けたJNK1と、空の発現ベクター(対照)またはMKK4もしくはMKK7をコードする発現ベクターをコトランスフェクトした。エピトープタグは、インフルエンザウイルスのヘマグルチニン蛋白質(HA)に由来する。JNK1は細胞溶解物の免疫沈降によって単離された。細胞は溶解緩衝液(20 mM Tris ( pH 7.4), 1% 登録商標TRITON X-100, 10%グリセロール、137 mM NaCl, 2 mM EDTA, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸ナトリウム、2 mMピロリン酸塩、1 mM PMSF、および10μg/mlロイペプチン)で溶解し、4℃で100,000 x gで15分間遠心した。エピトープタグを付けたプロテインキナーゼは、プロテインGセファロース(Pharmacia-LKB Biotechnology Inc.)に結合した抗HAモノクローナル抗体を用いて、4℃で3時間インキュベーションし、免疫沈降させた。免疫沈降物は、溶解緩衝液で3回洗った(Guptaら(1995) Science 267:389-393)。基質として[γ-32P]ATPおよびc-Junを用いて、免疫複合体でプロテインキナーゼ活性を測定した。リン酸化反応の産物は、SDS-PAGE後にオートラジオグラフィーによって可視化した。ERK2およびp38 MAPキナーゼは、共に発現されたMKK7によって活性化されなかった。対照実験では、ERK2とp38 MAPキナーゼが、それぞれの近縁MAPキナーゼキナーゼMKK1およびMKK6で活性化されることが示された。これとは対照に、MKK7はJNK1を活性化した。興味深いことに、同時に発現されたMKK7によるJNK1の活性化は、以前に記述されたJNK活性化因子MKK4によるものよりも、高レベルだった。これらのデータを合わせると、MKK7が培養細胞においてJNKの特異的な活性化因子として機能することが立証される。
【0131】
実施例26. MKK7によるJNKシグナル伝達経路の活性化
JNKシグナル伝達経路は、AP-1転写活性を調節することが知られている。(Whitmarsh (1996)上記)。MKK7の発現がAP-1の転写活性を上昇させるという仮説を検証するために、最小プロモーター要素の上流に3つのAP-1部位がクローニングされたルシフェラーゼレポーター遺伝子を用いたコトランスフェクション解析を行なった(RinconおよびFlavell (1994) EMBO J. 13:4370-4381)。ルシフェラーゼレポーター遺伝子発現は、レポータープラスミドpTRE-ルシフェラーゼ(Rincon (1994)上記)0.5μgと、βガラクトシダーゼ発現ベクターpCH110(Pharmacia-LKB)0.25μgとを用いたコトランスフェクション解析で測定された。GAL4融合蛋白質を用いた実験は、0.25μgのpGAL4-ATF2(残基1〜109)、0.5μgのレポータープラスミドpG5E1bLuc、および0.25μgのpCH110を用いて行なった(Guptaら(1995)上記)。プロテインキナーゼの効果は、0.3μgの空の発現ベクターまたはプロテインキナーゼ発現ベクターとのコトランスフェクションによって調べた。ERK2、p38、JNK1、MKK1、MKK3、MKK4、およびMKK6発現ベクターは、記述されている。細胞はトランスフェクション後36時間で回収した。細胞溶解物中のβガラクトシダーゼおよびルシフェラーゼの活性は、記述されたようにして測定した(Gupta (1995)上記)。MKK4、MKK7、またはJNK1が発現されても、AP-1レポーター遺伝子の発現に著しい変化は見られなかった(図16A)。これとは対照に、MKK7とJNK1が同時に発現すると、AP-1に依存するレポーター遺伝子の発現が上昇した。MKK4はMKK7よりも弱くJNKを活性化させるという観察と一致して、MKK4とJNKとが同時に発現すると、AP-1レポーター遺伝子の発現が、MKK7の場合よりも少なく、上昇した(図16A)。これらのデータを合わせると、MKK7がJNKシグナル伝達経路の活性化因子として機能することを示す。
【0132】
転写活性に対するMKK7の効果をさらに調べるために、転写因子ATF2に対するMKK7の影響を探った。以前の研究で、ATF2がJNKシグナル伝達経路の標的であることが示されている(van Damら(1995)上記;Guptaら(1995)上記;Livingstoneら(1995)上記)。JNKはATF2のNH2末端活性化ドメイン中の2つの部位(Thr-69およびThr-71)をリン酸化し、転写活性を上昇させる。ATF2の活性化ドメインの転写活性をモニターするために、GAL4融合蛋白質戦略を採用した(Gupta (1995)上記)。レポーター遺伝子の発現を測定すると、MKK4とJNK1が同時に発現すると、転写活性が上昇することが示された(図16B)。MKK7が発現してもレポーター遺伝子発現は同様なレベルで観察されたが、MKK7がJNK1と同時に発現すると、さらに大きな上昇が検出された。転写活性に対して、MKK4と比較してMKK7の方が強い効果を示すということは、JNK活性化に対するMKK7とMKK4の相対的な効果と一致している。レポーター遺伝子発現の上昇がATF2のリン酸化によって仲介されることを確認するために、ATF2リン酸化部位(Thr-69およびThr-71)をAlaに置換した効果を調べた。変異ATF2蛋白質は、MKK4、MKK7、またはJNK1によって調節されない(図16B)。これらのデータを合わせると、MKK7がJNKシグナル伝達経路の生理的な標的を調節し得ることを示す。
【0133】
用途
本発明のMKKポリペプチドおよびポリヌクレオチドは、MKKシグナル伝達経路を調節する試薬の同定に有用である。MKKシグナル伝達経路を調節する試薬は、MKK合成、MKKリン酸化またはMKK活性に対するその影響によって、同定できる。例えば、MKK活性に対する試薬の影響は、上述のインビトロキナーゼアッセイ法によって、測定できる。MKKは成分の反応を許すような条件下で、試験試薬の存在下および非存在下(対照)でインキュベートし、その後、試験試薬のキナーゼ活性に対する効果を測定する。MKKシグナル伝達経路を阻害する試薬は、MKKを介する疾病の治療に利用できる。MKKシグナル伝達経路を刺激する試薬は、組織におけるプログラムされた細胞死(アポトーシス)の誘導を含め、いくつもの方法に、使用できる。例えば、UVで損傷を受けた細胞の除去は、癌の予防に使用できる。
【0134】
一般に、MKKシグナル伝達経路を阻害する試薬の同定には、キナーゼアッセイ法(例えば、実施例3参照)が使用される。MKK基質および[γ-32P]ATPのような放射性標識を含む試験システムに、種々の濃度の試薬(例、1.0 nM〜100 mM)を添加する。適当な基質分子には、p38、JNK1、JNK2、またはATF2が含まれる。標識リン(例、[32P]または[33P]の基質への取り込みを決定し、試験試薬で得られた結果を対照の値と比較する。特に興味深いのは、[32P]の取り込みを約80%またはそれ以上阻害する試薬である。リン酸化は、リン酸化に依存する試薬、例えば、抗体を用いて調べることもできる。リン酸化依存性抗体は、活性化部位、Ser198およびThr202がリン酸化されたMKK7を用いて作製できる。これは、リン酸化されたSer198およびThr202を持つMKK7配列に対応する合成ペプチド(例えば、長さ約15アミノ酸)で動物を免疫して実施できる。そのような抗体の生産方法は、当技術分野で周知である。そのような抗体は、組織および細胞抽出物において活性化MKK7の検出(例、ウエスタンブロット)に役立ち、キットとして使用できる可能性がある。
【0135】
MKK合成に対する試薬の効果を検定するアッセイ法も、MKKシグナル伝達経路を阻害する化合物の同定に利用できる。MKK発現に対する試験試薬の影響は、例えば、MKKに特異的な抗体を用いたウエスタンブロット分析で測定される。抗体の結合は、オートラジオグラフィーまたは化学ルミネセンスによって可視化し、定量する。MKK mRNA発現に対する試験試薬の効果は、例えば、ポリヌクレオチドプローブを用いたノーザンブロット分析、またはポリメラーゼ連鎖反応によって、調べることができる。
【0136】
MKKシグナル伝達経路を阻害することが分かった試薬は、MKKを介する疾病の治療に使用できる。そのような試薬は、MKKシグナル伝達経路を阻害するために必要な分子の特異的な特徴を解明するための薬剤デザインにも、有用である。
【0137】
さらに、本発明はMKKを介するストレス関連および炎症性疾患の治療方法も提供する。この方法には、MKK機能を阻害する治療試薬の有効量の投与が含まれる。適当な試薬は、MKK活性または発現のいずれかを阻害する。投与する試薬の濃度は、適当な用量、投与経路、および治療する特定の症状を含め、いくつもの要素によって、決定される。適当な試薬用量は、通常の実験から個々の患者および治療する特定のMKKを介する疾患に必要な最適用量までを含め、当業者に周知の方法によって、決定される。投与に適当で治療に有効な特定の量は、当業者によって簡単に決定される(例、レミントンの薬学(Remington's Pharmaceutical Sciences) 第18版、Gennaro編、Mack Publishing Company、ペンシルベニア州イーストン、1990参照)。用量は約0.1〜10 mg/キロ/日の範囲に渡る可能性がある。
【0138】
本発明は、ストレス関連および炎症性疾患の急性治療および予防の方法を提供する。例えば、虚血性心疾患は、再灌流後の酸化ストレスおよび虚血の発現時に、治療されると想像される。さらに、虚血のリスクを持つ患者は、虚血症状の発現前に、治療できる。
【0139】
別の例では、MKK機能または活性を阻害する治療薬を投与して、TNFおよびIL-1を含む炎症促進型サイトカインの分泌を阻害することにより、炎症反応をコントロールする。
【0140】
ストレスに関連する増殖性疾患も、MKK機能または活性を阻害する治療試薬を投与することによって、本発明の方法により治療できる。そのような治療試薬は、単独または、例えば悪性腫瘍の治療における化学療法剤などの他の治療試薬と組合わせて、使用できる。実際に、本発明によって提供される治療試薬によるストレス活性化MKKのコントロールは、ストレスを誘導する他の治療戦略によって引き起こされる症状を調節できる。
【0141】
採用される治療試薬は、ポリヌクレオチド、ポリペプチド、およびアンチセンスオリゴヌクレオチドおよびリボザイムのような他の分子を含む、MKK機能または活性を阻害する化合物であり、本発明および当技術分野で周知の技術にしたがって製造できるものである。MKKのエピトープに結合するポリクローナルまたはモノクローナル抗体(その断片または誘導体を含む)も、治療試薬として採用できる。基質の結合および/またはリン酸化に関して、効率的にMKKを排除するまたはMKKと競合するMKKのドミナントネガティブ型は、プロテインキナーゼ活性を低下させるために使用できる。ドミナントネガティブ型は、当技術分野で周知の方法を用いて、上述(実施例13)のように、プロテインキナーゼの触媒ドメイン内に変異を導入することによって、作製できる。触媒性の残基は、すべてのMKKアイソフォームで保存されている。例えば、Lys76の変異は、MKK7の活性を阻害する。同様に、活性化リン酸化の保存された部位(Ser198、Thr202)の変異は、MKK7活性を阻害する。これらのMKK7のキナーゼ不活性型は、ドミナントネガティブ阻害剤として作用する。
【0142】
場合によっては、例えばアポトーシスの誘導のように、MKK活性の増加が望ましいことがある。本発明の方法を用いて、MKK機能または活性を増加させる能力のある試薬を同定できる。または、MKKを過剰発現して活性の増加が実現できる。MKKを介する疾病が、MKK発現の不足、または変異MKKポリペプチドの発現を伴う場合には、センスポリヌクレオチド配列(DNAコード鎖)またはMKKポリペプチドを細胞に導入して、正常なMKK活性を増大させることができる。必要ならば、これらの治療法は、投与様式(例、細胞の種類に特異的なウイルスベクターの使用)、または当技術分野で周知の方法により細胞の種類に特異的もしくは誘導可能なプロモーターを持つ組換え構築物にMKK7核酸を入れることによって、特定の細胞が標的となるようにする。例えば、MKK7核酸を含むベクターは、当技術分野で標準的な組換えDNA技術によって、作製できる。ベクターは、哺乳類細胞での複製および発現に使用される、プラスミド、ウイルス、または当技術分野で周知の他のベクターであり得る。MKK7核酸をコードする配列の発現は、哺乳類細胞、好ましくはヒト細胞において当技術分野で周知の任意のプロモーターでよい。そのようなプロモーターは、誘導性または構成性であり得る。そのようなプロモーターには、SV40初期プロモーター領域(Bernoistら、Nature 290:304, 1981);ラウス肉腫ウイルスの3'長い末端反復配列に含まれるプロモーター(Yamamotoら、Cell 22:787-797, 1988);ヘルペスチミジンキナーゼプロモーター(Wagnerら、Proc. Natl. Acad. Sci. USA 78:1441, 1981);またはメタロチオネイン遺伝子の調節配列(Brinsterら、Nature 296:39, 1988)が含まれるが、これらに限定されるわけではない。
【0143】
本発明の抗体は、注射または時間をかけた徐々の注入により、非経口的に投与できる。本発明のモノクローナル抗体は、静脈内、腹腔内、筋肉内、皮下、腔内、または経皮的に投与できる。
【0144】
本発明のポリペプチドまたは抗体の非経口投与用調製物には、無菌の水性または非水性溶液、懸濁液、および乳濁液が含まれる。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、およびオレイン酸エチルのような注射可能な有機エステルである。水性キャリアには、生理食塩水および緩衝媒体を含む、水、アルコール性/水性溶液、乳濁液、または懸濁液が含まれる。非経口賦形剤には、塩化ナトリウム溶液、リンゲルのブドウ糖、ブドウ糖および塩化ナトリウム、乳酸リンゲル液、または固定油が含まれる。静脈内賦形剤には、液体および栄養補充液、電解質補充液(リンゲルのブドウ糖に基づくものなど)、および同様なものが含まれる。例えば、抗菌剤、抗酸化剤、キレート剤、および不活性ガス、および同様なものなどの、保存剤および他の添加物も、存在する可能性がある。
【0145】
アンチセンス配列を含むポリヌクレオチド配列は、当技術分野で周知の様々な技術によって、治療用に投与できる。そのような治療法は、MKKポリヌクレオチドをMKKを介する疾病を持つ哺乳類の細胞中に導入することによって、その治療効果を発揮する。MKKポリヌクレオチドの送達は、遊離ポリヌクレオチド、またはキメラウイルスのような組換え発現ベクター、またはコロイド分散系を利用して実施できる。ヌクレオチド配列の治療用送達に特に好ましいのは、標的を定めたリポソームを利用することである。
【0146】
治療用試薬を、特定の組織にターゲッティングすることは、送達効率を上昇させるために望ましい。ターゲッティングは、投与経路を通じた受動的な機構によって、実現できる。特定の組織に対して積極的にターゲッティングすることもできる。リポソーム、コロイド懸濁液、およびウイルスベクターを利用すると、例えば、標的組織の成分の受容体として働く分子を入れるなど、治療用試薬を含む製剤の組成を変化させることによって、ターゲッティングが可能になる。例には、糖、糖脂質、ポリヌクレオチド、または蛋白質が含まれる。これらの分子は、治療用試薬に含まれることができる。または、例えば、この分子をコードするポリヌクレオチドを入れる、またはターゲッティング分子を提供するパッケージング系を利用することによって、間接的な方法によって、これらの分子を入れることもできる。本明細書に提供される教示を利用すれば、特定の組織に治療試薬を送達するために、どの分子や手順が有用であるか、当業者は理解するまたは確認するだろう。
【0147】
トランスジェニック動物
MKKポリペプチドは、トランスジェニック動物で発現することができる。これらの動物は、MKKの過剰発現または発現不足によって誘導または増悪される疾患の研究、およびMKKの発現または活性を調節する治療薬の開発のための、モデルシステムとなる。例えば、マウス中でドミナントネガティブで構成的に活性化された対立遺伝子を発現して、生理的機能を立証できる。
【0148】
トランスジェニック動物は、家畜(ブタ、ヤギ、ヒツジ、ウシ、ウマ、ウサギ、および同様なもの)、齧歯類(ラット、モルモット、およびマウスなど)、非ヒト霊長類(例えば、ヒヒ、サル、およびチンパンジー)、およびペット(例、イヌおよびネコ)であり得る。トランスジェニックマウスは、特に好ましい。
【0149】
トランスジェニック動物のファウンダー系統を作製するために、当技術分野で周知の任意の技術を用いて、MKKトランスジーンを動物に導入することができる。そのような技術には、前核マイクロインジェクション(米国特許第4,873,191号);レトロウイルスを介した生殖系統への遺伝子移入(Van der Puttenら、Proc. Natl. Acad. Sci. USA 82:6148, 1985);胚性肝細胞への遺伝子ターゲッティング(Thompsonら、Cell 56:313, 1989);および胚のエレクトロポレーション(Lo, Mol. Cell. Biol. 3:1803, 1983)が含まれるが、これらに限定されるわけではない。特に有用なのは、Yangら(Proc. Natl. Acad. Sci. USA 94:3004-3009, 1997)に記述される方法である。
【0150】
本発明は、すべての細胞にMKKトランスジーンを持つトランスジェニック動物、および一部の細胞にのみトランスジーンを持つ動物を提供する。すなわち、本発明は、モザイク動物を提供する。トランスジーンは、単一のトランスジーン、または例えば、逆方向もしくは同方向に縦につながったコンカテマーとして組み込まれ得る。トランスジーンは、特定のタイプの細胞に選択的に導入および活性化できる(Laskoら、Proc. Natl. Acad. Sci. USA 89:6232, 1992)。そのような細胞のタイプに特異的な活性化に必要な調節配列は、興味のある特定の細胞のタイプに依存し、当業者には明らかだろう。
【0151】
MKKトランスジーンが内因性MKK遺伝子の染色体部位に組み込まれることが望ましい場合には、遺伝子ターゲッティングが好ましい。簡単に述べると、そのような技術を使用する場合には、染色体配列との相同組換えによって内因性遺伝子に組み込まれ、その機能を破壊するために、内因性MKK遺伝子と相同なヌクレオチド配列を含むベクターがデザインされる。トランスジーンも、特定のタイプの細胞に選択的に導入され、そのタイプの細胞でのみ内因性MKK遺伝子を不活化することができる(Guら、Science 265:103, 1984)。そのような細胞のタイプに特異的な不活化に必要な調節配列は、興味のある特定の細胞のタイプに依存し、当業者には明らかだろう。これらの技術は、機能的なMKK遺伝子を持たない「ノックアウト」の調製に有用である。
【0152】
トランスジェニック動物が作製されたら、標準的な方法で、組換えMKK遺伝子の発現を解析できる。最初のスクリーニングは、サザンブロット分析またはPCR技術による、トランスジーンの組み込みが起きたかどうかの決定である可能性がある。トランスジェニック動物の組織中でのトランスジーンのmRNA発現のレベルは、動物から得た組織試料のノーザンブロット分析、インサイチューハイブリダイゼーション分析、およびRT-PCRを含むが、これらに限定されることのない技術を用いても、評価できる。MKK遺伝子発現組織の試料は、MKKトランスジーン産物に特異的な抗体を用いて免疫組織化学的にも、評価できる。
【0153】
トランスジェニック動物の作製および評価に使用できる技術の総説には、当業者はGordon (Intl. Rev. Cytol. 115:171-229, 1989)を参照し、例えば、Hoganら、(マウス胚の操作(Manipulating the Mouse Embryo), Cold Spring Harbor Press, Cold Spring Harbor, NY, 1986)、Krimpenfortら(Bio/Technology 9:86, 1991)、Palmiterら(Cell 41:343, 1985)、Kraemerら(初期哺乳類胚の遺伝子操作(Genetic Manipulation of the Early Mammalian Embryo), Cold Spring Harbor Press, Cold Spring Harbor, NY, 1985)、Hammer ら(Nature 315:680, 1985)、Purcelら(Science, 224:1281, 1986)、Wagner ら(米国特許第5,175,385号)、およびKrimpenfortら(米国特許第5,175,384号)から、さらに手引きを得ることができる可能性がある(最後の2つは、参照として本明細書に組み入れられる)。
【0154】
他の態様
本発明はその詳細な説明と関連して説明されてきたが、上述の説明は、添付の請求の範囲によって定義される本発明の範囲を明らかにするためのもので、それを制限する意図はないことを理解する必要がある。他の局面、利点、および修正は、請求の範囲内である。
【図面の簡単な説明】
【0155】
【図1】図1はMKK3(配列番号:2)、MKK4-α(配列番号:6)、ヒトMAPキナーゼキナーゼMEK1(配列番号:11)およびMEK2(配列番号:12)、ならびに酵母HOG1 MAPキナーゼキナーゼPBS2(配列番号:13)のアミノ酸配列を比較したものである。配列は、PILE-UPプログラム(バージョン7.2;Wisconsin Genetics Computer Group)を用いて比較した。蛋白質配列は、1文字コード(A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, TrpおよびY, Tyr)で示されている。PBS2配列は、NH2-(<)およびCOOH-(>)末端の両方で切断されている。整合を最適化するために配列に導入されたギャップは、ハイフンで示されている。同一の残基は、ピリオドで示されている。MEK中の活性化リン酸化の部位は、アステリスクで示されている。
【図2A】図2Aは、ヒトおよび酵母のMAPキナーゼキナーゼのメンバーの間の関係を示す樹状図である。樹状図は、算術平均を用いて、無加重のペアグループ法によって、作製された(PILE-UPプログラム)。ヒト(hu) MAPキナーゼキナーゼMEK1、MEK2、MEK3、およびMEK4;Sccharomyces cerevisiae(サッカロミセス・セレビジエ)(sc) MAPキナーゼキナーゼPBS2、MKK1、およびSTE7;およびSccharomyces pombe(サッカロミセス・ポンベ)(sp) MAPキナーゼキナーゼWIS1およびBYR1が示されている。
【図2B】図2Bは、MKKの間の関係を示す樹状図である。樹状図は、図2Aのようにして作製された。
【図3】図3は、ERK、p38、およびJNKシグナル伝達経路を模式図で表わしたものである。MEK1およびMEK2は、MAPキナーゼのERKサブグループの活性化因子である。MKK3およびMKK4は、p38 MAPキナーゼの活性化因子である。MKK4は、MAPキナーゼのp38およびJNKサブグループの両方の活性化因子であると同定されている。
【図4A】図4A〜4Dは、MKK3の核酸配列(配列番号:1)およびアミノ酸配列(配列番号:2)を表わしたものである。
【図4B】図4Aの続きを示す図である。
【図4C】図4Bの続きを示す図である。
【図4D】図4Cの続きを示す図である。
【図5A】図5A〜5Cは、MKK6の核酸配列(配列番号:3)およびアミノ酸配列(配列番号:4)を表わしたものである。
【図5B】図5Aの続きを示す図である。
【図5C】図5Bの続きを示す図である。
【図6A】図6A〜6Fは、MKK4αの核酸配列(配列番号:5)およびアミノ酸配列(配列番号:6)を表わしたものである。
【図6B】図6Aの続きを示す図である。
【図6C】図6Bの続きを示す図である。
【図6D】図6Cの続きを示す図である。
【図6E】図6Dの続きを示す図である。
【図6F】図6Eの続きを示す図である。
【図7A】図7A〜7Fは、MKK4βの核酸配列(配列番号:7)およびアミノ酸配列(配列番号:8)を表わしたものである。
【図7B】図7Aの続きを示す図である。
【図7C】図7Bの続きを示す図である。
【図7D】図7Cの続きを示す図である。
【図7E】図7Dの続きを示す図である。
【図7F】図7Eの続きを示す図である。
【図8A】図8A〜8Fは、MKK4γの核酸配列(配列番号:9)およびアミノ酸配列(配列番号:10)を表わしたものである。
【図8B】図8Aの続きを示す図である。
【図8C】図8Bの続きを示す図である。
【図8D】図8Cの続きを示す図である。
【図8E】図8Dの続きを示す図である。
【図8F】図8Eの続きを示す図である。
【図9A】図9は、PILE-UPプログラム(バージョン7.2;Wisconsin Genetics Computer Group)を用いて、MKK7(配列番号:18)の推定される一次構造を、hep(配列番号:21)、MAPキナーゼキナーゼMEK1(MKK1;配列番号:11)、MEK2(MKK2;配列番号:12)、MKK3(配列番号:2)、MKK4γ(配列番号:10)、MKK5(配列番号:22)、およびMKK6(配列番号:4)と比較したものである。整合を最適化するために配列に導入されたギャップは、ハイフン(-)で示されている。同一の残基は、ピリオド(.)で示されている。MAPキナーゼキナーゼ(2、27、37、および38)中の活性化リン酸化の部位は、アステリスク(*)で示されている。
【図9B】図9Aの続きを示す図である。
【図10A】図10A〜10Dは、MKK7の核酸配列(配列番号:17)およびアミノ酸配列(配列番号:18)を表わしたものである。
【図10B】図10Aの続きを示す図である。
【図10C】図10Bの続きを示す図である。
【図10D】図10Cの続きを示す図である。
【図11A】図11A〜11Dは、MKK7bの核酸配列(配列番号:19)およびアミノ酸配列(配列番号:20)を表わしたものである。
【図11B】図11Aの続きを示す図である。
【図11C】図11Bの続きを示す図である。
【図11D】図11Cの続きを示す図である。
【図12A】図12A〜12Bは、ヒトMKK7の核酸配列(配列番号:25)およびアミノ酸配列(配列番号:26)を表わしたものである。
【図12B】図12Aの続きを示す図である。
【図13A】図13A〜13Dは、マウスMKK7cの核酸配列(配列番号:27)およびアミノ酸配列(配列番号:28)を表わしたものである。
【図13B】図13Aの続きを示す図である。
【図13C】図13Bの続きを示す図である。
【図13D】図13Cの続きを示す図である。
【図14A】図14A〜14Dは、マウスMKK7dの核酸配列(配列番号:29)およびアミノ酸配列(配列番号:30)を表わしたものである。
【図14B】図14Aの続きを示す図である。
【図14C】図14Bの続きを示す図である。
【図14D】図14Cの続きを示す図である。
【図15A】図15A〜15Dは、マウスMKK7eの核酸配列(配列番号:31)およびアミノ酸配列(配列番号:32)を表わしたものである。
【図15B】図15Aの続きを示す図である。
【図15C】図15Bの続きを示す図である。
【図15D】図15Cの続きを示す図である。
【図16】図16Aは、細胞にAP-1レポータープラスミドpTRE-ルシフェラーゼと、MKK4、MKK7、JNK1、JNK1 (APF) の発現ベクターまたは対照ベクターとを、コトランスフェクトしたトランスフェクション解析のデータのグラフである。図16Bは、細胞にGAL4-ATF2融合ベクターと、MKK4、MKK7、JNK1、JNK1 (APF)の発現ベクターまたは対照ベクターとを、コトランスフェクトしたトランスフェクション解析のグラフである。
【技術分野】
【0001】
本発明は蛋白質キナーゼに関する。
【背景技術】
【0002】
マイトジェン活性化プロテイン(MAP)キナーゼは、細胞表面から核へのシグナル伝達の重要な仲介因子となっている。酵母では、SMK1、HOG1、MPK1、FUS3、およびKSS1を含む多数のMAPキナーゼが記述されている。哺乳類で同定されたMAPキナーゼは、細胞外シグナル制御MAPキナーゼ(ERK)、c-Junアミノ末端キナーゼ(JNK)、およびp38キナーゼ(Davis (1994) Trends Biochem. Sci. 19:470(非特許文献1))である。これらのMAPキナーゼアイソフォームは、スレオニンとチロシンの二重のリン酸化によって、活性化される。
【0003】
活性化転写因子2 (ATF2)、ATFa、およびcAMP応答配列結合蛋白質(CRE-BPa)は、多くの遺伝子のプロモーターにある類似配列に結合する関連した転写因子である(Ziff (1990) Trends in Genet. 6:69(非特許文献2))。これらの転写因子が結合すると、転写活性が上昇する。ATF2はオンコプロテインEla(LiuおよびGreen (1994) Nature 368:520(非特許文献3))、B型肝炎ウイルスX蛋白質(Maguireら(1991) Science 252:842(非特許文献4))、およびヒトT細胞白血病ウイルス1 tax蛋白質(WagnerおよびGreen (1993) Science 262:395(非特許文献5))を含むいくつかのウイルス蛋白質に結合する。またATF2は、癌抑制遺伝子産物Rb(Kimら(1992) Nature 358:331(非特許文献6))、高移動度群蛋白質HMG(I)Y(Duら(1993) Cell 74:887(非特許文献7))、ならびに転写因子である核NF- κB(Duら(1993) Cell 74:887(非特許文献7))およびc-Jun(BenbrookおよびJones (1990) Oncogene 5:295(非特許文献8))とも、相互作用する。
【非特許文献1】Davis (1994) Trends Biochem. Sci. 19:470
【非特許文献2】Ziff (1990) Trends in Genet. 6:69
【非特許文献3】LiuおよびGreen (1994) Nature 368:520
【非特許文献4】Maguireら(1991) Science 252:842
【非特許文献5】WagnerおよびGreen (1993) Science 262:395
【非特許文献6】Kimら(1992) Nature 358:331
【非特許文献7】Duら(1993) Cell 74:887
【非特許文献8】BenbrookおよびJones (1990) Oncogene 5:295
【発明の開示】
【0004】
本発明は、新しいグループのヒトマイトジェン活性化プロテインキナーゼキナーゼ(MKK)の同定と単離に基づく。本明細書に記述されるMKKアイソフォームMKK3、MKK6、MKK4(MKK4-α、-β、および-γを含む)、MKK7(マウスMKK7、ヒトMKK7、MKK7b、MKK7c、MKK7d、およびMKK7eを含む)は、セリン、スレオニン、およびチロシンキナーゼ活性を持つ。MKK3、MKK4、およびMKK6は、ヒトMAPキナーゼp38のThr180およびTyr182を特異的にリン酸化する。MKK4アイソフォームは、ヒトMAPキナーゼJNK(JNK1、JNK2、およびJNK5を含む)のThr183およびTyr185もリン酸化する。MKK7アイソフォームは、JNKのThr183およびTyr185をリン酸化する。
【0005】
したがって、本発明はヒトp38MAPキナーゼを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なヒトMKKポリペプチドを特徴とする。MKK3は、配列番号:2のアミノ酸配列を持つ。さらに本発明は、配列番号:4のアミノ酸配列を持ち、ヒトp38 MAPキナーゼを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つMKK6も含む。
【0006】
さらに本発明は、ヒトp38 MAPキナーゼおよびJNKを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なヒトMKKポリペプチドを特徴とする。MKK4アイソフォームのMKK4-αは、配列番号:6のアミノ酸配列を持つ。MKK4アイソフォームのMKK4-βは、配列番号:8のアミノ酸配列を持つ。MKK4アイソフォームのMKK4-γは、配列番号:10のアミノ酸配列を持つ。
【0007】
また、本発明はマイトジェン活性化プロテインキナーゼJNKを特異的にリン酸化するセリン、スレオニン、およびチロシンキナーゼ活性を持つ実質的に純粋なMKKポリペプチド(MKK7)をも特徴とする。MKKアイソフォームMKK7(マウス)およびMKK7(ヒト)は、それぞれ、配列番号:18および26のアミノ酸配列を持つ。MKK7アイソフォームのMKK7b、MKK7c、MKK7d、およびMKK7eは、それぞれ、配列番号:20、配列番号:28、配列番号:30および配列番号:32のアミノ酸配列を持つ。
【0008】
本明細書で使用される「マイトジェン活性化プロテインキナーゼキナーゼ」または「MKK」という用語は、ヒトマイトジェン活性化プロテインキナーゼのリン酸化および活性化という、特徴的な活性を持つ蛋白質キナーゼを意味する。MKKの例には、p38 MAPキナーゼのThr180およびTyr182を特異的にリン酸化し活性化するMKK3およびMKK6、p38 MAPキナーゼのThr180およびTyr182ならびにJNKのThr183およびTyr185を特異的にリン酸化し活性化するMKK4アイソフォーム、JNKのThr183およびTyr185を特異的にリン酸化するMKK7アイソフォームが含まれる。
【0009】
「MKK7」は、セリン、スレオニン、およびチロシンキナーゼ活性を持ち、マイトジェン活性化プロテイン(MAP)キナーゼJNKをリン酸化するがp38をリン酸化しない、マイトジェン活性化プロテインキナーゼキナーゼ(MKK)ポリペプチドの哺乳類アイソフォームである。
【0010】
本発明は、開示される特定のp38およびJNK MKK、ならびに本発明のMKKのポリヌクレオチドおよびアミノ酸配列から調製されるプローブまたは抗体を使用して同定および単離される近縁のMKKも含む。これは、例えば、ゲノム、cDNA、またはコンビナトリアルケミストリーのライブラリーを、開示されるMKKの核酸配列の全体または一部を持つプローブによってスクリーニングするなどの、標準的な技術を用いて実行できる。さらに本発明は、本明細書に記述されるMKKのアミノ酸配列の全体または一部を持つ合成ポリヌクレオチドも含む。
【0011】
「ポリペプチド」という用語は、長さまたは翻訳後修飾(例、グリコシル化またはリン酸化)に関わらず、任意のアミノ酸鎖を意味し、天然蛋白質ならびに合成または組換えのポリペプチドおよびペプチドを含む。
【0012】
ポリペプチドに関して用いる場合の「実質的に純粋」という用語は、天然に会合している蛋白質や天然に存在する有機分子を含まない、重量で少なくとも60%のポリペプチドを意味する。実質的に純粋なMKKポリペプチド(例、ヒト)は、重量で少なくとも75%、より好ましくは少なくとも90%、および最も好ましくは少なくとも99%、MKKポリペプチドである。実質的に純粋なMKKは、例えば、天然源からの抽出、MKKポリペプチドをコードする組換え核酸の発現、または蛋白質の化学合成によって得られる。純度は、例えば、カラムクロマトグラフィー、ポリアクリルアミドゲル電気泳動、またはHPLC分析のような、任意の適切な方法で測定できる。
【0013】
1つの局面において、本発明は、本発明のMKKをコードする単離ポリヌクレオチドを特徴とする。1つの態様では、ポリヌクレオチドは配列番号:1のヌクレオチド配列である。別の態様では、ポリヌクレオチドは、それぞれ配列番号:3、配列番号:5、配列番号:7、配列番号:9、配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、または配列番号:31のヌクレオチド配列である。
【0014】
本明細書で使用される「ポリヌクレオチド」とは、分離した断片、または、より大きな構築物の成分の形の、デオキシリボヌクレオチドまたはリボヌクレオチドの核酸配列を指す。本発明のポリペプチドの一部または全体をコードするDNAは、組換え転写ユニット中で発現できる合成遺伝子を提供するcDNA断片またはオリゴヌクレオチドから、組み立てることができる。本発明のポリヌクレオチド配列には、DNA、RNA、およびcDNA配列が含まれ、天然由来または当技術分野で公知の方法によって合成された合成配列であり得る。
【0015】
「単離された」ポリヌクレオチドとは、生物体の天然に存在するゲノムにおける配列から、何らかのやり方で分離された核酸分子である。したがって、「単離ポリヌクレオチド」という用語には、天然に存在しない任意の核酸分子が含まれる。したがって、この用語には、例えば、ベクター、自律的に複製するプラスミドもしくはウイルス、もしくは原核生物もしくは真核生物のゲノムDNAに組み込まれた組換えポリヌクレオチド、または他の配列とは独立した別個の分子として存在する組換えポリヌクレオチドが含まれる。また、別のポリペプチド配列をコードするハイブリッド遺伝子の一部である組換えDNAも含まれる。
【0016】
本発明の単離ポリヌクレオチド配列には、本明細書で説明するポリヌクレオチド配列に、ストリンジェントな条件下でハイブリダイズするポリヌクレオチド配列も含まれる。「ストリンジェントな条件」という用語は、本明細書に記述するような、ハイブリダイズするポリヌクレオチド配列間の特異性を保証するようなハイブリダイゼーション条件または、それよりも厳密な条件を意味する。当業者は、非特異的なハイブリダイゼーションの数を低下させ、相補性の高い配列のみが同定されるような、温度や塩濃度を含めたハイブリダイゼーション後の洗浄条件を選択することができる(Sambrookら(1989)「分子クローニング(Molecular Cloning)」第2版;Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY)。
【0017】
本発明の単離ポリヌクレオチド配列には、MKKをコードするポリヌクレオチドに相補的な配列(アンチセンス配列)も含まれる。アンチセンス核酸は、特定のmRNA分子の少なくとも一部に相補的なDNAまたはRNA分子である(Weintraub (1990) Scientific American 262:40)。本発明には、MKKポリペプチドの生産を阻害するすべてのアンチセンスポリヌクレオチドが含まれる。細胞中では、アンチセンス核酸は、対応するmRNAにハイブリダイズし、二重鎖分子を形成する。約15ヌクレオチドのアンチセンスオリゴマーは、簡単に合成され、標的MKK産生細胞に導入できるので、好ましい。アンチセンス法を用いて遺伝子の翻訳を阻害する方法は、当技術分野で公知であり、例えばMarcus-Sakura Anal. Biochem., 172:289 (1988)に記述されている。
【0018】
また、MKKのリボザイムヌクレオチド配列も、本発明に含まれる。リボザイムは、DNA制限エンドヌクレアーゼと同様な方法で、他の一本鎖RNAを特異的に切断する能力を持つRNA分子である。これらのRNAをコードするヌクレオチド配列を修飾することによって、RNA分子中の特異的なヌクレオチド配列を認識し、切断するように分子を操作できる(Cech (1988) J. Amer. Med. Assn. 260:3030)。この方法の大きな利点は、配列特異的であるため、特定の配列のmRNAのみが不活化されるということである。
【0019】
リボザイムには、テトラヒメナ型(Hasselhoff (1988) Nature 334:585)および「ハンマーヘッド」型の、2つの基本的なタイプがある。テトラヒメナ型リボザイムは、長さが4塩基の配列を認識するが、「ハンマーヘッド」型リボザイムは、長さが11〜18塩基の塩基配列を認識する。配列が長ければ長いほど、その配列が標的mRNA種のみに見られる可能性が高くなる。したがって、特定のmRNA種を不活化するためには、テトラヒメナ型リボザイムよりもハンマーヘッド型リボザイムの方が好ましく、短い認識配列よりも、18塩基の認識配列のほうが好ましい。
【0020】
MKKポリペプチドは、MKKポリペプチドのエピトープと免疫反応するか、結合する抗体を生産するためにも、使用できる。したがって、本発明の1つの局面は、本発明のMKKポリペプチドに対する抗体を特徴とする。本発明の抗体には、異なるエピトープ特異性を持つモノクローナル抗体のプールを含むポリクローナル抗体、および別個のモノクローナル抗体調製物が含まれる。モノクローナル抗体は、当技術分野で公知の方法によって、MKKポリペプチドの抗原含有断片から生産される(例えば、Kohlerら(1975) Nature 256:495参照)。
【0021】
本明細書で使用される「抗体」という用語は、エピトープ決定基に結合する能力のある、完全な分子、ならびにFa、F(ab')2、およびFvなどの、その断片を含む。MKKポリペプチドに特異的に結合する抗体は、完全なポリペプチドまたは目的の小さなペプチドを含む断片を、免疫抗原として使用して調製できる。動物の免疫に使用するポリペプチドまたはペプチドは、翻訳されたcDNA由来でも化学合成されたものでもよく、必要に応じてキャリア蛋白質に結合できる。ペプチドに化学的に結合されて一般的に使用されるキャリアには、ウシ血清アルブミンおよびチログロブリンが含まれる。その後、結合されたペプチドを用いて動物(例、マウス、ラットまたはウサギ)を免疫する。
【0022】
「特異的に結合する」分子(例、抗体)とは、例えばMKK7を含む生体試料などの試料中で、例えばMKK7のような特定のポリペプチドに結合するが、他の分子は実質的に認識しないか、または結合しない分子である。検出可能マーカーを含む化合物に結合した抗体(またはその断片)から成る構築物という表現には、化学的方法および組換え技術を含む、任意の技術により作製された構築物が含まれる。
【0023】
本発明は、MKKシグナル伝達経路の活性化を測定することによって、MKKを介する疾病のリスクを持つ患者を同定する方法も特徴とする。MKKシグナル伝達経路の活性化は、MKK合成;MKKアイソフォームの活性化;MKK基質p38またはJNKのアイソフォームの活性化;またはATF2、ATFa、CRE-BPaおよびc-Junのようなp38およびJNKの基質の活性化を、測定することにより決定できる。「JNK」または「JNKアイソフォーム」という用語には、JNK1、JNK2、およびJNK3が含まれる。本明細書で使用される「MKK基質」という用語には、MKKの基質およびMKKの基質の基質、例、p38、JNK、ATF2、およびc-Junも含まれる。
【0024】
1つの態様において、MKKシグナル伝達経路の活性化は、適当なMKKシグナル伝達経路基質(例、p38、JNKアイソフォーム、ATF2、ATFa、CRE-BPa、またはc-Junから選択される)の活性化を測定することによって決定される。MKK活性は、標識されたリン(例えば[32]Pまたは[33]P)の取り込みの割合を定量することによって決定される基質のリン酸化の割合によって測定される。これは、抗体のような、リン酸化に特異的な試薬を用いても測定できる。MKKの基質リン酸化の特異性は、p38の活性化、JNKの活性化、もしくはその両方を測定することによって、またはMKKリン酸化部位を持たない変異p38もしくはJNK分子を利用して調べられる。対照値と比較して基質のリン酸化が変化していることは、MKKシグナル伝達経路が変化したことを示し、患者のMKKを介する疾病のリスクが高まっていることを示す。MKKによるp38およびJNKの活性化は、MKKシグナル伝達の基質ATF2、またはATFaやCRE-BPaのような関連化合物と組み合わせたアッセイ法で検出できる。基質c-Junを用いても、活性化は検出できる。ATF2を解析に使用する際には、これは完全な蛋白質または完全な蛋白質の断片、例えば活性化ドメイン(残基1〜109、またはその一部)として存在する。ATF2は、MKK活性を測定する試験試料および[γ-32P]ATPと共に、ATF2がリン酸化されるのに十分な条件下でインキュベーションする。その後、ATF2を単離して、リン酸化の量を定量する。1つの特定の態様において、ATF2は免疫沈降によって単離され、SDS-PAGEによって解析され、オートラジオグラフィーによって検出される。
【0025】
別の態様において、MKKシグナル伝達経路の活性化は、試験試料中のMKK発現のレベルを測定することにより決定される。1つの特定の態様では、MKK発現のレベルは、ウエスタンブロット分析で測定される。試料中に存在する蛋白質は、ゲル電気泳動によって分別し、メンブレンに移し、MKKに対する標識抗体でプローブする。別の特定の態様では、MKK発現のレベルは、ノーザンブロット分析で測定する。細胞の総mRNAまたはリン酸化[ポリ(A)+]mRNAを試験試料から単離する。RNAは電気泳動によって分別し、メンブレンに移す。メンブレンは標識MKK cDNAでプローブする。別の態様いおいて、MKK発現は、発現したmRNAに対する定量的PCRによって測定される。
【0026】
本発明のMKKは、MKK活性を調節する試薬のスクリーニングに役立つ。MKKはリン酸化によって活性化される。したがって、1つの局面において、本発明は、試験試薬とMKKをインキュベートして、MKKの合成、リン酸化、機能、または活性に対する試験試薬の効果を測定することによって、MKK活性を調節する試薬を同定する方法を特徴とする。1つの態様において、試験試薬はMKKおよび[32]P-ATPと共にインキュベートされ、上述のようにMKKリン酸化の速度が決定される。別の態様では、MKKポリヌクレオチド発現ベクターによりトランスフェクトされた細胞と共に試験試薬をインキュベートし、上述のように、ノーザンブロット分析によってMKK転写に対する試験試薬の効果を測定する。別の態様では、MKK合成に対する試験試薬の効果は、MKKに対する抗体を用いてウエスタンブロット分析によって測定される。さらに別の態様では、MKK活性に対する試薬の効果は、試験試薬、[32]P-ATP、ならびにp38、JNK、およびATF2のうちの1つまたは複数を含むMKKシグナル伝達経路の基質と共に、MKKをインキュベートすることにより測定される。基質のリン酸化の割合は、上述のように決定される。
【0027】
「MKK活性の調節」という用語には、阻害または刺激効果が含まれる。
【0028】
本発明は、MKK活性を阻害する試薬のスクリーニングに特に有用である。そのような試薬は、例えば、炎症および酸化による損傷のような、MKKを介する疾病の治療または予防に役立つ。
【0029】
本発明はさらに、MKK活性を阻害する治療試薬の有効量を、必要としている患者に投与することにより、MKKを介する疾病を治療する方法を特徴とする。
【0030】
「MKKを介する疾病」とは、少なくとも一部は、MKKシグナル伝達経路の過剰な活性化の結果として起こる病的状態である。MKKシグナル伝達経路は、炎症およびストレスを含む、いくつかの要因により活性化される。MKKを介する疾病には、例えば、虚血性心疾患、熱または放射線(UV、X-線、γ、β等)による熱傷、腎不全、酸化ストレスまたはアルコールによる肝臓障害、呼吸障害症候群、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、およびその他の種類の炎症性疾病が含まれる。
【0031】
「治療試薬」とは、必要とする患者に投与された場合に、MKKを介する疾病に望ましい効果を与える任意の化合物または分子である。
【0032】
MKKを介する疾病には、増殖性疾患、特にストレスに関連した疾患がさらに含まれる。ストレスに関連したMKKを介する増殖性疾患の例には、乾癬、後天性免疫不全症候群、皮膚、骨髄、肺、肝臓、乳房、胃腸系、および尿生殖路の悪性腫瘍を含む、体の種々の組織の悪性腫瘍がある。好ましくは、治療試薬はMKKの活性または発現を阻害して、細胞の増殖を阻害するかアポトーシスを誘導する。
【0033】
「MKK活性を阻害する」治療試薬は、MKKを介するシグナル伝達経路に干渉する。例えば、治療試薬はMKKのプロテインキナーゼ活性を変化させたり、例えばMKK mRNAに結合できるアンチセンスポリヌクレオチドのように、MKKの転写または翻訳レベルを低下させたり、またはp38、JNK、もしくはATF2のMKKによるリン酸化を抑制することができ、その結果MKKを介するシグナル伝達経路を混乱させる。そのような試薬の例には、MKKポリペプチドに特異的に結合する抗体、およびMKKポリペプチド活性を競合阻害するMKKポリペプチド断片が含まれる。
【0034】
「MKK活性を増大させる」治療試薬は、MKKを介するシグナル伝達経路を補う。そのような試薬の例には、MKKポリペプチド自身が含まれ、これはMKKポリペプチドの発現不足、または変異MKKポリペプチドの発現に起因するMKKを介する疾病の場合に投与できる。さらに、MKKポリペプチドをコードするDNAの一部を、MKKポリペプチドの発現が不足している細胞に導入することもできる。
【0035】
「治療的有効量」とは、MKKを介する疾病に伴う症状を低下または予防するために十分な試薬の量である。
【0036】
本発明の方法によって同定されるMKKを介する疾病の治療のための治療試薬は、注射、注入、徐放性の注射または移植による非経口的方法を含む、当技術分野で公知のいくつもの方法で、患者の静脈内、腹腔内、筋肉内、皮下、または経皮的に投与される。表皮の疾患および上皮組織の疾患は、試薬の局所適用により治療される。試薬は、安定性と送達の効率を改善するために他の化合物と混合される(例、リポソーム、保存剤、またはジメチルスルホキシド(DMSO))。アンチセンス配列を含むポリヌクレオチド配列を、当技術分野で公知の技術を用いて治療用に投与し、MKKを介する疾病を患う患者の細胞中に導入することができる。これらの方法には、ウイルスベクター(例、レトロウイルス、アデノウイルス、ワクシニアウイルス、またはヘルペスウイルス)、コロイド分散、およびリポソームの使用が含まれる。
【0037】
本発明の材料は、MKKのレベルまたは活性の検出のためのキットの調製に、理想的に適している。したがって、本発明は、MKKに結合する抗体またはMKKポリヌクレオチドにハイブリダイズする核酸プローブ、および適当な緩衝液を含むキットを特徴とする。プローブまたはモノクローナル抗体は、MKKポリヌクレオチドまたは蛋白質への結合を検出するために標識することができる。1つの好ましい態様において、キットはMKKに対する標識抗体を特徴とする。
【0038】
特に定義しないかぎり、本明細書で使用される全ての技術および科学的用語は、本発明が属する技術分野の当業者が一般に理解するものと同一の意味を持つ。本発明の実施または試験には、本明細書に説明する方法および材料と類似または同等なものを使用することもできるが、適当な方法および材料を以下に記述する。本明細書に記載される出版物、特許出願、特許、および他の参考文献は全て、その全体が参照として組み入れられる。さらに、材料、方法、および実施例は説明のみのためであり、制限する意図はない。
【0039】
本発明の他の特徴および利点は、以下の詳細な説明および請求の範囲から明らかになると思われる。
【0040】
ヒトマイトジェン活性化プロテインキナーゼキナーゼ
本明細書に記述されるヒトMAPキナーゼキナーゼMKK3およびMKK4 (MKK3/4)、およびMKK7は、細胞表面から核までの特定の経路に沿った特異的なシグナルの伝達を仲介する。これらのシグナル伝達経路は、サイトカイン、UV照射、浸透圧ショック、および酸化ストレスのような要素によって開始する。MKK3/4、MKK6、およびMKK7が活性化されると、MAPキナーゼの活性化が起きる。p38はMKK3およびMKK4により活性化される。JNKはMKK4およびMKK7により活性化される。次に、p38およびJNKは、ATF2、ATFa、およびCRE-BPaのような関連する転写因子のグループを活性化する。次にこれらの転写因子は、特定の遺伝子の発現を活性化する。例えば、ATF2はヒトT細胞白血病ウイルス1(WagnerおよびGreen (1993) Science 262:395)、トランスフォーミング増殖因子b2(Kimら(1992)前記)、インターフェロンβ(Duら(1993) Cell 74:887)、およびE-セレクチン(DeLucaら(1994) J. Biol. Chem. 269:19193)の発現を活性化することが知られている。また、ATF2は、T細胞特異的エンハンサーの機能にも関係する(Georgopoulosら(1992) Mol. Cell. Biol. 12:747)。
【0041】
MAPキナーゼのJNKグループは、細胞が環境ストレスにさらされたり、炎症促進型サイトカインで細胞を処理すると活性化される(Guptaら(1994) EMBO J. 15:2760-2770; Derijardら(1991) Cell 76:1025-1037; Kyriakisら(1994) Nature 369:156-160; Slussら(1994) Mol. Cell. Biol. 14:8376-8384; Kallunkiら(1994) Genes & Dev. 8:2996-3007)。JNKシグナル伝達経路の標的には、転写因子ATF2およびc-jun(Whitmarsh & Davis (1996) J. Mol. Med. 74:589-607)が含まれる。これらの転写因子は、多くの遺伝子のプロモーター中のAP-1およびAP-1様部位に、ホモまたはヘテロダイマー複合体として結合するbZIPグループのメンバーである(Curran & Franza (1988) Cell 55:395-397)。JNKはATF2およびc-JunのNH2末端領域に結合し、各々の転写因子の活性化ドメイン内の2つの部位をリン酸化する(Derijardら(1994) Cell 76:1025-1037; van Damら(1995) EMBO J. 14:1798-1811; Livingstoneら(1995) EMBO J. 14:1785-1797)。このリン酸化によって、転写活性が上昇する(Whitmarsh、前記)。これらの生化学研究を合わせると、JNKシグナル伝達経路が、サイトカインおよび環境ストレスに応答したAP-1転写活性の調節に寄与していることが示される(Whitmarsh、前記)。JNKシグナル伝達経路がAP1転写活性の正常な調節に必要であることを示す遺伝的証拠が、この仮説を強く支持している(Yangら(1997) Proc. Natl. Acad. Sci. USA, 94:3004-3009)。
【0042】
JNKは、Thr-183およびTyr-185の二重のリン酸化によって活性化される(Derijard、前記)。MKK4(SEKIとしても知られる)は、JNKシグナル伝達経路の成分として同定された最初のMAPキナーゼキナーゼであった(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290; Sanchezら(1994) Nature 372:794-798)。MKK4がJNKをリン酸化し活性化することが生化学研究によって示されている(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290; Sanchezら(1994) Nature 372:794-798)。しかし、MKK4はp38 MAPキナーゼもリン酸化し活性化するので、MKK4の機能は、JNKシグナル伝達経路に限定されていない可能性がある(Derijardら(1995) Science 267:682-685; Linら(1995) Science 268:286-290)。MKK4がJNKおよびp38 MAPキナーゼの両方を活性化するというこの特異性により、サイトカインや環境ストレスによって処理された細胞における、これらのMAPキナーゼの同格の活性化を担うと考えられる機構が提供される(Davis (1994) Trends Biochem. Sci. 19:470-473)。しかし、この同格の活性化は、常に観察されるとは限らない。例えば、肝臓におけるJNKの活性化は、p38 MAPキナーゼ活性の低下と相関している(Mendelsonら(1996) Proc. Natl. Acad. Sci. USA 93:12908-12913)。これらのデータは、MKK4の性質は、インビボにおけるJNKの調節を説明するには不十分であることを示唆している。
【0043】
ヒトMKKの単離は、実施例1、実施例22、Derijardら((1995) Science 267:682-685、参照として本明細書に組み入れられる)、およびRaingeaudら((1995) Mol. Cell. Biol. 16:1247-1255)に記述されている。酵母PBS2配列の特徴的な領域を用いて、ポリメラーゼ連鎖反応(PCR)プライマーがデザインされた。これらのプライマーを用いてヒト脳mRNAを増幅すると、特異的な産物が形成されたので、これらをプラスミドベクターにクローニングし、配列の決定を行った。ヒトプロテインキナーゼをコードする2つの異なる相補的DNA(cDNA)が同定された:1つは36 kD蛋白質(MKK3)をコードし、1つは44 kD蛋白質(MKK4)をコードしていた。MKK4には、α、β、およびγと同定された、NH2末端が僅かに異なる3つのアイソフォームが含まれている。MKK3(配列番号:2)、MKK4-α(配列番号:6)、MKK4-β(配列番号:8)、およびMKK4-γ(配列番号:10)のアミノ酸配列は、図1に示されている。MKK3(図4)、MKK6(図5)、MKK4-α(図6)、MKK4-β(図7)、およびMKK4−γ(図8)の核酸およびアミノ酸配列も示されている。MKK6は、MKK3とのクロスハイブリダイゼーションによって、ヒトの骨格筋ライブラリーから単離された。N末端の違いを除いて、MKK6はMKK3と相同性が高い。存在するヒトMKK3およびMKK4の他のアイソフォームは、実施例1に説明する方法によって同定できる。
【0044】
これらのヒトMKKアイソフォームの発現を、8人の成人組織から単離されたmRNAのノーザン(RNA)ブロット分析によって調べた(実施例2)。両方のプロテインキナーゼとも、ヒトの組織で広範囲に発現されており、骨格筋組織で最も発現が多いことが分かった。
【0045】
MKK3の基質特異性は、エピトーグタグを付けた組換えMAPキナーゼ(JNK1、p38およびERK2)を基質として用いたインビトロのリン酸化解析で調べた(実施例3)。MKK3はp38をリン酸化したが、JNK1またはERK2はリン酸化しなかった。p38のリン酸化アミノ酸分析によって、ホスホスレオニンおよびホスホチロシンの存在が示された。p38の変異分析によって、Thr180およびTyr182のリン酸化部位を、それぞれAlaおよびPheによって置換すると、p38のリン酸化が阻止されることが示された。これらの結果は、MKK3がインビトロでp38 MAPキナーゼキナーゼとして働くことを立証するものである。
【0046】
MKK4の基質特異性のインビトロ実験は、実施例4に記載されている。[γ-32P]ATP、およびJNK1、p38、またはERK2と共にインキュベーションされたMKK4は、p38およびJNK1の両方をリン酸化することが分かった。MKK4によるJNKおよびp38の活性化もまた、MKK4を野生型または変異型JNK1またはp38とインキュベーションすることにより調べられた。p38の基質ATF2は、各解析に含まれていた。MKK4は、MKK3よりも自己リン酸化が少ないことが分かった。MKK4は、活性化されたMAPキナーゼの基質でもあることが分かった。MKK3とは異なり、MKK4はJNK1も活性化することが分かった。野生型JNK1とインキュベートされたMKK4は、ATF2のリン酸化を上昇させたが、変異型JNK1とインキュベーションした場合には上昇させなかった。これらの結果は、MKK4が、MAPキナーゼのJNKサブグループもリン酸化するp38 MAPキナーゼキナーゼであることを立証する。
【0047】
UV刺激を受けたMKK3によるインビボのp38の活性化は、実施例5に記載されている。MKK3を発現する細胞を、UV照射の存在下または非存在下で露出した。免疫沈降によってMKK3を単離し、基質のp38またはJNKを用いてプロテインキナーゼアッセイ法を行なった。一部のアッセイ法では、p38およびJNKの基質として、ATF2が含まれていた。非活性化培養COS細胞由来のMKK3は、MKK3の基礎活性による、p38 MAPキナーゼのリン酸化を低レベルで誘導した。UV照射した細胞のMKK3は、p38 MAPキナーゼのリン酸化を増加させたがJNK1のリン酸化は増加させなかった。p38活性の上昇は、基質としてATF2を含めたアッセイ法でも検出された。これらの結果は、MKK3がUV照射によって活性化されることを立証する。
【0048】
MKK3およびMKK4の発現のp38活性に対する効果は、COS-1細胞で調べられた(実施例6)。p38およびMEK1、MKK3、またはMKK4をコードするベクターで細胞をトランスフェクトした。細胞の一部は、EGFまたはUV照射の処理をした。免疫沈降によってp38を単離し、[γ-32P]ATPおよびATF2によって活性を解析した。ERK活性化因子MEK1の発現は、p38によるATF2のリン酸化を変化させなかった。これとは対照的に、MKK3またはMKK4の発現は、p38 MAPキナーゼの活性を上昇させた。MKK3およびMKK4によるp38の活性化は、UV照射細胞で観察されたものと類似しており、EGF処理細胞で検出されるものよりもはるかに強かった。このようなインビトロの結果は、MKK3とMKK4がインビボでp38を活性化させるという証拠となる。
【0049】
JNK1によるATF2の調節の可能性を調べるために、一連の実験が行なわれた。これらの実験は、参照として本明細書に組み入れられるGuptaら(1995) Science 267:389-393に記述されている。ATF2のリン酸化に対するUV照射の効果が、エピトープタグを付けたJNK1の存在下および非存在下でトランスフェクトされたCOS-1細胞において調べられた(実施例7)。細胞にUV照射をし、JNK1およびJNK2は、基質ATF2を用いたゲル内プロテインキナーゼアッセイ法によって可視化した。JNK1およびJNK2は、UV照射をしたトランスフェクト細胞および非トランスフェクト細胞で検出された;しかし、JNK1レベルは、トランスフェクト細胞で高かった。これらの結果は、ATF2がJNK1およびJNK2プロテインキナーゼの基質であり、これらのプロテインキナーゼはUV線に露出された細胞で活性化されることを示す。
【0050】
JNK1によるATF2のリン酸化部位は、欠失分析によって調べられた(実施例8)。連続的なNH2末端ドメイン欠失GST-ATF2融合蛋白質を作製し、UV照射細胞から単離されたJNK1によるリン酸化を調べた。その結果、ATF2のNH2末端ドメインのリン酸化には、JNK1はATF2の残基1〜60の存在を必要とすることが示された。
【0051】
JNK1の結合に必要なATF2の残基も同様に調べた。JNK1を、固定化したATF2と共にインキュベートし、結合しなかったJNK1を念入りに洗浄して除去し、結合したJNK1を[γ-32P]ATPとのインキュベーションによって検出した。結果は、JNK1による結合とリン酸化のためには、ATF2の残基20〜60が必要なことを示している。ATF2と55 kD JNK2プロテインキナーゼとの間の同様な結合相互作用も観察された。
【0052】
JNK1によるリン酸化は、ATF2の電気泳動の移動度を低下させることが示された(実施例9)。全長ATF2分子(残基1〜505)のリン酸化アミノ酸分析によって、JNKはThrおよびSer残基の両方をリン酸化することが示された。ThrおよびSerリン酸化の主要な部位は、それぞれNH2およびCOOH末端ドメインに存在していた。NH2末端のリン酸化部位は、リン酸化ペプチドマッピングと変異解析によって、Thr69およびThr71と同定された。これらのThrリン酸化部位は、ATF2内でJNK結合に必要なサブドメイン(残基20〜60)とは別の領域に存在している。
【0053】
ATF2のリン酸化で見られる電気泳動の移動度の低下を、さらに詳しく調べられた(実施例10)。JNK1を発現するCHO細胞に、UV照射、炎症促進型サイトカインであるインターロイキン-1(IL-1)処理、または血清処理をして、JNK1を活性化した。JNK1に活性化されたATF2の電気泳動の移動度の低下は、UV照射とIL-1処理をした細胞で観察された。血清で細胞を処理すると、これよりも小さな効果が見られた。これらの結果は、ATF2がJNK1のインビボでの基質であることを示す。
【0054】
野生型(Thr69、71)ATF2およびリン酸化欠損(Ala69、71)ATF2分子の性質に対する、UV照射の効果を調べた(実施例11)。UVに露出すると、内因性および過剰発現した野生型のATF2の両方の電気泳動の移動度が低下した。電気泳動の移動度のこの変化は、ATF2のリン酸化の増加を伴っていた。電気泳動の移動度のシフトおよびリン酸化の増加のいずれも、ATF2内でThr69およびThr71をAlaで置換すると阻止された。この変異により、インビボにおけるATF2のThr残基のリン酸化も阻止された。
【0055】
GAL4 DNA結合ドメイン、および野生型または変異型ATF2からなる融合蛋白質の転写活性を調べた(実施例12)。ATF2のThr69および/またはThr71における点変異は、野生型分子と比較してATF2の転写活性を有意に低下させ、これらの部位におけるリン酸化が、活性に生理的に関連していることを示した。
【0056】
JNK1がATF2のNH2末端活性化ドメインに結合することから(実施例8に記述)、触媒作用の不活性なJNK1分子が、野生型JNK1分子のドミナントな阻害剤として機能することが示唆された。この仮説は、触媒作用の不活性なJNK1分子のATF2機能に対する効果を調べて、検証された(実施例13)。触媒作用の不活性なJNK1変異体は、Thr183およびTyr185活性化リン酸化部位を、それぞれAlaおよびPheで置換して作製された(Ala183、Phe185、「ドミナントネガティブ」と呼ばれる)。野生型JNK1の発現は、血清刺激されたATF2の転写活性を僅かに上昇させた。これとは対照的に、ドミナントネガティブJNK1は、対照および血清刺激されたATF2活性の両方を阻害した。この阻害効果は、JNK1変異体がATF2活性化ドメインに非生産的に結合し、ATF2のリン酸化を効果的に阻害することによる。
【0057】
癌抑制遺伝子産物Rbは、ATF2に結合し、ATF2に刺激される遺伝子発現を上昇させる(Kimら(1992) Nature 358:331)。同様に、アデノウイルスのオンコプロテインE1AはATF2のDNA結合ドメインと会合し、ATF2のNH2末端活性化ドメインが必要な機構によって、ATF2に刺激される遺伝子発現を上昇させる(LiuおよびGreen (1994) Nature 368:520)。ATF2の転写活性は、ルシフェラーゼレポーター遺伝子システムを用いて、野生型または変異型ATF2分子を発現する、対照、Rb処理、およびE1A処理細胞において調べられた(実施例14)。RbとE1Aは、野生型および変異型ATF2の両方の、ATF2に刺激される遺伝子発現を上昇させることが分かった。しかし、変異型ATF2では、野生型ATF2よりも、レポーター遺伝子発現のレベルは低かった。これらの結果を合わせると、最大の転写活性を得るためには、ATF2リン酸化(Thr69およびThr71での)に加えて、RbまたはE1Aのいずれかが必要であることが示される。したがって、RbおよびE1AはATF2のリン酸化と協調して働き、転写活性を制御している。
【0058】
P38活性化の作用を調べ、p38 MAPキナーゼ経路とERKおよびJNKシグナル伝達経路との関係を確立するために、一連の実験が行われた(Raingeaudら(1995) J. Biol. Chem. 270:7420、参照として本明細書に組み入れられる)。まず、ERKおよび/またはJNKグループのMAPキナーゼの基質であることが示されている蛋白質と共にp38をインキュベートすることにより、p38の基質特異性を調べた(実施例15)。本発明者らは、MBP(Ericksonら(1990) J. Biol. Chem. 265:19728)、EGF-R(Northwoodら(1991) J. Biol. Chem. 266:15266)、細胞質ホスホリパーゼA2 (cPLA2) (Linら(1993) Cell 72:269)、c-Myc(Alvarezら(1991) J. Biol. Chem. 266:15277)、IκB、c-Jun、および野生型(Thr69、71)または変異(Ala69、71)ATF2のリン酸化を調べた。p38はMBPおよびEGF-Rをリン酸化し、それよりも低いレベルでIκBをリン酸化したが、他のERK基質はリン酸化せず、このことからp38の基質特異性が、ERKおよびJNKグループのMAPキナーゼとは異なることが示される。野生型ATF2はp38の優れた基質であるが、変異型ATF2(Ala69、71)はそうでないことが判明した。
【0059】
p38によるATF2のリン酸化は、ポリアクリルアミドゲル電気泳動において、ATF2の電気泳動の移動度のシフトを伴った。本発明者らは、p38によるATF2のリン酸化部位は、JNK1と同じであるという仮説を、Thr69およびThr71をAla (Ala69、71)で置換することにより、検証した。p38は変異ATF2をリン酸化しないことが分かったが、これはp38がATF2のNH2末端活性化ドメイン内のThr69およびThr71をリン酸化することを示す。
【0060】
JNK1またはp38を発現する細胞の抽出物を、エピトープのみ(GST)またはGST- ATF2(活性化ドメインを含む残基1〜109)とインキュベートして、ATF2に対するJNKとp38の結合の比較を行なった(実施例16)。結合したプロテインキナーゼは、ウエスタンブロット分析によって検出した。結果は、p38とJNKの両方がATF2の活性化ドメインに結合することを示す。
【0061】
EGFおよびホルボールエステルは、ERKシグナル伝達経路の強力な活性化因子であり(EganおよびWeinberg (1993) Nature 365:781)、MAPキナーゼのERKサブグループを最大限に活性化する。しかし、これらの処理は、JNKプロテインキナーゼの活性を僅かに上昇させるのみである(Derijardら(1994) 上記;Hibiら(1993)上記)。EGFまたはホルボールエステル、およびUV照射、浸透圧ショック、インターロイキン-1、腫瘍壊死因子、およびLPSのp38活性に対する効果を全て試験した(実施例17)。重要なことに、EGFおよびホルボールエステルは、p38のプロテインキナーゼ活性を中程度にしか上昇させなかったが、環境ストレス(UV照射および浸透圧ショック)は、p38とJNKの両方の活性を著しく上昇させた。p38とJNKのいずれも、炎症促進型サイトカイン(TNFおよびIL-1)または内毒素LPSで処理された細胞において活性化された。これらの結果を合わせると、p38は、JNKと同様に、ストレスに誘導されるシグナル伝達経路によって活性化されることが示される。
【0062】
ERKおよびJNKは、それぞれ、モチーフThr-Glu-TyrおよびThr-Pro-Tyr内の2重のリン酸化によって活性化される。これとは対照的に、p38は関連配列Thr-Glu-Tyrを持っている。このモチーフがp38の活性化に関連しているかどうかを調べるために、Thr-Gly-TyrをAla-Gly-Pheで置換した効果を調べた(実施例18)。野生型(Thr180、Tyr182)または変異型p38(Ala180、Phe182)を発現する細胞に対するUV照射の効果を調べた。抗ホスホチロシン抗体を用いたウエスタンブロット分析では、UV照射を行なうとp38のTyrリン酸化が上昇することが示された。Tyrリン酸化の上昇は、[γ-32P]リン酸塩標識細胞から単離されたp38のホスホ化アミノ酸分析によって、確認された。この分析では、UV照射がp38のThrリン酸化を上昇させることも示された。重要なことに、Thr180およびTyr182のリン酸化の上昇は、Ala180/Phe182変異によって、阻止された。この結果は、UV照射が二重のリン酸化によるp38の活性化を増加させることを示す。
【0063】
最近、マイトジェンに誘導される二重特異性を持つフォスファターゼMKP1およびPAC1によって、ERK活性が調節されていることが示された(Wardら(1994) Nature 367:651)。二重リン酸化によりp38が活性化される(実施例18)ため、p38も二重特異性ホスファターゼによって調節されているという可能性が生じる。本発明者らは、p38 MAPキナーゼ活性に対するMKP1およびPAC1の効果を調べた(実施例19)。ヒトMKP1およびPAC1を発現する細胞を、UV照射処理をしたものとしないものとで、p38活性を測定した。PAC1またはMKP1の発現は、p38活性を阻害することが分かった。MKP1の阻害効果は、PAC1よりも大きかった。これとは対照に、触媒作用の不活性な変異ホスファターゼ(変異PAC1 Cys257/Ser)でトランスフェクトされた細胞は、p38 MAPキナーゼを阻害しなかった。これらの結果は、p38が二重特異性ホスファターゼPAC1およびMKP1によって調節されることを示す。
【0064】
p38 MAPキナーゼの細胞内分布は、間接免疫蛍光顕微鏡法によって調べた(実施例20)。エピトープタグを付けたp38 MAPキナーゼは、M2モノクローナル抗体を用いて検出した。エピトープタグを付けたp38 MAPキナーゼでトランスフェクトされた細胞の特異的な染色は、細胞表面、細胞質、および核で観察された。UV照射後には、細胞表面および核のp38 MAPキナーゼには著しい変化は観察されなかったが、細胞質p38 MAPキナーゼの核周囲領域への局在の増加が検出された。
【0065】
高浸透圧培地によるJNKの活性化を研究するために、一連の実験を行った(実施例21)。これらの実験は、参照として本明細書に組み入れられるGalcheva-Gargovaら(1994) Science 265:806に報告された。エピトープタグを付けたJNK1を発現するCHO細胞は、0〜1000 mMソルビトールとインキュベートされ、基質c-Junを用いて免疫複合体キナーゼ解析によって、JNK1活性が測定された。100 mMソルビトールで1時間インキュベートされた細胞で、JNK1活性の上昇が観察された。300 mMソルビトールへの露出では、5分以内にJNK1活性の上昇が観察された。最大の活性は、浸透圧ショックの15〜30分後に観察され、この後は、JNK1活性が徐々に低下した。浸透圧ショックによるJNKの活性化は、野生型(Thr183、Tyr185)または変異(Ala183、Phe185)JNK1を発現する細胞で調べた。JNK1活性は、300 mMソルビトールの存在下または非存在下での15分のインキュベーション後に測定された。野生型JNK1を発現する細胞では、JNK1活性の上昇が見られたが、変異JNK1を発現する細胞では見られなかった。これらの結果は、高浸透圧培地に露出された哺乳類培養細胞において、JNKシグナル伝達経路が活性化されることを示す。
【0066】
上述の実験の結果は、図3に示されており、これはERK、p38、およびJNK MAPキナーゼシグナル伝達経路を模式的に表わしている。細胞をEGFまたはホルボールエステルで処理すると、ERKが強力に活性化される。これとは対照的に、p38はこのような条件では僅かに活性化されるのみである(実施例15)。しかし、UV照射、浸透圧ショック、および炎症性サイトカインは、p38活性を著しく上昇させる。ERKとp38の活性化パターンのこのような違いは、これらのMAPキナーゼが異なるシグナル伝達経路によって調節されていることを示唆する。これらのシグナル伝達経路が別のものであることの分子的基礎は、ERKを活性化するMAPキナーゼキナーゼ(MEK1およびMEK2)とp38(MKK3、MKK4、およびMKK6)が異なるという証明によって立証される。
【0067】
マウスおよびヒトMKK7の単離は、実施例22に記述されている。ショウジョウバエMAPキナーゼキナーゼhep配列の特徴的な領域を用いて、ポリメラーゼ連鎖反応(PCR)のプライマーがデザインされた。これらのプライマーを用いてマウス精巣mRNAを増幅すると、特異的な産物が形成され、これはプラスミドベクターにクローニングされ、配列決定が行なわれた。hepに関連する1つの配列が同定され、マウス精巣ライブラリーのスクリーニングに使用された。プロテインキナーゼをコードする5つのDNA(cDNA)が同定された:1つはMAPプロテインキナーゼキナーゼ(MKK7)をコードしていた。他は種々のスプライシング変異体をコードしていた:MKK7b(部分配列が図11に記載されている)、MKK7c(図13)、MKK7d(図14)、MKK7e(図15)。MKK7(配列番号:18)およびhep(配列番号:21)の推定アミノ酸配列は図9に示されており、MAPキナーゼキナーゼMEK1(配列番号:11)、MEK2(配列番号:12)、MKK3(配列番号:2)、MKK4(配列番号:10)、MKK5(配列番号:22)、およびMKK6(配列番号:4)と比較されている。ヒトMKK7は、全長(マウス)MKK7 cDNAプローブを用いてヒトcDNAライブラリーをスクリーニングして、同定された。同定された部分配列(3'末端を欠く)は、マウスMKK7cと相同である。
【0068】
MKK7およびMKK4アイソフォームの発現は、8つのマウス組織から単離されたポリA+ mRNAのノーザン(RNA)ブロット分析によって調べた(実施例23)。両方のプロテインキナーゼが、広範囲で発現されていることが判明した。
【0069】
MKK7の基質特異性は、エピトープタグを付けた組換えMAPキナーゼ(JNK1、p38、およびERK2)を基質としたインビトロリン酸化アッセイ法で調べた(実施例24)。MKK7はJNKをリン酸化したが、p38もERK2もリン酸化しなかった。MKK7は、p38およびJNK1によってリン酸化された。
【0070】
MKK7は、インビボでJNKプロテインキナーゼを特異的に活性化することが分かった(実施例25)。CHO細胞に、エピトープタグを付けたMAPキナーゼ(JNK1、p38、またはERK2)と、空の発現ベクターまたはMKK1、MKK4、MKK6、もしくはMKK7をコードする発現ベクターとを、コトランスフェクションし、リン酸化反応の産物を分析した。MKK7はJNK1しか活性化しなかったが、その程度はMKK4よりも高かった。
【0071】
MKK7がAP-1転写活性を上昇させるかどうかを検証するために、コトランスフェクション解析を実施した(実施例26)。MKK7とJNKとを共に発現させると、AP-1レポーター遺伝子発現が上昇し、それはMKK4およびJNKで見られる上昇よりも大きかった。レポーター遺伝子にATF2を使った場合も、同様な結果が観察された。また、MKK7単独で、ATF2の発現を増加させることができた(図16)。
【0072】
MKKアイソフォームは、MKK活性を調節する試薬のスクリーニングに有用である。実施例の後の用途の項には、MKK活性を阻害または活性化する能力のある試薬の同定方法が記述されている。
【0073】
ヒトMKKアイソフォームおよびMKKが仲介するシグナル伝達経路を見出すことは、MKKを介する疾患の治療にとって、臨床的意義がある。MKKアイソフォームの1つの用途は、MKK-MAPキナーゼ-ATF2経路の活性化を阻害または予防できる試薬をスクリーニングするための方法における使用である。
【0074】
実施例
以下の実施例は、本発明を例証するためのもので、制限するものではない。
【0075】
実施例1. MKKプロテインキナーゼ
MKK3およびMKK4の一次配列は、ヒト胎児脳ライブラリーから単離されたcDNAクローンの配列から、推定された。
【0076】
PBS2(Brewsterら(1993) Science 259:1760; Maedaら(1994) Nature 369:242)の配列に基づいて、プライマーTTYTAYGGNGCNTTYTTYATHGA(配列番号:14)およびATBCTYTCNGGNGCCATKTA(配列番号:15)がデザインされた。プライマーは、ヒト脳mRNAを鋳型としたPCR反応で使用された。PBS2関連プロテインキナーゼの断片をコードする2つの配列が同定された。全長ヒトcDNAクローンは、ヒト胎児脳ライブラリーのスクリーニングで単離された(Derijardら(1995) Science 267:682-685)。cDNAクローンは、アプライド バイオシステムズ(Applied Biosystems)モデル373Aを用いて配列を決定した。MKK3(2030塩基対(bp))およびMKK4(3576 bp)について得られた最長のクローンは、これらのプロテインキナーゼのコード領域全体を含んでいた。
【0077】
MKK3(配列番号:2)およびMKK4-α(配列番号:6)の一次構造は、図1に示されている。インフレームの停止コドンがMKK3 cDNAの5'非翻訳領域に存在するが、MKK4 cDNAの5'領域にはない。表示されたMKK4蛋白質配列は、2番目のインフレーム開始コドンから開始する。
【0078】
これらの配列を、ヒトMAPキナーゼMEK1(配列番号:11)およびMEK2(配列番号:12)(ZhengおよびGuan (1993) J. Biol. Chem. 268:11435)ならびに酵母MAPキナーゼキナーゼPBS2(配列番号:13)(BoguslawaskiおよびPolazzi (1987) Proc. Natl. Acad. Sci. USA 84:5848)と比較した(図1)。キナーゼとヒトMKK3(サブドメインIとXIの間)の同一性および類似性は、BESTFITプログラム(バージョン7.2、Wisconsin genetics Computer Group)によって計算した(同一性の割合/類似性の割合):MEK1, 41%/63%, MEK2, 41%/62%; MKK4α, 52%/73%; およびPBS2, 40%/59%。キナーゼとヒトMKK4αの同一性および類似性は以下のとおりであると計算された(同一性の割合/類似性の割合):MEK1, 44%/63%, MEK2, 45%/61%; MKK3, 52%/73%; およびPBS2, 44%/58%。
【0079】
MKK3およびMKK4γのcDNA配列は、それぞれ寄託番号L36719およびL36870でジーンバンク(GenBank)に寄託された。MKK4γcDNA配列には、異なるスプライシング部位からインビボで生成するMKK4αおよびMKK4βのcDNA配列が含まれている。当業者は、寄託されたcDNA配列から、MKK3およびMKK4アイソフォームのアミノ酸配列を簡単に決定できる。
【0080】
実施例2. 成人組織におけるMKK3およびMKK4 mRNAの発現
ヒトの心臓、脳、胎盤、肺、肝臓、筋肉、腎臓、および膵臓組織から単離されたポリアデニル化[ポリ(A)+]mRNA (2μg)を用いて、ノーザンブロット分析が行われた。変性アガロースゲル電気泳動によってmRNAは分別され、ナイロンメンブレンに移された。ブロットは、[α-32P]ATP(デオキシアデノシン3リン酸)(Amersham International PLC)を用いたランダムプライミングによって標識されたMKK3およびMKK4 cDNAでプローブした。MKK3およびMKK4は、調べたすべての組織で発現されていた;MKK3およびMKK4の発現が最高だったのは、骨格筋組織だった。
【0081】
ヒトと酵母のMAPキナーゼキナーゼグループのメンバー間の関係は、樹状図で示されている(図2)。MKK3/4は、ヒトMAPキナーゼキナーゼの独特なサブグループを形成する。
【0082】
実施例3. MKK3によるp38 MAPキナーゼのインビトロリン酸化
GST-JNK1およびGST-ERK2は記述されている(Derijardら(1994)上記;Guptaら(1995) Science 267: 389; WartmannおよびDavis (1994) J. Biol. Chem. 269:6695、各々は参照として本明細書に組み入れられる)。GST-p38 MAPキナーゼは、発現ベクターpGSTag(Dressierら(1992) Biotechniques 13:866)およびp38 MAPキナーゼcDNAのコード領域を含むPCR断片から、調製された。GST-MKK3およびMKK4は、pGEX3X(Pharmacia-LKB Biotechnology)およびMKK3およびMKK4 cDNAのコード領域を含むPCR断片から、調製された。GST融合蛋白質は、GSHアガロース(SmithおよびJohnson (1988) Gene 67:31)を用いたアフィニティークロマトグラフィーによって、精製された。発現ベクターpCMV-Flag-JNK1およびpCMV-MEK1は、記述されている(Derijardら(1994)上記;WartmannおよびDavis (1994)上記)。プラスミドpCMV-Flag-p38 MAPキナーゼは、発現ベクターpCMV5(Anderssonら(1989) J. Biol. Chem. 264:8222)およびp38 MAPキナーゼcDNAから調製された。MKK3およびMKK4の発現ベクターは、cDNAをpCDNA3(Invitrogen)のポリリンカーにサブクローニングして、調製された。Flagエピトープ(Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys(配列番号:16);Immunex、ワシントン州シアトル)は、挿入重複PCR(Hoら(1989) Gene 77:51)によって、キナーゼのコドン1と2の間に挿入された。
【0083】
プロテインキナーゼアッセイ法は、キナーゼ緩衝液(25 mM 4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸、pH 7.4、25 mM β-グリセロリン酸塩、25 mM MgCl2、2 mMジチオスレイトール、および0.1 mM オルトバナジン酸塩)中で実行された。組換えGST-MKK3は、[γ-32P]ATPおよび緩衝液、GST-JNK1、GST-p38 MAPキナーゼ、またはGST-ERK2とインキュベートされた。アッセイ法は、最終容量25μlの中に、1 μgの基質蛋白質および50 μM [γ-32P]ATP(10 Ci/mmol)を添加して開始された。25℃で30分後に、レムリ(Laemmli)サンプル緩衝液を添加して反応を終了させた。SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)後に、オートラジオグラフィーによって基質蛋白質のリン酸化を調べた。部分酸加水分解および薄層クロマトグラフィーによって、ホスホアミノ酸分析を行った(Derijardら(1994)上記;Alvarezら(1991) J. Biol. Chem. 266:15277)。すべてのグループでMKK3の自己リン酸化が観察された。MKK3は、p38 MAPキナーゼをリン酸化したが、JNK1またはERK2はしなかった。
【0084】
同様な挿入重複PCR手法を用いて、p38のThr180およびTyr182を、それぞれAlaおよびPheで置換した。すべてのプラスミドの配列は、アプライド バイオシステムズ(Applied Biosystems)モデル373Aを用いた自動シーケンシングによって、確認された。GST-MKK3は[γ-32P]ATPおよび緩衝液、野生型GST-p38 MAPキナーゼ(TGY)または変異GST-p38 MAPキナーゼ(AGF)とインキュベートされた。リン酸化された蛋白質は、SDS-PAGEで解析され、オートラジオグラフィーで検出された。野生型p38のリン酸化のみが観察された。
【0085】
実施例4. MKK4によるJNKおよびp38 MAPキナーゼのインビトロでのリン酸化および活性化
プロテインキナーゼアッセイ法は、実施例3のように行なわれた。組換えGST-MKK4は、[γ-32P]ATPおよび緩衝液、GST-JNK1、GST-p38 MAPキナーゼ、またはGST-ERK2とインキュベートされた。JNK1およびp38は、リン酸化され、またJNK1およびp38と共にインキュベートされたMKK4もリン酸化された。
【0086】
GST-MKK4は[γ-32P]ATPおよび緩衝液、野生型JNK1(Thr183、Tyr185)、または変異GST-JNK1(Ala183、Phe185)とインキュベートした。各インキュベーションには、JNK1の基質ATF2(Guptaら(1995)上記)も含まれていた。ATF2はMKK4および野生型JNK1の存在下で、リン酸化された。これらの結果は、MKK4がp38とJNK1の両方をリン酸化し、活性化することを立証する。
【0087】
実施例5. UV刺激を受けたMKK3によるp38 MAPキナーゼのリン酸化および活性化
エピトープタグを付けたMKK3を、ウシ胎仔血清(5%)(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で維持したCOS-1細胞で発現させた。細胞は、製造元(Gibco-BRL)の推奨する方法にしたがって、リポフェクタミン試薬を用いてトランスフェクトし、記述されたようにUV照射またはEGFで処理した(Derijardら(1994)上記)。
【0088】
細胞は、UV-Cの存在下または非存在下に露出した(40 J/m2)。細胞は溶解緩衝液(20 mM Tris, pH 7.4, 1% 登録商標TRITON X-100, 10%グリセロール、137 mM NaCl, 2 mM EDTA, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸ナトリウム、1 mMフェニルメチルスルホニルフルオリド、およびロイペプチン(10μg/ml))で溶解し、4℃で100,000 x gで15分間遠心した。MKK3は免疫沈降によって単離された。エピトープタグを付けたプロテインキナーゼは、プロテインGセファロース(Pharmacia-LKB Biotechnology)に結合したFlagエピトープ(IBI-Kodak)に対するM2抗体と、4℃で1時間インキュベートした。免疫沈降物は、溶解緩衝液で2回、キナーゼ緩衝液で2回洗った。
【0089】
プロテインキナーゼアッセイ法は、基質GST-p38 MAPキナーゼまたはJNK1を用いて行なった。一部のアッセイ法にはATF2が含まれていた。MKK3による基礎レベルのp38 MAPキナーゼリン酸化が観察された。UV照射を行なうと、p38 MAPキナーゼのリン酸化が増加したが、JNK1は増加しなかった。p38 MAPキナーゼ活性が増加すると、ATF2のリン酸化が増加した。
【0090】
実施例6. MKK3またはMKK4を発現する細胞におけるp38 MAPキナーゼの活性化
COS-1細胞はエピトープタグを付けたp38 MAPキナーゼと、空の発現ベクターまたはMEK1、MKK3、またはMKK4αをコードする発現ベクターでトランスフェクトした。培養物の一部は、UV照射(40 J/m2)または10 nM EGFで処理した。p38 MAPキナーゼは、モノクローナル抗体M2による免疫沈降で単離し、プロテインキナーゼ活性は、[γ-32P]ATPおよびATF2を基質として用いて、免疫複合体で測定された。リン酸化反応の産物は、SDS-PAGE後にオートラジオグラフィーによって可視化した。対照のMEK1またはEGF処理群ではATF2はリン酸化されなかったが、MKK3、MKK4、およびUV照射群ではリン酸化された。ATF2のMKK3およびMKK4によるリン酸化は、UV照射細胞から単離されたp38 MAPキナーゼで見られるものと類似していた。
【0091】
実施例7. JNK1およびJNK2によるATF2のリン酸化
COS-1細胞は、ウシ血清アルブミン(5%)(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で維持された。[32] Pによる代謝標識は、[32]オルトリン酸塩(2 mCi/ml)(Dupont-NEN)を添加したリン酸塩フリーのダルベッコ変法イーグル培地(Flow Laboratories Inc.)中で、3時間細胞をインキュベートして行なった。COS-1細胞は、エピトープタグを付けたJNK1の存在下(JNK1)または非存在下(モック)でトランスフェクトした。JNK1 cDNAをコードするプラスミド発現ベクターは、以前に記述されている(Derijardら(1994) Cell 76:1025、参照として本明細書に組み入れられる)。プラスミドDNAは、リポフェクタミン法(Gibco-BRL)によって、COS-1細胞にトランスフェクトした。48時間インキュベーションした後、培養物の一部に40 J/m2のUV照射を行ない、37℃で1時間インキュベートした。
【0092】
細胞は、20 mM Tris, pH 7.5, 25 mMβ-グリセロリン酸塩、10%グリセロール、1% 登録商標Triton X-100, 0.5% (w/v) デオキシコール酸塩、0.1% (w/v) SDS, 0.137 M NaCl, 2 mM ピロリン酸塩、1 mMオルトバナジン酸塩、2 mM EDTA, 10μg/mlロイペプチンおよび1 mM PMSF中で溶解された。可溶性抽出物は、4℃で20分間微量遠心して調製された。組換えJNK1を抗原として調製されたウサギ抗血清を用いた反応によって、JNK1免疫沈降物も調製された。
【0093】
ゲル内プロテインキナーゼアッセイ法は、細胞溶解物とJNK1免疫沈降物を用いて、SDS-PAGE後に、プロテインキナーゼを再生、ゲル中での基質(GST-ATF2、残基1〜505)の重合、および[γ-32P]ATPとのインキュベーションによって実行した(Derijardら(1994)上記)。[32P]リン酸塩の取り込みは、オートラジオグラフィーによって可視化し、ホスホールイメージャー(Phosphorimager)およびイメージクアント(ImageQuant)ソフトウェア(Molecular Dynamics Inc.、カリフォルニア州サニーベール)によって定量した。細胞溶解物には、UV照射細胞から調製された抽出物中でATF2をリン酸化する46 kDおよび55 kDのプロテインキナーゼが存在することが示された。46 kDおよび55 kDプロテインキナーゼは、それぞれJNK1およびJNK2であると同定された。
【0094】
実施例8. JNK1のATF2への結合およびNH2末端活性化ドメインのリン酸化
ATF2のJNK1によるリン酸化部位は、ATF2の連続的なNH2末端ドメイン欠失を作製して調べた。ATF2をコードするプラスミド発現ベクター(pECE-ATF2)(LiuおよびGreen (1994) および(1990))は、記述されている。GST-ATF2融合蛋白質の細菌の発現ベクターは、ポリメラーゼ連鎖反応(PCR)によるATF2 cDNA断片をpGEX-3X (Pharmacia-LKB Biotechnology Inc.)へサブクローニングして、作製された。作製されたすべてのプラスミドの配列は、アプライドバイオシステムズ(Applied Biosystems)モデル373Aを用いた自動シーケンシングによって確認された。GST-ATF2蛋白質は、記述されたようにして精製され(SmithおよびJohnson (1988) Gene 67:31)、SDS-PAGEで解析され、クーマシーブルーで染色された。GST-ATF2融合蛋白質には、残基1〜505、1〜349、350〜505、1〜109、20〜109、40〜109、および60〜109が含まれていた。
【0095】
UV照射細胞から単離されたJNK1によるGST-ATF2融合蛋白質のリン酸化は、免疫複合体キナーゼアッセイ法によって、調べられた。免疫複合体キナーゼアッセイ法は、Flagエピトープタグを付けたJNK1およびモノクローナル抗体M2 (IBI-Kodak)を用いて、Derijardら(1994)上記、に記述されたようにして行なわれた。免疫複合体蛋白質キナーゼアッセイ法は、組換えJNK1を抗原として調製されたウサギ抗血清を用いても、行なわれた。細胞は、20 mM Tris, pH 7.5, 10%グリセロール、1% 登録商標Triton X-100, 0.137 M NaCl, 25 mMβ-グリセロリン酸塩、2 mM EDTA, 1 mMオルトバナジン酸塩、2 mM ピロリン酸塩、10μg/mlロイペプチン、および1 mM PMSF中で溶解された。JNK1は、JNKに対するウサギポリクローナル抗体、またはFlagエピトープに対するM2モノクローナル抗体に結合した、プロテインG-セファロースを用いて免疫沈降させた。ビーズは溶解緩衝液で3回、キナーゼ緩衝液(20 mM Hepes, pH 7.6, 20 mM MgCl2, 25 mMβ-グリセロリン酸塩、100μMオルソバナジン酸ナトリウム、2 mMジチオスレイトール)で1回洗った。キナーゼアッセイ法は、30μlのキナーゼ緩衝液中で1 μgの基質、20 μMアデノシン3リン酸、および10μCiの[γ-32P]ATPを用いて、25℃で10分間行なった。反応は、レムリ(Laemmli)サンプル緩衝液を添加して停止し、産物はSDS-PAGE(10%ゲル)で解析した。JNK1は、残基1〜505、1〜349、1〜109、20〜109、および40〜109を持つGST-ATF2融合蛋白質をリン酸化するが、60〜109はしなかった。これらの結果は、JNKによるリン酸化には、ATF2の残基1〜60が必要であることを示す。
【0096】
固定したGST-ATF2融合蛋白質の結合は、Hibiら((1993) Genes Dev. 7:2135、参照として本明細書に組み入れられる)に記述されるようにして、固相キナーゼアッセイ法によって調べられた。UV照射細胞のJNK1は、GSH-アガロースに結合したGST-ATF2融合蛋白質とインキュベートした。アガロースビーズは、未結合のJNK1を取り除くために、念入りに洗った。結合したJNK1プロテインキナーゼによるGST-ATF2融合蛋白質のリン酸化は、[γ-32P]ATPを添加して調べた。JNK1は、残基1〜505、1〜349、1〜109、20〜109、および40〜109を持つGST-ATF2融合蛋白質に結合し、JNK1のATF2への結合には残基20〜60が必要であることが示された。
【0097】
実施例9. JNK1によるATF2のNH2末端活性化ドメインのThr69およびThr71のリン酸化
野生型(Thr69、71)およびリン酸化欠損(Ala69、71)ATF2分子の性質に対するUV照射の影響が調べられた。モックトランスフェクトおよびJNK1でトランスフェクトしたCOS細胞を、40 J/m2 UV照射の有無の処理をした。エピトープタグを付けたJNK1は、モノクローナル抗体M2による免疫沈降で単離された。GST- ATF2(残基1〜109)は、上述のように免疫複合体キナーゼアッセイ法で調べた。GST- ATF2は、SDS-PAGEによって他の蛋白質から分離し、クーマシーブルー染色した。GST- ATF2のリン酸化は、オートラジオグラフィーで検出した。JNK1トランスフェクト細胞は、ATF2をリン酸化したが、モックトランスフェクト細胞はしなかった。JNK1によるATF2のリン酸化は、UV照射をした細胞の方が多かった。JNK1によるATF2のリン酸化は、電気泳動の移動度の低下を伴っていた。
【0098】
別の実験では、全長ATF2(残基1〜505)、NH2末端断片(残基1〜109)、およびCOOH末端断片(残基95〜505)を含むGST融合蛋白質が、JNK1によってリン酸化され、ホスホアミノ酸分析によって、リン酸化部位が分析された。ホスホペプチドマッピングおよびホスホアミノ酸分析に使用した方法は、記述されている(Alvarezら(1991) J. Biol. Chem. 266:15277)。ペプチドマップの水平方向は電気泳動で、垂直方向はクロマトグラフィーである。ホスホペプチドマッピングおよび変異分析によって、リン酸化のNH2末端部位は、Thr69およびThr71であると同定された。上述のように、位置指定突然変異導入法を実行してThr69およびThr71をAlaに置換した。変異ATF2のリン酸化は観察されなかった。
【0099】
実施例10. JNK活性化ATF2の電気泳動移動度の低下
CHO細胞は、5%ウシ血清アルブミン(Gibco-BRL)を添加したハム(Ham)のF12培地で維持された。上述のようにして、細胞を標識し、JNK1によってトランスフェクトした。CHO細胞はUV-C(40 J/m2)、IL-1α(10 ng/ml)(Genzyme)、またはウシ胎仔血清(20%)(Gibco-BRL)で処理した。細胞を回収する前に、37℃で30分インキュベートした。ATF2の電気泳動の移動度は、SDS-PAGE後に蛋白質免疫ブロット分析によって調べた。UV、IL-1、および血清で処理された細胞で、ATF2の電気泳動の移動度の変化が観察された。これらの結果は、JNK1による活性化は、ATF2の電気泳動の移動度の変化を伴うことを示し、さらに、ATF2がJNK1のインビボの基質であることを示唆する。
【0100】
実施例11. JNKの活性化後のATF2リン酸化の増加
COS-1細胞は、上述のようにATF2発現ベクターの存在下(ATF2)または非存在下(対照)で、トランスフェクトされた(Haiら(1989)上記)。細胞を40 J /m2 UV-Cに露出した効果を調べた。照射後、細胞を37℃で0または30分(対照)および0、15、30、45分(ATF2)インキュベートし、回収した。SDS-PAGEでのATF2の電気泳動の移動度は、上述のように蛋白質免疫ブロット分析によって調べた。ATF2トランスフェクト細胞では、ATF2は2つの電気泳動移動度を示したが、対照細胞ではそうではなかった。
【0101】
40 J/m2 UV-Cの処理または非処理後、37℃で30分インキュベートした[32]P標識細胞において、野生型(Thr69、71)ATF2および変異型(Ala69、71)ATF2のリン酸化状態を調べた(Haiら(1989)上記)。ATF2蛋白質は免疫沈降で単離され、SDS-PAGEおよびオートラジオグラフィーで分析した。リン酸化ATF2蛋白質は、上述のようにホスホアミノ酸分析によって調べた。両方の型のATF2がホスホセリンを含んでいたが、野生型ATF2のみがホスホスレオニンを含んでいた。
【0102】
トリプシンホスホペプチドマッピングを用いて、インビトロでJNK1によってリン酸化されたATF2と、COS-1細胞中でリン酸化されたATF2とを比較した。インビボとインビトロでリン酸化されたATF2を等量含む試料(ミックス)でも、マップを作製した。ATF2のThr69およびThr71に変異が導入されると、UV照射細胞から単離されたATF2のマップで、2つのリン酸化トリプシンペプチドが失われる。これらのホスホペプチドは、Thr69およびThr71を含むモノリン酸化およびビスリン酸化ペプチドに対応する。このホスホペプチドのいずれも、インビトロでJNK1によってリン酸化されたATF2のマップで観察された。
【0103】
実施例12. リン酸化部位Thr69およびThr71の変異による、ATF2刺激遺伝子発現の阻害
上述のように、ATF2およびGAL4 DNA結合ドメインを含む融合蛋白質を、CHO細胞で発現させた。GAL4- ATF2融合蛋白質の活性は、レポータープラスミドpG5E1bLuc (Sethら(1992) J. Biol. Chem. 267:24796、参照として本明細書に組み入れられる) とのコトランスフェクション解析で測定した。このレポータープラスミドには、最小プロモーター要素およびホタルルシフェラーゼ遺伝子の上流に5つのGAL4部位がクローニングされている。トランスフェクションの効率は、βガラクトシダーゼを発現する対照プラスミドでモニターした(pCH110; Pharmacia-LKB Biotechnology)。細胞抽出物中で検出されるルシフェラーゼおよびβガラクトシダーゼの活性は、3回の実験の平均の活性比として測定された(Guptaら(1993) Proc. Natl. Acad. Sci. USA 90:3216、参照として本明細書に組み入れられる)。表1に示す結果は、転写活性にはThr69およびThr71のリン酸化が重要であることを示す。
【0104】
【表1】
【0105】
実施例13. ATF2機能に対するドミナントネガティブJNK1変異体の影響
野生型(Thr183、Tyr185)または変異(Ala183、Phe185)JNK1でトランスフェクトされた血清処理CHO細胞中の、ATF2リン酸化部位Thr69およびThr72における点変異の影響を、ルシフェラーゼレポータープラスミド系を用いて決定した。対照実験は、モックトランスフェクト細胞を用いて行なった。CHO細胞は、18時間血清除去した後、血清の存在下または非存在下で4時間インキュベートした。野生型ATF2が発現されると、血清に刺激されるATF2転写活性がわずかに上昇した。反対に、変異JNK1は対照および血清刺激ATF2活性の両方を阻害した。
【0106】
実施例14. ATF2に刺激される遺伝子発現に対する、癌抑制遺伝子産物RbおよびアデノウイルスオンコプロテインE1aの影響
癌抑制遺伝子産物RbおよびアデノウイルスオンコプロテインE1Aの発現のATF2転写活性に対する影響は、上述のように、ルシフェラーゼレポータープラスミドおよびGAL4- ATF2 (残基1〜505)を用いて調べた。細胞は、野生型(Thr69、71)または変異(Ala69、71)ATF2でトランスフェクトされた。GAL4- ATF2の非存在下では、ルシフェラーゼ活性に対するRbまたはE1Aの効果は、検出されなかった。野生型および変異ATF2のいずれでも、RbおよびE1AはATF2に刺激される遺伝子発現を増加させることが分かった。しかし、変異ATF2によるレポーター遺伝子発現のレベルは、野生型ATF2よりも低かった。これらの結果は、最大の転写活性を得るためには、ATF2のリン酸化(Thr69およびThr71)に加え、RbまたはE1Aが必要なことを示す。
【0107】
実施例15. p38 MAPキナーゼの基質特異性
p38 MAPキナーゼの基質リン酸化は、細菌で発現されたp38 MAPキナーゼを、IκB、cMyc、EGF-R、細胞質ホスフォリパーゼA2 (cPLA2)、c-Jun、および変異ATF2(Thr69、71)ならびにATP [γ-32P](Raingeaudら(1995) J. Biol. Chem 270:7420、参照として本明細書に組み入れられる)とインキュベーションして調べた。GST-IκBは、バルチモア(D. Baltiomore)博士(マサチューセッツ工科大学)から提供された。GST-cMyc (Alvarezら(1991) J. Biol. Chem. 266:15277)、GST-EGF-R(残基647〜688)(Kolandら(1990) Biochem. Biophys. Res. Commun. 166:90)、およびGST-c-Jun (Derijardら(1994)上記)は、記述されている。リン酸化反応は、30分後にLaemmliサンプル緩衝液を添加して、停止した。リン酸化蛋白質は、SDS-PAGEで分離し、オートラジオグラフィーで検出した。基質蛋白質のリン酸化率は、ホスホールイメージャー(PhosphorImager)(Molecular Dynamics Inc.) 分析によって定量した。ATF2、MBP、EGF-R、およびIκBの相対的リン酸化は、それぞれ、1.0、0.23、0.04、および0.001だった。
【0108】
実施例16. ATF2に対するp38 MAPキナーゼの結合
エピトープタグを付けたJNK1およびp38 MAPキナーゼを発現する細胞抽出物を、GSHアガロースに固定した、ATF2の活性化ドメイン(残基1〜109)を含むGST融合蛋白質とインキュベートした。上清を取り除き、アガロースを念入りに洗った。上清およびアガロース結合画分のウエスタンブロット分析は、以下のように行なった。蛋白質はSDS-PAGEによって分画し、電気泳動によってイモビロン(Immobilon)-Pメンブレンに移し、ホスホチロシン(PY20)およびフラッグエピトープ(M2)に対するモノクローナル抗体で、プローブした。免疫複合体は増幅化学ルミネセンス(Amersham International PLC)を用いて検出した。対照実験は、固定したGSTを用いて行なった。
【0109】
実施例17. 炎症促進型サイトカインおよび環境ストレスによるp38 MAPキナーゼおよびJNK1の活性化
ホルボールエステル、EGF、UV照射、浸透圧ストレス、IL-1、腫瘍壊死因子(TNF)、およびLPSの、p38 MAPキナーゼおよびJNK1の活性に対する影響は、ATP [γ-32P]およびATF2を基質として用いた免疫複合体タンパク質キナーゼアッセイ法によって、測定した。TBFαおよびIL-1αは、ゲンザイム社(Genzyme Corp.)のものである。リポ多糖(LPS)は、記述されたようにして(Mathisonら(1988) J. Clin. Invest. 81:1925)、凍結乾燥した細菌サルモネラ ミネソタ(Salmonella minesota)Re595から単離された。ホルボールミリステートアセテートは、シグマ(Sigma)のものだった。EGFはマウスの唾液腺から精製された(Davis (1988) J. Biol. Chem. 263:9462)。キナーゼアッセイ法は、p38およびJNKの免疫沈降物を用いて行なった。免疫複合体は、キナーゼ緩衝液(上述)で2回洗い、アッセイ法は、最終容量25μl中で、1μgのATF2および50μMの[γ-32P]ATP(10 Ci/mmol)を添加して開始した。反応は、30℃、30分後に、レムリ(Laemmli)サンプル緩衝液を添加して停止した。ATF2のリン酸化は、SDS-PAGE後にオートラジオグラフィーによって調べ、ATF2のリン酸化率は、ホスホールイメージャー(PhosphorImager)分析によって定量した。
【0110】
結果は表2に示されている。HeLa細胞を10 nMのホルボールミリステートアセテートに露出すると、p38とJNK1が非常に弱く活性化された。同様に、10 nMのEGFで処理しても、p38とJNK1を非常に弱く活性化したのみだった。これとは対照的に、40 J/m2 UV-C、300 mMソルビトール、10 ng/mlインターロイキン-1、および10 ng/ml TNFαで処理すると、p38とJNK1活性が強く活性化された。p38の活性に対するLPSの効果は、ヒトCD14を発現するCHO細胞を用いて調べた。CHO細胞を10 ng/ml LPSに露出しても、p38とJNK1活性は僅かに活性化されただけだった。
【0111】
【表2】
【0112】
実施例18. TyrおよびThrの二重リン酸化によるp38 MAPキナーゼの活性化
野生型(Thr180、Tyr182)または変異(Ala180、Phe182)p38 MAPキナーゼを発現するCOS-1細胞を、UV-C (40 J/m2)の照射の有無で処理した。細胞はUV照射の有無で処理後、30分で回収した。対照実験は、モックトランスフェクト細胞を用いて行なった。エピトープタグをつけたp38 MAPキナーゼの発現レベルおよび、p38 MAPキナーゼのTyrリン酸化状態は、モノクローナル抗体M2およびホスホチロシンモノクローナル抗体PY20を用いたウエスタンブロット分析によって調べた。免疫複合体は、増幅化学ルミネセンスによって検出した。
【0113】
野生型および変異p38は、同様なレベルで発現されていた。ウエスタンブロット分析では、UV照射によってp38のTyrリン酸化が増加したことが示された。Tyrリン酸化の増加は、[32P]リン酸塩標識細胞から単離されたp38のホスホアミノ酸分析でも確認された。この結果では、UV照射がp38のThrリン酸化を増加させたことも示された。TyrおよびThrのリン酸化の増加は、変異p38によって阻害された。野生型および変異型のp38は、免疫沈降によってCOS-1細胞から単離された。プロテインキナーゼ活性は、[γ-32P]ATPおよびGST-ATF2を基質として使用して、免疫複合体中で測定された。リン酸化GST- ATF2は、SDS-PAGE後にオートラジオグラフィーによって検出された。UV照射によって、野生型p38の活性が著しく上昇したが、変異p38は触媒作用が不活性であることが分かった。これらの結果は、p38がThr-Gly-Tyrモチーフ内の二重のリン酸化によって活性化されることを示す。
【0114】
実施例19. MAPキナーゼホスファターゼはp38 MAPキナーゼ活性化を阻害する
細胞を40 J/m2のUV-Cの照射の有無で処理した。対照実験は、モックトランスフェクトした細胞(対照)、および触媒作用が不活性の変異ホスファターゼmPAC1 (Cys257/Ser)およびヒトMKP1でトランスフェクトした細胞を用いて行なった。p38 MAPキナーゼの活性は、[γ-32P]ATPおよびGST- ATF2を基質として用いた免疫複合体プロテインキナーゼアッセイ法で測定した。PAC1またはMKP1の発現は、p38によるリン酸化を阻害することが分かったが、これはp38が両特異性を持つホスファターゼPAC1およびMKP1によって調節され得ることを示す。
【0115】
実施例20. p38 MAPキナーゼの細胞内分布
エピトープタグを付けたp38 MAPキナーゼは、COS細胞で発現された。細胞は、40 J/m2 のUV照射の有無で処理した後、37℃で60分間インキュベートした。p38 MAPキナーゼは、モノクローナル抗体M2を用いた間接免疫蛍光によって検出した。デジタルイメージング顕微鏡法によって像を得て、画像復元処理を行った。
【0116】
免疫組織化学
カバースリップ(22 mm x 22 mm No.1; ゴールドシールカバーグラス; Becton-Dickinson)は、0.1 N HCl中で10分間煮沸による前処理をして、蒸留水ですすぎ、オートクレーブして、0.01% ポリ-L-リジン(Sigma; ミズーリ州セントルイス)でコートした。カバースリップは、35 mmマルチウェル組織培養プレート(Becton Dickinson、英国)の底に置いた。トランスフェクトされたCOS-1細胞をカバースリップ上に直接播き、5%ウシ胎仔血清(Gibco-BRL)を添加したダルベッコ変法イーグル培地中で一晩接着させた。トランスフェクションの24時間後に、細胞を一度洗い、25 mM Hepes, pH 7.4、137 mM NaCl、6 mM KCl、1 mM MgCl2、1 mM CaCl2、10 mMグルコース中で37℃で30分間インキュベートした。リン酸緩衝生理食塩水で細胞を1度洗い、組織培養ウェルからカバースリップを取り出した。新しく調製したリン酸緩衝生理食塩水中の4%パラホルムアルデヒドで、22℃で15分間細胞を固定した。リン酸緩衝生理食塩水中の0.25% 登録商標Triton X-100で5分間処理して細胞を透過性にして、DWB溶液(150 mM NaCl、15 mMクエン酸ナトリウム、pH 7.0、2%ウマ血清、1%(w/v)ウシ血清アルブミン、0.05% 登録商標Triton X-100)で3回洗った。一次抗体(抗フラッグモノクローナル抗体M2、Eastman-Kodak Co.,コネチカット州ニューヘブン)は、DWBで250倍に希釈し、加湿環境下で22℃で1時間細胞に適用した。再び細胞を上述のように3回洗い、2次抗体であるフルオレセインイソチオシアネート結合ヤギ抗マウスIg(Kirkegaard & Perry Laboratories Inc.、メリーランド州ゲーサーズバーグ)は、250倍希釈して、加湿環境下で22℃で1時間適用した。その後、DWBで細胞を3回洗い、その後、免疫蛍光分析用に、ゲル−マウント(Gel-Mount)(Biomeda Corp.、カリフォルニア州フォスターシティ)によってスライド上にマウントした。対照実験は、観察された免疫蛍光の特異性を評価するために実行した。トランスフェクトされた細胞が、一次モノクローナル抗体M2の非存在下で染色されたトランスフェクト細胞や、モックトランスフェクトされた細胞では、蛍光は検出されなかった。
【0117】
デジタルイメージング顕微鏡法および画像復元
個々の細胞中の蛍光分布のデジタル画像は、既述のように、エピ蛍光用のゼイズ(Zeiss)IM-35顕微鏡上でニコン60x プラナポ(Planapo)対物レンズ(開口数=1.4)を用いて得た(Carringtonら(1990) 細胞生物学の非侵襲性技術(Non-invasive Techniques in Cell Biology)、Fosbett & Grinstein編、Wiley-Liss, NY; pp 53-72; Fayら(1989) J. Microsci. 153:133-149)。種々の焦平面の画像は、コンピュータ制御焦点機構および熱電流冷却電荷結合素子カメラ(モデル220;Photometrics Ltd.、アリゾナ州ツーソン)を用いて得た。試料の励起源への露出は、コンピュータ制御シャッターおよび波長選択システム(MVI、マサチューセッツ州エイボン)により決定した。電荷結合素子カメラおよび顕微鏡の機能は、マイクロコンピュータで制御され、カメラから得たデータはシリコン グラフィクス(Silicon Graphics)モデル4D/GTXワークステーション(カリフォルニア州マウンテンビュー)へ転送して画像処理をした。画像は、感度の不均一性および電荷結合素子検知器の暗電流のための補正をした。顕微鏡のぼやけの較正は、この装置の点広がり関数を、直径0.3μmの蛍光標識ラテックスビーズ(Molecular Probes Inc.)の0.125μm間隔の光学切片のシリーズとして測定することにより、決定した。画像復元アルゴリズムは、誤った問題の理論に基づいたもので、未処理画像よりも実質的に正確な細胞内の定量的色素密度値を得るものである(Carringtonら(1990)上記;Fayら(1989)上記)。画像処理後に、シリコン グラフィクス(Silicon Graphics)ワークステーション上のコンピュータグラフィックスソフトウェアを用いて、細胞の個々の光学切片を分析した。p38 MAPキナーゼは、細胞表面、細胞質、および核に観察された。照射後、細胞質p38の核周囲領域への局在が増加した。
【0118】
実施例21. 浸透圧ショックによるMKKシグナル伝達経路の活性化
CHO細胞を、プラスミドpCMV-Flag-Jnk1およびpRSV-Neoでコトランスフェクションした(Derijardら(1994)上記)。ジェネティシン(Gibco-BRL)を用いた選択によって、エピトープタグを付けたJnk1(Flag; Immunex Corp.)を発現する安定した細胞株を単離した。細胞を、0、100、150、300、600、または1000 mMソルビトールと37℃で1時間インキュベートした。細胞は溶解緩衝液(20 mM Tris, pH 7.4, 1% 登録商標TRITON X-100, 2 mM EDTA, 137 mM NaCl, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸塩、2 mMピロリン酸塩、10%グリセロール、1 mMフェニルメチルスルホニルフルオリド、および10μg/mlロイペプチン)中で回収し、4℃で100,000 x gで30分間遠心して可溶性抽出物を得た。エピトープタグを付けたJNK1は、モノクローナル抗体M2(Immunex Corp)を用いた免疫沈降によって単離した。免疫沈降物は、溶解緩衝液で念入りに洗った。免疫複合体キナーゼアッセイ法は、25μlの25 mM Hepes, pH 7.4, 25 mM MgCl2、25 mMβ-グリセロリン酸塩、2 mMジチオスレイトール、100μMオルトバナジン酸塩、および50μM ATP [γ-32P] (10μCi/mmol)中で、細菌で発現されたグルタチオン-S-トランスフェラーゼGST)融合c-Jun(残基1〜79)2.5μgを基質として使用して、行なった。c-Junのリン酸化はSDS-PAGE後に、オートラジオグラフィーおよびホスホールイメージャー(PhosphorImager)(Molecular Dynamics Inc.)分析によって調べた。すべての濃度のソルビトール処理で、JNK1の活性化が観察された。
【0119】
JNK1プロテインキナーゼ活性化の時間変化は、上述のように300 mMソルビトールを添加した培地中でインキュベートした細胞中で、測定された。ソルビトールへの露出後、5分以内にJNK1の活性の上昇が観察され、最大活性は15〜30分後に見られた。
【0120】
JNK1のリン酸化部位Thr183およびTyr185に変異を導入すると、浸透圧ショックによるJNK1プロテインキナーゼ活性の活性化が阻止された。CHO細胞をベクター、野生型JNK1(Thr183、Tyr185)、および変異JNK1(Ala183、Phe185)でトランスフェクトした。300 mMソルビトールの添加のある培地とない培地で15分間インキュベートしてから、上述のようにJNK1プロテインキナーゼ活性を測定した。JNK1の活性化は、野生型JNK1では見られたが、変異JNK1では見られなかった。
【0121】
実施例22. MKK7の分子クローニング
RT-PCRを用いて新規の哺乳類MAPキナーゼキナーゼの断片を同定した。このプロトコール用にデザインしたプライマーATNGCNGTNAARCARATG(配列番号:23)およびATNCKYTCNGGNGCCATRTA(配列番号:24)は、ショウジョウバエMAPキナーゼキナーゼhep(Gliseら(1995) Cell 83:451-461)の配列に基づいている。マウスの精巣mRNAは鋳型として使用された。マウス精巣mRNAのRT-PCR増幅後に、単一の生成物(461 bp)が検出された。配列分析により、このPCR産物が新規の哺乳類MAPキナーゼキナーゼの断片であることが同定された。全長マウスcDNAクローンは、マウス精巣ライブラリー(Stratagene Inc.)をスクリーニングして、単離された。このcDNAクローンの配列は、アプライドシステムズ(Applied Biosystems)モデル373Aを用いて決定した。配列分析により、7つのクローン群が、プロテインキナーゼと推定されるものをコードする単一の長いオープンリーディングフレームを含むと同定された(図9および図10;配列番号:17および配列番号:18)。インフレームの停止コドンがこれらのクローンの5'および3'領域に検出された。この配列は、プロテインキナーゼサブドメインI〜XIを含んでおり、MAPキナーゼキナーゼグループに関連している。この新規のプロテインキナーゼは、MKK7と命名された。MAPキナーゼキナーゼのサブドメインVIIIに存在するリン酸化を活性化する部位は、MKK7に保存されている。MKK7を他の哺乳類MAPキナーゼキナーゼグループメンバーと比較すると、MKK7がJNK活性化因子MKK4と近縁であることが示された。λファージライブラリーから単離された別の1つのcDNAクローンは、他の7つのクローンとは異なっていた。このクローンは、3'非翻訳領域およびMKK7のコード領域を含んでいたが、インフレームの停止コドンを欠く、異なる5'領域を持っていた。このクローンは、MKK7の異なるスプライシングを表わす(MKK7b;図11;配列番号:19)。MKK7b cDNAクローンは、その5'領域中に開始コドンを持たない;したがって、このcDNAは、単離された他のクローンと同じMKK7プロテインキナーゼをコードしている。しかし、MKK7b cDNAクローンが全長クローンでなければ、追加の5'配列がインフレームの開始コドンを含む可能性もある。もしそうならば、MKK7bはM-[?]-SPAPAPSQRAALQLPLANDGGSRSPSSESSPQHPTPPTRPRH- (配列番号:33)という配列をMKK7の開始メチオニン(図9)に融合させると予測される。ショウジョウバエのMAPキナーゼキナーゼhepはMKK7と実質的な配列類似性を持っているが、MKK7bのNH2末端部分は、hepプロテインキナーゼでは保存されていない。3つの別のクローンが、5'および3'領域が異なるMKK7のスプライシング変異体をコードしていた。これらのクローン(MKK7c(図13)、MKK7d(図14)、およびMKK7e(図15))は、5'および3'領域にインフレームの停止コドンが存在するため、全長である。
【0122】
全長のマウスMKK7 cDNAプローブで、ヒトcDNAライブラリーをスクリーニングした。単一のクローンが同定され、配列決定された。3'末端を持たない部分的MKK7配列が同定された(図12;配列番号:25、および配列番号:26)。配列は、マウスMKK7cと最も相同性が高かった。
【0123】
MKK7、MKK7b、hep、およびヒトMKK7のcDNAの配列は、それぞれ寄託番号U93030、U93031、U93032、AFOO319としてジーンバンク(Genbank)に寄託された。
【0124】
実施例23. MKK7の発現
MKK7の発現は、異なる組織から単離されたmRNAのノーザンブロット分析によって調べられた。分析は、異なる組織から単離され、変性アガロースゲル電気泳動により分画し、ナイロンメンブレン(Clontech)に移されたポリA+ mRNA(2μg)を用いて行なった。ブロットは、[α-32P]dATP(Amersham International PLC)を用いたランダムプライマーで標識したMKK4およびMKK7 cDNAでプローブした。
【0125】
MKK7はマウス組織で広範囲に発現されていることが分かった。精巣で2つのMKK7トランスクリプト(4.0 kbおよび1.6 kb)が検出されたのを除き、調べたすべての組織で、単一のMKK7トランスクリプト(約4.0 kb)が検出された。MKK7発現のレベルが最高だったのは、精巣だった。かなりのMKK7発現が、心臓、脳、肺、肝臓、および腎臓で観察された。これは、脳、肝臓、筋肉、心臓、および腎臓でかなりの量の発現が観察されるが、脳でレベルが最高であるMKK4の発現とは対照的だった。MKK4およびMKK7は、共に発現されるが、調べた組織によって、各MAPキナーゼキナーゼの相対的な量は、異なっていた。
【0126】
実施例24. インビトロにおける、MKK7によるJNKの特異的活性化
MKK7の特異性を調べるために、インビトロプロテインキナーゼアッセイ法を行なった。MKK7 cDNA(Eco RIおよびPvu II断片)をpGEX-5XL (Pharmacia-LKB) のEco RIおよびSma I部位にサブクローニングして、細菌のMKK7発現ベクターを調製した。アフィニティークロマトグラフィーによって、グルタチオン-S-トランスフェラーゼ(GST)融合蛋白質を精製した(SmithおよびJohnson (1988) Gene 67:31-40)。組換え蛋白質GST-ATF2 (Guptaら(1995) Science 267:389-393)、GST-cJun (Derijard (1994)上記)、GST-cMyc (Alvarezら(1991) J. Biol. Chem. 266:15277-15285)、GST-ERK2 (Sethら(1992) J. Biol. Chem. 267:24796-24804)、GST-p38 (Raingeaudら(1995) J. Biol. Chem. 270:7420-7426)、およびGST-JNK1 (Derijard (1994)上記)は記述されている。
【0127】
プロテインキナーゼアッセイ法は、キナーゼ緩衝液(25 mM 4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸塩(pH 7.4)、25 mM β-グリセロリン酸塩、25 mM MgCl2、2 mMジチオスレイトール、0.1 mM オルトバナジン酸塩)中で実行された。アッセイ法は、最終容量25μlの中に、1 μgの基質蛋白質および50 μM [γ-32P]ATP(10 Ci/mmol)を添加して開始された。25℃で30分後に、レムリ(Laemmli)サンプル緩衝液を添加して反応は終了した。SDS-ポリアクリルアミドゲル電気泳動(SDS-PAGE)後に、オートラジオグラフィーによって基質蛋白質のリン酸化を調べた。
【0128】
組換えMAPキナーゼは、基質ATP [γ-32P] を用いて、GST(対照)またはGST-MKK7とインキュベートされた。細菌から精製された組換えMKK7の自己リン酸化は観察されなかった。組換えMKK7をMAPキナーゼとインキュベートすると、MKK7はJNK1をリン酸化するが、p38とERK2はリン酸化しないことが示された。MKK7はp38とJNK1によってリン酸化された。MAPキナーゼによるMAPキナーゼキナーゼの逆方向のリン酸化の意義は不明であるが、同様な逆方向リン酸化はMKK4(Derijard (1995)上記)およびショウジョウバエJNK活性化因子hep (Sluss (1996)上記)を用いたキナーゼアッセイ法でも、検出されている。
【0129】
MKK7によるJNK1のリン酸化がプロテインキナーゼ活性を上昇させたかどうかを調べるために、JNK基質としてATF2を使った実験を行った。GST-MKK7は、プロテインキナーゼアッセイ法で組換えJNK1とインキュベートされた。JNK活性は、各アッセイ法にJNKの基質ATF2を含めて、測定された。ATF2はMKK7によってリン酸化されないが、JNK1によって弱くリン酸化される。JNK1とMKK7をインキュベートすると、JNK1がリン酸化され、ATF2のリン酸化が大きく上昇した。このデータは、MKK7がJNK1をリン酸化し、活性化することを示す。この結論を確認するために、JNKの二重のリン酸化のモチーフThr-Pro-TyrをAla-Pro-Pheで置換した効果を調べた。MKK7は変異JNK1 (APF)蛋白質をリン酸化しなかった。さらに、MKK7は、変異JNK1プロテインキナーゼによるATF2リン酸化を上昇させなかった。したがって、MKK7はインビトロにおけるJNKの活性化因子である。
【0130】
実施例25. インビボにおけるMKK7によるJNKの特異的活性化
インビボにおけるMKK7の特異性を調べるために、コトランスフェクション解析を行なった。CHO細胞は、ウシ胎仔血清(5%;Gibco-BRL)を添加したダルベッコ変法イーグル培地で維持された。細胞は、製造元(Gibco-BRL)の推奨する方法にしたがって、リポフェクタミン試薬を用いてトランスフェクトした(Derijardら(1994)上記)。細胞に、エピトープタグを付けたJNK1と、空の発現ベクター(対照)またはMKK4もしくはMKK7をコードする発現ベクターをコトランスフェクトした。エピトープタグは、インフルエンザウイルスのヘマグルチニン蛋白質(HA)に由来する。JNK1は細胞溶解物の免疫沈降によって単離された。細胞は溶解緩衝液(20 mM Tris ( pH 7.4), 1% 登録商標TRITON X-100, 10%グリセロール、137 mM NaCl, 2 mM EDTA, 25 mMβ-グリセロリン酸塩、1 mM オルトバナジン酸ナトリウム、2 mMピロリン酸塩、1 mM PMSF、および10μg/mlロイペプチン)で溶解し、4℃で100,000 x gで15分間遠心した。エピトープタグを付けたプロテインキナーゼは、プロテインGセファロース(Pharmacia-LKB Biotechnology Inc.)に結合した抗HAモノクローナル抗体を用いて、4℃で3時間インキュベーションし、免疫沈降させた。免疫沈降物は、溶解緩衝液で3回洗った(Guptaら(1995) Science 267:389-393)。基質として[γ-32P]ATPおよびc-Junを用いて、免疫複合体でプロテインキナーゼ活性を測定した。リン酸化反応の産物は、SDS-PAGE後にオートラジオグラフィーによって可視化した。ERK2およびp38 MAPキナーゼは、共に発現されたMKK7によって活性化されなかった。対照実験では、ERK2とp38 MAPキナーゼが、それぞれの近縁MAPキナーゼキナーゼMKK1およびMKK6で活性化されることが示された。これとは対照に、MKK7はJNK1を活性化した。興味深いことに、同時に発現されたMKK7によるJNK1の活性化は、以前に記述されたJNK活性化因子MKK4によるものよりも、高レベルだった。これらのデータを合わせると、MKK7が培養細胞においてJNKの特異的な活性化因子として機能することが立証される。
【0131】
実施例26. MKK7によるJNKシグナル伝達経路の活性化
JNKシグナル伝達経路は、AP-1転写活性を調節することが知られている。(Whitmarsh (1996)上記)。MKK7の発現がAP-1の転写活性を上昇させるという仮説を検証するために、最小プロモーター要素の上流に3つのAP-1部位がクローニングされたルシフェラーゼレポーター遺伝子を用いたコトランスフェクション解析を行なった(RinconおよびFlavell (1994) EMBO J. 13:4370-4381)。ルシフェラーゼレポーター遺伝子発現は、レポータープラスミドpTRE-ルシフェラーゼ(Rincon (1994)上記)0.5μgと、βガラクトシダーゼ発現ベクターpCH110(Pharmacia-LKB)0.25μgとを用いたコトランスフェクション解析で測定された。GAL4融合蛋白質を用いた実験は、0.25μgのpGAL4-ATF2(残基1〜109)、0.5μgのレポータープラスミドpG5E1bLuc、および0.25μgのpCH110を用いて行なった(Guptaら(1995)上記)。プロテインキナーゼの効果は、0.3μgの空の発現ベクターまたはプロテインキナーゼ発現ベクターとのコトランスフェクションによって調べた。ERK2、p38、JNK1、MKK1、MKK3、MKK4、およびMKK6発現ベクターは、記述されている。細胞はトランスフェクション後36時間で回収した。細胞溶解物中のβガラクトシダーゼおよびルシフェラーゼの活性は、記述されたようにして測定した(Gupta (1995)上記)。MKK4、MKK7、またはJNK1が発現されても、AP-1レポーター遺伝子の発現に著しい変化は見られなかった(図16A)。これとは対照に、MKK7とJNK1が同時に発現すると、AP-1に依存するレポーター遺伝子の発現が上昇した。MKK4はMKK7よりも弱くJNKを活性化させるという観察と一致して、MKK4とJNKとが同時に発現すると、AP-1レポーター遺伝子の発現が、MKK7の場合よりも少なく、上昇した(図16A)。これらのデータを合わせると、MKK7がJNKシグナル伝達経路の活性化因子として機能することを示す。
【0132】
転写活性に対するMKK7の効果をさらに調べるために、転写因子ATF2に対するMKK7の影響を探った。以前の研究で、ATF2がJNKシグナル伝達経路の標的であることが示されている(van Damら(1995)上記;Guptaら(1995)上記;Livingstoneら(1995)上記)。JNKはATF2のNH2末端活性化ドメイン中の2つの部位(Thr-69およびThr-71)をリン酸化し、転写活性を上昇させる。ATF2の活性化ドメインの転写活性をモニターするために、GAL4融合蛋白質戦略を採用した(Gupta (1995)上記)。レポーター遺伝子の発現を測定すると、MKK4とJNK1が同時に発現すると、転写活性が上昇することが示された(図16B)。MKK7が発現してもレポーター遺伝子発現は同様なレベルで観察されたが、MKK7がJNK1と同時に発現すると、さらに大きな上昇が検出された。転写活性に対して、MKK4と比較してMKK7の方が強い効果を示すということは、JNK活性化に対するMKK7とMKK4の相対的な効果と一致している。レポーター遺伝子発現の上昇がATF2のリン酸化によって仲介されることを確認するために、ATF2リン酸化部位(Thr-69およびThr-71)をAlaに置換した効果を調べた。変異ATF2蛋白質は、MKK4、MKK7、またはJNK1によって調節されない(図16B)。これらのデータを合わせると、MKK7がJNKシグナル伝達経路の生理的な標的を調節し得ることを示す。
【0133】
用途
本発明のMKKポリペプチドおよびポリヌクレオチドは、MKKシグナル伝達経路を調節する試薬の同定に有用である。MKKシグナル伝達経路を調節する試薬は、MKK合成、MKKリン酸化またはMKK活性に対するその影響によって、同定できる。例えば、MKK活性に対する試薬の影響は、上述のインビトロキナーゼアッセイ法によって、測定できる。MKKは成分の反応を許すような条件下で、試験試薬の存在下および非存在下(対照)でインキュベートし、その後、試験試薬のキナーゼ活性に対する効果を測定する。MKKシグナル伝達経路を阻害する試薬は、MKKを介する疾病の治療に利用できる。MKKシグナル伝達経路を刺激する試薬は、組織におけるプログラムされた細胞死(アポトーシス)の誘導を含め、いくつもの方法に、使用できる。例えば、UVで損傷を受けた細胞の除去は、癌の予防に使用できる。
【0134】
一般に、MKKシグナル伝達経路を阻害する試薬の同定には、キナーゼアッセイ法(例えば、実施例3参照)が使用される。MKK基質および[γ-32P]ATPのような放射性標識を含む試験システムに、種々の濃度の試薬(例、1.0 nM〜100 mM)を添加する。適当な基質分子には、p38、JNK1、JNK2、またはATF2が含まれる。標識リン(例、[32P]または[33P]の基質への取り込みを決定し、試験試薬で得られた結果を対照の値と比較する。特に興味深いのは、[32P]の取り込みを約80%またはそれ以上阻害する試薬である。リン酸化は、リン酸化に依存する試薬、例えば、抗体を用いて調べることもできる。リン酸化依存性抗体は、活性化部位、Ser198およびThr202がリン酸化されたMKK7を用いて作製できる。これは、リン酸化されたSer198およびThr202を持つMKK7配列に対応する合成ペプチド(例えば、長さ約15アミノ酸)で動物を免疫して実施できる。そのような抗体の生産方法は、当技術分野で周知である。そのような抗体は、組織および細胞抽出物において活性化MKK7の検出(例、ウエスタンブロット)に役立ち、キットとして使用できる可能性がある。
【0135】
MKK合成に対する試薬の効果を検定するアッセイ法も、MKKシグナル伝達経路を阻害する化合物の同定に利用できる。MKK発現に対する試験試薬の影響は、例えば、MKKに特異的な抗体を用いたウエスタンブロット分析で測定される。抗体の結合は、オートラジオグラフィーまたは化学ルミネセンスによって可視化し、定量する。MKK mRNA発現に対する試験試薬の効果は、例えば、ポリヌクレオチドプローブを用いたノーザンブロット分析、またはポリメラーゼ連鎖反応によって、調べることができる。
【0136】
MKKシグナル伝達経路を阻害することが分かった試薬は、MKKを介する疾病の治療に使用できる。そのような試薬は、MKKシグナル伝達経路を阻害するために必要な分子の特異的な特徴を解明するための薬剤デザインにも、有用である。
【0137】
さらに、本発明はMKKを介するストレス関連および炎症性疾患の治療方法も提供する。この方法には、MKK機能を阻害する治療試薬の有効量の投与が含まれる。適当な試薬は、MKK活性または発現のいずれかを阻害する。投与する試薬の濃度は、適当な用量、投与経路、および治療する特定の症状を含め、いくつもの要素によって、決定される。適当な試薬用量は、通常の実験から個々の患者および治療する特定のMKKを介する疾患に必要な最適用量までを含め、当業者に周知の方法によって、決定される。投与に適当で治療に有効な特定の量は、当業者によって簡単に決定される(例、レミントンの薬学(Remington's Pharmaceutical Sciences) 第18版、Gennaro編、Mack Publishing Company、ペンシルベニア州イーストン、1990参照)。用量は約0.1〜10 mg/キロ/日の範囲に渡る可能性がある。
【0138】
本発明は、ストレス関連および炎症性疾患の急性治療および予防の方法を提供する。例えば、虚血性心疾患は、再灌流後の酸化ストレスおよび虚血の発現時に、治療されると想像される。さらに、虚血のリスクを持つ患者は、虚血症状の発現前に、治療できる。
【0139】
別の例では、MKK機能または活性を阻害する治療薬を投与して、TNFおよびIL-1を含む炎症促進型サイトカインの分泌を阻害することにより、炎症反応をコントロールする。
【0140】
ストレスに関連する増殖性疾患も、MKK機能または活性を阻害する治療試薬を投与することによって、本発明の方法により治療できる。そのような治療試薬は、単独または、例えば悪性腫瘍の治療における化学療法剤などの他の治療試薬と組合わせて、使用できる。実際に、本発明によって提供される治療試薬によるストレス活性化MKKのコントロールは、ストレスを誘導する他の治療戦略によって引き起こされる症状を調節できる。
【0141】
採用される治療試薬は、ポリヌクレオチド、ポリペプチド、およびアンチセンスオリゴヌクレオチドおよびリボザイムのような他の分子を含む、MKK機能または活性を阻害する化合物であり、本発明および当技術分野で周知の技術にしたがって製造できるものである。MKKのエピトープに結合するポリクローナルまたはモノクローナル抗体(その断片または誘導体を含む)も、治療試薬として採用できる。基質の結合および/またはリン酸化に関して、効率的にMKKを排除するまたはMKKと競合するMKKのドミナントネガティブ型は、プロテインキナーゼ活性を低下させるために使用できる。ドミナントネガティブ型は、当技術分野で周知の方法を用いて、上述(実施例13)のように、プロテインキナーゼの触媒ドメイン内に変異を導入することによって、作製できる。触媒性の残基は、すべてのMKKアイソフォームで保存されている。例えば、Lys76の変異は、MKK7の活性を阻害する。同様に、活性化リン酸化の保存された部位(Ser198、Thr202)の変異は、MKK7活性を阻害する。これらのMKK7のキナーゼ不活性型は、ドミナントネガティブ阻害剤として作用する。
【0142】
場合によっては、例えばアポトーシスの誘導のように、MKK活性の増加が望ましいことがある。本発明の方法を用いて、MKK機能または活性を増加させる能力のある試薬を同定できる。または、MKKを過剰発現して活性の増加が実現できる。MKKを介する疾病が、MKK発現の不足、または変異MKKポリペプチドの発現を伴う場合には、センスポリヌクレオチド配列(DNAコード鎖)またはMKKポリペプチドを細胞に導入して、正常なMKK活性を増大させることができる。必要ならば、これらの治療法は、投与様式(例、細胞の種類に特異的なウイルスベクターの使用)、または当技術分野で周知の方法により細胞の種類に特異的もしくは誘導可能なプロモーターを持つ組換え構築物にMKK7核酸を入れることによって、特定の細胞が標的となるようにする。例えば、MKK7核酸を含むベクターは、当技術分野で標準的な組換えDNA技術によって、作製できる。ベクターは、哺乳類細胞での複製および発現に使用される、プラスミド、ウイルス、または当技術分野で周知の他のベクターであり得る。MKK7核酸をコードする配列の発現は、哺乳類細胞、好ましくはヒト細胞において当技術分野で周知の任意のプロモーターでよい。そのようなプロモーターは、誘導性または構成性であり得る。そのようなプロモーターには、SV40初期プロモーター領域(Bernoistら、Nature 290:304, 1981);ラウス肉腫ウイルスの3'長い末端反復配列に含まれるプロモーター(Yamamotoら、Cell 22:787-797, 1988);ヘルペスチミジンキナーゼプロモーター(Wagnerら、Proc. Natl. Acad. Sci. USA 78:1441, 1981);またはメタロチオネイン遺伝子の調節配列(Brinsterら、Nature 296:39, 1988)が含まれるが、これらに限定されるわけではない。
【0143】
本発明の抗体は、注射または時間をかけた徐々の注入により、非経口的に投与できる。本発明のモノクローナル抗体は、静脈内、腹腔内、筋肉内、皮下、腔内、または経皮的に投与できる。
【0144】
本発明のポリペプチドまたは抗体の非経口投与用調製物には、無菌の水性または非水性溶液、懸濁液、および乳濁液が含まれる。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、オリーブ油のような植物油、およびオレイン酸エチルのような注射可能な有機エステルである。水性キャリアには、生理食塩水および緩衝媒体を含む、水、アルコール性/水性溶液、乳濁液、または懸濁液が含まれる。非経口賦形剤には、塩化ナトリウム溶液、リンゲルのブドウ糖、ブドウ糖および塩化ナトリウム、乳酸リンゲル液、または固定油が含まれる。静脈内賦形剤には、液体および栄養補充液、電解質補充液(リンゲルのブドウ糖に基づくものなど)、および同様なものが含まれる。例えば、抗菌剤、抗酸化剤、キレート剤、および不活性ガス、および同様なものなどの、保存剤および他の添加物も、存在する可能性がある。
【0145】
アンチセンス配列を含むポリヌクレオチド配列は、当技術分野で周知の様々な技術によって、治療用に投与できる。そのような治療法は、MKKポリヌクレオチドをMKKを介する疾病を持つ哺乳類の細胞中に導入することによって、その治療効果を発揮する。MKKポリヌクレオチドの送達は、遊離ポリヌクレオチド、またはキメラウイルスのような組換え発現ベクター、またはコロイド分散系を利用して実施できる。ヌクレオチド配列の治療用送達に特に好ましいのは、標的を定めたリポソームを利用することである。
【0146】
治療用試薬を、特定の組織にターゲッティングすることは、送達効率を上昇させるために望ましい。ターゲッティングは、投与経路を通じた受動的な機構によって、実現できる。特定の組織に対して積極的にターゲッティングすることもできる。リポソーム、コロイド懸濁液、およびウイルスベクターを利用すると、例えば、標的組織の成分の受容体として働く分子を入れるなど、治療用試薬を含む製剤の組成を変化させることによって、ターゲッティングが可能になる。例には、糖、糖脂質、ポリヌクレオチド、または蛋白質が含まれる。これらの分子は、治療用試薬に含まれることができる。または、例えば、この分子をコードするポリヌクレオチドを入れる、またはターゲッティング分子を提供するパッケージング系を利用することによって、間接的な方法によって、これらの分子を入れることもできる。本明細書に提供される教示を利用すれば、特定の組織に治療試薬を送達するために、どの分子や手順が有用であるか、当業者は理解するまたは確認するだろう。
【0147】
トランスジェニック動物
MKKポリペプチドは、トランスジェニック動物で発現することができる。これらの動物は、MKKの過剰発現または発現不足によって誘導または増悪される疾患の研究、およびMKKの発現または活性を調節する治療薬の開発のための、モデルシステムとなる。例えば、マウス中でドミナントネガティブで構成的に活性化された対立遺伝子を発現して、生理的機能を立証できる。
【0148】
トランスジェニック動物は、家畜(ブタ、ヤギ、ヒツジ、ウシ、ウマ、ウサギ、および同様なもの)、齧歯類(ラット、モルモット、およびマウスなど)、非ヒト霊長類(例えば、ヒヒ、サル、およびチンパンジー)、およびペット(例、イヌおよびネコ)であり得る。トランスジェニックマウスは、特に好ましい。
【0149】
トランスジェニック動物のファウンダー系統を作製するために、当技術分野で周知の任意の技術を用いて、MKKトランスジーンを動物に導入することができる。そのような技術には、前核マイクロインジェクション(米国特許第4,873,191号);レトロウイルスを介した生殖系統への遺伝子移入(Van der Puttenら、Proc. Natl. Acad. Sci. USA 82:6148, 1985);胚性肝細胞への遺伝子ターゲッティング(Thompsonら、Cell 56:313, 1989);および胚のエレクトロポレーション(Lo, Mol. Cell. Biol. 3:1803, 1983)が含まれるが、これらに限定されるわけではない。特に有用なのは、Yangら(Proc. Natl. Acad. Sci. USA 94:3004-3009, 1997)に記述される方法である。
【0150】
本発明は、すべての細胞にMKKトランスジーンを持つトランスジェニック動物、および一部の細胞にのみトランスジーンを持つ動物を提供する。すなわち、本発明は、モザイク動物を提供する。トランスジーンは、単一のトランスジーン、または例えば、逆方向もしくは同方向に縦につながったコンカテマーとして組み込まれ得る。トランスジーンは、特定のタイプの細胞に選択的に導入および活性化できる(Laskoら、Proc. Natl. Acad. Sci. USA 89:6232, 1992)。そのような細胞のタイプに特異的な活性化に必要な調節配列は、興味のある特定の細胞のタイプに依存し、当業者には明らかだろう。
【0151】
MKKトランスジーンが内因性MKK遺伝子の染色体部位に組み込まれることが望ましい場合には、遺伝子ターゲッティングが好ましい。簡単に述べると、そのような技術を使用する場合には、染色体配列との相同組換えによって内因性遺伝子に組み込まれ、その機能を破壊するために、内因性MKK遺伝子と相同なヌクレオチド配列を含むベクターがデザインされる。トランスジーンも、特定のタイプの細胞に選択的に導入され、そのタイプの細胞でのみ内因性MKK遺伝子を不活化することができる(Guら、Science 265:103, 1984)。そのような細胞のタイプに特異的な不活化に必要な調節配列は、興味のある特定の細胞のタイプに依存し、当業者には明らかだろう。これらの技術は、機能的なMKK遺伝子を持たない「ノックアウト」の調製に有用である。
【0152】
トランスジェニック動物が作製されたら、標準的な方法で、組換えMKK遺伝子の発現を解析できる。最初のスクリーニングは、サザンブロット分析またはPCR技術による、トランスジーンの組み込みが起きたかどうかの決定である可能性がある。トランスジェニック動物の組織中でのトランスジーンのmRNA発現のレベルは、動物から得た組織試料のノーザンブロット分析、インサイチューハイブリダイゼーション分析、およびRT-PCRを含むが、これらに限定されることのない技術を用いても、評価できる。MKK遺伝子発現組織の試料は、MKKトランスジーン産物に特異的な抗体を用いて免疫組織化学的にも、評価できる。
【0153】
トランスジェニック動物の作製および評価に使用できる技術の総説には、当業者はGordon (Intl. Rev. Cytol. 115:171-229, 1989)を参照し、例えば、Hoganら、(マウス胚の操作(Manipulating the Mouse Embryo), Cold Spring Harbor Press, Cold Spring Harbor, NY, 1986)、Krimpenfortら(Bio/Technology 9:86, 1991)、Palmiterら(Cell 41:343, 1985)、Kraemerら(初期哺乳類胚の遺伝子操作(Genetic Manipulation of the Early Mammalian Embryo), Cold Spring Harbor Press, Cold Spring Harbor, NY, 1985)、Hammer ら(Nature 315:680, 1985)、Purcelら(Science, 224:1281, 1986)、Wagner ら(米国特許第5,175,385号)、およびKrimpenfortら(米国特許第5,175,384号)から、さらに手引きを得ることができる可能性がある(最後の2つは、参照として本明細書に組み入れられる)。
【0154】
他の態様
本発明はその詳細な説明と関連して説明されてきたが、上述の説明は、添付の請求の範囲によって定義される本発明の範囲を明らかにするためのもので、それを制限する意図はないことを理解する必要がある。他の局面、利点、および修正は、請求の範囲内である。
【図面の簡単な説明】
【0155】
【図1】図1はMKK3(配列番号:2)、MKK4-α(配列番号:6)、ヒトMAPキナーゼキナーゼMEK1(配列番号:11)およびMEK2(配列番号:12)、ならびに酵母HOG1 MAPキナーゼキナーゼPBS2(配列番号:13)のアミノ酸配列を比較したものである。配列は、PILE-UPプログラム(バージョン7.2;Wisconsin Genetics Computer Group)を用いて比較した。蛋白質配列は、1文字コード(A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, TrpおよびY, Tyr)で示されている。PBS2配列は、NH2-(<)およびCOOH-(>)末端の両方で切断されている。整合を最適化するために配列に導入されたギャップは、ハイフンで示されている。同一の残基は、ピリオドで示されている。MEK中の活性化リン酸化の部位は、アステリスクで示されている。
【図2A】図2Aは、ヒトおよび酵母のMAPキナーゼキナーゼのメンバーの間の関係を示す樹状図である。樹状図は、算術平均を用いて、無加重のペアグループ法によって、作製された(PILE-UPプログラム)。ヒト(hu) MAPキナーゼキナーゼMEK1、MEK2、MEK3、およびMEK4;Sccharomyces cerevisiae(サッカロミセス・セレビジエ)(sc) MAPキナーゼキナーゼPBS2、MKK1、およびSTE7;およびSccharomyces pombe(サッカロミセス・ポンベ)(sp) MAPキナーゼキナーゼWIS1およびBYR1が示されている。
【図2B】図2Bは、MKKの間の関係を示す樹状図である。樹状図は、図2Aのようにして作製された。
【図3】図3は、ERK、p38、およびJNKシグナル伝達経路を模式図で表わしたものである。MEK1およびMEK2は、MAPキナーゼのERKサブグループの活性化因子である。MKK3およびMKK4は、p38 MAPキナーゼの活性化因子である。MKK4は、MAPキナーゼのp38およびJNKサブグループの両方の活性化因子であると同定されている。
【図4A】図4A〜4Dは、MKK3の核酸配列(配列番号:1)およびアミノ酸配列(配列番号:2)を表わしたものである。
【図4B】図4Aの続きを示す図である。
【図4C】図4Bの続きを示す図である。
【図4D】図4Cの続きを示す図である。
【図5A】図5A〜5Cは、MKK6の核酸配列(配列番号:3)およびアミノ酸配列(配列番号:4)を表わしたものである。
【図5B】図5Aの続きを示す図である。
【図5C】図5Bの続きを示す図である。
【図6A】図6A〜6Fは、MKK4αの核酸配列(配列番号:5)およびアミノ酸配列(配列番号:6)を表わしたものである。
【図6B】図6Aの続きを示す図である。
【図6C】図6Bの続きを示す図である。
【図6D】図6Cの続きを示す図である。
【図6E】図6Dの続きを示す図である。
【図6F】図6Eの続きを示す図である。
【図7A】図7A〜7Fは、MKK4βの核酸配列(配列番号:7)およびアミノ酸配列(配列番号:8)を表わしたものである。
【図7B】図7Aの続きを示す図である。
【図7C】図7Bの続きを示す図である。
【図7D】図7Cの続きを示す図である。
【図7E】図7Dの続きを示す図である。
【図7F】図7Eの続きを示す図である。
【図8A】図8A〜8Fは、MKK4γの核酸配列(配列番号:9)およびアミノ酸配列(配列番号:10)を表わしたものである。
【図8B】図8Aの続きを示す図である。
【図8C】図8Bの続きを示す図である。
【図8D】図8Cの続きを示す図である。
【図8E】図8Dの続きを示す図である。
【図8F】図8Eの続きを示す図である。
【図9A】図9は、PILE-UPプログラム(バージョン7.2;Wisconsin Genetics Computer Group)を用いて、MKK7(配列番号:18)の推定される一次構造を、hep(配列番号:21)、MAPキナーゼキナーゼMEK1(MKK1;配列番号:11)、MEK2(MKK2;配列番号:12)、MKK3(配列番号:2)、MKK4γ(配列番号:10)、MKK5(配列番号:22)、およびMKK6(配列番号:4)と比較したものである。整合を最適化するために配列に導入されたギャップは、ハイフン(-)で示されている。同一の残基は、ピリオド(.)で示されている。MAPキナーゼキナーゼ(2、27、37、および38)中の活性化リン酸化の部位は、アステリスク(*)で示されている。
【図9B】図9Aの続きを示す図である。
【図10A】図10A〜10Dは、MKK7の核酸配列(配列番号:17)およびアミノ酸配列(配列番号:18)を表わしたものである。
【図10B】図10Aの続きを示す図である。
【図10C】図10Bの続きを示す図である。
【図10D】図10Cの続きを示す図である。
【図11A】図11A〜11Dは、MKK7bの核酸配列(配列番号:19)およびアミノ酸配列(配列番号:20)を表わしたものである。
【図11B】図11Aの続きを示す図である。
【図11C】図11Bの続きを示す図である。
【図11D】図11Cの続きを示す図である。
【図12A】図12A〜12Bは、ヒトMKK7の核酸配列(配列番号:25)およびアミノ酸配列(配列番号:26)を表わしたものである。
【図12B】図12Aの続きを示す図である。
【図13A】図13A〜13Dは、マウスMKK7cの核酸配列(配列番号:27)およびアミノ酸配列(配列番号:28)を表わしたものである。
【図13B】図13Aの続きを示す図である。
【図13C】図13Bの続きを示す図である。
【図13D】図13Cの続きを示す図である。
【図14A】図14A〜14Dは、マウスMKK7dの核酸配列(配列番号:29)およびアミノ酸配列(配列番号:30)を表わしたものである。
【図14B】図14Aの続きを示す図である。
【図14C】図14Bの続きを示す図である。
【図14D】図14Cの続きを示す図である。
【図15A】図15A〜15Dは、マウスMKK7eの核酸配列(配列番号:31)およびアミノ酸配列(配列番号:32)を表わしたものである。
【図15B】図15Aの続きを示す図である。
【図15C】図15Bの続きを示す図である。
【図15D】図15Cの続きを示す図である。
【図16】図16Aは、細胞にAP-1レポータープラスミドpTRE-ルシフェラーゼと、MKK4、MKK7、JNK1、JNK1 (APF) の発現ベクターまたは対照ベクターとを、コトランスフェクトしたトランスフェクション解析のデータのグラフである。図16Bは、細胞にGAL4-ATF2融合ベクターと、MKK4、MKK7、JNK1、JNK1 (APF)の発現ベクターまたは対照ベクターとを、コトランスフェクトしたトランスフェクション解析のグラフである。
【特許請求の範囲】
【請求項1】
セリン、スレオニン、およびチロシンキナーゼ活性を有し、マイトジェン活性化プロテイン (MAP)キナーゼJNKをリン酸化するがp38はリン酸化しない、実質的に純粋な哺乳類マイトジェン活性化プロテインキナーゼキナーゼ(MKK7)ポリペプチド。
【請求項2】
配列番号:18、配列番号:20、配列番号:26、配列番号:28、配列番号:30、または配列番号:32のアミノ酸配列を含む、請求項1記載のポリペプチド。
【請求項3】
請求項1記載のポリペプチドをコードする単離ポリヌクレオチド配列。
【請求項4】
配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、もしくは配列番号:31、もしくはその縮重変異体、または配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、もしくは配列番号:31の配列に完全に相補的なポリヌクレオチド配列、もしくはその縮重変異体の配列を含む、請求項3記載の単離ポリヌクレオチド配列。
【請求項5】
配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、配列番号:31の配列またはその相補配列に、ストリンジェントなハイブリダイゼーション条件下でハイブリダイズするポリヌクレオチド配列を含む、請求項3記載の単離ポリヌクレオチド配列。
【請求項6】
請求項3記載のポリヌクレオチド配列を含む組換え発現ベクター。
【請求項7】
請求項3記載のポリヌクレオチド配列を含む組換え宿主細胞。
【請求項8】
請求項1記載のポリペプチドに特異的に結合する精製抗体。
【請求項9】
請求項2記載のポリペプチドに特異的に結合する精製抗体。
【請求項10】
下記の段階を含む、生体試験試料中のマイトジェン活性化プロテインキナーゼキナーゼ(MKK7)の活性を測定する方法:
a) 試験試料を、請求項1記載のMKKポリペプチドのMKK基質および標識されたリン酸塩と共に、該基質がリン酸化されるのに十分な条件下でインキュベートする段階;および
b) 該基質への標識リン酸塩の取り込みの割合を決定する段階であって、該取り込みの割合がMKK活性の指標である段階。
【請求項11】
MKK基質が、JNK MAPキナーゼ、活性化転写因子-2(ATF-2)、ATFa、cAMP応答配列結合蛋白質(CRE-BPa)、およびc-Junからなる群より選択される、請求項10記載の方法。
【請求項12】
生体試験試料が哺乳類から得られた体液、細胞、または組織である、請求項10記載の方法。
【請求項13】
下記の段階を含む、生体試験試料におけるMKK7の合成を測定する方法:
a) 生体試料を得る段階;
b) 請求項1記載のMKK7ポリペプチドに特異的に結合する抗体と、該生体試料とを接触させる段階;および
c) MKK7ポリペプチドに結合した該抗体を検出する段階であって、MKK7合成のレベルが、結合した抗体の量によって決定される段階。
【請求項14】
下記の段階を含む、試験試料におけるMKK7の発現レベルを測定する方法:
a) 試験試料から総RNAまたはポリアデニル化RNAを単離する段階;
b) 請求項3記載のMKK7ポリヌクレオチドに特異的なポリヌクレオチドプローブと共にRNAをインキュベートする段階;
c) RNAにハイブリダイズした該プローブの量を決定する段階であって、MKK7の発現レベルが、RNAにハイブリダイズしたMKKプローブの量に直接に関連している段階。
【請求項15】
下記の段階を含む、MKK7活性を調節する試薬を同定する方法:
a) MKK7を含む試験試料を得る段階;
b) 試薬が存在しない場合に基質がリン酸化されるのに十分な条件下で、請求項1記載のMKKポリペプチドのMKK基質、一連の濃度の試薬、および標識リン酸塩と共に、該試験試料をインキュベートする段階;
c) 該基質のリン酸化を検出する段階;および
d) 対照と該試薬のMKK7活性に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7基質のリン酸化を調節できることが示される段階。
【請求項16】
MKK7基質がJNK、ATF2、ATFa、CRE-BPa、およびc-Junのうちの1つまたは複数である、請求項15記載の方法。
【請求項17】
調節がMKK7活性の阻害である、請求項15記載の方法。
【請求項18】
下記の段階を含む、MKK7合成を調節する試薬を同定する方法:
a) MKK7合成能力のある試料を提供する段階;
b) 試薬が存在しない場合にMKK7が合成される条件下で、一連の濃度の試薬と共に該試料をインキュベーションする段階;
c) 請求項1記載のMKK7ポリペプチドを検出する段階;および
d) 対照と該試薬のMKK7合成に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7を調節できることが示される段階。
【請求項19】
調節がMKK7合成の阻害である、請求項18記載の方法。
【請求項20】
下記の段階を含む、MKK7発現を調節する試薬を同定する方法:
a) MKK7発現能力のある試料を提供する段階;
b) 試薬の非存在下でMKK7が発現される条件下で、一連の濃度の試薬と共に該試料をインキュベーションする段階;
c) 試料から総RNAまたはポリアデニル化RNAを単離する段階;
d) 請求項3記載のMKK7核酸に特異的なポリヌクレオチドプローブと共にRNAをインキュベートする段階;および
e) 対照と該試薬のMKK7 RNA合成に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7発現を調節できることが示される段階。
【請求項21】
治療的に有効な量のMKK7活性を調節する試薬を患者に投与する段階を含む、患者におけるMKK7を介する疾患の治療方法。
【請求項22】
MKK7を介する疾患が、虚血性心疾患、腎不全、酸化性肝障害、呼吸障害症候群、熱および放射線による熱傷、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、および炎症性疾患からなる群より選択される、請求項21記載の方法。
【請求項23】
治療的に有効な量のMKK7ポリペプチドを患者に投与する段階を含む、患者におけるMKK7関連疾患の治療方法。
【請求項24】
MKK7関連疾患が、虚血性心疾患、腎不全、酸化性肝障害、呼吸障害症候群、熱および放射線による熱傷、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、または炎症性疾患である、請求項23記載の方法。
【請求項1】
セリン、スレオニン、およびチロシンキナーゼ活性を有し、マイトジェン活性化プロテイン (MAP)キナーゼJNKをリン酸化するがp38はリン酸化しない、実質的に純粋な哺乳類マイトジェン活性化プロテインキナーゼキナーゼ(MKK7)ポリペプチド。
【請求項2】
配列番号:18、配列番号:20、配列番号:26、配列番号:28、配列番号:30、または配列番号:32のアミノ酸配列を含む、請求項1記載のポリペプチド。
【請求項3】
請求項1記載のポリペプチドをコードする単離ポリヌクレオチド配列。
【請求項4】
配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、もしくは配列番号:31、もしくはその縮重変異体、または配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、もしくは配列番号:31の配列に完全に相補的なポリヌクレオチド配列、もしくはその縮重変異体の配列を含む、請求項3記載の単離ポリヌクレオチド配列。
【請求項5】
配列番号:17、配列番号:19、配列番号:25、配列番号:27、配列番号:29、配列番号:31の配列またはその相補配列に、ストリンジェントなハイブリダイゼーション条件下でハイブリダイズするポリヌクレオチド配列を含む、請求項3記載の単離ポリヌクレオチド配列。
【請求項6】
請求項3記載のポリヌクレオチド配列を含む組換え発現ベクター。
【請求項7】
請求項3記載のポリヌクレオチド配列を含む組換え宿主細胞。
【請求項8】
請求項1記載のポリペプチドに特異的に結合する精製抗体。
【請求項9】
請求項2記載のポリペプチドに特異的に結合する精製抗体。
【請求項10】
下記の段階を含む、生体試験試料中のマイトジェン活性化プロテインキナーゼキナーゼ(MKK7)の活性を測定する方法:
a) 試験試料を、請求項1記載のMKKポリペプチドのMKK基質および標識されたリン酸塩と共に、該基質がリン酸化されるのに十分な条件下でインキュベートする段階;および
b) 該基質への標識リン酸塩の取り込みの割合を決定する段階であって、該取り込みの割合がMKK活性の指標である段階。
【請求項11】
MKK基質が、JNK MAPキナーゼ、活性化転写因子-2(ATF-2)、ATFa、cAMP応答配列結合蛋白質(CRE-BPa)、およびc-Junからなる群より選択される、請求項10記載の方法。
【請求項12】
生体試験試料が哺乳類から得られた体液、細胞、または組織である、請求項10記載の方法。
【請求項13】
下記の段階を含む、生体試験試料におけるMKK7の合成を測定する方法:
a) 生体試料を得る段階;
b) 請求項1記載のMKK7ポリペプチドに特異的に結合する抗体と、該生体試料とを接触させる段階;および
c) MKK7ポリペプチドに結合した該抗体を検出する段階であって、MKK7合成のレベルが、結合した抗体の量によって決定される段階。
【請求項14】
下記の段階を含む、試験試料におけるMKK7の発現レベルを測定する方法:
a) 試験試料から総RNAまたはポリアデニル化RNAを単離する段階;
b) 請求項3記載のMKK7ポリヌクレオチドに特異的なポリヌクレオチドプローブと共にRNAをインキュベートする段階;
c) RNAにハイブリダイズした該プローブの量を決定する段階であって、MKK7の発現レベルが、RNAにハイブリダイズしたMKKプローブの量に直接に関連している段階。
【請求項15】
下記の段階を含む、MKK7活性を調節する試薬を同定する方法:
a) MKK7を含む試験試料を得る段階;
b) 試薬が存在しない場合に基質がリン酸化されるのに十分な条件下で、請求項1記載のMKKポリペプチドのMKK基質、一連の濃度の試薬、および標識リン酸塩と共に、該試験試料をインキュベートする段階;
c) 該基質のリン酸化を検出する段階;および
d) 対照と該試薬のMKK7活性に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7基質のリン酸化を調節できることが示される段階。
【請求項16】
MKK7基質がJNK、ATF2、ATFa、CRE-BPa、およびc-Junのうちの1つまたは複数である、請求項15記載の方法。
【請求項17】
調節がMKK7活性の阻害である、請求項15記載の方法。
【請求項18】
下記の段階を含む、MKK7合成を調節する試薬を同定する方法:
a) MKK7合成能力のある試料を提供する段階;
b) 試薬が存在しない場合にMKK7が合成される条件下で、一連の濃度の試薬と共に該試料をインキュベーションする段階;
c) 請求項1記載のMKK7ポリペプチドを検出する段階;および
d) 対照と該試薬のMKK7合成に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7を調節できることが示される段階。
【請求項19】
調節がMKK7合成の阻害である、請求項18記載の方法。
【請求項20】
下記の段階を含む、MKK7発現を調節する試薬を同定する方法:
a) MKK7発現能力のある試料を提供する段階;
b) 試薬の非存在下でMKK7が発現される条件下で、一連の濃度の試薬と共に該試料をインキュベーションする段階;
c) 試料から総RNAまたはポリアデニル化RNAを単離する段階;
d) 請求項3記載のMKK7核酸に特異的なポリヌクレオチドプローブと共にRNAをインキュベートする段階;および
e) 対照と該試薬のMKK7 RNA合成に対する効果を比較する段階であって、対照と比較した何らかの変化により、試薬がMKK7発現を調節できることが示される段階。
【請求項21】
治療的に有効な量のMKK7活性を調節する試薬を患者に投与する段階を含む、患者におけるMKK7を介する疾患の治療方法。
【請求項22】
MKK7を介する疾患が、虚血性心疾患、腎不全、酸化性肝障害、呼吸障害症候群、熱および放射線による熱傷、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、および炎症性疾患からなる群より選択される、請求項21記載の方法。
【請求項23】
治療的に有効な量のMKK7ポリペプチドを患者に投与する段階を含む、患者におけるMKK7関連疾患の治療方法。
【請求項24】
MKK7関連疾患が、虚血性心疾患、腎不全、酸化性肝障害、呼吸障害症候群、熱および放射線による熱傷、敗血症性ショック、慢性関節リウマチ、自己免疫疾患、または炎症性疾患である、請求項23記載の方法。
【図1】

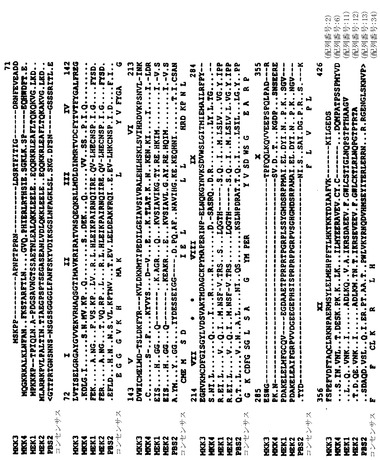
【図2A】
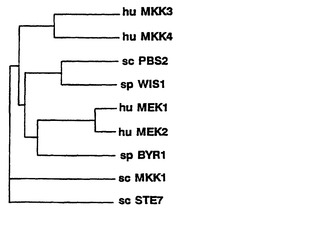
【図2B】
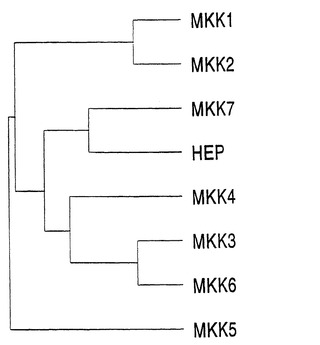
【図3】
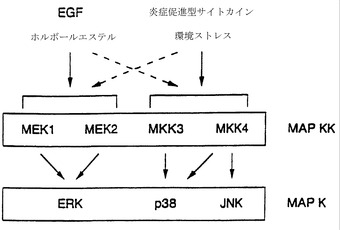
【図4A】

【図4B】

【図4C】

【図4D】

【図5A】

【図5B】

【図5C】

【図6A】

【図6B】

【図6C】

【図6D】

【図6E】

【図6F】

【図7A】

【図7B】

【図7C】

【図7D】

【図7E】

【図7F】

【図8A】

【図8B】

【図8C】

【図8D】

【図8E】

【図8F】

【図9A】

【図9B】
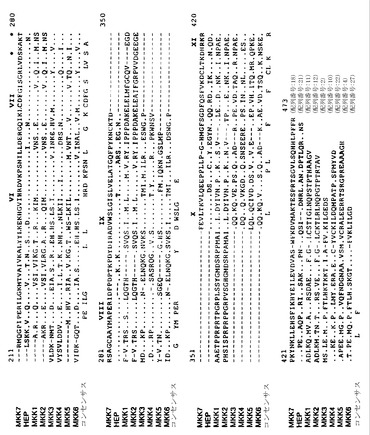
【図10A】

【図10B】

【図10C】

【図10D】

【図11A】

【図11B】

【図11C】

【図11D】

【図12A】

【図12B】

【図13A】

【図13B】

【図13C】

【図13D】
【図14A】

【図14B】

【図14C】

【図14D】

【図15A】

【図15B】

【図15C】

【図15D】

【図16】
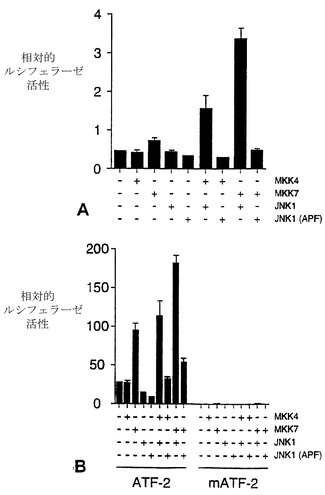
【図2A】
【図2B】
【図3】
【図4A】
【図4B】
【図4C】
【図4D】
【図5A】
【図5B】
【図5C】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図7F】
【図8A】
【図8B】
【図8C】
【図8D】
【図8E】
【図8F】
【図9A】
【図9B】
【図10A】
【図10B】
【図10C】
【図10D】
【図11A】
【図11B】
【図11C】
【図11D】
【図12A】
【図12B】
【図13A】
【図13B】
【図13C】
【図13D】
【図14A】
【図14B】
【図14C】
【図14D】
【図15A】
【図15B】
【図15C】
【図15D】
【図16】
【公開番号】特開2009−131261(P2009−131261A)
【公開日】平成21年6月18日(2009.6.18)
【国際特許分類】
【出願番号】特願2008−309904(P2008−309904)
【出願日】平成20年12月4日(2008.12.4)
【分割の表示】特願2000−502066(P2000−502066)の分割
【原出願日】平成10年7月7日(1998.7.7)
【出願人】(399093869)ユニバーシティー オブ マサチューセッツ (19)
【Fターム(参考)】
【公開日】平成21年6月18日(2009.6.18)
【国際特許分類】
【出願日】平成20年12月4日(2008.12.4)
【分割の表示】特願2000−502066(P2000−502066)の分割
【原出願日】平成10年7月7日(1998.7.7)
【出願人】(399093869)ユニバーシティー オブ マサチューセッツ (19)
【Fターム(参考)】
[ Back to top ]