
サクサグリプチンの結晶形態
本発明は、サクサグリプチン塩酸塩の新規な多形形態に関する。本発明は、サクサグリプチン塩酸塩の多形形態を作製する方法にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な多形形態のサクサグリプチン塩酸塩、これらの調製、およびそれらを含有する組成物に関する。
【背景技術】
【0002】
サクサグリプチン(1S,3S,5S)−2−[(2S)−2−アミノ−2−(3−ヒドロキシ−1−アダマンチル)アセチル]−2−アザビシクロ[3.1.0]ヘキサン−3−カルボニトリル、またはこの塩酸塩は、2型真性糖尿病、肥満または関連する疾患を治療するための治療薬である、経口活性のある可逆性のジペプチジルペプチダーゼ−4(DD4)阻害剤であり、例えばUS6395767B2、実施例60に開示されている。
【0003】
サクサグリプチンの特定の結晶形態、およびサクサグリプチン塩酸塩を含む特定の酸付加塩は、WO2008131149A2に開示されている。単一の化合物のいろいろな結晶形態の存在は、多形として知られ、いくつかの化合物および錯体および疑似多形体の特性である。多形形態は、それぞれ、異なる溶解性、異なる融点および/または異なるX線回折ピーク等の異なる物理的特性を有する。
【0004】
それぞれの多形体の溶解度は異なることがあるので、薬学的な多形体の存在を確認することは、予測可能な溶解性をもつ医薬組成物を提供するために重要である。すべての多形形態、疑似多形体および水和物を含む、薬剤のすべての固体状態の形態を調査すること、ならびにそれぞれの多形形態の安定性、溶解および流動の特性を測定することは望ましい。多形、および医薬品のための固体状態の特性の関連性を概観するために、例えばRolf Hilfiker、Polymorphism in the Pharmaceutical industry、Wiley−VCH 2006を参照されたい。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第6395767号明細書
【特許文献2】国際公開第2008/131149号
【非特許文献】
【0006】
【非特許文献1】Rolf Hilfiker、Polymorphism in the Pharmaceutical industry、Wiley−VCH 2006
【発明の概要】
【発明が解決しようとする課題】
【0007】
薬学的に有用な化合物の新規な多形形態の発見は、医薬品の性能特性を改善する新たな機会をもたらす。それは、例えば目標とする放出特性または他の所望の特性をもつ薬剤の医薬の剤形を設計するために利用可能な、製剤の科学者が有する材料のレパートリーを増やす。
【0008】
サクサグリプチン塩酸塩の公知の多形形態は、比較的高い水含有率を有するすべての水和した形態である。高水含有率の形態は、サクサグリプチンのような加水分解しがちな化合物がそのような形態で存在するときに低下した化学的安定性を示すことがあるように、特定の欠点を有する。そのうえ生薬の観点から、高い水含有率を有する大量の活性医薬成分は、妨害し合う、またはくっつき合う傾向があるので、時として、医薬組成物の製造のための配合工程において乏しい加工の挙動を有する。
【0009】
このように、公知の結晶形態の1つまたは複数の問題を回避する、サクサグリプチン塩酸塩の固体の形態が必要とされている。
【課題を解決するための手段】
【0010】
本発明によれば、1.5%重量/重量以下の水含有率を有し、好ましくは
a)形態I−Sと呼ぶ無水の形態
b)形態HT−Sと呼ぶ無水の形態
c)形態HT−IV−Sと呼ぶ無水の形態
d)形態IV−Sと呼ぶ無水の形態
のような実質的に純粋な形態の、好ましくは式
【0011】
【化1】

の新規な無水の形態のサクサグリプチン塩酸塩が提供される。
【0012】
水含有率は、カールフィッシャー法により測定される。
【0013】
本発明の文脈におけるサクサグリプチン塩酸塩の無水の形態は、25℃において相対湿度30%で24時間保管した後に、カールフィッシャー法により1.5%重量/重量以下の水含有率を示すサクサグリプチン塩酸塩の形態として定義される。
【図面の簡単な説明】
【0014】
【図1】サクサグリプチン塩酸塩の形態I−SのPXRDパターンを示すグラフである。
【図2】サクサグリプチン塩酸塩の形態I−SのFTIRスペクトルを示すグラフである。
【図3】サクサグリプチン塩酸塩の形態I−SのDSCを示すグラフである。
【図4】サクサグリプチン塩酸塩の形態HT−SのPXRDパターンを示すグラフである。
【図5】サクサグリプチン塩酸塩の形態HT−SのFTIRスペクトルを示すグラフである。
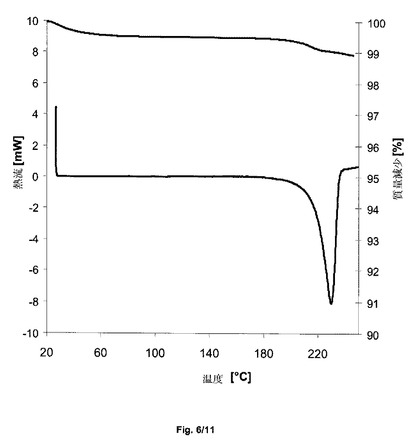
【図6】サクサグリプチン塩酸塩の形態HT−SのDSCを示すグラフである。
【図7】サクサグリプチン塩酸塩の形態HT−IV−SのPXRDパターンを示すグラフである。
【図8】サクサグリプチン塩酸塩の形態HT−IV−SのFTIRスペクトルを示すグラフである。
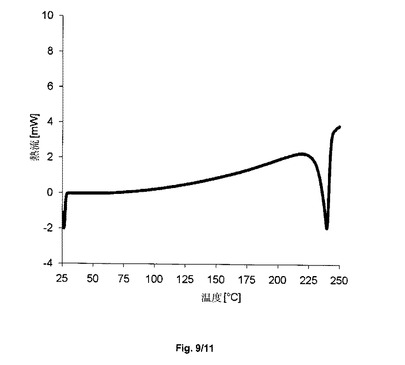
【図9】サクサグリプチン塩酸塩の形態HT−IV−SのDSCを示すグラフである。
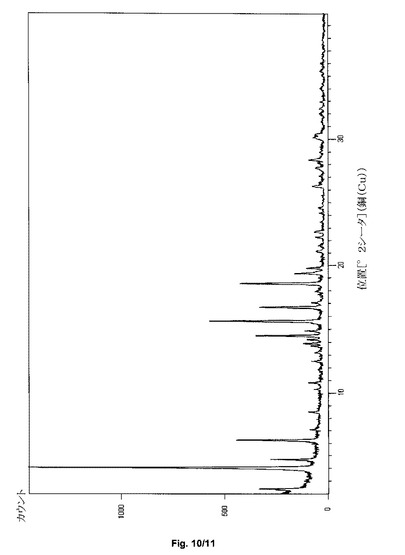
【図10】サクサグリプチン塩酸塩の形態IV−SのPXRDパターンを示すグラフである。
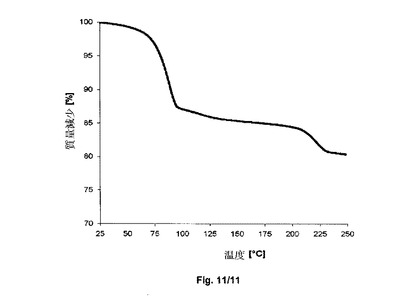
【図11】サクサグリプチン塩酸塩の形態IV−SのTGAを示すグラフである。
【発明を実施するための形態】
【0015】
遊離体として用いられるサクサグリプチン塩酸塩は、水を含有する公知の形態の一つ、例えば2頁のWO2008/131149A2において確認されるようなH2−1、H1.25−2またはH.75−3の形態である。
【0016】
形態I−S
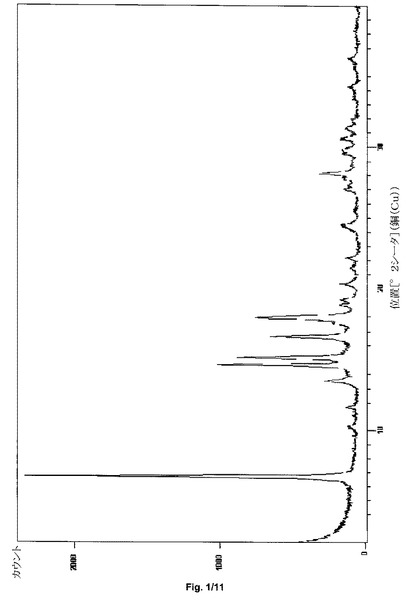
第1の態様において、本発明は、特に約13.5、24.5および28.1におけるさらなるピークを含む、約6.7、14.6、15.2、16.6および17.9±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態I−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態ISは、実質的に図1と一致するPXRDパターンをさらに特徴とし得る。
【0017】
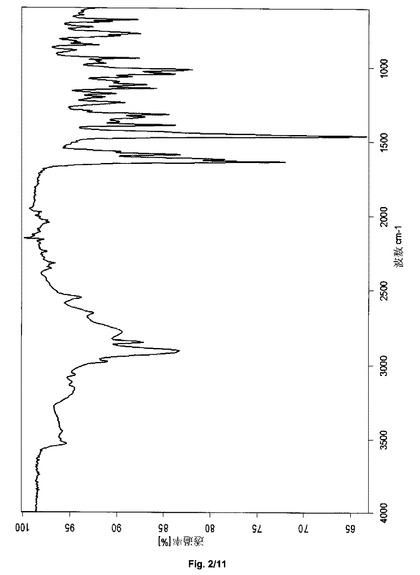
あるいは、サクサグリプチン塩酸塩の結晶形態I−Sは、2907、2853、1637、1589、1462、1391、1318、1045、1014および775+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態ISは、実質的に図2と一致するFTIRスペクトルをさらに特徴とし得る。
【0018】
形態I−Sの形態の無水の結晶性サクサグリプチン塩酸塩は、例えば実施例2に記載の通りに、形態I−Sの種結晶の存在下において、アルコール、好ましくはエタノールから、サクサグリプチン塩酸塩を結晶化することにより調製され得る。形態I−Sの種結晶は、驚くべきことに、サクサグリプチン遊離塩基の半水化物を有機溶媒中に溶解し、ハロゲン化アルキル二水和物により沈殿させ、沈殿したサクサグリプチン一塩酸塩の脱水物を除去し、冷却後に母液から結晶化することにより得られており、実施例1を参照されたい。結晶性のサクサグリプチン塩酸塩の形態I−Sは、この高い溶解速度を兼ね備えたその高い化学的安定性により、特に好ましい結晶形態である。
【0019】
形態HT−S
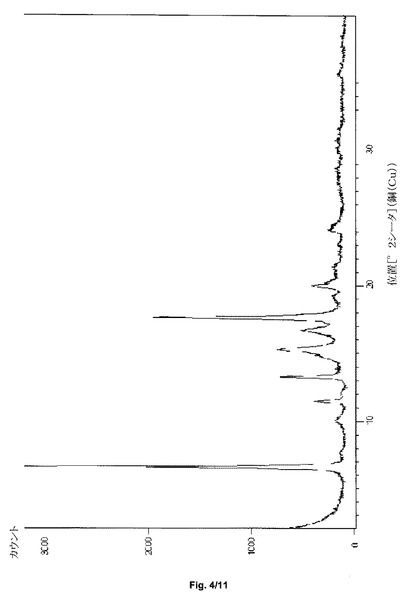
第2の態様において、本発明は、特に約11.5および15.3におけるさらなるピークを含む、約6.6、11.5、13.3、16.7および17.6±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態HT−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態HT−Sは、実質的に図4と一致するPXRDパターンをさらに特徴とし得る。
【0020】
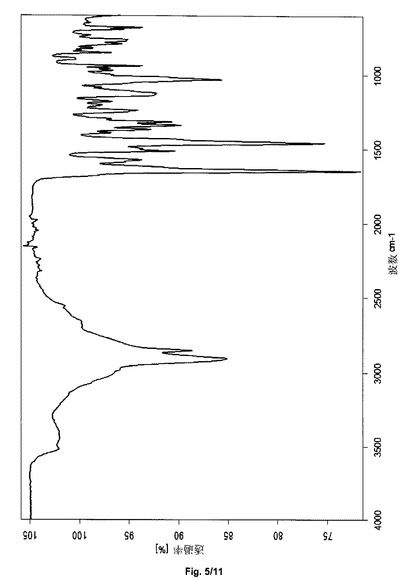
あるいは、サクサグリプチン塩酸塩の結晶形態HT−Sは、2906、2854、1649、1574、1513、1459、1338、1124、1032および851+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態HT−Sは、実質的に図5と一致するFTIRスペクトルをさらに特徴とし得る。
【0021】
HT−Sの形態の結晶性サクサグリプチン塩酸塩は、公知の形態のサクサグリプチン塩酸塩を約160℃から180℃に加熱し、形態HT−Sを単離することにより調製され得る。特に、形態HT−Sは、実施例3に記載の通りに調製され得る。
【0022】
形態HT−IV−S
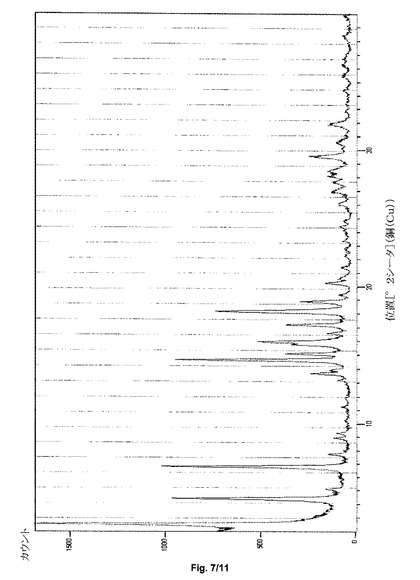
第3の態様において、本発明は、約2.6、4.5、6.8、14.6および18.1±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態HT−IV−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態HT−IV−Sは、実質的に図7と一致するPXRDパターンをさらに特徴とし得る。
【0023】
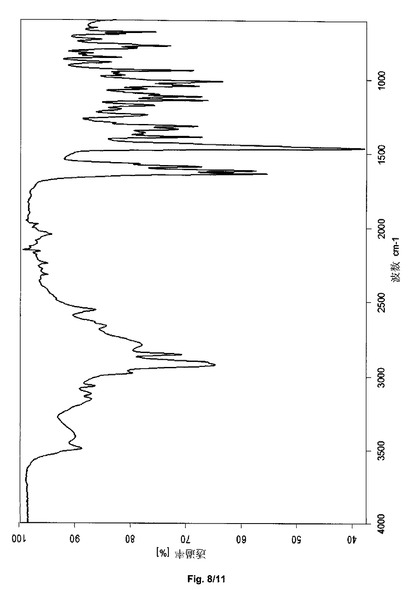
あるいは、サクサグリプチン塩酸塩の結晶形態HT−IV−Sは、3495、2921、1637、1616、1464、1242、1103、1013、940および774+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態HT−IV−Sは、実質的に図8と一致するFTIRスペクトルをさらに特徴とし得る。
【0024】
形態HT−IV−Sの結晶性サクサグリプチン塩酸塩は、結合した溶媒をサクサグリプチン−IV−Sから除去することにより調製され得る。特に、形態HT−IV−Sは、以下に記載の形態IV−Sを真空中で、例えば環境温度から約100℃で、例えば約80℃±10℃で、数時間、例えば3から24時間、乾燥することにより得られ得る。特に、形態HT−IV−Sは、実施例4に記載の通りに調製され得る。ある好ましい実施形態において、サクサグリプチン塩酸塩の形態HT−IV−Sは、
a)n−ブタノール中にサクサグリプチン一塩酸塩を溶解するステップ、
b)減圧下でn−ブタノールを除去して残留物を得るステップ、
c)2−メチル−2−ブタノールを添加してスラリーを得るステップ、
d)サクサグリプチン塩酸塩の溶媒和した結晶形態をそのスラリー中で形成させるステップ、
e)そのスラリーから溶媒和した結晶を除去するステップ、および
f)ステップe)から得られた溶媒和した結晶形態を乾燥して、無水の結晶性サクサグリプチン塩酸塩の形態HT−IVsを得るステップ
を含む方法で調製される。ステップf)における乾燥は、好ましくは、80℃+/−10℃で3−24時間行われる。
【0025】
形態IV−S
サクサグリプチンの形態IV−Sは、約2.4、4.1、4.7、6.3および15.6±0.2度の2シータのX線粉末回折反射を特徴とし得る。サクサグリプチンの形態IV−Sは、実質的に図1と一致するPXRDパターンをさらに特徴とし得る。
【0026】
形態IV−Sの形態の結晶性サクサグリプチン塩酸塩は、実施例5に記載の通りにサクサグリプチン塩酸塩をtert.アミルアルコール(2−メチル−2−ブタノール)から結晶化することにより調製され得るし、好ましくは調製される。サクサグリプチン塩酸塩のn−ブタノール中溶液は、乾燥するまで蒸発乾固されて、非晶質または弱い結晶性の残留物を生成する。tert.アミルアルコール中にその残留物を懸濁させるとすぐに、サクサグリプチン塩酸塩の新規な結晶形態IV−Sが形成され、これはサクサグリプチン塩酸塩のtert.アミルアルコール溶媒和物である。
【0027】
サクサグリプチン塩酸塩の形態IV−Sは、サクサグリプチン塩酸塩の形態HT−IV−Sの調製における価値ある中間体である。
【0028】
最も好ましくは、サクサグリプチン塩酸塩の溶液は、公知の形態の二水和物H2.1を用いて調製される。次いで、その結晶は単離され、約40%未満の相対湿度に保たれる。
【0029】
医薬の製剤および組成物
上述した通りの本発明のサクサグリプチン塩酸塩の結晶形態の任意の1つは、糖尿病、および関連する疾患を本発明に従って治療するときに使用する様々な医薬の製剤の中で用いられる。したがって、本発明は、上述した通りのサクサグリプチン塩酸塩の結晶形態の任意の1つ、および薬学的に許容できる担体を含む医薬組成物にも関する。
【0030】
サクサグリプチン塩酸塩の新規な結晶形態は、単独で、または1種もしくは複数種の抗糖尿病薬(糖尿病および関連する疾患を治療するために用いられる)、および/または1種もしくは複数種の他の治療薬と組み合わせて用いることができ、該治療薬は、単一剤形の同じ剤形による経口、または注射により投与され得る。
【0031】
新規な結晶形態の式Iの化合物と組み合わせて場合によって用いられ得る他の種類の抗糖尿病薬は、インスリン分泌促進物質、または好ましくはDP4阻害剤と異なる機構を有する他の抗糖尿病薬を含む、さらなる抗糖尿病薬または抗高血糖薬、抗高脂血症薬もしくは脂質調整薬であり、ビグアニジン、スルホニル尿素、グルコシダーゼ阻害剤、チアゾリジンジオン等のPPARγアゴニスト、SGLT2阻害剤、PARα/γ二重アンタゴニスト、aP’’阻害剤、グリコーゲンホスホリラーゼ阻害剤および/またはメグリチニド、ならびにインスリンおよび/またはグルカゴン様ペプチド−1(GLP−1)もしくはその模倣剤を含むことができる。
【0032】
本発明は、別の抗糖尿病薬および/または他の治療薬を含むまたは含まない、医薬の媒体または希釈剤と結びついた、新規な結晶形態、形態I−S、形態HT−S、形態HT−IVまたは形態IV−Sの式Iの化合物を含有する医薬組成物にも関する。その医薬組成物は、従来の固体または液体の媒体または希釈剤、および所望の投与の様式に適した種類の医薬の添加剤を用いて配合され得る。例えば、経口の経路により投与するためには、本発明の医薬組成物は、錠剤、カプセル、顆粒または粉末の形態であることができる。大人のための用量は、好ましくは1日あたり5mgから1000mgの間、好ましくは1日あたり5から100mgの間であり、単一の投与、または1日1から4回の別個の投与で投与され得る。
【0033】
本発明による結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、増量剤、甘味料、緩衝剤、滑剤、流動化剤、香料、潤滑剤、保存剤、界面活性剤、湿潤剤、バインダー、崩壊剤および増粘剤からなる群から選択される1種または複数の薬学的に許容できる賦形剤をさらに含むことができる。医薬組成物の分野において公知の他の賦形剤も用いられ得る。そのうえ、医薬組成物は、上述の群の構成要素の1つの中に、2種以上の賦形剤の組合せを含むこともできる。好ましくは、増量剤は甘味料でもある。
【0034】
一般的な錠剤は、充填剤等の1種または複数の賦形剤、場合によってバインダー、および場合によって崩壊剤を含有する。充填剤の例には、微結晶性セルロース等のセルロース誘導体、ラクトース、ショ糖、澱粉、アルファ化澱粉、ブドウ糖、マンニトール、フルクトース、キシリトール、ソルビトール、コーンスターチ、カルシウム塩、例えば炭酸カルシウム、リン酸カルシウム、リン酸二カルシウム等の無機塩、デキストリンもしくはデキストレート、マルトデキストリン圧縮糖および/または他の公知の充填剤もしくは増量剤が挙げられる。
【0035】
使用に適切なバインダーの例には、ヒドロキシプロピルセルロース、PVP、澱粉、ヒドロキシプロピルセルロース、酢酸セルロースの他に、カルナバワックス等のワックスのバインダー、ポリエチレンもしくは他の従来のバインダー剤またはこれらの混合物が挙げられる。
【0036】
崩壊剤の例には、クロスカルメロースナトリウム、クロスポビドン、澱粉、低置換度ヒドロキシプロピルセルロースおよび他の従来の崩壊剤が挙げられる。
【0037】
場合によって存在する潤滑剤には、例えば、ステアリン酸マグネシウム、ステアリン酸亜鉛、ステアリン酸カルシウム、タルク、カルナバワックス、ステアリン酸、パルミチン酸、ラウリル硫酸ナトリウムもしくは水素化された植物油脂もしくは他の公知の潤滑剤またはこれらの混合物が挙げられる。
【0038】
錠剤は、例えばUS2005/0266080に開示されている通りに、錠剤の核、および錠剤の核の上にコーティングされた内側シールコーティング層、錠剤の核の上の内部シールコーティングの上にコーティングされた本発明の結晶を含有する第2のコーティング層、および場合によって錠剤の第2のコーティング層の上にコーティングされた外側保護コーティング層を含んでコーティングされ得る。
【0039】
したがって、本発明は、US2005/0266080A1の段落[0014]から段落[0073]に記載のようなコーティングされた錠剤も提供し、US2005/0266080A1が「medicament」の用語を用いているときはいつでも、本発明の結晶性サクサグリプチン塩酸塩がUS2005/0266080A1の「medicament」の代わりに用いられ得ると理解され得る。言うまでもなく、特に、形態I−S、形態IV−Sまたは形態HT−IV−sを用いた場合において、すべてのステップは、一般に、比較的低い相対湿度の条件等の、多形転移を回避する条件下で行われる。
【0040】
本発明の新規な結晶形態を含有する経口投与のための一般的なカプセルは、例えばラクトース、クロスカルメロース、ステアリン酸マグネシウム、または例えばステアリルフマル酸ナトリウムを含有する。
【0041】
純度
本発明者らは、配合および貯蔵の工程中において、特に形態I−S、形態HT−IV−Sおよび形態IV−Sを安定化するための方法を見出した。
【0042】
したがって、本発明は、組成物中に存在する95%を超える形態I−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態I−Sとして安定的に存在する、形態I−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態I−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0043】
したがって、本発明は、組成物中に存在する95%を超える形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態HT−IV−Sとして安定的に存在する、形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態HT−IV−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0044】
したがって、本発明は、組成物中に存在する95%を超える形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態IV−Sとして安定的に存在する、形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態IV−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0045】
本明細書において定義される「安定的に存在」は、医薬組成物を180日間貯蔵した後でも、好ましくは2年間貯蔵した後でも、その医薬組成物中に最初に含まれていたサクサグリプチン塩酸塩の結晶形態が、示された期間の間貯蔵した後のサクサグリプチン塩酸塩の結晶形態としてまだ存在することを意味する。そのような組成物は、配合のステップ中における、空気の高い相対湿度等の湿潤な条件を回避することにより製造され得る。そのうえ、上記で確認した湿潤な条件は、本発明の医薬組成物を保存するための貯蔵の間において回避されるべきである。
【0046】
好ましい実施形態において、本発明の医薬組成物は、形態I−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を、サクサグリプチン塩酸塩の唯一の検出可能な形態として含む。医薬組成物中のサクサグリプチン塩酸塩の多形状態の分析は、当技術分野において公知の任意の適切な方法、例えばXRPDにより行われ得る。
【0047】
平衡湿度
形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、少なくとも180日間、好ましくは少なくとも2年間、50%未満の、好ましくは3%から50%の、より好ましくは10%から45%の、好ましくは15%から45%の、特により好ましくは15%から40%、または25%から35%の平衡相対湿度を示すことが好ましい。
【0048】
形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、少なくとも180日間、好ましくは少なくとも2年間、40%未満の、好ましくは3%から40%の、より好ましくは10%から40%の、好ましくは15%から40%の、特により好ましくは15%から40%、または25%から35%の平衡相対湿度を示すことが好ましい。
【0049】
結晶形態のサクサグリプチン塩酸塩を含む医薬組成物の平衡相対湿度は、以下の方法により一定温度において閉鎖系の中で湿度の平衡が成立した後に、試料、例えば、形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物の上方の空気中の相対湿度を%で決定することにより測定され、使用される装置は、BT−RS1型の湿度計を含む市販の測定容器、Rotronic AW−VCである。試料、例えば本発明の医薬組成物は、25+/−1℃の温度に恒温化された測定容器中に設置されるサンプリング皿の中に充填された後に、上記の容器は、閉鎖され密封される。相対湿度の平衡が成立し、その状態が、一般に、傾向の表示が消滅することにより示された後に、相対湿度の値を%で湿度計から読み取る。相対湿度は、本明細書に記載の通りに測定された医薬組成物の平衡相対湿度として定義される。容器の充填は、製造者の指示に従って上記の容器の完全な充填をもたらすような方法で行われ得る。試料が、経口懸濁液、すなわち液体の懸濁剤のための粉末または顆粒である場合において、上記の試料は、上述のサンプリング皿の中に直接に設置される。試料がカプセルである場合において、適切な数のカプセルが開封され、これらの内容物はサンプリング皿の中に充填される。試料が錠剤である場合において、適切な数の錠剤が乳鉢を用いることにより破砕され、サンプリング皿の中に充填される。平衡湿度が20%未満であると予測される場合において、本明細書において記載される通りの、測定前の試料の上述の調製および測定自体を、湿度計が備えられているグローブボックス中で行うことができ、約5%の相対湿度を、例えば乾燥した空気または窒素で流し出すことにより成立させることができる。本発明の医薬組成物の平衡相対湿度を測定するための上述の方法を、本明細書においてERH法とも呼ぶ。
【0050】
貯蔵条件
形態I−S、IV−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、比較的乾燥した環境の中で貯蔵され、好ましくは、貯蔵環境は、医薬組成物の寿命の間、比較的乾燥したままであることを確実にすることができる。
【0051】
好ましい一実施形態において、特に本発明による形態I−SおよびHT−IV−Sの化合物および組成物は、組成物の平衡相対湿度を、少なくとも180日間、より好ましくは少なくとも2年間、50%未満に、好ましくは10%から45%に、より好ましくは15%から40%に保つことが可能な容器の中で貯蔵される。これは、例えば、固く密封された容器を用いることにより、または組成物を比較的乾燥して保つための手段を容器に備えることにより達成され得る。
【0052】
したがって、別の好ましい実施形態において、本発明は、形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を含む容器にも関し、この容器は、組成物の平衡相対湿度を、少なくとも180日間、より好ましくは少なくとも2年間、40%未満に、好ましくは10%から40%に、より好ましくは15%から40%に保つことが可能である。これは、例えば、固く密封された容器を用いることにより、または組成物を比較的乾燥して保つための手段を容器に備えることにより達成され得る。
【0053】
このような乾燥手段は、例えば、例えばMINIPAXの商品名の下で市販されモレキュラーシーブ4オングストロームを2g含有する乾燥剤の袋、または例えばSORBITの商品名の下で入手可能でシリカゲル1gを含有する乾燥剤の缶、例えばDRICAPの商品名の下で入手可能でシリカゲル0.9gを含有する乾燥剤のカプセル、またはシリカゲル2gを含有する乾燥剤の栓であることができる。
【0054】
貯蔵容器
本明細書に記載の方法の様々なステップにおいて得られる生成物または中間生成物は、好ましくは、50%未満、好ましくは40%未満の環境相対湿度で貯蔵される。したがって、上記の生成物は、アルミニウムの樽またはドラム缶の中、Muller(登録商標)のドラム缶として市販されているようないわゆるNirosta(登録商標)のドラム缶の中に貯蔵され得る。上記のドラム缶は、気密にされ得る、例えば、それらの蓋に、密封リング等の密封手段を適用することにより空気遮断され得る。上記の生成物は、アルミニウム、または閉鎖物もしくは蓋に密封リング等の密封手段が設けられた上述のようなNirosta(登録商標)の材料から作製された容器の中にも貯蔵され得る。
【0055】
特に形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、50%未満の、好ましくは10%から45%の環境相対湿度で、本明細書に記載の通りの容器の中に包装または充填される。次に、上記の容器は、本明細書に記載の通りに固く閉鎖される。好ましくは、上記の容器は、例えば室温において、約20℃から30℃の温度等において、例えば約25℃において、長期間の間、例えば少なくとも6カ月、好ましくは少なくとも約24カ月の間、例えば少なくとも24カ月以下の間、例えば少なくとも約30カ月以下の間、約60カ月以下の間等、本発明の医薬組成物を安定的に貯蔵するために用いられる。
【0056】
形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、40%未満の、好ましくは10%から40%の環境相対湿度で、本明細書に記載の通りの容器の中に包装または充填される。次に、上記の容器は、本明細書に記載の通りに固く閉鎖される。好ましくは、上記の容器は、例えば室温において、約20℃から30℃の温度等において、例えば約25℃において、長期間の間、例えば少なくとも6カ月、好ましくは少なくとも約24カ月の間、例えば少なくとも24カ月以下の間、例えば少なくとも約30カ月以下の間、約60カ月以下の間等、本発明の医薬組成物を安定的に貯蔵するために用いられる。
【0057】
好ましい容器は、ボトル、例えばガラスもしくはプラスチックのボトル、例えば、例えばねじ込み式の閉鎖物を有する、securitainerとして知られているようなポリエチレンのボトルである、またはブリスター、例えばアルミニウムのブリスターもしくはストリップ、例えば2枚のアルミニウムの箔もしくはストリップからなるブリスター、またはAclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターである、または任意の他の適切な容器であってよい。より好ましくは、上記の容器は、空気遮断の容器等の気密容器である。
【0058】
好ましい容器は、アルミニウム膜で密封されたガラスまたはプラスチックのボトル、alu−aluのブリスターもしくはストリップ、またはAclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターである。本発明による容器は、本発明の医薬組成物を、本明細書に記載の通りの条件下で上記の容器の中に充填することにより得られる。
【0059】
好ましくは、乾燥手段と組み合わせた容器は、中でも形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物の平衡相対湿度を、少なくとも6カ月間、好ましくは少なくとも2年間、50%未満に、好ましくは10%から45%に維持することが可能である。好ましい実施形態において、その容器は、さらに、50%未満の、好ましくは10%から45%の相対湿度をもつ気体雰囲気を封入する。その容器に乾燥した気体雰囲気、例えば乾燥した空気、または乾燥した窒素ガスを備えることは、当技術分野に知られている通りに行われ得る。形態IV−Sに関して、乾燥手段と組み合わせた容器は、医薬組成物の平衡相対湿度を、少なくとも6カ月間、好ましくは少なくとも2年間、40%未満に、好ましくは10%から40%に維持することが可能である。好ましい実施形態において、その容器は、さらに、40%未満の、好ましくは10%から40%の相対湿度をもつ気体雰囲気を封入する。
【0060】
容器および乾燥手段の好ましい組合せは、乾燥剤のカプセルおよび/もしくは缶を含有しアルミニウム箔で密封されたポリエチレンボトル(PEボトル)、または乾燥剤の栓をもつガラスボトルである。
【0061】
組成物の調製
相対環境湿度について、および組成物の平衡相対湿度について、形態I−S、IV−Sまたは形態HT−IV−Sを含む本発明の医薬組成物の製造中において特別な注意をはらわなければならない。したがって、本発明は、
a)形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を、1種または複数の薬学的に許容できる賦形剤と、50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して40%未満の、好ましくは10から40%の相対湿度で混合するステップ、
b)ステップa)で得られた混合物を、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して50%未満の、好ましくは10%から40%の相対湿度で場合によって顆粒化するステップ、ならびに
c)ステップa)で得られた混合物、またはステップb)で得られた顆粒を、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して40%未満の、好ましくは10%から40%の相対湿度でさらに処理して、形態I−Sまたは形態HT−IV−Sまたは形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を得るステップ
を含む、形態I−S、IV−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を調製するための方法にも関する。得られた本発明の医薬組成物は、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の、より好ましくは15%から35%の、または20%から45%の平衡相対湿度、ならびに形態IV−Sに関して40%未満の、好ましくは10から40%の、より好ましくは15から30%の平衡相対湿度を示すことが好ましい。
【0062】
上述の通りに、ステップa)で得られた混合物、またはステップb)で得られた顆粒は、好ましくは、カプセルもしくは錠剤のような経口投与の形態、または経口懸濁液のための顆粒、または経口懸濁液のための粉末に加工される。
【0063】
好ましい実施形態において、約50%未満の、好ましくは10%から45%の平衡相対湿度を有する形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む得られた医薬組成物は、少なくとも6カ月間、医薬組成物の平衡相対湿度を約50%未満に、好ましくは10%から45%に維持することが可能な容器、例えば、医薬組成物の相対平衡湿度を50%未満に、好ましくは10%から45%に維持するのに十分な乾燥手段を場合によってさらに含むことがある上述の容器の中に充填される。
【0064】
形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物のための適切な貯蔵条件は、組成物を所望の形態に維持するために重要である。したがって、好ましくは、本発明の医薬組成物を貯蔵するために、少なくとも6カ月間、気体雰囲気を50%未満の、好ましくは10%から45%の相対湿度に維持することが可能な容器が用いられる。好ましくは、50%未満の、好ましくは10%から45%の相対湿度を有する気体雰囲気は、形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を安定化するために用いられ、形態IV−Sに関して40%未満の、好ましくは10%から40%の相対湿度を有する気体雰囲気が用いられる
本発明の医薬組成物を、本明細書に記載の容器中に充填した後に、上記の容器は、好ましくは、例えば固くまたは完全に密封されて、例えば任意の気体雰囲気が上記の容器の壁および/または閉鎖物を通って拡散することを防止する方法で固く閉鎖される。誘導密封により、および閉鎖物、例えばスクリュー式の閉鎖物を適用することにより、アルミニウム膜を上記のボトルのボトル開口部に適用することによるガラスもしくはプラスチックのボトルの密封等、または例えば公知の方法に類似した加熱密封による、alu−aluのブリスターもしくはストリップの、Aclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターの密封等の、上記の容器を固く密封するおよび/または閉鎖する方法が知られている。
【0065】
本明細書に記載されている方法の間に適用される温度は、好ましくは室温であり、例えば、約25℃等の、約20℃から約30℃の温度である。
【0066】
(実施例)
X線粉末回折パターン(XRPD)を、透過構造のシータ/シータの対のゴニオメータ、集束鏡によるCu−Kα1,2の照射(波長0.15419nm)、およびソリッドステートのPIXcel検出器を備えたPANalytical X’Pert PROの回折計により得た。パターンを、環境条件において、2°から40°の2θの角度範囲内で、1ステップあたり80sでステップ幅0.006°の2θを適用し、管電圧40kV、管電流40mAで記録した。2シータの値の一般的な精度は、約±0.2°の2シータの範囲内である。したがって、5.0°の2シータに現れる回折ピークは、標準的な条件下におけるほとんどのX線回折計により4.8から5.2°の2シータの間に現れることがある。
【0067】
赤外スペクトル(IR)を、Bruker Tensor 27のFTIR分光計で、MKII Golden Gate(商標)Single Reflection Diamond ATR(減衰全反射)のセルにより、4cm−1の分解能で収集した。スペクトルを収集するために、スパチュラ先端量の試料を、粉末の形態のダイアモンドの表面に塗布した。次いで、その試料をサファイア床でダイアモンド上に押し付け、スペクトルを記録した。清浄なダイアモンドのスペクトルをバックグラウンドのスペクトルとして用いた。波数の値の一般的な精度は、約±2cm−1の範囲内である。したがって、1716cm−1に現れる赤外のピークは、標準的な条件下におけるほとんどの赤外分光計により1714から1718cm−1の間に現れることがある。
【0068】
示差走査熱量測定(DSC)を、Pyrisのソフトウエアを用い、DSC 7(米国、コネチカット州、ノーウォーク、Perkin−Elmer)により行った。試料約4gmgを、25μlのAl皿の中に秤量した。乾燥した窒素をパージガスとして用いた(パージ、20mlmin−1)本明細書において用いられるときに、示差走査熱量測定により決定される「Tonset」の用語は、転移前のベースラインの、推定された転移の先端との交差に対応する温度を意味する。
【0069】
熱重量分析を、Windows(登録商標) NTのためのPyris Softwareを用いた熱重量分析システムTGA−7(米国、コネチカット州、ノーウォーク、Perkin−Elmer)、50μlの白金の皿、窒素のパージガス(試料のパージ:20mlmin−1、天秤のパージ:40mlmin−1)により行った。
【0070】
水分収着等温線を、SPS−11の水分収着分析計(ドイツ、ウルム、MD Mess−technik)により記録した。測定サイクルを、相対湿度(RH)0%で開始し、10%のステップでRH90%に、5%のステップでRH95%に増加させた。それぞれのステップについての平衡条件を、49minを超える一定質量±0.003%に設定した。温度は25±0.1℃であった。
【0071】
HPLCの検定を、以下の条件を適用することにより行った。
カラム:YMC−Ultra HT Pro C 18 3.0×50.0mm、2μm
溶離剤A:10mMスルファミン酸
溶離剤B:10mMのSAS250ml+アセトニトリル750ml
流量:0.64ml/min
温度:15℃
検出:210nmにおけるUV
勾配:
【0072】
【表1】
停止時間:15min
試料の濃度:約0.5mg/ml
溶媒:溶離剤A
注入体積:5μl
【実施例1】
【0073】
無水の形態I−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン遊離塩基の半水化物4.58gを、アセトン230ml中に溶解した。この溶液に、トリメチルクロロシラン2.2mlを撹拌の下で添加した。1時間後に、ゼラチン質の沈殿を含有する混合物を蒸発乾固した。固体の残留物にエタノール80mlを添加し、スラリーを、開放したフラスコ中で約1時間撹拌した。生成物を濾別し、tert.−ブチルメチルエーテルで洗浄し、次いで、真空オーブン中で乾燥して、サクサグリプチン一塩酸塩二水和物1.65gを得た。サクサグリプチン一塩酸塩二水和物の多形を、X線回折、FT−IRおよびDSCにより決定した。
【0074】
母液を、−5℃の冷蔵庫中に48時間入れた。驚くべきことに、さらなる結晶が母液から形成した。このように得られた結晶性の沈殿を濾別し、tert.−ブチルメチルエーテルで洗浄し、次いで、真空オーブン中で乾燥した。結晶を分析し、サクサグリプチン一塩酸塩1.22gであることがわかった。母液から得られた生成物は、形態I−Sと呼ぶ、サクサグリプチン一塩酸塩の無水の多形形態であることがわかった。
【0075】
HPLCによる純度:99.7面積%(最大の個々の不純物0.09%、全不純物0.33%)
サクサグリプチン一塩酸塩の形態I−Sの粉末X線回折パターンを、図1に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表1に示す。
【0076】
【表2】
【0077】
上記で得られた結晶形態I−Sのサクサグリプチン一塩酸塩は、2907、2853、1637、1589、1462、1391、1318、1045、1014および775cm−1(±2cm−1、図2)における吸収帯をもつ減衰全反射IRスペクトルを有する。
【0078】
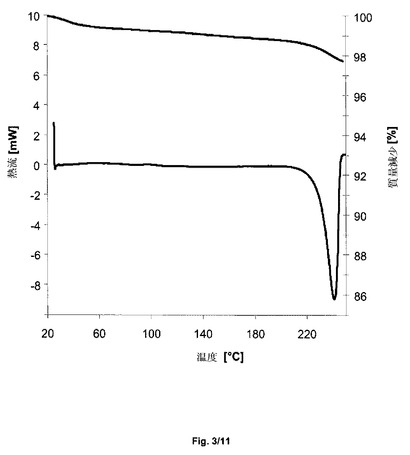
得られた結晶形態I−Sに、示差熱分析を受けさせた。図3(下の曲線)において参照され得る通りに、結晶形態I−Sは、著しい脱水の吸熱でなく、241℃におけるピーク(Tonset 230℃、加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【0079】
イオン性の塩素は、10.7%(理論10.08%)と決定された。
【0080】
得られた結晶形態I−Sの水含有率を、カールフィッシャーの装置を用いて1.2%重量/重量と決定した。25℃における相対湿度約50%において、形態1Sのサクサグリプチン塩酸塩は、約1.5%の水含有率を示す。この水は、25℃における相対湿度を0.5%に下げたときに完全に失われる。この過程は、サクサグリプチン塩酸塩を約50%の湿度に曝した場合において可逆的である。
【0081】
形態I−Sは、相対湿度50%未満において安定である。相対湿度50%を超えると、形態I−Sは、公知の二水和物H2−1に転移する。この転移は可逆的でない。
【実施例2】
【0082】
エタノールからのシーディングによる無水の形態I−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン一塩酸塩2.31gをエタノール62ml中に懸濁させ、生成した混合物を還流において加熱した。熱い溶液を濾過し、次いで実施例1の生成物でシードした。混合物を25℃に冷却し、次いで冷蔵庫の中に終夜設置した。白色固体を濾過により収集し、真空オーブン中で乾燥して、サクサグリプチン一塩酸塩の形態I−S0.78gを得た。
【実施例3】
【0083】
形態HT−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン一塩酸塩二水和物10mgを、窒素下において10°K/minで180℃に加熱した。形態HT−Sを得た。
【0084】
サクサグリプチン一塩酸塩の形態HT−Sの粉末X線回折パターンを、図4に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表2に示す。
【0085】
【表3】
【0086】
上記で得られたサクサグリプチン一塩酸塩の結晶形態HT−Sは、2906、2854、1649、1574、1513、1459、1338、1124、1032および851cm−1(±2cm−1、図5)の吸収帯をもつ減衰全反射のIRスペクトルを有する。
【0087】
得られた結晶形態HT−Sに、示差熱分析を受けさせた。図3(下の曲線)において参照され得る通りに、結晶形態HT−Sは、著しい脱水の吸熱でなく、230℃におけるピーク(加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【0088】
得られた結晶形態HT−Sは、熱重量分析に基づいて、0.5%重量/重量以下の水分の減少を示した(図6、上の線)。
【実施例4】
【0089】
形態HT−IV−Sの調製
ステップ1、IV−Sの調製
105mgのサクサグリプチン一塩酸塩二水和物の形態H2−1を、nブタノール11ml中に溶解した。得られた溶液を濾過し、溶媒を、ロータリーエバポレータを用いて約20mbarで真空により除去した。残留物に、tert.アミルアルコール4mlを添加し、懸濁液を得た。この懸濁液を3日間撹拌し、次いで結晶を吸引濾過により単離し、デシケータ中で相対湿度約30%に終夜保った。
収量:サクサグリプチン一塩酸塩の形態IV−S97mg
【0090】
ステップ2、HT−IV−Sの調製
結晶生成物のサクサグリプチン一塩酸塩IV−Sを、50℃の真空オーブン中で約10時間、約40℃の真空において乾燥した後に、約80℃で5時間乾燥して、結晶形態HT−IV Sを得た。
水(カールフィッシャー):0.7%
試料を、相対湿度45−49%で12時間貯蔵した。
水(カールフィッシャー):1.2%
結晶形態に変化は観測されなかった(FTIR)。
【0091】
サクサグリプチン一塩酸塩の形態HT−IV Sの粉末X線回折パターンを、図7に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表3に示す。
【0092】
【表4】
【0093】
上記で得られたサクサグリプチン塩酸塩の結晶形態HT−IV−Sは、3495、2921、1637、1616、1464、1242、1103、1013、940および774(±2cm−1、図8)の波数の吸収帯をもつ減衰全反射のIRスペクトルを有する。
【0094】
得られた結晶形態HT−IV−Sに、示差熱分析を受けさせた。図9において参照され得る通りに、結晶形態HT−IV−sは、著しい脱水の吸熱でなく、240℃におけるピーク(Tonset232℃、加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【実施例5】
【0095】
形態IV−Sのサクサグリプチン一塩酸塩の調製
105mgのサクサグリプチン一塩酸塩二水和物の形態H2−1を、nブタノール11ml中に溶解した。得られた溶液を濾過し、溶媒を、ロータリーエバポレータを用いて約20mbarで真空により除去した。残留物に、tert.アミルアルコール4mlを添加し、懸濁液を得た。この懸濁液を3日間撹拌し、次いで結晶を吸引濾過により単離し、デシケータ中で相対湿度約30%に終夜保った。
収量:97mg
【0096】
サクサグリプチン一塩酸塩の形態IV−Sの粉末X線回折パターンを、図10に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表4に示す。
【0097】
【表5】
【0098】
得られた結晶形態IV−Sに熱重量分析を受けさせた。図11において参照され得る通りに、結晶形態IV−Sは、100℃以下で開始する約12.8%の質量減少を示す。この質量減少は、水2.86molに相当する。
【0099】
水分収着分析は、水2.94molに相当する、25℃で相対湿度40%における13.1%の水含有率を示す。水分収着分析は、相対湿度0%において、相対湿度2.86%に相当する水含有率12.8%を示す。その過程は可逆的である。
【0100】
40%を超える相対湿度において、形態IV−Sは、公知の形態の二水和物H2−1に不可逆的に転移する。
【技術分野】
【0001】
本発明は、新規な多形形態のサクサグリプチン塩酸塩、これらの調製、およびそれらを含有する組成物に関する。
【背景技術】
【0002】
サクサグリプチン(1S,3S,5S)−2−[(2S)−2−アミノ−2−(3−ヒドロキシ−1−アダマンチル)アセチル]−2−アザビシクロ[3.1.0]ヘキサン−3−カルボニトリル、またはこの塩酸塩は、2型真性糖尿病、肥満または関連する疾患を治療するための治療薬である、経口活性のある可逆性のジペプチジルペプチダーゼ−4(DD4)阻害剤であり、例えばUS6395767B2、実施例60に開示されている。
【0003】
サクサグリプチンの特定の結晶形態、およびサクサグリプチン塩酸塩を含む特定の酸付加塩は、WO2008131149A2に開示されている。単一の化合物のいろいろな結晶形態の存在は、多形として知られ、いくつかの化合物および錯体および疑似多形体の特性である。多形形態は、それぞれ、異なる溶解性、異なる融点および/または異なるX線回折ピーク等の異なる物理的特性を有する。
【0004】
それぞれの多形体の溶解度は異なることがあるので、薬学的な多形体の存在を確認することは、予測可能な溶解性をもつ医薬組成物を提供するために重要である。すべての多形形態、疑似多形体および水和物を含む、薬剤のすべての固体状態の形態を調査すること、ならびにそれぞれの多形形態の安定性、溶解および流動の特性を測定することは望ましい。多形、および医薬品のための固体状態の特性の関連性を概観するために、例えばRolf Hilfiker、Polymorphism in the Pharmaceutical industry、Wiley−VCH 2006を参照されたい。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第6395767号明細書
【特許文献2】国際公開第2008/131149号
【非特許文献】
【0006】
【非特許文献1】Rolf Hilfiker、Polymorphism in the Pharmaceutical industry、Wiley−VCH 2006
【発明の概要】
【発明が解決しようとする課題】
【0007】
薬学的に有用な化合物の新規な多形形態の発見は、医薬品の性能特性を改善する新たな機会をもたらす。それは、例えば目標とする放出特性または他の所望の特性をもつ薬剤の医薬の剤形を設計するために利用可能な、製剤の科学者が有する材料のレパートリーを増やす。
【0008】
サクサグリプチン塩酸塩の公知の多形形態は、比較的高い水含有率を有するすべての水和した形態である。高水含有率の形態は、サクサグリプチンのような加水分解しがちな化合物がそのような形態で存在するときに低下した化学的安定性を示すことがあるように、特定の欠点を有する。そのうえ生薬の観点から、高い水含有率を有する大量の活性医薬成分は、妨害し合う、またはくっつき合う傾向があるので、時として、医薬組成物の製造のための配合工程において乏しい加工の挙動を有する。
【0009】
このように、公知の結晶形態の1つまたは複数の問題を回避する、サクサグリプチン塩酸塩の固体の形態が必要とされている。
【課題を解決するための手段】
【0010】
本発明によれば、1.5%重量/重量以下の水含有率を有し、好ましくは
a)形態I−Sと呼ぶ無水の形態
b)形態HT−Sと呼ぶ無水の形態
c)形態HT−IV−Sと呼ぶ無水の形態
d)形態IV−Sと呼ぶ無水の形態
のような実質的に純粋な形態の、好ましくは式
【0011】
【化1】
の新規な無水の形態のサクサグリプチン塩酸塩が提供される。
【0012】
水含有率は、カールフィッシャー法により測定される。
【0013】
本発明の文脈におけるサクサグリプチン塩酸塩の無水の形態は、25℃において相対湿度30%で24時間保管した後に、カールフィッシャー法により1.5%重量/重量以下の水含有率を示すサクサグリプチン塩酸塩の形態として定義される。
【図面の簡単な説明】
【0014】
【図1】サクサグリプチン塩酸塩の形態I−SのPXRDパターンを示すグラフである。
【図2】サクサグリプチン塩酸塩の形態I−SのFTIRスペクトルを示すグラフである。
【図3】サクサグリプチン塩酸塩の形態I−SのDSCを示すグラフである。
【図4】サクサグリプチン塩酸塩の形態HT−SのPXRDパターンを示すグラフである。
【図5】サクサグリプチン塩酸塩の形態HT−SのFTIRスペクトルを示すグラフである。
【図6】サクサグリプチン塩酸塩の形態HT−SのDSCを示すグラフである。
【図7】サクサグリプチン塩酸塩の形態HT−IV−SのPXRDパターンを示すグラフである。
【図8】サクサグリプチン塩酸塩の形態HT−IV−SのFTIRスペクトルを示すグラフである。
【図9】サクサグリプチン塩酸塩の形態HT−IV−SのDSCを示すグラフである。
【図10】サクサグリプチン塩酸塩の形態IV−SのPXRDパターンを示すグラフである。
【図11】サクサグリプチン塩酸塩の形態IV−SのTGAを示すグラフである。
【発明を実施するための形態】
【0015】
遊離体として用いられるサクサグリプチン塩酸塩は、水を含有する公知の形態の一つ、例えば2頁のWO2008/131149A2において確認されるようなH2−1、H1.25−2またはH.75−3の形態である。
【0016】
形態I−S
第1の態様において、本発明は、特に約13.5、24.5および28.1におけるさらなるピークを含む、約6.7、14.6、15.2、16.6および17.9±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態I−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態ISは、実質的に図1と一致するPXRDパターンをさらに特徴とし得る。
【0017】
あるいは、サクサグリプチン塩酸塩の結晶形態I−Sは、2907、2853、1637、1589、1462、1391、1318、1045、1014および775+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態ISは、実質的に図2と一致するFTIRスペクトルをさらに特徴とし得る。
【0018】
形態I−Sの形態の無水の結晶性サクサグリプチン塩酸塩は、例えば実施例2に記載の通りに、形態I−Sの種結晶の存在下において、アルコール、好ましくはエタノールから、サクサグリプチン塩酸塩を結晶化することにより調製され得る。形態I−Sの種結晶は、驚くべきことに、サクサグリプチン遊離塩基の半水化物を有機溶媒中に溶解し、ハロゲン化アルキル二水和物により沈殿させ、沈殿したサクサグリプチン一塩酸塩の脱水物を除去し、冷却後に母液から結晶化することにより得られており、実施例1を参照されたい。結晶性のサクサグリプチン塩酸塩の形態I−Sは、この高い溶解速度を兼ね備えたその高い化学的安定性により、特に好ましい結晶形態である。
【0019】
形態HT−S
第2の態様において、本発明は、特に約11.5および15.3におけるさらなるピークを含む、約6.6、11.5、13.3、16.7および17.6±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態HT−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態HT−Sは、実質的に図4と一致するPXRDパターンをさらに特徴とし得る。
【0020】
あるいは、サクサグリプチン塩酸塩の結晶形態HT−Sは、2906、2854、1649、1574、1513、1459、1338、1124、1032および851+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態HT−Sは、実質的に図5と一致するFTIRスペクトルをさらに特徴とし得る。
【0021】
HT−Sの形態の結晶性サクサグリプチン塩酸塩は、公知の形態のサクサグリプチン塩酸塩を約160℃から180℃に加熱し、形態HT−Sを単離することにより調製され得る。特に、形態HT−Sは、実施例3に記載の通りに調製され得る。
【0022】
形態HT−IV−S
第3の態様において、本発明は、約2.6、4.5、6.8、14.6および18.1±0.2度の2シータのX線粉末回折反射を特徴とし得る、形態HT−IV−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を提供する。サクサグリプチンの形態HT−IV−Sは、実質的に図7と一致するPXRDパターンをさらに特徴とし得る。
【0023】
あるいは、サクサグリプチン塩酸塩の結晶形態HT−IV−Sは、3495、2921、1637、1616、1464、1242、1103、1013、940および774+/−2cm−1の波数のピークを含む赤外スペクトルにより説明され得る。サクサグリプチン塩酸塩の形態HT−IV−Sは、実質的に図8と一致するFTIRスペクトルをさらに特徴とし得る。
【0024】
形態HT−IV−Sの結晶性サクサグリプチン塩酸塩は、結合した溶媒をサクサグリプチン−IV−Sから除去することにより調製され得る。特に、形態HT−IV−Sは、以下に記載の形態IV−Sを真空中で、例えば環境温度から約100℃で、例えば約80℃±10℃で、数時間、例えば3から24時間、乾燥することにより得られ得る。特に、形態HT−IV−Sは、実施例4に記載の通りに調製され得る。ある好ましい実施形態において、サクサグリプチン塩酸塩の形態HT−IV−Sは、
a)n−ブタノール中にサクサグリプチン一塩酸塩を溶解するステップ、
b)減圧下でn−ブタノールを除去して残留物を得るステップ、
c)2−メチル−2−ブタノールを添加してスラリーを得るステップ、
d)サクサグリプチン塩酸塩の溶媒和した結晶形態をそのスラリー中で形成させるステップ、
e)そのスラリーから溶媒和した結晶を除去するステップ、および
f)ステップe)から得られた溶媒和した結晶形態を乾燥して、無水の結晶性サクサグリプチン塩酸塩の形態HT−IVsを得るステップ
を含む方法で調製される。ステップf)における乾燥は、好ましくは、80℃+/−10℃で3−24時間行われる。
【0025】
形態IV−S
サクサグリプチンの形態IV−Sは、約2.4、4.1、4.7、6.3および15.6±0.2度の2シータのX線粉末回折反射を特徴とし得る。サクサグリプチンの形態IV−Sは、実質的に図1と一致するPXRDパターンをさらに特徴とし得る。
【0026】
形態IV−Sの形態の結晶性サクサグリプチン塩酸塩は、実施例5に記載の通りにサクサグリプチン塩酸塩をtert.アミルアルコール(2−メチル−2−ブタノール)から結晶化することにより調製され得るし、好ましくは調製される。サクサグリプチン塩酸塩のn−ブタノール中溶液は、乾燥するまで蒸発乾固されて、非晶質または弱い結晶性の残留物を生成する。tert.アミルアルコール中にその残留物を懸濁させるとすぐに、サクサグリプチン塩酸塩の新規な結晶形態IV−Sが形成され、これはサクサグリプチン塩酸塩のtert.アミルアルコール溶媒和物である。
【0027】
サクサグリプチン塩酸塩の形態IV−Sは、サクサグリプチン塩酸塩の形態HT−IV−Sの調製における価値ある中間体である。
【0028】
最も好ましくは、サクサグリプチン塩酸塩の溶液は、公知の形態の二水和物H2.1を用いて調製される。次いで、その結晶は単離され、約40%未満の相対湿度に保たれる。
【0029】
医薬の製剤および組成物
上述した通りの本発明のサクサグリプチン塩酸塩の結晶形態の任意の1つは、糖尿病、および関連する疾患を本発明に従って治療するときに使用する様々な医薬の製剤の中で用いられる。したがって、本発明は、上述した通りのサクサグリプチン塩酸塩の結晶形態の任意の1つ、および薬学的に許容できる担体を含む医薬組成物にも関する。
【0030】
サクサグリプチン塩酸塩の新規な結晶形態は、単独で、または1種もしくは複数種の抗糖尿病薬(糖尿病および関連する疾患を治療するために用いられる)、および/または1種もしくは複数種の他の治療薬と組み合わせて用いることができ、該治療薬は、単一剤形の同じ剤形による経口、または注射により投与され得る。
【0031】
新規な結晶形態の式Iの化合物と組み合わせて場合によって用いられ得る他の種類の抗糖尿病薬は、インスリン分泌促進物質、または好ましくはDP4阻害剤と異なる機構を有する他の抗糖尿病薬を含む、さらなる抗糖尿病薬または抗高血糖薬、抗高脂血症薬もしくは脂質調整薬であり、ビグアニジン、スルホニル尿素、グルコシダーゼ阻害剤、チアゾリジンジオン等のPPARγアゴニスト、SGLT2阻害剤、PARα/γ二重アンタゴニスト、aP’’阻害剤、グリコーゲンホスホリラーゼ阻害剤および/またはメグリチニド、ならびにインスリンおよび/またはグルカゴン様ペプチド−1(GLP−1)もしくはその模倣剤を含むことができる。
【0032】
本発明は、別の抗糖尿病薬および/または他の治療薬を含むまたは含まない、医薬の媒体または希釈剤と結びついた、新規な結晶形態、形態I−S、形態HT−S、形態HT−IVまたは形態IV−Sの式Iの化合物を含有する医薬組成物にも関する。その医薬組成物は、従来の固体または液体の媒体または希釈剤、および所望の投与の様式に適した種類の医薬の添加剤を用いて配合され得る。例えば、経口の経路により投与するためには、本発明の医薬組成物は、錠剤、カプセル、顆粒または粉末の形態であることができる。大人のための用量は、好ましくは1日あたり5mgから1000mgの間、好ましくは1日あたり5から100mgの間であり、単一の投与、または1日1から4回の別個の投与で投与され得る。
【0033】
本発明による結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、増量剤、甘味料、緩衝剤、滑剤、流動化剤、香料、潤滑剤、保存剤、界面活性剤、湿潤剤、バインダー、崩壊剤および増粘剤からなる群から選択される1種または複数の薬学的に許容できる賦形剤をさらに含むことができる。医薬組成物の分野において公知の他の賦形剤も用いられ得る。そのうえ、医薬組成物は、上述の群の構成要素の1つの中に、2種以上の賦形剤の組合せを含むこともできる。好ましくは、増量剤は甘味料でもある。
【0034】
一般的な錠剤は、充填剤等の1種または複数の賦形剤、場合によってバインダー、および場合によって崩壊剤を含有する。充填剤の例には、微結晶性セルロース等のセルロース誘導体、ラクトース、ショ糖、澱粉、アルファ化澱粉、ブドウ糖、マンニトール、フルクトース、キシリトール、ソルビトール、コーンスターチ、カルシウム塩、例えば炭酸カルシウム、リン酸カルシウム、リン酸二カルシウム等の無機塩、デキストリンもしくはデキストレート、マルトデキストリン圧縮糖および/または他の公知の充填剤もしくは増量剤が挙げられる。
【0035】
使用に適切なバインダーの例には、ヒドロキシプロピルセルロース、PVP、澱粉、ヒドロキシプロピルセルロース、酢酸セルロースの他に、カルナバワックス等のワックスのバインダー、ポリエチレンもしくは他の従来のバインダー剤またはこれらの混合物が挙げられる。
【0036】
崩壊剤の例には、クロスカルメロースナトリウム、クロスポビドン、澱粉、低置換度ヒドロキシプロピルセルロースおよび他の従来の崩壊剤が挙げられる。
【0037】
場合によって存在する潤滑剤には、例えば、ステアリン酸マグネシウム、ステアリン酸亜鉛、ステアリン酸カルシウム、タルク、カルナバワックス、ステアリン酸、パルミチン酸、ラウリル硫酸ナトリウムもしくは水素化された植物油脂もしくは他の公知の潤滑剤またはこれらの混合物が挙げられる。
【0038】
錠剤は、例えばUS2005/0266080に開示されている通りに、錠剤の核、および錠剤の核の上にコーティングされた内側シールコーティング層、錠剤の核の上の内部シールコーティングの上にコーティングされた本発明の結晶を含有する第2のコーティング層、および場合によって錠剤の第2のコーティング層の上にコーティングされた外側保護コーティング層を含んでコーティングされ得る。
【0039】
したがって、本発明は、US2005/0266080A1の段落[0014]から段落[0073]に記載のようなコーティングされた錠剤も提供し、US2005/0266080A1が「medicament」の用語を用いているときはいつでも、本発明の結晶性サクサグリプチン塩酸塩がUS2005/0266080A1の「medicament」の代わりに用いられ得ると理解され得る。言うまでもなく、特に、形態I−S、形態IV−Sまたは形態HT−IV−sを用いた場合において、すべてのステップは、一般に、比較的低い相対湿度の条件等の、多形転移を回避する条件下で行われる。
【0040】
本発明の新規な結晶形態を含有する経口投与のための一般的なカプセルは、例えばラクトース、クロスカルメロース、ステアリン酸マグネシウム、または例えばステアリルフマル酸ナトリウムを含有する。
【0041】
純度
本発明者らは、配合および貯蔵の工程中において、特に形態I−S、形態HT−IV−Sおよび形態IV−Sを安定化するための方法を見出した。
【0042】
したがって、本発明は、組成物中に存在する95%を超える形態I−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態I−Sとして安定的に存在する、形態I−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態I−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0043】
したがって、本発明は、組成物中に存在する95%を超える形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態HT−IV−Sとして安定的に存在する、形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態HT−IV−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0044】
したがって、本発明は、組成物中に存在する95%を超える形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩が形態IV−Sとして安定的に存在する、形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む上記の医薬組成物にも関し、特に、本発明は、形態IV−Sがサクサグリプチン塩酸塩の唯一の検出可能な結晶形態である、そのような医薬組成物に関する。
【0045】
本明細書において定義される「安定的に存在」は、医薬組成物を180日間貯蔵した後でも、好ましくは2年間貯蔵した後でも、その医薬組成物中に最初に含まれていたサクサグリプチン塩酸塩の結晶形態が、示された期間の間貯蔵した後のサクサグリプチン塩酸塩の結晶形態としてまだ存在することを意味する。そのような組成物は、配合のステップ中における、空気の高い相対湿度等の湿潤な条件を回避することにより製造され得る。そのうえ、上記で確認した湿潤な条件は、本発明の医薬組成物を保存するための貯蔵の間において回避されるべきである。
【0046】
好ましい実施形態において、本発明の医薬組成物は、形態I−Sと呼ぶサクサグリプチン塩酸塩の結晶形態を、サクサグリプチン塩酸塩の唯一の検出可能な形態として含む。医薬組成物中のサクサグリプチン塩酸塩の多形状態の分析は、当技術分野において公知の任意の適切な方法、例えばXRPDにより行われ得る。
【0047】
平衡湿度
形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、少なくとも180日間、好ましくは少なくとも2年間、50%未満の、好ましくは3%から50%の、より好ましくは10%から45%の、好ましくは15%から45%の、特により好ましくは15%から40%、または25%から35%の平衡相対湿度を示すことが好ましい。
【0048】
形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、少なくとも180日間、好ましくは少なくとも2年間、40%未満の、好ましくは3%から40%の、より好ましくは10%から40%の、好ましくは15%から40%の、特により好ましくは15%から40%、または25%から35%の平衡相対湿度を示すことが好ましい。
【0049】
結晶形態のサクサグリプチン塩酸塩を含む医薬組成物の平衡相対湿度は、以下の方法により一定温度において閉鎖系の中で湿度の平衡が成立した後に、試料、例えば、形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物の上方の空気中の相対湿度を%で決定することにより測定され、使用される装置は、BT−RS1型の湿度計を含む市販の測定容器、Rotronic AW−VCである。試料、例えば本発明の医薬組成物は、25+/−1℃の温度に恒温化された測定容器中に設置されるサンプリング皿の中に充填された後に、上記の容器は、閉鎖され密封される。相対湿度の平衡が成立し、その状態が、一般に、傾向の表示が消滅することにより示された後に、相対湿度の値を%で湿度計から読み取る。相対湿度は、本明細書に記載の通りに測定された医薬組成物の平衡相対湿度として定義される。容器の充填は、製造者の指示に従って上記の容器の完全な充填をもたらすような方法で行われ得る。試料が、経口懸濁液、すなわち液体の懸濁剤のための粉末または顆粒である場合において、上記の試料は、上述のサンプリング皿の中に直接に設置される。試料がカプセルである場合において、適切な数のカプセルが開封され、これらの内容物はサンプリング皿の中に充填される。試料が錠剤である場合において、適切な数の錠剤が乳鉢を用いることにより破砕され、サンプリング皿の中に充填される。平衡湿度が20%未満であると予測される場合において、本明細書において記載される通りの、測定前の試料の上述の調製および測定自体を、湿度計が備えられているグローブボックス中で行うことができ、約5%の相対湿度を、例えば乾燥した空気または窒素で流し出すことにより成立させることができる。本発明の医薬組成物の平衡相対湿度を測定するための上述の方法を、本明細書においてERH法とも呼ぶ。
【0050】
貯蔵条件
形態I−S、IV−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、比較的乾燥した環境の中で貯蔵され、好ましくは、貯蔵環境は、医薬組成物の寿命の間、比較的乾燥したままであることを確実にすることができる。
【0051】
好ましい一実施形態において、特に本発明による形態I−SおよびHT−IV−Sの化合物および組成物は、組成物の平衡相対湿度を、少なくとも180日間、より好ましくは少なくとも2年間、50%未満に、好ましくは10%から45%に、より好ましくは15%から40%に保つことが可能な容器の中で貯蔵される。これは、例えば、固く密封された容器を用いることにより、または組成物を比較的乾燥して保つための手段を容器に備えることにより達成され得る。
【0052】
したがって、別の好ましい実施形態において、本発明は、形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を含む容器にも関し、この容器は、組成物の平衡相対湿度を、少なくとも180日間、より好ましくは少なくとも2年間、40%未満に、好ましくは10%から40%に、より好ましくは15%から40%に保つことが可能である。これは、例えば、固く密封された容器を用いることにより、または組成物を比較的乾燥して保つための手段を容器に備えることにより達成され得る。
【0053】
このような乾燥手段は、例えば、例えばMINIPAXの商品名の下で市販されモレキュラーシーブ4オングストロームを2g含有する乾燥剤の袋、または例えばSORBITの商品名の下で入手可能でシリカゲル1gを含有する乾燥剤の缶、例えばDRICAPの商品名の下で入手可能でシリカゲル0.9gを含有する乾燥剤のカプセル、またはシリカゲル2gを含有する乾燥剤の栓であることができる。
【0054】
貯蔵容器
本明細書に記載の方法の様々なステップにおいて得られる生成物または中間生成物は、好ましくは、50%未満、好ましくは40%未満の環境相対湿度で貯蔵される。したがって、上記の生成物は、アルミニウムの樽またはドラム缶の中、Muller(登録商標)のドラム缶として市販されているようないわゆるNirosta(登録商標)のドラム缶の中に貯蔵され得る。上記のドラム缶は、気密にされ得る、例えば、それらの蓋に、密封リング等の密封手段を適用することにより空気遮断され得る。上記の生成物は、アルミニウム、または閉鎖物もしくは蓋に密封リング等の密封手段が設けられた上述のようなNirosta(登録商標)の材料から作製された容器の中にも貯蔵され得る。
【0055】
特に形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、50%未満の、好ましくは10%から45%の環境相対湿度で、本明細書に記載の通りの容器の中に包装または充填される。次に、上記の容器は、本明細書に記載の通りに固く閉鎖される。好ましくは、上記の容器は、例えば室温において、約20℃から30℃の温度等において、例えば約25℃において、長期間の間、例えば少なくとも6カ月、好ましくは少なくとも約24カ月の間、例えば少なくとも24カ月以下の間、例えば少なくとも約30カ月以下の間、約60カ月以下の間等、本発明の医薬組成物を安定的に貯蔵するために用いられる。
【0056】
形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物は、好ましくは、40%未満の、好ましくは10%から40%の環境相対湿度で、本明細書に記載の通りの容器の中に包装または充填される。次に、上記の容器は、本明細書に記載の通りに固く閉鎖される。好ましくは、上記の容器は、例えば室温において、約20℃から30℃の温度等において、例えば約25℃において、長期間の間、例えば少なくとも6カ月、好ましくは少なくとも約24カ月の間、例えば少なくとも24カ月以下の間、例えば少なくとも約30カ月以下の間、約60カ月以下の間等、本発明の医薬組成物を安定的に貯蔵するために用いられる。
【0057】
好ましい容器は、ボトル、例えばガラスもしくはプラスチックのボトル、例えば、例えばねじ込み式の閉鎖物を有する、securitainerとして知られているようなポリエチレンのボトルである、またはブリスター、例えばアルミニウムのブリスターもしくはストリップ、例えば2枚のアルミニウムの箔もしくはストリップからなるブリスター、またはAclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターである、または任意の他の適切な容器であってよい。より好ましくは、上記の容器は、空気遮断の容器等の気密容器である。
【0058】
好ましい容器は、アルミニウム膜で密封されたガラスまたはプラスチックのボトル、alu−aluのブリスターもしくはストリップ、またはAclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターである。本発明による容器は、本発明の医薬組成物を、本明細書に記載の通りの条件下で上記の容器の中に充填することにより得られる。
【0059】
好ましくは、乾燥手段と組み合わせた容器は、中でも形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物の平衡相対湿度を、少なくとも6カ月間、好ましくは少なくとも2年間、50%未満に、好ましくは10%から45%に維持することが可能である。好ましい実施形態において、その容器は、さらに、50%未満の、好ましくは10%から45%の相対湿度をもつ気体雰囲気を封入する。その容器に乾燥した気体雰囲気、例えば乾燥した空気、または乾燥した窒素ガスを備えることは、当技術分野に知られている通りに行われ得る。形態IV−Sに関して、乾燥手段と組み合わせた容器は、医薬組成物の平衡相対湿度を、少なくとも6カ月間、好ましくは少なくとも2年間、40%未満に、好ましくは10%から40%に維持することが可能である。好ましい実施形態において、その容器は、さらに、40%未満の、好ましくは10%から40%の相対湿度をもつ気体雰囲気を封入する。
【0060】
容器および乾燥手段の好ましい組合せは、乾燥剤のカプセルおよび/もしくは缶を含有しアルミニウム箔で密封されたポリエチレンボトル(PEボトル)、または乾燥剤の栓をもつガラスボトルである。
【0061】
組成物の調製
相対環境湿度について、および組成物の平衡相対湿度について、形態I−S、IV−Sまたは形態HT−IV−Sを含む本発明の医薬組成物の製造中において特別な注意をはらわなければならない。したがって、本発明は、
a)形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を、1種または複数の薬学的に許容できる賦形剤と、50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して40%未満の、好ましくは10から40%の相対湿度で混合するステップ、
b)ステップa)で得られた混合物を、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して50%未満の、好ましくは10%から40%の相対湿度で場合によって顆粒化するステップ、ならびに
c)ステップa)で得られた混合物、またはステップb)で得られた顆粒を、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の相対湿度で、形態IV−Sに関して40%未満の、好ましくは10%から40%の相対湿度でさらに処理して、形態I−Sまたは形態HT−IV−Sまたは形態IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を得るステップ
を含む、形態I−S、IV−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物を調製するための方法にも関する。得られた本発明の医薬組成物は、形態I−SおよびHT−IV−Sに関して50%未満の、好ましくは10%から45%の、より好ましくは15%から35%の、または20%から45%の平衡相対湿度、ならびに形態IV−Sに関して40%未満の、好ましくは10から40%の、より好ましくは15から30%の平衡相対湿度を示すことが好ましい。
【0062】
上述の通りに、ステップa)で得られた混合物、またはステップb)で得られた顆粒は、好ましくは、カプセルもしくは錠剤のような経口投与の形態、または経口懸濁液のための顆粒、または経口懸濁液のための粉末に加工される。
【0063】
好ましい実施形態において、約50%未満の、好ましくは10%から45%の平衡相対湿度を有する形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む得られた医薬組成物は、少なくとも6カ月間、医薬組成物の平衡相対湿度を約50%未満に、好ましくは10%から45%に維持することが可能な容器、例えば、医薬組成物の相対平衡湿度を50%未満に、好ましくは10%から45%に維持するのに十分な乾燥手段を場合によってさらに含むことがある上述の容器の中に充填される。
【0064】
形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を含む本発明の医薬組成物のための適切な貯蔵条件は、組成物を所望の形態に維持するために重要である。したがって、好ましくは、本発明の医薬組成物を貯蔵するために、少なくとも6カ月間、気体雰囲気を50%未満の、好ましくは10%から45%の相対湿度に維持することが可能な容器が用いられる。好ましくは、50%未満の、好ましくは10%から45%の相対湿度を有する気体雰囲気は、形態I−Sまたは形態HT−IV−Sと呼ぶ結晶形態のサクサグリプチン塩酸塩を安定化するために用いられ、形態IV−Sに関して40%未満の、好ましくは10%から40%の相対湿度を有する気体雰囲気が用いられる
本発明の医薬組成物を、本明細書に記載の容器中に充填した後に、上記の容器は、好ましくは、例えば固くまたは完全に密封されて、例えば任意の気体雰囲気が上記の容器の壁および/または閉鎖物を通って拡散することを防止する方法で固く閉鎖される。誘導密封により、および閉鎖物、例えばスクリュー式の閉鎖物を適用することにより、アルミニウム膜を上記のボトルのボトル開口部に適用することによるガラスもしくはプラスチックのボトルの密封等、または例えば公知の方法に類似した加熱密封による、alu−aluのブリスターもしくはストリップの、Aclar(登録商標)の箔およびアルミニウムの覆い箔を含むブリスターの密封等の、上記の容器を固く密封するおよび/または閉鎖する方法が知られている。
【0065】
本明細書に記載されている方法の間に適用される温度は、好ましくは室温であり、例えば、約25℃等の、約20℃から約30℃の温度である。
【0066】
(実施例)
X線粉末回折パターン(XRPD)を、透過構造のシータ/シータの対のゴニオメータ、集束鏡によるCu−Kα1,2の照射(波長0.15419nm)、およびソリッドステートのPIXcel検出器を備えたPANalytical X’Pert PROの回折計により得た。パターンを、環境条件において、2°から40°の2θの角度範囲内で、1ステップあたり80sでステップ幅0.006°の2θを適用し、管電圧40kV、管電流40mAで記録した。2シータの値の一般的な精度は、約±0.2°の2シータの範囲内である。したがって、5.0°の2シータに現れる回折ピークは、標準的な条件下におけるほとんどのX線回折計により4.8から5.2°の2シータの間に現れることがある。
【0067】
赤外スペクトル(IR)を、Bruker Tensor 27のFTIR分光計で、MKII Golden Gate(商標)Single Reflection Diamond ATR(減衰全反射)のセルにより、4cm−1の分解能で収集した。スペクトルを収集するために、スパチュラ先端量の試料を、粉末の形態のダイアモンドの表面に塗布した。次いで、その試料をサファイア床でダイアモンド上に押し付け、スペクトルを記録した。清浄なダイアモンドのスペクトルをバックグラウンドのスペクトルとして用いた。波数の値の一般的な精度は、約±2cm−1の範囲内である。したがって、1716cm−1に現れる赤外のピークは、標準的な条件下におけるほとんどの赤外分光計により1714から1718cm−1の間に現れることがある。
【0068】
示差走査熱量測定(DSC)を、Pyrisのソフトウエアを用い、DSC 7(米国、コネチカット州、ノーウォーク、Perkin−Elmer)により行った。試料約4gmgを、25μlのAl皿の中に秤量した。乾燥した窒素をパージガスとして用いた(パージ、20mlmin−1)本明細書において用いられるときに、示差走査熱量測定により決定される「Tonset」の用語は、転移前のベースラインの、推定された転移の先端との交差に対応する温度を意味する。
【0069】
熱重量分析を、Windows(登録商標) NTのためのPyris Softwareを用いた熱重量分析システムTGA−7(米国、コネチカット州、ノーウォーク、Perkin−Elmer)、50μlの白金の皿、窒素のパージガス(試料のパージ:20mlmin−1、天秤のパージ:40mlmin−1)により行った。
【0070】
水分収着等温線を、SPS−11の水分収着分析計(ドイツ、ウルム、MD Mess−technik)により記録した。測定サイクルを、相対湿度(RH)0%で開始し、10%のステップでRH90%に、5%のステップでRH95%に増加させた。それぞれのステップについての平衡条件を、49minを超える一定質量±0.003%に設定した。温度は25±0.1℃であった。
【0071】
HPLCの検定を、以下の条件を適用することにより行った。
カラム:YMC−Ultra HT Pro C 18 3.0×50.0mm、2μm
溶離剤A:10mMスルファミン酸
溶離剤B:10mMのSAS250ml+アセトニトリル750ml
流量:0.64ml/min
温度:15℃
検出:210nmにおけるUV
勾配:
【0072】
【表1】
停止時間:15min
試料の濃度:約0.5mg/ml
溶媒:溶離剤A
注入体積:5μl
【実施例1】
【0073】
無水の形態I−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン遊離塩基の半水化物4.58gを、アセトン230ml中に溶解した。この溶液に、トリメチルクロロシラン2.2mlを撹拌の下で添加した。1時間後に、ゼラチン質の沈殿を含有する混合物を蒸発乾固した。固体の残留物にエタノール80mlを添加し、スラリーを、開放したフラスコ中で約1時間撹拌した。生成物を濾別し、tert.−ブチルメチルエーテルで洗浄し、次いで、真空オーブン中で乾燥して、サクサグリプチン一塩酸塩二水和物1.65gを得た。サクサグリプチン一塩酸塩二水和物の多形を、X線回折、FT−IRおよびDSCにより決定した。
【0074】
母液を、−5℃の冷蔵庫中に48時間入れた。驚くべきことに、さらなる結晶が母液から形成した。このように得られた結晶性の沈殿を濾別し、tert.−ブチルメチルエーテルで洗浄し、次いで、真空オーブン中で乾燥した。結晶を分析し、サクサグリプチン一塩酸塩1.22gであることがわかった。母液から得られた生成物は、形態I−Sと呼ぶ、サクサグリプチン一塩酸塩の無水の多形形態であることがわかった。
【0075】
HPLCによる純度:99.7面積%(最大の個々の不純物0.09%、全不純物0.33%)
サクサグリプチン一塩酸塩の形態I−Sの粉末X線回折パターンを、図1に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表1に示す。
【0076】
【表2】
【0077】
上記で得られた結晶形態I−Sのサクサグリプチン一塩酸塩は、2907、2853、1637、1589、1462、1391、1318、1045、1014および775cm−1(±2cm−1、図2)における吸収帯をもつ減衰全反射IRスペクトルを有する。
【0078】
得られた結晶形態I−Sに、示差熱分析を受けさせた。図3(下の曲線)において参照され得る通りに、結晶形態I−Sは、著しい脱水の吸熱でなく、241℃におけるピーク(Tonset 230℃、加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【0079】
イオン性の塩素は、10.7%(理論10.08%)と決定された。
【0080】
得られた結晶形態I−Sの水含有率を、カールフィッシャーの装置を用いて1.2%重量/重量と決定した。25℃における相対湿度約50%において、形態1Sのサクサグリプチン塩酸塩は、約1.5%の水含有率を示す。この水は、25℃における相対湿度を0.5%に下げたときに完全に失われる。この過程は、サクサグリプチン塩酸塩を約50%の湿度に曝した場合において可逆的である。
【0081】
形態I−Sは、相対湿度50%未満において安定である。相対湿度50%を超えると、形態I−Sは、公知の二水和物H2−1に転移する。この転移は可逆的でない。
【実施例2】
【0082】
エタノールからのシーディングによる無水の形態I−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン一塩酸塩2.31gをエタノール62ml中に懸濁させ、生成した混合物を還流において加熱した。熱い溶液を濾過し、次いで実施例1の生成物でシードした。混合物を25℃に冷却し、次いで冷蔵庫の中に終夜設置した。白色固体を濾過により収集し、真空オーブン中で乾燥して、サクサグリプチン一塩酸塩の形態I−S0.78gを得た。
【実施例3】
【0083】
形態HT−Sのサクサグリプチン一塩酸塩の調製
サクサグリプチン一塩酸塩二水和物10mgを、窒素下において10°K/minで180℃に加熱した。形態HT−Sを得た。
【0084】
サクサグリプチン一塩酸塩の形態HT−Sの粉末X線回折パターンを、図4に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表2に示す。
【0085】
【表3】
【0086】
上記で得られたサクサグリプチン一塩酸塩の結晶形態HT−Sは、2906、2854、1649、1574、1513、1459、1338、1124、1032および851cm−1(±2cm−1、図5)の吸収帯をもつ減衰全反射のIRスペクトルを有する。
【0087】
得られた結晶形態HT−Sに、示差熱分析を受けさせた。図3(下の曲線)において参照され得る通りに、結晶形態HT−Sは、著しい脱水の吸熱でなく、230℃におけるピーク(加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【0088】
得られた結晶形態HT−Sは、熱重量分析に基づいて、0.5%重量/重量以下の水分の減少を示した(図6、上の線)。
【実施例4】
【0089】
形態HT−IV−Sの調製
ステップ1、IV−Sの調製
105mgのサクサグリプチン一塩酸塩二水和物の形態H2−1を、nブタノール11ml中に溶解した。得られた溶液を濾過し、溶媒を、ロータリーエバポレータを用いて約20mbarで真空により除去した。残留物に、tert.アミルアルコール4mlを添加し、懸濁液を得た。この懸濁液を3日間撹拌し、次いで結晶を吸引濾過により単離し、デシケータ中で相対湿度約30%に終夜保った。
収量:サクサグリプチン一塩酸塩の形態IV−S97mg
【0090】
ステップ2、HT−IV−Sの調製
結晶生成物のサクサグリプチン一塩酸塩IV−Sを、50℃の真空オーブン中で約10時間、約40℃の真空において乾燥した後に、約80℃で5時間乾燥して、結晶形態HT−IV Sを得た。
水(カールフィッシャー):0.7%
試料を、相対湿度45−49%で12時間貯蔵した。
水(カールフィッシャー):1.2%
結晶形態に変化は観測されなかった(FTIR)。
【0091】
サクサグリプチン一塩酸塩の形態HT−IV Sの粉末X線回折パターンを、図7に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表3に示す。
【0092】
【表4】
【0093】
上記で得られたサクサグリプチン塩酸塩の結晶形態HT−IV−Sは、3495、2921、1637、1616、1464、1242、1103、1013、940および774(±2cm−1、図8)の波数の吸収帯をもつ減衰全反射のIRスペクトルを有する。
【0094】
得られた結晶形態HT−IV−Sに、示差熱分析を受けさせた。図9において参照され得る通りに、結晶形態HT−IV−sは、著しい脱水の吸熱でなく、240℃におけるピーク(Tonset232℃、加熱速度10℃/分、ピンホールを開けたカプセル)のみを示す。
【実施例5】
【0095】
形態IV−Sのサクサグリプチン一塩酸塩の調製
105mgのサクサグリプチン一塩酸塩二水和物の形態H2−1を、nブタノール11ml中に溶解した。得られた溶液を濾過し、溶媒を、ロータリーエバポレータを用いて約20mbarで真空により除去した。残留物に、tert.アミルアルコール4mlを添加し、懸濁液を得た。この懸濁液を3日間撹拌し、次いで結晶を吸引濾過により単離し、デシケータ中で相対湿度約30%に終夜保った。
収量:97mg
【0096】
サクサグリプチン一塩酸塩の形態IV−Sの粉末X線回折パターンを、図10に示す。特徴的なXRPDの角度、d−間隔および相対強度を、表4に示す。
【0097】
【表5】
【0098】
得られた結晶形態IV−Sに熱重量分析を受けさせた。図11において参照され得る通りに、結晶形態IV−Sは、100℃以下で開始する約12.8%の質量減少を示す。この質量減少は、水2.86molに相当する。
【0099】
水分収着分析は、水2.94molに相当する、25℃で相対湿度40%における13.1%の水含有率を示す。水分収着分析は、相対湿度0%において、相対湿度2.86%に相当する水含有率12.8%を示す。その過程は可逆的である。
【0100】
40%を超える相対湿度において、形態IV−Sは、公知の形態の二水和物H2−1に不可逆的に転移する。
【特許請求の範囲】
【請求項1】
1.5%重量/重量以下の水含有率を有するサクサグリプチン塩酸塩の無水結晶形態。
【請求項2】
式
【化1】
の、請求項1に記載のサクサグリプチン塩酸塩。
【請求項3】
6.7±0.2、14.6±0.2、15.2±0.2、16.6±0.2および17.9±0.2度の2シータのピークを含み、特に24.5±0.2および28.2±0.2度の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または2907、2853、1637、1589、1462、1391、1318、1045、1014および775+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態I−S。
【請求項4】
6.6±0.2、13.3±0.2および17.6±0.2度の2シータのピークを含み、特に11.5±0.2および16.7±0.2度の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または2906、2854、1649、1574、1513、1459、1338、1124、1032および851+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態HT−S。
【請求項5】
2.6、4.5、6.8、14.6および18.1±0.2度の2シータのピークを含み、特に2.6±0.2および6.8±0.2の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または3495、2921、1637、1616、1464、1242、1103、1013、940および774+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態HT−IV−S。
【請求項6】
2.4、4.1、4.7、6.3および15.6±0.2度の2シータのピークを含むX線粉末回折パターンを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態IV−S。
【請求項7】
請求項1から6のいずれか一項に記載のサクサグリプチンの結晶形態を有効量で含み、好ましくは糖尿病および/または関連する疾患の治療に使用するための医薬組成物。
【請求項8】
サクサグリプチン塩酸塩を、サクサグリプチン塩酸塩の形態I−Sの種結晶の存在下でアルコールから結晶化させるステップを含む、請求項3に記載の結晶性サクサグリプチン塩酸塩の形態I−Sを調製する方法。
【請求項9】
サクサグリプチン塩酸塩の形態H2−1、H1.25−2またはH.75−3の公知の結晶形態を約160℃から180℃に加熱し、形態HT−Sを単離するステップを含む、請求項4に記載の結晶性サクサグリプチン塩酸塩の形態HT−Sを調製する方法。
【請求項10】
単独で、または1種もしくは複数種の抗糖尿病薬、および/または1種もしくは複数種の他の治療薬と組み合わせた、請求項1から6のいずれか一項に記載の結晶性サクサグリプチン塩酸塩の使用であって、該治療薬が、単一剤形の薬物の調製と同じ剤形による経口、または注射により投与され得る使用。
【請求項11】
組成物の平衡相対湿度が50%未満である、請求項1から6のいずれか一項に記載の結晶性サクサグリプチン塩酸塩を含む医薬組成物。
【請求項12】
組成物の平衡相対湿度が10%から40%である、請求項11に記載の医薬組成物。
【請求項13】
請求項5に記載のサクサグリプチン塩酸塩の形態HT−IV−Sを調製するための、請求項6に記載のサクサグリプチン塩酸塩の形態IV−Sの使用。
【請求項1】
1.5%重量/重量以下の水含有率を有するサクサグリプチン塩酸塩の無水結晶形態。
【請求項2】
式
【化1】
の、請求項1に記載のサクサグリプチン塩酸塩。
【請求項3】
6.7±0.2、14.6±0.2、15.2±0.2、16.6±0.2および17.9±0.2度の2シータのピークを含み、特に24.5±0.2および28.2±0.2度の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または2907、2853、1637、1589、1462、1391、1318、1045、1014および775+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態I−S。
【請求項4】
6.6±0.2、13.3±0.2および17.6±0.2度の2シータのピークを含み、特に11.5±0.2および16.7±0.2度の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または2906、2854、1649、1574、1513、1459、1338、1124、1032および851+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態HT−S。
【請求項5】
2.6、4.5、6.8、14.6および18.1±0.2度の2シータのピークを含み、特に2.6±0.2および6.8±0.2の2シータのピークをさらに含むX線粉末回折パターン、ならびに/または3495、2921、1637、1616、1464、1242、1103、1013、940および774+/−2cm−1の波数のピークを含む赤外スペクトルを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態HT−IV−S。
【請求項6】
2.4、4.1、4.7、6.3および15.6±0.2度の2シータのピークを含むX線粉末回折パターンを特徴とする、請求項1および2の少なくとも一項に記載のサクサグリプチン塩酸塩の結晶形態IV−S。
【請求項7】
請求項1から6のいずれか一項に記載のサクサグリプチンの結晶形態を有効量で含み、好ましくは糖尿病および/または関連する疾患の治療に使用するための医薬組成物。
【請求項8】
サクサグリプチン塩酸塩を、サクサグリプチン塩酸塩の形態I−Sの種結晶の存在下でアルコールから結晶化させるステップを含む、請求項3に記載の結晶性サクサグリプチン塩酸塩の形態I−Sを調製する方法。
【請求項9】
サクサグリプチン塩酸塩の形態H2−1、H1.25−2またはH.75−3の公知の結晶形態を約160℃から180℃に加熱し、形態HT−Sを単離するステップを含む、請求項4に記載の結晶性サクサグリプチン塩酸塩の形態HT−Sを調製する方法。
【請求項10】
単独で、または1種もしくは複数種の抗糖尿病薬、および/または1種もしくは複数種の他の治療薬と組み合わせた、請求項1から6のいずれか一項に記載の結晶性サクサグリプチン塩酸塩の使用であって、該治療薬が、単一剤形の薬物の調製と同じ剤形による経口、または注射により投与され得る使用。
【請求項11】
組成物の平衡相対湿度が50%未満である、請求項1から6のいずれか一項に記載の結晶性サクサグリプチン塩酸塩を含む医薬組成物。
【請求項12】
組成物の平衡相対湿度が10%から40%である、請求項11に記載の医薬組成物。
【請求項13】
請求項5に記載のサクサグリプチン塩酸塩の形態HT−IV−Sを調製するための、請求項6に記載のサクサグリプチン塩酸塩の形態IV−Sの使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2012−523395(P2012−523395A)
【公表日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願番号】特願2012−504028(P2012−504028)
【出願日】平成22年4月9日(2010.4.9)
【国際出願番号】PCT/EP2010/054692
【国際公開番号】WO2010/115974
【国際公開日】平成22年10月14日(2010.10.14)
【出願人】(305008042)サンド・アクチエンゲゼルシヤフト (54)
【Fターム(参考)】
【公表日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願日】平成22年4月9日(2010.4.9)
【国際出願番号】PCT/EP2010/054692
【国際公開番号】WO2010/115974
【国際公開日】平成22年10月14日(2010.10.14)
【出願人】(305008042)サンド・アクチエンゲゼルシヤフト (54)
【Fターム(参考)】
[ Back to top ]