
サブトラクション・ポリヌクレオチドの取得方法
【課題】ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチド(テスター特異的ポリヌクレオチド)を、容易に且つ短時間に、高効率に取得・増幅する方法を提供する。
【解決手段】(1)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか工程、(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程、を含むテスター特異的ポリヌクレオチドの増幅方法。
【解決手段】(1)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか工程、(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程、を含むテスター特異的ポリヌクレオチドの増幅方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを取得・増幅する方法に関する。
【背景技術】
【0002】
機能、性質の異なる細胞又は組織間においては、発現量の異なる遺伝子が存在しており、その細胞又は組織の機能解析において重要な役割を果たしている可能性がある。このような遺伝子を取得することは、その機能や発現機構の解明等に有用である。
【0003】
一方、機能、性質の異なる細胞又は組織間において、発現量の異なる遺伝子をクローニングする方法として、cDNA サブトラクション法がある。
このcDNA サブトラクション法は、以下の手順で行われる。
例えば癌細胞組織の特異的な機能、性質を解析する場合、
(1)先ず、癌細胞組織から調製した一本鎖RNA又はcDNA(テスター)と正常細胞組織から調製された過剰量の一本鎖RNA又はcDNA、若しくは二本鎖cDNA(ドライバー)とをハイブリダイゼーションさせる。この時、テスターとドライバーともに発現している遺伝子(ハウスキーピング遺伝子など)は二本鎖cDNA又はRNA-DNAハイブリッドを形成するが、テスターに特異的に発現している遺伝子由来のRNA又はcDNAはハイブリッドを形成せず、一本鎖RNA又はcDNAの状態である。
(2)次いで、ハイブリッドを形成しなかったテスターに特異的に発現している遺伝子由来の一本鎖RNA又はcDNAを、例えばハイドロキシアパタイト・クロマトグラフィー法(Hendrick, S.M.,Cohen, D.I., Nielsen, E.L. and Davis, M.M. (1984) Nature. 308, 149-153)、アビジン・ビオチン結合法(Duguin, J.R. and Dinauer, M.C. (1990) Nucleic Acid Research. 18, 2789-2792、特表平9−507021号公報)、ラテックス、ビーズ、カラム等を用いたオリゴ (dT)30固相法(Hara, E., Kato T., Nakada, S., Sekiya, L and Oda, K. (1991) Nucleic Acid Research. 19, 7097-7104、特開2001−269172号公報)、アビジン・ビオチン・磁気ビーズ法(特開2001−269172号公報、特開2002−253237号公報)等の方法により、ハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドと分離し、テスター特異的に発現している遺伝子を濃縮する。
【0004】
しかしながら、上記の方法は何れも物理的結合又は吸着を利用して、テスターに特異的に発現している遺伝子由来の一本鎖RNA又はcDNAとハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドとを分離するため、これらを厳密に分離することはできず、ハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドが混在してしまうという問題があった。その結果、上記の方法では、テスターおよびドライバー両方に発現している遺伝子(例えばハウスキーピング遺伝子など)の混入が多く、高効率にテスター特異的に発現している遺伝子を濃縮(取得)することはできなかった。また、上記方法は、操作が煩雑で作業工程が多く、時間と熟練を要するという問題もあった。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅する方法、当該方法により得られた(増幅された)ポリヌクレオチド、テスターにおける遺伝子変異を同定する方法、並びにこれらに用いられるキットを提供する。
【課題を解決するための手段】
【0006】
本発明は、以下の構成よりなる。
1.以下の工程を含む、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを増幅する方法;
(1)(i)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか、或いは(ii)テスター由来の一本鎖ポリヌクレオチドと、ドライバー由来の二本鎖ポリヌクレオチドとを混合して、熱変性後、ハイブリダイゼーションを行うかして、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成する工程、
(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、
(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び
(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程。
2.上記1の方法により増幅された(テスターに特異的に存在する)ポリヌクレオチドを同定することを特徴とするテスターにおける遺伝子変異を同定する方法。
【0007】
即ち、本発明者らは上記目的を達成すべく鋭意研究を重ねた結果、サブトラクティブハイブリダイゼーションにより形成された、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを、酵素処理により除去することにより、テスターでの存在量が別のドライバーでの存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅し得ることを見出し、本発明を完成させるに至った。
【発明の効果】
【0008】
本発明の方法により、テスターでの存在量がドライバーでの存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅することが可能であり、その結果、テスターにおける遺伝子変異を高精度に同定することが可能となる。
【図面の簡単な説明】
【0009】
【図1】本発明の取得方法又は増幅方法を示すチャート図である。
【0010】
【図2】本発明の一本鎖テスターcDNAの調製工程を模式的に示した図である。
【0011】
【図3】本発明の二本鎖ドライバーcDNAの調製工程を模式的に示した図である。
【0012】
【図4】本発明のテスターcDNAとドライバーcDNAとのハイブリダイゼーション工程〔工程(1)〕を模式的に示した図である。
【0013】
【図5】本発明のハイブリッド形成した二本鎖cDNAの除去工程〔工程(2)〕を模式的に示した図である。
【0014】
【図6】本発明のハイブリッド形成しなかった一本鎖テスター特異的cDNAの増幅工程〔工程(3)〕を模式的に示した図である。
【0015】
【図7】本発明のハイブリッド形成しなかった一本鎖ドライバーcDNAの除去工程〔工程(4)〕を模式的に示した図である。
【0016】
【図8】実施例で得られた、ヒト肝臓癌由来のHepG2細胞のcDNA(テスターcDNA)、ヒト正常肝臓組織由来のcDNA(ドライバーcDNA)及びサブトラクションcDNA(テスター特異的cDNA)をそれぞれ鋳型として、glyceraldehyde-3-phosphate dehydrogenase(GAPDH)遺伝子とalpha-fetoprotein(AFP)遺伝子とをPCR増幅して得られたPCR産物を電気泳動した結果である。
【発明を実施するための最良の形態】
【0017】
1.試料
本発明において、「ある試料(テスター)」及び「別の試料(ドライバー)」とは、両者がそれぞれ異なることを意味し、例えば(1)試料の種類は異なるが、試料の由来〔生体(個体・集団)、場所等〕は同一である場合、(2)試料の種類は同一であるが、試料の由来が異なる場合、(3)試料の種類及び試料の由来が異なる場合、(4)試料の種類及び試料の由来が同一である場合である。
より具体的には、例えば(1)同一個体(生体)における異なる種類の細胞、組織、器官、体液等、(2)異なる個体(生体・商品)間(特定個体と標準(正常)個体間)又は特定の個体(生体・商品)と集団間(特定個体と標準(正常)集団間)における同じ種類の細胞、組織、器官、体液等、(3)異なる個体間(罹患個体と標準(正常)個体間)又は特定個体と集団間(罹患個体と標準(正常)集団間)における異なる種類の細胞、組織、器官、体液(異常細胞と正常細胞、異常組織と正常組織、異常器官と正常器官、異常体液と正常体液等)等、(4)環境、状態等が異なる場合(薬剤投与前後、ストレス付加前後、病態の異なるステージ等)の同一個体(生体)における同一種類の細胞、組織、器官、体液等
【0018】
本発明が適用される試料の種類としては、この分野で用いられているものであれば良く、特に限定されない。このような試料としては、例えば細胞、組織、器官、体液(血液、血清、血漿、髄液、滑液、膵液、リンパ液等)、排泄物(尿、唾液、糞便等)、喀たん、膿、皮膚由来物等の生体由来試料;微生物(菌類、細菌類、ウイルス等);例えば環境試料(食品、飲料、水道水、海水、湖沼水、河川水、工場廃液、半導体用洗浄水、医療器具等を洗浄した後の洗浄液等);これらから得られる抽出液;及びこれらを水や通常この分野で用いられている緩衝液等(トリス緩衝液、グリシン緩衝液、リン酸緩衝液、ベロナール緩衝液、ホウ酸緩衝液、グッド緩衝液等)に適宜溶解させて再構成して得られた処理物等が挙げられる。
【0019】
本発明において、「ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多い」とは、(1)テスターには存在するがドライバーには存在しない場合や、(2)テスターとドライバーの両方に存在はしているものの、テスターでの存在量がドライバーでの存在量に比較して多い場合等を意味する。尚、「テスターに特異的に存在する」や「テスター特異的」も同じ意味である。
【0020】
本発明の方法により、取得・増幅される「ポリヌクレオチド」(即ち、「ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチド」)には、例えばmRNA等のRNA;当該mRNAに由来するcDNA、ゲノムDNA等のDNA等が含まれる。なかでも、本発明の方法は、mRNAに由来するcDNA、ゲノムDNA等のDNAを取得・増幅するのに有効であり、特にテスターでの存在量がドライバーでの存在量よりも多いmRNA由来のcDNAを取得・増幅するのに極めて有効である。
【0021】
2.本発明の方法
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチド(以下、テスター特異的ポリヌクレオチドと略記する。)を取得又は増幅する方法に関する。
本発明の当該ヌクレオチドの取得方法は、(1)テスター由来の一本鎖ポリヌクレオチドを用いてドライバー由来の一本鎖又は二本鎖ポリヌクレオチドとハイブリダイゼーションを行って、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成させること、及び(2)酵素処理により、ハイブリッド形成した二本鎖ポリヌクレオチドを除去することを特徴とする。
更に、当該二本鎖ポリヌクレオチドの除去後、ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅させることにより、テスターでの存在量がドライバーでの存在量よりも多いポリヌクレオチドを大量に得ることができる。
即ち、本発明の当該ヌクレオチドの増幅方法は、(1)テスター由来の一本鎖ポリヌクレオチドを用いてドライバー由来の一本鎖又は二本鎖ポリヌクレオチドとハイブリダイゼーションを行って、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成させること、(2)酵素処理により、ハイブリッド形成した二本鎖ポリヌクレオチドを除去すること、及び(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅することを特徴とする。
【0022】
2−1.本発明のテスター特異的ポリヌクレオチドの取得方法
(1)テスター由来の一本鎖ポリヌクレオチド
本発明におけるテスター由来のポリヌクレオチド(以下、テスターポリヌクレオチドと略記する。)は一本鎖ポリヌクレオチドである。当該一本鎖ポリヌクレオチドは、DNA及びRNAのどちらでも良いが、好ましくはDNAである。なかでも、ゲノムDNA及びテスターに存在するmRNAに由来するcDNA(換言すれば、テスターから抽出したmRNAを鋳型として合成されたcDNA)が好ましく、特にcDNAが好ましい。
【0023】
本発明の一本鎖テスターポリヌクレオチドは、前述した如き試料から適宜調製できる。一本鎖ポリヌクレオチドの調製は、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ、市販のキットを用いて行うこともできる。
このような方法としては、通常この分野で用いられている方法であれば特に限定されず、前述した如き試料から直接一本鎖ポリヌクレオチドを調製する方法〔例えば、Vieira J, Messing J. Methods in Enzymology. 1987;153:3-11;Krieg PA, Melton DA. Methods in Enzymology. 1987;155:397-415;Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Proc Natl Acad Sci U S A. 1984 Apr;81(7):2035-9等〕、前述した如き試料から一旦二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865;Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕、PCRプライマーの片側だけビオチン標識したプライマーを使用してPCR増幅し、それをマグネチック・ストレプトアビジン・ビーズと混合して一本鎖DNAを回収する方法等が何れも使用できる。また、市販のキットを用いて行うこともできる。ここで、二本鎖ポリヌクレオチドの調製も、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ〔例えば、Kazuo Maruyama and Sumio Sugano Gene, 138 ( 1994 ) 171-174;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 ( 1996 );Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000 Jun 15;28(12):E63;Shimada M, Hino F, Sagawa H, Mukai H, Asada K, Kato I. Rinsho Byori. 2002 May;50(5):528-32等〕、市販のキットを用いて行うこともできる。
尚、上記において、調製される一本鎖ポリヌクレオチド及び二本鎖ポリヌクレオチドは、テスター中に存在するポリヌクレオチドそのものでも、また、当該ポリヌクレオチドを鋳型として、例えばポリメラーゼ連鎖反応(PCR)等の自体公知の増幅方法により増幅された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドであってもよい。
【0024】
本発明においては、上記した如き一本鎖テスターヌクレオチドを得る方法のなかでも、二本鎖ポリヌクレオチドを調製した後、酵素処理等により一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865;Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕が好ましく、特に、テスター中に存在するポリヌクレオチドを鋳型として、例えばPCR等の自体公知の増幅方法により、当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅した後、酵素処理により一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕が好ましい。
従って、本発明の一本鎖テスターヌクレオチドを得る方法は、(1’)テスター由来の二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましく、特に(1”)テスター由来の二本鎖ポリヌクレオチドを増幅した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましい。
尚、上記において、酵素処理の際に用いられる酵素としては、通常この分野で用いられるものであれば良く特に限定されないが、例えばラムダ・エキソヌクレアーゼ等の二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素、例えば2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群(例えば5'突出末端を生じ得る制限酵素、3'突出末端を生じ得る制限酵素及びExonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素の組み合わせ等)等が挙げられる。尚、これら酵素の由来は特に限定されず、また、自体公知の方法で得られたもの及び市販されているものの何れも使用できる。
【0025】
また、上記した如き方法において使用される試薬類や反応条件等も自体公知の方法に準じて行えばよい。
分解酵素の使用量は、二本鎖ポリヌクレオチドの片側の鎖を充分に分解して一本鎖ポリヌクレオチドとし得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば酵素としてラムダ・エキソヌクレアーゼを用いる場合には、通常DNA2μgに対して0.1units〜100units、好ましくは0.5units〜50units、より好ましくは1units〜10unitsである。
当該酵素処理の条件(温度、時間、pH等)、即ち、二本鎖ポリヌクレオチドとラムダ・エキソヌクレアーゼとを反応(接触)させる際の条件は、通常25℃〜60℃、好ましくは30℃〜50℃、より好ましくは35℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜250mM、より好ましくは10mM〜100mMから適宜選択される。
【0026】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を用いる方法又は/及び加熱処理を行う方法により行えばよい。
反応停止剤を用いる方法としては、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液と、酵素処理を行って得られた反応液と混合させれる等により、当該反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
また、加熱処理により反応を停止する方法としては、酵素処理を行って得られた反応液を、通常60℃〜95℃、好ましくは70℃〜90℃、より好ましくは80℃〜85℃で処理すればよい。
反応停止剤による反応時間及び加熱処理時間は、通常5分〜60分、好ましくは10分〜30分、より好ましくは15分〜20分である。
上記の反応停止方法のうち、反応停止剤を用いる方法と加熱処理による方法の両者を併用するのが好ましい。
【0027】
尚、二本鎖ポリヌクレオチドとラムダ・エキソヌクレアーゼとを反応させる際には、通常この分野で用いられる活性化剤(例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウム塩に由来するマグネシウムイオン等の金属イオン等)、界面活性剤(例えばTriton-X100(ポリオキシエチレン(10)オクチルフェニルエーテル)等)、安定化剤(ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を存在させておいても良い。特に、マグネシウム塩及び界面活性を存在させるのが好ましい。尚、これらの使用量は通常この分野で使用される範囲から適宜選択し得、具体的には、マグネシウム塩の使用量としては、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMであり、界面活性剤の使用量としては、通常0.001〜1%(w/v)、好ましくは0.005〜0.5%(w/v)、より好ましくは0.01〜0.1%(w/v)である。
【0028】
更に、本発明で使用される一本鎖テスターポリヌクレオチドとしては、その5’末端又は/及び3’末端に、既知配列からなるアダプターが結合している(付加されている)ものが好ましく、特に当該アダプターは5’末端及び3’末端の両末端に結合(付加)させるのが好ましい。アダプターはいかなる配列を含んでいても良いが、その塩基数はそれ自体が単独でプライマーとして機能し得る程度が好ましい。従って、アダプターの塩基数は、通常15bp〜60bp、好ましくは20bp〜35bp、より好ましくは25bp〜30bpである。
尚、5’末端及び3’末端の両末端にアダプターを結合(付加)させる場合は、両末端のアダプターが相補的であると、一本鎖テスターポリヌクレオチド内でのアダプターハイブリッドを形成するおそれがあるので、相補的ではないアダプターを両末端に結合させるのが好ましい。また、当該アダプターは、ドライバー由来のポリヌクレオチド中に存在しない配列からなるものが好ましい。
このように一本鎖テスターポリヌクレオチドに上記した如きアダプターを結合(付加)させておくことにより、本発明のテスター特異的ポリヌクレオチドの取得方法で所得されたポリヌクレオチドを、ドライバー由来の(一本鎖)ポリヌクレオチドから容易に単離・精製することができる。
また、上記した如きアダプターは、後述する本発明のポリヌクレオチドの増幅方法において、本発明のテスター特異的ポリヌクレオチドの取得方法で所得されたポリヌクレオチド(ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチド)を増幅する際に有用である。即ち、当該アダプターを後述する工程(3)で行うPCR等の増幅反応用のプライマーが結合する既知の配列とすることにより、当該アダプターに結合するプライマーを用いて容易にテスター特異的ポリヌクレオチドを増幅することができる。
【0029】
一本鎖テスターポリヌクレオチドにアダプターを結合(付加)させる方法としては、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ、市販のキットを用いて行うこともできる。
このような方法としては、通常この分野で用いられている方法であれば特に限定されず(1)上記した如き方法により調製された一本鎖ポリヌクレオチドにアダプターを結合(付加)させる方法〔例えば、Oligo-capping:a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides, Kazuo Maruyama and Sumio Sugano, Gene, 138 (1994) 171-174に記載のオリゴ-キャッピング法;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 (1996)に記載のSMART法;High-efficiency full-length cDNA cloning by biotinylated CAP trapper, Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C., Genomics. 1996 Nov 1;37(3):327-36に記載のGene Trapper法等〕、(2)上記した如き方法において、調製された二本鎖ポリヌクレオチドにアダプターを結合(付加)させた後、これを一本鎖ポリヌクレオチドにする方法〔例えば、Determination of 5' ends of specific mRNAs by DNA ligase-dependent amplification, Bertling WM, Beier F, Reichenberger E., PCR Methods Appl. 1993 Oct;3(2):95-9に記載のRACE法等〕、(3)上記方法において、テスター中に存在するポリヌクレオチドを鋳型として、例えばアダプター配列に基づいて設計されたプライマーを用いたPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、アダプター(プライマー)が結合(付加)された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法等が挙げられる。
尚、上記方法(3)において、増幅されるポリヌクレオチドが二本鎖の場合は、当該二本鎖ポリヌクレオチドを上記した如き方法により一本鎖ポリヌクレオチドにする必要がある。この場合、使用する2種類のプライマーのうち片側のみの5’末端をリン酸化しておけば、増幅された二本鎖ポリヌクレオチドを二本鎖DNAの5’末端リン酸化DNA鎖を除去するラムダ・エキソヌクレアーゼ等で処理することにより、5’末端リン酸化プライマー側のcDNA鎖を除去することができ、容易にアダプターが結合した(付加された)一本鎖テスターポリヌクレオチドを得ることができる。
本発明においては、上記した如き一本鎖テスターポリヌクレオチドにアダプターを結合(付加)させる方法のなかでも、テスター中に存在するポリヌクレオチドを鋳型として、例えばアダプター配列に基づいて設計されたプライマーを用いたPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、アダプター(プライマー)が結合(付加)された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法が好ましい。
従って、本発明において、アダプターが結合した(付加された)一本鎖テスターヌクレオチドを得る方法は、(1’’’)テスター由来の二本鎖ポリヌクレオチドをアダプター配列に基づいて設計されたプライマーを用いて増幅した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましい。
【0030】
例えば以下の方法により、アダプターが結合した(付加された)一本鎖テスターポリヌクレオチドを得ることができる。
(1)自体公知の方法〔Gublerらの方法(Gulber, U. and Hoffman, B. J. (1983) Gene. 25, 263-269)等〕によってテスターから抽出したmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーを用い、cDNA挿入断片両端のベクター由来の既知配列で設計された5’末端がリン酸化されたプライマーとリン酸化されていないプライマーの2種のプライマーを作製し、これを用いてベクターに挿入されているcDNAを鋳型として、PCR法、LAMP法、ICAN法等によりテスター遺伝子由来の二本鎖cDNAを増幅する。次いで、増幅されたプライマー(アダプター)付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る〔Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕。
(2)既知配列(アダプター)で設計された5’末端がリン酸化されたプライマーとリン酸化されていないプライマーの2種のプライマーを作製し、これを用いてオリゴ・キャッピング法に従って5’末端及び3’末端にアダプター配列が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR法、LAMP法、ICAN法等により増幅し、次いで、増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る〔Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕。
(3)適当なプラスミドに目的とするポリヌクレオチドを挿入し、プラスミドのマルチクローニングサイトを利用して挿入されたポリヌクレオチド断片の片方を5'突出末端(例えばEcoR I等)とし、もう片方を3'突出末端(例えばPst I等)とした後、Exonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素で消化して一本鎖cDNAを得る〔Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕。
尚、上記方法のうち、(1)及び(2)が好ましい。
得られた一本鎖テスターポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0031】
(2)ドライバー由来のポリヌクレオチド
本発明におけるドライバー由来のポリヌクレオチド(以下、ドライバーポリヌクレオチドと略記する。)は一本鎖ポリヌクレオチドであっても二本鎖ポリヌクレオチドであってもどちらでも良いが、好ましくは二本鎖ポリヌクレオチドである。尚、ドライバーポリヌクレオチドが一本鎖である場合は、当然のことながら、当該一本鎖ドライバーポリヌクレオチドは、一本鎖テスターポリヌクレオチドと相補鎖となり得るものである。即ち、テスターポリヌクレオチドがセンス鎖である場合、ドライバーポリヌクレオチドはアンチセンス鎖であり、テスターポリヌクレオチドがアンチセンス鎖である場合、ドライバーポリヌクレオチドはセンス鎖である。
また、当該ドライバーポリヌクレオチドは、DNA及びRNAのどちらでも良いが、好ましくはDNAである。なかでも、ゲノムDNA及びテスターに存在するmRNAに由来するcDNA(換言すれば、テスターから抽出したmRNAを鋳型として合成されたcDNA)が好ましく、特にcDNAが好ましい。
【0032】
本発明のドライバーポリヌクレオチドも、上記した如き、テスターポリヌクレオチドを調製するために用いられる二本鎖ポリヌクレオチドを調製する方法、一本鎖ポリヌクレオチドを調製する方法等の通常この分野で用いられている自体公知の方法や、市販のキットを用いて得ることができる。
なかでも、ドライバー中に存在するポリヌクレオチドを鋳型として、例えばPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法が好ましい。
【0033】
また、本発明で使用されるドライバーポリヌクレオチドには、一本鎖テスターポリヌクレオチドと同様、その5’末端又は/及び3’末端に、既知配列からなるアダプターを結合(付加)させておいてもよいが、この場合、当該アダプターは、少なくとも一本鎖テスターポリヌクレオチドが有する、後述する工程(3)で行うPCR用のプライマーが結合する既知の配列とは異なるものである。換言すれば、後述する工程(3)で行うPCR用のプライマーが結合しない既知配列からなるアダプターであれば、当該ドライバーポリヌクレオチドに結合(付加)させておいてもよいが、当該ドライバーポリヌクレオチドには一本鎖テスターヌクレオチドに結合(付加)させたアダプターを含む既知配列を含まないか、或いはそれ自体が単独でプライマーとして機能し得る塩基数からなる当該アダプターの一部の配列を含まない〔即ち、アダプターを結合(付加)させない〕のが好ましい。また、相補的ではないアダプターを両末端に結合させるのが好ましい。
【0034】
例えば以下の方法により、ドライバーポリヌクレオチドを得ることができる。
(1)自体公知の方法〔Gublerらの方法(Gulber, U. and Hoffman, B. J. (1983) Gene. 25, 263-269)等〕によってドライバーから抽出したmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーを用い、cDNA挿入断片両端のベクター由来の既知配列で設計された2種のプライマーを作製し、これを用いてベクターに挿入されているcDNAを鋳型として、PCR法、LAMP法、ICAN法等によりドライバー遺伝子由来の二本鎖cDNAを増幅して、アダプターが結合した(付加された)二本鎖ドライバーcDNAを得る。
(2)上記(1)によって増幅されたアダプターが結合した(付加された)二本鎖cDNAの、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までを、挿入断片両側のマルチクローニングサイトの制限酵素部位を利用して適当な制限酵素で切断し、挿入断片のみからなる二本鎖ドライバーcDNA〔アダプターが結合(付加)していない二本鎖ドライバーcDNA〕を得る。尚、この方法は、テスター由来cDNAライブラリーとドライバー由来cDNAライブラリーとでcDNAライブラリーを構成するベクターが同系列(例えば、両方ともにpUC系プラスミドである場合等)である場合等、テスターポリヌクレオチドとドライバーポリヌクレオチドで同じプライマーを使用する場合(同じアダプターが結合している場合)に有用である。
【0035】
尚、上記の方法を用いる場合、以下の点で注意が必要である。
即ち、テスター由来cDNAライブラリーとドライバー由来cDNAライブラリーとでcDNAライブラリーを構成するベクターが同系列(例えば、両方ともにpUC系プラスミドである場合等)である場合等、テスターポリヌクレオチドとドライバーポリヌクレオチドで同じプライマーを使用する場合(同じアダプターが結合している場合)には、挿入されているcDNA断片の末端からプライマーまでの長さ、言い換えれば、プライマーの末端から挿入断片が結合しているプライマーを含めた鎖長が100bp以上であると、切断された挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの断片を後述する精製方法で除去することが困難となる。その場合、当該切断された断片がテスターに存在する相補部分とハイブリダイズして二本鎖ハイブリッドを形成し、後述する本発明の工程(2)においてテスターのアダプター領域(PCR用プライマーがアニールする領域)が除去されてしまい、工程(3)での目的の一本鎖テスター特異的ポリヌクレオチドの増幅に支障をきたす。従って、このような場合には、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの鎖長が100bp以下となるように、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの切断を複数の制限酵素を用いて行うか、切断された挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの断片を更に他の制限酵素を用いて切断することが好ましい。
(3)既知配列(アダプター)で設計された、5’末端がリン酸化された一本鎖テスターcDNAと相補鎖となる側のプライマーとリン酸化されていない一本鎖テスターcDNAと相同鎖となる側のプライマーの2種のプライマーを作製し〔調製される一本鎖テスターcDNAと相補鎖となるようにリン酸化プライマーと非リン酸化プライマーを設計する。言い換えれば、5’末端がリン酸化された一本鎖テスターcDNAと、これに相補的となる5’末端がリン酸化されていない一本鎖cDNAが生じるように2つのプライマー(リン酸化プライマーと非リン酸化プライマー)を設計する〕、これを用いてオリゴ・キャッピング法〔Maruyama, k. and Sugano, S. (1994) Gene. 138, 171-174等〕に従って5’末端及び3’末端に特定のプライマー(アダプター配列)が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR法、LAMP法、ICAN法等により増幅し、次いで、増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、一本鎖テスターcDNAと相補鎖となるcDNA〔即ち、アダプターが結合した(付加された)一本鎖ドライバーcDNA〕を得る。
(4)適当なプラスミドに目的とするポリヌクレオチドを挿入し、プラスミドのマルチクローニングサイトを利用して挿入されたポリヌクレオチド断片の片方を5'突出末端(例えばEcoR I等)とし、もう片方を3'突出末端(例えばPst I等)とした後、Exonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素で消化して一本鎖cDNAを得る〔Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕。
上記方法のうち(2)が好ましい。
尚、得られた一本鎖又は二本鎖ドライバーポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0036】
(3)テスター由来のポリヌクレオチドとドライバー由来のポリヌクレオチドとのハイブリダイゼーション〔工程(1)〕
上記の如くして調製された、(i)一本鎖テスターポリヌクレオチドと当該一本鎖テスターポリヌクレオチドと相補鎖となり得る一本鎖ドライバーポリヌクレオチドとの間、或いは(ii)一本鎖テスターポリヌクレオチドと二本鎖ドライバーポリヌクレオチドとの間で、ハイブリダイゼーション(所謂、サブトラクティブ・ハイブリダイゼーション)を行う。
このハイブリダイゼーションにより、ドライバーポリヌクレオチド中に相補的なものが存在する一本鎖テスターポリヌクレオチド〔即ち、テスターとドライバーの両方に存在するテスターポリヌクレオチド(テスター非特異的ポリヌクレオチド)〕は、テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)を形成する。一方、ドライバーポリヌクレオチド中に相補的なものが存在しない(或いは存在量が少ない)一本鎖テスターポリヌクレオチド(即ち、テスター特異的ポリヌクレオチド)は、ハイブリッドを形成せずに一本鎖のままで存在する。
【0037】
当該ハイブリダイゼーションは、通常この分野で用いられている自体公知のハイブリダイゼーション方法や、市販のキットを用いて行うことができる。
【0038】
例えば、上記の如くして調製した一本鎖テスターポリヌクレオチドと、当該テスターポリヌクレオチドに対して過剰量の一本鎖又は二本鎖ドライバーポリヌクレオチドを、適当量のナトリウム塩を含む緩衝液中で、要すれば熱変性後、適当な温度で適当な時間ハイブリダイゼーションを行い、テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)を形成させる。
上記において、使用されるドライバーポリヌクレオチドの量は、テスターポリヌクレオチドの量よりも多ければ良く、標的とするポリヌクレオチド(遺伝子)や使用目的等により異なるため一概には言えないが、使用されるドライバーポリヌクレオチドが一本鎖の場合のポリヌクレオチドの量(核酸量)は、テスターポリヌクレオチドの量(核酸量)の通常10〜5000倍、好ましくは50〜1000倍、より好ましくは100〜500倍である。また、使用されるドライバーポリヌクレオチドが二本鎖の場合のポリヌクレオチドの量(核酸量)は、テスターポリヌクレオチドの量(核酸量)の通常20〜10000倍、好ましくは100〜2000倍、より好ましくは200〜1000倍である。
使用されるナトリウム塩としては、通常この分野で用いられる、例えば塩化ナトリウム、クエン酸ナトリウム等が用いられ、また、その使用濃度も通常この分野で用いられる濃度範囲、例えば通常20mM〜2M、好ましくは50mM〜1.5M、より好ましくは100mM〜1Mから適宜選択される。
また、使用される緩衝液も、通常この分野で用いられるものは全て使用可能であり、例えば通常pH5〜9、好ましくはpH6〜8、より好ましくはpH6.5〜7.5の範囲で緩衝能を有する、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜1M、好ましくは5mM〜500mM、より好ましくは10mM〜100mMから適宜選択される。
上記において、熱変性は、通常90℃〜105℃、好ましくは93℃〜103℃、より好ましくは95℃〜100℃で、通常0.1分〜15分、好ましくは0.5分〜10分、より好ましくは1分〜5分行われる。尚、上記方法では、テスターポリヌクレオチドとドライバーポリヌクレオチドの共存下同時に熱変性をしているが、これらをそれぞれ別々に熱変性しても良い。この場合には、ナトリウム塩は特に必要ない。また、この場合には、熱変性した後、氷冷し、テスターポリヌクレオチドとドライバーポリヌクレオチドとを接触させてハイブリダイゼーションを行うのが好ましい。
ハイブリダイゼーションの条件としては、通常この分野で行われている方法に準じて行えば良く特に限定されないが、例えば上記した如きナトリウム塩の存在下、下限が通常30℃以上、好ましくは37℃以上、より好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下で、通常1分〜30時間、好ましくは30分〜20時間、より好ましくは1時間〜16時間行えばよい。
尚、上記方法において、ハイブリダーゼーション後に、更に上記した如き濃度範囲から選択されるドライバーポリヌクレオチドを追加してハイブリダイゼーションを繰り返してもよく、これにより、よりストリンジェントな条件でハイブリダイゼーションを行うことができる。
また、上記においては、通常この分野で使用される、例えば有機溶媒等の試薬類を用いることができる。
【0039】
(4)ハイブリッド形成した二本鎖ポリヌクレオチドの除去〔工程(2)〕
上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(即ち、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を酵素処理により除去する。
これにより、テスターポリヌクレオチドのうち、テスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチドを除去することができる。尚、二本鎖ドライバーポリヌクレオチドを用いた場合は、上記工程(1)において再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)も同時に除去される。
【0040】
本発明の上記工程(2)の酵素処理において用いられる酵素は、一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有するものであり、使用されるテスターポリヌクレオチド及びドライバーポリヌクレオチドの種類により異なる。例えばテスターポリヌクレオチド及びドライバーポリヌクレオチドが共にDNA(cDNA)である場合は、二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が使用され、テスターポリヌクレオチドとドライバーポリヌクレオチドのどちらか一方がDNA(cDNA)であって他方がRNAである場合は、DNA−RNAハイブリッドを優先的に分解し得る性質を有する分解酵素が使用される。また、テスターポリヌクレオチド及びドライバーポリヌクレオチドが共にRNAである場合は、二本鎖RNAを優先的に分解し得る性質を有するRNA分解酵素が使用される。このような分解酵素を用いて酵素処理することにより、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(及び再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を分解することができ、結果としてハイブリット形成した二本鎖ポリヌクレオチド(即ち、テスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチド)を除去することができる。
また、テスターポリヌクレオチドがDNA(cDNA)でドライバーポリヌクレオチドがRNAである場合には、DNA−RNAハイブリッド中のDNAを優先的に分解し得る性質を有するDNA分解酵素も使用でき、テスターポリヌクレオチドがRNAでドライバーポリヌクレオチドがDNA(cDNA)である場合は、DNA−RNAハイブリッド中のRNAを優先的に分解し得る性質を有するRNA分解酵素(例えばRNase H等)も使用できる。これらの分解酵素を用いて酵素処理行った場合には、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(及び再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)自体(全体)を分解することはできないが、ハイブリッド形成された二本鎖ポリヌクレオチド中のテスターポリヌクレオチドを分解することができるので、結果としてテスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチドを除去することができる。
上記した如き分解酵素のうち、二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が好ましく、このような分解酵素としては、例えば二本鎖特異的DNAヌクレアーゼ(Duplex-specific nuclease:DSN)等が挙げられる。当該酵素の由来については特に限定されないが、例えば海洋性甲殻類(エビ、カニ等)、ウニ等の海洋性無脊椎動物が挙げられる。なかでも海洋性甲殻類、特にカムチャッカカニ(Paralithodes camtschaticus)由来の二本鎖特異的DNAヌクレアーゼが好ましい。
尚、二本鎖特異的DNAヌクレアーゼは、自体公知の方法〔例えばShagin DA, Rebrikov DV, Kozhemyako VB, Altshuler IM, Shcheglov AS, Zhulidov PA, Bogdanova EA, Staroverov DB, Rasskazov VA, Lukyanov S. : Genome Research. 2002 Dec;12(12):1935-42〕に準じて製造することができ、また、市販品(例えばEvrogen社)を使用することもできる。
分解酵素の使用量は、ハイブリッド形成された二本鎖ポリヌクレオチドを充分に分解し得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合には、通常全ポリヌクレオチド 1μgに対して0.01u/μl〜10u/μl、好ましくは0.1u/μl〜5u/μl、より好ましくは0.5u/μl〜1u/μlである。
【0041】
当該酵素処理の条件(温度、時間、pH等)、即ち、ハイブリッド形成された二本鎖ポリヌクレオチドと分解酵素とを接触させる際の条件は、使用する分解酵素の種類によって異なるため一概には言えないが、通常使用する分解酵素が有する至適温度及び至適pH付近で、形成されたハイブリッド(又はハイブリッド中のテスターポリヌクレオチド)を充分分解し得る時間処理すればよい。
より具体的には、例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合には、下限が通常20℃以上、好ましくは30℃以上、より好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下で、通常pH6〜9、好ましくは6.5〜8.5、より好ましくは7〜8で、通常0.5分〜60分、好ましくは1分〜45分、より好ましくは5分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜100mM、より好ましくは10mM〜50mMから適宜選択される。
【0042】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
このような反応停止剤を当該反応液中に存在させるには、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液を酵素処理を行って得られた反応液と混合させればよい。
反応停止のための時間は、通常0.1分〜30分、好ましくは0.5分〜20分、より好ましくは1分〜10分である。
また、反応を停止する際の温度は特に限定されないが、50℃〜90℃、好ましくは60℃〜80℃、より好ましくは65℃〜75℃で行うのが好ましい。
【0043】
当該酵素処理は、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチドと上記した如き分解酵素とを接触させることにより実施される。
このような方法としては、最終的にハイブリッド形成された二本鎖ポリヌクレオチドと分解酵素とを接触し得るものであれば良く特に限定されないが、一般的には、上記した如き本発明の工程(1)を実施して得られたハイブリダイゼーション溶液に当該分解酵素を含有する溶液を添加混合することにより行われる。
上記において、分解酵素を含有させる溶液としては、当該分解酵素の分解作用を阻害しないものであればよく、水や上記した如き緩衝剤等の通常この分野で用いられる溶液が挙げられ、その使用濃度等も上記した如き濃度範囲から適宜選択すればよい。
ここで、当該溶液中には、分解酵素以外に、ナトリウム塩、通常この分野で用いられる活性化剤(マグネシウムイオン等の金属イオン等)、安定化剤(グリセロール、ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を含有させておいても良い。尚、これらナトリウム塩、活性化剤、安定化剤、防腐剤等は、分解酵素を含有する溶液とは別の上記した如き溶液に含有させておいても良く、この場合は、分解酵素を含む溶液とナトリウム塩、活性化剤、安定化剤又は防腐剤を含有する1種以上の溶液とをハイブリダイゼーション溶液に別々に添加することになる。なかでも、ナトリウム塩を含有させておくのが好ましい。
使用されるナトリウム塩としては、通常この分野で用いられる、例えば塩化ナトリウム、クエン酸ナトリウム等が用いられ、また、その使用濃度も通常この分野で用いられる濃度範囲、例えば通常20mM〜2M、好ましくは50mM〜1.5M、より好ましくは100mM〜1Mから適宜選択される。
【0044】
例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合、当該酵素処理〔本発明の工程(2)〕は以下の如くして実施される。
本発明の工程(1)を実施して得られたハイブリダイゼーション溶液に、二本鎖特異的DNAヌクレアーゼ含有溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、上記した如き条件で処理し(反応させ)て、当該ハイブリダイゼーション溶液中に存在するハイブリッド形成した二本鎖ポリヌクレオチド(テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド、又はこれと再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を分解除去する。
尚、上記において、活性化剤としてはマグネシウムイオンが好ましい。その由来としては、例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウムの塩が挙げられ、なかでも塩化マグネシウムが好ましい。また、マグネシウムの塩の使用量としては、二本鎖特異的DNAヌクレアーゼを充分活性化し得る量であればよく、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMである。
また、安定化剤としては例えばジチオスレイトール、メルカプトエタノール等のチオール化合物が好ましく、なかでもジチオスレイトールが好ましい。また、その使用量としては、二本鎖特異的DNAヌクレアーゼを安定化し得る量であればよく、通常0.01mM〜10mM、好ましくは0.1mM〜5mM、より好ましくは0.5 mM〜1mMである。
尚、本発明の工程(1)と(2)は、なるべく高い温度で実施するのが好ましい。例えば本発明の工程(1)と(2)は、下限が好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下でそれぞれ実施される。また、工程(1)に引き続いて工程(2)を実施する際には、上記した如き温度範囲から一旦下げること無く、上記した如き温度範囲を維持したまま、当該温度範囲から温度を一旦下げることなく実施されるのが好ましい。
【0045】
上記したように本発明の方法〔本発明の工程(1)及び工程(2)〕によって、テスター中に存在するポリヌクレオチドのうち、テスター非特異的ポリヌクレオチドを分解除去し、テスター特異的ポリヌクレオチドを取得することができる。
尚、本発明の工程(2)を実施して得られた反応液中には、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチド以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存しているが、共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドは、自体公知の単離(分離)処理又は精製処理により除去することができ、一本鎖テスター特異的ポリヌクレオチドのみを単離することができる。
このような方法としては、例えば一本鎖テスター特異的ポリヌクレオチドを、テスターcDNAを増幅したプライマー等のテスター特異的ポリヌクレオチドのみに存在するアダプター配列を基に設計したプライマーを使用してクレノウ・フラグメント(Klenow Fragment)で二本鎖にし、その後両端のアダプター配列中の制限酵素で切断して、適当なベクターに組み込み、テスター特異的ポリヌクレオチド(cDNA)のみを形質転換する方法、テスターcDNAを増幅したプライマー等のテスター特異的ポリヌクレオチドのみに存在するアダプター配列を基に設計したプライマーをビオチン標識して、テスター特異的ポリヌクレオチド(cDNA)とアニーリング後、ストレプトアビジン・ビーズ等の固相と結合させてテスター特異的ポリヌクレオチドを単離する方法等が挙げられる。
尚、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドとは、テスターポリヌクレオチドに相補的な一本鎖ドライバーポリヌクレオチドを用いた場合は、ドライバーに特異的な一本鎖ポリヌクレオチド及びテスター非特異的ポリヌクレオチドとハイブリッドを形成しなかったテスターとドライバーの両方に存在する一本鎖ドライバーポリヌクレオチドであり、二本鎖ドライバーポリヌクレオチドを用いた場合は、再会合せずに一本鎖として存在するドライバーに特異的な一本鎖ポリヌクレオチド(テスターポリヌクレオチドに対する相補鎖及び相同鎖の2種)及び再会合せずに一本鎖として存在するテスターとドライバーの両方に存在する一本鎖ドライバーポリヌクレオチド(テスターポリヌクレオチドに対する相補鎖及び相同鎖の2種)である。
【0046】
2−2.本発明のテスター特異的ポリヌクレオチドの増幅方法
かくして得られたテスター特異的ポリヌクレオチドを増幅することにより、テスター特異的ポリヌクレオチドを濃縮することができる。
【0047】
(1)ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドの増幅〔工程(3)〕
本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチドを大量に得ることができる。
即ち、本発明の工程(2)を実施した後に残存する一本鎖テスター特異的ポリヌクレオチドのみを実質的に増幅させる。
これにより、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しなかった場合でも、一本鎖テスター特異的ポリヌクレオチドと共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドは実質的に増幅されないので、一本鎖テスター特異的ポリヌクレオチドのみが濃縮される。
本発明において、一本鎖テスター特異的ポリヌクレオチドを増幅するとは、(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみを増幅する場合又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する場合を意味する。
【0048】
当該増幅は、本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチド〔(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみ又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖等〕を増幅することができるものであれば良く、通常この分野で用いられている自体公知のポリヌクレオチド増幅方法に従って実施できる。このような自体公知のポリヌクレオチド増幅方法としては、例えばPCR〔Oligo-capping:a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides, Kazuo Maruyama and Sumio Sugano, Gene, 138 (1994) 171-174に記載のオリゴ-キャッピング法;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 (1996)に記載のSMART法;High-efficiency full-length cDNA cloning by biotinylated CAP trapper, Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C., Genomics. 1996 Nov 1;37(3):327-36に記載のGene Trapper法;Determination of 5' ends of specific mRNAs by DNA ligase-dependent amplification, Bertling WM, Beier F, Reichenberger E., PCR Methods Appl. 1993 Oct;3(2):95-9に記載のRACE法〕、LAMP法〔Loop-mediated isothermal amplification of DNA, Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000 Jun 15;28(12):E63〕、ICAN法〔Development of the detection system for Mycobacterium tuberculosis DNA by using the isothermal DNA amplification method ICAN, Shimada M, Hino F, Sagawa H, Mukai H, Asada K, Kato I. Rinsho Byori. 2002 May;50(5):528-32〕等が挙げられ、なかでもPCRが好ましい。
尚、各種増幅条件(温度、時間、pH、サイクル数等)は上記した如き自体公知の方法に準じて行えばよく、また、使用される酵素類や試薬類等もこれら自体公知の方法で用いられているものが使用できる。
また、当該増幅は、市販のキットを用いて行うこともできる。
【0049】
当該増幅に使用されるプライマーは、本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチド〔(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみ又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖等〕を増幅することができるものであれば良く特に限定されない。
即ち、当該プライマーは、例えば、目的とする増幅方法及びその条件等に応じて、また、解離温度(Tm値)などを考慮して、適宜設計・作製すればよい。好ましくはプライマー配列としての特異性を維持するために必要な塩基数と考えられている、15塩基〜60塩基、好ましくは20塩基〜35塩基、より好ましくは25塩基〜30塩基の長さを有しているものがよい。
【0050】
例えば本発明の工程(2)を実施した後に、上記した如き単離(分離)処理又は精製処理により、一本鎖テスター特異的ポリヌクレオチドが単離されている場合には、これ以外のテスター非特異的ポリヌクレオチドやハイブリッドを形成しなかったドライバーポリヌクレオチドは共存していないので、どのようなプライマーも使用できる。
このようなプライマーとしては、例えば前述した如きアダプターが結合した(付加された)テスターポリヌクレオチドを用いた場合は、当該アダプター配列に基づいて設計・作製されたプライマー等が挙げられ、例えばcDNAライブラリーからテスターcDNAを調製した場合はcDNA挿入断片両端のベクター由来の配列に基づいて設計・作製された1種又は2種以上プライマー(即ち、前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプター配列又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド)等が挙げられる。
また、例えば本発明の工程(2)を実施した後に、単離(分離)処理又は精製処理を行わず、一本鎖テスター特異的ポリヌクレオチドが単離されていない場合には、テスター非特異的ポリヌクレオチドは共存していないものの、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。しかしながら、一本鎖テスター特異的ポリヌクレオチドにのみ存在し、ドライバー由来のポリヌクレオチド中に存在しない配列に基づいて設計・作製されたプライマーを用いることにより、一本鎖テスター特異的ポリヌクレオチドのみを増幅することができる。
このようなプライマーとしては、例えば前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプターに基づいて設計・作製された1種又は2種以上プライマー(即ち、前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプター配列又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド)等が挙げられる。
また、PCR等に使用する場合の2種以上のプライマーとしては、相補的ではないアダプター配列の一部若しくは全部、又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド、即ち、それぞれ異なる2種以上のプライマーが好ましい。
【0051】
当該プライマーは、標識物質で標識化されていてもよく、プライマーを標識物質で標識するために用いられる標識物質としては、放射性同位体(32P,33P,35S等)や酵素(アルカリホスファターゼ,西洋ワサビペルオキシダーゼ等)、蛍光物質〔Cyanine Dye系のCy3,Cy5(アマシャムバイオサイエンス株式会社)、フルオレセイン等〕、発光物質(Acridinium Esterを含む化学発光試薬等)、ビオチンなど公知の標識物質であれば何れも用いることができる。また、プライマーを標識物質で標識する方法も、例えばプライマーを合成する際に、放射性同位体や蛍光物質で標識されたヌクレオチドを取り込ませることによって、プライマーを標識する方法や、プライマーを合成した後、放射性同位体で標識する方法(ランダムプライマー法、ニックトランスレーション法、T4ポリヌクレオチド キナーゼによる5'−末端標識法、ターミナルデオキシヌクレオチドトランスフェラーゼを用いた3'−末端標識法、RNAラベリング法)、酵素分子を標識するプライマーに直接共有結合させる直接標識法等、リンカーアームを有するヌクレオチドを配列のオリゴヌクレオチドの一員として置換して、ヌクレオチドを蛍光物質で標識する方法、ヌクレオチドを発光標識又はビオチン標識する方法等、通常この分野で行われている自体公知の標識方法に従って行えばよい。
【0052】
本発明の工程(3)によって、一本鎖テスター特異的ポリヌクレオチドを増幅(濃縮)することができるが、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しない場合、即ち、本発明の工程(2)を実施して得られた、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドとハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドとが共存している反応液中の、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドを増幅する場合においては、本発明の工程(3)を実施して得られた反応液中には、増幅された、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチド以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。しかしながら、この場合には、後述する本発明の工程(4)における一本鎖ポリヌクレオチドを除去する工程により、増幅された一本鎖テスター特異的ポリヌクレオチドを単離することができる。
特に、後述する本発明の工程(4)における一本鎖ポリヌクレオチドを除去する工程が、酵素処理により一本鎖ポリヌクレオチドを除去する工程である場合には、酵素処理により一本鎖ポリヌクレオチドを特異的に分解除去して、増幅されたテスター特異的ポリヌクレオチドを容易に単離することができるため、本発明の工程(3)で増幅されたテスター特異的ポリヌクレオチドは二本鎖であるのが好ましい。
【0053】
増幅された二本鎖テスター特異的ポリヌクレオチドを得る方法としては、例えば上記した如き自体公知の増幅方法のうち、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する方法が挙げられる。
また、上記した如き自体公知の増幅方法のうち、一本鎖テスター特異的ポリヌクレオチドの相補鎖のみを増幅する場合には、増幅された一本鎖特異的ポリヌクレオチドの相補鎖を用いて、例えば以下の方法によって二本鎖テスター特異的ポリヌクレオチドを得ることができる。
(a)増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖を鋳型として、これに相補的なプライマーを用いて再度PCRを行って増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖に対する相補鎖を増幅させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法、(b)増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖を鋳型として、これに相補的なプライマーとクレノウ フラグメント等の重合酵素を用いて増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖に対する相補鎖をプライマー伸長させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法等。
これらの方法のなかでも、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する方法が好ましい。
尚、上記において、得られた二本鎖テスター特異的ポリヌクレオチドの二本鎖ハイブリットを形成させるには特別な処理は必要ではなく、例えば得られた一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖を含有する溶液を、通常30℃〜80℃、好ましくは37℃〜75℃、より好ましくは50℃〜70℃とすることによりこれらのハイブリットが形成される。
【0054】
上記したことから、本発明においては、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅するのが好ましい。
従って、本発明のテスター特異的ポリヌクレオチドの増幅方法は、(3’)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドとこれの相補鎖とを増幅してテスター由来の二本鎖ポリヌクレオチドを得る工程、を含んでなる方法が好ましい。
【0055】
例えば以下の方法により、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドを増幅することができる。
即ち、(a)本発明の工程(2)を実施して得られた反応液、要すれば更に単離処理又は精製処理により単離された一本鎖テスター特異的ポリヌクレオチドを含有する溶液に、上記した如きそれぞれ異なる2種のプライマーとデオキシリボヌクレオチド三リン酸(dATP、dCTP、dGTP及びdTTP)を適当量加え、得られた溶液を適当な温度で適当な時間加熱して熱変性する。(b)加熱後当該溶液を冷却して一本鎖テスター特異的ポリヌクレオチド(又は一本鎖テスター特異的ポリヌクレオチドと、増幅されたこれに相補的なポリヌクレオチド)にプライマーをアニールさせる。(c)次いで、適当な重合酵素を適当な温度で適当な時間作用させてプライマーを伸長させることによって、一本鎖テスター特異的ポリヌクレオチドに対する相補鎖(又は一本鎖テスター特異的ポリヌクレオチドに対する相補鎖と、増幅された、一本鎖テスター特異的ポリヌクレオチドに相補的なポリヌクレオチドに対する相補鎖)を合成(増幅)する。以上の(a)〜(c)の工程を適当回数(例えば通常5回〜60回、好ましくは10回〜50回、より好ましくは15回〜40回)繰り返すことによってハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖とを増幅する。
上記方法において、使用される試薬類の量、重合酵素の種類や量、反応条件等は上記した如き自体公知の方法に準じて適宜選択されればよい。
例えば、上記工程(a)の熱変性は、通常90℃〜105℃、好ましくは93℃〜103℃、より好ましくは95℃〜100℃で、通常0.01分〜10分、好ましくは0.05分〜5分、より好ましくは0.1分〜2分行われる。また、工程(b)のアニーリングは、通常30℃〜75℃、好ましくは40℃〜72℃、より好ましくは50℃〜68℃で、通常0.01分〜10分、好ましくは0.05分〜5分、より好ましくは0.1分〜2分行われる。上記工程(c)において使用される重合酵素としては、特に限定されず、この分野で使用される酵素が使用可能であるが、例えばDNAポリメラーゼI、クレノウ フラグメント、T4 DNAポリメラーゼ等のDNAポリメラーゼ、例えばTaqDNAポリメラーゼ、Tth DNA ポリメラーゼ、Pfu DNA ポリメラーゼ等の耐熱性酵素、逆転写酵素等が挙げられる。また、重合酵素を作用させる際の条件(温度、時間等)は、使用する重合酵素の種類等により異なるため一概には言えないが、例えばDNAポリメラーゼI等の非耐熱性酵素の場合は、通常20℃〜50℃、好ましくは25℃〜45℃、より好ましくは30℃〜37℃で、通常1分〜24時間、好ましくは5分〜12時間、より好ましくは30分〜4時間であり、例えばTaqDNAポリメラーゼ等の耐熱酵素の場合は、通常30℃〜85℃、好ましくは40℃〜80℃、より好ましくは50℃〜75℃で、通常0.01分〜20分、好ましくは0.05分〜15分、より好ましくは0.1分〜10分である。
尚、上記方法においては、(a)〜(c)の各工程後に、デオキシリボヌクレオチド三リン酸、プライマー、試薬類、重合酵素等を新たに添加してもよい。
なかでも、TaqDNAポリメラーゼ、Tth DNA ポリメラーゼ、Pfu DNA ポリメラーゼ等の耐熱性酵素を用いれば、(a)〜(c)の各工程後に、デオキシリボヌクレオチド三リン酸、プライマー、試薬類、重合酵素等を新たに添加する必要がなく、最初に全ての試薬類や重合酵素を共存させておくことができるので、特に好ましい。
得られた二本鎖テスター特異的ポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0056】
(2)ハイブリット形成しなかった一本鎖ドライバーポリヌクレオチドの除去〔工程(4)〕
上記したように、本発明の工程(3)によって増幅(濃縮)されたテスター特異的ポリヌクレオチド(一本鎖又は二本鎖)が、一本鎖ドライバーポリヌクレオチドと共存している場合、即ち、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しない場合は、本発明の工程(3)を実施して得られた反応液中には、増幅されたテスター特異的ポリヌクレオチド〔工程(2)でハイブリッドを形成しなかった、一本鎖テスター特異的ポリヌクレオチド〕以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。この場合には、当該一本鎖ドライバーポリヌクレオチドを除去することにより、増幅されたテスター特異的ポリヌクレオチド(一本鎖又は二本鎖)を単離することができる。
一本鎖ドライバーポリヌクレオチドを除去する方法としては、前述した如き自体公知の単離(分離)方法又は精製方法が挙げられる。
【0057】
また、増幅されたテスター特異的ポリヌクレオチドが二本鎖ポリヌクレオチドである場合には、一本鎖ポリヌクレオチドを分解するが二本鎖ポリヌクレオチドを分解しない性質を有する酵素を用いた酵素処理により、二本鎖テスター特異的ポリヌクレオチドと共存する一本鎖ポリヌクレオチド(一本鎖ドライバーポリヌクレオチド)を特異的に分解除去して、二本鎖テスター特異的ポリヌクレオチドを単離することもできる〔Convenient single-step, one tube purification of PCR products for direct sequencing. E Werle, C Schneider, M Renner, M Veolker, and W Fiehn, Nucleic Acids Res. 1994 October 11; 22(20): 4354-4355等〕。
【0058】
上記の方法のなかでも、酵素処理により一本鎖ポリヌクレオチドを除去(分解)する方法が好ましい。
従って、本発明において、一本鎖ドライバーポリヌクレオチドを除去する方法は、(4’)ハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程、を含んでなる方法が好ましい。
【0059】
当該酵素処理において用いられる酵素は、ハイブリッド形成された二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有するものであり、使用されるテスターポリヌクレオチド及びドライバーポリヌクレオチドの種類により異なる。
例えばテスターポリヌクレオチド及びドライバーポリヌクレオチドが共にDNA(cDNA)である場合は、例えば一本鎖特異的DNAヌクレアーゼ(エキソヌクレアーゼI、エキソヌクレアーゼIX等)等の二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が使用され、テスターポリヌクレオチド及びドライバーポリヌクレオチドが共にRNAである場合は、例えば一本鎖特異的RNAエンドヌクレアーゼ(リボヌクレアーゼ)(RNaseA、RNase IV、RNaseT1、RNaseT2、RNase II、RNase III、Rnase I等)等の二本鎖RNAを分解せず一本鎖RNAを優先的に分解し得る性質を有するRNA分解酵素が使用される。
このような分解酵素を用いて酵素処理することにより、上記した如き本発明の工程(3)において得られた二本鎖ポリヌクレオチド(二本鎖テスター特異的ポリヌクレオチド)と共存する一本鎖ドライバーポリヌクレオチド(一本鎖ドライバー特異的ポリヌクレオチド及び一本鎖ドライバー非特異的ポリヌクレオチド)を分解することができ、二本鎖ポリヌクレオチド(二本鎖テスター特異的ポリヌクレオチド)を単離することができる。
上記した如き分解酵素のうち、二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が好ましく、エキソヌクレアーゼIがより好ましい。
上記した如き分解酵素の由来については特に限定さず、通常この分野で用いられるものであれば使用可能である。尚、当該分解酵素は、自体公知の方法〔例えばLehman,IR., Nussbaum, A.L., J.Bio.Chem., 239, 2628-2636, 1964〕に準じて製造することができ、また、市販品(例えば New England Biolabs 社)を使用することもできる。
【0060】
分解酵素の使用量は、一本鎖ポリヌクレオチドを充分に分解し得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合には、通常テスターポリヌクレオチド 1 μg に対して0.1u/μl〜100u/μl、好ましくは0.5u/μl〜50u/μl、より好ましくは1u/μl〜10u/μlである。
当該酵素処理の条件(温度、時間、pH等)、即ち、一本鎖ポリヌクレオチドと分解酵素とを接触させる際の条件は、使用する分解酵素の種類によって異なるため一概には言えないが、通常使用する分解酵素が有する至適温度及び至適pH付近で、一本鎖ポリヌクレオチドを充分分解し得る時間処理すればよい。
より具体的には、例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合には、通常20℃〜60℃、好ましくは25℃〜50℃、より好ましくは30℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜250mM、より好ましくは10mM〜100mMから適宜選択される。
【0061】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を用いる方法又は/及び加熱処理を行う方法により行えばよい。
反応停止剤を用いる方法としては、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液と、酵素処理を行って得られた反応液と混合させれる等により、当該反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
また、加熱処理により反応を停止する方法としては、酵素処理を行って得られた反応液を、通常60℃〜90℃、好ましくは65℃〜85℃、より好ましくは70℃〜80℃で処理すればよい。
尚、反応停止剤による反応時間及び加熱処理時間は、通常1分〜60分、好ましくは5分〜30分、より好ましくは10分〜20分である。
上記の反応停止方法のうち、反応停止剤を用いる方法と加熱処理による方法の両者を併用するのが好ましい。
【0062】
当該酵素処理は、上記した如き本発明の工程(3)によって増幅(濃縮)されたテスター特異的ポリヌクレオチド及びそれと共存する一本鎖ドライバーポリヌクレオチドと上記した如き分解酵素とを接触させることにより実施される。
このような方法としては、最終的に増幅(濃縮)されたテスター特異的ポリヌクレオチド、共存する一本鎖ドライバーポリヌクレオチド及び分解酵素とを接触し得るものであれば良く特に限定されないが、一般的には、上記した如き本発明の工程(3)を実施して得られた反応液に当該分解酵素を含有する溶液を添加混合することにより行われる。
上記において、分解酵素を含有させる溶液としては、当該分解酵素の分解作用を阻害しないものであればよく、水や上記した如き緩衝剤等の通常この分野で用いられる溶液が挙げられ、その使用濃度等も上記した如き濃度範囲から適宜選択すればよい。
ここで、当該溶液中には、分解酵素以外に、通常この分野で用いられる活性化剤(マグネシウムイオン等の金属イオン等)、安定化剤(ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を含有させておいても良い。尚、これら活性化剤、安定化剤、防腐剤等は、分解酵素を含有する溶液とは別の上記した如き溶液に含有させておいても良く、この場合は、分解酵素を含む溶液と活性化剤、安定化剤又は防腐剤を含有する1種以上の溶液とを反応液に別々に添加することになる。
【0063】
例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合、当該酵素処理〔本発明の工程(4)〕は以下の如くして実施される。
本発明の工程(4)を実施して得られた反応液に、一本鎖特異的DNAヌクレアーゼ含有溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、上記した如き条件で処理し(反応させ)て、当該反応液中に存在するハイブリッド形成しなかった一本鎖ドライバーポリヌクレオチド(一本鎖ドライバー特異的ポリヌクレオチド及び一本鎖ドライバー非特異的ポリヌクレオチド)を分解除去する。
尚、上記において、活性化剤としてはマグネシウムイオンが好ましい。その由来としては、例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウム塩が挙げられ、なかでも塩化マグネシウムが好ましい。また、マグネシウム塩の使用量としては、一本鎖特異的DNAヌクレアーゼを充分活性化し得る量であればよく、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMである。
また、安定化剤としては例えばジチオスレイトール、メルカプトエタノール等のチオール化合物が好ましく、なかでもジチオスレイトールが好ましい。また、その使用量としては、一本鎖特異的DNAヌクレアーゼを安定化し得る量であればよく、通常0.01mM〜100mM、好ましくは0.05mM〜50mM、より好ましくは0.1mM〜10mMである。
【0064】
上記したように本発明の工程(4)によって、本発明の増幅方法〔本発明の工程(1)〜(3)〕で増幅されたテスター特異的ポリヌクレオチドを容易に単離することができる。
尚、工程(3)の増幅を行わずに、工程(2)で得られた一本鎖テスター特異的ポリヌクレオチドを、当該一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに相補的なプライマーとクレノウ フラグメント等の重合酵素を用いて一本鎖テスター特異的ポリヌクレオチドに対する相補鎖をプライマー伸長させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法等により二本鎖とした後に、本発明の工程(4)を行ってもよく、この場合には、テスター特異的ポリヌクレオチドは増幅(濃縮)されないものの、工程(2)で得られた一本鎖テスター特異的ポリヌクレオチドと共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを分解除去することができ、容易にテスター特異的ポリヌクレオチドのみを単離することができる。
【0065】
2−3.本発明の具体的方法
本発明の取得方法又は増幅方法は、一本鎖テスターポリヌクレオチドと一本鎖又は二本鎖ドライバーポリヌクレオチドとを用いて、図1に示されたチャートに従って実施することができる。
即ち、先ず、目的とするある試料から、自体公知の方法、好ましくはテスター由来の二本鎖ポリヌクレオチドを調製した後に酵素処理により一本鎖ポリヌクレオチドにする方法〔本発明の工程(1’)〕、より好ましくはテスター由来の二本鎖ポリヌクレオチドを増幅した後に当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする方法〔本発明の工程(1”)〕により、一本鎖テスターポリヌクレオチド、好ましくはアダプターが付加された一本鎖テスターポリヌクレオチドを調製する。一方、別の試料から、自体公知の方法により、二本鎖ドライバーポリヌクレオチド又は一本鎖テスターポリヌクレオチドに対して相補的な一本鎖ドライバーポリヌクレオチドを調製する。次いで、例えば以下の手順で本発明の取得方法又は増幅方法を実施する。
【0066】
(手順1)
(i)一本鎖テスターポリヌクレオチドと当該一本鎖テスターポリヌクレオチドと相補鎖となり得る一本鎖ドライバーポリヌクレオチドとの間、或いは(ii)一本鎖テスターポリヌクレオチドと二本鎖ドライバーポリヌクレオチドとの間で、ハイブリダイゼーションを行い、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチド(ドライバー非特異的ポリヌクレオチド)との二本鎖ポリヌクレオチド、又は当該ハイブリッドと再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチドをハイブリッド形成させる〔工程(1)〕。次いで、ハイブリッド形成された二本鎖ポリヌクレオチドを酵素処理により分解除去して、テスター非特異的ポリヌクレオチド又はこれと再会合したドライバー二本鎖ポリヌクレオチドを除去する〔工程(2)〕。更に要すれば、共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを、自体公知の単離(分離)方法又は精製方法等により除去する〔単離工程〕。以上の手順により一本鎖テスター特異的ポリヌクレオチドを取得又は単離することができる。(本発明の取得方法1)
【0067】
(手順2)
上記工程(1)及び(2)、要すれば単離工程を実施した後、取得又は単離された一本鎖テスター特異的ポリヌクレオチドを増幅させる〔工程(3)〕。これにより、テスター特異的ポリヌクレオチドのみを実質的に増幅させて、テスター特異的ポリヌクレオチドのみを大量に得ることができる。(本発明の増幅方法1)
【0068】
(手順3)
上記工程(1)及び(2)を実施した後、取得された一本鎖テスター特異的ポリヌクレオチドを増幅させる〔工程(3)〕。次いで、増幅されたテスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを除去する〔工程(4)〕。これにより、増幅されたテスター特異的ポリヌクレオチドを単離することができる。(本発明の増幅方法2)
【0069】
(手順4)
以下の手順により、より簡便にテスター特異的ポリヌクレオチドを増幅(濃縮)及び単離することができる。(本発明の増幅方法3)
即ち、上記工程(1)及び(2)を実施した後、取得された一本鎖ポリヌクレオチドとこれの相補鎖とを増幅させて二本鎖テスター特異的ポリヌクレオチドを得る〔工程(3’)〕。次いで、増幅された二本鎖テスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを酵素処理により分解除去し、二本鎖テスター特異的ポリヌクレオチドを単離する〔工程(4’)〕。
【0070】
(手順5)
上記工程(1)及び(2)を実施した後、取得された一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに対する相補鎖を合成して二本鎖テスター特異的ポリヌクレオチドを得る〔二本鎖工程〕。次いで、工程(4’)により、二本鎖テスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを酵素処理により分解除去する。この場合には、工程(2)で取得されたテスター特異的ポリヌクレオチドを容易に単離することができる。(本発明の取得方法2)
【0071】
(手順6)
上記工程(1)、(2)、及び単離工程を実施した後、上記二本鎖工程により、一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに対する相補鎖を合成して二本鎖テスター特異的ポリヌクレオチドを得る。(本発明の増幅方法4)
【0072】
本発明の取得方法としては、上記手順1が好ましい。また、本発明の増幅方法としては、上記手順2及び3が好ましく、手順3が特に好ましい。
【0073】
以下に、テスターポリヌクレオチドとしてアダプターが付加された一本鎖cDNAを用い、ドライバーポリヌクレオチドとして二本鎖cDNAを用いる場合を例に取り、本発明の方法を具体的に説明する。
【0074】
〔一本鎖テスターcDNAの調製〕
自体公知の方法によって目的とする試料から抽出されたmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーにおける、cDNA挿入断片両端のベクター由来の既知配列で設計された5’末端がリン酸化されたプライマーとこれとは異なる配列のリン酸化されていないプライマーの2種のプライマーを作製する。当該cDNAライブラリーを鋳型とし、作製した2種のプライマーを用いてPCR等によりテスター遺伝子由来の5’末端及び3’末端にアダプター配列が付加された二本鎖cDNAを増幅する。或いは、自体公知の方法により目的とする試料からmRNAを抽出する。既知配列で設計された5’末端がリン酸化されたプライマーとこれとは異なる配列のリン酸化されていないプライマーの2種のプライマーを作製する。抽出したmRNAを鋳型とし、作製した2種のプライマーを用いてオリゴ・キャッピング法に従って5’末端及び3’末端にアダプター配列が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR等により増幅する。
要すれば、増幅されたアダプター付加二本鎖cDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は/及び加熱処理を行うことによって当該酵素の反応を停止させて、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る。
尚、要すれば、得られた一本鎖テスターcDNAを、カラム精製等により精製する。
上記した方法の概略を図2に示す。
【0075】
〔二本鎖ドライバーcDNAの調製〕
自体公知の方法によって目的とする試料とは別の試料から抽出されたmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーにおける、cDNA挿入断片両端のベクター由来の既知配列で設計された、それぞれ配列が異なる2種のプライマーを作製する。当該cDNAライブラリーを鋳型とし、作製した2種のプライマーを用いてPCR等によりドライバー遺伝子由来の5’末端及び3’末端にアダプター配列が付加された二本鎖cDNAを増幅する。要すれば、増幅されたアダプター付加二本鎖cDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
増幅されたアダプター付加二本鎖cDNAの、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までを、挿入断片両側のマルチクローニングサイトを利用して切断し、挿入断片のみからなる二本鎖ドライバーcDNA〔アダプターが結合(付加)していない二本鎖ドライバーcDNA〕を得る。
尚、要すれば、得られた二本鎖ドライバーcDNAを、カラム精製等により精製する。
上記した方法の概略を図3に示す。
【0076】
〔テスターcDNAとドライバーcDNAとのハイブリダイゼーション:工程(1)〕
調製した一本鎖テスターcDNAと、当該一本鎖テスターcDNAに対して過剰量の二本鎖ドライバーcDNAを、適当量のナトリウム塩を含む緩衝液に添加混合した後、適当な温度で適当な時間加熱して熱変性を行う。次いで、適当な温度で適当な時間ハイブリダイゼーションを行い、テスターcDNAとこれに相補的なドライバーcDNAとの二本鎖cDNAをハイブリッド形成させ、一本鎖テスター特異的cDNA、一本鎖テスター非特異的cDNAとこれと相補的な一本鎖ドライバーcDNAとの二本鎖ハイブリッド、再会合した二本鎖ドライバー特異的cDNA、再会合した二本鎖ドライバー非特異的cDNA、再会合しなかった一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び再会合しなかった一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有するハイブリダイゼーション溶液を得る。
上記した方法の概略を図4に示す。
【0077】
〔ハイブリッド形成した二本鎖cDNAの除去:工程(2)〕
得られたハイブリダイゼーション溶液に、例えば二本鎖特異的DNAヌクレアーゼ等の二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素を含有する溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、通常20℃〜80℃、好ましくは30℃〜75℃、より好ましくは50℃〜70℃、通常pH6〜9、好ましくは6.5〜8.5、より好ましくは7〜8で、通常0.5分〜60分、好ましくは1分〜45分、より好ましくは5分〜30分処理し(反応させ)、当該ハイブリダイゼーション溶液中に含有するハイブリッド形成した二本鎖cDNA(一本鎖テスター非特異的cDNAとこれと相補的な一本鎖ドライバーcDNAとの二本鎖ハイブリッド、再会合した二本鎖ドライバー特異的cDNA及び再会合した二本鎖ドライバー非特異的cDNA)を分解し、その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は反応停止剤での処理及び加熱処理を行うことによって当該DNA分解酵素の反応を停止させて、一本鎖テスター特異的cDNA、再会合しなかった一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び再会合しなかった一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有する反応液を得る。
尚、要すれば、これらcDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
上記した方法の概略を図5に示す。
【0078】
〔ハイブリッド形成しなかった一本鎖テスター特異的cDNAの増幅:工程(3)〕
得られた反応液に、上記した一本鎖テスターcDNAの調製において用いたのと同じ配列の2種のプライマー(但し、何れも5’末端はリン酸化されていない)、デオキシリボヌクレオチド三リン酸(dATP、dCTP、dGTP及びdTTP)、例えばTaqDNAポリメラーゼ等の重合酵素及びPCR用の緩衝液を適当量加える。(a)得られた溶液を適当な温度で適当な時間加熱して熱変性処理を行う。次いで、(b)当該溶液を冷却して一本鎖テスター特異的cDNA(2サイクル目以降は、一本鎖テスター特異的cDNAと増幅されたこれの相補鎖)にプライマーをアニールさせる。その後、(c)当該溶液を適当な温度で適当な時間処理し、当該重合酵素の作用によりプライマーを伸長させて一本鎖テスター特異的cDNAに対する相補鎖(2サイクル目以降は、一本鎖テスター特異的cDNAに対する相補鎖と、増幅された、一本鎖テスター特異的cDNAの相補鎖に対する相補鎖)を合成(増幅)する。以上の(a)〜(c)の工程を適当回数繰り返すことによってハイブリッド形成しなかった一本鎖テスター特異的cDNAとこれの相補鎖とを増幅し、増幅された二本鎖テスター特異的cDNA、一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有する反応液を得る。
尚、要すれば、これらcDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
上記した方法の概略を図6に示す。
【0079】
〔ハイブリット形成しなかった一本鎖ドライバーcDNAの除去:工程(4)〕
得られた反応液に、一本鎖特異的DNAヌクレアーゼ等の二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素を含有する溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、通常20℃〜60℃、好ましくは25℃〜50℃、より好ましくは30℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理し(反応させ)当該反応液中に含有する一本鎖ドライバーcDNA〔一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)〕を分解する。その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は/及び加熱処理を行うことによって当該DNA分解酵素の反応を停止させる。
尚、要すれば、得られた二本鎖テスター特異的cDNAを、カラム精製等により精製する。
上記した方法の概略を図7に示す。
【0080】
2−4.本発明の遺伝子変異同定方法
本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドは、例えば以下の如き、テスターにおける遺伝子変異の同定等の各種同定及びスクリーニング等に使用される。
【0081】
(1)当該テスター特異的ポリヌクレオチドの塩基配列を自体公知の塩基配列決定法により決定することにより、変異遺伝子の塩基配列の同定や新規な遺伝子の配列の同定を行うことができる。
(2)変異遺伝子が病原性であるのか否かの同定
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(3)変異遺伝子のゲノムDNA上の位置の同定
ヒト、マウス、ラット等ゲノムの全DNA配列が解読されているものでは、データベース等を用いて当該全DNA配列と変異遺伝子の塩基配列とを比較することによってゲノムDNA上の変異遺伝子の位置が同定できる。また、全DNA配列が解読されていない場合は、例えばFISH法等の自体公知の方法によりゲノムDNA上の変異遺伝子の位置が同定できる。
(4)疾病の診断同定・予測・予防
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(5)疾病治療の効果の同定や治療時期の同定
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(6)当該テスター特異的ポリヌクレオチドをプローブとして用いてcDNAライブラリーやESTライブラリー等についてスクリーニングを行えば、ある機能又は形質発現に関与する変異遺伝子をスクリーニングすることができる。このような方法としては、例えば標識物質により標識されたテスター特異的ポリヌクレオチド又はその相補鎖の一部若しくは全部を含有するオリゴヌクレオチドをプローブとし、DNAチップ上に固定化された上記のライブラリーとハイブリダイゼーションを行う方法等、自体公知のスクリーニング方法〔Beck MT, Holle L, Chen WY. Biotechniques. 2001 Oct;31(4):782-4, 786〕等が挙げられる。
(7)当該テスター特異的ポリヌクレオチド(特にゲノムDNA又はcDNA)をクローニングし、サブトラクティブゲノムDNA又はcDNAライブラリーとする。正常(健常者)試料から抽出したゲノムDNA又はcDNAと異常(罹病)試料から抽出したゲノムDNA又はmRNA群をプローブとしてハイブリダイゼーションを行い、異常(罹病)試料のゲノムDNA又はmRNA群のみに反応したゲノムDNA又はcDNAが未知病原体由来である可能性がある。即ち、これにより、未知病原体を検出できる可能性がある。
(8)当該テスター特異的ポリヌクレオチド(特に、cDNA)を、例えばプラスミドベクター等の適当なベクターに挿入し、該ベクターを用いて大腸菌等を形質転換することにより、テスターに特異的な遺伝子をクローニングすることができる。
(9)当該テスター特異的ポリヌクレオチド(特に、cDNA)或いはクローニングされたDNAの塩基配列を決定し、その塩基配列に基づいて設計されたプライマーを用いて3'及び5'RACE法を行うことにより変異遺伝子の全長cDNAを得ることができる。
(10)当該プライマーを用いたTAIL-PCRを行うか、当該テスター特異的ポリヌクレオチドをプローブとして用いてゲノミックライブラリーについてスクリーニングを行えば、ある機能又は形質発現において特異的に働くプロモーター(変異遺伝子のプロモーター)をスクリーニングすることもできる。
(11)ハイスループットな特異的発現遺伝子の同定
本発明の方法で得られたテスター特異的ポリヌクレオチド(特に、cDNA)を用いてマイクロアレイ(チップ)を作製し、これを用いることによって、例えば上記の(1)〜(7)の同定・解析を迅速且つ簡便に行うことができる。
【0082】
従って、本発明の方法には、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドを用いた、上記した如き各種同定法やスクリーニング法が包含される。尚、当該同定法やスクリーニング法は、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドを用いる以外は、自体公知の方法に従って実施すれば良く、そこで使用される試薬類等も自体公知の方法で用いられるものが使用できる。
従って、本発明の遺伝子変異同定方法には、例えば以下の(1)〜(9)のから選ばれる少なくとも1つの工程を含むものが好ましい。
(1)得られたテスター特異的ポリヌクレオチドの塩基配列を決定する工程、
(2)ノーザンブロット法、RT-PCR法、リアルタイムPCR法等により、テスターとドライバーにおける得られたテスター特異的ポリヌクレオチド(特定の変異遺伝子;mRNA)の発現量を確認し、それらを比較する工程、
(3)全DNA配列と得られたテスター特異的ポリヌクレオチド(変異遺伝子)の塩基配列とを比較し、ゲノムDNA上の得られたテスター特異的ポリヌクレオチド(変異遺伝子)の位置を同定する工程、又はFISH法等の自体公知の方法によりゲノムDNA上の得られたテスター特異的ポリヌクレオチド(変異遺伝子)の位置を同定する工程、
(4)得られたテスター特異的ポリヌクレオチドをプローブとして用いてcDNAライブラリーやESTライブラリー等についてスクリーニングを行う工程、
(5)得られたテスター特異的ポリヌクレオチド(特にゲノムDNA又はcDNA)をクローニングし、サブトラクティブゲノムDNA又はcDNAライブラリーとし、正常(健常者)試料から抽出したゲノムDNA又はcDNAと異常(罹病)試料から抽出したゲノムDNA又はmRNA群をプローブとしてハイブリダイゼーションを行う工程、
(6)得られたテスター特異的ポリヌクレオチド(特に、cDNA)を、例えばプラスミドベクター等の適当なベクターに挿入し、該ベクターを用いて大腸菌等を形質転換することにより、テスターに特異的な遺伝子をクローニングする工程、
(7)得られたテスター特異的ポリヌクレオチドをプローブ又はプライマーとして用いて、変異遺伝子の全長cDNAを得る工程、
(8)得られたテスター特異的ポリヌクレオチドをプライマーとして用いたTAIL-PCRを行うか、当該テスター特異的ポリヌクレオチドをプローブとして用いてゲノミックライブラリーについてスクリーニングを行い、ある機能又は形質発現において特異的に働くプロモーター(変異遺伝子のプロモーター)をスクリーニングする工程、
(9)得られたテスター特異的ポリヌクレオチド(特に、cDNA)を用いてマイクロアレイ(チップ)を作製し、これを用いて、上記の(1)〜(7)を行う工程。
また、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチド又はその相補鎖の一部若しくは全部を含有するオリゴヌクレオチドからなるプローブやプライマー及び本発明の方法により得ることができるcDNAライブラリーをも包含する。
【0083】
3.本発明のキット
本発明のキットは、上記した如き本発明の取得方法、増幅方法又は同定方法を実施するために使用されるものである。
このようなキットとしては、a)少なくとも先述した如き本発明の工程(2)においてハイブリッド形成された二本鎖ポリヌクレオチド(即ち、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を除去するために用いられる、一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素、好ましくは二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素、より好ましくは二本鎖特異的DNAヌクレアーゼを含んでなるものであり、b)好ましくは、更に、先述した如き本発明の工程(1’)において調製されたテスター由来の二本鎖ポリヌクレオチドを一本鎖ポリヌクレオチドにするために用いられる、二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素或いは2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群、好ましくは二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素、より好ましくはラムダ・エキソヌクレアーゼを含んでなるものであり、c)より好ましくは、先述した如き本発明の工程(4)において二本鎖テスター特異的ポリヌクレオチドと共存する一本鎖ポリヌクレオチド(一本鎖ドライバーポリヌクレオチド)を特異的に分解にするために用いられる、二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素、好ましくは二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素、より好ましくは一本鎖特異的DNAヌクレアーゼ、更に好ましくはエキソヌクレアーゼIを含んでなるものである。
これら構成要件の好ましい態様と具体例は上で述べた通りである。
【0084】
更に、上記以外の試薬類を加えて、本発明のキットとすることもできる。このような試薬類としては、例えば以下の如きa)〜i)から選ばれる少なくとも1種が挙げられるが、これらに限定されない。
a)一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素用の緩衝液(例えばナトリウム塩、要すればマグネシウム塩及びチオール化合物を含有するHEPES等のグッド緩衝液)、
a’)一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素の反応を停止するための溶液(例えばキレート剤等の反応停止剤を含有する、例えば水や上記した如き緩衝剤)、
b)二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素或いは2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群用の緩衝液(例えばマグネシウム塩及び界面活性剤を含有するグリシン等の緩衝液)、
c)二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素用の緩衝液(例えばマグネシウム塩及びチオール化合物を含有するグリシン等の緩衝液)、
d)ハイブリダイゼーション用緩衝液(例えばナトリウム塩を含有するグッド緩衝液等)、
e)ポリヌクレオチド抽出用試薬(例えば粉砕用緩衝液、フェノール、クロロホルム、SDS、β−メルカプトエタノール等)、
f)PCR用試薬(例えば重合酵素、重合酵素用緩衝液、1種又は2種以上のプライマー、ヌクレオチド混合物等)、
g)電気泳動用試薬(アガロース又はポリアクリルアミドゲル、ローディング緩衝液、臭化エチジウム染色用試薬等)、
h)PCR反応後の余剰プライマーの除去、ミス・アニーリングにより増幅されてしまった鎖長の短いDNAの除去、ヌクレアーゼにより完全に消化されなかった短いDNAの除去等のために用いられるポリヌクレオチド単離(分離)又は精製用試薬(例えば精製用カラム等)、
i)アルコール沈澱用試薬(例えばエタノール溶液、イソプロパノール溶液、高分子キャリアー、酢酸ナトリウム溶液等)。
【0085】
また、前述した如き本発明の取得、増幅、同定方法での使用のための説明書等を含ませておいても良い。当該「説明書」とは、本発明の方法における特徴・原理・操作手順等が文章又は図表等により実質的に記載されている当該キットの取り扱い説明書、添付文書、或いはパンフレット(リーフレット)等を意味する。
【0086】
本発明の方法は、以下のような効果を奏する。
(1)テスターポリヌクレオチド及びドライバーポリヌクレオチドをcDNAライブラリーから作製できる。
従来の方法では、サブトラクションcDNAの作製工程で環状プラスミドを除去することができなかったため、テスターポリヌクレオチド及びドライバーポリヌクレオチド共にcDNAからしか作製できず、cDNA作製の手間や作製できるcDNA量に限界がある等の問題があった。一方、本発明方法では、工程(2)において一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素を用いた酵素処理により二本鎖ポリヌクレオチドを除去できるので、テスターポリヌクレオチド及びドライバーポリヌクレオチド共にcDNAライブラリー(プラスミドベクター)から作製できる。
(2)ハイブリダイゼーション(サブトラクティブ・ハイブリダイゼーション)は一回でよい。
従来の方法においては、テスターポリヌクレオチドは二本鎖であったため、再会合したテスターポリヌクレオチド間の二本鎖ハイブリッド形成がされてしまうので、ドライバーポリヌクレオチドを過剰量使用しても、高発現しているテスター非特異的ポリヌクレオチド(ハウスキーピング遺伝子)が残ってしまい、後のPCR増幅に悪影響を及ぼす場合がある。そのため、従来法では、ハイブリダイゼーションを2〜3回以上繰り返す必要があった。これに対して、本発明では、テスターポリヌクレオチドは一本鎖であるので、テスターポリヌクレオチド間の二本鎖ハイブリッドは形成されず、ハイブリダイゼーションは一回行えば充分である。
(3)一回の酵素処理でテスター中に存在するポリヌクレオチドのうち、テスター非特異的ポリヌクレオチドを分解除去することができる。
従来の物理的結合又は吸着によるサブトラクション法においては、サブトラクティブ・ハイブリダイゼーションの結果得られた一本鎖ポリヌクレオチドとハイブリッド形成した二本鎖ポリヌクレオチドとを、物理的結合又は吸着により分離していたため、実際にはこれらを厳密に分離することができず、サブトラクト効率低下の要因となっていた。しかしながら、本発明では酵素処理によりハイブリッド形成した二本鎖ポリヌクレオチドを除去するため、一回の酵素処理で二本鎖ポリヌクレオチドを高効率に除去することができ、効率良く一本鎖テスター特異的ポリヌクレオチドを得ることができる。
(4)テスター特異的ポリヌクレオチドを容易に濃縮できる。
一本鎖テスター特異的ポリヌクレオチドにのみ存在し、ドライバー由来のポリヌクレオチド中に存在しない配列に基づいて設計・作製されたプライマーを用いたPCR等を行えば、ドライバーポリヌクレオチドは増幅されず、テスター特異的ポリヌクレオチドのみが実質的に増幅されるので、テスター特異的ポリヌクレオチドを濃縮することができ、効率的なサブトラクト・ポリヌクレオチドを作製することができる。
(5)ドライバーポリヌクレオチドを容易に除去できる。
本発明の工程(4)により、テスター特異的ポリヌクレオチドに共存しているドライバーポリヌクレオチドを容易に除去することができる。特に、二本鎖テスター特異的ポリヌクレオチドと一本鎖ドライバーポリヌクレオチドが共存している場合には、酵素処理により容易に一本鎖ドライバーポリヌクレオチドを除去することができる。その結果、テスター特異的ポリヌクレオチドの密度をさらに高めることができるので、従来法で問題となっていたドライバーポリヌクレオチドの混入(残存)による影響がさらに低減され、高効率なサブトラクション・ポリヌクレオチドが作製できる。
【0087】
以下に実施例及び比較例を挙げて本発明を詳細に説明するが、本発明はこれらにより何等限定されるものではない。
【実施例】
【0088】
(1)ヒト肝臓癌由来のHep G2細胞からの全RNAの抽出
ISOGEN(ニッポンジーン社製)を用い、その添付文書に従いHep G2細胞(ヒューマンサイエンス研究資源バンク製)から全RNAを抽出した。
Hep G2細胞を225mL培養フラスコ(Nalge Nunc社製)4本に5×106細胞/cm2まで培養し、リン酸緩衝液(pH7.5)で洗浄した。ISOGEN(ニッポンジーン社製)を1培養フラスコに対して15mL入れて、細胞を剥がし、それぞれ、遠心チューブ(Nalge Nunc社製、Oak Riddge Centrifuge Tube,50mL)に移した。それぞれにクロロホルムを3mL入れ、攪拌後、高速遠心機(日立社製、SCR20B)で12000rpm、4℃、15分間遠心分離して、それぞれ、上清を10mL回収した。上清を新しい遠心チューブに移し、10mLのイソプロパノール(和光純薬工業社製)を加えて攪拌し、高速遠心機(日立社製、SCR20B)で12000rpm、4℃、15分間遠心分離した。上清を除去し、沈殿を70% エタノール(和光純薬工業社(株)製)で洗浄し、乾燥後、滅菌水に溶解して、分光光度計(Beckman社製、DU640)にてUV 260nmでRNA濃度を測定した。濃度測定後、滅菌水で2μg/μLに調製したRNA溶液を250μL(500μg)得た。
【0089】
(2)Hep G2細胞由来の全RNAからのPoly(A)+ RNAの抽出
上記(1)で得られたHep G2細胞全RNA 500μgを使用してOligotex-dT 30 Super(タカラバイオテク社製)を用い、その添付文書に従い、Poly(A)+ RNAを抽出した。
Hep G2細胞全RNA 250μL(500μg)に、2×Elution Buffer(2mM EDTA及び0.2% SDS含有20mM Tris-HCl緩衝液、pH7.5)を250μL加えて、混合した。その後、Oligotex-dT 30 Superを加えて、混合し、65℃、5分間加熱後、氷中に急冷した。5M 塩化ナトリウムを100μL加えて、37℃で、10分間インキュベートし、高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、5分間遠心分離した。上清を除去し、滅菌水を400μL加えて、混合し、65℃で、5分間加熱後、高速微量遠心機(日立社製、CF16RX)で、14500rpm、室温で、5分間遠心分離した。上清を回収し、5M 塩化ナトリウムを20μL及びエタノールを1mL加えて、混合し、高速微量遠心機(日立社製、CF16RX)で、14500rpm、4℃で、10分間遠心分離した。上清を除去し70% エタノールで洗浄し、乾燥後、滅菌水に溶解して、分光光度計(Beckman社製、DU640)にてUV 260nmでRNA濃度を測定した。
濃度測定後、滅菌水で0.25μg/μLに調製したPoly(A)+ RNA溶液を28μL(7μg)得た。
【0090】
(3)Hep G2細胞のcDNAライブラリーの作製
上記(2)で得られたHep G2細胞由来のPoly(A)+ RNA 5μgをとり、TimeSaver cDNA Synthesis Kit(Amersham社製)を使用して、その添付文書に従い作製した。ベクターはpNEB193(New England Biolabs 社製)を使用し、形質転換はDH5αを使用した。また、プラスミドライブラリーは、100 mlのLuria-Bertani’s broth(アンピシリンナトリウム 100 μg / ml を含む)で培養し後、回収した。
First-Strand Reaction Mixes(mersham社製)に、65℃、10分間加熱後氷中で急冷した、上記(2)で得られたHep G2 Poly(A)+ RNA 5μg(20μL)、Oligo-dT(24)-Pac I site Primer(5'-TTTTTTTAATTAATTTTTTTTTTTTTTTTTTTTTTTT-3'、シグマジェノシス社製、0.5 mg/ml) 1μL及びDTT Solution(Amersham社製) 1μLを混合し、37℃で、1時間インキュベートして、第一鎖反応(逆転写反応)を行った。Second-Strand Reaction Mixes(Amersham社製)に、第一鎖反応液を混合し、12℃で、30分インキュベート後、22℃、1時間インキュベートして第ニ鎖反応を行った。
次いで、65℃、10分加熱して、第ニ鎖反応を停止させ、SizeSep 400 Spun Columns(Amersham社製)にてPrimer及び低分子DNAを除去し、二本鎖cDNAを得た。
作製した二本鎖cDNA 100μLに、EcoR I/Not I adaptor(Amersham社製)5μL、PEG Buffer(Amersham社製)30μL、ATP Solution(Amersham社製、15mM)1μL及びT4 DNA Ligase(5units )(Amersham社製)1μLを加え、16℃で、1時間インキュベートした後、65℃で、10分加熱してT4 DNA Ligaseを失活させた。得られた反応液(アダプターを結合させたcDNAを含有する溶液)137μLに、フェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を140μL加えて、攪拌し、高速微量遠心機(日立社製、CF16RX)で、14500rpm、室温で、3分間遠心分離した。上清を回収し、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)7μL及びエタノール(和光純薬工業社製)350μLを加えて、混合して、高速微量遠心機(日立社製、CF16RX)で、14500rpm、4℃で、10分間遠心分離した。上清を除去し、70% エタノール(和光純薬工業社製)で洗浄して、乾燥後、滅菌水85μLに溶解した。滅菌水に溶解したcDNAに、10×NEB buffer1(New England Biolabs 社製)10μL、BSA溶液(New England Biolabs 社製)1μL及びPac I(New England Biolabs 社製、10units/μL)4μLを加えて、混合し、37℃で、2時時間インキュベートした後、65℃、20分間加熱し、Pac Iを失活させた。
得られた反応液(Pac Iで切断したcDNA含有溶液)を、SizeSep 400 Spun Columns(Amersham社製)により処理し、未結合のアダプター及び低分子DNAを除去し、全量150μLになるように滅菌水で調製した。これにより、5'末端がEcoR I、3'末端がPac Iの切断末端となったcDNAが得られた。
得られたcDNA 3μL(Poly(A)+ RNAに換算して約100ng)、EcoR I(ニッポンジーン社製)及びPac I(New England Biolabs 社製)で切断したpNEB193 DNA(New England Biolabs 社製)100ng(2μL)、及びDNA Ligation Kit Ver.2.1(タカラバイオテク社製)I液(Reaction Buffer)5μLを混合し、16℃で、1時間インキュベートした後、全量(10μL)をDH5α(100μL)に42℃で、30秒の熱ショックによって形質転換した。形質転換したcDNAライブラリーを100 mlのLuria-Bertani’s broth(アンピシリンナトリウム 100μg/mlを含む)で37℃、16時間培養し、一方向に挿入されたHep G2細胞プラスミドcDNAライブラリーを回収した。
得られたcDNAライブラリーの一部を滅菌水で10ng/μLに調製して、下記(4)のテスターcDNAの調製におけるPCR増幅反応の鋳型とした。
【0091】
(4)テスターcDNAの調製
上記(3)で調製したヒト肝臓癌由来のHepG2細胞のcDNAライブラリーを鋳型として、挿入断片cDNAをTOPOTAQ DNA Polmerase(Fidelity Systems 社製)を使用してPCR増幅した。
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
【0092】
・ヒト肝臓癌由来のHepG2細胞のcDNAライブラリー溶液:1μL(10ng)
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・M13/pUC Forwad Primer溶液:5μL
リン酸化プライマー(5’-P-CCAGTCACGACGTTGTAAAACG-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・M13/pUC Reverse Primer溶液:5μL
非リン酸化プライマー(5’-CACACAGGAAACAGCTATGACC-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:4μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
・滅菌水:30μL
【0093】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 30 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、44μLの滅菌水に溶解した。
次いで、溶解液に、10×Lambda Exonuclease Buffer(EPICENTRE 社製、25mM塩化マグネシウム及び0.1%Triton-X100含有670mMグリシン-KOH緩衝液、pH9.4)5μL及びLambda Exonuclease(EPICENTRE 社製、10units/μL)1μLを加えて、混合し、37℃、30分インキュベートした。次いで、更に80℃、15分インキュベートしてLambda Exonucleaseを失活させた。
これに、50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns (Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が1ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 ×37μg/mL ×(希釈倍率)/ 1000
【0094】
(5)ドライバーcDNAの調製
ヒト正常肝臓組織由来のcDNAライブラリー(BioChain 社製、cDNA Library: Human Adult Normal Tissue: Liver)を鋳型として、挿入断片cDNAをPCR増幅した。
ヒト正常肝臓組織由来のcDNAライブラリー溶液(BioChain 社製、cDNA Library: Human Adult Normal Tissue: Liver)1μL(10ng)を使用した以外は、上記(4)と同じ試薬を用いた。
各試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 30 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、10μLの滅菌水に溶解した。
得られたPCR増幅産物5μgを滅菌水で全量43μLに調製し、10×H Buffer(ニッポンジーン社製)5μL、EcoR I(ニッポンジーン社製、10units/μL)1μL及びXho I(ニッポンジーン社製、10units/μL)1μLを加えて、混合し、37℃で、2時間インキュベートして、挿入断片両側のマルチクローニング・サイトの制限酵素を使用してPCR Primerまでを切断した。その後、65℃で、20分間加熱して、制限酵素を失活させた。
次いで、これに50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns(Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が100ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 × 50μg/mL ×(希釈倍率)/ 1000
【0095】
(6)ハイブリダイゼーション
上記(4)で調製したテスターcDNA 1μL(1ng)及び上記(5)ドライバーcDNA 2μL(200ng)含有溶液 3μLに、1μLの4×Hybridization Buffer(2M NaCl含有200mM HEPES緩衝液、pH7.3)を加えて、混合し、これをサーマルサイクラー(MJ Research 社製、PTC-225)にて98℃で2分間熱変性した後、68℃で15時間、ハイブリダイゼーションした。
【0096】
(7)二本鎖特異的DNAヌクレアーゼ処理によるハイブリッド形成した二本鎖cDNAの除去
68℃に保温したハイブリダイゼーション溶液に、68℃に温めた5μL の2×DSN master Buffer(Evrogen 社製、10mM 塩化マグネシウム及び2mM DTT 100mM含有トリス塩酸緩衝液 pH8.0、)を加え、混合した後、68℃で10分間インキュベートした。これに、二本鎖特異的DNAヌクレアーゼ(Duplex-specific nuclease;Evrogen社製、1 kunitz-units/μL)1μLを加えて、68℃で30分、インキュベートした。
次いで、これに、Stop Solution(5mM EDTA水溶液)10μLを加えて、68℃で5分間インキュベートして二本鎖特異的DNAヌクレアーゼの反応を停止させた。
得られた反応液に、80μL の滅菌水及び等量(100μL)のフェノール:クロロホルム:イソアミルアルコール混合液(25:24:1)混合液(ニッポンジーン社製)を加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、31μLの滅菌水に溶解した。
【0097】
(8)ハイブリッド形成しなかったテスターcDNAのPCR増幅
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
【0098】
・上記(7)で調製した一本鎖テスターcDNA含有反応液:31μL
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・M13/pUC Forwad Primer溶液:5μL
リン酸化プライマー(5’-P-CCAGTCACGACGTTGTAAAACG-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・M13/pUC Reverse Primer溶液:5μL
非リン酸化プライマー(5’-CACACAGGAAACAGCTATGACC-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:4μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
【0099】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 35 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、44μLの滅菌水に溶解した。
【0100】
(9)Exonuclease I処理によるハイブリッド形成しなかった一本鎖ドライバーcDNAの除去
上記(8)で調製したPCR増幅産物(テスターcDNA増幅産物) 44μLに、10×Exonuclease I Buffer(New England Biolabs 社製、67mM 塩化マグネシウム及び100mM 2-メルカプトエタノール含有670mMグリシン-KOH緩衝液、pH9.5)5μL及びExonuclease I(New England Biolabs 社製、20units/μL)1μLを加えて、混合し、37℃、30分、インキュベートした。次いで、更に80℃、15分インキュベートしてExonuclease Iを失活させた。
これに、50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns (Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が10ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 × 50μg/mL ×(希釈倍率)/ 1000
【0101】
(10)サブトラクションcDNAの評価検討
上記(4)で得られたテスターcDNA(Hep G2 cDNA)、上記(5)で得られたドライバーcDNA(正常肝臓 cDNA)又は上記(9)で得られたテスター特異的cDNA(サブトラクションcDNA)10ngを鋳型として、高発現ハウスキーピング遺伝子である glyceraldehyde-3-phosphate dehydrogenase(GAPDH)(増幅鎖長:859bp)とHep G2特異的発現遺伝子であるalpha-fetoprotein(AFP)(増幅鎖長:931bp)をそれぞれPCR増幅した。
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ分注した。
【0102】
・cDAN含有溶液:1μL(10ng/μL cDNA含有)
上記(4)で調製した一本鎖テスターcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの、
上記(5)で調製した二本鎖ドライバーcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの、又は
上記(9)で得られたサブトラクションcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの。
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・Primer1溶液:1μL
GAPDH増幅用プライマー1(5’-GAGTACGTCGTGGAGTCCACTG-3’)又はAFP増幅用プライマー1(5’-GTACGGACATTCAGACTGCTG-3’)を10μMとなるように滅菌水に溶解したもの。
・Primer2溶液:1μL
GAPDH増幅用プライマー2(5’-CCTCACAGTTGCCATGTAGAC-3’)又はAFP増幅用プライマー2(5’-CATCCAGGAGAGCCAAGCATTG-3’)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:1μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
・滅菌水5μL
【0103】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 20秒 22 サイクル、72℃ 2分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR 増幅産物(20μL)10μLを、1×TAE Bufferを使用して1.5 % アガロースゲル(ニッポンジーン社製)で電気泳動後、エチジウムブロマイド染色により各遺伝子の増幅量を比較した。
その結果を図8に示す。尚、図中、レーン1は(4)で調製したテスターcDNA(ヒト肝臓癌由来のHepG2細胞のcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン2は(4)で調製したテスターcDNA(ヒト肝臓癌由来のHepG2細胞のcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果を、レーン3は(5)で調製したドライバーcDNA(ヒト正常肝臓組織由来のcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン4は(5)で調製したドライバーcDNA(ヒト正常肝臓組織由来のcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果を、レーン5は(9)で得られたテスター特異的cDNA(サブトラクションcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン6は(9)で得られたテスター特異的cDNA(サブトラクションcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果をそれぞれ示し、レーン7は分子量マーカー:100bp DNA Step Ladder (100-1.5kbp)(和光純薬工業(株)製)を用いた結果を示す。また、「←1500bp」は1500bpのDNAの泳動位置を、「←1000bp」は1000bpのDNAの泳動位置を、「←500bp」は500bpのDNAの泳動位置を、それぞれ示す。
【0104】
図8から明らかなように、高発現ハウスキーピング遺伝子であるGAPDHはHep G2 cDNA及び正常肝臓組織 cDNAの両者ともPCR増幅が確認できたが、サブトラクションcDNAでは確認できないことが判る。一方、Hep G2特異的発現遺伝子であるAFPは正常肝臓組織 cDNA において、PCR増幅が確認できないが、サブトラクション cDNAにおいてはHep G2 cDNAと同程度のPCR増幅を示した。
以上のことから、本発明によればテスター特異的cDNA(サブトラクション cDNA)を容易に且つ短時間に、高効率に取得し得ることが判る。
【技術分野】
【0001】
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを取得・増幅する方法に関する。
【背景技術】
【0002】
機能、性質の異なる細胞又は組織間においては、発現量の異なる遺伝子が存在しており、その細胞又は組織の機能解析において重要な役割を果たしている可能性がある。このような遺伝子を取得することは、その機能や発現機構の解明等に有用である。
【0003】
一方、機能、性質の異なる細胞又は組織間において、発現量の異なる遺伝子をクローニングする方法として、cDNA サブトラクション法がある。
このcDNA サブトラクション法は、以下の手順で行われる。
例えば癌細胞組織の特異的な機能、性質を解析する場合、
(1)先ず、癌細胞組織から調製した一本鎖RNA又はcDNA(テスター)と正常細胞組織から調製された過剰量の一本鎖RNA又はcDNA、若しくは二本鎖cDNA(ドライバー)とをハイブリダイゼーションさせる。この時、テスターとドライバーともに発現している遺伝子(ハウスキーピング遺伝子など)は二本鎖cDNA又はRNA-DNAハイブリッドを形成するが、テスターに特異的に発現している遺伝子由来のRNA又はcDNAはハイブリッドを形成せず、一本鎖RNA又はcDNAの状態である。
(2)次いで、ハイブリッドを形成しなかったテスターに特異的に発現している遺伝子由来の一本鎖RNA又はcDNAを、例えばハイドロキシアパタイト・クロマトグラフィー法(Hendrick, S.M.,Cohen, D.I., Nielsen, E.L. and Davis, M.M. (1984) Nature. 308, 149-153)、アビジン・ビオチン結合法(Duguin, J.R. and Dinauer, M.C. (1990) Nucleic Acid Research. 18, 2789-2792、特表平9−507021号公報)、ラテックス、ビーズ、カラム等を用いたオリゴ (dT)30固相法(Hara, E., Kato T., Nakada, S., Sekiya, L and Oda, K. (1991) Nucleic Acid Research. 19, 7097-7104、特開2001−269172号公報)、アビジン・ビオチン・磁気ビーズ法(特開2001−269172号公報、特開2002−253237号公報)等の方法により、ハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドと分離し、テスター特異的に発現している遺伝子を濃縮する。
【0004】
しかしながら、上記の方法は何れも物理的結合又は吸着を利用して、テスターに特異的に発現している遺伝子由来の一本鎖RNA又はcDNAとハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドとを分離するため、これらを厳密に分離することはできず、ハイブリッド形成した二本鎖cDNA又はRNA-DNAハイブリッドが混在してしまうという問題があった。その結果、上記の方法では、テスターおよびドライバー両方に発現している遺伝子(例えばハウスキーピング遺伝子など)の混入が多く、高効率にテスター特異的に発現している遺伝子を濃縮(取得)することはできなかった。また、上記方法は、操作が煩雑で作業工程が多く、時間と熟練を要するという問題もあった。
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅する方法、当該方法により得られた(増幅された)ポリヌクレオチド、テスターにおける遺伝子変異を同定する方法、並びにこれらに用いられるキットを提供する。
【課題を解決するための手段】
【0006】
本発明は、以下の構成よりなる。
1.以下の工程を含む、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを増幅する方法;
(1)(i)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか、或いは(ii)テスター由来の一本鎖ポリヌクレオチドと、ドライバー由来の二本鎖ポリヌクレオチドとを混合して、熱変性後、ハイブリダイゼーションを行うかして、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成する工程、
(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、
(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び
(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程。
2.上記1の方法により増幅された(テスターに特異的に存在する)ポリヌクレオチドを同定することを特徴とするテスターにおける遺伝子変異を同定する方法。
【0007】
即ち、本発明者らは上記目的を達成すべく鋭意研究を重ねた結果、サブトラクティブハイブリダイゼーションにより形成された、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを、酵素処理により除去することにより、テスターでの存在量が別のドライバーでの存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅し得ることを見出し、本発明を完成させるに至った。
【発明の効果】
【0008】
本発明の方法により、テスターでの存在量がドライバーでの存在量よりも多いポリヌクレオチドを、容易に且つ短時間に、高効率に取得・増幅することが可能であり、その結果、テスターにおける遺伝子変異を高精度に同定することが可能となる。
【図面の簡単な説明】
【0009】
【図1】本発明の取得方法又は増幅方法を示すチャート図である。
【0010】
【図2】本発明の一本鎖テスターcDNAの調製工程を模式的に示した図である。
【0011】
【図3】本発明の二本鎖ドライバーcDNAの調製工程を模式的に示した図である。
【0012】
【図4】本発明のテスターcDNAとドライバーcDNAとのハイブリダイゼーション工程〔工程(1)〕を模式的に示した図である。
【0013】
【図5】本発明のハイブリッド形成した二本鎖cDNAの除去工程〔工程(2)〕を模式的に示した図である。
【0014】
【図6】本発明のハイブリッド形成しなかった一本鎖テスター特異的cDNAの増幅工程〔工程(3)〕を模式的に示した図である。
【0015】
【図7】本発明のハイブリッド形成しなかった一本鎖ドライバーcDNAの除去工程〔工程(4)〕を模式的に示した図である。
【0016】
【図8】実施例で得られた、ヒト肝臓癌由来のHepG2細胞のcDNA(テスターcDNA)、ヒト正常肝臓組織由来のcDNA(ドライバーcDNA)及びサブトラクションcDNA(テスター特異的cDNA)をそれぞれ鋳型として、glyceraldehyde-3-phosphate dehydrogenase(GAPDH)遺伝子とalpha-fetoprotein(AFP)遺伝子とをPCR増幅して得られたPCR産物を電気泳動した結果である。
【発明を実施するための最良の形態】
【0017】
1.試料
本発明において、「ある試料(テスター)」及び「別の試料(ドライバー)」とは、両者がそれぞれ異なることを意味し、例えば(1)試料の種類は異なるが、試料の由来〔生体(個体・集団)、場所等〕は同一である場合、(2)試料の種類は同一であるが、試料の由来が異なる場合、(3)試料の種類及び試料の由来が異なる場合、(4)試料の種類及び試料の由来が同一である場合である。
より具体的には、例えば(1)同一個体(生体)における異なる種類の細胞、組織、器官、体液等、(2)異なる個体(生体・商品)間(特定個体と標準(正常)個体間)又は特定の個体(生体・商品)と集団間(特定個体と標準(正常)集団間)における同じ種類の細胞、組織、器官、体液等、(3)異なる個体間(罹患個体と標準(正常)個体間)又は特定個体と集団間(罹患個体と標準(正常)集団間)における異なる種類の細胞、組織、器官、体液(異常細胞と正常細胞、異常組織と正常組織、異常器官と正常器官、異常体液と正常体液等)等、(4)環境、状態等が異なる場合(薬剤投与前後、ストレス付加前後、病態の異なるステージ等)の同一個体(生体)における同一種類の細胞、組織、器官、体液等
【0018】
本発明が適用される試料の種類としては、この分野で用いられているものであれば良く、特に限定されない。このような試料としては、例えば細胞、組織、器官、体液(血液、血清、血漿、髄液、滑液、膵液、リンパ液等)、排泄物(尿、唾液、糞便等)、喀たん、膿、皮膚由来物等の生体由来試料;微生物(菌類、細菌類、ウイルス等);例えば環境試料(食品、飲料、水道水、海水、湖沼水、河川水、工場廃液、半導体用洗浄水、医療器具等を洗浄した後の洗浄液等);これらから得られる抽出液;及びこれらを水や通常この分野で用いられている緩衝液等(トリス緩衝液、グリシン緩衝液、リン酸緩衝液、ベロナール緩衝液、ホウ酸緩衝液、グッド緩衝液等)に適宜溶解させて再構成して得られた処理物等が挙げられる。
【0019】
本発明において、「ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多い」とは、(1)テスターには存在するがドライバーには存在しない場合や、(2)テスターとドライバーの両方に存在はしているものの、テスターでの存在量がドライバーでの存在量に比較して多い場合等を意味する。尚、「テスターに特異的に存在する」や「テスター特異的」も同じ意味である。
【0020】
本発明の方法により、取得・増幅される「ポリヌクレオチド」(即ち、「ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチド」)には、例えばmRNA等のRNA;当該mRNAに由来するcDNA、ゲノムDNA等のDNA等が含まれる。なかでも、本発明の方法は、mRNAに由来するcDNA、ゲノムDNA等のDNAを取得・増幅するのに有効であり、特にテスターでの存在量がドライバーでの存在量よりも多いmRNA由来のcDNAを取得・増幅するのに極めて有効である。
【0021】
2.本発明の方法
本発明は、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチド(以下、テスター特異的ポリヌクレオチドと略記する。)を取得又は増幅する方法に関する。
本発明の当該ヌクレオチドの取得方法は、(1)テスター由来の一本鎖ポリヌクレオチドを用いてドライバー由来の一本鎖又は二本鎖ポリヌクレオチドとハイブリダイゼーションを行って、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成させること、及び(2)酵素処理により、ハイブリッド形成した二本鎖ポリヌクレオチドを除去することを特徴とする。
更に、当該二本鎖ポリヌクレオチドの除去後、ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅させることにより、テスターでの存在量がドライバーでの存在量よりも多いポリヌクレオチドを大量に得ることができる。
即ち、本発明の当該ヌクレオチドの増幅方法は、(1)テスター由来の一本鎖ポリヌクレオチドを用いてドライバー由来の一本鎖又は二本鎖ポリヌクレオチドとハイブリダイゼーションを行って、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成させること、(2)酵素処理により、ハイブリッド形成した二本鎖ポリヌクレオチドを除去すること、及び(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅することを特徴とする。
【0022】
2−1.本発明のテスター特異的ポリヌクレオチドの取得方法
(1)テスター由来の一本鎖ポリヌクレオチド
本発明におけるテスター由来のポリヌクレオチド(以下、テスターポリヌクレオチドと略記する。)は一本鎖ポリヌクレオチドである。当該一本鎖ポリヌクレオチドは、DNA及びRNAのどちらでも良いが、好ましくはDNAである。なかでも、ゲノムDNA及びテスターに存在するmRNAに由来するcDNA(換言すれば、テスターから抽出したmRNAを鋳型として合成されたcDNA)が好ましく、特にcDNAが好ましい。
【0023】
本発明の一本鎖テスターポリヌクレオチドは、前述した如き試料から適宜調製できる。一本鎖ポリヌクレオチドの調製は、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ、市販のキットを用いて行うこともできる。
このような方法としては、通常この分野で用いられている方法であれば特に限定されず、前述した如き試料から直接一本鎖ポリヌクレオチドを調製する方法〔例えば、Vieira J, Messing J. Methods in Enzymology. 1987;153:3-11;Krieg PA, Melton DA. Methods in Enzymology. 1987;155:397-415;Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Proc Natl Acad Sci U S A. 1984 Apr;81(7):2035-9等〕、前述した如き試料から一旦二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865;Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕、PCRプライマーの片側だけビオチン標識したプライマーを使用してPCR増幅し、それをマグネチック・ストレプトアビジン・ビーズと混合して一本鎖DNAを回収する方法等が何れも使用できる。また、市販のキットを用いて行うこともできる。ここで、二本鎖ポリヌクレオチドの調製も、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ〔例えば、Kazuo Maruyama and Sumio Sugano Gene, 138 ( 1994 ) 171-174;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 ( 1996 );Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000 Jun 15;28(12):E63;Shimada M, Hino F, Sagawa H, Mukai H, Asada K, Kato I. Rinsho Byori. 2002 May;50(5):528-32等〕、市販のキットを用いて行うこともできる。
尚、上記において、調製される一本鎖ポリヌクレオチド及び二本鎖ポリヌクレオチドは、テスター中に存在するポリヌクレオチドそのものでも、また、当該ポリヌクレオチドを鋳型として、例えばポリメラーゼ連鎖反応(PCR)等の自体公知の増幅方法により増幅された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドであってもよい。
【0024】
本発明においては、上記した如き一本鎖テスターヌクレオチドを得る方法のなかでも、二本鎖ポリヌクレオチドを調製した後、酵素処理等により一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865;Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕が好ましく、特に、テスター中に存在するポリヌクレオチドを鋳型として、例えばPCR等の自体公知の増幅方法により、当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅した後、酵素処理により一本鎖ポリヌクレオチドにする方法〔例えば、Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕が好ましい。
従って、本発明の一本鎖テスターヌクレオチドを得る方法は、(1’)テスター由来の二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましく、特に(1”)テスター由来の二本鎖ポリヌクレオチドを増幅した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましい。
尚、上記において、酵素処理の際に用いられる酵素としては、通常この分野で用いられるものであれば良く特に限定されないが、例えばラムダ・エキソヌクレアーゼ等の二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素、例えば2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群(例えば5'突出末端を生じ得る制限酵素、3'突出末端を生じ得る制限酵素及びExonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素の組み合わせ等)等が挙げられる。尚、これら酵素の由来は特に限定されず、また、自体公知の方法で得られたもの及び市販されているものの何れも使用できる。
【0025】
また、上記した如き方法において使用される試薬類や反応条件等も自体公知の方法に準じて行えばよい。
分解酵素の使用量は、二本鎖ポリヌクレオチドの片側の鎖を充分に分解して一本鎖ポリヌクレオチドとし得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば酵素としてラムダ・エキソヌクレアーゼを用いる場合には、通常DNA2μgに対して0.1units〜100units、好ましくは0.5units〜50units、より好ましくは1units〜10unitsである。
当該酵素処理の条件(温度、時間、pH等)、即ち、二本鎖ポリヌクレオチドとラムダ・エキソヌクレアーゼとを反応(接触)させる際の条件は、通常25℃〜60℃、好ましくは30℃〜50℃、より好ましくは35℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜250mM、より好ましくは10mM〜100mMから適宜選択される。
【0026】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を用いる方法又は/及び加熱処理を行う方法により行えばよい。
反応停止剤を用いる方法としては、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液と、酵素処理を行って得られた反応液と混合させれる等により、当該反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
また、加熱処理により反応を停止する方法としては、酵素処理を行って得られた反応液を、通常60℃〜95℃、好ましくは70℃〜90℃、より好ましくは80℃〜85℃で処理すればよい。
反応停止剤による反応時間及び加熱処理時間は、通常5分〜60分、好ましくは10分〜30分、より好ましくは15分〜20分である。
上記の反応停止方法のうち、反応停止剤を用いる方法と加熱処理による方法の両者を併用するのが好ましい。
【0027】
尚、二本鎖ポリヌクレオチドとラムダ・エキソヌクレアーゼとを反応させる際には、通常この分野で用いられる活性化剤(例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウム塩に由来するマグネシウムイオン等の金属イオン等)、界面活性剤(例えばTriton-X100(ポリオキシエチレン(10)オクチルフェニルエーテル)等)、安定化剤(ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を存在させておいても良い。特に、マグネシウム塩及び界面活性を存在させるのが好ましい。尚、これらの使用量は通常この分野で使用される範囲から適宜選択し得、具体的には、マグネシウム塩の使用量としては、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMであり、界面活性剤の使用量としては、通常0.001〜1%(w/v)、好ましくは0.005〜0.5%(w/v)、より好ましくは0.01〜0.1%(w/v)である。
【0028】
更に、本発明で使用される一本鎖テスターポリヌクレオチドとしては、その5’末端又は/及び3’末端に、既知配列からなるアダプターが結合している(付加されている)ものが好ましく、特に当該アダプターは5’末端及び3’末端の両末端に結合(付加)させるのが好ましい。アダプターはいかなる配列を含んでいても良いが、その塩基数はそれ自体が単独でプライマーとして機能し得る程度が好ましい。従って、アダプターの塩基数は、通常15bp〜60bp、好ましくは20bp〜35bp、より好ましくは25bp〜30bpである。
尚、5’末端及び3’末端の両末端にアダプターを結合(付加)させる場合は、両末端のアダプターが相補的であると、一本鎖テスターポリヌクレオチド内でのアダプターハイブリッドを形成するおそれがあるので、相補的ではないアダプターを両末端に結合させるのが好ましい。また、当該アダプターは、ドライバー由来のポリヌクレオチド中に存在しない配列からなるものが好ましい。
このように一本鎖テスターポリヌクレオチドに上記した如きアダプターを結合(付加)させておくことにより、本発明のテスター特異的ポリヌクレオチドの取得方法で所得されたポリヌクレオチドを、ドライバー由来の(一本鎖)ポリヌクレオチドから容易に単離・精製することができる。
また、上記した如きアダプターは、後述する本発明のポリヌクレオチドの増幅方法において、本発明のテスター特異的ポリヌクレオチドの取得方法で所得されたポリヌクレオチド(ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチド)を増幅する際に有用である。即ち、当該アダプターを後述する工程(3)で行うPCR等の増幅反応用のプライマーが結合する既知の配列とすることにより、当該アダプターに結合するプライマーを用いて容易にテスター特異的ポリヌクレオチドを増幅することができる。
【0029】
一本鎖テスターポリヌクレオチドにアダプターを結合(付加)させる方法としては、通常この分野で用いられている自体公知の方法を適宜選択して用いることができ、市販のキットを用いて行うこともできる。
このような方法としては、通常この分野で用いられている方法であれば特に限定されず(1)上記した如き方法により調製された一本鎖ポリヌクレオチドにアダプターを結合(付加)させる方法〔例えば、Oligo-capping:a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides, Kazuo Maruyama and Sumio Sugano, Gene, 138 (1994) 171-174に記載のオリゴ-キャッピング法;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 (1996)に記載のSMART法;High-efficiency full-length cDNA cloning by biotinylated CAP trapper, Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C., Genomics. 1996 Nov 1;37(3):327-36に記載のGene Trapper法等〕、(2)上記した如き方法において、調製された二本鎖ポリヌクレオチドにアダプターを結合(付加)させた後、これを一本鎖ポリヌクレオチドにする方法〔例えば、Determination of 5' ends of specific mRNAs by DNA ligase-dependent amplification, Bertling WM, Beier F, Reichenberger E., PCR Methods Appl. 1993 Oct;3(2):95-9に記載のRACE法等〕、(3)上記方法において、テスター中に存在するポリヌクレオチドを鋳型として、例えばアダプター配列に基づいて設計されたプライマーを用いたPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、アダプター(プライマー)が結合(付加)された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法等が挙げられる。
尚、上記方法(3)において、増幅されるポリヌクレオチドが二本鎖の場合は、当該二本鎖ポリヌクレオチドを上記した如き方法により一本鎖ポリヌクレオチドにする必要がある。この場合、使用する2種類のプライマーのうち片側のみの5’末端をリン酸化しておけば、増幅された二本鎖ポリヌクレオチドを二本鎖DNAの5’末端リン酸化DNA鎖を除去するラムダ・エキソヌクレアーゼ等で処理することにより、5’末端リン酸化プライマー側のcDNA鎖を除去することができ、容易にアダプターが結合した(付加された)一本鎖テスターポリヌクレオチドを得ることができる。
本発明においては、上記した如き一本鎖テスターポリヌクレオチドにアダプターを結合(付加)させる方法のなかでも、テスター中に存在するポリヌクレオチドを鋳型として、例えばアダプター配列に基づいて設計されたプライマーを用いたPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、アダプター(プライマー)が結合(付加)された当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法が好ましい。
従って、本発明において、アダプターが結合した(付加された)一本鎖テスターヌクレオチドを得る方法は、(1’’’)テスター由来の二本鎖ポリヌクレオチドをアダプター配列に基づいて設計されたプライマーを用いて増幅した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、を含んでなる方法が好ましい。
【0030】
例えば以下の方法により、アダプターが結合した(付加された)一本鎖テスターポリヌクレオチドを得ることができる。
(1)自体公知の方法〔Gublerらの方法(Gulber, U. and Hoffman, B. J. (1983) Gene. 25, 263-269)等〕によってテスターから抽出したmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーを用い、cDNA挿入断片両端のベクター由来の既知配列で設計された5’末端がリン酸化されたプライマーとリン酸化されていないプライマーの2種のプライマーを作製し、これを用いてベクターに挿入されているcDNAを鋳型として、PCR法、LAMP法、ICAN法等によりテスター遺伝子由来の二本鎖cDNAを増幅する。次いで、増幅されたプライマー(アダプター)付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る〔Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕。
(2)既知配列(アダプター)で設計された5’末端がリン酸化されたプライマーとリン酸化されていないプライマーの2種のプライマーを作製し、これを用いてオリゴ・キャッピング法に従って5’末端及び3’末端にアダプター配列が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR法、LAMP法、ICAN法等により増幅し、次いで、増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る〔Higuchi RG, Ochman H. Nucleic Acid Research. 1989 Jul 25;17(14):5865等〕。
(3)適当なプラスミドに目的とするポリヌクレオチドを挿入し、プラスミドのマルチクローニングサイトを利用して挿入されたポリヌクレオチド断片の片方を5'突出末端(例えばEcoR I等)とし、もう片方を3'突出末端(例えばPst I等)とした後、Exonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素で消化して一本鎖cDNAを得る〔Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕。
尚、上記方法のうち、(1)及び(2)が好ましい。
得られた一本鎖テスターポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0031】
(2)ドライバー由来のポリヌクレオチド
本発明におけるドライバー由来のポリヌクレオチド(以下、ドライバーポリヌクレオチドと略記する。)は一本鎖ポリヌクレオチドであっても二本鎖ポリヌクレオチドであってもどちらでも良いが、好ましくは二本鎖ポリヌクレオチドである。尚、ドライバーポリヌクレオチドが一本鎖である場合は、当然のことながら、当該一本鎖ドライバーポリヌクレオチドは、一本鎖テスターポリヌクレオチドと相補鎖となり得るものである。即ち、テスターポリヌクレオチドがセンス鎖である場合、ドライバーポリヌクレオチドはアンチセンス鎖であり、テスターポリヌクレオチドがアンチセンス鎖である場合、ドライバーポリヌクレオチドはセンス鎖である。
また、当該ドライバーポリヌクレオチドは、DNA及びRNAのどちらでも良いが、好ましくはDNAである。なかでも、ゲノムDNA及びテスターに存在するmRNAに由来するcDNA(換言すれば、テスターから抽出したmRNAを鋳型として合成されたcDNA)が好ましく、特にcDNAが好ましい。
【0032】
本発明のドライバーポリヌクレオチドも、上記した如き、テスターポリヌクレオチドを調製するために用いられる二本鎖ポリヌクレオチドを調製する方法、一本鎖ポリヌクレオチドを調製する方法等の通常この分野で用いられている自体公知の方法や、市販のキットを用いて得ることができる。
なかでも、ドライバー中に存在するポリヌクレオチドを鋳型として、例えばPCR法、LAMP法、ICAN法等の自体公知の増幅方法により、当該ポリヌクレオチドに相補又は/及び相同なポリヌクレオチドを増幅する方法が好ましい。
【0033】
また、本発明で使用されるドライバーポリヌクレオチドには、一本鎖テスターポリヌクレオチドと同様、その5’末端又は/及び3’末端に、既知配列からなるアダプターを結合(付加)させておいてもよいが、この場合、当該アダプターは、少なくとも一本鎖テスターポリヌクレオチドが有する、後述する工程(3)で行うPCR用のプライマーが結合する既知の配列とは異なるものである。換言すれば、後述する工程(3)で行うPCR用のプライマーが結合しない既知配列からなるアダプターであれば、当該ドライバーポリヌクレオチドに結合(付加)させておいてもよいが、当該ドライバーポリヌクレオチドには一本鎖テスターヌクレオチドに結合(付加)させたアダプターを含む既知配列を含まないか、或いはそれ自体が単独でプライマーとして機能し得る塩基数からなる当該アダプターの一部の配列を含まない〔即ち、アダプターを結合(付加)させない〕のが好ましい。また、相補的ではないアダプターを両末端に結合させるのが好ましい。
【0034】
例えば以下の方法により、ドライバーポリヌクレオチドを得ることができる。
(1)自体公知の方法〔Gublerらの方法(Gulber, U. and Hoffman, B. J. (1983) Gene. 25, 263-269)等〕によってドライバーから抽出したmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーを用い、cDNA挿入断片両端のベクター由来の既知配列で設計された2種のプライマーを作製し、これを用いてベクターに挿入されているcDNAを鋳型として、PCR法、LAMP法、ICAN法等によりドライバー遺伝子由来の二本鎖cDNAを増幅して、アダプターが結合した(付加された)二本鎖ドライバーcDNAを得る。
(2)上記(1)によって増幅されたアダプターが結合した(付加された)二本鎖cDNAの、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までを、挿入断片両側のマルチクローニングサイトの制限酵素部位を利用して適当な制限酵素で切断し、挿入断片のみからなる二本鎖ドライバーcDNA〔アダプターが結合(付加)していない二本鎖ドライバーcDNA〕を得る。尚、この方法は、テスター由来cDNAライブラリーとドライバー由来cDNAライブラリーとでcDNAライブラリーを構成するベクターが同系列(例えば、両方ともにpUC系プラスミドである場合等)である場合等、テスターポリヌクレオチドとドライバーポリヌクレオチドで同じプライマーを使用する場合(同じアダプターが結合している場合)に有用である。
【0035】
尚、上記の方法を用いる場合、以下の点で注意が必要である。
即ち、テスター由来cDNAライブラリーとドライバー由来cDNAライブラリーとでcDNAライブラリーを構成するベクターが同系列(例えば、両方ともにpUC系プラスミドである場合等)である場合等、テスターポリヌクレオチドとドライバーポリヌクレオチドで同じプライマーを使用する場合(同じアダプターが結合している場合)には、挿入されているcDNA断片の末端からプライマーまでの長さ、言い換えれば、プライマーの末端から挿入断片が結合しているプライマーを含めた鎖長が100bp以上であると、切断された挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの断片を後述する精製方法で除去することが困難となる。その場合、当該切断された断片がテスターに存在する相補部分とハイブリダイズして二本鎖ハイブリッドを形成し、後述する本発明の工程(2)においてテスターのアダプター領域(PCR用プライマーがアニールする領域)が除去されてしまい、工程(3)での目的の一本鎖テスター特異的ポリヌクレオチドの増幅に支障をきたす。従って、このような場合には、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの鎖長が100bp以下となるように、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの切断を複数の制限酵素を用いて行うか、切断された挿入断片cDNAの両末端から増幅したプライマー(アダプター)までの断片を更に他の制限酵素を用いて切断することが好ましい。
(3)既知配列(アダプター)で設計された、5’末端がリン酸化された一本鎖テスターcDNAと相補鎖となる側のプライマーとリン酸化されていない一本鎖テスターcDNAと相同鎖となる側のプライマーの2種のプライマーを作製し〔調製される一本鎖テスターcDNAと相補鎖となるようにリン酸化プライマーと非リン酸化プライマーを設計する。言い換えれば、5’末端がリン酸化された一本鎖テスターcDNAと、これに相補的となる5’末端がリン酸化されていない一本鎖cDNAが生じるように2つのプライマー(リン酸化プライマーと非リン酸化プライマー)を設計する〕、これを用いてオリゴ・キャッピング法〔Maruyama, k. and Sugano, S. (1994) Gene. 138, 171-174等〕に従って5’末端及び3’末端に特定のプライマー(アダプター配列)が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR法、LAMP法、ICAN法等により増幅し、次いで、増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、5’末端リン酸化プライマー側のcDNA鎖を除去して、一本鎖テスターcDNAと相補鎖となるcDNA〔即ち、アダプターが結合した(付加された)一本鎖ドライバーcDNA〕を得る。
(4)適当なプラスミドに目的とするポリヌクレオチドを挿入し、プラスミドのマルチクローニングサイトを利用して挿入されたポリヌクレオチド断片の片方を5'突出末端(例えばEcoR I等)とし、もう片方を3'突出末端(例えばPst I等)とした後、Exonuclease III等の5'突出末端を認識する3'→5'Exonuclease活性を有する酵素で消化して一本鎖cDNAを得る〔Guo LH, Wu R. Methods in Enzymology. 1983;100:60-96等〕。
上記方法のうち(2)が好ましい。
尚、得られた一本鎖又は二本鎖ドライバーポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0036】
(3)テスター由来のポリヌクレオチドとドライバー由来のポリヌクレオチドとのハイブリダイゼーション〔工程(1)〕
上記の如くして調製された、(i)一本鎖テスターポリヌクレオチドと当該一本鎖テスターポリヌクレオチドと相補鎖となり得る一本鎖ドライバーポリヌクレオチドとの間、或いは(ii)一本鎖テスターポリヌクレオチドと二本鎖ドライバーポリヌクレオチドとの間で、ハイブリダイゼーション(所謂、サブトラクティブ・ハイブリダイゼーション)を行う。
このハイブリダイゼーションにより、ドライバーポリヌクレオチド中に相補的なものが存在する一本鎖テスターポリヌクレオチド〔即ち、テスターとドライバーの両方に存在するテスターポリヌクレオチド(テスター非特異的ポリヌクレオチド)〕は、テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)を形成する。一方、ドライバーポリヌクレオチド中に相補的なものが存在しない(或いは存在量が少ない)一本鎖テスターポリヌクレオチド(即ち、テスター特異的ポリヌクレオチド)は、ハイブリッドを形成せずに一本鎖のままで存在する。
【0037】
当該ハイブリダイゼーションは、通常この分野で用いられている自体公知のハイブリダイゼーション方法や、市販のキットを用いて行うことができる。
【0038】
例えば、上記の如くして調製した一本鎖テスターポリヌクレオチドと、当該テスターポリヌクレオチドに対して過剰量の一本鎖又は二本鎖ドライバーポリヌクレオチドを、適当量のナトリウム塩を含む緩衝液中で、要すれば熱変性後、適当な温度で適当な時間ハイブリダイゼーションを行い、テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)を形成させる。
上記において、使用されるドライバーポリヌクレオチドの量は、テスターポリヌクレオチドの量よりも多ければ良く、標的とするポリヌクレオチド(遺伝子)や使用目的等により異なるため一概には言えないが、使用されるドライバーポリヌクレオチドが一本鎖の場合のポリヌクレオチドの量(核酸量)は、テスターポリヌクレオチドの量(核酸量)の通常10〜5000倍、好ましくは50〜1000倍、より好ましくは100〜500倍である。また、使用されるドライバーポリヌクレオチドが二本鎖の場合のポリヌクレオチドの量(核酸量)は、テスターポリヌクレオチドの量(核酸量)の通常20〜10000倍、好ましくは100〜2000倍、より好ましくは200〜1000倍である。
使用されるナトリウム塩としては、通常この分野で用いられる、例えば塩化ナトリウム、クエン酸ナトリウム等が用いられ、また、その使用濃度も通常この分野で用いられる濃度範囲、例えば通常20mM〜2M、好ましくは50mM〜1.5M、より好ましくは100mM〜1Mから適宜選択される。
また、使用される緩衝液も、通常この分野で用いられるものは全て使用可能であり、例えば通常pH5〜9、好ましくはpH6〜8、より好ましくはpH6.5〜7.5の範囲で緩衝能を有する、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜1M、好ましくは5mM〜500mM、より好ましくは10mM〜100mMから適宜選択される。
上記において、熱変性は、通常90℃〜105℃、好ましくは93℃〜103℃、より好ましくは95℃〜100℃で、通常0.1分〜15分、好ましくは0.5分〜10分、より好ましくは1分〜5分行われる。尚、上記方法では、テスターポリヌクレオチドとドライバーポリヌクレオチドの共存下同時に熱変性をしているが、これらをそれぞれ別々に熱変性しても良い。この場合には、ナトリウム塩は特に必要ない。また、この場合には、熱変性した後、氷冷し、テスターポリヌクレオチドとドライバーポリヌクレオチドとを接触させてハイブリダイゼーションを行うのが好ましい。
ハイブリダイゼーションの条件としては、通常この分野で行われている方法に準じて行えば良く特に限定されないが、例えば上記した如きナトリウム塩の存在下、下限が通常30℃以上、好ましくは37℃以上、より好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下で、通常1分〜30時間、好ましくは30分〜20時間、より好ましくは1時間〜16時間行えばよい。
尚、上記方法において、ハイブリダーゼーション後に、更に上記した如き濃度範囲から選択されるドライバーポリヌクレオチドを追加してハイブリダイゼーションを繰り返してもよく、これにより、よりストリンジェントな条件でハイブリダイゼーションを行うことができる。
また、上記においては、通常この分野で使用される、例えば有機溶媒等の試薬類を用いることができる。
【0039】
(4)ハイブリッド形成した二本鎖ポリヌクレオチドの除去〔工程(2)〕
上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(即ち、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を酵素処理により除去する。
これにより、テスターポリヌクレオチドのうち、テスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチドを除去することができる。尚、二本鎖ドライバーポリヌクレオチドを用いた場合は、上記工程(1)において再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド(ハイブリッド)も同時に除去される。
【0040】
本発明の上記工程(2)の酵素処理において用いられる酵素は、一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有するものであり、使用されるテスターポリヌクレオチド及びドライバーポリヌクレオチドの種類により異なる。例えばテスターポリヌクレオチド及びドライバーポリヌクレオチドが共にDNA(cDNA)である場合は、二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が使用され、テスターポリヌクレオチドとドライバーポリヌクレオチドのどちらか一方がDNA(cDNA)であって他方がRNAである場合は、DNA−RNAハイブリッドを優先的に分解し得る性質を有する分解酵素が使用される。また、テスターポリヌクレオチド及びドライバーポリヌクレオチドが共にRNAである場合は、二本鎖RNAを優先的に分解し得る性質を有するRNA分解酵素が使用される。このような分解酵素を用いて酵素処理することにより、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(及び再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を分解することができ、結果としてハイブリット形成した二本鎖ポリヌクレオチド(即ち、テスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチド)を除去することができる。
また、テスターポリヌクレオチドがDNA(cDNA)でドライバーポリヌクレオチドがRNAである場合には、DNA−RNAハイブリッド中のDNAを優先的に分解し得る性質を有するDNA分解酵素も使用でき、テスターポリヌクレオチドがRNAでドライバーポリヌクレオチドがDNA(cDNA)である場合は、DNA−RNAハイブリッド中のRNAを優先的に分解し得る性質を有するRNA分解酵素(例えばRNase H等)も使用できる。これらの分解酵素を用いて酵素処理行った場合には、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチド(及び再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)自体(全体)を分解することはできないが、ハイブリッド形成された二本鎖ポリヌクレオチド中のテスターポリヌクレオチドを分解することができるので、結果としてテスターに特異的ではなくドライバーにも存在するテスター非特異的ポリヌクレオチドを除去することができる。
上記した如き分解酵素のうち、二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が好ましく、このような分解酵素としては、例えば二本鎖特異的DNAヌクレアーゼ(Duplex-specific nuclease:DSN)等が挙げられる。当該酵素の由来については特に限定されないが、例えば海洋性甲殻類(エビ、カニ等)、ウニ等の海洋性無脊椎動物が挙げられる。なかでも海洋性甲殻類、特にカムチャッカカニ(Paralithodes camtschaticus)由来の二本鎖特異的DNAヌクレアーゼが好ましい。
尚、二本鎖特異的DNAヌクレアーゼは、自体公知の方法〔例えばShagin DA, Rebrikov DV, Kozhemyako VB, Altshuler IM, Shcheglov AS, Zhulidov PA, Bogdanova EA, Staroverov DB, Rasskazov VA, Lukyanov S. : Genome Research. 2002 Dec;12(12):1935-42〕に準じて製造することができ、また、市販品(例えばEvrogen社)を使用することもできる。
分解酵素の使用量は、ハイブリッド形成された二本鎖ポリヌクレオチドを充分に分解し得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合には、通常全ポリヌクレオチド 1μgに対して0.01u/μl〜10u/μl、好ましくは0.1u/μl〜5u/μl、より好ましくは0.5u/μl〜1u/μlである。
【0041】
当該酵素処理の条件(温度、時間、pH等)、即ち、ハイブリッド形成された二本鎖ポリヌクレオチドと分解酵素とを接触させる際の条件は、使用する分解酵素の種類によって異なるため一概には言えないが、通常使用する分解酵素が有する至適温度及び至適pH付近で、形成されたハイブリッド(又はハイブリッド中のテスターポリヌクレオチド)を充分分解し得る時間処理すればよい。
より具体的には、例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合には、下限が通常20℃以上、好ましくは30℃以上、より好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下で、通常pH6〜9、好ましくは6.5〜8.5、より好ましくは7〜8で、通常0.5分〜60分、好ましくは1分〜45分、より好ましくは5分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜100mM、より好ましくは10mM〜50mMから適宜選択される。
【0042】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
このような反応停止剤を当該反応液中に存在させるには、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液を酵素処理を行って得られた反応液と混合させればよい。
反応停止のための時間は、通常0.1分〜30分、好ましくは0.5分〜20分、より好ましくは1分〜10分である。
また、反応を停止する際の温度は特に限定されないが、50℃〜90℃、好ましくは60℃〜80℃、より好ましくは65℃〜75℃で行うのが好ましい。
【0043】
当該酵素処理は、上記した如き本発明の工程(1)によりハイブリッド形成された二本鎖ポリヌクレオチドと上記した如き分解酵素とを接触させることにより実施される。
このような方法としては、最終的にハイブリッド形成された二本鎖ポリヌクレオチドと分解酵素とを接触し得るものであれば良く特に限定されないが、一般的には、上記した如き本発明の工程(1)を実施して得られたハイブリダイゼーション溶液に当該分解酵素を含有する溶液を添加混合することにより行われる。
上記において、分解酵素を含有させる溶液としては、当該分解酵素の分解作用を阻害しないものであればよく、水や上記した如き緩衝剤等の通常この分野で用いられる溶液が挙げられ、その使用濃度等も上記した如き濃度範囲から適宜選択すればよい。
ここで、当該溶液中には、分解酵素以外に、ナトリウム塩、通常この分野で用いられる活性化剤(マグネシウムイオン等の金属イオン等)、安定化剤(グリセロール、ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を含有させておいても良い。尚、これらナトリウム塩、活性化剤、安定化剤、防腐剤等は、分解酵素を含有する溶液とは別の上記した如き溶液に含有させておいても良く、この場合は、分解酵素を含む溶液とナトリウム塩、活性化剤、安定化剤又は防腐剤を含有する1種以上の溶液とをハイブリダイゼーション溶液に別々に添加することになる。なかでも、ナトリウム塩を含有させておくのが好ましい。
使用されるナトリウム塩としては、通常この分野で用いられる、例えば塩化ナトリウム、クエン酸ナトリウム等が用いられ、また、その使用濃度も通常この分野で用いられる濃度範囲、例えば通常20mM〜2M、好ましくは50mM〜1.5M、より好ましくは100mM〜1Mから適宜選択される。
【0044】
例えば分解酵素として二本鎖特異的DNAヌクレアーゼを用いる場合、当該酵素処理〔本発明の工程(2)〕は以下の如くして実施される。
本発明の工程(1)を実施して得られたハイブリダイゼーション溶液に、二本鎖特異的DNAヌクレアーゼ含有溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、上記した如き条件で処理し(反応させ)て、当該ハイブリダイゼーション溶液中に存在するハイブリッド形成した二本鎖ポリヌクレオチド(テスターポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド、又はこれと再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を分解除去する。
尚、上記において、活性化剤としてはマグネシウムイオンが好ましい。その由来としては、例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウムの塩が挙げられ、なかでも塩化マグネシウムが好ましい。また、マグネシウムの塩の使用量としては、二本鎖特異的DNAヌクレアーゼを充分活性化し得る量であればよく、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMである。
また、安定化剤としては例えばジチオスレイトール、メルカプトエタノール等のチオール化合物が好ましく、なかでもジチオスレイトールが好ましい。また、その使用量としては、二本鎖特異的DNAヌクレアーゼを安定化し得る量であればよく、通常0.01mM〜10mM、好ましくは0.1mM〜5mM、より好ましくは0.5 mM〜1mMである。
尚、本発明の工程(1)と(2)は、なるべく高い温度で実施するのが好ましい。例えば本発明の工程(1)と(2)は、下限が好ましくは50℃以上、更に好ましくは55℃以上、特に好ましくは60℃以上、最も好ましくは65℃以上であって、上限が通常80℃以下、好ましくは75℃以下、より好ましくは70℃以下でそれぞれ実施される。また、工程(1)に引き続いて工程(2)を実施する際には、上記した如き温度範囲から一旦下げること無く、上記した如き温度範囲を維持したまま、当該温度範囲から温度を一旦下げることなく実施されるのが好ましい。
【0045】
上記したように本発明の方法〔本発明の工程(1)及び工程(2)〕によって、テスター中に存在するポリヌクレオチドのうち、テスター非特異的ポリヌクレオチドを分解除去し、テスター特異的ポリヌクレオチドを取得することができる。
尚、本発明の工程(2)を実施して得られた反応液中には、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチド以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存しているが、共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドは、自体公知の単離(分離)処理又は精製処理により除去することができ、一本鎖テスター特異的ポリヌクレオチドのみを単離することができる。
このような方法としては、例えば一本鎖テスター特異的ポリヌクレオチドを、テスターcDNAを増幅したプライマー等のテスター特異的ポリヌクレオチドのみに存在するアダプター配列を基に設計したプライマーを使用してクレノウ・フラグメント(Klenow Fragment)で二本鎖にし、その後両端のアダプター配列中の制限酵素で切断して、適当なベクターに組み込み、テスター特異的ポリヌクレオチド(cDNA)のみを形質転換する方法、テスターcDNAを増幅したプライマー等のテスター特異的ポリヌクレオチドのみに存在するアダプター配列を基に設計したプライマーをビオチン標識して、テスター特異的ポリヌクレオチド(cDNA)とアニーリング後、ストレプトアビジン・ビーズ等の固相と結合させてテスター特異的ポリヌクレオチドを単離する方法等が挙げられる。
尚、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドとは、テスターポリヌクレオチドに相補的な一本鎖ドライバーポリヌクレオチドを用いた場合は、ドライバーに特異的な一本鎖ポリヌクレオチド及びテスター非特異的ポリヌクレオチドとハイブリッドを形成しなかったテスターとドライバーの両方に存在する一本鎖ドライバーポリヌクレオチドであり、二本鎖ドライバーポリヌクレオチドを用いた場合は、再会合せずに一本鎖として存在するドライバーに特異的な一本鎖ポリヌクレオチド(テスターポリヌクレオチドに対する相補鎖及び相同鎖の2種)及び再会合せずに一本鎖として存在するテスターとドライバーの両方に存在する一本鎖ドライバーポリヌクレオチド(テスターポリヌクレオチドに対する相補鎖及び相同鎖の2種)である。
【0046】
2−2.本発明のテスター特異的ポリヌクレオチドの増幅方法
かくして得られたテスター特異的ポリヌクレオチドを増幅することにより、テスター特異的ポリヌクレオチドを濃縮することができる。
【0047】
(1)ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドの増幅〔工程(3)〕
本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチドを大量に得ることができる。
即ち、本発明の工程(2)を実施した後に残存する一本鎖テスター特異的ポリヌクレオチドのみを実質的に増幅させる。
これにより、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しなかった場合でも、一本鎖テスター特異的ポリヌクレオチドと共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドは実質的に増幅されないので、一本鎖テスター特異的ポリヌクレオチドのみが濃縮される。
本発明において、一本鎖テスター特異的ポリヌクレオチドを増幅するとは、(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみを増幅する場合又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する場合を意味する。
【0048】
当該増幅は、本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチド〔(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみ又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖等〕を増幅することができるものであれば良く、通常この分野で用いられている自体公知のポリヌクレオチド増幅方法に従って実施できる。このような自体公知のポリヌクレオチド増幅方法としては、例えばPCR〔Oligo-capping:a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides, Kazuo Maruyama and Sumio Sugano, Gene, 138 (1994) 171-174に記載のオリゴ-キャッピング法;Diatchenko,L., et al. Proc. Natl. Acad. Sci. USA 91:6025-6030 (1996)に記載のSMART法;High-efficiency full-length cDNA cloning by biotinylated CAP trapper, Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C., Genomics. 1996 Nov 1;37(3):327-36に記載のGene Trapper法;Determination of 5' ends of specific mRNAs by DNA ligase-dependent amplification, Bertling WM, Beier F, Reichenberger E., PCR Methods Appl. 1993 Oct;3(2):95-9に記載のRACE法〕、LAMP法〔Loop-mediated isothermal amplification of DNA, Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000 Jun 15;28(12):E63〕、ICAN法〔Development of the detection system for Mycobacterium tuberculosis DNA by using the isothermal DNA amplification method ICAN, Shimada M, Hino F, Sagawa H, Mukai H, Asada K, Kato I. Rinsho Byori. 2002 May;50(5):528-32〕等が挙げられ、なかでもPCRが好ましい。
尚、各種増幅条件(温度、時間、pH、サイクル数等)は上記した如き自体公知の方法に準じて行えばよく、また、使用される酵素類や試薬類等もこれら自体公知の方法で用いられているものが使用できる。
また、当該増幅は、市販のキットを用いて行うこともできる。
【0049】
当該増幅に使用されるプライマーは、本発明の取得方法によって取得された一本鎖テスター特異的ポリヌクレオチド〔(1)一本鎖テスター特異的ポリヌクレオチドの相補鎖のみ又は(2)一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖等〕を増幅することができるものであれば良く特に限定されない。
即ち、当該プライマーは、例えば、目的とする増幅方法及びその条件等に応じて、また、解離温度(Tm値)などを考慮して、適宜設計・作製すればよい。好ましくはプライマー配列としての特異性を維持するために必要な塩基数と考えられている、15塩基〜60塩基、好ましくは20塩基〜35塩基、より好ましくは25塩基〜30塩基の長さを有しているものがよい。
【0050】
例えば本発明の工程(2)を実施した後に、上記した如き単離(分離)処理又は精製処理により、一本鎖テスター特異的ポリヌクレオチドが単離されている場合には、これ以外のテスター非特異的ポリヌクレオチドやハイブリッドを形成しなかったドライバーポリヌクレオチドは共存していないので、どのようなプライマーも使用できる。
このようなプライマーとしては、例えば前述した如きアダプターが結合した(付加された)テスターポリヌクレオチドを用いた場合は、当該アダプター配列に基づいて設計・作製されたプライマー等が挙げられ、例えばcDNAライブラリーからテスターcDNAを調製した場合はcDNA挿入断片両端のベクター由来の配列に基づいて設計・作製された1種又は2種以上プライマー(即ち、前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプター配列又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド)等が挙げられる。
また、例えば本発明の工程(2)を実施した後に、単離(分離)処理又は精製処理を行わず、一本鎖テスター特異的ポリヌクレオチドが単離されていない場合には、テスター非特異的ポリヌクレオチドは共存していないものの、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。しかしながら、一本鎖テスター特異的ポリヌクレオチドにのみ存在し、ドライバー由来のポリヌクレオチド中に存在しない配列に基づいて設計・作製されたプライマーを用いることにより、一本鎖テスター特異的ポリヌクレオチドのみを増幅することができる。
このようなプライマーとしては、例えば前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプターに基づいて設計・作製された1種又は2種以上プライマー(即ち、前述した如きドライバー由来のポリヌクレオチド中に存在しない配列からなるアダプター配列又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド)等が挙げられる。
また、PCR等に使用する場合の2種以上のプライマーとしては、相補的ではないアダプター配列の一部若しくは全部、又はその相補配列の一部若しくは全部を含有するオリゴヌクレオチド、即ち、それぞれ異なる2種以上のプライマーが好ましい。
【0051】
当該プライマーは、標識物質で標識化されていてもよく、プライマーを標識物質で標識するために用いられる標識物質としては、放射性同位体(32P,33P,35S等)や酵素(アルカリホスファターゼ,西洋ワサビペルオキシダーゼ等)、蛍光物質〔Cyanine Dye系のCy3,Cy5(アマシャムバイオサイエンス株式会社)、フルオレセイン等〕、発光物質(Acridinium Esterを含む化学発光試薬等)、ビオチンなど公知の標識物質であれば何れも用いることができる。また、プライマーを標識物質で標識する方法も、例えばプライマーを合成する際に、放射性同位体や蛍光物質で標識されたヌクレオチドを取り込ませることによって、プライマーを標識する方法や、プライマーを合成した後、放射性同位体で標識する方法(ランダムプライマー法、ニックトランスレーション法、T4ポリヌクレオチド キナーゼによる5'−末端標識法、ターミナルデオキシヌクレオチドトランスフェラーゼを用いた3'−末端標識法、RNAラベリング法)、酵素分子を標識するプライマーに直接共有結合させる直接標識法等、リンカーアームを有するヌクレオチドを配列のオリゴヌクレオチドの一員として置換して、ヌクレオチドを蛍光物質で標識する方法、ヌクレオチドを発光標識又はビオチン標識する方法等、通常この分野で行われている自体公知の標識方法に従って行えばよい。
【0052】
本発明の工程(3)によって、一本鎖テスター特異的ポリヌクレオチドを増幅(濃縮)することができるが、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しない場合、即ち、本発明の工程(2)を実施して得られた、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドとハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドとが共存している反応液中の、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドを増幅する場合においては、本発明の工程(3)を実施して得られた反応液中には、増幅された、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチド以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。しかしながら、この場合には、後述する本発明の工程(4)における一本鎖ポリヌクレオチドを除去する工程により、増幅された一本鎖テスター特異的ポリヌクレオチドを単離することができる。
特に、後述する本発明の工程(4)における一本鎖ポリヌクレオチドを除去する工程が、酵素処理により一本鎖ポリヌクレオチドを除去する工程である場合には、酵素処理により一本鎖ポリヌクレオチドを特異的に分解除去して、増幅されたテスター特異的ポリヌクレオチドを容易に単離することができるため、本発明の工程(3)で増幅されたテスター特異的ポリヌクレオチドは二本鎖であるのが好ましい。
【0053】
増幅された二本鎖テスター特異的ポリヌクレオチドを得る方法としては、例えば上記した如き自体公知の増幅方法のうち、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する方法が挙げられる。
また、上記した如き自体公知の増幅方法のうち、一本鎖テスター特異的ポリヌクレオチドの相補鎖のみを増幅する場合には、増幅された一本鎖特異的ポリヌクレオチドの相補鎖を用いて、例えば以下の方法によって二本鎖テスター特異的ポリヌクレオチドを得ることができる。
(a)増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖を鋳型として、これに相補的なプライマーを用いて再度PCRを行って増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖に対する相補鎖を増幅させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法、(b)増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖を鋳型として、これに相補的なプライマーとクレノウ フラグメント等の重合酵素を用いて増幅された一本鎖テスター特異的ポリヌクレオチドの相補鎖に対する相補鎖をプライマー伸長させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法等。
これらの方法のなかでも、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅する方法が好ましい。
尚、上記において、得られた二本鎖テスター特異的ポリヌクレオチドの二本鎖ハイブリットを形成させるには特別な処理は必要ではなく、例えば得られた一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖を含有する溶液を、通常30℃〜80℃、好ましくは37℃〜75℃、より好ましくは50℃〜70℃とすることによりこれらのハイブリットが形成される。
【0054】
上記したことから、本発明においては、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を増幅するのが好ましい。
従って、本発明のテスター特異的ポリヌクレオチドの増幅方法は、(3’)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドとこれの相補鎖とを増幅してテスター由来の二本鎖ポリヌクレオチドを得る工程、を含んでなる方法が好ましい。
【0055】
例えば以下の方法により、ハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドを増幅することができる。
即ち、(a)本発明の工程(2)を実施して得られた反応液、要すれば更に単離処理又は精製処理により単離された一本鎖テスター特異的ポリヌクレオチドを含有する溶液に、上記した如きそれぞれ異なる2種のプライマーとデオキシリボヌクレオチド三リン酸(dATP、dCTP、dGTP及びdTTP)を適当量加え、得られた溶液を適当な温度で適当な時間加熱して熱変性する。(b)加熱後当該溶液を冷却して一本鎖テスター特異的ポリヌクレオチド(又は一本鎖テスター特異的ポリヌクレオチドと、増幅されたこれに相補的なポリヌクレオチド)にプライマーをアニールさせる。(c)次いで、適当な重合酵素を適当な温度で適当な時間作用させてプライマーを伸長させることによって、一本鎖テスター特異的ポリヌクレオチドに対する相補鎖(又は一本鎖テスター特異的ポリヌクレオチドに対する相補鎖と、増幅された、一本鎖テスター特異的ポリヌクレオチドに相補的なポリヌクレオチドに対する相補鎖)を合成(増幅)する。以上の(a)〜(c)の工程を適当回数(例えば通常5回〜60回、好ましくは10回〜50回、より好ましくは15回〜40回)繰り返すことによってハイブリッド形成しなかった一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖とを増幅する。
上記方法において、使用される試薬類の量、重合酵素の種類や量、反応条件等は上記した如き自体公知の方法に準じて適宜選択されればよい。
例えば、上記工程(a)の熱変性は、通常90℃〜105℃、好ましくは93℃〜103℃、より好ましくは95℃〜100℃で、通常0.01分〜10分、好ましくは0.05分〜5分、より好ましくは0.1分〜2分行われる。また、工程(b)のアニーリングは、通常30℃〜75℃、好ましくは40℃〜72℃、より好ましくは50℃〜68℃で、通常0.01分〜10分、好ましくは0.05分〜5分、より好ましくは0.1分〜2分行われる。上記工程(c)において使用される重合酵素としては、特に限定されず、この分野で使用される酵素が使用可能であるが、例えばDNAポリメラーゼI、クレノウ フラグメント、T4 DNAポリメラーゼ等のDNAポリメラーゼ、例えばTaqDNAポリメラーゼ、Tth DNA ポリメラーゼ、Pfu DNA ポリメラーゼ等の耐熱性酵素、逆転写酵素等が挙げられる。また、重合酵素を作用させる際の条件(温度、時間等)は、使用する重合酵素の種類等により異なるため一概には言えないが、例えばDNAポリメラーゼI等の非耐熱性酵素の場合は、通常20℃〜50℃、好ましくは25℃〜45℃、より好ましくは30℃〜37℃で、通常1分〜24時間、好ましくは5分〜12時間、より好ましくは30分〜4時間であり、例えばTaqDNAポリメラーゼ等の耐熱酵素の場合は、通常30℃〜85℃、好ましくは40℃〜80℃、より好ましくは50℃〜75℃で、通常0.01分〜20分、好ましくは0.05分〜15分、より好ましくは0.1分〜10分である。
尚、上記方法においては、(a)〜(c)の各工程後に、デオキシリボヌクレオチド三リン酸、プライマー、試薬類、重合酵素等を新たに添加してもよい。
なかでも、TaqDNAポリメラーゼ、Tth DNA ポリメラーゼ、Pfu DNA ポリメラーゼ等の耐熱性酵素を用いれば、(a)〜(c)の各工程後に、デオキシリボヌクレオチド三リン酸、プライマー、試薬類、重合酵素等を新たに添加する必要がなく、最初に全ての試薬類や重合酵素を共存させておくことができるので、特に好ましい。
得られた二本鎖テスター特異的ポリヌクレオチドは、例えばフェノール/クロロホルム混合液、フェノール/クロロホルム/イソアミルアルコール混合液又は/及びクロロホルム/イソアミルアルコール混合液による抽出、アルコール沈澱、カラム精製、フィルター濾過等の自体公知の精製方法により精製するのが好ましい。
【0056】
(2)ハイブリット形成しなかった一本鎖ドライバーポリヌクレオチドの除去〔工程(4)〕
上記したように、本発明の工程(3)によって増幅(濃縮)されたテスター特異的ポリヌクレオチド(一本鎖又は二本鎖)が、一本鎖ドライバーポリヌクレオチドと共存している場合、即ち、本発明の工程(2)を実施した後に一本鎖テスター特異的ポリヌクレオチドを単離(分離)処理又は精製処理しない場合は、本発明の工程(3)を実施して得られた反応液中には、増幅されたテスター特異的ポリヌクレオチド〔工程(2)でハイブリッドを形成しなかった、一本鎖テスター特異的ポリヌクレオチド〕以外に、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドが共存している。この場合には、当該一本鎖ドライバーポリヌクレオチドを除去することにより、増幅されたテスター特異的ポリヌクレオチド(一本鎖又は二本鎖)を単離することができる。
一本鎖ドライバーポリヌクレオチドを除去する方法としては、前述した如き自体公知の単離(分離)方法又は精製方法が挙げられる。
【0057】
また、増幅されたテスター特異的ポリヌクレオチドが二本鎖ポリヌクレオチドである場合には、一本鎖ポリヌクレオチドを分解するが二本鎖ポリヌクレオチドを分解しない性質を有する酵素を用いた酵素処理により、二本鎖テスター特異的ポリヌクレオチドと共存する一本鎖ポリヌクレオチド(一本鎖ドライバーポリヌクレオチド)を特異的に分解除去して、二本鎖テスター特異的ポリヌクレオチドを単離することもできる〔Convenient single-step, one tube purification of PCR products for direct sequencing. E Werle, C Schneider, M Renner, M Veolker, and W Fiehn, Nucleic Acids Res. 1994 October 11; 22(20): 4354-4355等〕。
【0058】
上記の方法のなかでも、酵素処理により一本鎖ポリヌクレオチドを除去(分解)する方法が好ましい。
従って、本発明において、一本鎖ドライバーポリヌクレオチドを除去する方法は、(4’)ハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程、を含んでなる方法が好ましい。
【0059】
当該酵素処理において用いられる酵素は、ハイブリッド形成された二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有するものであり、使用されるテスターポリヌクレオチド及びドライバーポリヌクレオチドの種類により異なる。
例えばテスターポリヌクレオチド及びドライバーポリヌクレオチドが共にDNA(cDNA)である場合は、例えば一本鎖特異的DNAヌクレアーゼ(エキソヌクレアーゼI、エキソヌクレアーゼIX等)等の二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が使用され、テスターポリヌクレオチド及びドライバーポリヌクレオチドが共にRNAである場合は、例えば一本鎖特異的RNAエンドヌクレアーゼ(リボヌクレアーゼ)(RNaseA、RNase IV、RNaseT1、RNaseT2、RNase II、RNase III、Rnase I等)等の二本鎖RNAを分解せず一本鎖RNAを優先的に分解し得る性質を有するRNA分解酵素が使用される。
このような分解酵素を用いて酵素処理することにより、上記した如き本発明の工程(3)において得られた二本鎖ポリヌクレオチド(二本鎖テスター特異的ポリヌクレオチド)と共存する一本鎖ドライバーポリヌクレオチド(一本鎖ドライバー特異的ポリヌクレオチド及び一本鎖ドライバー非特異的ポリヌクレオチド)を分解することができ、二本鎖ポリヌクレオチド(二本鎖テスター特異的ポリヌクレオチド)を単離することができる。
上記した如き分解酵素のうち、二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素が好ましく、エキソヌクレアーゼIがより好ましい。
上記した如き分解酵素の由来については特に限定さず、通常この分野で用いられるものであれば使用可能である。尚、当該分解酵素は、自体公知の方法〔例えばLehman,IR., Nussbaum, A.L., J.Bio.Chem., 239, 2628-2636, 1964〕に準じて製造することができ、また、市販品(例えば New England Biolabs 社)を使用することもできる。
【0060】
分解酵素の使用量は、一本鎖ポリヌクレオチドを充分に分解し得る量であれば良く特に限定されない。具体的には、使用する分解酵素の種類によって異なるため一概には言えないが、例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合には、通常テスターポリヌクレオチド 1 μg に対して0.1u/μl〜100u/μl、好ましくは0.5u/μl〜50u/μl、より好ましくは1u/μl〜10u/μlである。
当該酵素処理の条件(温度、時間、pH等)、即ち、一本鎖ポリヌクレオチドと分解酵素とを接触させる際の条件は、使用する分解酵素の種類によって異なるため一概には言えないが、通常使用する分解酵素が有する至適温度及び至適pH付近で、一本鎖ポリヌクレオチドを充分分解し得る時間処理すればよい。
より具体的には、例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合には、通常20℃〜60℃、好ましくは25℃〜50℃、より好ましくは30℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理される。
また、上記した如きpH範囲を維持するために、通常この分野で用いられる緩衝剤が使用できる。このような緩衝液としては、上記した如きpH範囲で緩衝能を有するものであれば良く特に限定されないが、例えばトリス−塩酸緩衝液、グリシン緩衝液、グッド緩衝液(例えばHEPES、PIPES等)が挙げられる。使用濃度も通常この分野で用いられる濃度範囲、例えば通常1mM〜500mM、好ましくは5mM〜250mM、より好ましくは10mM〜100mMから適宜選択される。
【0061】
尚、上記した如き酵素処理を行った後、当該酵素の反応を停止するには、例えばEDTA等のキレート剤等の反応停止剤を用いる方法又は/及び加熱処理を行う方法により行えばよい。
反応停止剤を用いる方法としては、例えば水や上記した如き緩衝剤等に当該反応停止剤を含有させ、この反応停止剤含有溶液と、酵素処理を行って得られた反応液と混合させれる等により、当該反応停止剤を、酵素処理を行って得られた反応液中に存在させればよい。
また、加熱処理により反応を停止する方法としては、酵素処理を行って得られた反応液を、通常60℃〜90℃、好ましくは65℃〜85℃、より好ましくは70℃〜80℃で処理すればよい。
尚、反応停止剤による反応時間及び加熱処理時間は、通常1分〜60分、好ましくは5分〜30分、より好ましくは10分〜20分である。
上記の反応停止方法のうち、反応停止剤を用いる方法と加熱処理による方法の両者を併用するのが好ましい。
【0062】
当該酵素処理は、上記した如き本発明の工程(3)によって増幅(濃縮)されたテスター特異的ポリヌクレオチド及びそれと共存する一本鎖ドライバーポリヌクレオチドと上記した如き分解酵素とを接触させることにより実施される。
このような方法としては、最終的に増幅(濃縮)されたテスター特異的ポリヌクレオチド、共存する一本鎖ドライバーポリヌクレオチド及び分解酵素とを接触し得るものであれば良く特に限定されないが、一般的には、上記した如き本発明の工程(3)を実施して得られた反応液に当該分解酵素を含有する溶液を添加混合することにより行われる。
上記において、分解酵素を含有させる溶液としては、当該分解酵素の分解作用を阻害しないものであればよく、水や上記した如き緩衝剤等の通常この分野で用いられる溶液が挙げられ、その使用濃度等も上記した如き濃度範囲から適宜選択すればよい。
ここで、当該溶液中には、分解酵素以外に、通常この分野で用いられる活性化剤(マグネシウムイオン等の金属イオン等)、安定化剤(ジチオスレイトール、メルカプトエタノール等のチオール化合物等)、防腐剤等を含有させておいても良い。尚、これら活性化剤、安定化剤、防腐剤等は、分解酵素を含有する溶液とは別の上記した如き溶液に含有させておいても良く、この場合は、分解酵素を含む溶液と活性化剤、安定化剤又は防腐剤を含有する1種以上の溶液とを反応液に別々に添加することになる。
【0063】
例えば分解酵素として一本鎖特異的DNAヌクレアーゼを用いる場合、当該酵素処理〔本発明の工程(4)〕は以下の如くして実施される。
本発明の工程(4)を実施して得られた反応液に、一本鎖特異的DNAヌクレアーゼ含有溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、上記した如き条件で処理し(反応させ)て、当該反応液中に存在するハイブリッド形成しなかった一本鎖ドライバーポリヌクレオチド(一本鎖ドライバー特異的ポリヌクレオチド及び一本鎖ドライバー非特異的ポリヌクレオチド)を分解除去する。
尚、上記において、活性化剤としてはマグネシウムイオンが好ましい。その由来としては、例えば塩化マグネシウム、硫酸マグネシウム等のマグネシウム塩が挙げられ、なかでも塩化マグネシウムが好ましい。また、マグネシウム塩の使用量としては、一本鎖特異的DNAヌクレアーゼを充分活性化し得る量であればよく、通常0.1mM〜100mM、好ましくは0.5mM〜50mM、より好ましくは1mM〜10mMである。
また、安定化剤としては例えばジチオスレイトール、メルカプトエタノール等のチオール化合物が好ましく、なかでもジチオスレイトールが好ましい。また、その使用量としては、一本鎖特異的DNAヌクレアーゼを安定化し得る量であればよく、通常0.01mM〜100mM、好ましくは0.05mM〜50mM、より好ましくは0.1mM〜10mMである。
【0064】
上記したように本発明の工程(4)によって、本発明の増幅方法〔本発明の工程(1)〜(3)〕で増幅されたテスター特異的ポリヌクレオチドを容易に単離することができる。
尚、工程(3)の増幅を行わずに、工程(2)で得られた一本鎖テスター特異的ポリヌクレオチドを、当該一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに相補的なプライマーとクレノウ フラグメント等の重合酵素を用いて一本鎖テスター特異的ポリヌクレオチドに対する相補鎖をプライマー伸長させて、一本鎖テスター特異的ポリヌクレオチドとこれの相補鎖の二本鎖を得る方法等により二本鎖とした後に、本発明の工程(4)を行ってもよく、この場合には、テスター特異的ポリヌクレオチドは増幅(濃縮)されないものの、工程(2)で得られた一本鎖テスター特異的ポリヌクレオチドと共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを分解除去することができ、容易にテスター特異的ポリヌクレオチドのみを単離することができる。
【0065】
2−3.本発明の具体的方法
本発明の取得方法又は増幅方法は、一本鎖テスターポリヌクレオチドと一本鎖又は二本鎖ドライバーポリヌクレオチドとを用いて、図1に示されたチャートに従って実施することができる。
即ち、先ず、目的とするある試料から、自体公知の方法、好ましくはテスター由来の二本鎖ポリヌクレオチドを調製した後に酵素処理により一本鎖ポリヌクレオチドにする方法〔本発明の工程(1’)〕、より好ましくはテスター由来の二本鎖ポリヌクレオチドを増幅した後に当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする方法〔本発明の工程(1”)〕により、一本鎖テスターポリヌクレオチド、好ましくはアダプターが付加された一本鎖テスターポリヌクレオチドを調製する。一方、別の試料から、自体公知の方法により、二本鎖ドライバーポリヌクレオチド又は一本鎖テスターポリヌクレオチドに対して相補的な一本鎖ドライバーポリヌクレオチドを調製する。次いで、例えば以下の手順で本発明の取得方法又は増幅方法を実施する。
【0066】
(手順1)
(i)一本鎖テスターポリヌクレオチドと当該一本鎖テスターポリヌクレオチドと相補鎖となり得る一本鎖ドライバーポリヌクレオチドとの間、或いは(ii)一本鎖テスターポリヌクレオチドと二本鎖ドライバーポリヌクレオチドとの間で、ハイブリダイゼーションを行い、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチド(ドライバー非特異的ポリヌクレオチド)との二本鎖ポリヌクレオチド、又は当該ハイブリッドと再会合したドライバーポリヌクレオチドとドライバーポリヌクレオチドとの二本鎖ポリヌクレオチドをハイブリッド形成させる〔工程(1)〕。次いで、ハイブリッド形成された二本鎖ポリヌクレオチドを酵素処理により分解除去して、テスター非特異的ポリヌクレオチド又はこれと再会合したドライバー二本鎖ポリヌクレオチドを除去する〔工程(2)〕。更に要すれば、共存するハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを、自体公知の単離(分離)方法又は精製方法等により除去する〔単離工程〕。以上の手順により一本鎖テスター特異的ポリヌクレオチドを取得又は単離することができる。(本発明の取得方法1)
【0067】
(手順2)
上記工程(1)及び(2)、要すれば単離工程を実施した後、取得又は単離された一本鎖テスター特異的ポリヌクレオチドを増幅させる〔工程(3)〕。これにより、テスター特異的ポリヌクレオチドのみを実質的に増幅させて、テスター特異的ポリヌクレオチドのみを大量に得ることができる。(本発明の増幅方法1)
【0068】
(手順3)
上記工程(1)及び(2)を実施した後、取得された一本鎖テスター特異的ポリヌクレオチドを増幅させる〔工程(3)〕。次いで、増幅されたテスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを除去する〔工程(4)〕。これにより、増幅されたテスター特異的ポリヌクレオチドを単離することができる。(本発明の増幅方法2)
【0069】
(手順4)
以下の手順により、より簡便にテスター特異的ポリヌクレオチドを増幅(濃縮)及び単離することができる。(本発明の増幅方法3)
即ち、上記工程(1)及び(2)を実施した後、取得された一本鎖ポリヌクレオチドとこれの相補鎖とを増幅させて二本鎖テスター特異的ポリヌクレオチドを得る〔工程(3’)〕。次いで、増幅された二本鎖テスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを酵素処理により分解除去し、二本鎖テスター特異的ポリヌクレオチドを単離する〔工程(4’)〕。
【0070】
(手順5)
上記工程(1)及び(2)を実施した後、取得された一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに対する相補鎖を合成して二本鎖テスター特異的ポリヌクレオチドを得る〔二本鎖工程〕。次いで、工程(4’)により、二本鎖テスター特異的ポリヌクレオチドと共存している、ハイブリッドを形成しなかった一本鎖ドライバーポリヌクレオチドを酵素処理により分解除去する。この場合には、工程(2)で取得されたテスター特異的ポリヌクレオチドを容易に単離することができる。(本発明の取得方法2)
【0071】
(手順6)
上記工程(1)、(2)、及び単離工程を実施した後、上記二本鎖工程により、一本鎖テスター特異的ポリヌクレオチドを鋳型として、これに対する相補鎖を合成して二本鎖テスター特異的ポリヌクレオチドを得る。(本発明の増幅方法4)
【0072】
本発明の取得方法としては、上記手順1が好ましい。また、本発明の増幅方法としては、上記手順2及び3が好ましく、手順3が特に好ましい。
【0073】
以下に、テスターポリヌクレオチドとしてアダプターが付加された一本鎖cDNAを用い、ドライバーポリヌクレオチドとして二本鎖cDNAを用いる場合を例に取り、本発明の方法を具体的に説明する。
【0074】
〔一本鎖テスターcDNAの調製〕
自体公知の方法によって目的とする試料から抽出されたmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーにおける、cDNA挿入断片両端のベクター由来の既知配列で設計された5’末端がリン酸化されたプライマーとこれとは異なる配列のリン酸化されていないプライマーの2種のプライマーを作製する。当該cDNAライブラリーを鋳型とし、作製した2種のプライマーを用いてPCR等によりテスター遺伝子由来の5’末端及び3’末端にアダプター配列が付加された二本鎖cDNAを増幅する。或いは、自体公知の方法により目的とする試料からmRNAを抽出する。既知配列で設計された5’末端がリン酸化されたプライマーとこれとは異なる配列のリン酸化されていないプライマーの2種のプライマーを作製する。抽出したmRNAを鋳型とし、作製した2種のプライマーを用いてオリゴ・キャッピング法に従って5’末端及び3’末端にアダプター配列が付加されたcDNAを作製する。次いで、5’末端及び3’末端のアダプター配列でプライマーを作製して二本鎖cDNAをPCR等により増幅する。
要すれば、増幅されたアダプター付加二本鎖cDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
増幅されたアダプター付加二本鎖cDNAを、例えばラムダ・エキソヌクレアーゼ等の二本鎖DNAの5’末端リン酸化DNA鎖を除去する酵素により処理し、その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は/及び加熱処理を行うことによって当該酵素の反応を停止させて、5’末端リン酸化プライマー側のcDNA鎖を除去して、アダプターが結合した(付加された)一本鎖テスターcDNAを得る。
尚、要すれば、得られた一本鎖テスターcDNAを、カラム精製等により精製する。
上記した方法の概略を図2に示す。
【0075】
〔二本鎖ドライバーcDNAの調製〕
自体公知の方法によって目的とする試料とは別の試料から抽出されたmRNAを鋳型として作成されたcDNAライブラリー或いは市販のcDNAライブラリーにおける、cDNA挿入断片両端のベクター由来の既知配列で設計された、それぞれ配列が異なる2種のプライマーを作製する。当該cDNAライブラリーを鋳型とし、作製した2種のプライマーを用いてPCR等によりドライバー遺伝子由来の5’末端及び3’末端にアダプター配列が付加された二本鎖cDNAを増幅する。要すれば、増幅されたアダプター付加二本鎖cDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
増幅されたアダプター付加二本鎖cDNAの、挿入断片cDNAの両末端から増幅したプライマー(アダプター)までを、挿入断片両側のマルチクローニングサイトを利用して切断し、挿入断片のみからなる二本鎖ドライバーcDNA〔アダプターが結合(付加)していない二本鎖ドライバーcDNA〕を得る。
尚、要すれば、得られた二本鎖ドライバーcDNAを、カラム精製等により精製する。
上記した方法の概略を図3に示す。
【0076】
〔テスターcDNAとドライバーcDNAとのハイブリダイゼーション:工程(1)〕
調製した一本鎖テスターcDNAと、当該一本鎖テスターcDNAに対して過剰量の二本鎖ドライバーcDNAを、適当量のナトリウム塩を含む緩衝液に添加混合した後、適当な温度で適当な時間加熱して熱変性を行う。次いで、適当な温度で適当な時間ハイブリダイゼーションを行い、テスターcDNAとこれに相補的なドライバーcDNAとの二本鎖cDNAをハイブリッド形成させ、一本鎖テスター特異的cDNA、一本鎖テスター非特異的cDNAとこれと相補的な一本鎖ドライバーcDNAとの二本鎖ハイブリッド、再会合した二本鎖ドライバー特異的cDNA、再会合した二本鎖ドライバー非特異的cDNA、再会合しなかった一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び再会合しなかった一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有するハイブリダイゼーション溶液を得る。
上記した方法の概略を図4に示す。
【0077】
〔ハイブリッド形成した二本鎖cDNAの除去:工程(2)〕
得られたハイブリダイゼーション溶液に、例えば二本鎖特異的DNAヌクレアーゼ等の二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素を含有する溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、通常20℃〜80℃、好ましくは30℃〜75℃、より好ましくは50℃〜70℃、通常pH6〜9、好ましくは6.5〜8.5、より好ましくは7〜8で、通常0.5分〜60分、好ましくは1分〜45分、より好ましくは5分〜30分処理し(反応させ)、当該ハイブリダイゼーション溶液中に含有するハイブリッド形成した二本鎖cDNA(一本鎖テスター非特異的cDNAとこれと相補的な一本鎖ドライバーcDNAとの二本鎖ハイブリッド、再会合した二本鎖ドライバー特異的cDNA及び再会合した二本鎖ドライバー非特異的cDNA)を分解し、その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は反応停止剤での処理及び加熱処理を行うことによって当該DNA分解酵素の反応を停止させて、一本鎖テスター特異的cDNA、再会合しなかった一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び再会合しなかった一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有する反応液を得る。
尚、要すれば、これらcDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
上記した方法の概略を図5に示す。
【0078】
〔ハイブリッド形成しなかった一本鎖テスター特異的cDNAの増幅:工程(3)〕
得られた反応液に、上記した一本鎖テスターcDNAの調製において用いたのと同じ配列の2種のプライマー(但し、何れも5’末端はリン酸化されていない)、デオキシリボヌクレオチド三リン酸(dATP、dCTP、dGTP及びdTTP)、例えばTaqDNAポリメラーゼ等の重合酵素及びPCR用の緩衝液を適当量加える。(a)得られた溶液を適当な温度で適当な時間加熱して熱変性処理を行う。次いで、(b)当該溶液を冷却して一本鎖テスター特異的cDNA(2サイクル目以降は、一本鎖テスター特異的cDNAと増幅されたこれの相補鎖)にプライマーをアニールさせる。その後、(c)当該溶液を適当な温度で適当な時間処理し、当該重合酵素の作用によりプライマーを伸長させて一本鎖テスター特異的cDNAに対する相補鎖(2サイクル目以降は、一本鎖テスター特異的cDNAに対する相補鎖と、増幅された、一本鎖テスター特異的cDNAの相補鎖に対する相補鎖)を合成(増幅)する。以上の(a)〜(c)の工程を適当回数繰り返すことによってハイブリッド形成しなかった一本鎖テスター特異的cDNAとこれの相補鎖とを増幅し、増幅された二本鎖テスター特異的cDNA、一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)を含有する反応液を得る。
尚、要すれば、これらcDNAを、フェノール/クロロホルム/イソアミルアルコール混合液等による抽出処理、アルコール沈澱処理等に付す。
上記した方法の概略を図6に示す。
【0079】
〔ハイブリット形成しなかった一本鎖ドライバーcDNAの除去:工程(4)〕
得られた反応液に、一本鎖特異的DNAヌクレアーゼ等の二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素を含有する溶液と活性化剤及び安定化剤を含有する溶液とを添加混合し、通常20℃〜60℃、好ましくは25℃〜50℃、より好ましくは30℃〜40℃、通常pH7〜11、好ましくはpH8〜10.5、より好ましくは9〜10で、通常0.1分〜60分、好ましくは0.5分〜45分、より好ましくは1分〜30分処理し(反応させ)当該反応液中に含有する一本鎖ドライバーcDNA〔一本鎖ドライバー特異的cDNA(センス鎖とアンチセンス鎖の2種)及び一本鎖ドライバー非特異的cDNA(センス鎖とアンチセンス鎖の2種)〕を分解する。その後、例えばEDTA等のキレート剤等の反応停止剤での処理又は/及び加熱処理を行うことによって当該DNA分解酵素の反応を停止させる。
尚、要すれば、得られた二本鎖テスター特異的cDNAを、カラム精製等により精製する。
上記した方法の概略を図7に示す。
【0080】
2−4.本発明の遺伝子変異同定方法
本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドは、例えば以下の如き、テスターにおける遺伝子変異の同定等の各種同定及びスクリーニング等に使用される。
【0081】
(1)当該テスター特異的ポリヌクレオチドの塩基配列を自体公知の塩基配列決定法により決定することにより、変異遺伝子の塩基配列の同定や新規な遺伝子の配列の同定を行うことができる。
(2)変異遺伝子が病原性であるのか否かの同定
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(3)変異遺伝子のゲノムDNA上の位置の同定
ヒト、マウス、ラット等ゲノムの全DNA配列が解読されているものでは、データベース等を用いて当該全DNA配列と変異遺伝子の塩基配列とを比較することによってゲノムDNA上の変異遺伝子の位置が同定できる。また、全DNA配列が解読されていない場合は、例えばFISH法等の自体公知の方法によりゲノムDNA上の変異遺伝子の位置が同定できる。
(4)疾病の診断同定・予測・予防
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(5)疾病治療の効果の同定や治療時期の同定
例えばノーザンブロット法、RT-PCR法、リアルタイムPCR法等を用いることにより、テスター(異常(罹病)試料に由来するもの)とドライバー(正常(健常者)試料に由来するもの)における特定の変異遺伝子(mRNA)の発現量を確認し、比較することにより、当該特定の変異遺伝子(mRNA)が病原性であるか否かを同定することができる。
(6)当該テスター特異的ポリヌクレオチドをプローブとして用いてcDNAライブラリーやESTライブラリー等についてスクリーニングを行えば、ある機能又は形質発現に関与する変異遺伝子をスクリーニングすることができる。このような方法としては、例えば標識物質により標識されたテスター特異的ポリヌクレオチド又はその相補鎖の一部若しくは全部を含有するオリゴヌクレオチドをプローブとし、DNAチップ上に固定化された上記のライブラリーとハイブリダイゼーションを行う方法等、自体公知のスクリーニング方法〔Beck MT, Holle L, Chen WY. Biotechniques. 2001 Oct;31(4):782-4, 786〕等が挙げられる。
(7)当該テスター特異的ポリヌクレオチド(特にゲノムDNA又はcDNA)をクローニングし、サブトラクティブゲノムDNA又はcDNAライブラリーとする。正常(健常者)試料から抽出したゲノムDNA又はcDNAと異常(罹病)試料から抽出したゲノムDNA又はmRNA群をプローブとしてハイブリダイゼーションを行い、異常(罹病)試料のゲノムDNA又はmRNA群のみに反応したゲノムDNA又はcDNAが未知病原体由来である可能性がある。即ち、これにより、未知病原体を検出できる可能性がある。
(8)当該テスター特異的ポリヌクレオチド(特に、cDNA)を、例えばプラスミドベクター等の適当なベクターに挿入し、該ベクターを用いて大腸菌等を形質転換することにより、テスターに特異的な遺伝子をクローニングすることができる。
(9)当該テスター特異的ポリヌクレオチド(特に、cDNA)或いはクローニングされたDNAの塩基配列を決定し、その塩基配列に基づいて設計されたプライマーを用いて3'及び5'RACE法を行うことにより変異遺伝子の全長cDNAを得ることができる。
(10)当該プライマーを用いたTAIL-PCRを行うか、当該テスター特異的ポリヌクレオチドをプローブとして用いてゲノミックライブラリーについてスクリーニングを行えば、ある機能又は形質発現において特異的に働くプロモーター(変異遺伝子のプロモーター)をスクリーニングすることもできる。
(11)ハイスループットな特異的発現遺伝子の同定
本発明の方法で得られたテスター特異的ポリヌクレオチド(特に、cDNA)を用いてマイクロアレイ(チップ)を作製し、これを用いることによって、例えば上記の(1)〜(7)の同定・解析を迅速且つ簡便に行うことができる。
【0082】
従って、本発明の方法には、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドを用いた、上記した如き各種同定法やスクリーニング法が包含される。尚、当該同定法やスクリーニング法は、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチドを用いる以外は、自体公知の方法に従って実施すれば良く、そこで使用される試薬類等も自体公知の方法で用いられるものが使用できる。
従って、本発明の遺伝子変異同定方法には、例えば以下の(1)〜(9)のから選ばれる少なくとも1つの工程を含むものが好ましい。
(1)得られたテスター特異的ポリヌクレオチドの塩基配列を決定する工程、
(2)ノーザンブロット法、RT-PCR法、リアルタイムPCR法等により、テスターとドライバーにおける得られたテスター特異的ポリヌクレオチド(特定の変異遺伝子;mRNA)の発現量を確認し、それらを比較する工程、
(3)全DNA配列と得られたテスター特異的ポリヌクレオチド(変異遺伝子)の塩基配列とを比較し、ゲノムDNA上の得られたテスター特異的ポリヌクレオチド(変異遺伝子)の位置を同定する工程、又はFISH法等の自体公知の方法によりゲノムDNA上の得られたテスター特異的ポリヌクレオチド(変異遺伝子)の位置を同定する工程、
(4)得られたテスター特異的ポリヌクレオチドをプローブとして用いてcDNAライブラリーやESTライブラリー等についてスクリーニングを行う工程、
(5)得られたテスター特異的ポリヌクレオチド(特にゲノムDNA又はcDNA)をクローニングし、サブトラクティブゲノムDNA又はcDNAライブラリーとし、正常(健常者)試料から抽出したゲノムDNA又はcDNAと異常(罹病)試料から抽出したゲノムDNA又はmRNA群をプローブとしてハイブリダイゼーションを行う工程、
(6)得られたテスター特異的ポリヌクレオチド(特に、cDNA)を、例えばプラスミドベクター等の適当なベクターに挿入し、該ベクターを用いて大腸菌等を形質転換することにより、テスターに特異的な遺伝子をクローニングする工程、
(7)得られたテスター特異的ポリヌクレオチドをプローブ又はプライマーとして用いて、変異遺伝子の全長cDNAを得る工程、
(8)得られたテスター特異的ポリヌクレオチドをプライマーとして用いたTAIL-PCRを行うか、当該テスター特異的ポリヌクレオチドをプローブとして用いてゲノミックライブラリーについてスクリーニングを行い、ある機能又は形質発現において特異的に働くプロモーター(変異遺伝子のプロモーター)をスクリーニングする工程、
(9)得られたテスター特異的ポリヌクレオチド(特に、cDNA)を用いてマイクロアレイ(チップ)を作製し、これを用いて、上記の(1)〜(7)を行う工程。
また、本発明の方法により取得、増幅又は単離されたテスター特異的ポリヌクレオチド又はその相補鎖の一部若しくは全部を含有するオリゴヌクレオチドからなるプローブやプライマー及び本発明の方法により得ることができるcDNAライブラリーをも包含する。
【0083】
3.本発明のキット
本発明のキットは、上記した如き本発明の取得方法、増幅方法又は同定方法を実施するために使用されるものである。
このようなキットとしては、a)少なくとも先述した如き本発明の工程(2)においてハイブリッド形成された二本鎖ポリヌクレオチド(即ち、テスター非特異的ポリヌクレオチドとこれに相補的なドライバーポリヌクレオチドとの二本鎖ポリヌクレオチド)を除去するために用いられる、一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素、好ましくは二本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素、より好ましくは二本鎖特異的DNAヌクレアーゼを含んでなるものであり、b)好ましくは、更に、先述した如き本発明の工程(1’)において調製されたテスター由来の二本鎖ポリヌクレオチドを一本鎖ポリヌクレオチドにするために用いられる、二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素或いは2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群、好ましくは二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素、より好ましくはラムダ・エキソヌクレアーゼを含んでなるものであり、c)より好ましくは、先述した如き本発明の工程(4)において二本鎖テスター特異的ポリヌクレオチドと共存する一本鎖ポリヌクレオチド(一本鎖ドライバーポリヌクレオチド)を特異的に分解にするために用いられる、二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素、好ましくは二本鎖DNAを分解せず一本鎖DNAを優先的に分解し得る性質を有するDNA分解酵素、より好ましくは一本鎖特異的DNAヌクレアーゼ、更に好ましくはエキソヌクレアーゼIを含んでなるものである。
これら構成要件の好ましい態様と具体例は上で述べた通りである。
【0084】
更に、上記以外の試薬類を加えて、本発明のキットとすることもできる。このような試薬類としては、例えば以下の如きa)〜i)から選ばれる少なくとも1種が挙げられるが、これらに限定されない。
a)一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素用の緩衝液(例えばナトリウム塩、要すればマグネシウム塩及びチオール化合物を含有するHEPES等のグッド緩衝液)、
a’)一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素の反応を停止するための溶液(例えばキレート剤等の反応停止剤を含有する、例えば水や上記した如き緩衝剤)、
b)二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとする性質を有する酵素或いは2種以上の酵素を組み合わせることにより二本鎖ポリヌクレオチドの片側の核酸鎖を除去して一本鎖ポリヌクレオチドとすることができる酵素群用の緩衝液(例えばマグネシウム塩及び界面活性剤を含有するグリシン等の緩衝液)、
c)二本鎖ポリヌクレオチドを分解せず一本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素用の緩衝液(例えばマグネシウム塩及びチオール化合物を含有するグリシン等の緩衝液)、
d)ハイブリダイゼーション用緩衝液(例えばナトリウム塩を含有するグッド緩衝液等)、
e)ポリヌクレオチド抽出用試薬(例えば粉砕用緩衝液、フェノール、クロロホルム、SDS、β−メルカプトエタノール等)、
f)PCR用試薬(例えば重合酵素、重合酵素用緩衝液、1種又は2種以上のプライマー、ヌクレオチド混合物等)、
g)電気泳動用試薬(アガロース又はポリアクリルアミドゲル、ローディング緩衝液、臭化エチジウム染色用試薬等)、
h)PCR反応後の余剰プライマーの除去、ミス・アニーリングにより増幅されてしまった鎖長の短いDNAの除去、ヌクレアーゼにより完全に消化されなかった短いDNAの除去等のために用いられるポリヌクレオチド単離(分離)又は精製用試薬(例えば精製用カラム等)、
i)アルコール沈澱用試薬(例えばエタノール溶液、イソプロパノール溶液、高分子キャリアー、酢酸ナトリウム溶液等)。
【0085】
また、前述した如き本発明の取得、増幅、同定方法での使用のための説明書等を含ませておいても良い。当該「説明書」とは、本発明の方法における特徴・原理・操作手順等が文章又は図表等により実質的に記載されている当該キットの取り扱い説明書、添付文書、或いはパンフレット(リーフレット)等を意味する。
【0086】
本発明の方法は、以下のような効果を奏する。
(1)テスターポリヌクレオチド及びドライバーポリヌクレオチドをcDNAライブラリーから作製できる。
従来の方法では、サブトラクションcDNAの作製工程で環状プラスミドを除去することができなかったため、テスターポリヌクレオチド及びドライバーポリヌクレオチド共にcDNAからしか作製できず、cDNA作製の手間や作製できるcDNA量に限界がある等の問題があった。一方、本発明方法では、工程(2)において一本鎖ポリヌクレオチドを分解せずハイブリッド形成された二本鎖ポリヌクレオチドを優先的に分解し得る性質を有する酵素を用いた酵素処理により二本鎖ポリヌクレオチドを除去できるので、テスターポリヌクレオチド及びドライバーポリヌクレオチド共にcDNAライブラリー(プラスミドベクター)から作製できる。
(2)ハイブリダイゼーション(サブトラクティブ・ハイブリダイゼーション)は一回でよい。
従来の方法においては、テスターポリヌクレオチドは二本鎖であったため、再会合したテスターポリヌクレオチド間の二本鎖ハイブリッド形成がされてしまうので、ドライバーポリヌクレオチドを過剰量使用しても、高発現しているテスター非特異的ポリヌクレオチド(ハウスキーピング遺伝子)が残ってしまい、後のPCR増幅に悪影響を及ぼす場合がある。そのため、従来法では、ハイブリダイゼーションを2〜3回以上繰り返す必要があった。これに対して、本発明では、テスターポリヌクレオチドは一本鎖であるので、テスターポリヌクレオチド間の二本鎖ハイブリッドは形成されず、ハイブリダイゼーションは一回行えば充分である。
(3)一回の酵素処理でテスター中に存在するポリヌクレオチドのうち、テスター非特異的ポリヌクレオチドを分解除去することができる。
従来の物理的結合又は吸着によるサブトラクション法においては、サブトラクティブ・ハイブリダイゼーションの結果得られた一本鎖ポリヌクレオチドとハイブリッド形成した二本鎖ポリヌクレオチドとを、物理的結合又は吸着により分離していたため、実際にはこれらを厳密に分離することができず、サブトラクト効率低下の要因となっていた。しかしながら、本発明では酵素処理によりハイブリッド形成した二本鎖ポリヌクレオチドを除去するため、一回の酵素処理で二本鎖ポリヌクレオチドを高効率に除去することができ、効率良く一本鎖テスター特異的ポリヌクレオチドを得ることができる。
(4)テスター特異的ポリヌクレオチドを容易に濃縮できる。
一本鎖テスター特異的ポリヌクレオチドにのみ存在し、ドライバー由来のポリヌクレオチド中に存在しない配列に基づいて設計・作製されたプライマーを用いたPCR等を行えば、ドライバーポリヌクレオチドは増幅されず、テスター特異的ポリヌクレオチドのみが実質的に増幅されるので、テスター特異的ポリヌクレオチドを濃縮することができ、効率的なサブトラクト・ポリヌクレオチドを作製することができる。
(5)ドライバーポリヌクレオチドを容易に除去できる。
本発明の工程(4)により、テスター特異的ポリヌクレオチドに共存しているドライバーポリヌクレオチドを容易に除去することができる。特に、二本鎖テスター特異的ポリヌクレオチドと一本鎖ドライバーポリヌクレオチドが共存している場合には、酵素処理により容易に一本鎖ドライバーポリヌクレオチドを除去することができる。その結果、テスター特異的ポリヌクレオチドの密度をさらに高めることができるので、従来法で問題となっていたドライバーポリヌクレオチドの混入(残存)による影響がさらに低減され、高効率なサブトラクション・ポリヌクレオチドが作製できる。
【0087】
以下に実施例及び比較例を挙げて本発明を詳細に説明するが、本発明はこれらにより何等限定されるものではない。
【実施例】
【0088】
(1)ヒト肝臓癌由来のHep G2細胞からの全RNAの抽出
ISOGEN(ニッポンジーン社製)を用い、その添付文書に従いHep G2細胞(ヒューマンサイエンス研究資源バンク製)から全RNAを抽出した。
Hep G2細胞を225mL培養フラスコ(Nalge Nunc社製)4本に5×106細胞/cm2まで培養し、リン酸緩衝液(pH7.5)で洗浄した。ISOGEN(ニッポンジーン社製)を1培養フラスコに対して15mL入れて、細胞を剥がし、それぞれ、遠心チューブ(Nalge Nunc社製、Oak Riddge Centrifuge Tube,50mL)に移した。それぞれにクロロホルムを3mL入れ、攪拌後、高速遠心機(日立社製、SCR20B)で12000rpm、4℃、15分間遠心分離して、それぞれ、上清を10mL回収した。上清を新しい遠心チューブに移し、10mLのイソプロパノール(和光純薬工業社製)を加えて攪拌し、高速遠心機(日立社製、SCR20B)で12000rpm、4℃、15分間遠心分離した。上清を除去し、沈殿を70% エタノール(和光純薬工業社(株)製)で洗浄し、乾燥後、滅菌水に溶解して、分光光度計(Beckman社製、DU640)にてUV 260nmでRNA濃度を測定した。濃度測定後、滅菌水で2μg/μLに調製したRNA溶液を250μL(500μg)得た。
【0089】
(2)Hep G2細胞由来の全RNAからのPoly(A)+ RNAの抽出
上記(1)で得られたHep G2細胞全RNA 500μgを使用してOligotex-dT 30 Super(タカラバイオテク社製)を用い、その添付文書に従い、Poly(A)+ RNAを抽出した。
Hep G2細胞全RNA 250μL(500μg)に、2×Elution Buffer(2mM EDTA及び0.2% SDS含有20mM Tris-HCl緩衝液、pH7.5)を250μL加えて、混合した。その後、Oligotex-dT 30 Superを加えて、混合し、65℃、5分間加熱後、氷中に急冷した。5M 塩化ナトリウムを100μL加えて、37℃で、10分間インキュベートし、高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、5分間遠心分離した。上清を除去し、滅菌水を400μL加えて、混合し、65℃で、5分間加熱後、高速微量遠心機(日立社製、CF16RX)で、14500rpm、室温で、5分間遠心分離した。上清を回収し、5M 塩化ナトリウムを20μL及びエタノールを1mL加えて、混合し、高速微量遠心機(日立社製、CF16RX)で、14500rpm、4℃で、10分間遠心分離した。上清を除去し70% エタノールで洗浄し、乾燥後、滅菌水に溶解して、分光光度計(Beckman社製、DU640)にてUV 260nmでRNA濃度を測定した。
濃度測定後、滅菌水で0.25μg/μLに調製したPoly(A)+ RNA溶液を28μL(7μg)得た。
【0090】
(3)Hep G2細胞のcDNAライブラリーの作製
上記(2)で得られたHep G2細胞由来のPoly(A)+ RNA 5μgをとり、TimeSaver cDNA Synthesis Kit(Amersham社製)を使用して、その添付文書に従い作製した。ベクターはpNEB193(New England Biolabs 社製)を使用し、形質転換はDH5αを使用した。また、プラスミドライブラリーは、100 mlのLuria-Bertani’s broth(アンピシリンナトリウム 100 μg / ml を含む)で培養し後、回収した。
First-Strand Reaction Mixes(mersham社製)に、65℃、10分間加熱後氷中で急冷した、上記(2)で得られたHep G2 Poly(A)+ RNA 5μg(20μL)、Oligo-dT(24)-Pac I site Primer(5'-TTTTTTTAATTAATTTTTTTTTTTTTTTTTTTTTTTT-3'、シグマジェノシス社製、0.5 mg/ml) 1μL及びDTT Solution(Amersham社製) 1μLを混合し、37℃で、1時間インキュベートして、第一鎖反応(逆転写反応)を行った。Second-Strand Reaction Mixes(Amersham社製)に、第一鎖反応液を混合し、12℃で、30分インキュベート後、22℃、1時間インキュベートして第ニ鎖反応を行った。
次いで、65℃、10分加熱して、第ニ鎖反応を停止させ、SizeSep 400 Spun Columns(Amersham社製)にてPrimer及び低分子DNAを除去し、二本鎖cDNAを得た。
作製した二本鎖cDNA 100μLに、EcoR I/Not I adaptor(Amersham社製)5μL、PEG Buffer(Amersham社製)30μL、ATP Solution(Amersham社製、15mM)1μL及びT4 DNA Ligase(5units )(Amersham社製)1μLを加え、16℃で、1時間インキュベートした後、65℃で、10分加熱してT4 DNA Ligaseを失活させた。得られた反応液(アダプターを結合させたcDNAを含有する溶液)137μLに、フェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を140μL加えて、攪拌し、高速微量遠心機(日立社製、CF16RX)で、14500rpm、室温で、3分間遠心分離した。上清を回収し、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)7μL及びエタノール(和光純薬工業社製)350μLを加えて、混合して、高速微量遠心機(日立社製、CF16RX)で、14500rpm、4℃で、10分間遠心分離した。上清を除去し、70% エタノール(和光純薬工業社製)で洗浄して、乾燥後、滅菌水85μLに溶解した。滅菌水に溶解したcDNAに、10×NEB buffer1(New England Biolabs 社製)10μL、BSA溶液(New England Biolabs 社製)1μL及びPac I(New England Biolabs 社製、10units/μL)4μLを加えて、混合し、37℃で、2時時間インキュベートした後、65℃、20分間加熱し、Pac Iを失活させた。
得られた反応液(Pac Iで切断したcDNA含有溶液)を、SizeSep 400 Spun Columns(Amersham社製)により処理し、未結合のアダプター及び低分子DNAを除去し、全量150μLになるように滅菌水で調製した。これにより、5'末端がEcoR I、3'末端がPac Iの切断末端となったcDNAが得られた。
得られたcDNA 3μL(Poly(A)+ RNAに換算して約100ng)、EcoR I(ニッポンジーン社製)及びPac I(New England Biolabs 社製)で切断したpNEB193 DNA(New England Biolabs 社製)100ng(2μL)、及びDNA Ligation Kit Ver.2.1(タカラバイオテク社製)I液(Reaction Buffer)5μLを混合し、16℃で、1時間インキュベートした後、全量(10μL)をDH5α(100μL)に42℃で、30秒の熱ショックによって形質転換した。形質転換したcDNAライブラリーを100 mlのLuria-Bertani’s broth(アンピシリンナトリウム 100μg/mlを含む)で37℃、16時間培養し、一方向に挿入されたHep G2細胞プラスミドcDNAライブラリーを回収した。
得られたcDNAライブラリーの一部を滅菌水で10ng/μLに調製して、下記(4)のテスターcDNAの調製におけるPCR増幅反応の鋳型とした。
【0091】
(4)テスターcDNAの調製
上記(3)で調製したヒト肝臓癌由来のHepG2細胞のcDNAライブラリーを鋳型として、挿入断片cDNAをTOPOTAQ DNA Polmerase(Fidelity Systems 社製)を使用してPCR増幅した。
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
【0092】
・ヒト肝臓癌由来のHepG2細胞のcDNAライブラリー溶液:1μL(10ng)
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・M13/pUC Forwad Primer溶液:5μL
リン酸化プライマー(5’-P-CCAGTCACGACGTTGTAAAACG-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・M13/pUC Reverse Primer溶液:5μL
非リン酸化プライマー(5’-CACACAGGAAACAGCTATGACC-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:4μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
・滅菌水:30μL
【0093】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 30 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、44μLの滅菌水に溶解した。
次いで、溶解液に、10×Lambda Exonuclease Buffer(EPICENTRE 社製、25mM塩化マグネシウム及び0.1%Triton-X100含有670mMグリシン-KOH緩衝液、pH9.4)5μL及びLambda Exonuclease(EPICENTRE 社製、10units/μL)1μLを加えて、混合し、37℃、30分インキュベートした。次いで、更に80℃、15分インキュベートしてLambda Exonucleaseを失活させた。
これに、50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns (Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が1ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 ×37μg/mL ×(希釈倍率)/ 1000
【0094】
(5)ドライバーcDNAの調製
ヒト正常肝臓組織由来のcDNAライブラリー(BioChain 社製、cDNA Library: Human Adult Normal Tissue: Liver)を鋳型として、挿入断片cDNAをPCR増幅した。
ヒト正常肝臓組織由来のcDNAライブラリー溶液(BioChain 社製、cDNA Library: Human Adult Normal Tissue: Liver)1μL(10ng)を使用した以外は、上記(4)と同じ試薬を用いた。
各試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 30 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、10μLの滅菌水に溶解した。
得られたPCR増幅産物5μgを滅菌水で全量43μLに調製し、10×H Buffer(ニッポンジーン社製)5μL、EcoR I(ニッポンジーン社製、10units/μL)1μL及びXho I(ニッポンジーン社製、10units/μL)1μLを加えて、混合し、37℃で、2時間インキュベートして、挿入断片両側のマルチクローニング・サイトの制限酵素を使用してPCR Primerまでを切断した。その後、65℃で、20分間加熱して、制限酵素を失活させた。
次いで、これに50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns(Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が100ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 × 50μg/mL ×(希釈倍率)/ 1000
【0095】
(6)ハイブリダイゼーション
上記(4)で調製したテスターcDNA 1μL(1ng)及び上記(5)ドライバーcDNA 2μL(200ng)含有溶液 3μLに、1μLの4×Hybridization Buffer(2M NaCl含有200mM HEPES緩衝液、pH7.3)を加えて、混合し、これをサーマルサイクラー(MJ Research 社製、PTC-225)にて98℃で2分間熱変性した後、68℃で15時間、ハイブリダイゼーションした。
【0096】
(7)二本鎖特異的DNAヌクレアーゼ処理によるハイブリッド形成した二本鎖cDNAの除去
68℃に保温したハイブリダイゼーション溶液に、68℃に温めた5μL の2×DSN master Buffer(Evrogen 社製、10mM 塩化マグネシウム及び2mM DTT 100mM含有トリス塩酸緩衝液 pH8.0、)を加え、混合した後、68℃で10分間インキュベートした。これに、二本鎖特異的DNAヌクレアーゼ(Duplex-specific nuclease;Evrogen社製、1 kunitz-units/μL)1μLを加えて、68℃で30分、インキュベートした。
次いで、これに、Stop Solution(5mM EDTA水溶液)10μLを加えて、68℃で5分間インキュベートして二本鎖特異的DNAヌクレアーゼの反応を停止させた。
得られた反応液に、80μL の滅菌水及び等量(100μL)のフェノール:クロロホルム:イソアミルアルコール混合液(25:24:1)混合液(ニッポンジーン社製)を加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、31μLの滅菌水に溶解した。
【0097】
(8)ハイブリッド形成しなかったテスターcDNAのPCR増幅
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ5本に分注した。
【0098】
・上記(7)で調製した一本鎖テスターcDNA含有反応液:31μL
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・M13/pUC Forwad Primer溶液:5μL
リン酸化プライマー(5’-P-CCAGTCACGACGTTGTAAAACG-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・M13/pUC Reverse Primer溶液:5μL
非リン酸化プライマー(5’-CACACAGGAAACAGCTATGACC-3’)(シグマジェノシス社製)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:4μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
【0099】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 2分 35 サイクル、72℃ 5分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR増幅産物(100μL)を、0.6mLマイクロチューブに移して、これに、等量(100μL)のフェノール:クロロホルム:イソアミルアルコール(25:24:1)混合液(ニッポンジーン社製)を加え、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温で、3分間遠心分離後、水相(上清)を0.6mLマイクロチューブに移した。これに、エタ沈メイト(ニッポンジーン社製)1μL、5M NaCl(和光純薬工業社製)5μL及びエタノール(和光純薬工業社製)250μLを加えて、混合した。
高速微量遠心機(日立社製、CF16RX)で14500rpm、室温、5分間遠心分離後、上清を除去し、得られた沈殿物を70% エタノール(和光純薬工業社製)で洗浄後、乾燥させ、44μLの滅菌水に溶解した。
【0100】
(9)Exonuclease I処理によるハイブリッド形成しなかった一本鎖ドライバーcDNAの除去
上記(8)で調製したPCR増幅産物(テスターcDNA増幅産物) 44μLに、10×Exonuclease I Buffer(New England Biolabs 社製、67mM 塩化マグネシウム及び100mM 2-メルカプトエタノール含有670mMグリシン-KOH緩衝液、pH9.5)5μL及びExonuclease I(New England Biolabs 社製、20units/μL)1μLを加えて、混合し、37℃、30分、インキュベートした。次いで、更に80℃、15分インキュベートしてExonuclease Iを失活させた。
これに、50μL 滅菌水を加えて混合し、全量を100μLにして、CHROMA SPIN+TE-400 Columns (Clontech社製)を用いて、低速遠心機(TOMY 社製、LX-120)で700×g、室温で、5分間遠心分離して、低分子DNA及びPCRプライマーを除去した。その一部を分光光度計(Beckman社製、DU640)を用いて、UV 260nmで濃度を測定し、以下の式を用いてDNA濃度が10ng/μLとなるように滅菌水で希釈した。
DNA濃度(μg/μL)=OD260 × 50μg/mL ×(希釈倍率)/ 1000
【0101】
(10)サブトラクションcDNAの評価検討
上記(4)で得られたテスターcDNA(Hep G2 cDNA)、上記(5)で得られたドライバーcDNA(正常肝臓 cDNA)又は上記(9)で得られたテスター特異的cDNA(サブトラクションcDNA)10ngを鋳型として、高発現ハウスキーピング遺伝子である glyceraldehyde-3-phosphate dehydrogenase(GAPDH)(増幅鎖長:859bp)とHep G2特異的発現遺伝子であるalpha-fetoprotein(AFP)(増幅鎖長:931bp)をそれぞれPCR増幅した。
下記の試薬を混合し、その混合液20μLを0.2mL PCRチューブ分注した。
【0102】
・cDAN含有溶液:1μL(10ng/μL cDNA含有)
上記(4)で調製した一本鎖テスターcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの、
上記(5)で調製した二本鎖ドライバーcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの、又は
上記(9)で得られたサブトラクションcDNAをDNA濃度が10ng/μLとなるように滅菌水で希釈したもの。
・6mM MgCl2含有2×Amplification Buffer(Fidelity Systems 社製):50μL
・dNTPs溶液(各dNTP 10mM含有、Amersham 社製):5μL
・Primer1溶液:1μL
GAPDH増幅用プライマー1(5’-GAGTACGTCGTGGAGTCCACTG-3’)又はAFP増幅用プライマー1(5’-GTACGGACATTCAGACTGCTG-3’)を10μMとなるように滅菌水に溶解したもの。
・Primer2溶液:1μL
GAPDH増幅用プライマー2(5’-CCTCACAGTTGCCATGTAGAC-3’)又はAFP増幅用プライマー2(5’-CATCCAGGAGAGCCAAGCATTG-3’)を10μMとなるように滅菌水に溶解したもの。
・TOPOTAQ DNA Polymerase溶液:1μL
TOPOTAQ DNA Polymerase(Fidelity Systems 社製、3units/μL)1μLに1×Dilution buffer(Fidelity Systems 社製)3μLを加えて混合して4μLとしたもの。
・滅菌水5μL
【0103】
次いで、これを95℃ 2分、95℃ 20秒 ⇒ 60℃ 20秒 ⇒ 72℃ 20秒 22 サイクル、72℃ 2分でサーマルサイクラー(MJ Research 社製、PTC-225)にてPCR反応に付した。
得られたPCR 増幅産物(20μL)10μLを、1×TAE Bufferを使用して1.5 % アガロースゲル(ニッポンジーン社製)で電気泳動後、エチジウムブロマイド染色により各遺伝子の増幅量を比較した。
その結果を図8に示す。尚、図中、レーン1は(4)で調製したテスターcDNA(ヒト肝臓癌由来のHepG2細胞のcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン2は(4)で調製したテスターcDNA(ヒト肝臓癌由来のHepG2細胞のcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果を、レーン3は(5)で調製したドライバーcDNA(ヒト正常肝臓組織由来のcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン4は(5)で調製したドライバーcDNA(ヒト正常肝臓組織由来のcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果を、レーン5は(9)で得られたテスター特異的cDNA(サブトラクションcDNA)を鋳型としてGAPDH遺伝子をPCR増幅した結果を、レーン6は(9)で得られたテスター特異的cDNA(サブトラクションcDNA)を鋳型としてAFP遺伝子をPCR増幅した結果をそれぞれ示し、レーン7は分子量マーカー:100bp DNA Step Ladder (100-1.5kbp)(和光純薬工業(株)製)を用いた結果を示す。また、「←1500bp」は1500bpのDNAの泳動位置を、「←1000bp」は1000bpのDNAの泳動位置を、「←500bp」は500bpのDNAの泳動位置を、それぞれ示す。
【0104】
図8から明らかなように、高発現ハウスキーピング遺伝子であるGAPDHはHep G2 cDNA及び正常肝臓組織 cDNAの両者ともPCR増幅が確認できたが、サブトラクションcDNAでは確認できないことが判る。一方、Hep G2特異的発現遺伝子であるAFPは正常肝臓組織 cDNA において、PCR増幅が確認できないが、サブトラクション cDNAにおいてはHep G2 cDNAと同程度のPCR増幅を示した。
以上のことから、本発明によればテスター特異的cDNA(サブトラクション cDNA)を容易に且つ短時間に、高効率に取得し得ることが判る。
【特許請求の範囲】
【請求項1】
以下の工程を含む、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを増幅する方法;
(1)(i)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか、或いは(ii)テスター由来の一本鎖ポリヌクレオチドと、ドライバー由来の二本鎖ポリヌクレオチドとを混合して、熱変性後、ハイブリダイゼーションを行うかして、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成する工程、
(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、
(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び
(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程。
【請求項2】
工程(2)における酵素が、二本鎖特異的DNAヌクレアーゼである請求項1に記載の方法。
【請求項3】
工程(2)が、二本鎖特異的DNAヌクレアーゼを用いて、二本鎖ポリヌクレオチドを分解することにより、ハイブリット形成した二本鎖ポリヌクレオチドを除去するものである請求項1に記載の方法。
【請求項4】
二本鎖特異的DNAヌクレアーゼが、海洋性無脊椎動物由来の二本鎖特異的DNAヌクレアーゼである請求項2又は3に記載の方法。
【請求項5】
テスター由来の一本鎖ポリヌクレオチドが、(1’)テスター由来の二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、により得られたものである請求項1に記載の方法。
【請求項6】
テスター由来のポリヌクレオチド及びドライバー由来のポリヌクレオチドが、DNAである請求項1に記載の方法。
【請求項7】
テスター由来のポリヌクレオチドに対して過剰量のドライバー由来のポリヌクレオチドを使用する請求項1に記載の方法。
【請求項8】
工程(3)がPCRにより行われるものである請求項1に記載の方法。
【請求項9】
テスター由来のポリヌクレオチドが、その5’末端及び3’末端に、工程(3)で行うPCR用のプライマーが結合する既知の配列が結合されているものである請求項8に記載の方法。
【請求項10】
5’末端に結合させる既知配列と3’末端に結合させる既知配列とが相補的な配列ではないものである請求項9に記載の方法。
【請求項11】
当該既知配列が、ドライバー由来のポリヌクレオチド中に存在しない配列からなるものである請求項9に記載の方法。
【請求項12】
ドライバー由来のポリヌクレオチドが、工程(3)で行うPCR用のプライマーが結合する配列を有さないものである請求項9に記載の方法。
【請求項13】
工程(3)が(3’)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドとこれの相補鎖とを増幅してテスター由来の二本鎖ポリヌクレオチドを得る工程である、請求項1に記載の方法。
【請求項14】
工程(4)における酵素が、一本鎖特異的DNAヌクレアーゼである、請求項1に記載の方法。
【請求項15】
請求項1〜14に記載の方法により増幅されたある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを同定することを特徴とするテスターにおける遺伝子変異を同定する方法。
【請求項1】
以下の工程を含む、ある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを増幅する方法;
(1)(i)テスター由来の一本鎖ポリヌクレオチドと、当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となり得るドライバー由来の一本鎖ポリヌクレオチドとを混合してハイブリダイゼーションを行うか、或いは(ii)テスター由来の一本鎖ポリヌクレオチドと、ドライバー由来の二本鎖ポリヌクレオチドとを混合して、熱変性後、ハイブリダイゼーションを行うかして、テスター由来の一本鎖ポリヌクレオチドと当該テスター由来の一本鎖ポリヌクレオチドと相補鎖となるドライバー由来の一本鎖ポリヌクレオチドとの二本鎖ポリヌクレオチドを形成する工程、
(2)ハイブリッド形成した二本鎖ポリヌクレオチドを酵素処理により除去する工程、
(3)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドを増幅する工程、及び
(4)工程(1)でハイブリット形成しなかったドライバー由来の一本鎖ポリヌクレオチドを酵素処理により除去する工程。
【請求項2】
工程(2)における酵素が、二本鎖特異的DNAヌクレアーゼである請求項1に記載の方法。
【請求項3】
工程(2)が、二本鎖特異的DNAヌクレアーゼを用いて、二本鎖ポリヌクレオチドを分解することにより、ハイブリット形成した二本鎖ポリヌクレオチドを除去するものである請求項1に記載の方法。
【請求項4】
二本鎖特異的DNAヌクレアーゼが、海洋性無脊椎動物由来の二本鎖特異的DNAヌクレアーゼである請求項2又は3に記載の方法。
【請求項5】
テスター由来の一本鎖ポリヌクレオチドが、(1’)テスター由来の二本鎖ポリヌクレオチドを調製した後、当該二本鎖ポリヌクレオチドを酵素処理により一本鎖ポリヌクレオチドにする工程、により得られたものである請求項1に記載の方法。
【請求項6】
テスター由来のポリヌクレオチド及びドライバー由来のポリヌクレオチドが、DNAである請求項1に記載の方法。
【請求項7】
テスター由来のポリヌクレオチドに対して過剰量のドライバー由来のポリヌクレオチドを使用する請求項1に記載の方法。
【請求項8】
工程(3)がPCRにより行われるものである請求項1に記載の方法。
【請求項9】
テスター由来のポリヌクレオチドが、その5’末端及び3’末端に、工程(3)で行うPCR用のプライマーが結合する既知の配列が結合されているものである請求項8に記載の方法。
【請求項10】
5’末端に結合させる既知配列と3’末端に結合させる既知配列とが相補的な配列ではないものである請求項9に記載の方法。
【請求項11】
当該既知配列が、ドライバー由来のポリヌクレオチド中に存在しない配列からなるものである請求項9に記載の方法。
【請求項12】
ドライバー由来のポリヌクレオチドが、工程(3)で行うPCR用のプライマーが結合する配列を有さないものである請求項9に記載の方法。
【請求項13】
工程(3)が(3’)ハイブリッド形成しなかったテスター由来の一本鎖ポリヌクレオチドとこれの相補鎖とを増幅してテスター由来の二本鎖ポリヌクレオチドを得る工程である、請求項1に記載の方法。
【請求項14】
工程(4)における酵素が、一本鎖特異的DNAヌクレアーゼである、請求項1に記載の方法。
【請求項15】
請求項1〜14に記載の方法により増幅されたある試料(テスター)での存在量が別の試料(ドライバー)での存在量よりも多いポリヌクレオチドを同定することを特徴とするテスターにおける遺伝子変異を同定する方法。
【図1】


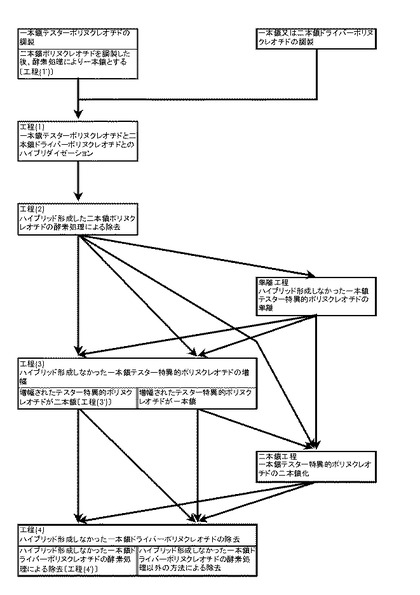
【図2】
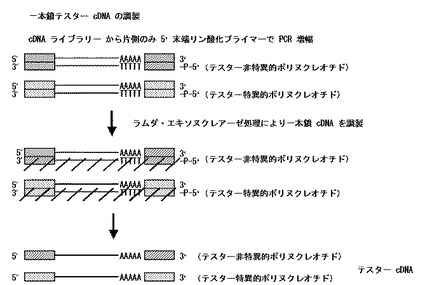
【図3】
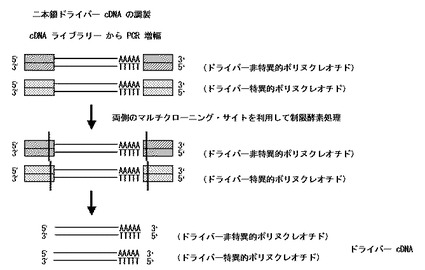
【図4】
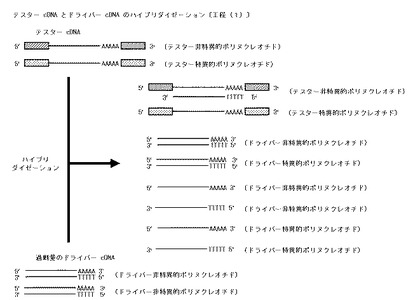
【図5】

【図6】
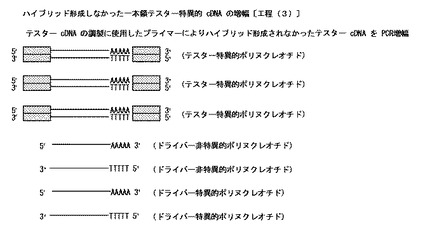
【図7】
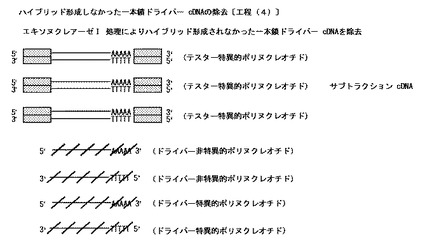
【図8】
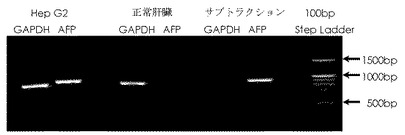
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−165755(P2012−165755A)
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2012−106482(P2012−106482)
【出願日】平成24年5月8日(2012.5.8)
【分割の表示】特願2007−505917(P2007−505917)の分割
【原出願日】平成18年2月27日(2006.2.27)
【出願人】(000252300)和光純薬工業株式会社 (105)
【Fターム(参考)】
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成24年5月8日(2012.5.8)
【分割の表示】特願2007−505917(P2007−505917)の分割
【原出願日】平成18年2月27日(2006.2.27)
【出願人】(000252300)和光純薬工業株式会社 (105)
【Fターム(参考)】
[ Back to top ]