
サラシア属植物由来の組成物、および該組成物の製造法
【課題】サラシア属植物由来の抽出物、該抽出物を含有する血糖値上昇抑制用医薬組成物および健康食品、さらに前記抽出物の品質評価方法を提供する。
【解決手段】脱硫酸コタラノールを高濃度で含有することにより、マルターゼ阻害活性の高いサラシア属植物由来の抽出物を得ることができる。
【解決手段】脱硫酸コタラノールを高濃度で含有することにより、マルターゼ阻害活性の高いサラシア属植物由来の抽出物を得ることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、サラシア属植物由来の組成物、特に脱硫酸コタラノールを含有する組成物、該組成物の利用および製造法に関する。
【背景技術】
【0002】
古来よりサラシア属植物由来の抽出物には糖尿病を予防する効果があることが知られており、インドやスリランカなどで伝承療法として用いられている。最近の研究により、サラシア属植物由来の抽出物には小腸に存在する糖質分解酵素(α−グルコシダーゼ)を強く阻害する作用があり、該抽出物を飲用すると食後の高い血糖濃度が低下することが明らかになっている。当該効果は、食事由来の糖の吸収が遅延することに基づいている。また、サラシア属植物由来の抽出物からサラシノール(特許文献1)、コタラノール(特許文献2)等の化合物が発見され、当該化合物がα−グルコシダーゼ阻害活性を有することが開示されている。
【0003】
これまでに、サラシア属植物由来の抽出物はスクロースを基質とする酵素(スクラーゼ)に対する強い阻害作用を有し、当該作用がα−グルコシダーゼ阻害の基礎になっていると考えられてきた(非特許文献1)。
【0004】
また、サラシア属植物は、品種、産地、保存状態などによって品質が変動するため、サラシア属植物由来の抽出物の品質評価方法が求められている。品質評価法として、マンギフェリンを指標とした間接的な方法(特許文献3)が開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許3030008号
【特許文献2】特開2000−86653号
【特許文献3】特許3386796号
【非特許文献】
【0006】
【非特許文献1】日本食品新素材研究会誌8(2)105−117(2005)
【発明の概要】
【発明が解決しようとする課題】
【0007】
食品にはデンプン由来の糖質が多く含まれているため、食事由来の糖の吸収を効果的に遅延させるには、マルターゼ活性を阻害することが重要と考えられた。さらに、マルターゼ阻害活性に着目した品質評価方法は存在しない。そこで、本発明は、高いマルターゼ阻害活性を有し、優れた血糖値上昇抑制効果を発揮する植物由来の抽出物、およびマルターゼ阻害に着目した植物由来の抽出物の品質評価方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記の課題を達成すべく、本発明者は、脱硫酸コタラノール含量を指標とする、マルターゼ阻害活性の高いサラシア属植物由来の抽出物の製造法を見出した。本発明は、かかる知見に基づいて完成されたものである。
【0009】
本発明の1つの側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物が提供される。本発明における脱硫酸コタラノールは、以下の式(I)で表される構造を有する。
【0010】
【化1】

【0011】
ここで、X−はカウンターイオンであって、任意の陰イオンを意味する。本発明においては、例えば、Cl−、Br−、I−、F−などのハロゲンイオン;BF4−などのルイス酸イオン;その他、酢酸イオン、トリフルオロ酢酸イオン、プロピオン酸イオン、安息香酸イオン等のカルボン酸イオン;リン酸イオン、シアン酸イオン、チオシアン酸イオン等の無機酸イオン;アルキルスルホン酸又はアルキルスルフィン酸イオン;などがあげられるが、これに限定されない。
【0012】
本発明におけるサラシノールとは、以下の式(II)で表される構造を有する。
【0013】
【化2】
【0014】
本発明における組成物とは、脱硫酸コタラノールとサラシノールを含んでいる限り特に限定されない。一つの態様として、当該組成物は、植物を原料として調製することができ、例えば、サラシア属の植物を原料としてもよい。また、本発明における組成物は、当該組成物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が、例えば以下
Y≧1.6X
の関係を有する組成物、あるいは、さらにY重量%とX重量%の合計(X+Y)が0.75重量%以上、特に1.05重量%以上であってよい。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0015】
本発明の一つの態様において、上記組成物の脱硫酸コタラノール含量(Y重量%)は、例えば0.46重量%以上であり、好ましくは0.78重量%以上であり、さらに好ましくは0.86重量%以上である。
【0016】
また、本発明における組成物は、マルターゼ阻害活性を有する。ここで、阻害活性とは、ある化合物または組成物による酵素活性の阻害の程度を示す指標を意味する。阻害活性は、例えば、酵素活性を50%阻害するのに必要な組成物濃度(IC50)として表すことができる。本発明におけるIC50は、Wistar系雄性ラットの小腸より得た酵素溶液を用いるマルターゼ活性阻害試験(本発明の試験例1(1)マルターゼ阻害活性試験)の条件において、例えば、84μg/mL以下、特に79μg/mLである。また、本発明の組成物は、例えば、水または熱水抽出により得ることができる。
【0017】
本発明の別の側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物を含んでなる、血糖値上昇抑制用医薬組成物または飲食品が提供される。前記飲食品として、例えば健康食品または機能性食品などが挙げられる。
【0018】
本発明の別の側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物の製造方法が提供される。前記製造方法は、例えば、サラシア属植物を水または熱水で処理して抽出物を得た後、特定の成分組成を有する前記抽出物を選択し、これを組成物とする工程を含む方法が本発明の一態様としてあげられる。前記特定の成分組成を有する抽出物とは、限定されないが、例えば、抽出物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が以下
Y≧1.6X
X+Y≧0.75
のいずれか一つの関係を有する抽出物をあげることができる。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0019】
また、本発明の一態様において、上記抽出物は一定以上のY重量%、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上を有する抽出物である。
【0020】
本発明の別の側面によれば、脱硫酸コタラノールとサラシノールを含む植物由来の抽出物の品質評価方法であって、前記抽出物の脱硫酸コタラノールの含量とサラシノールの含量に基づいて抽出物を評価する前記方法が提供される。上記側面の一態様において、前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を定量し、前記XおよびYが以下:
Y≧1.6X
X+Y≧0.75
のいずれか一つの関係を有する抽出物を選択する。
【0021】
植物由来の抽出物としては、例えばサラシア属植物由来の抽出物を使用することができる。この品質評価法によれば、α−グルコシダーゼ阻害活性、特にマルターゼ阻害活性の高い抽出物を容易に識別し、これを選択することができる。
【0022】
また、本発明の一態様において、上記抽出物は一定以上のY重量%、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上を有する抽出物である。
【発明の効果】
【0023】
本発明により、α−グルコシダーゼ、特にマルターゼ阻害活性の高い植物由来の組成物が提供される。当該組成物を医薬品または飲食品に使用すれば、食事中に豊富に含まれるデンプン由来の糖の吸収速度を効果的に遅延させ、高血糖濃度を効果的に低下させることができる。さらに、上記組成物の製造方法および品質評価方法によれば、植物由来の組成物を安定して製造することが可能になるため、組成物の品質の均一化および製造コストの低減を実現することができる。
【図面の簡単な説明】
【0024】
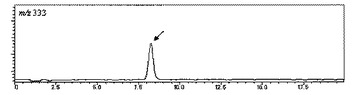
【図1】サラシノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
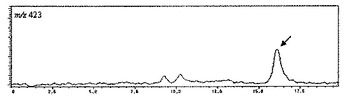
【図2】コタラノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
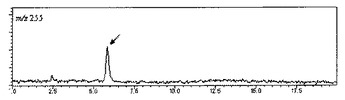
【図3】脱硫酸サラシノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
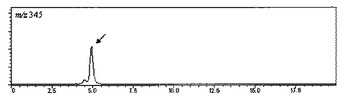
【図4】脱硫酸コタラノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
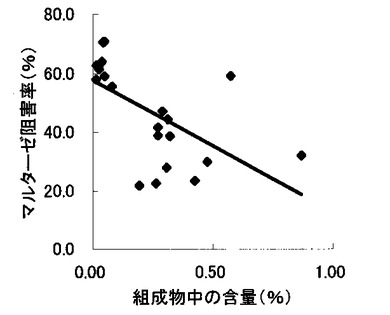
【図5】組成物中のサラシノール含量とマルターゼ阻害率との関係を示す図である。
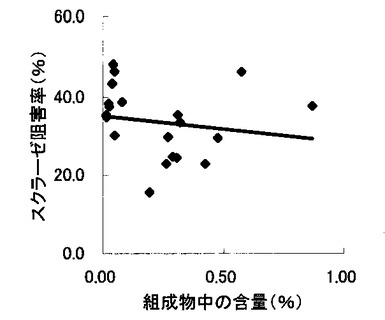
【図6】組成物中のサラシノール含量とスクラーゼ阻害率との関係を示す図である。
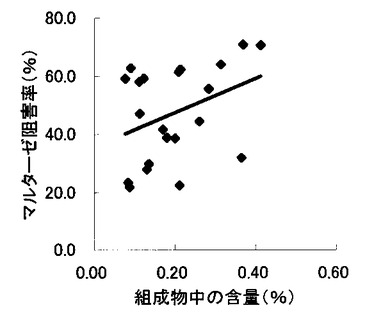
【図7】組成物中のコタラノール含量とマルターゼ阻害率との関係を示す図である。
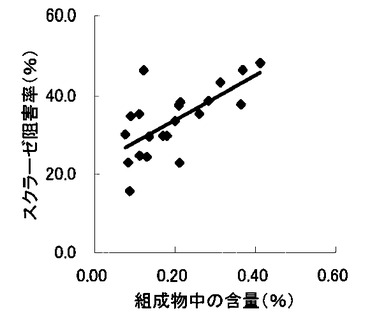
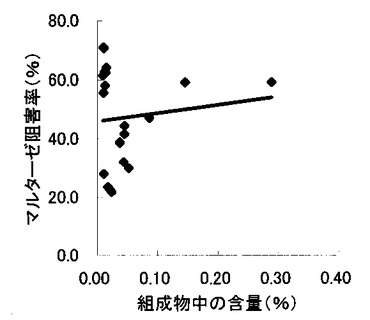
【図8】組成物中のコタラノール含量とスクラーゼ阻害率との関係を示す図である。
【図9】組成物中の脱硫酸サラシノール含量とマルターゼ阻害率との関係を示す図である。
【図10】組成物中の脱硫酸サラシノール含量とスクラーゼ阻害率との関係を示す図である。
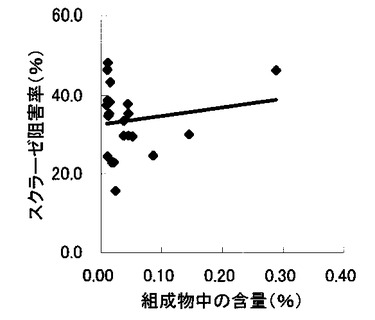
【図11】組成物中の脱硫酸コタラノール含量とマルターゼ阻害率との関係を示す図である。
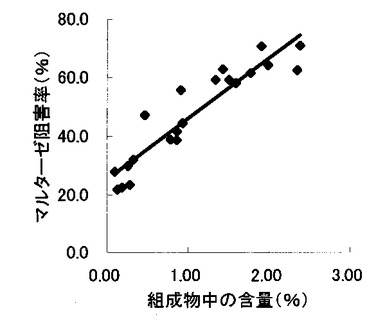
【図12】組成物中の脱硫酸コタラノール含量とスクラーゼ阻害率との関係を示す図である。
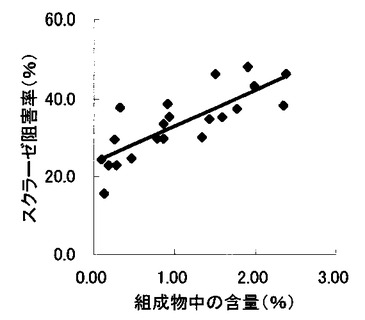
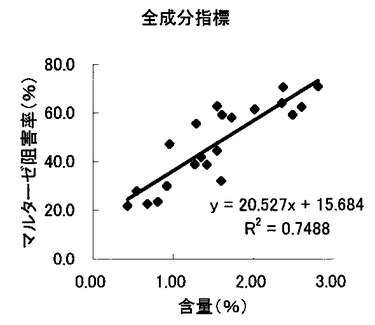
【図13】組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノール含量の合計とマルターゼ阻害率との関係を示す図である。
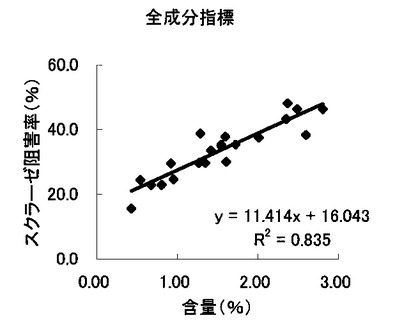
【図14】組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノール含量の合計とスクラーゼ阻害率との関係を示す図である。
【発明を実施するための形態】
【0025】
以下、本発明をさらに具体的に説明する。
本発明の1つの側面は、脱硫酸コタラノールおよびサラシノールを含む組成物に関する。本発明における脱硫酸コタラノールとは、コタラノールの硫酸基が脱離した式(I)の構造を有する化合物である。脱硫酸コタラノールは、例えば、以下のように式(III)の構造を有するコタラノールの脱硫酸化反応によりカウンターイオンがCH3OSO3−の脱硫酸コタラノールを得ることができる(HETEROCYCLES,Vol.75,No.6,1397−1405,2008を参照)。脱硫酸コタラノールのカウンターイオンは、他の任意のカウンターイオンに変換可能である。例えば、上記CH3OSO3−をカウンターイオンに持つ脱硫酸コタラノールを、他の陰イオンをカウンターイオンに持つ陰イオン交換樹脂に供することによって、脱硫酸コタラノールのカウンターイオンを交換する事ができる。例えば、陰イオン交換樹脂であるIRA400(Cl−形)樹脂とCH3OSO3−をカウンターイオンに持つ脱硫酸コタラノールを室温で12時間攪拌することによって、Cl−をカウンターイオンに持つ脱硫酸コタラノールを得ることができる。詳細は、G. Tanabe et al. /Bioorg. Med. Chem. 15(2007)3926−3937を参照。
【0026】
【化3】
【0027】
ここで、X−は先に定義した通りである。
脱硫酸コタラノールは、これまで上記のような合成的手法による入手方法が報告されているのみであった。本発明者は、植物抽出物に脱硫酸コタラノールが存在することを確認した。サラシノールは、式(II)の構造を有し、α−グルコシダーゼ阻害活性を有することが知られている。
【0028】
本発明の組成物は、脱硫酸コタラノールおよびサラシノールを含んでいれば特に限定されないが、好ましくは、植物抽出物を原料に使用することができる。原料の植物としては、例えば、サラシア属の植物があげられる。サラシア属の植物としては、例えば、サラシア・オブロンガ(Salacia oblonga)、サラシア・レティキュラータ(Salacia reticulata)、サラシア・キネンシス(Salacia chinensis)、サラシア・プリノイデス(Salacia prinoides)、サラシア・フルティコーサ(Salacia fruticosa)、サラシア・マクロスペルマ(Salacia macrosperma)などがあげられる。
【0029】
本願の発明者は、高いマルターゼ阻害活性を有する組成物は、脱硫酸コタラノールを多く含んでいることを初めて発見した。さらに、組成物中の脱硫酸コタラノールの含量は、他の活性成分:例えばサラシノール、コタラノール、脱硫酸サラシノール、の含量との関係で特定の比率で存在することを見出した。すなわち、脱硫酸コタラノール含量とその他の成分の含量との存在比から本発明の組成物を特定することができる。したがって、本発明の別の側面は、脱硫酸コタラノールと他の活性成分が特定の比率で存在する組成物に関する。一例として、脱硫酸コタラノール含量とサラシノール含量の組成物中の比率に基づいて本発明の組成物を特定する場合について以下に説明する。
【0030】
本発明の組成物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)の関係は、以下の式
Y≧αX
で表すことができる。前記Y重量%およびX重量%は、それぞれ、組成物中の脱硫酸コタラノール量(y)およびサラシノール量(x)の組成物全体の重量(z)にしめる重量パーセントを意味する。すなわち、前記Y重量%およびX重量%は以下の式により算出することができる。
【0031】
【化4】
【0032】
脱硫酸コタラノールおよびサラシノールの定量は、これらの化合物が定量できる限りにおいていずれの方法を用いてもよいが、例えば、一般的に知られた方法を使用するのが簡便である。このような方法として、例えば、高速液体クロマトグラフィー(HPLC)を利用した方法、酵素免疫測定法(ELISA)があげられる。
【0033】
あるいは、上記方法と異なる方法として、質量分析計などにより化合物を分析した際に生成する特徴的な擬分子イオンを指標とする方法がある。擬分子イオンとは、化合物に対して水素イオンが0もしくは1つ付加した分子イオン、または水素イオンが0もしくは1つ脱離した分子イオンを意味する。当該方法は、例えば、液体クロマトグラフィーと質量分析計を接続したLC−MSにより行うことができる。例えば、サラシノールを下記の分析条件−1に設定したLC−MSで分析すると、質量対電荷比(m/z)=333の擬分子イオンが生成する(保持時間8.2分)。当該擬分子イオンを選択的にモニタリングすれば、当該分子イオンのピーク面積を算出することができる。
【0034】
分析条件−1
<LC条件>
・カラム: Asahipak NH2P-50 (2.0 mm i.d.×150 mm、昭和電工株式会社)
・移動相: アセトニトリル:水=78:22 (v/v)
・流量: 0.2 mL/min
・カラム温度: 40℃
・注入量: 1.0 μL
<MS条件>
・イオン化: ESIネガティブモード
・ネブライズガス: 1.5 L/min
・窒素ガス圧: 0.15 MPa
・CDL 温度: 250℃
・ブロックヒーター温度: 200℃
・CDL 電圧: Constant-mode (-25V)
・Q array DS & RF voltage: Scan-mode
・SIM: m/z=333(サラシノール)、m/z=423(コタラノール)
一方、脱硫酸コタラノールを下記の分析条件−2に設定したLC−MSで分析すると、m/z=345の擬分子イオン(保持時間4.9分)が得られ、上記と同様にピーク面積を算出することができる。
【0035】
分析条件−2
<LC条件>
・カラム: Inertsil ODS-3 (2.1 mm i.d.×150 mm、GLサイエンス)
・移動相: 5 mM HFBA / MeOH=99:1 (v/v)
・流量: 0.2 mL/min
・カラム温度: 40℃
・注入量: 1.0 μL
<MS条件>
・イオン化: ESIポジティブモード
・ネブライズガス: 1.5 L/min
・窒素ガス圧: 0.15 MPa
・CDL 温度: 250℃
・ブロックヒーター温度: 250℃
・CDL 電圧: Constant-mode (-25V)
・Q array DS & RF voltage: Scan-mode
・SIM: m/z=255(脱硫酸サラシノール)、m/z=345(脱硫酸コタラノール)
上記の方法により測定した、サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの各擬分子イオンのピーク面積とそれぞれの標準品濃度との関係を表す検量線を作成すれば、組成物中の各化合物の含量を算出することができる。
【0036】
上記関係式中のαは、実験的に求めることができる。詳しくは、脱硫酸コタラノール(Y重量%)とサラシノール含量(X重量%)の比率とマルターゼ阻害活性との関係から導き出すことができる。αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。αの値は、脱硫酸コタラノールと対比する他の活性成分の種類によって変化することは、容易に理解できる。
【0037】
あるいは、上記方法において検量線を作成せず、組成物に含まれる各活性成分に対応する擬分子イオンの単位重量当たりのピーク面積比およびピーク面積の和を指標にすることもできる。例えば、脱硫酸コタラノールのm/z=345のピーク面積とサラシノールのm/z=333のピーク面積の比、およびこれらのピーク面積の和を指標にできる。この手法によれば、分析によって得られるピーク面積を直接使用するため、検量線を作成する上記方法に比べてより簡便で迅速に目的の組成物を選択することができる。
【0038】
本発明の別の側面は、組成物が上記Y≧1.6Xの関係を満たし、さらにX+Yが一定以上である組成物に関する。X+Yは、例えば0.75重量%以上であり、好ましくは1.05重量%以上、さらに好ましくは1.18重量%以上である。
【0039】
また、本発明の一態様として、上記組成物のY重量%は、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上である。
本発明の別の側面は、マルターゼ阻害活性が一定以上の組成物に関する。阻害活性は、例えば、マルターゼ活性を50%阻害するのに必要な組成物の濃度(IC50)で表すことができる。IC50は特に限定されないが、例えば、Wistar系雄性ラットの小腸より得た酵素溶液を用いるマルターゼ活性阻害試験(本発明の試験例1(1)マルターゼ阻害活性試験)の条件では84μg/mL以下、好ましくは79μg/mL以下、さらに好ましくは75μg/mL以下である。
【0040】
本発明の組成物は、特に限定されないが、例えば抽出物であってもよい。抽出物は、例えば、サラシア属の植物を水、熱水、有機溶媒、好ましくは水または熱水で処理することにより得ることができる。
【0041】
本発明の別の側面は、本発明の組成物を含んでなる血糖値上昇抑制用医薬組成物に関する。当該医薬組成物は、マルターゼ活性を効果的に阻害することにより、食事デンプン由来の糖の吸収を抑制することができるため、血糖値の上昇を抑制することができる。
【0042】
本発明の別の側面は、本発明の組成物を含んでなる飲食品に関する。飲食品とは、固体状、液状、ゲル状、その形式は特に制限されない。健康食品および機能性食品などが飲食品の例としてあげられる。飲食品の製造において本発明の組成物を添加する工程を設けてもよいし、飲食品の完成後に本発明の組成物を添加してもよい。
【0043】
上記医薬組成物、飲食品、健康食品および機能性食品は、糖尿病、糖尿病合併症および腎症の予防、治療または症状の軽減などに有効である。当該事項は、容易に理解できる。
前記医薬組成物および飲食品は、必要に応じ、従来公知の着色剤、保存剤、香料、風味剤、コーティング剤などの成分を配合して調製することもできる。
【0044】
また、本発明の医薬組成物および飲食品は、1以上の追加成分を配合して調製してもよい。追加成分の例としては、抗酸化剤、血糖降下剤、抗コレステロール剤、免疫賦活剤、ビタミン、アミノ酸、ペプチド、タンパク質、ミネラル分(鉄、亜鉛、マグネシウム、ヨードなど)、脂肪酸(EPA、DHAなど)などをあげることができる。
【0045】
ここで、抗酸化剤の例としては、特に限定はされないが、乾燥酵母、グルタチオン、リポ酸、ケルセチン、カテキン、コエンザイムQ10、エンゾジノール、プロアントシアニジン類、アントシアニジン、アントシアニン、カロチン類、リコピン、フラボノイド、リザベラトロール、イソフラボン類、亜鉛、メラトニン、および植物由来成分(例えば、イチョウ葉、月桃葉、ハイビスカス、またはそれらの抽出物)などが挙げられる。
【0046】
血糖降下剤の例としては、特に限定はされないが、難消化性デキストリン、α−リノレン酸、豆鼓エキス、小麦アルブミン、L−アラビノース、および植物由来成分(例えば、グアバ葉、桑葉、しょうが、アマチャヅル、オオムギ、キダチアロエ、セイヨウタンポポ、ダイダイ、チョウセンアザミ、ニンニク、ハトムギ、バナバ、ビルベリー、ブラックコホシュ、マコモ、杜仲葉、月見草、カイアポ、ニガウリ、マデグルシルまたはそれらの抽出物)などが挙げられる。
【0047】
抗コレステロール剤の例としては、特に限定はされないが、大豆タンパク質、リン脂質結合大豆ペプチド、キトサン、植物ステロール、植物ステロールエステル、植物スタノールエステル、難消化性デキストリン、アルギン酸ナトリウム、サイリウム種皮、アスタキサンチン、イノシトール、コエンザイムA、カルシウム、マグネシウム、カルニチン、シルクプロテイン、タウリン、メチオニン、α-リノレン酸、グアガム、コンドロイチン硫酸、大豆サポニン、および植物由来成分(例えば、アマチャヅル、アルファルファ、イチョウ、オオバコ、オオムギ、オーツ麦、オリーブ、ガジュツ、ギムネマ、キャッツクロー、クコ、クロレラ、スピルリナ、西洋サンザシ、唐辛子、ニンニク、ビルベリー、ベニバナ、ユッカ、ラフマ、アガリクス、紅麹、またはそれらの抽出物)などがあげられる。
【0048】
免疫賦活剤の例としては、特に限定はされないが、ラクトフェリン、アルギニン、トリプトファン、バリン、ロイシン、キチン、キトサン、および植物由来成分(例えば、アガリクス、冬虫夏草、アロエ、キダチアロエ、エキナセア、オウギ、キャッツクロー、クコ、スピルリナ、ハトムギ、紅花、マカ、マコモ、ラフマ、またはそれらの抽出物)などが挙げられる。
【0049】
ビタミンの例としては、特に限定はされないが、ビタミンA群に属するビタミン[例えば、レチナール、レチノール、レチノイン酸、カロチン、デヒドロレチナール、リコピンおよび薬理学的に許容されるそれらの塩類(例えば、酢酸レチノール、パルミチン酸レチノールなど)など]、ビタミンB群に属するビタミン[例えば、チアミン、チアミンジスルフィド、ジセチアミン、オクトチアミン、シコチアミン、ビスイブチアミン、ビスベンチアミン、プロスルチアミン、ベンフォチアミン、フルスルチアミン、リボフラビン、フラビンアデニンジヌクレオチド、ピリドキシン、ピリドキサール、ヒドロキソコバラミン、シアノコバラミン、メチルコバラミン、デオキシアデノコバラミン、葉酸、テトラヒドロ葉酸、ジヒドロ葉酸、ニコチン酸、ニコチン酸アミド、ニコチニックアルコール、パントテン酸、パンテノール、ビオチン、コリン、イノシトール、パンガミン酸およびそれらの薬理学的に許容されるこれらの塩類(例えば、塩酸チアミン、硝酸チアミン、塩酸ジセチアミン、塩酸フルスルチアミン、酪酸リボフラビン、フラビンアデニンジヌクレオチドナトリウム、塩酸ピリドキシン、リン酸ピリドキサール、リン酸ピリドキサールカルシウム、塩酸ヒドロキソコバラミン、酢酸ヒドロキソコバラミン、パントテン酸カルシウム、パントテン酸ナトリウムなど)など]、ビタミンC群に属するビタミン[アスコルビン酸及びその誘導体、エリソルビン酸及びその誘導体、および薬理学的に許容されるそれらの塩類(例えば、アスコルビン酸ナトリウム、エリソルビン酸ナトリウムなど)など]、ビタミンD群に属するビタミン[例えば、エルゴカルシフェロール、コレカルシフェロール、ヒドロキシコレカルシフェロール、ジヒドロキシコレカルシフェロール、ジヒドロタキステロール、および薬理学的に許容されるそれらの塩類など]、ビタミンE群に属するビタミン[例えば、トコフェロール及びその誘導体、ユビキノン誘導体及びそれらの薬理学的に許容される塩類(酢酸トコフェロール、ニコチン酸トコフェロール、コハク酸トコフェロール、コハク酸トコフェロールカルシウムなど)など]、その他のビタミン[例えば、カルニチン、フェルラ酸、γ−オリザノール、オロチン酸、ルチン(ビタミンP)、エリオシトリン、ヘスペリジン、および薬理学的に許容されるそれらの塩類(塩化カルニチンなど)など〕などが挙げられる。
【0050】
アミノ酸の例としては、特に限定はされないが、ロイシン、イソロイシン、バリン、メチオニン、トレオニン、アラニン、フェニルアラニン、トリプトファン、リジン、グリシン、アスパラギン、アスパラギン酸、セリン、グルタミン、グルタミン酸、プロリン、チロシン、システイン、ヒスチジン、オルニチン、ヒドロキシプロリン、ヒドロキシリジン、グリシルグリシン、アミノエチルスルホン酸(タウリン)、シスチン、または薬理学的に許容されるそれらの塩類(例えばアスパラギン酸カリウム、アスパラギン酸マグネシウム、塩酸システインなど)などが挙げられる。好ましい例は、バリン、ロイシンおよびイソロイシン等の分岐鎖アミノ酸、グルタチオン、システイン、グルタミン酸、グリシン、セリン、トリプトファン、チロシン、フェニルアラニン、ヒスチジン、メチオニン、スレオニン、リジン、シスチン、アルギニン、アラニン、アスパラギン酸、プロリン、アミノエチルスルホン酸である。
【0051】
本発明の組成物は、医薬組成物または飲食品(特に、機能性食品、健康食品、サプリメントなど)として継続的な摂取が行いやすいように、例えば顆粒剤(ドライシロップを含む)、カプセル剤(軟カプセル剤、硬カプセル剤)、錠剤(チュアブル剤などを含む)、散剤(粉末剤)、丸剤などの各種の固形製剤、または内服用液剤(液剤、懸濁剤、シロップ剤を含む)などの液状製剤などの形態で調製することができ、成分の安定性や摂取の簡便さの点からカプセル剤または錠剤の形態が好ましいが、特に限定されるものではない。
【0052】
カプセル剤または錠剤の形態の本発明の組成物は、医薬または食品として許容される公知の添加物を用いて製造することができ、医薬または食品の分野で採用されている通常の製剤化手法を適用することができる。例えば、錠剤は、各成分を処方に従って添加配合し、粉砕、造粒、乾燥、整粒および混合を行い、得られた調製混合物を打錠することによって調製することができる。
【0053】
製剤化のための添加物としては、限定はされないが、例えば、賦形剤、滑沢剤、結合剤、崩壊剤、流動化剤、分散剤、湿潤剤、防腐剤、粘稠剤、pH調整剤、着色剤、矯味矯臭剤、界面活性剤、溶解補助剤などが挙げられる。また、液剤の形態にする場合は、ペクチン、キサンタンガム、グアガムなどの増粘剤を配合することができる。また、コーティング剤を用いてコーティング錠剤にしたり、ペースト状の膠剤とすることもできる。さらに、他の形態に調製する場合であっても、従来の方法に従えばよい。
【0054】
さらに、本発明の組成物は、例えば、飲料、菓子類、パン類、スープ類などの各種飲食品またはその添加成分として;またはドッグフード、キャットフードなどの各種ペットフードまたはその添加成分として使用することができる。これらの飲食品の製造方法は、本発明の効果を損なわないものであれば特に限定されず、各用途で当業者によって使用されている方法に従えばよい。
【0055】
本発明の別の側面は、本発明の組成物の製造方法に関する。一態様として、例えば、以下の工程を含む製造方法があげられる:
工程1) サラシア属植物を水または熱水で処理すること;および
工程2) 抽出物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が以下の
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの関係を有する抽出物を選択し、これを組成物とする工程。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0056】
上記工程1は、サラシア属植物から有効成分を抽出する工程である。サラシア属植物としては、例えば、採取後に乾燥した幹、根、葉、または幹および根部の樹皮を裁断または粉砕したものを使用することができる。
【0057】
抽出は慣用の方法を適宜利用して行うことができ、例えば、連続抽出、浸漬抽出、向流抽出、超臨界抽出などの方法により行ってもよい。
抽出溶媒としては特に限定されないが、水、低級アルコール(メタノールおよびエタノール)、アセトンなどの親水性溶媒、またはそれらの混合溶媒を用いることが好ましく、特に水を使用することが好ましい。抽出時には加熱することが好ましく、例えば、抽出溶媒の還流温度で抽出を行うことができる。水を溶媒として使用する場合は、例えば、60〜110℃、好ましくは80〜90℃の温度下、例えば1〜24時間、好ましくは1〜4時間の抽出時間で抽出を行うことができる。
【0058】
本発明で使用する抽出物として、抽出液の原液、抽出液の濃縮液または抽出液濃縮物を乾燥して得られる固体を使用することができ、摂取の効率性の観点から固体化した濃縮物を使用するのが好ましい。抽出液の濃縮方法としては従来技術を適宜利用することができ、例えば、減圧乾燥法、凍結乾燥法、噴霧乾燥法などを行うことができる。
【0059】
サラシア属植物の抽出液は精製処理に付してもよい。精製処理としては、例えば、活性炭、イオン吸着樹脂などの吸着剤による処理、液−液向流分配処理などがあげられる。
サラシア属植物の抽出物は市販品を購入したものを使用してもよい。
【0060】
上記工程2は、工程1で得られた抽出物から、脱硫酸コタラノールとサラシノールの含量が一定の関係を満たす抽出物を選択し、これを組成物とする工程である。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。抽出物中の脱硫酸コタラノールとサラシノールの含量が以下:
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの条件を満たす植物由来の抽出物を選択し、これを高いマルターゼ阻害活性を有する組成物とすることができる。
【0061】
ここにおいて、αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。βは0.75以上であればよく、特に1.05以上であってよい。γは0.46以上、好ましくは0.78以上、さらに好ましくは0.86以上であってよい。
【0062】
別の側面において、本発明は、脱硫酸コタラノールとサラシノールを含む植物由来の抽出物の品質評価方法であって、評価対象となる抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)含量(Y重量%)およびサラシノール含量(X重量%)を定量し、YとXの関係が以下:
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの条件を満たす場合には、α−グルコシダーゼ阻害活性、特にマルターゼ阻害活性の高い植物由来の抽出物であると判断する。上記品質評価方法により、α−グルコシダーゼ阻害活性の高い植物由来の抽出物を選択することできる。
【0063】
ここにおいて、αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。βは0.75以上であればよく、特に1.05以上であってよい。γは0.46以上、好ましくは0.78以上、さらに好ましくは0.86以上であってよい。
【実施例】
【0064】
本発明をより具体的に説明するが、本発明の範囲はこれに限定されるものではない。
[実施例1] サラシノールおよびコタラノールの標準品の調製
自然乾燥させたタイ産サラシア・キネンシス幹部(15kg)を粉砕し、水(150L)を加えて95〜100℃で2時間、加熱抽出を行った。ろ過によりろ液を回収した。残渣に水(150L)を加え、上記と同様の操作を行った。上記2回の操作により得られたろ液を一つに合わせた。これを減圧濃縮させた後、噴霧乾燥させることによって熱水抽出物(収量1.41kg、収率9.37%)を得た。続いて当該熱水抽出物(1kg)にメタノール(10L)を加え、80℃で3時間処理し、ろ過によりろ液を回収した。残渣にメタノール(10L)を加え、上記と同様の操作を2回繰り返した。上記3回の操作により得られたメタノール抽出液を一つに合わせ、減圧下で溶媒を留去させ、メタノール抽出エキス(収量347.54g、収率3.26%)を得た。当該抽出エキス(303.8g)を蒸留水5Lに溶解し、これをDIAION-HP20樹脂(4kg、三菱化学株式会社)を用いたカラムクロマトグラフィーに供し、水溶出部(収量167.64g、収率1.80%)を得た。当該水溶出部(150gを)を蒸留水(7.5L)に溶かした後、Duolite A368S樹脂(OH形、3.75L、住化ケムテックス株式会社)を用いたカラムクロマトグラフィーに供し、蒸留水を通液後、酢酸水溶液(0.1N)を用いて酢酸溶出部(収量8.10g、収率0.10%)を得た。次に、当該酢酸溶出部(7.0g)を75%(v/v)アセトニトリル(700mL)に溶かした後、Chromatorex NH樹脂(210g、富士シリシア化学株式会社)を用いたカラムクロマトグラフィーに供し、75%(v/v)アセトニトリル溶出部(収量2.78g、収率0.039%)、50%(v/v)アセトニトリル溶出部(収量1.80g、収率0.025%)をそれぞれ得た。次にそれぞれをCOSMOSIL Sugar-D (20×250mm、ナカライテスク株式会社)を用いた高速液体クロマトグラフィーに供し、最終的にサラシノール(収量681.7mg、収率0.0094%)、コタラノール(収量230.4mg、収率0.0032%)を得た。両化合物の構造を1H−NMR、13C−NMRおよび質量分析計で解析した結果、以下の式(II)および式(III)の構造を有する化合物であることが確認された。これらをサラシノールの標準品およびコタラノールの標準品として使用した。
【0065】
【化5】
【0066】
[実施例2] 脱硫酸サラシノールおよび脱硫酸コタラノールの標準品の調製
サラシノール標準品(28mg)を5%(v/v)塩化水素含有メタノール(0.6mL)に溶解し、45℃で3時間反応させた。反応後、減圧下で溶媒を留去させて残渣を得た。当該残渣をKieselgel 60(70−230mesh、メルク株式会社)を用いたカラムクロマトグラフィーに供し、最終的に下記構造式に示される脱硫酸サラシノール(カウンターイオンとしてCH3OSO3−がイオン結合したもの、27mg)を得た。同様に、コタラノール(10mg)を5%(v/v)塩化水素含有メタノール(1.4mL)に溶解し、45℃で3時間反応させた。反応後、減圧下で溶媒を留去させて残渣を得た。当該残渣をKieselgel 60(70−230mesh、メルク株式会社)を用いたカラムクロマトグラフィーに供し、最終的に下記構造式に示される脱硫酸コタラノール(カウンターイオンとしてCH3OSO3−がイオン結合したもの、10mg)を得た。両化合物の構造を1H−NMR、13C−NMRおよび質量分析にかけた結果、以下の式(IV)および式(I)の構造を有する化合物であることが確認された。これらを脱硫酸サラシノールの標準品および脱硫酸コタラノールの標準品として使用した。
【0067】
【化6】
【0068】
ここで、X−は先に定義した通りである。
[実施例3] サラシア属植物由来の抽出物の調製
表1に示したサラシア属植物(No.1〜21)をそれぞれ小型超高速粉砕機WB−1(大阪ケミカル株式会社)で粉砕し、破砕粉末を得た。当該破砕粉末(2g)に蒸留水(20mL)を加え、95℃で120分間、撹拌しながら加熱還流による抽出を行った。その後、遠心分離(1500×g、室温、5分間)を行い、上清を回収した。残渣に蒸留水(20mL)を加え、95℃で120分間、上記と同様に加熱還流による抽出を行った。その後、遠心分離(1500×g、室温、5分間)を行い、上清を回収した。上記のようにして得られた上清を一つにして100mL容メスフラスコに移し、蒸留水で100mLにメスアップした。当該液(10mL)をメンブレンフィルター(孔径0.45μm)でろ過し、得られたろ液をサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの定量分析に使用した。一方、当該液の残り(90mL)は、減圧濃縮させた後、凍結乾燥させた。これをα−グルコシダーゼ阻害活性の測定用のサンプルとした。なお、抽出物の調製に用いた上記破砕粉末については、別に105℃、8時間での乾燥減量を測定し、抽出に用いた破砕粉末の乾燥重量を算出した。
【0069】
【表1】
【0070】
[実施例4] 組成物中の成分の定量分析
上記実施例3で調製した組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの定量分析は、高速液体クロマトグラフ質量分析装置(LC−MS)によって行った。
【0071】
LC部分は、システムコントローラ:CBM−20A、ポンプ:Shimadzu LC−20AB、紫外分光光度計検出器(UV):Shimadzu SPD−20A、カラムオーブン:CTO−20A、オートサンプラー:SIL−20A、から構成される(いずれも株式会社島津製作所)。MS部分は、Shimadzu LC−MS−2010EV(株式会社島津製作所)を用いた。
【0072】
サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールのMS部分よる検出は、各化合物に特徴的な擬分子イオンを選択的にモニタリングすることによって行った。各化合物の擬分子イオンのm/zは、サラシノール:m/z=333、コタラノール:m/z=423、脱硫酸サラシノール:m/z=255および脱硫酸コタラノール:m/z=345である。分析条件を以下に示す。
【0073】
分析条件−1:サラシノールおよびコタラノールの分析条件
<LC条件>
・カラム: Asahipak NH2P−50(2.0mmi.d.×150mm、昭和電工株式会社)
・移動相: アセトニトリル:水=78:22 (v/v)
・流量: 0.2mL/min
・カラム温度: 40℃
・注入量: 1.0μL
<MS条件>
・イオン化: ESIネガティブモード
・ネブライズガス: 1.5L/min
・窒素ガス圧: 0.15MPa
・CDL温度: 250℃
・ブロックヒーター温度: 200℃
・CDL電圧: Constant−mode(−25V)
・Q array DS & RF voltage: Scan−mode
・SIM: m/z=333(サラシノール)、m/z=423(コタラノール)
分析条件−2:脱硫酸サラシノールおよび脱硫酸コタラノールの分析条件
<LC条件>
・カラム: Inertsil ODS−3(2.1mmi.d.×150mm、ジーエルサイエンス株式会社)
・移動相: 5mM HFBA/MeOH=99:1 (v/v)
・流量: 0.2mL/min
・カラム温度: 40℃
・注入量: 1.0μL
<MS条件>
・イオン化: ESIポジティブモード
・ネブライズガス: 1.5L/min
・窒素ガス圧: 0.15MPa
・CDL温度: 250℃
・ブロックヒーター温度: 250℃
・CDL電圧: Constant−mode(−25V)
・Q array DS & RF voltage: Scan−mode
・SIM: m/z=255(脱硫酸サラシノール)、m/z=345(脱硫酸コタラノール)
分析結果は、表1のNo.1〜21のサンプル全てについて得ることができた。代表例として、表1のNo.1のサンプルのクロマトチャートを示す(図1〜4)。m/z=333(サラシノール)は8.2分、m/z=423(コタラノール)は16.1分、m/z=255(脱硫酸サラシノール)は5.8分、m/z=345(脱硫酸コタラノール)は4.9分の保持時間にピークが現れた。これらピークの保持時間は、各標準品のピークの保持時間と一致した。
【0074】
実施例1および2で調製した各標準品を用いて作成した検量線に基づいて、上記擬分子イオンのピーク面積からサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量を絶対検量線法にて算出した(表2)。なお、脱硫酸サラシノール、および脱硫酸コタラノールの標準品は、カウンターイオン(CH3OSO3−)を含んでいるため、以下の式によりカウンターイオンを含まない濃度に換算してこれらの検量線を作成した。
【0075】
A=B×255/366
A:[カウンターイオンを含まない脱硫酸サラシノール濃度]
B:[カウンターイオン(CH3OSO3−)を含む脱硫酸サラシノール濃度]
C=D×345/456
C:[カウンターイオンを含まない脱硫酸コタラノール濃度]
D:[カウンターイオン(CH3OSO3−)を含む脱硫酸コタラノール濃度]
【0076】
【表2−1】
【0077】
【表2−2】
【0078】
[試験例1] α−グルコシダーゼ活性阻害試験
(1)マルターゼ活性阻害試験
酵素溶液:Wistar系雄性ラットの小腸を摘出後、小腸内面の粘膜をスライドガラスを用いて採取した。採取した前記粘膜をマンニトール(50mM)を含有するトリス緩衝液(2mM、pH7.1)中でホモジナイズした後、塩化カルシウム(終濃度10mM)を添加し、混合した。得られた懸濁液を遠心分離(3000×g、4℃、5分間)し、上清を回収した。当該上清をさらに超遠心分離(27000×g、4℃、30分間)し、得られた沈殿をマレイン酸緩衝液(0.1M、pH6.0)に懸濁させ、回収した。当該懸濁液はα−グルコシダーゼを含んでいるため、当該懸濁液を酵素溶液として使用した。当該酵素溶液は、マルターゼ活性とスクラーゼ活性を有する。
【0079】
基質溶液:マルトース(ナカライテスク株式会社)をマレイン酸緩衝液(0.1M、pH6.0)に溶解し、マルトース基質溶液(74mM)を調製した。
試料溶液:実施例3で調製した各サンプルをジメチルスルホキシドおよびマレイン酸緩衝液(0.1M、pH6.0)に溶解し、試料溶液(240μg/mL、10%ジメチルスルホキシドを含む)を調製した。
【0080】
酵素反応:基質溶液(50μL)に試料溶液(25μL)を加えた後、酵素液(25μL)を加え、37℃で30分間反応させた。沸騰水浴中で2分間加熱し、酵素を失活させて反応を停止させた。これを評価サンプルとした。一方、酵素溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを酵素ブランク1とした。また、試料溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを試料ブランクとした。さらに、酵素溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)、および試料溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを酵素ブランク2とした。また、比較例として、α−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠(商標、武田薬品工業株式会社、1錠(200mg)あたりボグリボース0.3mg)についても同様の操作を行った。
【0081】
遊離グルコースの定量:反応を停止させた上記反応液中のD−グルコースの濃度をグルコースCIIテストワコー(和光純薬工業株式会社)により測定し、酵素反応によって生成したD−グルコース濃度を以下の式により算出した。
【0082】
[評価サンプルの生成D−グルコース濃度(A)]=[評価サンプルのD−グルコース濃度]−[酵素ブランク1のD−グルコース濃度]
[試料ブランクの生成D−グルコース濃度(B)]=[試料ブランクのD−グルコース濃度]−[酵素ブランク2のD−グルコース濃度]
さらに、上記のようにして算出した、評価サンプルの生成D−グルコース濃度および試料ブランクの生成D−グルコース濃度を下式にあてはめることによって、各サンプルのマルターゼ阻害率を算出した。
【0083】
マルターゼ阻害率(%)=100−{(A/B)×100}
結果を表3に示す。
【0084】
【表3】
【0085】
(2)スクラーゼ阻害活性試験
基質溶液:スクロース(ナカライテスク株式会社)をマレイン酸緩衝液(0.1M、pH6.0)に溶解し、スクロース基質溶液(74mM)を調製した。
【0086】
試料溶液:実施例3で調製した各サンプルをジメチルスルホキシドおよびマレイン酸緩衝液(0.1M、pH6.0)に溶解し、試料溶液(80μg/mL、10%ジメチルスルホキシドを含む)を調製した。
【0087】
当該基質溶液および試料溶液を用いる以外は、上記(1)の方法に従った。比較例として、α−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠についても同様の操作を行った。これにより各サンプルのスクラーゼ阻害率を算出した。結果を表4に示す。
【0088】
【表4】
【0089】
[試験例2]
上記試験例1(1)の試験法において、各試料溶液中のサンプル濃度を変化(2000、800、240、80、24μg/mL)させ、各サンプルのマルターゼ阻害活性IC50(マルターゼ活性を50%阻害する濃度)を測定した。また、比較例としてα−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠についても同様の操作を行った。結果を表5に示す。
【0090】
【表5】
【0091】
上記試験例1〜3の結果(表3〜5)を考慮すると、α−グルコシダーゼ阻害活性(マルターゼ阻害活性およびスクラーゼ阻害活性)は、サンプルごとにバラツキがあることが判明した。これは、原料となるサラシア植物の品質がその種類、生産国および樹齢や採取時期などによって異なることを示唆し、さらに品質が一定した原料の供給が極めて困難であることも示唆する。すなわち、最終製品のα−グルコシダーゼ阻害活性が一定しないことになり、ひいては目的の効果を十分に発揮できない製品が製造されることになるため、品質保証の観点から好ましくない。そこで、α−グルコシダーゼ阻害活性と各サンプル中に含まれるサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量との関係について以下のような検討を行った。
【0092】
表2〜4の結果に基づいて、上記サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量とマルターゼ阻害率もしくはスクラーゼ阻害率との関係を表す図を作成した(図5〜12)。さらに、各図のプロットに基づいて線形近似式および相関係数(r)を算出した(表6)。
【0093】
【表6】
【0094】
これらの結果から、サラシノールと脱硫酸サラシノール含量は、マルターゼ阻害率またはスクラーゼ阻害率のいずれとも相関はほとんどないことが判明した(図5〜8)。コタラノール含量は、スクラーゼ阻害率(図10)とある程度の相関があることが示唆されるものの、マルターゼ阻害率(図9)との相関は明らかでなかった。唯一、脱硫酸コタラノール含量のみがマルターゼ阻害率(図11)およびスクラーゼ阻害率(図12)の両方と高い相関があることが判明した。
【0095】
さらに、各サンプル中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量の合計とマルターゼ阻害率およびスクラーゼ阻害率との関係について検討を行った(図13および14)。当該検討結果から、上記4つの化合物によるマルターゼ阻害率は、脱硫酸コタラノール含量にほぼ依存していることが明らかになった。また、スクラーゼ阻害率に関しても、その大部分が脱硫酸コタラノール含量に依存していることが示された。
【0096】
以上の結果を鑑みると、本発明の組成物においては、脱硫酸コタラノールがα−グルコシダーゼ阻害活性の主な活性成分であることが明らかである。特に、マルターゼ阻害活性との高い相関が示された。本願出願時において、サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの単離された化合物は、α−グルコシダーゼ活性を同程度で阻害することが技術常識として知られている。このような技術常識を考慮すると、上記の結果は全くの予想外といえる。
【0097】
さらに、本発明者は、高いα−グルコシダーゼ阻害活性を有するサンプルを選択するための指標について検討を行った。各サンプル中の脱硫酸コタラノール含量、サラシノール含量、およびマルターゼ阻害活性(IC50)の関係を以下にまとめた(表7)。
【0098】
【表7−1】
【0099】
【表7−2】
【0100】
表7の結果から、IC50が特に低い(マルターゼ阻害活性が高いことを意味する)サンプル(表7の判定の欄に○があるもの)は、脱硫酸コタラノールとサラシノールの含量比(DesKo/Sa)が1.6以上であることが明らかとなった。さらに、サンプル中の脱硫酸コタラノールの存在量が0.09重量%以上であればマルターゼ阻害活性が認められ、さらに0.46重量%以上であれば極めて高いマルターゼ阻害活性が認められることが明らかとなった。このような指標を用いれば、サラシア属植物から組成物を調製した場合に、目的の効果を奏する組成物を迅速、容易に選択することができるため、組成物のα−グルコシダーゼ阻害活性の品質評価を容易に行うことができる。さらに、当該品質評価方法は、組成物のα−グルコシダーゼ阻害活性を任意の規格値に調整する用途にも適しているため、α−グルコシダーゼ阻害活性が均一な組成物を安定的に製造することができる。
【0101】
また、上記の0.09重量%および0.46重量%の脱硫酸コタラノールを含有する組成物は、それぞれサンプルNo.16および6に対応する。当該サンプルNo.16および6の製造に用いた原料の脱硫酸コタラノール含量は、表2から、それぞれ0.018重量%および0.055重量%である。すなわち、この値を指標にすれば、本発明の組成物を調製するために適した原料を選択することができる。これにより、高いα−グルコシダーゼ阻害活性、特にマルターゼ阻害活性を有する植物由来の組成物の製造を容易かつ効率的に行うこと、および組成物の品質評価をより容易に行うことができる。
【技術分野】
【0001】
本発明は、サラシア属植物由来の組成物、特に脱硫酸コタラノールを含有する組成物、該組成物の利用および製造法に関する。
【背景技術】
【0002】
古来よりサラシア属植物由来の抽出物には糖尿病を予防する効果があることが知られており、インドやスリランカなどで伝承療法として用いられている。最近の研究により、サラシア属植物由来の抽出物には小腸に存在する糖質分解酵素(α−グルコシダーゼ)を強く阻害する作用があり、該抽出物を飲用すると食後の高い血糖濃度が低下することが明らかになっている。当該効果は、食事由来の糖の吸収が遅延することに基づいている。また、サラシア属植物由来の抽出物からサラシノール(特許文献1)、コタラノール(特許文献2)等の化合物が発見され、当該化合物がα−グルコシダーゼ阻害活性を有することが開示されている。
【0003】
これまでに、サラシア属植物由来の抽出物はスクロースを基質とする酵素(スクラーゼ)に対する強い阻害作用を有し、当該作用がα−グルコシダーゼ阻害の基礎になっていると考えられてきた(非特許文献1)。
【0004】
また、サラシア属植物は、品種、産地、保存状態などによって品質が変動するため、サラシア属植物由来の抽出物の品質評価方法が求められている。品質評価法として、マンギフェリンを指標とした間接的な方法(特許文献3)が開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許3030008号
【特許文献2】特開2000−86653号
【特許文献3】特許3386796号
【非特許文献】
【0006】
【非特許文献1】日本食品新素材研究会誌8(2)105−117(2005)
【発明の概要】
【発明が解決しようとする課題】
【0007】
食品にはデンプン由来の糖質が多く含まれているため、食事由来の糖の吸収を効果的に遅延させるには、マルターゼ活性を阻害することが重要と考えられた。さらに、マルターゼ阻害活性に着目した品質評価方法は存在しない。そこで、本発明は、高いマルターゼ阻害活性を有し、優れた血糖値上昇抑制効果を発揮する植物由来の抽出物、およびマルターゼ阻害に着目した植物由来の抽出物の品質評価方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記の課題を達成すべく、本発明者は、脱硫酸コタラノール含量を指標とする、マルターゼ阻害活性の高いサラシア属植物由来の抽出物の製造法を見出した。本発明は、かかる知見に基づいて完成されたものである。
【0009】
本発明の1つの側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物が提供される。本発明における脱硫酸コタラノールは、以下の式(I)で表される構造を有する。
【0010】
【化1】
【0011】
ここで、X−はカウンターイオンであって、任意の陰イオンを意味する。本発明においては、例えば、Cl−、Br−、I−、F−などのハロゲンイオン;BF4−などのルイス酸イオン;その他、酢酸イオン、トリフルオロ酢酸イオン、プロピオン酸イオン、安息香酸イオン等のカルボン酸イオン;リン酸イオン、シアン酸イオン、チオシアン酸イオン等の無機酸イオン;アルキルスルホン酸又はアルキルスルフィン酸イオン;などがあげられるが、これに限定されない。
【0012】
本発明におけるサラシノールとは、以下の式(II)で表される構造を有する。
【0013】
【化2】
【0014】
本発明における組成物とは、脱硫酸コタラノールとサラシノールを含んでいる限り特に限定されない。一つの態様として、当該組成物は、植物を原料として調製することができ、例えば、サラシア属の植物を原料としてもよい。また、本発明における組成物は、当該組成物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が、例えば以下
Y≧1.6X
の関係を有する組成物、あるいは、さらにY重量%とX重量%の合計(X+Y)が0.75重量%以上、特に1.05重量%以上であってよい。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0015】
本発明の一つの態様において、上記組成物の脱硫酸コタラノール含量(Y重量%)は、例えば0.46重量%以上であり、好ましくは0.78重量%以上であり、さらに好ましくは0.86重量%以上である。
【0016】
また、本発明における組成物は、マルターゼ阻害活性を有する。ここで、阻害活性とは、ある化合物または組成物による酵素活性の阻害の程度を示す指標を意味する。阻害活性は、例えば、酵素活性を50%阻害するのに必要な組成物濃度(IC50)として表すことができる。本発明におけるIC50は、Wistar系雄性ラットの小腸より得た酵素溶液を用いるマルターゼ活性阻害試験(本発明の試験例1(1)マルターゼ阻害活性試験)の条件において、例えば、84μg/mL以下、特に79μg/mLである。また、本発明の組成物は、例えば、水または熱水抽出により得ることができる。
【0017】
本発明の別の側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物を含んでなる、血糖値上昇抑制用医薬組成物または飲食品が提供される。前記飲食品として、例えば健康食品または機能性食品などが挙げられる。
【0018】
本発明の別の側面によれば、脱硫酸コタラノールおよびサラシノールを含む組成物の製造方法が提供される。前記製造方法は、例えば、サラシア属植物を水または熱水で処理して抽出物を得た後、特定の成分組成を有する前記抽出物を選択し、これを組成物とする工程を含む方法が本発明の一態様としてあげられる。前記特定の成分組成を有する抽出物とは、限定されないが、例えば、抽出物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が以下
Y≧1.6X
X+Y≧0.75
のいずれか一つの関係を有する抽出物をあげることができる。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0019】
また、本発明の一態様において、上記抽出物は一定以上のY重量%、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上を有する抽出物である。
【0020】
本発明の別の側面によれば、脱硫酸コタラノールとサラシノールを含む植物由来の抽出物の品質評価方法であって、前記抽出物の脱硫酸コタラノールの含量とサラシノールの含量に基づいて抽出物を評価する前記方法が提供される。上記側面の一態様において、前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を定量し、前記XおよびYが以下:
Y≧1.6X
X+Y≧0.75
のいずれか一つの関係を有する抽出物を選択する。
【0021】
植物由来の抽出物としては、例えばサラシア属植物由来の抽出物を使用することができる。この品質評価法によれば、α−グルコシダーゼ阻害活性、特にマルターゼ阻害活性の高い抽出物を容易に識別し、これを選択することができる。
【0022】
また、本発明の一態様において、上記抽出物は一定以上のY重量%、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上を有する抽出物である。
【発明の効果】
【0023】
本発明により、α−グルコシダーゼ、特にマルターゼ阻害活性の高い植物由来の組成物が提供される。当該組成物を医薬品または飲食品に使用すれば、食事中に豊富に含まれるデンプン由来の糖の吸収速度を効果的に遅延させ、高血糖濃度を効果的に低下させることができる。さらに、上記組成物の製造方法および品質評価方法によれば、植物由来の組成物を安定して製造することが可能になるため、組成物の品質の均一化および製造コストの低減を実現することができる。
【図面の簡単な説明】
【0024】
【図1】サラシノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
【図2】コタラノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
【図3】脱硫酸サラシノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
【図4】脱硫酸コタラノールの擬分子イオンの選択イオンクロマトグラムを示す図である。
【図5】組成物中のサラシノール含量とマルターゼ阻害率との関係を示す図である。
【図6】組成物中のサラシノール含量とスクラーゼ阻害率との関係を示す図である。
【図7】組成物中のコタラノール含量とマルターゼ阻害率との関係を示す図である。
【図8】組成物中のコタラノール含量とスクラーゼ阻害率との関係を示す図である。
【図9】組成物中の脱硫酸サラシノール含量とマルターゼ阻害率との関係を示す図である。
【図10】組成物中の脱硫酸サラシノール含量とスクラーゼ阻害率との関係を示す図である。
【図11】組成物中の脱硫酸コタラノール含量とマルターゼ阻害率との関係を示す図である。
【図12】組成物中の脱硫酸コタラノール含量とスクラーゼ阻害率との関係を示す図である。
【図13】組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノール含量の合計とマルターゼ阻害率との関係を示す図である。
【図14】組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノール含量の合計とスクラーゼ阻害率との関係を示す図である。
【発明を実施するための形態】
【0025】
以下、本発明をさらに具体的に説明する。
本発明の1つの側面は、脱硫酸コタラノールおよびサラシノールを含む組成物に関する。本発明における脱硫酸コタラノールとは、コタラノールの硫酸基が脱離した式(I)の構造を有する化合物である。脱硫酸コタラノールは、例えば、以下のように式(III)の構造を有するコタラノールの脱硫酸化反応によりカウンターイオンがCH3OSO3−の脱硫酸コタラノールを得ることができる(HETEROCYCLES,Vol.75,No.6,1397−1405,2008を参照)。脱硫酸コタラノールのカウンターイオンは、他の任意のカウンターイオンに変換可能である。例えば、上記CH3OSO3−をカウンターイオンに持つ脱硫酸コタラノールを、他の陰イオンをカウンターイオンに持つ陰イオン交換樹脂に供することによって、脱硫酸コタラノールのカウンターイオンを交換する事ができる。例えば、陰イオン交換樹脂であるIRA400(Cl−形)樹脂とCH3OSO3−をカウンターイオンに持つ脱硫酸コタラノールを室温で12時間攪拌することによって、Cl−をカウンターイオンに持つ脱硫酸コタラノールを得ることができる。詳細は、G. Tanabe et al. /Bioorg. Med. Chem. 15(2007)3926−3937を参照。
【0026】
【化3】
【0027】
ここで、X−は先に定義した通りである。
脱硫酸コタラノールは、これまで上記のような合成的手法による入手方法が報告されているのみであった。本発明者は、植物抽出物に脱硫酸コタラノールが存在することを確認した。サラシノールは、式(II)の構造を有し、α−グルコシダーゼ阻害活性を有することが知られている。
【0028】
本発明の組成物は、脱硫酸コタラノールおよびサラシノールを含んでいれば特に限定されないが、好ましくは、植物抽出物を原料に使用することができる。原料の植物としては、例えば、サラシア属の植物があげられる。サラシア属の植物としては、例えば、サラシア・オブロンガ(Salacia oblonga)、サラシア・レティキュラータ(Salacia reticulata)、サラシア・キネンシス(Salacia chinensis)、サラシア・プリノイデス(Salacia prinoides)、サラシア・フルティコーサ(Salacia fruticosa)、サラシア・マクロスペルマ(Salacia macrosperma)などがあげられる。
【0029】
本願の発明者は、高いマルターゼ阻害活性を有する組成物は、脱硫酸コタラノールを多く含んでいることを初めて発見した。さらに、組成物中の脱硫酸コタラノールの含量は、他の活性成分:例えばサラシノール、コタラノール、脱硫酸サラシノール、の含量との関係で特定の比率で存在することを見出した。すなわち、脱硫酸コタラノール含量とその他の成分の含量との存在比から本発明の組成物を特定することができる。したがって、本発明の別の側面は、脱硫酸コタラノールと他の活性成分が特定の比率で存在する組成物に関する。一例として、脱硫酸コタラノール含量とサラシノール含量の組成物中の比率に基づいて本発明の組成物を特定する場合について以下に説明する。
【0030】
本発明の組成物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)の関係は、以下の式
Y≧αX
で表すことができる。前記Y重量%およびX重量%は、それぞれ、組成物中の脱硫酸コタラノール量(y)およびサラシノール量(x)の組成物全体の重量(z)にしめる重量パーセントを意味する。すなわち、前記Y重量%およびX重量%は以下の式により算出することができる。
【0031】
【化4】
【0032】
脱硫酸コタラノールおよびサラシノールの定量は、これらの化合物が定量できる限りにおいていずれの方法を用いてもよいが、例えば、一般的に知られた方法を使用するのが簡便である。このような方法として、例えば、高速液体クロマトグラフィー(HPLC)を利用した方法、酵素免疫測定法(ELISA)があげられる。
【0033】
あるいは、上記方法と異なる方法として、質量分析計などにより化合物を分析した際に生成する特徴的な擬分子イオンを指標とする方法がある。擬分子イオンとは、化合物に対して水素イオンが0もしくは1つ付加した分子イオン、または水素イオンが0もしくは1つ脱離した分子イオンを意味する。当該方法は、例えば、液体クロマトグラフィーと質量分析計を接続したLC−MSにより行うことができる。例えば、サラシノールを下記の分析条件−1に設定したLC−MSで分析すると、質量対電荷比(m/z)=333の擬分子イオンが生成する(保持時間8.2分)。当該擬分子イオンを選択的にモニタリングすれば、当該分子イオンのピーク面積を算出することができる。
【0034】
分析条件−1
<LC条件>
・カラム: Asahipak NH2P-50 (2.0 mm i.d.×150 mm、昭和電工株式会社)
・移動相: アセトニトリル:水=78:22 (v/v)
・流量: 0.2 mL/min
・カラム温度: 40℃
・注入量: 1.0 μL
<MS条件>
・イオン化: ESIネガティブモード
・ネブライズガス: 1.5 L/min
・窒素ガス圧: 0.15 MPa
・CDL 温度: 250℃
・ブロックヒーター温度: 200℃
・CDL 電圧: Constant-mode (-25V)
・Q array DS & RF voltage: Scan-mode
・SIM: m/z=333(サラシノール)、m/z=423(コタラノール)
一方、脱硫酸コタラノールを下記の分析条件−2に設定したLC−MSで分析すると、m/z=345の擬分子イオン(保持時間4.9分)が得られ、上記と同様にピーク面積を算出することができる。
【0035】
分析条件−2
<LC条件>
・カラム: Inertsil ODS-3 (2.1 mm i.d.×150 mm、GLサイエンス)
・移動相: 5 mM HFBA / MeOH=99:1 (v/v)
・流量: 0.2 mL/min
・カラム温度: 40℃
・注入量: 1.0 μL
<MS条件>
・イオン化: ESIポジティブモード
・ネブライズガス: 1.5 L/min
・窒素ガス圧: 0.15 MPa
・CDL 温度: 250℃
・ブロックヒーター温度: 250℃
・CDL 電圧: Constant-mode (-25V)
・Q array DS & RF voltage: Scan-mode
・SIM: m/z=255(脱硫酸サラシノール)、m/z=345(脱硫酸コタラノール)
上記の方法により測定した、サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの各擬分子イオンのピーク面積とそれぞれの標準品濃度との関係を表す検量線を作成すれば、組成物中の各化合物の含量を算出することができる。
【0036】
上記関係式中のαは、実験的に求めることができる。詳しくは、脱硫酸コタラノール(Y重量%)とサラシノール含量(X重量%)の比率とマルターゼ阻害活性との関係から導き出すことができる。αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。αの値は、脱硫酸コタラノールと対比する他の活性成分の種類によって変化することは、容易に理解できる。
【0037】
あるいは、上記方法において検量線を作成せず、組成物に含まれる各活性成分に対応する擬分子イオンの単位重量当たりのピーク面積比およびピーク面積の和を指標にすることもできる。例えば、脱硫酸コタラノールのm/z=345のピーク面積とサラシノールのm/z=333のピーク面積の比、およびこれらのピーク面積の和を指標にできる。この手法によれば、分析によって得られるピーク面積を直接使用するため、検量線を作成する上記方法に比べてより簡便で迅速に目的の組成物を選択することができる。
【0038】
本発明の別の側面は、組成物が上記Y≧1.6Xの関係を満たし、さらにX+Yが一定以上である組成物に関する。X+Yは、例えば0.75重量%以上であり、好ましくは1.05重量%以上、さらに好ましくは1.18重量%以上である。
【0039】
また、本発明の一態様として、上記組成物のY重量%は、例えば0.46重量%以上、好ましくは0.78重量%以上、さらに好ましくは0.86重量%以上である。
本発明の別の側面は、マルターゼ阻害活性が一定以上の組成物に関する。阻害活性は、例えば、マルターゼ活性を50%阻害するのに必要な組成物の濃度(IC50)で表すことができる。IC50は特に限定されないが、例えば、Wistar系雄性ラットの小腸より得た酵素溶液を用いるマルターゼ活性阻害試験(本発明の試験例1(1)マルターゼ阻害活性試験)の条件では84μg/mL以下、好ましくは79μg/mL以下、さらに好ましくは75μg/mL以下である。
【0040】
本発明の組成物は、特に限定されないが、例えば抽出物であってもよい。抽出物は、例えば、サラシア属の植物を水、熱水、有機溶媒、好ましくは水または熱水で処理することにより得ることができる。
【0041】
本発明の別の側面は、本発明の組成物を含んでなる血糖値上昇抑制用医薬組成物に関する。当該医薬組成物は、マルターゼ活性を効果的に阻害することにより、食事デンプン由来の糖の吸収を抑制することができるため、血糖値の上昇を抑制することができる。
【0042】
本発明の別の側面は、本発明の組成物を含んでなる飲食品に関する。飲食品とは、固体状、液状、ゲル状、その形式は特に制限されない。健康食品および機能性食品などが飲食品の例としてあげられる。飲食品の製造において本発明の組成物を添加する工程を設けてもよいし、飲食品の完成後に本発明の組成物を添加してもよい。
【0043】
上記医薬組成物、飲食品、健康食品および機能性食品は、糖尿病、糖尿病合併症および腎症の予防、治療または症状の軽減などに有効である。当該事項は、容易に理解できる。
前記医薬組成物および飲食品は、必要に応じ、従来公知の着色剤、保存剤、香料、風味剤、コーティング剤などの成分を配合して調製することもできる。
【0044】
また、本発明の医薬組成物および飲食品は、1以上の追加成分を配合して調製してもよい。追加成分の例としては、抗酸化剤、血糖降下剤、抗コレステロール剤、免疫賦活剤、ビタミン、アミノ酸、ペプチド、タンパク質、ミネラル分(鉄、亜鉛、マグネシウム、ヨードなど)、脂肪酸(EPA、DHAなど)などをあげることができる。
【0045】
ここで、抗酸化剤の例としては、特に限定はされないが、乾燥酵母、グルタチオン、リポ酸、ケルセチン、カテキン、コエンザイムQ10、エンゾジノール、プロアントシアニジン類、アントシアニジン、アントシアニン、カロチン類、リコピン、フラボノイド、リザベラトロール、イソフラボン類、亜鉛、メラトニン、および植物由来成分(例えば、イチョウ葉、月桃葉、ハイビスカス、またはそれらの抽出物)などが挙げられる。
【0046】
血糖降下剤の例としては、特に限定はされないが、難消化性デキストリン、α−リノレン酸、豆鼓エキス、小麦アルブミン、L−アラビノース、および植物由来成分(例えば、グアバ葉、桑葉、しょうが、アマチャヅル、オオムギ、キダチアロエ、セイヨウタンポポ、ダイダイ、チョウセンアザミ、ニンニク、ハトムギ、バナバ、ビルベリー、ブラックコホシュ、マコモ、杜仲葉、月見草、カイアポ、ニガウリ、マデグルシルまたはそれらの抽出物)などが挙げられる。
【0047】
抗コレステロール剤の例としては、特に限定はされないが、大豆タンパク質、リン脂質結合大豆ペプチド、キトサン、植物ステロール、植物ステロールエステル、植物スタノールエステル、難消化性デキストリン、アルギン酸ナトリウム、サイリウム種皮、アスタキサンチン、イノシトール、コエンザイムA、カルシウム、マグネシウム、カルニチン、シルクプロテイン、タウリン、メチオニン、α-リノレン酸、グアガム、コンドロイチン硫酸、大豆サポニン、および植物由来成分(例えば、アマチャヅル、アルファルファ、イチョウ、オオバコ、オオムギ、オーツ麦、オリーブ、ガジュツ、ギムネマ、キャッツクロー、クコ、クロレラ、スピルリナ、西洋サンザシ、唐辛子、ニンニク、ビルベリー、ベニバナ、ユッカ、ラフマ、アガリクス、紅麹、またはそれらの抽出物)などがあげられる。
【0048】
免疫賦活剤の例としては、特に限定はされないが、ラクトフェリン、アルギニン、トリプトファン、バリン、ロイシン、キチン、キトサン、および植物由来成分(例えば、アガリクス、冬虫夏草、アロエ、キダチアロエ、エキナセア、オウギ、キャッツクロー、クコ、スピルリナ、ハトムギ、紅花、マカ、マコモ、ラフマ、またはそれらの抽出物)などが挙げられる。
【0049】
ビタミンの例としては、特に限定はされないが、ビタミンA群に属するビタミン[例えば、レチナール、レチノール、レチノイン酸、カロチン、デヒドロレチナール、リコピンおよび薬理学的に許容されるそれらの塩類(例えば、酢酸レチノール、パルミチン酸レチノールなど)など]、ビタミンB群に属するビタミン[例えば、チアミン、チアミンジスルフィド、ジセチアミン、オクトチアミン、シコチアミン、ビスイブチアミン、ビスベンチアミン、プロスルチアミン、ベンフォチアミン、フルスルチアミン、リボフラビン、フラビンアデニンジヌクレオチド、ピリドキシン、ピリドキサール、ヒドロキソコバラミン、シアノコバラミン、メチルコバラミン、デオキシアデノコバラミン、葉酸、テトラヒドロ葉酸、ジヒドロ葉酸、ニコチン酸、ニコチン酸アミド、ニコチニックアルコール、パントテン酸、パンテノール、ビオチン、コリン、イノシトール、パンガミン酸およびそれらの薬理学的に許容されるこれらの塩類(例えば、塩酸チアミン、硝酸チアミン、塩酸ジセチアミン、塩酸フルスルチアミン、酪酸リボフラビン、フラビンアデニンジヌクレオチドナトリウム、塩酸ピリドキシン、リン酸ピリドキサール、リン酸ピリドキサールカルシウム、塩酸ヒドロキソコバラミン、酢酸ヒドロキソコバラミン、パントテン酸カルシウム、パントテン酸ナトリウムなど)など]、ビタミンC群に属するビタミン[アスコルビン酸及びその誘導体、エリソルビン酸及びその誘導体、および薬理学的に許容されるそれらの塩類(例えば、アスコルビン酸ナトリウム、エリソルビン酸ナトリウムなど)など]、ビタミンD群に属するビタミン[例えば、エルゴカルシフェロール、コレカルシフェロール、ヒドロキシコレカルシフェロール、ジヒドロキシコレカルシフェロール、ジヒドロタキステロール、および薬理学的に許容されるそれらの塩類など]、ビタミンE群に属するビタミン[例えば、トコフェロール及びその誘導体、ユビキノン誘導体及びそれらの薬理学的に許容される塩類(酢酸トコフェロール、ニコチン酸トコフェロール、コハク酸トコフェロール、コハク酸トコフェロールカルシウムなど)など]、その他のビタミン[例えば、カルニチン、フェルラ酸、γ−オリザノール、オロチン酸、ルチン(ビタミンP)、エリオシトリン、ヘスペリジン、および薬理学的に許容されるそれらの塩類(塩化カルニチンなど)など〕などが挙げられる。
【0050】
アミノ酸の例としては、特に限定はされないが、ロイシン、イソロイシン、バリン、メチオニン、トレオニン、アラニン、フェニルアラニン、トリプトファン、リジン、グリシン、アスパラギン、アスパラギン酸、セリン、グルタミン、グルタミン酸、プロリン、チロシン、システイン、ヒスチジン、オルニチン、ヒドロキシプロリン、ヒドロキシリジン、グリシルグリシン、アミノエチルスルホン酸(タウリン)、シスチン、または薬理学的に許容されるそれらの塩類(例えばアスパラギン酸カリウム、アスパラギン酸マグネシウム、塩酸システインなど)などが挙げられる。好ましい例は、バリン、ロイシンおよびイソロイシン等の分岐鎖アミノ酸、グルタチオン、システイン、グルタミン酸、グリシン、セリン、トリプトファン、チロシン、フェニルアラニン、ヒスチジン、メチオニン、スレオニン、リジン、シスチン、アルギニン、アラニン、アスパラギン酸、プロリン、アミノエチルスルホン酸である。
【0051】
本発明の組成物は、医薬組成物または飲食品(特に、機能性食品、健康食品、サプリメントなど)として継続的な摂取が行いやすいように、例えば顆粒剤(ドライシロップを含む)、カプセル剤(軟カプセル剤、硬カプセル剤)、錠剤(チュアブル剤などを含む)、散剤(粉末剤)、丸剤などの各種の固形製剤、または内服用液剤(液剤、懸濁剤、シロップ剤を含む)などの液状製剤などの形態で調製することができ、成分の安定性や摂取の簡便さの点からカプセル剤または錠剤の形態が好ましいが、特に限定されるものではない。
【0052】
カプセル剤または錠剤の形態の本発明の組成物は、医薬または食品として許容される公知の添加物を用いて製造することができ、医薬または食品の分野で採用されている通常の製剤化手法を適用することができる。例えば、錠剤は、各成分を処方に従って添加配合し、粉砕、造粒、乾燥、整粒および混合を行い、得られた調製混合物を打錠することによって調製することができる。
【0053】
製剤化のための添加物としては、限定はされないが、例えば、賦形剤、滑沢剤、結合剤、崩壊剤、流動化剤、分散剤、湿潤剤、防腐剤、粘稠剤、pH調整剤、着色剤、矯味矯臭剤、界面活性剤、溶解補助剤などが挙げられる。また、液剤の形態にする場合は、ペクチン、キサンタンガム、グアガムなどの増粘剤を配合することができる。また、コーティング剤を用いてコーティング錠剤にしたり、ペースト状の膠剤とすることもできる。さらに、他の形態に調製する場合であっても、従来の方法に従えばよい。
【0054】
さらに、本発明の組成物は、例えば、飲料、菓子類、パン類、スープ類などの各種飲食品またはその添加成分として;またはドッグフード、キャットフードなどの各種ペットフードまたはその添加成分として使用することができる。これらの飲食品の製造方法は、本発明の効果を損なわないものであれば特に限定されず、各用途で当業者によって使用されている方法に従えばよい。
【0055】
本発明の別の側面は、本発明の組成物の製造方法に関する。一態様として、例えば、以下の工程を含む製造方法があげられる:
工程1) サラシア属植物を水または熱水で処理すること;および
工程2) 抽出物中の脱硫酸コタラノールの含量(Y重量%)とサラシノールの含量(X重量%)が以下の
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの関係を有する抽出物を選択し、これを組成物とする工程。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。
【0056】
上記工程1は、サラシア属植物から有効成分を抽出する工程である。サラシア属植物としては、例えば、採取後に乾燥した幹、根、葉、または幹および根部の樹皮を裁断または粉砕したものを使用することができる。
【0057】
抽出は慣用の方法を適宜利用して行うことができ、例えば、連続抽出、浸漬抽出、向流抽出、超臨界抽出などの方法により行ってもよい。
抽出溶媒としては特に限定されないが、水、低級アルコール(メタノールおよびエタノール)、アセトンなどの親水性溶媒、またはそれらの混合溶媒を用いることが好ましく、特に水を使用することが好ましい。抽出時には加熱することが好ましく、例えば、抽出溶媒の還流温度で抽出を行うことができる。水を溶媒として使用する場合は、例えば、60〜110℃、好ましくは80〜90℃の温度下、例えば1〜24時間、好ましくは1〜4時間の抽出時間で抽出を行うことができる。
【0058】
本発明で使用する抽出物として、抽出液の原液、抽出液の濃縮液または抽出液濃縮物を乾燥して得られる固体を使用することができ、摂取の効率性の観点から固体化した濃縮物を使用するのが好ましい。抽出液の濃縮方法としては従来技術を適宜利用することができ、例えば、減圧乾燥法、凍結乾燥法、噴霧乾燥法などを行うことができる。
【0059】
サラシア属植物の抽出液は精製処理に付してもよい。精製処理としては、例えば、活性炭、イオン吸着樹脂などの吸着剤による処理、液−液向流分配処理などがあげられる。
サラシア属植物の抽出物は市販品を購入したものを使用してもよい。
【0060】
上記工程2は、工程1で得られた抽出物から、脱硫酸コタラノールとサラシノールの含量が一定の関係を満たす抽出物を選択し、これを組成物とする工程である。ここで、脱硫酸コタラノールの含量は、カウンターイオンを含めない含量である。抽出物中の脱硫酸コタラノールとサラシノールの含量が以下:
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの条件を満たす植物由来の抽出物を選択し、これを高いマルターゼ阻害活性を有する組成物とすることができる。
【0061】
ここにおいて、αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。βは0.75以上であればよく、特に1.05以上であってよい。γは0.46以上、好ましくは0.78以上、さらに好ましくは0.86以上であってよい。
【0062】
別の側面において、本発明は、脱硫酸コタラノールとサラシノールを含む植物由来の抽出物の品質評価方法であって、評価対象となる抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)含量(Y重量%)およびサラシノール含量(X重量%)を定量し、YとXの関係が以下:
Y≧αX
X+Y≧β
Y≧γ
のいずれか一つの条件を満たす場合には、α−グルコシダーゼ阻害活性、特にマルターゼ阻害活性の高い植物由来の抽出物であると判断する。上記品質評価方法により、α−グルコシダーゼ阻害活性の高い植物由来の抽出物を選択することできる。
【0063】
ここにおいて、αは1.6以上であればよく、好ましくは2.7以上、より好ましくは2.9以上であってよい。βは0.75以上であればよく、特に1.05以上であってよい。γは0.46以上、好ましくは0.78以上、さらに好ましくは0.86以上であってよい。
【実施例】
【0064】
本発明をより具体的に説明するが、本発明の範囲はこれに限定されるものではない。
[実施例1] サラシノールおよびコタラノールの標準品の調製
自然乾燥させたタイ産サラシア・キネンシス幹部(15kg)を粉砕し、水(150L)を加えて95〜100℃で2時間、加熱抽出を行った。ろ過によりろ液を回収した。残渣に水(150L)を加え、上記と同様の操作を行った。上記2回の操作により得られたろ液を一つに合わせた。これを減圧濃縮させた後、噴霧乾燥させることによって熱水抽出物(収量1.41kg、収率9.37%)を得た。続いて当該熱水抽出物(1kg)にメタノール(10L)を加え、80℃で3時間処理し、ろ過によりろ液を回収した。残渣にメタノール(10L)を加え、上記と同様の操作を2回繰り返した。上記3回の操作により得られたメタノール抽出液を一つに合わせ、減圧下で溶媒を留去させ、メタノール抽出エキス(収量347.54g、収率3.26%)を得た。当該抽出エキス(303.8g)を蒸留水5Lに溶解し、これをDIAION-HP20樹脂(4kg、三菱化学株式会社)を用いたカラムクロマトグラフィーに供し、水溶出部(収量167.64g、収率1.80%)を得た。当該水溶出部(150gを)を蒸留水(7.5L)に溶かした後、Duolite A368S樹脂(OH形、3.75L、住化ケムテックス株式会社)を用いたカラムクロマトグラフィーに供し、蒸留水を通液後、酢酸水溶液(0.1N)を用いて酢酸溶出部(収量8.10g、収率0.10%)を得た。次に、当該酢酸溶出部(7.0g)を75%(v/v)アセトニトリル(700mL)に溶かした後、Chromatorex NH樹脂(210g、富士シリシア化学株式会社)を用いたカラムクロマトグラフィーに供し、75%(v/v)アセトニトリル溶出部(収量2.78g、収率0.039%)、50%(v/v)アセトニトリル溶出部(収量1.80g、収率0.025%)をそれぞれ得た。次にそれぞれをCOSMOSIL Sugar-D (20×250mm、ナカライテスク株式会社)を用いた高速液体クロマトグラフィーに供し、最終的にサラシノール(収量681.7mg、収率0.0094%)、コタラノール(収量230.4mg、収率0.0032%)を得た。両化合物の構造を1H−NMR、13C−NMRおよび質量分析計で解析した結果、以下の式(II)および式(III)の構造を有する化合物であることが確認された。これらをサラシノールの標準品およびコタラノールの標準品として使用した。
【0065】
【化5】
【0066】
[実施例2] 脱硫酸サラシノールおよび脱硫酸コタラノールの標準品の調製
サラシノール標準品(28mg)を5%(v/v)塩化水素含有メタノール(0.6mL)に溶解し、45℃で3時間反応させた。反応後、減圧下で溶媒を留去させて残渣を得た。当該残渣をKieselgel 60(70−230mesh、メルク株式会社)を用いたカラムクロマトグラフィーに供し、最終的に下記構造式に示される脱硫酸サラシノール(カウンターイオンとしてCH3OSO3−がイオン結合したもの、27mg)を得た。同様に、コタラノール(10mg)を5%(v/v)塩化水素含有メタノール(1.4mL)に溶解し、45℃で3時間反応させた。反応後、減圧下で溶媒を留去させて残渣を得た。当該残渣をKieselgel 60(70−230mesh、メルク株式会社)を用いたカラムクロマトグラフィーに供し、最終的に下記構造式に示される脱硫酸コタラノール(カウンターイオンとしてCH3OSO3−がイオン結合したもの、10mg)を得た。両化合物の構造を1H−NMR、13C−NMRおよび質量分析にかけた結果、以下の式(IV)および式(I)の構造を有する化合物であることが確認された。これらを脱硫酸サラシノールの標準品および脱硫酸コタラノールの標準品として使用した。
【0067】
【化6】
【0068】
ここで、X−は先に定義した通りである。
[実施例3] サラシア属植物由来の抽出物の調製
表1に示したサラシア属植物(No.1〜21)をそれぞれ小型超高速粉砕機WB−1(大阪ケミカル株式会社)で粉砕し、破砕粉末を得た。当該破砕粉末(2g)に蒸留水(20mL)を加え、95℃で120分間、撹拌しながら加熱還流による抽出を行った。その後、遠心分離(1500×g、室温、5分間)を行い、上清を回収した。残渣に蒸留水(20mL)を加え、95℃で120分間、上記と同様に加熱還流による抽出を行った。その後、遠心分離(1500×g、室温、5分間)を行い、上清を回収した。上記のようにして得られた上清を一つにして100mL容メスフラスコに移し、蒸留水で100mLにメスアップした。当該液(10mL)をメンブレンフィルター(孔径0.45μm)でろ過し、得られたろ液をサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの定量分析に使用した。一方、当該液の残り(90mL)は、減圧濃縮させた後、凍結乾燥させた。これをα−グルコシダーゼ阻害活性の測定用のサンプルとした。なお、抽出物の調製に用いた上記破砕粉末については、別に105℃、8時間での乾燥減量を測定し、抽出に用いた破砕粉末の乾燥重量を算出した。
【0069】
【表1】
【0070】
[実施例4] 組成物中の成分の定量分析
上記実施例3で調製した組成物中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの定量分析は、高速液体クロマトグラフ質量分析装置(LC−MS)によって行った。
【0071】
LC部分は、システムコントローラ:CBM−20A、ポンプ:Shimadzu LC−20AB、紫外分光光度計検出器(UV):Shimadzu SPD−20A、カラムオーブン:CTO−20A、オートサンプラー:SIL−20A、から構成される(いずれも株式会社島津製作所)。MS部分は、Shimadzu LC−MS−2010EV(株式会社島津製作所)を用いた。
【0072】
サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールのMS部分よる検出は、各化合物に特徴的な擬分子イオンを選択的にモニタリングすることによって行った。各化合物の擬分子イオンのm/zは、サラシノール:m/z=333、コタラノール:m/z=423、脱硫酸サラシノール:m/z=255および脱硫酸コタラノール:m/z=345である。分析条件を以下に示す。
【0073】
分析条件−1:サラシノールおよびコタラノールの分析条件
<LC条件>
・カラム: Asahipak NH2P−50(2.0mmi.d.×150mm、昭和電工株式会社)
・移動相: アセトニトリル:水=78:22 (v/v)
・流量: 0.2mL/min
・カラム温度: 40℃
・注入量: 1.0μL
<MS条件>
・イオン化: ESIネガティブモード
・ネブライズガス: 1.5L/min
・窒素ガス圧: 0.15MPa
・CDL温度: 250℃
・ブロックヒーター温度: 200℃
・CDL電圧: Constant−mode(−25V)
・Q array DS & RF voltage: Scan−mode
・SIM: m/z=333(サラシノール)、m/z=423(コタラノール)
分析条件−2:脱硫酸サラシノールおよび脱硫酸コタラノールの分析条件
<LC条件>
・カラム: Inertsil ODS−3(2.1mmi.d.×150mm、ジーエルサイエンス株式会社)
・移動相: 5mM HFBA/MeOH=99:1 (v/v)
・流量: 0.2mL/min
・カラム温度: 40℃
・注入量: 1.0μL
<MS条件>
・イオン化: ESIポジティブモード
・ネブライズガス: 1.5L/min
・窒素ガス圧: 0.15MPa
・CDL温度: 250℃
・ブロックヒーター温度: 250℃
・CDL電圧: Constant−mode(−25V)
・Q array DS & RF voltage: Scan−mode
・SIM: m/z=255(脱硫酸サラシノール)、m/z=345(脱硫酸コタラノール)
分析結果は、表1のNo.1〜21のサンプル全てについて得ることができた。代表例として、表1のNo.1のサンプルのクロマトチャートを示す(図1〜4)。m/z=333(サラシノール)は8.2分、m/z=423(コタラノール)は16.1分、m/z=255(脱硫酸サラシノール)は5.8分、m/z=345(脱硫酸コタラノール)は4.9分の保持時間にピークが現れた。これらピークの保持時間は、各標準品のピークの保持時間と一致した。
【0074】
実施例1および2で調製した各標準品を用いて作成した検量線に基づいて、上記擬分子イオンのピーク面積からサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量を絶対検量線法にて算出した(表2)。なお、脱硫酸サラシノール、および脱硫酸コタラノールの標準品は、カウンターイオン(CH3OSO3−)を含んでいるため、以下の式によりカウンターイオンを含まない濃度に換算してこれらの検量線を作成した。
【0075】
A=B×255/366
A:[カウンターイオンを含まない脱硫酸サラシノール濃度]
B:[カウンターイオン(CH3OSO3−)を含む脱硫酸サラシノール濃度]
C=D×345/456
C:[カウンターイオンを含まない脱硫酸コタラノール濃度]
D:[カウンターイオン(CH3OSO3−)を含む脱硫酸コタラノール濃度]
【0076】
【表2−1】
【0077】
【表2−2】
【0078】
[試験例1] α−グルコシダーゼ活性阻害試験
(1)マルターゼ活性阻害試験
酵素溶液:Wistar系雄性ラットの小腸を摘出後、小腸内面の粘膜をスライドガラスを用いて採取した。採取した前記粘膜をマンニトール(50mM)を含有するトリス緩衝液(2mM、pH7.1)中でホモジナイズした後、塩化カルシウム(終濃度10mM)を添加し、混合した。得られた懸濁液を遠心分離(3000×g、4℃、5分間)し、上清を回収した。当該上清をさらに超遠心分離(27000×g、4℃、30分間)し、得られた沈殿をマレイン酸緩衝液(0.1M、pH6.0)に懸濁させ、回収した。当該懸濁液はα−グルコシダーゼを含んでいるため、当該懸濁液を酵素溶液として使用した。当該酵素溶液は、マルターゼ活性とスクラーゼ活性を有する。
【0079】
基質溶液:マルトース(ナカライテスク株式会社)をマレイン酸緩衝液(0.1M、pH6.0)に溶解し、マルトース基質溶液(74mM)を調製した。
試料溶液:実施例3で調製した各サンプルをジメチルスルホキシドおよびマレイン酸緩衝液(0.1M、pH6.0)に溶解し、試料溶液(240μg/mL、10%ジメチルスルホキシドを含む)を調製した。
【0080】
酵素反応:基質溶液(50μL)に試料溶液(25μL)を加えた後、酵素液(25μL)を加え、37℃で30分間反応させた。沸騰水浴中で2分間加熱し、酵素を失活させて反応を停止させた。これを評価サンプルとした。一方、酵素溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを酵素ブランク1とした。また、試料溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを試料ブランクとした。さらに、酵素溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)、および試料溶液の代わりにマレイン酸緩衝液(0.1M、pH6.0)を加えて上記と同じ反応操作を行ったものを酵素ブランク2とした。また、比較例として、α−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠(商標、武田薬品工業株式会社、1錠(200mg)あたりボグリボース0.3mg)についても同様の操作を行った。
【0081】
遊離グルコースの定量:反応を停止させた上記反応液中のD−グルコースの濃度をグルコースCIIテストワコー(和光純薬工業株式会社)により測定し、酵素反応によって生成したD−グルコース濃度を以下の式により算出した。
【0082】
[評価サンプルの生成D−グルコース濃度(A)]=[評価サンプルのD−グルコース濃度]−[酵素ブランク1のD−グルコース濃度]
[試料ブランクの生成D−グルコース濃度(B)]=[試料ブランクのD−グルコース濃度]−[酵素ブランク2のD−グルコース濃度]
さらに、上記のようにして算出した、評価サンプルの生成D−グルコース濃度および試料ブランクの生成D−グルコース濃度を下式にあてはめることによって、各サンプルのマルターゼ阻害率を算出した。
【0083】
マルターゼ阻害率(%)=100−{(A/B)×100}
結果を表3に示す。
【0084】
【表3】
【0085】
(2)スクラーゼ阻害活性試験
基質溶液:スクロース(ナカライテスク株式会社)をマレイン酸緩衝液(0.1M、pH6.0)に溶解し、スクロース基質溶液(74mM)を調製した。
【0086】
試料溶液:実施例3で調製した各サンプルをジメチルスルホキシドおよびマレイン酸緩衝液(0.1M、pH6.0)に溶解し、試料溶液(80μg/mL、10%ジメチルスルホキシドを含む)を調製した。
【0087】
当該基質溶液および試料溶液を用いる以外は、上記(1)の方法に従った。比較例として、α−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠についても同様の操作を行った。これにより各サンプルのスクラーゼ阻害率を算出した。結果を表4に示す。
【0088】
【表4】
【0089】
[試験例2]
上記試験例1(1)の試験法において、各試料溶液中のサンプル濃度を変化(2000、800、240、80、24μg/mL)させ、各サンプルのマルターゼ阻害活性IC50(マルターゼ活性を50%阻害する濃度)を測定した。また、比較例としてα−グルコシダーゼ阻害活性を有する医薬品であるベイスン錠についても同様の操作を行った。結果を表5に示す。
【0090】
【表5】
【0091】
上記試験例1〜3の結果(表3〜5)を考慮すると、α−グルコシダーゼ阻害活性(マルターゼ阻害活性およびスクラーゼ阻害活性)は、サンプルごとにバラツキがあることが判明した。これは、原料となるサラシア植物の品質がその種類、生産国および樹齢や採取時期などによって異なることを示唆し、さらに品質が一定した原料の供給が極めて困難であることも示唆する。すなわち、最終製品のα−グルコシダーゼ阻害活性が一定しないことになり、ひいては目的の効果を十分に発揮できない製品が製造されることになるため、品質保証の観点から好ましくない。そこで、α−グルコシダーゼ阻害活性と各サンプル中に含まれるサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量との関係について以下のような検討を行った。
【0092】
表2〜4の結果に基づいて、上記サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量とマルターゼ阻害率もしくはスクラーゼ阻害率との関係を表す図を作成した(図5〜12)。さらに、各図のプロットに基づいて線形近似式および相関係数(r)を算出した(表6)。
【0093】
【表6】
【0094】
これらの結果から、サラシノールと脱硫酸サラシノール含量は、マルターゼ阻害率またはスクラーゼ阻害率のいずれとも相関はほとんどないことが判明した(図5〜8)。コタラノール含量は、スクラーゼ阻害率(図10)とある程度の相関があることが示唆されるものの、マルターゼ阻害率(図9)との相関は明らかでなかった。唯一、脱硫酸コタラノール含量のみがマルターゼ阻害率(図11)およびスクラーゼ阻害率(図12)の両方と高い相関があることが判明した。
【0095】
さらに、各サンプル中のサラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの含量の合計とマルターゼ阻害率およびスクラーゼ阻害率との関係について検討を行った(図13および14)。当該検討結果から、上記4つの化合物によるマルターゼ阻害率は、脱硫酸コタラノール含量にほぼ依存していることが明らかになった。また、スクラーゼ阻害率に関しても、その大部分が脱硫酸コタラノール含量に依存していることが示された。
【0096】
以上の結果を鑑みると、本発明の組成物においては、脱硫酸コタラノールがα−グルコシダーゼ阻害活性の主な活性成分であることが明らかである。特に、マルターゼ阻害活性との高い相関が示された。本願出願時において、サラシノール、コタラノール、脱硫酸サラシノールおよび脱硫酸コタラノールの単離された化合物は、α−グルコシダーゼ活性を同程度で阻害することが技術常識として知られている。このような技術常識を考慮すると、上記の結果は全くの予想外といえる。
【0097】
さらに、本発明者は、高いα−グルコシダーゼ阻害活性を有するサンプルを選択するための指標について検討を行った。各サンプル中の脱硫酸コタラノール含量、サラシノール含量、およびマルターゼ阻害活性(IC50)の関係を以下にまとめた(表7)。
【0098】
【表7−1】
【0099】
【表7−2】
【0100】
表7の結果から、IC50が特に低い(マルターゼ阻害活性が高いことを意味する)サンプル(表7の判定の欄に○があるもの)は、脱硫酸コタラノールとサラシノールの含量比(DesKo/Sa)が1.6以上であることが明らかとなった。さらに、サンプル中の脱硫酸コタラノールの存在量が0.09重量%以上であればマルターゼ阻害活性が認められ、さらに0.46重量%以上であれば極めて高いマルターゼ阻害活性が認められることが明らかとなった。このような指標を用いれば、サラシア属植物から組成物を調製した場合に、目的の効果を奏する組成物を迅速、容易に選択することができるため、組成物のα−グルコシダーゼ阻害活性の品質評価を容易に行うことができる。さらに、当該品質評価方法は、組成物のα−グルコシダーゼ阻害活性を任意の規格値に調整する用途にも適しているため、α−グルコシダーゼ阻害活性が均一な組成物を安定的に製造することができる。
【0101】
また、上記の0.09重量%および0.46重量%の脱硫酸コタラノールを含有する組成物は、それぞれサンプルNo.16および6に対応する。当該サンプルNo.16および6の製造に用いた原料の脱硫酸コタラノール含量は、表2から、それぞれ0.018重量%および0.055重量%である。すなわち、この値を指標にすれば、本発明の組成物を調製するために適した原料を選択することができる。これにより、高いα−グルコシダーゼ阻害活性、特にマルターゼ阻害活性を有する植物由来の組成物の製造を容易かつ効率的に行うこと、および組成物の品質評価をより容易に行うことができる。
【特許請求の範囲】
【請求項1】
脱硫酸コタラノールおよびサラシノールを含む組成物。
【請求項2】
前記組成物がサラシア属植物由来の抽出物である、請求項1に記載の組成物。
【請求項3】
前記組成物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)が以下の式
Y≧1.6X
の関係を有する、
請求項1または2に記載の組成物。
【請求項4】
前記Y重量%が0.46以上である、請求項1〜3のいずれか1項に記載の組成物。
【請求項5】
前記Y重量%とX重量%の合計が0.75以上である、請求項1〜4のいずれか1項に記載の組成物。
【請求項6】
請求項1〜5のいずれか1項に記載の組成物を含んでなる飲食品。
【請求項7】
前記飲食品が健康食品または機能性食品である、請求項6に記載の飲食品。
【請求項8】
請求項1〜5のいずれか1項に記載の組成物を含んでなる、血糖値上昇抑制用医薬組成物。
【請求項9】
前記組成物が水または熱水抽出物である、請求項1〜5のいずれか1項に記載の組成物。
【請求項10】
脱硫酸コタラノールおよびサラシノールを含む組成物の製造方法であって、
サラシア属植物を水または熱水で処理して抽出物を得ること;
前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を測定すること;および
前記YとXが以下:
Y≧1.6X
X+Y≧0.75
Y≧0.46
のいずれか一つの関係を有する抽出物を選択し、これを前記組成物とすること;
を含んでなる、
前記組成物の製造方法。
【請求項11】
脱硫酸コタラノールおよびサラシノールを含む抽出物の品質評価方法であって、
前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を測定すること;および
前記XおよびYが以下:
Y≧1.6X
X+Y≧0.75
Y≧0.46
のいずれか一つの関係を有する抽出物を選択すること;
を含んでなる、
前記抽出物の品質評価方法。
【請求項1】
脱硫酸コタラノールおよびサラシノールを含む組成物。
【請求項2】
前記組成物がサラシア属植物由来の抽出物である、請求項1に記載の組成物。
【請求項3】
前記組成物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)が以下の式
Y≧1.6X
の関係を有する、
請求項1または2に記載の組成物。
【請求項4】
前記Y重量%が0.46以上である、請求項1〜3のいずれか1項に記載の組成物。
【請求項5】
前記Y重量%とX重量%の合計が0.75以上である、請求項1〜4のいずれか1項に記載の組成物。
【請求項6】
請求項1〜5のいずれか1項に記載の組成物を含んでなる飲食品。
【請求項7】
前記飲食品が健康食品または機能性食品である、請求項6に記載の飲食品。
【請求項8】
請求項1〜5のいずれか1項に記載の組成物を含んでなる、血糖値上昇抑制用医薬組成物。
【請求項9】
前記組成物が水または熱水抽出物である、請求項1〜5のいずれか1項に記載の組成物。
【請求項10】
脱硫酸コタラノールおよびサラシノールを含む組成物の製造方法であって、
サラシア属植物を水または熱水で処理して抽出物を得ること;
前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を測定すること;および
前記YとXが以下:
Y≧1.6X
X+Y≧0.75
Y≧0.46
のいずれか一つの関係を有する抽出物を選択し、これを前記組成物とすること;
を含んでなる、
前記組成物の製造方法。
【請求項11】
脱硫酸コタラノールおよびサラシノールを含む抽出物の品質評価方法であって、
前記抽出物中の脱硫酸コタラノール(カウンターイオンを含まない)の含量(Y重量%)とサラシノールの含量(X重量%)を測定すること;および
前記XおよびYが以下:
Y≧1.6X
X+Y≧0.75
Y≧0.46
のいずれか一つの関係を有する抽出物を選択すること;
を含んでなる、
前記抽出物の品質評価方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2010−202597(P2010−202597A)
【公開日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願番号】特願2009−51033(P2009−51033)
【出願日】平成21年3月4日(2009.3.4)
【出願人】(000125347)学校法人近畿大学 (389)
【出願人】(304026180)株式会社ダイアベティム (12)
【Fターム(参考)】
【公開日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願日】平成21年3月4日(2009.3.4)
【出願人】(000125347)学校法人近畿大学 (389)
【出願人】(304026180)株式会社ダイアベティム (12)
【Fターム(参考)】
[ Back to top ]