
シクロスポリン類似体の合成法
【課題】本発明は、構造的にシクロスポリンAに似ているシクロスポリン類似体の異性体混合物に関する。
【解決手段】本発明の混合物は、個々の異性体に比べて、そして天然及びその他の現在公知のシクロスポリン類及びシクロスポリン誘導体類に比べて、高い効果及び低い毒性を有する。本発明の実施形態は、ISATX247と呼ばれるシクロスポリンA類似体、及びそれらの誘導体類のシス及びトランス−異性体類に関するものである。ISATX247異性体の混合物は、天然及び現在公知のシクロスポリン類に比べてより高い効力とより低い毒性との組合わせを示す。ISATX247異性体及びアルキル化、アリール化、及び重水素化誘導体は、立体選択的経路によって合成され、その際特定の反応条件が立体選択性の程度を決める。
【解決手段】本発明の混合物は、個々の異性体に比べて、そして天然及びその他の現在公知のシクロスポリン類及びシクロスポリン誘導体類に比べて、高い効果及び低い毒性を有する。本発明の実施形態は、ISATX247と呼ばれるシクロスポリンA類似体、及びそれらの誘導体類のシス及びトランス−異性体類に関するものである。ISATX247異性体の混合物は、天然及び現在公知のシクロスポリン類に比べてより高い効力とより低い毒性との組合わせを示す。ISATX247異性体及びアルキル化、アリール化、及び重水素化誘導体は、立体選択的経路によって合成され、その際特定の反応条件が立体選択性の程度を決める。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は2001年10月19日に提出された米国特許出願第60/346,201号及び2002年4月5日に提出された米国特許出願第60/370,596号に基づく優先権を請求する。これら各出願の全ての開示は参考としてそのまま本明細書に組み込まれる。
【0002】
本発明は、シクロスポリンAに関係するシクロスポリン類似体の異性体混合物類に関するものである。これらの混合物は、個々の異性体に比べて、及び天然、及びその他の現在公知のシクロスポリン類及びシクロスポリン誘導体類に比べて、高い効果及び/又は低い毒性を有すると考えられる。さらに本発明は、シクロスポリンA類似体の異性体を生成する合成経路に関し、その際このような経路では特異的反応条件によって立体選択性の程度が種々変動する。
【0003】
下記の文献は本願に関連しており、本明細書の該当部分に、特許番号もしくは出願番号、又は著者名と発行年を括弧書きで記して、本明細書において参照する。
【0004】
【非特許文献1】ベネット(Bennett, W.M.)著、「新旧の免疫抑制剤の腎臓毒性」、Renal Failure,Vol.20, p.687-90(1998)。
【非特許文献2】ビールマン(J.-F. Biellmann)、ドュセプ(J.-B. Ducep)著、「ヘテロ原子によって置換されたアリルカルボアニオン及びベンジルカルボアニオン」、OrganicReactions、Vol.27,(ウィリー、ニューヨーク、1982), p.9。
【非特許文献3】カールセン(H.J. Carlsen)ら著、「四酸化ルテニウム触媒による有機化合物の酸化法の顕著な改良」,J. Org. Chem.,Vol.46, No.19, p.3736-3738 (1981)。
【非特許文献4】チャング(T. Chang)、ベネット(L.Z. Benet)、ヘバート(M.F. Hebert)著、「健康な志願者におけるシクロスポリンの薬物導体に与える水溶性ビタミンEの影響」,Clin.Pharmacol. Ther., Vol.59, p.297-303 (1996)。
【非特許文献5】コーリー(E.J. Corey)、デサイ(M.C. Desai)著、Tetrahedron Letters, Vol26, No.47,p.5747-8 (1985)。
【非特許文献6】エバール(M.K. Eberle)、ナニンガー(F. Nuninger)著、「シクロスポリンAの主代謝産物(OL−17)の合成」,J.Org. Chem., Vol.57, p.2689-2691 (1992)。
【非特許文献7】エーリンガ(E. Ehlinger)、マグナス(P. Magnus)著、「合成における珪素。10.(トリメチルシリル)アニオン:アルデヒド及びケトンをγ−ラクトンに変換するためのβ−アシルアニオン同等物」,J.Am. Chem. Soc., Vol.102, No.15, p.5004-5011(1980)。
【非特許文献8】フルマン(D.S. Fruman)、クリー(C.B. Klee)、ビーラ(B.E. Bierer)、ブラコフ(S.J. Burakoff)著、「T細胞におけるカルシニューリン燐酸活性はFK506及びシクロスポリンAによって阻害される」,Proc.Natl. Acad. Sci. USA, Vol.89, p.3686-90 (1992)。
【非特許文献9】グラネリ−ピペルノ(A. Granelli-Piperno)、アンドラス(L. Andrus)、スタインマン(R.M. Steinman)著、「刺激されたヒト細胞中のリンフォカイン及び非リンフォカインmRNAの濃度:シクロスポリンAの動力学、マイトジェン要求性及び効果」,J.Exp. Med., Vol163, p.922(1986)。
【非特許文献10】ハンソン(J.R. Hanson)著、「『アルコール類の保護』有機合成における保護基、2章」,p.24-25(シェフィールド・アカデミック・プレス、シェフィールド、英国、1999)。
【0005】
【非特許文献11】ヘバート(M.F. Hebert)、ロバーツ(J.P. Roberts)、プルークサリタノン(T. Prueksaritanont)、ベネット著、「リファンピンと同時投与されたシクロスポリンのバイオアベイラビリティは、肝酵素誘導により推測されるものより著しく低い」,Clin.Pharmacol. Ther., Vol.52, 453-7 (1992)。
【非特許文献12】ホフマン(R.W. Hoffmann)著、Angewandte Chemie International Edition, Vol.555(1982)。
【非特許文献13】ホフマン、ゼイ(H.-J Zei)著、「アルコールの立体選択的合成。8.クロチルボロネートを介するβ−メチルホモアリルアルコールのジアステレオ選択的合成」,J.Org. Chem., Vol46, p.1309-1314 (1981)。
【非特許文献14】ハードリク(P.F. Hurdlik)及びピーターソン(D. Peterson)著、「β−ヒドロキシシラン類の立体特異的オレフィン形成脱離反応」,J.Am. Chem. Soc., Vol97, No.6, p.1464-1468(1975)。
【非特許文献15】Y.イケダ、J.ウカイ、N.イケダ、H.ヤマモト著、「有機チタン及びリチウム試薬を使用した、アルデヒドからの(Z)−及び(E)−1,3−アルカジエン類の立体選択的合成」,Tetrahedron, Vol43, No.4, p.723-730(1987)。
【非特許文献16】コベル(Kobel)ら著、Europ. J. Applied Microbiology and Biotechnology, Vol.14,p.237-240 (1982)。
【非特許文献17】マクマリー(J. McMurry)著、「有機化学、5版」,(ブルックス/コール、パシフィック グローブ、2000), p.780-783。
【非特許文献18】リーツ(M.T. Reetz)著、「有機合成における有機チタン試薬」、(スプリンガー出版、ベルリン、1986), p.VII,148-149, 164-165。
【非特許文献19】リッチ(Rich)ら著、J. Med. Chem., Vol.29, p.978 (1986)。
【非特許文献20】ロウシュ(W.R. Roush)著、「『アリル有機金属類』、有機合成総覧」、ペルガモン プレス、第2巻、 p.1-53。
【0006】
【非特許文献21】シュライバー(S.L. Schreiber)、クラブトリー(G.R. Crabtree)著「シクロスポリンA及びFK506の作用メカニズム」,Immunol.Today, Vol.13, p.136-42 (1992)。
【非特許文献22】スケトリス(I. Sketris)、ヤツコフ(R. Yatscoff)、コウン(P. Keown)、カナファックス(D.M. Canafax)、ファースト(M.R.First)、ホルト(D.W. Holt)、シュローダ(T.J. Schroeder)、ライト(M. Wright)著、「腎臓移植におけるシクロスポリン使用の最適化」,、Clin.Biochem., Vol.28, p.195-211 (1995)。
【非特許文献23】スミス(M.B. Smith)及びマーチ(J. March)著「マーチの最新有機化学」、(ウィリー、ニューヨーク、2001),p.144-147。
【非特許文献24】ストレイトウィーザ(A. Streitwieser)、ヒースコック(C.H. Heathcock)著、「有機化学入門、2版」(マクミラン、ニューヨーク、1981),p.845-846。
【非特許文献25】スリヴァリス(J.A. Thliveris)、ヤツコフ(R.W. Yatscoff)、ルコウスキー(M.P. Lukowski)、コペランド(K.R.Copeland)、ジェファリー(J.R. Jeffery)、マーフィー(G.F. Murphy)著、「慢性シクロスポリン腎毒性:ウサギモデル」,Nephron, Vol.57, p.470-6 (1991)。
【非特許文献26】スリヴァリス、ヤツコフ、ミハチュ(M.J. Mihatsch)著、「慢性シクロスポリン誘導性腎毒性:ウサギモデル」,Transplantation, Vol.57, p.774-6(1994)。
【非特許文献27】トーマス(S.E. Thomas)著、「有機合成:ホウ素及びシリコーンの役割」(オックスフォード大学プレス、ニューヨーク、1991),p.84-87。
【非特許文献28】トレイバー(Traber)ら著,Helv. Chim. Acta, Vol.60, p.1247-1255 (1977)。
【非特許文献29】トレイバーら著,Helv. Chim. Acta, Vol.65,p.1655-1667 (1982)。
【非特許文献30】ツァイ(D.S. Tsai)、マッテソン(D.S. Matteson)著、「ピナコール(E)−1−トリメチルシリル−1−プロペン−3−ボロネートからの(Z)及び(E)末端ジエンの立体コントロールド合成」,TetrahedronLetters, Vol.22, No.29,p.2751-2752(1981)。
【0007】
【非特許文献31】ヴァランチン(H.A. Valantine)、シュローダ(J.S. Schroeder)著、「最近の心臓移植の進歩」[編集者のコメント],N. Engl. J. Med., Vol.333, No.10, p.660-1 (1995)。
【非特許文献32】フォン ワルトバーグ(von Wartburg)ら著, Progress in Allergy, Vol.38, p.28-45(1986)。
【非特許文献33】ウェンゲル(Wenger)著, Transpl. Proc.,Vol.15,付録1, p.2230 (1983)。
【非特許文献34】ウェンゲル著, Angew. Chem. Int. Ed., Vol.24, p.77 (1985)。
【非特許文献35】ウェンゲル著, Progress in the Chemistry of Organic Natural Products, Vol.50,p.123(1986)。
【非特許文献36】Y.ヤマモト、N.アサオ著, Chemical Reviews, p.2307(1993)。
【非特許文献37】ダン ヤング(Dan Yang)ら著、「非官能化オレフィンを触媒的非対称性エポキシ化するためのC2対称性キラルケトン」, J. Am.Chem. Soc., Vol118, p.491-492 (1996)。
【非特許文献38】ダン ヤングら著「触媒的酸化反応のための新規の環状ケトン」, J. Org. Chem., Vol.63, p.9888-9894(1998)。
【0008】
【特許文献1】米国特許第4,108,985号。
【特許文献2】米国特許第4,160,452号。
【特許文献3】米国特許第4,210,581号。
【特許文献4】米国特許第4,220,641号。
【特許文献5】米国特許第4,256,108号。
【特許文献6】米国特許第4,265,874号。
【特許文献7】米国特許第4,288,431号。
【特許文献8】米国特許第4,384,996号。
【特許文献9】米国特許第4,396,542号。
【特許文献10】米国特許第4,554,351号。
【特許文献11】米国特許第4,771,122号。
【特許文献12】米国特許第5,284,826号。
【特許文献13】米国特許第5,525,590号。
【特許文献14】欧州特許公開公報第0 034 567号。
【特許文献15】欧州特許公開公報第0 056 782号。
【特許文献16】国際特許公開公報WO86/02080。
【特許文献17】国際特許公開公報WO99/18120。
【0009】
上記の参照特許、特許出願及び出版物の各々の開示は参考としてそのまま本明細書に組み込まれる。
【背景技術】
【0010】
シクロスポリン誘導体類は、真菌Tolypocladium inflatum Gams種によって第二の代謝産物として産生され、11個のアミノ酸よりなる環式ポリペプチド類の一群を構成する。それらは、免疫担当リンパ球、特にT−リンパ球を、細胞周期のG0又はG1相において可逆的に阻害することが観察されている。シクロスポリン誘導体は、リンホカインの生成及び放出を可逆的に阻害することも観察されている(グラネリ−ピペルノら、1986)。多数のシクロスポリン誘導体が知られているが、シクロスポリンAが最も広く使用されている。シクロスポリンAの抑制効果は、T−細胞仲介性活性化事象の阻害に関連する。この抑制は、シクロスポリンがいたるところに存在する細胞内蛋白、シクロフィリンと結合することによって実現する。この錯体は、今度は酵素カルシニューリンのカルシウム−及びカルモジュリン依存性のセリン−スレオニンホスファターゼ活性を阻害する。カルシニューリンの阻害は、T−細胞活性化中のサイトカイン遺伝子(IL−2、IFN−γ、IL−4及びGM−CSF)の誘導に必要な、NFATp/c及びNF−κBなどの転写因子の活性化を妨害する。また、シクロスポリンはinvitroにおけるT−ヘルパー細胞によるリンホカイン生成を阻害し、胸腺の成熟CD8及びCD4細胞の発育を停止させる(グラネリ−ピペルノら、1986)。シクロスポリンのその他のinvitro特性には、IL−2生産T−リンパ球及び細胞傷害性T−リンパ球の阻害、活性化T−細胞によって放出されるIL−2の阻害、同種抗原及び外因性リンホカインに反応する静止T−リンパ球の阻害、IL−1生産の阻害、及びIL−2生産T−リンパ球のマイトジェン活性化の阻害がある(グラネリ−ピペルノ、1986)。
【0011】
シクロスポリンは、体液性免疫及び細胞仲介性免疫反応、例えば同種移植片拒絶、遅延型過敏症、実験的アレルギー性脳脊髄炎、フロイントアジュバント関節炎、グラフト対ホスト疾患などを抑制することが証明された、強力な免疫抑制剤である。これは、臓器移植後の臓器拒絶の予防;リウマチ性関節炎の治療;乾癬治療;及びI型糖尿病、クローン病、狼瘡などを含むその他の自己免疫病の治療に使用される。
【0012】
シクロスポリンが始めて発見されて以来、種々様々の天然シクロスポリンが分離され、同定され、多くの非天然シクロスポリンが全−又は半合成手段によって、又は改良培養法の適用によって調製された。こうしてシクロスポリンによって構成される群は、今や実質的な価値を有し、天然シクロスポリンAからZまで[トレイバーら(1977);トレイバーら(1982);コベルら(1982);及びフォンワルトバーグら(1986)を参照]並びに、次に記すような種々の非天然シクロスポリン誘導体及び人工又は合成シクロスポリン類を含む。これらにはジヒドロ−及びイソ−シクロスポリン類;誘導化シクロスポリン(例えば、−MeBmt−残基の3'−O−原子がアシル化されるか又はまた別の置換基が3−位置のサルコシル残基のα−炭素原子に導入されたもの);−MeBmt−残基が異性体型で存在するシクロスポリン類(例えば、−MeBmt−残基の位置6'及び7'を横切る構造がトランスよりもむしろシス型であるもの);及びウェンガー(R.Wenger)によって開発されたシクロスポリン全合成生産法などを利用して、ペプチド配列内の特異的部位に変形アミノ酸類を挿入したシクロスポリン類などがある(トレイバーら(1977)、トレイバーら(1982)及びコベルら(1982);米国特許第4,108,985号、第4,210,581号、第4,220,641号、第4,288,431号、第4,554,351号及び第4,396,542号;欧州特許公開公報第0034 567号及び第0 056 782号;国際特許公開公報WO86/02080;ウェンゲル(1983);ウェンゲル(1985);ウェンゲル(1986)などを参照)。1−位置の修飾されたアミノ酸を含むシクロスポリンA類似体は、リッチら(1986)によって報告されている。免疫抑制性、抗炎症性及び抗寄生虫シクロスポリンA類似体が、米国特許第4,384,996号;第4,771,122号;第5,284,826号;及び第5,525,590号に記載されており、これらの特許は全てサンド社に譲渡されている。その他のシクロスポリン類似体が、イソテクニカに譲渡されたWO99/18120に開示されている。用語Ciclosporin、ciclosporin、cyclosporine及びCyclosporineは同義であり、シクロスポリンを意味する。
【0013】
シクロスポリンA治療に関連する多くの副作用には、腎毒性、肝毒性、白内障誘発性、多毛症、パラセシス(parathesis)、及び歯肉過形成などがある(スケトリスら、1995)。これらのなかで腎毒性は、シクロスポリンA投与に起因するより深刻な用量関連性副作用の一つである。即時的に放出されるシクロスポリンA薬剤(例えばネオラル(商標)及びサンドインミューン(商標)など)は、速やかに遊離され、吸収されて血中薬剤濃度を高めることによって腎毒性及びその他の毒性副作用を起こし得る。その薬剤のピーク濃度が副作用と関係すると予想されている(ベネット、1998)。シクロスポリンAが腎臓損傷を起こす正確なメカニズムは知られていないが、腎臓中の血管収縮物質の濃度増加が求心性糸球体細動脈の血管収縮に導くと言われている。これは腎虚血、糸球体濾過率の低下を招き、長期にわたると、間質性線維症を起こす可能性がある。用量を減らすか又は他の免疫抑制剤に代えると、腎機能は改善する(ヴァランチン及びシュローダ、1995)。よって、有効かつ低毒性の免疫抑制剤が必要である。
【0014】
1−位置に修飾アミノ酸を含むシクロスポリン類似体が、WO99/18120に開示されており、この特許は本出願の譲渡人に譲渡され、そのまま本明細書に挿入されている。米国仮特許出願第60/346,201号もこの譲渡人に譲渡されており、ここで出願人は「ISATX247」と呼ばれる特に好ましいシクロスポリンA類似体を開示している。この類似体は、1−アミノ酸残基の修飾を除けば構造的にシクロスポリンAと同一である。出願人は、ISATX247のシス及びトランス異性体のある種の混合物が、天然に発生し、現在公知のシクロスポリン類に勝る高い効力及び/又は低毒性の組み合わせを有することを発見した。ISATX247のある種のアルキル化、アリール化、及び重水素化誘導体も開示された。
【0015】
一般的に、米国仮特許出願第60/346,201号に開示された混合物は、トランス異性体約10ないし90重量パーセント、及びシス異性体約90ないし10重量パーセントの範囲である;別の実施形態において、上記混合物は、約15ないし85重量パーセントのトランス異性体及び約85ないし15重量パーセントのシス異性体を含む;別の実施形態において、上記混合物は約25ないし75重量パーセントのトランス異性体及び約75ないし25重量パーセントのシス異性体を含む;また別の実施形態において、上記混合物は約35ないし65重量パーセントのトランス異性体及び約65ないし35重量パーセントのシス−異性体を含む;また別の実施形態において、上記混合物は約45ないし55重量パーセントのトランス異性体及び約55ないし45重量パーセントのシス異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントのトランス異性体及び約50ないし55重量パーセントのシス異性体を含むISATX247混合物である。これらの重量パーセンテージは、組成物の総重量を基準としている。言い換えれば、混合物は(E)−異性体を65重量パーセント及び(Z)異性体を35重量パーセント含み、その逆もあり得る。別の命名法では、シス異性体は(Z)-異性体と言うこともでき、トランス異性体は(E)−異性体と言うこともできる。
【0016】
よって、当該技術では、異性体ISATX247を含むシクロスポリン類似体の調製方法が必要である。個々の異性体に富む組成物、並びに上記2異性体の所望比を有する異性体混合物を生成する合成経路が必要である。ISATX247の誘導体の調製方法も必要である。
【発明の開示】
【0017】
発明の概要
シクロスポリン及びその類似体は、強力な免疫抑制活性を有する環状ポリペプチド群のメンバーである。これらの薬剤はそれらの免疫抑制活性、抗炎症性活性及び抗寄生虫活性に関する利点を提供する一方、シクロスポリンA治療に関しては腎毒性及び肝毒性などの副作用が多数ある。よって、薬理学的活性は天然化合物シクロスポリンAと同様であるが関連する毒性副作用はない、新しい免疫抑制剤が必要とされる。本発明は以下を提供する。
(1)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)アセチル−η−ハロシクロスポリンAを、トリアリールホスフィン、トリアルキルホスフィン、アリールアルキルホスフィン、及びトリアリールアルシンからなる群から選択される第一化合物と共に加熱して中間体を生成し;
b)前記中間体を、アセトアルデヒド、ホルムアルデヒド、重水素化ホルムアルデヒド、2−クロロベンズアルデヒド、及びベンズアルデヒドからなる群から選択される第二化合物と共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
c)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(2)アセチル−η−ハロシクロスポリンAがアセチル−η−ブロモシクロスポリンAである、項目1に記載の方法。
(3)アセチルシクロスポリンAを、N−ブロモスクシンイミド及びアゾ−ビス−イソブチロニトリルと共に還流することによって行われる臭素化反応を用いて、シクロスポリンAの1−アミノ酸残基の側鎖のη−炭素をハロゲン化する工程をさらに含む、項目1に記載の方法。
(4)第一化合物がトリフェニルホスフィンであり、中間体がアセチルシクロスポリンAのトリフェニルホスホニウムハリドである、項目1に記載の方法。
(5)第二化合物がホルムアルデヒドである、項目1又は4に記載の方法。
(6)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)アセチルシクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、アセチルシクロスポリンAアルデヒドをアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(7)ウィッティヒ反応に用いられるリンイリドが、トリフェニルホスフィン、トリアリールホスフィン、トリアルキルホスフィン及びアリールアルキルホスフィンからなる群から選択される、項目6に記載の方法。
(8)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が;
a)第一中間体アセチルシクロスポリンAの形成によってシクロスポリンAのβ−アルコールを保護し;
b)アセチルシクロスポリンAを酸化して第二中間体アセチルシクロスポリンAアルデヒドを形成し;
c)中間体アセチルシクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
d)アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(9)酸化工程が、オゾン、過マンガン酸カリ、四酸化ルテニウム、四酸化オスミウム、ポリマー支持四酸化オスミウム、及び塩化ルテニウムからなる群から選択される酸化剤で行われる、項目8に記載の方法。
(10)四酸化ルテニウム及び塩化ルテニウム酸化剤が、過ヨウ素酸塩及び次亜塩素酸塩からなる群から選択される補助酸化剤と共に使用される、項目9に記載の方法。
(11)四酸化ルテニウム及び塩化ルテニウム酸化剤がアセトニトリルと共に使用される、項目9に記載の方法。
(12)1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
(13)β−トリアルキルシリルアルコールの処理に使用される酸が、酢酸、硫酸及びルイス酸からなる群から選択される、項目12に記載の方法。
(14)1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;
c)前記アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(15)β−トリアルキルシリルアルコールの処理に用いる塩基が、水素化ナトリウム及び水素化カリウムからなる群から選択される、項目14に記載の方法。
(16)γ−(トリアルキルシリルアリル)ボロネートエステルがトリメチルシリルアリルボロネートエステルである、項目12又は14に記載の方法。
(17)E−γ−(トリアルキルシリルアリル)ジアルキルボランがE−γ−(トリメチルシリルアリル)−9−BBNである、項目12又は14に記載の方法。
(18)試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、項目12又は14記載の方法。
(19)β−トリアルキルシリルアルコールを酸で処理する工程が、ピーターソンのオレフィン化を含む、項目12に記載の方法。
(20)β−トリアルキルシリルアルコールを塩基で処理する工程が、ピーターソンのオレフィン化を含む、項目14に記載の方法。
(21)1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドをリチウム化アリルジフェニルホスフィンオキシドと反応させて、アセチル−(E)−1,3−ジエンを形成し;
b)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
(22)1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドを[3−(ジフェニルホスフィノ)アリル]チタンと反応させて、チタン含有中間体を形成し;
b)チタン含有中間体をエリスロ−α−付加物にし;
c)前記エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;
d)前記β−オキドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;
e)前記アセチル−(Z)−1,3−ジエンを塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(23)a)シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;
b)アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して、第一中間体アセチル−η−ブロモシクロスポリンAを生成し;
c)前記第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される中間体を生成し;
d)アセチルシクロスポリンAのトリフェニル又はトリアルキル塩(第二中間体トリフェニルホスホニウムブロミド)から生成するイリドを、ホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
e)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなるプロセスによってつくられる(E)及び(Z)−異性体混合物。
(24)アセチル−1,3−ジエンの処理に使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド及びカリウムアルコキシドからなる群から選択される、項目1、6、8、12、14、21、22及び23のいずれかの項に記載の方法。
(25)アセチル−η−ハロシクロスポリンAのトリフェニルホスホニウム塩を含む、組成物。
(26)ハリドが、ブロミド、クロリド、及びヨーディドからなる群から選択される、項目25に記載の組成物。
(27)アセチルシクロスポリンAのトリフェニルホスホニウムブロミドである、項目25に記載の組成物。
(28)シクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む、組成物。
(29)中間体がアセチルシクロスポリンAのトリフェニル−又はトリアルキルホスホニウムブロミドであり、前記中間体をホルムアルデヒドと共に撹拌する工程が、リチウムハリドの存在のもとで行われる、項目1に記載の方法。
(30)a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記アセチルシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(31)a)トリメチルシリル(TMS)シクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記TMSシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(32)オゾン分解工程が約−80℃ないし0℃の温度で行われる、項目30又は31に記載の方法。
(33)還元剤が、トリアルキルホスフィン、トリアリールホスフィン及びトリアルキルアミンからなる群から選択される、項目30又は31に記載の方法。
(34)還元剤が、アルキルアリールスルフィド、チオ硫酸塩、及びジアルキルスルフィドからなる群から選択される、項目30又は31に記載の方法。
(35)還元剤がジメチルスルフィドである、項目34に記載の方法。
(36)還元剤がトリブチルホスフィンである、項目33に記載の方法。
(37)還元剤がトリアルキルアミンである、項目33に記載の方法。
(38)還元剤がトリエチルアミンである、項目37に記載の方法。
(39)アセチルシクロスポリンAのオゾン分解に用いられる溶媒が低級アルコールである、項目30ないし38のいずれかの項に記載の方法。
(40)アルコールがメタノールである、項目39に記載の方法。
(41)オゾン分解に使用される溶媒が、ジクロロメタン、及びジクロロメタンと低級アルコールとの混液からなる群から選択される、項目31に記載の方法。
(42)低級アルコールがメタノールである、項目41に記載の方法。
(43)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)中間体アセチルシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハリド又はトリフェニルホスホニウムハリドからつくられたリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をアセチル1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(44)ホスホニウムハリドがホスホニウムブロミドである、項目43に記載の方法。
(45)ウィッティヒ反応が、テトラヒドロフラン及びトルエンからなる群から選択される溶媒中で行われ、前記溶媒はブリルリチウム、低級アルコキシドナトリウム、低級アルコキシドカリウム、及び炭酸塩からなる群から選択される化合物の存在下で、約−80℃ないし110℃の温度で使用される、項目43に記載の方法。
(46)低級アルコキシドカリウムがカリウム−tert−ブトキシドである、項目45に記載の方法。
(47)溶媒が、カリウム−tert−ブトキシドの存在下で、約−70℃ないし−100℃の温度で使用されるテトラヒドロフランである、項目46に記載の方法。
(48)ISATX247のE−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
(49)使用する酸が、酢酸、硫酸、又はルイス酸からなる群から選択される、項目48に記載の方法。
(50)酸がBF3である、項目48に記載の方法。
(51)ISATX247のZ−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してTMS−(Z)−1,3−ジエンを形成し;
c)前記TMS−(Z)−1,3−ジエンを脱保護して、ISATX247のZ−異性体を形成する
諸工程を含んでなる、前記方法。
(52)E−γ−(トリアルキルシリルアリル)ボランが:
a)アリルトリアルキルシランを塩基で脱プロトン化し;
b)脱プロトン化したアリルトリアルキルシランを、ジアルキルアルコキシボラン及びルイス酸と反応させる
諸工程によってつくられる、項目48又は51に記載の方法。
(53)ジアルキルアルコキシボランがジエチルメトキシボランであり、ルイス酸がBF3である、項目52に記載の方法。
(54)アリルトリアルキルシランがアリルトリメチルシランである、項目52に記載の方法。
(55)アリルトリアルキルシランの脱プロトン化に使用する塩基がブチルリチウムである、項目52に記載の方法。
(56)β−トリアルキルシリルアルコールを処理するために使用する塩基が、水酸化ナトリウム、水酸化カリウム、及びアルカリ低級アルコキシドからなる群から選択される、項目51に記載の方法。
(57)塩基がカリウムtert−ブトキシドである、項目56に記載の方法。
(58)脱保護が、塩酸、酢酸、クエン酸、ルイス酸、及びHFをベースとする試薬からなる群から選択される酸を用いて達成される、項目51に記載の方法。
(59)HF−ベースの試薬が、フッ化トリブチルアンモニウム及びフッ化カリウムからなる群から選択される、項目58に記載の方法。
(60)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、前記方法は、(E)−異性体及び(Z)−異性体が混合物中に所定の比率で存在するように、ISATX247の(E)及び(Z)−異性体をつくる合成経路を含み、前記合成経路が:
a)シクロスポリンAの1−アミノ酸のβアルコールを保護し;
b)保護シクロスポリンAを酸化して保護シクロスポリンAアルデヒドを生成し;
c)前記保護シクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、保護シクロスポリンAアルデヒドを保護1,3ジエンのE−及びZ−異性体の混合物に変換し;
d)前記保護1,3ジエンの脱保護により、E−及びZ−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(61)シクロスポリンAと、酢酸エステル、安息香酸エステル、置換安息香酸エステル、エーテル及びシリルエーテルからなる群から選択される試薬とを反応させて、保護シクロスポリンAを生成することにより、シクロスポリンAの1アミノ酸のβアルコールを保護する、項目60に記載の方法。
(62)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、前記方法は、(E)−異性体及び(Z)−異性体が混合物中に所定の比率で存在するように、ISATX247の(E)−異性体及び(Z)−異性体をつくる合成経路を含み、前記混合物中の異性体の比率は、混合物の総重量を基準として、約45ないし55重量パーセントの(E)−異性体から約55ないし45重量パーセントの(Z)−異性体までの範囲である、前記方法。
(63)1−アミノ酸残基が修飾されたシクロスポリンA類似体の所定の異性体混合物の調製方法であって:
a)ISATX247の(E)−異性体に富む第一材料をつくり;
b)ISATX247の(Z)−異性体に富む第二材料をつくり;
c)第一及び第二材料を、所望異性体組成物を与えるように設計された比率で混合する
諸工程を含んでなる、前記方法。
(64)a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβアルコールを保護し;
b)前記アセチルシクロスポリンAを、モノペルスルフェート、好ましくはオキソンで、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、7より高いpHで、アセチルシクロスポリンAエポキシドに変換し;
c)前記エポキシドを過ヨウ素酸又は過ヨウ素酸塩で、酸性条件下で分解する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(65)工程b及びcが一つの処理操作に組合わせられる、項目64に記載の方法。
(66)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)中間体TMSシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハリド又はトリフェニルホスホニウムハリドからつくられるリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をTMS−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)前記TMS−1,3−ジエンの(E)及び(Z)−異性体の混合物を酸で脱保護することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(67)ホスホニウムハリドがホスホニウムブロミドである、項目66に記載の方法。
(68)工程a)が、ナトリウム又はカリウム低級アルコキシド、又は炭酸塩の存在下で使用されるテトラヒドロフラン及び/又はトルエンを含む溶媒中で、約−80℃ないし110℃の温度で行われる、項目66に記載の方法。
(69)ナトリウム又はカリウム低級アルコキシドが、カリウム−tert−ブトキシドである、項目68に記載の方法。
(70)酸が、塩酸、酢酸、クエン酸、ルイス酸及びHF−ベースの試薬からなる群から選択される、項目66に記載の方法。
(71)ISATX247のE−異性体の立体選択的合成方法であって:
a)アセチルシクロスポリンAアルデヒドとE−γ−(トリアルキルシリルアリル)ボランとを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、アセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
(72)β−トリアルキルシリルアルコールを処理する工程が、さらにピーターソンのオレフィン化を含む、項目48、51又は71に記載の方法。
(73)ボラン試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、項目71に記載の方法。
(74)使用する酸が、酢酸、硫酸又はルイス酸からなる群から選択される、項目71に記載の方法。
(75)酸が硫酸である、項目74に記載の方法。
(76)1,3−ジエンの処理のために使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、項目71に記載の方法。
(77)塩基が、NH3及びジメチルアミンからなる群から選択される、項目76に記載の方法。
(78)アセチルシクロスポリン1,3−ジエンをISATX247に変換するための方法であって、前記アセチルシクロスポリン1,3−ジエンが、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、アルコキシドナトリウム、アルコキシドカリウム、及びNH3とヒドロキシルアミンとヒドラジンとジアルキルアミン類からなる群から選択されるアミン塩基からなる群から選択される塩基で処理される、前記方法。
(79)塩基がジメチルアミンである、項目78に記載の方法。
(80)ISATX247のZ−異性体の立体選択的合成法であって;
a)アセチルシクロスポリンAアルデヒドを、第一塩基の存在下で3−(ジメチルアミノ)プロピルトリフェニルホスホニウムハリドで処理して、アセチル−(Z)−オクテニルジメチルアミンを形成し;
b)前記アセチル−(Z)−オクテニルジメチルアミンを酸化試薬で処理して、アセチル−(Z)−オクテニルジメチルアミンオキシドを形成し;
c)前記アセチル−(Z)−オクテニルジメチルアミンオキシドをコープ脱離反応において加熱して、アセチル−(Z)−1,3−ジエンを形成し;
d)前記アセチル−(Z)−1,3−ジエンを第二塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(81)第一塩基がカリウム ヘキサメチルジシラチドである、項目80に記載の方法。
(82)酸化試薬がメタクロルペル安息香酸である、項目80に記載の方法。
(83)第二塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、項目80に記載の方法。
【0018】
本発明の実施形態は薬物学的に有効な、シクロスポリンA類似体のシス及びトランス異性体の幾つかの混合物を提供する。好ましい類似体は、ISATX247と呼ばれる。ISATX247異性体の混合物は、天然シクロスポリン及び現在公知のシクロスポリン類に比べてより高い効力とより低い毒性の組合わせを示す。
【0019】
本発明は、一部には、シクロスポリン類似体のある異性体混合物類がシクロスポリンAで生ずるような副作用をもたず、すぐれた免疫抑制作用を提供するという発見に基づいている。特に、1−アミノ酸残基を修飾したシクロスポリン類似体の約10:90ないし約90:10(トランス対シス)の範囲の異性体混合物類(すなわちシス−及びトランス−異性体両方の混合物)が予想外にすぐれた効果及び安全性をもたらすことを発見した。このような類似体の例は、WO90/18120に開示されており、重水素化及び非重水素化化合物を含んでいる。より詳細に述べれば、約45:55ないし約50:50(トランス対シス)の範囲、及び約50%ないし約55%トランス−及び約45%ないし約50%シス−の範囲の混合物が特に効率的であることが見いだされた。その上、これらの異性体混合物は、天然及びその他の現在公知のシクロスポリン及びシクロスポリン誘導体に比べてよりすぐれた効力とより低い毒性の組合わせを示すことが証明された。
【0020】
特に好ましい類似体(本明細書では「ISATX247」と名づける)は、分子の周囲の、1−アミノ酸残基に存在する修飾官能基を除けば構造的にシクロスポリンAと同様である。この特定の異性体類似体混合物の構造をシクロスポリンAの構造と比較して、図1A、1B、2A、2Bに示す。
【0021】
この異性体混合物は、とりわけ免疫抑制のために、そして種々の免疫障害、免疫病及び免疫疾患の予防、コントロール、緩和及び治療などのケアに使用できる。
【0022】
本発明の実施形態により、ISATX247異性体類(及びその誘導体類)は、選択性の程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)及び(Z)−異性体のどちらかに富む組成物を生成し、これらの組成物を、生成した混合物がこれら2異性体の所望比を有するように組合わせることもできる。或いは、生成した混合物が直接所望比を有するように立体選択的経路の反応条件を調節してもよい。ある混合物中の1異性体又は他の異性体のパーセンテージは、核磁気共鳴スペクトル法(NMR)又は当該技術で公知のその他の方法を使用して確認することができる。
【0023】
各経路は、一般的には不安定なアルコール官能基に保護基を適用して進む。一実施形態において、アルコールはアセテートとして保護され;他の実施形態において、保護基は安息香酸エステル類又はシリルエーテル類である。アセテート保護基は当該技術では一般的であるが、本明細書に記載される多くの典型的実施形態においては、安息香酸エステル又はシリルエーテルなどの保護基の使用によって、アセテート保護基に関係する幾つかの望ましくない副作用が回避されることを強調することは重要である。
【0024】
保護された化合物は、ウィッティヒ反応に参加するリン含有試薬を用いる経路や、有機金属試薬のメンバーとして無機元素を用いる経路などを含む、種々の立体選択的合成経路の前駆体としてはたらく。後者のタイプは、六員環遷移状態を通過して進行する。この場合、立体障害が配置的結果を決める。多くの有機金属試薬、例えばホウ素、珪素、チタン、リチウム及び硫黄などの無機元素を特徴とするものなどが使用できる。個々の異性体は、単一又は複数の前駆体からつくられる。
【0025】
立体選択的生成又は非立体選択的生成の如何にかかわらず、生成した混合物中の(E)対(Z)−異性体の比は、広範囲の数値をとり得る。例えば、混合物は約10ないし90パーセントの(E)−異性体から約90ないし10パーセントの(Z)−異性体までの範囲で構成される。その他の実施形態において、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)異性体を含むことができる;別の実施形態において、混合物は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体を含む;また別の実施形態において、混合物は約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体を含む;他の実施形態において、混合物は約45ないし55重量パーセントの(E)−異性体及び約55ないし45パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は約45ないし約50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセンテージは組成物の総重量を基準としており、(E)−異性体と(Z)−異性体との重量パーセントの合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体と35重量パーセントの(Z)−異性体を含むことができ、その逆でもよい。
【0026】
よって、一つの態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:アセチル−η−ハロシクロスポリンAをトリアルキルホスフィン、トリアリールホスフィン(例えばトリフェニルホスフィン)、アリールアルキルホスフィン、及びトリアリールアルシンと共に加熱して、中間体アセチルシクロスポリンAのホスホニウムハリドを生成し;中間体アセチルシクロスポリンAのホスホニウムハリドをホルムアルデヒドと共に、任意にリチウムハリドの存在下で撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基と共に処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法を提供する。
【0027】
また別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:中間体、例えば保護されたトリメチルシリル(TMS)シクロスポリンAアルデヒド又はアセチルシクロスポリンAアルデヒドを、前記中間体をウィッティヒ反応によって、任意にリチウムハリドの存在下でリンイリドと反応させることによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を、アセチル保護基の場合は塩基で、又はTMS−保護基の場合は例えば酸で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法に関するものである。
【0028】
さらにまた別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、E−異性体に富む混合物の生成方法であって、前記E−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと、トリアルキルシリルアリル硼酸エステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0029】
また別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、Z−異性体に富む混合物の生成方法であって、前記Z−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと、トリアルキルシリルアリル硼酸エステル及び(E−γ−トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0030】
もう一つの態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、E−異性体に富む混合物の生成方法であって、前記E−異性体に富む物質の立体選択的合成が;アセチルシクロスポリンAアルデヒドとリチウム化アリルジフェニルホスフィンオキシドとを反応させてアセチル−(E)−1,3−ジエンを形成し;アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0031】
さらに別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、Z−異性体に富む混合物の生成方法であって、前記Z−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと[3−(ジフェニルホスフィノ)アリル]チタンとを反応させてチタン含有中間体を形成し;チタン含有中間体がエリスロ−α−付加物になるまで反応を続け;エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;β−オキシドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する諸工程を含んでなる、前記方法を提供する。
【0032】
さらにまた別の態様において、シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して第一中間体アセチル−η−ブロモシクロスポリンAを生成し;第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される第二の中間体を生成し;アセチルシクロスポリンAのトリフェニル−又はトリアルキル塩から生成するイリド(第二中間体トリフェニルホスホニウムブロミド)をホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体混合物をつくり;アセチル−1,3−ジエンの(E)及び(Z)異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)異性体の混合物をつくる諸工程を含んでなるプロセスによってつくられる、(E)及び(Z)−異性体の混合物を提供する。
【0033】
本発明は、シクロスポリンAのβ−アルコールを保護し;シクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して第一中間体アセチル−η−ブロモシクロスポリンAを生成し;第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドから選択されるアセチルシクロスポリンAブロミドを生成する諸工程を含んでなるプロセスによってつくられた生成物である、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドを含む対象の組成物類にも関する。アセチルシクロスポリンAのトリフェニル又はトリアルキルホスホニウムブロミド誘導体を含む組成物類、及びシクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む組成物類も提供される。
【0034】
その他の態様において、本発明はシクロスポリンAアルデヒドを選択的に調製する方法であって、アセチルシクロスポリンA又はトリメチルシリル(TMS)シクロスポリンAの形成によってシクロスポリンAのβ−アルコールを保護し;アセチルシクロスポリンA又はTMSシクロスポリンAを、還元剤と共に用いる酸化剤としてのオゾンで酸化する諸工程を含んでなる、前記方法を提供する。
【0035】
その他の付加的態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:中間体アセチルシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハロゲニドからつくられたリンイリドとを、任意にリチウムハリドの存在下でウィッティヒ反応によって反応させることによって、前記中間体をアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによってISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法に関するものである。
【0036】
その他の態様において、本発明は、ISATX247のE−異性体の立体選択的合成方法であって、トリメチルシリル(TMS)シクロスポリンAアルデヒドをトリアルキルシリルボランと反応させてβ−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを処理して直接ISATX247のE−異性体を形成する諸工程を含んでなる、前記方法を提供する。
【0037】
発明のその他の態様は、ISATX247のZ−異性体の立体選択的合成方法であって、トリメチルシリル(TMS)シクロスポリンAアルデヒドをトリアルキルシリルアリルボランと反応させてβ-トリアルキルシリルアルコールを形成し;β-トリアルキルシリルアルコールを塩基で処理してTMS−1,3-ジエンを形成し;TMS−(Z)−1,3−ジエンを脱保護してISATX247のZ−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0038】
本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、(E)−異性体と(Z)−異性体とが所定の比で存在するようにISATX247の(E)−異性体及び(Z)−異性体つくる合成経路を含み、前記合成経路は:シクロスポリンAのβアルコールを保護し;保護されたシクロスポリンAを酸化して、第二中間体である保護されたシクロスポリンAアルデヒドを生成し;第二中間体である保護されたシクロスポリンAアルデヒドを、任意にリチウムハリドの存在下で、ウィッティヒ反応によってリンイリドと反応させることによって、前記第二中間体を保護された1,3ジエンのE−異性体及びZ−異性体の混合物に変換し;保護された1,3ジエンの脱保護によりE−及びZ−異性体の混合物をつくる諸工程を含んでなる、前記方法にも関する。
【0039】
本発明によって提供されるこのような混合物のその他の調製方法は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、混合物中に(E)−異性体と(Z)−異性体とが所定の比で存在するように、ISATX247の(E)−異性体及び(Z)−異性体をつくり、前記混合物中の異性体の比は、混合物の総重量を基準にして、(E)−異性体約45ないし55重量パーセント及び(Z)−異性体約55ないし45重量パーセントの範囲に及ぶ、前記方法も含む。
【0040】
本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の所定の異性体混合物の調製方法であって、ISATX247の(E)−異性体に富む第一材料をつくり;ISATX247の(Z)−異性体に富む第二材料をつくり;所望の異性体組成物を与えるように設計された比率で第一及び第二材料を混合する諸工程を含んでなる、前記方法も提供する。
【0041】
発明の詳細な説明
合成
シクロスポリン及びその類似体は、強力な免疫抑制活性を有する環式ポリペプチド群のメンバーである。これらの薬剤は、それらの免疫抑制性、抗炎症性及び抗寄生虫活性に関して利点を提供する一方、シクロスポリンA治療に関係した腎毒性及び肝毒性を含む多くの副作用がある。よって、薬理学的活性は天然化合物シクロスポリンAと同様であり、しかも関連する毒性副作用はない、という新しい免疫抑制剤が必要である。
【0042】
出願人は以前、「ISATX247」と呼ばれるシクロスポリンA類似体を開示した。この類似体は、1−アミノ酸残基が修飾されていることを除けばシクロスポリンAと同様の構造を有する。出願人は、ISATX247のシス及びトランス−異性体の幾つかの混合物が、天然及び現在公知のシクロスポリン類に勝る高い効力と低い毒性との組合わせを示すことを発見した。
【0043】
本発明の実施形態により、ISATX247異性体類(及びそれらの誘導体類)は、立体選択性程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)及び(Z)−異性体のどちらかに富む組成物を生成し、生成混合物が前記2異性体の所望比を有するように、これらの組成物を組み合わせることができる。或いは、立体選択的経路の反応条件を調節して、つくられた混合物が直接所望比を有するようにすることができる。
【0044】
本発明の、ISATX247と呼ばれる一免疫抑制剤シクロスポリン類似体の化学名は、シクロ{(E,Z)−(2S、3R、4R)−3−ヒドロキシ−4−メチル−2−(メチルアミノ)−6,8−ノナジエノイル}−L−2−アミノブチリル−N−メチル−グリシル−N−メチル−L−ロイシル−L−バリル−N−メチル−L−ロイシル−L−アラニル−D−アラニル−N−メチル−L−ロイシル−N−メチル−L−ロイシル−N−メチル−L−バリル}と化学的に記載される。その実験式はC63H111N11O12であり、約1214.85の分子量を有する。用語「ISATX247」は、この薬理学的活性化合物に与えられた商品名である。
【0045】
ISATX247の構造は、主として核磁気共鳴(NMR)分光法によって確認された。1H及び13Cスペクトルは両方とも、一次元及び二次元NMR実験系列を使用して設定され、シクロスポリンAの公知のNMR設定と比較された。ISATX247の(E)及び(Z)−異性体の絶対的設定は核オーバーハウザー効果(NOE)実験によって確認された。その他の証拠が、質量分析及び赤外スペクトルによって確認された。質量分析は分子量を確認し、この化合物の赤外スペクトルはシクロスポリンAの赤外スペクトルに非常に近いことが判明した。2化合物間に類似性があるため、後者の結果は予期されたものである。
【0046】
シクロスポリンAの構造を図1Aに示す。その構造は分子の環式ペプチド環を構成する11個のアミノ酸残基を確認するものである。これらの11個のアミノ酸残基は、環の頂上部中心に示されるアミノ酸(「1−アミノ酸」の基準標識を有する)から出発し、時計方向に増加するナンバーで標識化される。わかりやすいように第一アミノ酸を点線で四角く囲む。本明細書に記載される合成反応が起こる典型的部位であるので、1−アミノ酸残基の側鎖は化学的に書き表した。一般に、アミノ酸のカルボニル基に隣接する炭素は、α−炭素と標識付けられる。その鎖の下流方向、ペプチド環から離れる方向に隣接する炭素に、ギリシャアルファベット文字を順次付ける。シクロスポリンAの場合、図1Aに示すように、側鎖のβ−炭素はヒドロキシル基に結合し、側鎖のε−炭素とζ−炭素との間にはトランス方向の二重結合がある。
【0047】
シクロスポリンA構造のもう一つの略図を図1Bに示す。ここでは、前記分子の別の部分が点線で四角く囲まれる。この図は、本明細書に使用される命名法を明らかに示す:ここで用語「CsA」は四角く囲まれたシクロスポリンAの部分を指す。この命名法は、反応を記載するたびに分子の残りの部分を再記述することなく、合成反応が起こる領域(すなわち、図1B中の点線四角外に描かれた1−アミノ酸残基の側鎖)を示すという簡便な手段を提供する。側鎖のα−炭素とβ−炭素との間の結合は通常の長さであり、この図では用語「CsA」の定義をより明確にするために誇張されていることは当業者には当然のことである。
【0048】
上述のように、特に好ましいシクロスポリンA類似体はISATX247と呼ばれ、その2種類の立体異性体E(又はトランス)及びZ(又はシス)は、それぞれ図2A及び図2Bに示される。これらの立体異性体のシス又はトランス性は、側鎖のε及びζ−炭素間の二重結合の配置(すなわち、側鎖の末端にある二重結合に対して、ペプチド環により近い二重結合)を基準としている。
【0049】
立体化学的命名法について一言言及しなければならない。本明細書において、用語シスと(Z)とは交換可能に使用され、用語トランスと(E)とが交換可能に使用される。用語「エリスロ」及び「スレオ」の使用は、これらの意味に関して文献中に明らかな混乱があるため、最小限にとどめる。ホフマン及びゼイ著「アルコールの立体選択的合成。8.クロチルボロネートを介するβ−メチルホモアリルアルコールのジアステレオ選択的合成」J. Org. Chem., Vol.46, p.1309-1314(1981);ストレイトウィーザ及びヒースコック著、「有機化学入門、第2版」、(マクミラン、ニューヨーク、1981)、p.845-846;スミス及びマーチ著、「マーチの最新有機化学」、(ウィリー、ニューヨーク、2001)、p.144-147を参照されたい。本明細書において、スレオ/エリスロの用語を使用する数例では、ストレイトウィーザー及びヒースコックの約束事を適用する;その際「エリスロ」異性体は(R,S)及び(S,R)配置を、「スレオ」異性体は(R,R)及び(S,S)配置を指す。
【0050】
命名法に関する最後のコメントは、図2A及び図2Bに示される末端炭素−炭素二重結合に関するものである。別のナンバリングスキームにより、1−アミノ酸残基の側鎖の炭素類に、末端(θ)炭素から出発し、ペプチド環の方に戻るように標識ナンバーを付けてもよい。この系において、ISATX247異性体は有機化学の一般的命名法により1,3−ジエンと考えることができる。この場合、各二重結合は最小ナンバーの炭素によって決定される。
【0051】
図3−8に示される合成経路について述べる。本発明の実施形態により、異性体混合物が直接つくられる。その際、特定の合成経路の反応条件を調節して、混合物中の異性体の所望比を得る。或いは、シクロスポリンA類似体の2種類の幾何学的異性体のうちの一種類に富む組成物をつくり、それらの組成物を所定の比で合一して所望の混合物を得る。
【0052】
本発明の実施形態による合成経路の概観を図3に示す。ここでは、化学及び立体選択性による反応経路のグループ化が特に強調されている。図3を参照すると、ウィッティヒ反応を使用する合成経路が大体図の右手に示され、参照番号31が付けられる。一方、六員環遷移状態を形成すると考えられる有機金属試薬を利用する経路32及び33が、図の中央及び左側に示されている。いずれの合成経路も異性体類の混合物を与えるか、2異性体のうちの一つに富む組成物を生成する。
【0053】
本発明の実施形態は、所望の異性体混合物に到達する種々の方法を提供する。ここに開示する合成戦略のフレキシビリティ及び融通性が、図3の対称性及び非対称性によって一部反映される。各経路に共通する反応は、シクロスポリンA34の官能基の保護である;この典型的実施形態において、この反応はシクロスポリンA34からアセチルシクロスポリンA35への変換である。図3の非対称性は、チタン及びリチウム有機金属試薬経路の全てにおいて、前駆体としてアセチルシクロスポリンAアルデヒドを使用するが、リン含有ウィティッヒ反応経路が若干あることである。
【0054】
異性体を所望比で有する混合物を生成するように反応条件を調節した図3の合成経路は、概して、ウィティッヒ反応に関与する試薬としてリン含有試薬を使用する。その他の立体選択的経路は、無機元素類並びに、典型的に六員環遷移状態を通過する有機金属試薬のメンバー類を使用する。この遷移状態において、立体障害が配置的結果を決める。ホウ素、珪素、チタン、リチウム及び硫黄などの無機元素を特徴とする多数の有機金属試薬が、本発明に有用である。
【0055】
1対の異性体のうちの一つ又は他の一つどちらかに富む組成物を単一の前駆体からつくることがでる;又は、上記2組成物を異なる前駆体からつくることもできる。図3の立体選択的経路の一つ(経路32)において、選択した反応条件によって、単一の前駆体からISATX247の2異性体の両方が生成する。別の立体選択的経路(経路33)では、富化組成物の各々を生成するために2種類の前駆体が必要である。
【0056】
図3の反応を次に詳細に述べる。各経路に共通の反応は、1−アミノ酸残基の側鎖のβ−位置のアルコールの保護である。このような保護スキームは、第一官能基がその分子上のどこか別の部位にある第二(類似及び/又は同一の)官能基に対して行われる反応によって偶然に変形されるという有機合成では通常遭遇する問題である。前記スキームを実施するためには、第一官能基を保護基と反応させ、第二官能基で所望反応を行い、その後に第一官能基から保護基を除去する。
【0057】
保護基は有機合成においてはよく知られており、ハンソンが「有機合成における保護基(Protecting Groups in Organic Synthesis)」(シェフィールド・アカデミック・プレス、シェフィールド、英国、1999)の第2章(p.24-25)「アルコールの保護」において述べている。ハンソンは、ヒドロキシル基を、エステルかエーテルのどちらかに変換することによって保護する方法を教示している。おそらく酢酸エステルは、ヒドロキシル基を保護するために最もよく用いられるタイプの化学物質である。アセテート基を導入するために、広範囲の反応条件を取り得る。これらの試薬及び溶媒としては、無水酢酸及びピリジン;無水酢酸、ピリジン及びジメチルアミノピリジン(DMAP);無水酢酸及び酢酸ナトリウム;無水酢酸及びトルエン−p−スルホン酸、塩化アセチル、ピリジン及びDMAP;及びケテンがある。DMAPは、無水物から高い反応性を有するN−アシルピリジニウム塩を形成するため、有用なアシル化触媒である。
【0058】
本発明の一実施形態において、シクロスポリンA34のβ−アルコールは、塩化アセチル、酢酸エチル、又はこれらの組合わせと反応させて、化合物アセチルシクロスポリンA35を形成させることによって、アセテートとして保護される。別の実施形態において、β−アルコールは無水酢酸への求核付加を受け、アセチルシクロスポリンA35及び酢酸が形成される。これらの反応は、ジメチルアミノピリジン(DMAP)の存在のもとで行われ、無水酢酸の過剰は溶媒として作用する。これらの場合、接頭語「アセチル」は合成経路全体における、又はアセチル基が除去されるまでの間の名称として使用される。例えば、β−炭素にアセチル基を有する一経路の最後の中間体は、「アセチル−(E)−1,3−ジエン」と呼ばれる。
【0059】
アセチルシクロスポリンAの製法は、文献において十分確立されているが、酢酸エステル以外の保護基を使用してシクロスポリンA34の1−アミノ酸残基のβ−アルコールを保護することは、当業者には明らかである。これらの保護基には、安息香酸エステル類、置換安息香酸エステル類、エーテル類及びシリルエーテル類がある。ある反応条件のもとでは、アセテート保護基は脱離や加水分解などの不都合な副反応をおこしがちである。安息香酸エステル類、エーテル類及びシリルエーテル類は、同じ反応条件下でこのような副反応により強く抵抗することが多いため、アセテートの代わりにこのような保護基を使うことが有利であることが多い。アセチル基又はその他の保護基によって保護されたシクロスポリン又はシクロスポリン誘導体を、「保護−シクロスポリンA」と呼ぶ。同様に、上記の典型的経路における最後の中間体は、「アセチル−(E)−1,3−ジエン」の代わりに「保護−(E)−1,3−ジエン」と呼んでもよい。選択した保護基の性質は、反応系列のその後の工程の所望コースに影響を与えることがある。
【0060】
図3を参照すると、アセチルシクロスポリンA35は、この典型的経路において保護β−アルコールを有し、この化合物は数種の合成経路においてISATX247異性体を合成するための前駆体として役立つ。ウィッティヒ反応経路を先ず最初に論ずる。
【0061】
ウィッティヒ反応によるISATX247の(E)及び(Z)−異性体混合物の合成
ここに例示されるウィッティヒ反応経路は、図3の参照番号31によって確認される。方法1は、臭素中間体アセチル−η−ブロモシクロスポリン41を経て進むが、方法2は、出発点としてアセチルシクロスポリンAアルデヒド51を使用する。以下に記載する典型的方法はウィッティヒ反応を用い、混合立体化学的配置を有するアルケン官能基を導入する。
【0062】
本明細書に開示される典型的実施形態において、ISATX247の(E)及び(Z)−異性体混合物を合成するために用いられるウィッティヒ反応は、任意にリチウムハリドの存在下で行ってもよい。ウィッティヒ反応におけるリチウムハリドの存在は、生成する幾何異性体類の比に影響をもつことはよく知られており、したがってこのような化合物の添加は、ISATX247の(E)及び(Z)−異性体の所望混合物の生成に役立つ。
【0063】
方法1
本発明の一実施形態において、ISATX247の(E)及び(Z)−異性体混合物は、図4に示すようにつくられる。図4における波線(特に、化合物43と44を参照)の表示の使用は、例示的反応系列が(E)及び(Z)−異性体の混合物を生成することを示すものとする。一実施形態において、生成した(E)対(Z)−異性体のパーセント比は、(E)−異性体の約10ないし90パーセントから(Z)−異性体の約90ないし10パーセントまでの範囲であるが、これらの範囲は単なる例示であり、多くの他の範囲が可能である。例えば、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)−異性体を含むことができる。その他の実施形態において、混合物は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体;約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体;及び、約45ないし55重量パーセントの(E)−異性体及び約55ないし45パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセントは組成物の総重量を基準としており、(E)異性体及び(Z)異性体の重量の合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体及び35重量パーセントの(Z)−異性体を含むことができ、又はその逆でもよい。
【0064】
図4を参照すると、アセチルシクロスポリンAの1−アミノ酸残基の側鎖の末端η炭素を次の反応工程において臭素化する;すなわち、四塩化炭素などの溶媒中でアセチルシクロスポリンA35をN−ブロモスクシンイミド及びアゾ−ビス−イソブチロニトリルと共に還流し、中間体アセチル−η−ブロモシクロスポリンA41を生成する。N−ブロモスクシンイミドは、アリル水素を臭素で置換するためによく用いられる試薬であり、これは遊離基メカニズムによって行われると考えられている。この中間体41の製法は、エバール及びナニンガーによって「シクロスポリンAの主代謝産物(OL−17)の合成」(J. Org. Chem., Vol57, p.2686-2691(1992))で詳細に記載されている。
【0065】
新規の中間体、アセチルシクロスポリンAトリフェニルホスホニウムブロミド42は、アセチル−η−ブロモシクロスポリンA41から、この化合物をトルエンなどの溶媒中でトリフェニルホスフィンと共に加熱することによってつくることができる。
【0066】
新規の中間体42及びこれに類似するその他のものは、1−アミノ酸残基中に共役ジエン系を含む複数のシクロスポリンA類似体の合成における重要な中間体であると考えられる。例えば、トリフェニルホスフィンに加えて、トリアリールホスフィン類、トリアルキルホスフィン類、アリールアルキルホスフィン類、及びトリアリールアルシン類などの化合物をアセチル−η−ブロモシクロスポリンA41と反応させて42に類似するその他の活性化合物をつくることができる。
【0067】
再び図4を参照すると、アセチルシクロスポリンAのトリフェニルホスホニウムブロミド42と過剰のホルムアルデヒドをトルエン中で室温で撹拌することによって、アセチル−1,3−ジエン43の(E)及び(Z)−異性体混合物をつくることができる。ホルムアルデヒドの添加後、水酸化ナトリウムなどの塩基を滴下し、ジエン類の異性体混合物を酢酸エチルで抽出する。
【0068】
多数の有機化学の教科書が、ウィッティヒ反応を説明している。説明の一つは、特にマクマリーが「有機化学(Organic Chemistry)」、5版(ブルックス/コール、パシフィックグローブ、2000)、p.780-783で記載しているものである。ウィッティヒ反応は、ケトン又はアルデヒドをアルケンに変換するために利用される。このようなプロセスにおいて、リンイリド(ホスホランとも呼ばれる)をアルデヒド又はケトンと反応させると、ベタインと呼ばれる二極性中間体が得られる。一般的には、ベタイン中間体は分離されない;むしろ、それは四員環を経て自然に分解して、アルケンとトリフェニルホスフィンオキシドを与える。正味の結果は、元々はリンに結合していたR2C=基によってカルボニル酸素原子が置換されることである。
【0069】
非常に種々様々の試薬が上述の典型的ウィッティヒ反応試薬の代わりになり得ることは、当業者には明らかである。例えば、多数のアルキル、アリール、アルデヒド及びケトン化合物をホルムアルデヒドの代わりに使用すると、膨大な数のシクロスポリン誘導体がつくられる。出願人は、上記の合成をホルムアルデヒドで行い、また、ホルムアルデヒドの代わりにアセトアルデヒド、ジューテロ化ホルムアルデヒド、ジューテロ化アセトアルデヒド、2−クロロベンズアルデヒド、ベンズアルデヒド、及びブチルアルデヒドなどの化合物で行った。このようなウィッティヒ反応を、トリフェニルホスホニウム誘導体以外の化合物、例えばトリアリールホスフィン類、トリアルキルホスフィン類、アリールアルキルホスフィン類及びトリアリールアルシン類などで行ってもよい。水酸化ナトリウムを使用する代わりに、種々のその他の塩類、例えば炭酸ナトリウム、ブチルリチウム、ヘキシルリチウム、ナトリウムアミド、リチウムジイソプロピルアミドのようなリチウム障害性塩基、及びアルカリ金属アルコキシド類などを使用してもよい。これら種々の試薬が使用できる上に、反応は種々の有機溶媒中又は有機溶媒と水との混液中で、種々の塩、特にリチウムハリドの存在下で、種々の温度で行うことができる。上に列挙した因子の全ては、当業者によって合理的に選択され、形成される二重結合の立体化学、すなわちシス対トランス−異性体の比に所望の効果を与える。本発明の一実施形態において、ウィッティヒ反応は、テトラヒドロフラン及びトルエンからなる群から選択される溶媒中で行われ、その際その溶媒は、ブチルリチウム、ナトリウム低級アルコキシド、カリウム低級アルコキシド及びカルボネートから選択される化合物の存在下で、約−80℃ないし110℃の温度で使用される。カリウム低級オキシドは、カリウム−tert−ブトキシドでよい。さらに、溶媒は、カリウム−tert−ブトキシドの存在下で約−70℃ないし−100℃の温度で使用されるテトラヒドロフランでもよい。
【0070】
この合成の最終工程において、β−炭素上の保護基を下記の手順によって除去する。アセチル−(E)−1,3−ジエン及びアセチル−(Z)−1,3ジエンの混合物43をメタノールに溶解し、それから水を加える。炭酸カリウムのような塩基を加え、反応混合物を室温で撹拌する。使用できる炭酸カリウム以外の塩基として水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド及びカリウムアルコキシドなどがある。その後、酢酸エチルを使用して最終生成物であるISATX247の(E)及び(Z)−異性体混合物44を抽出する。
【0071】
方法2
ISATX247の(E)及び(Z)−異性体混合物を、ウィッティヒ反応戦略によって合成するまた別の反応経路においては、以下のように4工程の合成経路を使用する:1)方法1のようなβ−アルコールの保護、2)第一工程から生成したアセチル−シクロスポリンAを酸化してアルデヒドをつくる;3)ウィッティヒ反応;4)ウィッティヒ反応生成物を脱アシル化し、又は同等に、酢酸エステルを加水分解して、アルコールを回復させる。この反応系列は、図5で明らかにされる。
【0072】
この合成経路は、第一工程が酢酸エステル基でβ−アルコールを保護するという点で、図4のウィッティヒ反応経路と同様にして開始される。しかし、これら2経路は次の点で互いに異なる;方法2の次の工程は、アセチル−シクロスポリンA35をアルデヒド、すなわちアセチルシクロスポリンAアルデヒド51に変換する。この反応は、十分強い酸化剤を使用し、C=C結合を切断して2つの断片を生成する。アルケン切断は、当該技術では公知である。オゾンはおそらく最も一般的に使用される二重結合切断試薬であるが、その他の酸化剤、例えば過マンガン酸カリ(KMnO4)又は四酸化オスミウムも、二重結合切断を起こすことができる。
【0073】
本発明の実施形態によると、アセチル−シクロスポリンAは酸化剤としてのオゾンでアルデヒドに変換され、その後還元剤で処理されてアセチルシクロスポリンAアルデヒドを形成する。オゾン分解の工程は、約−80℃ないし0℃の温度範囲で行われる。オゾン分解中に使用する溶媒は、メタノールなどの低級アルコールである。還元剤は、トリブチルホスフィンのようなトリアルキルホスフィン、トリアリールホスフィン、トリエチルアミンのようなトリアルキルアミン、アルキルアリールスルフィド、チオ硫酸塩、又はジメチルスルフィドのようなジアルキルスルフィドでよい。還元剤としてトリブチルホスフィンを用いて処理する場合、その反応は用量調節反応であることは当業者には知られている。
【0074】
本発明のまた別の実施形態によると、シクロスポリンAのβアルコールをトリメチルシリル(TMS)基で保護し、酸化剤としてのオゾンで酸化し、その後還元剤で処理してTMSシクロスポリンAアルデヒドを形成する。オゾン分解工程は、約−80℃ないし0℃の温度範囲で行われる。オゾン分解中に使われる溶媒は、低級アルコールとジクロロメタンとの混液でよい。還元剤は、トリブチルホスフィンなどのトリアルキルホスフィン類、トリアリールホスフィン類、トリエチルアミンなどのトリアルキルアミン類、アルキルアリールスルフィド、チオ硫酸塩類又はジメチルスルフィドなどのジアルキルスルフィド類からなる群から選択される。還元剤としてのトリブチルホスフィンで処理する場合、その反応が用量調節されていることは当業者には知られている。
【0075】
付け加えると、シクロスポリンAアルデヒドは、シクロスポリンAのβ−アルコールをアセチルシクロスポリンAの形成によって保護し、その後そのアセチルシクロスポリンAを、アセトキシアセトン又はジアセトキシアセトンなどのケトンの存在下で、モノペル硫酸塩、好ましくはオキソンでアセチルシクロスポリンAエポキシドに変換する。この工程は、それらの反応条件下では不活性である有機溶媒、例えばアセトニトリル中で、又は水中で行われる。エチレンジアミン四酢酸二ナトリウム塩を加えて、存在する可能性のある重金属イオンを全て捕捉する。エポキシ化反応は、7より大きいpHで行うのが好ましい。このエポキシ化反応の後に、エポキシドを酸性条件下で過ヨウ素酸又は過ヨウ素酸塩によって酸化分解する。任意に酸化及び酸化分解を一つの処理操作に組み合わせてもよい。これらの反応については、ダンヤングらが「非官能化オレフィンを触媒的非対称性エポキシ化するためのC2対称性キラルケトン」J. Am. Chem.Soc., Vol.118, p.491-492(1996)及び「触媒的酸化反応のための新規の環状ケトン」J. Org. Chem., Vol.63,p.9888-9894(1998)において論じている。
【0076】
ルテニウム−ベースの酸化剤の使用が、カールセンらの「四酸化ルテニウム触媒による有機化合物の酸化法の顕著な改良」J. Org. Chem., Vol.46, No.19, p.3736-3738 (1981)で述べられている。カールセンらは、歴史的に、ルテニウム金属の費用が触媒法の発達の動機となったことを教示しており、最も人気のある触媒法は、化学量論的酸化剤として過ヨウ素酸塩か次亜塩素酸塩を使用している。これらの研究者はルテニウムの一般的使用では反応経過中に触媒活性が喪失することを見いだし、それはカルボン酸の存在によると仮定した。反応混合物にニトリル、特にアセトニトリルを加えると、CCl4/H2O/IO4−系においてアルケン類の酸化分解の速度及び程度が顕著に高まることが判明した。
【0077】
本発明の一実施形態によると、アセチルシクロスポリンAアルデヒド51は、次の手順でアセチルシクロスポリンA35から生成する;すなわち、アセチルシクロスポリンA35をアセトニトリルと水との混液に溶解し、それから最初に過ヨウ素酸ナトリウムを加え、次に塩化ルテニウム水加物を加える。アルデヒド51は酢酸エチルで抽出される。この酸化分解戦略によるアルデヒド51の合成は、以下に論ずる多くの立体選択的経路にとって重要であることに留意すべきであり、したがって読者はこの章を読み返して参考にすべきである。
【0078】
さらに、シクロスポリンAアルデヒドは、シクロスポリンAのβ−アルコールをアセチルシクロスポリンAの形成によって保護し、その後そのアセチルシクロスポリンAを、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、モノペル硫酸塩、より好ましくはオキソンで、アセチルシクロスポリンAエポキシドに変換する。この工程は、これらの反応条件下では不活性である有機溶媒、例えばアセトニトリル、又は水中で行われる。エチレンジアミン四酢酸二ナトリウム塩を加え、存在する可能性のある重金属イオンを全て捕捉する。エポキシ化反応は、7より大きいpHで行うのが好ましい。このエポキシ化反応の後に、エポキシドを酸性条件下で過ヨウ素酸又は過ヨウ素酸塩によって酸化分解する。酸化及び酸化分解は、一つの処理操作に組み合わせることもできる。これらの反応については、ダンヤングらが「非官能化オレフィンを触媒的非対称性エポキシ化のためのC2対称性キラルケトン」, J. Am. Chem.Soc., Vol.118, p.491-492 (1996)及び「触媒的酸化反応のための新規の環状ケトン」,J. Org. Chem., Vol,63,p.9888-9894 (1998)において論じている。
【0079】
方法2の第三工程は、方法1のそれと同様に、アルデヒド51をウィッティヒ反応によって(E)及び(Z)ジエンの混合物に変換することを含む。方法1と同様に、リンイリドをアルデヒドに加えるとベタインを生じ(これは分離しない)、その結果、アルデヒドのカルボニル酸素原子が、元々はリンに結合していたR2C=基によって置換される。ここでも、このようなウィッティヒ反応は、トリフェニルホスホニウム誘導体以外のリン含有化合物、例えばトリアリールホスフィン、トリアルキルホスフィン、アリールアルキルホスフィン及びトリアリールアルシンなどで、種々の温度で行うことができる。また種々の塩基性溶液及び溶媒の使用又は種々の無機塩の添加によって、新たに形成される二重結合の立体化学に影響を与えることもできる。
【0080】
一実施形態において、アセチルシクロスポリンAアルデヒド51をトルエンに溶解し、それに水酸化ナトリウム水溶液のような塩基を加える。それからアリルトリフェニルホスホニウムブロミド52を加え、反応物をしばらく撹拌する。生成したアセチル(E)及び(Z)−1,3−ジエン混合物53の処理は、ヘキサン及び/又は酢酸エチルによる抽出を含む;ここで、用語「処理(workup)」は、反応体、生成物、溶媒などの混合物から反応生成物を抽出及び/又は分離するプロセスを意味する。
【0081】
方法2の最終工程において、方法1の最終工程と同様に、β−炭素位置のアルコールを保護する酢酸エステル基を炭酸カリウムで除去すると、ISATX247の(E)及び(Z)−異性体混合物54が得られる。保護基の除去に使用できる炭酸カリウム以外の塩基としては、水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド、及びカリウムアルコキシドなどがある。
【0082】
ISATX247(E)及び(Z)−異性体のいずれかに富む組成物の有機金属経路による合成
本発明の実施形態によると、立体選択的合成経路は、珪素、ホウ素、チタン、硫黄、リン、及び/又はリチウムなどの元素類を含む無機試薬を使用することができる。これらの経路は六員環遷移状態を通過し、その際その六員環のメンバーの一つは、有機金属試薬からの無機元素である。幾つかの実施形態において、この遷移状態に関係する立体障害効果が反応の立体化学的結果に影響を与え得る。
【0083】
この開示において、2つの例示的立体選択的スキームが議論される。第一の立体選択的スキーム(方法3;図3の経路32としても示される)において、珪素含有化合物は脱離反応を受け、その脱離反応が酸性又は塩基性のいずれの条件において行われたかによって、(E)又は(Z)−異性体のいずれかを生成する。これはピーターソンのオレフィン化の一例である。第二の立体選択的スキーム(方法4;図3の経路33としても示される)において、各異性体は異なる前駆体から生成する。(Z)−異性体はチタン及びリン含有中間体から生成されるが、(E)−異性体はリチウム含有中間体を経て生成される。
【0084】
方法3
この経路は、アセチルシクロスポリンAアルデヒド51を経て進む。
同様な反応スキームは、ツァイ及びマッテソン著「ピナコール(E)−1−トリメチルシリル−1−プロペン−3−ボロネートからの(Z)及び(E)末端ジエン類の立体コントロールド合成」(Tetrahedron Letters, Vol.22, No.29, p.2751-2752(1981))において大体述べられている。この方法は、図6に示される。一般に、この合成は、トリメチルシリルアリルボロネートエステル試薬62をつくり、その後アセチルシクロスポリンAアルデヒド51を62で処理してβ−トリメチルシリルアルコール64を形成することを含む。このアルコールは、ホウ素含有遷移状態63を介して形成されると考えられている。ボロネートエステル類は、アリル硼酸化(allylboration)反応では緩徐に反応するので、E−γ-トリメチルシリルジエチルボラン又は9−(E−γ−トリメチルシリルアリル)−9−BBNなどの迅速反応性ボラン試薬の使用が有利であることは当業者には明らかである。β−トリメチルシリルアルコール64は、その後ピーターソンのオレフィン化を受けてアルケン、ここではジエン65かジエン67のいずれかをつくる。
【0085】
アルケンの形成は、脱離反応(オレフィン化)が酸性条件で行われるか塩基性条件で行われるかによって、異なる2経路のどちらかの経路を辿る。酸性条件下では、アンチ脱離が起こり(E)−異性体を形成し、塩基性条件下ではシス−脱離が起こり、(Z)−異性体が形成される。この合成経路の利用によって、同じ前駆体からどちらの異性体もつくることができることは当業者には明らかである。各脱離反応の生成物は、2異性体のどちらかの異性体に富む組成物を含む。一実施形態において、富む(又は富化)とはその組成物が約75重量パーセント以上の異性体を含むことを意味する。その他の実施形態において、富化組成物は1つの異性体を80、85及び90重量パーセント含むことができる。その後1異性体に富む組成物類を所定の比で組み合わせて、図10に示すような所望混合物を得ることができる。
【0086】
次に、図6の反応を、ホウ素含有試薬62の製法から始めて詳細に説明する。炭素−炭素結合形成反応体の合成における珪素試薬の使用に関する一般的研究は、エーリンガ及びマグナス著「合成における珪素。10.(トリメチルシリル)アリルアニオン:アルデヒド及びケトンをγ−ラクトンに変換するためのβ−アシルアニオン同等物」(J. Am. Chem. Soc., Vol.102, No.15, p.5004-5011 (1980))において論じられている。特に、これらの研究者は、(トリメチルシリル)アリルアニオンとアルデヒドとの反応について述べている。アニオンは、1当量のテトラメチルエチレンジアミン(TMEDA)を含む−76℃のテトラヒドロフラン中で、アリルトリメチルシランをsec−ブチルリチウムで脱プロトン化することによってつくることができる。
【0087】
アリルトリメチルシランの脱プロトン化(この工程は、図6には示されていない)は、ビールマン及びドュセプの「ヘテロ原子によって置換されたアリルカルボアニオン及びベンジルカルボアニオン」(Organic Reactions、第27巻(ウィリー、ニューヨーク、1982)、p.9)で述べられている。置換されたアリル系において、ヘテロ原子に対してアルファの位置にあるプロトンは、より塩基性の強い作用物質で除去される。種々のこのような作用物質が使用できるが、n−ブチルリチウムが多分最も一般的である。化学量のn−ブチルリチウムを、メタレーションをする予定の化合物と共に、テトラヒドロフラン(THF)を含む溶液中で使用する。温度は、通常0℃未満に維持する(−76℃未満の場合もよくある);その際n−ブチルリチウムは、その重合性のために低い反応性を有する。N,N,N',N'−テトラメチルエチレンジアミン(TMEDA)などのキレート剤を添加すると、その重合体は解離する。しかし、この反応は室温でも、TMEDAが存在しなくても起こり得る。
【0088】
アリルシランは、容易に脱プロトン化される。なぜならば、生成するアニオンは、隣接二重結合との共役によって安定化するだけでなく、近くのシリル基によっても安定化するからである。このアニオンは、求電子剤と、そのα炭素又はそのγ炭素を介して反応する。これらの反応の位置化学的及び立体化学的結末は幾つかの要因に依存するが、その最も重要な要因の一つは対イオンのアイデンティティである。トーマスの「有機合成:ホウ素及びシリコーンの役割」((オックスフォード大学プレス、ニューヨーク、1991)、p.84-87)によるアリルシラン類に関する議論を参照されたい。
【0089】
この反応スキームにおいて、脱プロトン化アリルシランはその後トリメチルボレートによる求電子捕獲を受け、中間体を生成する。この中間体はピナコールと反応すると、トランス−(トリメチルシリル)ボロネート化合物62を与える。ボロネート62は、「アリルボラン」(アリルボロネートエステル)とも呼ばれる。或いは、求電子捕獲に9−メトキシ−9−ジアルキルボランを使用するならば、ボロネート錯化合物が生成する。それを三フッ化ホウ素試薬(BF3Et2Oなど)によって脱メトキシル化すると、対応する9−(γ−トランス−トリメチルシリルアリル)−9−ジアルキルボランを生成することができる。
【0090】
アリルボランへのアルデヒドの付加について、S.E.トーマスが上記文献の34−35頁に述べている。アリルボランへのアルデヒド付加(この際アリルボランは、炭素−炭素二重結合の遠位端が非対称的に置換されている(「遠位(distal)」は、ホウ素原子から最も遠く離れていることを意味する))は、隣接する2つのキラル中心を含むホモアリルアルコールを生成する。(E)−アリルボラン類は、スレオ−ジアステレオ異性体を生じ、一方(Z)−アリルボラン類は、エリスロ−ジアステレオ異性体を生ずる。(E)−アリルボラン62とシクロスポリンAアルデヒド51との典型的反応が、図6に示される。ここでは、反応体をTHF溶液中で数日間撹拌した後、ホウ素中間体63が形成される。
【0091】
ホウ素中間体63(図6)中の参照番号69は、このホウ素位置でかなり多数の構造が可能であることを示すものである。例えば、ボロネート試薬62がトリアルキルシリルアリルボロネートエステルである場合、69の構造は、2個の酸素原子を含む五員環を構成する。62に使用するボロネート又はボラン試薬上の置換基が、63の構造中に存在する。
【0092】
アリルボランとアルデヒドを含む反応において得られる立体選択性は、ホウ素中間体63によって例示され、図6に描かれるような六員環いす形遷移状態によるものであると仮定されている。アルデヒドの2個のカルボニル原子(二重結合している炭素と酸素)だけが、六員環遷移のメンバーとなる;アルデヒドの残りは環を離れて延びる。六員環から離れて延びるアルデヒドのCsA部分は、その環に対してアキシアル配座というよりむしろエクアトリアル配座で存在すると仮定される。なぜならば、アキシアル配座はその置換基とアリルボラン62の酸素原子との間に好ましくない立体障害をおこすからである。図6において、(トリメチルシリル)アリルアニオンからのSiMe3基の位置がエクアトリアル位置を占めるように示されていることは当業者には明らかである。なぜならば、この例はアリルボランの(E)−ジアステレオマーから出発したからである。これとは異なり、開始アリルボランが(Z)−ジアステレオマーである場合は、SiMe3基はアキシアル位置に描かれる。
【0093】
或いは、上述の脱離反応とは反対に、酸脱離がシス−異性体を与え、塩基脱離がトランス−異性体を与えるようなエリスロ−シリルアルコールを形成することが考えられる。合成の最後には同じ生成物が得られることは、当業者にとって当然である。
【0094】
遷移状態の生成物63をトリエタノールアミンで処理すると、β−トリメチルシリルアルコール64が得られる。他方、(トリメチルシリルアリル)ジアルキルボランのアリルボレーション生成物は、NaOH/H2O2を使用する酸化又は水性処理でシリルアルコール64を与える。図6に描かれているアルコール64は、遷移状態のアリルボラン63が(E)−配置であったため、スレオ−ジアステレオ異性体である。ただし、Z−アリルボラン試薬から出発する場合は、その他のジアステレオ異性体がつくられることは当業者には明らかである。新しく作られたキラル中心におけるジアステレオ選択性は、この工程では確認されない;それは、この後の合成工程で、これらのキラル中心は脱離するからである。図6に示されるβ−トリメチルシリルアルコール64の構造は、出願人が分光法を用いて確認した。
【0095】
ピーターソンのオレフィン化として知られるアルケン合成法において、β−トリメチルシリルアルコール64からトリアルキルシリル基及びヒドロキシ基が除去され、アルケンが生成する;ここでは、鎖の2つの末端炭素の間にすでに二重結合が存在するため、ジエンが生成する。β−ヒドロキシシランのアルケンへの変換に関する議論が、S.E.トーマスの参考文献の68-69頁に記されている。この反応のより詳細な議論が、ハードリク及びピーターソンの「β−ヒドロキシシラン類の立体特異的オレフィン形成性脱離反応」J.Am. Chem. Soc.,Vol.97,No.6, p.1464-1468(1975)に記されている。
【0096】
図6を参照すると、アルコール64をジエンに変える脱離反応は、その反応が酸性条件で行われるか塩基性条件で行われるかによって、2つの異なるメカニズム経路を辿る。一方の経路はジエン65に導き、他方の経路はジエン67に導く。酸性条件下ではアンチ脱離が起き、一方塩基性条件下ではシン脱離が起きる。言い換えれば、β−ヒドロキシシランの脱離反応は立体特異的であり、酸−及び塩基−促進反応が反対の立体化学的コースをとる。酸促進反応のための典型的酸は、酢酸、硫酸及び種々のルイス酸などであり;典型的塩基は、水素化ナトリウム及び水素化カリウム又はカリウムtert−ブトキシドなどである。THF中水素化ナトリウムを使用する脱離反応は室温では緩徐で、水素化カリウムを使用する脱離反応はより速やかにあらわれるという場合もある。
【0097】
酸性条件下の脱離では、トリメチルシリル及びヒドロキシ基がアンチペリプラナー関係にあることが必要であるので、反応経路のこの工程で立体特異性が生ずる。反対に、塩基性条件下の脱離ではトリメチルシリル及びヒドロキシ基がシンペリプラナー関係にあることが必要である。後者の条件は、強い珪素−酸素結合及び中間体である四員環の形成を容易にする。この四員環は、ウィッティヒ反応の最終工程と同様の仕方で分解する。強い珪素−酸素結合はより弱い珪素−炭素結合にとって代わり、それはより弱い炭素−炭素π結合による強い炭素−酸素結合の置換を上回ることは当業者には明らかである。
【0098】
このように、β−ヒドロキシアルキルシランの立体特異的脱離の生成物は、アセチル−(E)−1,3−ジエン化合物67及びアセチル−(Z)−1,3−ジエン化合物65である。これまでに述べた方法のように、保護基は今やメタノール及び水中K2CO3による処理によって、これらのジエンの各々から除去される。これは1−アミノ酸残基のβ−炭素に結合した酢酸基を除去し、その炭素に結合する官能基をアルコールに戻す。保護基を除去するために使用できる炭酸カリウム以外の塩基類には、水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド、及びカリウムアルコキシドなどがある。
【0099】
この製造工程によって、合成は実質的に完了する。2異性体のうちのどちらか1つに富む組成物を混合し、混合物中の異性体類の所望比を得ることができる。「富む」とは、その異性体を少なくとも約75重量パーセント含む生成物を意味する;言い換えれば、その生成物が25重量パーセント以下の「望まない」異性体を含むことを意味する。望ましい薬理学的結果が得られるように混合物を設計する。
【0100】
方法4
この経路も、アセチルシクロスポリンAアルデヒド51を経て進む。
立体選択的異性体を生成するためのまた別のスキームを、図7−8に示す。この合成経路がこれまでに述べた経路と異なる点は、1)ISATX247の(E)−異性体を生成する合成経路が、(Z)−異性体のための中間体と異なる中間体を介して進むこと、及び2)これらの合成経路が、チタン及びリチウム含有試薬及び/又は中間体を使用することである。
【0101】
チタン試薬が有機合成では特に有用であることは知られている。なぜならば、それらはアルデヒド及びケトンとの反応において、位置−及び立体選択的であるからである。立体選択的化学におけるチタンの一般的性質は、リーツが「有機合成における有機チタン試薬」((スプリンガー出版、ベルリン、1986)、p.VII, 148-149及び164-165)で述べている。チタン試薬の電子的及び立体的アイデンティティが巧みに操作できるように、チタンリガンドの性質を変えることができ、多くのC−C結合形成反応の立体化学的結果が予測できると、ここには記されている。この化学的性質により、アキラル分子の2つのプロキラル中心の合体が、キラリティーの2つの中心を造り出す。立体選択的結果を決定する一般的ルールは、Z−配置のエノラート又はクロチル金属化合物が優先的にシン付加物を形成し、E−配置試薬はアンチ−ジアステレオマーを好んで形成することである。これらの傾向は、ここでもいす形を有する六員環状遷移状態を想定することによって説明できる。
【0102】
このタイプの立体選択的合成の特殊な例は、イケダらの「有機チタン及びリチウム試薬を使用した、アルデヒドから(Z)−及び(E)−1,3−アルカジエン類の立体選択的合成」(Tetrahedron, Vol.43, No,4,p.723-730(1987))に記載されている。この参考文献は、アリルジフェニルホスフィンを使用して[3−(ジフェニルホスフィノ)アリル]チタン試薬を生成し、この生成した試薬はその後アルデヒドと縮合し、その後ホスホニウム塩の形成によって、高度に位置選択的及び立体選択的な仕方で(Z)−1,3−アルカジエンを与えることを開示している。これに対して、リチウム化アリルジフェニルホスフィンオキシドはアルデヒドと縮合し、ここでも所望の立体選択性をもって直接(E)−1,3−アルカジエンを生成する。
【0103】
図7を参照すると、ISATX247の(Z)−異性体の合成は、(これまでのスキームと同様に)シクロスポリンA34からアセチルシクロスポリンAアルデヒド51を形成することから始まる。アリルジフェニルホスフィン71をt−BuLiのような強塩基で脱プロトン化し、その後生成物をチタンテトライソプロポキシドと反応させることによって、[3−(ジフェニルホスフィノ)アリル]チタン試薬72をつくる。遷移状態73は、理論的にはエリスロ−α−付加物74に移行すると考えられる。それは、その後74をヨードメタン(MeI)で処理することによって、β−オキシドホスホニウム塩75に変換できる。遷移状態73の存在が、少なくとも一部はこの合成経路の立体選択性をもたらすと考えられる。
【0104】
本明細書に概略した典型的方法によると、有機金属試薬の金属部位は、位置選択性をコントロールする存在でありうる(イケダ、p.725)。これは、図7のアルデヒド51がジフェニルホスフィノ化合物72とそのα位置で反応して、対応するα−付加物74を与えることを意味する。なぜならば、ジフェニルホスフィノ基のγ−炭素は、その金属(この場合はチタン)に配位しているからである。観察されるジエン生成物のZ選択性は、六員環遷移状態73を考慮することによって説明される。アルデヒド35のかさばったシクロスポリンA側鎖もジフェニルホスフィノ基も両方とも、遷移状態ではエクアトリアル位置を占めると考えられるので、エリスロα−付加物74が選択的に形成され、(Z)−1,3−ジエン76を生成する。
【0105】
ISATX247の(Z)−異性体がチタン遷移状態を経て形成される図7に描かれる反応経路とは異なり、(E)−異性体はこの方法では容易には生成しない。実際、この方法によって(E)−異性体を合成する試みは、概して低収率に終わることが報告されている。その代わり、図8に示すように、リチオ誘導体82は、アルデヒド51と反応してリチウム含有遷移状態83をつくり、それは約75:25より大きい範囲のE/Z比で1,3−ジエンを形成する。図7におけるように、反応生成物の高い立体選択性は、おそらく遷移状態83によるものである。この状態では、リチウム試薬82のビニル基及びアルデヒド51のシクロスポリンA側鎖はエクアトリアル位置を占め、それによって(E)−1,3−ジエン84を立体選択的に生成すると仮定される。前述のように、酢酸保護基に関する幾つかの不都合な副反応は、全ての立体選択的合成において、安息香酸エステル又はシリルエーテルなどの保護基の使用によって回避される。
【0106】
混合物の調製
前述のように、ISATX247のシス及びトランス−異性体のある種の混合物は、天然及び現在公知のシクロスポリンに勝る高い効力及び/又は低い毒性の組合わせを示すことが判明した。
【0107】
本発明の実施形態によると、ISATX247異性体(及びこれらの誘導体)は、立体選択性の程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)−異性体に富む第一の材料又は組成物、及び(Z)−異性体に富む第二の材料又は組成物を生成し、これらの材料を組み合わせて、生成した混合物が2異性体を所望比で有するようにすることができる。或いは、反応生成物を分離して(E)−異性体を単離し、富化することによって第一材料をつくり、反応生成物を分離して(Z)−異性体を単離し、富化することによって第二材料をつくることが考えられる。また別の実施形態においては、立体選択的経路の反応条件を調節して、生成混合物に直接所望比をもたらす。
【0108】
これらの原理は、図9A−C及び図10に説明される。図9A−Cには、(E)対(Z)−異性体の比がそれぞれ約65対35重量パーセント、50対50重量パーセント、及び35対65重量パーセントである仮定の合成反応が示される。もちろんこれらの比は例示的なものであり、説明の目的のために過ぎず、仮定のあらゆる数の組合わせが選択できる。生成混合物中の異なる異性体比を実現するために、図9Aの比を形成するために適用する反応条件は図9B及び図9Cのそれらと異なってよい、ということは当業者には当然である。各反応の条件を調節し、その場合に応じた2異性体の特定の比が形成される。
【0109】
異性体の混合物を生成する幾つかの合成経路とは異なり、異性体をまず最初に個々につくり、所定の比率で混合して所望比を得ることもできる。この概念は、図10に示される。ここでは、一立体選択的経路の生成物はこれら異性体のうちの1異性体に富み、生成物が約75重量パーセントより多い(E)異性体を含み、他の立体選択的経路の生成物は他の異性体に富み、この生成物は約75重量パーセントより多い(Z)異性体を含む。これらの数字も例示的であり、一実施形態において、立体選択的経路によって生成した所望異性体の純度は、約75重量パーセント以上であり得る。他の実施形態において、所望異性体はそれぞれ約80、85、90及び95重量パーセント以上を含みうる。
【0110】
これらの異性体を個々に合成した後、それらを混合して図10に示すように所望比を得ることができる。例示目的のために、図10では図9A−Cで用いた比率と同じ仮定的比率を選択する。図10を参照すると、(E)及び(Z)−異性体を混合して、それぞれ約65対35重量パーセント、50対50重量パーセント、及び35対65重量パーセントの(E)対(Z)−異性体比を有する3種類の混合物を得る。
【0111】
また別の実施形態において、ISATX247異性体の(E)及び(Z)−異性体の混合物を分離し、混合物中において、ある異性体が他の異性体より多いようにする。例えば、ディールス−アルダー反応を利用して、シス異性体を、アルケンとの反応により閉鎖環化合物に変換する。もしアルケンが単離可能の(例えば濾過可能の)基質に結合するならば、シス異性体はその混合物から実質的に除去され、トランス異性体に富む組成物が残る。シス異性体は閉鎖環化合物から加熱によって再構成され、シス異性体に富む組成物を生成する。このやり方で、シス及びトランス異性体が分離できる。
【0112】
実際、混合物を生成する方法の立体選択性の程度には無関係に、いかなる混合物中でも(E)対(Z)−異性体の比は広範囲の数値をとる。例えば、混合物は約10ないし90パーセントの(E)−異性体から約90ないし10パーセントの(Z)−異性体までを含む。その他の実施形態において、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)−異性体;又は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体;又は約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体;又は約45ないし55重量パーセントの(E)−異性体及び約55ないし45重量パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセントは組成物の総重量を基準にしており、(E)異性体及び(Z)異性体の重量パーセントの合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体及び35重量パーセントの(Z)−異性体を含むことがあり、その逆でもよい。
【0113】
混合物中のある異性体又はその他の異性体のパーセンテージは、核磁気共鳴(NMR)又は当該技術で公知のその他の方法を使用して確認できる。
【0114】
医薬組成物
本発明は、活性成分として本発明の混合物を含む医薬組成物の投与を含む、免疫抑制を必要とする患者の治療法にも関係する。この組合わせが対象とする適応症は、特に自己免疫病及び炎症性疾患、及び移植拒絶に関係するか、これに起因する異常などであり、例えば下記のような異常の治療(病因又は症状の改善、軽減、排除又は治癒を含む)又は防止(実質的又は完全な制限、予防又は回避を含む)を含む:
【0115】
a)急性臓器又は組織移植拒絶、例えば心臓、肺、心−肺同時、肝臓、腎臓、膵臓、皮膚、腸又は角膜移植片などのレシピエントの治療、特に、T細胞性拒絶、並びに例えば骨髄移植後のグラフト対ホスト疾患の予防及び/又は治療。
【0116】
b)移植臓器の慢性拒絶、特にグラフト血管疾患、例えば平滑筋細胞増殖による内膜肥厚や関連効果の結果としてのグラフト血管狭窄を特徴とするグラフト血管疾患の予防。
【0117】
c)臓器ドナーがレシピエントと異なる種である場合に起きる、臓器の急性、超急性、又は慢性拒絶、非常に特別なB細胞性拒絶又は抗体仲介性拒絶を含む、異種移植片拒絶。
【0118】
d)自己免疫病及び炎症性疾患、特に免疫学的又は自己免疫要素を含む病因を有する炎症性疾患、例えば関節炎(例えばリウマチ性関節炎、慢性進行性関節炎及び変形性関節炎)及びその他のリウマチ性疾患など。本発明の共力的組合わせを適用できる特異的自己免疫病は、自己免疫性血液疾患(溶血性貧血、再生不良性貧血、赤芽球ろう、特発性血小板減少症など)、全身性エリテマトーデス、多発性軟骨炎、強皮症、ウェーゲナー肉芽腫症、皮膚筋炎、慢性活性肝炎、重症筋無力症、乾癬、スティーヴン−ジョンソン症候群、特発性スプルー、(自己免疫性)炎症性腸疾患(例えば潰瘍性大腸炎及びクローン病を含む)、内分泌性眼障害、グレーブス病、サルコイドーシス、多発性硬化症、原発性胆汁性肝硬変、若年性糖尿病(I型糖尿病)、ぶどう膜炎(前部及び後部)、乾性角結膜炎及び春季角結膜炎、間質性肺線維症、乾癬性関節炎、糸球体腎炎(特発性ネフローゼ症候群又はリポイドネフローゼなどのネフローゼ症候群を伴う又は伴わない)、及び若年性皮膚筋炎を含む。皮膚の自己免疫病及び炎症性疾患も本発明の共力的組合わせを適用して治療及び予防できると考えられる;例えば乾癬、接触性皮膚炎、アトピー性皮膚炎、円形脱毛症、多形性紅斑、疱疹状皮膚炎、硬皮症、白斑、過敏性血管炎、蕁麻疹、水泡性類天疱瘡、エリテマトーデス、天疱瘡、後天性表皮水疱症、その他の炎症性又はアレルギー性皮膚障害などである;喘息、アレルギー及びじん肺症などの肺及び気道の炎症性異常も含まれる。
【0119】
本発明の異性体同族混合物はそれだけで、又は薬物学的担体と共に、それらを必要とする温血動物に投与される。薬物学的担体は固体でも液体でもよい。本発明の混合物は従来の毒性のない薬物学的に容認される担体、アジュバント及びビヒクルを含む投与単位の処方で、経口的、局所的、非経口的に、又は吸入スプレーによって、又は直腸内に投与される。ここに使用する用語「非経口的」とは、皮下注射、静脈内、筋肉内、胸骨内注射又は注入法を含む。
【0120】
本発明の混合物を含む医薬組成物は、例えば錠剤、トローチ、ロゼンジ、水性又は油性懸濁液、分散性粉末又は顆粒、乳濁液、硬又は軟カプセル、又はシロップ又はエリキシルのような経口的使用に適した形であるのが好ましい。経口使用のための組成物は、医薬組成物を製造する当該技術で公知の方法によってつくられ、そのような組成物は甘味料、香料、着色料及び保存料からなる群から選択される1種類以上の材料を含み、医薬品として洗練され、口当たりのよい製剤が提供される。活性成分を無毒性の薬物学的に容認される賦形剤と共に含む錠剤も、公知の方法によって製造される。使用する賦形剤は例えば、(1)炭酸カルシウム、乳糖、燐酸カルシウム又は燐酸ナトリウムなどの不活性希釈剤;(2)コーンスターチ又はアルギン酸などの顆粒化剤及び崩壊剤;(3)澱粉、ゼラチン又はアカシアなどの結合剤;及び(4)ステアリン酸マグネシウム、ステアリン酸又はタルクなどの滑剤である。これら錠剤は、コーティングしなくてもよいが、公知の方法によってコーティングし、消化管における崩壊及び吸収を遅らせ、それによって長時間にわたって持続的作用を提供することもできる。例えば、グリセリルモノステアレート又はグリセリルジステアレートなどの徐放性材料を使用してもよい。それらは、米国特許第4,256,108号;第4,160,452号;及び第4,265,874号に記載されている方法によってコーティングされ、浸透性治療錠を形成し、放出をコントロールする。
【0121】
ある場合には、経口使用のための処方は、ハードゼラチンカプセルの形で、その中に活性成分が炭酸カルシウム、燐酸カルシウム又はカオリンなどの不活性固体希釈剤と混合して含まれる。それらは、ソフトゼラチンカプセルの形でもよく、その中に活性成分が水、又はピーナツ油、液体パラフィン、又はオリーブ油などの油性メジウムと混合して含まれる。
【0122】
水性懸濁液は、活性成分を水性懸濁液の製造に適した賦形剤と混合して含むのが普通である。このような賦形剤としては:(1)カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガントガム及びアカシアゴムなどの懸濁剤;又は(2)分散剤又は湿潤剤、例えばレシチンなどの天然ホスファチド、ポリオキシエチレンステアレートなどのアルキレンオキシドと脂肪酸との縮合産物、ヘプタデカエチレンオキシセタノールなどのエチレンオキシドと長鎖脂肪アルコールとの縮合産物、ポリオキシエチレンソルビトールモノオレエートのような、脂肪酸及びヘキシトールから誘導される部分エステルとエチレンオキシドとの縮合産物、又はポリオキシエチレンソルビタン モノオレエートのような、脂肪酸及びヘキシトール無水物から誘導される部分エステルとエチレンオキシドとの縮合産物がある。
【0123】
水性懸濁液は、1種類以上の保存料、例えばエチル又はn−プロピルp−ヒドロキシベンゾエート;1種類以上の着色料;1種類以上の香料;及び1種類以上の甘味料、例えばスクロース、アスパルターム又はサッカリンを含むことができる。
【0124】
油性懸濁液は、活性成分をアラキス油、オリーブ油、ゴマ油又はココナツ油などの植物油、オメガ3脂肪酸を含む魚油、又は液体パラフィンなどの鉱油に懸濁させることによって処方される。油性懸濁液は、蜜蝋、ハードパラフィン又はセチルアルコールなどの濃化剤を含むことができる。甘味料及び香料を加えて、口当たりのよい経口製剤を提供することができる。これらの組成物は、アスコルビン酸などの抗酸化剤を加えることによって保存できる。
【0125】
分散性粉末及び顆粒は、水性懸濁液の調製に適する。それらは、分散剤又は湿潤剤、懸濁剤及び1種類以上の保存料と混合した活性成分を提供する。適切な分散剤又は湿潤剤及び懸濁剤の例はすでに上述されている。その他の賦形剤、例えば上記の甘味料、香料及び着色料も含むことができる。
【0126】
本発明の混合物を含む医薬組成物は、水中油型エマルションの形でもよい。油相は、オリーブ油又はアラキス油のような植物油、又は液体パラフィンのような鉱油又はそれらの混合物でよい。適切な乳化剤は、(1)アカシアゴム又はトラガントゴムのような天然のゴム類、(2)大豆油及びレシチンなどの天然のホスファチド類、(3)脂肪酸及びヘキシトール無水物から誘導されるエステル類又は部分エステル30、例えばソルビタンモノオレエートなど、(4)前記部分エステル類とエチレンオキシドとの縮合産物、例えばポリオキシエチレンソルビタンモノオレエート。乳化剤は、甘味料及び香料も含むことができる。
【0127】
シロップ類及びエリキシル類は、グリセロール、プロピレングリコール、ソルビタン、アスパルテーム又はスクロースなどの甘味料と共に処方される。このような処方は、緩和剤、保護剤、保存料及び香料及び着色剤も含むことができる。
【0128】
本発明の医薬組成物は、無菌の注射可能水溶液又は油性懸濁液の形でもよい。この懸濁液は、上述した適切な分散剤又は湿潤剤及び懸濁剤を使用して、公知の方法によって処方される。無菌の注射可能の製剤は、無毒性の非経口的投与可能の希釈液又は溶媒を使用した無菌の注射可能溶液又は懸濁液でもよく、例えば1,3−ブタンジオール溶液などである。使用できるビヒクル及び溶媒は水、リンゲル液及び等張塩化ナトリウム溶液である。その上、無菌の固定油が溶媒又は懸濁メジウムとして一般的に使用される。この目的のためには、合成モノ−又はジ−グリセリドを含む、あらゆるブランドの固定油も使用できる。さらに、オレイン酸などの脂肪酸も注射用製剤に使用できる。
【0129】
本発明の混合物は、薬剤の直腸投与用坐薬の形でも投与できる。これらの組成物は、薬剤を、普通の温度では固体であるが直腸温度では液体となり、そのため直腸で融解して薬剤を放出する、適切な非刺激性賦形剤と混合することによってつくられる。このような材料は、ココアバター及びポリエチレングリコール類である。
【0130】
局所的使用のためには、開示されたシクロスポリン類を含むクリーム、軟膏、ゲル、溶液又は懸濁液などが使われる。
【0131】
特に好ましい実施形態において、不活性成分として界面活性剤、エタノール、親油性及び/又は両極親和性溶媒を含む液体溶液を使用する。より詳細に述べれば、異性体類似体混合物と、非薬剤成分:d−アルファトコフェロールポリエチレングリコール1000琥珀酸(ビタミンE TPGS)、中鎖トリグリセリド(MCT)油、ツィーン40、及びエタノールとを含む経口マルチプルエマルション処方が用いられる。異性体類似体混合物と経口溶液の場合と同じ非薬剤成分とを含むソフトゼラチンカプセル(ゼラチン、グリセリン、水及びソルビトールを含む)も好適に使用される。
【0132】
1日に体重1kgあたり約0.05mgないし約50mgの桁の投与量、が上記の疾患の治療に有効である。投与量及び投与スケジュールは、使用する特定の異性体混合物、治療する症状、及び患者の年齢及び症状などの付加的要素によって変動する。好ましい投与量は、約0.5ないし約10mg/kg/日及び約0.1ないし約10mg/kg/日である。好ましい実施形態において、約2ないし約6mg/kg/日が1日2回経口投与される。特に好ましい実施形態においては約0.5ないし約3mg/kg/日を1日2回投与する。
【0133】
単回投与型を形成するために担体材料と組み合わせることができる活性成分量は、治療するホスト及び特定の投与法によって変動する。例えば、ヒトに経口投与するための製剤は、2.5mgないし2.5gの活性成分を適切かつ好都合な量の担体材料と組み合わせて含む。その量は、総組成物の約5ないし約95パーセントまでの間で変動する。単回投与型は、一般的に約5mgないし約500mgの活性成分を含む。好ましい実施形態においては、約50mgの異性体混合物を含む個々のカプセルを経口投与のために用いる。また別の好ましい実施形態においては、約50mg/mLの異性体混合物を含む経口溶液を経口投与のために使用する。
【0134】
しかし、いかなる特定の患者においても、その特異的投与量は使用する特異的化合物、年齢、体重、身体全体の健康、性別、食事、投与時間、投与経路、排泄速度、薬剤組合わせ及び治療を受ける特定の疾患又は異常の性質及び重症度、など種々の要素に左右される。
【0135】
方法論
Tolypocladium inflatum Gams真菌によって産生される環式ポリペプチドの一群であるシクロスポリン誘導体は、T−細胞仲介性反応に対するそれらの好ましい効果のために、免疫治療における使用が増加しつつある。シクロスポリン誘導体は、免疫担当リンパ球、特にT−リンパ球を可逆的に抑制し、リンホカインの産生及び放出を抑制する。この作用は主として、シクロスポリンA−誘起性カルシニューリン阻害によって仲介される。カルシニューリンは、細胞の細胞質に見いだされるホスファターゼ酵素である(シュライバー及びクラブトリー、1992)。シクロスポリンA又はシクロスポリンA誘導体の効果の指標は、カルシニューリンのホスファターゼ活性を阻害する能力である。カルシニューリン阻害アッセイは、その作用部位における薬剤活性を測定する試験法であり、そのアッセイはシクロスポリンA類似体の効力の最も正確で直接的なinvitro評価である(フルマンら、1992)。
【0136】
ISATX247は、分子のアミノ酸1残基上の官能基の新規の修飾を除けば、シクロスポリンAに類似したシクロスポリンA類似体である。我々は今や、invitroカルシニューリン阻害アッセイにおいて、ISATX247がシクロスポリンAの3倍までの効力を示すことを見いだした。
【0137】
薬力学的研究(in vivo及びin vitro)は、ISATX247がその他の存在するシクロスポリン化合物より効力が高いことを示した。シクロスポリン類似体の約10:90ないし約10:90(トランス−対シス−)の異性体混合物、特に50−55%Z−異性体及び45−50%E−異性体を有するISATX247の免疫抑制剤としての効果が(シクロスポリンAに比較して)、invitroカルシニューリン活性アッセイ、ラット心臓移植モデル、膵島細胞同種移植マウスモデル、マウスのコラーゲン誘起性関節炎モデル、及び/又はウサギの抗原誘起性関節炎モデルで証明されている。これらのデータは、これらの異性体混合物の効力が、シクロスポリンAの効力と同等か又はより大きく、したがって免疫調節性疾患の治療に有効であることを示している。
【0138】
腎毒性、肝毒性、白内障発生、多毛、パラセシス(parathesis)、歯肉増殖症などを含む、シクロスポリンA治療に関連する副作用が多数ある(スケトリスら、1995)。これらの中で、腎毒性はシクロスポリンA投与に起因するより深刻な用量関連性副作用の一つである。シクロスポリンAが腎障害をおこす正確なメカニズムは知られていない。しかし、腎臓の血管収縮性物質の濃度の増加が、求心性糸球体細動脈の局所的血管収縮を起こすことが予想される。これは虚血、糸球体濾過率の減少を起こし、長期にわたると間質性線維症を起こす。
【0139】
ISATX247の非臨床的安全性が多数の動物種で評価された。ラット、イヌ及び霊長類における反復経口投与による毒性研究により、ISATX247は耐容性がよく、免疫抑制と一致する効果をもたらすことが判明した。全ての種で認められた唯一の毒性作用は、下痢/軟便であった。
【0140】
ISATX247は、in vitro細菌逆突然変異及び染色体異常アッセイ、及びin vivoラット小核アッセイで証明されたように、突然変異誘発作用を示さない。発癌性の研究は、現在完了していない。ISATX247の生殖毒性試験が、妊娠ラット及びウサギで完了した。治療に関連する先天性異常又は変化はなかった。母体毒性を起こす用量では、相応の胚毒性が認められた。
【実施例】
【0141】
実施例1:シクロスポリンAのアセチル化
無水酢酸(140ミリリットル)をシクロスポリンA(50.0g、41.6mmol)に加え、混合物を室温でN2気流下で、シクロスポリンAの全てが溶解するまで撹拌した。ジメチルアミノピリジン(7.62g、62.4mmol)を加え、反応物を室温でN2気流下で3時間、又は反応が完了するまで撹拌した。反応混合物を5℃に冷やし、その後濾過した。冷却した固体をヘキサンで洗い、付加した無水酢酸を除去した。生成したペースト状固体を激しく撹拌した5%炭酸水素ナトリウム水溶液(1.5リットル)に徐々に移した。生成した懸濁液を、微細スラリーが生成し、CO2の発生が止むまで撹拌した。固体を濾過によって集め、濾液が中性pHを示すまで水で洗った。固体生成物を真空中で一晩乾燥すると(55℃)、生成物が着色固体として44.0g(85%)得られた。
【0142】
実施例2:実施例1からの生成物の酸化
アセトニトリル(320mL)及び水(80mL)をアセチルシクロスポリンA(42.97g、34.54mmol)に加え、材料の全てが溶解するまで混合物を撹拌した。過ヨウ素酸ナトリウム(14.77g、69.08mmol)を加え、その後塩化ルテニウム水加物(0.358g、1.73mmol)を加え、その後反応物を室温でN2気流下で3時間撹拌した。水(300mL)を加え、混合物を分液漏斗に移した。混合物を酢酸エチル(300mL、その後250mL)で2度抽出した。暗黒色の酢酸エチル抽出液を合一し、水250mLで、その後ブライン250mLで洗った。有機溶液をMgSO4上で乾燥し、溶媒を蒸発すると、緑色がかった黒い固体が生成した。その粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(29.1g、68%)が無色固体として得られた。
【0143】
実施例3:アセチルISATX247の製法
i)イリドのin situ生成
アセチルシクロスポリンAアルデヒド(31.84g、25.84mmol)を340mLトルエンに加え、材料が完全に溶解するまで混合物を撹拌した。生成した溶液に1ノルマル水酸化ナトリウム水溶液を340mL加えた。生成した混合物を激しく撹拌し、それからアリルトリフェニルホスホニウムブロミド(58.22g、151.90mmol)を加えた。反応物を室温で24時間撹拌し、追加的アリル トリフェニルホスホニウムブロミド(16.64g、43.42mmol)を加え、さらに24時間撹拌し続けた。混合物を分液漏斗に移し、トルエン相を分離した。水相をさらに、200mLのトルエンで抽出した。2つのトルエン抽出液を合一し、200mL脱イオン水及び200mL塩化ナトリウム飽和水溶液で逐次洗った。その溶液をMgSO4上で乾燥し、濾過し、トルエンを蒸発すると非常に粘稠なゲルが生成した。この物質を142mL酢酸エチルで処理し、微細スラリーが形成されるまで撹拌した。急速撹拌下でヘキサン(570mL)をゆっくりと加えた。撹拌を30分間続け、生成した懸濁液を濾過し、集めた固体を160mLの5:1ヘキサン/酢酸エチルで洗った。合一した濾液を回転蒸発器で濃縮すると粘稠な半固体が得られた。この物質を75mL酢酸エチルで処理し、微細スラリーが得られるまで撹拌した。ヘキサン(225mL)を急速撹拌下でゆっくりと加える。撹拌を30分間続け、それから生成した懸濁液を濾過し、集めた固体を100mLの5:1ヘキサン/酢酸エチルで洗った。濾液を回転蒸発器で濃縮すると、淡黄色固体が得られた。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液として使用してクロマトグラフィーにかけると、生成物(14.09g)が無色固体として得られた。
【0144】
ii)LiBrの存在下におけるプレフォームド イリドの生成及び反応
0℃に冷却したTHF(20mL)中アリルトリフェニルホスホニウムブロミド(7.67g、20mmol)の撹拌懸濁液に、テトラヒドロフラン中KOBut溶液(20mL、20mmol、1M溶液)を加えた。この温度で撹拌を30分間続け、THF中LiBr溶液(10mL、10mmol、1M溶液)を加えた。反応混合物を30分間撹拌し、THF(10mL)中アセチルCsA−アルデヒド溶液(4.93g、4mmol)溶液をカニューレによって加えた。室温で15分間撹拌後、反応混合物に飽和NH4Cl溶液(25mL)を加えて反応を停止した。上記のような処理及びクロマトグラフィーにより、アセチル化ISATX247が無色固体(3.5g)として生成した。
【0145】
実施例4:ISATX247の製法
アセチルISATX247(14.6g、11.62mmol)を340mLメタノールに溶解し、その後脱イオン水135mLを加えた。炭酸カリウム(13.36g、96.66mmol)を加え、混合物を室温で24ないし48時間、反応が完結するまで撹拌した。メタノールの大部分を蒸発し、それから250mL酢酸エチルを撹拌しながら加えた。10%クエン酸水溶液(120mL)をゆっくりと加え、酢酸エチル相を分離した。水相を追加200mL部分の酢酸エチルで抽出した。合一した酢酸エチル抽出液を150mL脱イオン水、100mL10%クエン酸水溶液及び150mL塩化ナトリウム飽和水溶液で逐次洗い、MgSO4上で乾燥した。酢酸エチルを蒸発すると、淡黄色固体が得られた。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液として用いてクロマトグラフィーにかけると、ISATX247(10.51g、75%)が無色固体として得られた。ISATX247は45−50%E−異性体及び50−55%Z−異性体を含む。
実施例1−4の生成物は、質量分析及び/又は核磁気共鳴分光法によって特徴づけられた。
【0146】
実施例5:アセチル−η−ブロモシクロスポリンAの製法
実施例1におけるようにつくられたアセチルシクロスポリンA(41.48g、33.3mmol)、N−ブロモスクシミド(10.39g、58.4mmol)及びアゾ−ビス−イソブチルニトリル(1.09g、6.67mmol)を四塩化炭素250mLに溶解し、生成した混合物を2.5時間加熱還流した。混合物を冷却し、溶媒を蒸発した。残留物を350mLジエチルエーテルで処理し、濾過して不溶性物質を除去した。濾液を150mL水及び150mLブラインで逐次洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗生成物をシリカゲル上でアセトン/ヘキサン(2:3)でクロマトグラフィーにかけるとアセチル−γ−ブロモシクロスポリンA28.57g(65%)が黄色固体として得られた。
【0147】
実施例6:アセチルシクロスポリンAのトリフェニルホスホニウムブロミドの製法
アセチル−γ−ブロモシクロスポリンA(28.55g、21.6mmol)及びトリフェニルホスフィン(7.55g、28.8mmol)をトルエン210mLに溶解し、生成した溶液を21時間、100℃に加熱した。溶液を冷却し、トルエンを蒸発した。生成した油性の半固体に250mLのヘキサン/エーテル(1:4)を加え、徹底的に混合し、溶媒を傾瀉して除去した。このプロセスを、150mLエーテルで3回繰り返した。残留物を50mL酢酸エチルに溶解し、220mLヘキサンで沈殿させた。生成した固体をその後濾過によって集めると、アセチルシクロスポリンAのトリフェニルホスホニウムブロミド22.5g(66%)が黄褐色固体として得られた。
【0148】
実施例7:ウィッティヒ反応
アセチルシクロスポリンのトリフェニルホスホニウムブロミド(100mg、0.06mmol)、過剰の37%ホルムアルデヒド(0.25mL)及びトルエン(2mL)を室温で速やかに撹拌した。1N水酸化ナトリウム水溶液(2mL)を滴下し、撹拌を3.5時間続けた。反応混合物を、酢酸エチル(20mL)及び水(10mL)で希釈した。酢酸エチル相を分離し、水(10mL)及びブライン(10mL)で逐次洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗物質をシリカゲル上で、アセトン/ヘキサン(2:3)でクロマトグラフィーにかけると、アセチルISATX247の(E)及び(Z)−異性体混合物70mg(88%)が無色固体として得られた。
【0149】
実施例8:ウィッティヒ反応生成物の脱アセチル化
実施例7から生成した異性体混合物(70mg、0.056mmol)をメタノール(5mL)に溶解し、水(1mL)を加えた。炭酸カリウム(75mg)を加え、反応物を室温で19時間撹拌した。メタノールの大部分を蒸発し、残留物に酢酸エチル15mLを加え、その後10%クエン酸水溶液10mLを加えた。酢酸エチル相を分離し、水相を追加的酢酸エチル10mLで抽出した。合一した酢酸エチル抽出液を、10mL水、10mL10%クエン酸水溶液及び10mLブラインで逐次洗い、その後硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗物質をシリカゲル上で、アセトン/ヘキサン(2:3)でクロマトグラフィーにかけると、ISATX247、37mg(54%)が約85%E−異性体及び約15%Z−異性体を含む無色固体として得られた。
実施例5−8における生成物は、質量分析及び/又は核磁気共鳴分光法によって特徴づけられた。
【0150】
実施例9:ISATX247の幾何異性体の製法
ISATX247のシス−及びトランス−異性体を下記の反応スキームを用いて独立的に合成した。反応系列は、アリルトリメチルシランの公知のメタレーション、トリメチルボレートによる求電子捕獲、及びその後の加水分解及びその後のエステル交換を含み、中間体トランス−(トリメチルシリル)アリルボロネートエステルを生成する。シクロスポリンアルデヒドのアリルボレーションはホウ素中間体を与え、それを金属イオン封鎖によって所望のβ-トリメチルシリルアルコールに変換する。新しいキラル中心の生成におけるジアステレオ選択性は、その後の段階でこれらの中心が除去されるため、この段階では決定しない。β−トリメチルシリルアルコールにおける2つの中心の相対的立体化学は予想通りアンチであり、トランス−(トリメチルシリル)ボロネート前駆体におけるトランス二重結合によるものであることに注目すべきである。
【0151】
β−トリメチルシリルアルコールの塩基促進性脱離(ハードリクら、1975)はアセチル−(Z)−1,3−ジエンに富む組成物を提供し、酸促進性脱離はアセチル−(E)−1,3−ジエンに富む組成物を与える。脱保護は、対応するジエンアルコール、ISATX247の(Z)及び(E)−異性体をそれぞれ与える。
【0152】
ジエンに至るまた別のアプローチは、アリルホスホランを使用する。アリルジフェニルホスフィンのメタレーション及びその後のTi(OPri)4によるトランスメタレーションは、チタン中間体を与える。アリルチタネーション及びその後の立体特異的脱離で、(Z)−ジエンに富む組成物が生成する。
【0153】
他方、アリルジフェニルホスフィンオキシドで同様な系列を行うと(図8)、主としてE−異性体が生成する(75%)。
【0154】
i)アセチルCsA−CHOのアリルボレーション:
(E)−1−トリメチルシリル−1−プロペン−3−ボロネートを前に報告された方法にしたがってつくった(イケダら、1987)。THF(3mL)中(E)−1−トリメチルシリル−1−プロペン−3−ボロネート(0.2g、0.832mmol)の撹拌溶液に、窒素気流下でアセチルシクロスポリンAアルデヒド(1.026g、0.832mmol)を加えた。反応混合物を高性能液体クロマトグラフィー(C−8カラム、逆相)によってモニターし、合計7日間撹拌した。その後トリエタノールアミン(0.196g、1.3mmol)を加え、さらに4日間撹拌し続けた。シリカゲルカラム上で精製すると、β−トリメチルシリルアルコールが得られた。MS(ES)m/z1368.9(M+Na+)。
【0155】
無水THF(1mL)中KH懸濁液(3.5mg、26.4μmol、無水ヘキサンで洗浄した30%鉱油分散液)に、β−トリメチルシリルアルコール(10mg、7.4micromol)を加え、室温で10分間撹拌した。反応混合物をジエチルエーテル(10mL)で希釈し、その後NaHCO3飽和溶液(2×5mL)で洗った。乾燥(Na2SO4)及び溶媒除去により、富化(Z)−アセチル−1,3−ジエンが得られた。MS(ES)m/z1294.8(M+K+)。
【0156】
ii)アセチルCsA−CHOのアリルチタン化:
無水THF(8mL)中アリルジフェニルホスフィン(0.54g、2.4mmol)の撹拌、冷却(−78℃)溶液に、t−BuLi(1.42mL、2.4mmol、1.7M溶液。ペンタン中)を加えた。赤煉瓦色の溶液をこの温度で15分間撹拌し、その後0℃で30分間撹拌した。それから再び−78℃に冷やし、Ti(OPri)4(0.71mL、2.4mmol)を加えた。その褐色溶液をこの温度で15分間撹拌し、それからTHF(10mL)中アセチルCsA−CHO(2.5g、2mmol)溶液をカニューレによって加えた。淡黄色溶液をさらに30分間撹拌し、それから一晩室温に温めた。この反応混合物に0℃でMeI(0.15mL、2.4mmol)を加えた。撹拌をこの温度で1時間続け、それから室温で2時間続けた。反応混合物を氷冷した1%HCl(100mL)に注いだ。水層をEtOAc(3×50mL)で抽出した。合一した有機抽出液を水(2×25mL)及びブライン(25mL)で洗った。溶媒を除去すると黄色固体が得られ、それをシリカゲルカラムでクロマトグラフィーにかけた。1:3アセトン−ヘキサン混合物で溶出すると、アセチルISATX247の(Z)−富化異性体が得られた。実施例4におけるように脱保護すると、ISATX247の(Z)−富化異性体が得られた(Z/E比、75:25)。
【0157】
実施例10:ISATX247異性体の(E)−富化混合物の製法
テトラヒドロフラン(5mL)中アリルジフェニルホスフィンオキシド(1mmol)及びヘキサメチルホスホルアミド(2mmol)の溶液に、−78℃で、n−ブチルリチウム(1mmol、ヘキサン中)を加えた。混合物を−78℃で30分間撹拌した。テトラヒドロフラン(7mL)中アセチルシクロスポリンAアルデヒド(0.8mmol)の溶液を加え、反応混合物を徐々に室温まで温め、それから18時間撹拌した。混合物を氷冷した1N塩酸(50mL)に注入し、それから酢酸エチルに抽出した。有機抽出液を水で洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。残留物をシリカゲル上で、25%アセトン/75%ヘキサンを溶出液としてクロマトグラフィーにかけると、アセチルISATX247の(E)及び(Z)−異性体混合物が得られた。実施例4におけるようにアセテート保護基を除去すると、ISATX247異性体の(E)−富化混合物が得られた。プロトンnmr分光法は、この混合物がISATX247の(E)−異性体75%及び(Z)−異性体25%からなることを示した。この反応は、シュロッセルの改良法(リュ−(R.Liu)、シュロッセル(M. Schlosser), Synlett, 1996, 1195)によっても行うことができる。THF(20mL)中アリルジフェニルホスフィンオキシド(1.21g、5mmol)の撹拌した冷却(−78℃)溶液に、n−BuLi(2mL、5mmol、2.5M溶液、ヘキサン中)を加えた。その赤色溶液を−78℃で40分間撹拌した。その後THF(12mL)中アセチルCsA−CHO(1.25g、1.02mmol)溶液をカニューレによって15分間にわたって加えた。反応混合物を室温で2時間撹拌した。上記のように処理及びクロマトグラフィーを行うと、アセチルISATX247(Z:E比40:60、1HNMR分析による)が得られた。
【0158】
実施例11:ベンゾイル保護シクロスポリンAの製法
シクロスポリンA(6.01g、5mmol)及び4−ジメチルアミノピリジン(305mg、2.5mmol)をピリジン(5mL)に溶解した。無水安息香酸(3.4g、15mmol)を加え、混合物を50℃で19時間撹拌した。追加的無水安息香酸(1.7g、7.5mmol)及びDMAP(305mg、2.5mmol)を加え、50℃でさらに24時間撹拌を続けた。無水安息香酸(0.85g、3.8mmol)を加え、反応物をさらに23時間撹拌した。反応混合物を撹拌しながらゆっくりと水中に注入した。沈殿したシクロスポリンA安息香酸を濾取し、水で洗った。集めたケーキを最小容量のメタノールに溶解し、10%クエン酸溶液に加え、1時間撹拌した。沈殿した生成物を濾過によって集め、濾液のpHが水のpHと等しくなるまで水で洗った。固体シクロスポリンA安息香酸を50℃で、真空下で乾燥すると無色固体が得られる。
【0159】
実施例12:トリエチルシリルエーテル−保護シクロスポリンAの製法
シクロスポリンA(3.606g、3mmol)を乾燥ピリジン(8mL)に溶解し、その後DMAP(122mg、1mmol)を加えた。反応混合物を0℃に冷やし、それからトリエチルシリルトリフルオロメタンスルホネート(3.6mmol)を滴下した。その混合物を放置して室温まで温め、一晩撹拌した。反応混合物を、撹拌しながらゆっくりと水中に注入した。沈殿したトリエチルシリルエーテルを濾別し、水で洗った。集めたケーキを最小容量のメタノールに溶解し、5%クエン酸溶液に加え、30分間撹拌した。沈殿した生成物を濾過によって集め、水で、濾液のpHが水のpHに達するまで洗った。固体トリエチルシリルエーテルを50℃で、真空下で乾燥すると無色固体が得られた。トリイソプロピルシリル及びtert−ブチルジメチルシリル保護基を、類似の方法によって導入することもできる。
【0160】
実施例13:カルシニューリン阻害アッセイを使用する免疫抑制活性
シクロスポリンA又はシクロスポリンA誘導体の効果の指標は、カルシニューリンのホスファターゼ活性を阻害する能力である。カルシニューリン阻害アッセイは、薬剤のその作用部位における活性を測定する。このようなアッセイは、シクロスポリンA類似体の効力の直接的invitro評価である(フルマンら、1992)。
【0161】
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)対シクロスポリンAの免疫抑制活性は、カルシニューリン(CN)阻害アッセイを使用して評価される。このアッセイの結果は、ISATX247(45−50%のZ−異性体及び50−55%のE−異性体)によるカルシニューリンホスファターゼ活性の阻害が、シクロスポリンAに比べて3倍より強力である(IC50により測定)ことを示す(図11)。
【0162】
種々の重水素化及び非重水素化異性体類似体混合物対シクロスポリンAの免疫抑制活性が、カルシニューリン(CN)阻害アッセイを用いて評価されている。これらの類似体の構造及び異性体組成は、図12に示される。図12において、「I4」は、ISATX2477の構造に対応する。I4−M2とは、実施例5−8に記載の方法によって生成されたISATX247を指す(この図では方法2である)。I4−D4は、実施例1−4に記載の方法によって生成された重水素化ISATX247を指す。I4−D2は、実施例5−8に記載の方法によって生成された重水素化ISATX247を指す。その他の異性体混合物も図に示されている。
【0163】
このアッセイの結果は、これらの異性体類似体混合物によるカルシニューリンホスファターゼ活性の阻害効果は、少なくともシクロスポリンAと同様であることを示す(IC50によって測定した)(図13)。CsAはシクロスポリンAを指す;Isocyclo4は、実施例1−4に記載の方法によって生成されたISATX247を指す。Isocyclo5は、図12のI5−M1に対応する。Isocyclo4−d4は、図12のI4−D4に対応する。Isocyclo5−d5は、図12のI5−D5に対応する。Isocyclo4−d2は、図12のI4−D2に対応する。Isocyclo4−M2は、図12のI4−M2に対応する。Isocyclo5−m2は、図12のI5−M5に対応する。
【0164】
実施例14:ラット心臓移植片モデルを使用する免疫抑制活性
異なる系統のラット間で移植された心臓の拒絶を阻止するISATX247(45−50%のE−異性体及び50−55%のZ−異性体)の効果を評価し、シクロスポリンAのそれと比較した。ラット心臓移植片モデルは、新しい免疫抑制剤のinvivo効力を評価する際に最もよく用いられるモデルである。それは、このモデルでは免疫拒絶により、持続的グラフト生存を実現することが難しいからである。
【0165】
この方法は、ウィスターラット(Wister Furth rats)からルイスラット(Lewis rats)への心臓の異所性移植(腹部大動脈及び下大静脈への)を含む。シクロスポリンA又は異性体類似体混合物のいずれかの腹腔内注射が移植片レシピエントに行われ、それは移植の3日前から始まり、移植後30日間続けられた。グラフト異機能が移植後30日の期間に認められた場合は、動物を殺した。動物が移植後30日より長く生きている場合は、試験及び対照試験を中止し、動物をグラフト異機能になるまで、又は移植後100日まで生存させた。
【0166】
レシピエント動物の各群の平均生存率を、表1にまとめる。これらの結果は最適量1.75mg/kg/日のISATX247(45−50%のE−異性体及び50−55%のZ−異性体)が、シクロスポリンAに比較して生存時間を約3倍延長することを示す。ISATX247を投与した多数の動物が、移植後100日目(投与中止後70日)にまだ機能するグラフトを有していた。これらのデータはこの異性体類似体混合物が、グラフト拒絶を阻止するための免疫抑制活性を有することを証明する。
【0167】
【表1】

【0168】
種々の重水素化及び非重水素化異性体類似体混合物(構造は図12に示される)が、異なる系統のラット間で移植された心臓の拒絶を阻止する効果も評価し、シクロスポリンAのそれと比較した。1.75mg/kg/日の用量を30日間投与した。表2に結果をまとめる。これらの結果は、1.75mg/kg/日の異性体混合物は、生存期間を少なくともシクロスポリンAと同様に延長することを示し、これらの異性体類似体混合物がグラフト拒絶阻止における免疫抑制活性を有することを証明した。
【0169】
【表2】
【0170】
実施例15:膵島細胞の同種移植術における免疫抑制活性
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)対シクロスポリンAの、マウスモデルにおける移植膵島細胞の生存を延長する能力を試験した。この研究は、CBA/Jマウスからの500個の膵島を糖尿病Balb/cマウスレシピエントの腎臓嚢に移植することを含む。
【0171】
移植後、ISATX247又はシクロスポリンAの投与量0(ビヒクル)、1.75、10、20又は25mg/kg/日を腹腔内注射(i.p.)によって30日間投与した。連続2日間17mmol/Lより高い血糖濃度を示す、という定義によるグラフト不全になるまで血糖値を毎日モニターした。
【0172】
その結果は、ISATX247が20mg/kg/日の投与量でグラフト生存期間を40%延長することを示している。投与量を増加すると、ISATX247の方がシクロスポリンAより毒性が小さいことも認められた。これは25mg/kg/日の投与量で特に明らかであった。
【0173】
【表3】
【0174】
実施例16:関節炎における免疫抑制活性
過去30年間に、ヒトリウマチ性関節炎の3動物モデルが詳細に試験され、新規の抗リウマチ薬の臨床前スクリーニング及び開発に広く用いられた。これらはアジュバント誘起性、コラーゲン誘起性、及び抗原誘起性関節炎モデルを含む。次の研究はマウスのコラーゲン誘起性関節炎モデル及びウサギの抗原誘起性関節炎モデル両方において、ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)の抗炎症性効果を評価するために計画された。これら2つのモデルに認められた組織病理学及び免疫病理学は、ヒト疾患における知見と似ている。両モデルにおいて、ISATX247の関節炎発生阻止効果(予防プロトコル)及び関節炎治療効果(治療プロトコル)を調べた。これらの研究は、請求の異性体類似体混合物が免疫抑制活性を有することを支持している。
【0175】
A.コラーゲン誘起性関節炎
ウィルス抗体をもたない状態の齢8ないし10週の雄DBA/1Lac J マウスを、フロイント完全アジュバント中に乳化したヒナドリII型コラーゲン100マイクログラムの皮下注射によって免疫した。ISATX247、シクロスポリンA、又はビヒクル(クレモフォルEL/エタノール72:28、容量/容量)を、生理食塩液で1−50倍に希釈した保存薬剤(0.25、0.5又は1mg/mL)を腹腔内注射によって毎日投与し、0(ビヒクル);ISATX247247では、125、250、又は500μg/マウス;シクロスポリンAでは250、又は500μg/マウスの濃度で与えた。予防プロトコルに割り当てられた動物(12匹/群)ではコラーゲンで免疫した日(0日目)から始めて、40日目に殺すまで投与した。治療プロトコルに割り当てられた動物(12匹/群)では、発病の日(〜28日目)に始めて、38日目に殺すまで投与した。
【0176】
評価パラメーターは、死亡率、血清クレアチニン、組織検査、及び結末の評価、例えば臨床的スコア(目視)、後足のむくみ、組織学的スコア、糜爛スコア、及び免疫組織学的検査などであった。
【0177】
糜爛スコア測定は盲検法で、中指の近位指節間(PIP)関節の矢状領域に糜爛(軟骨又は骨における炎症性組織の充満した、境界のはっきりした障害、と定義された)があるかないかによって行われた。このアプローチでは、同じ関節を比較することができた。これまでの研究では、未治療の関節炎動物の90%より多くの動物において、この関節の糜爛が明らかにされている。
【0178】
結果は、ISATX247多量投与群(500μg/マウス)の負の糜爛スコアが、ビヒクル投与群の負の糜爛スコアより有意に高いことを示した(p<0.05)。中程度量のISATX247(250μg/マウス)及び多量シクロスポリンA(500μg/マウス)治療群両方共ビヒクル投与群に比較して高い負の糜爛スコアを示した(p<0.1)。さらに、少量ISATX247(125μg/マウス)及び中程度量シクロスポリンA対照(250μg/マウス)治療群は、ビヒクル対照群に比較して有意に高いとは言えないまでも、より高い負の糜爛スコアを有した。
【0179】
関節の糜爛の発達を顕著に予防する唯一の治療は、500μg/マウスのISATX247であった。糜爛変化を示すPIP関節の比率が、ISATX247投与マウスの方がビヒクル対照群マウスに比較して有意に低いということは、ISATX247が疾患を変化させる性質を有することを証明するものである。
【0180】
B.抗原誘起性関節炎
特異的病原体のない状態で飼育されたニュージーランド白ウサギを、フロイント完全アジュバントと共に乳化した生理食塩液中卵白アルブミン10mgで免疫した。これは、首筋の数箇所に筋肉内及び皮下注射によって投与された。14日後、全ての動物で卵白アルブミン5mg及び生理食塩液中ヒト組換え形質転換増殖因子2(65ng)の1日おきの関節内注射が始められた。
【0181】
ISATX247、シクロスポリンA、又はビヒクル(クレモフォルEL/エタノール72:28、V/V)を、保存薬剤(ビヒクル中)を生理食塩液で1−ないし4倍に希釈した希釈液の皮下注射によって毎日投与し、0(ビヒクル);ISATX247では2.5、5.0、又は10mg/kg/日;シクロスポリンAでは5.0、10又は15mg/kg/日の濃度で与えた。予防プロトコルに割り当てられた動物(8/群)には、卵白アルブミン免疫の日(0日目)から始めて、42日目に殺すまで投与した。治療プロトコルに割り当てられた動物(8/群)では、発病の日(〜28日目)に始めて、42日目に殺すまで投与した。
【0182】
評価したパラメーターは、死亡率、体重、血清クレアチニン、組織検査、及び結末の評価、例えば膝関節の腫張、滑液カウント、肉眼による死後分析及び組織検査などを含む。
【0183】
ビヒクル対照動物と比較して、滑液の組織病理学スコアの顕著な減少が、ISATX247(P0.05)及びシクロスポリンA(P0.05)において、治療28日後(予防プロトコル)に認められた。これは滑液カウントの顕著な減少を伴った(ISATX247、P0.05;シクロスポリンA、P0.05)。確立された関節炎を有する動物の滑液組織病理学的スコアも、14日間のISATX247(P0.05)及びシクロスポリンA(P0TX05)治療後、ビヒクル対照に比較して有意に改善されることが判明した(治療プロトコル)。巨視的関節炎スコアの有意な減少が、ISATX247では明らかになったが(P=0.01)、シクロスポリンA治療動物では明らかでなかった。治療は耐容性がよく、血清クレアチニン分析又は死後組織検査で顕著な毒性は認められなかった。
【0184】
これらのデータは、ISATX247がウサギの抗体誘起性関節炎モデルにおいて、リウマチ性関節炎の治療及び予防においてシクロスポリンAと同等であるか、又は多分よりすぐれていることを示している。
【0185】
実施例17:薬物動態及び毒物動態特性
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)及びシクロスポリンAの薬物動態及び毒物動態パラメーターを、ウサギモデルで試験した。そのウサギは、シクロスポリンA腎臓毒性を研究するためのモデルとしても使われているが、ラットほどよく使われてはいない。研究では、ウサギに投与したシクロスポリンAは他の動物モデルでこれまでに報告されたものより低い用量、しかもヒトにおける治療範囲の少なくとも高い方の濃度以下でもある用量で、構造的及び機能的変化を引き起こすことが見いだされた(スリヴァリスら、1991,1994)。また、細管の細胞学的変化に加えて、間質性線維症及び動脈症の所見があることは、ウサギが腎臓毒性の研究により適したモデルであることを示唆する。なぜならば、これらの構造的変化は、ヒトで認められる腎臓毒性の顕著な特徴だからである。下記のスケジュールによって、ISATX247を最初の7日間静脈注射(i.v.)し、さらに23日間皮下注射した(s.c.)。
【0186】
【表4】
【0187】
認められたいかなる腎臓変化もISATX247効果によるもので、病原体によるものではないことを確実にするために、無病原体ウサギ(SPF)を使用した。1及び7日目に、薬剤を投与する前と、投与後0.5、1、2、4、8、12、18及び24時間後に血液サンプルを集め、薬物動態曲線を作成した。その他の評価パラメーターは、臨床的観察、体重、食餌消費量、血液検査値、臨床化学検査、肉眼的病理試験、及び選択された組織/臓器の組織病理学的検査などである。
【0188】
血液サンプルを、質量分析と高性能液体クロマトグラフィーとの組合わせ(LCMS)によって分析した。下の表5は、シクロスポリンA又はISATX247の10mg/kgを投与したウサギにおける平均薬物動態パラメーターをまとめたものである。
【0189】
【表5】
【0190】
10mg/kg/日を投与した雄ウサギでは、シクロスポリンAとISATX247との薬物動態パラメーターの間に統計的有意差はなかった。同量を投与した雌ウサギのISATX247の薬物動態パラメーターは、7日目の最大濃度を除けば雄ウサギで認められたパラメーターと有意に異ならなかった。
【0191】
ビヒクル対照、シクロスポリンA、又はISATX247を投与されたウサギの血液学的パラメーターにおいて有意な変化は認められなかった。下の表6に示すように、研究コース中、種々の群においてクレアチニン濃度に差が認められた。これらの差は、シクロスポリンAが、ビヒクル対照又はISATX247のいずれよりも有意に大きい負の効果を腎臓に与えることを示した。50%多い量、すなわち15mg/kg/日でさえ、10mg/kg/日シクロスポリンAに比べて、ISATX247は血清クレアチニン濃度の有意な増加を誘起しなかった。
【0192】
【表6】
【0193】
ビヒクル対照、10mg/kgシクロスポリンA、5mg/kgISATX247、又は10mg/kgISATX247を投与した全てのウサギの試験では、有意な異常はあらわれなかった。これは特に腎臓では事実であった;腎臓ではシクロスポリンA投与動物で通常見られる、間質性線維症(スリヴァリスら、1991、1994)の徴候は認められなかった。15mg/kgISATX247を投与した雄ウサギでは、精子形成の減少が認められた。この15mg/kg用量のISATX247投与で試験を完了した3匹の雌ウサギには、変化は認められなかった。
【0194】
実施例18:ISATX247の免疫抑制効果
カニクイザル(n=4)からの全血をISATX247又はシクロスポリンと共にインキュベートし、培養培地において種々のマイトジェンで刺激した。SG2M期の細胞で、トリチウム標識チミジンの挿入によって、及び増殖中の細胞核抗原(PCNA)発現のフローサイトメトリー分析によって、リンパ球の増殖を評価した。フローサイトメトリーを利用して、T細胞による細胞内サイトカインの産生及びTリンパ球活性化抗原の発現も評価した。その後EC50(最大効果の50%に達するために必要な薬剤濃度)をウィンノンリン(WinNonlin)(商標)ソフトウエアを用いて計算した。その結果は、下の表7に示されるEC50(ng/mL)からわかるように、リンパ球増殖、サイトカイン産生、及びT細胞表面抗原の発現は、シクロスポリンよりもISATX247によってより強力に阻止されることを示した。
【0195】
【表7】
【0196】
こうして、ex vivo全血アッセイにより、ISATX247が種々の免疫機能をシクロスポリンよりも2.3−6倍強く抑制することが判明した。
【0197】
実施例19:トリブチルアリルホスホニウムブロミドを使用するウィッティヒ反応
カリウムtertブトキシド(0.31g、2.8mmol)をテトラヒドロフラン20mLに溶解した。約−40℃で、テトラヒドロフラン3mLに溶解したトリブチルアリルホスホニウムブロミド(0.99g、3.1mmol)をゆっくり加えた。生成した黄色混合物を約−40℃で約10分間撹拌し、その後テトラヒドロフラン6mL中アセチルシクロスポリンAアルデヒド(1.5g、1.2mmol)溶液をゆっくり加えた。黄橙色反応混合物を1.5時間撹拌後、反応が完了した。反応を停止するために反応混合物を燐酸水溶液(1.2g、1.0mmol)に移した。生成水溶液をトルエン100mLで抽出し、その後トルエン50mLで抽出した。合一した有機層を水で洗い、減圧下で濃縮乾固した。生成物、アセチル化ISATX247がかすかに黄色の固体として約90%収率で得られた。異性体比は、約87%E−異性体及び約13%Z−異性体であった(1H−NMR分光法によって測定)。
【0198】
実施例20:トリブチルアリルホスホニウムブロミド及びリチウム塩基を使用するウィッティヒ反応
トリブチルアリルホスホニウムブロミド(1.38g、4.3mmol)をトルエン20mL及びテトラヒドロフラン3mLの混液に溶解した。約−78℃でブチルリチウム(ヘキサン中1.6M、2.43mL、3.9mmol)をゆっくり加えた。生成した黄色混合物を約−78℃で約10分間撹拌し、その後トルエン6mL中アセチルシクロスポリンAアルデヒド溶液(1.5g、1.2mmol)をゆっくり加えた。黄橙色の反応混合物を3.5時間撹拌後、その反応混合物を50mLトルエンと燐酸水溶液(0.25g、2.2mmol)との混液に移すことによって反応を停止した。生成した二相混合物を放置して周囲温度まで温め、その後二相を分離した。トルエン層を20mLの水で洗い、減圧下で濃縮乾固した。生成物、アセチル化ISATX247がかすかに黄色の固体として約80%収率で得られた。異性体比は、約70%E−異性体及び約30%Z−異性体であった(1H−NMR分光法によって測定)。
【0199】
実施例21:トリブチルアリルホスホニウムブロミド及びリチウム塩基を使用するウィッティヒ反応
上記のように、ただし約−40℃の温度でSAP018を作動する。実施例20の実験条件を反復した。このときは、反応温度約−40℃を使用した。これらの条件のもとで、分離した生成物、アセチル化ISATX247の異性体比は、1H−NMR分光法で測定して、E−異性体約74重量%及びZ−異性体約26重量%であった。
【0200】
実施例22:トリブチルアリルホスホニウムブロミドを使用するウィッティヒ反応
テトラヒドロフラン15mL中アセチルシクロスポリンAアルデヒド(1.5g、1.2mmol)及びトリブチルアリルホスホニウムブロミド(0.99g、3.1mmol)の溶液を約−80℃に冷却した。テトラヒドロフラン9mLに溶解したカリウムtert−ブトキシド(0.19g、1.7mmol)をゆっくりと加えた。反応を完了するために、生成した黄色混合物を約−80℃で1時間撹拌し、その後6mLのテトラヒドロフラン溶液をゆっくり加えた。黄橙色反応混合物を1.5時間撹拌後、反応は完了した。反応混合物の反応を停止するために、燐酸水溶液(0.15g、1.3mmol)を加えた。生成混合物を濃縮し、残留物をメタノール5mLに溶解した。その後、混合物を水5mLにゆっくり加えた。生成した沈殿を濾過し、4mLのメタノール/水(1/1)で洗い、真空中で乾燥した。生成物ISATX247が無色固体として約90%収率で得られた。異性体比は、約91重量%E−異性体及び9重量%Z−異性体であった(1H−NMR分光法によって測定)。
【0201】
実施例23:アセチルCsAのオゾン分解
メタノール200mL中アセチルシクロスポリンA(15g、12.1mmol)の溶液を、サンダーオゾン発生機を使って約1.1バールで、300L O2/時間の流量で、反応が完了するまで(約5分間)オゾン化した。溶液にアルゴンガスを通し、メタノールに溶解したジメチルスルフィドを加えた。還元を完全にするために、混合物を室温で一晩撹拌した。約50mLに濃縮後、その溶液を水500mLにゆっくりと加えた。生成した沈殿を濾過し、水60mLで洗い、真空中で乾燥した。生成物、アセチル化CsAアルデヒドが純度約98%の無色固体として約95%収率で得られた(HPLCによって測定)。
【0202】
実施例24:トリメチルシリル−保護シクロスポリンAの製法
シクロスポリンA(40g、1当量)を30℃で、ジクロロメタン(100ml)に溶解した。N,N−ビス−(トリメチルシリル)尿素(1.1当量)を加えた。30℃で5分撹拌後、p−トルエンスルホン酸(0.02当量)を加えた。反応混合物を反応が完了するまで還流加熱し、その後室温にまで冷やした。反応の完了は、薄層クロマトグラフィー(TLC)、高圧又は高性能液体クロマトグラフィー(HPLC)、又は質量分析(MS)によって確認された。半飽和状態の炭酸水素ナトリウム溶液(100ml)を加えた。水相を分離し、ジクロロメタンで再抽出した。合一した有機相を無水Na2SO4上で乾燥し、濾過した。溶媒を減圧下で除去すると、粗トリメチルシリル−保護シクロスポリンAが得られた。
【0203】
実施例25:トリメチルシリル−保護シクロスポリンAアルデヒドの製法
トリメチルシリル保護シクロスポリンA(5g、1当量)をジクロロメタン(50ml)に溶解した。溶液を約−78℃の温度に冷やし、その後オゾンをその溶液に、青色があらわれるまでぶくぶく通した。次に、その溶液が無色になるまでその溶液にアルゴンをぶくぶく通し、無色になった過剰のオゾンを除去した;この工程は過剰のオゾンを除去するために行われた。トリメチルアミン(5当量)を加え、反応混合物を室温で17時間撹拌した。水性処理後にトリメチルシリル−保護シクロスポリンAアルデヒドが得られた。
【0204】
実施例26:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の3:1混合物の製法
60分間あらかじめ撹拌した、トルエン(10ml)中カリウムtert−ブトキシド(3当量)及びアリルトリフェニルホスホニウムブロミド(2当量)の混合物に、トリメチルシリル−保護シクロスポリンAアルデヒド(1g、1当量)を加えた。この反応混合物を室温で1時間反応させた後に処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の3:1混合物(NMRによる)が得られた。
【0205】
実施例27:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:1混合物の製法
トリメチルシリル−保護シクロスポリンAアルデヒド(2.5g)を25mlトルエンに溶解し、1N水酸化ナトリウム水溶液(10当量)で処理した。反応混合物を激しく撹拌し、アリルトリフェニルホスホニウムブロミド(7.5当量、一部分ずつ)を加えた。室温で数時間反応後、反応混合物を処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:1混合物(NMRによる)が生成した。
【0206】
実施例28:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:2混合物の製法
トリメチルシリル−保護シクロスポリンAアルデヒド(1g)を、トルエン5mlに炭酸カルシウム(1.5当量)及びアリルトリフェニルホスホニウムブロミド(1.5当量)と共に溶解した。激しく撹拌しながら還流下で4時間反応させた後、その反応混合物を処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:2混合物(NMRによる)が生成した。
【0207】
実施例29:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:3混合物の製法
アリルトリブチルホスホニウムブロミド(3当量、アリルブロミド及びトリブチルホスフィンからつくられた)をTHF(3.5ml)に溶解した。トルエン(7.5ml)を加え、その後カリウムtert−ブトキシド(4当量)を加えた。室温で1時間撹拌後、溶液を約−30℃に冷却した。トリメチルシリル−保護シクロスポリンAアルデヒド(1g、1当量)のトルエン(5mL)溶液を滴下した。約−30℃で45分経過後、反応混合物を処理し、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:3混合物(NMRによる)を生成した。
【0208】
下記の2実施例、実施例30及び31は、アリルメタレーションに関するものである。
【0209】
実施例30:アセチル−保護シクロスポリンAβ−トリメチルシリルアルコールの製法
THF(15ml)中アリルトリメチルシラン(10.1当量)の溶液に、ブチルリチウム(ヘキサン中1.6M、10当量)を室温で加えた。30分間反応後、溶液を−75℃に冷却し、ジエチル−B−メトキシボラン(10.1当量)で処理した。1時間後、ホウ素トリフルオリドジエチルエーテル錯体(10.1当量)を加えると、B−(γ−トリメチルシリル−アリル)−ジエチルボラン試薬が生成した。1時間後、THF(15ml)中アセチル−保護シクロスポリンAアルデヒド(5g、1当量)溶液を滴下した。20分後、反応混合物を−10℃に温め、飽和NH4Cl水溶液を加えた。室温で1時間撹拌後、水(45ml)を加え、反応混合物を25ml酢酸エチルで3回抽出した。有機相を水(25ml)及び飽和NH4Cl水溶液(25ml)で逐次洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮した。粗生成物をクロマトグラフィーにかけ(シリカゲル、ジクロロメタン/メタノール又は酢酸エチル/ヘプタン)、アセチル−保護シクロスポリンAβ−トリメチルシリルアルコールを得た。
【0210】
実施例31:トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコールの製法
THF(15ml)中アリルトリメチルシラン(10.1当量)の溶液に、室温でブチルリチウム(ヘキサン中1.6M、10当量)を加えた。約30分間反応を進行させた後、溶液を−65℃に冷却し、ジエチル−B−メトキシボラン(10.1当量)で処理した。1時間後、ホウ素トリフルオリドジエチルエーテル錯体(10.1当量)を加え、B−(γ−トリメチルシリル−アリル)−ジエチルボラン試薬を生成した。1時間後、THF(15ml)中トリメチルシリル−保護シクロスポリンAアルデヒド(5g、1当量)の溶液を滴下した。15分後、反応混合物を10℃まで温め、飽和NH4Cl水溶液を加えた。室温で1時間[1時間]撹拌後、水(12.5ml)及び飽和NaHCO3(25ml)を加えた。反応混合物を25mlのメチル−t−ブチルエーテルで2回抽出した。有機相を水(2×25ml)で2回洗い、次に飽和NaCl水溶液(25ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮した。粗生成物をクロマトグラフィー(シリカゲル、ヘプタン/酢酸エチル)にかけ、トリメチルシリル−保護シクロスポリンAβ−トリメチルシリルアルコールを得た。
【0211】
下記の3実施例、実施例32、33及び34は、ピーターソン脱離反応に関するものである。
【0212】
実施例32:E−アセチル保護シクロスポリンAジエンの製法
アセチル保護シクロスポリンA β−トリメチルシリルアルコール(10g、1当量)を、THF(50ml)に溶解した。濃縮H2SO4(1.24ml、3当量)を加え、反応混合物を室温で20時間撹拌した。水(150ml)を加え、反応混合物をメチル−t−ブチルエーテル(200ml)で抽出した。水相をメチル−t−ブチルエーテル(150ml)で再抽出した。有機相を水(150ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、粗アセチル−保護シクロスポリンAジエン(アセチル−保護ISATX247)が得られた。その粗生成物をメチル−t−ブチルエーテル/THFから結晶化し、その後メチル−t−ブチルエーテル/DCMから再結晶すると、アセチル−保護シクロスポリンAジエン(アセチル保護ISATX247)がE及びZ二重結合異性体の99.97%:1−3%混合物として得られた(400MHzNMRによる。測定誤差2%)。
【0213】
E−アセチル−保護シクロスポリンAジエンの加水分解を次のように行った:アセチルシクロスポリンAジエン(4g、1当量)をメタノール(80ml)及び水(32ml)に溶解した。炭酸カリウム(3.65g、8.3当量)を加えた。室温で15時間撹拌した後、反応混合物を4時間40℃まで加熱した。反応混合物を減圧下で濃縮し、残留物を酢酸エチル(70ml)にとった。クエン酸水溶液15%(30ml)をゆっくりと加え、次に水(10ml)を加えた。水層を分離し、酢酸エチル(56ml)で再抽出した。有機相を水(30ml)、15%クエン酸溶液(40ml)及び飽和NaCl溶液(30ml)で洗った。有機層を合一し、Na2SO4上で乾燥し、減圧下で濃縮するとシクロスポリンAジエン(ISATX247)が二重結合異性体の98:2E/Z混合物として得られた(400MHz NMRによる。誤差約2−3%)。R.W.ホフマン著、Angewandte Chemie InternationalEdition, Vol.555(1982);W.R.ロウシュ著、「『アリル有機金属類』、有機合成総覧」、ペルガモン プレス、第2巻、p.1-53;及びY.ヤマモト、N.アサオ著、ChemicalReviews、p.2307(1993)を参照されたい。
【0214】
実施例33:Z−トリメチルシリル−保護シクロスポリンAジエンの製法及びZ−シクロスポリンAジエン(ISATX247)への変換
トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコール(2g、1当量)をTHF(20ml)に溶解した。溶液を0−2℃に冷却し、カリウムt−ブトキシド(4当量)を加えた。1.5時間の反応後、酢酸エチル(20ml)及び水(40ml)を加えた。水層を分離し、酢酸エチル(20ml)で再抽出した。有機相を飽和NaCl水溶液(20ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、Z−トリメチルシリル−保護シクロスポリンAジエン(トリメチルシリル−保護ISATX247)及びZ−シクロスポリンAジエン(ISATX247のZ−異性体)の混合物が得られた。粗生成物混合物をメタノール(溶液中10重量%)に溶解し、1M塩酸水溶液(1当量)を加えることによって、脱シリル化が完了した。室温で15分後に、水及び酢酸エチルを加えた。水層を分離し、酢酸エチルで再抽出した。有機相を飽和NaCl水溶液で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、シクロスポリンAジエン(ISATX247)がZ及びE二重結合異性体の94:6混合物として(NMRによる)生成した。
【0215】
実施例34:E−シクロスポリンAジエン(ISATX247)の製法
トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコール(500mg、1当量)をジクロロメタンに溶解した。この溶液を約0−2℃の範囲内に冷却し、ホウ素トリフルオリドジエチルエーテル錯体(5当量)で処理した。1時間後、水(20ml)及びジクロロメタン(20ml)を加えた。有機層を分離し、水(20ml)で洗い、Na2SO4上で乾燥し、濾過し、減圧下で濃縮すると、直接シクロスポリンAジエン(ISATX247)がE及びZ二重結合異性体の91:9混合物として(NMRによる)生成した。
【0216】
実施例35:トリメチルシリル−保護シクロスポリンAジエンの脱保護
トリメチルシリル−保護シクロスポリンAジエンをメタノールに溶解した(溶液中10重量%)。この溶液を1M塩酸水溶液(1当量)で処理した。室温で15分後、水と酢酸エチルを加えた。水層を分離し、酢酸エチルで再抽出した。有機相を飽和NaCl水溶液で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、シクロスポリンAジエン(ISATX247)が生成した。
【0217】
実施例36:アセチルシクロスポリンAのエポキシド化
アセチルシクロスポリンA(2.0g、1.61mmol)をアセトニトリル(30mL)に溶解した。1,3−ジアセトキシ−アセトン(0.14g、0.8mmol)を加え、その後0.0004Mエチレンジアミン四酢酸二ナトリウム塩(20mL)及び炭酸水素ナトリウム(0.405g、4.82mmol)水溶液を加えた。撹拌下の混合物に、オキソン(43.8%KHSO5)(2.23g、6.43mmol)を2時間にわたって一部分ずつ加えた。pH装置を使用して、1NNaOHを一定状態で添加し(総量6.4mL)、pHを8.2に維持した。冷水浴を用いて時々冷却することによって、温度を22−25℃に維持した。2.5時間後、反応混合物に数滴の重亜硫酸ナトリウム溶液を加えて反応を停止した。水(100mL)を加え、混合物をtert−ブチルメチルエーテル(100mL、その後75mL)で2回抽出した。その有機抽出液を塩化ナトリウムの希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると、粗アセチルシクロスポリンAエポキシド(1.92g、95%;HPLC:99.4%領域)が白色固体フォームとして得られた。
【0218】
実施例37:アセチルシクロスポリンAアルデヒドの製法
粗アセチルシクロスポリンAエポキシド(1.92g、1.52mmol)をアセトニトリル(25mL)に溶解した。水(20mL)を加え、その後過ヨウ素酸ナトリウム(489mg、2.28mmol)及び0.5M硫酸(3.05mL、1.52mmol)を加えた。反応混合物を40℃で18時間撹拌し、その後過剰の過ヨウ素酸ナトリウムを重亜硫酸ナトリウムの添加によって消滅させた。塩化ナトリウムの希水溶液(100mL)を加え、混合物をtert−ブチルメチルエーテル(各100mL)で2回抽出した。有機抽出液を塩化ナトリウム希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると、粗アセチルシクロスポリンAアルデヒド(1.74g、92%;HPLC:95.7%領域)が白色フォームとして得られる。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(1.41g、アセチルシクロスポリンAを基準にして71%;HPLC:100%領域)が白色固体フォームとして得られた。
【0219】
実施例38:ワンポット法を用いるアセチルシクロスポリンAアルデヒドの製法
アセチルシクロスポリンA(2.0g、1.61mmol)をアセトニトリル(30mL)に溶解した。1,3−ジアセトキシ−アセトン(0.084g、0.48mmol)を加え、その後0.0004Mエチレンジアミン四酢酸二ナトリウム塩(20mL)及び炭酸水素ナトリウム(0.405g、4.82mmol)水溶液を加えた。撹拌下の混合物に、オキソン(43.8%KHSO5)(1.67g、4.82mmol)を2時間にわたって一部分ずつ加えた。pH装置を使用して、1NNaOHを一定状態で添加し(総量3.4mL)、pHを8.2に維持した。温度は20−25℃に維持した。3.5時間後、0.5M硫酸(5mL、2.5mmol)を反応混合物に加え、その後数滴の濃硫酸を加え、pH1.3までにした。それから過ヨウ素酸ナトリウム(516mg、2.41mmol)を加え、反応混合物を室温で2時間、40℃で22時間撹拌した。水(100mL)を加え、混合物をtert−ブチルメチルエーテル(100mL、その後75mL)で2回抽出した。有機抽出液を塩化ナトリウム希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると粗アセチルシクロスポリンAアルデヒド(1.9g、96%;HPLC:83.4%領域)が白色フォームとして生成した。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(1.35g、アセチルシクロスポリンAを基準にして68%;HPLC:100%領域)が白色固体フォームとして得られた。
【0220】
実施例39(ISO);アセチルシクロスポリンAアルデヒドと3−ジメチルアミノプロピルトリフェニルリンイリデンとのウィッティヒ反応
1,3−ジエン類の立体選択的合成は、コリー及びデサイがTetrahedron Letters, Vol.26, No.47,p. 5747-8(1985)に記載している。この文献は、3−(ジメチルアミノ)プロピルトリフェニルホスホランをヘキサメチルジシラチドカリウムで処理することによって得られるイリドが、アルデヒドと共にウィッティヒ反応を受けて、選択的にZ−アルケニルジメチルアミンを形成し得ることを開示している。そのアミンのm−クロロペル安息香酸による酸化は、対応するN−オキシドを与え、それをその後加熱するとコープ脱離として知られる反応が起き、所望の1,3−ジエンが形成する。このウィッティヒ反応段階中に形成されるオレフィンの配置は、専らZ、又はシスである。
【0221】
同様に、ISATX247のZ−異性体は、アセチルシクロスポリンAアルデヒドと3−(ジメチルアミノ)−プロピルトリフェニルホスホニウムブロミドを、ヘキサメチルジシラチドカリウムで処理することによって得られるイリドとの反応によっても形成される。生成した中間体はその後酸化され、その後コープ脱離を受けて、アセチル−(Z)−ISATX247が生成する。塩基を用いて脱保護すると、(Z)−ISATX247が生成する。酸化試薬は、メタクロルペル安息香酸でよい。
【0222】
無水トルエン(20mL)中3−ジメチルアミノプロピルトリホスホニウムブロミド(2.5g、5.83mmol)の撹拌中の懸濁液に、ヘキサメチルジシラジドカリウム(11.6mL、5.8mmol、トルエン中0.5M溶液)を注射器によって加えた。室温で1時間撹拌後、その赤色溶液を遠心分離し、上澄液をカニューレによって反応フラスコに移した。その固体に無水トルエン(10mL)を加え、撹拌し、遠心分離した。上澄液を反応フラスコに移し、合一した赤色イリドにOAc−CsA−CHO(1.44g、1.17mmol)を加えた。撹拌を室温でさらに2時間続けると、色は淡黄色に変わった。反応混合物をEtOAc(50mL)で希釈し、その後飽和NaHCO3溶液(50mL)及びブライン(50mL)で洗った。乾燥及び溶媒除去をすると、淡黄色の固体が得られる。シリカゲルカラム上でクロマトグラフィーを行い、アセトン−ヘキサン混液(勾配:10ないし75%アセトン及び90ないし25%ヘキサン)で溶出すると、全てのリン関連不純物は除去される。アセトンでさらに溶出すると、所望の生成物が無色固体として提供された(1.28g、84%収率)。1HNMR(300 MHz、CDCl3):2.23(s,6H)、2.03(s,3H)。13C NMR(300MHz、CDCl3):129.33、126.95;MSm/z:1301(M+)、1324(M+Na+)。
【0223】
N−オキシドへの変換
ウィッティヒ反応によって得られたジメチルアミノ化合物(0.44g、0.34mmol)を、CHCl3(3mL)に溶解した撹拌冷溶液(0℃)に、CHCl3(2mL)中m−CPBA(0.07g、0.405mmol)の溶液を加えた。30分間撹拌後、ジメチルスルフィド(0.5mL)を加え、その後CH2Cl2(50mL)を加えた。NaHCO3溶液(25mL)及び水(25mL)で洗い、乾燥し、溶媒除去する処理を行うと固体(0.43g)が生成した。1HNMR(300MHz、CDCl3):3.19(s,3H)、3.18(s,3H)、2.03(s,3H)。13C NMR(300MHz、CDCl3):131.89、124.13;MSm/z:1340(M+Na+)。
【0224】
N−オキシドのコープ脱離。アセチルISATX247の(Z)−異性体の調製
N−オキシド(350mg)をそのまま撹拌し、真空下で2時間100℃で加熱した。これを、その後シリカゲルカラムを通過させた。アセトン−ヘキサン混液(勾配、5ないし25%アセトン及び95ないし75%ヘキサン)による溶出は、無色固体(314mg)を提供した。1HNMR(500MHz、CDCl3):6.49(dt,J=16.99、10.5Hz、1H);13C NMR(400MHz、CDCl3):132.20、131.09、129.70、116.85;MSm/z:1279(M+Na+)。
【0225】
ISATX247の(Z)−異性体
MeOH(4mL)中(Z)−アセチルISATX247(50mg)の溶液に、水(1.5mL)及びK2CO3(60mg)を加え、室温で48時間撹拌した。反応混合物から溶媒を除去し、EtOAc(20mL)で抽出した。有機層を水(10mL)及びブライン(10mL)で洗った。乾燥及び溶媒除去により無色固体が生成した。1HNMR(500MHz、CDCl3):6.58(dt,J=16.99、10.5Hz、1H);MS m/z:1236.8(M+Na+)。生成した化合物はISATX247のZ−異性体であった。NMRによる測定では、計り得るE−異性体は認められなかった。
【0226】
本発明の好ましい実施形態のみを詳細に開示し、上述したが、上記の教示に照らして、そして添付の請求の範囲内で、本発明の精神及び目的の範囲から逸脱することなく、本発明の多くの変形及び変更が可能であることは明らかである。
【図面の簡単な説明】
【0227】
【図1】図1Aは、分子の環式ペプチド環を構成する11個のアミノ酸残基を示すシクロスポリンAの構造、並びに1−アミノ酸残基の側鎖の構造を示す図である; 図1Bは、本明細書中に用いられる用語「CsA」の定義を特に強調したシクロスポリンAの構造を示すもう一つの図である;
【図2】図2Aは、ISATX247と呼ばれるシクロスポリンA類似体のE−異性体(又はトランス−異性体)の構造を示す図である;図2Bは、シクロスポリンA類似体ISATX247のZ−異性体(又はシス−異性体)の構造を示す図である;
【図3】図3は、本発明のシクロスポリン類似体をつくるために使用できる典型的合成経路の概観を示し、ここで立体選択的経路は反応条件によってグループ化される;
【図4】図4は、臭素化前駆体からISATX247の(E)及び(Z)−異性体の混合物を生成する合成経路を示す図である;
【図5】図5は、アルデヒド前駆体からISATX247の(E)及び(Z)−異性体の混合物を生成する別の合成経路を示す図である;
【図6】図6は、ISATX247の(E)又は(Z)−異性体どちらかに富む組成物をつくるために使用できる典型的立体選択的反応スキームを示し、その際いずれの異性体も同じ前駆体アルコールから合成できる;
【図7】図7は、ISATX247の(Z)−異性体に富む組成物の立体選択的合成の別の反応スキームを示す図である;
【図8】図8は、ISATX247の(E)−異性体に富む組成物の立体選択的合成の別の反応スキームを示す図である;
【図9A】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図9B】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図9C】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図10】図10は、ISATX247の(E)及び(Z)−異性体の混合物を生成するための典型的立体選択的経路を示す図であり、ここで2種類の異性体のうちの1種類の異性体に富む組成物を先ず第一につくり、その後所定の比率に合うように混合して所望比を得る;
【図11】図11は、ISATX247(E−異性体45−50%及びZ−異性体50−55%)によるカルシニューリンホスファターゼ活性阻害効果が、シクロスポリンAの阻害効果の3倍より強力であった(IC50によって測定)ことを示すアッセイ結果を提供する;
【図12】図12は、幾つかの重水素化及び非重水素化類似体異性体混合物の構造及び異性体組成を示す;
【図13】図13は、種々の重水素化及び非重水素化類似体異性体混合物によるカルシニューリンホスファターゼ活性の阻害効果が、シクロスポリンAに比較して少なくとも同様に強力であった(IC50によって測定)ことを示すアッセイ結果を提供する。
【技術分野】
【0001】
関連出願の相互参照
本出願は2001年10月19日に提出された米国特許出願第60/346,201号及び2002年4月5日に提出された米国特許出願第60/370,596号に基づく優先権を請求する。これら各出願の全ての開示は参考としてそのまま本明細書に組み込まれる。
【0002】
本発明は、シクロスポリンAに関係するシクロスポリン類似体の異性体混合物類に関するものである。これらの混合物は、個々の異性体に比べて、及び天然、及びその他の現在公知のシクロスポリン類及びシクロスポリン誘導体類に比べて、高い効果及び/又は低い毒性を有すると考えられる。さらに本発明は、シクロスポリンA類似体の異性体を生成する合成経路に関し、その際このような経路では特異的反応条件によって立体選択性の程度が種々変動する。
【0003】
下記の文献は本願に関連しており、本明細書の該当部分に、特許番号もしくは出願番号、又は著者名と発行年を括弧書きで記して、本明細書において参照する。
【0004】
【非特許文献1】ベネット(Bennett, W.M.)著、「新旧の免疫抑制剤の腎臓毒性」、Renal Failure,Vol.20, p.687-90(1998)。
【非特許文献2】ビールマン(J.-F. Biellmann)、ドュセプ(J.-B. Ducep)著、「ヘテロ原子によって置換されたアリルカルボアニオン及びベンジルカルボアニオン」、OrganicReactions、Vol.27,(ウィリー、ニューヨーク、1982), p.9。
【非特許文献3】カールセン(H.J. Carlsen)ら著、「四酸化ルテニウム触媒による有機化合物の酸化法の顕著な改良」,J. Org. Chem.,Vol.46, No.19, p.3736-3738 (1981)。
【非特許文献4】チャング(T. Chang)、ベネット(L.Z. Benet)、ヘバート(M.F. Hebert)著、「健康な志願者におけるシクロスポリンの薬物導体に与える水溶性ビタミンEの影響」,Clin.Pharmacol. Ther., Vol.59, p.297-303 (1996)。
【非特許文献5】コーリー(E.J. Corey)、デサイ(M.C. Desai)著、Tetrahedron Letters, Vol26, No.47,p.5747-8 (1985)。
【非特許文献6】エバール(M.K. Eberle)、ナニンガー(F. Nuninger)著、「シクロスポリンAの主代謝産物(OL−17)の合成」,J.Org. Chem., Vol.57, p.2689-2691 (1992)。
【非特許文献7】エーリンガ(E. Ehlinger)、マグナス(P. Magnus)著、「合成における珪素。10.(トリメチルシリル)アニオン:アルデヒド及びケトンをγ−ラクトンに変換するためのβ−アシルアニオン同等物」,J.Am. Chem. Soc., Vol.102, No.15, p.5004-5011(1980)。
【非特許文献8】フルマン(D.S. Fruman)、クリー(C.B. Klee)、ビーラ(B.E. Bierer)、ブラコフ(S.J. Burakoff)著、「T細胞におけるカルシニューリン燐酸活性はFK506及びシクロスポリンAによって阻害される」,Proc.Natl. Acad. Sci. USA, Vol.89, p.3686-90 (1992)。
【非特許文献9】グラネリ−ピペルノ(A. Granelli-Piperno)、アンドラス(L. Andrus)、スタインマン(R.M. Steinman)著、「刺激されたヒト細胞中のリンフォカイン及び非リンフォカインmRNAの濃度:シクロスポリンAの動力学、マイトジェン要求性及び効果」,J.Exp. Med., Vol163, p.922(1986)。
【非特許文献10】ハンソン(J.R. Hanson)著、「『アルコール類の保護』有機合成における保護基、2章」,p.24-25(シェフィールド・アカデミック・プレス、シェフィールド、英国、1999)。
【0005】
【非特許文献11】ヘバート(M.F. Hebert)、ロバーツ(J.P. Roberts)、プルークサリタノン(T. Prueksaritanont)、ベネット著、「リファンピンと同時投与されたシクロスポリンのバイオアベイラビリティは、肝酵素誘導により推測されるものより著しく低い」,Clin.Pharmacol. Ther., Vol.52, 453-7 (1992)。
【非特許文献12】ホフマン(R.W. Hoffmann)著、Angewandte Chemie International Edition, Vol.555(1982)。
【非特許文献13】ホフマン、ゼイ(H.-J Zei)著、「アルコールの立体選択的合成。8.クロチルボロネートを介するβ−メチルホモアリルアルコールのジアステレオ選択的合成」,J.Org. Chem., Vol46, p.1309-1314 (1981)。
【非特許文献14】ハードリク(P.F. Hurdlik)及びピーターソン(D. Peterson)著、「β−ヒドロキシシラン類の立体特異的オレフィン形成脱離反応」,J.Am. Chem. Soc., Vol97, No.6, p.1464-1468(1975)。
【非特許文献15】Y.イケダ、J.ウカイ、N.イケダ、H.ヤマモト著、「有機チタン及びリチウム試薬を使用した、アルデヒドからの(Z)−及び(E)−1,3−アルカジエン類の立体選択的合成」,Tetrahedron, Vol43, No.4, p.723-730(1987)。
【非特許文献16】コベル(Kobel)ら著、Europ. J. Applied Microbiology and Biotechnology, Vol.14,p.237-240 (1982)。
【非特許文献17】マクマリー(J. McMurry)著、「有機化学、5版」,(ブルックス/コール、パシフィック グローブ、2000), p.780-783。
【非特許文献18】リーツ(M.T. Reetz)著、「有機合成における有機チタン試薬」、(スプリンガー出版、ベルリン、1986), p.VII,148-149, 164-165。
【非特許文献19】リッチ(Rich)ら著、J. Med. Chem., Vol.29, p.978 (1986)。
【非特許文献20】ロウシュ(W.R. Roush)著、「『アリル有機金属類』、有機合成総覧」、ペルガモン プレス、第2巻、 p.1-53。
【0006】
【非特許文献21】シュライバー(S.L. Schreiber)、クラブトリー(G.R. Crabtree)著「シクロスポリンA及びFK506の作用メカニズム」,Immunol.Today, Vol.13, p.136-42 (1992)。
【非特許文献22】スケトリス(I. Sketris)、ヤツコフ(R. Yatscoff)、コウン(P. Keown)、カナファックス(D.M. Canafax)、ファースト(M.R.First)、ホルト(D.W. Holt)、シュローダ(T.J. Schroeder)、ライト(M. Wright)著、「腎臓移植におけるシクロスポリン使用の最適化」,、Clin.Biochem., Vol.28, p.195-211 (1995)。
【非特許文献23】スミス(M.B. Smith)及びマーチ(J. March)著「マーチの最新有機化学」、(ウィリー、ニューヨーク、2001),p.144-147。
【非特許文献24】ストレイトウィーザ(A. Streitwieser)、ヒースコック(C.H. Heathcock)著、「有機化学入門、2版」(マクミラン、ニューヨーク、1981),p.845-846。
【非特許文献25】スリヴァリス(J.A. Thliveris)、ヤツコフ(R.W. Yatscoff)、ルコウスキー(M.P. Lukowski)、コペランド(K.R.Copeland)、ジェファリー(J.R. Jeffery)、マーフィー(G.F. Murphy)著、「慢性シクロスポリン腎毒性:ウサギモデル」,Nephron, Vol.57, p.470-6 (1991)。
【非特許文献26】スリヴァリス、ヤツコフ、ミハチュ(M.J. Mihatsch)著、「慢性シクロスポリン誘導性腎毒性:ウサギモデル」,Transplantation, Vol.57, p.774-6(1994)。
【非特許文献27】トーマス(S.E. Thomas)著、「有機合成:ホウ素及びシリコーンの役割」(オックスフォード大学プレス、ニューヨーク、1991),p.84-87。
【非特許文献28】トレイバー(Traber)ら著,Helv. Chim. Acta, Vol.60, p.1247-1255 (1977)。
【非特許文献29】トレイバーら著,Helv. Chim. Acta, Vol.65,p.1655-1667 (1982)。
【非特許文献30】ツァイ(D.S. Tsai)、マッテソン(D.S. Matteson)著、「ピナコール(E)−1−トリメチルシリル−1−プロペン−3−ボロネートからの(Z)及び(E)末端ジエンの立体コントロールド合成」,TetrahedronLetters, Vol.22, No.29,p.2751-2752(1981)。
【0007】
【非特許文献31】ヴァランチン(H.A. Valantine)、シュローダ(J.S. Schroeder)著、「最近の心臓移植の進歩」[編集者のコメント],N. Engl. J. Med., Vol.333, No.10, p.660-1 (1995)。
【非特許文献32】フォン ワルトバーグ(von Wartburg)ら著, Progress in Allergy, Vol.38, p.28-45(1986)。
【非特許文献33】ウェンゲル(Wenger)著, Transpl. Proc.,Vol.15,付録1, p.2230 (1983)。
【非特許文献34】ウェンゲル著, Angew. Chem. Int. Ed., Vol.24, p.77 (1985)。
【非特許文献35】ウェンゲル著, Progress in the Chemistry of Organic Natural Products, Vol.50,p.123(1986)。
【非特許文献36】Y.ヤマモト、N.アサオ著, Chemical Reviews, p.2307(1993)。
【非特許文献37】ダン ヤング(Dan Yang)ら著、「非官能化オレフィンを触媒的非対称性エポキシ化するためのC2対称性キラルケトン」, J. Am.Chem. Soc., Vol118, p.491-492 (1996)。
【非特許文献38】ダン ヤングら著「触媒的酸化反応のための新規の環状ケトン」, J. Org. Chem., Vol.63, p.9888-9894(1998)。
【0008】
【特許文献1】米国特許第4,108,985号。
【特許文献2】米国特許第4,160,452号。
【特許文献3】米国特許第4,210,581号。
【特許文献4】米国特許第4,220,641号。
【特許文献5】米国特許第4,256,108号。
【特許文献6】米国特許第4,265,874号。
【特許文献7】米国特許第4,288,431号。
【特許文献8】米国特許第4,384,996号。
【特許文献9】米国特許第4,396,542号。
【特許文献10】米国特許第4,554,351号。
【特許文献11】米国特許第4,771,122号。
【特許文献12】米国特許第5,284,826号。
【特許文献13】米国特許第5,525,590号。
【特許文献14】欧州特許公開公報第0 034 567号。
【特許文献15】欧州特許公開公報第0 056 782号。
【特許文献16】国際特許公開公報WO86/02080。
【特許文献17】国際特許公開公報WO99/18120。
【0009】
上記の参照特許、特許出願及び出版物の各々の開示は参考としてそのまま本明細書に組み込まれる。
【背景技術】
【0010】
シクロスポリン誘導体類は、真菌Tolypocladium inflatum Gams種によって第二の代謝産物として産生され、11個のアミノ酸よりなる環式ポリペプチド類の一群を構成する。それらは、免疫担当リンパ球、特にT−リンパ球を、細胞周期のG0又はG1相において可逆的に阻害することが観察されている。シクロスポリン誘導体は、リンホカインの生成及び放出を可逆的に阻害することも観察されている(グラネリ−ピペルノら、1986)。多数のシクロスポリン誘導体が知られているが、シクロスポリンAが最も広く使用されている。シクロスポリンAの抑制効果は、T−細胞仲介性活性化事象の阻害に関連する。この抑制は、シクロスポリンがいたるところに存在する細胞内蛋白、シクロフィリンと結合することによって実現する。この錯体は、今度は酵素カルシニューリンのカルシウム−及びカルモジュリン依存性のセリン−スレオニンホスファターゼ活性を阻害する。カルシニューリンの阻害は、T−細胞活性化中のサイトカイン遺伝子(IL−2、IFN−γ、IL−4及びGM−CSF)の誘導に必要な、NFATp/c及びNF−κBなどの転写因子の活性化を妨害する。また、シクロスポリンはinvitroにおけるT−ヘルパー細胞によるリンホカイン生成を阻害し、胸腺の成熟CD8及びCD4細胞の発育を停止させる(グラネリ−ピペルノら、1986)。シクロスポリンのその他のinvitro特性には、IL−2生産T−リンパ球及び細胞傷害性T−リンパ球の阻害、活性化T−細胞によって放出されるIL−2の阻害、同種抗原及び外因性リンホカインに反応する静止T−リンパ球の阻害、IL−1生産の阻害、及びIL−2生産T−リンパ球のマイトジェン活性化の阻害がある(グラネリ−ピペルノ、1986)。
【0011】
シクロスポリンは、体液性免疫及び細胞仲介性免疫反応、例えば同種移植片拒絶、遅延型過敏症、実験的アレルギー性脳脊髄炎、フロイントアジュバント関節炎、グラフト対ホスト疾患などを抑制することが証明された、強力な免疫抑制剤である。これは、臓器移植後の臓器拒絶の予防;リウマチ性関節炎の治療;乾癬治療;及びI型糖尿病、クローン病、狼瘡などを含むその他の自己免疫病の治療に使用される。
【0012】
シクロスポリンが始めて発見されて以来、種々様々の天然シクロスポリンが分離され、同定され、多くの非天然シクロスポリンが全−又は半合成手段によって、又は改良培養法の適用によって調製された。こうしてシクロスポリンによって構成される群は、今や実質的な価値を有し、天然シクロスポリンAからZまで[トレイバーら(1977);トレイバーら(1982);コベルら(1982);及びフォンワルトバーグら(1986)を参照]並びに、次に記すような種々の非天然シクロスポリン誘導体及び人工又は合成シクロスポリン類を含む。これらにはジヒドロ−及びイソ−シクロスポリン類;誘導化シクロスポリン(例えば、−MeBmt−残基の3'−O−原子がアシル化されるか又はまた別の置換基が3−位置のサルコシル残基のα−炭素原子に導入されたもの);−MeBmt−残基が異性体型で存在するシクロスポリン類(例えば、−MeBmt−残基の位置6'及び7'を横切る構造がトランスよりもむしろシス型であるもの);及びウェンガー(R.Wenger)によって開発されたシクロスポリン全合成生産法などを利用して、ペプチド配列内の特異的部位に変形アミノ酸類を挿入したシクロスポリン類などがある(トレイバーら(1977)、トレイバーら(1982)及びコベルら(1982);米国特許第4,108,985号、第4,210,581号、第4,220,641号、第4,288,431号、第4,554,351号及び第4,396,542号;欧州特許公開公報第0034 567号及び第0 056 782号;国際特許公開公報WO86/02080;ウェンゲル(1983);ウェンゲル(1985);ウェンゲル(1986)などを参照)。1−位置の修飾されたアミノ酸を含むシクロスポリンA類似体は、リッチら(1986)によって報告されている。免疫抑制性、抗炎症性及び抗寄生虫シクロスポリンA類似体が、米国特許第4,384,996号;第4,771,122号;第5,284,826号;及び第5,525,590号に記載されており、これらの特許は全てサンド社に譲渡されている。その他のシクロスポリン類似体が、イソテクニカに譲渡されたWO99/18120に開示されている。用語Ciclosporin、ciclosporin、cyclosporine及びCyclosporineは同義であり、シクロスポリンを意味する。
【0013】
シクロスポリンA治療に関連する多くの副作用には、腎毒性、肝毒性、白内障誘発性、多毛症、パラセシス(parathesis)、及び歯肉過形成などがある(スケトリスら、1995)。これらのなかで腎毒性は、シクロスポリンA投与に起因するより深刻な用量関連性副作用の一つである。即時的に放出されるシクロスポリンA薬剤(例えばネオラル(商標)及びサンドインミューン(商標)など)は、速やかに遊離され、吸収されて血中薬剤濃度を高めることによって腎毒性及びその他の毒性副作用を起こし得る。その薬剤のピーク濃度が副作用と関係すると予想されている(ベネット、1998)。シクロスポリンAが腎臓損傷を起こす正確なメカニズムは知られていないが、腎臓中の血管収縮物質の濃度増加が求心性糸球体細動脈の血管収縮に導くと言われている。これは腎虚血、糸球体濾過率の低下を招き、長期にわたると、間質性線維症を起こす可能性がある。用量を減らすか又は他の免疫抑制剤に代えると、腎機能は改善する(ヴァランチン及びシュローダ、1995)。よって、有効かつ低毒性の免疫抑制剤が必要である。
【0014】
1−位置に修飾アミノ酸を含むシクロスポリン類似体が、WO99/18120に開示されており、この特許は本出願の譲渡人に譲渡され、そのまま本明細書に挿入されている。米国仮特許出願第60/346,201号もこの譲渡人に譲渡されており、ここで出願人は「ISATX247」と呼ばれる特に好ましいシクロスポリンA類似体を開示している。この類似体は、1−アミノ酸残基の修飾を除けば構造的にシクロスポリンAと同一である。出願人は、ISATX247のシス及びトランス異性体のある種の混合物が、天然に発生し、現在公知のシクロスポリン類に勝る高い効力及び/又は低毒性の組み合わせを有することを発見した。ISATX247のある種のアルキル化、アリール化、及び重水素化誘導体も開示された。
【0015】
一般的に、米国仮特許出願第60/346,201号に開示された混合物は、トランス異性体約10ないし90重量パーセント、及びシス異性体約90ないし10重量パーセントの範囲である;別の実施形態において、上記混合物は、約15ないし85重量パーセントのトランス異性体及び約85ないし15重量パーセントのシス異性体を含む;別の実施形態において、上記混合物は約25ないし75重量パーセントのトランス異性体及び約75ないし25重量パーセントのシス異性体を含む;また別の実施形態において、上記混合物は約35ないし65重量パーセントのトランス異性体及び約65ないし35重量パーセントのシス−異性体を含む;また別の実施形態において、上記混合物は約45ないし55重量パーセントのトランス異性体及び約55ないし45重量パーセントのシス異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントのトランス異性体及び約50ないし55重量パーセントのシス異性体を含むISATX247混合物である。これらの重量パーセンテージは、組成物の総重量を基準としている。言い換えれば、混合物は(E)−異性体を65重量パーセント及び(Z)異性体を35重量パーセント含み、その逆もあり得る。別の命名法では、シス異性体は(Z)-異性体と言うこともでき、トランス異性体は(E)−異性体と言うこともできる。
【0016】
よって、当該技術では、異性体ISATX247を含むシクロスポリン類似体の調製方法が必要である。個々の異性体に富む組成物、並びに上記2異性体の所望比を有する異性体混合物を生成する合成経路が必要である。ISATX247の誘導体の調製方法も必要である。
【発明の開示】
【0017】
発明の概要
シクロスポリン及びその類似体は、強力な免疫抑制活性を有する環状ポリペプチド群のメンバーである。これらの薬剤はそれらの免疫抑制活性、抗炎症性活性及び抗寄生虫活性に関する利点を提供する一方、シクロスポリンA治療に関しては腎毒性及び肝毒性などの副作用が多数ある。よって、薬理学的活性は天然化合物シクロスポリンAと同様であるが関連する毒性副作用はない、新しい免疫抑制剤が必要とされる。本発明は以下を提供する。
(1)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)アセチル−η−ハロシクロスポリンAを、トリアリールホスフィン、トリアルキルホスフィン、アリールアルキルホスフィン、及びトリアリールアルシンからなる群から選択される第一化合物と共に加熱して中間体を生成し;
b)前記中間体を、アセトアルデヒド、ホルムアルデヒド、重水素化ホルムアルデヒド、2−クロロベンズアルデヒド、及びベンズアルデヒドからなる群から選択される第二化合物と共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
c)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(2)アセチル−η−ハロシクロスポリンAがアセチル−η−ブロモシクロスポリンAである、項目1に記載の方法。
(3)アセチルシクロスポリンAを、N−ブロモスクシンイミド及びアゾ−ビス−イソブチロニトリルと共に還流することによって行われる臭素化反応を用いて、シクロスポリンAの1−アミノ酸残基の側鎖のη−炭素をハロゲン化する工程をさらに含む、項目1に記載の方法。
(4)第一化合物がトリフェニルホスフィンであり、中間体がアセチルシクロスポリンAのトリフェニルホスホニウムハリドである、項目1に記載の方法。
(5)第二化合物がホルムアルデヒドである、項目1又は4に記載の方法。
(6)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)アセチルシクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、アセチルシクロスポリンAアルデヒドをアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(7)ウィッティヒ反応に用いられるリンイリドが、トリフェニルホスフィン、トリアリールホスフィン、トリアルキルホスフィン及びアリールアルキルホスフィンからなる群から選択される、項目6に記載の方法。
(8)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が;
a)第一中間体アセチルシクロスポリンAの形成によってシクロスポリンAのβ−アルコールを保護し;
b)アセチルシクロスポリンAを酸化して第二中間体アセチルシクロスポリンAアルデヒドを形成し;
c)中間体アセチルシクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
d)アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(9)酸化工程が、オゾン、過マンガン酸カリ、四酸化ルテニウム、四酸化オスミウム、ポリマー支持四酸化オスミウム、及び塩化ルテニウムからなる群から選択される酸化剤で行われる、項目8に記載の方法。
(10)四酸化ルテニウム及び塩化ルテニウム酸化剤が、過ヨウ素酸塩及び次亜塩素酸塩からなる群から選択される補助酸化剤と共に使用される、項目9に記載の方法。
(11)四酸化ルテニウム及び塩化ルテニウム酸化剤がアセトニトリルと共に使用される、項目9に記載の方法。
(12)1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
(13)β−トリアルキルシリルアルコールの処理に使用される酸が、酢酸、硫酸及びルイス酸からなる群から選択される、項目12に記載の方法。
(14)1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;
c)前記アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(15)β−トリアルキルシリルアルコールの処理に用いる塩基が、水素化ナトリウム及び水素化カリウムからなる群から選択される、項目14に記載の方法。
(16)γ−(トリアルキルシリルアリル)ボロネートエステルがトリメチルシリルアリルボロネートエステルである、項目12又は14に記載の方法。
(17)E−γ−(トリアルキルシリルアリル)ジアルキルボランがE−γ−(トリメチルシリルアリル)−9−BBNである、項目12又は14に記載の方法。
(18)試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、項目12又は14記載の方法。
(19)β−トリアルキルシリルアルコールを酸で処理する工程が、ピーターソンのオレフィン化を含む、項目12に記載の方法。
(20)β−トリアルキルシリルアルコールを塩基で処理する工程が、ピーターソンのオレフィン化を含む、項目14に記載の方法。
(21)1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドをリチウム化アリルジフェニルホスフィンオキシドと反応させて、アセチル−(E)−1,3−ジエンを形成し;
b)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
(22)1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドを[3−(ジフェニルホスフィノ)アリル]チタンと反応させて、チタン含有中間体を形成し;
b)チタン含有中間体をエリスロ−α−付加物にし;
c)前記エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;
d)前記β−オキドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;
e)前記アセチル−(Z)−1,3−ジエンを塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(23)a)シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;
b)アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して、第一中間体アセチル−η−ブロモシクロスポリンAを生成し;
c)前記第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される中間体を生成し;
d)アセチルシクロスポリンAのトリフェニル又はトリアルキル塩(第二中間体トリフェニルホスホニウムブロミド)から生成するイリドを、ホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
e)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなるプロセスによってつくられる(E)及び(Z)−異性体混合物。
(24)アセチル−1,3−ジエンの処理に使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド及びカリウムアルコキシドからなる群から選択される、項目1、6、8、12、14、21、22及び23のいずれかの項に記載の方法。
(25)アセチル−η−ハロシクロスポリンAのトリフェニルホスホニウム塩を含む、組成物。
(26)ハリドが、ブロミド、クロリド、及びヨーディドからなる群から選択される、項目25に記載の組成物。
(27)アセチルシクロスポリンAのトリフェニルホスホニウムブロミドである、項目25に記載の組成物。
(28)シクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む、組成物。
(29)中間体がアセチルシクロスポリンAのトリフェニル−又はトリアルキルホスホニウムブロミドであり、前記中間体をホルムアルデヒドと共に撹拌する工程が、リチウムハリドの存在のもとで行われる、項目1に記載の方法。
(30)a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記アセチルシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(31)a)トリメチルシリル(TMS)シクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記TMSシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(32)オゾン分解工程が約−80℃ないし0℃の温度で行われる、項目30又は31に記載の方法。
(33)還元剤が、トリアルキルホスフィン、トリアリールホスフィン及びトリアルキルアミンからなる群から選択される、項目30又は31に記載の方法。
(34)還元剤が、アルキルアリールスルフィド、チオ硫酸塩、及びジアルキルスルフィドからなる群から選択される、項目30又は31に記載の方法。
(35)還元剤がジメチルスルフィドである、項目34に記載の方法。
(36)還元剤がトリブチルホスフィンである、項目33に記載の方法。
(37)還元剤がトリアルキルアミンである、項目33に記載の方法。
(38)還元剤がトリエチルアミンである、項目37に記載の方法。
(39)アセチルシクロスポリンAのオゾン分解に用いられる溶媒が低級アルコールである、項目30ないし38のいずれかの項に記載の方法。
(40)アルコールがメタノールである、項目39に記載の方法。
(41)オゾン分解に使用される溶媒が、ジクロロメタン、及びジクロロメタンと低級アルコールとの混液からなる群から選択される、項目31に記載の方法。
(42)低級アルコールがメタノールである、項目41に記載の方法。
(43)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)中間体アセチルシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハリド又はトリフェニルホスホニウムハリドからつくられたリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をアセチル1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(44)ホスホニウムハリドがホスホニウムブロミドである、項目43に記載の方法。
(45)ウィッティヒ反応が、テトラヒドロフラン及びトルエンからなる群から選択される溶媒中で行われ、前記溶媒はブリルリチウム、低級アルコキシドナトリウム、低級アルコキシドカリウム、及び炭酸塩からなる群から選択される化合物の存在下で、約−80℃ないし110℃の温度で使用される、項目43に記載の方法。
(46)低級アルコキシドカリウムがカリウム−tert−ブトキシドである、項目45に記載の方法。
(47)溶媒が、カリウム−tert−ブトキシドの存在下で、約−70℃ないし−100℃の温度で使用されるテトラヒドロフランである、項目46に記載の方法。
(48)ISATX247のE−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
(49)使用する酸が、酢酸、硫酸、又はルイス酸からなる群から選択される、項目48に記載の方法。
(50)酸がBF3である、項目48に記載の方法。
(51)ISATX247のZ−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してTMS−(Z)−1,3−ジエンを形成し;
c)前記TMS−(Z)−1,3−ジエンを脱保護して、ISATX247のZ−異性体を形成する
諸工程を含んでなる、前記方法。
(52)E−γ−(トリアルキルシリルアリル)ボランが:
a)アリルトリアルキルシランを塩基で脱プロトン化し;
b)脱プロトン化したアリルトリアルキルシランを、ジアルキルアルコキシボラン及びルイス酸と反応させる
諸工程によってつくられる、項目48又は51に記載の方法。
(53)ジアルキルアルコキシボランがジエチルメトキシボランであり、ルイス酸がBF3である、項目52に記載の方法。
(54)アリルトリアルキルシランがアリルトリメチルシランである、項目52に記載の方法。
(55)アリルトリアルキルシランの脱プロトン化に使用する塩基がブチルリチウムである、項目52に記載の方法。
(56)β−トリアルキルシリルアルコールを処理するために使用する塩基が、水酸化ナトリウム、水酸化カリウム、及びアルカリ低級アルコキシドからなる群から選択される、項目51に記載の方法。
(57)塩基がカリウムtert−ブトキシドである、項目56に記載の方法。
(58)脱保護が、塩酸、酢酸、クエン酸、ルイス酸、及びHFをベースとする試薬からなる群から選択される酸を用いて達成される、項目51に記載の方法。
(59)HF−ベースの試薬が、フッ化トリブチルアンモニウム及びフッ化カリウムからなる群から選択される、項目58に記載の方法。
(60)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、前記方法は、(E)−異性体及び(Z)−異性体が混合物中に所定の比率で存在するように、ISATX247の(E)及び(Z)−異性体をつくる合成経路を含み、前記合成経路が:
a)シクロスポリンAの1−アミノ酸のβアルコールを保護し;
b)保護シクロスポリンAを酸化して保護シクロスポリンAアルデヒドを生成し;
c)前記保護シクロスポリンAアルデヒドとリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、保護シクロスポリンAアルデヒドを保護1,3ジエンのE−及びZ−異性体の混合物に変換し;
d)前記保護1,3ジエンの脱保護により、E−及びZ−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(61)シクロスポリンAと、酢酸エステル、安息香酸エステル、置換安息香酸エステル、エーテル及びシリルエーテルからなる群から選択される試薬とを反応させて、保護シクロスポリンAを生成することにより、シクロスポリンAの1アミノ酸のβアルコールを保護する、項目60に記載の方法。
(62)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、前記方法は、(E)−異性体及び(Z)−異性体が混合物中に所定の比率で存在するように、ISATX247の(E)−異性体及び(Z)−異性体をつくる合成経路を含み、前記混合物中の異性体の比率は、混合物の総重量を基準として、約45ないし55重量パーセントの(E)−異性体から約55ないし45重量パーセントの(Z)−異性体までの範囲である、前記方法。
(63)1−アミノ酸残基が修飾されたシクロスポリンA類似体の所定の異性体混合物の調製方法であって:
a)ISATX247の(E)−異性体に富む第一材料をつくり;
b)ISATX247の(Z)−異性体に富む第二材料をつくり;
c)第一及び第二材料を、所望異性体組成物を与えるように設計された比率で混合する
諸工程を含んでなる、前記方法。
(64)a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβアルコールを保護し;
b)前記アセチルシクロスポリンAを、モノペルスルフェート、好ましくはオキソンで、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、7より高いpHで、アセチルシクロスポリンAエポキシドに変換し;
c)前記エポキシドを過ヨウ素酸又は過ヨウ素酸塩で、酸性条件下で分解する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
(65)工程b及びcが一つの処理操作に組合わせられる、項目64に記載の方法。
(66)1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:
a)中間体TMSシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハリド又はトリフェニルホスホニウムハリドからつくられるリンイリドとを、任意にリチウムハリドの存在下で、ウィッティヒ反応によって反応させることによって、前記中間体をTMS−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;
b)前記TMS−1,3−ジエンの(E)及び(Z)−異性体の混合物を酸で脱保護することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる
諸工程を含んでなる、前記方法。
(67)ホスホニウムハリドがホスホニウムブロミドである、項目66に記載の方法。
(68)工程a)が、ナトリウム又はカリウム低級アルコキシド、又は炭酸塩の存在下で使用されるテトラヒドロフラン及び/又はトルエンを含む溶媒中で、約−80℃ないし110℃の温度で行われる、項目66に記載の方法。
(69)ナトリウム又はカリウム低級アルコキシドが、カリウム−tert−ブトキシドである、項目68に記載の方法。
(70)酸が、塩酸、酢酸、クエン酸、ルイス酸及びHF−ベースの試薬からなる群から選択される、項目66に記載の方法。
(71)ISATX247のE−異性体の立体選択的合成方法であって:
a)アセチルシクロスポリンAアルデヒドとE−γ−(トリアルキルシリルアリル)ボランとを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、アセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
(72)β−トリアルキルシリルアルコールを処理する工程が、さらにピーターソンのオレフィン化を含む、項目48、51又は71に記載の方法。
(73)ボラン試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、項目71に記載の方法。
(74)使用する酸が、酢酸、硫酸又はルイス酸からなる群から選択される、項目71に記載の方法。
(75)酸が硫酸である、項目74に記載の方法。
(76)1,3−ジエンの処理のために使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、項目71に記載の方法。
(77)塩基が、NH3及びジメチルアミンからなる群から選択される、項目76に記載の方法。
(78)アセチルシクロスポリン1,3−ジエンをISATX247に変換するための方法であって、前記アセチルシクロスポリン1,3−ジエンが、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、アルコキシドナトリウム、アルコキシドカリウム、及びNH3とヒドロキシルアミンとヒドラジンとジアルキルアミン類からなる群から選択されるアミン塩基からなる群から選択される塩基で処理される、前記方法。
(79)塩基がジメチルアミンである、項目78に記載の方法。
(80)ISATX247のZ−異性体の立体選択的合成法であって;
a)アセチルシクロスポリンAアルデヒドを、第一塩基の存在下で3−(ジメチルアミノ)プロピルトリフェニルホスホニウムハリドで処理して、アセチル−(Z)−オクテニルジメチルアミンを形成し;
b)前記アセチル−(Z)−オクテニルジメチルアミンを酸化試薬で処理して、アセチル−(Z)−オクテニルジメチルアミンオキシドを形成し;
c)前記アセチル−(Z)−オクテニルジメチルアミンオキシドをコープ脱離反応において加熱して、アセチル−(Z)−1,3−ジエンを形成し;
d)前記アセチル−(Z)−1,3−ジエンを第二塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
(81)第一塩基がカリウム ヘキサメチルジシラチドである、項目80に記載の方法。
(82)酸化試薬がメタクロルペル安息香酸である、項目80に記載の方法。
(83)第二塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、項目80に記載の方法。
【0018】
本発明の実施形態は薬物学的に有効な、シクロスポリンA類似体のシス及びトランス異性体の幾つかの混合物を提供する。好ましい類似体は、ISATX247と呼ばれる。ISATX247異性体の混合物は、天然シクロスポリン及び現在公知のシクロスポリン類に比べてより高い効力とより低い毒性の組合わせを示す。
【0019】
本発明は、一部には、シクロスポリン類似体のある異性体混合物類がシクロスポリンAで生ずるような副作用をもたず、すぐれた免疫抑制作用を提供するという発見に基づいている。特に、1−アミノ酸残基を修飾したシクロスポリン類似体の約10:90ないし約90:10(トランス対シス)の範囲の異性体混合物類(すなわちシス−及びトランス−異性体両方の混合物)が予想外にすぐれた効果及び安全性をもたらすことを発見した。このような類似体の例は、WO90/18120に開示されており、重水素化及び非重水素化化合物を含んでいる。より詳細に述べれば、約45:55ないし約50:50(トランス対シス)の範囲、及び約50%ないし約55%トランス−及び約45%ないし約50%シス−の範囲の混合物が特に効率的であることが見いだされた。その上、これらの異性体混合物は、天然及びその他の現在公知のシクロスポリン及びシクロスポリン誘導体に比べてよりすぐれた効力とより低い毒性の組合わせを示すことが証明された。
【0020】
特に好ましい類似体(本明細書では「ISATX247」と名づける)は、分子の周囲の、1−アミノ酸残基に存在する修飾官能基を除けば構造的にシクロスポリンAと同様である。この特定の異性体類似体混合物の構造をシクロスポリンAの構造と比較して、図1A、1B、2A、2Bに示す。
【0021】
この異性体混合物は、とりわけ免疫抑制のために、そして種々の免疫障害、免疫病及び免疫疾患の予防、コントロール、緩和及び治療などのケアに使用できる。
【0022】
本発明の実施形態により、ISATX247異性体類(及びその誘導体類)は、選択性の程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)及び(Z)−異性体のどちらかに富む組成物を生成し、これらの組成物を、生成した混合物がこれら2異性体の所望比を有するように組合わせることもできる。或いは、生成した混合物が直接所望比を有するように立体選択的経路の反応条件を調節してもよい。ある混合物中の1異性体又は他の異性体のパーセンテージは、核磁気共鳴スペクトル法(NMR)又は当該技術で公知のその他の方法を使用して確認することができる。
【0023】
各経路は、一般的には不安定なアルコール官能基に保護基を適用して進む。一実施形態において、アルコールはアセテートとして保護され;他の実施形態において、保護基は安息香酸エステル類又はシリルエーテル類である。アセテート保護基は当該技術では一般的であるが、本明細書に記載される多くの典型的実施形態においては、安息香酸エステル又はシリルエーテルなどの保護基の使用によって、アセテート保護基に関係する幾つかの望ましくない副作用が回避されることを強調することは重要である。
【0024】
保護された化合物は、ウィッティヒ反応に参加するリン含有試薬を用いる経路や、有機金属試薬のメンバーとして無機元素を用いる経路などを含む、種々の立体選択的合成経路の前駆体としてはたらく。後者のタイプは、六員環遷移状態を通過して進行する。この場合、立体障害が配置的結果を決める。多くの有機金属試薬、例えばホウ素、珪素、チタン、リチウム及び硫黄などの無機元素を特徴とするものなどが使用できる。個々の異性体は、単一又は複数の前駆体からつくられる。
【0025】
立体選択的生成又は非立体選択的生成の如何にかかわらず、生成した混合物中の(E)対(Z)−異性体の比は、広範囲の数値をとり得る。例えば、混合物は約10ないし90パーセントの(E)−異性体から約90ないし10パーセントの(Z)−異性体までの範囲で構成される。その他の実施形態において、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)異性体を含むことができる;別の実施形態において、混合物は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体を含む;また別の実施形態において、混合物は約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体を含む;他の実施形態において、混合物は約45ないし55重量パーセントの(E)−異性体及び約55ないし45パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は約45ないし約50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセンテージは組成物の総重量を基準としており、(E)−異性体と(Z)−異性体との重量パーセントの合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体と35重量パーセントの(Z)−異性体を含むことができ、その逆でもよい。
【0026】
よって、一つの態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:アセチル−η−ハロシクロスポリンAをトリアルキルホスフィン、トリアリールホスフィン(例えばトリフェニルホスフィン)、アリールアルキルホスフィン、及びトリアリールアルシンと共に加熱して、中間体アセチルシクロスポリンAのホスホニウムハリドを生成し;中間体アセチルシクロスポリンAのホスホニウムハリドをホルムアルデヒドと共に、任意にリチウムハリドの存在下で撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基と共に処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法を提供する。
【0027】
また別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:中間体、例えば保護されたトリメチルシリル(TMS)シクロスポリンAアルデヒド又はアセチルシクロスポリンAアルデヒドを、前記中間体をウィッティヒ反応によって、任意にリチウムハリドの存在下でリンイリドと反応させることによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を、アセチル保護基の場合は塩基で、又はTMS−保護基の場合は例えば酸で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法に関するものである。
【0028】
さらにまた別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、E−異性体に富む混合物の生成方法であって、前記E−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと、トリアルキルシリルアリル硼酸エステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0029】
また別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、Z−異性体に富む混合物の生成方法であって、前記Z−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと、トリアルキルシリルアリル硼酸エステル及び(E−γ−トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0030】
もう一つの態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、E−異性体に富む混合物の生成方法であって、前記E−異性体に富む物質の立体選択的合成が;アセチルシクロスポリンAアルデヒドとリチウム化アリルジフェニルホスフィンオキシドとを反応させてアセチル−(E)−1,3−ジエンを形成し;アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0031】
さらに別の態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の、Z−異性体に富む混合物の生成方法であって、前記Z−異性体に富む物質の立体選択的合成が:アセチルシクロスポリンAアルデヒドと[3−(ジフェニルホスフィノ)アリル]チタンとを反応させてチタン含有中間体を形成し;チタン含有中間体がエリスロ−α−付加物になるまで反応を続け;エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;β−オキシドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する諸工程を含んでなる、前記方法を提供する。
【0032】
さらにまた別の態様において、シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して第一中間体アセチル−η−ブロモシクロスポリンAを生成し;第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される第二の中間体を生成し;アセチルシクロスポリンAのトリフェニル−又はトリアルキル塩から生成するイリド(第二中間体トリフェニルホスホニウムブロミド)をホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体混合物をつくり;アセチル−1,3−ジエンの(E)及び(Z)異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)異性体の混合物をつくる諸工程を含んでなるプロセスによってつくられる、(E)及び(Z)−異性体の混合物を提供する。
【0033】
本発明は、シクロスポリンAのβ−アルコールを保護し;シクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して第一中間体アセチル−η−ブロモシクロスポリンAを生成し;第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドから選択されるアセチルシクロスポリンAブロミドを生成する諸工程を含んでなるプロセスによってつくられた生成物である、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドを含む対象の組成物類にも関する。アセチルシクロスポリンAのトリフェニル又はトリアルキルホスホニウムブロミド誘導体を含む組成物類、及びシクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む組成物類も提供される。
【0034】
その他の態様において、本発明はシクロスポリンAアルデヒドを選択的に調製する方法であって、アセチルシクロスポリンA又はトリメチルシリル(TMS)シクロスポリンAの形成によってシクロスポリンAのβ−アルコールを保護し;アセチルシクロスポリンA又はTMSシクロスポリンAを、還元剤と共に用いる酸化剤としてのオゾンで酸化する諸工程を含んでなる、前記方法を提供する。
【0035】
その他の付加的態様において、本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、その合成経路が:中間体アセチルシクロスポリンAアルデヒドと、トリブチルアリルホスホニウムハロゲニドからつくられたリンイリドとを、任意にリチウムハリドの存在下でウィッティヒ反応によって反応させることによって、前記中間体をアセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物に変換し;アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによってISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなる、前記方法に関するものである。
【0036】
その他の態様において、本発明は、ISATX247のE−異性体の立体選択的合成方法であって、トリメチルシリル(TMS)シクロスポリンAアルデヒドをトリアルキルシリルボランと反応させてβ−トリアルキルシリルアルコールを形成し;β−トリアルキルシリルアルコールを処理して直接ISATX247のE−異性体を形成する諸工程を含んでなる、前記方法を提供する。
【0037】
発明のその他の態様は、ISATX247のZ−異性体の立体選択的合成方法であって、トリメチルシリル(TMS)シクロスポリンAアルデヒドをトリアルキルシリルアリルボランと反応させてβ-トリアルキルシリルアルコールを形成し;β-トリアルキルシリルアルコールを塩基で処理してTMS−1,3-ジエンを形成し;TMS−(Z)−1,3−ジエンを脱保護してISATX247のZ−異性体を形成する諸工程を含んでなる、前記方法に関するものである。
【0038】
本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、(E)−異性体と(Z)−異性体とが所定の比で存在するようにISATX247の(E)−異性体及び(Z)−異性体つくる合成経路を含み、前記合成経路は:シクロスポリンAのβアルコールを保護し;保護されたシクロスポリンAを酸化して、第二中間体である保護されたシクロスポリンAアルデヒドを生成し;第二中間体である保護されたシクロスポリンAアルデヒドを、任意にリチウムハリドの存在下で、ウィッティヒ反応によってリンイリドと反応させることによって、前記第二中間体を保護された1,3ジエンのE−異性体及びZ−異性体の混合物に変換し;保護された1,3ジエンの脱保護によりE−及びZ−異性体の混合物をつくる諸工程を含んでなる、前記方法にも関する。
【0039】
本発明によって提供されるこのような混合物のその他の調製方法は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の異性体混合物の調製方法であって、混合物中に(E)−異性体と(Z)−異性体とが所定の比で存在するように、ISATX247の(E)−異性体及び(Z)−異性体をつくり、前記混合物中の異性体の比は、混合物の総重量を基準にして、(E)−異性体約45ないし55重量パーセント及び(Z)−異性体約55ないし45重量パーセントの範囲に及ぶ、前記方法も含む。
【0040】
本発明は、1−アミノ酸残基が修飾されたシクロスポリンA類似体の所定の異性体混合物の調製方法であって、ISATX247の(E)−異性体に富む第一材料をつくり;ISATX247の(Z)−異性体に富む第二材料をつくり;所望の異性体組成物を与えるように設計された比率で第一及び第二材料を混合する諸工程を含んでなる、前記方法も提供する。
【0041】
発明の詳細な説明
合成
シクロスポリン及びその類似体は、強力な免疫抑制活性を有する環式ポリペプチド群のメンバーである。これらの薬剤は、それらの免疫抑制性、抗炎症性及び抗寄生虫活性に関して利点を提供する一方、シクロスポリンA治療に関係した腎毒性及び肝毒性を含む多くの副作用がある。よって、薬理学的活性は天然化合物シクロスポリンAと同様であり、しかも関連する毒性副作用はない、という新しい免疫抑制剤が必要である。
【0042】
出願人は以前、「ISATX247」と呼ばれるシクロスポリンA類似体を開示した。この類似体は、1−アミノ酸残基が修飾されていることを除けばシクロスポリンAと同様の構造を有する。出願人は、ISATX247のシス及びトランス−異性体の幾つかの混合物が、天然及び現在公知のシクロスポリン類に勝る高い効力と低い毒性との組合わせを示すことを発見した。
【0043】
本発明の実施形態により、ISATX247異性体類(及びそれらの誘導体類)は、立体選択性程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)及び(Z)−異性体のどちらかに富む組成物を生成し、生成混合物が前記2異性体の所望比を有するように、これらの組成物を組み合わせることができる。或いは、立体選択的経路の反応条件を調節して、つくられた混合物が直接所望比を有するようにすることができる。
【0044】
本発明の、ISATX247と呼ばれる一免疫抑制剤シクロスポリン類似体の化学名は、シクロ{(E,Z)−(2S、3R、4R)−3−ヒドロキシ−4−メチル−2−(メチルアミノ)−6,8−ノナジエノイル}−L−2−アミノブチリル−N−メチル−グリシル−N−メチル−L−ロイシル−L−バリル−N−メチル−L−ロイシル−L−アラニル−D−アラニル−N−メチル−L−ロイシル−N−メチル−L−ロイシル−N−メチル−L−バリル}と化学的に記載される。その実験式はC63H111N11O12であり、約1214.85の分子量を有する。用語「ISATX247」は、この薬理学的活性化合物に与えられた商品名である。
【0045】
ISATX247の構造は、主として核磁気共鳴(NMR)分光法によって確認された。1H及び13Cスペクトルは両方とも、一次元及び二次元NMR実験系列を使用して設定され、シクロスポリンAの公知のNMR設定と比較された。ISATX247の(E)及び(Z)−異性体の絶対的設定は核オーバーハウザー効果(NOE)実験によって確認された。その他の証拠が、質量分析及び赤外スペクトルによって確認された。質量分析は分子量を確認し、この化合物の赤外スペクトルはシクロスポリンAの赤外スペクトルに非常に近いことが判明した。2化合物間に類似性があるため、後者の結果は予期されたものである。
【0046】
シクロスポリンAの構造を図1Aに示す。その構造は分子の環式ペプチド環を構成する11個のアミノ酸残基を確認するものである。これらの11個のアミノ酸残基は、環の頂上部中心に示されるアミノ酸(「1−アミノ酸」の基準標識を有する)から出発し、時計方向に増加するナンバーで標識化される。わかりやすいように第一アミノ酸を点線で四角く囲む。本明細書に記載される合成反応が起こる典型的部位であるので、1−アミノ酸残基の側鎖は化学的に書き表した。一般に、アミノ酸のカルボニル基に隣接する炭素は、α−炭素と標識付けられる。その鎖の下流方向、ペプチド環から離れる方向に隣接する炭素に、ギリシャアルファベット文字を順次付ける。シクロスポリンAの場合、図1Aに示すように、側鎖のβ−炭素はヒドロキシル基に結合し、側鎖のε−炭素とζ−炭素との間にはトランス方向の二重結合がある。
【0047】
シクロスポリンA構造のもう一つの略図を図1Bに示す。ここでは、前記分子の別の部分が点線で四角く囲まれる。この図は、本明細書に使用される命名法を明らかに示す:ここで用語「CsA」は四角く囲まれたシクロスポリンAの部分を指す。この命名法は、反応を記載するたびに分子の残りの部分を再記述することなく、合成反応が起こる領域(すなわち、図1B中の点線四角外に描かれた1−アミノ酸残基の側鎖)を示すという簡便な手段を提供する。側鎖のα−炭素とβ−炭素との間の結合は通常の長さであり、この図では用語「CsA」の定義をより明確にするために誇張されていることは当業者には当然のことである。
【0048】
上述のように、特に好ましいシクロスポリンA類似体はISATX247と呼ばれ、その2種類の立体異性体E(又はトランス)及びZ(又はシス)は、それぞれ図2A及び図2Bに示される。これらの立体異性体のシス又はトランス性は、側鎖のε及びζ−炭素間の二重結合の配置(すなわち、側鎖の末端にある二重結合に対して、ペプチド環により近い二重結合)を基準としている。
【0049】
立体化学的命名法について一言言及しなければならない。本明細書において、用語シスと(Z)とは交換可能に使用され、用語トランスと(E)とが交換可能に使用される。用語「エリスロ」及び「スレオ」の使用は、これらの意味に関して文献中に明らかな混乱があるため、最小限にとどめる。ホフマン及びゼイ著「アルコールの立体選択的合成。8.クロチルボロネートを介するβ−メチルホモアリルアルコールのジアステレオ選択的合成」J. Org. Chem., Vol.46, p.1309-1314(1981);ストレイトウィーザ及びヒースコック著、「有機化学入門、第2版」、(マクミラン、ニューヨーク、1981)、p.845-846;スミス及びマーチ著、「マーチの最新有機化学」、(ウィリー、ニューヨーク、2001)、p.144-147を参照されたい。本明細書において、スレオ/エリスロの用語を使用する数例では、ストレイトウィーザー及びヒースコックの約束事を適用する;その際「エリスロ」異性体は(R,S)及び(S,R)配置を、「スレオ」異性体は(R,R)及び(S,S)配置を指す。
【0050】
命名法に関する最後のコメントは、図2A及び図2Bに示される末端炭素−炭素二重結合に関するものである。別のナンバリングスキームにより、1−アミノ酸残基の側鎖の炭素類に、末端(θ)炭素から出発し、ペプチド環の方に戻るように標識ナンバーを付けてもよい。この系において、ISATX247異性体は有機化学の一般的命名法により1,3−ジエンと考えることができる。この場合、各二重結合は最小ナンバーの炭素によって決定される。
【0051】
図3−8に示される合成経路について述べる。本発明の実施形態により、異性体混合物が直接つくられる。その際、特定の合成経路の反応条件を調節して、混合物中の異性体の所望比を得る。或いは、シクロスポリンA類似体の2種類の幾何学的異性体のうちの一種類に富む組成物をつくり、それらの組成物を所定の比で合一して所望の混合物を得る。
【0052】
本発明の実施形態による合成経路の概観を図3に示す。ここでは、化学及び立体選択性による反応経路のグループ化が特に強調されている。図3を参照すると、ウィッティヒ反応を使用する合成経路が大体図の右手に示され、参照番号31が付けられる。一方、六員環遷移状態を形成すると考えられる有機金属試薬を利用する経路32及び33が、図の中央及び左側に示されている。いずれの合成経路も異性体類の混合物を与えるか、2異性体のうちの一つに富む組成物を生成する。
【0053】
本発明の実施形態は、所望の異性体混合物に到達する種々の方法を提供する。ここに開示する合成戦略のフレキシビリティ及び融通性が、図3の対称性及び非対称性によって一部反映される。各経路に共通する反応は、シクロスポリンA34の官能基の保護である;この典型的実施形態において、この反応はシクロスポリンA34からアセチルシクロスポリンA35への変換である。図3の非対称性は、チタン及びリチウム有機金属試薬経路の全てにおいて、前駆体としてアセチルシクロスポリンAアルデヒドを使用するが、リン含有ウィティッヒ反応経路が若干あることである。
【0054】
異性体を所望比で有する混合物を生成するように反応条件を調節した図3の合成経路は、概して、ウィティッヒ反応に関与する試薬としてリン含有試薬を使用する。その他の立体選択的経路は、無機元素類並びに、典型的に六員環遷移状態を通過する有機金属試薬のメンバー類を使用する。この遷移状態において、立体障害が配置的結果を決める。ホウ素、珪素、チタン、リチウム及び硫黄などの無機元素を特徴とする多数の有機金属試薬が、本発明に有用である。
【0055】
1対の異性体のうちの一つ又は他の一つどちらかに富む組成物を単一の前駆体からつくることがでる;又は、上記2組成物を異なる前駆体からつくることもできる。図3の立体選択的経路の一つ(経路32)において、選択した反応条件によって、単一の前駆体からISATX247の2異性体の両方が生成する。別の立体選択的経路(経路33)では、富化組成物の各々を生成するために2種類の前駆体が必要である。
【0056】
図3の反応を次に詳細に述べる。各経路に共通の反応は、1−アミノ酸残基の側鎖のβ−位置のアルコールの保護である。このような保護スキームは、第一官能基がその分子上のどこか別の部位にある第二(類似及び/又は同一の)官能基に対して行われる反応によって偶然に変形されるという有機合成では通常遭遇する問題である。前記スキームを実施するためには、第一官能基を保護基と反応させ、第二官能基で所望反応を行い、その後に第一官能基から保護基を除去する。
【0057】
保護基は有機合成においてはよく知られており、ハンソンが「有機合成における保護基(Protecting Groups in Organic Synthesis)」(シェフィールド・アカデミック・プレス、シェフィールド、英国、1999)の第2章(p.24-25)「アルコールの保護」において述べている。ハンソンは、ヒドロキシル基を、エステルかエーテルのどちらかに変換することによって保護する方法を教示している。おそらく酢酸エステルは、ヒドロキシル基を保護するために最もよく用いられるタイプの化学物質である。アセテート基を導入するために、広範囲の反応条件を取り得る。これらの試薬及び溶媒としては、無水酢酸及びピリジン;無水酢酸、ピリジン及びジメチルアミノピリジン(DMAP);無水酢酸及び酢酸ナトリウム;無水酢酸及びトルエン−p−スルホン酸、塩化アセチル、ピリジン及びDMAP;及びケテンがある。DMAPは、無水物から高い反応性を有するN−アシルピリジニウム塩を形成するため、有用なアシル化触媒である。
【0058】
本発明の一実施形態において、シクロスポリンA34のβ−アルコールは、塩化アセチル、酢酸エチル、又はこれらの組合わせと反応させて、化合物アセチルシクロスポリンA35を形成させることによって、アセテートとして保護される。別の実施形態において、β−アルコールは無水酢酸への求核付加を受け、アセチルシクロスポリンA35及び酢酸が形成される。これらの反応は、ジメチルアミノピリジン(DMAP)の存在のもとで行われ、無水酢酸の過剰は溶媒として作用する。これらの場合、接頭語「アセチル」は合成経路全体における、又はアセチル基が除去されるまでの間の名称として使用される。例えば、β−炭素にアセチル基を有する一経路の最後の中間体は、「アセチル−(E)−1,3−ジエン」と呼ばれる。
【0059】
アセチルシクロスポリンAの製法は、文献において十分確立されているが、酢酸エステル以外の保護基を使用してシクロスポリンA34の1−アミノ酸残基のβ−アルコールを保護することは、当業者には明らかである。これらの保護基には、安息香酸エステル類、置換安息香酸エステル類、エーテル類及びシリルエーテル類がある。ある反応条件のもとでは、アセテート保護基は脱離や加水分解などの不都合な副反応をおこしがちである。安息香酸エステル類、エーテル類及びシリルエーテル類は、同じ反応条件下でこのような副反応により強く抵抗することが多いため、アセテートの代わりにこのような保護基を使うことが有利であることが多い。アセチル基又はその他の保護基によって保護されたシクロスポリン又はシクロスポリン誘導体を、「保護−シクロスポリンA」と呼ぶ。同様に、上記の典型的経路における最後の中間体は、「アセチル−(E)−1,3−ジエン」の代わりに「保護−(E)−1,3−ジエン」と呼んでもよい。選択した保護基の性質は、反応系列のその後の工程の所望コースに影響を与えることがある。
【0060】
図3を参照すると、アセチルシクロスポリンA35は、この典型的経路において保護β−アルコールを有し、この化合物は数種の合成経路においてISATX247異性体を合成するための前駆体として役立つ。ウィッティヒ反応経路を先ず最初に論ずる。
【0061】
ウィッティヒ反応によるISATX247の(E)及び(Z)−異性体混合物の合成
ここに例示されるウィッティヒ反応経路は、図3の参照番号31によって確認される。方法1は、臭素中間体アセチル−η−ブロモシクロスポリン41を経て進むが、方法2は、出発点としてアセチルシクロスポリンAアルデヒド51を使用する。以下に記載する典型的方法はウィッティヒ反応を用い、混合立体化学的配置を有するアルケン官能基を導入する。
【0062】
本明細書に開示される典型的実施形態において、ISATX247の(E)及び(Z)−異性体混合物を合成するために用いられるウィッティヒ反応は、任意にリチウムハリドの存在下で行ってもよい。ウィッティヒ反応におけるリチウムハリドの存在は、生成する幾何異性体類の比に影響をもつことはよく知られており、したがってこのような化合物の添加は、ISATX247の(E)及び(Z)−異性体の所望混合物の生成に役立つ。
【0063】
方法1
本発明の一実施形態において、ISATX247の(E)及び(Z)−異性体混合物は、図4に示すようにつくられる。図4における波線(特に、化合物43と44を参照)の表示の使用は、例示的反応系列が(E)及び(Z)−異性体の混合物を生成することを示すものとする。一実施形態において、生成した(E)対(Z)−異性体のパーセント比は、(E)−異性体の約10ないし90パーセントから(Z)−異性体の約90ないし10パーセントまでの範囲であるが、これらの範囲は単なる例示であり、多くの他の範囲が可能である。例えば、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)−異性体を含むことができる。その他の実施形態において、混合物は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体;約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体;及び、約45ないし55重量パーセントの(E)−異性体及び約55ないし45パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセントは組成物の総重量を基準としており、(E)異性体及び(Z)異性体の重量の合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体及び35重量パーセントの(Z)−異性体を含むことができ、又はその逆でもよい。
【0064】
図4を参照すると、アセチルシクロスポリンAの1−アミノ酸残基の側鎖の末端η炭素を次の反応工程において臭素化する;すなわち、四塩化炭素などの溶媒中でアセチルシクロスポリンA35をN−ブロモスクシンイミド及びアゾ−ビス−イソブチロニトリルと共に還流し、中間体アセチル−η−ブロモシクロスポリンA41を生成する。N−ブロモスクシンイミドは、アリル水素を臭素で置換するためによく用いられる試薬であり、これは遊離基メカニズムによって行われると考えられている。この中間体41の製法は、エバール及びナニンガーによって「シクロスポリンAの主代謝産物(OL−17)の合成」(J. Org. Chem., Vol57, p.2686-2691(1992))で詳細に記載されている。
【0065】
新規の中間体、アセチルシクロスポリンAトリフェニルホスホニウムブロミド42は、アセチル−η−ブロモシクロスポリンA41から、この化合物をトルエンなどの溶媒中でトリフェニルホスフィンと共に加熱することによってつくることができる。
【0066】
新規の中間体42及びこれに類似するその他のものは、1−アミノ酸残基中に共役ジエン系を含む複数のシクロスポリンA類似体の合成における重要な中間体であると考えられる。例えば、トリフェニルホスフィンに加えて、トリアリールホスフィン類、トリアルキルホスフィン類、アリールアルキルホスフィン類、及びトリアリールアルシン類などの化合物をアセチル−η−ブロモシクロスポリンA41と反応させて42に類似するその他の活性化合物をつくることができる。
【0067】
再び図4を参照すると、アセチルシクロスポリンAのトリフェニルホスホニウムブロミド42と過剰のホルムアルデヒドをトルエン中で室温で撹拌することによって、アセチル−1,3−ジエン43の(E)及び(Z)−異性体混合物をつくることができる。ホルムアルデヒドの添加後、水酸化ナトリウムなどの塩基を滴下し、ジエン類の異性体混合物を酢酸エチルで抽出する。
【0068】
多数の有機化学の教科書が、ウィッティヒ反応を説明している。説明の一つは、特にマクマリーが「有機化学(Organic Chemistry)」、5版(ブルックス/コール、パシフィックグローブ、2000)、p.780-783で記載しているものである。ウィッティヒ反応は、ケトン又はアルデヒドをアルケンに変換するために利用される。このようなプロセスにおいて、リンイリド(ホスホランとも呼ばれる)をアルデヒド又はケトンと反応させると、ベタインと呼ばれる二極性中間体が得られる。一般的には、ベタイン中間体は分離されない;むしろ、それは四員環を経て自然に分解して、アルケンとトリフェニルホスフィンオキシドを与える。正味の結果は、元々はリンに結合していたR2C=基によってカルボニル酸素原子が置換されることである。
【0069】
非常に種々様々の試薬が上述の典型的ウィッティヒ反応試薬の代わりになり得ることは、当業者には明らかである。例えば、多数のアルキル、アリール、アルデヒド及びケトン化合物をホルムアルデヒドの代わりに使用すると、膨大な数のシクロスポリン誘導体がつくられる。出願人は、上記の合成をホルムアルデヒドで行い、また、ホルムアルデヒドの代わりにアセトアルデヒド、ジューテロ化ホルムアルデヒド、ジューテロ化アセトアルデヒド、2−クロロベンズアルデヒド、ベンズアルデヒド、及びブチルアルデヒドなどの化合物で行った。このようなウィッティヒ反応を、トリフェニルホスホニウム誘導体以外の化合物、例えばトリアリールホスフィン類、トリアルキルホスフィン類、アリールアルキルホスフィン類及びトリアリールアルシン類などで行ってもよい。水酸化ナトリウムを使用する代わりに、種々のその他の塩類、例えば炭酸ナトリウム、ブチルリチウム、ヘキシルリチウム、ナトリウムアミド、リチウムジイソプロピルアミドのようなリチウム障害性塩基、及びアルカリ金属アルコキシド類などを使用してもよい。これら種々の試薬が使用できる上に、反応は種々の有機溶媒中又は有機溶媒と水との混液中で、種々の塩、特にリチウムハリドの存在下で、種々の温度で行うことができる。上に列挙した因子の全ては、当業者によって合理的に選択され、形成される二重結合の立体化学、すなわちシス対トランス−異性体の比に所望の効果を与える。本発明の一実施形態において、ウィッティヒ反応は、テトラヒドロフラン及びトルエンからなる群から選択される溶媒中で行われ、その際その溶媒は、ブチルリチウム、ナトリウム低級アルコキシド、カリウム低級アルコキシド及びカルボネートから選択される化合物の存在下で、約−80℃ないし110℃の温度で使用される。カリウム低級オキシドは、カリウム−tert−ブトキシドでよい。さらに、溶媒は、カリウム−tert−ブトキシドの存在下で約−70℃ないし−100℃の温度で使用されるテトラヒドロフランでもよい。
【0070】
この合成の最終工程において、β−炭素上の保護基を下記の手順によって除去する。アセチル−(E)−1,3−ジエン及びアセチル−(Z)−1,3ジエンの混合物43をメタノールに溶解し、それから水を加える。炭酸カリウムのような塩基を加え、反応混合物を室温で撹拌する。使用できる炭酸カリウム以外の塩基として水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド及びカリウムアルコキシドなどがある。その後、酢酸エチルを使用して最終生成物であるISATX247の(E)及び(Z)−異性体混合物44を抽出する。
【0071】
方法2
ISATX247の(E)及び(Z)−異性体混合物を、ウィッティヒ反応戦略によって合成するまた別の反応経路においては、以下のように4工程の合成経路を使用する:1)方法1のようなβ−アルコールの保護、2)第一工程から生成したアセチル−シクロスポリンAを酸化してアルデヒドをつくる;3)ウィッティヒ反応;4)ウィッティヒ反応生成物を脱アシル化し、又は同等に、酢酸エステルを加水分解して、アルコールを回復させる。この反応系列は、図5で明らかにされる。
【0072】
この合成経路は、第一工程が酢酸エステル基でβ−アルコールを保護するという点で、図4のウィッティヒ反応経路と同様にして開始される。しかし、これら2経路は次の点で互いに異なる;方法2の次の工程は、アセチル−シクロスポリンA35をアルデヒド、すなわちアセチルシクロスポリンAアルデヒド51に変換する。この反応は、十分強い酸化剤を使用し、C=C結合を切断して2つの断片を生成する。アルケン切断は、当該技術では公知である。オゾンはおそらく最も一般的に使用される二重結合切断試薬であるが、その他の酸化剤、例えば過マンガン酸カリ(KMnO4)又は四酸化オスミウムも、二重結合切断を起こすことができる。
【0073】
本発明の実施形態によると、アセチル−シクロスポリンAは酸化剤としてのオゾンでアルデヒドに変換され、その後還元剤で処理されてアセチルシクロスポリンAアルデヒドを形成する。オゾン分解の工程は、約−80℃ないし0℃の温度範囲で行われる。オゾン分解中に使用する溶媒は、メタノールなどの低級アルコールである。還元剤は、トリブチルホスフィンのようなトリアルキルホスフィン、トリアリールホスフィン、トリエチルアミンのようなトリアルキルアミン、アルキルアリールスルフィド、チオ硫酸塩、又はジメチルスルフィドのようなジアルキルスルフィドでよい。還元剤としてトリブチルホスフィンを用いて処理する場合、その反応は用量調節反応であることは当業者には知られている。
【0074】
本発明のまた別の実施形態によると、シクロスポリンAのβアルコールをトリメチルシリル(TMS)基で保護し、酸化剤としてのオゾンで酸化し、その後還元剤で処理してTMSシクロスポリンAアルデヒドを形成する。オゾン分解工程は、約−80℃ないし0℃の温度範囲で行われる。オゾン分解中に使われる溶媒は、低級アルコールとジクロロメタンとの混液でよい。還元剤は、トリブチルホスフィンなどのトリアルキルホスフィン類、トリアリールホスフィン類、トリエチルアミンなどのトリアルキルアミン類、アルキルアリールスルフィド、チオ硫酸塩類又はジメチルスルフィドなどのジアルキルスルフィド類からなる群から選択される。還元剤としてのトリブチルホスフィンで処理する場合、その反応が用量調節されていることは当業者には知られている。
【0075】
付け加えると、シクロスポリンAアルデヒドは、シクロスポリンAのβ−アルコールをアセチルシクロスポリンAの形成によって保護し、その後そのアセチルシクロスポリンAを、アセトキシアセトン又はジアセトキシアセトンなどのケトンの存在下で、モノペル硫酸塩、好ましくはオキソンでアセチルシクロスポリンAエポキシドに変換する。この工程は、それらの反応条件下では不活性である有機溶媒、例えばアセトニトリル中で、又は水中で行われる。エチレンジアミン四酢酸二ナトリウム塩を加えて、存在する可能性のある重金属イオンを全て捕捉する。エポキシ化反応は、7より大きいpHで行うのが好ましい。このエポキシ化反応の後に、エポキシドを酸性条件下で過ヨウ素酸又は過ヨウ素酸塩によって酸化分解する。任意に酸化及び酸化分解を一つの処理操作に組み合わせてもよい。これらの反応については、ダンヤングらが「非官能化オレフィンを触媒的非対称性エポキシ化するためのC2対称性キラルケトン」J. Am. Chem.Soc., Vol.118, p.491-492(1996)及び「触媒的酸化反応のための新規の環状ケトン」J. Org. Chem., Vol.63,p.9888-9894(1998)において論じている。
【0076】
ルテニウム−ベースの酸化剤の使用が、カールセンらの「四酸化ルテニウム触媒による有機化合物の酸化法の顕著な改良」J. Org. Chem., Vol.46, No.19, p.3736-3738 (1981)で述べられている。カールセンらは、歴史的に、ルテニウム金属の費用が触媒法の発達の動機となったことを教示しており、最も人気のある触媒法は、化学量論的酸化剤として過ヨウ素酸塩か次亜塩素酸塩を使用している。これらの研究者はルテニウムの一般的使用では反応経過中に触媒活性が喪失することを見いだし、それはカルボン酸の存在によると仮定した。反応混合物にニトリル、特にアセトニトリルを加えると、CCl4/H2O/IO4−系においてアルケン類の酸化分解の速度及び程度が顕著に高まることが判明した。
【0077】
本発明の一実施形態によると、アセチルシクロスポリンAアルデヒド51は、次の手順でアセチルシクロスポリンA35から生成する;すなわち、アセチルシクロスポリンA35をアセトニトリルと水との混液に溶解し、それから最初に過ヨウ素酸ナトリウムを加え、次に塩化ルテニウム水加物を加える。アルデヒド51は酢酸エチルで抽出される。この酸化分解戦略によるアルデヒド51の合成は、以下に論ずる多くの立体選択的経路にとって重要であることに留意すべきであり、したがって読者はこの章を読み返して参考にすべきである。
【0078】
さらに、シクロスポリンAアルデヒドは、シクロスポリンAのβ−アルコールをアセチルシクロスポリンAの形成によって保護し、その後そのアセチルシクロスポリンAを、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、モノペル硫酸塩、より好ましくはオキソンで、アセチルシクロスポリンAエポキシドに変換する。この工程は、これらの反応条件下では不活性である有機溶媒、例えばアセトニトリル、又は水中で行われる。エチレンジアミン四酢酸二ナトリウム塩を加え、存在する可能性のある重金属イオンを全て捕捉する。エポキシ化反応は、7より大きいpHで行うのが好ましい。このエポキシ化反応の後に、エポキシドを酸性条件下で過ヨウ素酸又は過ヨウ素酸塩によって酸化分解する。酸化及び酸化分解は、一つの処理操作に組み合わせることもできる。これらの反応については、ダンヤングらが「非官能化オレフィンを触媒的非対称性エポキシ化のためのC2対称性キラルケトン」, J. Am. Chem.Soc., Vol.118, p.491-492 (1996)及び「触媒的酸化反応のための新規の環状ケトン」,J. Org. Chem., Vol,63,p.9888-9894 (1998)において論じている。
【0079】
方法2の第三工程は、方法1のそれと同様に、アルデヒド51をウィッティヒ反応によって(E)及び(Z)ジエンの混合物に変換することを含む。方法1と同様に、リンイリドをアルデヒドに加えるとベタインを生じ(これは分離しない)、その結果、アルデヒドのカルボニル酸素原子が、元々はリンに結合していたR2C=基によって置換される。ここでも、このようなウィッティヒ反応は、トリフェニルホスホニウム誘導体以外のリン含有化合物、例えばトリアリールホスフィン、トリアルキルホスフィン、アリールアルキルホスフィン及びトリアリールアルシンなどで、種々の温度で行うことができる。また種々の塩基性溶液及び溶媒の使用又は種々の無機塩の添加によって、新たに形成される二重結合の立体化学に影響を与えることもできる。
【0080】
一実施形態において、アセチルシクロスポリンAアルデヒド51をトルエンに溶解し、それに水酸化ナトリウム水溶液のような塩基を加える。それからアリルトリフェニルホスホニウムブロミド52を加え、反応物をしばらく撹拌する。生成したアセチル(E)及び(Z)−1,3−ジエン混合物53の処理は、ヘキサン及び/又は酢酸エチルによる抽出を含む;ここで、用語「処理(workup)」は、反応体、生成物、溶媒などの混合物から反応生成物を抽出及び/又は分離するプロセスを意味する。
【0081】
方法2の最終工程において、方法1の最終工程と同様に、β−炭素位置のアルコールを保護する酢酸エステル基を炭酸カリウムで除去すると、ISATX247の(E)及び(Z)−異性体混合物54が得られる。保護基の除去に使用できる炭酸カリウム以外の塩基としては、水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド、及びカリウムアルコキシドなどがある。
【0082】
ISATX247(E)及び(Z)−異性体のいずれかに富む組成物の有機金属経路による合成
本発明の実施形態によると、立体選択的合成経路は、珪素、ホウ素、チタン、硫黄、リン、及び/又はリチウムなどの元素類を含む無機試薬を使用することができる。これらの経路は六員環遷移状態を通過し、その際その六員環のメンバーの一つは、有機金属試薬からの無機元素である。幾つかの実施形態において、この遷移状態に関係する立体障害効果が反応の立体化学的結果に影響を与え得る。
【0083】
この開示において、2つの例示的立体選択的スキームが議論される。第一の立体選択的スキーム(方法3;図3の経路32としても示される)において、珪素含有化合物は脱離反応を受け、その脱離反応が酸性又は塩基性のいずれの条件において行われたかによって、(E)又は(Z)−異性体のいずれかを生成する。これはピーターソンのオレフィン化の一例である。第二の立体選択的スキーム(方法4;図3の経路33としても示される)において、各異性体は異なる前駆体から生成する。(Z)−異性体はチタン及びリン含有中間体から生成されるが、(E)−異性体はリチウム含有中間体を経て生成される。
【0084】
方法3
この経路は、アセチルシクロスポリンAアルデヒド51を経て進む。
同様な反応スキームは、ツァイ及びマッテソン著「ピナコール(E)−1−トリメチルシリル−1−プロペン−3−ボロネートからの(Z)及び(E)末端ジエン類の立体コントロールド合成」(Tetrahedron Letters, Vol.22, No.29, p.2751-2752(1981))において大体述べられている。この方法は、図6に示される。一般に、この合成は、トリメチルシリルアリルボロネートエステル試薬62をつくり、その後アセチルシクロスポリンAアルデヒド51を62で処理してβ−トリメチルシリルアルコール64を形成することを含む。このアルコールは、ホウ素含有遷移状態63を介して形成されると考えられている。ボロネートエステル類は、アリル硼酸化(allylboration)反応では緩徐に反応するので、E−γ-トリメチルシリルジエチルボラン又は9−(E−γ−トリメチルシリルアリル)−9−BBNなどの迅速反応性ボラン試薬の使用が有利であることは当業者には明らかである。β−トリメチルシリルアルコール64は、その後ピーターソンのオレフィン化を受けてアルケン、ここではジエン65かジエン67のいずれかをつくる。
【0085】
アルケンの形成は、脱離反応(オレフィン化)が酸性条件で行われるか塩基性条件で行われるかによって、異なる2経路のどちらかの経路を辿る。酸性条件下では、アンチ脱離が起こり(E)−異性体を形成し、塩基性条件下ではシス−脱離が起こり、(Z)−異性体が形成される。この合成経路の利用によって、同じ前駆体からどちらの異性体もつくることができることは当業者には明らかである。各脱離反応の生成物は、2異性体のどちらかの異性体に富む組成物を含む。一実施形態において、富む(又は富化)とはその組成物が約75重量パーセント以上の異性体を含むことを意味する。その他の実施形態において、富化組成物は1つの異性体を80、85及び90重量パーセント含むことができる。その後1異性体に富む組成物類を所定の比で組み合わせて、図10に示すような所望混合物を得ることができる。
【0086】
次に、図6の反応を、ホウ素含有試薬62の製法から始めて詳細に説明する。炭素−炭素結合形成反応体の合成における珪素試薬の使用に関する一般的研究は、エーリンガ及びマグナス著「合成における珪素。10.(トリメチルシリル)アリルアニオン:アルデヒド及びケトンをγ−ラクトンに変換するためのβ−アシルアニオン同等物」(J. Am. Chem. Soc., Vol.102, No.15, p.5004-5011 (1980))において論じられている。特に、これらの研究者は、(トリメチルシリル)アリルアニオンとアルデヒドとの反応について述べている。アニオンは、1当量のテトラメチルエチレンジアミン(TMEDA)を含む−76℃のテトラヒドロフラン中で、アリルトリメチルシランをsec−ブチルリチウムで脱プロトン化することによってつくることができる。
【0087】
アリルトリメチルシランの脱プロトン化(この工程は、図6には示されていない)は、ビールマン及びドュセプの「ヘテロ原子によって置換されたアリルカルボアニオン及びベンジルカルボアニオン」(Organic Reactions、第27巻(ウィリー、ニューヨーク、1982)、p.9)で述べられている。置換されたアリル系において、ヘテロ原子に対してアルファの位置にあるプロトンは、より塩基性の強い作用物質で除去される。種々のこのような作用物質が使用できるが、n−ブチルリチウムが多分最も一般的である。化学量のn−ブチルリチウムを、メタレーションをする予定の化合物と共に、テトラヒドロフラン(THF)を含む溶液中で使用する。温度は、通常0℃未満に維持する(−76℃未満の場合もよくある);その際n−ブチルリチウムは、その重合性のために低い反応性を有する。N,N,N',N'−テトラメチルエチレンジアミン(TMEDA)などのキレート剤を添加すると、その重合体は解離する。しかし、この反応は室温でも、TMEDAが存在しなくても起こり得る。
【0088】
アリルシランは、容易に脱プロトン化される。なぜならば、生成するアニオンは、隣接二重結合との共役によって安定化するだけでなく、近くのシリル基によっても安定化するからである。このアニオンは、求電子剤と、そのα炭素又はそのγ炭素を介して反応する。これらの反応の位置化学的及び立体化学的結末は幾つかの要因に依存するが、その最も重要な要因の一つは対イオンのアイデンティティである。トーマスの「有機合成:ホウ素及びシリコーンの役割」((オックスフォード大学プレス、ニューヨーク、1991)、p.84-87)によるアリルシラン類に関する議論を参照されたい。
【0089】
この反応スキームにおいて、脱プロトン化アリルシランはその後トリメチルボレートによる求電子捕獲を受け、中間体を生成する。この中間体はピナコールと反応すると、トランス−(トリメチルシリル)ボロネート化合物62を与える。ボロネート62は、「アリルボラン」(アリルボロネートエステル)とも呼ばれる。或いは、求電子捕獲に9−メトキシ−9−ジアルキルボランを使用するならば、ボロネート錯化合物が生成する。それを三フッ化ホウ素試薬(BF3Et2Oなど)によって脱メトキシル化すると、対応する9−(γ−トランス−トリメチルシリルアリル)−9−ジアルキルボランを生成することができる。
【0090】
アリルボランへのアルデヒドの付加について、S.E.トーマスが上記文献の34−35頁に述べている。アリルボランへのアルデヒド付加(この際アリルボランは、炭素−炭素二重結合の遠位端が非対称的に置換されている(「遠位(distal)」は、ホウ素原子から最も遠く離れていることを意味する))は、隣接する2つのキラル中心を含むホモアリルアルコールを生成する。(E)−アリルボラン類は、スレオ−ジアステレオ異性体を生じ、一方(Z)−アリルボラン類は、エリスロ−ジアステレオ異性体を生ずる。(E)−アリルボラン62とシクロスポリンAアルデヒド51との典型的反応が、図6に示される。ここでは、反応体をTHF溶液中で数日間撹拌した後、ホウ素中間体63が形成される。
【0091】
ホウ素中間体63(図6)中の参照番号69は、このホウ素位置でかなり多数の構造が可能であることを示すものである。例えば、ボロネート試薬62がトリアルキルシリルアリルボロネートエステルである場合、69の構造は、2個の酸素原子を含む五員環を構成する。62に使用するボロネート又はボラン試薬上の置換基が、63の構造中に存在する。
【0092】
アリルボランとアルデヒドを含む反応において得られる立体選択性は、ホウ素中間体63によって例示され、図6に描かれるような六員環いす形遷移状態によるものであると仮定されている。アルデヒドの2個のカルボニル原子(二重結合している炭素と酸素)だけが、六員環遷移のメンバーとなる;アルデヒドの残りは環を離れて延びる。六員環から離れて延びるアルデヒドのCsA部分は、その環に対してアキシアル配座というよりむしろエクアトリアル配座で存在すると仮定される。なぜならば、アキシアル配座はその置換基とアリルボラン62の酸素原子との間に好ましくない立体障害をおこすからである。図6において、(トリメチルシリル)アリルアニオンからのSiMe3基の位置がエクアトリアル位置を占めるように示されていることは当業者には明らかである。なぜならば、この例はアリルボランの(E)−ジアステレオマーから出発したからである。これとは異なり、開始アリルボランが(Z)−ジアステレオマーである場合は、SiMe3基はアキシアル位置に描かれる。
【0093】
或いは、上述の脱離反応とは反対に、酸脱離がシス−異性体を与え、塩基脱離がトランス−異性体を与えるようなエリスロ−シリルアルコールを形成することが考えられる。合成の最後には同じ生成物が得られることは、当業者にとって当然である。
【0094】
遷移状態の生成物63をトリエタノールアミンで処理すると、β−トリメチルシリルアルコール64が得られる。他方、(トリメチルシリルアリル)ジアルキルボランのアリルボレーション生成物は、NaOH/H2O2を使用する酸化又は水性処理でシリルアルコール64を与える。図6に描かれているアルコール64は、遷移状態のアリルボラン63が(E)−配置であったため、スレオ−ジアステレオ異性体である。ただし、Z−アリルボラン試薬から出発する場合は、その他のジアステレオ異性体がつくられることは当業者には明らかである。新しく作られたキラル中心におけるジアステレオ選択性は、この工程では確認されない;それは、この後の合成工程で、これらのキラル中心は脱離するからである。図6に示されるβ−トリメチルシリルアルコール64の構造は、出願人が分光法を用いて確認した。
【0095】
ピーターソンのオレフィン化として知られるアルケン合成法において、β−トリメチルシリルアルコール64からトリアルキルシリル基及びヒドロキシ基が除去され、アルケンが生成する;ここでは、鎖の2つの末端炭素の間にすでに二重結合が存在するため、ジエンが生成する。β−ヒドロキシシランのアルケンへの変換に関する議論が、S.E.トーマスの参考文献の68-69頁に記されている。この反応のより詳細な議論が、ハードリク及びピーターソンの「β−ヒドロキシシラン類の立体特異的オレフィン形成性脱離反応」J.Am. Chem. Soc.,Vol.97,No.6, p.1464-1468(1975)に記されている。
【0096】
図6を参照すると、アルコール64をジエンに変える脱離反応は、その反応が酸性条件で行われるか塩基性条件で行われるかによって、2つの異なるメカニズム経路を辿る。一方の経路はジエン65に導き、他方の経路はジエン67に導く。酸性条件下ではアンチ脱離が起き、一方塩基性条件下ではシン脱離が起きる。言い換えれば、β−ヒドロキシシランの脱離反応は立体特異的であり、酸−及び塩基−促進反応が反対の立体化学的コースをとる。酸促進反応のための典型的酸は、酢酸、硫酸及び種々のルイス酸などであり;典型的塩基は、水素化ナトリウム及び水素化カリウム又はカリウムtert−ブトキシドなどである。THF中水素化ナトリウムを使用する脱離反応は室温では緩徐で、水素化カリウムを使用する脱離反応はより速やかにあらわれるという場合もある。
【0097】
酸性条件下の脱離では、トリメチルシリル及びヒドロキシ基がアンチペリプラナー関係にあることが必要であるので、反応経路のこの工程で立体特異性が生ずる。反対に、塩基性条件下の脱離ではトリメチルシリル及びヒドロキシ基がシンペリプラナー関係にあることが必要である。後者の条件は、強い珪素−酸素結合及び中間体である四員環の形成を容易にする。この四員環は、ウィッティヒ反応の最終工程と同様の仕方で分解する。強い珪素−酸素結合はより弱い珪素−炭素結合にとって代わり、それはより弱い炭素−炭素π結合による強い炭素−酸素結合の置換を上回ることは当業者には明らかである。
【0098】
このように、β−ヒドロキシアルキルシランの立体特異的脱離の生成物は、アセチル−(E)−1,3−ジエン化合物67及びアセチル−(Z)−1,3−ジエン化合物65である。これまでに述べた方法のように、保護基は今やメタノール及び水中K2CO3による処理によって、これらのジエンの各々から除去される。これは1−アミノ酸残基のβ−炭素に結合した酢酸基を除去し、その炭素に結合する官能基をアルコールに戻す。保護基を除去するために使用できる炭酸カリウム以外の塩基類には、水酸化ナトリウム、炭酸ナトリウム、ナトリウムアルコキシド、及びカリウムアルコキシドなどがある。
【0099】
この製造工程によって、合成は実質的に完了する。2異性体のうちのどちらか1つに富む組成物を混合し、混合物中の異性体類の所望比を得ることができる。「富む」とは、その異性体を少なくとも約75重量パーセント含む生成物を意味する;言い換えれば、その生成物が25重量パーセント以下の「望まない」異性体を含むことを意味する。望ましい薬理学的結果が得られるように混合物を設計する。
【0100】
方法4
この経路も、アセチルシクロスポリンAアルデヒド51を経て進む。
立体選択的異性体を生成するためのまた別のスキームを、図7−8に示す。この合成経路がこれまでに述べた経路と異なる点は、1)ISATX247の(E)−異性体を生成する合成経路が、(Z)−異性体のための中間体と異なる中間体を介して進むこと、及び2)これらの合成経路が、チタン及びリチウム含有試薬及び/又は中間体を使用することである。
【0101】
チタン試薬が有機合成では特に有用であることは知られている。なぜならば、それらはアルデヒド及びケトンとの反応において、位置−及び立体選択的であるからである。立体選択的化学におけるチタンの一般的性質は、リーツが「有機合成における有機チタン試薬」((スプリンガー出版、ベルリン、1986)、p.VII, 148-149及び164-165)で述べている。チタン試薬の電子的及び立体的アイデンティティが巧みに操作できるように、チタンリガンドの性質を変えることができ、多くのC−C結合形成反応の立体化学的結果が予測できると、ここには記されている。この化学的性質により、アキラル分子の2つのプロキラル中心の合体が、キラリティーの2つの中心を造り出す。立体選択的結果を決定する一般的ルールは、Z−配置のエノラート又はクロチル金属化合物が優先的にシン付加物を形成し、E−配置試薬はアンチ−ジアステレオマーを好んで形成することである。これらの傾向は、ここでもいす形を有する六員環状遷移状態を想定することによって説明できる。
【0102】
このタイプの立体選択的合成の特殊な例は、イケダらの「有機チタン及びリチウム試薬を使用した、アルデヒドから(Z)−及び(E)−1,3−アルカジエン類の立体選択的合成」(Tetrahedron, Vol.43, No,4,p.723-730(1987))に記載されている。この参考文献は、アリルジフェニルホスフィンを使用して[3−(ジフェニルホスフィノ)アリル]チタン試薬を生成し、この生成した試薬はその後アルデヒドと縮合し、その後ホスホニウム塩の形成によって、高度に位置選択的及び立体選択的な仕方で(Z)−1,3−アルカジエンを与えることを開示している。これに対して、リチウム化アリルジフェニルホスフィンオキシドはアルデヒドと縮合し、ここでも所望の立体選択性をもって直接(E)−1,3−アルカジエンを生成する。
【0103】
図7を参照すると、ISATX247の(Z)−異性体の合成は、(これまでのスキームと同様に)シクロスポリンA34からアセチルシクロスポリンAアルデヒド51を形成することから始まる。アリルジフェニルホスフィン71をt−BuLiのような強塩基で脱プロトン化し、その後生成物をチタンテトライソプロポキシドと反応させることによって、[3−(ジフェニルホスフィノ)アリル]チタン試薬72をつくる。遷移状態73は、理論的にはエリスロ−α−付加物74に移行すると考えられる。それは、その後74をヨードメタン(MeI)で処理することによって、β−オキシドホスホニウム塩75に変換できる。遷移状態73の存在が、少なくとも一部はこの合成経路の立体選択性をもたらすと考えられる。
【0104】
本明細書に概略した典型的方法によると、有機金属試薬の金属部位は、位置選択性をコントロールする存在でありうる(イケダ、p.725)。これは、図7のアルデヒド51がジフェニルホスフィノ化合物72とそのα位置で反応して、対応するα−付加物74を与えることを意味する。なぜならば、ジフェニルホスフィノ基のγ−炭素は、その金属(この場合はチタン)に配位しているからである。観察されるジエン生成物のZ選択性は、六員環遷移状態73を考慮することによって説明される。アルデヒド35のかさばったシクロスポリンA側鎖もジフェニルホスフィノ基も両方とも、遷移状態ではエクアトリアル位置を占めると考えられるので、エリスロα−付加物74が選択的に形成され、(Z)−1,3−ジエン76を生成する。
【0105】
ISATX247の(Z)−異性体がチタン遷移状態を経て形成される図7に描かれる反応経路とは異なり、(E)−異性体はこの方法では容易には生成しない。実際、この方法によって(E)−異性体を合成する試みは、概して低収率に終わることが報告されている。その代わり、図8に示すように、リチオ誘導体82は、アルデヒド51と反応してリチウム含有遷移状態83をつくり、それは約75:25より大きい範囲のE/Z比で1,3−ジエンを形成する。図7におけるように、反応生成物の高い立体選択性は、おそらく遷移状態83によるものである。この状態では、リチウム試薬82のビニル基及びアルデヒド51のシクロスポリンA側鎖はエクアトリアル位置を占め、それによって(E)−1,3−ジエン84を立体選択的に生成すると仮定される。前述のように、酢酸保護基に関する幾つかの不都合な副反応は、全ての立体選択的合成において、安息香酸エステル又はシリルエーテルなどの保護基の使用によって回避される。
【0106】
混合物の調製
前述のように、ISATX247のシス及びトランス−異性体のある種の混合物は、天然及び現在公知のシクロスポリンに勝る高い効力及び/又は低い毒性の組合わせを示すことが判明した。
【0107】
本発明の実施形態によると、ISATX247異性体(及びこれらの誘導体)は、立体選択性の程度が種々異なる立体選択的経路によって合成される。立体選択的経路は、(E)−異性体に富む第一の材料又は組成物、及び(Z)−異性体に富む第二の材料又は組成物を生成し、これらの材料を組み合わせて、生成した混合物が2異性体を所望比で有するようにすることができる。或いは、反応生成物を分離して(E)−異性体を単離し、富化することによって第一材料をつくり、反応生成物を分離して(Z)−異性体を単離し、富化することによって第二材料をつくることが考えられる。また別の実施形態においては、立体選択的経路の反応条件を調節して、生成混合物に直接所望比をもたらす。
【0108】
これらの原理は、図9A−C及び図10に説明される。図9A−Cには、(E)対(Z)−異性体の比がそれぞれ約65対35重量パーセント、50対50重量パーセント、及び35対65重量パーセントである仮定の合成反応が示される。もちろんこれらの比は例示的なものであり、説明の目的のために過ぎず、仮定のあらゆる数の組合わせが選択できる。生成混合物中の異なる異性体比を実現するために、図9Aの比を形成するために適用する反応条件は図9B及び図9Cのそれらと異なってよい、ということは当業者には当然である。各反応の条件を調節し、その場合に応じた2異性体の特定の比が形成される。
【0109】
異性体の混合物を生成する幾つかの合成経路とは異なり、異性体をまず最初に個々につくり、所定の比率で混合して所望比を得ることもできる。この概念は、図10に示される。ここでは、一立体選択的経路の生成物はこれら異性体のうちの1異性体に富み、生成物が約75重量パーセントより多い(E)異性体を含み、他の立体選択的経路の生成物は他の異性体に富み、この生成物は約75重量パーセントより多い(Z)異性体を含む。これらの数字も例示的であり、一実施形態において、立体選択的経路によって生成した所望異性体の純度は、約75重量パーセント以上であり得る。他の実施形態において、所望異性体はそれぞれ約80、85、90及び95重量パーセント以上を含みうる。
【0110】
これらの異性体を個々に合成した後、それらを混合して図10に示すように所望比を得ることができる。例示目的のために、図10では図9A−Cで用いた比率と同じ仮定的比率を選択する。図10を参照すると、(E)及び(Z)−異性体を混合して、それぞれ約65対35重量パーセント、50対50重量パーセント、及び35対65重量パーセントの(E)対(Z)−異性体比を有する3種類の混合物を得る。
【0111】
また別の実施形態において、ISATX247異性体の(E)及び(Z)−異性体の混合物を分離し、混合物中において、ある異性体が他の異性体より多いようにする。例えば、ディールス−アルダー反応を利用して、シス異性体を、アルケンとの反応により閉鎖環化合物に変換する。もしアルケンが単離可能の(例えば濾過可能の)基質に結合するならば、シス異性体はその混合物から実質的に除去され、トランス異性体に富む組成物が残る。シス異性体は閉鎖環化合物から加熱によって再構成され、シス異性体に富む組成物を生成する。このやり方で、シス及びトランス異性体が分離できる。
【0112】
実際、混合物を生成する方法の立体選択性の程度には無関係に、いかなる混合物中でも(E)対(Z)−異性体の比は広範囲の数値をとる。例えば、混合物は約10ないし90パーセントの(E)−異性体から約90ないし10パーセントの(Z)−異性体までを含む。その他の実施形態において、混合物は約15ないし85重量パーセントの(E)−異性体及び約85ないし15パーセントの(Z)−異性体;又は約25ないし75重量パーセントの(E)−異性体及び約75ないし25重量パーセントの(Z)−異性体;又は約35ないし65重量パーセントの(E)−異性体及び約65ないし35重量パーセントの(Z)−異性体;又は約45ないし55重量パーセントの(E)−異性体及び約55ないし45重量パーセントの(Z)−異性体を含む。また別の実施形態において、異性体混合物は、約45ないし50重量パーセントの(E)−異性体及び約50ないし55重量パーセントの(Z)−異性体を含むISATX247混合物である。これらの重量パーセントは組成物の総重量を基準にしており、(E)異性体及び(Z)異性体の重量パーセントの合計が100重量パーセントであることは当然である。言い換えれば、混合物は65重量パーセントの(E)−異性体及び35重量パーセントの(Z)−異性体を含むことがあり、その逆でもよい。
【0113】
混合物中のある異性体又はその他の異性体のパーセンテージは、核磁気共鳴(NMR)又は当該技術で公知のその他の方法を使用して確認できる。
【0114】
医薬組成物
本発明は、活性成分として本発明の混合物を含む医薬組成物の投与を含む、免疫抑制を必要とする患者の治療法にも関係する。この組合わせが対象とする適応症は、特に自己免疫病及び炎症性疾患、及び移植拒絶に関係するか、これに起因する異常などであり、例えば下記のような異常の治療(病因又は症状の改善、軽減、排除又は治癒を含む)又は防止(実質的又は完全な制限、予防又は回避を含む)を含む:
【0115】
a)急性臓器又は組織移植拒絶、例えば心臓、肺、心−肺同時、肝臓、腎臓、膵臓、皮膚、腸又は角膜移植片などのレシピエントの治療、特に、T細胞性拒絶、並びに例えば骨髄移植後のグラフト対ホスト疾患の予防及び/又は治療。
【0116】
b)移植臓器の慢性拒絶、特にグラフト血管疾患、例えば平滑筋細胞増殖による内膜肥厚や関連効果の結果としてのグラフト血管狭窄を特徴とするグラフト血管疾患の予防。
【0117】
c)臓器ドナーがレシピエントと異なる種である場合に起きる、臓器の急性、超急性、又は慢性拒絶、非常に特別なB細胞性拒絶又は抗体仲介性拒絶を含む、異種移植片拒絶。
【0118】
d)自己免疫病及び炎症性疾患、特に免疫学的又は自己免疫要素を含む病因を有する炎症性疾患、例えば関節炎(例えばリウマチ性関節炎、慢性進行性関節炎及び変形性関節炎)及びその他のリウマチ性疾患など。本発明の共力的組合わせを適用できる特異的自己免疫病は、自己免疫性血液疾患(溶血性貧血、再生不良性貧血、赤芽球ろう、特発性血小板減少症など)、全身性エリテマトーデス、多発性軟骨炎、強皮症、ウェーゲナー肉芽腫症、皮膚筋炎、慢性活性肝炎、重症筋無力症、乾癬、スティーヴン−ジョンソン症候群、特発性スプルー、(自己免疫性)炎症性腸疾患(例えば潰瘍性大腸炎及びクローン病を含む)、内分泌性眼障害、グレーブス病、サルコイドーシス、多発性硬化症、原発性胆汁性肝硬変、若年性糖尿病(I型糖尿病)、ぶどう膜炎(前部及び後部)、乾性角結膜炎及び春季角結膜炎、間質性肺線維症、乾癬性関節炎、糸球体腎炎(特発性ネフローゼ症候群又はリポイドネフローゼなどのネフローゼ症候群を伴う又は伴わない)、及び若年性皮膚筋炎を含む。皮膚の自己免疫病及び炎症性疾患も本発明の共力的組合わせを適用して治療及び予防できると考えられる;例えば乾癬、接触性皮膚炎、アトピー性皮膚炎、円形脱毛症、多形性紅斑、疱疹状皮膚炎、硬皮症、白斑、過敏性血管炎、蕁麻疹、水泡性類天疱瘡、エリテマトーデス、天疱瘡、後天性表皮水疱症、その他の炎症性又はアレルギー性皮膚障害などである;喘息、アレルギー及びじん肺症などの肺及び気道の炎症性異常も含まれる。
【0119】
本発明の異性体同族混合物はそれだけで、又は薬物学的担体と共に、それらを必要とする温血動物に投与される。薬物学的担体は固体でも液体でもよい。本発明の混合物は従来の毒性のない薬物学的に容認される担体、アジュバント及びビヒクルを含む投与単位の処方で、経口的、局所的、非経口的に、又は吸入スプレーによって、又は直腸内に投与される。ここに使用する用語「非経口的」とは、皮下注射、静脈内、筋肉内、胸骨内注射又は注入法を含む。
【0120】
本発明の混合物を含む医薬組成物は、例えば錠剤、トローチ、ロゼンジ、水性又は油性懸濁液、分散性粉末又は顆粒、乳濁液、硬又は軟カプセル、又はシロップ又はエリキシルのような経口的使用に適した形であるのが好ましい。経口使用のための組成物は、医薬組成物を製造する当該技術で公知の方法によってつくられ、そのような組成物は甘味料、香料、着色料及び保存料からなる群から選択される1種類以上の材料を含み、医薬品として洗練され、口当たりのよい製剤が提供される。活性成分を無毒性の薬物学的に容認される賦形剤と共に含む錠剤も、公知の方法によって製造される。使用する賦形剤は例えば、(1)炭酸カルシウム、乳糖、燐酸カルシウム又は燐酸ナトリウムなどの不活性希釈剤;(2)コーンスターチ又はアルギン酸などの顆粒化剤及び崩壊剤;(3)澱粉、ゼラチン又はアカシアなどの結合剤;及び(4)ステアリン酸マグネシウム、ステアリン酸又はタルクなどの滑剤である。これら錠剤は、コーティングしなくてもよいが、公知の方法によってコーティングし、消化管における崩壊及び吸収を遅らせ、それによって長時間にわたって持続的作用を提供することもできる。例えば、グリセリルモノステアレート又はグリセリルジステアレートなどの徐放性材料を使用してもよい。それらは、米国特許第4,256,108号;第4,160,452号;及び第4,265,874号に記載されている方法によってコーティングされ、浸透性治療錠を形成し、放出をコントロールする。
【0121】
ある場合には、経口使用のための処方は、ハードゼラチンカプセルの形で、その中に活性成分が炭酸カルシウム、燐酸カルシウム又はカオリンなどの不活性固体希釈剤と混合して含まれる。それらは、ソフトゼラチンカプセルの形でもよく、その中に活性成分が水、又はピーナツ油、液体パラフィン、又はオリーブ油などの油性メジウムと混合して含まれる。
【0122】
水性懸濁液は、活性成分を水性懸濁液の製造に適した賦形剤と混合して含むのが普通である。このような賦形剤としては:(1)カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガントガム及びアカシアゴムなどの懸濁剤;又は(2)分散剤又は湿潤剤、例えばレシチンなどの天然ホスファチド、ポリオキシエチレンステアレートなどのアルキレンオキシドと脂肪酸との縮合産物、ヘプタデカエチレンオキシセタノールなどのエチレンオキシドと長鎖脂肪アルコールとの縮合産物、ポリオキシエチレンソルビトールモノオレエートのような、脂肪酸及びヘキシトールから誘導される部分エステルとエチレンオキシドとの縮合産物、又はポリオキシエチレンソルビタン モノオレエートのような、脂肪酸及びヘキシトール無水物から誘導される部分エステルとエチレンオキシドとの縮合産物がある。
【0123】
水性懸濁液は、1種類以上の保存料、例えばエチル又はn−プロピルp−ヒドロキシベンゾエート;1種類以上の着色料;1種類以上の香料;及び1種類以上の甘味料、例えばスクロース、アスパルターム又はサッカリンを含むことができる。
【0124】
油性懸濁液は、活性成分をアラキス油、オリーブ油、ゴマ油又はココナツ油などの植物油、オメガ3脂肪酸を含む魚油、又は液体パラフィンなどの鉱油に懸濁させることによって処方される。油性懸濁液は、蜜蝋、ハードパラフィン又はセチルアルコールなどの濃化剤を含むことができる。甘味料及び香料を加えて、口当たりのよい経口製剤を提供することができる。これらの組成物は、アスコルビン酸などの抗酸化剤を加えることによって保存できる。
【0125】
分散性粉末及び顆粒は、水性懸濁液の調製に適する。それらは、分散剤又は湿潤剤、懸濁剤及び1種類以上の保存料と混合した活性成分を提供する。適切な分散剤又は湿潤剤及び懸濁剤の例はすでに上述されている。その他の賦形剤、例えば上記の甘味料、香料及び着色料も含むことができる。
【0126】
本発明の混合物を含む医薬組成物は、水中油型エマルションの形でもよい。油相は、オリーブ油又はアラキス油のような植物油、又は液体パラフィンのような鉱油又はそれらの混合物でよい。適切な乳化剤は、(1)アカシアゴム又はトラガントゴムのような天然のゴム類、(2)大豆油及びレシチンなどの天然のホスファチド類、(3)脂肪酸及びヘキシトール無水物から誘導されるエステル類又は部分エステル30、例えばソルビタンモノオレエートなど、(4)前記部分エステル類とエチレンオキシドとの縮合産物、例えばポリオキシエチレンソルビタンモノオレエート。乳化剤は、甘味料及び香料も含むことができる。
【0127】
シロップ類及びエリキシル類は、グリセロール、プロピレングリコール、ソルビタン、アスパルテーム又はスクロースなどの甘味料と共に処方される。このような処方は、緩和剤、保護剤、保存料及び香料及び着色剤も含むことができる。
【0128】
本発明の医薬組成物は、無菌の注射可能水溶液又は油性懸濁液の形でもよい。この懸濁液は、上述した適切な分散剤又は湿潤剤及び懸濁剤を使用して、公知の方法によって処方される。無菌の注射可能の製剤は、無毒性の非経口的投与可能の希釈液又は溶媒を使用した無菌の注射可能溶液又は懸濁液でもよく、例えば1,3−ブタンジオール溶液などである。使用できるビヒクル及び溶媒は水、リンゲル液及び等張塩化ナトリウム溶液である。その上、無菌の固定油が溶媒又は懸濁メジウムとして一般的に使用される。この目的のためには、合成モノ−又はジ−グリセリドを含む、あらゆるブランドの固定油も使用できる。さらに、オレイン酸などの脂肪酸も注射用製剤に使用できる。
【0129】
本発明の混合物は、薬剤の直腸投与用坐薬の形でも投与できる。これらの組成物は、薬剤を、普通の温度では固体であるが直腸温度では液体となり、そのため直腸で融解して薬剤を放出する、適切な非刺激性賦形剤と混合することによってつくられる。このような材料は、ココアバター及びポリエチレングリコール類である。
【0130】
局所的使用のためには、開示されたシクロスポリン類を含むクリーム、軟膏、ゲル、溶液又は懸濁液などが使われる。
【0131】
特に好ましい実施形態において、不活性成分として界面活性剤、エタノール、親油性及び/又は両極親和性溶媒を含む液体溶液を使用する。より詳細に述べれば、異性体類似体混合物と、非薬剤成分:d−アルファトコフェロールポリエチレングリコール1000琥珀酸(ビタミンE TPGS)、中鎖トリグリセリド(MCT)油、ツィーン40、及びエタノールとを含む経口マルチプルエマルション処方が用いられる。異性体類似体混合物と経口溶液の場合と同じ非薬剤成分とを含むソフトゼラチンカプセル(ゼラチン、グリセリン、水及びソルビトールを含む)も好適に使用される。
【0132】
1日に体重1kgあたり約0.05mgないし約50mgの桁の投与量、が上記の疾患の治療に有効である。投与量及び投与スケジュールは、使用する特定の異性体混合物、治療する症状、及び患者の年齢及び症状などの付加的要素によって変動する。好ましい投与量は、約0.5ないし約10mg/kg/日及び約0.1ないし約10mg/kg/日である。好ましい実施形態において、約2ないし約6mg/kg/日が1日2回経口投与される。特に好ましい実施形態においては約0.5ないし約3mg/kg/日を1日2回投与する。
【0133】
単回投与型を形成するために担体材料と組み合わせることができる活性成分量は、治療するホスト及び特定の投与法によって変動する。例えば、ヒトに経口投与するための製剤は、2.5mgないし2.5gの活性成分を適切かつ好都合な量の担体材料と組み合わせて含む。その量は、総組成物の約5ないし約95パーセントまでの間で変動する。単回投与型は、一般的に約5mgないし約500mgの活性成分を含む。好ましい実施形態においては、約50mgの異性体混合物を含む個々のカプセルを経口投与のために用いる。また別の好ましい実施形態においては、約50mg/mLの異性体混合物を含む経口溶液を経口投与のために使用する。
【0134】
しかし、いかなる特定の患者においても、その特異的投与量は使用する特異的化合物、年齢、体重、身体全体の健康、性別、食事、投与時間、投与経路、排泄速度、薬剤組合わせ及び治療を受ける特定の疾患又は異常の性質及び重症度、など種々の要素に左右される。
【0135】
方法論
Tolypocladium inflatum Gams真菌によって産生される環式ポリペプチドの一群であるシクロスポリン誘導体は、T−細胞仲介性反応に対するそれらの好ましい効果のために、免疫治療における使用が増加しつつある。シクロスポリン誘導体は、免疫担当リンパ球、特にT−リンパ球を可逆的に抑制し、リンホカインの産生及び放出を抑制する。この作用は主として、シクロスポリンA−誘起性カルシニューリン阻害によって仲介される。カルシニューリンは、細胞の細胞質に見いだされるホスファターゼ酵素である(シュライバー及びクラブトリー、1992)。シクロスポリンA又はシクロスポリンA誘導体の効果の指標は、カルシニューリンのホスファターゼ活性を阻害する能力である。カルシニューリン阻害アッセイは、その作用部位における薬剤活性を測定する試験法であり、そのアッセイはシクロスポリンA類似体の効力の最も正確で直接的なinvitro評価である(フルマンら、1992)。
【0136】
ISATX247は、分子のアミノ酸1残基上の官能基の新規の修飾を除けば、シクロスポリンAに類似したシクロスポリンA類似体である。我々は今や、invitroカルシニューリン阻害アッセイにおいて、ISATX247がシクロスポリンAの3倍までの効力を示すことを見いだした。
【0137】
薬力学的研究(in vivo及びin vitro)は、ISATX247がその他の存在するシクロスポリン化合物より効力が高いことを示した。シクロスポリン類似体の約10:90ないし約10:90(トランス−対シス−)の異性体混合物、特に50−55%Z−異性体及び45−50%E−異性体を有するISATX247の免疫抑制剤としての効果が(シクロスポリンAに比較して)、invitroカルシニューリン活性アッセイ、ラット心臓移植モデル、膵島細胞同種移植マウスモデル、マウスのコラーゲン誘起性関節炎モデル、及び/又はウサギの抗原誘起性関節炎モデルで証明されている。これらのデータは、これらの異性体混合物の効力が、シクロスポリンAの効力と同等か又はより大きく、したがって免疫調節性疾患の治療に有効であることを示している。
【0138】
腎毒性、肝毒性、白内障発生、多毛、パラセシス(parathesis)、歯肉増殖症などを含む、シクロスポリンA治療に関連する副作用が多数ある(スケトリスら、1995)。これらの中で、腎毒性はシクロスポリンA投与に起因するより深刻な用量関連性副作用の一つである。シクロスポリンAが腎障害をおこす正確なメカニズムは知られていない。しかし、腎臓の血管収縮性物質の濃度の増加が、求心性糸球体細動脈の局所的血管収縮を起こすことが予想される。これは虚血、糸球体濾過率の減少を起こし、長期にわたると間質性線維症を起こす。
【0139】
ISATX247の非臨床的安全性が多数の動物種で評価された。ラット、イヌ及び霊長類における反復経口投与による毒性研究により、ISATX247は耐容性がよく、免疫抑制と一致する効果をもたらすことが判明した。全ての種で認められた唯一の毒性作用は、下痢/軟便であった。
【0140】
ISATX247は、in vitro細菌逆突然変異及び染色体異常アッセイ、及びin vivoラット小核アッセイで証明されたように、突然変異誘発作用を示さない。発癌性の研究は、現在完了していない。ISATX247の生殖毒性試験が、妊娠ラット及びウサギで完了した。治療に関連する先天性異常又は変化はなかった。母体毒性を起こす用量では、相応の胚毒性が認められた。
【実施例】
【0141】
実施例1:シクロスポリンAのアセチル化
無水酢酸(140ミリリットル)をシクロスポリンA(50.0g、41.6mmol)に加え、混合物を室温でN2気流下で、シクロスポリンAの全てが溶解するまで撹拌した。ジメチルアミノピリジン(7.62g、62.4mmol)を加え、反応物を室温でN2気流下で3時間、又は反応が完了するまで撹拌した。反応混合物を5℃に冷やし、その後濾過した。冷却した固体をヘキサンで洗い、付加した無水酢酸を除去した。生成したペースト状固体を激しく撹拌した5%炭酸水素ナトリウム水溶液(1.5リットル)に徐々に移した。生成した懸濁液を、微細スラリーが生成し、CO2の発生が止むまで撹拌した。固体を濾過によって集め、濾液が中性pHを示すまで水で洗った。固体生成物を真空中で一晩乾燥すると(55℃)、生成物が着色固体として44.0g(85%)得られた。
【0142】
実施例2:実施例1からの生成物の酸化
アセトニトリル(320mL)及び水(80mL)をアセチルシクロスポリンA(42.97g、34.54mmol)に加え、材料の全てが溶解するまで混合物を撹拌した。過ヨウ素酸ナトリウム(14.77g、69.08mmol)を加え、その後塩化ルテニウム水加物(0.358g、1.73mmol)を加え、その後反応物を室温でN2気流下で3時間撹拌した。水(300mL)を加え、混合物を分液漏斗に移した。混合物を酢酸エチル(300mL、その後250mL)で2度抽出した。暗黒色の酢酸エチル抽出液を合一し、水250mLで、その後ブライン250mLで洗った。有機溶液をMgSO4上で乾燥し、溶媒を蒸発すると、緑色がかった黒い固体が生成した。その粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(29.1g、68%)が無色固体として得られた。
【0143】
実施例3:アセチルISATX247の製法
i)イリドのin situ生成
アセチルシクロスポリンAアルデヒド(31.84g、25.84mmol)を340mLトルエンに加え、材料が完全に溶解するまで混合物を撹拌した。生成した溶液に1ノルマル水酸化ナトリウム水溶液を340mL加えた。生成した混合物を激しく撹拌し、それからアリルトリフェニルホスホニウムブロミド(58.22g、151.90mmol)を加えた。反応物を室温で24時間撹拌し、追加的アリル トリフェニルホスホニウムブロミド(16.64g、43.42mmol)を加え、さらに24時間撹拌し続けた。混合物を分液漏斗に移し、トルエン相を分離した。水相をさらに、200mLのトルエンで抽出した。2つのトルエン抽出液を合一し、200mL脱イオン水及び200mL塩化ナトリウム飽和水溶液で逐次洗った。その溶液をMgSO4上で乾燥し、濾過し、トルエンを蒸発すると非常に粘稠なゲルが生成した。この物質を142mL酢酸エチルで処理し、微細スラリーが形成されるまで撹拌した。急速撹拌下でヘキサン(570mL)をゆっくりと加えた。撹拌を30分間続け、生成した懸濁液を濾過し、集めた固体を160mLの5:1ヘキサン/酢酸エチルで洗った。合一した濾液を回転蒸発器で濃縮すると粘稠な半固体が得られた。この物質を75mL酢酸エチルで処理し、微細スラリーが得られるまで撹拌した。ヘキサン(225mL)を急速撹拌下でゆっくりと加える。撹拌を30分間続け、それから生成した懸濁液を濾過し、集めた固体を100mLの5:1ヘキサン/酢酸エチルで洗った。濾液を回転蒸発器で濃縮すると、淡黄色固体が得られた。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液として使用してクロマトグラフィーにかけると、生成物(14.09g)が無色固体として得られた。
【0144】
ii)LiBrの存在下におけるプレフォームド イリドの生成及び反応
0℃に冷却したTHF(20mL)中アリルトリフェニルホスホニウムブロミド(7.67g、20mmol)の撹拌懸濁液に、テトラヒドロフラン中KOBut溶液(20mL、20mmol、1M溶液)を加えた。この温度で撹拌を30分間続け、THF中LiBr溶液(10mL、10mmol、1M溶液)を加えた。反応混合物を30分間撹拌し、THF(10mL)中アセチルCsA−アルデヒド溶液(4.93g、4mmol)溶液をカニューレによって加えた。室温で15分間撹拌後、反応混合物に飽和NH4Cl溶液(25mL)を加えて反応を停止した。上記のような処理及びクロマトグラフィーにより、アセチル化ISATX247が無色固体(3.5g)として生成した。
【0145】
実施例4:ISATX247の製法
アセチルISATX247(14.6g、11.62mmol)を340mLメタノールに溶解し、その後脱イオン水135mLを加えた。炭酸カリウム(13.36g、96.66mmol)を加え、混合物を室温で24ないし48時間、反応が完結するまで撹拌した。メタノールの大部分を蒸発し、それから250mL酢酸エチルを撹拌しながら加えた。10%クエン酸水溶液(120mL)をゆっくりと加え、酢酸エチル相を分離した。水相を追加200mL部分の酢酸エチルで抽出した。合一した酢酸エチル抽出液を150mL脱イオン水、100mL10%クエン酸水溶液及び150mL塩化ナトリウム飽和水溶液で逐次洗い、MgSO4上で乾燥した。酢酸エチルを蒸発すると、淡黄色固体が得られた。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液として用いてクロマトグラフィーにかけると、ISATX247(10.51g、75%)が無色固体として得られた。ISATX247は45−50%E−異性体及び50−55%Z−異性体を含む。
実施例1−4の生成物は、質量分析及び/又は核磁気共鳴分光法によって特徴づけられた。
【0146】
実施例5:アセチル−η−ブロモシクロスポリンAの製法
実施例1におけるようにつくられたアセチルシクロスポリンA(41.48g、33.3mmol)、N−ブロモスクシミド(10.39g、58.4mmol)及びアゾ−ビス−イソブチルニトリル(1.09g、6.67mmol)を四塩化炭素250mLに溶解し、生成した混合物を2.5時間加熱還流した。混合物を冷却し、溶媒を蒸発した。残留物を350mLジエチルエーテルで処理し、濾過して不溶性物質を除去した。濾液を150mL水及び150mLブラインで逐次洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗生成物をシリカゲル上でアセトン/ヘキサン(2:3)でクロマトグラフィーにかけるとアセチル−γ−ブロモシクロスポリンA28.57g(65%)が黄色固体として得られた。
【0147】
実施例6:アセチルシクロスポリンAのトリフェニルホスホニウムブロミドの製法
アセチル−γ−ブロモシクロスポリンA(28.55g、21.6mmol)及びトリフェニルホスフィン(7.55g、28.8mmol)をトルエン210mLに溶解し、生成した溶液を21時間、100℃に加熱した。溶液を冷却し、トルエンを蒸発した。生成した油性の半固体に250mLのヘキサン/エーテル(1:4)を加え、徹底的に混合し、溶媒を傾瀉して除去した。このプロセスを、150mLエーテルで3回繰り返した。残留物を50mL酢酸エチルに溶解し、220mLヘキサンで沈殿させた。生成した固体をその後濾過によって集めると、アセチルシクロスポリンAのトリフェニルホスホニウムブロミド22.5g(66%)が黄褐色固体として得られた。
【0148】
実施例7:ウィッティヒ反応
アセチルシクロスポリンのトリフェニルホスホニウムブロミド(100mg、0.06mmol)、過剰の37%ホルムアルデヒド(0.25mL)及びトルエン(2mL)を室温で速やかに撹拌した。1N水酸化ナトリウム水溶液(2mL)を滴下し、撹拌を3.5時間続けた。反応混合物を、酢酸エチル(20mL)及び水(10mL)で希釈した。酢酸エチル相を分離し、水(10mL)及びブライン(10mL)で逐次洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗物質をシリカゲル上で、アセトン/ヘキサン(2:3)でクロマトグラフィーにかけると、アセチルISATX247の(E)及び(Z)−異性体混合物70mg(88%)が無色固体として得られた。
【0149】
実施例8:ウィッティヒ反応生成物の脱アセチル化
実施例7から生成した異性体混合物(70mg、0.056mmol)をメタノール(5mL)に溶解し、水(1mL)を加えた。炭酸カリウム(75mg)を加え、反応物を室温で19時間撹拌した。メタノールの大部分を蒸発し、残留物に酢酸エチル15mLを加え、その後10%クエン酸水溶液10mLを加えた。酢酸エチル相を分離し、水相を追加的酢酸エチル10mLで抽出した。合一した酢酸エチル抽出液を、10mL水、10mL10%クエン酸水溶液及び10mLブラインで逐次洗い、その後硫酸マグネシウム上で乾燥し、溶媒を蒸発した。粗物質をシリカゲル上で、アセトン/ヘキサン(2:3)でクロマトグラフィーにかけると、ISATX247、37mg(54%)が約85%E−異性体及び約15%Z−異性体を含む無色固体として得られた。
実施例5−8における生成物は、質量分析及び/又は核磁気共鳴分光法によって特徴づけられた。
【0150】
実施例9:ISATX247の幾何異性体の製法
ISATX247のシス−及びトランス−異性体を下記の反応スキームを用いて独立的に合成した。反応系列は、アリルトリメチルシランの公知のメタレーション、トリメチルボレートによる求電子捕獲、及びその後の加水分解及びその後のエステル交換を含み、中間体トランス−(トリメチルシリル)アリルボロネートエステルを生成する。シクロスポリンアルデヒドのアリルボレーションはホウ素中間体を与え、それを金属イオン封鎖によって所望のβ-トリメチルシリルアルコールに変換する。新しいキラル中心の生成におけるジアステレオ選択性は、その後の段階でこれらの中心が除去されるため、この段階では決定しない。β−トリメチルシリルアルコールにおける2つの中心の相対的立体化学は予想通りアンチであり、トランス−(トリメチルシリル)ボロネート前駆体におけるトランス二重結合によるものであることに注目すべきである。
【0151】
β−トリメチルシリルアルコールの塩基促進性脱離(ハードリクら、1975)はアセチル−(Z)−1,3−ジエンに富む組成物を提供し、酸促進性脱離はアセチル−(E)−1,3−ジエンに富む組成物を与える。脱保護は、対応するジエンアルコール、ISATX247の(Z)及び(E)−異性体をそれぞれ与える。
【0152】
ジエンに至るまた別のアプローチは、アリルホスホランを使用する。アリルジフェニルホスフィンのメタレーション及びその後のTi(OPri)4によるトランスメタレーションは、チタン中間体を与える。アリルチタネーション及びその後の立体特異的脱離で、(Z)−ジエンに富む組成物が生成する。
【0153】
他方、アリルジフェニルホスフィンオキシドで同様な系列を行うと(図8)、主としてE−異性体が生成する(75%)。
【0154】
i)アセチルCsA−CHOのアリルボレーション:
(E)−1−トリメチルシリル−1−プロペン−3−ボロネートを前に報告された方法にしたがってつくった(イケダら、1987)。THF(3mL)中(E)−1−トリメチルシリル−1−プロペン−3−ボロネート(0.2g、0.832mmol)の撹拌溶液に、窒素気流下でアセチルシクロスポリンAアルデヒド(1.026g、0.832mmol)を加えた。反応混合物を高性能液体クロマトグラフィー(C−8カラム、逆相)によってモニターし、合計7日間撹拌した。その後トリエタノールアミン(0.196g、1.3mmol)を加え、さらに4日間撹拌し続けた。シリカゲルカラム上で精製すると、β−トリメチルシリルアルコールが得られた。MS(ES)m/z1368.9(M+Na+)。
【0155】
無水THF(1mL)中KH懸濁液(3.5mg、26.4μmol、無水ヘキサンで洗浄した30%鉱油分散液)に、β−トリメチルシリルアルコール(10mg、7.4micromol)を加え、室温で10分間撹拌した。反応混合物をジエチルエーテル(10mL)で希釈し、その後NaHCO3飽和溶液(2×5mL)で洗った。乾燥(Na2SO4)及び溶媒除去により、富化(Z)−アセチル−1,3−ジエンが得られた。MS(ES)m/z1294.8(M+K+)。
【0156】
ii)アセチルCsA−CHOのアリルチタン化:
無水THF(8mL)中アリルジフェニルホスフィン(0.54g、2.4mmol)の撹拌、冷却(−78℃)溶液に、t−BuLi(1.42mL、2.4mmol、1.7M溶液。ペンタン中)を加えた。赤煉瓦色の溶液をこの温度で15分間撹拌し、その後0℃で30分間撹拌した。それから再び−78℃に冷やし、Ti(OPri)4(0.71mL、2.4mmol)を加えた。その褐色溶液をこの温度で15分間撹拌し、それからTHF(10mL)中アセチルCsA−CHO(2.5g、2mmol)溶液をカニューレによって加えた。淡黄色溶液をさらに30分間撹拌し、それから一晩室温に温めた。この反応混合物に0℃でMeI(0.15mL、2.4mmol)を加えた。撹拌をこの温度で1時間続け、それから室温で2時間続けた。反応混合物を氷冷した1%HCl(100mL)に注いだ。水層をEtOAc(3×50mL)で抽出した。合一した有機抽出液を水(2×25mL)及びブライン(25mL)で洗った。溶媒を除去すると黄色固体が得られ、それをシリカゲルカラムでクロマトグラフィーにかけた。1:3アセトン−ヘキサン混合物で溶出すると、アセチルISATX247の(Z)−富化異性体が得られた。実施例4におけるように脱保護すると、ISATX247の(Z)−富化異性体が得られた(Z/E比、75:25)。
【0157】
実施例10:ISATX247異性体の(E)−富化混合物の製法
テトラヒドロフラン(5mL)中アリルジフェニルホスフィンオキシド(1mmol)及びヘキサメチルホスホルアミド(2mmol)の溶液に、−78℃で、n−ブチルリチウム(1mmol、ヘキサン中)を加えた。混合物を−78℃で30分間撹拌した。テトラヒドロフラン(7mL)中アセチルシクロスポリンAアルデヒド(0.8mmol)の溶液を加え、反応混合物を徐々に室温まで温め、それから18時間撹拌した。混合物を氷冷した1N塩酸(50mL)に注入し、それから酢酸エチルに抽出した。有機抽出液を水で洗い、硫酸マグネシウム上で乾燥し、溶媒を蒸発した。残留物をシリカゲル上で、25%アセトン/75%ヘキサンを溶出液としてクロマトグラフィーにかけると、アセチルISATX247の(E)及び(Z)−異性体混合物が得られた。実施例4におけるようにアセテート保護基を除去すると、ISATX247異性体の(E)−富化混合物が得られた。プロトンnmr分光法は、この混合物がISATX247の(E)−異性体75%及び(Z)−異性体25%からなることを示した。この反応は、シュロッセルの改良法(リュ−(R.Liu)、シュロッセル(M. Schlosser), Synlett, 1996, 1195)によっても行うことができる。THF(20mL)中アリルジフェニルホスフィンオキシド(1.21g、5mmol)の撹拌した冷却(−78℃)溶液に、n−BuLi(2mL、5mmol、2.5M溶液、ヘキサン中)を加えた。その赤色溶液を−78℃で40分間撹拌した。その後THF(12mL)中アセチルCsA−CHO(1.25g、1.02mmol)溶液をカニューレによって15分間にわたって加えた。反応混合物を室温で2時間撹拌した。上記のように処理及びクロマトグラフィーを行うと、アセチルISATX247(Z:E比40:60、1HNMR分析による)が得られた。
【0158】
実施例11:ベンゾイル保護シクロスポリンAの製法
シクロスポリンA(6.01g、5mmol)及び4−ジメチルアミノピリジン(305mg、2.5mmol)をピリジン(5mL)に溶解した。無水安息香酸(3.4g、15mmol)を加え、混合物を50℃で19時間撹拌した。追加的無水安息香酸(1.7g、7.5mmol)及びDMAP(305mg、2.5mmol)を加え、50℃でさらに24時間撹拌を続けた。無水安息香酸(0.85g、3.8mmol)を加え、反応物をさらに23時間撹拌した。反応混合物を撹拌しながらゆっくりと水中に注入した。沈殿したシクロスポリンA安息香酸を濾取し、水で洗った。集めたケーキを最小容量のメタノールに溶解し、10%クエン酸溶液に加え、1時間撹拌した。沈殿した生成物を濾過によって集め、濾液のpHが水のpHと等しくなるまで水で洗った。固体シクロスポリンA安息香酸を50℃で、真空下で乾燥すると無色固体が得られる。
【0159】
実施例12:トリエチルシリルエーテル−保護シクロスポリンAの製法
シクロスポリンA(3.606g、3mmol)を乾燥ピリジン(8mL)に溶解し、その後DMAP(122mg、1mmol)を加えた。反応混合物を0℃に冷やし、それからトリエチルシリルトリフルオロメタンスルホネート(3.6mmol)を滴下した。その混合物を放置して室温まで温め、一晩撹拌した。反応混合物を、撹拌しながらゆっくりと水中に注入した。沈殿したトリエチルシリルエーテルを濾別し、水で洗った。集めたケーキを最小容量のメタノールに溶解し、5%クエン酸溶液に加え、30分間撹拌した。沈殿した生成物を濾過によって集め、水で、濾液のpHが水のpHに達するまで洗った。固体トリエチルシリルエーテルを50℃で、真空下で乾燥すると無色固体が得られた。トリイソプロピルシリル及びtert−ブチルジメチルシリル保護基を、類似の方法によって導入することもできる。
【0160】
実施例13:カルシニューリン阻害アッセイを使用する免疫抑制活性
シクロスポリンA又はシクロスポリンA誘導体の効果の指標は、カルシニューリンのホスファターゼ活性を阻害する能力である。カルシニューリン阻害アッセイは、薬剤のその作用部位における活性を測定する。このようなアッセイは、シクロスポリンA類似体の効力の直接的invitro評価である(フルマンら、1992)。
【0161】
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)対シクロスポリンAの免疫抑制活性は、カルシニューリン(CN)阻害アッセイを使用して評価される。このアッセイの結果は、ISATX247(45−50%のZ−異性体及び50−55%のE−異性体)によるカルシニューリンホスファターゼ活性の阻害が、シクロスポリンAに比べて3倍より強力である(IC50により測定)ことを示す(図11)。
【0162】
種々の重水素化及び非重水素化異性体類似体混合物対シクロスポリンAの免疫抑制活性が、カルシニューリン(CN)阻害アッセイを用いて評価されている。これらの類似体の構造及び異性体組成は、図12に示される。図12において、「I4」は、ISATX2477の構造に対応する。I4−M2とは、実施例5−8に記載の方法によって生成されたISATX247を指す(この図では方法2である)。I4−D4は、実施例1−4に記載の方法によって生成された重水素化ISATX247を指す。I4−D2は、実施例5−8に記載の方法によって生成された重水素化ISATX247を指す。その他の異性体混合物も図に示されている。
【0163】
このアッセイの結果は、これらの異性体類似体混合物によるカルシニューリンホスファターゼ活性の阻害効果は、少なくともシクロスポリンAと同様であることを示す(IC50によって測定した)(図13)。CsAはシクロスポリンAを指す;Isocyclo4は、実施例1−4に記載の方法によって生成されたISATX247を指す。Isocyclo5は、図12のI5−M1に対応する。Isocyclo4−d4は、図12のI4−D4に対応する。Isocyclo5−d5は、図12のI5−D5に対応する。Isocyclo4−d2は、図12のI4−D2に対応する。Isocyclo4−M2は、図12のI4−M2に対応する。Isocyclo5−m2は、図12のI5−M5に対応する。
【0164】
実施例14:ラット心臓移植片モデルを使用する免疫抑制活性
異なる系統のラット間で移植された心臓の拒絶を阻止するISATX247(45−50%のE−異性体及び50−55%のZ−異性体)の効果を評価し、シクロスポリンAのそれと比較した。ラット心臓移植片モデルは、新しい免疫抑制剤のinvivo効力を評価する際に最もよく用いられるモデルである。それは、このモデルでは免疫拒絶により、持続的グラフト生存を実現することが難しいからである。
【0165】
この方法は、ウィスターラット(Wister Furth rats)からルイスラット(Lewis rats)への心臓の異所性移植(腹部大動脈及び下大静脈への)を含む。シクロスポリンA又は異性体類似体混合物のいずれかの腹腔内注射が移植片レシピエントに行われ、それは移植の3日前から始まり、移植後30日間続けられた。グラフト異機能が移植後30日の期間に認められた場合は、動物を殺した。動物が移植後30日より長く生きている場合は、試験及び対照試験を中止し、動物をグラフト異機能になるまで、又は移植後100日まで生存させた。
【0166】
レシピエント動物の各群の平均生存率を、表1にまとめる。これらの結果は最適量1.75mg/kg/日のISATX247(45−50%のE−異性体及び50−55%のZ−異性体)が、シクロスポリンAに比較して生存時間を約3倍延長することを示す。ISATX247を投与した多数の動物が、移植後100日目(投与中止後70日)にまだ機能するグラフトを有していた。これらのデータはこの異性体類似体混合物が、グラフト拒絶を阻止するための免疫抑制活性を有することを証明する。
【0167】
【表1】
【0168】
種々の重水素化及び非重水素化異性体類似体混合物(構造は図12に示される)が、異なる系統のラット間で移植された心臓の拒絶を阻止する効果も評価し、シクロスポリンAのそれと比較した。1.75mg/kg/日の用量を30日間投与した。表2に結果をまとめる。これらの結果は、1.75mg/kg/日の異性体混合物は、生存期間を少なくともシクロスポリンAと同様に延長することを示し、これらの異性体類似体混合物がグラフト拒絶阻止における免疫抑制活性を有することを証明した。
【0169】
【表2】
【0170】
実施例15:膵島細胞の同種移植術における免疫抑制活性
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)対シクロスポリンAの、マウスモデルにおける移植膵島細胞の生存を延長する能力を試験した。この研究は、CBA/Jマウスからの500個の膵島を糖尿病Balb/cマウスレシピエントの腎臓嚢に移植することを含む。
【0171】
移植後、ISATX247又はシクロスポリンAの投与量0(ビヒクル)、1.75、10、20又は25mg/kg/日を腹腔内注射(i.p.)によって30日間投与した。連続2日間17mmol/Lより高い血糖濃度を示す、という定義によるグラフト不全になるまで血糖値を毎日モニターした。
【0172】
その結果は、ISATX247が20mg/kg/日の投与量でグラフト生存期間を40%延長することを示している。投与量を増加すると、ISATX247の方がシクロスポリンAより毒性が小さいことも認められた。これは25mg/kg/日の投与量で特に明らかであった。
【0173】
【表3】
【0174】
実施例16:関節炎における免疫抑制活性
過去30年間に、ヒトリウマチ性関節炎の3動物モデルが詳細に試験され、新規の抗リウマチ薬の臨床前スクリーニング及び開発に広く用いられた。これらはアジュバント誘起性、コラーゲン誘起性、及び抗原誘起性関節炎モデルを含む。次の研究はマウスのコラーゲン誘起性関節炎モデル及びウサギの抗原誘起性関節炎モデル両方において、ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)の抗炎症性効果を評価するために計画された。これら2つのモデルに認められた組織病理学及び免疫病理学は、ヒト疾患における知見と似ている。両モデルにおいて、ISATX247の関節炎発生阻止効果(予防プロトコル)及び関節炎治療効果(治療プロトコル)を調べた。これらの研究は、請求の異性体類似体混合物が免疫抑制活性を有することを支持している。
【0175】
A.コラーゲン誘起性関節炎
ウィルス抗体をもたない状態の齢8ないし10週の雄DBA/1Lac J マウスを、フロイント完全アジュバント中に乳化したヒナドリII型コラーゲン100マイクログラムの皮下注射によって免疫した。ISATX247、シクロスポリンA、又はビヒクル(クレモフォルEL/エタノール72:28、容量/容量)を、生理食塩液で1−50倍に希釈した保存薬剤(0.25、0.5又は1mg/mL)を腹腔内注射によって毎日投与し、0(ビヒクル);ISATX247247では、125、250、又は500μg/マウス;シクロスポリンAでは250、又は500μg/マウスの濃度で与えた。予防プロトコルに割り当てられた動物(12匹/群)ではコラーゲンで免疫した日(0日目)から始めて、40日目に殺すまで投与した。治療プロトコルに割り当てられた動物(12匹/群)では、発病の日(〜28日目)に始めて、38日目に殺すまで投与した。
【0176】
評価パラメーターは、死亡率、血清クレアチニン、組織検査、及び結末の評価、例えば臨床的スコア(目視)、後足のむくみ、組織学的スコア、糜爛スコア、及び免疫組織学的検査などであった。
【0177】
糜爛スコア測定は盲検法で、中指の近位指節間(PIP)関節の矢状領域に糜爛(軟骨又は骨における炎症性組織の充満した、境界のはっきりした障害、と定義された)があるかないかによって行われた。このアプローチでは、同じ関節を比較することができた。これまでの研究では、未治療の関節炎動物の90%より多くの動物において、この関節の糜爛が明らかにされている。
【0178】
結果は、ISATX247多量投与群(500μg/マウス)の負の糜爛スコアが、ビヒクル投与群の負の糜爛スコアより有意に高いことを示した(p<0.05)。中程度量のISATX247(250μg/マウス)及び多量シクロスポリンA(500μg/マウス)治療群両方共ビヒクル投与群に比較して高い負の糜爛スコアを示した(p<0.1)。さらに、少量ISATX247(125μg/マウス)及び中程度量シクロスポリンA対照(250μg/マウス)治療群は、ビヒクル対照群に比較して有意に高いとは言えないまでも、より高い負の糜爛スコアを有した。
【0179】
関節の糜爛の発達を顕著に予防する唯一の治療は、500μg/マウスのISATX247であった。糜爛変化を示すPIP関節の比率が、ISATX247投与マウスの方がビヒクル対照群マウスに比較して有意に低いということは、ISATX247が疾患を変化させる性質を有することを証明するものである。
【0180】
B.抗原誘起性関節炎
特異的病原体のない状態で飼育されたニュージーランド白ウサギを、フロイント完全アジュバントと共に乳化した生理食塩液中卵白アルブミン10mgで免疫した。これは、首筋の数箇所に筋肉内及び皮下注射によって投与された。14日後、全ての動物で卵白アルブミン5mg及び生理食塩液中ヒト組換え形質転換増殖因子2(65ng)の1日おきの関節内注射が始められた。
【0181】
ISATX247、シクロスポリンA、又はビヒクル(クレモフォルEL/エタノール72:28、V/V)を、保存薬剤(ビヒクル中)を生理食塩液で1−ないし4倍に希釈した希釈液の皮下注射によって毎日投与し、0(ビヒクル);ISATX247では2.5、5.0、又は10mg/kg/日;シクロスポリンAでは5.0、10又は15mg/kg/日の濃度で与えた。予防プロトコルに割り当てられた動物(8/群)には、卵白アルブミン免疫の日(0日目)から始めて、42日目に殺すまで投与した。治療プロトコルに割り当てられた動物(8/群)では、発病の日(〜28日目)に始めて、42日目に殺すまで投与した。
【0182】
評価したパラメーターは、死亡率、体重、血清クレアチニン、組織検査、及び結末の評価、例えば膝関節の腫張、滑液カウント、肉眼による死後分析及び組織検査などを含む。
【0183】
ビヒクル対照動物と比較して、滑液の組織病理学スコアの顕著な減少が、ISATX247(P0.05)及びシクロスポリンA(P0.05)において、治療28日後(予防プロトコル)に認められた。これは滑液カウントの顕著な減少を伴った(ISATX247、P0.05;シクロスポリンA、P0.05)。確立された関節炎を有する動物の滑液組織病理学的スコアも、14日間のISATX247(P0.05)及びシクロスポリンA(P0TX05)治療後、ビヒクル対照に比較して有意に改善されることが判明した(治療プロトコル)。巨視的関節炎スコアの有意な減少が、ISATX247では明らかになったが(P=0.01)、シクロスポリンA治療動物では明らかでなかった。治療は耐容性がよく、血清クレアチニン分析又は死後組織検査で顕著な毒性は認められなかった。
【0184】
これらのデータは、ISATX247がウサギの抗体誘起性関節炎モデルにおいて、リウマチ性関節炎の治療及び予防においてシクロスポリンAと同等であるか、又は多分よりすぐれていることを示している。
【0185】
実施例17:薬物動態及び毒物動態特性
ISATX247(45−50%のE−異性体及び50−55%のZ−異性体)及びシクロスポリンAの薬物動態及び毒物動態パラメーターを、ウサギモデルで試験した。そのウサギは、シクロスポリンA腎臓毒性を研究するためのモデルとしても使われているが、ラットほどよく使われてはいない。研究では、ウサギに投与したシクロスポリンAは他の動物モデルでこれまでに報告されたものより低い用量、しかもヒトにおける治療範囲の少なくとも高い方の濃度以下でもある用量で、構造的及び機能的変化を引き起こすことが見いだされた(スリヴァリスら、1991,1994)。また、細管の細胞学的変化に加えて、間質性線維症及び動脈症の所見があることは、ウサギが腎臓毒性の研究により適したモデルであることを示唆する。なぜならば、これらの構造的変化は、ヒトで認められる腎臓毒性の顕著な特徴だからである。下記のスケジュールによって、ISATX247を最初の7日間静脈注射(i.v.)し、さらに23日間皮下注射した(s.c.)。
【0186】
【表4】
【0187】
認められたいかなる腎臓変化もISATX247効果によるもので、病原体によるものではないことを確実にするために、無病原体ウサギ(SPF)を使用した。1及び7日目に、薬剤を投与する前と、投与後0.5、1、2、4、8、12、18及び24時間後に血液サンプルを集め、薬物動態曲線を作成した。その他の評価パラメーターは、臨床的観察、体重、食餌消費量、血液検査値、臨床化学検査、肉眼的病理試験、及び選択された組織/臓器の組織病理学的検査などである。
【0188】
血液サンプルを、質量分析と高性能液体クロマトグラフィーとの組合わせ(LCMS)によって分析した。下の表5は、シクロスポリンA又はISATX247の10mg/kgを投与したウサギにおける平均薬物動態パラメーターをまとめたものである。
【0189】
【表5】
【0190】
10mg/kg/日を投与した雄ウサギでは、シクロスポリンAとISATX247との薬物動態パラメーターの間に統計的有意差はなかった。同量を投与した雌ウサギのISATX247の薬物動態パラメーターは、7日目の最大濃度を除けば雄ウサギで認められたパラメーターと有意に異ならなかった。
【0191】
ビヒクル対照、シクロスポリンA、又はISATX247を投与されたウサギの血液学的パラメーターにおいて有意な変化は認められなかった。下の表6に示すように、研究コース中、種々の群においてクレアチニン濃度に差が認められた。これらの差は、シクロスポリンAが、ビヒクル対照又はISATX247のいずれよりも有意に大きい負の効果を腎臓に与えることを示した。50%多い量、すなわち15mg/kg/日でさえ、10mg/kg/日シクロスポリンAに比べて、ISATX247は血清クレアチニン濃度の有意な増加を誘起しなかった。
【0192】
【表6】
【0193】
ビヒクル対照、10mg/kgシクロスポリンA、5mg/kgISATX247、又は10mg/kgISATX247を投与した全てのウサギの試験では、有意な異常はあらわれなかった。これは特に腎臓では事実であった;腎臓ではシクロスポリンA投与動物で通常見られる、間質性線維症(スリヴァリスら、1991、1994)の徴候は認められなかった。15mg/kgISATX247を投与した雄ウサギでは、精子形成の減少が認められた。この15mg/kg用量のISATX247投与で試験を完了した3匹の雌ウサギには、変化は認められなかった。
【0194】
実施例18:ISATX247の免疫抑制効果
カニクイザル(n=4)からの全血をISATX247又はシクロスポリンと共にインキュベートし、培養培地において種々のマイトジェンで刺激した。SG2M期の細胞で、トリチウム標識チミジンの挿入によって、及び増殖中の細胞核抗原(PCNA)発現のフローサイトメトリー分析によって、リンパ球の増殖を評価した。フローサイトメトリーを利用して、T細胞による細胞内サイトカインの産生及びTリンパ球活性化抗原の発現も評価した。その後EC50(最大効果の50%に達するために必要な薬剤濃度)をウィンノンリン(WinNonlin)(商標)ソフトウエアを用いて計算した。その結果は、下の表7に示されるEC50(ng/mL)からわかるように、リンパ球増殖、サイトカイン産生、及びT細胞表面抗原の発現は、シクロスポリンよりもISATX247によってより強力に阻止されることを示した。
【0195】
【表7】
【0196】
こうして、ex vivo全血アッセイにより、ISATX247が種々の免疫機能をシクロスポリンよりも2.3−6倍強く抑制することが判明した。
【0197】
実施例19:トリブチルアリルホスホニウムブロミドを使用するウィッティヒ反応
カリウムtertブトキシド(0.31g、2.8mmol)をテトラヒドロフラン20mLに溶解した。約−40℃で、テトラヒドロフラン3mLに溶解したトリブチルアリルホスホニウムブロミド(0.99g、3.1mmol)をゆっくり加えた。生成した黄色混合物を約−40℃で約10分間撹拌し、その後テトラヒドロフラン6mL中アセチルシクロスポリンAアルデヒド(1.5g、1.2mmol)溶液をゆっくり加えた。黄橙色反応混合物を1.5時間撹拌後、反応が完了した。反応を停止するために反応混合物を燐酸水溶液(1.2g、1.0mmol)に移した。生成水溶液をトルエン100mLで抽出し、その後トルエン50mLで抽出した。合一した有機層を水で洗い、減圧下で濃縮乾固した。生成物、アセチル化ISATX247がかすかに黄色の固体として約90%収率で得られた。異性体比は、約87%E−異性体及び約13%Z−異性体であった(1H−NMR分光法によって測定)。
【0198】
実施例20:トリブチルアリルホスホニウムブロミド及びリチウム塩基を使用するウィッティヒ反応
トリブチルアリルホスホニウムブロミド(1.38g、4.3mmol)をトルエン20mL及びテトラヒドロフラン3mLの混液に溶解した。約−78℃でブチルリチウム(ヘキサン中1.6M、2.43mL、3.9mmol)をゆっくり加えた。生成した黄色混合物を約−78℃で約10分間撹拌し、その後トルエン6mL中アセチルシクロスポリンAアルデヒド溶液(1.5g、1.2mmol)をゆっくり加えた。黄橙色の反応混合物を3.5時間撹拌後、その反応混合物を50mLトルエンと燐酸水溶液(0.25g、2.2mmol)との混液に移すことによって反応を停止した。生成した二相混合物を放置して周囲温度まで温め、その後二相を分離した。トルエン層を20mLの水で洗い、減圧下で濃縮乾固した。生成物、アセチル化ISATX247がかすかに黄色の固体として約80%収率で得られた。異性体比は、約70%E−異性体及び約30%Z−異性体であった(1H−NMR分光法によって測定)。
【0199】
実施例21:トリブチルアリルホスホニウムブロミド及びリチウム塩基を使用するウィッティヒ反応
上記のように、ただし約−40℃の温度でSAP018を作動する。実施例20の実験条件を反復した。このときは、反応温度約−40℃を使用した。これらの条件のもとで、分離した生成物、アセチル化ISATX247の異性体比は、1H−NMR分光法で測定して、E−異性体約74重量%及びZ−異性体約26重量%であった。
【0200】
実施例22:トリブチルアリルホスホニウムブロミドを使用するウィッティヒ反応
テトラヒドロフラン15mL中アセチルシクロスポリンAアルデヒド(1.5g、1.2mmol)及びトリブチルアリルホスホニウムブロミド(0.99g、3.1mmol)の溶液を約−80℃に冷却した。テトラヒドロフラン9mLに溶解したカリウムtert−ブトキシド(0.19g、1.7mmol)をゆっくりと加えた。反応を完了するために、生成した黄色混合物を約−80℃で1時間撹拌し、その後6mLのテトラヒドロフラン溶液をゆっくり加えた。黄橙色反応混合物を1.5時間撹拌後、反応は完了した。反応混合物の反応を停止するために、燐酸水溶液(0.15g、1.3mmol)を加えた。生成混合物を濃縮し、残留物をメタノール5mLに溶解した。その後、混合物を水5mLにゆっくり加えた。生成した沈殿を濾過し、4mLのメタノール/水(1/1)で洗い、真空中で乾燥した。生成物ISATX247が無色固体として約90%収率で得られた。異性体比は、約91重量%E−異性体及び9重量%Z−異性体であった(1H−NMR分光法によって測定)。
【0201】
実施例23:アセチルCsAのオゾン分解
メタノール200mL中アセチルシクロスポリンA(15g、12.1mmol)の溶液を、サンダーオゾン発生機を使って約1.1バールで、300L O2/時間の流量で、反応が完了するまで(約5分間)オゾン化した。溶液にアルゴンガスを通し、メタノールに溶解したジメチルスルフィドを加えた。還元を完全にするために、混合物を室温で一晩撹拌した。約50mLに濃縮後、その溶液を水500mLにゆっくりと加えた。生成した沈殿を濾過し、水60mLで洗い、真空中で乾燥した。生成物、アセチル化CsAアルデヒドが純度約98%の無色固体として約95%収率で得られた(HPLCによって測定)。
【0202】
実施例24:トリメチルシリル−保護シクロスポリンAの製法
シクロスポリンA(40g、1当量)を30℃で、ジクロロメタン(100ml)に溶解した。N,N−ビス−(トリメチルシリル)尿素(1.1当量)を加えた。30℃で5分撹拌後、p−トルエンスルホン酸(0.02当量)を加えた。反応混合物を反応が完了するまで還流加熱し、その後室温にまで冷やした。反応の完了は、薄層クロマトグラフィー(TLC)、高圧又は高性能液体クロマトグラフィー(HPLC)、又は質量分析(MS)によって確認された。半飽和状態の炭酸水素ナトリウム溶液(100ml)を加えた。水相を分離し、ジクロロメタンで再抽出した。合一した有機相を無水Na2SO4上で乾燥し、濾過した。溶媒を減圧下で除去すると、粗トリメチルシリル−保護シクロスポリンAが得られた。
【0203】
実施例25:トリメチルシリル−保護シクロスポリンAアルデヒドの製法
トリメチルシリル保護シクロスポリンA(5g、1当量)をジクロロメタン(50ml)に溶解した。溶液を約−78℃の温度に冷やし、その後オゾンをその溶液に、青色があらわれるまでぶくぶく通した。次に、その溶液が無色になるまでその溶液にアルゴンをぶくぶく通し、無色になった過剰のオゾンを除去した;この工程は過剰のオゾンを除去するために行われた。トリメチルアミン(5当量)を加え、反応混合物を室温で17時間撹拌した。水性処理後にトリメチルシリル−保護シクロスポリンAアルデヒドが得られた。
【0204】
実施例26:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の3:1混合物の製法
60分間あらかじめ撹拌した、トルエン(10ml)中カリウムtert−ブトキシド(3当量)及びアリルトリフェニルホスホニウムブロミド(2当量)の混合物に、トリメチルシリル−保護シクロスポリンAアルデヒド(1g、1当量)を加えた。この反応混合物を室温で1時間反応させた後に処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の3:1混合物(NMRによる)が得られた。
【0205】
実施例27:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:1混合物の製法
トリメチルシリル−保護シクロスポリンAアルデヒド(2.5g)を25mlトルエンに溶解し、1N水酸化ナトリウム水溶液(10当量)で処理した。反応混合物を激しく撹拌し、アリルトリフェニルホスホニウムブロミド(7.5当量、一部分ずつ)を加えた。室温で数時間反応後、反応混合物を処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:1混合物(NMRによる)が生成した。
【0206】
実施例28:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:2混合物の製法
トリメチルシリル−保護シクロスポリンAアルデヒド(1g)を、トルエン5mlに炭酸カルシウム(1.5当量)及びアリルトリフェニルホスホニウムブロミド(1.5当量)と共に溶解した。激しく撹拌しながら還流下で4時間反応させた後、その反応混合物を処理すると、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:2混合物(NMRによる)が生成した。
【0207】
実施例29:ウィッティヒ反応による、トリメチルシリル−保護シクロスポリンAジエンのZ対E二重結合異性体の1:3混合物の製法
アリルトリブチルホスホニウムブロミド(3当量、アリルブロミド及びトリブチルホスフィンからつくられた)をTHF(3.5ml)に溶解した。トルエン(7.5ml)を加え、その後カリウムtert−ブトキシド(4当量)を加えた。室温で1時間撹拌後、溶液を約−30℃に冷却した。トリメチルシリル−保護シクロスポリンAアルデヒド(1g、1当量)のトルエン(5mL)溶液を滴下した。約−30℃で45分経過後、反応混合物を処理し、トリメチルシリル−保護シクロスポリンAジエンのZ及びE二重結合異性体の約1:3混合物(NMRによる)を生成した。
【0208】
下記の2実施例、実施例30及び31は、アリルメタレーションに関するものである。
【0209】
実施例30:アセチル−保護シクロスポリンAβ−トリメチルシリルアルコールの製法
THF(15ml)中アリルトリメチルシラン(10.1当量)の溶液に、ブチルリチウム(ヘキサン中1.6M、10当量)を室温で加えた。30分間反応後、溶液を−75℃に冷却し、ジエチル−B−メトキシボラン(10.1当量)で処理した。1時間後、ホウ素トリフルオリドジエチルエーテル錯体(10.1当量)を加えると、B−(γ−トリメチルシリル−アリル)−ジエチルボラン試薬が生成した。1時間後、THF(15ml)中アセチル−保護シクロスポリンAアルデヒド(5g、1当量)溶液を滴下した。20分後、反応混合物を−10℃に温め、飽和NH4Cl水溶液を加えた。室温で1時間撹拌後、水(45ml)を加え、反応混合物を25ml酢酸エチルで3回抽出した。有機相を水(25ml)及び飽和NH4Cl水溶液(25ml)で逐次洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮した。粗生成物をクロマトグラフィーにかけ(シリカゲル、ジクロロメタン/メタノール又は酢酸エチル/ヘプタン)、アセチル−保護シクロスポリンAβ−トリメチルシリルアルコールを得た。
【0210】
実施例31:トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコールの製法
THF(15ml)中アリルトリメチルシラン(10.1当量)の溶液に、室温でブチルリチウム(ヘキサン中1.6M、10当量)を加えた。約30分間反応を進行させた後、溶液を−65℃に冷却し、ジエチル−B−メトキシボラン(10.1当量)で処理した。1時間後、ホウ素トリフルオリドジエチルエーテル錯体(10.1当量)を加え、B−(γ−トリメチルシリル−アリル)−ジエチルボラン試薬を生成した。1時間後、THF(15ml)中トリメチルシリル−保護シクロスポリンAアルデヒド(5g、1当量)の溶液を滴下した。15分後、反応混合物を10℃まで温め、飽和NH4Cl水溶液を加えた。室温で1時間[1時間]撹拌後、水(12.5ml)及び飽和NaHCO3(25ml)を加えた。反応混合物を25mlのメチル−t−ブチルエーテルで2回抽出した。有機相を水(2×25ml)で2回洗い、次に飽和NaCl水溶液(25ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮した。粗生成物をクロマトグラフィー(シリカゲル、ヘプタン/酢酸エチル)にかけ、トリメチルシリル−保護シクロスポリンAβ−トリメチルシリルアルコールを得た。
【0211】
下記の3実施例、実施例32、33及び34は、ピーターソン脱離反応に関するものである。
【0212】
実施例32:E−アセチル保護シクロスポリンAジエンの製法
アセチル保護シクロスポリンA β−トリメチルシリルアルコール(10g、1当量)を、THF(50ml)に溶解した。濃縮H2SO4(1.24ml、3当量)を加え、反応混合物を室温で20時間撹拌した。水(150ml)を加え、反応混合物をメチル−t−ブチルエーテル(200ml)で抽出した。水相をメチル−t−ブチルエーテル(150ml)で再抽出した。有機相を水(150ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、粗アセチル−保護シクロスポリンAジエン(アセチル−保護ISATX247)が得られた。その粗生成物をメチル−t−ブチルエーテル/THFから結晶化し、その後メチル−t−ブチルエーテル/DCMから再結晶すると、アセチル−保護シクロスポリンAジエン(アセチル保護ISATX247)がE及びZ二重結合異性体の99.97%:1−3%混合物として得られた(400MHzNMRによる。測定誤差2%)。
【0213】
E−アセチル−保護シクロスポリンAジエンの加水分解を次のように行った:アセチルシクロスポリンAジエン(4g、1当量)をメタノール(80ml)及び水(32ml)に溶解した。炭酸カリウム(3.65g、8.3当量)を加えた。室温で15時間撹拌した後、反応混合物を4時間40℃まで加熱した。反応混合物を減圧下で濃縮し、残留物を酢酸エチル(70ml)にとった。クエン酸水溶液15%(30ml)をゆっくりと加え、次に水(10ml)を加えた。水層を分離し、酢酸エチル(56ml)で再抽出した。有機相を水(30ml)、15%クエン酸溶液(40ml)及び飽和NaCl溶液(30ml)で洗った。有機層を合一し、Na2SO4上で乾燥し、減圧下で濃縮するとシクロスポリンAジエン(ISATX247)が二重結合異性体の98:2E/Z混合物として得られた(400MHz NMRによる。誤差約2−3%)。R.W.ホフマン著、Angewandte Chemie InternationalEdition, Vol.555(1982);W.R.ロウシュ著、「『アリル有機金属類』、有機合成総覧」、ペルガモン プレス、第2巻、p.1-53;及びY.ヤマモト、N.アサオ著、ChemicalReviews、p.2307(1993)を参照されたい。
【0214】
実施例33:Z−トリメチルシリル−保護シクロスポリンAジエンの製法及びZ−シクロスポリンAジエン(ISATX247)への変換
トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコール(2g、1当量)をTHF(20ml)に溶解した。溶液を0−2℃に冷却し、カリウムt−ブトキシド(4当量)を加えた。1.5時間の反応後、酢酸エチル(20ml)及び水(40ml)を加えた。水層を分離し、酢酸エチル(20ml)で再抽出した。有機相を飽和NaCl水溶液(20ml)で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、Z−トリメチルシリル−保護シクロスポリンAジエン(トリメチルシリル−保護ISATX247)及びZ−シクロスポリンAジエン(ISATX247のZ−異性体)の混合物が得られた。粗生成物混合物をメタノール(溶液中10重量%)に溶解し、1M塩酸水溶液(1当量)を加えることによって、脱シリル化が完了した。室温で15分後に、水及び酢酸エチルを加えた。水層を分離し、酢酸エチルで再抽出した。有機相を飽和NaCl水溶液で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、シクロスポリンAジエン(ISATX247)がZ及びE二重結合異性体の94:6混合物として(NMRによる)生成した。
【0215】
実施例34:E−シクロスポリンAジエン(ISATX247)の製法
トリメチルシリル−保護シクロスポリンA β−トリメチルシリルアルコール(500mg、1当量)をジクロロメタンに溶解した。この溶液を約0−2℃の範囲内に冷却し、ホウ素トリフルオリドジエチルエーテル錯体(5当量)で処理した。1時間後、水(20ml)及びジクロロメタン(20ml)を加えた。有機層を分離し、水(20ml)で洗い、Na2SO4上で乾燥し、濾過し、減圧下で濃縮すると、直接シクロスポリンAジエン(ISATX247)がE及びZ二重結合異性体の91:9混合物として(NMRによる)生成した。
【0216】
実施例35:トリメチルシリル−保護シクロスポリンAジエンの脱保護
トリメチルシリル−保護シクロスポリンAジエンをメタノールに溶解した(溶液中10重量%)。この溶液を1M塩酸水溶液(1当量)で処理した。室温で15分後、水と酢酸エチルを加えた。水層を分離し、酢酸エチルで再抽出した。有機相を飽和NaCl水溶液で洗った。合一した有機相をNa2SO4上で乾燥し、濾過し、減圧下で濃縮すると、シクロスポリンAジエン(ISATX247)が生成した。
【0217】
実施例36:アセチルシクロスポリンAのエポキシド化
アセチルシクロスポリンA(2.0g、1.61mmol)をアセトニトリル(30mL)に溶解した。1,3−ジアセトキシ−アセトン(0.14g、0.8mmol)を加え、その後0.0004Mエチレンジアミン四酢酸二ナトリウム塩(20mL)及び炭酸水素ナトリウム(0.405g、4.82mmol)水溶液を加えた。撹拌下の混合物に、オキソン(43.8%KHSO5)(2.23g、6.43mmol)を2時間にわたって一部分ずつ加えた。pH装置を使用して、1NNaOHを一定状態で添加し(総量6.4mL)、pHを8.2に維持した。冷水浴を用いて時々冷却することによって、温度を22−25℃に維持した。2.5時間後、反応混合物に数滴の重亜硫酸ナトリウム溶液を加えて反応を停止した。水(100mL)を加え、混合物をtert−ブチルメチルエーテル(100mL、その後75mL)で2回抽出した。その有機抽出液を塩化ナトリウムの希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると、粗アセチルシクロスポリンAエポキシド(1.92g、95%;HPLC:99.4%領域)が白色固体フォームとして得られた。
【0218】
実施例37:アセチルシクロスポリンAアルデヒドの製法
粗アセチルシクロスポリンAエポキシド(1.92g、1.52mmol)をアセトニトリル(25mL)に溶解した。水(20mL)を加え、その後過ヨウ素酸ナトリウム(489mg、2.28mmol)及び0.5M硫酸(3.05mL、1.52mmol)を加えた。反応混合物を40℃で18時間撹拌し、その後過剰の過ヨウ素酸ナトリウムを重亜硫酸ナトリウムの添加によって消滅させた。塩化ナトリウムの希水溶液(100mL)を加え、混合物をtert−ブチルメチルエーテル(各100mL)で2回抽出した。有機抽出液を塩化ナトリウム希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると、粗アセチルシクロスポリンAアルデヒド(1.74g、92%;HPLC:95.7%領域)が白色フォームとして得られる。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(1.41g、アセチルシクロスポリンAを基準にして71%;HPLC:100%領域)が白色固体フォームとして得られた。
【0219】
実施例38:ワンポット法を用いるアセチルシクロスポリンAアルデヒドの製法
アセチルシクロスポリンA(2.0g、1.61mmol)をアセトニトリル(30mL)に溶解した。1,3−ジアセトキシ−アセトン(0.084g、0.48mmol)を加え、その後0.0004Mエチレンジアミン四酢酸二ナトリウム塩(20mL)及び炭酸水素ナトリウム(0.405g、4.82mmol)水溶液を加えた。撹拌下の混合物に、オキソン(43.8%KHSO5)(1.67g、4.82mmol)を2時間にわたって一部分ずつ加えた。pH装置を使用して、1NNaOHを一定状態で添加し(総量3.4mL)、pHを8.2に維持した。温度は20−25℃に維持した。3.5時間後、0.5M硫酸(5mL、2.5mmol)を反応混合物に加え、その後数滴の濃硫酸を加え、pH1.3までにした。それから過ヨウ素酸ナトリウム(516mg、2.41mmol)を加え、反応混合物を室温で2時間、40℃で22時間撹拌した。水(100mL)を加え、混合物をtert−ブチルメチルエーテル(100mL、その後75mL)で2回抽出した。有機抽出液を塩化ナトリウム希水溶液(100mL)で洗い、合一し、Na2SO4上で乾燥し、濃縮すると粗アセチルシクロスポリンAアルデヒド(1.9g、96%;HPLC:83.4%領域)が白色フォームとして生成した。粗生成物をシリカゲル上で、40%アセトン/60%ヘキサンを溶出液としてクロマトグラフィーにかけると、生成物(1.35g、アセチルシクロスポリンAを基準にして68%;HPLC:100%領域)が白色固体フォームとして得られた。
【0220】
実施例39(ISO);アセチルシクロスポリンAアルデヒドと3−ジメチルアミノプロピルトリフェニルリンイリデンとのウィッティヒ反応
1,3−ジエン類の立体選択的合成は、コリー及びデサイがTetrahedron Letters, Vol.26, No.47,p. 5747-8(1985)に記載している。この文献は、3−(ジメチルアミノ)プロピルトリフェニルホスホランをヘキサメチルジシラチドカリウムで処理することによって得られるイリドが、アルデヒドと共にウィッティヒ反応を受けて、選択的にZ−アルケニルジメチルアミンを形成し得ることを開示している。そのアミンのm−クロロペル安息香酸による酸化は、対応するN−オキシドを与え、それをその後加熱するとコープ脱離として知られる反応が起き、所望の1,3−ジエンが形成する。このウィッティヒ反応段階中に形成されるオレフィンの配置は、専らZ、又はシスである。
【0221】
同様に、ISATX247のZ−異性体は、アセチルシクロスポリンAアルデヒドと3−(ジメチルアミノ)−プロピルトリフェニルホスホニウムブロミドを、ヘキサメチルジシラチドカリウムで処理することによって得られるイリドとの反応によっても形成される。生成した中間体はその後酸化され、その後コープ脱離を受けて、アセチル−(Z)−ISATX247が生成する。塩基を用いて脱保護すると、(Z)−ISATX247が生成する。酸化試薬は、メタクロルペル安息香酸でよい。
【0222】
無水トルエン(20mL)中3−ジメチルアミノプロピルトリホスホニウムブロミド(2.5g、5.83mmol)の撹拌中の懸濁液に、ヘキサメチルジシラジドカリウム(11.6mL、5.8mmol、トルエン中0.5M溶液)を注射器によって加えた。室温で1時間撹拌後、その赤色溶液を遠心分離し、上澄液をカニューレによって反応フラスコに移した。その固体に無水トルエン(10mL)を加え、撹拌し、遠心分離した。上澄液を反応フラスコに移し、合一した赤色イリドにOAc−CsA−CHO(1.44g、1.17mmol)を加えた。撹拌を室温でさらに2時間続けると、色は淡黄色に変わった。反応混合物をEtOAc(50mL)で希釈し、その後飽和NaHCO3溶液(50mL)及びブライン(50mL)で洗った。乾燥及び溶媒除去をすると、淡黄色の固体が得られる。シリカゲルカラム上でクロマトグラフィーを行い、アセトン−ヘキサン混液(勾配:10ないし75%アセトン及び90ないし25%ヘキサン)で溶出すると、全てのリン関連不純物は除去される。アセトンでさらに溶出すると、所望の生成物が無色固体として提供された(1.28g、84%収率)。1HNMR(300 MHz、CDCl3):2.23(s,6H)、2.03(s,3H)。13C NMR(300MHz、CDCl3):129.33、126.95;MSm/z:1301(M+)、1324(M+Na+)。
【0223】
N−オキシドへの変換
ウィッティヒ反応によって得られたジメチルアミノ化合物(0.44g、0.34mmol)を、CHCl3(3mL)に溶解した撹拌冷溶液(0℃)に、CHCl3(2mL)中m−CPBA(0.07g、0.405mmol)の溶液を加えた。30分間撹拌後、ジメチルスルフィド(0.5mL)を加え、その後CH2Cl2(50mL)を加えた。NaHCO3溶液(25mL)及び水(25mL)で洗い、乾燥し、溶媒除去する処理を行うと固体(0.43g)が生成した。1HNMR(300MHz、CDCl3):3.19(s,3H)、3.18(s,3H)、2.03(s,3H)。13C NMR(300MHz、CDCl3):131.89、124.13;MSm/z:1340(M+Na+)。
【0224】
N−オキシドのコープ脱離。アセチルISATX247の(Z)−異性体の調製
N−オキシド(350mg)をそのまま撹拌し、真空下で2時間100℃で加熱した。これを、その後シリカゲルカラムを通過させた。アセトン−ヘキサン混液(勾配、5ないし25%アセトン及び95ないし75%ヘキサン)による溶出は、無色固体(314mg)を提供した。1HNMR(500MHz、CDCl3):6.49(dt,J=16.99、10.5Hz、1H);13C NMR(400MHz、CDCl3):132.20、131.09、129.70、116.85;MSm/z:1279(M+Na+)。
【0225】
ISATX247の(Z)−異性体
MeOH(4mL)中(Z)−アセチルISATX247(50mg)の溶液に、水(1.5mL)及びK2CO3(60mg)を加え、室温で48時間撹拌した。反応混合物から溶媒を除去し、EtOAc(20mL)で抽出した。有機層を水(10mL)及びブライン(10mL)で洗った。乾燥及び溶媒除去により無色固体が生成した。1HNMR(500MHz、CDCl3):6.58(dt,J=16.99、10.5Hz、1H);MS m/z:1236.8(M+Na+)。生成した化合物はISATX247のZ−異性体であった。NMRによる測定では、計り得るE−異性体は認められなかった。
【0226】
本発明の好ましい実施形態のみを詳細に開示し、上述したが、上記の教示に照らして、そして添付の請求の範囲内で、本発明の精神及び目的の範囲から逸脱することなく、本発明の多くの変形及び変更が可能であることは明らかである。
【図面の簡単な説明】
【0227】
【図1】図1Aは、分子の環式ペプチド環を構成する11個のアミノ酸残基を示すシクロスポリンAの構造、並びに1−アミノ酸残基の側鎖の構造を示す図である; 図1Bは、本明細書中に用いられる用語「CsA」の定義を特に強調したシクロスポリンAの構造を示すもう一つの図である;
【図2】図2Aは、ISATX247と呼ばれるシクロスポリンA類似体のE−異性体(又はトランス−異性体)の構造を示す図である;図2Bは、シクロスポリンA類似体ISATX247のZ−異性体(又はシス−異性体)の構造を示す図である;
【図3】図3は、本発明のシクロスポリン類似体をつくるために使用できる典型的合成経路の概観を示し、ここで立体選択的経路は反応条件によってグループ化される;
【図4】図4は、臭素化前駆体からISATX247の(E)及び(Z)−異性体の混合物を生成する合成経路を示す図である;
【図5】図5は、アルデヒド前駆体からISATX247の(E)及び(Z)−異性体の混合物を生成する別の合成経路を示す図である;
【図6】図6は、ISATX247の(E)又は(Z)−異性体どちらかに富む組成物をつくるために使用できる典型的立体選択的反応スキームを示し、その際いずれの異性体も同じ前駆体アルコールから合成できる;
【図7】図7は、ISATX247の(Z)−異性体に富む組成物の立体選択的合成の別の反応スキームを示す図である;
【図8】図8は、ISATX247の(E)−異性体に富む組成物の立体選択的合成の別の反応スキームを示す図である;
【図9A】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図9B】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図9C】図9A−Cは、ISATX247の(E)及び(Z)−異性体の混合物を生成する典型的合成経路を示す図であり、前記2異性体が特定の例示的比率で生成するように各反応条件は調節される;
【図10】図10は、ISATX247の(E)及び(Z)−異性体の混合物を生成するための典型的立体選択的経路を示す図であり、ここで2種類の異性体のうちの1種類の異性体に富む組成物を先ず第一につくり、その後所定の比率に合うように混合して所望比を得る;
【図11】図11は、ISATX247(E−異性体45−50%及びZ−異性体50−55%)によるカルシニューリンホスファターゼ活性阻害効果が、シクロスポリンAの阻害効果の3倍より強力であった(IC50によって測定)ことを示すアッセイ結果を提供する;
【図12】図12は、幾つかの重水素化及び非重水素化類似体異性体混合物の構造及び異性体組成を示す;
【図13】図13は、種々の重水素化及び非重水素化類似体異性体混合物によるカルシニューリンホスファターゼ活性の阻害効果が、シクロスポリンAに比較して少なくとも同様に強力であった(IC50によって測定)ことを示すアッセイ結果を提供する。
【特許請求の範囲】
【請求項1】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法。
【請求項2】
β−トリアルキルシリルアルコールの処理に使用される酸が、酢酸、硫酸及びルイス酸からなる群から選択される、請求項1に記載の方法。
【請求項3】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;
c)前記アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項4】
β−トリアルキルシリルアルコールの処理に用いる塩基が、水素化ナトリウム及び水素化カリウムからなる群から選択される、請求項3に記載の方法。
【請求項5】
γ−(トリアルキルシリルアリル)ボロネートエステルがトリメチルシリルアリルボロネートエステルである、請求項1又は3に記載の方法。
【請求項6】
E−γ−(トリアルキルシリルアリル)ジアルキルボランがE−γ−(トリメチルシリルアリル)−9−BBNである、請求項1又は3に記載の方法。
【請求項7】
試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、請求項1又は3記載の方法。
【請求項8】
β−トリアルキルシリルアルコールを酸で処理する工程が、ピーターソンのオレフィン化を含む、請求項1に記載の方法。
【請求項9】
β−トリアルキルシリルアルコールを塩基で処理する工程が、ピーターソンのオレフィン化を含む、請求項3に記載の方法。
【請求項10】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドをリチウム化アリルジフェニルホスフィンオキシドと反応させて、アセチル−(E)−1,3−ジエンを形成し;
b)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項11】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドを[3−(ジフェニルホスフィノ)アリル]チタンと反応させて、チタン含有中間体を形成し;
b)チタン含有中間体をエリスロ−α−付加物にし;
c)前記エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;
d)前記β−オキドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;
e)前記アセチル−(Z)−1,3−ジエンを塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項12】
a)シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;
b)アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して、第一中間体アセチル−η−ブロモシクロスポリンAを生成し;
c)前記第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される中間体を生成し;
d)アセチルシクロスポリンAのトリフェニル又はトリアルキル塩(第二中間体トリフェニルホスホニウムブロミド)から生成するイリドを、ホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
e)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなるプロセスによってつくられる(E)及び(Z)−異性体混合物。
【請求項13】
アセチル−1,3−ジエンの処理に使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド及びカリウムアルコキシドからなる群から選択される、請求項1、3、10、11及び12のいずれかの項に記載の方法。
【請求項14】
アセチル−η−ハロシクロスポリンAのトリフェニルホスホニウム塩を含む、組成物。
【請求項15】
ハリドが、ブロミド、クロリド、及びヨーディドからなる群から選択される、請求項14に記載の組成物。
【請求項16】
アセチルシクロスポリンAのトリフェニルホスホニウムブロミドである、請求項14に記載の組成物。
【請求項17】
シクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む、組成物。
【請求項18】
a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記アセチルシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項19】
a)トリメチルシリル(TMS)シクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記TMSシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項20】
オゾン分解工程が約−80℃ないし0℃の温度で行われる、請求項18又は19に記載の方法。
【請求項21】
還元剤が、トリアルキルホスフィン、トリアリールホスフィン及びトリアルキルアミンからなる群から選択される、請求項18又は19に記載の方法。
【請求項22】
還元剤が、アルキルアリールスルフィド、チオ硫酸塩、及びジアルキルスルフィドからなる群から選択される、請求項18又は19に記載の方法。
【請求項23】
還元剤がジメチルスルフィドである、請求項22に記載の方法。
【請求項24】
還元剤がトリブチルホスフィンである、請求項21に記載の方法。
【請求項25】
還元剤がトリアルキルアミンである、請求項21に記載の方法。
【請求項26】
還元剤がトリエチルアミンである、請求項25に記載の方法。
【請求項27】
アセチルシクロスポリンAのオゾン分解に用いられる溶媒が低級アルコールである、請求項18ないし26のいずれかの項に記載の方法。
【請求項28】
アルコールがメタノールである、請求項27に記載の方法。
【請求項29】
オゾン分解に使用される溶媒が、ジクロロメタン、及びジクロロメタンと低級アルコールとの混液からなる群から選択される、請求項19に記載の方法。
【請求項30】
低級アルコールがメタノールである、請求項29に記載の方法。
【請求項31】
ISATX247のE−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキ
ルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、ISATX247のE−
異性体を形成する
諸工程を含んでなる、前記方法。
【請求項32】
使用する酸が、酢酸、硫酸、又はルイス酸からなる群から選択される、請求項31に記載の方法。
【請求項33】
酸がBF3である、請求項31に記載の方法。
【請求項34】
ISATX247のZ−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してTMS−(Z)−1,3−ジエンを形成し;
c)前記TMS−(Z)−1,3−ジエンを脱保護して、ISATX247のZ−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項35】
E−γ−(トリアルキルシリルアリル)ボランが:
a)アリルトリアルキルシランを塩基で脱プロトン化し;
b)脱プロトン化したアリルトリアルキルシランを、ジアルキルアルコキシボラン及びルイス酸と反応させる
諸工程によってつくられる、請求項31又は34に記載の方法。
【請求項36】
ジアルキルアルコキシボランがジエチルメトキシボランであり、ルイス酸がBF3である、請求項35に記載の方法。
【請求項37】
アリルトリアルキルシランがアリルトリメチルシランである、請求項35に記載の方法。
【請求項38】
アリルトリアルキルシランの脱プロトン化に使用する塩基がブチルリチウムである、請求項35に記載の方法。
【請求項39】
β−トリアルキルシリルアルコールを処理するために使用する塩基が、水酸化ナトリウム、水酸化カリウム、及びアルカリ低級アルコキシドからなる群から選択される、請求項34に記載の方法。
【請求項40】
塩基がカリウムtert−ブトキシドである、請求項39に記載の方法。
【請求項41】
脱保護が、塩酸、酢酸、クエン酸、ルイス酸、及びHFをベースとする試薬からなる群から選択される酸を用いて達成される、請求項34に記載の方法。
【請求項42】
HF−ベースの試薬が、フッ化トリブチルアンモニウム及びフッ化カリウムからなる群から選択される、請求項41に記載の方法。
【請求項43】
a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβアルコールを保護し;
b)前記アセチルシクロスポリンAを、モノペルスルフェート、好ましくはオキソンで、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、7より高いpHで、アセチルシクロスポリンAエポキシドに変換し;
c)前記エポキシドを過ヨウ素酸又は過ヨウ素酸塩で、酸性条件下で分解する諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項44】
工程b及びcが一つの処理操作に組合わせられる、請求項64に記載の方法。
【請求項45】
ISATX247のE−異性体の立体選択的合成方法であって:
a)アセチルシクロスポリンAアルデヒドとE−γ−(トリアルキルシリルアリル)ボランとを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、アセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項46】
β−トリアルキルシリルアルコールを処理する工程が、さらにピーターソンのオレフィン化を含む、請求項31、34又は45に記載の方法。
【請求項47】
ボラン試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、請求項45に記載の方法。
【請求項48】
使用する酸が、酢酸、硫酸又はルイス酸からなる群から選択される、請求項45に記載の方法。
【請求項49】
酸が硫酸である、請求項48に記載の方法。
【請求項50】
1,3−ジエンの処理のために使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、請求項45に記載の方法。
【請求項51】
塩基が、NH3及びジメチルアミンからなる群から選択される、請求項50に記載の方法。
【請求項52】
アセチルシクロスポリン1,3−ジエンをISATX247に変換するための方法であって、前記アセチルシクロスポリン1,3−ジエンが、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、アルコキシドナトリウム、アルコキシドカリウム、及びNH3とヒドロキシルアミンとヒドラジンとジアルキルアミン類からなる群から選択されるアミン塩基からなる群から選択される塩基で処理される、前記方法。
【請求項53】
塩基がジメチルアミンである、請求項52に記載の方法。
【請求項54】
ISATX247のZ−異性体の立体選択的合成法であって;
a)アセチルシクロスポリンAアルデヒドを、第一塩基の存在下で3−(ジメチルアミノ)プロピルトリフェニルホスホニウムハリドで処理して、アセチル−(Z)−オクテニルジメチルアミンを形成し;
b)前記アセチル−(Z)−オクテニルジメチルアミンを酸化試薬で処理して、アセチル−(Z)−オクテニルジメチルアミンオキシドを形成し;
c)前記アセチル−(Z)−オクテニルジメチルアミンオキシドをコープ脱離反応において加熱して、アセチル−(Z)−1,3−ジエンを形成し;
d)前記アセチル−(Z)−1,3−ジエンを第二塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項55】
第一塩基がカリウムヘキサメチルジシラチドである、請求項54に記載の方法。
【請求項56】
酸化試薬がメタクロルペル安息香酸である、請求項54に記載の方法。
【請求項57】
第二塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、請求項54に記載の方法。
【請求項58】
明細書に記載の発明。
【請求項1】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理してアセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理してISATX247の(E)−異性体を形成する諸工程を含んでなる、前記方法。
【請求項2】
β−トリアルキルシリルアルコールの処理に使用される酸が、酢酸、硫酸及びルイス酸からなる群から選択される、請求項1に記載の方法。
【請求項3】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドと、γ−(トリアルキルシリルアリル)ボロネートエステル及びE−γ−(トリアルキルシリルアリル)ジアルキルボランからなる群から選択される試薬とを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してアセチル−(Z)−1,3−ジエンを形成し;
c)前記アセチル−(Z)−1,3−ジエンを塩基で処理してISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項4】
β−トリアルキルシリルアルコールの処理に用いる塩基が、水素化ナトリウム及び水素化カリウムからなる群から選択される、請求項3に記載の方法。
【請求項5】
γ−(トリアルキルシリルアリル)ボロネートエステルがトリメチルシリルアリルボロネートエステルである、請求項1又は3に記載の方法。
【請求項6】
E−γ−(トリアルキルシリルアリル)ジアルキルボランがE−γ−(トリメチルシリルアリル)−9−BBNである、請求項1又は3に記載の方法。
【請求項7】
試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、請求項1又は3記載の方法。
【請求項8】
β−トリアルキルシリルアルコールを酸で処理する工程が、ピーターソンのオレフィン化を含む、請求項1に記載の方法。
【請求項9】
β−トリアルキルシリルアルコールを塩基で処理する工程が、ピーターソンのオレフィン化を含む、請求項3に記載の方法。
【請求項10】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のE−異性体富化混合物の生成方法であって、前記E−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドをリチウム化アリルジフェニルホスフィンオキシドと反応させて、アセチル−(E)−1,3−ジエンを形成し;
b)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247の(E)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項11】
1−アミノ酸残基が修飾されたシクロスポリンA類似体のZ−異性体富化混合物の生成方法であって、前記Z−異性体富化物質の立体選択的合成が:
a)アセチルシクロスポリンAアルデヒドを[3−(ジフェニルホスフィノ)アリル]チタンと反応させて、チタン含有中間体を形成し;
b)チタン含有中間体をエリスロ−α−付加物にし;
c)前記エリスロ−α−付加物をヨードメタン処理によってβ−オキシドホスホニウム塩に変換し;
d)前記β−オキドホスホニウム塩をアセチル−(Z)−1,3−ジエンに変換し;
e)前記アセチル−(Z)−1,3−ジエンを塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項12】
a)シクロスポリンAのβ−アルコールを保護してアセチルシクロスポリンAを形成し;
b)アセチルシクロスポリンAの1−アミノ酸残基の側鎖のη−炭素を臭素化して、第一中間体アセチル−η−ブロモシクロスポリンAを生成し;
c)前記第一中間体アセチル−η−ブロモシクロスポリンAを、トリフェニルホスフィン及びトリアルキルホスフィンからなる群から選択される試薬と共に加熱して、アセチルシクロスポリンAのトリフェニル−及びトリアルキルホスホニウムブロミドからなる群から選択される中間体を生成し;
d)アセチルシクロスポリンAのトリフェニル又はトリアルキル塩(第二中間体トリフェニルホスホニウムブロミド)から生成するイリドを、ホルムアルデヒドと共に撹拌することによって、アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物をつくり;
e)前記アセチル−1,3−ジエンの(E)及び(Z)−異性体の混合物を塩基で処理することによって、ISATX247の(E)及び(Z)−異性体の混合物をつくる諸工程を含んでなるプロセスによってつくられる(E)及び(Z)−異性体混合物。
【請求項13】
アセチル−1,3−ジエンの処理に使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド及びカリウムアルコキシドからなる群から選択される、請求項1、3、10、11及び12のいずれかの項に記載の方法。
【請求項14】
アセチル−η−ハロシクロスポリンAのトリフェニルホスホニウム塩を含む、組成物。
【請求項15】
ハリドが、ブロミド、クロリド、及びヨーディドからなる群から選択される、請求項14に記載の組成物。
【請求項16】
アセチルシクロスポリンAのトリフェニルホスホニウムブロミドである、請求項14に記載の組成物。
【請求項17】
シクロスポリンAのβ−トリメチルシリルアルコール誘導体を含む、組成物。
【請求項18】
a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記アセチルシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項19】
a)トリメチルシリル(TMS)シクロスポリンAを形成することによって、シクロスポリンAのβ−アルコールを保護し;
b)前記TMSシクロスポリンAを酸化剤としてのオゾンで酸化し、その後還元剤で処理する
諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項20】
オゾン分解工程が約−80℃ないし0℃の温度で行われる、請求項18又は19に記載の方法。
【請求項21】
還元剤が、トリアルキルホスフィン、トリアリールホスフィン及びトリアルキルアミンからなる群から選択される、請求項18又は19に記載の方法。
【請求項22】
還元剤が、アルキルアリールスルフィド、チオ硫酸塩、及びジアルキルスルフィドからなる群から選択される、請求項18又は19に記載の方法。
【請求項23】
還元剤がジメチルスルフィドである、請求項22に記載の方法。
【請求項24】
還元剤がトリブチルホスフィンである、請求項21に記載の方法。
【請求項25】
還元剤がトリアルキルアミンである、請求項21に記載の方法。
【請求項26】
還元剤がトリエチルアミンである、請求項25に記載の方法。
【請求項27】
アセチルシクロスポリンAのオゾン分解に用いられる溶媒が低級アルコールである、請求項18ないし26のいずれかの項に記載の方法。
【請求項28】
アルコールがメタノールである、請求項27に記載の方法。
【請求項29】
オゾン分解に使用される溶媒が、ジクロロメタン、及びジクロロメタンと低級アルコールとの混液からなる群から選択される、請求項19に記載の方法。
【請求項30】
低級アルコールがメタノールである、請求項29に記載の方法。
【請求項31】
ISATX247のE−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキ
ルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、ISATX247のE−
異性体を形成する
諸工程を含んでなる、前記方法。
【請求項32】
使用する酸が、酢酸、硫酸、又はルイス酸からなる群から選択される、請求項31に記載の方法。
【請求項33】
酸がBF3である、請求項31に記載の方法。
【請求項34】
ISATX247のZ−異性体の立体選択的合成方法であって:
a)トリメチルシリル(TMS)シクロスポリンAアルデヒドをE−γ−(トリアルキルシリルアリル)ボランと反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを塩基で処理してTMS−(Z)−1,3−ジエンを形成し;
c)前記TMS−(Z)−1,3−ジエンを脱保護して、ISATX247のZ−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項35】
E−γ−(トリアルキルシリルアリル)ボランが:
a)アリルトリアルキルシランを塩基で脱プロトン化し;
b)脱プロトン化したアリルトリアルキルシランを、ジアルキルアルコキシボラン及びルイス酸と反応させる
諸工程によってつくられる、請求項31又は34に記載の方法。
【請求項36】
ジアルキルアルコキシボランがジエチルメトキシボランであり、ルイス酸がBF3である、請求項35に記載の方法。
【請求項37】
アリルトリアルキルシランがアリルトリメチルシランである、請求項35に記載の方法。
【請求項38】
アリルトリアルキルシランの脱プロトン化に使用する塩基がブチルリチウムである、請求項35に記載の方法。
【請求項39】
β−トリアルキルシリルアルコールを処理するために使用する塩基が、水酸化ナトリウム、水酸化カリウム、及びアルカリ低級アルコキシドからなる群から選択される、請求項34に記載の方法。
【請求項40】
塩基がカリウムtert−ブトキシドである、請求項39に記載の方法。
【請求項41】
脱保護が、塩酸、酢酸、クエン酸、ルイス酸、及びHFをベースとする試薬からなる群から選択される酸を用いて達成される、請求項34に記載の方法。
【請求項42】
HF−ベースの試薬が、フッ化トリブチルアンモニウム及びフッ化カリウムからなる群から選択される、請求項41に記載の方法。
【請求項43】
a)アセチルシクロスポリンAを形成することによって、シクロスポリンAのβアルコールを保護し;
b)前記アセチルシクロスポリンAを、モノペルスルフェート、好ましくはオキソンで、ケトン、好ましくは活性化ケトン、より好ましくはアセトキシアセトン又はジアセトキシアセトンの存在下で、7より高いpHで、アセチルシクロスポリンAエポキシドに変換し;
c)前記エポキシドを過ヨウ素酸又は過ヨウ素酸塩で、酸性条件下で分解する諸工程を含んでなる、シクロスポリンAアルデヒドの調製方法。
【請求項44】
工程b及びcが一つの処理操作に組合わせられる、請求項64に記載の方法。
【請求項45】
ISATX247のE−異性体の立体選択的合成方法であって:
a)アセチルシクロスポリンAアルデヒドとE−γ−(トリアルキルシリルアリル)ボランとを反応させて、β−トリアルキルシリルアルコールを形成し;
b)前記β−トリアルキルシリルアルコールを酸で処理して、アセチル−(E)−1,3−ジエンを形成し;
c)前記アセチル−(E)−1,3−ジエンを塩基で処理して、ISATX247のE−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項46】
β−トリアルキルシリルアルコールを処理する工程が、さらにピーターソンのオレフィン化を含む、請求項31、34又は45に記載の方法。
【請求項47】
ボラン試薬がE−γ−(トリメチルシリルアリル)ジエチルボランである、請求項45に記載の方法。
【請求項48】
使用する酸が、酢酸、硫酸又はルイス酸からなる群から選択される、請求項45に記載の方法。
【請求項49】
酸が硫酸である、請求項48に記載の方法。
【請求項50】
1,3−ジエンの処理のために使用する塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、請求項45に記載の方法。
【請求項51】
塩基が、NH3及びジメチルアミンからなる群から選択される、請求項50に記載の方法。
【請求項52】
アセチルシクロスポリン1,3−ジエンをISATX247に変換するための方法であって、前記アセチルシクロスポリン1,3−ジエンが、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、アルコキシドナトリウム、アルコキシドカリウム、及びNH3とヒドロキシルアミンとヒドラジンとジアルキルアミン類からなる群から選択されるアミン塩基からなる群から選択される塩基で処理される、前記方法。
【請求項53】
塩基がジメチルアミンである、請求項52に記載の方法。
【請求項54】
ISATX247のZ−異性体の立体選択的合成法であって;
a)アセチルシクロスポリンAアルデヒドを、第一塩基の存在下で3−(ジメチルアミノ)プロピルトリフェニルホスホニウムハリドで処理して、アセチル−(Z)−オクテニルジメチルアミンを形成し;
b)前記アセチル−(Z)−オクテニルジメチルアミンを酸化試薬で処理して、アセチル−(Z)−オクテニルジメチルアミンオキシドを形成し;
c)前記アセチル−(Z)−オクテニルジメチルアミンオキシドをコープ脱離反応において加熱して、アセチル−(Z)−1,3−ジエンを形成し;
d)前記アセチル−(Z)−1,3−ジエンを第二塩基で処理して、ISATX247の(Z)−異性体を形成する
諸工程を含んでなる、前記方法。
【請求項55】
第一塩基がカリウムヘキサメチルジシラチドである、請求項54に記載の方法。
【請求項56】
酸化試薬がメタクロルペル安息香酸である、請求項54に記載の方法。
【請求項57】
第二塩基が、水酸化ナトリウム、炭酸ナトリウム、炭酸カリウム、ナトリウムアルコキシド、カリウムアルコキシド、及びNH3とヒドロキシルアミンとヒドラジンと低級ジアルキルアミンとからなる群から選択されるアミン塩基からなる群から選択される、請求項54に記載の方法。
【請求項58】
明細書に記載の発明。
【図9A】

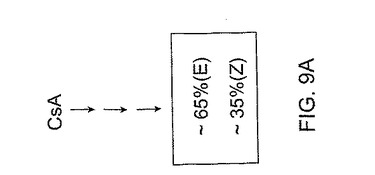
【図9B】
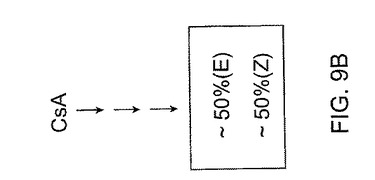
【図9C】
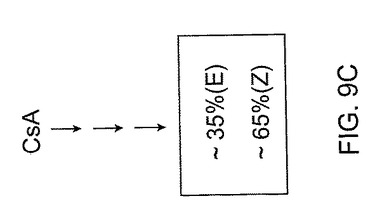
【図10】
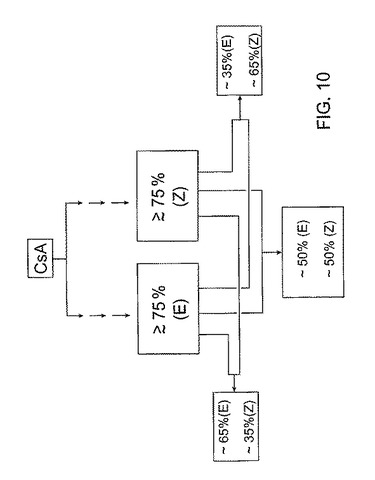
【図1】
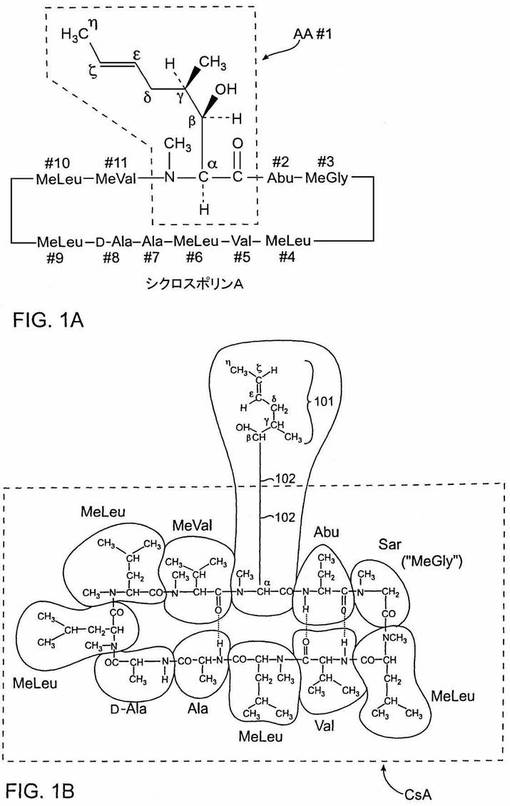
【図2】

【図3】
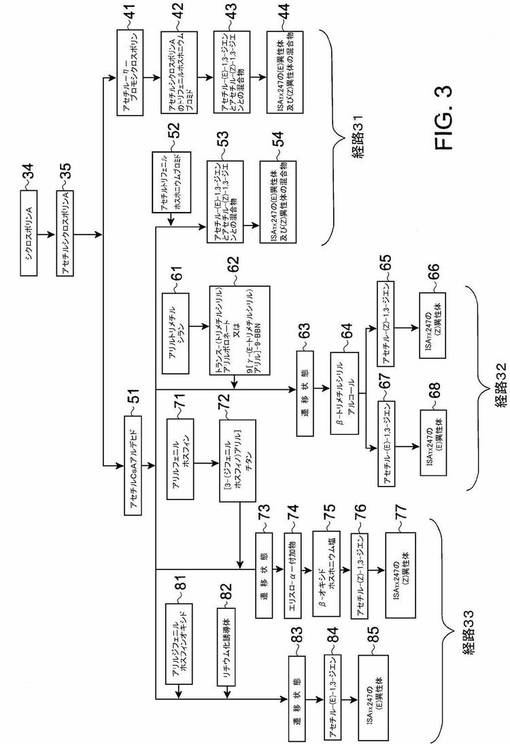
【図4】
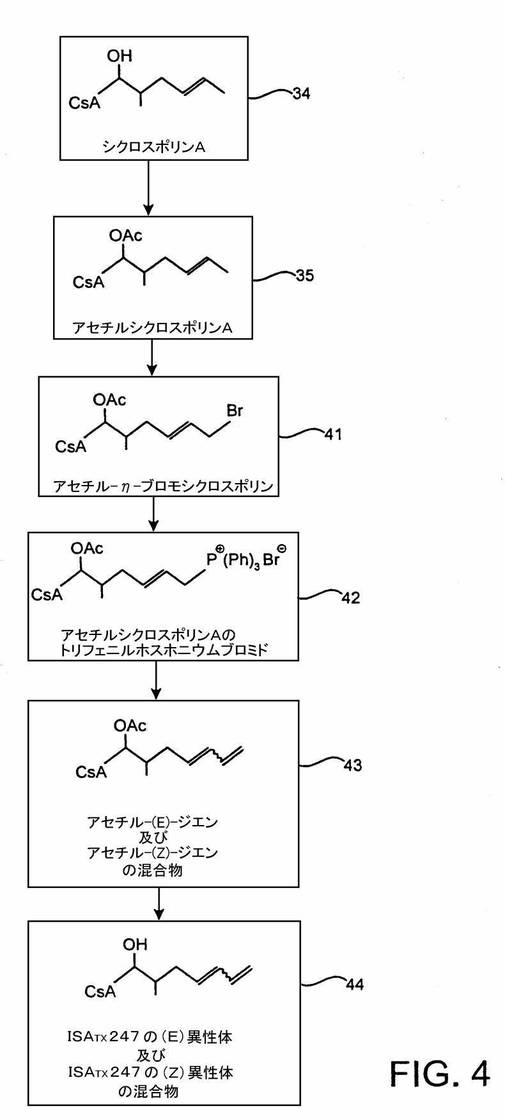
【図5】
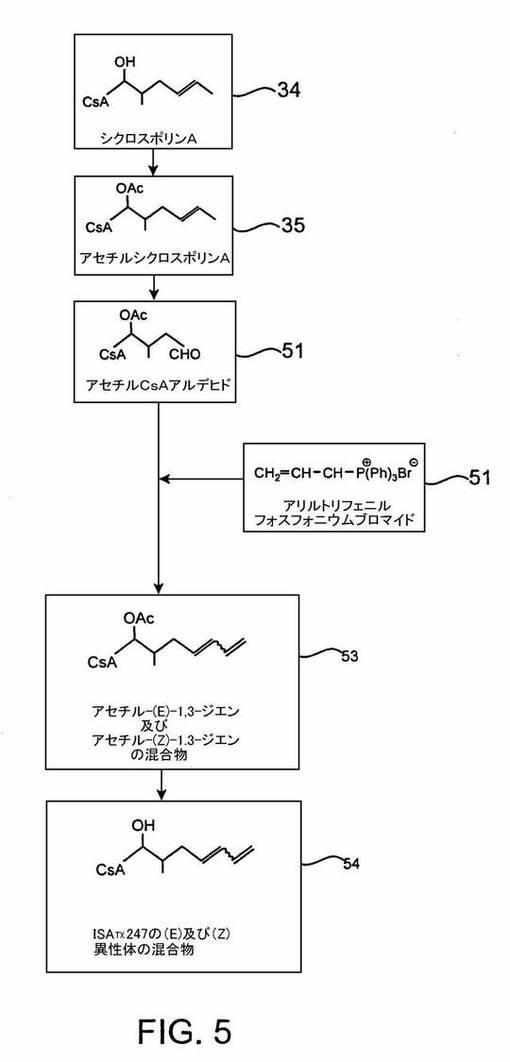
【図6】
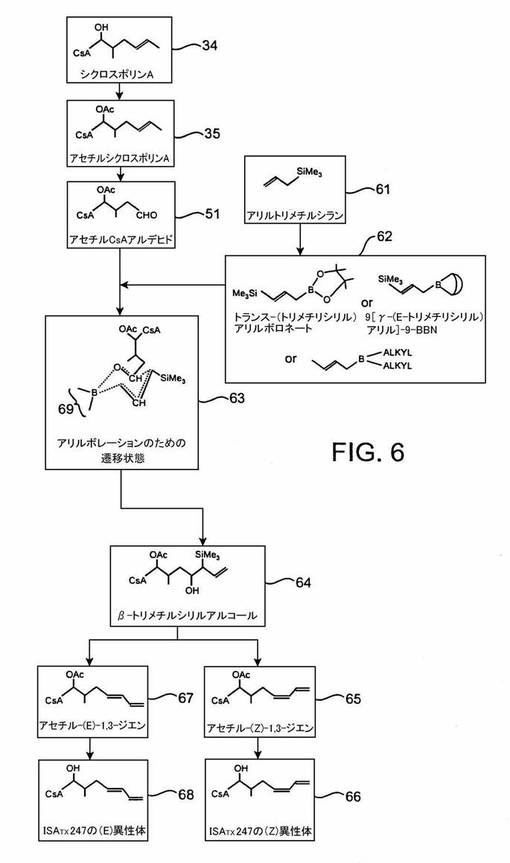
【図7】
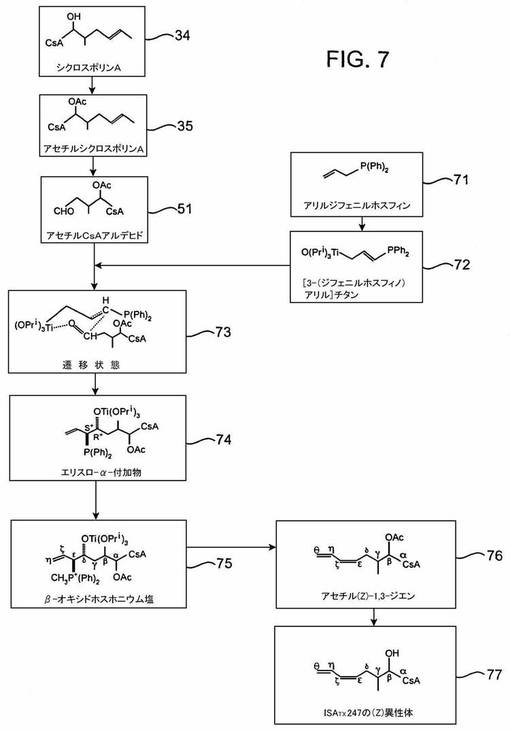
【図8】
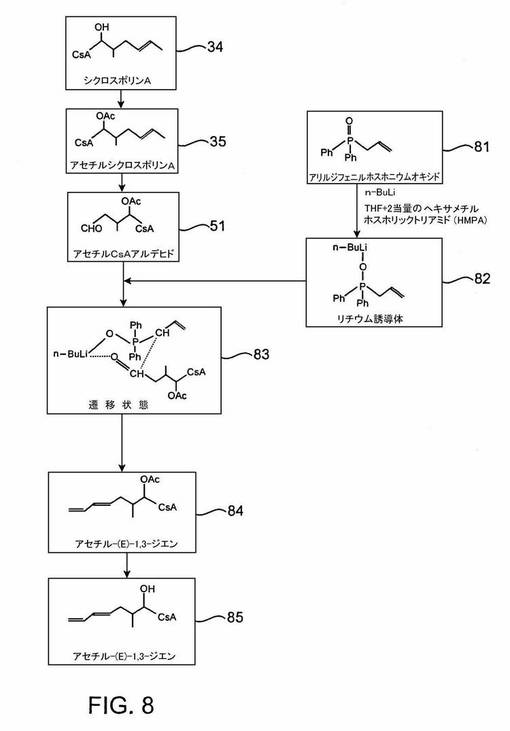
【図11】
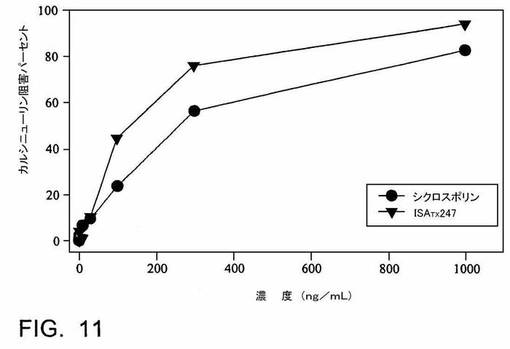
【図12】
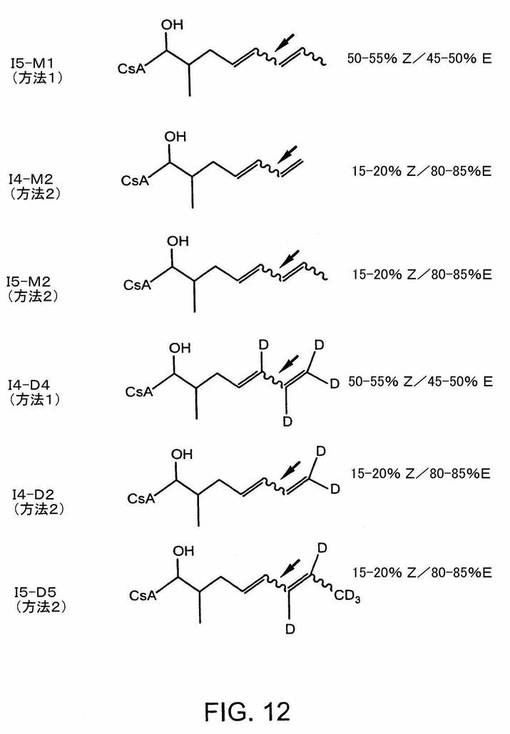
【図13】
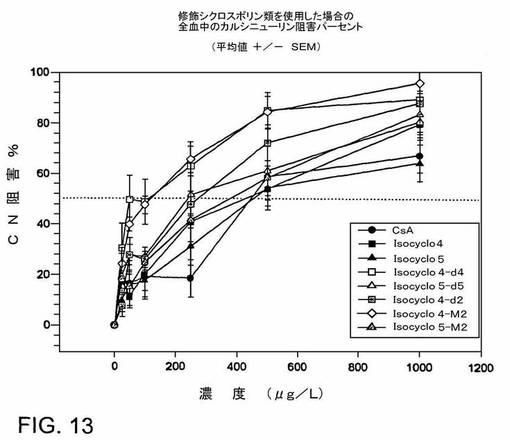
【図9B】
【図9C】
【図10】
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図11】
【図12】
【図13】
【公開番号】特開2009−46515(P2009−46515A)
【公開日】平成21年3月5日(2009.3.5)
【国際特許分類】
【出願番号】特願2008−271446(P2008−271446)
【出願日】平成20年10月21日(2008.10.21)
【分割の表示】特願2003−536264(P2003−536264)の分割
【原出願日】平成14年10月17日(2002.10.17)
【出願人】(500132720)アイソテクニカ インコーポレイテッド (10)
【Fターム(参考)】
【公開日】平成21年3月5日(2009.3.5)
【国際特許分類】
【出願日】平成20年10月21日(2008.10.21)
【分割の表示】特願2003−536264(P2003−536264)の分割
【原出願日】平成14年10月17日(2002.10.17)
【出願人】(500132720)アイソテクニカ インコーポレイテッド (10)
【Fターム(参考)】
[ Back to top ]