
シクロパミン類似体の使用方法
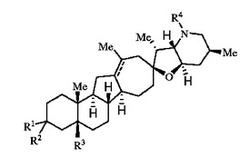
【課題】 ヘッジホッグ経路のより強力な阻害剤を提供すること。
【解決手段】 下記式を有するシクロパミンの類似体を用いて、ヘッジホッグ経路(hedgehog pathway)によってもたらされる過剰増殖疾患及び様々な症状を処置することを特徴とする。

【解決手段】 下記式を有するシクロパミンの類似体を用いて、ヘッジホッグ経路(hedgehog pathway)によってもたらされる過剰増殖疾患及び様々な症状を処置することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
この出願は、2006年12月28日に出願されたUSSN60/878,018及び2007年6月1日に出願されたUSSN60/941,596の優先権を主張し、これらの出願はそれらのすべてが参照によって本願明細書に組み込まれる。
【政府提供資金】
【0002】
本明細書に記載の幾つかの研究(some of the work)は、NIH/NCIによって与えられた認可番号K23CA107040の政府支援の下で行われた。(合衆国連邦)政府は本発明の特定の権利を有し得る。
【背景技術】
【0003】
本発明は、一般的に、ヘッジホッグ経路に対抗(アンタゴナイズ)し、かつ、シクロパミン類似体を用いて様々な症状を処置するための方法に関する。
【0004】
ある種の癌におけるヘッジホッグ経路の抑制は、腫瘍成長の抑制に帰着することが知られている。例えば、抗ヘッジホッグ抗体は、ヘッジホッグ経路の機能をアンタゴナイズし、かつ、腫瘍の成長を抑制することが知られている。ヘッジホッグ経路の低分子抑制もまた、いくつかの癌タイプで細胞死に帰着することが知られている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
この分野の研究は、主に、ヘッジホッグ経路生物学の解明及び新規のヘッジホッグ経路阻害剤ないし抑制剤(inhibitors)の発見に注力されている。ヘッジホッグ経路の阻害剤が特定されてはいるが、ヘッジホッグ経路のより強力な阻害剤を特定する必要がある。
【0006】
2006年3月9日に公開され、本願と同じ譲受人に譲渡されたPCT公開公報(WO2006/026430)は、多種多様なシクロパミン類似体を開示し、それらシクロパミンのA又はB環の不飽和に注視している。本願においては、驚くべきことに、強力な類似体は完全に飽和したA及びB環を含んでいる。
【課題を解決するための手段】
【0007】
本発明は、ヘッジホッグ経路(hedgehog pathway)によってもたらされる(mediated)過剰増殖(hyperproliferative)疾患及び症状を処置するための方法に関する。
【0008】
一視点において、本発明は過剰増殖疾患を処置するための方法に関する。本方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;R4の上記式中、各Wは夫々独立してジラジカルであり;
各qは夫々1、2、3、4、5若しくは6において独立であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
ある実施態様において、R1はスルホンアミドである。
【0009】
症状は、皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される。
【0010】
別の視点において、本発明はヘッジホッグ経路(hedgehog pathway)によってもたらされる(mediated)症状を処置するための方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;各qは、夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
ある実施態様において、R1はスルホンアミドである。
【0011】
症状は、皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される。ある実施例は、小細胞肺癌、膵臓癌、髄芽腫、多発性髄芽腫、白血病、骨髄異形成症候群、非ホジキンリンパ腫、及びホジキン病を含む。化合物は、経口的、静脈内、又は、局所的に投与されてよい。
【0012】
別の視点において、本発明は、被験者ないし患者においてヘッジホッグ経路に対抗(アンタゴナイズ)する方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;R4の上記式中、各Wは、夫々独立してジラジカルであり;各qは夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
【0013】
上記の実施態様において、化合物は下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である。
【0014】
別の視点において、本発明は、被験者ないし患者においてヘッジホッグ経路に対抗(アンタゴナイズ)する方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは独立して1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは独立してH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成し;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)であり、ここに各Rは独立してH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
R7及びR7´の夫々がHであり;又は
R7及びR7´が一緒になって=Oを形成し;
R8及びR9は夫々Hであるか、又は、R8及びR9は一緒になってボンド(結合)を形成し;及び
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1はヒドロキシルとなることはできず、かつ、R2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1は酢酸となることはできず、かつ、R2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR8がHである場合;R1及びR2が一緒になって=Oとなることができず;及び
R3、R4、R8、R9がHであり、かつR7及びR7´がHである場合;R1及びR2がHとなることができない。
ある実施態様において、R1はスルホンアミドである。
【発明を実施するための形態】
【0015】
本願明細書で用いた用語の定義は、化学及び薬学分野における各用語として認識された現在の技術水準の定義を組み込むことを意図する。適切な場所に、実例(exemplification)を提供する。定義は、特定の場合に限定される場合を除き、個々に又は大きな群の一部のいずれかとして、この明細書を通して使用された用語に適用する。
【0016】
本願明細書で使用されるように、各表現ないし語句(expression)の定義、例えば、アルキル、m、nなどは、それが任意の構造(式)の中で1回以上出現した場合、同一の構造(式)における他の場所のその定義からは独立であることを意図する。
【0017】
用語「アシルアミノ」(acylamino)は、一般式:
によって表すことができる部分ないし部位(半部分:moiety)を意味し、
式中、R50及びR54は、水素、アルキル、アルケニル若しくは‐(CH2)m‐R61を表し、
式中、R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環(polycycle)を表し、そして、mは、ゼロ若しくは1から8までの範囲内の整数である。
【0018】
用語「アルケニル」及び「アルキニル」は、長さ及び置換可能な点で、上記アルキルに類似するが、少なくとも1つの二重若しくは三重結合を夫々含有する不飽和脂肪族基を意味する。
【0019】
用語「アルコキシル」若しくは「アルコキシ」は、そこに結合した酸素ラジカルを有する、上記で定義したアルキル基を意味する。代表的なアルコキシル基は、メトキシ、エトキシ、プロピルオキシ、tert‐ブトキシ及び同様のものを含む。
【0020】
用語「アルキル」は、直鎖アルキル基、分枝状鎖のアルキル基、シクロアルキル(脂環式)基、アルキル置換シクロアルキル基、及びシクロアルキル置換アルキル基を含む飽和脂肪族基のラジカルを意味する。ある実施態様では、直鎖若しくは分岐状鎖のアルキルは、30以下の炭素原子を主要骨格ないしバックボーン(backbone)に有する(例えば、C1‐C30が直鎖であり、C3‐C30が分岐状鎖である)、20以下(の炭素原子を主要骨格に有する)。同様に、あるシクロアルキルは、3‐10の炭素原子をその環状構造に有し、その他は5、6若しくは7の炭素を環状構造に有する。
【0021】
用語「アルキルチオ」は、そこに結合した硫黄ラジカルを有する、上記で定義したアルキル基を意味する。ある実施態様では、「アルキルチオ」の部分ないし部位は、‐S‐アルキル、‐S‐アルケニル、‐S‐アルキニル及び‐S‐(CH2)m‐R61の1つによって表され、式中、mおよびR61は、上記で定義される。代表的なアルキルチオ基は、メチルチオ、エチルチオ及び同様のものを含む。
【0022】
用語「アミド」は、アミノ置換カルボニルとして当該技術で認識(art recognized)され、そして一般式:
によって表すことができる部分ないし部位を含み、式中、R50及びR51は、夫々独立して、水素、アルキル、アルケニル、‐(CH2)m‐R61を表し、又は、R50及びR51は、それらが結合するN原子と一緒になって、4から8までの原子を環状構造に有するヘテロ環を完成し、R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環を表し、そしてmはゼロ若しくは1から8までの範囲内の整数である。本発明のアミドのある実施態様は、不安定でありうるイミドを含まないだろう。
【0023】
用語「アミン」及び「アミノ」は、当該技術で認識され、そして非置換及び置換アミンの両方を意味し、例えば、一般式:
によって表すことができる部分であり、式中、R50、R51及びR52は、夫々独立して、水素、アルキル、アルケニル又は(CH2)m‐R61を表し、又は、R50及びR51は、それらが結合するN原子と一緒になって、4から8までの原子を環状構造に有するヘテロ環を完成し;R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環を表し、そしてmはゼロ若しくは1から8までの範囲内の整数である。したがって、用語「アルキルアミン」は、そこに結合した置換若しくは非置換アルキルを有する、すなわち、少なくともR50及びR51の一つはアルキル基である、上記で定義したアミン基を含む。
【0024】
本願明細書で使用した用語「アラルキル」は、アリール基(例えば、芳香族若しくはヘテロ芳香族基)で置換されたアルキル基を意味する。
【0025】
本願明細書で使用された用語「アリール」は、0から4までのヘテロ原子を含むことができる5、6及び7員単環芳香族基、例えば、ベンゼン、アントラセン、ナフタレン、ピレン、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、トリアゾール、ピラゾール、ピリジン、ピラジン、ピリダジン及びピリミジンを含み、そして同様のものを含む。環状構造にヘテロ原子を有するそれらのアリール基を、「アリールヘテロ環」若しくは「ヘテロ芳香族」と称することもできる。芳香族環は1つ以上の環状位置(ring positions)を上記置換基、例えば、ハロゲン、アジド、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アルコキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、スルホンアミド、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分(moieties)、‐CF3(トリフルオロメタン)、−CN(シアン)若しくは同様のもので置換することができる。用語「アリール」は、2以上の炭素が2つの隣接する環に共通する(その環は、「縮合環」である)、2つ以上のサイクリック環を有する多環の環状システムも含み、ここで、少なくとも、1の環は、芳香族であり、例えば、他のサイクリック環は、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/またはヘテロ環であり得る。
【0026】
用語「ブレンステッド酸」(Bronsted acid)は、水素イオン(プロトン)供与体ないしドナー(donor)として作用することができる任意の物質(substance)を意味する。
【0027】
用語「カルボキシル」は、一般式:
によって表すことができるような部分ないし部位を含み、式中、X50は、ボンド(結合)であるか、若しくは、酸素または硫黄を表し、そして、R55及びR56の夫々は、独立して、水素、アルキル、アルケニル、−(CH2)m−R61、又は薬学的に許容可能な塩を表し、ここで、mおよびR61は、上記で定義されている。
【0028】
用語「ジラジカル」は、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール及びヘテロアラルキル基からなる一連の二価群の任意のものを意味する。例えば、
は、アルキルジラジカルであり、
もアルキルジラジカルであり、
は、アラルキルジラジカルであり、
は(アルキル)へテロアラルキルジラジカルである。典型的な例は、一般的な構造(CH2)xのアルキレン、ここでXは1‐6である、及び、2‐6の炭素原子及び1つ以上の二重若しくは三重結合を有するアルケニレン及びアルキニレンリンカーに対応するもの、3‐8員環を有するシクロアルキレン基、及び
とその異性体のようなアラルキル基、ここで、1の開放原子価(open valence)はアリール環の上で、1(の開放原子価)はアルキル部の上である、を含む。
【0029】
“有効量”は化合物の量を意味し、大体の所望の効果(例えば、処置される疾患のための臨床的に許容可能な基準(clinically acceptable standards)に従う、細胞増殖の比率及び/又は細胞の生存率における変化)をもたらす所望の投与計画(dosage regimen)の一部として投与されるときの化合物の量である。
【0030】
本願明細書で使用される用語「ハロアルキル(haloalkyl)」は、1から全水素の任意のところがハライドで置き換えられたアルキル基を意味する。「パーハロアルキル(perhaloalkyl)」は、全ての水素がハライドで置き換えられた[アルキル基]である。
【0031】
本願明細書で使用される用語「ヘテロ原子」は、炭素又は水素以外の任意の元素の原子を意味する。ヘテロ原子の例は、ホウ素、窒素、酸素、リン、硫黄及びセレニウムを含む。
【0032】
用語「ヘテロ環」若しくは「ヘテロ環基」は、環状構造が1から4のヘテロ原子を含む3から10員環構造(幾つかの例では、3から7員環)を意味する。ヘテロ環は、多環(polycycles)でもあり得る。ヘテロ環基は、例えば、チオフェン、チアントレン、フラン、ピラン、イソベンゾフラン、クロメン、キサンテン、フェノキサチイン、ピロール、イミダゾール、ピラゾール、イソチアゾール、イソキサゾール(ないしイソオキサゾール)、ピリジン、ピラジン、ピリミジン、ピリダジン、インドリジン、イソインドール、インドール、インダゾール、プリン、キノリジン、イソキノリン、キノリン、フタラジン、ナフチリジン、キノキサリン、キナゾリン、シンノリン、プテリジン、カルバゾール、カルボリン、フェナントリジン、アクリジン、ピリミジン、フェナントロリン、フェナジン、フェナルサジン、フェノチアジン、フラザン、フェノキサジン、ピロリジン、オキソラン、チオラン、オキサゾール、ピペリジン、ピペラジン、モルフォリン、ラクトン、アゼチジノン及びピロリジノンのようなラクタム、スルタム、スルトンおよび同様のものを含む。ヘテロ環は、1つ以上の環状位置(ring positions)を上記置換基、例えば、ハロゲン、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分(moieties)、‐CF3、‐CN又は同様のもので置換することができる。
【0033】
本発明の化合物との関係における用語「分離された」(isolated)は、化合物が細胞若しくは生命体内に存在せず、化合物が典型的には自然界においてそれに付いているいくつかのまたは、全ての化合物から引き離されたことを意味する。
【0034】
用語「ルイス酸」は、電子対の受容体として作用することができる任意の物質を意味する。
【0035】
炭素の数が他に特定されていなければ、本願明細書で使用される「低級アルキル」は、上記で定義したアルキル基を意味するが、その主要骨格の構造において、1から10の炭素を有する、いくつかの実施態様では、1から6の炭素原子を有するアルキル基を意味する。同様に、「低級アルケニル」及び「低級アルキニル」は、同様の鎖の長さを有する。あるアルキル基は、低級アルキルである。いくつかの実施態様では、アルキルとして本願明細書でデザインされた置換基は、低級アルキルである。
【0036】
本願明細書で使用されるように、用語「ニトロ」は、−NO2を意味し、用語「ハロゲン」は、‐F、‐Cl、‐Br若しくは‐Iを意味し、用語「スルフヒドリル」は、‐SHを意味し、用語「ヒドロキシル」は、‐OHを意味し、そして、用語「スルホニル」は、‐SO2‐を意味する。
【0037】
用語「オキソ」は、カルボニル酸素(=0)を意味する。
【0038】
用語「ポリシクリル」若しくは「多環基」は、2以上の炭素が2の隣接する環、例えば、その環は「縮合環」である、に共通する2つ以上の環(例えば、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/またはヘテロ環)を意味する。隣接していない原子を通して連結した環を、「架橋(bridged)」環と名付けた。多環の環の夫々は、上記のような置換基、例えば、ハロゲン、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分、‐CF3、−CN、又は同様のもので置換することができる。
【0039】
本発明の化合物との関係における用語「エピメリカルに純粋(epimerically pure)」は、化合物が、R3が結合した不斉中心の立体配置が反転した化合物の立体異性体を実質的に含まない(substantially free of)ことを意味する。例えば、以下の式:
(ここで、R1、R2、R3、R4、R7、R7´、R8及びR9は下記に定義される)
によって表されるエピメリカルに純粋な化合物は、以下の式:
(ここで、R1、R2、R3、R4、R7、R7´、R8及びR9は下記に定義される)
によって表される化合物を実質的に含まない。すなわち、エピメリカルに純粋な化合物は、質量で約20%未満、質量で約15%未満、質量で約10%未満、質量で約5%未満、若しくは、質量で約3%未満の、その化合物に関するR3が結合した不斉中心の立体配置が反転した立体異性体化合物を含む。
【0040】
本願明細書で使用される表現「保護基」は、望まない化学的な変換から潜在的な反応性の官能基を保護する一時的な置換基を意味する。そのような保護基の例は、カルボン酸のエステル、アルコールのシリルエーテル、そして、アルデヒド及びケトンのアセタール及びケタール夫々を含む。保護基化学の分野は、概説されている(Greene, T. W.; Wuts, P.G.M., Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991)。いくつかのケースでは、保護化された官能基及び保護基は、一緒になって、1部分ないし部位を意味する。例えば、下に示した断片ないしフラグメントは、しばしば、ベンジルカルボナートとして称される、すなわち、保護された(下線が引かれた)O(酸素)は、カルボナートの一部を構成する。
【0041】
同様に、保護化されたN(窒素)がカルバメートの一部を構成している下記に示した断片ないしフラグメントは、ベンジルカルバメートとして称される。
【0042】
本願明細書で使用される用語「糖」は、1以上のピラノース若しくはフラノース環を含有する天然の若しくは非天然の単糖、二糖若しくは多糖を意味する。糖は、エーテル結合若しくはアルキルの架橋(alkyl linkage)を介して本発明のステロイド系アルカロイドに共有結合的に結合することができる。ある実施態様では、糖の部分ないし部位は、サッカライドリング(saccharide ring)のアノマー中心において、本発明のステロイド系アルカロイドに共有結合的に結合することができる。糖は、リブロース、アラビノース、キシロース、リキソース、アロース、アルトロース、グルコース、マンノース、グロース、イドース、ガラクトース、タロース、グルコース及びトレハロースを含むことができるが、限定されない。
【0043】
本願明細書で使用される用語「スルホンアミド(sulfonamido)」ないし「スルホンアミド(sulfonamide)」は、以下の式:
ここで、R50は、上記で定義されている、のいずれかを有する部分を含む。
【0044】
用語「トリフリル」、「トシル」、「メシル」及び「ノナフリル」は、トリフルオロメタンスルホニル、p‐トルエンスルホニル、メタンスルホニル及びノナフルオロブタンスルホニル基を夫々意味する。用語「トリフレート」、「トシレート」、「メシレート」及び「ノナフレート」は、トリフルオロメタンスルホン酸エステル、p‐トルエンスルホン酸エステル、メタンスルホン酸エステル、ノナフルオロブタンスルホン酸エステル官能基、及びその基を含む分子を夫々意味する。
【0045】
用語「チオキソ」は、カルボニル硫黄(carbonyl sulfur, =S)を意味する。
【0046】
「置換」若しくは、「置換された」とは、そのような置換が、置換された原子及び置換基の許容原子価(permitted valence)に従う、また、置換は安定した、例えば、再編成、環化、脱離などによって自発的に変形を起こさない化合物になるという暗黙の条件(implicit proviso)を含むと理解されるだろう。
【0047】
本発明のある化合物は、幾何学的な、若しくは立体異性の形状で存在することができる。本発明は、シス‐及びトランス‐異性体、R‐及びS‐鏡像異性体、ジアステレオマー、(D)‐異性体、(L)‐異性体、そのラセミ混合物及びその他の混合物を含む、全ての化合物が発明の範囲内に入るものと考えられる。さらに、不斉炭素原子は、アルキル基のような置換基に存在できる。全てのそのような異性体及びその混合物は、本発明に含まれることが意図される。
【0048】
上で述べたように、当該化合物のある実施態様は、アミノ、アルキルアミノのような塩基性の官能基を含むことができ、したがって、薬学的に許容可能な酸と、薬学的に許容可能な塩を形成することができる。この点において、用語「薬学的に許容可能な塩」は、本発明の化合物の比較的毒性でない、本発明の化合物の無機及び有機酸の添加塩を意味する。これらの塩は、投与媒体のインサイチュ(in situ)において若しくは剤形製造工程内において、また、個々に、精製されたフリーベースフォーム(free base form)の発明の化合物と、適切な有機若しくは無機酸とを反応し、後の精製の間にそのように形成された塩を分離することによって調製することができる。代表的な塩は、臭化水素酸塩、塩酸塩、硫酸塩、硫酸水素塩(bisulfate)、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート(tosylate)、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチレート(napthylate)、メシル酸塩、グルコヘプトン酸塩、ラクトビオン酸及びラウリルスルホン酸塩(laurylsulphonate salts)、そして同様のものを含む。(例えば、Berge, et al.”Pharmaceutical Salts“ J. Pharm. Sci. (1977) 66:1-19参照)。
【0049】
本発明の化合物の薬学的に許容可能な塩は、例えば、無毒性の有機若しくは無機酸からなる、化合物の慣用的な無毒性の塩若しくは4級アンモニウム塩を含む。例えば、そのような慣用的な無毒性の塩は、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸、硝酸及び同様のもののような無機酸からなるそれらを含み、そして、塩は、酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パルミチン酸、マレイン酸、ヒドロキシマレイン酸(hydroxymaleic)、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸(salicyclic)、スルファニル酸(sulfanilic)、2‐アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、イソチオニック(isothionic)および同様のもののような有機酸から調製された塩を含む。
【0050】
他のケースでは、本発明の化合物は、1以上の酸性の官能基を含むことができ、そして、従って、薬学的に許容可能な塩基と薬学的に許容可能な塩を作ることができる。これらの例における用語「薬学的に許容可能な塩」は、比較的に無毒性の無機及び有機塩基を添加した本発明の化合物の塩を意味する。これらの塩は、同様に、インサイチュ(in situ)における投与媒体内若しくは剤形製造工程内で調製することができ、若しくは、薬学的に許容可能な金属カチオンの水酸化物、炭酸化物若しくは炭酸水素化物と、アンモニア若しくは薬学的に許容可能な1級、2級、3級若しくは4級の有機アミンとのように、遊離酸形状にある精製した化合物と適した塩基とを個々に反応することによって調製することができる。代表的なアルカリ(alkaliないしalkaline)土類塩は、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム及びアルミニウム塩、そして、同様のものを含む。塩基添加塩の形成に有用な代表的な有機アミンは、エチルアミン、ジエチルアミン、エチレンジアミン、エタノールアミン、ジエタノールアミン、ピペラジン、及び同様のものを含む。(例えば、Berge, et al., supra参照)
【0051】
ステロイド系アルカロイド化合物の合成
上記環拡大ステロイド系アルカロイド誘導体は、自然発生のステロイド系アルカロイド若しくはその合成類似体から直接調製できる。ある例では、ステロイド系アルカロイド出発物質は、シクロパミン若しくはジャービン(jervine)があり得る。これらのステロイド系アルカロイドは、商業的に購入でき若しくはバケイソウ(Veratrum Californicum)から抽出できる。手短には、本発明の方法は、適切な出発ステロイド系アルカロイド誘導体のシクロプロパン化、続いて、シクロプロピル誘導体の環拡大再編成のステップを含む。いくつかの例では、分子上に存在する反応性官能基をシクロプロパン化の前に適切に保護すること又は他に変換することが望ましいだろう。例えば、R1に存在するアルコール及び融合したフラノ‐ピペリジン環上に存在する2級の窒素は、両方ともシクロプロパン化の前に保護化できる。ある実施態様では、効率的にアルカロイドに加え、そしてアルカロイドから取り除かれ、合成プロセスにおける中間体の取り扱い性質を改善して産出し、形成された合成中間体の効率的な精製を可能にする保護基が望ましいだろう。
【0052】
酸素保護基の例は、ギ酸、酢酸、クロロ酢酸、ジクロロ酢酸、トリクロロ酢酸、ピバロアート(pivaloate)、ベンゾアート、アルキルカルボナート、アルケニルカルボナート、アリールカルボナート、アラルキルカルボナート(例えば、ベンジルカルボナート)、2,2,2‐トリクロロエチルカルボナート、アルコキシメチルエーテル、アラルコキシメチルエーテル、アルキルチオメチルエーテル、アラルキルチオエーテル、アリールチオエーテル、トリアルキルシリルエーテル、アルキルアリールシリルエーテル、ベンジルエーテル、アリールメチルエーテル及びアリルエーテルを含むが、これらに限定されない。
【0053】
窒素保護基の例は、ホルミル、クロロアセチル、トリクロロアセチル、トリフルオロアセチル、フェニルアセチル、ベンゾイル、ベンズアミド、アルキルカルバメート、アラルキルカルバメート(例えば、ベンジルカルバメート)、アリールカルバメート、アリル、アラルキル、アルコキシメチル、アラルコキシメチル、N‐2‐シアノエチル、ジアリールホスフィンアミド、ジアルキルホスフィンアミダート、ジアリールホスフィンアミダート及びトリアルキルシリルを含むが、これらに限定されない。
【0054】
本発明の方法に使用できる更なる保護基は、Green, T. W.; Wuts, P. G., Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. 1999.に開示されている。
【0055】
種々のシクロプロパネート剤が、ステロイド系アルカロイドをシクロプロパン化することに使用できる。カルベノイドと称される1,1‐ハロアルキル金属複合体及び反応種は、通常、オレフィンをシクロプロパン化することに使用される。これらの試薬は、ジヨードアルカン若しくはジアゾアルカンと、Et2Zn、iBu3Al、サマリウム、銅、ロジウム若しくはパラジウムのような金属若しくは有機金属種を使用して典型的に作られる。ある実施態様では、Et2Zn及びジヨードメタンが、1,1‐ハロアルキル金属種を生成することに使用される。
【0056】
1,1‐ハロアルキル亜鉛複合体の反応性及び取扱いの容易さは、酸のような、ある種の試薬の添加によって変更できる。1,1‐ハロアルキル亜鉛種への酸の添加は、アルキル亜鉛混合塩を生成すると信じられている。下記の例では、ビアリールリン酸が、推定上のハロアルキル亜鉛リン酸シクロプロパネート剤を生成するために、ジヨードメタン及びジエチル亜鉛と組み合わされる。種々のリン酸が、推定上のハロアルキル亜鉛リン酸を生成するために使用できる。
【0057】
カルボニルに結合したオレフィンと反応して、CH2若しくはCH‐アルキル若しくはCH‐アリール基を加えるために硫黄イリドを利用する、そして、ジアゾメタン及びエチルジアゾアセテートのようなジアゾアルキル及びα‐ジアゾ‐カルボニル化合物の金属触媒分解を利用する方法のように、他の知られたシクロプロプロパン化方法も使用できる、すなわち、これらの方法は、アルキル、アリール、アルコキシカルボニル(‐COOR)若しくはアシル置換基を有するシクロプロパンを迅速に提供する。更なるシクロプロパネート剤は、Masalov, et al., Organic Letters (2004) Vol. 6, pp. 2365-2368 及び Hansen, et al., Chem. Comm. (2006) 4838- 4840.に開示されている。
【0058】
シクロプロピル環は、置換できるし、また置換しなくてもよい。シクロプロピル環が置換されたケースでは、シクロプロパンのメチレンに付した基は、再編成及び環拡大の後、D環の上に設置される。
【0059】
シクロプロパン化反応は、非プロトン性の溶媒の中で行うことができる。適切な溶媒は、ジエチルエーテル、1,2‐ジメトキシエタン、ジグリム、t‐ブチルメチルエーテル、テトラヒドロフラン及び同様のもののようなエーテル、クロロホルム、ジクロロメタン、ジクロロエタン及び同様のもののようなハロゲン化溶媒、ベンゼン、キシレン、トルエン、ヘキサン、ペンタン及び同様のもののような脂肪族若しくは芳香族の炭化水素溶媒、酢酸エチル、アセトン及び2‐ブタノンのようなエステル及びケトン、アセトニトリル、ジメチルスルフォキシド、ジメチルホルムアミド及び同様のもののような非プロトン性極性溶媒、又は2以上の溶媒の組み合わせを含む。ある実施態様では、ジクロロメタンは、ジアルキル亜鉛及びジヨードメタンが使用された場合に、シクロプロパン化のために使用された溶媒である。
【0060】
下記の実施例では、シクロプロパネート剤を含有する溶液を、最初にジエチル亜鉛の溶液にリン酸の溶液を加え、続いて、その反応溶液へのジヨードメタンの添加によって調製する。次に、シクロプロパン化の基質を、この溶液に加える。あるいは、シクロプロパネート剤は、試薬の添加の順番を変えることによってシクロプロパン化の基質の存在中に調製できる。ある実施態様では、ジアルキル亜鉛の溶液に最初にリン酸を加え、続いて、シクロプロパン化の基質の添加、そして最後にジハロアルカンを加えることによってシクロプロパン化反応を行う。この方法を使用して、シクロプロパネート剤は、管理条件下で生成され、そして、直ちにシクロプロパン化の基質と反応する。
【0061】
シクロプロパン化ステロイド系アルカロイドコア(core)の合成に続き、当該技術分野内で知られた種々の官能基付与反応を使用して、その化合物を誘導体化することができる。代表的な例は、アルケニルハライド又はアリールハライドのカップリング反応、酸化、還元、求核剤による還元、求電子剤による還元、ペリ環状反応、ラジカル反応、保護基の設置、保護基の除去及び同様のものを含む。
【0062】
ルイス若しくはブレンステッド酸の存在中に、シクロプロピル類似体は、1の炭素によって拡大しているD環を有するステロイド系アルカロイド類似体を生成するため、再編成及び環拡大を起こす。
【0063】
シクロプロパン化及び環拡大は、2ステップ1反応容器のプロセス若しくは2ステップ2反応容器のプロセスで行うことができる。シクロプロパン化及び環拡大を同一の反応容器で行う場合、環拡大再編成を開始するために使用した酸は、シクロプロパン化反応の完了後に加えられる。ある条件下では、ステロイド系アルカロイドのシクロプロパン化の過程の中で生成された亜鉛の塩は、それ自身が、環拡大再編成を触媒するルイス酸として作用できる。シクロプロパン化の後に生成された亜鉛の塩の反応性は、更なる活性のルイス酸を生成する酸の添加によって変更できる。
【0064】
下記の実施例のように、メタンスルホン酸を、シクロプロパン化の完了後、シクロプロパン化の反応容器に加える。更なる適切な酸の例は、亜鉛の塩、炭素化合物、マグネシウムの塩、チタンの塩、インジウムの塩、アルミニウムの塩、スズの塩、ランタンの塩、トリフルオロメタンスルホン酸、ジアリールオキシリン酸、酢酸、HClを含むが、これらに限定されない。本発明のある実施態様では、使用されるルイス酸は、亜鉛の塩若しくはBF3である。
【0065】
これらの環拡大類似体は、当該技術分野内で知られた種々の官能基付与反応を使用して、さらに官能基化できる。代表的な例は、アルケニルハライド若しくはアリールハライドのパラジウムカップリング反応、酸化、還元、求核剤による還元、求電子剤による還元、ペリ環状反応、ラジカル反応、保護基の設置、保護基の除去及び同様のものを含む。
医薬組成物
【0066】
1以上の薬学的に許容可能な担体(添加物)及び/または希釈液を用いて、本願明細書に開示の化合物は、投与に最適な組成物に処方される。本発明の医薬組成物は、特に固形状若しくは液状の投薬に処方することができ、以下に適用されたそれらを含む:(1)経口投与、例えば、水薬(水性または 非水性溶液 若しくは 懸濁物)、例えば、頬側、舌下、全身吸収を標的とした錠剤、カプセル、ボーラス(boluses)、散剤、顆粒剤、舌用のペースト剤、(2)非経口投与、例えば、皮下、筋肉、静脈、硬膜外注射によって、例えば、無菌溶液または懸濁液、若しくは持続性放出処方液として、(3)外的適用、例えば、皮膚用のクリーム、軟膏、放出制御パッチ若しくはスプレーとして、(4)膣用若しくは直腸用、例えば、ペッサリー、クリーム若しくは気泡として、(5)舌下用(6)眼用(7)経皮用(8)肺用(pulmonarily)、(9)経鼻用。
【0067】
本発明の医薬組成物に含めることができる適切な水性または 非水性担体の例は、水、エタノール、ポリオール(polyols)(グリセロール、プロピレングリコール、ポリエチレングリコール及び同様のもののような)及びその適切な混合溶液、オリーブ油のような野菜油、そして、オレイン酸エチルのような注射可能な有機エステルを含む。適した流動性は、例えば、レシチンのようなコーティング物質の使用によって、分散の場合における要求された粒子サイズの調節によって、及び界面活性剤の使用によって調節できる。
【0068】
これらの組成物は、保存料のような補助剤、湿潤剤、乳化剤、分散剤、潤滑剤及び/または抗酸剤を含有することもできる。本願明細書に開示の化合物上の微生物の活動の防止は、例えば、パラベン、クロロブタノール、ソルビン酸フェノール及び同様のもののような種々の抗菌及び抗真菌性剤の含有によって保障できる。糖、塩化ナトリウム及び同様のもののような等張剤(isotonic agents)を化合物に含むことも望ましい。加えて、注射可能な医薬形(pharmaceutical form)の遷延吸収を、モノステアリン酸アルミニウム及びゼラチンのような吸収を遅らせる薬剤の含有によってもたらすことができる。
【0069】
これらの処方薬若しくは組成物の調製方法は、(本発明の)化合物と、担体及び任意の1以上の付属成分とを結合物(association)にするステップを含む。一般的に、処方薬は、一様にまた本質的に、液状の担体若しくは細かく分割した固形の担体、または両方で(本発明の)化合物を結合物(association)にすること、そして、次に必要であれば製造物を成形することによって調製される。
【0070】
本願明細書に開示の化合物をヒトや動物に医薬品として投与する場合、それ自身で、または、例えば、薬学的に許容可能な担体との組み合わせにおいて約0.1から99%、若しくは約10から50%、若しくは約10から40%、若しくは約10から30%、若しくは約10から20%、若しくは約10から15%の活性成分を含む医薬組成物として与えることができる。
【0071】
(本発明の)医薬組成物における活性成分の実際の投与量レベルは、患者に毒性を与えずに、特定の患者の所望の治療反応を達成する効果がある活性成分の量、組成及び投与モードを得るように、変えることができる。
【0072】
選択された投与量レベルは、使用する(本発明の)特定の化合物の活性、そのエステル、塩であるか若しくはアミドであるか、投与経路、投与時間、使用されている特定の化合物の排泄若しくは代謝の割合、吸収割合及び範囲、処置の持続時間、使用する特定の化合物と組みあわせて使用する他の薬剤、化合物及びまたは物質、年齢、性別、体重、体調、通常健康状態、処置を受ける患者の前の医療履歴及び医療分野で良く知られた同様の要素を含む種々の要素に依存するであろう。
【0073】
一般的に、本願明細書に開示の化合物の適切な一日投与量は、治療効果を生じる効果がある最低の量の化合物の量であろう。そのような効果量は、一般的に上記要素に依存する。一般的に、患者への(本発明の)化合物の経口、静脈内、皮下投与量は、指示した効果のために使用した場合、1日に、体重1キログラムあたり、約0.0001から約200mg、若しくは約0.001から約100mg、若しくは約0.01から約100mg、若しくは約0.1から約100mgまで、若しくは約1から約50mgまでの範囲であろう。
【0074】
化合物は、毎日、一日おき、週に3回、週に2回、1週間毎、又は2週間毎に投与できる。投薬スケジュールは、“ドラッグホリデー(drug holiday)”を含むことができる、すなわち、薬物の2週間投与に1週間休み、若しくは3週間投与に1週間休み、若しくは4週間投与に1週間休みなどが可能であり、又は、ドラッグホリデー無しで連続した薬物の投与ができる。化合物は、経口的に、静脈内に、腹腔内に、局所的に、経皮的に、筋肉内に、皮下に、鼻腔内に、舌下に、又は、任意の他の経路によって投与できる。
【0075】
この処置を受けるものは、霊長類、特にヒト、及びウマ、ウシ、ブタ及びヒツジ、すなわち一般的な家畜及びペットを含む、必要がある任意の動物である。
処置の方法
【0076】
ヘッジホッグシグナルは、発生(development)の多くの段階、特に、左右対称性の形成において必須である。ヘッジホッグシグナルの欠損若しくは減少は、発生上の欠陥及び奇形の多発を導き、最も顕しいそれの1つは、単眼症である。
【0077】
多くの腫瘍および増殖性の状態が、ヘッジホッグ経路に依存していることが示されてきた。そのような細胞の成長及び生存は、本願明細書に開示の化合物で処置することによって影響を与えることができる。最近、散発性の基底細胞腫(Xie et al. (1998) Nature 391 : 90-2)及び中枢神経系の原始的な神経外胚葉性腫瘍(Reifenberger et al. (1998) Cancer Res 58: 1798-803)において、活性化ヘッジホッグ経路変異が、発生することが報告されている。ヘッジホッグ経路の非制御の活性化は、膵臓癌、食道癌、胃癌を含む胃腸管(GI tract)癌(Berman et al. (2003) Nature 425: 846-51, Thayer et al. (2003) Nature 425: 851-56)、肺癌(Watkins et al. (2003) Nature 422: 313-317), 前立腺癌(Karhadkar et al. (2004) Nature 431 : 707-12, Sheng et al. (2004) Molecular Cancer 3: 29-42, Fan et al. (2004) Endocrinology 145: 3961-70)、乳癌(Kubo et al. (2004) Cancer Research 64: 6071-74, Lewis et al. (2004) Journal of Mammary Gland Biology and Neoplasia 2: 165-181)及び肝細胞癌(Sicklick et al. (2005) ASCO conference, Mohini et al. (2005) AACR conference)のような多くの癌タイプにおいて示されている。
【0078】
例えば、ヘッジホッグ経路の小分子阻害(small molecule inhibition)は、基底細胞癌(Williams, et al., 2003 PNAS 100: 4616-21)、髄芽腫(Berman et al., 2002 Science 297: 1559-61)、膵臓癌(Berman et al., 2003 Nature 425: 846-51)、消化器系の癌(Berman et al., 2003 Nature 425: 846-51, PCT公開公報 WO 05/013800)、食道癌(Berman et al., 2003 Nature 425: 846-51)、肺癌(Watkins et al., 2003. Nature 422: 313-7)、および前立腺癌(Karhadkar et al., 2004. Nature 431: 707-12)の成長を阻害することが示されている。
【0079】
加えて、例えば、乳癌(Kubo et al., 2004. Cancer Research 64: 6071-4)、肝細胞癌(Patil et al., 2005. 96th Annual AACR conference, 要約#2942, Sicklick et al., 2005. ASCO annual meeting, 要約 #9610)、造血器腫瘍(Watkins and Matsui, unpublished results)、基底[細胞]癌(Bale & Yu, 2001. Human Molec. Genet. 10:757-762, Xie et al., 1998 Nature 391: 90-92)、髄芽腫(Pietsch et al., 1997. Cancer Res. 57: 2085-88)、胃癌(Ma et al., 2005 Carcinogenesis May 19, 2005 (Epub))など、多くの癌タイプは、ヘッジホッグ経路の非制御活性化を有することが示されている。加えて、調査者は、ヘッジホッグ経路の小分子の抑制が乾癬の症候ないし症状を回復することが示されていることを見つけた(Tas, et al., Dermatology (2004) 209: 126-131)。実施例に示すように、本願明細書に開示する化合物は、ヘッジホッグ経路を調節することを示し、選択した化合物は腫瘍成長を阻害することを示した。従って、これらの化合物は、種々の癌のような様々な過剰増殖疾患を処置することに有用であると信じられる。
【0080】
本明細書に開示の方法を用いて処置できる過剰増殖疾患は、肺がん(小細胞肺癌及び非小細胞肺癌を含む)、肺系(pulmonary system)の他の癌、髄芽腫及び他の脳癌、膵臓癌、基底細胞癌、乳癌、前立腺癌及び他の尿生殖器癌、胃腸間質性腫瘍(GIST)及び消化管の他の癌、結腸癌、結腸直腸癌、卵巣癌、造血系(多発性骨髄腫、急性リンパ性白血病、急性骨髄性白血病、慢性骨髄性白血病、慢性リンパ性白血病、ホジキンリンパ腫、及び非ホジキンリンパ腫、並びに骨髄異形成症候群)の癌、真性多血症、ワルデンシュトレームマクログロブリン血症、重鎖病、軟部肉腫(線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、扁平上皮癌、基底細胞癌、メラノーマ及び他の皮膚癌など)、腺癌、汗腺癌、乳頭癌、乳頭状腺癌、スタデノカルチノーマ(stadenocarcinoma)、髄様癌、気管支原性肺癌、腎細胞癌、肝細胞癌、胆管癌、繊毛癌、セミノーマ、胚性癌腫、ウィルムス腫瘍、子宮頚癌、子宮癌、精巣癌、膀胱癌、及び他の尿生殖器癌、上皮癌、グリオーマ、アストロサイトーマ、髄芽腫、頭蓋咽頭腫、上衣腫、松果体腫、血管芽腫、聴神経腫瘍、乏突起細胞腫、髄膜腫、神経芽細胞腫、網膜芽細胞腫、子宮内膜癌、濾胞性リンパ腫、びまん性大細胞型B細胞リンパ腫、マントル細胞リンパ腫、肝細胞癌、甲状腺癌、胃癌、食道癌、頭頚部癌、小細胞癌、本態性血小板血症、原発性骨髄線維症、好酸球増加症候群、全身性肥満細胞症、家族性過好酸球増加症(familiar hypereosinophilia)、慢性好酸球性白血病、甲状腺癌、神経内分泌癌、及びカルチノイド腫瘍を含む。追加的な疾患は、ゴーリン症候群及び乾癬を含む。
【0081】
この処置を受ける被験者ないし患者は、必要とされる任意の動物であり、霊長類、特に、ヒト、他の動物(ウマ、ウシ、ブタ及びヒツジなど);並びに家禽及び一般的なペットを含む。
【0082】
本明細書に開示のヘッジホッグ阻害剤は、他の癌処置と組合せができる。例えば、それらは、外科処置;放射線;バイオセラピー(biotherapeutics)(インターフェロン、サイトカイン、例えば、インターフェロンα、インターフェロンγ、及び腫瘍壊死因子、造血成長因子、単クローン性血清療法、ワクチン及び免疫賦活薬);抗体(例えば、アバスチン、エルビタックス、リツキサン、及びベキサール);内分泌療法(ペプチドホルモン、副腎皮質ステロイド、エストロゲン、アンドロゲン及びアロマターゼ阻害剤を含む);抗エストロゲン薬(例えば、タモキシフェン、ラロキシフェン、及びメゲストロール);LHRH作用薬(例えば、ゴスクリクリン(goscrclin)及びリュープロリド酢酸塩);抗アンドロゲン薬(例えば、フルタミド及びビカルタミド);遺伝子治療;骨髄移植;光力学治療(例えば、ベルトポルフィン(vertoporfin)(BPD−MA)、フタロシアニン、フォトセンシタイザーPc4、及びデメトキシ‐ハイポクレリン(Demethoxy-hypocrellin)A(2BA−2−DMHA));並びに化学療法と組合せることができる。
【0083】
化学療法の例は、ゲムシタビン、メトトレキサート、タキソール、メルカプトプリン、チオグアニン、ヒドロキシウレア、シタラビン、シクロホスファミド、イホスファミド、ニトロソ尿素、シスプラチン、カルボプラチン、ミトマイシン、ダカルバジン、プロカルビジン(procarbizine)、エトポシド、プレドニゾロン、デキサメタゾン、シタルビン、キャンパセシン(campathecins)、ブレオマイシン、ドキソルビシン、イダルビシン、ダウノルビシン、ダクチノマイシン、プリカマイシン、ミトキサントロン、アスパラギナーゼ、ビンブラスチン、及びビノレルビンを含む。付加的な薬剤(additional agents)は、ナイトロジェンマスタード(例えば、シクロホスファミド、イホスファミド、トロホスファミド、クロラムブシル、エストラムスチン、及びメルファラン)、ニトロソ尿素(例えば、カルムスチン(BCNU)及びロムスチン(CCNU))、アルキルスルホナート(alkylsulphonates)(例えば、ブスルファン及びトレオスルファン)、トリアゼン(例えば、ダカルバジン及びテモゾロミド)、化合物を含有する白金(例えば、シスプラチン、カルボプラチン、及びオキサリプラチン)、ビンカアルカロイド(例えば、ビンクリスチン、ビンブラスチン、ビンデシン、及びビノレルビン)、タキソイド(例えば、パクリタキセル及びドセタキソール)、エピポドフィリン(epipodophyllins)(例えば、エトポシド、テニポシド、トポテカン、9−アミノカンプトテシン、カンプトイリノテカン、クリスナトール(Crisnatol)、マイトマイシンC、及びマイトマイシンC)、抗代謝物(anti-metabolites)、DHFR阻害剤(例えば、メトトレキサート及びトリメトレキサート)、IMPデヒドロゲナーゼ阻害剤(例えば、ミコフェノール酸、チアゾフリン、リバビリン、及びEICAR)、リボヌクレオチド還元酵素阻害剤(例えば、ヒドロキシウレア及びデフェロキサミン)、ウラシル類似体(例えば、フルオロウラシル、フロクスウリジン、ドキシフルリジン、ラチトレキサド(Ratitrexed)、及びカペシタビン)、シトシン類似体(例えば、シタラビン(ara C)、シトシンアラビノシド、及びフルダラビン)、プリン類似体(例えば、メルカプトプリン及びチオグアニン)、ビタミンD3類似体(例えば、EB1089、CB1093、及びKH1060)、イソプレニル化阻害剤(例えば、ロバスタチン)、ドーパミン系神経毒(dopaminergic neurotoxins)(例えば、1−メチル−4−フェニルピリジニウムイオン)、細胞周期阻害剤(例えば、スタウロスポリン)、アクチノマイシン(例えば、アクチノマイシンD及びダクチノマイシン)、ブレオマイシン(例えば、ブレオマイシンA2、ブレオマイシンB2、及びペプロマイシン)、アントラサイクリン(例えば、ダウノルビシン、ドキソルビシン(アドリアマイシン)、イダルビシン、エピルビシン、ピラルビシン、ゾルビシン、及びミトキサントロン)、MDR阻害剤(例えば、べラパミル)、Ca2+ATPase阻害剤(例えば、タプシガルジン)、イマチニブ、サリドマイド、レナリドマイド、エルロチニブ、ゲフィチニブ、ソラフェニブ、及びスニチニブ、並びにボルテゾミブを含むプロテアソーム阻害剤を含む。
【0084】
本明細書に開示のヘッジホッグ阻害剤が他の処置(例えば、付加的な治療又は放射線若しくは外科と)との組合せで投与されるとき、各薬剤又は治療の投与量ないし用量は、ほとんどの場合、単独の薬剤療法における対応する投与量ないし用量より低い。また、一般的に、本明細書に記載のヘッジホッグ阻害剤及び第二の治療剤は同一の医薬組成物で投与されるべきではなく、異なる物理的及び化学的特徴のために、異なる経路で投与されてよい。例えば、ある化合物は経口投与が可能であり、一方、第二の治療(剤)は静脈内投与される。同一の医薬組成物において可能である、投与の方法及び投与の妥当性の決定は、熟練した臨床医の知識の範囲内でよい。初回投与は、当該技術分野で知られている確立されたプロトコールにしたがって為されることができ、次いで、観察される効果に基づいて、投与量、投与の方法及び投与の時間は、熟練した臨床医によって変更できる。
【0085】
ヘッジホッグ阻害剤及び第二の治療剤及び/又は放射線は、増殖疾患の性質、患者の状態、及び第二の治療剤及び又は放射線の実際の選択に依存して、同時的に(例えば、同時に、本質的に同時に若しくは同一の処置プロトコール内で)又は連続して(つまり、順次に、任意の時間間隔を空けて)投与されてよい。
【0086】
ヘッジホッグ阻害剤及び第二の治療剤及び/又は放射線が同時に又は本質的に同時に投与されないのであれば、投与の最適な順番は異なった状況に応じて異なってよい。よって、ある状況では、ヘッジホッグ阻害剤は最初に投与され、次いで、第二の治療剤及び/又は放射線が投与され;別の状況では、第二の治療剤及び/又は放射線は最初に投与され、次いで、ヘッジホッグ阻害剤が投与される。この代替の投与は、単一の処置プロトコールの間に繰り返されてよい。投与の順番の決定及び処置プロトコールの間の各治療剤の投与の反復回数は、処置される疾患及び患者の状況の評価後に、熟練した臨床医の知識の範囲内でよい。例えば、第二の治療剤及び/又は放射線は、特に、細胞傷害性薬物であれば、最初に投与されてよく、次いで、ヘッジホッグ阻害剤の投与による処置が続き、そこで有用だと判断されれば、第二の治療剤及び/又は放射線によって処置プロトコールが終了するまで続けられる。
実施例
【0087】
現に、一般的に開示する発明は、単に、本発明のある視点及び実施態様を説明する目的のために含まれ、そして発明を限定することを意図しない、以下の実施例を参照することによってさらに容易に理解されるであろう。
実施例1
ステップA
【0088】
再結晶化されたシクロパミン2(14.1g, 34.0mmol, 1当量)を、無水DCM(70mL)および無水MeOH(29mL)に溶解する。澄んだ溶液を冷却し、そしてトリエチルアミン(10.4g, 102.7mmol, 3当量)、続いて、クロロギ酸ベンジル(benzyl chloroformate, 6.20g, 36.3mmol, 1.1当量)を加える。添加が完了した後、溶液をアイスバス内で30分間攪拌する。3部(portions)のクロロギ酸ベンジル(3x0.35g, 3.46mmol, 0.03当量)を3時間かけて加える。温度を20℃未満に維持しつつ、反応を水(71mL)で徐々に抑える。層が安定して分離する前に、混合物を15分間攪拌する。有機層を硫酸ナトリウム上で乾燥し、濾過する。合併した濾液を、無水ピリジン(30mL)で緩衝し、濃縮し、そして、更なる無水ピリジン(43mL)で溶媒を交換し、そして濃縮する。
【0089】
化合物のピリジン(43mL)溶液を、更なる無水ピリジン(85mL)でさらに希釈する。トリメチルアセチルクロリド(8.3g, 68.7mmol, 2当量)を反応混合溶液に徐々に加え、反応溶液を45℃まで加熱する。反応溶液を45℃で30分間攪拌する。反応溶液を冷却し、[反応を]無水MeOH(4.5mL)の添加によって抑える。抑制した反応混合溶液を室温で40分間攪拌し、そして次にトルエン(97mL)で希釈し、そして水(35mL)及び10wt%の炭酸ナトリウム水溶液(100mL)で順番に処置する。勢い良く攪拌した後、層を分離し、そして有機層を水で2回(2x100mL)洗浄し、硫酸ナトリウム上で乾燥し、そして濾過する。濾過ケーキをトルエン(49mL)でリンスし、そして破棄する。合併した濾液を濃縮し、そして溶媒を濃縮でトルエン(145mL)に交換し、そしてさらに乾燥状態まで濃縮する。生成物をトルエン及びヘプタンから再結晶化する。結晶化産物を吸引濾過によって分離し、冷たいヘプタンで洗浄し、そして恒量まで乾燥して、15.1gの所望の生成物を得る。
ステップB
【0090】
ビス(2,6‐ジメチルフェニル)リン酸塩(10.65g, 34.8mmol, 3.1当量)を無水DCM(42mL)から濃縮によって乾燥し、そして窒素雰囲気下で保持する。次に、リン酸塩を無水DCM(110mL)に再溶解する。別のフラスコに、純粋なジエチル亜鉛(4.17g, 33.8mmol, 3.0当量)の無水DCM溶液(35 mL)を調製し、そして−25℃まで冷却する。−10℃以下に温度を維持しつつ、リン酸塩溶液を1時間かけて徐々にジエチル亜鉛が入った容器に移す。澄んだエチル亜鉛リン酸塩溶液を0℃まで温め、そして15分間攪拌した。0〜5℃の間に反応温度を維持しつつ、ジヨードメタン(9.25g, 34.5mmoles, 3.0当量)を、エチル亜鉛リン酸塩溶液に徐々に加える。添加が完了した後、亜鉛カルベノイド溶液をさらに20分間攪拌する。
【0091】
別のフラスコに、化合物3(7.20g, 11.4mmol, 1当量)を無水DCM(36mL)に溶解し、反応フラスコに移す。添加が完了した後、アイスバスを取り除き、そして反応混合溶液を室温まで温める。6時間後、フラスコの内容物を−53℃まで冷却する。反応温度を−45℃未満に維持しつつ、メタンスルホン酸(3.38g, 35.2mmol, 3.1当量)の無水DCM溶液(3mL)を加える。10分後、反応温度を−40℃未満に維持しつつ、モルフォリン(20g, 230mmol, 20当量)を反応混合溶液に加える。反応溶液を、室温までオーバーナイトで温める。モルフォリンの塩を濾過によって取り除き、そして濾過ケーキをDCM(22mL)でリンスする。合併した濾液を2N塩酸水溶液(2x140mL)、5%炭酸水素ナトリウム水溶液(140mL)、5%炭酸水素ナトリウム水溶液(70mL)及び5%亜硫酸水素ナトリウム水溶液(70ml)、そして鹹水(144mL)で洗浄する。有機層を硫酸マグネシウム上で乾燥し、そして濾過する。乾燥状態にすることなく、DCM溶液を濃縮し、そして溶媒をメタノール(280mL)で交換する。懸濁物をアイスバスで冷却し、そして40分間攪拌する。固形物を濾過によって分離し、冷たいメタノールで2回(2x25mL)洗浄し、そして恒量まで乾燥して、5.94gの所望の生成物を得る。
ステップC
【0092】
丸底フラスコの中で、化合物4(11.67g, 18.1mmol, 1当量)及びウェットカーボン上(に担持した)20%水酸化パラジウム(2.40g, 1.71mmol, 0.09当量)を窒素雰囲気下に置き、そしてEtOAc(酢酸エチル, 115mL)及びトルエン(60mL)で希釈する。溶液を、窒素で(3X)減圧吸引/パージサイクル(evacuation/purge cycles)で脱気し、そしてその工程を水素で繰り返す。懸濁物を勢い良く室温で1.5時間攪拌する。水素雰囲気を、窒素で置き換える。エチレンジアミン(0.57g, 9.5mmol, 0.52当量)を反応に加え、そして結果として生じる混合溶液を20分間攪拌する。その溶液を窒素下で濾過し、そして濾過溶液を2%(wt/wt)エチレンジアミン水溶液(125mL)で、次に水(130mL)で洗浄し、そして次に、硫酸マグネシウム上で乾燥する。乾燥剤を濾過によって取り除き、そして濾過液を減圧下で乾燥状態まで濃縮する。残った固形物は、トルエン(2x55mL)で回転吸引機上で追跡(chased)し、そして結果として生じる物質を更に精製することなく次のステップで使用する。
【0093】
前のステップからの物質を無水DCM(26mL)に溶解する。反応温度を−10から−25℃の間に維持しつつ、結果として生じる澄んだ溶液を1MのDIBALのDCM(65mL, 65mmol, 3.6当量)溶液に加える。30分後、0℃以下に反応温度を維持して、反応をアセトン(13mL)で抑える。抑制した反応混合溶液を17分間攪拌した後、それを冷たい攪拌された20%(wt/wt)ロッシェル塩水溶液(200mL)が入ったフラスコに分画して(in portions)加える。結果として生じるゼラチン状の懸濁液を室温で15時間攪拌する。攪拌した後、きれいな層を分離し、そして水性の層をDCM(30mL)で逆抽出する。合併した有機層を水(60mL)で洗浄し、そして硫酸ナトリウム上で乾燥する。乾燥剤を濾過によって取り除き、そして廃棄する。濾過液を減圧下で濃縮し、そして溶媒をトルエンに交換する(225mLを分画して添加)。結果として生じる溶液を懸濁液(50mL)まで、さらに濃縮し、そしてヘプタン(115mL)で希釈する。結果として生じる混合溶液を均質に変化するまで加熱(92℃)する。溶液を12時間かけて徐々に15℃まで冷却し、そして次に、更なる16時間保持する。結晶化物を吸引濾過によって分離し、ヘプタン(2x75mL)で洗浄し、そして恒量まで乾燥し、7.70gの所望の生成物を得る。
【0094】
丸底フラスコを、ホモアリリックアルコール(homo-allylic alcohol, 7.50g, 17.6mmol, 1当量)、アルミニウムトリ‐tert‐ブトキシド(6.10g, 24.8mmol, 1.4当量)、無水トルエン(115mL)及び2‐ブタノン(90g, 1.24mol, 7当量)で順番にチャージ(charged)する。懸濁液を窒素雰囲気下で75℃まで16時間加熱する。次に、反応温度を49℃まで冷却する。20%(w/w)酒石酸カリウムナトリウム水溶液(226g)を攪拌した懸濁液に加える。懸濁液を室温で3.5時間攪拌する。層を分離する。有機層を20%ロッシェル塩水溶液(2x250mL)及び水(225mL)で洗浄し、次に硫酸ナトリウム上で乾燥し、そして濾過する。残留物をトルエン(30mL)でリンスし、そして廃棄する。合併した有機物を乾燥状態まで濃縮する。2−プロパノール(250 mL, 分画して添加)から44gの最終溶液質量までの濃縮によって、残りの反応溶媒を物質から取り除く。最終溶液質量を41gにする2‐プロパノールからヘプタン(275mL, 分画して添加)への溶媒の交換は、所望の生成物を完全に沈殿させる。懸濁液を更なるヘプタン(40mL)で希釈し、室温で1時間攪拌し、そして濾過する。生成物をn‐ヘプタン(17mL)で洗浄し、そして乾燥し、5.4gの所望の生成物を得る。
ステップD
【0095】
丸底フラスコを、出発物質(110mg, 0.26mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(106mg)でチャージする。固形物をピリジン(4mL)に懸濁する。懸濁液を窒素雰囲気下(1気圧)に置き、そして混合溶液をオーバーナイトで室温で攪拌する。その反応混合溶液を、セライト(登録商標)を介して濾過し、そして濾過液を真空中で濃縮する。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、93mgの所望の化合物を得る。([M+H] = 426.6m/z)。
実施例2
ステップA
【0096】
シクロパミン2(5.02g, 12.2mmol, 1.0当量)を無水ピリジン(25mL)に溶解する。DMAP(300mg, 2.44mmol, 0.2当量)及びトリエチルアミン(5.5mL, 39.1mmol, 3.2当量)、続いて、BtO‐Cbz(10.5g, 39.1mmol, 3.2当量)を加え、そして40℃で2時間加熱する。混合溶液を室温まで冷まし、30mLの水で処置し、均質溶液を得るために加熱し、そして室温まで冷却する。形成された白い沈殿物を濾過によって収集し、濾過ケーキを水で数回(3x50mL)洗浄し、そして空気中で乾燥し、6.75gの所望の生成物を得るためのトルエン/ヘプタン(1:9, 70mL)から結晶化された9.53gの未精製物質を得る。
ステップB
【0097】
反応温度を−8℃未満に維持しつつ、−20℃の5.0mlのジエチル亜鉛(572mg, 482μL, 4.63mmol, 3.00当量)DCM溶液に、ビス‐(2,6‐ジエチルフェニル)リン酸塩(1.42g, 4.63mmol, 3.00当量)DCM溶液(15mL)を加える。ビスカルボキシルベンジルシクロパミン(BisCBzcyclopamine, 1.05g, 1.54mmol, 1.0当量)のDCM溶液(10mL)を加える前に、溶液を15分間0℃で熟成し、純粋なジヨードメタン(1.24g, 374μL, 3.00当量)を加え、15分間0℃で熟成する。冷却バスを室温の水バス(water bath)によって置き換え、そして室温で4.5時間維持する。混合溶液をドライアイス‐アセトンバスで−76℃まで冷却し、そして反応温度を−74℃未満に維持しつつ、メタンスルホン酸DCM溶液(0.6mL 50% v/v溶液 4.63mmol, 3.0当量)で滴下処置する。混合溶液を15‐20分間熟成し、そして反応温度を−65℃未満に維持しつつ、モルフォリン(2.69g, 2.70mL, 20当量)の滴下で抑える。冷却バスを取り除き、反応混合溶液を16‐18時間攪拌し、白い沈殿物を濾過除去し、そして濾過液を2.0MのHCl(2x20mL)、飽和炭酸水素ナトリウム(2x20mL)、水(2x20mL)及び鹹水(20mL)で連続的に洗浄する。硫酸マグネシウム上で乾燥し、真空中で乾燥状態まで濃縮し、そして未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc 17:3→4:1)によって精製し、924mg(1.33mmol, 86%)の所望の生成物を得る。
ステップC
【0098】
化合物7(4.05g, 5.83mmol, 1当量)のEtOAc:トルエン溶液(2:1, 60mL)の溶液に、カーボン上(に担持した)20%水酸化パラジウム(823mg, 0.583mmol, 0.1当量)を加える。フラスコを、3回、吸引し、そして水素で満たす。混合溶液を水素雰囲気下で1時間攪拌する。純粋なエチレンジアミン(0.38mL)を加え、1時間攪拌し、そして触媒を濾過除去する。濾過ケーキを2回、EtOAc:トルエン(2:1, 12 mL)で洗浄する。合併した濾液を、2%エチレンジアミン水溶液(3x20mL)で洗浄し、硫酸ナトリウム上で乾燥し、そして真空中で濃縮し、白い結晶化固形物として2.46gを得る。
ステップD
【0099】
丸底フラスコを、ホモアリリックアルコール8(7.50g, 17.6mmol, 1当量)、アルミニウムトリ-tert-ブトキシド(6.10 g, 24.8 mmol, 1.4当量)、無水トルエン(115 mL)、及び2−ブタノン(90 g, 1.24 mol, 7当量)で連続的にチャージする。懸濁液を窒素雰囲気下で75℃まで16時間加熱する。次に、反応溶液を49℃まで冷却する。20%(w/w)酒石酸カリウムナトリウム(226g)水溶液を攪拌した懸濁液に加える。懸濁液を室温で3.5時間攪拌する。層を分離する。有機層を20%ロッシェル塩水溶液(2x250mL)及び水(225mL)で洗浄し、次に、硫酸ナトリウム上で乾燥し、そして濾過する。残留物をトルエン(30mL)でリンスし、そして廃棄する。合併した有機物を乾燥状態まで濃縮する。残りの反応溶媒を2−プロパノール(250mL, 分画して添加)から最終溶液質量の44gまでの濃縮によって物質から取り除く。最終溶液質量を41gにする2‐プロパノールからヘプタン(275mL, 分画して添加)への溶媒の交換は、所望の生成物を完全に沈殿させる。懸濁液を更なるヘプタン(40mL)で希釈し、室温で1時間攪拌し、そして濾過する。生成物をn‐ヘプタン(17mL)で洗浄し、そして乾燥し、5.4gの所望の生成物を得る。
ステップE
【0100】
丸底フラスコを、出発物質(110mg, 0.26mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(106mg)でチャージする。固形物をピリジン(4mL)に懸濁する。懸濁液を窒素雰囲気下(1気圧)に置き、そして混合溶液をオーバーナイトで室温で攪拌する。反応混合溶液を、セライト(登録商標)を介して濾過し、そして濾過液を真空中で濃縮する。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、93mgの所望の化合物を得る。([M+H] = 426.6m/z)。
実施例3
【0101】
密封管の中に、ケトン6(85mg, 0.199mmol, 1当量)をチャージし、そしてトリエチレングリコール(2mL)、続いて、ヒドラジン一水和物(hydrazine monohydrate, 500mg, 10mmol, 50当量)及び炭酸カリウム(138mg, 1mmol, 5当量)を加えた。管を密封し、反応溶液を150℃で16時間加熱した。反応溶液を室温まで冷却し、そして水を加えた。残留物をクロロホルムで(3X)抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、そして乾燥状態まで濃縮する。無色の油をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 96:4)を使用して精製した。精製分画をプールし、そして乾燥状態まで濃縮する。結果として生じる油をMTBEに溶解し、水(2回)、2NのNaOH、そして次に鹹水で洗浄した。合併した有機層をNa2SO4上で乾燥し、濾過し、そして蒸発させ、64mgの所望の物質を白い気泡として得る([M+H] = 412.7 m/z)。
実施例4
【0102】
密封管を化合物5(223mg, 0.52mmol, 1当量)及びDMF(1mL)でチャージした。2−ブロモプロパン(1.3g, 10.5mmol, 20当量)及びNa2CO3(73mg, 0.68mmol, 1.3当量)を加え、そしてフラスコを密封し、そして50℃まで加熱した。〜70%の変換が観察された時点で16時間、混合溶液を攪拌した。更なる[2−ブロモプロパン](0.26 g, 2.12 mmol, 4当量)を加えた。反応溶液を2時間攪拌し、そして更なる2−ブロモプロパン(0.13 g, 1.1 mmol, 2当量)を加えた。反応溶液を1時間攪拌した。反応溶液を室温まで冷却し、そして水を加えた。残留物をMTBE(3X)で抽出した。有機層を合併し、鹹水で洗浄し、Na2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。白い気泡をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 99:1)を使用して精製し、206mgの白い気泡としてのN‐イソプロピル誘導体を得た。
【0103】
N‐イソプロピル誘導体(205mg, 0.44mmol, 1当量)を4‐メトキシピリジン(1.5 mL)に溶解した。フラスコを不活性環境下に置き、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 40mg)を加えた。フラスコを密封し、水素で3回パージ(purge)し、そして1気圧水素下に16時間置いた。セライト(登録商法)を反応混合溶液に加えた。セライト(登録商標)の小さなパッドを介して混合溶液を濾過し、そしてEtOAcで洗浄した。有機層を1N塩酸水溶液で(2x)、次に水で洗浄した。有機層をNa2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて、34mgの未精製物を得た。水性の層を2NのKOHで中和し、DCMで(3X)抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして、初期の34mgの未精製物と組み合わせた。未精製物質をヘキサン/EtOAc(6:4)のシリカゲルフラッシュクロマトグラフィーを使用して精製し、80mgの所望の生成物を得た。([M+H] = 468.7 m/z)。
実施例5
【0104】
丸底フラスコの中で、化合物6(88 mg, 0.21 mmol, 1当量)を無水THF(1ml)に溶解した。混合溶液を0℃まで冷却した。ピリジン(84μL, 1mmol, 5当量)及び過酸化ベンゾイル(benzoylperoxide, 150mg, 0.62mmol, 3当量)を連続的に加えた。均質な混合溶液を室温まで2時間かけて徐々に温め、オーバーナイトで室温で攪拌した。飽和炭酸水素ナトリウム(NaHCO3)を加えることによって反応を抑えた。残留物をMTBEで抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして減圧下で濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc(9:1から4:1)を使用して精製し、白い気泡としてN‐O誘導体の生成物(60 mg, 0.11 mmol)を得た。2mLのMeOHにこの気泡を溶解し、続いて2NのKOH水溶液(0.4mL)を[加えた]。反応混合溶液を1時間攪拌した。ほとんどのMeOHを窒素流動下(under a stream of nitrogen)で蒸発させ、1NのHCl(500μL)を加えた。物質をDCM(3X)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして減圧下で濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc(88:12→1:1))で精製し、11mgの所望の生成物を得た。([M+H] = 442.5 m/z)。
実施例6
ステップA
【0105】
丸底フラスコの中で、化合物6(89mg, 0.209 mol, 1当量)及びN‐(ベンジルオキシカルボニル)‐アミノアセトアルデヒド(N-(benzyloxycarbonyl)-aminoacetaldehyde, 148mg,0.85mmol, 4当量)をDCM(2mL)に溶解した。トリアセトキシ水素化ホウ素ナトリウム(Sodium triacetoxyborohydride, 177mg, 0.85mmol, 4当量)を加え、そして反応溶液を3時間室温で攪拌した。混合溶液を飽和NaHCO3水溶液に注ぎ、そして残留物をDCM(3X)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて気泡の固形物(247 mg)を得た。未精製物をEtOAc(2mL)に溶解し、そして4MのHCl(156μL)で処置した。30分後、白い沈殿物が徐々に形成された。結果として生じる泥状物を15分間攪拌した。濾過により、120mgの白い固形物を得た。物質をEtOAcに溶解し、飽和NaHCO3水溶液で処置した。有機層を収集し、水性の層をEtOAc(2X)で抽出した。合併した有機層を鹹水で洗浄し、Na2SO4上で乾燥した。濾過及び蒸発により、所望の中間体を得た。この物質を精製することなく次のステップで使用した。
ステップB
【0106】
ステップAからの全ての物質をEtOAc(3mL)に溶解し、そして10%のPd/C(30mg, ウェット, Aldrich Degussa type ElOl)で処置した。フラスコを密封し、そして水素で3回パージ(purged)し、そして1気圧水素下にオーバーナイトで置いた。16時間後、混合溶液をセライト(登録商標)の小さなパッドを介して精製し、EtOAcで洗浄し、白い気泡としての52mgのアミンを得た。
ステップC
【0107】
アミン14(52mg, 0.11mmol, 1当量)が入った丸底フラスコを、1H‐テトラゾール‐5‐酢酸(1H-tetrazole-5-acetic acid, 21mg, 0.166mmol, 1.5当量)、DCM(2ml)、EDCI(42mg, 0.22mmol, 2当量)及びN,N‐ジイソプロピルエチルアミン(N,N-diisopropylethylamine, 57mg, 0.44mmol, 4当量)でチャージした。結果として生じる黄色い溶液を室温で4時間攪拌した。飽和NaHCO3水溶液の添加によって反応を抑え、そして残留物をDCM(3X)で抽出した。合併した有機層をNa2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて、62mgのオフホワイトの固形物を得た。この物質をシリカゲルフラッシュクロマトグラフィー(MeOH/DCM, 5:95→10:90)を使用して精製し、31mgの所望の生成物を得た。([M+H] = 579.7 m/z)。
実施例7
【0108】
丸底フラスコを出発物質(47mg, 0.110mmol, 1当量)及び炭酸カリウム(150mg, 1.09mmol, 10当量)でチャージした。固形物を2mlのDCMに懸濁した。ヨードメタン(14μL, 0.22mmol, 2当量)を加え、そして混合溶液を2[時間]室温で攪拌した。TLC(DCM/MeOH 95:5)は、>90%の完了を示した。ヨードメタン(14 μL, 0.22 mmol, 2当量)を反応混合溶液に加え、2時間攪拌した。反応混合溶液に水を加えた。層を分離させ、有機物を乾燥し、乾燥状態まで濃縮した。残留物をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 100:0→98:2)を使用して精製し、34mgの所望の生成物を得た。([M+H] = 440.5 m/z)。
実施例8
【0109】
丸底フラスコを出発物質(59mg, 0.126mmol, 1当量)及び炭酸カリウム(350mg, 2.5mmol, 20当量)でチャージした。固形物を3mlのDCMに懸濁した。反応溶液をヨードメタンでチャージ(80μL, 1.29mmol, 10当量)し、混合溶液をオーバーナイトで室温で攪拌した。反応混合溶液を水でチャージした。有機層を分離し、水性の層をDCMで逆抽出した。合併した有機層を乾燥し、乾燥状態まで濃縮した。残留物をDCM/MeOH(95:5→90:10)のシリカゲルフラッシュクロマトグラフィーを使用して精製し、52mgの所望の生成物を得た。([M+H] = 639.5 m/z)。
実施例9
ステップA
【0110】
丸底フラスコの中で、化合物5(50mg, 0.12mmol, 1当量)及びN‐(t‐ブトキシカルボニル)‐アミノアセトアルデヒド(N-(t-butoxycarbonyl)-aminoacetaldehyde, 6mg, 0.38mmol, 3.1当量)をDCM(2mL)に溶解した。トリアセトキシ水素化ホウ素ナトリウム(8mg, 0.38mmol, 3.1当量)を加え、そして反応溶液を2時間室温で攪拌した。混合溶液を飽和NaHCO3水溶液に注ぎ、残留物をDCM(3x)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて気泡の固形物(95mg)を得た。未精製物質を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 1:1)を使用して精製し、55mgの化合物18を得た。
ステップB
【0111】
丸底フラスコを出発物質18(800mg, 1.4mmol, 1当量)でチャージした。固形物をDCM及びTFAの溶液(10mL, 1:1)に溶解した。溶液を45分間室温で攪拌した。反応溶液を10%炭酸ナトリウム溶液とDCMの間で分画した。有機層を分離し、そして10%炭酸ナトリウムで洗浄した。有機層を乾燥状態まで濃縮した。残留物を更なる精製無しに次のステップで使用した。
ステップC
【0112】
丸底フラスコをTHF/ACN(1:1, 4mL)に溶解した出発物質(300mg, 0.64mmol, 1当量)でチャージした。反応溶液を37%ホルムアルデヒド水溶液(240μL, 3.22mmol, 5当量)及びシアノ水素化ホウ素ナトリウム(sodium cyanoborohydride, 64mg, 1mmol, 1.6当量)でチャージした。混合溶液を30分間室温で攪拌した。次に、反応溶液を飽和炭酸水素ナトリウム水溶液とDCMの間で分画した。有機層を分離、乾燥し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95→10:90)を使用して精製し、所望の物質を得た。
ステップD
【0113】
丸底フラスコを出発物質20(30mg, 0.06mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(30mg)でチャージした。固形物をピリジン(2mL)に懸濁した。懸濁液を水素雰囲気下に置き、混合溶液をオーバーナイトで室温で攪拌した。反応混合溶液をセライト(登録商標)上で濾過し、そして濾過液を乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95→10:90)を使用して精製し、所望の物質を得た。([M+H] = 497.7 m/z)。
実施例10
【0114】
丸底フラスコを出発物質(85 mg, 0.20mmol, 1当量)でチャージし、DCM(4ml)に溶解した。反応溶液を、N‐(2‐オキソエチル)アセトアミド(N-(2-oxoethyl)acetamide, 80mg, 0.70mmol, 3.5当量)及びトリアセトキシ水素化ホウ素ナトリウム(Sodium triacetoxyborohydride, 177mg, 0.85mmol, 4当量)でチャージした。混合溶液を1時間室温で攪拌した。反応溶液を飽和炭酸水素ナトリウム水溶液とDCMの間で分画した。有機物を分離し、乾燥し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、所望の物質を得た。([M+H] = 511.7 m/z)。
実施例11
【0115】
N‐(2‐オキソエチル)アセトアミドの替わりに、N‐メチル‐N‐(2‐オキソエチル)アセトアミド(N-methyl-N-(2-oxoethyl)acetamide)を使用して化合物22を実施例9に記載の製法に従って合成した。([M+H] = 525.7 m/z)。
実施例12
【0116】
N‐(2‐オキソエチル)アセトアミドの替わりにN‐(2‐オキソエチル)‐3‐フェニルプロパンアミド(N-(2-oxoethyl)-3-phenylpropanamide)を使用して、化合物23を実施例10に記載の製法に従って合成した。([M+H] = 601.8 m/z)。
実施例13
【0117】
N‐(2‐オキソエチル)アセトアミドの替わりにN‐メチル‐N‐(2‐オキソエチル)3‐フェニルプロパンアミドを使用して、化合物23[sic. 24]を実施例10に記載の製法に従って合成した。([M+H] 615.9 m/z)。
実施例14
ステップA
【0118】
丸底フラスコを化合物6(4.23g, 9.94mmol, 1当量)及びTHF(60mL)でチャージした。トリエチルアミン(6.92mL, 49.7mmol, 5.0当量)及びクロロギ酸ベンジル(1.54mL, 10.93mmol, 1.1当量)を加え、そして混合溶液を1時間室温で攪拌した。反応混合溶液を飽和炭酸水素水溶液(100mL)とEtOAc(100mL)の間で分画した。層を分離させ、有機物を乾燥(Na2SO4)し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 2:98→14:86)を使用して精製し、3.75gの物質を得た。
ステップB
【0119】
0℃のセリウムトリクロリド7水和物(cerium trichloride heptahydrate, 260mg, 0.69mmol, 1.3当量)のMeOH溶液(10ml)を水素化ホウ素ナトリウム(24mg, 0.65mmol, 1.2当量)で処置し、15分間攪拌し、そして次に−78℃まで冷却した。ケトン26(300mg, 0.54 mmol, 1当量)のTHF溶液(10ml)を加え、混合溶液を1時間攪拌し、そして次に室温まで温めた。水(50ml)及びEtOAc(50ml)を加え、混合し、そして層を分画した。有機層を収集し、鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、白い残留物まで濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)を使用して精製し、235mgの3‐βアルコール27を得た。
ステップC
【0120】
化合物27(235mg, 0.42mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 50mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として130mgの化合物25を得た。([M+H] = 427.4 m/z)。
実施例15
ステップA
【0121】
−78℃のケトン26(300mg, 0.54mmol, 1当量)のTHF溶液(10ml)をK‐セレクトリド(K-Selectride:Potassium tri-sec-butylborohydride, 0.58ml, 0.58mmol, 1.1当量, 登録商標)で処置し、そして60分間攪拌した。メタノール(1ml)を加え、そして溶液を室温まで温めた。水(50ml)及びEtOAc(50ml)を加え、混合し、そして層を分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして白い残留物まで濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:14)で精製し、170mgの純粋な3‐αアルコール29を得た。
ステップB
【0122】
化合物29(170 mg, 0.30 mmol, 1当量)を攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 35mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして、室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として76mgの化合物28を得た。([M+H] = 427.4 m/z)。
実施例16
ステップA
【0123】
化合物27(100mg, 0.18mmol, 1当量)及びベンジルトリエチルアンモニウムクロリド(benzyltriethylammonium chloride, 8mg, 0.36mmol, 0.2当量)をDCM(5ml)に溶解し、ジメチル硫酸(130μL, 1.43mmol, 8当量)及び50%水酸化カリウム水溶液(0.5ml)と共に室温で18時間勢い良く攪拌した。混合溶液を水(30ml)とEtOAc(30ml)の間で分画し、そして次に有機層を鹹水で洗浄し、硫酸ナトリウム上で乾燥し、そして、澄んだ油まで濃縮した。未精製のエーテルをシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 3:7→9:113)によって精製し、澄んだ油として75mgのメチルエーテルを得た。
ステップB
【0124】
化合物31(66mg, 0.115mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として22mgの化合物30を得た。([M+H] = 441.4m/z)。
実施例17
ステップA
【0125】
化合物27(100mg, 0.18mmol, 1当量)をDCM(5ml)に溶解し、4‐ジメチルアミノピリジン(4-dimethylaminopyridine, 4mg, 0.35mmol, 0.2当量)、N,N‐ジイソプロピルエチルアミン(N,N-diisopropylethylamine, 0.15ml, 0.9mmol, 5当量)及び無水酢酸(0.070ml, 0.72mmol, 4当量)を加えた。12時間室温で攪拌した後、溶液をEtOAc(30ml)と5%炭酸水素ナトリウム水溶液(15ml)の間で分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして澄んだ油まで濃縮した。未精製のエステルをシリカゲルクロマトグラフィー(エーテル/ヘキサン 3:7→9:113)によって精製し、澄んだ油として100mgのエステルを得た。
ステップB
【0126】
化合物33(100mg, 0.18mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として45mgの化合物32を得た。([M+H] = 469.4 m/z)。
実施例18
【0127】
化合物27の替わりに、化合物29を使用して化合物34を実施例16に記載の製法に従って合成した。([M+H] = 441.4 m/z)。
実施例19
【0128】
化合物27の替わりに、化合物29を使用して化合物34[sic. 35]を実施例17に記載の製法に従って合成した。([M+H] = 469.4 m/z)
実施例20
ステップA
【0129】
化合物26(185mg, 0.3mmol, 1当量)のエタノール溶液(5ml)をヒドロキシルアミン塩酸塩(140 mg, 2 mmol, 6当量)、酢酸ナトリウム(160 mg, 2 mmol, 6当量)および水(0.5 mL)で処置し、そして混合溶液を室温で1時間攪拌した。混合溶液をEtOAcと水(夫々、50ml)の間で分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして白い残留物まで濃縮した。未精製の生成物をシリカゲルクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)によって精製し、193mgのオキシム37を得た。
ステップB
【0130】
化合物37(65mg, 0.113mmol)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末、シス及びトランスオキシム異性体として15mgの化合物36を得た。([M+H] = 440.3 m/z)。
実施例21
ステップA
【0131】
化合物27(42mg, 0.075mmol, 1当量)をDCM(5ml)に溶解し、4‐ジメチルアミノピリジン(2mg, 0.02mmol, 0.2当量)、N‐Cbzグリシン(23mg, 0.110mmol, 1.5当量)及びジイソプロピルカルボジイミド(0.023ml, 0.150mmol, 2当量)を加えた。12時間室温で攪拌した後、溶液をEtOAc(30ml)と5%炭酸水素ナトリウム水溶液(15ml)の間で分画した。有機層を鹹水で洗浄し、硫酸ナトリウム上で乾燥し、そして澄んだ油まで濃縮した。未精製のエステルをシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)によって精製し、澄んだ油として35mgのエステルを得た。
ステップB
【0132】
化合物39(235mg, 0.42mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 50mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として17mgの所望の生成物を得た。([M+H] = 452.4m/z)。
実施例22
【0133】
化合物27の替わりに、化合物29を使用して化合物40を実施例21に記載の製法に従って合成した。([M+H] = 452.4 m/z).
実施例23
【0134】
N‐(2‐オキソエチル)アセトアミドの替わりに、N‐(2‐オキソエチル)‐2‐フェニルアセトアミドを使用して化合物41を実施例10に記載の製法に従って合成した。([M+H] = 587.7 m/z)。
実施例24
ステップA
【0135】
丸底フラスコをアルコール29(7.60g, 13.53mmol, 1当量)でチャージし、DCM(115mL)に溶解した。反応溶液をトリエチルアミン(8.21g, 81mmol, 6.0当量)でチャージした。混合溶液を0℃まで冷却し、メタンスルホニルクロリド(methanesulfonylchloride, 1.86g, 16.2mmol, 1.2当量)でチャージした。30分後、反応混合溶液を飽和炭酸水素ナトリウム水溶液とEtOAcの間で分画した。有機層を分離し、硫酸ナトリウム上で乾燥し、そして乾燥状態まで濃縮した。残留物を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 10→25%)を使用して精製し、所望の物質メシル酸塩(メシラート)を得た。
【0136】
丸底フラスコをメシル酸塩(9.1g, 14.22mmol, 1当量)でチャージし、50mlのDMPUに溶解した。反応溶液をアジ化ナトリウム(4.62g, 71.1mmol, 5.0当量)でチャージし、60℃まで加熱した。17時間混合溶液を攪拌した。次に、反応混合溶液を室温まで冷却し、そして水でチャージした。30分間混合溶液を攪拌した。混合溶液を減圧下で濾過し、水でリンスし、そして空気乾燥し、そして精製すること無しに直接次のステップに使用した。
ステップB
【0137】
丸底フラスコをアジド43(8.35g, 14.23mmol, 1当量)でチャージし、THF(120mLを加えた。次に、反応溶液をトリフェニルホスフィン(11.2g, 42.7mmol, 3.0当量)でチャージした。混合溶液を50℃まで加熱し、20時間攪拌した。次に、反応混合溶液を室温まで冷却し、そして溶媒を減圧下で取り除いた。残留物を、シリカゲルフラッシュクロマトグラフィー(メタノール/DCM 10%→20%)を使用して精製し、アミンを得た。
【0138】
丸底フラスコをアミン(5.10 g, 9.09 mmol, 1当量)でチャージし、DCM(60 mL)に溶解した。反応をN,N‐ジイソプロピルエチルアミン(5.88g, 45.5mmol, 5.0当量)でチャージした。混合溶液を0℃まで冷却し、メタンスルホニルクロリド(2.08g, 18.2mmol, 2.0)でチャージした。30分後、反応混合溶液を飽和炭酸水素ナトリウム水溶液とEtOAcの間で分画した。有機層を収集し、硫酸ナトリウム上で乾燥し、そして乾燥状態まで濃縮した。残留物を、シリカゲルフラッシュクロマトグラフィー(EtOAc /ヘキサン 10→30%)を使用して精製し、Cbzで保護化されたメタンスルホンアミドを得た。
ステップC
【0139】
丸底フラスコをCbzで保護化されたメタンスルホンアミド(5.37g, 8.41mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(1.0g)でチャージした。固形物を2−プロパノール(50mL)に懸濁した。懸濁液を水素雰囲気下に置き、そして混合溶液を4時間25℃で攪拌した。次に、反応混合溶液をセライト(登録商標)上で濾過し、濾過液を乾燥状態まで濃縮した。次に、残留物を、シリカゲルフラッシュクロマトグラフィー(DCM/メタノール 0→5%)を使用して精製し、所望の生成物を得た。[M+H] = 505.6 m/z。
化合物42の代替合成
【0140】
再結晶化されたシクロパミン(2.07g)を適正な大きさの反応容器にチャージし、不活性環境下に置く。EtOAc(7.6g)、トリエチルアミン(1.53g)及びDMAP(307mg)を順番に加える。懸濁液を40℃まで温める。内部温度を45℃未満に維持しつつ、Cbz‐OBtを90分かけて3部(portions)で加える。反応混合溶液を40℃で90分間攪拌する。メタノール(26.4g)を徐々に反応混合溶液に加える間、温度を維持する。結果として生じる懸濁液を室温まで冷却し、少なくとも15時間攪拌する。未精製の製造物を濾過によって収集し、メタノール(5g)でリンスする。白い固形物を減圧下で恒量まで乾燥し、ヘプタン(30.3g)及びトルエン(3.2g)から再結晶化し、化合物24a(3.0g)を得る。
【0141】
固形のビス(2,6‐ジメチルフェニル)水素リン酸塩及び化合物24aを前乾燥し、そして窒素雰囲気下に置く。純粋なジエチル亜鉛(722mg)をDCM(9.0g)が入った適正な大きさの反応容器にチャージする。リン酸塩のDCM溶液(17.9gの中に1.83g)及びIPI‐332690(3.6 gの中に1.34 g)を順番に25℃以下で加える。ジヨードメタン(1.58g)をチャージし、反応溶液を28℃で4‐6時間攪拌する。反応溶液を−45℃まで冷却し、メタンスルホン酸DCM溶液(1.5gの中に566mg)をチャージする。15分後、モルフォリン(1.711g)を加え、そして混合溶液を室温までオーバーナイトで温める。有機層を2N塩酸(2x13.6g)、次に4.8wt%炭酸ナトリウム(水溶液)、4.8wt%亜硫酸ナトリウム(水溶液)及び4.8wt%鹹水(夫々13.6g)で順番に洗浄する。有機層を乾燥し、濾過し、4gまで濃縮し、イソプロパノール(4g)で希釈する。メタノール(9.3g)を徐々に添加することによって生成物を溶液から結晶化する。メタノールリンス(2.6g)、濾過及び乾燥で1.09gの化合物24b(79%の分離収量)を得る。
【0142】
ジョンソンマッセイ Pd/C 触媒 A−305038−5(890mg)、続いて化合物24b(2.24g)を適正な大きさの反応容器にチャージする。反応容器をN2でパージ(purged)し、トルエン(21.8g)及び2−プロパノール(6.7g)を順番に加える。システムを脱気し、そして窒素雰囲気下に置き、そしてその工程を水素で繰り返す。システムを勢い良く攪拌し、そして水素雰囲気(blanket)を1気圧で4−5時間維持する。反応をTLC若しくはHPLCのいずれかでモニターする。未完了であったら、反応を不活性化し、追加の触媒(145mg)をチャージし、そして水素雰囲気を更に1時間繰り返す。エチレンジアミン(12.9mg)をチャージし、そして混合溶液を15分間攪拌した。触媒を濾過によって取り除き、トルエン:IPA(3:1)でリンスする。濾液及びリンス[液]を濃縮し、溶媒をトルエンに交換する。生成物をトルエン(19.0g)及びヘプタン(18.0g)から結晶化し、白い結晶化固形物(1.34g, 98%収量)として化合物24cを得る。
【0143】
化合物24c(644mg)、続いてアルミニウム‐t‐ブトキシド(525mg)、トルエン(8.34g, 15ボリューム)及び2‐ブタノン(7.83g, 15ボリューム)を適正な大きさの反応容器にチャージする。酸素を取り除くためにフラスコの内容物を減圧吸引/窒素パージ(purge)サイクルで脱気し、反応混合溶液を勢い良く攪拌しつつ、75℃で16‐18時間加熱する。反応を水性のロッシェル塩(10.3gの水の中2.6 g)の添加によって抑え、混合溶液を勢い良く1時間45℃で攪拌する。水性及び有機層を分画する。トルエン(2.9g)及びEtOAc(2.9g)の混合溶液で水性の層を逆抽出する。有機層を合併し、新鮮なロッシェル塩溶液(10.3gの水の中2.6g)、次に水(12.9g)で洗浄する。結果として生じる有機層を硫酸ナトリウム(1.97 g)上で乾燥し、濾過し、そして真空中で濃縮する。最初にIPA(6.5g)、次にヘプタン(7.7g)のチャージ及び濃縮溶媒交換を経由して、生成物を結晶化する。濃ヘプタン泥状物(〜2.7g)をオーバーナイトで攪拌し、固形物を濾過によって収集する。真空吸引乾燥により化合物24d(550mg)を85%の収量で得る。
【0144】
エノン24d(459mg)及びジョンソン‐マッセイのカーボン上(に担持した)5%パラジウム(A503023-5, 101mg)を適正な大きさのマルチネック反応容器にチャージする。容器を窒素でパージ(purged)し、3‐ピコリン(2.2g)を溶媒としてチャージする。攪拌を開始し、容器を、窒素を使用して最初の脱気をし、そして次に大気圧の水素下で8時間攪拌する。反応の終了時に、触媒を0.2ミクロンメディアを介した濾過によって取り除き、ACN(1.4ml)でリンスする。機械的な攪拌、内部温度プローブ、及び窒素雰囲気を備えたクリーンないし清潔な反応容器の中で、濾過液及びリンス[液]を合併する。クエン酸(3.7g)の水(9.2ml)溶液を反応容器に30℃以下でチャージし、20℃、次に0℃でクエン酸塩として、溶液からIPI‐335589を徐々に結晶化する。結晶化物を吸引濾過によって回収し、水(3.7ml)で洗浄する。乾燥後、水和物(3-5wt% 水)として、89.5%収量(622mg)で、90:1に近いβ:αの割合で、クエン酸塩24eを分離する。
【0145】
上記クエン酸塩24e(1.50g)を、2‐メチルテトラヒドロフラン(7.7g)及び1M炭酸ナトリウム(9.0ml)と共に、適正な大きさの反応器にチャージする。添加漏斗(addition funnel)を介してクロロギ酸ベンジル(454mg)の2‐メチルテトラヒドロフラン(300mg)溶液を加え、反応を1−2時間、環境温度にする。反応が完了したとき、攪拌を止め、層を分離し、そして有機層を水で2回(2x6g)洗浄する。有機層を硫酸ナトリウム(3g)上で乾燥し、濾過し、そして濃縮する。新鮮な2‐メチルテトラヒドロフラン(6.5g)からの濃縮によって残りの水を更に減らし、無水2‐メチルテトラヒドロフランの溶液として物質を次の反応に移す。
【0146】
市販の1M K‐セレクトリドのTHF(1.20g)を乾燥した反応容器に窒素雰囲気下でチャージし、そして無水2‐メチルテトラヒドロフラン(2.10g)で希釈し、そして−65℃に冷却する。次に、内部温度を−65℃±5℃に調節するために、溶液24f(0.41g)の2−メチルテトラヒドロフラン(1.5g)溶液を徐々に反応容器に加える。反応溶液を2時間攪拌し、そしておよそ1時間かけて−20℃まで温め、そして更に1時間攪拌する。反応をHPLCでモニターし、未完了であった反応を追加のK‐セレクトリドで完了にする。低温度でメタノール(0.33g)、次に、−20℃で3M水酸化ナトリウム(2.4g)、5℃以下で15%過酸化水素水(1.04g)で反応を抑え、次に、オーバーナイトで、環境温度で攪拌する。層を分画し、そして有機層を1M水酸化ナトリウム水溶液(2ml)、0.5MのNa2SO3水溶液(2ml)、及び塩酸でpH3に調節した水(2ml)で洗浄する。有機層を硫酸ナトリウム(0.82g)上で乾燥し、濾過し、そして濃縮する。生成物24g(0.457g)をDCM(0.9g)から再濃縮し、次の反応に使用する。
【0147】
上記生成物24g(1.36g)を無水DCM(18.1g)で適正な大きさの反応容器にチャージし、不活性環境下に置き、そして−20℃まで冷却する。トリエチルアミン(0.61mg)をチャージし、続いて、メタンスルホニルクロリド(373mg)の無水DCM(30mg)を徐々に添加する。反応溶液を1時間−20℃で攪拌する。反応をHPLCによってモニターする。未完了の反応を追加のメタンスルホニルクロリドで完了にする。完了していた場合、反応を水(13.6g)で抑え、そして放温する。層を分離し、有機層を2.5wt%炭酸水素ナトリウム(13.8g)及び、次に水(10.9g)で洗浄する。有機層を硫酸ナトリウム(4g)上で乾燥し、濾過し、そして濃縮する。t‐ブチルメチルエーテル(10.9ml)及び次にl,3‐ジメチル‐3,4,5,6‐テトラヒドロ‐2(1H)ピリミジノン(DMPU, 4.7ml)のチャージ及び濃縮を経由して、生成溶液を溶媒交換する。DMPU溶液を直接次の反応に使用する。
【0148】
アジ化ナトリウム(0.74g)を適正な大きさの反応容器にチャージする。生成物24h(1.46 g)のDMPU(5.9g)溶液を反応容器にチャージし、更なるDMPU(1.9g)でリンスする。反応全体において窒素スイープ(sweep)を維持したままで、懸濁液を60℃まで15時間加熱する。反応溶液を環境温度まで冷却し、そしてMTBE(11.7g)で希釈する。有機溶液を2%生理食塩(3x8g)で3回洗浄し、硫酸ナトリウム(4.4g)上で乾燥し、濾過し、そして濃縮する。生成物をTHF(6.4g)から濃縮し、直接次の反応に使用する。
【0149】
未精製の生成物24i(1.34g)をTHF(12.6g)で溶解し、そして適切な大きさの反応溶液に移す。トリフェニルホスフィン(0.70g)及び水(0.44g)をチャージし、そして反応溶液を55℃まで15‐24時間加熱する。完了したとき、反応溶液を環境温度まで冷却し、硫酸マグネシウム(1.4g)で乾燥し、濾過し、そして濃縮する。試薬に基く不純物を取り除くために、固形物を溶解し、3部(portions)のDCM(3x9g)から濃縮し、そしてDCM/MeOH/Et3N勾配を使用したシリカゲルクロマトグラフィーによって精製する。プールした分画を乾燥状態まで濃縮し、DCM(6.8g)に溶解し、そして再び乾燥状態まで濃縮し、次のステップで使用する非晶質の固形物(1.12g)を得る。
【0150】
生成物24j(1.09g)を無水DCM(15.8 g)で溶解し、適正な大きさの反応容器に移し、窒素雰囲気下に置く。溶液を0℃まで冷却する。温度を5℃未満の間に維持しつつ、N,N‐ジイソプロピルエチルアミン(357mg)及び純粋なメタンスルホニルクロリド(0.165ml)を順番にチャージする。反応をHPLCによってモニターする。未完了の反応を追加のメタンスルホニルクロリドで完了にする。0.4M炭酸水素ナトリウム(11.4 g)水溶液で反応を抑え、環境温度まで温める。層を分離し、水性の層をDCM(5.8g)で逆抽出する。合併した有機層を硫酸マグネシウム(0.55g)上で乾燥し、濾過し、そして濃縮する。残りのDCMを取り除くために生成物24kを溶解し、2‐プロパノール(4.0 g)から除去し、次の反応に直接使用する。
【0151】
アルドリッチ デグサ タイプ ElOl NE/W 10% Pd/C(249mg)を適正な大きさの反応容器にチャージし、そして窒素雰囲気下に置く。24k(1.24g)の2−プロパノール(9.8g)溶液を反応容器にチャージする。システムを脱気し、そして窒素雰囲気下に置き、そしてその工程を水素で繰り返す。反応溶液を1気圧の水素下で、環境温度で8時間攪拌する。不活性環境を容器に戻し、そして2−プロパノール(0.5g)中に泥状化した触媒(125mg)の2度目のチャージを反応に加える。反応混合溶液を脱気し、そして窒素雰囲気下に置き、その工程を水素で繰り返す。反応溶液を1気圧の水素下でさらに15時間、環境温度で攪拌する。反応をHPLCによってモニターする。未完了の反応を追加の触媒及び水素で処置する。完了した場合、反応溶液を濾過し、蒸気活性炭(200mg)で処置し、そして再び濾過する。溶液を部分濃縮によって乾燥し、反応容器に移し、理論的収量に基く0.09Mまで無水2−プロパノールで希釈する。2−プロパノールの1.25M塩酸溶液(1.64g)を20分間かけてチャージする。塩酸塩を穏やかな攪拌で徐々に結晶化させ、濾過によって分離する。結晶化物を2−プロパノール(2.5g)で洗浄し、真空吸引乾燥し、1:1のIPA溶媒和化合物として化合物42(916mg, 80%収率)を得る。
実施例25
ステップA
【0152】
丸底フラスコをアミン42(1.1g, 2.1mmol, 1当量)、無水テトラヒドロフラン(10ml)、及びピリジン(880uL, 10.9mmol, 5当量)でチャージした。冷却した(0℃)混合溶液を過酸化ベンゾイル(1.6g, 6.5mmol, 3当量)で処理した。混合溶液を2時間0℃で、次にオーバーナイトで25℃で攪拌した。反応混合溶液をMTBEで希釈し、飽和NaHCO3水溶液と1NのNaOHの混合溶液で層が分離するまで洗浄した。有機層を収集し、水性の層をMTBEで1回再抽出した。合併した有機層を鹹水で洗浄し、Na2SO4上で乾燥し、濾過し、乾燥状態まで濃縮した。未精製の油を5mlのCH2Cl2に溶解し、SiO2(40g)カラムの上に乗せ、ヘキサン/EtOAc(10%から50%)から溶出し、ベンゾイル誘導体48(380mg)を得た。([M+H] = 625.4 m/z)
ステップB
【0153】
丸底フラスコを[ベンゾイル誘導体]48(374mg, 0.6mmol, 1当量)及びMeOH(5mL)でチャージした。2NのKOH(0.3mL, 0.6mmol, 1当量)の存在下で、溶液を25℃に処置した。混合溶液を3時間攪拌した。溶媒を減圧下で取り除いた。MTBEを残留物に加え、1NのHClで中和した。層を切り、水性の層を2部(portions)のCH2Cl2で抽出した。合併した有機層をNa2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。未精製物質(380mg)をCH2Cl2で溶解し、SiO2(12g)カラムの上に乗せ、ヘキサン/EtOAc(0%から100%)で溶出し、ヒドロキシルアミン47を得た。物質をt−BuOH/7%H2Oから凍結乾燥し、白い粉末として213mgの[ヒドロキシルアミン]47を得た([M+H] = 521.4 m/z)。
実施例26
ステップA
【0154】
ドライヤーで乾燥したフラスコを、無水CH2Cl2(5mL)及びベンジルアルコール(785uL, 7.58mmol, 1.3当量)でチャージした。冷却した(0℃)溶液をクロロスルホニルイソシアネート(506uL, 5.83mmol, 1当量)で処理した。次に、DMAP(1.4g, 11.6mmol, 2当量)を加え、そして混合溶液を1時間25℃で攪拌した。DMAPの溶解が完了した後、短い期間、反応溶液は、澄んでいた。次に、白い泥状の沈殿物が形成した。混合溶液をCH2Cl2(30mL)で希釈し、そして3部(portions)(夫々30mL)の水で洗浄した。有機層をNa2SO4上で乾燥し、濾過し、そして、乾燥状態まで蒸発させた。所望の白い固形物51を精製することなしに次のステップに用いた。
ステップB
【0155】
丸底フラスコを生成物52(30mg, 0.053mmol, 1当量)及び生成物51(18mg, 0.053mmol, 1当量)でチャージした。両方の試薬をCH2Cl2(2mL)に溶解し、溶液を25℃で攪拌した。未精製の物質をSiO2カラム(4g)に乗せ、ヘキサン/EtOAc(0%から50%)で溶出し、16mgのスルファモイル化(sulfamoylated)誘導体53を得た([M+Na] = 796.4 m/z)。
ステップC
【0156】
丸底フラスコを生成物53(16mg, 0.021mmol, 1当量)及び11mgの10% Pd/C(ウェット, Aldrich Degussa type ElOl)でチャージした。物質を2−プロパノール(3mL)に懸濁した。フラスコを密封し、そして水素で3回パージ(purged)し、そしてオーバーナイトで1気圧の水素下に置いた。泥状物を0.2ミクロンアクロディスク(Acrodisc)を介して濾過し、2−プロパノールで洗浄し、そして溶媒を減圧下で取り除いた。残留物を、CH2Cl2/MeOH(5%から10%)で溶出するSiO2カラム(1g)によって精製した。主要生成物をt‐BuOH/7%H2Oから凍結乾燥し、9mgのスルファミド50を得た([M+H] = 506.4m/z)。
実施例27
ステップA
【0157】
丸底フラスコをシクロパミン4‐エン‐3‐オン(cyclopamine 4-en-3-one, 3.5g, 8.5mmol, 1当量)及びピリジン(70mL)でチャージした。反応容器をPd/C(10% Pd, 500mg)でチャージした。反応溶液を1気圧の水素下に置いた。3.5時間後、LCMSは出発物質の完全な消耗を示した。触媒をアクロディスク0.2ミクロンフィルター上で濾過除去し、そしてトルエンで洗浄した。溶媒をトルエン(2x10mL)で共沸除去によって取り除いた。所望の物質56、3.5g([M+H] = 412.5m/z)を、そのままで次のステップに使用した。
ステップB
【0158】
丸底フラスコを生成物56(1.2g, 2.8mmol, 1当量)、CH2Cl2(10mL)及びトリエチルアミン(1.9mL, 14.2mmol, 5当量)でチャージした。冷却した(0℃)溶液をCBz‐Cl(440uL, 2.8mmol, 1当量)で処理した。1時間後、LCMSは出発物質の完全な消耗を示した。混合溶液を水で希釈した。層を切り、そして有機層を水で2回洗浄した。有機層を硫酸ナトリウム上で乾燥し、濾過し、そして乾燥状態まで濃縮した。生成物をヘキサン/EtOAc(0から20%)で溶出するカラムクロマトグラフィー(SiO2, 40g)によって精製し、ケトン生成物57(891mg)を得た([M+Na] = 468.4m/z)。
ステップC
【0159】
丸底フラスコの中で、ケトン生成物57を2回CH2Cl2で共沸蒸留し、そして真空下で1時間乾燥した。窒素下において、ケトン2(693mg, 1.27mmol, 1当量)を無水THF(20 mL)に溶解し、そして溶液を−78℃まで冷却した。1MのK‐セレクトリドのTHF溶液(1.9mL, 1.9mmol, 1.5当量)を滴下で加えた。1時間後、反応をTLCによって完了した。反応を2.6mlの5NのNaOHの添加、続いて2.6mLの30%wtのH2O2の緩徐添加によって抑えた。結果として生じる混合溶液をオーバーナイトで攪拌した。混合溶液を水とEtOAcの間で分画した。水性の層をEtOAcで逆抽出した。合併した有機[層]を最初に水(ごく一部の塩化アンモニウムで緩衝された)、次に鹹水で洗浄した。有機[層]を乾燥し、濾過し、そして未精製の気泡(840 mg)まで濃縮した。未精製の物質をCH2Cl2に溶解し、SiO2カラム(4Og)に乗せ、ヘキサン/EtOAc(0から50%)で溶出し、アルコール生成物58(565 mg)を得た。
ステップD
【0160】
窒素下の丸底フラスコの中で、アルコール生成物58(530mg, 0.98mmol, 1当量)を5mLの無水CH2Cl2及びトリエチルアミン(800uL, 5.81mmol, 6当量)に溶解した。反応混合溶液を0℃まで冷却し、Ms‐Cl(112uL, 1.45mmol, 1.5当量)を滴下で加えた。混合溶液を0℃で30分間攪拌した。TLC(ヘキサン:EtOAc, 7:3)は、〜70%の変換を示した。70μLのトリエチルアミン(70uL, 0.5当量)及びMs‐Cl(10uL, 0.1当量)を反応容器にチャージした。90分後、飽和炭酸水素塩の溶液をチャージし、残留物をCH2Cl2で抽出した。有機層を水で洗浄し、乾燥し、そしてオフホワイトの気泡まで濃縮した。物質をCH2Cl2に溶解し、そしてヘキサン/酢酸エチル(0%から50%)で溶出するSiO2(40g)で精製し、メシラート生成物59(430mg)を得た。
ステップE
【0161】
丸底フラスコの中で、メシラート生成物59(420mg, 0.67mmol, 1当量)を2mLのDMPUに溶解した。溶液を60℃で5時間アジ化ナトリウム(218mg, 3.4mmol, 5当量)で処理した。混合溶液を25℃まで冷却し、次に氷水に注ぎ、白い固形物を得た。化合物をMTBEで抽出した(3回)。合併した有機層を水(2X)、次に鹹水で洗浄した。有機層をNa2SO4上で乾燥し、濾過し、そして、白い気泡(342mg)まで濃縮した。所望の物質60を、そのままで次のステップに使用した。
ステップF
【0162】
コンデンサを備えた丸底フラスコの中で、アジド60(336mg, 0.58mmol, 1当量)を7mLのTHF及び140μLの水に溶解し、トリフェニルホスフィン(462mg, 1.76mmol, 3当量)で処理した。混合溶液を70℃までオーバーナイトで加熱した。TLC(ヘキサン/EtOAc, 7:3)は、反応が完了したことを確認した。反応溶液を乾燥状態まで濃縮した。未精製の物質をCH2Cl2に溶解し、12gのSiO2の上に乗せ、そしてCH2Cl2/MeOH(0から20%)で溶出し、アミン61(254mg)を得た。
ステップG
【0163】
窒素下の丸底フラスコの中で、アミン61(248mg, 0.45mmol, 1当量)を7mLの無水CH2Cl2及びN,N‐ジイソプロピルエチルアミン(237uL, 0.91mmol, 2当量)に溶解した。反応混合溶液を0℃まで冷却し、Ms‐Cl(70uL, 1.45mmol, 1.5当量)を滴下で加えた。混合溶液を0℃で2時間攪拌した。TLC(ヘキサン/EtOAc, 7:3)は、少量のアミンを示した。混合溶液を10μLのMs‐Cl(0.2当量)でチャージし、そして25℃まで1時間温めた。反応混合溶液をCH2Cl2、次にNaHCO3の飽和溶液で希釈した。層を切った。水性の層を一部(portion)のCH2Cl2で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。未精製物(326mg)をSiO2カラム(12g)に加え、ヘキサン/EtOAc(0から50%)で溶出し、スルホンアミド62(256mg)を得た。
ステップH
【0164】
丸底フラスコをスルホンアミド62(250mg, 0.4mmol, 1当量)及び50mgの10% Pd/C(ウェット, Aldrich Degussa type ElOl lot 08331KC)でチャージした。物質をEtOAc(5mL)に懸濁した。フラスコを密封し、そして水素で3回パージ(purged)し、そして1気圧の水素下で攪拌した。3時間後、変換が観察されたが、多くの出発物質はそのままであった。泥状物を0.2ミクロンのアクロディスク(Acrodisc)を介して濾過し、2−プロパノールで洗浄した。濾過液を54mgの触媒を加えた条件の反応に再び供した。反応は、3時間後に完了した。泥状物を0.2ミクロンアクロディスクを介して濾過し、2‐プロパノールで洗浄し、そして溶媒を乾燥状態まで濃縮した。未精製物質(200 mg)をSiO2カラム(12g)の上に乗せ、そして化合物をCH2Cl2/MeOH勾配(0 から10%)を使用して溶出し、遊離アミンを得た。物質をt‐BuOH/7%H2Oから凍結乾燥し、白い粉末として175mgの生成物55を得た([M+H] = 491.3 m/z)。
実施例28
細胞培養におけるヘッジホッグ経路の阻害
【0165】
ヘッジホッグ経路特異的な癌細胞死滅効果は、次の検定を使用して確認することができる。C3H10T1/2細胞は、ソニックヘッジホッグペプチド(Shh-N)に接触した場合に、骨芽細胞に変異する。変異した、すなわち、これらの骨芽細胞は、酵素学的な検定で測定することができる高レベルのアルカリフォスファターゼ(AP)を生成する(Nakamura et al., 1997 BBRC (1997) 237: 465)。C3H10T1/2の骨芽細胞への変異(Shh依存的なイベント)を妨げる化合物は、従って、AP生産の減少によって同定することができる(van der Horst et al., Bone (2003) 33: 899)。検定の詳細は、以下に開示する。変異検定の概略結果(阻害のEC50)を下の表1に示す。
検定プロトコル
細胞培養
【0166】
マウス胎性中胚葉繊維芽細胞のC3H10T1/2細胞(ATCCから得た)を、10%加熱不活性化FBS(Hyclone)、50ユニット/mlのペニシリン、及び50ug/mlのストレプトマイシン(Gibco/Invitrogen)を補充した基本MEM培地(Gibco/Invitrogen)内で、37℃で、5%CO2の空気雰囲気中で培養した。
アルカリフォスファターゼ検定
【0167】
C3H10T1/2細胞を96ウェル[プレート]に8x103細胞/ウェルの密度で蒔いた。細胞は、コンフルエンスまで成長した(72時間)。ソニックヘッジホッグ(sonic Hedgehog, 250ng/ml)及び/または化合物処理の後、細胞を溶解緩衝液(50mMトリス pH7.4, 0.1%トライトンXlOO)に溶解し、プレートを超音波処理し、そしてライセートを0.2μmPVDFプレート(Corning)を介してスピンした(spun)。40μLのライセートを、1mg/ml p‐ニトロフェニルリン酸塩を含むアルカリ緩衝溶液(Sigma)内でAP活性について検定した。30分間37℃でインキュベートした後、プレートをエンビション(Envision)プレートリーダ上で、405nmで解読した(read)。全タンパク質を、ピアス(Pierce)からのBCAタンパク質アッセーキットで、製造者の説明書に従って数量化した。AP活性を、全タンパク質量に対して規格化した。「A」は、IC50が20nM未満であることを示し、「B」はIC50が20〜100nMであることを示し、「C」は、IC50が>100nMであることを示すことを意味する。
表1−阻害のための概略EC50
実施例29
膵臓癌モデル
【0168】
化合物42の活性をヒト膵臓モデルでさらにテストした、すなわち、BXPC‐3細胞をマウスの右足の側面の皮下の中に移植した。腫瘍移植後、42日目に、マウスを、ビヒクル(担体:Vehicle)(30%HPBCD)若しくは化合物42のいずれかを受ける2群に無作為に区分した。化合物42を40mg/kg/日で経口投与した。25日間の毎日の投与を受けた後、ビヒクルコントロールと比べた場合、化合物42は、統計学的に腫瘍の成長量を40%減少させた(p=0.0309)。実験の最後に、HH経路遺伝子のq‐RT‐PCR解析によって標的に対する反応を評価するために、最終投与の4時間後、腫瘍を回収した。ヒトGli‐1の解析では、変調がなかった。マウスのGli‐1のmRNAレベルの解析は、ビヒクル処置群と比べた場合、化合物を処置した群において強い低下(抑制)制御となった。ヒトの腫瘍細胞ではないが、マウスの細胞のヘッジホッグ経路の抑制は、ヘッジホッグ阻害剤の一つの効果が腫瘍‐間質相互作用(tumor-stroma interaction)に影響を及ぼすことを示す。
実施例30
髄芽腫モデル
【0169】
化合物42の活性も髄芽腫の遺伝子導入マウスモデルで評価した。腫瘍サプレッサーPatched 1(Ptch1)及びHypermethylated in Cancer 1(Hic1)における機能欠損変異のヘテロ接合であるマウスは、自発性の骨芽腫を発生する。ヒト髄芽腫に似て、これらの腫瘍は、残りのHic1アレルの完全な過剰メチル化のプロモーターを表し、そして野生型Ptch1アレルの発現の欠損を表す。皮下の移植片として継代された場合、これらの細胞は、激しく成長し、そしてヘッジホッグ経路依存的である。経口的な化合物の投与の効果を評価するため、及び活性と血漿及び腫瘍における薬物暴露とを関連付けるために、このモデルを採用した。ドーズ投与(dose administration)の8時間後のGli‐1 mRNAの発現の減少によって測定されたものとして、化合物42の単ドーズ(single dose)の経口投与(PO)は、皮下に移植された腫瘍におけるHH経路の量依存的(dose-dependent)な低下制御を導いた。
【0170】
化合物の毎日(QD)の経口投与(PO)は、投与量依存的な腫瘍の成長の阻害を導き、多量の投与ではあからさまな腫瘍退縮が観察された。腫瘍成長を50%阻害する(ED50)ための、およその効果的な毎日の経口的な投与量は、4mg/kgである。動物が毎日(QD)の[投与]を21日間処置された場合、処置の休止の後、長期間の生存(>60日)が観察され、腫瘍の再成長がほとんどなかった。これは、ヘッジホッグ阻害剤化合物42がヘッジホッグ経路と遺伝子突然変異によるヘッジホッグ経路に依存する腫瘍の成長との両者を抑制することを実証する。
実施例31
肺癌モデル
【0171】
ヒトSCLC腫瘍モデルにおける化合物42の活性をテストするために、LX22細胞を右足の側面の皮下に移植した。LX22は、化学的に未処置の患者由来のSCLCの主要な異種移植モデルであり、マウスからマウスへの継代によって維持されている。この腫瘍は、臨床設定に近似するということにおいてエトポシド(etoposide)/カルボプラチン(carboplatin)化学療法に反応する。LX22は、化学療法の処置の間退行し、寛解期を経て、そして次に再発し始める。LX22モデルにおいて、化合物単試薬活性及びそれの化学抵抗性再発を調節する能力をテストした。腫瘍移植後、32日目に、マウスをビヒクル(30%HPBCD)、当該化合物又はエトポシド及びカルボプラチンの組み合わせ化学療法(E/P)を受ける3群に無作為にグループ分けした。化合物42を40mg/kg/日の投与量で経口投与し、そして16連続投与の後、処置群とビヒクル群の間に測定可能な違いがなかった。腫瘍移植後、34、35、36、及び48日目に、エトポシドを12mg/kgで静脈に投与し、また、34、41及び48日目に、カルボプラチンを60mg/kgで静脈に投与した。50日目に、E/P処置マウスをさらにビヒクル(30%HPBCD)又は化合物追跡処置を受ける[群]に無作為に区分した。化合物をMTD(最大耐性量)40mg/kg/日で複数回、経口的に投与し、そして35の連続投与の後、ビヒクル群に比べて、処置群において腫瘍の再発に実質的な遅延があった (p=0.0101)。
実施例32
多発性骨髄腫
【0172】
ヒト多発性骨髄腫の細胞株(cell lines)(NCI−H929及びKMS12)並びにMM患者由来の初代の臨床骨髄標本(primary clinical bone marrow specimens)を用いて、インビトロ(in vitro)で多発性骨髄腫細胞(MM)の成長を抑制する化合物42の性能を試験した。細胞は化合物で96時間処置され、洗浄されて、次いで、メチルセルロースに培養された。腫瘍コロニーは、処置の後の細胞増殖の可能性の指標として、10〜21日後に定量化された。細胞株又は初代の患者の標本の処置は、未処置の対照区と比較して、減少した細胞成長の結果となった。未処置の対照区は100%の細胞成長を示し、臨床サンプルと同様に処置された各細胞株は、約25%未満の成長を示した。
実施例33
急性骨髄性白血病及び骨髄異形成症候群
【0173】
急性骨髄性白血病(AML、細胞株U937)及び骨髄異形成症候群(MDS、細胞株KG1及びKG1a)の患者由来のヒト細胞株のインビトロ(in vitro)成長を抑制する化合物42の性能を研究した。各々の細胞株は化合物42(1.0uM)で72時間処理され、続いてメチルセルロースに培養された。下記表にまとめたように、それら細胞株の成長は化合物42によって抑制された。
表2.AML及びMDSにおける細胞成長の抑制
実施例34
非ホジキンリンパ腫(NHL)及びホジキン病(HD)
【0174】
非ホジキンリンパ腫(細胞株RL及びJeko-1)及びホジキン病(細胞株L428)の患者由来のヒト細胞株のインビトロ(in vitro)成長を抑制する化合物42の性能を研究した。各々の細胞株は化合物42(1.0uM)で72時間処理され、続いてメチルセルロースに培養された。下記表にまとめたように、それら細胞株の成長は化合物42によって抑制される。
表3.HD及びNHLにおける細胞成長の抑制
実施例35
プレB細胞急性リンパ性白血病
【0175】
Gli反応性ルシフェラーゼリポーターが一時的に細胞に形質移入された、一過性形質転換アッセイ(transient transfection assay)を用い、プレB細胞急性リンパ性白血病の3つの細胞株(REH、RS4−11及びNalm−6)に対する化合物42(1uM)の性能が研究された。化合物42での処置は、ビヒクル処置の対照区と比較して、ルシフェラーゼの活性を抑制した(表4)。これは、化合物42がヘッジホッグ経路の効果的なアンタゴニストであることを実証した。
表4.ルシフェラーゼ活性の抑制
【0176】
また、それら細胞株のうち2つ(の細胞株)の成長における化合物42の効果を72時間インビトロ(in vitro)で処置して研究した。処置後、細胞は洗浄されて、メチルセルロースに培養された。コロニー形成の抑制はほとんど無かったが、以降のコロニーの再培養(replating)は顕著な細胞成長の抑制を実証した(表5)。
表5.すべて(の細胞)における細胞成長の抑制
引用による繰込み
【0177】
本願明細書に引用された全てのU.S.特許及びU.S.公開特許出願は、これにより、引用によって本願明細書に繰み込み記載される。
等価物
【0178】
当業者は、通例のルーチン的実験を行うだけで、本願明細書に開示された本発明の特定の実施態様の多くの等価物を認識し、又は確認することができるであろう。そのような等価物は、以下の特許請求の範囲の請求項によって包含されることを意図する。
【技術分野】
【0001】
この出願は、2006年12月28日に出願されたUSSN60/878,018及び2007年6月1日に出願されたUSSN60/941,596の優先権を主張し、これらの出願はそれらのすべてが参照によって本願明細書に組み込まれる。
【政府提供資金】
【0002】
本明細書に記載の幾つかの研究(some of the work)は、NIH/NCIによって与えられた認可番号K23CA107040の政府支援の下で行われた。(合衆国連邦)政府は本発明の特定の権利を有し得る。
【背景技術】
【0003】
本発明は、一般的に、ヘッジホッグ経路に対抗(アンタゴナイズ)し、かつ、シクロパミン類似体を用いて様々な症状を処置するための方法に関する。
【0004】
ある種の癌におけるヘッジホッグ経路の抑制は、腫瘍成長の抑制に帰着することが知られている。例えば、抗ヘッジホッグ抗体は、ヘッジホッグ経路の機能をアンタゴナイズし、かつ、腫瘍の成長を抑制することが知られている。ヘッジホッグ経路の低分子抑制もまた、いくつかの癌タイプで細胞死に帰着することが知られている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
この分野の研究は、主に、ヘッジホッグ経路生物学の解明及び新規のヘッジホッグ経路阻害剤ないし抑制剤(inhibitors)の発見に注力されている。ヘッジホッグ経路の阻害剤が特定されてはいるが、ヘッジホッグ経路のより強力な阻害剤を特定する必要がある。
【0006】
2006年3月9日に公開され、本願と同じ譲受人に譲渡されたPCT公開公報(WO2006/026430)は、多種多様なシクロパミン類似体を開示し、それらシクロパミンのA又はB環の不飽和に注視している。本願においては、驚くべきことに、強力な類似体は完全に飽和したA及びB環を含んでいる。
【課題を解決するための手段】
【0007】
本発明は、ヘッジホッグ経路(hedgehog pathway)によってもたらされる(mediated)過剰増殖(hyperproliferative)疾患及び症状を処置するための方法に関する。
【0008】
一視点において、本発明は過剰増殖疾患を処置するための方法に関する。本方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;R4の上記式中、各Wは夫々独立してジラジカルであり;
各qは夫々1、2、3、4、5若しくは6において独立であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
ある実施態様において、R1はスルホンアミドである。
【0009】
症状は、皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される。
【0010】
別の視点において、本発明はヘッジホッグ経路(hedgehog pathway)によってもたらされる(mediated)症状を処置するための方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;各qは、夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
ある実施態様において、R1はスルホンアミドである。
【0011】
症状は、皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される。ある実施例は、小細胞肺癌、膵臓癌、髄芽腫、多発性髄芽腫、白血病、骨髄異形成症候群、非ホジキンリンパ腫、及びホジキン病を含む。化合物は、経口的、静脈内、又は、局所的に投与されてよい。
【0012】
別の視点において、本発明は、被験者ないし患者においてヘッジホッグ経路に対抗(アンタゴナイズ)する方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;R4の上記式中、各Wは、夫々独立してジラジカルであり;各qは夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり、
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
ある実施態様において、R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもない。
ある実施態様において、R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく、及びR1及びR2は一緒になってC=Oでない。
【0013】
上記の実施態様において、化合物は下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である。
【0014】
別の視点において、本発明は、被験者ないし患者においてヘッジホッグ経路に対抗(アンタゴナイズ)する方法に関する。当該方法は、下記式:
を有する化合物又はその薬学的に許容可能な塩の有効量を被験者ないし患者(subject)に投与することを含み;
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは独立して1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは独立してH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;
又は、同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成し;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)であり、ここに各Rは独立してH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
R7及びR7´の夫々がHであり;又は
R7及びR7´が一緒になって=Oを形成し;
R8及びR9は夫々Hであるか、又は、R8及びR9は一緒になってボンド(結合)を形成し;及び
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1はヒドロキシルとなることはできず、かつ、R2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1は酢酸となることはできず、かつ、R2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR8がHである場合;R1及びR2が一緒になって=Oとなることができず;及び
R3、R4、R8、R9がHであり、かつR7及びR7´がHである場合;R1及びR2がHとなることができない。
ある実施態様において、R1はスルホンアミドである。
【発明を実施するための形態】
【0015】
本願明細書で用いた用語の定義は、化学及び薬学分野における各用語として認識された現在の技術水準の定義を組み込むことを意図する。適切な場所に、実例(exemplification)を提供する。定義は、特定の場合に限定される場合を除き、個々に又は大きな群の一部のいずれかとして、この明細書を通して使用された用語に適用する。
【0016】
本願明細書で使用されるように、各表現ないし語句(expression)の定義、例えば、アルキル、m、nなどは、それが任意の構造(式)の中で1回以上出現した場合、同一の構造(式)における他の場所のその定義からは独立であることを意図する。
【0017】
用語「アシルアミノ」(acylamino)は、一般式:
によって表すことができる部分ないし部位(半部分:moiety)を意味し、
式中、R50及びR54は、水素、アルキル、アルケニル若しくは‐(CH2)m‐R61を表し、
式中、R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環(polycycle)を表し、そして、mは、ゼロ若しくは1から8までの範囲内の整数である。
【0018】
用語「アルケニル」及び「アルキニル」は、長さ及び置換可能な点で、上記アルキルに類似するが、少なくとも1つの二重若しくは三重結合を夫々含有する不飽和脂肪族基を意味する。
【0019】
用語「アルコキシル」若しくは「アルコキシ」は、そこに結合した酸素ラジカルを有する、上記で定義したアルキル基を意味する。代表的なアルコキシル基は、メトキシ、エトキシ、プロピルオキシ、tert‐ブトキシ及び同様のものを含む。
【0020】
用語「アルキル」は、直鎖アルキル基、分枝状鎖のアルキル基、シクロアルキル(脂環式)基、アルキル置換シクロアルキル基、及びシクロアルキル置換アルキル基を含む飽和脂肪族基のラジカルを意味する。ある実施態様では、直鎖若しくは分岐状鎖のアルキルは、30以下の炭素原子を主要骨格ないしバックボーン(backbone)に有する(例えば、C1‐C30が直鎖であり、C3‐C30が分岐状鎖である)、20以下(の炭素原子を主要骨格に有する)。同様に、あるシクロアルキルは、3‐10の炭素原子をその環状構造に有し、その他は5、6若しくは7の炭素を環状構造に有する。
【0021】
用語「アルキルチオ」は、そこに結合した硫黄ラジカルを有する、上記で定義したアルキル基を意味する。ある実施態様では、「アルキルチオ」の部分ないし部位は、‐S‐アルキル、‐S‐アルケニル、‐S‐アルキニル及び‐S‐(CH2)m‐R61の1つによって表され、式中、mおよびR61は、上記で定義される。代表的なアルキルチオ基は、メチルチオ、エチルチオ及び同様のものを含む。
【0022】
用語「アミド」は、アミノ置換カルボニルとして当該技術で認識(art recognized)され、そして一般式:
によって表すことができる部分ないし部位を含み、式中、R50及びR51は、夫々独立して、水素、アルキル、アルケニル、‐(CH2)m‐R61を表し、又は、R50及びR51は、それらが結合するN原子と一緒になって、4から8までの原子を環状構造に有するヘテロ環を完成し、R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環を表し、そしてmはゼロ若しくは1から8までの範囲内の整数である。本発明のアミドのある実施態様は、不安定でありうるイミドを含まないだろう。
【0023】
用語「アミン」及び「アミノ」は、当該技術で認識され、そして非置換及び置換アミンの両方を意味し、例えば、一般式:
によって表すことができる部分であり、式中、R50、R51及びR52は、夫々独立して、水素、アルキル、アルケニル又は(CH2)m‐R61を表し、又は、R50及びR51は、それらが結合するN原子と一緒になって、4から8までの原子を環状構造に有するヘテロ環を完成し;R61は、アリール、シクロアルキル、シクロアルケニル、ヘテロ環若しくは多環を表し、そしてmはゼロ若しくは1から8までの範囲内の整数である。したがって、用語「アルキルアミン」は、そこに結合した置換若しくは非置換アルキルを有する、すなわち、少なくともR50及びR51の一つはアルキル基である、上記で定義したアミン基を含む。
【0024】
本願明細書で使用した用語「アラルキル」は、アリール基(例えば、芳香族若しくはヘテロ芳香族基)で置換されたアルキル基を意味する。
【0025】
本願明細書で使用された用語「アリール」は、0から4までのヘテロ原子を含むことができる5、6及び7員単環芳香族基、例えば、ベンゼン、アントラセン、ナフタレン、ピレン、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、トリアゾール、ピラゾール、ピリジン、ピラジン、ピリダジン及びピリミジンを含み、そして同様のものを含む。環状構造にヘテロ原子を有するそれらのアリール基を、「アリールヘテロ環」若しくは「ヘテロ芳香族」と称することもできる。芳香族環は1つ以上の環状位置(ring positions)を上記置換基、例えば、ハロゲン、アジド、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アルコキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、スルホンアミド、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分(moieties)、‐CF3(トリフルオロメタン)、−CN(シアン)若しくは同様のもので置換することができる。用語「アリール」は、2以上の炭素が2つの隣接する環に共通する(その環は、「縮合環」である)、2つ以上のサイクリック環を有する多環の環状システムも含み、ここで、少なくとも、1の環は、芳香族であり、例えば、他のサイクリック環は、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/またはヘテロ環であり得る。
【0026】
用語「ブレンステッド酸」(Bronsted acid)は、水素イオン(プロトン)供与体ないしドナー(donor)として作用することができる任意の物質(substance)を意味する。
【0027】
用語「カルボキシル」は、一般式:
によって表すことができるような部分ないし部位を含み、式中、X50は、ボンド(結合)であるか、若しくは、酸素または硫黄を表し、そして、R55及びR56の夫々は、独立して、水素、アルキル、アルケニル、−(CH2)m−R61、又は薬学的に許容可能な塩を表し、ここで、mおよびR61は、上記で定義されている。
【0028】
用語「ジラジカル」は、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール及びヘテロアラルキル基からなる一連の二価群の任意のものを意味する。例えば、
は、アルキルジラジカルであり、
もアルキルジラジカルであり、
は、アラルキルジラジカルであり、
は(アルキル)へテロアラルキルジラジカルである。典型的な例は、一般的な構造(CH2)xのアルキレン、ここでXは1‐6である、及び、2‐6の炭素原子及び1つ以上の二重若しくは三重結合を有するアルケニレン及びアルキニレンリンカーに対応するもの、3‐8員環を有するシクロアルキレン基、及び
とその異性体のようなアラルキル基、ここで、1の開放原子価(open valence)はアリール環の上で、1(の開放原子価)はアルキル部の上である、を含む。
【0029】
“有効量”は化合物の量を意味し、大体の所望の効果(例えば、処置される疾患のための臨床的に許容可能な基準(clinically acceptable standards)に従う、細胞増殖の比率及び/又は細胞の生存率における変化)をもたらす所望の投与計画(dosage regimen)の一部として投与されるときの化合物の量である。
【0030】
本願明細書で使用される用語「ハロアルキル(haloalkyl)」は、1から全水素の任意のところがハライドで置き換えられたアルキル基を意味する。「パーハロアルキル(perhaloalkyl)」は、全ての水素がハライドで置き換えられた[アルキル基]である。
【0031】
本願明細書で使用される用語「ヘテロ原子」は、炭素又は水素以外の任意の元素の原子を意味する。ヘテロ原子の例は、ホウ素、窒素、酸素、リン、硫黄及びセレニウムを含む。
【0032】
用語「ヘテロ環」若しくは「ヘテロ環基」は、環状構造が1から4のヘテロ原子を含む3から10員環構造(幾つかの例では、3から7員環)を意味する。ヘテロ環は、多環(polycycles)でもあり得る。ヘテロ環基は、例えば、チオフェン、チアントレン、フラン、ピラン、イソベンゾフラン、クロメン、キサンテン、フェノキサチイン、ピロール、イミダゾール、ピラゾール、イソチアゾール、イソキサゾール(ないしイソオキサゾール)、ピリジン、ピラジン、ピリミジン、ピリダジン、インドリジン、イソインドール、インドール、インダゾール、プリン、キノリジン、イソキノリン、キノリン、フタラジン、ナフチリジン、キノキサリン、キナゾリン、シンノリン、プテリジン、カルバゾール、カルボリン、フェナントリジン、アクリジン、ピリミジン、フェナントロリン、フェナジン、フェナルサジン、フェノチアジン、フラザン、フェノキサジン、ピロリジン、オキソラン、チオラン、オキサゾール、ピペリジン、ピペラジン、モルフォリン、ラクトン、アゼチジノン及びピロリジノンのようなラクタム、スルタム、スルトンおよび同様のものを含む。ヘテロ環は、1つ以上の環状位置(ring positions)を上記置換基、例えば、ハロゲン、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分(moieties)、‐CF3、‐CN又は同様のもので置換することができる。
【0033】
本発明の化合物との関係における用語「分離された」(isolated)は、化合物が細胞若しくは生命体内に存在せず、化合物が典型的には自然界においてそれに付いているいくつかのまたは、全ての化合物から引き離されたことを意味する。
【0034】
用語「ルイス酸」は、電子対の受容体として作用することができる任意の物質を意味する。
【0035】
炭素の数が他に特定されていなければ、本願明細書で使用される「低級アルキル」は、上記で定義したアルキル基を意味するが、その主要骨格の構造において、1から10の炭素を有する、いくつかの実施態様では、1から6の炭素原子を有するアルキル基を意味する。同様に、「低級アルケニル」及び「低級アルキニル」は、同様の鎖の長さを有する。あるアルキル基は、低級アルキルである。いくつかの実施態様では、アルキルとして本願明細書でデザインされた置換基は、低級アルキルである。
【0036】
本願明細書で使用されるように、用語「ニトロ」は、−NO2を意味し、用語「ハロゲン」は、‐F、‐Cl、‐Br若しくは‐Iを意味し、用語「スルフヒドリル」は、‐SHを意味し、用語「ヒドロキシル」は、‐OHを意味し、そして、用語「スルホニル」は、‐SO2‐を意味する。
【0037】
用語「オキソ」は、カルボニル酸素(=0)を意味する。
【0038】
用語「ポリシクリル」若しくは「多環基」は、2以上の炭素が2の隣接する環、例えば、その環は「縮合環」である、に共通する2つ以上の環(例えば、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/またはヘテロ環)を意味する。隣接していない原子を通して連結した環を、「架橋(bridged)」環と名付けた。多環の環の夫々は、上記のような置換基、例えば、ハロゲン、アルキル、アラルキル、アルケニル、アルキニル、シクロアルキル、ヒドロキシル、アミノ、ニトロ、スルフヒドリル、イミノ、アミド、スルホナート、ホスフィナート、カルボニル、カルボキシル、シリル、エーテル、アルキルチオ、スルホニル、ケトン、アルデヒド、エステル、ヘテロ環、芳香族若しくはヘテロ芳香族部分、‐CF3、−CN、又は同様のもので置換することができる。
【0039】
本発明の化合物との関係における用語「エピメリカルに純粋(epimerically pure)」は、化合物が、R3が結合した不斉中心の立体配置が反転した化合物の立体異性体を実質的に含まない(substantially free of)ことを意味する。例えば、以下の式:
(ここで、R1、R2、R3、R4、R7、R7´、R8及びR9は下記に定義される)
によって表されるエピメリカルに純粋な化合物は、以下の式:
(ここで、R1、R2、R3、R4、R7、R7´、R8及びR9は下記に定義される)
によって表される化合物を実質的に含まない。すなわち、エピメリカルに純粋な化合物は、質量で約20%未満、質量で約15%未満、質量で約10%未満、質量で約5%未満、若しくは、質量で約3%未満の、その化合物に関するR3が結合した不斉中心の立体配置が反転した立体異性体化合物を含む。
【0040】
本願明細書で使用される表現「保護基」は、望まない化学的な変換から潜在的な反応性の官能基を保護する一時的な置換基を意味する。そのような保護基の例は、カルボン酸のエステル、アルコールのシリルエーテル、そして、アルデヒド及びケトンのアセタール及びケタール夫々を含む。保護基化学の分野は、概説されている(Greene, T. W.; Wuts, P.G.M., Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991)。いくつかのケースでは、保護化された官能基及び保護基は、一緒になって、1部分ないし部位を意味する。例えば、下に示した断片ないしフラグメントは、しばしば、ベンジルカルボナートとして称される、すなわち、保護された(下線が引かれた)O(酸素)は、カルボナートの一部を構成する。
【0041】
同様に、保護化されたN(窒素)がカルバメートの一部を構成している下記に示した断片ないしフラグメントは、ベンジルカルバメートとして称される。
【0042】
本願明細書で使用される用語「糖」は、1以上のピラノース若しくはフラノース環を含有する天然の若しくは非天然の単糖、二糖若しくは多糖を意味する。糖は、エーテル結合若しくはアルキルの架橋(alkyl linkage)を介して本発明のステロイド系アルカロイドに共有結合的に結合することができる。ある実施態様では、糖の部分ないし部位は、サッカライドリング(saccharide ring)のアノマー中心において、本発明のステロイド系アルカロイドに共有結合的に結合することができる。糖は、リブロース、アラビノース、キシロース、リキソース、アロース、アルトロース、グルコース、マンノース、グロース、イドース、ガラクトース、タロース、グルコース及びトレハロースを含むことができるが、限定されない。
【0043】
本願明細書で使用される用語「スルホンアミド(sulfonamido)」ないし「スルホンアミド(sulfonamide)」は、以下の式:
ここで、R50は、上記で定義されている、のいずれかを有する部分を含む。
【0044】
用語「トリフリル」、「トシル」、「メシル」及び「ノナフリル」は、トリフルオロメタンスルホニル、p‐トルエンスルホニル、メタンスルホニル及びノナフルオロブタンスルホニル基を夫々意味する。用語「トリフレート」、「トシレート」、「メシレート」及び「ノナフレート」は、トリフルオロメタンスルホン酸エステル、p‐トルエンスルホン酸エステル、メタンスルホン酸エステル、ノナフルオロブタンスルホン酸エステル官能基、及びその基を含む分子を夫々意味する。
【0045】
用語「チオキソ」は、カルボニル硫黄(carbonyl sulfur, =S)を意味する。
【0046】
「置換」若しくは、「置換された」とは、そのような置換が、置換された原子及び置換基の許容原子価(permitted valence)に従う、また、置換は安定した、例えば、再編成、環化、脱離などによって自発的に変形を起こさない化合物になるという暗黙の条件(implicit proviso)を含むと理解されるだろう。
【0047】
本発明のある化合物は、幾何学的な、若しくは立体異性の形状で存在することができる。本発明は、シス‐及びトランス‐異性体、R‐及びS‐鏡像異性体、ジアステレオマー、(D)‐異性体、(L)‐異性体、そのラセミ混合物及びその他の混合物を含む、全ての化合物が発明の範囲内に入るものと考えられる。さらに、不斉炭素原子は、アルキル基のような置換基に存在できる。全てのそのような異性体及びその混合物は、本発明に含まれることが意図される。
【0048】
上で述べたように、当該化合物のある実施態様は、アミノ、アルキルアミノのような塩基性の官能基を含むことができ、したがって、薬学的に許容可能な酸と、薬学的に許容可能な塩を形成することができる。この点において、用語「薬学的に許容可能な塩」は、本発明の化合物の比較的毒性でない、本発明の化合物の無機及び有機酸の添加塩を意味する。これらの塩は、投与媒体のインサイチュ(in situ)において若しくは剤形製造工程内において、また、個々に、精製されたフリーベースフォーム(free base form)の発明の化合物と、適切な有機若しくは無機酸とを反応し、後の精製の間にそのように形成された塩を分離することによって調製することができる。代表的な塩は、臭化水素酸塩、塩酸塩、硫酸塩、硫酸水素塩(bisulfate)、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート(tosylate)、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチレート(napthylate)、メシル酸塩、グルコヘプトン酸塩、ラクトビオン酸及びラウリルスルホン酸塩(laurylsulphonate salts)、そして同様のものを含む。(例えば、Berge, et al.”Pharmaceutical Salts“ J. Pharm. Sci. (1977) 66:1-19参照)。
【0049】
本発明の化合物の薬学的に許容可能な塩は、例えば、無毒性の有機若しくは無機酸からなる、化合物の慣用的な無毒性の塩若しくは4級アンモニウム塩を含む。例えば、そのような慣用的な無毒性の塩は、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸、硝酸及び同様のもののような無機酸からなるそれらを含み、そして、塩は、酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パルミチン酸、マレイン酸、ヒドロキシマレイン酸(hydroxymaleic)、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸(salicyclic)、スルファニル酸(sulfanilic)、2‐アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、イソチオニック(isothionic)および同様のもののような有機酸から調製された塩を含む。
【0050】
他のケースでは、本発明の化合物は、1以上の酸性の官能基を含むことができ、そして、従って、薬学的に許容可能な塩基と薬学的に許容可能な塩を作ることができる。これらの例における用語「薬学的に許容可能な塩」は、比較的に無毒性の無機及び有機塩基を添加した本発明の化合物の塩を意味する。これらの塩は、同様に、インサイチュ(in situ)における投与媒体内若しくは剤形製造工程内で調製することができ、若しくは、薬学的に許容可能な金属カチオンの水酸化物、炭酸化物若しくは炭酸水素化物と、アンモニア若しくは薬学的に許容可能な1級、2級、3級若しくは4級の有機アミンとのように、遊離酸形状にある精製した化合物と適した塩基とを個々に反応することによって調製することができる。代表的なアルカリ(alkaliないしalkaline)土類塩は、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム及びアルミニウム塩、そして、同様のものを含む。塩基添加塩の形成に有用な代表的な有機アミンは、エチルアミン、ジエチルアミン、エチレンジアミン、エタノールアミン、ジエタノールアミン、ピペラジン、及び同様のものを含む。(例えば、Berge, et al., supra参照)
【0051】
ステロイド系アルカロイド化合物の合成
上記環拡大ステロイド系アルカロイド誘導体は、自然発生のステロイド系アルカロイド若しくはその合成類似体から直接調製できる。ある例では、ステロイド系アルカロイド出発物質は、シクロパミン若しくはジャービン(jervine)があり得る。これらのステロイド系アルカロイドは、商業的に購入でき若しくはバケイソウ(Veratrum Californicum)から抽出できる。手短には、本発明の方法は、適切な出発ステロイド系アルカロイド誘導体のシクロプロパン化、続いて、シクロプロピル誘導体の環拡大再編成のステップを含む。いくつかの例では、分子上に存在する反応性官能基をシクロプロパン化の前に適切に保護すること又は他に変換することが望ましいだろう。例えば、R1に存在するアルコール及び融合したフラノ‐ピペリジン環上に存在する2級の窒素は、両方ともシクロプロパン化の前に保護化できる。ある実施態様では、効率的にアルカロイドに加え、そしてアルカロイドから取り除かれ、合成プロセスにおける中間体の取り扱い性質を改善して産出し、形成された合成中間体の効率的な精製を可能にする保護基が望ましいだろう。
【0052】
酸素保護基の例は、ギ酸、酢酸、クロロ酢酸、ジクロロ酢酸、トリクロロ酢酸、ピバロアート(pivaloate)、ベンゾアート、アルキルカルボナート、アルケニルカルボナート、アリールカルボナート、アラルキルカルボナート(例えば、ベンジルカルボナート)、2,2,2‐トリクロロエチルカルボナート、アルコキシメチルエーテル、アラルコキシメチルエーテル、アルキルチオメチルエーテル、アラルキルチオエーテル、アリールチオエーテル、トリアルキルシリルエーテル、アルキルアリールシリルエーテル、ベンジルエーテル、アリールメチルエーテル及びアリルエーテルを含むが、これらに限定されない。
【0053】
窒素保護基の例は、ホルミル、クロロアセチル、トリクロロアセチル、トリフルオロアセチル、フェニルアセチル、ベンゾイル、ベンズアミド、アルキルカルバメート、アラルキルカルバメート(例えば、ベンジルカルバメート)、アリールカルバメート、アリル、アラルキル、アルコキシメチル、アラルコキシメチル、N‐2‐シアノエチル、ジアリールホスフィンアミド、ジアルキルホスフィンアミダート、ジアリールホスフィンアミダート及びトリアルキルシリルを含むが、これらに限定されない。
【0054】
本発明の方法に使用できる更なる保護基は、Green, T. W.; Wuts, P. G., Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. 1999.に開示されている。
【0055】
種々のシクロプロパネート剤が、ステロイド系アルカロイドをシクロプロパン化することに使用できる。カルベノイドと称される1,1‐ハロアルキル金属複合体及び反応種は、通常、オレフィンをシクロプロパン化することに使用される。これらの試薬は、ジヨードアルカン若しくはジアゾアルカンと、Et2Zn、iBu3Al、サマリウム、銅、ロジウム若しくはパラジウムのような金属若しくは有機金属種を使用して典型的に作られる。ある実施態様では、Et2Zn及びジヨードメタンが、1,1‐ハロアルキル金属種を生成することに使用される。
【0056】
1,1‐ハロアルキル亜鉛複合体の反応性及び取扱いの容易さは、酸のような、ある種の試薬の添加によって変更できる。1,1‐ハロアルキル亜鉛種への酸の添加は、アルキル亜鉛混合塩を生成すると信じられている。下記の例では、ビアリールリン酸が、推定上のハロアルキル亜鉛リン酸シクロプロパネート剤を生成するために、ジヨードメタン及びジエチル亜鉛と組み合わされる。種々のリン酸が、推定上のハロアルキル亜鉛リン酸を生成するために使用できる。
【0057】
カルボニルに結合したオレフィンと反応して、CH2若しくはCH‐アルキル若しくはCH‐アリール基を加えるために硫黄イリドを利用する、そして、ジアゾメタン及びエチルジアゾアセテートのようなジアゾアルキル及びα‐ジアゾ‐カルボニル化合物の金属触媒分解を利用する方法のように、他の知られたシクロプロプロパン化方法も使用できる、すなわち、これらの方法は、アルキル、アリール、アルコキシカルボニル(‐COOR)若しくはアシル置換基を有するシクロプロパンを迅速に提供する。更なるシクロプロパネート剤は、Masalov, et al., Organic Letters (2004) Vol. 6, pp. 2365-2368 及び Hansen, et al., Chem. Comm. (2006) 4838- 4840.に開示されている。
【0058】
シクロプロピル環は、置換できるし、また置換しなくてもよい。シクロプロピル環が置換されたケースでは、シクロプロパンのメチレンに付した基は、再編成及び環拡大の後、D環の上に設置される。
【0059】
シクロプロパン化反応は、非プロトン性の溶媒の中で行うことができる。適切な溶媒は、ジエチルエーテル、1,2‐ジメトキシエタン、ジグリム、t‐ブチルメチルエーテル、テトラヒドロフラン及び同様のもののようなエーテル、クロロホルム、ジクロロメタン、ジクロロエタン及び同様のもののようなハロゲン化溶媒、ベンゼン、キシレン、トルエン、ヘキサン、ペンタン及び同様のもののような脂肪族若しくは芳香族の炭化水素溶媒、酢酸エチル、アセトン及び2‐ブタノンのようなエステル及びケトン、アセトニトリル、ジメチルスルフォキシド、ジメチルホルムアミド及び同様のもののような非プロトン性極性溶媒、又は2以上の溶媒の組み合わせを含む。ある実施態様では、ジクロロメタンは、ジアルキル亜鉛及びジヨードメタンが使用された場合に、シクロプロパン化のために使用された溶媒である。
【0060】
下記の実施例では、シクロプロパネート剤を含有する溶液を、最初にジエチル亜鉛の溶液にリン酸の溶液を加え、続いて、その反応溶液へのジヨードメタンの添加によって調製する。次に、シクロプロパン化の基質を、この溶液に加える。あるいは、シクロプロパネート剤は、試薬の添加の順番を変えることによってシクロプロパン化の基質の存在中に調製できる。ある実施態様では、ジアルキル亜鉛の溶液に最初にリン酸を加え、続いて、シクロプロパン化の基質の添加、そして最後にジハロアルカンを加えることによってシクロプロパン化反応を行う。この方法を使用して、シクロプロパネート剤は、管理条件下で生成され、そして、直ちにシクロプロパン化の基質と反応する。
【0061】
シクロプロパン化ステロイド系アルカロイドコア(core)の合成に続き、当該技術分野内で知られた種々の官能基付与反応を使用して、その化合物を誘導体化することができる。代表的な例は、アルケニルハライド又はアリールハライドのカップリング反応、酸化、還元、求核剤による還元、求電子剤による還元、ペリ環状反応、ラジカル反応、保護基の設置、保護基の除去及び同様のものを含む。
【0062】
ルイス若しくはブレンステッド酸の存在中に、シクロプロピル類似体は、1の炭素によって拡大しているD環を有するステロイド系アルカロイド類似体を生成するため、再編成及び環拡大を起こす。
【0063】
シクロプロパン化及び環拡大は、2ステップ1反応容器のプロセス若しくは2ステップ2反応容器のプロセスで行うことができる。シクロプロパン化及び環拡大を同一の反応容器で行う場合、環拡大再編成を開始するために使用した酸は、シクロプロパン化反応の完了後に加えられる。ある条件下では、ステロイド系アルカロイドのシクロプロパン化の過程の中で生成された亜鉛の塩は、それ自身が、環拡大再編成を触媒するルイス酸として作用できる。シクロプロパン化の後に生成された亜鉛の塩の反応性は、更なる活性のルイス酸を生成する酸の添加によって変更できる。
【0064】
下記の実施例のように、メタンスルホン酸を、シクロプロパン化の完了後、シクロプロパン化の反応容器に加える。更なる適切な酸の例は、亜鉛の塩、炭素化合物、マグネシウムの塩、チタンの塩、インジウムの塩、アルミニウムの塩、スズの塩、ランタンの塩、トリフルオロメタンスルホン酸、ジアリールオキシリン酸、酢酸、HClを含むが、これらに限定されない。本発明のある実施態様では、使用されるルイス酸は、亜鉛の塩若しくはBF3である。
【0065】
これらの環拡大類似体は、当該技術分野内で知られた種々の官能基付与反応を使用して、さらに官能基化できる。代表的な例は、アルケニルハライド若しくはアリールハライドのパラジウムカップリング反応、酸化、還元、求核剤による還元、求電子剤による還元、ペリ環状反応、ラジカル反応、保護基の設置、保護基の除去及び同様のものを含む。
医薬組成物
【0066】
1以上の薬学的に許容可能な担体(添加物)及び/または希釈液を用いて、本願明細書に開示の化合物は、投与に最適な組成物に処方される。本発明の医薬組成物は、特に固形状若しくは液状の投薬に処方することができ、以下に適用されたそれらを含む:(1)経口投与、例えば、水薬(水性または 非水性溶液 若しくは 懸濁物)、例えば、頬側、舌下、全身吸収を標的とした錠剤、カプセル、ボーラス(boluses)、散剤、顆粒剤、舌用のペースト剤、(2)非経口投与、例えば、皮下、筋肉、静脈、硬膜外注射によって、例えば、無菌溶液または懸濁液、若しくは持続性放出処方液として、(3)外的適用、例えば、皮膚用のクリーム、軟膏、放出制御パッチ若しくはスプレーとして、(4)膣用若しくは直腸用、例えば、ペッサリー、クリーム若しくは気泡として、(5)舌下用(6)眼用(7)経皮用(8)肺用(pulmonarily)、(9)経鼻用。
【0067】
本発明の医薬組成物に含めることができる適切な水性または 非水性担体の例は、水、エタノール、ポリオール(polyols)(グリセロール、プロピレングリコール、ポリエチレングリコール及び同様のもののような)及びその適切な混合溶液、オリーブ油のような野菜油、そして、オレイン酸エチルのような注射可能な有機エステルを含む。適した流動性は、例えば、レシチンのようなコーティング物質の使用によって、分散の場合における要求された粒子サイズの調節によって、及び界面活性剤の使用によって調節できる。
【0068】
これらの組成物は、保存料のような補助剤、湿潤剤、乳化剤、分散剤、潤滑剤及び/または抗酸剤を含有することもできる。本願明細書に開示の化合物上の微生物の活動の防止は、例えば、パラベン、クロロブタノール、ソルビン酸フェノール及び同様のもののような種々の抗菌及び抗真菌性剤の含有によって保障できる。糖、塩化ナトリウム及び同様のもののような等張剤(isotonic agents)を化合物に含むことも望ましい。加えて、注射可能な医薬形(pharmaceutical form)の遷延吸収を、モノステアリン酸アルミニウム及びゼラチンのような吸収を遅らせる薬剤の含有によってもたらすことができる。
【0069】
これらの処方薬若しくは組成物の調製方法は、(本発明の)化合物と、担体及び任意の1以上の付属成分とを結合物(association)にするステップを含む。一般的に、処方薬は、一様にまた本質的に、液状の担体若しくは細かく分割した固形の担体、または両方で(本発明の)化合物を結合物(association)にすること、そして、次に必要であれば製造物を成形することによって調製される。
【0070】
本願明細書に開示の化合物をヒトや動物に医薬品として投与する場合、それ自身で、または、例えば、薬学的に許容可能な担体との組み合わせにおいて約0.1から99%、若しくは約10から50%、若しくは約10から40%、若しくは約10から30%、若しくは約10から20%、若しくは約10から15%の活性成分を含む医薬組成物として与えることができる。
【0071】
(本発明の)医薬組成物における活性成分の実際の投与量レベルは、患者に毒性を与えずに、特定の患者の所望の治療反応を達成する効果がある活性成分の量、組成及び投与モードを得るように、変えることができる。
【0072】
選択された投与量レベルは、使用する(本発明の)特定の化合物の活性、そのエステル、塩であるか若しくはアミドであるか、投与経路、投与時間、使用されている特定の化合物の排泄若しくは代謝の割合、吸収割合及び範囲、処置の持続時間、使用する特定の化合物と組みあわせて使用する他の薬剤、化合物及びまたは物質、年齢、性別、体重、体調、通常健康状態、処置を受ける患者の前の医療履歴及び医療分野で良く知られた同様の要素を含む種々の要素に依存するであろう。
【0073】
一般的に、本願明細書に開示の化合物の適切な一日投与量は、治療効果を生じる効果がある最低の量の化合物の量であろう。そのような効果量は、一般的に上記要素に依存する。一般的に、患者への(本発明の)化合物の経口、静脈内、皮下投与量は、指示した効果のために使用した場合、1日に、体重1キログラムあたり、約0.0001から約200mg、若しくは約0.001から約100mg、若しくは約0.01から約100mg、若しくは約0.1から約100mgまで、若しくは約1から約50mgまでの範囲であろう。
【0074】
化合物は、毎日、一日おき、週に3回、週に2回、1週間毎、又は2週間毎に投与できる。投薬スケジュールは、“ドラッグホリデー(drug holiday)”を含むことができる、すなわち、薬物の2週間投与に1週間休み、若しくは3週間投与に1週間休み、若しくは4週間投与に1週間休みなどが可能であり、又は、ドラッグホリデー無しで連続した薬物の投与ができる。化合物は、経口的に、静脈内に、腹腔内に、局所的に、経皮的に、筋肉内に、皮下に、鼻腔内に、舌下に、又は、任意の他の経路によって投与できる。
【0075】
この処置を受けるものは、霊長類、特にヒト、及びウマ、ウシ、ブタ及びヒツジ、すなわち一般的な家畜及びペットを含む、必要がある任意の動物である。
処置の方法
【0076】
ヘッジホッグシグナルは、発生(development)の多くの段階、特に、左右対称性の形成において必須である。ヘッジホッグシグナルの欠損若しくは減少は、発生上の欠陥及び奇形の多発を導き、最も顕しいそれの1つは、単眼症である。
【0077】
多くの腫瘍および増殖性の状態が、ヘッジホッグ経路に依存していることが示されてきた。そのような細胞の成長及び生存は、本願明細書に開示の化合物で処置することによって影響を与えることができる。最近、散発性の基底細胞腫(Xie et al. (1998) Nature 391 : 90-2)及び中枢神経系の原始的な神経外胚葉性腫瘍(Reifenberger et al. (1998) Cancer Res 58: 1798-803)において、活性化ヘッジホッグ経路変異が、発生することが報告されている。ヘッジホッグ経路の非制御の活性化は、膵臓癌、食道癌、胃癌を含む胃腸管(GI tract)癌(Berman et al. (2003) Nature 425: 846-51, Thayer et al. (2003) Nature 425: 851-56)、肺癌(Watkins et al. (2003) Nature 422: 313-317), 前立腺癌(Karhadkar et al. (2004) Nature 431 : 707-12, Sheng et al. (2004) Molecular Cancer 3: 29-42, Fan et al. (2004) Endocrinology 145: 3961-70)、乳癌(Kubo et al. (2004) Cancer Research 64: 6071-74, Lewis et al. (2004) Journal of Mammary Gland Biology and Neoplasia 2: 165-181)及び肝細胞癌(Sicklick et al. (2005) ASCO conference, Mohini et al. (2005) AACR conference)のような多くの癌タイプにおいて示されている。
【0078】
例えば、ヘッジホッグ経路の小分子阻害(small molecule inhibition)は、基底細胞癌(Williams, et al., 2003 PNAS 100: 4616-21)、髄芽腫(Berman et al., 2002 Science 297: 1559-61)、膵臓癌(Berman et al., 2003 Nature 425: 846-51)、消化器系の癌(Berman et al., 2003 Nature 425: 846-51, PCT公開公報 WO 05/013800)、食道癌(Berman et al., 2003 Nature 425: 846-51)、肺癌(Watkins et al., 2003. Nature 422: 313-7)、および前立腺癌(Karhadkar et al., 2004. Nature 431: 707-12)の成長を阻害することが示されている。
【0079】
加えて、例えば、乳癌(Kubo et al., 2004. Cancer Research 64: 6071-4)、肝細胞癌(Patil et al., 2005. 96th Annual AACR conference, 要約#2942, Sicklick et al., 2005. ASCO annual meeting, 要約 #9610)、造血器腫瘍(Watkins and Matsui, unpublished results)、基底[細胞]癌(Bale & Yu, 2001. Human Molec. Genet. 10:757-762, Xie et al., 1998 Nature 391: 90-92)、髄芽腫(Pietsch et al., 1997. Cancer Res. 57: 2085-88)、胃癌(Ma et al., 2005 Carcinogenesis May 19, 2005 (Epub))など、多くの癌タイプは、ヘッジホッグ経路の非制御活性化を有することが示されている。加えて、調査者は、ヘッジホッグ経路の小分子の抑制が乾癬の症候ないし症状を回復することが示されていることを見つけた(Tas, et al., Dermatology (2004) 209: 126-131)。実施例に示すように、本願明細書に開示する化合物は、ヘッジホッグ経路を調節することを示し、選択した化合物は腫瘍成長を阻害することを示した。従って、これらの化合物は、種々の癌のような様々な過剰増殖疾患を処置することに有用であると信じられる。
【0080】
本明細書に開示の方法を用いて処置できる過剰増殖疾患は、肺がん(小細胞肺癌及び非小細胞肺癌を含む)、肺系(pulmonary system)の他の癌、髄芽腫及び他の脳癌、膵臓癌、基底細胞癌、乳癌、前立腺癌及び他の尿生殖器癌、胃腸間質性腫瘍(GIST)及び消化管の他の癌、結腸癌、結腸直腸癌、卵巣癌、造血系(多発性骨髄腫、急性リンパ性白血病、急性骨髄性白血病、慢性骨髄性白血病、慢性リンパ性白血病、ホジキンリンパ腫、及び非ホジキンリンパ腫、並びに骨髄異形成症候群)の癌、真性多血症、ワルデンシュトレームマクログロブリン血症、重鎖病、軟部肉腫(線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、扁平上皮癌、基底細胞癌、メラノーマ及び他の皮膚癌など)、腺癌、汗腺癌、乳頭癌、乳頭状腺癌、スタデノカルチノーマ(stadenocarcinoma)、髄様癌、気管支原性肺癌、腎細胞癌、肝細胞癌、胆管癌、繊毛癌、セミノーマ、胚性癌腫、ウィルムス腫瘍、子宮頚癌、子宮癌、精巣癌、膀胱癌、及び他の尿生殖器癌、上皮癌、グリオーマ、アストロサイトーマ、髄芽腫、頭蓋咽頭腫、上衣腫、松果体腫、血管芽腫、聴神経腫瘍、乏突起細胞腫、髄膜腫、神経芽細胞腫、網膜芽細胞腫、子宮内膜癌、濾胞性リンパ腫、びまん性大細胞型B細胞リンパ腫、マントル細胞リンパ腫、肝細胞癌、甲状腺癌、胃癌、食道癌、頭頚部癌、小細胞癌、本態性血小板血症、原発性骨髄線維症、好酸球増加症候群、全身性肥満細胞症、家族性過好酸球増加症(familiar hypereosinophilia)、慢性好酸球性白血病、甲状腺癌、神経内分泌癌、及びカルチノイド腫瘍を含む。追加的な疾患は、ゴーリン症候群及び乾癬を含む。
【0081】
この処置を受ける被験者ないし患者は、必要とされる任意の動物であり、霊長類、特に、ヒト、他の動物(ウマ、ウシ、ブタ及びヒツジなど);並びに家禽及び一般的なペットを含む。
【0082】
本明細書に開示のヘッジホッグ阻害剤は、他の癌処置と組合せができる。例えば、それらは、外科処置;放射線;バイオセラピー(biotherapeutics)(インターフェロン、サイトカイン、例えば、インターフェロンα、インターフェロンγ、及び腫瘍壊死因子、造血成長因子、単クローン性血清療法、ワクチン及び免疫賦活薬);抗体(例えば、アバスチン、エルビタックス、リツキサン、及びベキサール);内分泌療法(ペプチドホルモン、副腎皮質ステロイド、エストロゲン、アンドロゲン及びアロマターゼ阻害剤を含む);抗エストロゲン薬(例えば、タモキシフェン、ラロキシフェン、及びメゲストロール);LHRH作用薬(例えば、ゴスクリクリン(goscrclin)及びリュープロリド酢酸塩);抗アンドロゲン薬(例えば、フルタミド及びビカルタミド);遺伝子治療;骨髄移植;光力学治療(例えば、ベルトポルフィン(vertoporfin)(BPD−MA)、フタロシアニン、フォトセンシタイザーPc4、及びデメトキシ‐ハイポクレリン(Demethoxy-hypocrellin)A(2BA−2−DMHA));並びに化学療法と組合せることができる。
【0083】
化学療法の例は、ゲムシタビン、メトトレキサート、タキソール、メルカプトプリン、チオグアニン、ヒドロキシウレア、シタラビン、シクロホスファミド、イホスファミド、ニトロソ尿素、シスプラチン、カルボプラチン、ミトマイシン、ダカルバジン、プロカルビジン(procarbizine)、エトポシド、プレドニゾロン、デキサメタゾン、シタルビン、キャンパセシン(campathecins)、ブレオマイシン、ドキソルビシン、イダルビシン、ダウノルビシン、ダクチノマイシン、プリカマイシン、ミトキサントロン、アスパラギナーゼ、ビンブラスチン、及びビノレルビンを含む。付加的な薬剤(additional agents)は、ナイトロジェンマスタード(例えば、シクロホスファミド、イホスファミド、トロホスファミド、クロラムブシル、エストラムスチン、及びメルファラン)、ニトロソ尿素(例えば、カルムスチン(BCNU)及びロムスチン(CCNU))、アルキルスルホナート(alkylsulphonates)(例えば、ブスルファン及びトレオスルファン)、トリアゼン(例えば、ダカルバジン及びテモゾロミド)、化合物を含有する白金(例えば、シスプラチン、カルボプラチン、及びオキサリプラチン)、ビンカアルカロイド(例えば、ビンクリスチン、ビンブラスチン、ビンデシン、及びビノレルビン)、タキソイド(例えば、パクリタキセル及びドセタキソール)、エピポドフィリン(epipodophyllins)(例えば、エトポシド、テニポシド、トポテカン、9−アミノカンプトテシン、カンプトイリノテカン、クリスナトール(Crisnatol)、マイトマイシンC、及びマイトマイシンC)、抗代謝物(anti-metabolites)、DHFR阻害剤(例えば、メトトレキサート及びトリメトレキサート)、IMPデヒドロゲナーゼ阻害剤(例えば、ミコフェノール酸、チアゾフリン、リバビリン、及びEICAR)、リボヌクレオチド還元酵素阻害剤(例えば、ヒドロキシウレア及びデフェロキサミン)、ウラシル類似体(例えば、フルオロウラシル、フロクスウリジン、ドキシフルリジン、ラチトレキサド(Ratitrexed)、及びカペシタビン)、シトシン類似体(例えば、シタラビン(ara C)、シトシンアラビノシド、及びフルダラビン)、プリン類似体(例えば、メルカプトプリン及びチオグアニン)、ビタミンD3類似体(例えば、EB1089、CB1093、及びKH1060)、イソプレニル化阻害剤(例えば、ロバスタチン)、ドーパミン系神経毒(dopaminergic neurotoxins)(例えば、1−メチル−4−フェニルピリジニウムイオン)、細胞周期阻害剤(例えば、スタウロスポリン)、アクチノマイシン(例えば、アクチノマイシンD及びダクチノマイシン)、ブレオマイシン(例えば、ブレオマイシンA2、ブレオマイシンB2、及びペプロマイシン)、アントラサイクリン(例えば、ダウノルビシン、ドキソルビシン(アドリアマイシン)、イダルビシン、エピルビシン、ピラルビシン、ゾルビシン、及びミトキサントロン)、MDR阻害剤(例えば、べラパミル)、Ca2+ATPase阻害剤(例えば、タプシガルジン)、イマチニブ、サリドマイド、レナリドマイド、エルロチニブ、ゲフィチニブ、ソラフェニブ、及びスニチニブ、並びにボルテゾミブを含むプロテアソーム阻害剤を含む。
【0084】
本明細書に開示のヘッジホッグ阻害剤が他の処置(例えば、付加的な治療又は放射線若しくは外科と)との組合せで投与されるとき、各薬剤又は治療の投与量ないし用量は、ほとんどの場合、単独の薬剤療法における対応する投与量ないし用量より低い。また、一般的に、本明細書に記載のヘッジホッグ阻害剤及び第二の治療剤は同一の医薬組成物で投与されるべきではなく、異なる物理的及び化学的特徴のために、異なる経路で投与されてよい。例えば、ある化合物は経口投与が可能であり、一方、第二の治療(剤)は静脈内投与される。同一の医薬組成物において可能である、投与の方法及び投与の妥当性の決定は、熟練した臨床医の知識の範囲内でよい。初回投与は、当該技術分野で知られている確立されたプロトコールにしたがって為されることができ、次いで、観察される効果に基づいて、投与量、投与の方法及び投与の時間は、熟練した臨床医によって変更できる。
【0085】
ヘッジホッグ阻害剤及び第二の治療剤及び/又は放射線は、増殖疾患の性質、患者の状態、及び第二の治療剤及び又は放射線の実際の選択に依存して、同時的に(例えば、同時に、本質的に同時に若しくは同一の処置プロトコール内で)又は連続して(つまり、順次に、任意の時間間隔を空けて)投与されてよい。
【0086】
ヘッジホッグ阻害剤及び第二の治療剤及び/又は放射線が同時に又は本質的に同時に投与されないのであれば、投与の最適な順番は異なった状況に応じて異なってよい。よって、ある状況では、ヘッジホッグ阻害剤は最初に投与され、次いで、第二の治療剤及び/又は放射線が投与され;別の状況では、第二の治療剤及び/又は放射線は最初に投与され、次いで、ヘッジホッグ阻害剤が投与される。この代替の投与は、単一の処置プロトコールの間に繰り返されてよい。投与の順番の決定及び処置プロトコールの間の各治療剤の投与の反復回数は、処置される疾患及び患者の状況の評価後に、熟練した臨床医の知識の範囲内でよい。例えば、第二の治療剤及び/又は放射線は、特に、細胞傷害性薬物であれば、最初に投与されてよく、次いで、ヘッジホッグ阻害剤の投与による処置が続き、そこで有用だと判断されれば、第二の治療剤及び/又は放射線によって処置プロトコールが終了するまで続けられる。
実施例
【0087】
現に、一般的に開示する発明は、単に、本発明のある視点及び実施態様を説明する目的のために含まれ、そして発明を限定することを意図しない、以下の実施例を参照することによってさらに容易に理解されるであろう。
実施例1
ステップA
【0088】
再結晶化されたシクロパミン2(14.1g, 34.0mmol, 1当量)を、無水DCM(70mL)および無水MeOH(29mL)に溶解する。澄んだ溶液を冷却し、そしてトリエチルアミン(10.4g, 102.7mmol, 3当量)、続いて、クロロギ酸ベンジル(benzyl chloroformate, 6.20g, 36.3mmol, 1.1当量)を加える。添加が完了した後、溶液をアイスバス内で30分間攪拌する。3部(portions)のクロロギ酸ベンジル(3x0.35g, 3.46mmol, 0.03当量)を3時間かけて加える。温度を20℃未満に維持しつつ、反応を水(71mL)で徐々に抑える。層が安定して分離する前に、混合物を15分間攪拌する。有機層を硫酸ナトリウム上で乾燥し、濾過する。合併した濾液を、無水ピリジン(30mL)で緩衝し、濃縮し、そして、更なる無水ピリジン(43mL)で溶媒を交換し、そして濃縮する。
【0089】
化合物のピリジン(43mL)溶液を、更なる無水ピリジン(85mL)でさらに希釈する。トリメチルアセチルクロリド(8.3g, 68.7mmol, 2当量)を反応混合溶液に徐々に加え、反応溶液を45℃まで加熱する。反応溶液を45℃で30分間攪拌する。反応溶液を冷却し、[反応を]無水MeOH(4.5mL)の添加によって抑える。抑制した反応混合溶液を室温で40分間攪拌し、そして次にトルエン(97mL)で希釈し、そして水(35mL)及び10wt%の炭酸ナトリウム水溶液(100mL)で順番に処置する。勢い良く攪拌した後、層を分離し、そして有機層を水で2回(2x100mL)洗浄し、硫酸ナトリウム上で乾燥し、そして濾過する。濾過ケーキをトルエン(49mL)でリンスし、そして破棄する。合併した濾液を濃縮し、そして溶媒を濃縮でトルエン(145mL)に交換し、そしてさらに乾燥状態まで濃縮する。生成物をトルエン及びヘプタンから再結晶化する。結晶化産物を吸引濾過によって分離し、冷たいヘプタンで洗浄し、そして恒量まで乾燥して、15.1gの所望の生成物を得る。
ステップB
【0090】
ビス(2,6‐ジメチルフェニル)リン酸塩(10.65g, 34.8mmol, 3.1当量)を無水DCM(42mL)から濃縮によって乾燥し、そして窒素雰囲気下で保持する。次に、リン酸塩を無水DCM(110mL)に再溶解する。別のフラスコに、純粋なジエチル亜鉛(4.17g, 33.8mmol, 3.0当量)の無水DCM溶液(35 mL)を調製し、そして−25℃まで冷却する。−10℃以下に温度を維持しつつ、リン酸塩溶液を1時間かけて徐々にジエチル亜鉛が入った容器に移す。澄んだエチル亜鉛リン酸塩溶液を0℃まで温め、そして15分間攪拌した。0〜5℃の間に反応温度を維持しつつ、ジヨードメタン(9.25g, 34.5mmoles, 3.0当量)を、エチル亜鉛リン酸塩溶液に徐々に加える。添加が完了した後、亜鉛カルベノイド溶液をさらに20分間攪拌する。
【0091】
別のフラスコに、化合物3(7.20g, 11.4mmol, 1当量)を無水DCM(36mL)に溶解し、反応フラスコに移す。添加が完了した後、アイスバスを取り除き、そして反応混合溶液を室温まで温める。6時間後、フラスコの内容物を−53℃まで冷却する。反応温度を−45℃未満に維持しつつ、メタンスルホン酸(3.38g, 35.2mmol, 3.1当量)の無水DCM溶液(3mL)を加える。10分後、反応温度を−40℃未満に維持しつつ、モルフォリン(20g, 230mmol, 20当量)を反応混合溶液に加える。反応溶液を、室温までオーバーナイトで温める。モルフォリンの塩を濾過によって取り除き、そして濾過ケーキをDCM(22mL)でリンスする。合併した濾液を2N塩酸水溶液(2x140mL)、5%炭酸水素ナトリウム水溶液(140mL)、5%炭酸水素ナトリウム水溶液(70mL)及び5%亜硫酸水素ナトリウム水溶液(70ml)、そして鹹水(144mL)で洗浄する。有機層を硫酸マグネシウム上で乾燥し、そして濾過する。乾燥状態にすることなく、DCM溶液を濃縮し、そして溶媒をメタノール(280mL)で交換する。懸濁物をアイスバスで冷却し、そして40分間攪拌する。固形物を濾過によって分離し、冷たいメタノールで2回(2x25mL)洗浄し、そして恒量まで乾燥して、5.94gの所望の生成物を得る。
ステップC
【0092】
丸底フラスコの中で、化合物4(11.67g, 18.1mmol, 1当量)及びウェットカーボン上(に担持した)20%水酸化パラジウム(2.40g, 1.71mmol, 0.09当量)を窒素雰囲気下に置き、そしてEtOAc(酢酸エチル, 115mL)及びトルエン(60mL)で希釈する。溶液を、窒素で(3X)減圧吸引/パージサイクル(evacuation/purge cycles)で脱気し、そしてその工程を水素で繰り返す。懸濁物を勢い良く室温で1.5時間攪拌する。水素雰囲気を、窒素で置き換える。エチレンジアミン(0.57g, 9.5mmol, 0.52当量)を反応に加え、そして結果として生じる混合溶液を20分間攪拌する。その溶液を窒素下で濾過し、そして濾過溶液を2%(wt/wt)エチレンジアミン水溶液(125mL)で、次に水(130mL)で洗浄し、そして次に、硫酸マグネシウム上で乾燥する。乾燥剤を濾過によって取り除き、そして濾過液を減圧下で乾燥状態まで濃縮する。残った固形物は、トルエン(2x55mL)で回転吸引機上で追跡(chased)し、そして結果として生じる物質を更に精製することなく次のステップで使用する。
【0093】
前のステップからの物質を無水DCM(26mL)に溶解する。反応温度を−10から−25℃の間に維持しつつ、結果として生じる澄んだ溶液を1MのDIBALのDCM(65mL, 65mmol, 3.6当量)溶液に加える。30分後、0℃以下に反応温度を維持して、反応をアセトン(13mL)で抑える。抑制した反応混合溶液を17分間攪拌した後、それを冷たい攪拌された20%(wt/wt)ロッシェル塩水溶液(200mL)が入ったフラスコに分画して(in portions)加える。結果として生じるゼラチン状の懸濁液を室温で15時間攪拌する。攪拌した後、きれいな層を分離し、そして水性の層をDCM(30mL)で逆抽出する。合併した有機層を水(60mL)で洗浄し、そして硫酸ナトリウム上で乾燥する。乾燥剤を濾過によって取り除き、そして廃棄する。濾過液を減圧下で濃縮し、そして溶媒をトルエンに交換する(225mLを分画して添加)。結果として生じる溶液を懸濁液(50mL)まで、さらに濃縮し、そしてヘプタン(115mL)で希釈する。結果として生じる混合溶液を均質に変化するまで加熱(92℃)する。溶液を12時間かけて徐々に15℃まで冷却し、そして次に、更なる16時間保持する。結晶化物を吸引濾過によって分離し、ヘプタン(2x75mL)で洗浄し、そして恒量まで乾燥し、7.70gの所望の生成物を得る。
【0094】
丸底フラスコを、ホモアリリックアルコール(homo-allylic alcohol, 7.50g, 17.6mmol, 1当量)、アルミニウムトリ‐tert‐ブトキシド(6.10g, 24.8mmol, 1.4当量)、無水トルエン(115mL)及び2‐ブタノン(90g, 1.24mol, 7当量)で順番にチャージ(charged)する。懸濁液を窒素雰囲気下で75℃まで16時間加熱する。次に、反応温度を49℃まで冷却する。20%(w/w)酒石酸カリウムナトリウム水溶液(226g)を攪拌した懸濁液に加える。懸濁液を室温で3.5時間攪拌する。層を分離する。有機層を20%ロッシェル塩水溶液(2x250mL)及び水(225mL)で洗浄し、次に硫酸ナトリウム上で乾燥し、そして濾過する。残留物をトルエン(30mL)でリンスし、そして廃棄する。合併した有機物を乾燥状態まで濃縮する。2−プロパノール(250 mL, 分画して添加)から44gの最終溶液質量までの濃縮によって、残りの反応溶媒を物質から取り除く。最終溶液質量を41gにする2‐プロパノールからヘプタン(275mL, 分画して添加)への溶媒の交換は、所望の生成物を完全に沈殿させる。懸濁液を更なるヘプタン(40mL)で希釈し、室温で1時間攪拌し、そして濾過する。生成物をn‐ヘプタン(17mL)で洗浄し、そして乾燥し、5.4gの所望の生成物を得る。
ステップD
【0095】
丸底フラスコを、出発物質(110mg, 0.26mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(106mg)でチャージする。固形物をピリジン(4mL)に懸濁する。懸濁液を窒素雰囲気下(1気圧)に置き、そして混合溶液をオーバーナイトで室温で攪拌する。その反応混合溶液を、セライト(登録商標)を介して濾過し、そして濾過液を真空中で濃縮する。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、93mgの所望の化合物を得る。([M+H] = 426.6m/z)。
実施例2
ステップA
【0096】
シクロパミン2(5.02g, 12.2mmol, 1.0当量)を無水ピリジン(25mL)に溶解する。DMAP(300mg, 2.44mmol, 0.2当量)及びトリエチルアミン(5.5mL, 39.1mmol, 3.2当量)、続いて、BtO‐Cbz(10.5g, 39.1mmol, 3.2当量)を加え、そして40℃で2時間加熱する。混合溶液を室温まで冷まし、30mLの水で処置し、均質溶液を得るために加熱し、そして室温まで冷却する。形成された白い沈殿物を濾過によって収集し、濾過ケーキを水で数回(3x50mL)洗浄し、そして空気中で乾燥し、6.75gの所望の生成物を得るためのトルエン/ヘプタン(1:9, 70mL)から結晶化された9.53gの未精製物質を得る。
ステップB
【0097】
反応温度を−8℃未満に維持しつつ、−20℃の5.0mlのジエチル亜鉛(572mg, 482μL, 4.63mmol, 3.00当量)DCM溶液に、ビス‐(2,6‐ジエチルフェニル)リン酸塩(1.42g, 4.63mmol, 3.00当量)DCM溶液(15mL)を加える。ビスカルボキシルベンジルシクロパミン(BisCBzcyclopamine, 1.05g, 1.54mmol, 1.0当量)のDCM溶液(10mL)を加える前に、溶液を15分間0℃で熟成し、純粋なジヨードメタン(1.24g, 374μL, 3.00当量)を加え、15分間0℃で熟成する。冷却バスを室温の水バス(water bath)によって置き換え、そして室温で4.5時間維持する。混合溶液をドライアイス‐アセトンバスで−76℃まで冷却し、そして反応温度を−74℃未満に維持しつつ、メタンスルホン酸DCM溶液(0.6mL 50% v/v溶液 4.63mmol, 3.0当量)で滴下処置する。混合溶液を15‐20分間熟成し、そして反応温度を−65℃未満に維持しつつ、モルフォリン(2.69g, 2.70mL, 20当量)の滴下で抑える。冷却バスを取り除き、反応混合溶液を16‐18時間攪拌し、白い沈殿物を濾過除去し、そして濾過液を2.0MのHCl(2x20mL)、飽和炭酸水素ナトリウム(2x20mL)、水(2x20mL)及び鹹水(20mL)で連続的に洗浄する。硫酸マグネシウム上で乾燥し、真空中で乾燥状態まで濃縮し、そして未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc 17:3→4:1)によって精製し、924mg(1.33mmol, 86%)の所望の生成物を得る。
ステップC
【0098】
化合物7(4.05g, 5.83mmol, 1当量)のEtOAc:トルエン溶液(2:1, 60mL)の溶液に、カーボン上(に担持した)20%水酸化パラジウム(823mg, 0.583mmol, 0.1当量)を加える。フラスコを、3回、吸引し、そして水素で満たす。混合溶液を水素雰囲気下で1時間攪拌する。純粋なエチレンジアミン(0.38mL)を加え、1時間攪拌し、そして触媒を濾過除去する。濾過ケーキを2回、EtOAc:トルエン(2:1, 12 mL)で洗浄する。合併した濾液を、2%エチレンジアミン水溶液(3x20mL)で洗浄し、硫酸ナトリウム上で乾燥し、そして真空中で濃縮し、白い結晶化固形物として2.46gを得る。
ステップD
【0099】
丸底フラスコを、ホモアリリックアルコール8(7.50g, 17.6mmol, 1当量)、アルミニウムトリ-tert-ブトキシド(6.10 g, 24.8 mmol, 1.4当量)、無水トルエン(115 mL)、及び2−ブタノン(90 g, 1.24 mol, 7当量)で連続的にチャージする。懸濁液を窒素雰囲気下で75℃まで16時間加熱する。次に、反応溶液を49℃まで冷却する。20%(w/w)酒石酸カリウムナトリウム(226g)水溶液を攪拌した懸濁液に加える。懸濁液を室温で3.5時間攪拌する。層を分離する。有機層を20%ロッシェル塩水溶液(2x250mL)及び水(225mL)で洗浄し、次に、硫酸ナトリウム上で乾燥し、そして濾過する。残留物をトルエン(30mL)でリンスし、そして廃棄する。合併した有機物を乾燥状態まで濃縮する。残りの反応溶媒を2−プロパノール(250mL, 分画して添加)から最終溶液質量の44gまでの濃縮によって物質から取り除く。最終溶液質量を41gにする2‐プロパノールからヘプタン(275mL, 分画して添加)への溶媒の交換は、所望の生成物を完全に沈殿させる。懸濁液を更なるヘプタン(40mL)で希釈し、室温で1時間攪拌し、そして濾過する。生成物をn‐ヘプタン(17mL)で洗浄し、そして乾燥し、5.4gの所望の生成物を得る。
ステップE
【0100】
丸底フラスコを、出発物質(110mg, 0.26mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(106mg)でチャージする。固形物をピリジン(4mL)に懸濁する。懸濁液を窒素雰囲気下(1気圧)に置き、そして混合溶液をオーバーナイトで室温で攪拌する。反応混合溶液を、セライト(登録商標)を介して濾過し、そして濾過液を真空中で濃縮する。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、93mgの所望の化合物を得る。([M+H] = 426.6m/z)。
実施例3
【0101】
密封管の中に、ケトン6(85mg, 0.199mmol, 1当量)をチャージし、そしてトリエチレングリコール(2mL)、続いて、ヒドラジン一水和物(hydrazine monohydrate, 500mg, 10mmol, 50当量)及び炭酸カリウム(138mg, 1mmol, 5当量)を加えた。管を密封し、反応溶液を150℃で16時間加熱した。反応溶液を室温まで冷却し、そして水を加えた。残留物をクロロホルムで(3X)抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、そして乾燥状態まで濃縮する。無色の油をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 96:4)を使用して精製した。精製分画をプールし、そして乾燥状態まで濃縮する。結果として生じる油をMTBEに溶解し、水(2回)、2NのNaOH、そして次に鹹水で洗浄した。合併した有機層をNa2SO4上で乾燥し、濾過し、そして蒸発させ、64mgの所望の物質を白い気泡として得る([M+H] = 412.7 m/z)。
実施例4
【0102】
密封管を化合物5(223mg, 0.52mmol, 1当量)及びDMF(1mL)でチャージした。2−ブロモプロパン(1.3g, 10.5mmol, 20当量)及びNa2CO3(73mg, 0.68mmol, 1.3当量)を加え、そしてフラスコを密封し、そして50℃まで加熱した。〜70%の変換が観察された時点で16時間、混合溶液を攪拌した。更なる[2−ブロモプロパン](0.26 g, 2.12 mmol, 4当量)を加えた。反応溶液を2時間攪拌し、そして更なる2−ブロモプロパン(0.13 g, 1.1 mmol, 2当量)を加えた。反応溶液を1時間攪拌した。反応溶液を室温まで冷却し、そして水を加えた。残留物をMTBE(3X)で抽出した。有機層を合併し、鹹水で洗浄し、Na2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。白い気泡をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 99:1)を使用して精製し、206mgの白い気泡としてのN‐イソプロピル誘導体を得た。
【0103】
N‐イソプロピル誘導体(205mg, 0.44mmol, 1当量)を4‐メトキシピリジン(1.5 mL)に溶解した。フラスコを不活性環境下に置き、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 40mg)を加えた。フラスコを密封し、水素で3回パージ(purge)し、そして1気圧水素下に16時間置いた。セライト(登録商法)を反応混合溶液に加えた。セライト(登録商標)の小さなパッドを介して混合溶液を濾過し、そしてEtOAcで洗浄した。有機層を1N塩酸水溶液で(2x)、次に水で洗浄した。有機層をNa2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて、34mgの未精製物を得た。水性の層を2NのKOHで中和し、DCMで(3X)抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして、初期の34mgの未精製物と組み合わせた。未精製物質をヘキサン/EtOAc(6:4)のシリカゲルフラッシュクロマトグラフィーを使用して精製し、80mgの所望の生成物を得た。([M+H] = 468.7 m/z)。
実施例5
【0104】
丸底フラスコの中で、化合物6(88 mg, 0.21 mmol, 1当量)を無水THF(1ml)に溶解した。混合溶液を0℃まで冷却した。ピリジン(84μL, 1mmol, 5当量)及び過酸化ベンゾイル(benzoylperoxide, 150mg, 0.62mmol, 3当量)を連続的に加えた。均質な混合溶液を室温まで2時間かけて徐々に温め、オーバーナイトで室温で攪拌した。飽和炭酸水素ナトリウム(NaHCO3)を加えることによって反応を抑えた。残留物をMTBEで抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして減圧下で濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc(9:1から4:1)を使用して精製し、白い気泡としてN‐O誘導体の生成物(60 mg, 0.11 mmol)を得た。2mLのMeOHにこの気泡を溶解し、続いて2NのKOH水溶液(0.4mL)を[加えた]。反応混合溶液を1時間攪拌した。ほとんどのMeOHを窒素流動下(under a stream of nitrogen)で蒸発させ、1NのHCl(500μL)を加えた。物質をDCM(3X)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして減圧下で濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(ヘキサン/EtOAc(88:12→1:1))で精製し、11mgの所望の生成物を得た。([M+H] = 442.5 m/z)。
実施例6
ステップA
【0105】
丸底フラスコの中で、化合物6(89mg, 0.209 mol, 1当量)及びN‐(ベンジルオキシカルボニル)‐アミノアセトアルデヒド(N-(benzyloxycarbonyl)-aminoacetaldehyde, 148mg,0.85mmol, 4当量)をDCM(2mL)に溶解した。トリアセトキシ水素化ホウ素ナトリウム(Sodium triacetoxyborohydride, 177mg, 0.85mmol, 4当量)を加え、そして反応溶液を3時間室温で攪拌した。混合溶液を飽和NaHCO3水溶液に注ぎ、そして残留物をDCM(3X)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて気泡の固形物(247 mg)を得た。未精製物をEtOAc(2mL)に溶解し、そして4MのHCl(156μL)で処置した。30分後、白い沈殿物が徐々に形成された。結果として生じる泥状物を15分間攪拌した。濾過により、120mgの白い固形物を得た。物質をEtOAcに溶解し、飽和NaHCO3水溶液で処置した。有機層を収集し、水性の層をEtOAc(2X)で抽出した。合併した有機層を鹹水で洗浄し、Na2SO4上で乾燥した。濾過及び蒸発により、所望の中間体を得た。この物質を精製することなく次のステップで使用した。
ステップB
【0106】
ステップAからの全ての物質をEtOAc(3mL)に溶解し、そして10%のPd/C(30mg, ウェット, Aldrich Degussa type ElOl)で処置した。フラスコを密封し、そして水素で3回パージ(purged)し、そして1気圧水素下にオーバーナイトで置いた。16時間後、混合溶液をセライト(登録商標)の小さなパッドを介して精製し、EtOAcで洗浄し、白い気泡としての52mgのアミンを得た。
ステップC
【0107】
アミン14(52mg, 0.11mmol, 1当量)が入った丸底フラスコを、1H‐テトラゾール‐5‐酢酸(1H-tetrazole-5-acetic acid, 21mg, 0.166mmol, 1.5当量)、DCM(2ml)、EDCI(42mg, 0.22mmol, 2当量)及びN,N‐ジイソプロピルエチルアミン(N,N-diisopropylethylamine, 57mg, 0.44mmol, 4当量)でチャージした。結果として生じる黄色い溶液を室温で4時間攪拌した。飽和NaHCO3水溶液の添加によって反応を抑え、そして残留物をDCM(3X)で抽出した。合併した有機層をNa2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて、62mgのオフホワイトの固形物を得た。この物質をシリカゲルフラッシュクロマトグラフィー(MeOH/DCM, 5:95→10:90)を使用して精製し、31mgの所望の生成物を得た。([M+H] = 579.7 m/z)。
実施例7
【0108】
丸底フラスコを出発物質(47mg, 0.110mmol, 1当量)及び炭酸カリウム(150mg, 1.09mmol, 10当量)でチャージした。固形物を2mlのDCMに懸濁した。ヨードメタン(14μL, 0.22mmol, 2当量)を加え、そして混合溶液を2[時間]室温で攪拌した。TLC(DCM/MeOH 95:5)は、>90%の完了を示した。ヨードメタン(14 μL, 0.22 mmol, 2当量)を反応混合溶液に加え、2時間攪拌した。反応混合溶液に水を加えた。層を分離させ、有機物を乾燥し、乾燥状態まで濃縮した。残留物をシリカゲルフラッシュクロマトグラフィー(DCM/MeOH 100:0→98:2)を使用して精製し、34mgの所望の生成物を得た。([M+H] = 440.5 m/z)。
実施例8
【0109】
丸底フラスコを出発物質(59mg, 0.126mmol, 1当量)及び炭酸カリウム(350mg, 2.5mmol, 20当量)でチャージした。固形物を3mlのDCMに懸濁した。反応溶液をヨードメタンでチャージ(80μL, 1.29mmol, 10当量)し、混合溶液をオーバーナイトで室温で攪拌した。反応混合溶液を水でチャージした。有機層を分離し、水性の層をDCMで逆抽出した。合併した有機層を乾燥し、乾燥状態まで濃縮した。残留物をDCM/MeOH(95:5→90:10)のシリカゲルフラッシュクロマトグラフィーを使用して精製し、52mgの所望の生成物を得た。([M+H] = 639.5 m/z)。
実施例9
ステップA
【0110】
丸底フラスコの中で、化合物5(50mg, 0.12mmol, 1当量)及びN‐(t‐ブトキシカルボニル)‐アミノアセトアルデヒド(N-(t-butoxycarbonyl)-aminoacetaldehyde, 6mg, 0.38mmol, 3.1当量)をDCM(2mL)に溶解した。トリアセトキシ水素化ホウ素ナトリウム(8mg, 0.38mmol, 3.1当量)を加え、そして反応溶液を2時間室温で攪拌した。混合溶液を飽和NaHCO3水溶液に注ぎ、残留物をDCM(3x)で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、綿を介して濾過し、そして蒸発させて気泡の固形物(95mg)を得た。未精製物質を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 1:1)を使用して精製し、55mgの化合物18を得た。
ステップB
【0111】
丸底フラスコを出発物質18(800mg, 1.4mmol, 1当量)でチャージした。固形物をDCM及びTFAの溶液(10mL, 1:1)に溶解した。溶液を45分間室温で攪拌した。反応溶液を10%炭酸ナトリウム溶液とDCMの間で分画した。有機層を分離し、そして10%炭酸ナトリウムで洗浄した。有機層を乾燥状態まで濃縮した。残留物を更なる精製無しに次のステップで使用した。
ステップC
【0112】
丸底フラスコをTHF/ACN(1:1, 4mL)に溶解した出発物質(300mg, 0.64mmol, 1当量)でチャージした。反応溶液を37%ホルムアルデヒド水溶液(240μL, 3.22mmol, 5当量)及びシアノ水素化ホウ素ナトリウム(sodium cyanoborohydride, 64mg, 1mmol, 1.6当量)でチャージした。混合溶液を30分間室温で攪拌した。次に、反応溶液を飽和炭酸水素ナトリウム水溶液とDCMの間で分画した。有機層を分離、乾燥し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95→10:90)を使用して精製し、所望の物質を得た。
ステップD
【0113】
丸底フラスコを出発物質20(30mg, 0.06mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(30mg)でチャージした。固形物をピリジン(2mL)に懸濁した。懸濁液を水素雰囲気下に置き、混合溶液をオーバーナイトで室温で攪拌した。反応混合溶液をセライト(登録商標)上で濾過し、そして濾過液を乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95→10:90)を使用して精製し、所望の物質を得た。([M+H] = 497.7 m/z)。
実施例10
【0114】
丸底フラスコを出発物質(85 mg, 0.20mmol, 1当量)でチャージし、DCM(4ml)に溶解した。反応溶液を、N‐(2‐オキソエチル)アセトアミド(N-(2-oxoethyl)acetamide, 80mg, 0.70mmol, 3.5当量)及びトリアセトキシ水素化ホウ素ナトリウム(Sodium triacetoxyborohydride, 177mg, 0.85mmol, 4当量)でチャージした。混合溶液を1時間室温で攪拌した。反応溶液を飽和炭酸水素ナトリウム水溶液とDCMの間で分画した。有機物を分離し、乾燥し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(MeOH/DCM 5:95)を使用して精製し、所望の物質を得た。([M+H] = 511.7 m/z)。
実施例11
【0115】
N‐(2‐オキソエチル)アセトアミドの替わりに、N‐メチル‐N‐(2‐オキソエチル)アセトアミド(N-methyl-N-(2-oxoethyl)acetamide)を使用して化合物22を実施例9に記載の製法に従って合成した。([M+H] = 525.7 m/z)。
実施例12
【0116】
N‐(2‐オキソエチル)アセトアミドの替わりにN‐(2‐オキソエチル)‐3‐フェニルプロパンアミド(N-(2-oxoethyl)-3-phenylpropanamide)を使用して、化合物23を実施例10に記載の製法に従って合成した。([M+H] = 601.8 m/z)。
実施例13
【0117】
N‐(2‐オキソエチル)アセトアミドの替わりにN‐メチル‐N‐(2‐オキソエチル)3‐フェニルプロパンアミドを使用して、化合物23[sic. 24]を実施例10に記載の製法に従って合成した。([M+H] 615.9 m/z)。
実施例14
ステップA
【0118】
丸底フラスコを化合物6(4.23g, 9.94mmol, 1当量)及びTHF(60mL)でチャージした。トリエチルアミン(6.92mL, 49.7mmol, 5.0当量)及びクロロギ酸ベンジル(1.54mL, 10.93mmol, 1.1当量)を加え、そして混合溶液を1時間室温で攪拌した。反応混合溶液を飽和炭酸水素水溶液(100mL)とEtOAc(100mL)の間で分画した。層を分離させ、有機物を乾燥(Na2SO4)し、そして乾燥状態まで濃縮した。未精製物質を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 2:98→14:86)を使用して精製し、3.75gの物質を得た。
ステップB
【0119】
0℃のセリウムトリクロリド7水和物(cerium trichloride heptahydrate, 260mg, 0.69mmol, 1.3当量)のMeOH溶液(10ml)を水素化ホウ素ナトリウム(24mg, 0.65mmol, 1.2当量)で処置し、15分間攪拌し、そして次に−78℃まで冷却した。ケトン26(300mg, 0.54 mmol, 1当量)のTHF溶液(10ml)を加え、混合溶液を1時間攪拌し、そして次に室温まで温めた。水(50ml)及びEtOAc(50ml)を加え、混合し、そして層を分画した。有機層を収集し、鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、白い残留物まで濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)を使用して精製し、235mgの3‐βアルコール27を得た。
ステップC
【0120】
化合物27(235mg, 0.42mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 50mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として130mgの化合物25を得た。([M+H] = 427.4 m/z)。
実施例15
ステップA
【0121】
−78℃のケトン26(300mg, 0.54mmol, 1当量)のTHF溶液(10ml)をK‐セレクトリド(K-Selectride:Potassium tri-sec-butylborohydride, 0.58ml, 0.58mmol, 1.1当量, 登録商標)で処置し、そして60分間攪拌した。メタノール(1ml)を加え、そして溶液を室温まで温めた。水(50ml)及びEtOAc(50ml)を加え、混合し、そして層を分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして白い残留物まで濃縮した。未精製物をシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:14)で精製し、170mgの純粋な3‐αアルコール29を得た。
ステップB
【0122】
化合物29(170 mg, 0.30 mmol, 1当量)を攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 35mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして、室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として76mgの化合物28を得た。([M+H] = 427.4 m/z)。
実施例16
ステップA
【0123】
化合物27(100mg, 0.18mmol, 1当量)及びベンジルトリエチルアンモニウムクロリド(benzyltriethylammonium chloride, 8mg, 0.36mmol, 0.2当量)をDCM(5ml)に溶解し、ジメチル硫酸(130μL, 1.43mmol, 8当量)及び50%水酸化カリウム水溶液(0.5ml)と共に室温で18時間勢い良く攪拌した。混合溶液を水(30ml)とEtOAc(30ml)の間で分画し、そして次に有機層を鹹水で洗浄し、硫酸ナトリウム上で乾燥し、そして、澄んだ油まで濃縮した。未精製のエーテルをシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 3:7→9:113)によって精製し、澄んだ油として75mgのメチルエーテルを得た。
ステップB
【0124】
化合物31(66mg, 0.115mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として22mgの化合物30を得た。([M+H] = 441.4m/z)。
実施例17
ステップA
【0125】
化合物27(100mg, 0.18mmol, 1当量)をDCM(5ml)に溶解し、4‐ジメチルアミノピリジン(4-dimethylaminopyridine, 4mg, 0.35mmol, 0.2当量)、N,N‐ジイソプロピルエチルアミン(N,N-diisopropylethylamine, 0.15ml, 0.9mmol, 5当量)及び無水酢酸(0.070ml, 0.72mmol, 4当量)を加えた。12時間室温で攪拌した後、溶液をEtOAc(30ml)と5%炭酸水素ナトリウム水溶液(15ml)の間で分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして澄んだ油まで濃縮した。未精製のエステルをシリカゲルクロマトグラフィー(エーテル/ヘキサン 3:7→9:113)によって精製し、澄んだ油として100mgのエステルを得た。
ステップB
【0126】
化合物33(100mg, 0.18mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(5ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として45mgの化合物32を得た。([M+H] = 469.4 m/z)。
実施例18
【0127】
化合物27の替わりに、化合物29を使用して化合物34を実施例16に記載の製法に従って合成した。([M+H] = 441.4 m/z)。
実施例19
【0128】
化合物27の替わりに、化合物29を使用して化合物34[sic. 35]を実施例17に記載の製法に従って合成した。([M+H] = 469.4 m/z)
実施例20
ステップA
【0129】
化合物26(185mg, 0.3mmol, 1当量)のエタノール溶液(5ml)をヒドロキシルアミン塩酸塩(140 mg, 2 mmol, 6当量)、酢酸ナトリウム(160 mg, 2 mmol, 6当量)および水(0.5 mL)で処置し、そして混合溶液を室温で1時間攪拌した。混合溶液をEtOAcと水(夫々、50ml)の間で分画した。有機層を鹹水(30ml)で洗浄し、硫酸ナトリウム上で乾燥し、そして白い残留物まで濃縮した。未精製の生成物をシリカゲルクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)によって精製し、193mgのオキシム37を得た。
ステップB
【0130】
化合物37(65mg, 0.113mmol)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 20mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末、シス及びトランスオキシム異性体として15mgの化合物36を得た。([M+H] = 440.3 m/z)。
実施例21
ステップA
【0131】
化合物27(42mg, 0.075mmol, 1当量)をDCM(5ml)に溶解し、4‐ジメチルアミノピリジン(2mg, 0.02mmol, 0.2当量)、N‐Cbzグリシン(23mg, 0.110mmol, 1.5当量)及びジイソプロピルカルボジイミド(0.023ml, 0.150mmol, 2当量)を加えた。12時間室温で攪拌した後、溶液をEtOAc(30ml)と5%炭酸水素ナトリウム水溶液(15ml)の間で分画した。有機層を鹹水で洗浄し、硫酸ナトリウム上で乾燥し、そして澄んだ油まで濃縮した。未精製のエステルをシリカゲルフラッシュクロマトグラフィー(エーテル/ヘキサン 2:3→1:1)によって精製し、澄んだ油として35mgのエステルを得た。
ステップB
【0132】
化合物39(235mg, 0.42mmol, 1当量)を、攪拌棒及びゴム弁付のフラスコ内でEtOAc(7ml)に溶解した。溶液を窒素でスパージ(sparged)し、10%のPd/C(ウェット, Aldrich Degussa type ElOl, 50mg)を加えた。混合溶液を窒素で、そして次に水素ガスでスパージ(sparged)し、そして室温で3時間攪拌した。次に、混合溶液を窒素でスパージ(sparged)し、0.45μmのポリエチレン膜を介して濾過し、そして澄んだ油まで濃縮した。油をシリカゲルフラッシュクロマトグラフィー(NH40H(水溶液)/MeOH/DCM 0.5:2:97.5→0.5:6:93.5)によって精製し、白い粉末として17mgの所望の生成物を得た。([M+H] = 452.4m/z)。
実施例22
【0133】
化合物27の替わりに、化合物29を使用して化合物40を実施例21に記載の製法に従って合成した。([M+H] = 452.4 m/z).
実施例23
【0134】
N‐(2‐オキソエチル)アセトアミドの替わりに、N‐(2‐オキソエチル)‐2‐フェニルアセトアミドを使用して化合物41を実施例10に記載の製法に従って合成した。([M+H] = 587.7 m/z)。
実施例24
ステップA
【0135】
丸底フラスコをアルコール29(7.60g, 13.53mmol, 1当量)でチャージし、DCM(115mL)に溶解した。反応溶液をトリエチルアミン(8.21g, 81mmol, 6.0当量)でチャージした。混合溶液を0℃まで冷却し、メタンスルホニルクロリド(methanesulfonylchloride, 1.86g, 16.2mmol, 1.2当量)でチャージした。30分後、反応混合溶液を飽和炭酸水素ナトリウム水溶液とEtOAcの間で分画した。有機層を分離し、硫酸ナトリウム上で乾燥し、そして乾燥状態まで濃縮した。残留物を、シリカゲルフラッシュクロマトグラフィー(EtOAc/ヘキサン 10→25%)を使用して精製し、所望の物質メシル酸塩(メシラート)を得た。
【0136】
丸底フラスコをメシル酸塩(9.1g, 14.22mmol, 1当量)でチャージし、50mlのDMPUに溶解した。反応溶液をアジ化ナトリウム(4.62g, 71.1mmol, 5.0当量)でチャージし、60℃まで加熱した。17時間混合溶液を攪拌した。次に、反応混合溶液を室温まで冷却し、そして水でチャージした。30分間混合溶液を攪拌した。混合溶液を減圧下で濾過し、水でリンスし、そして空気乾燥し、そして精製すること無しに直接次のステップに使用した。
ステップB
【0137】
丸底フラスコをアジド43(8.35g, 14.23mmol, 1当量)でチャージし、THF(120mLを加えた。次に、反応溶液をトリフェニルホスフィン(11.2g, 42.7mmol, 3.0当量)でチャージした。混合溶液を50℃まで加熱し、20時間攪拌した。次に、反応混合溶液を室温まで冷却し、そして溶媒を減圧下で取り除いた。残留物を、シリカゲルフラッシュクロマトグラフィー(メタノール/DCM 10%→20%)を使用して精製し、アミンを得た。
【0138】
丸底フラスコをアミン(5.10 g, 9.09 mmol, 1当量)でチャージし、DCM(60 mL)に溶解した。反応をN,N‐ジイソプロピルエチルアミン(5.88g, 45.5mmol, 5.0当量)でチャージした。混合溶液を0℃まで冷却し、メタンスルホニルクロリド(2.08g, 18.2mmol, 2.0)でチャージした。30分後、反応混合溶液を飽和炭酸水素ナトリウム水溶液とEtOAcの間で分画した。有機層を収集し、硫酸ナトリウム上で乾燥し、そして乾燥状態まで濃縮した。残留物を、シリカゲルフラッシュクロマトグラフィー(EtOAc /ヘキサン 10→30%)を使用して精製し、Cbzで保護化されたメタンスルホンアミドを得た。
ステップC
【0139】
丸底フラスコをCbzで保護化されたメタンスルホンアミド(5.37g, 8.41mmol, 1当量)及びカーボン上(に担持した)10%パラジウム(1.0g)でチャージした。固形物を2−プロパノール(50mL)に懸濁した。懸濁液を水素雰囲気下に置き、そして混合溶液を4時間25℃で攪拌した。次に、反応混合溶液をセライト(登録商標)上で濾過し、濾過液を乾燥状態まで濃縮した。次に、残留物を、シリカゲルフラッシュクロマトグラフィー(DCM/メタノール 0→5%)を使用して精製し、所望の生成物を得た。[M+H] = 505.6 m/z。
化合物42の代替合成
【0140】
再結晶化されたシクロパミン(2.07g)を適正な大きさの反応容器にチャージし、不活性環境下に置く。EtOAc(7.6g)、トリエチルアミン(1.53g)及びDMAP(307mg)を順番に加える。懸濁液を40℃まで温める。内部温度を45℃未満に維持しつつ、Cbz‐OBtを90分かけて3部(portions)で加える。反応混合溶液を40℃で90分間攪拌する。メタノール(26.4g)を徐々に反応混合溶液に加える間、温度を維持する。結果として生じる懸濁液を室温まで冷却し、少なくとも15時間攪拌する。未精製の製造物を濾過によって収集し、メタノール(5g)でリンスする。白い固形物を減圧下で恒量まで乾燥し、ヘプタン(30.3g)及びトルエン(3.2g)から再結晶化し、化合物24a(3.0g)を得る。
【0141】
固形のビス(2,6‐ジメチルフェニル)水素リン酸塩及び化合物24aを前乾燥し、そして窒素雰囲気下に置く。純粋なジエチル亜鉛(722mg)をDCM(9.0g)が入った適正な大きさの反応容器にチャージする。リン酸塩のDCM溶液(17.9gの中に1.83g)及びIPI‐332690(3.6 gの中に1.34 g)を順番に25℃以下で加える。ジヨードメタン(1.58g)をチャージし、反応溶液を28℃で4‐6時間攪拌する。反応溶液を−45℃まで冷却し、メタンスルホン酸DCM溶液(1.5gの中に566mg)をチャージする。15分後、モルフォリン(1.711g)を加え、そして混合溶液を室温までオーバーナイトで温める。有機層を2N塩酸(2x13.6g)、次に4.8wt%炭酸ナトリウム(水溶液)、4.8wt%亜硫酸ナトリウム(水溶液)及び4.8wt%鹹水(夫々13.6g)で順番に洗浄する。有機層を乾燥し、濾過し、4gまで濃縮し、イソプロパノール(4g)で希釈する。メタノール(9.3g)を徐々に添加することによって生成物を溶液から結晶化する。メタノールリンス(2.6g)、濾過及び乾燥で1.09gの化合物24b(79%の分離収量)を得る。
【0142】
ジョンソンマッセイ Pd/C 触媒 A−305038−5(890mg)、続いて化合物24b(2.24g)を適正な大きさの反応容器にチャージする。反応容器をN2でパージ(purged)し、トルエン(21.8g)及び2−プロパノール(6.7g)を順番に加える。システムを脱気し、そして窒素雰囲気下に置き、そしてその工程を水素で繰り返す。システムを勢い良く攪拌し、そして水素雰囲気(blanket)を1気圧で4−5時間維持する。反応をTLC若しくはHPLCのいずれかでモニターする。未完了であったら、反応を不活性化し、追加の触媒(145mg)をチャージし、そして水素雰囲気を更に1時間繰り返す。エチレンジアミン(12.9mg)をチャージし、そして混合溶液を15分間攪拌した。触媒を濾過によって取り除き、トルエン:IPA(3:1)でリンスする。濾液及びリンス[液]を濃縮し、溶媒をトルエンに交換する。生成物をトルエン(19.0g)及びヘプタン(18.0g)から結晶化し、白い結晶化固形物(1.34g, 98%収量)として化合物24cを得る。
【0143】
化合物24c(644mg)、続いてアルミニウム‐t‐ブトキシド(525mg)、トルエン(8.34g, 15ボリューム)及び2‐ブタノン(7.83g, 15ボリューム)を適正な大きさの反応容器にチャージする。酸素を取り除くためにフラスコの内容物を減圧吸引/窒素パージ(purge)サイクルで脱気し、反応混合溶液を勢い良く攪拌しつつ、75℃で16‐18時間加熱する。反応を水性のロッシェル塩(10.3gの水の中2.6 g)の添加によって抑え、混合溶液を勢い良く1時間45℃で攪拌する。水性及び有機層を分画する。トルエン(2.9g)及びEtOAc(2.9g)の混合溶液で水性の層を逆抽出する。有機層を合併し、新鮮なロッシェル塩溶液(10.3gの水の中2.6g)、次に水(12.9g)で洗浄する。結果として生じる有機層を硫酸ナトリウム(1.97 g)上で乾燥し、濾過し、そして真空中で濃縮する。最初にIPA(6.5g)、次にヘプタン(7.7g)のチャージ及び濃縮溶媒交換を経由して、生成物を結晶化する。濃ヘプタン泥状物(〜2.7g)をオーバーナイトで攪拌し、固形物を濾過によって収集する。真空吸引乾燥により化合物24d(550mg)を85%の収量で得る。
【0144】
エノン24d(459mg)及びジョンソン‐マッセイのカーボン上(に担持した)5%パラジウム(A503023-5, 101mg)を適正な大きさのマルチネック反応容器にチャージする。容器を窒素でパージ(purged)し、3‐ピコリン(2.2g)を溶媒としてチャージする。攪拌を開始し、容器を、窒素を使用して最初の脱気をし、そして次に大気圧の水素下で8時間攪拌する。反応の終了時に、触媒を0.2ミクロンメディアを介した濾過によって取り除き、ACN(1.4ml)でリンスする。機械的な攪拌、内部温度プローブ、及び窒素雰囲気を備えたクリーンないし清潔な反応容器の中で、濾過液及びリンス[液]を合併する。クエン酸(3.7g)の水(9.2ml)溶液を反応容器に30℃以下でチャージし、20℃、次に0℃でクエン酸塩として、溶液からIPI‐335589を徐々に結晶化する。結晶化物を吸引濾過によって回収し、水(3.7ml)で洗浄する。乾燥後、水和物(3-5wt% 水)として、89.5%収量(622mg)で、90:1に近いβ:αの割合で、クエン酸塩24eを分離する。
【0145】
上記クエン酸塩24e(1.50g)を、2‐メチルテトラヒドロフラン(7.7g)及び1M炭酸ナトリウム(9.0ml)と共に、適正な大きさの反応器にチャージする。添加漏斗(addition funnel)を介してクロロギ酸ベンジル(454mg)の2‐メチルテトラヒドロフラン(300mg)溶液を加え、反応を1−2時間、環境温度にする。反応が完了したとき、攪拌を止め、層を分離し、そして有機層を水で2回(2x6g)洗浄する。有機層を硫酸ナトリウム(3g)上で乾燥し、濾過し、そして濃縮する。新鮮な2‐メチルテトラヒドロフラン(6.5g)からの濃縮によって残りの水を更に減らし、無水2‐メチルテトラヒドロフランの溶液として物質を次の反応に移す。
【0146】
市販の1M K‐セレクトリドのTHF(1.20g)を乾燥した反応容器に窒素雰囲気下でチャージし、そして無水2‐メチルテトラヒドロフラン(2.10g)で希釈し、そして−65℃に冷却する。次に、内部温度を−65℃±5℃に調節するために、溶液24f(0.41g)の2−メチルテトラヒドロフラン(1.5g)溶液を徐々に反応容器に加える。反応溶液を2時間攪拌し、そしておよそ1時間かけて−20℃まで温め、そして更に1時間攪拌する。反応をHPLCでモニターし、未完了であった反応を追加のK‐セレクトリドで完了にする。低温度でメタノール(0.33g)、次に、−20℃で3M水酸化ナトリウム(2.4g)、5℃以下で15%過酸化水素水(1.04g)で反応を抑え、次に、オーバーナイトで、環境温度で攪拌する。層を分画し、そして有機層を1M水酸化ナトリウム水溶液(2ml)、0.5MのNa2SO3水溶液(2ml)、及び塩酸でpH3に調節した水(2ml)で洗浄する。有機層を硫酸ナトリウム(0.82g)上で乾燥し、濾過し、そして濃縮する。生成物24g(0.457g)をDCM(0.9g)から再濃縮し、次の反応に使用する。
【0147】
上記生成物24g(1.36g)を無水DCM(18.1g)で適正な大きさの反応容器にチャージし、不活性環境下に置き、そして−20℃まで冷却する。トリエチルアミン(0.61mg)をチャージし、続いて、メタンスルホニルクロリド(373mg)の無水DCM(30mg)を徐々に添加する。反応溶液を1時間−20℃で攪拌する。反応をHPLCによってモニターする。未完了の反応を追加のメタンスルホニルクロリドで完了にする。完了していた場合、反応を水(13.6g)で抑え、そして放温する。層を分離し、有機層を2.5wt%炭酸水素ナトリウム(13.8g)及び、次に水(10.9g)で洗浄する。有機層を硫酸ナトリウム(4g)上で乾燥し、濾過し、そして濃縮する。t‐ブチルメチルエーテル(10.9ml)及び次にl,3‐ジメチル‐3,4,5,6‐テトラヒドロ‐2(1H)ピリミジノン(DMPU, 4.7ml)のチャージ及び濃縮を経由して、生成溶液を溶媒交換する。DMPU溶液を直接次の反応に使用する。
【0148】
アジ化ナトリウム(0.74g)を適正な大きさの反応容器にチャージする。生成物24h(1.46 g)のDMPU(5.9g)溶液を反応容器にチャージし、更なるDMPU(1.9g)でリンスする。反応全体において窒素スイープ(sweep)を維持したままで、懸濁液を60℃まで15時間加熱する。反応溶液を環境温度まで冷却し、そしてMTBE(11.7g)で希釈する。有機溶液を2%生理食塩(3x8g)で3回洗浄し、硫酸ナトリウム(4.4g)上で乾燥し、濾過し、そして濃縮する。生成物をTHF(6.4g)から濃縮し、直接次の反応に使用する。
【0149】
未精製の生成物24i(1.34g)をTHF(12.6g)で溶解し、そして適切な大きさの反応溶液に移す。トリフェニルホスフィン(0.70g)及び水(0.44g)をチャージし、そして反応溶液を55℃まで15‐24時間加熱する。完了したとき、反応溶液を環境温度まで冷却し、硫酸マグネシウム(1.4g)で乾燥し、濾過し、そして濃縮する。試薬に基く不純物を取り除くために、固形物を溶解し、3部(portions)のDCM(3x9g)から濃縮し、そしてDCM/MeOH/Et3N勾配を使用したシリカゲルクロマトグラフィーによって精製する。プールした分画を乾燥状態まで濃縮し、DCM(6.8g)に溶解し、そして再び乾燥状態まで濃縮し、次のステップで使用する非晶質の固形物(1.12g)を得る。
【0150】
生成物24j(1.09g)を無水DCM(15.8 g)で溶解し、適正な大きさの反応容器に移し、窒素雰囲気下に置く。溶液を0℃まで冷却する。温度を5℃未満の間に維持しつつ、N,N‐ジイソプロピルエチルアミン(357mg)及び純粋なメタンスルホニルクロリド(0.165ml)を順番にチャージする。反応をHPLCによってモニターする。未完了の反応を追加のメタンスルホニルクロリドで完了にする。0.4M炭酸水素ナトリウム(11.4 g)水溶液で反応を抑え、環境温度まで温める。層を分離し、水性の層をDCM(5.8g)で逆抽出する。合併した有機層を硫酸マグネシウム(0.55g)上で乾燥し、濾過し、そして濃縮する。残りのDCMを取り除くために生成物24kを溶解し、2‐プロパノール(4.0 g)から除去し、次の反応に直接使用する。
【0151】
アルドリッチ デグサ タイプ ElOl NE/W 10% Pd/C(249mg)を適正な大きさの反応容器にチャージし、そして窒素雰囲気下に置く。24k(1.24g)の2−プロパノール(9.8g)溶液を反応容器にチャージする。システムを脱気し、そして窒素雰囲気下に置き、そしてその工程を水素で繰り返す。反応溶液を1気圧の水素下で、環境温度で8時間攪拌する。不活性環境を容器に戻し、そして2−プロパノール(0.5g)中に泥状化した触媒(125mg)の2度目のチャージを反応に加える。反応混合溶液を脱気し、そして窒素雰囲気下に置き、その工程を水素で繰り返す。反応溶液を1気圧の水素下でさらに15時間、環境温度で攪拌する。反応をHPLCによってモニターする。未完了の反応を追加の触媒及び水素で処置する。完了した場合、反応溶液を濾過し、蒸気活性炭(200mg)で処置し、そして再び濾過する。溶液を部分濃縮によって乾燥し、反応容器に移し、理論的収量に基く0.09Mまで無水2−プロパノールで希釈する。2−プロパノールの1.25M塩酸溶液(1.64g)を20分間かけてチャージする。塩酸塩を穏やかな攪拌で徐々に結晶化させ、濾過によって分離する。結晶化物を2−プロパノール(2.5g)で洗浄し、真空吸引乾燥し、1:1のIPA溶媒和化合物として化合物42(916mg, 80%収率)を得る。
実施例25
ステップA
【0152】
丸底フラスコをアミン42(1.1g, 2.1mmol, 1当量)、無水テトラヒドロフラン(10ml)、及びピリジン(880uL, 10.9mmol, 5当量)でチャージした。冷却した(0℃)混合溶液を過酸化ベンゾイル(1.6g, 6.5mmol, 3当量)で処理した。混合溶液を2時間0℃で、次にオーバーナイトで25℃で攪拌した。反応混合溶液をMTBEで希釈し、飽和NaHCO3水溶液と1NのNaOHの混合溶液で層が分離するまで洗浄した。有機層を収集し、水性の層をMTBEで1回再抽出した。合併した有機層を鹹水で洗浄し、Na2SO4上で乾燥し、濾過し、乾燥状態まで濃縮した。未精製の油を5mlのCH2Cl2に溶解し、SiO2(40g)カラムの上に乗せ、ヘキサン/EtOAc(10%から50%)から溶出し、ベンゾイル誘導体48(380mg)を得た。([M+H] = 625.4 m/z)
ステップB
【0153】
丸底フラスコを[ベンゾイル誘導体]48(374mg, 0.6mmol, 1当量)及びMeOH(5mL)でチャージした。2NのKOH(0.3mL, 0.6mmol, 1当量)の存在下で、溶液を25℃に処置した。混合溶液を3時間攪拌した。溶媒を減圧下で取り除いた。MTBEを残留物に加え、1NのHClで中和した。層を切り、水性の層を2部(portions)のCH2Cl2で抽出した。合併した有機層をNa2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。未精製物質(380mg)をCH2Cl2で溶解し、SiO2(12g)カラムの上に乗せ、ヘキサン/EtOAc(0%から100%)で溶出し、ヒドロキシルアミン47を得た。物質をt−BuOH/7%H2Oから凍結乾燥し、白い粉末として213mgの[ヒドロキシルアミン]47を得た([M+H] = 521.4 m/z)。
実施例26
ステップA
【0154】
ドライヤーで乾燥したフラスコを、無水CH2Cl2(5mL)及びベンジルアルコール(785uL, 7.58mmol, 1.3当量)でチャージした。冷却した(0℃)溶液をクロロスルホニルイソシアネート(506uL, 5.83mmol, 1当量)で処理した。次に、DMAP(1.4g, 11.6mmol, 2当量)を加え、そして混合溶液を1時間25℃で攪拌した。DMAPの溶解が完了した後、短い期間、反応溶液は、澄んでいた。次に、白い泥状の沈殿物が形成した。混合溶液をCH2Cl2(30mL)で希釈し、そして3部(portions)(夫々30mL)の水で洗浄した。有機層をNa2SO4上で乾燥し、濾過し、そして、乾燥状態まで蒸発させた。所望の白い固形物51を精製することなしに次のステップに用いた。
ステップB
【0155】
丸底フラスコを生成物52(30mg, 0.053mmol, 1当量)及び生成物51(18mg, 0.053mmol, 1当量)でチャージした。両方の試薬をCH2Cl2(2mL)に溶解し、溶液を25℃で攪拌した。未精製の物質をSiO2カラム(4g)に乗せ、ヘキサン/EtOAc(0%から50%)で溶出し、16mgのスルファモイル化(sulfamoylated)誘導体53を得た([M+Na] = 796.4 m/z)。
ステップC
【0156】
丸底フラスコを生成物53(16mg, 0.021mmol, 1当量)及び11mgの10% Pd/C(ウェット, Aldrich Degussa type ElOl)でチャージした。物質を2−プロパノール(3mL)に懸濁した。フラスコを密封し、そして水素で3回パージ(purged)し、そしてオーバーナイトで1気圧の水素下に置いた。泥状物を0.2ミクロンアクロディスク(Acrodisc)を介して濾過し、2−プロパノールで洗浄し、そして溶媒を減圧下で取り除いた。残留物を、CH2Cl2/MeOH(5%から10%)で溶出するSiO2カラム(1g)によって精製した。主要生成物をt‐BuOH/7%H2Oから凍結乾燥し、9mgのスルファミド50を得た([M+H] = 506.4m/z)。
実施例27
ステップA
【0157】
丸底フラスコをシクロパミン4‐エン‐3‐オン(cyclopamine 4-en-3-one, 3.5g, 8.5mmol, 1当量)及びピリジン(70mL)でチャージした。反応容器をPd/C(10% Pd, 500mg)でチャージした。反応溶液を1気圧の水素下に置いた。3.5時間後、LCMSは出発物質の完全な消耗を示した。触媒をアクロディスク0.2ミクロンフィルター上で濾過除去し、そしてトルエンで洗浄した。溶媒をトルエン(2x10mL)で共沸除去によって取り除いた。所望の物質56、3.5g([M+H] = 412.5m/z)を、そのままで次のステップに使用した。
ステップB
【0158】
丸底フラスコを生成物56(1.2g, 2.8mmol, 1当量)、CH2Cl2(10mL)及びトリエチルアミン(1.9mL, 14.2mmol, 5当量)でチャージした。冷却した(0℃)溶液をCBz‐Cl(440uL, 2.8mmol, 1当量)で処理した。1時間後、LCMSは出発物質の完全な消耗を示した。混合溶液を水で希釈した。層を切り、そして有機層を水で2回洗浄した。有機層を硫酸ナトリウム上で乾燥し、濾過し、そして乾燥状態まで濃縮した。生成物をヘキサン/EtOAc(0から20%)で溶出するカラムクロマトグラフィー(SiO2, 40g)によって精製し、ケトン生成物57(891mg)を得た([M+Na] = 468.4m/z)。
ステップC
【0159】
丸底フラスコの中で、ケトン生成物57を2回CH2Cl2で共沸蒸留し、そして真空下で1時間乾燥した。窒素下において、ケトン2(693mg, 1.27mmol, 1当量)を無水THF(20 mL)に溶解し、そして溶液を−78℃まで冷却した。1MのK‐セレクトリドのTHF溶液(1.9mL, 1.9mmol, 1.5当量)を滴下で加えた。1時間後、反応をTLCによって完了した。反応を2.6mlの5NのNaOHの添加、続いて2.6mLの30%wtのH2O2の緩徐添加によって抑えた。結果として生じる混合溶液をオーバーナイトで攪拌した。混合溶液を水とEtOAcの間で分画した。水性の層をEtOAcで逆抽出した。合併した有機[層]を最初に水(ごく一部の塩化アンモニウムで緩衝された)、次に鹹水で洗浄した。有機[層]を乾燥し、濾過し、そして未精製の気泡(840 mg)まで濃縮した。未精製の物質をCH2Cl2に溶解し、SiO2カラム(4Og)に乗せ、ヘキサン/EtOAc(0から50%)で溶出し、アルコール生成物58(565 mg)を得た。
ステップD
【0160】
窒素下の丸底フラスコの中で、アルコール生成物58(530mg, 0.98mmol, 1当量)を5mLの無水CH2Cl2及びトリエチルアミン(800uL, 5.81mmol, 6当量)に溶解した。反応混合溶液を0℃まで冷却し、Ms‐Cl(112uL, 1.45mmol, 1.5当量)を滴下で加えた。混合溶液を0℃で30分間攪拌した。TLC(ヘキサン:EtOAc, 7:3)は、〜70%の変換を示した。70μLのトリエチルアミン(70uL, 0.5当量)及びMs‐Cl(10uL, 0.1当量)を反応容器にチャージした。90分後、飽和炭酸水素塩の溶液をチャージし、残留物をCH2Cl2で抽出した。有機層を水で洗浄し、乾燥し、そしてオフホワイトの気泡まで濃縮した。物質をCH2Cl2に溶解し、そしてヘキサン/酢酸エチル(0%から50%)で溶出するSiO2(40g)で精製し、メシラート生成物59(430mg)を得た。
ステップE
【0161】
丸底フラスコの中で、メシラート生成物59(420mg, 0.67mmol, 1当量)を2mLのDMPUに溶解した。溶液を60℃で5時間アジ化ナトリウム(218mg, 3.4mmol, 5当量)で処理した。混合溶液を25℃まで冷却し、次に氷水に注ぎ、白い固形物を得た。化合物をMTBEで抽出した(3回)。合併した有機層を水(2X)、次に鹹水で洗浄した。有機層をNa2SO4上で乾燥し、濾過し、そして、白い気泡(342mg)まで濃縮した。所望の物質60を、そのままで次のステップに使用した。
ステップF
【0162】
コンデンサを備えた丸底フラスコの中で、アジド60(336mg, 0.58mmol, 1当量)を7mLのTHF及び140μLの水に溶解し、トリフェニルホスフィン(462mg, 1.76mmol, 3当量)で処理した。混合溶液を70℃までオーバーナイトで加熱した。TLC(ヘキサン/EtOAc, 7:3)は、反応が完了したことを確認した。反応溶液を乾燥状態まで濃縮した。未精製の物質をCH2Cl2に溶解し、12gのSiO2の上に乗せ、そしてCH2Cl2/MeOH(0から20%)で溶出し、アミン61(254mg)を得た。
ステップG
【0163】
窒素下の丸底フラスコの中で、アミン61(248mg, 0.45mmol, 1当量)を7mLの無水CH2Cl2及びN,N‐ジイソプロピルエチルアミン(237uL, 0.91mmol, 2当量)に溶解した。反応混合溶液を0℃まで冷却し、Ms‐Cl(70uL, 1.45mmol, 1.5当量)を滴下で加えた。混合溶液を0℃で2時間攪拌した。TLC(ヘキサン/EtOAc, 7:3)は、少量のアミンを示した。混合溶液を10μLのMs‐Cl(0.2当量)でチャージし、そして25℃まで1時間温めた。反応混合溶液をCH2Cl2、次にNaHCO3の飽和溶液で希釈した。層を切った。水性の層を一部(portion)のCH2Cl2で抽出した。合併した有機層を水で洗浄し、Na2SO4上で乾燥し、濾過し、そして乾燥状態まで濃縮した。未精製物(326mg)をSiO2カラム(12g)に加え、ヘキサン/EtOAc(0から50%)で溶出し、スルホンアミド62(256mg)を得た。
ステップH
【0164】
丸底フラスコをスルホンアミド62(250mg, 0.4mmol, 1当量)及び50mgの10% Pd/C(ウェット, Aldrich Degussa type ElOl lot 08331KC)でチャージした。物質をEtOAc(5mL)に懸濁した。フラスコを密封し、そして水素で3回パージ(purged)し、そして1気圧の水素下で攪拌した。3時間後、変換が観察されたが、多くの出発物質はそのままであった。泥状物を0.2ミクロンのアクロディスク(Acrodisc)を介して濾過し、2−プロパノールで洗浄した。濾過液を54mgの触媒を加えた条件の反応に再び供した。反応は、3時間後に完了した。泥状物を0.2ミクロンアクロディスクを介して濾過し、2‐プロパノールで洗浄し、そして溶媒を乾燥状態まで濃縮した。未精製物質(200 mg)をSiO2カラム(12g)の上に乗せ、そして化合物をCH2Cl2/MeOH勾配(0 から10%)を使用して溶出し、遊離アミンを得た。物質をt‐BuOH/7%H2Oから凍結乾燥し、白い粉末として175mgの生成物55を得た([M+H] = 491.3 m/z)。
実施例28
細胞培養におけるヘッジホッグ経路の阻害
【0165】
ヘッジホッグ経路特異的な癌細胞死滅効果は、次の検定を使用して確認することができる。C3H10T1/2細胞は、ソニックヘッジホッグペプチド(Shh-N)に接触した場合に、骨芽細胞に変異する。変異した、すなわち、これらの骨芽細胞は、酵素学的な検定で測定することができる高レベルのアルカリフォスファターゼ(AP)を生成する(Nakamura et al., 1997 BBRC (1997) 237: 465)。C3H10T1/2の骨芽細胞への変異(Shh依存的なイベント)を妨げる化合物は、従って、AP生産の減少によって同定することができる(van der Horst et al., Bone (2003) 33: 899)。検定の詳細は、以下に開示する。変異検定の概略結果(阻害のEC50)を下の表1に示す。
検定プロトコル
細胞培養
【0166】
マウス胎性中胚葉繊維芽細胞のC3H10T1/2細胞(ATCCから得た)を、10%加熱不活性化FBS(Hyclone)、50ユニット/mlのペニシリン、及び50ug/mlのストレプトマイシン(Gibco/Invitrogen)を補充した基本MEM培地(Gibco/Invitrogen)内で、37℃で、5%CO2の空気雰囲気中で培養した。
アルカリフォスファターゼ検定
【0167】
C3H10T1/2細胞を96ウェル[プレート]に8x103細胞/ウェルの密度で蒔いた。細胞は、コンフルエンスまで成長した(72時間)。ソニックヘッジホッグ(sonic Hedgehog, 250ng/ml)及び/または化合物処理の後、細胞を溶解緩衝液(50mMトリス pH7.4, 0.1%トライトンXlOO)に溶解し、プレートを超音波処理し、そしてライセートを0.2μmPVDFプレート(Corning)を介してスピンした(spun)。40μLのライセートを、1mg/ml p‐ニトロフェニルリン酸塩を含むアルカリ緩衝溶液(Sigma)内でAP活性について検定した。30分間37℃でインキュベートした後、プレートをエンビション(Envision)プレートリーダ上で、405nmで解読した(read)。全タンパク質を、ピアス(Pierce)からのBCAタンパク質アッセーキットで、製造者の説明書に従って数量化した。AP活性を、全タンパク質量に対して規格化した。「A」は、IC50が20nM未満であることを示し、「B」はIC50が20〜100nMであることを示し、「C」は、IC50が>100nMであることを示すことを意味する。
表1−阻害のための概略EC50
実施例29
膵臓癌モデル
【0168】
化合物42の活性をヒト膵臓モデルでさらにテストした、すなわち、BXPC‐3細胞をマウスの右足の側面の皮下の中に移植した。腫瘍移植後、42日目に、マウスを、ビヒクル(担体:Vehicle)(30%HPBCD)若しくは化合物42のいずれかを受ける2群に無作為に区分した。化合物42を40mg/kg/日で経口投与した。25日間の毎日の投与を受けた後、ビヒクルコントロールと比べた場合、化合物42は、統計学的に腫瘍の成長量を40%減少させた(p=0.0309)。実験の最後に、HH経路遺伝子のq‐RT‐PCR解析によって標的に対する反応を評価するために、最終投与の4時間後、腫瘍を回収した。ヒトGli‐1の解析では、変調がなかった。マウスのGli‐1のmRNAレベルの解析は、ビヒクル処置群と比べた場合、化合物を処置した群において強い低下(抑制)制御となった。ヒトの腫瘍細胞ではないが、マウスの細胞のヘッジホッグ経路の抑制は、ヘッジホッグ阻害剤の一つの効果が腫瘍‐間質相互作用(tumor-stroma interaction)に影響を及ぼすことを示す。
実施例30
髄芽腫モデル
【0169】
化合物42の活性も髄芽腫の遺伝子導入マウスモデルで評価した。腫瘍サプレッサーPatched 1(Ptch1)及びHypermethylated in Cancer 1(Hic1)における機能欠損変異のヘテロ接合であるマウスは、自発性の骨芽腫を発生する。ヒト髄芽腫に似て、これらの腫瘍は、残りのHic1アレルの完全な過剰メチル化のプロモーターを表し、そして野生型Ptch1アレルの発現の欠損を表す。皮下の移植片として継代された場合、これらの細胞は、激しく成長し、そしてヘッジホッグ経路依存的である。経口的な化合物の投与の効果を評価するため、及び活性と血漿及び腫瘍における薬物暴露とを関連付けるために、このモデルを採用した。ドーズ投与(dose administration)の8時間後のGli‐1 mRNAの発現の減少によって測定されたものとして、化合物42の単ドーズ(single dose)の経口投与(PO)は、皮下に移植された腫瘍におけるHH経路の量依存的(dose-dependent)な低下制御を導いた。
【0170】
化合物の毎日(QD)の経口投与(PO)は、投与量依存的な腫瘍の成長の阻害を導き、多量の投与ではあからさまな腫瘍退縮が観察された。腫瘍成長を50%阻害する(ED50)ための、およその効果的な毎日の経口的な投与量は、4mg/kgである。動物が毎日(QD)の[投与]を21日間処置された場合、処置の休止の後、長期間の生存(>60日)が観察され、腫瘍の再成長がほとんどなかった。これは、ヘッジホッグ阻害剤化合物42がヘッジホッグ経路と遺伝子突然変異によるヘッジホッグ経路に依存する腫瘍の成長との両者を抑制することを実証する。
実施例31
肺癌モデル
【0171】
ヒトSCLC腫瘍モデルにおける化合物42の活性をテストするために、LX22細胞を右足の側面の皮下に移植した。LX22は、化学的に未処置の患者由来のSCLCの主要な異種移植モデルであり、マウスからマウスへの継代によって維持されている。この腫瘍は、臨床設定に近似するということにおいてエトポシド(etoposide)/カルボプラチン(carboplatin)化学療法に反応する。LX22は、化学療法の処置の間退行し、寛解期を経て、そして次に再発し始める。LX22モデルにおいて、化合物単試薬活性及びそれの化学抵抗性再発を調節する能力をテストした。腫瘍移植後、32日目に、マウスをビヒクル(30%HPBCD)、当該化合物又はエトポシド及びカルボプラチンの組み合わせ化学療法(E/P)を受ける3群に無作為にグループ分けした。化合物42を40mg/kg/日の投与量で経口投与し、そして16連続投与の後、処置群とビヒクル群の間に測定可能な違いがなかった。腫瘍移植後、34、35、36、及び48日目に、エトポシドを12mg/kgで静脈に投与し、また、34、41及び48日目に、カルボプラチンを60mg/kgで静脈に投与した。50日目に、E/P処置マウスをさらにビヒクル(30%HPBCD)又は化合物追跡処置を受ける[群]に無作為に区分した。化合物をMTD(最大耐性量)40mg/kg/日で複数回、経口的に投与し、そして35の連続投与の後、ビヒクル群に比べて、処置群において腫瘍の再発に実質的な遅延があった (p=0.0101)。
実施例32
多発性骨髄腫
【0172】
ヒト多発性骨髄腫の細胞株(cell lines)(NCI−H929及びKMS12)並びにMM患者由来の初代の臨床骨髄標本(primary clinical bone marrow specimens)を用いて、インビトロ(in vitro)で多発性骨髄腫細胞(MM)の成長を抑制する化合物42の性能を試験した。細胞は化合物で96時間処置され、洗浄されて、次いで、メチルセルロースに培養された。腫瘍コロニーは、処置の後の細胞増殖の可能性の指標として、10〜21日後に定量化された。細胞株又は初代の患者の標本の処置は、未処置の対照区と比較して、減少した細胞成長の結果となった。未処置の対照区は100%の細胞成長を示し、臨床サンプルと同様に処置された各細胞株は、約25%未満の成長を示した。
実施例33
急性骨髄性白血病及び骨髄異形成症候群
【0173】
急性骨髄性白血病(AML、細胞株U937)及び骨髄異形成症候群(MDS、細胞株KG1及びKG1a)の患者由来のヒト細胞株のインビトロ(in vitro)成長を抑制する化合物42の性能を研究した。各々の細胞株は化合物42(1.0uM)で72時間処理され、続いてメチルセルロースに培養された。下記表にまとめたように、それら細胞株の成長は化合物42によって抑制された。
表2.AML及びMDSにおける細胞成長の抑制
実施例34
非ホジキンリンパ腫(NHL)及びホジキン病(HD)
【0174】
非ホジキンリンパ腫(細胞株RL及びJeko-1)及びホジキン病(細胞株L428)の患者由来のヒト細胞株のインビトロ(in vitro)成長を抑制する化合物42の性能を研究した。各々の細胞株は化合物42(1.0uM)で72時間処理され、続いてメチルセルロースに培養された。下記表にまとめたように、それら細胞株の成長は化合物42によって抑制される。
表3.HD及びNHLにおける細胞成長の抑制
実施例35
プレB細胞急性リンパ性白血病
【0175】
Gli反応性ルシフェラーゼリポーターが一時的に細胞に形質移入された、一過性形質転換アッセイ(transient transfection assay)を用い、プレB細胞急性リンパ性白血病の3つの細胞株(REH、RS4−11及びNalm−6)に対する化合物42(1uM)の性能が研究された。化合物42での処置は、ビヒクル処置の対照区と比較して、ルシフェラーゼの活性を抑制した(表4)。これは、化合物42がヘッジホッグ経路の効果的なアンタゴニストであることを実証した。
表4.ルシフェラーゼ活性の抑制
【0176】
また、それら細胞株のうち2つ(の細胞株)の成長における化合物42の効果を72時間インビトロ(in vitro)で処置して研究した。処置後、細胞は洗浄されて、メチルセルロースに培養された。コロニー形成の抑制はほとんど無かったが、以降のコロニーの再培養(replating)は顕著な細胞成長の抑制を実証した(表5)。
表5.すべて(の細胞)における細胞成長の抑制
引用による繰込み
【0177】
本願明細書に引用された全てのU.S.特許及びU.S.公開特許出願は、これにより、引用によって本願明細書に繰み込み記載される。
等価物
【0178】
当業者は、通例のルーチン的実験を行うだけで、本願明細書に開示された本発明の特定の実施態様の多くの等価物を認識し、又は確認することができるであろう。そのような等価物は、以下の特許請求の範囲の請求項によって包含されることを意図する。
【特許請求の範囲】
【請求項1】
過剰増殖疾患を処置するための薬物(medicament)の調製における式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化1】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは夫々1、2、3、4、5若しくは6において独立であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項2】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、請求項1の使用。
【請求項3】
R1はスルホンアミドである、請求項1の使用。
【請求項4】
症状が皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される、請求項1乃至3のいずれか一項の使用。
【請求項5】
ヘッジホッグ経路によってもたらされる(mediated)症状を処置するための薬物(medicament)の調製のための式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化2】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;
各qは、夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項6】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、
請求項5の使用。
【請求項7】
R1はスルホンアミドである、請求項5の使用。
【請求項8】
前記症状は小細胞肺癌である、請求項5の使用。
【請求項9】
前記症状は膵臓癌である、請求項5の使用。
【請求項10】
前記症状は髄芽腫である、請求項5の使用。
【請求項11】
前記症状は多発性髄芽腫、白血病、骨髄異形成症候群、非ホジキンリンパ腫、及びホジキン病からなる群から選択される、請求項5の使用。
【請求項12】
前記化合物は経口的に投与される、請求項5乃至7のいずれか一項の使用。
【請求項13】
前記化合物は静脈内に投与される、請求項5乃至7のいずれか一項の使用。
【請求項14】
前記化合物は局所的に投与される、請求項5乃至7のいずれか一項の使用。
【請求項15】
患者のヘッジホッグ経路に対抗(アンタゴナイズ)するための薬物(medicament)の調製における式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化3】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;
各qは夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項16】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、請求項15の使用。
【請求項17】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項1、5又は15のいずれか一項の使用。
【化4】
【請求項18】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項4の使用。
【化5】
【請求項19】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項6乃至11のいずれか一項の使用。
【化6】
【請求項20】
患者のヘッジホッグ経路に対抗(アンタゴナイズ)するための薬物(medicament)の調製における式(2)の化合物又はその薬学的に許容可能な塩の使用であって、式(2)の該化合物は下記式を有し、
【化7】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは独立して1、2、3、4、5若しくは6であり;
X−はハライドであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)であり;
各Rは独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
R7及びR7´の夫々がHであり;又はR7及びR7´が一緒になって=Oを形成し;
R8及びR9はHであるか、又は、R8及びR9は一緒になってボンドを形成し;及び
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1はヒドロキシルとなることはできず、かつR2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1は酢酸となることはできず、かつR2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7がH2である場合;R1及びR2が一緒になって=Oとなることができず;及び
R3、R4、R8、R9がHであり、かつR7及びR7´がHである場合;R1及びR2がHとなることができない。
【請求項21】
R1はスルホンアミドである、請求項20の使用。
【請求項1】
過剰増殖疾患を処置するための薬物(medicament)の調製における式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化1】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは夫々1、2、3、4、5若しくは6において独立であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項2】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、請求項1の使用。
【請求項3】
R1はスルホンアミドである、請求項1の使用。
【請求項4】
症状が皮膚癌、中枢神経系の癌、消化管の癌、肺系の癌、尿生殖器癌、乳癌、肝細胞癌、脳癌、及び造血系の癌からなる群から選択される、請求項1乃至3のいずれか一項の使用。
【請求項5】
ヘッジホッグ経路によってもたらされる(mediated)症状を処置するための薬物(medicament)の調製のための式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化2】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;
各qは、夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項6】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、
請求項5の使用。
【請求項7】
R1はスルホンアミドである、請求項5の使用。
【請求項8】
前記症状は小細胞肺癌である、請求項5の使用。
【請求項9】
前記症状は膵臓癌である、請求項5の使用。
【請求項10】
前記症状は髄芽腫である、請求項5の使用。
【請求項11】
前記症状は多発性髄芽腫、白血病、骨髄異形成症候群、非ホジキンリンパ腫、及びホジキン病からなる群から選択される、請求項5の使用。
【請求項12】
前記化合物は経口的に投与される、請求項5乃至7のいずれか一項の使用。
【請求項13】
前記化合物は静脈内に投与される、請求項5乃至7のいずれか一項の使用。
【請求項14】
前記化合物は局所的に投与される、請求項5乃至7のいずれか一項の使用。
【請求項15】
患者のヘッジホッグ経路に対抗(アンタゴナイズ)するための薬物(medicament)の調製における式(1)の化合物又はその薬学的に許容可能な塩の使用であって、式(1)の該化合物は下記式を有し、
【化3】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、若しくは=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは、夫々独立してジラジカルであり;
各qは夫々独立して、1、2、3、4、5若しくは6であり;
X−はハライドであり;
各Rは、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくは、アラルキルであり;
各R5は、夫々独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)である。
【請求項16】
R2、R3及びR4がHである場合に、R1はヒドロキシル又は糖でもなく;及び
R4がヒドロキシルである場合に、R1は糖又はヒドロキシルでもなく;及び
R4がヒドロキシルである場合に、R1及びR2は一緒になってC=Oでない、請求項15の使用。
【請求項17】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項1、5又は15のいずれか一項の使用。
【化4】
【請求項18】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項4の使用。
【化5】
【請求項19】
前記化合物は、下記式の化合物からなる群から選択されるか、又は、それの薬学的に許容可能な塩である、請求項6乃至11のいずれか一項の使用。
【化6】
【請求項20】
患者のヘッジホッグ経路に対抗(アンタゴナイズ)するための薬物(medicament)の調製における式(2)の化合物又はその薬学的に許容可能な塩の使用であって、式(2)の該化合物は下記式を有し、
【化7】
式中、R1はH、アルキル、−OR、アミノ、スルホンアミド、スルファミド、−OC(O)R5、−N(R5)C(O)R5、若しくは糖であり;
R2はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ニトリル、若しくはヘテロシクロアルキルであり;
又は、R1とR2は一緒になって=O、=S、=N(OR)、=N(R)、=N(NR2)、=C(R)2を形成し;
R3はH、アルキル、アルケニル、若しくはアルキニルであり;
R4はH、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、ハロアルキル、−OR5、−C(O)R5、−CO2R5、−SO2R5、−C(O)N(R5)(R5)、−[C(R)2]q−R5、−[(W)−N(R)C(O)]qR5、−[(W)−C(O)]qR5、−[(W)−C(O)O]qR5、−[(W)−OC(O)]qR5、−[(W)−SO2]qR5、−[(W)−N(R5)SO2]qR5、−[(W)−C(O)N(R5)]qR5、−[(W)−O]qR5、−[(W)−N(R)]qR5、−W−NR53+X−若しくは−[(W)−S]qR5であり;
R4の上記式中、各Wは独立してジラジカルであり;
各qは独立して1、2、3、4、5若しくは6であり;
X−はハライドであり;
各R5は、独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、ヘテロシクロアルキル、アラルキル、ヘテロアリール、ヘテロアラルキル、若しくは−[C(R)2]p−R6(但し、pは0−6)であり;又は同一の置換基のR5の任意の2つは一緒になって、N、O、S及びPから選択される0−3へテロ原子を含有する4−8員の任意に置換された環を形成することができ;
各R6は、独立して、ヒドロキシル、−N(R)COR、−N(R)C(O)OR、−N(R)SO2R、−C(O)N(R)2、−OC(O)N(R)(R)、−SO2N(R)(R)、−N(R)(R)、−COOR、−C(O)N(OH)(R)、−OS(O)2OR、−S(O)2OR、−OP(O)(OR)(OR)、−NP(O)(OR)(OR)、若しくは−P(O)(OR)(OR)であり;
各Rは独立して、H、アルキル、アルケニル、アルキニル、アリール、シクロアルキル、若しくはアラルキルであり;
R7及びR7´の夫々がHであり;又はR7及びR7´が一緒になって=Oを形成し;
R8及びR9はHであるか、又は、R8及びR9は一緒になってボンドを形成し;及び
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1はヒドロキシルとなることはできず、かつR2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7及びR7´が一緒になって=Oを形成する場合;R1は酢酸となることはできず、かつR2はHとなることができず;
R3、R4、R8、R9がHであり、かつR7がH2である場合;R1及びR2が一緒になって=Oとなることができず;及び
R3、R4、R8、R9がHであり、かつR7及びR7´がHである場合;R1及びR2がHとなることができない。
【請求項21】
R1はスルホンアミドである、請求項20の使用。
【公表番号】特表2010−514797(P2010−514797A)
【公表日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願番号】特願2009−544277(P2009−544277)
【出願日】平成19年12月27日(2007.12.27)
【国際出願番号】PCT/US2007/088995
【国際公開番号】WO2008/083252
【国際公開日】平成20年7月10日(2008.7.10)
【出願人】(506418758)インフィニティ・ディスカバリー・インコーポレイテッド (13)
【氏名又は名称原語表記】INFINITY DISCOVERY, INC.
【出願人】(504457614)ジョンズ ホプキンス ユニバーシティ (2)
【Fターム(参考)】
【公表日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願日】平成19年12月27日(2007.12.27)
【国際出願番号】PCT/US2007/088995
【国際公開番号】WO2008/083252
【国際公開日】平成20年7月10日(2008.7.10)
【出願人】(506418758)インフィニティ・ディスカバリー・インコーポレイテッド (13)
【氏名又は名称原語表記】INFINITY DISCOVERY, INC.
【出願人】(504457614)ジョンズ ホプキンス ユニバーシティ (2)
【Fターム(参考)】
[ Back to top ]