
シグナルペプチド、それをコードするDNA配列、該配列を含む発現構築物、プラスミド及び微生物細胞並びに組み換えタンパク質の発酵的製造方法
【課題】新規のシグナルペプチドを提供する。
【解決手段】シグナルペプチドであって、切断部位前の最後の3つのアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチドによって解決される。
【解決手段】シグナルペプチドであって、切断部位前の最後の3つのアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチドによって解決される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、組み換えタンパク質の製造のためのシグナルペプチドに関する。
【背景技術】
【0002】
組み換えタンパク質の大容量の経済的な製造は、バイオテクノロジー産業と医薬品産業にとってますます重要となっている。一般に、組み換えタンパク質は、哺乳類細胞か又は微生物系のいずれかにおいて製造される。微生物系は、こうすることで組み換えタンパク質がより短時間でかつ少ないコストで製造できるという利点を、哺乳類細胞培養に対して有している。従って組み換えタンパク質の製造のためには、とりわけ細菌、有利にはエシェリキア属の細菌、特に有利にはE.コリが適している。E.コリでは、組み換えタンパク質は、原則的に、種々の様式で製造することができる:
1. 可溶性タンパク質として細胞内産生;
2. 封入体("インクルージョンボディー")として細胞内産生;
3. ペリプラズム又は栄養培地への分泌。
【0003】
組み換えタンパク質の製造方法は、その際常に、2つの部分に分かれる。第一の部分は、粗産物をもたらす発酵である。粗産物とは、この場合には、組み換えタンパク質と、更にくわえて不純物となる宿主固有のタンパク質とを含有する発酵成果物を指す。製造方法の第二の部分は、粗産物から出発する組み換えタンパク質の精製を含む。
【0004】
組み換えタンパク質の労力と費用は、発酵直後に組み換えタンパク質と並んで宿主タンパク質を含む混合物として存在する粗産物の製造コストの他に、実質的に、所望の組み換えタンパク質へと粗産物を精製するコストによっても決められる。精製は、たいていの場合に、クロマトグラフィー法による複数の段階を介して行われる。その際、部分的に免疫原となるか又は毒性がある不純物としての宿主タンパク質の除去精製が重要な役割を担う。
【0005】
E.コリにおけるタンパク質の分泌は、たいていの場合に、いわゆるsec経路(Driessen他(1998年))を介して行われる。この系は、細菌固有のタンパク質の排出を担っている。このタンパク質のための遺伝子は、5′末端に、それぞれいわゆるシグナル配列を有する。タンパク質合成において、該配列はシグナルペプチドに翻訳され、そして細胞質膜を通じたタンパク質の分泌をもたらす。分泌の後に、シグナルペプチドは、シグナルペプチダーゼといった酵素によって開裂され、成熟タンパク質が遊離される。
【0006】
sec系は、組換体、例えば異種タンパク質の分泌のためにも使用することができる(Lee他著、Methods in Molecular Biology 308,2005年)。そのために、産生されるべき組み換えタンパク質のための組み換え遺伝子を、シグナル配列と結合させる("読み枠内融合"(in frame Fusion))。これは、シグナルペプチド−タンパク質融合物の産生をもたらす。シグナル配列によってコードされるシグナルペプチドは、細胞質膜を通ってペリプラズムへの組み換えタンパク質の分泌を、細菌固有のsec系によって媒介する。その際、シグナルペプチドは、シグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中に得られる。その後に組み換えタンパク質は、ペリプラズムから精製することができる。
【0007】
分泌は、他の製造方法と比較して、組み換えタンパク質が、直接的に、本来の可溶性の正しく折り畳まれたタンパク質として得られ、"封入体"法に対して、収率の高い損失を伴う工程である変性と再度の復元を行う必要が無いという利点を有する。更に、この場合に粗産物は、細胞内の可溶性産物と比較して僅かな宿主タンパク質で汚染されているに過ぎない。それというのも細菌のペリプラズムは、細胞質よりもはるかに少ない宿主タンパク質しか含まないからである。
【0008】
規定の条件下で又は規定の細菌株において、組み換えタンパク質は、ペリプラズムから栄養培地中に遊離され(例えばRay他(2002年);EP0338410号B1;Nagahari他(1985年);Yang他(1998年);EP0677109号B1)、そしてそこから精製することができる。
【0009】
タンパク質の栄養培地中への分泌は、ペリプラズムへの分泌と比較して、タンパク質がその際にさらに純粋な形で存在するという更なる利点を有する。更に、最初の精製段階として、ペリプラズムの面倒な調製又は細胞の分解は必要なく、全細胞のはるかに容易な再現可能に行える分離が必要とされるだけである。
【0010】
言及したように、産生されるべきタンパク質の分泌のために、それをコードする遺伝子をシグナル配列と結合させるため、産生されるべきタンパク質はまずはシグナル配列によってコードされるシグナルペプチドとの融合物として産生されることとなる。このシグナルペプチドは、産生されるタンパク質の分泌をもたらす。
【0011】
シグナルペプチドは3つの領域から構成されている:N末端のN領域(1〜5アミノ酸)は、一般に、正電荷を有する1つ以上のアミノ酸を有する。中央に位置するH領域は、大抵は、多くが疎水性である7〜15個のアミノ酸からなる。一般に、3〜7個のアミノ酸を含むC領域は、切断部位前の位置−1と位置−3に、主に中性の短鎖アミノ酸(A,G,S,T,C)を有する。
【0012】
種々のシグナル配列と属するシグナルペプチドは、先行技術において記載されており、例えばphoA、ompA、pelB、ompF、ompT、lamB、malE、スタフィロコッカスのプロテインA、StII(ChoiとLee(2004年);EP0396612号B1)である。
【0013】
種々の菌株、例えばクレブシエラ・オキシトカ(Klebsiella oxytoca)(クレブシエラ・ニューモニアエ(Klebsiella pneumoniae)M5a1)由来のシクロデキストリン−グリコシルトランスフェラーゼ(CGTアーゼ)のシグナルペプチド並びにそれらのE.コリ菌株におけるCGTアーゼの分泌のための使用は、US5395927号に記載されている。
【0014】
同様に、組み換えタンパク質、例えばヒルジン誘導体を、E.コリ菌株中で、組み換えタンパク質のための遺伝子とCGTアーゼのシグナル配列とを融合させることで産生させ、分泌させることができることが記載されている(EP0448093号B1)。特定のヒルジン誘導体の場合には、振盪フラスコ培養において250mg/lの収率がもたらされ、発酵においては2.63g/lの収率がもたらされる。EP0448093号B1は、しかしながらまた、別の組み換えタンパク質では、25mg/lまでの収率しか得られないことも記載している。従って、CGTアーゼのシグナルペプチドは、全ての別の公知のシグナルペプチドも同様に、任意の組み換えタンパク質の分泌を同等の高さの収率で媒介することができない。各組み換えタンパク質は固有のDNA配列によってコードされ、かつ特にシグナル配列と組み換えタンパク質をコードする配列との間の接合部位でのDNA配列がそれと異なるので、一般に、各組み換えタンパク質について最適なシグナルペプチドを見出さねばならない。
【特許文献1】Lee他著、Methods in Molecular Biology 308,2005年
【特許文献2】EP0338410号B1
【特許文献3】EP0677109号B1
【特許文献4】EP0396612号B1
【特許文献5】US5395927号
【非特許文献1】EP0448093号B1
【発明の開示】
【発明が解決しようとする課題】
【0015】
本発明の課題は、新規のシグナルペプチドを提供することである。
【課題を解決するための手段】
【0016】
前記課題は、シグナルペプチドであって、切断部位前の最後の3つのアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチドによって解決される。
【0017】
有利には、該シグナルペプチドは、アミノ酸配列:MKRNRFFNTSAAIAISIALQIFFPSASAFA(配列番号1)を有するか、又は配列番号1と比較して、1〜10個の、有利には1〜5個の、特に有利には1〜3個のアミノ酸が、切断部位前の最後の3個のアミノ酸を除いて改変されているアミノ酸配列を有する。
【0018】
前記のように、シグナルペプチドは、規定の群のアミノ酸(例えば荷電した又は疎水性の又は短鎖の)を有する種々の領域からなる。従って、当業者は、アミノ酸を他の匹敵する特性を有するアミノ酸によって交換することによって、特性が変化していない新規のシグナルペプチドを作製することができる。
【0019】
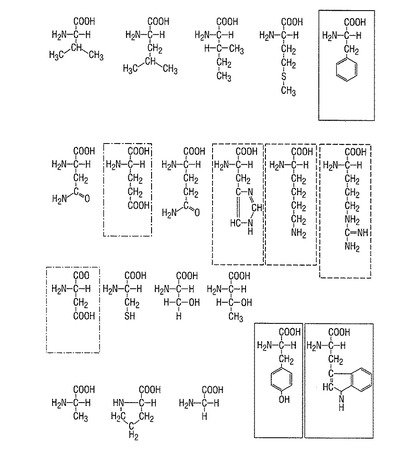
前記の理由から、本発明によるシグナルペプチドとしては、配列番号2と比較して、1〜10個の、特に有利には1〜5個の、特に有利には1〜3個のアミノ酸が改変されたものを察することができる。有利には、それは、類似の生化学的特性を有するアミノ酸、例えば塩基性アミノ酸(リジン、アルギニン、ヒスチジン)を、塩基性アミノ酸と交換すること、酸性アミノ酸(アスパラギン酸、グルタミン酸、アスパラギン、グルタミン)を酸性アミノ酸と交換すること、疎水性アミノ酸を疎水性アミノ酸と交換することなどである。アミノ酸の生化学的特性に関する概要を、図1に示す。
【0020】
更に本発明は、本発明によるシグナルペプチドをコードするシグナル配列に関する。
【0021】
該配列は、切断部位を有するシグナルペプチドをコードし、該切断部位前のその最後の3個のアミノ酸がアラニン−フェニルアラニン−アラニンであることを特徴とする。
【0022】
有利には、該配列は、DNA配列
【化1】

(配列番号2)並びに縮重された遺伝子コードに基づき配列番号1のアミノ酸配列をコードする全てのDNA配列を有するシグナル配列である。
【0023】
スクリーニングの範囲において、CGTアーゼのシグナル配列(MKRNRFFNTSAAIAISIALNTFFCSMQTIA、配列番号3)の改変によって得られる種々のシグナル配列を、その特性に関して比較した。驚くべきことに、本発明によるシグナル配列は、CGTアーゼのシグナル配列又は他のシグナル配列よりも、より広範囲の組み換えタンパク質をより高収率で宿主細胞中で産生させ、分泌させるために適していることが判明した。
【0024】
本発明によるDNA配列は、遺伝子合成によって又は相応のオリゴヌクレオチドのライゲーションによって、当業者に公知の方法によって得ることができる。本発明によるDNA配列は、読み枠内で(in frame)、当業者に公知の方法(例えばLee他(2005年)と同様に)に従って、産生されるべき組み換えタンパク質の遺伝子と結合され、それをベクター中に導入することができる。
【0025】
有利には、このシグナル配列と組み換え遺伝子との組合せ物は、E.コリにおいて機能的な発現シグナル(プロモーター、転写開始部位、翻訳開始部位、リボソーム結合部位)を備える。プロモーターとしては、当業者に公知のあらゆるプロモーター、例えば一方では誘導可能なプロモーター、例えばlacプロモーター、tacプロモーター、trcプロモーター、λPLプロモーター、araプロモーター又はtetプロモーター又はそれらから誘導される配列が適している。他方では、構成的プロモーター、例えばGAPDHプロモーターを使用することによって構成的な発現を行うこともできる。しかしながら、通常は、産生されるべき組み換えタンパク質の遺伝子と結合されたプロモーターを使用することもできる。
【0026】
従って、本発明は、発現シグナルと、本発明によるシグナル配列と、産生されるべき組み換えタンパク質をコードする読み枠内で結合された組み換え遺伝子とを含む発現構築物にも関する。
【0027】
この本発明による発現構築物は、当業者に公知の方法を使用して宿主細胞に導入される。それは、例えばベクター上で、例えばプラスミド上で、例えば公知の発現ベクターの誘導体、例えばpUC18、pBR322、pACYC184、pASK−IBA3又はpET上で行われる。プラスミドのための選択マーカーとしては、例えばアンピシリン、テトラサイクリン、クロラムフェニコール、カナマイシン又は別の抗生物質に対する耐性をコードする遺伝子が適している。
【0028】
従って、本発明によるシグナル配列又は本発明による発現構築物を有するプラスミドも本発明の対象である。
【0029】
組み換えタンパク質は、有利には異種タンパク質である。有利には、組み換えタンパク質は、工業的アプローチで使用されるタンパク質又は医薬品作用物質として使用されるタンパク質(バイオロジクス、生体医薬品)である。係るタンパク質は、例えばヒルジン、インスリン、インターフェロン、例えばα−インターフェロン又はβ−インターフェロン(例えばインターフェロンα2b)、抗体もしくは抗体フラグメント(例えばFabフラグメント、scFv)又は別の結合タンパク質もしくは酵素、例えばCGTアーゼである。
【0030】
本発明による発現構築物は、当業者に公知の方法に従って微生物細胞(宿主細胞)中に導入される。結果として、本発明による発現構築物は、更にプラスミドとして宿主細胞中に存在しても、又は宿主細胞の染色体中に組み込まれてもよい。
【0031】
従って、本発明によるシグナル配列又は本発明による発現構築物又は本発明によるプラスミドを有する微生物細胞も本発明の対象である。
【0032】
宿主細胞は、腸内細菌科からの細菌株の細胞、有利にはエシェリキア・コリの種の菌株である。特に、エシェリキア(E.)・コリ菌株であって、本発明による発現構築物での形質転換後に発酵によって、本発明による発現構築物で形質転換されたE.コリW3110(ATCC27325)よりも高濃度の組み換えタンパク質をペリプラズム中もしくは栄養培地中に示すという点で優れているE.コリ菌株が好ましい。
【0033】
殊に好ましくは、その菌株は、以下のE.コリ菌株:
− BLR:Ray他(2002年)、Novagenで購入できる
− K802=CGSC*5610:Yang他(1998年)
− WCM105:EP0338410号B1に従って製造できる
− MM28=CGSC*#5892:Nagahari他(1985年)
− RV308=ATCC**31608;EP0677109号B1
− RR1:ATCC**31434:Nagahari他(1985年)
である。
* 大腸菌ストックセンター(E.coli Genetic Stock Center)CGSC(830 Kline Biology Tower,MCD Biology Department,266 Whitney Ave.,PO box 208103,Yale University,New Haven)から購入できる
** LGC Promochem(Mercatorstr.51,46485 Wesel,ドイツ在)から購入できる
産生されたタンパク質の分泌は、宿主細胞のsec装置によって行われる。引き続きペリプラズムへの分泌の後に、シグナルペプチダーゼ(例えばE.コリではLepB)によって本発明によるシグナルペプチドは開裂されて、所望の組み換えタンパク質が生ずる。
【0034】
従って、本発明は、本発明による発現構築物を有する宿主細胞によって発酵培地中で組み換えタンパク質を発酵的に製造するための方法にも関する。前記方法は、本発明による宿主菌株を発酵培地中で培養し、該宿主菌株が組み換えタンパク質を読み枠内でのシグナルペプチド−タンパク質融合物の形で産生し、その際、該シグナルペプチドは、本発明によるシグナルペプチドでありかつ該シグナルペプチドは、細胞質膜を通じてペリプラズム中にシグナルペプチド−タンパク質融合物を分泌した場合にシグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中もしくは発酵培地中で得られ、その組み換えタンパク質を発酵後に精製することを特徴としている。
【0035】
組み換えタンパク質は、発酵において、ペリプラズム中に又は有利には発酵培地中に分泌される。
【0036】
組み換えタンパク質は、宿主細胞のペリプラズムからか又は有利には発酵培地から細胞の分離によって精製することができる。
【0037】
組み換えタンパク質の本発明による製造のための微生物菌株の発酵は、有利には完全培地又は最少塩培地中で行われる。これらの培地は、文献から公知である。
【0038】
炭素源としては、原則的に、あらゆる使用可能な糖、糖アルコール、有機酸もしくはそれらの塩、デンプン加水分解物、蜜ろう又は別の物質を使用することができる。その際、グルコース又はグリセリンを使用することが好ましい。また複数の種々異なる炭素源を組み合わせて供給することも可能である。窒素源としては、尿素、アンモニア及びそれらの塩、硝酸塩並びに他のN源を用いることができる。可能な窒素源には、複合アミノ酸混合物、例えば酵母エキス、ペプトン、麦芽エキス、大豆ペプトン、カザミノ酸、コーンスティープリカー及びNZアミン(例えばKerry Bio−Science,Chicago,USA)も該当する。
【0039】
更に、該培地に、ビタミン、塩、酵母エキス、アミノ酸及び微量元素のような他の成分を添加してよく、それによって細胞増殖が向上する。
【0040】
菌株のインキュベートは、有利には好気的培養条件下で、16〜150時間の時間にわたって、かつその都度の菌株に最適な増殖温度の範囲で行われる。
【0041】
最適な温度範囲としては、15〜55℃が好ましい。特に、28〜37℃の温度が好ましい。
【0042】
菌株の培養は、振盪フラスコ又は発酵器において実施することができ、その際、容量に関する制限は与えられていない。その培養は、回分法、流加法又は連続法で実施することができる。
【0043】
その際、組み換えタンパク質の発現は、構成的に、すなわち非誘導的にも、又は物理的もしくは生理学的な刺激による誘導によって行われる。発現は、例えばプロモーターを誘導する物質、例えばlacプロモーターもしくはtacプロモーターの場合にはラクトースもしくはIPTGの添加によって誘導することができる。
【0044】
ペリプラズムもしくは培養培地からのタンパク質の精製は、当業者に公知の方法に従って、例えば細胞の破壊又は分離、クロマトグラフィー的な精製、タンパク質の複合体化、濾過又は沈降によって行うことができる。
【0045】
次の実施例は、本発明を更に説明するために用いられるものである。
【実施例】
【0046】
実施例1:本発明によるシグナル配列及び本発明によるベクターの作製
出発プラスミドとして、プラスミドpCGTを以下のように作製する:
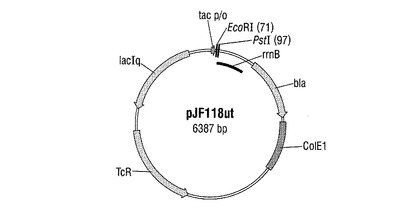
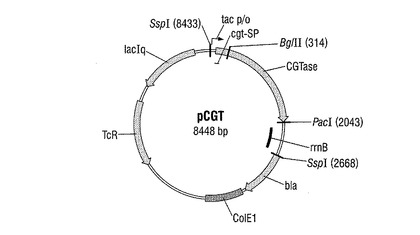
配列番号4を有し、クレブシエラ・ニューモニアエM5a1由来のシクロデキストリン−グリコシルトランスフェラーゼ(CGTアーゼ)遺伝子を有するDNA断片(Genbank番号M15264)を、遺伝子合成によって製造した。このDNA断片を、発現ベクターpJF118ut(図2)中にクローニングした。該ベクターは、DSMZ−ドイツ微生物細胞培養収集館(Braunschweig)で番号DSM18596として寄託されている。pJF118utは、公知の発現ベクターpKK223−3(Amersham Pharmacia Biotech)の誘導体であり、β−ラクタマーゼ遺伝子とテトラサイクリン耐性遺伝子の他に、さらに同様にプラスミド上に存在するLacIq遺伝子産物によって再始動されかつ例えばD−ラクトースもしくはイソプロピル−β−D−チオガラクトピラノシド(IPTG)のようなインデューサーによってスイッチオンすることができるtacプロモーターをも有する。プラスミドpJF118utを、制限酵素EcoRIで完全に切断し、かつそれぞれ直鎖状DNA断片の5′末端で突出した塩基を、S1ヌクレアーゼで分解した。前記のようにして予備調製されたベクターDNA分子を、CGTアーゼを含むDNA断片(配列番号4)とT4−リガーゼを使用してライゲーションした。菌株DH5αを、ライゲーションバッチでCaCl2法に従って形質転換し、その際、アンピシリン(100mg/l)によってプラスミド保有細胞について選択した。アンピシリン耐性形質転換体から、再びプラスミドを単離し、そして制限分析によって調査した。前記のようにして作製された、CGTアーゼ遺伝子の発現がtacプロモーターの制御下にあるプラスミドを、pCGTと呼称した(図3)。
【0047】
CGTアーゼのためのシグナル配列に融合されたCGTアーゼのための遺伝子を除去した:そのために8448bpの大きさのプラスミドを、制限酵素SspI及びPacIによって部分消化によって当業者に公知の方法に従って切断した。6390bpの大きさの断片を単離し、そしてクレノウ酵素で処理し、それによって末端を平滑化させた。2058bpの大きさの断片を除去した。
【0048】
次いで、以下の4つのDNA断片を遺伝子合成によって製造した:
phoA−IFNα2b 配列番号5
【化2】
ompA−IFNα2b 配列番号6
【化3】
cgt−IFNα2b 配列番号7
【化4】
AFA−IFNα2b 配列番号8
【化5】
【0049】
全てのDNA断片は、tacプロモーター領域と、インターフェロンα2bのための遺伝子と、4つの種々のシグナル配列(太字で示した)とを有する。これらの4つの種々のシグナル配列は、以下の4つの種々のシグナルペプチドをコードし、その際、最初の3つのシグナルペプチド(配列番号9、10、3)は、先行技術から公知であり、四番目のシグナルペプチド(配列番号2)は、本発明によるものである:
phoA: MKQSTIALALLPLLFTPVTKA
配列番号9
ompA: MKKTAIAIAVALAGFATVAQA
配列番号10
cgt: MKRNRFFNTSAAIAISIALNTFFCSMQTIA
配列番号3
AFA: MKRNRFFNTSAAIAISIALQIFFPSASAFA
配列番号2
前記のクローニングによって以下の4つのプラスミドが得られた:
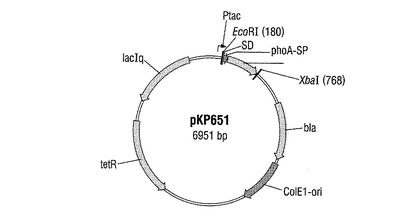
− pKP651(phoA−シグナル配列) 図4
− pKP652(ompA−シグナル配列) 図5
− pKPIFN(cgt−シグナル配列) 図6
− pBaBIFN1(AFA:本発明によるシグナル配列) 図7
これらのプラスミドを、公知の方法によってE.コリ菌株DH5αに導入した。本発明による基株DH5α/pBaBIFN1は、DSMZ(ドイツ微生物細胞培養収集館、D−38142 Braunschweig)で番号DSM18343としてブタペスト条約に従って寄託された。
【0050】
実施例2:本発明によるシグナル配列を使用することによるインターフェロン産生の増大
プラスミドpKP651(phoA−シグナル配列)、pKP652(ompA−シグナル配列)、pKPIFN(cgt−シグナル配列)及びpBaBIFN1(本発明によるAFA−シグナル配列)(実施例1を参照のこと)を、菌株WCM105(EP0338410号B1に従って製造できる)中に通常の方法(CaCl2−形質転換によって)に従って導入した。プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。
【0051】
以下の菌株が得られた:
− WCM105/pKP651(phoA−シグナル配列)
− WCM105/pKP652(ompA−シグナル配列)
− WCM105/pKPIFN(cgt−シグナル配列)
− WCM105/pBaBIFN1(AFA−シグナル配列)
得られた菌株中でのインターフェロンα2bの産生を調査した。そのために、それらの菌株を、100mg/Lのアンピシリンと1%のグルコースとを有する10mlのLB培地中で30℃において培養した。600nmでの光学密度(OD600)が0.5になったら、インターフェロンα2bの産生を、IPTG(イソプロピルチオガラクトシド)を0.5mMまで添加することによって誘導する。培養上清において、24時間後、48時間後及び72時間後で、形成されかつ分泌されたインターフェロンを、SDSゲル中でのタンパク質の分離と抗インターフェロン特異抗体でのイムノブロットでの検出によって以下のようにして定量化した。
【0052】
それぞれ1μl(48時間及び72時間)もしくは5μl(24時間)の上清を、サンプルバッファーと混合した(2×Tris SDS−サンプルバッファー(Invitrogen カタログ番号LC2676):0.125Mのトリス塩酸(pH6.8)、4%(w/v)のSDS、20%(v/v)のグリセリン、0.005%(v/v)のブロモフェノールブルー、5%のβ−メルカプトエタノール)。更に、定義された量のインターフェロンα2bを、スタンダードとして一緒に施与した。タンパク質の変性を、100℃に5分間加熱し、氷上で2分間冷却し、そして遠心分離することによって実施した。それらのタンパク質を、電気泳動によって、12%のNuPAGE(登録商標)Bis−Tris−Gel(Invitrogen カタログ番号NP0341)中で、1×MES含有ランニングバッファー(Invitrogen カタログ番号NP0002)を用いて分離させた(電気泳動パラメータ:200Vで40分間)。イムノブロットによる検出と定量化を、以下の仕様に従って実施した:
【0053】
湿式ブロッティング法での転写:
モジュール:Amersham:Hoefer TE 22 Mini Tank Transfer Unit、コード番号:80−6204−26。
【0054】
メンブレン:ニトロセルロースメンブレン(Schleicher&Schuell,BA85,硝酸セルロース(E),0.45μmの細孔サイズ)
Whatmanフィルタとニトロセルロースメンブレンを、適切な大きさに切断し、そして発泡物品(スポンジ)で転写バッファー(Invitrogen カタログ番号LC3675)に気泡なく染み込ませた。
【0055】
積層の構成:黒い格子、カソードとの接続、各3mm厚を有する2つのスポンジ、Whatmanペーパー、SDSポリアクリルアミドゲル、NCメンブレン、Whatman、6mm厚を有する1つのスポンジ、白い格子、アノードとの接続
転写条件:I=200mAの一定の電流、U=無制限、運転時間60分
【0056】
プレハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー中で該メンブレンをインキュベートする。
室温で30分間振り動かす。
【0057】
一次抗体のハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー+0.15μg/ml(→3.75μg)の抗ヒトIFNΑ抗体(Pepro Tech EC,Biozolを経由した カタログ番号:500−P32A)中で該メンブレンをインキュベートする。
【0058】
室温で90分間又は一晩振り動かす。
【0059】
洗浄
1×PBSと一緒に室温で10秒間振り動かし、バッファーを捨てる。
1×PBSと一緒に室温で15分間2回振り動かし、バッファーを捨てる。
【0060】
二次抗体のハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー+25μl(1:1000)のヤギ抗ウサギIgGセイヨウワサビペルオキシダーゼコンジュゲート(HRP)(Southern Biotech、Biozolを経由した カタログ番号4050−05)中で該メンブレンをインキュベートする。
【0061】
室温で60分間振り動かす。
【0062】
洗浄
1×PBSと一緒に室温で10秒間振り動かし、バッファーを捨てる。
1×PBSと一緒に室温で15分間2回振り動かし、バッファーを捨てる。
【0063】
化学発光による検出
Lumi−Lightウェスタンブロッティング基質(Roche,カタログ番号2015200)を準備する:Lumi−Lightルミノール/エンハンサー溶液とLumi−Light安定ペルオキシド溶液とを1:1の比率で混合する:NCメンブレン当たり3ml。
【0064】
ブロットを室温でLumi−Lightウェスタンブロッティング基質と一緒に5分間インキュベートし、過剰の水気を切り、メンブレンをラップフィルムで覆って、直ちにX線フィルム(Kodak、X−OMAT)を載せ、2分間暴露し、現像し、そして固定する。シグナルが弱い場合に、暴露をより長時間にわたり繰り返す。
【0065】
バッファー
プレハイブリダイゼーションバッファー:1×PBS中の5%脱脂粉乳
10×PBS:100mMのNaH2PO4、1.5MのNaCl、NaOHでpH7.5、0.5%のTriton100
1×PBS:10×PBSを完全脱塩水で1:10希釈したもの
【0066】
定量化
定量的評価は、Biorad社製のGS−800 Calibrated Densitometerでイムノブロットをスキャンすることによって、Quantity One 1−D分析ソフト(Biorad)を用いて、施与されたスタンダードと比較することによって実施した。
【0067】
図9は、前記の実施例からのイムノブロットを示している。第1表において、定量化されたインターフェロンα2bの収率をまとめる:
【表1】
【0068】
第1表:それぞれ使用されたシグナル配列の点で異なる種々のプラスミドを用いて24時間後、48時間後又は72時間後に得られたインターフェロンα2bの収率
これらの結果は、配列番号2の本発明によるシグナル配列が、産生されるべきタンパク質の収率及び分泌に関して、別のシグナル配列より優れていることを一義的に示している。
【0069】
実施例3:本発明によるシグナル配列の挿入によって改善されたCGTアーゼ産生プラスミドの作製
プラスミドpCGT(実施例1を参照)は、CGTアーゼのためのシグナル配列と読み枠内で融合されたCGTアーゼのための遺伝子を有する。このシグナル配列を、ここで本発明によるシグナル配列と交換した。
【0070】
そのために8448bpの大きさのプラスミドを、制限酵素SspI及びBglIIによって部分消化によって当業者に公知の方法に従って切断した。8119bpの大きさの断片を単離し、そしてクレノウ酵素で処理し、それによって末端を平滑化させた。CGTアーゼのシグナル配列と、約150bpのCGTアーゼ遺伝子の5末端とを有する329bpの大きさの断片を除去した。
【0071】
前記の断片と配列に関して、CGTアーゼのシグナル配列が本発明によるシグナル配列と交換されていること以外は同一である以下の329bpの大きさのDNA断片を遺伝子合成によって製造した:
【化6】
【0072】
このDNA断片を、単離された8119bpの大きさの断片と、当業者に公知の方法に従ってライゲーションした。それによって得られたプラスミドpCM207AFAを、配列決定によって配列の精度について調査した。
【0073】
実施例4:本発明によるシグナル配列を使用することによるCGTアーゼ産生の改善
プラスミドpCM703AFAとプラスミドpCGT(実施例3を参照)を、以下の菌株に通常の方法に従って(例えばCaCl2法によって)形質転換により導入した:
− K802=CGSC*5610:Yang他(1998年)
− WCM105:EP0338410号B1に従って製造できる
プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。それによって以下の本発明による菌株:
− WCM105/pCM703AFA
− K802/pCM703AFA
及び以下の対照菌株:
− WCM105/pCGT
− K802/pCGT
が得られた。
【0074】
これらの菌株を、シクロデキストリン−グリコシルトランスフェラーゼの製造のために使用し、そして1%のグルコースと100mg/Lのアンピシリンを有する10mlのLB培地中で30℃において培養した。ODが0.5となったら、シクロデキストリン−グリコシルトランスフェラーゼの産生を、IPTG(イソプロピルチオガラクトシド)を0.5mMまで添加することによって誘導した。
【0075】
該菌株の培養上清において、シクロデキストリン−グリコシルトランスフェラーゼの収率を以下の活性試験によって測定した:
試験バッファー:5mMのトリス塩酸バッファー(>pH6.5)、5mMのCaSO4・2H2O
基質:試験バッファー(pH6.5)中の10%のNoredux溶液
試験バッチ:1mlの基質溶液+1mlの遠心分離された培養上清(5分、12000rpm)+3mlのメタノール
反応温度:40℃
酵素試験:
・ 溶液を予熱する(約5分、40℃で)。
【0076】
・ 酵素溶液を基質溶液に添加する;迅速に混合する(渦ミキサ(Whirl−Mixer))。
【0077】
・ 40℃で3分間インキュベートする。
【0078】
・ 酵素反応をメタノールの添加によって停止させる;迅速に混合する(渦ミキサ)。
【0079】
・ バッチを氷上で冷却する(約5分間)。
【0080】
・ 遠心分離(5分、12000rpm)をし、そして澄明な上清をピペットで取り出す。
【0081】
・ 生じたCDをHPLCによって分析する。
【0082】
酵素活性:A=G*V1*V2/(t*MG)(ユニット/ml)
A=活性
G=CDの含有率(mg/l)=試験バッチ:単位表面×104/標準溶液(10mg/ml)/単位表面
V1=希釈係数/試験バッチ(→5)
V2=希釈係数/酵素溶液
t=反応時間(分)(→3)
MG=分子量(g/モル)(CD→973)
1ユニット=1マイクロモルの産物/分
第2表は、本発明による菌株の高められた特異的なシクロデキストリン−グリコシルトランスフェラーゼ収率を示している。
【0083】
【表2】
【0084】
第2表:種々の菌株におけるシクロデキストリン−グリコシルトランスフェラーゼの収率
実施例5:本発明によるシグナル配列を使用することによるヒルジン産生の改善
特許EP0448093号B1に記載されるプラスミドpCMT203を、使用されるシグナル配列を本発明によるシグナル配列AFAと交換することで改変させた。この交換は、実施例1及び実施例3と同様に実施した。得られたプラスミドをpCMT203AFAと呼称した。pCMT203及びpCMT203AFAを、菌株WCM105(EP0338410号B1に従って製造できる)中に、通常の方法による形質転換によって(例えばCaCl2−形質転換によって)導入した。
【0085】
プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。
【0086】
以下の菌株が得られた:
− WCM105/pCMT203
− WCM105/pCMT203AFA
両方の菌株を、EP0448093号B1に記載されるように、10lの発酵器中で培養し、そして得られたヒルジンを、EP0448093号B1に記載されるように、IPTGの添加45時間後に定量化した。
【0087】
第3表中の結果は、本発明によるシグナル配列を使用することでヒルジンの高められた収率がもたらされることを示している。
【0088】
【表3】
【0089】
第3表:種々の菌株における、IPTGの添加45時間後での10lの発酵におけるヒルジンの収率(AT−U/ml及びg/L)
実施例6:本発明によるシグナル配列を使用することによる、機能的なFab抗体フラグメントの産生の改善
本実施例は、良好に特徴付けられた抗リゾチーム抗体D1.3のFabフラグメントの改善された製造を記載する。
【0090】
抗リゾチームFabフラグメントの遺伝子のクローニングと発現のための出発ベクターとして、プラスミドpJF118ut(実施例1を参照)を使用した。このプラスミド中に、2つの連続した工程において、抗リゾチームFabフラグメントの重鎖(VH−CH1−ドメイン)もしくは軽鎖(VL−CL−ドメイン)についての両方の読み枠を、それぞれシグナル配列を含めてクローニングした。そのために以下のように実施した:
配列番号12を有するDNA断片(重鎖)を、遺伝子合成によって製造し、そして該DNA断片は、E.コリのompA遺伝子のシグナル配列と、Fabフラグメントの重鎖(VH−CH1)のための読み枠とからなる遺伝子融合物を含む。この読み枠直後に6個のヒスチジンコドンが引き続き、従って融合タンパク質のC末端を形成している。このヒスチジンタグによって、後に完全に会合されたFabフラグメントの簡単な精製が親和性クロマトグラフィーによって可能となる。このDNA断片を、制限酵素EcoRI及びPstIで切断し、そして同じ制限酵素で切断されている発現ベクターpJF118utとライゲーションした。このクローニングから得られた、重鎖のための遺伝子の発現がtacプロモーターの制御下にあるプラスミドを、pHC−Anti−Lysozymと呼称した。
【0091】
配列番号13を有するDNA断片(軽鎖)を、同様に遺伝子合成によって製造し、そして該DNA断片は、配列番号3に記載されるCGTアーゼのシグナルペプチドをコードするDNA配列(配列番号7において太字で示した)と、Fabフラグメントの軽鎖(VL−CL)のための読み枠とからなる遺伝子融合物を含む。このDNA断片を、まず、制限酵素PstIで切断し、引き続き同じ制限酵素で切断されているベクターpHC−Anti−Lysozymとライゲーションした。こうして得られたプラスミドを、pFab−Anti−Lysozym(図9)と呼称した。前記のようにして、重鎖と軽鎖についてのそれぞれの読み枠からなる、tacプロモーターの制御下にある人工オペロンを作製した。従って、インデューサー(例えばIPTG)の添加によって、両方の遺伝子の同期発現が可能である。
【0092】
本発明によるプラスミドpFab−Anti−LysozymVLAFA及びpFab−Anti−LysozymVHAFAの製造のために、実施例1に記載されるのと同様にして、軽鎖のためのシグナル配列(pFab−Anti−LysozymVLAFA)か又は重鎖のためのシグナル配列(pFab−Anti−LysozymLHAFA)かのいずれかを、配列番号2の本発明によるシグナル配列と交換した。
【0093】
抗リゾチームFabフラグメントの製造ために、菌株WCM105(実施例4を参照)を、プラスミドpFab−Anti−Lysozym、pFab−Anti−LysozymVLAFAもしくはpFab−Anti−LysozymVHAFAで、CaCl2法に従って形質転換させた。プラスミド保有細胞についての選択は、アンピシリン(100mg/l)によって実施した。
【0094】
抗リゾチームFabフラグメントの製造を、10l規模で実施した。製造は、10lの撹拌槽型発酵器中で実施した。6lの培地FM4(1.5g/lのKH2PO4;5g/lの(NH4)2SO4;0.3g/lのMgSO4×7H2O;0.05g/lのCaCl2×2H2O、0.075g/lのFeSO4×7H2O;1g/lのNa3クエン酸塩×2H2O;0.5g/lのNaCl;1ml/lの微量元素溶液(0.15g/lのNa2MoO4×2H2O;2.5g/lのNa3BO3;0.7g/lのCoCl2×6H2O;0.25g/lのCuSO4×5H2O;1.6g/lのMnCl2×4H2O;0.3g/lのZnSO4×7H2O);5mg/lのビタミンB1;3g/lのフィトン;1.5g/lの酵母エキス;10g/lのグルコース;100mg/lのアンピシリン)で満たされた発酵器に、1:10の比率で、一晩同じ培地中で培養された前培養を植えた。発酵の間に、30℃の温度に調整し、そしてpH値をNH4OHもしくはH3PO4を計量供給することによって7.0の値に一定に保った。グルコースは、発酵にわたって計量供給され、その際、培地中の最大グルコース濃度を10g/l未満にすることに努めた。発現の誘導は、対数増殖期の終わりにイソプロピル−β−D−チオガラクトピラノシド(IPTG)を0.1mMで添加することによって実施した。
【0095】
72時間の発酵後に、試料を取りだし、細胞を培養培地の遠心分離によって分離した。培養上清からの抗リゾチームFabフラグメントの精製は、Skerra(1994年、Gene 141,79−84)に記載される親和性クロマトグラフィーによって実施した。
【0096】
精製された抗リゾチームFabフラグメントの定量化並びに活性の測定は、抗原としてリゾチームを用いたELISA試験(Skerra,1994年、Gene 141,79−84)によって実施した。
【0097】
第4表において、それぞれ20mlの培養上清から72時間の発酵後に単離することができた機能的な抗リゾチームFabフラグメントの収率を列記する。
【0098】
第4表:72時間の発酵後の培養上清中での抗リゾチームFabフラグメントの収率
【表4】
【図面の簡単な説明】
【0099】
【図1】図1は、アミノ酸の概要を示している。
【図2】図2は、プラスミドpJF118utのプラスミド地図を示している。
【図3】図3は、プラスミドpCGTのプラスミド地図を示している。
【図4】図4は、プラスミドpKP651のプラスミド地図を示している。
【図5】図5は、プラスミドpKP652のプラスミド地図を示している。
【図6】図6は、プラスミドpKPIFNのプラスミド地図を示している。
【図7】図7は、プラスミドpBaBIFN1のプラスミド地図を示している。
【図8】図8は、プラスミドpFab−Anti−Lysozymのプラスミド地図を示している。
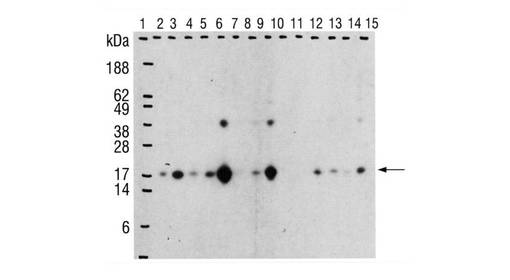
【図9】図9は、インターフェロンα2bを産生する細胞(実施例2を参照)の培養上清が分析されるイムノブロットを示している。 レーン1:サイズ基準 レーン2とレーン3:20ngと40ngのインターフェロンα2b レーン4〜6:24時間後、48時間後、72時間後の、プラスミドpBaBIFN1を有する培養の上清 レーン7〜9:24時間後、48時間後、72時間後の、プラスミドpKPIFNを有する培養の上清 レーン10〜12:24時間後、48時間後、72時間後の、プラスミドpKP651を有する培養の上清 レーン13〜15:24時間後、48時間後、72時間後の、プラスミドpKP652を有する培養の上清 24時間の場合に、それぞれ5μlが施与され、48時間及び72時間の場合に、1μlが施与された。 それらの細胞は、シグナル配列において相違するインターフェロンα2b−発現プラスミドを有する。形成されたインターフェロンα2bを、抗体によって検出した(矢印)。本発明によるシグナル配列の使用は、インターフェロンα2b産生の向上をもたらす。
【技術分野】
【0001】
本発明は、組み換えタンパク質の製造のためのシグナルペプチドに関する。
【背景技術】
【0002】
組み換えタンパク質の大容量の経済的な製造は、バイオテクノロジー産業と医薬品産業にとってますます重要となっている。一般に、組み換えタンパク質は、哺乳類細胞か又は微生物系のいずれかにおいて製造される。微生物系は、こうすることで組み換えタンパク質がより短時間でかつ少ないコストで製造できるという利点を、哺乳類細胞培養に対して有している。従って組み換えタンパク質の製造のためには、とりわけ細菌、有利にはエシェリキア属の細菌、特に有利にはE.コリが適している。E.コリでは、組み換えタンパク質は、原則的に、種々の様式で製造することができる:
1. 可溶性タンパク質として細胞内産生;
2. 封入体("インクルージョンボディー")として細胞内産生;
3. ペリプラズム又は栄養培地への分泌。
【0003】
組み換えタンパク質の製造方法は、その際常に、2つの部分に分かれる。第一の部分は、粗産物をもたらす発酵である。粗産物とは、この場合には、組み換えタンパク質と、更にくわえて不純物となる宿主固有のタンパク質とを含有する発酵成果物を指す。製造方法の第二の部分は、粗産物から出発する組み換えタンパク質の精製を含む。
【0004】
組み換えタンパク質の労力と費用は、発酵直後に組み換えタンパク質と並んで宿主タンパク質を含む混合物として存在する粗産物の製造コストの他に、実質的に、所望の組み換えタンパク質へと粗産物を精製するコストによっても決められる。精製は、たいていの場合に、クロマトグラフィー法による複数の段階を介して行われる。その際、部分的に免疫原となるか又は毒性がある不純物としての宿主タンパク質の除去精製が重要な役割を担う。
【0005】
E.コリにおけるタンパク質の分泌は、たいていの場合に、いわゆるsec経路(Driessen他(1998年))を介して行われる。この系は、細菌固有のタンパク質の排出を担っている。このタンパク質のための遺伝子は、5′末端に、それぞれいわゆるシグナル配列を有する。タンパク質合成において、該配列はシグナルペプチドに翻訳され、そして細胞質膜を通じたタンパク質の分泌をもたらす。分泌の後に、シグナルペプチドは、シグナルペプチダーゼといった酵素によって開裂され、成熟タンパク質が遊離される。
【0006】
sec系は、組換体、例えば異種タンパク質の分泌のためにも使用することができる(Lee他著、Methods in Molecular Biology 308,2005年)。そのために、産生されるべき組み換えタンパク質のための組み換え遺伝子を、シグナル配列と結合させる("読み枠内融合"(in frame Fusion))。これは、シグナルペプチド−タンパク質融合物の産生をもたらす。シグナル配列によってコードされるシグナルペプチドは、細胞質膜を通ってペリプラズムへの組み換えタンパク質の分泌を、細菌固有のsec系によって媒介する。その際、シグナルペプチドは、シグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中に得られる。その後に組み換えタンパク質は、ペリプラズムから精製することができる。
【0007】
分泌は、他の製造方法と比較して、組み換えタンパク質が、直接的に、本来の可溶性の正しく折り畳まれたタンパク質として得られ、"封入体"法に対して、収率の高い損失を伴う工程である変性と再度の復元を行う必要が無いという利点を有する。更に、この場合に粗産物は、細胞内の可溶性産物と比較して僅かな宿主タンパク質で汚染されているに過ぎない。それというのも細菌のペリプラズムは、細胞質よりもはるかに少ない宿主タンパク質しか含まないからである。
【0008】
規定の条件下で又は規定の細菌株において、組み換えタンパク質は、ペリプラズムから栄養培地中に遊離され(例えばRay他(2002年);EP0338410号B1;Nagahari他(1985年);Yang他(1998年);EP0677109号B1)、そしてそこから精製することができる。
【0009】
タンパク質の栄養培地中への分泌は、ペリプラズムへの分泌と比較して、タンパク質がその際にさらに純粋な形で存在するという更なる利点を有する。更に、最初の精製段階として、ペリプラズムの面倒な調製又は細胞の分解は必要なく、全細胞のはるかに容易な再現可能に行える分離が必要とされるだけである。
【0010】
言及したように、産生されるべきタンパク質の分泌のために、それをコードする遺伝子をシグナル配列と結合させるため、産生されるべきタンパク質はまずはシグナル配列によってコードされるシグナルペプチドとの融合物として産生されることとなる。このシグナルペプチドは、産生されるタンパク質の分泌をもたらす。
【0011】
シグナルペプチドは3つの領域から構成されている:N末端のN領域(1〜5アミノ酸)は、一般に、正電荷を有する1つ以上のアミノ酸を有する。中央に位置するH領域は、大抵は、多くが疎水性である7〜15個のアミノ酸からなる。一般に、3〜7個のアミノ酸を含むC領域は、切断部位前の位置−1と位置−3に、主に中性の短鎖アミノ酸(A,G,S,T,C)を有する。
【0012】
種々のシグナル配列と属するシグナルペプチドは、先行技術において記載されており、例えばphoA、ompA、pelB、ompF、ompT、lamB、malE、スタフィロコッカスのプロテインA、StII(ChoiとLee(2004年);EP0396612号B1)である。
【0013】
種々の菌株、例えばクレブシエラ・オキシトカ(Klebsiella oxytoca)(クレブシエラ・ニューモニアエ(Klebsiella pneumoniae)M5a1)由来のシクロデキストリン−グリコシルトランスフェラーゼ(CGTアーゼ)のシグナルペプチド並びにそれらのE.コリ菌株におけるCGTアーゼの分泌のための使用は、US5395927号に記載されている。
【0014】
同様に、組み換えタンパク質、例えばヒルジン誘導体を、E.コリ菌株中で、組み換えタンパク質のための遺伝子とCGTアーゼのシグナル配列とを融合させることで産生させ、分泌させることができることが記載されている(EP0448093号B1)。特定のヒルジン誘導体の場合には、振盪フラスコ培養において250mg/lの収率がもたらされ、発酵においては2.63g/lの収率がもたらされる。EP0448093号B1は、しかしながらまた、別の組み換えタンパク質では、25mg/lまでの収率しか得られないことも記載している。従って、CGTアーゼのシグナルペプチドは、全ての別の公知のシグナルペプチドも同様に、任意の組み換えタンパク質の分泌を同等の高さの収率で媒介することができない。各組み換えタンパク質は固有のDNA配列によってコードされ、かつ特にシグナル配列と組み換えタンパク質をコードする配列との間の接合部位でのDNA配列がそれと異なるので、一般に、各組み換えタンパク質について最適なシグナルペプチドを見出さねばならない。
【特許文献1】Lee他著、Methods in Molecular Biology 308,2005年
【特許文献2】EP0338410号B1
【特許文献3】EP0677109号B1
【特許文献4】EP0396612号B1
【特許文献5】US5395927号
【非特許文献1】EP0448093号B1
【発明の開示】
【発明が解決しようとする課題】
【0015】
本発明の課題は、新規のシグナルペプチドを提供することである。
【課題を解決するための手段】
【0016】
前記課題は、シグナルペプチドであって、切断部位前の最後の3つのアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチドによって解決される。
【0017】
有利には、該シグナルペプチドは、アミノ酸配列:MKRNRFFNTSAAIAISIALQIFFPSASAFA(配列番号1)を有するか、又は配列番号1と比較して、1〜10個の、有利には1〜5個の、特に有利には1〜3個のアミノ酸が、切断部位前の最後の3個のアミノ酸を除いて改変されているアミノ酸配列を有する。
【0018】
前記のように、シグナルペプチドは、規定の群のアミノ酸(例えば荷電した又は疎水性の又は短鎖の)を有する種々の領域からなる。従って、当業者は、アミノ酸を他の匹敵する特性を有するアミノ酸によって交換することによって、特性が変化していない新規のシグナルペプチドを作製することができる。
【0019】
前記の理由から、本発明によるシグナルペプチドとしては、配列番号2と比較して、1〜10個の、特に有利には1〜5個の、特に有利には1〜3個のアミノ酸が改変されたものを察することができる。有利には、それは、類似の生化学的特性を有するアミノ酸、例えば塩基性アミノ酸(リジン、アルギニン、ヒスチジン)を、塩基性アミノ酸と交換すること、酸性アミノ酸(アスパラギン酸、グルタミン酸、アスパラギン、グルタミン)を酸性アミノ酸と交換すること、疎水性アミノ酸を疎水性アミノ酸と交換することなどである。アミノ酸の生化学的特性に関する概要を、図1に示す。
【0020】
更に本発明は、本発明によるシグナルペプチドをコードするシグナル配列に関する。
【0021】
該配列は、切断部位を有するシグナルペプチドをコードし、該切断部位前のその最後の3個のアミノ酸がアラニン−フェニルアラニン−アラニンであることを特徴とする。
【0022】
有利には、該配列は、DNA配列
【化1】
(配列番号2)並びに縮重された遺伝子コードに基づき配列番号1のアミノ酸配列をコードする全てのDNA配列を有するシグナル配列である。
【0023】
スクリーニングの範囲において、CGTアーゼのシグナル配列(MKRNRFFNTSAAIAISIALNTFFCSMQTIA、配列番号3)の改変によって得られる種々のシグナル配列を、その特性に関して比較した。驚くべきことに、本発明によるシグナル配列は、CGTアーゼのシグナル配列又は他のシグナル配列よりも、より広範囲の組み換えタンパク質をより高収率で宿主細胞中で産生させ、分泌させるために適していることが判明した。
【0024】
本発明によるDNA配列は、遺伝子合成によって又は相応のオリゴヌクレオチドのライゲーションによって、当業者に公知の方法によって得ることができる。本発明によるDNA配列は、読み枠内で(in frame)、当業者に公知の方法(例えばLee他(2005年)と同様に)に従って、産生されるべき組み換えタンパク質の遺伝子と結合され、それをベクター中に導入することができる。
【0025】
有利には、このシグナル配列と組み換え遺伝子との組合せ物は、E.コリにおいて機能的な発現シグナル(プロモーター、転写開始部位、翻訳開始部位、リボソーム結合部位)を備える。プロモーターとしては、当業者に公知のあらゆるプロモーター、例えば一方では誘導可能なプロモーター、例えばlacプロモーター、tacプロモーター、trcプロモーター、λPLプロモーター、araプロモーター又はtetプロモーター又はそれらから誘導される配列が適している。他方では、構成的プロモーター、例えばGAPDHプロモーターを使用することによって構成的な発現を行うこともできる。しかしながら、通常は、産生されるべき組み換えタンパク質の遺伝子と結合されたプロモーターを使用することもできる。
【0026】
従って、本発明は、発現シグナルと、本発明によるシグナル配列と、産生されるべき組み換えタンパク質をコードする読み枠内で結合された組み換え遺伝子とを含む発現構築物にも関する。
【0027】
この本発明による発現構築物は、当業者に公知の方法を使用して宿主細胞に導入される。それは、例えばベクター上で、例えばプラスミド上で、例えば公知の発現ベクターの誘導体、例えばpUC18、pBR322、pACYC184、pASK−IBA3又はpET上で行われる。プラスミドのための選択マーカーとしては、例えばアンピシリン、テトラサイクリン、クロラムフェニコール、カナマイシン又は別の抗生物質に対する耐性をコードする遺伝子が適している。
【0028】
従って、本発明によるシグナル配列又は本発明による発現構築物を有するプラスミドも本発明の対象である。
【0029】
組み換えタンパク質は、有利には異種タンパク質である。有利には、組み換えタンパク質は、工業的アプローチで使用されるタンパク質又は医薬品作用物質として使用されるタンパク質(バイオロジクス、生体医薬品)である。係るタンパク質は、例えばヒルジン、インスリン、インターフェロン、例えばα−インターフェロン又はβ−インターフェロン(例えばインターフェロンα2b)、抗体もしくは抗体フラグメント(例えばFabフラグメント、scFv)又は別の結合タンパク質もしくは酵素、例えばCGTアーゼである。
【0030】
本発明による発現構築物は、当業者に公知の方法に従って微生物細胞(宿主細胞)中に導入される。結果として、本発明による発現構築物は、更にプラスミドとして宿主細胞中に存在しても、又は宿主細胞の染色体中に組み込まれてもよい。
【0031】
従って、本発明によるシグナル配列又は本発明による発現構築物又は本発明によるプラスミドを有する微生物細胞も本発明の対象である。
【0032】
宿主細胞は、腸内細菌科からの細菌株の細胞、有利にはエシェリキア・コリの種の菌株である。特に、エシェリキア(E.)・コリ菌株であって、本発明による発現構築物での形質転換後に発酵によって、本発明による発現構築物で形質転換されたE.コリW3110(ATCC27325)よりも高濃度の組み換えタンパク質をペリプラズム中もしくは栄養培地中に示すという点で優れているE.コリ菌株が好ましい。
【0033】
殊に好ましくは、その菌株は、以下のE.コリ菌株:
− BLR:Ray他(2002年)、Novagenで購入できる
− K802=CGSC*5610:Yang他(1998年)
− WCM105:EP0338410号B1に従って製造できる
− MM28=CGSC*#5892:Nagahari他(1985年)
− RV308=ATCC**31608;EP0677109号B1
− RR1:ATCC**31434:Nagahari他(1985年)
である。
* 大腸菌ストックセンター(E.coli Genetic Stock Center)CGSC(830 Kline Biology Tower,MCD Biology Department,266 Whitney Ave.,PO box 208103,Yale University,New Haven)から購入できる
** LGC Promochem(Mercatorstr.51,46485 Wesel,ドイツ在)から購入できる
産生されたタンパク質の分泌は、宿主細胞のsec装置によって行われる。引き続きペリプラズムへの分泌の後に、シグナルペプチダーゼ(例えばE.コリではLepB)によって本発明によるシグナルペプチドは開裂されて、所望の組み換えタンパク質が生ずる。
【0034】
従って、本発明は、本発明による発現構築物を有する宿主細胞によって発酵培地中で組み換えタンパク質を発酵的に製造するための方法にも関する。前記方法は、本発明による宿主菌株を発酵培地中で培養し、該宿主菌株が組み換えタンパク質を読み枠内でのシグナルペプチド−タンパク質融合物の形で産生し、その際、該シグナルペプチドは、本発明によるシグナルペプチドでありかつ該シグナルペプチドは、細胞質膜を通じてペリプラズム中にシグナルペプチド−タンパク質融合物を分泌した場合にシグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中もしくは発酵培地中で得られ、その組み換えタンパク質を発酵後に精製することを特徴としている。
【0035】
組み換えタンパク質は、発酵において、ペリプラズム中に又は有利には発酵培地中に分泌される。
【0036】
組み換えタンパク質は、宿主細胞のペリプラズムからか又は有利には発酵培地から細胞の分離によって精製することができる。
【0037】
組み換えタンパク質の本発明による製造のための微生物菌株の発酵は、有利には完全培地又は最少塩培地中で行われる。これらの培地は、文献から公知である。
【0038】
炭素源としては、原則的に、あらゆる使用可能な糖、糖アルコール、有機酸もしくはそれらの塩、デンプン加水分解物、蜜ろう又は別の物質を使用することができる。その際、グルコース又はグリセリンを使用することが好ましい。また複数の種々異なる炭素源を組み合わせて供給することも可能である。窒素源としては、尿素、アンモニア及びそれらの塩、硝酸塩並びに他のN源を用いることができる。可能な窒素源には、複合アミノ酸混合物、例えば酵母エキス、ペプトン、麦芽エキス、大豆ペプトン、カザミノ酸、コーンスティープリカー及びNZアミン(例えばKerry Bio−Science,Chicago,USA)も該当する。
【0039】
更に、該培地に、ビタミン、塩、酵母エキス、アミノ酸及び微量元素のような他の成分を添加してよく、それによって細胞増殖が向上する。
【0040】
菌株のインキュベートは、有利には好気的培養条件下で、16〜150時間の時間にわたって、かつその都度の菌株に最適な増殖温度の範囲で行われる。
【0041】
最適な温度範囲としては、15〜55℃が好ましい。特に、28〜37℃の温度が好ましい。
【0042】
菌株の培養は、振盪フラスコ又は発酵器において実施することができ、その際、容量に関する制限は与えられていない。その培養は、回分法、流加法又は連続法で実施することができる。
【0043】
その際、組み換えタンパク質の発現は、構成的に、すなわち非誘導的にも、又は物理的もしくは生理学的な刺激による誘導によって行われる。発現は、例えばプロモーターを誘導する物質、例えばlacプロモーターもしくはtacプロモーターの場合にはラクトースもしくはIPTGの添加によって誘導することができる。
【0044】
ペリプラズムもしくは培養培地からのタンパク質の精製は、当業者に公知の方法に従って、例えば細胞の破壊又は分離、クロマトグラフィー的な精製、タンパク質の複合体化、濾過又は沈降によって行うことができる。
【0045】
次の実施例は、本発明を更に説明するために用いられるものである。
【実施例】
【0046】
実施例1:本発明によるシグナル配列及び本発明によるベクターの作製
出発プラスミドとして、プラスミドpCGTを以下のように作製する:
配列番号4を有し、クレブシエラ・ニューモニアエM5a1由来のシクロデキストリン−グリコシルトランスフェラーゼ(CGTアーゼ)遺伝子を有するDNA断片(Genbank番号M15264)を、遺伝子合成によって製造した。このDNA断片を、発現ベクターpJF118ut(図2)中にクローニングした。該ベクターは、DSMZ−ドイツ微生物細胞培養収集館(Braunschweig)で番号DSM18596として寄託されている。pJF118utは、公知の発現ベクターpKK223−3(Amersham Pharmacia Biotech)の誘導体であり、β−ラクタマーゼ遺伝子とテトラサイクリン耐性遺伝子の他に、さらに同様にプラスミド上に存在するLacIq遺伝子産物によって再始動されかつ例えばD−ラクトースもしくはイソプロピル−β−D−チオガラクトピラノシド(IPTG)のようなインデューサーによってスイッチオンすることができるtacプロモーターをも有する。プラスミドpJF118utを、制限酵素EcoRIで完全に切断し、かつそれぞれ直鎖状DNA断片の5′末端で突出した塩基を、S1ヌクレアーゼで分解した。前記のようにして予備調製されたベクターDNA分子を、CGTアーゼを含むDNA断片(配列番号4)とT4−リガーゼを使用してライゲーションした。菌株DH5αを、ライゲーションバッチでCaCl2法に従って形質転換し、その際、アンピシリン(100mg/l)によってプラスミド保有細胞について選択した。アンピシリン耐性形質転換体から、再びプラスミドを単離し、そして制限分析によって調査した。前記のようにして作製された、CGTアーゼ遺伝子の発現がtacプロモーターの制御下にあるプラスミドを、pCGTと呼称した(図3)。
【0047】
CGTアーゼのためのシグナル配列に融合されたCGTアーゼのための遺伝子を除去した:そのために8448bpの大きさのプラスミドを、制限酵素SspI及びPacIによって部分消化によって当業者に公知の方法に従って切断した。6390bpの大きさの断片を単離し、そしてクレノウ酵素で処理し、それによって末端を平滑化させた。2058bpの大きさの断片を除去した。
【0048】
次いで、以下の4つのDNA断片を遺伝子合成によって製造した:
phoA−IFNα2b 配列番号5
【化2】
ompA−IFNα2b 配列番号6
【化3】
cgt−IFNα2b 配列番号7
【化4】
AFA−IFNα2b 配列番号8
【化5】
【0049】
全てのDNA断片は、tacプロモーター領域と、インターフェロンα2bのための遺伝子と、4つの種々のシグナル配列(太字で示した)とを有する。これらの4つの種々のシグナル配列は、以下の4つの種々のシグナルペプチドをコードし、その際、最初の3つのシグナルペプチド(配列番号9、10、3)は、先行技術から公知であり、四番目のシグナルペプチド(配列番号2)は、本発明によるものである:
phoA: MKQSTIALALLPLLFTPVTKA
配列番号9
ompA: MKKTAIAIAVALAGFATVAQA
配列番号10
cgt: MKRNRFFNTSAAIAISIALNTFFCSMQTIA
配列番号3
AFA: MKRNRFFNTSAAIAISIALQIFFPSASAFA
配列番号2
前記のクローニングによって以下の4つのプラスミドが得られた:
− pKP651(phoA−シグナル配列) 図4
− pKP652(ompA−シグナル配列) 図5
− pKPIFN(cgt−シグナル配列) 図6
− pBaBIFN1(AFA:本発明によるシグナル配列) 図7
これらのプラスミドを、公知の方法によってE.コリ菌株DH5αに導入した。本発明による基株DH5α/pBaBIFN1は、DSMZ(ドイツ微生物細胞培養収集館、D−38142 Braunschweig)で番号DSM18343としてブタペスト条約に従って寄託された。
【0050】
実施例2:本発明によるシグナル配列を使用することによるインターフェロン産生の増大
プラスミドpKP651(phoA−シグナル配列)、pKP652(ompA−シグナル配列)、pKPIFN(cgt−シグナル配列)及びpBaBIFN1(本発明によるAFA−シグナル配列)(実施例1を参照のこと)を、菌株WCM105(EP0338410号B1に従って製造できる)中に通常の方法(CaCl2−形質転換によって)に従って導入した。プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。
【0051】
以下の菌株が得られた:
− WCM105/pKP651(phoA−シグナル配列)
− WCM105/pKP652(ompA−シグナル配列)
− WCM105/pKPIFN(cgt−シグナル配列)
− WCM105/pBaBIFN1(AFA−シグナル配列)
得られた菌株中でのインターフェロンα2bの産生を調査した。そのために、それらの菌株を、100mg/Lのアンピシリンと1%のグルコースとを有する10mlのLB培地中で30℃において培養した。600nmでの光学密度(OD600)が0.5になったら、インターフェロンα2bの産生を、IPTG(イソプロピルチオガラクトシド)を0.5mMまで添加することによって誘導する。培養上清において、24時間後、48時間後及び72時間後で、形成されかつ分泌されたインターフェロンを、SDSゲル中でのタンパク質の分離と抗インターフェロン特異抗体でのイムノブロットでの検出によって以下のようにして定量化した。
【0052】
それぞれ1μl(48時間及び72時間)もしくは5μl(24時間)の上清を、サンプルバッファーと混合した(2×Tris SDS−サンプルバッファー(Invitrogen カタログ番号LC2676):0.125Mのトリス塩酸(pH6.8)、4%(w/v)のSDS、20%(v/v)のグリセリン、0.005%(v/v)のブロモフェノールブルー、5%のβ−メルカプトエタノール)。更に、定義された量のインターフェロンα2bを、スタンダードとして一緒に施与した。タンパク質の変性を、100℃に5分間加熱し、氷上で2分間冷却し、そして遠心分離することによって実施した。それらのタンパク質を、電気泳動によって、12%のNuPAGE(登録商標)Bis−Tris−Gel(Invitrogen カタログ番号NP0341)中で、1×MES含有ランニングバッファー(Invitrogen カタログ番号NP0002)を用いて分離させた(電気泳動パラメータ:200Vで40分間)。イムノブロットによる検出と定量化を、以下の仕様に従って実施した:
【0053】
湿式ブロッティング法での転写:
モジュール:Amersham:Hoefer TE 22 Mini Tank Transfer Unit、コード番号:80−6204−26。
【0054】
メンブレン:ニトロセルロースメンブレン(Schleicher&Schuell,BA85,硝酸セルロース(E),0.45μmの細孔サイズ)
Whatmanフィルタとニトロセルロースメンブレンを、適切な大きさに切断し、そして発泡物品(スポンジ)で転写バッファー(Invitrogen カタログ番号LC3675)に気泡なく染み込ませた。
【0055】
積層の構成:黒い格子、カソードとの接続、各3mm厚を有する2つのスポンジ、Whatmanペーパー、SDSポリアクリルアミドゲル、NCメンブレン、Whatman、6mm厚を有する1つのスポンジ、白い格子、アノードとの接続
転写条件:I=200mAの一定の電流、U=無制限、運転時間60分
【0056】
プレハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー中で該メンブレンをインキュベートする。
室温で30分間振り動かす。
【0057】
一次抗体のハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー+0.15μg/ml(→3.75μg)の抗ヒトIFNΑ抗体(Pepro Tech EC,Biozolを経由した カタログ番号:500−P32A)中で該メンブレンをインキュベートする。
【0058】
室温で90分間又は一晩振り動かす。
【0059】
洗浄
1×PBSと一緒に室温で10秒間振り動かし、バッファーを捨てる。
1×PBSと一緒に室温で15分間2回振り動かし、バッファーを捨てる。
【0060】
二次抗体のハイブリダイゼーション
25mlのプレハイブリダイゼーションバッファー+25μl(1:1000)のヤギ抗ウサギIgGセイヨウワサビペルオキシダーゼコンジュゲート(HRP)(Southern Biotech、Biozolを経由した カタログ番号4050−05)中で該メンブレンをインキュベートする。
【0061】
室温で60分間振り動かす。
【0062】
洗浄
1×PBSと一緒に室温で10秒間振り動かし、バッファーを捨てる。
1×PBSと一緒に室温で15分間2回振り動かし、バッファーを捨てる。
【0063】
化学発光による検出
Lumi−Lightウェスタンブロッティング基質(Roche,カタログ番号2015200)を準備する:Lumi−Lightルミノール/エンハンサー溶液とLumi−Light安定ペルオキシド溶液とを1:1の比率で混合する:NCメンブレン当たり3ml。
【0064】
ブロットを室温でLumi−Lightウェスタンブロッティング基質と一緒に5分間インキュベートし、過剰の水気を切り、メンブレンをラップフィルムで覆って、直ちにX線フィルム(Kodak、X−OMAT)を載せ、2分間暴露し、現像し、そして固定する。シグナルが弱い場合に、暴露をより長時間にわたり繰り返す。
【0065】
バッファー
プレハイブリダイゼーションバッファー:1×PBS中の5%脱脂粉乳
10×PBS:100mMのNaH2PO4、1.5MのNaCl、NaOHでpH7.5、0.5%のTriton100
1×PBS:10×PBSを完全脱塩水で1:10希釈したもの
【0066】
定量化
定量的評価は、Biorad社製のGS−800 Calibrated Densitometerでイムノブロットをスキャンすることによって、Quantity One 1−D分析ソフト(Biorad)を用いて、施与されたスタンダードと比較することによって実施した。
【0067】
図9は、前記の実施例からのイムノブロットを示している。第1表において、定量化されたインターフェロンα2bの収率をまとめる:
【表1】
【0068】
第1表:それぞれ使用されたシグナル配列の点で異なる種々のプラスミドを用いて24時間後、48時間後又は72時間後に得られたインターフェロンα2bの収率
これらの結果は、配列番号2の本発明によるシグナル配列が、産生されるべきタンパク質の収率及び分泌に関して、別のシグナル配列より優れていることを一義的に示している。
【0069】
実施例3:本発明によるシグナル配列の挿入によって改善されたCGTアーゼ産生プラスミドの作製
プラスミドpCGT(実施例1を参照)は、CGTアーゼのためのシグナル配列と読み枠内で融合されたCGTアーゼのための遺伝子を有する。このシグナル配列を、ここで本発明によるシグナル配列と交換した。
【0070】
そのために8448bpの大きさのプラスミドを、制限酵素SspI及びBglIIによって部分消化によって当業者に公知の方法に従って切断した。8119bpの大きさの断片を単離し、そしてクレノウ酵素で処理し、それによって末端を平滑化させた。CGTアーゼのシグナル配列と、約150bpのCGTアーゼ遺伝子の5末端とを有する329bpの大きさの断片を除去した。
【0071】
前記の断片と配列に関して、CGTアーゼのシグナル配列が本発明によるシグナル配列と交換されていること以外は同一である以下の329bpの大きさのDNA断片を遺伝子合成によって製造した:
【化6】
【0072】
このDNA断片を、単離された8119bpの大きさの断片と、当業者に公知の方法に従ってライゲーションした。それによって得られたプラスミドpCM207AFAを、配列決定によって配列の精度について調査した。
【0073】
実施例4:本発明によるシグナル配列を使用することによるCGTアーゼ産生の改善
プラスミドpCM703AFAとプラスミドpCGT(実施例3を参照)を、以下の菌株に通常の方法に従って(例えばCaCl2法によって)形質転換により導入した:
− K802=CGSC*5610:Yang他(1998年)
− WCM105:EP0338410号B1に従って製造できる
プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。それによって以下の本発明による菌株:
− WCM105/pCM703AFA
− K802/pCM703AFA
及び以下の対照菌株:
− WCM105/pCGT
− K802/pCGT
が得られた。
【0074】
これらの菌株を、シクロデキストリン−グリコシルトランスフェラーゼの製造のために使用し、そして1%のグルコースと100mg/Lのアンピシリンを有する10mlのLB培地中で30℃において培養した。ODが0.5となったら、シクロデキストリン−グリコシルトランスフェラーゼの産生を、IPTG(イソプロピルチオガラクトシド)を0.5mMまで添加することによって誘導した。
【0075】
該菌株の培養上清において、シクロデキストリン−グリコシルトランスフェラーゼの収率を以下の活性試験によって測定した:
試験バッファー:5mMのトリス塩酸バッファー(>pH6.5)、5mMのCaSO4・2H2O
基質:試験バッファー(pH6.5)中の10%のNoredux溶液
試験バッチ:1mlの基質溶液+1mlの遠心分離された培養上清(5分、12000rpm)+3mlのメタノール
反応温度:40℃
酵素試験:
・ 溶液を予熱する(約5分、40℃で)。
【0076】
・ 酵素溶液を基質溶液に添加する;迅速に混合する(渦ミキサ(Whirl−Mixer))。
【0077】
・ 40℃で3分間インキュベートする。
【0078】
・ 酵素反応をメタノールの添加によって停止させる;迅速に混合する(渦ミキサ)。
【0079】
・ バッチを氷上で冷却する(約5分間)。
【0080】
・ 遠心分離(5分、12000rpm)をし、そして澄明な上清をピペットで取り出す。
【0081】
・ 生じたCDをHPLCによって分析する。
【0082】
酵素活性:A=G*V1*V2/(t*MG)(ユニット/ml)
A=活性
G=CDの含有率(mg/l)=試験バッチ:単位表面×104/標準溶液(10mg/ml)/単位表面
V1=希釈係数/試験バッチ(→5)
V2=希釈係数/酵素溶液
t=反応時間(分)(→3)
MG=分子量(g/モル)(CD→973)
1ユニット=1マイクロモルの産物/分
第2表は、本発明による菌株の高められた特異的なシクロデキストリン−グリコシルトランスフェラーゼ収率を示している。
【0083】
【表2】
【0084】
第2表:種々の菌株におけるシクロデキストリン−グリコシルトランスフェラーゼの収率
実施例5:本発明によるシグナル配列を使用することによるヒルジン産生の改善
特許EP0448093号B1に記載されるプラスミドpCMT203を、使用されるシグナル配列を本発明によるシグナル配列AFAと交換することで改変させた。この交換は、実施例1及び実施例3と同様に実施した。得られたプラスミドをpCMT203AFAと呼称した。pCMT203及びpCMT203AFAを、菌株WCM105(EP0338410号B1に従って製造できる)中に、通常の方法による形質転換によって(例えばCaCl2−形質転換によって)導入した。
【0085】
プラスミド保有菌株についての選択は、アンピシリン(100mg/L)によって実施した。
【0086】
以下の菌株が得られた:
− WCM105/pCMT203
− WCM105/pCMT203AFA
両方の菌株を、EP0448093号B1に記載されるように、10lの発酵器中で培養し、そして得られたヒルジンを、EP0448093号B1に記載されるように、IPTGの添加45時間後に定量化した。
【0087】
第3表中の結果は、本発明によるシグナル配列を使用することでヒルジンの高められた収率がもたらされることを示している。
【0088】
【表3】
【0089】
第3表:種々の菌株における、IPTGの添加45時間後での10lの発酵におけるヒルジンの収率(AT−U/ml及びg/L)
実施例6:本発明によるシグナル配列を使用することによる、機能的なFab抗体フラグメントの産生の改善
本実施例は、良好に特徴付けられた抗リゾチーム抗体D1.3のFabフラグメントの改善された製造を記載する。
【0090】
抗リゾチームFabフラグメントの遺伝子のクローニングと発現のための出発ベクターとして、プラスミドpJF118ut(実施例1を参照)を使用した。このプラスミド中に、2つの連続した工程において、抗リゾチームFabフラグメントの重鎖(VH−CH1−ドメイン)もしくは軽鎖(VL−CL−ドメイン)についての両方の読み枠を、それぞれシグナル配列を含めてクローニングした。そのために以下のように実施した:
配列番号12を有するDNA断片(重鎖)を、遺伝子合成によって製造し、そして該DNA断片は、E.コリのompA遺伝子のシグナル配列と、Fabフラグメントの重鎖(VH−CH1)のための読み枠とからなる遺伝子融合物を含む。この読み枠直後に6個のヒスチジンコドンが引き続き、従って融合タンパク質のC末端を形成している。このヒスチジンタグによって、後に完全に会合されたFabフラグメントの簡単な精製が親和性クロマトグラフィーによって可能となる。このDNA断片を、制限酵素EcoRI及びPstIで切断し、そして同じ制限酵素で切断されている発現ベクターpJF118utとライゲーションした。このクローニングから得られた、重鎖のための遺伝子の発現がtacプロモーターの制御下にあるプラスミドを、pHC−Anti−Lysozymと呼称した。
【0091】
配列番号13を有するDNA断片(軽鎖)を、同様に遺伝子合成によって製造し、そして該DNA断片は、配列番号3に記載されるCGTアーゼのシグナルペプチドをコードするDNA配列(配列番号7において太字で示した)と、Fabフラグメントの軽鎖(VL−CL)のための読み枠とからなる遺伝子融合物を含む。このDNA断片を、まず、制限酵素PstIで切断し、引き続き同じ制限酵素で切断されているベクターpHC−Anti−Lysozymとライゲーションした。こうして得られたプラスミドを、pFab−Anti−Lysozym(図9)と呼称した。前記のようにして、重鎖と軽鎖についてのそれぞれの読み枠からなる、tacプロモーターの制御下にある人工オペロンを作製した。従って、インデューサー(例えばIPTG)の添加によって、両方の遺伝子の同期発現が可能である。
【0092】
本発明によるプラスミドpFab−Anti−LysozymVLAFA及びpFab−Anti−LysozymVHAFAの製造のために、実施例1に記載されるのと同様にして、軽鎖のためのシグナル配列(pFab−Anti−LysozymVLAFA)か又は重鎖のためのシグナル配列(pFab−Anti−LysozymLHAFA)かのいずれかを、配列番号2の本発明によるシグナル配列と交換した。
【0093】
抗リゾチームFabフラグメントの製造ために、菌株WCM105(実施例4を参照)を、プラスミドpFab−Anti−Lysozym、pFab−Anti−LysozymVLAFAもしくはpFab−Anti−LysozymVHAFAで、CaCl2法に従って形質転換させた。プラスミド保有細胞についての選択は、アンピシリン(100mg/l)によって実施した。
【0094】
抗リゾチームFabフラグメントの製造を、10l規模で実施した。製造は、10lの撹拌槽型発酵器中で実施した。6lの培地FM4(1.5g/lのKH2PO4;5g/lの(NH4)2SO4;0.3g/lのMgSO4×7H2O;0.05g/lのCaCl2×2H2O、0.075g/lのFeSO4×7H2O;1g/lのNa3クエン酸塩×2H2O;0.5g/lのNaCl;1ml/lの微量元素溶液(0.15g/lのNa2MoO4×2H2O;2.5g/lのNa3BO3;0.7g/lのCoCl2×6H2O;0.25g/lのCuSO4×5H2O;1.6g/lのMnCl2×4H2O;0.3g/lのZnSO4×7H2O);5mg/lのビタミンB1;3g/lのフィトン;1.5g/lの酵母エキス;10g/lのグルコース;100mg/lのアンピシリン)で満たされた発酵器に、1:10の比率で、一晩同じ培地中で培養された前培養を植えた。発酵の間に、30℃の温度に調整し、そしてpH値をNH4OHもしくはH3PO4を計量供給することによって7.0の値に一定に保った。グルコースは、発酵にわたって計量供給され、その際、培地中の最大グルコース濃度を10g/l未満にすることに努めた。発現の誘導は、対数増殖期の終わりにイソプロピル−β−D−チオガラクトピラノシド(IPTG)を0.1mMで添加することによって実施した。
【0095】
72時間の発酵後に、試料を取りだし、細胞を培養培地の遠心分離によって分離した。培養上清からの抗リゾチームFabフラグメントの精製は、Skerra(1994年、Gene 141,79−84)に記載される親和性クロマトグラフィーによって実施した。
【0096】
精製された抗リゾチームFabフラグメントの定量化並びに活性の測定は、抗原としてリゾチームを用いたELISA試験(Skerra,1994年、Gene 141,79−84)によって実施した。
【0097】
第4表において、それぞれ20mlの培養上清から72時間の発酵後に単離することができた機能的な抗リゾチームFabフラグメントの収率を列記する。
【0098】
第4表:72時間の発酵後の培養上清中での抗リゾチームFabフラグメントの収率
【表4】
【図面の簡単な説明】
【0099】
【図1】図1は、アミノ酸の概要を示している。
【図2】図2は、プラスミドpJF118utのプラスミド地図を示している。
【図3】図3は、プラスミドpCGTのプラスミド地図を示している。
【図4】図4は、プラスミドpKP651のプラスミド地図を示している。
【図5】図5は、プラスミドpKP652のプラスミド地図を示している。
【図6】図6は、プラスミドpKPIFNのプラスミド地図を示している。
【図7】図7は、プラスミドpBaBIFN1のプラスミド地図を示している。
【図8】図8は、プラスミドpFab−Anti−Lysozymのプラスミド地図を示している。
【図9】図9は、インターフェロンα2bを産生する細胞(実施例2を参照)の培養上清が分析されるイムノブロットを示している。 レーン1:サイズ基準 レーン2とレーン3:20ngと40ngのインターフェロンα2b レーン4〜6:24時間後、48時間後、72時間後の、プラスミドpBaBIFN1を有する培養の上清 レーン7〜9:24時間後、48時間後、72時間後の、プラスミドpKPIFNを有する培養の上清 レーン10〜12:24時間後、48時間後、72時間後の、プラスミドpKP651を有する培養の上清 レーン13〜15:24時間後、48時間後、72時間後の、プラスミドpKP652を有する培養の上清 24時間の場合に、それぞれ5μlが施与され、48時間及び72時間の場合に、1μlが施与された。 それらの細胞は、シグナル配列において相違するインターフェロンα2b−発現プラスミドを有する。形成されたインターフェロンα2bを、抗体によって検出した(矢印)。本発明によるシグナル配列の使用は、インターフェロンα2b産生の向上をもたらす。
【特許請求の範囲】
【請求項1】
組み換えタンパク質との切断部位を有するシグナルペプチドであって、その切断部位前の最後の3個のアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチド。
【請求項2】
請求項1記載のシグナルペプチドであって、アミノ酸配列:MKRNRFFNTSAAIAISIALQIFFPSASAFA(配列番号1)を有するか、又は配列番号1と比較して、1〜10個の、有利には1〜5個の、特に有利には1〜3個のアミノ酸が、切断部位前の最後の3個のアミノ酸を除いて改変されているアミノ酸配列を有することを特徴とするシグナルペプチド。
【請求項3】
請求項1又は2記載のシグナルペプチドをコードするDNA配列。
【請求項4】
請求項3記載のDNA配列であって、塩基配列:
【化1】
(配列番号2)並びに縮重された遺伝子コードに基づき、配列番号1のアミノ酸配列をコードするあらゆる塩基配列を特徴するDNA配列。
【請求項5】
発現シグナルと、請求項3又は4に記載のDNA配列と、読み枠内で結合された組み換えタンパク質をコードする組み換え遺伝子とを含む発現構築物。
【請求項6】
請求項3又は4記載のDNA配列又は請求項5記載の発現構築物を有するプラスミド。
【請求項7】
請求項3又は4記載のDNA配列又は請求項5記載の発現構築物又は請求項6記載のプラスミドを有する微生物細胞。
【請求項8】
請求項7記載の微生物細胞であって、それが腸内細菌科の細菌株の細胞、有利にはエシェリキア・コリの種の菌株の細胞であることを特徴とする微生物細胞。
【請求項9】
組み換えタンパク質の発酵的製造方法において、請求項7又は8記載の微生物細胞を、発酵培地中で培養し、該微生物細胞が組み換えタンパク質をシグナルペプチド−タンパク質融合物の形で産生し、その際、該シグナルペプチドは、請求項1又は2記載のシグナルペプチドでありかつ該シグナルペプチドは、細胞質膜を通じてペリプラズム中にシグナルペプチド−タンパク質融合物を分泌した場合に、シグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中もしくは発酵培地中で得られ、その組み換えタンパク質を発酵後に精製することを特徴とする方法。
【請求項10】
請求項9記載の方法において、組み換えタンパク質が、工業的アプローチで使用されるタンパク質又は医薬作用物質として使用されるタンパク質であることを特徴とする方法。
【請求項1】
組み換えタンパク質との切断部位を有するシグナルペプチドであって、その切断部位前の最後の3個のアミノ酸が、アラニン−フェニルアラニン−アラニン(AFA)であることを特徴とするシグナルペプチド。
【請求項2】
請求項1記載のシグナルペプチドであって、アミノ酸配列:MKRNRFFNTSAAIAISIALQIFFPSASAFA(配列番号1)を有するか、又は配列番号1と比較して、1〜10個の、有利には1〜5個の、特に有利には1〜3個のアミノ酸が、切断部位前の最後の3個のアミノ酸を除いて改変されているアミノ酸配列を有することを特徴とするシグナルペプチド。
【請求項3】
請求項1又は2記載のシグナルペプチドをコードするDNA配列。
【請求項4】
請求項3記載のDNA配列であって、塩基配列:
【化1】
(配列番号2)並びに縮重された遺伝子コードに基づき、配列番号1のアミノ酸配列をコードするあらゆる塩基配列を特徴するDNA配列。
【請求項5】
発現シグナルと、請求項3又は4に記載のDNA配列と、読み枠内で結合された組み換えタンパク質をコードする組み換え遺伝子とを含む発現構築物。
【請求項6】
請求項3又は4記載のDNA配列又は請求項5記載の発現構築物を有するプラスミド。
【請求項7】
請求項3又は4記載のDNA配列又は請求項5記載の発現構築物又は請求項6記載のプラスミドを有する微生物細胞。
【請求項8】
請求項7記載の微生物細胞であって、それが腸内細菌科の細菌株の細胞、有利にはエシェリキア・コリの種の菌株の細胞であることを特徴とする微生物細胞。
【請求項9】
組み換えタンパク質の発酵的製造方法において、請求項7又は8記載の微生物細胞を、発酵培地中で培養し、該微生物細胞が組み換えタンパク質をシグナルペプチド−タンパク質融合物の形で産生し、その際、該シグナルペプチドは、請求項1又は2記載のシグナルペプチドでありかつ該シグナルペプチドは、細胞質膜を通じてペリプラズム中にシグナルペプチド−タンパク質融合物を分泌した場合に、シグナルペプチドと組み換えタンパク質との間の切断部位で開裂され、そして所望の組み換えタンパク質がペリプラズム中もしくは発酵培地中で得られ、その組み換えタンパク質を発酵後に精製することを特徴とする方法。
【請求項10】
請求項9記載の方法において、組み換えタンパク質が、工業的アプローチで使用されるタンパク質又は医薬作用物質として使用されるタンパク質であることを特徴とする方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2008−73044(P2008−73044A)
【公開日】平成20年4月3日(2008.4.3)
【国際特許分類】
【出願番号】特願2007−248133(P2007−248133)
【出願日】平成19年9月25日(2007.9.25)
【出願人】(390008969)ワッカー ケミー アクチエンゲゼルシャフト (417)
【氏名又は名称原語表記】Wacker Chemie AG
【住所又は居所原語表記】Hanns−Seidel−Platz 4, D−81737 Muenchen, Germany
【Fターム(参考)】
【公開日】平成20年4月3日(2008.4.3)
【国際特許分類】
【出願日】平成19年9月25日(2007.9.25)
【出願人】(390008969)ワッカー ケミー アクチエンゲゼルシャフト (417)
【氏名又は名称原語表記】Wacker Chemie AG
【住所又は居所原語表記】Hanns−Seidel−Platz 4, D−81737 Muenchen, Germany
【Fターム(参考)】
[ Back to top ]