
シスタチンCおよびその遺伝子、抗シスタチンC抗体、ならびに、ネコ腎症の診断用キット、診断方法

【課題】ネコ由来のシスタチンCに特異的な抗体を提供し、さらにはそれを用いることでネコの腎症を迅速かつ簡便に診断できる方法、キットを提供する。
【解決手段】特定な配列からなるアミノ酸配列を有するタンパク質、ならびに、当該タンパク質をコードする構造遺伝子であって、好ましくは特定な配列からなる塩基配列を有する構造遺伝子。ネコ由来シスタチンCに特異的に結合する抗体であって、好ましくは、タンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである抗体。抗体を含むネコ腎症の診断用キット、ならびに、本発明の抗体を用いたネコ腎症の診断方法。
【解決手段】特定な配列からなるアミノ酸配列を有するタンパク質、ならびに、当該タンパク質をコードする構造遺伝子であって、好ましくは特定な配列からなる塩基配列を有する構造遺伝子。ネコ由来シスタチンCに特異的に結合する抗体であって、好ましくは、タンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである抗体。抗体を含むネコ腎症の診断用キット、ならびに、本発明の抗体を用いたネコ腎症の診断方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ネコに由来するシスタチンCおよびそれをコードする遺伝子に関する。また本発明は、ネコに由来するシスタチンCに対する抗体およびそれを用いたネコ腎症の診断用キット、診断方法にも関する。
【背景技術】
【0002】
近年、少子化に伴い、ペットを飼う世帯は増加の一途をたどっている。しかしながら、ペットの性質に即した飼い方がなされていないケースも少なくはない。特に、偏食の結果、ペットが糖尿病などの成人病的症状を引き起こしてしまい、ペットを獣医に通院させるケースまで見られる。
【0003】
このような現状から、近年はペットの診断に関する事業が拡大しつつある。仮に、ペットの腎症を早期に発見することができれば、獣医師は、飼い主によるペットの飼い方、特に食事の与え方について改善を指導できるようになる。一般に、腎症のマーカーの1つとして、シスタチンC(CysC)が挙げられる。
【0004】
シスタチンCは、たとえばヒト由来の場合には、分子量13000Daの塩基性低分子タンパクである。ヒト由来のシスタチンCは、ヒトの全身の細胞で産生されており、細胞内外の環境変化にはほとんど影響を受けないで一定の産生量で細胞外に分泌され、近年では、糖尿病性腎症などの早期診断の指標として有用であるとする報告例がみられる
しかしながら、ネコ由来のシスタチンCに関しては、当該タンパクに特異的な抗体が存在しないどころ、当該タンパクのアミノ酸配列すら解明されていないのが現状である。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Journal of Veterinary Internal Medicine. 22(5): 1111-1117, 2008
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、上記課題を解決するためになされたものであって、その目的とするところは、ネコ由来のシスタチンCに特異的な抗体を提供し、さらにはそれを用いることでネコの腎症を迅速かつ簡便に診断できる方法、キットを提供することである。
【課題を解決するための手段】
【0007】
本発明者は、鋭意研究の結果、ネコの遺伝子の中でシスタチンCをコードする構造遺伝子を初めて特定し、当該構造遺伝子からネコ由来のシスタチンCを発現させ、そのアミノ酸配列も解析した。さらにはネコ由来シスタチンCに特異的な抗体を作製し、本発明を完成するに至った。すなわち、本発明は以下のとおりである。
【0008】
本発明は、配列番号1で表わされるアミノ酸配列を有するタンパク質を提供する。
本発明はまた、上述した本発明のタンパク質をコードする構造遺伝子についても提供する。本発明の構造遺伝子は、配列番号2で表わされる塩基配列を有することが好ましい。
【0009】
本発明はさらに、ネコ由来シスタチンCに特異的に結合する抗体についても提供する。本発明の抗体は、上述した本発明のタンパク質を抗原として、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものであることが、好ましい。
【0010】
本発明は、上述した本発明の抗体を含むネコ腎症の診断用キットについても提供する。
本発明は、上述した本発明の抗体を用いたネコ腎症の診断方法についても提供する。
【発明の効果】
【0011】
本発明によれば、従来と比較して格段に迅速かつ簡便にネコ腎症を診断することができるようになる。
【図面の簡単な説明】
【0012】
【図1】ネコ由来のCysCのアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。
【図2】ネコ由来のCysC遺伝子のcDNAの塩基配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。
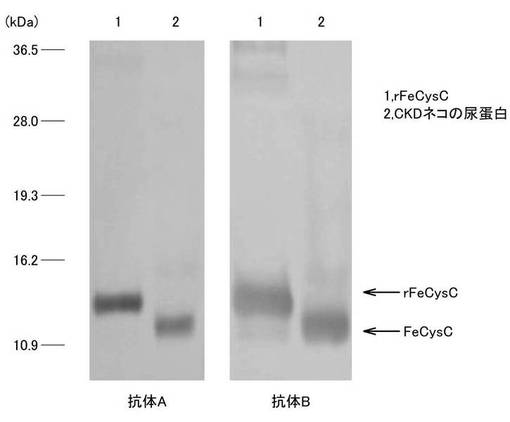
【図3】実験例3におけるネコのnativeなCysCに対する抗体A、Bの特異性の実験結果を示す写真である。
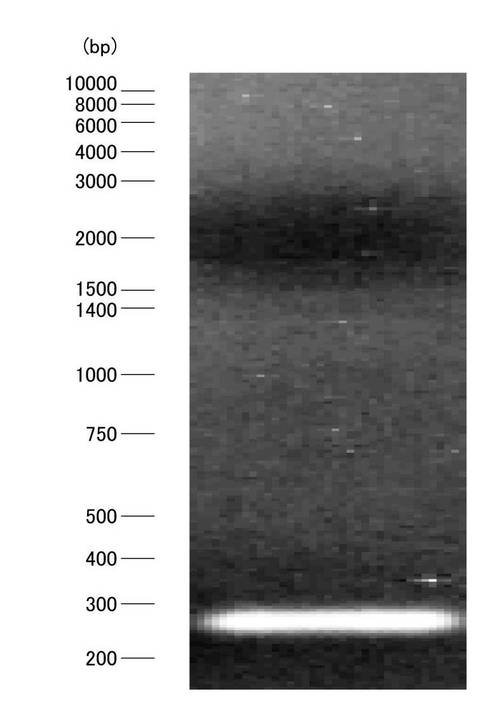
【図4】実験例1におけるfirst-strand cDNAのPCRの結果を示す電気泳動写真である。
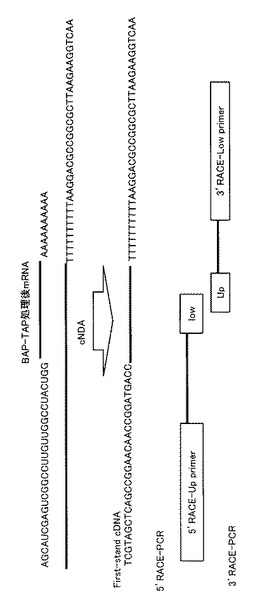
【図5】実験例1で使用したキットによって合成できる2本鎖DNAとプライマーとの位置関係を模式的に示す図である。
【図6】実験例1における5’RACE−PCRの結果を示す電気泳動写真である。
【図7】実験例1における3’RACE−PCRの結果を示す電気泳動写真である。
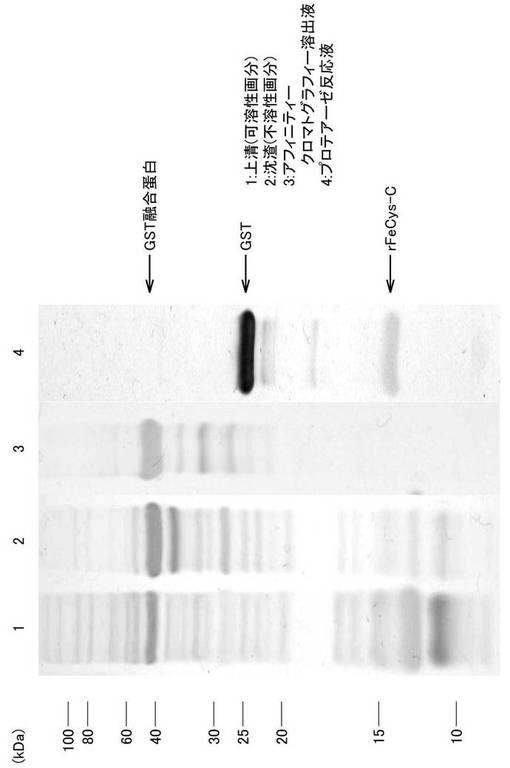
【図8】実験例2におけるGST融合タンパク発現後のSDS−PAGEの結果を示す写真である。
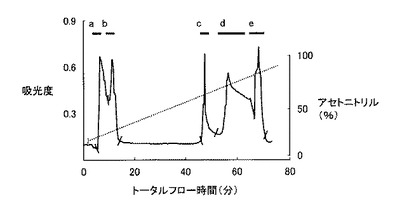
【図9】実験例2におけるHPLCの結果得られたクロマトパターンを示すグラフであり、左側の縦軸は波長220nmでの吸光度、右側の縦軸はアセトニトリル濃度(%)、横軸は時間(分)を示している。
【図10】実験例2におけるHPLCの主要画分a, b, c, d, eについてのSDS−PAGEの結果を示す写真である。
【発明を実施するための形態】
【0013】
本発明によれば、配列番号1で表わされるアミノ酸配列を有するタンパク質が提供される。本発明者は、ネコの遺伝子の中でシスタチンCをコードする構造遺伝子(本明細書中において「CysC遺伝子」と呼称する。)を初めて特定し、当該CysC遺伝子によりネコ由来のシスタチン(本明細書中において「CysC」と呼称する。)のアミノ酸配列も初めて解析した。配列番号1に示されるアミノ酸配列を有する本発明のタンパク質が、今回、本発明者によって初めてアミノ酸配列が特定されたネコ由来のシスタチンCである。
【0014】
ここで、図1は、配列番号1で表わされる本発明のタンパク質(ネコのCysC)のアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。図1中、四角で囲っている部分は、各動物種間で共通するアミノ酸配列である。配列番号1で表わされる本発明のタンパク質のアミノ酸数は全長で147個であり、ヒト、サルおよびブタのアミノ酸数は146個、ウシのアミノ酸数は148個であり、ラットのアミノ酸数140個と近似した値であり、また、他動物種間で保存されていた構造アミノ酸システインの位置も数も同様である。詳細は実験例2において後述するが、本発明のタンパク質のアミノ酸配列は、他動物種のCysCのアミノ酸配列との平均相同性は69.15%であり、他動物種(ヒト、ウシ、ブタおよびラット)間のCysCのアミノ酸配列の相同性は、62.22〜97.26%の範囲に分布していることからすると、本発明のタンパク質はネコ由来のCysCであると考えられる。
【0015】
本発明のネコ由来のCysCは、人工的な合成により好適に得ることができる。今回、本発明者は、ネコ由来CysCの構造遺伝子(CysC遺伝子)の塩基配列(配列番号2で表わされる塩基配列)を初めて見出した。本発明は、ネコ由来シスタチンをコードする構造遺伝子についても提供するものであり、この構造遺伝子は、配列番号2で表わされる塩基配列を有することが、好ましい。すなわち、本発明の構造遺伝子は、配列番号2で表わされる塩基配列をエキソンとして含んでいるのであれば、上記以外の塩基配列をイントロンとして含んでいてもよい。
【0016】
ここで、図2は、配列番号2で表わされる本発明の構造遺伝子(ネコのCysC遺伝子)を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。図2中、四角で囲っている部分は、各動物種間で共通する塩基配列である。CysC遺伝子の核酸長は、ヒト、サルおよびブタが441 base、ウシでは447 base、ラットは423 baseであったのに対し、配列番号2で表わされる本発明のネコのCysC遺伝子ではその長さは444 baseであった。また、配列番号2で表わされる本発明のネコのCysC遺伝子の塩基配列と他動物種のCysC遺伝子の塩基配列との相同性は平均で77.69%であり、他動物種(ヒト、サル、ウシ、ブタおよびラット)間のCysC遺伝子の塩基配列の相同性は、67.21〜96.71%の範囲に分布していることからすると、本発明の構造遺伝子はネコ由来のCysC遺伝子であると考えられる。
【0017】
本発明は、ネコ由来のCysCに特異的に結合する新規な抗体についても提供する。本発明者は、詳細は実験例3にて後述するように、上述した本発明のネコ由来のCysC遺伝子からネコ由来のCysCを発現させ、これを抗原として抗体を産生し得る細胞を作製した。このような細胞株は新規なものであり、出願人らは、今回、平成21年12月1日付けで独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行った(受領番号:FERM AP-21877、FERM AP-21878)。
【0018】
本発明の抗体は、好ましくは、上述した本発明のタンパク質を抗原として、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである。ここで、図3は、ネコのnativeなCysCに対し本発明の抗体が特異的に結合した実験結果を示す写真である。詳細は実験例3として後述するが、図3に示される抗体Aは細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)により産生されたIgG1のκ鎖のアイソタイプのモノクローナル抗体であり、抗体Bは細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたIgG2aのκ鎖のアイソタイプの抗体である。図3に示されるように、本発明の抗体は、ネコのnativeなCysCに対し特異的に結合し得るものであることが分かる。
【0019】
本発明は、さらに上述した本発明の抗体を利用したネコ腎症の診断方法、診断キットについても提供するものである。本発明の抗体は、ネコ腎症のマーカーであるCysCに特異的に結合し得るものであるため、たとえばネコの尿をサンプルとして用いて、当該ネコが腎症に罹っているか否か、従来と比較して迅速かつ簡便に診断することが可能となる。本発明の診断キットは、本発明の抗体以外に、たとえばウェル、色原性基質溶液、反応停止液、洗浄液、標準溶液などを含むことができる。
【0020】
<実験例>
以下、実験例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【0021】
<実験例1:CysC遺伝子の特定>
(1)供試動物
本実験例においては、実験動物施設にて維持されている血液生化学および尿生化学検査において異常が認められない、10歳齢の雄の日本ネコ1頭を使用した。このネコの飼養条件は、12時間昼、12時間夜とし猫用ケージにて飼育し、1日1回の給餌による自由採食、自由飲水とした。
【0022】
(2)ネコ白血球からのTotal RNAの抽出
まず、EDTA採血管を用いて供試動物の外頸静脈からネコ血液を採取した。採血された5mlの血液をコニカルチューブに移し、3000×rpmで5分間遠心後、バフィーコート(白血球層)を分離させた。次に、QIAamp RNA Blood Kit(QIAGEN)を用いて、添付のプロトコールに従ってTotal RNAを抽出した。得られたTotal RNAは使用時まで4℃で保存した。
【0023】
次に、Oligotex(商標)-dT30 Super mRNA Purification Kit(タカラバイオ株式会社)を用いて、添付のプロトコルに従ってTotal RNAからmRNAを分離精製した。具体的には、まず、60μlのTotal RNAを70μlの2×Binding Bufferおよび14μlのOligotex(商標)-dT30と混和した後、サーマルクライマー(PC801、ASTEC)で70℃、3分間加温した。加温後、mRNAとOligotex(商標)-dT30 Superとのハイブリダイゼーションを室温、10分間放置により行った。反応溶液の入ったカラムを15700×gで5分間遠心分離し、Wash Buffer 350μlで懸濁後、付属のスピンカラムセットのカップに移し、15700×gで30秒間遠心分離し、再びWash Buffer 350μlで懸濁後、15700×gで30秒間遠心分離した。カラム内のOligotex(商標)-dT30を、あらかじめ70℃に加温されたRNase free H2O 30μlで懸濁し、付属の新しいスピンカラム用遠心チューブを用いてmRNAを溶出させた。この操作を2回繰り返し、得られた溶液をmRNA溶液とした。
【0024】
次に、得られたmRNA溶液とfirst-strand cDNA Synthesis Kit(GEヘルスケアバイオサイエンス)を用い、添付のプロトコールに従ってfirst-strand cDNAを作製した。具体的には、まず、30μlのmRNAをサーマルクライマーにて65℃で10分間加温した後、氷上で2分間急冷した。その後、11μlのBulk first-strand reaction-mix、1μlのDTT solutionおよび1μlのランダムヘキサマーを添加した。その溶液をサーマルクライマーにて37℃で1時間加温し、得られた溶液をfirst-strand cDNAとした。
【0025】
(3)ネコ由来CysC遺伝子の中間領域の塩基配列の決定
明らかにされている動物種のmRNAの塩基配列の中で高度に保存されている領域の塩基配列を基に、以下の塩基配列を有するネコ由来CysC遺伝子の特異的プライマーを設計した。
【0026】
・上流側プライマー1:5'-SGWSRGCGATWCAACAAR-3'(配列番号3)
・下流側プライマー1:5'-CTGRCAGSTGGAYTTCRM-3'(配列番号4)
なお、上記塩基配列において、SはGまたはC、WはAまたはT、RはAまたはG、YはCまたはT、MはAまたはCをそれぞれ表している。
【0027】
このように設計された上流側プライマー1および下流側プライマー1を用い、first-strand cDNAをPCRで増幅させた。ここで、図4は、first-strand cDNAのPCRの結果を示す電気泳動写真である。アガロース電気泳動によりPCR産物の理論長付近に出現したバンドを確認後、アニーリング温度を理想的な条件60℃に調整し、図4に示すような単一のバンドが得られた。電気泳動で得られた単一のバンドをアガロースゲルから切り出し、DNAを抽出した。DNA抽出は、QIAquick Gel Extraction Kit(QIAGEN)を用いて、添付のプロトコールに従って行った。切り出されたDNAバンドについて、ゲルの重量を測定し、3倍量のQGバッファーを添加し、50℃の恒温槽(TR-2A、ASONE)内で10分間加温し、ゲルを完全に溶解させた後、ゲルと同量のイソプロパノールを添加し、よく混和させた。DNA溶液を、キットに付属のカラムがセットされた2mlのコレクションチューブに加え、室温で13400×g、1分間遠心分離した。その後、コレクションチューブ内の濾液を捨てた後、再びカラムに0.75mlのPEバッファーを添加し、室温で15700×g、1分間遠心分離にて洗浄後、濾液を除去し、更に1分間遠心分離した。その後、カラムを新しい1.5mlのマイクロチューブにセットし、EBバッファー50μlを添加後、室温で1分間放置し、15700×gで1分間遠心分離により抽出液を回収した。
【0028】
次に、TOPO TA Cloning Kit(Invitrogen)およびpGEM-T Easy Vector System(Promega)を用いて、添付のプロトコールに従って得られたDNAを処理した。具体的には、まず、3μlの保存されたPCR産物、1μlのpGEM-T Easy Vector、1μlのT4 DNA Ligase(3 Weiss units/μl)、および2×Rapid Ligation Buffer、5μlのT4 DNA Ligaseを500μlのエッペンドルフチューブ内で混和させ、4℃で一晩インキュベートしてライゲートさせた。得られた反応液を、さらに大腸菌にトランスフォーメーションさせた。2.5μlのライゲーション反応液を、E.coli JM109 Compitent cells(タカラバイオ株式会社)に添加し、氷上に静置後、42℃の恒温槽で45秒間Heat Shockを与え、ただちに2分間急冷した。更に、反応液に、450μlのS.O.C培地(2% Tryptone、0.5% Yeast Extract、10mM NaCl、2.5mM KCl、10mM MgCl2、10mM MgSO4、20mM glucose)を緩やかに添加した後、振盪培養器(PERSONA-11、TAITEC)を用いて37℃で90分間150rpmの速度で振盪培養された。培養後の大腸菌浮遊液を、DMSOで溶解した20mg/ml X-gal(タカラバイオ株式会社)20μlおよび100mM Isopropyl-β-D-thigalactopyranoside(IPTG)100μlが塗布されたLB寒天平板培地(タカラバイオ株式会社)に100μlずつコンラージ棒で均一に塗り広げ、培養器(IS62、TAITEC)を用いて37℃で培養した。18時間後、白いコロニーのみを滅菌爪楊枝で釣菌し、5mg/mlのアンピシリンが添加された3mlのLB液体培地に接種し、37℃で24時間培養した。培養後、QIAPrep Spin Mini Kit50(QIAGEN)を用いて、添付のプロトコールに従って大腸菌のプラスミドを抽出した。得られたプラスミドを、制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりライゲーションの有無を確認した。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約260 baseの塩基配列(配列番号5)が明らかになった。
【0029】
(4)Oligo-Capping法を用いた完全長mRNAの作成
上述した方法でネコ白血球から分離した1〜5μgのmRNAを、40UのRNasin Ribonuclease Inhibitor(Promega)および0.5UのBacterial Alkarine Phosphatase(BAP:タカラバイオ株式会社)を含むBAP buffer中で混和させ、37℃で60分間反応させた。酵素反応後、BAP処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイト(WAKO)を用いて沈澱した。BAP処理したmRNAを、さらに、60UのRNasin、8.0UのTobacco Acid Pyrophosphatase(TAP:WAKO)およびTAP bufferに混和し、37℃で60分間反応させた。酵素反応終了後、BAP-TAP処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイトを用いて濃縮した。BAP-TAP処理したmRNAに、100ngの合成Oligo-RNA(5’-AGCAUCGAGUCGGCCUUGUUGGCCUACUGG-3’:配列番号6)を添加し、65℃で5分間反応後、40UのRNasinおよび50UのT4 RNA ligase(タカラバイオ株式会社)を含むligation bufferに混和し、20℃で3時間反応させた。酵素反応終了後、RNA ligation処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイト(WAKO)を用いて濃縮後、40UのRNasinおよび10UのRNase FreeのDNase I(タカラバイオ株式会社)を含むDNase bufferに混和させ、37℃で10分間反応させた。酵素反応終了後、得られたmRNA溶液に、フェノール・クロロホルム抽出を行い、エタチンメイト(WAKO)で濃縮し、first-strand cDNA Synthesis Kit(GEヘルスケアバイオサイエンス)を用いて、添付のプロトコールに従いoligo(dT)Primerとして5’-AACTGGAAGAATTCGCGGCCGCAGGAAT18-3’(配列番号7)を用い、AMV Reverse transcriptase、first-strand bufferを加え、42℃60分でfirst-strand DNAを合成した(図5)。
【0030】
(5)5’RACE−PCR法
上述の方法で作成されたdsDNAを5’RACE−PCR法により増幅させた。5’RACE−PCR法に用いるプライマーは、上流側プライマーは付加したRNA adaptorの配列を基に、下流側プライマーは既に決定した中間領域の塩基配列を基に、それぞれ以下の塩基配列を有するように設計した。
【0031】
・5’RACE−上流側プライマー:5'-AGCATCGAGTCGGCCTTGTTG-3'(配列番号8)
・5’RACE−下流側プライマー:5'-TTCATCCCAGCCACGACCTGCTTTC-3'(配列番号9)
このように設計されたプライマーを用いて、95℃2分1サイクル、95℃1分、60℃1分、72℃1分を30サイクル、72℃10分1サイクルの条件でPCRを行った。図6は、5’RACE−PCRの結果を示す電気泳動写真であり、図6に示されるように得られたPCR産物は二本のバンドとして確認された。QIAquick Gel Extraction Kit(QIAGEN)を用いてPCR産物からDNAを抽出し、再度PCRを行った後、TOPO TA Cloning Kit(Invitrogen)、pGEM-T Easy Vector System(Promega)および E.coli JM109 Compitent cells(タカラバイオ株式会社)を用いてライゲーションおよびトランスフォーメーションを行った。培養後のE.coli JM109より、QIAPrep Spin Mini Kit50(QIAGEN)を用いてプラスミドを抽出し、得られたプラスミドを制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりDNA断片の挿入を確認した。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約350 baseの塩基配列(配列番号10)が明らかになった。
【0032】
(6)3’RACE−PCR法
上述した方法で作成されたdsDNAを3’RACE−PCR法により増幅した。3’RACE−PCR法に用いるプライマーは、上流側プライマーは既に決定した中間領域の塩基配列を基に、下流側プライマーは3’端に付加したDNA adaptorの配列を基に、それぞれ以下の塩基配列を有するように設計した。
【0033】
・3’RACE−上流側プライマー:5'-GCTCTTTCCAGATATACACTGTACCCT-3'(配列番号11)
・3’RACE−下流側プライマー:5'-AGAATTCGCGGCCGCAGGAATT-3'(配列番号12)
このように設計されたプライマーを用いて、95℃2分1サイクル、95℃1分、55℃1分、72℃1分を30サイクル、72℃10分1サイクルの条件でPCRを行った。図7は、3’RACE−PCRの結果を示す電気泳動写真であり、図7に示されるように得られたPCR産物は完全に単一のバンドとして確認された。さらに、得られたPCR産物について、TOPO TA Cloning Kit(Invitrogen)、pGEM-T Easy Vector System(Promega)および E.coli JM109 Compitent cells(タカラバイオ株式会社)を用いてライゲーションおよびトランスフォーメーションを行った。培養後のE.coli JM109より、QIAPrep Spin Mini Kit50(QIAGEN)を用いてプラスミドを抽出し、得られたプラスミドを制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりDNA断片の挿入が確認された。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約390 baseの塩基配列(配列番号13)が明らかになった。
【0034】
(7)得られたcDNA全体における塩基配列の解析
以上より得られた塩基配列を基に配列全体を構築した(配列番号14)。配列番号14で示される完全長の核酸の配列は796 baseであり、中にCysCをコードしていた。配列番号14に示す塩基配列のうち59番目〜502番目がCysCをコードする構造遺伝子(CysC遺伝子:配列番号2)であることが分かった。
【0035】
図2は、得られたネコのCysC遺伝子のcDNAの塩基配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。図中、互いに共通する塩基配列部分を四角で囲んで示している。CysC遺伝子の核酸長は、ヒト、サルおよびブタが441 base、ウシでは447 base、ラットは423 baseであったのに対し、今回得られたネコのCysC遺伝子ではその長さは444 baseであった。また、表1に示されるように、他動物種(ヒト、サル、ウシ、ブタおよびラット)間のCysC遺伝子の塩基配列の相同性は、67.21〜96.71%の範囲に分布していたのに対し、今回得られたネコのCysC遺伝子の塩基配列と他動物種のCysC遺伝子の塩基配列との相同性は、平均で77.69%であり、得られた塩基配列はネコのCysC遺伝子であることが明らかになった。
【0036】
【表1】

【0037】
<実験例2:ネコ由来CysCの合成>
(1)ネコ由来CysCのアミノ酸配列の解析
実験例1で得られたネコ由来のCysC遺伝子の塩基配列(配列番号2)からアミノ酸配列を翻訳し、ネコ由来のCysCのアミノ酸配列(配列番号1)を解析した。図1は、得られたネコのCysCのアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。図中、互いに共通するアミノ酸配列部分を四角で囲んで示している。結果、ネコ由来のCysCのアミノ酸数は全長で147個であり、ヒト、サルおよびブタのアミノ酸数は146個、ウシのアミノ酸数は148個であり、ラットのアミノ酸数140個と近似した値であった。また、他動物種間で保存されていた構造アミノ酸システインの位置も数も同様であった。また、表2に示されるように、相同性に関しては、他動物種(ヒト、ウシ、ブタおよびラット)間のCysCのアミノ酸配列の相同性は、62.22〜97.26%の範囲に分布していたのに対し、今回得られたネコのCysCのアミノ酸配列と他動物種との平均相同性は69.15%であった。したがって、得られたアミノ酸配列はネコ由来のCysCであることが明らかになった。
【0038】
【表2】
【0039】
(2)GST融合タンパクを用いた組み換え型タンパクの発現および精製
シグナルペプチド領域と考えられる部分を除いた、ネコ由来のCysCタンパク領域のみの核酸を増幅するためにPCRを行った。プライマーは、上流側プライマーは5’末端にEcoRIの制限酵素サイトを付加し、下流側プライマーは3’末端にXhoIの制限酵素サイトを付加し、それぞれ以下の塩基配列を有するように設計した。
【0040】
・上流側プライマー:5'-CACGAATTCACCGGCAGGAGAAACAACAAG-3'(配列番号15)
・下流側プライマー:5'-CACCTCGAGTTATGCATCCTGGCAGCTGGACTTCACCAG-3'(配列番号16)
上述したような上流側プライマーおよび下流側プライマーを用いて、95℃2分1サイクル、95℃1分、75℃1分、72℃1分30サイクル、72℃10分1サイクルの条件でPCRを行った。PCR産物をアガロースゲル電気泳動した後、アガロースゲルからDNA抽出した。DNA抽出溶液をフェノールと等量混合した後、15700×gで5分間遠心分離後、核酸が含まれる水層を分離した。分離した水層にクロロホルムを等量混合し、15700×gで5分間遠心分離した後、上清を分離した。次に、分離後の溶液に2.5倍量の100%エタノールを添加し、−80℃で30分静置し、その後15700×gで5分間遠心分離した後、上清を除去し、沈渣を得た。沈渣に70%エタノールを添加し、15700×gで5分間遠心分離後、上清を除去し、PCR産物の濃縮試料を得た。PCR産物の濃縮試料に、5μlのEcoRI(タカラバイオ株式会社)、5μlのXhoI(タカラバイオ株式会社)、5μlのH.Buffer(500mM Tris-HCl, pH7.5、100mM MgCl2、10mM Dithiothreitol、1000mM NaCl)および35μlのRNase free H2Oを混和した。また、5μl(2.5μg)のpGEX6P-1(GEヘルスケアバイオサイエンス)を、5μlのEcoRI、5μlのXhoI、5μlのH.Bufferおよび30μlのRNase free H2Oと混和した。各溶液を37℃で1晩インキュベートすることで制限酵素処理した後、アガロースゲル電気泳動を行い、QIAquick Gel Extraction Kit(QIAGEN)を使用して、各DNAバンドの抽出を行った。DNA Ligation Kit(タカラバイオ株式会社)を用いてライゲーションを行った。すなわち、5μlのLigation Mix、1μlの制限酵素処理されたCysCのcDNA溶液および4μlのpGEX6P-1を混和し、16℃で1晩静置し、CysCのcDNAをライゲーションさせたplasmid vector(pGEX-CysC)が作出された。さらに、このpGEX-CysC溶液2.5μlを、E.coli JM109 Competent Cells(タカラバイオ株式会社)25μlに添加し、氷上で30分間静置し、次に42℃の恒温槽で45秒間Heat Shockを与え、直ちに2分間氷冷後SOC培地(2% Tryptone、0.5% Yeast extract、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM Glucose)250μlを緩やかに加え、37℃1時間保温した。pGEX-CysCがトランスフェクトされた大腸菌溶液100μlをアンピシリン加LB培地にそれぞれ塗布し、37℃で1晩静置後、コロニーを釣菌し、アンピシリン加LB液体培地1.2mlに混和後、37℃で1晩培養した。培養後の液体培地は、13400×gで1分間遠心分離後、上清を完全に除去し、得られた沈渣から、QIAPrep Spin Mini Kit50(QIAGEN)を用いて、pGEX-CysCの抽出を行った。このpGEX-CysCを、アガロースゲル電気泳動法により確認した。また、pGEX-CysCのサブクローニングの成否は、T7プライマーを用いてDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用した塩基配列解析により行った。
【0041】
(3)GST融合タンパク発現の確認
pGEX-CysCがトランスフェクトされた大腸菌を、37℃、1晩LB培地で培養後、100μlをIsopropyl-β-D-thiogalactopyranoside(IPTG:0.1mM)20μlと混和し、30℃で約2時間振盪培養(BR40-LF、TAITEC)した。振盪培養後の大腸菌溶液を、15700×gで1分間遠心分離後、上清を除去し、沈渣に可溶化液(50μlの50mM Tris-HCl、100μlの1×RIPA Lysis Buffer(Up State)、140μlのProtease Inhibitor、710μlのH2O)30μlを加え可溶化後、15700×gで5分遠心分離し、上清と沈渣とに分けた。上清30μlに、30μlの2×SB溶液(2% SDS、40% Glycerol、0.6% BPB、25mM Tris-HCl Buffer(pH6.8、20℃))および1μlの2MEを加え、95℃で3分間加温した。沈渣にSB溶液20μlを加え、超音波破砕機(UR-20P、TOMY SEIKO CO,LTD)で5秒間破砕後、95℃で3分間加温した。その後、上清および沈渣について、SDS−PAGEにてGST融合タンパク発現の確認およびGST融合タンパクの大腸菌での可溶性を確認した。
【0042】
(4)SDS−PAGE法
SDS−PAGEは、コンパクトPAGE(AE-7300、ATTO)を用いてLaemmliの方法に準拠し、これに以下に示す修正を加え実施した。すなわち、分離ゲルの組成は、15% Acrylamide、0.2% N,N-Methylene-bis-Acrylamide、0.1% SDS、375mM Tris-HCl buffer(pH8.8、20℃)とした。ゲルは2・4連ゲル作製器(AE-7344、ATTO)を用いて作製した。電極緩衝液の組成は、0.1% SDS、129mM Glycine、25mM Tris(pH8.3、20℃)とした。泳動用試料(SB)の組成は、1% SDS、20% Glycerol、0.3% BPB、12.5mM Tris-HCl Buffer(pH6.8、20℃)とした。また、マーカーとして、プレステインドSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)もしくはSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)を用いた。泳動は、Tris-Gly/PAGE Highモードで30分泳動した後、Tris-Gly/PAGE Lowモードにして、下部イオン界面がゲル下端から1〜2mm上方の位置に移動したときに終了した。SDS−PAGE終了後のゲルについて、Oakle法に準拠した銀染色法を実施した。具体的には、ゲルを30%ethanol、10% acetic acid溶液にて固定後、洗浄し、20% ethanolに5分間2回浸漬した。20% ethanol除去後、5%glutaraldehyde溶液にて4分間反応させ、純水で洗浄後、20% ethanolに4分間2回浸漬した。その後、純水で洗浄し、アンモニア性硝酸銀溶液にて5分間反応させ、純水で洗浄後、0.005%citric acid、0.019%formaldehyde溶液で発色させた。発色確認後のゲルについて、20%ethanol、10% acetic acid溶液にて5分間固定し、20%ethanolに5分間2回浸漬後、写真を撮影した。なお、銀染色法はすべて遮光条件下にて実施した。
【0043】
(5)GST融合タンパクの発現誘導と単離
His−Tag付きGST融合タンパクの発現が確認された大腸菌を、アンピシリン加LB寒天培地に塗布し、コロニーを釣菌後、3mlのアンピシリン加LB液体培地に加え1晩37℃で振盪培養した。続いて、その培養液3mlを、アンピシリン加LB液体培地250mlに加え、37℃で約150分振盪培養後、2.5mlの0.1mM IPTGを添加し、約2時間、30℃で振盪培養した。GST融合タンパク発現誘導後の培養液を6000×gで15分間遠心分離した沈渣を、Phosphate buffer saline(PBS:140mM NaCl、2.7mM KCl、10mM Na2PO4、1.8mM KH2PO4、pH7.3)に懸濁し、超音波破砕機で20秒間×5回破砕後、Triton X100を最終濃度1%になるように加え、室温で撹拌しながら30分間放置後、9300×gで20分間遠心分離し、上清と沈渣をSDS−PAGEにて分析した。泳動の結果、GST融合タンパクは上清および沈渣の両分画に含まれていたが、可用性分画の方が利便性に優れているのでアフィニティークロマトグラフィーには上清を用いた(図8)。
【0044】
(6)アフイニティークロマトグラフィー法
上述のようにして得られた上清について、GSTrap HP column(GEヘルスケアバイオサイエンス)を用いてアフイニティークロマトグラフィーを行った。上清を結合バッファーであるPBSにて透析した後、カラムに添加し結合バッファーでよく洗浄し、10mM reduced glutathione加50mM Tris-HCl(pH8.0)で溶出させた。なお、カラム操作はペリスタポンプ(SJ-1211L、ATTO)を用い、流速0.5ml/minで添加した。また、溶出液の吸光度は紫外部吸光度モニター(AC-5100L、ATTO)を用いて吸光波長220nmでモニターされ、記録計(R-01A、RIKADENKI)で記録された。溶出液のSDS−PAGE像は図8に示されたとおり、GST融合タンパクをメインバンドとして複数のバンドが確認された。得られたGST融合タンパク溶出液2mlに、濃度が1mMになるようにDTTを添加し混和後、分子量13kDaカットの透析膜(UC30-32-100、三光純薬株式会社)に入れられ150mM NaCl、1mM EDTA加50mM Tris-HCl(pH7.5)2Lを用いて約6時間透析された。透析後のGST融合タンパク溶出液について、DC Protein Assay(Bio-Rad)を用いてタンパク定量後、タンパク量200μgに対しPreScission Protease(GEヘルスケアバイオサイエンス)1μlを添加して混和し、4℃で6時間以上反応させた。酵素切断後のSDS−PAGE像では、切断されたGSTとネコの組み替え型CysC(rFeCysC)が確認された。さらに、この溶液を高速液体クロマトグラフィー(HPLC)用の試料とした。
【0045】
(7)HPLC法
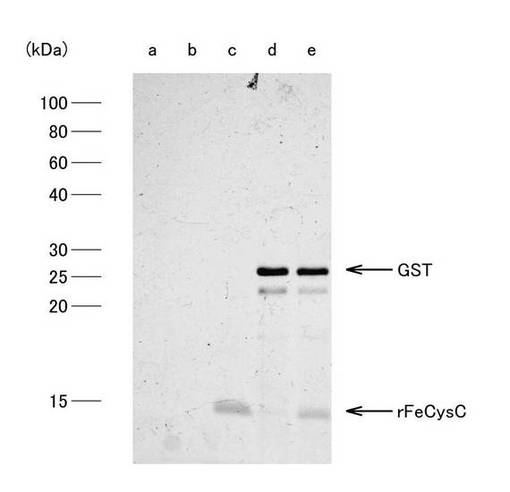
HPLCシステムは、システムコントローラー(SCL-10A VP、Shimadzu)、送液ユニット(LC-10AD VP、Shimadzu)、紫外部分光高度計(SPD-10A VP、Shimadzu)、カラムオーブン(CTO-10A VP、Shimadzu)および脱気ユニット(DGU-14A、Shimadzu)から構成され、カラムはMightysilRP-18 GP250-4.6(関東化学)を使用した。HPLCの分離条件は、移動相の流速を1ml/min、試料添加量を400μlとし、0.1% trifluoroacetic acid(TFA)溶液で平衡化させたカラムに、0.1%TFA加acetonitrile溶液を用いてacetonitrile濃度0〜80%のライナーグラジエントにして行った。なお、溶出液は、吸光波長220nmで吸光度をモニターし、検出されたピークを分取し、濃縮遠心機(CC-181、TOMY)にて1時間遠心分離後、凍結乾燥機(FDU-540、EYELA)にて乾燥させた後−20℃で保存した。クロマトパターンは、図9に示すとおりで、だいたい5個のピークa, b, c, d, eに分離され、それぞれの溶出された分画のタンパク組成は、SDS−PAGE法により分析された。図10は、HPLCの主要画分a, b, c, d, eについてのSDS−PAGEの結果を示す写真である。分析の結果、図10に示されるように、c分画に単独で溶出された。
【0046】
<実験例3:抗体産生ハイブリドーマ、抗rFeCysC抗体の作製>
実験例2で合成したタンパクを組み替え型ネコCysC(rFeCysC)の抗原としてモノクローナル抗体を作製するにあたり、まずは抗体産生ハイブリドーマを作製した。
【0047】
(1)抗体産生ハイブリドーマの作製
(1−1)免疫法
免疫法は、精製rFeCysCを抗原としてBalb/cマウスの後肢肉球(footpad)の皮下に注射することにより行った。免疫は5日間隔で4回行い、初回から第3回目までの免疫は抗原溶液100μl(1mg/ml)とアジュバントを等量混合させてエマルジョン化させた抗原液200μl(50μg/foot)を、また、最終免疫では抗原溶液20μl(10μg/foot)のみを用いて行った。また、アジュバントは初回免疫ではAdjuvant Complete Freund(和光純薬工業株式会社)を、第2から3回目の免疫ではAdjuvant Incomplete Freund(和光純薬工業株式会社)を用いた。
【0048】
(1−2)細胞融合
最終免疫から3日後、膝窩リンパ節を摘出し、リンパ球を回収後、GenomONE-CF(石原産業株式会社)を用いて、細胞融合を行った。また、ミエローマ細胞としてはP3X63-Ag8.653(大日本住友製薬株式会社)を用いた。融合方法は添付のプロトコールに従って行った。具体的には、まず、リンパ球とミエローマ細胞とを細胞数が5:1の比率になるように混合し、1000rpm、4℃で5分間遠心した後、上清を除去した。そこに、氷冷した融合用緩衝液をリンパ球108cellsあたり1ml添加し、均一に懸濁した後、氷冷したHVJ-Envelope懸濁液を細胞混合液1mlあたり25μl添加した。細胞懸濁液を氷上で5分間静置した後、1000rpm、4℃5分間遠心し、上清を除去せずに細胞がペレット化した状態のまま37℃で15分間インキュベートした。
【0049】
インキュベート終了後、37℃に加温した増殖用培地をリンパ球108cells当たり50ml加え、懸濁後、96穴プレート(96 Well Cell Culture Plate:Greiner bio-one)に100μl/wellで播種した。なお、増殖用培地としてRPMI1640(Invitrogen)にペニシリンG(PG;明治製薬株式会社)10万IU/ml、ストレプトマイシン(SM;明治製薬株式会社)100mg/ml、7.5% Bri Clone(IL-6、ヒト、ブライクローン;Cat. No. BR-001、大日本住友製薬株式会社)、10% 非働化ウシ胎仔血清(FBS;株式会社ニチレイ)を加えたものを用い、添加、懸濁の際は穏やかに操作した。24時間培養後、培養培地を上記の増殖用培地に2% HAT(Invitrogen)を添加したHAT培地に交換した。
【0050】
(2)抗体産生ハイブリドーマのスクリーニング
得られたハイブリドーマについて、細胞融合から1週間後にELISA法を用いた一次スクリーニングを行い、この結果、反応陽性となったwellのハイブリドーマのみをWestern blotting法を用いた二次スクリーニングで確認した。
【0051】
(2−1)一次スクリーニング
rFeCysCを抗原としたELISA法を用いて、抗体産生ハイブリドーマの一次スクリーニングを行った。ELISAプレートとしては、96 Well ELISA Microplate(Greiner bio-one)を使用した。また、プレートの洗浄には自動洗浄機(Auto Mini Washer AMW-8、バイオテック株式会社)を用い、洗浄液としてはPBS(1.37M NaCl、27mM KCl、100mM Na2HPO4、18mM KH2PO4、pH7.4、25℃)を使用した。固相として、PBSで3μg/mlに調整したrFeCysCを50μl/wellでプレートに添加し、4℃で一晩反応させた。固相反応終了後、プレートの抗原液を捨て、ブロッキング液として0.5% Bovine Serum Albumin(BSA;和光純薬工業株式会社)を添加したPBSを150μl/wellで加え、37℃で60分間反応させた。ブロッキング反応終了後、プレートを1回洗浄し、一次抗体として各ハイブリドーマ培養の培養上清を50μl/wellで加え、37℃で60分間反応させた。一次抗体反応終了後、プレートを1回洗浄し、二次抗体として0.1% BSAを添加したPBSで1000倍に希釈したペルオキシダーゼ標識抗マウスIgG抗体(SIGMA-ALDRICH)を50μl/wellで加え、37℃で60分間反応させた。二次抗体反応終了後、プレートを3回洗浄し、基質液として0.04% o-フェニレンジアミン、0.04% H2O2を添加したPBSを150μl/wellで加え、室温、遮光下で30〜60分間反応された。基質反応終了後、3M H2SO4を反応停止液として50μl/wellで加え、1分間振盪後、Microplate Reader(Model 550、BIO-RAD)で波長490nmにおける吸光度を測定した。吸光度の高かった陽性wellの細胞を、24穴プレート(24 Well Cell Culture Plate;Greiner bio-one)に移して培養した。
【0052】
(2−2)二次スクリーニング
rFeCysCを抗原としたWestern blotting法で確認し、抗体産生ハイブリドーマの二次スクリーニングを行った。Lowryの方法に基づき、DC Protein Assay Kit(BIO-RAD)を用いて、Microplate Readerで波長655nmにおける吸光度を測定し、タンパク質を定量した。検量線はBSAを用いて作製した。Western blotting法はTowbinらの方法に準拠し、以下のように実施した。転写膜はポリビニリデンジフルオリド(PVDF)膜(BIO-RAD)を使用した。PVDF膜は100% methanolに10秒間、さらに転写用電極buffer(25mM Tris-HCl(pH8.3、20℃)、192mM glycine、5% methanol)に30分間浸潤し、泳動に供した。転写装置の組み立ては、陽極電極板上に下から順に濾紙(BIO-RAD)、PVDF膜、SDS−PAGE終了後のゲル、濾紙を重層し、その上に陰極電極板を固定した。なお、濾紙は予め電極bufferに2〜3分浸しておいた。転写条件は1.9mA/cm2の定電流で60分間とした。転写終了後のPVDF膜は10mM Tris-HCl(pH7.5、20℃)、140mM NaCl、0.01% Tween20(TBST)に0.5% BSAを加え、室温で60分間振盪し、ブロッキング操作を行った。ブロッキング終了後、TBSTで5分間、2回振盪洗浄し、一次抗体として細胞の培養上清を用い、室温で90分振盪反応させた。一次抗体反応終了後、TBSTで5分間、2回振盪洗浄した後、TBSTで1000倍希釈したペルオキシダーゼ標識抗マウスIgG抗体を、室温で60分間振盪反応させた。二次抗体反応終了後、TBSTで5分間、2回振盪洗浄し、0.06% 3,3-diaminobenzidine tetra-hydrochloride、0.03% H2O2、50mM Tris-HCl(pH7.6、20℃)を基質反応液として使用し、1〜5分間反応させた。基質反応終了後、水洗し反応を停止させた後、乾燥して保存した。反応陽性を示したハイブリドーマについては後述する限界希釈法によりクローニングを行った。
【0053】
(3)クローニング
ハイブリドーマのクローニングには限界希釈法を用いた。具体的には、スクリーニング後のハイブリドーマを2cells/100μlとなるようにHAT培地で希釈し、100μl/wellで96穴プレートに播種した。ハイブリドーマはセミコンフルエントになったところで24穴プレートに拡大培養し、再びセミコンフルエントになるまで培養した後、二次スクリーニングと同様にrFeCysCを抗原としたWestern blotting法で確認した。このクローニング操作を2回行った。また、ハイブリドーマを長期間継代培養することにより抗体産生能が減少するのを防ぐため、クローニング毎に細胞凍結保存液(セルバンカー(BLC-1)、十慈フィールド株式会社)を用いて保存した。
【0054】
(4)抗体産生ハイブリドーマの大量培養および抗rFeCysC・mAbの採取と精製
クローニングが終了したハイブリドーマを、浮遊細胞培養フラスコ(フィルタートップSCフラスコ250ml 75cm2;Greiner bio-one)を用いて大量培養した。なお、培養は37℃、5% CO2、5日間、CO2インキュベーター(十慈フィールド株式会社)で行い、培地としてはHAT培地を用いた。大量培養されたハイブリドーマを無血清RPMIで懸濁し、ヌードマウス(Balb/c-nu)の腹腔内に2×107cells/headで投与した。投与してから10〜20日後、腹水を採取した。ヌードマウスから採取した腹水を室温で1時間、あるいは4℃で一晩静置した後、3000rpm、4℃で5分間遠心し、腹水中のフィブリン、ハイブリドーマ、赤血球などを除去した。分離した上清を50%の硫安で塩析させた。具体的には、氷上で撹拌しながら上清と等量の飽和硫酸アンモニウム溶液を徐々に滴下し、滴下後さらに1時間撹拌した。これを10000rpm、4℃で10分間遠心し、沈殿物を20mM リン酸ナトリウムbuffer(pH7.0)に溶解した。塩析後のグロブリン溶液を、20mM リン酸ナトリウムbuffer(pH7.0)で平衡化したSephadex G-25 Fine(GEヘルスケアバイオサイエンス)カラム(内径1.5cm、長さ30cm)を用いて脱塩した。クロマトグラフィーの流速をペリスタポンプ(SJ-1211L、ATTO)で0.5ml/minに調節した。脱塩後のグロブリン溶液を、エコカラム(内径2.5cm、長さ10.0cm:BIO-RAD)に充填したProtein G Sepharose 4 Fast Flow(GEヘルスケアバイオサイエンス)を用いたアフィニティークロマトグラフィー法により精製した。具体的には、脱塩後のグロブリン溶液を20mM リン酸ナトリウムbuffer(pH7.0)で平衡化されたカラムに流速0.5ml/minで添加し、その後カラムを100mM glycine(pH3.0)で溶出させた。溶出液は直ちに10分の1量の1M Tris-HCl(pH9.0)で中和した。精製後の溶出液を50mM 酢酸アンモニウム(pH7.0)で平衡化させたSephadex G-25 Fineカラム(内径2cm、長さ30cm)で脱塩させた後、Freeze Dryer(FDU540、EYELA東京理化器械株式会社)を用いて凍結乾燥し、−20℃で保存した。
【0055】
(5)アイソタイプの決定
Mouse Monoclonal Isotyping Kit(コスモバイオ株式会社)を用い、方法は添付のプロトコールに従って、得られた抗rFeCysC・mAbのアイソタイプの決定を行った。具体的には、抗rFeCysC・mAbサンプルをdevelopment tubeに150μl加え、室温で30秒間インキュベートした後、撹拌した。そこに、isotyping stripを入れ、さらに室温で10〜15分間インキュベートした後、classとsubclassを読み取った。抗rFeCysC・mAbサンプルとしては、2回目のクローニングが終了したハイブリドーマの培養上清を1% BSAを添加したPBSで10倍に希釈したものを用いた。モノクローナル抗体は2種類得られ、一方の抗体AのアイソタイプはIgG1のκ鎖、もう一方の抗体BのアイソタイプはIgG2aのκ鎖であった。
【0056】
(6)ネコのnativeなCysCに対する特異性
抗体A、Bについて、慢性腎疾患(Chronic Kidney disease:CKD)のネコの尿タンパクを抗原としたWestern blotting法を用いて、ネコのnativeなCysCに対する特異性を確認した。Western blotting法は上述の方法と同様に実施した。ただし、SDS−PAGEの泳動用尿タンパクサンプルは、ネコの尿タンパクを2-MercaptoethanolでSS結合が切断したものを用いた。図3は、ネコのnativeなCysCに対する抗体A、Bの特異性の実験結果を示す写真であり、それぞれレーン1はrFeCysC、レーン2はCKDネコの尿タンパクを示している。図3に示されるように、抗体A、Bともに、nativeなCysCとも特異的に反応することが確認された。
【0057】
今回開示された実施の形態および実験例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【技術分野】
【0001】
本発明は、ネコに由来するシスタチンCおよびそれをコードする遺伝子に関する。また本発明は、ネコに由来するシスタチンCに対する抗体およびそれを用いたネコ腎症の診断用キット、診断方法にも関する。
【背景技術】
【0002】
近年、少子化に伴い、ペットを飼う世帯は増加の一途をたどっている。しかしながら、ペットの性質に即した飼い方がなされていないケースも少なくはない。特に、偏食の結果、ペットが糖尿病などの成人病的症状を引き起こしてしまい、ペットを獣医に通院させるケースまで見られる。
【0003】
このような現状から、近年はペットの診断に関する事業が拡大しつつある。仮に、ペットの腎症を早期に発見することができれば、獣医師は、飼い主によるペットの飼い方、特に食事の与え方について改善を指導できるようになる。一般に、腎症のマーカーの1つとして、シスタチンC(CysC)が挙げられる。
【0004】
シスタチンCは、たとえばヒト由来の場合には、分子量13000Daの塩基性低分子タンパクである。ヒト由来のシスタチンCは、ヒトの全身の細胞で産生されており、細胞内外の環境変化にはほとんど影響を受けないで一定の産生量で細胞外に分泌され、近年では、糖尿病性腎症などの早期診断の指標として有用であるとする報告例がみられる
しかしながら、ネコ由来のシスタチンCに関しては、当該タンパクに特異的な抗体が存在しないどころ、当該タンパクのアミノ酸配列すら解明されていないのが現状である。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Journal of Veterinary Internal Medicine. 22(5): 1111-1117, 2008
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、上記課題を解決するためになされたものであって、その目的とするところは、ネコ由来のシスタチンCに特異的な抗体を提供し、さらにはそれを用いることでネコの腎症を迅速かつ簡便に診断できる方法、キットを提供することである。
【課題を解決するための手段】
【0007】
本発明者は、鋭意研究の結果、ネコの遺伝子の中でシスタチンCをコードする構造遺伝子を初めて特定し、当該構造遺伝子からネコ由来のシスタチンCを発現させ、そのアミノ酸配列も解析した。さらにはネコ由来シスタチンCに特異的な抗体を作製し、本発明を完成するに至った。すなわち、本発明は以下のとおりである。
【0008】
本発明は、配列番号1で表わされるアミノ酸配列を有するタンパク質を提供する。
本発明はまた、上述した本発明のタンパク質をコードする構造遺伝子についても提供する。本発明の構造遺伝子は、配列番号2で表わされる塩基配列を有することが好ましい。
【0009】
本発明はさらに、ネコ由来シスタチンCに特異的に結合する抗体についても提供する。本発明の抗体は、上述した本発明のタンパク質を抗原として、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものであることが、好ましい。
【0010】
本発明は、上述した本発明の抗体を含むネコ腎症の診断用キットについても提供する。
本発明は、上述した本発明の抗体を用いたネコ腎症の診断方法についても提供する。
【発明の効果】
【0011】
本発明によれば、従来と比較して格段に迅速かつ簡便にネコ腎症を診断することができるようになる。
【図面の簡単な説明】
【0012】
【図1】ネコ由来のCysCのアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。
【図2】ネコ由来のCysC遺伝子のcDNAの塩基配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。
【図3】実験例3におけるネコのnativeなCysCに対する抗体A、Bの特異性の実験結果を示す写真である。
【図4】実験例1におけるfirst-strand cDNAのPCRの結果を示す電気泳動写真である。
【図5】実験例1で使用したキットによって合成できる2本鎖DNAとプライマーとの位置関係を模式的に示す図である。
【図6】実験例1における5’RACE−PCRの結果を示す電気泳動写真である。
【図7】実験例1における3’RACE−PCRの結果を示す電気泳動写真である。
【図8】実験例2におけるGST融合タンパク発現後のSDS−PAGEの結果を示す写真である。
【図9】実験例2におけるHPLCの結果得られたクロマトパターンを示すグラフであり、左側の縦軸は波長220nmでの吸光度、右側の縦軸はアセトニトリル濃度(%)、横軸は時間(分)を示している。
【図10】実験例2におけるHPLCの主要画分a, b, c, d, eについてのSDS−PAGEの結果を示す写真である。
【発明を実施するための形態】
【0013】
本発明によれば、配列番号1で表わされるアミノ酸配列を有するタンパク質が提供される。本発明者は、ネコの遺伝子の中でシスタチンCをコードする構造遺伝子(本明細書中において「CysC遺伝子」と呼称する。)を初めて特定し、当該CysC遺伝子によりネコ由来のシスタチン(本明細書中において「CysC」と呼称する。)のアミノ酸配列も初めて解析した。配列番号1に示されるアミノ酸配列を有する本発明のタンパク質が、今回、本発明者によって初めてアミノ酸配列が特定されたネコ由来のシスタチンCである。
【0014】
ここで、図1は、配列番号1で表わされる本発明のタンパク質(ネコのCysC)のアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。図1中、四角で囲っている部分は、各動物種間で共通するアミノ酸配列である。配列番号1で表わされる本発明のタンパク質のアミノ酸数は全長で147個であり、ヒト、サルおよびブタのアミノ酸数は146個、ウシのアミノ酸数は148個であり、ラットのアミノ酸数140個と近似した値であり、また、他動物種間で保存されていた構造アミノ酸システインの位置も数も同様である。詳細は実験例2において後述するが、本発明のタンパク質のアミノ酸配列は、他動物種のCysCのアミノ酸配列との平均相同性は69.15%であり、他動物種(ヒト、ウシ、ブタおよびラット)間のCysCのアミノ酸配列の相同性は、62.22〜97.26%の範囲に分布していることからすると、本発明のタンパク質はネコ由来のCysCであると考えられる。
【0015】
本発明のネコ由来のCysCは、人工的な合成により好適に得ることができる。今回、本発明者は、ネコ由来CysCの構造遺伝子(CysC遺伝子)の塩基配列(配列番号2で表わされる塩基配列)を初めて見出した。本発明は、ネコ由来シスタチンをコードする構造遺伝子についても提供するものであり、この構造遺伝子は、配列番号2で表わされる塩基配列を有することが、好ましい。すなわち、本発明の構造遺伝子は、配列番号2で表わされる塩基配列をエキソンとして含んでいるのであれば、上記以外の塩基配列をイントロンとして含んでいてもよい。
【0016】
ここで、図2は、配列番号2で表わされる本発明の構造遺伝子(ネコのCysC遺伝子)を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。図2中、四角で囲っている部分は、各動物種間で共通する塩基配列である。CysC遺伝子の核酸長は、ヒト、サルおよびブタが441 base、ウシでは447 base、ラットは423 baseであったのに対し、配列番号2で表わされる本発明のネコのCysC遺伝子ではその長さは444 baseであった。また、配列番号2で表わされる本発明のネコのCysC遺伝子の塩基配列と他動物種のCysC遺伝子の塩基配列との相同性は平均で77.69%であり、他動物種(ヒト、サル、ウシ、ブタおよびラット)間のCysC遺伝子の塩基配列の相同性は、67.21〜96.71%の範囲に分布していることからすると、本発明の構造遺伝子はネコ由来のCysC遺伝子であると考えられる。
【0017】
本発明は、ネコ由来のCysCに特異的に結合する新規な抗体についても提供する。本発明者は、詳細は実験例3にて後述するように、上述した本発明のネコ由来のCysC遺伝子からネコ由来のCysCを発現させ、これを抗原として抗体を産生し得る細胞を作製した。このような細胞株は新規なものであり、出願人らは、今回、平成21年12月1日付けで独立行政法人 産業技術総合研究所 特許生物寄託センターに寄託を行った(受領番号:FERM AP-21877、FERM AP-21878)。
【0018】
本発明の抗体は、好ましくは、上述した本発明のタンパク質を抗原として、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)または細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである。ここで、図3は、ネコのnativeなCysCに対し本発明の抗体が特異的に結合した実験結果を示す写真である。詳細は実験例3として後述するが、図3に示される抗体Aは細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)により産生されたIgG1のκ鎖のアイソタイプのモノクローナル抗体であり、抗体Bは細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたIgG2aのκ鎖のアイソタイプの抗体である。図3に示されるように、本発明の抗体は、ネコのnativeなCysCに対し特異的に結合し得るものであることが分かる。
【0019】
本発明は、さらに上述した本発明の抗体を利用したネコ腎症の診断方法、診断キットについても提供するものである。本発明の抗体は、ネコ腎症のマーカーであるCysCに特異的に結合し得るものであるため、たとえばネコの尿をサンプルとして用いて、当該ネコが腎症に罹っているか否か、従来と比較して迅速かつ簡便に診断することが可能となる。本発明の診断キットは、本発明の抗体以外に、たとえばウェル、色原性基質溶液、反応停止液、洗浄液、標準溶液などを含むことができる。
【0020】
<実験例>
以下、実験例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【0021】
<実験例1:CysC遺伝子の特定>
(1)供試動物
本実験例においては、実験動物施設にて維持されている血液生化学および尿生化学検査において異常が認められない、10歳齢の雄の日本ネコ1頭を使用した。このネコの飼養条件は、12時間昼、12時間夜とし猫用ケージにて飼育し、1日1回の給餌による自由採食、自由飲水とした。
【0022】
(2)ネコ白血球からのTotal RNAの抽出
まず、EDTA採血管を用いて供試動物の外頸静脈からネコ血液を採取した。採血された5mlの血液をコニカルチューブに移し、3000×rpmで5分間遠心後、バフィーコート(白血球層)を分離させた。次に、QIAamp RNA Blood Kit(QIAGEN)を用いて、添付のプロトコールに従ってTotal RNAを抽出した。得られたTotal RNAは使用時まで4℃で保存した。
【0023】
次に、Oligotex(商標)-dT30 Super mRNA Purification Kit(タカラバイオ株式会社)を用いて、添付のプロトコルに従ってTotal RNAからmRNAを分離精製した。具体的には、まず、60μlのTotal RNAを70μlの2×Binding Bufferおよび14μlのOligotex(商標)-dT30と混和した後、サーマルクライマー(PC801、ASTEC)で70℃、3分間加温した。加温後、mRNAとOligotex(商標)-dT30 Superとのハイブリダイゼーションを室温、10分間放置により行った。反応溶液の入ったカラムを15700×gで5分間遠心分離し、Wash Buffer 350μlで懸濁後、付属のスピンカラムセットのカップに移し、15700×gで30秒間遠心分離し、再びWash Buffer 350μlで懸濁後、15700×gで30秒間遠心分離した。カラム内のOligotex(商標)-dT30を、あらかじめ70℃に加温されたRNase free H2O 30μlで懸濁し、付属の新しいスピンカラム用遠心チューブを用いてmRNAを溶出させた。この操作を2回繰り返し、得られた溶液をmRNA溶液とした。
【0024】
次に、得られたmRNA溶液とfirst-strand cDNA Synthesis Kit(GEヘルスケアバイオサイエンス)を用い、添付のプロトコールに従ってfirst-strand cDNAを作製した。具体的には、まず、30μlのmRNAをサーマルクライマーにて65℃で10分間加温した後、氷上で2分間急冷した。その後、11μlのBulk first-strand reaction-mix、1μlのDTT solutionおよび1μlのランダムヘキサマーを添加した。その溶液をサーマルクライマーにて37℃で1時間加温し、得られた溶液をfirst-strand cDNAとした。
【0025】
(3)ネコ由来CysC遺伝子の中間領域の塩基配列の決定
明らかにされている動物種のmRNAの塩基配列の中で高度に保存されている領域の塩基配列を基に、以下の塩基配列を有するネコ由来CysC遺伝子の特異的プライマーを設計した。
【0026】
・上流側プライマー1:5'-SGWSRGCGATWCAACAAR-3'(配列番号3)
・下流側プライマー1:5'-CTGRCAGSTGGAYTTCRM-3'(配列番号4)
なお、上記塩基配列において、SはGまたはC、WはAまたはT、RはAまたはG、YはCまたはT、MはAまたはCをそれぞれ表している。
【0027】
このように設計された上流側プライマー1および下流側プライマー1を用い、first-strand cDNAをPCRで増幅させた。ここで、図4は、first-strand cDNAのPCRの結果を示す電気泳動写真である。アガロース電気泳動によりPCR産物の理論長付近に出現したバンドを確認後、アニーリング温度を理想的な条件60℃に調整し、図4に示すような単一のバンドが得られた。電気泳動で得られた単一のバンドをアガロースゲルから切り出し、DNAを抽出した。DNA抽出は、QIAquick Gel Extraction Kit(QIAGEN)を用いて、添付のプロトコールに従って行った。切り出されたDNAバンドについて、ゲルの重量を測定し、3倍量のQGバッファーを添加し、50℃の恒温槽(TR-2A、ASONE)内で10分間加温し、ゲルを完全に溶解させた後、ゲルと同量のイソプロパノールを添加し、よく混和させた。DNA溶液を、キットに付属のカラムがセットされた2mlのコレクションチューブに加え、室温で13400×g、1分間遠心分離した。その後、コレクションチューブ内の濾液を捨てた後、再びカラムに0.75mlのPEバッファーを添加し、室温で15700×g、1分間遠心分離にて洗浄後、濾液を除去し、更に1分間遠心分離した。その後、カラムを新しい1.5mlのマイクロチューブにセットし、EBバッファー50μlを添加後、室温で1分間放置し、15700×gで1分間遠心分離により抽出液を回収した。
【0028】
次に、TOPO TA Cloning Kit(Invitrogen)およびpGEM-T Easy Vector System(Promega)を用いて、添付のプロトコールに従って得られたDNAを処理した。具体的には、まず、3μlの保存されたPCR産物、1μlのpGEM-T Easy Vector、1μlのT4 DNA Ligase(3 Weiss units/μl)、および2×Rapid Ligation Buffer、5μlのT4 DNA Ligaseを500μlのエッペンドルフチューブ内で混和させ、4℃で一晩インキュベートしてライゲートさせた。得られた反応液を、さらに大腸菌にトランスフォーメーションさせた。2.5μlのライゲーション反応液を、E.coli JM109 Compitent cells(タカラバイオ株式会社)に添加し、氷上に静置後、42℃の恒温槽で45秒間Heat Shockを与え、ただちに2分間急冷した。更に、反応液に、450μlのS.O.C培地(2% Tryptone、0.5% Yeast Extract、10mM NaCl、2.5mM KCl、10mM MgCl2、10mM MgSO4、20mM glucose)を緩やかに添加した後、振盪培養器(PERSONA-11、TAITEC)を用いて37℃で90分間150rpmの速度で振盪培養された。培養後の大腸菌浮遊液を、DMSOで溶解した20mg/ml X-gal(タカラバイオ株式会社)20μlおよび100mM Isopropyl-β-D-thigalactopyranoside(IPTG)100μlが塗布されたLB寒天平板培地(タカラバイオ株式会社)に100μlずつコンラージ棒で均一に塗り広げ、培養器(IS62、TAITEC)を用いて37℃で培養した。18時間後、白いコロニーのみを滅菌爪楊枝で釣菌し、5mg/mlのアンピシリンが添加された3mlのLB液体培地に接種し、37℃で24時間培養した。培養後、QIAPrep Spin Mini Kit50(QIAGEN)を用いて、添付のプロトコールに従って大腸菌のプラスミドを抽出した。得られたプラスミドを、制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりライゲーションの有無を確認した。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約260 baseの塩基配列(配列番号5)が明らかになった。
【0029】
(4)Oligo-Capping法を用いた完全長mRNAの作成
上述した方法でネコ白血球から分離した1〜5μgのmRNAを、40UのRNasin Ribonuclease Inhibitor(Promega)および0.5UのBacterial Alkarine Phosphatase(BAP:タカラバイオ株式会社)を含むBAP buffer中で混和させ、37℃で60分間反応させた。酵素反応後、BAP処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイト(WAKO)を用いて沈澱した。BAP処理したmRNAを、さらに、60UのRNasin、8.0UのTobacco Acid Pyrophosphatase(TAP:WAKO)およびTAP bufferに混和し、37℃で60分間反応させた。酵素反応終了後、BAP-TAP処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイトを用いて濃縮した。BAP-TAP処理したmRNAに、100ngの合成Oligo-RNA(5’-AGCAUCGAGUCGGCCUUGUUGGCCUACUGG-3’:配列番号6)を添加し、65℃で5分間反応後、40UのRNasinおよび50UのT4 RNA ligase(タカラバイオ株式会社)を含むligation bufferに混和し、20℃で3時間反応させた。酵素反応終了後、RNA ligation処理されたmRNA溶液を、フェノール・クロロホルム抽出し、エタチンメイト(WAKO)を用いて濃縮後、40UのRNasinおよび10UのRNase FreeのDNase I(タカラバイオ株式会社)を含むDNase bufferに混和させ、37℃で10分間反応させた。酵素反応終了後、得られたmRNA溶液に、フェノール・クロロホルム抽出を行い、エタチンメイト(WAKO)で濃縮し、first-strand cDNA Synthesis Kit(GEヘルスケアバイオサイエンス)を用いて、添付のプロトコールに従いoligo(dT)Primerとして5’-AACTGGAAGAATTCGCGGCCGCAGGAAT18-3’(配列番号7)を用い、AMV Reverse transcriptase、first-strand bufferを加え、42℃60分でfirst-strand DNAを合成した(図5)。
【0030】
(5)5’RACE−PCR法
上述の方法で作成されたdsDNAを5’RACE−PCR法により増幅させた。5’RACE−PCR法に用いるプライマーは、上流側プライマーは付加したRNA adaptorの配列を基に、下流側プライマーは既に決定した中間領域の塩基配列を基に、それぞれ以下の塩基配列を有するように設計した。
【0031】
・5’RACE−上流側プライマー:5'-AGCATCGAGTCGGCCTTGTTG-3'(配列番号8)
・5’RACE−下流側プライマー:5'-TTCATCCCAGCCACGACCTGCTTTC-3'(配列番号9)
このように設計されたプライマーを用いて、95℃2分1サイクル、95℃1分、60℃1分、72℃1分を30サイクル、72℃10分1サイクルの条件でPCRを行った。図6は、5’RACE−PCRの結果を示す電気泳動写真であり、図6に示されるように得られたPCR産物は二本のバンドとして確認された。QIAquick Gel Extraction Kit(QIAGEN)を用いてPCR産物からDNAを抽出し、再度PCRを行った後、TOPO TA Cloning Kit(Invitrogen)、pGEM-T Easy Vector System(Promega)および E.coli JM109 Compitent cells(タカラバイオ株式会社)を用いてライゲーションおよびトランスフォーメーションを行った。培養後のE.coli JM109より、QIAPrep Spin Mini Kit50(QIAGEN)を用いてプラスミドを抽出し、得られたプラスミドを制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりDNA断片の挿入を確認した。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約350 baseの塩基配列(配列番号10)が明らかになった。
【0032】
(6)3’RACE−PCR法
上述した方法で作成されたdsDNAを3’RACE−PCR法により増幅した。3’RACE−PCR法に用いるプライマーは、上流側プライマーは既に決定した中間領域の塩基配列を基に、下流側プライマーは3’端に付加したDNA adaptorの配列を基に、それぞれ以下の塩基配列を有するように設計した。
【0033】
・3’RACE−上流側プライマー:5'-GCTCTTTCCAGATATACACTGTACCCT-3'(配列番号11)
・3’RACE−下流側プライマー:5'-AGAATTCGCGGCCGCAGGAATT-3'(配列番号12)
このように設計されたプライマーを用いて、95℃2分1サイクル、95℃1分、55℃1分、72℃1分を30サイクル、72℃10分1サイクルの条件でPCRを行った。図7は、3’RACE−PCRの結果を示す電気泳動写真であり、図7に示されるように得られたPCR産物は完全に単一のバンドとして確認された。さらに、得られたPCR産物について、TOPO TA Cloning Kit(Invitrogen)、pGEM-T Easy Vector System(Promega)および E.coli JM109 Compitent cells(タカラバイオ株式会社)を用いてライゲーションおよびトランスフォーメーションを行った。培養後のE.coli JM109より、QIAPrep Spin Mini Kit50(QIAGEN)を用いてプラスミドを抽出し、得られたプラスミドを制限酵素(EcoRI)で処理後、アガロースゲル電気泳動法によりDNA断片の挿入が確認された。また、T7プライマーを用い、さらにDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用して、塩基配列の解析を行った結果、約390 baseの塩基配列(配列番号13)が明らかになった。
【0034】
(7)得られたcDNA全体における塩基配列の解析
以上より得られた塩基配列を基に配列全体を構築した(配列番号14)。配列番号14で示される完全長の核酸の配列は796 baseであり、中にCysCをコードしていた。配列番号14に示す塩基配列のうち59番目〜502番目がCysCをコードする構造遺伝子(CysC遺伝子:配列番号2)であることが分かった。
【0035】
図2は、得られたネコのCysC遺伝子のcDNAの塩基配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysC遺伝子の塩基配列と比較して示す図である。図中、互いに共通する塩基配列部分を四角で囲んで示している。CysC遺伝子の核酸長は、ヒト、サルおよびブタが441 base、ウシでは447 base、ラットは423 baseであったのに対し、今回得られたネコのCysC遺伝子ではその長さは444 baseであった。また、表1に示されるように、他動物種(ヒト、サル、ウシ、ブタおよびラット)間のCysC遺伝子の塩基配列の相同性は、67.21〜96.71%の範囲に分布していたのに対し、今回得られたネコのCysC遺伝子の塩基配列と他動物種のCysC遺伝子の塩基配列との相同性は、平均で77.69%であり、得られた塩基配列はネコのCysC遺伝子であることが明らかになった。
【0036】
【表1】
【0037】
<実験例2:ネコ由来CysCの合成>
(1)ネコ由来CysCのアミノ酸配列の解析
実験例1で得られたネコ由来のCysC遺伝子の塩基配列(配列番号2)からアミノ酸配列を翻訳し、ネコ由来のCysCのアミノ酸配列(配列番号1)を解析した。図1は、得られたネコのCysCのアミノ酸配列を、既に知られているヒト、サル、ウシ、ブタおよびラットのCysCのアミノ酸配列と比較して示す図である。図中、互いに共通するアミノ酸配列部分を四角で囲んで示している。結果、ネコ由来のCysCのアミノ酸数は全長で147個であり、ヒト、サルおよびブタのアミノ酸数は146個、ウシのアミノ酸数は148個であり、ラットのアミノ酸数140個と近似した値であった。また、他動物種間で保存されていた構造アミノ酸システインの位置も数も同様であった。また、表2に示されるように、相同性に関しては、他動物種(ヒト、ウシ、ブタおよびラット)間のCysCのアミノ酸配列の相同性は、62.22〜97.26%の範囲に分布していたのに対し、今回得られたネコのCysCのアミノ酸配列と他動物種との平均相同性は69.15%であった。したがって、得られたアミノ酸配列はネコ由来のCysCであることが明らかになった。
【0038】
【表2】
【0039】
(2)GST融合タンパクを用いた組み換え型タンパクの発現および精製
シグナルペプチド領域と考えられる部分を除いた、ネコ由来のCysCタンパク領域のみの核酸を増幅するためにPCRを行った。プライマーは、上流側プライマーは5’末端にEcoRIの制限酵素サイトを付加し、下流側プライマーは3’末端にXhoIの制限酵素サイトを付加し、それぞれ以下の塩基配列を有するように設計した。
【0040】
・上流側プライマー:5'-CACGAATTCACCGGCAGGAGAAACAACAAG-3'(配列番号15)
・下流側プライマー:5'-CACCTCGAGTTATGCATCCTGGCAGCTGGACTTCACCAG-3'(配列番号16)
上述したような上流側プライマーおよび下流側プライマーを用いて、95℃2分1サイクル、95℃1分、75℃1分、72℃1分30サイクル、72℃10分1サイクルの条件でPCRを行った。PCR産物をアガロースゲル電気泳動した後、アガロースゲルからDNA抽出した。DNA抽出溶液をフェノールと等量混合した後、15700×gで5分間遠心分離後、核酸が含まれる水層を分離した。分離した水層にクロロホルムを等量混合し、15700×gで5分間遠心分離した後、上清を分離した。次に、分離後の溶液に2.5倍量の100%エタノールを添加し、−80℃で30分静置し、その後15700×gで5分間遠心分離した後、上清を除去し、沈渣を得た。沈渣に70%エタノールを添加し、15700×gで5分間遠心分離後、上清を除去し、PCR産物の濃縮試料を得た。PCR産物の濃縮試料に、5μlのEcoRI(タカラバイオ株式会社)、5μlのXhoI(タカラバイオ株式会社)、5μlのH.Buffer(500mM Tris-HCl, pH7.5、100mM MgCl2、10mM Dithiothreitol、1000mM NaCl)および35μlのRNase free H2Oを混和した。また、5μl(2.5μg)のpGEX6P-1(GEヘルスケアバイオサイエンス)を、5μlのEcoRI、5μlのXhoI、5μlのH.Bufferおよび30μlのRNase free H2Oと混和した。各溶液を37℃で1晩インキュベートすることで制限酵素処理した後、アガロースゲル電気泳動を行い、QIAquick Gel Extraction Kit(QIAGEN)を使用して、各DNAバンドの抽出を行った。DNA Ligation Kit(タカラバイオ株式会社)を用いてライゲーションを行った。すなわち、5μlのLigation Mix、1μlの制限酵素処理されたCysCのcDNA溶液および4μlのpGEX6P-1を混和し、16℃で1晩静置し、CysCのcDNAをライゲーションさせたplasmid vector(pGEX-CysC)が作出された。さらに、このpGEX-CysC溶液2.5μlを、E.coli JM109 Competent Cells(タカラバイオ株式会社)25μlに添加し、氷上で30分間静置し、次に42℃の恒温槽で45秒間Heat Shockを与え、直ちに2分間氷冷後SOC培地(2% Tryptone、0.5% Yeast extract、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM Glucose)250μlを緩やかに加え、37℃1時間保温した。pGEX-CysCがトランスフェクトされた大腸菌溶液100μlをアンピシリン加LB培地にそれぞれ塗布し、37℃で1晩静置後、コロニーを釣菌し、アンピシリン加LB液体培地1.2mlに混和後、37℃で1晩培養した。培養後の液体培地は、13400×gで1分間遠心分離後、上清を完全に除去し、得られた沈渣から、QIAPrep Spin Mini Kit50(QIAGEN)を用いて、pGEX-CysCの抽出を行った。このpGEX-CysCを、アガロースゲル電気泳動法により確認した。また、pGEX-CysCのサブクローニングの成否は、T7プライマーを用いてDye Deoxy Terminator Cycle Sequencing Kit(Applied Biosystems)およびApplied Biosystems 3130xl Genetic Analyzer(Applied Biosystems)を使用した塩基配列解析により行った。
【0041】
(3)GST融合タンパク発現の確認
pGEX-CysCがトランスフェクトされた大腸菌を、37℃、1晩LB培地で培養後、100μlをIsopropyl-β-D-thiogalactopyranoside(IPTG:0.1mM)20μlと混和し、30℃で約2時間振盪培養(BR40-LF、TAITEC)した。振盪培養後の大腸菌溶液を、15700×gで1分間遠心分離後、上清を除去し、沈渣に可溶化液(50μlの50mM Tris-HCl、100μlの1×RIPA Lysis Buffer(Up State)、140μlのProtease Inhibitor、710μlのH2O)30μlを加え可溶化後、15700×gで5分遠心分離し、上清と沈渣とに分けた。上清30μlに、30μlの2×SB溶液(2% SDS、40% Glycerol、0.6% BPB、25mM Tris-HCl Buffer(pH6.8、20℃))および1μlの2MEを加え、95℃で3分間加温した。沈渣にSB溶液20μlを加え、超音波破砕機(UR-20P、TOMY SEIKO CO,LTD)で5秒間破砕後、95℃で3分間加温した。その後、上清および沈渣について、SDS−PAGEにてGST融合タンパク発現の確認およびGST融合タンパクの大腸菌での可溶性を確認した。
【0042】
(4)SDS−PAGE法
SDS−PAGEは、コンパクトPAGE(AE-7300、ATTO)を用いてLaemmliの方法に準拠し、これに以下に示す修正を加え実施した。すなわち、分離ゲルの組成は、15% Acrylamide、0.2% N,N-Methylene-bis-Acrylamide、0.1% SDS、375mM Tris-HCl buffer(pH8.8、20℃)とした。ゲルは2・4連ゲル作製器(AE-7344、ATTO)を用いて作製した。電極緩衝液の組成は、0.1% SDS、129mM Glycine、25mM Tris(pH8.3、20℃)とした。泳動用試料(SB)の組成は、1% SDS、20% Glycerol、0.3% BPB、12.5mM Tris-HCl Buffer(pH6.8、20℃)とした。また、マーカーとして、プレステインドSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)もしくはSDS−PAGEスタンダード(Broad)マーカー(BIO-RAD)を用いた。泳動は、Tris-Gly/PAGE Highモードで30分泳動した後、Tris-Gly/PAGE Lowモードにして、下部イオン界面がゲル下端から1〜2mm上方の位置に移動したときに終了した。SDS−PAGE終了後のゲルについて、Oakle法に準拠した銀染色法を実施した。具体的には、ゲルを30%ethanol、10% acetic acid溶液にて固定後、洗浄し、20% ethanolに5分間2回浸漬した。20% ethanol除去後、5%glutaraldehyde溶液にて4分間反応させ、純水で洗浄後、20% ethanolに4分間2回浸漬した。その後、純水で洗浄し、アンモニア性硝酸銀溶液にて5分間反応させ、純水で洗浄後、0.005%citric acid、0.019%formaldehyde溶液で発色させた。発色確認後のゲルについて、20%ethanol、10% acetic acid溶液にて5分間固定し、20%ethanolに5分間2回浸漬後、写真を撮影した。なお、銀染色法はすべて遮光条件下にて実施した。
【0043】
(5)GST融合タンパクの発現誘導と単離
His−Tag付きGST融合タンパクの発現が確認された大腸菌を、アンピシリン加LB寒天培地に塗布し、コロニーを釣菌後、3mlのアンピシリン加LB液体培地に加え1晩37℃で振盪培養した。続いて、その培養液3mlを、アンピシリン加LB液体培地250mlに加え、37℃で約150分振盪培養後、2.5mlの0.1mM IPTGを添加し、約2時間、30℃で振盪培養した。GST融合タンパク発現誘導後の培養液を6000×gで15分間遠心分離した沈渣を、Phosphate buffer saline(PBS:140mM NaCl、2.7mM KCl、10mM Na2PO4、1.8mM KH2PO4、pH7.3)に懸濁し、超音波破砕機で20秒間×5回破砕後、Triton X100を最終濃度1%になるように加え、室温で撹拌しながら30分間放置後、9300×gで20分間遠心分離し、上清と沈渣をSDS−PAGEにて分析した。泳動の結果、GST融合タンパクは上清および沈渣の両分画に含まれていたが、可用性分画の方が利便性に優れているのでアフィニティークロマトグラフィーには上清を用いた(図8)。
【0044】
(6)アフイニティークロマトグラフィー法
上述のようにして得られた上清について、GSTrap HP column(GEヘルスケアバイオサイエンス)を用いてアフイニティークロマトグラフィーを行った。上清を結合バッファーであるPBSにて透析した後、カラムに添加し結合バッファーでよく洗浄し、10mM reduced glutathione加50mM Tris-HCl(pH8.0)で溶出させた。なお、カラム操作はペリスタポンプ(SJ-1211L、ATTO)を用い、流速0.5ml/minで添加した。また、溶出液の吸光度は紫外部吸光度モニター(AC-5100L、ATTO)を用いて吸光波長220nmでモニターされ、記録計(R-01A、RIKADENKI)で記録された。溶出液のSDS−PAGE像は図8に示されたとおり、GST融合タンパクをメインバンドとして複数のバンドが確認された。得られたGST融合タンパク溶出液2mlに、濃度が1mMになるようにDTTを添加し混和後、分子量13kDaカットの透析膜(UC30-32-100、三光純薬株式会社)に入れられ150mM NaCl、1mM EDTA加50mM Tris-HCl(pH7.5)2Lを用いて約6時間透析された。透析後のGST融合タンパク溶出液について、DC Protein Assay(Bio-Rad)を用いてタンパク定量後、タンパク量200μgに対しPreScission Protease(GEヘルスケアバイオサイエンス)1μlを添加して混和し、4℃で6時間以上反応させた。酵素切断後のSDS−PAGE像では、切断されたGSTとネコの組み替え型CysC(rFeCysC)が確認された。さらに、この溶液を高速液体クロマトグラフィー(HPLC)用の試料とした。
【0045】
(7)HPLC法
HPLCシステムは、システムコントローラー(SCL-10A VP、Shimadzu)、送液ユニット(LC-10AD VP、Shimadzu)、紫外部分光高度計(SPD-10A VP、Shimadzu)、カラムオーブン(CTO-10A VP、Shimadzu)および脱気ユニット(DGU-14A、Shimadzu)から構成され、カラムはMightysilRP-18 GP250-4.6(関東化学)を使用した。HPLCの分離条件は、移動相の流速を1ml/min、試料添加量を400μlとし、0.1% trifluoroacetic acid(TFA)溶液で平衡化させたカラムに、0.1%TFA加acetonitrile溶液を用いてacetonitrile濃度0〜80%のライナーグラジエントにして行った。なお、溶出液は、吸光波長220nmで吸光度をモニターし、検出されたピークを分取し、濃縮遠心機(CC-181、TOMY)にて1時間遠心分離後、凍結乾燥機(FDU-540、EYELA)にて乾燥させた後−20℃で保存した。クロマトパターンは、図9に示すとおりで、だいたい5個のピークa, b, c, d, eに分離され、それぞれの溶出された分画のタンパク組成は、SDS−PAGE法により分析された。図10は、HPLCの主要画分a, b, c, d, eについてのSDS−PAGEの結果を示す写真である。分析の結果、図10に示されるように、c分画に単独で溶出された。
【0046】
<実験例3:抗体産生ハイブリドーマ、抗rFeCysC抗体の作製>
実験例2で合成したタンパクを組み替え型ネコCysC(rFeCysC)の抗原としてモノクローナル抗体を作製するにあたり、まずは抗体産生ハイブリドーマを作製した。
【0047】
(1)抗体産生ハイブリドーマの作製
(1−1)免疫法
免疫法は、精製rFeCysCを抗原としてBalb/cマウスの後肢肉球(footpad)の皮下に注射することにより行った。免疫は5日間隔で4回行い、初回から第3回目までの免疫は抗原溶液100μl(1mg/ml)とアジュバントを等量混合させてエマルジョン化させた抗原液200μl(50μg/foot)を、また、最終免疫では抗原溶液20μl(10μg/foot)のみを用いて行った。また、アジュバントは初回免疫ではAdjuvant Complete Freund(和光純薬工業株式会社)を、第2から3回目の免疫ではAdjuvant Incomplete Freund(和光純薬工業株式会社)を用いた。
【0048】
(1−2)細胞融合
最終免疫から3日後、膝窩リンパ節を摘出し、リンパ球を回収後、GenomONE-CF(石原産業株式会社)を用いて、細胞融合を行った。また、ミエローマ細胞としてはP3X63-Ag8.653(大日本住友製薬株式会社)を用いた。融合方法は添付のプロトコールに従って行った。具体的には、まず、リンパ球とミエローマ細胞とを細胞数が5:1の比率になるように混合し、1000rpm、4℃で5分間遠心した後、上清を除去した。そこに、氷冷した融合用緩衝液をリンパ球108cellsあたり1ml添加し、均一に懸濁した後、氷冷したHVJ-Envelope懸濁液を細胞混合液1mlあたり25μl添加した。細胞懸濁液を氷上で5分間静置した後、1000rpm、4℃5分間遠心し、上清を除去せずに細胞がペレット化した状態のまま37℃で15分間インキュベートした。
【0049】
インキュベート終了後、37℃に加温した増殖用培地をリンパ球108cells当たり50ml加え、懸濁後、96穴プレート(96 Well Cell Culture Plate:Greiner bio-one)に100μl/wellで播種した。なお、増殖用培地としてRPMI1640(Invitrogen)にペニシリンG(PG;明治製薬株式会社)10万IU/ml、ストレプトマイシン(SM;明治製薬株式会社)100mg/ml、7.5% Bri Clone(IL-6、ヒト、ブライクローン;Cat. No. BR-001、大日本住友製薬株式会社)、10% 非働化ウシ胎仔血清(FBS;株式会社ニチレイ)を加えたものを用い、添加、懸濁の際は穏やかに操作した。24時間培養後、培養培地を上記の増殖用培地に2% HAT(Invitrogen)を添加したHAT培地に交換した。
【0050】
(2)抗体産生ハイブリドーマのスクリーニング
得られたハイブリドーマについて、細胞融合から1週間後にELISA法を用いた一次スクリーニングを行い、この結果、反応陽性となったwellのハイブリドーマのみをWestern blotting法を用いた二次スクリーニングで確認した。
【0051】
(2−1)一次スクリーニング
rFeCysCを抗原としたELISA法を用いて、抗体産生ハイブリドーマの一次スクリーニングを行った。ELISAプレートとしては、96 Well ELISA Microplate(Greiner bio-one)を使用した。また、プレートの洗浄には自動洗浄機(Auto Mini Washer AMW-8、バイオテック株式会社)を用い、洗浄液としてはPBS(1.37M NaCl、27mM KCl、100mM Na2HPO4、18mM KH2PO4、pH7.4、25℃)を使用した。固相として、PBSで3μg/mlに調整したrFeCysCを50μl/wellでプレートに添加し、4℃で一晩反応させた。固相反応終了後、プレートの抗原液を捨て、ブロッキング液として0.5% Bovine Serum Albumin(BSA;和光純薬工業株式会社)を添加したPBSを150μl/wellで加え、37℃で60分間反応させた。ブロッキング反応終了後、プレートを1回洗浄し、一次抗体として各ハイブリドーマ培養の培養上清を50μl/wellで加え、37℃で60分間反応させた。一次抗体反応終了後、プレートを1回洗浄し、二次抗体として0.1% BSAを添加したPBSで1000倍に希釈したペルオキシダーゼ標識抗マウスIgG抗体(SIGMA-ALDRICH)を50μl/wellで加え、37℃で60分間反応させた。二次抗体反応終了後、プレートを3回洗浄し、基質液として0.04% o-フェニレンジアミン、0.04% H2O2を添加したPBSを150μl/wellで加え、室温、遮光下で30〜60分間反応された。基質反応終了後、3M H2SO4を反応停止液として50μl/wellで加え、1分間振盪後、Microplate Reader(Model 550、BIO-RAD)で波長490nmにおける吸光度を測定した。吸光度の高かった陽性wellの細胞を、24穴プレート(24 Well Cell Culture Plate;Greiner bio-one)に移して培養した。
【0052】
(2−2)二次スクリーニング
rFeCysCを抗原としたWestern blotting法で確認し、抗体産生ハイブリドーマの二次スクリーニングを行った。Lowryの方法に基づき、DC Protein Assay Kit(BIO-RAD)を用いて、Microplate Readerで波長655nmにおける吸光度を測定し、タンパク質を定量した。検量線はBSAを用いて作製した。Western blotting法はTowbinらの方法に準拠し、以下のように実施した。転写膜はポリビニリデンジフルオリド(PVDF)膜(BIO-RAD)を使用した。PVDF膜は100% methanolに10秒間、さらに転写用電極buffer(25mM Tris-HCl(pH8.3、20℃)、192mM glycine、5% methanol)に30分間浸潤し、泳動に供した。転写装置の組み立ては、陽極電極板上に下から順に濾紙(BIO-RAD)、PVDF膜、SDS−PAGE終了後のゲル、濾紙を重層し、その上に陰極電極板を固定した。なお、濾紙は予め電極bufferに2〜3分浸しておいた。転写条件は1.9mA/cm2の定電流で60分間とした。転写終了後のPVDF膜は10mM Tris-HCl(pH7.5、20℃)、140mM NaCl、0.01% Tween20(TBST)に0.5% BSAを加え、室温で60分間振盪し、ブロッキング操作を行った。ブロッキング終了後、TBSTで5分間、2回振盪洗浄し、一次抗体として細胞の培養上清を用い、室温で90分振盪反応させた。一次抗体反応終了後、TBSTで5分間、2回振盪洗浄した後、TBSTで1000倍希釈したペルオキシダーゼ標識抗マウスIgG抗体を、室温で60分間振盪反応させた。二次抗体反応終了後、TBSTで5分間、2回振盪洗浄し、0.06% 3,3-diaminobenzidine tetra-hydrochloride、0.03% H2O2、50mM Tris-HCl(pH7.6、20℃)を基質反応液として使用し、1〜5分間反応させた。基質反応終了後、水洗し反応を停止させた後、乾燥して保存した。反応陽性を示したハイブリドーマについては後述する限界希釈法によりクローニングを行った。
【0053】
(3)クローニング
ハイブリドーマのクローニングには限界希釈法を用いた。具体的には、スクリーニング後のハイブリドーマを2cells/100μlとなるようにHAT培地で希釈し、100μl/wellで96穴プレートに播種した。ハイブリドーマはセミコンフルエントになったところで24穴プレートに拡大培養し、再びセミコンフルエントになるまで培養した後、二次スクリーニングと同様にrFeCysCを抗原としたWestern blotting法で確認した。このクローニング操作を2回行った。また、ハイブリドーマを長期間継代培養することにより抗体産生能が減少するのを防ぐため、クローニング毎に細胞凍結保存液(セルバンカー(BLC-1)、十慈フィールド株式会社)を用いて保存した。
【0054】
(4)抗体産生ハイブリドーマの大量培養および抗rFeCysC・mAbの採取と精製
クローニングが終了したハイブリドーマを、浮遊細胞培養フラスコ(フィルタートップSCフラスコ250ml 75cm2;Greiner bio-one)を用いて大量培養した。なお、培養は37℃、5% CO2、5日間、CO2インキュベーター(十慈フィールド株式会社)で行い、培地としてはHAT培地を用いた。大量培養されたハイブリドーマを無血清RPMIで懸濁し、ヌードマウス(Balb/c-nu)の腹腔内に2×107cells/headで投与した。投与してから10〜20日後、腹水を採取した。ヌードマウスから採取した腹水を室温で1時間、あるいは4℃で一晩静置した後、3000rpm、4℃で5分間遠心し、腹水中のフィブリン、ハイブリドーマ、赤血球などを除去した。分離した上清を50%の硫安で塩析させた。具体的には、氷上で撹拌しながら上清と等量の飽和硫酸アンモニウム溶液を徐々に滴下し、滴下後さらに1時間撹拌した。これを10000rpm、4℃で10分間遠心し、沈殿物を20mM リン酸ナトリウムbuffer(pH7.0)に溶解した。塩析後のグロブリン溶液を、20mM リン酸ナトリウムbuffer(pH7.0)で平衡化したSephadex G-25 Fine(GEヘルスケアバイオサイエンス)カラム(内径1.5cm、長さ30cm)を用いて脱塩した。クロマトグラフィーの流速をペリスタポンプ(SJ-1211L、ATTO)で0.5ml/minに調節した。脱塩後のグロブリン溶液を、エコカラム(内径2.5cm、長さ10.0cm:BIO-RAD)に充填したProtein G Sepharose 4 Fast Flow(GEヘルスケアバイオサイエンス)を用いたアフィニティークロマトグラフィー法により精製した。具体的には、脱塩後のグロブリン溶液を20mM リン酸ナトリウムbuffer(pH7.0)で平衡化されたカラムに流速0.5ml/minで添加し、その後カラムを100mM glycine(pH3.0)で溶出させた。溶出液は直ちに10分の1量の1M Tris-HCl(pH9.0)で中和した。精製後の溶出液を50mM 酢酸アンモニウム(pH7.0)で平衡化させたSephadex G-25 Fineカラム(内径2cm、長さ30cm)で脱塩させた後、Freeze Dryer(FDU540、EYELA東京理化器械株式会社)を用いて凍結乾燥し、−20℃で保存した。
【0055】
(5)アイソタイプの決定
Mouse Monoclonal Isotyping Kit(コスモバイオ株式会社)を用い、方法は添付のプロトコールに従って、得られた抗rFeCysC・mAbのアイソタイプの決定を行った。具体的には、抗rFeCysC・mAbサンプルをdevelopment tubeに150μl加え、室温で30秒間インキュベートした後、撹拌した。そこに、isotyping stripを入れ、さらに室温で10〜15分間インキュベートした後、classとsubclassを読み取った。抗rFeCysC・mAbサンプルとしては、2回目のクローニングが終了したハイブリドーマの培養上清を1% BSAを添加したPBSで10倍に希釈したものを用いた。モノクローナル抗体は2種類得られ、一方の抗体AのアイソタイプはIgG1のκ鎖、もう一方の抗体BのアイソタイプはIgG2aのκ鎖であった。
【0056】
(6)ネコのnativeなCysCに対する特異性
抗体A、Bについて、慢性腎疾患(Chronic Kidney disease:CKD)のネコの尿タンパクを抗原としたWestern blotting法を用いて、ネコのnativeなCysCに対する特異性を確認した。Western blotting法は上述の方法と同様に実施した。ただし、SDS−PAGEの泳動用尿タンパクサンプルは、ネコの尿タンパクを2-MercaptoethanolでSS結合が切断したものを用いた。図3は、ネコのnativeなCysCに対する抗体A、Bの特異性の実験結果を示す写真であり、それぞれレーン1はrFeCysC、レーン2はCKDネコの尿タンパクを示している。図3に示されるように、抗体A、Bともに、nativeなCysCとも特異的に反応することが確認された。
【0057】
今回開示された実施の形態および実験例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【特許請求の範囲】
【請求項1】
配列番号1で表わされるアミノ酸配列を有するタンパク質。
【請求項2】
請求項1に記載のタンパク質をコードする構造遺伝子。
【請求項3】
配列番号2で表わされる塩基配列を有する構造遺伝子。
【請求項4】
ネコ由来シスタチンCに特異的に結合する抗体。
【請求項5】
請求項1に記載されたタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)により産生されたものである、請求項4に記載の抗体。
【請求項6】
請求項1に記載されたタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである、請求項4に記載の抗体。
【請求項7】
請求項4〜6のいずれかに記載の抗体を含む、ネコ腎症の診断用キット。
【請求項8】
請求項4〜6のいずれかに記載の抗体を用いた、ネコ腎症の診断方法。
【請求項1】
配列番号1で表わされるアミノ酸配列を有するタンパク質。
【請求項2】
請求項1に記載のタンパク質をコードする構造遺伝子。
【請求項3】
配列番号2で表わされる塩基配列を有する構造遺伝子。
【請求項4】
ネコ由来シスタチンCに特異的に結合する抗体。
【請求項5】
請求項1に記載されたタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb1(受領番号:FERM AP-21877)により産生されたものである、請求項4に記載の抗体。
【請求項6】
請求項1に記載されたタンパク質を抗原とし、細胞株Mouse-Mouse hybridoma CysC mAb2(受領番号:FERM AP-21878)により産生されたものである、請求項4に記載の抗体。
【請求項7】
請求項4〜6のいずれかに記載の抗体を含む、ネコ腎症の診断用キット。
【請求項8】
請求項4〜6のいずれかに記載の抗体を用いた、ネコ腎症の診断方法。
【図1】


【図2】

【図5】
【図9】
【図3】
【図4】
【図6】

【図7】
【図8】
【図10】
【図2】
【図5】
【図9】
【図3】
【図4】
【図6】
【図7】
【図8】
【図10】
【公開番号】特開2011−125267(P2011−125267A)
【公開日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願番号】特願2009−286711(P2009−286711)
【出願日】平成21年12月17日(2009.12.17)
【出願人】(598041566)学校法人北里研究所 (180)
【出願人】(000135036)ニプロ株式会社 (583)
【Fターム(参考)】
【公開日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願日】平成21年12月17日(2009.12.17)
【出願人】(598041566)学校法人北里研究所 (180)
【出願人】(000135036)ニプロ株式会社 (583)
【Fターム(参考)】
[ Back to top ]