
シランカップリング剤及びその製造方法
【課題】基質との結合力が高く、しかも350℃以上の雰囲気に4時間以上曝露しても、これら化合物による改質表面の接触角の低下が見られないというほどの優れた耐熱性、離型性、及び防汚性を有するシランカップリング剤を提供する。
【解決手段】本発明のシランカップリング剤は下記一般式(1)で表される。

(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【解決手段】本発明のシランカップリング剤は下記一般式(1)で表される。
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なシランカップリング剤及びその製造方法に関する。
【背景技術】
【0002】
近年、特にシランカップリング剤を離型剤として用い、母型に形成された微細パターンを再現性・解像性良く被転写部材に転写する技術の研究が盛んに行われている。
転写は、母型の微細パターン上に形成した離型剤層上に被転写部材を覆った後、離型剤層から被転写部材を剥離して行われるが、金属を真空蒸着によって、あるいは樹脂を熱硬化させて被転写部材として離型剤層上に設ける場合、離型剤としては高温耐熱性であることが要求される。しかしながら、従来から知られている離型剤にはこの要求に応えられるものがなかった。
【0003】
そこで、本発明者等は、先に、特に耐熱性及び離型性の点に優れた下記一般式(A)で表されるビフェニルアルキル基を有するシランカップリング剤を開発した(特許文献1参照)。
【0004】
【化1】
(式(A)中、RfはF(CF2)nのペルフルオロアルキル基を示し、nは1〜14の整数を示す。)
【0005】
上記一般式(A)で表されるシランカップリング剤を用いると、上述のような真空蒸着あるいは熱硬化のような高温プロセスによって被転写部材を形成する場合でも、1μm未満の高さでアスペクト比が2以上あるような超微細な突起群を含むような母型のパターンを、再現性・解像性良く被転写部材に転写することができる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2008/108438号
【発明の概要】
【発明が解決しようとする課題】
【0007】
ところで、離型剤層を形成する際には、母型表面にシランカップリング剤を含む液を塗布した後に、シランカップリング剤を強固に固定させるように、加熱処理等が行われる。
しかしながら、例えば、微細パターン表面に離型剤としてシランカップリング剤層が形成された母型を金型として繰り返し使用して、パターンが転写された被転写部材を数多く作製していくと、離型剤が徐々に微細パターン表面から剥離してしまうことがある。
そこで、このような問題が発生しないような、母型の基質との結合力が高いシランカップリング剤の出現が期待されている。
【0008】
本発明は、上記事情に鑑みてなされたものであり、その第一の課題は基質との結合力の高いシランカップリング剤を提供することにある。さらに第二の課題は耐熱性、離型性、及び防汚性の良好な、特に350℃以上の温度においても、これら化合物による改質表面接触角の低下が見られない等の優れた物性を有するシランカップリング剤を提供することにある。
【課題を解決するための手段】
【0009】
本発明者等は、特定の一般式で表されるビフェニルアルキル基を有するシランカップリング剤によれば上記課題を解決できることを見出し、本発明を完成させた。より具体的には、本発明は下記のとおりである。
【0010】
[1] 下記一般式(1)で表されるビフェニルアルキル基を有するシランカップリング剤。
【化2】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【0011】
[2] 下記一般式(2)
【化3】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化4】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化5】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化6】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化7】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化8】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させて、下記一般式(8)
【化9】
(式(8)中、nは3〜4の整数を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得る、シランカップリング剤の製造方法。
【0012】
[3] 下記一般式(8)
【化10】
(式(8)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Yはハロゲン原子を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させて、下記式(9)
【化11】
で表される4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得る、シランカップリング剤の製造方法。
【0013】
[4] 下記一般式(1)で表されるシランカップリング剤と溶剤とを含む離型剤。
【化12】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【発明の効果】
【0014】
本発明のシランカップリング剤は、基質との結合力が高く、しかも350℃以上の雰囲気に4時間以上曝露しても、これら化合物による改質表面の接触角の低下が見られないというほどの優れた耐熱性、離型性、及び防汚性を有するものであり、格別の効果と有用性を有する。
【図面の簡単な説明】
【0015】
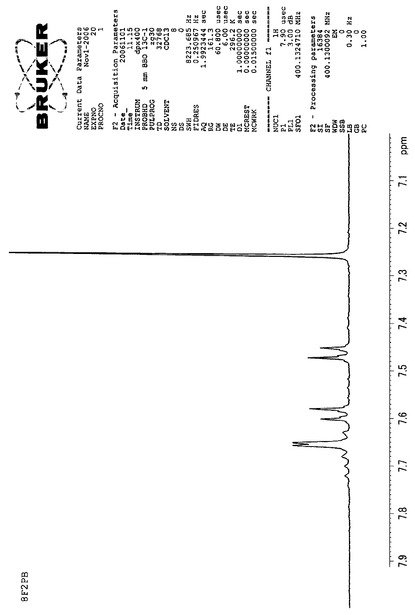
【図1】8F2PBのNMRスペクトルである(実施例1)。
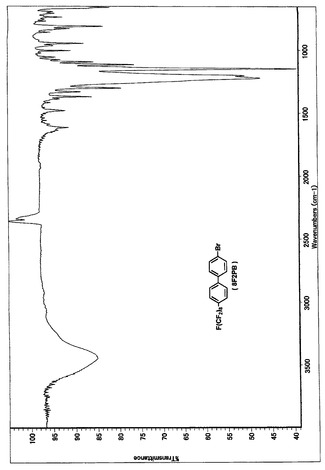
【図2】8F2PBのIRスペクトルである(実施例1)。
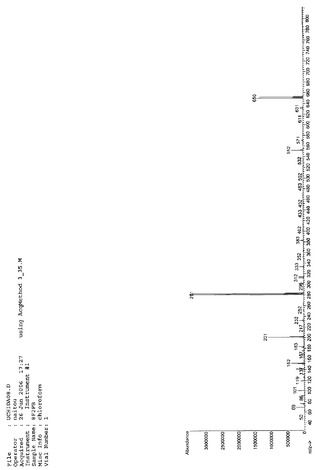
【図3】8F2PBのMassスペクトルである(実施例1)。
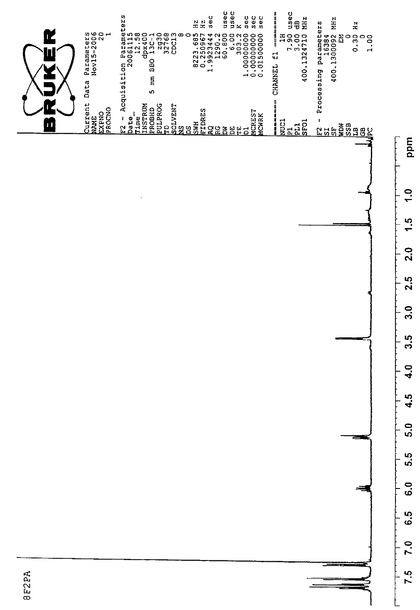
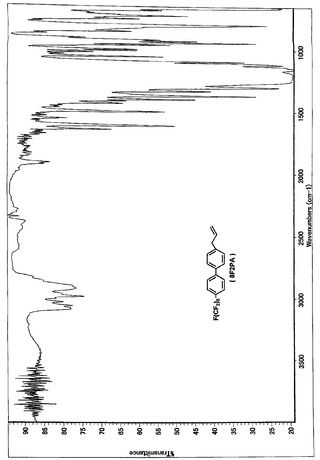
【図4】8F2PAのNMRスペクトルである(実施例1)。
【図5】8F2PAのIRスペクトルである(実施例1)。
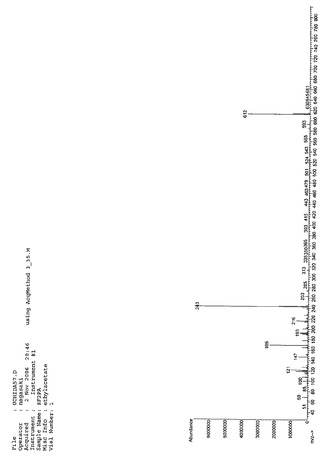
【図6】8F2PAのMassスペクトルである(実施例1)。
【図7】10F2PBのNMRスペクトルである(実施例3)。
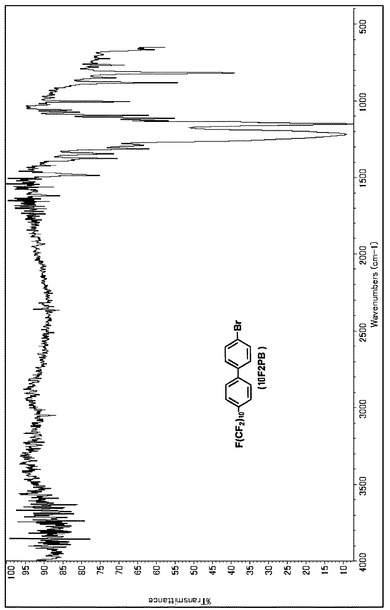
【図8】10F2PBのIRスペクトルである(実施例3)。
【図9】10F2PBのMassスペクトルである(実施例3)。
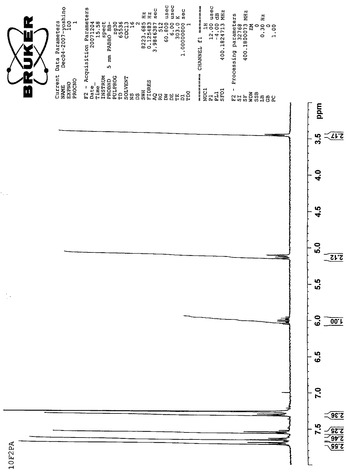
【図10】10F2PAのNMRスペクトルである(実施例3)。
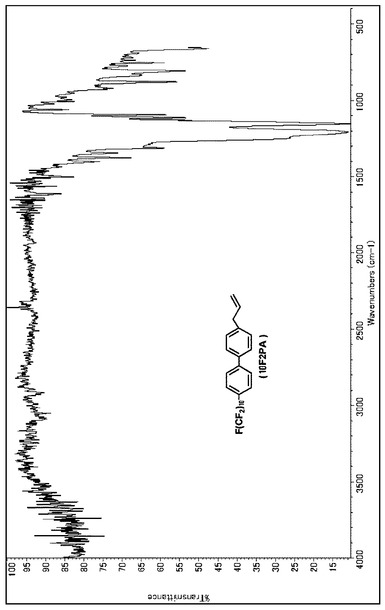
【図11】10F2PAのIRスペクトルである(実施例3)。
【図12】10F2PAのMassスペクトルである(実施例3)。
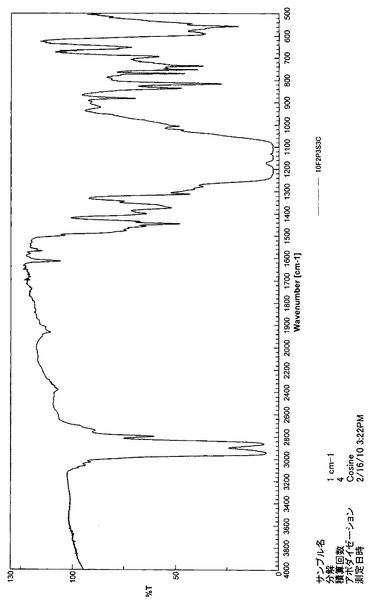
【図13】10F2P3S3CのIRスペクトルである(実施例3)。
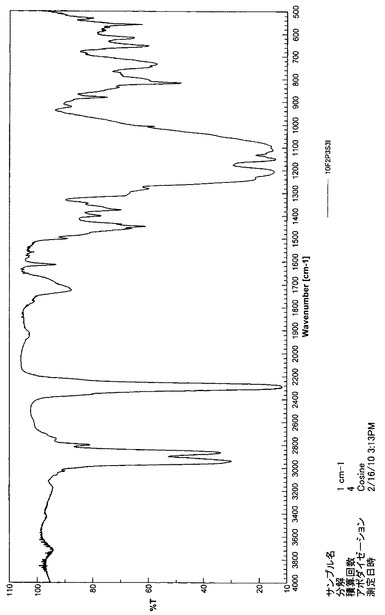
【図14】10F2P3S3IのIRスペクトルである(実施例4)。
【発明を実施するための形態】
【0016】
本発明のシランカップリング剤は下記一般式(1)で表されるものである。
【0017】
【化13】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【0018】
上記一般式(1)で表されるシランカップリング剤のうち、Xがハロゲン原子であるものは、下記のようにして製造することができる。
【0019】
すなわち、下記一般式(2)
【化14】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化15】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化16】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化17】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化18】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化19】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させることにより、下記一般式(8)
【化20】
(式(8)中、nは3〜4の整数を示す。)
で表される本発明の(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得ることができる。
【0020】
また、上記一般式(1)で表されるシランカップリング剤のうち、Xがイソシアナト基であるものは、下記のようにして製造することができる。
【0021】
すなわち、上記(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させることにより、下記式(9)
【化21】
で表される本発明の4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得ることができる。
【実施例】
【0022】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれらの実施例により限定されるものではない。
【0023】
[実施例1]
F(CF2)8(C6H4)2CH2CH2CH2SiCl3[8F2P3S3C]の合成
【0024】
・F(CF2)8(C6H4)2Br[8F2PB]の合成
【化22】
【0025】
500mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉63.6g(1.0mol)、4,4’−ジブロモビフェニル62.4g(200mmol)、ペルフルオロオクチルヨージド115g(210mmol)、溶媒としてジメチルスルホキシド(DMSO)300mlを加え、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体(粗製8F2PB)とを濾別し、DMSOで洗浄した。続いて酢酸エチルを溶媒に用い、銅粉と白色固体の混合物とをソックスレー抽出し、粗製8F2PBを得た。抽出液を飽和食塩水で洗浄し、さらに抽出液を硫酸マグネシウムで脱水し、酢酸エチルを減圧除去した。残留物を減圧蒸留して、留出物を得た。
得られた留出物についてMassスペクトルを分析した結果、m/z(分子量)651により、8F2PBであると同定した。NMR、IR、Massの各スペクトルを図1、図2、図3に示す。
【0026】
収量 68.0g(104mmol)
収率 52%
沸点 130−135℃/30Pa
性状 白色固体
【0027】
・F(CF2)8(C6H4)2CH2CH=CH2[8F2PA]の合成
【化23】
【0028】
1Lナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液32.6ml(90.0mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液58.4ml(45.0mmol)を加え、氷冷下で1時間撹拌した。その後、750mlのジエチルエーテルに溶解させた8F2PB 19.5gg(30.0mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅2.0g(10.5mmol)を加えた後、アリルブロミド18.2g(150mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらにヘキサンを展開溶剤としてシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
Massスペクトルを分析した結果、m/z(分子量)612により、8F2PAであると同定した。NMR、IR、Massの各スペクトルを図4、図5、図6に示す。
【0029】
収量 14.7g(24.0mmol)
収率 80%
沸点 130−135℃/20Pa
性状 白色固体
【0030】
・F(CF2)8(C6H4)2CH2CH2CH2SiCl3[8F2P3S3C]の合成
【化24】
【0031】
100ml肉厚ガラスアンプル管に撹拌子を入れ、ここに8F2PA 2.00g(3.27mmol)、THF10mlを採取し、窒素雰囲気に置換し、外部を液体窒素で冷却し、トリクロロシラン1.0g(8.2mmol)、触媒の0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、生じた黒色粉末状沈殿(Pt)を濾別除去した。濾液中からTHF、トリメクロロシランを減圧留去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)747により、8F2P3S3Cであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2P3S3Cのものとほぼ同じであった。
【0032】
収量 2.32g
収率 95%
性状 白色固体
【0033】
[実施例2]
F(CF2)8(C6H4)2CH2CH2CH2Si(NCO)3[8F2P3S3I]の合成
【化25】
【0034】
実施例1で得られた8F2P3S3C 2.0g(2.7mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀2.00g(13.3mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
Massスペクトルを分析した結果、m/z(分子量)767により、8F2P3S3Iであると同定した。また、NMRスペクトルをとったところ、実施例4の10F2P3S3Iのものとほぼ同じであった。
【0035】
収量 1.97g(2.5mmol)
収率 93%
性状 白色固体
【0036】
[実施例3]
F(CF2)10(C6H4)2CH2CH2CH2SiCl3[10F2P3S3C]の合成
【0037】
・F(CF2)10(C6H4)2Br[10F2PB]の合成
【化26】
【0038】
500mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉63.6g(1.0mol)、4,4’−ジブロモビフェニル64.0g(205mmol)、ペルフルオロデシルヨージド137g(212mmol)、溶媒としてDMSO350mlを加えた後、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体(粗製10F2PB)とを濾別し、DMSOで洗浄した。続いて酢酸エチルを溶媒に用い、銅粉と白色固体との混合物をソックスレー抽出し、粗製10F2PBを得た。抽出液を飽和食塩水で洗浄し、さらに抽出液を硫酸マグネシウムで脱水し、酢酸エチルを減圧除去した。残留物を減圧蒸留し、さらにクロロホルム可溶部からクロロホルムを減圧除去し、白色固体として10F2PBを得た。
Massスペクトルを分析した結果、m/z(分子量)751により、10F2PBであると同定した。NMR、IR、Massの各スペクトルを図7、図8、図9に示す。
【0039】
収量 59.4g(79.1mmol)
収率 39%
沸点 140−145℃/15Pa
性状 白色固体
【0040】
・F(CF2)10(C6H4)2CH2CH=CH2[10F2PA]の合成
【化27】
【0041】
1Lナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液30.6ml(84.5mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液52.6ml(40.5mmol)を加え、氷冷下で1時間撹拌した。その後、750mlのジエチルエーテルに溶解させた10F2PB 21.0gg(27.9mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅1.74g(9.14mmol)を加えた後、アリルブロミド26.9g(222mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらにヘキサンを展開溶剤としてシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
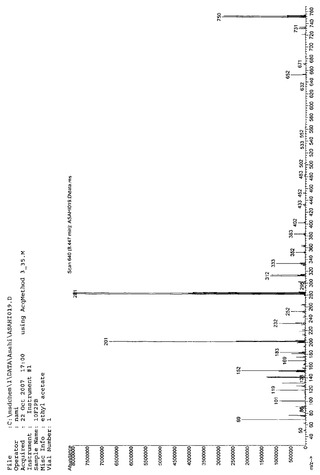
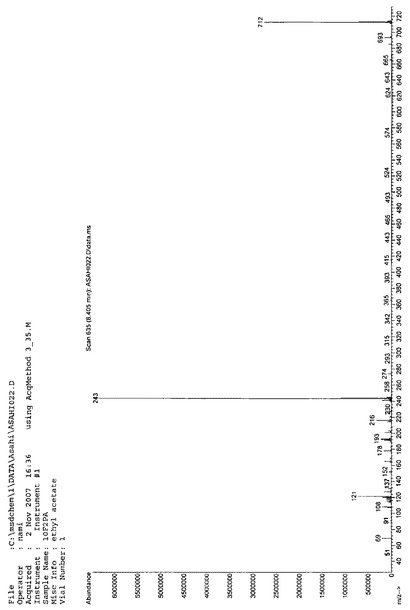
Massスペクトルを分析した結果、m/z(分子量)712により、10F2PAであると同定した。NMR、IR、Massの各スペクトルを図10、図11、図12に示す。
【0042】
収量 16.5g(23.2mmol)
収率 83%
沸点 115−120℃/15Pa
性状 白色固体
【0043】
・F(CF2)10(C6H4)2CH2CH2CH2SiCl3[10F2P3S3C]の合成
【化28】
【0044】
100ml肉厚ガラスアンプル管に撹拌子を入れ、ここに10F2PA 2.00g(3.07mmol)、THF10mlを採取し、窒素雰囲気に置換し、外部を液体窒素で冷却し、トリクロロシラン1.0g(8.2mmol)、触媒の0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、生じた黒色粉末状沈殿(Pt)を濾別除去した。濾液中からTHF、トリメクロロシランを減圧留去した。
【0045】
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)847により、10F2P3S3Cであると同定した。
また、10F2P3S3CのIRスペクトルを図13に示す。1100〜1200cm−1の大きな吸収はC−F伸縮振動を示しているが、2290cm−1付近の吸収ピークがなく、−N=C=Oがないことを示している。
また、10F2P3S3CのNMRスペクトルは、以下の通りであった(Hは、下線プロトンの積分比を示す。)。
0.9113−0.9389ppm:多重線 −CH2−Si 2H
1.8588ppm:多重線 −CH2−CH2−Si 2H
2.7162ppm:多重線 −CH2−CH2−CH2−Si 2H
7.2960−7.7147ppm:ベンゼン環プロトン 8H
【0046】
収量 2.35g
収率 98%
性状 白色固体
【0047】
[実施例4]
F(CF2)10(C6H4)2CH2CH2CH2Si(NCO)3[10F2P3S3I]の合成
【化29】
【0048】
実施例3で得られた10F2P3S3C 2.30g(2.71mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀2.00g(13.3mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)867により、10F2P3S3Iであると同定した。
また、10F2P3S3IのIRスペクトルを図14に示す。1100〜1200cm−1の大きな吸収はC−F伸縮振動を示しており、2290cm−1の吸収ピークが−N=C=Oによる特徴的なピークである。
また、10F2P3S3IのNMRスペクトルは、以下の通りであった(Hは、下線プロトンの積分比を示す。)。
0.9199ppm:多重線 −CH2−Si 2H
1.8439ppm:多重線 −CH2−CH2−Si 2H
2.7115ppm:多重線 −CH2−CH2−CH2−Si 2H
7.2935−7.6991ppm:ベンゼン環プロトン 8H
【0049】
収量 2.30g
収率 97%
性状 白色固体
【0050】
[実施例5]
F(CF2)12(C6H4)2CH2CH2CH2SiCl3[12F2P3S3C]の合成
【0051】
・F(CF2)12(C6H4)2Br[12F2PB]の合成
【化30】
【0052】
100mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉6.4g(0.1mol)、4,4’−ジブロモビフェニル6.3g(20mmol)、ペルフルオロドデシルヨージド15.7g(21mmol)、溶媒としてDMSO30mlを加え、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体12F2PB(粗製)とを濾別し、DMSOで洗浄した。続いて、酢酸エチルを溶媒に用い、銅粉と白色固体との混合物をソックスレー抽出し、粗製12F2PBを得た。抽出した溶液を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後、酢酸エチルを減圧除去した。残留物を減圧蒸留し、さらにクロロホルム可溶部からクロロホルムを減圧除去し、白色固体として12F2PBを得た。
Massスペクトルを分析した結果、m/z(分子量)851により、12F2PBであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2PBのものとほぼ同じであった。
【0053】
収量 8.5g(10mmol)
収率 50%
沸点 145−150℃/18Pa
性状 白色固体
【0054】
・F(CF2)12(C6H4)2CH2CH=CH2[12F2PA]の合成
【化31】
【0055】
100mlナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液3.3ml(9.2mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液5.8ml(4.5mmol)を加え、氷冷下で1時間撹拌した。その後、20mlのジエチルエーテルに溶解させた12F2PB 2.0g(2.4mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅0.2g(1.0mmol)を加えた後、アリルブロミド1.3ml(10.7mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらに展開溶媒にヘキサンを用いたシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
Massスペクトルを分析した結果、m/z(分子量)812により、12F2PAであると同定した。
【0056】
収量 1.5g(1.85mmol)
収率 80%
沸点 135−140℃/20Pa
性状 白色固体
【0057】
・F(CF2)12(C6H4)2CH2CH2CH2SiCl3[12F2P3S3C]の合成
【化32】
【0058】
50ml肉厚ガラスアンプル管に撹拌子を入れ、窒素雰囲気に置換し、外部を液体窒素で冷却した状態でTHF20ml、12F2PA 1.00g(1.23mmol)、トリクロロシラン1.0g(7.4mmol)、触媒として0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、内容物から黒色粉末状固体(Pt)を濾別除去した。濾液中の揮発成分であるTHF、トリメクロロシランを減圧留去した。
得られた化合物について、Massスペクトルを分析した結果、m/z(分子量)947により、12F2P3S3Cであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2P3S3Cのものとほぼ同じであった。
【0059】
収量 1.05g
収率 90%
性状 白色固体
【0060】
[実施例6]
F(CF2)12(C6H4)2CH2CH2CH2Si(NCO)3[12F2P3S3I]の合成
【化33】
【0061】
実施例5で得られた12F2P3S3C 1.05g(1.1mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀1.5g(10mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)967により、12F2P3S3Iであると同定した。また、NMRスペクトルをとったところ、実施例4の10F2P3S3Iのものとほぼ同じであった。
【0062】
収量 0.96g
収率 90%
性状 白色固体
【0063】
[実施例7]
以下に、物性の測定について詳述する。物性の測定は、基質として、ガラスを用いて行った。
(ガラスの洗浄)
スライドガラス(マツナミ製S−7214)を1N水酸化カリウム水溶液(pH>9)に2時間浸した後に取り出し、蒸留水で十分に洗浄した。その後、スライドガラスをデシケーター中で乾燥し、次の表面改質に使用した。
(改質溶液の調製)
実施例4で合成した10F2P3S3Iのシランカップリング剤を用い、THFを溶媒とした3%溶液を準備した。
(ガラスの表面改質)
200ml広口受器に前記の方法で洗浄済みのスライドガラスを入れ、窒素置換を行った。これに対して、上記の改質溶液を広口受器に加え、改質溶液中にスライドガラスを完全に浸し、7時間加熱還流を行った。冷却後、取り出したガラスをTHFで洗浄し、さらに室温で水に5分間浸漬した後、室温で一昼夜自然乾燥した。
【0064】
(改質ガラスの接触角の測定)
シランカップリング剤として用いた10F2P3S3Iの改質ガラスに対する水の接触角を測定した。接触角の測定は、ニック製LSE−B100L型接触角測定装置を使用し、0.9μlの水滴を水平なガラス板上に滴下して接触角を測定する液滴法を用いた。
【0065】
(10F2P3S3Iを用いた改質ガラスの耐熱性試験)
上記改質ガラスそのものの接触角は115.1度であった。
次に、上記改質ガラスを130℃にて30分間のキュアリングを行ったものの接触角は、111.7度であった。
引き続いて、350℃で30分間の熱暴露を行ってから接触角を測定したところ、107.4度であり、耐熱性があることが分かった。
さらに続けて、350℃で120分間の熱暴露を行ってから接触角を測定したところ、96.7度であった(ここまでで、350℃での熱暴露合計時間は2時間30分である)。
続けて、350℃にて60分間の熱暴露を行ってから接触角を測定したところ、90.9度であった(ここまでで、350℃の熱暴露合計時間は合計3時間30分である)。
以上の接触角の値の変化状況から、10F2P3S3Iは十分耐熱性があり、また、接触角のデータは満足できる離型性があると言える。
【0066】
以上のデータに示した水との接触角が高いということは、表面自由エネルギーが低いことを示しており、離型性及び防汚性が高いことを示している。
なお、具体的なデータは示さないが、特開2004−107274号公報と同様に、本発明のシランカップリング剤は、耐酸性、耐酸化性も高いものである。
【産業上の利用可能性】
【0067】
以上説明したように、本発明のペルフルオロアルキル基及びビフェニルアルキル基を有するシランカップリング剤は、耐熱性、耐久性、離型性、及び防汚性がいずれも高いものである。
また、特に、NCO基及びハロゲン基は基質の水酸基との結合性が高いことから、改質表面がモノレーヤー(分子一層)であることも加わって、精密な離型処理が可能な優れた耐熱耐久離型剤として、一般の離型剤としてばかりでなく、ミクロなパターニングを施したSOGのようなシリコン材料、グラシーカーボンのような炭素材料、金属、石英、ニッケル電鋳等に最も適した離型剤となり得る上に、繰り返し転写しても離型剤の剥離を生じない利点を有するものである。
また、近い将来大きな発展が見込まれるナノインプリント用耐熱耐久離型剤として、現段階では最も優れた離型剤となる。
もちろん、耐熱性の高い撥水撥油性表面改質剤として、例えば、電子レンジ内でも使用可能な防汚性のガラス容器等の表面改質剤として使用できる。
また、いずれのシランカップリング剤も、基質や粉体の表面に使用でき、それらの表面を改質できるものである。
さらに、融点が300℃以上の耐熱性プラスチック、エンジニアリングプラスチックス等に対する離型剤、カップリング剤として格別の効果と有用性を有するものである。
【技術分野】
【0001】
本発明は、新規なシランカップリング剤及びその製造方法に関する。
【背景技術】
【0002】
近年、特にシランカップリング剤を離型剤として用い、母型に形成された微細パターンを再現性・解像性良く被転写部材に転写する技術の研究が盛んに行われている。
転写は、母型の微細パターン上に形成した離型剤層上に被転写部材を覆った後、離型剤層から被転写部材を剥離して行われるが、金属を真空蒸着によって、あるいは樹脂を熱硬化させて被転写部材として離型剤層上に設ける場合、離型剤としては高温耐熱性であることが要求される。しかしながら、従来から知られている離型剤にはこの要求に応えられるものがなかった。
【0003】
そこで、本発明者等は、先に、特に耐熱性及び離型性の点に優れた下記一般式(A)で表されるビフェニルアルキル基を有するシランカップリング剤を開発した(特許文献1参照)。
【0004】
【化1】
(式(A)中、RfはF(CF2)nのペルフルオロアルキル基を示し、nは1〜14の整数を示す。)
【0005】
上記一般式(A)で表されるシランカップリング剤を用いると、上述のような真空蒸着あるいは熱硬化のような高温プロセスによって被転写部材を形成する場合でも、1μm未満の高さでアスペクト比が2以上あるような超微細な突起群を含むような母型のパターンを、再現性・解像性良く被転写部材に転写することができる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2008/108438号
【発明の概要】
【発明が解決しようとする課題】
【0007】
ところで、離型剤層を形成する際には、母型表面にシランカップリング剤を含む液を塗布した後に、シランカップリング剤を強固に固定させるように、加熱処理等が行われる。
しかしながら、例えば、微細パターン表面に離型剤としてシランカップリング剤層が形成された母型を金型として繰り返し使用して、パターンが転写された被転写部材を数多く作製していくと、離型剤が徐々に微細パターン表面から剥離してしまうことがある。
そこで、このような問題が発生しないような、母型の基質との結合力が高いシランカップリング剤の出現が期待されている。
【0008】
本発明は、上記事情に鑑みてなされたものであり、その第一の課題は基質との結合力の高いシランカップリング剤を提供することにある。さらに第二の課題は耐熱性、離型性、及び防汚性の良好な、特に350℃以上の温度においても、これら化合物による改質表面接触角の低下が見られない等の優れた物性を有するシランカップリング剤を提供することにある。
【課題を解決するための手段】
【0009】
本発明者等は、特定の一般式で表されるビフェニルアルキル基を有するシランカップリング剤によれば上記課題を解決できることを見出し、本発明を完成させた。より具体的には、本発明は下記のとおりである。
【0010】
[1] 下記一般式(1)で表されるビフェニルアルキル基を有するシランカップリング剤。
【化2】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【0011】
[2] 下記一般式(2)
【化3】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化4】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化5】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化6】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化7】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化8】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させて、下記一般式(8)
【化9】
(式(8)中、nは3〜4の整数を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得る、シランカップリング剤の製造方法。
【0012】
[3] 下記一般式(8)
【化10】
(式(8)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Yはハロゲン原子を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させて、下記式(9)
【化11】
で表される4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得る、シランカップリング剤の製造方法。
【0013】
[4] 下記一般式(1)で表されるシランカップリング剤と溶剤とを含む離型剤。
【化12】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【発明の効果】
【0014】
本発明のシランカップリング剤は、基質との結合力が高く、しかも350℃以上の雰囲気に4時間以上曝露しても、これら化合物による改質表面の接触角の低下が見られないというほどの優れた耐熱性、離型性、及び防汚性を有するものであり、格別の効果と有用性を有する。
【図面の簡単な説明】
【0015】
【図1】8F2PBのNMRスペクトルである(実施例1)。
【図2】8F2PBのIRスペクトルである(実施例1)。
【図3】8F2PBのMassスペクトルである(実施例1)。
【図4】8F2PAのNMRスペクトルである(実施例1)。
【図5】8F2PAのIRスペクトルである(実施例1)。
【図6】8F2PAのMassスペクトルである(実施例1)。
【図7】10F2PBのNMRスペクトルである(実施例3)。
【図8】10F2PBのIRスペクトルである(実施例3)。
【図9】10F2PBのMassスペクトルである(実施例3)。
【図10】10F2PAのNMRスペクトルである(実施例3)。
【図11】10F2PAのIRスペクトルである(実施例3)。
【図12】10F2PAのMassスペクトルである(実施例3)。
【図13】10F2P3S3CのIRスペクトルである(実施例3)。
【図14】10F2P3S3IのIRスペクトルである(実施例4)。
【発明を実施するための形態】
【0016】
本発明のシランカップリング剤は下記一般式(1)で表されるものである。
【0017】
【化13】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【0018】
上記一般式(1)で表されるシランカップリング剤のうち、Xがハロゲン原子であるものは、下記のようにして製造することができる。
【0019】
すなわち、下記一般式(2)
【化14】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化15】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化16】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化17】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化18】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化19】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させることにより、下記一般式(8)
【化20】
(式(8)中、nは3〜4の整数を示す。)
で表される本発明の(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得ることができる。
【0020】
また、上記一般式(1)で表されるシランカップリング剤のうち、Xがイソシアナト基であるものは、下記のようにして製造することができる。
【0021】
すなわち、上記(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させることにより、下記式(9)
【化21】
で表される本発明の4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得ることができる。
【実施例】
【0022】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれらの実施例により限定されるものではない。
【0023】
[実施例1]
F(CF2)8(C6H4)2CH2CH2CH2SiCl3[8F2P3S3C]の合成
【0024】
・F(CF2)8(C6H4)2Br[8F2PB]の合成
【化22】
【0025】
500mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉63.6g(1.0mol)、4,4’−ジブロモビフェニル62.4g(200mmol)、ペルフルオロオクチルヨージド115g(210mmol)、溶媒としてジメチルスルホキシド(DMSO)300mlを加え、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体(粗製8F2PB)とを濾別し、DMSOで洗浄した。続いて酢酸エチルを溶媒に用い、銅粉と白色固体の混合物とをソックスレー抽出し、粗製8F2PBを得た。抽出液を飽和食塩水で洗浄し、さらに抽出液を硫酸マグネシウムで脱水し、酢酸エチルを減圧除去した。残留物を減圧蒸留して、留出物を得た。
得られた留出物についてMassスペクトルを分析した結果、m/z(分子量)651により、8F2PBであると同定した。NMR、IR、Massの各スペクトルを図1、図2、図3に示す。
【0026】
収量 68.0g(104mmol)
収率 52%
沸点 130−135℃/30Pa
性状 白色固体
【0027】
・F(CF2)8(C6H4)2CH2CH=CH2[8F2PA]の合成
【化23】
【0028】
1Lナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液32.6ml(90.0mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液58.4ml(45.0mmol)を加え、氷冷下で1時間撹拌した。その後、750mlのジエチルエーテルに溶解させた8F2PB 19.5gg(30.0mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅2.0g(10.5mmol)を加えた後、アリルブロミド18.2g(150mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらにヘキサンを展開溶剤としてシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
Massスペクトルを分析した結果、m/z(分子量)612により、8F2PAであると同定した。NMR、IR、Massの各スペクトルを図4、図5、図6に示す。
【0029】
収量 14.7g(24.0mmol)
収率 80%
沸点 130−135℃/20Pa
性状 白色固体
【0030】
・F(CF2)8(C6H4)2CH2CH2CH2SiCl3[8F2P3S3C]の合成
【化24】
【0031】
100ml肉厚ガラスアンプル管に撹拌子を入れ、ここに8F2PA 2.00g(3.27mmol)、THF10mlを採取し、窒素雰囲気に置換し、外部を液体窒素で冷却し、トリクロロシラン1.0g(8.2mmol)、触媒の0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、生じた黒色粉末状沈殿(Pt)を濾別除去した。濾液中からTHF、トリメクロロシランを減圧留去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)747により、8F2P3S3Cであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2P3S3Cのものとほぼ同じであった。
【0032】
収量 2.32g
収率 95%
性状 白色固体
【0033】
[実施例2]
F(CF2)8(C6H4)2CH2CH2CH2Si(NCO)3[8F2P3S3I]の合成
【化25】
【0034】
実施例1で得られた8F2P3S3C 2.0g(2.7mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀2.00g(13.3mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
Massスペクトルを分析した結果、m/z(分子量)767により、8F2P3S3Iであると同定した。また、NMRスペクトルをとったところ、実施例4の10F2P3S3Iのものとほぼ同じであった。
【0035】
収量 1.97g(2.5mmol)
収率 93%
性状 白色固体
【0036】
[実施例3]
F(CF2)10(C6H4)2CH2CH2CH2SiCl3[10F2P3S3C]の合成
【0037】
・F(CF2)10(C6H4)2Br[10F2PB]の合成
【化26】
【0038】
500mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉63.6g(1.0mol)、4,4’−ジブロモビフェニル64.0g(205mmol)、ペルフルオロデシルヨージド137g(212mmol)、溶媒としてDMSO350mlを加えた後、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体(粗製10F2PB)とを濾別し、DMSOで洗浄した。続いて酢酸エチルを溶媒に用い、銅粉と白色固体との混合物をソックスレー抽出し、粗製10F2PBを得た。抽出液を飽和食塩水で洗浄し、さらに抽出液を硫酸マグネシウムで脱水し、酢酸エチルを減圧除去した。残留物を減圧蒸留し、さらにクロロホルム可溶部からクロロホルムを減圧除去し、白色固体として10F2PBを得た。
Massスペクトルを分析した結果、m/z(分子量)751により、10F2PBであると同定した。NMR、IR、Massの各スペクトルを図7、図8、図9に示す。
【0039】
収量 59.4g(79.1mmol)
収率 39%
沸点 140−145℃/15Pa
性状 白色固体
【0040】
・F(CF2)10(C6H4)2CH2CH=CH2[10F2PA]の合成
【化27】
【0041】
1Lナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液30.6ml(84.5mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液52.6ml(40.5mmol)を加え、氷冷下で1時間撹拌した。その後、750mlのジエチルエーテルに溶解させた10F2PB 21.0gg(27.9mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅1.74g(9.14mmol)を加えた後、アリルブロミド26.9g(222mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらにヘキサンを展開溶剤としてシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
Massスペクトルを分析した結果、m/z(分子量)712により、10F2PAであると同定した。NMR、IR、Massの各スペクトルを図10、図11、図12に示す。
【0042】
収量 16.5g(23.2mmol)
収率 83%
沸点 115−120℃/15Pa
性状 白色固体
【0043】
・F(CF2)10(C6H4)2CH2CH2CH2SiCl3[10F2P3S3C]の合成
【化28】
【0044】
100ml肉厚ガラスアンプル管に撹拌子を入れ、ここに10F2PA 2.00g(3.07mmol)、THF10mlを採取し、窒素雰囲気に置換し、外部を液体窒素で冷却し、トリクロロシラン1.0g(8.2mmol)、触媒の0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、生じた黒色粉末状沈殿(Pt)を濾別除去した。濾液中からTHF、トリメクロロシランを減圧留去した。
【0045】
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)847により、10F2P3S3Cであると同定した。
また、10F2P3S3CのIRスペクトルを図13に示す。1100〜1200cm−1の大きな吸収はC−F伸縮振動を示しているが、2290cm−1付近の吸収ピークがなく、−N=C=Oがないことを示している。
また、10F2P3S3CのNMRスペクトルは、以下の通りであった(Hは、下線プロトンの積分比を示す。)。
0.9113−0.9389ppm:多重線 −CH2−Si 2H
1.8588ppm:多重線 −CH2−CH2−Si 2H
2.7162ppm:多重線 −CH2−CH2−CH2−Si 2H
7.2960−7.7147ppm:ベンゼン環プロトン 8H
【0046】
収量 2.35g
収率 98%
性状 白色固体
【0047】
[実施例4]
F(CF2)10(C6H4)2CH2CH2CH2Si(NCO)3[10F2P3S3I]の合成
【化29】
【0048】
実施例3で得られた10F2P3S3C 2.30g(2.71mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀2.00g(13.3mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)867により、10F2P3S3Iであると同定した。
また、10F2P3S3IのIRスペクトルを図14に示す。1100〜1200cm−1の大きな吸収はC−F伸縮振動を示しており、2290cm−1の吸収ピークが−N=C=Oによる特徴的なピークである。
また、10F2P3S3IのNMRスペクトルは、以下の通りであった(Hは、下線プロトンの積分比を示す。)。
0.9199ppm:多重線 −CH2−Si 2H
1.8439ppm:多重線 −CH2−CH2−Si 2H
2.7115ppm:多重線 −CH2−CH2−CH2−Si 2H
7.2935−7.6991ppm:ベンゼン環プロトン 8H
【0049】
収量 2.30g
収率 97%
性状 白色固体
【0050】
[実施例5]
F(CF2)12(C6H4)2CH2CH2CH2SiCl3[12F2P3S3C]の合成
【0051】
・F(CF2)12(C6H4)2Br[12F2PB]の合成
【化30】
【0052】
100mlナス型フラスコを窒素雰囲気に置換し、銅ブロンズ粉6.4g(0.1mol)、4,4’−ジブロモビフェニル6.3g(20mmol)、ペルフルオロドデシルヨージド15.7g(21mmol)、溶媒としてDMSO30mlを加え、140℃で40時間加熱撹拌した。溶液を室温まで冷却し、吸引濾過により過剰の銅粉と白色固体12F2PB(粗製)とを濾別し、DMSOで洗浄した。続いて、酢酸エチルを溶媒に用い、銅粉と白色固体との混合物をソックスレー抽出し、粗製12F2PBを得た。抽出した溶液を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後、酢酸エチルを減圧除去した。残留物を減圧蒸留し、さらにクロロホルム可溶部からクロロホルムを減圧除去し、白色固体として12F2PBを得た。
Massスペクトルを分析した結果、m/z(分子量)851により、12F2PBであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2PBのものとほぼ同じであった。
【0053】
収量 8.5g(10mmol)
収率 50%
沸点 145−150℃/18Pa
性状 白色固体
【0054】
・F(CF2)12(C6H4)2CH2CH=CH2[12F2PA]の合成
【化31】
【0055】
100mlナス型フラスコを窒素置換し、氷冷下に冷却した後、2.76M n−ブチルリチウム ヘキサン溶液3.3ml(9.2mmol)を加え、続いて0.77M i−プロピルマグネシウムブロミド/THF溶液5.8ml(4.5mmol)を加え、氷冷下で1時間撹拌した。その後、20mlのジエチルエーテルに溶解させた12F2PB 2.0g(2.4mmol)を滴下し、氷冷下で1時間撹拌した。触媒としてヨウ化銅0.2g(1.0mmol)を加えた後、アリルブロミド1.3ml(10.7mmol)を滴下し、室温で67時間撹拌後、飽和塩化アンモニウム水溶液を沈殿が生じなくなるまで加え、反応を止めた。反応溶液を酢酸エチルで抽出し、有機層を硫酸マグネシウムで脱水後、溶媒を減圧留去した。得られた黄色固体を減圧蒸留し、さらに展開溶媒にヘキサンを用いたシリカゲルカラムクトマトグラフィー(ワコーゲルC−300)により精製した。
Massスペクトルを分析した結果、m/z(分子量)812により、12F2PAであると同定した。
【0056】
収量 1.5g(1.85mmol)
収率 80%
沸点 135−140℃/20Pa
性状 白色固体
【0057】
・F(CF2)12(C6H4)2CH2CH2CH2SiCl3[12F2P3S3C]の合成
【化32】
【0058】
50ml肉厚ガラスアンプル管に撹拌子を入れ、窒素雰囲気に置換し、外部を液体窒素で冷却した状態でTHF20ml、12F2PA 1.00g(1.23mmol)、トリクロロシラン1.0g(7.4mmol)、触媒として0.1M H2PtCl6/THF溶液0.1ml(0.01mmol)を採取した。アンプル管内部の試薬類を完全に固化させた後、真空ポンプを用いて40Paに減圧し、その状態で酸素バーナーにてガラスアンプル管を溶封した。これを室温に戻した後、80℃で2時間、さらに100℃で20時間撹拌した。放冷後開管し、内容物から黒色粉末状固体(Pt)を濾別除去した。濾液中の揮発成分であるTHF、トリメクロロシランを減圧留去した。
得られた化合物について、Massスペクトルを分析した結果、m/z(分子量)947により、12F2P3S3Cであると同定した。また、NMRスペクトルをとったところ、実施例3の10F2P3S3Cのものとほぼ同じであった。
【0059】
収量 1.05g
収率 90%
性状 白色固体
【0060】
[実施例6]
F(CF2)12(C6H4)2CH2CH2CH2Si(NCO)3[12F2P3S3I]の合成
【化33】
【0061】
実施例5で得られた12F2P3S3C 1.05g(1.1mmol)を100mlナスフラスコに取り、THF20mlに溶解し、シアン酸銀1.5g(10mmol)を加えて沸点で撹拌し、加熱還流操作を5時間行った。室温まで冷却し、生じた塩化銀、未反応のシアン酸銀を濾別除去した。濾液中の揮発成分を減圧除去した。
得られた化合物についてMassスペクトルを分析した結果、m/z(分子量)967により、12F2P3S3Iであると同定した。また、NMRスペクトルをとったところ、実施例4の10F2P3S3Iのものとほぼ同じであった。
【0062】
収量 0.96g
収率 90%
性状 白色固体
【0063】
[実施例7]
以下に、物性の測定について詳述する。物性の測定は、基質として、ガラスを用いて行った。
(ガラスの洗浄)
スライドガラス(マツナミ製S−7214)を1N水酸化カリウム水溶液(pH>9)に2時間浸した後に取り出し、蒸留水で十分に洗浄した。その後、スライドガラスをデシケーター中で乾燥し、次の表面改質に使用した。
(改質溶液の調製)
実施例4で合成した10F2P3S3Iのシランカップリング剤を用い、THFを溶媒とした3%溶液を準備した。
(ガラスの表面改質)
200ml広口受器に前記の方法で洗浄済みのスライドガラスを入れ、窒素置換を行った。これに対して、上記の改質溶液を広口受器に加え、改質溶液中にスライドガラスを完全に浸し、7時間加熱還流を行った。冷却後、取り出したガラスをTHFで洗浄し、さらに室温で水に5分間浸漬した後、室温で一昼夜自然乾燥した。
【0064】
(改質ガラスの接触角の測定)
シランカップリング剤として用いた10F2P3S3Iの改質ガラスに対する水の接触角を測定した。接触角の測定は、ニック製LSE−B100L型接触角測定装置を使用し、0.9μlの水滴を水平なガラス板上に滴下して接触角を測定する液滴法を用いた。
【0065】
(10F2P3S3Iを用いた改質ガラスの耐熱性試験)
上記改質ガラスそのものの接触角は115.1度であった。
次に、上記改質ガラスを130℃にて30分間のキュアリングを行ったものの接触角は、111.7度であった。
引き続いて、350℃で30分間の熱暴露を行ってから接触角を測定したところ、107.4度であり、耐熱性があることが分かった。
さらに続けて、350℃で120分間の熱暴露を行ってから接触角を測定したところ、96.7度であった(ここまでで、350℃での熱暴露合計時間は2時間30分である)。
続けて、350℃にて60分間の熱暴露を行ってから接触角を測定したところ、90.9度であった(ここまでで、350℃の熱暴露合計時間は合計3時間30分である)。
以上の接触角の値の変化状況から、10F2P3S3Iは十分耐熱性があり、また、接触角のデータは満足できる離型性があると言える。
【0066】
以上のデータに示した水との接触角が高いということは、表面自由エネルギーが低いことを示しており、離型性及び防汚性が高いことを示している。
なお、具体的なデータは示さないが、特開2004−107274号公報と同様に、本発明のシランカップリング剤は、耐酸性、耐酸化性も高いものである。
【産業上の利用可能性】
【0067】
以上説明したように、本発明のペルフルオロアルキル基及びビフェニルアルキル基を有するシランカップリング剤は、耐熱性、耐久性、離型性、及び防汚性がいずれも高いものである。
また、特に、NCO基及びハロゲン基は基質の水酸基との結合性が高いことから、改質表面がモノレーヤー(分子一層)であることも加わって、精密な離型処理が可能な優れた耐熱耐久離型剤として、一般の離型剤としてばかりでなく、ミクロなパターニングを施したSOGのようなシリコン材料、グラシーカーボンのような炭素材料、金属、石英、ニッケル電鋳等に最も適した離型剤となり得る上に、繰り返し転写しても離型剤の剥離を生じない利点を有するものである。
また、近い将来大きな発展が見込まれるナノインプリント用耐熱耐久離型剤として、現段階では最も優れた離型剤となる。
もちろん、耐熱性の高い撥水撥油性表面改質剤として、例えば、電子レンジ内でも使用可能な防汚性のガラス容器等の表面改質剤として使用できる。
また、いずれのシランカップリング剤も、基質や粉体の表面に使用でき、それらの表面を改質できるものである。
さらに、融点が300℃以上の耐熱性プラスチック、エンジニアリングプラスチックス等に対する離型剤、カップリング剤として格別の効果と有用性を有するものである。
【特許請求の範囲】
【請求項1】
下記一般式(1)で表されるビフェニルアルキル基を有するシランカップリング剤。
【化1】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【請求項2】
下記一般式(2)
【化2】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化3】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化4】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化5】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化6】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化7】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させて、下記一般式(8)
【化8】
(式(8)中、nは3〜4の整数を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得る、シランカップリング剤の製造方法。
【請求項3】
下記一般式(8)
【化9】
(式(8)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Yはハロゲン原子を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させて、下記式(9)
【化10】
で表される4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得る、シランカップリング剤の製造方法。
【請求項4】
下記一般式(1)で表されるシランカップリング剤と溶剤とを含む離型剤。
【化11】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【請求項1】
下記一般式(1)で表されるビフェニルアルキル基を有するシランカップリング剤。
【化1】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【請求項2】
下記一般式(2)
【化2】
で表される4,4’−ジブロモビフェニルを、下記式(3)
【化3】
(式(3)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示す。)
で表されるペルフルオロアルキルヨージドと極性溶媒中で、銅ブロンズ粉触媒を用いて反応させて、下記一般式(4)
【化4】
で表される4−ペルフルオロアルキル−4’−ブロモビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−ブロモビフェニルを、下記式(5)
【化5】
(式(5)中、yは1〜2の整数を示す。)
で表されるアルケニルブロミドと極性溶媒中で、ヨウ化銅触媒を用いて反応させて、下記一般式(6)
【化6】
で表される4−ペルフルオロアルキル−4’−アルケニルビフェニルを合成し、
次いで、該4−ペルフルオロアルキル−4’−アルケニルビフェニルを、下記式(7)
【化7】
(式(7)中、Yはハロゲン原子を示す。)
で表されるトリハロゲン化シランと有機溶媒中で、塩化白金酸触媒を用いて反応させて、下記一般式(8)
【化8】
(式(8)中、nは3〜4の整数を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを得る、シランカップリング剤の製造方法。
【請求項3】
下記一般式(8)
【化9】
(式(8)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Yはハロゲン原子を示す。)
で表される(4−ペルフルオロアルキルビフェニル)アルキルトリハロゲン化シランを有機溶媒中で、シアン酸銀と反応させて、下記式(9)
【化10】
で表される4−ペルフルオロアルキルビフェニル)アルキルトリイソシアナトシランを得る、シランカップリング剤の製造方法。
【請求項4】
下記一般式(1)で表されるシランカップリング剤と溶剤とを含む離型剤。
【化11】
(式(1)中、RfはF(CF2)mのペルフルオロアルキル基を示し、mは4〜14の整数を示し、nは3〜4の整数を示し、Xはハロゲン原子又はイソシアナト基を示す。)
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2011−184304(P2011−184304A)
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願番号】特願2010−47882(P2010−47882)
【出願日】平成22年3月4日(2010.3.4)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構、「ナノインプリント用の高耐熱性離型剤の開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(803000115)学校法人東京理科大学 (545)
【Fターム(参考)】
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願日】平成22年3月4日(2010.3.4)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構、「ナノインプリント用の高耐熱性離型剤の開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(803000115)学校法人東京理科大学 (545)
【Fターム(参考)】
[ Back to top ]