
シロリムスのテトラゾール誘導体のワンポット合成
【課題】大規模でのゾタロリムス及び他のラパマイシン誘導体を得るためのワンポット合成法の提供。
【解決手段】乾燥ラパマイシンを酢酸イソプロピルに溶解する。冷却及び2,6−ルチジンの添加の後に、トリフルオロメタンスルホン酸無水物を−30℃で徐々に加える。塩をろ過により除去する。テトラゾールを、続いて第3級塩基ジイソプロピルエチルアミンを加える。室温でインキュベートした後、生成物を濃縮し、THF/ヘプタンを溶離剤として用いてシリカゲルカラムにより精製する。生成物を収集し、濃縮し、アセトン/ヘプタンカラムを用いて精製する。生成物を含む画分を濃縮する。生成物をt−BMEに溶解し、ヘプタンで沈殿させる。固体をアセトンに溶解し、ブチルヒドロキシトルエンで処理し、溶液を濃縮する。この工程をアセトンを用いて2回繰り返して溶媒を除去する。乾燥の前に0.5%のBHTなどの少なくとも1つの安定化剤を加える。
【解決手段】乾燥ラパマイシンを酢酸イソプロピルに溶解する。冷却及び2,6−ルチジンの添加の後に、トリフルオロメタンスルホン酸無水物を−30℃で徐々に加える。塩をろ過により除去する。テトラゾールを、続いて第3級塩基ジイソプロピルエチルアミンを加える。室温でインキュベートした後、生成物を濃縮し、THF/ヘプタンを溶離剤として用いてシリカゲルカラムにより精製する。生成物を収集し、濃縮し、アセトン/ヘプタンカラムを用いて精製する。生成物を含む画分を濃縮する。生成物をt−BMEに溶解し、ヘプタンで沈殿させる。固体をアセトンに溶解し、ブチルヒドロキシトルエンで処理し、溶液を濃縮する。この工程をアセトンを用いて2回繰り返して溶媒を除去する。乾燥の前に0.5%のBHTなどの少なくとも1つの安定化剤を加える。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
[0001][関連出願に対する相互参照]
【0002】
[0002]適用せず。
【0003】
[0003][連邦政府により委託された研究又は開発に関する陳述]
【0004】
[0004]適用せず。
【0005】
[0005][コンパクトディスクで提出した資料の参照による組み込み]
【0006】
[0006]適用せず。
【0007】
[0007][技術分野]
【0008】
[0008]本発明は、ラパマイシンの類似体を合成する新規な方法に関する。該類似体は、抗増殖及び免疫調節への適用に有用である。
【0009】
[0009][発明の背景]
【0010】
[0010]緒言
【0011】
[0011]シロリムス
【0012】
[0012]モアイが見下ろしているとき、カナダ探検隊は1964年に土壌を掘って、強力な免疫抑制、抗真菌及び抗細胞増殖分子を産生する真菌を発見した。真菌は、イースター島からカナダの研究室にSuren Sehgalの手により届けられた。彼は、1972年に真菌ストレプトミセスハイグロスコピカス(Streptomyces hygroscopicus)の精製化合物の特性を解明したが、この知見は捨てられ、企業の優先事項の犠牲となった。Sehgalは、1987年に研究を復活させ、化合物を免疫抑制剤として開発した。今日、ラパマイシン(イースター島原住民がその祖国を思い出す名称であるラパヌイ(Rapa Nui)にちなんで命名された)は、臓器移植のリスク及びステントの副作用を低減するために用いられており、抗腫瘍医薬品として研究されている。
【0013】
[0013]ラパマイシンは、シロリムスとしても知られており、in vitro及びin vivoで、特にカンジダアルビカンス(Candida albicans)に対して、真菌の増殖を抑制する大環状トリエン抗生物質である(Bakerら、1978、Sehgal、1975、Sehgal、1976、Sehgalら、1975、Vezinaら、1975)。シロリムス単独(Surendra、1989)又はピシバニルとの併用(Eng、1983)は、抗腫瘍活性を有することが示された。1977年に、シロリムスがアレルギー性脳脊髄炎(多発性硬化症のモデル)、アジュバント関節炎及びリウマチ関節炎の実験的モデルにおいて免疫抑制剤として有効であることが示された(Martelら、1977)。シロリムスはまた、IgE様抗体の形成を効果的に抑制する(Martelら、1977)。その構造を下(VI)に示す。
【0014】
[0014]ABT−578[40−エピ−(1−テトラゾリル)−ラパマイシン]は、今日ではゾタロリムスとしてよりよく知られており、シロリムスから誘導された半合成マクロライドトリエン抗生物質である。ゾタロリムスは、その前駆体ゾタロリムスと同様に、T細胞リンパ球の増殖の強力な抑制剤である。ゾタロリムスは、再狭窄を最小限にするために心血管ステント、特に薬剤溶出ステント(DES)を被覆することに例外的適用を見い出された(Mollisonら、2003)。ゾタロリムスは、両方がN−1異性体である、主要なピラン(10位における6員環の異性体;1)及び副次的なオキセパン異性体(9位における7員環の異性体;2)の2つの異性体として存在する(Mollison、2000)。
【0015】
[0015]ラパマイシンの他の化学修飾が試みられた。これらは、ラパマイシンのモノ及びジエステル誘導体(Caufield、1992)、ラパマイシンの27−オキシム(Failli、1992a)、ラパマイシンの40−オキソ類似体(Caufield、1991)、二環式ラパマイシン(Kao、1992a)、ラパマイシン二量体(Kao、1992b)、ラパマイシンのシリルエーテル(Failli、1992b)並びにアリールスルホン酸及びスルファミン酸(Failli、1993)の調製などである。
【0016】
[0016]その抗真菌、免疫抑制及び抗腫瘍活性に加えて、シロリムスは、動物モデルにおける新生内膜の増殖並びにヒトにおける再狭窄率を低減させる。シロリムスはまた、リウマチ関節炎の治療用の薬剤としてのその選択を支持する特性である抗炎症作用を示す。エベロリムス及び特にゾタロリムスなどのシロリムスの類似体を被覆したステントは、臨床試験において再狭窄を予防するのに有効である。
【0017】
[0017]ステント及び他の埋込み医療デバイス
【0018】
[0018]ステントは、種々の疾患及び状態、特にアテローム性動脈硬化疾患に起因する血管又は管路の径の重大な減少を治療するために用いられ、しばしば血管形成術後に用いられる。ステントは、動脈に頻繁に用いられるが、静脈、胆管、食道、気管、大気管支、尿管及び尿道などの他の構造にも用いられる。ステントは、英国の歯科医であるCharles Stent(1845〜1901年)の新しいアイディアである。
【0019】
[0019]有害な内腔の狭窄の治療に有効であるが、血管ステントは、医学的に皮肉な例において、治療のためにそれらが使用される状態を再形成するおそれもある。ステントは、内腔の内側の厚い内皮組織、すなわち新生内膜の成長をもたらし得る。成長の程度は様々であるが、新生内膜は、成長して血管内腔を閉塞することができ、これは再狭窄の典型的なタイプである。
【化1】

【化2】
【0020】
[0020]ゾタロリムスの以前の合成
【0021】
[0021]Mollisonは、シロリムスからゾタロリムスを生成させるいくつかの方法を示した(Mollison、2000)。例えば、シロリムスのC−40ヒドロキシルをトリフラートの形成により活性化し、次いで、トリフラートをカラムクロマトグラフィーにより精製する。トリフラートの精製の間、クロマトグラフィー中の水の存在により、活性化された中間体のうちの一部はシロリムス及びそのエピマーであるエピ−シロリムスに逆戻りする。次いで、精製トリフラートを第2のステップにおいてテトラゾールと反応させて、シロリムスの40−エピ−テトラゾール誘導体、すなわち、ゾタロリムスを生成させる。次いで、粗生成物をカラムクロマトグラフィーにより精製する。しかし、この精製によってさえも、最終生成物は、シロリムス及びエピ−シロリムスの混入物を含む可能性がある。
【0022】
[0025][発明の概要]
【0023】
[0026]本発明は、ラパマイシンの誘導体をワンポットで実施する方法を提供し、そのような方法により調製された、抗酸化剤を含む組成物を提供する。
【0024】
[0027]第1の態様において、本発明は、式I
【化3】
の分子を調製する方法であって、
第1のステップ(a)において式II
【化4】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化5】
の分子を生成させ、次いで、第2のステップ(b)において式IIIの分子を式IV
【化6】
の分子と反応させる、方法を提供する。
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【0025】
[0028]方法のステップ(a)は、2,6−ジメチルピリジン又はジイソプロピルエチルアミンなどの非求核塩基の存在下で実施する。ステップ(a)はまた、酢酸イソプロピル又はジクロロメタンなどの溶媒中で実施する。いくつかの実施形態において、ジクロロメタンを、ステップ(b)の前又はステップ(b)の間に酢酸イソプロピルに交換する。
【0026】
[0029]式IVによって表される分子において、R10はHであってよく、R9はH、メチル又はフェニルである。いくつかの実施形態において、R9及びR10は、Hである。
【0027】
[0030]ステップ(b)は、非プロトン性溶媒などの溶媒の存在下で実施する。非プロトン性溶媒は、例えば、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、ジメチルアミン、N,N−ジメチルホルムアミド又は炭酸プロピレンであってよい。いくつかの実施形態において、ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン、アセトニトリル、ジメチルアミン又はN,N−ジメチルホルムアミドのいずれかの存在下で実施する。いくつかの実施形態において、ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの中で実施する。
【0028】
[0031]特定の実施形態において、式IIによって表される分子で、R1は=Oであり、R2はHであり、R3は=Oであり、R4はHである。
【0029】
[0032]他の態様において、ゾタロリムスは、ラパマイシンから本発明の新規な方法により提供される。
【0030】
[0033]他の態様において、本発明は、抗酸化剤と組み合わされた、本発明の方法により調製される分子の組成物を提供する。そのような抗酸化剤としては、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などがある。1つの実施形態において、抗酸化剤は3,5−ジ−tert−4−ブチルヒドロキシトルエンである。
【0031】
[0034]他の態様において、本発明は、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などの抗酸化剤を用いて調合される、本発明の方法により調製されるゾタロリムスの組成物を提供する。1つの実施形態において、抗酸化剤が3,5−ジ−tert−4−ブチルヒドロキシトルエンである。
【0032】
[0035][発明の詳細な説明]
【0033】
[0036]本発明は、ゾタロリムスを生成させ、以前の方法のシロリムス及びエピ−シロリムス混入物を実質的に除去し、医薬品を調製するより効果的な方法である、C−40位におけるシロリムスのテトラゾール類似体を調製するワンポット法を提供する。この方法において、トリフラートを、2,6−ルチジン又は2,6−ジ−tert−ブチルピリジン若しくは2,4,6−コリジンのような他の置換ピリジン、ピリジン、又はヒューニッヒ塩基ジイソプロピルアミン(DIEA)のような非求核塩基の存在下で溶媒としての酢酸イソプロピル(IPAc)又はジクロロメタン(DCM)中で生成させる。IPAcをトリフラートの生成時に溶媒として用いる場合、塩をろ過し、トリフラート溶液をDIEAの存在下でテトラゾールと反応させることができる。DCMをトリフラートの生成時に溶媒として用いる場合、溶媒をIPAcに切り替える。その後、テトラゾールとのSN2反応をDIEAを塩基として用いてIPAc及びテトラゾール中で行わせる。溶媒の除去後の粗生成物を、THF/ヘプタン中、続いてヘプタン/アセトン中カラムクロマトグラフィーにより精製する。精製された生成物は、t−ブチルメチルエーテル(t−BME)/ヘプタンで処理することにより固体として分離することができる。そのように得られたゾタロリムスは、室温で不安定であるが、BHT(2,6−ジ−t−ブチル−4−メチルフェノール、ブチル化ヒドロキシトルエン)、2,6−ジ−t−ブチル−4−エチルフェノール(DEP)、2,6−ジ−t−ブチル−4−メトキシフェノール(DMP)などの抗酸化剤の添加により安定化することができる。
【0034】
[0037]ワンポット法のいくつかの重要な利点は以下の通りである。
1.以前の方法において最終生成物中の混入物の重要な源であったトリフラートの精製を不要とすること、
2.方法の粗生成物をTHF:ヘプタン中で精製することによる、SN2反応中に生成するシロリムス及びエピ−シロリムス副生成物レベルのさらなる低下、
3.容易に回収し、再使用することができる非プロトン性溶媒をSN2反応に使用し、それにより、以前の方法によりもたらされる費用及び環境上の問題を低減すること、
4.t−BMEに溶解し、ヘプタンを加えることによる、又は逆添加法(reverse−addition procedure)による生成物の容易な分離及び精製、
5.抗酸化剤を加えることによる、不純物のない生成物の容易な安定化、及び
6.アセトニトリル又はアセトニトリル:水における凍結乾燥による容易な分離。
【0035】
[0038]定義
【0036】
[0039]「プロドラッグ」は、in vivoで例えば、血液中の加水分解により親化合物に速やかに変換される化合物を意味する。十分な解説が利用可能である(Higuchi及びStella、1987;Roche、1987)。
【0037】
[0040]「薬学的に許容できるプロドラッグ」は、適切な医学的判断の範囲内で、過度の毒性、刺激及びアレルギー反応を伴わずにヒト及び下等哺乳類の組織との接触に使用することに適しており、妥当なベネフィット/リスク比に対応しており、それらの意図した用途並びに可能な場合本発明の化合物の両性イオンの形態で有効である、本発明の化合物のプロドラッグを意味する。本発明の特に好ましい薬学的に許容できるプロドラッグは、本発明の化合物のC−31ヒドロキシル基のプロドラッグエステルである。
【0038】
[0041]「プロドラッグエステル」は、生理的条件下で加水分解されるいくつかのエステル形成基のいずれかを意味する。プロドラッグエステル基の例としては、アセチル、エタノイル、ピバロイル、ピバロイルオキシメチル、アセトキシメチル、フタリジル、メトキシメチル、インダニル等、並びに本発明の化合物のC−31ヒドロキシル基への天然又は非天然由来アミノ酸の結合により得られるエステル基などが挙げられる。
【0039】
[0042]「治療用物質」は、適切な用量で対象に適切に投与したとき、対象への有用な効果を有するあらゆる物質を意味する。
【0040】
[0043]置換基又は可変基(例えば、アリール、アルコキシ、R1、R2、R3、R5、R6等)が式中に複数回出現する場合、各出現時のそのような可変基又は置換基の定義は、特に示さない限り、他のあらゆる出現時のその定義と無関係である。本発明の化合物の成分中に存在する置換基及び/又は可変基の組合せは、そのような組合せが安定な化合物をもたらす場合にのみ許容できる。
【0041】
[0044]R1(例えば、(H,OR6))のような置換基の定義に用いる括弧に入った名称は、関連原子の両原子価に置換基を反映させることを意図する。本発明は特定の異性体に限定されず、括弧内の部分の順序は特定の配置を示唆するものでない。
【0042】
[0045]「アシロキシ」は、−OC(O)−(アルキル)及び−OC(O)−(アリール)を意味する。
【0043】
[0046]「アルケニル」(単独又は組合せ)は、1つ又は複数の二重結合を有するアルキルラジカルを意味する。そのようなアルケニルラジカルの例は、エテニル、プロペニル、1−ブテニル、シス−2−ブテニル、トランス−2−ブテニル、イソ−ブチレニル、シス−2−ペンテニル、トランス−2−ペンテニル、3−メチル−1−ブテニル、2,3−ジメチル−2−ブテニル、1−ペンテニル、1−ヘキセニル、1−オクテニル、デセニル、ドデセニル、テトラデセニル、ヘキサデセニル、シス及びトランス−9−オクタデセニル、1,3−ペンタジエニル、2,4−ペンタジエニル、2,3−ペンタジエニル、1,3−ヘキサジエニル、2,4−ヘキサジエニル、5,8,11,14−エイコサテトラエニル及び9,12,15−オクタデカトリエニルを含むが、これらに限定されない。
【0044】
[0047]「アルコキシル」は、酸素に結合したアルキル基を意味する。
【0045】
[0048]「アルキル」(単独又は組合せ)は、1〜約22個の炭素原子、約1〜約18個の炭素原子又は約1〜約12個の炭素原子を含む直鎖又は枝分れ鎖アルキルラジカルを意味する。そのようなラジカルの例は、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソ−アミル、ヘキシル、オクチル、ノニル、デシル、ドデシル、テトラデシル、ヘキサデシル、オクタデシル及びエイコシルを含むが、これらに限定されない。
【0046】
[0049]「アルキルシクロアルケニル」及び「アルケニルシクロアルケニル」は、上で定義したようなアルキル又はアルケニルラジカルで置換されている、上で定義したようなシクロアルケニルラジカルを意味する。アルキルシクロアルケニル及びアルケニルシクロアルケニルラジカルの例は、1−メチル−2−シクロペンチル、1−ヘキシル−2−シクロペンテニル、1−エチル−2−シクロヘキセニル、1−ブチル−2−シクロヘキセニル、1−(9−オクタデセニル)−2−シクロヘキセニル及び1−(2−ペンテニル)−2−シクロヘキセニルを含むが、これらに限定されない。
【0047】
[0050]「アルキルシクロアルキル」及び「アルケニルシクロアルキル」は、上で定義したようなアルキル又はアルケニルラジカルで置換されている、上で定義したようなシクロアルキルラジカルを意味する。アルキルシクロアルキル及びアルケニルシクロアルキルラジカルの例は、2−エチルシクロブチル、1−メチルシクロペンチル、1−ヘキシルシクロペンチル、1−メチルシクロヘキシル、1−(9−オクタデセニル)シクロペンチル及び1−(9−オクタデセニル)シクロヘキシルを含むが、これらに限定されない。
【0048】
[0051]「アルキニル」(単独又は組合せ)は、1つ又は複数の三重結合を有するアルキルラジカルを意味する。そのようなアルキニル基の例は、エチニル、プロピニル(プロパルギル)、1−ブチニル、1−オクチニル、9−オクタデシニル、1,3−ペンタジイニル、2,4−ペンタジイニル、1,3−ヘキサジイニル及び2,4−ヘキサジイニルを含むが、これらに限定されない。
【0049】
[0052]「アミノ」は、−NH2、−N(アルキル)2、−NH(アルキル)、−N(アリール)2及び−NH(アリール)を意味する。
【0050】
[0053]「アラルキル」(単独又は組合せ)は、ベンジル、2−フェニルエチルなどの、1個の水素原子が上で定義したようなアリールラジカルで置換されている上で定義したようなアルキル又はシクロアルキルラジカルを意味する。
【0051】
[0054]「アリール」(単独又は組合せ)は、フェニル、p−トリル、4−メトキシフェニル、4−(tert−ブトキシ)フェニル、4−フルオロフェニル、4−クロロフェニル、4−ヒドロキシフェニル、1−ナフチル、2−ナフチル等のような、アルキル、シクロアルキル、シクロアルケニル、アリール、複素環、アルコキシアリール、アルカリル、アルコキシ、ハロゲン、ヒドロキシ、アミン、シアノ、ニトロ、アルキルチオ、フェノキシ、エーテル、トリフルオロメチル等から選択される1つ又は複数の置換基を場合によって有するフェニル又はナフチルラジカルを意味する。
【0052】
[0055]「シクロアルケニル」(単独又は組合せ)は、1つ又は複数の二重結合を有するシクロアルキルラジカルを意味する。シクロアルケニルラジカルの例は、シクロペンテニル、シクロヘキセニル、シクロオクテニル、シクロペンタジエニル、シクロヘキサジエニル及びシクロオクタジエニルを含むが、これらに限定されない。
【0053】
[0056]「シクロアルケニルアルキル」は、上で定義したようなシクロアルケニルラジカルで置換されている、上で定義したようなアルキルラジカルを意味する。シクロアルケニルアルキルラジカルの例は、2−シクロヘキセン−1−イルメチル、1−シクロペンテン−1−イルメチル、2−(1−シクロヘキセン−1−イル)エチル、3−(1−シクロペンテン−1−イル)プロピル、1−(1−シクロヘキセン−1−イルメチル)ペンチル、1−(1−シクロペンテン−1−イル)ヘキシル、6−(1−シクロヘキセン−1−1−イル)ヘキシル、1−(1−シクロペンテン−1−イル)ノニル及び1−(1−シクロヘキセン−1−イル)ノニルを含むが、これらに限定されない。
【0054】
[0057]「シクロアルキル」(単独又は組合せ)は、3〜約10個、好ましくは3〜約8個、最も好ましくは3〜約6個の炭素原子を含むシクロアルキルラジカルを意味する。そのようなシクロアルキルラジカルの例は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル及びペルヒドロナフチルを含むが、これらに限定されない。
【0055】
[0058]「シクロアルキルアルキル」は、上で定義したようなシクロアルキルで置換されている、上で定義したようなアルキルラジカルを意味する。シクロアルキルアルキルラジカルの例は、シクロヘキシルメチル、シクロペンチルメチル、(4−イソプロピルシクロヘキシル)メチル、(4−t−ブチル−シクロヘキシル)メチル、3−シクロヘキシルプロピル、2−シクロヘキシルメチルペンチル、3−シクロペンチルメチルヘキシル、1−(4−ネオペンチルシクロヘキシル)メチルヘキシル及び1−(4−イソプロピルシクロヘキシル)メチルヘプチルを含むが、これらに限定されない。
【0056】
[0059]「シクロアルキルシクロアルキル」は、上で定義したような他のシクロアルキルラジカルで置換されている、上で定義したような他のシクロアルキルラジカルを意味する。シクロアルキルシクロアルキルラジカルの例は、シクロヘキシルシクロペンチル及びシクロヘキシルシクロヘキシルを含むが、これらに限定されない。
【0057】
[0060]「ハロゲン」は、フルオロ、クロロ、ブロモ及びヨードを含む。
【0058】
[0061]「複素環」は、飽和又は不飽和であり、炭素原子並びにN、O及びSからなる群から独立に選択される1〜3個のヘテロ原子からなり、窒素及び硫黄ヘテロ原子が酸化されていてよく、窒素へテロ原子が第四級化されていてよく、複素環がベンゼン環に縮合した二環の群を含む、安定な5〜7員単環若しくは二環式又は安定な7〜10員二環式複素環を含む。複素環は、安定な構造をもたらすヘテロ原子又は炭素原子に結合していてよい。複素環の要素の例は、ピペリジル、ピペリジニル、ピペラジニル、ピリミジニル、ピリダジニル、オキサゾリル、フリル及びチエニルを含む。複素環は、ヘテロ原子に結合している炭素原子が、へテロ原子により、C1〜C6アルキル、アリール、ヒドロキシル、C1〜C6アルコキシ、アシルオキシ、アミノ、N−アシルアミノ、ニトロ及びハロゲンであってよい1〜4つの構成員により直接置換されないような仕方で置換されていてよい。
【0059】
[0062]「複素環」は、環内に、炭素に加えて、少なくとも1つの他の種類の原子を含む環構造を意味する。最も一般的な他の種類の原子は、窒素、酸素及び硫黄を含む。複素環の例は、ピロリジニル、ピペリジル、イミダゾリジニル、テトラヒドロフリル、テトラヒドロチエニル、フリル、チエニル、ピリジル、キノリル、イソキノリル、ピリダジニル、ピラジニル、インドリル、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル、ピリジニル、ベンズオキサジアゾリル、ベンゾチアジアゾリル、トリアゾリル及びテトラゾリル基を含むが、これらに限定されない。
【0060】
[0063]「ケトン」は、−C(O)−を意味する。
【0061】
[0064]「N−アシルアミノ」は、−NHC(O)−(アルキル)及び−NHC(O)−(アリール)を意味する。
【0062】
[0065]「窒素含有複素環」は、環の2つの炭素及び1つの窒素が15員大環状リガンドの一部でもある環構造を意味する。環10構造は、約2〜約20又は約4〜約10個の炭素原子を含むことができ、置換又は非置換、部分的又は完全に不飽和又は飽和であってよく、15員大環状リガンドの一部でもない環の一部に窒素、酸素及び/又は硫黄原子を含むこともできる。
【0063】
[0066]「飽和、部分的に飽和又は不飽和環」は、環の2個の原子が15員大環状リガンドの一部でもある縮合環構造を意味する。環構造は、約3〜約20個の炭素原子又は約5〜約10個の炭素原子を含むことができ、炭素に加えて1又は複数個の他の種類の原子も含むことができる。最も一般的な他の種類の原子は、窒素、酸素及び硫黄である。環構造は、1つよりも多い環を含むこともできる。
【0064】
[0067]「飽和、部分的に飽和又は不飽和環構造」は、環の1つの炭素が15員大環状リガンドの一部でもある環構造を意味する。環構造は、約3〜約20個又は約5〜約10個の炭素原子を含むことができ、窒素、酸素及び/又は硫黄原子を含むこともできる。
【0065】
[0068]発明の実施
【0066】
[0069]式I
【化7】
の分子を調製するために、
式II
【化8】
[式中、
R1は、=O(H,H)又は(H,OH)であり、
R2及びR5は、独立にH、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6又は−C(=S)OR6であり、
R3は、=O若しくはOR5であるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H又はC1〜C4アルキルであり、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基又は複素環基であり、
R7及びR8は、独立にH、C1〜C6アルキルであるか、又はR7及びR8は、統合すると=Oである]をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化9】
の分子を生成させて、ステップ(a)を完結させ、
次いで、それを式IV
【化10】
[式中、
R9及びR10は、独立にH、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ又はその組合せである]
の分子と反応させて、ステップ(b)を完結させる。
【0067】
[0070]ステップ(a)
【0068】
[0071]式IIの分子において、R1は好ましくはOであり、R2は好ましくはHであり、R3は好ましくはOであり、R4は好ましくはHである。
【0069】
[0072]塩基 ステップ(a)は、非求核塩基、好ましくは2,6−ジメチルピリジン又はジイソプロピルエチルアミンの存在下で実施する。
【0070】
[0073]溶媒 このステップはまた、酢酸イソプロピル又はジクロロメタンなどの溶媒の存在下で実施する。溶媒がジクロロメタンである場合、それは、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換することができる。
【0071】
[0074]ステップ(b)
【0072】
[0075]式IVの分子において、R10は好ましくはHであり、R9はH、メチル及びフェニルからなる群から選択されるものであり、より好ましくは、R9及びR10はHである。
【0073】
[0076]溶媒 ステップ(b)も溶媒、好ましくは非プロトン性溶媒の存在下で実施する。非プロトン性溶媒の例としては、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、ジメチルアミン、N,N−ジメチルホルムアミド及び炭酸プロピレンなどがある。他の非プロトン性溶媒を用いることができる。好ましい非プロトン性溶媒は、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン、アセトニトリル、ジメチルアミン及びN,N−ジメチルホルムアミドなどであり、最も好ましいものは、ジイソプロピルエチルアミンと酢酸イソプロピル又はジクロロメタンのいずれかである。
【0074】
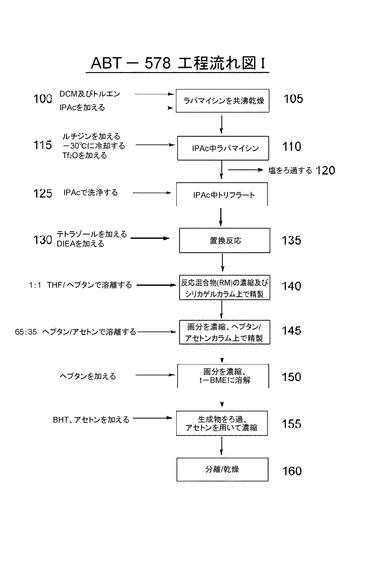
[0077]スキーム2に本発明の好ましい実施形態の概要を示す。図1にゾタロリムスを調製するためのワンポット法におけるステップを概説する流れ図を示す。第1の実施形態において、シロリムス(市販又は記載されたように生成させた(Paivaら、1991;Sehgalら、1975;Vezinaら、1975))をDCM:トルエン(1:2など)100に溶解する。反応混合物を濃縮して乾燥させ105、共沸乾燥工程105を好ましくはDCM:トルエンを用いてさらに1〜5回、より好ましくは2〜4回、最も好ましくは2回繰り返す。得られる泡様固体をIPAcに溶解し110、次いで、2,6−ルチジンを加える115。溶液を−30℃に冷却する115。次いで、トリフルオロメタンスルホン酸無水物を徐々に溶液に加える115。反応混合物を撹拌した後、溶液を窒素中でろ過する。回収された塩120をIPAcで洗浄する125。塩に1−H−テトラゾール及びDIEAを加える130。反応混合物を室温(例えば、22〜25℃)で撹拌し135、次いで、濃縮する。粗反応混合物を、例えばシリカゲルカラムを用いて精製し、例えば1:1 THF:ヘプタンを用いて溶離させる140。画分をN−1異性体(N−2異性体より遅く溶離する)についてモニターし、プールし、濃縮して、油を生成させる。油を最小限のDCMに溶解し、溶液を、例えば、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加える145。カラムを例えば、65:35 ヘプタン:アセトンで溶離させ、画分を純生成物についてモニターし、プールし、濃縮する150。
【化11】
【0075】
[0078]次いで、精製生成物をt−BMEに溶解し、次いで、溶液を激しく撹拌しながら、n−ヘプタンを徐々に加えて、沈殿を生成させる150。沈殿固体を5〜10℃で撹拌し、ろ過し、ヘプタンで再び洗浄し、窒素を用いて漏斗上で乾燥する。生成物をアセトンに溶解し、BHTで処理する155。溶液を濃縮し、アセトンに溶解し、次いで濃縮し、乾燥した。次いで、生成物を真空中47℃で乾燥する160。
【0076】
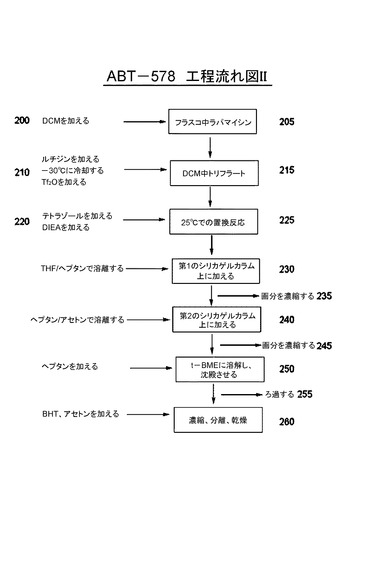
[0079]第2の好ましい実施形態において、図2に示す流れ図で、シロリムスをDCMに溶解する200、205。次いで、2,6−ルチジンを加え、溶液を−30℃に冷却し、トリフルオロメタンスルホン酸無水物を徐々に加える210。反応混合物215を混合し、テトラゾールを加え、次いで、DIEAを加える220。反応混合物を約25℃でインキュベートし225、例えば、1:1 THF:n−ヘプタン(体積/体積)で調製したシリカゲルカラム上に加える230。粗反応混合物を1:1 THF:n−ヘプタンを用いて精製する。生成物を含む画分を収集し、濃縮する235。濃縮固体を最小限のDCMに溶解し、例えば、70:30 n−ヘプタン:アセトン中に充填したシリカゲルカラム240上に加える。カラムを溶離させ、純生成物を含む画分を濃縮する245。精製生成物をt−BMEに溶解し、n−ヘプタンに徐々に加える250。沈殿固体をろ過し、n−ヘプタンで洗浄し、乾燥する255。BHTを固体に加え、固体をアセトンに溶解し、ろ過し、濃縮する260。残留物をアセトンで2回処理し260、毎回濃縮して乾燥する。次いで、生成物を真空中で乾燥する260。
【0077】
[0080]第3の実施形態において、シロリムス(ラパマイシン)をジクロロメタンに溶解する。2,6−ルチジンを加え、溶液を−30℃に冷却する。トリフルオロメタンスルホン酸無水物を徐々に加える。反応物を撹拌した後、溶液を10℃に加温する。反応溶液を濃縮し、残留物をIPACに溶解する。1−H−テトラゾールを、続いて、DIEAを加え、反応混合物を22〜25℃で撹拌する。次いで、溶液を濃縮し、例えば、1:1 THF:ヘプタンで溶離するシリカゲルカラム上で精製した。N−1異性体を含む画分を収集し、プールし、濃縮する。得られた油を最小限のDCMに溶解し、例えば、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加える。カラムをヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮する。濃縮物をt−BMEに溶解し、激しく撹拌しながらn−ヘプタンに徐々に加える。次いで、沈殿物を5〜10℃で1時間以下撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥する。BHTを固体に加え、混合物をアセトンに溶解する。次いで、溶液をフィルターに通し、濃縮する。残留物をさらに2回アセトンで処理し、毎回濃縮して乾燥する。最終生成物を真空中50℃で乾燥する。
【0078】
[0081]本発明を遂行するために、本発明の方法において種々の試薬に代えることができる。例えば、トリフラートを調製するために、2,6−ジ−t−ブチルピリジン及びDIEAを2,6−ルチジンの代わりに用いることができる。ピリジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジン及び4−ジメチルアミノピリジン(DMAP)などの他の置換ピリジン、N−メチルモルホリン並びに当業者に明らかである他のものを含む他の塩基をこのステップで用いることができる。様々な溶媒、塩基(DIEAの代わり)及び求核剤(テトラゾールの代わり)も本発明の方法に用いることができる。例を下の表1に示す。
【表1】
【0079】
[0082]塩基及び溶媒 1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン(DBU)、炭酸カリウム(K2CO3)、4−ジメチルアミノピリジン(DMAP)及びカリウムtert−ブトキシド(KOtBu)などの強塩基は、広範な分解をもたらし、一般的にN−2異性体の生成に有利であり、したがって、好ましくない。ルチジン、TEA及びNMMなどのより弱い塩基は、SN2反応を遅くし、約1:1の比でN−1及びN−2異性体の両方が生成する。IPAc、DME、ジオキサン及びTHFなどの非プロトン性溶媒は、十分に機能し、N−1異性体に有利であり、好ましい。DCMを用いることにより、約1:1の比の異性体が得られる。DMA及びDMFのような非プロトン性極性溶媒は、反応生成物の分解をもたらす。
【0080】
[0083]温度 分解が通常認められるが、加熱することにより反応を加速することができる。しかし、反応は通常4時間までに又はそれより早く完結する。したがって、反応混合物をより早期に処理して、分解を最小限にすることができる。SN2置換反応を加速するのに好ましい温度は、20〜35℃、好ましくは22〜33℃、より好ましくは25〜33℃、最も好ましくは28〜30℃を含む。
【0081】
[0084]置換求核剤 一般的に、5−置換テトラゾールをテトラゾールの代わりに用いることができる。例えば、テトラゾールの代わりに用いるとき、5−メチルテトラゾール及び5−フェニルテトラゾールが、好ましい。5−置換テトラゾールは、N−2異性体の生成に有利であるため、好ましい。
【0082】
[0085]抗酸化剤 ワンポット法により調製したゾタロリムスを安定化するために、抗酸化剤を用いることができる。それらは、組成物中に約1重量%まで、より好ましくは0.05%〜0.75%、3,5−ジ−tert−4−ブチルヒドロキシトルエン(BHT)の場合には0.5%存在していてよい。抗酸化剤の例としては、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などがある。好ましくは、抗酸化剤はBHTである。
【0083】
[0086] [実施例]
【0084】
[0087]実施例1 ろ過を伴うジクロロメタン−トルエン 酢酸イソプロピルワンポット法(1)
【0085】
[0088]この実施例では、ゾタロリムスは、ジクロロメタン、トルエン及び酢酸イソプロピルを用いてワンポット法でラパマイシンから調製した。次いで、調製物を精製し、濃縮し、乾燥した。次いで、精製生成物をその1H、13CNMR共鳴により、COSY、ROESY、TOCSY、HSQC及びHMBCスペクトルから確認した。
【0086】
[0089]ラパマイシン(10g)をジクロロメタン(DCM、25ml)及びトルエン(50ml)に溶解した。反応混合物を濃縮して乾燥した。この共沸乾燥工程は、DCM/トルエンを用いて2回繰り返した。泡様固体を酢酸イソプロピル(IPAc、65ml)に溶解し、2,6−ルチジン(3.2ml)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(2.8ml)を10分間に徐々に加えた。反応混合物を30分間撹拌し、次いで、窒素雰囲気中でろ過した。塩をIPAc(10ml)で洗浄した。1−H−テトラゾール(2.3g)を、続いて、ジイソプロピルエチルアミン(DIEA、7.4ml)を加えた。反応混合物を室温で6時間撹拌し、次いで、濃縮した。粗反応混合物を1:1 THF/ヘプタンで溶離するシリカゲルカラム(350g)上で精製した。後に溶離させた生成物を含む画分(主としてN−1異性体)を収集し、濃縮した。濃縮油を最小限のDCMに溶解し、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加えた。カラムを65:35 ヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮した。
【0087】
[0090]次いで、精製生成物をt−ブチルメチルエーテル(t−BME、13.5g)に溶解し、激しく撹拌しながらn−ヘプタン(53g)を徐々に加えた。沈殿固体を5〜10℃で2時間撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥して、3.2gの湿った生成物を得た。固体(1.0g)をアセトン(10ml)に溶解し、2,6−ジ−tert−ブチル−4−エチルフェノール(DEP、0.2%)で処理した。溶液を濃縮し、アセトン(10ml)に溶解し、濃縮して乾燥した。生成物を真空中47℃で18時間乾燥して、0.83gのゾタロリムスを得た。生成物をその1H、13CNMR共鳴により、そのCOSY、ROESY、TOCSY、HSQC及びHMBCスペクトルから特性決定した。
【0088】
[0091]1H−NMR(DMSO−d6,括弧の位置):ppm 0.73(Me,43);0.81(Me,49);0.84(Me,46);0.89(Me,48);0.98(Me,45);1.41、1.05(CH2,24);1.18、1.10(CH2,36);1.52(CH,37);1.53(CH2,12 & 42);1.59、1.30(CH2,5);1.41、1.67(CH2,4);1.11、1.73(CH2,38);1.21、1.83(CH2,15);1.21、1.83(CH2,13);1.62(Me,44);1.73(Me,47);1.76(CH,35);1.60、2.09(CH2,3);1.93、2.21(CH2,41);2.05(CH,11);2.22(CH,23);2.47(CH,25);2.40、2.77(CH2,33);3.06(OCH3,50);3.16(OCH3,51);3.22、3.44(CH2,6);3.29(OCH3,52);3.29(CH,31);3.60(CH,39)、3.62(CH,16);3.89(CH,27);4.01(CH,14);4.02(CH,28);4.95(CH,2);5.02(CH,34);5.10(=CH,30);5.17(CH,40);5.24(OH,28);5.46(=CH,22);6.09(=CH,18);6.15(=CN,21);6.21(=CH,20);6.42(=CH,19);6.42(OH,10)、9.30(CH,53)。
【0089】
[0092]13C NMR(DMSO−d6,括弧の位置):ppm 10.4(Me,44);13.1(Me,47);13.6(Me,46);14.5(Me,49);15.5(Me,43 & 48);20.3(CH2,4);21.6(Me,45);24.4(CH2,4);26.2(CH2,12);26.4(CH2,3);26.8(CH2,41);27.2(CH2,42);29.6(CH2,13);31.6(CH2,38)、31.7(CH,37);32.9(CH,35);34.8(CH,11);35.2(CH,23);38.2(CH2,36);39.1(CH,25);39.4(CH2,33);39.6(CH2,24)、40.0(CH2,15);43.4(CH2,6);45.2(CH,31);50.6(CH,2);55.4(OCH3,50);55.8(OCH3,52);57.0(OCH3,52);55.9(CH,40);66.2(CH,14);73.4(CH,34);75.6(CH,28);77.4(CH,39);82.3(CH,16);85.7(CH,27);99.0(CH,10);125.3(=CH,30);127.0(=CH,18 & 19);130.4(=CH,21);132.2(=CH,20);137.2(=CMe,29);137.7(=CMe,17);139.2(=CH,22);144.6(CH,53);167.0(C=O,8);169.1(C=O,1);199.0(C=O,9);207.5(C=O,32);210.7(C=O,26)。
【0090】
[0093]実施例2 ジクロロメタン−酢酸イソプロピルワンポット法(2)
【0091】
[0094]この実施例では、ゾタロリムスは、ジクロロメタン及び酢酸イソプロピルを用いてワンポット法でラパマイシンから調製した。次いで、化合物を精製し、濃縮し、乾燥した。
【0092】
[0095]ラパマイシン(10g)をジクロロメタン(DCM、100g)に溶解した。2,6−ルチジン(2.92g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(4.62g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、15分以内に10℃に加温した。反応溶液を濃縮した。残留物をIPAc(55g)に溶解した。1−H−テトラゾール(2.68g)を、続いて、ジイソプロピルエチルアミン(DIEA、7.08g)を加えた。反応混合物を室温で6時間撹拌し、次いで、濃縮した。粗反応混合物をシリカゲルカラム(360g)上で精製し、1:1 THF:ヘプタンで溶離させた。後に溶離させた生成物を含む画分(主としてN−1)を収集し、濃縮した。濃縮油を最小限のDCMに溶解し、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム(180g)上に加えた。カラムを65:35 ヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮した。
【0093】
[0096]精製生成物をt−ブチルメチルエーテル(t−BME、23g)に溶解し、激しく撹拌しながらn−ヘプタン(80g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.015g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回処理し(20g)、毎回濃縮して乾燥した。次いで、生成物を真空中50℃以下で18時間乾燥して、2.9gのゾタロリムスを得た。
【0094】
[0097]実施例3 ジクロロメタンワンポット法(3)
【0095】
[0098]この例では、ゾタロリムスは、ジクロロメタンを用いてワンポット法でラパマイシンから調製した。次いで、実施例2で述べたように、化合物を精製し、濃縮し、乾燥した。
【0096】
[0099]ラパマイシン(7.5g)をDCM(30g)に溶解した。2,6−ルチジン(1.76g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(2.89g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、次いで、ラパマイシンの存在を分析して、反応における消費を測定した。1−H−テトラゾール(1.44g)を、続いて、DIEA(5.29g)を加えた。反応混合物を室温で6時間撹拌し、次いで、1:1 THF:n−ヘプタン(体積/体積)中に調製したシリカゲル(270g)カラム上に直接加えた。粗反応混合物を1:1 THF:n−ヘプタンで精製した。後に溶離させた生成物を含む画分を収集し、濃縮した。濃縮固体を最小限のDCMに溶解し、70:30 n−ヘプタン:アセトン中で充填したシリカゲルカラム(135g)上に加えた。カラムを70:30 n−ヘプタン:アセトンで溶離させ、薄層クロマトグラフィー(TLC)により同定された、純生成物を含む画分を濃縮した。
【0097】
[0100]精製生成物をt−BME(9g)に溶解し、10±10℃で激しく撹拌しながらn−ヘプタン(36g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、n−ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.006g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回(各20g)処理し、毎回濃縮して乾燥した。生成物を真空中50℃以下で18時間以下乾燥して、2.5gのゾタロリムスを得た。
【0098】
[0101]上の方法は、ステップ1aにおいて非求核剤としての2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジン(2,3,5−トリメチルピリジン)の存在下でラパマイシンを用いて実施した場合、許容できる純度であったが、より低い収率のゾタロリムスを生成させた。
【0099】
[0102]実施例4 ワンポット合成法により調製したゾタロリムスの高圧液体クロマトグラフィー(HPLC)精製
【0100】
[0103]この実施例では、ゾタロリムスを本発明のワンポット合成法を用いてラパマイシンから調製し(DCMを用いて)、次いで、HPLCを用いた精製のさらなるラウンドに供した。
【0101】
[0104]ラパマイシン(3.75g)をジクロロメタン(DCM、15g)に溶解し、次いで、2,6−ルチジン(0.88g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(1.45g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、次いで、1−H−テトラゾール(0.72g)を、続いて、DIEA(2.65g)を加えた。反応混合物を25℃で6時間撹拌し、次いで、ヘプタン70:30 n−ヘプタン:アセトン中に調製したシリカゲル(115g)カラム上に直接加えた。粗反応混合物を70:30 n−ヘプタン:アセトンで精製した。生成物を含む画分を収集し、濃縮した。
【0102】
[0105]濃縮固体をアセトニトリル−水に溶解した。C−18TechniKromカラム(5cm×25cm)上に加え、0.1%BHTを含む64:36 アセトニトリル−水で溶離させた。画分を逆相(RP)−HPLCにより分析し、生成物画分をプールし、濃縮して、アセトニトリルを除去した。生成物を酢酸エチル又は酢酸イソプロピルで抽出し、乾燥し(硫酸ナトリウム)、濃縮した。
【0103】
[0106]精製生成物をt−BME(4.5g)に溶解し、約10℃で激しく撹拌しながらn−ヘプタン(18g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、n−ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.005g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回処理し(20g)、毎回濃縮して乾燥した。生成物を真空中50℃以下で18時間以下乾燥して、1.2gの高品質のゾタロリムスを得た。
【0104】
[0107]実施例5 ワンポット合成法により調製したゾタロリムスの安定性分析
【0105】
[0108]この実施例では本発明の方法により調製されたゾタロリムスを抗酸化剤を用いて安定化できることを示す。
【0106】
[0109]ワンポット法により調製したゾタロリムスのいくつかのロットは、時間の経過とともに力価を著しく失った。力価損失は温度上昇でより高かったが、不純物プロファイルの明らかな変化はなかった。表2に種々の時間間隔及び温度条件でのゾタロリムスのロットの力価を示す。例えば、室温(25℃)の密閉容器中でさえも、力価は3カ月間にわたり95.1%から69.8%に低下した。この損失は、40℃で保管した場合、密閉容器中で悪化し、ほんの2カ月間で96.0%から37.4%になった。
【0107】
[0110]その後の調査により、この力価の損失は複数の分解生成物をもたらす分子の酸化的分解に起因していたことが明らかになった。この酸化を防止するためにフェノール系抗酸化剤を用いて安定性試験を実施したところ、BHTが適切な化合物と特定された。表3及び4に種々の濃度(重量/重量で表した%)及び温度でBHTを用いた安定性データを示す。例えば、40℃では、0.5%BHTは約3カ月間にわたり力価を初期の96.5%から95.9%の最終力価まで維持したが、BHTの非存在下での同様の条件下では、力価は96%からほんの2カ月後に37.4%に激減した。
【表2】
【表3】
【表4】
【0108】
[0111]これらの安定性試験により、ゾタロリムスの純度、力価及び安定性を維持するためには、BHTのような抗酸化剤の添加が非常に重要であることが確認される。
【0109】
[0112]実施例6 ゾタロリムス平衡異性体の分離及び特性解析
【0110】
[0113]C−18又はフェニルカラム上のゾタロリムスの逆相分析により、7員環オキセパン(2)異性体と比較して早く溶離した主要な異性体は、6員環ピラン形であり、副次的成分であるオキセパン異性体は3〜4分後に溶離したことが示された。順相HPLC(シリカゲル−YMC Co.Ltd;京都、日本)では、2つの形態はベースライン分離を示さなかった。しかし、オキセパン形はピラン形の直前に溶離した。
【0111】
[0114]この平衡を実証するために、pH4の逆相フェニルカラム上へのゾタロリムスの複数回のHPLC注入により、各形態を分離した。次いで、分離された各形態を再注入して、種々の間隔でそれらの平衡を試験した。試験により、ピラン形は3〜4日で平衡状態に到達したが、オキセパン形(副次的成分)はほぼ6日後でさえ完全には平衡に達しなかったことが示された。試験中、開環酸(open ring acid)のある程度の生成があった。表5(緩衝液を含む)及び表6(緩衝液を含まない)に示すこの試験の結果は、2つの形態は平衡状態にあり、ピラン形が熱力学的により安定であることを明確に示している。
【0112】
[0115]アセトニトリル/水の溶媒混合物中で非緩衝条件下でも試験を実施した。非緩衝媒体中水中66%アセトニトリルを用いてC−18 Altimaカラム(Alltech Associates,Inc.;Deerfield、IL)上へのゾタロリムスの複数回の注入を行った。ピラン及びオキセパン形を収集した。これらの形態をC−18カラム上に再注入して、種々の間隔で平衡比を試験した。表4及び5に示すこれらのデータから、2異性体間の平衡化が速やかであり、約7〜8時間以内に完結することが示唆された。これらの所見は、ゾタロリムスは約10:1のピラン(1)対オキセパン(2)形の平衡混合物で存在することを確認するものであった。
【表5】
【表6】
【0113】
[0116]前記の詳細な説明及び付随する実施例は、単に説明のためのものであって、添付の特許請求の範囲及びそれらの同等物によってのみ規定される発明の範囲に対する制限と解釈すべきでないと理解される。開示された実施形態の様々な変更及び修正は、当業者には明らかであろう。化学構造、置換基、誘導体、中間体、合成、調合及び/又は本発明の使用の方法に関するものを制限なしに含む、そのような変更及び修正は、その精神及び範囲から逸脱することなく行うことができる。
【0114】
[0117]特に示さない限り、すべての参考文献は、参照によりそれらの全部が本明細書に組み込まれる。
【0115】
[0118]参考文献
Baker, H., A. Sidorowicz, S.N. Sehgal, and C. Vezina. 1978. Rapamycin (AY−22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J Antibiot (Tokyo). 31:539−45.
Caufield. US Patent No. 5,023,262. 1991. Hydrogenated Rapamycin Derivatives.
Caufield. WO 92/05179. 1992. Carboxylic Acid Esters of Rapamycin.
Eng. US Patent No. 4,401,653.1983. Combination of Rapamycin and Picibanil for the Treatment of Tumors.
Failli. EPO 467606. 1992a. Rapamycin Derivatives.
Failli. US Patent No. 5,120,842. 1992b. Silyl Ethers of Rapamycin.
Failli. US Patent No. 5,177,203. 1993. Rapamycin 42−Sulfonatcs and 42−(N−Carboalkoxy) Sulfamatcs Useful as Imunosuppressie Agents.
Higuchi, T., and V. Stella. 1987. Pro−drugs as Novel Delivery systems.
Hughes, P., J. Musser, M. Conklin, and R. Russo. 1992. The isolation, synthesis and characterization of an isomeric form of rapamycin. Tetrahedron Lttrs. 33:4739−4742.
Kao. US Patent No. 5,120,725. 1992a. Bicyclic Rapamycins.
Kao. US Patent No. 5,120,727. 1992b. Rapamycin Dimers.
Martel, R.R., J. Klicius, and S. Galet. 1977. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 55:48−51.
Mollison, K. US Patent No. 6,015,815. 2000. Tetrazole−containing rapamycin analogs with shortened half−lives.
Mollison, K., A. LeCaptain, S. Burke, K. Cromack, P. Tarcha, Y.−C.J. Chen, and J. Toner. US Patent Application Publication 20030129215. 2003. Medical devices containing rapamycin analogs
Paiva, N.L., A.L. Demain, and M.F. Roberts. 1991. Incorporation of acetate, propionate, and methionine into rapamycin by Streptomyces hygroscopicus. J Nat Prod. 54:167−77.
Roche, E. 1987. Bioreversible Carriers in Drug Design. American Pharmaceutical Association and Pergamon Press.
Sehgal, S.N. US Patent No. 3,929,992. 1975. Rapamycin and Process of Preparation.
Sehgal, S.N. US Patent No. 3,993,749. 1976. Rapamycin and Process of Preparation.
Sehgal, S.N., H. Baker, and C. Vezina. 1975. Rapamycin (AY−22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 28:727−32.
Surendra. US Patent No. 4,885,171. 1989. Use of Rapamycin in Treatment of Certain Tumors.
Vezina, C., A. Kudelski, and S.N. Sehgal. 1975. Rapamycin (AY−22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 28:721−6.
【図面の簡単な説明】
【0116】
【図1】本発明によるゾタロリムスを調製するワンポット法の実施形態の流れ図を示す。
【図2】本発明によるゾタロリムスを調製するワンポット法の実施形態の流れ図を示す。
【発明の詳細な説明】
【0001】
[0001][関連出願に対する相互参照]
【0002】
[0002]適用せず。
【0003】
[0003][連邦政府により委託された研究又は開発に関する陳述]
【0004】
[0004]適用せず。
【0005】
[0005][コンパクトディスクで提出した資料の参照による組み込み]
【0006】
[0006]適用せず。
【0007】
[0007][技術分野]
【0008】
[0008]本発明は、ラパマイシンの類似体を合成する新規な方法に関する。該類似体は、抗増殖及び免疫調節への適用に有用である。
【0009】
[0009][発明の背景]
【0010】
[0010]緒言
【0011】
[0011]シロリムス
【0012】
[0012]モアイが見下ろしているとき、カナダ探検隊は1964年に土壌を掘って、強力な免疫抑制、抗真菌及び抗細胞増殖分子を産生する真菌を発見した。真菌は、イースター島からカナダの研究室にSuren Sehgalの手により届けられた。彼は、1972年に真菌ストレプトミセスハイグロスコピカス(Streptomyces hygroscopicus)の精製化合物の特性を解明したが、この知見は捨てられ、企業の優先事項の犠牲となった。Sehgalは、1987年に研究を復活させ、化合物を免疫抑制剤として開発した。今日、ラパマイシン(イースター島原住民がその祖国を思い出す名称であるラパヌイ(Rapa Nui)にちなんで命名された)は、臓器移植のリスク及びステントの副作用を低減するために用いられており、抗腫瘍医薬品として研究されている。
【0013】
[0013]ラパマイシンは、シロリムスとしても知られており、in vitro及びin vivoで、特にカンジダアルビカンス(Candida albicans)に対して、真菌の増殖を抑制する大環状トリエン抗生物質である(Bakerら、1978、Sehgal、1975、Sehgal、1976、Sehgalら、1975、Vezinaら、1975)。シロリムス単独(Surendra、1989)又はピシバニルとの併用(Eng、1983)は、抗腫瘍活性を有することが示された。1977年に、シロリムスがアレルギー性脳脊髄炎(多発性硬化症のモデル)、アジュバント関節炎及びリウマチ関節炎の実験的モデルにおいて免疫抑制剤として有効であることが示された(Martelら、1977)。シロリムスはまた、IgE様抗体の形成を効果的に抑制する(Martelら、1977)。その構造を下(VI)に示す。
【0014】
[0014]ABT−578[40−エピ−(1−テトラゾリル)−ラパマイシン]は、今日ではゾタロリムスとしてよりよく知られており、シロリムスから誘導された半合成マクロライドトリエン抗生物質である。ゾタロリムスは、その前駆体ゾタロリムスと同様に、T細胞リンパ球の増殖の強力な抑制剤である。ゾタロリムスは、再狭窄を最小限にするために心血管ステント、特に薬剤溶出ステント(DES)を被覆することに例外的適用を見い出された(Mollisonら、2003)。ゾタロリムスは、両方がN−1異性体である、主要なピラン(10位における6員環の異性体;1)及び副次的なオキセパン異性体(9位における7員環の異性体;2)の2つの異性体として存在する(Mollison、2000)。
【0015】
[0015]ラパマイシンの他の化学修飾が試みられた。これらは、ラパマイシンのモノ及びジエステル誘導体(Caufield、1992)、ラパマイシンの27−オキシム(Failli、1992a)、ラパマイシンの40−オキソ類似体(Caufield、1991)、二環式ラパマイシン(Kao、1992a)、ラパマイシン二量体(Kao、1992b)、ラパマイシンのシリルエーテル(Failli、1992b)並びにアリールスルホン酸及びスルファミン酸(Failli、1993)の調製などである。
【0016】
[0016]その抗真菌、免疫抑制及び抗腫瘍活性に加えて、シロリムスは、動物モデルにおける新生内膜の増殖並びにヒトにおける再狭窄率を低減させる。シロリムスはまた、リウマチ関節炎の治療用の薬剤としてのその選択を支持する特性である抗炎症作用を示す。エベロリムス及び特にゾタロリムスなどのシロリムスの類似体を被覆したステントは、臨床試験において再狭窄を予防するのに有効である。
【0017】
[0017]ステント及び他の埋込み医療デバイス
【0018】
[0018]ステントは、種々の疾患及び状態、特にアテローム性動脈硬化疾患に起因する血管又は管路の径の重大な減少を治療するために用いられ、しばしば血管形成術後に用いられる。ステントは、動脈に頻繁に用いられるが、静脈、胆管、食道、気管、大気管支、尿管及び尿道などの他の構造にも用いられる。ステントは、英国の歯科医であるCharles Stent(1845〜1901年)の新しいアイディアである。
【0019】
[0019]有害な内腔の狭窄の治療に有効であるが、血管ステントは、医学的に皮肉な例において、治療のためにそれらが使用される状態を再形成するおそれもある。ステントは、内腔の内側の厚い内皮組織、すなわち新生内膜の成長をもたらし得る。成長の程度は様々であるが、新生内膜は、成長して血管内腔を閉塞することができ、これは再狭窄の典型的なタイプである。
【化1】
【化2】
【0020】
[0020]ゾタロリムスの以前の合成
【0021】
[0021]Mollisonは、シロリムスからゾタロリムスを生成させるいくつかの方法を示した(Mollison、2000)。例えば、シロリムスのC−40ヒドロキシルをトリフラートの形成により活性化し、次いで、トリフラートをカラムクロマトグラフィーにより精製する。トリフラートの精製の間、クロマトグラフィー中の水の存在により、活性化された中間体のうちの一部はシロリムス及びそのエピマーであるエピ−シロリムスに逆戻りする。次いで、精製トリフラートを第2のステップにおいてテトラゾールと反応させて、シロリムスの40−エピ−テトラゾール誘導体、すなわち、ゾタロリムスを生成させる。次いで、粗生成物をカラムクロマトグラフィーにより精製する。しかし、この精製によってさえも、最終生成物は、シロリムス及びエピ−シロリムスの混入物を含む可能性がある。
【0022】
[0025][発明の概要]
【0023】
[0026]本発明は、ラパマイシンの誘導体をワンポットで実施する方法を提供し、そのような方法により調製された、抗酸化剤を含む組成物を提供する。
【0024】
[0027]第1の態様において、本発明は、式I
【化3】
の分子を調製する方法であって、
第1のステップ(a)において式II
【化4】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化5】
の分子を生成させ、次いで、第2のステップ(b)において式IIIの分子を式IV
【化6】
の分子と反応させる、方法を提供する。
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【0025】
[0028]方法のステップ(a)は、2,6−ジメチルピリジン又はジイソプロピルエチルアミンなどの非求核塩基の存在下で実施する。ステップ(a)はまた、酢酸イソプロピル又はジクロロメタンなどの溶媒中で実施する。いくつかの実施形態において、ジクロロメタンを、ステップ(b)の前又はステップ(b)の間に酢酸イソプロピルに交換する。
【0026】
[0029]式IVによって表される分子において、R10はHであってよく、R9はH、メチル又はフェニルである。いくつかの実施形態において、R9及びR10は、Hである。
【0027】
[0030]ステップ(b)は、非プロトン性溶媒などの溶媒の存在下で実施する。非プロトン性溶媒は、例えば、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、ジメチルアミン、N,N−ジメチルホルムアミド又は炭酸プロピレンであってよい。いくつかの実施形態において、ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン、アセトニトリル、ジメチルアミン又はN,N−ジメチルホルムアミドのいずれかの存在下で実施する。いくつかの実施形態において、ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの中で実施する。
【0028】
[0031]特定の実施形態において、式IIによって表される分子で、R1は=Oであり、R2はHであり、R3は=Oであり、R4はHである。
【0029】
[0032]他の態様において、ゾタロリムスは、ラパマイシンから本発明の新規な方法により提供される。
【0030】
[0033]他の態様において、本発明は、抗酸化剤と組み合わされた、本発明の方法により調製される分子の組成物を提供する。そのような抗酸化剤としては、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などがある。1つの実施形態において、抗酸化剤は3,5−ジ−tert−4−ブチルヒドロキシトルエンである。
【0031】
[0034]他の態様において、本発明は、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などの抗酸化剤を用いて調合される、本発明の方法により調製されるゾタロリムスの組成物を提供する。1つの実施形態において、抗酸化剤が3,5−ジ−tert−4−ブチルヒドロキシトルエンである。
【0032】
[0035][発明の詳細な説明]
【0033】
[0036]本発明は、ゾタロリムスを生成させ、以前の方法のシロリムス及びエピ−シロリムス混入物を実質的に除去し、医薬品を調製するより効果的な方法である、C−40位におけるシロリムスのテトラゾール類似体を調製するワンポット法を提供する。この方法において、トリフラートを、2,6−ルチジン又は2,6−ジ−tert−ブチルピリジン若しくは2,4,6−コリジンのような他の置換ピリジン、ピリジン、又はヒューニッヒ塩基ジイソプロピルアミン(DIEA)のような非求核塩基の存在下で溶媒としての酢酸イソプロピル(IPAc)又はジクロロメタン(DCM)中で生成させる。IPAcをトリフラートの生成時に溶媒として用いる場合、塩をろ過し、トリフラート溶液をDIEAの存在下でテトラゾールと反応させることができる。DCMをトリフラートの生成時に溶媒として用いる場合、溶媒をIPAcに切り替える。その後、テトラゾールとのSN2反応をDIEAを塩基として用いてIPAc及びテトラゾール中で行わせる。溶媒の除去後の粗生成物を、THF/ヘプタン中、続いてヘプタン/アセトン中カラムクロマトグラフィーにより精製する。精製された生成物は、t−ブチルメチルエーテル(t−BME)/ヘプタンで処理することにより固体として分離することができる。そのように得られたゾタロリムスは、室温で不安定であるが、BHT(2,6−ジ−t−ブチル−4−メチルフェノール、ブチル化ヒドロキシトルエン)、2,6−ジ−t−ブチル−4−エチルフェノール(DEP)、2,6−ジ−t−ブチル−4−メトキシフェノール(DMP)などの抗酸化剤の添加により安定化することができる。
【0034】
[0037]ワンポット法のいくつかの重要な利点は以下の通りである。
1.以前の方法において最終生成物中の混入物の重要な源であったトリフラートの精製を不要とすること、
2.方法の粗生成物をTHF:ヘプタン中で精製することによる、SN2反応中に生成するシロリムス及びエピ−シロリムス副生成物レベルのさらなる低下、
3.容易に回収し、再使用することができる非プロトン性溶媒をSN2反応に使用し、それにより、以前の方法によりもたらされる費用及び環境上の問題を低減すること、
4.t−BMEに溶解し、ヘプタンを加えることによる、又は逆添加法(reverse−addition procedure)による生成物の容易な分離及び精製、
5.抗酸化剤を加えることによる、不純物のない生成物の容易な安定化、及び
6.アセトニトリル又はアセトニトリル:水における凍結乾燥による容易な分離。
【0035】
[0038]定義
【0036】
[0039]「プロドラッグ」は、in vivoで例えば、血液中の加水分解により親化合物に速やかに変換される化合物を意味する。十分な解説が利用可能である(Higuchi及びStella、1987;Roche、1987)。
【0037】
[0040]「薬学的に許容できるプロドラッグ」は、適切な医学的判断の範囲内で、過度の毒性、刺激及びアレルギー反応を伴わずにヒト及び下等哺乳類の組織との接触に使用することに適しており、妥当なベネフィット/リスク比に対応しており、それらの意図した用途並びに可能な場合本発明の化合物の両性イオンの形態で有効である、本発明の化合物のプロドラッグを意味する。本発明の特に好ましい薬学的に許容できるプロドラッグは、本発明の化合物のC−31ヒドロキシル基のプロドラッグエステルである。
【0038】
[0041]「プロドラッグエステル」は、生理的条件下で加水分解されるいくつかのエステル形成基のいずれかを意味する。プロドラッグエステル基の例としては、アセチル、エタノイル、ピバロイル、ピバロイルオキシメチル、アセトキシメチル、フタリジル、メトキシメチル、インダニル等、並びに本発明の化合物のC−31ヒドロキシル基への天然又は非天然由来アミノ酸の結合により得られるエステル基などが挙げられる。
【0039】
[0042]「治療用物質」は、適切な用量で対象に適切に投与したとき、対象への有用な効果を有するあらゆる物質を意味する。
【0040】
[0043]置換基又は可変基(例えば、アリール、アルコキシ、R1、R2、R3、R5、R6等)が式中に複数回出現する場合、各出現時のそのような可変基又は置換基の定義は、特に示さない限り、他のあらゆる出現時のその定義と無関係である。本発明の化合物の成分中に存在する置換基及び/又は可変基の組合せは、そのような組合せが安定な化合物をもたらす場合にのみ許容できる。
【0041】
[0044]R1(例えば、(H,OR6))のような置換基の定義に用いる括弧に入った名称は、関連原子の両原子価に置換基を反映させることを意図する。本発明は特定の異性体に限定されず、括弧内の部分の順序は特定の配置を示唆するものでない。
【0042】
[0045]「アシロキシ」は、−OC(O)−(アルキル)及び−OC(O)−(アリール)を意味する。
【0043】
[0046]「アルケニル」(単独又は組合せ)は、1つ又は複数の二重結合を有するアルキルラジカルを意味する。そのようなアルケニルラジカルの例は、エテニル、プロペニル、1−ブテニル、シス−2−ブテニル、トランス−2−ブテニル、イソ−ブチレニル、シス−2−ペンテニル、トランス−2−ペンテニル、3−メチル−1−ブテニル、2,3−ジメチル−2−ブテニル、1−ペンテニル、1−ヘキセニル、1−オクテニル、デセニル、ドデセニル、テトラデセニル、ヘキサデセニル、シス及びトランス−9−オクタデセニル、1,3−ペンタジエニル、2,4−ペンタジエニル、2,3−ペンタジエニル、1,3−ヘキサジエニル、2,4−ヘキサジエニル、5,8,11,14−エイコサテトラエニル及び9,12,15−オクタデカトリエニルを含むが、これらに限定されない。
【0044】
[0047]「アルコキシル」は、酸素に結合したアルキル基を意味する。
【0045】
[0048]「アルキル」(単独又は組合せ)は、1〜約22個の炭素原子、約1〜約18個の炭素原子又は約1〜約12個の炭素原子を含む直鎖又は枝分れ鎖アルキルラジカルを意味する。そのようなラジカルの例は、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソ−アミル、ヘキシル、オクチル、ノニル、デシル、ドデシル、テトラデシル、ヘキサデシル、オクタデシル及びエイコシルを含むが、これらに限定されない。
【0046】
[0049]「アルキルシクロアルケニル」及び「アルケニルシクロアルケニル」は、上で定義したようなアルキル又はアルケニルラジカルで置換されている、上で定義したようなシクロアルケニルラジカルを意味する。アルキルシクロアルケニル及びアルケニルシクロアルケニルラジカルの例は、1−メチル−2−シクロペンチル、1−ヘキシル−2−シクロペンテニル、1−エチル−2−シクロヘキセニル、1−ブチル−2−シクロヘキセニル、1−(9−オクタデセニル)−2−シクロヘキセニル及び1−(2−ペンテニル)−2−シクロヘキセニルを含むが、これらに限定されない。
【0047】
[0050]「アルキルシクロアルキル」及び「アルケニルシクロアルキル」は、上で定義したようなアルキル又はアルケニルラジカルで置換されている、上で定義したようなシクロアルキルラジカルを意味する。アルキルシクロアルキル及びアルケニルシクロアルキルラジカルの例は、2−エチルシクロブチル、1−メチルシクロペンチル、1−ヘキシルシクロペンチル、1−メチルシクロヘキシル、1−(9−オクタデセニル)シクロペンチル及び1−(9−オクタデセニル)シクロヘキシルを含むが、これらに限定されない。
【0048】
[0051]「アルキニル」(単独又は組合せ)は、1つ又は複数の三重結合を有するアルキルラジカルを意味する。そのようなアルキニル基の例は、エチニル、プロピニル(プロパルギル)、1−ブチニル、1−オクチニル、9−オクタデシニル、1,3−ペンタジイニル、2,4−ペンタジイニル、1,3−ヘキサジイニル及び2,4−ヘキサジイニルを含むが、これらに限定されない。
【0049】
[0052]「アミノ」は、−NH2、−N(アルキル)2、−NH(アルキル)、−N(アリール)2及び−NH(アリール)を意味する。
【0050】
[0053]「アラルキル」(単独又は組合せ)は、ベンジル、2−フェニルエチルなどの、1個の水素原子が上で定義したようなアリールラジカルで置換されている上で定義したようなアルキル又はシクロアルキルラジカルを意味する。
【0051】
[0054]「アリール」(単独又は組合せ)は、フェニル、p−トリル、4−メトキシフェニル、4−(tert−ブトキシ)フェニル、4−フルオロフェニル、4−クロロフェニル、4−ヒドロキシフェニル、1−ナフチル、2−ナフチル等のような、アルキル、シクロアルキル、シクロアルケニル、アリール、複素環、アルコキシアリール、アルカリル、アルコキシ、ハロゲン、ヒドロキシ、アミン、シアノ、ニトロ、アルキルチオ、フェノキシ、エーテル、トリフルオロメチル等から選択される1つ又は複数の置換基を場合によって有するフェニル又はナフチルラジカルを意味する。
【0052】
[0055]「シクロアルケニル」(単独又は組合せ)は、1つ又は複数の二重結合を有するシクロアルキルラジカルを意味する。シクロアルケニルラジカルの例は、シクロペンテニル、シクロヘキセニル、シクロオクテニル、シクロペンタジエニル、シクロヘキサジエニル及びシクロオクタジエニルを含むが、これらに限定されない。
【0053】
[0056]「シクロアルケニルアルキル」は、上で定義したようなシクロアルケニルラジカルで置換されている、上で定義したようなアルキルラジカルを意味する。シクロアルケニルアルキルラジカルの例は、2−シクロヘキセン−1−イルメチル、1−シクロペンテン−1−イルメチル、2−(1−シクロヘキセン−1−イル)エチル、3−(1−シクロペンテン−1−イル)プロピル、1−(1−シクロヘキセン−1−イルメチル)ペンチル、1−(1−シクロペンテン−1−イル)ヘキシル、6−(1−シクロヘキセン−1−1−イル)ヘキシル、1−(1−シクロペンテン−1−イル)ノニル及び1−(1−シクロヘキセン−1−イル)ノニルを含むが、これらに限定されない。
【0054】
[0057]「シクロアルキル」(単独又は組合せ)は、3〜約10個、好ましくは3〜約8個、最も好ましくは3〜約6個の炭素原子を含むシクロアルキルラジカルを意味する。そのようなシクロアルキルラジカルの例は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル及びペルヒドロナフチルを含むが、これらに限定されない。
【0055】
[0058]「シクロアルキルアルキル」は、上で定義したようなシクロアルキルで置換されている、上で定義したようなアルキルラジカルを意味する。シクロアルキルアルキルラジカルの例は、シクロヘキシルメチル、シクロペンチルメチル、(4−イソプロピルシクロヘキシル)メチル、(4−t−ブチル−シクロヘキシル)メチル、3−シクロヘキシルプロピル、2−シクロヘキシルメチルペンチル、3−シクロペンチルメチルヘキシル、1−(4−ネオペンチルシクロヘキシル)メチルヘキシル及び1−(4−イソプロピルシクロヘキシル)メチルヘプチルを含むが、これらに限定されない。
【0056】
[0059]「シクロアルキルシクロアルキル」は、上で定義したような他のシクロアルキルラジカルで置換されている、上で定義したような他のシクロアルキルラジカルを意味する。シクロアルキルシクロアルキルラジカルの例は、シクロヘキシルシクロペンチル及びシクロヘキシルシクロヘキシルを含むが、これらに限定されない。
【0057】
[0060]「ハロゲン」は、フルオロ、クロロ、ブロモ及びヨードを含む。
【0058】
[0061]「複素環」は、飽和又は不飽和であり、炭素原子並びにN、O及びSからなる群から独立に選択される1〜3個のヘテロ原子からなり、窒素及び硫黄ヘテロ原子が酸化されていてよく、窒素へテロ原子が第四級化されていてよく、複素環がベンゼン環に縮合した二環の群を含む、安定な5〜7員単環若しくは二環式又は安定な7〜10員二環式複素環を含む。複素環は、安定な構造をもたらすヘテロ原子又は炭素原子に結合していてよい。複素環の要素の例は、ピペリジル、ピペリジニル、ピペラジニル、ピリミジニル、ピリダジニル、オキサゾリル、フリル及びチエニルを含む。複素環は、ヘテロ原子に結合している炭素原子が、へテロ原子により、C1〜C6アルキル、アリール、ヒドロキシル、C1〜C6アルコキシ、アシルオキシ、アミノ、N−アシルアミノ、ニトロ及びハロゲンであってよい1〜4つの構成員により直接置換されないような仕方で置換されていてよい。
【0059】
[0062]「複素環」は、環内に、炭素に加えて、少なくとも1つの他の種類の原子を含む環構造を意味する。最も一般的な他の種類の原子は、窒素、酸素及び硫黄を含む。複素環の例は、ピロリジニル、ピペリジル、イミダゾリジニル、テトラヒドロフリル、テトラヒドロチエニル、フリル、チエニル、ピリジル、キノリル、イソキノリル、ピリダジニル、ピラジニル、インドリル、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル、ピリジニル、ベンズオキサジアゾリル、ベンゾチアジアゾリル、トリアゾリル及びテトラゾリル基を含むが、これらに限定されない。
【0060】
[0063]「ケトン」は、−C(O)−を意味する。
【0061】
[0064]「N−アシルアミノ」は、−NHC(O)−(アルキル)及び−NHC(O)−(アリール)を意味する。
【0062】
[0065]「窒素含有複素環」は、環の2つの炭素及び1つの窒素が15員大環状リガンドの一部でもある環構造を意味する。環10構造は、約2〜約20又は約4〜約10個の炭素原子を含むことができ、置換又は非置換、部分的又は完全に不飽和又は飽和であってよく、15員大環状リガンドの一部でもない環の一部に窒素、酸素及び/又は硫黄原子を含むこともできる。
【0063】
[0066]「飽和、部分的に飽和又は不飽和環」は、環の2個の原子が15員大環状リガンドの一部でもある縮合環構造を意味する。環構造は、約3〜約20個の炭素原子又は約5〜約10個の炭素原子を含むことができ、炭素に加えて1又は複数個の他の種類の原子も含むことができる。最も一般的な他の種類の原子は、窒素、酸素及び硫黄である。環構造は、1つよりも多い環を含むこともできる。
【0064】
[0067]「飽和、部分的に飽和又は不飽和環構造」は、環の1つの炭素が15員大環状リガンドの一部でもある環構造を意味する。環構造は、約3〜約20個又は約5〜約10個の炭素原子を含むことができ、窒素、酸素及び/又は硫黄原子を含むこともできる。
【0065】
[0068]発明の実施
【0066】
[0069]式I
【化7】
の分子を調製するために、
式II
【化8】
[式中、
R1は、=O(H,H)又は(H,OH)であり、
R2及びR5は、独立にH、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6又は−C(=S)OR6であり、
R3は、=O若しくはOR5であるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H又はC1〜C4アルキルであり、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基又は複素環基であり、
R7及びR8は、独立にH、C1〜C6アルキルであるか、又はR7及びR8は、統合すると=Oである]をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化9】
の分子を生成させて、ステップ(a)を完結させ、
次いで、それを式IV
【化10】
[式中、
R9及びR10は、独立にH、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ又はその組合せである]
の分子と反応させて、ステップ(b)を完結させる。
【0067】
[0070]ステップ(a)
【0068】
[0071]式IIの分子において、R1は好ましくはOであり、R2は好ましくはHであり、R3は好ましくはOであり、R4は好ましくはHである。
【0069】
[0072]塩基 ステップ(a)は、非求核塩基、好ましくは2,6−ジメチルピリジン又はジイソプロピルエチルアミンの存在下で実施する。
【0070】
[0073]溶媒 このステップはまた、酢酸イソプロピル又はジクロロメタンなどの溶媒の存在下で実施する。溶媒がジクロロメタンである場合、それは、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換することができる。
【0071】
[0074]ステップ(b)
【0072】
[0075]式IVの分子において、R10は好ましくはHであり、R9はH、メチル及びフェニルからなる群から選択されるものであり、より好ましくは、R9及びR10はHである。
【0073】
[0076]溶媒 ステップ(b)も溶媒、好ましくは非プロトン性溶媒の存在下で実施する。非プロトン性溶媒の例としては、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、ジメチルアミン、N,N−ジメチルホルムアミド及び炭酸プロピレンなどがある。他の非プロトン性溶媒を用いることができる。好ましい非プロトン性溶媒は、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン、アセトニトリル、ジメチルアミン及びN,N−ジメチルホルムアミドなどであり、最も好ましいものは、ジイソプロピルエチルアミンと酢酸イソプロピル又はジクロロメタンのいずれかである。
【0074】
[0077]スキーム2に本発明の好ましい実施形態の概要を示す。図1にゾタロリムスを調製するためのワンポット法におけるステップを概説する流れ図を示す。第1の実施形態において、シロリムス(市販又は記載されたように生成させた(Paivaら、1991;Sehgalら、1975;Vezinaら、1975))をDCM:トルエン(1:2など)100に溶解する。反応混合物を濃縮して乾燥させ105、共沸乾燥工程105を好ましくはDCM:トルエンを用いてさらに1〜5回、より好ましくは2〜4回、最も好ましくは2回繰り返す。得られる泡様固体をIPAcに溶解し110、次いで、2,6−ルチジンを加える115。溶液を−30℃に冷却する115。次いで、トリフルオロメタンスルホン酸無水物を徐々に溶液に加える115。反応混合物を撹拌した後、溶液を窒素中でろ過する。回収された塩120をIPAcで洗浄する125。塩に1−H−テトラゾール及びDIEAを加える130。反応混合物を室温(例えば、22〜25℃)で撹拌し135、次いで、濃縮する。粗反応混合物を、例えばシリカゲルカラムを用いて精製し、例えば1:1 THF:ヘプタンを用いて溶離させる140。画分をN−1異性体(N−2異性体より遅く溶離する)についてモニターし、プールし、濃縮して、油を生成させる。油を最小限のDCMに溶解し、溶液を、例えば、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加える145。カラムを例えば、65:35 ヘプタン:アセトンで溶離させ、画分を純生成物についてモニターし、プールし、濃縮する150。
【化11】
【0075】
[0078]次いで、精製生成物をt−BMEに溶解し、次いで、溶液を激しく撹拌しながら、n−ヘプタンを徐々に加えて、沈殿を生成させる150。沈殿固体を5〜10℃で撹拌し、ろ過し、ヘプタンで再び洗浄し、窒素を用いて漏斗上で乾燥する。生成物をアセトンに溶解し、BHTで処理する155。溶液を濃縮し、アセトンに溶解し、次いで濃縮し、乾燥した。次いで、生成物を真空中47℃で乾燥する160。
【0076】
[0079]第2の好ましい実施形態において、図2に示す流れ図で、シロリムスをDCMに溶解する200、205。次いで、2,6−ルチジンを加え、溶液を−30℃に冷却し、トリフルオロメタンスルホン酸無水物を徐々に加える210。反応混合物215を混合し、テトラゾールを加え、次いで、DIEAを加える220。反応混合物を約25℃でインキュベートし225、例えば、1:1 THF:n−ヘプタン(体積/体積)で調製したシリカゲルカラム上に加える230。粗反応混合物を1:1 THF:n−ヘプタンを用いて精製する。生成物を含む画分を収集し、濃縮する235。濃縮固体を最小限のDCMに溶解し、例えば、70:30 n−ヘプタン:アセトン中に充填したシリカゲルカラム240上に加える。カラムを溶離させ、純生成物を含む画分を濃縮する245。精製生成物をt−BMEに溶解し、n−ヘプタンに徐々に加える250。沈殿固体をろ過し、n−ヘプタンで洗浄し、乾燥する255。BHTを固体に加え、固体をアセトンに溶解し、ろ過し、濃縮する260。残留物をアセトンで2回処理し260、毎回濃縮して乾燥する。次いで、生成物を真空中で乾燥する260。
【0077】
[0080]第3の実施形態において、シロリムス(ラパマイシン)をジクロロメタンに溶解する。2,6−ルチジンを加え、溶液を−30℃に冷却する。トリフルオロメタンスルホン酸無水物を徐々に加える。反応物を撹拌した後、溶液を10℃に加温する。反応溶液を濃縮し、残留物をIPACに溶解する。1−H−テトラゾールを、続いて、DIEAを加え、反応混合物を22〜25℃で撹拌する。次いで、溶液を濃縮し、例えば、1:1 THF:ヘプタンで溶離するシリカゲルカラム上で精製した。N−1異性体を含む画分を収集し、プールし、濃縮する。得られた油を最小限のDCMに溶解し、例えば、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加える。カラムをヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮する。濃縮物をt−BMEに溶解し、激しく撹拌しながらn−ヘプタンに徐々に加える。次いで、沈殿物を5〜10℃で1時間以下撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥する。BHTを固体に加え、混合物をアセトンに溶解する。次いで、溶液をフィルターに通し、濃縮する。残留物をさらに2回アセトンで処理し、毎回濃縮して乾燥する。最終生成物を真空中50℃で乾燥する。
【0078】
[0081]本発明を遂行するために、本発明の方法において種々の試薬に代えることができる。例えば、トリフラートを調製するために、2,6−ジ−t−ブチルピリジン及びDIEAを2,6−ルチジンの代わりに用いることができる。ピリジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジン及び4−ジメチルアミノピリジン(DMAP)などの他の置換ピリジン、N−メチルモルホリン並びに当業者に明らかである他のものを含む他の塩基をこのステップで用いることができる。様々な溶媒、塩基(DIEAの代わり)及び求核剤(テトラゾールの代わり)も本発明の方法に用いることができる。例を下の表1に示す。
【表1】
【0079】
[0082]塩基及び溶媒 1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン(DBU)、炭酸カリウム(K2CO3)、4−ジメチルアミノピリジン(DMAP)及びカリウムtert−ブトキシド(KOtBu)などの強塩基は、広範な分解をもたらし、一般的にN−2異性体の生成に有利であり、したがって、好ましくない。ルチジン、TEA及びNMMなどのより弱い塩基は、SN2反応を遅くし、約1:1の比でN−1及びN−2異性体の両方が生成する。IPAc、DME、ジオキサン及びTHFなどの非プロトン性溶媒は、十分に機能し、N−1異性体に有利であり、好ましい。DCMを用いることにより、約1:1の比の異性体が得られる。DMA及びDMFのような非プロトン性極性溶媒は、反応生成物の分解をもたらす。
【0080】
[0083]温度 分解が通常認められるが、加熱することにより反応を加速することができる。しかし、反応は通常4時間までに又はそれより早く完結する。したがって、反応混合物をより早期に処理して、分解を最小限にすることができる。SN2置換反応を加速するのに好ましい温度は、20〜35℃、好ましくは22〜33℃、より好ましくは25〜33℃、最も好ましくは28〜30℃を含む。
【0081】
[0084]置換求核剤 一般的に、5−置換テトラゾールをテトラゾールの代わりに用いることができる。例えば、テトラゾールの代わりに用いるとき、5−メチルテトラゾール及び5−フェニルテトラゾールが、好ましい。5−置換テトラゾールは、N−2異性体の生成に有利であるため、好ましい。
【0082】
[0085]抗酸化剤 ワンポット法により調製したゾタロリムスを安定化するために、抗酸化剤を用いることができる。それらは、組成物中に約1重量%まで、より好ましくは0.05%〜0.75%、3,5−ジ−tert−4−ブチルヒドロキシトルエン(BHT)の場合には0.5%存在していてよい。抗酸化剤の例としては、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール又は2−tert−ブチル−4−ヒドロキシアニソール及びフマル酸などがある。好ましくは、抗酸化剤はBHTである。
【0083】
[0086] [実施例]
【0084】
[0087]実施例1 ろ過を伴うジクロロメタン−トルエン 酢酸イソプロピルワンポット法(1)
【0085】
[0088]この実施例では、ゾタロリムスは、ジクロロメタン、トルエン及び酢酸イソプロピルを用いてワンポット法でラパマイシンから調製した。次いで、調製物を精製し、濃縮し、乾燥した。次いで、精製生成物をその1H、13CNMR共鳴により、COSY、ROESY、TOCSY、HSQC及びHMBCスペクトルから確認した。
【0086】
[0089]ラパマイシン(10g)をジクロロメタン(DCM、25ml)及びトルエン(50ml)に溶解した。反応混合物を濃縮して乾燥した。この共沸乾燥工程は、DCM/トルエンを用いて2回繰り返した。泡様固体を酢酸イソプロピル(IPAc、65ml)に溶解し、2,6−ルチジン(3.2ml)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(2.8ml)を10分間に徐々に加えた。反応混合物を30分間撹拌し、次いで、窒素雰囲気中でろ過した。塩をIPAc(10ml)で洗浄した。1−H−テトラゾール(2.3g)を、続いて、ジイソプロピルエチルアミン(DIEA、7.4ml)を加えた。反応混合物を室温で6時間撹拌し、次いで、濃縮した。粗反応混合物を1:1 THF/ヘプタンで溶離するシリカゲルカラム(350g)上で精製した。後に溶離させた生成物を含む画分(主としてN−1異性体)を収集し、濃縮した。濃縮油を最小限のDCMに溶解し、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム上に加えた。カラムを65:35 ヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮した。
【0087】
[0090]次いで、精製生成物をt−ブチルメチルエーテル(t−BME、13.5g)に溶解し、激しく撹拌しながらn−ヘプタン(53g)を徐々に加えた。沈殿固体を5〜10℃で2時間撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥して、3.2gの湿った生成物を得た。固体(1.0g)をアセトン(10ml)に溶解し、2,6−ジ−tert−ブチル−4−エチルフェノール(DEP、0.2%)で処理した。溶液を濃縮し、アセトン(10ml)に溶解し、濃縮して乾燥した。生成物を真空中47℃で18時間乾燥して、0.83gのゾタロリムスを得た。生成物をその1H、13CNMR共鳴により、そのCOSY、ROESY、TOCSY、HSQC及びHMBCスペクトルから特性決定した。
【0088】
[0091]1H−NMR(DMSO−d6,括弧の位置):ppm 0.73(Me,43);0.81(Me,49);0.84(Me,46);0.89(Me,48);0.98(Me,45);1.41、1.05(CH2,24);1.18、1.10(CH2,36);1.52(CH,37);1.53(CH2,12 & 42);1.59、1.30(CH2,5);1.41、1.67(CH2,4);1.11、1.73(CH2,38);1.21、1.83(CH2,15);1.21、1.83(CH2,13);1.62(Me,44);1.73(Me,47);1.76(CH,35);1.60、2.09(CH2,3);1.93、2.21(CH2,41);2.05(CH,11);2.22(CH,23);2.47(CH,25);2.40、2.77(CH2,33);3.06(OCH3,50);3.16(OCH3,51);3.22、3.44(CH2,6);3.29(OCH3,52);3.29(CH,31);3.60(CH,39)、3.62(CH,16);3.89(CH,27);4.01(CH,14);4.02(CH,28);4.95(CH,2);5.02(CH,34);5.10(=CH,30);5.17(CH,40);5.24(OH,28);5.46(=CH,22);6.09(=CH,18);6.15(=CN,21);6.21(=CH,20);6.42(=CH,19);6.42(OH,10)、9.30(CH,53)。
【0089】
[0092]13C NMR(DMSO−d6,括弧の位置):ppm 10.4(Me,44);13.1(Me,47);13.6(Me,46);14.5(Me,49);15.5(Me,43 & 48);20.3(CH2,4);21.6(Me,45);24.4(CH2,4);26.2(CH2,12);26.4(CH2,3);26.8(CH2,41);27.2(CH2,42);29.6(CH2,13);31.6(CH2,38)、31.7(CH,37);32.9(CH,35);34.8(CH,11);35.2(CH,23);38.2(CH2,36);39.1(CH,25);39.4(CH2,33);39.6(CH2,24)、40.0(CH2,15);43.4(CH2,6);45.2(CH,31);50.6(CH,2);55.4(OCH3,50);55.8(OCH3,52);57.0(OCH3,52);55.9(CH,40);66.2(CH,14);73.4(CH,34);75.6(CH,28);77.4(CH,39);82.3(CH,16);85.7(CH,27);99.0(CH,10);125.3(=CH,30);127.0(=CH,18 & 19);130.4(=CH,21);132.2(=CH,20);137.2(=CMe,29);137.7(=CMe,17);139.2(=CH,22);144.6(CH,53);167.0(C=O,8);169.1(C=O,1);199.0(C=O,9);207.5(C=O,32);210.7(C=O,26)。
【0090】
[0093]実施例2 ジクロロメタン−酢酸イソプロピルワンポット法(2)
【0091】
[0094]この実施例では、ゾタロリムスは、ジクロロメタン及び酢酸イソプロピルを用いてワンポット法でラパマイシンから調製した。次いで、化合物を精製し、濃縮し、乾燥した。
【0092】
[0095]ラパマイシン(10g)をジクロロメタン(DCM、100g)に溶解した。2,6−ルチジン(2.92g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(4.62g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、15分以内に10℃に加温した。反応溶液を濃縮した。残留物をIPAc(55g)に溶解した。1−H−テトラゾール(2.68g)を、続いて、ジイソプロピルエチルアミン(DIEA、7.08g)を加えた。反応混合物を室温で6時間撹拌し、次いで、濃縮した。粗反応混合物をシリカゲルカラム(360g)上で精製し、1:1 THF:ヘプタンで溶離させた。後に溶離させた生成物を含む画分(主としてN−1)を収集し、濃縮した。濃縮油を最小限のDCMに溶解し、65:35 ヘプタン:アセトン中に充填したシリカゲルカラム(180g)上に加えた。カラムを65:35 ヘプタン:アセトンで溶離させ、純生成物を含む画分を濃縮した。
【0093】
[0096]精製生成物をt−ブチルメチルエーテル(t−BME、23g)に溶解し、激しく撹拌しながらn−ヘプタン(80g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.015g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回処理し(20g)、毎回濃縮して乾燥した。次いで、生成物を真空中50℃以下で18時間乾燥して、2.9gのゾタロリムスを得た。
【0094】
[0097]実施例3 ジクロロメタンワンポット法(3)
【0095】
[0098]この例では、ゾタロリムスは、ジクロロメタンを用いてワンポット法でラパマイシンから調製した。次いで、実施例2で述べたように、化合物を精製し、濃縮し、乾燥した。
【0096】
[0099]ラパマイシン(7.5g)をDCM(30g)に溶解した。2,6−ルチジン(1.76g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(2.89g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、次いで、ラパマイシンの存在を分析して、反応における消費を測定した。1−H−テトラゾール(1.44g)を、続いて、DIEA(5.29g)を加えた。反応混合物を室温で6時間撹拌し、次いで、1:1 THF:n−ヘプタン(体積/体積)中に調製したシリカゲル(270g)カラム上に直接加えた。粗反応混合物を1:1 THF:n−ヘプタンで精製した。後に溶離させた生成物を含む画分を収集し、濃縮した。濃縮固体を最小限のDCMに溶解し、70:30 n−ヘプタン:アセトン中で充填したシリカゲルカラム(135g)上に加えた。カラムを70:30 n−ヘプタン:アセトンで溶離させ、薄層クロマトグラフィー(TLC)により同定された、純生成物を含む画分を濃縮した。
【0097】
[0100]精製生成物をt−BME(9g)に溶解し、10±10℃で激しく撹拌しながらn−ヘプタン(36g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、n−ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.006g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回(各20g)処理し、毎回濃縮して乾燥した。生成物を真空中50℃以下で18時間以下乾燥して、2.5gのゾタロリムスを得た。
【0098】
[0101]上の方法は、ステップ1aにおいて非求核剤としての2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジン(2,3,5−トリメチルピリジン)の存在下でラパマイシンを用いて実施した場合、許容できる純度であったが、より低い収率のゾタロリムスを生成させた。
【0099】
[0102]実施例4 ワンポット合成法により調製したゾタロリムスの高圧液体クロマトグラフィー(HPLC)精製
【0100】
[0103]この実施例では、ゾタロリムスを本発明のワンポット合成法を用いてラパマイシンから調製し(DCMを用いて)、次いで、HPLCを用いた精製のさらなるラウンドに供した。
【0101】
[0104]ラパマイシン(3.75g)をジクロロメタン(DCM、15g)に溶解し、次いで、2,6−ルチジン(0.88g)を加えた。溶液をアセトニトリル−ドライアイス浴中で−30℃に冷却し、トリフルオロメタンスルホン酸無水物(1.45g)を10分間に徐々に加えた。反応混合物を20分間撹拌し、次いで、1−H−テトラゾール(0.72g)を、続いて、DIEA(2.65g)を加えた。反応混合物を25℃で6時間撹拌し、次いで、ヘプタン70:30 n−ヘプタン:アセトン中に調製したシリカゲル(115g)カラム上に直接加えた。粗反応混合物を70:30 n−ヘプタン:アセトンで精製した。生成物を含む画分を収集し、濃縮した。
【0102】
[0105]濃縮固体をアセトニトリル−水に溶解した。C−18TechniKromカラム(5cm×25cm)上に加え、0.1%BHTを含む64:36 アセトニトリル−水で溶離させた。画分を逆相(RP)−HPLCにより分析し、生成物画分をプールし、濃縮して、アセトニトリルを除去した。生成物を酢酸エチル又は酢酸イソプロピルで抽出し、乾燥し(硫酸ナトリウム)、濃縮した。
【0103】
[0106]精製生成物をt−BME(4.5g)に溶解し、約10℃で激しく撹拌しながらn−ヘプタン(18g)に徐々に加えた。沈殿固体を5〜10℃で1時間以下撹拌し、ろ過し、n−ヘプタンで洗浄し、窒素を用いて漏斗上で乾燥した。BHT(0.005g)を固体に加えた。固体をアセトン(20g)に溶解し、フィルターに通し、濃縮した。残留物をアセトンで2回処理し(20g)、毎回濃縮して乾燥した。生成物を真空中50℃以下で18時間以下乾燥して、1.2gの高品質のゾタロリムスを得た。
【0104】
[0107]実施例5 ワンポット合成法により調製したゾタロリムスの安定性分析
【0105】
[0108]この実施例では本発明の方法により調製されたゾタロリムスを抗酸化剤を用いて安定化できることを示す。
【0106】
[0109]ワンポット法により調製したゾタロリムスのいくつかのロットは、時間の経過とともに力価を著しく失った。力価損失は温度上昇でより高かったが、不純物プロファイルの明らかな変化はなかった。表2に種々の時間間隔及び温度条件でのゾタロリムスのロットの力価を示す。例えば、室温(25℃)の密閉容器中でさえも、力価は3カ月間にわたり95.1%から69.8%に低下した。この損失は、40℃で保管した場合、密閉容器中で悪化し、ほんの2カ月間で96.0%から37.4%になった。
【0107】
[0110]その後の調査により、この力価の損失は複数の分解生成物をもたらす分子の酸化的分解に起因していたことが明らかになった。この酸化を防止するためにフェノール系抗酸化剤を用いて安定性試験を実施したところ、BHTが適切な化合物と特定された。表3及び4に種々の濃度(重量/重量で表した%)及び温度でBHTを用いた安定性データを示す。例えば、40℃では、0.5%BHTは約3カ月間にわたり力価を初期の96.5%から95.9%の最終力価まで維持したが、BHTの非存在下での同様の条件下では、力価は96%からほんの2カ月後に37.4%に激減した。
【表2】
【表3】
【表4】
【0108】
[0111]これらの安定性試験により、ゾタロリムスの純度、力価及び安定性を維持するためには、BHTのような抗酸化剤の添加が非常に重要であることが確認される。
【0109】
[0112]実施例6 ゾタロリムス平衡異性体の分離及び特性解析
【0110】
[0113]C−18又はフェニルカラム上のゾタロリムスの逆相分析により、7員環オキセパン(2)異性体と比較して早く溶離した主要な異性体は、6員環ピラン形であり、副次的成分であるオキセパン異性体は3〜4分後に溶離したことが示された。順相HPLC(シリカゲル−YMC Co.Ltd;京都、日本)では、2つの形態はベースライン分離を示さなかった。しかし、オキセパン形はピラン形の直前に溶離した。
【0111】
[0114]この平衡を実証するために、pH4の逆相フェニルカラム上へのゾタロリムスの複数回のHPLC注入により、各形態を分離した。次いで、分離された各形態を再注入して、種々の間隔でそれらの平衡を試験した。試験により、ピラン形は3〜4日で平衡状態に到達したが、オキセパン形(副次的成分)はほぼ6日後でさえ完全には平衡に達しなかったことが示された。試験中、開環酸(open ring acid)のある程度の生成があった。表5(緩衝液を含む)及び表6(緩衝液を含まない)に示すこの試験の結果は、2つの形態は平衡状態にあり、ピラン形が熱力学的により安定であることを明確に示している。
【0112】
[0115]アセトニトリル/水の溶媒混合物中で非緩衝条件下でも試験を実施した。非緩衝媒体中水中66%アセトニトリルを用いてC−18 Altimaカラム(Alltech Associates,Inc.;Deerfield、IL)上へのゾタロリムスの複数回の注入を行った。ピラン及びオキセパン形を収集した。これらの形態をC−18カラム上に再注入して、種々の間隔で平衡比を試験した。表4及び5に示すこれらのデータから、2異性体間の平衡化が速やかであり、約7〜8時間以内に完結することが示唆された。これらの所見は、ゾタロリムスは約10:1のピラン(1)対オキセパン(2)形の平衡混合物で存在することを確認するものであった。
【表5】
【表6】
【0113】
[0116]前記の詳細な説明及び付随する実施例は、単に説明のためのものであって、添付の特許請求の範囲及びそれらの同等物によってのみ規定される発明の範囲に対する制限と解釈すべきでないと理解される。開示された実施形態の様々な変更及び修正は、当業者には明らかであろう。化学構造、置換基、誘導体、中間体、合成、調合及び/又は本発明の使用の方法に関するものを制限なしに含む、そのような変更及び修正は、その精神及び範囲から逸脱することなく行うことができる。
【0114】
[0117]特に示さない限り、すべての参考文献は、参照によりそれらの全部が本明細書に組み込まれる。
【0115】
[0118]参考文献
Baker, H., A. Sidorowicz, S.N. Sehgal, and C. Vezina. 1978. Rapamycin (AY−22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J Antibiot (Tokyo). 31:539−45.
Caufield. US Patent No. 5,023,262. 1991. Hydrogenated Rapamycin Derivatives.
Caufield. WO 92/05179. 1992. Carboxylic Acid Esters of Rapamycin.
Eng. US Patent No. 4,401,653.1983. Combination of Rapamycin and Picibanil for the Treatment of Tumors.
Failli. EPO 467606. 1992a. Rapamycin Derivatives.
Failli. US Patent No. 5,120,842. 1992b. Silyl Ethers of Rapamycin.
Failli. US Patent No. 5,177,203. 1993. Rapamycin 42−Sulfonatcs and 42−(N−Carboalkoxy) Sulfamatcs Useful as Imunosuppressie Agents.
Higuchi, T., and V. Stella. 1987. Pro−drugs as Novel Delivery systems.
Hughes, P., J. Musser, M. Conklin, and R. Russo. 1992. The isolation, synthesis and characterization of an isomeric form of rapamycin. Tetrahedron Lttrs. 33:4739−4742.
Kao. US Patent No. 5,120,725. 1992a. Bicyclic Rapamycins.
Kao. US Patent No. 5,120,727. 1992b. Rapamycin Dimers.
Martel, R.R., J. Klicius, and S. Galet. 1977. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 55:48−51.
Mollison, K. US Patent No. 6,015,815. 2000. Tetrazole−containing rapamycin analogs with shortened half−lives.
Mollison, K., A. LeCaptain, S. Burke, K. Cromack, P. Tarcha, Y.−C.J. Chen, and J. Toner. US Patent Application Publication 20030129215. 2003. Medical devices containing rapamycin analogs
Paiva, N.L., A.L. Demain, and M.F. Roberts. 1991. Incorporation of acetate, propionate, and methionine into rapamycin by Streptomyces hygroscopicus. J Nat Prod. 54:167−77.
Roche, E. 1987. Bioreversible Carriers in Drug Design. American Pharmaceutical Association and Pergamon Press.
Sehgal, S.N. US Patent No. 3,929,992. 1975. Rapamycin and Process of Preparation.
Sehgal, S.N. US Patent No. 3,993,749. 1976. Rapamycin and Process of Preparation.
Sehgal, S.N., H. Baker, and C. Vezina. 1975. Rapamycin (AY−22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 28:727−32.
Surendra. US Patent No. 4,885,171. 1989. Use of Rapamycin in Treatment of Certain Tumors.
Vezina, C., A. Kudelski, and S.N. Sehgal. 1975. Rapamycin (AY−22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 28:721−6.
【図面の簡単な説明】
【0116】
【図1】本発明によるゾタロリムスを調製するワンポット法の実施形態の流れ図を示す。
【図2】本発明によるゾタロリムスを調製するワンポット法の実施形態の流れ図を示す。
【特許請求の範囲】
【請求項1】
式I
【化1】
の分子を調製する方法であって、
(a)式II
【化2】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化3】
の分子を生成させるステップと、
(b)式IIIの分子を式IV
【化4】
の分子と反応させるステップと、
を含む方法
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項2】
ワンポットで実施する、請求項1に記載の方法。
【請求項3】
ステップ(a)を非求核塩基の存在下で実施する、請求項1に記載の方法。
【請求項4】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項3に記載の方法。
【請求項5】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項4に記載の方法。
【請求項6】
ステップ(a)を溶媒の存在下で実施する、請求項3に記載の方法。
【請求項7】
前記溶媒が酢酸イソプロピル又はジクロロメタンを含む、請求項6に記載の方法。
【請求項8】
前記溶媒が、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換されるジクロロメタンを含む、請求項6に記載の方法。
【請求項9】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項1に記載の方法。
【請求項10】
R9及びR10がHである、請求項1に記載の方法。
【請求項11】
ステップ(b)を溶媒の存在下で実施する、請求項1に記載の方法。
【請求項12】
前記溶媒が非プロトン性溶媒を含む、請求項11に記載の方法。
【請求項13】
前記非プロトン性溶媒が、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、N,N−ジメチルホルムアミド又は炭酸プロピレンを含む、請求項12に記載の方法。
【請求項14】
ステップ(b)をジイソプロピルエチルアミン並びに酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン及びアセトニトリルからなる群から選択される溶媒の存在下で実施する、請求項1に記載の方法。
【請求項15】
ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの存在下で実施する、請求項1に記載の方法。
【請求項16】
R1が=Oであり、
R2がHであり、
R3が=Oであり、
R4がHである、
請求項1に記載の方法。
【請求項17】
式V
【化5】
の分子を調製する方法であって、
(a)式VI
【化6】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式VII
【化7】
の分子を生成させるステップと、
(b)式VIIの分子を式IV
【化8】
の分子と反応させるステップと、
を含む方法
[式中、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項18】
ワンポットで実施する、請求項17に記載の方法。
【請求項19】
ステップ(a)を非求核塩基の存在下で実施する、請求項17に記載の方法。
【請求項20】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項19に記載の方法。
【請求項21】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項20に記載の方法。
【請求項22】
ステップ(a)を溶媒の存在下で実施する、請求項20に記載の方法。
【請求項23】
前記溶媒が酢酸イソプロピル又はジクロロメタンを含む、請求項22に記載の方法。
【請求項24】
前記溶媒が、ジクロロメタンを含み、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換される、請求項22に記載の方法。
【請求項25】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項17に記載の方法。
【請求項26】
R9及びR10がHである、請求項17に記載の方法。
【請求項27】
ステップ(b)を溶媒の存在下で実施する、請求項17に記載の方法。
【請求項28】
前記溶媒が非プロトン性溶媒を含む、請求項27に記載の方法。
【請求項29】
前記非プロトン性溶媒が、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、N,N−ジメチルホルムアミド又は炭酸プロピレンを含む、請求項28に記載の方法。
【請求項30】
ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの存在下で実施する、請求項17に記載の方法。
【請求項31】
式III
【化9】
の分子を調製する方法であって、
式II
【化10】
の分子をトリフルオロメタンスルホン酸無水物と反応させること、
を含む方法
[式中、R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oである]。
【請求項32】
トリフルオロメタンスルホン酸無水物との反応を非求核塩基の存在下で実施する、請求項31に記載の方法。
【請求項33】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項32に記載の方法。
【請求項34】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項33に記載の方法。
【請求項35】
式I
【化11】
の分子を調製する方法であって、
式III
【化12】
の分子を式IV
【化13】
の分子と反応させること、
を含む方法
[式中、R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項36】
式IVの分子を用いて、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N−メチルイミダゾール又は4−ジメチルアミノピリジンの存在下で実施する、請求項35に記載の方法。
【請求項37】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項35に記載の方法。
【請求項38】
R9及びR10がHである、請求項35に記載の方法。
【請求項39】
式VII
【化14】
の分子を調製する方法であって、
式VI
【化15】
の分子をトリフルオロメタンスルホン酸無水物と反応させること、
を含む方法。
【請求項40】
非求核塩基の存在下で実施する、請求項39に記載の方法。
【請求項41】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項40に記載の方法。
【請求項42】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項41に記載の方法。
【請求項43】
式V
【化16】
の分子を調製する方法であって、
式VI
【化17】
の分子を式IV
【化18】
の分子と反応させること、
を含む方法
[式中、R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項44】
式IVの分子を用いて、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N−メチルイミダゾール又は4−ジメチルアミノピリジンの存在下で実施する、請求項43に記載の方法。
【請求項45】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項43に記載の方法。
【請求項46】
R9及びR10が両方ともHである、請求項43に記載の方法。
【請求項47】
式I
【化19】
の分子及び抗酸化剤を含む組成物
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oである]。
【請求項48】
前記抗酸化剤が、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール、2−tert−ブチル−4−ヒドロキシアニソール若しくはフマル酸又はその組合せを含む、請求項47に記載の組成物。
【請求項49】
式V
【化20】
の分子
及び抗酸化剤を含む組成物。
【請求項50】
前記抗酸化剤が3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール、2−tert−ブチル−4−ヒドロキシアニソール、フマル酸、,6−ジ−t−ブチル−4−エチルフェノール、2,6−ジ−t−ブチル−4−メトキシフェノール又はその組合せを含む、請求項49に記載の組成物。
【請求項1】
式I
【化1】
の分子を調製する方法であって、
(a)式II
【化2】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式III
【化3】
の分子を生成させるステップと、
(b)式IIIの分子を式IV
【化4】
の分子と反応させるステップと、
を含む方法
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項2】
ワンポットで実施する、請求項1に記載の方法。
【請求項3】
ステップ(a)を非求核塩基の存在下で実施する、請求項1に記載の方法。
【請求項4】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項3に記載の方法。
【請求項5】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項4に記載の方法。
【請求項6】
ステップ(a)を溶媒の存在下で実施する、請求項3に記載の方法。
【請求項7】
前記溶媒が酢酸イソプロピル又はジクロロメタンを含む、請求項6に記載の方法。
【請求項8】
前記溶媒が、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換されるジクロロメタンを含む、請求項6に記載の方法。
【請求項9】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項1に記載の方法。
【請求項10】
R9及びR10がHである、請求項1に記載の方法。
【請求項11】
ステップ(b)を溶媒の存在下で実施する、請求項1に記載の方法。
【請求項12】
前記溶媒が非プロトン性溶媒を含む、請求項11に記載の方法。
【請求項13】
前記非プロトン性溶媒が、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、N,N−ジメチルホルムアミド又は炭酸プロピレンを含む、請求項12に記載の方法。
【請求項14】
ステップ(b)をジイソプロピルエチルアミン並びに酢酸イソプロピル、ジクロロメタン、1,2−ジメトキシエタン、テトラヒドロフラン及びアセトニトリルからなる群から選択される溶媒の存在下で実施する、請求項1に記載の方法。
【請求項15】
ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの存在下で実施する、請求項1に記載の方法。
【請求項16】
R1が=Oであり、
R2がHであり、
R3が=Oであり、
R4がHである、
請求項1に記載の方法。
【請求項17】
式V
【化5】
の分子を調製する方法であって、
(a)式VI
【化6】
の分子をトリフルオロメタンスルホン酸無水物と反応させて、式VII
【化7】
の分子を生成させるステップと、
(b)式VIIの分子を式IV
【化8】
の分子と反応させるステップと、
を含む方法
[式中、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項18】
ワンポットで実施する、請求項17に記載の方法。
【請求項19】
ステップ(a)を非求核塩基の存在下で実施する、請求項17に記載の方法。
【請求項20】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項19に記載の方法。
【請求項21】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項20に記載の方法。
【請求項22】
ステップ(a)を溶媒の存在下で実施する、請求項20に記載の方法。
【請求項23】
前記溶媒が酢酸イソプロピル又はジクロロメタンを含む、請求項22に記載の方法。
【請求項24】
前記溶媒が、ジクロロメタンを含み、ステップ(b)の前又はステップ(b)中に酢酸イソプロピルに交換される、請求項22に記載の方法。
【請求項25】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項17に記載の方法。
【請求項26】
R9及びR10がHである、請求項17に記載の方法。
【請求項27】
ステップ(b)を溶媒の存在下で実施する、請求項17に記載の方法。
【請求項28】
前記溶媒が非プロトン性溶媒を含む、請求項27に記載の方法。
【請求項29】
前記非プロトン性溶媒が、ペルフルオロヘキサン、α,α,α−トリフルオロトルエン、ペンタン、ヘキサン、シクロヘキサン、メチルシクロヘキサン、デカヒドロナフタレン、四塩化炭素、ジオキサン、フルオロトリクロロメタン、ベンゼン、トルエン、トリエチルアミン、二硫化炭素、ジイソプロピルエーテル、ジエチルエーテル、t−ブチルメチルエーテル、クロロホルム、酢酸エチル、1,2−ジメトキシエタン、2−メトキシエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、テトラヒドロフラン、塩化メチレン、ピリジン、2−ブタノン、アセトン、ヘキサメチルホスホルアミド、N−メチルピロリジノン、ニトロメタン、ジメチルホルムアミド、アセトニトリル、スルホラン、ジメチルスルホキシド、ジイソプロピルエチルアミン、酢酸イソプロピル、ジクロロメタン、N,N−ジメチルホルムアミド又は炭酸プロピレンを含む、請求項28に記載の方法。
【請求項30】
ステップ(b)をジイソプロピルエチルアミン及び酢酸イソプロピル又はジクロロメタンのいずれかの存在下で実施する、請求項17に記載の方法。
【請求項31】
式III
【化9】
の分子を調製する方法であって、
式II
【化10】
の分子をトリフルオロメタンスルホン酸無水物と反応させること、
を含む方法
[式中、R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oである]。
【請求項32】
トリフルオロメタンスルホン酸無水物との反応を非求核塩基の存在下で実施する、請求項31に記載の方法。
【請求項33】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項32に記載の方法。
【請求項34】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項33に記載の方法。
【請求項35】
式I
【化11】
の分子を調製する方法であって、
式III
【化12】
の分子を式IV
【化13】
の分子と反応させること、
を含む方法
[式中、R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oであり、
R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項36】
式IVの分子を用いて、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N−メチルイミダゾール又は4−ジメチルアミノピリジンの存在下で実施する、請求項35に記載の方法。
【請求項37】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項35に記載の方法。
【請求項38】
R9及びR10がHである、請求項35に記載の方法。
【請求項39】
式VII
【化14】
の分子を調製する方法であって、
式VI
【化15】
の分子をトリフルオロメタンスルホン酸無水物と反応させること、
を含む方法。
【請求項40】
非求核塩基の存在下で実施する、請求項39に記載の方法。
【請求項41】
前記非求核塩基が、置換ピリジン、ジイソプロピルエチルアミン又はピリジンを含む、請求項40に記載の方法。
【請求項42】
前記置換ピリジンが、2,6−ルチジン、2,6−ジ−tert−ブチルピリジン又は2,4,6−コリジンを含む、請求項41に記載の方法。
【請求項43】
式V
【化16】
の分子を調製する方法であって、
式VI
【化17】
の分子を式IV
【化18】
の分子と反応させること、
を含む方法
[式中、R9及びR10は、H、アルケニル、アルケニルシクロアルケニル、アルケニルシクロアルキル、アルキル、アルキルシクロアルケニル、アルキルシクロアルキル、アルキニル、アラルキル、アリール、シクロアルケニル、シクロアルキル、シクロアルキルアルキル、シクロアルキルシクロアルキル、シクロアルケニルアルキル、ヘテロシクリル、アザ、アミド、アンモニウム、オキサ、チア、スルホニル、スルフィニル、スルホンアミド、ホスホリル、ホスフィニル、ホスフィノ、ホスホニウム、ケト、エステル、アルコール、カルバメート、尿素、チオカルボニル、ホウ酸、ボラン、ボラザ、シリル、シロキシ、シラザ及びその組合せからなる群から独立に選択される]。
【請求項44】
式IVの分子を用いて、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N−メチルイミダゾール又は4−ジメチルアミノピリジンの存在下で実施する、請求項43に記載の方法。
【請求項45】
R10がHであり、R9がH、メチル及びフェニルからなる群から選択される、請求項43に記載の方法。
【請求項46】
R9及びR10が両方ともHである、請求項43に記載の方法。
【請求項47】
式I
【化19】
の分子及び抗酸化剤を含む組成物
[式中、
R1は、=O(H,H)及び(H,OH)からなる群から選択され、
R2及びR5は、H、−C(=O)R6、−C(=O)OR6、−C(=O)NHR6及び−C(=S)OR6からなる群から独立に選択され、
R3は、=O及びOR5からなる群から選択されるか、又はR2及びR3は、統合すると式A−C(R7)(R8)−O−Bの部分を形成することができ、Aは炭素28に結合した酸素への結合であり、Bは上で定義したように炭素28への結合であり、
R4は、H及びC1〜C4アルキルからなる群から選択され、
R6は、C1〜C10アルキル、C3〜C6シクロアルキル、アリール基及び複素環基からなる群から選択され、
R7及びR8は、H、C1〜C6アルキルからなる群から独立に選択されるか、又はR7及びR8は、統合すると=Oである]。
【請求項48】
前記抗酸化剤が、3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール、2−tert−ブチル−4−ヒドロキシアニソール若しくはフマル酸又はその組合せを含む、請求項47に記載の組成物。
【請求項49】
式V
【化20】
の分子
及び抗酸化剤を含む組成物。
【請求項50】
前記抗酸化剤が3,5−ジ−tert−4−ブチルヒドロキシトルエン、DL−α−トコフェロール、没食子酸プロピル、パルミチン酸アスコビル、3−tert−ブチル−4−ヒドロキシアニソール、2−tert−ブチル−4−ヒドロキシアニソール、フマル酸、,6−ジ−t−ブチル−4−エチルフェノール、2,6−ジ−t−ブチル−4−メトキシフェノール又はその組合せを含む、請求項49に記載の組成物。
【図1】

【図2】
【図2】
【公開番号】特開2013−32367(P2013−32367A)
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−212775(P2012−212775)
【出願日】平成24年9月26日(2012.9.26)
【分割の表示】特願2008−545933(P2008−545933)の分割
【原出願日】平成18年12月12日(2006.12.12)
【出願人】(507316642)アボット ラボラトリーズ (18)
【Fターム(参考)】
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−212775(P2012−212775)
【出願日】平成24年9月26日(2012.9.26)
【分割の表示】特願2008−545933(P2008−545933)の分割
【原出願日】平成18年12月12日(2006.12.12)
【出願人】(507316642)アボット ラボラトリーズ (18)
【Fターム(参考)】
[ Back to top ]