
ジアザベンゾ[デ]アントラセン−3−オン化合物、及び、PARPの阻害方法
【課題】ジアザベンゾ[デ]アントラセン−3−オン化合物、及び、PARPの阻害方法の提供。
【解決手段】本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法に関する。
【解決手段】本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法に関する。
【背景技術】
【0002】
PARP(EC2.4.2.30)は、PARS(ポリ(ADP−リボース)合成酵素)又はADPRT(NAD:タンパク質(ADP−リボシル)トランスフェラーゼ(重合性))としても知られ、116kDaの主要な核タンパク質である。これは主に、ほとんど全ての真核生物に存在する。この酵素によりポリ(ADP−リボース)が合成され、これは、NAD由来の200個を超えるADP−リボース単位から構成され得る分岐鎖ポリマーである。ポリ(ADP−リボース)のタンパク質受容体は、DNAの一体性維持に直接的又は間接的に関与している。これには、ヒストン、トポイソメラーゼ、DNA及びRNAポリメラーゼ、DNAリガーゼ並びにCa2+及びMg+依存性エンドヌクレアーゼが含まれる。
【0003】
PARPタンパク質は、多くの組織に、最も顕著には免疫系、心臓、脳及び胚株細胞内に高レベルで発現する。正常な生理学的条件下では、PARP活性は非常に小さい。しかし、DNAの損傷によって、PARPは500倍にまで急激に活性化される。PARPに起因する多くの機能の中でも、主な役割は、ADP−リボシル化によるDNA修復の促進及び多数のDNA修復タンパク質の調整である。PARPが活性化されると、NADレベルが著しく減少する。多くの内因性及び外因性因子がDNAを損傷させてかつPARPを活性化させるということが報告されているが、例えばショック、卒中及び炎症において、報告されているinvivoでの種々の疾患症状に関与する主な原因は、酸化窒素(NO)と過酸化物との組み合わせによって形成される過酸化亜硝酸であると考えられる。
【0004】
また、3−アミノベンズアミドなどのPARP阻害剤は、通常は例えば過酸化水素又はγ線に応答して、DNA修復に影響を与えることも知られている(非特許文献1)。特に、Cristovaoらは、過酸化水素で処理された白血球内においてDNA鎖のPARP依存性回復が停止することを観察した。
【0005】
核酵素ポリ(ADP−リボース)ポリメラーゼ−1(PARP−1)は、塩基除去修復及び他の細胞プロセスの促進において重要な役割を果たしている。PARP−1は分子DNAニックのセンサとして作用し、一重鎖DNAの切断を検出しかつ適切な修復酵素を集めるということが提唱されている。PARP−1は、酵素のアミノ末端DNA結合ドメイン中の2つのジンクフィンガーを介してDNA鎖切断部分に結合し、その活性はDNA結合に依存する。この酵素は、基質NAD+から、PARP−1自身も含む受容体タンパク質へのADP−リボースの移動を触媒するホモダイマーとして作用している。これによって多量の負電荷を帯びたPARポリマーが形成され、DNA鎖及びクロマチンタンパク質の静電的反発が生じて、これにより、損傷した鎖に塩基除去修復複合体が接近してDNAが修復される。鎖切断によって最初に活性化された後はPARP−1はDNAから遊離し、ポリマーはPARグリコヒドロラーゼにより分解されて、PARP−1酵素は次のDNA結合及び活性化において使用することができる(非特許文献2)。
【0006】
PARP阻害剤は、放射線増感された低酸素腫瘍細胞において共力剤又は増強剤として有効であることが報告されている。またPARP阻害剤は、放射線療法後の致死性DNA損傷からの腫瘍細胞の回復の阻害において、おそらくはそのDNA修復阻害能に起因して、共力剤として有効であることが報告されている(特許文献1〜3)。
【0007】
ガン治療に用いるための、及び、虚血や内毒素ストレス後における細胞損傷の抑制に用いるための、化学増強剤でもあり放射線増強剤でもあるPARP阻害剤の開発には、大きな関心が集まっている。特に、強力なPARP−1阻害剤を用いた臨床前研究ではテモゾロマイド細胞傷害性の増強が観察されるが、これは塩基除去修復の阻害及びその後の細胞傷害(N7−メチルグアニン及びN3−メチルアデニンの不完全なプロセシングによる)を反映する。現在、一連の臨床前データによって、テモゾロマイドの細胞傷害性は、PARP阻害剤をin vitro又はin vivoのいずれかで共投与することにより増強されるということが示されている(非特許文献2)。
【0008】
DNAメチル化剤であるテモゾロマイドによってDNA損傷が誘発されるが、この損傷は、O6−アルキルグアニンアルキルトランスフェラーゼ(ATase)及びポリ(ADP−リボース)ポリメラーゼ−1(PARP−1)依存性塩基除去修復により修復される。テモゾロマイドは、神経膠腫及び悪性黒色腫の治療において用いられる、経口投与用の単官能性DNAアルキル化剤である。テモゾロマイドは速やかに吸収され、自発的に分解して、活性モノメチルトリアゼンである5−(3−メチル−1−トリアゼノ)イミダゾール−4−カルボキサミドを形成する。モノメチルトリアゼンからはいくつかのDNAメチル化産物が形成されるが、主な種は、N7−メチルグアニン(70%)、N3−メチルアデニン(9%)及びO6−メチルグアニン(5%)である。O6−アルキルグアニンアルキルトランスフェラーゼによって修復されない場合、O6−メチルグアニンは、DNA複製の際にチミンとの誤対合により細胞傷害性を有する。この誤対合は、ミスマッチ修復タンパク質及び除去されたチミンによって、娘鎖上で認識される。しかし、メチル付加物のAtaseが媒介する除去によって親鎖中の元のO6−メチルグアニンヌクレオチドが修復されなければ、チミンは再び挿入され得る。チミン除去が繰り返されることにより無駄に時間が経過することによって、かつ、未修復のO6−メチルグアニンヌクレオチドが結合することによって、鎖が切断された状態が持続し、ミスマッチ修復系のMutS分岐によってG2−M細胞周期停止及びアポトーシスの開始についてのシグナルが送られる。テモゾロマイドによって形成される多量のN7−メチルグアニン及びN3−メチルアデニンヌクレオチドアルキル化産物は、塩基除去修復によって迅速に修復される(非特許文献2)。
【0009】
PARP阻害剤による化学増感は、テモゾロマイドの場合に制限されない。概して細胞傷害性薬剤は、又は、放射線は、PARP−1の活性化を誘発する可能性があり、PARP−1の阻害剤は、化学療法及び放射線照射によるDNA損傷及び細胞傷害効果を増強する可能性があることが示されている(非特許文献3)。DNA損傷剤に応答するPARP−1媒介DNA修復によって、腫瘍における薬剤耐性のメカニズムが示され、上記酵素が阻害されると、イオン化放射線の活性及びいくつかの細胞傷害性抗腫瘍剤(テモゾロマイド及びトポテカンを含む)が増強されることが示されている。Sutoらは、特許文献4で、イオン化放射線又は化学療法薬剤が腫瘍細胞に及ぼす致死性効果を増強する際に用いられるいくつかのイソキノリン類を開示している。非特許文献4において、腫瘍細胞をアルキル化剤で併用治療した場合の、PARP活性の阻害、腫瘍細胞の増殖低下及び顕著な共力剤的効果が開示されている。このことから、PARP−1は、DNAを損傷させるガン治療を促進するための非常に重要な治療標的である。
【0010】
多数の公知のPARP阻害剤が、非特許文献5及び6中に記載されている。しかし、上述した方法において、これらのPARP阻害剤を使用すると望ましくない副作用が同時に起こるため、これらのPARP阻害剤の有効な使用は制限されている。非特許文献7を参照。
【0011】
上記に加えて、PARP阻害剤は、以下の国際特許出願に開示されかつ記載されている:特許文献5〜15。従来技術についての包括的な概説は、Li及びZhangにより非特許文献8に報告されている。
【0012】
腫瘍細胞をイオン化放射線に対して放射線増感させることによるか又は腫瘍細胞を化学療法薬剤の細胞傷害性効果に対して化学増感させることによるかのいずれかによって細胞傷害性剤の致死性を増強するというPARP阻害剤の能力は、とりわけ、特許文献16〜18;非特許文献2及び9〜13;Griffin R.J.らによる特許文献19;及び、Helleday Tらによる特許文献14に報告されている。
【0013】
末梢神経障害の誘発は、化学治療薬剤による治療が制限される一般的な要因である(非特許文献14)。化学療法により誘発される神経障害は、多くの従来の化学療法(例、タキソール、ビンクリスチン、シスプラチン)及び新たな化学療法(例、ベルケイド、エポチロン)を用いた場合に生じる副作用である。結果として、用いた物質に応じて、純感覚性かつ痛みを伴う神経障害(シスプラチン、オキサリプラチン、カルボプラチンの使用)又は自律神経系の関与を伴う又は伴わない混合型の感覚運動神経障害(ビンクリスチン、タキソール、スラミンの使用)が生じ得る。神経毒性は、用いる薬剤の総蓄積用量及び種類に依存する。単一の薬剤を適用した後でも神経障害が起こる場合もある。症候から完全に回復しない場合もあり、機能を再開するのに長期間の再生が必要とされる。今日まで、化学療法により誘発される神経障害を確実に予防又は治療することができる薬品は存在しない。
【0014】
副作用が非常に小さく、かつ、イオン化放射線及び/又は化学療法薬剤が腫瘍細胞に及ぼす致死効果を増強する効果的かつ強力なPARP阻害剤が依然として必要とされている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】米国特許第5,032,617号
【特許文献2】米国特許第5,215,738号
【特許文献3】米国特許第5,041,653号
【特許文献4】米国特許第5,177,075号
【特許文献5】国際特許WO00/42040
【特許文献6】国際特許WO00/39070
【特許文献7】国際特許WO00/39104
【特許文献8】国際特許WO99/11623
【特許文献9】国際特許WO99/11628
【特許文献10】国際特許WO99/11622
【特許文献11】国際特許WO99/59975
【特許文献12】国際特許WO99/11644
【特許文献13】国際特許WO99/11945
【特許文献14】国際特許WO99/11649
【特許文献15】国際特許WO99/59973
【特許文献16】米国特許出願公開第2002/0028815号
【特許文献17】米国特許出願公開第2003/0134843号
【特許文献18】米国特許出願公開第2004/0067949号
【特許文献19】国際特許WO98/33802
【特許文献20】国際特許WO2005/012305
【非特許文献】
【0016】
【非特許文献1】Cristovaoら,“Effect of a Poly(ADP−Ribose) Polymerase Inhibitor on DNA Breakage and Cytotoxicity Induced by Hydrogen Peroxide and γ−Radiation,”Terato.,Carcino.,and Muta., 16:219−27(1996)
【非特許文献2】Plummer ERら,11(9)Clin.Cancer Res.3402(2005)
【非特許文献3】Kockら,45 J.Med.Chem.4961(2002)
【非特許文献4】Weltinら,“Effect of 6(5H)−Phenanthridinone, an Inhibitor of Poly(ADP−ribose) Polymerase,on Cultured Tumor Cells”, Oncol.Res.,6:9,399−403(1994)
【非特許文献5】Banasikら,“Specific Inhibitors of Poly(ADP−Ribose) Synthetase and Mono(ADP−Ribosyl)−Transferase”, J.Biol Chem.,267:3,1569−75(1992)
【非特許文献6】Banasikら,“Inhibitors and Activators of ADP−Ribosylation Reactions”,Molec.Cell.Biochem.,138,185−97(1994)
【非特許文献7】Milamら,“Inhibitors of Poly(Adenosine Diphosphate−Ribose)Synthesis; Effect on Other Metabolic Processes,”Science,223,589−91(1984)
【非特許文献8】Li and Zhang, Drugs 2001,4(7): 804−812(PharmaPress Ltd ISSN 1369−7056)
【非特許文献9】White AWら,14 Bioorg.&Med.Chem Letts.2433(2004)
【非特許文献10】Canon Koch SSら,45 J.Med.Chem.4961(2002)
【非特許文献11】Skalitsky DJら,46 J.Med.Chem.210(2003)
【非特許文献12】Farmer Hら,434 Nature 917(14 April 2005)
【非特許文献13】Tikhe JGら,47 J.Med.Chem.5467 (2004)
【非特許文献14】Quasthoff and Hartung,J.Neurology,249,9−17(2002)
【発明の概要】
【発明が解決しようとする課題】
【0017】
(発明の要旨)
本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法を提供する。
【課題を解決するための手段】
【0018】
また、本発明は、
以下のグループIの化合物から選択されるジアザベンゾ[デ]アントラセン−3−オン化合物:
【0019】
【化1】

【0020】
【化2】
【0021】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物
を提供する。
【0022】
また、本発明は、(i)治療上有効量のグループIの化合物及び(ii)医薬品に許容される担体を含む医薬組成物にも関する。
【0023】
本発明は、ポリ(ADP−リボース)ポリメラーゼ(PARP)のin vitro及び/又はin vivoでのポリメラーゼ活性を阻害する化合物、並びに:開示された化合物を含む組成物を提供する。
【0024】
本発明は、溶液、細胞、組織、器官又は器官系で、ポリ(ADP−リボース)ポリメラーゼ(PARP)のin vitro及び/又はin vivoでのポリメラーゼ活性を阻害、制限及び/又は制御する方法を提供する。一実施形態において、本発明は、ヒトなどの哺乳類において、局所的又は全身的でのいずれかでPARP活性を制限又は阻害する方法を提供する。
【0025】
本発明は、PARP産生の増加により悪化するか又はPARP産生の増加に関与する疾患、症候群及び/又は症状の治療及び/又は予防方法を提供する。これらの方法は、本発明の化合物を、上記治療又は予防を必要とするヒトの細胞、組織、器官又は器官系に適用又は投与することを含む。
【0026】
他の実施形態において、本発明の化合物及び組成物は、ヒトなどの動物において壊死又はアポトーシス、大脳虚血及び灌流損傷又は神経変性疾患による細胞の損傷又は細胞死を治療又は予防するのに用いることができる。
【0027】
他の実施形態において、本発明の化合物及び組成物は、細胞の寿命及び増殖能力を拡張するのに用いることができる、これに関連する疾患を治療又は予防するのに用いることができる。
【0028】
他の実施形態において、本発明は、ガンの影響を治療若しくは予防若しくは緩和する方法、並びに/又は、腫瘍細胞若しくは低酸素腫瘍細胞を放射線増感させて腫瘍細胞の放射線療法に対する感受性を高めることによって、放射線療法後の非常に致死性の高いDNA損傷から腫瘍細胞が回復するのを妨げる方法に用いることができる。この実施形態の方法は、腫瘍細胞を特異的かつ優先的に放射線増感することによって、腫瘍細胞の放射線療法に対する感受性を非腫瘍細胞よりも高めることに関する。
【0029】
また、本発明は、イオン化放射線及び/又は化学療法薬剤の腫瘍細胞への細胞傷害性効果を増強することにより、ガンの影響を治療、予防及び/又は緩和するためのグループIのジアザベンゾ[デ]アントラセン−3−オン化合物を提供する。
【0030】
一実施形態において、本発明は、
ガン及び/又は腫瘍を治療するための化学増感方法であって、
腫瘍又はガン細胞を、グループIの細胞傷害性効果を有するジアザベンゾ[デ]アントラセン−3−オン化合物と接触させること、及び、腫瘍又はガン細胞を、さらに抗ガン剤と接触させることを含む
ことを特徴とする方法
を提供する。
【0031】
本発明は、
哺乳類、特にヒトにおいてガンを治療するための化学増感方法であって、
哺乳類に、グループIから選択されるジアザベンゾ[デ]アントラセン−3−オン化合物を投与することを含む
ことを特徴とする方法
を提供する。
【0032】
本発明の一実施形態において、本発明の化学増感方法に用いられる化合物は以下である。
【0033】
【化3】
【0034】
他の実施形態において、本発明は、第1の用量の少なくとも1つのグループIの化合物が、それを必要とする患者に単回で又は繰り返して投与され、有効量の化学増感を提供できる時間の経過後、引き続いて、第2の用量の少なくとも1つの化学療法薬剤が、上記患者に単回又は繰り返して投与される、化学増感方法を提供する。
【0035】
他の実施形態において、本発明は、医薬品に許容される遊離塩基、塩、水和物、エステル、溶媒和物、プロドラッグ、代謝物、立体異性体及びこれらの混合物からなる群より選択される形態の化学増感ジアザベンゾ[デ]アントラセン−3−オン誘導体を含む医薬調製物を提供する。他の実施形態によれば、医薬調製物は、医薬品に許容される担体、及び、必要に応じて化学療法薬剤をさらに含む。そのような化学療法薬剤の例を以下に記載するが、これらに限定されない。
【0036】
本発明の他の実施形態によれば、化学増感化合物及び化学療法薬剤は、実質的に同時に投与される。
【0037】
本発明の他の実施形態によれば、化学療法薬剤は、テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、タキソイド、ダクチノマイシン、ダノルビシン(danorubicin)、4’−デオキシドキソルビシン、ブレオマイシン、ピルカマイシン(pilcamycin)、マイトマイシン、ネオマイシン及びゲンタマイシン、エトポシド、4−OHシクロフォスファミド、白金配位錯体、トポテカン、その治療上有効なアナログ及び誘導体、並びに、これらの混合物からなる群より選択される。好ましい態様によれば、化学療法薬剤はテモゾロマイドである。
【0038】
一実施形態によれば、本発明は、グループIから選択される化学増感有効量の少なくとも1つのジアザベンゾ[デ]アントラセン−3−オン化合物を含む医薬組成物を提供する。他の態様において、医薬組成物は以下を含む。
【0039】
【化4】
【発明の効果】
【0040】
本発明の化合物は、副作用が非常に小さく、かつ、イオン化放射線及び/又は化学療法薬剤が腫瘍細胞に及ぼす致死効果を増強する効果的かつ強力なPARP阻害剤としての効果を奏する。
【図面の簡単な説明】
【0041】
【図1】経口用化合物4i+TMZによる、脳内に黒色腫を有するマウスの生存率の増大
【図2】B6D2F1マウスにおけるB16皮下黒色腫に対する化合物4i+TMZの効果
【発明を実施するための形態】
【0042】
(発明の詳細な説明)
定義
本発明は、動物又は哺乳類において本明細書中に記載のいずれかの疾患又は障害を治療するための医薬の製造における本発明の化合物の使用に関する。
【0043】
本明細書中で用いる「アルキル」は、指定された数の炭素原子を含む分岐鎖又は非分岐鎖の飽和炭化水素鎖を意味する。例えば、C1−C6直鎖又は分岐鎖アルキル炭化水素鎖は、1〜6個の炭素原子を含み、特に示さない限り、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、tert−ブチル、n−ペンチル、n−ヘキシルなどの置換基が挙げられるが、これらに限定されない。
【0044】
「アルケニル」は、指定された数の炭素原子を含む分岐鎖又は非分岐鎖の不飽和炭化水素鎖を意味する。例えば、C2−C6直鎖又は分岐鎖アルケニル炭化水素鎖は、少なくとも1つの二重結合を有する2〜6個の炭素原子を有し、特に示さない限り、例えば、エテニル、プロペニル、イソプロペニル、ブテニル、イソブテニル、tert−ブテニル、n−ペンテニル、n−ヘキセニルなどの置換基が挙げられるが、これらに限定されない。
【0045】
「アルコキシ」は、基−OR(式中、Rは、本明細書中で定義するアルキルである)を意味する。また、Rは、1〜6個の炭素原子を含む分岐鎖又は非分岐鎖飽和炭化水素鎖であってもよい。
【0046】
本明細書中で接頭語として用いる「シクロ」は、閉環により特徴付けられる構造をいう。
【0047】
「ハロ」は、特に示さない限り、少なくとも1つのフルオロ、クロロ、ブロモ又はヨード部位を示す。
【0048】
「アミノ」化合物は、アミン(NH2)及び置換されたアミノを含む。
【0049】
「Ar」、「アリール」又は「ヘテロアリール」は、置換された又は未置換の、特に環状又は縮合環式環である部位を意味し、単環式、二環式又は三環式の炭素環式又はヘテロ環式環が挙げられ、上記環は、1〜5個の位置が未置換であるか又は次のもので置換されている:ハロ、ハロアルキル、ヒドロキシル、ニトロ、トリフルオロメチル、C1−C6直鎖又は分岐鎖アルキル、C2−C6直鎖又は分岐鎖アルケニル、C1−C6アルコキシ、C2−C6アルケニルオキシ、フェノキシ、ベンジルオキシ、アミノ、チオカルボニル、エステル、チオエステル、シアノ、イミノ、アルキルアミノ、アミノアルキル、スルフヒドリル、チオアルキル及びスルホニル:ここで、環の大きさはそれぞれ5〜8員である:ヘテロ環式環は、O、N又はSからなる群より選択される1〜4個のヘテロ原子を含む:芳香族又は第三級アルキルアミンは、必要に応じて、対応するN−オキシドに酸化される。ヘテロアリールは、他の環に結合していても、上記環のヘテロ原子及び/又は炭素原子を介して置換されていてもよい。アリール又はヘテロアリール部位には、フェニル、ベンジル、ナフチル、ピロリル、ピロリジニル、ピリジニル、ピリミジニル、プリニル、キノリル、イソキノリル、フリル、チオフェニル、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル及びチエニルが挙げられるが、これらに限定されない。
【0050】
「フェニル」には、必要に応じて以下からなる群より選択される置換基でモノ置換又は複数置換された全ての可能な異性体フェニル基を含む:アミノ、トリフルオロメチル、C1−C6直鎖又は分岐鎖アルキル、C2−C6直鎖又は分岐鎖アルケニル、カルボニル、チオカルボニル、エステル、チオエステル、アルコキシ、アルケノキシ、シアノ、ニトロ、イミノ、アルキルアミノ、アミノアルキル、スルフヒドリル、チオアルキル、スルホニル、ヒドロキシ、ハロ、ハロアルキル、NR2(式中、R2は、水素、(C1−C6)直鎖又は分岐鎖アルキル、(C3−C6)直鎖又は分岐鎖アルケニル又はアルキニル、及び、(C1−C4)架橋アルキル(上記架橋アルキルは、NR1の窒素から出発して、上記アルキル又はアルケニル鎖の炭素原子のうち1つで終結するヘテロ環式環を形成し、ここで、上記ヘテロ環式環は、必要に応じてAr基に縮合する)からなる群より選択される)。
【0051】
必要に応じて少なくとも1つのヘテロ原子を含むシクロアルキルには、C5又はC6環などの飽和C3−C8環が挙げられ、ここで、O、N、Sから選択される1〜4個のヘテロ原子が上記環の炭素原子を必要に応じて置換していてもよい。必要に応じて少なくとも1つのヘテロ原子を含むシクロアルキルは、上記のように、少なくとも1つの5員又は6員のアリール又はヘテロアリールで置換されても、あるいは、これに縮合していてもよい。ヘテロ原子を含むその他のシクロアルキルには、ピロリジニル、イミダゾリジニル、ピラゾリジニル、ピペリジニル、ピペラジニル、モルホリノ及びチオモルホリノが挙げられる。
【0052】
用語「神経変性疾患」には、アルツハイマー病、パーキンソン病及びハンチントン病が挙げられるが、これらに限定されない。
【0053】
用語「神経性傷害」は、神経組織に対する任意の損傷及びそれから生じる任意の障害又は死を意味する。神経性傷害の原因は、代謝性、毒性、神経毒性、医原性、熱的又は化学的なものがあり、虚血、低酸素症、脳血管傷害、外傷、手術、圧力、脳圧排(mass effect)、出血、放射線、血管攣縮、神経変性疾患、感染、パーキンソン病、筋萎縮性側索硬化症(ALS)、ミエリン化/脱ミエリン化プロセス、痙攣、認知障害、グルタミン酸異常及びそのいずれかの副作用が挙げられるが、これらに限定されない。
【0054】
用語「神経保護性」は、神経性傷害を低減、停止又は緩和する効果、及び、神経性傷害に罹患した神経組織を保護、蘇生又は復活させる効果を意味する。
【0055】
用語「神経変性の予防」は、神経変性疾患の予防、及び、神経変性疾患に既に罹患した又はその症候を有する患者におけるさらなる神経変性の予防を含む。
【0056】
用語「治療」は、以下を意味する:
(i)疾患、障害及び/又は症状に罹っているかもしれないが、未だ罹患していると診断されていない動物において疾患、障害又は症状が起こるのを予防すること;
(ii)疾患、障害又は症状を阻害する、すなわち、その進行を止めること;及び
(iii)疾患、障害又は症状を緩和する、すなわち、疾患、障害及び/又は症状の後退を生じさせること。
【0057】
用語「虚血損傷及び灌流損傷及び神経変性疾患から生じる神経系組織の損傷」には、血管卒中並びに全身及び局所的虚血において見られる神経毒性による損傷などの損傷が挙げられる。
【0058】
用語「虚血」は、動脈血流入の閉塞による局在化した組織貧血に関する。全身虚血は、脳全体への血流が一定の期間にわたって停止する条件下で起こり、心停止から生じ得るものなどである。局所虚血は、脳の一部が、その正常な血液供給が欠乏している条件下で起こり、大脳血管の血栓塞栓性閉塞、外傷性の頭部損傷、浮腫及び脳腫瘍から生じ得る。
【0059】
用語「心血管疾患」は、心筋梗塞、狭心症、血管又は心筋虚血、及び、当業者に知られているであろう関連症状に関し、心臓又は脈管構造の不全又は組織損傷が挙げられ、特に、PARP活性化に関する組織損傷が挙げられるが、これに限定されない。
【0060】
本明細書中で用いる用語「放射線増感剤」は、治療上有効な量で動物に投与して、電磁放射線に放射線増感させる細胞の感度を増大させるような、かつ/又は、電磁放射線で治療可能な疾患の治療を促進するような、低分子量分子などの分子であると定義される。電磁放射線で治療可能な疾患には、新生物疾患、良性及び悪性腫瘍、並びにガン細胞が挙げられる。本明細書中に列挙されていない他の疾患の電磁放射線治療もまた、本発明で意図される。本明細書中で用いる用語「電磁放射線」及び「放射線」には、10−20〜100メートルの波長を有する放射線が挙げられるが、これらに限定されない。本発明の好ましい実施形態は、γ線(10−20〜10−13m)、X線(10−11〜10−9m)、紫外線(10nm〜400nm)、可視光線(400nm〜700nm)、赤外線(700nm〜1.0mm)又はマイクロ波放射線(1mm〜30cm)の電磁放射線を用いる。
【0061】
本明細書中で用いる用語「化学増感剤」は、化学療法薬剤の抗腫瘍活性を増強する本発明の化合物の能力を意味する。そのような化学増感は、例えば、所定用量の化学療法薬剤の腫瘍増殖遅延若しくは停止効果を増大させる際に、又は、用量を減らしても抗腫瘍効力を維持できることによって化学療法薬剤の副作用特性を改善する際に、有用である。
【0062】
(本発明の医薬用途)
本発明は、核酵素ポリ(アデノシン5’−ジホスホ−リボース)ポリメラーゼ[「ポリ(ADP−リボース)ポリメラーゼ」又は「PARP」、ADPRT(NAD:タンパク質(ADP−リボシルトランスフェラーゼ(重合性)))、pADPRT(ポリ(ADP−リボース)トランスフェラーゼ)及びPARS(ポリ(ADP−リボース)合成酵素)とも呼ばれる]を阻害するための化合物、方法及び医薬組成物を提供する。さらに、本発明は、壊死又はアポトーシスに起因する細胞の損傷又は死;例えば、大脳虚血性卒中、頭部外傷又は脊椎損傷などの、虚血損傷及び再灌流損傷から生じる神経系組織の損傷;例えば、パーキンソン病又はアルツハイマー病及び多発性硬化症などの、神経学的障害及び神経変性疾患を予防及び/又は治療するための:血管卒中を予防又は治療するための:例えば、心筋梗塞などの心血管障害を治療又は予防するための:例えば、加齢性筋退化、AIDS及び他の免疫老化疾患、関節炎、アテローム性動脈硬化、毛細血管拡張性運動失調症、悪液質、ガン、複製老化に関与する骨格筋の変性疾患、糖尿病(真性糖尿病など)、炎症性腸障害(大腸炎及びクローン病など)、急性膵炎、粘膜炎、出血性ショック、内臓動脈閉塞ショック、多臓器不全(腎臓、肝臓、腎臓、肺、網膜、膵臓及び/又は骨格筋系のいずれかを含むものなど)、急性自己免疫甲状腺炎、筋ジストロフィー、骨関節炎、骨粗鬆症、慢性及び急性疼痛(神経障害性疼痛など)、腎不全、網膜虚血、敗血性ショック(内毒素性ショックなど)、局所的及び/又は遠隔内皮細胞不全(内部依存性弛緩応答及び接着分子の上方調節により認識されるものなど)、炎症及び皮膚加齢などの他の症状及び/又は障害を治療するための:例えば、酸化剤、炎症誘発性媒介物及び/又はサイトカインの産生における一般的な媒介物として、並びに、白血球の浸潤、カルシウムイオンの過負荷、リン脂質の過酸化、一酸化窒素代謝の損傷及び/又はATP産生の低減における一般的な媒介物として、細胞の寿命及び増殖能力を拡張するための;老化細胞の遺伝子発現を変えるための:あるいは、低酸素腫瘍細胞を放射線増感させるための、本発明のPARP阻害剤の使用方法を提供する。
【0063】
本発明の化合物は、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷を治療又は予防することができ;局所虚血、心筋梗塞及び灌流損傷に続くものを含む神経系又は心血管組織損傷を緩和することができ;PARP活性により引き起こされるか又は悪化する種々の疾患及び症状を治療することができ;細胞の寿命又は増殖能力を拡張又は増大させることができ;老化細胞の遺伝子発現を変えることができ;かつ、細胞を放射線増感させることができる。一般に、PARP活性の阻害は、細胞をエネルギー欠乏から保護し、神経系細胞の場合にはニューロンを不可逆的損傷から保護して、これによって神経を保護する。理論上、PARPの活性化は、フリーラジカル及びNOの産生だけでなく、おそらくまだ発見されていない他の細胞傷害(excitotoxic)メカニズムにおいて主要な役割を果たしている可能性があると考えられている。
【0064】
上記理由により、本発明は、さらに、治療上有効量の上記化合物を、PARP活性を阻害するのに十分な量で投与して、壊死又はアポトーシスに起因する細胞の損傷又は死を治療又は予防するための、NMDA傷害により媒介されない神経活性に影響を与えるための、NMDA傷害により媒介される神経活性に影響を与えるための、虚血損傷及び灌流損傷、神経学的障害及び神経変性疾患から生じる神経系組織の損傷を治療するための;血管卒中を予防又は治療するための;心血管障害を治療又は予防するための;加齢性筋退化、AIDS及び他の免疫老化疾患、関節炎、アテローム性動脈硬化、毛細血管拡張性運動失調症、悪液質、ガン、複製老化に関与する骨格筋の変性疾患、糖尿病、頭部外傷、免疫老化、炎症性腸障害(大腸炎及びクローン病など)、筋ジストロフィー、骨関節炎、骨粗鬆症、慢性及び/又は急性疼痛(神経障害性疼痛など)、腎不全、網膜虚血、敗血性ショック(内毒素性ショックなど)及び皮膚加齢などの他の症状及び/又は障害を治療するための;細胞の寿命及び増殖能力を拡張するための;老化細胞の遺伝子発現を変えるための;あるいは、低酸素腫瘍細胞を放射線増感させるための、方法に関する。また、本発明は、動物における疾患及び症状の治療であって、上記動物に、治療上有効量の上記で特定した化合物を投与することを含む治療に関する。
【0065】
本発明は、動物において神経学的障害を治療、予防又は阻害する方法であって、上記動物に、治療上有効量の上記で特定した化合物を投与することを含む、方法に関する。他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。さらなる他の実施形態は、脱髄性疾患及び神経学的障害が神経変性に関する場合である。他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の好ましい実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0066】
他の実施形態は、動物に有効量の本発明の化合物を投与することにより、上記動物においてPARP活性化に関連する狭心症、心筋梗塞、心血管虚血及び心血管組織損傷などの心血管疾患を治療、予防又は阻害する方法である。
【0067】
また、本発明は、動物において神経学的障害を治療、予防又は阻害するために、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷を治療、予防又は阻害するための、PARP活性を阻害するための本発明の化合物の使用を意図する。
【0068】
他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。
【0069】
他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。さらに他の実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0070】
また、本発明は、本明細書中に記載の動物において疾患及び障害のいずれかを治療するための医薬の製造における、本発明の化合物の使用を意図する。
【0071】
他の実施形態において、疾患又は障害は、神経学的障害である。
【0072】
他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。他の実施形態は、灌流損傷が血管卒中である場合である。他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。
【0073】
さらに他の実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0074】
本発明の他の実施形態において、急性網膜虚血又は急性血管卒中と診断されたヒトは、すぐに、断続的又は連続的な静脈内投与により、本発明の化合物を、単回用量で又は一組の分割用量の化合物として非経口的に投与される。この最初の治療の後、ヒトが示す神経学的症候に応じて、ヒトは、必要に応じて、同じ又は異なる本発明の化合物を、別の非経口投与用の形態で服用してもよい。本発明の化合物は、化合物を含む生体適合性の生分解性ポリマーマトリックスデリバリー系の埋め込みにより、又は、脳の梗塞領域に化合物を直接投与するための硬膜下ポンプにより、断続的又は連続的投与によって投与することができる。
【0075】
他の実施形態において、本発明は、例えば酸化剤の産生の一般的な媒介物として、炎症誘発性の媒介物及び/若しくはサイトカインとして、かつ/又は、白血球浸潤、カルシウムイオン過負荷、リン脂質過酸化、酸化窒素代謝の低下及び/若しくはATP産生の減少の一般的な媒介物として本発明の化合物を使用する際に、細胞の寿命及び増殖能力を拡張する方法を提供する。
【0076】
さらに、本発明の方法は、ガンを治療するのに、及び、腫瘍細胞を放射線増感させるのに用いることができる。用語「ガン」は、広く解釈される。本発明の化合物は、「抗ガン剤」であってよく、上記用語は、「抗腫瘍細胞増殖剤」及び「抗新生物剤」もまた包含する。例えば、本発明の方法は、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍などのガンにおける、ガンの治療、及び、腫瘍細胞の放射線増感において有用である。
【0077】
また、本発明の方法は、有効量のテモゾロマイド及び本発明の化合物を用いる哺乳類のガンの治療であり得る。ガンは、黒色腫、リンパ腫及び多形神経膠芽腫であってよい。
【0078】
放射線増感剤は、電磁放射線の傷害性効果に対するガン細胞の感度を増大させることが知られている。放射線増感剤の作用様式のいくつかのメカニズムが文献中に提唱されており、以下のものが挙げられる:低酸素細胞放射線増感剤(例、2−ニトロイミダゾール化合物、及びベンゾトリアジンジオキシド化合物)が、低酸素組織の再酸化を促進する及び/又は有害な酸素ラジカルの発生を触媒する;非低酸素細胞の放射線増感剤(例、ハロゲン化ピリミジン類)は、DNA塩基のアナログであってよく、ガン細胞のDNAに優先的に取り込まれて、放射線により誘発されるDNA分子の切断を促進する及び/又は正常なDNA修復メカニズムを阻止する;並びに、考えられる種々の他の作用メカニズムについて、疾患の治療における放射線増感剤であるとして仮定されている。
【0079】
多くのガン治療プロトコルは、現在、X線の電磁放射線照射により活性化される放射線増感剤を用いる。X線により活性化される放射線増感剤の例には以下のものが挙げられるが、これらに限定されない:メトロニダゾール、ミソニダゾール(misonidazole)、デスメチルミソニダゾール(desmethylmisonidazole)、ピモニダゾール(pimonidazole)、エタニダゾール(etanidazole)、ニモラゾール(nimorazole)、マイトマイシンC、RSU 1069、SR 4233、EO9、RB 6145、ニコチンアミド、5−ブロモデオキシウリジン(BUdR)、5−ヨードデオキシウリジン(IUdR)、ブロモデオキシシチジン、フルオロデオキシウリジン(FudR)、ヒドロキシウレア、シスプラチン、並びに、その治療上有効なアナログ及び誘導体。
【0080】
ガンの光学力学治療(PDT)は、増感剤の放射線活性化剤などの可視光を用いる。光学力学的放射線増感剤の例には以下が挙げられるが、これらに限定されない:ヘマトポルフィリン誘導体、フォトフリン、ベンゾポルフィリン誘導体、NPe6、スズエチオポルフィリン(tin etioporphyrin)SnET2、フェオボルビド(pheoborbide)−a、バクテリオクロロフィル−a、ナフタロシアニン、フタロシアニン、亜鉛フタロシアニン、並びに、その治療上有効なアナログ及び誘導体。
【0081】
放射線増感剤は、治療上有効量の1つ又はそれ以上の以下の他の化合物(これらに限定されない)と組み合わせて投与することができる:放射線増感剤の標的細胞への取り込みを促進する化合物;治療薬、栄養及び/又は酸素の標的細胞への流入を制御する化合物;さらに放射線が存在する場合又は存在しない場合に腫瘍に作用する化学療法薬剤;あるいは、ガン又は他の疾患を治療するための他の治療上有効な化合物。放射線増感剤と組み合わせて用いることができる治療剤の例には以下が挙げられるが、これらに限定されない:5−フルオロウラシル、ロイコボリン、5’−アミノ−5’デオキシチミジン、酸素、カーボジェン(carbogen)、赤血球輸血、パーフルオロカーボン(例、フルオゾル−DA(Fluosol−DA))、2,3−DPG、BW12C、カルシウムチャネル阻害剤、ペントキシフィリン、抗脈管新生化合物、ヒドララジン及びLBSO。放射線増感剤と組み合わせて用いることができる化学療法薬剤の例には以下が挙げられるが、これらに限定されない:アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、並びに、その治療上有効なアナログ及び誘導体。
【0082】
本発明は、化学療法により誘発される末梢神経障害の治療手段を提供する。本発明の1つの態様によれば、本発明の化合物は、少なくとも1つの化学療法薬剤の投与の前に又はそれと同時に投与されて、神経障害の症候の進行を予防するか、又は、上記症候の重篤性を緩和する。さらなる態様によれば、本発明の化合物は、少なくとも1つの化学療法薬剤の投与後に投与されて、患者の神経障害の症候を治療するか、又は上記症候の重篤性を緩和する。他の態様において、本発明は、哺乳類において腫瘍細胞の増殖を抑制、遅延又は停止させる方法であって、化学療法薬剤の投与を含み、さらに、グループIの化合物を、上記化学療法薬剤の抗腫瘍活性を増強するのに十分な量で投与することを含む、方法を提供する。
【0083】
他の実施形態において、本発明の化合物は、PARP阻害剤として作用し、他の化学療法薬剤の細胞傷害性効果を化学的に増強することにより、ガンを治療又は予防する。
【0084】
本発明は、腫瘍細胞へのイオン化放射線の細胞傷害性効果を増強することにより、ガンの影響を治療、予防及び/又は緩和するためのグループIの化合物、その誘導体、並びに、これらの化合物を含む組成物を提供する。
【0085】
他の実施形態において、本発明は、化学療法薬剤の腫瘍細胞への細胞傷害性効果を増強することによりガンの影響を治療、予防及び/又は緩和するための、本願明細書に記載の化合物、その誘導体、及び、これらの化合物を含む組成物を提供する。
【0086】
他の実施形態において、本発明の方法は、ガンを治療するのに、及び、腫瘍細胞を化学増感させるのに用いることができる。本明細書中で用いる用語「ガン」は、広く定義される。本発明の化合物は、「抗ガン剤」の効果を増強することができ、上記用語はまた、「抗腫瘍細胞増殖剤」、「化学療法薬剤」、「細胞増殖抑制剤」、「細胞傷害性剤」及び「抗新生物剤」も包含する。
【0087】
一実施形態において、本発明の方法は、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍などのガンにおける、ガンの治療、及び、腫瘍細胞の放射線増感又は化学増感に有用である。
【0088】
本発明は、腫瘍及び/又はガン細胞の治療のための化学増感方法であって、上記ガン細胞をグループIのジアザベンゾ[デ]アントラセン−3−オン化合物と接触させること、及び、上記ガン細胞を抗ガン剤にさらに接触させることを含む、方法を提供する。
【0089】
本発明の特定の実施形態には、グループIに示すジアザベンゾ[デ]アントラセン−3−オン化合物、並びに、その中性及び/又は塩の形態、並びに、適切な場合にはそのエナンチオマー及びラセミ体混合物が挙げられる。
【0090】
本発明の化合物は1つ又はそれ以上の不斉中心を有していてもよく、従って、立体異性体混合物(ラセミ体及び非ラセミ体)として、あるいは個別のエナンチオマー又はジアステレオマーとして製造することができる。個別の立体異性体は、光学活性な出発物質を用いることによって、又は、合成のいくつかの適切な段階で中間体のラセミ体若しくは非ラセミ体混合物を分割することによって、又は、グループIの化合物を分割することによって、得ることができる。個別の立体異性体並びに立体異性体の混合物(ラセミ体及び非ラセミ体)は、本発明の範囲に含まれることが理解される。
【0091】
本発明の化合物は、遊離塩基の形態で、医薬品に許容される塩、医薬品に許容される水和物、医薬品に許容されるエステル、医薬品に許容される溶媒和物、医薬品に許容されるプロドラッグ、医薬品に許容される代謝物の形態で、かつ、医薬品に許容される立体異性体の形態で有用である。これらの形態は全て本発明の範囲内である。
【0092】
「医薬品に許容される塩」、「水和物」、「エステル」又は「溶媒和物」は、所望の薬理学的活性を有するが生物学的な意味やそれ以外の意味で望ましくない、本発明の化合物の塩、水和物、エステル又は溶媒和物を意味する。有機酸は、塩、水和物、エステル又は溶媒和物、例えば、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、重硫酸塩、スルファミン酸塩、硫酸塩、ナフチル酸塩、酪酸塩、クエン酸塩、ショウノウ酸塩、ショウノウスルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、エタンスルホン酸、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、ヘミ硫酸ヘプタン酸塩、ヘキサン酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、トシル酸塩及びウンデカン酸塩などを製造するのに用いることができる。無機塩は、塩、水和物、エステル又は溶媒和物、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩及びチオシアン酸塩などの製造に用いることができる。
【0093】
適切な塩基塩、水和物、エステル又は溶媒和物の例には、アンモニアの水酸化物、炭酸塩及び重炭酸塩、ナトリウム塩、リチウム塩及びカリウム塩などのアルカリ金属塩、カルシウム塩及びマグネシウム塩などのアルカリ土類金属塩、アルミニウム塩及び亜鉛塩が挙げられる。
【0094】
また、塩、水和物、エステル又は溶媒和物は、有機塩基によっても形成することができる。本発明の化合物の医薬品に許容される塩基付加塩、水和物、エステル又は溶媒和物の形成に適切な有機塩基には、非毒性でかつ塩、水和物、エステル又は溶媒和物を形成するのに十分に強いものが挙げられる。例えば、そのような有機塩基の分類には、モノ−、ジ−及びトリアルキルアミン、例えば、メチルアミン、ジメチルアミン、トリエチルアミン及びジシクロヘキシルアミン;モノ−、ジ−又はトリヒドロキシアルキルアミン、例えば、モノ−、ジ−及びトリエタノールアミン;アミノ酸、例えば、アルギニン及びリジン;グアニジン;N−メチル−グルコサミン;N−メチル−グルカミン;L−グルタミン;N−メチル−ピペラジン;モルホリン;エチレンジアミン;N−ベンジル−フェネチルアミン;(トリヒドロキシメチル)アミノエタンなどを挙げることができる。例えば,“Pharmaceutical Salt”J.Pharm.Sci,66:1,1−19(1977)を参照されたい。従って、塩基性窒素含有基は、以下に挙げられる剤で4級化することができる:塩化、臭化及びヨウ化メチル、塩化、臭化及びヨウ化エチル、塩化、臭化及びヨウ化プロピル、並びに、塩化、臭化及びヨウ化ブチルなどの低級アルキルハロゲン化物;硫酸ジメチル、ジエチル、ブチル及びジアミルなどの硫酸ジアルキル;塩化、臭化及びヨウ化デシル、塩化、臭化及びヨウ化ラウリル、塩化、臭化及びヨウ化ミリスチル、並びに、塩化、臭化及びヨウ化ステアリルなどの長鎖ハロゲン化物;並びに、ベンジル及びフェネチルブロミドなどのアラルキルハロゲン化物。
【0095】
塩基性化合物の酸付加塩、水和物、エステル又は溶媒和物は、本発明の化合物の遊離塩基を水溶液又はアルコール水溶液に溶解するか、あるいは、適切な酸又は塩基を含む他の適切な溶媒に溶解し、溶液を蒸発させて塩を単離することにより製造することができる。あるいは、本発明の化合物の遊離塩基を酸と反応させることができ、また、酸基を有する本発明の化合物を塩基と反応させることができ、その結果、反応物が有機溶媒中に得られ、この場合、塩が直接分離するか、又は、溶液を濃縮することによって塩を得ることができる。
【0096】
「医薬品に許容されるプロドラッグ」は、生体内で変換された後でその薬理学的効果を発揮するような、本発明の化合物の誘導体を意味する。プロドラッグは、化学的安定性の改善、患者受容性及びコンプライアンスの改善、バイオアベイラビリティーの改善、作用期間の延長、器官選択性の改善、処方の改善(例、水溶性の増大)及び/又は副作用(例、毒性)の低減を目的として処方される。プロドラッグは、Burgers Medicinal Chemistry and Drug Chemistry,Fifth Ed,Vol.1,pp.172−178,949−982(1995)に記載のような上記分野で公知の方法を用いて、本発明の化合物から容易に製造することができる。例えば、本発明の化合物は、1つ又はそれ以上のヒドロキシ基又はカルボキシ基をエステルに転化することにより、プロドラッグに変換することができる。
【0097】
「医薬品に許容される代謝物」は、代謝性変換を受ける薬剤を意味する。体内に入った後、大半の薬剤は、これらの物理的特性及び生物的効果を変化させ得る化学反応の基質である。化合物の極性に概して影響を与えるこれらの代謝性変換は、薬物が体内で分散して体内から排泄される経路を変える。しかし、治療上の効果のために薬物の代謝が必要とされる場合もある。例えば、抗代謝物クラスの抗ガン剤は、ガン細胞に輸送された後で活性型に転化される必要がある。多くの薬剤は何らかの代謝性変換を受けるため、薬物代謝に関与する生化学的反応は多数かつ広範囲にわたる。薬物代謝の主要な部位は肝臓であるが、他の組織が関与する場合もある。
【0098】
本発明の医薬組成物
また、本発明は、(i)治療上有効量のジアザベンゾ[デ]アントラセン−3−オン誘導体の化合物、及び、(ii)医薬品に許容される担体を含む医薬組成物に関する。
【0099】
本発明の化合物の好ましい使用及び投与の実施形態に関する上記論述は、本発明の医薬組成物にも適用される。
【0100】
本明細書中で用いる用語「医薬品に許容される担体」には、任意の担体、希釈剤、賦形剤、懸濁剤、滑剤、アジュバント、ビヒクル、デリバリーシステム、乳化剤、崩壊剤、吸着剤、保存剤、界面活性剤、着色料、香料又は甘味料が挙げられる。
【0101】
これらの目的のため、本発明の組成物は、経口的に、非経口的に、吸入スプレー、吸収、吸着により、局所的に、経直腸的に、経鼻的に、経頬的に、経膣的に、脳室内に、医薬品に許容される従来の非毒性担体を含む投与調製物中におけるインプラントレザバーによって、又は、任意の他の簡便な投与形態により、投与することができる。本明細書中で用いる用語、非経口的には、皮下、静脈内、筋肉内、腹膜内、くも膜下腔内、脳室内、胸骨内及び頭蓋内注射又は灌流技術が挙げられる。
【0102】
非経口的に投与される場合、組成物は、単回投与の無菌の注射可能な形態(溶液、懸濁液又はエマルション)であり、これは、好ましくは、患者の血液と医薬品に許容される担体と等張性である。このような無菌の注射可能な形態の例には、無菌の注射可能な水性又は油性の懸濁液が挙げられる。これらの懸濁液は、上記分野で公知の技術に従って、適切な分散剤又は湿潤剤及び懸濁剤を用いて処方することができる。また、無菌の注射可能な形態は、非毒性の非経口的に受容され得る希釈剤又は溶液中の無菌の注射可能な溶液又は懸濁液、例えば、1,3−ブタンジオール溶液である。許容されるビヒクル及び溶液としては、水、生理食塩水、リンガー液、デキストロース溶液、等張性の塩化ナトリウム溶液及びハンクス溶液を用いることができる。さらに、無菌の不揮発性油が、溶液又は懸濁媒体として通常使用される。この目的のために、任意の無刺激性の不揮発性油を用いることができ、合成モノ又はジグリセリド、コーン油、綿実油、ピーナツ油及びゴマ油が挙げられる。オレイン酸エチル、ミリスチン酸イソプロピル及びオレイン酸などの脂肪酸、並びに、オリーブ油及びヒマシ油、特にこれらのポリオキシエチル化体を含むグリセリド誘導体は、注射可能薬剤の製造に有用である。また、これらの油性溶液又は懸濁液は、長鎖アルコール希釈剤又は分散剤を含んでいてよい。
【0103】
無菌の生理食塩水は担体として好ましく、化合物は、予期可能なあらゆる用途のための溶液とするのに十分に水溶性である場合が多い。担体は、溶解度、等張性及び化学的安定性を増強する物質、例えば、抗酸化剤、緩衝剤及び保存剤などの添加剤を少量含んでいてもよい。
【0104】
経鼻又は経頬投与に適切な調製物(自己推進性の粉体払い出し用調製物)は、約0.1%〜約5%w/wの、例えば1%w/wの活性成分を含んでいてもよい。従って、本発明のヒト用医薬用途の調製物は、活性成分を、医薬品に許容される担体と、必要に応じて他の治療上の成分と共に含む。
【0105】
経口的に投与される場合、組成物は、通常、上記分野で公知の従来の装置及び技術を用いて、錠剤、カシェ剤、散剤、顆粒、ビーズ、チュアブル錠剤、カプセル、液体、水性懸濁液又は溶液などの単回投与形態又は類似の投与形態に処方されるであろう。このような調製物は、代表的には、固体、半固体又は液体の担体を含む。担体としては、例えば、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アラビアゴム、リン酸カルシウム、鉱油、カカオバター、カカオ油、アルギン酸塩、トラガカント、ゼラチン、シロップ、メチルセルロース、ポリオキシエチレンソルビタンモノラウレート、ヒドロキシ安息香酸メチル、ヒドロキシ安息香酸プロピル、タルク、ステアリン酸マグネシウムなどが挙げられる。
【0106】
本発明の組成物は、好ましくは、グループIの化合物の単回又は分割用量を含むカプセル又は錠剤として投与される。好ましくは、組成物は、単回又は複数回投与用の無菌の溶液、懸濁液又はエマルションとして投与される。錠剤は、ラクトース及びコーンスターチなどの担体、並びに/又は、ステアリン酸マグネシウムなどの滑剤を含んでいてもよい。カプセルは、ラクトース及び乾燥コーンスターチを含む希釈剤を含んでもよい。
【0107】
錠剤は、活性成分を、必要に応じて1つ又はそれ以上の補助成分と共に圧縮又は成形することにより製造することができる。圧縮錠剤は、適切な機械中で、活性成分を、散剤又は顆粒などの自由に流動する形態で圧縮し、必要に応じて、結合剤、滑剤、不活性な希釈剤、界面活性剤又は分散剤と共に混合することにより製造できる。成形錠剤は、適切な機械で、粉末活性成分と、不活性な液状希釈剤で湿らせた適切な担体との混合物を成形することにより製造することができる。
【0108】
また、本発明の化合物は、坐剤の形態で経直腸的に投与することができる。これらの組成物は、室温では固体であるが直腸温では液体であり、直腸で融解して薬剤を放出できる適切な非刺激性の賦形剤と共に混合することによって製造することができる。このような材料には、カカオバター、ミツロウ及びポリエチレングリコールが挙げられる。
【0109】
また、本発明の組成物及び方法は、制御放出技術を適用することができる。このため、例えば、本発明の化合物を、一定期間で制御放出させる目的で疎水性ポリマーマトリックスに配合してもよい。次いで、本発明の組成物は、頻繁に投与しなおす必要なく、長期間にわたってPARP阻害剤の有効濃度を提供するために、固体インプラント、又は、外用パッチに成形することができる。このような制御放出フィルムは上記分野で周知である。特に好ましいものは、経皮デリバリーシステムである。本発明に用いることができる、この目的に一般的に用いられる他の例証されるポリマーには、外用又は内服で用いることができる非分解性エチレン−酢酸ビニルコポリマー、分解性乳酸−グリコール酸コポリマーが挙げることができる。ポリ(ヒドロキシエチルメタクリレート)又はポリ(ビニルアルコール)などの特定のヒドロゲルもまた有用であり得るが、上記したような他のポリマー放出システムより放出サイクルが短い。
【0110】
好ましい実施形態において、担体は、適当な時間放出特性及び放出動力学を有する固体生分解性ポリマー又は生分解性ポリマーの混合物である。また、本発明の組成物は、本発明の化合物の有効な濃度を、頻繁に投与しなおす必要なく長期間にわたって提供するのに適切な固体インプラントに成形することができる。本発明の組成物は、当業者に公知の任意の適切な様式で生分解性ポリマー又はポリマー混合物に配合することができ、生分解性ポリマーと共に均一なマトリックスを形成することができるか、又は、所定の方法でポリマー中に封入することができるか、又は、固体インプラントに成形することができる。
【0111】
一実施形態において、生分解性ポリマー又はポリマー混合物を使用して、本発明の医薬組成物を含む軟質の「デポー剤」を形成することができ、これは、流動性の液体として例えば注射により投与できるが、注射部位の周辺の局所で医薬組成物を維持することができるほど十分な粘性を保つ。このように形成されたデポー剤の分解時間は、選択したポリマー及びその分子量に依存して、数日から数年の間で変動し得る。ポリマー組成物を注射可能な形態で用いることにより、切開する必要がなくなる場合がある。いずれにしても、可撓性又は流動性のデリバリー用「デポー剤」は、周囲組織に外傷をほとんどつけずに、体内での空間の形状に適合するであろう。本発明の医薬組成物は治療上有効な量で用いられ、この量は、所望の放出特性、増感効果を発揮するために要する医薬組成物の濃度、及び、治療のために医薬組成物を放出するべき期間の長さに依存するであろう。
【0112】
本発明の化合物は、組成物中において治療上効果的な量で用いられる。組成物は、滅菌することができ、並びに/又は、保存剤、安定化剤、ウェル化剤若しくは乳化剤、溶液促進剤、浸透圧を調整するための塩及び/若しくは緩衝剤などのアジュバントを含んでいてよい。さらに、これらはまた、本明細書中に記載の特定の化学療法薬剤などの他の治療上有益な物質を含んでいてよいが、これらに限定されない。組成物は、従来の混合、顆粒化又はコーティング方法に従って製造され、約0.1〜75重量%,好ましくは1〜50重量%の本発明の化合物を含む。
【0113】
中枢神経系標的として有効な治療であるためには、本発明の化合物は、末梢から投与された場合に血液−脳関門を容易に通過すべきである。血液−脳関門を通過できない化合物は、脳室経路又は脳への投与に適切な他のデリバリーシステムにより、効果的に投与することができる。
【0114】
医薬用途には、治療上の効果を達成するために必要な活性成分の量は、特定の化合物、投与の経路、治療される哺乳類及び治療される特定の障害又は疾患により変動するであろう。本明細書中の上記に記載の症状のいずれかに罹患した又はおそらく罹患しているであろう哺乳類についての本発明の化合物又はその薬理学的に許容される塩の適切な全身用量は、活性成分化合物の約0.1mg/kg〜約100mg/kgであり、最も好ましい用量は約1〜約10mg/kgである。
【0115】
しかし、任意の特定の患者についての特定の用量レベルは、用いる特定の化合物の活性、年齢、体重、全身の健康状態、性別、食事、投与時間、排泄速度、薬剤の組み合わせ、及び、治療される特定の疾患の重篤性、及び、投与形態を含む種々の因子に依存するであろうことが理解される。
【0116】
通常の技術を有する医師又は獣医師は、治療薬が投与される症状の治療の予防又は治療用の化合物の有効量を、容易に決定しかつ処方するであろうことが理解される。例えば、その作業中において、医師又は獣医師は、適切であると考えられる場合には、静脈内ボーラスを用い、続いて、静脈内灌流を実施することができる、及び、非経口的又は経口的に繰り返し投与することができる。活性成分は単独で投与することもできるが、活性成分を調製物として提供することが好ましい。
【0117】
本発明の組成物を含む投与形態を製造する場合、化合物は、ゼラチン、α化デンプンなどの結合剤;例えば、水素添加植物油、ステアリン酸などの滑剤;ラクトース、マンノース及びスクロースなどの希釈剤;カルボキシメチルセルロース及びナトリウムデンプングリコレートなどの崩壊剤;ポビドン、ポリビニルアルコールなどの懸濁剤;二酸化ケイ素などの吸収剤;メチルパラベン、プロピルパラベン及び安息香酸ナトリウムなどの保存剤;ラウリルスルホン酸ナトリウム、ポリソルベート80などの界面活性剤;F.D.&C.染料及びレーキなどの着色料;着香料;及び甘味料などの従来の賦形剤と混合することもできる。
【0118】
本発明は、本明細書中に記載の動物におけるいずれかの疾患又は障害を治療するための医薬の製造における、グループIの化合物の使用に関する。一実施形態において、本発明の化合物は、ガンを治療するのに使用される。好ましい実施形態において、本発明の化合物は、イオン化放射線の細胞傷害性効果を増強するのに用いられる。上記実施形態において、本発明の化合物は、放射線増感剤として作用する。別の好ましい実施形態において、本発明の化合物は、化学療法薬剤の細胞傷害性効果を増強するのに用いられる。上記実施形態において、本発明の化合物は、化学増感剤として作用する。
【0119】
DNAを損傷するよう作用する医薬品に許容される化学療法薬剤はいずれも、本発明の化学療法薬剤として適切である。特に、本発明は、化学治療上有効量の少なくとも1つの以下の化学療法薬剤の使用を意図するが、これに限定されない:テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、その治療上有効なアナログ及び誘導体、並びに、これらの混合物。好ましい態様によれば、化学療法薬剤はテモゾロマイドである。
【0120】
本明細書中に含まれる開示は、本発明の化合物及び組成物が、腫瘍及び/又はガン細胞の化学療法薬剤に対する放射線増感及び/又は化学増感などにより、ガンを治療及び/又は予防するのに有用であることを示す。
【0121】
本発明のジアザベンゾ[デ]アントラセン−3−オン化合物は、米国第60/644,584号(その全体は、全ての目的のために参照として援用される)に開示された出発物質及び方法を用いて合成することができる。
【0122】
本発明の方法及び医薬組成物に用いるPARP阻害剤のうちのいくつかは、刊行物に図示された一般的な合成経路および例示を用いて、有機化学の標準的な技術により、容易に製造することができる。例えば、Wuら, “The Protective Effect of GPI 18078, a Novel Water Soluble Poly(ADP−Ribose) Polymerase Inhibitor in Myocardial Ischemia−Reprefusion Injury, Experimental Biology,“FASEB, April 11−15 (2003); Wuら,“Myocardial Protection and Anti−Inflammatory Effect of GPI 15427, a Novel Water Soluble Poly(ADP−Ribose) Polymerase Inhibitor: Comparison with GPI 6150, Experimental Biology,” FASEB, April 11−15 (2003); Kalishら, “Design, Synthesis and SAR of PARP−1 Inhibitors, ISMC Meeting, Barcelona,”September 4, 2002; Xuら,“Design and Synthesis of Novel Potent Poly(ADP−Ribose) Polymerase (PARP) Inhibitors, 224th ACS National Meeting,” Boston, August 18−23 (2002); Williamsら, “Intravenous Delivery of GPI 15427/C and GPI 16539/C, Potent Water−Soluble PARP Inhibitors, Reduces Infarct Volume Following Permanent and Transient Focal Cerebral Ischemia, Soceity for Neuroscience,” Orlando FL, October (2002); Tentori L,ら,“Systemic administration of the PARP−1 inhibitor GPI 15427 increases the anti−tumor activity of temozolomide against metastatic melanoma,” Medical Science Monitor, Vol. 9, supplement 1, 34 (2003); Tentoriら, “Poly(ADP−Ribose) Polymerase Inhibitor to Increase Temozolomide Efficacy Against Melanoma, Glioma and Lymphoma at the CNS Site,” AACR poster, April (2003); Sutoら, “Dihydroiso−quinolinones: The Design and Synthesis of a New Series of Potent Inhibitors of Poly(ADP−ribose) Polymerase,” Anticancer Drug Des., 6:107−17 (1991);並びに、米国特許第6,348,475号、同第6,545,011号、米国再発行特許第36,397号、米国特許第6,380,211号、同第6,235,748号、同第6,121,278号、同第6,197,785号、同第6,380,193号、同第6,346,536号、同第6,514,983号、同第6,306,889号、同第6,387,902号、同第6,201,020号及び同第6,291,425号並びに米国特許出願第10/853,714号(上記特許、特許出願及び刊行物の全ての内容は、これらが本明細書中に全文が記載されているのと同様に、本明細書中で援用される)を参照されたい。
【0123】
本発明の化合物は、以下のスキーム1に例示される従来の様式で製造することができる。出発誘導体は、化学文献で公知であり、当業者に公知のプロセスで入手可能である。
【0124】
【化5】
【0125】
一般的手順A:7−ブロモメチル−9−オキソキサンテン−l−カルボン酸メチルエステルの製造
【0126】
N−ブロモスクシンイミド、臭素及び臭化錯体、例えば臭化ピリジニウムを含むブロモ化剤を用いることによって、7−メチル−9−オキソキサンテン−1−カルボン酸メチルエステル(1)を7−ブロモメチル−9−オキソキサンテン−l−カルボン酸メチルエステル(2)に転化することができる。適切な溶液には、塩素化炭化水素、極性非プロトン性溶液及び種々のエーテルが挙げられるが、これらに限定されない。温度は、一般的に0〜100℃の間であり、50〜70℃の範囲が好ましい。
【0127】
実施例1:
過酸化ベンゾイル(10g、0.041mol)を含む還流四塩化炭素(10L)中に化合物5(400g、1.49mol)を含む溶液を撹拌している中に、NBS(292g、1.64mol)を、数回に分けて45分間かけて添加した。得られた混合物を12時間還流し、次に一晩かけて室温まで冷却した。沈殿したものをろ過し、得られた固形物を水(1.2L)で十分に洗浄し、乾燥させ、322gの化合物6を白色固体で得た(62%)。
【0128】
実施例2:
四塩化炭素(400mL)中に化合物1(1.97g、7.3mmol、1.00eq)を含む溶液に、N−ブロモコハク酸イミド(1.44g、8.1mmol、1.10eq)及び過酸化ベンゾイル(45mg、0.2mmol、触媒)の触媒量を添加した。反応混合物を加熱、6時間還流し、次に、室温まで冷却した。得られた白色沈殿物を真空ろ過により単離した。残った溶媒は除去し、ろ過固形物は酢酸エチル及びヘキサンで2度再結晶化し、白色固体2(1.15g,45%)を得た。
1H−NMR(400MHz,CDCl3)8.27(d,J=2.5Hz,1H),7.72−7.80(m,2H),7.57(dd,J=8.5及び1.1Hz,1H),7.48(d,J=8.5Hz,1H),7.33(dd,J=7.0及び1.1Hz,1H),4.57(s,2H),4.00(s,3H).13C−NMR(400MHz,CDCl3)31.97,53.06,118.63,118.71,119.63,121.56,122.81,126.83,134.15,134.18,134.50,135.95,155.31,155.87,169.72,175.48.
【0129】
一般的手順B:置換9−オキソキサンテン−1−カルボン酸メチルエステルの調製
化合物2中の第一臭化物(primary bromide)は、第一級及び第二級アミンを含む求核剤によって容易に置換され、これは炭酸カリウムなどの非反応性塩基種の存在下で好ましく実施される。これらの変換にはジメチルホルムアミドやアセトニトリルなどの極性及び非プロトン性のものが最適であるが、その他の溶媒中でも上記反応を実施することができる。温度範囲は0〜100℃であってよく、50〜80℃が好ましいであろう。
【0130】
実施例1:
化合物2(3.47g,10.0mmol,1.00eq)のジメチルホルムアミド(100mL)溶液に、炭酸カリウム(13.82g,100.0mmol,10.00eq)及び第二級アミン(10mmol,1eq)を添加する。反応混合物を70℃で6時間加熱し、次に室温まで冷却する。反応混合物に水(100mL)を加え、続いて酢酸エチル(200mL)を加える。有機層を回収し、水で洗浄し、続いて塩水で洗浄し、次に硫酸ナトリウム又は硫酸マグネシウムで乾燥させる。溶媒を真空において除去し、残渣を、溶離液として酢酸エチル及びヘキサンを使用してカラムクロマトグラフィーで精製し、生成物3を50〜90%の収率で得る。
【0131】
実施例2:
化合物2(1.53g,4.4mmol,1.00eq)のアセトニトリル(50mL)溶液に、炭酸カリウム(1.2g,8.7mmol,2.00eq)及び1−メチルピペラジン(0.51mL,4.6mmol,1.05eq)を添加した。次に反応混合物を加熱し一晩還流した。室温まで冷却した後、固体をろ過にて除去し、有機層は蒸発させて、油状残渣を得た。この物質を酢酸エチル(150ml)に溶解し、1N HCl(150ml)で抽出した。有機層は廃棄し、水層のpHを6N水酸化ナトリウムで9より大きくなるよう調整した。次に、生成物を酢酸エチル(100ml)で2回に分けて抽出し、次いでこれらを合わせて、水及び塩水で連続して洗浄し、硫酸マグネシウムで乾燥させた。全ての溶媒を真空において除去し、白色固体の3aを得た(0.95g,59%)。
3aの1H−NMR(400MHz,CDCl3):8.18(d,J=2.5Hz,1H),7.71−7.75(m,2H),7.56(dd,J=8.5及び1.1Hz,1H),7.45(d,J=8.5Hz,1H),7.32(dd,J=7.0及び1.1Hz,1H),4.04(s,3H),3.58(s,2H),2.45(br,8H),2.27(s,3H).
【0132】
一般的手順C:
10−アミノメチル−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン誘導体の調製
ベンゾピラノ[4,3,2−デ]フタラジン環を形成する化合物3におけるケトン及びメチルエステルの環化反応はヒドラジンを使用して実施することができ、10−アミノメチル−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン誘導体4を高収率で得る。溶媒としてはエタノールが好ましいが、反応は本溶媒に限定されない。温度は0〜120℃の範囲であってよく、70〜90℃が最も望ましい。
【0133】
実施例1:
化合物3(5mmol)の無水エタノール(10ml)溶液に、無水ヒドラジンのエタノール(1ml)溶液を室温で滴下する。滴下終了後、溶液を加熱し、一晩還流する。一度室温まで冷却し、氷冷水(100mL)を加え、白色固体を沈殿させる。固体を真空ろ過で回収し、水及びエタノールで連続して洗浄し、真空において乾燥させ、白色固体4(収率40〜85%)を得る。
【0134】
実施例2:
7−(1,4−ジオキサ−8−アザ−スピロ[4.5]デカ−8−イルメチル)−9−オキソ−9Η−キサンテン−1−カルボン酸メチルエステル、3n(1.6g,3.91mmol,1eq)のメタノール(55ml)溶液を80℃まで加熱し、撹拌して全物質を溶解させた。これに、ヒドラジン一水和物(20ml,過剰量)を10分かけて滴下した。反応混合物を加熱し還流を一晩行い、重い白色沈殿物を形成させた。この溶液を室温まで冷却し、生成物を真空ろ過で単離した。少量の水、エタノール及びペンタンで連続して洗浄し、真空において乾燥して4nを高収率で得た(1.4g,92%)。
1H−NMR(DMSO−d6,300MHz):1.62(t,J=5.0,4H),2.40−2.50(m,4H),3.55(s,2H),3.85(s,4H),7.34(d,J=9.4Hz,1H),7.47(d,J=7.5Hz,1H),7.67−7.70(m,1H),7.86−7.92(m,2H),7.98(s,1H),12.62(s,1H).
【0135】
化合物4a:10−(4−イソプロピル−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0136】
【化6】
【0137】
一般的手順B及びCに従って、化合物2及び1−プロピル−ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4aを得た。
MS(ES+):377.1H−NMR(CDCl3,300MHz):0.90−1.00(m,6H),2.25−2.50(m,8H),2.55−2.60(m,1H),3.34(s,2H),7.35(d,1H),7.46(d,1H),7.68(dd,1H),7.80−7.95(m,2H),7.95−8.05(m,1H),12.63(s,1H).Anal.Calcd.for C22H24N4O2:C,70.19;H,6.43;N,14.88.Found:C,70.09;H,6.51;N,14.77.
【0138】
化合物4b:10−[4−(2−メトキシ−エチル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2−ジアゾ−ベンゾ[デ]アントラセン−3−オン
【0139】
【化7】
【0140】
一般的手順B及びCに従って、化合物2及び1−(2−メトキシエチル)ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4bを得た。
MS(ES+):393.1H−NMR(DMSO−d6,300MHz):2.30−2.49(m,10H),3.22(s,3H),3.30−3.45(m,2H),3.50(m,2H),7.30−7.35(m,1H),7.40−7.48(m,1H),7.65−7.70(m,1H),7.85−7.95(m,2H),7.95−8.05(m,1H),12.63(s,1H).Anal.Calcd.forC22H24N4O3:C,67.33;H,6.16;N,14.28.Found:C,67.35;H,6.16;N,14.45.
【0141】
化合物4c:10−(4−ピリミジン−2−イル−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0142】
【化8】
【0143】
一般的手順B及びCに従って、化合物2及び1−(2−ピリミジル)ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4cを得た。
MS(ES+):413.1H−NMR(DMSO−d6,300MHz):3.28−3.34(m,4H),3.60(s,2H),3.70−3.78(m,4H),6.60−6.62(m,1H),7.38−7.40(m,1H),7.50−7.60(m,1H),7.70−7.74(m,1H),7.80−7.95(m,2H),8.05−8.10(m,1H),8.30−8.40(m,2H),12.64(s,1H).Anal.Calcd.for C23H20N6O2:C,66.98;H,4.89;N,20.38.Found:C,67.03;H,4.88;N,20.15.
【0144】
化合物4d:10−(3−オキソ−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0145】
【化9】
【0146】
一般的手順B及びCに従って、化合物2及びピペラジン−2−オンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4dを得た。
MS(ES+):349;1H−NMR(DMSO−d6,300MHz):2.58−2.62(m,2H),2.94(s,2H),3.16−3.20(m,2H),3.63(s,2H),7.30−7.35(m,1H),7.40−7.48(m,1H),7.65−7.70(m,1H),7.85−7.95(m,2H),7.95−8.05(m,1H).
【0147】
化合物4e:10−[4−(2−ピロリジン−1−イル−エチル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2,ジアザ−ベンゾ[デ]アントラセン−3−オン
【0148】
【化10】
【0149】
一般的手順B及びCに従って、化合物2及び1−(2−ピロリジン−1−イル−エチル)−ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4eを得た。
1H−NMR(DMSO−d6,300MHz):1.64(m,4H),2.30−2.55(m,16H),3.52(s,2H),7.30−7.40(m,1H),7.45−7.50(m,1H),7.70−7.75(m,1H),7.80−7.90(m,2H),8.00−8.05(m,1H).Anal.Calcd.for C25H29N5O2−(0.7H2O):C,67.61;H,6.90;N,15.77;Found:C,67.25;H,6.81;N,15.67.
【0150】
化合物4f:10−[4−(3−ジメチルアミノ−プロピル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0151】
【化11】
【0152】
一般的手順B及びCに従って、化合物2及びジメチル−(3−ピペラジン−1−イル−プロピル)−アミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4fを得た。
1H−NMR(DMSO−d6,300MHz):1.45−1.55(m,2H),2.09(s,6H),2.10−2.40(m,12H),3.52(s,2H),7.30−7.40(9m,1H),7.40−7.50(m,1H),7.70−7.75(m,1H),7.80−7.95(m,2H),8.01(s,1H).
【0153】
化合物4g:10−{[(2−ジメチルアミノ−エチル)−エチル−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0154】
【化12】
【0155】
一般的手順B及びCに従って、化合物2及びN,N−ジメチル−N’−エチル−エタン−1,2−ジアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4gを得た。
1H−NMR(DMSO−d6,300MHz):0.90(t,J=7.2Hz,6H),1.00(t,J=6.8Hz,3H),2.41(dd,J=14.3及び7.2Hz,4H),2.45−2.55(m,6H),3.62(s,2H),7.35(d,J=8.6Hz,1H),7.49(dd,J=8.3及び2.3Hz,1H),7.69(dd,J=7.1及び2.4Hz,1H),7.86−7.93(m,2H),8.03(d,J=2.3Hz,1H)
【0156】
化合物4h:10−{[(2−ジエチルアミノ−エチル)−メチル−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0157】
【化13】
【0158】
一般的手順B及びCに従って、化合物2及びN,N−ジエチル−N’−メチル−エタン−1,2−ジアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4hを得た。
1H−NMR(DMSO−d6,300MHz):12.63(s,1H),8.00(d,J=1.9Hz,1H),7.91−7.80(m,2H),7.70(dd,J=7.1及び2.0Hz,1H),7.49(dd,J=8.6及び2.0Hz,1H),7.36(d,J=8.5Hz,1H),3.55(s,2H),3.88(m,4H),2.47(q,J=7.0Hz,4H),2.17(s,3H),0.93(t,J=7.0Hz,6H).Anal.Calcd.for C22H26N4O2:C,69.82;H,6.92;N,14.80;Found:C,69.56;H,6.95;N,14.60.
【0159】
化合物4i:10−(4−ヒドロキシ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0160】
【化14】
【0161】
一般的手順B及びCに従って、化合物2及びピペリジン−4−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4iを得た。
1H−NMR(DMSO−d6,300MHz):12.60(s,1H),7.96(d,J=1.9Hz,1H),7.91−7.84(m,2H),7.66(dd,J=6.9及び2.3Hz,1H),7.43(dd,J=8.6及び2.1Hz,1H),7.32(d,J=8.6Hz,1H),4.54(d,J=4.2Hz,1H),3.48(s,2H),3.46(m,1H),2.65(m,2H),2.05(m,2H),1.68(m,2H),1.40(m,2H).Anal.Calcd.for C20H19N3O3:C,68.75;H,5.48;N,12.03;Found:C,68.66;H,5.48;N,12.13.
【0162】
化合物4j:10−{[エチル−(2−ヒドロキシ−エチル)−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0163】
【化15】
【0164】
一般的手順B及びCに従って、化合物2及び2−エチルアミノ−エタノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4jを得た。
1H−NMR(DMSO−d6,300MHz):1.00(t,J=6.4Hz,3H),2.45−2.55(m,4H),3.49(dd,J=12.0及び5.5Hz,2H),3.64(s,2H),4.39(t,J=5.1Hz,1H),7.35(d,J=8.5Hz,1H),7.50(dd,J=8.8及び6.5Hz,1H),7.69(dd,J=6.5及び4.3Hz,1H),7.86−7.91(m,2H),8.02(d,J=2.0,1H).
【0165】
化合物4k:10−[(ジイソプロピルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0166】
【化16】
【0167】
一般的手順B及びCに従って、化合物2及びジイソプロピルアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4kを得た。
1H−NMR(DMSO−d6,300MHz):1.01(d,J=6.3Hz,12H),2.93−3.04(m,2H),3.66(s,2H),7.34(d,J=8.4Hz,1H),7.51(dd,J=9.2及び1.9Hz,1H),7.69(dd,J=6.5及び2.7Hz,1H),7.86−7.91(m,2H),8.09(d,J=1.9Hz,1H).
【0168】
化合物4l:10−(3−ヒドロキシ−ピロリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0169】
【化17】
【0170】
一般的手順B及びCに従って、化合物2及ピロリジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4lを得た。
1H−NMR(DMSO−d6,300MHz):1.50−1.60(m,1H),1.95−2.05(m,1H),2.31−2.35(m,1H),2.55−2.65(m,1H),2.68−2.74(m,1H),3.62(d,J=4.2Hz,2H),4.18−4.25(m,1H),4.72(d,J=4.5Hz,1H),7.35(d,J=8.6Hz,1H),7.46−7.49(m,1H),7.70(dd,J=6.9及び2.2Hz,1H),7.87−7.91(m,2H),8.00(d,J=1.6Hz,1H).Anal.Calcd.for C19H17N3O3:C,68.05;H,5.11;N,12.53;Found:C,67.80;H,5.11;N,12.49.
【0171】
化合物4m:10−[4−(2−ヒドロキシ−エチル)−ピペリジン−1−イルメチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0172】
【化18】
【0173】
一般的手順B及びCに従って、化合物2及び2−ピペリジン−4−イル−エタノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4mを得た。
1H−NMR(DMSO−d6,300MHz):1.12−1.17(m,2H),1.34−1.38(m,3H),1.61(d,J=12Hz,1H),1.92(t,J=11Hz,2H),2.80(d,J=10.6Hz,2H),3.42−3.48(m,4H),4.34(t,J=5.2,1H),7.33(d,J=8.8Hz,1H),7.43−7.46(m,1H),7.68(dd,J=6.9及び2.5Hz,1H),7.86−7.98(m,3H).Anal.Calcd.for C22H23N3O3:C,70.01;H,6.14;N,11.13;Found:C,69.82;H,6.12;N,11.08.
【0174】
化合物4n:10−(1,4−ジオキサ−8−アザ−スピロ[4.5]デカ−8−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0175】
【化19】
【0176】
一般的手順B及びCに従って、化合物2及び1,4−ジオキサ−8−アザ−スピロ[4.5]デカンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4nを得た。
1H−NMR(DMSO−d6,300MHz):1.62(t,J=5.0,4H),2.40−2.50(m,4H),3.55(s,2H),3.85(s,4H),7.34(d,J=9.4Hz,1H),7.47(d,J=7.5Hz,1H),7.67−7.70(m,1H),7.86−7.92(m,2H),7.98(s,1H),12.62(s,1H).Anal.Calcd.for C22H21N3O4:C,67.41;H,5.43;N,10.98;Found:C,67.15;H,5.30;N,11.03.
【0177】
化合物4o:10−(3−ヒドロキシ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0178】
【化20】
【0179】
一般的手順B及びCに従って、化合物2及びピペリジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4oを得た。
1H−NMR(DMSO−d6,300MHz):1.00−1.12(m,1H),1.30−1.50(m,1H),1.52−1.95(m,4H),2.66−2.83(m,2H),3.45−3.60(m,3H),4.60(d,J=5.0Hz,1H),7.34−7.48(m,2H),7.68−7.70(dd,J=6.8及び2.2Hz,1H),7.87−8.00(m,3H).Anal.Calcd.for C20H19N3O3:C,68.75;H,5.48;N,12.03;Found:C,68.85;H,5.48;N,12.10.
【0180】
化合物4p:10−(3−ヒドロキシ−アゼチジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0181】
【化21】
【0182】
一般的手順B及びCに従って、化合物2及びアゼチジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4pを得た。
1H−NMR(DMSO−d6,300MHz):2.79(t,J=6.9Hz,2H),3.51(t,J=6.3Hz,2H),3.61(s,2H),4.23(dd,J=12.9及び6.3Hz,1H),5.33(d,J=6.5Hz,1H),7.33(d,J=8.4Hz,1H),7.71−7.44(m,1H),7.68(dd,J=6.8,2.6,1H),7.86−7.96(m,3H),12.62(bs,1H).Anal.Calcd.for C18H15N3O3−(0.5H2O):C,65.45;H,4.88;N,12.72;Found:C,65.06;H,4.60;N,13.03.
【0183】
化合物4q:10−[(2−モルフォリン−4−イル−エチルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0184】
【化22】
【0185】
一般的手順B及びCに従って、化合物2及び2−モルフォリン−4−イル−エチルアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4qを得た。
1H−NMR(DMSO−d6,300MHz):2.16(bs,1H),2.34(bs,4H),2.40(t,J=6.6Hz,2H),2.60(t,J=6.1Hz,2H),3.56(t,J=4.6Hz,4H),3.76(s,2H),7.33(d,J=8.1Hz,1H),7.48(dd,J=8.3及び1.8Hz,1H),7.68(dd,J=6.6及び2.3,1H),7.88−7.93(m,2H),8.01(d,J=1.5,1H),12.63(bs,1H).Anal.Calcd.for C21H22N4O3−(0.75H2O):C,64.35;H,6.04;N,14.29;Found:C,64.35;H,5.91;N,14.26.
【0186】
化合物4r:10−[(4−ヒドロキシ−シクロヘキシルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0187】
【化23】
【0188】
一般的手順B及びCに従って、化合物2及びtrans−4−アミノ−シクロヘキサノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4rを得た。
1H−NMR(DMSO−d6,300MHz):1.03−1.16(m,4H),1.75−1.90(m,4H),2.30−2.40(m,1H),3.75(s,2H),4.48(d,J=3.4,1H),7.31(d,J=8.1Hz,1H),7.48(dd,J=8.5及び2.1Hz,1H),7.67(dd,J=6.8及び2.4Hz,1H,)7.85−7.92(m,2H),8.02(d,J=1.9Hz,1H).Anal.Calcd.for C21H21N3O3−(0.5H2O)・(0.05N2H4):C,67.44;H,5.98;N,11.61;Found:C,67.56;H,5.74;N,11.68.
【0189】
化合物4s:10−(4−オキソ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0190】
【化24】
【0191】
化合物4n(100mg,0.24mmol)を酢酸(3ml)中で撹拌し、そこへ濃塩酸(0.6ml,過剰量)を室温において添加した。反応物を90℃で1時間加熱し、次に室温まで冷却した。生成物を1N NaOHでpH11〜12に塩基性化した後、酢酸エチルで抽出し単離した。有機分は硫酸マグネシウムで乾燥させ、真空において濃縮して白色固体4sを得た(60mg,71%)。
1H−NMR(DMSO−d6,300MHz):2.37(t,J=2.4Hz,4H),2.73(t,J=2.7Hz,4H),3.69(s,2H),7.38(d,J=8.5Hz,1H),7.53(dd,J=9.0及び6.1Hz),7.70(dd,J=7.1及び4.7Hz,1H),7.86−7.94(m,2H),8.06(d,J=2.4Hz,1H).
【0192】
本発明の化合物の製造の他の様式、変形又は順序は、当業者に容易に明らかとなろう。
【0193】
本発明の化合物は、遊離塩基の形態で、可能であれば塩基塩の形態で、及び、付加塩の形態で、並びに、遊離酸の形態で有用であろう。これら全ての形態は本発明の範囲内である。実用上は、塩形態の使用は塩基形態の使用と同等である。本発明の範囲内の医薬品に許容される塩は、塩酸及び硫酸などの無機酸;並びに、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸などの有機酸から誘導されるもの(例えば、塩酸塩、スルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩がそれぞれ挙げられる)、又は、適切な有機及び無機塩基などの塩基から誘導されるものが挙げられる。本発明の化合物との医薬品に許容される塩基付加塩の例には、非毒性でありかつそのような塩を形成するのに十分に強い有機塩基が挙げられる。これらの有機塩基及びその使用は、当業者に容易に理解される。単に例として挙げるものであるが、このような有機塩基には、メチルアミン、ジエチルアミン及びトリエチルアミンなどのモノ、ジ及びトリアルキルアミン類;モノ、ジ及びトリエタノールアミンなどのモノ、ジ又はトリヒドロキシアルキルアミン類;アルギニン及びリジンなどのアミノ酸類;グアニジン;N−メチルグルコサミン;N−メチルグルカミン;L−グルタミン;N−メチルピペラジン;モルホリン;エチレンジアミン;N−ベンジルフェネチルアミン;トリス(ヒドロキシメチル)アミノエタン;などが挙げられる。
【0194】
塩基性化合物の酸付加塩は以下のように製造することができる:本発明の化合物の遊離塩基を、水溶液若しくはアルコール水溶液又は適切な酸若しくは塩基を含む他の適切な溶液に溶解し、溶液を蒸発させることにより塩を単離するか、あるいは、反応物が有機溶液中に存在するように、本発明の化合物の遊離塩基を酸と反応させることによって、また、酸基を有する本発明の化合物を塩基と反応させることによって、塩を直接分離するか又は溶液の濃縮により得ることができる。
【0195】
本発明の化合物は、薬理学的活性を示すので、医薬として有用である。さらに、上記化合物は、中枢神経系及び心血管系活性を示す。
【0196】
(PARPアッセイ)
1.IC50
PARP阻害剤化合物のIC50を決定する簡便な方法は、Trevigan(メリーランド州ゲーサーズバーグ)の精製組み換えヒトPARPを用いるPARPアッセイであり、以下のとおりである:PARP酵素アッセイを、氷上で、100mMのTris−HCl(pH8.0)、1mMのMgCl2、28mMのKCl、28mMのNaCl、0.1mg/mlのDNaseI活性化ニシン精子DNA(Sigma,ミズーリ州)、3.0マイクロモル[3H]ニコチンアミドアデニンジヌクレオチド(470mci/ミリモル)、7マイクログラム/mlのPARP酵素及び試験する種々の濃度の化合物からなる100マイクロリットルの容積で開始する。反応を、25℃で混合物をインキュベートすることにより開始する。15分間インキュベートした後、氷冷20%(w/v)トリクロロ酢酸500マイクロリットルを添加することにより反応を終結させる。生成した沈殿をグラスファイバーフィルター(Packard Unifilter−GF/B)上に移し、エタノールで3回洗浄する。フィルターを乾燥した後、シンチレーション計数により放射線活性を測定する。
【0197】
本発明の化合物は、この阻害アッセイにおいてIC50が数nM〜20μMの範囲という強力な酵素活性を有することが見出された。
【0198】
上記PARPアッセイを用い、以下の化合物についておおよそのIC50値を得た。
【0199】
【表1】
【0200】
【表2】
【0201】
2.mRNA老化細胞における変化した遺伝子発現の測定
遺伝子発現変化はヒト繊維芽細胞BJ細胞を用いて測定することができ、上記細胞を細胞集団倍加数(Population Doubling)(PDL)94で通常の増殖培地上に播種し、次いで、低血漿培地に変えて、Linskensら,Nucleic acids Res.,23,3244−3251(1995)に記載の生理学的条件を反映させる。0.5%仔ウシ血清を添加したDMEM/199培地を用いる。細胞を13日間にわたって毎日処理する。PARP阻害剤の投与に使用する溶液を用いて、又は、これを用いずに、コントロール細胞を処理する。未処理の老化コントロール細胞及び若いコントロール細胞を、比較のために試験する。処理した細胞及びコントロール細胞からPCT公開第96/13610号に記載の技術に従ってRNAを調製し、ノーザンブロッティングを実施する。老化関連遺伝子に特異的なプローブを分析し、処理し、コントロール細胞を比較する。結果の分析において、遺伝子発現の最低レベルを任意に1に設定して、比較用のための基準とする。皮膚における加齢性変化に特に関連する3つの遺伝子は、コラーゲン、コラゲナーゼ及びエラスチンである。WestArch.Derm.130,87−95(1994)。PARP阻害剤で処理した細胞のエラスチン発現は、コントロール細胞と比較して顕著に増加するものと予測される。エラスチン発現は、若い細胞において老化細胞よりも顕著に高いはずであるため、PARP阻害剤での処理によって老化細胞でのエラスチン発現レベルは非常に若い細胞における同レベルと同等まで変化するはずである。同様に、PARP阻害剤での処理により、コラゲナーゼ及びコラーゲン発現において有益な効果がみられるはずである。
【0202】
3.老化細胞におけるタンパク質の変化した遺伝子発現の測定
遺伝子発現変化は、PDLが95〜100のBJ細胞を約105個用い、上記細胞を15cmディッシュに平板培養することによって測定できる。増殖培地は、10%仔ウシ血清を添加したDMEM/199である。細胞をPARP阻害剤(100μg/1mLの培地)で毎日24時間処理する。WO99/11645を参照されたい。細胞をリン酸バッファー(PBS)で洗浄し、次いで、4%パラホルムアルデヒドで5分間透過可能にした後、PBSで洗浄し、100%冷メタノールで10分間処理する。メタノールを除去し、細胞をPBSで洗浄した後、10%血清で処理して、非特異的な抗体結合をブロックする。約1mLの適切な市販抗体溶液(1:500希釈、Vector)を細胞に添加し、混合物を1時間インキュベートする。細胞をPBSで3回洗浄する。二次抗体であるビオチン化ヤギ抗マウスIgG(1mL)を、アルカリホスファターゼ結合ストレプトアビジンを含む溶液1mL及び1mLのNBT剤(Vector)と共に添加する。細胞を洗浄し、遺伝子発現の変化を比色法により観察する。PARP阻害剤で処理した老化細胞において、老化に特異的な4つの遺伝子−コラーゲンI、コラーゲンIII、コラゲナーゼ及びインターフェロンγを観察した結果、インターフェロンγ発現の低下が示され、他の3つの遺伝子の発現レベルには変化がみられなかったが、このことは、PARP阻害剤が、老化に特異的な遺伝子の発現を変化させることができるということを示す。
【0203】
4.細胞の増殖能力及び寿命の拡張又は増加
細胞の増殖能力及び寿命を拡張するための本方法の有効性を示すため、ヒト繊維芽細胞細胞株(細胞集団倍加数(PDL)が23であるWl38、又は、PDLが71であるBJ細胞のいずれか)を解凍し、T75フラスコに平板培養し、通常の培地(10%仔ウシ血清を添加したDMEM/M199)中で約1週間増殖させることによって(このとき細胞はコンフルエントである)、培養物を細分用に準備する。細分に際して、培地を吸引し、細胞をリン酸緩衝生理食塩水(PBS)で洗浄し、次いでトリプシン化する。Coulterカウンターを用いて細胞を計数し、10%仔ウシ血清及び種々の量(0.10μM及びImM:DMEM/M199培地中の100倍(IOOX)原液より)のPARP阻害剤を添加したDMEM/199培地を入れた6ウェル組織培養プレート中で、1cm2当たり細胞105個の密度で平板培養する。この方法を、細胞が細分を停止するように見えるまで7日毎に繰り返す。未処理の(コントロール)細胞は、培地中約40日後に老化に達し、細分を停止する。
【0204】
(テモゾロマイドの投与)
実施例1:化合物4i及びテモゾロマイドの経口投与は、CNS部位に悪性腫瘍を有するマウスの生存を増大させる。
【0205】
頭蓋内移植の手順は、TentoriLら, “Effects of single or split exposure of leukemic cells to temozolomide, combined with poly(ADP−ribose) polymerase inhibitors on cell growth, chromosomal aberrations and base excision repair components,” Cancer Chemother Pharmacol, 47, 361−9 (2001)に記載のように実施する。マウス黒色腫B16細胞(104個)を、雄B6D2F1(C57BL/6×DBA/2)マウスに頭蓋内(ic)注射した。治療のタイミングを決定するため、脳での腫瘍増殖の組織学的評価を、腫瘍の曝露後1〜5日に行った。
【0206】
カリウムを含まない70mM PBSに化合物4iを溶解し、テモゾロマイド(TMZ)投与の1時間前に経口投与した。TMZをジメチルスルホキシド(40mg/ml)中に溶解し、生理食塩水(5mg/ml)で希釈し、100mg/Kgの用量で5日にわたって腹腔内投与した。化合物4iを用いて経口栄養補給により、10mg/kg/日又は40mg/kg/日の用量で、1日1回5日間にわたってマウスを処置した。生存中央値(median survival times)(MST)を決定し、寿命(ILS)の増加の割合を、([処置したマウスのMST(日)/コントロールマウスのMST(日)]−l)×100として算出した。処置の効力を、処置群とコントロール群との間の生存曲線の比較により評価した。
【0207】
B16黒色腫を有するマウスでは、結果は、化合物4i及びTMZの組み合わせで処置した群の平均生存時間が、TMZを単一の剤として服用した動物で観察された平均生存時間よりも顕著に高かったことを示した(図I及び表II)。
【0208】
表II.脳内にB16黒色腫を有するマウスの生存率
【0209】
【表3】
【0210】
実施例2:化合物4iの投与による、皮下黒色腫ガンモデルにおけるテモゾロマイド効果の増強
【0211】
TMZ±化合物4iによる処置の効力を、マウスにおいて皮下(s.c.)増殖黒色腫についても評価した。この目的のため、B16細胞(2.5×105個)を動物の脇腹に皮下接種した。腫瘍をノギスで測定し、容積を、式:[(幅)2×長さ]/2に従って算出した。曝露の6日後、腫瘍結節の容積が100〜150mm3に達したときに、薬剤処置を開始した。化合物4i(40mg/kg経口)を、テモゾロマイド(100mg/kg腹腔内)の20分前に、1日1回5日間にわたって投与した。黒色腫増殖を、腫瘍結節を3日毎に3週間にわたって測定することによって観察した。
【0212】
化合物4i及びTMZによる併用治療によって、B16黒色腫の増殖が顕著に減少した(P<0.01、9日〜23日、TMZ単独に対して)(図II)。
【0213】
実施例3:ラットにおいてシスプラチンで誘発される神経障害における、交感神経透過速度(SNCV)
【0214】
本発明の化合物の神経保護効果を、ラットにおいてシスプラチンで誘発される神経障害モデルで示した。神経透過速度の変化は、化学毒性物質が誘発する末梢神経障害の高感度な測定であることがよく実証されている。化合物4iは、シスプラチンでの慢性的な治療により誘発される神経透過速度の欠損を低減することが示された。
【0215】
この実験において、雌Wistar Hannoverラットに、神経障害を誘発する用量のシスプラチン(2mg/kg腹腔内;週2回を4週間)を、化合物4i(毎日40mg/kg、経口)と共に及び化合物4iなしで投与した。ラットにおいて、ベースライン(シスプラチン投与前)及び処置後の尾部神経での交感神経透過速度(SNCV)の変化を観察した。さらに、後根神経節及び坐骨神経標本の後根神経節神経上における形態計測分析(体細胞、核及び核小体の大きさ)を、病理解剖学的に評価した。
【0216】
処置期間の最初と最後に、各動物の尾部において、Cavalettiら, “Protective Effects of glutathione on cisplatin neurotoxicity in rats,” Int. J. Radiation Oncology, 29, 771−776 (1994) and Trediciら, “Low−Dose Glutathione administration, in the prevention of cisplatin−induced peripheral neuropathy in rats,” Neuro toxicology, 15, 701−704 (1994)に既に記載されているように、SNCV測定を測定した。尾神経における逆方向性のSNCVを、記録用環状電極を尾の遠位に配置することにより評価し、ここで、刺激環状電極は、記録点から5cm及び10cm離して配置した。神経刺激後の2箇所で記録された電位の反応時間を決定し(ピークからピーク)、これに従って神経透過速度を計算した。
【0217】
犠牲にした動物から得た各群に由来するラットの左側L5後根神経節(DRG)を、既に報告されたプロトコル[Cavalettiら;Trediciら]に従って処理し、樹脂に埋め込み、光学顕微鏡及び電子顕微鏡での観察並びに形態計測に用いた。厚み1μmの半薄切片で、画像分析ソフトウェア(Image J,NIH)を用いて、DRGニューロンの細胞体、核及び核小体の断面積の形態測定を行った。
【0218】
神経透過速度の相違及び実験中に後根神経節神経で得られた形態計測データの相違を、分散分析(ANOVA)及びTukey−Kramer後検定(有意性レベルをp<0.05に設定)を用いて、統計学的に評価した。
【0219】
化合物4iの共投与によって、慢性的なシスプラチン治療による尾神経透過速度の損傷における統計学的に顕著な減少が誘発されるということが見出された(表3及び4)。
【0220】
表3:実験終了後におけるSNCV(m/秒)
【0221】
【表4】
【0222】
CDDP=シスプラチン
【0223】
表4:統計学的分析(一方向ANOVA)
【0224】
【表5】
【0225】
DRG形態計測
【0226】
DRGニューロンにおける形態計測の研究により、化合物4iを有するDRGニューロンの細胞体の大きさのみについて顕著な効果が明らかとなった。表5は形態計測の結果を示し、統計学的データを表6(細胞体)及び表7(核)に列挙する。
【0227】
表5:DRGの形態計測(μm2)
【0228】
【表6】
【0229】
Nu=核、Nucl=核小体
【0230】
表6:細胞体
【0231】
【表7】
【0232】
表7:核小体
【0233】
【表8】
【0234】
上記では本発明について説明しているが、多くの方法によって改変可能な場合があるということは明らかであろう。このような改変は、本発明の趣旨及び範囲内であるとみなされ、このような改変は全て、添付の特許請求の範囲の範囲内に包含されるものである。
【0235】
(参照による援用)
本明細書中に記載する全ての刊行物、特許及び付与前特許出願公開は、任意の及び全ての目的のために、あたかもそれぞれの個別の刊行物及び特許出願が参照として援用されていることが示されているかのように、特別にかつ個別に本明細書中に参照として援用される。不一致がある場合、本開示が優先する。
【技術分野】
【0001】
本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法に関する。
【背景技術】
【0002】
PARP(EC2.4.2.30)は、PARS(ポリ(ADP−リボース)合成酵素)又はADPRT(NAD:タンパク質(ADP−リボシル)トランスフェラーゼ(重合性))としても知られ、116kDaの主要な核タンパク質である。これは主に、ほとんど全ての真核生物に存在する。この酵素によりポリ(ADP−リボース)が合成され、これは、NAD由来の200個を超えるADP−リボース単位から構成され得る分岐鎖ポリマーである。ポリ(ADP−リボース)のタンパク質受容体は、DNAの一体性維持に直接的又は間接的に関与している。これには、ヒストン、トポイソメラーゼ、DNA及びRNAポリメラーゼ、DNAリガーゼ並びにCa2+及びMg+依存性エンドヌクレアーゼが含まれる。
【0003】
PARPタンパク質は、多くの組織に、最も顕著には免疫系、心臓、脳及び胚株細胞内に高レベルで発現する。正常な生理学的条件下では、PARP活性は非常に小さい。しかし、DNAの損傷によって、PARPは500倍にまで急激に活性化される。PARPに起因する多くの機能の中でも、主な役割は、ADP−リボシル化によるDNA修復の促進及び多数のDNA修復タンパク質の調整である。PARPが活性化されると、NADレベルが著しく減少する。多くの内因性及び外因性因子がDNAを損傷させてかつPARPを活性化させるということが報告されているが、例えばショック、卒中及び炎症において、報告されているinvivoでの種々の疾患症状に関与する主な原因は、酸化窒素(NO)と過酸化物との組み合わせによって形成される過酸化亜硝酸であると考えられる。
【0004】
また、3−アミノベンズアミドなどのPARP阻害剤は、通常は例えば過酸化水素又はγ線に応答して、DNA修復に影響を与えることも知られている(非特許文献1)。特に、Cristovaoらは、過酸化水素で処理された白血球内においてDNA鎖のPARP依存性回復が停止することを観察した。
【0005】
核酵素ポリ(ADP−リボース)ポリメラーゼ−1(PARP−1)は、塩基除去修復及び他の細胞プロセスの促進において重要な役割を果たしている。PARP−1は分子DNAニックのセンサとして作用し、一重鎖DNAの切断を検出しかつ適切な修復酵素を集めるということが提唱されている。PARP−1は、酵素のアミノ末端DNA結合ドメイン中の2つのジンクフィンガーを介してDNA鎖切断部分に結合し、その活性はDNA結合に依存する。この酵素は、基質NAD+から、PARP−1自身も含む受容体タンパク質へのADP−リボースの移動を触媒するホモダイマーとして作用している。これによって多量の負電荷を帯びたPARポリマーが形成され、DNA鎖及びクロマチンタンパク質の静電的反発が生じて、これにより、損傷した鎖に塩基除去修復複合体が接近してDNAが修復される。鎖切断によって最初に活性化された後はPARP−1はDNAから遊離し、ポリマーはPARグリコヒドロラーゼにより分解されて、PARP−1酵素は次のDNA結合及び活性化において使用することができる(非特許文献2)。
【0006】
PARP阻害剤は、放射線増感された低酸素腫瘍細胞において共力剤又は増強剤として有効であることが報告されている。またPARP阻害剤は、放射線療法後の致死性DNA損傷からの腫瘍細胞の回復の阻害において、おそらくはそのDNA修復阻害能に起因して、共力剤として有効であることが報告されている(特許文献1〜3)。
【0007】
ガン治療に用いるための、及び、虚血や内毒素ストレス後における細胞損傷の抑制に用いるための、化学増強剤でもあり放射線増強剤でもあるPARP阻害剤の開発には、大きな関心が集まっている。特に、強力なPARP−1阻害剤を用いた臨床前研究ではテモゾロマイド細胞傷害性の増強が観察されるが、これは塩基除去修復の阻害及びその後の細胞傷害(N7−メチルグアニン及びN3−メチルアデニンの不完全なプロセシングによる)を反映する。現在、一連の臨床前データによって、テモゾロマイドの細胞傷害性は、PARP阻害剤をin vitro又はin vivoのいずれかで共投与することにより増強されるということが示されている(非特許文献2)。
【0008】
DNAメチル化剤であるテモゾロマイドによってDNA損傷が誘発されるが、この損傷は、O6−アルキルグアニンアルキルトランスフェラーゼ(ATase)及びポリ(ADP−リボース)ポリメラーゼ−1(PARP−1)依存性塩基除去修復により修復される。テモゾロマイドは、神経膠腫及び悪性黒色腫の治療において用いられる、経口投与用の単官能性DNAアルキル化剤である。テモゾロマイドは速やかに吸収され、自発的に分解して、活性モノメチルトリアゼンである5−(3−メチル−1−トリアゼノ)イミダゾール−4−カルボキサミドを形成する。モノメチルトリアゼンからはいくつかのDNAメチル化産物が形成されるが、主な種は、N7−メチルグアニン(70%)、N3−メチルアデニン(9%)及びO6−メチルグアニン(5%)である。O6−アルキルグアニンアルキルトランスフェラーゼによって修復されない場合、O6−メチルグアニンは、DNA複製の際にチミンとの誤対合により細胞傷害性を有する。この誤対合は、ミスマッチ修復タンパク質及び除去されたチミンによって、娘鎖上で認識される。しかし、メチル付加物のAtaseが媒介する除去によって親鎖中の元のO6−メチルグアニンヌクレオチドが修復されなければ、チミンは再び挿入され得る。チミン除去が繰り返されることにより無駄に時間が経過することによって、かつ、未修復のO6−メチルグアニンヌクレオチドが結合することによって、鎖が切断された状態が持続し、ミスマッチ修復系のMutS分岐によってG2−M細胞周期停止及びアポトーシスの開始についてのシグナルが送られる。テモゾロマイドによって形成される多量のN7−メチルグアニン及びN3−メチルアデニンヌクレオチドアルキル化産物は、塩基除去修復によって迅速に修復される(非特許文献2)。
【0009】
PARP阻害剤による化学増感は、テモゾロマイドの場合に制限されない。概して細胞傷害性薬剤は、又は、放射線は、PARP−1の活性化を誘発する可能性があり、PARP−1の阻害剤は、化学療法及び放射線照射によるDNA損傷及び細胞傷害効果を増強する可能性があることが示されている(非特許文献3)。DNA損傷剤に応答するPARP−1媒介DNA修復によって、腫瘍における薬剤耐性のメカニズムが示され、上記酵素が阻害されると、イオン化放射線の活性及びいくつかの細胞傷害性抗腫瘍剤(テモゾロマイド及びトポテカンを含む)が増強されることが示されている。Sutoらは、特許文献4で、イオン化放射線又は化学療法薬剤が腫瘍細胞に及ぼす致死性効果を増強する際に用いられるいくつかのイソキノリン類を開示している。非特許文献4において、腫瘍細胞をアルキル化剤で併用治療した場合の、PARP活性の阻害、腫瘍細胞の増殖低下及び顕著な共力剤的効果が開示されている。このことから、PARP−1は、DNAを損傷させるガン治療を促進するための非常に重要な治療標的である。
【0010】
多数の公知のPARP阻害剤が、非特許文献5及び6中に記載されている。しかし、上述した方法において、これらのPARP阻害剤を使用すると望ましくない副作用が同時に起こるため、これらのPARP阻害剤の有効な使用は制限されている。非特許文献7を参照。
【0011】
上記に加えて、PARP阻害剤は、以下の国際特許出願に開示されかつ記載されている:特許文献5〜15。従来技術についての包括的な概説は、Li及びZhangにより非特許文献8に報告されている。
【0012】
腫瘍細胞をイオン化放射線に対して放射線増感させることによるか又は腫瘍細胞を化学療法薬剤の細胞傷害性効果に対して化学増感させることによるかのいずれかによって細胞傷害性剤の致死性を増強するというPARP阻害剤の能力は、とりわけ、特許文献16〜18;非特許文献2及び9〜13;Griffin R.J.らによる特許文献19;及び、Helleday Tらによる特許文献14に報告されている。
【0013】
末梢神経障害の誘発は、化学治療薬剤による治療が制限される一般的な要因である(非特許文献14)。化学療法により誘発される神経障害は、多くの従来の化学療法(例、タキソール、ビンクリスチン、シスプラチン)及び新たな化学療法(例、ベルケイド、エポチロン)を用いた場合に生じる副作用である。結果として、用いた物質に応じて、純感覚性かつ痛みを伴う神経障害(シスプラチン、オキサリプラチン、カルボプラチンの使用)又は自律神経系の関与を伴う又は伴わない混合型の感覚運動神経障害(ビンクリスチン、タキソール、スラミンの使用)が生じ得る。神経毒性は、用いる薬剤の総蓄積用量及び種類に依存する。単一の薬剤を適用した後でも神経障害が起こる場合もある。症候から完全に回復しない場合もあり、機能を再開するのに長期間の再生が必要とされる。今日まで、化学療法により誘発される神経障害を確実に予防又は治療することができる薬品は存在しない。
【0014】
副作用が非常に小さく、かつ、イオン化放射線及び/又は化学療法薬剤が腫瘍細胞に及ぼす致死効果を増強する効果的かつ強力なPARP阻害剤が依然として必要とされている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】米国特許第5,032,617号
【特許文献2】米国特許第5,215,738号
【特許文献3】米国特許第5,041,653号
【特許文献4】米国特許第5,177,075号
【特許文献5】国際特許WO00/42040
【特許文献6】国際特許WO00/39070
【特許文献7】国際特許WO00/39104
【特許文献8】国際特許WO99/11623
【特許文献9】国際特許WO99/11628
【特許文献10】国際特許WO99/11622
【特許文献11】国際特許WO99/59975
【特許文献12】国際特許WO99/11644
【特許文献13】国際特許WO99/11945
【特許文献14】国際特許WO99/11649
【特許文献15】国際特許WO99/59973
【特許文献16】米国特許出願公開第2002/0028815号
【特許文献17】米国特許出願公開第2003/0134843号
【特許文献18】米国特許出願公開第2004/0067949号
【特許文献19】国際特許WO98/33802
【特許文献20】国際特許WO2005/012305
【非特許文献】
【0016】
【非特許文献1】Cristovaoら,“Effect of a Poly(ADP−Ribose) Polymerase Inhibitor on DNA Breakage and Cytotoxicity Induced by Hydrogen Peroxide and γ−Radiation,”Terato.,Carcino.,and Muta., 16:219−27(1996)
【非特許文献2】Plummer ERら,11(9)Clin.Cancer Res.3402(2005)
【非特許文献3】Kockら,45 J.Med.Chem.4961(2002)
【非特許文献4】Weltinら,“Effect of 6(5H)−Phenanthridinone, an Inhibitor of Poly(ADP−ribose) Polymerase,on Cultured Tumor Cells”, Oncol.Res.,6:9,399−403(1994)
【非特許文献5】Banasikら,“Specific Inhibitors of Poly(ADP−Ribose) Synthetase and Mono(ADP−Ribosyl)−Transferase”, J.Biol Chem.,267:3,1569−75(1992)
【非特許文献6】Banasikら,“Inhibitors and Activators of ADP−Ribosylation Reactions”,Molec.Cell.Biochem.,138,185−97(1994)
【非特許文献7】Milamら,“Inhibitors of Poly(Adenosine Diphosphate−Ribose)Synthesis; Effect on Other Metabolic Processes,”Science,223,589−91(1984)
【非特許文献8】Li and Zhang, Drugs 2001,4(7): 804−812(PharmaPress Ltd ISSN 1369−7056)
【非特許文献9】White AWら,14 Bioorg.&Med.Chem Letts.2433(2004)
【非特許文献10】Canon Koch SSら,45 J.Med.Chem.4961(2002)
【非特許文献11】Skalitsky DJら,46 J.Med.Chem.210(2003)
【非特許文献12】Farmer Hら,434 Nature 917(14 April 2005)
【非特許文献13】Tikhe JGら,47 J.Med.Chem.5467 (2004)
【非特許文献14】Quasthoff and Hartung,J.Neurology,249,9−17(2002)
【発明の概要】
【発明が解決しようとする課題】
【0017】
(発明の要旨)
本発明は、ポリ(ADP−リボース)ポリメラーゼ(「PARP」)を阻害するジアザベンゾ[デ]アントラセン−3−オン化合物、これらの化合物を含む組成物、並びに、本明細書中に記載の症状の影響を治療、予防及び/又は緩和するためのこれらのPARP阻害剤の使用方法を提供する。
【課題を解決するための手段】
【0018】
また、本発明は、
以下のグループIの化合物から選択されるジアザベンゾ[デ]アントラセン−3−オン化合物:
【0019】
【化1】
【0020】
【化2】
【0021】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物
を提供する。
【0022】
また、本発明は、(i)治療上有効量のグループIの化合物及び(ii)医薬品に許容される担体を含む医薬組成物にも関する。
【0023】
本発明は、ポリ(ADP−リボース)ポリメラーゼ(PARP)のin vitro及び/又はin vivoでのポリメラーゼ活性を阻害する化合物、並びに:開示された化合物を含む組成物を提供する。
【0024】
本発明は、溶液、細胞、組織、器官又は器官系で、ポリ(ADP−リボース)ポリメラーゼ(PARP)のin vitro及び/又はin vivoでのポリメラーゼ活性を阻害、制限及び/又は制御する方法を提供する。一実施形態において、本発明は、ヒトなどの哺乳類において、局所的又は全身的でのいずれかでPARP活性を制限又は阻害する方法を提供する。
【0025】
本発明は、PARP産生の増加により悪化するか又はPARP産生の増加に関与する疾患、症候群及び/又は症状の治療及び/又は予防方法を提供する。これらの方法は、本発明の化合物を、上記治療又は予防を必要とするヒトの細胞、組織、器官又は器官系に適用又は投与することを含む。
【0026】
他の実施形態において、本発明の化合物及び組成物は、ヒトなどの動物において壊死又はアポトーシス、大脳虚血及び灌流損傷又は神経変性疾患による細胞の損傷又は細胞死を治療又は予防するのに用いることができる。
【0027】
他の実施形態において、本発明の化合物及び組成物は、細胞の寿命及び増殖能力を拡張するのに用いることができる、これに関連する疾患を治療又は予防するのに用いることができる。
【0028】
他の実施形態において、本発明は、ガンの影響を治療若しくは予防若しくは緩和する方法、並びに/又は、腫瘍細胞若しくは低酸素腫瘍細胞を放射線増感させて腫瘍細胞の放射線療法に対する感受性を高めることによって、放射線療法後の非常に致死性の高いDNA損傷から腫瘍細胞が回復するのを妨げる方法に用いることができる。この実施形態の方法は、腫瘍細胞を特異的かつ優先的に放射線増感することによって、腫瘍細胞の放射線療法に対する感受性を非腫瘍細胞よりも高めることに関する。
【0029】
また、本発明は、イオン化放射線及び/又は化学療法薬剤の腫瘍細胞への細胞傷害性効果を増強することにより、ガンの影響を治療、予防及び/又は緩和するためのグループIのジアザベンゾ[デ]アントラセン−3−オン化合物を提供する。
【0030】
一実施形態において、本発明は、
ガン及び/又は腫瘍を治療するための化学増感方法であって、
腫瘍又はガン細胞を、グループIの細胞傷害性効果を有するジアザベンゾ[デ]アントラセン−3−オン化合物と接触させること、及び、腫瘍又はガン細胞を、さらに抗ガン剤と接触させることを含む
ことを特徴とする方法
を提供する。
【0031】
本発明は、
哺乳類、特にヒトにおいてガンを治療するための化学増感方法であって、
哺乳類に、グループIから選択されるジアザベンゾ[デ]アントラセン−3−オン化合物を投与することを含む
ことを特徴とする方法
を提供する。
【0032】
本発明の一実施形態において、本発明の化学増感方法に用いられる化合物は以下である。
【0033】
【化3】
【0034】
他の実施形態において、本発明は、第1の用量の少なくとも1つのグループIの化合物が、それを必要とする患者に単回で又は繰り返して投与され、有効量の化学増感を提供できる時間の経過後、引き続いて、第2の用量の少なくとも1つの化学療法薬剤が、上記患者に単回又は繰り返して投与される、化学増感方法を提供する。
【0035】
他の実施形態において、本発明は、医薬品に許容される遊離塩基、塩、水和物、エステル、溶媒和物、プロドラッグ、代謝物、立体異性体及びこれらの混合物からなる群より選択される形態の化学増感ジアザベンゾ[デ]アントラセン−3−オン誘導体を含む医薬調製物を提供する。他の実施形態によれば、医薬調製物は、医薬品に許容される担体、及び、必要に応じて化学療法薬剤をさらに含む。そのような化学療法薬剤の例を以下に記載するが、これらに限定されない。
【0036】
本発明の他の実施形態によれば、化学増感化合物及び化学療法薬剤は、実質的に同時に投与される。
【0037】
本発明の他の実施形態によれば、化学療法薬剤は、テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、タキソイド、ダクチノマイシン、ダノルビシン(danorubicin)、4’−デオキシドキソルビシン、ブレオマイシン、ピルカマイシン(pilcamycin)、マイトマイシン、ネオマイシン及びゲンタマイシン、エトポシド、4−OHシクロフォスファミド、白金配位錯体、トポテカン、その治療上有効なアナログ及び誘導体、並びに、これらの混合物からなる群より選択される。好ましい態様によれば、化学療法薬剤はテモゾロマイドである。
【0038】
一実施形態によれば、本発明は、グループIから選択される化学増感有効量の少なくとも1つのジアザベンゾ[デ]アントラセン−3−オン化合物を含む医薬組成物を提供する。他の態様において、医薬組成物は以下を含む。
【0039】
【化4】
【発明の効果】
【0040】
本発明の化合物は、副作用が非常に小さく、かつ、イオン化放射線及び/又は化学療法薬剤が腫瘍細胞に及ぼす致死効果を増強する効果的かつ強力なPARP阻害剤としての効果を奏する。
【図面の簡単な説明】
【0041】
【図1】経口用化合物4i+TMZによる、脳内に黒色腫を有するマウスの生存率の増大
【図2】B6D2F1マウスにおけるB16皮下黒色腫に対する化合物4i+TMZの効果
【発明を実施するための形態】
【0042】
(発明の詳細な説明)
定義
本発明は、動物又は哺乳類において本明細書中に記載のいずれかの疾患又は障害を治療するための医薬の製造における本発明の化合物の使用に関する。
【0043】
本明細書中で用いる「アルキル」は、指定された数の炭素原子を含む分岐鎖又は非分岐鎖の飽和炭化水素鎖を意味する。例えば、C1−C6直鎖又は分岐鎖アルキル炭化水素鎖は、1〜6個の炭素原子を含み、特に示さない限り、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、tert−ブチル、n−ペンチル、n−ヘキシルなどの置換基が挙げられるが、これらに限定されない。
【0044】
「アルケニル」は、指定された数の炭素原子を含む分岐鎖又は非分岐鎖の不飽和炭化水素鎖を意味する。例えば、C2−C6直鎖又は分岐鎖アルケニル炭化水素鎖は、少なくとも1つの二重結合を有する2〜6個の炭素原子を有し、特に示さない限り、例えば、エテニル、プロペニル、イソプロペニル、ブテニル、イソブテニル、tert−ブテニル、n−ペンテニル、n−ヘキセニルなどの置換基が挙げられるが、これらに限定されない。
【0045】
「アルコキシ」は、基−OR(式中、Rは、本明細書中で定義するアルキルである)を意味する。また、Rは、1〜6個の炭素原子を含む分岐鎖又は非分岐鎖飽和炭化水素鎖であってもよい。
【0046】
本明細書中で接頭語として用いる「シクロ」は、閉環により特徴付けられる構造をいう。
【0047】
「ハロ」は、特に示さない限り、少なくとも1つのフルオロ、クロロ、ブロモ又はヨード部位を示す。
【0048】
「アミノ」化合物は、アミン(NH2)及び置換されたアミノを含む。
【0049】
「Ar」、「アリール」又は「ヘテロアリール」は、置換された又は未置換の、特に環状又は縮合環式環である部位を意味し、単環式、二環式又は三環式の炭素環式又はヘテロ環式環が挙げられ、上記環は、1〜5個の位置が未置換であるか又は次のもので置換されている:ハロ、ハロアルキル、ヒドロキシル、ニトロ、トリフルオロメチル、C1−C6直鎖又は分岐鎖アルキル、C2−C6直鎖又は分岐鎖アルケニル、C1−C6アルコキシ、C2−C6アルケニルオキシ、フェノキシ、ベンジルオキシ、アミノ、チオカルボニル、エステル、チオエステル、シアノ、イミノ、アルキルアミノ、アミノアルキル、スルフヒドリル、チオアルキル及びスルホニル:ここで、環の大きさはそれぞれ5〜8員である:ヘテロ環式環は、O、N又はSからなる群より選択される1〜4個のヘテロ原子を含む:芳香族又は第三級アルキルアミンは、必要に応じて、対応するN−オキシドに酸化される。ヘテロアリールは、他の環に結合していても、上記環のヘテロ原子及び/又は炭素原子を介して置換されていてもよい。アリール又はヘテロアリール部位には、フェニル、ベンジル、ナフチル、ピロリル、ピロリジニル、ピリジニル、ピリミジニル、プリニル、キノリル、イソキノリル、フリル、チオフェニル、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル及びチエニルが挙げられるが、これらに限定されない。
【0050】
「フェニル」には、必要に応じて以下からなる群より選択される置換基でモノ置換又は複数置換された全ての可能な異性体フェニル基を含む:アミノ、トリフルオロメチル、C1−C6直鎖又は分岐鎖アルキル、C2−C6直鎖又は分岐鎖アルケニル、カルボニル、チオカルボニル、エステル、チオエステル、アルコキシ、アルケノキシ、シアノ、ニトロ、イミノ、アルキルアミノ、アミノアルキル、スルフヒドリル、チオアルキル、スルホニル、ヒドロキシ、ハロ、ハロアルキル、NR2(式中、R2は、水素、(C1−C6)直鎖又は分岐鎖アルキル、(C3−C6)直鎖又は分岐鎖アルケニル又はアルキニル、及び、(C1−C4)架橋アルキル(上記架橋アルキルは、NR1の窒素から出発して、上記アルキル又はアルケニル鎖の炭素原子のうち1つで終結するヘテロ環式環を形成し、ここで、上記ヘテロ環式環は、必要に応じてAr基に縮合する)からなる群より選択される)。
【0051】
必要に応じて少なくとも1つのヘテロ原子を含むシクロアルキルには、C5又はC6環などの飽和C3−C8環が挙げられ、ここで、O、N、Sから選択される1〜4個のヘテロ原子が上記環の炭素原子を必要に応じて置換していてもよい。必要に応じて少なくとも1つのヘテロ原子を含むシクロアルキルは、上記のように、少なくとも1つの5員又は6員のアリール又はヘテロアリールで置換されても、あるいは、これに縮合していてもよい。ヘテロ原子を含むその他のシクロアルキルには、ピロリジニル、イミダゾリジニル、ピラゾリジニル、ピペリジニル、ピペラジニル、モルホリノ及びチオモルホリノが挙げられる。
【0052】
用語「神経変性疾患」には、アルツハイマー病、パーキンソン病及びハンチントン病が挙げられるが、これらに限定されない。
【0053】
用語「神経性傷害」は、神経組織に対する任意の損傷及びそれから生じる任意の障害又は死を意味する。神経性傷害の原因は、代謝性、毒性、神経毒性、医原性、熱的又は化学的なものがあり、虚血、低酸素症、脳血管傷害、外傷、手術、圧力、脳圧排(mass effect)、出血、放射線、血管攣縮、神経変性疾患、感染、パーキンソン病、筋萎縮性側索硬化症(ALS)、ミエリン化/脱ミエリン化プロセス、痙攣、認知障害、グルタミン酸異常及びそのいずれかの副作用が挙げられるが、これらに限定されない。
【0054】
用語「神経保護性」は、神経性傷害を低減、停止又は緩和する効果、及び、神経性傷害に罹患した神経組織を保護、蘇生又は復活させる効果を意味する。
【0055】
用語「神経変性の予防」は、神経変性疾患の予防、及び、神経変性疾患に既に罹患した又はその症候を有する患者におけるさらなる神経変性の予防を含む。
【0056】
用語「治療」は、以下を意味する:
(i)疾患、障害及び/又は症状に罹っているかもしれないが、未だ罹患していると診断されていない動物において疾患、障害又は症状が起こるのを予防すること;
(ii)疾患、障害又は症状を阻害する、すなわち、その進行を止めること;及び
(iii)疾患、障害又は症状を緩和する、すなわち、疾患、障害及び/又は症状の後退を生じさせること。
【0057】
用語「虚血損傷及び灌流損傷及び神経変性疾患から生じる神経系組織の損傷」には、血管卒中並びに全身及び局所的虚血において見られる神経毒性による損傷などの損傷が挙げられる。
【0058】
用語「虚血」は、動脈血流入の閉塞による局在化した組織貧血に関する。全身虚血は、脳全体への血流が一定の期間にわたって停止する条件下で起こり、心停止から生じ得るものなどである。局所虚血は、脳の一部が、その正常な血液供給が欠乏している条件下で起こり、大脳血管の血栓塞栓性閉塞、外傷性の頭部損傷、浮腫及び脳腫瘍から生じ得る。
【0059】
用語「心血管疾患」は、心筋梗塞、狭心症、血管又は心筋虚血、及び、当業者に知られているであろう関連症状に関し、心臓又は脈管構造の不全又は組織損傷が挙げられ、特に、PARP活性化に関する組織損傷が挙げられるが、これに限定されない。
【0060】
本明細書中で用いる用語「放射線増感剤」は、治療上有効な量で動物に投与して、電磁放射線に放射線増感させる細胞の感度を増大させるような、かつ/又は、電磁放射線で治療可能な疾患の治療を促進するような、低分子量分子などの分子であると定義される。電磁放射線で治療可能な疾患には、新生物疾患、良性及び悪性腫瘍、並びにガン細胞が挙げられる。本明細書中に列挙されていない他の疾患の電磁放射線治療もまた、本発明で意図される。本明細書中で用いる用語「電磁放射線」及び「放射線」には、10−20〜100メートルの波長を有する放射線が挙げられるが、これらに限定されない。本発明の好ましい実施形態は、γ線(10−20〜10−13m)、X線(10−11〜10−9m)、紫外線(10nm〜400nm)、可視光線(400nm〜700nm)、赤外線(700nm〜1.0mm)又はマイクロ波放射線(1mm〜30cm)の電磁放射線を用いる。
【0061】
本明細書中で用いる用語「化学増感剤」は、化学療法薬剤の抗腫瘍活性を増強する本発明の化合物の能力を意味する。そのような化学増感は、例えば、所定用量の化学療法薬剤の腫瘍増殖遅延若しくは停止効果を増大させる際に、又は、用量を減らしても抗腫瘍効力を維持できることによって化学療法薬剤の副作用特性を改善する際に、有用である。
【0062】
(本発明の医薬用途)
本発明は、核酵素ポリ(アデノシン5’−ジホスホ−リボース)ポリメラーゼ[「ポリ(ADP−リボース)ポリメラーゼ」又は「PARP」、ADPRT(NAD:タンパク質(ADP−リボシルトランスフェラーゼ(重合性)))、pADPRT(ポリ(ADP−リボース)トランスフェラーゼ)及びPARS(ポリ(ADP−リボース)合成酵素)とも呼ばれる]を阻害するための化合物、方法及び医薬組成物を提供する。さらに、本発明は、壊死又はアポトーシスに起因する細胞の損傷又は死;例えば、大脳虚血性卒中、頭部外傷又は脊椎損傷などの、虚血損傷及び再灌流損傷から生じる神経系組織の損傷;例えば、パーキンソン病又はアルツハイマー病及び多発性硬化症などの、神経学的障害及び神経変性疾患を予防及び/又は治療するための:血管卒中を予防又は治療するための:例えば、心筋梗塞などの心血管障害を治療又は予防するための:例えば、加齢性筋退化、AIDS及び他の免疫老化疾患、関節炎、アテローム性動脈硬化、毛細血管拡張性運動失調症、悪液質、ガン、複製老化に関与する骨格筋の変性疾患、糖尿病(真性糖尿病など)、炎症性腸障害(大腸炎及びクローン病など)、急性膵炎、粘膜炎、出血性ショック、内臓動脈閉塞ショック、多臓器不全(腎臓、肝臓、腎臓、肺、網膜、膵臓及び/又は骨格筋系のいずれかを含むものなど)、急性自己免疫甲状腺炎、筋ジストロフィー、骨関節炎、骨粗鬆症、慢性及び急性疼痛(神経障害性疼痛など)、腎不全、網膜虚血、敗血性ショック(内毒素性ショックなど)、局所的及び/又は遠隔内皮細胞不全(内部依存性弛緩応答及び接着分子の上方調節により認識されるものなど)、炎症及び皮膚加齢などの他の症状及び/又は障害を治療するための:例えば、酸化剤、炎症誘発性媒介物及び/又はサイトカインの産生における一般的な媒介物として、並びに、白血球の浸潤、カルシウムイオンの過負荷、リン脂質の過酸化、一酸化窒素代謝の損傷及び/又はATP産生の低減における一般的な媒介物として、細胞の寿命及び増殖能力を拡張するための;老化細胞の遺伝子発現を変えるための:あるいは、低酸素腫瘍細胞を放射線増感させるための、本発明のPARP阻害剤の使用方法を提供する。
【0063】
本発明の化合物は、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷を治療又は予防することができ;局所虚血、心筋梗塞及び灌流損傷に続くものを含む神経系又は心血管組織損傷を緩和することができ;PARP活性により引き起こされるか又は悪化する種々の疾患及び症状を治療することができ;細胞の寿命又は増殖能力を拡張又は増大させることができ;老化細胞の遺伝子発現を変えることができ;かつ、細胞を放射線増感させることができる。一般に、PARP活性の阻害は、細胞をエネルギー欠乏から保護し、神経系細胞の場合にはニューロンを不可逆的損傷から保護して、これによって神経を保護する。理論上、PARPの活性化は、フリーラジカル及びNOの産生だけでなく、おそらくまだ発見されていない他の細胞傷害(excitotoxic)メカニズムにおいて主要な役割を果たしている可能性があると考えられている。
【0064】
上記理由により、本発明は、さらに、治療上有効量の上記化合物を、PARP活性を阻害するのに十分な量で投与して、壊死又はアポトーシスに起因する細胞の損傷又は死を治療又は予防するための、NMDA傷害により媒介されない神経活性に影響を与えるための、NMDA傷害により媒介される神経活性に影響を与えるための、虚血損傷及び灌流損傷、神経学的障害及び神経変性疾患から生じる神経系組織の損傷を治療するための;血管卒中を予防又は治療するための;心血管障害を治療又は予防するための;加齢性筋退化、AIDS及び他の免疫老化疾患、関節炎、アテローム性動脈硬化、毛細血管拡張性運動失調症、悪液質、ガン、複製老化に関与する骨格筋の変性疾患、糖尿病、頭部外傷、免疫老化、炎症性腸障害(大腸炎及びクローン病など)、筋ジストロフィー、骨関節炎、骨粗鬆症、慢性及び/又は急性疼痛(神経障害性疼痛など)、腎不全、網膜虚血、敗血性ショック(内毒素性ショックなど)及び皮膚加齢などの他の症状及び/又は障害を治療するための;細胞の寿命及び増殖能力を拡張するための;老化細胞の遺伝子発現を変えるための;あるいは、低酸素腫瘍細胞を放射線増感させるための、方法に関する。また、本発明は、動物における疾患及び症状の治療であって、上記動物に、治療上有効量の上記で特定した化合物を投与することを含む治療に関する。
【0065】
本発明は、動物において神経学的障害を治療、予防又は阻害する方法であって、上記動物に、治療上有効量の上記で特定した化合物を投与することを含む、方法に関する。他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。さらなる他の実施形態は、脱髄性疾患及び神経学的障害が神経変性に関する場合である。他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の好ましい実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0066】
他の実施形態は、動物に有効量の本発明の化合物を投与することにより、上記動物においてPARP活性化に関連する狭心症、心筋梗塞、心血管虚血及び心血管組織損傷などの心血管疾患を治療、予防又は阻害する方法である。
【0067】
また、本発明は、動物において神経学的障害を治療、予防又は阻害するために、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷を治療、予防又は阻害するための、PARP活性を阻害するための本発明の化合物の使用を意図する。
【0068】
他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。
【0069】
他の実施形態は、灌流損傷が血管卒中である場合である。さらに他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。さらに他の実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0070】
また、本発明は、本明細書中に記載の動物において疾患及び障害のいずれかを治療するための医薬の製造における、本発明の化合物の使用を意図する。
【0071】
他の実施形態において、疾患又は障害は、神経学的障害である。
【0072】
他の実施形態において、神経学的障害は、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳損傷に関連する脳卒中、局所虚血、全身虚血、灌流損傷、脱髄性疾患及び神経変性に関する神経学的障害からなる群より選択される。他の実施形態は、灌流損傷が血管卒中である場合である。他の実施形態は、末梢神経障害がギラン・バレー症候群により引き起こされる場合である。
【0073】
さらに他の実施形態は、脱髄性疾患が多発性硬化症である場合である。他の実施形態は、神経変性に関連する神経学的障害が、アルツハイマー病、パーキンソン病及び筋萎縮性側索硬化症からなる群より選択される場合である。
【0074】
本発明の他の実施形態において、急性網膜虚血又は急性血管卒中と診断されたヒトは、すぐに、断続的又は連続的な静脈内投与により、本発明の化合物を、単回用量で又は一組の分割用量の化合物として非経口的に投与される。この最初の治療の後、ヒトが示す神経学的症候に応じて、ヒトは、必要に応じて、同じ又は異なる本発明の化合物を、別の非経口投与用の形態で服用してもよい。本発明の化合物は、化合物を含む生体適合性の生分解性ポリマーマトリックスデリバリー系の埋め込みにより、又は、脳の梗塞領域に化合物を直接投与するための硬膜下ポンプにより、断続的又は連続的投与によって投与することができる。
【0075】
他の実施形態において、本発明は、例えば酸化剤の産生の一般的な媒介物として、炎症誘発性の媒介物及び/若しくはサイトカインとして、かつ/又は、白血球浸潤、カルシウムイオン過負荷、リン脂質過酸化、酸化窒素代謝の低下及び/若しくはATP産生の減少の一般的な媒介物として本発明の化合物を使用する際に、細胞の寿命及び増殖能力を拡張する方法を提供する。
【0076】
さらに、本発明の方法は、ガンを治療するのに、及び、腫瘍細胞を放射線増感させるのに用いることができる。用語「ガン」は、広く解釈される。本発明の化合物は、「抗ガン剤」であってよく、上記用語は、「抗腫瘍細胞増殖剤」及び「抗新生物剤」もまた包含する。例えば、本発明の方法は、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍などのガンにおける、ガンの治療、及び、腫瘍細胞の放射線増感において有用である。
【0077】
また、本発明の方法は、有効量のテモゾロマイド及び本発明の化合物を用いる哺乳類のガンの治療であり得る。ガンは、黒色腫、リンパ腫及び多形神経膠芽腫であってよい。
【0078】
放射線増感剤は、電磁放射線の傷害性効果に対するガン細胞の感度を増大させることが知られている。放射線増感剤の作用様式のいくつかのメカニズムが文献中に提唱されており、以下のものが挙げられる:低酸素細胞放射線増感剤(例、2−ニトロイミダゾール化合物、及びベンゾトリアジンジオキシド化合物)が、低酸素組織の再酸化を促進する及び/又は有害な酸素ラジカルの発生を触媒する;非低酸素細胞の放射線増感剤(例、ハロゲン化ピリミジン類)は、DNA塩基のアナログであってよく、ガン細胞のDNAに優先的に取り込まれて、放射線により誘発されるDNA分子の切断を促進する及び/又は正常なDNA修復メカニズムを阻止する;並びに、考えられる種々の他の作用メカニズムについて、疾患の治療における放射線増感剤であるとして仮定されている。
【0079】
多くのガン治療プロトコルは、現在、X線の電磁放射線照射により活性化される放射線増感剤を用いる。X線により活性化される放射線増感剤の例には以下のものが挙げられるが、これらに限定されない:メトロニダゾール、ミソニダゾール(misonidazole)、デスメチルミソニダゾール(desmethylmisonidazole)、ピモニダゾール(pimonidazole)、エタニダゾール(etanidazole)、ニモラゾール(nimorazole)、マイトマイシンC、RSU 1069、SR 4233、EO9、RB 6145、ニコチンアミド、5−ブロモデオキシウリジン(BUdR)、5−ヨードデオキシウリジン(IUdR)、ブロモデオキシシチジン、フルオロデオキシウリジン(FudR)、ヒドロキシウレア、シスプラチン、並びに、その治療上有効なアナログ及び誘導体。
【0080】
ガンの光学力学治療(PDT)は、増感剤の放射線活性化剤などの可視光を用いる。光学力学的放射線増感剤の例には以下が挙げられるが、これらに限定されない:ヘマトポルフィリン誘導体、フォトフリン、ベンゾポルフィリン誘導体、NPe6、スズエチオポルフィリン(tin etioporphyrin)SnET2、フェオボルビド(pheoborbide)−a、バクテリオクロロフィル−a、ナフタロシアニン、フタロシアニン、亜鉛フタロシアニン、並びに、その治療上有効なアナログ及び誘導体。
【0081】
放射線増感剤は、治療上有効量の1つ又はそれ以上の以下の他の化合物(これらに限定されない)と組み合わせて投与することができる:放射線増感剤の標的細胞への取り込みを促進する化合物;治療薬、栄養及び/又は酸素の標的細胞への流入を制御する化合物;さらに放射線が存在する場合又は存在しない場合に腫瘍に作用する化学療法薬剤;あるいは、ガン又は他の疾患を治療するための他の治療上有効な化合物。放射線増感剤と組み合わせて用いることができる治療剤の例には以下が挙げられるが、これらに限定されない:5−フルオロウラシル、ロイコボリン、5’−アミノ−5’デオキシチミジン、酸素、カーボジェン(carbogen)、赤血球輸血、パーフルオロカーボン(例、フルオゾル−DA(Fluosol−DA))、2,3−DPG、BW12C、カルシウムチャネル阻害剤、ペントキシフィリン、抗脈管新生化合物、ヒドララジン及びLBSO。放射線増感剤と組み合わせて用いることができる化学療法薬剤の例には以下が挙げられるが、これらに限定されない:アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、並びに、その治療上有効なアナログ及び誘導体。
【0082】
本発明は、化学療法により誘発される末梢神経障害の治療手段を提供する。本発明の1つの態様によれば、本発明の化合物は、少なくとも1つの化学療法薬剤の投与の前に又はそれと同時に投与されて、神経障害の症候の進行を予防するか、又は、上記症候の重篤性を緩和する。さらなる態様によれば、本発明の化合物は、少なくとも1つの化学療法薬剤の投与後に投与されて、患者の神経障害の症候を治療するか、又は上記症候の重篤性を緩和する。他の態様において、本発明は、哺乳類において腫瘍細胞の増殖を抑制、遅延又は停止させる方法であって、化学療法薬剤の投与を含み、さらに、グループIの化合物を、上記化学療法薬剤の抗腫瘍活性を増強するのに十分な量で投与することを含む、方法を提供する。
【0083】
他の実施形態において、本発明の化合物は、PARP阻害剤として作用し、他の化学療法薬剤の細胞傷害性効果を化学的に増強することにより、ガンを治療又は予防する。
【0084】
本発明は、腫瘍細胞へのイオン化放射線の細胞傷害性効果を増強することにより、ガンの影響を治療、予防及び/又は緩和するためのグループIの化合物、その誘導体、並びに、これらの化合物を含む組成物を提供する。
【0085】
他の実施形態において、本発明は、化学療法薬剤の腫瘍細胞への細胞傷害性効果を増強することによりガンの影響を治療、予防及び/又は緩和するための、本願明細書に記載の化合物、その誘導体、及び、これらの化合物を含む組成物を提供する。
【0086】
他の実施形態において、本発明の方法は、ガンを治療するのに、及び、腫瘍細胞を化学増感させるのに用いることができる。本明細書中で用いる用語「ガン」は、広く定義される。本発明の化合物は、「抗ガン剤」の効果を増強することができ、上記用語はまた、「抗腫瘍細胞増殖剤」、「化学療法薬剤」、「細胞増殖抑制剤」、「細胞傷害性剤」及び「抗新生物剤」も包含する。
【0087】
一実施形態において、本発明の方法は、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍などのガンにおける、ガンの治療、及び、腫瘍細胞の放射線増感又は化学増感に有用である。
【0088】
本発明は、腫瘍及び/又はガン細胞の治療のための化学増感方法であって、上記ガン細胞をグループIのジアザベンゾ[デ]アントラセン−3−オン化合物と接触させること、及び、上記ガン細胞を抗ガン剤にさらに接触させることを含む、方法を提供する。
【0089】
本発明の特定の実施形態には、グループIに示すジアザベンゾ[デ]アントラセン−3−オン化合物、並びに、その中性及び/又は塩の形態、並びに、適切な場合にはそのエナンチオマー及びラセミ体混合物が挙げられる。
【0090】
本発明の化合物は1つ又はそれ以上の不斉中心を有していてもよく、従って、立体異性体混合物(ラセミ体及び非ラセミ体)として、あるいは個別のエナンチオマー又はジアステレオマーとして製造することができる。個別の立体異性体は、光学活性な出発物質を用いることによって、又は、合成のいくつかの適切な段階で中間体のラセミ体若しくは非ラセミ体混合物を分割することによって、又は、グループIの化合物を分割することによって、得ることができる。個別の立体異性体並びに立体異性体の混合物(ラセミ体及び非ラセミ体)は、本発明の範囲に含まれることが理解される。
【0091】
本発明の化合物は、遊離塩基の形態で、医薬品に許容される塩、医薬品に許容される水和物、医薬品に許容されるエステル、医薬品に許容される溶媒和物、医薬品に許容されるプロドラッグ、医薬品に許容される代謝物の形態で、かつ、医薬品に許容される立体異性体の形態で有用である。これらの形態は全て本発明の範囲内である。
【0092】
「医薬品に許容される塩」、「水和物」、「エステル」又は「溶媒和物」は、所望の薬理学的活性を有するが生物学的な意味やそれ以外の意味で望ましくない、本発明の化合物の塩、水和物、エステル又は溶媒和物を意味する。有機酸は、塩、水和物、エステル又は溶媒和物、例えば、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、重硫酸塩、スルファミン酸塩、硫酸塩、ナフチル酸塩、酪酸塩、クエン酸塩、ショウノウ酸塩、ショウノウスルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、エタンスルホン酸、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、ヘミ硫酸ヘプタン酸塩、ヘキサン酸塩、2−ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、トシル酸塩及びウンデカン酸塩などを製造するのに用いることができる。無機塩は、塩、水和物、エステル又は溶媒和物、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩及びチオシアン酸塩などの製造に用いることができる。
【0093】
適切な塩基塩、水和物、エステル又は溶媒和物の例には、アンモニアの水酸化物、炭酸塩及び重炭酸塩、ナトリウム塩、リチウム塩及びカリウム塩などのアルカリ金属塩、カルシウム塩及びマグネシウム塩などのアルカリ土類金属塩、アルミニウム塩及び亜鉛塩が挙げられる。
【0094】
また、塩、水和物、エステル又は溶媒和物は、有機塩基によっても形成することができる。本発明の化合物の医薬品に許容される塩基付加塩、水和物、エステル又は溶媒和物の形成に適切な有機塩基には、非毒性でかつ塩、水和物、エステル又は溶媒和物を形成するのに十分に強いものが挙げられる。例えば、そのような有機塩基の分類には、モノ−、ジ−及びトリアルキルアミン、例えば、メチルアミン、ジメチルアミン、トリエチルアミン及びジシクロヘキシルアミン;モノ−、ジ−又はトリヒドロキシアルキルアミン、例えば、モノ−、ジ−及びトリエタノールアミン;アミノ酸、例えば、アルギニン及びリジン;グアニジン;N−メチル−グルコサミン;N−メチル−グルカミン;L−グルタミン;N−メチル−ピペラジン;モルホリン;エチレンジアミン;N−ベンジル−フェネチルアミン;(トリヒドロキシメチル)アミノエタンなどを挙げることができる。例えば,“Pharmaceutical Salt”J.Pharm.Sci,66:1,1−19(1977)を参照されたい。従って、塩基性窒素含有基は、以下に挙げられる剤で4級化することができる:塩化、臭化及びヨウ化メチル、塩化、臭化及びヨウ化エチル、塩化、臭化及びヨウ化プロピル、並びに、塩化、臭化及びヨウ化ブチルなどの低級アルキルハロゲン化物;硫酸ジメチル、ジエチル、ブチル及びジアミルなどの硫酸ジアルキル;塩化、臭化及びヨウ化デシル、塩化、臭化及びヨウ化ラウリル、塩化、臭化及びヨウ化ミリスチル、並びに、塩化、臭化及びヨウ化ステアリルなどの長鎖ハロゲン化物;並びに、ベンジル及びフェネチルブロミドなどのアラルキルハロゲン化物。
【0095】
塩基性化合物の酸付加塩、水和物、エステル又は溶媒和物は、本発明の化合物の遊離塩基を水溶液又はアルコール水溶液に溶解するか、あるいは、適切な酸又は塩基を含む他の適切な溶媒に溶解し、溶液を蒸発させて塩を単離することにより製造することができる。あるいは、本発明の化合物の遊離塩基を酸と反応させることができ、また、酸基を有する本発明の化合物を塩基と反応させることができ、その結果、反応物が有機溶媒中に得られ、この場合、塩が直接分離するか、又は、溶液を濃縮することによって塩を得ることができる。
【0096】
「医薬品に許容されるプロドラッグ」は、生体内で変換された後でその薬理学的効果を発揮するような、本発明の化合物の誘導体を意味する。プロドラッグは、化学的安定性の改善、患者受容性及びコンプライアンスの改善、バイオアベイラビリティーの改善、作用期間の延長、器官選択性の改善、処方の改善(例、水溶性の増大)及び/又は副作用(例、毒性)の低減を目的として処方される。プロドラッグは、Burgers Medicinal Chemistry and Drug Chemistry,Fifth Ed,Vol.1,pp.172−178,949−982(1995)に記載のような上記分野で公知の方法を用いて、本発明の化合物から容易に製造することができる。例えば、本発明の化合物は、1つ又はそれ以上のヒドロキシ基又はカルボキシ基をエステルに転化することにより、プロドラッグに変換することができる。
【0097】
「医薬品に許容される代謝物」は、代謝性変換を受ける薬剤を意味する。体内に入った後、大半の薬剤は、これらの物理的特性及び生物的効果を変化させ得る化学反応の基質である。化合物の極性に概して影響を与えるこれらの代謝性変換は、薬物が体内で分散して体内から排泄される経路を変える。しかし、治療上の効果のために薬物の代謝が必要とされる場合もある。例えば、抗代謝物クラスの抗ガン剤は、ガン細胞に輸送された後で活性型に転化される必要がある。多くの薬剤は何らかの代謝性変換を受けるため、薬物代謝に関与する生化学的反応は多数かつ広範囲にわたる。薬物代謝の主要な部位は肝臓であるが、他の組織が関与する場合もある。
【0098】
本発明の医薬組成物
また、本発明は、(i)治療上有効量のジアザベンゾ[デ]アントラセン−3−オン誘導体の化合物、及び、(ii)医薬品に許容される担体を含む医薬組成物に関する。
【0099】
本発明の化合物の好ましい使用及び投与の実施形態に関する上記論述は、本発明の医薬組成物にも適用される。
【0100】
本明細書中で用いる用語「医薬品に許容される担体」には、任意の担体、希釈剤、賦形剤、懸濁剤、滑剤、アジュバント、ビヒクル、デリバリーシステム、乳化剤、崩壊剤、吸着剤、保存剤、界面活性剤、着色料、香料又は甘味料が挙げられる。
【0101】
これらの目的のため、本発明の組成物は、経口的に、非経口的に、吸入スプレー、吸収、吸着により、局所的に、経直腸的に、経鼻的に、経頬的に、経膣的に、脳室内に、医薬品に許容される従来の非毒性担体を含む投与調製物中におけるインプラントレザバーによって、又は、任意の他の簡便な投与形態により、投与することができる。本明細書中で用いる用語、非経口的には、皮下、静脈内、筋肉内、腹膜内、くも膜下腔内、脳室内、胸骨内及び頭蓋内注射又は灌流技術が挙げられる。
【0102】
非経口的に投与される場合、組成物は、単回投与の無菌の注射可能な形態(溶液、懸濁液又はエマルション)であり、これは、好ましくは、患者の血液と医薬品に許容される担体と等張性である。このような無菌の注射可能な形態の例には、無菌の注射可能な水性又は油性の懸濁液が挙げられる。これらの懸濁液は、上記分野で公知の技術に従って、適切な分散剤又は湿潤剤及び懸濁剤を用いて処方することができる。また、無菌の注射可能な形態は、非毒性の非経口的に受容され得る希釈剤又は溶液中の無菌の注射可能な溶液又は懸濁液、例えば、1,3−ブタンジオール溶液である。許容されるビヒクル及び溶液としては、水、生理食塩水、リンガー液、デキストロース溶液、等張性の塩化ナトリウム溶液及びハンクス溶液を用いることができる。さらに、無菌の不揮発性油が、溶液又は懸濁媒体として通常使用される。この目的のために、任意の無刺激性の不揮発性油を用いることができ、合成モノ又はジグリセリド、コーン油、綿実油、ピーナツ油及びゴマ油が挙げられる。オレイン酸エチル、ミリスチン酸イソプロピル及びオレイン酸などの脂肪酸、並びに、オリーブ油及びヒマシ油、特にこれらのポリオキシエチル化体を含むグリセリド誘導体は、注射可能薬剤の製造に有用である。また、これらの油性溶液又は懸濁液は、長鎖アルコール希釈剤又は分散剤を含んでいてよい。
【0103】
無菌の生理食塩水は担体として好ましく、化合物は、予期可能なあらゆる用途のための溶液とするのに十分に水溶性である場合が多い。担体は、溶解度、等張性及び化学的安定性を増強する物質、例えば、抗酸化剤、緩衝剤及び保存剤などの添加剤を少量含んでいてもよい。
【0104】
経鼻又は経頬投与に適切な調製物(自己推進性の粉体払い出し用調製物)は、約0.1%〜約5%w/wの、例えば1%w/wの活性成分を含んでいてもよい。従って、本発明のヒト用医薬用途の調製物は、活性成分を、医薬品に許容される担体と、必要に応じて他の治療上の成分と共に含む。
【0105】
経口的に投与される場合、組成物は、通常、上記分野で公知の従来の装置及び技術を用いて、錠剤、カシェ剤、散剤、顆粒、ビーズ、チュアブル錠剤、カプセル、液体、水性懸濁液又は溶液などの単回投与形態又は類似の投与形態に処方されるであろう。このような調製物は、代表的には、固体、半固体又は液体の担体を含む。担体としては、例えば、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アラビアゴム、リン酸カルシウム、鉱油、カカオバター、カカオ油、アルギン酸塩、トラガカント、ゼラチン、シロップ、メチルセルロース、ポリオキシエチレンソルビタンモノラウレート、ヒドロキシ安息香酸メチル、ヒドロキシ安息香酸プロピル、タルク、ステアリン酸マグネシウムなどが挙げられる。
【0106】
本発明の組成物は、好ましくは、グループIの化合物の単回又は分割用量を含むカプセル又は錠剤として投与される。好ましくは、組成物は、単回又は複数回投与用の無菌の溶液、懸濁液又はエマルションとして投与される。錠剤は、ラクトース及びコーンスターチなどの担体、並びに/又は、ステアリン酸マグネシウムなどの滑剤を含んでいてもよい。カプセルは、ラクトース及び乾燥コーンスターチを含む希釈剤を含んでもよい。
【0107】
錠剤は、活性成分を、必要に応じて1つ又はそれ以上の補助成分と共に圧縮又は成形することにより製造することができる。圧縮錠剤は、適切な機械中で、活性成分を、散剤又は顆粒などの自由に流動する形態で圧縮し、必要に応じて、結合剤、滑剤、不活性な希釈剤、界面活性剤又は分散剤と共に混合することにより製造できる。成形錠剤は、適切な機械で、粉末活性成分と、不活性な液状希釈剤で湿らせた適切な担体との混合物を成形することにより製造することができる。
【0108】
また、本発明の化合物は、坐剤の形態で経直腸的に投与することができる。これらの組成物は、室温では固体であるが直腸温では液体であり、直腸で融解して薬剤を放出できる適切な非刺激性の賦形剤と共に混合することによって製造することができる。このような材料には、カカオバター、ミツロウ及びポリエチレングリコールが挙げられる。
【0109】
また、本発明の組成物及び方法は、制御放出技術を適用することができる。このため、例えば、本発明の化合物を、一定期間で制御放出させる目的で疎水性ポリマーマトリックスに配合してもよい。次いで、本発明の組成物は、頻繁に投与しなおす必要なく、長期間にわたってPARP阻害剤の有効濃度を提供するために、固体インプラント、又は、外用パッチに成形することができる。このような制御放出フィルムは上記分野で周知である。特に好ましいものは、経皮デリバリーシステムである。本発明に用いることができる、この目的に一般的に用いられる他の例証されるポリマーには、外用又は内服で用いることができる非分解性エチレン−酢酸ビニルコポリマー、分解性乳酸−グリコール酸コポリマーが挙げることができる。ポリ(ヒドロキシエチルメタクリレート)又はポリ(ビニルアルコール)などの特定のヒドロゲルもまた有用であり得るが、上記したような他のポリマー放出システムより放出サイクルが短い。
【0110】
好ましい実施形態において、担体は、適当な時間放出特性及び放出動力学を有する固体生分解性ポリマー又は生分解性ポリマーの混合物である。また、本発明の組成物は、本発明の化合物の有効な濃度を、頻繁に投与しなおす必要なく長期間にわたって提供するのに適切な固体インプラントに成形することができる。本発明の組成物は、当業者に公知の任意の適切な様式で生分解性ポリマー又はポリマー混合物に配合することができ、生分解性ポリマーと共に均一なマトリックスを形成することができるか、又は、所定の方法でポリマー中に封入することができるか、又は、固体インプラントに成形することができる。
【0111】
一実施形態において、生分解性ポリマー又はポリマー混合物を使用して、本発明の医薬組成物を含む軟質の「デポー剤」を形成することができ、これは、流動性の液体として例えば注射により投与できるが、注射部位の周辺の局所で医薬組成物を維持することができるほど十分な粘性を保つ。このように形成されたデポー剤の分解時間は、選択したポリマー及びその分子量に依存して、数日から数年の間で変動し得る。ポリマー組成物を注射可能な形態で用いることにより、切開する必要がなくなる場合がある。いずれにしても、可撓性又は流動性のデリバリー用「デポー剤」は、周囲組織に外傷をほとんどつけずに、体内での空間の形状に適合するであろう。本発明の医薬組成物は治療上有効な量で用いられ、この量は、所望の放出特性、増感効果を発揮するために要する医薬組成物の濃度、及び、治療のために医薬組成物を放出するべき期間の長さに依存するであろう。
【0112】
本発明の化合物は、組成物中において治療上効果的な量で用いられる。組成物は、滅菌することができ、並びに/又は、保存剤、安定化剤、ウェル化剤若しくは乳化剤、溶液促進剤、浸透圧を調整するための塩及び/若しくは緩衝剤などのアジュバントを含んでいてよい。さらに、これらはまた、本明細書中に記載の特定の化学療法薬剤などの他の治療上有益な物質を含んでいてよいが、これらに限定されない。組成物は、従来の混合、顆粒化又はコーティング方法に従って製造され、約0.1〜75重量%,好ましくは1〜50重量%の本発明の化合物を含む。
【0113】
中枢神経系標的として有効な治療であるためには、本発明の化合物は、末梢から投与された場合に血液−脳関門を容易に通過すべきである。血液−脳関門を通過できない化合物は、脳室経路又は脳への投与に適切な他のデリバリーシステムにより、効果的に投与することができる。
【0114】
医薬用途には、治療上の効果を達成するために必要な活性成分の量は、特定の化合物、投与の経路、治療される哺乳類及び治療される特定の障害又は疾患により変動するであろう。本明細書中の上記に記載の症状のいずれかに罹患した又はおそらく罹患しているであろう哺乳類についての本発明の化合物又はその薬理学的に許容される塩の適切な全身用量は、活性成分化合物の約0.1mg/kg〜約100mg/kgであり、最も好ましい用量は約1〜約10mg/kgである。
【0115】
しかし、任意の特定の患者についての特定の用量レベルは、用いる特定の化合物の活性、年齢、体重、全身の健康状態、性別、食事、投与時間、排泄速度、薬剤の組み合わせ、及び、治療される特定の疾患の重篤性、及び、投与形態を含む種々の因子に依存するであろうことが理解される。
【0116】
通常の技術を有する医師又は獣医師は、治療薬が投与される症状の治療の予防又は治療用の化合物の有効量を、容易に決定しかつ処方するであろうことが理解される。例えば、その作業中において、医師又は獣医師は、適切であると考えられる場合には、静脈内ボーラスを用い、続いて、静脈内灌流を実施することができる、及び、非経口的又は経口的に繰り返し投与することができる。活性成分は単独で投与することもできるが、活性成分を調製物として提供することが好ましい。
【0117】
本発明の組成物を含む投与形態を製造する場合、化合物は、ゼラチン、α化デンプンなどの結合剤;例えば、水素添加植物油、ステアリン酸などの滑剤;ラクトース、マンノース及びスクロースなどの希釈剤;カルボキシメチルセルロース及びナトリウムデンプングリコレートなどの崩壊剤;ポビドン、ポリビニルアルコールなどの懸濁剤;二酸化ケイ素などの吸収剤;メチルパラベン、プロピルパラベン及び安息香酸ナトリウムなどの保存剤;ラウリルスルホン酸ナトリウム、ポリソルベート80などの界面活性剤;F.D.&C.染料及びレーキなどの着色料;着香料;及び甘味料などの従来の賦形剤と混合することもできる。
【0118】
本発明は、本明細書中に記載の動物におけるいずれかの疾患又は障害を治療するための医薬の製造における、グループIの化合物の使用に関する。一実施形態において、本発明の化合物は、ガンを治療するのに使用される。好ましい実施形態において、本発明の化合物は、イオン化放射線の細胞傷害性効果を増強するのに用いられる。上記実施形態において、本発明の化合物は、放射線増感剤として作用する。別の好ましい実施形態において、本発明の化合物は、化学療法薬剤の細胞傷害性効果を増強するのに用いられる。上記実施形態において、本発明の化合物は、化学増感剤として作用する。
【0119】
DNAを損傷するよう作用する医薬品に許容される化学療法薬剤はいずれも、本発明の化学療法薬剤として適切である。特に、本発明は、化学治療上有効量の少なくとも1つの以下の化学療法薬剤の使用を意図するが、これに限定されない:テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α,β,γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、その治療上有効なアナログ及び誘導体、並びに、これらの混合物。好ましい態様によれば、化学療法薬剤はテモゾロマイドである。
【0120】
本明細書中に含まれる開示は、本発明の化合物及び組成物が、腫瘍及び/又はガン細胞の化学療法薬剤に対する放射線増感及び/又は化学増感などにより、ガンを治療及び/又は予防するのに有用であることを示す。
【0121】
本発明のジアザベンゾ[デ]アントラセン−3−オン化合物は、米国第60/644,584号(その全体は、全ての目的のために参照として援用される)に開示された出発物質及び方法を用いて合成することができる。
【0122】
本発明の方法及び医薬組成物に用いるPARP阻害剤のうちのいくつかは、刊行物に図示された一般的な合成経路および例示を用いて、有機化学の標準的な技術により、容易に製造することができる。例えば、Wuら, “The Protective Effect of GPI 18078, a Novel Water Soluble Poly(ADP−Ribose) Polymerase Inhibitor in Myocardial Ischemia−Reprefusion Injury, Experimental Biology,“FASEB, April 11−15 (2003); Wuら,“Myocardial Protection and Anti−Inflammatory Effect of GPI 15427, a Novel Water Soluble Poly(ADP−Ribose) Polymerase Inhibitor: Comparison with GPI 6150, Experimental Biology,” FASEB, April 11−15 (2003); Kalishら, “Design, Synthesis and SAR of PARP−1 Inhibitors, ISMC Meeting, Barcelona,”September 4, 2002; Xuら,“Design and Synthesis of Novel Potent Poly(ADP−Ribose) Polymerase (PARP) Inhibitors, 224th ACS National Meeting,” Boston, August 18−23 (2002); Williamsら, “Intravenous Delivery of GPI 15427/C and GPI 16539/C, Potent Water−Soluble PARP Inhibitors, Reduces Infarct Volume Following Permanent and Transient Focal Cerebral Ischemia, Soceity for Neuroscience,” Orlando FL, October (2002); Tentori L,ら,“Systemic administration of the PARP−1 inhibitor GPI 15427 increases the anti−tumor activity of temozolomide against metastatic melanoma,” Medical Science Monitor, Vol. 9, supplement 1, 34 (2003); Tentoriら, “Poly(ADP−Ribose) Polymerase Inhibitor to Increase Temozolomide Efficacy Against Melanoma, Glioma and Lymphoma at the CNS Site,” AACR poster, April (2003); Sutoら, “Dihydroiso−quinolinones: The Design and Synthesis of a New Series of Potent Inhibitors of Poly(ADP−ribose) Polymerase,” Anticancer Drug Des., 6:107−17 (1991);並びに、米国特許第6,348,475号、同第6,545,011号、米国再発行特許第36,397号、米国特許第6,380,211号、同第6,235,748号、同第6,121,278号、同第6,197,785号、同第6,380,193号、同第6,346,536号、同第6,514,983号、同第6,306,889号、同第6,387,902号、同第6,201,020号及び同第6,291,425号並びに米国特許出願第10/853,714号(上記特許、特許出願及び刊行物の全ての内容は、これらが本明細書中に全文が記載されているのと同様に、本明細書中で援用される)を参照されたい。
【0123】
本発明の化合物は、以下のスキーム1に例示される従来の様式で製造することができる。出発誘導体は、化学文献で公知であり、当業者に公知のプロセスで入手可能である。
【0124】
【化5】
【0125】
一般的手順A:7−ブロモメチル−9−オキソキサンテン−l−カルボン酸メチルエステルの製造
【0126】
N−ブロモスクシンイミド、臭素及び臭化錯体、例えば臭化ピリジニウムを含むブロモ化剤を用いることによって、7−メチル−9−オキソキサンテン−1−カルボン酸メチルエステル(1)を7−ブロモメチル−9−オキソキサンテン−l−カルボン酸メチルエステル(2)に転化することができる。適切な溶液には、塩素化炭化水素、極性非プロトン性溶液及び種々のエーテルが挙げられるが、これらに限定されない。温度は、一般的に0〜100℃の間であり、50〜70℃の範囲が好ましい。
【0127】
実施例1:
過酸化ベンゾイル(10g、0.041mol)を含む還流四塩化炭素(10L)中に化合物5(400g、1.49mol)を含む溶液を撹拌している中に、NBS(292g、1.64mol)を、数回に分けて45分間かけて添加した。得られた混合物を12時間還流し、次に一晩かけて室温まで冷却した。沈殿したものをろ過し、得られた固形物を水(1.2L)で十分に洗浄し、乾燥させ、322gの化合物6を白色固体で得た(62%)。
【0128】
実施例2:
四塩化炭素(400mL)中に化合物1(1.97g、7.3mmol、1.00eq)を含む溶液に、N−ブロモコハク酸イミド(1.44g、8.1mmol、1.10eq)及び過酸化ベンゾイル(45mg、0.2mmol、触媒)の触媒量を添加した。反応混合物を加熱、6時間還流し、次に、室温まで冷却した。得られた白色沈殿物を真空ろ過により単離した。残った溶媒は除去し、ろ過固形物は酢酸エチル及びヘキサンで2度再結晶化し、白色固体2(1.15g,45%)を得た。
1H−NMR(400MHz,CDCl3)8.27(d,J=2.5Hz,1H),7.72−7.80(m,2H),7.57(dd,J=8.5及び1.1Hz,1H),7.48(d,J=8.5Hz,1H),7.33(dd,J=7.0及び1.1Hz,1H),4.57(s,2H),4.00(s,3H).13C−NMR(400MHz,CDCl3)31.97,53.06,118.63,118.71,119.63,121.56,122.81,126.83,134.15,134.18,134.50,135.95,155.31,155.87,169.72,175.48.
【0129】
一般的手順B:置換9−オキソキサンテン−1−カルボン酸メチルエステルの調製
化合物2中の第一臭化物(primary bromide)は、第一級及び第二級アミンを含む求核剤によって容易に置換され、これは炭酸カリウムなどの非反応性塩基種の存在下で好ましく実施される。これらの変換にはジメチルホルムアミドやアセトニトリルなどの極性及び非プロトン性のものが最適であるが、その他の溶媒中でも上記反応を実施することができる。温度範囲は0〜100℃であってよく、50〜80℃が好ましいであろう。
【0130】
実施例1:
化合物2(3.47g,10.0mmol,1.00eq)のジメチルホルムアミド(100mL)溶液に、炭酸カリウム(13.82g,100.0mmol,10.00eq)及び第二級アミン(10mmol,1eq)を添加する。反応混合物を70℃で6時間加熱し、次に室温まで冷却する。反応混合物に水(100mL)を加え、続いて酢酸エチル(200mL)を加える。有機層を回収し、水で洗浄し、続いて塩水で洗浄し、次に硫酸ナトリウム又は硫酸マグネシウムで乾燥させる。溶媒を真空において除去し、残渣を、溶離液として酢酸エチル及びヘキサンを使用してカラムクロマトグラフィーで精製し、生成物3を50〜90%の収率で得る。
【0131】
実施例2:
化合物2(1.53g,4.4mmol,1.00eq)のアセトニトリル(50mL)溶液に、炭酸カリウム(1.2g,8.7mmol,2.00eq)及び1−メチルピペラジン(0.51mL,4.6mmol,1.05eq)を添加した。次に反応混合物を加熱し一晩還流した。室温まで冷却した後、固体をろ過にて除去し、有機層は蒸発させて、油状残渣を得た。この物質を酢酸エチル(150ml)に溶解し、1N HCl(150ml)で抽出した。有機層は廃棄し、水層のpHを6N水酸化ナトリウムで9より大きくなるよう調整した。次に、生成物を酢酸エチル(100ml)で2回に分けて抽出し、次いでこれらを合わせて、水及び塩水で連続して洗浄し、硫酸マグネシウムで乾燥させた。全ての溶媒を真空において除去し、白色固体の3aを得た(0.95g,59%)。
3aの1H−NMR(400MHz,CDCl3):8.18(d,J=2.5Hz,1H),7.71−7.75(m,2H),7.56(dd,J=8.5及び1.1Hz,1H),7.45(d,J=8.5Hz,1H),7.32(dd,J=7.0及び1.1Hz,1H),4.04(s,3H),3.58(s,2H),2.45(br,8H),2.27(s,3H).
【0132】
一般的手順C:
10−アミノメチル−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン誘導体の調製
ベンゾピラノ[4,3,2−デ]フタラジン環を形成する化合物3におけるケトン及びメチルエステルの環化反応はヒドラジンを使用して実施することができ、10−アミノメチル−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン誘導体4を高収率で得る。溶媒としてはエタノールが好ましいが、反応は本溶媒に限定されない。温度は0〜120℃の範囲であってよく、70〜90℃が最も望ましい。
【0133】
実施例1:
化合物3(5mmol)の無水エタノール(10ml)溶液に、無水ヒドラジンのエタノール(1ml)溶液を室温で滴下する。滴下終了後、溶液を加熱し、一晩還流する。一度室温まで冷却し、氷冷水(100mL)を加え、白色固体を沈殿させる。固体を真空ろ過で回収し、水及びエタノールで連続して洗浄し、真空において乾燥させ、白色固体4(収率40〜85%)を得る。
【0134】
実施例2:
7−(1,4−ジオキサ−8−アザ−スピロ[4.5]デカ−8−イルメチル)−9−オキソ−9Η−キサンテン−1−カルボン酸メチルエステル、3n(1.6g,3.91mmol,1eq)のメタノール(55ml)溶液を80℃まで加熱し、撹拌して全物質を溶解させた。これに、ヒドラジン一水和物(20ml,過剰量)を10分かけて滴下した。反応混合物を加熱し還流を一晩行い、重い白色沈殿物を形成させた。この溶液を室温まで冷却し、生成物を真空ろ過で単離した。少量の水、エタノール及びペンタンで連続して洗浄し、真空において乾燥して4nを高収率で得た(1.4g,92%)。
1H−NMR(DMSO−d6,300MHz):1.62(t,J=5.0,4H),2.40−2.50(m,4H),3.55(s,2H),3.85(s,4H),7.34(d,J=9.4Hz,1H),7.47(d,J=7.5Hz,1H),7.67−7.70(m,1H),7.86−7.92(m,2H),7.98(s,1H),12.62(s,1H).
【0135】
化合物4a:10−(4−イソプロピル−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0136】
【化6】
【0137】
一般的手順B及びCに従って、化合物2及び1−プロピル−ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4aを得た。
MS(ES+):377.1H−NMR(CDCl3,300MHz):0.90−1.00(m,6H),2.25−2.50(m,8H),2.55−2.60(m,1H),3.34(s,2H),7.35(d,1H),7.46(d,1H),7.68(dd,1H),7.80−7.95(m,2H),7.95−8.05(m,1H),12.63(s,1H).Anal.Calcd.for C22H24N4O2:C,70.19;H,6.43;N,14.88.Found:C,70.09;H,6.51;N,14.77.
【0138】
化合物4b:10−[4−(2−メトキシ−エチル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2−ジアゾ−ベンゾ[デ]アントラセン−3−オン
【0139】
【化7】
【0140】
一般的手順B及びCに従って、化合物2及び1−(2−メトキシエチル)ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4bを得た。
MS(ES+):393.1H−NMR(DMSO−d6,300MHz):2.30−2.49(m,10H),3.22(s,3H),3.30−3.45(m,2H),3.50(m,2H),7.30−7.35(m,1H),7.40−7.48(m,1H),7.65−7.70(m,1H),7.85−7.95(m,2H),7.95−8.05(m,1H),12.63(s,1H).Anal.Calcd.forC22H24N4O3:C,67.33;H,6.16;N,14.28.Found:C,67.35;H,6.16;N,14.45.
【0141】
化合物4c:10−(4−ピリミジン−2−イル−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0142】
【化8】
【0143】
一般的手順B及びCに従って、化合物2及び1−(2−ピリミジル)ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4cを得た。
MS(ES+):413.1H−NMR(DMSO−d6,300MHz):3.28−3.34(m,4H),3.60(s,2H),3.70−3.78(m,4H),6.60−6.62(m,1H),7.38−7.40(m,1H),7.50−7.60(m,1H),7.70−7.74(m,1H),7.80−7.95(m,2H),8.05−8.10(m,1H),8.30−8.40(m,2H),12.64(s,1H).Anal.Calcd.for C23H20N6O2:C,66.98;H,4.89;N,20.38.Found:C,67.03;H,4.88;N,20.15.
【0144】
化合物4d:10−(3−オキソ−ピペラジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0145】
【化9】
【0146】
一般的手順B及びCに従って、化合物2及びピペラジン−2−オンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4dを得た。
MS(ES+):349;1H−NMR(DMSO−d6,300MHz):2.58−2.62(m,2H),2.94(s,2H),3.16−3.20(m,2H),3.63(s,2H),7.30−7.35(m,1H),7.40−7.48(m,1H),7.65−7.70(m,1H),7.85−7.95(m,2H),7.95−8.05(m,1H).
【0147】
化合物4e:10−[4−(2−ピロリジン−1−イル−エチル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2,ジアザ−ベンゾ[デ]アントラセン−3−オン
【0148】
【化10】
【0149】
一般的手順B及びCに従って、化合物2及び1−(2−ピロリジン−1−イル−エチル)−ピペラジンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4eを得た。
1H−NMR(DMSO−d6,300MHz):1.64(m,4H),2.30−2.55(m,16H),3.52(s,2H),7.30−7.40(m,1H),7.45−7.50(m,1H),7.70−7.75(m,1H),7.80−7.90(m,2H),8.00−8.05(m,1H).Anal.Calcd.for C25H29N5O2−(0.7H2O):C,67.61;H,6.90;N,15.77;Found:C,67.25;H,6.81;N,15.67.
【0150】
化合物4f:10−[4−(3−ジメチルアミノ−プロピル)−ピペラジン−1−イルメチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0151】
【化11】
【0152】
一般的手順B及びCに従って、化合物2及びジメチル−(3−ピペラジン−1−イル−プロピル)−アミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4fを得た。
1H−NMR(DMSO−d6,300MHz):1.45−1.55(m,2H),2.09(s,6H),2.10−2.40(m,12H),3.52(s,2H),7.30−7.40(9m,1H),7.40−7.50(m,1H),7.70−7.75(m,1H),7.80−7.95(m,2H),8.01(s,1H).
【0153】
化合物4g:10−{[(2−ジメチルアミノ−エチル)−エチル−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0154】
【化12】
【0155】
一般的手順B及びCに従って、化合物2及びN,N−ジメチル−N’−エチル−エタン−1,2−ジアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4gを得た。
1H−NMR(DMSO−d6,300MHz):0.90(t,J=7.2Hz,6H),1.00(t,J=6.8Hz,3H),2.41(dd,J=14.3及び7.2Hz,4H),2.45−2.55(m,6H),3.62(s,2H),7.35(d,J=8.6Hz,1H),7.49(dd,J=8.3及び2.3Hz,1H),7.69(dd,J=7.1及び2.4Hz,1H),7.86−7.93(m,2H),8.03(d,J=2.3Hz,1H)
【0156】
化合物4h:10−{[(2−ジエチルアミノ−エチル)−メチル−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0157】
【化13】
【0158】
一般的手順B及びCに従って、化合物2及びN,N−ジエチル−N’−メチル−エタン−1,2−ジアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4hを得た。
1H−NMR(DMSO−d6,300MHz):12.63(s,1H),8.00(d,J=1.9Hz,1H),7.91−7.80(m,2H),7.70(dd,J=7.1及び2.0Hz,1H),7.49(dd,J=8.6及び2.0Hz,1H),7.36(d,J=8.5Hz,1H),3.55(s,2H),3.88(m,4H),2.47(q,J=7.0Hz,4H),2.17(s,3H),0.93(t,J=7.0Hz,6H).Anal.Calcd.for C22H26N4O2:C,69.82;H,6.92;N,14.80;Found:C,69.56;H,6.95;N,14.60.
【0159】
化合物4i:10−(4−ヒドロキシ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0160】
【化14】
【0161】
一般的手順B及びCに従って、化合物2及びピペリジン−4−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4iを得た。
1H−NMR(DMSO−d6,300MHz):12.60(s,1H),7.96(d,J=1.9Hz,1H),7.91−7.84(m,2H),7.66(dd,J=6.9及び2.3Hz,1H),7.43(dd,J=8.6及び2.1Hz,1H),7.32(d,J=8.6Hz,1H),4.54(d,J=4.2Hz,1H),3.48(s,2H),3.46(m,1H),2.65(m,2H),2.05(m,2H),1.68(m,2H),1.40(m,2H).Anal.Calcd.for C20H19N3O3:C,68.75;H,5.48;N,12.03;Found:C,68.66;H,5.48;N,12.13.
【0162】
化合物4j:10−{[エチル−(2−ヒドロキシ−エチル)−アミノ]−メチル}−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0163】
【化15】
【0164】
一般的手順B及びCに従って、化合物2及び2−エチルアミノ−エタノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4jを得た。
1H−NMR(DMSO−d6,300MHz):1.00(t,J=6.4Hz,3H),2.45−2.55(m,4H),3.49(dd,J=12.0及び5.5Hz,2H),3.64(s,2H),4.39(t,J=5.1Hz,1H),7.35(d,J=8.5Hz,1H),7.50(dd,J=8.8及び6.5Hz,1H),7.69(dd,J=6.5及び4.3Hz,1H),7.86−7.91(m,2H),8.02(d,J=2.0,1H).
【0165】
化合物4k:10−[(ジイソプロピルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0166】
【化16】
【0167】
一般的手順B及びCに従って、化合物2及びジイソプロピルアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4kを得た。
1H−NMR(DMSO−d6,300MHz):1.01(d,J=6.3Hz,12H),2.93−3.04(m,2H),3.66(s,2H),7.34(d,J=8.4Hz,1H),7.51(dd,J=9.2及び1.9Hz,1H),7.69(dd,J=6.5及び2.7Hz,1H),7.86−7.91(m,2H),8.09(d,J=1.9Hz,1H).
【0168】
化合物4l:10−(3−ヒドロキシ−ピロリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0169】
【化17】
【0170】
一般的手順B及びCに従って、化合物2及ピロリジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4lを得た。
1H−NMR(DMSO−d6,300MHz):1.50−1.60(m,1H),1.95−2.05(m,1H),2.31−2.35(m,1H),2.55−2.65(m,1H),2.68−2.74(m,1H),3.62(d,J=4.2Hz,2H),4.18−4.25(m,1H),4.72(d,J=4.5Hz,1H),7.35(d,J=8.6Hz,1H),7.46−7.49(m,1H),7.70(dd,J=6.9及び2.2Hz,1H),7.87−7.91(m,2H),8.00(d,J=1.6Hz,1H).Anal.Calcd.for C19H17N3O3:C,68.05;H,5.11;N,12.53;Found:C,67.80;H,5.11;N,12.49.
【0171】
化合物4m:10−[4−(2−ヒドロキシ−エチル)−ピペリジン−1−イルメチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0172】
【化18】
【0173】
一般的手順B及びCに従って、化合物2及び2−ピペリジン−4−イル−エタノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4mを得た。
1H−NMR(DMSO−d6,300MHz):1.12−1.17(m,2H),1.34−1.38(m,3H),1.61(d,J=12Hz,1H),1.92(t,J=11Hz,2H),2.80(d,J=10.6Hz,2H),3.42−3.48(m,4H),4.34(t,J=5.2,1H),7.33(d,J=8.8Hz,1H),7.43−7.46(m,1H),7.68(dd,J=6.9及び2.5Hz,1H),7.86−7.98(m,3H).Anal.Calcd.for C22H23N3O3:C,70.01;H,6.14;N,11.13;Found:C,69.82;H,6.12;N,11.08.
【0174】
化合物4n:10−(1,4−ジオキサ−8−アザ−スピロ[4.5]デカ−8−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0175】
【化19】
【0176】
一般的手順B及びCに従って、化合物2及び1,4−ジオキサ−8−アザ−スピロ[4.5]デカンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4nを得た。
1H−NMR(DMSO−d6,300MHz):1.62(t,J=5.0,4H),2.40−2.50(m,4H),3.55(s,2H),3.85(s,4H),7.34(d,J=9.4Hz,1H),7.47(d,J=7.5Hz,1H),7.67−7.70(m,1H),7.86−7.92(m,2H),7.98(s,1H),12.62(s,1H).Anal.Calcd.for C22H21N3O4:C,67.41;H,5.43;N,10.98;Found:C,67.15;H,5.30;N,11.03.
【0177】
化合物4o:10−(3−ヒドロキシ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0178】
【化20】
【0179】
一般的手順B及びCに従って、化合物2及びピペリジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4oを得た。
1H−NMR(DMSO−d6,300MHz):1.00−1.12(m,1H),1.30−1.50(m,1H),1.52−1.95(m,4H),2.66−2.83(m,2H),3.45−3.60(m,3H),4.60(d,J=5.0Hz,1H),7.34−7.48(m,2H),7.68−7.70(dd,J=6.8及び2.2Hz,1H),7.87−8.00(m,3H).Anal.Calcd.for C20H19N3O3:C,68.75;H,5.48;N,12.03;Found:C,68.85;H,5.48;N,12.10.
【0180】
化合物4p:10−(3−ヒドロキシ−アゼチジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0181】
【化21】
【0182】
一般的手順B及びCに従って、化合物2及びアゼチジン−3−オールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4pを得た。
1H−NMR(DMSO−d6,300MHz):2.79(t,J=6.9Hz,2H),3.51(t,J=6.3Hz,2H),3.61(s,2H),4.23(dd,J=12.9及び6.3Hz,1H),5.33(d,J=6.5Hz,1H),7.33(d,J=8.4Hz,1H),7.71−7.44(m,1H),7.68(dd,J=6.8,2.6,1H),7.86−7.96(m,3H),12.62(bs,1H).Anal.Calcd.for C18H15N3O3−(0.5H2O):C,65.45;H,4.88;N,12.72;Found:C,65.06;H,4.60;N,13.03.
【0183】
化合物4q:10−[(2−モルフォリン−4−イル−エチルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0184】
【化22】
【0185】
一般的手順B及びCに従って、化合物2及び2−モルフォリン−4−イル−エチルアミンから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4qを得た。
1H−NMR(DMSO−d6,300MHz):2.16(bs,1H),2.34(bs,4H),2.40(t,J=6.6Hz,2H),2.60(t,J=6.1Hz,2H),3.56(t,J=4.6Hz,4H),3.76(s,2H),7.33(d,J=8.1Hz,1H),7.48(dd,J=8.3及び1.8Hz,1H),7.68(dd,J=6.6及び2.3,1H),7.88−7.93(m,2H),8.01(d,J=1.5,1H),12.63(bs,1H).Anal.Calcd.for C21H22N4O3−(0.75H2O):C,64.35;H,6.04;N,14.29;Found:C,64.35;H,5.91;N,14.26.
【0186】
化合物4r:10−[(4−ヒドロキシ−シクロヘキシルアミノ)−メチル]−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0187】
【化23】
【0188】
一般的手順B及びCに従って、化合物2及びtrans−4−アミノ−シクロヘキサノールから調製した。エタノールで結晶化させることにより生成物を精製し、白色固体4rを得た。
1H−NMR(DMSO−d6,300MHz):1.03−1.16(m,4H),1.75−1.90(m,4H),2.30−2.40(m,1H),3.75(s,2H),4.48(d,J=3.4,1H),7.31(d,J=8.1Hz,1H),7.48(dd,J=8.5及び2.1Hz,1H),7.67(dd,J=6.8及び2.4Hz,1H,)7.85−7.92(m,2H),8.02(d,J=1.9Hz,1H).Anal.Calcd.for C21H21N3O3−(0.5H2O)・(0.05N2H4):C,67.44;H,5.98;N,11.61;Found:C,67.56;H,5.74;N,11.68.
【0189】
化合物4s:10−(4−オキソ−ピペリジン−1−イルメチル)−2H−7−オキサ−1,2−ジアザ−ベンゾ[デ]アントラセン−3−オン
【0190】
【化24】
【0191】
化合物4n(100mg,0.24mmol)を酢酸(3ml)中で撹拌し、そこへ濃塩酸(0.6ml,過剰量)を室温において添加した。反応物を90℃で1時間加熱し、次に室温まで冷却した。生成物を1N NaOHでpH11〜12に塩基性化した後、酢酸エチルで抽出し単離した。有機分は硫酸マグネシウムで乾燥させ、真空において濃縮して白色固体4sを得た(60mg,71%)。
1H−NMR(DMSO−d6,300MHz):2.37(t,J=2.4Hz,4H),2.73(t,J=2.7Hz,4H),3.69(s,2H),7.38(d,J=8.5Hz,1H),7.53(dd,J=9.0及び6.1Hz),7.70(dd,J=7.1及び4.7Hz,1H),7.86−7.94(m,2H),8.06(d,J=2.4Hz,1H).
【0192】
本発明の化合物の製造の他の様式、変形又は順序は、当業者に容易に明らかとなろう。
【0193】
本発明の化合物は、遊離塩基の形態で、可能であれば塩基塩の形態で、及び、付加塩の形態で、並びに、遊離酸の形態で有用であろう。これら全ての形態は本発明の範囲内である。実用上は、塩形態の使用は塩基形態の使用と同等である。本発明の範囲内の医薬品に許容される塩は、塩酸及び硫酸などの無機酸;並びに、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸などの有機酸から誘導されるもの(例えば、塩酸塩、スルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩がそれぞれ挙げられる)、又は、適切な有機及び無機塩基などの塩基から誘導されるものが挙げられる。本発明の化合物との医薬品に許容される塩基付加塩の例には、非毒性でありかつそのような塩を形成するのに十分に強い有機塩基が挙げられる。これらの有機塩基及びその使用は、当業者に容易に理解される。単に例として挙げるものであるが、このような有機塩基には、メチルアミン、ジエチルアミン及びトリエチルアミンなどのモノ、ジ及びトリアルキルアミン類;モノ、ジ及びトリエタノールアミンなどのモノ、ジ又はトリヒドロキシアルキルアミン類;アルギニン及びリジンなどのアミノ酸類;グアニジン;N−メチルグルコサミン;N−メチルグルカミン;L−グルタミン;N−メチルピペラジン;モルホリン;エチレンジアミン;N−ベンジルフェネチルアミン;トリス(ヒドロキシメチル)アミノエタン;などが挙げられる。
【0194】
塩基性化合物の酸付加塩は以下のように製造することができる:本発明の化合物の遊離塩基を、水溶液若しくはアルコール水溶液又は適切な酸若しくは塩基を含む他の適切な溶液に溶解し、溶液を蒸発させることにより塩を単離するか、あるいは、反応物が有機溶液中に存在するように、本発明の化合物の遊離塩基を酸と反応させることによって、また、酸基を有する本発明の化合物を塩基と反応させることによって、塩を直接分離するか又は溶液の濃縮により得ることができる。
【0195】
本発明の化合物は、薬理学的活性を示すので、医薬として有用である。さらに、上記化合物は、中枢神経系及び心血管系活性を示す。
【0196】
(PARPアッセイ)
1.IC50
PARP阻害剤化合物のIC50を決定する簡便な方法は、Trevigan(メリーランド州ゲーサーズバーグ)の精製組み換えヒトPARPを用いるPARPアッセイであり、以下のとおりである:PARP酵素アッセイを、氷上で、100mMのTris−HCl(pH8.0)、1mMのMgCl2、28mMのKCl、28mMのNaCl、0.1mg/mlのDNaseI活性化ニシン精子DNA(Sigma,ミズーリ州)、3.0マイクロモル[3H]ニコチンアミドアデニンジヌクレオチド(470mci/ミリモル)、7マイクログラム/mlのPARP酵素及び試験する種々の濃度の化合物からなる100マイクロリットルの容積で開始する。反応を、25℃で混合物をインキュベートすることにより開始する。15分間インキュベートした後、氷冷20%(w/v)トリクロロ酢酸500マイクロリットルを添加することにより反応を終結させる。生成した沈殿をグラスファイバーフィルター(Packard Unifilter−GF/B)上に移し、エタノールで3回洗浄する。フィルターを乾燥した後、シンチレーション計数により放射線活性を測定する。
【0197】
本発明の化合物は、この阻害アッセイにおいてIC50が数nM〜20μMの範囲という強力な酵素活性を有することが見出された。
【0198】
上記PARPアッセイを用い、以下の化合物についておおよそのIC50値を得た。
【0199】
【表1】
【0200】
【表2】
【0201】
2.mRNA老化細胞における変化した遺伝子発現の測定
遺伝子発現変化はヒト繊維芽細胞BJ細胞を用いて測定することができ、上記細胞を細胞集団倍加数(Population Doubling)(PDL)94で通常の増殖培地上に播種し、次いで、低血漿培地に変えて、Linskensら,Nucleic acids Res.,23,3244−3251(1995)に記載の生理学的条件を反映させる。0.5%仔ウシ血清を添加したDMEM/199培地を用いる。細胞を13日間にわたって毎日処理する。PARP阻害剤の投与に使用する溶液を用いて、又は、これを用いずに、コントロール細胞を処理する。未処理の老化コントロール細胞及び若いコントロール細胞を、比較のために試験する。処理した細胞及びコントロール細胞からPCT公開第96/13610号に記載の技術に従ってRNAを調製し、ノーザンブロッティングを実施する。老化関連遺伝子に特異的なプローブを分析し、処理し、コントロール細胞を比較する。結果の分析において、遺伝子発現の最低レベルを任意に1に設定して、比較用のための基準とする。皮膚における加齢性変化に特に関連する3つの遺伝子は、コラーゲン、コラゲナーゼ及びエラスチンである。WestArch.Derm.130,87−95(1994)。PARP阻害剤で処理した細胞のエラスチン発現は、コントロール細胞と比較して顕著に増加するものと予測される。エラスチン発現は、若い細胞において老化細胞よりも顕著に高いはずであるため、PARP阻害剤での処理によって老化細胞でのエラスチン発現レベルは非常に若い細胞における同レベルと同等まで変化するはずである。同様に、PARP阻害剤での処理により、コラゲナーゼ及びコラーゲン発現において有益な効果がみられるはずである。
【0202】
3.老化細胞におけるタンパク質の変化した遺伝子発現の測定
遺伝子発現変化は、PDLが95〜100のBJ細胞を約105個用い、上記細胞を15cmディッシュに平板培養することによって測定できる。増殖培地は、10%仔ウシ血清を添加したDMEM/199である。細胞をPARP阻害剤(100μg/1mLの培地)で毎日24時間処理する。WO99/11645を参照されたい。細胞をリン酸バッファー(PBS)で洗浄し、次いで、4%パラホルムアルデヒドで5分間透過可能にした後、PBSで洗浄し、100%冷メタノールで10分間処理する。メタノールを除去し、細胞をPBSで洗浄した後、10%血清で処理して、非特異的な抗体結合をブロックする。約1mLの適切な市販抗体溶液(1:500希釈、Vector)を細胞に添加し、混合物を1時間インキュベートする。細胞をPBSで3回洗浄する。二次抗体であるビオチン化ヤギ抗マウスIgG(1mL)を、アルカリホスファターゼ結合ストレプトアビジンを含む溶液1mL及び1mLのNBT剤(Vector)と共に添加する。細胞を洗浄し、遺伝子発現の変化を比色法により観察する。PARP阻害剤で処理した老化細胞において、老化に特異的な4つの遺伝子−コラーゲンI、コラーゲンIII、コラゲナーゼ及びインターフェロンγを観察した結果、インターフェロンγ発現の低下が示され、他の3つの遺伝子の発現レベルには変化がみられなかったが、このことは、PARP阻害剤が、老化に特異的な遺伝子の発現を変化させることができるということを示す。
【0203】
4.細胞の増殖能力及び寿命の拡張又は増加
細胞の増殖能力及び寿命を拡張するための本方法の有効性を示すため、ヒト繊維芽細胞細胞株(細胞集団倍加数(PDL)が23であるWl38、又は、PDLが71であるBJ細胞のいずれか)を解凍し、T75フラスコに平板培養し、通常の培地(10%仔ウシ血清を添加したDMEM/M199)中で約1週間増殖させることによって(このとき細胞はコンフルエントである)、培養物を細分用に準備する。細分に際して、培地を吸引し、細胞をリン酸緩衝生理食塩水(PBS)で洗浄し、次いでトリプシン化する。Coulterカウンターを用いて細胞を計数し、10%仔ウシ血清及び種々の量(0.10μM及びImM:DMEM/M199培地中の100倍(IOOX)原液より)のPARP阻害剤を添加したDMEM/199培地を入れた6ウェル組織培養プレート中で、1cm2当たり細胞105個の密度で平板培養する。この方法を、細胞が細分を停止するように見えるまで7日毎に繰り返す。未処理の(コントロール)細胞は、培地中約40日後に老化に達し、細分を停止する。
【0204】
(テモゾロマイドの投与)
実施例1:化合物4i及びテモゾロマイドの経口投与は、CNS部位に悪性腫瘍を有するマウスの生存を増大させる。
【0205】
頭蓋内移植の手順は、TentoriLら, “Effects of single or split exposure of leukemic cells to temozolomide, combined with poly(ADP−ribose) polymerase inhibitors on cell growth, chromosomal aberrations and base excision repair components,” Cancer Chemother Pharmacol, 47, 361−9 (2001)に記載のように実施する。マウス黒色腫B16細胞(104個)を、雄B6D2F1(C57BL/6×DBA/2)マウスに頭蓋内(ic)注射した。治療のタイミングを決定するため、脳での腫瘍増殖の組織学的評価を、腫瘍の曝露後1〜5日に行った。
【0206】
カリウムを含まない70mM PBSに化合物4iを溶解し、テモゾロマイド(TMZ)投与の1時間前に経口投与した。TMZをジメチルスルホキシド(40mg/ml)中に溶解し、生理食塩水(5mg/ml)で希釈し、100mg/Kgの用量で5日にわたって腹腔内投与した。化合物4iを用いて経口栄養補給により、10mg/kg/日又は40mg/kg/日の用量で、1日1回5日間にわたってマウスを処置した。生存中央値(median survival times)(MST)を決定し、寿命(ILS)の増加の割合を、([処置したマウスのMST(日)/コントロールマウスのMST(日)]−l)×100として算出した。処置の効力を、処置群とコントロール群との間の生存曲線の比較により評価した。
【0207】
B16黒色腫を有するマウスでは、結果は、化合物4i及びTMZの組み合わせで処置した群の平均生存時間が、TMZを単一の剤として服用した動物で観察された平均生存時間よりも顕著に高かったことを示した(図I及び表II)。
【0208】
表II.脳内にB16黒色腫を有するマウスの生存率
【0209】
【表3】
【0210】
実施例2:化合物4iの投与による、皮下黒色腫ガンモデルにおけるテモゾロマイド効果の増強
【0211】
TMZ±化合物4iによる処置の効力を、マウスにおいて皮下(s.c.)増殖黒色腫についても評価した。この目的のため、B16細胞(2.5×105個)を動物の脇腹に皮下接種した。腫瘍をノギスで測定し、容積を、式:[(幅)2×長さ]/2に従って算出した。曝露の6日後、腫瘍結節の容積が100〜150mm3に達したときに、薬剤処置を開始した。化合物4i(40mg/kg経口)を、テモゾロマイド(100mg/kg腹腔内)の20分前に、1日1回5日間にわたって投与した。黒色腫増殖を、腫瘍結節を3日毎に3週間にわたって測定することによって観察した。
【0212】
化合物4i及びTMZによる併用治療によって、B16黒色腫の増殖が顕著に減少した(P<0.01、9日〜23日、TMZ単独に対して)(図II)。
【0213】
実施例3:ラットにおいてシスプラチンで誘発される神経障害における、交感神経透過速度(SNCV)
【0214】
本発明の化合物の神経保護効果を、ラットにおいてシスプラチンで誘発される神経障害モデルで示した。神経透過速度の変化は、化学毒性物質が誘発する末梢神経障害の高感度な測定であることがよく実証されている。化合物4iは、シスプラチンでの慢性的な治療により誘発される神経透過速度の欠損を低減することが示された。
【0215】
この実験において、雌Wistar Hannoverラットに、神経障害を誘発する用量のシスプラチン(2mg/kg腹腔内;週2回を4週間)を、化合物4i(毎日40mg/kg、経口)と共に及び化合物4iなしで投与した。ラットにおいて、ベースライン(シスプラチン投与前)及び処置後の尾部神経での交感神経透過速度(SNCV)の変化を観察した。さらに、後根神経節及び坐骨神経標本の後根神経節神経上における形態計測分析(体細胞、核及び核小体の大きさ)を、病理解剖学的に評価した。
【0216】
処置期間の最初と最後に、各動物の尾部において、Cavalettiら, “Protective Effects of glutathione on cisplatin neurotoxicity in rats,” Int. J. Radiation Oncology, 29, 771−776 (1994) and Trediciら, “Low−Dose Glutathione administration, in the prevention of cisplatin−induced peripheral neuropathy in rats,” Neuro toxicology, 15, 701−704 (1994)に既に記載されているように、SNCV測定を測定した。尾神経における逆方向性のSNCVを、記録用環状電極を尾の遠位に配置することにより評価し、ここで、刺激環状電極は、記録点から5cm及び10cm離して配置した。神経刺激後の2箇所で記録された電位の反応時間を決定し(ピークからピーク)、これに従って神経透過速度を計算した。
【0217】
犠牲にした動物から得た各群に由来するラットの左側L5後根神経節(DRG)を、既に報告されたプロトコル[Cavalettiら;Trediciら]に従って処理し、樹脂に埋め込み、光学顕微鏡及び電子顕微鏡での観察並びに形態計測に用いた。厚み1μmの半薄切片で、画像分析ソフトウェア(Image J,NIH)を用いて、DRGニューロンの細胞体、核及び核小体の断面積の形態測定を行った。
【0218】
神経透過速度の相違及び実験中に後根神経節神経で得られた形態計測データの相違を、分散分析(ANOVA)及びTukey−Kramer後検定(有意性レベルをp<0.05に設定)を用いて、統計学的に評価した。
【0219】
化合物4iの共投与によって、慢性的なシスプラチン治療による尾神経透過速度の損傷における統計学的に顕著な減少が誘発されるということが見出された(表3及び4)。
【0220】
表3:実験終了後におけるSNCV(m/秒)
【0221】
【表4】
【0222】
CDDP=シスプラチン
【0223】
表4:統計学的分析(一方向ANOVA)
【0224】
【表5】
【0225】
DRG形態計測
【0226】
DRGニューロンにおける形態計測の研究により、化合物4iを有するDRGニューロンの細胞体の大きさのみについて顕著な効果が明らかとなった。表5は形態計測の結果を示し、統計学的データを表6(細胞体)及び表7(核)に列挙する。
【0227】
表5:DRGの形態計測(μm2)
【0228】
【表6】
【0229】
Nu=核、Nucl=核小体
【0230】
表6:細胞体
【0231】
【表7】
【0232】
表7:核小体
【0233】
【表8】
【0234】
上記では本発明について説明しているが、多くの方法によって改変可能な場合があるということは明らかであろう。このような改変は、本発明の趣旨及び範囲内であるとみなされ、このような改変は全て、添付の特許請求の範囲の範囲内に包含されるものである。
【0235】
(参照による援用)
本明細書中に記載する全ての刊行物、特許及び付与前特許出願公開は、任意の及び全ての目的のために、あたかもそれぞれの個別の刊行物及び特許出願が参照として援用されていることが示されているかのように、特別にかつ個別に本明細書中に参照として援用される。不一致がある場合、本開示が優先する。
【特許請求の範囲】
【請求項1】
以下から選択される化合物:
【化1】
【化2】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物。
【請求項2】
以下である:
【化3】
:ことを特徴とする請求項1に記載の化合物。
【請求項3】
動物において、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷、ニューロンが媒介する組織の損傷又は疾患、虚血損傷及び灌流損傷及び神経学的障害、心血管障害、心臓バイパス手術から生じる神経系組織の損傷、冠状動脈バイパス手術後の神経系の欠損による鬱及び認知低下の治療、関節炎、糖尿病、毛細血管拡張性運動失調症、悪液質、複製老化に関与する骨格筋の変性疾患、炎症性腸障害、炎症、痛風、慢性疼痛、急性疼痛、神経障害性疼痛、神経性傷害、末梢神経損傷、腎不全、網膜虚血、敗血性ショック、出血性ショック、多発性硬化症、細胞の寿命又は増殖能力に関連する疾患又は障害、並びに、細胞の老化により誘発される又は悪化する疾患又は症状からなる群より選択される疾患又は症状を治療する方法であって、
前記動物に、以下からなる群より選択される化合物:
【化4】
【化5】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物の有効量を投与することを含む
ことを特徴とする方法。
【請求項4】
神経学的障害が、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳卒中及び脱髄性疾患からなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項5】
心血管障害が、心血管組織損傷、冠状動脈疾患、心筋梗塞、狭心症、心原性ショック、冠状動脈バイパス手術、心停止及び心肺蘇生からなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項6】
細胞の老化により誘発される又は悪化する疾患又は疾患症状が、皮膚加齢、アルツハイマー病、アテローム性動脈硬化、骨関節炎、骨粗鬆症、筋ジストロフィー、加齢性筋退化、免疫老化及びエイズからなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項7】
前記大脳虚血又は灌流損傷が、心停止及び心肺蘇生後の大脳損傷である
ことを特徴とする請求項3に記載の方法。
【請求項8】
敗血性ショックが内毒素性ショックである
ことを特徴とする請求項3に記載の方法。
【請求項9】
腸障害が大腸炎である
ことを特徴とする請求項3に記載の方法。
【請求項10】
前記腸障害がクローン病である
ことを特徴とする請求項3に記載の方法。
【請求項11】
放射線療法を必要とする哺乳類において、腫瘍細胞を放射線増感する方法であって、
前記哺乳類に、以下から選択される化合物:
【化6】
【化7】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物を、前記腫瘍細胞を前記放射線療法の効果に増感させるのに十分な量で投与することを含む
ことを特徴とする方法。
【請求項12】
化合物が以下である:
【化8】
:ことを特徴とする請求項11に記載の方法。
【請求項13】
腫瘍細胞が、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞ガン)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍からなる群より選択される
ことを特徴とする請求項11に記載の方法。
【請求項14】
前記哺乳類がヒトである
ことを特徴とする請求項11に記載の方法。
【請求項15】
化学療法を必要とする哺乳類に、以下から選択される化合物:
【化9】
【化10】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物を、前記腫瘍細胞を少なくとも1つの化学療法薬剤の効果に増感させるのに十分な量で投与することにより、前記哺乳類において腫瘍細胞を化学増感することを含む
ことを特徴とする治療方法。
【請求項16】
前記化学増感化合物が以下である:
【化11】
:ことを特徴とする請求項15に記載の方法。
【請求項17】
前記哺乳類がヒトである
ことを特徴とする請求項15に記載の方法。
【請求項18】
第1に、前記化合物の投与後に時間をおいて、有効量の化学増感を提供すること、及び、第2に、前記哺乳類に、医薬上有効用量の前記化学療法薬剤を投与することをさらに含む
ことを特徴とする請求項15に記載の方法。
【請求項19】
前記化合物が、前記化合物及び医薬品に許容される担体、希釈剤又は賦形剤を含む医薬組成物で投与される
ことを特徴とする請求項15に記載の方法。
【請求項20】
治療有効用量の前記化学療法薬剤を前記哺乳類に投与することをさらに含み、前記化学増感化合物と前記化学療法薬剤とが実質的に同時に投与される
ことを特徴とする請求項15に記載の方法。
【請求項21】
前記化学療法薬剤が、テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α、β、γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、これらの治療上有効なアナログ及び誘導体並びにこれらの混合物からなる群より選択される
ことを特徴とする請求項15に記載の方法。
【請求項22】
前記化学療法薬剤がテモゾロマイドである
ことを特徴とする請求項21に記載の方法。
【請求項23】
以下からなる群より選択される少なくとも1つの化合物:
【化12】
【化13】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物の治療上有効量、並びに、医薬品に許容される担体、希釈剤又は賦形剤を含む
ことを特徴とする医薬組成物。
【請求項24】
化合物が以下である:
【化14】
:ことを特徴とする
請求項23記載の医薬組成物。
【請求項25】
化学療法上有効量の少なくとも1つの化学療法薬剤をさらに含み、前記化学療法薬剤が、タキソイド、テモゾロマイド、ダクチノマイシン、ダノルビシン、ドキソルビシン、4’−デオキシドキソルビシン、ブレオマイシン、ピルカマイシン、マイトマイシン、ネオマイシン及びゲンタマイシン、エトポシド、4−OHシクロフォスファミド、白金配位錯体並びにこれらの混合物から選択される
ことを特徴とする請求項23記載の医薬組成物。
【請求項26】
前記化学療法薬剤がテモゾロマイドである
ことを特徴とする請求項25記載の医薬組成物。
【請求項1】
以下から選択される化合物:
【化1】
【化2】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物。
【請求項2】
以下である:
【化3】
:ことを特徴とする請求項1に記載の化合物。
【請求項3】
動物において、壊死又はアポトーシスに起因する細胞の損傷又は死から生じる組織損傷、ニューロンが媒介する組織の損傷又は疾患、虚血損傷及び灌流損傷及び神経学的障害、心血管障害、心臓バイパス手術から生じる神経系組織の損傷、冠状動脈バイパス手術後の神経系の欠損による鬱及び認知低下の治療、関節炎、糖尿病、毛細血管拡張性運動失調症、悪液質、複製老化に関与する骨格筋の変性疾患、炎症性腸障害、炎症、痛風、慢性疼痛、急性疼痛、神経障害性疼痛、神経性傷害、末梢神経損傷、腎不全、網膜虚血、敗血性ショック、出血性ショック、多発性硬化症、細胞の寿命又は増殖能力に関連する疾患又は障害、並びに、細胞の老化により誘発される又は悪化する疾患又は症状からなる群より選択される疾患又は症状を治療する方法であって、
前記動物に、以下からなる群より選択される化合物:
【化4】
【化5】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物の有効量を投与することを含む
ことを特徴とする方法。
【請求項4】
神経学的障害が、肉体的損傷又は疾患状態により引き起こされる末梢神経障害、外傷による脳損傷、脊椎への肉体的損傷、脳卒中及び脱髄性疾患からなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項5】
心血管障害が、心血管組織損傷、冠状動脈疾患、心筋梗塞、狭心症、心原性ショック、冠状動脈バイパス手術、心停止及び心肺蘇生からなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項6】
細胞の老化により誘発される又は悪化する疾患又は疾患症状が、皮膚加齢、アルツハイマー病、アテローム性動脈硬化、骨関節炎、骨粗鬆症、筋ジストロフィー、加齢性筋退化、免疫老化及びエイズからなる群より選択される
ことを特徴とする請求項3に記載の方法。
【請求項7】
前記大脳虚血又は灌流損傷が、心停止及び心肺蘇生後の大脳損傷である
ことを特徴とする請求項3に記載の方法。
【請求項8】
敗血性ショックが内毒素性ショックである
ことを特徴とする請求項3に記載の方法。
【請求項9】
腸障害が大腸炎である
ことを特徴とする請求項3に記載の方法。
【請求項10】
前記腸障害がクローン病である
ことを特徴とする請求項3に記載の方法。
【請求項11】
放射線療法を必要とする哺乳類において、腫瘍細胞を放射線増感する方法であって、
前記哺乳類に、以下から選択される化合物:
【化6】
【化7】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物を、前記腫瘍細胞を前記放射線療法の効果に増感させるのに十分な量で投与することを含む
ことを特徴とする方法。
【請求項12】
化合物が以下である:
【化8】
:ことを特徴とする請求項11に記載の方法。
【請求項13】
腫瘍細胞が、ACTH産生腫瘍、急性リンパ球性白血病、急性非リンパ球性白血病、副腎皮質ガン、膀胱ガン、脳ガン、乳ガン、子宮頚部ガン、慢性リンパ球性白血病、慢性骨髄球性白血病、直腸結腸ガン、皮膚T−細胞リンパ腫、子宮内膜ガン、食道ガン、ユーイング肉腫、胆嚢ガン、毛様細胞白血病、頭頚部ガン、ホジキンリンパ腫、カポジ肉腫、腎臓ガン、肝臓ガン、肺ガン(小細胞及び/又は非小細胞ガン)、悪性腹膜滲出、悪性胸膜滲出、黒色腫、中皮腫、多発性骨髄腫、神経芽細胞腫、非ホジキンリンパ腫、骨肉腫、卵巣ガン、卵巣(胚細胞)ガン、前立腺ガン、膵臓ガン、陰茎ガン、網膜芽細胞腫、皮膚ガン、軟組織肉腫、扁平上皮細胞ガン腫、胃ガン、精巣ガン、甲状腺ガン、栄養膜新生物、子宮ガン、膣ガン、外陰部ガン及びウィルムス腫瘍からなる群より選択される
ことを特徴とする請求項11に記載の方法。
【請求項14】
前記哺乳類がヒトである
ことを特徴とする請求項11に記載の方法。
【請求項15】
化学療法を必要とする哺乳類に、以下から選択される化合物:
【化9】
【化10】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物を、前記腫瘍細胞を少なくとも1つの化学療法薬剤の効果に増感させるのに十分な量で投与することにより、前記哺乳類において腫瘍細胞を化学増感することを含む
ことを特徴とする治療方法。
【請求項16】
前記化学増感化合物が以下である:
【化11】
:ことを特徴とする請求項15に記載の方法。
【請求項17】
前記哺乳類がヒトである
ことを特徴とする請求項15に記載の方法。
【請求項18】
第1に、前記化合物の投与後に時間をおいて、有効量の化学増感を提供すること、及び、第2に、前記哺乳類に、医薬上有効用量の前記化学療法薬剤を投与することをさらに含む
ことを特徴とする請求項15に記載の方法。
【請求項19】
前記化合物が、前記化合物及び医薬品に許容される担体、希釈剤又は賦形剤を含む医薬組成物で投与される
ことを特徴とする請求項15に記載の方法。
【請求項20】
治療有効用量の前記化学療法薬剤を前記哺乳類に投与することをさらに含み、前記化学増感化合物と前記化学療法薬剤とが実質的に同時に投与される
ことを特徴とする請求項15に記載の方法。
【請求項21】
前記化学療法薬剤が、テモゾロマイド、アドリアマイシン、カンプトテシン、カルボプラチン、シスプラチン、ダウノルビシン、ドセタキセル、ドキソルビシン、インターフェロン(α、β、γ)、インターロイキン2、イリノテカン、パクリタキセル、トポテカン、これらの治療上有効なアナログ及び誘導体並びにこれらの混合物からなる群より選択される
ことを特徴とする請求項15に記載の方法。
【請求項22】
前記化学療法薬剤がテモゾロマイドである
ことを特徴とする請求項21に記載の方法。
【請求項23】
以下からなる群より選択される少なくとも1つの化合物:
【化12】
【化13】
:並びに、その医薬品に許容される塩、水和物、エステル、溶媒和物及びこれらの混合物の治療上有効量、並びに、医薬品に許容される担体、希釈剤又は賦形剤を含む
ことを特徴とする医薬組成物。
【請求項24】
化合物が以下である:
【化14】
:ことを特徴とする
請求項23記載の医薬組成物。
【請求項25】
化学療法上有効量の少なくとも1つの化学療法薬剤をさらに含み、前記化学療法薬剤が、タキソイド、テモゾロマイド、ダクチノマイシン、ダノルビシン、ドキソルビシン、4’−デオキシドキソルビシン、ブレオマイシン、ピルカマイシン、マイトマイシン、ネオマイシン及びゲンタマイシン、エトポシド、4−OHシクロフォスファミド、白金配位錯体並びにこれらの混合物から選択される
ことを特徴とする請求項23記載の医薬組成物。
【請求項26】
前記化学療法薬剤がテモゾロマイドである
ことを特徴とする請求項25記載の医薬組成物。
【図1】

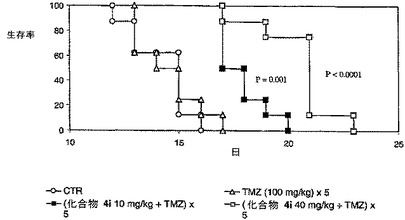
【図2】
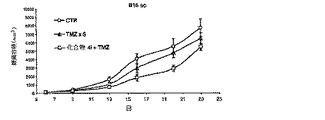
【図2】
【公開番号】特開2012−233015(P2012−233015A)
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2012−195313(P2012−195313)
【出願日】平成24年9月5日(2012.9.5)
【分割の表示】特願2007−552227(P2007−552227)の分割
【原出願日】平成18年1月19日(2006.1.19)
【出願人】(509316350)エーザイ インク. (2)
【Fターム(参考)】
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成24年9月5日(2012.9.5)
【分割の表示】特願2007−552227(P2007−552227)の分割
【原出願日】平成18年1月19日(2006.1.19)
【出願人】(509316350)エーザイ インク. (2)
【Fターム(参考)】
[ Back to top ]